Embed Size (px)
DESCRIPTION
RNA sampler : un nouvel algorithme basé sur l’échantillonnage pour la prédiction de la structure secondaire commune & l’alignement structural. Par : Karima BELKAHLA Zahra FONDOU Amel LOUHIBI Professeur : Abdoulay Banéro DIALLO. BIF 7001. 1.Introduction. - PowerPoint PPT Presentation
Citation preview

RNA samplerRNA sampler: un nouvel algorithme basé sur : un nouvel algorithme basé sur l’échantillonnage pour la prédiction de la l’échantillonnage pour la prédiction de la
structure secondaire commune & l’alignement structure secondaire commune & l’alignement structuralstructural
Par : Karima BELKAHLA
Zahra FONDOU
Amel LOUHIBI
Professeur : Abdoulay Banéro DIALLO
BIF 7001BIF 7001

1.Introduction L’ARN, est composé de ribose, de phosphate,
d'adénine, de cytosine, de guanine et d'uracile. les ARN servent d’intermédiaire dans la synthèse
protéique avec les ARN messagers. Il existe néanmoins d’autres types d’ARN, issus de la
transcription, mais qui ne subissent pas de traduction. Ces ARN sont fonctionnels par eux-mêmes, sans
codercoder pour une protéinepour une protéine. Pour les distinguer des ARN messagers, on les appelle
des ARN non-codants (ARNnc).

Elles sont souvent caractérisées par l’évolution des structures secondaires conservées qui sont critiques à leurs fonctions.
La prédiction des structures secondaires d’ARN, est l’un des grands problèmes challenges de la bioinformatique.
Mfold et RNAfold utilisent la programmation dynamique, pour calculer la structure secondaire d’ARN, avec un minimum d’énergie libre pour une seule séquence.
PKNOTS est une extension de Mfold, pour la prédiction des stucture d’ARN avec noeuds.
Une approche plus fiable consiste à utiliser l'analyse comparative pour prédire les structures secondaires d’ARN consensus connexes à partir de plusieurs séquences.
Une stratégie est d’aligner les séquences fiables en premier, puis établir l’alignement.
RNAalifold utilise la stabilité thermodinamique et la covariation de séquences ensemble pour prédire la structure communes des alignements.
La matrice de poids max (MWM) une approche graphique etait introduite pour prédire les structures secondaires communes autorisant les noeuds.
Une autre stratégie (Sankoff 1985) est de simultanément aligner et répertorier les séquences d’ARN (sa compléxité, non pratique pour plus 2 séquences).
Foldalign et Dynalign rendent l’algo de Sankoff plus pratique pour les séquences courtes mais reste toujours lent.
Une troisième stratégie est de prédire les structures des séquences individuelles séparemment et ensuite aligner les structures (implémentée dans RNAshapes & MARNA).
•comRNA utilse un différent shéma appliqué dans les approches des théories de graphes pour comparer et trouver les brins conservés des séquences multiples, ensuite assemble les blocks de brins conservés pour former les structures consensus ou les noeuds sont autorisés.
•Nous présentons un nouveau algorithme, de prédiction de structures communes dans les séquences multiples non alignées, qui adopte l’idéé d’assemblage de brins de comRNA et combine les probabilités de pairage de bases intraséquenceprobabilités de pairage de bases intraséquence & prob d’alignement de base interséquenceprob d’alignement de base interséquence pour mesurer la conservation de brins.

Emploi procédure itérative, pour l’alignement probabiliste de paire de brins compatibles & met à jour les prob basées sur les structures en brin, jusqu’a convergence.
Extension algo pour séquences multiples, par méthode basée sur cohérence.
L’algo n’a pas de limite pour prédiction des noeuds. Dans tests approfondis sur données de séquences réelles, il est meilleurs que d’autres progs, du point de vue sensibilité & spécificité avec une vitesse raisonable.

2.METHODE

La structure secondaire d’ARN :1) Tiges (stems) : Empilage (stacking) des paires de bases A-U,G-C et G-U2) Boucles (Loops) : régions simples brins.

Principe:La prédiction de la structure secondaire commune d’ARN pour plusieurs séquences peut être simplement de trouver les tiges conservées compatibles entre les séquences.•Les paires de tiges conservées = un bon pairage des bases dans leurs séquences individuelles, mais aussi s’alignent bien entre elles (entre les séquences).•Mesure de la conservation des tiges = En combinant les probabilités de pairages des bases intra séquences et les probabilités d’alignements des bases inter séquences .

L’algorithme RNA sampler:
Entrée : Deux séquences et plus d’ARN
Sortie : Structure secondaire d’ARN commune entre les séquences.
Principalement en deux étapes:
Initialisation
Itération
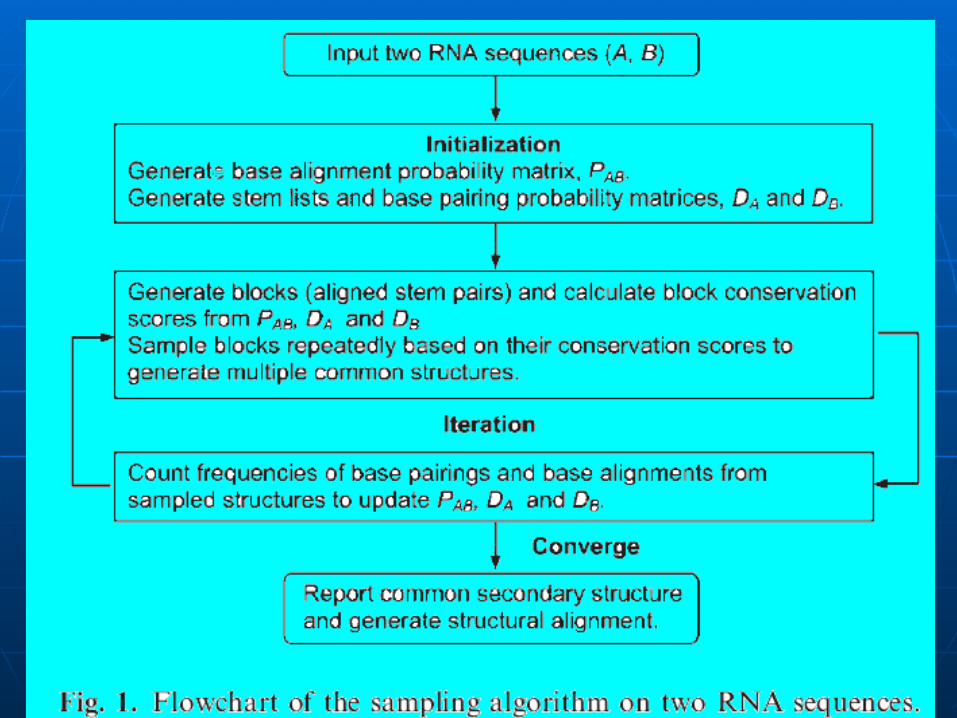

A) Étape d’initialisation:1.Calcule des probabilités d’alignements de bases PAB(ai,bk) ou
PAB(aj,bl) : Les probabilités initiales de l’alignement des bases possibles entre deux séquences
en utilisant une méthode basée sur la fonction de partition.
2. Calcule des probabilités de pairage des bases DA(ai,aj) ou DB(bk,bl):
Les probabilités initiales de pairage des bases pour toutes les paires de bases possibles dans chaque séquence.
RNAfold (une méthode basée sur la fonction de partition) CONTRAfold (posterior probabilities)
3. Générer la liste de toutes les tiges possibles pour chaque séquence :
sl : Une longueur minimum d’une tige (3 par défaut) ou 4 pour les longues séquences ou des séquences avec pseudo-nœud.
Aucune boucle ni bulge n’est permis dans une tige. La combinaison variable de ses tiges peut former une structure d’ARN différente.
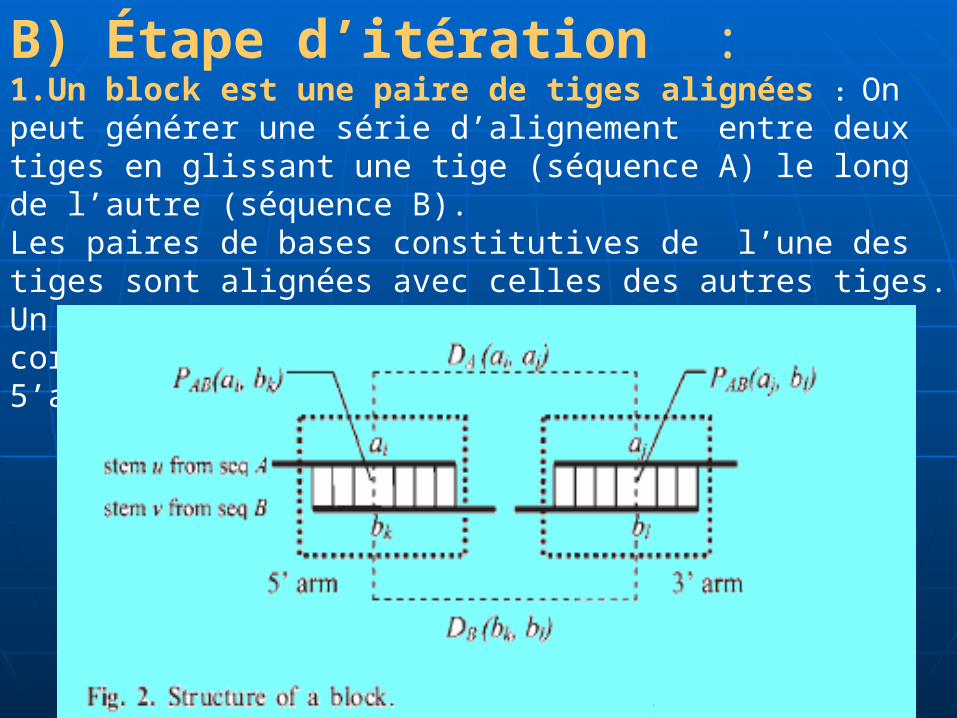
B) Étape d’itération :1.Un block est une paire de tiges alignées : On peut générer une série d’alignement entre deux tiges en glissant une tige (séquence A) le long de l’autre (séquence B).Les paires de bases constitutives de l’une des tiges sont alignées avec celles des autres tiges. Un alignement consiste à deux moitiés correspondantes avec des largeurs égales. The 5’arm et 3’arm
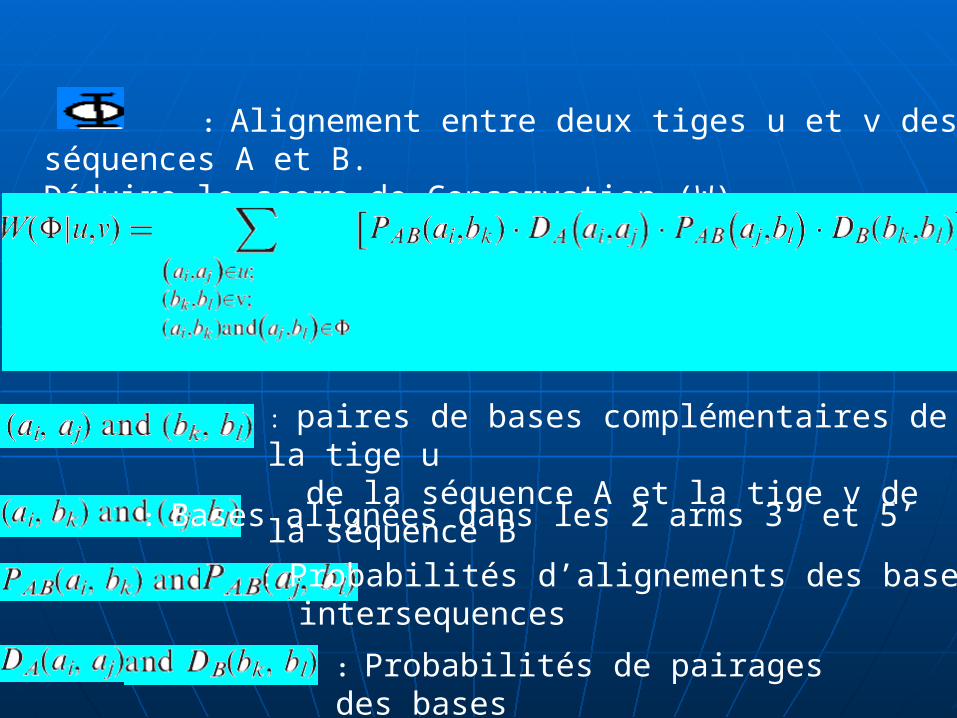
: Alignement entre deux tiges u et v des séquences A et B.Déduire le score de Conservation (W).
: paires de bases complémentaires de la tige u de la séquence A et la tige v de la séquence B
: Bases alignées dans les 2 arms 3’ et 5’
: Probabilités d’alignements des bases intersequences
: Probabilités de pairages des bases intrasequences
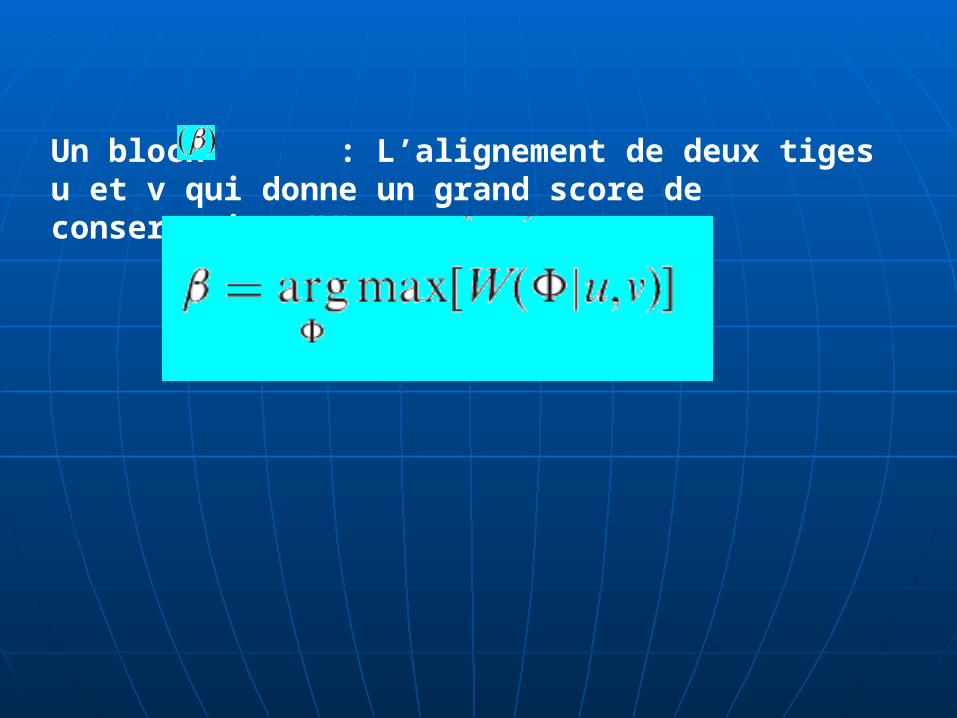
Un block : L’alignement de deux tiges u et v qui donne un grand score de conservation (W)

2. Générer une liste de blocks:Une liste complète de blocks est produite en alignant chaque tige de la séquence A, aux différentes tiges de la séquence B.Les blocks se composant des paires de tiges avec de meilleurs scores de conservations réciproques sont considérés .
Un paramètre d : la distance maximum de décalage entre les positions alignées des deux tiges .But : Réduire la complexité informatique

3.Échantillon de blocks compatibles pour générer la structure commune:Approche d’échantillonnage probabiliste : sélectionner les blocks compatibles basés sur leurs scores de conservation et les assemblés dans une structure commune.La chance pour que le block B soit échantillonner est défini avec la probabilité:
Les blocks avec un haut score de conservation ont plus de chance à être sélectionnés.L’échantillonnage est répété S fois (S est la taille de l’échantillon et S structures communes sont générées dans chaque itération)

4. Mise à jour itératives des probabilités d’alignements et de pairages des bases:Calculer la fréquence d’apparition des paires de bases dans S (structures communes) pour chaque séquence, qui nous donne une nouvelle probabilité de pairage.

Calculer la fréquence d’alignement des bases dans S(structure commune) entre deux séquences, qui nous donne une nouvelle probabilité d’alignement :

Les étapes 2, 3 et 4 sont répétées pour des itérations multiples. Les probabilités de pairage et d’alignements des bases sont misent à jour, elles sont employées pour recalculer les scores de conservation des blocks et de leurs probabilités à être prélever dans l'itération suivante.
les blocks qui sont constitués des tiges conservées obtiennent des scores de conservation intensifiés. Ils ont des chances de plus en plus élevées d'être prélevés, alors que d'autres blocks tendent à avoir leurs scores de conservation atténués. Les structures prélevées tendent à converger dans des ensembles de blocks conservés compatibles.
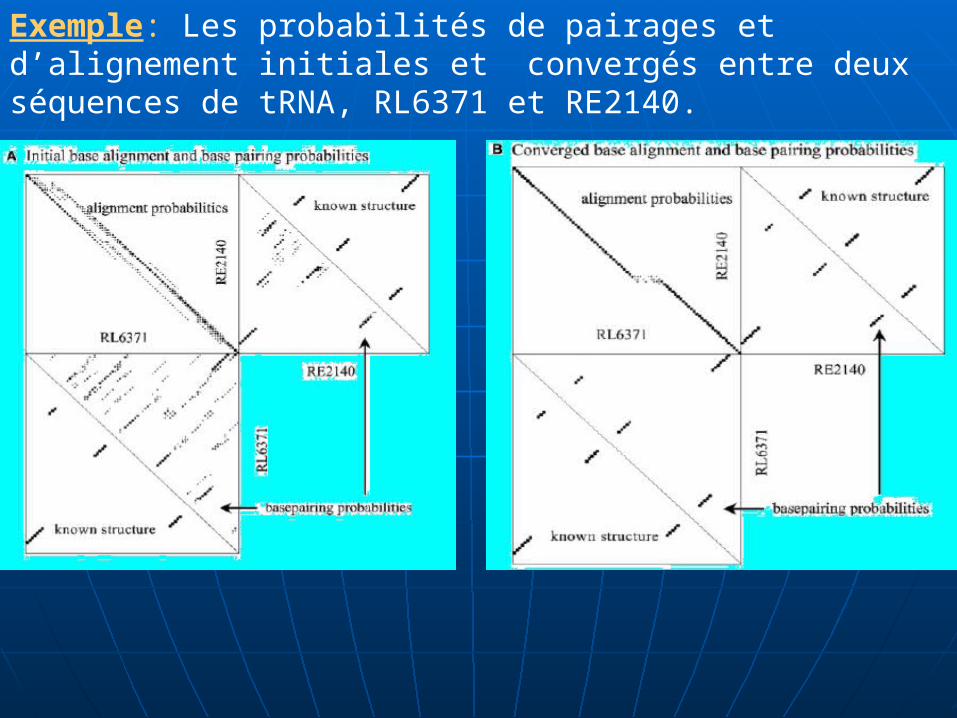
Exemple: Les probabilités de pairages et d’alignement initiales et convergés entre deux séquences de tRNA, RL6371 et RE2140.

C) Structure commune entre deux séquences:Un algorithme vorace est utilisé pour assembler les blocks convergés dans la structure consensus finale.Les blocks avec haut score de conservation sont sélectionnés aucun block ne reste
ClustalW pour aligner les régions simples brins, qui sont assemblées avec les blocks conservés pour générer l’alignement structural final.

D) De deux séquences d’ARN à plusieurs:
Étape d’initialisation (idem)
Étape d’itération :•Échantillon de la structure commune entre toutes les paires de séquences.•Une méthode basée sur la cohérence est appliquée pour les mise à jour des probabilités et le recalcule des scores de conservations.•Dans chaque itération , S structures sont échantillonnés entre chaque paire de séquence.•Chaque séquence est impliquée dans (N-1)S structures échantillons.
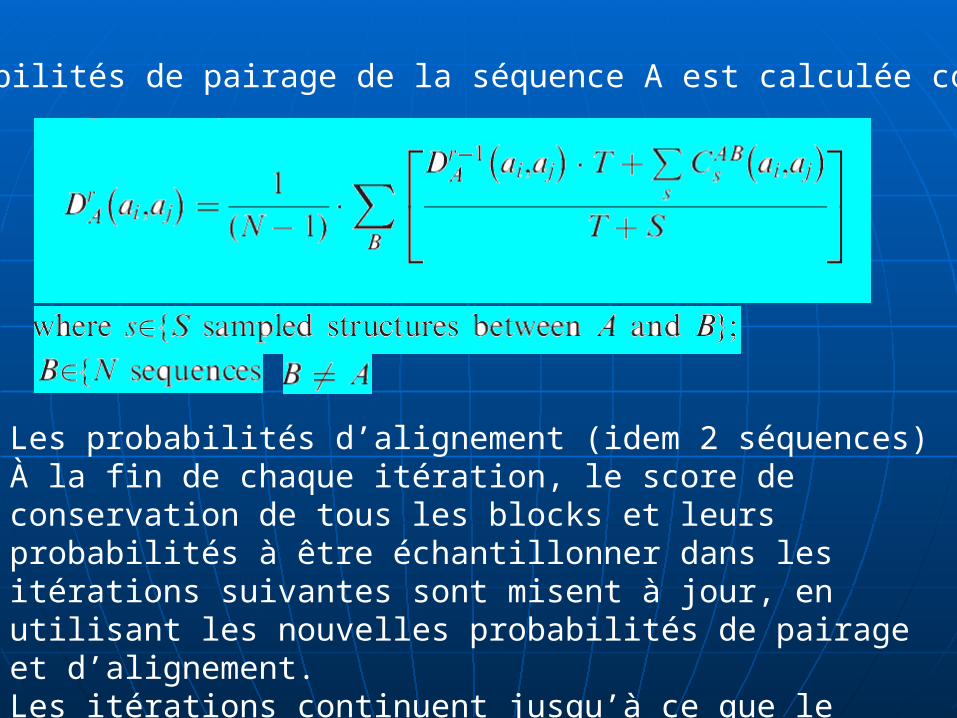
Les probabilités d’alignement (idem 2 séquences)À la fin de chaque itération, le score de conservation de tous les blocks et leurs probabilités à être échantillonner dans les itérations suivantes sont misent à jour, en utilisant les nouvelles probabilités de pairage et d’alignement. Les itérations continuent jusqu’à ce que le nombre des structures échantillonnées entre chaque paire de séquences converges.
Les probabilités de pairage de la séquence A est calculée comme suit:

E. La structure commune des séquences multiplesla structure commune finale partagée par des séquences multiples est rapportée en assemblant des ensembles de tiges conservées compatibles en utilisant un algorithme vorace.Tous les blocks entre les paires de séquences sont rangés, en se basant sur leurs scores finals de conservation.Le block avec un haut score de conservation devient une graine(seed), et tous les blocks qui contiennent une des tiges dans le block graine sont collectés.Une nouvelle tige à partir du block avec un haut score de conservation dans le sous-enssemble est ajoutée à la graine.Le processus se répète aucun block ne reste.ClustalW pour aligner les tiges conservées et les régions simples brins, qui sont assemblées pour donner un alignement structural final.

3. RESULTATS L’algorithme est implémenté en langage C
L’algorithme est testé dans différents ensembles de données,
contenant deux ou plusieurs séquences réelles
Sa performance a été comparée à d’autres méthodes de
prédiction de structures secondaires comme:
- CARNAC - Stemloc
- ARNalifold - FoldalignM

RESULTATS Le coefficient de corrélation (CC) définit par Mathews et
Turner, est utilisé pour évalué la précision de la prédiction
qui est estimée comme la moyenne géométrique de la
sensibilité et la spécificité :
CC = √(SEN . SPE)
0 ≤ cc ≤ 1
SEN : est la fraction de la paire de bases rèelles qui sont
prédits correctement
SPE : est fraction de la paire de bases prédits réellement.

3.1 Prédiction des structures communes
L’algorithme a été testé sur des données réelles
- sur 10 motifs d’ARN régulateur
ou - sur des familles du gène ncRNA
Extraits de la base de données Rfam.
Rfam : base de données de protéines
Y compris ces données:
Cobolamin, gcvT, glmS, purine, REN, sbox THI,
ARNt, U1 et yyb P-ykoY

Prédiction des structures communes
Ces ensembles de données ont été utilisées dans d’autres
études

Prédiction des structures communes
Pour la famille d’ARN de la B.D. Rfam, l’alignement a été
corrigé manuellement et aussi les structures communes
correspondantes qui sont utilisées pour déterminer exactement
les paires de base dans des séquences individuelles

Prédiction des structures communes
1. L’algorithme d’échantillonnage d’ARN est testé sur
un ensemble de deux séquences, générées à partir
de toutes les séquences uniques dans l’alignement
de chaque famille d’ARN.
Le test effectué consistait à calculer (cc)
La moyenne calculée cc = 0.60 dans un interval
[0.42..0.82]

Prédiction des structures communes
Cette valeur est très proche de la moyenne de la
méthode Dynalign cc=0.61, qui était la plus
performante par rapport à d’autres méthodes
La performance des programmes Stemloc et
RNAalifold est comparable a celle de l’algorithme
par échantillonnage d’ARN dans certains familles et
mauvaise dans d’autres.
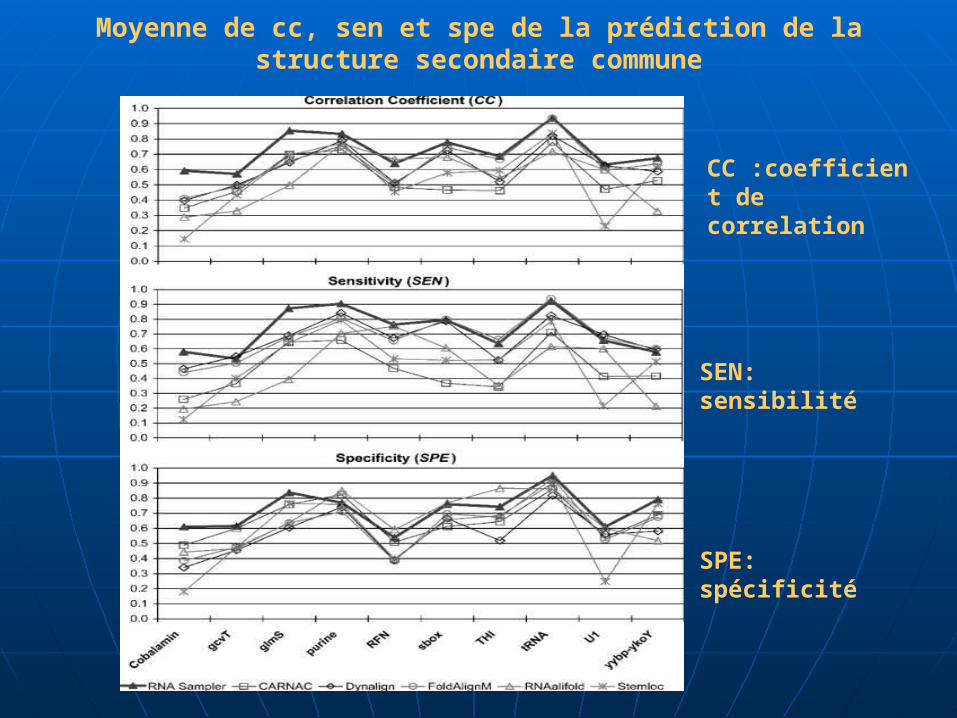
Moyenne de cc, sen et spe de la prédiction de la structure secondaire commune
CC :coefficient de correlation
SEN: sensibilité
SPE: spécificité

Graphe
Comparaison entre les différentes méthodes en calculant la
moyenne de CC, SEN et SPE sur des ensembles de 10 familles
d’ARN de séquences-multiples

Prédiction des structures communes
L’algorithme d’échantillonnage
Dynalign
Stemloc
RNAalifold
Toutes ces méthodes donnent un (cc) meilleur que celui de la
méthode CARNAC

Prédiction des structures communes
Entre tous les programmes testés la méthode
d’échantillonnage donne une meilleure performance et une
vitesse plus rapide

Prédiction des structures communes
2. l’algorithme d’échantillonnage et les autres méthodes ont été
testés sur un ensemble de 10 familles d’ARN à multiple
séquences
Pour chaque famille d’ARN des ensembles de 100
séquences sont générées
Cinq séquences uniques sont sélectionnées aléatoirement à
partir de la BD Rfam de la graine d’alignement

Prédiction des structures communes
La précision de la prédiction par échantionnage d’ARN est
nettement améliorée sur plusieurs rapport par rapport à deux
séquences
En général, la méthode de prédiction par échantillonnage
d’ARN, donne une meilleure prédiction dans les dix familles
d’ARN avec une moyenne de :
CC = 0.72, SEN = 0.73, SPE = 0.72

Prédiction des structures communes
Cette méthode est la plus performante parmi toutes
les autres méthodes testées.
CARNAC : prédit seulement les structures
partiellement correctes, ce qui conduit à une faible
sensibilité et relativement à une bonne spécificité

Prédiction des structures communes
Stemloc: sa faible performance est donc due partiellement à la
faible valeur de (-nf) utilisé comme option dans le test
(-nf=100)
Les valeurs supérieures de (-nf) exigent plus d’espace
mémoire, ce qui rendu l’exécution plus lente et souvent la
mémoire ‘crasch ’

Prédiction des structures communes
RNAalifold : exige un alignement fiable pour une
bonne prédiction
(clustalw est utilisé pour l’alignement)
Ce programme a une bonne performance dans
l’ensemble de famille RFN qui a une identification
de séquence très élevée, et une faible performance
dans une faible identification de séquences.

Prédiction des structures communes
La méthode de prédiction par échantillonnage, a une bonne
performance dans la famille RFN et donne de bons résultats

Prédiction des structures communes
Dynalign donne la même ou légèrement une meilleure
sensibilité que la méthode de prédiction par échantillonnage
dans les familles de :
qcvT, sbox, yybp-ykoY et U1
Mais la méthode de prédiction montre une meilleure
sensibilité et en générale une performance dans d’autres
familles
Elle est plus rapide que Dynalign

Prédiction des structures communes
FoldalignM: a la 2ème meilleure performance après la méthode
de prédiction, et montre une aussi bonne prédiction sur les
familles:
Sbox, THI, RNAt et yybp-ykoY
Une faible sensibilité dans les autres familles

Prédiction des structures communes
La prédiction a été évaluée au niveau de la tige quand
la sensibilité (SEN) est la fraction des véritables tiges
qui se chevauchent avec les tiges prédits, et la
spécificité (SPE) est la fraction des tiges prédits qui
se chevauchent avec les tiges réelles .

Prédiction des structures communes
Avec ces mesures la moyenne CC, SEN et SPE la prédiction
échantillonnage d’ARN des dix familles augmentent de 0.72,
0.73, 0.72 au niveau de la paire de base jusqu’à 0.77, 0.80,
0.76 respectivement.

Prédiction des structures communes
Les autres méthodes sont presque similaires au niveau de la
paire de base.

3.2 Prédiction d’alignements structuraux
Emploi et extension de méthode proposée dans étude ultérieure de l’alignement structural de deux séquences pour évaluer la perfomance de RNA Sampler sur un alignement structural de séquence multiple à travers un large rang de séquences identités.

Utiliser score des sommes des paires (SPS) la fraction des paires de bases alignées dans l’alignement référence, correctement aligné dans la prédiction d’alignement pour mesurer l’exactitude des alignements structuraux à un niveau de paires de bases.
L’index de conservation structural(SCI) calculé par RNAz, utilisé pour mesurer l’info de la structure conservée, contenue dans l’alignement prédit.(0 & 1)

Pour chacunes des 10 familles d’ARN, Génération aléatoire des ensembles de séquences multiples qui couvrent éventuellement un large éventail de paires de séquences identités significatives (entre 40% & 80%).
<40% famille ARNt. >80% famille de U1.

Utiliser l’alignement résultat de Rfarm, et les structures communes comme références pour comparer les alignements et les prédictions de structures (RNA Sampler et FoldalignM), ainsi que les résultats de RNAalifold(entrée:align de ClustalW)
Déterminer CC, SPS et SCI comme fonctions de la paire de séquences identités principales pour chaque famille d’ARN.
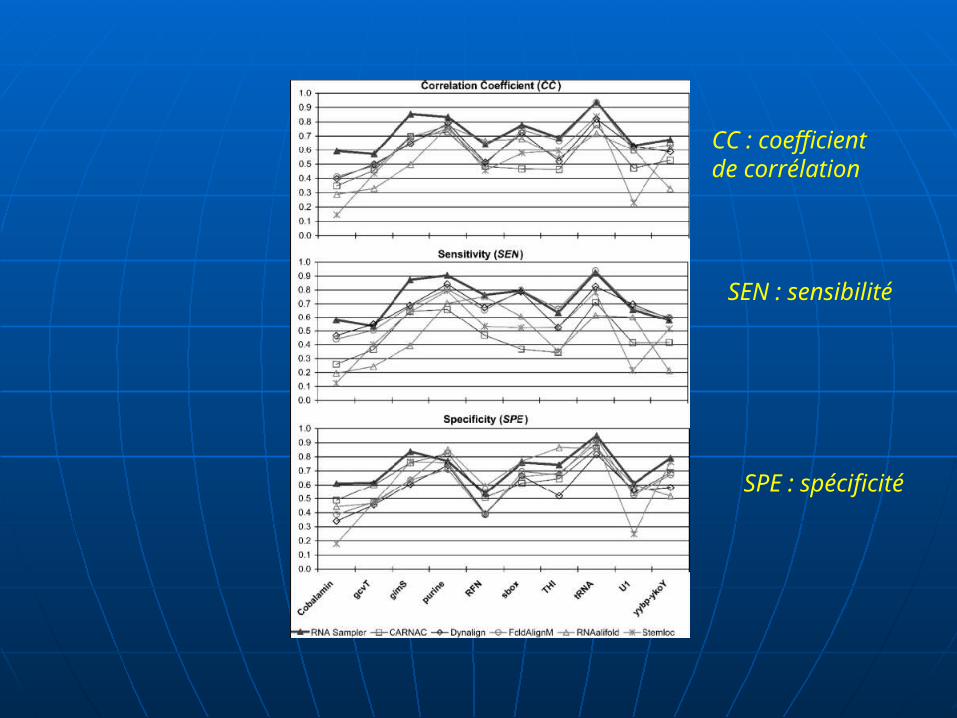
CC : coefficient de corrélation
SEN : sensibilité
SPE : spécificité

Déterminer CC, SPS et SCI comme fonctions de la principale identité de paires de séquences, pour chaque famille d’ARN.
Le résultat de CC, montre que ARNsampler donne en général une meilleure prédiction de structure que FoldalignM par 10% à 20%.
D’autres part, la séquence identité entière varie exeptionnellement au dessus de 40% sur l’ensemble de données de ARNt.

ARNsampler donne nettement de meilleures prédictions que RNAalifold dans l'ensemble de l'identité (au dessous de 80%), spécialement dans les régions à basse identité.
et donne de bonnes prédictions sur RNAalifold comme séquence fixe des identités au-dessus de 80%, les ensembles U1.

Les courbes SPS de RNAsampler, FoldalignM et ClustalW montrent une tendance similaire: les scores SPS ont tendance à diminuer avec la baisse des séquences identités.

Bien que les alignements structurels par RNAsampler et FoldalignM ne correspond pas tout à la référence Rfam alignements, la SCI donne de hauts scores proches de ceux des alignements de référence dans l'ensemble de l'identité de gamme, ce qui indique que leurs alignements ont en effet pris la structure de l'information conservée.
RNAsampler fait le mieux que FoldalignM, avec des scores SCI plus élevés dans les 40% -70% identité gamme.

3.3 Prédiction des structures de Noeuds
RNA sampler peut prédire les structures à noeuds, si l’utilisateur autorise la formation de croisements de blocks de brins.
Paramétre X : nbre max de croisements autorisés dans structure.
RNAsampler testé sur des strctures de noeuds connues (séquences chefs de mRNA de 10 bactéries opéron α et 8 bactéries S15)
RNAsampler ne prédit correctement que 46% des paires connues sur l’ensemble d’opérons α.
Mais 2 brins formant le noyau de la structure de noeuds etaient correctement prédits.
Les séquences chefs S15 peuvent former 2 structures alternatives:
1. Une contient des noeuds.2. L’autre est une structure de boucles de
tiges sans noeuds.
Au niveau des paires de bases, RNAsampler a donné une sensibilité de 0.74% et 0.76% pour les 2 alternatives de structures.
Il manquait qlques paires de base pour les longues tiges puisque les gonflements (ernies) et les boucles n’etaient pas autorisés dans les tiges.
Au niveau des tiges, RNAsampler prédit correctement ttes les tiges complétes des structures à noeuds est aussi dans une alternative de structure de boucle de tige quand les croisement sont autorisés.

Les performances de RNAsampler sur ces ensembles de données sont moins bons que comRNA, mais meilleurs que les autres prgs testés dans l’étude de comRNA.
Le temps d’exécution de RNAsampler sur ces données est de 4 à 10mn.

3.4 complexité de l’algorithme
La vitesse de l’algorithme est limité par l’étape d’itération.
Pour chaque itération, il prend O(m.n)pour générer m.n blocks possibles en comparant tous les m brins de la séquence A, et tous les n brins de la séquence B.

complexité de l’algorithme
Le paramètre d est introduit, est le maximum de la
distance autorisée dans le block entre deux tiges, et la
contrainte de recueillir uniquement les tiges paires
avec un meilleur score réciproque de conservation
Réduit le nombre total des blocks des échantillons de
m.n à n (m≤n).

complexité de l’algorithme
La procédure d’échantillonnage prend O(m) temps
pour éliminer les blocks en conflit en comparant les
blocks avec celui choisi.
Cependant la complexité du temps pour prédire une
structure commune entre deux séquences est de
l’ordre de O(m².r.S)
(r:nbre total d’itération, S:taille de échantillon ie nbre
de structure échantillonné pour chaque itération).

complexité de l’algorithme
Pour 1 ensemble de séq multiples, RNA sampler échantillonne les structures communes entre ttes les paires de séq et la compléxité du temps devient O(m2.r.S.N2) (N:nbre de séquences)
Sur une données de 5 séq(long de 70 à 200nt), RNAsampler prend 6-180s pour compléter la prédiction de structure.

complexité de l’algorithme
CARNAC, RNAalifold et Stemloc ont un temps d’execution meilleur ou similaire à RNAsampler, mais ce dernier est plus exacte.
RNAsampler s’exécute plus rapidement que Dynalign et FoldalignM et donne les prédictions les plus éxactes.





























