Embed Size (px)
Citation preview

A
Me
Ea
b
c
i
HAD
MMMI
1
nmlelLfnHg
n
l2
1h
Revue du rhumatisme 80 (2013) 157–160
Disponible en ligne sur
www.sciencedirect.com
rticle original
aladie de Kimura et maladie de Behc et au sein de la même famille :xiste-t-il une association génétique?�
ldad Ben-Chetrit a,∗, Isabelle Touitoub, Yakov Felligc, Mouna Barat-Houarib
Unité de rhumatologie, département de médecine interne, université médicale Hadassah-Hebrew, 12000 Jérusalem, IsraëlUnité médicale des maladies auto-inflammatoires, laboratoire de génétique, hôpital Arnaud-de-Villeneuve, 34295 Montpellier, FranceService d’anatomopathologie, université médicale Hadassah-Hebrew, Jérusalem, Israël
n f o a r t i c l e
istorique de l’article :ccepté le 10 avril 2012isponible sur Internet le 24 juillet 2012
ots clés :aladie de Behc etaladie de Kimura
L-10
r é s u m é
Contexte. – La maladie de Behc et (MB) et la maladie de Kimura (MK) sont deux maladies inflammatoiresfréquentes au Japon mais rares en Israël. Récemment, nous avons observé une famille au sein de laquelletrois membres de la fratrie étaient atteints d’une MB et un de leur frère avait une MK. Cette associationfamiliale soulève la question d’un facteur génétique prédisposant commun à ces deux maladies.Objectif. – L’objectif de cette étude a été de décrire cette famille et de rechercher un allèle prédisposantcommun à la MB et à la MK, au sein du gène de l’IL-10 dans la mesure où plusieurs polymorphismesmononucléotidiques (SNP) de ce gène sont connus pour être associés à la MB.Méthodes. – Trois membres de la fratrie atteints de MB et un de leur frère atteint de MK ont fait l’objetd’un examen clinique. L’ADN génomique a été préparé à partir d’échantillons sanguins prélevés chez les
neuf membres de la famille. L’ADN a été génotypé pour les variations de séquences de six SNP sur le gènede l’IL-10.Conclusions. – Les SNP du gène de l’IL-10, connus comme facteurs de susceptibilité pour la MB, n’étaientpas associés à la MB dans notre famille. La question d’une susceptibilité génétique commune à la MK età la MB nécessite de nouvelles études.© 2012 Société Française de Rhumatologie. Publié par Elsevier Masson SAS. Tous droits réservés.
. Introduction
La maladie de Behc et (MB) est une maladie inflammatoire chro-ique et récidivante, caractérisée par une aphtose bipolaire, desanifestations cutanées et une uvéite [1,2]. Elle peut aussi toucher
es vaisseaux sanguins et être à l’origine de thromboses vasculairest d’une vascularite. D’autres organes peuvent être atteints, commee système nerveux central, les articulations et le tube digestif.’étiologie exacte de la MB reste inconnue. Toutefois, l’agrégationamiliale, la prédominance de la maladie chez les patients origi-aires du bassin méditerranéen et d’Asie, l’association avec le gèneLA-B51, sont autant d’éléments soulignant l’importance du facteur
énétique [3].La maladie de Kimura (MK) est une maladie inflammatoire chro-ique d’origine inconnue qui se présente habituellement sous la
DOI de l’article original : http://dx.doi.org/10.1016/j.jbspin.2012.04.001.� Ne pas utiliser, pour citation, la référence franc aise de cet article, maisa référence anglaise de Joint Bone Spine (http://dx.doi.org/10.1016/j.jbspin.012.04.001).∗ Auteur correspondant.
Adresse e-mail : [email protected] (E. Ben-Chetrit).
169-8330/$ – see front matter © 2012 Société Française de Rhumatologie. Publié par Elsttp://dx.doi.org/10.1016/j.rhum.2012.06.006
forme d’adénopathies cervicales unilatérales et indolores ou demasses sous-cutanées dans la région cervico-céphalique [4–7]. Laplupart des cas de cette maladie rare ont été rapportés en Asiedu Sud-Est. Si la physiopathologie de la MK reste inconnue, diversmécanismes ont été impliqués, de type allergique, traumatique ouauto-immun. À ce jour, aucun contexte génétique n’a été impliquédans la MK.
Les formes familiales de MB ont été rapportées à plusieursreprises [7,8] de même que des cas comportant plusieurs maladesau sein de la même famille [9,10]. Mais il n’a pas été rapporté jusqu’àprésent d’association familiale entre MB et MK.
Dans ce travail, nous décrivons une famille au sein de laquelletrois membres de la fratrie étaient atteints d’une MB et un autrefrère était atteint d’une MK. Ce dernier était l’un des deux jumeauxdizygotes dont la sœur jumelle avait une MB. Dans la mesure où lesdeux maladies ne sont pas exceptionnelles en Asie du Sud-Est etau Japon, et qu’elles sont survenues ici dans une même famille, etdans la mesure où des polymorphismes mononucléotidiques (SNP)ont été décrits en association avec la MB [11,12], nous nous sommes
demandés si ces maladies pouvaient avoir des allèles prédisposantsen commun au sein de ce locus. Nous rapportons donc brièvementles caractéristiques cliniques de ces quatre patients et les résultatsde notre étude génétique.evier Masson SAS. Tous droits réservés.
158 E. Ben-Chetrit et al. / Revue du rhumatisme 80 (2013) 157–160
Fig. 1. Ségrégation familiale des haplotypes polymorphismes mononucléotidiques(SNP) de l’IL-10, construite avec le logiciel GNAlogique (disponible sur le sitehm6
2
2
aeedssfiae
2
1rsuabftgsvldpCMta(vun
2
a
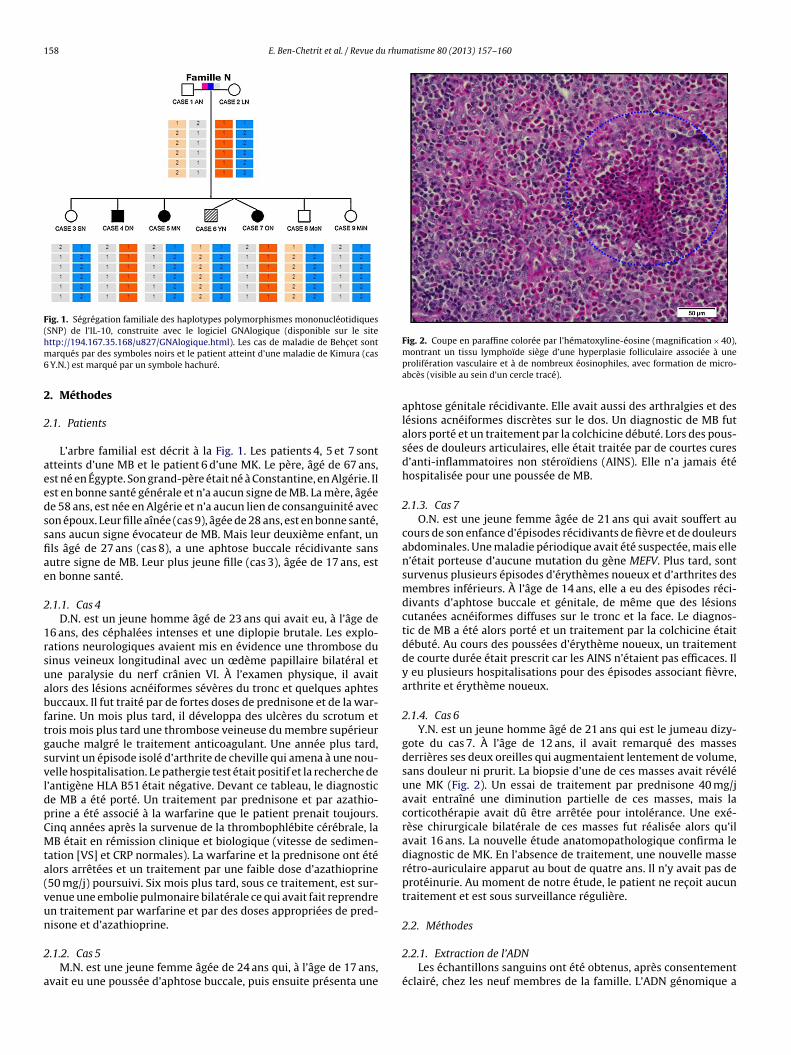
Fig. 2. Coupe en paraffine colorée par l’hématoxyline-éosine (magnification × 40),
ttp://194.167.35.168/u827/GNAlogique.html). Les cas de maladie de Behc et sontarqués par des symboles noirs et le patient atteint d’une maladie de Kimura (casY.N.) est marqué par un symbole hachuré.
. Méthodes
.1. Patients
L’arbre familial est décrit à la Fig. 1. Les patients 4, 5 et 7 sonttteints d’une MB et le patient 6 d’une MK. Le père, âgé de 67 ans,st né en Égypte. Son grand-père était né à Constantine, en Algérie. Ilst en bonne santé générale et n’a aucun signe de MB. La mère, âgéee 58 ans, est née en Algérie et n’a aucun lien de consanguinité avecon époux. Leur fille aînée (cas 9), âgée de 28 ans, est en bonne santé,ans aucun signe évocateur de MB. Mais leur deuxième enfant, unls âgé de 27 ans (cas 8), a une aphtose buccale récidivante sansutre signe de MB. Leur plus jeune fille (cas 3), âgée de 17 ans, estn bonne santé.
.1.1. Cas 4D.N. est un jeune homme âgé de 23 ans qui avait eu, à l’âge de
6 ans, des céphalées intenses et une diplopie brutale. Les explo-ations neurologiques avaient mis en évidence une thrombose duinus veineux longitudinal avec un œdème papillaire bilatéral etne paralysie du nerf crânien VI. À l’examen physique, il avaitlors des lésions acnéiformes sévères du tronc et quelques aphtesuccaux. Il fut traité par de fortes doses de prednisone et de la war-arine. Un mois plus tard, il développa des ulcères du scrotum etrois mois plus tard une thrombose veineuse du membre supérieurauche malgré le traitement anticoagulant. Une année plus tard,urvint un épisode isolé d’arthrite de cheville qui amena à une nou-elle hospitalisation. Le pathergie test était positif et la recherche de’antigène HLA B51 était négative. Devant ce tableau, le diagnostice MB a été porté. Un traitement par prednisone et par azathio-rine a été associé à la warfarine que le patient prenait toujours.inq années après la survenue de la thrombophlébite cérébrale, laB était en rémission clinique et biologique (vitesse de sedimen-
ation [VS] et CRP normales). La warfarine et la prednisone ont étélors arrêtées et un traitement par une faible dose d’azathioprine50 mg/j) poursuivi. Six mois plus tard, sous ce traitement, est sur-enue une embolie pulmonaire bilatérale ce qui avait fait reprendren traitement par warfarine et par des doses appropriées de pred-isone et d’azathioprine.
.1.2. Cas 5M.N. est une jeune femme âgée de 24 ans qui, à l’âge de 17 ans,
vait eu une poussée d’aphtose buccale, puis ensuite présenta une
montrant un tissu lymphoïde siège d’une hyperplasie folliculaire associée à uneprolifération vasculaire et à de nombreux éosinophiles, avec formation de micro-abcès (visible au sein d’un cercle tracé).
aphtose génitale récidivante. Elle avait aussi des arthralgies et deslésions acnéiformes discrètes sur le dos. Un diagnostic de MB futalors porté et un traitement par la colchicine débuté. Lors des pous-sées de douleurs articulaires, elle était traitée par de courtes curesd’anti-inflammatoires non stéroïdiens (AINS). Elle n’a jamais étéhospitalisée pour une poussée de MB.
2.1.3. Cas 7O.N. est une jeune femme âgée de 21 ans qui avait souffert au
cours de son enfance d’épisodes récidivants de fièvre et de douleursabdominales. Une maladie périodique avait été suspectée, mais ellen’était porteuse d’aucune mutation du gène MEFV. Plus tard, sontsurvenus plusieurs épisodes d’érythèmes noueux et d’arthrites desmembres inférieurs. À l’âge de 14 ans, elle a eu des épisodes réci-divants d’aphtose buccale et génitale, de même que des lésionscutanées acnéiformes diffuses sur le tronc et la face. Le diagnos-tic de MB a été alors porté et un traitement par la colchicine étaitdébuté. Au cours des poussées d’érythème noueux, un traitementde courte durée était prescrit car les AINS n’étaient pas efficaces. Ily eu plusieurs hospitalisations pour des épisodes associant fièvre,arthrite et érythème noueux.
2.1.4. Cas 6Y.N. est un jeune homme âgé de 21 ans qui est le jumeau dizy-
gote du cas 7. À l’âge de 12 ans, il avait remarqué des massesderrières ses deux oreilles qui augmentaient lentement de volume,sans douleur ni prurit. La biopsie d’une de ces masses avait révéléune MK (Fig. 2). Un essai de traitement par prednisone 40 mg/javait entraîné une diminution partielle de ces masses, mais lacorticothérapie avait dû être arrêtée pour intolérance. Une exé-rèse chirurgicale bilatérale de ces masses fut réalisée alors qu’ilavait 16 ans. La nouvelle étude anatomopathologique confirma lediagnostic de MK. En l’absence de traitement, une nouvelle masserétro-auriculaire apparut au bout de quatre ans. Il n’y avait pas deprotéinurie. Au moment de notre étude, le patient ne rec oit aucuntraitement et est sous surveillance régulière.
2.2. Méthodes
2.2.1. Extraction de l’ADNLes échantillons sanguins ont été obtenus, après consentement
éclairé, chez les neuf membres de la famille. L’ADN génomique a

E. Ben-Chetrit et al. / Revue du rhumatisme 80 (2013) 157–160 159
Tableau 1Séquences des amorces et localisations.
Nom de l’amorce Séquence (5′→3′) Taille desfragmentsamplifiés (pb)
IL10-5′UTR-P1F CCCAGGTAGAGCAACACTCC 265IL10-5′UTR-P1R GCTTCTGTGGCTGGAGTCTAAIL10-5′UTR-P2F TATCCAGCCTCCATGGAATC 633IL10-5′UTR-P2R GAGAAATAATTGGGTCCCCCIL10-int1P1F GCCATGGGTTTGGTGAGTTA 703IL10-int1P1R GCTGCAGGAAGAACAAAAGGIL10-int2P1F TCAAGTTCATTCTCCTTTTGTTCT 232IL10-int12P1R TGGATGTGCTGAGTTAACATCTTIL10-int3P1F TACGGCGCTGTGTAAGTAGCA 433IL10-int13P1R TTGGGCCCTTCTTAGAGCTT
Fm
ésR
2
(udSl(c3U
GGel
3
TeramasdMs
ls[cs
iden
tifi
cati
on
des
pol
ymor
ph
ism
es
mon
onu
cléo
tid
iqu
es
(SN
P)
des
gèn
es
de
l’IL-
10, l
ocal
isat
ion
s
(sel
on
NG
0120
88.1
et
NM
0005
72.2
Ref
SeqG
ene)
et
gén
otyp
es
dan
s
la
fam
ille
N.
atio
nN
G
0120
88.1
NM
0005
72.2
(ATG
=
+1)
Loca
lisa
tion
Var
ian
tal
léli
que
3888
.1
3888
.2
3888
.3
3888
.4
3888
.5
3888
.6
3888
.7
3888
.8
3888
.9
g.39
43A
>G
c.-1
117A
>G
5′ UTR
G
AG
AA
AG
AG
AG
AA
AG
AA
AG
g.42
06C
>T
c.-8
54C
>T
5′ UTR
T
CT
CT
CT
CC
CT
TT
CC
TT
CT
g.44
33C
>A
c.-6
27C
>A
5′ UTR
A
CA
CA
CA
CC
CA
AA
CC
AA
CA
g.55
29G
>T
c.16
5+30
5G>T
Intr
on
1
T
GT
GT
GT
GG
GT
TT
GG
TT
GT
g.61
95G
>A
c.22
5+56
G>A
Intr
on
2
A
AG
AG
AG
GG
AG
AA
GG
AA
AG
g.66
07T>
C
c.37
8+19
T>C
Intr
on
3
C
TC
TC
TC
CC
TC
TT
CC
TT
TC
ig. 3. Description du gène de l’IL-10 et des localisations des polymorphismesononucléotidiques (SNP).
té préparé à partir de chaque échantillon (15 mL de sang totalur EDTA) en utilisant le kit QIAamp DNA Blood maxi (Qiagen,oyaume-Uni).
.2.2. Études des polymorphismes mononucléotidiquesL’ADN a été génotypé pour six SNP du gène de l’IL-10
Tableaux 1 et 2, Fig. 3). L’amplification par PCR a été réalisée entilisant le Promega PCR Master Mix, selon les recommandationsu fabricant (Promega, Madison, WI, États-Unis). Le génotypage desNP (Tableau 1) de tous les membres de la famille a été réalisé à’aide de terminateurs d’élongation du kit BigDye Terminator v3.1BDT v3.1) Cycle Sequencing, selon les recommandations du fabri-ant, suivi d’une électrophorèse des amplicons sur analyseur ABI100XL Genetic Analyzer (Applied Biosystems, Foster City, États-nis).
Les haplotypes ont été construits en utilisant le logicielNAlogique (disponible sur le site http://194.167.35.168/u827/NAlogique.html). La ségrégation familiale des haplotypes futnsuite réalisée dans le but de rechercher une association entrees haplotypes de la MB et ceux de la MK.
. Résultats
Les résultats de l’analyse génétique sont exposés dans leableau 2 et sur la Fig. 1. Deux des patients atteints de MB (D.N.t O.N., cas 4 et 7) étaient homozygotes pour les allèles de réfé-ence de tous les SNP à l’exception de rs1800896. L’autre patienttteint de MB (M.N., cas 5) n’avait qu’un seul haplotype com-un (haplotype gris sur la Fig. 1) avec les deux autres patients
tteints de MB (D.N. et O.N.). Cet haplotype était hérité du pèreain (A.N., cas 1) et était aussi présent chez deux membres sainse la fratrie (S.N. et Mi.N., cas 3 et 9). Le patient M.N. atteint deB (cas 5) avait deux haplotypes en commun avec ces deux sujets
ains (S.N. et Mi.N.).Le patient ayant une MK (Y.N., cas 6) était homozygote pour tous
es allèles variants définis comme des allèles à risque pour la MB
elon les deux études d’association portant sur le génome (GWAS)11,12]. De plus, le sujet sain Mo.N. (cas 8) avait deux haplotypes enommun avec le patient atteint de MK. Donc les SNP de l’IL-10 neont pas associés avec la MB ou la MK dans cette famille. Table
au
2N
um
éros
d’
Nu
mér
od
’iden
tifi
crs 18
0089
6
1800
871
1800
872
3024
490
1518
111
1554
286

1 u rhum
4
aopMdstfapc[apc
laMtLddiLvqtLslS
(nLslaph
1an
[
[
[
60 E. Ben-Chetrit et al. / Revue d
. Discussion
La famille que nous avons décrite ici est unique par plusieursspects. À l’inverse de certains articles concernant la MB familialeù les patients sont du même sexe, ici les deux sexes sont atteintsar la MB [9,10]. Dans la plupart des études ayant rapporté desB familiales, un des deux parents est atteint. Ici, trois membres
e la famille ont une MB alors que leurs parents non consanguinsont parfaitement indemnes. Les familles précédemment rappor-ées étaient porteuses de HLA-B51 ce qui n’est pas le cas dans laamille que nous rapportons ici. Il est notable qu’aucun des sujetstteints de MB dans notre famille n’ait eu d’atteinte oculaire. Lalupart d’entre eux avaient une atteinte cutanéomuqueuse et arti-ulaire, ce qui va dans le sens de la remarque faite par Tunc et al.13] selon laquelle il existe plusieurs formes de présentation, l’unessociant les atteintes cutanées et articulaires. Cette observationousserait à utiliser le terme de syndrome de Behc et plutôt queelui de maladie de Behc et.
La caractéristique unique de notre famille réside dans’association d’un patient atteint d’une MK et de trois patientstteints d’une MB au sein de la même fratrie. Le patient atteint d’uneK est le jumeau dizygote d’un patient atteint de MB. La coexis-
ence de la MB et de la MK au sein d’une même famille est intrigante.es deux maladies ont certains points en commun. Elles sont touteseux rares en Occident et relativement « endémiques » en Asieu Sud-Est et au Japon. Elles ont toutes deux une composante
nflammatoire atteignant la peau avec une évolution récidivante.’exérèse chirurgicale des lésions de la MK est habituellement sui-ie d’une récidive et la corticothérapie les fait disparaître. Il sembleu’au cours de la MK, la peau présente une hypersensibilité auxraumatismes comme dans le cas du pathergie test de la MB [14].e fait que ces deux maladies soient rares en Israël et surviennent auein d’une même famille doit faire envisager, sur le plan théorique,’existence de facteurs génétiques ou environnementaux communs.a fréquence au Japon va dans le même sens.
Récemment, deux études d’association portant sur le génomeGWAS) réalisées chez des patients turcs, du Moyen-Orient et japo-ais ont montré que plusieurs SNP sont associés à la MB [11,12].es SNP les plus fréquents sont situés sur le gène HLA-B51. Leseconds SNP en fréquence sont situés sur le gène de l’IL-10. Dansa mesure où HLA-B51 n’était pas présent dans notre famille, nousvons décidé d’effectuer le génotypage des membres de la familleour six SNP de IL-10 et d’analyser la ségrégation familiale desaplotypes selon ces deux maladies.
Notre étude génétique montre que les haplotypes (SNP de l’IL-0) n’étaient pas différents au sein de la fratrie entre les sujetstteints de MB, les sujets sains et le sujet atteint de MK. Il estotable que le patient ayant une MK a deux haplotypes identiques
[
[
atisme 80 (2013) 157–160
composés des allèles à risque pour la MB alors que deux autresmembres de la fratrie, qui sont eux atteints d’une MB, étaient homo-zygotes pour les allèles de référence. Nos résultats ne confirmentpas que le gène de l’IL-10 soit un gène de susceptibilité pour la MBet ne montre pas de facteur génétique commun entre MB et MKdans cette famille. Une des explications de l’absence d’associationentre les SNP de l’IL-10 et la MB est le fait que la famille est origi-naire d’Afrique du Nord, une région qui n’a pas été testée dans lesGWAS [11]. De plus, dans la mesure où dans cette famille tous lescas de MB ont débuté avant l’âge de 17 ans, il est possible que leterrain génétique des formes familiales et/ou des formes à débutjuvénile de la MB soient différents de celui des MB de l’adulte.
Déclaration d’intérêts
Les auteurs déclarent ne pas avoir de conflits d’intérêts en rela-tion avec cet article.
Financement : cette étude a rec u le soutien financier des amiscanadiens de l’Université hébraïque.
Références
[1] Sakane T, Takeno M, Suzuki N, et al. Behcet’s disease. N Engl J Med1999;341:1284–91.
[2] Yazici H, Yurdakul S, Hamuryudan V. Behcet’s disease. Curr Opin Rheumatol2001;13:18–22.
[3] Piga M, Mathieu A. Genetic susceptibility to Behcet’s disease: role ofgenes belonging to the MHC region. Rheumatology (Oxford) 2011;50:299–310.
[4] Sun QF, Xu DZ, Pan SH, et al. Kimura disease: review of the literature. InternMed J 2008;38:668–72.
[5] Varshney MK, Kumar A, Khan SA, et al. Kimura disease of extremity: unusualmanifestation in a long bone. Joint Bone Spine 2008;75:492–4 [Epub 2008 May23].
[6] Kyung-Jin S, Kwang-Bok L. Kimura’s disease occurred in the whole arm. JointBone Spine 2008;75:76–7.
[7] Abdel-Aziz AH, Fairburn EA. Familial Behcet’s syndrome. Cutis1978;21:649–52.
[8] Chamberlain MA. A family study of Behcet’s syndrome. Ann Rheum Dis1978;37:459–65.
[9] Woodrow JC, Graham DR, Evans CC. Behcet’s syndrome in HLA-identicalsiblings. Br J Rheumatol 1990;29:225–7.
10] Villanueva JL, Gonzalez-Dominguez J, Gonzalez-Fernandez R, et al. HLA anti-gen familial study in complete Behcet’s syndrome affecting three sisters. AnnRheum Dis 1993;52:155–79.
11] Remmers EF, Cosan F, Kirino Y, et al. Genome-wide association study identifiesvariants in the MHC class I, IL10, and IL23R-IL12RB2 regions associated withBehc et’s disease. Nat Genet 2010;42:698–702.
12] Mizuki N, Meguro A, Ota M, et al. Genome-wide association studies iden-tify IL23R-IL12RB2 and IL10 as Behc et’s disease susceptibility loci. Nat Genet2010;42:703–6.
13] Tunc R, Keyman E, Melikoglu M, et al. Target organ associations in Turkishpatients with Behc et’s disease: a cross sectional study by exploratory factoranalysis. J Rheumatol 2002;29:2393–6.
14] Barnes CG. History and diagnosis. In: Yazici Y, Yazici H, editors. Behcet’s syn-drome. New York: Springer; 2010. p. 7–34.















![Lesaeintesvasculaires aucoursdelamaladiede$ Behçet ... · Risque$de$récidives$$des$thromboses$ veineuses$au$cours$de$la$maladie$de$ Behçet$ [1] JK. Ahn et al. Clin Rheumatol 2008](https://img.pdfslide.fr/doc/110x75/5e627a2a2a5a2c122d7716fe/lesaeintesvasculaires-aucoursdelamaladiede-behet-risquedercidivesdesthromboses.jpg)













