Embed Size (px)
Citation preview

Geoffrey MASUYER Décembre 2004
Master 2 Pro Expression Génique et Protéines Recombinantes
PROTEASOME
Inhibition de la dégradation de p53 par leProtéasome
– Interaction MDM2-p53
Résumé
La croissance et le métabolisme normaux des cellules sont dépendants non
seulement de la présence et de l’activation de protéines critiques, mais également de
leur dégradation en temps voulu, permettant le bon déroulement du cycle cellulaire.
Un acteur clé dans la dégradation des protéines de la cellule est le protéasome 26S.
La majorité des dégradations de protéines cellulaires sont réalisées par la voie
ubiquitine-protéasome. Le suppresseur de tumeur p53 est l’un des principaux
médiateurs de l’arrêt du cycle cellulaire et initiateur de l’apoptose en réponse à des
dommages cellulaires. Ainsi le contrôle de la stabilité de p53 et de son activation
représente une approche intéressante en vue d’une thérapie anti-cancéreuse. La
régulation de p53 dans la cellule repose essentiellement sur son régulateur négatif
MDM2 avec lequel p53 interagit. Un moyen d’empêcher la dégradation de p53 par le
protéasome est de cibler l’interaction p53-MDM2 par des antagonistes de MDM2.
Mots clés : protéasome, inhibition du protéasome, interaction p53-MDM2, Nutlin

2
Introduction
L’homéostasie des protéines est critique pour les processus biologiques qui
sont fondamentaux pour la survie des cellules cancéreuses. Ainsi, l’un des principaux
points d’intérêts de la recherche contre le cancer est de cibler la régulation de la
production et la destruction des protéines. En particulier les facteurs qui favorisent la
prolifération et autres caractéristiques particulières des cellules malignes.
Récemment, il est devenu largement admis que la destruction ordonnée des
protéines responsables de la régulation du cycle cellulaire est cruciale pour le
contrôle des phénomènes cellulaires associés au cancer. La voie ubiquitine-
protéasome qui intervient dans 80% de la dégradation de toutes les protéines
cellulaires est le principal mécanisme de dégradation des protéines, incluant celles
jouant un rôle dans le cycle cellulaire [1•]. En conséquence, le protéasome apparaît
comme une cible attractive pour la thérapie cancéreuse. Cette étude introduit tout
d’abord les caractéristiques principales du protéasome ainsi que son rôle dans la
cellule. Ensuite l’intérêt de l’inhibition du protéasome et plus particulièrement la
responsabilité de p53 et son interaction avec MDM2 seront discutés.
Le protéasome
Le protéasome 26S est un complexe enzymatique multicatalytique exprimé
dans le noyau et le cytoplasme de toutes les cellules eucaryotes. Sa fonction
première est de dégrader les protéines, ses substrats incluent entre autres les
suppresseurs de tumeurs, les régulateurs du cycle cellulaire, les facteurs de
transcription et les protéines anti-apoptotiques [2]. Quand la dégradation de ces
protéines est interrompue, les effets sont potentiellement vastes, particulièrement
dans les cellules cancéreuses se développant rapidement et nécessitant des
protéines favorisant la croissance pour soutenir la mitose incontrôlée, caractéristique
du développement des tumeurs.
La voie ubiquitine-protéasome
Pour qu’une protéine soit reconnue par le protéasome, un petit peptide,
l’ubiquitine, doit d’abord être attaché à la protéine cible. Ce processus intervient

3
Figure 1: Ubiquitination des protéines ciblées pour le protéasome. [3•]Une enzyme activatrice de l’ubiquitine (E1) se lie à l’ubiquitine, qui est alors transféré àune enzyme conjugatrice de l’ubiquitine (E2); une ubiquitine-ligase (E3) aide le transfertde l’ubiquitine à la cible substrat. Plusieurs molécules d’ubiquitine sont attachées auxprotéines avant leur reconnaissance et dégradation par le protéasome 26S; avant ladégradation, la chaîne d’ubiquitine est détachée, permettant à l’ubiquitine libre d’êtrerecyclée.
Figure 2: Représentation schématique du protéasome 26S [2]

4
grâce à une cascade d’enzymes (E1, E2, et E3) (figure 1, [3•]) qui active l’ubiquitine
libre et la dirige vers la protéine cible. E1 active et transfère l’ubiquitine sur la
protéine porteuse E2. E2 présente l’ubiquitine à E3. E3 se lie à la protéine cible et
interagit avec E2 pour attacher l’ubiquitine à la protéine cible de façon covalente. Ce
processus est répété plusieurs fois, créant une chaîne poly-ubiquitine qui va pouvoir
être reconnue pour la destruction par le protéasome. Actuellement une seule
protéine E1 est connue, alors qu’il existe une vingtaine d’enzymes E2 et encore un
plus large nombre de ligases E3. Cette hiérarchie permet des interactions très
spécifiques qui sont essentielles à la forte coordination que nécessite la dégradation
des protéines cellulaires. Cette voie par laquelle les protéines sont ubiquitinées et
dégradées par le protéasome est critique à la fois pour la cellule mais également
pour la réponse inflammatoire et la présentation des antigènes.
Structure
Le complexe multiprotéique du protéasome consiste d’un corps principal 20S
qui est associé avec une ou deux particules régulatrices 19S (figure 2, [2]). Chaque
sous-unité 19S est capable de se lier à la chaîne poly-ubiquitine et de la détacher de
la protéine substrat. Ce substrat est alors dénaturé et introduit dans le cœur
protéolytique. La particule 20S est un cylindre composé de quatre anneaux empilés,
elle possède une faible activité catalytique dans son milieu cellulaire [3•]. Les deux
anneaux extérieurs (appelés « α ») se complexent avec les particules régulatrices
19S, formant un canal étroit à travers lequel seules les protéines dénaturées peuvent
passer. La chambre catalytique est formée par les deux anneaux β internes, chacun
contenant trois sites actifs. Ces sites diffèrent dans leur spécificité et activité et sont
nommés d’après les enzymes qui présentent des activités protéolytiques similaires.
Ces sites sont ainsi dénommés chymotrypsique, trypsique, et PGPH pour post-
glutamyl peptide hydrolase-like. Les protéines sont dégradées par le cœur
protéolytique d’une façon progressive, générant des peptides de 3 à 25 acides
aminés de long qui peuvent servir à la présentation des antigènes.

5
Classe de protéines Protéine Fonction protéique
Cyclines et protéines associées Cyclines A, B, D, E Progression du cycle cellulaireCycline-dépendante kinase inhibiteurs Régulation de l'activité cycline
Suppresseur de tumeur p53 Facteur de transcriptionOncogènes c-fos/c-jun Facteur de transcription
c-myc Facteur de transcriptionN-myc Facteur de transcription
Protéines inhibitrices IκB Inhibiteur de NF-κBp130 Inhibiteur de E2F-1
Enzymes cdc 25 phosphatase CDK1/cycline B phosphataseTyrosine amino transferase (TAT) Métabolisme de la tyrosineTopoisomérase I DNA supercoilingTopoisomérase IIα DNA supercoiling
Table 1: Fonction des protéines relatives au cycle cellulaire dégradées par le protéasome [1•]

6
Inhibition du protéasome
Le protéasome 26S est donc responsable de la dégradation des protéines chez
les eucaryotes et parmi elles plusieurs protéines responsables du cycle cellulaire
(Table 1, [1•]).
Une indication du rôle important que joue le protéasome dans ce processus est
que l’arrêt des fonctions du protéasome stoppe la division cellulaire. Aussi les
cellules tumorales apparaissent plus sensitives à la perte d’activité du protéasome et
des études montrent que ces cellules sont plus sensibles à l’apoptose [1•]. Le
premier inhibiteur du protéasome à être entré en phase clinique est le Bortezomib [2],
de la famille des peptides analogues au boronate, et qui permet l’inhibition de
l’activité chymotrypsine du protéasome en se liant au site correspondant. La capacité
thérapeutique de l’inhibition du protéasome repose dans la possibilité de stabiliser
les protéines qui inhibent la survie cellulaire et la progression du cycle cellulaire [16].
Une telle protéine, p53, à été mise en évidence par MacLaren et al [11••].
Interaction p53-MDM2
Une autre approche allant dans ce sens est d’inhiber la dégradation d’une cible
importante du cycle cellulaire en amont de sa dégradation par le protéasome. Depuis
la découverte de ses puissantes activités, suppression de la croissance et apoptose,
le suppresseur de tumeur p53 a été largement étudié. D’autant plus qu’il est
majoritairement contrôlé par un seul régulateur, MDM2 (pour mouse double minute
2) qui s’y attache et contrôle négativement son activité et sa stabilité [8•]. Ainsi, des
antagonistes de MDM2 capables de séparer les deux protéines sont supposés
stabiliser et activer le suppresseur de tumeur, induisant l’arrêt du cycle cellulaire et la
mort programmée des cellules cancéreuses.
Le suppresseur de tumeur p53 joue un rôle clé dans la protection contre le
développement de cancer du à diverses formes de stress cellulaire [14•]. Ce stress
engendre des modifications post-traductionnelles, c’est à dire la phosphorylation des
sérines 15 et 20 de p53. Ceci entraîne la dissociation de MDM2 et permet donc la
tétramérisation de p53 qui va pouvoir réguler la transcription. p53 est un facteur de
transcription efficace qui régule de multiples gènes impliqués dans le contrôle du
cycle cellulaire, l’apoptose et l’antiangiogénèse.
7
MDM2
Ub
p53
MDM2
p53
MDM2
26S
CytoplasmeNoyau
dégradation
p53
apoptose
p53
MDM2
stresscellulaire p53
MDM2
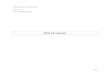
Figure 3: Schématisation du rôle de MDM2 dans la voie de p53
Figure 4: Représentation de l’interaction p53-MDM2, structure cristallographiqueréalisée par Kussie et al (Science 1996) [10]En vert, MDM2, et en jaune le domaine de transactivation de p53. En évidence les3 acides aminés critiques F19, L26 et W23.

8
La concentration en p53 dans la cellule est soumise à un contrôle fort par
MDM2. En effet, MDM2 est une phosphoprotéine qui joue le rôle d’ubiquitine ligase
pour p53, elle permet entre autre l’exportation de p53 du noyau vers le cytoplasme et
son ubiquitinilation, favorisant ainsi sa dégradation par le protéasome 26S [14•].
(Figure 3, 4). De plus il existe une boucle de régulation entre p53 et MDM2,
puisqu’une forte concentration en p53 favorise la transcription de MDM2.
Inhibition du protéasome et importance de p53
MacLaren et al [11••] ont étudié l’apoptose p-53-dépendante induite par
l’inhibition du protéasome chez des cellules épithéliales mammaires. Pour cela
l’inhibition du protéasome par un peptide aldéhyde inhibiteur spécifique (MG132) est
observée sur une lignée cellulaire murine (KIM-2). Cette lignée cellulaire représente
un bon model in vitro pour étudier le développement des cellules épithéliales
mammaires car elle possède les marqueurs de différentiation spécifiques à ce type
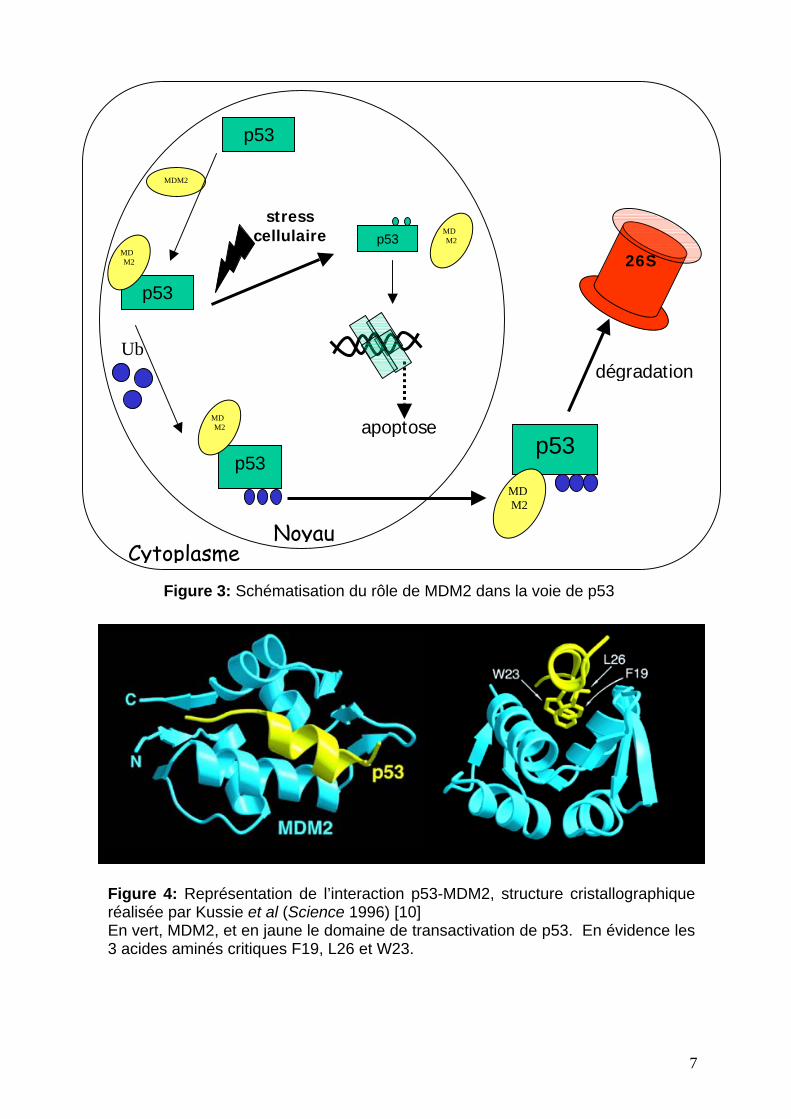
de cellules. La figure 5 représente les résultats obtenus pour l’examen du traitement
de ces cellules par MG132 pendant 24 heures. L’apoptose est analysée par le test à
l’annexin V. Les résultats montrent que des cellules apoptotiques apparaissent après
4h de traitement. Le niveau d’apoptose augmente jusqu’à 18-20 h où il atteint un
maximum de 20%. Ceci corrèle avec le temps de division cellulaire qui est de 24h
dans cette lignée. Par ailleurs, d’autres inhibiteurs du protéasome 26 S sont testés et
présentent le même type de résultats, permettant de conclure que l’initiation de
l’apoptose est due au blocage de la dégradation des protéines par le protéasome
26S.
De plus il est reconnu que p53 est impliquée dans les signaux d’induction de
l’apoptose [14•]. Son importance dans ce phénomène au niveau de l’inhibition du
protéasome est donc étudiée. Des cellules ES contenant p53 sauvage et d’autres
étant nulles pour p53 ont été obtenues et traitées avec MG132, inhibiteur du
protéasome (plus particulièrement au niveau de l’activité chymotrypsique). Ces
cellules sont ensuite analysées comme précédemment. Les résultats de la figure 6
montrent que l’apoptose est très fortement augmentée dans les cellules exprimant
p53 sauvage comparées aux p53 nulles. De plus le même test a été réalisé dans une
lignée cellulaire exprimant une p53 mutée, montrant un faible de taux d’apoptose
9
ES wt p53 ES p53 nulle
Figure 5: Mesure de l’Annexin V au cours du temps sur les cellules KIM-2. Les cellulessont traitées avec MG132 pendant 24h. Les échantillons sont récoltés et soumis à uneanalyse à l’Annexin V. Les résultats montrés sont la moyenne de trois expériences, etsont exprimés comme pourcentage des cellules positives à l’Annexin V. [11]
Figure 6: Dépendance à p53 de l’apoptose induite par l’inhibition du protéasome.Des cellules ES contenant p53 sauvage ou non (nulle) sont traitées au MG132 àdifférentes concentrations. Une analyse par Annexin V est ensuite réalisée. [11]

10
dans ces cellules. Ces résultats confirment l’importance de p53 sauvage dans
l’induction de l’apoptose, et donc l’intérêt d’inhiber sa dégradation par le protéasome.
Ciblage de l’interaction p53-MDM2
Les différentes approches
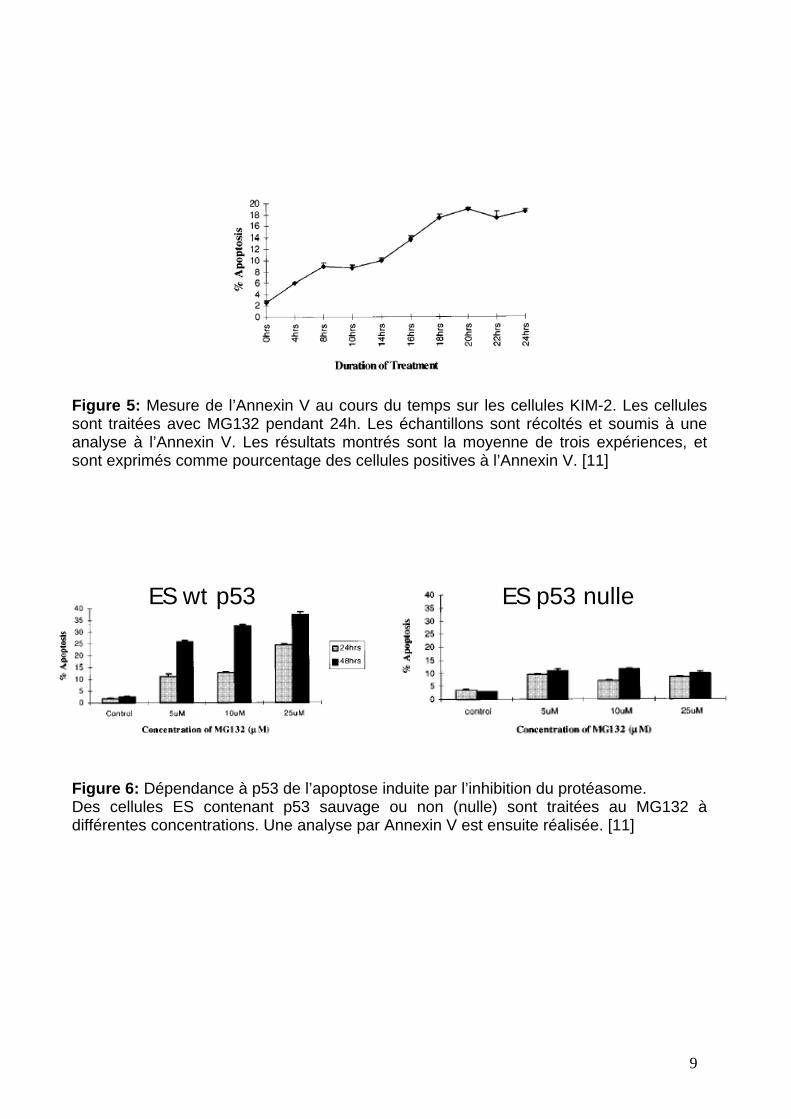
La découverte que la liaison p53-MDM2 implique une interaction de 3 résidus
acides aminés critiques de p53 avec une poche hydrophobe bien définie à la surface
de MDM2 (figure 4) a donné l’espoir de pouvoir identifier des inhibiteurs
pharmacologiques de cette interaction [13••]. Différentes approches ont été utilisées
pour valider l’interaction p53-MDM2 comme cible médicamenteuse. Celles-ci incluent
la micro-injection d’anticorps monoclonaux dirigés contre le site de liaison de p53 sur
MDM2 [4,5], l’inhibition de l’expression de MDM2 par des oligonucléotides antisens
[7,17•], ou encore la micro-injection ou expression intracellulaire de petites protéines
de fusion [15]. Toutes ces études ont démontré que le blocage de la liaison p53-
MDM2 peut perturber la boucle de régulation de p53, provoquant l’accumulation et
l’activation de la voie de p53. Cependant, ces expériences ont principalement utilisé
des outils protéiques (peptides ou protéines), qui ont donc des applications limitées,
particulièrement pour le besoin d’études en cellules vivantes ou des modèles
animaux de cancer.
L’une des exceptions est cependant l’approche utilisant les oligonucléotides
antisens. Les études utilisant cette technique ont montré que des oligonucléotides à
phosphorothioate (OPT) dirigés contre MDM2 peuvent supprimer efficacement
l’expression de MDM2, permettant ainsi l’accumulation et l’activation de p53. Les
conséquences de cette activation ont été examinées dans plusieurs souris
xenogreffes modèles de cancer humain [17•]. Les observations ont confirmé une
suppression de la croissance des tumeurs, améliorée par la combinaison avec des
médicaments génotoxiques reconnus. De plus, d’une façon surprenante, toutes les
études impliquant l’utilisation d’oligonucléotides antisens ont démontré des effets de
suppression de la croissance des tumeurs, c’est à dire aussi bien chez les cellules
cancéreuses exprimant p53 sauvage que mutée. Ces observations compliquent
l’interprétation des résultats mais laissent penser qu’il existe une voie indépendante
de p53 liant à l’apoptose et activée par MDM2.
11
Figure 7: Structure et mode de liaison des inhibiteurs de MDM2. [13]A gauche, la structure cristallographique de MDM2 avec l’inhibiteur Nutlin-2A droite, recouvrement de la Nutlin-2 (Carbone en blanc, Azote en bleu, Oxygène en enrouge, et Bromine en marron) avec la chaîne peptidique Phe19, Trp23 et Leu26.
Figure 8: Analyse de l’inhibition de la liaison p53-MDM2 par Biacore. [13]

12
Un aspect important afin de valider l’interaction p53-MDM2 comme cible du cancer
est d’évaluer les conséquences de l’activation de p53 dans les tissus normaux
proliférant. Il a été montré que l’activation de la voie de p53 peut induire une forte
toxicité dans les tissus à fort taux de prolifération. Cette toxicité peut être causée par
les deux fonctions principales de p53 [14•], c’est à dire l’arrêt du cycle cellulaire et
l’apoptose, et peut ainsi limiter toute fenêtre thérapeutique impliquant l’activation de
p53.
Inhibiteurs pharmacologiques de l’interaction p53-MDM2
De nombreux efforts sont dirigés sur le développement d’antagonistes de
MDM2 pouvant se diriger directement sur la liaison protéine-protéine. Les premières
molécules chimiques ayant démontré une activité anti-tumorale, les « chalcones »,
peuvent se lier à la poche hydrophobique de MDM2 et activent la voie de p53 mais
causent également de nombreux effets indésirables, limitant leurs applications [9].
Des dérivés de ces molécules sont également a l’étude.
Plus récemment, des molécules chimiques, les « Nutlins », antagonistes de
l’interaction p53-MDM2 ont été identifiées par criblage d’une banque de molécules
synthétiques [13••]. Ce sont des dérivés de cis-imidazoline qui peuvent se lier
étroitement dans la poche p53 de MDM2 grâce a une forte similitude avec
l’interaction entre MDM2 et les 3 acides aminés critiques de p53 (figure 7).
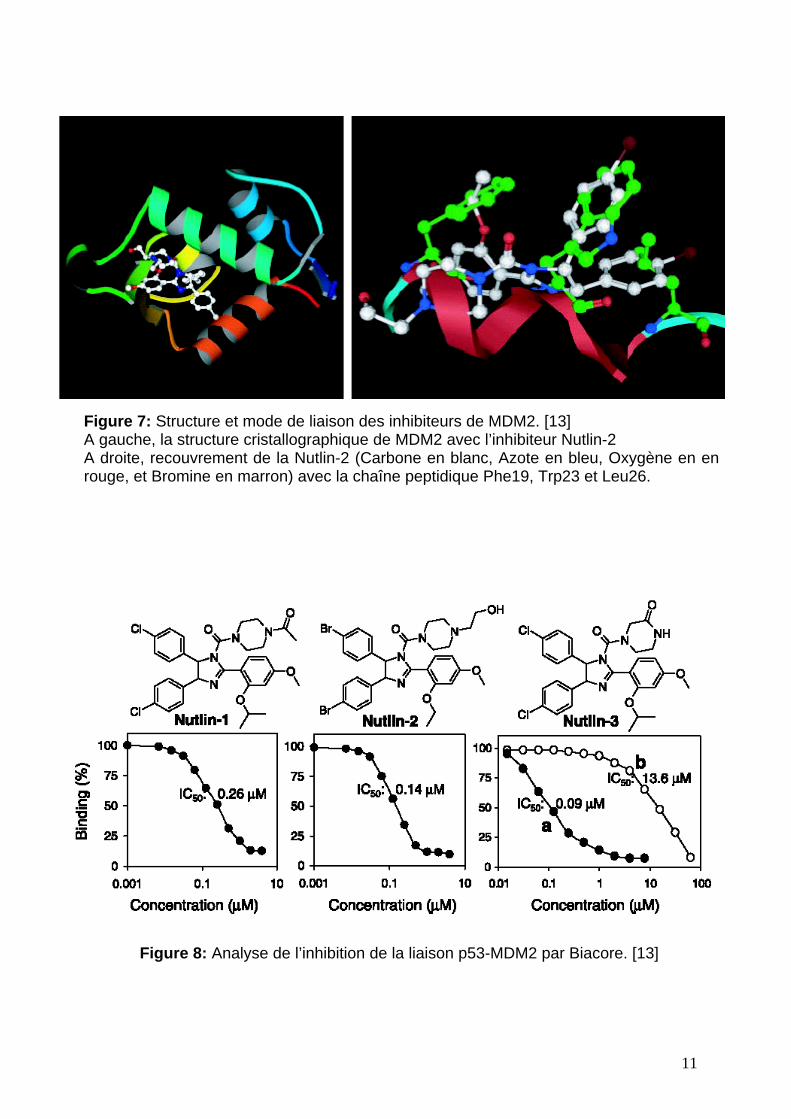
La capacité de liaison de ces composés est analysée par résonance
plasmonique de surface, grâce à l’équipement Biacore, dans un format de
compétition en solution (figure 8). Chaque composé est incubé, à une gamme de
concentrations différentes, avec une MDM2 humaine recombinante (exprimée dans
E.coli) puis injecté à la surface d’une puce où est fixée p53. p53 est obtenue par
expression dans E.coli et immobilisée grâce à un tag histidine. La liaison est
mesurée à l’équilibre et calculée par rapport au pourcentage maximum de liaison,
déterminé lui par l’interaction p53-MDM2 sans inhibiteur. Les résultats montrent que
ces composés délogent p53 de MDM2 pour une moyenne de concentration variant
de 100 à 300 nM. Ces imidazolines sont synthétisées en mélange racémique à partir
duquel les énantiomères peuvent être séparés par colonne chirale. Ceci a permis de
démontrer qu’il existe une différence d’activité entre les énantiomères de Nutlin, plus
13
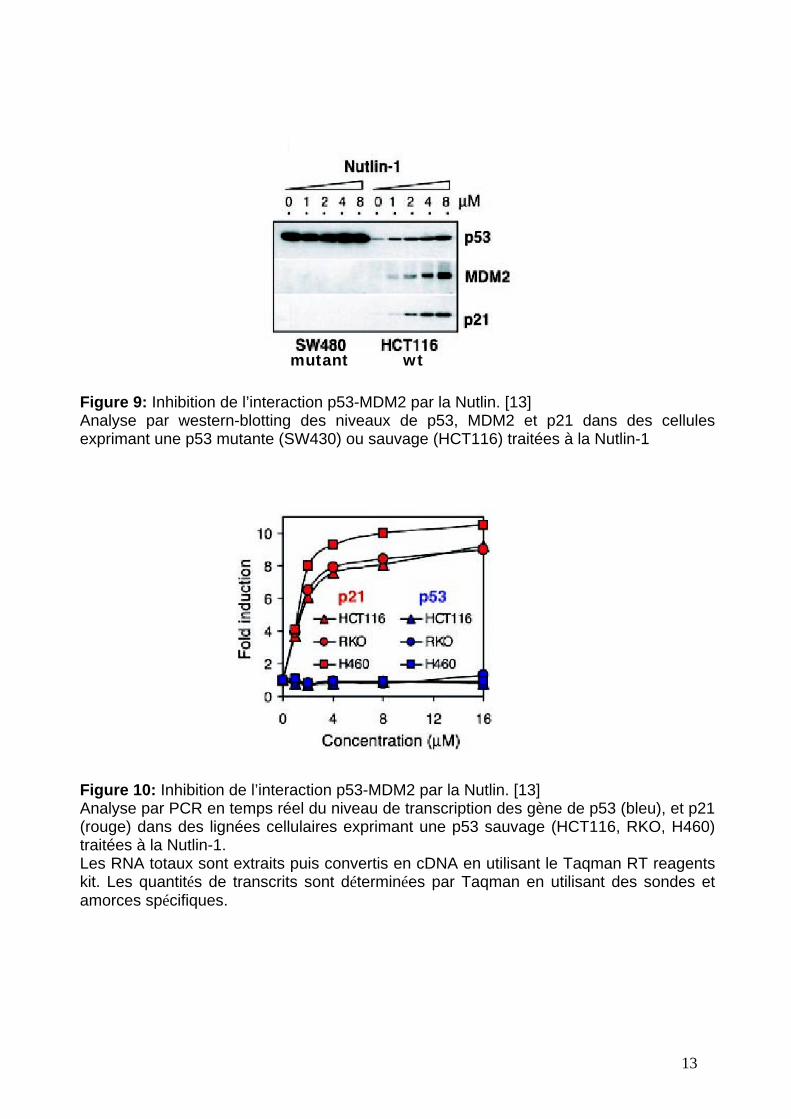
Figure 9: Inhibition de l’interaction p53-MDM2 par la Nutlin. [13]Analyse par western-blotting des niveaux de p53, MDM2 et p21 dans des cellulesexprimant une p53 mutante (SW430) ou sauvage (HCT116) traitées à la Nutlin-1
Figure 10: Inhibition de l’interaction p53-MDM2 par la Nutlin. [13]Analyse par PCR en temps réel du niveau de transcription des gène de p53 (bleu), et p21(rouge) dans des lignées cellulaires exprimant une p53 sauvage (HCT116, RKO, H460)traitées à la Nutlin-1.Les RNA totaux sont extraits puis convertis en cDNA en utilisant le Taqman RT reagentskit. Les quantités de transcrits sont déterminées par Taqman en utilisant des sondes etamorces spécifiques.
mutant wt

14
particulièrement pour la Nutlin-3 (figure 8c) où une différence de 150 peut être
observée entre ses deux formes.
Afin de déterminer si l’inhibition de l’interaction p53-MDM2 par ces analogues
d’imidazoline se traduit par l’activation de la voie cellulaire de p53, des cellules
cancéreuses exprimant p53 sauvage sont étudiées [13••] (HCT116 et RKO). Par
ailleurs, des lignées cellulaires exprimant une p53 mutante sont utilisées comme
contrôle négatif (SW480, MDA-MB-435, PC3). Tout d’abord les effets des inhibiteurs
de MDM2 sur les niveaux cellulaires de p53, MDM2 et p21 sont examinés (figure 9).
Il faut préciser que p21 est une cible de transcription majeure de p53 activée. Les
deux différentes lignées cellulaires, exprimant p53 mutée ou sauvage, sont incubées
pendant 8h avec de la Nutlin-1. Les lysats cellulaires sont ensuite analysés par
western-blotting en utilisant des anticorps spécifiques pour p53, MDM2 et p21. Les
résultats montrent que le traitement à la Nutlin-1 a généré une augmentation des
concentrations des trois protéines, dépendante de la concentration en inhibiteur chez
les cellules exprimant p53 sauvage. A l’opposé, les cellules exprimant p53 mutée ne
montrent aucun changement dans les concentrations en protéines, si ce n’est une
plus forte quantité initiale de p53. Les mêmes observations sont faites dans des
autres lignées cellulaires, en liaison avec le statut de p53. Ainsi, ces résultats
confirment que p53 sauvage s’accumule dans la cellule après un traitement à la
Nutlin-1 et provoque une augmentation des taux de MDM2 et p21, conformément au
cycle d’activation de la voie p53.
Afin de confirmer que l’accumulation de p53 est due à une inhibition de la
dégradation de la protéine plutôt qu’à une augmentation du niveau d’expression du
gène p53, l’expression des gènes p53 et p21 est mesurée par PCR en temps réel
(Taqman), dans 3 lignées cellulaires exprimant p53 sauvage et traitées à la Nutlin-1
(figure 10). Les résultats montrent que la transcription de p21 augmente de manière
dépendante de la concentration en inhibiteur et en accord avec l’augmentation de la
concentration en p53, son activateur. Par contre, la transcription du gène p53 n’est
pas affectée par le traitement à la Nutlin-1. Ces données indiquent que les Nutlins
régulent p53 par un mécanisme post-traductionnel. Il peut être remarqué qu’aucun
témoin négatif n’est utilisé pour cette expérience. Ceci peut s’expliquer par le fait que
la p53 mutée est inactive, et ne permettrait donc pas l’activation de la transcription de
p21, essentielle à l’expérience. Aussi p53 garderait elle-même sa concentration de
base et ne subirait pas de changement au niveau transcriptionnel.

15
Figure 11: Inhibition de l’interaction p53-MDM2 par la Nutlin. [13]Analyse de l’activité antiproliférative et cytotoxique de la Nutlin-1 mesurée par le test MTTdans des lignées cellulaires cancéreuses exprimant une p53 sauvage (bleu: HCT116,RKO, SJSA-1) ou mutante (jaune: MDA-MB-435, SW480). Les cellules sont traitéespendant 5 jours à différentes concentrations.
Figure 12: Activité antitumorale in vivo des inhibiteurs de MDM2. [13]Des souris nues (10 animaux par groupe de dose) portant des xénogreffes sous-cutanées de cancer humain (SJSA-1) d’un volume moyen de 185 mm3 reçoivent 200mg/kg d’une dose orale de Nutlin-3, 2 fois/jour ou une 10 mg/kg de doxorubicine enintraveineuse une fois par semaine pendant 3 semaines. Le volume des tumeurs estmesuré périodiquement pendant la durée du traitement.En carré noir: Nutlin-3 et carré blanc: contrôleEn rond noir: Doxorubicin et rond blanc: contrôle

16
Ensuite, l’effet de l’inhibiteur de MDM2 sur la croissance et la viabilité des cellules
cancéreuses est observé (figure 11). Cinq lignées cellulaires différentes, exprimant
p53 mutée ou sauvage, sont incubées avec de la Nutlin-1 pendant 5 jours. La
viabilité des cellules est mesurée avec le MTT assay. Cette méthode utilise une
coloration au 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, et peut
être mesurée par spectrophotométrie. Les résultats montrent une activité
antiproliférative et cytotoxique concentration-dépendante, et en corrélation avec le
statut de p53 dans les différentes lignées cellulaires. La valeur IC50 des cellules avec
p53 sauvage (1,4 à 1,8 µM) est sensiblement plus faible que celle des autres cellules
avec p53 mutante (13 à 21 µM), démontrant que la voie de p53 ne peut être activée
que dans les cellules exprimant la p53 sauvage.
Finalement, la capacité de ces inhibiteurs de l’interaction p53-MDM2 à
supprimer la croissance de tumeur xénogreffe établie dans des souris est étudiée
(figure 12). Pour cette expérience, la Nutlin-3 est utilisée ainsi que la lignée cellulaire
SJSA-1 d’osteosarcoma humain. La Nutlin-3 est bien tolérée par administration orale
à 200 mg/kg, deux fois par jour pendant 20 jours, permettant d’atteindre des
concentrations dans le plasma au-dessus de son IC90 déterminé dans la lignée
cellulaire correspondante (3,5 µM). Le traitement à la Nutlin-3 chez les souris portant
des tumeurs de 100 à 300 mm3 résulte dans 90% des cas à l’inhibition de la
croissance des tumeurs, en fonction du mode d’administration. De plus, les souris ne
présentent pas de perte de poids ou autre anormalité après la fin du traitement
[13••].
Conclusion
L’interaction entre le suppresseur de tumeur clé p53 et son régulateur négatif
MDM2 est un point d’intérêt dans la recherche de médicament contre le cancer
depuis une dizaine d’années. De nombreuses études ont prouvé le concept que la
disruption de cette interaction protéine-protéine peut activer la voie de p53 dans les
cellules cancéreuses. L’identification de molécules chimiques de synthèse
antagonistes de MDM2, a permis de réaliser des études in vivo et de renforcer la
notion de l’interaction p53-MDM2 comme une stratégie potentiellement viable pour
traiter le cancer. Cependant de nombreuses questions subsistent concernant le vrai

17
mécanisme de ces molécules pour la thérapie contre le cancer. Aussi, bien qu’au
moins 50% des tumeurs humaines retiennent leur p53 sauvage, et de ce fait doivent
être sensibles à une thérapie d’activation de p53, le taux de réponse peut être limité
par des problèmes intervenant à d’autres niveaux de la voie de p53. Actuellement, il
est reconnu que les tumeurs qui répondent le mieux à une thérapie par des
antagonistes de MDM2 sont celles possédant p53 et une amplification du gène
MDM2. Dans ces cellules, la surexpression de MDM2 est la seule aberration et donc
la restauration de la fonction de p53 doit permettre une forte réponse apoptotique.
Des études pré cliniques en cours se concentrent sur le rôle du statut de MDM2 et la
nature génétique des autres membres de la voie de p53 en réponse à l’activation de
p53.

18
Références
1. • Adams J. Proteasome inhibition: a novel approach to cancer therapy.
Trends in Molecular Medicine 2002, 8: No. 4 (Suppl.)
2. Adams J. The development of proteasome inhibitors as anticancer drugs.Cancer Cell 2003, 5: 417-421
3. • Adams J. The proteasome: structure, function, and role in the cell. Cancer
Treatment Review 2003; 29 (Suppl. 1): 3–9
4. Blaydes JP, Gire V, Rowson JM, Wynford-Thomas D. Tolerance of high levelsof wild-type p53 in transformed epithelial cells dependent on auto-regulation by mdm-2. Oncogene 1997, 14: 1859–1868
5. Bottger A, Bottger V, Sparks A, Liu WL, Howard SF, Lane DP. Design of asynthetic Mdm2-binding mini protein that activates the p53 response invivo. Curr Biol 1997, 7: 860– 869
6. Burger AM, Seth AK. The ubiquitin-mediated protein degradation pathway incancer: therapeutic implications. European Journal of Cancer 2004, 40: 2217–
2229
7. Geiger T, Husken D, Weiler J, Natt F, Woods-Cook KA, Hall J, Fabbro D.
Consequences of the inhibition of Hdm2 expression in humanosteosarcoma cells using antisense oligonucleotides. Anticancer Drug Des
2000, 15: 423–430
8. • Klein C, Vassilev LT. Targeting the p53–MDM2 interaction to treat cancer.
British Journal of Cancer 2004, 91: 1415 – 1419
9. Kumar SK, Hager E, Pettit C, Gurulingappa H, Davidson NE, Khan SR. Design,synthesis, and evaluation of novel boronic–chalcone derivatives asantitumor agents. J Med Chem 2003 46: 2813–2815
10. Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, Pavletich
NP. Structure of the MDM2 oncoprotein bound to the tumor suppressortransactivation domain. Science, 274: 948-953
11. •• MacLaren AP, Chapman RS, Wyllie AH, Watson CJ. p53-dependent
apoptosis induced by proteasome inhibition in mammary epithelial cells.Cell Death and Differentiation 2001, 8: 210-218

19
12. Pickart CM. Mechanisms underlaying ubiquitination. Annual Review of
Biochemistry 2001, 70: 503-533
13. •• Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N,
Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. In Vivo Activation of the p53Pathway by Small-Molecule Antagonists of MDM2. Science 2004, 303: 844-
848
14. • Vousden KH, Lu X. Live or let die: the cell’s response to p53. Nature
Reviews Cancer 2002, 2: 594-604
15. Wasylyk C, Salvi R, Argentini M, Dureuil C, Delumeau I, Abecassis J, Debussche
L, Wasylyk B. p53 mediated death of cells overexpressing MDM2 by aninhibitor of MDM2 interaction with p53. Oncogene 1999. 18: 1921–1934
16. Williams SA, McConkey DJ. The Proteasome Inhibitor Bortezomib Stabilizes aNovel Active Form of p53 in Human LNCaP-Pro5 Prostate Cancer Cells.Cancer Research 2003, 63: 7338–7344
17. • Zhang Z, Wang H, Agrawal S, Zhang R. Antisens therapy targeting MDM2
oncogene in prostate cancer: Effects on proliferation, apoptosis, multiplegene expression, and chemotherapy. PNAS 2003, 100: 11636-11641