Embed Size (px)
Citation preview

IPH/ EPI REPORTS Nr. 2000 - 012
Institut Scientifique de la Santé Publique Service d' Epidémiologie
RESEAU DE SURVEILLANCE DE LA MALADIE DE CREUTZFELDT-JAKOB EN
BELGIQUE
rapport annuel 1999
Pour la Commission Creutzfeldt -Jakob
S. QUOILIN

Rapport Annuel Commission CJD 2
IPH/ EPI REPORTS Nr. 2000 - 012
Réseau de surveillance de la Maladie de Creutzfeldt -Jakob
Service d'Epidémiologie, 2000; Bruxelles
Institut Scientifique de la Santé Publique, IPH/EPI REPORTS N° 2000 - 012
Numéro de dépôt: D/2000/2505/42
RESEAU DE SURVEILLANCE DE LA MALADIE DE CREUTZFELDT-JAKOB EN BELGIQUE
Institut Scientifique de la Santé Publique Service d'Epidémiologie 14, rue Juliette Wytsman 1050 Bruxelles ( 32 2 642 57 85 fax. 32 2 642 54 10 email: [email protected] http://www.iph.fgov.be/epidemio/

Rapport Annuel Commission CJD 3
IPH/ EPI REPORTS Nr. 2000 - 012
INTRODUCTION .................................................................................................................................. 4
COMMISSION CJD ..............................................................................................................................5
A. ORIGINE ........................................................................................................................................... 5 B. FONCTIONNEMENT............................................................................................................................ 5 C. ORGANISATION DU RESEAU DE SURVEILLANCE................................................................................... 6 D. ACTIVITES EN 1999........................................................................................................................... 7
1. Réunion annuelle .......................................................................................................................... 7 2. Collecte des données ..................................................................................................................... 7 3. List server..................................................................................................................................... 7 4. Site internet................................................................................................................................... 7 5. Réunion européenne...................................................................................................................... 7 6. Rédaction de recommandations pour les hôpitaux.......................................................................... 8 7. Autres ........................................................................................................................................... 8
GENERALITES..................................................................................................................................... 9
A. PROTEINES PRIONS ........................................................................................................................... 9 B. MECANISME PATHOGENE .................................................................................................................. 9 C. TRANSMISSION ............................................................................................................................... 10
MALADIE DE CREUTZFELDT-JAKOB..........................................................................................12
A. CJD ............................................................................................................................................... 12 1. Description clinique.................................................................................................................... 12 2. Examens diagnostiques ............................................................................................................... 14 3. Traitement .................................................................................................................................. 16
B. SITUATION EN BELGIQUE ................................................................................................................ 16 1. Caractéristiques épidémiologiques .............................................................................................. 16 2. Caractéristiques cliniques ........................................................................................................... 19 3. Caractéristiques géographiques .................................................................................................. 20
C. SITUATION EN EUROPE .................................................................................................................... 23 1. Suivi épidémiologique ................................................................................................................. 23 2. Résultats épidémiologiques ......................................................................................................... 23
ENCEPHALOPATHIE SPONGIFORME BOVINE .......................................................................... 26
A. SITUATION EN BELGIQUE ................................................................................................................ 26 B. SITUATION EN EUROPE .................................................................................................................... 27
PROBLEMES DE SANTE PUBLIQUE..............................................................................................29
A. RISQUE ET FACTEURS DE RISQUE ..................................................................................................... 29 1. Risque......................................................................................................................................... 29 2. Facteurs de risque....................................................................................................................... 29
B. EXPOSITION PROFESSIONNELLE ....................................................................................................... 30 C. TRANSFUSION SANGUINE................................................................................................................. 30 D. PRODUITS PHARMACEUTIQUES ........................................................................................................ 31 E. AUTRES .......................................................................................................................................... 31
CONCLUSION..................................................................................................................................... 32
ANNEXES ............................................................................................................................................ 33
A. CRITERES DIAGNOSTIQUES ET CLASSIFICATION ................................................................................ 33 1. Forme sporadique....................................................................................................................... 33 2. Forme variante ........................................................................................................................... 33
B. MATERIEL A RISQUE SPECIFIE (MRS)............................................................................................... 34 C. SITES INTERNET.............................................................................................................................. 34 D. MEMBRES DE LA COMMISSION CJD................................................................................................. 34
BIBLIOGRAPHIE...............................................................................................................................37

Rapport Annuel Commission CJD 4
Introduction La maladie de Creutzfeldt -Jakob (CJD) est une encéphalopathie spongiforme qui se caractérise par la détériorat ion rapide des fonctions cérébrales. Elle est décrite depuis près d’un siècle et a une incidence attendue de 1/1,000,000 habitants/an. Cette incidence attendue est également admise pour notre pays.
Trois formes de la maladie étaient décrites (familiale, s poradique et iatrogène) quant en octobre 1995, une quatrième forme fut mise en évidence pour la première fois en Grande -Bretagne; la nouvelle variante qui est aujourd’hui appelée variante (vCJD). Elle se distingue par quelques particularités anatomopatholo giques et surtout par la tranche d’âge touchée.
La maladie de Creutzfeldt -Jakob reste une maladie rare à laquelle on ne s’intéresse qu’en raison du lien de la forme variante avec l’encéphalopathie spongiforme bovine (ESB). La possible, très large, exposit ion de la population à ce nouvel agent, la protéine prion, suggère un risque épidémique qui à l’heure actuelle ne peut toujours pas être écarté.

Rapport Annuel Commission CJD 5
Commission CJD
A. Origine
Le lien possible entre la vCJD et l’ESB, lien établi en Grande -Bretagne en 1995, a forcé les autorités nationales et internationales à émettre des recommandations et à mettre en place des mesures de protection de la population.
L’épidémie de ESB en Grande -Bretagne et l’incrimination de l’utilisation de farines contenant des protéines animales dans l’alimentation du bétail ont forcé les autorités belges à prendre les mesures suivantes au niveau animal :
ü Interdiction d’importation du bétail britannique entre 1990 et 1999. La Belgique a suivi la recommandation de l’Union européenne qui a levé l’interdiction en août 1999.
ü Interdiction de l’utilisation de matériel à risque spécifié (voir définition en annexe) dans la chaîne alimentaire animale ou humaine depuis février 1998.
ü Interdiction d’utilisation de farines animales dans l’alimentatio n du bétail depuis 1994.
ü Système de surveillance de l’apparition de cas de ESB dans le bétail belge.
ü Mesures d’élimination des troupeaux en cas d’identification d’un cas en leur sein.
ü Recherche de l’origine de la contamination de chaque cas.
Les autorit és sanitaires belges se sont aussi interrogées sérieusement sur le risque d’épidémie de vCJD dans notre pays. La difficulté d’établir ce risque a induit la prise des mesures suivantes :
ü Elaboration d’un système de surveillance des différentes formes de la maladie de Creutzfeldt -Jakob via la création d’une Commission.
ü Sensibilisation des médecins belges à cette maladie et à l’importance de leur rôle dans un système de surveillance.
ü Participation à un projet européen de concertation.
La Commission CJD a ét é créée en 1998 à l’initiative du Ministère de la Santé Publique et est l’une des commissions du Conseil Supérieur de l’Hygiène.
B. Fonctionnement
La Commission est composée de représentants du Ministère de la Santé Publique, de 7 universités belges et de l’Institut Scientifique de la Santé Publique (I.S.P.).
La Commission rend des avis.
Elle a rédigé le document d’informations générales ∗, établi les procédures du système de surveillance et opté pour la réalisation d’une enquête épidémiologique sur base du questionnaire européen.
La Commission assure un suivi de l’apparition de cas de cette maladie via le Groupe Technique. Le Groupe Technique rassemble, sur base volontaire, les médecins issus des 7 centres universitaires et de l’ISP. Il se réunit aussi souvent que nécessaire pour discuter des nouveaux cas, effectuer le suivi épidémiologique ou discuter de recommandations en matière de santé publique.
La coordination des activités et le secrétariat de la Commission sont assurés par l’ISP.
∗ Dossier technique de la Commission CJD, disponible auprès de l’ISP.

Rapport Annuel Commission CJD 6
Une collabora tion étroite sera assurée avec les médecins traitants (neurologues et généralistes).
La diffusion des informations scientifiques ainsi que des résultats émanant du groupe de travail sera effectuée régulièrement vers les personnes concernées.
Le fonctionne ment du réseau de surveillance repose sur l’identification correcte des cas et donc sur la réalisation systématique des autopsies des patients suspects de Creutzfeldt -Jakob. Afin de lever le frein éventuel que représente le coût des autopsies, le fonctionn ement du réseau inclut un remboursement des autopsies qui est accordé aux centres universitaires de référence selon un montant forfaitaire.
C. Organisation du réseau de surveillance
Le réseau de surveillance fonctionne grâce à la participation des généra listes qui réfèrent les patients qui présentent des symptômes suggestifs vers les spécialistes en neurologie. Lorsque ceux-ci on établit un diagnostic possible de CJD, ils peuvent référer les patients vers l’un des 7 centres universitaires dit de référence . Ces centres offrent une aide à la confirmation du diagnostic. Cinq parmi eux sont habilités à réaliser des autopsies (voir la liste en annexe ). A l’heure actuelle, seul l’examen neuro -anatomopathologique autorise une confirmation du diagnostic.
N.B. : La Commission confirme que les autopsies des patients suspects de CJD doivent être réalisées dans ces centres car
ü Elle doivent se faire dans des conditions d’hygiène et d’asepsie que tous les centres périphériques ne peuvent assurer.
ü Le coût du déplaceme nt du spécialiste est aussi important, spécialistes qui par ailleurs n’ont pas le temps d’effectuer ces déplacements.
Il y a donc lieu d’expliquer clairement aux familles des patients quels sont les objectifs de l’autopsie (confirmer le diagnostic, défin ir les implications éventuelles au niveau génétique mais également les aspects de santé publique) et que celle -ci n’entraînera aucun coût à leur charge.
L’Institut Scientifique de la Santé Publique est en relation avec les 7 centres universitaires dans le cadre de la Commission CJD et exerce les rôles de coordination des activités et de secrétariat de la Commission. Après accord de ces centres de référence, dans le cadre de l’enquête épidémiologique, il peut être amené à prendre contact avec les autres pa rtenaires que sont les neurologues et les médecins généralistes.
Schéma qui présente les liens entre les différents partenaires du réseau
E
Neurologues
7 Centres de référence
Généralistes
Institut Scientifique de la Santé Publique
Ministère d e la Santé Publique - Conseil supérieur de l’Hygiène - Commission CJD

Rapport Annuel Commission CJD 7
D. Activités en 1999
1. Réunion annuelle
La réunion annuelle de la Commission s’est tenue le mardi 14 décembre 1999. Le but de cette réunion est d’établir la synthèse de la situation de la CJD en Belgique et le bilan des activités de l’année écoulée.
Au cours de cette réunion, il a été décidé d’organiser une journée scientifique autour du thème de la CJD et ESB. Elle se tiendra le samedi 18 novembre 2000. Tout renseignement complémentaire peut être obtenu auprès de l’I.S.P.
2. Collecte des données
Les sept centres de référence en matière de CJD transmettent régulièrement un paquet minimum d’informations à l’ISP qui se charge de les traiter.
Selon le mode de fonctionnement du réseau de surveillance, l’ensemble des autopsies effectuées dans le cadre d’une suspicion de maladie de Creutzfeldt -Jakob ont été remboursées.
De janvier 98 à septem bre 99, 27 autopsies ont ainsi été remboursées pour un total de 21 cas confirmés ou probables de CJD.
Outre cette récolte d’informations minimales, la commission avait accepté le principe de participer à une enquête cas -témoins selon le modèle britannique. Dans le cadre d’une mise en commun européenne des données, recueillir une information sur une dizaine de cas par an prenait un sens. L’objectif de cette étude est d’essayer d’identifier des facteurs de risque d’exposition à cette maladie ou facteurs de ri sque de la développer. L’enquête cas -témoins débutera pour les cas identifiés à partir du premier janvier 2000 grâce au renfort de l’UIA dont une infirmière pourra prendre du temps pour rendre visite à la famille des patients et à des témoins.
3. List server
Le « list server » est une adresse mail commune à un groupe de personnes qui peuvent ainsi communiquer aisément. L’objectif est de communiquer rapidement des informations d’intérêt général et à caractère scientifique ou pratique. Ce list server est ou vert à un nombre restreint de participants à savoir les membres de la Commission CJD et tout groupe de travail et de recherche dans les domaines de l’ESB ou CJD en Belgique. Il suffit d’envoyer les références à l’I.S.P. qui rajoutera les adresses qui serai ent proposées. Chaque membre de la Commission peut évidemment proposer des adresses supplémentaires.
4. Site internet
Le site internet de l’ISP comporte à ce jour les rapports annuels de la Commission. Le contenu de ce site sera étoffé progressivement.
De nombreux sites CJD existent et les références de deux sites intéressants sont données en annexe (point C).
5. Réunion européenne
En 1999, le groupe européen Biomed II s’est réuni à deux reprises. Le but de ces réunions est de suivre l’évolution de la mala die de Creutzfeldt -Jakob dans les différents pays européens et de faire le bilan des recherches cliniques et diagnostiques.
Si dans un premier temps, il apparaît essentiel d’ajuster les connaissances et établir un consensus à ce sujet, la maladie de Creut zfeldt-Jakob reste un maladie marginale sauf si on convient qu’en raison de son lien avec l’ESB, la variante peut constituer un problème de santé publique. Pour cette raison, les problèmes de santé publique (recommandations pour la

Rapport Annuel Commission CJD 8
prévention de la transmi ssion dans les hôpitaux, risque des transfusions sanguines, … ) seront traités au cours des prochaines réunions.
Par ailleurs, un forum de discussion européen autour de l’utilisation de la protéine 14 -3-3 comme critère diagnostic de la maladie de Creutzfel dt-Jakob a permis la mise en place de standards quant à la méthode et dans l’interprétation des résultats.
6. Rédaction de recommandations pour les hôpitaux
En raison des trois facteurs suivants : risque de transmission iatrogène de la maladie, inconnue q uant à la contamination exacte de la population par l’agent de l’ESB et grande résistance de cet agent aux méthodes de désinfection habituelle, la Section Hygiène Hospitalière du Conseil Supérieur de l’Hygiène a désiré émettre des recommandations pour la prévention de la transmission de CJD dans les hôpitaux. Un comité d’experts issus de cette section et de la Commission CJD s’est réuni afin d’écrire ces recommandations. Le document est dans sa phase de finalisation.
Ce texte sera diffusé par la Section Hy giène Hospitalière du Conseil Supérieur de l’Hygiène à l’ensemble des hôpitaux mais il est disponible sur demande auprès de l’I.S.P. et sera prochainement placé sur le site internet.
7. Autres
Le prochain thème abordé par un comité d’experts sera le risqu e lié à la transfusion sanguine.

Rapport Annuel Commission CJD 9
Généralités
A. Protéines Prions
La protéine Prion a été identifiée comme agent causal des encéphalopathies spongiformes transmissibles par Prusiner en 1982.
La protéine Prion, PrPc, protéine normale du cerveau, est enco dée et transcrite du gène prnp qui est situé sur le chromosome 20. Le gène de la protéine prions contient 253 acides aminés.
La fonction physiologique de cette protéine n’est pas encore bien connue. On pense que la déficience en cette protéine pourrait me ner à la neurotoxicité via la diminution de la résistance neuronale au stress oxydatif et autres mécanismes associés.
La PrPsc, protéine anormale, est transcrite du même gène et a donc la même séquence d’acides aminés que la protéine normale.
Les protéin es normales et anormales présentent la même séquence d’acides aminés, les mêmes chaînes latérales d’hydrates de carbone et la même terminaison glycolipidique. La seule différence entre ces protéines résulte en leur structure tridimensionnelle. Cette variat ion confère aux protéines anormales une résistance à l’action des protéases.
Une protéine normale exposée aux protéases est complètement dégradée tandis qu’une protéine anormale libère un fragment appelé PrP 27 -30.
La terminaison glycolipidique permet aux protéines de s’ancrer à la surface des membranes cellulaires où se trouvent donc la majorité des protéines normales. Par contre, les PrPsc s’accumulent à l’intérieur de la cellule.
Les protéines normales sont abondantes dans le cerveau (neurones, astrocy tes et cellules gliales) mais on les retrouve également dans les poumons, la rate, les muscles et les lymphocytes B et T.
Dans la maladie de Creutzfeldt -Jakob, il a été mis en évidence l’existence de 4 modèles de protéines. Si le type 4 se retrouve dans l a forme variante, les types 1 à 3 sont présents dans les autres formes de la maladie. ( 1)
B. Mécanisme pathogène
Le mécanisme pathogène doit être explicité à trois niveaux :
1. Ingestion de l’agent et dissémination
2. Transformation de la protéine
3. Atteinte des cellules cérébrales
1. Ingestion de l’agent et dissémination
Chez la vache, le mécanisme pourrait être le suivant :
Les protéines anormales sont ingérées. Leur résistance à l’acidité de l’estomac et aux enzymes intestinales leur permet d’atteindre les plaques de Peyer de l’iléon termi nal où elles sont absorbées. Ensuite, via le système lymphatique, elle gagne la rate. A ce niveau, deux théories prévalent. Soit elles remontent vers le cerveau via le système nerveux périphérique soit elles s’y hissent toujours via le système lymphatiques où les lymphocytes B et T constitueraient ensuite le moyen de franchir la barrière hémato -encéphalique.
Ceci a été démontré de manière expérimentale. Lorsque l’on infecte du bétail avec 100 gr de tissu cérébral de souris infectée, on retrouve l’agent au niveau de l’iléon distal après 6 mois pour disparaître après 22 à 26 mois et enfin réapparaître ensuite. Après 35 mois, juste avant l’apparition des signes cliniques, on le retrouve dans le cerveau, la moelle épinière, les ganglions cervicaux et thoracique s et dans la moelle osseuse ( 2).

Rapport Annuel Commission CJD 10
Chez l’h omme non plus, le mécanisme n’est pas clairement identifié. Les protéines PrPsc ont été identifiées dans les amygdales et dans l’appendice. Le tissu lymphoréticulaire serait donc également impliqué dans la pathogénèse de cette maladie. A l’intérieur de ce processus, les lymphocytes circulants auraient un rôle essentiel.
2. Transformation de la protéine
Puisque les formes normales et anormales sont constituées de la même séquence d’acides aminés, la forme anormale provient d’une altération post -transcriptio nnelle. Cette altération modifie sa conformation ce qui lui confère des propriétés physio -chimiques différentes. La PrPsc devient résistante aux protéases et s’accumulent sous forme de dépôts amyloïdes.
Le mécanisme par lequel cette structure est altérée n ’est pas encore clairement identifié.
• Origine exogène pure
c.-à-d. par ingestion d’une dose infectante de PrPsc suffisante.
• Transformation de la protéine normale en protéine anormale (3)
La proximité de l’agent, la protéine PrPsc exogène, induirait directement la transformation des protéines normales en protéines anormales.
Ou
L’action d’une troisième protéine, protéine chaperonne, qui serait une enzyme et qui en présence d’une protéine PrPsc exogène, induirait la transformation des protéines normales en protéines anormales ( 4).
3. Atteinte des cellules cérébrales
Quel est le mécanisme exact au niveau cé rébral ?
• Soit les PrPsc s’accumulent dans les astrocytes qui ne peuvent plus assurer leur fonction. Quant un grand nombre d’astrocytes sont étouffés par les protéines anormales, les neurones meurent.
• Soit la PrPsc agit directement sur les neurones.
La progression de la maladie est en tous les cas due à l’accumulation continue de protéines anormales dans le cerveau.
C. Transmission
1. Lien entre ESB et vCJD
Le lien entre l’ESB et la vCJD mis en évidence en 1997 au cours d’une étude expérimentale (30) a été formellement confirmé par une expérience sur des souris transgéniques ( 5).
L’expérience peut être résumée de la façon suivante : des chercheurs ont administré des prions de l’ESB et de la vCJD à des souris transgéniques. Elles ont développé les mêmes symptômes neurologiques après une période d’incub ation de 250 jours. Tandis que les souris infectées par l’agent de la tremblante du mouton, ont développé une maladie bien différente.
La voie orale serait la voie de transmission. Elle est, en tous les cas, utilisée pour infecter les animaux de manière e xpérimentale.
La voie parentérale a également été utilisée avec succès dans ce cadre.
2. Transmission iatrogène
Bien que la CJD soit une maladie dégénérative et qu’il n’existe pas de preuve que l’agent puisse se transmettre de personne à personne lors d e contacts étroits ou lors de certaines activités à risque, il apparaît que CJD soit transmissible sous certaines conditions.
Ce mécanisme n’est pas encore bien compris bien que le consensus soit obtenu pour considérer la protéine PrPsc comme l’agent infe ctieux.

Rapport Annuel Commission CJD 11
Chez l’homme, on connaît surtout la transmission iatrogène survenue à la suite de certaines interventions chirurgicales comme des greffes de dure -mère et de cornée ou lors de l’injection d’hormones de croissance extraites de cadavre.
Par ailleurs , en raison de l’identification de protéines anormales au niveau des amygdales et de l’appendice chez l’homme et en raison de la résistance de la protéine anormale aux mesures de décontamination habituelle du matériel chirurgical, il n’est pas exclu que l’ on puisse craindre un tel mode de transmission pour la vCJD.
3. Transmission verticale
Notons encore qu’un risque de transmission verticale, de la mère à l’enfant, à été suggéré mais non confirmé, à la suite d’un cas suspect en Grande -Bretagne.

Rapport Annuel Commission CJD 12
Maladie de Creutzfeldt-Jakob
A. CJD
1. Description clinique
La maladie de Creutzfeldt -Jakob appartient au groupe des encéphalopathies spongiformes subaiguë. Elles sont décrites comme
subaiguës en raison de l’évolution rapide de la maladie qui évolue inexorablem ent vers la mort.
spongiformes en raison des lésions spongieuses identifiées au cours de l’examen neuropathologiques.
Le tableau ci -dessous présente les encéphalopathies spongiformes transmissibles
Sporadique Maladie de Creutzfeldt -Jakob (80 à 85% des ca s)
Héréditaires Syndrome de Gerstmann -Sträussler -Scheinker
Maladie de Creutzfeldt -Jakob (10 à 15% des cas)
Insomnie fatale familiale
Acquises Maladie de Creutzfeldt -Jakob iatrogène
Kuru
Variante Creutzfeldt -Jakob
Toutes ces formes ont des caractéristiq ues communes :
ü Longues périodes d’incubation,
ü Evolution progressive de la gravité,
ü Décès inéluctable,
ü Pas de réponse immunitaire, lésions dégénératives non inflammatoires au niveau du SNC.
Il existe donc quatre formes de CJD :
Sporadique,
Familiale,
Iatrogène,
Variante.
La définition des formes sporadique et variante se trouve en annexe.
Un aperçu global des caractéristiques de ces 4 formes sont présentées dans le tableau ci -dessous ( 4).

Rapport Annuel Commission CJD 13
Sporadique Héréditaire Iatrogène Variante
Fréquence 90% 10% / /
Age moyen 60 ans 34-50 ans Variable par définition 28 ans
Facteurs de risque Prédisposition génétique Maladie autosomique dominante Hormones de croissance, Greffe de dure-mère ou cornée, Intervention neuro-chirurgicale, …
Vivre en Grande-Bretagne (ESB) Codon 129 méthionine – homozygote Age (moyenne 28 ans)
Létalité 100% 100% 100% 100%
Temps survie après apparition 1er symptômes
6 à 12 mois Plus long 22 mois à 13 ans environ 12 mois
Symptômes Diminution des capacités intellectuelles jusqu'à la démence.
Idem Idem Les symptômes sont identiques mais la première phase de la maladie est marquée par les symptômes psychiatriques.
Symptômes neurologiques: Ataxie locomotrice Mouvements involontaires Dysarthrie, myoclonie Chorée et mouvements athétosiques
Les autres symptômes surviennent plus tard.
Hypertonie musculaire Syndrome pyramidal bilatéral Cécité corticale Epilepsie Mutisme, coma et décès
Test diagnostic EEG: activité paroxystique
LCR: protéines cérébrales spécifiques
EEG est souvent normal
Idem
Test de confirmation Autopsie cérébrale Autopsie cérébrale donne des lésions d'aspects différents et constitue le seul examen de certitude.
Immunohistochimie ou immunoblot pour mise en évidence de PrPsc
Idem
RMN

Rapport Annuel Commission CJD 14
2. Examens diagnostiques
2.a EEG et RMN
L’EEG (6) et de plus en plus la RMN sont des éléments essentiels pour poser un diagnostic correct de la maladie de Creutzfeldt-Jakob.
L’image de RMN donne un signal de haute densité au niveau thalamus postérieur (7-10). En ce domaine, il existe un projet de recherche européen auquel participe une équipe de la KUL.
2.b Protéine 14-3-3
« La protéine 14-3-3 est probablement une protéine synaptique qui est principalement produite par les neurones. Dans la maladie de Creutzfeldt-Jakob, cette protéine peut être détectée de manière spécifique dans le LCR. Elle est alors le reflet de la mort neuronale qui caractérise la maladie de Creutzfeldt-Jakob.
Le laboratoire de Neurobiologie de l’UIA (voir coordonnées en annexe) recherche la présence de protéine 14-3-3 à la demande du médecin traitant qui inclut la maladie de Creutzfeldt-Jakob dans son diagnostic différentiel. A cet effet, il est demandé un bref rapport sur l’évolution clinique du patient. La protéine est détectée selon un protocole de détection immunologique via western blot.
En 1999, le laboratoire de l’UIA a reçu 57 échantillons de LCR. Sur base des données cliniques et anatomopathologiques complémentaires, 46 d’entre eux (81%) ont été classés comme cas possibles de CJD.
Pour 40 patients, l’évolution clinique et neuropathologique ont été transmises (70%).
La protéine a été détectée dans 17 des échantillons de LCR. Un diagnostic définitif de CJD a été posé dans 9 cas. Pour 5 autres cas, il y a un diagnostic probable car l’autopsie n’a pas été effectuée (3 patients) ou car les données ne sont pas encore disponibles (2 patients).
La protéine a été détectée chez trois patients pour lesquels un autre diagnostic a été posé (encéphalopathie à herpès simplex, encéphalopathie métabolique ou démence diffuse Lewy Body).
Chez les 23 patients restants, la protéine n’a pu être détectée et dans tous les cas un diagnostic autre que CJD a été posé.
Ce test de détection de la protéine 14-3-3, utilisé parmi des patients avec un diagnostic possible de CJD, a une sensibilité de 100% et une spécificité de 89%. »
Par M. Bart Van Everbroeck, UIA
Cette protéine normale du cerveau n’est donc pas spécifique de CJD mais sa présence doit être replacée dans un éventuel cadre suggestif de la maladie.
Plusieurs pays européens réalisent ces dosages et leurs résultats sont mis en commun afin d’améliorer les capacité et critère diagnostiques.
Par ailleurs, si la sensibilité du test dans des cas de forme sporadique est très élevée, il n’en va pas de même dans les cas de forme variante. La sensibilité est de 52% pour une spécificité de 91% (Travaux de Alison Green, données présentées au cours de la rencontre européenne, avril 2000).
2.c Autopsie
A l’heure actuelle, la confirmation du diagnostic ne se fait que sur base de l’examen neuro-pathologique. La biopsie cérébrale ne peut être utile que si elle contient un champs de lésions spécifiques.
Les lésions neurologiques caractéristiques sont :
Aspect spongiforme,
Perte neuronale,

Rapport Annuel Commission CJD 15
Prolifération astrocytaire et gliose,
Avec des dépôts de PrPsc et une absence d’image inflammatoire.
En plus de ces lésions caractéristiques, l’identification de 4 types moléculaires de la protéine PrPsc a permis un diagnostic de variante lors des autopsies du cerveau. Tous les cas de CJD sporadiques ou iatrogéniques sont de type 1-3 tandis que tous ceux de la forme variante sont de type 4.
2.d Biopsie tonsillaire
Les protéines prions se répliquent dans le tissu lympho-réticulaire avant d’entamer l’invasion du SNC. Dans une phase précoce de la maladie, la biopsie de tissu lymphoréticulaire a donc été suggérée comme moyen de diagnostic de la maladie. Une étude démontre que l’analyse par western blot ou immunohistochimie était positive pour toutes les biopsies issues de patients atteints de vCJD tandis qu’elle ne l’était pas pour les contrôles (11, 12, 13).
Ces biopsies peuvent être réalisées sur la rate, l’appendice ou des ganglions lymphatiques.
La généralisation de cet examen comme test de dépistage n’est toutefois pas admise au niveau du groupe européen. Tout d’abord car aucune solution de traitement ne pourrait être apportée aux patients. Il s’agit donc d’abord d’une question d’éthique. Ensuite, la PrPsc ne serait présente dans les amygdales et appendices que très tôt après le contact infectant et de manière provisoire. Comme test de dépistage, on risque de sous-estimer l’importance du phénomène.
Remarques :
Si l’agent infectant se trouve dans ces organes de 2 à 3 ans avant le début de la phase clinique de la maladie, une contamination iatrogène via les interventions chirurgicales peut et/ou a pu se produire. Si la population a été exposée à l’agent, eu égard à la période d’incubation, on peut craindre la survenue d’un réel problème de santé publique au cours des années à venir.
2.f Analyse génétique
Il existe un polymorphisme sur le codon 129 dans la population normale : 40% de Méthionine-Méthionine (M-M), 10% de Valine-Valine (V-V) et 50% de Méthionine-Valine (M-V). Cette répartition varie en fonction de l’âge et un peu en fonction des pays (4).
Cette séquence induit-elle une plus grande sensibilité ou diminue-t-elle la période d’incubation que certains estiment s’étaler entre 6 et 20 ans ?
Des études antérieures avaient déjà montré une représentation plus importante d’homozygotes M-M parmi les patients atteints de la forme sporadique (69%) par rapport à la population générale (39%). Il est d’autant plus troublant de constater que tous les cas de la variante CJD sont porteurs du génotype M-M sur le codon 129.
La détermination du génotype du codon 129 est un élément important dans un cas de CJD car il est un facteur de susceptibilité et un déterminant du phénotype clinique.
Dans la GSS, il y a une mutation sur le gène de la protéine prions en position 102 où une leucine remplace une proline.
Dans l’Insomnie Familiale Fatale comme dans la forme familiale de Creutzfeldt-Jakob, il y a une mutation en position 178. La même mutation est donc cause de deux syndromes différents. La distinction va se produire en fonction de la présence d’une méthionine ou d’une valine sur le codon 129. On aura alors respectivement une IFF ou une CJD.
L’identification du génotype est donc très importante bien que les résultats ne reposent encore que sur de faibles échantillons de population. Il faudra réaliser cette analyse sur l’ensemble des cas de CJD et sur des échantillons plus larges dans la population générale.
La mise en commun et la confrontation des données provenant des différents pays européens est à ce niveau aussi un élément essentiel.

Rapport Annuel Commission CJD 16
3. Traitement
Il n’existe toujours pas de traitement de cette pathologie bien qu’il existe des voies de recherche (14).
Ces voies se basent sur la meilleure connaissance du mécanisme pathologique et s’orientent selon trois axes :
Diminuer la production de PrPsc
Prévenir l’altération post-transcriptionnelle de la PrPc
Prévenir la neurotoxicité de la PrPsc et les dépôts amyloïdes
Des expériences réalisées in vitro ne sont pas encore concluantes.
B. Situation en Belgique
1. Caractéristiques épidémiologiques
1.a Nombre de cas
Les chiffres présentés pour la période 1988-1997 sont issus de l’étude rétrospective (15) menée par le Professeur P. Cras et son équipe. Ensuite, les chiffres proviennent de la surveillance active mise en route en 1998.
Remarque :
Les résultats de 1998 qui apparaissent dans ce rapport ne sont pas identiques à ceux exposés dans le rapport précédent. Les caractéristiques démographiques et cliniques d’un patient décédé en 1998, et pour lequel un diagnostic final n’a été posé qu’en 1999, ont été ajoutées. Il n’y a toutefois pas de différence en termes démographiques ou cliniques à pointer.
Le diagnostic final, pour ce patient qui présentait des lésions hautement suspectes à l’examen neuropathologique, a été donné par examen immuno-histochimique qui a montré un immuno-marquage pour la protéine prion à l’aide de l’anticorps monoclonal KG9.
Le nombre de nouveaux cas pour 1998 se monte donc à 12 cas confirmés et 2 cas probables.
Nombre de cas confirmés et probables des différentes formes de CJD en Belgique entre 1988 et 1999
1988 1989 1990 1991 1992 1993 1994 1995 1996 1997 1998 1999 Total
Sporadic 5 4 5 7 4 5 5 6 6 10 14 13 84
Familial 0 0 0 0 1 1 0 0 0 1 0 0 3
Iatrogenic 0 0 0 0 0 0 0 0 0 0 0 0 0
v CJD 0 0 0 0 0 0 0 0 0 0 0 0 0
5 4 5 7 5 6 5 6 6 11 14 13 87
En 1999, il y eut 22 patients référés pour suspicion de CJD dans les 7 centres universitaires de référence. Pour 7 patients, l’autopsie a permis d’exclure le diagnostic de CJD, le diagnostic pour 2 patients est encore en cours, 3 patients sont des cas probables et 10 patients sont des cas qui ont été confirmés par un examen neuropathologique.
En 1999, les 13 cas sont de forme sporadique.
En Belgique, entre 1988 et 1999, tous les cas sont de forme sporadique à l’exception de 3 cas de forme familiale identifiés en 1992, 1993 et 1997. Aucun cas de forme iatrogène ou variante n’a été identifié à ce jour.
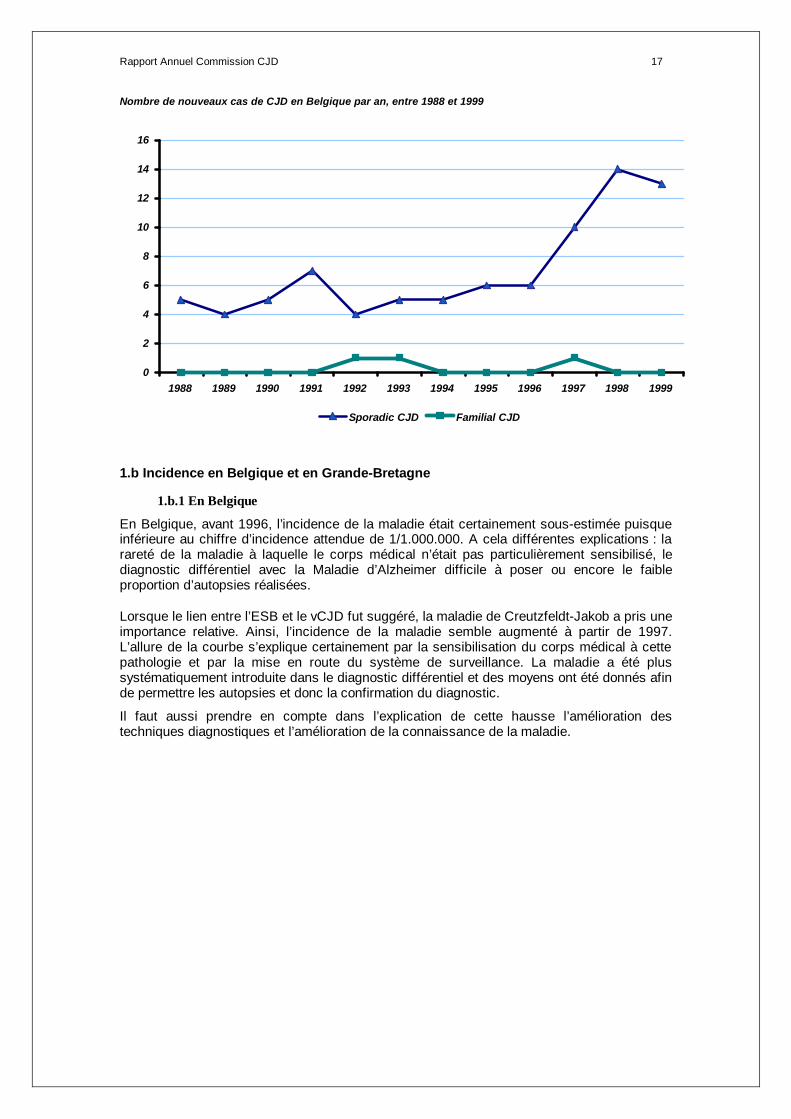
Rapport Annuel Commission CJD 17
Nombre de nouveaux cas de CJD en Belgique par an, entre 1988 et 1999
0
2
4
6
8
10
12
14
16
1988 1989 1990 1991 1992 1993 1994 1995 1996 1997 1998 1999
Sporadic CJD Familial CJD
1.b Incidence en Belgique et en Grande-Bretagne
1.b.1 En Belgique
En Belgique, avant 1996, l’incidence de la maladie était certainement sous-estimée puisque inférieure au chiffre d’incidence attendue de 1/1.000.000. A cela différentes explications : la rareté de la maladie à laquelle le corps médical n’était pas particulièrement sensibilisé, le diagnostic différentiel avec la Maladie d’Alzheimer difficile à poser ou encore le faible proportion d’autopsies réalisées.
Lorsque le lien entre l’ESB et le vCJD fut suggéré, la maladie de Creutzfeldt-Jakob a pris une importance relative. Ainsi, l’incidence de la maladie semble augmenté à partir de 1997. L’allure de la courbe s’explique certainement par la sensibilisation du corps médical à cette pathologie et par la mise en route du système de surveillance. La maladie a été plus systématiquement introduite dans le diagnostic différentiel et des moyens ont été donnés afin de permettre les autopsies et donc la confirmation du diagnostic.
Il faut aussi prendre en compte dans l’explication de cette hausse l’amélioration des techniques diagnostiques et l’amélioration de la connaissance de la maladie.
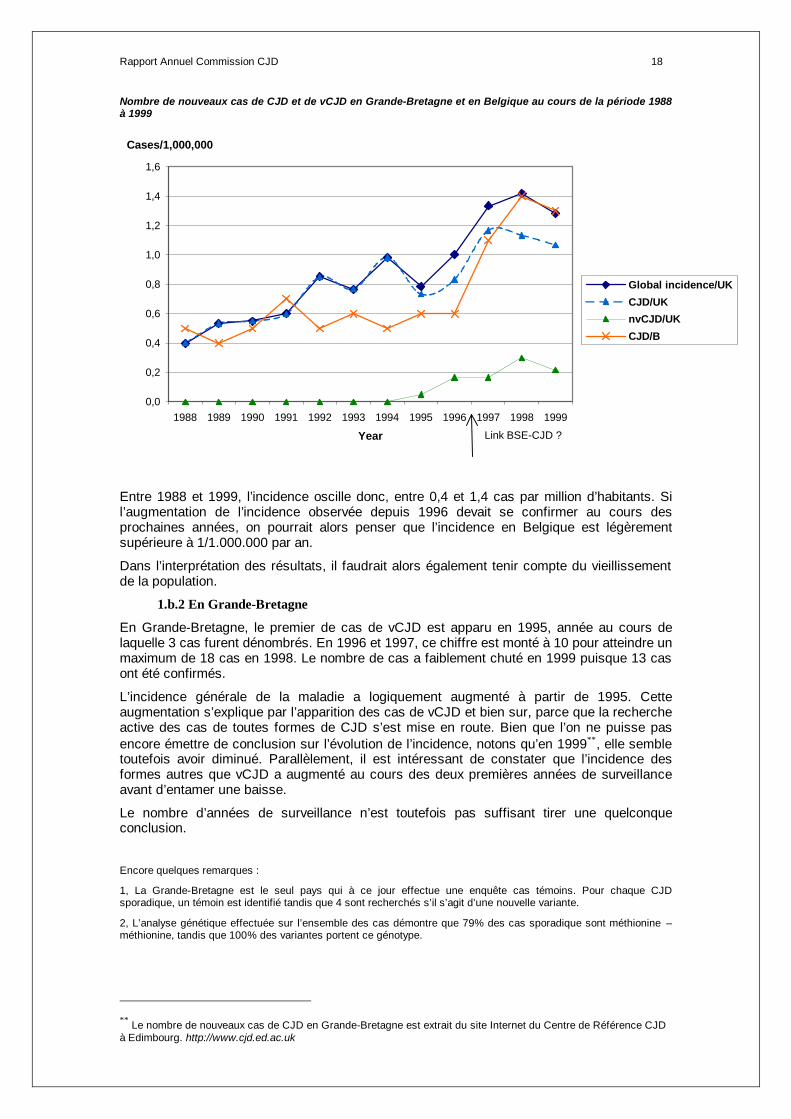
Rapport Annuel Commission CJD 18
Nombre de nouveaux cas de CJD et de vCJD en Grande-Bretagne et en Belgique au cours de la période 1988 à 1999
0,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
1,6
1988 1989 1990 1991 1992 1993 1994 1995 1996 1997 1998 1999
Year
Cases/1,000,000
Global incidence/UK
CJD/UKnvCJD/UK
CJD/B
Link BSE-CJD ?
Entre 1988 et 1999, l’incidence oscille donc, entre 0,4 et 1,4 cas par million d’habitants. Si l’augmentation de l’incidence observée depuis 1996 devait se confirmer au cours des prochaines années, on pourrait alors penser que l’incidence en Belgique est légèrement supérieure à 1/1.000.000 par an.
Dans l’interprétation des résultats, il faudrait alors également tenir compte du vieillissement de la population.
1.b.2 En Grande-Bretagne
En Grande-Bretagne, le premier de cas de vCJD est apparu en 1995, année au cours de laquelle 3 cas furent dénombrés. En 1996 et 1997, ce chiffre est monté à 10 pour atteindre un maximum de 18 cas en 1998. Le nombre de cas a faiblement chuté en 1999 puisque 13 cas ont été confirmés.
L’incidence générale de la maladie a logiquement augmenté à partir de 1995. Cette augmentation s’explique par l’apparition des cas de vCJD et bien sur, parce que la recherche active des cas de toutes formes de CJD s’est mise en route. Bien que l’on ne puisse pas encore émettre de conclusion sur l’évolution de l’incidence, notons qu’en 1999∗∗, elle semble toutefois avoir diminué. Parallèlement, il est intéressant de constater que l’incidence des formes autres que vCJD a augmenté au cours des deux premières années de surveillance avant d’entamer une baisse.
Le nombre d’années de surveillance n’est toutefois pas suffisant tirer une quelconque conclusion.
Encore quelques remarques :
1, La Grande-Bretagne est le seul pays qui à ce jour effectue une enquête cas témoins. Pour chaque CJD sporadique, un témoin est identifié tandis que 4 sont recherchés s’il s’agit d’une nouvelle variante.
2, L’analyse génétique effectuée sur l’ensemble des cas démontre que 79% des cas sporadique sont méthionine – méthionine, tandis que 100% des variantes portent ce génotype.
∗∗ Le nombre de nouveaux cas de CJD en Grande-Bretagne est extrait du site Internet du Centre de Référence CJD à Edimbourg. http://www.cjd.ed.ac.uk
Rapport Annuel Commission CJD 19
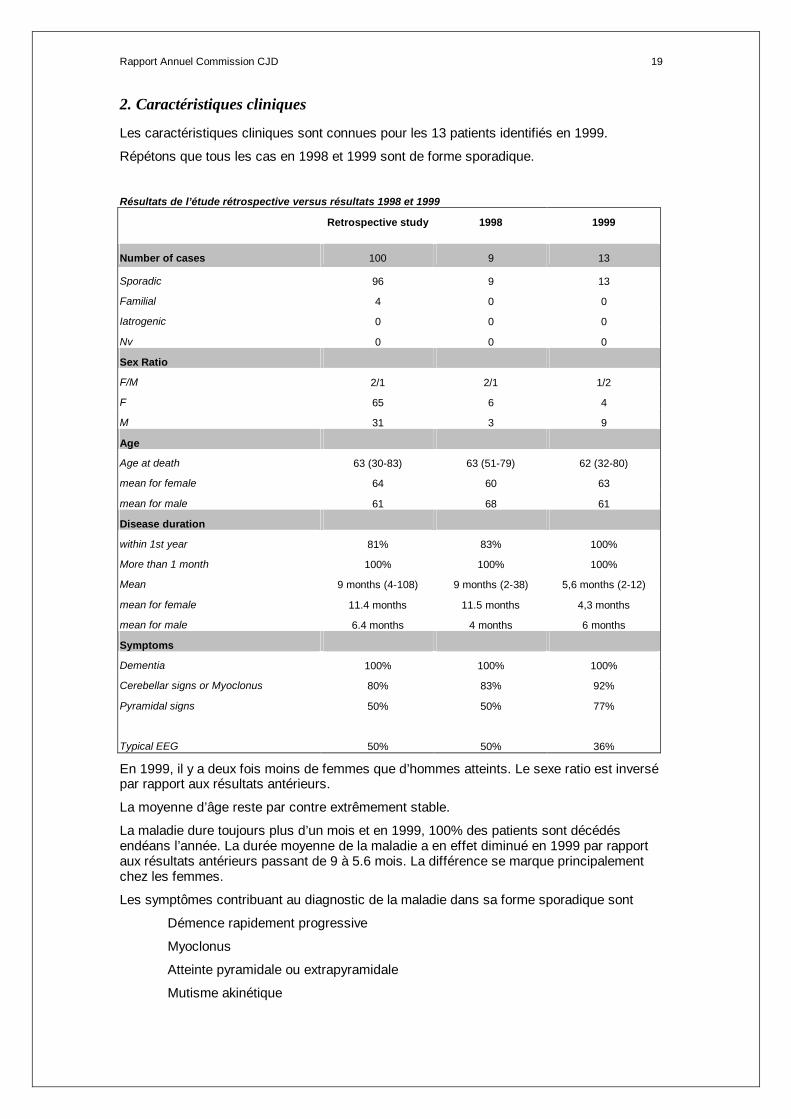
2. Caractéristiques cliniques
Les caractéristiques cliniques sont connues pour les 13 patients identifiés en 1999.
Répétons que tous les cas en 1998 et 1999 sont de forme sporadique.
Résultats de l’étude rétrospective versus résultats 1998 et 1999
Retrospective study 1998 1999
Number of cases 100 9 13
Sporadic 96 9 13
Familial 4 0 0
Iatrogenic 0 0 0
Nv 0 0 0
Sex Ratio
F/M 2/1 2/1 1/2
F 65 6 4
M 31 3 9
Age
Age at death 63 (30-83) 63 (51-79) 62 (32-80)
mean for female 64 60 63
mean for male 61 68 61
Disease duration
within 1st year 81% 83% 100%
More than 1 month 100% 100% 100%
Mean 9 months (4-108) 9 months (2-38) 5,6 months (2-12)
mean for female 11.4 months 11.5 months 4,3 months
mean for male 6.4 months 4 months 6 months
Symptoms
Dementia 100% 100% 100%
Cerebellar signs or Myoclonus 80% 83% 92%
Pyramidal signs 50% 50% 77%
Typical EEG 50% 50% 36%
En 1999, il y a deux fois moins de femmes que d’hommes atteints. Le sexe ratio est inversé par rapport aux résultats antérieurs.
La moyenne d’âge reste par contre extrêmement stable.
La maladie dure toujours plus d’un mois et en 1999, 100% des patients sont décédés endéans l’année. La durée moyenne de la maladie a en effet diminué en 1999 par rapport aux résultats antérieurs passant de 9 à 5.6 mois. La différence se marque principalement chez les femmes.
Les symptômes contribuant au diagnostic de la maladie dans sa forme sporadique sont
Démence rapidement progressive
Myoclonus
Atteinte pyramidale ou extrapyramidale
Mutisme akinétique
Rapport Annuel Commission CJD 20
La démence rapidement progressive se manifeste dans 100% des cas, les pourcentages de la présence des autres symptômes varient quelque peu. Ainsi, en 1999, les signes pyramidaux sont présents dans 77% des cas et le myoclonus dans 92% des cas.
Les données sur le mutisme akinétique sont inconnues dans 7 cas sur 13 et ne figurent donc pas dans le tableau. Signalons toutefois que dans les 7 cas connus, le signe est présent chez trois patients.
En terme d’examens complémentaires, notons que
L’EEG n’a été contributif à l’établissement du diagnostic que dans 36% des cas en 1999.
Parmi les 13 patients pour lesquels il y eut un dosage de la protéine 14-3-3, 11 ont un résultat positif.
De même, l’analyse génétique a été réalisée chez 10 patients. Le génotype est méthionine-méthionine chez 6 patients soit 60%, 2 sont du génotype valine-valine et 2 également du génotype méthionine- valine. Le nombre d’analyses est trop faible pour oser une comparaison mais notons toutefois que ces résultats sont du même ordre de grandeur que ceux déjà publiés. La représentation d’homozygotes M-M parmi les patients atteints de la forme sporadique (69%) est plus importante par rapport à la répartition dans la population générale (39%) (16).
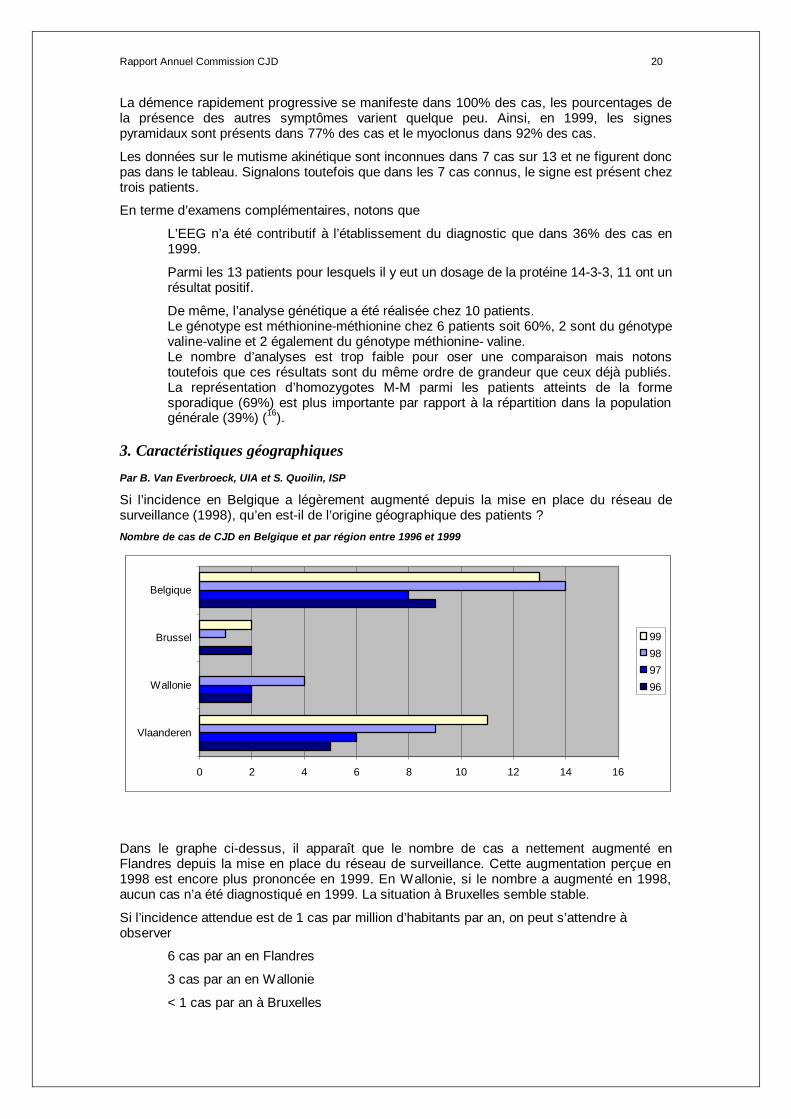
3. Caractéristiques géographiques
Par B. Van Everbroeck, UIA et S. Quoilin, ISP
Si l’incidence en Belgique a légèrement augmenté depuis la mise en place du réseau de surveillance (1998), qu’en est-il de l’origine géographique des patients ? Nombre de cas de CJD en Belgique et par région entre 1996 et 1999
0 2 4 6 8 10 12 14 16
Vlaanderen
Wallonie
Brussel
Belgique
99989796
Dans le graphe ci-dessus, il apparaît que le nombre de cas a nettement augmenté en Flandres depuis la mise en place du réseau de surveillance. Cette augmentation perçue en 1998 est encore plus prononcée en 1999. En Wallonie, si le nombre a augmenté en 1998, aucun cas n’a été diagnostiqué en 1999. La situation à Bruxelles semble stable.
Si l’incidence attendue est de 1 cas par million d’habitants par an, on peut s’attendre à observer
6 cas par an en Flandres
3 cas par an en Wallonie
< 1 cas par an à Bruxelles
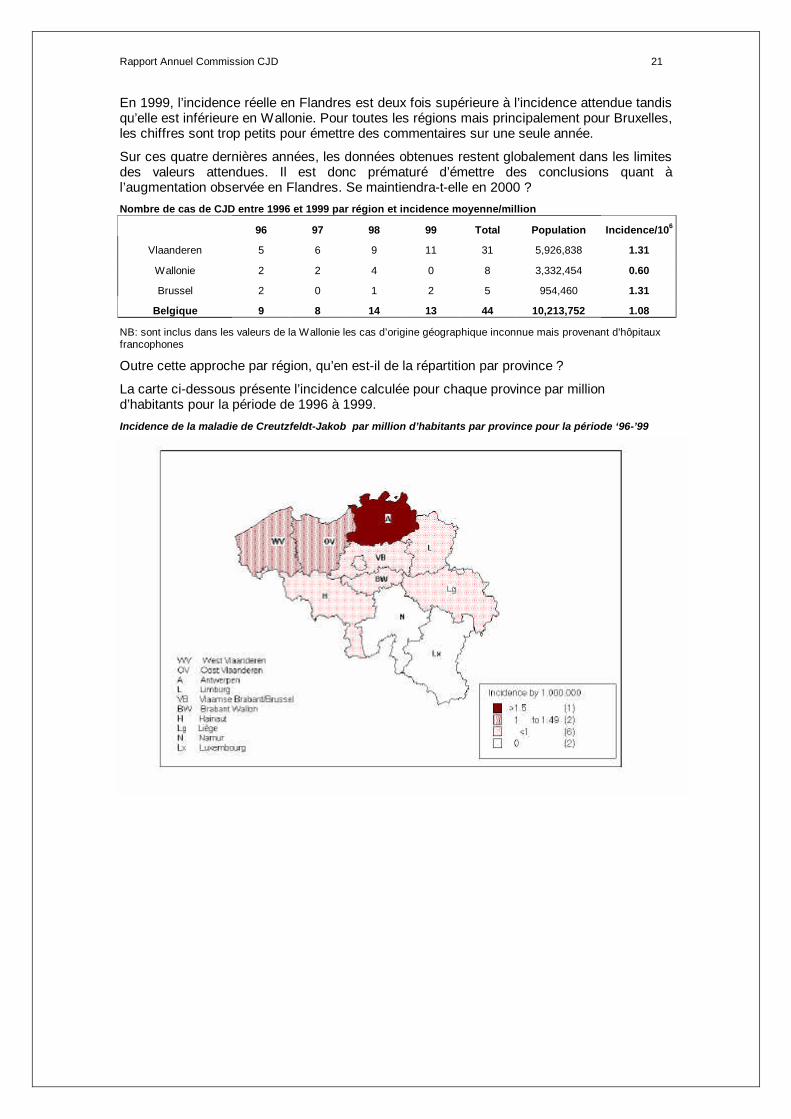
Rapport Annuel Commission CJD 21
En 1999, l’incidence réelle en Flandres est deux fois supérieure à l’incidence attendue tandis qu’elle est inférieure en Wallonie. Pour toutes les régions mais principalement pour Bruxelles, les chiffres sont trop petits pour émettre des commentaires sur une seule année.
Sur ces quatre dernières années, les données obtenues restent globalement dans les limites des valeurs attendues. Il est donc prématuré d’émettre des conclusions quant à l’augmentation observée en Flandres. Se maintiendra-t-elle en 2000 ? Nombre de cas de CJD entre 1996 et 1999 par région et incidence moyenne/million
96 97 98 99 Total Population Incidence/106
Vlaanderen 5 6 9 11 31 5,926,838 1.31
Wallonie 2 2 4 0 8 3,332,454 0.60
Brussel 2 0 1 2 5 954,460 1.31
Belgique 9 8 14 13 44 10,213,752 1.08
NB: sont inclus dans les valeurs de la Wallonie les cas d’origine géographique inconnue mais provenant d'hôpitaux francophones
Outre cette approche par région, qu’en est-il de la répartition par province ?
La carte ci-dessous présente l’incidence calculée pour chaque province par million d’habitants pour la période de 1996 à 1999. Incidence de la maladie de Creutzfeldt-Jakob par million d’habitants par province pour la période ‘96-’99

Rapport Annuel Commission CJD 22
A l’observation de cette carte, il apparaît nettement un gradient sud-nord. Sur base de l’incidence calculée pour les 4 dernières années, on peut observer que les trois provinces les plus touchées sont, de manière croissante, les provinces de West Vlaanderen (incidence entre 1 et 1.49/1.000.000), Oost Vlaanderen (incidence entre 1 et 1.49/1.000.000) et Antwerpen (>1.5 /1.000.000).
La province d’Antwerpen semble être plus atteinte que les autres avec, notamment, des incidences de 3.05/1.000.000 en 1998 et 1.98/1.000.000 en 1999 alors que le nombre de cas attendu par an devrait être de 1.6.
De même, la province de Oost Vlaanderen a connu un doublement de l’incidence attendue en 1999 avec 2.94/1.000.000. Il s’agit de l’incidence la plus élevée en 1999.
Dans les provinces de Namur et de Luxembourg aucun cas n’aurait été détecté au cours des 4 dernières années.
Les autres provinces comptent moins d’un cas par million d’habitants par an.
Cette situation est-elle anormale ?
Répondant à l’adage du « qui cherche, trouve », la mise en place, depuis 1998, du système de surveillance a engendré une augmentation non significative de l’incidence pour le pays.
En ce qui concerne la répartition géographique, il est trop tôt pour juger de la réalité du gradient géographique.
Différents facteurs peuvent expliquer la tendance aujourd’hui observée:
1. Accès aux autopsies
Existe-t-il des facteurs culturels expliquant un consentement plus aisé des familles aux autopsies, une sensibilité plus marquée du public médical en Flandres ?
Existe-t-il un accès plus aisé aux hôpitaux de référence en Flandres ?
Pour évaluer ce point, on peut faire appel aux nombres de patients référés pour suspicion de CJD. En 1998 et 1999, seules les provinces du Brabant wallon, de Namur et du Luxembourg n’en ont pas référés. Et intervient ici le second facteur dont il faut tenir compte dans l’explication. Il s’agit de la densité de population.
2. Densité de population
Ces trois provinces sont en effet aussi les moins peuplées. Il faudrait alors un recul de 2.5 à 4 ans pour voir apparaître un cas.
Afin d’éclaircir cette question, une analyse statistique plus élaborée sera réalisée sur base des données de 1998, 1999 et 2000.
La poursuite de la surveillance au cours des prochaines années permettra aussi de mieux cerner ces tendances. Si celles-ci devaient se maintenir, voire s’accentuer, il serait alors nécessaire de mettre en oeuvre les moyens pour les expliquer.

Rapport Annuel Commission CJD 23
C. Situation en Europe
Pour rappel, la concertation des différents pays de l’Union européenne autour de la surveillance de la Maladie de Creutzfeldt-Jakob se fait en deux groupes. Le premier groupe rassemble des pays comme l’Allemagne, la France, l’Espagne, La Grande-Bretagne, l’Italie, la Suisse, … c.-à-d. les pays qui ont été, dans un premier temps, les plus concernés par la ESB et CJD. Il existe depuis 1995. Il fait partie d’un programme de l’UE appelé Biomed I. Afin d’étendre ces rencontres à l’ensemble des pays de l’UE, un second groupe, toujours organisé par le centre de référence d’Edinburgh, a été créé en 1998 et fait partie du programme Biomed II. La Belgique participe à ce second groupe de concertation.
1. Suivi épidémiologique
De manière résumée, on peut dire que les différents pays de l’Union européenne ont mis en place un système de collecte de l’information que ce soit par contact entre les différents spécialistes (ex. : Grèce), par obligation légale (ex. : Suède), par un système de surveillance propre (ex. : Belgique). Seul le Portugal éprouve quelques difficultés à mettre en place le système de surveillance.
2. Résultats épidémiologiques
Tous les pays de l’Union européenne ont déclaré ont moins un cas de CJD y compris l’Islande où a été effectuée une recherche active et rétrospective de cas.
La forme la plus courante est la forme sporadique. Seul, Israël connaît une proportion plus importante de forme familiale qui touche une population originaire de Libye.
Trois pays déclarent des cas de la forme variante : la Grande-Bretagne dénombre 53 cas entre 1995 et 1999, la France a identifié un second cas en 1999 et l’Irlande un premier cas au cours de la même année.
Entre 1995 et 1999, on compte un total de 56 cas de forme variante.
L’incidence observée dans 9 pays de l’Union européenne et Israël pour 1998 et 1999 est présentée dans le tableau ci-dessous.
Incidence des cas probables ou confirmés de CJD (par 1.000.000) pour 9 pays européens et Israël en 1998 et 1999
Great-Britain
Belgium Denmark Finland Israel Italy Norway Sweden Portugal
1998 1,4 1,4 1,8 0.8 2.8 1 0,5 1,8 ?
1999 1,3 1,3 0,4 1.2 2 1,4 0,2 1,6 0,3
Les résultats des pays qui font partie du groupe européen Biomed II sont présentés ci-dessous.
2.a Danemark
Depuis 1997, tout cas suspect de la Maladie de Creutzfeldt-Jakob est soumis à la déclaration obligatoire au Danemark. Le pays dénombre 9 cas confirmés en 1997, 10 cas en 1998 et 2 cas confirmés en 1999.
Tous sont de forme sporadique.
2.b Finlande
En 1998, il y eut 4 cas confirmés tandis qu’il y en a 6 en 1999.
Tous sont de forme sporadique.

Rapport Annuel Commission CJD 24
2.c Grande-Bretagne
En 1999, on dénombre un total de 77 cas de CJD pour 161 patients référés. Parmi les cas confirmés et probables, on note 56 cas de forme sporadique, 6 cas de forme iatrogène et 13 cas de forme variante.
Entre 1995 et 1999, le nombre total de forme variante s’élève donc à 53 cas.
L’âge des cas de vCJD est compris entre 15 et 54 ans avec une moyenne au moment du décès à 29 ans. Le sex ratio est proche de 1. La durée moyenne de la maladie est de 14 mois. Tous les cas étaient méthionine-méthionine.
Par ailleurs, il faut noter qu’une étude a permis le passage en revue de 640 dossiers médicaux de patients décédés entre 15 et 44 ans de cause neurologique entre 1979 et 1996 et ce, à la recherche de diagnostics manqués de CJD. Il ressort de cette étude qu’aucun cas n’a été mal classé. On peut donc estimer que l’augmentation des cas de CJD et de la variante n’est pas simplement dû à la mise en place d’un système de surveillance (17).
Différentes équipes de chercheurs continuent à tenter des estimations de l’ampleur que pourrait prendre l’épidémie de vCJD eu égard aux caractéristiques de l’agent infectieux, à l’étendue de l’exposition aux prions, à la période d’incubation de la maladie ou encore à l’évolution de l’incidence ou de la mortalité de la maladie. Les estimations sont encore très étendues allant de quelques dizaines de cas à plusieurs millions (18, 19, 20).
2.d Grèce
Depuis 1997, seuls 13 cas ont été confirmés. En 1999, il y a 4 cas confirmés et tous de forme sporadique. L’incidence pour le pays est donc de 0,88/1.000.000. Il faut noter que l’incidence pour la seule île crétoise grimpe jusqu’à 2,85/1.000.000. Aucune explication n’a pu encore être donnée.
2.e Irlande
CJD est devenue une maladie à déclaration obligatoire en 1996. Entre 1980 et 1999, ils dénombrent 31 cas dont 29 de forme sporadique, 1 variante et 1 insomnie fatale. Sur base d’une étude rétrospective réalisée sur les cas survenus avant 1997, l’incidence de la maladie est de 0,31/1.000.000 habitants.
En 1996, il y avait 3 cas confirmés dont 2 sporadiques et 1 insomnie familiale fatale. En 1998, il y avait 6 cas confirmés et de forme sporadique. En 1999, il n’y a que deux cas, si l’un est de forme sporadique, l’autre est une forme variante.
Une surveillance active a démarré en juillet 1999.
L’Irlande compte le deuxième cas hors Royaume-Uni de vCJD. Parmi les facteurs de risque, il faut noter que ce patient a vécu en Grande-Bretagne entre 1989 et 1995.
2.f Islande
Une étude rétrospective a été menée pour la période 1960 et 1997 et a permis de mettre en évidence l’existence de 3 cas confirmés de CJD soit une incidence de 0,5/1.000.000. Le dernier cas date de 1997. En 1999, il y aurait eu un cas mais il doit encore être confirmé.
2.g Israël
Au cours de l’année 1998, 7 cas de Creutzfeldt-Jakob dont 2 sporadiques et 5 de la forme familiale ont été diagnostiqués. En 1999, il y a 5 cas de forme familiale. Ces chiffres sont en diminution par rapport aux années précédentes. Les formes familiales ont toujours été nombreuses dans ce pays en raison de la survenue de cette maladie au sein d’un groupe ethnique déterminé. Une analyse du polymorphisme du codon 129 a été réalisée et montre une majorité de méthionine-méthionine.

Rapport Annuel Commission CJD 25
2.h Norvège
Maladie à déclaration obligatoire depuis 1997. En 1998, il y eut 2 cas de forme sporadique et en 1999, trois cas ont été référés dont 1 pour lequel le diagnostic final n’est pas encore connu.
2.i Portugal
En 1999, il y eut 4 patients référés dont 2 sont des cas probables de forme sporadique. Au cours de cette même année, l’ESB a atteint plus de 350 vaches (1.200.000 têtes de bétail dans le pays). Il faut souligner que les mesures de protection de la chaîne agroalimentaire n’ont été prises qu’en 1998.
2.j Suède
En 1997, 11 cas sporadiques et 2 probables.
En 1998, 11 cas sporadiques et 4 probables.
En 1999, 7 cas sporadiques.
2.k Autres
Il faut noter que la France, qui dénombre aussi des cas de forme iatrogène, compte deux cas de forme variante. Chez ces deux cas, aucun facteur de risque particulier n’a pu être mis en évidence. Ils ne s’étaient jamais rendu en Grande-Bretagne et n’avaient été soumis à aucun risque de transmission iatrogène.
En Italie, depuis 1993, 675 patients ont été référés pour suspicion de CJD et 358 d’entre eux ont été confirmés. La majorité des cas sont des formes sporadiques (83,5%). Il y a encore 49 formes familiales et 2 formes iatrogènes mais pas de forme variante. En 1999, il y eut 63 cas sporadiques pour 9 cas familiaux.
Rapport Annuel Commission CJD 26
Encéphalopathie Spongiforme Bovine Selon les estimations de 400.000 à plus de 700.000 vaches infectées par l’ESB seraient entrées en Grande-Bretagne dans la chaîne alimentaire avant que la situation ne soit connue (21). Il faut aussi tenir compte des animaux infectés qui à l’heure actuelle entrent encore dans la chaîne alimentaire. Si un dépistage actif n’est pas réalisé, ceci concerne un certain nombre de bêtes en période d’incubation et un certain nombre de bêtes qui présentent les premiers symptômes et ne sont pas déclarés par les éleveurs qui ont appris à reconnaître la maladie.
On peut donc présumer qu’un grand nombre de personnes ont été exposées à l’agent infectieux.
A. Situation en Belgique
Sur base de la quantité de viande britannique importée dans notre pays au cours de la période critique (1986-1995) et sur base du nombre de cas de ESB en Belgique, il est raisonnable de présumer que le risque pour la population belge est faible. L’estimation du risque reste toutefois difficile à établir avec précision.
Sur base des chiffres remis par le « National Creutzfeldt-Jakob Disease Surveillance Unit » d’Edinburgh, on peut estimer que les exportations de viande britannique, sous différentes formes, vers la Belgique, au cours de la période critique, n’excèdent pas 5% du total exporté vers les différents pays de l’Union européenne. Quant aux farines et autres granulés destinés au bétail, les exportations avoisinent les 15% de ce total.
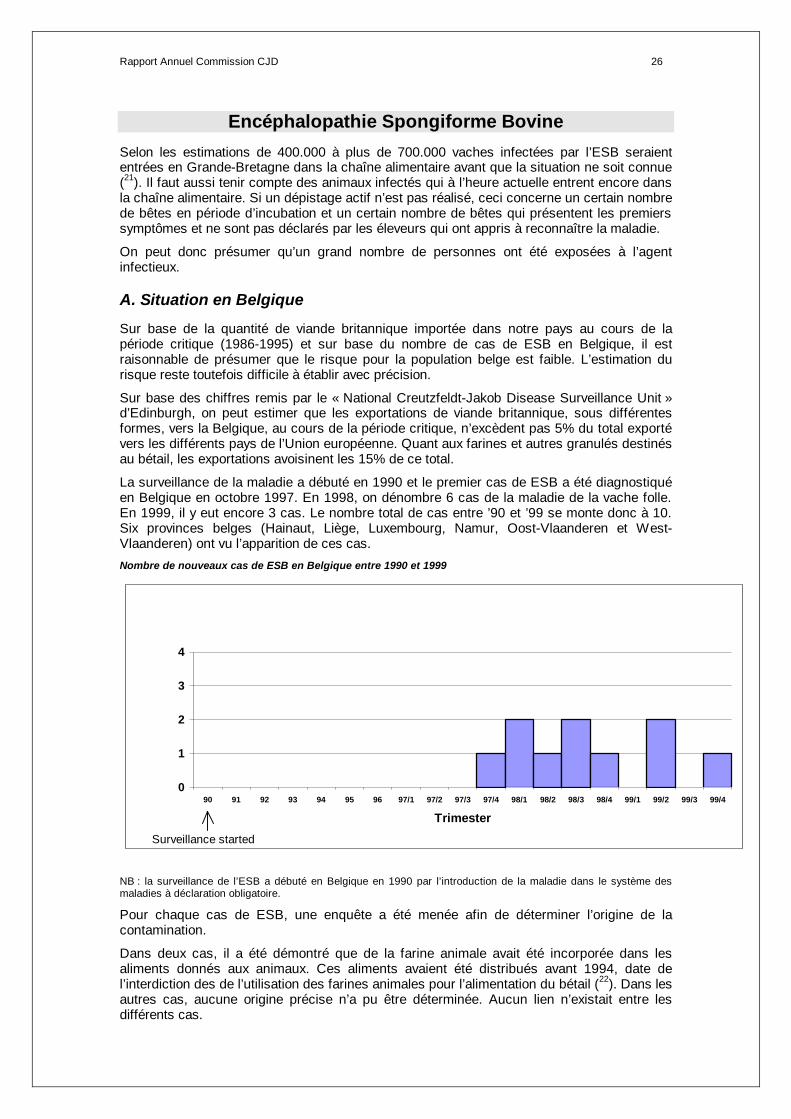
La surveillance de la maladie a débuté en 1990 et le premier cas de ESB a été diagnostiqué en Belgique en octobre 1997. En 1998, on dénombre 6 cas de la maladie de la vache folle. En 1999, il y eut encore 3 cas. Le nombre total de cas entre ’90 et ’99 se monte donc à 10. Six provinces belges (Hainaut, Liège, Luxembourg, Namur, Oost-Vlaanderen et West-Vlaanderen) ont vu l’apparition de ces cas. Nombre de nouveaux cas de ESB en Belgique entre 1990 et 1999
0
1
2
3
4
90 91 92 93 94 95 96 97/1 97/2 97/3 97/4 98/1 98/2 98/3 98/4 99/1 99/2 99/3 99/4
Trimester
Surveillance started
NB : la surveillance de l’ESB a débuté en Belgique en 1990 par l’introduction de la maladie dans le système des maladies à déclaration obligatoire.
Pour chaque cas de ESB, une enquête a été menée afin de déterminer l’origine de la contamination.
Dans deux cas, il a été démontré que de la farine animale avait été incorporée dans les aliments donnés aux animaux. Ces aliments avaient été distribués avant 1994, date de l’interdiction des de l’utilisation des farines animales pour l’alimentation du bétail (22). Dans les autres cas, aucune origine précise n’a pu être déterminée. Aucun lien n’existait entre les différents cas.
Rapport Annuel Commission CJD 27
Les hypothèses de la contamination peuvent être les suivantes (23):
• cas sporadiques sans cause identifiable,
• contamination croisée entre des farines destinées à d’autres types d’animaux et qui contiennent encore des protéines animales et celles, destinées notamment aux bovins, qui n’en contiennent plus. Contamination survenue pendant le processus de fabrication, de stockage ou de distribution,
• animaux contaminés avant juillet 94 date de l’interdiction de l’utilisation des farines animales,
• ou malgré l’interdiction, utilisation de farines animales.
En raison de la période d’incubation moyenne estimée à 5 ans chez les bovins, on peut penser que le nombre de cas qui seront identifiés dans les années à venir devrait être en diminution eu égard aux mesures prises en 1994. Il faut toutefois remarquer que l’on se réfère à une moyenne et que la distribution réelle des valeurs de la période d’incubation va de 2 à 20 ans !
A ce jour, il n’existe encore aucune certitude quant au risque de transmission verticale entre la vache et le veau.
B. Situation en Europe
L’interdiction d’exportation de boeuf britannique par l’Union européenne date de 1990. Cet embargo a été levé en août 1999.
Afin de diagnostiquer plus précocement la maladie dans le cheptel bovin mais également de mieux en suivre l’évolution, des tests de diagnostic ont été développés.
La Commission européenne a homologué trois tests de détection rapide et a approuvé un projet de surveillance de l’ESB via l’utilisation de ces tests. A partir de 2001, les Etats-membres devront contrôler un échantillon du cheptel et notamment tous les animaux morts dans des conditions suspectes.
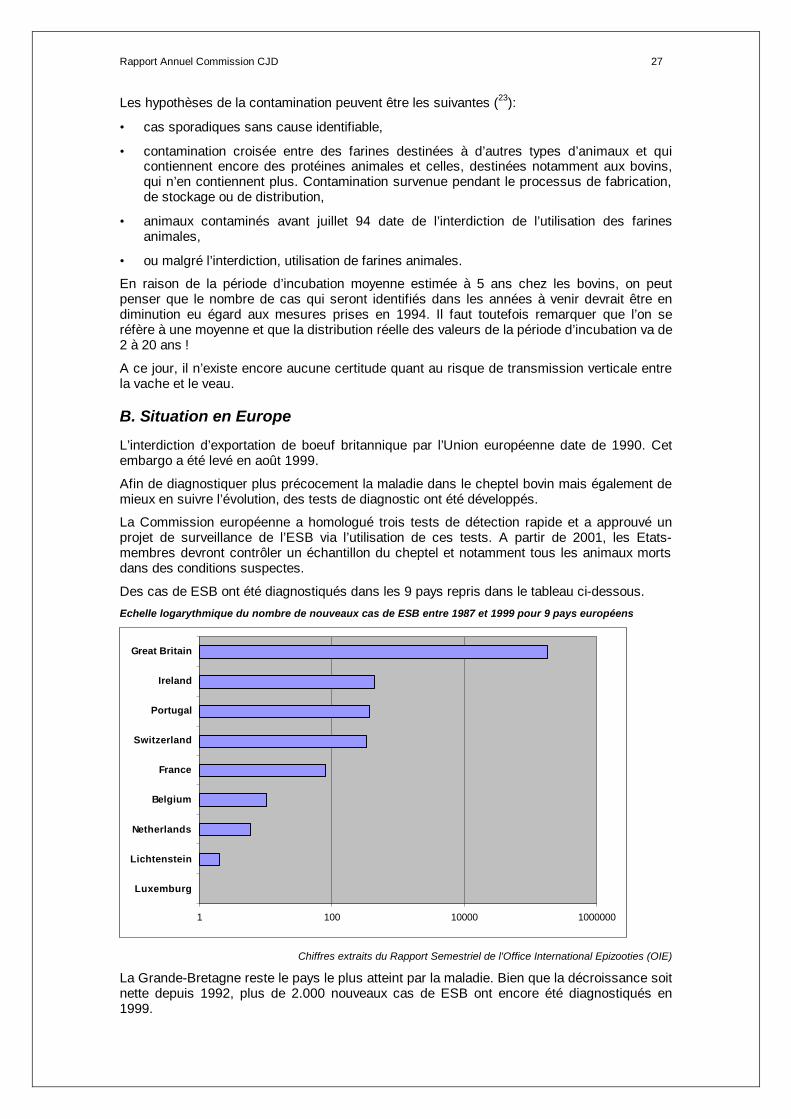
Des cas de ESB ont été diagnostiqués dans les 9 pays repris dans le tableau ci-dessous. Echelle logarythmique du nombre de nouveaux cas de ESB entre 1987 et 1999 pour 9 pays européens
Chiffres extraits du Rapport Semestriel de l'Office International Epizooties (OIE)
La Grande-Bretagne reste le pays le plus atteint par la maladie. Bien que la décroissance soit nette depuis 1992, plus de 2.000 nouveaux cas de ESB ont encore été diagnostiqués en 1999.
1 100 10000 1000000
Luxemburg
Lichtenstein
Netherlands
Belgium
France
Switzerland
Portugal
Ireland
Great Britain
Rapport Annuel Commission CJD 28
Outre la Grande-Bretagne, le pays le plus touché en 1999 est le Portugal. Ensuite, viennent en ordre croissant l’Irlande, la France et la Suisse.
Si la Grande-Bretagne connaît une décroissance des cas, le Portugal, la Suisse, la France et l’Irlande connaissent encore en 1999 une augmentation du nombre de cas.
Il faut également noter que des cas importés ont été identifiés en Allemagne (6 en ‘92, ‘94 et ‘97), Canada (1 en 93), Danemark (1 en ‘92), Italie (2 en ‘94), Iles Falkland (1 en ‘89) et Oman (2 en ’89).
La classification peut être faite sur base de l’incidence :
> 2 cas / 1.000.000 de bovins = Irlande, Suisse, Portugal
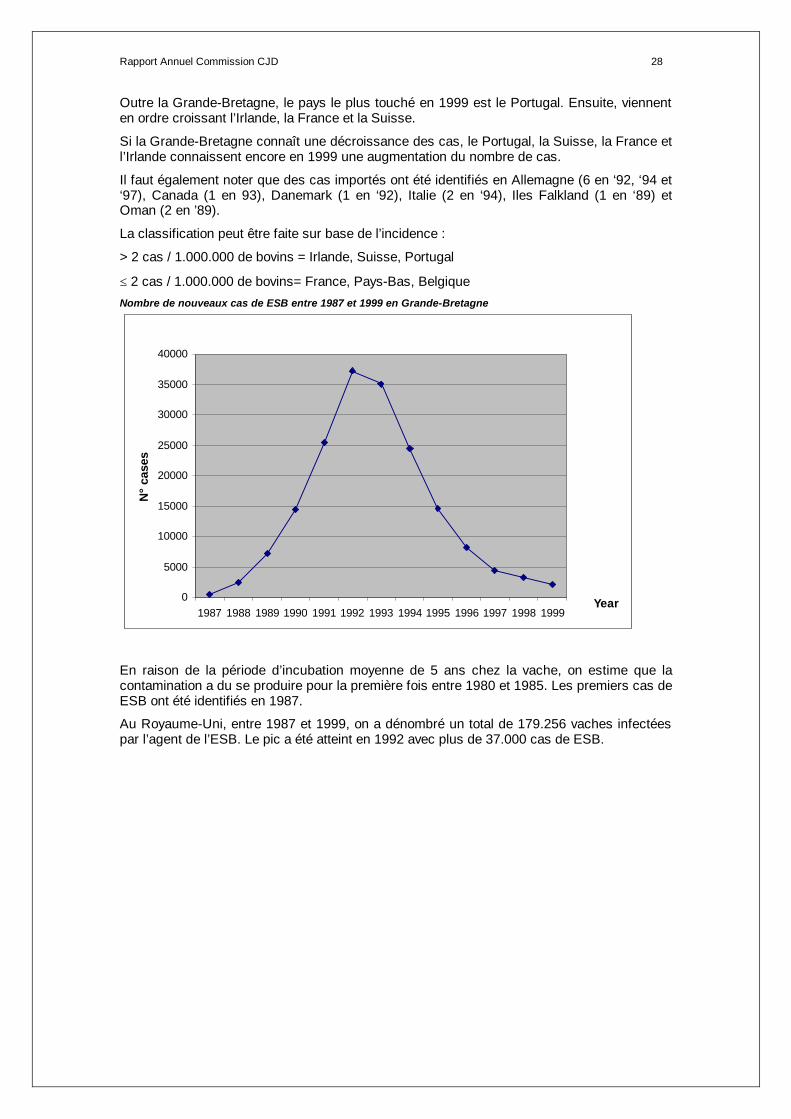
≤ 2 cas / 1.000.000 de bovins= France, Pays-Bas, Belgique Nombre de nouveaux cas de ESB entre 1987 et 1999 en Grande-Bretagne
En raison de la période d’incubation moyenne de 5 ans chez la vache, on estime que la contamination a du se produire pour la première fois entre 1980 et 1985. Les premiers cas de ESB ont été identifiés en 1987.
Au Royaume-Uni, entre 1987 et 1999, on a dénombré un total de 179.256 vaches infectées par l’agent de l’ESB. Le pic a été atteint en 1992 avec plus de 37.000 cas de ESB.
0
5000
10000
15000
20000
25000
30000
35000
40000
1987 1988 1989 1990 1991 1992 1993 1994 1995 1996 1997 1998 1999Year
N°
case
s

Rapport Annuel Commission CJD 29
Problèmes de santé publique
A. Risque et facteurs de risque
1. Risque
En raison du lien établi entre l’ESB et la vCJD et de la probable très large exposition de la population, la maladie de Creutzfeldt-Jakob doit être considérée comme un problème de santé publique potentiel.
A ce jour, il est toujours excessivement hasardeux de se lancer dans des estimations de la potentielle survenue d’une épidémie de vCJD. Un certain nombre d’estimation ont été tentées et les limites sont toujours aussi hasardeuses, allant de quelques dizaines de cas à quelques millions.
A la fin de l’année 1998, une augmentation du nombre de décès a fait craindre un changement de la pente de la courbe d’incidence. Si cette accélération ne s’est pas confirmée en 1999, la situation n’est pas encore rassurante car les prédictions sur l’épidémie dépendent de la durée d’incubation et celle-ci n’est pas encore clairement définie car de longue durée.
Par ailleurs, il reste encore beaucoup d’inconnues pour juger exactement de l‘ampleur possible : la voie de contamination, la dose infectante, la barrière des espèces, l’effet du génotype (24).
Et au-delà de la dose infectante, il ne faut pas oublier que celle-ci varie en fonction du tissu d’origine. Le tissu le plus infectant est évidemment le tissu cérébral.
2. Facteurs de risque
Plusieurs études ont été menées afin d’identifier des facteurs de risque (pour les formes autres que la forme variante).
Une étude menée en Australie tend à démontrer que les personnes qui subissent plus de trois interventions chirurgicales ont deux fois plus de risque de développer une CJD sporadique (25). Cette même étude met en évidence le même risque pour les personnes vivant à proximité des fermes ou employés dans celles-ci. Ce constat n’est pas confirmé par d’autres études.
Une étude a été réalisée pour déterminer si la zone d’habitation à proximité d’une entreprise qui produit de la nourriture pour bétail pouvait constituer un risque de développer la variante. Cette hypothèse n’a pas été confirmée (26).
Le risque lié aux interventions chirurgicales a également été mis en évidence dans une étude allemande (27). Cette même étude souligne l’importance du stress comme agent déclencheur de la maladie et qui peut aussi modifier l’évolution de la maladie.
En ce qui concerne la forme vCJD, les trois facteurs de risque admis sont :
1. Vivre en Grande-Bretagne
2. Codon 129 méthionine – homozygote
3. Age (moyenne 28 ans)
Tous les patients vCJD testé jusqu’à présent présentaient un codon 129 homozygote méthionine -méthionine au niveau du gène PrP. Il apparaît donc que les sujets homozygotes ont une période d’incubation plus courte et /ou une susceptibilité plus grande à la maladie.
Puisque cette association méthionine -méthionine est la moins répandue dans la population, il n’est donc pas exclu que la majorité des cas ne se déclarent que plus tard.

Rapport Annuel Commission CJD 30
B. Exposition professionnelle
Il n’y a pas de certitudes quant à la transmission de la maladie de Creutzfeldt-Jakob par des contacts étroits de personne à personne ou par exposition professionnelle.
1. Milieu hospitalier
Si les informations à ce sujet restent fragmentaires, la transmission a été décrite dans certaines situations telles greffe de dure-mère, de cornée ou l’injection d’hormones de croissance. On connaît aussi la résistance des prions aux méthodes de décontamination.
Il faut donc considérer certaines professions et plus spécifiquement certains actes techniques comme étant à risque.
En Belgique, la Section Hygiène Hospitalière du Conseil Supérieur de l’Hygiène a établi en collaboration avec la Commission Creutzfeldt-Jakob un document de recommandations pour la prévention de la transmission de l’agent des EST en milieu hospitalier∗∗∗.
2. Autres
Ces aspects sont évidemment applicables aux catégories professionnelles exposées au risque de l’ESB.
Des études (28) ont démontré qu’il n’y avait pas d’augmentation de la mortalité pour la maladie de Creutzfeldt-Jakob ou tout autre forme de démence chez les bouchers, fermiers et vétérinaires. D’autres constatent une augmentation significative du nombre de cas de CJD chez des travailleurs journaliers ou des fermiers dans les fermes où il y avait eu un cas de ESB, il s’agissait de la forme classique et non de la variante (2).
C. Transfusion sanguine
A propos de la transmission par le sang de nombreuses questions se posent. La transfusion de sang complet, de composants ou de dérivés sanguins n’a jamais démontré être un risque de transmission de la maladie. Toutefois, de manière expérimentale, l’inoculation intracérébrale de composants sanguins a démontré une certaine infectivité.
Par ailleurs, dans les formes variantes, on sait que la protéine PrPsc se réplique dans le système lymphoréticulaire et que les lymphocytes B seraient l’élément qui permet le passage de la barrière hémato-encéphalique.
Si la transfusion sanguine ne paraît pas être chargée de risque, on ne peut l’exclure totalement. Alors une fois de plus, il reste la même préoccupation : quel niveau de risque paraît tolérable ?
A l’heure actuelle, en Belgique, seuls sont exclus des dons de sang les personnes qui mentionnent l’existence d’un cas dans la famille ou qu’ils ont subi une greffe de cornée ou de dure-mère.
Aux USA, la Food and Drug Administration a tranché pour une mesure de prévention. Toutes les personnes qui ont séjourné plus de 6 mois en Grande-Bretagne entre 1980 et 1996 sont écartées des dons de sang. Ceci représenterait de 2 à 3% des donneurs et ne devrait pas avoir de conséquences sur la disponibilité de sang.
En Grande-Bretagne depuis 1989, toutes les personnes ayant reçu des hormones de croissance, ont été exclues des dons de sang. Afin de ne négliger aucun risque, la leucodépletion est envisagée comme technique systématique de décontamination. La réduction systématique du nombre de globules blancs du sang destiné à être transfusé est aussi une mesure obligatoire en Suisse depuis septembre 1999 (29).
La recherche se penche sur le développement de tests diagnostiques mais il reste encore quelques inconnues à éclaircir : à quel moment la protéine passe-t-elle dans le sang et à quelle concentration est-elle détectable ?
∗∗∗ Pour tout renseignement, prendre contact avec S. Quoilin. ISP. Voir adresse en annexe.

Rapport Annuel Commission CJD 31
La prise de mesures d’exclusion des dons de sang à l’égard des personnes ayant séjournés en Grande-Bretagne doit être précédée par la comparaison du risque entre transmettre la maladie de Creutzfeldt-Jakob et provoquer une pénurie de sang.
D. Produits pharmaceutiques
Certains produits pharmaceutiques contiennent des substances d’origine humaine ou animale. Faisant suite à la directive 97/534 de la Commission européenne sur l’utilisation du matériel à risque, un arrêté Royal du 22.12.97 instaure un contrôle pour ces produits.
Pour chaque substance, les firmes pharmaceutiques doivent compléter un formulaire qui contient un certain nombre d’informations quant au type de tissus, pays et/ou centre d’origine, . . ., etc. Les formulaires sont renvoyés vers l’Institut Scientifique de la Santé Publique qui constitue une base de données et exerce un contrôle sécurité.
E. Autres
En Grande-Bretagne, d’autres formes de risque ont été envisagées comme par exemple, la réutilisation de lentilles de contact. Les opticiens ne peuvent plus utiliser une même paire de lentilles pour des essais chez des clients.
Les transplantations peuvent également être perçues comme un risque. Celui-ci apparaît faible mais des chercheurs se penchent néanmoins sur le développement de test de dépistage.

Rapport Annuel Commission CJD 32
Conclusion On ne connaît pas la dose minimale infectante,
On ne connaît toujours pas les différentes voies de transmission,
On ne connaît toujours pas exactement la période d’incubation,
On ne connaît pas encore le mécanisme d’infection avec précision,
On ne connaît pas encore la prévalence d’exposition au risque,
On ne connaît pas encore le risque des personnes exposées de développer la maladie,
On sait le lien entre ESB et vCJD.
Il y a donc lieu de persévérer dans la surveillance et dans la recherche afin de mieux connaître et le potentiel problème de santé publique et la maladie.
Mais au-delà des mesures prises pour le problème spécifique de l’ESB et donc de vCJD, il est impératif d’envisager le problème sous un angle plus global. Dans le domaine de l’agroalimentaire ou de l’environnement, la santé du public sera-t-elle un jour aussi importante que le profit ? Quels mécanismes de protection et de contrôle seront mis en place pour éviter excès et égarement ?

Rapport Annuel Commission CJD 33
Annexes
A. Critères diagnostiques et classification
du programme de surveillance européen
1. Forme sporadique
I. Démence progressive rapide
II. A. Myoclonus
B. Problèmes cérébelleux ou visuels
C. Signes pyramidaux ou extrapyramidaux
D. Mutisme akinétique
III. A. Perturbation caractéristique de l’EEG
B. Présence de protéine 14-3-3 dans le liquide céphalorachidien
Cas confirmé : uniquement par l’examen neuropathologique ou immunohistochimie
Cas probable : critère I plus deux des signes de la liste II et le IIIA
Ou
cas qui répond à la définition du cas possible plus le critère IIIB
Cas possible : critère I plus deux signes de la liste II et une durée de la maladie de moins de deux ans
2. Forme variante
I. A. Désordre neuropsychiatrique progressif
B. Durée de la maladie supérieur à 6 mois
C. Examens de routine éliminent autres possibilités
D. Pas d’exposition iatrogénique possible
II. A. Symptômes psychiatriques précoces*
B. Sensations douloureuses persistantes**
C. Ataxie
D. Myoclonus ou chorée ou dystonie
E. Démence
III. A. EEG anormal *** mais pas caractéristique de la forme sporadique ou EEG non réalisé
B. Signal dense bilatéral au niveau du thalamus à la RMN
IV. Biopsie des amygdales positive (uniquement admis par les Britanniques)
Cas confirmé : IA et confirmation à l’examen neuropathologique ****
Cas probable : critère I plus quatre des signes de la liste II et le IIIA et IIIB
Ou
critère I et IV
Cas possible : critère I plus quatre signes de la liste II et IIIA

Rapport Annuel Commission CJD 34
* Dépression, anxiété, apathie, repli, hallucinations
** Franches douleurs ou simples dysesthésies
*** Un Complexe périodique triphasique généralisé par seconde
**** Aspect spongiforme, large dépôts de PrP sous forme de plaques au niveau du cerveau et cervelet
B. Matériel à risque spécifié (MRS)
Organes et tissus dans lesquels se concentre l’agent de l’ESB.
Les MRS définis dans la loi sont
a) le crâne y compris la cervelle, les yeux, les amygdales, la moelle épinière et l’iléon :
de bovins âgés de plus de 12 mois ;
d’ovins et de caprins âgés de plus de 12 mois ou qui présente une incisive permanente ayant percé la gencive ;
b) la rate d’ovins et de caprins de tous âges
C. Sites Internet
Institut Scientifique de la Santé Publique : www.iph.fgov.be/epidemio
Universitaire Instelling Antwerpen : neurobio-www.uia.ac.be/neurobio/CJD/
National Creutzfeldt-Jakob Disease Surveillance Unit : www.euroCJD.ed.ac.uk
D. Membres de la Commission CJD
Institut Scientifique de la Santé Publique (Coordination et secrétariat de la Commission) 14, rue Juliette Wytsman 1050 Bruxelles Dr Sophie Quoilin 02/642.57.85 [email protected] Dr Frank Van Loock 02/642.50.26 [email protected] Dr André Stroobant 02/642.52.33 [email protected]
Universitaire Instelling Antwerpen (Centre de référence effectuant des autopsies) Patrick Cras, MD, PhD Dept of Neurology Lab of Neurobiology University of Antwerp

Rapport Annuel Commission CJD 35
University Hospital of Antwerp Born Bunge Fdn 32 3 821 34 23 hosp 32 3 820 26 04 lab 32 3 825 54 67 fax hosp 32 3 820 26 69 fax lab [email protected]
Université de Liège (Centre de référence effectuant des autopsies) Prof. Pastoret (Président de la Commission) Faculté de Médecine Vétérinaire Service Immunologie-vaccinologie B43b 20, boulevard de Colonster 4000 Liège Tél.: 04/366,42,60 fax: 04/366,42,61 Dr Deprez CHU Sart Tilman Service de neuropathologie 4000 Liège Tél.: 04/366.72.54 Fax: 04/366.74.99
Universiteit Gent Prof. Dr. Jan De Bleecker Dr P. Santens Neurologie, U.Z. Gent 185, de Pintelaan 9000 Gent Tél.: 09/240.21.11 Fax: 09/240.49.71
Université Catholique Louvain (Centre de référence effectuant des autopsies) Prof. Ch. Sindic Prof. C. Godfraind Cliniques Universitaires Saint Luc 10, avenue hippocrate 1200 Bruxelles 02/764.10.82 (Secrétariat) 02/764.11.11 (Centrale); bip 1083
Katholiek Universiteit Leuven (Centre de référence effectuant des autopsies) Prof. Carton Prof. Sciot Prof. Robberecht Gasthuisberg 49, Herestraat 3000 Leuven Tél.: 016/33.22.11 Fax: 016/34.42.85
Vrije Universiteit Brussel (Centre de référence effectuant des autopsies)Prof. Edinger Dr Michotte AZ VUB 101, Laarbeeklaan 1090 Brussel Tél.: 02/477.41.11 Fax: 02/477.63.81

Rapport Annuel Commission CJD 36
Université Libre de Bruxelles Dr Hildebrand Hôpital Erasme 808, route de Lennik 1070 Bruxelles Tél.: 02/555.31.11 Fax: 02/555.39.42

Rapport Annuel Commission CJD 37
Bibliographie 1. De Bleser D, Plum J. Transmissible spongiform encephalopathies or Prion protein
diseases and public health. Arch Public Health 1998; 56: 169-186.
2. Anonymous Advisory Committee on Dangerous Pathogens – TSE Agents. UK Website: http://www.official-documents.co.uk. 2000;
3. Mestel R. Putting prions to the test. Science 1996; 273: 184-189.
4. Johnson RT, Gibbs CJ. Creutzfeldt Jakob disease and related transmissible spongiform encephalopathies. The New England Journal of Medecine 1998; 1994-2004.
5. Scott M. Compelling transgenetic evidence for transmission of bovine spongiform encephalopathy prions to humans. PNAS 2000; 96: 1-6.
6. Will RG, Zeidler M, Stewart GE, Macleod MA, Ironside JW, et al. Diagnosis of New Variant Creutzfeldt-Jakob Disease. Ann Neurol 2000; 47: 575-582.
7. Zeidler M, Sellar R, Collie DA, Knight R, Stewart G, et al. The pulvinar sign on magnetic resonance imaging in variant Creutzfeldt-Jakob disease. The Lancet 2000; 355: 1412-1418.
8. Urbach H, Klisch J, Wolf HK, Brechtelsbauer D, Gass S, Solymosi L. MRI in sporadic Creutzfeldt-Jakob disease: correlation with clinical and neuropathological data. Neuroradiology 1998; 40/2: 65-70.
9. Demaerel Ph, Heiner L, Robberecht W, Sciot R, Wilms G. Diffusion-weighted MRI in sporadic Creutzfeldt-Jakob disease. Neurology 1999; 52/1: 205-208.
10. Bahn MM, Kido DK, Lin W, Pearlman AL. Brain magnetic resonance diffusion abnormalities in Creutzfeldt-Jakob disease. Arch Neurol 1997; 54/11: 1411-1415.
11. Hill A, Butterworth R, Joiner S, et al. Investigation of variant CJD and other human prion diseases with tonsil biopsy samples. The Lancet 0 AD/1/16; 353: 183-189.
12. Kmietowicz Z. New variant CJD found in tonsils. BMJ 1999; 318: 215
13. Hilton DA, Fathers E, Edwards Ph, Ironside JW, Zajicek J. Prion immunoreactivity in appendix before clinical onset of variant Creutzfeldt-Jakob disease. The Lancet 1998; 352: 703-704.
14. Knight R. Therapeutic possibilities in CJD: patents 1996-1999. Exp.Opin.Ther.Patents 2000; 10/1: 1-9.
15. Pals P. A retrospective study of Creutzfeldt-Jakob disease in Belgium. European Journal of Epidemiology 1999; 15: 517-519.

Rapport Annuel Commission CJD 38
16. Windl O, Dempster M, Estibeiro JP, et al. Genetic basis of Creutzfeldt-Jakob disease in the United Kingdom: a systematic analysis of predisposing mutations and allelic variation in the PRNP gene. Hum Genet 1996; 98/3: 259-264.
17. Mejeed A. Extent of misclassification of death from Creutzfeldt-Jakob disease in England 1979-96: retrospective examination of clinical records. BMJ 2000; 320: 145-147.
18. Bonn D. New predictions for total vCJD mortality lower than before. The Lancet 2000; 356: 570
19. Andrews NJ, Farrington CP, Cousens, et al. Incidence of variant Creurtzfeldt-Jakob disease in the UK. The Lancet 2000; 356: 481-482.
20. Ghani AC, Donnelly CA, Ferguson NM, Anderson RM. Assessment of the prevalence of vCJD through testing tonsils and appendices for abnormal prion protein. Proc R Soc Lond B Biol Sci 2000;
21. Morabia A. Ethics of ignorance: lessons from the epidemiological assessment of the bovine spongiform encephalopathy ("mad cow disease") epidemic. Perspectives in Biology and Medecine 1998; 42: 259-266.
22. Ministère des Classes Moyennes et de l'Agriculture. Rapport d'activités 1998. Report 1998;
23. Saegerman C, Dechamps P, Vanopdenbosch E, Roels S, Petroff K, et al. Epidémiosurveillance de l'encéphalopathie spongiforme bovine en Belgique : bilan de l'année 1998. Ann.Méd.Vét. 2000; 143: 423-436.
24. Alpérovitch A. Le nouveau variant de la maladie de Creutzfeldt-Jakob: quelques questions sans réponse. Rev.Epidém.et Santé Publ. 1999; 47: 313-314.
25. Kmietowicz Z. Surgery increases risk of sporadic CJD. BMJ 1999; 318: 625
26. Cousens, Linsell, Smith et al. Geographical distribution of variant CJD in the UK (excluding Nothern Ireland). The Lancet 1999; 353: 18-21.
27. Laske C. The effect of stress on the onset and progression of Creutzfeldt-Jakob disease: Results of a German pilot case-control study. European Journal of Epidemiology 1999; 15: 631-635.
28. Aylin P. Mortality from dementia in occupations at risk of exposure to bovine spongiform encephalopathy: analysis of death registrations. BMJ 2000; 318: 1044-1045.
29. Fischer S. Conséquence de la maladie de la vache folle: la Suisse rend le sang plus sûr. Médecine et Hygiène 1999; 573.
30. Collee,J.; Bradley,R. BSE: a decade on-part 2. The Lancet 1997, 349: 715-721.





























