Embed Size (px)
Citation preview

Cours BMG1- L2 UPMC
Partie Génétique
Frédéric [email protected]
18h de cours (12 fois 90 minutes)
Organisation des génomes et structure des gènes (3 cours)Variabilité des génomes- Polymorphisme génétique (3 cours)
Cartographies génétiques et physiques (6 cours)

Cartographies génétiques et localisation des gènes
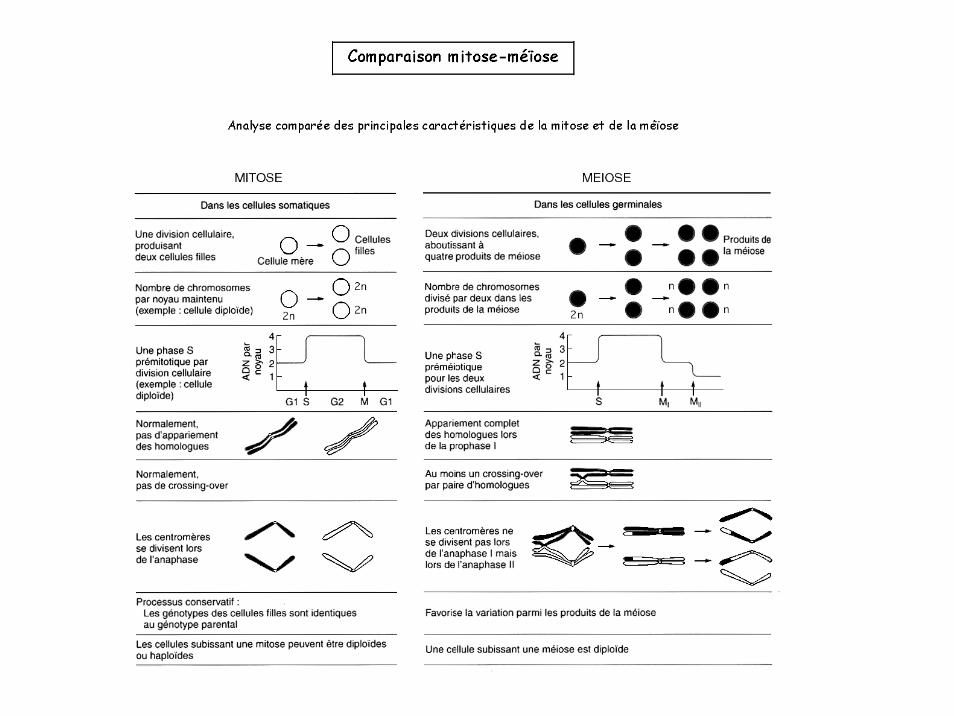
I. Transmissions des caractères et brassage génétique.1- Cycles de vie2- Méïose et crossing over3- Ségrégation des gènes
II. Cartographie génétique.
III. Cartographie et Recherche de gènes candidats chez l’Homme.
IV. Comparaison cartes génétiques/ cartes physiques.
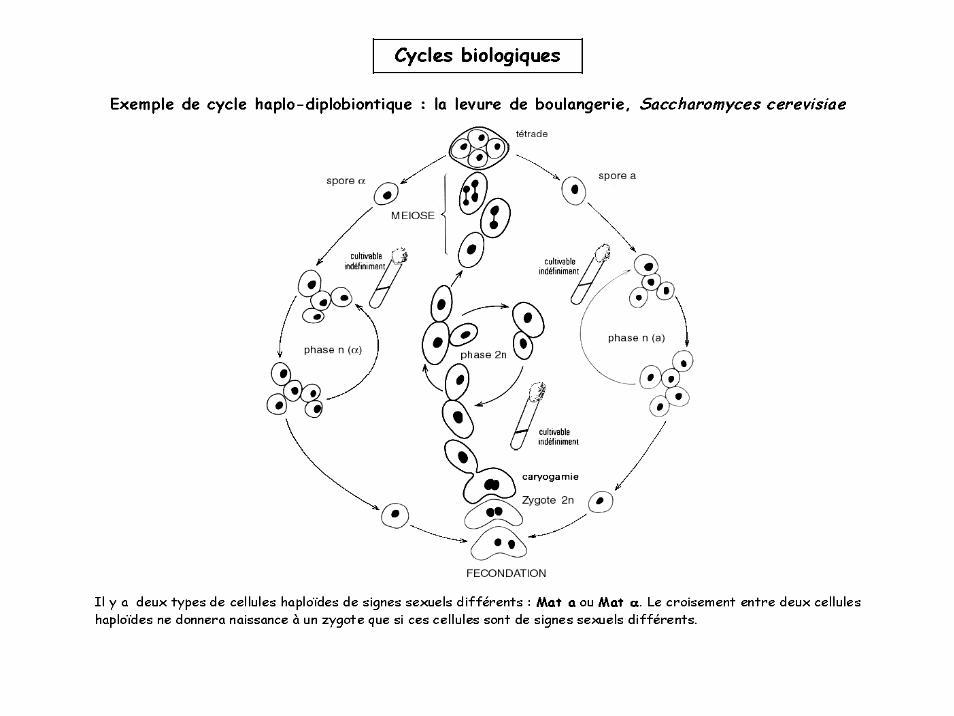
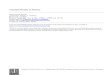
Cycles biologiques
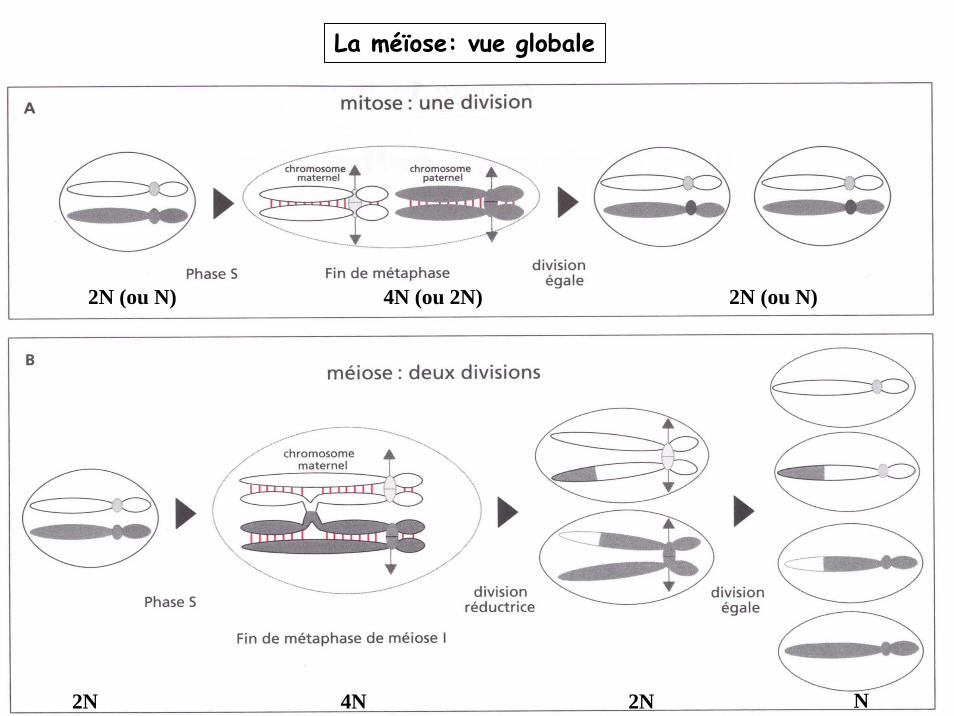
La méïose: vue globale
2N (ou N) 4N (ou 2N) 2N (ou N)
2N 4N 2N N
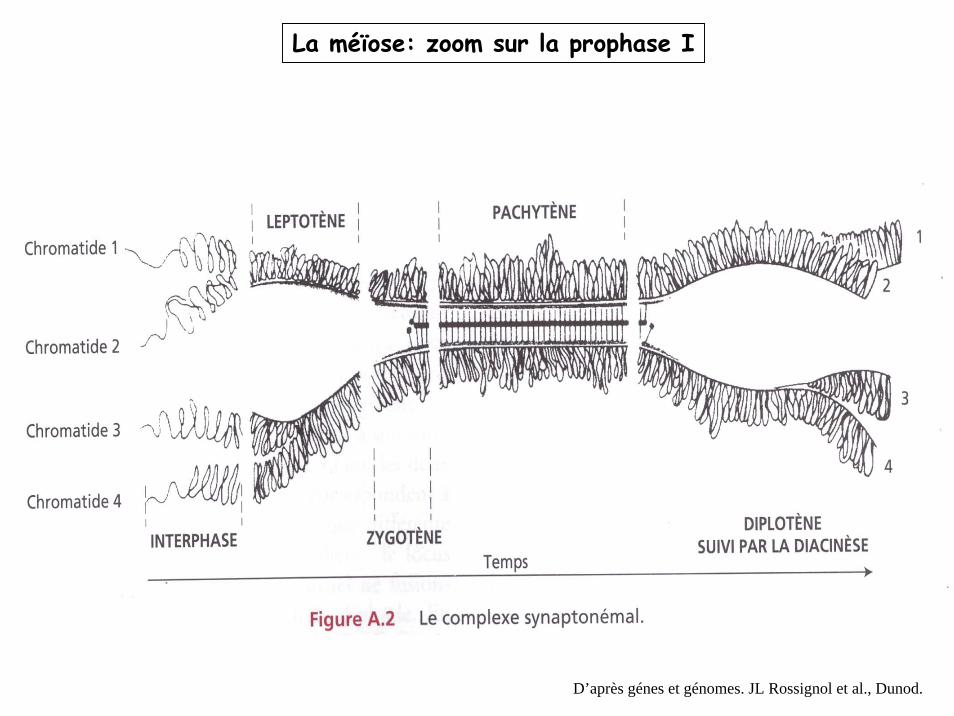
La méïose: zoom sur la prophase I
D’après génes et génomes. JL Rossignol et al., Dunod.
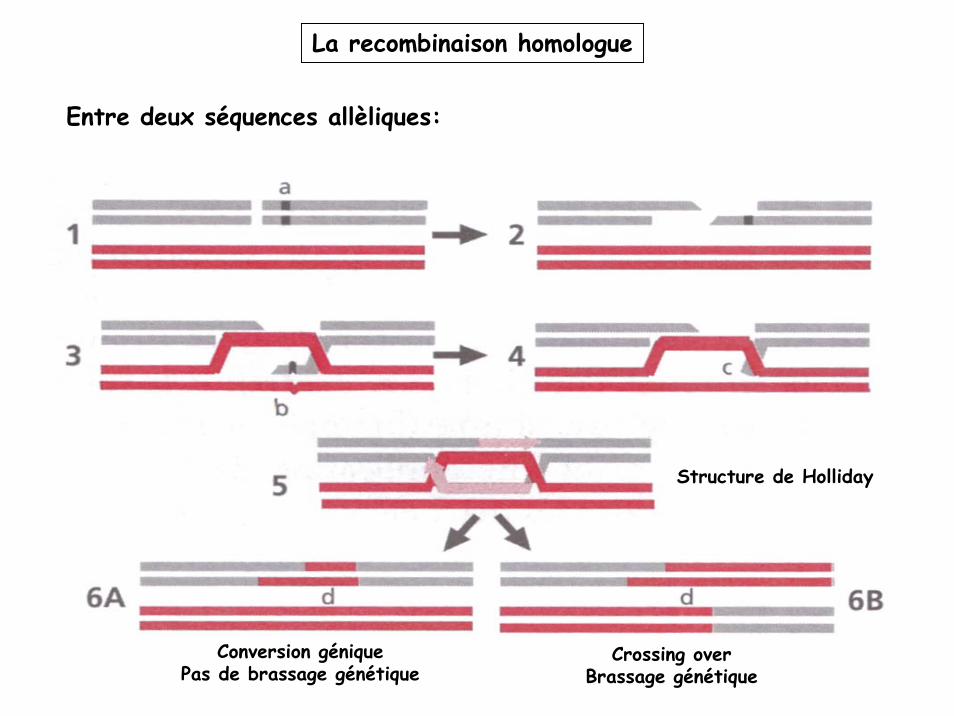
La recombinaison homologue
Entre deux séquences allèliques:
Structure de Holliday
Conversion géniquePas de brassage génétique
Crossing overBrassage génétique
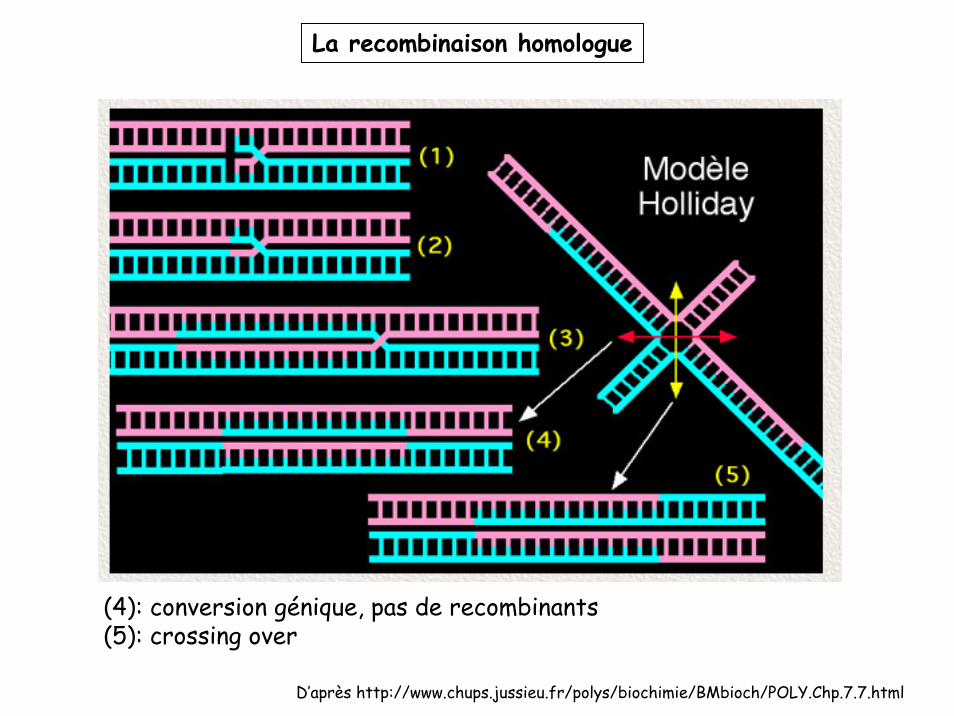
La recombinaison homologue
D’après http://www.chups.jussieu.fr/polys/biochimie/BMbioch/POLY.Chp.7.7.html
(4): conversion génique, pas de recombinants(5): crossing over
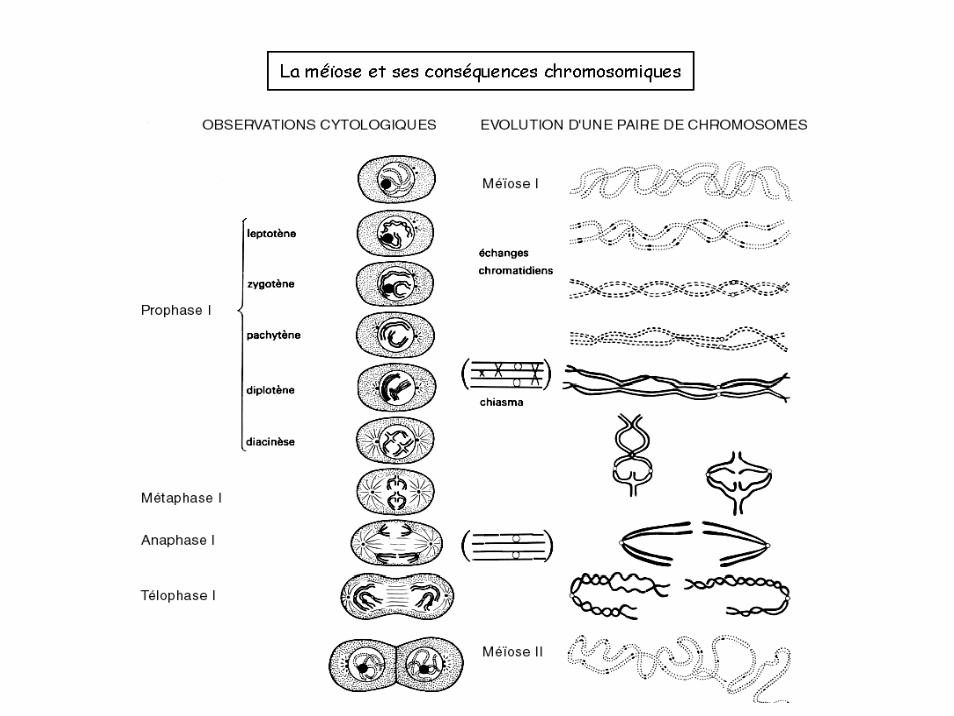
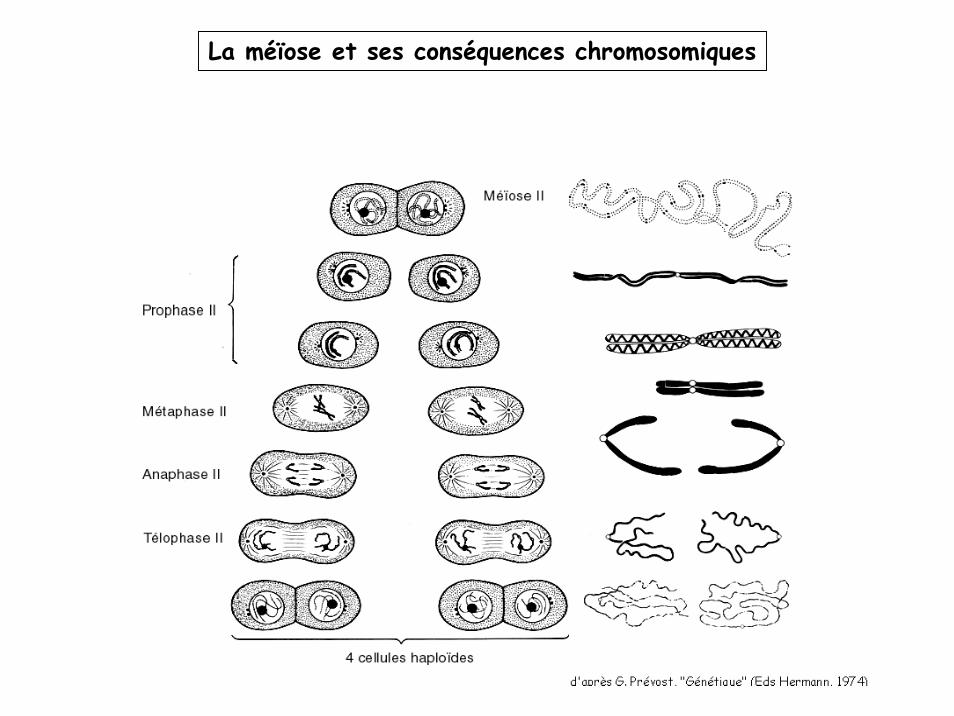
La méïose et ses conséquences chromosomiques
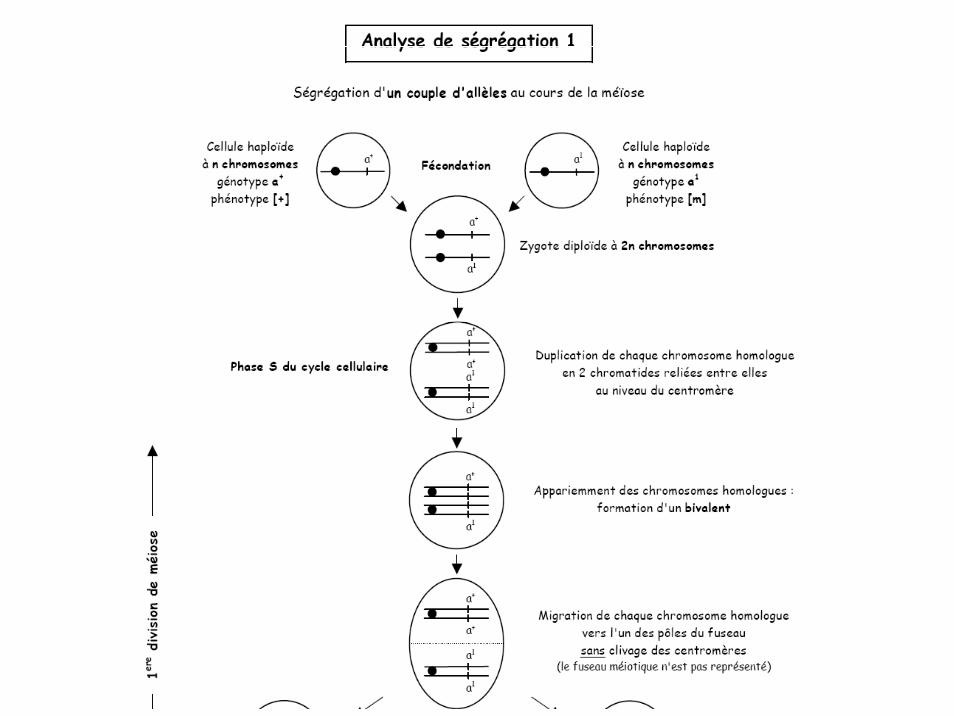
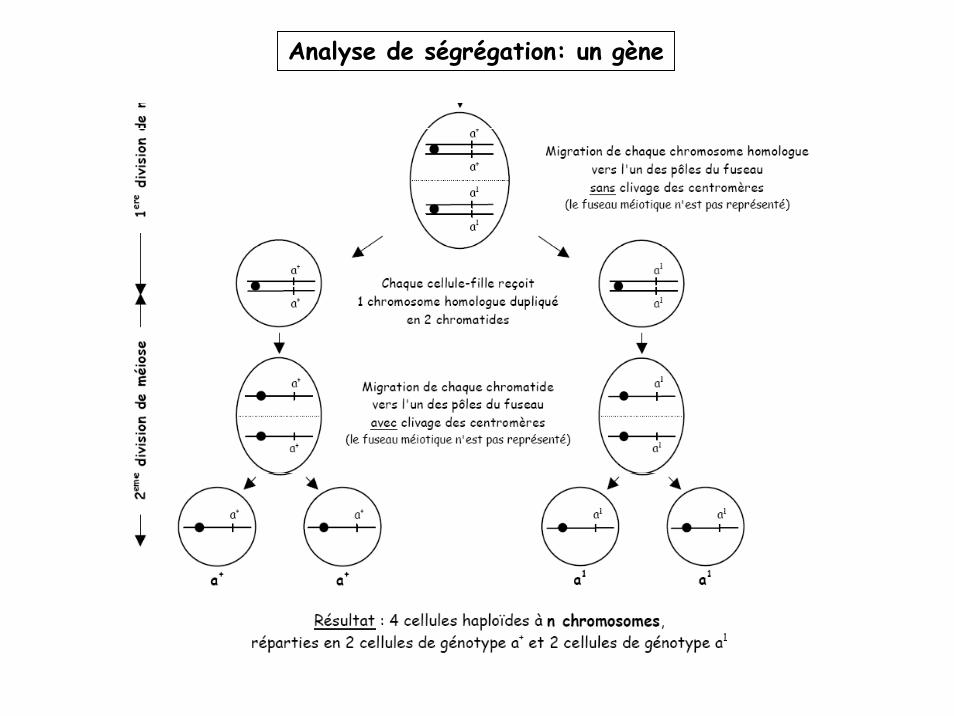
Analyse de ségrégation: un gène
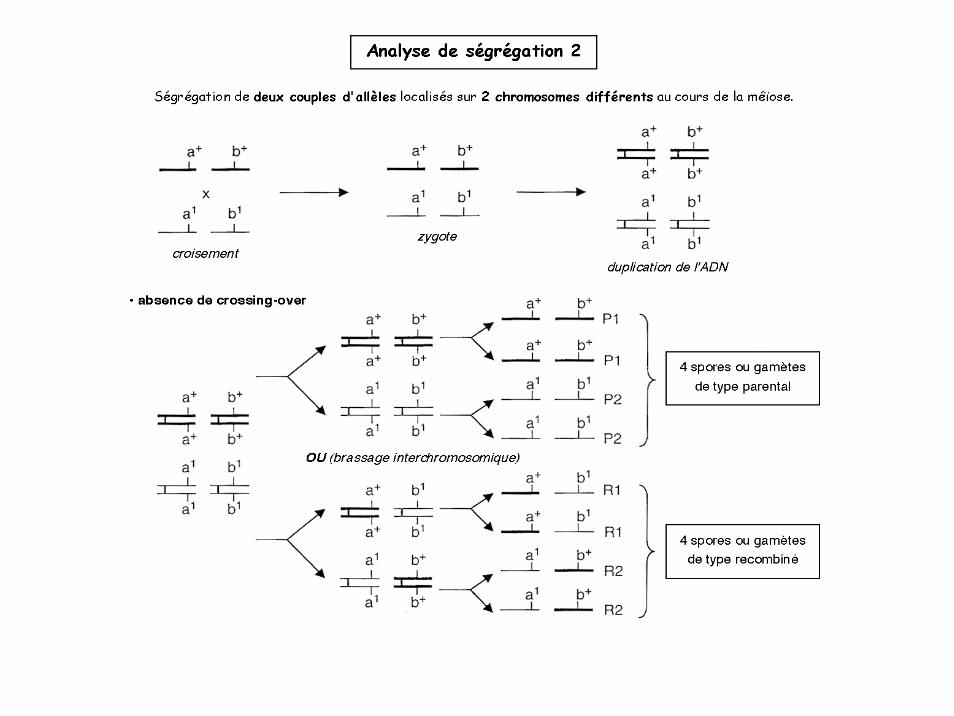
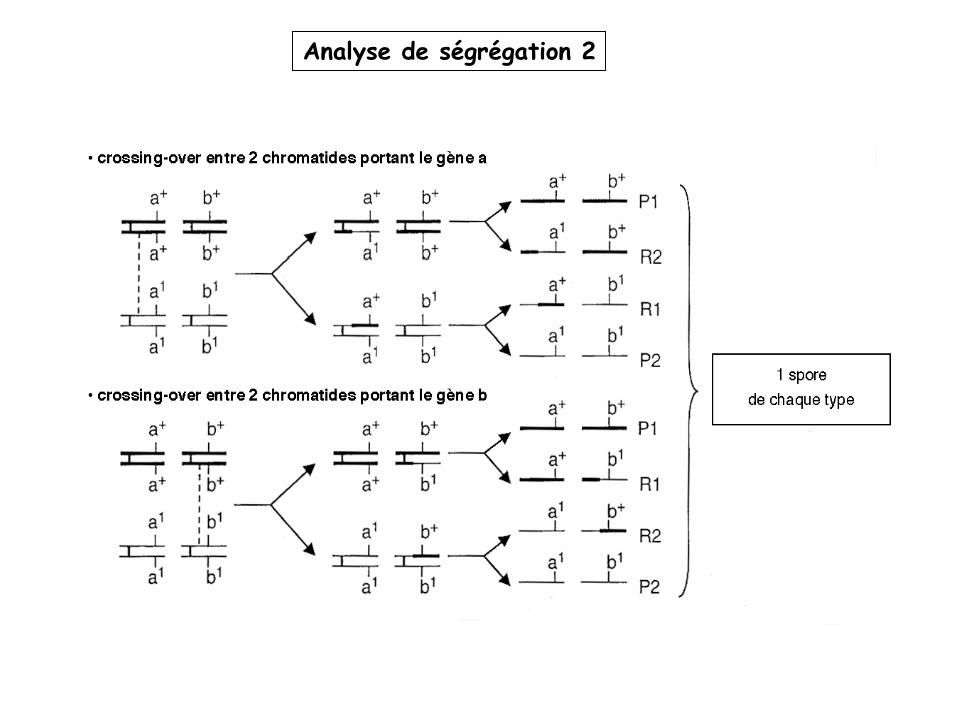
Analyse de ségrégation 2
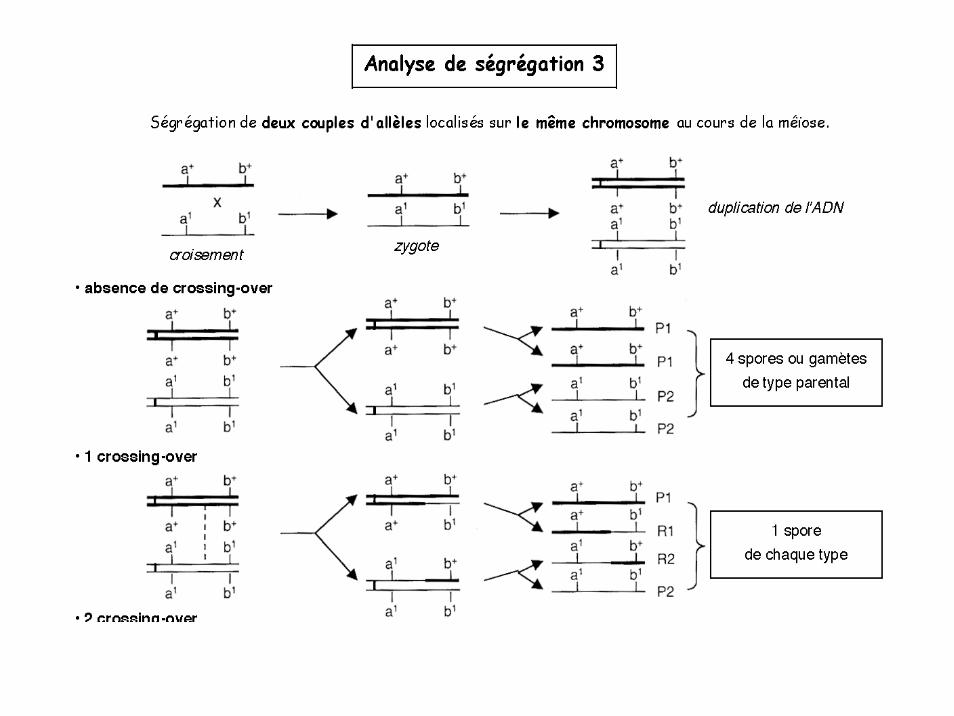
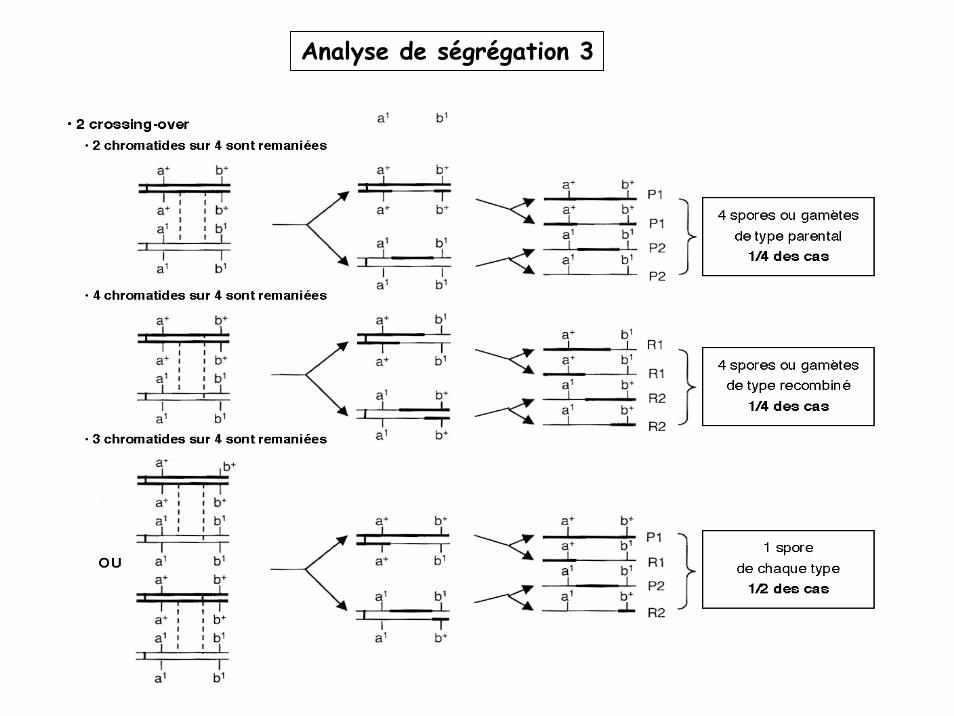
Analyse de ségrégation 3
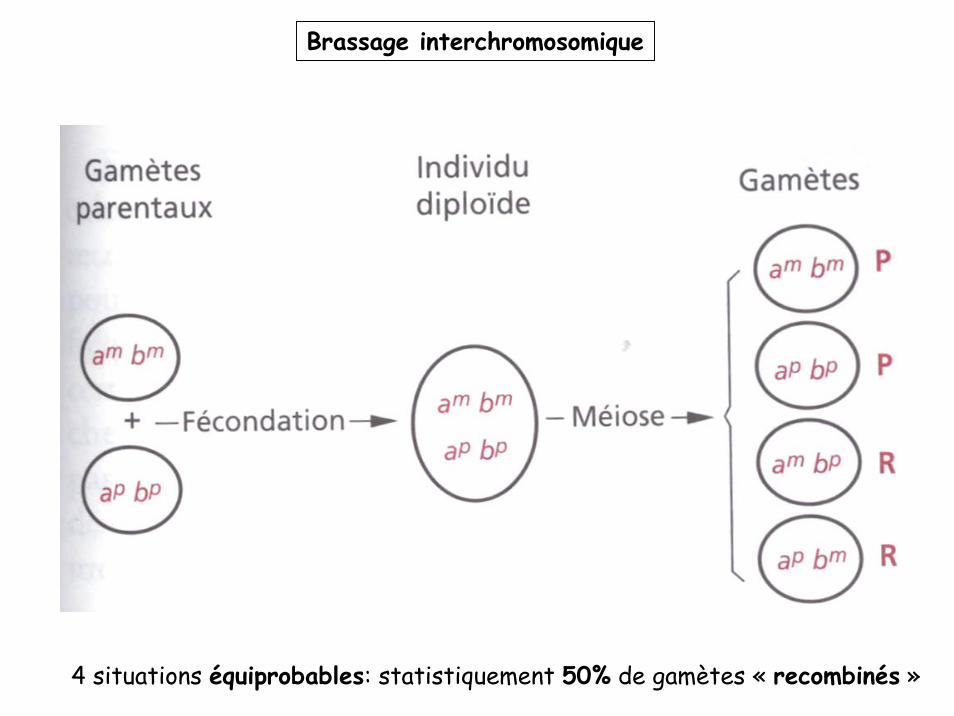
Brassage interchromosomique
4 situations équiprobables: statistiquement 50% de gamètes « recombinés »

Brassage intrachromosomique
Crossing over rares = le % de gamètes R sera d’autant plus faible que les gènes seront proches.

Cartographies génétiques et localisation des gènes
I. Transmissions des caractères et brassage génétique.
II. Cartographie génétique.1- Notion de marqueur génétique2- Pourcentage de recombinaison et distance génétique3- Construction de cartes génétiques
III. Cartographie et Recherche de gènes candidats chez l’Homme.
IV. Comparaison cartes génétiques/ cartes physiques.

Marqueurs génétiques
Cartographie génétique: établir les distances génétiques entre des marqueurs en exploitant le pourcentage de recombinaisons à la méïose.
Le marqueur idéal: Fréquemment présent à l’état hétérozygote (pour pouvoir détecter les événements de recombinaison), détectable sans ambiguité = marqueurs très polymorphes, si possible co-dominants.
Marqueurs morphologiques (phénotypiques): Caractères morphologiques ou phénotypiques (couleur des poils, capacité à poussé en milieu carencé, etc…). faciles à mesurer mais peu polymorphes, rarement co-dominants, ambigues (interfèrent entre eux).
Marqueurs moléculaires: Polymorphisme au niveau de l’ADN (SNP, microsatellites, RFLP,…). Nombreux, très polymorphes, co-dominants, non ambigues mais requièrent la biologie moléculaire pour être détectés.
Une fois localisés les uns par rapport aux autres, les marqueurs vont permettre de localiser des gènes d’intérêt, de nouveaux marqueurs ou de rechercher les régions génomiques associées à un marqueur phénotypique (recherche de gènes candidats).

Distance génétique
Distance génétique entre deux locus = fréquence de recombinaison (θ) entre ces locus = nombre de produits de méïose recombinés/ nombre total= R/(P+R)
L’unité est le centimorgan (cM).1 cM = 1% de produits recombinés = un « segment » de chromatine sur lequel la probabilité de crossing over par méïose est de 1%.
Indépendance génétique: on dit que deux gènes sont indépendants quand leur distance génétique est de 50 cM (R=P=50%). Ceci peut signifier que les deux gènes sont sur des chromosomes différents (brassage interchromosomique) ou très éloignés sur le même chromosome.
Liaison génétique: on dit que deux gènes sont liés quand leur distance génétique est inférieure à 50 cM (P>50%, R<50%). Ceci signifie que les gènes sont proches sur le même chromosome: la probabilité d’avoir un crossing over entre les deux gènes est d’autant plus faible qu’ils sont proches.
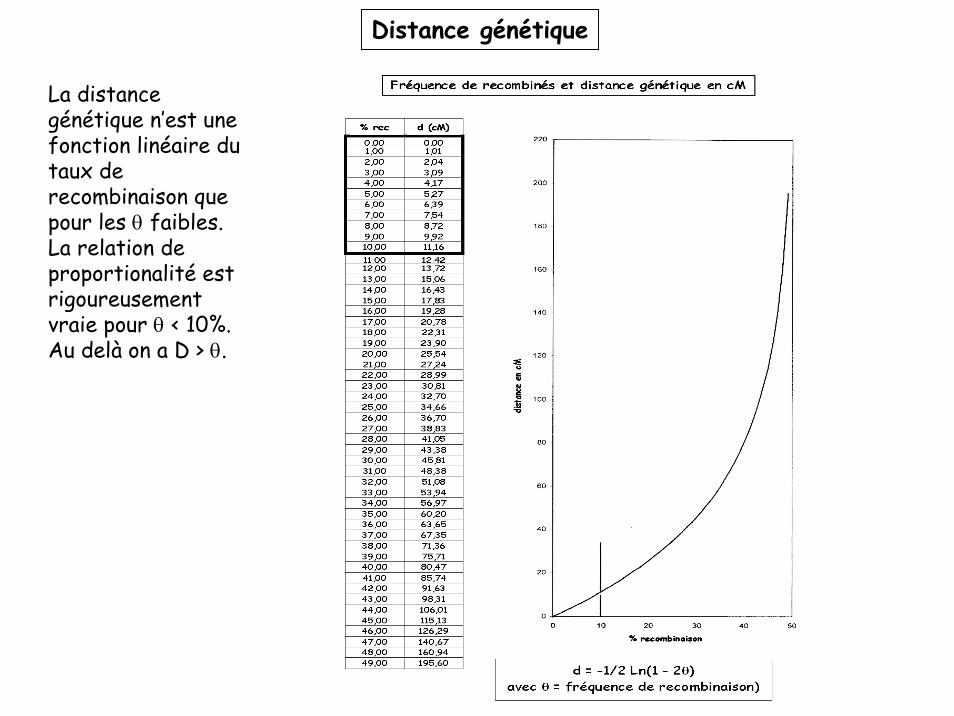
Distance génétique
La distance génétique n’est une fonction linéaire du taux de recombinaison que pour les θ faibles.La relation de proportionalité est rigoureusement vraie pour θ < 10%.Au delà on a D > θ.

Distance génétique
La distance génétique n’est une fonction linéaire du taux de recombinaison que pour les θfaibles.
Pourquoi?Cas 1: les deux marqueurs sont proches (<10 cM), il ne se produit jamais plus d’un crossing over par méïose entre les deux. Tous les crossing over conduisent à l’apparition de produits recombinés. D = θ
Cas 2: les deux marqueurs sont plus éloignés (10 < D < 50 cM), il peut se produire des crossing over multiples entre les deux. Dans certains cas, deux crossing over affectant les deux mêmes chromatides peuvent restaurer un génotype parental (voir cours ségrégation). La fréquence de produits recombinés sous-estime la distance réelle entre les deux marqueurs. D > θ
Cas 3: Les deux marqueurs sont tellement éloignés (D > 50cM) qu’il se produit toujours au moins un crossing over par méïose. Les deux marqueurs apparaissent indépendants (θ = 50%) et on ne peut pas calculer la distance génétique réelle. D > θ
Conclusion:Pour construire des cartes génétiques précises, il faut un grand nombre de marqueurs proches deux à deux.

Cartographie génétique
Exemple 1: Analyse directe des produits de méïose chez une espèce haplodiplobiontique.
Exemple 1-A: distance entre deux marqueurs moléculaires (les gènes THR4 et MAT) chez la levure Saccharomyces cerevisiae.
La distance génétique entre THR4 et MAT est estimée à au moins 20,3 cM.THR4 et MAT sont liés (sur le même chromosome).
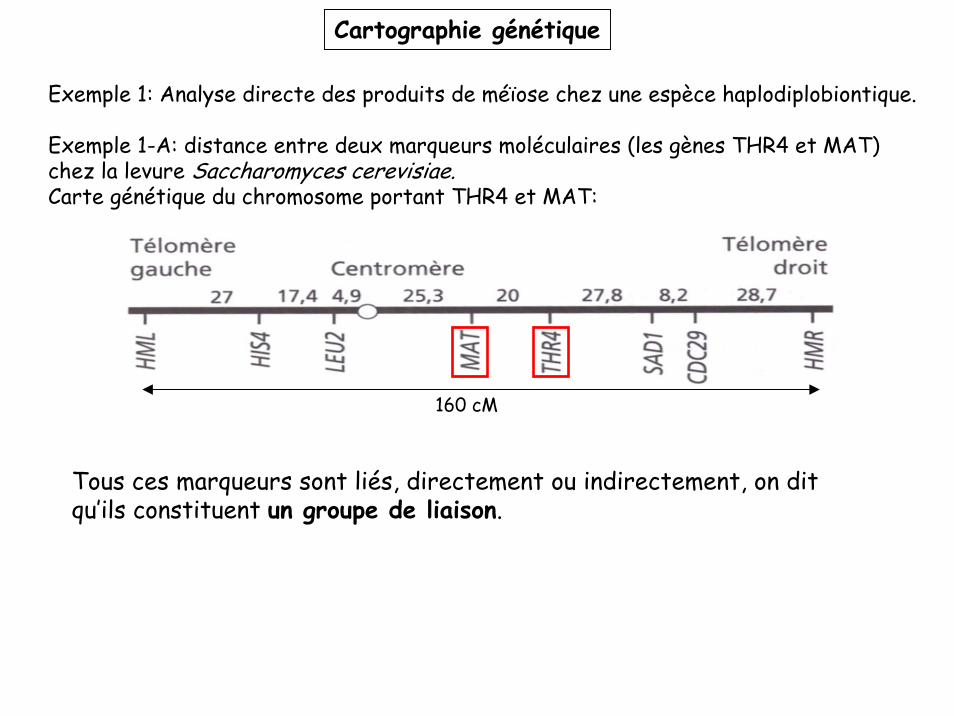
Cartographie génétique
Exemple 1: Analyse directe des produits de méïose chez une espèce haplodiplobiontique.
Exemple 1-A: distance entre deux marqueurs moléculaires (les gènes THR4 et MAT) chez la levure Saccharomyces cerevisiae.Carte génétique du chromosome portant THR4 et MAT:
160 cM
Tous ces marqueurs sont liés, directement ou indirectement, on dit qu’ils constituent un groupe de liaison.
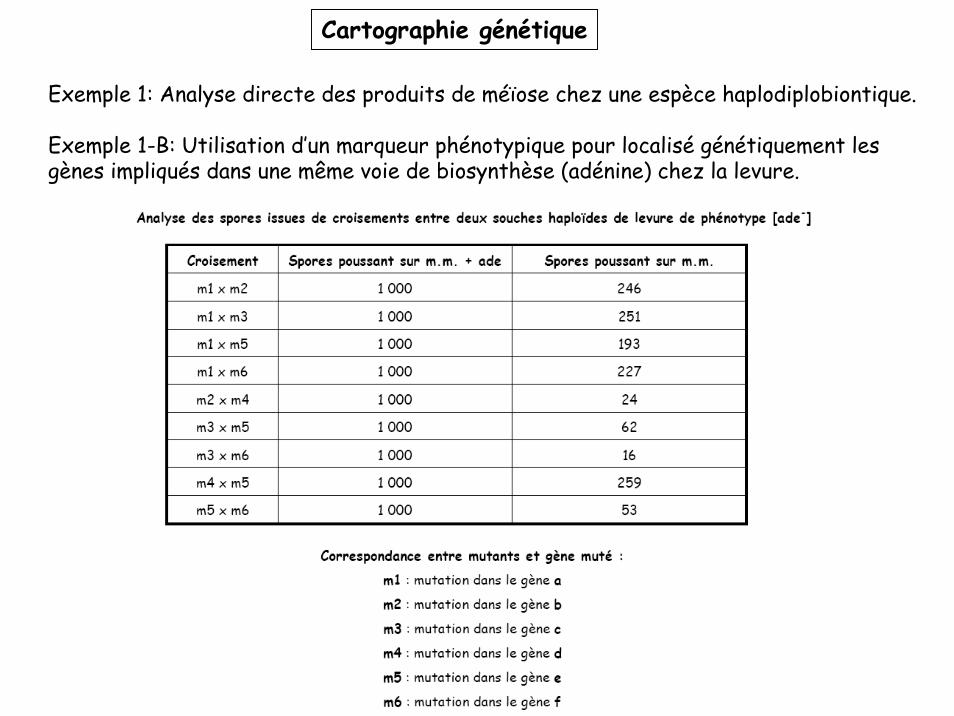
Cartographie génétique
Exemple 1: Analyse directe des produits de méïose chez une espèce haplodiplobiontique.
Exemple 1-B: Utilisation d’un marqueur phénotypique pour localisé génétiquement les gènes impliqués dans une même voie de biosynthèse (adénine) chez la levure.
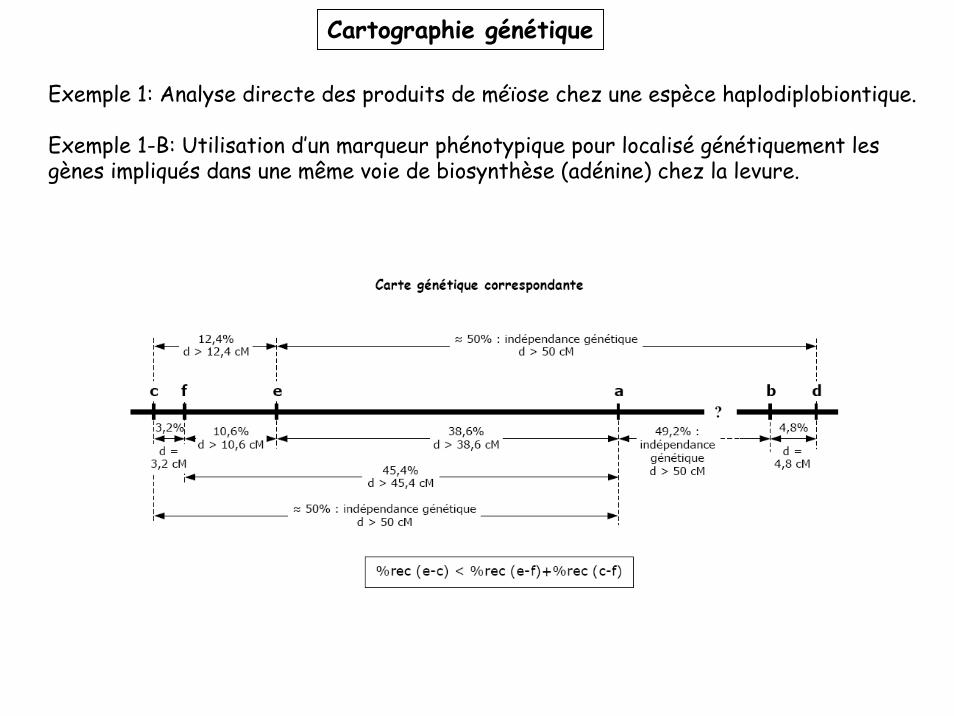
Cartographie génétique
Exemple 1: Analyse directe des produits de méïose chez une espèce haplodiplobiontique.
Exemple 1-B: Utilisation d’un marqueur phénotypique pour localisé génétiquement les gènes impliqués dans une même voie de biosynthèse (adénine) chez la levure.

Cartographie génétique
Cartographie chez les espèces diplobiontiques.
Problème: les génotypes et phénotypes des produits de méïose (gamètes) ne sont pas facilement accessibles.
Solution: analyse génétique des descendants diploïdes
1- Utiliser des lignées pures: homozygotes pour les caractères d’intérêt mais avec des allèles différents (générations P1 et P2).
2- Accoupler les individus des deux lignées: création d’hétérozygotes pour les caractères d’intérêt (génération F1).
3- Accoupler un individu F1 avec une des deux lignées parentales, obtention de la génération F2 qui donne accès à la ségrégation des allèles dans les gamètes de F1.Ce procédé s’appelle un back-cross.
Dans le cas d’un caractère mutant récessif, on croise F1 avec la lignée parentale mutante, on parle de test-cross. Dans ce cas particulier, le phénotype des F2 donne directement le génotype des gamètes issues de F1.
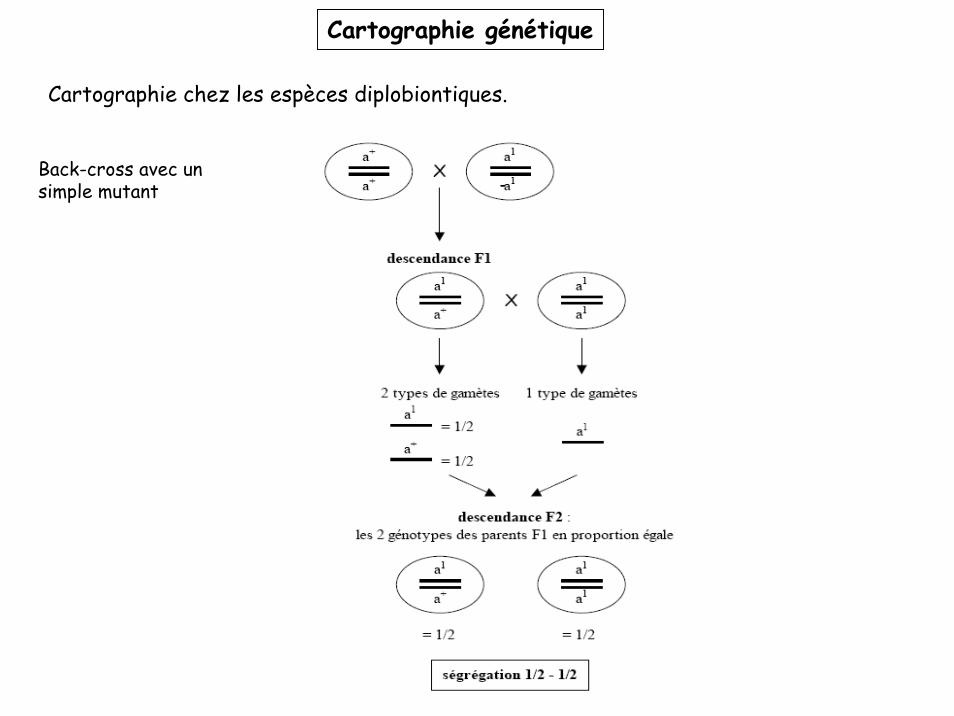
Cartographie génétique
Cartographie chez les espèces diplobiontiques.
Back-cross avec un simple mutant
Cartographie génétique
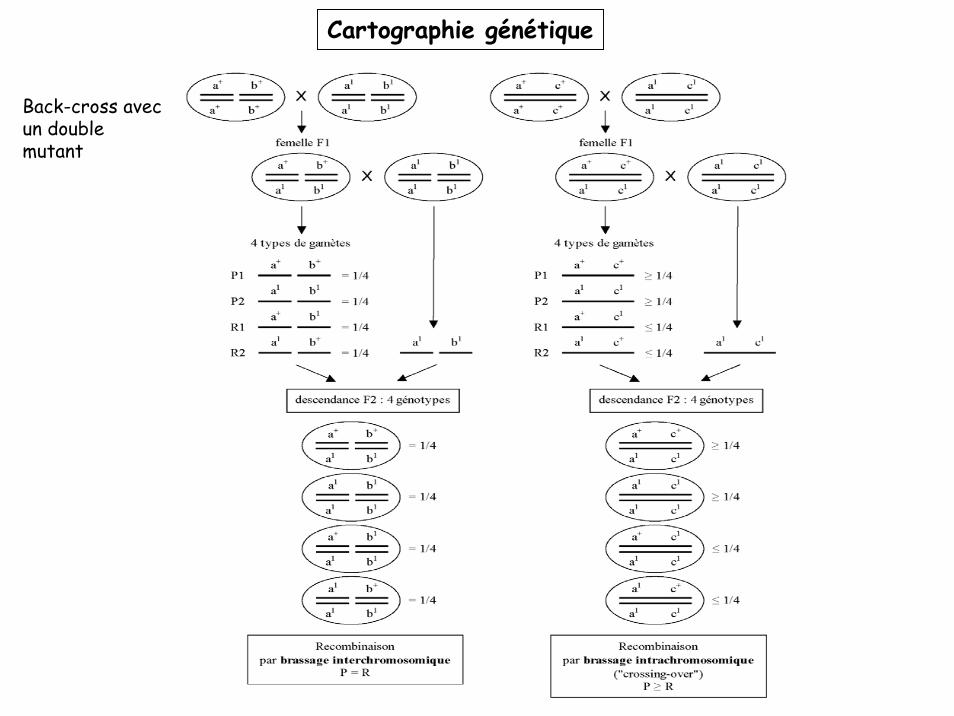
Back-cross avec un double mutant
Cartographie génétique
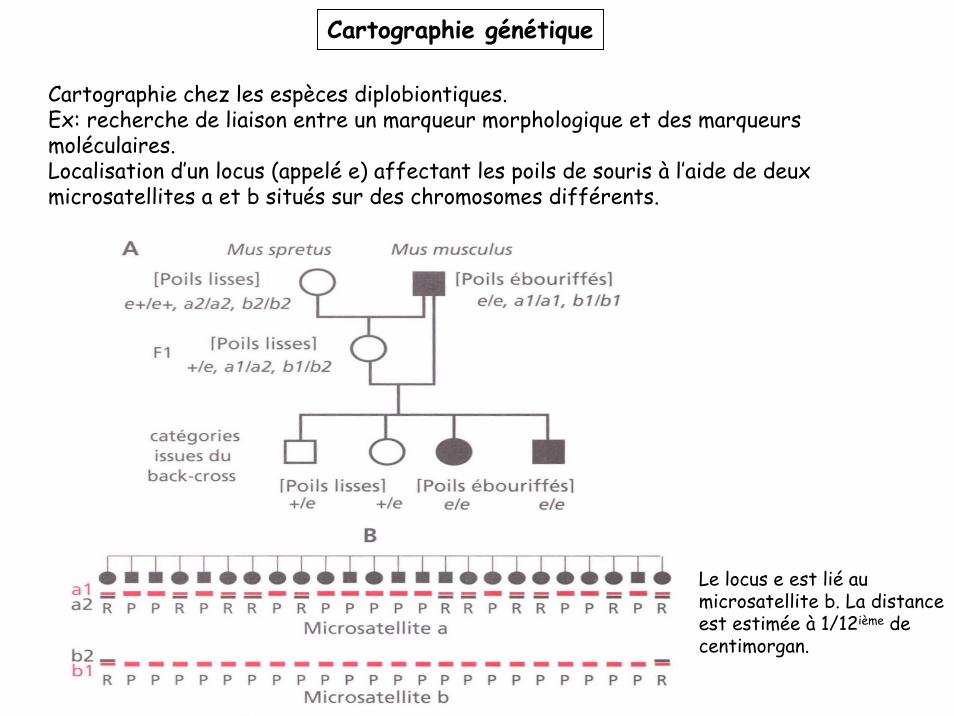
Cartographie chez les espèces diplobiontiques.Ex: recherche de liaison entre un marqueur morphologique et des marqueurs moléculaires. Localisation d’un locus (appelé e) affectant les poils de souris à l’aide de deux microsatellites a et b situés sur des chromosomes différents.
Le locus e est lié au microsatellite b. La distance est estimée à 1/12ième de centimorgan.

Cartographies génétiques et localisation des gènes
I. Transmissions des caractères et brassage génétique.
II. Cartographie génétique.
III. Cartographie et Recherche de gènes candidats chez l’Homme.
1- Problèmes spécifiques2- Analyse de pedigree3- LOD score et autres méthodes
IV. Comparaison cartes génétiques/ cartes physiques.

Cartographie génétique chez l’Homme
Problèmes de la génétique humaine:
Pas de lignées pures.
Faible nombre de descendants diploïdes: faible valeur statistique des mesures.
Backcross difficilement réalisables.
Manipulations génotypiques et phénotypiques limitées.
Conclusion: Le généticien humain aime les grandes familles et les unions consanguines.

Cartographie génétique chez l’Homme
Analyses des pedigree pour:
déterminer le mode de transmission des caractères et donc sur quel type de chromosome se trouvent les mutations causales du caractère étudié.
Extrapoler le génotype des individus et identifier les individus potentiellement les plus informatifs
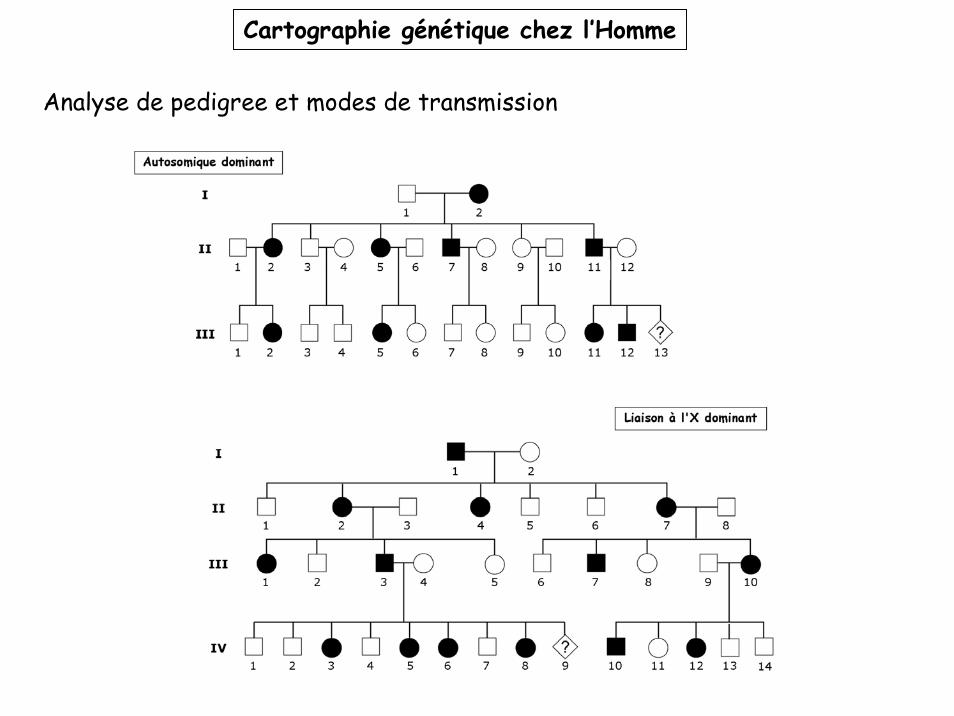
Cartographie génétique chez l’Homme
Analyse de pedigree et modes de transmission
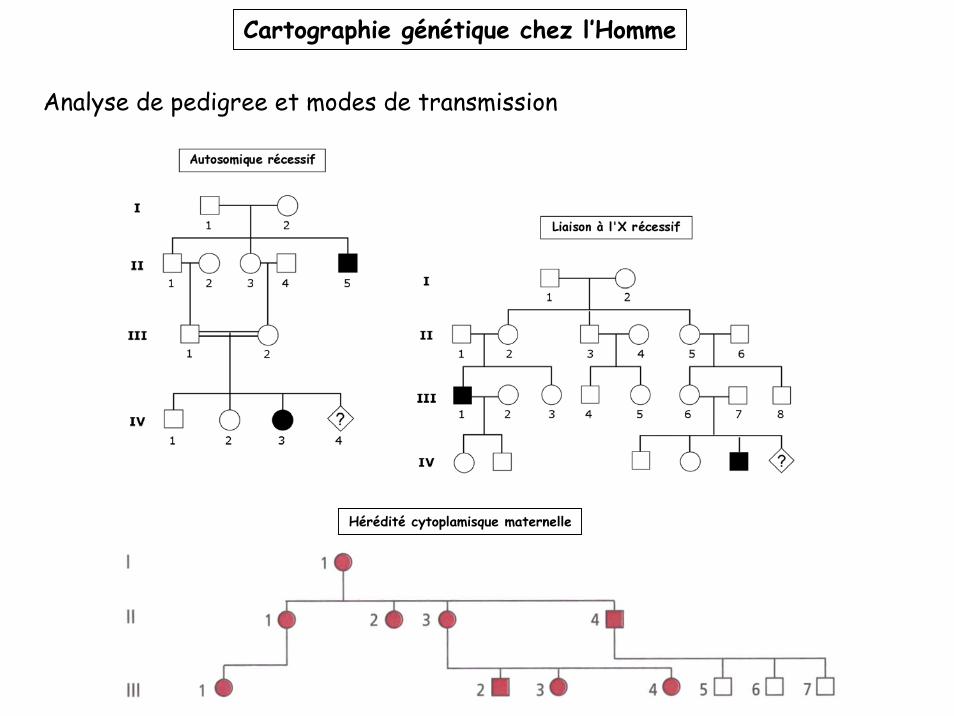
Cartographie génétique chez l’Homme
Analyse de pedigree et modes de transmission
Hérédité cytoplamisque maternelle

Cartographie génétique chez l’Homme
Cartographie génétique et méthode des LOD scores
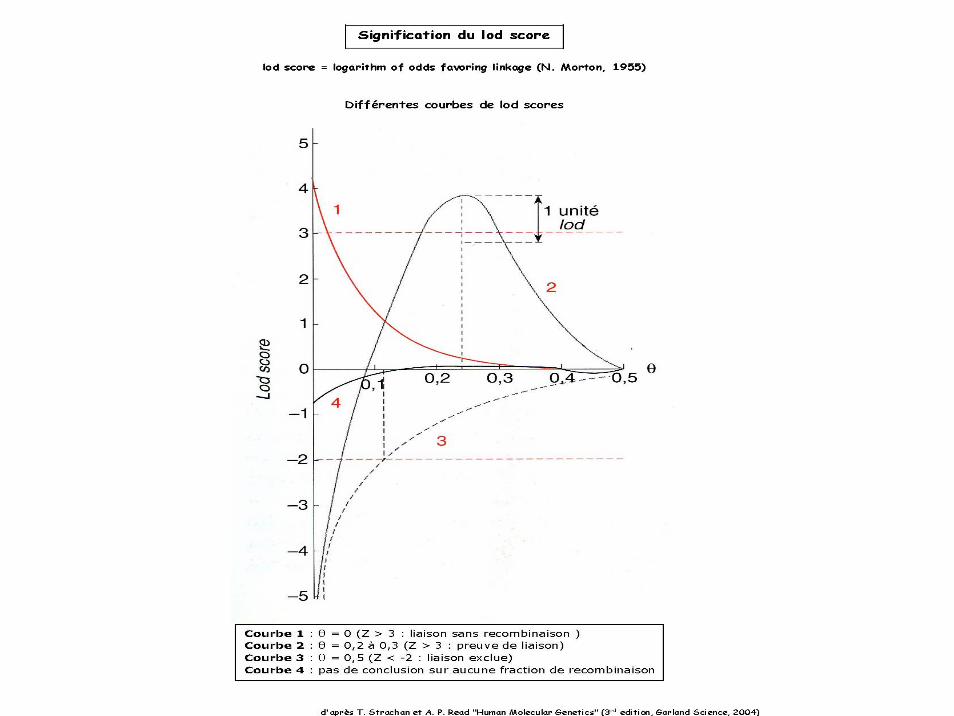
Le LOD score (noté Z) est un indicateur statistique servant à estimer la probabilité de laison de deux marqueurs sur des populations de petits effectifs.
Il s’agit du logarithme du rapport de vraisemblance (probabilité notée L) entre deux hypothèses:Hypothèse 1: les marqueurs étudiés sont génétiquement liés (θ < 0.5).Hypothèse 2: les marqueurs sont indépendants (θ = 0.5).
Ainsi, Z(θ) = Log10 [L(θ<0.5)/L(θ=0.5)]
Z(θ) = 3 signifie donc que l’hypothèse de marqueurs liés génétiquement et distants de θ est mille fois plus probable que l’hypothèse d’indépendance.
Classiquement,l ’hypothèse d’indépendance est rejetée si Z(θ) est égal ou supérieur à 3.L’hypothèse de liaison est rejetée si Z(θ) est égal ou inférieur à –2.Entre –2 et 3, on ne peut pas conclure.
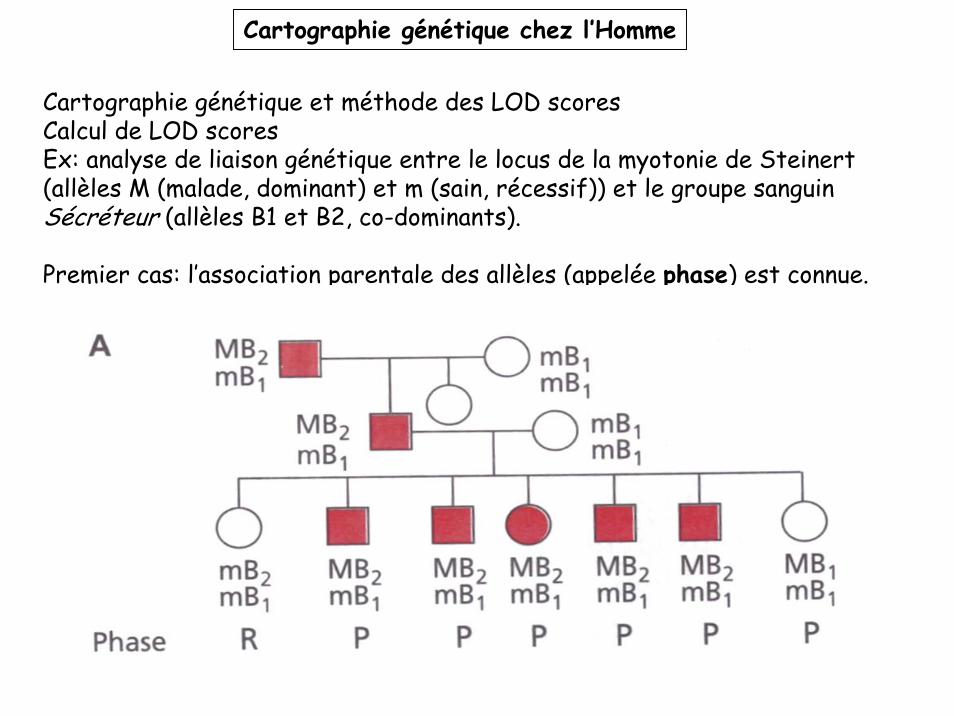
Cartographie génétique chez l’Homme
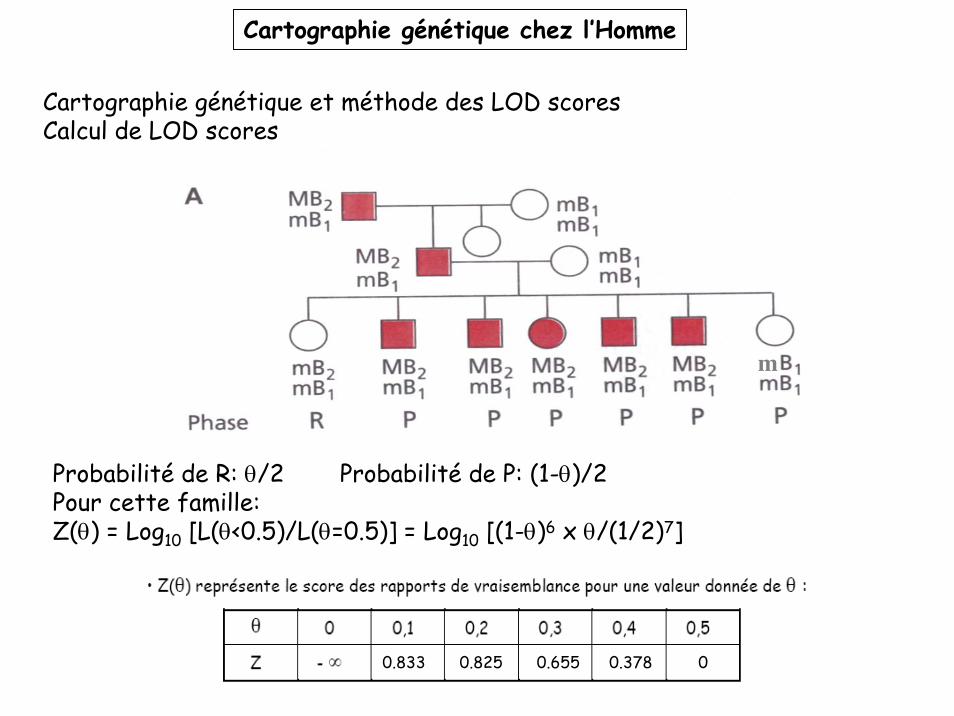
Cartographie génétique et méthode des LOD scoresCalcul de LOD scoresEx: analyse de liaison génétique entre le locus de la myotonie de Steinert (allèles M (malade, dominant) et m (sain, récessif)) et le groupe sanguin Sécréteur (allèles B1 et B2, co-dominants).
Premier cas: l’association parentale des allèles (appelée phase) est connue.
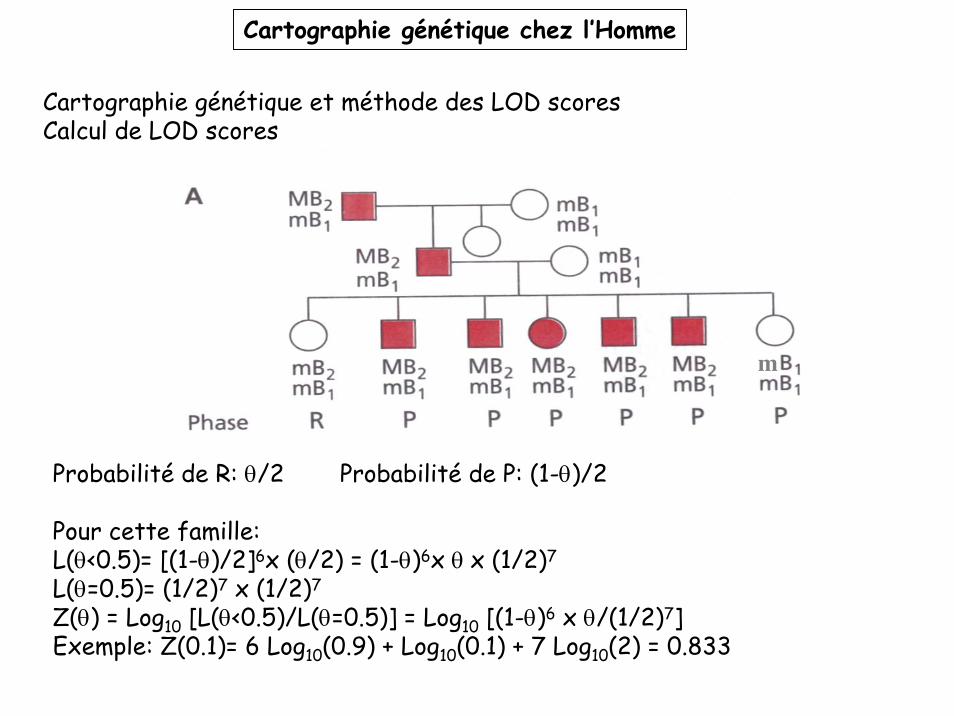
Cartographie génétique chez l’Homme
Cartographie génétique et méthode des LOD scoresCalcul de LOD scores
Probabilité de R: θ/2 Probabilité de P: (1-θ)/2
Pour cette famille:L(θ<0.5)= [(1-θ)/2]6x (θ/2) = (1-θ)6x θ x (1/2)7
L(θ=0.5)= (1/2)7 x (1/2)7
Z(θ) = Log10 [L(θ<0.5)/L(θ=0.5)] = Log10 [(1-θ)6 x θ/(1/2)7]Exemple: Z(0.1)= 6 Log10(0.9) + Log10(0.1) + 7 Log10(2) = 0.833
m
Cartographie génétique chez l’Homme
Cartographie génétique et méthode des LOD scoresCalcul de LOD scores
Probabilité de R: θ/2 Probabilité de P: (1-θ)/2Pour cette famille:Z(θ) = Log10 [L(θ<0.5)/L(θ=0.5)] = Log10 [(1-θ)6 x θ/(1/2)7]
0.833 0.825 0.655 0.378 0
m
Cartographie génétique chez l’Homme
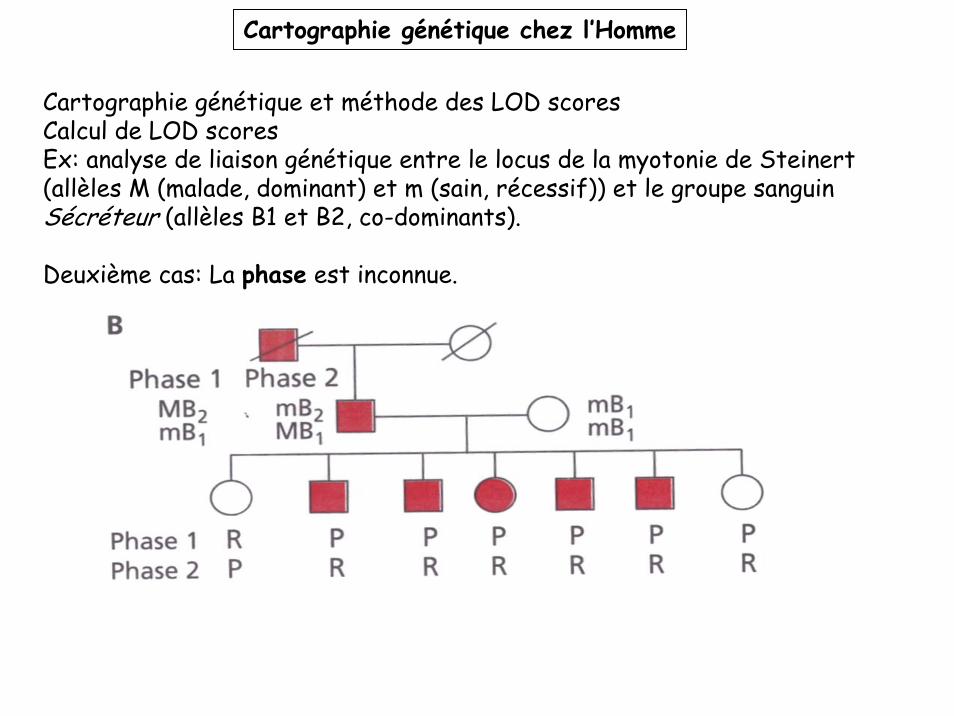
Cartographie génétique et méthode des LOD scoresCalcul de LOD scoresEx: analyse de liaison génétique entre le locus de la myotonie de Steinert (allèles M (malade, dominant) et m (sain, récessif)) et le groupe sanguin Sécréteur (allèles B1 et B2, co-dominants).
Deuxième cas: La phase est inconnue.
Cartographie génétique chez l’Homme
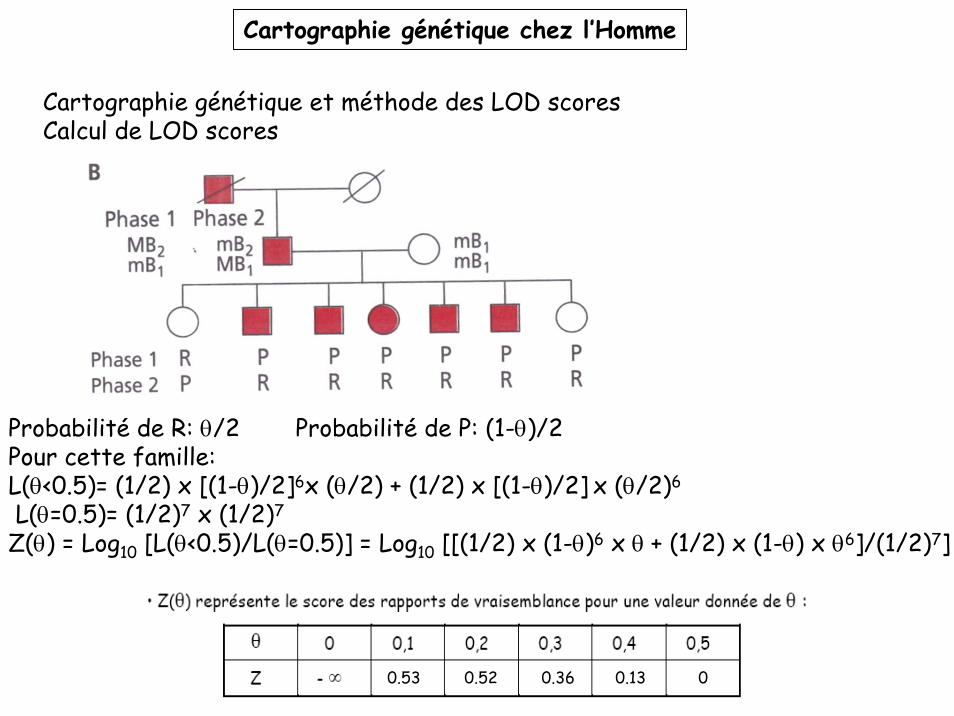
Cartographie génétique et méthode des LOD scoresCalcul de LOD scores
Probabilité de R: θ/2 Probabilité de P: (1-θ)/2Pour cette famille:L(θ<0.5)= (1/2) x [(1-θ)/2]6x (θ/2) + (1/2) x [(1-θ)/2] x (θ/2)6
L(θ=0.5)= (1/2)7 x (1/2)7
Z(θ) = Log10 [L(θ<0.5)/L(θ=0.5)] = Log10 [[(1/2) x (1-θ)6 x θ + (1/2) x (1-θ) x θ6]/(1/2)7]
0.53 0.52 0.36 0.13 0

Cartographie génétique chez l’Homme
Cartographie génétique et méthode des LOD scoresCalcul de LOD scores
On peut améliorer le LOD score en additionant différentes familles pour augmenter le nombre d’individus, à condition que:
Les familles soient informatives (au moins un des deux partents double hétérozygote pour les marqueurs utilisés)
Elles comportent au moins deux enfants
Le phénotype (dans le cas d’une maladie par exemple) doit être clair et homogène entre tous les individus atteints de la cohorte.
Dans l’exemple précédent (myotonie de Steinert et groupe sanguin) la liaison a été démontrée en rajoutant des familles, avec θmax = 0.07 et Z(0.07) = 4.
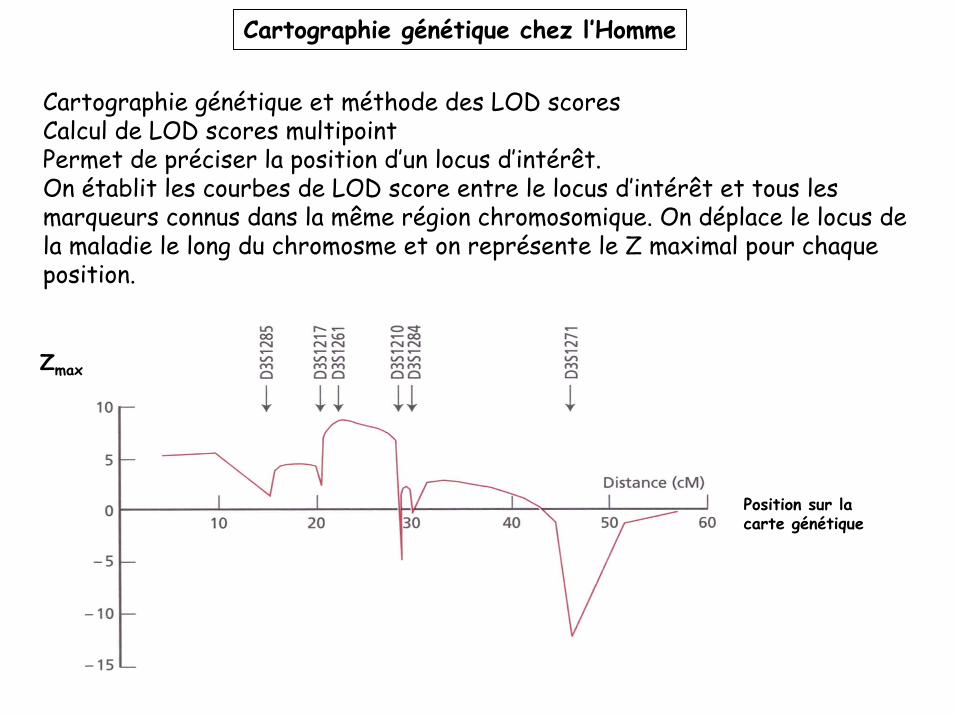
Cartographie génétique chez l’Homme
Cartographie génétique et méthode des LOD scoresCalcul de LOD scores multipointPermet de préciser la position d’un locus d’intérêt.On établit les courbes de LOD score entre le locus d’intérêt et tous les marqueurs connus dans la même région chromosomique. On déplace le locus de la maladie le long du chromosme et on représente le Z maximal pour chaque position.
Zmax
Position sur la carte génétique
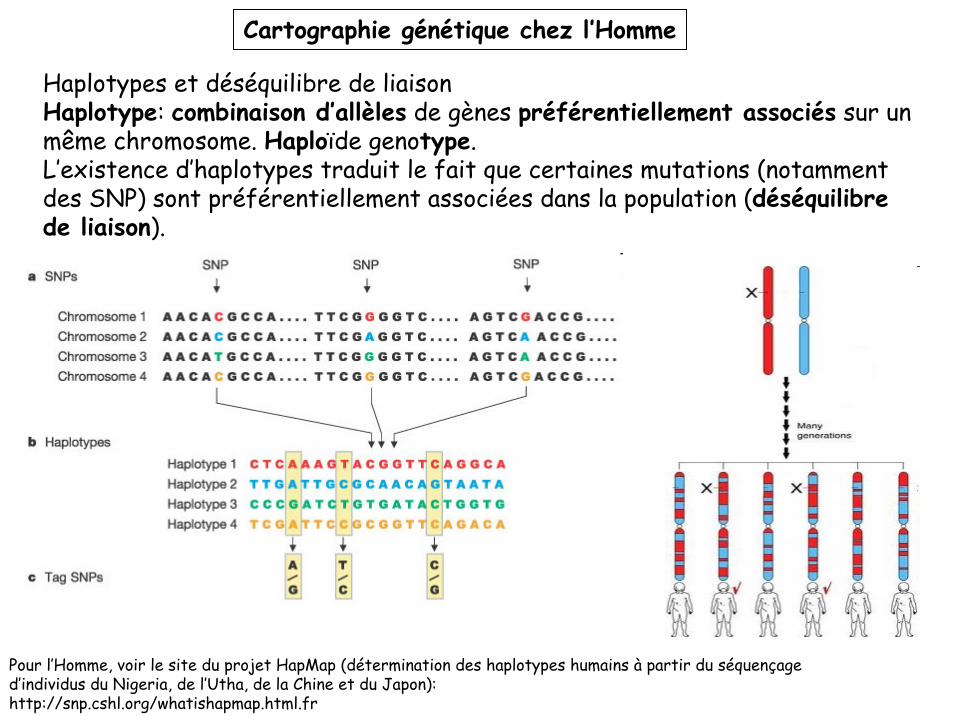
Cartographie génétique chez l’Homme
Haplotypes et déséquilibre de liaisonHaplotype: combinaison d’allèles de gènes préférentiellement associés sur un même chromosome. Haploïde genotype.L’existence d’haplotypes traduit le fait que certaines mutations (notamment des SNP) sont préférentiellement associées dans la population (déséquilibre de liaison).
Pour l’Homme, voir le site du projet HapMap (détermination des haplotypes humains à partir du séquençage d’individus du Nigeria, de l’Utha, de la Chine et du Japon):http://snp.cshl.org/whatishapmap.html.fr
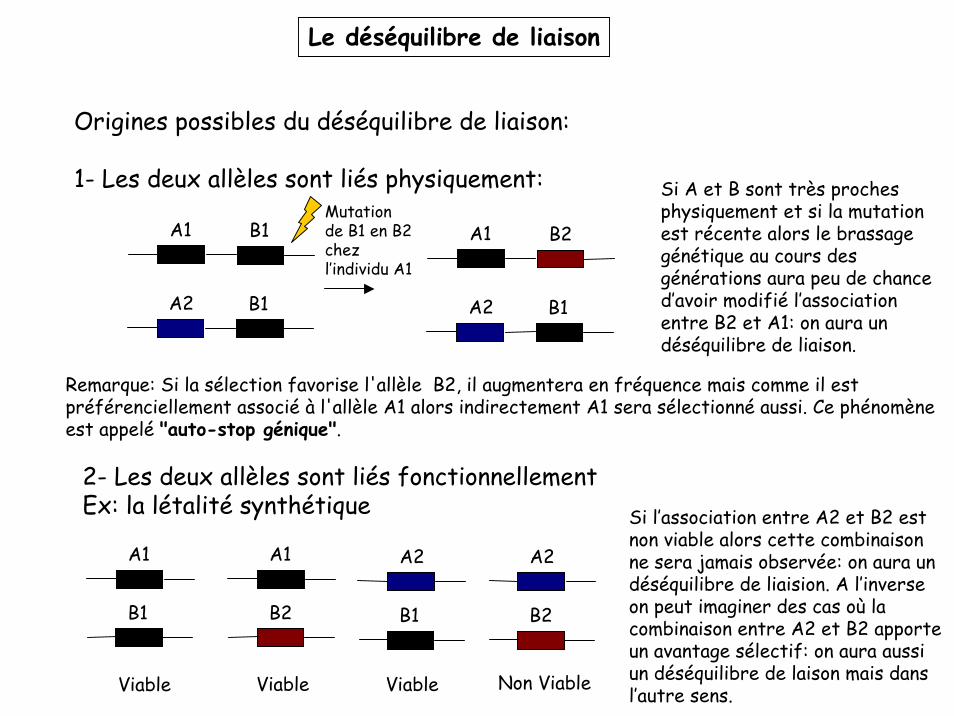
Le déséquilibre de liaison
Origines possibles du déséquilibre de liaison:
1- Les deux allèles sont liés physiquement:
2- Les deux allèles sont liés fonctionnellementEx: la létalité synthétique
A1 B1
A2 B1
Mutation de B1 en B2 chez l’individu A1
A1 B2
A2 B1
Si A et B sont très proches physiquement et si la mutation est récente alors le brassage génétique au cours des générations aura peu de chance d’avoir modifié l’association entre B2 et A1: on aura un déséquilibre de liaison.
A1
B1
Viable
A1
B2
A2
B1
A2
B2
Viable Non ViableViable
Si l’association entre A2 et B2 est non viable alors cette combinaison ne sera jamais observée: on aura un déséquilibre de liaision. A l’inverse on peut imaginer des cas où la combinaison entre A2 et B2 apporte un avantage sélectif: on aura aussi un déséquilibre de laison mais dans l’autre sens.
Remarque: Si la sélection favorise l'allèle B2, il augmentera en fréquence mais comme il est préférenciellement associé à l'allèle A1 alors indirectement A1 sera sélectionné aussi. Ce phénomène est appelé "auto-stop génique".
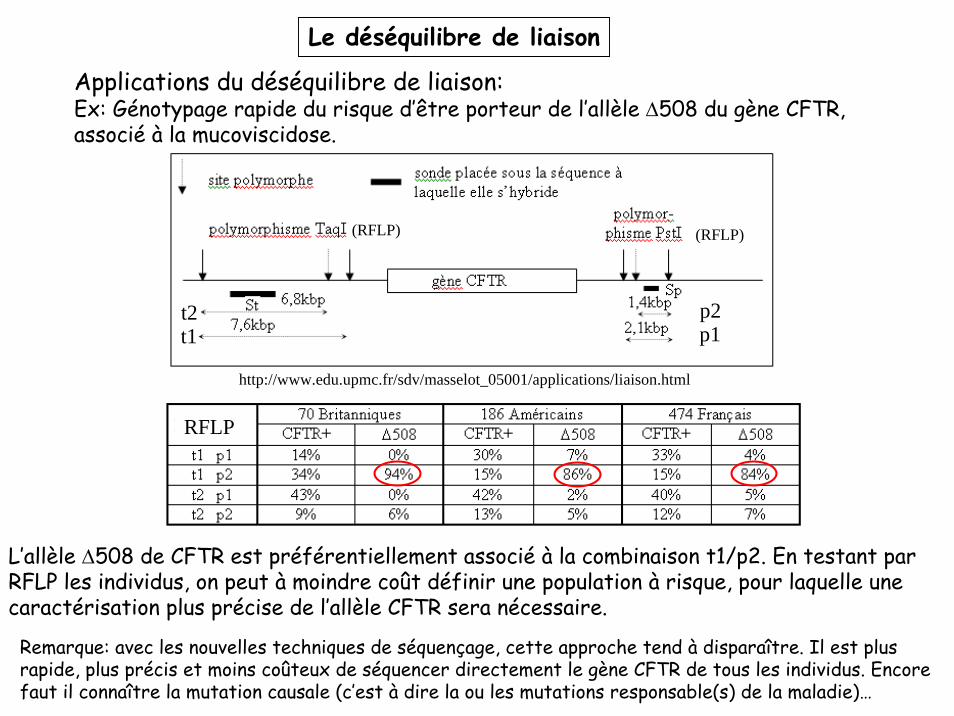
Le déséquilibre de liaison
Applications du déséquilibre de liaison:Ex: Génotypage rapide du risque d’être porteur de l’allèle Δ508 du gène CFTR, associé à la mucoviscidose.
(RFLP) (RFLP)
t1t2
p1p2
http://www.edu.upmc.fr/sdv/masselot_05001/applications/liaison.html
RFLP
L’allèle Δ508 de CFTR est préférentiellement associé à la combinaison t1/p2. En testant par RFLP les individus, on peut à moindre coût définir une population à risque, pour laquelle une caractérisation plus précise de l’allèle CFTR sera nécessaire.
Remarque: avec les nouvelles techniques de séquençage, cette approche tend à disparaître. Il est plus rapide, plus précis et moins coûteux de séquencer directement le gène CFTR de tous les individus. Encore faut il connaître la mutation causale (c’est à dire la ou les mutations responsable(s) de la maladie)…
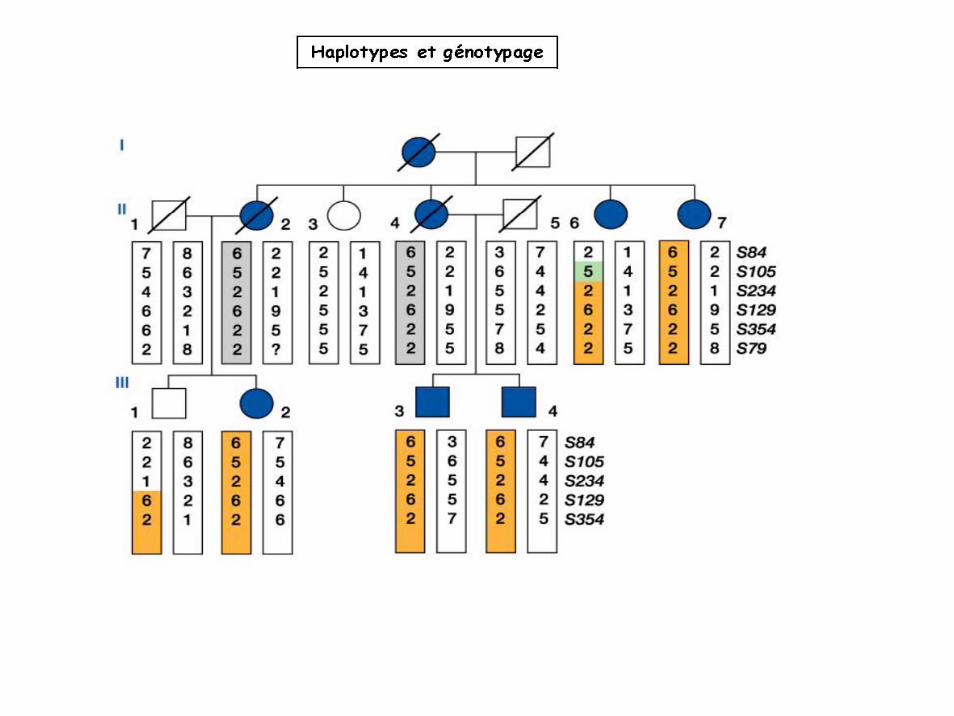
Cartographie génétique chez l’Homme
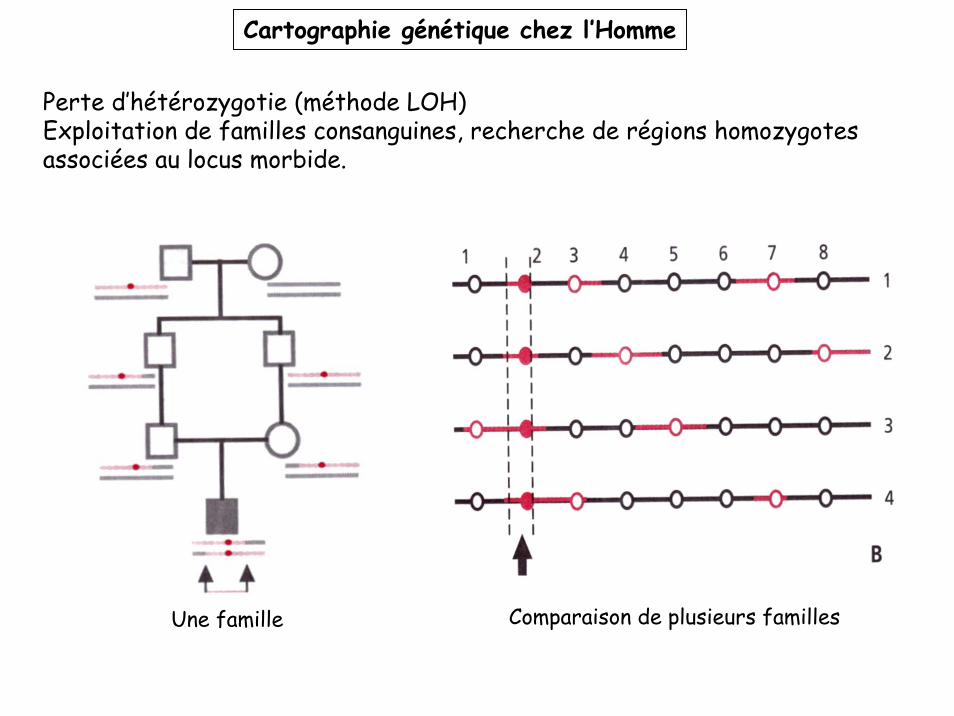
Perte d’hétérozygotie (méthode LOH)Exploitation de familles consanguines, recherche de régions homozygotes associées au locus morbide.
Une famille Comparaison de plusieurs familles
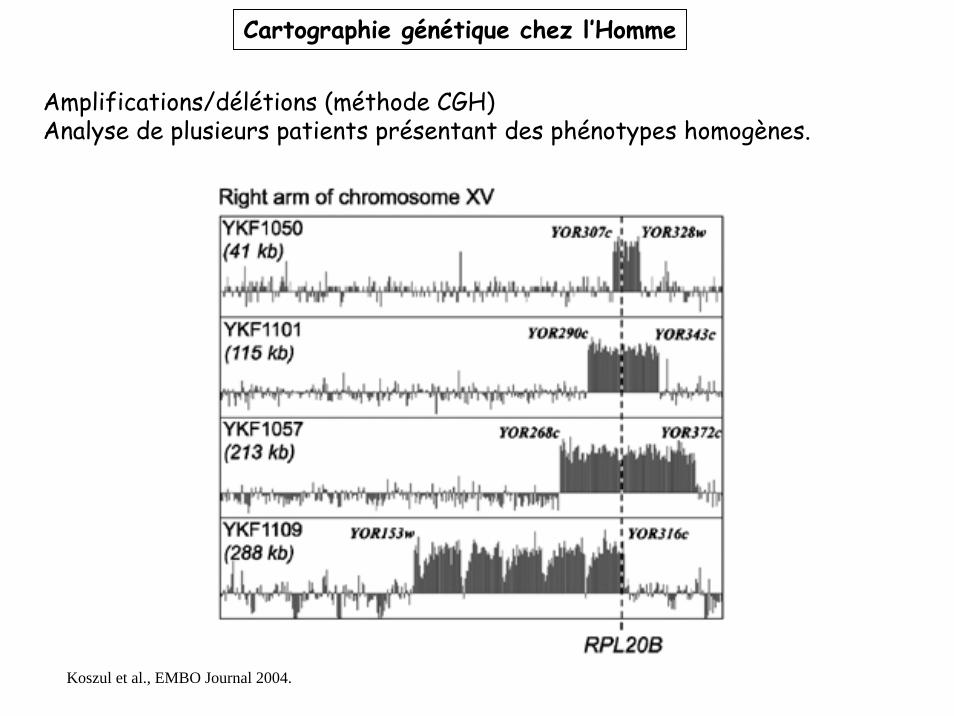
Koszul et al., EMBO Journal 2004.
Cartographie génétique chez l’Homme
Amplifications/délétions (méthode CGH)Analyse de plusieurs patients présentant des phénotypes homogènes.

Cartographies génétiques et localisation des gènes
I. Transmissions des caractères et brassage génétique.
II. Cartographie génétique.
III. Cartographie et Recherche de gènes candidats chez l’Homme.
IV. Comparaison cartes génétiques/ cartes physiques.1- Cartographie physique2- Correspondance cartes génétiques/ cartes physiques3- Recherche de gènes candidats

Cartographie physique
Cartographie physique: établir les distances physiques entre les gènes.
Distance physique: elle se mesure en paires de bases (et kilo-, mega ou giga parires de bases).
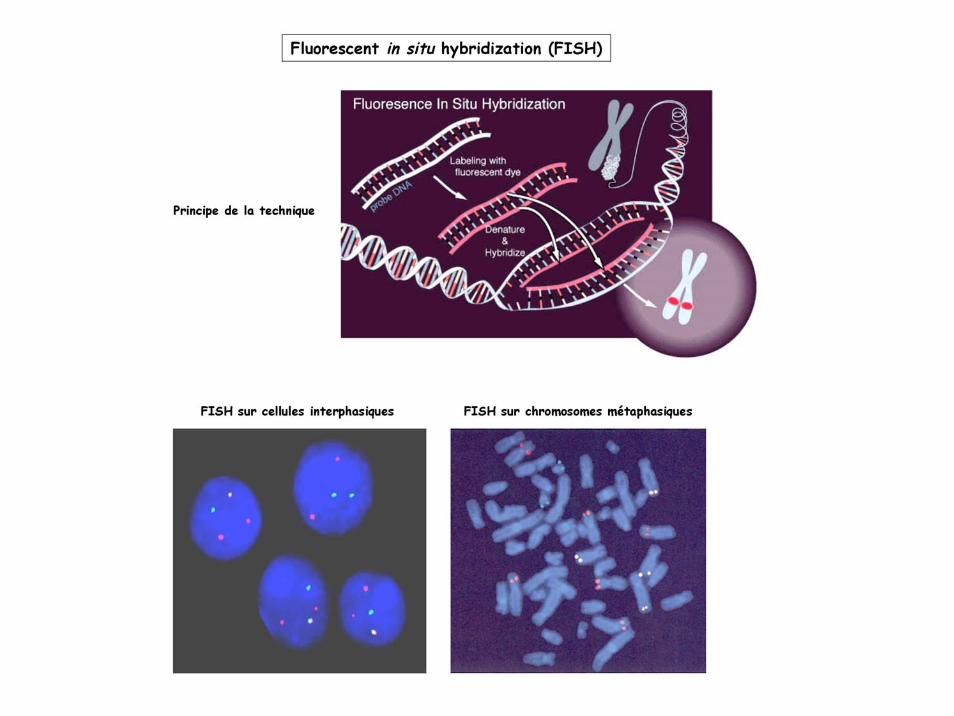
Techniques de cartographie physique:Cytogénétique moléculaire (ex: caryotypes et FISH)Hybrides cellulaires somatiques interspécifiquesPeignage moléculaireElectrophorèse en champ pulséCartographie par contig ou marche sur le chromosome
La carte physique ultime est obtenue par séquençage et assemblage complet de la séquence du génome (voir début du cours BMG1).
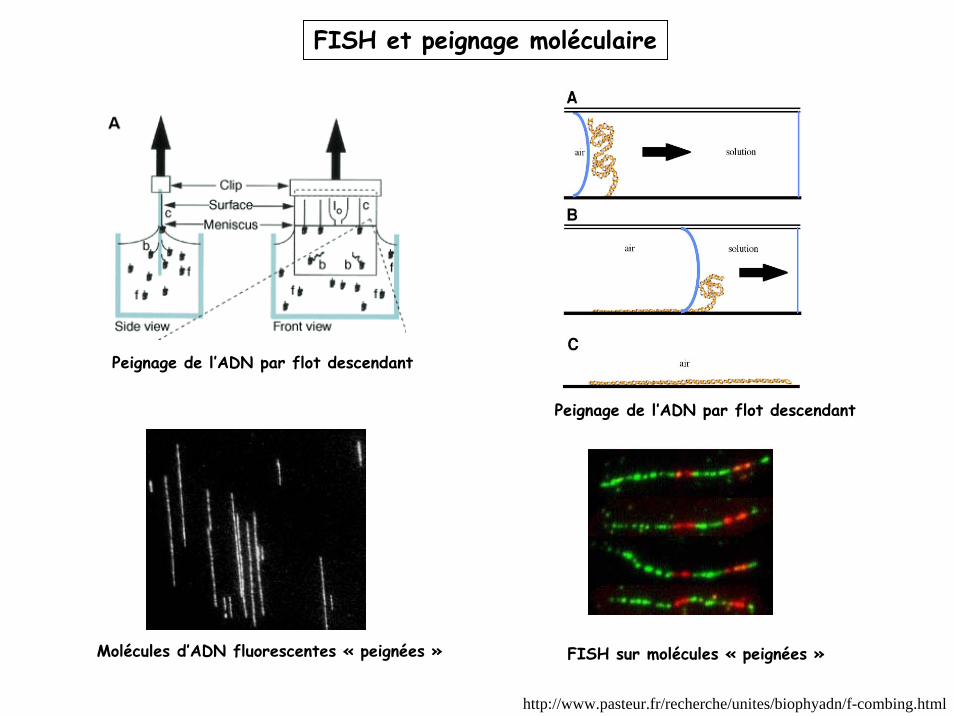
http://www.pasteur.fr/recherche/unites/biophyadn/f-combing.html
FISH et peignage moléculaire
Molécules d’ADN fluorescentes « peignées » FISH sur molécules « peignées »
Peignage de l’ADN par flot descendant
Peignage de l’ADN par flot descendant
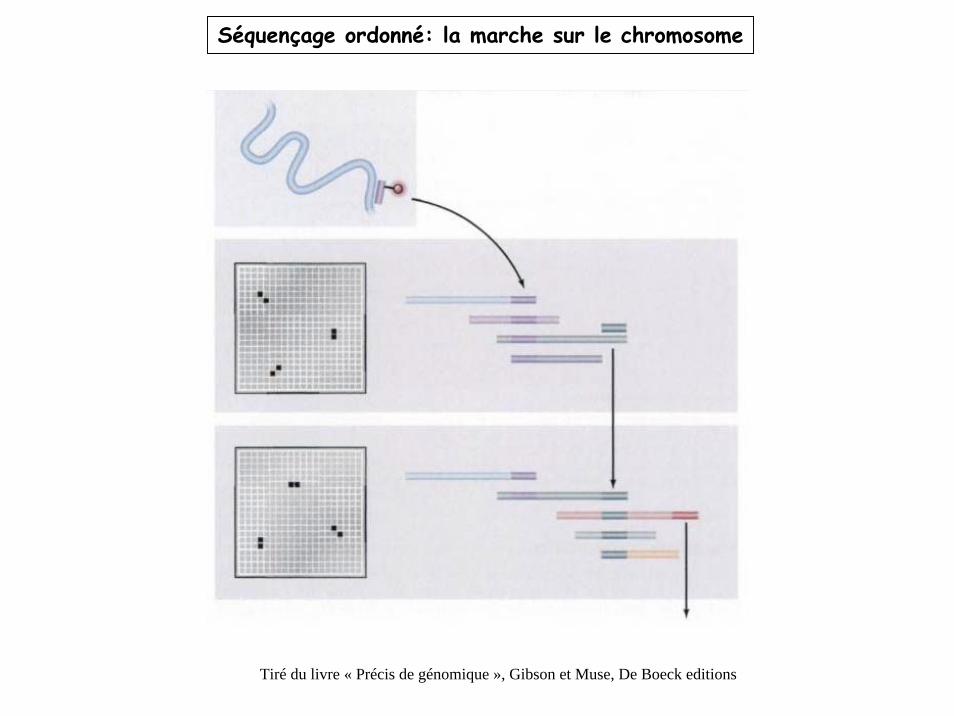
Séquençage ordonné: la marche sur le chromosome
Tiré du livre « Précis de génomique », Gibson et Muse, De Boeck editions
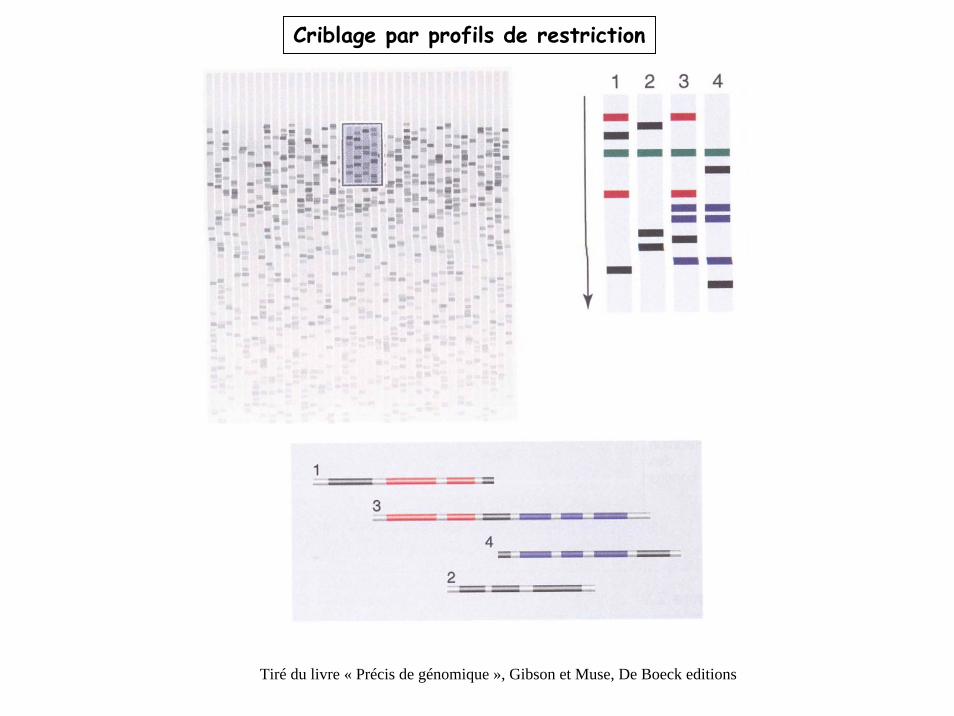
Criblage par profils de restriction
Tiré du livre « Précis de génomique », Gibson et Muse, De Boeck editions
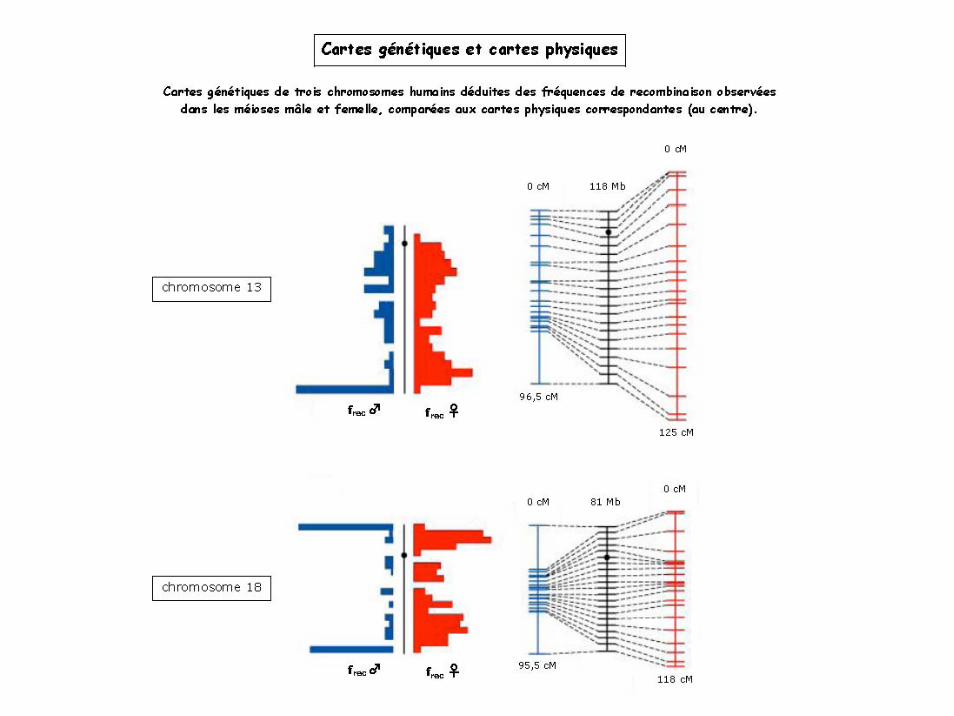

Cartographies génétiques et cartes physiques
Leurs différences viennent du fait que le taux de recombinaison n’est pas constant le long des chromosomes:
Très faible aux centromères
Très élevé au niveau des télomères et des séquences répétées
Chez les mammifères, les recombinaisons sont plus fréquentes dans les méïoses femelles.
La correspondance entre distance génétique et distance physique est donc très variable. On estime qu’un centimorgan équivaut à entre 1 et 10 000 kb (kilopaires de bases).
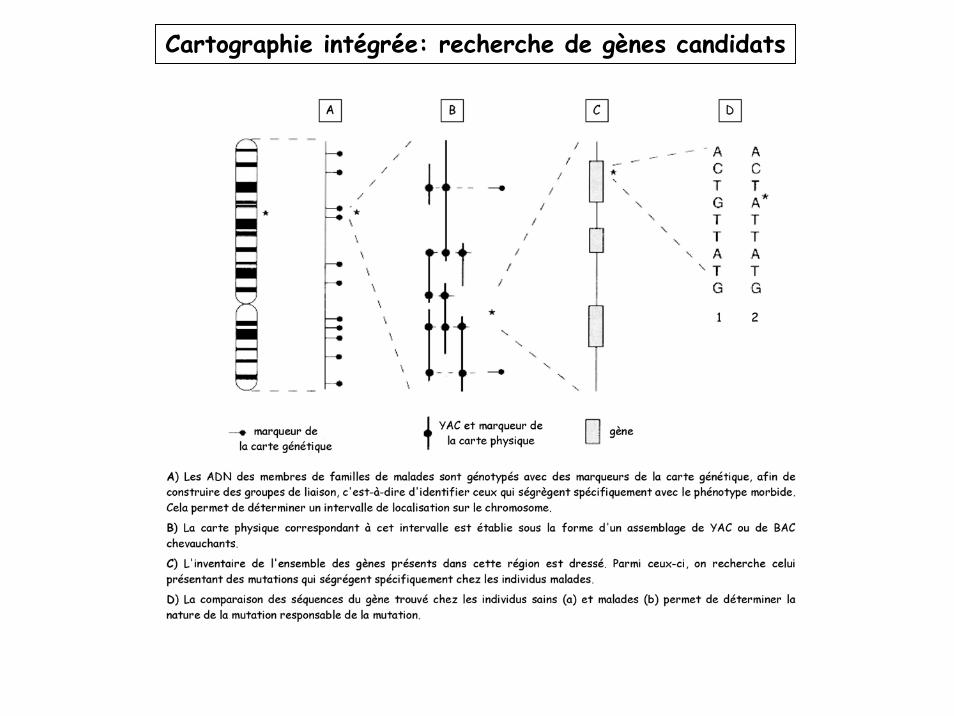
Cartographie intégrée: recherche de gènes candidats
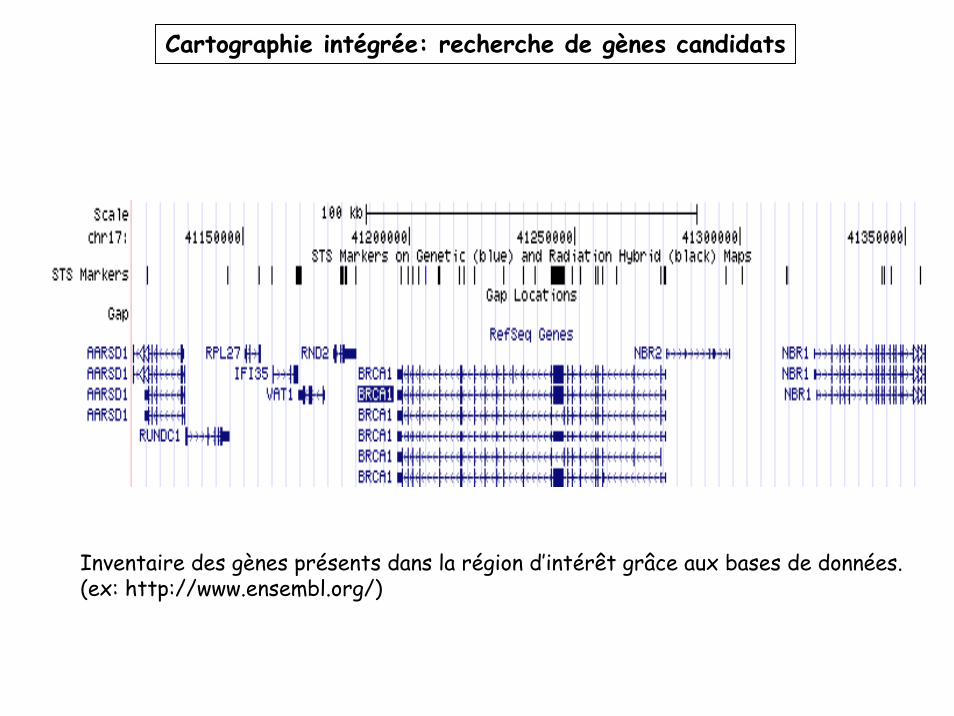
Inventaire des gènes présents dans la région d’intérêt grâce aux bases de données.(ex: http://www.ensembl.org/)
Cartographie intégrée: recherche de gènes candidats

Un cas réel: identification d’une relation entre l’exon 3 de la calpaïne et le diabète de type 2 non insulino-dépendant (NIDDM1) (Horikawa et al., Nature genetics 2000):
Cartographie intégrée: recherche de gènes candidats
Tiré du livre « Précis de génomique », Gibson et Muse, De Boeck editions
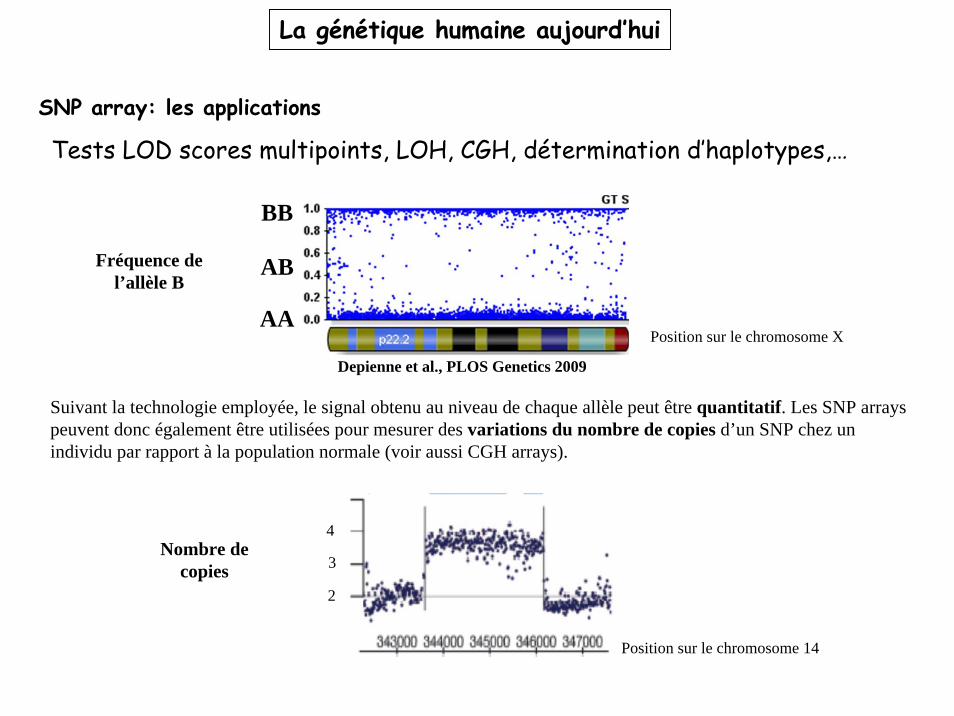
SNP array: les applications
Tests LOD scores multipoints, LOH, CGH, détermination d’haplotypes,…
Suivant la technologie employée, le signal obtenu au niveau de chaque allèle peut être quantitatif. Les SNP arrays peuvent donc également être utilisées pour mesurer des variations du nombre de copies d’un SNP chez un individu par rapport à la population normale (voir aussi CGH arrays).
Position sur le chromosome 14
2
3
4
Fréquence de l’allèle B
Position sur le chromosome X
BB
AB
AA
Depienne et al., PLOS Genetics 2009
Nombre de copies
La génétique humaine aujourd’hui
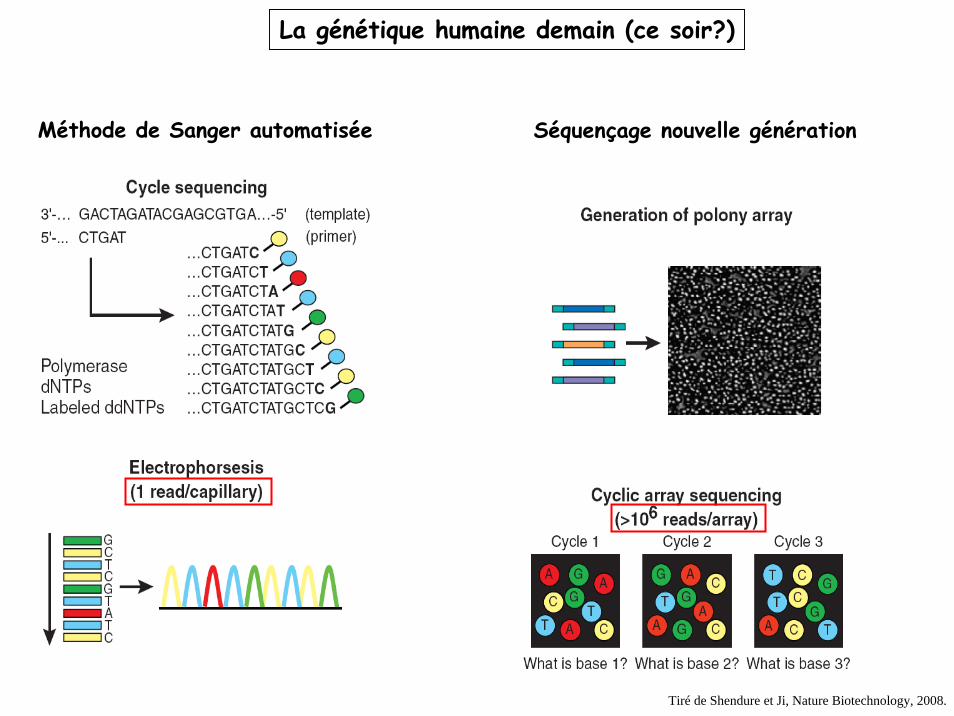
La génétique humaine demain (ce soir?)
Méthode de Sanger automatisée Séquençage nouvelle génération
Tiré de Shendure et Ji, Nature Biotechnology, 2008.

1990 10
1995 100
1999 5000
2002 20 000
2005 250 000
2007 1 000 000
2008 40 000 000
Nombre de séquences analysables en une seule expérience
1990 16 ans
2002 6 ans
2008 6 mois
Temps nécessaire au séquençage d’un génome humain
La génétique humaine demain (ce soir?)
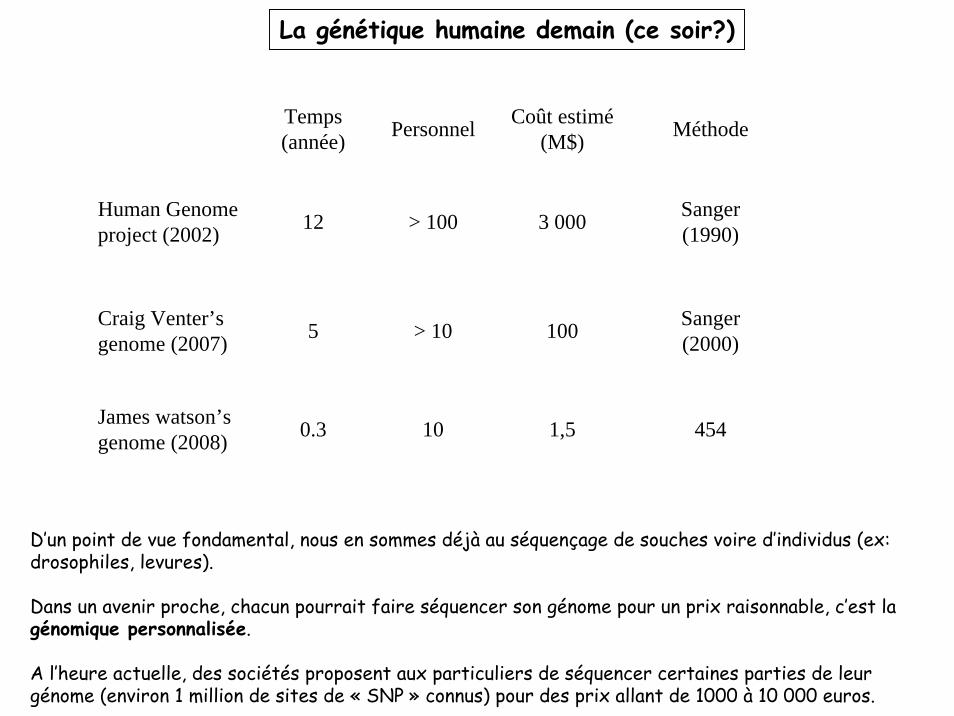
La génétique humaine demain (ce soir?)
Human Genome project (2002)
Craig Venter’s genome (2007)
James watson’s genome (2008)
Temps (année) Personnel Coût estimé
(M$)
12
5
0.3
Méthode
Sanger (1990)
Sanger (2000)
454
> 100
> 10
10
3 000
100
1,5
D’un point de vue fondamental, nous en sommes déjà au séquençage de souches voire d’individus (ex: drosophiles, levures).
Dans un avenir proche, chacun pourrait faire séquencer son génome pour un prix raisonnable, c’est lagénomique personnalisée.
A l’heure actuelle, des sociétés proposent aux particuliers de séquencer certaines parties de leur génome (environ 1 million de sites de « SNP » connus) pour des prix allant de 1000 à 10 000 euros.

Principales limitations
Quels que soient les progrès techniques de la génétique moléculaire:
La plupart des caractères (et des maladies humaines) ont un déterminisme multigénique: il n’y a pas une seule mutation causale mais tout un contexte génétique qui prédispose au développement de la maladie.
Une grande partie des caractères (et donc des maladies) sont influencés par l’environnement et l’histoire de vie des individus. La génétique n’aura donc pas toujours (rarement?) un caractère prédictif absolu.
C’est le cas par exemple de 90% des cancers, de l’autisme et de la plupart des cas d’obésité, qui sont clairement complexes au niveau génétique et fortement influencés par l’environnement. Pour ces maladies (exceptés les quelques cas à déterminisme génétique simple), on peut au mieux définir un indice génétique de prédisposition.
Les approches de génétique humaine modernes combinent donc des analyses génomiques globales avec la prise en compte d’un maximum de paramètres environnementaux et historiques. Elles utilisent les compétences des généticiens, des mathématiciens (analyses multivariées) et des informaticiens (algorithmes prédictifs d’apprentissage à partir de données hétérogènes complexes).

What would you do if you could sequence everything?
Kahvejian, Quackenbush et ThompsonNature Biotech. 2008
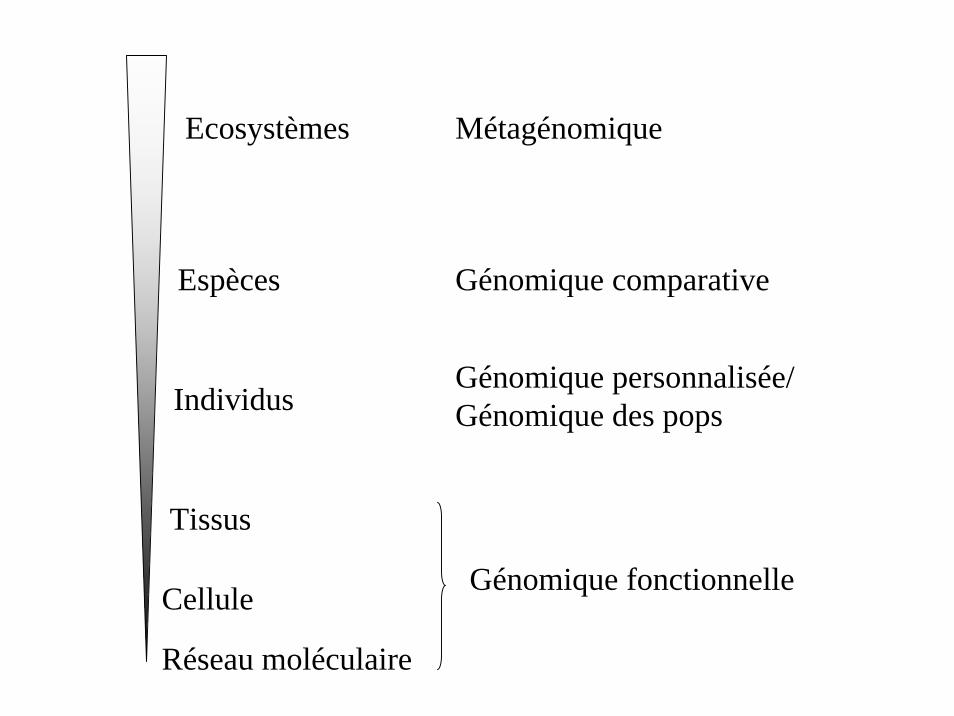
Réseau moléculaire
Cellule
Tissus
Individus
Espèces
Ecosystèmes
Génomique fonctionnelle
Génomique personnalisée/Génomique des pops
Génomique comparative
Métagénomique