Embed Size (px)
Citation preview

Lignes directrices pour l’application d’un système
de management de la qualité dans les laboratoires d’analyse des drogues
Centre international de Vienne, Boîte postale 500, 1400 Vienne (Autriche)Téléphone: (+43-1) 26060-0, Télécopie: (+43-1) 26060-5866, www.unodc.org
Notre engagement: la qualité et l’amélioration continue*0981417*
Publication des Nations UniesImprimé en Autriche
Numéro de vente: F.09.XI.10
ST/NAR/37
12 USDISBN 978-92-1-248169-2
V.09-81417 — Septembre 2009 — 350

Crédits photo: Bibliothèque de l’UNODC; UNODC/Ioulia Kondratovitch; Alessandro Scotti.

Section scientifique et du laboratoireOFFICE DES NATIONS UNIES CONTRE LA DROGUE ET LE CRIME
Vienne
Lignes directrices pour l’application d’un système
de management de la qualité dans les laboratoires d’analyse des drogues
Notre engagement: la qualité et l’amélioration continue
NATIONS UNIESNew York, 2009

Remerciements
Le présent manuel a été élaboré par la Section scientifique et du laboratoire de l’Office des Nations Unies contre la drogue et le crime (UNODC), sous la direction du juge Tettey. Nous remercions chaleureusement toutes les personnes qui y ont participé (équipe en charge: Iphigenia Naidis et Satu Turpeinen).
La Section scientifique et du laboratoire tient à remercier le docteur Pirjo Lillsunde, de l’Unité de recherche sur les drogues de l’Institut finlandais de santé publique, qui a mis au point la première version du présent manuel.
La Section scientifique et du laboratoire adresse également ses remerciements aux membres du Comité permanent du Programme international de contrôle de la qualité de l’UNODC, Dr Robert Anderson, Dr Robert Bramley, Dr David Clarke et Dr Pirjo Lillsunde, qui ont examiné et finalisé le texte*.
Enfin, la Section scientifique et du laboratoire remercie le Comité du laboratoire de la Coopération européenne d’accréditation pour ses précieux commentaires.
*Les coordonnées des personnes susmentionnées peuvent être obtenues auprès de la Section scientifique et du laboratoire de l’UNODC [Boîte postale 500, 1400 Vienne (Autriche)].
PUBLICATION DES NATIONS UNIESNuméro de vente: F.09.XI.10
ISBN 978-92-1-248169-2
ST/NAR/37
La présente publication n’a pas été revue par les services d’édition.

iii
Table des matières Pages
1. AVANT-PROPOS: OBJET DU MANUEL. . . . . . . . . . . . . . . . . . . . . . . . . 1
2. INTRODUCTION . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4
3. SYSTÈME DE MANAGEMENT DE LA QUALITÉ. . . . . . . . . . . . . . . . 5 3.1. Politique qualité. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6 3.2. Manuel qualité . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6
4. EXIGENCES EN MATIÈRE D’ADMINISTRATION. . . . . . . . . . . . . . . . 8 4.1. Organisation du laboratoire. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 8 4.2. Maîtrise de la documentation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 9 4.3. Revue des demandes des clients . . . . . . . . . . . . . . . . . . . . . . . . . . . 10 4.4. Sous-traitance du travail d’analyse. . . . . . . . . . . . . . . . . . . . . . . . . . 11 4.5. Achat de services et de fournitures . . . . . . . . . . . . . . . . . . . . . . . . . 11 4.6. Services aux clients. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11 4.7. Réclamations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11 4.8. Actions correctives et préventives . . . . . . . . . . . . . . . . . . . . . . . . . . 11 4.9. Contrôle des enregistrements/chaîne de responsabilité . . . . . . . . . . 12 4.10. Audits internes. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14 4.11. Revues de direction. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14
5. EXIGENCES TECHNIQUES . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16 5.1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16 5.2. Personnel . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16 5.3. Installations et conditions ambiantes . . . . . . . . . . . . . . . . . . . . . . . . 17 5.4. Santé et sécurité . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 19 5.5. Méthodes d’essai, validation des méthodes et procédures d’essai . 20 5.6. Équipement . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 25 5.7. Normes de référence, matériaux et réactifs . . . . . . . . . . . . . . . . . . . 26 5.8. Manipulation des objets d’essai . . . . . . . . . . . . . . . . . . . . . . . . . . . 29 5.9. Rapport sur les résultats . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 32 5.10. Contrôle de la qualité, tests d’aptitude et comparaisons interlaboratoires . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 33
Bibliographie . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 35
Annexe Modèle de manuel qualité. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 37


1
1. Avant-propos: objet du manuel
Les services de détection et de répression ainsi que d’autres clients des laboratoires d’analyse des drogues, comme la police, les douanes, les procureurs et les avocats de la défense, ont besoin de résultats d’analyse qui soient:
Fiables; "
Valides; "
Fondés sur des procédures normalisées; "
Compatibles avec les résultats obtenus par d’autres laboratoires; "
Conformes aux normes admises comme preuve par les différents systèmes "judiciaires, administratifs et juridiques;
Obtenus de manière effective et efficiente dans les délais requis; "
D’un bon rapport qualité-prix. "
La qualité des analyses et des résultats de ces laboratoires a des conséquences importantes pour le système de justice, la détection et la répression ainsi que pour la prévention du crime et la politique de santé, de même que pour l’harmonisation internationale et l’échange et la coordination au niveau mondial d’informations et de données sur les drogues [1].
La compatibilité et l’acceptation des résultats de laboratoire entre pays sont facilitées par leur conformité à la norme ISO/CEI 17025:2005 (ISO 17025) [2], à la norme inter-nationale de qualité des laboratoires d’étalonnages et d’essai et aux recommandations connexes de la Conférence internationale sur l’agrément des laboratoires d’essai (ILAC G19:2002) applicables aux laboratoires de police scientifique [3]. La conformité à la norme ISO 17025 assure également la conformité aux exigences de la norme ISO 9001 concernant les laboratoires d’analyse de drogues. Cependant, la conformité à la norme ISO 9001 seule ne signifie pas la conformité à la norme ISO 17025, la norme ISO 9001 ne traitant pas de la compétence technique des laboratoires d’essai.
Les lignes directrices ci-dessous, qui concernent spécifiquement les laboratoires d’analyse des drogues, sont fondées sur les principes généraux établis par la norme ISO 17025. Elles prennent également en compte les Principes recommandés d’assurance de qualité et de bonnes pratiques de laboratoire de l’UNODC [4], les

2 Lignes directrices pour l’application d’un système de management de la qualité
recommandations du SWGDRUG [5], les exigences de l’American Society of Crime Laboratory Directors’ Laboratory Accreditation Manual [6], les lignes directrices SOFT/AAFS intitulées Forensic Toxicology Laboratory Guidelines [7] et les lignes directrices européennes pour les laboratoires de dépistage de drogues sur le lieu de travail [8, 9].
L’attachement à la qualité est le principe de base d’un laboratoire de criminalistique. Le présent manuel vise à fournir des lignes directrices aux laboratoires de crimina- li stique pour qu’ils atteignent un niveau élevé de qualité, utilisent les techniques appropriées pour trouver les “réponses” et s’améliorent en permanence. Il est conçu comme un manuel pratique et aborde des domaines qui ne sont pas explicitement traités en profondeur par la norme ISO 17025.
Le présent manuel se propose de servir d’introduction aux systèmes de management de la qualité des laboratoires et de fournir des lignes directrices pratiques aux autorités nationales et aux analystes pour les aider à appliquer ces systèmes en se fondant sur les meilleures pratiques de laboratoire. La “meilleure pratique” peut varier selon les laboratoires en raison de différences dans le niveau des ressources dont ils disposent et dans les prescriptions légales applicables. Par exemple, à l’échelle internationale, on peut considérer que la meilleure pratique repose sur l’utilisation de la LC-SM-SM, mais de nombreux laboratoires ne possèdent pas l’instrumentation nécessaire pour cette technique, ce qui ne les empêche pas d’obtenir de bons résultats. De même, certains pays exigent des analyses quantitatives des drogues ou l’identification d’isomères spécifiques, d’autres non. L’expression “meilleure pratique” doit donc être entendue comme la meilleure pratique réalisable dans un laboratoire et un pays donnés, pour autant que les travaux effectués répondent aux exigences indiquées dans le présent document. Il existe différentes manières d’obtenir le résultat final et ces lignes directrices aideront les directeurs de laboratoire à garantir que la leur se situe à un niveau international.
Le respect par tous les laboratoires d’analyse des drogues des lignes directrices qui suivent est essentiel pour garantir l’harmonisation de leur performance à l’échelle mondiale. Ces lignes directrices peuvent également servir de guide non seulement aux laboratoires, mais aussi aux auditeurs et inspecteurs lors de l’évaluation pendant le processus d’accréditation.
L’accréditation externe selon la norme ISO 17025 est le processus par lequel un organisme d’accréditation autorisé reconnaît formellement qu’un laboratoire est com-pétent pour effectuer des tâches spécifiques. Au cours du processus d’accréditation, le laboratoire doit démontrer que sa direction, ses opérations, son personnel, ses procédures, son équipement, ses installations et ses procédures en matière de sûreté, de santé et de sécurité satisfont aux normes de qualité établies. Bien que cela puisse exiger l’investissement de ressources importantes, l’obtention de la certification ISO 17025 accroît la confiance des directeurs de laboratoires, des autorités de détection et de répression et des autres clients quant à la performance du laboratoire. L’accréditation externe est par conséquent un objectif à rechercher lors de l’élabora-

Lignes directrices pour l’application d’un système de management de la qualité 3
tion d’un système de management de la qualité (SMQ); de fait, de nombreux laboratoires d’essai sont désormais accrédités dans plusieurs pays.
À l’avenir, il se peut que seuls les laboratoires d’analyse des drogues agréés soient autorisés à effectuer des analyses de toxicologie médico-légale ou à exercer en tant que laboratoires de police criminelle.
L’UNODC aide les laboratoires à instaurer et à mettre en œuvre un système de management de la qualité de diverses manières, notamment en leur fournissant des échantillons de référence des substances placées sous contrôle, en élaborant des manuels sur les méthodes recommandées, en leur offrant des opportunités de formation, en leur permettant de participer au programme pour les exercices collaboratifs internationaux (ICE) et en facilitant l’échange d’informations, de matériaux et de données [10].
Des exemples sont donnés dans tout le manuel pour clarifier les différents points abordés. Ceux-ci sont précédés des termes “par exemple”. Les exemples donnés en l’occurrence ne sont pas exhaustifs et visent unique-ment à illustrer les différents points abordés.

4
2. Introduction
Les laboratoires qui suivent les lignes directrices pratiques présentées dans ce document doivent satisfaire aux exigences de la norme ISO 17025. Ces lignes directrices sont fondées sur des considérations théoriques mais aussi sur l’expérience pratique acquise au cours du processus d’accréditation des laboratoires. Elles visent à assurer l’identification correcte des substances, à faire en sorte que les processus entrepris résistent à un examen juridique et à offrir des garanties pour protéger les droits des individus. Le but des lignes directrices est d’améliorer la qualité des services de laboratoire et de définir des critères communs d’assurance qualité.
Le présent manuel met l’accent à la fois sur les exigences techniques (importance d’une formation adéquate du personnel, installations d’essais et conditions environ-nementales appropriées, validation des méthodes, accès à l’équipement requis étalons de référence, contrôle de la qualité et communication des résultats) et sur les exi-gences en matière de management (contrôle des documents, satisfaction des besoins des clients, actions préventives et correctives, la nécessité d’audits réguliers de la conformité au système de management de la qualité et amélioration permanente de la performance du laboratoire).
Les lignes directrices s’appliquent à toutes les étapes clefs du processus de dépistage des drogues, de la collecte et de l’analyse d’échantillons à l’interprétation et à la présentation des résultats. Les laboratoires qui exécutent toutes ces étapes doivent veiller à ce que les lignes directrices énoncées dans le présent document soient intégralement respectées. Lorsque des clients exécutent une ou plusieurs étapes du processus au sein de leur organisation (par exemple la collecte d’échantillons ou l’interprétation/la revue des résultats), le laboratoire est tenu de veiller à ce que le client comprenne toutes les implications de ces activités dans le processus de dépistage des drogues.

5
3. Système de management de la qualité
Le laboratoire doit avoir un système de management de la qualité (SMQ) couvrant toutes ses activités, y compris l’échantillonnage, l’analyse et la communication des résultats, que ces activités soient effectuées au sein de l’installation principale du laboratoire, dans des installations mobiles ou temporaires ou en des sites extérieurs tels qu’un laboratoire clandestin, au bord d’une route ou sur le lieu de saisies importantes de drogues. Le système d’assurance de qualité comprend tous les documents se rapportant aux politiques, systèmes et procédures appliqués par le laboratoire ainsi qu’aux instructions, le cas échéant, qui visent à garantir la qualité des résultats, à assurer le respect des exigences juridictionnelles et réglementaires et à satisfaire les besoins des clients.
La hiérarchie du système de management de la qualité est présentée à la figure 1.
Figure 1. Hiérarchie du système de management de la qualité
Code de
déontologie
Norme ISO/IEC
17025
Exigences
des clients
Instructions
et recommandations
extérieures au
laboratoire (par exemple
normes internationales)
Prescriptions
législatives et
juridictionnelles et
autres exigences
réglementaires et
institutionnelles
MANUEL QUALITÉ
POLITIQUE QUALITÉ
SySTèME dE MANAgEMENT dE LA QUALITÉ
Politiques, systèmes,
procédures, instructions,
enregistrements etc.

6 Lignes directrices pour l’application d’un système de management de la qualité
3.1. Politique qualité [11]
Le laboratoire doit avoir un exposé concis de la direction générale indiquant la volonté de cette dernière d’adhérer aux principes de qualité dans tous les aspects du travail du laboratoire. Ce document doit également comporter les éventuels codes de pratique ou de déontologie qui s’appliquent. Cette déclaration constitue la politique qualité du laboratoire [4].
L’ensemble du personnel, du directeur de rang le plus élevé aux assistants, est tenu d’adhérer à cette politique qualité.
L’exposé de la politique doit comprendre au moins les points suivants:
Déclaration par la direction de son engagement à l’égard de la qualité et "de l’amélioration permanente de la performance du laboratoire;
Une déclaration de la direction indiquant que la qualité du travail du "laboratoire est adaptée à son objectif, dans ce sens qu’il résistera à tout examen scientifique et juridique et remplira les conditions convenues avec le client en matière budgétaire et en termes de délais;
Une déclaration de la direction par laquelle celle-ci s’engage à fournir les "ressources nécessaires pour gérer et appliquer le système qualité;
Une déclaration indiquant que tous les membres du personnel ont un rôle "à jouer pour faire en sorte que le travail du laboratoire soit effectué conformément aux exigences d’ISO 17025 et d’autres codes de pratique applicables au travail en question, ainsi qu’à toutes les prescriptions juridictionnelles, réglementaires et de sécurité pertinentes spécifiées dans le système de management de la qualité du laboratoire;
Le personnel responsable du système de management de la qualité doit avoir directement accès au niveau hiérarchique le plus élevé de la direction au sujet de la politique qualité du laboratoire.
3.2. Manuel qualité
Le laboratoire doit disposer d’un manuel décrivant succinctement le système de management de la qualité, indiquant l’organisation et le format des documents ainsi que les rôles et responsabilités des membres du personnel chargés d’administrer le système et les procédures techniques. Le manuel qualité doit également documenter les aspects administratifs, organisationnels et scientifiques du travail du laboratoire qui sont nécessaires à sa bonne administration.
La documentation doit être facile à se procurer dans le laboratoire et accessible à l’ensemble du personnel concerné. Elle doit être constamment révisée et mise à jour pour que tous changements des circonstances soient pris en compte.

Lignes directrices pour l’application d’un système de management de la qualité 7
L’utilisation de procédures documentées garantit que les travaux effectués sont contrô-lés et répondent aux objectifs fixés, que les exigences des normes internationales de qualité sont respectées et que les résultats des analyses de matériaux saisis ou d’échantillons biologiques peuvent être utilisés avec confiance dans toute procédure judiciaire ultérieure.

8
4. Exigences en matière d’administration
4.1. Organisation du laboratoire
Le manuel qualité doit préciser les éléments suivants:
L’organisation et la structure administrative du laboratoire et sa place au "sein de toute organisation plus grande ainsi que les relations entre le management de la qualité, les opérations techniques et les services d’appui. En outre, si le laboratoire fait partie d’une organisation plus vaste, les responsabilités de toute personne de cette organisation associée participant aux travaux du laboratoire ou pouvant avoir une influence sur ces travaux devraient être définies;
La personne légalement responsable du travail du laboratoire en cas d’action "en justice (par exemple, par un client). Cette responsabilité légale peut incomber, par exemple, au directeur de l’organisation à laquelle est rattaché le laboratoire, au directeur du laboratoire ou aux scientifiques;
La responsabilité, l’autorité et les relations entre tous les membres du "personnel qui dirigent, exécutent ou vérifient les travaux influant sur la qualité des résultats;
Les membres du personnel qui ont une autorité managériale et technique "en matière de management de la qualité, leurs responsabilités spécifiques, leurs suppléants désignés et les moyens mis à leur disposition pour s’acquitter de leurs fonctions. Il doit comprendre:
Un responsable de la qualité qui, indépendamment de ses autres –fonctions et responsabilités, aurait la responsabilité et le pouvoir de faire en sorte que le système qualité soit mis en œuvre et suivi à tout moment, aurait un accès direct au plus haut niveau de la direction où sont prises les décisions concernant la politique ou les ressources du laboratoire; éditerait et tiendrait à jour le manuel qualité; contrôlerait les pratiques du laboratoire; validerait les nouvelles procédures techniques; sélection-nerait, formerait et évaluerait les auditeurs internes; recommanderait des activités de formation pour améliorer la qualification du personnel du laboratoire; et proposerait des améliorations du système qualité;

Lignes directrices pour l’application d’un système de management de la qualité 9
Un directeur technique ou le personnel chargé d’opérations techniques –spécifiques, la fourniture des ressources nécessaires pour assurer la qualité requise de ces opérations, l’enquête sur les problèmes et leur résolution et l’évaluation des dossiers d’étalonnage et d’entretien des instruments.
Les dispositions prises pour assurer que la direction et le personnel du "laboratoire ne subissent pas de pressions et d’influences internes et externes commerciales, financières et autres indues, susceptibles de nuire à la qualité de leur travail et qu’ils ne sont pas associés à des activités qui auraient pour effet de diminuer la confiance dans la compétence, l’impartialité, le jugement ou l’intégrité opérationnelle du laboratoire. La réputation d’un laboratoire est plus facile à perdre qu’à rétablir;
Les garanties pour assurer la protection des informations confidentielles et "le respect des droits de propriété des clients, y compris les procédures de sauvegarde et de stockage des données et la transmission des résultats;
Les dispositions adoptées pour la supervision du personnel effectuant le "travail de laboratoire par les membres du personnel ayant eux-mêmes démontré leur compétence dans ce domaine.
4.2. Maîtrise de la documentation
Un système de management de la qualité (SMQ) ne peut fonctionner efficacement que si les politiques, systèmes, procédures et méthodes sont documentés et tenus à jour. La maîtrise de la documentation est le mécanisme par lequel les documents du système de management de la qualité sont créés, modifiés, examinés, approuvés, diffusés et archivés de manière que l’ensemble du personnel utilise les dernières versions autorisées.
Le directeur qualité doit veiller à ce que tous les aspects du travail du laboratoire soient documentés dans le SMQ et que les nouveaux documents soient établis par des personnes compétentes et autorisées par le personnel désigné avant d’être publiés. Il doit également veiller à ce que tous les documents soient périodiquement revus et, le cas échéant, révisés de manière à tenir compte des changements de circonstances et incorporer la meilleure pratique de laboratoire. Une fois approuvés, le directeur qualité doit faire le nécessaire pour qu’ils soient mis à la disposition de tout le personnel sur le lieu de travail. Lorsque des modifications ont été apportées à des documents existants, le directeur qualité doit s’assurer qu’elles sont mises en évidence dans les versions les plus récentes.
Tous les documents du SMQ doivent être identifiés de manière unique et porter le nom/la signature de la personne qui les a autorisés. Chaque page d’un document

10 Lignes directrices pour l’application d’un système de management de la qualité
doit être numérotée individuellement “page x de y pages” et comporter l’identifiant unique du document du SMQ la date d’émission et la version. Le système d’identi-fication des pages limite au minimum le risque d’omission non détectée de pages courantes et de rétention non détectées de pages obsolètes.
Si les documents sont tenus et distribués électroniquement, ils doivent être accessibles en lecture seulement et ne pouvoir être modifiés que par le personnel autorisé.
Il faudrait tenir une liste principale portant la date de publication et, le cas échéant, un enregistrement complet de toutes les versions, les dates auxquelles elles ont été établies, le nom de la personne habilitée et la liste de distribution. Elle pourrait être complétée par une feuille jointe à chaque document et donnant les mêmes informations.
Le laboratoire peut accepter que des modifications mineures soient apportées à la main aux documents imprimés pour corriger des fautes de frappe, mais ces modifi-cations doivent être apportées à l’encre permanente, datées et autorisées. Une version révisée doit toutefois être publiée dès que possible.
Les documents non valides ou périmés doivent être rapidement retirés de manière à éviter leur utilisation accidentelle. Une copie de chaque document périmé doit être conservée à des fins légales ou de sauvegarde des connaissances et porter une marque appropriée (par exemple la mention “non valide”).
4.3. Revue des demandes des clients
Le laboratoire doit avoir des procédures pour garantir que les exigences des clients sont correctement définies, documentées et comprises et qu’il dispose des moyens permettant d’y répondre avant d’accepter (par contrat) de faire le travail. C’est ce qui est appelé “Revue des contrats” dans la norme ISO 17025. L’accord peut être écrit ou oral, mais s’il est oral il doit par la suite être confirmé par écrit. Si le laboratoire n’a pas les moyens nécessaires, il doit essayer de convenir avec le client des tâches qu’il pourrait effectuer ou confier à un sous traitant avant d’entreprendre des travaux quelconques. Tout accord révisé doit également être documenté.
Le contrat doit être revu par le client et le laboratoire. Si de nouvelles exigences sont formulées par le client, ou si le laboratoire n’est pas en mesure de respecter l’accord initial ou les nouvelles exigences du client, les parties doivent en discuter et conclure un contrat révisé. Il faudrait également garder des traces de toutes les communications entre le client et le laboratoire au sujet du travail effectué. Ce sera un moyen d’assurer la compréhension commune des exigences, des responsabilités et du travail à effectuer par le laboratoire, ses clients et toutes les autres parties concernées.

Lignes directrices pour l’application d’un système de management de la qualité 11
4.4. Sous-traitance du travail d’analyse
Si le laboratoire fait appel à un tiers (sous-traitant) pour effectuer le travail en son nom, il doit avoir des politiques et procédures documentées assurant que ce tiers est compétent pour effectuer le travail.
4.5. Achat de services et de fournitures
Le laboratoire doit avoir une politique et une ou plusieurs procédure(s) pour la sélection et l’achat des services, des réactifs et des produits consommables suscep-tibles d’influer sur la qualité de ses travaux, ainsi qu’une procédure pour la réception et le stockage des réactifs et des produits consommables et pour garantir que les services, les réactifs et les produits consommables sont conformes aux spécifications techniques du système de management de la qualité avant d’être achetés ou utilisés.
Le laboratoire doit garder une trace des mesures prises pour vérifier la conformité et les résultats de ces vérifications.
4.6. Service aux clients
Le laboratoire doit s’assurer qu’il comprend les besoins de ses clients et les tenir informés de la progression de son travail. Il doit aussi demander à ses clients un retour d’information sur sa performance. Les réactions, tant négatives que positives, sont importantes pour améliorer les services du laboratoire. La confidentialité du client doit à tout moment être respectée.
4.7. Réclamations
Le laboratoire doit avoir un système pour traiter efficacement les réclamations des clients qui comporterait l’obligation d’informer les clients de toutes mesures prises pour régler le problème et éviter sa répétition. Il faudrait conserver des dossiers de toutes les réclamations et actions correctives et les mettre à profit pour améliorer le management de la qualité dans le laboratoire. Le fait de ne pas répondre aux réclamations des clients peut nuire à la relation client-laboratoire.
4.8. Actions correctives et préventives
Lorsque le travail effectué par le laboratoire n’est pas conforme à son système de management de la qualité (par exemple s’il s’écarte d’un mode opératoire ou des

12 Lignes directrices pour l’application d’un système de management de la qualité
exigences des clients), la norme ISO 17025 parle de “non-conformité”. Le labora-toire doit avoir des systèmes (par exemple, vérification du travail au sein du laboratoire, dossier ou audit du SMQ, information en retour du personnel/client) pour reconnaître quand une non-conformité s’est produite et comment la gérer. Ce processus doit comporter une enquête sur les causes de la non-conformité, l’évaluation de son importance, l’information du client si nécessaire et l’autorisation de modifier, livrer ou rappeler le travail non conforme. Tout doit être fait pour détecter les non-conformités avant la remise des résultats au client, car la communication de résultats incorrects peut porter gravement atteinte à la relation laboratoire-client et conduire à des erreurs judiciaires. Si la non-conformité se reproduit sans que des mesures plus générales aient été adoptées (par exemple, modification du SMQ ou perfectionnement du personnel), des actions préventives doivent être autorisées au niveau approprié pour éviter la répétition du problème.
Toutes les non-conformités, actions correctives et actions préventives doivent être enregistrées. L’efficacité des actions correctives doit être vérifiée par le personnel autorisé afin de garantir que le travail est désormais conforme aux exigences du SMQ/client. Il faudrait également suivre les actions préventives pour s’assurer (par exemple par un audit) que les non conformités ne se répètent pas.
La reconnaissance des non-conformités et la mise en œuvre d’actions correctives et préventives sont essentielles pour l’amélioration continue de l’efficacité de la performance du laboratoire.
4.9. Contrôle des enregistrements/chaîne de responsabilité
Le laboratoire doit avoir des systèmes pour la création, l’identification, la gestion, le stockage, le mouvement/la transmission, le retrait et d’élimination de tous les enregistrements, sur support papier ou sous forme électronique.
Tous les enregistrements du laboratoire sur support papier doivent être facilement lisibles, identifiables sans ambiguïté (porter par exemple la date, l’auteur et le nombre de pages) et rédigés à l’encre permanente. Le crayon papier ne doit pas être utilisé. Aucun enregistrement ne doit être supprimé. Les modifications et corrections apportées à la main ne doivent pas occulter les données initiales et doivent être signées/paraphées et datées. Des mesures doivent également être prises pour sauvegarder les enregistrements électroniques originaux (par exemple en créant des sauvegardes de fichiers informatiques), identifier toute modification éventuelle (par exemple au moyen d’un journal d’audit électronique que certains fabricants proposent avec le logiciel) et assurer leur intégrité et confidentialité.
Tous les enregistrements doivent être systématiquement archivés pour permettre une consultation facile. En même temps, ils doivent être traités comme confidentiels, la

Lignes directrices pour l’application d’un système de management de la qualité 13
protection des données et les prescriptions légales relatives au droit à la vie privée, notamment, doivent être respectées et l’accès à ces enregistrements doit être limité au personnel autorisé.
Les enregistrements peuvent être subdivisés en enregistrements qualitatifs et enregistrements techniques.
Les enregistrements qualitatifs comprennent les rapports d’audit, les essais d’aptitude, les retours d’information des clients, les actions correctives et préventives et les revues de direction. Les enregistrements doivent être identifiables sans ambi-guïté (porter par exemple la date, le nom de l’auteur et le nombre de pages) et gardés en un lieu sûr et réglementé accessible au seul personnel autorisé. La durée de conservation de ces enregistrements (par exemple, compte tenu des prescriptions légales) doit être définie. Les enregistrements à éliminer doivent être traités comme des déchets confidentiels et incinérés ou déchiquetés.
Les enregistrements techniques comprennent tous les documents relatifs aux essais, y compris les formulaires de soumission des échantillons, les documents de la chaîne de responsabilité, les notes de travail (y compris les dessins et diagrammes), les photographies, les enregistrements de conversations téléphoniques, les spectres, données d’étalonnage et autres données du contrôle de la qualité, les paramètres opérationnels des instruments et les sorties d’imprimante, les rapports, déclarations, et autres, les registres de maintenance des instruments et les dossiers de formation, de compétence et d’autorisation du personnel. Ces enregistrements doivent être effectués au moment où le travail est exécuté.
Chaque donnée enregistrée doit pouvoir être reliée à l’analyste/examinateur et, le cas échéant, à un essai ou à un résultat identifié sans ambiguïté. Il doit ressortir clairement de l’enregistrement qui a effectué toutes les phases de l’analyse/examen et quand chaque étape de l’analyse/examen a eu lieu (par exemple au moyen des dates pertinentes). Tous les enregistrements doivent contenir suffisamment d’infor-mations pour permettre de tracer l’audit pour montrer qui a fait quoi et quand.
Les conclusions essentielles (par exemple les odeurs et les observations visuelles comme les tests colorimétriques qui ne peuvent pas être confirmées indépendamment plus tard à partir de l’enregistrement, les calculs et les transferts de données qui ne font pas partie d’un processus électronique validé) doivent être vérifiées, de préférence par une deuxième personne autorisée.
Le laboratoire doit avoir des procédures documentées pour la revue globale des enregistrements par les personnes autorisées. Chaque enregistrement doit indiquer que ces contrôles et revues ont été effectués, quand et par qui. Ces mentions peuvent être apportées de différentes manières (par exemple par une note en regard de chaque résultat, une note ajoutée au résumé des résultats ou une déclaration à cet effet jointe aux enregistrements). Si l’auditeur ou le vérificateur de l’essai récuse un point quelconque de l’enregistrement initial, la ou les raison(s) de son désaccord et les mesures prises en conséquence doivent être consignées.

14 Lignes directrices pour l’application d’un système de management de la qualité
En général, les enregistrements nécessaires pour étayer les conclusions doivent être tels qu’en l’absence de l’analyste ou de l’examinateur un autre analyste/examinateur puisse évaluer ce qui avait été fait, interpréter les données [12] et, le cas échéant, refaire le travail. Quand le résultat d’un essai ou une observation est rejeté, la (les) raison(s) doive(nt) en être indiquée(s). Cette information est nécessaire pour qu’un autre analyste puisse comprendre comment l’essai a été géré.
Les enregistrements techniques doivent être conservés dans un lieu sûr et protégé pour prévenir les dommages, la détérioration, l’accès non autorisé ou la perte, pendant une période qui dépend des besoins du client et des règlements pertinents en vigueur dans le pays. La durée de conservation peut aussi dépendre de la nature de l’infraction en cause, de sorte que les enregistrements concernant des infractions graves, telles que homicide et trafic de drogues, sont conservés plus longtemps que ceux relatifs à des infractions mineures, telles que la simple possession d’une subs-tance placée sous contrôle. Cela signifie que certains enregistrements peuvent être conservés indéfiniment, détruits à l’issue de la procédure judiciaire ou être soumis à des dispositions intermédiaires. Les enregistrements voués à la destruction doivent être traités comme des déchets confidentiels et être incinérés ou déchiquetés.
4.10. Audits internes
Le laboratoire doit effectuer périodiquement, conformément à une procédure et à un calendrier prédéfinis, un audit de toutes ses activités (par exemple de chaque méthode d’essai) pour vérifier que toutes ses opérations sont conformes aux exigences du SMQ. Les audits doivent être organisés par le directeur qualité au moins une fois par an et exécutés et consignés par un personnel qualifié formé, indépendant, chaque fois que les ressources le permettent, de l’activité soumise à l’audit. Lorsque les conclusions de l’audit jettent un doute sur l’efficacité des opérations ou sur la correction ou la validité du travail du laboratoire, ces conclusions doivent être discutées par l’auditeur, le directeur qualité et le personnel concerné et des actions correctives appropriées doivent être adoptées. Le laboratoire doit rapidement informer les clients par écrit si l’audit révèle que les résultats étaient incorrects.
Le secteur d’activités faisant l’objet de l’audit, les conclusions de l’audit et les actions correctives doivent être enregistrés. Les audits futurs de la même activité doivent évaluer l’efficacité des actions correctives adoptées afin d’identifier les enseignements tirés et, partant, d’améliorer la performance du laboratoire.
4.11. Revues de direction
Le directeur qualité doit organiser une revue annuelle du SMQ et des activités d’essais pour s’assurer qu’elles demeurent appropriées et efficaces ou identifier tous

Lignes directrices pour l’application d’un système de management de la qualité 15
changements et améliorations nécessaires. La revue doit être effectuée par la haute direction de l’organisation à laquelle sont rattachés le laboratoire, le directeur qualité et les autres membres du personnel désignés. La revue et toutes actions recommandées doivent être enregistrées et les actions menées dans un délai approprié convenu par le personnel chargé de la revue et le directeur qualité, compte tenu des ressources disponibles.
La revue de direction devrait porter sur les points suivants:
Changements dans le volume et le type de travail; "
Activités de contrôle de la qualité, ressources et formation du personnel et "autres facteurs pertinents;
Pertinence des politiques et procédures; "
Rapports du personnel d’encadrement; "
Résultats d’audits internes récents; "
Évaluations effectuées par des organismes extérieurs; "
Informations en retour des clients; "
Réclamations; "
Résultats des essais de comparaison interlaboratoires ou des tests d’aptitude; "
Actions correctives et préventives; "
Recommandations pour améliorer la performance du laboratoire. "

16
5. Exigences techniques
5.1. Introduction
De nombreux facteurs, tels que le personnel, les installations et les conditions ambiantes, les méthodes d’essai, la validation des méthodes, l’équipement, les étalons de référence, l’échantillonnage et la manipulation des objets d’essai, contribuent à l’exactitude et à la fiabilité des résultats et déterminent également dans une large mesure l’incertitude de la mesure. Ces facteurs doivent tous être pris en compte au moment de l’élaboration des méthodes et des procédures, de la formation du personnel, du choix et de l’utilisation de l’équipement.
5.2. Personnel
Le personnel est l’atout le plus précieux d’un laboratoire. Ce dernier doit favoriser un climat qui incite les employés à améliorer leurs connaissances et leurs compétences, à se développer en tant que personnes et à mettre pleinement en valeur leur potentiel.
Le laboratoire ne doit avoir recours qu’à des collaborateurs qui sont sous contrat avec lui, y compris le personnel temporaire. La direction du laboratoire doit veiller à ce que le personnel possède le niveau d’études, la formation, l’expérience, les connaissances, les qualifications et les aptitudes requises (c’est-à-dire la compétence) pour effectuer les travaux confiés au laboratoire [13], qu’il soit convenablement encadré et travaille conformément au SMQ du laboratoire.
Le laboratoire doit avoir des politiques et des procédures permettant d’identifier les besoins de formation et d’assurer la formation du personnel pour l’aider à atteindre et maintenir un niveau de compétence (par exemple par des programmes structurés de formation sur place, la participation à des réunions, conférences et ateliers scientifiques, des cours de formation technique, des cours sur le fonctionnement et la maintenance des instruments dispensés par des commerciaux, des réunions, cours et séminaires techniques sur place et la formation continue). Tout au long de la formation, le personnel doit être plus étroitement encadré et l’efficacité de sa formation contrôlée et évaluée. Lorsqu’il offre une formation particulière à un

Lignes directrices pour l’application d’un système de management de la qualité 17
essai ou à une technique donnée, le laboratoire doit prévoir des critères d’acceptation (par exemple l’observation des essais ou des analyses pertinents par un agent expérimenté, le rendement satisfaisant dans l’analyse des échantillons de contrôle de la qualité ou d’assurance de la qualité et la corrélation entre les résultats obtenus par les analystes et ceux obtenus par d’autres employés formés dans ce domaine). Le cas échéant, les programmes de formation doivent également comprendre un module sur la présentation des pièces à conviction devant les tribunaux.
Le laboratoire doit tenir un dossier sur la formation, les qualifications et les certificats de formation des membres du personnel ainsi que sur la liste des tâches qu’ils sont habilités et autorisés à effectuer (par exemple les types d’essais qu’ils peuvent réa-liser, les rapports d’essais qu’ils sont habilités à délivrer, les avis et les interprétations qu’ils peuvent émettre et les types particuliers d’équipement qu’ils peuvent faire fonctionner). Ces informations doivent pouvoir être facilement consultées par le personnel et la date à laquelle la compétence et l’habilitation ont été confirmées doit être consignée de manière que tous les membres du personnel comprennent clairement l’étendue des tâches qui leur sont confiées et le champ de leurs responsabilités.
Chaque membre du personnel doit disposer d’une description du poste dont il est convenu avec son supérieur hiérarchique désigné, qui doit préciser ses responsabi-lités, ses obligations et ses compétences.
(La norme ISO 17025 ne traite pas des spécifications professionnelles (diplômes, qualifications, connaissances, aptitudes, compétences, etc.) requises pour des postes particuliers. Des recommandations à cet égard figurent toutefois dans d’autres docu-ments [4, 13].)
5.3. Installations et conditions ambiantes
Les types d’échantillons analysés par le laboratoire (matériaux saisis ou spécimens biologiques ou les deux), les effectifs du personnel et la charge de travail projetée influent sur les besoins de superficie, d’entreposage et de sécurité.
Les installations et les conditions ambiantes du laboratoire doivent être de nature à garantir la conformité des travaux réalisés aux normes de qualité requises. Des précautions particulières doivent être prises lorsque des analyses sont effectuées hors du laboratoire et non dans ses installations principales. Les facteurs à prendre en compte comprennent: l’espace, la sûreté, la santé et la sécurité du personnel, le contrôle de la température et de l’humidité, l’éclairage, l’aération et la ventilation, en plus des services de base essentiels (par exemple l’électricité, le gaz, l’eau, le téléphone et les connexions informatiques, les paillasses de laboratoire, les armoires de sécurité, les réfrigérateurs et les congélateurs).

18 Lignes directrices pour l’application d’un système de management de la qualité
Les installations du laboratoire doivent permettre l’exécution correcte du travail et, si les conditions ambiantes sont susceptibles d’affecter les résultats, elles doivent être spécifiées, documentées et contrôlées (par exemple la température de stockage des échantillons). Le matériel sensible à l’environnement doit être installé dans des zones où l’accès est réduit et les microbalances doivent être protégées des vibrations et de la corrosion chimique. Un espace suffisant et approprié doit être alloué à chaque activité/fonction et employé. Des locaux spéciaux doivent être prévus pour le stockage des grandes quantités de substances dangereuses, tels que les gaz comprimés, les solvants et les produits chimiques dangereux, afin de protéger la santé et d’assurer la sécurité du personnel. En outre, un local approprié doit être réservé à l’entreposage des pièces à conviction afin d’empêcher leur perte, leur détérioration ou leur contamination et de maintenir leur intégrité et identité, tant avant qu’après leur examen.
Les zones du laboratoire doivent être suffisamment propres et ordonnées pour réduire au minimum les risques de contamination et pour que la qualité du travail effectué ne soit pas compromise. Il devrait donc y avoir une séparation spatiale effective des activités incompatibles (par exemple l’examen des substances saisies en vrac et l’analyse de traces de drogues des spécimens biologiques) qui ne devraient pas avoir lieu dans les mêmes installations. Des mesures devraient également être prises pour prévenir les risques de contamination croisée (par exemple entre deux différentes saisies en vrac ou entre des matériaux et des échantillons de référence). Ces mesures pourraient consister à réglementer l’accès à certaines zones du laboratoire, le transport des échantillons et le partage de l’équipement (par exemple, les spécimens biologiques dangereux ouverts et la verrerie sale ne devraient pas transiter dans des zones non protégées, et le matériel en verre qui sert à analyser les matériaux saisis (contenant de fortes concentrations de drogues) ne devrait pas être utilisé pour l’analyse de traces d’échantillons biologiques).
L’accès aux laboratoires d’analyse des drogues et aux zones où sont entreposées les pièces à conviction doit être réglementé et contrôlé en toutes circonstances afin d’éviter tout vol ou intervention. Des contrôles devraient être effectués tant aux points d’entrée que de sortie des installations et entre les différentes zones réglementées (par exemple au moyen de clefs ou de cartes magnétiques distribuées au personnel autorisé à des fins spécifiques). De cette façon, le personnel non autorisé ne sera pas en mesure de manipuler des échantillons ou d’avoir accès aux zones où des drogues, des spécimens ou des enregistrements sont conservés.
Le laboratoire doit tenir à jour la liste de tout le personnel autorisé à pénétrer dans les zones réglementées du laboratoire. Cette liste devrait être revue et mise à jour régulièrement. Les personnes non autorisées qui doivent accéder aux zones réglemen-tées (par exemple les autres employés du laboratoire, les clients, les ingénieurs de maintenance, les agents d’entretien, le personnel administratif et les visiteurs) doivent être accompagnées en permanence par des personnes autorisées et il faut tenir un registre de toutes les entrées et sorties.

Lignes directrices pour l’application d’un système de management de la qualité 19
5.4. Santé et sécurité
La politique de sécurité du laboratoire doit être définie dans un manuel décrivant les procédures à suivre pour assurer la sécurité et la santé du personnel, éviter les accidents et ne pas exposer le personnel à des risques sanitaires. Ces procédures doivent reposer sur l’évaluation des risques de toutes les activités et des systèmes de sûreté documentés du travail (par exemple la nécessité de manipuler des produits chimiques dangereux, tels que ceux utilisés pour pulvériser sous hotte les plaques de chromatographie sur couche mince).
Des informations détaillées sur les éléments suivants devraient figurer dans le manuel:
Personnel chargé de la sécurité (par exemple l’agent de sûreté, l’agent de "sécurité biologique, l’agent de sécurité incendie, le personnel de premier secours). Ces responsabilités peuvent être confiées à plusieurs personnes ou à un seul membre du personnel;
Procédures d’urgence et personnes à contacter (par exemple que faire en "cas d’incendie, de fuite de produits chimiques, de lésions corporelles);
Formation du personnel (par exemple les exercices incendie et les premiers "secours);
Emplacement des infrastructures et équipements (par exemple les lavabos, "les douches d’urgence, les armoires de premiers secours, les solutions ophtalmiques, les armoires de sécurité/hottes d’évacuation, les appareils autoclaves, les extincteurs d’incendie, le lieu de stockage des solvants et produits chimiques, le site d’élimination des déchets, des produits chimiques, des aiguilles et des substances radioactives, la signalisation de sécurité/ de danger, les informations sur les sorties de secours, l’emplacement des équipements de sécurité et les numéros de téléphone d’urgence);
Équipements de protection (par exemple les blouses de laboratoire, les gants "jetables, les lunettes de protection, les masques de protection, les protections acoustiques et les badges de protection contre les radiations);
Hygiène et sûreté générales du laboratoire (par exemple le nettoyage et "la désinfection des surfaces, le verrouillage automatique des appareils biologiquement contaminés, le port d’équipements de protection, l’interdiction de manger, de boire et de fumer dans le laboratoire, l’interdiction de porter des vêtements de laboratoire dans les zones stériles désignées et l’interdiction de travailler seul dans les laboratoires);
Risques biologiques spécifiques (par exemple les enceintes de sécurité "microbiologique, la vaccination du personnel, l’élimination sûre des déchets cliniques, la stérilisation des instruments et le port d’équipements de protection);
Risques de radioactivité (par exemple référence aux règlements concernant "la manipulation des substances radioactives).

20 Lignes directrices pour l’application d’un système de management de la qualité
5.5. Méthodes d’essai, validation des méthodes [14] et procédures d’essai
Le laboratoire doit utiliser des méthodes et procédures appropriées pour toutes ses activités: échantillonnage, manipulation, transport et stockage des pièces à conviction, utilisation des équipements, essais, évaluation et interprétation des résultats, et présentation de rapports. Les méthodes et procédures doivent être à jour, entièrement consignées par écrit et pouvoir être facilement consultées par le personnel concerné.
La documentation des méthodes doit préciser:
Le nom/numéro de référence de la méthode; "
Le champ d’application de la méthode (par exemple les analytes, la matrice, "la gamme de concentration, les interférences connues);
La théorie et le principe de la méthode; "
Un résumé des paramètres de validation [14] et l’indication de l’emplacement/ "du numéro d’identification du fichier contenant les données de validation;
Les produits chimiques, appareils et équipements requis, y compris les "spécifications techniques;
Les étalons/matériaux de référence requis; les conditions ambiantes néces- "saires (par exemple la température de la pièce) et toute période de stabilisa-tion nécessaire (par exemple le temps d’équilibrage d’une chromatographie sur couche mince);
Une description étape par étape de la procédure, y compris: "
Toutes les précautions particulières à prendre (par exemple, en matière –de santé et de sécurité);
Les exigences relatives à l’échantillonnage, l’étiquetage, l’emballage, le –transport et l’entreposage des échantillons;
La préparation des échantillons, matériaux de référence, échantillons de –contrôle et solutions d’étalonnage;
Les exigences en matière de contrôle et d’étalonnage des équipements –(par exemple la réalisation d’un échantillon de référence, le réglage et l’étalonnage d’un spectromètre de masse);
Le processus d’analyse/la procédure d’essai et le contrôle de la qualité –(par exemple l’utilisation de blancs, de contrôles et de calibrateurs);
L’enregistrement et le traitement des résultats (par exemple les calculs, –la préparation des courbes et graphiques d’étalonnage), y compris les critères et/ou les exigences en matière d’acceptation et de rejet (par exemple, si les résultats se situent en dehors de la gamme d’étalonnage ou si les contrôles de qualité donnent des résultats inacceptables);

Lignes directrices pour l’application d’un système de management de la qualité 21
Les exigences relatives à la présentation des résultats; –
Les exigences relatives à l’incertitude* de la méthode. –
(De plus amples informations concernant l’incertitude sont données ailleurs [14, 15].)
Lorsqu’une procédure d’identification ou de quantification d’une drogue repose sur plus d’une méthode, la manière dont ces méthodes sont liées doit être expliquée (par exemple, à l’aide d’un graphique ou d’une description textuelle), de même que la combinaison, l’évaluation et la présentation des résultats.
Tout écart par rapport à ces protocoles et procédures ne doit être autorisée que s’il est justifié, autorisé et accepté par le client, le cas échéant, et documenté.
Développement et validation des méthodes
Les méthodes et procédures appliquées doivent de préférence reposer sur des travaux publiés dans des revues scientifiques visées par des spécialistes (par exemple Forensic Science International, Journal of Analytical Toxicology, Journal of Chromatography et les publications de l’UNODC portant la cote ST/NAR s’inspirant de publications scientifiques) et tenir compte des besoins du client. À défaut de méthodes publiées disponibles, celles développées par le laboratoire peuvent être admises à condition d’être pertinentes et de répondre aux besoins du client. Dans ce cas, les mesures qui ont été prises doivent être consignées de manière suffisamment détaillée afin de permettre à une autre personne convenablement qualifiée de comprendre la méthode suivie et les résultats obtenus.
Toutes les méthodes, y compris les méthodes non normalisées/élaborées par le laboratoire, doivent être “validées” ou “vérifiées” pour démontrer leur adéquation aux objectifs recherchés et fonctionneront dans l’environnement opérationnel du laboratoire. Toutes les validations et vérifications doivent être effectuées selon la procédure agréée du laboratoire et être entièrement documentée.
La validation est la confirmation par un examen et la production d’éléments objectifs que les exigences particulières pour un usage spécifique prévu sont satisfaites. La validation doit être aussi complète qu’il est nécessaire. Des orientations sur ce qu’il convient de faire dans différentes circonstances et les différentes méthodes à appliquer figurent dans de nombreuses publications (par exemple Guidelines on Validation of Analytical Methodology and Calibration of Equipment used for Testing
* Le mot “incertitude” signifie “doute”, et donc au sens le plus large du terme “incertitude de mesure” signifie qu’il existe un doute quant à la validité du résultat d’une mesure ainsi qu’un doute quant à l’exactitude du résultat. L’incertitude de mesure comprend, en général, de nombreuses composantes. Le laboratoire devrait s’efforcer d’identifier tous les éléments d’incertitude, d’en faire une estimation raison-nable et de garantir que le formulaire sur lequel sont reportés les résultats ne donne pas une impression fausse de l’incertitude.

22 Lignes directrices pour l’application d’un système de management de la qualité
of Illicit Drugs in Seized Materials and Biological Specimens [Lignes directrices pour la validation de la méthode analytique, et l’étalonnage du matériel utilisé pour l’analyse des drogues illicites dans les matériaux saisis et les spécimens biologiques] de l’UNODC, et les références qui y sont mentionnées). La vérification est similaire à la validation à ceci près qu’elle n’intervient que lorsque la méthode a déjà été validée ailleurs et qu’il a été démontré qu’elle satisfait aux exigences requises lorsqu’elle est utilisée par le personnel du laboratoire. La question de la validation est examinée en détail dans le manuel de l’UNODC [14].
Lorsque des ordinateurs ou des équipements automatisés sont utilisés pour l’acquisi-tion, le traitement, l’enregistrement, la notification, le stockage ou la récupération des données d’essai ou d’étalonnage, le laboratoire doit veiller à ce que le logiciel utilisé soit décrit suffisamment en détail et ait été correctement validé.
Le laboratoire doit tenir un fichier du personnel participant aux travaux de validation, noter les dates, les résultats obtenus et la procédure utilisée et comprendre une déclaration indiquant si la méthode convient pour son usage et la signature autorisant/approuvant son emploi.
Lorsqu’un laboratoire introduit une méthode qui a été validée par d’autres, il doit commencer par démontrer la fiabilité de la procédure sur place en se basant sur les caractéristiques de performance de la procédure publiée dans la littérature. Une trace de cette procédure de vérification de la performance doit être conservée pour référence.
Lorsqu’une procédure d’identification ou de quantification d’une drogue repose sur plusieurs méthodes, chacune doit être validée/vérifiée.
Composition d’une série d’échantillons/de lots analytiques
L’analyse d’une série d’échantillons ou d’un lot analytique consiste à analyser en même temps un mélange d’échantillons, généralement des échantillons d’essai, de contrôle et des calibrateurs. La composition d’un lot analytique dépend de la métho-dologie qui sera suivie et de l’objet de l’analyse. Trois types de méthodologies sont envisagés: immunodosages (pour les spécimens biologiques), analyses qualitatives et analyses quantitatives. En règle générale, dans un lot, 10 % de tous les échantillons devraient correspondre à des calibrateurs et à des échantillons de contrôle. Cela s’applique à tous les types d’analyses [4].
Immunodosages
Chaque lot d’échantillons analysé par immunodosage doit contenir au moins un échantillon de contrôle négatif (par exemple un échantillon d’urine ne contenant pas de drogue), un ou plusieurs échantillons dont l’un doit contenir l’analyte à la concentration maximale autorisée et au moins un échantillon

Lignes directrices pour l’application d’un système de management de la qualité 23
de contrôle positif. Ils sont généralement inclus dans les kits commerciaux d’immunodosages.
Analyses qualitatives
L’identification qualitative d’une drogue ou d’un métabolite doit reposer sur une comparaison directe des données analytiques relatives au spécimen soumis et des données correspondantes obtenues pour un étalon de référence analysé en même temps dans les mêmes conditions. Pour cette raison, les lots d’échan-tillons qui doivent être soumis à une analyse qualitative doivent comporter des étalons de référence pour chacun des analytes de drogues dont on soupçonne la présence.
Analyses quantitatives
Les lots d’échantillons soumis à une analyse quantitative doivent tous contenir des blancs, des solutions d’étalonnage et des étalons de contrôle. Six étalons sont recommandés, à des concentrations comprises dans les limites de référence utilisées dans le processus de validation décrit ci-dessous [14]. Si la concen-tration de l’analyte d’un échantillon est supérieure à la fourchette des concen-trations représentée, l’échantillon doit être dilué et analysé une nouvelle fois. Une autre méthode consiste à analyser d’autres solutions d’étalonnage afin d’englober la fourchette des concentrations mesurées de l’analyte des spécimens fournis, mais elle est moins couramment utilisée.
Acceptation des résultats d’analyse
Les critères spécifiques de ce qui constitue un essai positif doivent être établis et clairement spécifiés dans la procédure méthodologique. Ces critères doivent également indiquer les conditions dans lesquelles les résultats sont acceptables pour les échantillons de contrôle de la qualité. Avant de pouvoir dire qu’un échantillon contient une ou plusieurs drogues, il faut en analyser deux parties distinctes, chacune par au moins deux méthodes différentes validées. Ces deux méthodes devraient reposer sur des principes scientifiques, dont une devrait si possible fournir des indications sur la structure chimique de l’analyte (par exemple, IR, SM, ou méthodes en tandem telles que GC-MS).
En outre, avant qu’un échantillon puisse être déclaré positif, les résultats des analyses doivent être vérifiés par au moins deux membres autorisés du personnel (générale-ment, l’analyste et un scientifique confirmé), qui connaissent bien les méthodes d’analyse. La vérification devrait porter sur l’examen des résultats des essais, des résultats du contrôle de la qualité, de tous les documents relatifs à la manipulation des échantillons (par exemple la chaîne de responsabilité et les calculs) et de toutes les informations consignées par écrit (par exemple les données reportées sur des feuilles de calcul).

24 Lignes directrices pour l’application d’un système de management de la qualité
Utilisation de cartes de contrôle dans l’analyse quantitative
Une méthode analytique est sous contrôle statistique lorsque ses résultats s’inscrivent constamment dans les limites de contrôle établies. La conformité avec le contrôle statistique peut être assurée graphiquement au moyen de cartes de contrôle (par exemple les cartes de Shewhart et de Cusum). Ces cartes sont utiles pour les métho-des d’analyse courantes sujettes à des erreurs systématiques ou à une variabilité accrue. Sur une carte de contrôle, les résultats des essais sont indiqués par rapport au temps. Si la méthode d’analyse est sous contrôle statistique, tous les résultats se trouvent en deçà des limites de contrôle prédéterminées, qui sont aussi généralement indiquées sur la carte:
Limite de surveillance, qui correspond à ± 2 écarts types par rapport à la "moyenne.
Limite de réglage, qui correspond à ± 3 écarts types par rapport à la "moyenne.
On peut donner comme exemple un graphique des concentrations mesurées des solutions d’échantillons de contrôle en fonction du temps. Dans ce cas, tous les résultats se trouvent sur la valeur vraie ou à proximité et en distribution normale. Cependant, même si la méthode est sous contrôle statistique, on peut s’attendre qu’environ 5 % des résultats se situent au-delà de la limite de surveillance. Si une valeur observée se situe en dehors de la limite de réglage, des mesures immédiates doivent être prises pour en identifier la cause et adopter des actions correctives.
Outre qu’elles montrent les résultats isolés qui peuvent s’écarter de la valeur vraie, les cartes de ce type montrent de manière évidente si la moyenne diffère de la valeur vraie (biais) ou s’il y a une tendance régulière qui fait que les valeurs s’en écartent dans une direction donnée.
Compétence de l’analyste
Les laboratoires devraient établir une procédure pour identifier les essais ou analyses peu fréquents. Dans ce cas, deux méthodes permettent d’établir la compétence de l’analyste, qui sont aussi valides l’une que l’autre:
L’analyse régulière des échantillons de contrôle et l’utilisation de cartes de "contrôle pendant les périodes où les spécimens soumis ne sont pas analysés;
La revérification avant l’essai ou l’analyse en question au moyen d’au moins "un matériau de référence approprié, suivie de la répétition de l’essai ou de l’analyse du spécimen visé.

Lignes directrices pour l’application d’un système de management de la qualité 25
5.6. Équipement
Les laboratoires doivent assurer la fiabilité et la performance de l’équipement utilisé. Les matériels et logiciels nécessaires pour le travail effectué doivent être adaptés aux buts visés et devraient de préférence être disponibles dans le laboratoire. Lorsqu’un laboratoire utilise un équipement autre que le sien, celui-ci doit être conforme aux exigences du SMQ. Le laboratoire doit tenir un inventaire de l’équipement et consigner les informations concernant son emplacement, la date d’achat, l’historique du service et la maintenance. Les principales pièces d’équipement (par exemple des instruments tels que des spectromètres) devraient avoir leur propre journal pour assurer la bonne tenue de ces renseignements.
Le personnel doit être formé à l’utilisation de l’équipement et autorisé à l’utiliser uniquement quand il a été jugé compétent. La formation et l’autorisation doivent être documentées dans les dossiers du personnel.
Lorsque l’équipement est acheté, les spécifications du fabricant doivent être au minimum conformes aux exigences du laboratoire. Il faut le vérifier au moment de l’installation afin de garantir sa conformité aux spécifications du fabricant (vérification de la performance, tâche généralement effectuée par l’ingénieur chargé par le fournis-seur de l’installation, opération également appelée “essai de réception” ou “validation de l’équipement”). Si un équipement est déplacé par la suite, l’essai de réception doit être répété et sa performance certifiée si nécessaire (par exemple une balance déplacée devra être réétalonnée).
Le matériel doit en outre être contrôlé régulièrement pendant son usage, conformé-ment aux procédures documentées, pour montrer que sa performance demeure acceptable (par exemple, un instrument pourrait être vérifié avant l’analyse de chaque lot d’échantillons afin de s’assurer de son bon fonctionnement). Il peut s’agir, selon l’appareil visé, du contrôle des températures, des pressions de gaz, du réglage et de l’étalonnage, etc. Les échantillons d’essai peuvent également être analysés aux fins de vérification. Des actions correctives doivent être mises en œuvre en cas de besoin. Les instructions à jour concernant l’utilisation et l’entretien de l’équipement (par exemple les notices abrégées rédigées par le laboratoire à partir des manuels d’utilisation ainsi que les manuels d’utilisation fournis par le fabricant) doivent pouvoir être facilement consultées par le personnel autorisé et être conservées, de préférence, à proximité de l’appareillage.
Les équipements dont les propriétés affectent significativement les résultats doivent être étalonnés selon un calendrier et des procédures consignées par écrit mises à la disposition des utilisateurs autorisés. Tout équipement sous contrôle du laboratoire doit être étiqueté pour indiquer le statut de l’étalonnage (par exemple la date du dernier étalonnage et la date ou les critères d’échéance du prochain étalonnage) et éviter tout risque de confusion avec les équipements non étalonnés. Les instruments de première importance (tels que les balances, les thermomètres, les pipettes) doivent

26 Lignes directrices pour l’application d’un système de management de la qualité
être identifiés de façon unique et les registres d’étalonnage, y compris les certificats, s’ils existent, doivent être conservés.
Les membres du personnel en charge de l’équipement devraient veiller à ce que les échantillons de contrôle, les étalonnages et les blancs enregistrés soient régulièrement analysés et à ce que les spécifications de performance soient à jour. Tous les étalon-nages, entretiens et maintenances doivent être rapportés par écrit soit par le personnel du laboratoire, soit par un organisme extérieur. Les éléments devant être consignés sont notamment les suivants:
L’identification unique de l’élément d’équipement et du système de données; "
Le nom du fabricant, l’identification de type et le numéro de série; "
L’emplacement actuel de l’équipement, le cas échéant; "
Les contrôles de performance; "
Les manuels d’utilisation élaborés par le laboratoire; "
Les instructions du fabricant, si elles sont disponibles, ou les coordonnées "du lieu où elles se trouvent;
Les dates, les résultats et les copies de rapports et de certificats de "l’ensemble des étalonnages, critères d’acceptation et date prévue du prochain étalonnage;
Le plan de maintenance, s’il y a lieu, et l’entretien effectué à ce jour; "
Tous les dégâts, dysfonctionnements, modifications ou réparations de "l’équipement.
Les défauts identifiés doivent être portés à l’attention de la personne responsable de l’équipement et des actions correctives adoptées. Si la défaillance est importante (par exemple, si la source de lumière d’un spectromètre est instable ou si une pompe à vide ne retient pas suffisamment de pression à vide), l’appareil doit être retiré du service tant que le défaut n’a pas été supprimé. La manière dont celui-ci a été corrigé, de même que l’heure et la date, doivent être enregistrés dans la fiche de l’appareil. De même, un équipement qui a été l’objet d’une mauvaise manutention au point qu’il donne des résultats suspects ou qui s’est révélé défectueux doit être mis hors service. Il doit être isolé afin d’empêcher son utilisation ou être clairement étiqueté ou marqué comme étant hors service jusqu’à ce qu’il ait été réparé et qu’un étalonnage ou un essai ait montré qu’il fonctionne correctement. Le laboratoire doit examiner l’effet de la défaillance et instituer la procédure de “maîtrise des travaux non conformes” (voir le paragraphe 4.8.).
5.7. Normes de référence, matériaux et réactifsLes étalons de référence, les matériaux et les réactifs doivent correspondre à la méthode d’analyse utilisée et aux spécifications de qualité requises par la méthode.

Lignes directrices pour l’application d’un système de management de la qualité 27
Le nombre de lots/échantillons et de réactifs doit être consigné par écrit et ces derniers doivent être analysés pour en garantir la fiabilité.
Les étiquettes des normes et réactifs doivent porter les informations suivantes:
Leur nom; "
Leur concentration, le cas échéant; "
Leur date de préparation et/ou de péremption; "
L’identité du préparateur; "
Les conditions d’entreposage, le cas échéant; "
Un avis de danger, le cas échéant. "
Le laboratoire doit tenir un registre des préparations des solutions de réactifs qui doit être conservé dans un lieu approprié. Chaque fois qu’une telle solution est préparée, la personne responsable de la préparation doit inscrire dans ce registre la date, le poids et le volume des ingrédients effectivement employés et sa signature. Ces renseignements sont nécessaires pour retracer d’éventuelles sources d’erreurs dans une analyse. Attribuer à chaque lot de réactifs un identifiant unique peut être utile à cet égard.
Les normes de référence, les étalons de travail, les étalons de référence élaborés par le laboratoire, les réactifs et les autres matériaux doivent être correctement entreposés afin d’assurer leur stabilité et leur intégrité. La date de péremption indiquée par le fournisseur/fabricant indique la période pendant laquelle l’étalon/ le réactif/le matériau peut être utilisé, à moins qu’une vérification opérée après cette date ne prouve qu’ils sont toujours utilisables. Lorsqu’il y a lieu, la date d’ouverture et de péremption des récipients doit également être enregistrée.
Il importe de noter que les étalons et les réactifs peuvent poser problème après leur réception par le laboratoire. Dès qu’un récipient est ouvert, un risque existe que son contenu soit contaminé ou que sa composition soit modifiée. Le contenu d’un flacon qui n’est pas hermétiquement rescellé et fermé est exposé à l’air et sa teneur en humidité peut augmenter ou diminuer de même que son absorption de gaz carbo-nique ou d’autres vapeurs contaminantes. L’usage d’une spatule ou d’une pipette contaminée pour effectuer des prélèvements ou introduire des produits ou d’autres réactifs dans un flacon de solvant et pour reverser les quantités inutilisées dans les flacons de réactifs doit être fermement découragé. Les solvants utilisés pour le rinçage des instruments (par exemple les seringues d’un microlitre employées en chromatographie en phase gazeuse) doivent être changés fréquemment. Tout le per-sonnel participant à ce travail doit être suffisamment formé et bien connaître les exigences de la qualité pour être conscient de l’extrême soin qu’il faut apporter pour préserver l’intégrité de ces matériaux
La traçabilité des normes de référence doit être assurée, dans la mesure du possible, ce qui signifie que les normes du laboratoire doivent pouvoir être reliées directement

28 Lignes directrices pour l’application d’un système de management de la qualité
à des normes nationales ou internationales. Un matériau de référence certifié peut être utilisé le cas échéant [4]. Un matériau de référence certifié est un étalon de référence, généralement obtenu dans le commerce, dont la concentration de l’analyte a été certifiée par analyse, et qui est accompagné par un certificat ou un autre document délivré par un organisme de certification ou qui peut être traçable à un certificat ou autre document délivré par un tel organisme. Cependant, peu de matériaux de référence certifiés contenant des substances sous contrôle international sont disponibles, à l’exception notamment des étalons d’alcool.
En l’absence de matériau de référence certifié, il faut utiliser des étalons de référence commerciaux. Ces derniers sont généralement accompagnés d’une description de leur identité chimique, de leur pureté et de leur concentration (par exemple d’une “authentification et d’un certificat d’analyse”). Néanmoins, il est recommandé au laboratoire de vérifier de façon indépendante leur identité et leur pureté (ou concen-tration) avant de les utiliser (par exemple au moyen de comparaisons interlabora-toires ou intralaboratoires à partir d’un étalon de référence utilisé précédemment).
Les étalons de référence requis pour chaque procédure/méthode utilisée par le laboratoire doivent être documentés et consignés dans un dossier disponible dans le laboratoire. Ils doivent être appropriés pour l’essai effectué (par exemple, la pureté de la drogue doit être connue avec précision, et pour l’analyse des drogues dans les spécimens biologiques, les étalons devraient contenir l’analyte à des concentrations faibles). Un registre doit indiquer leur origine, la date de leur acquisition et la quantité détenue par le laboratoire, car ces informations peuvent être requises par les autorités nationales de contrôle des drogues. Lorsque des étalons de substances placées sous contrôle doivent être importés ou exportés, les autorisations d’impor-tation/exportation doivent être obtenues de l’autorité nationale compétente [16].
La méthode de préparation des étalons de travail à partir des étalons de référence originaux et la vérification du produit final doivent être consignées et figurer dans un dossier.
Les étalons de référence internes (par exemple, préparés à partir de matériaux saisis) devraient être vérifiés, dans la mesure où cela est techniquement et économiquement faisable, par comparaison avec des matériaux de référence certifiés, des étalons disponibles dans le commerce ou les échantillons de référence [17] fournis par l’UNODC.
L’obtention et la conservation des étalons de référence doivent être confiées à une personne spécialement désignée pour tenir un registre central de ces substances. Ce registre doit mentionner toutes les substances et préparations de référence officielles, de même que tous les étalons de référence non officiels de diverses sources extérieures, ainsi que tous les étalons de référence secondaires ou étalons de travail préparés au laboratoire.
Les calibrateurs, préparés à partir des étalons de référence ou obtenus dans le com-merce, servent à étalonner le dosage. Dans la mesure du possible, ceux qui doivent

Lignes directrices pour l’application d’un système de management de la qualité 29
servir à analyser des échantillons biologiques doivent être préparés dans une matrice analogue à celle des échantillons. Initialement, il conviendrait de travailler avec un nombre suffisant de calibrateurs pour déterminer les caractéristiques de la courbe d’étalonnage: un blanc et au moins cinq points d’étalonnage sont recommandés. La stabilité de la courbe d’étalonnage devrait être vérifiée dans les conditions de laboratoire par l’adjonction d’échantillons de contrôle à la fois positifs et négatifs.
Les échantillons de contrôle sont préparés à partir des étalons de référence et servent à déterminer la linéarité et la stabilité d’une détermination quantitative au cours du temps. Ils devraient être achetés ou pris dans un ensemble d’échantillons analysés précédemment et pesés ou mesurés séparément. Dans la mesure du possible, ils doivent avoir une matrice analogue à celle des échantillons et des solutions d’étalonnage.
5.8. Manipulation des objets d’essai
Échantillonnage, étiquetage et emballage
Le laboratoire doit avoir des procédures documentées relatives à l’échantillonnage des matériaux et des spécimens biologiques saisis, à la fois sur les lieux où ils sont prélevés et au laboratoire, qui doivent pouvoir être consultées sur place. La procédure d’échantillonnage doit garantir que l’élément prélevé pour analyse est représentatif de l’ensemble.
Le plan d’échantillonnage et/ou la procédure d’échantillonnage, l’identification de l’échantillonneur et les conditions ambiantes, s’il y a lieu, doivent être enregistrés.
Pour l’échantillonnage des matériaux saisis, les plans et les procédures d’échantillonnage publiés par des organisations internationalement reconnues, tels que l’UNODC [4], le Réseau européen des instituts de police scientifique (ENFSI) [18] ou le Groupe de travail scientifique sur l’analyse des drogues saisies (SWGDRUG) [5], sont recommandés. Pour l’échantillonnage des spécimens biologiques, il est recommandé d’utiliser les plans et procédures d’échantillonnage publiés par l’UNODC [4] ou les lignes directrices européennes pour les tests présomptifs de drogues sur le lieu de travail [8].
Les principes généraux sont les suivants:
Si la substance à analyser se trouve dans un petit nombre d’emballages "(généralement dix ou moins), un échantillon doit être prélevé dans chacun. Si le nombre d’emballages est plus important, soit ils doivent être homogénéisés, soit plusieurs échantillons doivent être prélevés dans la totalité des emballages;
S’il y a une quantité plus importante d’emballages, étant donné qu’il est "impossible de prélever un échantillon dans chacun, une stratégie devrait

30 Lignes directrices pour l’application d’un système de management de la qualité
être élaborée pour veiller à ce que les échantillons prélevés soient représenta-tifs de la totalité des emballages. Si les emballages diffèrent par leur contenu, ils doivent d’abord être séparés en groupes homogènes et un plan d’échantillonnage approprié doit ensuite être élaboré pour chaque groupe.
Le plan d’échantillonnage peut reposer sur une source non statistique (par exemple une directive de la direction du laboratoire ou une injonction judiciaire) ou une source statistique (par exemple, hypergéométrique ou bayésienne), mais devrait être pratique et facile à réaliser par des non-scientifiques; il ne devrait pas exiger de travail supplémentaire de la part du laboratoire et être facile à expliquer et à défendre devant un tribunal.
Le laboratoire doit fournir des lignes directrices pour garantir que les objets d’essai ont été correctement prélevés, étiquetés, emballés, conservés et stockés avant leur soumission au laboratoire. L’étiquetage doit être conçu aux fins d’identification unique des échantillons et des sous-échantillons et de leur lien avec leur origine première. Il est important qu’ils soient emballés de telle sorte que seules les personnes autorisées y aient accès afin d’éviter leur perte ou endommagement pendant le transit.
Réception, manipulation et élimination par le laboratoire
Le laboratoire doit disposer d’une procédure écrite réglementant les modalités d’acceptation et de rejet des matériaux saisis et/ou de spécimens biologiques. Des exigences spécifiques devraient être établies concernant les matériaux, les échantillons ou les instruments (par exemple les aiguilles) susceptibles de constituer une menace pour la santé ou la sécurité du personnel. Les documents accompagnant les objets d’essai doivent se référer à chaque objet et fournir suffisamment de données pour que le laboratoire identifie le type de travail demandé. Il doit exister un niveau acceptable de concordance entre tous les détails figurant sur les échantillons étiquetés et ceux figurant sur la notice d’accompagnement ou le formulaire de présentation des objets soumis.
Tous les matériaux et spécimens doivent être identifiés à l’aide d’un numéro unique et les données correspondantes enregistrées dans le registre du laboratoire. Les personnes présentant des objets d’essai au laboratoire doivent recevoir un reçu daté et signé mentionnant tous les matériaux ou spécimens soumis. Les rapports ultérieurs sur ces objets d’essai doivent être clairement identifiés au moyen du même numéro de dossier.
Au laboratoire, une personne autorisée doit recevoir et vérifier soigneusement les échantillons soumis et les documents qui les accompagnent. Une ou plusieurs personnes identifiées devraient également être autorisées à rejeter partiellement ou totalement les spécimens qui ne sont pas conformes à la procédure d’acceptation du laboratoire. Elles doivent informer le directeur du laboratoire de ces rejets. Les actions prises pour y remédier doivent toujours être consignées.

Lignes directrices pour l’application d’un système de management de la qualité 31
La procédure d’acceptation du laboratoire doit envisager un certain nombre de défauts, tels que ceux énumérés ci-après, qui doivent être considérés comme des motifs de rejet s’ils ne peuvent être résolus:
Erreur ou illisibilité du nom, du numéro d’identification ou de toute autre "information portée sur l’étiquette attachée à l’échantillon;
Présence de plus d’une étiquette sur l’échantillon; "
Absence d’étiquette sur l’échantillon ou le spécimen; "
Répétition du numéro de l’échantillon ou du spécimen; "
Incohérence entre les échantillons reçus et les échantillons listés sur les "documents qui les accompagnent;
Étanchéisation insuffisante susceptible d’altérer l’intégrité de l’échantillon; "
Preuve de falsification; "
Absence d’échantillon ou fuite excessive du flacon. "
Le laboratoire doit disposer d’une procédure établie pour sécuriser l’entreposage des échantillons, tant avant qu’après leur analyse, pour corréler les échantillons à d’autres données qui les accompagnent (par exemple la demande d’analyse), pour identifier les sous-échantillons préparés et pour montrer la progression de l’analyse, la date de publication du rapport d’analyse et la date et les moyens utilisés par la suite en vue de l’élimination de tous les spécimens restants après analyse. Le système doit être conçu et géré de façon à garantir l’impossibilité de confondre physiquement les objets ou la référence qui y est faite dans les enregistrements ou les autres documents du laboratoire.
Le laboratoire doit également disposer de procédures établies et d’installations appropriées pour éviter la détérioration, la perte ou l’endommagement des échan-tillons lors du stockage, de la manutention et de l’analyse.
Lorsque des échantillons biologiques sont présentés en double exemplaire, l’un doit être utilisé pour l’analyse et l’autre stocké et congelé à des fins d’analyse plus poussée si nécessaire.
Lorsque des objets d’essai sont conservés par le laboratoire après analyse, ils doivent être stockés pendant un délai spécifié par la juridiction ou le client.
Le laboratoire doit disposer de procédures documentées en vue de l’élimination des substances et échantillons placés sous contrôle international qui doivent être confor-mes aux lois et procédures applicables dans la juridiction concernée. Il est nécessaire de conserver une trace écrite de toute substance éliminée.
Si des objets d’essai sont recueillis par le laboratoire pour être acheminés vers une destination ultérieure (par exemple vers un tribunal en tant que pièces à conviction),

32 Lignes directrices pour l’application d’un système de management de la qualité
il convient d’inscrire le moment où le spécimen a été remis et le nom de la personne qui l’a reçu dans un registre, qui doit être signé par cette personne.
Les échantillons tels que le sang et l’urine doivent être considérés comme présentant un risque biologique et être éliminés conformément aux lois et procédures applicables.
5.9. Rapport sur les résultatsLes résultats des essais effectués par le laboratoire doivent être rapportés de manière exacte, claire, non ambiguë, objective et conformément aux exigences du client. Le rapport d’essai doit être conçu de manière à tenir compte de chaque type d’analyse effectué et éviter le risque d’incompréhension ou d’utilisation malencontreuse.
Si les résultats doivent être présentés en justice, chaque rapport d’essai doit comporter au moins les indications suivantes:
Le titre du rapport; "
L’indication unique de l’étude et de chaque objet ou échantillon; "
Le nom de l’organisme ou de l’individu ayant présenté l’objet ou "l’échantillon;
La date de réception de l’échantillon par le laboratoire; "
La date d’élaboration du rapport d’essai; "
L’indication de chaque page du rapport d’essai; "
La description et l’identification non ambiguë de l’objet soumis à l’essai; "
Les résultats de l’essai; "
Le (les) nom(s), fonction(s) et signature(s), ou une identification équiva- "lente, de la (des) personne(s) autorisant le rapport d’essai.
S’il y a lieu, une déclaration selon laquelle les résultats ne se rapportent qu’aux objets soumis à l’essai ou à l’étalonnage peut également être apportée.
Terminologie
Lorsqu’une substance particulière a été identifiée conformément aux protocoles du laboratoire, cela doit être mentionné au moyen de l’expression “l’échantillon/le spéci-men contient” ou des termes “l’essai a révélé la présence de la substance en question”. Les termes “négatif” ou “non détecté” peuvent être utilisés pour indiquer l’absence d’un ou de plusieurs analytes. Il est préférable d’utiliser l’expression “non détecté” car elle

Lignes directrices pour l’application d’un système de management de la qualité 33
indique que les substances considérées étaient absentes dans les limites du ou des essais effectués, c’est-à-dire qu’elles étaient inférieures à la limite de détection.
Si la substance détectée est supérieure à la limite de détection mais est inférieure à la limite de quantification du dosage (généralement considérée comme le point le plus bas de la courbe d’étalonnage), elle peut être indiquée dans le rapport comme ayant été détectée sous “quantités traces”.
Dans les cas de dosages/essais présomptifs, tout résultat inférieur à la valeur limite désignée doit être communiqué comme “non détecté”. Les résultats supérieurs à la valeur limite doivent être déclarés en tant que teneurs détectées au moyen d’une méthode appropriée et ensuite déclarés “positifs”.
Toutes les unités employées dans le rapport doivent être conformes aux exigences du client.
Avis et interprétations
Lorsque des avis et interprétations sont donnés, le laboratoire doit formuler par écrit les bases sur lesquelles reposent les avis et interprétations émis. Les avis et interpréta-tions doivent être clairement signalés comme tels dans un rapport d’essai.
Résultats obtenus auprès de sous-traitants
Lorsque le rapport d’essai contient des résultats d’essais effectués par des sous-traitants, ces résultats doivent être clairement indiqués.
Amendements aux rapports d’essai
Les amendements de fond à un rapport d’essai après son émission doivent exclusi-vement faire l’objet d’un nouveau document portant la mention “Supplément au rapport d’essai” et citer le numéro de référence de l’original. Lorsqu’il est nécessaire d’émettre un nouveau rapport d’essai, celui-ci doit comporter une identification unique et faire mention de l’original qu’il remplace.
5.10. Contrôle de la qualité, tests d’aptitude et comparaisons interlaboratoires
Chaque essai doit être effectué à un niveau approprié de contrôle de la qualité. Les échantillons de contrôle de la qualité doivent être analysés selon les procédures

34 Lignes directrices pour l’application d’un système de management de la qualité
définies, à la fréquence voulue. Lorsque des cartes de contrôle sont utilisées, la performance dépassant les critères acceptables doit être enregistrée.
Les résultats du nouvel essai des échantillons doivent révéler une mesure acceptable de concordance avec les analyses originales.
Le laboratoire devrait participer à des tests d’aptitude et à des comparaisons interlaboratoires, par exemple le programme pour les exercices collaboratifs inter-nationaux (ICE) de l’UNODC, qui est décrit dans une publication distincte [10].
Lorsque des anomalies ou des possibilités d’amélioration sont identifiées, une procédure doit être mise en place en vue de la mise en œuvre de mesures appro-priées. Les améliorations et les actions correctives adoptées doivent être enregistrées. Il doit aussi exister un système efficace permettant de relier la performance des tests d’aptitude au contrôle régulier de la qualité.

Lignes directrices pour l’application d’un système de management de la qualité 35
Bibliographie
1. Commission des stupéfiants des Nations Unies, Résolution 50/4, Amélioration de la qualité et de la performance des laboratoires d’analyse de drogues, 2007.
2. Organisation internationale de normalisation/Commission électrotechnique inter nationale, ISO/CEI 17025:2005 Prescriptions générales concernant la compétence des laboratoires d’étalonnages et d’essai.
3. Conférence internationale sur l’agrément des laboratoires d’essai (ILAC), ILAC-G19:2002 Guidelines for Forensic Science Laboratories.
4. Office des Nations Unies contre la drogue et le crime, Principes recommandés d’assurance de qualité et de bonnes pratiques de laboratoire, STR/NAR/25, 1995.
5. Scientific Working Group for the Analysis of Seized Drugs, Scientific Working Group for the Analysis of Seized Drugs (SWGDRUG) Recommendations, 2008.
6. American Society of Crime Laboratory Directors/Laboratory Accreditation Board (ASCLD/LAB), Legacy Accreditation Manual, 2008.
7. Society of Forensic Toxicologists/American Academy of Forensic Sciences (SOFT/AAS), Forensic Toxicology Laboratory Guidelines, 2006.
8. European Workplace Drug Testing Society (EWDTS), European Laboratory Guidelines for Legally Defensible Workplace Drug Testing, 2002.
9. London Toxicology Group Workplace Drug Testing Forum, UK Laboratory Guidelines for Legally Defensible Workplace Drug Testing, 2001.
10. Office des Nations Unies contre la drogue et le crime, Brochure sur le Programme international d’assurance de la qualité, 1998, et Protocole pour les exercices collabo-ratifs internationaux dans le cadre du programme international d’assurance de la qualité, 1998.
11. Organisation internationale de normalisation/Commission électrotechnique inter nationale, ISO/CEI 17025:2005 Prescriptions générales concernant la compétence des laboratoires d’étalonnages et d’essai, paragraphe 4.2.2.
12. Conférence internationale sur l’agrément des laboratoires d’essai (ILAC), ILAC-G19:2002 Guidelines for Forensic Science Laboratories, paragraphe 4.12.2.1.a.
13. Office des Nations Unies contre la drogue et le crime, Compétences requises et équipement de base pour un laboratoire de stupéfiants, 2009.
14. Office des Nations Unies contre la drogue et le crime, Validation of analytical methodology and calibration of equipment used for testing of illicit drugs in seized materials and biological specimen, 2009.
15. Eurachem, Quantifying Uncertainty in Analytical Measurement, deuxième édition, 2000.

36 Lignes directrices pour l’application d’un système de management de la qualité
16. Organe international de contrôle des stupéfiants (OICS), Lignes directrices pour l’importation et l’exportation d’étalons de référence de drogues et de précurseurs, 2007.
17. Office des Nations Unies contre la drogue et le crime, Directives pour les demandes d’étalons/échantillons de drogues placées sous contrôle international.
18. Réseau européen des instituts de police scientifique (ENFSI), Guidelines on Represen-tative Drug Sampling, 2007.
Des informations supplémentaires sur les documents cités en référence peuvent être obtenues sur les sites Web suivants (au 2 décembre 2008):
ASCLD/LAB www.ascld-lab.org/
ENFSI www.enfsi.eu/
EURACHEM www.eurachem.org/
EWDTS www.ewdts.org/
ILAC www.ilac.org/
ISO www.iso.org/iso/home.htm
LTG http://ltg.uk.net/
OICS www.incb.org/incb/index.html
SOFT/AAS www.soft-tox.org/
SWGDRUG www.swgdrug.org/
UNODC www.unodc.org/

37
Annexe. Modèle de manuel qualité
IntroductionLe manuel qualité est le document de plus haut niveau et le plus générique, qui énonce les politiques, procédures et pratiques générales du laboratoire en matière de qualité. Il doit indiquer la structure de la documentation utilisée dans le SMQ. Il devrait également comprendre ou mentionner les procédures d’appui, y compris les procédures techniques. Dans une large mesure, les dispositions du manuel qualité sont régies par les exigences de la norme de qualité à laquelle adhère le laboratoire, le plus souvent la norme ISO/CEI 17025:2005(F), sur laquelle reposent les présentes lignes directrices.
Objectif du modèle Le modèle présenté ici a pour vocation de servir de cadre pour la préparation d’un manuel qualité. Lorsque c’est possible, des indications sont données sur le contenu des sections du manuel qualité et quelques suggestions sont faites. Il n’est ni possible ni souhaitable de proposer un manuel complet, car le processus même de création d’un manuel est un exercice pédagogique formateur pour tout le personnel du laboratoire. Tous les laboratoires n’ont ni les mêmes besoins ni les mêmes moyens, et il faut en tenir compte dans les détails de leur propre SMQ individualisé et le manuel qualité, qui en décrit le fonctionnement.
Comment utiliser le modèleLes sections du présent modèle devraient être complétées compte tenu des conseils donnés dans les lignes directrices. Il n’y a pas de manière “correcte” de le faire. Les détails donnés dans ce document sont suffisamment souples pour permettre aux laboratoires d’imposer leur caractère propre au document final, pour autant que chacune des exigences énoncées dans la norme ISO 17025 soit satisfaite.
Les titres des différentes sections du manuel qualité constituent donc un canevas. Des précisions sur le SMQ peuvent être insérées dans les espaces appropriés. Il se peut que certaines sections ne soient pas pertinentes et il suffit alors de l’indiquer. Inversement, un laboratoire peut avoir besoin d’introduire des sections supplémentaires qui ne figurent pas dans l’ébauche générale proposée.
Les notes explicatives sur les informations devant être ajoutées au manuel sont indiquées en italique dans le modèle.

38 Lignes directrices pour l’application d’un système de management de la qualité
MANUEL QUALITÉ POUR LES LABORATOIRES D’ANALYSE DES DROGUES
Modèle
Nom de l’Organisation: _______________________________________________________
Nom, acronyme du Département/de la Section: ____________________________________
Adresse: ____________________________________________________________________
____________________________________________________________________________
Version: ___________________________________________________________________ Numéro de la version actuelle
Valable depuis: _____________________________________________________________ Date d’émission de la version actuelle
Remplace la version: ________________________________________________________ Numéro de la version précédente
Exemplaires: _______________________________________________________________ Nombre d’exemplaires de la version actuelle
Distribution/Emplacement: ____________________________________________________ Copies papier/par exemple, laboratoire
Version électronique: ________________________________________________________ Référence du fichier contenant le manuel du laboratoire
Approuvé par: ___________________________ Signature:____________________________ Nom, par exemple, Directeur du laboratoire
Personnes responsables: ___________________ Signature: _________________________ Nom, par exemple, Directeur du laboratoire

Lignes directrices pour l’application d’un système de management de la qualité 39
Nom du laboratoire:Révision: Date de révision: Page: 3/9
Manuel qualité
Table des matières
Mention des améliorations apportées au système de management de la qualité
Déclaration politique de la haute direction du laboratoire énonçant sa volonté de respecter la politique de qualité
1. INTRODUCTION: OBJECTIF DU MANUEL QUALITÉ
2. INFORMATIONS SUR L’ORGANISATION
3. SYSTÈME D’ADMINISTRATION DE LA QUALITÉ
4. EXIGENCES EN MATIÈRE D’ADMINISTRATION
4.1. Organisation du laboratoire
4.2. Maîtrise de la documentation
4.3. Revue des demandes des clients
4.4. Sous-traitance des essais
4.5. Achat de services et de fournitures
4.6. Services aux clients
4.7. Réclamations
4.8. Actions correctives et actions préventives
4.9. Maîtrise des enregistrements/chaîne de traçabilité
4.10. Audits internes
4.11. Revues de direction
5. EXIGENCES TECHNIQUES
5.1. Introduction
5.2. Personnel
5.3. Installations et conditions ambiantes
5.4. Santé et sécurité
5.5. Méthodes d’essai, validation des méthodes et procédures
5.6. Équipement
5.7. Normes de référence, matériaux et réactifs
5.8. Manipulation des objets d’essai
5.9. Rapport sur les résultats
5.10. Contrôle de la qualité, tests d’aptitude et comparaisons interlaboratoires
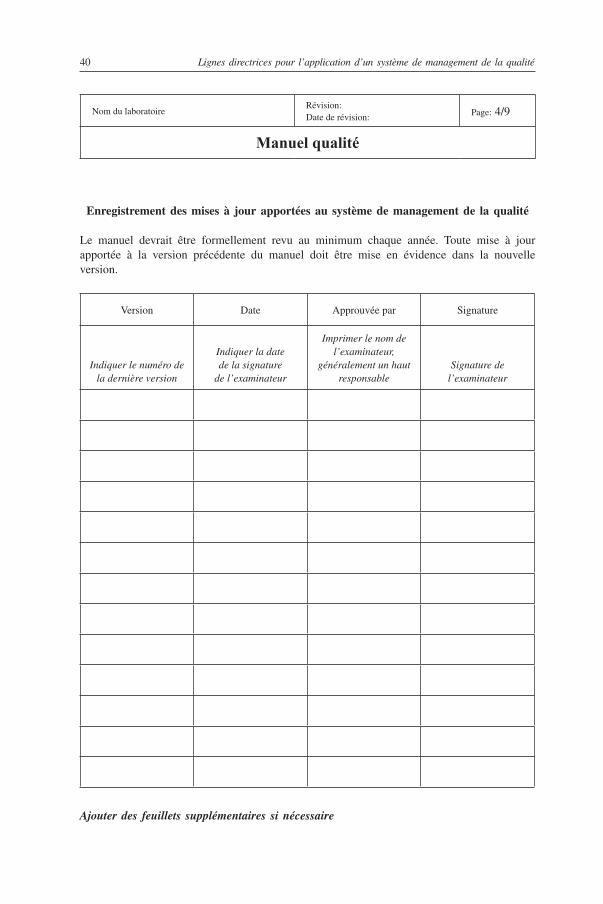
40 Lignes directrices pour l’application d’un système de management de la qualité
Nom du laboratoireRévision: Date de révision: Page: 4/9
Manuel qualité
Enregistrement des mises à jour apportées au système de management de la qualité
Le manuel devrait être formellement revu au minimum chaque année. Toute mise à jour apportée à la version précédente du manuel doit être mise en évidence dans la nouvelle version.
Version Date Approuvée par Signature
Indiquer le numéro de la dernière version
Indiquer la date de la signature
de l’examinateur
Imprimer le nom de l’examinateur,
généralement un haut responsable
Signature de l’examinateur
Ajouter des feuillets supplémentaires si nécessaire

Lignes directrices pour l’application d’un système de management de la qualité 41
Nom du laboratoireRévision: Date de révision: Page: 5/9
Manuel qualité
Déclaration d’engagement
J’ai lu le manuel qualité et m’engage à suivre les exigences du système de management de la qualité.
Date Nom Signature
Indiquer la date de la signature de chaque membre du personnel
Imprimer le nom de chaque membre du personnel
Signature de chaque membre du personnel
Ajouter des feuillets supplémentaires si nécessaire

42 Lignes directrices pour l’application d’un système de management de la qualité
Nom du laboratoireRévision: Date de révision: Page: 6/9
Manuel qualité
1. INTRODUCTION: OBJECTIF DU MANUEL QUALITÉ
Cette section devrait expliquer brièvement (en un ou plusieurs paragraphes) ce qu’est le manuel qualité et pourquoi il est nécessaire.
2. INFORMATIONS SUR l’ORGANISATION
Décrivez la structure et la politique du laboratoire ou du département etc., en indiquant sa situation générale et sa structure, son statut juridique, ses rôles, ses attributions, ses valeurs, sa vision, ses objectifs, sa stratégie, ses finances et son budget, etc., le cas échéant.
Donnez aussi ses coordonnées:
Adresse:Téléphone:Télécopie:Courrier électronique:Site Web:
3. SYSTÈME DE MANAGEMENT DE LA QUALITÉ
3.1. Déclaration sur la politique de qualité
Insérez une déclaration concise du laboratoire (en général ne dépassant pas une page), datée et signée. Voir le paragraphe 3.1 des Lignes directrices.
3.2 Manuel qualité
Insérez ici une déclaration sur le manuel qualité si cela n’a pas été déjà fait.
4. EXIGENCES EN MATIÈRE D’ADMINISTRATION
4.1. Organisation du laboratoire
Cette section sur l’organisation et l’administration devrait définir le cadre général et le mandat de l’organisation. Des diagrammes sont utiles pour montrer les aspects organisation-nels et les rôles des principaux responsables, y compris le mode d’interaction entre le Direc-teur qualité et les autres membres du personnel. Il est recommandé que les responsabilités susmentionnées soient exercées par différents membres du personnel. Néanmoins, selon les ressources disponibles, certaines tâches pourront être déléguées à la même personne.
Insérez un organigramme montrant la structure de gestion du laboratoire, par exemple:

Lignes directrices pour l’application d’un système de management de la qualité 43
Pour les sections 4.2 à 4.11 suivantes, ébauchez les politiques et pratiques suivies par votre laboratoire en faisant référence aux documents SMQ pertinents.
4.2. Maîtrise de la documentation
Indiquez comment est organisée votre maîtrise de la documentation (par exemple, liste récapitula-tive/registre de la documentation en pièce jointe). Voir le paragraphe 4.2 des Lignes directrices.
4.3. Revue des demandes des clients
Voir le paragraphe 4.3 des Lignes directrices.
4.4. Sous-traitance des essais
Voir le paragraphe 4.4 des Lignes directrices.
4.5. Achat de services et de fournitures
Voir le paragraphe 4.5 des Lignes directrices.
4.6. Services aux clients
Voir le paragraphe 4.6 des Lignes directrices.
4.7. Réclamations
Décrivez la politique appliquée par votre laboratoire pour répondre aux demandes et aux réclamations des clients, régler les problèmes rencontrés, appliquer des actions préventives et correctives et évaluer leur efficacité. Voir le paragraphe 4.7 des Lignes directrices.
4.8. Actions correctives et préventives
Voir le paragraphe 4.8 des Lignes directrices.
Nom du laboratoireRévision: Date de révision: Page: 7/9
Manuel qualité
Directeur du laboratoire
Chef analyste Chef technicien Directeur qualité

44 Lignes directrices pour l’application d’un système de management de la qualité
Nom du laboratoireRévision: Date de révision: Page: 8/9
Manuel qualité
4.9. Maîtrise des enregistrements/traçabilité
Décrivez la procédure de maîtrise des enregistrements et la chaîne de traçabilité en vigueur au laboratoire. Voir le paragraphe 4.9 des Lignes directrices.
4.10. Audit interne
Indiquez votre système de contrôles périodiques (par exemple, annuels) ou d’audits pour montrer que toutes les procédures techniques et administratives en place pour garantir la qualité sont effectivement mises en œuvre, respectées et enregistrées. Voir le paragraphe 4.10 des Lignes directrices.
4.11. Revues de direction
Les éléments à prendre en compte par la revue de direction doivent être spécifiés. Voir le paragraphe 4.11 des Lignes directrices.
5. EXIGENCES TECHNIQUES
Décrivez, pour les chapitres 5.2 à 5.10, les politiques et pratiques suivies par votre laboratoire en vous reportant aux documents du système de management de la qualité.
5.1. Introduction
Voir le chapitre 5.1 des Lignes directrices.
5.2. Personnel
Indiquez quels dossiers personnels sont tenus et comment ils sont mis à jour. Indiquez le titre de chaque emploi (nom et attributions du titulaire actuel, responsabilités, qualifications, etc., de préférence en annexe); niveau d’autorisation et de compétence pour approuver les documents, autorisations des tâches, formations suivies. Décrivez les exigences générales en matière de formation du personnel, de formation individuelle et de développement personnel, indiquez les réunions prévues pour discuter du travail du laboratoire et d’autres mécanismes pour la communication interne. Voir le paragraphe 5.2 des Lignes directrices.
5.3. Installations et conditions ambiantes
Décrivez ici, par exemple, le contrôle de la sécurité, de la température et de l’humidité. Voir le chapitre 5.3 des Lignes directrices.
5.4. Santé et sécurité
Décrivez les procédures préconisées pour assurer la santé et la sécurité, les mesures prises pour assurer aux employés un environnement de travail sûr, etc. Voir le paragraphe 5.4 des Lignes directrices.

Lignes directrices pour l’application d’un système de management de la qualité 45
Nom du laboratoireRévision: Date de révision: Page: 9/9
Manuel qualité
5.5. Méthodes d’essai, validation des méthodes et procédures
Voir le paragraphe 5.5 des Lignes directrices. Voir également le manuel de l’UNODC intitulé “Validation of analytical methodology and calibration of equipment used for testing of illicit drugs in seized materials and biological specimen” [Validation de la méthodologie analytique et de l’étalonnage de l’équipement utilisé pour les analyses de drogues illicites dans les matériaux saisis et les spécimens biologiques], 2009.
5.6. Équipement
Décrivez les pratiques de votre laboratoire en matière d’identification et de tenue de registres du matériel, etc. Voir le paragraphe 5.6 des Lignes directrices.
5.7. Normes de référence, matériaux et réactifs
Esquissez la politique et la pratique concernant la préparation, l’étiquetage, l’entreposage et l’utilisation des étalons de référence, des étalons de travail, des réactifs, etc. Voir le para-graphe 5.7 des Lignes directrices.
5.8. Manipulation des objets d’essai
Indiquez où se trouvent les informations relatives aux procédures en place, comme celles qui concernent la réception des échantillons, l’identification des échantillons par rapport aux demandes d’analyse, et qui montrent le progrès de l’analyse, le stockage des échantillons, la publication de rapports, l’élimination des échantillons, etc. Voir le paragraphe 5.8 des Lignes directrices.
5.9. Notification des résultats
Décrivez le format et le contenu des rapports, la procédure d’autorisation de leur publication et leur communication par le laboratoire, etc. Voir le paragraphe 5.9 des Lignes directrices.
5.10. Contrôle de la qualité, tests d’aptitude et comparaisons interlaboratoires
Décrivez comment le contrôle de la qualité est effectué et évalué à l’aide des matériaux de référence, des cartes de contrôle, des échantillons de contrôle de la qualité, des tests d’aptitude, des comparaisons interlaboratoires, etc. Voir le paragraphe 5.10 des Lignes directrices.




Crédits photo: Bibliothèque de l’UNODC; UNODC/Ioulia Kondratovitch; Alessandro Scotti.

Lignes directrices pour l’application d’un système
de management de la qualité dans les laboratoires d’analyse des drogues
Centre international de Vienne, Boîte postale 500, 1400 Vienne (Autriche)Téléphone: (+43-1) 26060-0, Télécopie: (+43-1) 26060-5866, www.unodc.org
Notre engagement: la qualité et l’amélioration continue*0981417*
Publication des Nations UniesImprimé en Autriche
Numéro de vente: F.09.XI.10
ST/NAR/37
12 USDISBN 978-92-1-248169-2
V.09-81417 — Décembre 2009 — 350