Embed Size (px)
Citation preview

IMAGERIE DES HISTIOCYTOSES
LANGERHANSIENNES DE L’ADULTE
C. Lacombe¹, C. Viala², M. Lewin¹, M Svrcek³, P Cervera³, L. Monnier-Cholley¹, A. Belkacem¹,
C. Pradel¹, L. Arrivé¹, J. Cabane² et JM. Tubiana¹
¹ Service de radiologie, hôpital Saint-Antoine, Paris² Service de médecine interne, hôpital Saint-Antoine, Paris³ Service d’anatomopathologie, hôpital Saint-Antoine, Paris

INTRODUCTION
• Les histiocytoses constituent un groupe de maladies chroniques hétérogènes caractérisées par une atteinte multiviscérale et pluritissulaire. Elles sont divisées en 3 groupes:
– Histiocytoses langerhansiennes (objet du poster)
– Histiocytoses non langerhansiennes : les plus connues sont la maladie de Whipple, d’origine infectieuse, les maladies de Gaucher et de Niemann-Pick, héréditaires, et la maladied’Erdheim-Chester, à expression sporadique, d’étiologie inconnue
– Histiocytoses malignes, très rares. Elles se rapprochent plutôt de la famille des hémopathies malignes.

DEFINITIONS• L’histiocytose à cellules de Langerhans (HL), également appelée
histiocytose X ou granulomatose à cellules de Langerhans regroupe plusieurs syndromes dont la caractéristique commune est histologique : infiltration tissulaire par des cellules de Langerhans organisées en granulomes.
• On distingue cliniquement 3 formes– Formes unitissulaires, localisées, de relativement bon pronostic, intéressant surtout l’adulte– Formes pluritissulaires, multifocales (syndrome de Hand-Schüller-Christian) concernant le grand enfant ou l’adolescent, mais également l’adulte, de novo, ou dans le cadre d’une forme pédiatrique passée à la chronicité.– Formes disséminées (syndrome de Letterer-Siwe) qui sont plutôt des formes pédiatriques (nourrisson), généralement gravissimes.
• La sémiologie radiologique pour un même organe est la même, que la forme soit uni ou pluritissulaire.

PATHOGENIE
• L’origine de l’HL est inconnue, sans agent étiologique identifié. Néanmoins, il existe un lien étroit entre le tabagisme et la forme pulmonaire isolée d’HL, suggérant un rôle pathogène du tabac.
• Plusieurs équipes considèrent l’HL comme une maladie clonale, bénigne dans la plupart des cas, mais l’hypothèse immunitaire n’est pas exclue.
• Certains facteurs impliqués dans la pathogénie de l’HL ont étéindividualisés : – Les cellules de Langerhans secrètent du GM-CSF, facteur essentiel
pour la prolifération cellulaire. – L’interleukine 17, dont il existe des récepteurs partout dans
l’organisme, apparaît également impliquée dans la maladie, expliquant ainsi le caractère polymorphe des manifestations.
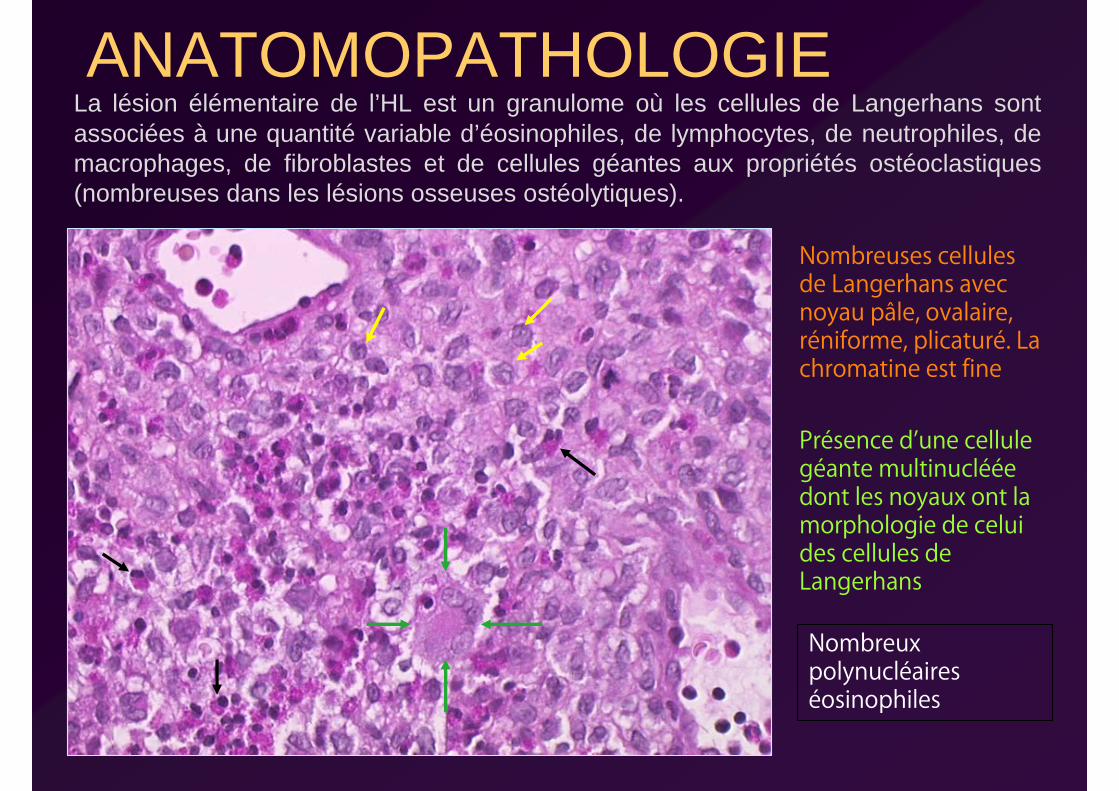
ANATOMOPATHOLOGIELa lésion élémentaire de l’HL est un granulome où les cellules de Langerhans sont associées à une quantité variable d’éosinophiles, de lymphocytes, de neutrophiles, de macrophages, de fibroblastes et de cellules géantes aux propriétés ostéoclastiques (nombreuses dans les lésions osseuses ostéolytiques).
Nombreuses cellules de Langerhans avec noyau pâle, ovalaire, réniforme, plicaturé. La chromatine est fine
Présence d’une cellule géante multinucléée dont les noyaux ont la morphologie de celui des cellules de Langerhans
Nombreux polynucléaires éosinophiles
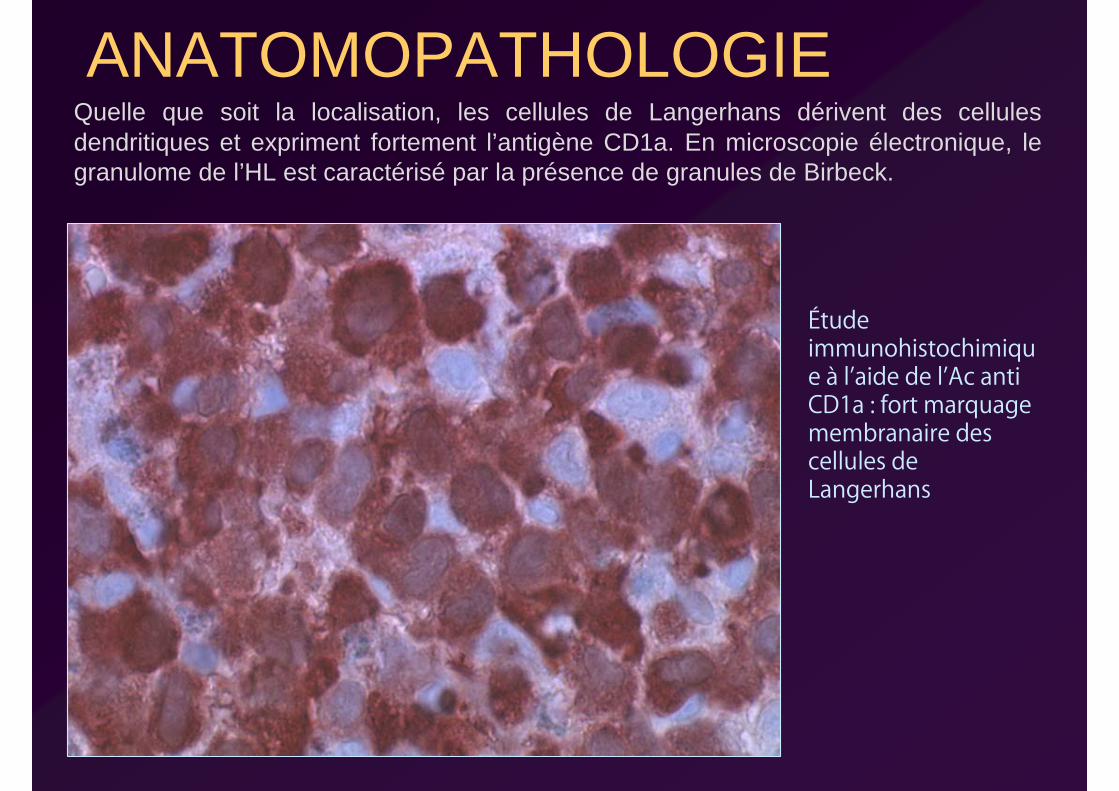
ANATOMOPATHOLOGIEQuelle que soit la localisation, les cellules de Langerhans dérivent des cellules dendritiques et expriment fortement l’antigène CD1a. En microscopie électronique, le granulome de l’HL est caractérisé par la présence de granules de Birbeck.
Étude immunohistochimique à l’aide de l’Ac anti CD1a : fort marquage membranaire des cellules de Langerhans

CLINIQUE• L’histiocytose langerhansienne est une maladie touchant essentiellement l’enfant et
l’adulte jeune, avec, chez l’adulte, une survenue de novo, ou le passage à la chronicité d’une forme pédiatrique.
• Âge médian à la survenue du 1er symptôme : 27 ans.
• Sex-ratio de 2H / 1F.
• Incidence: 1 pour 200000 par an.
• Pratiquement tout l’organisme peut être intéressé par la maladie, mais les organes les plus fréquemment atteints sont l’os, la peau, la post hypophyse, la sphère ORL, le foie, le poumon, la moelle, les ganglions, la rate, le système nerveux central.
NB : chez les plus de 60 ans, les localisations cutanéomuqueuses sont les plus fréquentes
• Présentation éminemment variable, allant d’un granulome unique, cutané ou osseux à une atteinte multiviscérale. Les atteintes plurifocales peuvent survenir simultanément ou non.
• Aspect comparable de l’imagerie pour une même topographie, que l’atteinte soit unique ou multiple
• Évolution variable et actuellement imprévisible, avec une régression spontanée possible ou un passage à la chronicité.
• Acutisation très rare chez l’adulte.

IMAGERIE DES DIFFERENTES LOCALISATIONS

Granulome éosinophile osseux• Forme d’histiocytose langerhansienne la plus fréquente
(70% des cas), la plus focale et la moins agressive
• Lésion tissulaire de la médullaire osseuse, ostéolytique, pouvant s’étendre aux parties molles adjacentes, unique dans 80 à 90% des cas, mais pouvant concerner tous les os.
• Localisations préférentielles– prédilection pour les os plats– crâne dans 43% des cas (os temporal++)– mandibule, fémur, sternum, bassin, vertèbres et côtes– extrémités exceptionnellement concernées– os longs : atteinte le plus souvent diaphysaire

Granulome éosinophile osseux• Clinique :
– lésion asymptomatique ou douloureuse, avec ou sans tuméfaction des parties molles en regard.
– Une fracture pathologique est possible.
• Complications par extension locale : – compression médullaire dans les atteintes vertébrales – atteinte cochléo-vestibulaire, exophtalmie ou compression du nerf
optique dans les atteintes du rocher– avulsion dentaire dans les atteintes mandibulaires
• Diagnostic différentiel :La rapidité d’évolution et les caractères agressif et lytique font
souvent évoquer une néoplasie maligne (sarcome d’Ewing), un lymphome, ou une ostéite

Granulome éosinophile osseuxTraitements :- abstention thérapeutique dans les formes peu invalidantes
(régression spontanée possible)
- curetage concomitant de la biopsie à visée diagnostique dans les lésions osseuses isolées. La rechute après curetage est possible, mais l’évolution est le plus souvent de bon pronostic.
- fixation ou greffe osseuse en cas de lésion vertébrale instable.
- infiltration locale de corticoïdes à visée antalgique possible dans les lésions douloureuses.
- Une radiothérapie peut être proposée dans les lésions présentant un risque en cas d’extension locale (contact orbitaire).
- Les traitements généraux (corticothérapie, indométhacine,biphosphonates ou chimiothérapie cytotoxique) sont plutôt l’apanage des formes osseuses multifocales symptomatiques.

OS LONGS
• RADIOGRAPHIE STANDARD– Lésions ostéolytiques aux contours bien limités, avec ou sans condensation
périphérique, « à l’emporte pièce », intéressant également la corticale.– Évolution rapide à la phase initiale, parfois en quelques semaines.– Réaction périostée inconstante– Persistance possible d’un fragment osseux intact au sein de la lésion lytique :
séquestre
• TDM– Confirme le diagnostic d’une lésion osseuse détruisant la corticale et les parties
molles adjacentes. Rehaussement variable après injection de produit de contraste– Précise l’étendue des lésions– Permet de guider la biopsie nécessaire au diagnostic
• IRM– La lésion lytique apparaît en hyposignal T1, hypersignal hétérogène T2 et STIR.
Rehaussement marqué après Gd.– Œdème périosté, médullaire, et des tissus mous au contact. Extension possible
aux tissus mous adjacents, parfois très délabrante
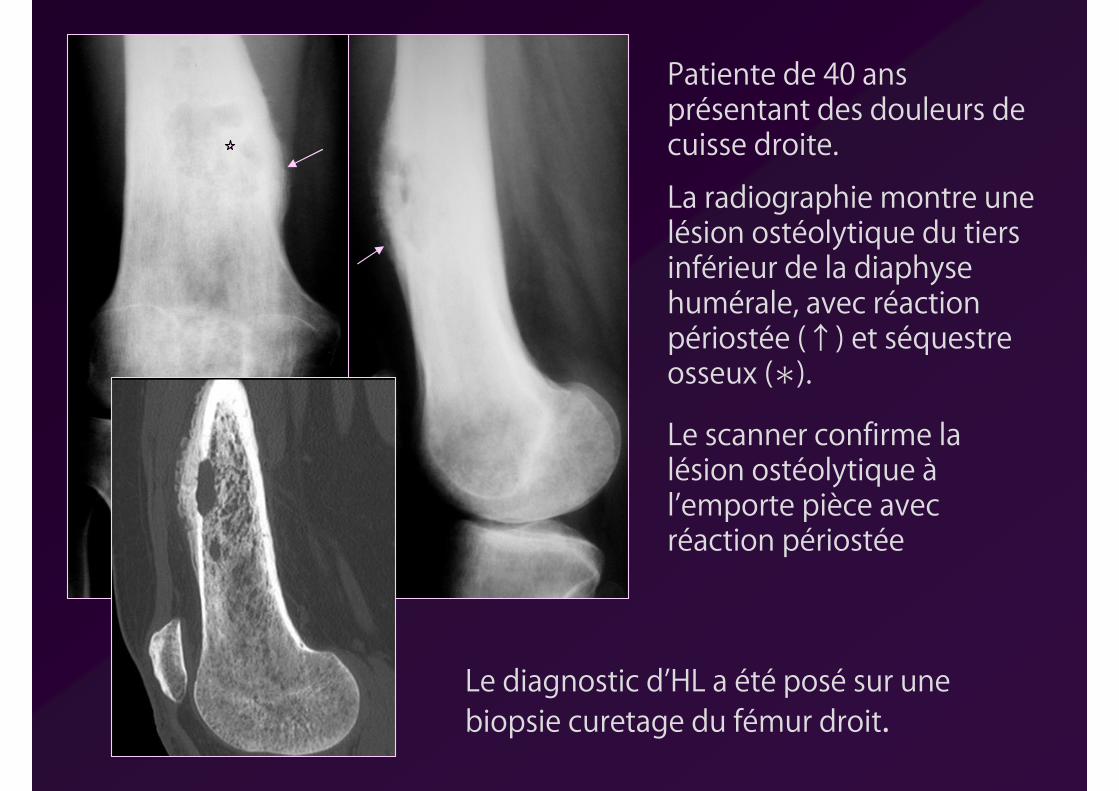
Patiente de 40 ans présentant des douleurs de cuisse droite.
La radiographie montre une lésion ostéolytique du tiers inférieur de la diaphyse humérale, avec réaction périostée (↑) et séquestre osseux (∗).
Le diagnostic d’HL a été posé sur une biopsie curetage du fémur droit.
Le scanner confirme la lésion ostéolytique àl’emporte pièce avec réaction périostée
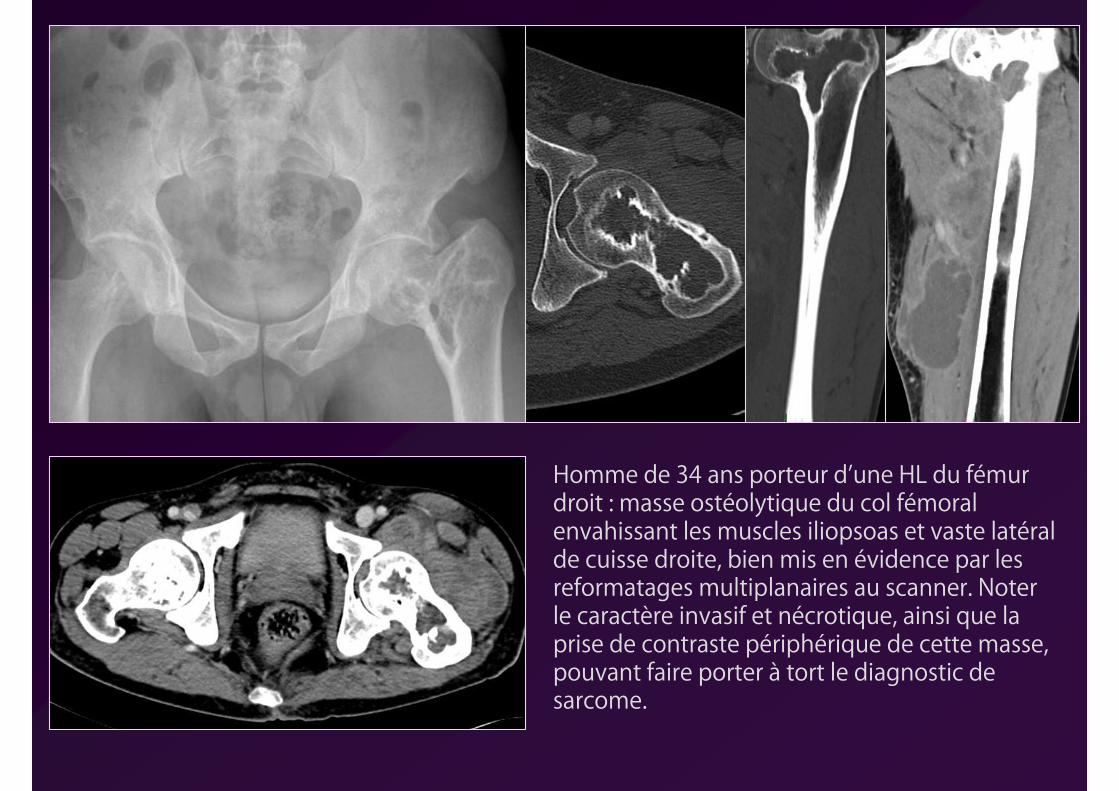
Homme de 34 ans porteur d’une HL du fémur droit : masse ostéolytique du col fémoral envahissant les muscles iliopsoas et vaste latéral de cuisse droite, bien mis en évidence par les reformatages multiplanaires au scanner. Noter le caractère invasif et nécrotique, ainsi que la prise de contraste périphérique de cette masse, pouvant faire porter à tort le diagnostic de sarcome.

RACHIS
• A l’étage rachidien, le granulome éosinophile de l’histiocytose àcellules de Langerhans donne des atteintes corporéales, avec une prédilection pour rachis dorsal. Typiquement, on observe un tassement vertébral respectant le mur postérieur et les disques intervertébraux, donnant à la vertèbre un aspect caractéristique appelé «vertebra plana». En cas de signes neurologiques, une extension épidurale peut-être observée au scanner et en IRM, avec présence de fuseaux paravertébraux. L’évolution se fait généralement vers la reconstitution ad integrum de la vertèbre, avec disparition des signes neurologiques éventuels.
• Dans les atteintes cervicales sévères, on peut observer des fractures pathologiques aux conséquences potentiellement dramatiques et des extensions aux parties molles sous formes de fuseaux périvertébraux
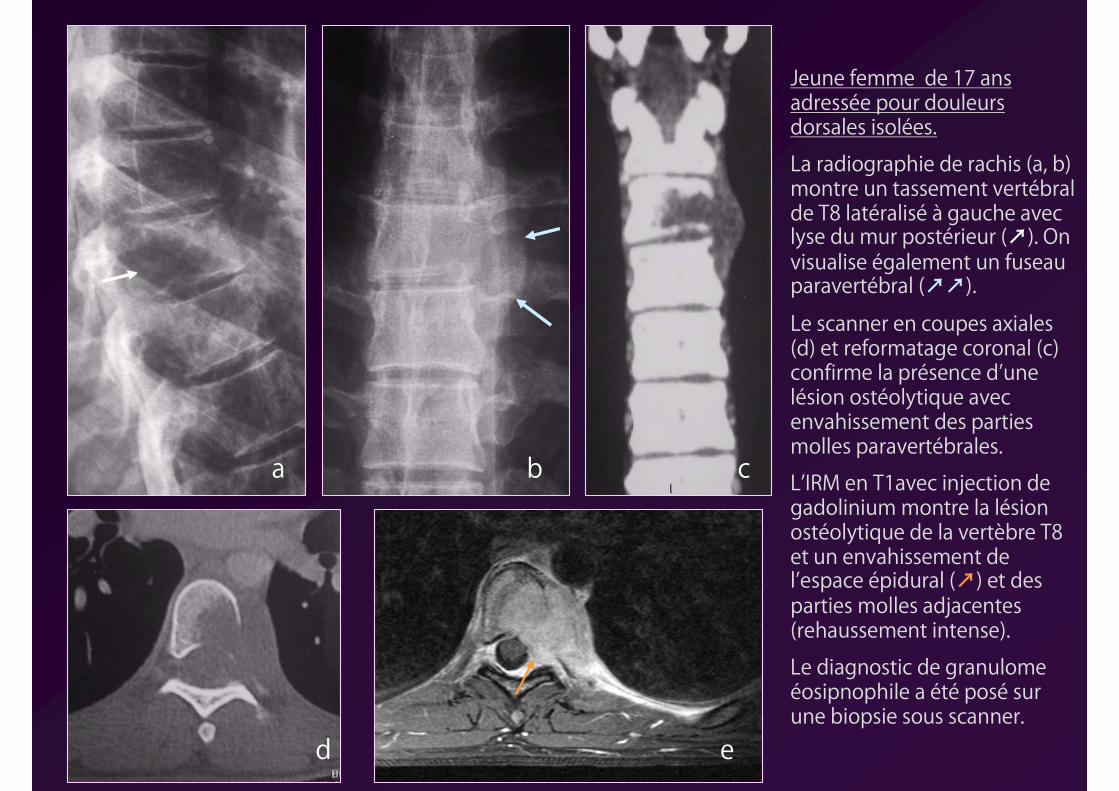
a b c
d e
Jeune femme de 17 ans adressée pour douleurs dorsales isolées.
La radiographie de rachis (a, b) montre un tassement vertébral de T8 latéralisé à gauche avec lyse du mur postérieur (↗). On visualise également un fuseau paravertébral (↗↗).
Le scanner en coupes axiales (d) et reformatage coronal (c) confirme la présence d’une lésion ostéolytique avec envahissement des parties molles paravertébrales.
L’IRM en T1avec injection de gadolinium montre la lésion ostéolytique de la vertèbre T8 et un envahissement de l’espace épidural (↗) et des parties molles adjacentes (rehaussement intense).
Le diagnostic de granulome éosipnophile a été posé sur une biopsie sous scanner.
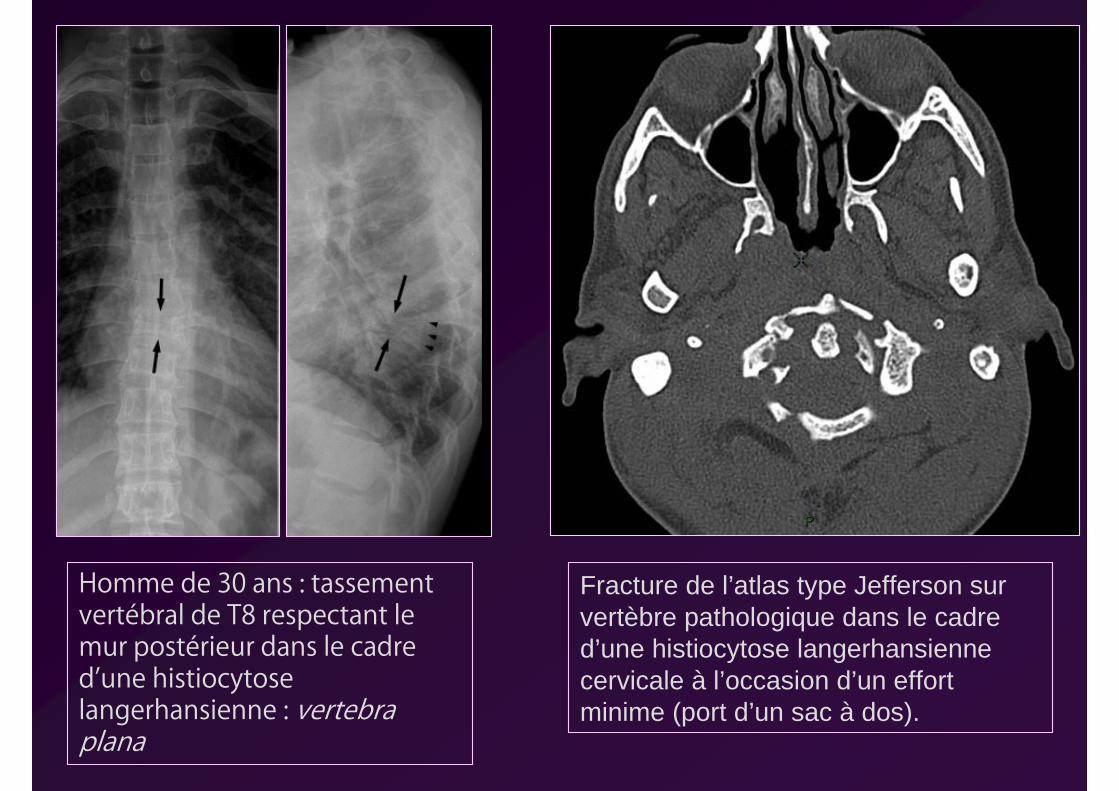
Homme de 30 ans : tassement vertébral de T8 respectant le mur postérieur dans le cadre d’une histiocytose langerhansienne : vertebra plana
Fracture de l’atlas type Jefferson sur vertèbre pathologique dans le cadre d’une histiocytose langerhansienne cervicale à l’occasion d’un effort minime (port d’un sac à dos).
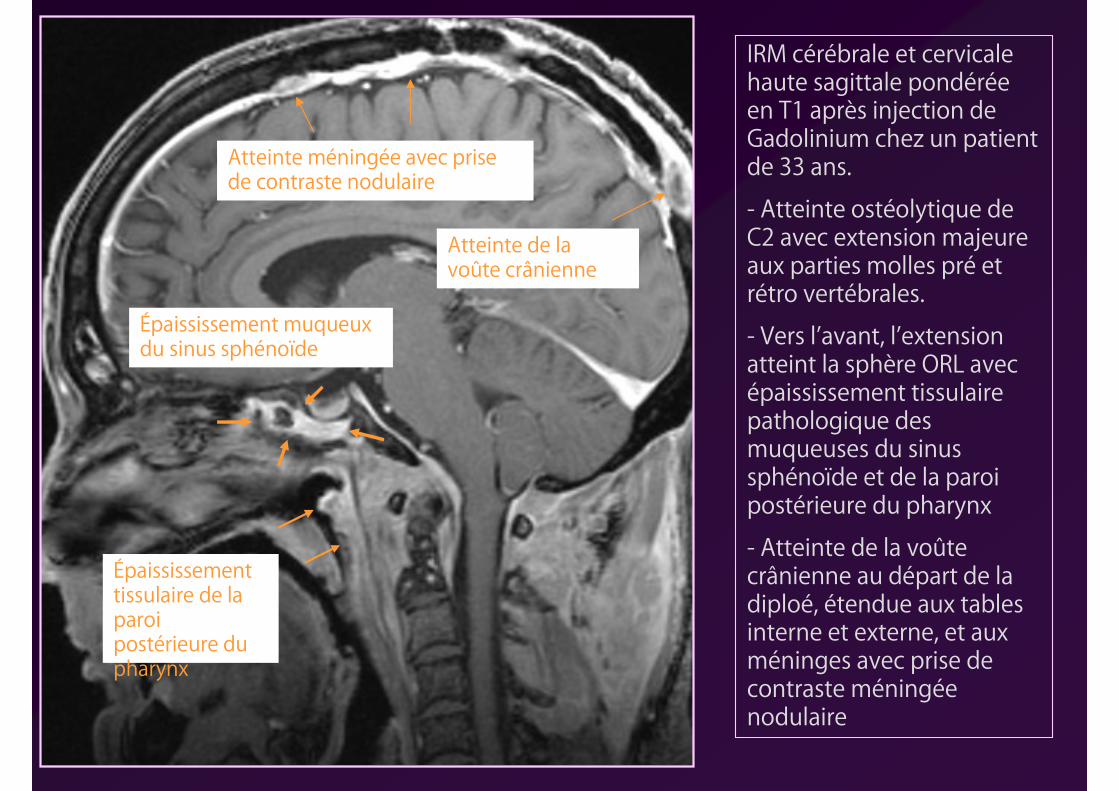
IRM cérébrale et cervicale haute sagittale pondérée en T1 après injection de Gadolinium chez un patient de 33 ans.
- Atteinte ostéolytique de C2 avec extension majeure aux parties molles pré et rétro vertébrales.
- Vers l’avant, l’extension atteint la sphère ORL avec épaississement tissulaire pathologique des muqueuses du sinus sphénoïde et de la paroi postérieure du pharynx
- Atteinte de la voûte crânienne au départ de la diploé, étendue aux tables interne et externe, et aux méninges avec prise de contraste méningée nodulaire
Épaississement muqueux du sinus sphénoïde
Épaississement tissulaire de la paroi postérieure du pharynx
Atteinte de la voûte crânienne
Atteinte méningée avec prise de contraste nodulaire

CRÂNE
• Les atteintes de la base du crâne, de la voûte crânienne et des sinus sont les plus fréquentes dans l’HL (90% des atteintes osseuses de l’adulte). Les os les plus atteints sont l’os temporal, l’orbite, la voûte crânienne et les os de la face extraorbitaires.
• Sur la voûte crânienne, le granulome éosinophile se manifeste également sous la forme d’une lésion lytique «à l’emporte pièce», développée dans la diploé. L’extension aux tables internes et externes peut se faire de manière différente, et donner un aspect biseauté à la lésion. Les lésions peuvent être multiples et confluer, prenant un aspect en carte de géographie. Une extension méningée est possible.
• Les atteintes sinusiennes ou de l’os temporal peuvent donner lieu à des extensions des parties molles parfois très délabrantes, pseudotumorales, pouvant entraîner des complications à type decophose ou de diplopie.
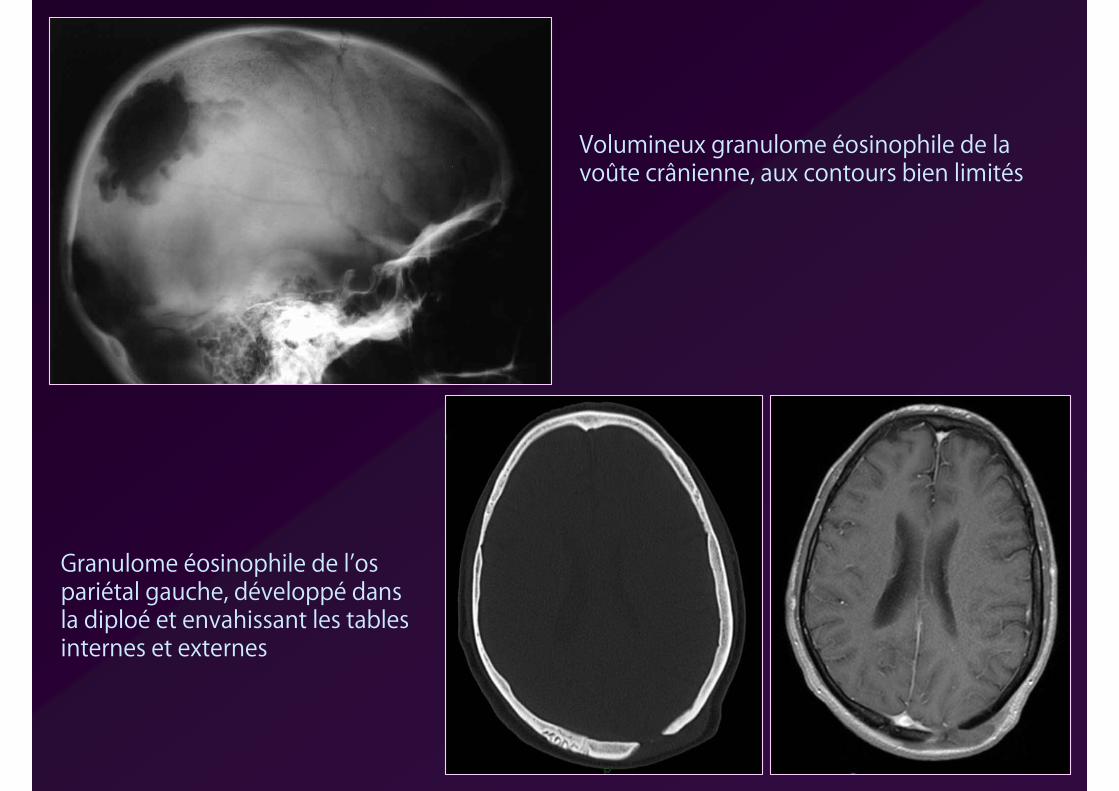
Volumineux granulome éosinophile de la voûte crânienne, aux contours bien limités
Granulome éosinophile de l’os pariétal gauche, développé dans la diploé et envahissant les tables internes et externes
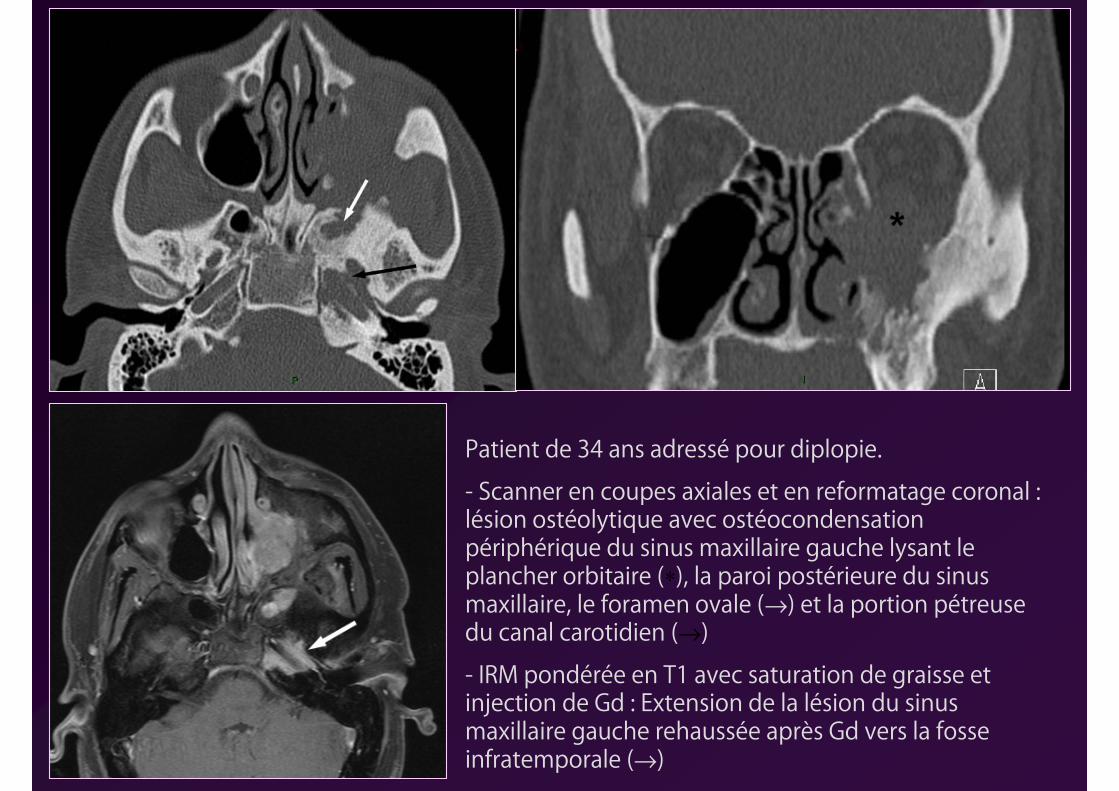
Patient de 34 ans adressé pour diplopie.
- Scanner en coupes axiales et en reformatage coronal : lésion ostéolytique avec ostéocondensation périphérique du sinus maxillaire gauche lysant le plancher orbitaire (∗), la paroi postérieure du sinus maxillaire, le foramen ovale (→) et la portion pétreuse du canal carotidien (→)
- IRM pondérée en T1 avec saturation de graisse et injection de Gd : Extension de la lésion du sinus maxillaire gauche rehaussée après Gd vers la fosse infratemporale (→)
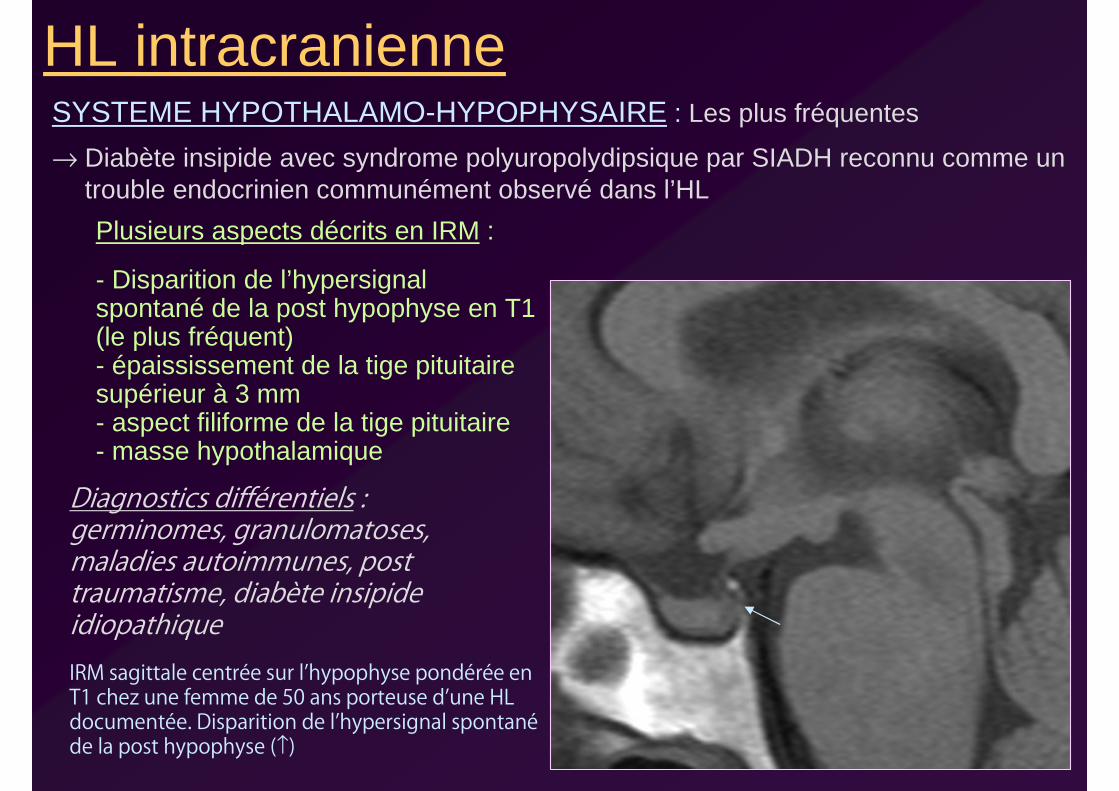
HL intracranienneSYSTEME HYPOTHALAMO-HYPOPHYSAIRE : Les plus fréquentes
→ Diabète insipide avec syndrome polyuropolydipsique par SIADH reconnu comme un trouble endocrinien communément observé dans l’HL
IRM sagittale centrée sur l’hypophyse pondérée en T1 chez une femme de 50 ans porteuse d’une HL documentée. Disparition de l’hypersignal spontanéde la post hypophyse (↑)
Plusieurs aspects décrits en IRM :
- Disparition de l’hypersignal spontané de la post hypophyse en T1 (le plus fréquent)- épaississement de la tige pituitaire supérieur à 3 mm- aspect filiforme de la tige pituitaire- masse hypothalamique
Diagnostics différentiels : germinomes, granulomatoses, maladies autoimmunes, post traumatisme, diabète insipide idiopathique
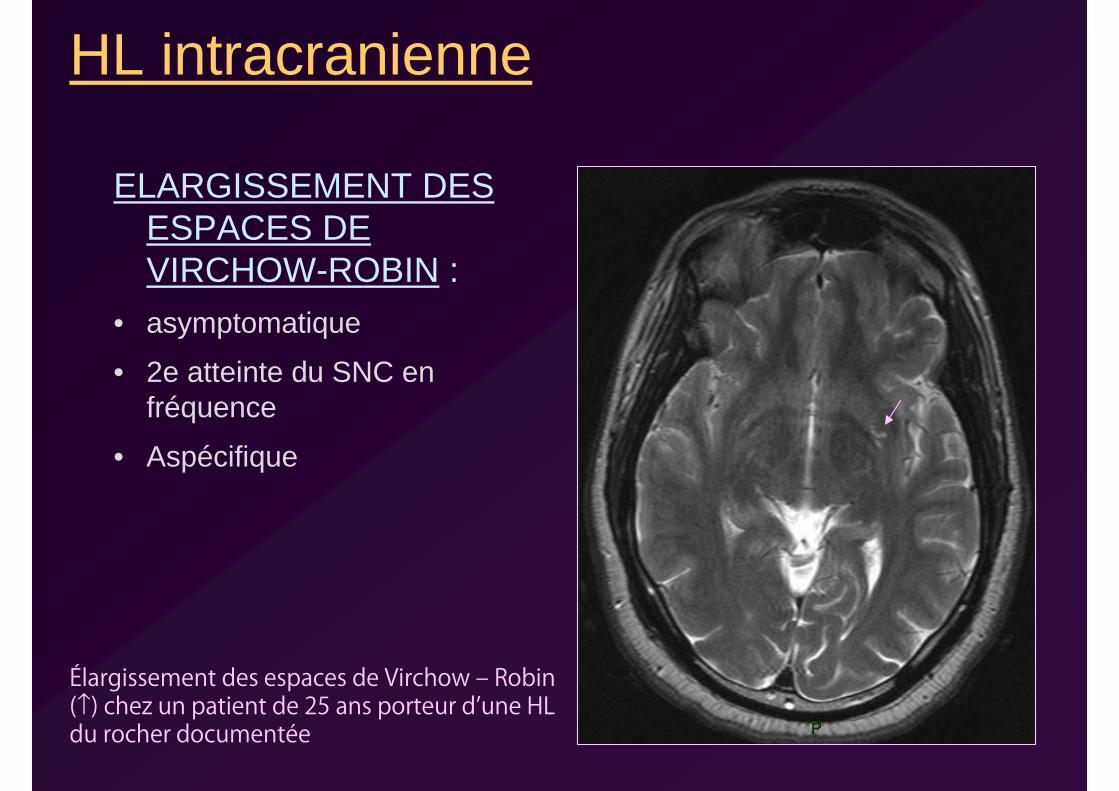
HL intracranienne
ELARGISSEMENT DES ESPACES DE VIRCHOW-ROBIN :
• asymptomatique
• 2e atteinte du SNC en fréquence
• Aspécifique
Élargissement des espaces de Virchow ‒ Robin (↑) chez un patient de 25 ans porteur d’une HL du rocher documentée
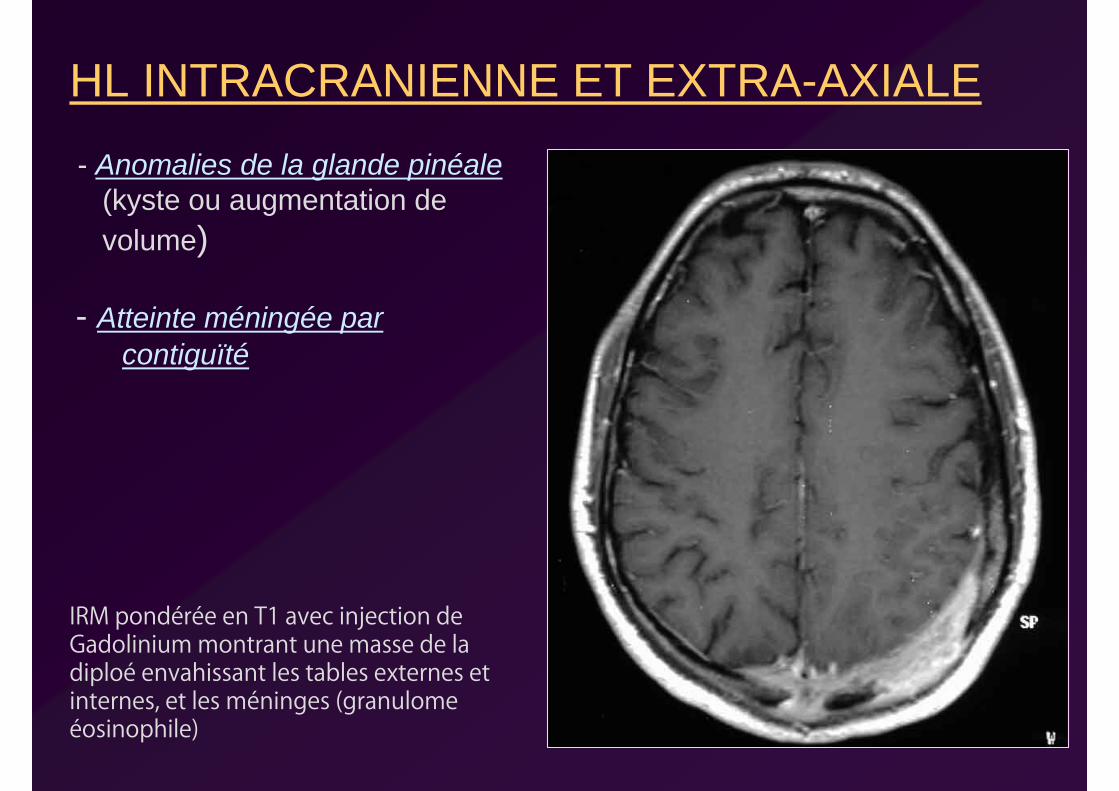
HL INTRACRANIENNE ET EXTRA-AXIALE
- Anomalies de la glande pinéale(kyste ou augmentation de volume)
- Atteinte méningée par contiguïté
IRM pondérée en T1 avec injection de Gadolinium montrant une masse de la diploé envahissant les tables externes et internes, et les méninges (granulome éosinophile)

HL DU SYSTÈME NERVEUX CENTRAL
Anomalies sous tentorielles à type de leucoencéphalopathie pontique ou cérébelleuse,
Souvent associées à une atteinte de la substance grise (noyaux dentelés) :
IRM : hypersignaux linéaires ou en plages en T2 et FLAIR, +/- rehaussés après injection de Gadolinium.
Les patients porteurs de ce type de lésions présentent des tableaux neurologiques variables en fonction de la sévérité de l’atteinte, allant de l’absence de symptôme à l’ataxie sévère, avec une évolution pouvant être fatale.Le traitement des formes neurologiques sévères est encore mal codifié.
• Diagnostic différentiel : atrophie multisystémique, encéphalomyélite, encéphalite, syndromes paranéoplasiques
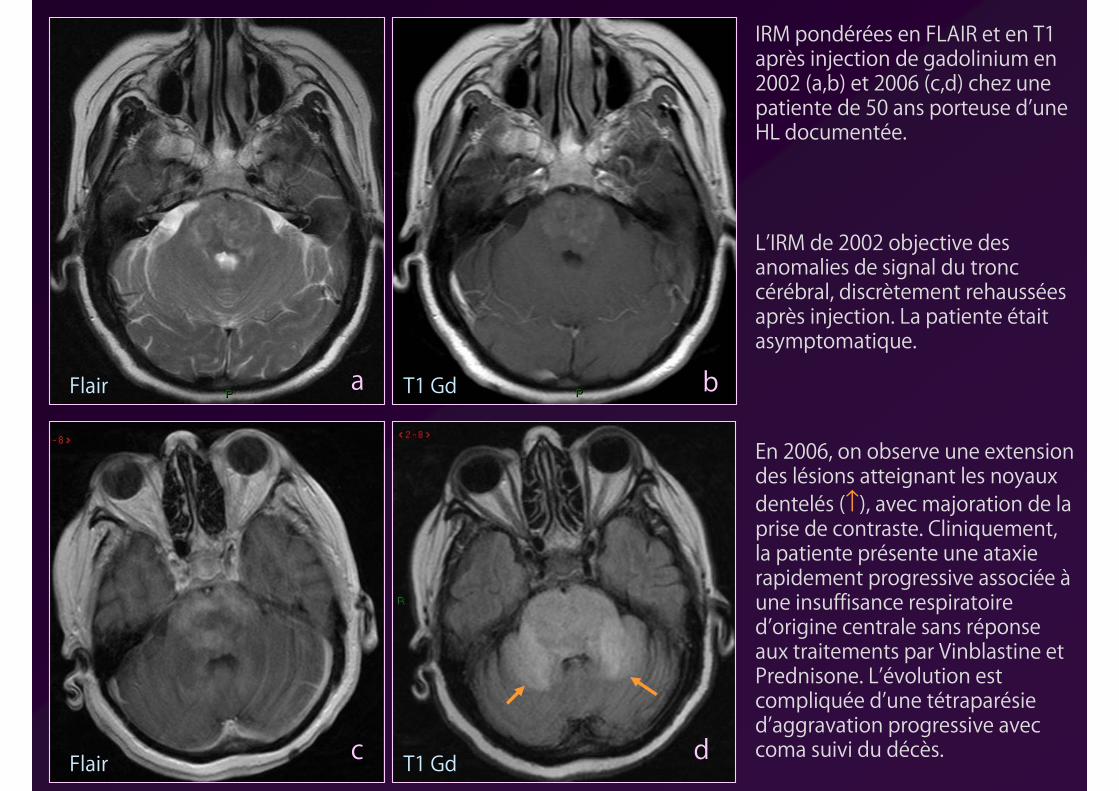
a b
c d
IRM pondérées en FLAIR et en T1 après injection de gadolinium en 2002 (a,b) et 2006 (c,d) chez une patiente de 50 ans porteuse d’une HL documentée.
L’IRM de 2002 objective des anomalies de signal du tronc cérébral, discrètement rehaussées après injection. La patiente était asymptomatique.
En 2006, on observe une extension des lésions atteignant les noyaux dentelés (↑), avec majoration de la prise de contraste. Cliniquement, la patiente présente une ataxie rapidement progressive associée àune insuffisance respiratoire d’origine centrale sans réponse aux traitements par Vinblastine et Prednisone. L’évolution est compliquée d’une tétraparésied’aggravation progressive avec coma suivi du décès.
Flair
Flair
T1 Gd
T1 Gd

HL PULMONAIRE• Forme clinique particulière le plus souvent limitée aux poumons, touchant l’adulte fumeurdes 2 sexes entre 20 et 40 ans.
• Présentation variable :– 25% des cas : asymptomatique– 50% des cas : toux sèche, dyspnée d’effort, rarement pneumothorax
– 30% des cas : signes généraux associés ( fièvre, asthénie, amaigrissement)
• Évolution imprévisible :– 50% : amélioration spontanée ou sous traitement corticoïde (arrêt tabac)– stabilisation– aggravation évoluant vers insuffisance respiratoire terminale (seul traitement : greffe pulmonaire)– Peut récidiver sur poumon greffé
• Imagerie :- Association de nodules plus ou moins excavés et de KYSTES confluents d’âges différents, à parois épaisses, pouvant évoluer vers la fibrose et la destruction parenchymateuse- Répartition épargnant relativement les bases
- Diagnostics différentiels : lymphangioleïomyomatose, emphysème, fibrose
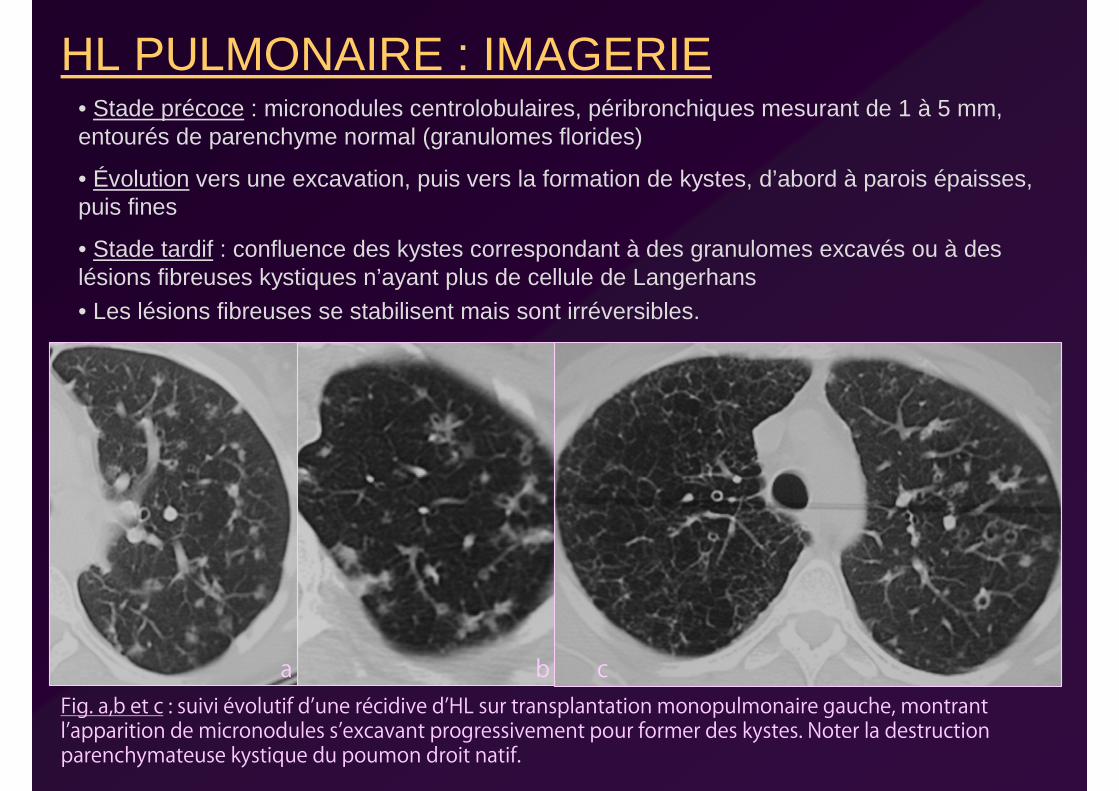
HL PULMONAIRE : IMAGERIE• Stade précoce : micronodules centrolobulaires, péribronchiques mesurant de 1 à 5 mm, entourés de parenchyme normal (granulomes florides)
• Évolution vers une excavation, puis vers la formation de kystes, d’abord à parois épaisses, puis fines
• Stade tardif : confluence des kystes correspondant à des granulomes excavés ou à des lésions fibreuses kystiques n’ayant plus de cellule de Langerhans
• Les lésions fibreuses se stabilisent mais sont irréversibles.
Fig. a,b et c : suivi évolutif d’une récidive d’HL sur transplantation monopulmonaire gauche, montrant l’apparition de micronodules s’excavant progressivement pour former des kystes. Noter la destruction parenchymateuse kystique du poumon droit natif.
a b c
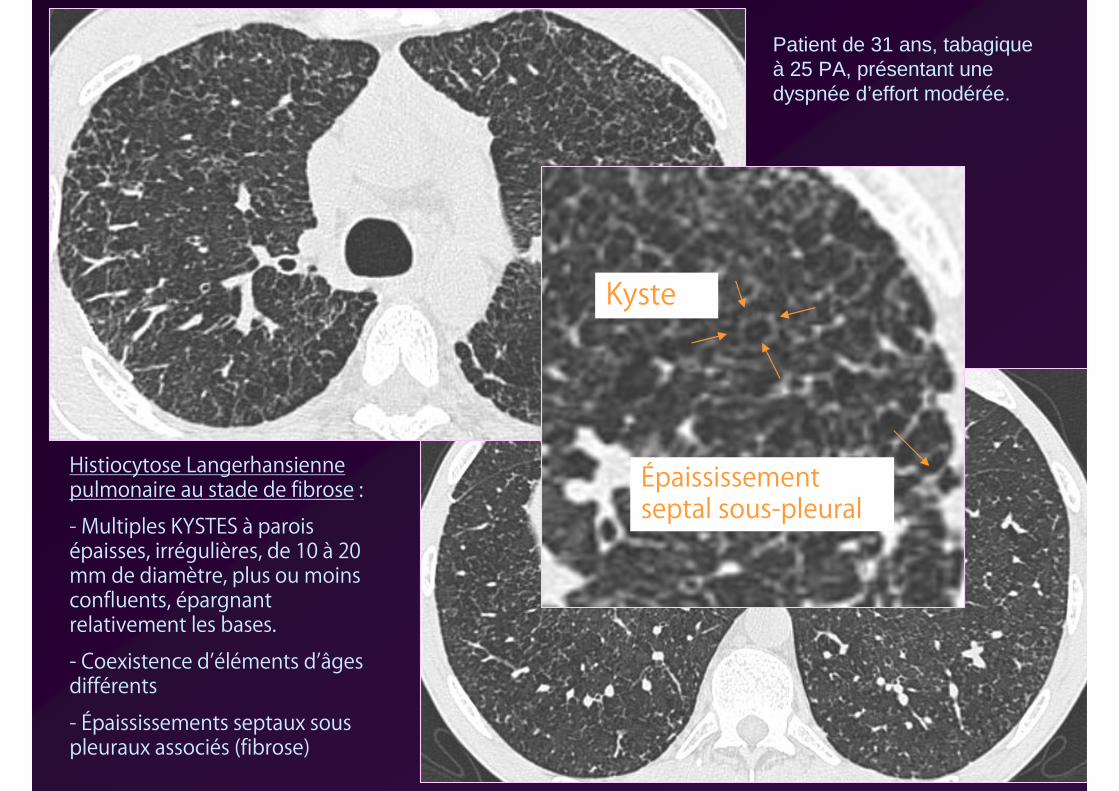
Patient de 31 ans, tabagique à 25 PA, présentant une dyspnée d’effort modérée.
Histiocytose Langerhansienne pulmonaire au stade de fibrose :
- Multiples KYSTES à parois épaisses, irrégulières, de 10 à 20 mm de diamètre, plus ou moins confluents, épargnant relativement les bases.
- Coexistence d’éléments d’âges différents
- Épaississements septaux sous pleuraux associés (fibrose)
Kyste
Épaississement septal sous-pleural

HL HEPATIQUE
• L’atteinte hépatique de l’HL est rare et est caractérisée par une infiltration histiocytaire périportale évoluant vers la fibrose et la cirrhose. Elle survient généralement chez des patients ayant d’autres localisations.
• Quelques cas de cholangite sclérosante ont été décrits chez l’adulte porteur d’une HL (atteinte plus fréquente chez l’enfant)
• L’imagerie est variable, en fonction de la sévérité de l’atteinte, classée en 4 stades histologiques : prolifératif, granulomateux, xanthomateux et fibreux.
– Formes xanthomateuse : Infiltration périportale hyperéchogène en échographie, hypodense, de tonalité graisseuse au scanner, et de signal graisseux en IRM (stéatose focale)
– Formes avec cholangite sclérosante : alternance de sténoses et de dilatations des voies biliaires en cholangiographie par IRM
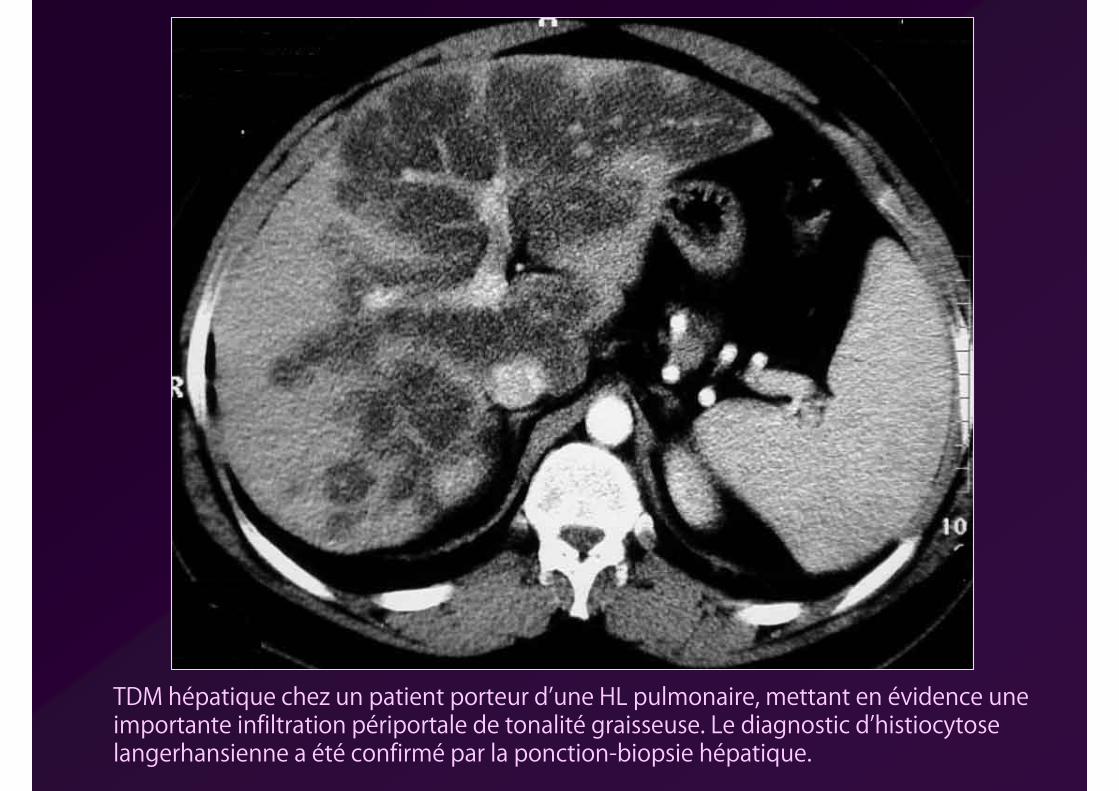
TDM hépatique chez un patient porteur d’une HL pulmonaire, mettant en évidence une importante infiltration périportale de tonalité graisseuse. Le diagnostic d’histiocytose langerhansienne a été confirmé par la ponction-biopsie hépatique.

AUTRES LOCALISATIONS
D’autres localisations plus rares sont possibles :
• Cutanées : lésions érythémato-squammeuses fréquentes
• Hématologiques, plus rares que chez l’enfant : adénopathies, splénomégalie.
• Tube digestif (entéropathie exsudative)
• Muqueuses génitales et ovaires

CONCLUSION• L’histiocytose Langerhansienne est une maladie ubiquitaire bénigne pouvant revêtir de multiples formes, dont l’évolution est imprévisible.
• La localisation est unique ou multiple, localisée ou extensive.
• Le pronostic varie en fonction de la sévérité et du site de l’atteinte.
• Elle doit être évoquée en imagerie devant des lésion osseuses lytiques, parfois localement évoluées, une atteinte du système hypothalamo-hypophysaire, des images de leucoencéphalopathie pontique ou cérébelleuse, et/ou une atteinte kystique pulmonaire.
• Les diagnostics différentiels varient selon les localisations :
- néoplasie maligne ou ostéite pour les atteintes osseuses, et de la base du crâne,
- leucoencéphalopathies pour les atteintes du SNC,
- germinomes, granulomatoses et pathologies autoimmunes pour les atteintes de l’axe hypothalamo-hypophysaire,
- fibrose, emphysème et lymphangioléiomyomatose pour les atteintes pulmonaires
• Quelque soit la localisation, le diagnostic de certitude est histologique.
• Le traitement varie selon les localisations et la sévérité de l’atteinte.

BIBLIOGRAPHIE
1. The French Langerhans’ Cell Histiocytosis Study Group. A multicentre retrospective survey of Langerhans’ cell histiocytosis : 348 cases observed between 1983 and1993.
2. Tazi A. Histiocytoses. In: Kahn MF, Peltier AP, Meyer O et Piette JC. Maladies et syndromes systémiques. Paris. Médecine-science Flammarion, 2000; 1077-1098.
3. Meyer JS, Harty MP, Mahboubi S, Heyman S, et Al. Langerhans cell histiocytosis :presentation and evolution of radiologic findings with clinical correlation.Radiographics 1995 ;15 :1135-1146.
4. Azouz EM, Saigal G, Rodriguez MM, Podda A. Langerhans’ cell histiocytosis :pathology, imaging and treatment of skeletal involvement. Pediatr Radiol 2005 ; 35 : 103-115.
5. Prayer D, Grois N, Prosch H, et Al. MR Imaging presentation of intracranial disease associated with langerhans cell histiocytosis. Am J Neuroradiol 2004 ; 25 :880-891.
6. Abbott GF, Rosado-de-Christenson ML, Franks TJ, Frazier AA et Galvin JR. PulmonaryLangerhans cell histiocytosis. Radiographics 2004; 24:821-841
7. König CW, Pfannenberg C, Trübenbach, Remy C et Al. MR cholangiography in the diagnosis of sclerosing cholangitis in Langerhans’ cell histiocytosis. Eur. Radiol. 2001; 11: 2516-2520.






