Embed Size (px)
Citation preview

JULES ADÉ
CLONAGE ET CARACTÉRISATION D'UN GÈNE DE CORRECTION DES MÉSAPPARIEMENTS D'ADN
(AtMSH2) CHEZ ARABIDOPSIS THALIANA (Lm) HEYNH.
These presentde
à la Faculte des études supérieures de l'Université Laval
pour l'obtention du grade de Philosophiae Doctor (Ph.i).)
Département de Phytologie FACULTÉ DES SCIENCES DE L'AGRICULTURE ET DE L'ALIMENTATION
UNIVERSITE LAVAL QUÉBEC
O Jules Ad&, 1999

National Library Bibliothèque nationale du Canada
Acquisitions and Acquisitions et Bibliographie Services services bibliographiques
395 Wellington Street 395, nm Wellington ûttawaOfU K 1 A W OItawaON K 1 A W Canada CaMda
The author has granted a non- L'auteur a accordé une licence non exclusive licence aiiowing the exclusive permettant à la National Library of Canada to Bibliothèque nationale du Canada de reproduce, loan, distn'bute or seiI reproduire, prêter, distribuer ou copies of this thesis in microfoxm, vendre des copies de cette thèse sous paper or electronic formats. la forme de microfiche/film, de
reproduction sur papier ou sur format électronique.
The author retains ownership of the L'auteur conserve la propriété du copyright in this thesis. Neither the droit d'auteur qui protège cette thèse. thesis nor substantial extracts fiom it Ni la thèse ni des extraits substantiels may be printed or otherwise de celle-ci ne doivent être imprimés reproduced without the author's ou autrement reproduits sans son permission. autorisation,

RÉSUMÉ COURT
Cette étude nous a permis de cloner et de caractériser le gbne AtMSH2, une composante importante du système de correction des mésappariements d'ADN chez Arabidopsis. C'est un gène simple copie situé sur le chromosome III dVArabidopsis et dont l'expression est si faible qu'elle n'a et6 ddtectable que dans des suspensions cellulaires en croissance mitotique exponentielle. L'expression de AtMSH2 chez E. coli a fourni des &idences suggerant que ce gène est bel et bien implique dans la correction des m6sappariements. En effet, cette expression entraîne un phénotype mutateur et fait apparaître une activité de liaison préférentielle B l'ADN m6sappari6. Finalement, cette Btude nous a permis de caractériser la premier9 famille de transposons Foldback chez Arabidopsis, dont un des membres est insert3 dans la région 3' de AtMSH2 chez certains écotypes.

RÉSUMÉ LONG
Au cours de la réplication de I'ADN et de la recombinaison génétique, des mésappariements d'ADN ont lieu et doivent être corrigés. Sinon, ces erreurs entraîneront des mutations qui seront transmises aux générations suivantes. Des systèmes de correction des mésappariements ont été identifiés chez une large gamme d'organismes allant des procaryotes aux eucaryotes. Plusieurs études suggèrent que les composantes et les mécanismes de correction des mesappariements ont et6 hautement conservés au cours de l'évolution.
Chez les eucaryotes, en plus de jouer un rôle primordial dans la correction des dommages causes a I'ADN, le gbne MSH2 (MutS Homologue-2) serait implique dans le contrdle de la specificite des Bvdnements de recombinaison. Spdcifiquement, l'inactivation de ce gbne facilite les Bchanges entre espbces apparentdes. En am6lioration des plantes, un accroissement de ce type d'&change génétique constituerait un gain appréciable car il permettrait de faciliter grandement l'exploitation de la biodiversitd v6gétale.

Dans le but de mieux comprendre la correction des mésappariements d'ADN et la recombinaison g6n6tique chez les plantes, nous avons cloné le premier homologue végétal de MSW, celui d8Arabidopsis thaliana. Les clones d'ADNc et genomique ont et6 isolés. Une forte identite existe entre le gène d8Arabidopsis et les gènes MSH2 clonds chez d'autres eucaryotes. AtMSU2 est un gbne simple copie, situ6 sur le chromosome 3 d8Arabidopsis et exprime à faible niveau dans les jeunes plantules.
Notre deuxibme objectif &ait d'étudier la fonction de ce gbne. Pour ce faire, nous avons pu montrer que l'expression de la p ro the AtMSH2 dans des bactéries engendre un phénotype mutateur (accroissement du taux de mutations spontanées). Cette observation indique que la proteine vdg6tale est fonctionnelle et qu'elle interfere avec le système bactérien de correction des mésappariements. D'autre part, nous avons également montre sur gel de retardement que la prot6ine AtMSH2 a une affinité plus grande pour l'ADN mésapparid, une caractéristique qui est tout à fait conforme à son rôle dans la reconnaissance des rnésappariements.
Dans un troisibme volet, des comparaisons de sequences entre les allèles AtMSH2 chez les ecotypes Landsberg erecta et Columbia ont révW la présence d'un élément transposable 196 pb au-delà du codon stop chez Landsberg erecta. Une fouille informatique dans les banques de sequences nous a permis d'identifier six nouveaux éléments de la même famille. Cette famille que nous avons nommée ~~Hairpinm constitue la première famille des aFoldback Transposonsm chez Arabidopsis thaliana. Ils sont présents en 5 à 10 copies dans différents écotypes d'Arabidopsis. Finalement, nous avons ddmontré que leur mobilité dans le génome dlArabidopsis est recente.
En conclusion, les rbsultats de cette Btude sugghrent que le processus de correction des m6sappariements d'ADN est conservé chez les plantes. De plus, l'apparente conservation de fonction permet d'espérer que des modifications apportées à ce système faciliteront I'introgression de ghnes en provenance d'espèces sauvages.

AVANT-PROPOS
J'aimerais exprimer ma profonde reconnaissance mon directeur de these, le professeur François J. Belrile pour ses directives, ses conseils judicieux et constructifs qui ont permis de compldter cette thhe. Je le remercie également pour son soutien moral, sa bienveillance et son enthousiasme constant.
J'aimerais remercier Dr. Marie-Pascale Doutriaux pour sa collaboration dans le cadre de ce projet. et pour m'avoir permis de sbjourner dans son laboratoire B l'Institut de Biotechnologie des Plantes (Univenit6 Pans-Sud, France). J'ai été très touché par son hospitalit6 au cours de mon séjour.
Je remercie Dr. Serge Laberge qui, malgr6 toutes ses occupations a effectué la pré- lecture de cette these en vue de son amdlioration. J'aimerais remercier aussi Dr. Armand Seguin et Dr. Gregory G. Brown pour avoir accepté d'évaluer cette thèse. Mes remerciements vont également au professeur Daniel Dostaler pour tout le soutien qu'il m'a apporte depuis mon entrée dans le programme jusqu' à cette phase finale.
Je remercie tous mes coll&gues de laboratoire pour cet esprit d'équipe qui a toujours été la <arbgle d'or. primant sur toute autre considération.
Je tiens à remercier le Programme Canadien de Bourses de la Francophonie pour le financement de mes études gradubes.
J'adresse mes sincares remerciements B mes parents qui n'ont ménagé aucun effort pour m'apporter leur soutien indefectible pendant toute la durée de ma formation.
J'aimerais rendre un hommage exceptionnel & mon epouse Florence et à mes enfants Jaurbs et Marlbne. Vos sacrifices sont inestimables! Que ce travail soit pour vous une compensation symbolique.
Enfin, que tous ceux qui m'ont entour6 pendant certains moments pénibles trouvent ici l'expression de ma profonde gratitude.

Je dédie cette thèse à:
- mon épouse Florence - mon fiis Jaurès et ma fille Marfène
Pour toutes ces années de séparation et de sacrifice

vii
TABLE DES MATIÈRES
. . RÉSUMÉ COURT ................................................................................................................. II
... RÉSUMÉ LONG .................................................................................................................. III
................................................................................................................. AVANT-PROPOS v . . ................................................................................................... TABLE DES MATI~RES VII
LISTE DES TABLEAUX ...................................................................................................... x ........................................................................................................ LISTE DES FIGURES xi
... USTE DES ABR~/IATIONS ........................................................................................... XIII
CHAPITRE I : REVUE DE LITTÉRATURE .................................................................... 3
1 .l Introduction ................................................................................................................ 4 1.2 Correction des m6sappariements chez les procaryotes : le
systhme MutHLS de E.coli. ................................................................................... 5 1.2.1 Modele de correction des m6sappariements chez E. coli ......................... 5 1.2.2 Phénotypes des mutants ................................................................................. 8
1.3 Correction des mesappariements chez les eucaryotes. ................................... 9 1.3.1 Homologues eucaryotiques de MutS ............................................................ 9 1.3.2 Homologues eucaryotiques de MutL ............................... : .......................... 1 2 1.3.3 Mécanisme de correction des mesappariements chez les
....................................................................................................... eucaryotes -1 3 1.4 Correction des m6sappariements et recombinaison génétique ................... 15 1.5 Hypothbses de recherche et objectifs ............................................................... 1 8
CHAPITRE II : Four mismatch repair paralogues coexist in Arabidopsis
thaliana: AtMSH2, AtMSH3, AtMSH6- 1 and AtMSHô-2.. ............... .2 0
Résum4 du manuscrit ................................................................................................... 23
Abstract ........................................................................................................................... 24
2.1 Introduction .............................................................................................................. 25 .......................................................................................... 2.2 Materials and methods 28

2.2.1 Growth of ceIl suspension ............................................................................. 28 2.2.2 RNA isolation and Northem blot analysis ................................................... 28
................................ 2.2.3 Genomic DNA isolation and Southern blot analysis 28 2.2.4 Radiolabeled probes ...................................................................................... 28 2.2.5 Reverse transcription and ?CR .................................................................... 29
............................................................................ 2.2.6 Isolation of AtMSH2 cDNA 29 ........................ 2.2.7 Isolation of the AtMSH3 and AtMSHô cDNA sequences 30
2.2.8 Isolation of AtMSH3 cornplete coding sequence ...................................... 30 ............................ 2.2.9 Isolation of the AtMSH6-2 complete coding sequence 30
2.2.10 Oligonucleotides ........................................................................................... 31 2.2.1 1 Mapping of AtMSH2 and AtMSH6-2 ......................................................... 32 2.2.1 2 P hy logenetic analyses ................................................................................. 32
2.3 Results ............................................................................................................ 3 3 ......................... 2.3.1 Isolation of the AtMH2. AtMSH3 and AtMSH6-2 cDNAs 33
2.3.2 Genetic mapping ............................................................................................. 38 2.3.3 Deduced amino acid sequence comparison ............................................. 38 2.3.4 Expression studies ......................................................................................... -42
2.4 Discussion ............................................................................................................... 44
...................................................................................................... Acknowledgements 47
CHAPITRE III : Functional analysis of the Arabidopsis thaliana rnismatch
............................................................................... repair gene MSH2 -54
.................................................................................................. Résumé du manuscrit -56 Abstract ........................................................................................................................... 57 3.1 Introduction .............................................................................................................. 58 3.2 Materials and methods .......................................................................................... 61
................................................................................................. 3.2.1 fluctuation test 61 3.2.2 Expression and partial purification of AtMSH2 protein ............................ 62 . . ......................................................................................... 3.2.3 DNA Bnding assay 62
......................................................................................... 3.3 Resutts and discussion -64 3.3.1 AtMSH2 protein is mutagenic in E . cdi ....................................................... 64 3.3.2 Expression of AtMSH2 protein ..................................................................... 66 3.3.3 Mismatch affhity of recombinant AtMSH2 protein .................................... 67

ix
References ..................................................................................................................... 70
CHAPITRE IV : Hairpin Elements. the fint family of Foldback Transposons
............................................................. (FTs) in Arabidopsis haliana 74
.................................................................................................. Résume du manuscrit. 77 Summary ........................................................................................................................ 78 4.1 Introduction .............................................................................................................. 79
...................................................................................................................... 4.2 Results 81
4.2.1 The Landsberg erecta allele of the AtMSH2 gene contains a transposon-like sequence in its 3' region .............................. 81
4.2.2 Hairpin-1 is a member of a family of Foldback ........................................................... transposons in Arabidopsis thaliana 81
4.2.3 Hairpin elements are present in low copy number in the ...................................................................................... Arabidopsis genome 83
4.2.4 Hairpin elements are useful indicators of the phylogenetic .................................................................. relationships between ecotypes -87
4.3 Discussion ............................................................................................................... 90 ..................................................................................... 4.4 Experimental procedures -93
4.4.1 Plant material ................................................................................................... 93 4.4.2 PCR amplification ............................................................................................ 93
................................................ 4.4.3 DNA isolation and Southem hybridization -93 4.4.4 Radiolabelled probe ....................................................................................... 94
4.4.5 DNA sequence analysis ................................................................................ 94 ......................................................................................................... Acknowledgments 94
..................................................................................................................... References 95
CHAPITRE V : DISCUSSION GÉNÉRALE ET CONCLUSION ................................ 97
LISTE COMPL~TE DES OUVRAGES CITES .......................................................... 1 02

LISTE DES TABLEAUX
CHAPITRE II:
Table 1: Percent amino-acid identity of the hurnan, yeast and Arabidopsis ................................................................... Msh2, Msh3 and Msh6 sequences .39
CHAPITRE III:
.................... Table 1 : Effect of AtMSH2 gene expression on mutation rates in E. coli 65
CHAPITRE IV:
................................................................ Table 1 : Characteristics of H a m elements.. -85

LISTE DES FIGURES
CHAPITRE 1:
Figure 1 : Modele de correction des mésappariements chez E. coli .............................. .6
Figure 2: Interaction de MSH3 ou MSH6 avec MSHP au coun de la correction des me3sappanements .................................................................. 1 1
Figure 3: Modele de correction des mesappariements chez les eucaryotes.. .......... .14
Figure 4: Mesappariements générés au cours de la recombinaison ........................... 16
CHAPITRE II:
Figure 1 : Amino-acid alignment of the human, yeast and Arabidopsis Msh2, Msh3 and Msh6 C-terminal conserveci regions ................................... 34
Figure 2: Polymorphisms between the Landsberg erecta and ............................................................................... Columbia alleles of A t m 2 36
Figure 3: Southem blot analysis of the genomic AtMSH3 ............................................................................................ and AtMSW-2 loci.. ..37
Figure 4: Phylogenetic analysis of the 197 aligned amino-acid from the conserved region of al1 available Msh2,
................................................................................ Msh3 and Msh6 sequences 41
Figure 5: Northem blot analysis of the MSH genes expression ................................................................... in Arabidopsis suspension cuitures 43

CHAPITRE III:
figure 1: Mismatch binding activity of the AtMSH2 protein ............................................ 68
CHAPITRE IV:
Figure 1 : Multiple sequence alignment of Hairpîn elements ......................................... 82
Figure 2: Predicted DNA secondary structure of the consensus Hairpin sequence (a) and the Hairpin-1 element (b) ..................................................... û4
............... Figure 3: Copy nurnber of Haitpin elements in four ecotypes of A . thaliana 86
Figure 4: Sunrey of different Arabidopsis thaliana ecotypes for the presence of Haîtpin- 1 at the AtMSH2 locus .................................................... -89

LISTE DES ABRÉVIATIONS
ADN, DNA ADNc, cDNA ARN, RNA ARNm, mRNA ATP CAPS BAC
bp, pb cm CM CTAB OMS0 dNTP EDTA FT
g fmole g/l; g 1-1 GM h HTH IPTG IR IR-IO IR-OD Kcal mol-1 kD kb 1 LB MMR
mg
acide désoxyribonucleique ADN compl6mentaire acide ribonucl6ique ARN messager adenosine 5'-triphosphate cleaved amplified polymorphic sequence chromosome artificiel de bactérie paire de bases centimètre centiMorgan bromure d'hexad6cyltrimethyl-ammonium sulfoxide de dim6thyl
desoxy nbonucléotide triphosphate acide éthyl8nedinitrilotétraacetique foldback transposon force gravitationnelle
femtomoles gramme par litre milieu de germination heure hélice-tour-hdlice isopropyl thyogalactoside répétition invers& (inverted repeat)
domaine ii??eme des IR (IR-inner domain) domaine externe des IR (IR-outer domain) kilocalories par mole kilodaltons kilobases litre milieu Luria-Bertani mismatch repair milligramme

min MITE m l M L m PA MF NAA
ng N J NTP PAGE PCR pmole PMSF RACE RFLP R I rRNA S D S sec S M S S C TBE
minute miniature inverted-repeat transposable element millilitre maximum Iikelihood millirnolaire maximum parsimony acide naphtalene acétique nanogramme neighbor joining nucl6otide triphospahte polyacrylamide gel electrophoresis polymerase chain reaction picornole phenylmethylsulfonil fluoride rapid amplification of cDNA end restriction fragment length polymorphism recombinant in bred ARN ribosomique dodécyl sulfate de sodium seconde milieu de suspension tampon citrate de sodium Tris-borate degr6 celsius micro Curie microgramme microlitre micromolaire

INTRODUCTION GÉNÉRALE
La recombinaison gh6tique est un processus qui permet les échanges d'informations gh8tiques. Elle joue un rôle très important en amélioration des plantes puisqu'elle g&nbre de nouvelles combinaisons de g h e s dont les plus intéressantes sont retenues pour les besoins d'une agriculture plus performante. En effet, il est quasi-impossible de retrouver dans une meme espace v6g6tale. toutes les caractéristiques 'idéales' pouvant assurer, de façon stable et continue, une production maximale tout en faisant face à toutes les adversites du milieu. Ainsi, a-t- on souvent recours à des croisements dans le but de géndrer de nouvel:es combinaisons de gènes rendant les nouveaux cultivars plus performants que les parents. Dans cette optique, des esphces sauvages ayant des caractéristiques intéressantes sont parfois croisées avec des espèces d'intérêt agronomique afin de créer des hybrides intersp6cifiques. Mais malgr6 les progrès marqués qui ont été faits en matibre de production d'hybrides interspbcifiques, la faible fréquence (ou l'absence) d16changes g&n&iques entre chromosomes homeologues (séquences similaires mais non identiques) repr6sente un facteur limitant dans l'exploitation de cette précieuse biodiversité génétique. En effet, elle rend difficile une introgression efficace de gènes en provenance d'espbces sauvages. L'un des défis à relever par la science dans ce domaine est de faire sauter, selon les besoins, cette barrière à la recombinaison entre espèces vdg6tales apparentbes.
Aussi bien chez les procaryotes que chez les eucaryotes, il a et6 clairement mis en évidence ces dernieres anndes, une corrélation Btroite entre l'altération du système de correction des m6sappariements d'ADN et la perte de la sp6cificité des événements de recombinaison g&n&ique (Rayssiguier et ab, 1989; Selva et al., 1995; de Wind et al., 1995). En d'autres termes, l'inactivation de ce systeme entraîne une augmentation de recombinaison entre séquences d'ADN homéologues.
Or, eu Bgard B ce qui pr&c&de, la possibilit6 de moduler la sp&cificitd des événements de recombinaison pourrait avoir un impact trbs significatif en

amélioration des plantes. Notamment. l'inactivation du systbme de correction des mésappariements d'ADN chez les plantes pourrait favoriser des Bchanges génétiques chez les hybrides intersp6cifiques en modulant la spécificité des interactions intergénomiques. Cela faciliterait Itintrogression de gbnes sans l'introduction de caractères indesirables, phénomène connu sous le terne de alinkage draga (Young et Tanksley, 1989). A long terme, une caractérisation moléculaire du systhne de correction des mesappariements d'ADN pourrait même permettre d'envisager la possibilit6 de stimuler ou de réprimer la recombinaison géndtique dans des regions chromosomiques d1int6rét, ou de ddvelopper des systèmes efficaces de recombinaison homologue chez les plantes. Mais malgr6 ces applications potentielles en amMoration des plantes, au moment où ces travaux ont été inities, aucun des gènes impliques dans la correction des mesappariements d'ADN n'avait fait l'objet d16tude chez les plantes.
La présente étude a donc ét6 initide dans le but d'acquérir une meilleure compréhension du processus de correction des mésappariements d'ADN et de la recombinaison chez les plantes, en particulier chez Arabidopsis thaliana.

CHAPITRE I
REVUE DE LITTÉRATURE

1.1 Introduction
La synthèse de I'ADN, bien qu'étant un mécanisme trhs hautement precis, demeure néanmoins un processus imparfait. En effet. des mesappariements de bases surviennent quotidiennement dans I'ADN de tout organisme comme le résultat de plusieurs mécanismes, notamment les erreurs au cours de la replication de I'ADN, les dommages causds h I'ADN par des agents physiques ou chimiques, la deamination spontan6e de la 5-methylcytosine et la recombinaison génétique entre séquences d'ADN similaires mais non identiques. Si ces erreurs ne sont pas corrigées avant la prochaine ronde de réplication, elles sont fixées dans le génome et transmises aux g6n6rations suivantes, compromettant ainsi la stabilite du génome. Beaucoup de ces alt6rations bloqueraient la transmission de l'information génétique à la géneration suivante. D'autres erreurs, si elles n'étaient pas corrigées, se perpétueraient dans le génome de la descendance et produiraient des changements inacceptables dans les protéines et les enzymes nécessaires au maintien de la vie cellulaire.
L'une des évidences indiquant l'importance de la correction des mésappariements d'ADN est l'acquisition, au cours de I'evolution, de la fonction 3'05' exmuctéase de I'ADt-4 polymerase permettant l'élimination et le remplacement des bases rnésappariees au cours de la réplication par le processus d'édition. En dépit de cette capacité de I'ADN polymérase, certaines erreurs introduites au cours de la réplication de I'ADN lui échappent. A ces erreurs. il faudra ajouter celles liées aux processus autres que la réplication. Les cellules ont alors élabore une machinerie complexe pour minimiser t'effet de de ces dégâts causés à I'ADN. Les systèmes de correction des mésappariements ont été identifiés chez une large gamme d'organismes (Modrich, 1991 ; Reenan et Kolodner, 1 992a; Varlet et al., 1994). Cependant, l'organisme chez lequel le mecanisme de correction des rnésappariements d'ADN a et6 le plus caracteris6 est la bacterie E. coli (Su et Modrich, 1986).

1.2 Correction de8 m6sappiriements chez les procaryotes : le systbme MutHLS de Ecoli.
Le système de correction des mesappariements d'ADN est connu comme jouant deux rôles majeurs dans la cellule. D'une part, il assure la correction des erreurs survenues au cours de la r4plication de I'ADN et d'autre part, il ernpache la recombinaison entre sdquences d'ADN divergentes (Modnch et Lahue, 1996).
1.2.1 Modhle de correction des mbappariements chez E. coli.
Le système MutHLS de E. COS est le mieux caracteris6 de tous les systbmes de correction des mbsappariements et a pu être reconstitué in vitro a partir de ses composantes purifides (Lahue et al., 1989; Modrich, 1991). Chez E. coli, les composantes centrales de reconnaissance des mesappariements sont MutS, un polypeptide de 97 kD qui reconnaît les m4sappariements in vitro et MutL, un polypeptide de 70 kD qui se dimérise et interagit avec MutS pour accroître la stabilité du complexe MutS-ADN. La formation du complexe MutS-MutL-ADN est indispensable pour activer la protéine MutH, une endonucléase à spécificité monocatenaire de 25 kD, qui reconnaît et coupe le brin nouvellement synthétise qui, par définition, contient les erreurs de réplication. Les principales étapes de ce processus sont présentdes à la Figure 1.
Dans tous les cas, le processus de correction des mésappariements requiert en plus des trois prot6ines majeures (MutS, MutL et MutH), sept autres protéines (Modrich, 1991) : I'ADN hélicase II, fa protéine de liaison a I'ADN simple brin (SSB), I'exonucléase 1, I'exonucl6ase VII, I'exonuclease RecJ, I'holoenzyme de I'ADN polymérase III et I'ADN ligase. L'intervention de ces sept prot6ines est restreinte aux étapes de l'excision et de la resynthbse de I'ADN (Figure 1). L'ATP est indispensable comme cofacteur. La region excisée peut varier de quelques centaines de bases B 1 kb et parfois davantage (Modrich, 1991); d'ou le qualificatif de 4ong patch mismatch repairm (correction de longs segments de mésappariements) associe B ce systbme. Enfin, les étapes de resynthese et de ligation viennent achever le processus.
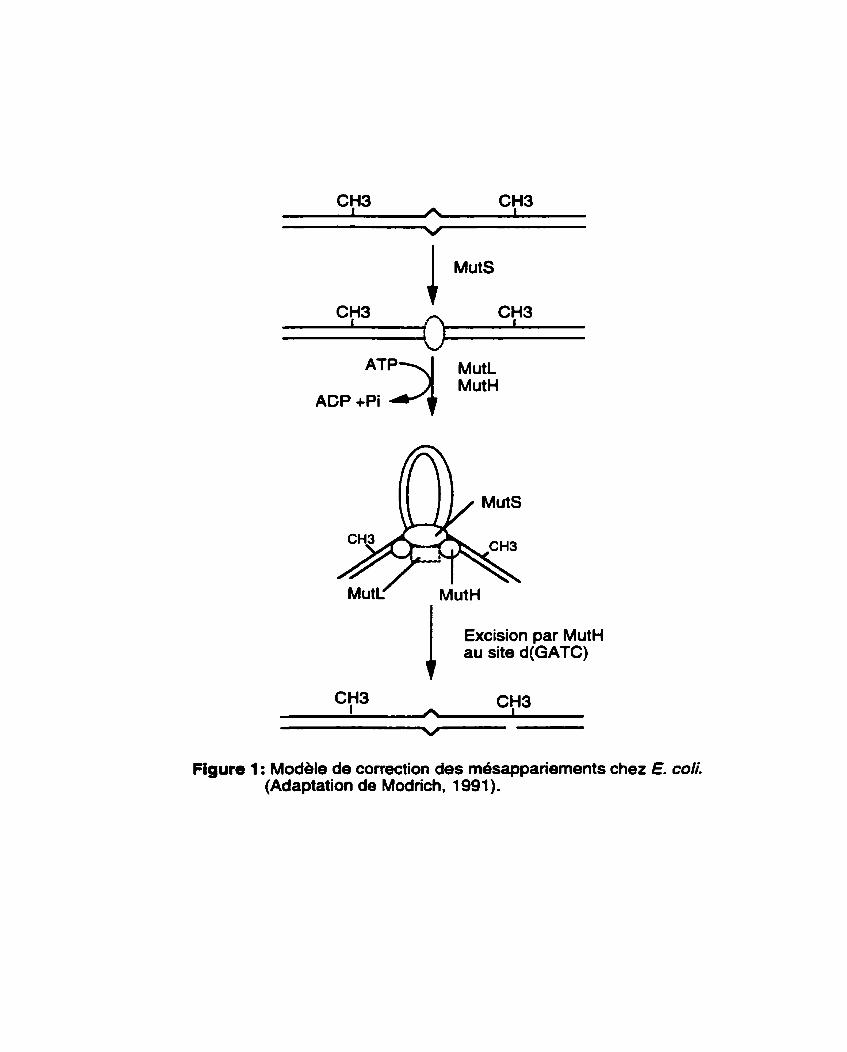
ACP +Pi *"q Ma
1 Excision par Mut" au site d(GATC)
Figure 1 : Modele de correction des mdsappanements chez E. d i . (Adaptation de Modrich, 1 991).

Pami toutes les protdines irnpliqudes dans la correction de t'ADN, seule MutS est capable d'une interaction specifique avec I'ADN en absence de toutes les autres protéines, expliquant ainsi le grand int6rêt qu'a suscite 1'6tude de cette proteine (Modrich, 1991). MutS se lie activement B I'ADN rn6sapparie en absence de cofacteurs (Su and Modrich, 1986) mais en prdsence de I'ATP, elle promouvoie la formation de structures en boucle a (Figure 1). Haber et Walker (1991) ont montre que I1alt&ation du domaine de liaison a I'ATP de MutS résulte en un phhotype rnutateur. Quant au produit du g h e Mut#, il est responsable de la reconnaissance et de la spécificitb de l'excision du brin B corriger (Kramer et al., 1984; Lahue et al., 1989; Langle-Ronault et al., 1987). Contrairement a MutS et MutH, aucune activité individuelle n'est attribue0 B MutL bien qu'elle interagit avec MutS et est requise pour l'activation de MutH. L'une des hypothèses avancées est que MutL agirait comme une interface protéine-proteine entre MutS et MutH.
La reconnaissance du nouveau brin en vue de la correction des erreurs de réplication est possible grâce B la méthylation de l'adénine au niveau des séquences GATC. Se rdfdrant a cette methylation. le système MutHLS de E. coliest qualifié de méthyl-ddpendant~. Ainsi, les portions d'ADN portant des séquences GATC méthylées au niveau des deux brins ne sont pas un bon substrat pour la correction (Hennan et Modrich, 1981 ; Marinus et al., 1984). Puisque la m6thylation de I'ADN est une modification post-replicationnelle, le nouveau brin est temporairement à I'Btat non méthyle et c'est cette absence transitoire de methylation qui cible la correction au nouveau brin. Des études récentes ont montré que la présence d'une simple sequence GATC hémimethylée est suffisante pour déterminer la specificitb du brin h corriger (Modrich, 1991 ; Chi et Kolodner, 1994a). D'autres groupes de recherche (Nicolaides et al., 1994; Horii et al., 1994) ont montre qu'une coupure pr6-existante sur un brin est suffisante pour déterminer la specificité de la correction des m6sappariements. Le rôle de MutH est donc principalement de fournir cette coupure monocat6naire au bon endroit (sur le brin hémirn6thyl6 B l'endroit d'un GATC).
Le systeme MutHLS reconnaît et comge toutes les bases simples mesappariees à l'exception de C-C. L'efficacitd de la correction depend de la nature du mesappariement et peut être influencée Bgalement par le contexte dans lequel se trouvent les mhappariements (Jones et al., 1987). Parmi les 8 types de rn&appariements, seul C-C est refractaire au systeme MutHLS. G-T, A-C, A-A et G- G sont de bons substrats alors que TT, T-C et A-G sont conigds avec une efficacit6

variable qui depend du contexte de la sequence (Dohet et al. 1985; Fazakerley et al., 1986; Jones et al., 1987. Su et al., 1989). De plus, ce systeme corrige les petites insertions/def6tions non appariees, mais il ne peut pas reconnaître efficacement celles qui ont plus de 4 bases non-appari6es.
1.2.2 Phhotypes des mutants
L'inactivation de MutH, MutL ou MutS donne lieu à un accroissement de 100 à 1000 fois du taux de mutations spontanees de la cellule (COX, 1976). Cet accroissement est comparable au taux d'erreurs de replication de l'holoenzyme de I'ADN polymérase observe in vitro (Fersht et Knill-Jones, 1981 ; Loeb et Kunkel, 1982). La majorité des mutations accumulees dans ces souches mutantes sont des transitions et des delétions d'une base, auxquelles s'ajoute un faible pourcentage de transversions (Choy et Fowler, 1985; Leong et al., 1986; Rewinsky et Marinus, 1987). Cette gamme de mutations reflhte bien les erreurs d'incorporation de I'ADN poiymérase au cours de la replication.
En plus de ce phenotype mutateur, les cellules déficientes pour ce système sont tres sensibles aux agents alkylants (René et al., 1988; Rydberg, 1978). En effet, de tels agents (par exemple fa nitrosamine) transfhnt un groupement méthyle ou éthyle à des positions critiques de certains nucl6otides. les obligeant a contracter une liaison hydrogène supl6rnentaire. II en résulte un appariement avec une base induite. De telles cellules montrent aussi une instabilité des microsatellites qui résulterait d'un ccderapagem de la polymérase au cours de la PCR. Ce dérapage engendre autour d'une forme majoritaire n unités de r6pétitions1 des formes a nkl, n e , nk3, etc. unites de rbpétitions. Les distorsions ainsi causees sur le nouveau brin sont en principe corrigees par le systhme de correction des mesappariements de la cellule en vue d'assurer la stabilit6 dans la longueur des microsatellites. En l'absence d'un systbme de correction fonctionnel, ces derapages ne sont pas corrigés et il en résulte une instabilite des microsatellites. Les mutants du systhme de correction des m6sappariements presentent 6galement un accroissement de la frequence de recombinaison homéologue (Rayssiguier et ai., 1989). Ce dernier point sera abordé en detail plus tard dans ce chapitre.

1.3 Correction de8 m61sapparlements chez 1.8 eucaryotes.
Eu égard de l'importance du systhme de correction des m6sappariements dans la physiologie de la cellule, il serait surprenant que cette machinerie ne soit pas conservde chez les eucaryotes. Aussi, des gbnes impliques dans la correction des mésappariements d'ADN ont Btd identifiés chez plusieurs eucaryotes (Kramer et ab, 1989a; Reenari et Kolodner, 1992a; Fishel et al., 1993; Leach et al., 1993; New et al., 1 993; Bronner et al., 1 994; Papadopoulos et al., 1 994).
1 .3.l Homologues eucaryotiques de MutS
Chez les eucaryotes, la duplication de gènes et la sp6cialisation de fonction ont conduit à la présence de plusieurs homologues de MutS. Par exemple chez la levure, six homologues de MUtS (MSHI, MSH2, MSH3, MSH4, MSHS et MSH6; Reenan et Kolodner, 1992a; New et al., 1993; Ross-Macdonald et Roeder, 1994; Hollingsworth et al., 1995; laccarino et al., 1996) ont ét6 identifids, chacun présentant une homologie significative avec MutS. Tout comme MutS, ces homologues prdsentent tous un domaine de liaison aux NTPs et un domaine helice- tour-helice très bien conservds.
MSH1 est une protéine mitochondriale de 109 kD impliquée dans la stabilisation du génome mitochondrial (Reenan et Kolodner, 1992a). Elle se lie aux simple bases mésappariées et aux petites insertions/délétions de 2-4 bases. Tout comme la protéine bacterienne MUS, MSH1 a une activité dependante de I'ATP mais à un degré beaucoup moindre. Chi et Kolodner (1994b) ont montre qu'en présence de I'ATP, on assiste A une augmentation de 60% de la capacité de la protéine MSH1 à discriminer I'het6roduplex OTT de I'hornoduplexe WC.
Des analyses gdnétiques et biochimiques récentes chez la levure suggbrent que MSH2 est l'homologue nucldaire majeur de MutS (Reenan et Kolodner, 1992a.b; Miret et al., 1993). La pmt6ine MSHP de 109 kD diffbre remarquablement de MSHl et MutS. En effet, contrairement à la proteine bacterienne et MSHI qui ne se lient pas aux insertions/d81étions de plus de 4 bases, MSHP est capable de se lier avec une forte affinit6 aux hMroduplex contenant des insertions/d6létions de 12 a 14 nucléotides palindromiques (Alani et al., 1995). Les h&t&oduplex non palindromiques de 1-1 2 nucl6otides sont reconnus avec une affinit6 interniddiaire

croissante en fonction de la taille de l'insertion, alors que les mesappariements de type G K sont reconnus avec une plus faible affinitd.
D'autres analyses @netiques chez la levure ont révélé que les mutants msh2 sont déficients dans la correction des mesappariements (Reenan et Kolodner, 1 992b, Alani et al., 1994). Ainsi, les mutants msh2 presentent une augmentation du taux de mutations spontanées, une augmentation du taux de recombinaison hom6ologue, un taux élevé des insertions/del&ions de 2 à 4 bases (Reenan et Kolodner, 1992b; Strand et al., 1993) et une destabilisation des microsatellites. Chez les mammifères, l'inactivation de MSH2 est aussi à l'origine de certains types de cancers du colon (Fishel et al., 1993).
Les proteines MSH3 et MSH6 de Saccharomyces cerevisiae sont deux autres homologues de MutS impliquees dans la correction des mésappariements d'ADN et la stabilisation du g6nome. D'aprBs les études de Johnson et al. (1 996). MSHP fonctionne avec MSH3 ou MSH6 selon la nature des rnésappariements d'ADN (Figure 2). Dans les cellules de levure, MSH2 et MSH3 coopèrent pour la correction des insertions/déletions de 2 B 4 bases alors que MSH2 et MSHG prochdent à la correction des bases simples mésappari6es. L'absence de MSH3 ou de MSH6 peut être compensee par la pr6sence de l'une ou l'autre de ces deux protéines (Johnson et al., 1996). Des mutations dans les gènes MSH3 et MSH6 donnent lieu B des phénotypes mutateurs similaires aux mutants msh2 mais avec une moindre intensité (Marsischky et al., 1996, Johnson et al., 1996).
Enfin, MSH4 et MSHS sont les seuls homologues de MutS qui ne sont pas impliqu6s dans la correction des mesappariements BADN (Hollingsworth et ab, 1995; Ross-Macdonald et Roeder, 1994). Ils ont été clonés sur la base d'homologie de séquences et, comme les autres homologues de MutS, possèdent les domaines de liaison à l'ADN et aux NTPs à leur extr6mit6 C-terminale. Des mutants msh4 ou msh5 montrent une diminution du taux de viabilit6 des spores et une augmentation du taux de non-disjonction des chromosomes A la m6iose I (Hollingsworth et a/-, 1995). Les doubles mutants msh4msh5 ont permis de montrer que ces deux gènes sont dans le mdme groupe Bpistasique dont le r6le est de faciliter les crossing-over interhomologues au cours de la m6iose.
Au total, plusieurs homologues de MutS existent chez les eucaryotes et se repartissent dans trois catdgories fonctionnelles : MSHl pour la correction au
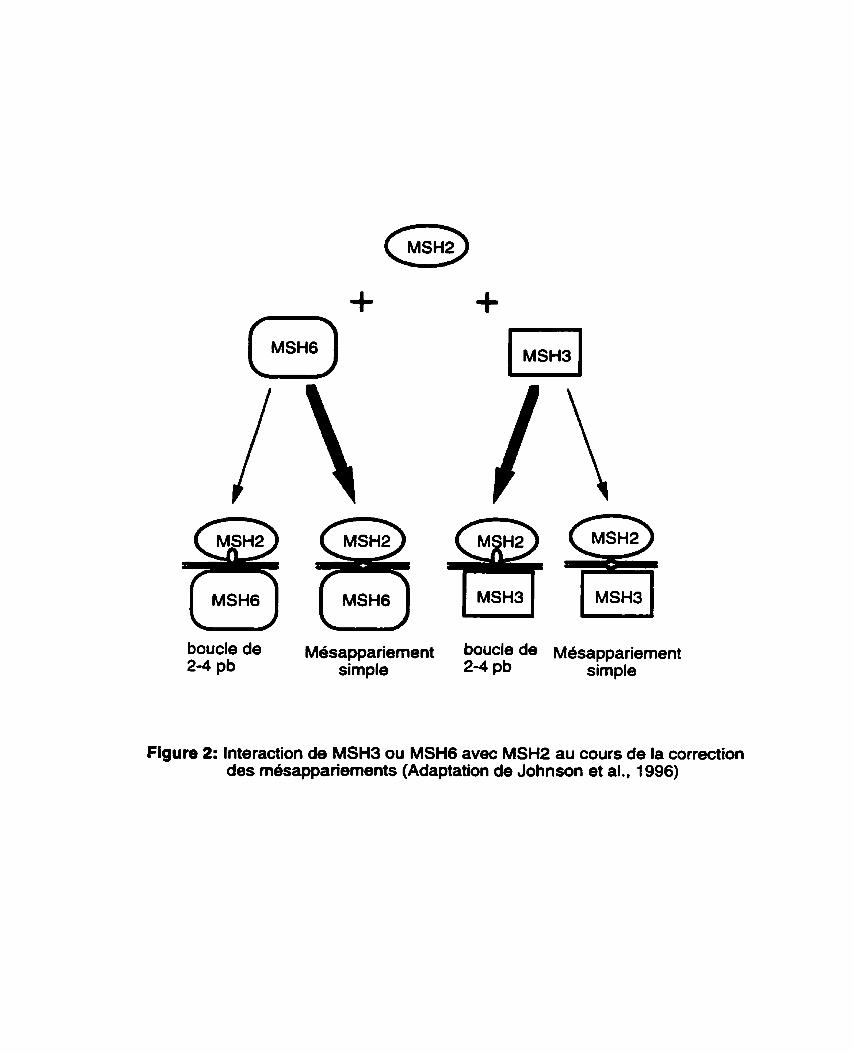
boucle de Mesappariement boucle de Mesappariement 2-4 pb simple 2-4 pb simple
Figure 2: Interaction de MSH3 ou MSH6 avec MSH2 au cours de la correction des m6sappariements (Adaptation de Johnson et al., 1996)

niveau mitochondrial, MSH2, MSH3 et MSH6 interviennent dans la correction au niveau nucldaire, alors que M W 4 et MSHS sont impliqu6s dans la mbiose.
1.3.2 Homologues eucaryotiques de MutL
Tout comme MutS, le gBne MutL possbde plusieurs homologues eucaryotiques. Par exemple chez Saccharomyces cerevisiae, quatre homologues de MutL ont Bt6 identifiés (PMS1, MLH1, MLH2 et MLH3; [Flores-Rozas et Kolodner, 1998; Kramer et al., 1989a; Prolla et al., 1994bl) alors qu'on connait présent trois homologues de MulK chez l'humain (PMS1, PMS2 et MLH1). Ces homologues de MutL sont impliques dans la correction des mésappariements et leur inactivation donne lieu à des phénotypes mutateurs. Les phénotypes associes aux mutants mlhl de la levure sont très similaires a ceux de pmsl, et puisque les phénotypes des doubles mutants mlhllpmsl sont les mQmes que ceux de l'une ou l'autre mutation simple, on en a déduit que les deux gènes sont dans le même groupe épistasique (Modrich et Lahue, 1996).
II convient aussi de mentionner que les phhotypes observes chez les doubles mutants pms l lmlh 1 sont presque identiques à ceux observes chez les mutants msh2, confirmant I'hypothbse selon laquelle MSH2, PMSl et MLH l sont dans le meme groupe Bpistasique et interagissent au cours de la correction des mesappariements (Bishop et al., 1987, 1989; Kramer et al., 1989b; Strand et al., 1993; Alani et a/., 1994; Prolla et al., 1994a). Cette hypothèse a été confirmée par des études ultérieures montrant que les protéines MSH2, PMS1 et MLHl forment un complexe avec les dupiexes d'oIigonucléotides contenant des m4sappariernents GK (Prolla et al., 1994b).
En plus de jouer un rôle dans la correction des mbsappariements d'ADN, certains homologues eucaryotiques de MutL sont impliqu6s dans la rnbiose. Par exemple, chez la souris, les mutants pms2 de sexe male sont stériles alors que les mutants mlh 1 sont st6riles quelque soit leur sexe (Baker et al., 1995, 1996).

1.3.3 M6canisrne de correction des rn6sappariements chez les eucaryotes
Le processus de correction des m6sappariements d'ADN chez les eucaryotes est très similaire h celui des procaryotes à quelques exceptions près. Ainsi, contrairement aux procaryotes, c'est en partenariat que les homologues eucaryotiques de MutS vont reconnaître les mdsappariements d'ADN de façon efficace.
Lorsqu'il s'agit d'un simple mésappariement baseibase, le complexe MSHP-MSH6 vient se lier h I'ADN mdsapparié avec une grande affinité. Par contre, si I'on est en présence d'insertion/d6l&ion, ce r6le est joue par le complexe MSH2-MSH3 (Figure 2). La formation du complexe ADN-MSH2-MSH3 ou ADN-MSH2-MSH6 est la première etape du processus de correction des mesappariements chez les eucaryotes (Figure 3). La deuxième etape de la rdaction consiste en la formation du complexe d'hétérodimere MLH1-PMSt qui vient se lier au premier complexe mentionne ci-dessus. Ce complexe d'heterodimère marque une autre différence par rapport au système MutHLS de E. coli ou I'on a un complexe d'homodimère MutL- MutL. II convient aussi de mentionner que dans le cas de l'humain, le complexe d'heterodimbre est forme par MLHl et PMS2. Des études récentes effectuées par Flores-Rozas et Kolodner (1998) suggbrent un second complexe d'h6terodim6re MLH 1 /MLH3 qui peut remplacer partiellement le complexe MLH W M S 1 dans la correction des mésappariements reconnus par le complexe MSH2/MSH3. Puisqu' aucun homologue eucaryotique de MutH n'a été identifié jusqu'à présent, on suppose qu'une endonucléase monocaténaire eucaryotique de type MutH se lierait au complexe et procède à l'excision du brin contenant le mésappariement. Ensuite les étapes de la resynthese et de la ligation viennent achever le processus de correction (Figure 3).
Chez E. coli, si I'on admet que la sp6cificit6 du brin B corriger est procurée par la méthylation, plusieurs hypothbses sont avancees chez les eucaryotes puisque cette modification n'intervient pas dans toutes les espbces (Modrich et Lahue, 1996). D'une part, l'existence d'une simple cassure d'ADN suffirait pour cibler la coffection des mdsappariements chez la drosophile et les mammiferes. D'autre part, certaines Btudes ont sugg6r6 que chez les eucaryotes, la mdthylation de I'ADN pourrait ddtenniner la spécificite du brin & corriger (Hare et Taylor, 1985). Cependant, si cela peut 8tre le cas chez certains eucaryotes, il semblerait qu'un tel signal est à exclure
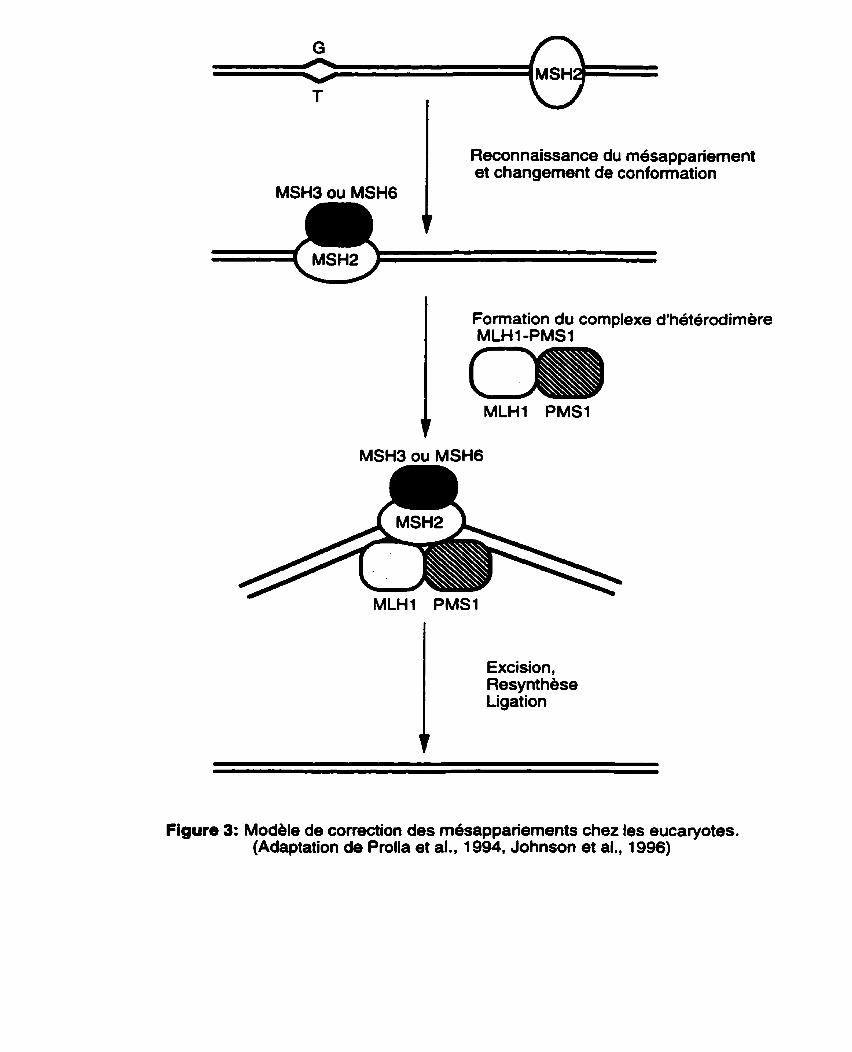
Reconnaissance du mesappariement et changement de conformation
l
Formation du complexe d'héterodimere MLH1 -PMS1
Excision, Resynthbse Ligation
Figure 3: Modele de correction des m6sappanements chez les eucaryotes. (Adaptation de Prolla et al., 1994, Johnson et al., 1996)

dans le cas de la levure et de la drosophile dont les génomes ne sont pas sujets B de telles modifications (Proffitt et al., 1984). La troisième hypothdse postule l'existence de proteines associ6es à chacun des brins de la double hélice d'ADN, et qui ségregeraient lors du passage de la fourche de réplication (Modrich et Lahue, 1 996).
1.4 Correction des misappariements et recombinaison gbnitique
La recombinaison homologue a fréquemment lieu entre sequences d'ADN identiques alors que la recombinaison homeologue est definie comme étant des échanges génétiques entre séquences d'ADN similaires mais non identiques.
Des études chez plusieurs organismes ont montré que les divergences de sequences reduisent la frequence des recombinaisons (de Wind et al., 1995; Fishel et Kolodner, 1995; Rayssiguier et al., 1989). Les limites d'homéologie en terme de pourcentage de divergence ne sont pas définies précisement cause de fa réponse variable a I'homéologie observée dans differents systèmes expérimentaux.
La recombinaison entre deux sequences d'ADN homéologues forme des hétéroduplex contenant des mésappariements d'ADN (Figure 4). Les mésappariements ainsi formés sont corrigés par le système de correction de l'organisme concerné. Les résultats de nombreuses études supportent l'idée selon laquelle les intermédiaires de la recombinaison contenant de multiples mesappariements sont des cibles pour les protéines de correction des mésappariements, lesquelles agissent pour empêcher l'achèvement des événements de recombinaison impliquant des sequences homéologues (Radman, 1989) ou modifier leur issue (Worth et al., 1994).
Par exemple Rayssiguier et ses collaborateurs (1 989) ont montre que chez E. coli et Salmonella typhimurium, lesquels presentent une divergence de 20% au niveau des sequences d'ADN, le taux de recombinaison entre sequences hom6ologues est réduit de 1000 fois par rapport au taux de recombinaison entre des sequences homologues. Par contre, des mutations dans MutS, MutL ou MutH, éliminent cette suppression de la recombinaison entre sdquences homéologues prouvant ainsi que les protéines de correction des mdsappariements agissent comme une barribre à la recombinaison entre séquences divergentes au cours de la conjugaison. L'effet
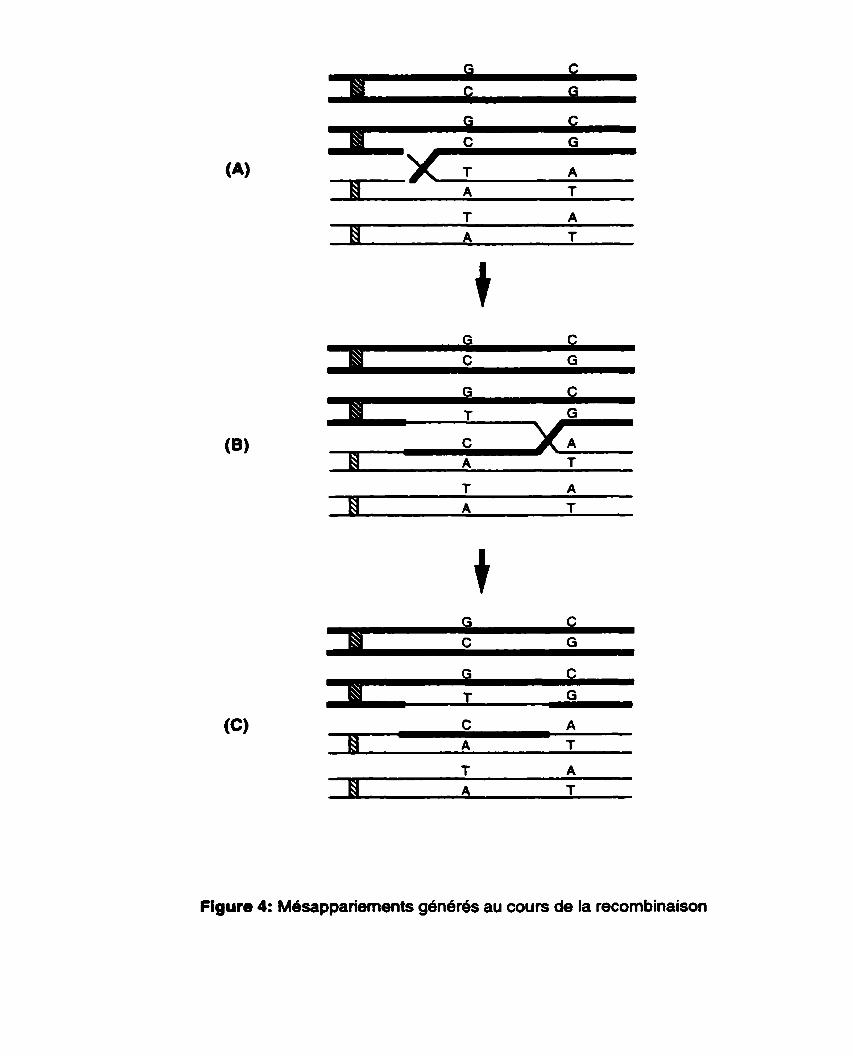
Figure 4: Mdsappariements generes au cours de la recombinaison

majeur de MutS et Mut1 serait de bloquer la migration des branches lors des échanges hom6ologues (Radman, 1989).
Chez ta levure, Selva et ses collaborateurs (1 995) ont utilisé le gene SPT15 de Saccharomyces cerevisiae et son homologue TBP chez Schizosaccharomyces pombe, des genes qui présentent une divergence nucleotidique de 25%' pour montrer le rôle de la correction des mésappariements dans la recombinaison. Dans chacun des deux gènes, ils ont introduit une mutation qui inactive le gène et seufe une recombinaison peut rendre l'un ou l'autre gene fonctionnel. Chez les individus sauvages MSH2, ils ont noté une diminution de 150 à 180 fois du taux de recombinaison homeologue (SPT15xTBP) comparé au taux de recombinaison homologue (SPTISxSPTIS). D'autre part, ces auteurs ont noté une augmentation de 17 fois du taux de recombinaison homéologue entre ces deux genes chez des individus mshZ compares aux individus mshP, prouvant ainsi que le gene MSH2 est implique dans la suppression de la recombinaison homéologue; ce résultat est la confirmation de l'hypothèse selon laquelle le système de correction des rnésappariements agit comme une barrière aux échanges génetiques entre séquences d'ADN divergentes.
Les cellules des mammifères montrent aussi une discrimination vis-à-vis la recombinaison homéologue. Chez la souris, de Wind et al. (1995) ont démontré que deux lignées de cellules qui montraient une réduction de recombinaison de 50 fois à cause d'une divergence nucléotidique de 0.6%. sont devenues très permissives pour la recombinaison après une mutation dans le gène MSH2. A prion, on pourrait dégager de ce résultat que l'augmentation du taux de recombinaison homéologue chez les mammifères msh2- est aussi élevée que chez les procaryotes contrairement a la faible augmentation observée chez la levure. Ce résultat montre que chez les mammifères, MSH2 est impliqué dans la prévention de la recombinaison entre séquences homéologues.
Bien que les protéines de correction des mésappariements accèdent clairement aux mésappariements générés lors des échanges de brins et influencent le cours des événements subs6quents, les traits marquants des mécanismes en cause ne sont pas bien connus. L'une des possibilit6s est que la migration des branches est arrêt6e (Radman, 1989). Selon cette hypothèse, les protéines de correction des mésappariements agissent pour empêcher ou même arrêter le progrès des échanges de brins a l'intérieur des hetéroduplex Les évidences supportant cette

notion viennent d'études biochimiques (Worth et al., 1994) et génétiques (Alani et al., 1994) indiquant que les activités de MutS et MutL chez E. coli, et de MSH2 et PMS1 chez la levure, inhibent la migration des branches dans les hétéroduplex. Une seconde possibilit6 est que la reconnaissance des mésappariements stimule la résolution des intermédiaires de la recombinaison aux sites de mésappariements (ou proche de ces sites) et agit ainsi pour empecher les échanges (Alani et al., 1994). La troisième possibilité suggère une stimulation de l'excision des mésappariements conduisant à la destruction de l'événement de recombinaison.
1.5 Hypothèses de recherche et objectifs
De ce qui précède, il ressort que les gènes impliques dans la correction des mesappariements d'ADN ont été bien conservés au cours de l'évolution à travers les différents organismes. Parmi ces genes, MSH2 (MutS chez E. coli) joue un rôle central dans le processus de correction des m6sappariements et détermine la spécificité des événements de recombinaison. Les motifs les plus conservés dans ces gènes sont les domaines fonctionnels à savoir le domaine de liaison à l'ADN et le domaine de liaison aux NTPs. Cette conservation de mécanisme et de fonction du système de correction des mbsappariements d'ADN, des procaryotes aux eucaryotes supérieurs, suggère très fortement qu'il s'agit d'un mecanisme présent dans tout organisme vivant.
Nous avons postulé alors que chez les plantes, l'homologue du gène MSHZ, la composante centrale de ce système (Reenan et Kolodner, 1992b), joue un rôle semblable dans la correction des mésappanements d'ADN et interviendrait dans la recombinaison. Les deux principaux objectifs vises dans cette étude sont:
1. Cloner et caractériser l'homologue du gene MSH2 chez Arabidopsis thaliana.
2. Démontrer que le produit de ce gène est fonctionnel.
Dans le premier volet de ce projet (chapitre II) nous avons rapporte le clonage, et la caracterisation du gene AtMSH2 ainsi que de trois de ses paralogues chez Arabidopsis thaliana. Dans un deuxième volet (chapitre III), l'étude fonctionnelle du produit de ce gene a été realisbe. Ensuite, le troisihme volet (chapitre IV) a été consacre à la caractérisation d'une famille d1e16ments transposables jamais

identifiée auparavant chez Arabidopsis thaliana et dont l'un des membres est insér6 dans la région 3' du gène AtMSH2 chez certains écotypes. Enfin, dans un chapitre de discussion gbnerale, nous avons résumé les principaux acquis de ces travaux et les perspectives qu'ils offrent tant en recherche fondamentale qu'appliquée.

CHAPITRE II
Four mismatch repair paralogues coexist in Arabidopsis thaliana: AtMSH2, AtMSH3, AtMSH6- 1 and AtMSH6-2.

Note : Ce chapitre rddige sous forme de publication rapporte le clonage et une caractérisation d6taiMe de quatre homologues de MutS impliqu6s dans la correction des rn6sappariements d'ADN chez Arabidopsis thaliana. II est le fruit d'un travail de collaboration. Parmi les quatres gènes rapportés, trois ont été clonés (AtMSH2, AtMSH3 et AtMSH6-2) alors que le quatrieme (AtMSH6- 1 ) provient du projet de sequençage du génome dlArabidopsis. Dans le cadre de cette these, notre laboratoire a fait le clonage de AtMSH2 et d'un fragment de AtMSH6-2 de meme que la cartographie des différents ghes. Dr. Marie-Pascale Doutriaux a réalise le clonage des gènes AtMSH3 et AtMSH6-2 ainsi que l'expression des differents gènes. Le volet de l'analyse phylog6n6tique a 616 r6alisé par Dr. Herv6 Phillipe.

Four mismatch repair paralogues coexist in Arabidopsis thaliana: A tMSH2, A tMSH3, A tMSH6- 1 and A tMSH6-2.
Jules AdéO, François BelzileO, HewB Philippe & Marie-Pascale Doutriaux*
O Département de Phytologie, 1243 Pavillon Marchand, Université Laval, Québec, Canada G1 K 7P4
* Laboratoire de Biologie Cellulaire (CNRS URA 2227), Bât. 444, Université Paris- Sud, 91 405 Orsay cedex, France
Institut de Biotechnologie des Plantes (CNRS UMR 8618). Bât. 630, Université Paris-Sud, 91405 Orsay, France
Mol Gen Genet (1999) 262: 239-249

Résumé du manuscrit
Nous avons utilise des amorces dégénérées basées sur des homologies de séquence entre des homologues connus de MutS pour isoler trois cDNA de MSH chez Arabidopsis thaliana (6cotype Columbia) qui sont membres des familles de genes eucaryotiques MSH2, MSH3 et MSH6. Les séquences genomiques de deux de ces gènes (AtMSH2 et AtMSH6-2) ont été isolées et déteminees, alors que la séquence génomique de AtMSH3 provenait du projet de séquençage daArabidopsis de même que celle d'un autre homologue distinct de AtMSH6 (AtMSH6-1). L'analyse comparée de la sequence génomique de AtMSH2 de Landsberg erecta (décrite ici) et celle précédemment décrite chez Columbia a révél6 la presence de plusieurs polymorphismes incluant la présence d'un Blement transposable dans la région 3' non transcrite de l'allèle de Landsberg erecta. Arabidopsis est également le premier organisme qui montre une telle divergence de deux genes AtMSH6. divergence fortement confirmée par l'analyse phylogénétique. L'hybridation Southern a révélé que les trois genes que nous avons isoles sont présents en simple copie et la cartographie indique que AtMSH2 et AtMSH6 sont tous deux situés sur le chromosome 3. Finalement, l'expression de ces trois genes n'a été observée que dans des suspensions cellulaires dlArabidopsis thaliana. Cette suspension cellulaire est activement en division mitotique après le repiquage et c'est à cette étape que les genes AtMSH sont le plus fortement exprimés.

Abstract
By using degenerate oligonucleotides based on the sequence homology between known MutS homologues, three MSH cDNA belonging to the MSH2, MSH3 and MSH6 families, as defined in eukaryotes, have been isolated from Arabidopsis thaliana (ecotype Columbia). Genomic sequences for two of these genes (AtMSH2 and AtMSH6-2) were also isolated and detemined, whereas the genomic sequence of AtMSH3 was obtained through the Arabidopsis sequencing project as was the sequence of another and distinct AtMçH6 homologue (AtMSH6-1). Comparative analysis of the AtMSH2 Landsberg erecta genornic sequence (reported here) and the previously descnbed AtMSH2 Columbia allele revealed several polymorphisrns including the presence of a small, transposon-like element in the 3' untranscribed region of the former allele. Also, Arabidopsis is the first organism showing such divergence of two AtMSH6 genes, a divergence which is strongly ascertained by sequence data and phylogenetic analysis. Southem hybridization revealed that the three genes we isolated are single-copy and genetic mapping indicated that AtMSH2 and AtMSH6-2 both reside on chromosome III. Finally, expression of these three genes could only be obsewed when taking advantage of a cell suspension of Arabidopsis thaliana. This cell suspension is actively dividing after subculture and this is when the AtMSH genes are most strongly expressed.

2.1 Introduction
The mismatch repair system (MMR) is essential to genetic stability, as it protects the genome against arising mutations and regulates recombination between related DNA sequences. MMR is responsible for the recognition and processing of mispaired bases that are spontaneously produced in the DNA as a consequence of replication errors, genetic recombination or deamination of 5 me-cytosines (Kolodner 1996). Repair of replication errors contributes to the conservation of the original information carried by the DNA. On the other hand, the fine tuning of the length of recombination intermediates (heteroduplex) and their editing or not by the MMR define the degree to which recombination may occur between homologous but non-identical DNA sequences (Vulic et al. 1997). Recognition of mispaired bases in the heteroduplex region triggers the aborlion of recombination events and prevents rearrangements between DNA sequences that are tco divergent (Rayssiguier et al. 1 989).
The MutHLS mismatch repair system in Escherichia coli is by far the best characterized and derives its name from the three genes required to initiate MMR (MutH, MutL, MutS). The MutS protein is responsible for the detection of mismatches, and its binding determines further processing and repair of mismatch- containing DNA molecules by the other components of the MMR. The MutH protein allows discrimination of the newly replicated DNA strand as it is transiently undermethylated at adenines in GATC sequences. Association of the MutL protein with MutS bound to the mismatch stimulates endonucleolytic cleavage of the unmethylated GATC sequence by MutH. Exonucleolytic degradation then proceeds to remove a stretch of up to 1000 bases around the mismatched base, followed by gap repair synthesis and ligation of the correct DNA sequence (Modrich and Lahue 1996). In the absence of a functional MMR system, bacteria show mutator or en hanced recombination proficiency phenotypes (Cox 1 976, Feinstein and Low 1 986, Rayssiguier et al. 1989).
Our current knowledge shows that the general features of the bacterial MMR seem to be rather well conserved across al1 living organisms: mismatch repair activity or mismatch repair genes have been invariably avidenced when studied in a wide variety of eukaryotes (Modrich and Lahue 1996). Mismatch repair can be assayed following transfection of artificially constructed heteroduplex DNAs into yeast

(Bishop et al. 1989, Kramer et al. 1989) and mammalian cells (Hare and Taylor, 1985; Folger et al. 1985; Brown and Jiricny 1988) or after incubation into cell free extracts of Drosophila (Holmes et a1.1990; Bhui-Kaur et al. 1998), Xenopus (Varlet et al. 1996) or human (Holmes et a1.1990; Thomas et al. 1991). This mismatch repair activity is found abolished in ail available mutants deficient for the rnismatch repair functions (Parsons et al. 1993; Umar et al. 1994; Luhr et al. 1998). Finally,
homologues of MutS and MutL have been isolated from eukaryotes, but their number suggests a higher level of MMR cornplexity or the involvement of more specialized processes. While in bacteria the MutS proteins seem to belong to two different lineages (MutS-l and MutS-Il as defined by Eisen, 1998) which are not necessarily both present in every bacterial species, gene duplication and functional specialization have led to the divergence of many MutS homologues in eukaryotes. Six MutS homologues coexist in yeast: Msh2, Msh3 and Msh6 are involved in nuclear MMR, Msh4 and Msh5 participate in meiotic recombination and finally, Mshl is invoved in mitochondrïal MMR (Reenan and Kolodner 1992; Ross-Macdonald and Roeder 1994; Hollingsworth et al. 1995; Marsischky et al. 1996). Msh2, Msh3 and Msh6 homologues have since then been found in different organisms, including mammals, Drosophila, Neurospora and Arabidopsis (for review , see Kolodner 1996). These eukaryotic MSH genes were classified as belonging to the MutS-l ( M S H I ; MSH2; MSH3; MSHG) or the MutS-Il Iineage ( M W 4 and MSHS) (Eisen, 1 998).
The current model for mismatch repair in eukaryotes is that Msh2 interacts with either Msh3 or Msh6 to form complexes with different recognition specifities: Msh2/3 complexes show a greater affinity for small insertions/deletions and Msh2/6 for single base mismatches (Marsischky et al. 1996; Kolodner 1996). This model is clearly supported by genetical and biochemichal data (Reenan and Kolodner 1992b; Drummond et al. 1995; Palornbo et al. 1995; Marsischky et al. 1996; Acharya et al. 1996; Genschel et al. 1998). Reminiscent of the mutS phenotype in bacteria, yeast msh2 mutants exhibit a rnutator phenotype as do the msh3msh6 double mutants (Marsischky et al. 1996). In human, it is now well established that MMR deficiencies can lead to some hereditary cancer predisposition syndromes (Modrich and Lahue 1996). These cancers are associated with genetic instability, a phenotype that can be detected as an increased mutation rate in reporter genes or
in tracts of short repeated DNA sequences. Such microsatellite instabilities presumably result from a slippage of the replication machinery which generates

short insertions/deletions that would nonnally be recognized and repaired by the MMR. They are specifically obseived in msh2, msh3 or msh6 tumor cells or gennline mutations (Modrich and Lahue 1996; Risinger et al. 1996; Akiyama et al. 1997;
Miyaki et al. 1997). Genetic variability and cancer susceptibility are also dramatically increased in mice canying nuIl mutations of the MSH2 or MSH6 genes (de Wind et al. 1995; Reitmair et al. 1995; Edelmann et al. 1997).
As well as its role in surveying replication fidelity, the MMR is also involved in regulating genetic recombination between homologous but non-identical DNA sequences (Rayssiguier et al. 1989). If the outcome of recombination depends on the formation of an heteroduplex intermediate, the presence of mismatches in the heteroduplex makes it an obvious target for the MMR. A fvnctional MMR acting upon the mismatches can destabilize the heteroduplex thus impeding recombination between homeologous DNA sequences. Studies in bactena, yeast and mouse cells have al1 shown that mutations affecting components of the MMR can increase many fold the amount of recombination between divergent DNA sequences (Rayssiguier et al. 1989; Selva et al. 1995; de Wind et al. 1995; Datta et al. 1997).
In plants, not much is known about misrnatch repair. Plant cell extracts from pea can repair misrnatched oligonucleotides (Cerovic et al. 1991) and an MSH2 homologue was recently isolated from Arabidopsis thaliana (Culligan and Hays 1997). With the airn of gaining insights into the role and activity of mismatch repair in plants, we have isolated homologuss of MSH2, MSH3 and MSH6 in Arabidopsis thaliana. Here, we provide a detailed characterization of these genes including studies on their expression which is found to be detectable only in a mitotic cell suspension of A rabidopsis.

2.2 Materials and methods
2.2.1 Growth of cell suspension
The cell suspension (ecotype Columbia) was initiated by Axelos et al. (1 992) and is continuously propagated by weekly subculture (1.5mV25ml) in Gamborg's B-5 basal medium (G-5893, SIGMA), 30gA sucrose, 200mg/l NAA. The cell suspension grows under agitation in a culture room with a 15h photoperiod. Haivested plant material was stored at -70°C before RNA extraction.
2.2.2 RNA isolation and Northern blot analysis
Total RNA from the cell suspension was extracted in the presence of TRIZOL (Gibco BRL) after homogenizing the cells in liquid Np. POI~A+ RNA were isolated using the
Dynabeads mRNA direct kit (DYNAL). ? o l y ~ + RNA was separated in 1% agarose/fomaldehyde gels after denaturation (Sambrook et al. 1 989). Gels were transfered ont0 Nylon Hybond N+ membranes (Amersham) by capillary blotting. ARer hybridization to radiolabeled probes, the filters were washed in O.lXSSC, 0.1 %SDS at 62°C and autoradiographed.
2.2.3 Genomic DNA isolation and Southern blot analysis
Genomic DNA was extracted from the cell suspension according to Dellaporta et al. (1 983). Enzymatic digestion and DNA migration were done using standard techniques. DNA was transfered ont0 Nylon Hybond N+ membranes (Amersham) by capillary blotting. Genomic DNA sequences were isolated from a previously constnicted Sau3Al partial genomic library (Doutriaux et al. 1998).
2.2.4 Radiolabelcd probes
32~-radioabelling of the probes was carried out with the Stratagene Pnme it II kit. Hybridization with 32~-radiolabeled probes corresponding to the complete coding regions of the bean translation elongation factor EF-lalpha (pCHA0041; Axelos et

al. 1989), AtRAD51 (Doutriaux et al. 1998) genes and 285 ribosomal RNA (Arabidopsis Biological Resource Center) were perfomed at 62'C according to
Church and Gilbert (1984)
2.2.5 Reverse transcription and PCR
One pg of total RNA was reverse transcribed by the MMLV reverse transcriptase after priming with random oligonucleotides and in the presence of dNTPs. Using 2 different sets of degenerate oligonucleotides (1pM each primer), PCR was perfomed using Crst strand cDNA or genomic DNA in a final volume of 100pl. in the presence of dNTPs (0.2mM). 1xPCR buffer and Taqpolymerase (2 units). PCR parameters were either: for setl oligonucleotides (touchdown PCR), 3 rounds of 3 cycles each (94 OC- 1 min; 45 OC, 41 OC and 37 OC- 2 min; 72 OC- 1 min) followed by 35 cycles (94 OC- 30 sec; 48 OC- 30 sec; 72 OC- 30 sec) and finally 10 min at 72 OC or for set2 oligonucleotides, 94°C -5 min. followed by 30 cycles of (95°C-40 sec; 45°C- 1 min; 72°C-1 min). The amplification products At23, At24 for setl, and S5. S8 for set2 were subcloned and sequenced. Of these clones, At24 (654 bp, derived from genomic amplification) was homologous to MSH2, S5 (351 bp) was homologous to M S H 3 and At23 (623 bp) and 58 (351 bp) were identical (except for the presence of introns in At23 which was amplified from genomic DNA) and homologous to MSHG.
2.2.6 isolation of AtMSH2 cDNA
To obtain a cDNA clone, ten pools of 10,000 clones each from library CD446 (ecotype Columbia, provided by the Arabidopsis Biological Resource Center) were plated on 15 cm Petri dishes. The amplified phages were collected in 3 ml SM buffer (10 mM NaCI, 1 mM MgS04.7H20, 50 mM Tris-HCI pH 7.5, 2% gelatin) of which 1 pl
was used to perform PCR with the primers MSH2-1 and MSH2-2 specific for AtMSH2. One of the positive pools was used to generate ten pools of one thousand clones each. PCR was used to identify positive pools of 1,000 phages from which two replicate filters were lifted. Two positive plaques were identified following hybridization with the At24 insert and in vivo excision (Stratagene) was used to obtain a pfasmid version of one of the cloned cDNAs.

2.2.7 Isolation of the AtMSH3 and AtMSH6 cDNA sequences
Complete cDNA sequences were isolated according to the Marathon cDNA amplification kit procedure (Clontech). In brief: double stranded cDNA was produced by reverse transcription of 2pg po l y~+ RNA from the cell suspension culture of Arabidopsis. Adaptors were ligated on each side of the cDNA. The ligated cDNA was used as a template for 5' and 3' RACE PCR reactions in the presence of primers specific for the adaptor on one side (AP1 and AP2), and specific for the targeted gene on the other side (see below), as defined from the previously isolated consensus regions S5 and S8. A 5' and a 3' fragment that overlap were produced for each gene.
2.2.8 Isolation of AtMSH3 complete coding sequence
PCR performed on the ligated cDNA with primers 636 and API for the 5' RACE PCR was followed by a second round of amplification with the nested pnmers AP2 and S525 which produced a 2720 bp DNA fragment. Another primer (S51) was designed closer to the 5' border and permitted the detemination of 99bp upstream of the ATG initiation codon. For the 3' RACE PCR, a first PCR reaction was performed with primers AP1 and 635, followed by a second round of amplification, using the nested primers AP2 and S523 which produced a DNA fragment of 890 bp. Both DNA fragments were subcloned into pGEM-T and sequenced. Since PCR amplification using the Expand Long Template PCR System (Boe hringer- Mannheim) produced errors in the sequence. new oligonucleotides were designed to re-isolate these sequences by PCR, but with the high fidelity DNA polymerase Pfu. PCR with primers 1S5 and S53 amplified a 1244 bp fragment (cloned into pUC18ISmal). PCR with primers S52 and 255 amplified a 2104bp fragment (cloned into pUC18/Smal). These two clones were ligated after digestion by BamHl for which a unique site is present in the overlapping region. The complete reconstituted AtMSH3 coding sequence is 3246 bp long.
2.2.9 Isolation of the AtMSH6-2 complete coding sequence
The same procedure allowed the isolation of the AtMSH6-2 cDNA. For the 5' RACE PCR, primers 638 and AP1 allowed the amplification of a 2889 DNA fragment. Primer S81 hetped define the 142 bp upstream of the ATG initiation codon. On the

3' side, RACE PCR was initially performed with pnmers S823 and API , and then with the nested primers 637 and AP2, to produce a 774 bp DNA fragment. As for AtMSH3, these fragments were cloned and sequenced. Due to PCR amplification replication errors, re-isolation of this DNA sequence using the high fidelity Pfu polymerase and newly designed primers 1S8 and S83 (for the 5' side, 2182 bp
clone 43 in pUC18/Smal), and pnmers S82 and 2S8 (for the 3' side, 1379 bp clone 62 in pUC18/Smal) was canied out. Clones 43 and 62 were digested by Xmnl for which a unique site is present in the overiapping region, and ligated. The complete reconstituted AtMSH6-2 coding sequence is 3330bp. An AtMSH6-2 genomic sequence was also isolated from a genomic DNA library constituted after partial SaulllAl digestion of DNA from the Arabidopsis cell suspension. 8062 bp were sequenced that covered the AtMSH6-2 gene and showed precise colinearity with the cDNA.
2.2.1 0 Oligonucleotides
MSH degenerate primers: Set1 : MMRl (CGTGGATCCTCAClGGICCNAA(C/T)ATG GG); MMRP (GGTGAAlT CGTGGAA(A/G)TGIGTNGC(A/G)AA) Set2 (as in Reenan and Kolodner, 1992): MMR3 (CTGGATCCACIGGICCIAA(CWATG); MMR4 (CTGGATCC(A/G)TA(A/G)TGIG TI (A/G)C(A/G)AA.
AtMSH2 specific primers: MSH2-1 (TCCAClTACATCCGCCAGGTTGATG); MSH2-2 (ATGCTCACATATAG CCCAAGCTAAACC) MSH2-3 (AAACTTGTGAGCTCGCTCT GCCCC).
AtMSH3 specific pnmers: 1 S5 (ATCCCGGGATGGGCAAGCAAAAGCAGCAGACGA); PSS(ATCCCGGGTCAA AATGAACAAGlTGGTmAGTC); S53 (GACAAAGA GCGAAATGAGGCCCCTTGG) ; S52 (GCCACATCTGACTGTTCAAGCCCTCGC); S51 (GGATCGGGTACTGGGTllT GAGTGTGAGG); S525 (AGGlTCTGAlTATGTGTG ACGCTiTAClTA); S523 (TCAG ACAGTATCCAGCATGGCAG AAGTA) ; 635 (GCACG TGCTTGATGGTGTTTTCAC); 636 (TGCTAGTGCCTCTTGCAAGCTCAT).
AtMSH6-2 specific primers: 1 S8 (ATCCCGGGATGCAGCGCCAGAGATCGATi" TTGT); 2S8 (ATCCCGGGTTATT TGGGAACACAGTAAGAGGArr); S82 (GCGTTCGA TCATCAGCCTCTGTGTTGC);

S83 (CGCTATCTATGGCTGCTKGAATGAG); S81 (CGTCGCCT'rTAGCATCCCC ITCCITCAC); 637 (GACAGCGTCAGTTCTTCAGAATGC); 638 (TCTCTACCAGGTG ACGAAAAACCG); S823 (GCTTGGCGCATCTAATA GAATCATGACAGG).
2.2.1 1 Mapping of AtMSH2 and AtMSH6-2
Primers MSH2-1 and MSH2-3 were used to arnplify a 1.3 kb segment of AtMSH2 from ecotypes Landsberg erecta and Columbia. A polymorphic Mbol site was identified by sequence analysis and used to score 96 Recombinant lnbred (RI) lines resulting from a cross between Landsberg erecta and Columbia (Lister and Dean, 1993). For AtMçHG, a RFLP between these two ecotypes was observed following digestion of genomic DNA with Hindlll and hybridization with a PCR product of 2kb This polymorphism was scored on a subset (24) of the RI lines mentioned previously. The MapMaker program (Lander et al. 1987) was used to detemine the map position of the AtMSH2 and AtMSH6-2 genes.
2.2.1 2 Phylogenetic analyses
Alignment of the sequences was carried out visually with the help of the ED program of the MUST package version 1.0 (Philippe, 1993). Phylogenetic trees were constructed with maximum likelihood (ML), maximum parsimony (MP) and distance based methods (Neighbor Joining, NJ) with the programs PROTML version 2.3 (Adachi and Hasegawa, 1996), PAUP version 3.1 (Swofford, 1993) and NJ in the MUST package version 1 .O (Philippe, 1993), respectively. The distances were cornputed with the substitution model of Kimura (Kimura, 1983). MP trees were obtained by 100 random addition heuristic search replicates and ML trees by the quick add OTUs search. with the JTT model of amino acid substitution and retaining the 500 top ranking trees (options -jf -q -n 500). Since it is important to take into account among-site rates variation for inferring phylogeny (Yang, 1996). these 500 trees were further analysed with the PUZZLE program (Strimmer and von Haeseler, 1996) as user trees with 8 Gamma rate categories. Bootstrap proportions were calculated by the analysis of 1000 replicates for MP and NJ analysis. For ML analysis, bootstrap proportions were computed by using the RELL method (Kishino and Hasegawa, 1989) because of computing time limitations.

2.3 Results
2.3.1 Isolation of the AtMU2, AtMSH3 and AtMSH6-2 cDNAs
Based upon a comparison of consewed amino-acid sequences of known MutS- related proteins from various species, one set (setl) of degenerate oligonucleotides was designed while another (set2) was used that had been previously described (Reenan and Kolodner 1992b). PCR amplifications were perforrned using either Arabidopsis (ecotype Columbia) genomic DNA or first strand cDNA as a template w hich allowed the isolation of consensus reg ions for three potential homologous mutS genes. At24 (654 bp), At23 (623bp), S5 (351 bp) and S8 (351) were cloned and sequence analysis indicated that they were homologous respectively to MSH2 (At24), MSH3 (SS) and MSH6 (At23, S8), three of the MSH genes described in yeast. After the design of oligonucleotides specific for the genes of interest. different approaches were followed to isolate their cornplete cDNA sequences. AtMSH2 was isolated from a cDNA library, after successive rounds of selection of positive clones by PCR; AtMSH3 and AtMSH6-2were isolated following the Marathon cDNA amplification procedure which relies on 5' and 3' RACE-PCR.
The AtMSH2 cDNA clone is 3039 bp long. contains an open reading frame of 281 1
nucleotides which is identical to that reported recently by Culligan and Hays (1997). The predicted protein is 937 amino acids long for a predicted rnolecular weight of 105.5 kDa. The reconstituted AtMSH3 sequence is 3553 bp long and contains a 3246 bp long open reading frame with untranslated regions of 99 bp (5') and 144 bp (3'). The cDNA encodes a putative protein of 1081 amino acids (predicted molecular weight of 117.8 kD). The AtMSH6-2 sequence is 3701 bp long and contains an open reading frarne encoding 1 109 arnino acids (predicted rnolecular weight of 122.5 kD); its coding region starts 141 bp from the 5' end and the poly (A) tail starts 106 bp downstream from the TAA stop codon. A short sequence (351 bp) identical to the AtMSfl6-2 consensus region has previously been described by Culligan and Hays (1 997).
Along the predicted protein sequences, the typical Msh functional domains can be defined in the C terminal end of the translated genes (Figure 1). Like other members of the MutS family, AtMsh2, AtMsh3 and AtMsh6-2 present the four characteristic


motifs (A, 6, C, D; see Fig. 1) of an NTP-binding domain, as defined by Gorbalenya
and Koonin (1990) for the superfamily of UvrA-related proteins. The second
conseived domain containing the amino-acid residues essential for the formation of the Helix-Tum-Helix structure (HTH; see Fig. 1) is also present in the Arabidopsis Msh proteins (Ohlendorf et al. 1983).
Genomic clones sequences were determined for both AtMSH2 and AtMSH6-2. The AtMSH2 genomic clone which we report here was isolated frorn the ecotype Landsberg erecta (GenBank accession AF109243) and it shows several differences relative to the previously reported genomic clone of the Columbia allele (Culligan and Hays, 1997; GenBank accession AF003005). While the number and position of al1 12 introns are identical in both alletes, numerous polymorphisms are seen both in the coding and non-coding regions (see Figure 2). Along the 13 exons. a total of 11 single base substitutions are observed of which six are neutral whereas five lead to a change in the amino acid sequence. None of these changes occurs at a position which is consenred among the eukaryotic MSH2 genes. The most striking difference between the two alleles, however, is the presence of a 239 bp insertion located 196 bp after the stop codon in the 3' untranscribed region of the Landsberg
erecta allele. This insertion is flanked by a direct duplication of 5 bp and bears many but not al1 of the features of a miniature inverted-repeat transposabfe element (MITE), a class of small transposable elements recently reported in plants (Bureau and Wessler, 1994b). This element is different from the Emigrant element, the only
MITE reported to date in Arabidopsis (Casacuberta et al., 1 W8), and will be described elsewhere in detail (J. Ade and F. Belzile, unpublished).
A 8062 bp genomic region (Columbia ecotype) covering the AtMSH6-2 gene was also defined and revealed the presence of 16 introns scattered along the sequenced region. The genomic region of AtMSH3 has been completed recently through the Arabidopsis sequencing project. The 11 exons of this gene are found within a 5.5 kb stretch of the BAC clone M7J2 (GenBank accession AL022197). Southern blot hybridization of Arabidopsis restricted DNA with probes corresponding to the genomic consensus regions for AtMSH3 and AtMSH6-2 genes indicates that they are single copy genes and do not cross-hybridize (see Figure 3). The sizes of the detected fragments always correlated exactly with their expected sizes whenever they could be determined from available sequence information. Surprisingly, a fourth MSH gene was encountered in the course of the Arabidopsis

Base substitution O O 14 Insertion/deletion > O 0 +5
Figure 2: Polymorphisms between the Landsberg erecta and Columbia alleles of AtMSH2. In this diagram, exons are shown as open rectangles whereas introns are drawn as v-shaped lines separating exons. The position of the start (ATG) and stop (TGA) codons as well as that of each of the 11 polymorphisms (al1 single base substitutions) located within the coding region are indicated above the gene. Position 1 refers to the first base of the genomic sequence of this allele (GenBank accession AF109243). Substitutions which lead to a change in amino acid sequence are indicated with an asterisk ('). The nature (number of base substitutions or length of insertion/deletion) of polymorphisms located in introns is indicated below the diagram. A 239 bp miniature inverted- repeat transposable element (MITE-like) insertion (hatched box) flanked by a 5 bp duplication (0 ) at the insertion site was found uniquely in the 3' region of the Landsberg erecta allele.
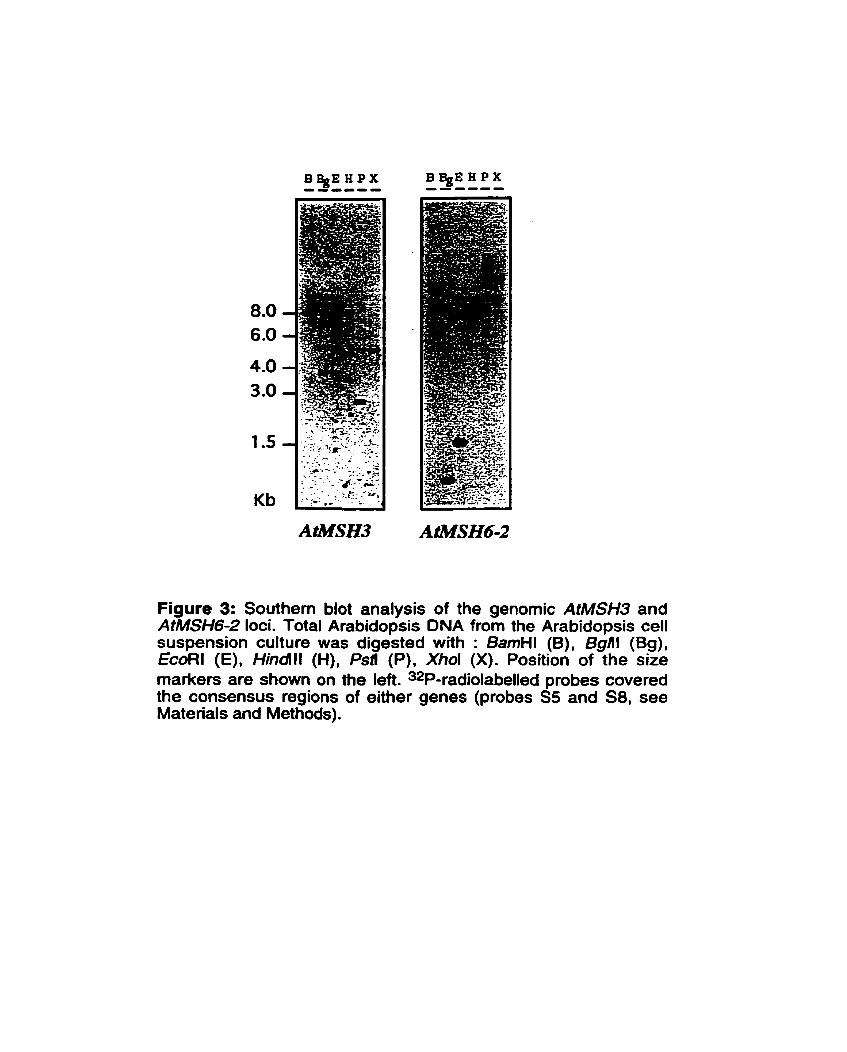
Figure 3: Southem blot analysis of the genomic AtMSH3 and AtMSH6-2 loci. Total Arabidopsis DNA from the Arabidopsis cell suspension culture was digested with : BamHl (B), Bgill (Bg), EcoRl (E), Hindlll (H), Psfi (P), Xhol (X). Position of the size markers are shown on the left. 32P-radiolabelled probes covered the consensus regions of either genes (probes SS and S8, see Materials and Methods).

genome sequencing project (ID ATAF1308, product narne Tl 0M 13.8). Sequence cornparisons indicate that this gene is related to the MSH6 family, since it was the
first AtMSH6 to be released in the databases we have named it AtMSH6-1 in this study. Its full genomic sequence comprises 19 introns of which only two coincide with introns of the AtMSH6-2 genornic sequence (data not shown).
2.3.2 Genetic mapping
Chromosomal positions were determined for the AtMSH2, AtMSH6-2 and AtMSH3 genes. In the case of AtMSH2. a CAPS marker was developed based on a polymorphic Mbol site present in Landsberg erecta DNA but absent from Columbia DNA. This marker was used to follow the segregation at this locus among a population of 96 recombinant inbred lines (L. erecta x Columbia). AtMSH2 was found to reside on the top am of Arabidopsis chromosome l l l at 1.1 CM from locus m105. For AtMSH6-2, a RFLP mapping approach was used on a subset of 24 RI lines and this indicated that AtMSH6-2 was also located on chromosome III and cosegregates with AB13. For AtMSH3, both CAPS and RFLP approaches were unsuccessful due to a lack of detectable polymorphism between the mapping ecotypes. However, the location of the recently sequenced BAC clone (M7J2) containing the A t W 3 gene indicates that it is located on the top of chromosome IV (closest marker PG19). AtMSH6-1 also resides on chromosome IV based on the location of BAC clone Tl 0M13 (closest marker GTl48).
2.3.3 Deduced amino acid sequence cornparison
Initially, the alignement was restricted to the conserved region which comprises the four NTP-binding domains in the C terminal region of the proteins (roughly 250 amino acids, see Fig.1). As a general observation, the different AtMsh deduced protein sequences are more similar to their human counterparts than to the yeast ones. Over the conserved region, AtMsh2 is 71 % identical to the hurnan Msh2 protein, AtMsh3 is 59 % identical to the human Msh3, AtMsh6-1 and AtMsh6-2 are respectively 55 % and 54 % identical to the human Msh6 (see Table 1). While these levels of identity are the highest observed, the Arabidopsis consensus sequences also share more identities with their other respective orthologues than with any paralogous Msh family members. The two Arabidopsis Msh6 amino acid

Table 1: Percent amino-acid identity of the human, yeast and Arabidopsis Msh2, Msh3 and Msh6 sequences. Values were calculated based on the 197 alignable residues as described in Fig. 1. Values in parentheses were calculated for the entirely aligned proteins using CLUSTALW.

sequences differ from each other, but are still closer to each other (58 % identities) than to the human or yeast Msh6. The three proteins described in this study are clearly not members of the Mshl or Msh4 and Msh5 families, the rnitochondrial and meiotic MutS homologues. Comparing the full length proteins corroborates these obseivations, although alignement with CLUSTALW over their complete sequences should be considered less accurate since no further manual refinement was done. This analysis shows that AtMsh2 presents 40 YO identities (61 % similarities) to the entire human Msh2 protein; AtMsh3 is 36 % identical (54 % similar) to the human Msh3 and AtMsh6-2 is 29 % identical (44 % similar) to its human counterpart (31 % identities and 48 % similarities were found between the human Msh6 and AtMsh6- 1). In al1 instances, the levels of identity andor similarity mentioned above are the highest amongst al1 the other combinations of compared proteins presented in Table 1. Furthenore, along the aligned protein sequences, some amino acid motifs or positions are found specifically consewed within each of the three Msh families and the Arabidopsis proteins also present these specific patterns (data not shown).
A phylogenetic study was perfotmed using 197 unambiguously aligned amino acids at or around the consensus region in the C-terminal end (as in Fig. 1) . All available Msh2, 3 and 6 sequences were analysed using a maximum Iikelihood (ML) method which takes into account among-site rate variation through a gamma law. The pattern of the tree comforts the classification established according to the degree of identity (Figure 4). The three groups consisting of Msh2, Msh3 or Msh6 homologs are distinctly defined, and the Arabidopsis Msh sequences we isolated are firmly branched to their respective groups. The evolutionary rate is lowest for Msh2, and highest for Msh3 and Msh6 proteins. In al1 three Msh subgroups, plant and animals (except for the Drosophila Msh2) tend to group together and separated from fungi. The occurence of two intraspecies Msh6 homologues evolving independently may be restricted to Arabidopsis (or plants), since divergence of these two genes seems to have occured after the divergence of plants and animals.
Conservation of few intron positions between the two AtMSHG genes reinforces this observation: if only strictly aligned intron positions are taken into account, two introns are found at the exact same poslion in both AtMSH6 genes (introns 5 and 14 of AtMSH6-2 / introns 7 and 15 of AtMSiYG-l), of which one position is also conserved in AtMSH3 (intron 14 of AtMSH6-2 / intron 15 of AtMSH6-1 / 10 of
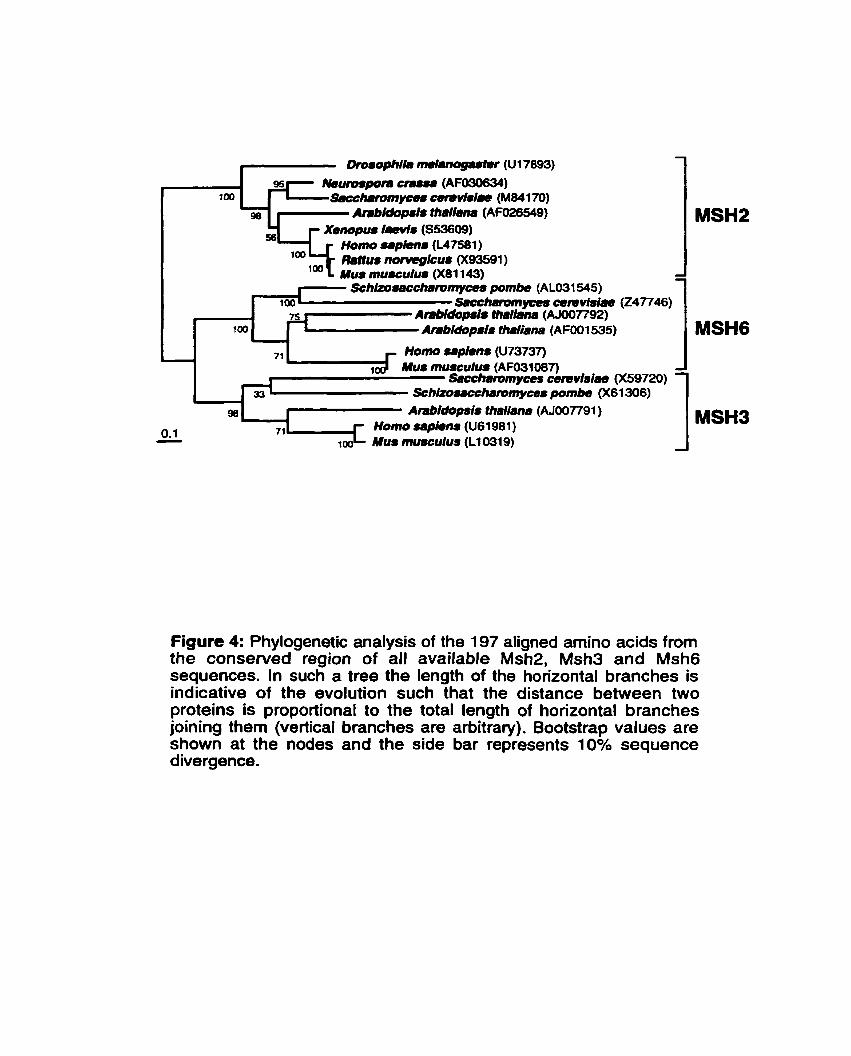
Xenopus I W s (S53609) Homo sapkns (L47581) MUS norvcgicus (X93591) Mus murculus (X81143)
Sch~saecharvmyces pombe (AL031 545) S8ccharomyces ccrisvfsise (Z4T74ô)
llrsbIdbpd~ îhdlibm (AJ007792) AmbMopsIs thaliana (AFûû1535)
Homo mpkns (U73737) MUS muscu~us (~~031087)
Saccharomyces cemviuise (X59720) Schbsaccharumycsr pombe (X6 1 306)
ArablâopsIs thallana (AJOOï791)
O. 1 Homo 8apiens (U6198 1 ) - (LI 031 9)
Figure 4: Phylogenetic analysis of the 197 aligned amino acids from the conserved region of all available Msh2, Msh3 and Msh6 sequences. In such a tree the length of the horizontal branches is indicative of the evolution such that the distance between two proteins is proportional to the total length of horizontal branches joining them (vertical branches are arbitrary). Bootstrap values are shown at the nodes and the side bar represents 10% sequence divergence.

AfMSH3). None of these intron positions are also shared by AtMSH2 or any other MSHG.
2.3.4 Expression studies
Expression of the different AtMSH genes was assessed by Northem blot analysis perfomed with polyA+ RNA (see Figure 5). The size and the low expression level of these genes imposed the extraction of polyA' RNA as they migrate to the same region as the 28s RNA and thus do not focalize enough to produce a noticeable signal. Since AtMSH genes are very poorly expressed in plant tissues (data not shown), we took advantage of an Arabidopsis thaliana cell suspension(Axelos et al. 1992). This cell suspension is mitotic: the cells grow exponentially for the first 5 days following inoculation, before entenng the stationary phase after which the number of growing cells, as measured by their ability to fom protoplasts, starts to decrease (data not shown). Northem blot analysis identified rnRNAs of approximately 3.4 kb for AtMSH2, 3.5kb for AtMSH3 and 3.7 kb for AtMSH6-2 which is in accordance with the sizes predicted from the isolated cDNAs (Fig. 5). At day 2, when the cells are in the early exponentional growth phase, the AtMSH6-2 transcript is expressed at a higher level than at day 8 ; the same is true for AtMSH2 and AtMSH3, albeit to a lesser extent. These data are to be correlated with the AtRAD51 expression pattern, which is higher at day 2 than at day 8 as expected (Doutriaux et al, 1998). The probes used covered the 5' regions of the three genes, outside the consensus regions, and were chosen so that they would not cross-hybridize with the different MSH genes. This is confinned by the fact that a single band of the expected size was detected with each probe.
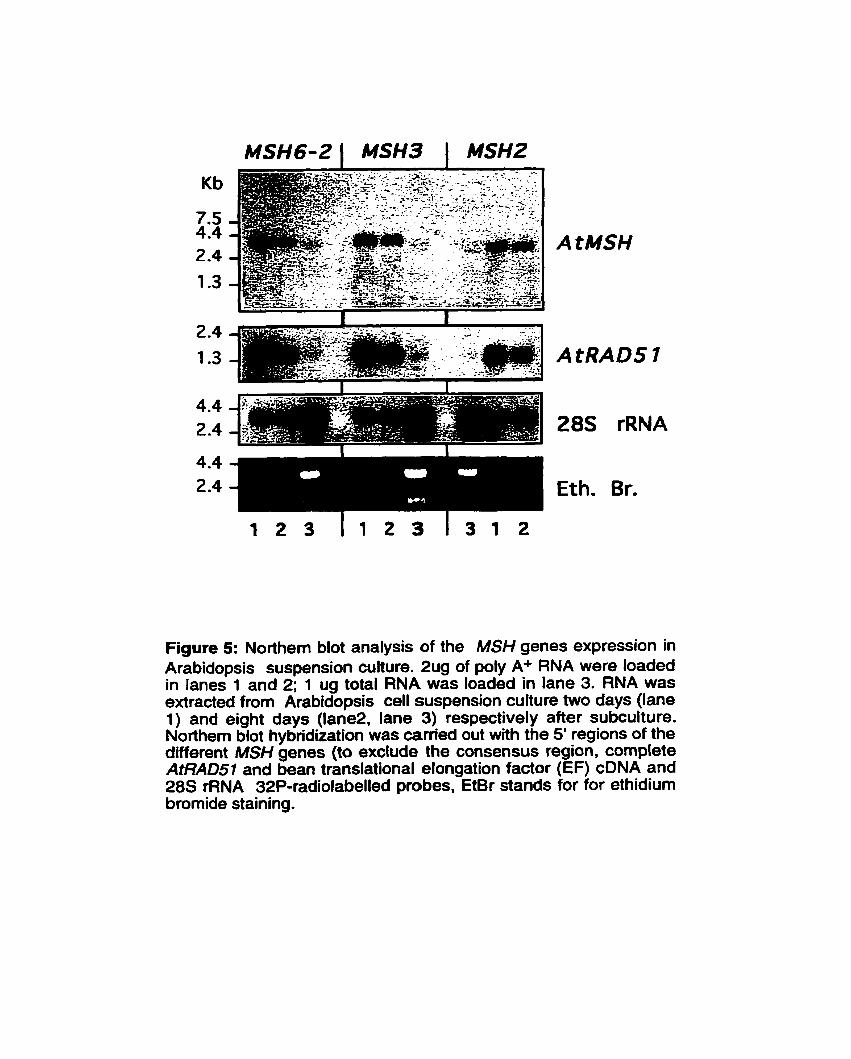
Figure 5: Northem blot analysis of the MSH genes expression in Arabidopsis suspension culture. 2ug of poly A+ RNA were loaded in lanes 1 and 2; 1 ug total RNA was loaded in lane 3. RNA was extracted from Arabidopsis cell suspension culture two days (lane 1) and eight days (lane2. lane 3) respectively after subculture. Northem blot hybtïdization was camed out with the 5' regions of the different MSH genes (to exclude the consensus region, complete AtRAD51 and bean translational elongation factor (EF) cDNA and 28s rRNA 32P-radiolabelled probes, EtBr stands for for ethidium bromide staining.

2.4 Discussion
On the basis of their conservation with MSH genes from other species, we have isolated three Arabidopsis homologues of the mutS gene, known to be essential for the repair of DNA mismatches in Escherichia coli. Taking into account another MSH gene encountered during the course of the Arabidopsis genome sequencing project, four MSH genes are now known in this plant species: AtMSH2, AtMSH3 and two AtMSH6 (-1 and -2). These four genes share the sequence characteristics of al1 MSH family members. The length and molecular weights of the predicted proteins are similar to other MutS homologues. In their C-terminal region, two highly conserved motifs, the NTP-binding domain and a Helix-Turn-Helix domain are found. From sequence comparisons, we classify them as belonging to the Msh2, Msh3 and Msh6 families. At the genomic level, the threeMSH genes we isolated are unique and detected as single bands with restriction enzymes that do not recognize any site in the probed region. Nevertheless, another MSHG homologue exists in Arabidopsis, MSHG- 1, detected du ring the systematic sequencing of Arabidopsis chromosome IV (Johnson et al. 1997). This gene is different from AtMSH6-2 in ternis of sequence, chromosomal location, intron distribution; furthemore, it is not detected with a probe covering the consewed region of AtMSH6-2.
Sequence comparisons of the consewed C-terminal region of the Msh proteins, as
well as over their entire length, clearly allow us to designate the four AtMSH genes of Arabidopsis as belonging ?O the MSH2, MSHG or MSH3 farnilies. Such comparisons also gave rise to other important obse~ations. It appears that the levels of identity are much higher among the Msh2 orthologous proteins than is the case among the Msh3 or Msh6 orthologues. A common idea is that the more interactions proteins are involved in, the lower their evolutionary rate (Dickerson, 1971). As a functional misrnatch repair systern relies on Msh2 binding either to Msh3 or Msh6 followed by an interaction with the Mlhl/Pmsl complex, Msh2 interacts with at least three proteins while Msh3 and Msh6 only bind to Msh2 (Prolla et al. 1994; Acharya et al. 1996). Double hybrid expenments also identified PCNA and Ex01 as partners of the yeast or human Msh2 proteins (Umar et al. 1996; Tishkoff et al. 1997; Gu et al. 1998). Such an experimental approach has not been reported for Msh3 and Msh6 and therefore we cannot exclude that other proteins interact with these gene products. However, al1 indications are in favor of Msh2 being at the center of a

cornplex protein network, which might more severely restrict sequence fluctuations
of this protein.
A phylogenetic study, including al1 available eukaryotic Msh (2, 3 and 6) sequences, confined the previous assignment of each of the four Arabidopsis Msh predicted proteins to the three different Msh families. Analysing the phylogeny of the Msh proteins is complex due to the heterogeneity of evolutionary rates which can produce tree reconstruction artefacts, al1 the more because the number of nucleotides used is low. The difficuky to reconstruct Msh phylogeny is illustrated by the fact that the monophyly of fungi Msh6 sequences is only recovered when among-site variation is taken into account (data not shown). Variation of evolutionary rates is not only observed between paralogues but also between species within each paralogue (see for example the branch lengths of fungi on Fig. 4). The most likely artefact is the long branch attraction phenomenon (Felsenstein, 1978). which generally results in the incorrect early emergence of fast-evolving sequences (Philippe and Laurent, in press). For instance, the Msh2 sequence of Drosophila, which emerges at the base of this group far from other Metazoa. is very likely misplaced because of this phenomenon. Similarly, although fungi are known to be the closest relatives of animals (Baldauf and Palmer 1993), the phylogenies based on Msh always found that animals are closely related to plants and not to fungi which could be due to a higher evolutionary rate of MMR proteins in fungi. lnterestingly the same observation, i.e. an increased evolutionary rate of fungi and Drosophila, has been previously reported in a Rad51 phylogenetic analysis (Yeager Stassen et al. 1997). Finally. the finding that two intron positions are common to both Arabidopsis MSH6 genes and that one of these is also coincident with an intron position in AtMSH3 may argue in favor of the relatedness of these two families.
The occurence of two intraspecies Msh6 homologues evolving independently is unexpected and may be restricted to Arabidopsis (or plants) since divergence of these two genes occured after the separation of plants and animals. Nevertheless. one rnay wonder about the situation in other eukaryotes. As its genome has been completely sequenced, there is no doubt about the uniqueness of MSH6 in S. cerevîsiae. Although a unique MSH6 gene has been described for human and mouse, this cannot be considered definitive as long as the human genome has not been totally sequenced. Despite considerable effort to identify al1 human MSH genes, MSH4 and MSH5, the rneiotic mutS homologues have only recently been discovered (Paquis-Flucklinger et al. 1 997; Her et al. 1998). Except for the particular

case of Sarcophyton glaucum (Pont-Kingdon et al. 1998). no MSHl gene has yet been found in any higher eukaryotes, but we are aware that in the view of the rate and spectrum of mitochondrial mutations in human cells, its existence remains questionable (Khrapo et al. 1997). In fact, uniqueness of the MSH6 genes is greatly supported by the persistence of a specific and similar phenotype when Msh6 is defective in either S. cerevisiae or mammals (Marsichsky et al. 1996; Edelmann et al. 1997). Phylogenetic analysis is also in favor of a plantonly duplication of two Msh6 proteins evolving independently. In the absence of expression or functional studies, it is clear that we cannot yet discuss the effective occurrence of an AtMsh6-1 protein in A rabidopsis. However, the phylogenetic analysis suggests that it is a functional protein, otherwise it would have probably exhibited mutations in the known functional domains. Whether Msh6-2 and AtMsh6-1 coexist in the same tissues or not, are redundant or have acquired different specialized functions will have to be assessed.
The three MSH genes we describe are expressed in Arabidupsis: cDNA clones were successfully obtained and mRNA specific to each gene were detected in Northem blot experiments. The AtMSH2, AtMSH3 and AtMSH6-2 transcripts differ in size and their estimated size correlates with the lengths of the detenined cDNA sequences. All three genes are expressed in a cell suspension derived from Arabidopsis thaliana, slightly more in the exponential growth phase than in the stationary phase. In cornparison, we also find a mvch higher level of AtRAD51 transcripts in the day 2 cells than in the day 8 cells; AtRAD51 has been shown previously to be regulated in S-phase (Doutriaux et al. 1998). Precise assessrnent of the phase of induction in the cell cycle would require cell synchronization. In S. cerevisiae, early S-phase induction has been described for the MSH2 and M S H 6 genes while, MSH3 transcription was found to be constitutive during the cell cycle (Kramer et al. 1996). In Escherichia coli, MutS was also found to be depleted in stationary phase cultures (Feng et al. 1996). Overall, these data support the idea that MSH genes are expressed at a time when cells divide actively and thus replicate their DNA.

Acknowledgements
We thank Catherine Bergounioux, Najat Takvorian and Michael Hodges for valuable comments on the manuscript and Roland Boyer for photographic work. This work was supported in part (M-P. D. and H. P.) by CNRS, université Paris XI, Biogemma and Danone, and in part (J. A. and F. B.) by NSERC (Canada). J. A. also wishes to acknowledge a fellowship from the Programme Canadien de Bourses de la Francophonie m.

References
Acharya S, Wilson T, Gradia S, Kane MF, Guenette S. Marsischky GT, Kolodner R, Fishel R (1996) hMSH2 forms specific mispair-binding complexes with hMSH3 and hMSH6. Proc Natl Acad Sci U S A 93 : 13629-13634
Adachi J, Hasegawa M (1996) PROTML: Maximum Likelihood Interference of Protein Phylogeny. lnstitute of Statistical Mathematics. Tokyo
Akiyama Y, Sato Hl Yamada T, Nagasaki H, Tsuchiya A, Abe R. Yuasa Y (1997) Gerrn-line mutation of the hMSH6/GTBP gene in an atypical hereditary nonpolyposis colorectal cancer kindred. Cancer Res 57 : 3920-3923
Axelos M, Bardet C, Liboz T, Le Van Thai A, Curie Cl Lescure B (1989) The gene family encoding the Arabidopsis thaliana translation elongation factor EF-1 alpha: molecular cloning, characterization and expression. Mol Gen Genet 21 9 : 106-1 12
Axelos M, Curie C, Mazzolini L, Bardet C, Lescure B (1992) A protocol for transient gene expression in Arabidopsis thaliana protoplasts isolated from cell suspension culture. Plant Physiol Biochem 30 : 1-6
Baldauf SL, Palmer JD (1993) Animals and fungi are each other's closest relatives. Science 257 : 74-76
Bhui-Kaur A, Goodman MF, Tower J (1998) DNA mismatch repair catalyzed by extracts of mitotic. postmitotic, and senescent Drosophila tissues and involvement of mei-9 gene function for full activity. Mol Cell Biol 18 : 1436-1 443
Bishop DU, Andersen J. Kofodner RD (1989) Specificity of rnismatch repair following transformation of Saccharomyces cerevisiae with heteroduplex plasmid DNA. Proc Natl Acad Sci U S A 86 : 3713-3717
Brown TC, Jiricny J (1 988) Different baselbase mispairs are corrected with different efficiencies and specificities in monkey kidney cells. Cell 54 : 705-71 1
Bureau TE, Wessler SR (1994) Stowaway: A new family of inverted-repeat elements associated with the genes of both monocotyledonous and dicotylenodous plants. Plant Cell 6 : 907-91 6
Casacuberta E, Casacuberta JM, Puigdomenech P, Monfort A (1 998) Presence of a miniature inverted-repeat transposable element (MITES) in the genome of Arabidopsis thaliana: characterization of the Emigrant family of elements. Plant J 16 : 79-85

Cerovic G, Bozin Dl Dimitrijevic 6 (1991) Mismatch-specific DNA breakdown in nuclear extract from tobacco (Nicotiana tabacum) callus. Plant Mol Biol 17 :
887-894 Church GM, Gilbert W (1984) Genomic sequencing. Proc Natl Acad Sci USA 81 :
1991 -1 995 Culligan KM, Hays JB (1997) DNA misrnatch repair in plants. An Arabidopsis
thaliana gene that predicts a protein belonging to the MSH2 subfamily of eukaryotic MutS homologs. Plant Physiol 115 : 833-839
Cox EC (1976) Bacterial mutator genes and the control of spontaneous mutation. Ann Rev Genet 10 : 1 35-1 56
Datta A, Hendrix Ml Lipsitch M, Jinks-Robertson S (1997) Dual roles for DNA sequence identity and the mismatch repair system in the regulation of mitotic crossing-over in yeast. Proc Natl Acad Sci U S A 94 : 9757-9762
Dellaporta SL, Wood J, Hicks JB (1983) A plant DNA minipreparation: version II. Plant Mol Biol Reporter 1 : 19-21
Dickerson RE (1971) The structures of cytochrome c and the rates of molecular evolution. J Mol Evof 1 : 2645
Doutriaux MP, Couteau F, White C (1998) Isolation and characterisation of the RAD51 and DMCl homologs from Arabidopsis thaliana. Mol Gen Genet 257 : 283-29 1
Drummond JT, Li GM, Longley MJ, Modrich P 1995) Isolation of an hMSH2-pl60 heterodimer that restores DNA mismatch repair to tumor cells. Science 268 :
l909-l912 Edelmann W, Yang K, Umar A, Heyer J, Lau K, Fan K. Liedtke W, Cohen PE, Kane
MF, Lipford JR, Yu N, Crouse GF, Pollard JW, Kunkel T, Lipkin Ml Kolodner R, Kucherlapati R (1997) Mutation in the misrnatch repair gene Msh6 causes cancer susceptibility. Cell 91 : 467-477
Eisen JA (1998) A phylogenornic study of the MutS family of proteins. Nucleic Acids R e s 26 : 4291 -4300
Feinstein SI, Low KB (1986) Hyper-recombining recipient strains in bacterial conjugation. Genetics 1 13 : 13-33
Felsenstein J (1978) Cases in which parsimony or compatibility methods will be positively misleading. Syst Zool 27 : 401-410
Feng G, Tsui HCT, Winkler ME (1996) Depletion of the cellular amounts of the MutS and MutH methyl-directed mismatch repair proteins in stationary-phase Escherichia coli K-12 cells. J Bacteriol 178 : 2388-2396

Folger KR, Thomas K, Capecchi MR (1985) Efficient correction of mismatched bases in plasmid heteroduplexes injected into cultured mammalian cell nuclei. Mol
Cell Biol 5 : 70-74 Genschel J, Littman SJ, Drummond JT, Modrich P (1998) Isolation of MutSbeta from
human cells and comparison of the mismatch repair specificities of MutSbeta and MutSalpha. J Biol Chem 273 : 19895-19901
Gorbalenya AE, Koonin EV (1990) Superfamily of UvrA-related NTP-binding proteins: implications for rational classification of recombinationallrepair systems. J Mol Bi01213 : 583-591
Gu L, Hong Y, McCulloch S. Watanabe H, Li GM (1998) ATP-dependent interaction of human mismatch repair proteins and dual role of PCNA in mismatch repair. Nucleic Acids Res 26 : 1 1 73-1 1 78
Hare JT, Taylor JH (1985) One role for DNA methylation in vertebrate cells is strand discrimination in mismatch repair. Proc Natl Acad Sci U S A 82 : 7350-7354
Her C, Doggett NA (1 998) Cloning, structural characterization, and chromosomal localization of the human orthologue of Saccharomyces cerevisiae MSH5 gene. Genomics 52 : 50-61
Hollingsworth NM, Ponte L, Halsey C (1995) MSH5, a novel MutS homolog, facilitates meiotic reciprocal recombination between homoloys in Saccharomyces cerevisiae but not mismatch repair. Genes and Dev 9 : 1728- 1739
Holmes J Jr, Clark S, Modrich P (1990) Strand-specific mismatch correction in nudear extracts of human and Drosophila melanogaster cell lines. Proc Natl Acad Sci U S A 87 : 5837-5841
Johnson AF, de la Bastide M, Lodhi M, Hoffman J, Hasegawa A, Gnoj L, Gottesman T, Granat S, Hameed A, Kaplan N, Schutz K, Shohdy N, Van Keuren K, Parnell L, Dedhia N, Martienssen R, McCombie W (1997) The sequence of the Arabidopsis thaliana T l 0M13 BAC. ACCESSION AF001308, GenBanWEMBL
Khrapo K, Coller HA, André PC, Li XC, Hanekamp JS, Thilly WG (1998) Mitochondrial mutational spectra in human cells and tissues. Proc Natl Acad Sci U S A 94 : 13798-1 3803
Kimura M (1983) The neutral theory of molecular evolution. Cambridge. Cambridge University Press
Kishino H, Hasegawa M (1989) Evaluation of the maximum likelihood estimate of the evolutionary tree topologies from DNA sequence data, and the branching order in hominoidea. J Mol Evol29 : 170-1 79

Kolodner RD (1 996) Biochemistry and genetics of eukaryotic mismatch repair genes. Genes and dev 10 : 1433-1442
Kramer W, Fartmann B, Ringbeck EC (1996) Transcription of mutS and mutL- homologous genes in Saccharomyces cerevisiae during the cell cycle. Mol Gen Genet 252 : 275-283
Lander ES, Green Pl Abrahamson J, Barlow A, Daly MJ, Linclon SE, Noewbum L (1 987) MAPMAKER: An interactive computer package for constructing pnmary genetic linkage maps of experimental and natural populations. Genomics 1 : 174-1 81
Lister C, Dean C (1993) Recombinant inbred lines for mapping RFLP and phenotypic markers in Arabidopsis thaliana. Plant J 4 : 745-750
Luhr 6, Scheller J, Meyer P, Kramer W (1998) Analysis of in vivo correction of defined mismatches in the DNA mismatch repair mutants msh2, msh3 and msh6 of Saccharomyces cerevisiae. Mol Gen Genet 257 : 362-367
Marsischky GT, Filosi N, Kane MF, Kolodner R (1996) Redundancy of Saccharomyces cerevisiae MSH3 and MSH6 in MSHBdependent mismatch repair. Genes and Dev 10 : 407-420
Miyaki M. Konishi M, Tanaka K. Kikuchi-Yanoshita R, Muraoka M, Yasuno M, lgan T, Koike M, Chiba M, Mori T (1997) Gemline mutation of MSH6 as the cause of hereditary nonpolyposis colorectal cancer. Nat Genet 17 : 271 -272
Modrich P, Lahue R (1996) Mismatch repair in replication fidelity, genetic recombination and cancer biology. Annu Rev Biochem 65 : 101-133
Ohlendorf DH, Anderson WF, Matthews BW (1983) Many gene-regulatory proteins appear to have a similar alpha-helical fold that binds DNA and evolved from a common precursor. J Mol Evol 19 : 109-1 14
Palombo F, Gallinari P, laccarino 1, Lettieri T, Hughes M, D'Arrigo A, Truong O, Hsuan JJ, Jiricny J (1995) GTBP, a 160-kilodalton protein essential for mismatch-binding activity in human cells. Science 268 : 191 2-1 91 4
Paquis-Flucklinger V. Santucci-Darmanin S. Paul R, Turc-Carel C, Desnuelle C (1 997) Cloning and expression analysis of a meiosis-specific MutS homolog: the human MSH4 gene. Genomics 44 : 188-194
Parsons R, Li GM, Longley MJ, Fang WH, Papadopoulos N, Jen J, de la Chapelle A, Kinzler KW, Vogelstein B. Modrich P (1993) Hypermutability and mismatch repair deficiency in RER+ tumor cells. Ce1175 :1227-36
Philippe H (1993) MUST, a computer package of Management Utilities for Sequences and Trees. Nucleic Acids Res 21 : 5264-5272

Philippe H, Laurent J (in press) How good are deep phylogenetic trees? Curr Opin Genet Dev
Pont-Kingdon G, Okada NA, Macfarlana JL, Beagfey CT, Watkins-Sims CD,Cavalier-Smith T, Clark-Walker GD, Wolstenholme DR (1 998) Mitochondrial DNA of the coral Sarcophyton glaucum contains a gene for a homologue of bacterial MutS: a possible case of gene transfer from the nucleus to the mitochondnon. J Mol Evol46 : 419-431
Prolla TA, Pang Q. Alani El Kolodner RD, Liskay RM (1994) MLH1, PMS1, and MSHP interactions during the initiation of DNA mismatch repair in yeast. Science 265 : 1091 -1 093
Rayssiguier Cl Thaler DS, Radrnan M (1989) The barrier to recombination between Escherichia coli and Salmonella typhimurium is disrupted in mismatch -repair mutants. Nature 342 : 396401
Reenan RA, Kolodner RD (1992a) Isolation and characterization of two Saccharomyces cerevisiae genes encoding homologs of the bacterial HexA and MutS mismatch repair proteins. Genetics 132 : 963-973
Reenan RA, Kolodner RD (1992b) Characterization of insertion mutations in the Saccharomyces cerevisiae MSHl and MSH2 genes:evidence for separate mitochondrial and nuclear functions. Genetics 132 : 975-985
Reitmair AH, Schmits R, Ewel A, Bapat 6, Redston M, Mitri Al Waterhouse P, Mittmcker HW, Wakeham A, Liu B, Thornason A, Griesser H, Gallinger SI Ballhausen WG, Fishel R, Mak TW (1995) MSH2 deficient mice are viable and susceptible to lymphoid tumours. Nat Genet 11 : 64-70
Risinger JI, Umar A, Boyd J, Berchuck A, Kunkel TA, Banett JC (1996) Mutation of MSH3 in endometrial cancer and evidence for its functional role in heteroduplex repair. Nat Genet 1 4 : 1 02-5
Ross-Macdonald P, Roeder GS (1 994) Mutation of a meiosis-specific MutS homolog decreases crossing over but not mismatch correction. Cell 79: 1069-1 080
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular Cloning: A Laboratory Manual. Second Edition (Cold Spring Harbor Laboratory Press, New York)
Selva EM, New L, Crouse GF, Lahue RS (1995) Mismatch correction acts as a barrier to homeologous recombination in Saccharomyces cerevisiae. 139 : 1175-1 188
Strimmer K, von Haeseler A (1996) Quartet punling: a quartet maximum likelihood method for reconstructing tree topologies. Mol Biol Evoll3 : 964-969
Swofford DL (1 993) Illinois Natural History Survey, Champaign Illinois

Thomas DG, Roberts JD, Kunkel TA (1991) Heteroduplex repair in extracts of human HeLa cells. J Biol Chem 266 : 3744-3751
Tishkoff DX, Boerger AL, Bertrand Pl Filosi N, Gaida GM, Kane MF, Kolodner RD (1 997) Identification and characterization of Saccharomyces cerevisiae EX01 , a gene encoding an exonuclease that interacts with MSHP. Proc Natl Acad Sci U S A 94 : 7487-7492
Umar A, Boyer JC, Thomas DG, Nguyen DC, Risinger JI, Boyd J, lonov Y, Perucho M, Kunkel TA (1994) Defective mismatch repair in extracts of colorectal and endometrial cancer cell lines exhibiting microsatellite instability. J Biol Chern 259 : 1-4
Umar A, Buerrneyer AB, Simon J, Thomas DC, Clark AB, Liskay RM, Kunkel TA (1996) Requirement for PCNA in DNA mismatch repair at a step preceding DNA synthesis. Cell 97 : 505-51 4
Varlet 1, Canard B, Brooks P. Cerovic G, Radman M (1996) Mismatch repair in Xenopus egg extracts: DNA strand breaks act as signals rather than excision points. Proc Natl Acad Sci U S A 93 :IO1 56-1 01 61
Vulic M. Dioniso F, Taddei F, Radman M (1997) Molecular keys to speciation: DNA polymorphism and the control of genetic exchange in enterobacteria. Proc Natl Acad Sci USA 94 : 9763-9767
de Wind N, Dekker M. Berns A, Radrnan M, te Riele H (1995) Inactivation of the mouse Msh2 gene results in mismatch repair deficiency, methylation tolerance, hyperrecombination, and predisposition to cancer. Cell 82 : 321-330
Yang Z (1996) Among-site rate variation and Rs impact on phylogenetic analyses. Trends Ecol Evol 11 : 367-370
Yeager Stassen N, Logsdon JM, Vora GJ, Offenberg HH. Palmer JD, Zolan ME (1 997) Isolation and characterization of RA 05 1 orthologs from Coprinus cinereus and Lycopersicon esculentum, and phylogenetic analysis of eukaryotic recA hornologs. Cun Genet 31 : 144-1 57.

CHAPITRE III
Functional analysis of the Arabidopsis thaliana mismatch repair gene MSH2.

Functional analysis of the Arabidopsis thaliana mismatch repair gene MSH2.
Jules Ad6 and François J. Belzile
Département de phytologie, Pavillon C.-E. Marchand, Université Laval, Ste- Foy, Canada, G 1 K 7P4.
Manusrcrit soumis à "Theplant Journala

Résumé du manuscrit
Le gène MSHZ d'Arabidopsis thaliana (AtMSH2) code pour une protéine qui est membre d'une famille de proteines très conservées ( MutS Homologues, MSH ) qui sont impliquees dans la correction des rnésappariements d'ADN. Des analyses de séquences avaient suggéré que ce gene simple copie est un homologue de MSH2, un gene jouant un rôle central dans la correction des mésappariements chez les eucaryotes. Cependant. aucune étude fonctionnelle n'a encore démontré un tel rôle pour la protéine AtMSH2. Dans cet article, nous avons montré que la protéine AtMSH2 possède des propri6tds caracteristiques des protéines de correction des mésappariements caractérisées auparavant. D'une part, la surexpression de cette protéine chez E. coli conduit à un phénotype mutateur semblable à celui rapporté pour d'autres homologues fonctionnels. D'autre part, des essais sur gels de retardement ont révélé que la protéine AtMSH2 a une affinité dix fois plus grande pour I'ADN contenant un nucl6otide mesapparié vs. I'ADN parfaitement apparié. Ces résultats constituent la première évidence expérimentale que AtMSH2 est bel et bien un homologue fonctionnel de MutS chez Arabidopsis thaliana.

Abstract
The Arabidopsis thaliana MSH2 (AtMSH2) gene encodes a protein which belongs to a family of highly consewed proteins (MUS Homologues, MSH) involved in DNA mismatch repair. Sequence analyses strongly suggest that this single copy gene is indeed a homologue of MSH2, a gene known to play a central role in eukaryotic mismatch repair. No functional studies, however, have yet demonstrated such a role for the AtMSH2 protein. In this report, we show that the AtMSH2 protein has functional attributes characteristic of previously characterized mismatch repair proteins. Firstty, overexpression of this protein in E. coli leads to a mutator phenotype similar to that reported previously for known functional homologues. Secondly, gel retardation assays revealed that the AtMSH2 protein has a 10-fold greater affinity for DNA containing a single pair of mismatched nucleotides vs. perfectly matched DNA. These results provide the first experimental evidence that AtMSH2 is indeed a functional homologue of MutS in Arabidopsis thaliana.

3.1 Introduction
DNA mismatches occur in al1 organisms as a result of several processes: emrs during DNA replication, DNA damage induced by spontaneous deamination of 5- methylcytosine, DNA damage by physical or chemical agents and genetic recornbination between hornologous but non identical sequences (Modrich, 1991). If they are not corrected before the next round of DNA replication, these mismatches result in mutations which can lead to the accumulation of deleterious mutations and eventually cell death. Therefore, the repair of DNA mismatches is essential to the maintenance of genome stability and cell viability.
DNA mismatch repair systems have been identified in prokaryotic as well as in eu karyotic organisms (Dohet et al., 1 985; Kolodner et al., 1 994; Kramer et a/. , 1 984; de Wind et a/., 1995). The best characterized of al1 mismatch repair systems is the E. coli MutHLS or methyl-directed mismatch repair system (Modrich, 1991). As its narne indicates, this system involves three major gene products, the MutS, MutL and MutH proteins (Lu et aL, 1983). Collectively, these proteins ensure the recognition of rnisrnatched base pairs and the excision of the erroneous base(s) and surrounding nucleotides. The excised segment might Vary from a few hundred nucfeotides up to 1 kb, hence the appellation of long patch mismatch repair.
Among al1 mismatch repair proteins, only MutS is capable of specific interaction with misrnatched DNA in the absence of other proteins and is thus responsible for the recognition of such anomalies. The MutH gene encodes an endonuclease which is responsible for the creation of a single strand nick in hemimethylated or unrnethylated DNA. Unlike MutS and MutH, no activity has been attributed to MutL although it is thought to be a protein-protein interface between MutS and Mutt-i. Thus the current model regarding the initiation of mismatch repair in E. coli proposes the following: MutS protein recognizes and binds to a mismatched base pair, MutL protein then joins the complex fonned by MutS and the mismatched DNA, finally the MutH endonuclease is activated by the MutS-MutL-misrnatch complex and introduces a nick in the DNA. M e r these steps, the action of several cellular enzymes (helicase II, exonucleases and DNA polymerase III) leads to the creation of a single strand gap which is then repaired to restore a fully complementary DNA duplex (Friedberg et al., 1 995).

The mismatch repair machinery haî been well consewed during evolution. Several research groups have identified eukaryotic homologues of MutS and MutL (Bronner et al., 1994; Fishel et al., 1993; Leach et al., 1993; New et al., 1993; Papadopoulos
et al., 1 994; Reenan and Kolodner, 1 992a). Clearly, however, the eukaryotic repair system is more complex as exemplified by the situation in yeast where six homologues of MutS ( M W 1 -6; [Hollingsworth et al., 1995; laccarino et al., 1996; New et al., 1993; Reenan et al., 1992a; Ross-Macdonald and Roeder, 19941) and four homologues of MutL (MLHI, MLH2, MLH3 and PMS1; [Flore-Rozas and Kolodner, 1998; Kramer et al., 1989; Prolla et al., 1994aJ) have been identified that present significant homology to their bacterial counterparts. An obvious challenge in eukaryotes is the assignment of precise roles to each of these homologues.
Recent biochemical and genetic studies have shown that, of the various eukaryotic Mots homologues, MSH2 plays a central role in the recognition of DNA mismatches. Indeed, the initial recognition of mismatched DNA is perfomed by protein complexes composed of either MSH2fMSH3 or MSH2/MSH6; the former ensuring the recognition of insertionsldeletions and the latter of single mispaired bases (Alani, 1996; laccarino et al., 1996; Johnson et al., 1996). Although misrnatch recognition in vivo likely relies on such protein complexes, it has been documented that MSH2 alone can bind rnismatched DNA. For example, the yeast and human MSH2 proteins have been shown to bind mispaired bases as well as insertion mismatches of less than 14 bp (Alani et al., 1995; Fishel et al., 1994; Prolla et al., 1994b).
Genetic analyses in yeast, mice and humans have revealed that msh2 mutants were deficient in mismatch repair (Alani et al., 1994; Reenan and Kolodner, 1992b) and therefore displayed an increased rate of spontaneous mutations (Eshfeman et al., 1993; Reenan and Kolodner, 1992b; Strand et al., 1993). A mutator phenotype has also be observed when expressing heterologous components of the rnismatch repair systern in a wild-type host. For example, an increased mutation rate (up to 200-fold) was seen upon expression of the Streptococcus pneumoniae HexA gene (a homolog of Mut9 in E. coli (Prudhomme et al., 1991 .).
Despite the importance of the MMR system in maintaining the stability of the genome over time, it is only very recently that the first plant homologs of the MutS gene have been cloned in Arabidopsis thaliana (Culligan and Hays, 1 997; Ade et al., in press). Until now, however, no functional analysis of such homologs have been reported. In

this paper, we report the first data to indicate that the Arabidopsis thaliana MSH2 (AtMSH2) gene product has functional attributes characteristic of al1 characterized MutS homologs. Firstly, we show that the Arabidopsis thaliana MSH2 protein is mutagenic when overexpressed in wild-type bacteria and, secondly , t hat this protein preferentially binds to DNA containing mismatched nucleotides, a hallmark feature of MutS and its homologs.

3.2 Materials and methods
3.2.1 Fluctuation test
In order to examine the consequences of expressing AtMSH2 protein in wild-type bacteria, we performed a fluctuation test. To achieve this, the Arabidopsis thaliana MSH2 cDNA was ligated in frame in the Non site of the expression vector pET28a (Novagen) and used to transfomi E. coli strain BL21 (DE3). Two sets of experiment were done. A first set was performed at 37OC. Since preliminary experiments had shown a large variation in the nurnber of viable cells in saturated cultures grown at 37*C, we intercepted cultures in the exponential growth phase. Therefore, twenty individual colonies were selected and grown at 37°C in 3 ml LE3 medium supplemented with 30 pg m l - Kanamycin and 0.5 mM isopropyl-P-D-
thiogalactopyranoside (IPTG) until cultures reached exponential phase (OD6oo around 0.5). The titer of each culture was determined in a separate experiment by plating successive dilutions from each culture. To determine the number of rifampicin resistant colonies in each culture, about 2 x 108 cells were plated on LB agar plus 30 pg mm Kan and 100 pg ml-1 rifampicin. Plates were kept at 37°C overnight and resistant colonies were counted. The second set of experiment was carried out at 28OC. Fourty individual colonies were selected and grown to saturation at 28OC ovemight in 3 ml LB medium supplemented with 30 pg ml-1 Kanamycin and 0.5 mM IPTG. The rifampicin resistant colonies were determined as above &y plating approximately 8 x IO8 cells on LB agar plus 30 pg ml-' Kan and 100 pg ml-' rifampicin. To determine the mutation rate (p) in both cases, we used the maximum likelihood method for smaller counts (Lea and Coulson, 1949) and solved the following equation rn=pn, where m is the maximal likelihood estimate of the zero class with no mutant and n the number of cells. As controls, we performed parallel experiments using the expression vector alone with the same treatrnents as described above.

3.2.2 Expression and partial puriticlition of AtMSH2 protein -
As described earlier, the AtMSH2 cDNA was ligated in frarne in the pET28a expression vector. This construct was used to transfomi E. coli strain BL21 (DE3). LB medium containing an ovemight culture of transformed E. coli in a ratio of t100 was grown at 28°C until the OD600 reached 0.6. The culture was then induced with 0.5 mM IPTG and grown for an additional 6 hours at 28OC. Parallel to this, a control culture of transformed E. coli was grown in the same conditions without induction. One liter of each culture (induced and uninduced) was pelleted by centrifugation at 8,000 g for 10 min and resuspended in 10 ml of lysis buffer (20 rnM NaH2P04, 300 mM NaCi, 5 mM lmidazol pH 8.0) supplemented with 0.5 rnM phenylmethylsulfonil fluoride (PMSF). Cells were broken by two passes through a French Press Cell. The lysed cells were centrifuged at 35,000 g for 30 min at 4'C to remove cellular debris. Supernatant was applied to NiNTA agarose pre-equilibrated with lysis buffer plus PMSF. The column was washed with 3 ml of wash buffer (20 mM NaH2P04, 300 mM NaCI, 20 mM Imidazol pH 8.0) and eluted in 3 ml of elution buffer (20 mM NaH2P04, 300 mM NaCI, 150 mM lmidazol pH 8.0). Three ml of purified protein were concentrated using the Ultrafree-15 Centrifuga1 Filter Device (Millipore) to reduce the volume to 300 pl.
3.2.3 DNA Binding alrsay
Three 24-mer oligonucleotides (#1: CTTATTGCTGGAGnGACAGCTGC; #2: GCAGCTGTCAACTCCAGCAATAAG and #3: GCAGCTGTCÇACTCCAGCAAT AAG) were synthesized such that #i and #2 fom a perfectly matched homoduplex, while #1 and #3 fom a single G/T mismatch heteroduplex after annealing. Ten pmoles of oligonucleotide #1 was labelled using T4-polynucleotide kinase and [y-32P]ATP in a
20 pl reaction volume according to the manufacturer's specifications. The unincorporated label was removed using Microspin 6-25 Columns (Pharmacia). Five pmoles of labelled #1 oligonucleotide were mixed with either 5 pmoles of unlabelled oligonucleotide #2 or #3 in a 20 FI volume. Then, each mixture was heated to 94OC and gradually cooled to 50°C over a period of 2 h and kept at 50% for an additional 12 h for efficient annealing. The annealed duplexes were ethanol precipitated by adding 1/10 volume of 3M sodium acetate and 2 volumes ethanol. The pellet is resuspended in water and stored at -20°C.

For the binding reaction, 1.5 pl of 1 Ox reaction buffer (1 00 mM Tris-HCI pH 7.5; 10 mM EDTA; 0.5 mM DTT; 500 mM KCI; 50% glycerol) was mixed with 5 pg of poly(deoxyinosine-deoxycytosine) and either 500 ng of proteins (from induced or uninduced cells) or no protein (negative control). The reactions were incubated at roorn temperature for 10 min, followed by the addition of 40 fmoles of labelleled homoduplex or heteroduplex and water to a final volume of 15 pl. The reactions were incubated on ice for 30 min before adding 2 pl of 10x loading buffer (50% glycerol) and loading ont0 a 5% non-denaturing polyacrylamide gel. The gel was run at 25 mA at 4OC (for 2 h 30 min) in 0 . 2 5 ~ TBE until the bromophenol blue (loaded in adjacent wells) had migrated 3/4 of the length of the gel. The gel was then fixed in 7% acetic acid for 15 min and dried before being exposed on film for 96 h at -80°C.

3.3 Results and discussion
3.3.1 AtMSH2 protein is mutagenic in E. coli
To investigate the function of the Arabidopsis AtMSH2 gene, a putative mismatch repair gene, the AtMHS2 cDNA was inserted in a prokaryotic expression vector and introduced in wild type E. coli. We wished to examine if this expression could result in negative complementation and thus produce an increased mutation rate (see materials and rnethods). We measured the forward mutation rate (resistance to rifampicin) in bacterial cells which were either expressing or not expressing the AtMSH2 protein. The results of this experirnent are presented in Table 1. As depicted in this table, at 3 P C , cells containing the vector alone (pET28a) or the pET28a-MSH2 construct under inducing conditions (with IPTG) exhibited mutation rates which were not significantly different (1.33M.67 x 10-9 and 2.07k0.84 x 1 Ow9, respectively). In both cases, the results observed were very similar to the riP mutation rate (1.4 x 10'9) reported by Fishel et al. (1 993) and suggested that the expression of AtMSH2 in E. coli was without effect.
In contrast, however, when the cells were grown at 28OC, a different picture emerged. E. coli cells containing the pET28a-MSH2 construct and grown in the presence of IPTG showed an increased mutation rate (8.411.8 x 10-10) compared to the vector alone (1.75W.77 x IO-'*) (Table 1). Thus, at 28OC, the expression of AtMSH2 protein in wild type E. coli generated approximately 5-fold more mutations per cell division compared to the control used in this assay. These results suggest that AtMSHZ causes a mutator phenotype in E. coli, possibly by negative complementation.
This obsenration is in agreement with previous studies investigating the role of Muts hornologs in E. coli. These have consistently shown similar mutator phenotypes. For example, Prudhomme et al. (1 991) expressed the Streptococcus pneumoniae HexA gene while Fishel et al. (1993) expressed the human MSH2 gene in E. coli. In both cases, an increase in the frequency of mutations was seen upon expression of such MutS hornologs in a heterologous host. It has been hypothesized that the MutS
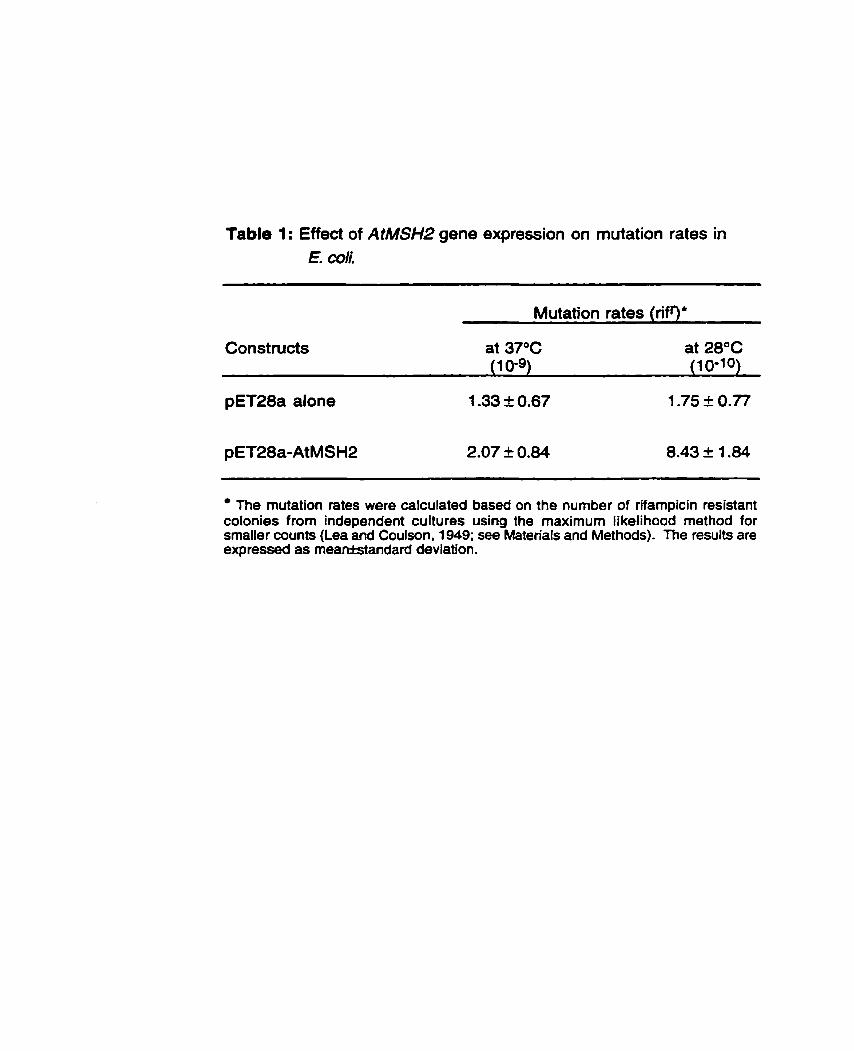
Table 1 : Effect of AtMSH2 gene expression on mutation rates in E. Coli.
Mutation rates (rifg '
Constructs at 37°C at 28°C (1 0-9) (1 0-10)
The mutation rates were calculated based on the number of rifampicin resistant colonies from independent cultures using the maximum Iikelihood method for srnaller counts (Lea and Coulson, 1949; see Materials and Methods). The results are expressed as meanistandard deviation.

homologs (HexA or hMSH2) interact with mismatched nucleotides but are incapable of interacting with the other proteins acting in the repair pathway (MutL and MutH) (Prudhomme et al., 1991). These foreign proteins thereby interfere with endogenous mismatch repair by competing with MutS for binding to mismatches and preventing repair at these sites. The magnitude of the increase in mutation rate was similar to that seen with the human protein (-8.5-fold) while it was lower than tha? observed with the S. pneumoniae protein (up to 200-fold). It is conceivable that eukaryotic MSHP alone (in the absence of MSH3 and MSH6) has a lower affinity for mismatched DNA whereas prokaryotic HexA, which likely acts alone, can bind strongly to mismatched DNA on its own. Our results therefore suggest that the Arabidopsis AtMSH2 protein has the ability to interact with mismatched DNA in E. coli.
As will be described below, the lack of mutator effect at 37OC may have been due to the lack of soluble AtMSH2 protein as the vast majority of fusion protein was found in insoluble form (inclusion bodies). Lower temperatures (such as 28°C) are known to favor the expression of fusion protein in a soluble form. We propose that, at 2a°C, a sufficient amount of AtMSH2 is present in soluble fom to impede mismatch repair in the bacterial cell.
3.3.2 Expression of AtMSH2 protein
Preliminary expression studies had shown that AtMSH2 was expressed at high levels in E. coli at 37OC but that the protein was sequestered in inclusion bodies (data not shown). Although easy to purify, the protein in these inclusion bodies failed to retain any biological activity (as measured by the DNA binding test described below) following its solubilization through a cycle of denaturation and renaturation. Similarly, we were unable to find alternative growth conditions in which high levels of soluble protein could be obtained. We assumed, however, that the increased mutation rate observed for cells grown at 2a°C was indicative of the presence of active and presumably soluble protein. Cells were therefore grown under these conditions and crude protein extracts were loaded ont0 NiNTA columns. Unfortunately, we were unable to achieve a very high degree of enrichment for the his-tagged AtMSH2 protein. Rather, a mixture of proteins was eluted from the

column and no significant difference between partially purified extracts from induced and control cultures could be seen (data not shwon).
3.3.3 Mismatch affinity of recombinant AtMSH2 protein.
The DNA mismatch binding activity of the partially purified protein extracts described above was assessed by using a gel retardation assay. The affinity of each extract for homoduplex or heteroduplex DNA was measured by its ability to bind to DNA and slow its migration through the gel matrix. As illustrated in Figure 1, in the absence of protein (fanes 1 and 2), no retardation of duplex migration is observed. A protein extract from uninduced cells (lanes 5 and 6) also failed to bind either with the perfectly matched homoduplex or with the G/T-mismatched heteroduplex. Sirnilarly, protein extracts purified in the same fashion from cells containing only the vector (pET28a) failed to show any binding to either homoduplex or heteroduplex (data not shown). However, protein extract obtained from induced cells (lanes 3 and 4) displayed a retarded complex with heteroduplex DNA as well as a weaker one with homoduplex DNA. A quantitative analysis of the intensity of the two signals revealed that binding to heteroduplex DNA was approximately 10-fold greater than binding to hornodu plex.
These results show that AtMSH2 protein can bind DNA and that its affinity for a mismatched duplex is 10-fold greater than for homoduplex. This is in agreement with previous findings which revealed that Saccharomyces cerevisiae MSHP protein (Alani et ai., 1995) and human MSH2 protein (Fishel et al., 1994; Whitehouse et al., 1996, 1997) can both bind DNA duplexes containing mismatched nucleotides. Although current models of DNA mismatch repair in eukaryotes suggest that recognition of mismatches in vivo is carried out by protein complexes composed of MSH2/MSH6 or MSH2/MSH3 (Alani, 1996; Johnson et al., 1996), it has been shown that MSH2 alone has misrnatch binding properties in the absence of other partners (Whitehouse et al., 1996; Alani et al., 1995). Our results also indicate that AtMSH2 protein can bind DNA in the absence of mismatches although to a lesser degree. Similar results were obtained by Alani et ai. (1995) who have shown that the yeast MSH2 protein exhibits at least two modes of binding to DNA: first, the formation of nonspecific, unstable complexes and second, the formation of specific, stable complexes that are dependent on the presence of mismatched bases in the
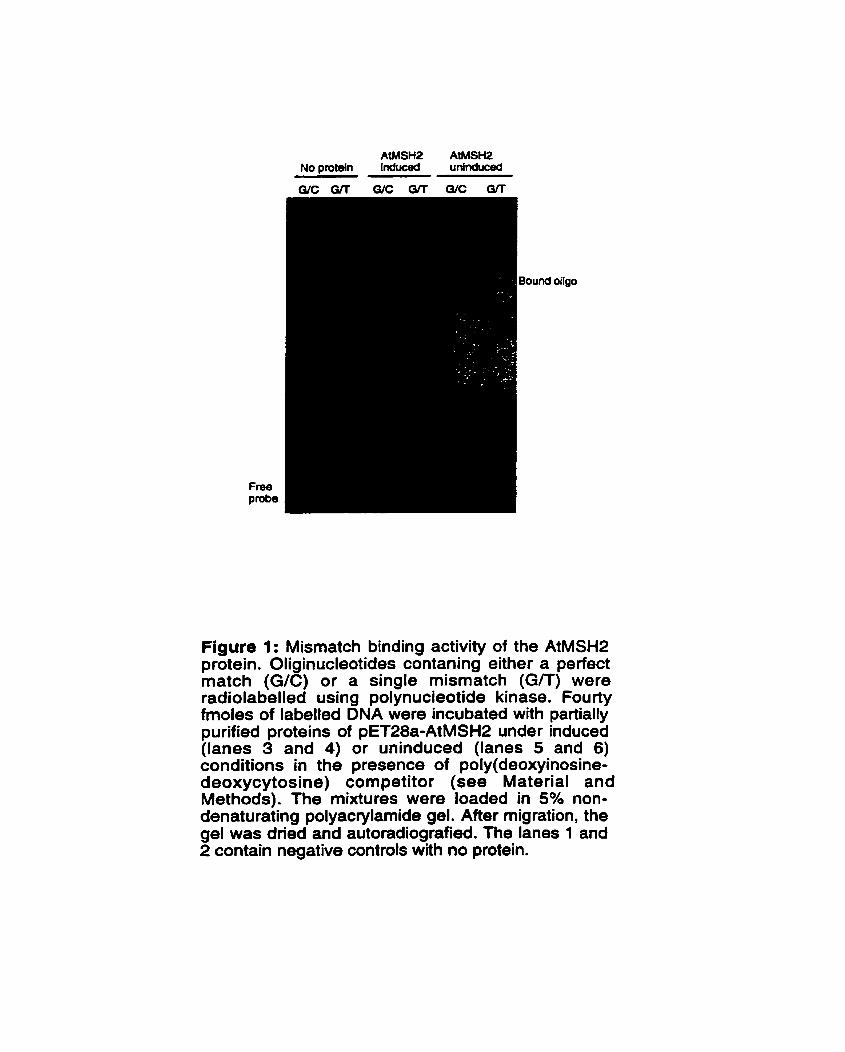
und
Figure 1: Mismatch binding activity of the AtMSH2 protein. Oliginucleotides contaning either a perfect match (G/C) or a single mismatch (GR) were radiolabelled using polynucieotide kinase. Fourty fmoles of labelled DNA were incubated with partially purified proteins of pET28a-AtMSH2 under induced (lanes 3 and 4) or uninduced (lanes 5 and 6) conditions in the presence of poly(deoxyinosine- deoxycytosine) competitor (se8 Material and Methods). The mixtures were loaded in 5% non- denaturating polyacrylamide gel. After migration, the gel was dried and autoradiografied. The lanes 1 and 2 contain negative controls with no protein.

DNA substrates. Presumably, the weaker affinity of AtMSH2 for homodupiex DNA reflects the first mode of binding and its increased affin* for heteroduplex DNA reflects the second more stable interaction with mismatched DNA.
Taken together, the data presented here are the first to indicate that AtMSH2 is indeed a functional homologue of MutS in Arabidopsis thaliana. Efforts are currently under way to identify mutants or transgenic lines in which the AfMSH2 gene has been inactivated so as to directly address the function of this gene in planta.
Acknowledgments
The authors would like to thank Dr. E. Lindsay and D. Spertini for helpful discussions. J. Ad6 was supported by a graduate scholarship from the "Programme Canadien de Bourses de la Francophonie" (Govemrnent of Canada). This work was supported by a research grant from the Natural Sciences and Engineering Research Council of Canada to F. Belzile.

Ref erences
Ade, J., Belzile, J.F., Philippe, H. and Doutriaux, M-P. (1999) Four mismatch repair paralogues coexist in Arabidopsis thaliana: AtMSH2, AtMSH3, AtMSH6- 1 and AtMSH6-2. Mol. Gen. Genet. 262:239-249.
Alani, E. (1996) The Saccharomyces cerevisiae Msh2 and Msh6 proteins form a complex that specifically binds to duplex oligonucleotides containing mismatched DNA base pairs. Mol. Cell. Biol. 16, 5604-561 5.
Alani, E., Chi, N-W. and Kolodner, R. (1995) The Saccharomyces cerevisiae Msh2 protein specifically binds to duplex oligonucleotides containing mismatched DNA base pairs and insertions. Genes & Dev9, 234-247.
Alani, E., Reenan, R.A.G. and Kolodner R. (1994) Interaction between mismatch repair and genetic recombination in Saccharomyces cerevisiae. Genetics 137, 1 9-39.
Bronner, C.E., Baker, S.M., Morrison, P X , Warren, O., Smith, L.G., Lescoe, M.K., Kane, M., Earabino, Cm, Lipford, J., Lindblom, A., Tannegard, P., Bollag, R.J., Godwin, A., Ward, D.C., Nordenskjold, M., Fishel, R., Kolodner, R. and Liskay, R.M. (1994) Mutation in the DNA mismatch repair gene homologue hML H 1 is associated with hereditaiy non-polyposis colon cancer. Nature 368, 258-261.
Culligan, K. and Hays, J.B. (1997) DNA mismatch repair in plants: An Arabidopsis thaliana gene that predicts a protein belonging to the MSH2 subfamily of eukaryotic MutS hornologs. Plant Physiol. 1 1 5 , 833-839.
Dohet, C., Wagner, R. and Radman, M. (1985) Repair of defined single base- pair mismatches in Escherichia coli. Proc Nat1 Acad Sci USA 82. 503-505.
Eshleman, J.R., Lang, E.Z., Bowerfind, O.K., Parsons, R., Vogelstein, B., Willson, J.K., Veigl, M.L., Sedwick, W.O. and Markowitt, S.D. (1993) lncreased mutation rate at the hprt locus accompanies microsatellite instability in colon cancer. Oncogene 10, 33-37.
Fishel, R., Ewel, A., and Lescoe, M.K. (1994) Purified human MSH2 protein binds to DNA contaning mismatched nucfeotides. Cancer Res., 54, 553905542.
Fishel, R., Lescoe, M.K., Rao, M.R.S., Copeland, N.G., Jenkins, N.A., Garber, J., Kane, M. and Kolodner, RD. (1993) The human mutator

gene homolog M S H 2 and its association with hereditary nonpolyposis colon cancer. Ce11 75, 1 027-1 038.
Flores-Rozas, Hm and Kolodner, R.D. (1 998) The Saccharomyces cerevisiae MLH3 gene functions in MSH3-dependent suppression of frameshift mutations. Proc. Natl. Acad. Sci. USA. 95, 1 2404- 1 2409.
Friedberg, E.C., Walker, G.C. and Siede, W. (1995).DNA repair and mutagenesis. ASM Press, Washington, D.C.
Hollingsworth, N.M., Ponte, Lm and Halsey, Cm (1995) MSHS, a novel MutS Hornolog, facilitates meiotic reciprocal recombination between hornologs in Saccharomyces cerevisiae but not mismatch repair. Genes & Dev 9, 1728- 1739.
laccarino, L., Palombo, F., Drummond, J., Totty, NsFs, Hsuan, J.J., Modrich, P. and Jiricny, J. (1996) MSH6, a Saccharomyces cerevisiae protein that binds to mismatches as a heterodimer with MSHP. Current Biology 6, 484-486.
Johnson, RE., Kovvali, O.K., Prakash, L. and Prakash, S. (1996) Requierement of the yeast MSH3 and MSH6 genes for MSH2 dependent genomic stability. J. Biol. Chem. 271, 7285-7288.
Kolodner, RD., Hall, NA., Lipford, J., Kane, M.F., Rao, M.R.S., Morrison, P., Wirth, Lm, Finan, P.J., Burn, J s , Chapman, P., Earabino, Cs, Merchant, E. and Bishop, D.T. (1994) Structure of the human MSH2 locus and analysis of two Muir-Torre kindreds for msh2 mutations. Genomics 24, 51 6-526.
Kramer, B., Kramer, W. and Fritz, H.J. (1 984) Different base/base mismatches are corrected with different etficiencies by the rnethyl-directed mismatch repair system of E. coli. Cell38, 879-887.
Kramer, W., Kramer, B., Williamson, M.S. and Fogel, S. (1989). Cloning and nucleotide sequence of DNA mismatch repair gene P M S l from Saccharomyces cerevisiae: homology of PMS 1 to prokaryotic MutL and HexB. J Bacteriol. 171, 533905346.
Lea, D.E. and Coulson, As (1949) The distribution of numbers of mutants in bacterial populations. J. Genet. 49, 264-285.
Leach, F.S., Nicolaides, N.C., Papadopoulos, N., Liu, B., Jen, J., Parsons, R., Peltomaki, P., Sistonen, P., Aaltonen, L.A., Nystrom- Lahti, M., Guan, X.Y., Zhang, J., Meltzer, P.S., Yu, J.W., Kao, F.T., Chen, D.J., Cerosaletti, K.M., Fournier, R.E.K., Todd, S., Lewis, Tm,

Leach, R.J., Naylor, S.L., Weissenbach, J., Mecklin, J.P., Jarvinen, Hm, Petersen, G.M., Hamilton, SR., Green, J., Jass, J., Watson, P., Lynch, H.T., Trent, J.M., de la Chapelle, A., Kinzler, K.W. and Vogelstein, B. (1993) Mutations of a MutS homolog in hereditary nonpolyposis colorectal cancer. Ce11 75, 121 5-1 225.
Lu, A.L., Clark, S. and Modrich, P. (1983) Methyl-directed repair of DNA base pair mismatches in vitro. Proc. Natl. Acad. Sci. USA. 80, 4639-4643.
Modrich, P. (1991) Mechanism and biological effects of mismatch repair. Ann Rev Genet 25, 229-253.
Modrich, P. and Lahue, R. (1996) Mismatch repair in replication fidelity, genetic recombination and cancer biology. Annu Rev Biochem 65, 101 -1 33.
New, Lm, Liu, K. and Crouse, G.F. (1993) The yeast gene MSH3 defines a new class of eukaryotic MutS homologues. Mol Gen Genet 239, 97-1 08.
Papadopoulos, N., Nicolaides, N.C., Wei, Y.F., Ruben, S.M., Carter, K.C., Rosen, C.A., Haseltine, W.A., Fleischrnann, RD., Fraser, C.M., Adams, M.D., Venter, J.C., Hamilton, SR., Petersen, G.M.,Watson, P., Lynch, H.T., Peltomaki, P., Mecklin, J.P., de la Chapelle, A., Kinzler, K.W. and Vogelstein, B. (1994) Mutation of a mutL homolog in hereditary colon cancer. Science 263, 1625-1 628.
Prolla, T.A., Christie, D.M. and Liskay, R.M. (1994a) Dual requirement in yeast DNA rnismatch repair for MLHl and PMS1, two homologs of the bacterial mutL gene. Mol Ce11 Bi01 14, 407-41 5.
Prolla, T.A., Pang, Q., Alani, E., Kolodner, R. and Liskay, R.M. (1994b) MLH1, PMS1, and MSH2 interactions during the initiation of DNA mismatch repair in yeast. Science 265, 1 091 - 1 093.
Prudhomme, M., Mejean, V., Martin, B. and Claverys, J-P. (1991) Mismatch repair genes of Streptococcus pneumoniae: HexA confers a mutator phenotype in Escherichia coli by negative complementation. J. Bacteriol. 173, 71 96-7203.
Reenan, R.A.G. and Kolodner, R. (1992a) Isolation and characterization of two Saccharomyces cerevisiae genes encoding homologs of bacterial HexA and MutS mismatch repair proteins. Genetics 132, 963-973.
Reenan, R.A.G. and Kolodner, R. (1 992b) Characterization of insertion mutations in the Saccharomyces cerevisiae MSHI and MSH2 genes: evidence for separate mitochondrial and nuclear functions. Genetics 132, 975- 985.

Ross-Macdonald, P. and Roeder, G.S. (1994) Mutation of a meiosis-specific MutS homolog decreases crossing over but not mismatch correction. Ce// 79. 1069-1 080.
Strand, M., Prolla, T.A., Lyskay, R.M. and Petes, T. (1993) Destabilization of tracts of simple repetitive DNA in yeast by mutations affecting DNA mismatch repair. Nature 365, 274-276.
Whitehouse, A., Taylor, G.R., Deeble, J., Philips, SmEwVw, Meredith, Mm and Markham, A.F. (1996) A carboxy terminal domain of the hMSH2 gene product is sufficient for binding specific mismatched oligonucleotides. Biochem Biophys Res Commun 225, 289-295.
Whitehouse, A., Deeble, J., Taylor, G.R., Guillou, P.J., Philips, SmEmVw, Meredith, M. and Markham, A.F. (1997) Mapping the minimal domain of hMSH2 sufficient for binding mismatched oligonucleotides. Biochem Biophys Res Commun 232, 1 O- 1 3.
de Wind, Nw, Dekker, M., Berns, A., Radman, MW and Riele, H. (1995) Inactivation of the mouse Msh2 gene results in mismatch repair deficiency, methyl tolerance, hyperrecombination, and predisposition to cancer. Ce11 82, 321 -330.

CHAPITRE IV
Hairpin Elements, the first family of Foldback Transposons (FTs) in Arabidopsis thaliana

Note : Comme mentionné dans le chapitre 1, les deux principaux objectifs de cette thèse sont, d'une part de cloner et caract6riser le gène MSH2 chez Arabidopis thaliana, et d'autre part de montrer que le produit de ce gène est fonctionnel. Les deux volets précedents (chapitres 2 et 3) ont comble ces objectifs. Mais parallèlement à nos travaux, une autre équipe à isole l'allèle AtMSH2 chez Columbia (cf. chapitre 2). La comparaison des deux allèles nous a révélé la presence d'un Bl6rnent transposable dans celui de Landsberg erecta. Le présent chapitre, rédigée sous forme de publication, est consacré à la caract6risation de ce transposon.

Hairpin Elements, the first family of Foldback Transposons (FTs) in Arabidopsis thaliana.
Jules Ad6 and François J. Belzile
Département de phytologie, Pavillon C.-E. Marchand, Université Laval, Ste- Foy, Canada, G1 K 7P4.
Running title: Foldback Transposons in Arabidopsis. Key words: Arabidopsis / Foldback Transposons 1 Hairpin elements / Phylogeny I Transposable elements.
The Plant Joumel (1 999) 19. 59 1 -597

Résume du manuscrit
Nous avons identifié chez Arabidopsis thaliana une nouvelle famille d'éléments transposables nornm6e Haitpin. Ces éléments possbdent toutes les caractéristiques structurales des transposons ~Foldbackm (FTs) décrits pour la première fois chez la drosophile et très récemment chez les solanac6es. Les éléments Hairpin sont membres de la première famille de fTs identifide chez Arabidopsis thaliana, et même la première famille de FTs de type 3 décite dans le règne végétal. Contrairement aux FTs d&crits pr6cédemrnent, Hairpin paraît être une famille homogène aussi bien par la taille (238k7 pb) que par la structure. Les éléments Hairpin sont disperses dans le gdnome dlArabidopsis et l'hybridation Southem a révélé qu'ils sont présents en un nombre de copies relativement faible. Finalement, nous avons discute de 11utilit6 potentielle de ces éléments pour étudier les relations phylog&&iques entre écotypes dlArabidopsis.

Summary
We report here the identification in Arabidopsis thaliana of a new family of transposable elements named Hajrpin. These elements bear al1 the structural characteristics of Foldback Transposons (FTs) first described in Drosophila and very recently in Solanaceae. Hairpin elements are the first family of FTs reported in Arabidopsis thaliana and the first family of FTs of type 3 to be described in the plant kingdom. In contrast to previous FTs described, Hairpin appears to be a homogeneous family in size (238k7 bp) as well as in structure. Hairpin elements are dispersed in the Arabidopsis genome and Southem hybndization revealed that they are present in relatively low copy number. Finally, we discuss the potential usefulness of these elements to study the phylogenetic relationships between Arabidopsis ecotypes.

4.1 Introduction
In the last three decades, our view of transposable elements has changed entirely; from genetic oddities found in a limited number of organisms to the present recognition that they are ubiquitous and can constitute a very significant portion of an organism's genorne. Key characteristics of these genetic entities make them interesting objects of inquiry. Contrary to the vast majority of sequences found in a genome, they are mobile and their insertion into a gene or its vicinity can alter this gene's function (Banville and Boie, 1 989; Baumruker et al., 1 988; Grandbastien, 1992; Maichele et a/., 1993). In addition to this mutagenic activity, they can act to modify the structure of a genome through inversions, deletions and translocations (Collins and Rubin, 1984). In view of these properties, it has been hypothesized that these elements participate in the shaping and reshaping of a genome particularly in times of stress (Walbot, 1992).
Based on their structure and mechanism of transposition, numerous classes of transposable elements have been described (Grandbastien, 1992). Among these, Foldback elements present a unique structure because when denatured and allowed to reanneal under dilute DNA conditions favoring intramolecular renaturation, the inverted repeat sequences fold back rapidly, yielding a hairpin structure. Foldback Transposons as a whole form a very heterogeneous group as they Vary considerably in size and structure (Rebatchouk and Narita, 1997; Truett et
al., 1981).
Typically, Fold back Transposons are cornposed of two inverted repeats (1 R*) separated by a middle domain (M). Each IR contains two distinct sequence domains: an outer domain (IR-OD) composed of a series of imperfect direct repeats and an inner domain (IR-ID) that consists of a different non-repeating nucleotide sequence which is AT-rich (Hoffman-Liberrnann et al., 1989). Rebatchouk and Narita (1 997) referred to the elements of this classical structure as type 1; type 2 elements lack IR- 10s and type 3 lack IRODs. Therefore, Foldback Transposons of type 3 have two IR- IDs separated by a middle segment (M). Like most transposable elements, Foldback Transposons are generally flanked by direct repeats which correspond to a duplication of the sequence at the insertion site.
In this paper, "IR" is used as an abbreviation for "inverted repeat" rather than "IVR' used by Hoffman- Libennann et al. (1989) and Rebatchouk and Nanta (1997).

Very recently , the Solanaceae Foldback Transposons ( SoFT) have been described in tomato and potato (Rebatchouk and Narita, 1997) suggesting that Foldback Transposons are ubiquitous among eukaryotes. Within the SoFT family, SoFTl and SoFT2 have IR-ODs consisting of tandemly arranged subrepeats whereas the IR-IDs are non-repetitive and AT-rich; they are classified as type 1 . In contrast, SoFT3 has IRs consisting of only IR-ODs and therefore is a type 2 element.
Representatives of most of the known transposable element classes have been reported in Arabidopsis thaliana despite its small genome size. But until now, no Foldback Transposons had been identified. In this paper, we repott the first family of Foldback Transposons in Arabidopsis thaliana and these have been named Hairpin. The structural characteristics of Hairpin elements are consistent with their being Foldback Transposons of type 3: they consist of AT-rich IR-IDs which have the potential to form stable DNA secondary structure and which are separated by a short rniddle segment (6-7 bp). Our results indicate that Hairpin elements are present in the genome of Arabidopsis thaliana in relatively low copy number. Finally, we illustrate and discuss how the presence/absence of these elements can be used to study phylogenetic relationships between Arabidopsis ecotypes.

4.2 Results
4.2.1 The Landsberg erecta allele of the AtMSHZ gene contains s transposon-like sequence in its 3' region.
Sequence cornparison of the Arabidopsis rnismatch repair gene AtMSH2, from ecotype Landsberg erecta (Ler; AF109243). recently cloned in our laboratory, with the corresponding Columbia allele (Col; AF003005) revealed the presence of an additional 244 bp in the Ler allele. This insertion is located in the 3' region of the gene, 196 bp downstream from the stop codon. Upon closer inspection, it was noticed that this insertion resembled a transposable element. Indeed, a 239 bp sequence is flanked by a direct duplication of a 5 bp sequence (CTTAG) which is present in single copy in the Col allele at the site of insertion. Also, the termini of this insertion sequence appeared to be inverted repeats (IR) : the first five nucleotides are perfect IR and, allowing various mismatches or gaps, the IR nature of the temini coutd be extended further. As this element is very short (239 bp), extremely AT-rich (75.7%) and contains no significant open reading frame, it is likely a non- autonomous element.
4.2.2 Halrpin-1 is a member of a family of Foldback transposons in Arabidopsis thaliana.
To further characterize this insertion, we examined sequence databases for homologous sequences. Thes8 revealed six additional highly homologous sequences in the Arabidopsis genome. No significant matches were found in other organisms. Sequence identity between these related sequences ranged between 79 and 92% (among al1 pairwise comparisons) indicating that these sequences are closely related (Figure 1). As can be seen in this figure, sequence identity is high throughout these elements and allows one to derive a consensus sequence which is also shown in Figure 1.
Of the seven elements. only Hairpin-1 presents a perfect direct duplication (CTTAG) at the insertion site (Figure l ) , a sequence which is found in single copy in the Col allele of the AtMSH2 gene. The other six elements are however flanked by sequences which not only are imperfect direct repeats but also closely resemble the


direct repeat flanking Hairpin- 1. For example, Hairpin-4 is bordered on its left by
CTTAG and on its right by CTAAG. A consensus for the sequences flanking Hairpin elernents yields ClTAG on the left side and CTAAA on the right side. Considering both sides simultaneously, a single consensus sequence can be obtained and expressed as C10T14T7A13G8 where the numerical indices give the number of occurences of this base at a given position among the 14 sequences. This apparent conservation of the sequences ftanking Hairpin insertions may indicate an insertion site preference for this family of transposons.
A secondary structure analysis perfonned on the various Hairpin elements and the consensus showed that the consensus sequence has the potential to form an aimost perfect hairpin (Figure 2a). The left half of the element is almost a perfect IR relative to the right half. This striking structural feature is also conseived, albeit to a slightly lesser degree, in the individual elements themselves as is illustrated for Hairpin-1 (Figure 2b). Because of this feature, we have named the elements of this family of transposon-like sequences "Hairpin" elements (Hairpin-1 to -7).
The main features of these H a i ~ i n elements are summarized in Table 1. They form a very homogeneous family as al1 are small (238 f 7 bp), AT-rich (77 t 3%) and closely related to the consensus sequence (83.2-91.4% identity). In al1 cases, the left and right IR are almost identical in size (1 10-123 bp) and al1 show the potential to form a very stable hairpin structure (AG values range between -54.0 to -92.8 Kcal mol-'). Finally, the Hairpin elements are located at variable positions in the genome of Arabidopsis thaliana: Hairpin-2, -4, -5 and -6 were located on different and widely dispersed BAC clones from chromosome 2, Hairpin-3 and -7 were located on different BAC clones from chromosome 5, white Hairpin- 1 was located on chromosome 3 (Table 1 ). This result suggests that Hairpin elements are dispersed in the genome of Arabidopsis theliana.
4.2.3 Hairpin elements are preaent in low copy number in the Arabidopsis genome.
To determine the number of copies of Hairpin elements in the Arabidopsis genome, we perfomed a Southem hybridization using Hairpin-1 as a probe on genomic DNA from different ecotypes of Arabidopsis (Figure 3). As the enzymes used (Hindlll and EcoRI) do not cut within the characterized Hairpin elements, each fragment

(a) Consensus Energy: AG=-91.1 kcal mol-'
(b) Hairpin-1 Energy: AG=-82.2 kcal mol-1
Figure 2: Predicted DNA secondary structure of the consensus Hairpin sequence (a) and of the Hairpin-1 element (b). These structures were generated using the FOLD program of UWGCG. The energy values indicate the stability of these structures.
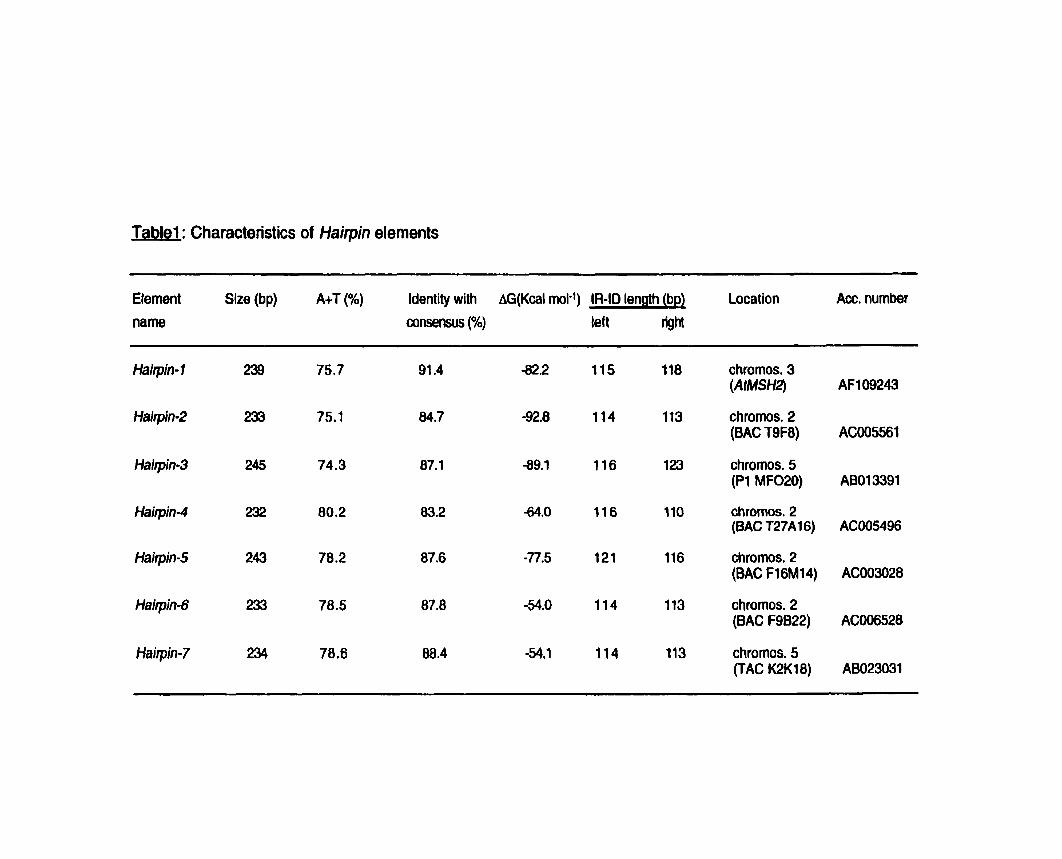
Table1 : Characteristics of Hairpin elements
Efement Size (bp) A+T (%) ldentity with AG(Kcal mol-') IR-ID length (bp) Location Acc. number
name consensus (%) teft @lm
Haitpin- 1 239 75.7 91.4 -82.2 115 118 chromos. 3 (AtMSH2)
Hairpin-2 233 75.1 84.7 -92.8 114 113 chromos. 2 (BAC T9F8)
Hairpin-3 245 74.3 87.1 89.1 116 123 chromos. 5 (Pl MF020)
Hajrpin-4 232 80.2 83.2 44.0 116 110 chromos. 2 (BAC T27Al6)
Hairpin-5 243 78.2 87.6 -77.5 121 116 chromos. 2 (BAC F16Ml4)
Haiipin-6 233 78.5 87.8 -54.0 114 113 chromos. 2 (BAC F9B22)
Hairpin-7 234 78.6 88.4 -54.1 114 113 chromos. 5 (TAC K2K18)
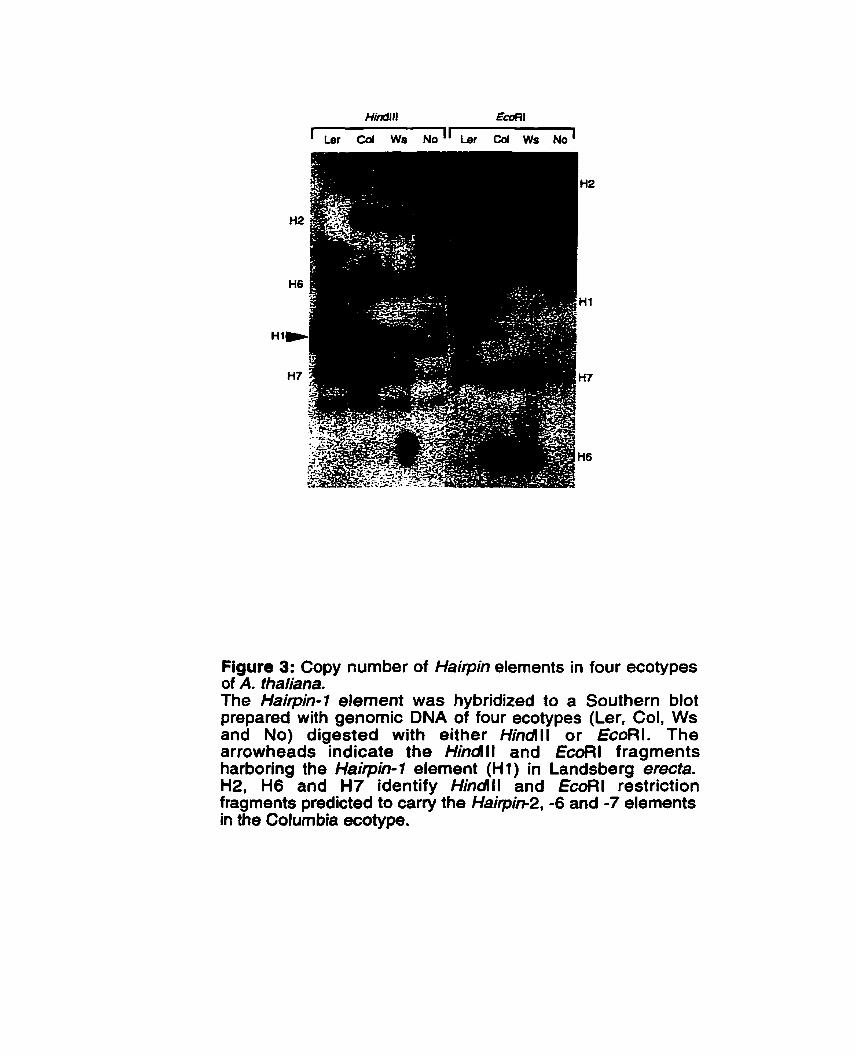
Figure 3: Copy number of Hairpin elements in four ecotypes of A. fhaliana. The Hairpin-1 element was hybridized to a Southern blot prepared with genomic DNA of four ecotypes (Ler. Col, Ws and No) digested with either Hindll or EcoRI. The arrowheads indicate the Hindll and EcoRl fragments harboring the Hairpin-1 element (Hl) in Landsberg erecta. H2, H6 and H7 identify Hindlll and EcoRl restriction fragments predicted to carry the Haipim2, -6 and -7 elements in the Columbia ecotype.

obsewed likely indicates a different copy of the element. In three of the four ecotypes examined (Landsberg erecta, Columbia and Wassifewskija), a total of five copies were observed. In Nossen, this number may be greater (approximately ten copies) although partial digestion could also explain in part the observed increase in the nurnber of bands. In some cases, the elements seem to be at the same locus in different ecotypes as indicated by shared bands (Figure 3). Restriction fragment polymorphisms are seen however and suggest either that the elements are not at the same locus in the different ecotypes or, altematively, that there are differences in the location of the adjacent restriction sites. In the case of the Hairpin-7 element, which is located in the 3' region of the AtMSH2 gene in ecotype Landsberg erecta, it resides on a 3 kb Hindlll fragment and on a 4 kb EcoRl fragment (both indicated by an arrowhead in Figure 3). Fragments of the same size are not seen in the other three ecotypes suggesting that the location of the Hairpin-1 element is not the same in al1 ecotypes or that a copy of this element is not present in the other ecotypes. Also, we have compared the restriction fragments obseived on the Southem for the Col ecotype with those predicted based on the genornic sequence data (BAC clones; also derived from this ecotype). In three cases, Hairpin-2, -6 and -7, restriction fragments of the expected size are found suggesting that these elements are detected using the Hairpin-1 element as probe. Fragments consistent with the detection of the other three elements (Hairpin-3, -4 and -5) were not observed. lnterestingly the three elernents for which restriction fragments of the expected size were observed were those which share the greatest sequence identity with Hairppin- 1. This suggests that Southern hybridization allows the detection of only the most closely-related Hairpin elements.
4.2.4 Hairpin elements are useful indicators of the phylogenetic relafjonships between ecotypes
The presence of Hairpin-1 in one ecotype (Ler) and its absence in another (Col) suggested that this element had likely transposed since the divergence of these two ecotypes. The lack of an excision footprint in the Col allele of AtMSH2 suggested that Hairpin-1 had inserted into the Ler allele rather than been lost from the Col allele. To verify this hypothesis, we examined the 3' region of the AtMSH2 gene in a collection of twenty-five ecotypes of Arabidopsis. We synthesized two primers flanking Hairpin-1 and these were used to amplify this region of the AtMSH2 gene in

the different ecotypes. A PCR product of 341 bp (indicating the presence of the Hairpin-7 element) was obtained in only three ecotypes (Ler, Cvi-O and Bla-1), while the twenty-two other ecotypes produced a PCR product of 97 bp indicative of the absence of Hairpin-1 at this locus (Figure 4). The sequence of this 97 bp product was detennined for three ecotypes (No, Nd and Ws) and in each case, it was identical to the Col allele. No sign of somatic excision was seen among the three ecotypes harboring the Hairpin- 1 insertion as no 97-bp product was detected in these ecotypes. These results strongly suggest that Hairpin-7 transposed into the AtMSH2 3' region in an ancestor common to the Ler, Cvi-O and Bla-1 ecotypes.

1kb Ler Coi Ws No
Figure 4: Survey of different Arabidopsis thaliana ecotypes for the presence of Hairpin-7 at the AtMSH2 locus. The 3' region of AtMSH2 was examined by PCR for the presence of Hairpin-1. The first lane contains a molecular weight rnarker (1 kb DNA ladder, Gibco-BRL). In the following lanes, results are shown for four (Ler, Col, Ws and No) of the 25 ecotypes examined. A 341 pb product is seen in the presence of Hairpin-1 while a 97 bp product is obsewed in its absence.

4.3 Discussion
In this paper, we report the characterization of a novel family of mobile elements, named Hairpin. in Arabidopsis thaliana. The seven family members identified to date are highly homologous but differ significantly from al1 previously described mobile elements in Arabidopsis in ternis of both sequence and secondary structure. The most striking structural feature characterizing the rnemben of this family is that the left half of each element is almost a perfect inverted repeat relative to the right haif. Therefore, each element can fold back to form a stable hairpin structure separated by a short middle segment of 6-7 bp. This characteristic is unique among the Arabidopsis transposable elements described to date. These features led us to classify the Hairpin elements as members of the Foldback type of transposable elements first described in Drosophila (Potter et al., 1 980).
Foldback transposons represent a structurally diverse group of transposons which have been divided into three subclasses according to their structure (Hoffman- Lieberrnann et al., 1989; Rebatchouk and Narita, 1997). The Hairpin elements we describe here are cleariy of type 3. Such elements are characterized by AT-rich inverted repeats (IVR-ID according to the language used by HoMan-Liebermann et al., 1989) separated by a middle segment of variable length which can be as short as a few nucleotides. Hairpin elements match this description perfectly. To the best of OUT knowledge, Hairpin is the first family of Foldback elements reported in Arabidopsis thaliana and the fint Foldback element of type 3 ever to be reported in the plant kingdom. In contrast to most Foldback elements described previously (Potter et al., 1980; Rebatchouk and Narita, 1997; Truett et al.. 1981), Hairpin elements appear to be a very homogeneous family in terms of their size (238k7 bp) and structure.
Apart from Hairpin-1, no other element of this family is flanked by perfect direct repeats. As for the other elements, they are flanked by sequences which can be described as imperfect direct repeats. Such apparently degenerate repeats have been docurnented in other studies (Bureau and Wessler, 1992; Rebatchouk and Narita, 1997). Thus, al1 Haïpin elements are flanked by sequences which resemble a consensus sequence (CTTAG), in which the third and fifth positions are the most variable. This bears a striking resemblance with the CTNAG insertion site of the Tc4

transposable element of C. elegans which also presents a foldback structure (Yuan et aL, 1991). This similarity may be indicatative of a shared transposition medianism between Hairpin and Tc4 elements. Most importantly, however, the obseived consewation in the' flanking DNA suggests that Hairpin elements do not insert randomly into the genome but rather show a preference for insertion at particular sites.
Southem hybridization revealed the presence of Hairpin elements in relatively low copy number. it is likely that the number of copies estimated based on the Southem data (five copies in three of the four ecotypes examined) represents an underestimate. Already, a computer survey of the available sequence data revealed at least six Hairpin elements in ecotype Columbia. Given the current status of the sequencing project (approxirnately 45% of the sequence completed as of March 1999) and assuming an even distribution of these elements throughout the genome, the number of copies could ultimately reach some 15 copies. This is quite similar to what has been reported previously for the Tatl (2 to 10 copies) and Athila (-30 copies) elements (Pelernan et al., 1991; Pélissier et ab, 1995; Wright and Voytas, 1998).
Interestingly, our data suggest a relatively recent transposition of Hairpin elements in the evolutionary history of Arabidopsis. Only three ecotypes (Ler, Cvi-O, Bla-1) showed the presence of the Hairpin-1 element at the AtMSH2 locus while the other 22 ecotypes examined were shown to lack this insertion. This argues in favor of an event which occurred after the divergence of most ecotypes but which precedes the divergence which led to the three aforernentioned ecotypes.
The fact that only Ler, Cvi-O and Bla-1 ecotypes possess the Hairpin-1 element at the AtMSH2 locus strongly suggests that these are cfosely related and derive from a common ancestor in which the insertion occuned. This is in apparent contradiction with previously published phylogenetic data obtained based on microsatellite markers in which these same three ecotypes were studied (Loridon et al., 1998). In the latter study, Cvi-O was proposed to be quite distantly related to Ler and Bla-1. We propose that the presence of a common insertion event in these three ecotypes strongly suggests a shared cornmon ancestor as identity by descent seems by far the most likely cause for the presence of the same insertion.

Finally, we did not detect signs of somatic excision of Hairpin-1 out of the AtMSH2 locus in any of the three ecotypes which harbor the insertion allele. This may be taken to indicate that either somatic excision events occur at a low frequency (and were simply not detected) or, alternatively, that Hairpin elements are no longer active transposons. If transposition does still occur, however, it must rely on a source of transposase which has yet to be identified since the Hairpin elements lack a coding capacity and clearly cannot be the source of a hypothetical transposase.

4.4 Experimental procedures
4.4.1 Plant material
The plants used in this study were Arabidopsis thaliana ecotypes Ag-O, BeO, Bla-1, Bs-f , Bu-O, Chi-O, Col-4, Cvi-O, En-1 , Est4 , In-O, Kas4 , Ko-2, Ler-O, Lu-1, Mh-O, Ms- O, Mt-O, Mv-O, Nd, No, Nok-O, Pog-O, Ws and Yo-O. These were kindly provided by Arabidopsis Biological Resources Center (ABRC, Ohio). After sterilization, seeds were pfated on Germination Medium (GM) and kept at 4OC for 72 h. Plants were grown in a plant tissue culture chamber at 25°C under 16 h day length. Two weeks- old seedling were harvested for DNA extraction.
4.4.2 PCR amplification
Two specific oligonucleotides corresponding to sequences flanking the Hairpin-1 element in Landsberg erecta ecotype (#1: 5'-AlllTGCCTATTAGAATTCTTGAT-3' and #2: 5'-ACATTGGAATTCAAAATGGCTCTTC-3') were used to perform amplification in 50 pl reactions containing lx PCR buffer (Pharrnacia), 1 pM each primer, 0.2 mM dNTPs and 2 units of Taq DNA polyrnerase. Genomic DNA from the twenty-five ecotypes (listed above) were used as template for PCR. The following amplification parameters were used : 94°C for 5 min followed by 30 cycles of (94°C for 30 sec, 50°C for 30 sec, 72°C for 1 min) and finally 10 min at 72°C. The PCR products were separated on a 2% agarose gel.
4.4.3 DNA isolation and Southern hybridization
Genomic DNA was extracted from seedlings of the four different ecotypes (Ler, Col, Ws, No) of Arabidopsis thaliana using a modified CTAB protocol described in Dubois et al. (1998). Two micrograms of genomic DNA were digested with Hindlll and EcoRl (separately). Electrophoresis was performed in a 0.8% agarose gel and the DNA transferred ont0 Gene Screen Plus (Dupont) nylon membrane under alkaline conditions. Prehybridization and hybridization were carried out in buffer

containing 6x SSC, 5x DenhardYs solution, 100 pg ml-' of denatured salmon spem DNA, 0.5% SDS at 60°C for 16 h. The membrane was washed twice in 5x SSC, 1% SDS for 15 min at room temperature, followed by two washes in 2x SSC, 1 % SDS for 30 min at 60°C. The washed membrane was exposed on film at -80°C for 24 h.
4.4.4 Radiolabelled probe
The probe was labeled with Taq DNA polymerase. Ten nanograms of the PCR product from ecotype Landsberg erecta and obtained with pnmers #1 and #2 were used as template to perform a radioactive labeling reaction in a 50 pl volume containing l x PCR buffer (Pharmacia). 0.5 pM each primer. 20 pM (dNTPs-dATP), 50 pCi 32P[a-dATP] and 1 unit of Taq DNA polyrnerase. The amplification
parameters were 94OC for 5 min followed by 5 cycles of (94°C for 30 sec, 50°C for 30 sec, 72OC for 1 min) and finally 10 min at 72OC.
4.4.5 DNA sequence snelysis
Database searches and sequence analyses were performed using the GCG software package (Devereux et al., 1984). The programs GAP, BESTFIT, FASTA and PILEUP of this software were used for sequence similarity searches and alignment. Secondary structure was predicted using the FOLD program of the same software.
Acknowledgments
J. Adé was supported by a graduate scholarship from the 'Programme Canadien de Bourses de la Francophonie" (Government of Canada). This work was supported by a research grant from the Natural Sciences and Engineering Research Council of Canada to F. Belzile.

References
Banville, D. and Boie, Y. (1989) Retroviral long terminal repeat is the promoter of the gene encoding the tumor-associated calcium-binding protein oncornodulin in the rat. J. Mol, Biol. 207, 481490.
Baumruker, T., Gehe, C. and Horak, 1. (1988) Insertion of a retrotransposon within the 3' end of a mouse gene provide a new functional polyadenylation signal. Nucl. Acids Res., 1 6, 7241 -725 1 .
Bureau, T.E. and Wessler, S,R. (1992) Tourist. A large family of small inverted repeat elements frequently associated with Maize genes. Plant Cell, 4, 1283- 1294.
Collins, M. and Rubin, G.M. (1984) Structure of chromosomal rearrangements induced by the FB transposable elements in Drosophila. Nature, 308, 323- 327.
Devereux, J., Haeberli, P. and Srnithies, 0. (1984) A comprehensive set of sequence analysis programs for the VAX. Nucl. Acids Res., 12. 387-395.
Dubois, P., Cutler, S. and Belzile, F.J. (1998) Regional insertional mutagenesis on chromosome III of Arabidopsis thaliana using the maize Ac element. Plant J. 13, 141 -1 51.
Grandbastien, M.A. (1992) Retroelements in higher plants. Trends. Genet. 8, 1 03-1 08.
Hoffman-Liebermann, B., Liebermann, D. and Cohen, S. (1989) TU Elements and Puppy Sequences. In Mobile DNA. (Berg, D.E. and Howe, M.M., eds). American Society for Microbiology, Washington, D.C., pp. 575-592.
Loridon, K., Cournoyer, B., Goubely, C., Depeiges, A. and Picard, O. (1 998) Length polymorphism and allele structure of tnnucleotide microsatellites in natural accessions of Arabidopsis thaliana. Theor. Appl. Genet 97, 591 -604.
Maichele, A.J., Farwell, N.J. and Chamberlain, J.S. (1993) A 82 repeat insertion generates altemate structure of the mouse muscle y-phosphorylase kinase gene. Genomics 16, 1 39-1 49.
Peleman, J., Cottyn, B. Van Camp, W., Van Montagu, M. and Inze, D. (1991) Transcient occurence of extrachromosomal DNA of an Arabidopsis thaliana transposon-like element. Tat 1. Proc. Nat/ Acad. Sci. USA, 88, 361 8- 3622.

Pélissier, T., Tutois, S., Deragon. J.M., Tourmente, S., Genestier, S. and Picard, G. (1995) Athilia, a new retroelement from Arabidopsis thaliana. Plant. Mol. Biol. 29, 441 -45 1 .
Potter, S., Truett, M., Phillips, M. and Maher, A. (1980) Eukaryotic transposable genetic elements with inverted terminal repeats. Cell, 20, 639- 647.
Rebatchouk, D. and Narita, J.O. (1997) Foldback transposable elements in plants. Plant Mol. Biol. 34, 831-835.
Truett, M.A., Jones, R.S. and Potter, S.S. (1981) Unusual structure of the FB family of transposable elements in Drosophila melanogaster. Cell, 24, 753- 763.
Walbot, V. (1992) Reactivation of Mutator transposable elements of maize by ultraviolet light. Mol. Gen. Genet. 234, 353-360.
Wright, D.A. and Voytas, D.F. (1998) Potential retroviruses in plants: Tatl is related to a group of Arabidopsis thaliana. Ty3Igypsy retrot ransposons t hat encode envelope-li ke proteins. Genetics, 1 49, 703-71 5.
Yuan, J.Y., Finney, M., Tsung, N. and Horvitz, H.R. (1991) Tc4, a Caenorhabditis elegans transposable element with an unusual fold-back structure. Proc. Nat1 Acad. Sci. USA, 88, 333403338.

CHAPITRE V
DISCUSSION GÉNÉRALE ET CONCLUSION
Comme nous l'avions mentionné au début de cette étude, la synthèse de I'ADN, bien qu'étant un processus d'une grande fidélité, demeure néanmoins un processus imparfait. En effet, malgré la fonction d'édition de I'ADN polymérase lui permettant d'éliminer les bases incorrectes incorporées au cours de la synthèse par l'activité 3'-5' exonucléase, certaines erreurs lui échappent. À ces erreurs, il faudra ajouter le fait que la structure de I'ADN peut être même modifiée par l'action des rayons ultraviolets provenant du soleil, des rayons X et de nombreuses substances chimiques. En d'autres termes, les organismes sont constamment soumis à toute sorte d'agents mutagènes pouvant modifier leur ADN. Or les séquences d'ADN porteuses de l'information génétique se doivent d'être fidèlement reproduites au cours des nombreuses divisions cellulaires, ceci étant essentiel à la suwie de l'espèce et de l'individu. C'est ainsi que les cellules se sont dotées, au cours de l'évolution, d'un système de correction des m6sappariements d'ADN tout aussi complexe qu'efficace pour se premunir de tels dangers.
La présente Btude initiée dans le cadre général de la compréhension des mécanismes de correction des mésappariements d'ADN et de la recombinaison génétique chez les plantes, en particulier chez Arabidopsis thaliana, avait deux

objectifs principaux. Dans un premier temps, cloner et caractériser le gène MSW de Arabidopsis, ensuite, montrer que le produit de ce gene est fonctionnel.
Comme il a ét6 mentionné dans le chapitre Il chez les eucaryotes, c'est en partenariat que les homologues de MutS reconnaissent les mesappariements d'ADN (Alani, 1996; Johnson et al., 1996). Au centre des deux complexes de
reconnaissance initiale de ces lésions dans I'ADN, se retrouve toujours ta protdine MSHP (Johnson et al., 1996). D'où l'emphase particulière portée sur le gène MSH2 au cours de la présente étude.
Les résultats présentés au chapitre II ont montre que le premier objectif de ce travail a été atteint. En effet, la composante centrale du système de correction des mésappariements d'ADN, le gène AtMSH2 a et6 clone chez Arabidopsis thaliana. II s'agit d'un gène simple copie situé sur le chromosome III daArabidopsis. La présence en une seule copie de ce gene est d'une signification particulière sur le plan fonctionnel, en ce sens qu'une inactivation de ce gène, par mutation par exemple, donnera lieu à un phénotype non ambigue, puisqu'il n'y aura pas d'autres copies pouvant compenser l'absence du gène inactivé. Bien que la RT-PCR ait prouvé la présence des ARNm de AtMSH2, même à partir dlARN total de jeunes plantules, les seules conditions où il a été possible de détecter un messager par analyse northem ont été l'utilisation dlARNm provenant d'une suspension cetlulaire dlArabidopsis en phase de croissance exponentielle (Axelos et al., 1992). Dans de telles conditions, les cellules sont constamment en division mitotique active où elles répliquent leur ADN. Ces résultats sont tout à fait conformes aux attentes des homologues de MutS impliqués dans la correction des mésappariements d'ADN chez les plantes, en particulier AtMSH2. En effet, c'est dans les organes et au cours des stades où la réplication de I'ADN est plus intense qu'il y aura potentiellement le plus de correction des erreurs d'incorporation de la polymérase. Cela fait abstraction des autres conditions où les organismes sont soumis des agents mutagènes pouvant endommager leurs ADN. ou au cours de recombinaisons homéologues ou l'on s'attend Bgalement à une forte expression de ces g8nes.
L'analyse phylogénétique incluant AtMSH2, mais aussi trois autres homologues de MutS chez Arabiodpsis (AtMSH3, AtMSH6-1, AtMSH6-2) a clairement placé AtMSH2 parmi ses orthologues des autres espéces, confirmant une fois de plus son identité. Un autre point interessant de cette analyse phylogénétique est que parmi les quatre paralogues de MutS chez Arabiodpsis, AtMSH2 est celui qui a le moins

évolué, laissant ainsi présager de son rôle central dans le mécanisme de correction des rnésappariements d'ADN chez Arabidopsis, oii il devra interagir avec plusieurs autres partenaires si les fonctions sont conservées (Prolla et al., 1994; Johnson et
al., 1996). La règle genérale en la matière étant que, si une protéine devra interagir avec plusieurs partenaires, moins elle a la liberté d'6voluer. Cette analyse phylogénétique suggère tres fortement une activité fonctionnelle pour chacun de ces paralogues de MutS chez Arabidopsis, en particulier AtMSH2.
Le troisième chapitre de cette thèse nous a permis de mettre en évidence la fonction du produit de ce gène. En effet, que ce soit grâce à la mutagenèse dans E. coli ou sur gel de retardement, nous avons pu montrer que la proteine AtMSH2 est capable d'interagir avec de l'ADN mésapparié, ce qui est conforme à sa fonction de correction des mésappairements d'ADN.
Les résultats présentés dans ce troisième chapitre constituent la première preuve expérimentale où une fonction a été démontrée pour un gène de correction des mésappariements d'ADN chez les plantes. Si l'on étend la conservation de fonction au point où la correction des mesappariements chez les plantes est responsable de la suppression de recombinaisons entre séquences apparentées, alors on peut espérer qu'une modification apportée à ce système pourrait accroître significativement la contribution des croisements interspécifiques en amélioration des plantes. Actuellement, le manque de recombinaison, ou leur faible fréquence, constitue une contrainte majeure pour une introgression plus efficace des caractères agronomiques intéressants dans les espèces cultivées.
Un cas particulièrement édifiant existe chez le blé, ou l'absence d'appariement et de recombinaison entre régions homéologues a été établi, et ce phenornene est sous un contrôle génétique. Cette situation est due à l'action du gène Ph1 décrit pour ta première fois au cours des années 1950s (Riley et Chapman, 1958). Chez les mutants ph1, l'appariement et la recombinaison entre chromosomes homeologues sont beaucoup plus permissifs. Les mécanismes par lesquels Ph1 détermine la specificite de l'appariement et de la recombinaison ne sont pas tres bien connus. Jusqu'à date, la plupart des modèles proposés supposent que Ph1 agit au niveau chromosomique, soit pour une reconnaissance mutuelle, soit pour l'établissement d'une association préméiotique entre chromosomes homologues favorisant ainsi l'appariement et la recombinaison (Feldman, 1993). Une étude récente effectuée par Luo et ses collaborateurs (1996) suggère très clairement que

Ph1 agit plutôt directement au niveau de la reconnaissance des hetéroduplex d'ADN. En effet, ces auteurs ont construit des chromosomes hybrides 1A/1A" constitués de segments intercallaires du chromosome 1A du blé panifiable Triticum aestivum et celui 1Am de T. monococcum. Chez les individus où le gène Ph1 est fonctionnel, la recombinaison entre de tels chromosomes hybrides et le chromosome normal 1 A de T. aestivum est limitée aux régions d'homologie; aucune recombinaison n'a été observ6e dans les regions homéologues. Par contre, chez les mutants p h l , une recombinaison s'étendant à tout le chromosome a été observée indépendamment de I'homéologie.
Ce phénomène relie à Ph1 rappele ce qui a été observe chez les mutants de correction des mesappairements d'ADN tant chez les procaryotes (Rayssiguier et al., 1989) que chez eucaryotes (Selva et al., 1995; de Wind et al., 1995) [cf. chapitre Il. Eu égard a ce qui précède, il apparait alors très attrayant et tres prometteur de proposer que le système de correction des mésappariements d'ADN détermine également la spécificité de la recombinaison chez les plantes. Puisqu'au terme de cette Btude on a pu mettre en évidence la conservation de fonction du systeme de correction des mésappairements chez les plantes, il serait tres intéressant que des travaux futurs puissent valider l'hypothèse de l'implication de ce systeme dans la recombinaison génetique chez les plantes en vue d'une application en amélioration des plantes.
Le troisième volet de cette thèse (chapitre IV) a été au-delà des objectifs initiaux de ce projet de recherche. II nous a permis d'identifier et de caractériser la première famille d'éléments transposable de type Foldback chez Arabidopsis thaliana, et qui par ailleurs s'est révélée être la première famille de Transposon Foldback de type 3 identifiée dans tout le règne vegétal: les Hairpin. De plus, on a pu démontrer qu'il y
a eu une transposition dans un passé relativement récent. En effet, la présence de l'élément Hairpin-1 (un membre de cette famille) dans le locus AtMSH2 de Landsberg erecta après la divergence de ce dernier de Columbia, qui est un écotype trés proche (chapitre IV) montre clairement que ces éléments ont transposé dans un pas& pas tres lointain.
Au-delà de son caractère novateur, ce volet (Chapitre IV) revêt également un impact potentiel significatif pour des analyses phylogénétiques entre les Bcotypes d'Arabidopsis. En effet, nos résultats suggèrent très fortement que les écotypes Ler, Cvi-O et Bla-1 dérivent d'un même ancêtre commun dont ils ont reçu l'élément

Hairpin-7. Ceci semble contredire des Btudes phylogénétiques antérieures basées sur des marqueurs microsatellites (London et al., 1998). lesquelles présentent Cvi-O comme un écotype distant de Ler et Bla-1. La présence de l'élément Hairpin-1 au
locus AtMSH2 de chacun de ces trois ecotypes indique clairement une identité par descendance. Or l'identité par descendance est un critère beaucoup plus fiable qu'un marqueur microsatellite pour expliquer les relations de parenté. En effet, elle réflhte la transmission de caracthres des parents aux descendants alors que les polymorphismes de microsatellites ont rapport B la longueur de sequences d'ADN. L'examen de la pr6sence des autres 6lements Hairpin (et ceux à identifier) à des loci particuliers pourrait donc être un outil très intéressant pour des études phylogénétiques.
En conclusion, ces travaux constituent une première incursion dans un domaine jusque là complètement inexploré chez les vdgétaux : le processus de correction des mésappariements. Les premiers résultats suggèrent que ce processus est très largement conservé chez les eucaryotes, y compris chez les plantes. De plus, l'apparente conservation de fonction permet d'esp8rer que des modifications apportées au systbme de correction des mesappariements faciliteront I'introgression de g h e s en provenance d'esp6ces sauvages. Aussi, ces travaux nous ont-ils permis de caract6riser la première famille de ~Foldback Transposons. chez A rabidopsis thaliana.

102
LISTE COMPLETE DES OUVRAGES CITÉS
Acharya, S., Wilson, T., Gradia, S., Kane, M.F., Guerrette, S., Marsischky, G.T., Kolodner, R. and Fishel, R. (1996) hMSH2 forms specific mispair-binding complexes with hMSH3 and hMSH6. Proc Nat1 Acad Sei USA 93, 13629-1 3634.
Adachi, J. and Hasegawa, M. (1996) PROTML: Maximum Likelihood Interference of Protein Phylogeny. lnstitute of Statistical Mathematics, Tokyo.
Ade, J., Belzile, J.F., Philippe, H. and Doutriaux, Y-P. (in press) Four mismatch repair paralogues coexist in Arabidopsis thaliana: AtMSH2. AtMSH3, AtMSH6-1 and AtMSH6-2. Mol. Gen. Genet. 262: 239-249.
Akiyama, Y., Sato, H., Yamada, T., Nagasaki, H., Tsuchiya, A., Abe, R. and Yuasa, Y. (1997) Germ-line mutation of the hMSHG/GTBP gene in an atypical hereditary nonpolyposis colorectal cancer kindred. Cancer Res 57, 392093923.
Alani, E. (1996) The Saccharomyces cerevisiae Msh2 and Msh6 proteins form a cornplex that specifically binds ta duplex oligonucleotides containing mismatched DNA base pairs. Mol. Cell. Biol. 1 6 , 5604-561 5.
Alani, E., Chi, N-W. and Kolodner, R. (1 995) The Saccharomyces cerevisiae Msh2 protein specifically binds to duplex oligonucleotides containing mismatched DNA base pairs and insertions. Genes & Dev9, 234-247.
Alani, E., Reenan, R.A.G. and Kolodner R. (1994) Interaction between rnismatch repair and genetic recombination in Saccharomyces cerevisiae. Genetics 137, 19-39.
Axelos, M., Bardet, C., Liboz, T., Le Van Thai, A., Curie, C. and Lescure, B. (1989) The gene farnily encoding the Arabidopsis thaliana translation elongation factor EF-1 alpha: molecular cloning, characterization and expression. Mol Gen Genet 21 9, 1 06-1 1 2.
Axelos, M., Curie, C., Mauolini, L., Bardet, C. and Lescure, B. (1992) A protocol for transient gene expression in Arabidopsis thaliana protoplasts isolated from cell suspension culture. Plant Physiol Biochem 30, 1 -6.

Baker, S.M., Bronner, C.E., Zhang, L., Plug, ArnWrn, Robatzek, M., Warren, G., Elliott, ErnA., Yu, Jrn, Ashley, T., Arnheim, N., Flavell, KA. and Liskay, R.M. (1995) Male mice defective in the DNA mismatch repair gene PMS2 exhibit abnormal chromosome synapsis in meiosis. Ce11 82, 309-31 9.
Baker, SrnM., Plug, ArnWrn, Prolla, T.A., Bronner, C.E., Harris, A.C., Yao, X., Christie, D.M., Monell, C., Arnheim, N., Bradley A., Ashley, T. and Liskay, RrnMrn (1996) Involvement of mouse Mlhl in DNA mismatch repair and meiotic crossing over. Nat. Genet. 13, 336-42.
Baldauf, S.L. and Palmer, J.D. (1993) Animals and fungi are each other's cfosest relatives. Science 257, 74-76.
Banville, D. and Boie, Y. (1989) Retroviral long terminal repeat is the promoter of the gene encoding the tumor-associated calcium-binding protein oncornodulin in the rat. J. Mol. Biol. 207, 481-490.
Baumruker, T., Gehe, C. and Horak, 1. (1988) Insertion of a retrotransposon within the 3' end of a mouse gene provide a new functional polyadenylation signal. Nucl. Acids Res., 1 6, 724 1 -725 1 .
Bhui-Kaur, A., Goodman, MrnF. and Tower, J. (1998) DNA rnismatch repair catalyzed by extracts of mitotic, postmitotic, and senescent Drosophila tissues and involvement of mei-9 gene function for full activity. Mol Cell Bioll8. 1436- 1 443.
Bishop, DrnKrn, Andersen, J. and Kolodner, RmDw (1989) Specificity of mismatch repair following transformation of Saccharomyces cerevisiae with heteroduplex plasmid DNA. Proc Nat1 Acad Sci USA 86, 371 3-371 7.
Bishop, D.K., Williamson, M.S., Fogel, S. and Kolodner, RD. (1987) The role of heteroduplex correction in gene conversion in Saccha romyces cerevisiae. Nature 328: 362-364.
Bronner, C.E., Baker, SM., Morrison, P.T., Warren, G., Smith, LOS, Lescoe, M.K., Kane, M., Earabino, C., Lipford, J., Lindblom, Arn, Tannegard, P., Bollag, R.J., Godwin, A., Ward, D.C., Nordenskjold, M., Fishel, R., Kolodner, R. and Liskay, R.M. (1994) Mutation in the DNA rnismatch repair gene homologue hMLHl is associated with hereditary non-polyposis colon cancer. Nature 368, 258-261.

Brown, T.C. and Jiricny, J. ( 1 988) Different baselbase mispairs are corrected with different efficiencies and specificities in monkey kidney cells. Ce11 54,705- 711.
Bureau, T.E. and Wessler, S.R. (1992) Tourist. A large family of small inverted repeat elements frequently associated with Maize genes. Plant CeIl, 4, 1283- 1294.
Bureau, T.E. and Wessler, S.R. (1994) Stowaway: A new family of inverted- repeat elements associated with the genes of both monocotyledonous and dicotylenodous pants. Plant Ce11 6, 907-9 1 6.
Casacuberta, E., Casacuberta, J.M., Puigdomenech, P. and Monfort, A. (1 998) Presence of a miniature inverted-repeat transposable element (MITES) in the genome of Arabidopsis thaliana: characterization of the Emigrant family of elernents. Plant J 16, 79-85.
Cerovic, G., Bozin, 0. and Dimitrijevic, B. (1991) Mismatch-specific DNA breakdown in nuclear extract from tobacco (Nicotiana tabacum) callus. Plant Mol Bi01 17, 887-894.
Chi, N. and Kolodner, R.D. (1994a) Purification and characterization of Mshl , a yeast mitochondrial protein that binds to DNA mismatches. J. Biol. Chem. 269, 29984-29992.
Chi, N. and Kolodner, RD. (1994b) The effect of DNA mismatches on the ATPase activity of Mshl, a protein in yeast mitochondria that recognizes DNA mismatches. J. Biol. Chem. 269, 29993-29997.
Choy, H., E. and Fowler, R.G. (1985) The specificity of base-pair substitution induced by the mutL and mutS mutators in E. coli. Mutat. Res. 142, 93-97.
Church, G.M. and Gilbert, W. (1984) Genomic sequencing. Proc Nat1 Acad Sci USA 81, 1991-1995.
Collins, M. and Rubin, G.M. (1984) Structure of chromosomal rearrangements induced by the FB transposable elements in Drosophila. Nature, 308, 323- 327.
Cox, E.C. (1976) Bacterial mutator genes and the control of spontaneous mutation. Ann Rev Genet 10, 135-1 56.
Culligan, K.M. and Hays, J.B. (1997) DNA mismatch repair in plants. An Arabidopsis thaliana gene that predicts a protein belonging to the MSHP subfamily of eukaryotic MutS homologs. Plant Physiol115, 833-839
Datta, A., Hendrix, M., Lipsitch, M. and Jinks-Robertson, S. (1997) Dual roles for DNA sequence identity and the mismatch repair system in the

regulation of mitotic crossing-over in yeast. Proc Natl Acad Sci USA 94, 9757- 9762.
Dellaporta, S.L., Wood, J. and Hicks, J.B. (1983) A plant DNA minipreparation: version II. Plant Mol Bi01 Reporter 1 , 1 9-21.
Devereux, J., Haeberli, P. and Srnithies, 0. (1984) A comprehensive set of sequence analysis programs for the VAX. Nucl. Acids Res., 12, 387-395.
Dickerson, RE. (1971) The structures of cytochrome c and the rates of molecular evolution. J Mol Evol 1 , 26-45.
Dohet, C., Wagner, R. and Radman, M. (1985) Repair of defined single base- pair mismatches in Eschenchia coli. Proc Natl Acad Sci USA 82, 503-505.
Doutriaux, M.P., Couteau, F. and White, C. (1998) isolation and characterisation of the RAD51 and DMCl homologs from Arabidopsis thaliana. Mol Gen Genet 257, 283-291,
Drummond, J.T., Li, G.M., Longley, M.J. and Modrich, P. (1995) Isolation of an hMSH2-pl60 heterodimer that restores DNA mismatch repair to tumor cells. Science 268, 1909-1912,
Dubois, P., Cutler, S. and Belzile, F.J. (1998) Regional insertional mutagenesis on chromosome III of Arabidopsis thaliana using the rnaize A c element. Plant J. 13, 141 -1 51.
Edelmann, W., Yang, K., Umar, A., Heyer, J., Lau, K., Fan, K., Liedtke, W., Cohen, P.E., Kane, M.F., Liptord, J.R., Yu, N., Crouse, G.F., Pollard, J.W., Kunkel, T., Lipkin, M., Kolodner, R. and Kucherlapati, R. (1997) Mutation in the mismatch repair gene Msh6 causes cancer susceptibility. Cell91, 467-477,
Eisen, J.A. (1998) A phylogenomic study of the MutS family of proteins. Nucleic Acids Res 26, 429 1 -4300.
Eshleman, J.R., Lang, E.Z., Bowerfind, O.K., Parsons, R., Vogelstein, B., Willson, J.K., Veigl, M.L., Sedwick, W.D. and Markowitz, S.D. (1 993) lncreased mutation rate at the hprt locus accompanies microsatellite instability in colon cancer. Oncogene 10, 33-37.
Fazakerley , G.V., Quignard, E., Woisard, A., Guschlbauer, W., and van der Marel, G.A. (1986) Structures of rnismatched base pairs in DNA and t heir recognition by Escherichia coli mismatch repair system. EMBO J. 5.3697- 3703.
Feinstein, S.1. and Low, K.B. (1 986) Hyper-recombining recipient strains in bacterial conjugation. Genetics 1 13, 13-33.

Felsenstein, J. (1 978) Cases in which parsimony or compatibility methods will be positively misleading. Syst Zoo1 27, 401 4 1 0.
Feng, Gw, Tsui, H.C.T. and Winkler, MwE. (1996) Depletion of the cellular amounts of the MutS and MutH methyl-directed mismatch repair proteins in stationary-phase Escherichia coli K-12 cells. J Bacterio1 1 78, 238892396.
Fersht, A.R. and Knill-Jones, J.W. (1981) DNA polymerase accuracy and spontaneous mutation rates: Frequencies of purine-purine, purine-pynmidine, and pyrimidine-pyrimidine mismatches during DNA replication. Proc. Natl. Acad. Sci. USA. 78, 4251 -4255.
Fishel, R., Ewel, A., and Lescoe, M.K. (1994) Purified human MSH2 protein binds to DNA contaning mismatched nucieotides. Cancer Res., 54, 553905542.
Fishel, R. and Kolodner, R.D. (1995) Identification of mismatch repair genes and their role in the development of cancer. Curr. Opin. Genet. Dev. 5, 382- 395.
Fishel, R., Lescoe, M.K., Rao, M.R.S., Copeland, N.G., Jenkins, N.A., Garber, J., Kane, M. and Kolodner, RD. (1993) The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Ce11 75, 1027-1 038.
Flores-Rozas, H. and Kolodner, RD. (1 998) The Saccharomyces cerevisiae ML H3 gene functions in MSH3-dependent suppression of frameshift mutations. Proc. Natl. Acad. Sci. USA. 95, 12404-1 2409.
Folger, K.R., Thomas, K. and Capecchi, M.R. (1985) Efficient correction of mismatched bases in plasmid heteroduplexes injected into cultured rnammalian cell nuclei. Mo! Ce11 Bi01 5, 70-74.
Friedberg, EwC., Walker, G.C. and Siede, W. (1995).DNA repair and mutagenesis. ASM Press, Washington, D.C.
Genschel, J., Littman, S.J., Drummond. J.T. and Modrich. P. (1998) Isolation of MutSbeta from human cells and comparison of the misrnatch repair specificities of MutSbeta and MutSalpha. J Bi01 Chem 273, 1 9895-1 9901 .
Gorbalenya, AwE. and Koonin E.V. (1990) Superfamily of UvrA-related NTP- binding proteins: implications for rational classification of recombinationaVrepair systems. J Mol Bi01 21 3, 583-591.
Grandbastien, M.A. (1992) Retroelements in higher plants. Trends. Genet . 8, 103-1 08.

Gu, L., Hong, Y., McCulloch, S., Watanabe, Hm, Li, G.I. (1998) ATP- dependent interaction of human rnismatch repair proteins and dual role of PCNA in mismatch repair. Nucleic Acids Res 26, 1 1 73-1 1 78.
Haber, L.T. and Walker, G.C. (1991) Altering the conserved nucleotide binding motif in the Salmonella typhimurium MutS mismatch repair protein affect both its ATPase and rnismacth binding activities. EMBO J. 10, 2707-271 5.
Hare, J.T. and Taylor, J.H. (1985) One role for DNA methylation in vertebrate cells is strand discrimination in rnismatch repair. Proc Natl Acad Sci USA 82 735007354.
Her, C. and Doggett, N.A. (1998) Cloning, structural characterization, and chromosomal localization of the human orthologue of Saccharomyces cerevisiae MSHS gene. Genomics 52, 50-61.
Herman, G.E. and Modrich, P. (1981) Escherichia coli K-12 clones that overproduce dam methy lase are hype mutable. J. Bacteriol. 1 45, 644-646.
Hoffman-Liebermann, B., Liebermann, D. and Cohen, S. (1989) TU Elements and Puppy Sequences. In Mobile DNA. (Berg, D.E. and Howe, M.M., eds). American Society for Microbiology, Washington, D.C., pp. 575-592.
Hollingsworth, N.M., Ponte, L. and Halsey, C. (1995) MSHS, a novel MutS homolog, facilitates meiotic reciprocal recombination between homologs in Saccharomyces cerevisiae but not mismatch repair. Genes and Dev9 1728- 1739.
Holmes, J.Jr., Clark, S. and Modrich, P. (1990) Strand-specific mismatch correction in nuclear extracts of hurnan and Drosophila melanogaster cell lines. Proc Natl Acad Sci USA 87, 5837-5841.
Horii, A., Han, H.J., Sasaki, S., Shimada, M. and Nakamura, Y. (1994) Cloning, characterization and chromosomal assignrnent of the human genes homologous to yeast PMSI, a member of mismatch repair genes. Biochem. Siophys. Res. Comm. 204, 1257-1 264.
laccarino, L., Palombo, F., Drummond, J., Totty, N.F., Hsuan, J.J.,
Modrich, P. end Jiricny, J. (1996) MSH6, a Saccharomyces cerevisiae protein that binds to mismatches as a heterodimer with MSH2. Current Biology 6, 484-486.
Johnson, A.F., de la Bastide, M., Lodhi, M., Hoffman, J., Hasegawa, A., Gnoj, L., Gottesman, T., Granat, S., Hameed, A., Kaplan, N., Schutz, K., Shohdy, N., Van Keuren, K., Parnell, L., Dedhia, N.,

Martienssen, Re and McCombie, W. (1997) The sequence of the Arabidopsis thaliana Tl 0M13 BAC. ACCESSION AF001308.
Johnson, R.E., Kovvali, G.K., Prakash, L. and Prakash, S. (1996) Requierement of the yeast MSH3 and MSH6 genes for MSH2 dependent genomic stability. J. Biol. Chem. 271 , 728507288.
Jones, M., Wagner, R. and Radman, M. (1987) Repair of a mismatch is influenced by the base composition of the surrounding nucleotide sequence : Genetics 1 15, 605-61 0.
Khrapo, K., Coller, H.A., André, P.C., Li, X.C., Hanekamp, J.S., Thilly, W.G. (1 998) Mitochondrial mutational spectra in human cells and tissues. Proc Nat1 Acad Sci USA 94, 1 3798-1 3803.
Kimura, M. (1983) The neutral theory of molecular evolution. Cambridge. Cambridge University Press.
Kishino, Hm, Hasegawa, M. (1989) Evaluation of the maximum likelihood estimate of the evolutionary tree topologies from DNA sequence data, and the branching order in hominoidea. J Mol Evo129, 170-1 79.
Kolodner, RD. (1996) Biochemistry and genetics of eukaryotic mismatch repair genes. Genes and Dev 10, 1433-1 442.
Kolodner, R.D., Hall, N.R., Lipford, J., Kane, M.F., Rao, M.R.S., Morrison, P., Wirth, L., Finan, P.J., Burn, J., Chapman, P., Earabino, Cm, Merchant, E. and Bishop, D.T. (1994) Structure of the human MSH2 locus and analysis of two Muir-Torre kindreds for msh2 mutations. Genomics 24, 51 6-526.
Kramer, W., Fartmann, B. and Ringbeck, E.C. (1996) Transcription of MutS and MutL- homologous genes in Saccharomyces cerevisiae during the cell cycle. Mol Gen Genet 252, 275-283.
Kramer, B., Kramer, W. and Fritz, H.J. (1 984) Different baselbase mismatches are corrected with different efficiencies by the methyl-directed mismatch repair system of E. coli. Cell 38, 879-887.
Kramer, W., Kramer, B., Williamson, M.S. and Fogel, S. (1989a). Cloning and nucleotide sequence of DNA mismatch repair gene PMS1 from Saccharomyces cerevisiae: homology of PMSl to prokaryotic MutL and HexB. J Bacteriol. 171, 5339-5346.
Kramer, B., Kramer, W., Williamson, M.S. and Fogel, S. (1989b)
Heteroduplex DNA correction in Saccharomyces cerevisiae is mismatch specific and requires functional PMS genes. Mol. Cell. Biol. 9, 443204440.

Lahue, R.S., Au, K.G. and Modrich, P. (1989) DNA mismatch correction in a defined system. Science 245, 1 60-1 64.
Cander, E.S., Green, P., Abraharnson, J., Barlow, A., Daly, M.J., Linclon, S.E. and Noewburn, L. (1 987) MAPMAKER: An interactive cornputer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics 1, 1 74-1 8 1 .
Langle-Rouault, F., Maenhaut, M.G. and Radman, M. (1987) GATC sequences, DNA nicks and the MutH function in Escherichia coli mismatch mismatch repair. EMBO J. 6, 1 121 -1 127.
Lea, D.E. and Coulson, A. (1949) The distribution of numbers of mutants in bacterial populations. J. Genet. 49, 264-285.
Leach, F.S., Nicolaides, N.C., Papadopoulos, N., Liu, B., Jen, J., Parsons, R., Peltomaki, P., Sistonen, P., Aaltonen, L.A., Nystrom- Lahti, M., Guan, X.Y., Zhang, J., Meltzer, P.S., Yu, J.W., Kao, F.T., Chen, D.J., Cerosaletti, K.M., Fournier, R.E.K., Todd, S., Lewis, T., Leach, R.J., Naylor, S.L., Weissenbach, J., Meciclin, J.P., Jarvinen, H., Petersen, G.M., Hamilton, S.R., Green, J., Jass, J., Watson, P., Lynch, H.T., Trent, J.M., de la Chapelle, A., Kinzler, K.W. and Vogelstein, B. (1993) Mutations of a MutS homolog in hereditary nonpolyposis colorectal cancer. Ce11 75, 121 5-1 225.
Leang, P.M., Hsia. H.C. and Miller, J.H. (1986) Analysis of spontaneous base substitutions generated in mismatch-repair-deficient strains of Escherichia coli. J. Bacteriol. 1 68, 41 2-41 6.
Lister, C. and Dean, C. (1993) Recombinant inbred lines for mapping RFLP and p henotypic markers in Arabidopsis thaliana. Plant J 4, 745-750.
Loeb, L.A. and Kunkel, T.A. (1982) Fidelity of DNA synthesis. Annu. Rev. Biochem. 51, 429-457.
Loridon, K., Cournoyer, B., Goubely, C., Depeiges, A. and Picard, O. (1 998) Length polymorphism and allete structure of trinucleotide microsatellites in natural accessions of Arabidopsis thaliana. Theor. Appl. Genet 97, 591-604.
Lu, A.L., Clark, S. and Modrich, P. (1983) Methyl-directed repair of DNA base pair mismatches in vitro. Proc. Natl. Acad. Sci. USA. 80, 46394643.
Luhr, B., Scheller, J., Meyer, P. and Kramer, W. (1998) Analysis of in vivo correction of defined mismatches in the DNA mismatch repair mutants msh2, msh3 and msh6 of Saccharomyces cerevisiae. Mol Gen Genet 257, 362-367.

Maichele, A.J., Farwell, N.J. and Chamberlain, J.S. (1993) A 82 repeat insertion generates altemate structure of the mouse muscle y-phosphorylase
kinase gene. Genomics 1 6, 1 39-1 49.
Marinus, MG., Poteete, A. and Arraj, J.A. (1984) Correlation of DNA adenine methylase activity in Escherichia con K-12. Gene 28, 1 23-1 25.
Marsischky, G.T., Filosi, Ne, Kane, M.F. and Kolodner, R. (1996) Redundancy of Saccharomyces cerevisiae MSH3 and MSH6 in MSH2- dependent mismatch repair. Genes and Dev 1 O, 407-420.
Miret, J.J., Milla, M.G. and Lahue, R.S. (1993) Characterisation of a DNA mismatch binding activity in yeast extracts. J. Biol. Chem. 268: 3507-35 1 3.
Miyaki, M., Konishi, M., Tanaka, K., Kikuchi-Yanoshita, R., Muraoka, M., Yasuno, M., Igari, T., Koike, M., Chiba, M. and Mori, T. (1997) Germline mutation of MSH6 as the cause of hereditary nonpolyposis colorectal cancer. Nat Genet 17, 271 -272.
Modrich, P. (1991) Mechanism and biological effects of rnismatch repair. Ann Rev Genet 25, 229-253.
Modrich, P. and Lahue, R. (1996) Mismatch repair in replication fidelity, genetic recombination and cancer biology. Annu Rev Bochem 65, 1 01 -1 33.
New, L.. Liu, K. and Crouse, G.F. (1993) The yeast gene MSH3 defines a new class of eukaryotic MutS homologues. Mol Gen Genet 239, 97-1 08.
Nicolaides, N.C., Papadopoulos, N., Liu, B., Wei, Y., Carter, K.C., Ruben, S.M., Rosen, C.A., Haseltine, W.A., Fleischmann, R.D., Fraser,C.M., Adams, M.D., Venter, J.C., Dunlop, M.G., Hamilton, SR., Petersen, G.M., de la Chapelle, A., Vogelstein, B. and Kinzler, K. (1994) Mutation of two PMS homologues in hereditary nonpolyposis colon cancer. Nature 371, 75-80.
Ohlendorf, D.H., Anderson, W.F. and Matthews, B.W. (1983) Many gene- regulatory proteins appear to have a similar alpha-helical fold that binds DNA and evolved from a common precursor. J Mol Evol19, 109-1 14.
Palombo, F., Gallinari, P., laccarino, I., Lettieri, T., Hughes, M., D'Arrigo, A., Truong, O., Hsuan, J.J. and Jiricny, J. (1995) GTBP, a 160-kilodalton protein essential for mismatch-binding activity in human cells. Science268, 1912-1914.
Papadopoulos, N., Nicolaides, N.C., Wei, Y.F., Ruben, SM., Carter, K.C., Rosen, C.A., Haseltine, W.A., Fleischmann, R.D., Fraser, C.M., Adams, M.D., Venter, J.C., Hamilton, S.R., Petersen,

G.M.,Watson, P., Lynch, H.T., Peltomaki, P., Mecklin, J.P., de la Chapelle, A., Kinzler, K.W. and Vogelstein, B. (1994) Mutation of a mutL homolog in hereditary colon cancer. Science 263, 1625-1 628.
Paquis-Flucklinger, V., Santucci-Darmanin, S., Paul, R., Turc-Carel, C. and Desnuelle, C. (1997) Cloning and expression analysis of a rneiosis- specific MutS homolog: the human MSH4 gene. Genomics 44, 18û-194.
Parsons, R., Li, G.M., Longley, M.J., Fang, W.H., Papadopoulos, N., Jen, J., de la Chapelle, A., Kinzler, K.W., Vogelstein, B. and Modrich, P. (1993) Hypermutability and mismatch repair deficiency in RER+ tumor cells. Ce11 75,1227-36.
Peleman, J., Cottyn, B. Van Camp, W., Van Montagu, M. and Inze, D. (1 991 ) Transcient occurence of extrachromosomal DNA of an Arabidopsis thaliana transposon-like element, Tat 1. Proc. Nat1 Acad. Sci. USA, 88, 361 8- 3622.
Pilissier, T., Tutois, S., Deragon. J.M., Tourmente, S., Genestier, S. and Picard, 0. (1995) Athilia, a new retroelement from Arabidopsis thaliana. Plant. Mol. Biol. 29, 441 -451.
Philippe, H. (1993) MUST, a computer package of Management Utilities for Sequences and Trees. Nucleic Acids Res 21, 5264-5272.
Philippe, H. and Laurent, J. (in press) How good are deep phylogenetic trees? Curr Opin Genet Dev.
Pont-Kingdon, Ga, Okada, N.A., Macfarlane, J.L., Beagley, C.T., Watkins-Sims, C.D., Cavalier-Smith, Tm, Clark-Walker, G.D. and Wolstenholme, D.R. (1 998) Mitochondrial DNA of the coral Sarcophyton glaucum contains a gene for a homologue of bacterial Mots: a possible case of gene transfer from the nucleus to the mitochondrion. J Mol Evol46, 41 9-431.
Potter, S., Truett, M., Phillips, M. and Yeher, A. (1980) Eukaryotic transposable genetic efements with inverted terminal repeats. Cell, 20, 639- 647.
Proffitt J.H., Davie, J.R., Swinton, D. and Hattman, S. (1984) 5- Methylcytosine is not detectable in Saccharomyces cerevisiae DNA. Mol. Cell. Biol. 4, 985-988
Prolla, T.A., Christie, D.M. and Liskay, R.M. (1994a) Dual requirement in yeast DNA mismatch repair for MLHl and PMS1, two homologs of the bacterial mutL gene. Mal Ce11 Biol 14, 407-41 5.

Prolla, T.A., Pang, Q., Ahni, E., Kolodner, R. and Liskay, R.M. (1994b) MLH1, PMSI, and MSH2 interactions during the initiation of DNA mismatch
repair in yeast. Science 265, 1 091 -1 093. Prudhomme, M., Melean, V., Martin, B. and Claverys, J-P. (1991)
M ismatch repair genes of Streptococcus pneumoniae: HexA confen a mutator phenotype in Escherichia coli by negative complementation. J. Bactenol. 1 73, 71 96-7203.
Radman, M. (1 989) Mismatch repair and the fidelity of genetic recombination. Genome, 31, 68-73.
Rayssiguier, C., Thaler, D.S. and Radman, M. (1989) The barrier to recombination between Escherichia coli and Salmonella typhimurium is disnipted in mismatch -repair mutants. Nature 342, 396-401.
Rebatchouk, D. and Narita, J.O. (1997) Foldback transposable elements in plants. Plant Mol. Biol. 34, 83 1 -835.
Reenan, RA. and Kolodner, R.D. (1992a) Isolation and characterization of two Saccharomyces cerevisiae genes encoding homologs of the bacterial HexA and MutS mismatch repair proteins. Genetics 132, 963-973.
Reenan, RA. and Kolodner, R.D. (1992b) Characterization of insertion mutations in the Saccharomyces cerevisiae MSHl and MSH2 genes:evidence for separate mitochondrial and nuclear functions. Genetics 132, 975-985.
Reitmair, A.H., Schmits, R., Ewel, A., Bapat, B., Redston, M., Mitri, A., Waterhouse, P., Mittrücker, H.W., Wakeham, A., Liu, B., Thomason, A., Griesser, H., Gallinger, S., Ballhausen, W.G., Fishel, R. and Mak, T.W. (1995) MSH2 deficient mice are viable and susceptible to lymphoid tumours. Nat Genet 11, 64-70.
René, B., Auclair, C. and Paoletti, C. (1988) Frameshift lesions induced by oxazolopyridocarbazoles are recognized by misrnatch repair system in Eschenchia coli. Mutat. Res. 1 93, 269-273.
Rewinski, C. and Marinus, M.G. (1987) Mutation spectrurn in Escherichia coli DNA mismatch repair deficient (mutH) strain. Nucleic Acids Res. 15, 8205- 821 5.
Risinger, J.I., Uma,r A., Boyd, J., Berchuck, A., Kunkel, T.A., Barrett, J.C. (1996) Mutation of MSH3 in endometrial cancer and evidence for its functional role in heteroduplex repair. Nat Genet 14, 102-5.

Ross-Macdonald, P. and Roeder, O.S. (1994) Mutation of a meiosis-specific Mots homolog decreases crossing over but not misrnatch correction. Ce11 79, 1 069- 1 080.
Rydberg, B. (1 978). Bromouracil mutagenesis and mismatch repair in mutator strains of Escherichia coli. Mutat. Res. 52, 1 1-24.
Truett, M.A., Jones, R.S. and Potter, S.S. (1981) Unusual structure of the FB famil y of transposable elements in Orosophila melanogaster. Ce//, 24, 753- 763.
Sambrook, J., Fritsch, E.F. and Maniatis, T. (1989) Molecular Cloning: A Laboratory Manual. Second Edition (Cold Spring Harbor Laboratory Press, New York).
Selva, E.M., New, L., Crouse, G.F. and Lahue, R.S. (1995) Mismatch correction acts as a barrier to homeologous recombination in Saccharomyces cerevisiae. 139, 1 175-1 188.
Strand, M., Prolla, T.A., Lyskay, R.M. and Petes, T. (1993) Destabilization of tracts of simple repetitive DNA in yeast by mutations affecting DNA mismatch repair. Nature 365, 274-276.
Strimmer, K. and von Haeseler, A. (1996) Quartet puzzling: a quartet maximum likelihood method for reconstructing tree topologies. Mol Bi01 Evol 13, 964-969.
Su, S-S., Grilley, M., Thresher, R., Griffith, J. and Modrich. P. (1989). Gap formation is associated with methyl directed mismatch correction under conditions of restricted DNA synthesis. Genome 31, 104-1 1 1.
Su. S-S and Modrich, P. (1986) Escherichia coli mutSencoded protein binds to mismached DNA base pairs. Proc. Nat/. Acad. Sci. USA 83, 5057-5061.
Swofford, D.L. (1 993) Illinois Natural History Survey, Champaign Illinois Thomas, D.C., Roberts, J.O. and Kunkel, T.A. (1991) Heteroduplex repair in
extracts of human HeLa cells. J Bi01 Chem 266, 3744-3751. Tishkoff, D.X., Boerger, A.L., Bertrand, P., Filosi, N., Gaida, G.M., Kane,
M.F. and Kolodner, R.D. (1997) Identification and characterization of Saccharomyces cerevisiae EXO1, a gene encoding an exonuclease that interacts with MSH2. Frac Nat1 Acad Sci USA 94, 7487-7492
Umar, A., Boyer, J-C., Thomas, D.C., Nguyen, D.C., Risinger, J.I., Boyd, J., lonov, Y., Perucho, M. and Kunkel, T.A. (1994) Defective misrnatch repair in extracts of colorectal and endometrial cancer cell lines exhibiting microsatellite instability. J Biol Chem 259, 1 -4.

Umar, A., Buerrneyer, A.B., Simon, J O y Thomas, D.C., Clark, A.B., Liskay, R.M. and Kunkel, T.A. (1996) Requirement for PCNA in DNA misrnatch repair at a step preceding DNA synthesis. Ce1197, 505-514.
Varlet, I., Canard, B., Brooks, P., Cerovic, G. and Radman, M. (1996) Mismatch repair in Xenopus egg extracts: DNA strand breaks act as signals rather than excision points. Proc Nafl Acad Sci USA 93, 1 01 56-1 01 61 .
Varlet, I., Pallard, C., Radman, M., Moreau, J. and de Wind, N. (1994) Cloning and expression of the Xenopus and mouse Msh2 DNA mismatch repair genes. Nucl. Acids Res. 22, 572305728.
Vulic, M., Dioniso, Fq Taddei, F. and Radman, M. (1997) Molecular keys to speciation: DNA polymorphism and the control of genetic exchange in enterobactena. Proc Nat1 Acad Sci USA 94, 9763-9767.
Walbot, V. (1992) Reactivation of Mutator transposable elements of maize by ultraviolet light. Mol. Gen. Genet. 234, 353-360.
Whitehouse, A., Taylor, G.R., Deeble, J., Philips, S.E.V., Meredith, M. and Markham, A.F. (1996) A carboxy terminal domain of the hMSH2 gene product is sufficient for binding specific mismatchad oligonucleotides. Biochem Biophys Res Commun 225, 289-295.
Whitehouse, A., Deeble, JOy Taylor, G.R., Guillou, P.J., Philips, S.E.V., Meredith, M. and Markham, A.F. (1997) Mapping the minimal domain of hMSH2 sufficient for binding mismatched oligonucleotides. Biochem Biophys Res Commun 232, 1 0- 1 3.
de Wind, N., Dekker, M., Berns, A., Radman, M. and te Riele, H. (1995) Inactivation of the mouse Msh2 gene results in mismatch repair deficiency, methylation tolerance, hyperrecombination, and predisposition to cancer. Ce11 82, 321 -330.
Worth, L.J., Clark, S., Radman, M. and Modrich, P. (1994) Mismatch repair proteins MutS and MutL inhibit RecA-catalysed strand transfer between diverged DNAs. Pro. Nafl. Acad. Sci. USA 91: 3238-3241.
Wright, D.A. and Voytas, D.F. (1998) Potential retroviruses in plants: Tatl is related to a group of Arabidopsis thaliana. Ty3/gypsy retrotransposons that encode envelope-like proteins. Genetics, 149, 703-71 5.
Yang, 2. (1996) Among-site rate variation and its impact on phylogenetic analyses. Trends Eco1 Evol 1 1 , 367-370.
Yeager Stassen, N., Logsdon, J.M., Vora, G.J., Offenberg, H.H., Palmer, J.O., Zolan, M.E. (1 997) Isolation and characterization of RA D5 1 orthologs

from Coprinus cinereus and Lycopersicon esculentum, and phylogenetic analysis of eukaryatic recA_hornologs. Curr Genet 31, 144-1 57.
Young, N.D. and Tanksley, S.D. (1989) RFLP analysis of the size of chromosomal segments retained around the Tm02 locus of tomato during backcross breeding. Theor. Appl. Genet. 77, 353-359.
Yuan, J.Y., Finney, M., Tsung, N. and Horvitz, H.R. (1991) Tc4, a Caenorhabditis elegans transposable element with an unusual fold-back structure. Proc. Nat1 Acad. Sci. USA, 88, 33343338.

![Ppt0000012 [Lecture seule] · E. coli: proportion de souches invasives résistantes aux carbapénèmes, Europe, 2009 - 2010 % d’isolats de E. coli résistants aux carbapénèmes,](https://img.pdfslide.fr/doc/110x75/5ed7b987498700329150e49f/ppt0000012-lecture-seule-e-coli-proportion-de-souches-invasives-rsistantes.jpg)



























