Embed Size (px)
Citation preview

UE9 – Immunopathologie et ImmunointerventionPr P Gasque
Date : 20/03/2018 Plage horaire : 14h-16h
Promo : DFGSM2 2017/2018
Ronéistes :ARNAUD Eulalie CLAIN Raphaëlle
Enseignant : Pr Gasque
Mécanismes physiopathologiques des maladies auto-immunes (MAI) - Partie 1
I. I. Introduction: Définition et concepts clés
II. II. Deux types de MAI (SO et NSO)
III. III. Mécanismes de tolérance (Centrale et périphérique des
lymphocytes T et B)
IV. IV. Rupture de la tolérance et autoréactivitéA. Modification de l’autoantigène (médicament)
B. Réaction croisée entre pathogène et Ag du soi
C. Libération des Ags séquestrés (et modifiées)
V. V. Dysfonctionnement des T regs
VI. VI. Autres facteurs favorisant l’auto-immunité
VII. VII. Mécanismes d’action des autoanticorpsA. Modulation de l’activité des récepteurs
B. Cytotoxicité
VIII. VIII. Mécanismes d’action des cellules T cytotoxiques
IX. IX. Traitements des MAI
Les diapos sont à la fois en anglais et en français afin de nous familiariser avec les terminologies anglaises. Objectif : obtenir les bases de l’auto-immunité et les illustrer avec certaines pathologies tels que le lupus, la polyarthrite rhumatoïde (PAR) ou encore la sclérose en plaque (SEP).On a laissé les parties VII, VIII et IX (même si Gasque n’a pas traité cela aujourd’hui) pour avoir le coursen entier. Il continuera la suite au prochain cours !! (tout a été traité cette année)

Ce 1er cours va jeter les bases et les éléments cellulaires ou moléculaires qui contribuent aux maladiesauto-immunes.Dans un 1er temps on va voir les notions de cette auto-immunité, avec des notions sur les auto-antigèneset des notions sur qu'est-ce qui fait qu'on va avoir une maladie autoimmune. (MAI)Attention: souvent on fait référence à des auto-anticorps mais il n'y a pas que des auto-anticorps qui sontà l'origine des MAI mais on a aussi des lymphocytes T qui sont également à leur origine.C'est souvent les 2: les auto-anticorps et les lymphocytes T auto-réactifs contre nos propres auto- antigènes qui vont contribuer à la pathologie autoimmune.
Des exemples de maladies auto-immunes :
- Le vitiligo: maladie qui va atteindre la peau et qui va toucher particulièrement les mélanocytes. Lephénomène de dépigmentation visible sur cette jeune patiente est donc dû dans cette pathologie auto- immune à la destruction des mélanocytes (qui produisent de la mélanine, ensuite absorbée par les kératinocytes, qui et qui justifie la coloration brune de la peau). Ce n’est pas la MAI la plus douloureuse au sens analgésique du terme, mais elle a un fort impact sur la vie sociale du patient.
On a des mélanocytes, qui sont entre l'épiderme et le derme. Ces cellules contiennent de la mélanine, responsable de la pigmentation de notre peau. (Attention, il n'y a pas plus de mélanocytes pour les peaux brunes ou moins de mélanocytes pour les peaux blanches, mais c'est selon qu'ils produisent plus ou moins de mélanine.)
Dans cette pathologie, on a des lymphocytes T qui vont venir tuer les mélanocytes qui ne seront plus capables de produire de la mélanine. Cette mélanine est un pigment brun ou rouge qui va venir se déposer dans les noyaux de nos kératinocytes pour protéger nos kératinocytes des rayons UV. Ceux-ci sont extrèmement nécrosant, ils peuvent provoquer l'apoptose des kératinocytes. Mais si les noyaux des kératinocytes sont recouverts de mélanine elle va absorber les UV et protéger l'ADN de ces rayonnements qui pourraient être à l'origine de cancers.
La protéine reconnue est la tyrosinase, c'est une enzyme capable de synthétiser de la mélanine à partir de la L-DOPA. C'est une enzyme majoritaire des mélanocytes.La sclérose en plaque va atteindre le SNC. On a une destruction de la gaine de myéline avec appartion de plaques. D'où le nom « sclérose en plaque » car la moelle est sclérosée. La sclérose en plaque concerne la myeline produite par les oligodendrocytes et pas celles du SNP. Les cellules qui myélinisent le SNP sont les cellules de Schwann, qui concerne une autre pathologie auto-immune appelée le syndrome de Guillain Barré.
Les oligodendrocytes sont détruits par les lymphocytes T et également par des auto-anticorps. Ces lymphocytes T et auto-anticorps reconnaissant une protéine majoritaire de l'oligodendrocyte : la MBP (Myelin Basic Protein).La MOG (Myelin Oligodendrocyte Glycoprotein) est une autre protéine cible dans la sclérose en plaque.

- Dans la myasthénie (Myastenia Gravis), il existe des anticorps dirigés contre un récepteur del’acétylcholine (NT qui favorise le mouvement) au niveau de la jonction neuro-musculaire qui va empêcher signal nerveux vers la plaque musculaire et engendrer une perte de la motricité
- Le Diabète de Type I, important à distinguer du diabète de type II (qui n’est pas une MAI,)s’explique par une production d’anticorps contre l’hormone insuline qui est produite par les cellules bêta du pancréas. Il y’a donc destruction de ces cellules : perte de synthèse d’insuline : diabète,perte d’absorption du glucose.
- Le Lupus (=SLE) va affecter essentiellement la peau, donc ça se voit assez facilement chez les patientsatteints. Tous les organes sont touchés car la cible du lupus c’est l’ADN et les protéines nucléaires exprimées dans toutes les cellules. On a chez ces patients des auto-anticorps contre la molécule d'ADN qui vont créer un complexe Ac/Ag, l'activation du complément, et la destruction du tissu.
Tous les organes peuvent être le siège de mécanismes auto-immuns comme on peut le voir sur la diapo ci- contre, l’objectif n’étant pas de faire de manière exhaustive le catalogue de toutes ces pathologies mais de comprendre les mécanismes essentiels.
Il y a une très forte association entre les maladies infectieuses et l'autoimmunité. On sait que si on a par exemple une infection des poumons par des bactéries, on va avoir l'accumulation de débris dérivés de nos cellules. On va avoir l'accumulation de cellules en apoptose. Le pire des scénarios serait que ces cellules en apoptose ne soient pas éliminées et s'accumulent sous forme de cellules nécrosées. Si de l'ADN est libéré par ces cellules nécrosées, inéluctablement notre système immunitaire va trouver cet ADN qui est accumulé de manière anormale. L'ADN n'est pas censé être à l'extérieur d'une cellule. Ici si il est libéré, notre SI qui ne l'a jamais vu va considérer que c'est un Ag et va lutter contre lui de la même manière qu'il peut lutter contre des bactéries.C'est pour cela qu'on peut avoir un processus d'initiation d'une MAI contre nos propres constituants sur la base d'une maladie infectieuse.

Ici on a des neutrophiles qui vont mourir en apoptose. C'est le cas de ce patient avec une infection par aspergillus dans le cerveau. Ces neutrophiles qui sont censés venir lutter contre cet aspergillus vont rapidement mourir dans le cerveau (où ils ne sont pas censés être présents) et cette mort des neutrophiles va libérer beaucoup de constituants dérivés de nos propres cellules et contribuer à ce complexe auto-immun.
Attention : ce n'est pas parce qu'on a une maladie infectieuse qu'on aura nécessairement une pathologie auto-immune.Fort heureusement il existe des mécanismes de tolérance pour lutter contre ces auto-anticorps ou lymphocytes T autoréactifs dirigés contre nos propres constituants.
Malheureusement si l'infection est trop importante et si elle perdure notre système de tolérance ne pourra pas exercer son activité de tolérance et notre SI va générer des auto-anticorps et lymphocytes T auto- réactifs.
Si on reprend le schéma classique d'initiation d'une réponse immunitaire. :
Ici on a notre cellule présentatrice d'Ag, notre lymphocyte T, notre lymphocyte B, et notre cellule en nécrose qui est la source de l'auto-antigène.
Fort heureusement on aura des mécanismes régulateurs qui vont bloquer les lymphocytes T et B (produisent des auto-AC) et bloquer la présentation de l’Ag

I. Introduction: Définition et concepts clésL’auto-immunité concerne toute réaction immunitaire cellulaire (lymphocytes T ou B autoréactifs)
ou humorale (production d’auto-anticorps) développée vis-à-vis des propres constituants de l’organisme.
Les réponses auto-immunes sont dirigés contre des éléments (antigène) du soi appelé auto-antigène (Exemples, acides nucléiques, AN/lupus ou encore récepteur de l’acétylcholine, neurotransmetteur: Myastenia gravis)L’autoimmunité peut être spécifique d’un organe (MAI SO) (thyroïde, hashimoto) ou systémique (MAI NSO, lupus)Il existe normalement des mécanismes de tolérance immunitaire mais qui font défauts dans les MAI.Les MAI sont des pathologies chroniques (Exple: sclérose en plaques): on peut contrôler mais ne pasguérir d’une MAI (si pas contrôlée elle peut s’étendre à d’autres tissus)
Du point de vue historique, le 1er qui a émis l'hypothèse qu'il y aurait un système auto-immun dirigé contre nos propres Ag c'est Paul Ehrlich qui travaillait sur l'immunité auto-infectieuse.Il a parlé d'une situation d'horreur où son propre SI serait dirigé contre ses propres cellules. Il a émis l'hypothèse de « horror autotoxicus », à savoir un mécanisme immunitaire dirigé contre nos propres cellules
Concept n°1 : L’immunité T et B antivirale est identique à la réponse auto-immune dirigé contre le soi.
Ici un schéma pour rappeler quels sont les mécanismes cellulaires et moléculaire de l'auto-immunité.Ici c'est un schéma classique où on a un Ag qui est phagocyté par un macrophage. Ensuite ce macrophage va présenter des portions de cet Ag (ici en rouge) aux lymphocytes T.
On aura besoin de 2 signaux pour l'engagement du SI : MHC II, CD4 ; et CD80/86, CD28.
Ce qui va permettre l'activation du lymphocyte T en soit Th2 soit Th1, et permettre via la génèse d'interleukine 6 la production et l'activation des lymphocytes B qui sont spécifique de cet Ag via la reconnaissance par le B-cell receptor avec la production d'Ac.
On a le rôle clé de molécules telles que TNFα produit par la cellule présentatrice d'Ag (CPAG = svt les macrophages) pour activer les lymphocytes T ; le rôle clé de l'interféron α et g. Retenir que ce soit un virus ou une bactérie, la réaction immunitaire se déroulera de la même manière. Les réactions immune & auto-immune emploient les mêmes moyens (très important)
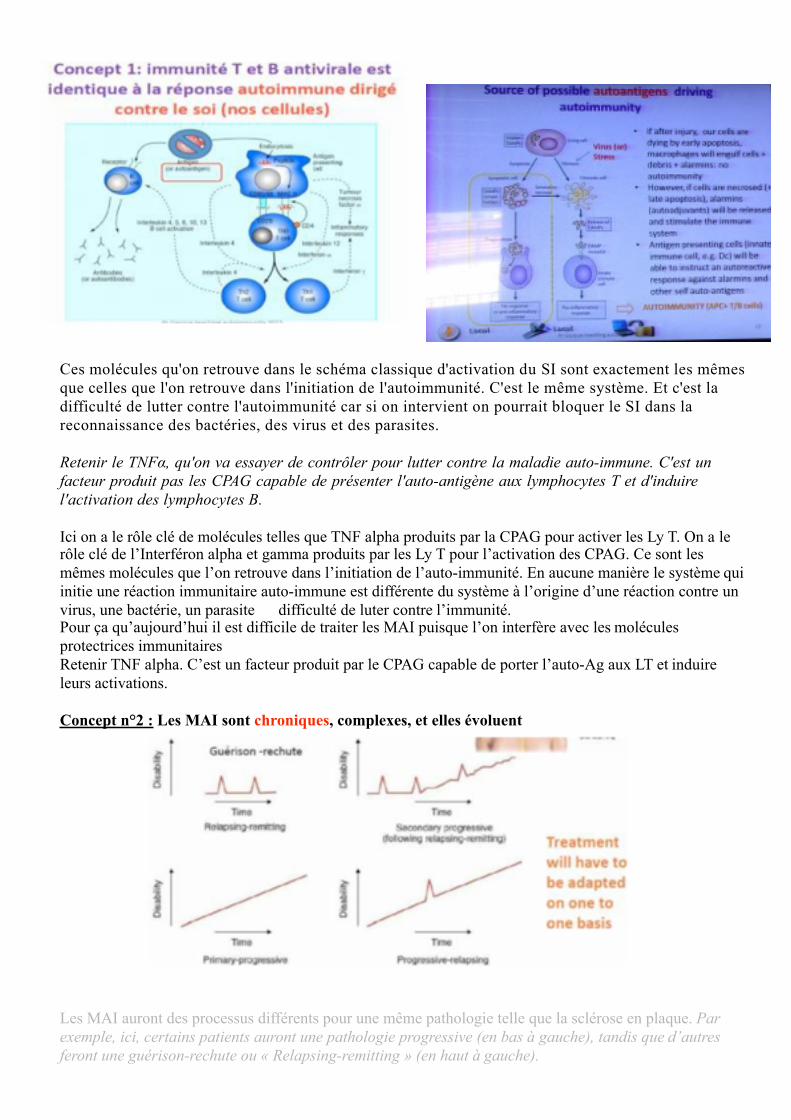
Ces molécules qu'on retrouve dans le schéma classique d'activation du SI sont exactement les mêmes que celles que l'on retrouve dans l'initiation de l'autoimmunité. C'est le même système. Et c'est la difficulté de lutter contre l'autoimmunité car si on intervient on pourrait bloquer le SI dans la reconnaissance des bactéries, des virus et des parasites.
Retenir le TNFα, qu'on va essayer de contrôler pour lutter contre la maladie auto-immune. C'est un facteur produit pas les CPAG capable de présenter l'auto-antigène aux lymphocytes T et d'induire l'activation des lymphocytes B.
Ici on a le rôle clé de molécules telles que TNF alpha produits par la CPAG pour activer les Ly T. On a le rôle clé de l’Interféron alpha et gamma produits par les Ly T pour l’activation des CPAG. Ce sont les mêmes molécules que l’on retrouve dans l’initiation de l’auto-immunité. En aucune manière le système qui initie une réaction immunitaire auto-immune est différente du système à l’origine d’une réaction contre un virus, une bactérie, un parasite difficulté de luter contre l’immunité.Pour ça qu’aujourd’hui il est difficile de traiter les MAI puisque l’on interfère avec les molécules protectrices immunitairesRetenir TNF alpha. C’est un facteur produit par le CPAG capable de porter l’auto-Ag aux LT et induire leurs activations.
Concept n°2 : Les MAI sont chroniques, complexes, et elles évoluent
Les MAI auront des processus différents pour une même pathologie telle que la sclérose en plaque. Par exemple, ici, certains patients auront une pathologie progressive (en bas à gauche), tandis que d’autres feront une guérison-rechute ou « Relapsing-remitting » (en haut à gauche).

Il est important de comprendre qu’on est sur la même MAI, ce qui signifie que les mécanismes pathologiques ne sont pas nécessairement identiques : ils dépendent de la concentration d’anticorps, de l’épitope généré, si il y a une seule ou plusieurs cibles et même de la barrière hémato-encéphalique pour cette MAI. Le type de mécanisme va jouer un rôle déterminant dans le traitement.
Il n’y a pas qu’un seul auto-Ag responsable de la MAI de la SEP mais que rapidement il y aura plusieurs auto-Ag intervenant dans la SEP.Dans la SEP, on a l’oligodendrocyte capable de myélinisé les axones. Un des auto-Ag de la SEP est en effet les protéines de la Myéline mais rapidement dans la SEP on a une destruction des axones. Ceux-ci libèrent des protéines axonales telles que les neurofilmentes et deviennent également des auto-Ag. Donc dans la SEP il n’y a pas que des auto-Ac et des LT dirigés contre l’oligodendrocyte mais des auto-Ac et des Ly contre les composés tels que l’axone.Tableau pas à retenir (pas ici)Contribution de tous les auto-Ag et auto-Ac SEP qui est d’autant plus difficile à traiter.Important à ce stade qu’il y a plusieurs auto-Ag et pas qu’un seul (même pour une pathologie auto-immune) !!!!!
Concept 3 : La cible à l’origine de la pathologie n’est pas nécessairement la seule cible de la pathologie.
Il n’y a pas qu’un seul auto-Ag responsable de la MAI de la SEP mais que rapidement il y aura plusieursauto-Ag intervenant dans la SEP.Dans la SEP, on a l’oligodendrocyte capable de myélinisé les axones. Un des auto-Ag de la SEP est en effet les protéines de la Myéline mais rapidement dans la SEP on a une destruction des axones. Ceux-ci libèrent des protéines axonales telles queles ‘neurofilmentes’ et deviennentégalement des auto-Ag. Donc dans laSEP il n’y a pas que des auto-Ac et des LT dirigés contre l’oligodendrocyte mais des auto- Ac et des Ly contre les composés tels que l’axone.Tableau pas à retenir (pas ici) Contribution de tous les auto-Ag et auto-Ac SEP qui est d’autantplus difficile à traiter. Important à ce stade qu’il y a plusieurs auto-Ag et pas qu’unseul (même pour une pathologie auto-immune) !!!!!
Même si on est atteint d’une pathologie auto-immune qui pourrait concerner un seul type cellulaire, le système immunitaire peut ensuite rapidement reconnaître d’autres auto-antigènes et on a alors des atteintes multi-organes.
Ex : La sclérose en plaque. Ce sont des LTs autoréactifs et des auto-anticorps dirigés contre cette cellule (en jaune) : l’oligodendrocyte, car les LTs et les auto-anticorps sont dirigés contre la myéline. Sauf que lorsque l’axone (ici en orange) va être lésé, il deviendra antigénique.Il y aura alors dans la sclérose en plaque, des auto-anticorps et des LTs autoréactifs contre l’axone, bien que ça ne soit pas l’origine de la MAI.

Concept 4 : Les mécanismes : Pourquoi nos cellules sont-elles reconnues comme des auto-antigènes ?
L’hypothèse de travail des groupes de recherche :A la base, il y a une infection particulièrement sévère (ex: par le virus de Chikungunya) et qui peut aboutir dans certains tissus à de nombreux débris cellulaires.
Ces derniers vont contribuer à exacerber le système immunitaire. Comme dit à l’introduction le terme« pathogène » ne désigne pas uniquement virus, bactérie ou parasite mais aussi l’ADN, les lipides oxydés, les protéines amyloïdes, ou encore la protéine prion qui est normale mais qui peut être potentiellement pathogène.
Lorsque tout cela est libéré à côté d’un virus, le système immunitaire n’arrive plus à faire la différence. On aboutit à un scénario unique, à savoir que l’infection sera éliminée mais le patient aura établi une auto- immunité contre ses propres composés.
Cependant cette hypothèse est très difficile à vérifier car au moment où le patient développe sa MAI et que l’on recherche le lien infectieux avec celle-ci, l’infection a déjà été éliminée.
On sait donc qu’il existe un lien fort entre infection et MAI mais c’est très difficile à prouver. Sauf dans certains cas comme à la Réunion, où 38-40% de la population a été touchée par le virus Chikungunya et 10% de cette population, surtout chez les personnes âgées, qui sont aujourd’hui atteint de MAI.
Pourquoi nos cellules deviennent des auto-Ag et libèrent des auto-Ag ?Prenons l’exemple de l’ADN. Dans une cellule viable, il est à l’intérieur de notre noyau et le système immunitaire ne verra pas l’ADN. Si une cellule va en apoptose, elle ne perméabilisera pas sa membrane. Elle va se dégrader à l’intérieur. Elles seront phagocytées par les macrophages. Si infection trop importante ou stress trop importante, les cellules au lieu d’être en apoptose vont se nécrosées libération de l’ADNqui va se présenter au CPA qui à son tour présentera l’auto-Ag aux Ly T genèse d’une pathologie auto-immune. Si nécrose trop importante présentation de l’auto-Ag à l’origine de la MAI.
Chaque seconde, 2000 cellules meurent dans l’organisme milliards chaque jour. Si ils libèrent de l’ADN risque d’avoir une pathologie auto-immune mais meurent par apoptose et seront éliminer par les
macrophages avant que le système immunitaire les ai vu.
Les neurofilaments et l’ADN peuvent être des cibles dans la SEP

I. II. Deux types de MAI (SO ET NSO)
Je suis obligé de vous dire qu’il existe une classification, mais je ne l’aime pas du tout car il y a trop d’exception pour la garder.Les MAI spécifiques d’organe (MAI SO) c’est soit des auto-anticorps soit des LTs autoréactifs qui reconnaissent un auto-antigène exprimé uniquement dans un organe donné, dans une cellule donnée. Par exemple, les autos anticorps dirigés contre : - les ilots de Langerhans dans le diabète de type 1
- la thyroïde : Basedow, Hashimoto- la surrénale, provoquant une insuffisance rénale lente
A l’inverse, il y a les MAI non spécifiques d’un organe, systémiques (NSO) c'est-à-dire que plusieurs organes comme par exemple la Polyarthrite Rhumatoïde (PAR) peuvent être impacté par la MAI. Il n’aime pas cette classification puisque dans la PAR, l’organe c’est l’articulation. Comme plusieurs articulations sont touchées, les spécialistes ont considéré que c’était une NSO.Elle vaut pour le lupus puisque en effet on a une MAI contre l’ADN qui se retrouve dans tous les tissusMAI NSO.Info : Pour lui la PAR est une MAI SO mais dans les ouvrages c’est une MAI NSO ne tombera pas àl’examen pour cette différence !!!!!
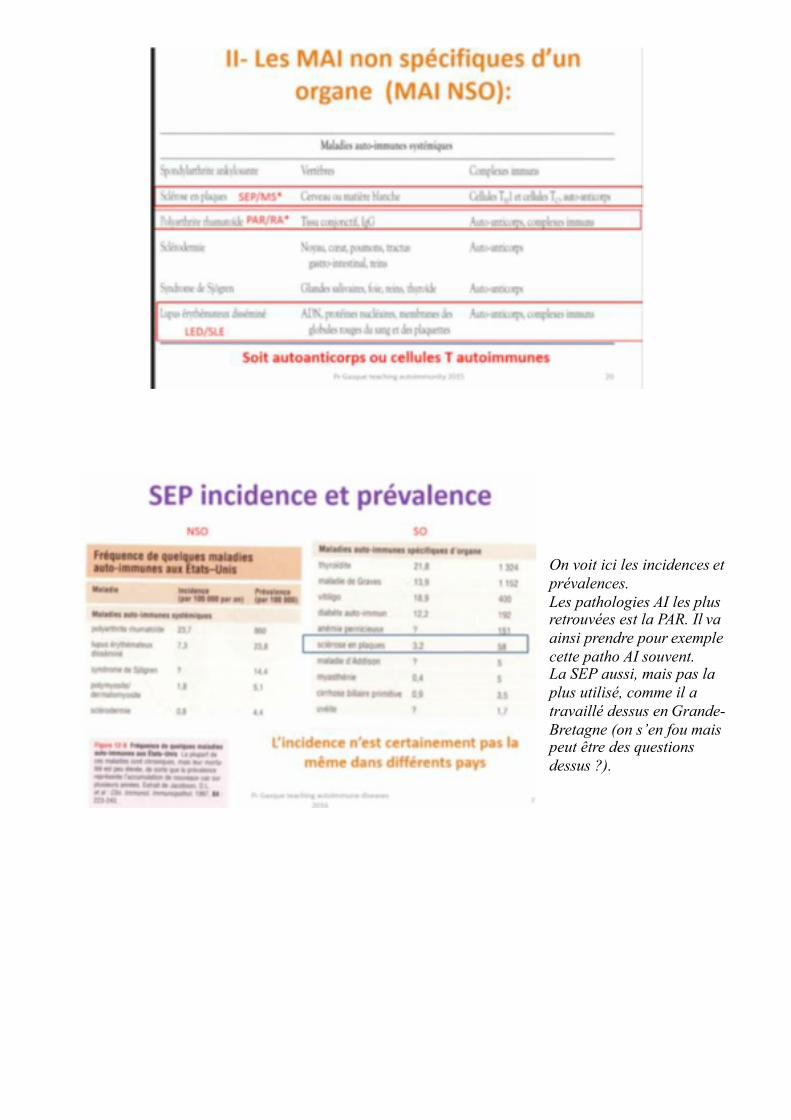
On voit ici les incidences et prévalences.Les pathologies AI les plus retrouvées est la PAR. Il va ainsi prendre pour exemple cette patho AI souvent.La SEP aussi, mais pas la plus utilisé, comme il a travaillé dessus en Grande- Bretagne (on s’en fou mais peut être des questions dessus ?).

Pour cette SEP, les pays nordiques, Grande-Bretagne, USA, Norvège, Allemagne sont plus impactés.Une des raisons qui explique le fait qu’il y en a plus dans certains pays sont :-un facteur environnemental, une raison viral dont le virus de l’EPB pour la SEP- les molécules HLA qui présentent l’Ag. Certains auront plus d’affinités pour des auto- Ag.
Les auto-Ac ont une spécificité contre des antigènes ubiquitaires cad distribués systématiquement dans plusieurs tissus, organes ou cellules.Les autoantigènes (souvent intracellulaires) doivent être accessibles : ex: nécrose/lyse des cellules cibles.Les LTs autoréactifs sont également impliqués.
L’exemple qui est souvent pris pour les MAI NSO, c’est celui du bas, le lupus ou LED pour Lupusérythémateux disséminé.Il lit les exemples en rougePour la sclérose en plaque, c’est là qu’il n’est pas d’accord avec la classification, car c’est écrit cerveau (donc ça serait spécifique d’organe) mais il y a aussi la moelle épinière.Pour la polyarthrite rhumatoïde, elle touche uniquement les articulations mais elle est classée dans NSO. Il n’est pas important de connaitre cette classification.
Après les tableaux de la diapo ne sont bien sûr pas à apprendre, c’est pour illustrer le propos que quasiment toutes les protéines ou composés connus aujourd’hui sont potentiellement des auto-antigènes. C’est toute la complexité dans le diagnostic d’un patient qui souffre d’une MAI, c’est de savoir quel est l’auto-antigène mis en jeu pour pouvoir contrôler sa pathologie. Inéluctablement, il y a de fortes chances qu’il n’y est pas qu’un seul auto-antigène, pas qu’un seul auto-Ac, pas qu’un seul LT autoréactifs mais de multiples cibles. C’est pour cela que les traitements utilisés sont relativement systémiques dans leurs mécanismes d’action pour essayer de contrôler le plus largement possible la réaction auto-immune.

Cependant d’autres cibles des MAI concernent les molécules auto-immunes elles-mêmes. Aujourd’hui, on sait que les patients sont porteurs d’une MAI, lorsqu’ils génèrent des Ac contre la molécule C1q, ce qui empêche le complément d’éliminer les cellules en apoptose et qui exacerbe encore plus la pathologie.C1q se fixe sur la cellule apoptotique pour initier la phagocytose. Si cette dernière n’a pas lieu, cette cellule apoptotique opsonisée va se nécroser. Les cellules immunitaires vont réagir contre la cellule nécrosée mais également contre C1q On peut donc détecter chez des patients lupiques des anticorps anti-C1q).
III. Mécanismes de tolérance (centrales et périphériques deslymphocytes T et B)
Ils sont très importants comme ils peuvent entrainer des pathologies AI avec le nombre important de cellules mourantes par jour. Dans 99% des cas, cette auto-immunité sera contrôlée par des mécanismes de tolérance afin d’empêcher les LTs autoréactifs de persister.L’exemple pris montre comment notre système immunitaire permet d’éliminer un LT autoréactif:Soit par un phénomène de tolérance central : cad qu’il sera éliminé à l’intérieur même du tissu où il est généré. Mais même si il gagne le sang, qu’on considère comme un organe périphérique, ils seront également éliminés.
Deux niveaux d’induction de la tolérance des lymphocytes :
Central : dans le site où les cellules sont produites-Les lymphocytes T: Thymus, au contact des cellules épithéliales.-Les lymphocytes B: Moelle Osseuse.
Périphérique (dans le sang, les ganglions/rate et autres tissus)Ces lymphocytes auto-réactifs produits seront donc détruit au niveau central ou périphérique.
Tolérance centrale :
L’antigène du soi est présent dans les organes producteurs, ici dans le thymus. Les LT autoréactifs seront donc activés dès leurs productions. Cette activation sera perçue comme étant anormale et les cellules épithéliales du thymus déclencheront l’apoptose de ces cellules autoréactives et elles seront éliminées. Normalement, elles n’ont pas le droit d’être activées et de proliférer dans le thymus, elles y sont juste produites.A l’inverse, les 2 LTs de gauche reconnaissent par exemple un virus. Le virus n’étant pas présent dans le thymus, les LTs vont donc poursuivre leur maturation. Si ils détectent le virus dans le sang, à un temps T1, ils pourront alors proliférer en ayant échappés au mécanisme de tolérance et de sélection au niveau du thymus.Pour les LB, c’est au niveau de la MO. Même principe, élimination des LB auto-réactif par apoptose, mort programmée.

Tolérance périphérique :
Dans le cas d’un antigène infectieux, lorsque la cellule présente l’antigène (en bleu), cette présentation doit se faire aux LTs via 2 signaux: le signal médié par le T-cell Receptor et celui médié par les molécules de coactivation. CD80/86 véhiculés par la cellule présentant l’Ag vont envoyer un co-signal. Les 2 signaux, CD28-dépendant et TCR-dépendant vont aboutir à la prolifération de ces LTs. Il s’agit de l’immunité classique.CMH de classe 2 c’est un LT CD4 et CMH de classe 1 c’est un LT CD8.Si la cellule présentant l’Ag présente un Ag du Soi, elle n’aura pas augmenté sa régulation des molécules CD80/86, qui ne seront donc pas exprimées. In fine, il n’y aura qu’un seul signal transmis aux LTs (qui doivent forcément percevoir les 2 signaux, il le répète), celui du TCR mais pas celui du CD28. La cellule T autoréactive va rentrer en apoptose ou devenir anergique (elle ne sera pas capable de proliférer).
Donc les LT auto-réactif contre un Ag du soi vont mourir dans le cadre de la présentation de l’Ag du soi et il y aura absence d’auto-immunité par élimination du LT. C’est le phénomène de tolérance périphérique au niveau du sang (répéter et répéter pour bien prendre en compte l’importance quoi….).
Pour les LB se sont d’autres mécanismes mais qui répondent au même schéma, avec un mécanisme detolérance centrale dans la MO et un mécanisme de tolérance périphérique dans le sang.Les AC via leurs fragments Fc (cristallisable et non pas constant au niveau C-terminal) sont reconnus pardes récepteurs de type Fcgamma2b exprimé par les LB. Cet Ag du soi sera aussi reconnu par le B-cell receptor (spécifique de l’Ag du soi). Ce sont les 2 signaux : Fcgamma2b et BCR qui vont induire un signal d’apoptose pour les LB.Ceci est à la base de la tolérance de l’organisme vis-à-vis de ses propres Ag. Toute rupture de cette tolérance entraîne une auto-immunisation.
IV. (Mécanisme de) Rupture de la tolérance et autoréactivité
« Dans l’intérêt de ce cours, je présenterai que 3 scénarios »Malheureusement dans 1% des cas, il y aura une rupture de la tolérance. Il n’y aura pas d’élimination desLTs et LB autoréactifs.Plusieurs mécanismes concourent à la levée de la tolérance et l’induction de l’auto-réactivité : - modification de l’auto-ag du soi (cas du médicament),- réaction croisée entre un pathogène et un antigène du soi,- libération et reconnaissance des auto-antigènes modifiés qui sont normalement séquestrés dans les cellules.

A. Modification de l’auto-antigène (médicament)
Dans les traitements inhibiteurs de l’agrégation des plaquettes.Le médicament est un antigène, c’est une molécule chimique. Notre système immunitaire n’a jamais été éduqué pour reconnaitre un médicament pour la 1ere fois comme une molécule du Soi. Naturellement, il va le considérer comme une molécule étrangère. Il est donc reconnu par un anticorps: en se fixant sur les cellules sanguines (plaquettes), il modifie leur antigénicité (complexe Ag-Ac, réaction du complément) et provoque une réaction immunitaire contre ces Ag modifiés et destruction des cellules.
Les plaquettes ont un rôle extrêmement important dans le contrôle de la coagulation.L’absence de contrôle de la coagulation entraine des anémies hémolytiques, thrombocytopéniques car chute du nombre de plaquettes.La deuxième cause liant un médicament à une maladie auto-immune:
La fixation du médicament induit la formation d’un néo-épitope antigénique reconnu comme étranger. L’auto-Ac reconnait cet auto-Ag et induit une réaction immune.
Partie gauche du schéma:Dans le premier cas, un patient qui prend un ttt anti-agrégant et que ce ttt consiste en la fixation du médicament sur les récepteurs les plaquettes. Ici exemple IIb-IIIa. La fixation de ce médicament sur ce récepteur qui n’est pas naturel va être reconnue par un auto-Ag dirigé contre le médicament. Cela va entrainer une élimination des plaquettes puisque cela a créé un complexe Ag-Ac. Soit le complément (voie classique ici) détruit la plaquette, soit ce sont les macrophages. Thrombocytopénie avec une chute du taux de plaquette
Partie droite du schéma :On peut voir ce néo-épitope. Le médicament se fixe au niveau du même récepteur, mais lui n’est pas reconnu comme antigène. En se fixant, il change la conformation des récepteurs et crée un domaine (épitope, site Agénique nouveau) qui sera reconnu par l’auto-Ac comme antigénique. Ce domaine n’est pas classiquement observé. Pareil, il y aura une élimination soit par le complexe Ag-Ac soit par les macrophages.La conséquence est la même: une thrombocytopénie anémique.
Réponse à une question: un même médicament n’induira pas une maladie auto-immune à tous les patients, seulement certains seront touchés.

B. Développement d’une réaction croisée ‘mimicry‘ entre un antigèneétranger et un auto-antigène du soi
Le système immunitaire va lutter contre une bactérie ou un virus, mais qui possède une protéine bactérienne ou virale qui ressemble beaucoup (molecular mimicry car l’Ag bactérien va mimer l’Ag du soi) à l’un de nos propres protéines. L’Ag étranger présente une homologie avec l’Ag du soi. Cela entraîne la productiond’AC qui reconnaissent et causent la destruction des deux antigènes : on parle de réaction croisée.
Le virus exprime un épitope (en rouge) (une séquence d’acide aminé), qui est commun avec un épitope présent dans une protéine du Soi. Celui-ci va induire une réponse anti-virale.En effet l’épitope présenté par les cellules dendritiques via les molécules HLA de classe 2 (en vert) vont être reconnu via le TCR.La molécule co-stimulatrice n’est pas présentée sur le schéma mais elle est là activation des LT dirigécontre le virus, notamment contre cet épitope.
C’est un des arguments de la sclérose en plaque, avec les infections à EBV.Certains des lymphocytes anti-EBV se mettent à reconnaître des protéines de la myéline, générant la sclérose en plaque par destruction de la myéline.
Imaginons que 90% de la salle a été infecté par EBV. L’EBV va induire la production de LTs. Dans 99%des cas, aucun d’entre vous n’aura de MAI sauf que chez certains patients ces LTs vont réagir contre la MBP. On voit ici le lien : maladie infectieuse qui induit une MAI. Dans aucun cas, la MBP n’a produit une MAI, c’est à travers le virus.
Or les LTs ne sont pas censé croiser la MBP dans le cerveau, il faut d’autres éléments confondants pour que cette MBP soit libérée dans le sang à un moment donné, pour que le LT soit activé et qu’il se mette à réagir contre le MBP. C’est pour cela qu’il n’y a pas de lien de cause à effet obligatoire dans l’auto-immunité : il faut plusieurs événements.
Exemple : les syndromes post-streptococciques arthritogènes, appelés SRA. On fait une infection à streptocoque, on génère des AC contre ce streptocoque, mais malheureusement ces AC vont aussi reconnaître nos propres protéines.

Exemple d’homologie de séquence entre la MBP et la protéine de la rougeole : la protéine virale P3 a une séquence en acides aminés (en bleu) qui est identique à celle que l’on retrouve dans les résidus de la protéine MBP (myelin basic protein) à partir de la position 61. Si vous avez un Lymphocyte T réactif contre la rougeole et contre cette séquence, il pourrait (conditionnel) engendrer une réponse contre les protéines de la myéline et l’oligodendrocyte. Il y a une longue liste d’agents pathogènes qui présentent des analogies de séquences (en bleu) avec vos propres protéines (le récepteur de l’insuline, de l’acétylcholine...).Ces 2Aa différents ne sont pas suffisants pour qur le LT réagisse contre cette myéline. A 3 ou 4 différents par exemple permettra la réaction du LT qui tuera les oligodendrocytes. Ce qui contribue à un processus auto-immmun contre la myéline et donc contre la SEP.
Autre exemple : Syndrome de Guillain-Barré.Ca c’est quand on a des auto-Ac dirigé contre une protéolipide de la myéline exprimé au niveau des nerfs périphériques (la SEP est au niveau central pas périphérique).La plus forte association entre les Ac contre la myéline périphérique serait due à une réinfection des patients par cette bactérie : campylobacter jejuni. En effet cette bactérie si elle se retrouve au niveau de l’intestin peut proliférer et aboutir à une genèse d’auto-Ac contre cette bactérie, notamment contre un composé lipidique du LPS de la bactérie qui ressemble beaucoup aux protéolipides exprimés parles nerfs périphériques. Même si on génère des Ac contre campylobacter, malheureusement ces Ac vont reconnaître la myéline périphérique et donner le Guillain-Barré.

C. Libération et reconnaissance des auto-antigènes séquestrés etmodifiés (théorie des clones interdits)
Antigènes séquestrés
Une infection virale pourrait induire la libération de molécules du soi autrement séquestrés dans les cellules (protéines du cytosquelette (vimentine), protéines et AN nucléaires (ADN) et des mitochondries (cardiolipin) donc normalement non-reconnaissable par notre système immunitaire.Les cellules infectées induites en apoptose ou en nécrose pourraient libérer les auto-antigènes séquestrés qui seront alors reconnus par notre système immunitaire, et qui génèreront une auto-immunité.Souvent on dit que ces Ag sont des DAMP (Danger Associated Molecular Pattern).
Rq : PNN pas sensé ê au niv synoviale mais ds sang et lors infect°, nécrose ou atteinte patho : infiltration PNN dans synovie
Les 3 évènements sont obligatoires pour développer une MAI !!!!!
Autre exemple de la PAR qui sera bien décrite dans un autre cours.Aujourd’hui on a un test permettant de découvrir la forme citrunillé (Arg -> Citrulline) des auto-Ac dirigécontre les formes citrunillés. C’est le test de la mesure des anti CCP, auto-Ac dirigé contre le Cyclic Citrullinated Peptide (CCP). Cette technique permet en utilisant un peptide citrunillé de détecter si le patient présente des auto-Ac capables de reconnaître ce peptide citrunillé. Si le taux est élevé, le patient a une PAR. Les protéines sont en général non citrunillées & sont reconnnues comme auto-Ag
Pour comprendre cet Ag séquestré, on prend l’exemple de l’ADN et du lupus déjà pris.
Dans une cellule normale l’ADN est séquestré dans le noyau. Même si la cellule est en apoptose, l’ADN reste à l’intérieur du noyau et sera dégradé. La cellule sera phagocyté et il n’y aura pas d’inflammation, pas d’auto-immunité.A l’inverse lors d’une infection virale si cet ADN est libéré il y aura une présentation par une cellule dendritique, uns instruction des LT auto-réactif, une genèse d’auto-Ac contre la molécule ADN et la genèse du lupus.

Exemple, très connu chez l’animal, l’infection au virus coxsackie CB3: Il est connu pour infecter les myocytes car ces cellules expriment le récepteur. Le système immunitaire va donc venir l’éliminer mais il reconnaîtra également une protéine libérée en quantité importante par les cellules lésées : la myosine. Or celle-ci n’est pas censée être vue par le système immunitaire car contenue dans les myocytes. Il y a donc la genèse de ce qu’on appelle des myosin specific T cell, soit de type Th1, soit de type Th2 qui vont en plus induire des auto-Ac contre la myosine et on aboutit à une MAI. Cela mènera alors à une myocardite avec destruction du tissu cardiaque à cause de ces lymphocytes T auto- réactifs.
V. Dysfonctionnement du système immunitaire :
Même si on a une rupture de la tolérance par les 3 mécanismes énoncés plus haut, on a le rôle de ces cellules que l’on appelle des Ly irégulateurs qui vont inhiber la réponse T par voie de conséquence B également de l’activité des LB. En effet ce sont des cellules capables de bloquer l’activité des LT que ce soit des Ly Th0 (dans sang non activé), Th1, Th2, Th17.A la base si on a une CPA et un LT naïf, ce LT va s’activer et selon le contexte de l’activation, du parasite, de la nature du pathogène ils vont générer :- Ly Th1 : identifiable puisque produisent beaucoup d’Interférons gamma + IL2- Ly Th2 : ceux-là sont essentiellement impliqué dans l’asthme et reconnaissable par l’IL4, IL6 (impliqués
dans la production des LB et des auto-Ac) --> allergies +++
- Ly Th17 : dans les pathologies AI, reconnaissables par l’Interleukine 17.

Quelque soit la nature des différents LT, les LT régulateurs vont inhiber toutes ces réponses. Ils bloquent l’activation des Th1, Th2 et Th17 bloque l’auto-immunité par 2 mécanismes :-le premier est capable de produire des Ly T régulateurs, des cytokines IL4, IL6, TGFβ (Cytokines anti- inflammatoire) qui vont inhiber les LT Th1, Th2, Th17 mais également inhiber l’expression de l’activation des CPA avec une chute des molécules CD40, CD80 (B71), CD86 (B72), IL12 et une augmentation de la production d’IL10. Ces facteurs vont concourir à une inhibition de la capacité des CPA à activer les LT. Ca mécanisme à distance.-le second est un mécanisme par contact direct où le LT régulateurs entrent en contact avec une CPA etbloque son processus d’activation. diminution expression CD80, CD40, CD46 ->
phénomène d’anergie (disparition allergie)
Donc soit il le fait à distance en produisant des facteurs solubles ou soit il le fait directement en venant au contact avec la CPA et inhiber sn activation.
VI. Autres facteurs favorisants l’auto-immunitéCe sont des facteurs que l’on a du mal à étudier car il faudrait passer au modèle animal pour les étudier. Hors, ces modèles n’ont pas la même complexité que chez l’homme. On n’a pas de preuves expérimentales pour confirmer ces facteurs chez l’homme.
- L’âge. Il va jouer un rôle important. Les maladies auto-immunes surviennent chez le jeune adulte, mais la fréquence des auto-ACs chez le sujet sain augmente avec l’âge : développement d’une auto-immunité naturelle.
- Le sexe : prédominance féminine (rôle hormonal notamment les œstrogènes). Fréquence 2 à 10 fois plusimportante chez la femme, en fonction de la MAIBien que l’on ait beaucoup de mal à savoir exactement pourquoi, il y a une prédominance des maladies auto-immunes chez la femme. On suppose que c’est dû aux hormones car l’œstradiol favorise l’auto- immunité, mais on ne sait pas par quel mécanisme.
- Facteurs génétiques : les jumeaux homozygotes expriment la même maladie AI. Lien avec le typage HLA :certains typages HLA particuliers (HLA-B27, HLA-BR1) sont souvent associés à des MAI. Molécules HLA = présentation des antigènes et auto-antigènes.
- Association de maladies auto-immunes : Un même sujet qui aura tendance à accumuler plusieurs MAI, avec une tendance à faire encore d'autres MAI: ce sujet a un dysfonctionnement général du système

immunitaire (SI) ; avec notamment des mécanismes de tolérance sévèrement perturbés.- Infections virales et bactériennes et parasitaires : activation du SI et accumulation de débris (auto- antigènes). Exemples de la sclérose en plaque, du Guillain-Barré…Association entre maladies infectieuses et maladies auto-immunes : Des bactéries sont capables de tuer nos cellules, de libérer ces antigènes du soi, qui vont être reconnus par notre SI comme pas normales.
- Certains traitements par des médicaments
Mr Gasque s’est arrêté ici dans ce ronéo. On vous laisse la suite
VII. Mécanismes d'action des auto-anticorpsLes auto-AC peuvent agir par différents mécanismes.
A. Modulation de l’activité des récepteurs
Plusieurs scénarios sont possibles : blocage ou stimulation.
· Blocage fonctionnel : → Diabète I : l’auto-Ac bloque le récepteur à insuline. L'insuline ne pourra plus se fixer sur son récepteur et ne pourra plus induire le phénomène de capture du glucose.
· Myasthénie : l’auto-anticorps bloque le récepteur nicotinique de l’acétylcholine situé au niveau des muscles. L'acétylcholine ne peut plus agir, la conduction nerveuse ne se fait plus et peut aboutir à un handicap.
· Stimulation fonctionnelle : exemple de l'auto-immunité de Basedow : l’auto-Ac se fixe sur le récepteur à TSH au niveau de la thyroïde et le stimule en permanence (agoniste du rcpt) : surproduction d’hormones thyroïdiennes.
A gauche l'action classique de la TSH libérée par l'hypophyse, avec stimulation de la production des hormones thyroïdiennes.
A droite l'auto-Ac se fixe au récepteur, et par sa nature bivalente engage 2 récepteurs à TSH et provoque une surproduction d'hormones thyroïdiennes avec toutes les conséquences que cela engendre.
B. Cytotoxicité
directe : destruction de la cellule médiée :- par le complément : la voie classique du complément est engagéeExemple de l’anémie hémolytique auto-immune (AHAI) : le complément se fixe sur le GR grâce à l’auto- AC, il est activé par voie classique, aboutissant à la formation du complexe d’attaque membranaire (CAM) qui lyse le GR.- par les cellules K natural killer (fonction importante : ADCC : Antibody-dependent cell cytotoxicity) Exemple de la thyroïdite auto-immune où les AC vont induire une cytotoxicité via les cellules NK (voir plus

indirecte : par opsonisation des anticorps sur la cible.Exemple du purpura thrombopénique auto-immun (PTAI) : les auto-AC fixés sur la plaquette jouent le rôle d’opsonine qui favorise la phagocytose par les macrophages qui vont reconnaître cette plaquette recouverte d'auto-ACs via leurs récepteurs Fc.→ Donc phagocytose extrêmement importante des plaquettes qui donne des purpuras.
FcγR : Récepteur pour les IgG (gamma) et la partie Fc : cristallisable.Récepteur γ : pour les Ig G Récepteur ε : pour les Ig E Récepteur δ : pour les Ig D Récepteur α : pour les Ig A Récepteur µ: pour les Ig M
- Les anticorps sont des opsonines reconnues par les récepteurs Fc (domaine des Acs) des macrophages - Les macrophages sont activés et phagocytent (ingèrent) les cibles- Destruction des GRs (anémie) et plaquettes (thrombocytopénie)
Mécanismes de l’ADCC (TRÈS IMPORTANT)
Voici l'exemple d'une cellule recouverte par des auto-AC . Ce complexe est re connu par les cellules NK qui expriment des récepteurs aux Fc des immunoglobulines qu'on appelle des FcγReceptor 3 (ou CD16). On a donc une reconnaissance via les CD16.Quand on a au moins 2 Ig engageant 2 récepteurs CD16, cela active les cellules NK, qui vont se dégranuler afin de libérer de la perforine, des granzymes, et des Fas ligands qui sont impliqués dans l'apoptose.
Cas particulier des vascularites (Immuncomplexes, complexe antigène anticorps) :
Dans le lupus, l'auto-AC n'est pas directement dirigé contre la cellule mais est dirigé contre de l'ADN. Celaentraîne la formation d'immuncomplexes (= complexes immuns, multimériques, circulants, à gauche du cadre B). L'immuncomplexe n'est pas naturellement dirigé contre la cellule ! Mais il va s'accumuler dans les vaisseaux et donc induit une activation du complément. Le complément ainsi activé s'attaque aux cellules endothéliales. Mais attention : il se dépose de manière non spécifique au contact des cellules endothéliales.
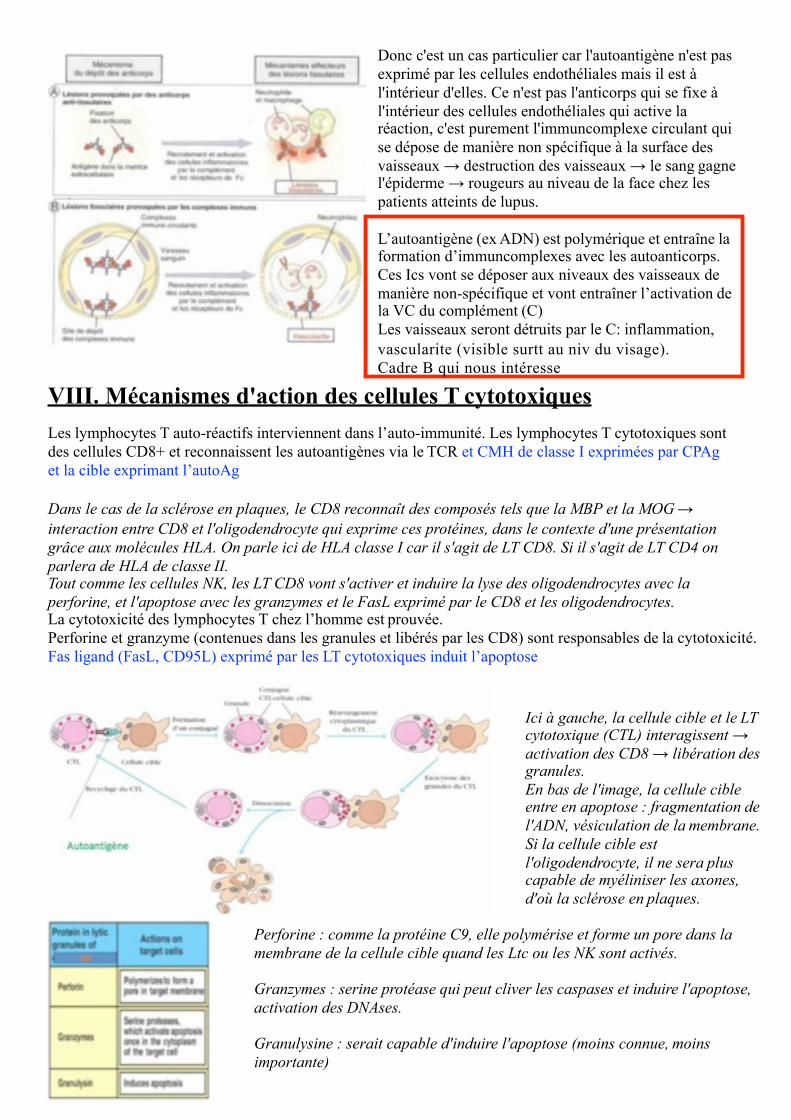
Donc c'est un cas particulier car l'autoantigène n'est pas exprimé par les cellules endothéliales mais il est à l'intérieur d'elles. Ce n'est pas l'anticorps qui se fixe à l'intérieur des cellules endothéliales qui active la réaction, c'est purement l'immuncomplexe circulant qui se dépose de manière non spécifique à la surface des vaisseaux → destruction des vaisseaux → le sang gagne l'épiderme → rougeurs au niveau de la face chez les patients atteints de lupus.
L’autoantigène (ex ADN) est polymérique et entraîne la formation d’immuncomplexes avec les autoanticorps. Ces Ics vont se déposer aux niveaux des vaisseaux de manière non-spécifique et vont entraîner l’activation de la VC du complément (C)Les vaisseaux seront détruits par le C: inflammation,vascularite (visible surtt au niv du visage). Cadre B qui nous intéresse
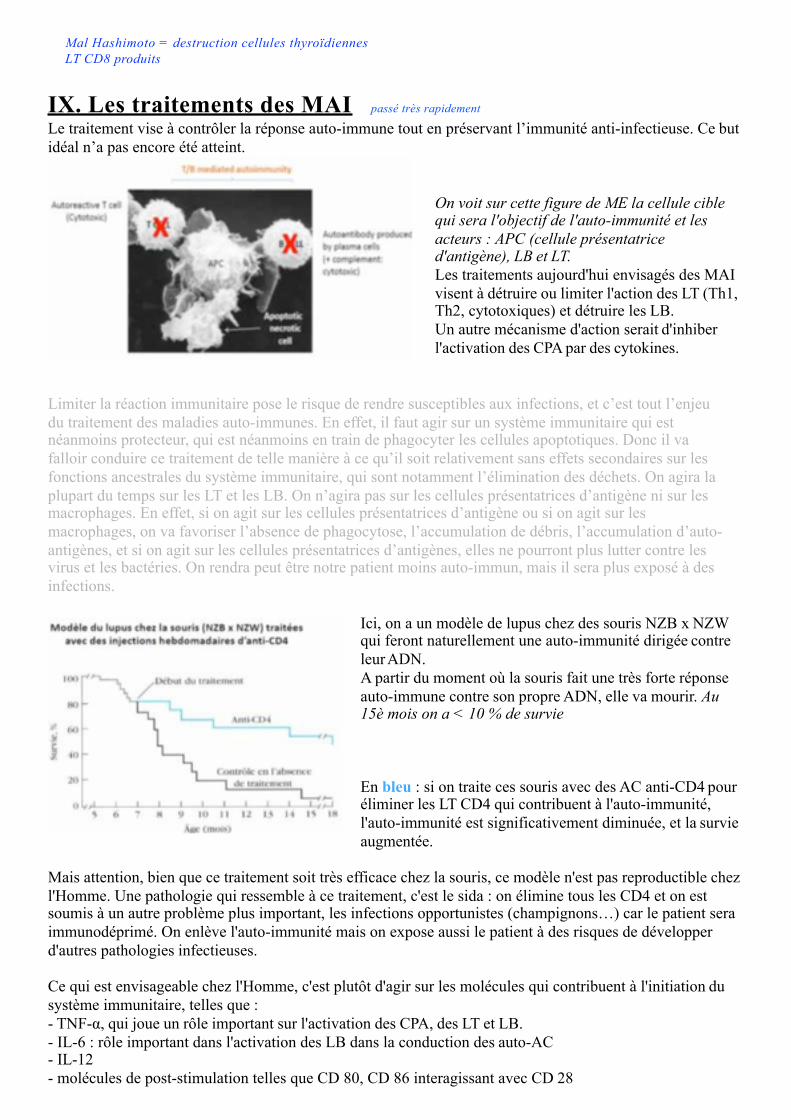
VIII. Mécanismes d'action des cellules T cytotoxiquesLes lymphocytes T auto-réactifs interviennent dans l’auto-immunité. Les lymphocytes T cytotoxiques sont des cellules CD8+ et reconnaissent les autoantigènes via le TCR et CMH de classe I exprimées par CPAg et la cible exprimant l’autoAg
Dans le cas de la sclérose en plaques, le CD8 reconnaît des composés tels que la MBP et la MOG →interaction entre CD8 et l'oligodendrocyte qui exprime ces protéines, dans le contexte d'une présentation grâce aux molécules HLA. On parle ici de HLA classe I car il s'agit de LT CD8. Si il s'agit de LT CD4 on parlera de HLA de classe II.Tout comme les cellules NK, les LT CD8 vont s'activer et induire la lyse des oligodendrocytes avec laperforine, et l'apoptose avec les granzymes et le FasL exprimé par le CD8 et les oligodendrocytes. La cytotoxicité des lymphocytes T chez l’homme est prouvée.Perforine et granzyme (contenues dans les granules et libérés par les CD8) sont responsables de la cytotoxicité. Fas ligand (FasL, CD95L) exprimé par les LT cytotoxiques induit l’apoptose
Ici à gauche, la cellule cible et le LT cytotoxique (CTL) interagissent → activation des CD8 → libération des granules.En bas de l'image, la cellule cible entre en apoptose : fragmentation de l'ADN, vésiculation de la membrane. Si la cellule cible est l'oligodendrocyte, il ne sera plus capable de myéliniser les axones, d'où la sclérose en plaques.
Perforine : comme la protéine C9, elle polymérise et forme un pore dans la membrane de la cellule cible quand les Ltc ou les NK sont activés.
Granzymes : serine protéase qui peut cliver les caspases et induire l'apoptose, activation des DNAses.
Granulysine : serait capable d'induire l'apoptose (moins connue, moinsimportante)
Mal Hashimoto = destruction cellules thyroïdiennes LT CD8 produits
IX. Les traitements des MAI passé très rapidement
Le traitement vise à contrôler la réponse auto-immune tout en préservant l’immunité anti-infectieuse. Ce but idéal n’a pas encore été atteint.
On voit sur cette figure de ME la cellule cible qui sera l'objectif de l'auto-immunité et les acteurs : APC (cellule présentatrice d'antigène), LB et LT.Les traitements aujourd'hui envisagés des MAI visent à détruire ou limiter l'action des LT (Th1, Th2, cytotoxiques) et détruire les LB.Un autre mécanisme d'action serait d'inhiber l'activation des CPA par des cytokines.
Limiter la réaction immunitaire pose le risque de rendre susceptibles aux infections, et c’est tout l’enjeu du traitement des maladies auto-immunes. En effet, il faut agir sur un système immunitaire qui est néanmoins protecteur, qui est néanmoins en train de phagocyter les cellules apoptotiques. Donc il va falloir conduire ce traitement de telle manière à ce qu’il soit relativement sans effets secondaires sur les fonctions ancestrales du système immunitaire, qui sont notamment l’élimination des déchets. On agira la plupart du temps sur les LT et les LB. On n’agira pas sur les cellules présentatrices d’antigène ni sur les macrophages. En effet, si on agit sur les cellules présentatrices d’antigène ou si on agit sur les macrophages, on va favoriser l’absence de phagocytose, l’accumulation de débris, l’accumulation d’auto-antigènes, et si on agit sur les cellules présentatrices d’antigènes, elles ne pourront plus lutter contre les virus et les bactéries. On rendra peut être notre patient moins auto-immun, mais il sera plus exposé à des infections.
Ici, on a un modèle de lupus chez des souris NZB x NZW qui feront naturellement une auto-immunité dirigée contre leur ADN.A partir du moment où la souris fait une très forte réponse auto-immune contre son propre ADN, elle va mourir. Au 15è mois on a < 10 % de survie
En bleu : si on traite ces souris avec des AC anti-CD4 pour éliminer les LT CD4 qui contribuent à l'auto-immunité, l'auto-immunité est significativement diminuée, et la survie augmentée.
Mais attention, bien que ce traitement soit très efficace chez la souris, ce modèle n'est pas reproductible chez l'Homme. Une pathologie qui ressemble à ce traitement, c'est le sida : on élimine tous les CD4 et on est soumis à un autre problème plus important, les infections opportunistes (champignons…) car le patient sera immunodéprimé. On enlève l'auto-immunité mais on expose aussi le patient à des risques de développer d'autres pathologies infectieuses.
Ce qui est envisageable chez l'Homme, c'est plutôt d'agir sur les molécules qui contribuent à l'initiation du système immunitaire, telles que :- TNF-α, qui joue un rôle important sur l'activation des CPA, des LT et LB.- IL-6 : rôle important dans l'activation des LB dans la conduction des auto-AC - IL-12- molécules de post-stimulation telles que CD 80, CD 86 interagissant avec CD 28

L'objectif des traitements des MAI est donc de bloquer ces molécules, grâce à des anticorps, des facteurs solubles, pour contrôler le mécanisme d'action de ces substances ; mais il suffit d'arrêter le traitement pour que le patient retrouve son immunité. C’est de la biothérapie
Ces substances sont représentées dans ce tableau :
TNF-α : les anticorps dirigés contre le TNF-α pourront être utilisés notamment dans le cas de la PAR. Cependant, si cela met le patient à risque de développer des maladies infectieuses, on arrête le traitement et on attend qu'il retrouve une immunité correcte pour lutter contre la maladie.On peut utiliser la fraction soluble du récepteur au TNF, qui va se fixer au TNF et empêcher son action.
TNF-αR-Ig : On fabrique de manière artificielle une molécule qui représente uniquement la forme soluble du TNF-αR (rcpt) et qui est capable de bloquer le TNF-α. Si on injecte cette molécule à un patient, le R soluble va bloquer le TNF-α. Sauf que ce R sous sa forme soluble n'est pas stable, on va donc le coupler à une Ig pour le stabiliser. En effet, l'Ig a une demi-vie très importante, elle est bivalente et capable de résister aux protéases. On associe donc la fonction d'une Ig ayant une longue demi-vie au TNF-αR : on obtient donc une TNF-αR-Ig = le domaine d'une Ig avec 2 récepteurs TNF-αR.Cette technique marche très bien pour la PAR mais est très coûteuse (faut le produire par des cellules de manière recombinante, vérifier qu’on n’injecte pas du LPS si on a produit ça dans une cellule bactérienne…) donc utilisée que dans les grandes firmes.
IL-1Ra : (forme soluble) Nous le produisons déjà naturellement. « Ra » désigne Récepteur antagoniste. Cette forme soluble du R à l'IL-1 vient se fixer à l'IL-1 et l'empêche d'interagir avec les R fixés à la surface des cellules pour activer ces cellules. Cette technique est utilisée pour la PAR.
Nous verrons plus spécifiquement l'action des intégrines α4, des AC dirigés contre cette intégrine, dans le cas de la sclérose en plaques.
Interféron β : Dans la colonne CIBLE, on a un « ? », car on s'est rendu compte de manière empirique que lorsqu'on traitait des patients pour une infection virale avec de l'interféron β (car son action est une lutte antivirale), si ce patient avait une MAI on voyait que cette auto-immunité était probablement contrôlée par l'interféron β. De manière empirique, on sait aujourd'hui que l'interféron β est un agent capable de réguler le système immunitaire, mais on ne connaît pas vraiment les mécanismes d'action.L’interféron est très utilisé dans la sclérose en plaques sans que l’on sache réellement pourquoi (parce que paradoxalement l’interféron induit l’activation du système immunitaire). L’interféron, en plus de son activité inflammatoire et activatrice du système immunitaire, aurait des activités immunosuppressives. De manière, empirique, on a déterminé que si l’on traite un patient avec de l’interféron-béta, il ira beaucoup mieux, et on diminuera ses phénomènes de rechute. C’est donc une molécule très utilisée aujourd’hui.

Questions/ des élèves / Réponses 2015-2016 :
Comment se comporte le R soluble ?→ Si on imagine que TNFα va stimuler un LT dans sa capacité à proliférer, le R soluble se fixe sur le TNFα, donc celui-ci n'est plus capable d'agir en se fixant sur les R membranaires des LT. Il y a compétition entre le R soluble et le R membranaire, donc plus de transmission du signal entre le TNFα et les LT exprimant le R membranaire car on aura bloqué l'interaction ligand-R membranaire.
Question inaudible sur le même sujet→ Donc il n'évolue plus de manière extracellulaire, il sera toujours bloqué. Mais ça ne veut pas dire que si on arrête le traitement, le TNFα ne sera plus produit. Si on arrête le traitement, on aura plus la forme soluble du R donc le TNFα agira sur la forme membranaire. Le problème de l'auto-immunité, c'est qu'on est obligés d'inhiber le système immunitaire qui a pour fonction de nous protéger de manière générale, donc si on l'inhibe il faut trouver un équilibre bénéfices/risques.
Est-ce que le TNFαR-Ig ne permettrait pas de créer des complexes entre TNF circulant et TNFαR-Ig et ainsi se comporter comme un immuncomplexe induisant une activation du système du complément ; comme dans une vascularite telle que le lupus ?→ La logique voudrait que ça fonctionne comme ça, ça a créé des problèmes dans les premiers développements de la molécule. Donc les biotechnologies ont muté le domaine des Ig responsable de l'activation de la voie classique du complément.
CTLA4-Ig : Cette molécule se fixe sur CD80 et CD86. C'est la même chose, on utilise cette molécule pour bloquer l'interaction de CD80 et CD86 exprimés par les CPA et les LT,

ANNALES
2013 session 1 33- Concernant les maladies auto-immunes A. Elles sont dues à des dysfonctionnements du système immunitaire inné et adaptatif B. Ce sont des maladies systémiques C. Ce sont des maladies chroniques dégénératives D. Les autoanticorps et les cellules T auto-immunes en sont les principaux effecteurs E. Elles ne touchent que le sujet âgé
34- Concernant les mécanismes de rupture de la tolérance immunitaire A. Ils peuvent être induits par certains traitements médicamenteux B. Ils peuvent être secondaires à certaines infections C. Un défaut d’expression des TLRs par les cellules phagocytaires est souvent la cause des maladies
auto-immunes D. La perte de la fonction T suppressive est une des causes de la rupture de tolérance immunitaire E. Une hyperactivité intrinsèque des lymphocytes B auto-réactifs peut en être la cause d’une rupture de
tolérance immunitaire
35- Concernant la sclérose en plaque A. C’est une maladie auto-immune qui touche uniquement le SNC B. C’est une pathologie qui conduit à la perte de motricité C. Elle est due à une réponse auto-immune dirigée spécifiquement contre les protéine du cytosquelette
des neurones D. C’est une maladie caractérisée par des périodes de rechutes liée à une variation dans la nature de
l’autoantigène E. Elle se caractérise par une inflammation du SNC
2014 session 1
26. Concernant les maladies auto-immunes :
A. Elles font intervenir des auto-antigènes.
B. Ce sont des pathologies chroniques.
C. Elles sont toujours spécifiques d’un type cellulaire
D. Elles sont initiées par des modifications post- transcriptionnelles des éléments du soi
E. Aucune des propositions ci-dessus.

27. Concernant la tolérance immunitaire:
A. Elle prévient l’activation de lymphocytes auto-réactifs.
B. Elle implique les molécules co-stimulatrices CD80 et CD86.
C. Elle est entièrement acquise dans les organes lymphoïdes primaires.
D. Elle fait intervenir des cellules dendritiques.
E. Aucune des propositions ci-dessus.
28. La rupture de tolérance immunitaire peut être provoquée :
A. par la reconnaissance d’un auto-antigène modifié.
B. par une réaction croisée entre antigène du soi et antigène exogène.
C. par des autogreffes.
D. par des traitements médicamenteux.
E. Aucune des propositions ci-dessus.
29. Concernant la sclérose en plaques:
A. C’est une maladie auto-immune limitée au système nerveux central.
B. C’est une maladie chronique dont la nature de l’autoantigène peut varier.
C. Elle conduit à la destruction des gaines de myéline.
D. Elle se traduit par l’infiltration de macrophages auto-réactifs.
E. Aucune des propositions ci-dessus.

2015 session 1
54. La citrullination du collagène à l’origine de l’arthrite rhumatoïde :
A. Implique une enzyme qui convertit l’acide aminé aspartate en citrulline.
B. implique une déiminase produite par les neutrophiles.
C. contribue à la production d’autoanticorps qui reconnaissent la forme native du collagène.
D. est un processus physiologique impliqué dans la différentiation des fibroblastes.
E. est un événement pouvant être concomitant à une infection virale.
55. La maladie auto-immune de la sclérose en plaques :
A. est induite exclusivement par des autoanticorps neutralisants.
B. implique la lyse des oligodendrocytes par l’action conjointe des autoanticorps et du complément.
C. implique des autoanticorps dirigés contre la MBL.
D. implique des lymphocytes CD8+ auto-réactifs et l’action cytotoxique de la perforine.
E. est observée uniquement dans l’hémisphère nord.
2015 session 2
27. Concernant les maladies auto-immunes :
A. Elles font intervenir des auto-antigènes.
B. Ce sont des pathologies chroniques.
C. Elles sont plus fréquentes chez le sujet âgé.
D. Elles sont toujours spécifiques d’un type cellulaire
E. Aucune des propositions ci-dessus.

33. Concernant la tolérance immunitaire:
A. Elle prévient l’activation de lymphocytes auto-réactifs.
B. Elle implique les molécules co-stimulatrices CD80 et CD86.
C. Elle est entièrement acquise dans les organes lymphoïdes primaires.
D. Elle fait intervenir des cellules dendritiques.
E. Aucune des propositions ci-dessus.
34. La rupture de tolérance immunitaire peut être provoquée :
A. par la reconnaissance d’un auto-antigène modifié.
B. par une réaction croisée entre antigène du soi et antigène exogène.
C. par des autogreffes.
D. par des traitements médicamenteux.
E. Aucune des propositions ci-dessus.
35. Concernant la sclérose en plaques:
A. C’est une maladie auto-immune limitée au système nerveux périphérique.
B. C’est une maladie chronique dont la nature de l’autoantigène peut varier.
C. Elle conduit à la destruction des gaines de myéline.
D. Elle se traduit par l’infiltration de macrophages auto-réactifs.
E. Aucune des propositions ci-dessus.

2016 session 1-2 (mm question)
59. Concernant la modification des antigènes du soi séquestrés et modifiées à l’origine de l’arthrite rhumatoïde :
A. Elle implique une enzyme qui convertit l’acide aminé arginine en citrulline.
B. Elle implique une déiminase produite par les neutrophiles.
C. Elle contribue à la production d’autoanticorps qui peuvent reconnaitre la forme modifiée (citrullinée) de la vimentine.
D. C’est un processus physiologique.
E. C’est un événement pouvant être concomitant à une infection virale.
2017 : session 1-2 (mm question)
55 - La citrullination de la vimentine à l’origine de l’arthrite rhumatoïde
A. implique une enzyme qui convertit l’AA aspartate en citrulline
B. implique une déiminase produite par les neutrophiles
C. contribue à la production d’autoantiicorps qui reconnaissent la forme native de la vimentine
D. est un processus physiologique impliqué dans la différentiation des fibroblastes
E. est un événement pouvant être concomitant à une infection virale





























