Embed Size (px)
Citation preview

N° 2005-ISAL-0012 Année 2005
THESE
Rôle des gangliosides dans les perturbations de la prolifération des péricytes
rétiniens et des cellules mésangiales rénales : implication dans le développement
de la rétinopathie et de la néphropathie diabétiques
Présentée devant
L'institut National des Sciences Appliquées de Lyon
Pour obtenir
Le grade de Docteur en biochimie
Par Elodie MASSON
Ingénieur Agronome (ENSA de Rennes)
DEA de Biologie et Production Animales (Université de Rennes / ENSAR)
Soutenue publiquement le 2 février 2005 devant la commission d'examen
Jury:
Dr Jean-Claude MICHALSKI (rapporteur)
Pr Philippe ZAOUI (rapporteur)
Dr Jacques PORTOUKALIAN
Dr Nicolas WIERNSPERGER
Pr Michel LAGARDE
Dr Samer EL BAWAB
Thèse préparée au sein de l'Unité Thématique de Microangiopathie Diabétique MERCK Santé /
INSERM UMR 585, à l'INSA de Lyon.


Ecoles doctorales SIGLE ECOLE DOCTORALE NOM ET COORDONNEES DU RESPONSABLE
CHIMIE DE LYON M. Denis SINOU Université Claude Bernard Lyon 1 Lab Synthèse Asymétrique UMR UCB/CNRS 5622 Bât 308 2ème étage 43 bd du 11 novembre 1918 69622 VILLEURBANNE Cedex Tél : 04.72.44.81.83 [email protected]
E2MC
ECONOMIE, ESPACE ET MODELISATION DES COMPORTEMENTS
M. Alain BONNAFOUS Université Lyon 2 14 avenue Berthelot MRASH Laboratoire d’Economie des Transports 69363 LYON Cedex 07 Tél : 04.78.69.72.76 [email protected]
E.E.A.
ELECTRONIQUE, ELECTROTECHNIQUE, AUTOMATIQUE
M. Daniel BARBIER INSA DE LYON Laboratoire Physique de la Matière Bâtiment Blaise Pascal 69621 VILLEURBANNE Cedex Tél : 04.72.43.64.43 [email protected]
E2M2
EVOLUTION, ECOSYSTEME, MICROBIOLOGIE, MODELISATION http://biomserv.univ-lyon1.fr/E2M2
M. Jean-Pierre FLANDROIS UMR 5558 Biométrie et Biologie Evolutive Equipe Dynamique des Populations Bactériennes Faculté de Médecine Lyon-Sud Laboratoire de Bactériologie BP 1269600 OULLINS Tél : 04.78.86.31.50 [email protected]
EDIIS
INFORMATIQUE ET INFORMATION POUR LA SOCIETE http://www.insa-lyon.fr/ediis
M. Lionel BRUNIE INSA DE LYON EDIIS Bâtiment Blaise Pascal 69621 VILLEURBANNE Cedex Tél : 04.72.43.60.55 [email protected]
EDISS
INTERDISCIPLINAIRE SCIENCES-SANTEhttp://www.ibcp.fr/ediss
M. Alain Jean COZZONE IBCP (UCBL1) 7 passage du Vercors 69367 LYON Cedex 07 Tél : 04.72.72.26.75 [email protected]
MATERIAUX DE LYON http://www.ec-lyon.fr/sites/edml
M. Jacques JOSEPH Ecole Centrale de Lyon Bât F7 Lab. Sciences et Techniques des Matériaux et des Surfaces 36 Avenue Guy de Collongue BP 163 69131 ECULLY Cedex Tél : 04.72.18.62.51 [email protected]
Math IF
MATHEMATIQUES ET INFORMATIQUE FONDAMENTALE http://www.ens-lyon.fr/MathIS
M. Franck WAGNER Université Claude Bernard Lyon1 Institut Girard Desargues UMR 5028 MATHEMATIQUES Bâtiment Doyen Jean Braconnier Bureau 101 Bis, 1er étage 69622 VILLEURBANNE Cedex Tél : 04.72.43.27.86 [email protected]
MEGA
MECANIQUE, ENERGETIQUE, GENIE CIVIL, ACOUSTIQUE http://www.lmfa.ec-lyon.fr/autres/MEGA/index.html
M. François SIDOROFF Ecole Centrale de Lyon Lab. Tribologie et Dynamique des Systêmes Bât G8 36 avenue Guy de Collongue BP 163 69131 ECULLY Cedex Tél :04.72.18.62.14 [email protected]

Novembre 2003 INSTITUT NATIONAL DES SCIENCES APPLIQUEES DE LYON Directeur : STORCK A. Professeurs : AMGHAR Y. LIRIS AUDISIO S. PHYSICOCHIMIE INDUSTRIELLE BABOT D. CONT. NON DESTR. PAR RAYONNEMENTS IONISANTS BABOUX J.C. GEMPPM*** BALLAND B. PHYSIQUE DE LA MATIERE BAPTISTE P. PRODUCTIQUE ET INFORMATIQUE DES SYSTEMES MANUFACTURIERS BARBIER D. PHYSIQUE DE LA MATIERE BASKURT A. LIRIS BASTIDE J.P. LAEPSI**** BAYADA G. MECANIQUE DES CONTACTS BENADDA B. LAEPSI**** BETEMPS M. AUTOMATIQUE INDUSTRIELLE BIENNIER F. PRODUCTIQUE ET INFORMATIQUE DES SYSTEMES MANUFACTURIERS BLANCHARD J.M. LAEPSI**** BOISSE P. LAMCOS BOISSON C. VIBRATIONS-ACOUSTIQUE BOIVIN M. (Prof. émérite) MECANIQUE DES SOLIDES BOTTA H. UNITE DE RECHERCHE EN GENIE CIVIL - Développement Urbain BOTTA-ZIMMERMANN M. (Mme) UNITE DE RECHERCHE EN GENIE CIVIL - Développement Urbain BOULAYE G. (Prof. émérite) INFORMATIQUE BOYER J.C. MECANIQUE DES SOLIDES BRAU J. CENTRE DE THERMIQUE DE LYON - Thermique du bâtiment BREMOND G. PHYSIQUE DE LA MATIERE BRISSAUD M. GENIE ELECTRIQUE ET FERROELECTRICITE BRUNET M. MECANIQUE DES SOLIDES BRUNIE L. INGENIERIE DES SYSTEMES D’INFORMATION BUFFIERE J-Y. GEMPPM*** BUREAU J.C. CEGELY* CAMPAGNE J-P. PRISMA CAVAILLE J.Y. GEMPPM*** CHAMPAGNE J-Y. LMFA CHANTE J.P. CEGELY*- Composants de puissance et applications CHOCAT B. UNITE DE RECHERCHE EN GENIE CIVIL - Hydrologie urbaine COMBESCURE A. MECANIQUE DES CONTACTS COURBON GEMPPM COUSIN M. UNITE DE RECHERCHE EN GENIE CIVIL - Structures DAUMAS F. (Mme) CENTRE DE THERMIQUE DE LYON - Energétique et Thermique DJERAN-MAIGRE I. UNITE DE RECHERCHE EN GENIE CIVIL DOUTHEAU A. CHIMIE ORGANIQUE DUBUY-MASSARD N. ESCHIL DUFOUR R. MECANIQUE DES STRUCTURES DUPUY J.C. PHYSIQUE DE LA MATIERE EMPTOZ H. RECONNAISSANCE DE FORMES ET VISION ESNOUF C. GEMPPM*** EYRAUD L. (Prof. émérite) GENIE ELECTRIQUE ET FERROELECTRICITE FANTOZZI G. GEMPPM*** FAVREL J. PRODUCTIQUE ET INFORMATIQUE DES SYSTEMES MANUFACTURIERS FAYARD J.M. BIOLOGIE FONCTIONNELLE, INSECTES ET INTERACTIONS FAYET M. (Prof. émérite) MECANIQUE DES SOLIDES FAZEKAS A. GEMPPM FERRARIS-BESSO G. MECANIQUE DES STRUCTURES FLAMAND L. MECANIQUE DES CONTACTS FLEURY E. CITI FLORY A. INGENIERIE DES SYSTEMES D’INFORMATIONS FOUGERES R. GEMPPM*** FOUQUET F. GEMPPM*** FRECON L. (Prof. émérite) REGROUPEMENT DES ENSEIGNANTS CHERCHEURS ISOLES GERARD J.F. INGENIERIE DES MATERIAUX POLYMERES GERMAIN P. LAEPSI**** GIMENEZ G. CREATIS** GOBIN P.F. (Prof. émérite) GEMPPM*** GONNARD P. GENIE ELECTRIQUE ET FERROELECTRICITE GONTRAND M. PHYSIQUE DE LA MATIERE GOUTTE R. (Prof. émérite) CREATIS** GOUJON L. GEMPPM*** GOURDON R. LAEPSI****. GRANGE G. (Prof. émérite) GENIE ELECTRIQUE ET FERROELECTRICITE GUENIN G. GEMPPM*** GUICHARDANT M. BIOCHIMIE ET PHARMACOLOGIE GUILLOT G. PHYSIQUE DE LA MATIERE

GUINET A. PRODUCTIQUE ET INFORMATIQUE DES SYSTEMES MANUFACTURIERS GUYADER J.L. VIBRATIONS-ACOUSTIQUE GUYOMAR D. GENIE ELECTRIQUE ET FERROELECTRICITE HEIBIG A. MATHEMATIQUE APPLIQUEES DE LYON JACQUET-RICHARDET G. MECANIQUE DES STRUCTURES JAYET Y. GEMPPM*** JOLION J.M. RECONNAISSANCE DE FORMES ET VISION JULLIEN J.F. UNITE DE RECHERCHE EN GENIE CIVIL - Structures JUTARD A. (Prof. émérite) AUTOMATIQUE INDUSTRIELLE KASTNER R. UNITE DE RECHERCHE EN GENIE CIVIL - Géotechnique KOULOUMDJIAN J. (Prof. émérite) INGENIERIE DES SYSTEMES D’INFORMATION LAGARDE M. BIOCHIMIE ET PHARMACOLOGIE LALANNE M. (Prof. émérite) MECANIQUE DES STRUCTURES LALLEMAND A. CENTRE DE THERMIQUE DE LYON - Energétique et thermique LALLEMAND M. (Mme) CENTRE DE THERMIQUE DE LYON - Energétique et thermique LAREAL P (Prof. émérite) UNITE DE RECHERCHE EN GENIE CIVIL - Géotechnique LAUGIER A. (Prof. émérite) PHYSIQUE DE LA MATIERE LAUGIER C. BIOCHIMIE ET PHARMACOLOGIE LAURINI R. INFORMATIQUE EN IMAGE ET SYSTEMES D’INFORMATION LEJEUNE P. UNITE MICROBIOLOGIE ET GENETIQUE LUBRECHT A. MECANIQUE DES CONTACTS MASSARD N. INTERACTION COLLABORATIVE TELEFORMATION TELEACTIVITE MAZILLE H. (Prof. émérite) PHYSICOCHIMIE INDUSTRIELLE MERLE P. GEMPPM*** MERLIN J. GEMPPM*** MIGNOTTE A. (Mle) INGENIERIE, INFORMATIQUE INDUSTRIELLE MILLET J.P. PHYSICOCHIMIE INDUSTRIELLE MIRAMOND M. UNITE DE RECHERCHE EN GENIE CIVIL - Hydrologie urbaine MOREL R. (Prof. émérite) MECANIQUE DES FLUIDES ET D’ACOUSTIQUES MOSZKOWICZ P. LAEPSI**** NARDON P. (Prof. émérite) BIOLOGIE FONCTIONNELLE, INSECTES ET INTERACTIONS NAVARRO Alain (Prof. émérite) LAEPSI**** NELIAS D. LAMCOS NIEL E. AUTOMATIQUE INDUSTRIELLE NORMAND B. GEMPPM NORTIER P. DREP ODET C. CREATIS** OTTERBEIN M. (Prof. émérite) LAEPSI**** PARIZET E. VIBRATIONS-ACOUSTIQUE PASCAULT J.P. INGENIERIE DES MATERIAUX POLYMERES PAVIC G. VIBRATIONS-ACOUSTIQUE PECORARO S. GEMPPM PELLETIER J.M. GEMPPM*** PERA J. UNITE DE RECHERCHE EN GENIE CIVIL - Matériaux PERRIAT P. GEMPPM*** PERRIN J. INTERACTION COLLABORATIVE TELEFORMATION TELEACTIVITE PINARD P. (Prof. émérite) PHYSIQUE DE LA MATIERE PINON J.M. INGENIERIE DES SYSTEMES D’INFORMATION PONCET A. PHYSIQUE DE LA MATIERE POUSIN J. MODELISATION MATHEMATIQUE ET CALCUL SCIENTIFIQUE PREVOT P. INTERACTION COLLABORATIVE TELEFORMATION TELEACTIVITE PROST R. CREATIS** RAYNAUD M. CENTRE DE THERMIQUE DE LYON - Transferts Interfaces et Matériaux REDARCE H. AUTOMATIQUE INDUSTRIELLE RETIF J-M. CEGELY* REYNOUARD J.M. UNITE DE RECHERCHE EN GENIE CIVIL - Structures RICHARD C. LGEF RIGAL J.F. MECANIQUE DES SOLIDES RIEUTORD E. (Prof. émérite) MECANIQUE DES FLUIDES ROBERT-BAUDOUY J. (Mme) (Prof. émérite) GENETIQUE MOLECULAIRE DES MICROORGANISMES ROUBY D. GEMPPM*** ROUX J.J. CENTRE DE THERMIQUE DE LYON – Thermique de l’Habitat RUBEL P. INGENIERIE DES SYSTEMES D’INFORMATION SACADURA J.F. CENTRE DE THERMIQUE DE LYON - Transferts Interfaces et Matériaux SAUTEREAU H. INGENIERIE DES MATERIAUX POLYMERES SCAVARDA S. (Prof. émérite) AUTOMATIQUE INDUSTRIELLE SOUIFI A. PHYSIQUE DE LA MATIERE SOUROUILLE J.L. INGENIERIE INFORMATIQUE INDUSTRIELLE THOMASSET D. AUTOMATIQUE INDUSTRIELLE THUDEROZ C. ESCHIL – Equipe Sciences Humaines de l’Insa de Lyon UBEDA S. CENTRE D’INNOV. EN TELECOM ET INTEGRATION DE SERVICES VELEX P. MECANIQUE DES CONTACTS VERMANDE P. (Prof émérite) LAEPSI VIGIER G. GEMPPM*** VINCENT A. GEMPPM*** VRAY D. CREATIS** VUILLERMOZ P.L. (Prof. émérite) PHYSIQUE DE LA MATIERE

Directeurs de recherche C.N.R.S. : BERTHIER Y. MECANIQUE DES CONTACTS CONDEMINE G. UNITE MICROBIOLOGIE ET GENETIQUE COTTE-PATAT N. (Mme) UNITE MICROBIOLOGIE ET GENETIQUE ESCUDIE D. (Mme) CENTRE DE THERMIQUE DE LYON FRANCIOSI P. GEMPPM*** MANDRAND M.A. (Mme) UNITE MICROBIOLOGIE ET GENETIQUE POUSIN G. BIOLOGIE ET PHARMACOLOGIE ROCHE A. INGENIERIE DES MATERIAUX POLYMERES SEGUELA A. GEMPPM*** VERGNE P. LaMcos Directeurs de recherche I.N.R.A. : FEBVAY G. BIOLOGIE FONCTIONNELLE, INSECTES ET INTERACTIONS GRENIER S. BIOLOGIE FONCTIONNELLE, INSECTES ET INTERACTIONS RAHBE Y. BIOLOGIE FONCTIONNELLE, INSECTES ET INTERACTIONS Directeurs de recherche I.N.S.E.R.M. : KOBAYASHI T. PLM PRIGENT A.F. (Mme) BIOLOGIE ET PHARMACOLOGIE MAGNIN I. (Mme) CREATIS** * CEGELY CENTRE DE GENIE ELECTRIQUE DE LYON ** CREATIS CENTRE DE RECHERCHE ET D’APPLICATIONS EN TRAITEMENT DE L’IMAGE ET DU SIGNAL ***GEMPPM GROUPE D'ETUDE METALLURGIE PHYSIQUE ET PHYSIQUE DES MATERIAUX ****LAEPSI LABORATOIRE D’ANALYSE ENVIRONNEMENTALE DES PROCEDES ET SYSTEMES INDUSTRIELS

REMERCIEMENTS
Les travaux de thèse présentés dans ce manuscrit ont été menés au sein de l’Unité Thématique de
Microangiopathie Diabétique (UTMD) Merck Santé / INSERM UMR 585 co-dirigée par le Docteur
Nicolas WIERNSPERGER et le Professeur Michel LAGARDE. Je leur suis reconnaissante de
m'avoir accueillie au sein de leur unité de recherche.
Je remercie en premier lieu le Docteur Samer EL BAWAB pour m'avoir encadrée dans ce travail
de thèse et pour avoir accompagné mes premières armes dans le monde de la recherche en me
faisant partager son expérience et ses connaissances scientifiques, et aussi pour m'avoir supportée
durant ces années, dans tous les sens du terme.
Je témoigne ma gratitude au Professeur Michel LAGARDE pour sa bienveillance à mon égard,
ainsi que pour l'attention et l'intérêt qu'il a porté à mes travaux en tant que co-directeur de thèse.
Je remercie le Docteur Nicolas WIERNSPERGER pour l'intérêt qu'il a accordé à mon travail
malgré son caractère biochimique et pour avoir financé mes travaux par l'intermédiaire de Merck
Santé.
J'exprime ensuite mon estime et mes remerciements aux membres de mon jury :
Le Professeur Philippe ZAOUI et le Docteur Jean-Claude MICHALSKI qui m'ont fait l'honneur
de juger ce travail de thèse et de me faire ainsi bénéficier de leur expertise dans les domaines
respectifs de la néphropathie diabétique et des gangliosides.
Le Docteur Jacques PORTOUKALIAN pour m'avoir fait l'honneur d'accepter la présidence de ce
jury et m'avoir aimablement apporté une aide technique dans le domaine des gangliosides.
Je remercie également le Docteur Daniel Ruggiero pour avoir encadrer, avec toute sa sympathie,

ma première année de thèse.
Je remercie le Professeur Philippe LEGRAND pour les encouragements, les conseils et l'attention
qu'il m'a témoignés depuis ma sortie d'école tout comme pendant mon cursus à l'ENSAR ainsi que
pour m'avoir encouragée à choisir cette formation "par la recherche", et je l'espère aussi "pour la
recherche", qu'est la thèse.
Je tiens aussi à témoigner mon amitié à feu l'équipe Merck qui m'a accompagnée au quotidien:
Karen, compagne de thèse, je lui souhaite beaucoup de bonheur; Pierre, ancien collègue thésard à
qui je souhaite bonne chance pour l'avenir; Lysiane que je remercie vivement pour son aide
précieuse et sa gentillesse; Anne-Marie et sa bonne humeur, Brigitte que je remercie pour sa
sympathie et son soutien logistique très appréciable (c'est surtout quand elle n'est plus là qu'on s'en
rend compte), et Corinne que je remercie pour la sympathie et les encouragements qu'elle m'a
témoignés.
Je remercie enfin l'ensemble des membres du laboratoire "Physiopathologie des Lipides et
Membranes" INSA/INSERM pour la sympathie ou l'aide qu'ils m'ont témoignées durant ces trois
années, notamment Patrick, Martine, Madeleine et Madeleine, Evelyne et Catherine, Véronique,
André, Salim et l'indispensable informaticien Fabien. J'adresse particulièrement mes remerciements
aux anciens thésards (les Dr Caroline, Laurent, Pierre, Sandrine, Olivier, Hiba) qui m'ont fait
profiter de leur expérience, et mes encouragements et vœux de réussite aux futurs docteurs (Nelly,
Saïda, Bader, Caroline, Deborah).
Je n'oublie pas M. Raymond Brut, de la société STA des abattoirs de Corbas, que je remercie pour
sa serviabilité et sa bonne humeur matinale.
J'adresse pour finir mon attachement et mes remerciements à
Mes parents, grâce auxquels j'en suis arrivée là aujourd'hui.
Jérôme, pour son écoute, son soutien essentiel et son aide.
Ma famille et mes amis pour leur écoute et leurs encouragements très précieux.

TABLE DES MATIERES
AVANT-PROPOS ....................................................................................................... 1
ABREVIATIONS ........................................................................................................ 3
RESUME ...................................................................................................................... 6
ABSTRACT ................................................................................................................. 7
INTRODUCTION ....................................................................................................... 8
ETUDE BIBLIOGRAPHIQUE ............................................................................... 11
LE DIABETE ...............................................................................................................................12
I. LE DIABETE SUCRE 12
1. Les differents types de diabéte.......................................................................................13
1.1. Le diabète de type 1 ...............................................................................................13
1.2. Le diabète de type 2 ...............................................................................................14
2. Les complications liées au diabète.................................................................................15
2.1. Les complications aiguës .......................................................................................15
2.2. Les complications chroniques ................................................................................15 2.2.1 La macroangiopathie diabétique ........................................................................16 2.2.2 La microangiopathie diabétique.........................................................................16
II. PHYSIOPATHOLOGIE DE LA RETINOPATHIE ET DE LA NEPHROPATHIE DIABETIQUES 17
1. Les microvaisseaux au sein de leur structure.................................................................18
1.1. La rétine..................................................................................................................18 1.1.1 L’œil et la rétine.................................................................................................18 1.1.2 Les microvaisseaux............................................................................................19 1.1.3 Les cellules.........................................................................................................20

(a) Les cellules endothéliales 20 (b) Les péricytes 21
1.2. Les reins .................................................................................................................21 1.2.1 Le système urinaire et les reins..........................................................................21 1.2.2 Les glomérules, site de filtration........................................................................24 1.2.3 Les cellules.........................................................................................................25
(a) Les cellules endothéliales 25 (b) Les podocytes, cellules epithéliales spécialisées 25 (c) Les cellules mésangiales 26
2. Deux pathologies évolutives ..........................................................................................27
2.1. Rétinopathie diabétique..........................................................................................28 2.1.1 Les différentes phases de la maladie..................................................................28
(a) Phase non-proliférative 28 (b) Phase pré-proliférative 28 (c) Phase proliférative 28
2.1.2 Les facteurs de risque.........................................................................................29 2.1.3 Le traitement ......................................................................................................29
2.2. Néphropathie diabétique (ND) ...............................................................................29 2.2.1 Les différents stades de la maladie ....................................................................29
(a) ND silencieuse et hyperfiltration glomérulaire 30 (b) ND débutante et microalbuminurie 30 (c) ND confirmée et protéinurie 30 (d) Insuffisance rénale terminale 31
2.2.2 Les facteurs de risque.........................................................................................31 2.2.3 Le traitement ......................................................................................................31
3. Altérations structurales et fonctionnelles.......................................................................32
3.1. Membrane basale et matrice extracellulaire...........................................................32
3.2. Altérations cellulaires.............................................................................................34 3.2.1 Cellules microvasculaires rétiniennes................................................................34
(a) Les péricytes 34 (b) Les cellules endothéliales 35
3.2.2 Cellules glomérulaires .......................................................................................36 (a) Les cellules mésangiales 36 (b) Les podocytes 38 (c) Cellules épithéliales tubulaires 38 (d) Cellules endothéliales 39
3.3. Modifications des paramètres sanguins..................................................................39
3.4. Modifications des parois capillaires.......................................................................39
III. LES MECANISMES 40
1. L'hypothèse hémodynamique ........................................................................................41
2. La voie des polyols ........................................................................................................41
3. L'activation de la pkc .....................................................................................................42
4. La glycosylation non enzymatique ................................................................................43
4.1. Formation des AGE................................................................................................43
4.2. Caractérisation et mesure .......................................................................................45

4.3. Mécanismes d’action et conséquences pathologiques ...........................................45 4.3.1 Glycation des protéines à durée de vie courte : protéines circulantes,
intracellulaires et acides nucléiques...................................................................46 4.3.2 Glycation des protéines de la matrice ................................................................47 4.3.3 Interaction avec des récepteurs membranaires spécifiques ...............................48
(a) Macrophage "scavenger" récepteurs 48 (b) Complexe AGE-récepteur 48 (c) RAGE 50
4.4. AGE et rétinopathie diabétique ..............................................................................52
4.5. AGE et néphropathie diabétique ............................................................................54
5. La voie des hexosamines ...............................................................................................56
5.1. Définition ...............................................................................................................56
5.2. Mécanismes d’action..............................................................................................58 5.2.1 Apport de précurseurs glycosidiques .................................................................59 5.2.2 O-glycosylation simple de protéines..................................................................59
5.3. Conséquences pathologiques .................................................................................61 5.3.1 Insulino-résistance .............................................................................................61 5.3.2 Néphropathie diabétique ....................................................................................62
6. Le stress oxydant ...........................................................................................................64
LES GANGLIOSIDES ................................................................................................................66
I. STRUCTURE ET LOCALISATION 66
1. Structure.........................................................................................................................66
1.1. Les glycosphingolipides.........................................................................................66
1.2. Les gangliosides .....................................................................................................68
2. Localisation....................................................................................................................69
2.1. Localisation tissulaire.............................................................................................69
2.2. Localisation cellulaire ............................................................................................69
II. LE METABOLISME DES GANGLIOSIDES 71
1. Biosynthèse....................................................................................................................71
1.1. Les premières étapes: formation du précurseur commun, le lactosylcéramide......72
1.2. La chaîne d'assemblage ..........................................................................................72
2. Catabolisme ...................................................................................................................73
3. Régulation du metabolisme ...........................................................................................75
3.1. Equipement enzymatique et disponibilité des substrats.........................................75
3.2. Régulation au niveau transcriptionnel....................................................................76
3.3. Mécanismes de régulation épigénétiques...............................................................76 3.3.1 Au niveau post-transcriptionnel .........................................................................76 3.3.2 Rétrocontrôle négatif .........................................................................................77 3.3.3 Le pH .................................................................................................................78
III. FONCTIONS 78

1. Double fonction des gangliosides ..................................................................................79
1.1. Reconnaissance cellulaire ......................................................................................79 1.1.1 Propriétés antigéniques ......................................................................................80 1.1.2 Reconnaissance par des protéines de liaison aux hydrates de carbone..............81 1.1.3 Modulation des récepteurs intégrines ................................................................81 1.1.4 Interaction entre gangliosides et GSL................................................................82 1.1.5 Interaction avec des virus, bactéries ou toxines.................................................82
1.2. Modulation du signal transmembranaire................................................................83 1.2.1 Régulation des récepteurs à tyrosine kinase ......................................................83 1.2.2 Modulation de la protéine kinase C ...................................................................86
2. Importance biologique / physiologique des gangliosides ..............................................87
2.1. Développement, différenciation cellulaire et tissulaire, embryogenèse.................87
2.2. Oncogénèse ............................................................................................................88
2.3. Prolifération cellulaire............................................................................................89
IV. GANGLIOSIDES ET DIABETE 92
1. Gangliosides et auto-immunité ......................................................................................93
2. Gangliosides et microangiopathie diabetique ................................................................94
3. Gangliosides et insulino-resistance................................................................................95
MATERIEL ET METHODES................................................................................. 97
I. CULTURES CELLULAIRES 98
1. Péricytes rétiniens de bœuf ............................................................................................98
1.1. Isolement des microvaisseaux et mise en culture...................................................98
1.2. Passages et traitements .........................................................................................100
2. Cellules mésangiales rénales de rat .............................................................................100
2.1. Isolement des glomérules et mise en culture........................................................100
2.2. Passages et traitements .........................................................................................102
II. TRAITEMENT DES CELLULES 102
1. Les AGE ......................................................................................................................102
1.1. Préparation des AGE............................................................................................103
1.2. Traitement ............................................................................................................103
2. La glucosamine ............................................................................................................103
3. Gangliosides exogènes.................................................................................................104
4. Anticorps anti-gangliosides .........................................................................................104
5. PPMP ...........................................................................................................................104
III. TRANSFECTION DES CELLULES AVEC LES SIRNA 105
IV. ANIMAUX DIABETIQUES 107

V. ANALYSE DE LA PROLIFERATION CELLULAIRE 107
1. Comptage des cellules .................................................................................................107
2. Dosage des protéines ...................................................................................................107
3. Mesure des quantités d'ADN .......................................................................................108
4. Analyse du cycle cellulaire ..........................................................................................109
5. Mesure de l’expression des protéines inhibitrices du cycle cellulaire.........................109
VI. ANALYSE DES GANGLIOSIDES 110
1. Marquage métabolique ................................................................................................110
2. Extraction, purification, séparation et détection des gangliosides...............................110
2.1. Extraction et séparation des gangliosides.............................................................110
2.2. Révélation et identification des gangliosides .......................................................111
3. Dosage des activités enzymatiques..............................................................................114
VII. ANALYSE STATISTIQUE DES RESULTATS 116
RESULTATS-DISCUSSION ................................................................................. 117
LES AGE DIMINUENT LA PROLIFERATION DES PERICYTES ET DES CELLULES MESANGIALES: IMPLICATION DES GANGLIOSIDES .................................................119
I. EFFETS DES AGE SUR LA PROLIFERATION CELLULAIRE 119
II. EFFETS DES AGE SUR LE METABOLISME DES GANGLIOSIDES 121
1. Le profil basal en gangliosides ....................................................................................121
1.1. Effet des AGE sur la composition en gangliosides des péricytes ........................122
1.2. Effet des AGE sur la composition en gangliosides des cellules mésangiales......124
2. Effet des AGE sur l’activité des enzymes de biosynthèse des gangliosides................126
III. IMPLICATION DES GANGLIOSIDES DANS LA DIMINUTION DE LA PROLIFERATION CELLULAIRE CAUSEE PAR LES AGE 128
1. Effet d’un traitement par des gangliosides exogènes sur la prolifération des péricytes et des cellules mésangiales ..............................................................................................128
2. Effet d’un traitement avec les AGE en présence d’anticorps dirigés contre les gangliosides de la série-a .............................................................................................130
3. Effet d’un cotraitement AGE et PPMP........................................................................131
4. Effet de la transfection avec un SiRNA de GM3 synthase..........................................133
IV. DISCUSSION 134
L’ACTIVATION DE LA VOIE DES HEXOSAMINES DIMINUE LA PROLIFERATION DES PERICYTES ET DES CELLULES MESANGIALES : IMPLICATION DES GANGLIOSIDES.......................................................................................................................138

I. EFFETS DE LA GLUCOSAMINE SUR LA PROLIFERATION CELLULAIRE 139
II. EFFETS DE LA GLUCOSAMINE SUR LE METABOLISME DES GANGLIOSIDES 140
1. Effet de la glucosamine sur le profil en gangliosides des péricytes ............................141
2. Effet de la glucosamine sur le profil en gangliosides des cellules mésangiales ..........141
III. EFFET DE LA GLUCOSAMINE SUR L’ACTIVITE GM3 SYNTHASE 144
IV. IMPLICATION DES GANGLIOSIDES DANS LA DIMINUTION DE PROLIFERATION CELLULAIRE CAUSEE PAR LA GLUCOSAMINE 146
1. Effet d’un traitement par les gangliosides exogènes sur la prolifération des péricytes et des cellules mésangiales ..............................................................................................146
2. Effet d’un cotraitement glucosamine et PPMP............................................................148
V. ETUDE DES MECANISMES CONDUISANT A L’INHIBITION DE PROLIFERATION DES CELLULES MESANGIALES PAR LA GLUCOSAMINE 150
1. Effet de la glucosamine sur l’hypertrophie des RMC..................................................150
2. Effet de la glucosamine sur le cycle cellulaire ............................................................152
2.1. Analyse du cycle par FACS .................................................................................152
2.2. Effet de la glucosamine sur l’expression des inhibiteurs de kinase cycline-dépendante............................................................................................................153
3. Effet de la glucosamine sur le profil en gangliosides ..................................................155
4. Effet de GM2 et GM1 sur l’hypertrophie et le cycle cellulaire des RMC...................156
4.1. Effet des gangliosides sur l’hypertrophie.............................................................156
4.2. Effet des gangliosides sur le cycle cellulaire .......................................................157
VI. DISCUSSION 159
METABOLISME DES GANGLIOSIDES DU CORTEX RENAL DE SOURIS DB/DB...164
I. MESURE DES ACTIVITES GM3 ET GD3 SYNTHASE 164
II. MESURE DES TAUX DE GM3 165
III. DISCUSSION 166
CONCLUSION ET PERSPECTIVES .................................................................. 168
REFERENCES BIBLIOGRAPHIQUES.............................................................. 174
PUBLICATIONS..................................................................................................... 197

INDEX DES FIGURES
Figure 1: évolution des cas de diabète dans le monde. 12
Figure 2: structure de l'œil en coupe. 18
Figure 3:représentation schématique de la structure de la rétine. 19
Figure 4: structure d'un capillaire rétinien. 20
Figure 5: structure du système urinaire et du rein. 22
Figure 6: structure d'un néphron. 23
Figure 7: structure d'un corpuscule rénal et de la membrane de filtration glomérulaire. 25
Figure 8: coupe d'un glomérule rénal. 26
Figure 9: comportement biphasique des cellules mésangiales rénales au cours de la néphropathie diabétique. 37
Figure 10: différentes voies de formation des AGE. 44
Figure 11: le complexe récepteur aux AGE. 49
Figure 12: signalisation consécutive à la liaison des AGE au récepteur RAGE. 51
Figure 13: voie de biosynthèse des hexosamines. 57
Figure 14: mécanisme proposé pour l’induction de la voie des hexosamines par l’hyperglycémie. 58
Figure 15: différentes protéines O-glycosylées connues dans la cellule. 60
Figure 16: mécanisme d’action de la glucosamine proposé pour la surproduction de matrice extracellulaire. 63
Figure 17 : le stress oxydant, lien unificateur entre les différents mécanismes biochimiques issus de l'hyperglycémie.65
Figure 18 : structure d'un glycosphingolipide et position dans la membrane plasmique. 68
Figure 19 : structure d'un ganglioside. 69
Figure 20 : structure d'un microdomaine membranaire. 71
Figure 21 : schéma de biosynthèse des gangliosides. 73
Figure 22 : catabolisme des gangliosides. 74

Figure 23 : récapitulatif des différentes fonctions occupées par les gangliosides. 80
Figure 24 : mécanisme de l'interaction du GM1 avec la toxine cholérique. 83
Figure 25 : modulation de l'activité de différents récepteurs aux facteurs de croissance par les gangliosides. 84
Figure 26 : processus apoptotique impliquant le GD3 (SM, sphingomyélinase; ST8, sialyltransférase 8: GD3
synthase). 91
Figure 27 : mécanisme d'action proposé pour expliquer l'implication du GM3 dans l'insulino-résistance. 96
Figure 28 : croissance des péricytes depuis un microvaisseau rétinien (A) Péricytes rétiniens (B). 99
Figure 29 : croissance des cellules mésangiales à partir d'un glomérule rénal (A) Cellules mésangiales (B). 101
Figure 30 : mécanisme de l'interférence ARN. 105
Figure 31 : Exemple de plaque HPTLC révélée par autoradiographie (A) puis par révélation colorimétrique au
résorcinol (B). 112
Figure 32 : immunuodétection du GM2. 113
Figure 33 : détection du GM1 par la toxine cholérique. 114
Figure 34 : principe du dosage des activités enzymatiques GM3 et GD3 synthases. 115
Figure 35 : Les AGE inhibent la prolifération des péricytes et des cellules mésangiales. 120
Figure 36 : Profil en gangliosides des péricytes rétiniens et des cellules mésangiales rénales. 122
Figure 37 : Les AGE modifient le profil en gangliosides des péricytes rétiniens. 123
Figure 38 : Les AGE augmentent la synthèse du GM2 dans les péricytes. 124
Figure 39 : Les AGE modifient le profil en gangliosides des cellules mésangiales. 125
Figure 40 : Les AGE augmentent la synthèse du GM2 et du GM1 dans les cellules mésangiales. 126
Figure 41 : Les AGE augmentent l'activité GM3 synthase et diminuent l'activité GD3 synthase. 127
Figure 42 : L’ajout exogène de gangliosides de la série a diminue la prolifération des BRP. 129
Figure 43 : L’ajout exogène de gangliosides de la série a diminue la prolifération des RMC. 130
Figure 44 : L’utilisation d’anticorps anti-GM2 et anti-GM1 protège partiellement de l’effet anti-prolifératif des
AGE. 131
Figure 45 : L’utilisation de PPMP réduit l’inhibition de prolifération causée par les AGE dans les péricytes. 132

Figure 46 : La transfection avec un SiRNA spécifique de la GM3 synthase protège partiellement les RMC de
l’inhibition de prolifération causée par les AGE. 133
Figure 47 : La glucosamine diminue la prolifération des péricytes et des cellules mésangiales. 140
Figure 48 : La glucosamine modifie le profil en gangliosides des péricytes rétiniens. 141
Figure 49 : La glucosamine modifie le profil en gangliosides des cellules mésangiales. 143
Figure 50 : La glucosamine modifie l'activité GM3 synthase des péricytes et des cellules mésangiales. 145
Figure 51 : Le traitement par le GM1 et le GD3 diminue la prolifération des BRP. 147
Figure 52 : Le traitement par les GM2, GM1 et GD1a diminue la prolifération des RMC. 148
Figure 53 : L’utilisation de PPMP réduit l’inhibition de prolifération causée par la glucosamine sur les péricytes.149
Figure 54 : La glucosamine inhibe la croissance des cellules mésangiales. 151
Figure 55 : Le traitement avec la glucosamine induit l’hypertrophie des cellules mésangiales. 152
Figure 56 : La glucosamine bloque le cycle cellulaire des RMC de façon dose-dépendante. 153
Figure 57 : La glucosamine augmente l’expression de p21Waf1/Cip1. 154
Figure 58 : La glucosamine modifie le profil en gangliosides des RMC. 155
Figure 59 : GM2 et GM1 induisent l’hypertrophie des cellules mésangiales. 156
Figure 60 : GM2 et GM1 bloquent le cycle cellulaire des RMC en phase G0/G1. 157
Figure 61 : GM2 et GM1 augmentent l’expression de p21Waf1/Cip1. 158
Figure 62 : Les activités GM3 et GD3 synthase sont modifiées dans le cortex rénal de souris db/db. 165
Figure 63 : Le taux de GM3 est augmenté dans le cortex rénal de souris db/db. 166
Figure 64 : Bilan des modifications du métabolisme des gangliosides dans les péricytes et les cellules mésangiales en
réponse aux AGE et à la glucosamine, et dans les souris db/db. 172


AVANT-PROPOS
Les travaux menés au cours de ces trois années de thèse ont fait l'objet de communications
scientifiques : publications acceptées ou en révision, résumés ou posters ainsi que d'un brevet.
PUBLICATIONS :
MASSON E., TRONCY L., RUGGIERO D., WIERNSPERGER N., LAGARDE M., EL BAWAB
S. a-Series gangliosides mediate the effects of Advanced Glycation End-products on pericyte and
mesangial cell proliferation: a common mediator for retinal and renal microangiopathy ?. Diabetes
2005 Jan;54(1):220-7.
MASSON E., WIERNSPERGER N., LAGARDE M., EL BAWAB S. Involvement of gangliosides
in glucosamine-induced proliferation decrease of retinal pericytes. Accepté pour publication dans
Glycobiology 2004 Dec 29 (Advance Access).
MASSON E., WIERNSPERGER N., LAGARDE M., EL BAWAB S. Glucosamine induces cell
cycle arrest and hypertrophy of mesangial cells: implication of gangliosides. Accepté pour
publication dans Biochemical Journal 17 January 2005 (Immediate Publication).
MASSON E., RUGGIERO D., WIERNSPERGER N., LAGARDE M., EL BAWAB S. Advanced
glycation end-products decrease pericyte and mesangial cell proliferation: involvement of the a-
series gangliosides. Diabetes abstract book, June 2003, 63rd Scientific Sessions, New Orleans,
Louisiana.
1

COMMUNICATIONS AFFICHEES :
MASSON E., RUGGIERO D., WIERNSPERGER N., LAGARDE M., EL BAWAB S. Advanced
glycation end-products decrease pericyte and mesangial cell proliferation: involvement of the a-
series gangliosides, Congrès de Lipidomique, GERLI (Groupe d'Etude et de Recherche sur les
Lipides et les Lipoprotéines), Paris, septembre 2003.
MASSON E., RUGGIERO D., WIERNSPERGER N., LAGARDE M.. Ganglioside profile of
microvascular retinal and glomerular cells in the scope of diabetic microangiopathy, GERLI,
Nantes, avril 2002.
BREVET :
MASSON Elodie, RUGGIERO Daniel, TONCY Lysiane, WIERNSPERER Nicolas, LAGARDE
Michel, EL BAWAB Samer. GM3 synthase, comme cible thérapeutique dans les complications
microvasculaires du diabète. Brevet français n° 04 04971 déposé le 7 mai 2004.
2
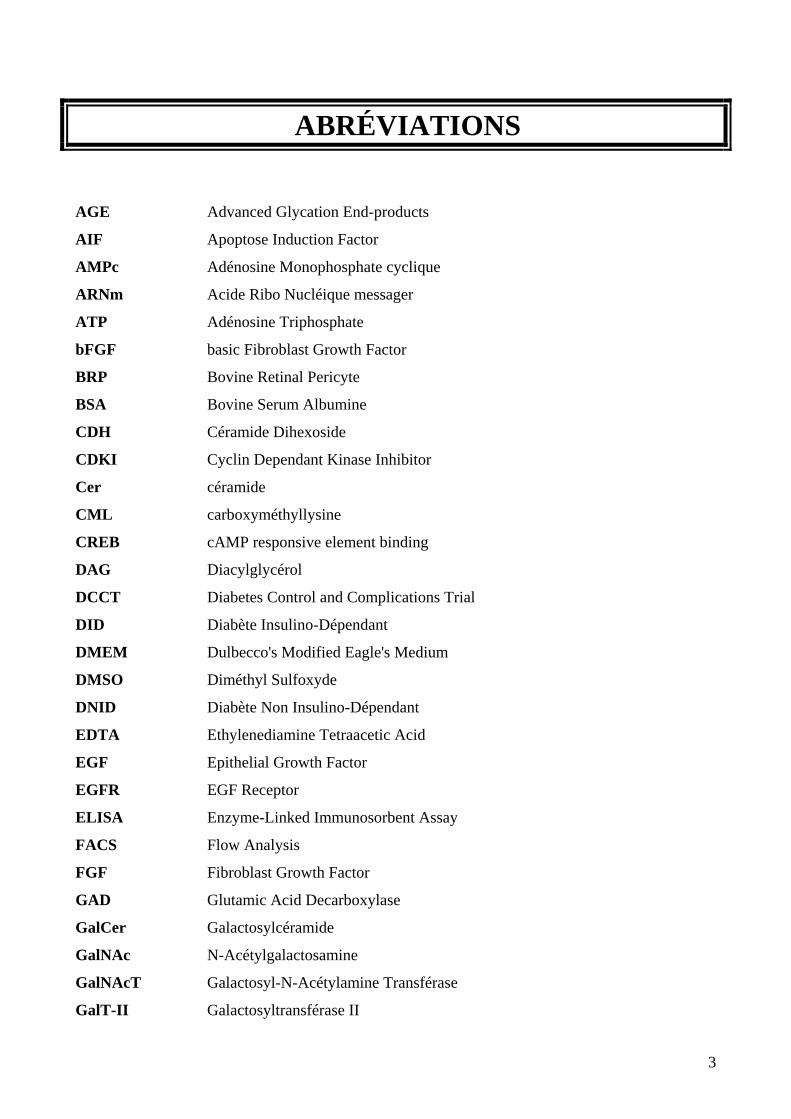
ABRÉVIATIONS
AGE Advanced Glycation End-products
AIF Apoptose Induction Factor
AMPc Adénosine Monophosphate cyclique
ARNm Acide Ribo Nucléique messager
ATP Adénosine Triphosphate
bFGF basic Fibroblast Growth Factor
BRP Bovine Retinal Pericyte
BSA Bovine Serum Albumine
CDH Céramide Dihexoside
CDKI Cyclin Dependant Kinase Inhibitor
Cer céramide
CML carboxyméthyllysine
CREB cAMP responsive element binding
DAG Diacylglycérol
DCCT Diabetes Control and Complications Trial
DID Diabète Insulino-Dépendant
DMEM Dulbecco's Modified Eagle's Medium
DMSO Diméthyl Sulfoxyde
DNID Diabète Non Insulino-Dépendant
EDTA Ethylenediamine Tetraacetic Acid
EGF Epithelial Growth Factor
EGFR EGF Receptor
ELISA Enzyme-Linked Immunosorbent Assay
FACS Flow Analysis
FGF Fibroblast Growth Factor
GAD Glutamic Acid Decarboxylase
GalCer Galactosylcéramide
GalNAc N-Acétylgalactosamine
GalNAcT Galactosyl-N-Acétylamine Transférase
GalT-II Galactosyltransférase II
3

GAPDH Glyceraldéhyde-3-Phosphate Dehydrogénase
GD Disialoganglioside
GFAT Glutamine Fructose-6-P Amidotransférase
GlcN Glucosamine
GlcN-6-P Glucosamine-6-P
GlcNAc N-Acétylglucosamine
GM Monosialoganglioside
GOLD Glyoxal-lysine dimer
GPI Glycosyl-Phosphatidylinositol
GSL Glycosphingolipide
GT Trisialoganglioside
HBSS Hanks' Balanced Salt Solution
HDL High Density Lipoprotein
HEPES Acide (Hydroxy-2-Ethyl)-4-Pipérazinyl-1-2-Ethansulfonique
HGF Hepatocyte Growth Factor
HPLC High Pression Liquid Chromatography
HPTLC High Performance Thin Layer Chromatography
ICA Islet Cell Antibodies
ICAM-1 Intercellular Adhesion Molecule-1
Ig Immunogloguline (G, M)
IGF1 Insulin Growth Factor 1
IL-1 Interleukine
IRS1 Insuline Receptor Substrate 1
Laccer Lactosylcéramide
LDL Low Density Lipoprotein
MAG Myelin Associated Glycoprotein
MAM Membranes Associées aux Mitochondries
MAP-kinase Mitogen Activated Protein kinase
MOLD Methyl glyoxal-lysine dimer
MRS Macrophage Scavenger Receptor
NADPH Nicotinamide Adenine Dinucleotide Phosphate
ND Néphropathie Diabétique
NeuAc Acide N-Acétylneuraminique
NeuGc Acide N-Glycolylneuraminique
NF-κB Nuclear Factor-κB
4

NGF Nerve Growth Factor
OGT O-GlNAc Transférase
OMS Organisation Mondiale de la Santé
PAF Platelet Activating Factor
PAI-I Plasminogen Activator Inhibitor
PBS Phosphate-Buffered Saline
PDGF Platelet Derived Growth Factor
PDGFR PDGF Receptor
PDMP DL-threo-1-Decanoylamino-3-morpholino-1-propanol
PG Prostaglandine (E1, H2, I2…)
PKA Protein Kinase (A, C, Cβ1…)
PLC Phospholipase C
PPMP DL-threo-1-Phenyl-2-palmitoylamino-3-morpholino-1-propanol
RAGE AGE Receptor
RD Rétinopathie Diabétique
RMC Renal Mesangial Cell
ROS Reactive Oxygen Species
SAP Sphingolipid Activator Protein
SBP Sugar Binding Protein
SiRNA Small interfering Ribo Nucleic Acid
ST Sialyltransférases (IV et V)
STZ Streptozotocine
SVF Serum de Veau Foetal
TGFβ Transforming Growth Factor β
TLCK Tosyle de Lysinechlorométhylcétone
TNF-α Tumor Necrosis Factor-α
tPA Tissular Plasminogen Activator
TxA2 Thromboxane A2
UDP Uridine Diphosphate
VCAM Vascular Cell Adhesion Molecule
VEGF Vascular Endothelial Growth Factor
VLDL Very Low Density Lipoprotein
VPF Vascular Permeability Factor
5


RESUME
La perte des péricytes rétiniens est une des premières altérations de la rétinopathie diabétique.
L’arrêt du cycle cellulaire, l’hypertrophie puis la disparition des cellules mésangiales rénales sont
des caractéristiques typiques de la néphropathie diabétique. La voie de la glycation (formation des
produits avancés de glycation ou AGE) et la voie de l’hexosamine sont deux hypothèses
biochimiques proposées pour expliquer les altérations cellulaires des microcomplications du
diabète. D’autre part, bien que les gangliosides aient souvent été décrits pour moduler la
prolifération cellulaire, leur rôle potentiel dans les perturbations de prolifération caractéristiques de
la rétinopathie et de la néphropathie diabétiques a été très peu étudié. L’objectif de ce travail de
thèse a été de déterminer les effets des AGE et d’une activation de la voie de l’hexosamine, mimée
par la glucosamine, sur la prolifération et le métabolisme des gangliosides des péricytes et de
cellules mésangiales puis d’établir l’implication des gangliosides dans ces processus de régulation
de la prolifération.
Nos résultats ont montré que les AGE et la glucosamine inhibent la prolifération des deux types
cellulaires étudiés. De plus, ils ont révélé que la glucosamine bloque le cycle cellulaire et induit
l’hypertrophie des cellules mésangiales. D’autre part, les AGE comme la glucosamine sont capables
de modifier le profil en gangliosides des cellules, en modulant les activités de leurs enzymes de
biosynthèse. Enfin, nos observations suggèrent que les gangliosides sont impliqués dans l’inhibition
de la prolifération et l’hypertrophie causée par les AGE et la glucosamine dans les péricytes et les
cellules mésangiales. Elles présentent ainsi les gangliosides comme un nouveau mécanisme
d’action des AGE et de la voie de l’hexosamine et permettent de proposer les gangliosides, en
particulier ceux de la série-a, et la GM3 synthase, comme un mécanisme potentiel commun au
développement de la rétinopathie et de la néphropathie diabétiques.
Ce travail est un des premiers à suggérer l’implication des gangliosides dans les complications
microvasculaires du diabète que sont la rétinopathie et la néphropathie diabétiques. Par
l’intermédiaire des gangliosides, il ouvre des perspectives thérapeutiques nouvelles et communes
pour le traitement des deux microcomplications.
6

ABSTRACT
Loss of retinal pericyte is one of the earliest alteration of diabetic retinopathy. Cell cycle arrest,
hypertrophy then death of renal mesangial cells are typical hallmarks of diabetic nephropathy.
Glycation (advanced glycation end-products, AGE, formation) and the hexosamine pathway are two
biochemical hypotheses proposed to explain cellular alteration occurring during diabetic vascular
complication. On the other hand, although gangliosides have often been described as modulators of
cellular proliferation, very few studies have explored their potential role in cell proliferation
alteration of diabetic retinopathy and nephropathy. The aim of the present study was to determine
the effects of AGE and hexosamine pathway activation, mimicked by glucosamine, on pericyte and
mesangial cell proliferation and ganglioside metabolism and then to establish the implication of
gangliosides in these regulation of proliferation process. Our result showed that AGE and
glucosamine inhibit the proliferation of both cell types studied. Moreover, they revealed that
glucosamine blocks the cell cycle and induces hypertrophy of mesangial cells. On the other hand,
both AGE and glucosamine are able to affect cellular ganglioside profile by modulating their
biosynthetic enzyme activities. Finally, our observations suggested that gangliosides are implicated
in the inhibition of cell proliferation and hypertrophy caused by AGE and glucosamine in pericyte
and mesangial cells. Thus, they present gangliosides as a novel mechanism of action of AGE and
the hexosamine pathway and lead to propose gangliosides, especially a-series gangliosides and
GM3 synthase, as a potential common mechanism of diabetic retinopathy and nephropathy
development. This work is one of the first suggesting the implication of gangliosides in diabetic
retinopathy and nephropathy. Through gangliosides, it offers new therapeutic prospects common to
both microvascular complications.
7

INTRODUCTION
8


L’hyperglycémie est le facteur initial essentiel menant aux complications microvasculaires
du diabète. Les mécanismes de la toxicité du glucose au niveau des tissus ciblés par ces
complications sont multiples. La formation des produits avancés de glycation (AGE) est connue
pour jouer un rôle important dans le développement de la rétinopathie et de la néphropathie
diabétiques. La voie des hexosamines, et notamment l’accumulation de glucosamine, est également
de plus en plus souvent mise en cause dans le développement des complications
microangiopathiques, en particulier de la néphropathie.
De nombreuses études ont mis en avant le rôle crucial des péricytes et des cellules
mésangiales, respectivement dans le développement de la rétinopathie et de la néphropathie
diabétiques. La perte des péricytes rétiniens est en effet une des premières altérations structurales
observées au cours de la rétinopathie diabétique. Après une première phase précoce et limitée de
prolifération, l’arrêt de croissance, l’hypertrophie et la disparition des cellules mésangiales sont des
évènements caractéristiques de la néphropathie diabétique. Il apparaît donc que la régulation de la
prolifération de ces deux types cellulaires est un processus pathologique clé des complications
microvasculaires. Cependant, les mécanismes soutenant ces altérations sont encore mal connus.
Les gangliosides sont connus pour jouer un rôle dans la différenciation cellulaire, la
régulation de la croissance ou l’apoptose. Des altérations des taux de gangliosides ont été associées
au processus d’oncogénèse ou à des maladies héréditaires comme la maladie de Farber, même si
peu de choses sont connues des mécanismes ou de la signification biologique de ces altérations.
Cependant, les modifications des gangliosides et leur implication potentielle dans la
microangiopathie diabétique, pathologie où la croissance cellulaire est également sévèrement
affectée, ont été très peu étudiées. Pourtant, il a été récemment décrit que les AGE pouvaient
induire des altérations spécifiques de glycoprotéines (Rellier N. et al., 1997) et de glycolipides
(Natalizio A. et al., 2001). De plus, il a été montré que la voie de l’hexosamine pouvait réguler de
nombreuses enzymes et voies biochimiques ainsi que fournir des intermédiaires glycosylés pour la
biosynthèse de glycoconjugués dont les gangliosides. Il est donc probable que le métabolisme des
gangliosides soit affecté par l’environnement diabétique.
L’objectif de ce travail était de déterminer : i) les effets de deux voies biochimiques du
diabète, les AGE et la voie des hexosamines, sur la prolifération des péricytes rétiniens et des
cellules mésangiales rénales, ii) les effets des AGE et des hexosamines sur le métabolisme des
gangliosides et iii) le rôle des gangliosides dans les processus de perturbation de prolifération
exercée par les AGE et la voie des hexosamines.
Cette étude a été menée dans une optique comparative à la fois des deux voies biochimiques, AGE
9

et voie des hexosamines, et des deux complications microvasculaires, rétinopathie et néphropathie
diabétiques.
Le manuscrit s'articule comme suit:
• une "étude bibliographique" ayant pour objet de présenter les données nécessaires à la
compréhension du travail, de son contexte et de ses enjeux. La première partie est consacrée à la
rétinopathie et à la néphropathie diabétiques alors que la deuxième traite des gangliosides,
• une seconde partie décrivant les matériel et méthodes utilisés au cours du travail expérimental,
• les "résultats et discussion" qui exposent et discutent les résultats obtenus selon une première
partie traitant des AGE et une seconde de la glucosamine,
• et pour finir, les conclusions et perspectives.
10

ETUDE BIBLIOGRAPHIQUE
11


LE DIABETE
I. LE DIABETE SUCRE
Le diabète est une maladie chronique qui prend les proportions d'une véritable épidémie (Figure
1). Elle touche aujourd'hui 150 millions de personnes, sur tous les continents et de tous les âges, soit
environ 3% de la population mondiale. Elle est responsable de 9% de la mortalité totale, tuant
chaque année 4 millions de malades. A l'horizon 2025, en l'absence de prévention primaire, le
nombre de diabétiques aura plus que doublé selon les prévisions de l'OMS, plaçant le diabète parmi
les principales causes d'incapacités et de décès.
0
50
100
150
200
250
300
350
1995 2000 2025
mill
ions
de
mal
ades
pays développés
pays en voie dedéveloppementmonde
0
50
100
150
200
250
300
350
1995 2000 2025
mill
ions
de
mal
ades
pays développés
pays en voie dedéveloppementmondemonde
Figure 1: évolution des cas de diabète dans le monde. Source OMS
La maladie entraîne en effet des complications graves, voire mortelles, essentiellement d'origine
vasculaire, conséquences d'un taux de glucose sanguin en permanence anormalement élevé.
12

L'hyperglycémie chronique est la principale caractéristique du diabète, l'origine de toutes les
complications et la base du diagnostic. Un patient est considéré diabétique s'il présente deux
glycémies à jeun supérieures à 1,26 g/l (~ 7mmol/l). Cette glycémie oscille normalement entre 0,8 à
jeun et 1,4 g/l après un repas grâce à un système régulateur dont l'insuline est l'un des principaux
acteurs. Seule hormone hypoglycémiante de l'organisme, elle est produite par les cellules β des îlots
de Langerhans du pancréas. Elle stimule l'absorption du glucose sanguin par les tissus dits insulino-
dépendants (foie, muscle squelettique et tissu adipeux) et son stockage sous forme de glycogène. De
plus, elle inhibe les voies de production hépatique de glucose (néoglucogénèse et glycogénolyse).
Elle intervient enfin dans la régulation du métabolisme lipidique en inhibant la lipolyse des
triglycérides adipocytaires et en facilitant leur synthèse, ainsi que celle de protéines dans le foie et
le tissu adipeux. Dans le cas du diabète, un défaut de sécrétion de l'insuline et/ou une diminution de
sensibilité des tissus à son action ne permet plus une régulation adaptée du taux de glucose sanguin
qui augmente conduisant à l'hyperglycémie. Il existe deux types de diabète : le diabète de type 1 et
le diabète de type 2, partageant de nombreuses complications mais qui diffèrent par leurs causes ou
leur épidémiologie et correspondent à des mécanismes pathogéniques différents.
1. LES DIFFERENTS TYPES DE DIABETE
1.1.Le diabète de type 1
Le diabète de type 1, auparavant appelé diabète insulino-dépendant (DID), représente environ
10% des cas de diabète mondiaux. Il apparaît le plus souvent chez l'enfant et le jeune adulte, c'est
pourquoi il est aussi appelé "diabète juvénile". Les symptômes classiques les plus manifestes sont
une sécrétion excessive d'urine (polyurie), une sensation de soif (polydipsie) ainsi qu'une perte de
poids, d'où son autre dénomination de "diabète maigre".
C'est une maladie auto-immune conduisant à la destruction sélective et progressive des cellules
β pancréatiques, productrices d'insuline. Cette destruction résulte de la production d'auto-
anticorps dirigés contre des antigènes des cellules β (gangliosides, insuline, GAD : glutamic acid
decarboxylase). Elle semble apparaître chez des sujets génétiquement prédisposés, c'est à dire
possédant des gènes de susceptibilité liés au système HLA mais le processus auto-immun serait
déclenché par un facteur environnemental encore mal connu. Il pourrait correspondre à une
infection virale (par coxsackie B4 ou la rubéole in utéro) ou bien à un fragment de lactalbumine
bovine contenu dans le lait et mal digéré par le tube digestif immature du nourrisson. Ce processus
de destruction entraîne une carence en insuline absolue et définitive responsable de l'apparition
d'une hyperglycémie chronique permanente.
13

Le pancréas étant incapable de produire l'insuline, la survie de ces malades dépend entièrement
d'injection quotidienne de cette hormone, d'où son nom de diabète insulino-dépendant. Ce
traitement est associé à un régime alimentaire en glucose finement contrôlé.
1.2.Le diabète de type 2
Le diabète de type 2, anciennement diabète non insulino-dépendant (DNID) est de loin la forme
de la maladie la plus fréquente puisqu'elle représente 90% des cas mondiaux. C'est un véritable
problème de santé publique, touchant notamment 1,5 millions de français auxquels il faut ajouter
environ 300 000 malades qui s'ignorent. L'installation de la maladie est en effet insidieuse et les
symptômes sont souvent moins marqués que dans le diabète de type 1 pouvant même être
inexistants au début. Le diagnostic ne sera alors posé que plusieurs années après l'apparition de la
maladie (en moyenne 8 ans), alors que l'effondrement de la production d'insuline par les cellules β
et les complications existent déjà. Le diabète de type 2 est aussi appelé "diabète mature" car il
survient le plus souvent chez l'adulte, autour de la cinquantaine, sa prévalence augmentant avec
l'âge. Cependant, il apparaît, depuis quelques années, de manière inquiétante, chez l'adolescent et
l'enfant. Les sujets atteints sont généralement en surcharge pondérale, c'est pourquoi il est
également fait référence à cette maladie en tant que "diabète gras".
L'hyperglycémie des diabétiques de type 2 est la conséquence de deux grands mécanismes
physiopathologiques. Le premier correspond à une diminution de la sensibilité tissulaire à
l'action de l'insuline (insulino-résistance) touchant les tissus périphériques que sont le muscle, le
tissu adipeux et le foie. Cette résistance découle d'une altération de la signalisation de l'insuline qui
toucherait notamment le nombre de récepteurs à l'insuline et/ou leur affinité pour l'hormone et le
nombre de transporteurs membranaires dépendants de l'insuline qui permettent l'entrée du glucose
dans les cellules. Le deuxième phénomène consiste en une anomalie de l'insulino-sécrétion. La
production d'insuline est tout d’abord augmentée pour palier son inefficacité et l'hyperinsulinémie
permet dans un premier temps de maintenir une glycémie normale. Plus la maladie progresse et plus
la sensibilité à l'insuline baisse. L'hypersécrétion d'insuline ne suffit alors plus à compenser
l'insulino-résistance ce qui se manifeste à la fois par une hyperinsulinémie et une hyperglycémie.
Peu à peu, les cellules β deviennent moins sensibles au stimulus glucose. De plus, leur nombre et
leur masse diminuent à cause de la toxicité du glucose. La sécrétion d'insuline finit alors par
diminuer et les diabétiques de type 2 deviennent à terme, comme les diabétiques de type 1,
insulinopéniques. Avec cette baisse de l'insulino-sécrétion, l'hyperglycémie se fait plus sévère. Ces
désordres du métabolisme glucidique sont associés à des troubles importants du métabolisme
lipidique résultant en une dyslipidémie avec augmentation des triglycérides, acides gras libres, LDL
et baisse du HDL cholestérol. Les causes initiales de ce diabète sont mal connues et certainement
14

multiples, facteurs génétiques et environnementaux agissant de concert. Les facteurs génétiques
sont cruciaux puisque le diabète de type 2 est une maladie polygénique largement familiale. Les
facteurs environnementaux, comme le manque d'exercice physique et l'excès de poids, jouent
également un rôle majeur. 80% des diabétiques de type 2 présentent en effet un excès pondéral.
L'obésité, principalement la graisse androïde, favorise l'insulino-résistance et ainsi l'apparition du
diabète qui est d'ailleurs la complication la plus répandue de l'obésité. Chez un grand nombre
d'individus, le diabète de type 2 est lié au syndrome métabolique ou "syndrome X" associant excès
de poids voire obésité, résistance à l'insuline, dyslipidémie et hypertension artérielle, qui constitue
une situation à risque pour le développement de ce type de diabète. Dans la plupart des pays,
l'augmentation du nombre de personnes diabétiques résulte de changements sociaux tels
l'urbanisation, la sédentarisation et la baisse d'activité physique, le changement de régime
alimentaire…. Cette augmentation est particulièrement marquée dans les pays en voie de
développement.
Le traitement de ces malades passe tout d'abord par une réduction de la charge pondérale grâce à un
régime alimentaire approprié et un exercice physique accru. Il peut être associé à la prise d'anti-
diabétiques oraux (biguanides insulino-sensibilisateurs ou sulfamides hypoglycémiants stimulant
l'insulino-sécrétion). Si la survie des diabétiques de type 2 ne dépend pas de l'injection d'insuline, au
moins au départ, un tiers d'entre eux y ont quand même recours pour faire baisser leur glycémie.
Des stratégies indépendantes de la glycémie comme le contrôle de la pression artérielle, du profil
lipidique ou le blocage de l'angiotensine 2 peuvent également être mises en place.
2. LES COMPLICATIONS LIEES AU DIABETE
2.1.Les complications aiguës
Les comas hypoglycémiques ou hyperosmolaires ainsi que les acidoses cétosiques constituent les
principales complications aiguës du diabète. Elles sont causées par un mauvais contrôle glycémique
ou liées à la prise d'un traitement inadapté. Ces évènements conduisent fréquemment à
l'hospitalisation et mettent parfois en jeu le pronostic vital. Cependant, elles se raréfient grâce à la
meilleure prise en charge, l'éducation et l'auto-surveillance des diabétiques.
2.2.Les complications chroniques
Les diabétiques développent, en fonction de la gravité et de la durée de la maladie, des
complications chroniques d'origine vasculaire ayant principalement pour cause une normalisation
insuffisante et inconstante de la glycémie. La mise en place de ces complications est favorisée par le
15

caractère quasi asymptomatique de l'hyperglycémie modérée qui empêche une prise en charge
précoce. L'hyperglycémie crée insidieusement des altérations structurales et fonctionnelles des
gros et des petits vaisseaux conduisant respectivement à la macroangiopathie et la
microangiopathie.
2.2.1 La macroangiopathie diabétique
Ce terme désigne l’ensemble des lésions artérielles consécutives au diabète, qu’il soit de type 1 ou
de type 2, allant d'une altération discrète de l'intima à l'obstruction complète de certains vaisseaux
qui entraînent une ischémie du territoire périphérique correspondant. Elles consistent en fait en une
athérosclérose accélérée et aggravent par conséquent le risque d’accidents cardio-vasculaires. Ces
macrocomplications sont majoritairement représentées par les maladies coronaires mais aussi par
des accidents vasculaires cérébraux et des artérites des membres inférieurs. Les lésions
athéromateuses des diabétiques se caractérisent par leur incidence élevée et leur précocité, leur
caractère souvent asymptomatique et leur localisation multiple ainsi que par leur gravité. Ces
cardiopathies sont en effet la première cause de mortalité chez les diabétiques (50% des décès de
patients diabétiques dans les pays industrialisés, source OMS). Le risque de mortalité cardio-
vasculaire est accru d’un facteur 3 à 5 chez les malades du diabète comparé au reste de la
population et 15% des infarctus, angioplasties et pontages en France sont dus au diabète de type 2.
La pathogénèse des macrocomplications met en jeu trois facteurs principaux : des anomalies
lipidiques (en particulier des modifications quantitatives et qualitatives des lipoprotéines), des
anomalies de l’hémostase (hyperactivité plaquettaire et état procoagulant) et des modifications
pariétales (épaississement et perte de compliance de la paroi vasculaire).
2.2.2 La microangiopathie diabétique
Les complications microvasculaires consistent en des altérations structurales, telles qu’un
épaississement de la membrane basale, et fonctionnelles comme une augmentation de la
perméabilité des petits vaisseaux (artérioles, veinules et capillaires) de diamètre inférieur à 30 µm.
Il est admis que l’hyperglycémie est le facteur initial essentiel menant à ces complications
microvasculaires. Deux études épidémiologiques, l'une américaine (DCCT) (1993), l'autre
européenne (UKPDS) (1998a) ont en effet révélé qu’un contrôle strict de la glycémie permet de
retarder l’apparition et la progression des microangiopathies. Si ces lésions atteignent l'ensemble de
l'organisme, elles ne se manifestent cliniquement qu’au niveau de certains tissus et organes
particulièrement affectés. Il s’agit de la rétine (rétinopathie), du rein (néphropathie) et du système
nerveux (neuropathie).
16

• La rétinopathie diabétique touche environ 40 % des diabétiques. Son apparition est
dépendante et progressive en fonction de l'ancienneté du diabète. Elle conduit à une baisse
progressive de l’acuité visuelle pouvant aller jusqu'à la perte de la vue. Elle est d'ailleurs la
deuxième cause de cécité acquise dans les pays industrialisés après la dégénérescence maculaire
liée à l’âge et est par exemple responsable en France de 500 à 1000 cécités par an.
• La néphropathie diabétique est une complication un peu moins fréquente (30 % des patients
après 15 ans de diabète) mais très lourde de conséquences. L'atteinte rénale est en effet un important
facteur de surmortalité des diabétiques, notamment cardio-vasculaire. Les lésions et
dysfonctionnements rénaux évoluent plus ou moins rapidement vers une hypertension artérielle, une
réduction de la fonction de filtration glomérulaire amenant dans les stades terminaux à l'insuffisance
rénale. La néphropathie diabétique s'affirme d'ailleurs progressivement comme la cause la plus
fréquente d'insuffisance rénale terminale et est la principale cause de mise en dialyse et de
transplantation en Europe du Nord et aux USA. Son apparition semble atteindre un plateau après 15
à 20 ans d'évolution du diabète.
Ces deux premières complications microvasculaires, très souvent associées chez les malades,
intéressent notre étude et seront plus précisément décrites par la suite.
• La neuropathie diabétique est probablement la complication la plus courante du diabète.
Jusqu’à 50 % des diabétiques en souffrent à des degrés divers. C’est une maladie complexe
associant des perturbations à la fois des fibres nerveuses et des microvaisseaux qui leur sont
associés touchant aussi bien les nerfs périphériques que le système nerveux autonome. Elle conduit
à une perte de sensibilité (douleur, perception thermique) et à des lésions des membres inférieurs
(perte de réflexes moteurs, paralysie…) ainsi qu'à des troubles de la régulation du rythme cardiaque
ou de la motricité digestive et vésicale. Le pied diabétique est une conséquence ultime de
l’évolution pathologique au niveau des nerfs et des vaisseaux conduisant à une ulcération et bien
souvent à l’amputation.
II. PHYSIOPATHOLOGIE DE LA RETINOPATHIE ET DE LA
NEPHROPATHIE DIABETIQUES
Les microvaisseaux: artérioles, veinules et en particulier les capillaires qui les unissent, sont le siège
des échanges entre le sang et les tissus. Ils assurent l'apport de substrats métaboliques et d'oxygène
17

aux cellules ainsi que l'épuration des déchets cellulaires des tissus qu'ils irriguent. Leur intégrité est
donc nécessaire au bon fonctionnement des organes et des tissus. La paroi de ces microvaisseaux
est composée d'une couche mince de cellules endothéliales reposant sur une membrane basale,
éventuellement entourée d'autres types cellulaires, parfois spécifiques du tissu comme c'est la cas
pour la rétine ou le rein.
1. LES MICROVAISSEAUX AU SEIN DE LEUR STRUCTURE
1.1.La rétine
1.1.1 L’œil et la rétine
Le globe oculaire peut être divisé en trois couches concentriques (Figure 2) : la tunique fibreuse
(sclérotique et cornée) qui lui donne sa forme et protège ses parties internes, la tunique vasculaire
(choroïde, corps ciliaire et iris) qui l'irrigue en partie et la tunique nerveuse, la plus interne, que
constitue la rétine. Celle-ci recouvre les trois quarts postérieurs de l'œil et a pour fonction de
recevoir les signaux visuels focalisés par la cornée et ajustés par le cristallin, puis de les traiter
avant de les transmettre au cerveau via le nerf optique.
Cornée
CristallinHumeur aqueuse
Corps vitré
Tunique fibreuse
Tunique vasculaire
Tunique nerveuse
Cornée
CristallinHumeur aqueuse
Corps vitré
Tunique fibreuse Tunique fibreuse
Tunique vasculaire Tunique vasculaire
Tunique nerveuse Tunique nerveuse
Figure 2: structure de l'œil en coupe
(d'après http://www.bioinformatics.org/œil-couleur/dossier/anatomie.html)
18

La rétine est formée d'un épithélium pigmentaire et d'une partie neurale (Figure 3). Cette dernière
présente trois couches distinctes de neurones séparées par deux zones où se retrouvent les contacts
synaptiques (couches plexiformes) : les photorécepteurs (étage de réception), les cellules bipolaires
et les cellules ganglionnaires (étages de transmission). Entre ces éléments se trouvent des éléments
de soutien et de nutrition comme les cellules névrogliques, les limitantes et les cellules de Müller.
Lit vasculaire choroïdien
Epithélium pigmentaire
Photorécepteurs
Limitante externe
Limitante interne
Cellules ganglionnaires
Plexiforme interne
Nucléaire interne(cellules bipolaires)
Plexiforme externe
Nucléaire externe
Étage de transmission
Étage de réception
Lit microvasculaire rétinien:
veinuleartèriole
capillaires
Lit vasculaire choroïdien
Epithélium pigmentaire
Photorécepteurs
Limitante externe
Limitante interne
Cellules ganglionnaires
Plexiforme interne
Nucléaire interne(cellules bipolaires)
Plexiforme externe
Nucléaire externe
Étage de transmission
Étage de réception
Lit microvasculaire rétinien:
veinuleartèriole
capillaires
Lit vasculaire choroïdien
Epithélium pigmentaire
Photorécepteurs
Limitante externe
Limitante interne
Cellules ganglionnaires
Plexiforme interne
Nucléaire interne(cellules bipolaires)
Plexiforme externe
Nucléaire externe
Étage de transmission
Étage de réception
Lit microvasculaire rétinien:
veinuleartèriole
capillaires
Figure 3:représentation schématique de la structure de la rétine
La rétine est un tissu particulièrement irrigué et le seul endroit du corps où il est possible
d'examiner directement les vaisseaux sanguins qui traversent sa surface antérieure (examen du fond
de l'œil). L'artère rétinienne centrale se ramifie et s'étale en éventail pour irriguer la surface
antérieure de la rétine. Ce lit vasculaire est inégalement réparti dans 2 régions, majoritairement à la
surface de la rétine mais également enfoui dans la couche de cellules bipolaires.
1.1.2 Les microvaisseaux
Les capillaires rétiniens sont constitués de deux types cellulaires (Figure 4). Les cellules
endothéliales forment une monocouche du côté luminal reposant sur la membrane basale. Les
péricytes, enfouis dans cette membrane basale, enveloppent les microvaisseaux. La membrane se
19

compose principalement de collagène type IV, de protéoglycanes et de glycoprotéines comme la
laminine et la fibronectine. Cellules endothéliales et péricytes se répartissent selon un ratio 1 pour 1.
Ceci constitue une particularité de la paroi vasculaire rétinienne, les péricytes étant dans la plupart
des autres tissus en très large minorité. Une deuxième singularité repose sur l'existence de jonctions
serrées qui relient les cellules adjacentes de l'endothélium, prévenant ainsi les fuites de certaines
substances. Cellules endothéliales, péricytes et cellules neuronales (cellules de Müller), forment,
avec la membrane basale, la barrière hémato-rétinenne et sont donc responsables de la
perméabilité capillaire. Cette barrière étanche contrôle en effet les échanges entre le sang et les
espaces extra-vasculaires par voie transcellulaire active. Elle protège ainsi les éléments nerveux de
la rétine de certains facteurs circulants et permet de maintenir un environnement biochimique
particulier dans le compartiment extravasculaire.
cellule endothéliale
péricyte
Figure 4: structure d'un capillaire rétinien. Schéma et photo en microscopie
1.1.3 Les cellules
(a) Les cellules endothéliales
Outre la formation de la barrière hémato-rétinenne, les cellules endothéliales exercent de
nombreuses fonctions, participant à la régulation de phénomènes physiologiques. Elles jouent en
effet un rôle important dans la régulation du tonus vasculaire en sécrétant des agents vaso-
dilatateurs comme la prostaglandine PGI2 et le monoxyde d'azote ou des agents vaso-constricteurs
tels que les endothélines. Les cellules endothéliales interviennent également dans la régulation de la
fibrinolyse en produisant l'activateur du plasminogène tissulaire (tPA) et l'inhibiteur de l'activateur
de plasminogène (PAI-I). D'autre part, elles peuvent produire plusieurs facteurs de croissance
membrane basale
lumière du capillaire
cellule endothéliale
péricyte
membrane basale
lumière du capillairelumière du capillaire
20

comme le "fibroblast growth factor" (FGF) qui stimule d'ailleurs la prolifération de péricytes en
culture. Enfin, les cellules endothéliales sécrètent des composants de la membrane basale tels que la
fibronectine, le collagène IV ou la laminine (Kohner E.M. et al., 1994).
(b) Les péricytes
La fonction des péricytes est moins claire. Bien qu'ils soient enchâssés dans la membrane basale, ils
interagissent étroitement avec les cellules environnantes. Ils émettent des prolongements
cytoplasmiques permettant des contacts directs avec les cellules endothéliales par des jonctions
gap, des molécules d'adhésion ou des glycoconjugués de surface, ou encore par la sécrétion de
facteurs solubles (Ruggiero D. et al., 1997). Ils joueraient ainsi un rôle important dans la régulation
du métabolisme et de la croissance des cellules endothéliales. Des expériences de co-culture
permettant les contacts cellule-cellule ont montré que les péricytes inhibaient la croissance des
cellules endothéliales, en activant le TGFβ (Orlidge A. et al., 1987). Il a également été suggéré que
les péricytes pourraient stimuler la production de prostacycline des cellules endothéliales ou les
protéger contre le stress oxydant (Yamagishi S. et al., 1993). D'autre part, les péricytes serviraient à
maintenir l'intégrité structurale des microvaisseaux en supportant les cellules endothéliales. Il a
été observé qu’une diminution de la couverture de péricytes s’accompagnait d’une augmentation de
segments de capillaires acellulaires, correspondant à une perte des cellules endothéliales (Hammes
H.P. et al., 2002). Les péricytes répondent au PDGF et une carence dans ce facteur de croissance
pourrait perturber le recrutement des péricytes vers leur destination finale. Lindahl et al. ont montré
qu’une perte de péricytes chez des souris déficientes en PDGF conduisait à une dilatation des
capillaires et à des hémorragies (Lindahl P. et al., 1997). Les péricytes possèdent de plus une
activité contractile proche de celle des cellules musculaires dont ils pourraient dériver.
Contractions et dilatations permettent de moduler le diamètre des vaisseaux et le tonus
vasculaire et donc de réguler le débit sanguin. Comme les cellules endothéliales, ils synthétisent
des constituants de la membrane basale, participent à la régulation du tonus vasculaire en produisant
par exemple des prostacyclines vasodilatatrices et interviennent également dans le contrôle de
l'hémostase en sécrétant la trombospondine, molécule d'adhésion des plaquettes à l'endothélium, et
le PAI-I.
1.2.Les reins
1.2.1 Le système urinaire et les reins
Avec les deux uretères, la vessie et l'urètre, les reins constituent le système urinaire qui prend en
charge l'élimination des déchets de l'organisme (Figure 5). Les reins règlent la composition et le
volume sanguin en évacuant sous forme d'urine une quantité sélective de déchets issus du sang
21

(substances toxiques et substances essentielles excédentaires, notamment les déchets azotés issus du
catabolisme des protéines, les ions tels Na+, H+, le dioxyde de carbone ou l'eau). Ils assurent ainsi
l'homéostasie hydro-électrique. Ils régulent également la pression sanguine. Ils assurent de cette
façon le maintien de l'homéostasie de l'organisme en adaptant la quantité et la composition de
l'urine produite en fonction des circonstances comme l'alimentation par exemple. Le rein comporte
une région externe appelée cortex, une région interne appelée medulla ainsi qu'une cavité centrale,
le bassinet, où s'écoule l'urine avant d'être évacuée vers la vessie par l'uretère. Les reins, de par leur
fonction de filtration du sang, renferment un grand nombre de vaisseaux sanguins issus des
ramifications très développées de l'artère et de la veine rénales.
medulla
cortex
bassinet
medulla
cortex
bassinet
Figure 5: structure du système urinaire et du rein (d'après
http://www.doctissimo.fr/html/sante/atlas/atlas_sysurogenital_78a.htm)
Cortex et medulla sont traversés par environ 1 million d'unités élémentaires de filtration du sang,
appelées néphrons (Figure 6). C'est au niveau de ces unités structurales et fonctionnelles que se
déroulent les processus menant à la formation de l'urine. Le néphron est constitué de deux parties:
un corpuscule rénal qui filtre le liquide et un tubule rénal dans lequel s'écoule le liquide filtré. Le
corpuscule est lui-même composé d'une masse de capillaires artériels, appelée glomérule,
entourée d'une structure épithéliale à double paroi, en forme de coupe, appelée capsule glomérulaire
ou de Bowman. Le sang pénètre dans le glomérule par une artériole afférente qui se divise pour
former le réseau enchevêtré de capillaires qui constitue le glomérule. Les capillaires glomérulaires
22
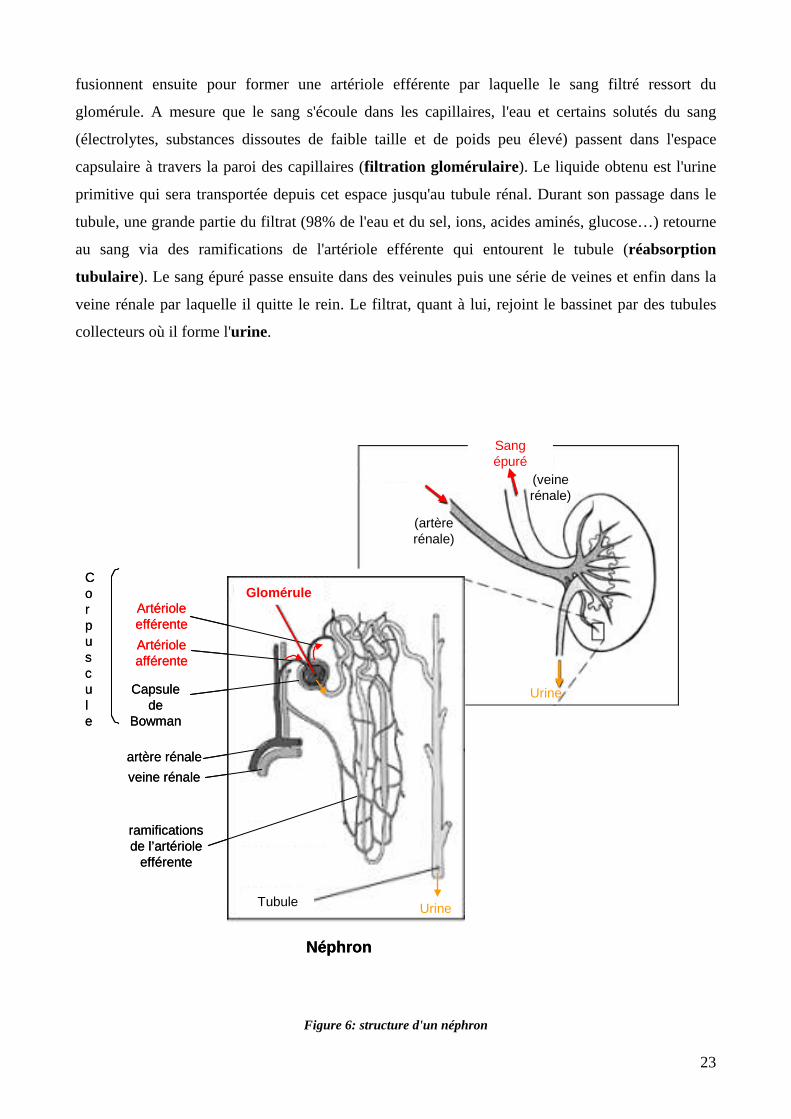
fusionnent ensuite pour former une artériole efférente par laquelle le sang filtré ressort du
glomérule. A mesure que le sang s'écoule dans les capillaires, l'eau et certains solutés du sang
(électrolytes, substances dissoutes de faible taille et de poids peu élevé) passent dans l'espace
capsulaire à travers la paroi des capillaires (filtration glomérulaire). Le liquide obtenu est l'urine
primitive qui sera transportée depuis cet espace jusqu'au tubule rénal. Durant son passage dans le
tubule, une grande partie du filtrat (98% de l'eau et du sel, ions, acides aminés, glucose…) retourne
au sang via des ramifications de l'artériole efférente qui entourent le tubule (réabsorption
tubulaire). Le sang épuré passe ensuite dans des veinules puis une série de veines et enfin dans la
veine rénale par laquelle il quitte le rein. Le filtrat, quant à lui, rejoint le bassinet par des tubules
collecteurs où il forme l'urine.
Figure 6: structure d'un néphron
Artériole afférente
Artériole efférente
Néphron
Urine
Capsule de
Bowman
GloméruleCorpuscule
ramifications de l’artériole
efférente
Urine
Sang épuré
Tubule
(veine rénale)
(artère rénale)
veine rénaleartère rénale
Artériole afférente
Artériole efférente
Néphron
Urine
Capsule de
Bowman
GloméruleCorpuscule
ramifications de l’artériole
efférente
Urine
Sang épuré
Tubule
(veine rénale)
(artère rénale)
veine rénaleartère rénale
23

1.2.2 Les glomérules, site de filtration
Le glomérule, bouquet de capillaires, est le siège de l'activité de filtration du rein (figure 7A). Il
se compose de 20 à 40 boucles de capillaires soutenus par un tissu mésangial intercapillaire, ou
mesangium, qui comprend des éléments à la fois cellulaires (cellules mésangiales) et acellulaires
(matrice extracellulaire mésangiale). L'endothélium des capillaires, le feuillet viscéral de la capsule
glomérulaire et la membrane basale qui les sépare, forment la membrane glomérulaire (Figure
7B) qui agit comme un filtre, laissant passer certaines substances en provenance du sang et en
arrêtant d'autres en fonction de leur taille et de leur charge. L'endothélium des capillaires se
compose d'une couche simple de cellules endothéliales avec des fenestrations (lamina fenestrae)
qui empêche le passage des cellules du sang. Le feuillet viscéral de la capsule est formé de cellules
épithéliales spécialisées appelées podocytes. Elles se prolongent par de nombreuses ramifications
en forme de pied qui recouvrent la membrane basale de manière discontinue. Les espaces laissés
représentent les fentes ou pores de filtration. Cet épithélium empêche le passage des protéines de
taille moyenne. La membrane basale glomérulaire est une couche dense de matériel
extracellulaire (lamina densa) située entre l'endothélium et l'épithélium. Elle est en continuité avec
la matrice mésangiale. Elles contiennent toutes deux collagène type IV, protéoglycanes, laminine et
fibronectine. Les groupes sulfates portés par un grand nombre de ces protéines confèrent une charge
anionique à la membrane basale qui contribue à la perméabilité sélective et dépendante de la charge
des microvaisseaux glomérulaires. Elle empêche également le passage des plus grosses protéines.
La barrière de filtration glomérulaire peut être considérée comme une membrane perforée par des
pores et recouverte de charges négatives. La filtration se fait donc au niveau des capillaires selon un
chemin extracellulaire, à travers l'endothélium (probablement via les fenestrations), la membrane
basale et l'épithélium (probablement via les pores de filtration) jusqu'à l'espace capsulaire. La
composition du filtrat dépend, en plus de facteurs hémodynamiques, principalement de la
perméabilité de la membrane glomérulaire, et à la fois la taille et la charge des molécules
influencent leur passage à travers cette membrane. Les petites molécules (eau, urée, glucose…)
passent librement au travers et plus le poids moléculaire augmente, plus le passage des particules à
travers les pores est entravé. La filtration glomérulaire est ainsi une ultrafiltration, produisant un
filtrat quasiment exempt de macromolécules.
24

Figure 7: structure d'un corpuscule rénal et de la membrane de filtration glomérulaire
1.2.3 Les cellules
(a) Les cellules endothéliales
Les cellules endothéliales forment une monocouche bordant la lumière capillaire. Elles assurent
différentes fonctions comme le transport de molécules plasmatiques ou le contrôle du tonus
vasculaire en synthétisant des médiateurs vasoactifs, comme le NO ou les endothélines, qui peuvent
notamment agir sur les cellules de type musculaire voisines. En formant un endothélium fenestré,
elles participent également à la barrière de filtration glomérulaire (Trevisan R. et al., 1997).
(b) Les podocytes, cellules epithéliales spécialisées
Il existe deux types de cellules épithéliales dans le glomérule: les cellules de l'épithélium pariétal
(feuillet externe) de la capsule de Bowman et celles de l'épithélium viscéral (feuillet interne)
appelées podocytes. Les podocytes, contrairement aux cellules mésangiales ou endothéliales, ne
sont pas enfouis dans la membrane basale glomérulaire mais la recouvrent. Ce sont des cellules
polarisées qui développent des ramifications en forme de pied attachées à la membrane basale du
glomérule alors que le corps cellulaire flotte dans la capsule de Bowman. Ces ramifications
recouvrent la membrane basale glomérulaire de manière discontinue, laissant entre elles des pores
de filtration. Les podocytes sont impliqués dans différentes fonctions du glomérule. Tout d'abord,
ils constituent avec les cellules endothéliales la paroi des capillaires et participent à la formation de
la membrane basale glomérulaire et potentiellement à sa dégradation. De plus, les "pieds" des
podocytes et plus précisément les fentes laissées entre eux (environ 10% de la surface de la
membrane basale glomérulaire), sont un composant majeur de la barrière de filtration
glomérulaire. Ils joueraient un rôle crucial dans la sélectivité dépendante de la taille des
molécules. Comme le mesangium, les podocytes pourraient également, grâce à leur cytosquelette
podocyte
mesangium
endothelium fenestré
endothelium fenestré
membrane basale
BARRIERE
DE
FILTRATION
urine primitive
feuillet interne
sangcapillaire
podocytes
glomérule
Capsule de Bowman
A B
podocyte
mesangium
endothelium fenestré
endothelium fenestré
membrane basale
BARRIERE
DE
FILTRATION
urine primitive
feuillet interne
sangcapillaire
podocytes
glomérule
Capsule de Bowman
A B
25
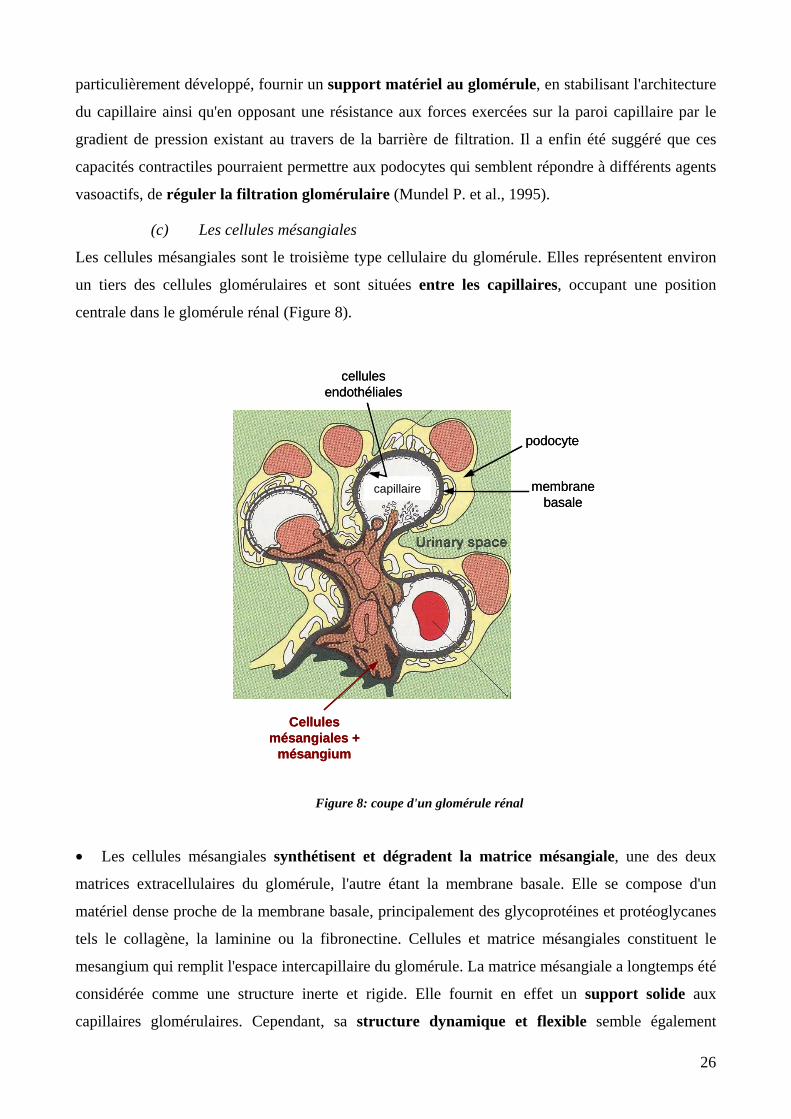
particulièrement développé, fournir un support matériel au glomérule, en stabilisant l'architecture
du capillaire ainsi qu'en opposant une résistance aux forces exercées sur la paroi capillaire par le
gradient de pression existant au travers de la barrière de filtration. Il a enfin été suggéré que ces
capacités contractiles pourraient permettre aux podocytes qui semblent répondre à différents agents
vasoactifs, de réguler la filtration glomérulaire (Mundel P. et al., 1995).
(c) Les cellules mésangiales
Les cellules mésangiales sont le troisième type cellulaire du glomérule. Elles représentent environ
un tiers des cellules glomérulaires et sont situées entre les capillaires, occupant une position
centrale dans le glomérule rénal (Figure 8).
membrane basale
Cellules mésangiales +
mésangium
podocyte
cellules endothéliales
capillaire membrane basale
Cellules mésangiales +
mésangium
podocyte
cellules endothéliales
capillaire
Figure 8: coupe d'un glomérule rénal
• Les cellules mésangiales synthétisent et dégradent la matrice mésangiale, une des deux
matrices extracellulaires du glomérule, l'autre étant la membrane basale. Elle se compose d'un
matériel dense proche de la membrane basale, principalement des glycoprotéines et protéoglycanes
tels le collagène, la laminine ou la fibronectine. Cellules et matrice mésangiales constituent le
mesangium qui remplit l'espace intercapillaire du glomérule. La matrice mésangiale a longtemps été
considérée comme une structure inerte et rigide. Elle fournit en effet un support solide aux
capillaires glomérulaires. Cependant, sa structure dynamique et flexible semble également
26

largement influencer la physiologie des cellules mésangiales à la fois en conditions normales et
dans des états pathologiques, au cours desquels la matrice mésangiale est souvent modifiée. De
plus, la matrice mésangiale permet des connexions entre les cellules mésangiales et la membrane
basale, avec laquelle elle est en continuité.
• Les cellules mésangiales maintiennent par ailleurs des contacts directs avec les cellules
endothéliales par des prolongements cytoplasmiques, au niveau de zones dites juxtaglomérulaires,
où la membrane basale et les podocytes recouvrent l'endothélium de manière discontinue. Ces
contacts étroits entre la surface de filtration endothéliale et le mesangium ont d'ailleurs conduit à
suggérer que les cellules mésangiales elles-mêmes puissent être impliquées dans les processus de
filtration glomérulaire et capter des macromolécules, comme par exemple des complexes immuns.
• Les cellules mésangiales sont particulièrement riches en éléments contractiles et parfois
considérées comme des cellules musculaires lisses modifiées ou des péricytes vasculaires
contractiles. Elles sont capables de se contracter ou de se relâcher en réponse à différents agents
vasoactifs. Elles pourraient de ce fait réguler le diamètre des capillaires et la filtration
glomérulaire en exerçant une action mécanique par contraction-relaxation sur les matrices
extracellulaires et l'endothélium. Par ailleurs, une expansion du mesangium, qui survient dans
certaines situations pathologiques comme le diabète, peut également affecter la surface de filtration
du glomérule en diminuant le diamètre des capillaires.
• Enfin, des études de culture cellulaire ont montré que les cellules mésangiales synthétisent
diverses enzymes et facteurs (rénine, protéinase, Plasminogen-Activator et PAI), hormones et
cytokines (erythropoïétine, Il, PDGF, IGF1, auxquels elles peuvent répondre de manière autocrine)
et lipides bioactifs (PAF, PG) (Mene P. et al., 1989)
Le terme de "myofibroblaste" est parfois utilisé pour décrire la nature indéterminée des cellules
mésangiales, partageant à la fois des caractères structuraux et fonctionnels des cellules musculaires
lisses et des fibroblastes. Cependant, leur importance dans la fonction glomérulaire est de plus en
plus claire, surtout lorsque le glomérule est endommagé.
2. DEUX PATHOLOGIES EVOLUTIVES Rétinopathie et néphropathie diabétiques correspondent à un ensemble d’altérations anatomiques et
fonctionnelles s’aggravant progressivement. Les manifestations cliniques apparaissent au fil des
différentes phases qui définissent la maladie, évoluant à terme vers la cécité et l’insuffisance rénale
terminale, respectivement. Cette évolution vers un handicap majeur et définitif peut être freinée par
une intervention médicale précoce.
27

2.1.Rétinopathie diabétique
2.1.1 Les différentes phases de la maladie
La rétinopathie diabétique regroupe les manifestations rétiniennes spécifiques du diabète et
représente un ensemble anatomo-clinique et fonctionnel qui évolue progressivement vers des
troubles de la vision jusqu’au stade ultime de cécité. Cette évolution est classée selon différentes
phases.
(a) Phase non-proliférative
Une des premières manifestations cliniques de la rétinopathie diabétique consiste en une dilatation
des microvaisseaux. Elle est la conséquence d’altérations structurales comme l’épaississement de
la membrane basale, la perte des péricytes et les altérations des cellules endothéliales ainsi que
d’une augmentation du débit sanguin, qui diminuent la résistance des capillaires (Forrester J.V. et
al., 1997). Les microanévrismes consécutifs conduisent à l’apparition de multiples hémorragies.
Les exsudats font également partie des lésions précoces de la rétinopathie diabétique et
correspondent à des fuites de constituants plasmatiques à travers la paroi des microvaisseaux
anormaux. Ils sont la conséquence d’une augmentation de la perméabilité vasculaire et favorisent
l’apparition d’œdèmes. Cette étape qui ne se manifeste généralement pas par une perte de vision,
est très courante et demeure en l’état chez la plupart des patients pendant de nombreuses années
(Dowler J.G.F. et al., 1997).
(b) Phase pré-proliférative
L’occlusion des capillaires est une caractéristique de cette phase pré-proliférative qui conduit à des
zones d’ischémie. Elle est associée à des modifications des microvaisseaux intrarétiniens comme
des dilatations, la formation de boucles, qui pourraient représenter des tentatives de
revascularisation de la rétine ischémiée, prélude à la formation de nouveaux vaisseaux. L’occlusion
des capillaires est favorisée par un profil sanguin pro-thrombotique (augmentation de l’activité
plaquettaire et fibrinolytique, adhésion de leucocytes ou monocytes circulants à l’endothélium…)
(Forrester J.V. et al., 1997). Cette phase intermédiaire constitue un risque majeur d’évolution vers la
phase proliférative de la rétinopathie diabétique et indique la nécessité d’une intervention rapide
pour freiner l’évolution de la maladie.
(c) Phase proliférative
Lorsque les zones de non-perfusion résultant de l’occlusion des capillaires sont trop importantes, la
prolifération des cellules endothéliales est stimulée dans les zones adjacentes pour former des
nouveaux microvaisseaux et tenter de revasculariser la rétine ischémiée. La production locale de
facteurs de croissance comme le VEGF serait responsable de ce phénomène d’angiogénèse
28

(Forrester J.V. et al., 1997). Ces nouveaux microvaisseaux sont dilatés, fragiles et perméables,
favorisant ainsi l’apparition d’hémorragies. D’abord formés dans la rétine, ils gagnent
progressivement la surface et s’attachent à la face postérieure du vitré, dans lequel ils peuvent
même pénétrer et provoquer des hémorragies. Le vitré se rétracte en exerçant une traction
importante sur la rétine qui se décolle. Cette étape est la plus significative en terme d’atteinte
visuelle et si les microvaisseaux néoformés ne sont pas traités, ils risquent d’occasionner la perte de
la vue (Kohner E.M., 1994).
2.1.2 Les facteurs de risque
L’hyperglycémie apparaît comme une cause majeure de la rétinopathie diabétique. Cette idée est
soutenue par des données du DCCT (Diabetes Control and Complications Trial) qui indiquent
qu’un contrôle strict de la glycémie peut retarder l’apparition et/ou améliorer la sévérité de cette
pathologie (1993). L’hypertension constitue également un facteur aggravant. La durée du diabète
est aussi un facteur prédictif important de la rétinopathie diabétique.
2.1.3 Le traitement
Le traitement préventif de la maladie passe par un contrôle optimisé à la fois de la glycémie et de
la pression sanguine. Des études menées sur les patients diabétiques de type I et de type II ont
révélé qu’un traitement intensif de l’hyperglycémie réduisait de 54 et 25 %, respectivement
l’incidence de la rétinopathie (Fong D.S. et al., 2003). Le traitement de l’hypertension représente
également un moyen d’enrayer la progression de la rétinopathie diabétique. Une étude clinique a
notamment montré que la baisse de la pression sanguine chez les diabétiques de type II permettait
de réduire la progression de la rétinopathie (1998b). Le principal traitement de la rétinopathie
diabétique déclarée est resté inchangé depuis quasiment 50 ans. Il consiste à éliminer les parties de
rétine malades par photocoagulation au laser ou par cryothérapie. Les microvaisseaux
nouvellement formés, particulièrement dangereux pour la vision, répondent cependant bien à la
photocoagulation. Des méthodes de microchirurgie se sont de plus développées, permettant aux
patients de récupérer une partie des capacités visuelles qu’ils avaient perdues.
2.2.Néphropathie diabétique (ND)
2.2.1 Les différents stades de la maladie
La néphropathie diabétique est généralement considérée comme une dégradation de la fonction
rénale depuis une albuminurie normale jusqu’à une insuffisance rénale terminale passant par des
états intermédiaires marqués notamment par une microalbuminurie. L’évolution se fait
29

progressivement d’une phase à l’autre, à une vitesse très variable selon l’individu. La progression
de la maladie peut également être stoppée à un stade donné où le malade restera toute sa vie, voire
même régresser.
(a) ND silencieuse et hyperfiltration glomérulaire
Cette première étape (d'une durée de 8 à 12 ans en moyenne) correspond à une augmentation du
taux de filtration glomérulaire qui s’accompagne d’une augmentation du volume des reins et
d’une hypertrophie glomérulaire. Elle est la conséquence d’une augmentation de la pression
sanguine intraglomérulaire et d’une augmentation de la surface de filtration. Cette dernière est en
effet augmentée dans les stades précoces de la ND suite à un épaississement de la membrane basale
(Trevisan R. et al., 1997). Cette hyperfiltration est partiellement réversible par un contrôle strict de
la glycémie et de la tension, mettant en particulier en jeu l'angiotensine 2.
(b) ND débutante et microalbuminurie
La microalbuminurie est le premier signe clinique de la ND. Le taux normal d’excrétion
d’albumine est < 20 mg/jour et la microalbuminurie correspond à un taux allant de 30 à 300
mg/jour. Ces taux ne sont pas détectables par les méthodes classiques et nécessitent des techniques
plus sensibles alors que les lésions rénales sont déjà importantes. Une excrétion d’albumine
persistante et plus élevée (> 300 mg/jour), détectable par l’utilisation de bandelettes, correspond à la
macroalbuminurie.
La microalbuminurie est un facteur clé de la néphropathie diabétique. Les patients passent
premièrement par cette étape avant de développer une néphropathie plus avancée et la
microalbuminurie peut ainsi représenter un précieux marqueur de l’atteinte rénale débutante.
Les malades souffrant de microalbuminurie sont à risque pour développer une protéinurie même si
tous n’évolueront pas vers cette aggravation de la maladie. Plus la microalbuminurie s’est
développée tôt après la naissance du diabète et plus les risques de progression vers une protéinurie
sont forts (Krowleski A.S. et al., 1997) La microalbuminurie est également liée à une dysfonction
endothéliale généralisée et représente un marqueur de risque cardiovasculaire. La microalbuminurie
est due à une augmentation de la perméabilité des capillaires consécutive à une augmentation
de la pression sanguine dans le glomérule ainsi qu’à une perte de sélectivité de filtration de la
barrière glomérulaire. La filtration glomérulaire reste accrue à ce stade.
(c) ND confirmée et protéinurie
Au fur et à mesure que la barrière de filtration glomérulaire se dégrade en perdant sa sélectivité
(perte des charges négatives et augmentation de la taille des pores), la microalbuminurie progresse
en macroalbuminurie. La sélectivité initiale pour l’albumine disparaît et l’excrétion de protéines
30

devient non sélective, concernant notamment les IgG, c’est la protéinurie. Elle est accompagnée
d’une hypertension artérielle qui accélère l’augmentation de l’albuminurie et le déclin du taux de
filtration glomérulaire. La fonction rénale se dégrade et les patients atteints par la protéinurie
présentent de très forts risques de perte de la fonction rénale.
(d) Insuffisance rénale terminale
Elle correspond au stade ultime de la maladie où la fonction rénale s’effondre avec moins de 10 %
de néphrons fonctionnels, nécessitant le recours à des traitements épurateurs extrarénaux. Cette
perte de la fonction rénale est notamment due à l’expansion du mesangium qui conduit à la
sclérose diffuse des glomérules qui s'étend à la fibrose des structures tubulo-intersticielles. Le
risque de dégradation de la fonction rénale est alors quantitativement lié au niveau de protéinurie.
Parallèlement à la ND, d’autres complications se mettent en place. Les atteintes cardio-vasculaires
sont importantes et les diabétiques de type 2 décèdent souvent d’accident cardio-vasculaire avant
d’avoir atteint le stade d’insuffisance rénale terminale. L’artériopathie ou la neuropathie sont
également fréquentes chez les patients atteints par la ND. Enfin, la rétinopathie diabétique est
largement associée à la ND, à tel point qu’elle est par exemple systématique au stade de la ND
confirmée chez les diabétiques de type 1.
2.2.2 Les facteurs de risque
Tous les diabétiques ne développent pas de ND. Comme pour la rétinopathie, la glycémie joue un
rôle majeur et semble être le facteur principal influençant le passage d’une albuminurie normale à
une microalbuminurie. Plusieurs études cliniques ont montré qu’une amélioration du contrôle
glycémique diminuait le risque de développer une microalbuminurie ou d’évoluer vers la
néphropathie avérée. D’autres facteurs de risque semblent également entrer en ligne de compte
comme l’hypertension artérielle, le tabagisme ou un taux élevé de LDL. L’hyperlipidémie
pourrait favoriser la progression de la maladie (Krowleski A.S. et al., 1997). De plus, il est
démontré qu’un facteur de susceptibilité génétique, notamment une prédisposition à l’hypertension,
détermine en partie le risque de développer une ND.
2.2.3 Le traitement
Le traitement de la ND repose largement sur un contrôle strict de la glycémie. Il peut réduire ou
même normaliser la microalbuminurie et l’hyperfiltration glomérulaire. La baisse de la tension
artérielle est aussi un objectif du traitement par des antihypertenseurs, comme des inhibiteurs de
l’enzyme de conversion. Ceux-ci diminueraient la pression sanguine et l’hypertension glomérulaire
31

ainsi que la protéinurie et ralentiraient la diminution du taux de filtration glomérulaire. La réduction
de l’apport alimentaire en protéines réduirait également les altérations de la ND (Walker J.D. et al.,
1994). De plus, la réduction de l’hyperlipidémie semble améliorer le pronostic de la ND. Enfin,
l’utilisation de produits anti-AGE (produits avancés de glycation, formés en conditions
hyperglycémiques et impliqués dans le développement des complications microangiopathiques, cf
partie 5), comme l’aminoguanidine, pourrait constituer un traitement efficace.
Lorsque la fonction rénale est perdue, le recours à l’hémodialyse ou la dialyse péritonéale est
nécessaire. A un stade ultime de la maladie, la transplantation devient la solution de choix. Elle
peut concerner le rein ou combiner le rein et le pancréas. Plus récente et prometteuse, la greffe
d’îlots de Langerhans pourrait devenir une alternative.
3. ALTERATIONS STRUCTURALES ET FONCTIONNELLES Durant le rétinopathie et la néphropathie diabétiques, la structure et la fonction des microvaisseaux
sont progressivement et fortement altérées. Ces anomalies se manifestent à différents niveaux :
3.1.Membrane basale et matrice extracellulaire
L’épaississement des membranes basales est une des premières altérations observées lors de la
microangiopathie diabétique. Il atteint le système vasculaire de nombreux tissus (peau, muscle,
cerveau, cœur…) et en particulier la rétine et les glomérules et tubules rénaux. Cet épaississement
résulte d’une augmentation de la synthèse et du dépôt de composants de la matrice comme le
collagène IV et la fibronectine. Au contraire, la synthèse de protéoglycanes sulfatés, chargés
négativement, est diminuée, modifiant ainsi en plus la composition chimique des membranes
basales. En parallèle, les processus de dégradation de la matrice sont altérés, notamment par une
diminution de l’expression et de l’activité de collagenase et de metalloprotéinases. De plus, un
phénomène de glycation des protéines de la matrice se produit dans les conditions
hyperglycémiques du diabète. Cette modification leur confère notamment une résistance aux
protéases, limitant ainsi leur dégradation. La conjonction d’une augmentation de la production des
protéines matricielles et d’une diminution de leur dégradation altère le turnover des composants de
la membrane basale et concoure à leur accumulation (Tsilibary E.C., 2003).
Au niveau du glomérule rénal, l’augmentation de production de protéines matricielles affecte
également la matrice mésangiale. La biosynthèse du collagène IV et de la fibronectine est
augmentée alors que celle des protéoglycanes sulfatés est au contraire diminuée. L'augmentation
de la synthèse de matrice mésangiale est largement responsable de l'expansion du mesangium,
observée après quelques années de diabète. Ce phénomène est étroitement lié à la détérioration de la
32

fonction glomérulaire. Une corrélation entre l'expansion mésangiale et les manifestations cliniques
de la néphropathie diabétique comme l'albuminurie, l'hypertension ou la diminution du taux de
filtration glomérulaire a été mise en évidence. Associée à un épaississement de la membrane basale,
l’expansion du mesangium est responsable du grossissement du glomérule rénal, considéré comme
un caractère majeur de la néphropathie diabétique. Il semble que cette surproduction de protéines
matricielles soit en partie causée par une augmentation de l’expression du TGFβ, facteur pro-
sclérosant. Celle-ci a été montrée à la fois dans des cellules mésangiales soumises à de fortes
concentrations de glucose et dans des modèles d’animaux diabétiques. L’utilisation d’anticorps
anti-TGFβ in vitro et in vivo a par ailleurs permis de démontrer que l’induction de ce facteur de
croissance était nécessaire à l’hypertrophie rénale et à l’accumulation de matrice extracellulaire
(Sharma K. et al., 1996; Ziyadeh F.N. et al., 1994). D’autres facteurs de croissance comme le
PDGF, les produits avancés de glycation ou bien l’angiotensine II ont également été impliqués dans
la stimulation de la synthèse de protéines matricielles, certainement via une induction du TGFβ
(Wolf G. et al., 1999).
Les altérations structurales des membranes basales ont des conséquences fonctionnelles importantes
participant aux dysfonctionnements vasculaires :
• Les matrices extracellulaires jouent un rôle très important dans le comportement cellulaire et
ainsi, les modifications de la membrane basale vont moduler ce comportement. Le métabolisme des
cellules vasculaires est modifié et leur migration, leur adhésion ou leur croissance sont affectées. Il
a notamment été montré que la glycation de la matrice rétinienne diminuait l’adhésion et la
prolifération des péricytes alors qu’elle augmentait la prolifération des cellules endothéliales
pouvant ainsi favoriser l’angiogénèse caractéristique des stades tardifs de la rétinopathie diabétique
(Beltramo E. et al., 2002; Kalfa T.A. et al., 1995). L’épaississement de la membrane basale peut
également perturber les contacts importants qui existent entre les cellules, par exemple entre
cellules endothéliales et péricytes. Par ailleurs, Makino at al. ont suggéré que la survie et la mort
des cellules mésangiales pouvaient être régulées par la matrice extracellulaire via les intégrines
(Makino H. et al., 2000).
• Les membranes basales déterminent en partie la perméabilité vasculaire. L’épaississement et
les modifications de composition de la membrane basale glomérulaire par exemple se traduisent par
une diminution des charges négatives et ainsi une perte de sélectivité de la filtration. De plus, il a
été montré que la glycation des protéines de la membrane basale la rend plus poreuse aux
macromolécules (Boyd-White J. et al., 1996). Combinées à une hypertension, ces modifications de
la membrane basale conduisent notamment à une fuite de protéines plasmatiques vers l’espace
extra-vasculaire : passage d’albumine dans l’urine (microalbuminurie) ou d’exsudats dans la
rétine.
33

• Au niveau rénal, dans les phases précoces du diabète, l'augmentation de production de la
membrane basale cause un élargissement du glomérule et une augmentation de la surface de
filtration. Au fur et à mesure que la néphropathie diabétique évolue, le mesangium empiète peu à
peu sur la membrane basale glomérulaire, diminuant ainsi la surface de filtration. Plus le volume de
glomérule occupé par le mesangium augmente, plus la surface de filtration capillaire diminue.
Associée à une ischémie secondaire à l’occlusion des artérioles afférentes, l’expansion mésangiale
aboutit à l’occlusion de la lumière capillaire et diminue progressivement le taux de filtration
glomérulaire. Cette expansion du mesangium conduit à terme à la sclérose globale du glomérule
et à la perte de la fonction rénale. L’épaississement de la membrane basale au niveau du tubule
participe à la fibrose tubulo-intersticielle qui contribue également à la perte de la fonction rénale.
3.2.Altérations cellulaires
L’environnement diabétique affecte fortement le métabolisme des cellules microvasculaires, et ce
en fonction du type cellulaire considéré. Leur croissance est généralement perturbée et leur survie
peut être mise en jeu. Le fonctionnement des microvaisseaux est alors sévèrement dégradé par les
bouleversements de leur structure cellulaire.
3.2.1 Cellules microvasculaires rétiniennes
(a) Les péricytes
La perte des péricytes est, avec l’épaississement de la membrane basale, une des premières
manifestations histologiques de la rétinopathie diabétique. La disparition précoce des péricytes
abaisse le ratio péricytes/cellules endothéliales de 1 pour 1 à 1 pour 10 après plusieurs années de
diabète mal contrôlé. Ce phénomène est important car il a été montré qu’en conditions normales,
plus ce ratio est fort et plus le turnover cellulaire est lent (Kohner E.M. et al., 1994).
Plusieurs travaux suggèrent que les péricytes disparaissent par un phénomène d’apoptose. La
présence de péricytes apoptotiques a en effet été mise en évidence dans des rétines de patients
diabétiques (Mizutani M. et al., 1996). Romeo et al. ont montré que le diabète in situ et de fortes
concentrations de glucose in vitro induisaient l’apoptose des péricytes en activant NF-κB (Romeo
G. et al., 2002). L'apoptose des péricytes rétiniens a également été décrite en réponse aux AGE
(Denis U. et al., 2002). De plus, une surexpression des protéines Bax, pro-apoptotique (Podesta F.
et al., 2000), et caspase-3, protéase impliquée dans l’apoptose (Li W. et al., 1999), a été mesurée
dans des péricytes de diabétiques. D’autre part, des auto-anticorps dirigés contre les péricytes ont
été trouvés chez des patients diabétiques, mettant ainsi en cause des phénomènes immunologiques
dans la disparition de ces cellules (Attawia M.A. et al., 1999; Nayak R.C. et al., 2003).
Les péricytes jouant un rôle majeur dans la structure et dans la fonction des microvaisseaux, leur
34

disparition va largement participer aux altérations caractéristiques de la rétinopathie diabétique. La
perte des péricytes va tout d’abord fragiliser l’architecture de la paroi capillaire et favoriser la
dilatation des capillaires. De plus, en l’absence de l’activité contractile exercée par les péricytes,
le tonus vasculaire diminue et le débit sanguin augmente consécutivement. Les forces de
cisaillement générées par l’augmentation du débit sanguin fragilisent d’autant plus l’endothélium
qui devient plus perméable à différents constituants plasmatiques et permet la formation de dépôts
extravasculaires, ainsi que l’apparition de microanévrismes et d’hémorragies, caractéristiques de
la rétinopathie diabétique. La perte des péricytes précède la dégénération des microvaisseaux et la
protection qu’ils exercent sur les microvaisseaux rétiniens a plusieurs fois été suggérée. Hammes et
al. ont par exemple rapporté qu’une diminution du nombre de péricytes chez des souris PDGF+/-
conduisait à une augmentation des capillaires acellulaires par perte des cellules endothéliales après
induction du diabète (Hammes H.P. et al., 2002). D’autre part, ils ont rapporté que cette déficience
en péricytes induit une augmentation du nombre de microvaisseaux nouvellement formés en
réponse à une hypoxie suggérant l’implication des péricytes dans la phase proliférative de la
rétinopathie diabétique. La perte des péricytes favorise également la prolifération des cellules
endothéliales et le processus de néovascularisation, formation de microvaisseaux exempts de
péricytes plus fragiles et sujets aux hémorragies. Cette différence d’effet des péricytes sur
l’endothélium pourrait être liée au taux du facteur angiogénique VEGF, plutôt faible dans les stades
précoces et élevé dans la phase proliférative. Enge et al. ont reproduit des altérations
caractéristiques du diabète au niveau de microvaisseaux rétiniens de souris déficientes en péricytes
par ablation du PDGF présentant ainsi la perte des péricytes comme un événement pathogène causal
dans la rétinopathie diabétique (Enge M. et al., 2002) .
(b) Les cellules endothéliales
Les péricytes exerçant un rôle régulateur sur les cellules endothéliales, leur disparition affecte
sérieusement le métabolisme et la croissance de ces dernières. La perte de contact avec les péricytes
semble prédisposer à une prolifération endothéliale. Généralement, les cellules endothéliales
soumises à un environnement diabétique présentent en effet une multiplication accélérée conduisant
à la formation de nouveaux microvaisseaux. L’hypoxie consécutive aux altérations de la
rétinopathie diabétique est le principal facteur causant l’angiogénèse. Nomura et al. ont rapporté
qu’une augmentation de l’expression du VEGF, facteur angiogénique, par les péricytes et les
cellules endothéliales elle-mêmes pourrait être responsable de l’augmentation de prolifération des
cellules endothéliales en réponse à l’hypoxie (Nomura M. et al., 1995). De nombreux autres
facteurs de croissance ont été impliqués dans les phénomènes d’angiogénèse, notamment FGF,
TGFβ ou IGF-1 (Forrester J.V. et al., 1997). Cependant, certains microvaisseaux voient leur
nombre de cellules endothéliales diminuer, ce qui conduit à la formation de capillaires acellulaires
35

ou "fantômes". De plus, la morphologie et la fonction des cellules endothéliales sont modifiées au
cours de la rétinopathie diabétique. Les jonctions cellulaires, normalement serrées, deviennent
d’adhérence intermédiaire suite à une baisse des taux d’une protéine majeure dans ces structures,
l’occludine.
Ces altérations cellulaires affectent l'intégrité des microvaisseaux et ont des conséquences
fonctionnelles graves. La prolifération excessive des cellules endothéliales peut localement oblitérer
les capillaires et favoriser l’apparition d’ischémies. Les capillaires acellulaires sont eux-aussi
associés à des zones d’hypoxie. Combinée à la disparition des péricytes, la dégradation des contacts
intercellulaires au niveau de l'endothélium contribue à la rupture de la barrière hémato-
rétinienne et à de petites hémorragies ou des fuites de macromolécules dans l’espace
extravasculaire. Les microvaisseaux nouvellement formés sont plus fragiles et donc sujets à ces
évènements.
3.2.2 Cellules glomérulaires
(a) Les cellules mésangiales
Des études sur des modèles d'animaux diabétiques ainsi que des expériences de culture cellulaire en
conditions hyperglycémiques, ont révélé une réponse biphasique des cellules mésangiales à
l'environnement diabétique (Wolf G., 2000) (Figure 9). Dans un premier temps, les cellules suivent
une prolifération limitée, en sortant d'un état de quasi-quiescence pour entrer dans le cycle
cellulaire et effectuer 1 à 2 mitoses. Elles sont ensuite rapidement arrêtées en phase G1 du cycle
cellulaire puis s’hypertrophient. Les cellules mésangiales augmentent leur synthèse protéique en
entrant en phase G1 mais ne se divisent pas. Cet arrêt du cycle cellulaire apparaît comme un des
mécanismes responsables de l'hypertrophie des cellules mésangiales. De plus, il semble qu’une
réduction de l’activité des protéases intracellulaires lors de l’état diabétique contribue également à
l’accumulation de protéines et donc à l’hypertrophie cellulaire.
36

cellule mésangiale normale
Prolifération initiale
Hypertrophie + synthèse accrue de
protéines matricielles
Néphropathie diabétique
DIABETE
cellule mésangiale normale
Prolifération initiale
Hypertrophie + synthèse accrue de
protéines matricielles
Néphropathie diabétique
DIABETE
Figure 9: comportement biphasique des cellules mésangiales rénales au cours de la néphropathie diabétique
(d'après Wolf G., 2000)
L'induction des protéines p21cip1 et p27kip1, inhibitrices des complexes cycline/kinase cycline-
dependante (CDKI), complexes nécessaires à l'accomplissement du cycle cellulaire, semble
primordiale dans ce blocage. Une augmentation de l’expression de p21Cip1 ou p27Kip1 a d’ailleurs été
montrée aussi bien in vitro dans des cellules mésangiales soumises à de fortes concentrations de
glucose (Kuan C.J. et al., 1998) qu’in vivo dans les glomérules de souris db/db (Wolf G. et al.,
1998) ou de rats STZ (Kuan C.J. et al., 1998). Les CDKI p21Cip1 et p27Kip1 semblent également être
des régulateurs de l'hypertrophie mésangiale. Wolf et al. ont par exemple montré que des cellules
mésangiales déficientes en p27Kip1 ne s’hypertrophient pas en réponse au glucose contrairement aux
cellules natives (Wolf G., 2000). De plus, Shankland et al. ont rapporté que des souris diabétiques
dont le gène codant pour p21Cip1 a été invalidé ne développent pas d’hypertrophie glomérulaire à la
différence des souris sauvages (al Douahji M. et al., 1999).
Là encore, différents facteurs de croissance sont en cause. L’augmentation de l’expression de
nombreux facteurs de croissance a été montrée à la fois in vitro et in vivo en conditions diabétiques.
Il paraît clair que ces facteurs de croissance, en particulier le TGFβ mais aussi le PDGF, l’HGF,
l’IGF-I, l’angiotensine II ou l’endothéline-1 sont des médiateurs de l’hypertrophie rénale et de
l’accumulation de protéines de la matrice extracellulaire (Wolf G. et al., 1999). Le TGFβ est connu
pour médier l’hypertrophie des cellules mésangiales induite par de fortes concentrations de glucose.
De plus, il semble que ce facteur de croissance induit par l’hyperglycémie, les AGE ou
l’angiotensine II, stimule l’expression des CDKI p21Cip1 et p27Kip1 pour conduire à l’arrêt de
prolifération et à l’hypertrophie cellulaire (Wolf G., 2000). Il a également été montré qu’une forte
concentration de glucose inhibe la prolifération de cellules mésangiales via une augmentation de
37

l’expression du TGFβ (Kolm-Litty V. et al., 1998; Wolf G. et al., 1992). Le PDGF pourrait quant à
lui stimuler la prolifération précoce des cellules mésangiales (Wolf G. et al., 1999). Ces mêmes
facteurs peuvent également stimuler la synthèse de matrice extracellulaire et la phase d'hypertrophie
des cellules mésangiales est associée à une augmentation de la synthèse et du dépôt de protéines de
la matrice extracellulaire, conduisant à l'expansion du mesangium. Cette expansion mésangiale a
des conséquences fonctionnelles très importantes, comme il a déjà été décrit, liée à l’albuminurie,
l’hypertension et la diminution du taux de filtration glomérulaire. Les phénomènes d'apoptose
pourraient contribuer aux anomalies tardives de la néphropathie diabétique. Bien que l'apoptose des
cellules glomérulaires ne soit pas connue comme un caractère de la maladie, une augmentation de
l'apoptose des cellules mésangiales a été montrée. Elle pourrait constituer un mécanisme de
réparation visant à normaliser le nombre de cellules mésangiales (Griffin S.V. et al., 2003). Dans
les stades finaux de la néphropathie diabétique, le glomérule est complètement sclérosé et les
cellules mésangiales disparaissent.
(b) Les podocytes
L’expansion mésangiale et l’épaississement de la membrane basale glomérulaire ont très souvent
été associés à la dysfonction rénale. Plus récemment, les altérations des podocytes ont été
considérées comme contribuant à la pathogénèse de la néphropathie diabétique. Des études ont
montré une diminution du nombre ou de la densité de podocytes chez des diabétiques de type I
(Bjorn S.F. et al., 1995; Steffes M.W. et al., 2001) ou de type II (Dalla V.M. et al., 2003;
Pagtalunan M.E. et al., 1997). Des changements structuraux tels une hypertrophie, un
élargissement ou une perte des pieds des podocytes ont également été décrits (Obineche E.N. et
al., 2001; Pagtalunan M.E. et al., 1997). Il est probable que le CDKI p27Kip1 (Bjorn S.F. et al., 1995;
Hoshi S. et al., 2002) et le TGFβ (Bjorn S.F. et al., 1995; Nicholas S.B., 2003) soient impliqués
dans l’atteinte des podocytes.
Les podocytes font partie intégrante de la barrière de filtration glomérulaire et leur altération est en
partie responsable de la microalbuminurie puis de la protéinurie. La perte des podocytes met à nu
certaines zones de la membrane et favorise la sclérose du glomérule.
(c) Cellules épithéliales tubulaires
Les cellules épithéliales tubulaires subissent également une hypertrophie importante. Associée à
un épaississement de la membrane basale tubulaire, elle est largement responsable de
l'hypertrophie rénale observée durant la néphropathie diabétique (Wolf G. et al., 1999). Les
phénomènes d’hypertrophie qui surviennent dans le glomérule ou le tubule affectent très fortement
la fonction rénale en précédant toujours la sclérose du glomérule et la fibrose tubulointersticielle.
Un phénomène de transdifférenciation des cellules épithéliales tubulaires en myofibroblastes
38

s’opère également au cours de la néphropathie diabétique et semble important dans la fibrose
tubulointersticielle. Les cellules épithéliales perdent leur morphologie et forment des fibres de
stress. Les adhésions cellulaires sont perturbées et la migration et l’invasion des cellules sont
augmentées. Le TGFβ, les produits avancés de glycation et l’angiotensine II ont été décrits pour
induire cette différenciation (Lan H.Y., 2003).
(d) Cellules endothéliales
Les cellules endothéliales glomérulaires sont quiescentes mais conservent la capacité de proliférer
en fonction de l'équilibre entre facteurs pro-angiogéniques comme le VEGF facteurs et anti-
angiogéniques. Comme dans la rétine, la réponse aux lésions pourrait conduire à la formation de
nouveaux microvaisseaux ou à la perte de la microvasculature contribuant à la sclérose du
glomérule et à la perte de la fonction rénale (Griffin S.V. et al., 2003). Des modifications de forme
ou une diminution de la relaxation ont également été rapportées, pouvant être en cause dans la
microalbuminurie (Trevisan R. et al., 1997).
3.3.Modifications des paramètres sanguins
Les paramètres sanguins sont largement modifiés au cours du diabète. Les modifications
hémodynamiques consistent en une augmentation du flux et de la pression sanguine notamment
à cause de la perte des péricytes rétiniens et des podocytes qui assurent en partie le tonus des
microvaisseaux. Des modifications rhéologiques interviennent également: augmentation de la
viscosité sanguine, diminution de la déformabilité des globules rouges et blancs et augmentation
des capacités d’adhésion des leucocytes à l’endothélium. Cette adhésion des leucocytes, activant un
processus inflammatoire, est favorisée par une augmentation des taux de molécules d’adhésion
comme ICAM-1 à la surface de l’endothélium. Enfin, il existe des troubles importants de
l’hémostase avec une diminution de l’activité fibrinolytique et une hyperaggrégabilité
plaquettaire. L’ensemble de ces paramètres contribue à endommager l’endothélium.
L’augmentation du débit sanguin favorise l’hyperfiltration et le profil thrombotique du sang
contribue à l’occlusion des capillaires.
Ces modifications hémodynamiques sont à la base d’une des hypothèses proposées pour expliquer
la mise en place des complications microvasculaires.
3.4.Modifications des parois capillaires
Dans les yeux ou les reins de modèles d’animaux diabétiques, une augmentation de la
perméabilité vasculaire à l’albumine a été décrite après induction du diabète (Pugliese G. et al.,
1989). Cliniquement, les défauts de perméabilité vasculaire se manifestent par la fuite de
fluoresceine des capillaires rétiniens lors d’angiographie et par le passage d’albumine des
39

capillaires glomérulaires retrouvée dans l’urine. Les altérations des membranes basales, des cellules
de la paroi vasculaire ainsi que des paramètres sanguins agissent de concert pour perturber la
barrière hémato-rétinienne et la barrière de filtration glomérulaire.
• L’épaississement et les modifications de composition des membranes basales, notamment la
diminution des charges négatives ou la glycation, conduisent à la perte de sélectivité des
membranes qui deviennent poreuses à des substances dont elles bloquent habituellement le passage.
• La disparition des éléments de soutien que sont les péricytes rétiniens et les podocytes au cours
du diabète fragilise la paroi capillaire. La perte de contact entre les péricytes et les cellules
endothéliales, dont les jonctions serrées se désorganisent, perturbe l’étanchéité de l’endothélium.
• L’augmentation du débit et la pression sanguine qui survient au niveau du rein et de la rétine en
conditions diabétiques accentue les forces de cisaillement qui s’exercent sur l’endothélium, ce qui
favorise la vasodilatation, l’augmentation de la pression transglomérulaire et l’hyperfiltration au
travers de la paroi vasculaire.
• La perméabilité vasculaire est également due à la production de facteur spécifique comme le
VEGF ou VPF (vascular permeability factor), produits par les cellules endothéliales ou les
macrophages et qui se fixe sur des récepteurs endothéliaux (Forrester J.V. et al., 1997).
Tous ces éléments conduisent à une augmentation de la perméabilité vasculaire qui favorise la
progression des microcomplications. Au niveau rénal, elle engendre une microalbuminurie puis un
protéinurie et une perte de la fonction rénale. Au niveau rétinien, elle favorise les fuites de
macromolécules vers l’espace extravasculaire et l’apparition d’œdème, ainsi que le dépôt de
molécules circulantes, notamment des lipoprotéines, dans la paroi vasculaire et la membrane basale
où viennent alors s’accumuler les macrophages.
Il apparaît que la majorité des altérations structurales et fonctionnelles causées par l'environnement
diabétique touche de manière similaire les microvaisseaux rétiniens et glomérulaires et sont donc
communes à la rétinopathie et à la néphropathie diabétiques, deux microcomplications qui évoluent
en parallèle chez les patients diabétiques. Cependant, il existe des spécificités tissulaires dans les
réponses au stress hyperglycémique : le risque occulaire est un risque de rupture vasculaire et
d'hémorragie alors que le risque rénal consiste en l'évolution vers la sclérose glomérulaire.
III. LES MECANISMES
L'hyperglycémie est aujourd'hui connue comme la principale cause des complications
microvasculaires du diabète. Cependant, les mécanismes de la toxicité du glucose ne sont pas
40

encore clairement déterminés. Le glucose et ses métabolites étant utilisés par de nombreuses voies
dans la cellule, les effets néfastes de l'hyperglycémie reposent sans nul doute sur de multiples
mécanismes. Plusieurs hypothèses ont été formulées pour expliquer les altérations microvasculaires
du diabète. Elles interviennent probablement de concours dans la genèse de la maladie. La première
hypothèse est hémodynamique et concerne la physiologie de la microcirculation et du flux sanguin.
Cinq hypothèses biochimiques ont par ailleurs été proposées concernant des modifications aiguës,
répétées mais réversibles (la voie des polyols, l'activation de la PKC et la voie de l'hexosamine) ou
des changements cumulatifs et irréversibles (la glycosylation non enzymatique et l'apparition d'un
stress oxydant).
1. L'HYPOTHESE HEMODYNAMIQUE Les facteurs hémodynamiques sont largement modifiés au cours du diabète et l'hyperglycémie
conduirait à la perte de la régulation du tonus vasculaire caractérisée par une vasodilatation
chronique des microvaisseaux ainsi que par une augmentation consécutive de la pression et du débit
sanguin microvasculaires (Zatz R. et al., 1986a). L'hypothèse hémodynamique présente cette
augmentation de la pression sanguine comme une cause primaire des complications
microangiopathiques. Le stress hémodynamique (action mécanique, forces de cisaillement)
endommagerait l'endothélium et provoquerait une réponse adaptative de la paroi vasculaire
consistant en un épaississement de la membrane basale. Celui-ci, associé à l'hypertension, augmente
la perméabilité de la paroi vasculaire et induit une hyperfiltration. Le stress hémodynamique peut
également conduire à une prolifération accrue des cellules, notamment mésangiales et endothéliales
qui, associée à l'épaississement de membrane basale peut favoriser l'obstruction des
microvaisseaux. L'ensemble de ces modifications altère fortement la fonction microvasculaire et les
perturbations hémodynamiques ont été mises en cause dans le développement de la rétinopathie
(Kohner E.M. et al., 1995) comme de la néphropathie (Trevisan R. et al., 1997; Zatz R. et al.,
1986b) diabétiques.
2. LA VOIE DES POLYOLS L'hyperglycémie peut augmenter la métabolisation du glucose excédentaire par une voie alternative
à la glycolyse, saturée, la voie des polyols. Celle-ci fait intervenir successivement deux enzymes :
l'aldose réductase et la sorbitol déshydrogénase. La première, activée seulement en présence de
fortes concentrations de glucose du fait de sa faible affinité pour ce substrat, réduit le glucose en
sorbitol en utilisant le NADPH comme donneur d'hydrogène. La seconde enzyme oxyde le sorbitol
41

en fructose en utilisant le NAD+ comme cofacteur. L'activation de cette voie conduit à une
accumulation intracellulaire de sorbitol et de fructose ainsi qu'à une modification des rapports
NADPH/NADP+ et NADH+/NAD+ qui perturbent le fonctionnement cellulaire de plusieurs
manières (Sheetz M.J. et al., 2002).
Le sorbitol formé se dégrade lentement et diffuse difficilement à travers les membranes. Il
s'accumule donc et peut altérer les cellules de certains tissus via un stress osmotique. De plus, les
concentrations importantes en glucose et sorbitol pourraient entrer en compétition pour la captation
d'autres osmolytes comme par exemple le myo-inositol. La déplétion intracellulaire en myo-inositol
entraînerait une diminution de la synthèse des phosphoinositides, de la production de DAG et
consécutivement de l'activation de la PKC ou de l'activité de la pompe Na+/K+ ATPase. Par ailleurs,
l'altération du potentiel redox semble jouer un rôle important dans les effets néfastes de l'activation
de la voie des polyols. La formation du sorbitol s'accompagne en effet d'une baisse des ressources
en NADPH, au détriment d'autres réactions nécessitant également ce cofacteur. La glutathion
réductase par exemple, nécessite des niveaux élevés de NADPH pour régénérer du glutathion
réduit. La baisse de son activité suite à une déplétion en NADPH entrave le cycle redox du
glutathion et contribue ainsi à générer un stress oxydant. La déplétion en NADPH peut également
interférer avec la formation de NO par la NO synthase, qui utilise également ce cofacteur.
L'utilisation d'inhibiteurs de l'aldose réductase ont permis expérimentalement de prévenir les
altérations de la rétinopathie (Kador P.F. et al., 1990) et de la néphropathie (Mauer S.M. et al.,
1989) diabétiques.
3. L'ACTIVATION DE LA PKC L'activation de la PKC est un second mécanisme proposé pour expliquer le développement des
complications microvasculaires du diabète. Une augmentation des taux de glucose conduit en effet
à une augmentation de la glycolyse qui stimule la synthèse de novo de diacylglycérol (DAG) à
partir du glyceraldéhyde-3-P qu'elle produit. Cette élévation des taux de DAG entraîne alors une
activation de la PKC. Par ailleurs, l'activation de la PKC peut aussi être consécutive au stress
oxydant ou à l'interaction des AGE avec RAGE (cf partie suivante). Les protéines kinases C sont
des molécules très importantes de la signalisation intracellulaire qui peuvent réguler diverses
fonctions vasculaires comme la perméabilité, la contractibilité des vaisseaux ou le flux sanguin. La
PKC peut être impliquée dans l'induction de facteurs de croissance ou affecter des voies de
signalisation comme les MAP kinases ou NF-κB. Une activation de certaines isoformes de PKC a
été décrite dans la rétine ou les glomérules de rats diabétiques et plusieurs travaux ont rapporté que
l'utilisation d'inhibiteurs de PKC permet de prévenir des dysfonctionnements de la rétinopathie et de
42

la néphropathie diabétiques comme l'hyperperméabilité ou l'expansion mésangiale (Sheetz M.J. et
al., 2002).
4. LA GLYCOSYLATION NON ENZYMATIQUE La formation des produits avancés de glycation ou AGE (Advanced Glycation End products) figure
parmi les modifications irréversibles causées par l'hyperglycémie. La présence des AGE est
étroitement reliée à l'hyperglycémie et leur accumulation a été montrée au cours du diabète. De
nombreuses études ont permis de mettre en cause les AGE dans le développement des
complications microangiopathiques du diabète.
4.1.Formation des AGE
Les sucres réducteurs, comme le glucose, peuvent réagir, de façon covalente et non
enzymatique, avec les groupements amines des protéines, lipides ou acides nucléiques par une
série de réactions regroupées sous le terme de glycosylation non enzymatique ou glycation. Ce
processus est également appelé réaction de Maillard, du nom de celui qui, début 1900, a découvert
que les acides aminés chauffés en présence de sucres réducteurs réagissaient pour former une
couleur caractéristique jaune-brun. Durant les premières étapes de la réaction, le groupement
aldéhyde d'un sucre réducteur, sous sa forme linéaire, réagit avec le groupement amine de certains
acides aminés, phospholipides ou molécules d'ADN pour former une base de Schiff (Figure 10).
Cette base de Schiff, ou produit de glycation précoce, se réarrange ensuite pour se stabiliser en
produit d'Amadori. Ces premières étapes de la glycation sont rapides et réversibles. La formation de
ces produits intermédiaires peu stables dépend de la concentration en substrats, notamment en sucre
réducteur, et est donc augmentée au cours du diabète. Notons que le glucose a un taux de glycation
relativement lent, au contraire d'autres sucres réducteurs intracellulaires comme le glucose 6-P ou le
fructose. La formation des AGE dépend aussi du temps de contact entre les substrats.
L'hémoglobine glyquée est d'ailleurs un produit d'Amadori utilisé chez le diabétique comme
indicateur de la glycémie des 6 à 12 semaines précédentes. Les produits de glycation intermédiaires
que sont les produits d'Amadori conduisent ensuite aux produits avancés de glycation par une série
de réarrangements. Ils peuvent subir une fragmentation oxydative pour former des produits dits de
glycoxydation comme la carboxyméthyllysine (CML) ou la pentosidine, AGE parmi les mieux
caractérisés. Une fragmentation non oxydative peut également avoir lieu et conduire à la formation
de produits intermédiaires de réaction, les α-dicarbonyles (ou α-oxoaldéhydes) comme le 3-
désoxyglucosone ou les glyoxal et méthylglyoxal. Ces produits, hautement réactifs, vont enfin
réagir, de manière irréversible, avec des protéines à durée de vie longue, pour donner naissance à
43

des composés chimiquement stables, les AGE. Les AGE peuvent également se former à partir de la
réaction d'un dicarbonyle avec un produit d'Amadori ou de la condensation de deux produits
d'Amadori, qui réagissent ensuite avec des groupements amine. Certains AGE ainsi obtenus sont
capables de former des ponts intermoléculaires ou "cross-link" dans certaines protéines. D'autres
voies que la réaction de Maillard peuvent donner lieu à la formation de dicarbonyles. La
fragmentation des trioses P, dérivés de la glycolyse, l'oxydation de l'acétone au cours du
métabolisme des corps cétoniques ou la peroxydation lipidique, peuvent par exemple conduire à la
formation de méthylglyoxal. D'autre part, des composés dicarbonyles peuvent également se former
suite à une auto-oxydation de sucres réducteurs comme le glucose. Les AGE dérivés du
méthylglyoxal, carboxyéthyllysine ou MOLD sont d'ailleurs des produits majeurs observés lors du
diabète. (Singh R. et al., 2001).
Figure 10: différentes voies de formation des AGE, d'après Singh et al. (Singh R. et al., 2001)
Ces modifications irréversibles de protéines par les sucres se produisent normalement durant le
processus de vieillissement mais la formation des AGE est largement accélérée au cours du diabète.
Elle se produit généralement dans l'organisme, particulièrement en conditions hyperglycémiques,
44

mais les AGE peuvent également provenir de sources exogènes comme le tabac ou l'alimentation.
Les aliments, en particulier par le chauffage qui induit un brunissement non enzymatique ou
réaction de Maillard, pourraient constituer une source significative d'AGE (Goldberg T. et al.,
2004).
4.2.Caractérisation et mesure
La glycosylation non enzymatique est un processus multiple et complexe, aboutissant à la formation
d'une grande diversité de structure d'AGE, tant par la diversité des dicarbonyles issus du
métabolisme cellulaire, que par celle des acides aminés avec lesquels ils réagissent. De plus, les
protéines glyquées peuvent subir des pontages intra ou intermoléculaires. Ainsi les AGE désignent
un ensemble très hétérogène et complexe de molécules par conséquent assez difficiles à
identifier. Une série de preuves expérimentales permet aujourd'hui de penser que les AGE
s'accumulent au cours du diabète. Cependant, il n'existe pas de méthode ni d'unité de mesure
établies et universelles. Le manque de standard interne rend la mesure d'autant plus difficile et
n’exclu pas un certain biais. Les techniques les plus couramment utilisées pour détecter les AGE
sont l'HPLC, l'ELISA et l'immunohistochimie. La nature chimique des AGE tels qu’ils se forment
naturellement in vivo n’est que partiellement connue. Il existe cependant un nombre croissant
d’AGE structurellement définis qui se sont révélés augmentés dans des tissus diabétiques. C’est le
cas notamment de la CML et de la pentosidine. Des études ont montré que leurs concentrations sont
environ doublées au cours du diabète et corrélées à la sévérité des complications microvasculaires.
Des concentrations augmentées de pyrraline ont également été détectées dans le plasma et la peau
de patients diabétiques (Portero-Otin M. et al., 1995). Le dicarbonyle 3-DG a été mesuré en
concentrations importantes dans le plasma de patients diabétiques particulièrement ceux présentant
un déficit rénal (Singh R. et al., 2001). Enfin, les sels d’imidazolium GOLD et des molécules
"cross-link" ont été identifiés dans la cornée et les érythrocytes de patients diabétiques. L'étude de
modèles d'animaux diabétiques a montré que les concentrations en AGE augmentent dès quelques
semaines après le déclenchement du diabète. Cette augmentation semble être systémique, touchant
à la fois le plasma, les reins, la peau, les tissus vasculaires…(Singh R. et al., 2001).
4.3.Mécanismes d’action et conséquences pathologiques
Les processus de glycation perturbent la conformation des molécules ciblées, principalement les
protéines, mais aussi les lipides ou les acides nucléiques. Ils induisent la formation de ponts intra
et intermoléculaires qui participent certainement largement aux dommages causés par les AGE. La
structure, la fonction ou la reconnaissance des molécules glyquées sont ainsi largement altérées. La
formation des AGE a été mise en cause dans le développement de la macroangiopathie et des
45

complications microangiopathiques du diabète. Par ailleurs, les AGE semblent impliqués dans des
pathologies aussi disparates que l'arthrite rhumatoïde, la maladie d'Alzheimer ou l'atteinte rénale
terminale. Trois mécanismes généraux sont mis en avant pour expliquer les changements
pathologiques causés par la formation des AGE. Premièrement, la formation rapide d'AGE sur
des protéines circulantes ou intracellulaires, dégrade leur fonction. Deuxièmement, la glycation
des protéines de la matrice extracellulaire altère ses propriétés. Enfin, l'interaction entre les
AGE extracellulaires et des récepteurs membranaires spécifiques peut modifier l'expression de
certains gènes.
4.3.1 Glycation des protéines à durée de vie courte : protéines circulantes, intracellulaires
et acides nucléiques
Les protéines circulantes sont fortement touchées par les phénomènes de glycation car elles se
trouvent en contact direct avec le glucose sanguin.
• La glycation s'opère notamment sur l'apoprotéine B et les LDL. La présence de groupements
amine sur certains phospholipides comme phosphatidylserine ou phosphatidyléthanolamine, fournit
des sites appropriés où le glucose peut réagir pour former des AGE. Les taux d'AGE-ApoB sont
environ 4 fois plus importants chez les patients diabétiques. Les AGE augmentent la susceptibilité
des LDL à l'oxydation, c'est pourquoi la glycation est souvent suivie d'une oxydation des LDL
(Bucala R. et al., 1993). La clairance de ces LDL modifiées se trouve ralentie par rapport aux LDL
natives. En diminuant leur élimination et facilitant leur accumulation dans la paroi vasculaire, la
glycation des LDL pourraient donc activement contribuer aux dyslipidémies, courantes chez les
diabétiques et caractérisées par une augmentation du taux de LDL. Les HDL et les VLDL sont
également glyquées au cours du diabète ce qui altère leur fonction de transport du cholestérol et
d'élimination des triglycérides. Ces dyslipidémies prédisposent les diabétiques à l'athérosclérose.
• L'hémoglobine est largement sujette à la glycation. Cette modification augmente sa viscosité à
l'intérieur des globules rouges et exerce des effets négatifs sur le relarguage de l'oxygène.
• Les immunoglobulines, principalement les IgG et les IgM peuvent être glyquées (Danze P.M.
et al., 1987). Leur activité est ainsi modifiée, ce qui participerait à la diminution de la résistance à
l'infection observée chez les diabétiques.
• Les globules rouges peuvent également subir une glycation. Elle a été corrélée à une
diminution de leur fluidité membranaire et de leur déformabilité (Miller J.A. et al., 1980) et à une
adhérence accrue à l’endothélium par l’intermédiaire de récepteurs aux AGE ce qui contribue au
dysfonctionnement de l’endothélium (Wautier J.L. et al., 1994).
Des AGE se forment aussi directement à l'intérieur des cellules. Ce phénomène semble d'ailleurs
être plus rapide qu'en milieu extracellulaire car les sucres intracellulaires augmentés au cours du
46

diabète (glucose 6-P, fructose, métabolisés en dicarbonyles comme le méthylglyoxal) sont
largement plus réactifs que le glucose. bFGF est la principale protéine modifiée par les AGE, dans
les cellules endothéliales notamment. Cette modification réduit d'ailleurs son activité mitogénique
de 70 % (King G.L. et al., 1996). Dans la cellule, la glycation peut également toucher les
nucléotides des acides nucléiques. L'exposition de l'ADN à de fortes concentrations de glucose
cause une glycation des groupes amines des nucléotides. Des cellules endothéliales humaines
exposées à de fortes concentrations de glucose ont révélé une augmentation des cassures dans les
simples brins d'ADN et une diminution du taux de réparation de l'ADN (Lorenzi M. et al., 1986).
4.3.2 Glycation des protéines de la matrice
La durée d'exposition des protéines au glucose étant un des facteurs principaux gouvernant la
formation des AGE, ces derniers se forment et s'accumulent en premier lieu sur des macromolécules
à durée de vie longue. Les protéines de la matrice extracellulaire, par leur faible taux de
renouvellement, sont donc particulièrement touchées par la glycation, d'autant qu'elles se trouvent
en contact direct avec le glucose sanguin.
• Les AGE induisent des changements sur de nombreux composants de la matrice comme le
collagène, la fibronectine, la laminine ou la vitronectine. De plus, ces protéines matricielles
glyquées peuvent former des ponts intermoléculaires avec les molécules matricielles adjacentes. La
glycation de la membrane basale diminue sa solubilité et sa digestion par des enzymes. Des
études in vitro ont montré que le collagène glyqué et lié par des ponts intermoléculaires à d'autres
composants glyqués de la matrice, est plus résistant à la dégradation enzymatique. L'accumulation
d'AGE dans la matrice extracellulaire peut ainsi conduire à l'épaississement de membrane basale
caractéristique du diabète.
• De plus, la formation des AGE perturbe l'auto-assemblage géométrique et ordonné des
protéines de la membrane basale. Par exemple, la glycation de la laminine, de la vitronectine et
du collagène peut sérieusement altérer les charges des molécules, perturber leurs interactions et leur
capacité à s'assembler en agrégats tridimensionnels. Ces perturbations s'accompagnent d'une
augmentation de la taille des pores intermoléculaires, qui peuvent, associées à l'altération des
protéoglycanes anioniques, dégrader la barrière de filtration charge-sélective de la matrice et
provoquer des fuites de protéines circulantes au travers de la paroi vasculaire.
• Les ponts intermoléculaires formés par les AGE peuvent également lier les protéines de la
matrice avec les protéines plasmatiques comme les LDL. L'accumulation de ce matériel sur la paroi
des microvaisseaux pourrait progressivement causer le rétrécissement de la lumière vasculaire et à
terme l'occlusion des microvaisseaux.
• La formation d'AGE au niveau de la matrice extracellulaire interfère également dans les
47

interactions matrice-cellules. Par exemple, il a été montré que l'adhésion et l'étalement des
cellules endothéliales sont altérés par la glycation du collagène IV et de la laminine. De plus, la
glycation des composants de la membrane basale peut également affecter la fonction des cellules
vasculaires en jouant sur la transduction du signal via des récepteurs comme les intégrines (King
G.L. et al., 1996).
La formation d'AGE au niveau de la matrice extracellulaire, notamment la membrane basale,
perturbe donc ses propriétés physico-chimiques et sa fonction ce qui se répercute sur la fonction des
microvaisseaux eux-mêmes, dont l’élasticité ou les propriétés de filtration sont par exemple
altérées.
4.3.3 Interaction avec des récepteurs membranaires spécifiques
Différentes molécules se liant aux AGE ont été décrites. Il s’agit des récepteurs scavenger, des
récepteurs aux AGE : AGE-R1, AGE-R2 et AGE-R3 et du récepteur RAGE. Certains sont plutôt
avancés comme servant à promouvoir les effets des AGE sur les cellules et les tissus, d’autres
servant à les limiter.
(a) Macrophage "scavenger" récepteurs
Les MRS (macrophage scavenger receptor) de la classe A (type I et II) (Radoff S. et al., 1988) ainsi
que de la classe B (CD36) (Ohgami N. et al., 2001) ont été décrits pour se lier aux AGE. Ils
constitueraient un système de captation et de dégradation des AGE par les macrophages (Araki
N. et al., 1995). Ces récepteurs pourraient être impliqués dans la macroangiopathie diabétique en
internalisant les LDL modifiées par la glycation et en formant des cellules spumeuses s’accumulant
sur la paroi des microvaisseaux (Sakata N. et al., 2001).
(b) Complexe AGE-récepteur
Trois protéines ont été identifiées : une première de 60 kDa (p60) aussi appelée AGE-R1, une
seconde de 90 kDa (p90) ou AGE-R2 et la galectine-3 ou AGE-R3 qui forment le complexe
récepteur aux AGE. Stitt et al. ont montré, dans des cellules endothéliales microvasculaires
rétiniennes, que les différents éléments AGE-R1, -R2 et -R3 de ce complexe sont enrichis au sein
des cavéoles de la membrane plasmique, qui serait le lieu de leur assemblage et de leur interaction
avec les AGE (Figure 11). AGE-R2 ne semble pas lier directement les AGE mais pourrait être
impliqué dans la transduction du signal par un mécanisme de phosphorylation (Stitt A.W. et al.,
2000b). L’expression de ce complexe est positivement régulée par une exposition aux AGE (Stitt
A.W. et al., 1999).
48

Figure 11: le complexe récepteur aux AGE d'après Stitt et al. (Stitt A.W. et al., 2000b)
Le complexe AGE-R est plus souvent décrit commune une voie de détoxification des AGE,
médiant leur captation, leur endocytose et leur dégradation. Ce système semble être prédominant à
la surface des macrophages mais existe également sur d’autres cellules endothéliales, mésangiales
ou neuronales (Vlassara H., 2001). Des études récentes ont montré leur rôle protecteur dans les
complications diabétiques. Tout d’abord, il a été rapporté que les AGE liés à ce complexe sont
internalisés et dirigés vers des endosomes et des structures de type lysosomes (Stitt A.W. et al.,
2000b). Iacobini et al. ont par ailleurs montré que des souris déficientes en galectine 3 (AGE-R3)
développent une glomérulopathie diabétique accélérée (Iacobini C. et al., 2003). Ils ont ensuite
précisé que cette déficience en AGE-R3 est associée à une susceptibilité accrue aux troubles rénaux
induits par une infusion d’AGE (expansion mésangiale, protéinurie et sclérose glomérulaire) ainsi
qu’à une augmentation des taux et de la signalisation des AGE (Iacobini C. et al., 2004). D’autre
part, il a été décrit que des cellules mésangiales murines surexprimant AGE-R1 présentent une
liaison, une captation et une dégradation accrue des AGE. De plus, la voie de signalisation de
RAGE conduisant à une atteinte inflammatoire est inhibée dans ces cellules (Lu C. et al., 2004).
D’ailleurs, alors que l’expression de récepteurs des AGE comme RAGE est augmentée en
conditions diabétiques, l’expression de AGE-R1 est diminuée chez des souris diabétiques avec
atteinte rénale grave (He C.J. et al., 2000) ou des patients souffrant de néphropathie diabétique (He
C.J. et al., 2001), ce qui soutient leur rôle de protection contre les effets des AGE. Cependant, ce
complexe récepteur a aussi été impliqué dans une transduction de signal. Chibber et al. ont par
exemple montré que les effets toxiques exercés par les AGE sur des péricytes rétiniens ou des
49

cellules endothéliales en culture peuvent être reversés par l’utilisation d’anticorps anti-p60 et -p90
(Chibber R. et al., 1997) alors que Doi et al. ont montré que ces mêmes composants du complexe
AGE-R peuvent être impliqués dans l’expression de PDGF (Doi T. et al., 1992).
(c) RAGE
Le récepteur RAGE est le mieux caractérisé des récepteurs aux AGE, membre de la superfamille
des immunoglobulines. Il est de plus en plus considéré comme un médiateur de la signalisation
intracellulaire des AGE et il semble qu’une grande partie des effets délétères des AGE soient
médiés par une interaction avec RAGE. Il est présent sur différents types cellulaires comme les
macrophages, lymphocytes, cellules endothéliales, cellules musculaires lisses, cellules mésangiales
ou neuronales, péricytes. Son expression est augmentée au cours du diabète (Wautier J.L. et al.,
2001). La présence de RAGE a été décrite dans les cavéoles (Lisanti M.P. et al., 1994).
• La fixation des AGE au RAGE a été décrite pour induire une cascade de signalisation
impliquant le stress oxydant par la production de radicaux libres, l’activation de protéines comme
p21ras et des MAP-kinases (ERK1/ERK2) (Lander H.M. et al., 1997), et de NF-κB qui induit
finalement l’expression de différents gènes (Figure 12). Il a été montré que la NADPH oxydase est
impliquée dans la production de produits dérivés de l’oxygène et la génération d’un stress oxydant
induites par RAGE (Wautier M.P. et al., 2001). Cependant, le mécanisme de couplage de RAGE à
l’induction d’un stress oxydant n’est pas connu. L’implication de p38 MAP-kinase dans cette
cascade de signalisation a également été décrite (Yeh C.H. et al., 2001). Parmi les protéines dont
l’expression est induite par RAGE, des cytokines pro-inflammatoires comme le TNF-α ou
l’interleukine-1 (Il-1), ou des molécules d’adhésion comme VCAM, ont jusqu’à présent été
identifiées (Singh R. et al., 2001).
50

RAGE
AGE
Stress oxydant
P21ras/MAP kinase
activation NF-κB
Expression de gènes
- Cytokines pro-inflamatoires(TNF-α, IL-1, IGF-1)
- Vasoconstricteurs (endothéline-1)- Pro-coagulants (thrombomoduline
PAI-1, facteur tissulaire…)
(NADPH oxydase)
- Molécules d’adhésion(VCAM)
RAGE
AGE
RAGE
AGE
Stress oxydant
P21ras/MAP kinase
activation NF-κB
Expression de gènes
- Cytokines pro-inflamatoires(TNF-α, IL-1, IGF-1)
- Vasoconstricteurs (endothéline-1)- Pro-coagulants (thrombomoduline
PAI-1, facteur tissulaire…)
(NADPH oxydase)
- Molécules d’adhésion(VCAM)
Figure 12: signalisation consécutive à la liaison des AGE au récepteur RAGE
Au niveau des macrophages par exemple, l’interaction des AGE avec RAGE conduit suivant cette
cascade de signalisation à la libération de TNF-α et d’Il-1 qui vont agir sur l’endothélium en
causant des dysfonctionnements vasculaires notamment une augmentation de la perméabilité, et sur
les cellules mésangiales, augmentant la synthèse de matrice (Mullarkey C.J. et al., 1994). De plus,
la fixation d’AGE circulants (globules rouges ou LDL glyquées) sur l’endothélium par
l’intermédiaire de RAGE augmente également la perméabilité vasculaire et favorise la dysfonction
endothéliale (Wautier J.L. et al., 1996). L’induction d’un processus inflammatoire consécutif à
l’interaction des AGE avec RAGE, combiné à une accumulation de lipoprotéines, accélère
l’athérosclérose (Singh R. et al., 2001).
L’interaction entre les AGE et le récepteur RAGE a été impliquée dans les altérations de la
microangiopathie diabétique, comme suggéré dans les paragraphes suivants.
• Trois éléments capables d'induire la transcription du gène de RAGE ont été identifiés : les AGE
eux-mêmes, le TNF-α et la 17β-estradiol. Le feed-back positif exercé par les AGE sur l'expression
51

de RAGE peut exacerber les effets délétères des AGE. Récemment, deux nouveaux variants de la
forme membranaire connue ont été découverts : une forme membranaire tronquée du côté N-
terminal et une forme soluble tronquée du côté C-terminal et libérée dans le milieu. Cette dernière
forme (esRAGE pour endogenous secretory RAGE) pourrait exercer un rôle protecteur sur les
cellules en capturant les AGE circulants dans le milieu extracellulaire (Sakurai S. et al., 2003).
• RAGE peut fixer différents ligands, ce qui lui confère un rôle potentiel dans différentes
pathologies : l'amphotérine, protéine impliquée dans la migration cellulaire et l’invasion des cellules
tumorales, le peptide β amyloïde qui s'accumule notamment lors de la maladie d’Alzheimer, et
certains membres de la famille des calgranulines qui présentent des propriétés pro-inflammatoires
(Wendt T. et al., 2003).
4.4.AGE et rétinopathie diabétique
Un grand nombre de travaux menés à la fois in vitro et in vivo ainsi que des études cliniques
indiquent clairement que les AGE jouent un rôle pathogène important dans la rétinopathie
diabétique.
Hammes et al. ont montré que les AGE s’accumulent dans la rétine au cours du diabète
(Hammes H.P. et al., 1999) et une augmentation des concentrations de fructosyl-lysine, de
carboxyméthyl-lysine ainsi que d’AGE dérivés du glyoxal et du méthylglyoxal a été mesurée dans
la rétine de rats STZ (Karachalias N. et al., 2003). Une accumulation d’AGE, fonction de la durée
du diabète, a également été décrite dans les péricytes et les cellules endothéliales de microvaisseaux
rétiniens ainsi que dans la membrane basale, chez des rats STZ (Stitt A.W. et al., 1997). De plus, la
même étude a permis de montrer que les AGE étaient colocalisés avec les récepteurs AGE-R1 et
AGE-R2. L’existence du récepteur RAGE a par ailleurs été décrite dans les cellules
microvasculaires rétiniennes, cellules endothéliales et péricytes (Yamagishi S. et al., 2002a;
Yonekura H. et al., 2003) et une colocalisation entre les AGE et ce récepteur RAGE a été décrite au
niveau de la rétine (Soulis T. et al., 1997). Chez des diabétiques de type 2, un fort taux d’AGE dans
le sérum a été corrélé à la sévérité de la rétinopathie diabétique (Aso Y. et al., 2000).
Le rôle des AGE dans la pathogenèse de la rétinopathie diabétique est renforcé par les résultats
d’études basées sur l’utilisation d’inhibiteurs d’AGE. L’utilisation d’aminoguanidine, un inhibiteur
de la formation des AGE, a permis de limiter l’accumulation d’AGE au niveau des capillaires et de
prévenir la prolifération des cellules endothéliales et la perte des péricytes ainsi que d’atténuer
l’apparition de capillaires acellulaires ou de microanévrismes chez des rats diabétiques (Hammes
H.P. et al., 1991). Le traitement de rats diabétiques avec l’aminoguanidine a également permis de
protéger les capillaires rétiniens de l’épaississement de membrane basale caractéristique de la
microangiopathie diabétique (Gardiner T.A. et al., 2003). Cependant il est possible que cette
52

protection révèle l’implication d’autres voies car l’aminoguanidine n’est pas un inhibiteur
totalement spécifique des AGE mais présente aussi des propriétés anti-oxydantes (Stitt A.W.,
2003).
Les AGE agissent à différents niveaux pour contribuer aux altérations cellulaires et tissulaires de la
rétinopathie diabétique :
• Ils perturbent la prolifération des cellules microvasculaires rétiniennes. Plusieurs études ont
montré que les AGE sont toxiques pour les péricytes. In vitro, le traitement de péricytes rétiniens
avec des AGE diminue significativement leur prolifération (Chibber R. et al., 1997; Ruggiero-
Lopez D. et al., 1997; Yamagishi S. et al., 1995). Cette action anti-proliférative des AGE pourrait
passer par un effet récepteur car le blocage de l’interaction entre les AGE et leurs récepteurs par
l’utilisation d’anticorps ou d’oligonucléotides anti-sens permet de protéger les péricytes (Chibber R.
et al., 1997; Yamagishi S. et al., 1995). Des travaux antérieurs de notre équipe ont de plus montré
que les AGE induisent l’apoptose de péricytes rétiniens via une cascade de signalisation impliquant
notamment l’induction d’un stress oxydant, une augmentation des taux de DAG et de céramides et
l’activation de caspases (Denis U. et al., 2002). D’autres travaux ont également révélé l’apoptose
des péricytes traités avec d’autres types d’AGE (Chen B.H. et al., 2003; Yamagishi S. et al., 2002a)
ou avec du méthylglyoxal (Kim J. et al., 2004). Les AGE pourraient ainsi être en cause dans la perte
des péricytes qui survient lors de la rétinopathie diabétique. En ce qui concerne les cellules
endothéliales, plusieurs études ont montré que leur prolifération était augmentée en réponse aux
AGE (Chibber R. et al., 1997; Ruggiero-Lopez D. et al., 1997).
• La glycation affecte les protéines de la matrice extracellulaire. Ces modifications ont des
conséquences à deux niveaux. D’une part, les AGE induisent la formation de ponts inter-
moléculaires entre les protéines de la matrice comme le collagène type IV, la laminine ou la
fibronectine, qui deviennent plus résistantes à la dégradation. Les AGE participeraient de cette
manière à l’épaississement de la membrane basale (Wautier J.L. et al., 2001). La glycation
perturbe de plus l’auto-assemblage des protéines de la membrane basale qui perd alors de sa
sélectivité de filtration. D’autre part, la glycation des protéines matricielles modifie le
comportement cellulaire. Il a notamment été montré que l’adhésion et la viabilité des péricytes
sont réduites (Beltramo E. et al., 2002; Beltramo E. et al., 2003; Stitt A.W. et al., 2004) alors que la
prolifération des cellules endothéliales est augmentée sur une matrice glyquée (Kalfa T.A. et al.,
1995).
• Les AGE sont également capables d’induire l’expression de VEGF, facteur angiogénique et
de perméabilité très important dans la rétinopathie diabétique. Une augmentation de l’expression
de l’ARNm du VEGF dans les cellules épithéliales rétiniennes a été décrite chez des souris (Treins
53

C. et al., 2001) et des rats (Stitt A.W. et al., 2000a) infusés avec des AGE. Une augmentation de la
fuite d’albumine à travers les microvaisseaux rétiniens, attribuée à une augmentation de
l’expression de VEGF, a également été mesurée dans des rats infusés avec des AGE (Stitt A.W. et
al., 2000a).
• Il semble enfin que les AGE puissent participer à une réponse pro-inflammatoire. Il a été
récemment montré que l’endothélium microvasculaire rétinien exposé aux AGE in vitro et in vivo,
augmente l’expression de la molécule d’adhésion ICAM-1 et l’adhésion de leucocytes (Stitt A.W.,
2003).
4.5.AGE et néphropathie diabétique
De même que pour la rétinopathie diabétique, un grand nombre de travaux permettent de penser que
les AGE sont en cause dans le développement de la néphropathie diabétique. Une augmentation des
concentrations de fructosyl-lysine, de carboxyméthyl- et carboxyéthyl-lysine ainsi que d’AGE
dérivés du glyoxal et du méthylglyoxal a été mesurée dans les glomérules rénaux de rats STZ
(Karachalias N. et al., 2003). La détection des AGE à l’aide d’anticorps spécifiques a révélé leur
présence dans les zones d’expansion mésangiale et dans la membrane basale glomérulaire dans les
reins de rats diabétiques (Shikata K. et al., 1995). Une augmentation des taux d'AGE et notamment
de carboxyméthyl-lysine, a également été mesurée dans les reins ou les lésions glomérulaires au
niveau de la matrice mésangiale ou de la paroi capillaire, de patients diabétiques (Horie K. et al.,
1997; Niwa T. et al., 1997).
Il a également été rapporté que l’infusion de rats avec des AGE induit une augmentation du volume
glomérulaire, un épaississement de la membrane basale et une expansion mésangiale indiquant une
sclérose glomérulaire, ainsi qu’une perte d’albumine et de protéines dans l’urine. Le co-traitement
de ces animaux avec de l’aminoguanidine, inhibiteur de la formation des AGE, a permis de réduire
ces altérations structurales et fonctionnelles (Vlassara H. et al., 1994). L’utilisation d’un inhibiteur
ALT-946 de la formation de ponts intermoléculaires, plus spécifique, a également permis de réduire
l'albuminurie chez des animaux diabétiques (Forbes J.M. et al., 2001).
Yamamoto et al. ont de plus montré que des souris doublement transgéniques, diabétiques et
surexprimant le récepteur RAGE humain, présentent un tableau d’altération rénale aggravé
(élargissement du rein, hypertrophie glomérulaire, albuminurie, expansion mésangiale, et sclérose
glomérulaire ) comparé aux souris diabétiques sans le transgène RAGE (Yamamoto Y. et al., 2001).
De plus, la blocage de RAGE dans des souris db/db s’est révélé prévenir les altération structurales
et fonctionnelles du rein (Wendt T. et al., 2003). Une co-localisation entre les AGE et ce récepteur
RAGE a d’ailleurs été décrite au niveau du glomérule rénal (Soulis T. et al., 1997) et une
surexpression de RAGE a été montrée dans des reins de patients diabétiques (Wendt T. et al.,
54

2003).
Enfin, chez des diabétiques de type 2, un fort taux d’AGE dans le sérum a été corrélé à la sévérité
de la néphropathie diabétique (Aso Y. et al., 2000).
Différents effets des AGE ont été décrits qui concourent aux altérations cellulaires et tissulaires de
la néphropathie diabétique :
• La glycation des protéines de la matrice extracellulaire, en les rendant moins sensibles à la
dégradation protéolytique notamment par la formation de ponts intermoléculaires, participe à
l’épaississement des membranes basales et à l’expansion du mesangium. Il a d’ailleurs été
montré que la cassure de ces ponts par un agent spécifique, le ALT-711, diminue l’accumulation de
matrice au niveau du rein (Forbes J.M. et al., 2001). De plus, la glycation perturbe l’assemblage des
différents composants protéiques, augmente la taille des pores inter-moléculaires et diminue la
charge négative des protéoglycanes contribuant ainsi à l’augmentation de la perméabilité et à la
perte de sélectivité de la barrière de filtration glomérulaire (Wautier J.L. et al., 2001).
L’utilisation d’inhibiteurs de ponts intermoléculaires permet également de limiter le développement
de l’albuminurie (Forbes J.M. et al., 2003).
• Les AGE contribuent également au développement de la néphropathie diabétique en induisant
l’expression de différentes cytokines pro-inflammatoires et facteurs de croissance. En particulier, le
TGFβ semble être un médiateur important des effets des AGE. Il a par exemple été rapporté que
l’utilisation d’aminoguanidine chez des rats STZ permet de limiter la surexpression du TGFβ et du
PDGF ainsi que de réduire le dépôt de collagène IV dans les membranes (Kelly D.J. et al., 2001).
De plus, Yang et al. ont montré une augmentation des taux d’ARNm du TGFβ, du collagène IV et
de la laminine, ainsi qu’une hypertrophie glomérulaire dans des souris ayant reçu des injections
d’AGE pendant 4 semaines. Ils ont également observé que ces altérations caractéristiques de la
néphropathie diabétique sont réduites par la co-administration d’aminoguanidine (Yang C.W. et al.,
1994). La même équipe a d’ailleurs montré que ces effets pourraient être médiés par le PDGF (Doi
T. et al., 1992). D’autre part, il a été montré que les AGE peuvent induire la production de VEGF et
de molécule d’adhésion des monocytes (MCP-1) par des cellules mésangiales humaines favorisant
ainsi le processus inflammatoire (Yamagishi S. et al., 2002b).
• Un effet direct des AGE sur la prolifération des cellules mésangiales rénales a également été
montré. La culture de cellules mésangiales murines en présence de protéines de sérum glyquées
s’est révélée inhiber la prolifération cellulaire (Ziyadeh F.N. et al., 1993). De plus, Yamagishi et al.
ont montré qu’un traitement avec les AGE est capable d’induire l’apoptose de cellules mésangiales
humaines (Yamagishi S. et al., 2002b).
• D’autre part, Oldfield et al. ont décrit que l’exposition de cellules épithéliales tubulaires aux
55

AGE induit leur différenciation en myofibroblastes suggérant ainsi le rôle des AGE dans la fibrose
tubulo-intersticielle qui survient lors de la néphropathie diabétique. Ils ont de plus montré que cet
effet peut être neutralisé par des anticorps dirigé contre le récepteur RAGE ou le TGFβ suggérant
ainsi une signalisation des AGE via une interaction avec RAGE et l’induction de ce facteur de
croissance, particulièrement important dans la néphropathie diabétique (Oldfield M.D. et al., 2001).
Il faut noter que les reins sont le site principal d’élimination des AGE de la circulation. Lorsque la
fonction rénale est altérée au cours du diabète, la clairance des AGE s’en trouve amoindrie ce qui
favorise l’aggravation des lésions notamment rénales.
5. LA VOIE DES HEXOSAMINES
5.1.Définition
En 1991, l’équipe de Marshall découvre dans les adipocytes une nouvelle voie métabolique médiant
la désensibilisation induite par le glucose du système de transport du glucose insulino-dépendant
(Marshall S. et al., 1991). Cette voie, qui nécessite la présence de glucose, d’insuline et de
glutamine et qui met en jeu une glutamine amidotransférase est la voie des hexosamines :
Une fois entré dans les cellules via le système de transport, le glucose est rapidement phosphorylé
en glucose-6-P et converti en fructose-6-P. Alors que la majorité de ce fructose-6-P est catabolisé
par la glycolyse, une partie est convertie en glucosamine-6-P par la glutamine:fructose-6-P
amidotransférase (GFAT), la première enzyme limitante de la voie de biosynthèse des
hexosamines. La glucosamine-6-P est ensuite rapidement transformée puis activée en UDP-N-
acétylglucosamine (UDP-GlcNAc), précurseur d’autres sucres aminés, produits de la voie des
hexosamines, comme le CMP-NAc acide neuraminique. Ces produits sont ensuite utilisés pour la
biosynthèse de glycoconjugués : glycoprotéines et glycolipides comme les gangliosides, ou
protéoglycanes (Marshall S., 2002) (Figure 13).
56
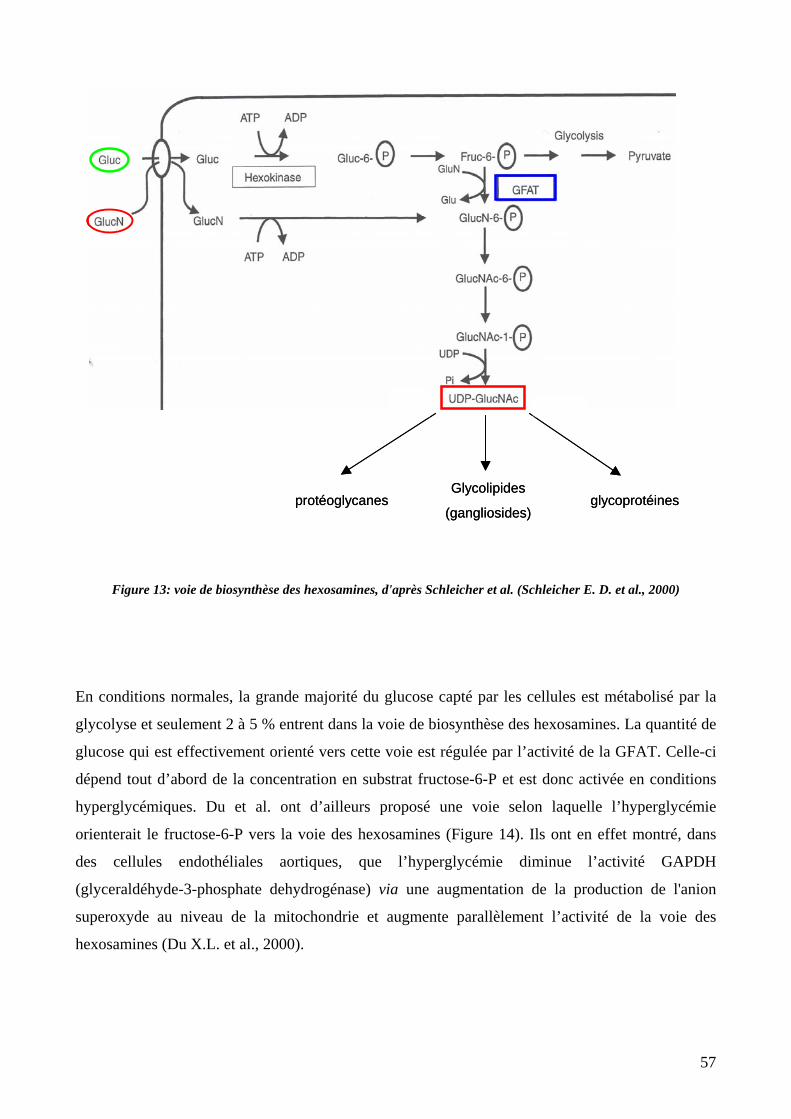
protéoglycanesGlycolipides
(gangliosides)glycoprotéinesprotéoglycanes
Glycolipides
(gangliosides)glycoprotéines
Figure 13: voie de biosynthèse des hexosamines, d'après Schleicher et al. (Schleicher E. D. et al., 2000)
En conditions normales, la grande majorité du glucose capté par les cellules est métabolisé par la
glycolyse et seulement 2 à 5 % entrent dans la voie de biosynthèse des hexosamines. La quantité de
glucose qui est effectivement orienté vers cette voie est régulée par l’activité de la GFAT. Celle-ci
dépend tout d’abord de la concentration en substrat fructose-6-P et est donc activée en conditions
hyperglycémiques. Du et al. ont d’ailleurs proposé une voie selon laquelle l’hyperglycémie
orienterait le fructose-6-P vers la voie des hexosamines (Figure 14). Ils ont en effet montré, dans
des cellules endothéliales aortiques, que l’hyperglycémie diminue l’activité GAPDH
(glyceraldéhyde-3-phosphate dehydrogénase) via une augmentation de la production de l'anion
superoxyde au niveau de la mitochondrie et augmente parallèlement l’activité de la voie des
hexosamines (Du X.L. et al., 2000).
57

Figure 14: mécanisme proposé pour l’induction de la voie des hexosamines par l’hyperglycémie,
d'après Du et al. (Du X.-L. et al., 2000)
La GFAT est également inhibée selon un rétro-contrôle négatif par son produit UDP-GlcNAc et
activée par phosphorylation dépendante de l’AMPc. Le flux à travers la voie des hexosamines
dépend aussi de la quantité de l’enzyme dans la cellule et de l’expression du gène de la GFAT. Sa
transcription pourrait être régulée par le facteur Sp1, l’EGF, le glucose et la glucosamine elle-
même. Il existerait deux isoformes de la GFAT, 1 et 2 (Schleicher E.D. et al., 2000). L’expression
de la GFAT a été mesurée dans les tissus humains à l’aide d’un anti-sérum spécifique de la protéine
et d’une sonde dirigée contre son ARNm. Des taux d’expression élevé de la protéine et de son
ARNm ont été mesurés dans les adipocytes et le muscle squelettique, sièges de l’insulino-
résistance, ainsi que dans les cellules musculaires lisses de microvaisseaux. Dans le tissu rénal, la
GFAT s’est révélée majoritairement tubulaire en conditions normales alors qu’elle était également
présente dans les cellules glomérulaires épithéliales et mésangiales issues de reins de patients
atteints de néphropathie diabétique. Ceci suggère que l’expression de la GFAT peut être induite par
le diabète dans des tissus cibles des microcomplications (Nerlich A.G. et al., 1998). Notons que la
GFAT a également été détectée dans la rétine (Mazlen R.G. et al., 1970).
5.2.Mécanismes d’action
A partir de l’UDP-GlcNAc généré par la voie des hexosamines, deux voies aux fonctions
différentes se distinguent. La manière dont les cellules régulent ces deux voies et coordonnent
58

l’utilisation du précurseur commun qu’est l’UDP-GlcNAc reste à déterminer.
5.2.1 Apport de précurseurs glycosidiques
L’UDP-GlcNAc issue de la voie des hexosamines peut être convertie en d’autres UDP-N-
acétylhexosamines qui sont les précurseurs glycosidiques aminés de la biosynthèse des
glycoconjugués. Il s’agit notamment de l’UDP-N-acétylgalactosamine, de l’acide CMP-N-
acétylneuraminique (acide sialique) ou de la N-mannosamine. Ces résidus peuvent être transportés
jusqu’au réticulum endoplasmique où ils sont utilisés pour glycosyler des protéines, notamment.
Cette glycosylation est généralement multiple sous forme de chaîne glycosidique. Les
glycoconjugués complexes ainsi formés sont le plus souvent destinés à être insérés au niveau de la
membrane plasmique à la surface des cellules ou bien à être secrétés. Ce mécanisme n’est donc pas
directement considéré comme une voie de signalisation cellulaire.
5.2.2 O-glycosylation simple de protéines
Le résidu UDP-GlcNAc formé par la voie des hexosamines peut être utilisé pour modifier de façon
covalente des protéines selon une liaison O-glycosidique (O-glycosylation) avec des résidus
thréonine et serine. Ce processus, contrairement au précédent, concerne, non pas des protéines de
surface ou secrétées, mais des protéines cytosoliques ou nucléaires. Il consiste en l’addition d’un
résidu monosaccharidique unique et non d’une chaîne oligosaccharidique. C’est un processus
dynamique et réversible qui varie en réponse à des stimuli physiologiques. Marshall et al.
présentent cette voie de signalisation des hexosamines comme un mécanisme gluco-sensible par
lequel le glucose pourrait agir directement sur le fonctionnement cellulaire (Marshall S., 2002).
Le processus dynamique et inductible de O-glycosylation est assuré par les activités régulées
d’enzymes cytosoliques et nucléaires : la O-GlcNAc transférase (OGT) et la O-GlcNAcase. La
première gère l’attachement du résidu O-GlcNAc sur les protéines et est régulée par O-
glycosylation elle-même et par phosphorylation. Son activité est inhibée par l’UDP, résidu de la
réaction de transfert et activée par l’UDP-GlcNAC dont le taux dépend lui-même notamment des
concentrations de glucose. La O-GlcNAcase qui catalyse l’enlèvement du résidu O-GlcNAc est
quant à elle une β-N-acétylglucosaminidase (Wells L. et al., 2001). Il a été montré que la délétion
de l’activité OGT est létale pour des cellules embryonnaires de souris, soulignant ainsi l’importance
de la O-glycosylation (Shafi R. et al., 2000). Cette modification se retrouve d’ailleurs dans toutes
les cellules eucaryotes (Comer F.I. et al., 2000).
La liste des protéines cytosoliques et nucléaires modifiées par O-glycosylation est toujours
croissante (Figure 15). Ces protéines sont généralement très importantes pour le fonctionnement
cellulaire contrôlant l’expression génique, la croissance ou la division cellulaire, les activités
59

enzymatiques ou l’intégrité structurale du cytosquelette. Les nucléoporines, qui gouvernent le
transport de macromolécules vers l’intérieur et l’extérieur du noyau, sont par exemple parmi les
premières protéines O-glycosylées structurellement caractérisées. Le rôle de cette modification
n’est pas encore clairement établi. Au niveau du noyau, l’ARN polymérase II est aussi connue pour
être modifiée par O-glycosylation. Cette modification pourrait induire des changements structuraux
altérant la conformation de l’enzyme et ainsi son interaction avec d’autres composants de la
machinerie de transcription. La O-glycosylation cible également un certain nombre de protéines du
cytosquelette et semble varier au cours du cycle cellulaire (Comer F.I. et al., 2000). Il a par ailleurs
été montré que la O-glycosylation joue un rôle dans le turnover et la transactivation de facteurs de
transcription comme par exemple Sp1. Ce facteur de transcription ubiquiste a été décrit pour être
fortement modifié par O-glycosylation (Jackson S.P. et al., 1988). Yang et al. ont rapporté que la O-
glycosylation de Sp1 inhibe son activité transcriptionnelle (Yang X. et al., 2001). Au contraire, Du
et al. ont montré que l’hyperglycémie induit une augmentation de la O-glycosylation de Sp1,
résultant en une augmentation de son activité promotrice (Du X.L. et al., 2000). De plus, il a été
montré que la O-glycosylation de cette protéine la protège contre la dégradation par le protéasome
(Han I. et al., 1997). La O-glycosylation des facteurs de transcription implique ainsi potentiellement
la voie des hexosamines dans la régulation de l’expression d’un grand nombre de gènes.
(Sp1, c-fos, AP1…)
(Src, c-myc, p53…)
(Tau, βamyloïde, cytokeratine, vinculine,
clathrine…)
(UDP-GlcNActransférase)
(Sp1, c-fos, AP1…)
(Src, c-myc, p53…)
(Tau, βamyloïde, cytokeratine, vinculine,
clathrine…)
(UDP-GlcNActransférase)
Figure 15: différentes protéines O-glycosylées connues dans la cellule d'après Comer et al. (Comer F.I. et al., 2000)
60

La O-glycosylation apparaît comme aussi répandue et dynamique que la phosphorylation et il
semble y avoir une certaine réciprocité entre les deux mécanismes. Griffith et al. ont par exemple
montré que l’inhibition de kinases augmente le taux de protéines O-glycosylées alors que leur
activation le diminue (Griffith L.S. et al., 1999). Sur plusieurs protéines, O-GlcNAc et O-phosphate
occupent les mêmes sites ou bien des sites adjacents et toutes les protéines O-glycosylées décrites
existent également à l’état phosphorylé. Si des exemples d’exclusion mutuelle de ces deux
modifications existent, elles pourraient tout de même se trouver en combinaison sur certaines
protéines et la régulation post-traductionnelle des protéines pourrait bien mettre en jeu une action
concertée de ces deux mécanismes (Comer F.I. et al., 2000).
Après la découverte de la modification par O-glycosylation d’un très grand nombre de protéines il y
a une vingtaine d’année, il est aujourd’hui important et prometteur d’élucider le rôle biologique de
la voie de signalisation des hexosamines dans les différentes cellules et tissus et de découvrir les
liens entre cette voie et certaines pathologies. Quelques éléments de réponse ont déjà été obtenus,
particulièrement en ce qui concerne le diabète.
5.3.Conséquences pathologiques
5.3.1 Insulino-résistance
L’insulino-résistance est le premier contexte pathologique dans lequel la voie des hexosamines a été
mise en cause (Marshall S. et al., 1991). Depuis, un nombre important de résultats a confirmé l’idée
que cette voie joue un rôle dans les mécanismes de l’insulino-résistance. Par exemple, la
surexpression de la GFAT dans le muscle et le tissu adipeux de souris a révélé que ces animaux
développent un hyperinsulinémie et une insulino-résistance (Hebert L.F., Jr. et al., 1996). Les
mêmes résultats ont été obtenus chez des souris surexprimant la GFAT au niveau des cellules β
pancréatiques (Tang J. et al., 2000). Il a également été décrit que le pancréas de rats rendus
diabétiques par l’injection de streptozotocine présente une augmentation des protéines O-
glycosylées associée à une perte des cellules β (Akimoto Y. et al., 2000). Enfin, la surexpression de
la GFAT dans des cellules β pancréatiques a été associée à une détérioration de la fonction de ces
cellules avec un défaut de sécrétion d’insuline stimulée par le glucose (Kaneto H. et al., 2001).
Vosseller et al. ont par ailleurs montré que l’augmentation de la O-glycosylation, par l’utilisation
d’un inhibiteur de O-GlcNAcase dans des adipocytes, cause une insulino-résistance, associée à une
augmentation de la modification par O-glycosylation des protéines impliquées dans la signalisation
de l’insuline comme IRS1 (Vosseller K. et al., 2002). Chen et al. ont également proposé un
mécanisme pour expliquer l’insulino-résistance induite par la voie des hexosamines mettant en jeu
61

la O-glycosylation d’une protéine impliquée dans la translocation dépendante de l’insuline de
GLUT4 (Chen G. et al., 2003).
5.3.2 Néphropathie diabétique
La voie des hexosamines a déjà été mise en cause à plusieurs reprises dans les altérations
caractéristiques de la néphropathie diabétique, et en particulier dans la surproduction de matrice
extracellulaire. Un certain nombre d’observations faites sur des cellules mésangiales permet de
proposer un mécanisme selon lequel la glucosamine induit une augmentation de la production de
protéines matricielles (Figure 16).
• Kolm-Litty et al. ont montré, dans des cellules mésangiales glomérulaires porcines, que la
glucosamine est capable de mimer, de manière plus efficace, les effets d’une forte concentration de
glucose : inhibition de prolifération cellulaire, induction du TGFβ, cytokine pro-sclérotique
importante dans la néphropathie diabétique, et augmentation consécutive de la production de
matrice. De plus, ces effets du glucose se sont révélés inhibés par l’utilisation d’azaserine, un
inhibiteur de GFAT ou d’oligonucléotides anti-sens dirigés contre cette enzyme. Enfin, ils ont
précisé que la glucosamine induit une augmentation du taux d’ARNm du TGFβ ainsi que de la
production de la protéine suggérant une régulation au niveau transcriptionnel (Kolm-Litty V. et al.,
1998). Weigert et al. ont confirmé l’implication de la voie des hexosamines et de la GFAT dans
l’induction du TGFβ en montrant qu’une surexpression de la GFAT dans des cellules mésangiales
induit une augmentation de l’expression du TGFβ et de la fibronectine (Weigert C. et al., 2003). De
plus, Singh et al. ont rapporté que les effets de la glucosamine et du TGFβ sur la surproduction de
protéines matricielles ne sont pas additifs et que l’utilisation d’anticorps dirigés contre le TGFβ
bloque la synthèse de fibronectine induite par la glucosamine (Singh L.P. et al., 2004).
Par ailleurs, plusieurs travaux de Singh et al. ont suggéré que cette augmentation de synthèse de
protéines matricielles par la voie des hexosamines implique les protéines kinases A et C. Ils ont tout
d’abord montré que la glucosamine et le TGFβ augmente la production de laminine et de
fibronectine ainsi que les activités PKC et PKA et que l’utilisation d’inhibiteurs de ces kinases
bloque l’augmentation de production de laminine induite par la glucosamine ou le TGFβ (Singh
L.P. et al., 2000; Singh L.P. et al., 2004). De plus, ils ont décrit que le glucose et la glucosamine
(Singh L.P. et al., 2001), ainsi que le TGFβ (Singh L.P. et al., 2004) augmentent la phosphorylation
et l’activité nucléaire du facteur de transcription CREB (cAMP Responsive Element Binding), qui
pourrait réguler le promoteur du gène de la fibronectine. L’ensemble de ces résultats a conduit
l’équipe à proposer que les effets du TGFβ sur l’augmentation de production de protéines
62
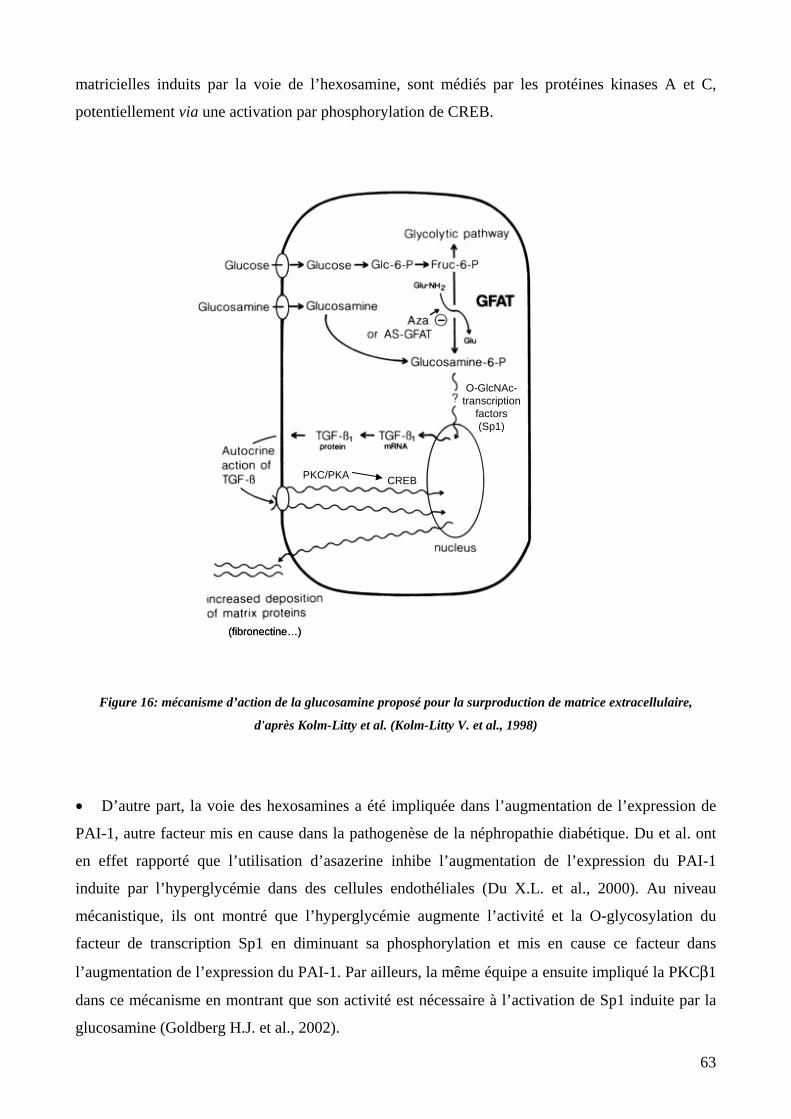
matricielles induits par la voie de l’hexosamine, sont médiés par les protéines kinases A et C,
potentiellement via une activation par phosphorylation de CREB.
PKC/PKA CREB
O-GlcNAc-transcription
factors (Sp1)
(fibronectine…)
PKC/PKA CREB
O-GlcNAc-transcription
factors (Sp1)
(fibronectine…)
Figure 16: mécanisme d’action de la glucosamine proposé pour la surproduction de matrice extracellulaire,
d'après Kolm-Litty et al. (Kolm-Litty V. et al., 1998)
• D’autre part, la voie des hexosamines a été impliquée dans l’augmentation de l’expression de
PAI-1, autre facteur mis en cause dans la pathogenèse de la néphropathie diabétique. Du et al. ont
en effet rapporté que l’utilisation d’asazerine inhibe l’augmentation de l’expression du PAI-1
induite par l’hyperglycémie dans des cellules endothéliales (Du X.L. et al., 2000). Au niveau
mécanistique, ils ont montré que l’hyperglycémie augmente l’activité et la O-glycosylation du
facteur de transcription Sp1 en diminuant sa phosphorylation et mis en cause ce facteur dans
l’augmentation de l’expression du PAI-1. Par ailleurs, la même équipe a ensuite impliqué la PKCβ1
dans ce mécanisme en montrant que son activité est nécessaire à l’activation de Sp1 induite par la
glucosamine (Goldberg H.J. et al., 2002).
63

6. LE STRESS OXYDANT Le stress oxydant est la conséquence d'un déséquilibre entre la production de radicaux libres et leur
destruction par des systèmes de défenses anti-oxydantes. Les radicaux libres peuvent engendrer des
dommages importants sur la structure et le métabolisme cellulaire en dégradant de nombreuses
cibles : protéines, lipides et acides nucléiques.
La production de radicaux libres est augmentée au cours du diabète. L'hyperglycémie peut induire
un stress oxydant consécutivement à différents mécanismes, précédemment décrits: fragmentation
oxydative des produits d'Amadori et interaction des AGE avec RAGE, auto-oxydation du glucose et
altération du potentiel redox par la voie des polyols. De plus, les capacités anti-oxydantes sont
réduites chez les diabétiques (Ceriello A. et al., 1997).
L'équipe de Brownlee a présenté le stress oxydant comme le facteur inducteur de toutes les
hypothèses biochimiques jusqu'à présent proposées pour expliquer le développement des
complications microvasculaires du diabète (Figure 17). Ils ont effet montré dans des cellules
endothéliales aortiques bovines, que la production d'anion superoxyde par la mitochondrie en
réponse à l'hyperglycémie activait la voie des hexosamines, via une inhibition de la glyceraldéhyde-
3-P deshydrogénase (Du X.L. et al., 2000). De plus, ils ont décrit que la normalisation de la
production de dérivés actifs de l'oxygène par différents agents permettait de prévenir l'activation de
la protéine kinase C, la formation des AGE, l'accumulation de sorbitol et l'activation de NF-κB
induites par l'hyperglycémie (Nishikawa T. et al., 2000). Cet anion superoxyde proviendrait d'une
augmentation du gradient de protons généré par la chaîne respiratoire mitochondriale suite à une
production accrue de cofacteur NADH résultant de l'oxydation du pyruvate par la glycolyse activée.
64
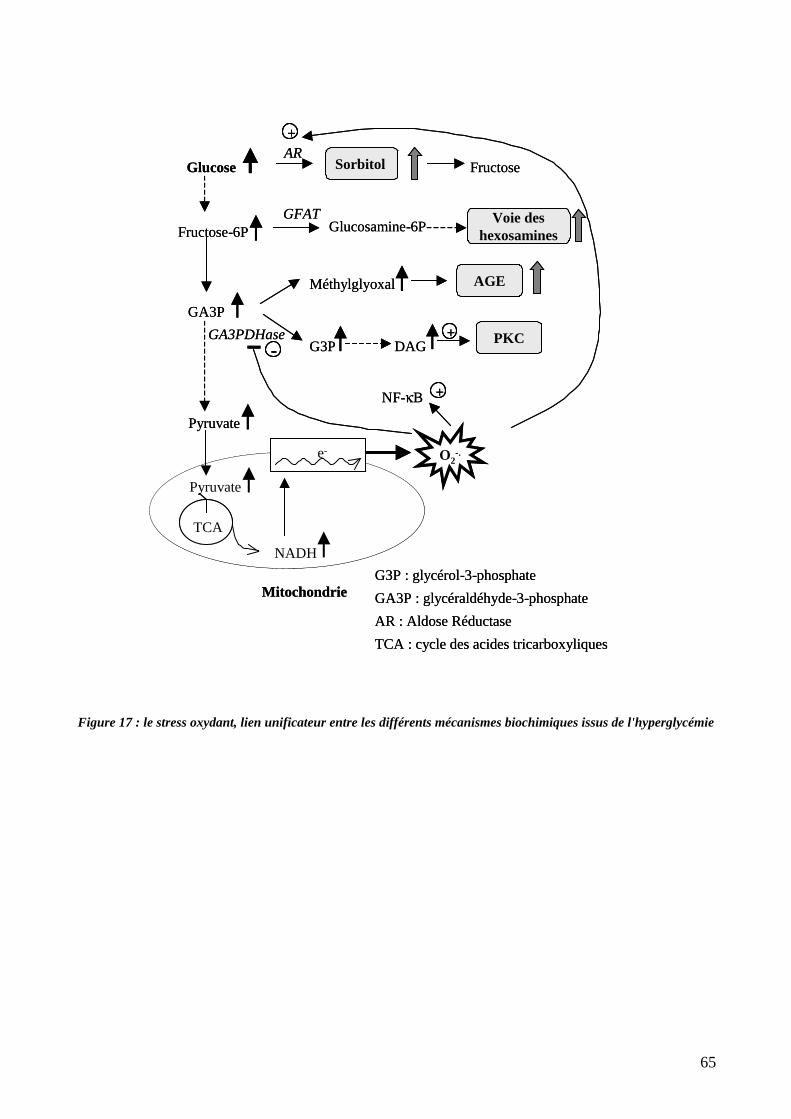
TCA
NADH
e-
Mitochondrie
O2-.
GA3PDHase
AGEMéthylglyoxal
Fructose Sorbitol AR
Pyruvate
Pyruvate
GA3P
Glucose
G3P : glycérol-3-phosphateGA3P : glycéraldéhyde-3-phosphateAR : Aldose RéductaseTCA : cycle des acides tricarboxyliques
PKCG3P DAG
Fructose-6PGFAT
Glucosamine-6P Voie des hexosamines
+
+
+NF-κB
-
TCA
NADH
e-
Mitochondrie
O2-.
GA3PDHase
AGEMéthylglyoxal
Fructose Sorbitol AR
Pyruvate
Pyruvate
GA3P
Glucose
G3P : glycérol-3-phosphateGA3P : glycéraldéhyde-3-phosphateAR : Aldose RéductaseTCA : cycle des acides tricarboxyliques
PKCG3P DAG
Fructose-6PGFAT
Glucosamine-6P Voie des hexosamines
++
++
++NF-κB
--
Figure 17 : le stress oxydant, lien unificateur entre les différents mécanismes biochimiques issus de l'hyperglycémie
65

LES GANGLIOSIDES
I. STRUCTURE ET LOCALISATION
1. STRUCTURE
1.1.Les glycosphingolipides
Les gangliosides appartiennent à une famille hétérogène de lipides appelée glycosphingolipides
(GSL), présents chez tous les eucaryotes, vertébrés et animaux inférieurs, ainsi que minoritairement
dans les plantes, mais rarement dans les bactéries. Les GSL sont des composés amphiphatiques de
la membrane plasmique faisant partie du glycocalix qui la recouvre. Ils sont constitués d'une
structure céramide hydrophobe ancrée dans la bicouche lipidique et d'une chaîne de sucres,
hydrophile, exposée au milieu extracellulaire (Figure 18). Le céramide se compose lui-même
d'un alcool aminé, la base sphingoïde, relié au niveau de son groupement amine à un acide gras
généralement à longue chaîne par une liaison amide. La chaîne de sucres est quant à elle liée par
une liaison glycosidique, au niveau de la fonction alcool primaire du céramide (Figure 18).
Les GSL présentent une énorme hétérogénéité de structure, tant dans la partie céramide que dans la
chaîne glycosidique.
66

Figure 18 : structure d'un glycosphingolipide et position dans la membrane plasmique
• Plus de 60 bases sphingoïdes et 300 chaînes oligosaccharidiques différentes ont été caractérisées
jusqu'à présent, offrant par combinaison des centaines de structures théoriquement possibles. La
base sphingoïde peut varier en longueur, saturation, hydroxylation mais la principale présente chez
les mammifères est la sphingosine (18:1), ou quelquefois ses dérivés comme la sphinganine. L'acide
gras peut également varier en longueur, saturation ou hydroxylation. Chez les mammifères, il est
généralement long (≥C16) et saturé. Les conséquences fonctionnelles de l'hétérogénéité du
céramide sont encore mal connues. Il semble toutefois que la partie lipidique puisse influencer la
localisation et les fonctions des GSL dans la membrane plasmique, probablement par interaction
directe avec le cholestérol, les phospholipides ou les domaines transmembranaires de protéines.
• La chaîne oligosaccharidique peut contenir de un à une dizaine de résidus et comprend des
hexoses (essentiellement du glucose, galactose, fucose), des N-acétylhexosamines (galactosamine,
glucosamine) et de l'acide sialique. Un groupement sulfate peut également y être additionné.
Partie lipidique = ceramide
O
OOH
OH
OOH
OHO
HO
OHO
OH
OHOH
NH
CH3OOH
O
OHN
OH
O
H3C
HO
H3C
Base sphingoïde
Acide gras
Partie oligosaccharidique
membrane plasmique
cytosol
milieu extracellulaire
glycosphingolipide
Partie lipidique = ceramide
O
OOH
OH
OOH
OHO
HO
OHO
OH
OHOH
NH
CH3OOH
O
OHN
OH
O
H3C
HO
H3CO
OOH
OH
OOH
OHO
HO
OHO
OH
OHOH
NH
CH3OOH
O
OHN
OH
O
H3C
HO
H3C
Base sphingoïde
Acide gras
Partie oligosaccharidique
membrane plasmique
cytosol
milieu extracellulaire
glycosphingolipide
membrane plasmique
cytosol
milieu extracellulaire
glycosphingolipide
67

Cependant, malgré un grand nombre de combinaisons possibles, seul un nombre assez restreint de
GSL différents sont effectivement retrouvés dans les organismes. Ils sont regroupés en séries,
définies par la composition et l'enchaînement de la chaîne oligosaccharidique qui fonde la
classification des GSL. On distingue:
- les GSL neutres dont la chaîne oligosaccharidique ne contient que des sucres neutres:
glucosylcéramide, galactosylcéramide, lactosylcéramide, céramide trihexoside et les séries
globo et lacto dont les structures de base sont respectivement GalNAcβ1,3-Galα1,4-Galβ1,4-
Glcβ1,1-Cer et Galβ1,3-GlcNAcβ1,3-Galβ1,4-Glcβ1,1-Cer.
- les sulfato-GSL comprenant notamment le lactosyl-céramide-sulfate qui semblerait d'ailleurs
représenter un facteur de risque potentiel pour le diabète de type I.
- les gangliosides qui contiennent un ou plusieurs acides sialiques chargés négativement dans
leur chaîne glycanniques et dont la structure de base est Galβ1,3-GalNAcβ1,4-Galβ1,4-
Glcβ1,1-Cer.
1.2.Les gangliosides
Les gangliosides sont des GSL dont le céramide est substitué par une chaîne glycannique contenant
de l'acide sialique ou acide neuraminique (Figure 19). Ce résidu est généralement sous la forme
d'acide N-acétylneuraminique (NeuAc) ou N-glycolylneuraminique (NeuGc) mais il existe environ
30 dérivés obtenus par exemple par O-acétylation de ces derniers. L'ensemble des acides
neuraminiques est regroupé sous le nom d'acide sialique.
Ces sialoglycosphingolipides sont dénommés par des abréviations correspondant au nombre de
résidus acides sialiques présents dans la molécule (GM pour monosialoganglioside, GD pour
disialoganglioside, GT pour trisialoganglioside…) et leur ordre de migration en chromatographie
(GM3, GM2, GM1…). Cette classification, appelée nomenclature de Svennerholm, est la plus
couramment utilisée et celle qui sera employée dans ce manuscrit, pour plus de simplicité.
Cependant, une nouvelle nomenclature se fondant sur la connaissance détaillée des séquences
glycanniques a été recommandée par la commission de nomenclature biochimique (IUPAC-IUB,
1977). L'abréviation Cer pour céramide est précédée du préfixe lac ou, si la chaîne est plus
complexe, ose indicé du nombre de résidus glucidiques présents et précédé des lettres Gg pour la
série ganglio. La position de l'acide sialique substitué latéralement est indiquée par un chiffre
romain et l'anomérie de liaison par α ou β. C'est ainsi que le GM3 devient II α-NeuAc-Lac-Cer et le
GM1 II α-NeuAc-GgOse4-Cer.
68

glucosegalactose
galactose N-acétylgalactosamine
acide
N-acétylneuraminique
glucosegalactose
galactose N-acétylgalactosamine
acide
N-acétylneuraminique
Figure 19 : structure d'un ganglioside
2. LOCALISATION
2.1.Localisation tissulaire
Les gangliosides sont largement exprimés dans les tissus des mammifères, selon un profil
spécifique de l'espèce, de l'organe ou du tissu considéré, de la cellule, ainsi que du stade de
développement de l'organe ou de différenciation de la cellule. Ils sont particulièrement abondants
dans les tissus nerveux et le cerveau où ils ont d'ailleurs été découverts dans les années 40 par
Klenk. On les retrouve cependant quasiment dans tous les tissus de l'organisme, principalement
sous forme de GM3 et de GD3, et notamment dans les cellules vasculaires, bien qu'en faible
quantité. Le profil varie selon la localisation des cellules mais le GM3 semble toutefois le
ganglioside prédominant dans les cellules vasculaires jusqu'à maintenant étudiées. Une dizaine de
gangliosides différents de source inconnue ont également été détectés dans le sérum, liés à des
lipoprotéines hépatiques (Senn H.J. et al., 1989).
2.2.Localisation cellulaire
Les gangliosides, et plus généralement les GSL, sont majoritairement présents dans le feuillet
externe de la membrane plasmique bien qu'ils ne constituent qu'une fraction minoritaire (5 %) des
lipides membranaires. De plus en plus d'études tendent à prouver l'existence de microdomaines
membranaires enrichis en GSL et gangliosides. Des observations microscopiques, l'utilisation de
molécules marquées ou encore d'anticorps spécifiques ont permis de montrer que les gangliosides se
regroupent dans des fragments particuliers de membrane plasmique, appelés radeaux lipidiques
(rafts). Ces microdomaines possèdent la particularité d'être insolubles dans le Triton X100 à basse
69

température (4°C), propriété exploitée pour leur extraction. Ils présentent une composition lipidique
et protéique bien particulières: riches en cholestérol, sphingomyéline, en protéine à pied d'ancrage
glycosyl-phosphatidylinositol (GPI) et en protéines impliquées dans la signalisation telles la famille
src tyrosine kinase. Les microdomaines pourraient jouer le rôle de plate-forme d’attachement pour
ces protéines. Au sein des radeaux lipidiques, les GSL et les gangliosides s'accumulent dans des
invaginations appelées cavéoles caractérisées par la présence d'une protéine spécifique, la
cavéoline, et contenant en particulier des protéines G, Ras (Figure 20). L'assemblage des
gangliosides et plus généralement des GSL dans ces microdomaines semble être à la base de leurs
fonctions. Les rafts sont particulièrement riches en molécules fonctionnelles impliquées dans la
transduction du signal et les gangliosides qui y sont associés peuvent interagir avec ces protéines
(récepteurs ou protéines kinases) pour moduler leur fonction. De plus, il a été montré que les GSL
peuvent affecter la formation et la fonction des microdomaines dans des cellules de carcinome de
poumon (Inokuchi J.I. et al., 2000) et que l'addition de GM1 à des cellules CHO-K1 modifie la
distribution des protéines à pied d'ancrage GPI dans la membrane plasmique (Crespo P.M. et al.,
2002). Les GSL contribuent aussi aux propriétés physico-chimiques des microdomaines et les
cavéoles sont d'ailleurs des lieux d'endocytose.
Cela étant, il semble qu'un certain nombre de GSL soient également présents dans le cytosol. Par
exemple, la fraction soluble du cerveau contiendrait environ 5 % des gangliosides totaux de
l'organe, de composition semblable à celle des gangliosides membranaires. Certains gangliosides
ont été retrouvés associés à la vimentine, un composant des filaments intracellulaires
intermédiaires, ce qui correspondrait peut-être à une voie de recyclage (Kotani M. et al., 1994).
D'autre part, les gangliosides sont également présents dans les membranes internes comme
l'appareil de Golgi, le réticulum endoplasmique, les endosomes et lysosomes, où ils sont
synthétisés, transportés ou dégradés. Enfin, il a été récemment montré que des gangliosides se
trouveraient dans des membranes associées aux mitochondries, ou MAM, un sous-compartiment du
réticulum endoplasmique étroitement associé à la mitochondrie (Ardail D. et al., 2003).
70
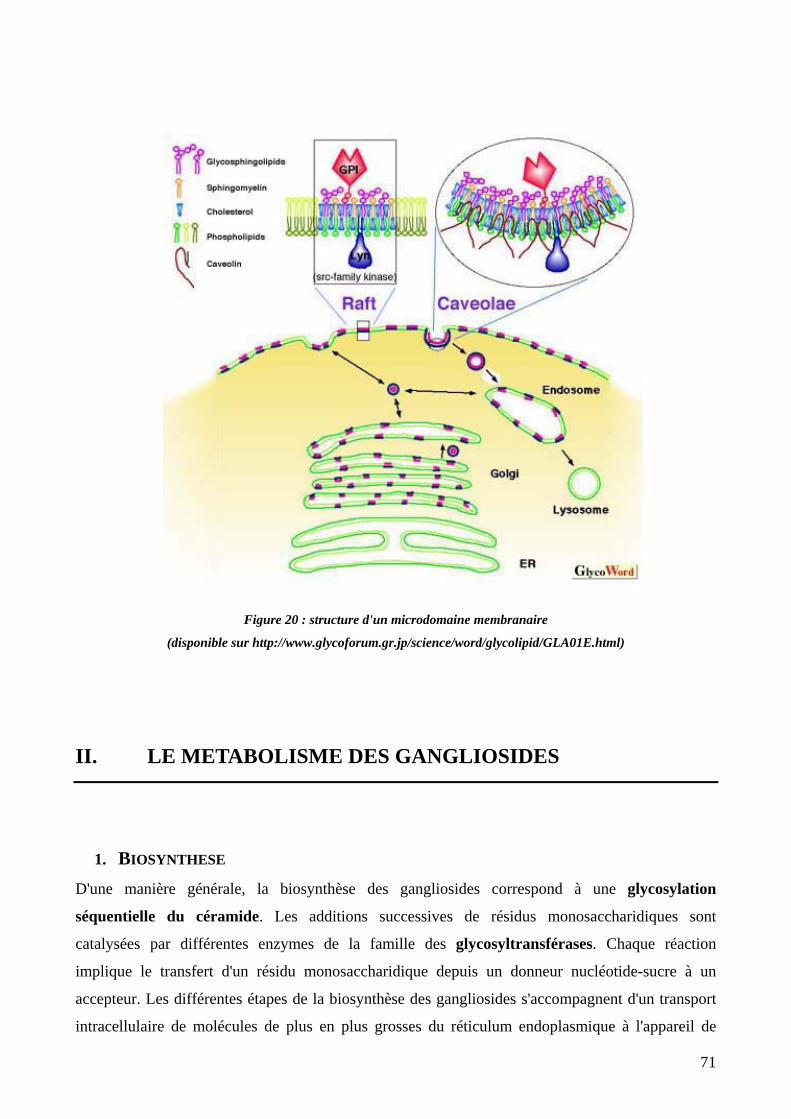
Figure 20 : structure d'un microdomaine membranaire
(disponible sur http://www.glycoforum.gr.jp/science/word/glycolipid/GLA01E.html)
II. LE METABOLISME DES GANGLIOSIDES
1. BIOSYNTHESE D'une manière générale, la biosynthèse des gangliosides correspond à une glycosylation
séquentielle du céramide. Les additions successives de résidus monosaccharidiques sont
catalysées par différentes enzymes de la famille des glycosyltransférases. Chaque réaction
implique le transfert d'un résidu monosaccharidique depuis un donneur nucléotide-sucre à un
accepteur. Les différentes étapes de la biosynthèse des gangliosides s'accompagnent d'un transport
intracellulaire de molécules de plus en plus grosses du réticulum endoplasmique à l'appareil de
71

Golgi puis à la membrane plasmique.
1.1.Les premières étapes: formation du précurseur commun, le lactosylcéramide
Le céramide formé dans le réticulum endoplasmique est d'abord transporté jusqu'à la face
cytosolique de l'appareil de Golgi par des voies assez mal connues mettant en jeu des vésicules
membranaires ou alternativement une protéine de transfert du céramide, récemment identifiée. La
glucosylcéramide synthase lui transfert alors un résidu glucose depuis l'UDP-glucose. Toutes les
autres enzymes de glycosylation de la biosynthèse des gangliosides se trouvant du côté luminal, le
glucosylcéramide ainsi formé est transloqué, probablement à l'aide d'une protéine de type flippase.
A ce niveau, une galactosyltransférase ajoute un résidu galactose sur le glucosylcéramide pour
former le lactosylcéramide, précurseur commun de tous les gangliosides, mis à part le GM4
synthétisé directement à partir du glucosylcéramide (Degroote S. et al., 2004).
1.2.La chaîne d'assemblage
Les résidus monosacharidiques suivants sont alors transférés à la chaîne glycannique grandissante
par étapes précisément ordonnées au niveau de la face luminale du Golgi. Le lactosylcéramide et
ses dérivés sialylés, GM3, GD3 et GT3, obtenus par l'action de sialyltransférases, servent de
précurseurs pour la biosynthèse des gangliosides complexes des séries 0, a, b, c (Figure 21). Ces
différentes séries sont caractérisées par la l'absence (série-0), ou la présence d'un (série-a), deux
(série-b) ou trois (série-c) acides sialiques liés sur le premier résidu galactose.
Les sialyltransférases I, II et III catalysant les premières étapes de la biosynthèse des gangliosides
(formation du GM3, GD3 et GT3) présentent une spécificité importante pour leur substrat. Les
quantités relatives des glycolipides précurseurs Laccer, GM3, GD3 et GT3 semblent déterminer en
partie les taux de gangliosides des séries 0, a, b et c qui en dérivent respectivement et uniquement.
En revanche, les glycosylations successives de ces précurseurs sont catalysées par des
glycosyltransférases à faible spécificité, la N-acétylgalactosamine transférase (GalNAcT) et la
galactosyltransférase II (GalT-II) ainsi que les deux sialyltransférases ST-IV et ST-V. Elles
transfèrent un résidu saccharidique sur des accepteurs qui diffèrent uniquement par le nombre
d'acides sialiques liés au galactose, c'est à dire par leur série. Des données récentes suggèrent que
les précurseurs sont formés dans le Golgi proximal et distal alors que la synthèse des gangliosides
plus complexes se limiterait au Golgi distal (Kolter T. et al., 2002). Roseman a le premier proposé
que la biosynthèse des chaînes oligosaccharidiques pourrait être catalysée par des systèmes de
multiglycosyltransférases (Roseman S., 1970). L’existence de ces complexes fonctionnels a été
confirmée par la co-immunoprécipitation de GalNAcT et GalT-II, formant un complexe qui accepte
GM3 comme substrat pour libérer directement GM1 (Giraudo C.G. et al., 2001). Ceci pourrait
72

notamment expliquer pourquoi le cerveau, par exemple, contient de grandes quantités de GM1 et
GD1a mais peu de GM2.
Une fois synthétisés, les gangliosides gagnent leur destination principale, la membrane plasmique,
vraisemblablement via un transport vésiculaire.
Les glycosyltransférases présentes et actives dans l'appareil de Golgi varient en fonction du type
cellulaire, du stade de développement, ou d'un état pathologique (Degroote S. et al., 2004). Cet
équipement enzymatique conditionne la composition en gangliosides de la cellule. Les GM3, GD3
et GT3 synthases revêtent une importance particulière car elles gouvernent respectivement l’entrée
dans la voie de biosynthèse des séries-a, -b, et -c.
Figure 21 : schéma de biosynthèse des gangliosides,
d'après Van Echten G. (Van Echten G. et al, 1993)
2. CATABOLISME Avant d'être dégradés, les gangliosides présents dans la membrane plasmique sont tout d'abord
internalisés par endocytose et acheminés par des vésicules "coatées" jusqu'aux endosomes (Figure
22). De là, une partie peut être réinsérée dans la membrane plasmique ou rejoindre l'appareil de
Golgi mais la majeure partie est acheminée vers les lysosomes, lieu de leur dégradation.
SialyltransferaseIV
SialyltransferaseV
Cer
Cer CerGM3
(Lactosylceramide
CDH)
Glucosylceramide(CMH)
Cer
GM2
Cer
GM1
Cer
GD1a
Cer
GD3
Cer
GD2
CerGD1b
Cer
GT1b
Cer
GT3
Cer
GT2
CerGT1c
Cer
GQ1c
Sialyltransferase I Sialyltransferase II Sialyltransferase IIIGalNAc-transferase
Galactosyl-transferase II
CerGA2
CerGA1
Cer
GM1b
Cer Cer Cer Cer
GT1a GQ1b GP1cGD1
Série-a Série-b Série-cSérie asialo (série-0)
Galactosyltransferase I
CerCeramide
Glucosylceramide synthase Galactosylceramide
Cer
Cer
GM4
Sulfatide
glucose
galactose
acide sialique
N-acetylgalactosamine
sulfate
Cer
Ceramidetrihexoside
Cer
GlobosideLactoside
Cer
SialyltransferaseIV
SialyltransferaseV
CerCer
CerCerCer CerCerCerCerGM3
(Lactosylceramide
CDH)(Lactosylceramide
CDHLactosylceramide
CDH)
Glucosylceramide(CMH)
Glucosylceramide(CMH)
CerCer
GM2
Cer
GM1
CerCer
GD1a
CerCerCerCer
GD3
CerCer
GD2
CerCerGD1b
CerCer
GT1b
CerCerCerCer
GT3
CerCer
GT2
CerCerGT1c
CerCer
GQ1c
Sialyltransferase I Sialyltransferase II Sialyltransferase IIIGalNAc-transferase
Galactosyl-transferase II
CerCerGA2
CerCerGA1
CerCer
GM1b
Cer Cer CerCer CerCer
GT1a GQ1b GP1cGD1
Série-a Série-b Série-cSérie asialo (série-0)
Galactosyltransferase I
CerCerCeramide
Glucosylceramide synthase Galactosylceramide
CerCer
Cer
GM4CerCer
GM4
Sulfatide
glucose
galactose
acide sialique
N-acetylgalactosamine
sulfate
glucoseglucose
galactosegalactose
acide sialiqueacide sialique
N-acetylgalactosamineN-acetylgalactosamine
sulfate
CerCer
Ceramidetrihexoside
CerCerCer
GlobosideLactoside
CerCer
73

Les gangliosides y sont dégradés par l'action conjuguée de différentes enzymes. Des
exoglycosidases spécifiques enlèvent séquentiellement les résidus monosaccharidiques. L’action de
ces hydrolases est facilitée, pour les gangliosides à chaîne de sucres courte, par la présence de petits
cofacteurs protéiques appelés SAP (Sphingolipid Activator Proteins), comme par exemple le
"GM2-activator", qui dégagent les gangliosides de la membrane des lysosomes pour faciliter leur
accessibilité aux enzymes (van Echten G. et al., 1993). La chaîne de sucres entière peut également
être détachée par une céramide glycanase. Enfin, l’acide gras du céramide peut être enlevé par
l’action d’une céramide-N-désacylase.
Par ailleurs, des sialidases catalysent le détachement de l’acide sialique des gangliosides et des
glycoprotéines. Trois sialidases ont été identifiées chez les mammifères, différant par leurs
propriétés enzymatiques et leur localisation subcellulaire : lysosomale (Neu1), cytosolique (Neu 2)
ou membranaire (Neu3). Cette dernière, localisée dans la membrane plasmique où les taux d’autres
glycosydases sont très faibles, est spécifique des gangliosides. Elle pourrait jouer un rôle important
dans la signalisation membranaire en modulant le taux des gangliosides de la membrane plasmique
(Sasaki A. et al., 2003).
Les gangliosides présents dans la membrane plasmique sont régulièrement dégradés par la cellule.
Les études concernant leur taux de renouvellement donnent des demi-vies allant de quelques jours à
plusieurs semaines, suivant le système ou la méthode utilisés.
Lysosome
Endosome
Appareil de Golgi
Coated pit
Coated vesicleUncoated vesicleMembrane plasmique
Lysosome
Endosome
Appareil de Golgi
Coated pit
Coated vesicleUncoated vesicleMembrane plasmique
Lysosome
Endosome
Appareil de Golgi
Coated pit
Coated vesicleUncoated vesicleMembrane plasmique
Figure 22 : catabolisme des gangliosides
74

3. REGULATION DU METABOLISME L'équilibre entre biosynthèse et catabolisme des gangliosides est précisément contrôlé dans la
cellule et les déséquilibres ont des conséquences fortes, dont certaines sont bien identifiées.
Lorsqu'un défaut dans les processus de dégradation survient, suite à une déficience génétique
affectant une glycosidase ou une protéine activatrice, le ganglioside substrat de cette enzyme
s'accumule et est stocké dans les lysosomes. Ce déséquilibre ou gangliosidose, occasionne des
pathologies héréditaires neurodégénératives sérieuses. Les maladies de Tay-Sachs et Sandhoff sont
par exemple deux formes d'une déficience de l'enzyme β-hexosaminidase conduisant à une
accumulation de GM2.
A l'inverse, une activité diminuée des enzymes de biosynthèse est probablement à l'origine des
chaînes glycanniques incomplètes observées dans certaines cellules transformées. En effet, il a été
montré que des cellules transformées par des virus oncogènes ou par irradiation sont carencées en
gangliosides complexes à cause d'un diminution d'activité de glycosyltransférase spécifique
(Fishman P.H. et al., 1976).
La régulation des enzymes de biosynthèse des gangliosides est aussi particulièrement importante au
cours du développement. Des changements majeurs dans les quantités et le profil en gangliosides du
cerveau ont été observés au cours du développement, correspondant à des changements dans les
activités des glycosyltransférases. L’incapacité à synthétiser les gangliosides durant cette période
critique résulterait en des troubles neurologiques sévères (Fishman P.H. et al., 1976).
L’expression différentielle des gangliosides dans les tissus et les changements observés durant le
développement ou les pathologies comme l’oncogenèse apparaît donc comme un point clé de la
biologie des gangliosides. Il est donc particulièrement important de comprendre les mécanismes de
la régulation des enzymes du métabolisme des gangliosides, qui restent pourtant encore assez mal
connus.
3.1.Equipement enzymatique et disponibilité des substrats
Les premiers éléments qui gouvernent le métabolisme des gangliosides tiennent aux caractéristiques
même des enzymes : taux, spécificité, paramètres cinétiques comme le Km et organisation des
enzymes dans le Golgi. Un certain nombre de glycosyltransférases partageant les mêmes substrats,
une compétition s’opère. La disponibilité en substrats joue donc un rôle important dans la
détermination du profil en gangliosides. Il a par exemple été montré que la transformation de lignée
cellulaire de mélanome exprimant GM3 et GD3 avec l’ADNc de la GM2/GD2 synthase convertit
les cellules en transformants exprimant essentiellement du GM2. Les taux élevés de GM2/GD2
synthase ont dirigé dans ce modèle l’utilisation du GM3 vers la biosynthèse des gangliosides de la
série-a (Lloyd K.O. et al., 1998). L'expression et l'activité relatives des différentes
75

glycosyltransférases gouvernent le profil en gangliosides des différents types cellulaires.
3.2.Régulation au niveau transcriptionnel
La régulation du métabolisme des gangliosides se joue probablement principalement à ce niveau.
Des expériences ont révélé une forte corrélation entre la quantité des GSL, l’activité des
glycosyltransférases et leur expression (Ruan S. et al., 1999; Watanabe R. et al., 1998). Le clonage
assez récent de la plupart des glycosyltransférases a permis de fournir des informations sur la
régulation de l’activité des enzymes au niveau transcriptionnel. Furukawa at al. ont par exemple
montré que le gène codant pour la GM2/GD2 synthase humaine présente 3 sites d’initiation de la
transcription avec des séquences consensus pour la liaison des facteurs de transcription EGR-1,
HNE-5 et Sp1 (Furukawa K. et al., 1996). De même, le fragment 5’ de la GM3 synthase humaine a
été caractérisé. Huit facteurs de transcription pouvant se lier à la région proximale ou être recrutés
comme coactivateurs ont été identifiés, parmi lesquels CREB, PPAR ou Stat 6 (Zeng G. et al.,
2003). Enfin, le promoteur de la GD3 synthase de rat a également été analysé, révélant des sites
consensus pour la liaison de facteurs de transcription comme Sp1, AP-1, NFkB, C/EBP et TFIID
(Zeng G. et al., 1998). Cette régulation par des promoteurs pourrait être responsable de la spécificité
cellulaire de l'expression des gangliosides mais peu de preuves directes ont été apportées pour le
moment. Très récemment, il a été décrit dans des cellules HL60, que l'expression de la GM3
synthase est positivement régulée par le facteur de transcription CREB activé par la voie de
signalisation PKC/ERKs (Chung T.W. et al., 2004). Une étude de Tagami et al. a par ailleurs
montré que l'activité GM3 synthase et le taux d'ARNm de la protéine étaient augmentés dans des
adipocytes rendus insulino-résistant par le TNFα. Les auteurs suggèrent que la transcription du
gène de la GM3 synthase pourrait être activée par la signalisation du TNFα via le récepteur au
TNF1 (Tagami S. et al., 2002).
3.3.Mécanismes de régulation épigénétiques
En dehors de la régulation au niveau génétique, quelques observations suggèrent l'existence de
différents mécanismes régulateurs des enzymes du métabolisme des gangliosides.
3.3.1 Au niveau post-transcriptionnel
• Un certain nombre de travaux indiquent que l'activité des glycosyltransférases pourrait être
régulée par des mécanismes de phosphorylation / déphosphorylation. Par exemple, Scheideler et
Dawson ont montré qu'une inhibition de la synthèse d'AMPc par un agent pharmacologique
diminue l'activité GM2 synthase alors qu'une accumulation intracellulaire d'AMPc produit l'effet
inverse. De plus, ils ont révélé que l'ajout d'AMPc à des microsomes de cerveau de rat induit une
76

phosphorylation de la GM2 synthase et double consécutivement son activité, révélant ainsi un rôle
direct de l'AMPc et de la phosphorylation dans les changements d'activité de l'enzyme (Scheideler
M.A. et al., 1986). D'autre part, Gu et al. ont montré, in vitro, que la protéine kinase C inhibe
l'activité de préparations purifiées de sialyltransférases I et IV de rat, en phosphorylant
respectivement les résidus thréonine et serine. Ils ont également montré que cette diminution
d'activité peut être reversée en traitant les sialytransférases phosphorylées par une phosphatase,
renforçant ainsi, par sa réversibilité, l'importance de ce mécanisme de phosphorylation /
déphosphorylation comme régulateur de l'activité de l'enzyme (Gu X. et al., 1995). Enfin, la même
équipe rapporte que les changements dans la composition en gangliosides des cellules NG108-15,
observés lors de leur différenciation, peuvent être induits par les systèmes de phosphorylation liés
aux protéines kinases C et A. Ainsi, la phosphorylation de la GalNAcT augmenterait son activité
tandis que celle de la ST-IV la diminuerait (Bieberich E. et al., 1998). Bien que tous ces travaux
suggèrent l'existence de la phosphorylation comme facteur de régulation de l'activité des
glycosyltransférases, cette phosphorylation et sa signification en terme d'activité de l'enzyme n'ont
pas encore été directement montrés in situ.
• Par ailleurs, un autre type de modification covalente des glycosyltransférases, la N-
glycosylation, a été mise en cause dans la régulation de leur activité. Dans un premier temps,
l'analyse de l'ADNc cloné de glycosyltransférases comme la GD3 ou la GM2 synthase a permis de
prédire la présence de plusieurs sites potentiels pour la N-glycosylation. La signification de ces sites
a été clarifiée par différents travaux. Martina et al. ont montré, en utilisant une glycosidase sur des
cellules COS7 transfectées avec la GD3 synthase de poulet, que la forme déglycosylée de l’enzyme
était moins stable que la forme native. De plus, ils ont montré qu’un traitement avec un inhibiteur
de N-glycosylation bloque l’activité de l’enzyme et provoque sa rétention au niveau du reticulum
endoplasmique (Martina J.A. et al., 1998). L’importance de la N-glycosylation dans le trafic et la
localisation subcellulaire des glycosyltransférases a été confirmée par les travaux de Bieberich et al.
qui ont montré que la glycosylation augmente le "turnover" et perturbe le transport de la GD3
synthase depuis le reticulum endoplasmique jusqu’à l’appareil de Golgi (Bieberich E. et al., 2000).
En revanche, les travaux d’Haraguchi et al. ont révélé que la déglycosylation de la GM2 synthase
diminue son activité sans affecter sa localisation subcellulaire (Haraguchi M. et al., 1995). Il est
difficile de statuer sur la signification biologique de la N-glycosylation des glycosyltransférases du
fait de la variabilité des observations faites en fonction du type cellulaire.
3.3.2 Rétrocontrôle négatif
Un rétrocontrôle négatif de la biosynthèse des gangliosides par les gangliosides eux-même a été
observé in vitro. Ainsi, la GM2 synthase est inhibée par le GM3 et plus efficacement par les
77

gangliosides avec 2 ou 3 acides sialiques dans leur structure (Nores G.A. et al., 1984). Cette
observation a ensuite été confirmée par Yusuf et al. qui a montré que la GM2 synthase est
préférentiellement inhibée par le GD1a puis, moins fortement, par le GM1 et le GM2, mais
également par le GT1b. De même, l’activité GD3 synthase est inhibée le plus efficacement par le
GQ1b puis par le GT1b mais aussi par le GD1a (Yusuf H.K. et al., 1987). Ces résultats mettent en
évidence l’existence d’un rétrocontrôle négatif de la biosynthèse des gangliosides de la série-a et de
la série-b, préférentiellement par les produits finaux respectifs de la série mais qui ne semble pas
être totalement spécifique. Bien qu’une telle régulation n’ait jamais été mise en évidence in vivo,
elle paraît fort probable du fait de la co-localisation de nombreuses étapes de la biosynthèse des
gangliosides dans un même compartiment cellulaire.
3.3.3 Le pH
Le pH de l’environnement des enzymes pourrait également jouer un rôle dans la régulation de leur
activité. Un "shift" réversible de la série-a vers la série-b a été observé dans des cellules neuronales
murines lorsque le pH est abaissé de 7.4 à 6.2 (Iber H. et al., 1990). Cette observation pourrait
s’expliquer par la différence de pH optimum des premières enzymes catalysant la biosynthèse des
gangliosides de la série-a (GalNAcT) et de la série-b (ST-II).
Il semble donc que la composition en gangliosides des différents tissus soit le résultat d’une
expression sélective et d’une régulation active des enzymes spécifiques responsables de leur
métabolisme.
III. FONCTIONS
Le rôle fonctionnel des gangliosides se clarifie progressivement par le recours à différentes
approches in vitro : addition de gangliosides exogènes, blocage de la biosynthèse des gangliosides
par des inhibiteurs spécifiques ou des moyens biomoléculaires, utilisation de mutants déficients en
certains gangliosides ou au contraire introduction de nouveaux gangliosides dans les cellules par
transfection de l’ADNc de leur enzyme de biosynthèse. Plus récemment, le clonage de nombreuses
glycosyltransférases et le développement de techniques consistant à invalider des gènes précis dans
des organismes entiers (animaux KO) a permis d’obtenir des informations sur le rôle des
gangliosides in vivo. L'ensemble des observations faites jusqu'à présent a révélé que les
78

gangliosides exercent une double fonction : la reconnaissance cellulaire et la modulation du
signal transmembranaire. Ces deux fonctions leur confèrent un rôle majeur dans la prolifération,
la différenciation et la migration cellulaire et ainsi dans le développement et l’organisation des
tissus ou dans les phénomènes d’oncogénèse.
1. DOUBLE FONCTION DES GANGLIOSIDES
1.1.Reconnaissance cellulaire
Par leur localisation à la surface de la membrane plasmique et leur distribution ubiquiste et
spécifique, les gangliosides sont bien placés pour intervenir dans les phénomènes de reconnaissance
cellulaire. Ils possèdent des propriétés antigéniques et sont impliqués à la fois dans la
reconnaissance cellule-cellule et cellule-matrice par l’intermédiaire de différents types
d’interaction : avec des glycoprotéines, des récepteurs aux intégrines ou avec d’autres GSL. D’autre
part, les gangliosides ont été depuis longtemps décrits comme récepteurs pour différents virus,
bactéries ou toxines (Figure 23).
79
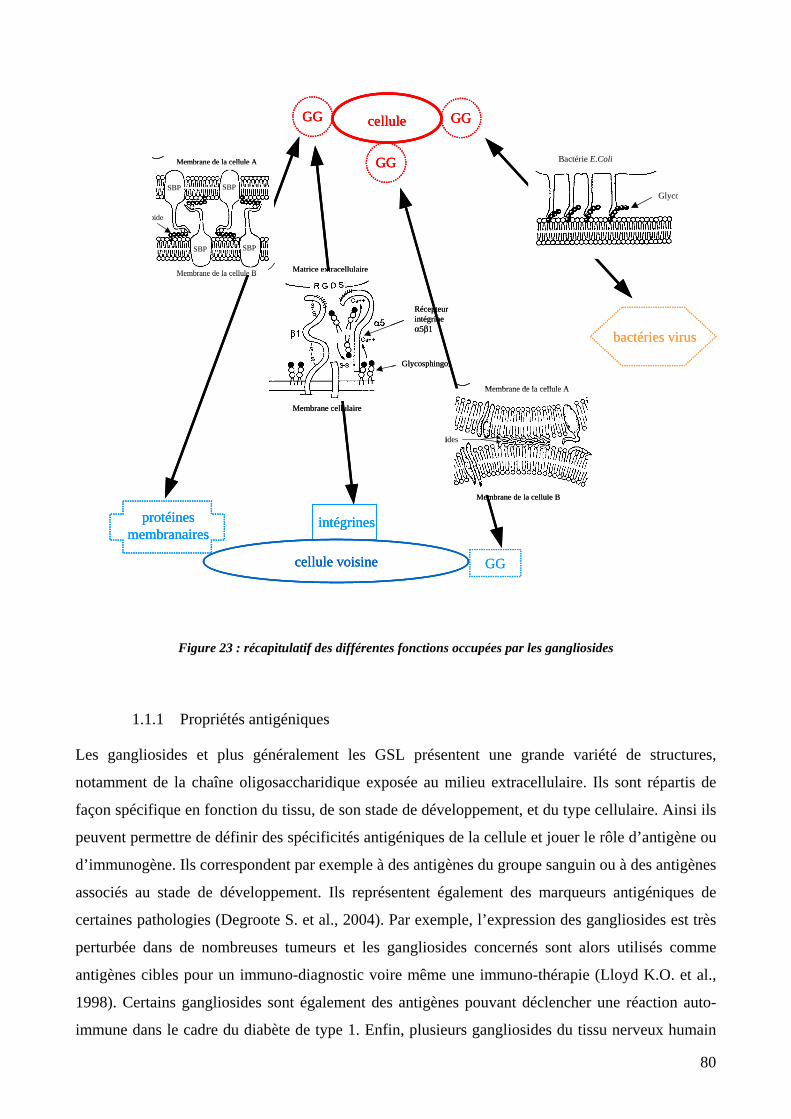
Figure 23 : récapitulatif des différentes fonctions occupées par les gangliosides
1.1.1 Propriétés antigéniques
Les gangliosides et plus généralement les GSL présentent une grande variété de structures,
notamment de la chaîne oligosaccharidique exposée au milieu extracellulaire. Ils sont répartis de
façon spécifique en fonction du tissu, de son stade de développement, et du type cellulaire. Ainsi ils
peuvent permettre de définir des spécificités antigéniques de la cellule et jouer le rôle d’antigène ou
d’immunogène. Ils correspondent par exemple à des antigènes du groupe sanguin ou à des antigènes
associés au stade de développement. Ils représentent également des marqueurs antigéniques de
certaines pathologies (Degroote S. et al., 2004). Par exemple, l’expression des gangliosides est très
perturbée dans de nombreuses tumeurs et les gangliosides concernés sont alors utilisés comme
antigènes cibles pour un immuno-diagnostic voire même une immuno-thérapie (Lloyd K.O. et al.,
1998). Certains gangliosides sont également des antigènes pouvant déclencher une réaction auto-
immune dans le cadre du diabète de type 1. Enfin, plusieurs gangliosides du tissu nerveux humain
celluleGG
bactéries virus
GG
GG Bactérie E.Coli
Glyco
ides
Membrane de la cellule A
Membrane de la cellule B
SBP SBP
SBP SBP
pide
Membrane de la cellule A
Membrane de la cellule B
protéines membranaires
GG
intégrines
Glycosphingol
Membrane cellulaire
Matrice extracellulaire
Récepteurintégrineα5β1
cellule voisine
cellulecelluleGGGG
bactéries virus
GGGG
GGGG Bactérie E.Coli
Glyco
ides
Membrane de la cellule A
Membrane de la cellule B
SBP SBP
SBP SBP
pide
Membrane de la cellule A
Membrane de la cellule B
protéines membranaires
protéines membranaires
GG
intégrinesintégrines
Glycosphingol
Membrane cellulaire
Matrice extracellulaire
Récepteurintégrineα5β1
cellule voisinecellule voisine
80

ont été décrits comme des antigènes dans des neuropathies auto-immunes (Yoshino H. et al., 1993).
1.1.2 Reconnaissance par des protéines de liaison aux hydrates de carbone
Quelques travaux suggèrent l'existence de molécules membranaires se liant aux hydrates de carbone
(SBP, sugar binding protein). Différents types de SBP, plus ou moins bien identifiées, ont été
décrites pour se lier aux gangliosides. Schnaar et al. ont par exemple montré qu'une molécule
présente dans la membrane de cerveau de rat, essentielle dans le développement du système
nerveux central, se lie spécifiquement au GT1b (Tiemeyer M. et al., 1989). La même équipe a
également mis en évidence la liaison d'une lectine (MAG, myelin associated glycoprotein) médiant
certaines interactions entre la myéline et les cellules neuronales, avec les gangliosides (Schnaar
R.L. et al., 1998). De la même manière, Hattori et al. ont révélé la présence à la surface de
"granulosa cell" immatures, d'une protéine servant de site de liaison au GM3, en soulignant le rôle
potentiel de cette interaction dans la différenciation des cellules (Hattori M. et al., 1995). Une sialo-
adhésine présente sur les macrophages et impliquée dans les interactions cellulaires au niveau des
tissus hématopoïétiques a également été décrite pour reconnaître spécifiquement une séquence
oligosaccharidique contenant de l'acide sialique dans les gangliosides ou bien les glycoprotéines
(Crocker P.R. et al., 1991). L'interaction entre le GM1 et la galectine 1 a par ailleurs été décrite dans
une lignée cellulaire de neuroblastome, en suggérant l'importance de cette interaction dans la
différentiation et l'inhibition de contact de ces cellules (Kopitz J. et al., 1998).
1.1.3 Modulation des récepteurs intégrines
Les gangliosides ont souvent été impliqués dans les interactions entre cellules et matrice
extracellulaire soit par liaison directe avec des protéines matricielles soit par interaction avec les
récepteurs intégrines. Il a été suggéré que les gangliosides puissent jouer un rôle de récepteurs pour
la fibronectine, glycoprotéine médiant une grande variété de phénomènes d'adhésion cellulaire, et
ce d'autant plus efficacement que le nombre d'acides sialiques contenu dans le ganglioside est élevé
(Yamada K.M. et al., 1983). Le GD3 a aussi été décrit pour interagir avec le collagène ou la
laminine (Nakano J. et al., 1999). Les travaux de Wang et al. ont montré que le GT1b interagit
directement avec l'intégrine α5β1, prévenant ainsi son interaction avec la fibronectine et inhibant
l'attachement et la migration de keratinocytes sur une matrice fibronectine. De plus, ils ont suggéré
que l'interaction entre le GT1b et α5β1 inhibe la voie de signalisation intégrine-kinase liée aux
intégrines – PKB – Akt (Wang X.Q. et al., 2001b). Au contraire, Zheng et al. ont décrit que la forte
adhésion à la fibronectine d'une lignée cellulaire de carcinome mammaire murin est due à un
support fonctionnel du GM3 sur le récepteur intégrine α5β1 (Zheng M. et al., 1993).
81

1.1.4 Interaction entre gangliosides et GSL
Alternativement à une interaction entre les gangliosides et des protéines, Hakomori et al. ont
proposé la possibilité d'une interaction entre un GSL et un autre GSL, complémentaire, exprimé à la
surface d'une cellule voisine, par l'intermédiaire de leur chaîne hydrate de carbone. L'utilisation de
liposomes contenant du GM3 a permis de montrer une interaction avec une phase solide "coatée"
avec le globoside Gb4 ou le lactosylcéramide, notamment, qui présenteraient des structures
complémentaires au GM3 (Kojima N. et al., 1991). De plus, une interaction a été découverte entre
le GM3 fortement exprimé à la surface de cellules de mélanomes murin et le lactosylcéramide
exprimé à la surface des cellules endothéliales, soulignant ainsi le rôle potentiel de cette interaction
dans les métastases de mélanomes (Hakomori S. et al., 1995). Ces interactions gangliosides-GSL,
rapides mais moins solides que les liaisons gangliosides-protéines, semblent intervenir dans des
phénomènes de reconnaissance cellulaire mais aussi d'adhésion ou de migration indépendants des
systèmes fibronectine/intégrines ou lectines.
1.1.5 Interaction avec des virus, bactéries ou toxines
Les gangliosides étant capables d'interagir avec des sucres ou des protéines, ils ont souvent été
décrits comme les cibles de divers pathogènes qui les utilisent en tant que site d'attachement. Les
gangliosides ont par exemple été décrits comme récepteurs pour le virus de la grippe (Suzuki Y.,
1994). Le GM1 serait de plus capable de lier une glycoprotéine de l'enveloppe du HIV (McAlarney
T. et al., 1994) alors que plusieurs souches de rotavirus bovins seraient capables de se lier aux GM2
et GD1a (Delorme C. et al., 2001). Les microorganismes pourraient posséder des lectines
spécifiques capables de reconnaître et de lier certains résidus des gangliosides membranaires de la
cellule hôte. Il semble d'ailleurs que la reconnaissance soit dirigée vers une portion interne plutôt
que terminale de la chaîne oligosaccharidique voire même vers la portion céramide. Les
gangliosides sont également utilisés comme des récepteurs par différentes toxines, dont la plus
connue est la toxine cholérique, enterotoxine produite par Vibrio cholerae, interagissant
spécifiquement avec le GM1. Cette propriété est d’ailleurs souvent exploitée pour l’identification
du GM1. Le mécanisme de cette interaction est bien connu et représenté en Figure 24. La liaison de
la sous-unité B de la toxine cholérique au GM1 induit un changement de conformation, une
dissociation de la molécule et l'entrée de la sous-unité A dans la membrane cellulaire. Il s'ensuit
l'activation de l'adénylate cyclase et la production d'AMPc qui pourrait médier les effets biologiques
de la toxine cholérique (Fishman P.H. et al., 1976). D'autres toxines bactériennes peuvent
également se lier aux gangliosides: la toxine tétanique (GD1b) (VAN HEYNINGEN W.E. et al.,
1961), la toxine botulinique (GT1b et GQ1b) (Simpson L.L. et al., 1971) ou la δ toxine produite par
Clostridium perfringens (GM2).
82

Figure 24 : mécanisme de l'interaction du GM1 avec la toxine cholérique
(disponible sur http://www.glycoforum.gr.jp/science/word/glycolipid/GLB02E.html)
1.2.Modulation du signal transmembranaire
Les gangliosides sont connus pour moduler la transduction du signal, en interagissant avec des
protéines membranaires impliquées dans des cascades de signalisation. Deux systèmes majeurs de
signalisation transmembranaire en action dans la majorité des cellules eucaryotes ont été décrits
pour être modulés par les gangliosides, ou leurs produits de dégradation : les protéines kinases
associées à des récepteurs aux facteurs de croissance et la PKC.
1.2.1 Régulation des récepteurs à tyrosine kinase
Ce système de signalisation est de loin le plus étudié en terme de régulation par les gangliosides.
Depuis la première association découverte entre une inhibition de la prolifération cellulaire en
présence de gangliosides et une action au niveau du récepteur à l’EGF, presque sinon tous les
récepteurs de facteurs de croissance ont été décrits pour être modulés par les gangliosides. La
Figure 25 récapitule de manière non exhaustive les effets des gangliosides sur les principaux
récepteurs de facteur de croissance étudiés dans différents types cellulaires.
83

EGFR IR
Figure 25 : modulation de l'activité de différents récepteurs aux facteurs de croissance par les gangliosides
• L'inhibition de l’activité du récepteur à l’EGF (EGFR) par les gangliosides a souvent été décrite.
Bremer et al. ont par exemple montré que le GM3 et le GM1 sont capables d’inhiber la
phosphorylation de l’EGFR stimulée par l’EGF et ainsi de diminuer la croissance de lignées
cellulaires KB et A431 (Bremer E.G. et al., 1986). De même, Mirkin et al. ont révélé que le
traitement avec les gangliosides GM3, GT1b et GD1a des cellules de neuroblastomes inhibe la
phosphorylation de l’EGFR (Mirkin B.L. et al., 2002). Contrairement à cet effet inhibiteur du GM3,
un produit issu de sa dégradation, le dé-N-acetylGM3, semble stimuler l'activité tyrosine kinase
associée à l'EGFR et ainsi la croissance de cellules 3T3 (Hanai N. et al., 1988). Des approches
opposées consistant à utiliser des mutants déficients en gangliosides (Weis F.M. et al., 1990), à
transfecter une sialidase diminuant les taux de GM3 (Meuillet E.J. et al., 1999) ou à traiter les
cellules avec un inhibiteur de glucosylcéramide synthase (Meuillet E.J. et al., 2000) ont révélé une
augmentation de l’autophosphorylation de l'EGFR et une croissance cellulaire plus forte,
confirmant ainsi l'action inhibitrice des gangliosides sur l'EGFR. En revanche, Liu et al. ont montré
que le GD1a augmente à la fois l’autophosphorylation de l’EGFR induite par l’EGF et son activité
GM3, GM1, GD1a, GT1b GM3,
GM2, GM1
PDGFR
FGFR
GM1, GM2, GD1a, GD1b, GD3, GT1b
GM3, GM2, GM1
GM3GD1a
NGFR
GM1
-
-
-- +++
EGFR IR
GM3, GM1, GD1a, GT1b GM3,
GM2, GM1
PDGFR
FGFR
GM1, GM2, GD1a, GD1b, GD3, GT1b
GM3, GM2, GM1
GM3GD1a
NGFR
GM1
-
-
-- +++
84

tyrosine kinase dans des fibroblastes (Liu Y. et al., 2004).
• Concernant le récepteur au PDGF (PDGFR), Yates et al. ont montré que les gangliosides GM1,
GD1a, GD1b, GD3 et GT1b mais pas GM3 inhibent sa phosphorylation à la fois dans des cellules
3T3 (Van Brocklyn J. et al., 1993) et dans des cellules dérivées de gliome humain (Yates A.J. et al.,
1995). Hynds et al. ont confirmé cette inhibition de la phosphorylation du PDGFR par GM1, GM2,
GD1a et GT1b dans des cellules de neuroblastome humain (Hynds D.L. et al., 1995). Plusieurs
études ont également montré que les gangliosides, et en particulier GM2 et GM1, inhibent la
phosphorylation du PDGFR et la cascade de signalisation consécutive, notamment la stimulation de
la PLC et l’élévation du Ca2+ cytosolique, inhibant ainsi l’effet prolifératif du PDGF (Sachinidis A.
et al., 1996; Saqr H.E. et al., 1995; Yates A.J. et al., 1995).
• Par ailleurs, Bremer et al. ont décrit l'inhibition de la fonction du récepteur au FGF et la
diminution de croissance de fibroblastes de hamster par le GM3 (Bremer E.G. et al., 1982). Au
contraire, Slevin et al. ont montré que la croissance de cellules endothéliales aortiques bovines
induite par le bFGF est inhibée par le GM2 et le GM1 alors qu’elle est augmentée par le GM3. Le
GM2 et le GM1 se sont avérés modifier la liaison du bFGF à son récepteur et inhiber la voie de
signalisation mitogène conduisant à l’activation des PLCγ, MAPK et PKC (Slevin M. et al., 1999).
Etonnamment, le GM1 a été décrit pour se lier au FGF2 et renforcer son activité mitogène ce qui
suggérerait que le GM1 puisse agir comme un corécepteur du FGF2 (Rusnati M. et al., 2002).
• Le GM1 s'est également révélé activer le récepteur au NGF en synergie avec le facteur de
croissance, stimulant ainsi la différenciation de cellules PC12 en présence de NGF (Mutoh T. et al.,
1995).
• Enfin, il a été rapporté que l’incubation de cellules avec du GM3 exogène diminue
l’autophosphorylation du récepteur à l’insuline stimulé par son ligand (Nojiri H. et al., 1991). De
même, Tagami et al. ont montré que l’incubation d’adipocytes avec du GM3 supprime la
phosphorylation du récepteur à l’insuline et de son substrat IRS1 (Tagami S. et al., 2002). De plus,
Sasaki et al. ont révélé qu’une accumulation de GM2 et GM1 suite à la surexpression d’une
sialidase dans des souris transgéniques diminue significativement la phosphorylation du récepteur
en réponse à l’insuline ainsi que de son substrat IRS1 (Sasaki A. et al., 2003). Ces travaux sont
d’ailleurs d’une importance cruciale dans le cadre du diabète et seront discutés par la suite.
Les mécanismes par lesquels les gangliosides modulent l’activité de ces récepteurs de facteurs de
croissance à tyrosine kinase sont encore assez mal connus. Il a été montré que l’efficacité à inhiber
la phosphorylation de l’EGFR et la croissance de neuroblastomes, par exemple, différe en fonction
du ganglioside considéré, indiquant une relative spécificité de structure dans l’action des
gangliosides et suggérant que des modifications différentielles du profil en gangliosides pourraient
85

jouer un rôle dans la modulation de la croissance cellulaire (Mirkin B.L. et al., 2002). Bien que
quelques travaux suggèrent une perturbation de la liaison du facteur de croissance à son récepteur
(Liu Y. et al., 2004; Slevin M. et al., 1999), le mécanisme d’action des gangliosides ne semble
impliquer, dans la plupart des cas, ni une diminution du nombre de récepteurs, ni une inhibition de
la liaison du ligand. Le plus souvent, la diminution d’activité du récepteur est décrite comme la
conséquence d’une diminution de l’autophosphorylation du récepteur et de son activité tyrosine
kinase conduisant à une inhibition de la cascade de signalisation qui en découle normalement. Il a
été suggéré que cette inhibition de phosphorylation puisse être la conséquence d’une perturbation de
la dimérisation du récepteur requise pour l'activité tyrosine kinase, comme par exemple pour
l’EGRF ou le PDGFR (Hynds D.L. et al., 1995; Van Brocklyn J. et al., 1993; Yates A.J. et al.,
1995). Il a par ailleurs été avancé que le GM3 pourrait interagir directement avec l’EGFR en se
liant à un site présent sur le domaine extracellulaire du récepteur, distinct du site de liaison de
l’EGF (Miljan E.A. et al., 2002). Wang et al. vont également dans ce sens, en précisant que la
glycosylation du récepteur est nécessaire pour l’interaction avec le GM3 (Wang X.Q. et al., 2001a).
Certains auteurs ont également proposé que les gangliosides puissent agir sur la distribution des
récepteurs aux facteurs de croissance au niveau des microdomaines (Mitsuda T. et al., 2002; Wang
X.Q. et al., 2002).
1.2.2 Modulation de la protéine kinase C
La PKC est une enzyme ubiquiste, dépendante du Ca2+ et des phospholipides, jouant un rôle clé
dans la transduction du signal transmembranaire et impliquée dans de nombreux processus tels que
la prolifération ou la différenciation cellulaires. Kreutter et al. ont montré lors d'essais in vitro que
l'activité PKC est fortement inhibée par un mélange de gangliosides cérébraux, d'autant plus
efficacement que le nombre d'acides sialiques présents dans la molécule est élevé. Ils suggèrent que
l'inhibition exercée par les gangliosides n'est pas due à une interaction avec le substrat, une
compétition avec les phospholipides ou une chélation du Ca2+ mais plutôt à une action directe sur
l'enzyme (Kreutter D. et al., 1987). Cette inhibition de l'activité PKC par le GM3, GD3 et le GT1b a
été confirmée par l'équipe de Katoh qui a attribué cet effet à une perturbation de l'association de la
PKC ou de son substrat avec les phospholipides membranaires (Katoh N., 1995). Kanda et al. ont
également montré que le GM3 exogène supprime l'activité PKC dans des cellules endothéliales
microvasculaires du cerveau (Kanda T. et al., 1993).
D'autres protéines kinases ont été décrites pour être modulées par les gangliosides. C'est le cas par
exemple de la protéine kinase A positivement régulée par le GM3 dans les cellules endothéliales
(Kanda T. et al., 1993). Une protéine kinase spécifiquement inhibée par les gangliosides a de plus
été purifiée du cerveau de cochon d'Inde alors qu'un système de phosphorylation de protéines
86

stimulé par les gangliosides a été détecté à la surface de cellules de neuroblastome (Chan K.F.,
1988).
Les gangliosides présentent donc clairement une double activité, l'une médiant les interactions de la
cellule avec son environnement et l'autre modulant la fonction de protéines membranaires et
régulant la transduction du signal transmembranaire. Ces deux fonctions sont liées et confèrent aux
gangliosides une importance majeure dans divers processus biologiques, évoqués ci-dessous. Il est
important de noter que les intermédiaires de biosynthèse ou bien les métabolites des gangliosides,
c'est à dire le céramide, la sphingosine, la sphingosine 1-P, les lyso-gangliosides…, peuvent eux
aussi inhiber ou activer de nombreux processus intracellulaires et sont connus comme des
médiateurs importants dans l'apoptose, la prolifération….
2. IMPORTANCE BIOLOGIQUE / PHYSIOLOGIQUE DES GANGLIOSIDES
2.1.Développement, différenciation cellulaire et tissulaire, embryogenèse
L'importance capitale des gangliosides dans le développement embryonnaire et la différenciation
des tissus a été mise en lumière avec la mort précoce à 7.5 jours d'embryons de souris KO pour la
glucosylcéramide synthase, totalement déficients en gangliosides (Yamashita T. et al., 1999).
Les gangliosides sont connus pour intervenir dans les phénomènes de reconnaissance cellule-cellule
essentiels pour l'embryogenèse et la morphogenèse. Leur rôle dans la différenciation a été montré in
vitro, particulièrement pour le système nerveux. Tout d’abord, l’interaction entre les gangliosides et
des protéines impliquées dans le développement du tissu nerveux ou la différenciation des cellules
neuronales a été plusieurs fois décrites (Schnaar R.L. et al., 1998; Tiemeyer M. et al., 1989). De
plus, il a été montré que la transfection de cellules Neuro2a de souris avec l'ADNc de la GD3
synthase induit une croissance des neurites et la différenciation cholinergique de ces cellules
(Kojima N. et al., 1994). Il a également été rapporté que l'expression des gangliosides de cellules de
la rétine neuronale varie fortement au cours de leur différenciation (Panzetta P. et al., 2000). Par
ailleurs, l'augmentation des taux de GM3, par ajout exogène ou blocage de la synthèse des
gangliosides plus complexes, a provoqué la différenciation de cellules HL-60 en
monocytes/macropahges ou granulocytes (Nojiri H. et al., 1986; Zeng G. et al., 1995).
Le rôle des gangliosides dans le développement du système nerveux est renforcé par le fait que le
profil en gangliosides varie de manière importante au cours du développement et que les
gangliosidoses, comme par exemple la maladie de Tay-Sachs qui correspond à une accumulation de
GM2, causent des dysfonctionnements neurologiques sévères chez les patients atteints.
87

Cependant, les observations tirées de l'étude de souris KO pour différents gènes de
glycosyltransférases sont mitigées. Par exemple, la souris KO pour la GM2/GD2 synthase
déficiente en gangliosides complexes, développée par l'équipe de Furukawa est viable et ne présente
pas de changements morphologiques apparents (Takamiya K. et al., 1996). Ce résultat surprenant
pourrait s'expliquer, selon les auteurs, par une compensation des fonctions des gangliosides
complexes par les gangliosides plus simples (GM3 et GD3) toujours exprimés dans l'animal.
Cependant, la conductivité nerveuse de cette souris et la régénération après une atteinte nerveuse
sont affectées, ce qui souligne l'importance des gangliosides complexes dans le maintien et la
réparation du tissu nerveux. De plus, cette souris présente un défaut de spermatogénèse et une
stérilité mâle, révélant un nouveau rôle des gangliosides.
2.2.Oncogénèse
L’expression des gangliosides est généralement aberrante dans les cellules transformées et les
tumeurs, tant quantitativement que qualitativement avec l’apparition de nouveaux gangliosides dans
le profil. Ainsi, des mélanocytes de la peau, exprimant initialement du GM3 en majorité,
synthétisent de grandes quantités de GD3 après transformation (Carubia J.M. et al., 1984).
L'analyse de carcinomes de cou et de tête humains a révélé la présence de GM2, GD2, GM1, GD1a
ou GT1b alors que les tissus normaux expriment un profil plus simple avec une majorité de GM3 et
GD3 (Bolot G. et al., 1998). La même équipe a également découvert un ganglioside spécifique
associé à cette tumeur, GalNAc-GM1 (Bolot G. et al., 1999).
Ces modifications sont suffisamment nettes et caractéristiques pour faire des gangliosides des
antigènes de tumeurs, utilisés pour le diagnostic ou le traitement. Des thérapies prometteuses
utilisant des anticorps anti-gangliosides ou bien des vaccins, comme par exemple un vaccin à base
de GM2, ganglioside exprimé dans de nombreux mélanomes, sont d'ailleurs en cours de
développement.
Les mécanismes par lesquels les gangliosides sont impliqués dans la cancérogenèse restent obscurs.
• Il semble que les tumeurs synthétisent et libèrent des gangliosides dans leur
microenvironnement qui pourraient favoriser leur développement en supprimant les réponses
immunitaires de l'organisme (Li R. et al., 1996). Il a récemment été montré que le GM3 et le GD3,
purifiés à partir de mélanome humain, inhibent la différenciation et induisent l'apoptose de cellules
dendritiques (Peguet-Navarro J. et al., 2003).
• Les gangliosides pourraient aussi avoir une importance dans la radiosensibilité des cellules
tumorales ainsi que dans leur capacité métastatique. Zebda et al. ont montré que des cellules de
mélanomes humains à fort pouvoir métastatique présentent des défauts de biosynthèse des
gangliosides, n'exprimant que du GM3 en faibles quantités (Zebda N. et al., 1995). La sensibilité
88

aux radiations de cellules de mélanome humain M4Be a d’autre part été inversement corrélée à la
quantité totale de gangliosides ainsi qu'à la présence de GD3. De plus, l'incubation de cellules
radiosensibles avec du GM1 augmente la proportion de cellules résistantes et diminue leur potentiel
métastatique (Thomas C.P. et al., 1997).
• Les gangliosides ont été décrits pour moduler les interactions, l’adhésion ou la motilité
cellulaires et pourraient ainsi participer aux propriétés invasives des cellules cancéreuses. Merzak et
al. ont montré que les gangliosides, et particulièrement le GD3, favorisent l'adhésion de trois lignées
cellulaires dérivées de gliome humain à la fibronectine, le collagène I, la vitronectine et la laminine
(Merzak A. et al., 1995). Nakano et al. ont également suggéré que les gangliosides pourraient
augmenter le potentiel métastatique de cellules de mélanome en renforçant l'attachement des
cellules aux protéines de la matrice extracellulaire, par interaction directe ou par l'intermédiaire des
récepteurs intégrines (Nakano J. et al., 1999). L'interaction entre le GM3 et le
gangliotriaosylcéramide ou LacCer joue un rôle dans l'adhésion de cellules de mélanomes murins
B16 aux cellules endothéliales et a été avancé comme le facteur initiateur des métastases de ces
mélanomes (Hakomori S. et al., 1995).
Au contraire, Nojiri et al. ont proposé un rôle thérapeutique du GM3 dans le cancer. Ils ont montré
que le GM3 induit une différenciation terminale, culminant par la mort par apoptose de cellules de
carcinome humain et qu’un traitement de tumeurs de souris nude avec du GM3 diminue leur
croissance (Nojiri H. et al., 2002). De même, Aoki et al. ont très récemment montré une diminution
de la motilité et des capacités invasives de cellules de carcinome rénal en diminuant les taux de
GM2, prédominant dans ces cellules et en faisant apparaître du GM3 par inhibition de la GM2
synthase par la technique des SiRNA (Aoki H. et al., 2004).
2.3.Prolifération cellulaire
L’implication des gangliosides dans la régulation de la prolifération cellulaire est maintenant bien
établie.
• De multiples études, basées sur l’utilisation de gangliosides exogènes, ont permis de déterminer
leurs effets in vitro. Quelques travaux indiquent un effet prolifératif des gangliosides : stimulation
de la croissance de cellules musculaires lisses par GM1 et GM2 (Gouni-Berthold I. et al., 2001) ou
augmentation de la prolifération de fibroblastes par GD1a (Liu Y. et al., 2004), mais le plus
souvent, les gangliosides sont décrits pour inhiber la croissance cellulaire. Citons par exemple
l’action anti-proliférative exercée par le GM3 sur des cellules endothéliales microvasculaires,
rapportée par Alessandri et al. (Alessandri G. et al., 1992). Il a également été montré que la
prolifération de cellules de neuroblastome humain est inhibée par les GM3, GM1, GD1a et GT1b
(Mirkin B.L. et al., 2002) ou que le GM3 et le GM1 diminuent la croissance de lignées cellulaires
89

KB ou A431 (Bremer E.G. et al., 1986). Une inhibition de prolifération des cellules vasculaires de
muscle lisse a aussi été observée en réponse aux GM3, GM2 et GM1 (Sachinidis A. et al., 1996).
Tsuboi et al. ont décrit les gangliosides comme des inhibiteurs endogènes de la croissance dans des
cellules mésangiales glomérulaires (Tsuboi N. et al., 2001). Ils ont en effet montré que les cellules
sécrètent des gangliosides dans le milieu de culture, en majorité du GM3, qui inhibent la
prolifération lorsqu’ils sont utilisés pour traiter les cellules. Ils spéculent que ces gangliosides
pourraient être sécrétés par des cellules mésangiales activées sous certains conditions pathologiques
comme un mécanisme de défense pour contrecarrer une prolifération excessive stimulée par des
facteurs de croissance.
Ces observations ont été confirmées plus récemment par des techniques de transfection. Mitsuda et
al. ont notamment montré que la surexpression du GM1 par transfection des ADNc des enzymes
GM2 et GM1 synthases dans des cellules 3T3 diminue leur croissance (Mitsuda T. et al., 2002).
Ces effets sont la plupart du temps attribués à une interaction des gangliosides avec les récepteurs
de facteurs de croissance, comme cela a été décrit précédemment.
Cependant d’autres mécanismes ont été proposés pour expliquer l’effet des gangliosides sur la
prolifération cellulaire :
• Nakatsuji et al. ont décrit une action des gangliosides sur le cycle cellulaire. Ils ont rapporté que
le GM3 diminue la prolifération de cellules neurales en bloquant leur cycle cellulaire et induisant
l’apoptose, en partie via un inhibiteur de kinase cycline-dépendante p27Kip1 (Nakatsuji Y. et al.,
2001). Une étude très récente a confirmé l’existence d’un effet des gangliosides sur le cycle
cellulaire. Les auteurs ont en effet révélé que la surexpression de la GD3 synthase inhibe la
prolifération de cellules vasculaires de muscle lisse, en diminuant l’expression de protéines
associées au cycle cellulaire, cycline E et CDK2, et en induisant un inhibiteur du cycle cellulaire,
p21 (Moon S. K. et al., 2004).
• Par ailleurs, il semble que les gangliosides soient aussi impliqués dans l’induction de l’apoptose.
Le GM3 a par exemple été décrit pour induire l’apoptose de cellules neurales (Nakatsuji Y. et al.,
2001). L’apoptose de monocytes a également été provoquée par un traitement avec du GM3 et du
GD3 purifiés à partir de mélanomes humains (Peguet-Navarro J. et al., 2003). Il a de plus été
rapporté que le GD3 induit l’apoptose d’oligodendrocytes (Simon B.M. et al., 2002) ou de cellules
musculaires lisses aortiques (Bhunia A.K. et al., 2002). Plusieurs travaux ont permis d’expliciter le
rôle du GD3 dans la propagation du signal apoptotique généré par le récepteur CD95 (Fas) (Figure
26). La fixation d’un ligand à ce récepteur au niveau des complexes de mort conduit à l’activation
de deux voies apoptotiques : une cascade protéolytique médiée par les caspases et une voie
lipidique induisant l’activation de la sphingomyélinase acide et la libération de céramide (Malisan
90

F. et al., 1999). L’équipe de Testi a montré que l’accumulation de céramide conduit à la synthèse de
GD3 et que ce GD3 perturbe le potentiel transmembranaire de la mitochondrie. Ils ont de plus
montré que la surexpression de la GD3 synthase provoque à elle-seule directement l’apoptose alors
que l’inhibition de la synthèse de GD3 par des oligonucléotides anti-sens prévient l’apoptose
induite par CD95 (De Maria R. et al., 1997). Ils ont enfin rapporté que la transition de perméabilité
de la mitochondrie induite par le GD3 s’accompagne d’une libération de cytochrome c, de facteurs
induisant l’apoptose (AIF) et de l’activation de la caspase 9 (Rippo M.R. et al., 2000). Ces effets
n’étaient pas induits par d’autres gangliosides que le GD3. Ces résultats ont été confirmés par
Kristal et al. (Kristal B.S. et al., 1999) et Garcia-Ruiz et al. (Garcia-Ruiz C. et al., 2000) dans des
mitochondries isolées et des hépatocytes. Il semble donc que la mitochondrie soit la cible du GD3
dans l’induction du processus apoptotique. Le trafic intracellulaire de ce ganglioside à la
mitochondrie a d’ailleurs été décrit dans des hépatocytes (Garcia-Ruiz C. et al., 2002).
Figure 26 : processus apoptotique impliquant le GD3
(SM, sphingomyélinase; ST8, sialyltransférase 8: GD3 synthase)
d'après Tomassini B. et al (Tomassini B. et al., 2002)
Colell et al. ont cependant proposé un mécanisme alternatif par lequel le GD3 réprimerait
l’activation de NF-κB, facteur associé à une voie de survie (Colell A. et al., 2001). D’autre part,
Garofalo et al. ont montré que l’induction du système CD95/Fas provoque l’association du GM3
91

avec la caspase 8 au niveau des complexes de mort, suggérant un rôle structural des gangliosides
dans cette voie apoptotique (Garofalo T. et al., 2003).
Notons pour finir que les gangliosides ont déjà été impliqués dans les phénomènes d’angiogénèse.
Ziche et al. les ont décrits comme des promoteurs de l’angiogénèse dans la cornée de lapin. Ils ont
en effet montré que les GM1, GT1b ou GD3, contrairement au GM3, stimulent la
néovascularisation induite par des facteurs angiogéniques comme la prostaglandine E1 (PGE1) ou
le bFGF, ainsi que le nombre et la croissance des vaisseaux nouvellement formés, (Ziche M. et al.,
1989; Ziche M. et al., 1992). Ils ont également montré que ces gangliosides agissent en synergie
avec la PGE1 et le bFGF pour améliorer la survie, la croissance et la migration des cellules
endothéliales (De Cristan G. et al., 1990). Une étude menée sur des cellules de gliome humain a de
plus révélé que le GD3 stimule la production de VEGF, suggérant ainsi son rôle angiogénique
potentiel (Koochekpour S. et al., 1996). Enfin, Slevin et al. ont rapporté que le GM3 augmente la
prolifération de cellules endothéliales induite par le bFGF au contraire de GM2 et GM1, proposant
les gangliosides comme modulateurs de l’angiogénèse (Slevin M. et al., 1999).
Par leur double fonction de sites de reconnaissance cellulaire et de modulateurs du signal
transmembranaire, les gangliosides sont impliqués dans une grande variété de fonctions biologiques
dans l’organisme. Les plus connues ont été décrites précédemment mais les gangliosides ont déjà
été associés à d’autres processus comme par exemple la spermatogenèse, les lésions auditives, les
neuropathies ou l’arthrose. Il est fort à parier que d’autres rôles et mécanismes d’action sont encore
à découvrir. Les gangliosides ont été mis en cause dans différentes pathologies, essentiellement le
cancer et certaines maladies neurodégénératives, mais peu de recherches ont été faites sur leur rôle
potentiel dans le diabète.
IV. GANGLIOSIDES ET DIABETE
Des altérations des glycoconjugués et de leur métabolisme ont été décrites au cours du diabète.
• In vivo, différents travaux ont montré des modifications d’activité des enzymes du métabolisme
des glycoprotéines, ainsi une augmentation de plusieurs glycosidases dans le sérum de patients
diabétiques (Serrano M.A. et al., 1983). Des modifications sélectives de la chaîne de sucres de
92

glycoprotéines, avec une diminution des galactose, fucose et acide sialique, ont été décrites dans des
cellules microvasculaires rétiniennes en réponse aux AGE et associées à des modifications
d’activité des glycosyltransférases et glycosidases correspondantes, confirmées dans des rétines de
rat diabétique (Rellier N. et al., 1997; Rellier N. et al., 1999).
• Une forte augmentation de l’activité sialidase ainsi qu’une diminution de l’activité
sialyltransférase et de la concentration d’acide sialique lié ont également été mesurées dans le
cortex rénal de rats STZ (Cohen-Forterre L. et al., 1990). Le taux d’acide sialique plasmatique est
d’ailleurs bien connu pour être augmenté dans le diabète. Une augmentation a été mesurée chez les
diabétiques de type 1 et corrélée à la présence et la sévérité de la rétinopathie (Crook M.A. et al.,
2001). De plus, une accumulation d’acide sialique a également été mesurée dans le sérum et la paroi
des capillaires glomérulaires de patients atteints de néphropathie diabétique (Tomino Y. et al.,
1987; Tomino Y. et al., 1988).
Cependant, peu d’études se sont intéressées jusqu’à présent aux glycolipides ou plus
particulièrement aux gangliosides dans le cadre du diabète. S’ils sont clairement impliqués dans les
phénomènes d’auto-immunité caractéristiques du diabète de type 1, leur rôle dans les complications
diabétiques n’a été que rarement envisagé. Des études récentes les associant aux mécanismes de
l’insulino-résistance ravivent pourtant l’intérêt d’étudier les gangliosides dans cette pathologie.
1. GANGLIOSIDES ET AUTO-IMMUNITE
Le diabète de type 1 est la conséquence d’une destruction auto-immnune des cellules β
pancréatiques productrices d’insuline. Ce phénomène est caractérisé par la présence dans la
circulation, d’auto-anticorps et de lymphocytes T auto-réactifs dirigés contre de multiples
autoantigènes présents au niveau des îlots pancréatiques. Certains autoantigènes sont de nature
protéiques et d’autres ont été identifiés comme étant des gangliosides. Des anticorps circulants
dirigés contre le GT3 (Gillard B.K. et al., 1989) et le GD3 (Tiberti C. et al., 1995) ont par exemple
été détectés chez des diabétiques de type 1 récemment diagnostiqués. Le ganglioside GM2-1 (Cer-
glucose-galactosamine-galactose-acide sialique), exprimé spécifiquement dans les îlots
pancréatiques, a également été identifié comme la cible d’autoanticorps du groupe ICA (Islet Cell
Antibodies), eux-aussi spécifiques des îlots pancréatiques (Dotta F. et al., 1996).
93

2. GANGLIOSIDES ET MICROANGIOPATHIE DIABETIQUE Quelques travaux décrivent des modifications du profil en gangliosides en réponse à un
environnement diabétique, in vivo ou in vitro.
Une perte spécifique des gangliosides de la série-c a été décrite dans le pancréas et le cristallin de
rats STZ (Saito M. et al., 1999; Saito M. et al., 2000). Sanchez et al. ont quant à eux observé une
modification des gangliosides du foie chez des rats STZ. Ces modifications concernent le profil en
gangliosides avec une augmentation du GM3, GM1, GD1b et GT1b ainsi que leurs enzymes de
biosynthèse puisque l’activité GM2/GD2 synthase est augmentée alors que les activités des
galactosyltransférase-II et sialyltransférase-IV sont diminuées. Ils ont de plus montré que ces effets
du diabète sur le métabolisme des gangliosides sont normalisés par un traitement des animaux à
l’insuline (Sanchez S.S. et al., 2000).
Un petit nombre d’études suggère l’implication potentielle des gangliosides dans les complications
microangiopathiques:
Zador et al. ont révélé une accumulation de glucosylcéramide et de GM3 dans les reins
hypertrophiés de rats STZ. De plus, ils ont montré qu’un traitement des animaux diabétiques avec
un inhibiteur de glucosylcéramide synthase, D-PDMP, ramene le poids de leurs reins à un niveau
normal, suggérant ainsi le rôle des GSL et du GM3 dans l’hypertrophie rénale caractéristique de la
néphropathie diabétique (Zador I.Z. et al., 1993).
Au contraire, une équipe coréenne a montré une diminution des taux d’acides sialiques et des
gangliosides totaux, en particulier du GM3 dans les glomérules de rats diabétiques STZ. Ils
suggèrent que cette appauvrissement en GM3 pourrait conduire à une diminution de la charge
anionique de la barrière glomérulaire et ainsi contribuer à la perte de sélectivité de la filtration et à
la protéinurie consécutive, observée au cours de la néphropathie (Kwak D.H. et al., 2003). La même
équipe a très récemment montré que de fortes concentrations de glucose stimulent la prolifération
de cellules mésangiales, effet reproduit par un traitement avec le PDMP. De plus, les fortes
concentrations de glucose diminuaient fortement les taux de GM3 et l’activité GM3 synthase. Les
auteurs présument que la diminution des taux de GM3 pourrait contribuer à la stimulation de
prolifération cellulaire induite par l’hyperglycémie et participer ainsi à l’hypertrophie rénale qui
survient lors du diabète (Rho Y.I. et al., 2004).
Enfin, une étude réalisée dans notre laboratoire a mis en évidence des modifications spécifiques du
profil en gangliosides de cellules microvasculaires rétiniennes soumises à un traitement avec les
AGE. Natalizio et al. ont montré une augmentation des taux de tous les gangliosides dans les
cellules endothéliales et une augmentation des taux de tous les gangliosides exceptée une
diminution des taux de GD3 dans les péricytes, en réponse aux AGE. Ces résultats sont les premiers
94

à révéler des modifications des gangliosides par l’environnement diabétique dans des cellules
rétiniennes particulièrement touchées au cours du diabète, et à souligner ainsi le rôle potentiel des
gangliosides dans la rétinopathie diabétique (Natalizio A. et al., 2001).
3. GANGLIOSIDES ET INSULINO-RESISTANCE Des études récentes menées notamment sur des souris génétiquement modifiées renforcent
vivement l'importance des gangliosides dans le diabète en suggérant leur rôle dans l’insulino-
résistance.
• Tagami et al. ont les premiers apporté des preuves soutenant l’implication du GM3 dans
l’acquisition de l’insulino-résistance (Tagami S. et al., 2002). Ils ont tout d’abord montré que
l’induction de l’insulino-résistance dans des adipocytes murins 3T3 par traitement avec de faibles
concentrations de TNFα s’accompagne d’une augmentation du taux de GM3 cellulaire. Celle-ci est
liée à une régulation positive de l’activité GM3 synthase ainsi que du taux d’ARNm de l’enzyme,
suggérant une activation de la biosynthèse du GM3 par le TNFα au niveau transcriptionnel. Cette
augmentation des taux d’ARNm de la GM3 synthase a d’ailleurs été également observée dans le
tissu adipeux de modèles d’animaux diabétiques, rat Zucker ou souris ob/ob. Les auteurs ont ensuite
montré que l’incubation des cellules en présence de GM3 mime les effets du TNFα sur la
signalisation de l’insuline, c’est à dire inhibe la phosphorylation du récepteur à l’insuline et de son
substrat IRS-1 ainsi que la captation du glucose en réponse à l’insuline. De plus, ils ont rapporté que
l’inhibition de la synthèse du GM3 par le D-PDMP, un inhibiteur de glucosylcéramide synthase,
normalise les défauts de phosphorylation du récepteur à l’insuline et de IRS-1 induits par le TNFα.
Ils suggèrent ainsi que l’augmentation de la biosynthèse du GM3 induite par le TNFα pourrait
participer à la mise en place de l’insulino-résistance caractéristique du diabète de type 2. Les
auteurs proposent un mécanisme d’action du GM3 mettant en jeu les microdomaines, illustré par la
figure 27. La voie de signalisation métabolique de l’insuline, contrairement à la voie du signal de
croissance (Shc-Ras-MAPK), nécessite l’internalisation du récepteur au niveau des microdomaines
et l’interaction avec son substrat. Dans les adipocytes rendus insulino-résistants par le TNFα, ce
processus est perturbé alors que le signal de croissance n’est pas affecté. Le récepteur à l’insuline
semble être déplacé à l’extérieur des microdomaines. Le GM3 pourrait participer à ces changements
structuraux et fonctionnels des microdomaines qui sous-tendent le phénomène d’insulino-
résistance.
95

GM3GM3
Figure 27 : mécanisme d'action proposé pour expliquer l'implication du GM3 dans l'insulino-résistance
(d'après http://www.glycoforum.gr.jp/science/word/glycopathology/GD-B03E.html)
• L’implication du GM3 dans l’insulino-résistance a été confirmée un an plus tard par Yamashita
et al. à l’aide d’une souris KO pour le gène de la GM3 synthase (Yamashita T. et al., 2003). Ils ont
en effet rapporté que cet animal, incapable de synthétiser le GM3, présente une sensibilité accrue à
l’insuline, liée à une augmentation de la phosphorylation du récepteur dans le muscle squelettique.
Ces souris mutantes étaient également protégées contre une insulino-résistance induite par un
régime hyperlipidémique.
• Enfin, la surexpression d’une sialidase (Neu3) a été décrite pour atténuer la signalisation de
l’insuline, en inhibant la phosphorylation du récepteur à l’insuline et de son substrat, dans un
modèle de souris transgénique (Sasaki A. et al., 2003). Les auteurs ont mesuré une augmentation du
taux des gangliosides GM1 et GM2, résistants à la Neu3, qui deviennent majoritaires dans
différents tissus. Ils ont également montré que ces deux gangliosides inhibent la phosphorylation du
récepteur à l’insuline in vitro et ont donc suggéré que la surexpression de la sialidase Neu3 inhibe la
signalisation de l’insuline au moins en partie via une accumulation de GM1 et GM2.
96


MATERIEL ET METHODES
97


I. CULTURES CELLULAIRES
Les expériences ont été menées sur des cultures primaires et non des lignées de péricytes rétiniens
et de cellules mésangiales rénales afin d'être le plus proche possible de la physiologie des cellules
natives. Pour éviter toute dégénérescence, les cellules ont de plus été utilisées pendant un nombre
de passages restreint (P2 pour les BRP, P5-P15 pour les RMC). Cependant, une limitation de ces
modèles tient au fait que la croissance et le renouvellement des péricytes et des cellules mésangiales
dans les microvaisseaux in vivo est très faible. Ces choix ne permettent pas de cultiver des cellules
en grande quantité et nécessitent, surtout pour les péricytes, des mises en culture très fréquentes.
D’autre part, ceci amène une certaine hétérogénéité entre les expériences, en particulier dans le cas
des péricytes rétiniens, pour lesquels les cellules étaient issues à chaque préparation de bœufs
différents.
1. PERICYTES RETINIENS DE BŒUF Les péricytes rétiniens sont obtenus à partir des microvaisseaux issus de rétine de bœuf.
1.1.Isolement des microvaisseaux et mise en culture
Des yeux de bœuf, fraîchement énucléés, sont obtenus auprès des abattoirs Cibevial (Corbas,
France) et transportés jusqu’au laboratoire sur de la glace. Les yeux sont d’abord nettoyés de tout
tissu attenant et placés dans du tampon PBS froid (Sigma, Saint-Quentin Fallavier, France). Toutes
les manipulations ultérieures s’effectuent à 4 °C et de manière stérile, sous une hotte à flux
laminaire.
Les yeux sont rapidement plongés dans une solution d’éthanol à 70 % puis rincés dans du tampon
HBSS sans Mg2+ ni Ca2+ (Sigma). Ils sont ensuite découpés 5 mm postérieurement à la cornée afin
d’éliminer le cristallin et l’humeur vitrée à l’aide d’une pince fine et de faire apparaître la rétine.
Celle-ci est alors délicatement décollée du globe oculaire, sectionnée au niveau du nerf optique puis
placée dans une boite de Pétri contenant une solution d’HBSS. Cette solution, préparée à partir de
HBSS sans Mg2+ ni Ca2+, contient 10 mM d’HEPES, 100 U/ml de pénicilline et 100 µg/ml de
streptomycine (Sigma). Après 10 min d'oxygénation par un mélange O2/CO2 (95/5, v/v), de la BSA
est ajoutée à une concentration finale de 0,5 % et le pH est ajusté à 7,5 avec quelques gouttes de
NaOH concentré (en suivant le virage de l'indicateur rouge phénol présent dans l'HBSS).
Les rétines sont ensuite débarrassées d’éventuelles cellules pigmentaires contaminantes et placées
98

par lot de deux dans 10 ml de solution HBSS propre. Elles sont découpées aux ciseaux en petits
fragments avant d’être homogénéisées (Dounce 15 ml, piston A) et centrifugées (5 min, 300g). Le
culot, enrichi en microvaiseaux est repris par 5 ml d’une solution enzymatique de collagénase-
dispase. Cette solution, préparée dans du tampon HBSS sans Mg2+ ni Ca2+, contient 1 mg/ml de
collagénase-dispase (Boehringer-Mannheim), 10 mM d’HEPES, 100 U/ml de pénicilline et 100
µg/ml de streptomycine. Son pH est ajusté à 7,4 après 10 minutes d’oxygénation et avant addition
de TLCK (147 ng/ml) et de DNAse (200 U/ml) (Sigma). La digestion enzymatique s’effectue à 37
°C pendant 20 minutes sous agitation douce. Les homogénats de rétines sont régulièrement
resuspendus par retournement des tubes afin de disperser les agrégats.
La suspension est ensuite passée sur un filtre de porosité 40 µm. Les fragments de microvaisseaux
retenus sur le filtre sont récupérés dans 10 ml de solution HBSS puis centrifugés (5 min, 300 g). Le
culot est repris dans 3 ml de milieu de culture pour péricytes, c'est à dire du DMEM (Sigma)
contenant 10 % de SVF (Gibco, Invitrogen Corporation, N.Y., USA), 2 mM de glutamine, 100
U/ml de pénicilline et 100 µg/ml de streptomycine. Les fragments de microvaisseaux sont enfin
ensemencés dans une boite de Pétri de 6 cm de diamètre "coatée" avec de la fibronectine
(incubation préalable des boites avec 1,5 ml d'une solution de fibronectine à 57 µg/ml dans du
DMEM pendant 1 à 2 heures à 37°C). Après ensemencement, les boites sont placées dans un
incubateur à 37 °C sous atmosphère humide contrôlée contenant 5 % de CO2. Après environ 4
heures d’incubation, le milieu contenant les débris de microvaisseaux n’ayant pas adhéré à la
matrice est éliminé et remplacé par 3 ml de milieu de culture frais. La croissance des péricytes hors
des microvaisseaux apparaît au bout de 48 heures (Figure 28A). Le milieu de culture est remplacé
tous les deux jours et les cellules atteignent la confluence en une dizaine de jours.
A BA B
Figure 28 : croissance des péricytes depuis un microvaisseau rétinien (A)
Péricytes rétiniens (B)
99

Les pericytes se présentent sous la forme de cellules polygonales irrégulières, avec émission de
pseudopodes et croissant de manière non jointive (Figure 28B). Ces cellules ont été caractérisées au
laboratoire par étude de leur morphologie en microscopie à contraste de phase, et par
immunodétection à l'aide d'un anticorps anti-α1-actine et d'un anticorps 3G5 spécifique des
péricytes (Nayak R.C. et al., 1988). Cette technique permet d’obtenir des cultures d’une pureté
proche de 100 %, de manière reproductible (Lecomte M. et al., 1996).
1.2.Passages et traitements
• Une fois la culture arrivée à confluence, les cellules sont rincées 2 fois avec 3 ml de PBS puis
trypsinées pendant environ 5 minutes à 37 °C par 1 ml d’une solution de trypsine à 0,5 % contenant
0,2 % d’EDTA (Sigma). Quand les cellules commencent à se décoller, la trypsine est inactivée par
2 ml de PBS contenant 10 % de SVF. Les cellules sont alors récupérées puis centrifugées pendant 3
min à 300g. Elles sont ensuite réensemencées dans un rapport 1:3 (1 boite 6 cm passée dans une
boite 10 cm) dans des boites "coatées" avec de la fibronectine.
• Les cellules sont cultivées jusqu'au deuxième passage (P2), passage pendant lequel elles sont
utilisées et traitées pour les différentes expériences.
2. CELLULES MESANGIALES RENALES DE RAT Les cellules mésangiales rénales sont obtenues à partir de glomérules rénaux de jeunes rats mâles
Wistar (Charles River, L'Arbresle, France).
2.1.Isolement des glomérules et mise en culture
Les glomérules rénaux sont isolés par une technique de tamisage différentiel.
Le rat est euthanasié au CO2 puis les manipulations ultérieures s’effectuent de manière stérile, sous
une hotte à flux laminaire. L'abdomen du rat est tout d'abord nettoyé à l'aide d'une compresse
imbibée d'éthanol 70 % puis ouvert pour prélever les reins. Ceux-ci sont rapidement plongés dans
une solution d’éthanol à 70 % puis rincés dans du tampon HBSS sans Mg2+ ni Ca2+. Les reins sont
alors nettoyés en enlevant le tissu conjonctif et la capsule fibro-adipeuse qui les entoure. Ils sont
ensuite de nouveau rincés dans du HBSS et le cortex est prélevé à l'aide d'un scalpel.
Le cortex est découpé en fragments avant d'être broyé à l'aide d'un piston sur un tamis de type mesh
60 (230 µm). Le tamis est ensuite rincé avec environ 50 ml de solution d'HBSS (identique à celle
utilisée pour l'isolement de microvaisseaux rétiniens). Le filtrat obtenu est récupéré et passé sur un
deuxième tamis, de type mesh 200 (73,7 µm). Il est de nouveau broyé avec un piston et le tamis est
rincé avec 50 ml de solution d'HBSS. Le filtrat est récupéré et passé sur un tamis de type Falcon 70
100

µm. Le rétentat est récupéré en rinçant le filtre avec environ 15 ml de solution d'HBSS.
Le rétentat ainsi obtenu, enrichi en glomérules, est centrifugé 10 min à 300g. Le culot est repris
dans du milieu de culture contenant du DMEM, 20 % de SVF, 2 mM de glutamine, 100 U/ml de
pénicilline et 100 µg/ml de streptomycine et ensemencé dans des boîtes de Pétri préalablement
"coatées" avec de la fibronectine (les glomérules obtenus à partir de 2 reins sont ensemencés dans 4
boîtes de 6 cm ). Après ensemencement, les boîtes sont placées dans un incubateur à 37°C sous
atmosphère humide contrôlée contenant 5 % de CO2. Après environ 3-4 jours de période
d'adhésion, le milieu contenant les glomérules n’ayant pas adhéré à la matrice est éliminé et
remplacé par du milieu de culture frais. Les cellules mésangiales poussent hors du glomérule après
environ 3 semaines (Figure 29A), après les cellules endothéliales qui sont progressivement
éliminées. Le milieu de culture des cellules mésangiales, c'est à dire du DMEM contenant 15 % de
SVF, 2 mM de glutamine, 100 U/ml de pénicilline et 100 µg/ml de streptomycine est alors remplacé
tous les deux jours.
A BA B
Figure 29 : croissance des cellules mésangiales à partir d'un glomérule rénal (A)
Cellules mésangiales (B)
Les cellules mésangiales sont identifiés sur la base de critères morphologiques (cellules irrégulières
prenant une forme allongée à confluence, Figure 29B) et par immunodétection, absence de
marquage pour le facteur VIII et la cytokératine et marquage du cytosquelette très important pour
l'actine.
101

2.2.Passages et traitements
• Une fois la culture arrivée à confluence, les cellules sont rincées 2 fois avec 3 ml de PBS puis
trypsinées par incubation avec 1 ml d'une solution de trypsine à 0,5 % contenant 0,2 % d’EDTA
pendant environ 5 minutes à 37 °C. Quand les cellules commencent à se décoller, l'action de la
trypsine est inactivée par ajout de 2 ml de PBS contenant 10 % de SVF. Les cellules sont alors
récupérées puis centrifugées pendant 3 min à 300g. Elles sont ensuite filtrées sur un tamis Falcon de
40 µm afin d'éliminer les glomérules et débris, puis réensemencées dans un rapport 1:5 dans des
boites préalablement "coatées" avec de la fibronectine. Les cellules sont cultivées pendant 5
passages avant d'être utilisées pour les expériences, afin de se débarrasser des glomérules et des
cellules endothéliales contaminantes et jusqu'àu 15ème passage pour éviter toute dégénérescence.
Les cellules mésangiale sont donc utilisées et traitées pour les expériences entre P5 et P15.
• Les cellules mésangiales peuvent être congelées après collecte par trypsination, dans un milieu
de culture contenant 20 % de SVF et 5 % de DMSO, dans l'azote liquide. La décongélation doit être
rapide et les cellules rincées avant d'être réensemencées.
II. TRAITEMENT DES CELLULES
1. LES AGE Les effets des AGE in vivo sont difficiles à reproduire in vitro car les AGE représentent un
ensemble complexe et hétérogène de molécules tant par la variété des produits d’Amadori et des
dicarbonyles que par celle des protéines impliquées dans les différentes réactions menant à leur
formation.
Dans cette étude, nous avons choisi de former des AGE à partir de l’incubation du méthylglyoxal et
d’albumine de sérum de bœuf car un certain nombre de travaux permettent de penser que les AGE
dérivés du méthylglyoxal, carboxyéthyllysine ou MOLD, sont des produits majeurs observés lors
du diabète. Tout d’abord, il a été montré que le méthylglyoxal est augmenté dans le sang de patients
diabétiques (McLellan A.C. et al., 1994). Ce dicarbonyle peut être formé de différentes manières :
décomposition des intermédiaires trioses phosphate, dégradation oxydative des sucres ou des
lipides, dégradation des bases de Schiff ou produits d’Amadori (Degenhardt T.P. et al., 1998; Singh
R. et al., 2001). Degenhardt et al. ont décrit MOLD et GOLD, AGE respectivement issus du
méthylglyoxal et du glyoxal, comme les modifications chimiques majeures observées dans les
102

protéines tissulaires de patients diabétiques, à des concentrations 10 à 50 fois supérieures à la
pentosidine par exemple (Degenhardt T.P. et al., 1998). De plus, une augmentation des
concentrations des AGE dérivés du méthylglyoxal, notamment la carboxyéthyl-lysine, a été
mesurée dans la rétine et les glomérules rénaux de rats STZ (Karachalias N. et al., 2003).
1.1.Préparation des AGE
Les AGE méthylglyoxal sont obtenus en incubant 7,2 mg/ml de BSA avec 100 mM de
méthylglyoxal (concentrations finales) pendant 50 h à 37 °C. Cette concentration de méthylglyoxal
a déjà été décrite pour induire des modifications sur les résidus arginine et lysine de la BSA et
conduire à la formation de différents AGE (Westwood M.E. et al., 1994). La solution de "BSA
contrôle" est obtenue en incubant de l’albumine dans les mêmes conditions mais sans
méthylglyoxal. Afin d’éliminer les sels et le méthylglyoxal libre n’ayant pas réagi, les solutions
d'AGE et de BSA contrôle sont ensuite filtrées sur colonnes de Sephadex G-25 M (colonnes PD10,
Amersham Pharmacia Biotech) et éluées par du DMEM selon les indications du fabricant. Les
solutions sont enfin stérilisées (filtre Acrodisc 0,2 µm) et aliquotées pour être conservées à –20 °C
jusqu’à utilisation.
1.2.Traitement
Dans le but d'étudier les effets à long terme d'une exposition des cellules à l'environnement
diabétique, les péricytes et les cellules mésangiales sont traités de manière chronique durant un
passage entier soit respectivement 7 et 4 jours, avec une concentration finale de 3 µM d’AGE ou de
BSA-contrôle.
2. LA GLUCOSAMINE La glucosamine exogène est rapidement captée par les cellules via les transporteurs au glucose et
phosphorylée par l'hexokinase en glucosamine-6-P, entrant ainsi sélectivement dans la voie de
l'hexosamine juste an aval de l'enzyme limitante GFAT (Schleicher E.D. et al., 2000). Pour ces
raisons, la glucosamine est utilisée expérimentalement pour mimer une suractivation de la voie de
l'hexosamine.
• Afin d'étudier les effets à long terme d'une exposition des cellules à l'environnement diabétique,
les péricytes et les cellules mésangiales sont cultivées pendant un passage entier, soit
respectivement 7 et 4 jours, en présence de 2 ou 5 mM de glucosamine (Sigma), concentrations
finales.
• Les cellules mésangiales ont également été traitées pendant un temps plus court (48 heures)
103

et avec des concentrations plus faibles (0,5 mM) afin d'étudier des mécanismes d'action spécifiques
de la glucosamine.
3. GANGLIOSIDES EXOGENES Les gangliosides, ainsi que leurs précurseurs non sialylés, Glucosylcéramide et Lactosylcéramide
(Matreya, Biovalley, Marne la Vallée, France), utilisés comme contrôles, sont utilisés sous forme de
complexe avec la BSA afin de faciliter leur incorporation dans la cellule. Pour cela, une solution de
DMEM contenant 10 mM d'Hepes, pH 7,4 et 0,34 mg/ml de BSA délipidée est préparée puis
stérilisée par filtration. Après évaporation du solvant chloroforme/méthanol 2:1, les gangliosides
sont repris dans cette solution selon un rapport 1:1 avec la BSA.
Pour déterminer les effets des gangliosides exogènes sur la prolifération des BRP et des RMC, les
cellules sont cultivées en plaque 96 puits en présence de concentrations croissantes (10-100 µM) de
ces complexes glycolipides- BSA pendant un passage.
Dans le cas de l'étude des effets des gangliosides GM2 et GM1 sur l'hypertrophie et le cycle
cellulaire des cellules mésangiales, les cellules sont cultivées dans des boites de Pétri de 10 cm en
présence de 60 µM de ces complexes glycolipides-BSA, pendant 48 heures.
L'ajout exogène de gangliosides est une technique largement utilisée pour déterminer leur rôle
notamment dans la régulation de la prolifération cellulaire. Cependant, il semble que seule une
petite partie de ces gangliosides exogènes s'incorpore effectivement dans la membrane plasmique
(Bremer E.G. et al., 1984). De plus, les espèces de gangliosides exogènes apportées peuvent
potentiellement être métabolisées au sein de la cellule en d'autres gangliosides plus ou moins
complexes si bien qu'il est délicat d'attribuer les effets à une espèce de ganglioside en particulier.
4. ANTICORPS ANTI-GANGLIOSIDES Des anticorps ont été utilisés pour bloquer l'effet potentiel des gangliosides de la série-a. Les
cellules sont cultivées en plaque de 96 puits, en présence de 0,5 µg par puits d’anticorps
monoclonal anti-GM3 (Cosmobio), ou de 5 µg par puits d'anticorps polyclonal anti-GM2
(Calbiochem), anti-GM1 (Matreya) ou d’IgG de lapin utilisée comme contrôle.
5. PPMP Le DL-threo-1-Phenyl-2-palmitoylamino-3-morpholino-1-propanol ou PPMP (Sigma) est un
104
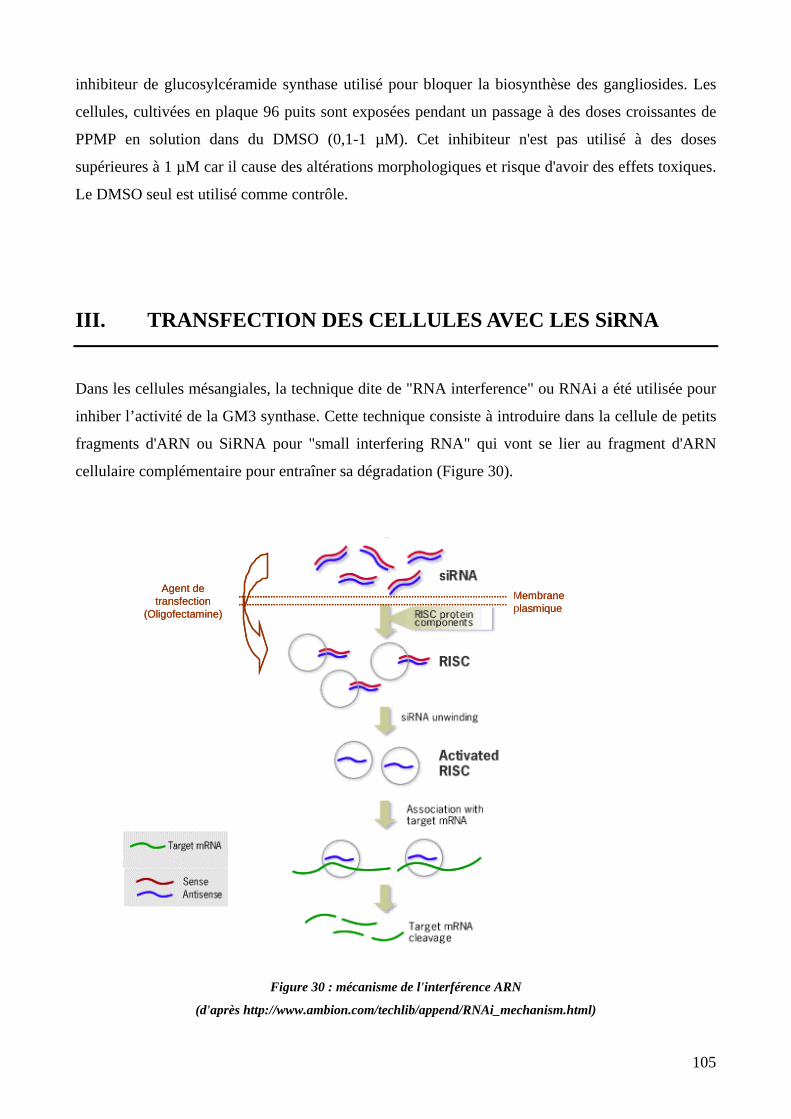
inhibiteur de glucosylcéramide synthase utilisé pour bloquer la biosynthèse des gangliosides. Les
cellules, cultivées en plaque 96 puits sont exposées pendant un passage à des doses croissantes de
PPMP en solution dans du DMSO (0,1-1 µM). Cet inhibiteur n'est pas utilisé à des doses
supérieures à 1 µM car il cause des altérations morphologiques et risque d'avoir des effets toxiques.
Le DMSO seul est utilisé comme contrôle.
III. TRANSFECTION DES CELLULES AVEC LES SiRNA
Dans les cellules mésangiales, la technique dite de "RNA interference" ou RNAi a été utilisée pour
inhiber l’activité de la GM3 synthase. Cette technique consiste à introduire dans la cellule de petits
fragments d'ARN ou SiRNA pour "small interfering RNA" qui vont se lier au fragment d'ARN
cellulaire complémentaire pour entraîner sa dégradation (Figure 30).
Membrane plasmique
Agent de transfection
(Oligofectamine)
Membrane plasmique
Agent de transfection
(Oligofectamine)
Figure 30 : mécanisme de l'interférence ARN
(d'après http://www.ambion.com/techlib/append/RNAi_mechanism.html)
105
• Préparation des SiRNA : les SiRNA ont été designés contre la séquence ADNc du gène de la
GM3 synthase de rat. La séquence antisens utilisée était la suivante :
GGGUUAUUCUGAACAUGUUtt (Ambion, Huntingdon, UK). Les SiRNA sont tout d'abord
repris dans de l'eau ultrapure sans RNAse pour obtenir une solution stock à 20 µM. Les conditions
de transfection sont les suivantes, données à titre d'exemple pour un puits de plaque 6 puits et une
concentration finale de transfection de 400 nM :
500 µl800 µl11 µl4 µl165 µl20 µl
Milieu de culture 3X
DMEMOpti-MEMAgent de transfection
Oligofectamine (Invitrogen
Corporation)
Opti-MEMSolution stock d’oligonucléotide
(20 µM)
185 µl : 5-10 min à température ambiante
Dilution des SiRNA
15 µl : 5-10 min à température ambiante
Dilution de l’Oligofectamine
200 µl : 15-20 min à température ambiante
Homogénéisation douce SiRNA + agent de transfection
1 ml : 4h à 37 °Ctransfection
culture
500 µl800 µl11 µl4 µl165 µl20 µl
Milieu de culture 3X
DMEMOpti-MEMAgent de transfection
Oligofectamine (Invitrogen
Corporation)
Opti-MEMSolution stock d’oligonucléotide
(20 µM)
185 µl : 5-10 min à température ambiante
Dilution des SiRNA
15 µl : 5-10 min à température ambiante
Dilution de l’Oligofectamine
200 µl : 15-20 min à température ambiante
Homogénéisation douce SiRNA + agent de transfection
1 ml : 4h à 37 °Ctransfection
culture
• Transfection des cellules : les cellules sont transfectées dans des plaques 6 puits à environ 30 %
de confluence afin d'optimiser le rendement de transfection sans pour autant atteindre la confluence
au terme de la période de traitement. Elles sont tout d'abord rincées avec du DMEM puis les 200 µl
de la solution de SiRNA combiné à l'agent de transfection sont ajoutés dans le puits avec 800 µl de
DMEM, soit un volume final de transfection de 1 ml. Les cellules sont placées dans l'incubateur
pendant 4 heures à 37 °C. A l'issue de l'incubation, 500 µl de milieu 3X sont rajoutés dans le puits.
Ce milieu de culture ne contient pas d'agents anti-bactériens (pénicilline/streptomycine) qui pourrait
perturber la transfection. L'activité GM3 synthase est mesurée 72 h après la transfection.
Alternativement, les cellules sont traitées avec 3 µM de BSA ou AGE, 24 h après la transfection et
106

la prolifération est évaluée au bout de 3 jours.
IV. ANIMAUX DIABETIQUES
Les souris diabétiques db/db et leur contrôle db/m (Charles River) sont sacrifiées par décapitation
après euthanasie à l'isoflurane (Minerve). Les reins sont ensuite prélevés, nettoyés et le cortex est
prélevé. Les fragments de cortex sont ensuite disséqués et homogénéisés mécaniquement à l'aide
d'un Potter de 7 ml (verre/téflon) dans un tampon composé d'Hepes 25 mM, d'EDTA 1 mM, de
Triton 0,1 % et de 10 µl/ml de cocktail d'inhibiteurs de protéases.
V. ANALYSE DE LA PROLIFERATION CELLULAIRE
Plusieurs techniques ont été utilisées pour étudier la prolifération des péricytes et des cellules
mésangiales en réponse à différents traitements.
1. COMPTAGE DES CELLULES Les cellules sont tout d’abord collectées par trypsination. Le culot est rincé 2 fois dans du PBS puis
repris dans un volume donné de ce tampon (environ 1 ml pour 105 cellules). Une aliquote de 10 µl
est prélevée et ajoutée à 10 µl de bleu trypan. 10 µl de ce mélange sont alors utilisés pour compter
les cellules dans une chambre hématimétrique.
2. DOSAGE DES PROTEINES
• Après collecte par trypsination, le culot de cellules est rincé 2 fois avec du PBS et repris dans
un volume donné de ce tampon (environ 1 ml pour 105 cellules). Uns aliquote de 10 µl est alors
prélevés et ajoutés à 50 µl de tampon de lyse contenant 0,1 N de NaOH et 0,05 % de triton X100.
Les cellules sont lysées à 4 °C pendant une nuit. Une gamme étalon de 0 à 12 µg de BSA est
107

réalisée dans 50 µl de tampon de lyse. Les protéines ont ensuite été dosées par la technique
colorimétrique de Bradford (Compton S.J. et al., 1985). Le volume de lysat est ajusté à 800 µl avec
de l’eau ultrapure et 200 µl de réactif Bio-Rad sont ajoutés. 200 µl de la solution sont transférés
dans une plaque de 96 puits et l’absorption est lue à 620 nm. La quantification est faite par rapport à
la gamme étalon à l’aide du logiciel Biolise.
• Pour les expériences menées en plaque de 96 puits, la technique de dosage des protéines BCA
(Pierce) a été utilisée pour doser les protéines directement dans les puits, sans collecte préalable des
cellules. Les cellules sont tout d’abord rincées 3 fois par du PBS froid puis lysées pendant 30
minutes sous agitation à 37 °C dans 50 µl de tampon de lyse RIPA contenant 1 % de NP40, 0,5 %
de désoxycholate de sodium, 0,1 % de SDS, 10 µl/ml de cocktail d’inhibiteurs de protéases et du
PBS 10 mM. Les cellules lysées sont ensuite incubées avec 200 µl par puits de réactif de Pierce
reconstitué selon les recommandations du fournisseur, pendant 30 min sous agitation à 37 °C.
L’absorption est mesurée à 562 nm et la quantification est faite par rapport à la gamme étalon (de 0
à 15 µg de BSA par puits) à l’aide du logiciel Biolise. Pour les expériences menées dans les plaques
de 6 puits, 600 µl de tampon de lyse sont ajoutés dans les puits et 50 µl de lysats sont utilisés pour
le dosage des protéines selon le même protocole. Etant donné que la quantité totale de protéines
était corrélée au nombre de cellules, le dosage de protéines a été utilisé, dans certaines expériences
notamment celles menées en puits, pour estimer la prolifération cellulaire à la place du comptage
des cellules pour des raisons de praticité.
3. MESURE DES QUANTITES D'ADN La technique du CyQuant a été utilisée pour mesurer les quantités d'ADN. Les cellules sont
ensemencées dans des plaques de 96 puits à fond blanc de type Optilux. A la fin de la période de
traitement, les cellules sont rincées 3 fois avec du PBS froid. La plaque de 96 puits est alors placée
à –70 °C pour faire éclater les cellules et faciliter l'entrée du composé de marquage fluorescent. Une
fois la plaque revenue à température ambiante, 200 µl de réactif, reconstitué selon les
recommandations du fabricant, sont ajoutés à chaque puits. Les cellules sont ensuite incubées
pendant 1 heure à température ambiante. La plaque est enfin agitée 2 min avant de mesurer la
fluorescence à une longueur d'onde d'excitation de 480 nm et une longueur d'onde d'émission à 520
nm.
108

4. ANALYSE DU CYCLE CELLULAIRE Après collecte des cellules par trypsination et rinçages avec du PBS, le culot de cellules est
resuspendu avec soin dans du PBS (environ 75 µl pour 106 cellules) et les cellules sont fixées en
ajoutant goutte à goutte de l’éthanol 100 % à -20 °C de manière à ce qu’il soit à 70 % au final. Le
mélange doit être "vortexé" tout au long de la fixation à l’éthanol. Les cellules sont incubées dans
cette solution d’éthanol pendant une nuit à 4 °C. Elles sont ensuite rincées en remplissant les tubes
de PBS et centrifugées 5-10 min, 300g à 4 °C. Le culot est ensuite repris dans 100 µl de RNAse à 1
mg/ml et incubé pendant 10-30 min afin que seul l’ADN soit analysé par la suite, puis 400 µl de
PBS sont ajoutés. Environ 30 min avant l’analyse, l’ADN des cellules est marqué par ajout de 10 µl
d’iodure de propidium à 2 mg/ml qui s’intercale au niveau de l’ADN. Le cycle cellulaire est enfin
analysé par cytométrie en flux à l’aide d’un FACSCAN et du logiciel CellQuest (Becton Dickinson,
Franklin Lakes, N.J., USA).
5. MESURE DE L’EXPRESSION DES PROTEINES INHIBITRICES DU CYCLE
CELLULAIRE A la fin de la période de traitement, les cellules sont rincées avec du PBS puis incubées pendant 20
min à 4 °C avec du tampon de lyse RIPA. Les cellules sont alors collectées par grattage et le
matériel insoluble est éliminé par centrifugation à 10,000g pendant 5 min à 4 °C. Les protéines du
surnageant sont ensuite dosées et utilisées pur réaliser l'essai. Une quantité égale de protéines de
chaque échantillon est chauffé à 100 °C pendant 5 min en présence d'un tampon Laemli afin de
dénaturer les protéines. Les échantillons ainsi préparés sont ensuite déposés sur un gel SDS-
polyacrylamide 12 % avec un standard de poids moléculaire et les protéines sont séparées par
électrophorèse à 100 V. Elles sont ensuite transférées sur une membrane PVDF à 80 V pendant 30
min.
La membrane PVDF est bloquée dans du PBS-Tween (PBST) contenant 5 % de lait pendant 30 min
puis incubée avec l'anticorps primaire pendant 1 heure à température ambiante. Les anticorps
primaires monoclonaux sont préparés dans une solution de PBST 5 % BSA à 1 µg/ml pour
p21Waf1/Cip1 et à 1:2000 pour p27Kip1 (BD Biosciences, Le Pont de Claix, France). La membrane est
rincée 3 fois 10 min dans du PBST-BSA 5 % puis incubée pendant 45 min avec l'anticorps
secondaire anti-souris couplée à la peroxydase (Calbiochem) à une dilution de 1:2000. Après
rinçages, le signal est visualisé par chimioluminescence. La membrane est ensuite révélée avec un
anticorps anti-β actine afin de contrôler l'équivalence du dépôt et du transfert des protéines de
chaque échantillon. Pour cela, après plusieurs rinçages, la membrane PVDF est de nouveau bloquée
109

dans le PBST-BSA 5 % pendant plusieurs heures puis incubée avec un anticorps primaire anti-β
actine 1:5000 dans la solution de blocage pendant 1 heure. Après rinçages, l'anticorps secondaire est
ajouté pendant 45 min et le signal est révélé de même par chimioluminescence. Le signal est
quantifié par densitométrie à l'aide d'une caméra Image Master VDS-CL et du logiciel Image Quant
(Amersham Pharmacia Biotech). Les résultats obtenus pour l'expression de p21Waf1/Cip1 et p27Kip1
sont corrigés par le signal de la β actine pour chaque échantillon.
VI. ANALYSE DES GANGLIOSIDES
1. MARQUAGE METABOLIQUE Le marquage métabolique des gangliosides cellulaires est réalisé avec du galactose marqué au 14C.
Cet ose a été choisi car il est rapidement incorporé et métabolisé par la cellule et est présent, ainsi
que ses métabolites comme le glucose, dans tous les gangliosides, permettant ainsi un marquage
efficace et global. La veille de la collecte des cellules, 0,2 µCi/ml, ou 1 µCi/ml pour les expériences
nécessitant un marquage à plus forte activité spécifique, de 14C-[U]-galactose (329,5 mCi/mmol)
(PerkinElmer Life Sciences, Boston, MA) est ajouté au milieu de culture. Les cellules sont ainsi
incubées pendant 16 heures.
2. EXTRACTION, PURIFICATION, SEPARATION ET DETECTION DES GANGLIOSIDES
2.1.Extraction et séparation des gangliosides
Au terme de la période de traitement et de marquage métabolique, les cellules sont récupérées par
trypsination, rincées avec du PBS et centrifugées pendant 5 min à 300g. Le culot est ensuite repris
dans 2 ml de chloroforme/méthanol (1:1, v/v) et 1 µg de GT1b est ajouté à chaque échantillon. Ce
ganglioside, absent du profil des péricytes et des cellules mésangiales, est utilisé comme standard
interne. Les tubes sont vigoureusement "vortexés" et l'extraction des lipides se poursuit pendant une
nuit à 4°C. Les tubes sont alors centrifugés (1000 g, 5min) et le solvant contenant les lipides totaux
est récupéré. Le culot est ré-extrait deux fois par 1 ml du même mélange de solvant. Les phases
organiques sont rassemblées et évaporées à sec sous un flux d'azote. Le résidu est repris dans 3 ml
110

de chloroforme/méthanol (1:1, v/v), auxquels est ajouté 1 ml de PBS 1 mM. Les tubes sont
"vortexés" et les phases sont séparées par centrifugation (1000 g, 5min). La phase supérieure
contenant les gangliosides est récupérée et la phase inférieure est ré-extraite deux fois par 2 ml de
méthanol/PBS 1 mM (1:1, v/v). Les trois phases supérieures sont rassemblées et évaporées sous
flux d'azote. Les gangliosides sont repris dans 2 ml de méthanol/PBS 10 mM (1:1, v/v) pour être
déposés sur une colonne de silice C18 (Waters Corporation, Milford, MA), lavée par du méthanol et
équilibrée dans du méthanol/PBS 10 mM (1:1, v/v). Tous les composants polaires, tels que les sels
et les sucres sont éliminés par 10 ml d'eau. Les gangliosides sont ensuite élués par 6ml de méthanol
et 4ml de chloroforme/méthanol (2:1, v/v). Le solvant est évaporé et les gangliosides purifiés sont
repris dans un petit volume de chloroforme/méthanol (2:1, v/v) (30 µl) pour être déposés sur une
plaque HPTLC (gel de silice 60, épaisseur 0,2 mm, support plaque en verre). Le dépôt est réalisé
par un déposeur automatique de plaque (Automatic TLC Sampler III) à l'aide du logiciel Wincats
(Camag). Des standards commerciaux de gangliosides sont également déposés sur la plaque. Les
gangliosides sont enfin séparés par migration sur la plaque HPTLC dans un mélange de
chloroforme/méthanol/CaCl2 0,2% (55:45:10, v/v/v).
Alternativement, une autre technique d'extraction est utilisée pour extraire les gangliosides du
cortex rénal, plus longue mais permettant une meilleure purification à partir de tissus (Dreyfus H. et
al., 1997). Les lipides totaux des homogénats de cortex sont tout d'abord extraits 3 fois avec 6 ml de
chloroforme/méthanol 1:1 puis repris dans du chloroforme après évaporation du solvant. Les
différentes classes lipidiques sont ensuite séparées par chromatographie sur une colonne de silice 60
(Merck) équilibrée dans du chloroforme. 12 ml de chloroforme permettent d'éluer les lipides neutres
puis 1,5 ml de chloroforme : méthanol : acétone: acide acétique : eau (52:8:8:18:4, par volume)
suivi de 8 ml de chloroforme : méthanol (4:1, v/v) permettent d'éluer les phospholipides et
sphingolipides. Enfin, les gangliosides sont élués avec 2,5 ml de chloroforme : méthanol (2:3, v/v),
2,5 ml de chloroforme : méthanol : eau (65:25:4, v/v/v), 2,5 ml de chloroforme : méthanol : eau
(60:35:8, v/v/v) puis 2,5 ml de chloroforme : méthanol : eau (48:35:10, v/v/v). Le solvant est
évaporé et les gangliosides sont repris afin d'être déposés sur plaque HPTLC et séparés par
migration.
2.2.Révélation et identification des gangliosides
Les gangliosides ainsi séparés sont alors révélés par deux techniques : autoradiographie et
révélation colorimétrique. Les plaques sont tout d'abord placées au contact d'un écran de type
Phosphoscreen pendant 2 jours. L'écran impressionné est ensuite lu à l'aide d'un Storm 820
(Molecular Dynamics, Amersham Pharmacia Biotech, Piscataway, NJ). D'autre part, la plaque
HPTLC est vaporisée avec un mélange de résorcinol/HCl/Cu2+ (10 ml de résorcinol à 1,5 % (w/v
111
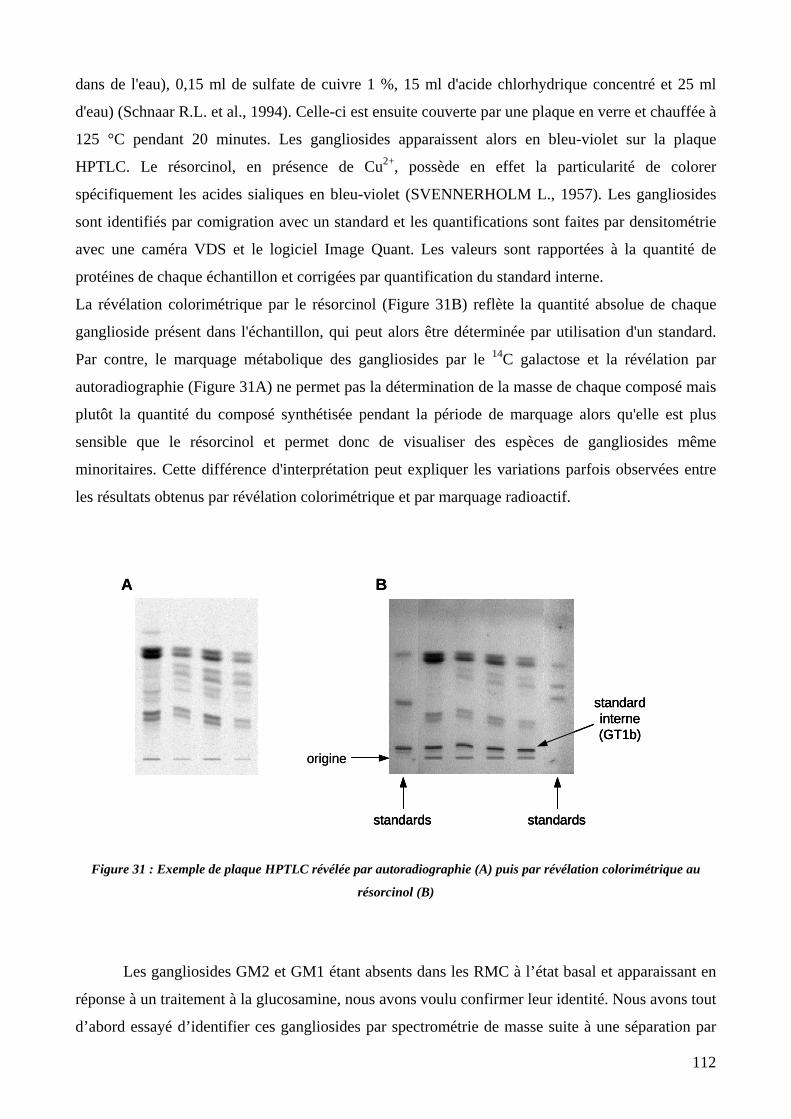
dans de l'eau), 0,15 ml de sulfate de cuivre 1 %, 15 ml d'acide chlorhydrique concentré et 25 ml
d'eau) (Schnaar R.L. et al., 1994). Celle-ci est ensuite couverte par une plaque en verre et chauffée à
125 °C pendant 20 minutes. Les gangliosides apparaissent alors en bleu-violet sur la plaque
HPTLC. Le résorcinol, en présence de Cu2+, possède en effet la particularité de colorer
spécifiquement les acides sialiques en bleu-violet (SVENNERHOLM L., 1957). Les gangliosides
sont identifiés par comigration avec un standard et les quantifications sont faites par densitométrie
avec une caméra VDS et le logiciel Image Quant. Les valeurs sont rapportées à la quantité de
protéines de chaque échantillon et corrigées par quantification du standard interne.
La révélation colorimétrique par le résorcinol (Figure 31B) reflète la quantité absolue de chaque
ganglioside présent dans l'échantillon, qui peut alors être déterminée par utilisation d'un standard.
Par contre, le marquage métabolique des gangliosides par le 14C galactose et la révélation par
autoradiographie (Figure 31A) ne permet pas la détermination de la masse de chaque composé mais
plutôt la quantité du composé synthétisée pendant la période de marquage alors qu'elle est plus
sensible que le résorcinol et permet donc de visualiser des espèces de gangliosides même
minoritaires. Cette différence d'interprétation peut expliquer les variations parfois observées entre
les résultats obtenus par révélation colorimétrique et par marquage radioactif.
A B
origine
standards
standard interne (GT1b)
standards
A B
origine
standards
standard interne (GT1b)
standardsstandards
standard interne (GT1b)
standards
Figure 31 : Exemple de plaque HPTLC révélée par autoradiographie (A) puis par révélation colorimétrique au
résorcinol (B)
Les gangliosides GM2 et GM1 étant absents dans les RMC à l’état basal et apparaissant en
réponse à un traitement à la glucosamine, nous avons voulu confirmer leur identité. Nous avons tout
d’abord essayé d’identifier ces gangliosides par spectrométrie de masse suite à une séparation par
112

HPLC en utilisant une colonne LiChrosorb-NH2 par élution avec des gradients de mélanges
acétonitrile/tampon phosphate. Cependant, des problèmes de quantité de matériel et de pureté
d’échantillon n’ont pas permis d’obtenir des résultats concluants. L'identité du GM2 et du GM1 a
alors été confirmée par immunocytochimie en utilisant un anticorps spécifique dirigé contre GM2
(don généreux du Dr J. Portoukalian) et la sous-unité β de la toxine cholérique, spécifique du GM1.
• Les gangliosides des cellules mésangiales traitées ou non avec 5 mM de glucosamine sont
déposés en double de façon identique sur une plaque HPTLC à support en aluminium (Merck).
Après la séparation des gangliosides par migration, la plaque est découpée en deux aux ciseaux
pour donner deux parties identiques. Une partie est révélée par pulvérisation au résorcinol alors que
l'autre est utilisée pour l'immnuodétection. Celle-ci est tout d'abord rapidement immergée dans une
solution de polyisobutylmétacrylate à 0,4 % dans l'hexane afin de fixer la silice. La plaque est
ensuite bloquée dans du PBS-BSA 1 % pendant 2 heures puis incubée à température ambiante avec
l'anticorps primaire anti-GM2. Après rinçage, la plaque est incubée avec un anticorps secondaire
biotinylé pendant 1 heure puis un complexe streptavidine-peroxydase est ajouté après rinçage. Au
bout de 45 min, le signal peroxydase est visualisé avec une solution préparée en dissolvant 2 mg de
4-chloronaphtol dans 3 ml de méthanol, 7 ml de tampon et 5 µl de H2O2 à 30 %. Comme le montre
la Figure 32, cette expérience nous a permit de confirmer l'identité du GM2, en comparant les
parties de plaque révélées au résorcinol et par immunodétection.
Immunodétection (anticorps anti-GM2) révélation au résorcinol
GM2 standard
GM2 échantillon
Glucosamine (mM)0 50 5
GM2 standard
Immunodétection (anticorps anti-GM2) révélation au résorcinol
GM2 standard
GM2 échantillon
Glucosamine (mM)0 50 5
GM2 standard
Figure 32 : immunuodétection du GM2
113
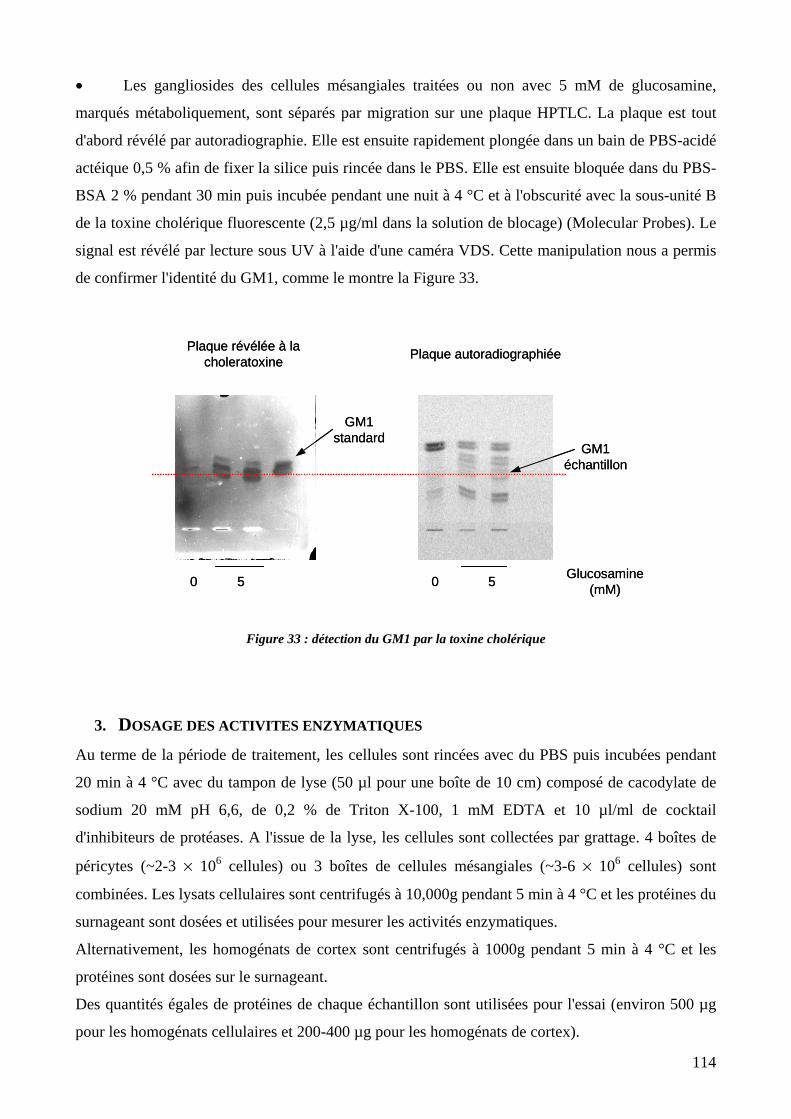
• Les gangliosides des cellules mésangiales traitées ou non avec 5 mM de glucosamine,
marqués métaboliquement, sont séparés par migration sur une plaque HPTLC. La plaque est tout
d'abord révélé par autoradiographie. Elle est ensuite rapidement plongée dans un bain de PBS-acidé
actéique 0,5 % afin de fixer la silice puis rincée dans le PBS. Elle est ensuite bloquée dans du PBS-
BSA 2 % pendant 30 min puis incubée pendant une nuit à 4 °C et à l'obscurité avec la sous-unité B
de la toxine cholérique fluorescente (2,5 µg/ml dans la solution de blocage) (Molecular Probes). Le
signal est révélé par lecture sous UV à l'aide d'une caméra VDS. Cette manipulation nous a permis
de confirmer l'identité du GM1, comme le montre la Figure 33.
Plaque révélée à la choleratoxine Plaque autoradiographiée
Glucosamine (mM)0 50 5
GM1 standard
GM1 échantillon
Plaque révélée à la choleratoxine Plaque autoradiographiée
Glucosamine (mM)0 50 5
GM1 standard
GM1 échantillon
Figure 33 : détection du GM1 par la toxine cholérique
3. DOSAGE DES ACTIVITES ENZYMATIQUES Au terme de la période de traitement, les cellules sont rincées avec du PBS puis incubées pendant
20 min à 4 °C avec du tampon de lyse (50 µl pour une boîte de 10 cm) composé de cacodylate de
sodium 20 mM pH 6,6, de 0,2 % de Triton X-100, 1 mM EDTA et 10 µl/ml de cocktail
d'inhibiteurs de protéases. A l'issue de la lyse, les cellules sont collectées par grattage. 4 boîtes de
péricytes (~2-3 × 106 cellules) ou 3 boîtes de cellules mésangiales (~3-6 × 106 cellules) sont
combinées. Les lysats cellulaires sont centrifugés à 10,000g pendant 5 min à 4 °C et les protéines du
surnageant sont dosées et utilisées pour mesurer les activités enzymatiques.
Alternativement, les homogénats de cortex sont centrifugés à 1000g pendant 5 min à 4 °C et les
protéines sont dosées sur le surnageant.
Des quantités égales de protéines de chaque échantillon sont utilisées pour l'essai (environ 500 µg
pour les homogénats cellulaires et 200-400 µg pour les homogénats de cortex).
114

Les activités des sialyltranséfrases GM3 synthase et GD3 synthase sont déterminées grâce à
l'incorporation d'acide N-acétyl neuraminique marqué au 14C (CMP-[14C]-NeuAc) sur un substrat
accepteur. Celui-ci est le CDH (lactosylcéramide) pour le dosage de la GM3 synthase et le GM3
pour le dosage la GD3 synthase. Les activités enzymatiques sont estimées en fonction de la quantité
de produit radioactif formé (Figure 34).
CMP-[14C]-NeuAc CMP
(GM3) (GD3)
Gal Gluc CeramideNeuAc Gal Gluc CeramideNeuAc[14C]NeuAc
CMP-[14C]-NeuAc CMP
(CDH) (GM3)
Gal Gluc Ceramide Gal Gluc Ceramide[14C]NeuAcGM3 synthase
GD3 synthase
CMP-[14C]-NeuAc CMP
(GM3) (GD3)
Gal Gluc CeramideNeuAc Gal Gluc CeramideNeuAc Gal Gluc CeramideNeuAc[14C]NeuAc Gal Gluc CeramideNeuAc[14C]NeuAc
CMP-[14C]-NeuAc CMP
(CDH) (GM3)
Gal Gluc Ceramide Gal Gluc Ceramide[14C]NeuAcGM3 synthase
GD3 synthase
Figure 34 : principe du dosage des activités enzymatiques GM3 et GD3 synthases
Les échantillons sont mélangés à un volume égal de tampon de réaction contenant en concentrations
finales : 0,1 mM de lactosylcéramide ou de GM3 (Matreya, Biovalley, Marne la Vallée, France)
pour les activités GM3 et GD3 synthase, respectivement, 4 µCi/ml de [sialic-4,5,6,7,8,9-14C]CMP-
acide sialique (325.2 mCi/mmol) (PerkinElmer Life Sciences), 100 µM de CMP-acide sialique
(Sigma), 10 mM MgCl2, 0,2 % Triton X-100, et 100 mM de cacodylate de sodium pH 6,6. Les
mélanges réactionnels sont ensuite incubés à 37 °C pendant 150 min sous agitation. La réaction est
stoppée en déposant les échantillons sur une colonne de silice 60 (Merck) préalablement lavée au
méthanol puis équilibrée à l'eau, afin de séparer l'excès de substrats des produits de réaction. Le
substrat n'ayant pas réagi est éliminé avec 4 ml d'eau puis les gangliosides sont élués avec 2 ml de
chloroforme/méthanol (1:1, v/v). Le solvant est évaporé et les gangliosides sont repris dans 30 µl de
chloroforme/méthanol 2:1 puis déposés sur plaque de chromatographie sur couche mince, avec des
standards, pour être séparés par migration dans un mélange chloroforme/méthanol/CaCl2 0,2 %
(55:45:10, v/v/v). La révélation des produits se fait par autoradiographie après impression d'un
écran Phosphoscreen pendant 10 jours. Les gangliosides sont également révélés par pulvérisation de
résorcinol afin d'identifier les bandes par rapport aux standards. Les quantifications sont faites par
115

densitométrie avec le logiciel Image Quant. Les activités enzymatiques sont exprimées en pmoles
de produit formées par heure et par mg de protéines.
VII. ANALYSE STATISTIQUE DES RESULTATS
Les valeurs présentées correspondent aux moyennes ± SEM de n expériences indépendantes.
Dans les études menées sur culture cellulaire, le test non paramétrique pour valeurs appariées de
Wilcoxon a été choisi pour comparer les différences entre groupes. Pour les expériences réalisées
sur les souris, le test t de Student a été utilisé. Les résultats sont jugés statistiquement significatifs
pour p<0,05.
116


RESULTATS-DISCUSSION
117


CONTEXTE ET OBJECTIFS
L'hyperglycémie est à l'origine des complications, notamment microvasculaires, du diabète.
Le glucose exerce ses effets toxiques via un certain nombre de voies biochimiques décrites, dont la
formation des produits avancé de glycation (AGE) et la voie des hexosamines. L'utilisation
d'inhibiteurs de la formation des AGE permet en effet de limiter le développement de la rétinopathie
(Hammes H.P. et al., 1991)et de la néphropathie (Forbes J.M. et al., 2001) chez des animaux
diabétiques. De même l'utilisation d'un inhibiteur de la voie des hexosamines permet de prévenir
par exemple
La prolifération cellulaire est largement affectée au cours du diabète, particulièrement celle des
péricytes au niveau rétinien et celle des cellules mésangiales au niveau rénal.
Par ailleurs, les gangliosides sont connus pour jouer un rôle dans la régulation de la
prolifération cellulaire.
L’objectif de ce travail de thèse a été de déterminer les effets d’un environnement
diabétique sur la prolifération cellulaire et le rôle potentiel des gangliosides dans ces
processus.
La multiplicité des voies par lesquelles l'hyperglycémie peut générer des effets toxiques
amenuise l'efficacité des interventions ciblées contre une seule de ces voies. Dans cette étude,
l’environnement diabétique a été mimé à la fois par un traitement avec les AGE et avec la
glucosamine afin de mettre en parallèle les conséquences de la formation des produits avancés de
glycation et de l’activation de la voie des hexosamines et d'identifier ainsi des mécanismes
intracellulaires communs par lesquels ces deux voies biochimiques induites par l'hyperglycémie
exerceraient leurs effets.
De plus, chez les patients diabétiques, rétinopathie et néphropathie sont souvent associées et
plusieurs caractéristiques pathologiques sont communes à ces deux complications, qu'il serait
profitable de pouvoir traiter en même temps. C’est pourquoi nos travaux ont été menés à la fois sur
des péricytes rétiniens et sur des cellules mésangiales rénales, dans une optique comparative
rétinopathie-néphropathie et de recherche de mécanismes communs aux deux microcomplications.
118


LES AGE DIMINUENT LA PROLIFERATION DES
PERICYTES ET DES CELLULES MESANGIALES:
IMPLICATION DES GANGLIOSIDES
Les AGE ont été largement impliqués lors d'études in vitro et in vivo, dans le développement des
complications microvasculaires du diabète que sont la rétinopathie et la néphropathie. Leur
accumulation a été montrée notamment dans le rein et la rétine de patients diabétiques. Différentes
équipes ont montré que les AGE affectent la croissance et la viabilité à la fois des péricytes
(Chibber R. et al., 1997; Denis U. et al., 2002; Yamagishi S. et al., 2002a) et des cellules
mésangiales (Yamagishi S. et al., 2002b; Ziyadeh F.N. et al., 1993). Cependant, les mécanismes
d'action de la toxicité des AGE n'ont pas été totalement identifiés.
D'autre part, de précédents travaux réalisés dans le laboratoire ont révélé que les AGE peuvent agir
sur le métabolisme des glycoconjugués et notamment des gangliosides de cellules microvasculaires
rétiniennes (Natalizio A. et al., 2001).
L'objectif de cette première étude a été de déterminer l'effet des AGE sur la prolifération cellulaire
et le profil en gangliosides, ceci dans une approche comparative péricytes-cellules mésangiales.
Ensuite, nous avons étudié l’hypothèse d’un lien potentiel entre modifications de la croissance
cellulaire et métabolisme des gangliosides.
I. EFFETS DES AGE SUR LA PROLIFERATION
CELLULAIRE
Péricytes rétiniens et cellules mésangiales rénales ont été cultivés durant un passage (7 et 4 jours,
respectivement) en présence de 3µM de BSA contrôle ou d’AGE. Au terme de cette période de
traitement, la prolifération cellulaire a été évaluée par comptage des cellules et par dosage des
protéines totales.
La Figure 35A montre que, dans les péricytes, les AGE diminuent le nombre de cellules de
33 % par rapport au nombre de cellules témoin. Cet effet délétère des AGE se manifeste également
119
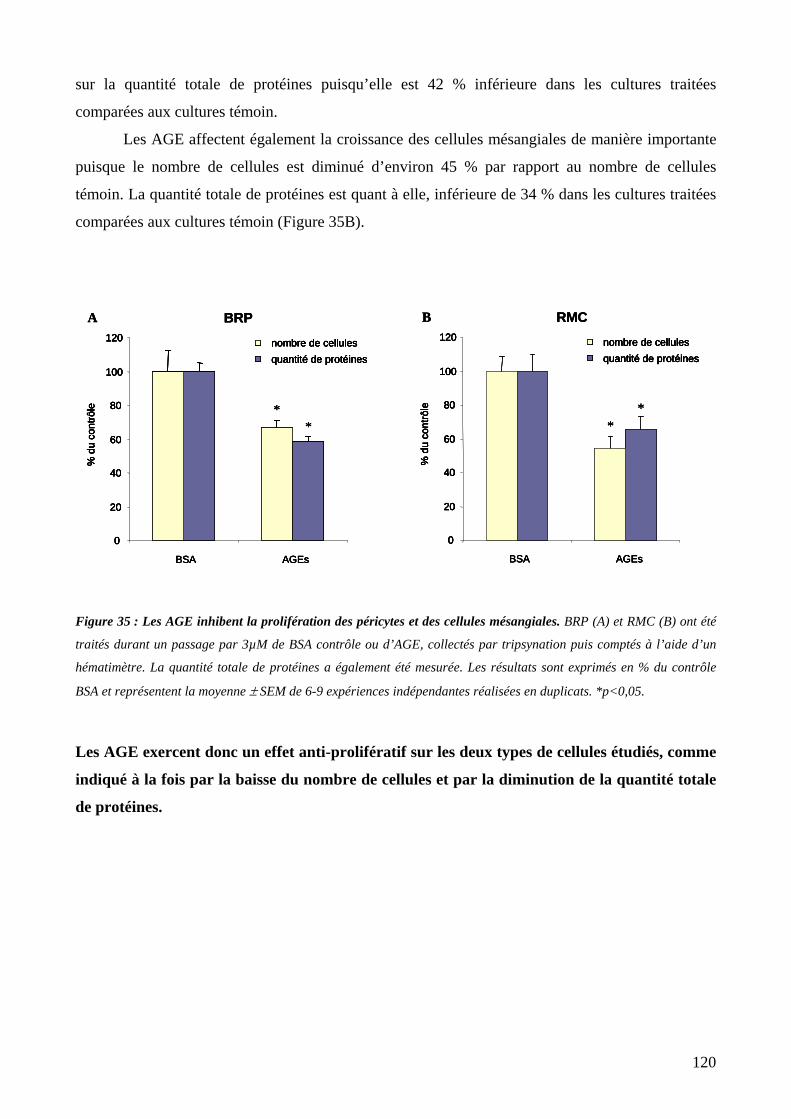
sur la quantité totale de protéines puisqu’elle est 42 % inférieure dans les cultures traitées
comparées aux cultures témoin.
Les AGE affectent également la croissance des cellules mésangiales de manière importante
puisque le nombre de cellules est diminué d’environ 45 % par rapport au nombre de cellules
témoin. La quantité totale de protéines est quant à elle, inférieure de 34 % dans les cultures traitées
comparées aux cultures témoin (Figure 35B).
BSA AGEs
0
20
40
60
80
100
120
%du
con
trôle
nombre de cellulesquantité de protéines
**
A BRP
BSA AGEs
0
20
40
60
80
100
120
%du
con
trôle
nombre de cellulesquantité de protéines
**
B RMC
BSA AGEs
0
20
40
60
80
100
120
%du
con
trôle
nombre de cellulesquantité de protéines
**
A BRP
BSA AGEs
0
20
40
60
80
100
120
%du
con
trôle
BSA AGEs
0
20
40
60
80
100
120
%du
con
trôle
0
20
40
60
80
100
120
%du
con
trôle
nombre de cellulesquantité de protéinesnombre de cellulesnombre de cellulesquantité de protéinesquantité de protéines
**
A BRP
BSA AGEs
0
20
40
60
80
100
120
%du
con
trôle
nombre de cellulesquantité de protéines
**
B RMC
BSA AGEs
0
20
40
60
80
100
120
%du
con
trôle
BSA AGEs
0
20
40
60
80
100
120
%du
con
trôle
0
20
40
60
80
100
120
%du
con
trôle
nombre de cellulesquantité de protéinesnombre de cellulesnombre de cellulesquantité de protéinesquantité de protéines
**
B RMC
Figure 35 : Les AGE inhibent la prolifération des péricytes et des cellules mésangiales. BRP (A) et RMC (B) ont été
traités durant un passage par 3µM de BSA contrôle ou d’AGE, collectés par tripsynation puis comptés à l’aide d’un
hématimètre. La quantité totale de protéines a également été mesurée. Les résultats sont exprimés en % du contrôle
BSA et représentent la moyenne ± SEM de 6-9 expériences indépendantes réalisées en duplicats. *p<0,05.
Les AGE exercent donc un effet anti-prolifératif sur les deux types de cellules étudiés, comme
indiqué à la fois par la baisse du nombre de cellules et par la diminution de la quantité totale
de protéines.
120

II. EFFETS DES AGE SUR LE METABOLISME DES
GANGLIOSIDES
1. LE PROFIL BASAL EN GANGLIOSIDES Les deux modèles cellulaires ont été traités pendant un passage avec 3 µM de BSA contrôle ou
d’AGE puis les gangliosides ont été extraits, purifiés, séparés par HPTLC et révélés à la fois au
résorcinol et par autoradiographie grâce à un marquage métabolique préalable au 14C-galactose.
L'analyse a permis de déterminer d'une part la composition en gangliosides des BRP et RMC à l'état
basal et d'autre part l'effet des AGE sur ce profil en gangliosides.
Comme l'illustre la Figure 36A, le profil basal en gangliosides est spécifique du type cellulaire. Les
gangliosides se présentent sous la forme de doublets, correspondant probablement à des différences
dans les chaînes hydrophobes formant le céramide pour une même chaîne glycosidique. Les
gangliosides sont ici identifiés par comigration avec un standard. Dans les péricytes, les
gangliosides majoritaires sont le GM3 (63 % de tous les gangliosides détectés) et le GM1 (9 %)
parmi les gangliosides de la série-a (Figure 36B), et le GD3 (28 %) parmi les gangliosides de la
série-b. Le profil en gangliosides des cellules mésangiales diffère par une faible proportion de GD3
(5 % de tous les gangliosides détectés) et parmi la série-a, par la présence de GD1a (20 %), le GM3
restant largement majoritaire (75 %). Ces résultats sont en accord avec la composition des cellules
microvasculaires déjà caractérisées, où le GM3 est prédominant.
121

Figure 36 : Profil en gangliosides des péricytes rétiniens et des cellules mésangiales rénales. Les gangliosides des
BRP et des RMC ont été extraits puis purifiés. Ils ont ensuite été séparés sur plaque HPTLC et révélés au résorcinol.
Les différentes espèces ont été identifiées par co-migration avec des standards.
1.1.Effet des AGE sur la composition en gangliosides des péricytes
Comme représenté sur la Figure 37A, les AGE induisent des modifications spécifiques du profil en
gangliosides présents dans les péricytes. Les quantités de GM3 et GM1 augmentent d’environ 40 %
par rapport au contrôle BSA, alors que celle de GD3 diminue de 24 %, en réponse aux AGE. Des
résultats similaires ont été obtenus par autoradiographie (Figure 37B).
GM3
GD3
GT1b(standard interne)
RMCBRP
GM1
GT1b(standard interne) standardsstandards
GD1a
GM2
A B
Laccer GM3 GT3
GM2 GD2 GT2
GM1 GD1b GT1c
GD1a GT1b GQ1c
GT1a GQ1b GP1c
GD3
série-a série-b série-c
GM3
GT1b(standard interne)
RMCBRP
GM1
GD3
GT1b(standard interne) standardsstandards
GD1a
GM2
GM3
GT1b(standard interne)
GT1b(standard interne)
RMCBRP
GM1
GD3
GT1b(standard interne) standardsstandards
GD1aGD1a
GM2
A B
Laccer GM3 GT3
GM2 GD2 GT2
GM1 GD1b GT1c
GD1a GT1b GQ1c
GT1a GQ1b GP1c
GD3
série-a série-b série-c
Laccer GM3 GT3
GM2 GD2 GT2
GM1 GD1b GT1c
GD1a GT1b GQ1c
GT1a GQ1b GP1c
GD3
série-a série-b série-c
122
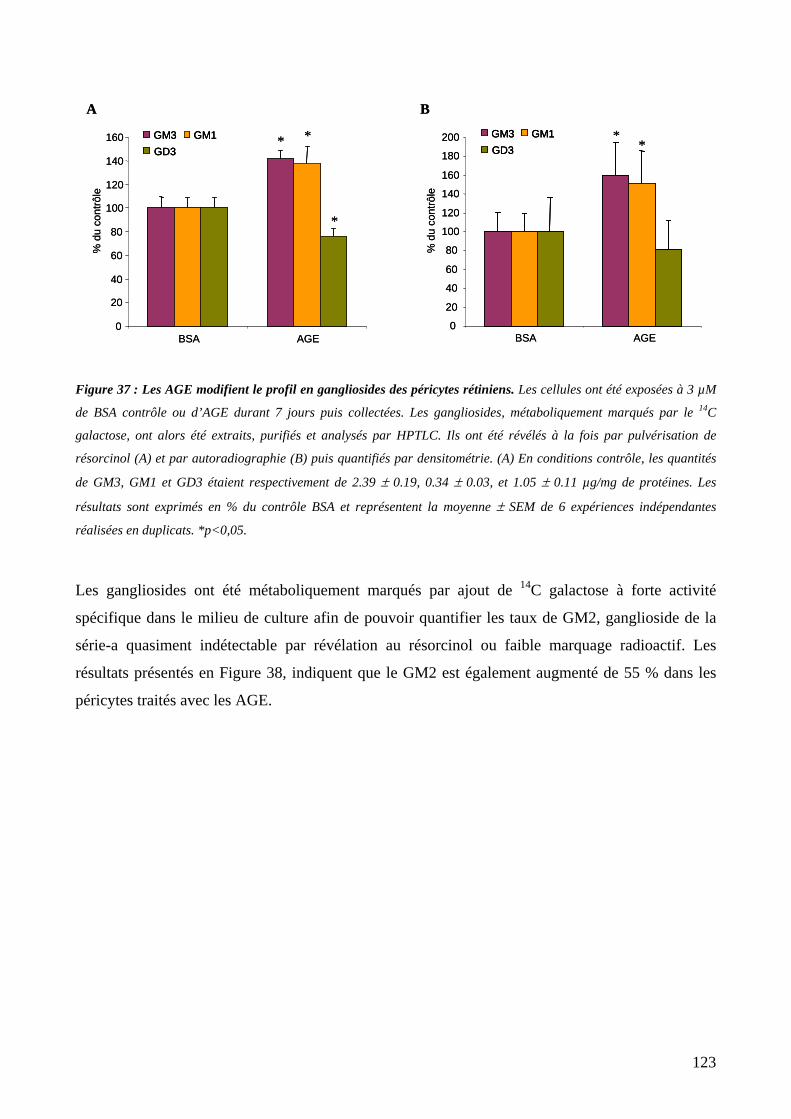
0
20
40
60
80
100
120
140
160
180
200
% d
u co
ntrô
le
GM3 GM1GD3
BSA AGE0
20
40
60
80
100
120
140
160
BSA AGE
% d
u co
ntrô
le
GM3 GM1GD3
* *
*
A B
**
0
20
40
60
80
100
120
140
160
180
200
% d
u co
ntrô
le
GM3 GM1GD3GM3 GM1GD3
BSA AGE0
20
40
60
80
100
120
140
160
BSA AGE
% d
u co
ntrô
le
GM3 GM1GD3GM3 GM1GD3
* *
*
A B
**
Figure 37 : Les AGE modifient le profil en gangliosides des péricytes rétiniens. Les cellules ont été exposées à 3 µM
de BSA contrôle ou d’AGE durant 7 jours puis collectées. Les gangliosides, métaboliquement marqués par le 14C
galactose, ont alors été extraits, purifiés et analysés par HPTLC. Ils ont été révélés à la fois par pulvérisation de
résorcinol (A) et par autoradiographie (B) puis quantifiés par densitométrie. (A) En conditions contrôle, les quantités
de GM3, GM1 et GD3 étaient respectivement de 2.39 ± 0.19, 0.34 ± 0.03, et 1.05 ± 0.11 µg/mg de protéines. Les
résultats sont exprimés en % du contrôle BSA et représentent la moyenne ± SEM de 6 expériences indépendantes
réalisées en duplicats. *p<0,05.
Les gangliosides ont été métaboliquement marqués par ajout de 14C galactose à forte activité
spécifique dans le milieu de culture afin de pouvoir quantifier les taux de GM2, ganglioside de la
série-a quasiment indétectable par révélation au résorcinol ou faible marquage radioactif. Les
résultats présentés en Figure 38, indiquent que le GM2 est également augmenté de 55 % dans les
péricytes traités avec les AGE.
123
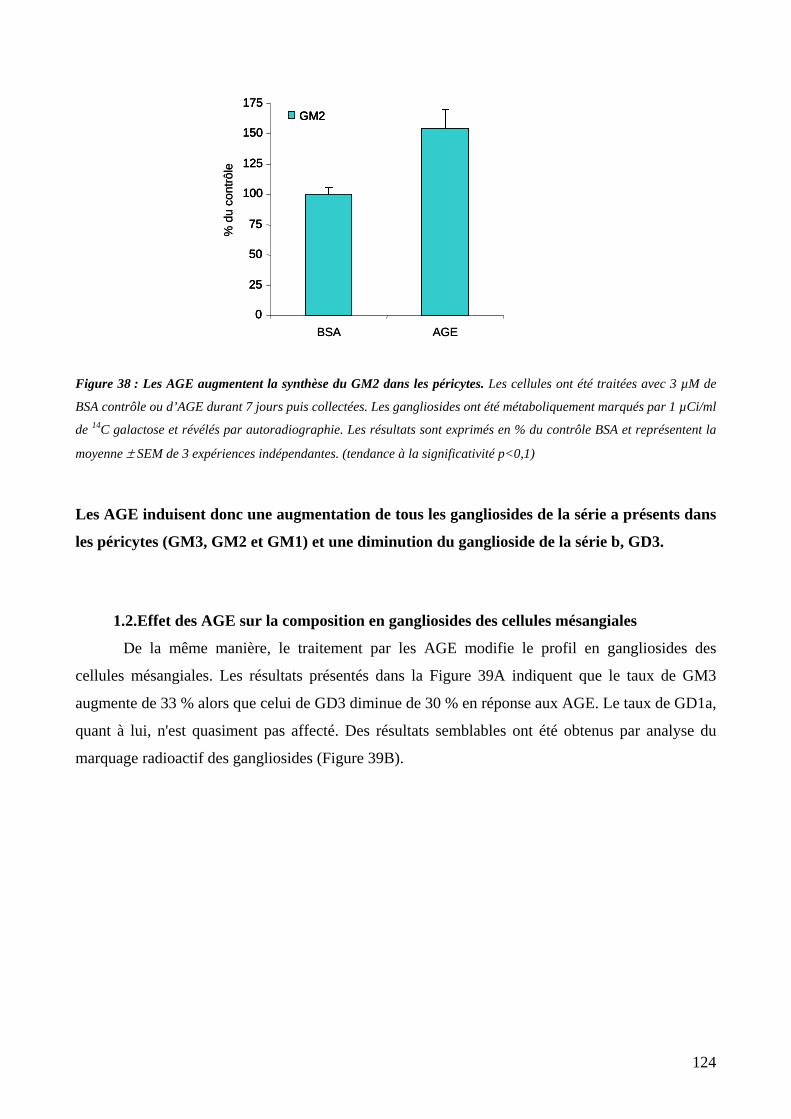
BSA AGE
%du
con
trôle
GM2
0
25
50
75
100
125
150
175
BSA AGE
%du
con
trôle
GM2GM2
0
25
50
75
100
125
150
175
0
25
50
75
100
125
150
175
Figure 38 : Les AGE augmentent la synthèse du GM2 dans les péricytes. Les cellules ont été traitées avec 3 µM de
BSA contrôle ou d’AGE durant 7 jours puis collectées. Les gangliosides ont été métaboliquement marqués par 1 µCi/ml
de 14C galactose et révélés par autoradiographie. Les résultats sont exprimés en % du contrôle BSA et représentent la
moyenne ± SEM de 3 expériences indépendantes. (tendance à la significativité p<0,1)
Les AGE induisent donc une augmentation de tous les gangliosides de la série a présents dans
les péricytes (GM3, GM2 et GM1) et une diminution du ganglioside de la série b, GD3.
1.2.Effet des AGE sur la composition en gangliosides des cellules mésangiales
De la même manière, le traitement par les AGE modifie le profil en gangliosides des
cellules mésangiales. Les résultats présentés dans la Figure 39A indiquent que le taux de GM3
augmente de 33 % alors que celui de GD3 diminue de 30 % en réponse aux AGE. Le taux de GD1a,
quant à lui, n'est quasiment pas affecté. Des résultats semblables ont été obtenus par analyse du
marquage radioactif des gangliosides (Figure 39B).
124
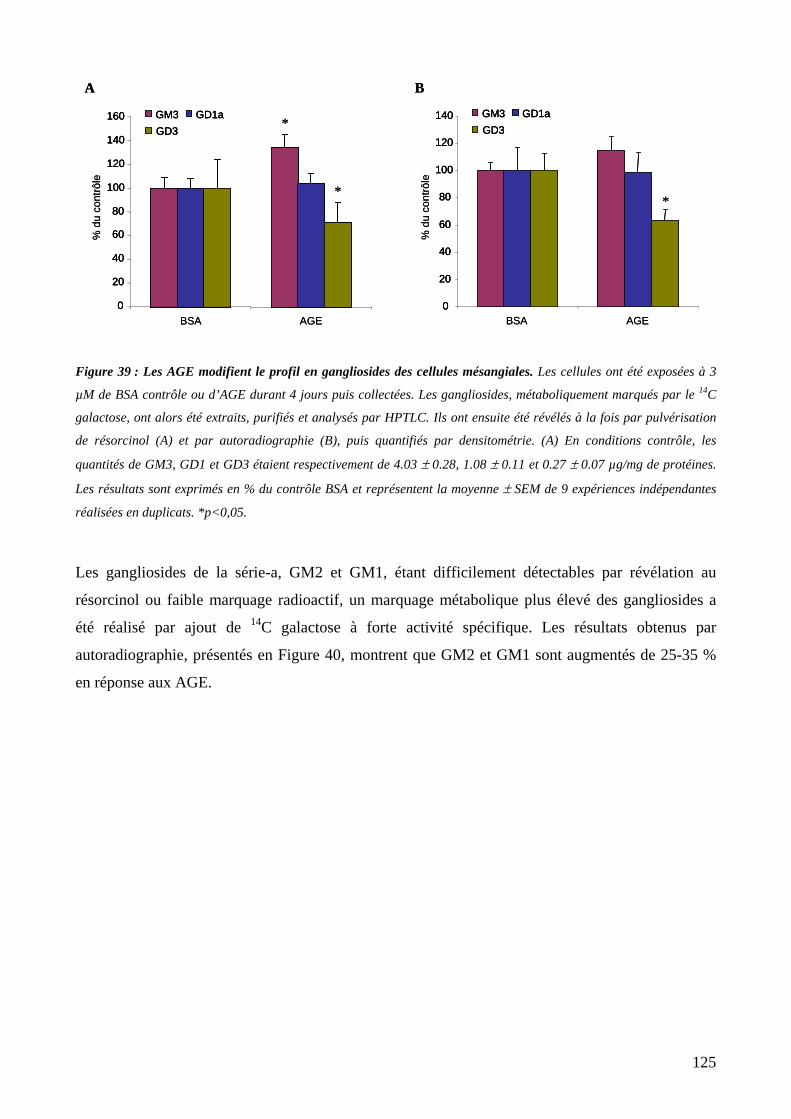
A B
0
20
40
60
80
100
120
140
160
BSA AGE
%du
con
trôle
GM3 GD1aGD3 *
*
0
20
40
60
80
100
120
140
BSA AGE
%du
con
trôle
GM3 GD1aGD3
*
A B
0
20
40
60
80
100
120
140
160
0
20
40
60
80
100
120
140
160
BSA AGE
%du
con
trôle
GM3 GD1aGD3GM3 GD1aGD3 *
*
0
20
40
60
80
100
120
140
0
20
40
60
80
100
120
140
BSA AGE
%du
con
trôle
GM3 GD1aGD3GM3 GD1aGD3
*
Figure 39 : Les AGE modifient le profil en gangliosides des cellules mésangiales. Les cellules ont été exposées à 3
µM de BSA contrôle ou d’AGE durant 4 jours puis collectées. Les gangliosides, métaboliquement marqués par le 14C
galactose, ont alors été extraits, purifiés et analysés par HPTLC. Ils ont ensuite été révélés à la fois par pulvérisation
de résorcinol (A) et par autoradiographie (B), puis quantifiés par densitométrie. (A) En conditions contrôle, les
quantités de GM3, GD1 et GD3 étaient respectivement de 4.03 ± 0.28, 1.08 ± 0.11 et 0.27 ± 0.07 µg/mg de protéines.
Les résultats sont exprimés en % du contrôle BSA et représentent la moyenne ± SEM de 9 expériences indépendantes
réalisées en duplicats. *p<0,05.
Les gangliosides de la série-a, GM2 et GM1, étant difficilement détectables par révélation au
résorcinol ou faible marquage radioactif, un marquage métabolique plus élevé des gangliosides a
été réalisé par ajout de 14C galactose à forte activité spécifique. Les résultats obtenus par
autoradiographie, présentés en Figure 40, montrent que GM2 et GM1 sont augmentés de 25-35 %
en réponse aux AGE.
125
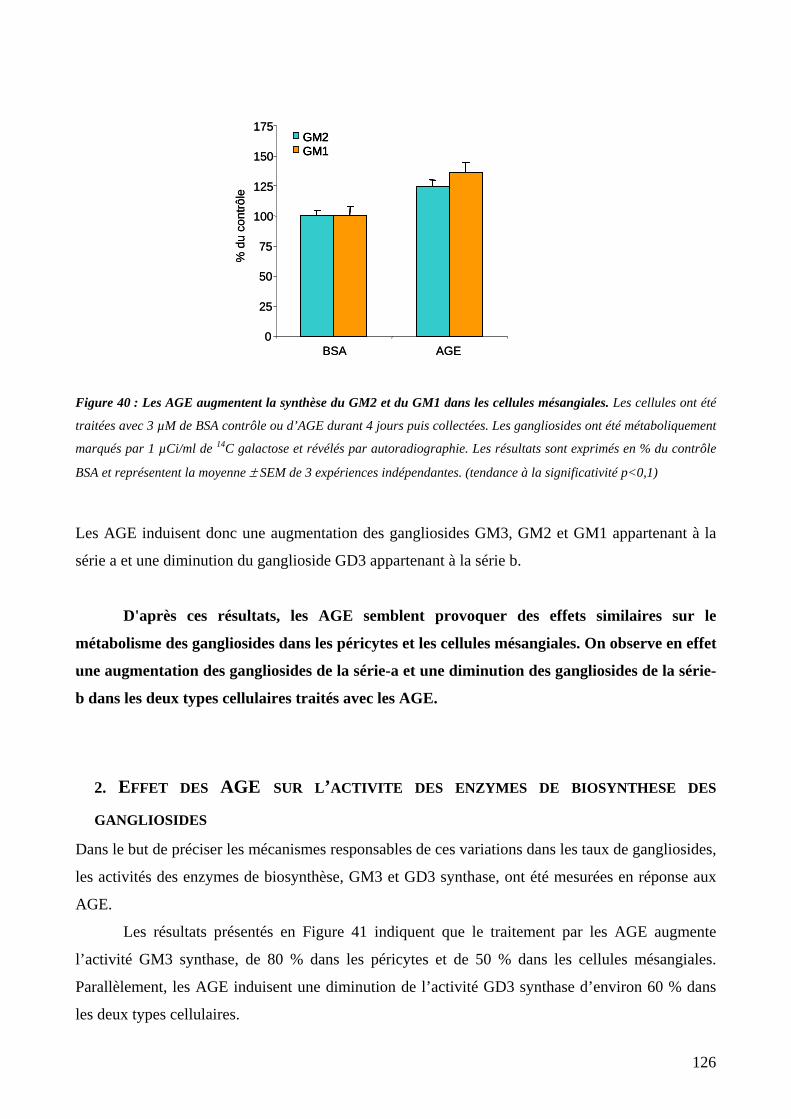
BSA AGE
%du
con
trôle
0
25
50
75
100
125
150
175GM2GM1
BSA AGE
%du
con
trôle
0
25
50
75
100
125
150
175GM2GM1GM2GM1
Figure 40 : Les AGE augmentent la synthèse du GM2 et du GM1 dans les cellules mésangiales. Les cellules ont été
traitées avec 3 µM de BSA contrôle ou d’AGE durant 4 jours puis collectées. Les gangliosides ont été métaboliquement
marqués par 1 µCi/ml de 14C galactose et révélés par autoradiographie. Les résultats sont exprimés en % du contrôle
BSA et représentent la moyenne ± SEM de 3 expériences indépendantes. (tendance à la significativité p<0,1)
Les AGE induisent donc une augmentation des gangliosides GM3, GM2 et GM1 appartenant à la
série a et une diminution du ganglioside GD3 appartenant à la série b.
D'après ces résultats, les AGE semblent provoquer des effets similaires sur le
métabolisme des gangliosides dans les péricytes et les cellules mésangiales. On observe en effet
une augmentation des gangliosides de la série-a et une diminution des gangliosides de la série-
b dans les deux types cellulaires traités avec les AGE.
2. EFFET DES AGE SUR L’ACTIVITE DES ENZYMES DE BIOSYNTHESE DES
GANGLIOSIDES Dans le but de préciser les mécanismes responsables de ces variations dans les taux de gangliosides,
les activités des enzymes de biosynthèse, GM3 et GD3 synthase, ont été mesurées en réponse aux
AGE.
Les résultats présentés en Figure 41 indiquent que le traitement par les AGE augmente
l’activité GM3 synthase, de 80 % dans les péricytes et de 50 % dans les cellules mésangiales.
Parallèlement, les AGE induisent une diminution de l’activité GD3 synthase d’environ 60 % dans
les deux types cellulaires.
126
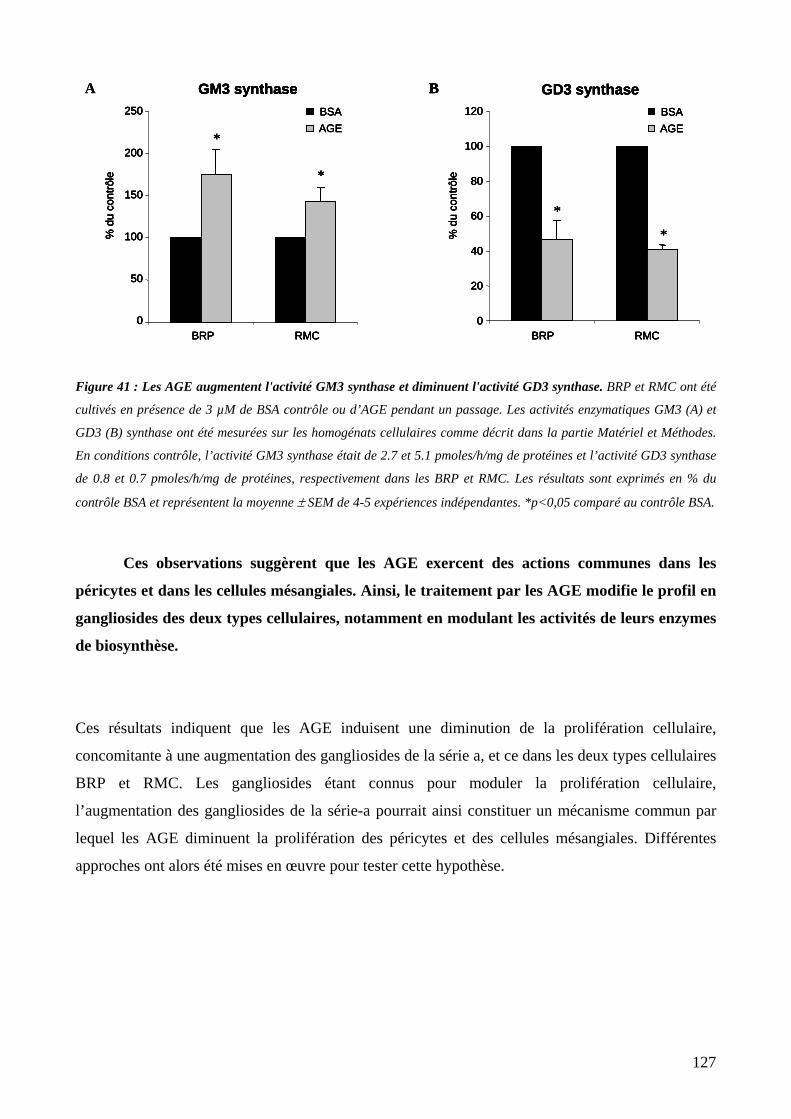
A%
du c
ontrô
le
*
*
0
50
100
150
200
250
BRP RMC
BSAAGE
GM3 synthase B
0
20
40
60
80
100
120
**
%du
con
trôle
BSAAGE
BRP RMC
GD3 synthaseA%
du c
ontrô
le
*
*
0
50
100
150
200
250
BRP RMC
BSAAGE
GM3 synthase B
0
20
40
60
80
100
120
**
%du
con
trôle
BSAAGE
BRP RMC
GD3 synthase%
du c
ontrô
le
*
*
0
50
100
150
200
250
BRP RMC
BSAAGE
GM3 synthase%
du c
ontrô
le
*
*
0
50
100
150
200
250
BRP RMC
BSAAGE
GM3 synthase B
0
20
40
60
80
100
120
**
%du
con
trôle
BSAAGE
BRP RMC
GD3 synthaseB
0
20
40
60
80
100
120
**
%du
con
trôle
BSAAGE
BRP RMC
GD3 synthase
Figure 41 : Les AGE augmentent l'activité GM3 synthase et diminuent l'activité GD3 synthase. BRP et RMC ont été
cultivés en présence de 3 µM de BSA contrôle ou d’AGE pendant un passage. Les activités enzymatiques GM3 (A) et
GD3 (B) synthase ont été mesurées sur les homogénats cellulaires comme décrit dans la partie Matériel et Méthodes.
En conditions contrôle, l’activité GM3 synthase était de 2.7 et 5.1 pmoles/h/mg de protéines et l’activité GD3 synthase
de 0.8 et 0.7 pmoles/h/mg de protéines, respectivement dans les BRP et RMC. Les résultats sont exprimés en % du
contrôle BSA et représentent la moyenne ± SEM de 4-5 expériences indépendantes. *p<0,05 comparé au contrôle BSA.
Ces observations suggèrent que les AGE exercent des actions communes dans les
péricytes et dans les cellules mésangiales. Ainsi, le traitement par les AGE modifie le profil en
gangliosides des deux types cellulaires, notamment en modulant les activités de leurs enzymes
de biosynthèse.
Ces résultats indiquent que les AGE induisent une diminution de la prolifération cellulaire,
concomitante à une augmentation des gangliosides de la série a, et ce dans les deux types cellulaires
BRP et RMC. Les gangliosides étant connus pour moduler la prolifération cellulaire,
l’augmentation des gangliosides de la série-a pourrait ainsi constituer un mécanisme commun par
lequel les AGE diminuent la prolifération des péricytes et des cellules mésangiales. Différentes
approches ont alors été mises en œuvre pour tester cette hypothèse.
127

III. IMPLICATION DES GANGLIOSIDES DANS LA
DIMINUTION DE LA PROLIFERATION CELLULAIRE CAUSEE
PAR LES AGE
1. EFFET D’UN TRAITEMENT PAR DES GANGLIOSIDES EXOGENES SUR LA
PROLIFERATION DES PERICYTES ET DES CELLULES MESANGIALES De nombreuses données de la littérature montrent que les gangliosides, et en particulier ceux de la
série-a, inhibent la croissance cellulaire. L’utilisation de gangliosides exogènes est courante dans
ces études et il a été rapporté qu’au moins une partie de ces gangliosides exogènes apportés aux
cellules dans le milieu de culture s’incorpore dans la membrane plasmique (Bremer E.G. et al.,
1984). L’effet sur la prolifération cellulaire des différents gangliosides de la série-a, augmentés en
réponse aux AGE, a donc été étudié.
Dans ce but, les cellules ont été traitées par des doses croissantes de GM3, GM2, GM1 ainsi que de
leurs précurseurs non sialylés, Glucosylcéramide et Lactosylcéramide, utilisés comme contrôles,
sous forme de complexe avec la BSA. La prolifération cellulaire a ensuite été évaluée par dosage de
la quantité totale de protéines et par mesure de la quantité d’ADN.
La figure 42 montre que les gangliosides testés inhibent la prolifération des BRP, de manière
dose-dépendante, alors que Glucosylcéramide et Lactosylcéramide sont sans effet. L’inhibition de
prolifération est observée à partir d’une concentration de 50 µM et semble plus efficace avec les
gangliosides GM2 et GM1, atteignant 25 % à une concentration de 100 µM. Les résultats obtenus
en utilisant le test du CyQuant (Figure 42B) sont conformes bien que plus modérés.
128

BA
% d
u co
ntrô
le
0
20
40
60
80
100
120
140
contrôle GM3 GM2 GM1 GlucCer LacCer
10µM 50µM 100µM
% d
u co
ntrô
le
0
20
40
60
80
100
120
140
contrôle GM3 GM2 GM1 GlucCer LacCer
10µM 50µM 100µM
** **
** *
**
BA
% d
u co
ntrô
le
0
20
40
60
80
100
120
140
contrôle GM3 GM2 GM1 GlucCer LacCer
10µM 50µM 100µM
% d
u co
ntrô
le
0
20
40
60
80
100
120
140
contrôle GM3 GM2 GM1 GlucCer LacCer
10µM 50µM 100µM
BA
% d
u co
ntrô
le
0
20
40
60
80
100
120
140
0
20
40
60
80
100
120
140
contrôle GM3 GM2 GM1 GlucCer LacCer
10µM10µM 50µM50µM 100µM100µM
% d
u co
ntrô
le
0
20
40
60
80
100
120
140
contrôle GM3 GM2 GM1 GlucCer LacCer
10µM 50µM 100µM10µM10µM 50µM50µM 100µM100µM
** **
** *
**
Figure 42 : L’ajout exogène de gangliosides de la série a diminue la prolifération des BRP. Différentes doses de
complexes glycolipides-BSA ont été ajoutées au milieu de culture pendant un passage. Les protéines totales (A) et
l'ADN (B) ont ensuite été mesurés. Les résultats sont exprimés en % du contrôle et représentent la moyenne ± SEM de 5
expériences indépendantes réalisées en triplicats. *p<0,05 comparé au contrôle.
Dans les cellules mésangiales, les gangliosides testés exercent également un effet anti-
prolifératif contrairement aux Glucosylcéramide et Lactosylcéramide qui restent sans effet (Figure
43). L’effet est observé à partir d’une concentration de 50 µM et l’inhibition de prolifération atteint
25-30 % à 100 µM. Les résultats obtenus par le test du CyQuant confirment cet effet d’inhibition de
la prolifération exercé par les gangliosides exogènes.
129
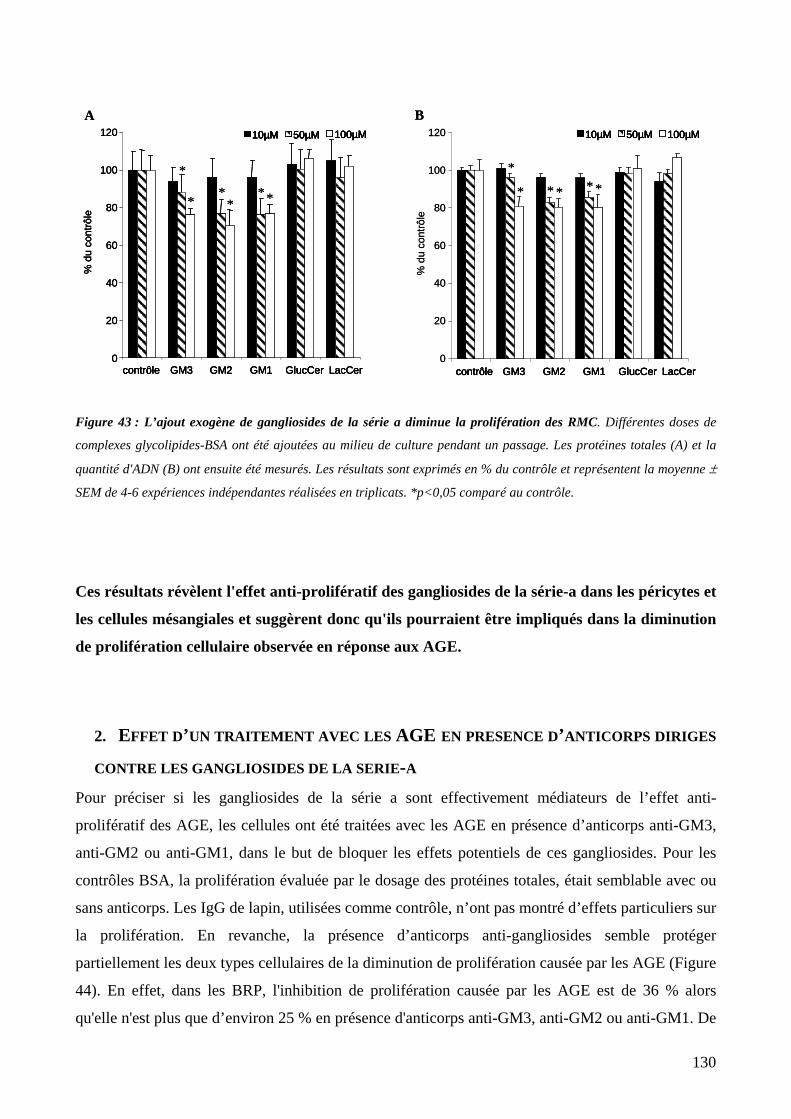
BA
% d
u co
ntrô
le
10µM 50µM 100µM
% d
u co
ntrô
le
10µM 50µM 100µM
contrôle GM3 GM2 GM1 GlucCer LacCer contrôle GM3 GM2 GM1 GlucCer LacCer0
20
40
60
80
100
120
0
20
40
60
80
100
120
***
**
*****
*
*
BA
% d
u co
ntrô
le
10µM 50µM 100µM10µM10µM 50µM50µM 100µM100µM
% d
u co
ntrô
le
10µM 50µM 100µM
contrôle GM3 GM2 GM1 GlucCer LacCer contrôle GM3 GM2 GM1 GlucCer LacCer0
20
40
60
80
100
120
% d
u co
ntrô
le
10µM 50µM 100µM10µM10µM 50µM50µM 100µM100µM
contrôle GM3 GM2 GM1 GlucCer LacCer contrôle GM3 GM2 GM1 GlucCer LacCercontrôle GM3 GM2 GM1 GlucCer LacCer contrôle GM3 GM2 GM1 GlucCer LacCer0
20
40
60
80
100
120
0
20
40
60
80
100
120
***
**
*****
*
*
Figure 43 : L’ajout exogène de gangliosides de la série a diminue la prolifération des RMC. Différentes doses de
complexes glycolipides-BSA ont été ajoutées au milieu de culture pendant un passage. Les protéines totales (A) et la
quantité d'ADN (B) ont ensuite été mesurés. Les résultats sont exprimés en % du contrôle et représentent la moyenne ±
SEM de 4-6 expériences indépendantes réalisées en triplicats. *p<0,05 comparé au contrôle.
Ces résultats révèlent l'effet anti-prolifératif des gangliosides de la série-a dans les péricytes et
les cellules mésangiales et suggèrent donc qu'ils pourraient être impliqués dans la diminution
de prolifération cellulaire observée en réponse aux AGE.
2. EFFET D’UN TRAITEMENT AVEC LES AGE EN PRESENCE D’ANTICORPS DIRIGES
CONTRE LES GANGLIOSIDES DE LA SERIE-A Pour préciser si les gangliosides de la série a sont effectivement médiateurs de l’effet anti-
prolifératif des AGE, les cellules ont été traitées avec les AGE en présence d’anticorps anti-GM3,
anti-GM2 ou anti-GM1, dans le but de bloquer les effets potentiels de ces gangliosides. Pour les
contrôles BSA, la prolifération évaluée par le dosage des protéines totales, était semblable avec ou
sans anticorps. Les IgG de lapin, utilisées comme contrôle, n’ont pas montré d’effets particuliers sur
la prolifération. En revanche, la présence d’anticorps anti-gangliosides semble protéger
partiellement les deux types cellulaires de la diminution de prolifération causée par les AGE (Figure
44). En effet, dans les BRP, l'inhibition de prolifération causée par les AGE est de 36 % alors
qu'elle n'est plus que d’environ 25 % en présence d'anticorps anti-GM3, anti-GM2 ou anti-GM1. De
130

même dans les cellules mésangiales, l'inhibition de prolifération passe de 35 % avec les AGE seuls
à environ 15 % en présence des anti-GM2 et anti-GM1, l’anticorps anti-GM3 ne présentant pas
d’effet significatif.
A
% d
u co
ntrô
le
0
20
40
60
80
100
120
BSA
AGE
contrôle IgG anti GM3 anti GM1anti GM2
B
% d
u co
ntrô
le0
20
40
60
80
100
120
BSA
AGE
contrôle IgG anti GM3 anti GM1anti GM2
BRP RMC
* *
*
A
% d
u co
ntrô
le
0
20
40
60
80
100
120
BSA
AGE
contrôle IgG anti GM3 anti GM1anti GM2
B
% d
u co
ntrô
le0
20
40
60
80
100
120
BSA
AGE
contrôle IgG anti GM3 anti GM1anti GM2
BRP RMCA
% d
u co
ntrô
le
0
20
40
60
80
100
120
BSA
AGE
contrôle IgG anti GM3 anti GM1anti GM2
B
% d
u co
ntrô
le0
20
40
60
80
100
120
BSA
AGE
contrôle IgG anti GM3 anti GM1anti GM2
A
% d
u co
ntrô
le
0
20
40
60
80
100
120
BSA
AGE
contrôle IgG anti GM3 anti GM1anti GM2
A
% d
u co
ntrô
le
0
20
40
60
80
100
120
BSA
AGE
BSA
AGE
contrôle IgG anti GM3 anti GM1anti GM2contrôle IgG anti GM3 anti GM1anti GM2
B
% d
u co
ntrô
le0
20
40
60
80
100
120
BSA
AGE
contrôle IgG anti GM3 anti GM1anti GM2
B
% d
u co
ntrô
le0
20
40
60
80
100
120
BSA
AGE
BSA
AGE
contrôle IgG anti GM3 anti GM1anti GM2contrôle IgG anti GM3 anti GM1anti GM2
BRP RMC
* *
*
Figure 44 : L’utilisation d’anticorps anti-GM2 et anti-GM1 protège partiellement de l’effet anti-prolifératif des
AGE. BRP (A) et RMC (B) ont été traitées avec 3 µM de BSA contrôle ou d’AGE pendant un passage en présence ou en
absence de 0.5 µg par puits d’anticorps monoclonal anti-GM3 ou 5 µg par puits d'anticorps polyclonal anti-GM2, anti-
GM1 ou d’IgG. Après rinçage et lyse des cellules, les protéines totales ont été mesurées. Les résultats sont exprimés en
% du contrôle BSA et représentent la moyenne ± SEM de 5-6 expériences indépendantes réalisées en triplicats.
*p<0,05 comparé aux cellules traitées avec AGE seul.
Combinés avec l’effet des gangliosides exogènes, ces résultats suggèrent que, dans les péricytes
et les cellules mésangiales, les gangliosides de la série-a, en particulier GM2 et GM1, sont des
médiateurs de l’inhibition de prolifération causée par les AGE.
3. EFFET D’UN COTRAITEMENT AGE ET PPMP Une approche complémentaire a été envisagée pour soutenir l’implication des gangliosides de la
série-a dans les effets des AGE. Le PPMP, un inhibiteur de glucosylcéramide synthase, a été utilisé
pour bloquer l’augmentation des gangliosides causée par les AGE.
Ainsi, BRP et RMC ont été traités par les AGE en présence de concentrations croissantes de PPMP
puis la prolifération a été évaluée par dosage des protéines totales.
Dans les BRP, l'ajout de PPMP au traitement AGE permet de réduire l'inhibition de
prolifération causée par les AGE. En effet, la quantité de protéines est 26 % plus importante dans
131
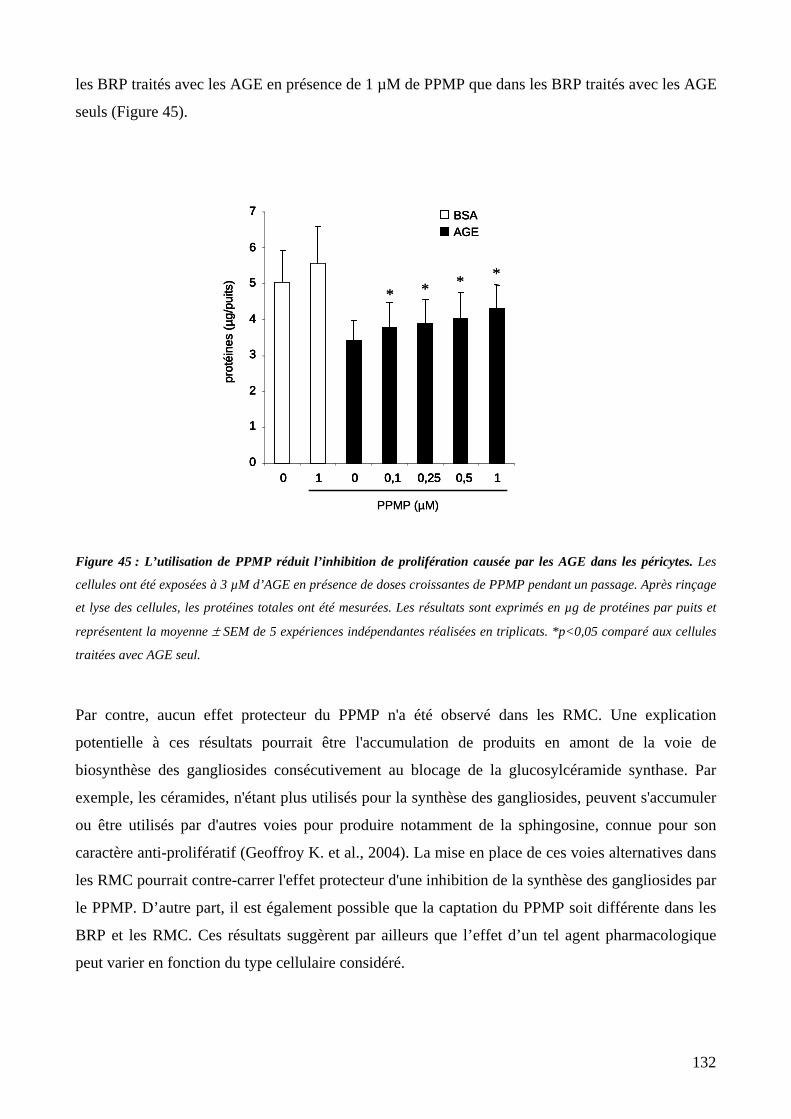
les BRP traités avec les AGE en présence de 1 µM de PPMP que dans les BRP traités avec les AGE
seuls (Figure 45).
PPMP (µM)
prot
éine
s(µ
g/pu
its)
0
1
2
3
4
5
6
7
* * *
BSAAGE
*
0 1 0 0,1 0,25 0,5 1
PPMP (µM)
prot
éine
s(µ
g/pu
its)
0
1
2
3
4
5
6
7pr
otéi
nes
(µg/
puits
)
0
1
2
3
4
5
6
7
0
1
2
3
4
5
6
7
* * *
BSAAGEBSAAGE
*
0 1 0 0,1 0,25 0,5 10 1 0 0,1 0,25 0,5 10 1 0 0,1 0,25 0,5 1
Figure 45 : L’utilisation de PPMP réduit l’inhibition de prolifération causée par les AGE dans les péricytes. Les
cellules ont été exposées à 3 µM d’AGE en présence de doses croissantes de PPMP pendant un passage. Après rinçage
et lyse des cellules, les protéines totales ont été mesurées. Les résultats sont exprimés en µg de protéines par puits et
représentent la moyenne ± SEM de 5 expériences indépendantes réalisées en triplicats. *p<0,05 comparé aux cellules
traitées avec AGE seul.
Par contre, aucun effet protecteur du PPMP n'a été observé dans les RMC. Une explication
potentielle à ces résultats pourrait être l'accumulation de produits en amont de la voie de
biosynthèse des gangliosides consécutivement au blocage de la glucosylcéramide synthase. Par
exemple, les céramides, n'étant plus utilisés pour la synthèse des gangliosides, peuvent s'accumuler
ou être utilisés par d'autres voies pour produire notamment de la sphingosine, connue pour son
caractère anti-prolifératif (Geoffroy K. et al., 2004). La mise en place de ces voies alternatives dans
les RMC pourrait contre-carrer l'effet protecteur d'une inhibition de la synthèse des gangliosides par
le PPMP. D’autre part, il est également possible que la captation du PPMP soit différente dans les
BRP et les RMC. Ces résultats suggèrent par ailleurs que l’effet d’un tel agent pharmacologique
peut varier en fonction du type cellulaire considéré.
132
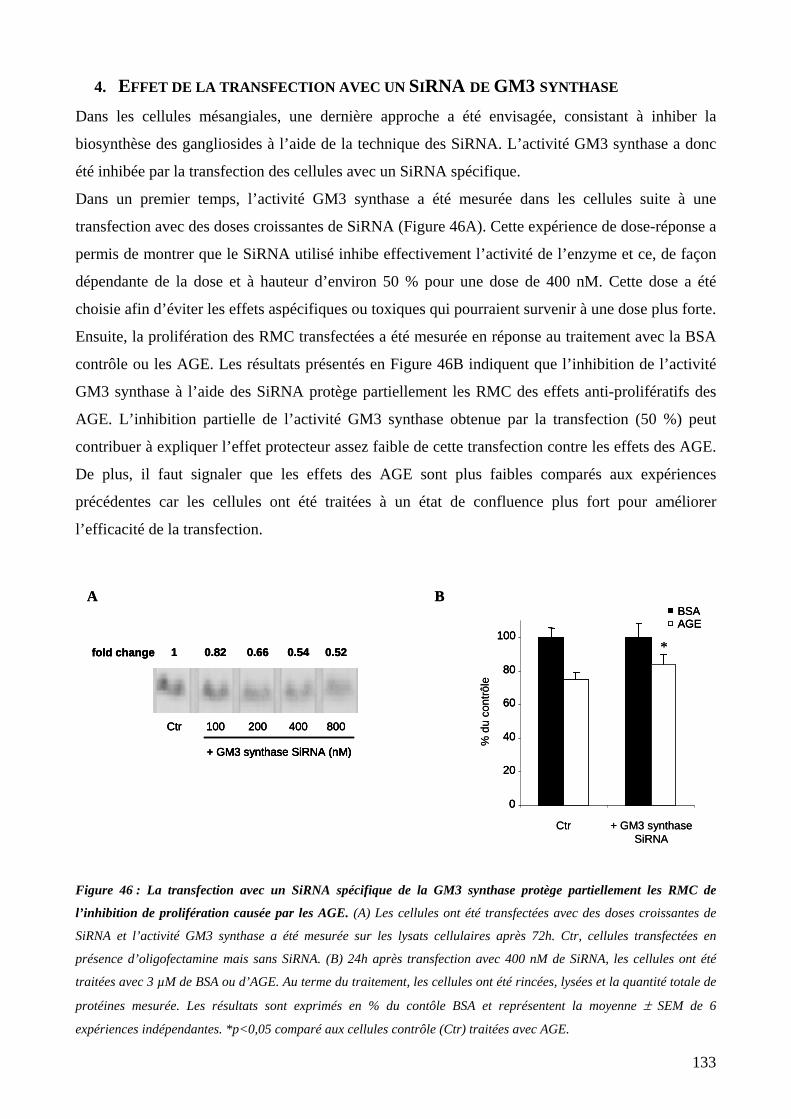
4. EFFET DE LA TRANSFECTION AVEC UN SIRNA DE GM3 SYNTHASE Dans les cellules mésangiales, une dernière approche a été envisagée, consistant à inhiber la
biosynthèse des gangliosides à l’aide de la technique des SiRNA. L’activité GM3 synthase a donc
été inhibée par la transfection des cellules avec un SiRNA spécifique.
Dans un premier temps, l’activité GM3 synthase a été mesurée dans les cellules suite à une
transfection avec des doses croissantes de SiRNA (Figure 46A). Cette expérience de dose-réponse a
permis de montrer que le SiRNA utilisé inhibe effectivement l’activité de l’enzyme et ce, de façon
dépendante de la dose et à hauteur d’environ 50 % pour une dose de 400 nM. Cette dose a été
choisie afin d’éviter les effets aspécifiques ou toxiques qui pourraient survenir à une dose plus forte.
Ensuite, la prolifération des RMC transfectées a été mesurée en réponse au traitement avec la BSA
contrôle ou les AGE. Les résultats présentés en Figure 46B indiquent que l’inhibition de l’activité
GM3 synthase à l’aide des SiRNA protège partiellement les RMC des effets anti-prolifératifs des
AGE. L’inhibition partielle de l’activité GM3 synthase obtenue par la transfection (50 %) peut
contribuer à expliquer l’effet protecteur assez faible de cette transfection contre les effets des AGE.
De plus, il faut signaler que les effets des AGE sont plus faibles comparés aux expériences
précédentes car les cellules ont été traitées à un état de confluence plus fort pour améliorer
l’efficacité de la transfection.
% d
u co
ntrô
le
BA
200 400 800Ctr 100
+ GM3 synthase SiRNA (nM)
fold change 1 0.82 0.66 0.54 0.52
Ctr + GM3 synthase SiRNA
BSAAGE
0
20
40
60
80
100*
% d
u co
ntrô
le
BA
200 400 800Ctr 100
+ GM3 synthase SiRNA (nM)
fold change 1 0.82 0.66 0.54 0.52
200 400 800Ctr 100
+ GM3 synthase SiRNA (nM)+ GM3 synthase SiRNA (nM)
fold change 1 0.82 0.66 0.54 0.52
Ctr + GM3 synthase SiRNA
BSAAGEBSAAGE
0
20
40
60
80
100
0
20
40
60
80
100*
Figure 46 : La transfection avec un SiRNA spécifique de la GM3 synthase protège partiellement les RMC de
l’inhibition de prolifération causée par les AGE. (A) Les cellules ont été transfectées avec des doses croissantes de
SiRNA et l’activité GM3 synthase a été mesurée sur les lysats cellulaires après 72h. Ctr, cellules transfectées en
présence d’oligofectamine mais sans SiRNA. (B) 24h après transfection avec 400 nM de SiRNA, les cellules ont été
traitées avec 3 µM de BSA ou d’AGE. Au terme du traitement, les cellules ont été rincées, lysées et la quantité totale de
protéines mesurée. Les résultats sont exprimés en % du contôle BSA et représentent la moyenne ± SEM de 6
expériences indépendantes. *p<0,05 comparé aux cellules contrôle (Ctr) traitées avec AGE.
133

Ces observations confirment clairement l’implication de la GM3 synthase et des gangliosides
de la série-a dans l’inhibition de prolifération des cellules mésangiales causée par les AGE.
IV. DISCUSSION
Cette première partie du travail expérimental porte sur les AGE, produits s’accumulant dans un
environnement diabétique. Ces produits de glycation avancés se retrouvent notamment dans le rein
et la rétine de patients diabétiques et sont connus pour jouer un rôle important dans le
développement de la rétinopathie et la néphropathie diabétiques. Les AGE utilisés ont été préparés
par modification de l'albumine par le méthylglyoxal. Ces AGE-méthylglyoxal ont été observés dans
les protéines tissulaires chez le diabétique et sont donc représentatifs d'AGE s'accumulant in vivo au
cours du diabète (Degenhardt T.P. et al., 1998; Shamsi F.A. et al., 1998). Par ailleurs, le traitement
avec les AGE a été réalisé à une concentration déjà décrite pour induire des modifications du
métabolisme et de la prolifération cellulaire (Chibber R. et al., 1997; Yamagishi S. et al., 2002b;
Yamagishi S. et al., 2002a), notamment par notre équipe (Denis U. et al., 2002; Natalizio A. et al.,
2001).
Dans un premier temps, nous avons évalué l'effet des AGE sur la prolifération des BRP et
des RMC. Les résultats ont montré que le traitement avec les AGE inhibe la prolifération des deux
types cellulaires d'intérêt. Ces observations sont en accord avec celles de précédentes études
utilisant d'autres types d'AGE à la fois dans les péricytes (Chibber R. et al., 1997; Yamagishi S. et
al., 2002a) et les cellules mésangiales (Yamagishi S. et al., 2002b; Ziyadeh F.N. et al., 1993). Nous
n’avons pas exploré ici les mécanismes responsables de la diminution de prolifération mesurée.
Cependant, Denis et al. ont montré que les AGE sont capables d’induire l’apoptose des péricytes
rétiniens, même si ce processus n’est pas responsable à lui-seul de l’inhibition de prolifération
exercée par les AGE sur les péricytes (Denis U. et al., 2002).
Dans un deuxième temps, l'effet des AGE sur le profil en gangliosides a été étudié. Les
résultats indiquent que le traitement avec les AGE provoque une augmentation des gangliosides de
la série-a (GM3, GM2, GM1) parallèlement à une diminution des gangliosides de la série-b (GD3),
et ce dans les deux types cellulaires. Ces modifications peuvent s'expliquer par l'augmentation de
l'activité GM3 synthase et la diminution de l'activité GD3 synthase observées en réponse aux AGE.
134

L’augmentation de l’activité GM3 synthase se fait très certainement via une augmentation du Vmax
de la réaction plutôt que du Km (résultats non présentés). Ces résultats révèlent que les AGE sont
capables d’induire des effets opposés sur ces deux sialyltransférases et suggèrent qu’ils peuvent
réguler spécifiquement l'activité des enzymes de biosynthèse des gangliosides. Cependant ces
mécanismes de régulation n'ont pas encore été étudiés. Certaines données de la littérature nous
permettent tout de même de formuler une hypothèse. Il a en effet été montré que la liaison des AGE
au RAGE génère un stress oxydant capable d’activer des facteurs de transcription parmi lesquels
Sp1 (Du X.L. et al., 2000). Par ailleurs des analyses du promoteur du gène de la GM3 synthase
humaine ont révélé que sa transcription était positivement régulée par le facteur Sp1 (Zeng G. et al.,
2003). L'expression de RAGE ayant été décrite dans les BRP (Yamagishi S. et al., 2002a; Yonekura
H. et al., 2003) et dans les RMC (Vlassara H., 2001), il est envisageable que les AGE modulent
l'activité des enzymes GM3 et GD3 synthase en régulant leur expression génétique via la voie de
signalisation de RAGE.
Les investigations menées par la suite ont visé à vérifier si les gangliosides de la série-a,
dont les taux se trouvent augmentés en réponse aux AGE, peuvent être des médiateurs des effets
anti-prolifératifs de ces AGE.
Tout d'abord, nous avons étudié l’effet des gangliosides exogènes sur la prolifération des
deux types cellulaires. Le traitement des cellules par les gangliosides GM3, GM2 et GM1 induit une
diminution de la croissance des BRP et des RMC. Ces observations révèlent un effet anti-
prolifératif des gangliosides testés dans nos deux modèles cellulaires. L'inhibition de la prolifération
atteint un maximum de 25-30 % pour une dose de 100 µM. Cet effet relativement faible des
gangliosides exogènes peut s'expliquer par des données sur leur comportement au sein de la cellule.
Ainsi, même si l'addition exogène de gangliosides est utilisée dans de nombreuses études pour
déterminer leur rôle dans la régulation de la prolifération cellulaire, leur comportement dans la
cellule est assez mal connu. Cependant, un certain nombre de travaux anciens ont étudié leur
intégration dans la membrane cellulaire. Bremer et al. ont par exemple montré en utilisant des
gangliosides marqués au tritium, qu'environ 10-20 % des gangliosides ajoutés au milieu de culture
s'associent à la cellule et que, au bout de 48 h, 70 % de ces gangliosides associés sont effectivement
insérés dans la bicouche lipidique. Ces proportions n'étaient pas différentes quelque soit le
ganglioside testé (Bremer E.G. et al., 1984).
Une approche complémentaire a ensuite consisté à utiliser des anticorps dirigés contre les
gangliosides de la série a, GM3, GM2 et GM1, et destinés à bloquer leur action. Le traitement des
cellules avec les AGE en présence de ces anticorps s'est révélé protéger partiellement les BRP et les
RMC de la chute de prolifération causée par les AGE. Ces résultats supportent l'implication des
135

gangliosides dans les effets anti-prolifératifs des AGE. Le caractère partiel de la protection exercée
par les anticorps peut provenir du fait que ces anticorps n'accèdent pas complètement au
compartiment de gangliosides actifs. D'autre part, cela peut également signifier que d'autres voies
sont en cause. Par exemple, de précédents travaux menés au laboratoire ont permis de proposer une
cascade impliquant le stress oxydant, l'accumulation de céramides et les caspases pour expliquer
l'apoptose des péricytes en réponse aux AGE (Denis U. et al., 2002).
Par ailleurs, le PPMP, un inhibiteur de la glucosylcéramide synthase, a été utilisé pour
bloquer l'augmentation de la biosynthèse des gangliosides de la série-a qui s’opère en réponse aux
AGE. L’addition de PPMP a permis de protéger partiellement les BRP de la chute de prolifération
induite par les AGE. Là encore, ces observations soutiennent l’hypothèse du rôle de médiateurs des
effets des AGE des gangliosides. La protection partielle exercée par le PPMP suggère, comme déjà
évoqué, l'existence d'autres voies d'action des AGE sur la prolifération cellulaire.
Enfin, nous avons eu recours à la technique des SiRNA pour inhiber spécifiquement
l’activité GM3 synthase et donc l’augmentation de la biosynthèse des gangliosides observée en
réponse aux AGE. La transfection des cellules mésangiales avec un SiRNA spécifique de la GM3
synthase a partiellement protégé les cellules de la diminution de prolifération causée par les AGE.
Ce résultat confirme de manière plus directe l’implication des gangliosdies dans l’inhibition de
prolifération causée par les AGE.
L'ensemble des résultats obtenus par ces différentes approches, conforte l'hypothèse que les
gangliosides de la série-a peuvent effectivement participer à l'effet anti-prolifératif des AGE sur les
BRP et les RMC. Notons que les gangliosides ont déjà été décrits comme inhibiteurs endogènes de
la croissance de cellules mésangiales (Tsuboi N. et al., 2001). Ce rôle est en plus conforté par de
nombreux travaux montrant que les gangliosides, particulièrement de la série-a, diminuent la
prolifération de différents types cellulaires, en interagissant avec la signalisation de facteurs de
croissance. Par exemple, Bremer et al. ont montré que l'ajout exogène de GM3 et GM1 diminue la
prolifération de lignées cellulaires humaines de carcinome d’épiderme en inhibant la
phosphorylation du récepteur à l'EGF (Bremer E.G. et al., 1986). De plus, une croissance accélérée
et une autophosphorylation accrue du récepteur à l'EGF ont été montrées dans des lignées
cellulaires transfectées avec un gène codant une sialidase, diminuant le taux de GM3 (Meuillet E.J.
et al., 1999). Les mêmes observations ont été faites dans des lignées cellulaires issues d'ovaires de
hamster et présentant un défaut dans la biosynthèse des gangliosides (Weis F.M. et al., 1990). De la
même manière, différents travaux ont indiqué que GM1 et GM2 diminuent la croissance cellulaire
en inhibant la phosphorylation ou la dimérisation du récepteur au PDGF (Hynds D.L. et al., 1995;
Van Brocklyn J. et al., 1993). Enfin, des lignées cellulaires Swiss 3T3 surexprimant le GM1 ont
136

révélé une prolifération ralentie et une diminution de la phosphorylation du récepteur au PDGF
(Mitsuda T. et al., 2002).
En conclusion, les résultats de cette première partie suggèrent que les gangliosides de
la série-a sont des médiateurs de l’effet anti-prolifératif des AGE à la fois dans les péricytes et
dans les cellules mésangiales. De plus, l'augmentation des gangliosides de la série-a apparaît
comme un nouveau mécanisme d'action potentiel des AGE, commun aux deux types
cellulaires.
137


L’ACTIVATION DE LA VOIE DES HEXOSAMINES
DIMINUE LA PROLIFERATION DES PERICYTES
ET DES CELLULES MESANGIALES :
IMPLICATION DES GANGLIOSIDES
Un nombre grandissant de travaux suggère l'implication de la voie des hexosamines dans la
néphropathie diabétique. Précisément, cette voie semble jouer un rôle important dans les altérations
des cellules mésangiales, comme la surproduction de matrice extracellulaire. Cependant, son rôle
dans les perturbations de la prolifération de ces cellules a été peu étudié. Il existe également très peu
d'études, sinon aucune, s'intéressant à l'implication de la voie des hexosamines dans la rétinopathie
diabétique, bien qu'un grand nombre de caractéristiques pathologiques soient communes à la
néphropathie et à la rétinopathie.
Par ailleurs, l'addition d'un résidu monosaccharidique, la N-acétylglucosamine (GlcNAc), produit
de la voie de l'hexosamine, est connue pour modifier de façon covalente des protéines cytosoliques
ou nucléaires et réguler ainsi leur activité. Les cibles potentielles de cette O-glycosylation étant
nombreuses, la voie des hexosamines pourrait interférer avec de multiples voies biochimiques. De
plus, l’UDP-GlcNAc peut être utilisée comme précurseur du CMP-acide sialique ou de l’UDP-N-
acétylgalactosamine (UDP-GalNAc) qui sont des substrats pour la biosynthèse des gangliosides.
Ainsi, ces voies métaboliques soulèvent la possibilité d'une connexion entre la voie des
hexosamines et les gangliosides.
L'objet de cette deuxième partie expérimentale a été de déterminer l'effet d'une activation de la voie
des hexosamines, mimée par un traitement avec la glucosamine, sur la prolifération et le profil en
gangliosides des péricytes et des cellules mésangiales ainsi que d'éclaircir le lien potentiel entre ces
deux éléments.
138

I. EFFETS DE LA GLUCOSAMINE SUR LA
PROLIFERATION CELLULAIRE
Péricytes et cellules mésangiales ont été cultivés durant un passage (7 et 4 jours, respectivement) en
milieu contrôle ou en présence de 2 ou 5 mM de glucosamine. A l’issue de cette période de
traitement, la prolifération cellulaire a été évaluée par comptage des cellules ainsi que par dosage
des protéines totales.
Les résultats présentés en Figure 47A montrent que la glucosamine inhibe la prolifération
des péricytes de manière dose-dépendante. A une concentration de 5 mM, le nombre de cellules
traitées est diminué de 41 % par rapport au nombre de cellules témoin. De même, la quantité de
protéines est 47 % inférieure dans les cultures traitées à la glucosamine comparée aux cultures
témoin.
La glucosamine affecte encore plus fortement la prolifération des cellules mésangiales, de manière
dose-dépendante. Un traitement avec la glucosamine diminue le nombre de cellules de 62 % et 77
% par rapport au contrôle pour des concentrations respectivement de 2 mM et 5 mM. La quantité
totale de protéines diminue également de façon dose-dépendante puisqu’elle est inférieure de 43%
et 60 % avec 2 et 5 mM, respectivement, par rapport aux cultures contrôle (Figure 47B). Il faut par
ailleurs noter que la quantité de protéines est affectée dans des proportions moindres que le nombre
de cellules. Ces résultats suggèrent une hypertrophie des cellules mésangiales en réponse à la
glucosamine, point sur lequel nous reviendrons dans la suite de l'étude.
139
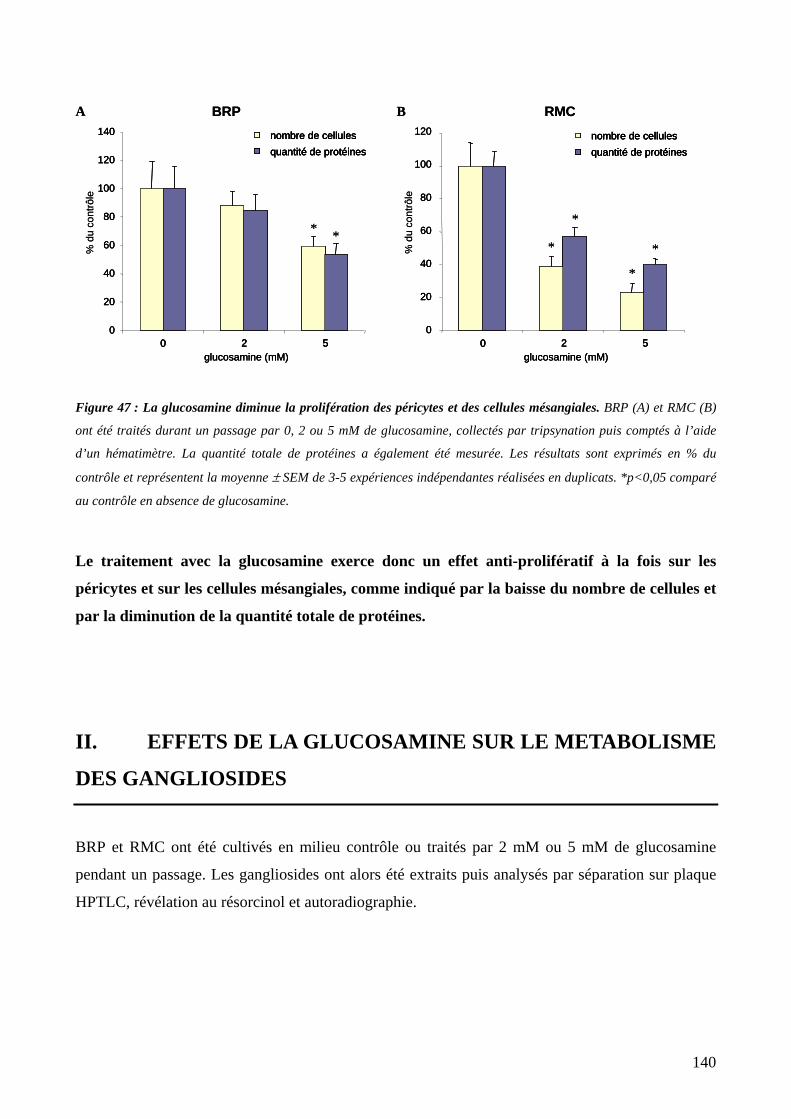
%du
con
trôle
nombre de cellulesquantité de protéines
* *
A BRP
%du
con
trôle
nombre de cellulesquantité de protéines
*
*
B RMC
0
20
40
60
80
100
120
140
0 2 5glucosamine (mM)
0
20
40
60
80
100
120
0 2 5glucosamine (mM)
*
*%du
con
trôle
nombre de cellulesquantité de protéinesnombre de cellulesnombre de cellulesquantité de protéinesquantité de protéines
* *
A BRP
%du
con
trôle
nombre de cellulesquantité de protéinesnombre de cellulesnombre de cellulesquantité de protéinesquantité de protéines
*
*
B RMC
0
20
40
60
80
100
120
140
0
20
40
60
80
100
120
140
0 2 5glucosamine (mM)
0 2 50 2 5glucosamine (mM)
0
20
40
60
80
100
120
0
20
40
60
80
100
120
0 2 5glucosamine (mM)
0 2 50 2 5glucosamine (mM)
*
*
Figure 47 : La glucosamine diminue la prolifération des péricytes et des cellules mésangiales. BRP (A) et RMC (B)
ont été traités durant un passage par 0, 2 ou 5 mM de glucosamine, collectés par tripsynation puis comptés à l’aide
d’un hématimètre. La quantité totale de protéines a également été mesurée. Les résultats sont exprimés en % du
contrôle et représentent la moyenne ± SEM de 3-5 expériences indépendantes réalisées en duplicats. *p<0,05 comparé
au contrôle en absence de glucosamine.
Le traitement avec la glucosamine exerce donc un effet anti-prolifératif à la fois sur les
péricytes et sur les cellules mésangiales, comme indiqué par la baisse du nombre de cellules et
par la diminution de la quantité totale de protéines.
II. EFFETS DE LA GLUCOSAMINE SUR LE METABOLISME
DES GANGLIOSIDES
BRP et RMC ont été cultivés en milieu contrôle ou traités par 2 mM ou 5 mM de glucosamine
pendant un passage. Les gangliosides ont alors été extraits puis analysés par séparation sur plaque
HPTLC, révélation au résorcinol et autoradiographie.
140
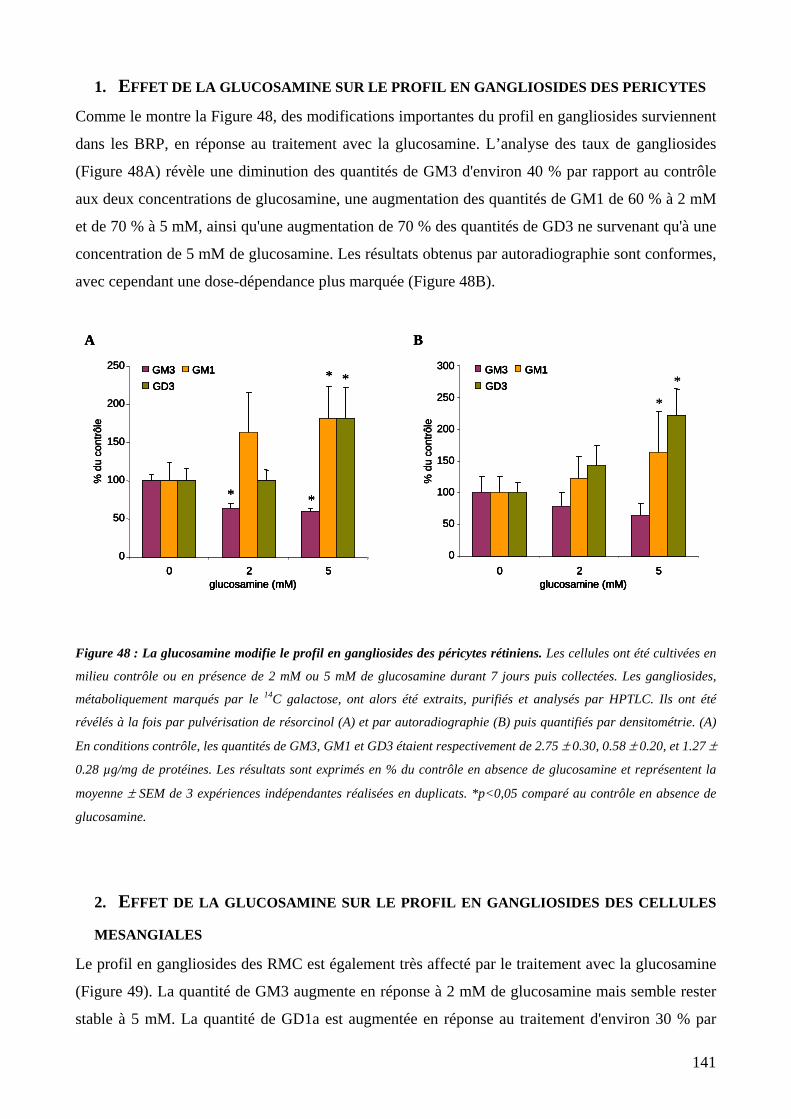
1. EFFET DE LA GLUCOSAMINE SUR LE PROFIL EN GANGLIOSIDES DES PERICYTES Comme le montre la Figure 48, des modifications importantes du profil en gangliosides surviennent
dans les BRP, en réponse au traitement avec la glucosamine. L’analyse des taux de gangliosides
(Figure 48A) révèle une diminution des quantités de GM3 d'environ 40 % par rapport au contrôle
aux deux concentrations de glucosamine, une augmentation des quantités de GM1 de 60 % à 2 mM
et de 70 % à 5 mM, ainsi qu'une augmentation de 70 % des quantités de GD3 ne survenant qu'à une
concentration de 5 mM de glucosamine. Les résultats obtenus par autoradiographie sont conformes,
avec cependant une dose-dépendance plus marquée (Figure 48B).
% d
u co
ntrô
le
GM3 GM1GD3
B
% d
u co
ntrô
le
GM3 GM1GD3
* *
*
A
0
50
100
150
200
250
0 2 5glucosamine (mM)
0
50
100
150
200
250
300*
0 2 5glucosamine (mM)
**
% d
u co
ntrô
le
GM3 GM1GD3
B
% d
u co
ntrô
le
GM3 GM1GD3
* *
*
A
0
50
100
150
200
250
0 2 5glucosamine (mM)
0
50
100
150
200
250
300*
0 2 5glucosamine (mM)
% d
u co
ntrô
le
GM3 GM1GD3GM3 GM1GD3
B
% d
u co
ntrô
le
GM3 GM1GD3GM3 GM1GD3
* *
*
A
0
50
100
150
200
250
0 2 5glucosamine (mM)
0 2 5glucosamine (mM)
0
50
100
150
200
250
300*
0 2 5glucosamine (mM)
0 2 5glucosamine (mM)
**
Figure 48 : La glucosamine modifie le profil en gangliosides des péricytes rétiniens. Les cellules ont été cultivées en
milieu contrôle ou en présence de 2 mM ou 5 mM de glucosamine durant 7 jours puis collectées. Les gangliosides,
métaboliquement marqués par le 14C galactose, ont alors été extraits, purifiés et analysés par HPTLC. Ils ont été
révélés à la fois par pulvérisation de résorcinol (A) et par autoradiographie (B) puis quantifiés par densitométrie. (A)
En conditions contrôle, les quantités de GM3, GM1 et GD3 étaient respectivement de 2.75 ± 0.30, 0.58 ± 0.20, et 1.27 ±
0.28 µg/mg de protéines. Les résultats sont exprimés en % du contrôle en absence de glucosamine et représentent la
moyenne ± SEM de 3 expériences indépendantes réalisées en duplicats. *p<0,05 comparé au contrôle en absence de
glucosamine.
2. EFFET DE LA GLUCOSAMINE SUR LE PROFIL EN GANGLIOSIDES DES CELLULES
MESANGIALES Le profil en gangliosides des RMC est également très affecté par le traitement avec la glucosamine
(Figure 49). La quantité de GM3 augmente en réponse à 2 mM de glucosamine mais semble rester
stable à 5 mM. La quantité de GD1a est augmentée en réponse au traitement d'environ 30 % par
141

rapport au contrôle à 2 mM et 50 % à 5 mM. Le GD3, quant à lui, devient indétectable dès 2 mM de
glucosamine. L'autoradiographie confirme la disparition du GD3 et indique en revanche une
diminution du GM3 radioactif par rapport au contrôle (Figure 49C). Ceci suggère qu’une quantité
moindre de GM3 a été synthétisée dans les cellules traitées pendant le "pulse" de marquage
métabolique, ce qui est d’ailleurs conforme à la diminution d’activité GM3 synthase mesurée en
réponse à la glucosamine (paragraphe suivant). Cette diminution de flux ne se manifeste cependant
pas immédiatement sur les quantités de GM3, probablement à cause de la masse importante que
représente ce ganglioside.
Les taux de gangliosides GM1 et surtout GM2 subissent des variations beaucoup plus
spectaculaires (Figure 49B). Ces espèces, quasiment indétectables dans les cellules témoin,
atteignent 2.37 et 0.70 µg/mg de protéines, représentant ainsi respectivement 24 % et 7 % de tous
les gangliosides détectés dans les cellules traitées avec 5 mM de glucosamine. Cette forte
augmentation de GM2 et GM1 est également nettement visible par analyse des gangliosides
marqués (Figure 49D). Etant donné que ces deux gangliosides sont absents dans les cellules
contrôle, nous avons voulu vérifier leur identité. Après séparation sur plaque HPTLC, les
gangliosides ont été révélés par immunocytochimie en utilisant un anticorps spécifique dirigé contre
GM2 (don généreux du Dr J. Portoukalian) et la sous-unité β de la toxine cholérique, spécifique du
GM1.
142
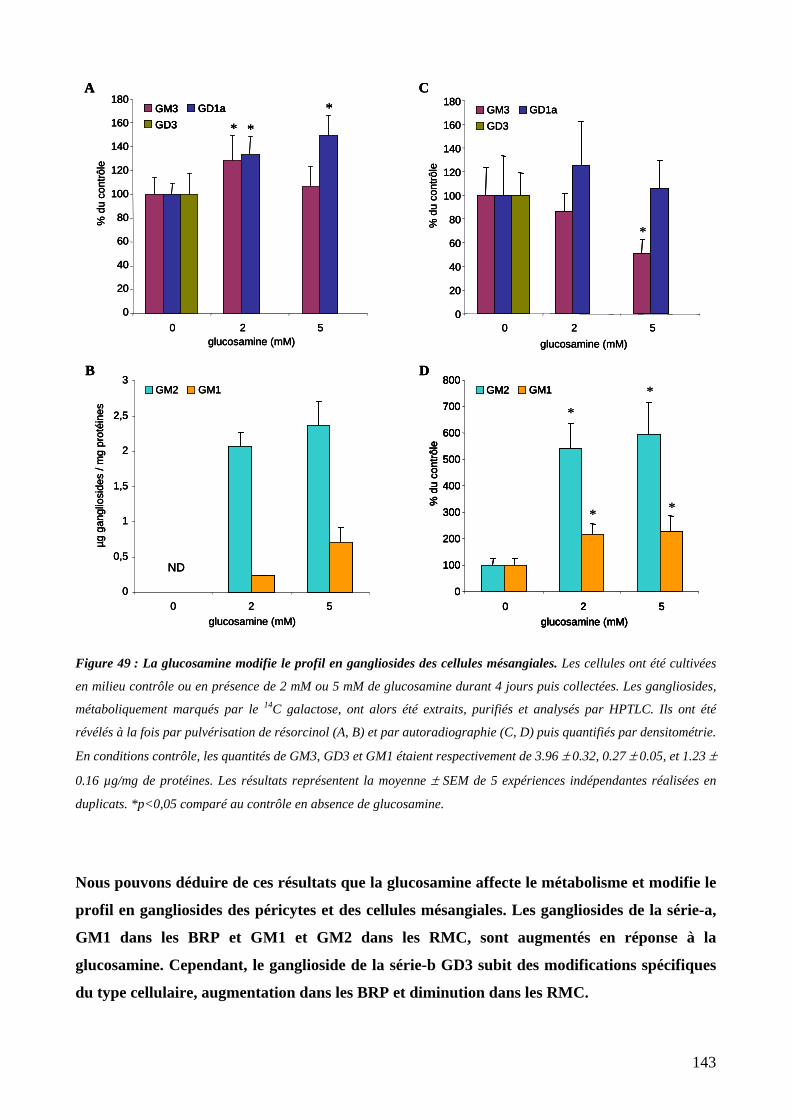
0
20
40
60
80
100
120
140
160
180
0 2 5glucosamine (mM)
%du
con
trôle
GM3 GD1aGD3
0 2 5glucosamine (mM)
GM2 GM1
GM3 GD1aGD3
0
20
40
60
80
100
120
140
160
180
0 2 5glucosamine (mM)
%du
con
trôle
0
100
200
300
400
500
600
700
800GM2 GM1
0 2 5glucosamine (mM)
%du
con
trôle
A C
B D
* **
0
0,5
1
1,5
2
2,5
3
µg g
angl
iosi
des
/ mg
prot
éine
s
ND
*
*
*
*
*
0
20
40
60
80
100
120
140
160
180
0 2 5glucosamine (mM)
%du
con
trôle
GM3 GD1aGD3
0 2 5glucosamine (mM)
GM2 GM1
GM3 GD1aGD3
0
20
40
60
80
100
120
140
160
180
0 2 5glucosamine (mM)
%du
con
trôle
0
100
200
300
400
500
600
700
800GM2 GM1
0 2 5glucosamine (mM)
%du
con
trôle
A C
B D
* **
0
0,5
1
1,5
2
2,5
3
µg g
angl
iosi
des
/ mg
prot
éine
s
ND
0
20
40
60
80
100
120
140
160
180
0 2 5glucosamine (mM)
%du
con
trôle
GM3GM3 GD1aGD1aGD3GD3
0 2 5glucosamine (mM)
GM2 GM1
GM3 GD1aGD3
0
20
40
60
80
100
120
140
160
180
0 2 5glucosamine (mM)
%du
con
trôle
0
100
200
300
400
500
600
700
800
0
100
200
300
400
500
600
700
800GM2 GM1GM2 GM1
0 2 5glucosamine (mM)
%du
con
trôle
0 2 5glucosamine (mM)
%du
con
trôle
A C
B D
* **
0
0,5
1
1,5
2
2,5
3
µg g
angl
iosi
des
/ mg
prot
éine
s
ND
*
*
*
*
*
Figure 49 : La glucosamine modifie le profil en gangliosides des cellules mésangiales. Les cellules ont été cultivées
en milieu contrôle ou en présence de 2 mM ou 5 mM de glucosamine durant 4 jours puis collectées. Les gangliosides,
métaboliquement marqués par le 14C galactose, ont alors été extraits, purifiés et analysés par HPTLC. Ils ont été
révélés à la fois par pulvérisation de résorcinol (A, B) et par autoradiographie (C, D) puis quantifiés par densitométrie.
En conditions contrôle, les quantités de GM3, GD3 et GM1 étaient respectivement de 3.96 ± 0.32, 0.27 ± 0.05, et 1.23 ±
0.16 µg/mg de protéines. Les résultats représentent la moyenne ± SEM de 5 expériences indépendantes réalisées en
duplicats. *p<0,05 comparé au contrôle en absence de glucosamine.
Nous pouvons déduire de ces résultats que la glucosamine affecte le métabolisme et modifie le
profil en gangliosides des péricytes et des cellules mésangiales. Les gangliosides de la série-a,
GM1 dans les BRP et GM1 et GM2 dans les RMC, sont augmentés en réponse à la
glucosamine. Cependant, le ganglioside de la série-b GD3 subit des modifications spécifiques
du type cellulaire, augmentation dans les BRP et diminution dans les RMC.
143

III. EFFET DE LA GLUCOSAMINE SUR L’ACTIVITE GM3
SYNTHASE
Les gangliosides de la série-a étant augmentés dans les deux types cellulaires en réponse à 5 mM de
glucosamine, l'activité GM3 synthase a été mesurée afin d'éclaircir les mécanismes sous-tendant ces
variations.
Les résultats présentés en Figure 50A montrent que le traitement avec la glucosamine induit
une augmentation de l'activité GM3 synthase d'environ 160 % dans les BRP. L'augmentation de
l'activité de cette enzyme limitante pour la biosynthèse de l’ensemble des gangliosides, est
compatible avec l'augmentation des taux de GM1 et de GD3 observée en réponse à la glucosamine.
En revanche, une diminution du taux de GM3 a été mesurée malgré cette augmentation de l'activité
GM3 synthase. GM3 étant le précurseur des gangliosides plus complexes, ceci suggère qu'il a été
métabolisé en GM1 et GD3, tous les deux fortement augmentés en réponse au traitement.
L'augmentation de la biosynthèse de GM3 consécutive à une augmentation de l'activité GM3
synthase ne suffirait pas à compenser sa consommation pour la biosynthèse accrue de GM1 et GD3.
Contrairement à ce qui est attendu, l'activité GM3 synthase est diminuée d'environ 50 %
dans les RMC traitées à la glucosamine (Figure 50B). On observe cette diminution d'activité malgré
une augmentation massive des taux de GM2 et de GM1, produits indirects de cette enzyme. Une
explication possible serait que ces produits, fortement augmentés, exercent un rétrocontrôle négatif
sur la GM3 synthase puisqu'un rétrocontrôle négatif de l’activité des enzymes de biosynthèse des
gangliosides a été décrite in vitro (Yusuf H.K. et al., 1987). Une analyse cinétique aurait été
nécessaire pour vérifier cette hypothèse. L’activité GD3 synthase n’a pas été mesurée en réponse à
la glucosamine. Les modifications des gangliosides des RMC étant similaires suite au traitement
avec la glucosamine et avec les AGE (augmentation des gangliosides de la série-a et diminution du
GD3), il est probable que l’activité GD3 synthase soit diminuée en réponse à la glucosamine
comme elle l’est en réponse aux AGE.
144
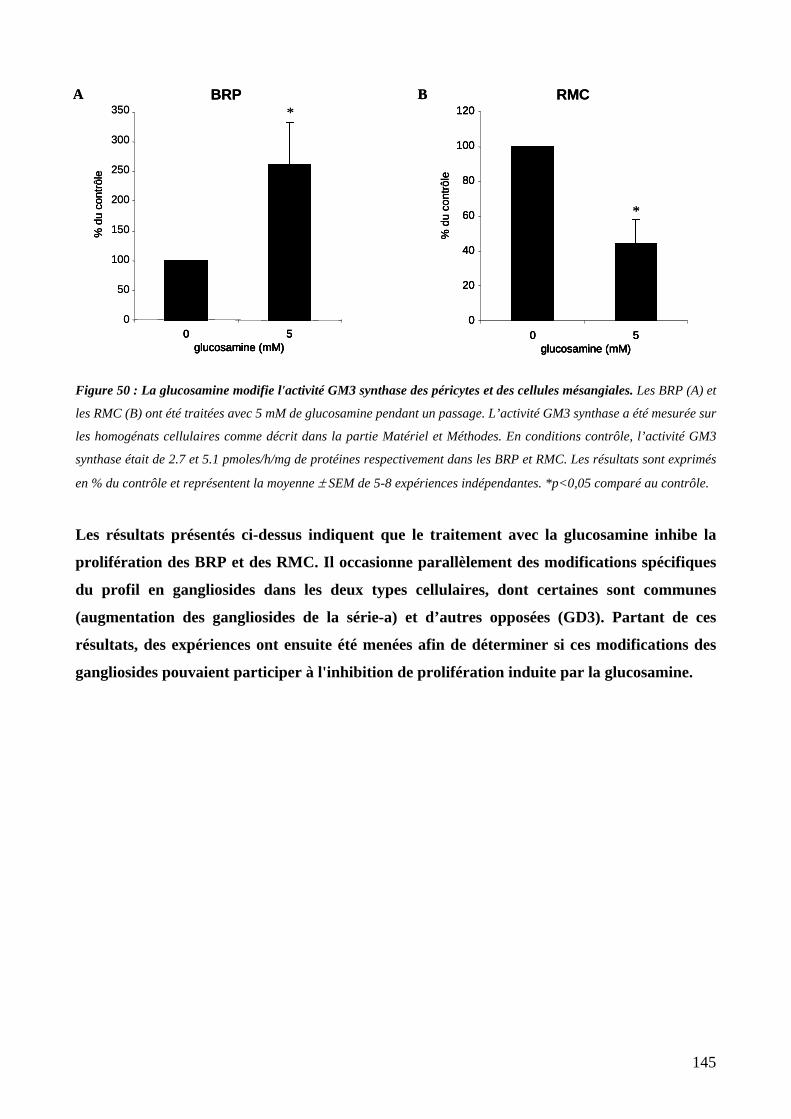
A BRP B RMC*
0
50
100
150
200
250
300
350%
du c
ontrô
le
0 5glucosamine (mM)
0
20
40
60
80
100
120
%du
con
trôle
0 5glucosamine (mM)
*
A BRP B RMC*
0
50
100
150
200
250
300
350%
du c
ontrô
le
0 5glucosamine (mM)
%du
con
trôle
0 5glucosamine (mM)
0 5glucosamine (mM)
0
20
40
60
80
100
120
0
20
40
60
80
100
120
%du
con
trôle
0 5glucosamine (mM)
%du
con
trôle
0 5glucosamine (mM)
0 5glucosamine (mM)
*
Figure 50 : La glucosamine modifie l'activité GM3 synthase des péricytes et des cellules mésangiales. Les BRP (A) et
les RMC (B) ont été traitées avec 5 mM de glucosamine pendant un passage. L’activité GM3 synthase a été mesurée sur
les homogénats cellulaires comme décrit dans la partie Matériel et Méthodes. En conditions contrôle, l’activité GM3
synthase était de 2.7 et 5.1 pmoles/h/mg de protéines respectivement dans les BRP et RMC. Les résultats sont exprimés
en % du contrôle et représentent la moyenne ± SEM de 5-8 expériences indépendantes. *p<0,05 comparé au contrôle.
Les résultats présentés ci-dessus indiquent que le traitement avec la glucosamine inhibe la
prolifération des BRP et des RMC. Il occasionne parallèlement des modifications spécifiques
du profil en gangliosides dans les deux types cellulaires, dont certaines sont communes
(augmentation des gangliosides de la série-a) et d’autres opposées (GD3). Partant de ces
résultats, des expériences ont ensuite été menées afin de déterminer si ces modifications des
gangliosides pouvaient participer à l'inhibition de prolifération induite par la glucosamine.
145

IV. IMPLICATION DES GANGLIOSIDES DANS LA
DIMINUTION DE PROLIFERATION CELLULAIRE CAUSEE PAR
LA GLUCOSAMINE
1. EFFET D’UN TRAITEMENT PAR LES GANGLIOSIDES EXOGENES SUR LA
PROLIFERATION DES PERICYTES ET DES CELLULES MESANGIALES Les gangliosides ont été décrits comme des modulateurs, et le plus souvent des inhibiteurs, de la
croissance cellulaire. L’effet sur la prolifération cellulaire des différents gangliosides augmentés en
réponse à la glucosamine a donc été étudié.
Pour cela, des concentrations croissantes de gangliosides ainsi que de leurs précurseurs non sialylés,
Glucosylcéramide et Lactosylcéramide, utilisés comme contrôles, ont été ajoutées au milieu de
culture, sous la forme de complexe avec la BSA. La prolifération a ensuite été évaluée par dosage
de la quantité totale de protéines et par mesure de la quantité d’ADN.
La Figure 51 montre que les gangliosides testés inhibent la prolifération des BRP, de
manière dose-dépendante, alors que Glucosylcéramide et Lactosylcéramide sont sans effet.
L’inhibition de prolifération, dose-dépendante atteint environ 25 % à une concentration de 100 µM.
Les résultats obtenus par la technique du CyQuant sont conformes (Figure 51B) bien qu’atténués.
146
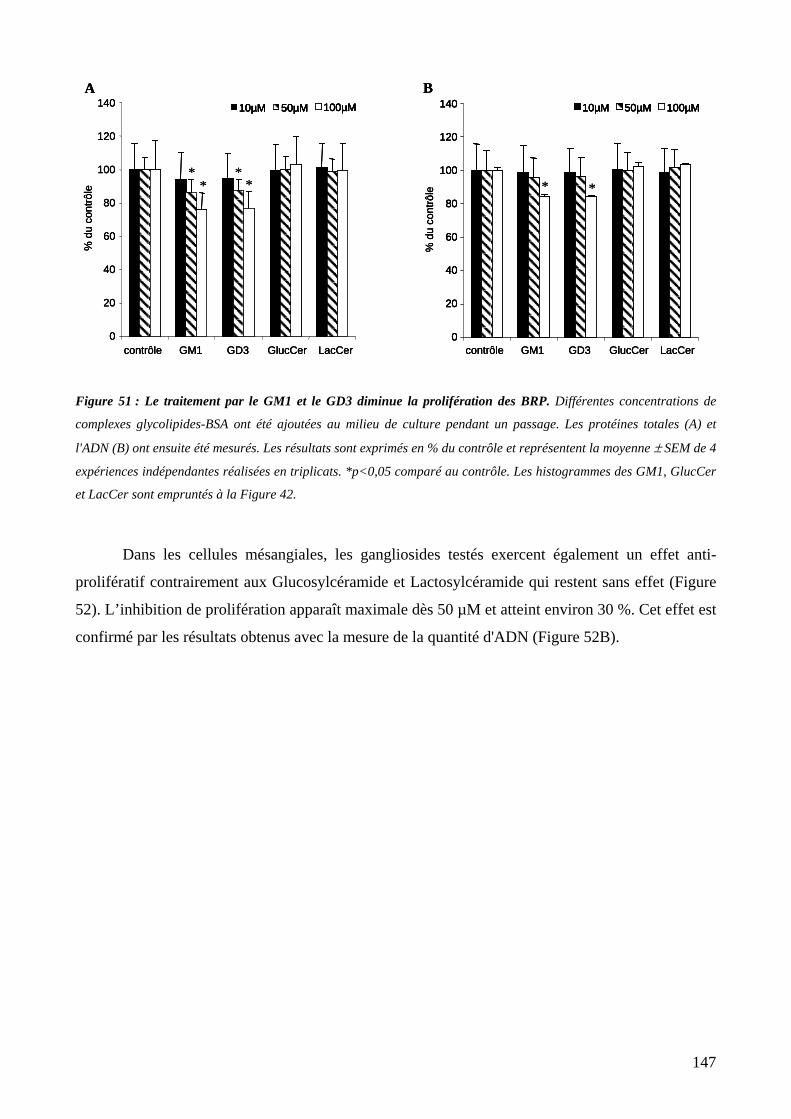
BA%
du
cont
rôle
0
20
40
60
80
100
120
140
contrôle GM1 GD3 GlucCer LacCer
10µM 50µM 100µM
% d
u co
ntrô
le
0
20
40
60
80
100
120
140 10µM 50µM 100µM
contrôle GM1 GD3 GlucCer LacCer
* ** * * *
BA%
du
cont
rôle
0
20
40
60
80
100
120
140
contrôle GM1 GD3 GlucCer LacCer
10µM 50µM 100µM
% d
u co
ntrô
le
0
20
40
60
80
100
120
140 10µM 50µM 100µM
contrôle GM1 GD3 GlucCer LacCer
BA%
du
cont
rôle
0
20
40
60
80
100
120
140
contrôle GM1 GD3 GlucCer LacCer
10µM 50µM 100µM10µM10µM 50µM50µM 100µM100µM
% d
u co
ntrô
le
0
20
40
60
80
100
120
140
0
20
40
60
80
100
120
140 10µM 50µM 100µM10µM10µM 50µM50µM 100µM100µM
contrôle GM1 GD3 GlucCer LacCer
* ** * * *
Figure 51 : Le traitement par le GM1 et le GD3 diminue la prolifération des BRP. Différentes concentrations de
complexes glycolipides-BSA ont été ajoutées au milieu de culture pendant un passage. Les protéines totales (A) et
l'ADN (B) ont ensuite été mesurés. Les résultats sont exprimés en % du contrôle et représentent la moyenne ± SEM de 4
expériences indépendantes réalisées en triplicats. *p<0,05 comparé au contrôle. Les histogrammes des GM1, GlucCer
et LacCer sont empruntés à la Figure 42.
Dans les cellules mésangiales, les gangliosides testés exercent également un effet anti-
prolifératif contrairement aux Glucosylcéramide et Lactosylcéramide qui restent sans effet (Figure
52). L’inhibition de prolifération apparaît maximale dès 50 µM et atteint environ 30 %. Cet effet est
confirmé par les résultats obtenus avec la mesure de la quantité d'ADN (Figure 52B).
147

A%
du
cont
rôle
10µM 50µM 100µM
contrôle GM2 GM1 GD1a GlucCer LacCer0
20
40
60
80
100
120B
% d
u co
ntrô
le
0
20
40
60
80
100
120 10µM 50µM 100µM
contrôle GM2 GM1 GD1a GlucCer LacCer
* * ***
* **** **
A%
du
cont
rôle
10µM 50µM 100µM10µM10µM 50µM50µM 100µM100µM
contrôle GM2 GM1 GD1a GlucCer LacCercontrôle GM2 GM1 GD1a GlucCer LacCer0
20
40
60
80
100
120B
% d
u co
ntrô
le
0
20
40
60
80
100
120B
% d
u co
ntrô
le
0
20
40
60
80
100
120 10µM 50µM 100µM10µM10µM 50µM50µM 100µM100µM
contrôle GM2 GM1 GD1a GlucCer LacCercontrôle GM2 GM1 GD1a GlucCer LacCer
* * ***
* **** **
Figure 52 : Le traitement par les GM2, GM1 et GD1a diminue la prolifération des RMC. Différentes doses de
complexes glycolipides-BSA ont été ajoutées au milieu de culture pendant un passage. Les protéines totales (A) et la
quantité d'ADN (B) ont ensuite été mesurées. Les résultats sont exprimés en % du contrôle et représentent la moyenne ±
SEM de 4-6 expériences indépendantes réalisées en triplicats. *p<0,05 comparé au contrôle. Les histogrames des
GM2, GM1, GlucCer et LacCer sont empruntés à la Figure 43.
Ces résultats révèlent un effet anti-prolifératif des gangliosides augmentés suite au traitement
avec la glucosamine dans les péricytes et les cellules mésangiales. Ils suggèrent ainsi qu'ils
pourraient être impliqués dans la diminution de prolifération cellulaire observée en réponse à
la glucosamine.
2. EFFET D’UN COTRAITEMENT GLUCOSAMINE ET PPMP Le PPMP, un inhibiteur de glucosylcéramide synthase, a été utilisé pour bloquer l’augmentation des
gangliosides causée par le traitement et vérifier ainsi leur implication dans les effets de la
glucosamine.
BRP et RMC ont été traités avec la glucosamine en présence de doses croissantes de PPMP puis la
prolifération a été évaluée par dosage des protéines totales.
Dans les BRP, l'ajout de PPMP permet de réduire l'inhibition de prolifération causée par la
glucosamine. En effet, la quantité de protéines est 36 % plus importante dans les BRP traités avec la
glucosamine en présence de 1 µM de PPMP que dans les BRP traités avec la glucosamine seule
148
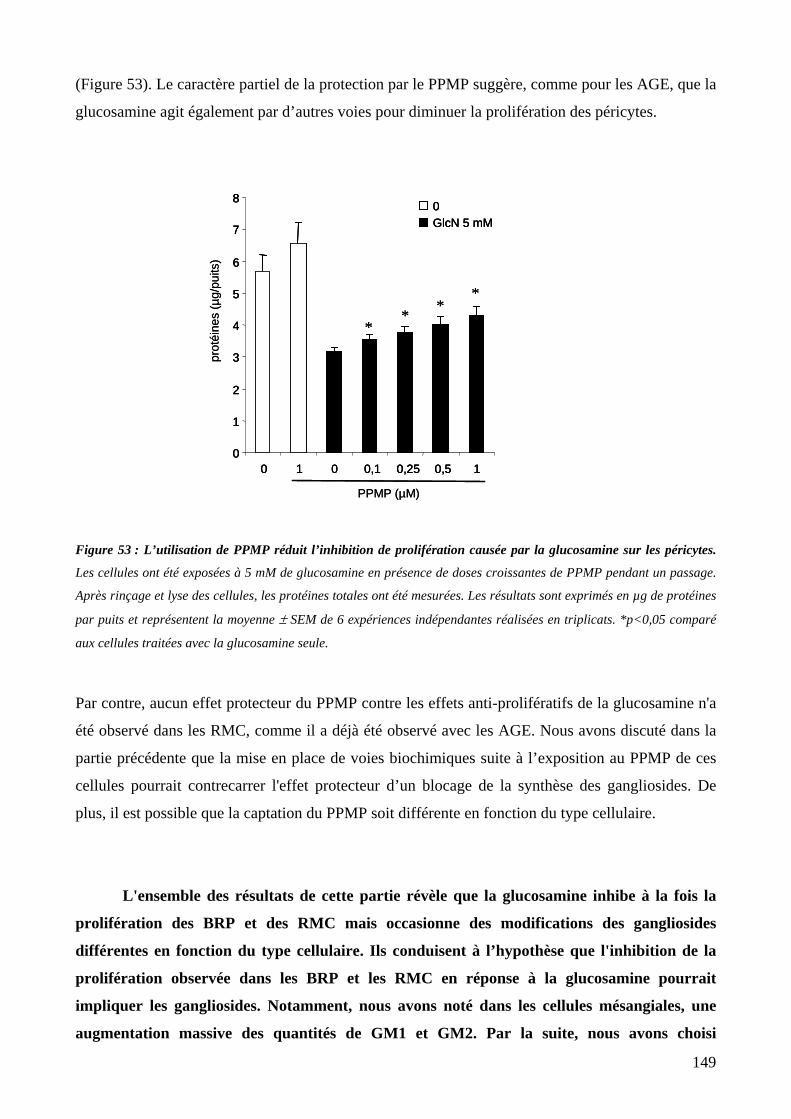
(Figure 53). Le caractère partiel de la protection par le PPMP suggère, comme pour les AGE, que la
glucosamine agit également par d’autres voies pour diminuer la prolifération des péricytes.
prot
éine
s(µ
g/pu
its)
PPMP (µM)
0GlcN 5 mM
* **
*
0
1
2
3
4
5
6
7
8
0 1 0 0,1 0,25 0,5 1
prot
éine
s(µ
g/pu
its)
PPMP (µM)
0GlcN 5 mM0GlcN 5 mM
* **
*
0
1
2
3
4
5
6
7
8
0 1 0 0,1 0,25 0,5 10
1
2
3
4
5
6
7
8
0 1 0 0,1 0,25 0,5 1
Figure 53 : L’utilisation de PPMP réduit l’inhibition de prolifération causée par la glucosamine sur les péricytes.
Les cellules ont été exposées à 5 mM de glucosamine en présence de doses croissantes de PPMP pendant un passage.
Après rinçage et lyse des cellules, les protéines totales ont été mesurées. Les résultats sont exprimés en µg de protéines
par puits et représentent la moyenne ± SEM de 6 expériences indépendantes réalisées en triplicats. *p<0,05 comparé
aux cellules traitées avec la glucosamine seule.
Par contre, aucun effet protecteur du PPMP contre les effets anti-prolifératifs de la glucosamine n'a
été observé dans les RMC, comme il a déjà été observé avec les AGE. Nous avons discuté dans la
partie précédente que la mise en place de voies biochimiques suite à l’exposition au PPMP de ces
cellules pourrait contrecarrer l'effet protecteur d’un blocage de la synthèse des gangliosides. De
plus, il est possible que la captation du PPMP soit différente en fonction du type cellulaire.
L'ensemble des résultats de cette partie révèle que la glucosamine inhibe à la fois la
prolifération des BRP et des RMC mais occasionne des modifications des gangliosides
différentes en fonction du type cellulaire. Ils conduisent à l’hypothèse que l'inhibition de la
prolifération observée dans les BRP et les RMC en réponse à la glucosamine pourrait
impliquer les gangliosides. Notamment, nous avons noté dans les cellules mésangiales, une
augmentation massive des quantités de GM1 et GM2. Par la suite, nous avons choisi
149

d'explorer plus en détails les mécanismes spécifiques par lesquels la glucosamine inhibe la
prolifération des RMC et la place de ces deux gangliosides dans ces mécanismes.
V. ETUDE DES MECANISMES CONDUISANT A
L’INHIBITION DE PROLIFERATION DES CELLULES
MESANGIALES PAR LA GLUCOSAMINE
Pour préciser ces mécanismes, nous avons réalisé des études plus détaillées en utilisant deux
concentrations de glucosamine, une faible (0,5 mM) et une forte (5 mM).
1. EFFET DE LA GLUCOSAMINE SUR L’HYPERTROPHIE DES RMC L’hypertrophie des cellules mésangiales est un processus qui contribue à l’expansion mésangiale
caractéristique de la néphropathie diabétique. Nous avons étudié ici ce phénomène en réponse à un
traitement avec la glucosamine.
Dans un premier temps, les cinétiques de croissance des RMC ont été déterminées en réponse à un
traitement avec 0,5 ou 5 mM de glucosamine. Le nombre de cellules ainsi que la quantité de
protéines ont été mesurés, permettant ainsi le calcul du rapport protéines totales / nombre de
cellules. Ce rapport est un moyen bien établi de mesurer l'hypertrophie cellulaire.
Les résultats présentés en Figure 54 indiquent que la glucosamine diminue fortement la croissance
des RMC. L’effet, visible dès 48 h, correspond à une diminution du nombre de cellules de 56 %
pour une concentration de 0,5 mM et de 85 % pour une concentration de 5 mM au terme du
traitement (Figure 54A). Cet effet est également révélé par une baisse de la quantité totale de
protéines, mais dans une moindre mesure (Figure 54B). Par conséquent, l’augmentation du rapport
protéines totales/nombre de cellules semble indiquer que la glucosamine induit l'hypertrophie des
cellules mésangiales (Figure 54C).
150
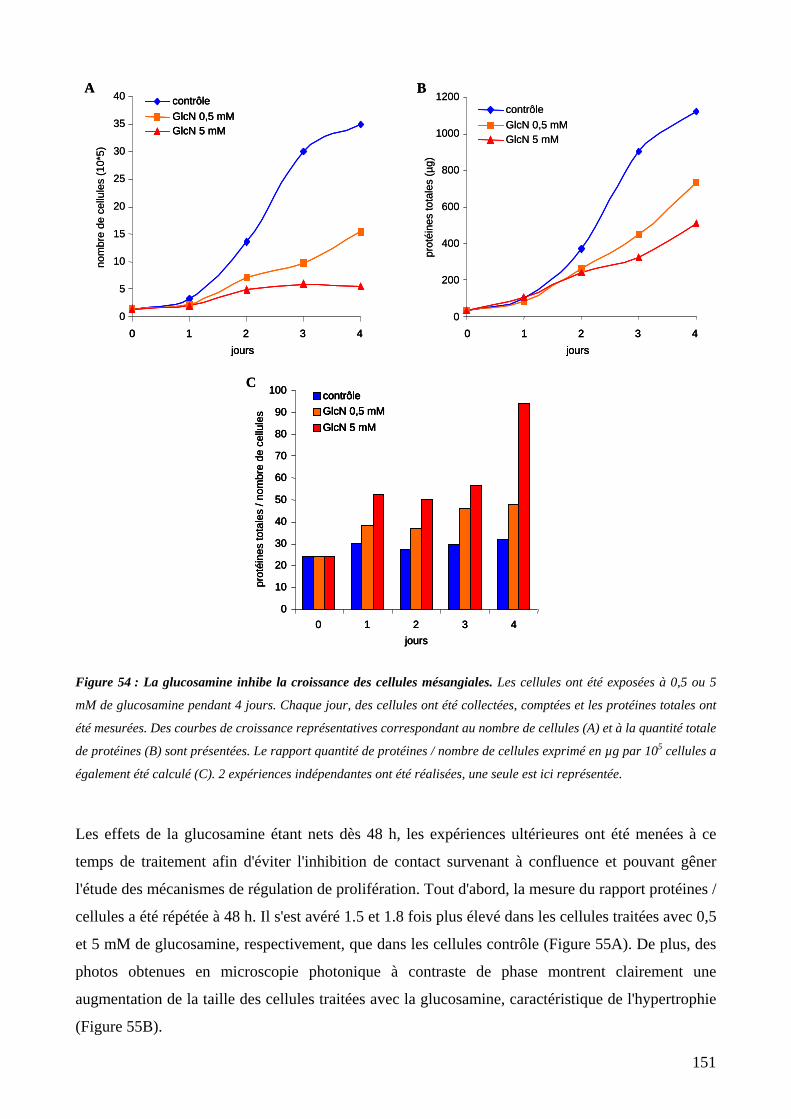
A B
0
5
10
15
20
25
30
35
40
jours0 1 2 3 4
nom
bre
de c
ellu
les
(10*
5)contrôleGlcN 0,5 mMGlcN 5 mM
0
200
400
600
800
1000
1200
prot
éine
s to
tale
s(µ
g)
jours0 1 2 3
C
4
0
10
20
30
40
50
60
70
80
90
100
0 1 2 3 4jours
prot
éine
s to
tale
s/n
ombr
ede
cel
lule
s
contrôleGlcN 0,5 mMGlcN 5 mM
contrôleGlcN 0,5 mMGlcN 5 mM
A B
0
5
10
15
20
25
30
35
40
jours0 1 2 3 4
jours0 1 2 3 4
nom
bre
de c
ellu
les
(10*
5)contrôleGlcN 0,5 mMGlcN 5 mM
contrôleGlcN 0,5 mMGlcN 5 mM
0
200
400
600
800
1000
1200
prot
éine
s to
tale
s(µ
g)
jours0 1 2 3
jours0 1 2 3
C
0
10
20
30
40
50
60
70
80
90
100
0 1 2 3 4jours
prot
éine
s to
tale
s/n
ombr
ede
cel
lule
s
contrôleGlcN 0,5 mMGlcN 5 mM
0
10
20
30
40
50
60
70
80
90
100
0 1 2 3 4jours
prot
éine
s to
tale
s/n
ombr
ede
cel
lule
s
contrôleGlcN 0,5 mMGlcN 5 mM
contrôleGlcN 0,5 mMGlcN 5 mM
contrôleGlcN 0,5 mMGlcN 5 mM
44
Figure 54 : La glucosamine inhibe la croissance des cellules mésangiales. Les cellules ont été exposées à 0,5 ou 5
mM de glucosamine pendant 4 jours. Chaque jour, des cellules ont été collectées, comptées et les protéines totales ont
été mesurées. Des courbes de croissance représentatives correspondant au nombre de cellules (A) et à la quantité totale
de protéines (B) sont présentées. Le rapport quantité de protéines / nombre de cellules exprimé en µg par 105 cellules a
également été calculé (C). 2 expériences indépendantes ont été réalisées, une seule est ici représentée.
Les effets de la glucosamine étant nets dès 48 h, les expériences ultérieures ont été menées à ce
temps de traitement afin d'éviter l'inhibition de contact survenant à confluence et pouvant gêner
l'étude des mécanismes de régulation de prolifération. Tout d'abord, la mesure du rapport protéines /
cellules a été répétée à 48 h. Il s'est avéré 1.5 et 1.8 fois plus élevé dans les cellules traitées avec 0,5
et 5 mM de glucosamine, respectivement, que dans les cellules contrôle (Figure 55A). De plus, des
photos obtenues en microscopie photonique à contraste de phase montrent clairement une
augmentation de la taille des cellules traitées avec la glucosamine, caractéristique de l'hypertrophie
(Figure 55B).
151
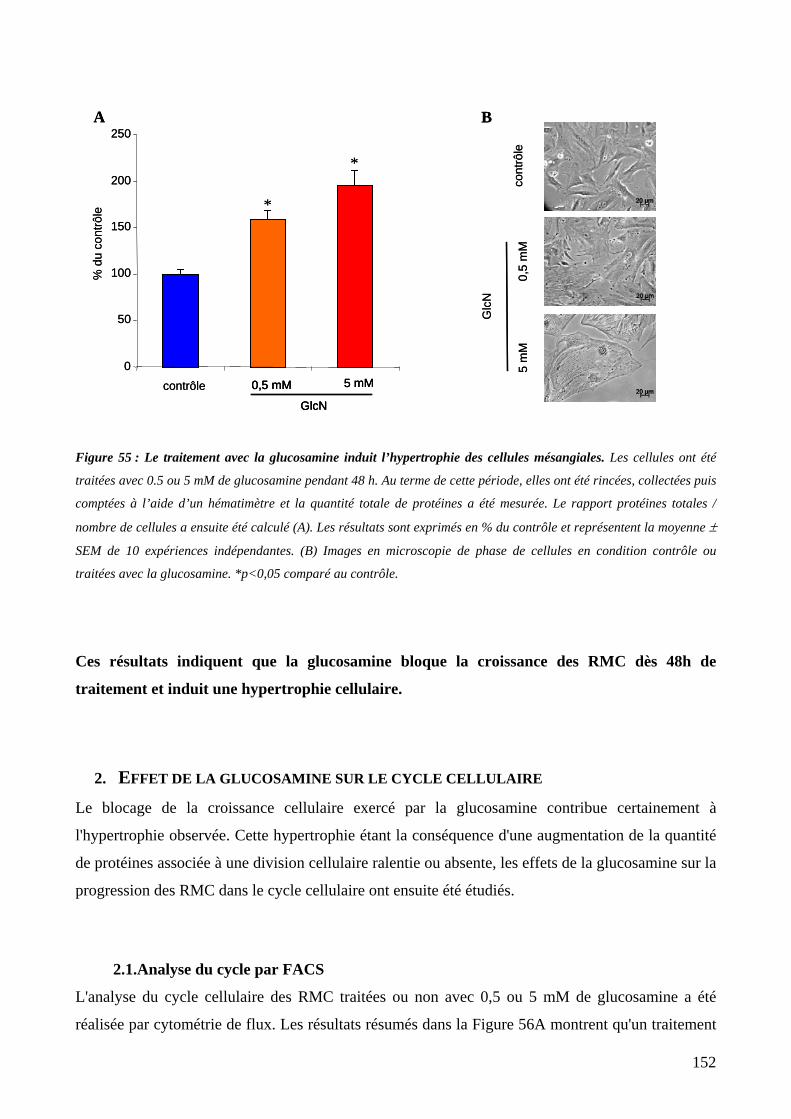
*
*
0
50
100
150
200
250
contrôle
% d
u co
ntrô
le
0,5 mM 5 mM
GlcN
A B
cont
rôle
0,5
mM
5m
M
Glc
N
*
*
0
50
100
150
200
250
contrôle
% d
u co
ntrô
le
0,5 mM 5 mM
GlcN
0,5 mM 5 mM
GlcN
A B
cont
rôle
0,5
mM
5m
M
Glc
N
Figure 55 : Le traitement avec la glucosamine induit l’hypertrophie des cellules mésangiales. Les cellules ont été
traitées avec 0.5 ou 5 mM de glucosamine pendant 48 h. Au terme de cette période, elles ont été rincées, collectées puis
comptées à l’aide d’un hématimètre et la quantité totale de protéines a été mesurée. Le rapport protéines totales /
nombre de cellules a ensuite été calculé (A). Les résultats sont exprimés en % du contrôle et représentent la moyenne ±
SEM de 10 expériences indépendantes. (B) Images en microscopie de phase de cellules en condition contrôle ou
traitées avec la glucosamine. *p<0,05 comparé au contrôle.
Ces résultats indiquent que la glucosamine bloque la croissance des RMC dès 48h de
traitement et induit une hypertrophie cellulaire.
2. EFFET DE LA GLUCOSAMINE SUR LE CYCLE CELLULAIRE Le blocage de la croissance cellulaire exercé par la glucosamine contribue certainement à
l'hypertrophie observée. Cette hypertrophie étant la conséquence d'une augmentation de la quantité
de protéines associée à une division cellulaire ralentie ou absente, les effets de la glucosamine sur la
progression des RMC dans le cycle cellulaire ont ensuite été étudiés.
2.1.Analyse du cycle par FACS
L'analyse du cycle cellulaire des RMC traitées ou non avec 0,5 ou 5 mM de glucosamine a été
réalisée par cytométrie de flux. Les résultats résumés dans la Figure 56A montrent qu'un traitement
152
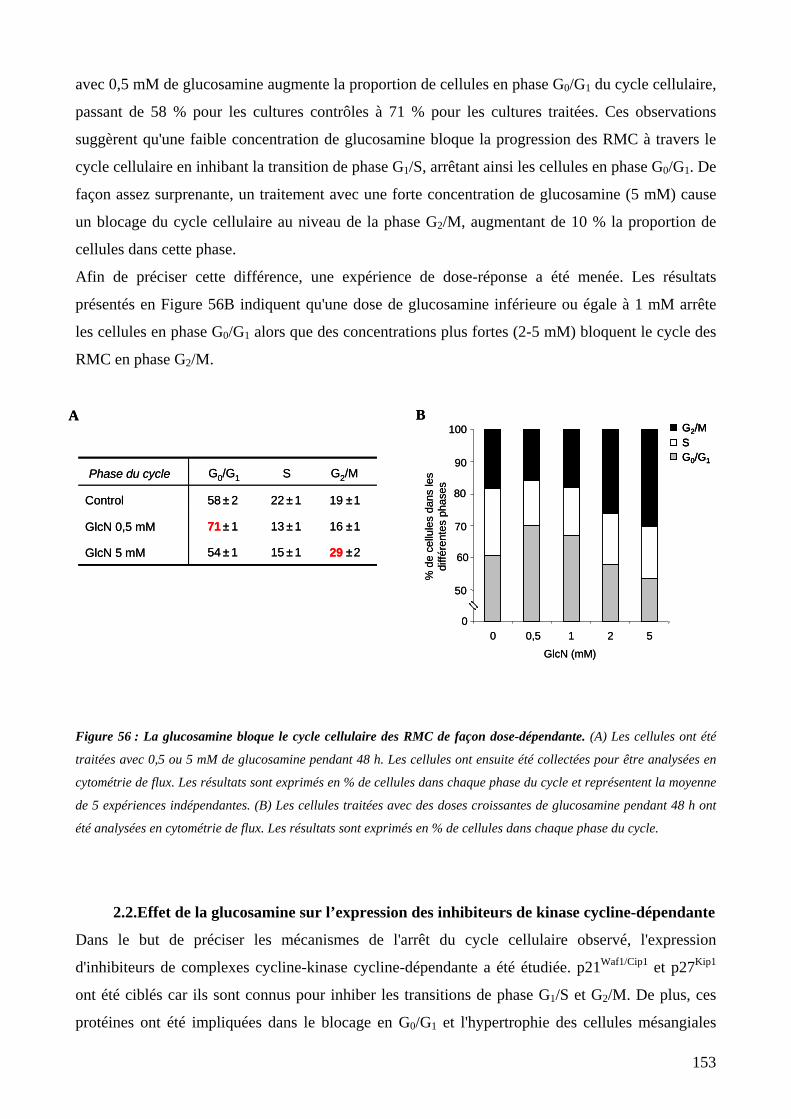
avec 0,5 mM de glucosamine augmente la proportion de cellules en phase G0/G1 du cycle cellulaire,
passant de 58 % pour les cultures contrôles à 71 % pour les cultures traitées. Ces observations
suggèrent qu'une faible concentration de glucosamine bloque la progression des RMC à travers le
cycle cellulaire en inhibant la transition de phase G1/S, arrêtant ainsi les cellules en phase G0/G1. De
façon assez surprenante, un traitement avec une forte concentration de glucosamine (5 mM) cause
un blocage du cycle cellulaire au niveau de la phase G2/M, augmentant de 10 % la proportion de
cellules dans cette phase.
Afin de préciser cette différence, une expérience de dose-réponse a été menée. Les résultats
présentés en Figure 56B indiquent qu'une dose de glucosamine inférieure ou égale à 1 mM arrête
les cellules en phase G0/G1 alors que des concentrations plus fortes (2-5 mM) bloquent le cycle des
RMC en phase G2/M.
0
50
60
70
80
90
100
0 0,5 1 2 5
% d
e ce
llule
s da
ns le
s di
ffére
ntes
pha
ses
GlcN (mM)
G2/MSG0/G1
A B
Phase du cycle
Control 58 ± 2 22 ± 1 19 ±1
GlcN 0,5 mM 71 ± 1 13 ± 1 16 ±1
GlcN 5 mM 54 ± 1 15 ± 1 ±2
Figure 56 : La glucosamine bloque le cycle cellulaire des RMC de façon dose-dépendante. (A) Les cellules ont été
traitées avec 0,5 ou 5 mM de glucosamine pendant 48 h. Les cellules ont ensuite été collectées pour être analysées en
cytométrie de flux. Les résultats sont exprimés en % de cellules dans chaque phase du cycle et représentent la moyenne
de 5 expériences indépendantes. (B) Les cellules traitées avec des doses croissantes de glucosamine pendant 48 h ont
été analysées en cytométrie de flux. Les résultats sont exprimés en % de cellules dans chaque phase du cycle.
2.2.Effet de la glucosamine sur l’expression des inhibiteurs de kinase cycline-dépendante
Dans le but de préciser les mécanismes de l'arrêt du cycle cellulaire observé, l'expression
d'inhibiteurs de complexes cycline-kinase cycline-dépendante a été étudiée. p21Waf1/Cip1 et p27Kip1
ont été ciblés car ils sont connus pour inhiber les transitions de phase G1/S et G2/M. De plus, ces
protéines ont été impliquées dans le blocage en G0/G1 et l'hypertrophie des cellules mésangiales
29
G0/G1 S G2/M
0
50
60
70
80
90
100
0 0,5 1 2 5
% d
e ce
llule
s da
ns le
s di
ffére
ntes
pha
ses
GlcN (mM)
G2/MSG0/G1
G2/MSG0/G1
A B
Phase du cycle
Control 58 ± 2 22 ± 1 19 ±1
GlcN 0,5 mM ± 1 13 ± 1 16 ±171
GlcN 5 mM 54 ± 1 15 ± 1 ±229
G0/G1 S G2/M
153
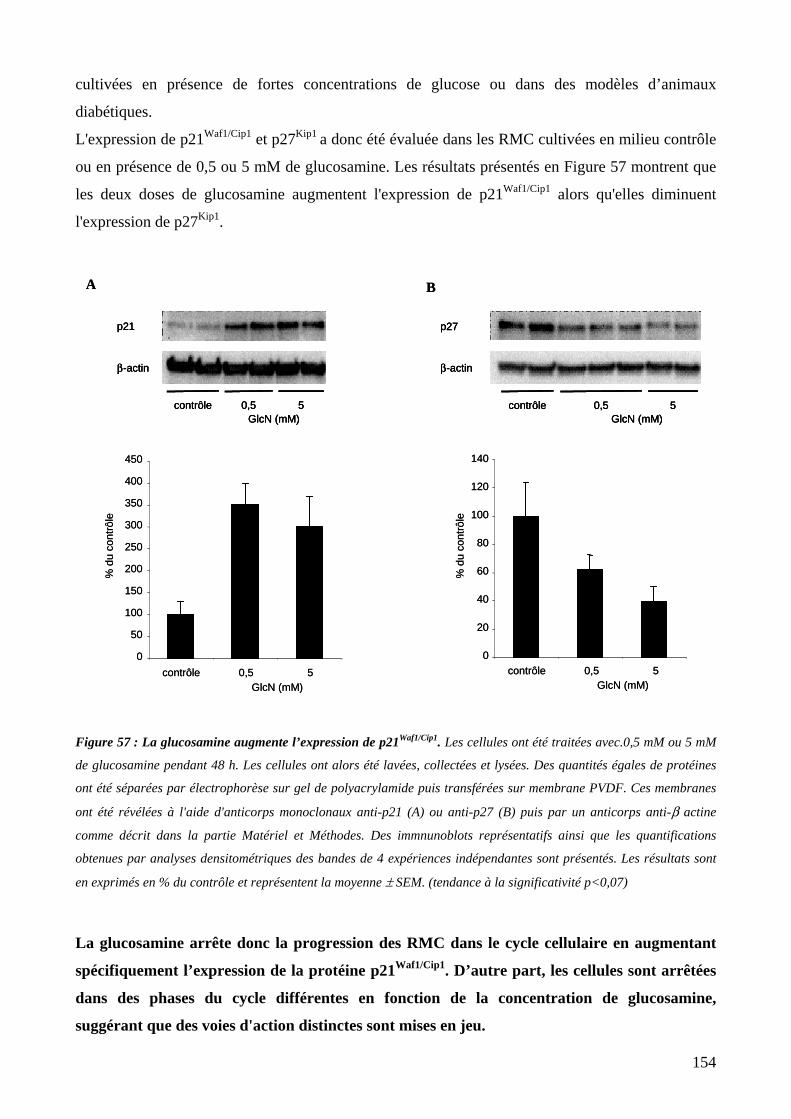
cultivées en présence de fortes concentrations de glucose ou dans des modèles d’animaux
diabétiques.
L'expression de p21Waf1/Cip1 et p27Kip1 a donc été évaluée dans les RMC cultivées en milieu contrôle
ou en présence de 0,5 ou 5 mM de glucosamine. Les résultats présentés en Figure 57 montrent que
les deux doses de glucosamine augmentent l'expression de p21Waf1/Cip1 alors qu'elles diminuent
l'expression de p27Kip1.
A B
β-actin
p21
contrôle 0,5 5GlcN (mM)
β-actin
p27
contrôle 0,5 5GlcN (mM)
0
50
100
150
200
250
300
350
400
450
contrôle 0,5 5
% d
u co
ntrô
le
GlcN (mM)
0
20
40
60
80
100
120
140%
du
cont
rôle
contrôle 0,5 5GlcN (mM)
A B
β-actin
p21
contrôle 0,5 5GlcN (mM)
β-actin
p21
contrôle 0,5 5GlcN (mM)
β-actin
p27
contrôle 0,5 5GlcN (mM)
β-actin
p27
contrôle 0,5 5GlcN (mM)
0
50
100
150
200
250
300
350
400
450
contrôle 0,5 5
% d
u co
ntrô
le
GlcN (mM)
0
20
40
60
80
100
120
140%
du
cont
rôle
contrôle 0,5 5GlcN (mM)
Figure 57 : La glucosamine augmente l’expression de p21Waf1/Cip1. Les cellules ont été traitées avec.0,5 mM ou 5 mM
de glucosamine pendant 48 h. Les cellules ont alors été lavées, collectées et lysées. Des quantités égales de protéines
ont été séparées par électrophorèse sur gel de polyacrylamide puis transférées sur membrane PVDF. Ces membranes
ont été révélées à l'aide d'anticorps monoclonaux anti-p21 (A) ou anti-p27 (B) puis par un anticorps anti-β actine
comme décrit dans la partie Matériel et Méthodes. Des immnunoblots représentatifs ainsi que les quantifications
obtenues par analyses densitométriques des bandes de 4 expériences indépendantes sont présentés. Les résultats sont
en exprimés en % du contrôle et représentent la moyenne ± SEM. (tendance à la significativité p<0,07)
La glucosamine arrête donc la progression des RMC dans le cycle cellulaire en augmentant
spécifiquement l’expression de la protéine p21Waf1/Cip1. D’autre part, les cellules sont arrêtées
dans des phases du cycle différentes en fonction de la concentration de glucosamine,
suggérant que des voies d'action distinctes sont mises en jeu.
154
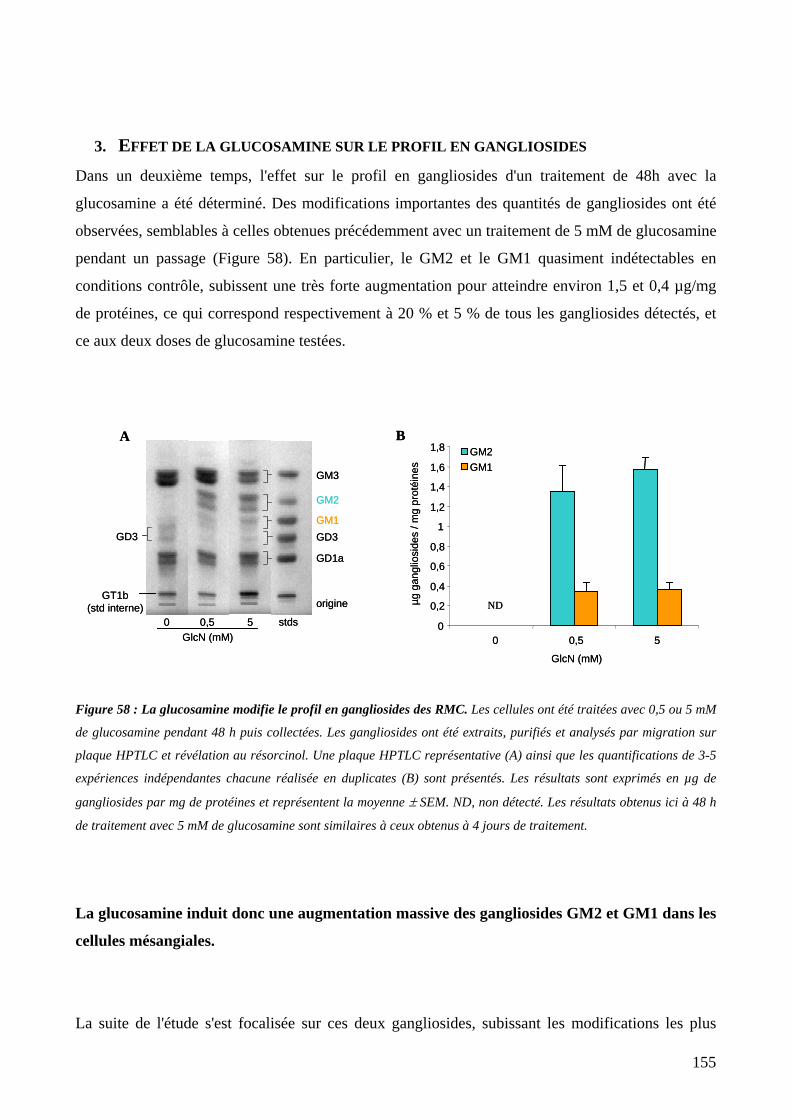
3. EFFET DE LA GLUCOSAMINE SUR LE PROFIL EN GANGLIOSIDES Dans un deuxième temps, l'effet sur le profil en gangliosides d'un traitement de 48h avec la
glucosamine a été déterminé. Des modifications importantes des quantités de gangliosides ont été
observées, semblables à celles obtenues précédemment avec un traitement de 5 mM de glucosamine
pendant un passage (Figure 58). En particulier, le GM2 et le GM1 quasiment indétectables en
conditions contrôle, subissent une très forte augmentation pour atteindre environ 1,5 et 0,4 µg/mg
de protéines, ce qui correspond respectivement à 20 % et 5 % de tous les gangliosides détectés, et
ce aux deux doses de glucosamine testées.
GlcN (mM)stds50 0,5
GT1b(std interne) origine
GM1
GM2
GM3
GD3
GD1a
GD3
0
0,2
0,4
0,6
0,8
1
1,2
1,4
1,6
1,8
0 0,5 5
GlcN (mM)
µg g
angl
iosi
des
/ mg
prot
éine
s
GM2GM1
A B
ND
GlcN (mM)stds50 0,5
GT1b(std interne) origine
GM1
GM2
GM3
GD3
GD1a
GD3
0
0,2
0,4
0,6
0,8
1
1,2
1,4
1,6
1,8
0 0,5 5
GlcN (mM)
µg g
angl
iosi
des
/ mg
prot
éine
s
GM2GM1
A B
ND
Figure 58 : La glucosamine modifie le profil en gangliosides des RMC. Les cellules ont été traitées avec 0,5 ou 5 mM
de glucosamine pendant 48 h puis collectées. Les gangliosides ont été extraits, purifiés et analysés par migration sur
plaque HPTLC et révélation au résorcinol. Une plaque HPTLC représentative (A) ainsi que les quantifications de 3-5
expériences indépendantes chacune réalisée en duplicates (B) sont présentés. Les résultats sont exprimés en µg de
gangliosides par mg de protéines et représentent la moyenne ± SEM. ND, non détecté. Les résultats obtenus ici à 48 h
de traitement avec 5 mM de glucosamine sont similaires à ceux obtenus à 4 jours de traitement.
La glucosamine induit donc une augmentation massive des gangliosides GM2 et GM1 dans les
cellules mésangiales.
La suite de l'étude s'est focalisée sur ces deux gangliosides, subissant les modifications les plus
155
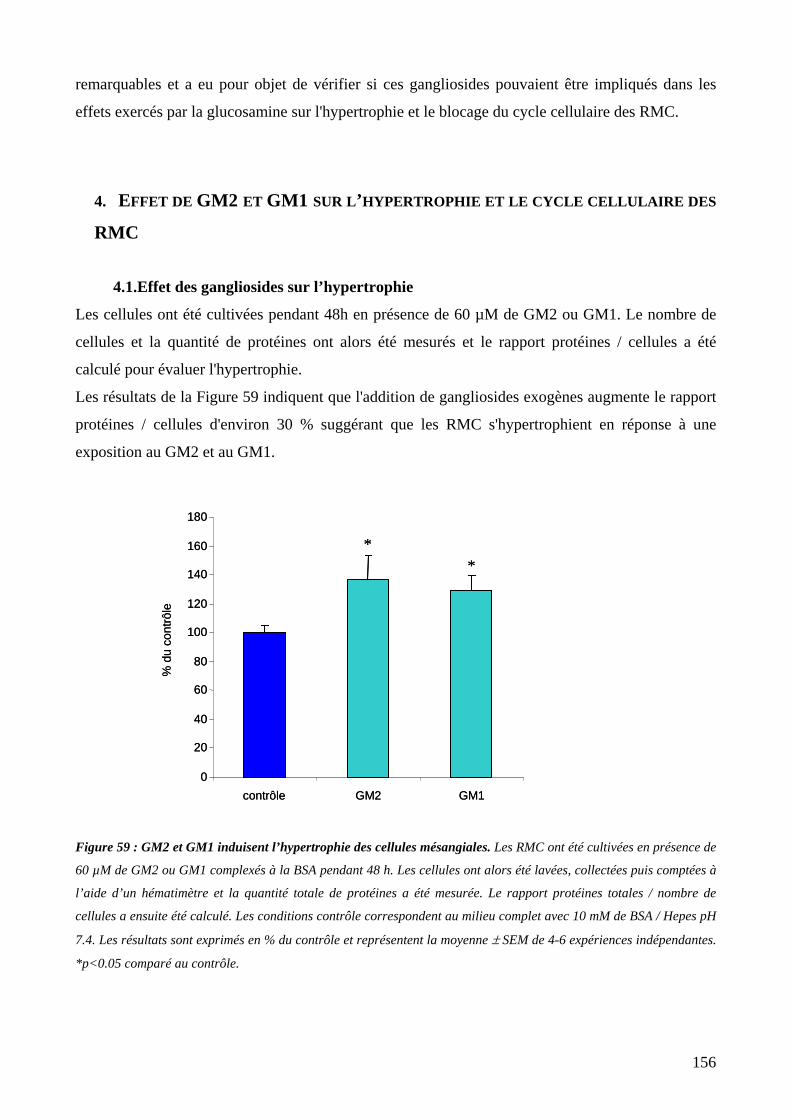
remarquables et a eu pour objet de vérifier si ces gangliosides pouvaient être impliqués dans les
effets exercés par la glucosamine sur l'hypertrophie et le blocage du cycle cellulaire des RMC.
4. EFFET DE GM2 ET GM1 SUR L’HYPERTROPHIE ET LE CYCLE CELLULAIRE DES
RMC
4.1.Effet des gangliosides sur l’hypertrophie
Les cellules ont été cultivées pendant 48h en présence de 60 µM de GM2 ou GM1. Le nombre de
cellules et la quantité de protéines ont alors été mesurés et le rapport protéines / cellules a été
calculé pour évaluer l'hypertrophie.
Les résultats de la Figure 59 indiquent que l'addition de gangliosides exogènes augmente le rapport
protéines / cellules d'environ 30 % suggérant que les RMC s'hypertrophient en réponse à une
exposition au GM2 et au GM1.
**
% d
u co
ntrô
le
0
20
40
60
80
100
120
140
160
180
contrôle GM2 GM1
**
% d
u co
ntrô
le
0
20
40
60
80
100
120
140
160
180
contrôle GM2 GM1
Figure 59 : GM2 et GM1 induisent l’hypertrophie des cellules mésangiales. Les RMC ont été cultivées en présence de
60 µM de GM2 ou GM1 complexés à la BSA pendant 48 h. Les cellules ont alors été lavées, collectées puis comptées à
l’aide d’un hématimètre et la quantité totale de protéines a été mesurée. Le rapport protéines totales / nombre de
cellules a ensuite été calculé. Les conditions contrôle correspondent au milieu complet avec 10 mM de BSA / Hepes pH
7.4. Les résultats sont exprimés en % du contrôle et représentent la moyenne ± SEM de 4-6 expériences indépendantes.
*p<0.05 comparé au contrôle.
156
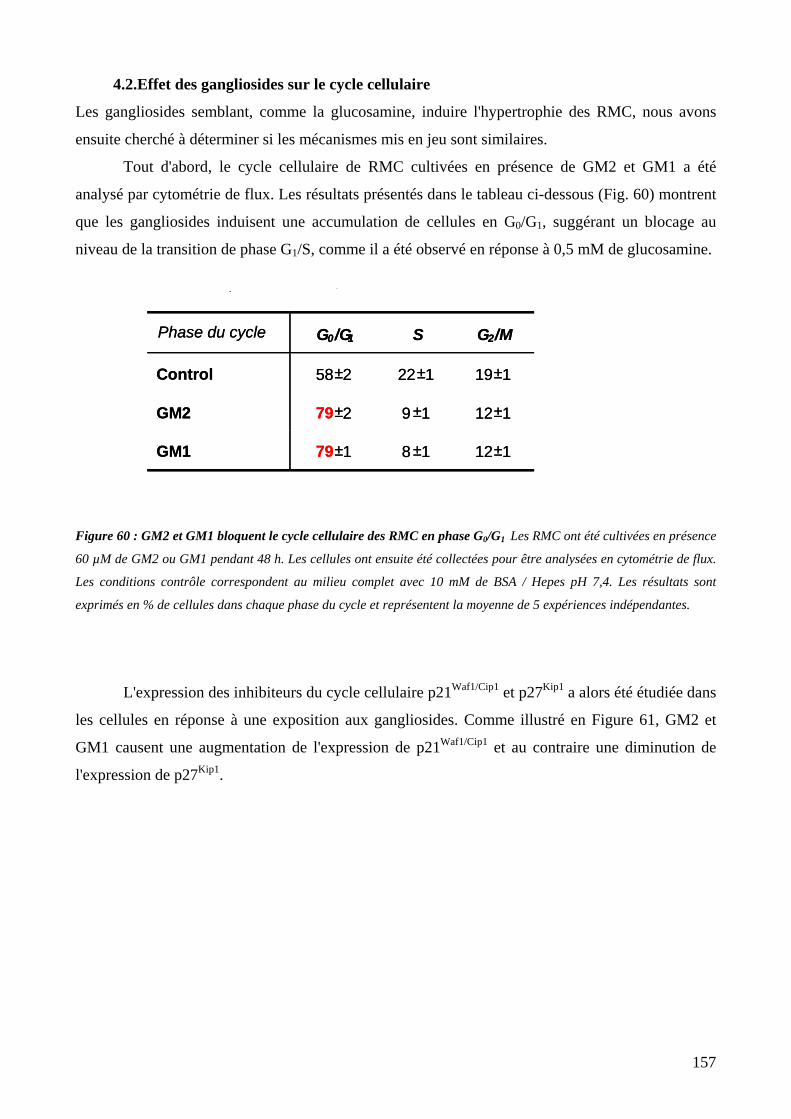
4.2.Effet des gangliosides sur le cycle cellulaire
Les gangliosides semblant, comme la glucosamine, induire l'hypertrophie des RMC, nous avons
ensuite cherché à déterminer si les mécanismes mis en jeu sont similaires.
Tout d'abord, le cycle cellulaire de RMC cultivées en présence de GM2 et GM1 a été
analysé par cytométrie de flux. Les résultats présentés dans le tableau ci-dessous (Fig. 60) montrent
que les gangliosides induisent une accumulation de cellules en G0/G1, suggérant un blocage au
niveau de la transition de phase G1/S, comme il a été observé en réponse à 0,5 mM de glucosamine.
G0/G1 S G2/M
Control 58±2 22±1 19±1
GM2 79±2 9 ±1 12±1
GM1 1 8 ±1 12±1
Figure 60 : GM2 et GM1 bloquent le cycle cellulaire des RMC en phase G0/G1 Les RMC ont été cultivées en présence
60 µM de GM2 ou GM1 pendant 48 h. Les cellules ont ensuite été collectées pour être analysées en cytométrie de flux.
Les conditions contrôle correspondent au milieu complet avec 10 mM de BSA / Hepes pH 7,4. Les résultats sont
exprimés en % de cellules dans chaque phase du cycle et représentent la moyenne de 5 expériences indépendantes.
L'expression des inhibiteurs du cycle cellulaire p21Waf1/Cip1 et p27Kip1 a alors été étudiée dans
les cellules en réponse à une exposition aux gangliosides. Comme illustré en Figure 61, GM2 et
GM1 causent une augmentation de l'expression de p21Waf1/Cip1 et au contraire une diminution de
l'expression de p27Kip1.
79±
Phase du cycle G0/G1 S G2/M
Control 58±2 22±1 19±1
GM2 2 9 ±1 12±179±
GM1 1 8 ±1 12±179±
Phase du cycle
157

A B
β-actin
p21
contrôle GM2 GM1
0
50
100
150
200
250
contrôle GM2 GM1
% d
u co
ntrô
le
0
20
40
60
80
100
120
140
% d
u co
ntrô
lecontrôle GM2 GM1
β-actin
p27
contrôle GM2 GM1
A B
β-actin
p21
contrôle GM2 GM1
0
50
100
150
200
250
contrôle GM2 GM1contrôle GM2 GM1
% d
u co
ntrô
le
0
20
40
60
80
100
120
140
% d
u co
ntrô
lecontrôle GM2 GM1contrôle GM2 GM1
β-actin
p27
contrôle GM2 GM1
β-actin
p27
contrôle GM2 GM1
Figure 61 : GM2 et GM1 augmentent l’expression de p21Waf1/Cip1. Les RMC ont été cultivées en présence 60 µM de
GM2 ou GM1 pendant 48 h. Les cellules ont alors été rincées, collectées et lysées. Des quantités égales de protéines ont
été séparées par électrophorèse sur gel de polyacrylamide puis transférées sur membrane PVDF. Ces membranes ont
été révélées à l'aide d'anticorps monoclonaux anti-p21 (A) ou anti-p27 (B) puis par un anticorps anti-β actine comme
décrit dans la partie Matériel et Méthodes. Des immnunoblots représentatifs ainsi que les quantifications obtenues par
analyses densitométriques des bandes de 4 expériences indépendantes sont présentés. Les résultats sont exprimés en %
du contrôle et représentent la moyenne ± SEM. (tendance à la significativité p<0,07)
Ces observations indiquent que la glucosamine à faible dose et les gangliosides GM2 et GM1
induisent l'hypertrophie et inhibent la prolifération des RMC par des mécanismes similaires,
en augmentant l'expression de p21Waf1/Cip1 et en bloquant les cellules en phase G0/G1 du cycle
cellulaire. Elles suggèrent que les gangliosides GM2 et GM1, fortement augmentés en réponse
à la glucosamine, pourraient médier les effets de la glucosamine sur les cellules mésangiales.
158

VI. DISCUSSION
Cette deuxième partie a consisté à explorer la voie des hexosamines, une des hypothèses
biochimiques proposée pour expliquer les complications microvasculaires du diabète, en particulier
la néphropathie. Elle consiste à convertir le fructose-6-P issu du glucose en glucosamine-6-P
(GlcN-6-P) via une enzyme spécifique et limitante, activée en conditions hyperglycémiques, la
glutamine: fructose-6-P-amidotransférase (GFAT). La GlcN-6-P est ensuite rapidement convertie et
activée en UDP-N-acétylglucosamine (UDP-GlcNAc), précurseur des sucres aminés nécessaires à
la biosynthèse des glycoprotéines et glycolipides et résidu utilisé pour la O-glycosylation des
protéines.
L’expression de la GFAT a été décrite dans les cellules épithéliales et mésangiales glomérulaires de
reins de patients diabétiques atteints de néphropathie (Nerlich A.G. et al., 1998). Cette voie est
souvent mise en cause dans le développement des caractéristiques pathologiques de la néphropathie
diabétique comme par exemple l’augmentation de la production de TGFβ et consécutivement de
protéines de la matrice extracellulaire (Schleicher E.D. et al., 2000). Une activité GFAT a de plus
été mesurée dans la rétine, et les quantités d'hexosamines sont augmentées dans les tissus rétiniens
d'homme et de rat diabétiques, soulignant ainsi la possibilité d'un rôle de la voie des hexosamines
dans la rétinopathie (Heath H. et al., 1967; Mazlen R.G. et al., 1970).
L’UDP-N-acétylglucosamine est un des principaux métabolites issus de la voie des hexosamines.
La glucosamine exogène est rapidement captée par les cellules via les transporteurs au glucose et
phosphorylée par l'hexokinase, entrant ainsi sélectivement dans la voie des hexosamines an aval de
l'enzyme limitante GFAT (Schleicher E.D. et al., 2000). Pour ces raisons, la glucosamine est
utilisée expérimentalement pour mimer une suractivation de la voie des hexosamines. Cependant, la
quantité de glucosamine exogène effectivement captée par les cellules varie certainement en
fonction du type cellulaire considéré. Ainsi, des différences de concentration intracellulaire en
glucosamine peuvent exister entre deux types cellulaires cultivés en présence de concentrations
extracellulaires de glucosamine identiques. Dans cette optique, même si certains résultats ont été
observés dans les deux modèles cellulaires étudiés, il faut rester prudent sur les analogies entre BRP
et RMC. Par ailleurs, la glucosamine est un agent pharmacologique puissant pouvant être toxique à
forte dose ou à long terme. Il est donc important de garder à l'esprit, lors de l'interprétation des
résultats obtenus, que ces effets toxiques potentiels peuvent masquer, dans les cellules, les effets
spécifiques de la glucosamine.
Dans un premier temps, nous avons étudié les effets d’un traitement avec la glucosamine sur
159

la prolifération cellulaire. Les résultats ont montré que la glucosamine diminue la prolifération des
péricytes et encore plus fortement celle des cellules mésangiales, de manière dose-dépendante.
Ensuite, nous avons déterminé les effets de la glucosamine sur le métabolisme des
gangliosides. Dans les BRP, les taux de GM1 et de GD3 sont augmentés, au détriment du GM3,
dont le taux diminue en réponse au traitement. Dans les RMC, une augmentation du taux des
gangliosides de la série-a, et particulièrement une très forte augmentation du GM2 et du GM1, a été
mesurée, au détriment du GD3, dont le taux est nettement diminué en réponse à la glucosamine.
Les mécanismes par lesquels la glucosamine peut affecter le métabolisme des gangliosides n’ont
pas été précisément explorés. Cependant, il est connu que la N-acétyl glucosamine peut modifier,
par une O-glycosylation simple, l’activité d’une grande variété de protéines et d’enzymes. Par
exemple, il a été montré que l’activité du facteur de transcription Sp1 peut être positivement
modulée par la liaison d’un résidu GlcNAc (Goldberg H.J. et al., 2002). Plusieurs enzymes du
métabolisme des gangliosides, notamment GM3, GD3 et GM2 synthases, présentent des sites de
liaisons potentiels à ce facteur de transcription dans leur promoteur. Il est donc possible que la
glucosamine puisse modifier les activités d’enzymes de biosynthèse des gangliosides en régulant
leur expression. Par ailleurs, il semble que les enzymes du métabolisme des gangliosides puissent
être régulées par phosphorylation (Bieberich E. et al., 1998; Gu X. et al., 1995). Or, sur certaines
protéines, O-glucosamine et O-phosphate peuvent occuper les mêmes sites (Wells L. et al., 2001). Il
est donc envisageable que ces enzymes soient directement régulées par O-glycosylation. Ces modes
de régulation pourraient expliquer l’augmentation de l’activité de la GM3 synthase observée dans
les BRP en réponse à la glucosamine. D’autre part, la glucosamine peut être convertie en différents
métabolites, notamment en acide sialique, composant caractéristique de tous les gangliosides. De
plus, la GlucNAc peut être isomérisée en N-acétylgalactosamine (GalNAc), pouvant elle-même être
incorporée dans la structure de certains gangliosides, tels le GM2 et le GM1. Il est donc probable
que la glucosamine affecte le métabolisme des gangliosides par action de masse, en fournissant des
quantités importantes de précurseurs glycosidiques utilisés comme substrats pour leur biosynthèse.
La glucosamine affecte le métabolisme des gangliosides dans les deux types cellulaires d’intérêt.
Cependant, alors que le traitement inhibe à la fois la prolifération des BRP et des RMC, les
modifications du profil en gangliosides observées en réponse à la glucosamine se sont révélées
différentes en fonction du type cellulaire considéré. Il est possible que l’augmentation des
gangliosides de la série-a (GM1 dans les BRP et GM2 et GM1 dans les RMC), facteur commun aux
deux types cellulaires, soit en partie responsable de l’inhibition de prolifération induite pat le
traitement avec la glucosamine. Il est aussi envisageable que les différents gangliosides agissent,
éventuellement selon des mécanismes distincts, en fonction du type cellulaire pour concourir à la
diminution de prolifération observée. Il a déjà été évoqué dans la partie précédente qu’un certain
160

nombre de gangliosides exercent un effet anti-prolifératif via une interaction avec la voie de
signalisation de facteurs de croissance. Par ailleurs, le GD3 principalement, a été décrit comme un
médiateur intracellulaire de l’apoptose, plus précisément pour son rôle dans la transition de
perméabilité mitochondriale, le relarguage de cytochrome c et l’activation de caspase.
L’augmentation importante de GD3 observée dans les BRP pourrait ainsi jouer un rôle primordial
dans la diminution de prolifération causée par la glucosamine, en participant à l’induction d’un
processus apoptotique. Les résultats préliminaires que nous avons obtenus en mesurant l’apoptose
dans les péricytes traités à la glucosamine (mesure de l’activité caspase 3 et détection d’une
fragmentation de l’ADN) ne vont cependant pas dans ce sens. Enfin, les gangliosides ont déjà été
impliqués, bien que plus rarement, dans la régulation du cycle cellulaire. Nakatsuji et al. ont par
exemple montré que le GM3 induit un arrêt du cycle cellulaire des astrocytes en partie via une
augmentation de l’expression de p27Kip1 (Nakatsuji Y. et al., 2001).
Dans la suite de l’étude, nous avons choisi de nous intéresser plus en détail aux mécanismes
par lesquels la glucosamine, par l’intermédiaire des gangliosides, peut inhiber la prolifération
cellulaire. Ce travail, plus mécanistique, a été mené uniquement dans les RMC.
Premièrement, les courbes de croissance des RMC en réponse à la glucosamine ont montré
un blocage assez rapide de la prolifération cellulaire. Réalisées à la fois sur le nombre de cellules et
la quantité de protéines, elles ont également mis en évidence une hypertrophie des cellules traitées.
Cette hypertrophie a également été révélée par l’augmentation de taille des cellules, aisément
remarquable par simple observation au microscope. L’hypertrophie des cellules mésangiales est une
caractéristique pathologique typique de la néphropathie diabétique. Ce phénomène a été décrit à la
fois in vivo dans les reins de rats rendus diabétiques par injection de streptozotocine ou de souris
db/db et in vitro dans des cellules mésangiales cultivées en présence de fortes concentrations de
glucose (Wolf G., 2000). Il est particulièrement important car, associé à une hypersécrétion de
protéines de la matrice extracellulaire, il participe au grossissement puis à la sclérose du glomérule,
altérations majeures de la néphropathie diabétique, étroitement corrélées aux manifestations
cliniques comme l’albuminurie, l’hypertension ou la diminution du taux de filtration glomérulaire.
D’autre part, il a été montré que cette hypertrophie est consécutive à un arrêt des cellules
mésangiales en phase G1 du cycle cellulaire. C’est pourquoi nous avons ensuite étudié le cycle
cellulaire des RMC traitées avec la glucosamine. Les résultats ont montré qu’un traitement avec une
concentration de 0,5 mM de glucosamine induit un blocage des cellules en phase G0/G1 du cycle
cellulaire, en accord avec les observations déjà faites in vivo et in vitro. Ces résultats sont les
premiers, à notre connaissance, à montrer que la glucosamine peut bloquer le cycle cellulaire et
induire l’hypertrophie des cellules mésangiales. Par contre, il s’est avéré qu’un traitement avec 5
161

mM de glucosamine cause un blocage en phase G2/M. Ces observations montrent clairement qu’une
faible et une forte concentration de glucosamine exercent des effets différents sur les RMC. Ces
différences pourraient s’expliquer à la lumière de récents travaux de Marshal et al. dans les
adipocytes (Marshall S. et al., 2004). Ces auteurs ont en effet montré que des concentrations de
glucosamine assez élevées (>2 mM) causaient une accumulation massive de GlcN-6-P, un produit
intermédiaire de la voie de l’hexosamine, accompagnée par une chute du taux d’ATP. Au contraire,
ces effets n’étaient pas observés à de plus faibles concentrations de glucosamine (0,25 mM) ou bien
à des fortes concentrations de glucose (20 mM). Ces observations ont été faites avec des adipocytes
et il est délicat de les extrapoler aux cellules mésangiales. Cependant, nous pouvons émettre
l’hypothèse qu’à 5 mM de glucosamine, d’autres mécanismes absents à des doses plus faibles, se
mettent en place, tels la déplétion en ATP, exerçant des effets délétères sur les cellules et
conduisant au blocage en phase G2/M. Il est alors probable que l’utilisation de faibles
concentrations de glucosamine soit plus adaptée pour révéler les effets spécifiques de la
glucosamine et découvrir les effets de l’hyperglycémie médiés par la voie de l’hexosamine.
Pour préciser les mécanismes responsables de l’arrêt du cycle cellulaire observé suite aux
traitements avec la glucosamine, l’expression de deux inhibiteurs de kinases cycline–dépendantes
p21Waf1/Cip1 et p27Kip1 a été évaluée. Les résultats ont montré que l’expression de p21Waf1/Cip1 est
environ triplée en réponse à la glucosamine, suggérant ainsi son rôle dans l’arrêt du cycle cellulaire
des RMC. Par contre, l’expression de p27Kip1 est plutôt diminuée par le traitement avec la
glucosamine. Une expression diminuée ou inchangée de cette protéine a déjà été décrite dans les
glomérules de modèles d’animaux diabétiques (Griffin S.V. et al., 2003; Kuan C.J. et al., 1998),
ainsi que dans des cellules mésangiales cultivées en présence de fortes concentrations de
glucose(Griffin S.V. et al., 2003). Ces résultats suggèrent que les effets de la glucosamine sur le
cycle cellulaire des RMC sont spécifiquement médiés par p21Waf1/Cip1. D’autre part, p21Waf1/Cip1 a
été décrit comme un régulateur clé de l’hypertrophie. Fan et al. ont montré que l’inhibition de cette
protéine à l’aide d’oligonucléotides antisens permet de diminuer l’hypertrophie de cellules
mésangiales induites par l’hyperglycémie et l’IGF-I (Fan Y.P. et al., 2004). De plus, in vivo, des
souris diabétiques dont le gène de p21Waf1/Cip1 a été invalidé n’ont pas développé d’hypertrophie
glomérulaire (al Douahji M. et al., 1999). Ces travaux suggèrent que l’augmentation de l’expression
de p21Waf1/Cip1 pourrait également participer aux mécanismes conduisant à l’hypertrophie des
cellules mésangiales observée ici en réponse à la glucosamine. Par ailleurs, étant donné que les
effets de la glucosamine sur les RMC sont similaires à ceux de fortes concentrations de glucose déjà
décrits dans la littérature, nous pouvons penser que les effets de l’hyperglycémie sur la croissance et
l’hypertrophie des cellules mésangiales sont, au moins en partie, médiés par la voie de
l’hexosamine et p21Waf1/Cip1.
162

Deuxièmement, nous avons déterminé l’effet sur le profil en gangliosides d’un traitement de
48h avec une concentration faible ou une concentration forte de glucosamine. Les modifications se
sont révélées similaires à celles déjà observées suite à un traitement pendant un passage, avec en
particulier une augmentation massive de GM2 et GM1. Nous avons ensuite cherché à savoir si ces
gangliosides peuvent être des médiateurs de la glucosamine, en étudiant l’effet de GM2 et GM1 sur
l’hypertrophie et le cycle cellulaire des RMC. Le traitement avec ces deux gangliosides occasionne
en effet une augmentation du rapport protéines/cellules, marqueur de l’hypertrophie. De plus,
l’analyse du cycle cellulaire des RMC exposées à GM2 ou GM1 a révélé un blocage en phase
G0/G1, associé à une augmentation de l’expression de p21Waf1/Cip1. Ainsi, le GM2 et le GM1
exogènes reproduisent l’effet d’une faible concentration de glucosamine sur l’hypertrophie et l’arrêt
du cycle cellulaire des RMC. Ces observations suggèrent que ces gangliosides sont effectivement
en cause dans les effets de la glucosamine sur les RMC et proposent la modulation des taux de
gangliosides comme un nouveau mécanisme d’action de la voie de l’hexosamine. Comme il a déjà
été signalé, très peu d’études, à part celle de Nakatsuji et al. montrant l’effet du GM3 sur l’arrêt du
cycle cellulaire des astrocytes (Nakatsuji Y. et al., 2001), ont impliqué les gangliosides dans le
cycle cellulaire. Nos résultats suggèrent non seulement le rôle de GM2 et GM1 dans le blocage du
cycle cellulaire, mais indiquent également que les gangliosides pourraient participer aux
mécanismes conduisant à l’hypertrophie cellulaire.
163

METABOLISME DES GANGLIOSIDES DU
CORTEX RENAL DE SOURIS db/db
Les résultats précédemment obtenus in vitro suggèrent que les gangliosides sont impliqués dans les
perturbations de prolifération cellulaire causées par les AGE ou une activation de la voie de
l’hexosamine. Ils soulignent ainsi le rôle potentiel des gangliosides dans les complications
microangiopathiques du diabète. Nous avons donc cherché à déterminer si le métabolisme des
gangliosides pouvait également être affecté par un environnement diabétique in vivo. Pour cela, les
activités GM3 et GD3 synthase ont été mesurées et le profil en gangliosides a été analysé sur des
homogénats de cortex de souris contrôle db/m et de souris diabétiques db/db de 11 semaines. Ces
souris obèses développent un profil diabétique type II et présentent une complication rénale
importante, proche de celle observée chez l’Homme.
I. MESURE DES ACTIVITES GM3 ET GD3 SYNTHASE
Le cortex rénal a été isolé, fragmenté puis homogénéisé. Les activités enzymatiques ont été
mesurées sur ces homogénats comme décrit dans la partie Matériel et méthodes.
Les résultats de la Figure 62 montrent que l’activité GM3 synthase est augmentée d’environ 80 %
alors que l’activité GD3 synthase est diminuée d’environ 50 % dans le cortex rénal des souris
diabétiques comparées aux souris contrôle.
164
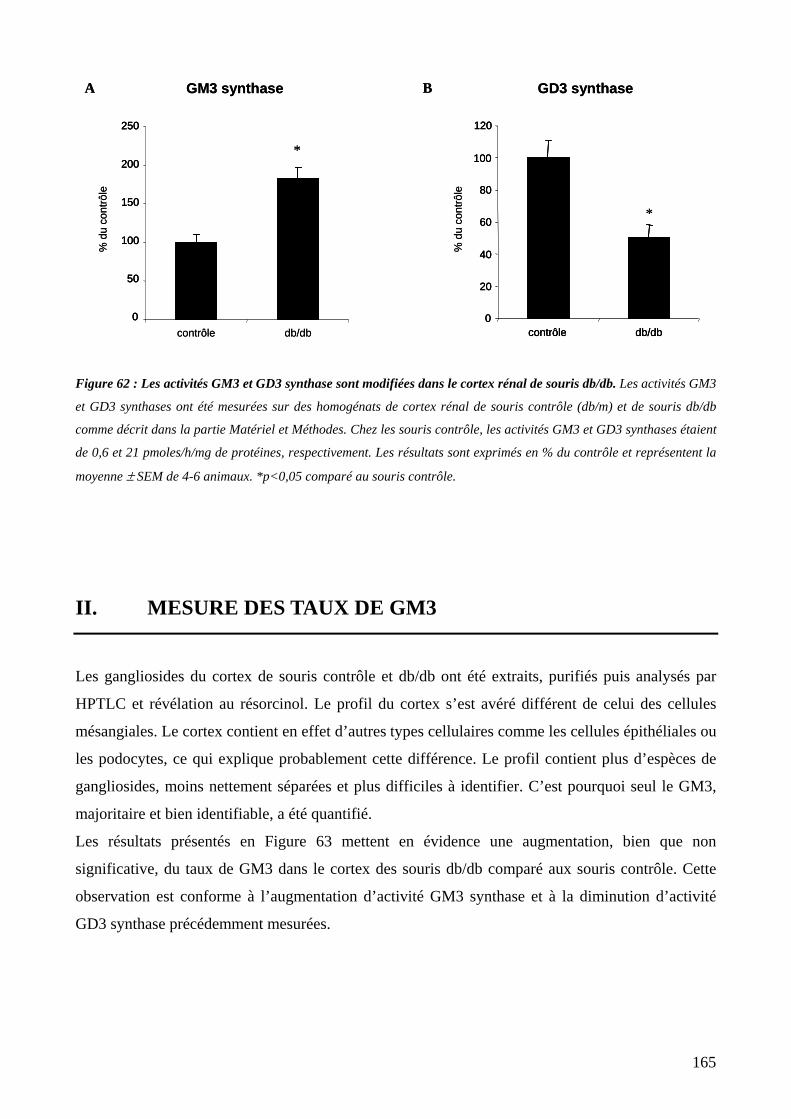
A GM3 synthase B GD3 synthase
%du
con
trôle
contrôle db/db0
50
100
150
200
250
% d
u co
ntrô
le
*
0
20
40
60
80
100
120
contrôle db/db
*
A GM3 synthase B GD3 synthase
%du
con
trôle
contrôle db/db0
50
100
150
200
250
0
50
100
150
200
250
% d
u co
ntrô
le
*
0
20
40
60
80
100
120
0
20
40
60
80
100
120
contrôle db/dbcontrôle db/db
*
Figure 62 : Les activités GM3 et GD3 synthase sont modifiées dans le cortex rénal de souris db/db. Les activités GM3
et GD3 synthases ont été mesurées sur des homogénats de cortex rénal de souris contrôle (db/m) et de souris db/db
comme décrit dans la partie Matériel et Méthodes. Chez les souris contrôle, les activités GM3 et GD3 synthases étaient
de 0,6 et 21 pmoles/h/mg de protéines, respectivement. Les résultats sont exprimés en % du contrôle et représentent la
moyenne ± SEM de 4-6 animaux. *p<0,05 comparé au souris contrôle.
II. MESURE DES TAUX DE GM3
Les gangliosides du cortex de souris contrôle et db/db ont été extraits, purifiés puis analysés par
HPTLC et révélation au résorcinol. Le profil du cortex s’est avéré différent de celui des cellules
mésangiales. Le cortex contient en effet d’autres types cellulaires comme les cellules épithéliales ou
les podocytes, ce qui explique probablement cette différence. Le profil contient plus d’espèces de
gangliosides, moins nettement séparées et plus difficiles à identifier. C’est pourquoi seul le GM3,
majoritaire et bien identifiable, a été quantifié.
Les résultats présentés en Figure 63 mettent en évidence une augmentation, bien que non
significative, du taux de GM3 dans le cortex des souris db/db comparé aux souris contrôle. Cette
observation est conforme à l’augmentation d’activité GM3 synthase et à la diminution d’activité
GD3 synthase précédemment mesurées.
165
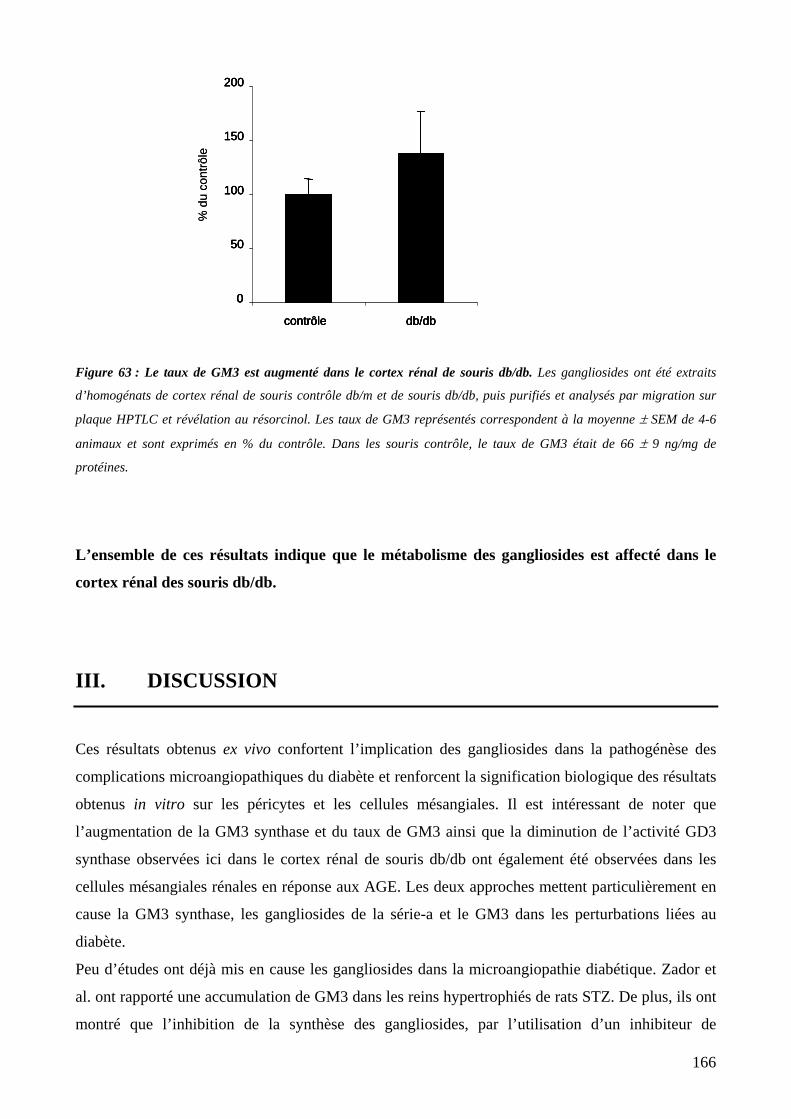
% d
u co
ntrô
le
contrôle db/db
0
50
100
150
200
% d
u co
ntrô
le
contrôle db/db
0
50
100
150
200
contrôle db/dbcontrôle db/db
0
50
100
150
200
0
50
100
150
200
Figure 63 : Le taux de GM3 est augmenté dans le cortex rénal de souris db/db. Les gangliosides ont été extraits
d’homogénats de cortex rénal de souris contrôle db/m et de souris db/db, puis purifiés et analysés par migration sur
plaque HPTLC et révélation au résorcinol. Les taux de GM3 représentés correspondent à la moyenne ± SEM de 4-6
animaux et sont exprimés en % du contrôle. Dans les souris contrôle, le taux de GM3 était de 66 ± 9 ng/mg de
protéines.
L’ensemble de ces résultats indique que le métabolisme des gangliosides est affecté dans le
cortex rénal des souris db/db.
III. DISCUSSION
Ces résultats obtenus ex vivo confortent l’implication des gangliosides dans la pathogénèse des
complications microangiopathiques du diabète et renforcent la signification biologique des résultats
obtenus in vitro sur les péricytes et les cellules mésangiales. Il est intéressant de noter que
l’augmentation de la GM3 synthase et du taux de GM3 ainsi que la diminution de l’activité GD3
synthase observées ici dans le cortex rénal de souris db/db ont également été observées dans les
cellules mésangiales rénales en réponse aux AGE. Les deux approches mettent particulièrement en
cause la GM3 synthase, les gangliosides de la série-a et le GM3 dans les perturbations liées au
diabète.
Peu d’études ont déjà mis en cause les gangliosides dans la microangiopathie diabétique. Zador et
al. ont rapporté une accumulation de GM3 dans les reins hypertrophiés de rats STZ. De plus, ils ont
montré que l’inhibition de la synthèse des gangliosides, par l’utilisation d’un inhibiteur de
166

glucosylcéramide synthase, permet de ramener le poids des reins de ces animaux diabétiques à un
niveau contrôle (Zador I.Z. et al., 1993). Une augmentation des taux d’ARNm de la GM3 synthase
a par ailleurs été mesurée dans le tissu adipeux de rats Zucker fa/fa (Tagami S. et al., 2002). Ces
données de la littérature obtenues sur différents modèles d’animaux diabétiques ainsi que ceux que
nous avons obtenus chez des souris db/db associent la GM3 synthase et les gangliosides de la série-
a à l’état diabétique.
167

CONCLUSION ET PERSPECTIVES
168


L’objectif de ce travail était de déterminer : i) les effets de deux voies biochimiques du
diabète, les AGE et la voie des hexosamines, sur la prolifération des péricytes rétiniens et des
cellules mésangiales rénales, ii) les effets des AGE et des hexosamines sur le métabolisme des
gangliosides et iii) le rôle des gangliosides dans les processus de régulation de prolifération exercés
par les AGE et la voie des hexosamines. Nos résultats ont permis de montrer que les AGE et la
glucosamine, produit de la voie des hexosamines, inhibent la prolifération et affectent le
métabolisme des gangliosides à la fois dans les péricytes rétiniens et dans les cellules mésangiales
rénales. De plus, ils suggèrent que ces modifications du métabolisme des gangliosides pourraient
médier en partie les effets anti-prolifératifs des AGE et de la glucosamine. Si les gangliosides sont
souvent associés à des pathologies comme le cancer ou certaines maladies neurodégénératives, ce
travail est un des premiers à suggérer leur implication dans les complications microvasculaires du
diabète que sont la rétinopathie et la néphropathie diabétiques. Il ouvre un certain nombre de
perspectives, notamment thérapeutiques, et soulève des questions appelant des travaux
complémentaires à la fois en terme de mécanismes et de pertinence physiopathologique.
La glycation et la voie des hexosamines sont deux hypothèses biochimiques proposées pour
expliquer les altérations cellulaires et tissulaires causées par l’hyperglycémie. Différents modes
d’action ont été mis en évidence qui peuvent occasionner un effet délétère sur les cellules. Les AGE
sont connus pour agir notamment selon une cascade de signalisation impliquant un récepteur
membranaire, RAGE, le stress oxydant et l’activation de différents facteurs de croissance ou
cytokines. De plus, la formation des AGE altère la fonction des protéines et, au niveau des protéines
matricielles, perturbe le comportement cellulaire. La glucosamine, produit majeur de la voie des
hexosamines, peut quant à elle servir de précurseur pour la biosynthèse de glycoconjugués
importants pour les fonctions cellulaires. De plus, le résidu UDP-Nacétylglucosamine est utilisé
pour O-glycosyler un grand nombre de protéines, de structure, nucléaires ou facteurs de
transcription, et ainsi réguler leur fonction. Nous avons observé lors de cette étude que les AGE et
la glucosamine inhibent la prolifération des péricytes rétiniens et des cellules mésangiales
rénales. De plus, nos observations ont révélé que la glucosamine bloque le cycle cellulaire et
induit l’hypertrophie des cellules mésangiales. Ces perturbations de croissance cellulaire sont en
accord avec celles observées in vivo sur les péricytes rétiniens et les cellules mésangiales rénales.
En effet, la perte des péricytes rétiniens est une des altérations précoces de la rétinopathie
diabétique qui participe largement aux dysfonctions liées à la pathologie et à leur évolution vers des
troubles de la vue. L’arrêt de croissance, l’hypertrophie et finalement la disparition des cellules
mésangiales rénales sont des caractéristiques de la néphropathie diabétique, en partie responsables
de la sclérose glomérulaire et de la perte de la fonction rénale. La prévention des altérations de ces
169

deux types cellulaires serait un moyen d’enrayer la progression de la rétinopathie et de la
néphropathie diabétiques. Ces résultats soutiennent ainsi l’implication des AGE et de la
glucosamine dans les altérations cellulaires caractéristiques de ces deux complications
microvasculaires.
Notre étude met pour la première fois en cause la glucosamine dans le blocage du cycle cellulaire et
l’hypertrophie des cellules mésangiales. In vivo, l’hyperglycémie caractéristique du diabète, ainsi
qu’une régulation de l’enzyme limitante GFAT, donnent lieu à une modulation de la quantité de
glucosamine effectivement formée. Dans notre étude en revanche, l’utilisation directe de la
glucosamine exogène permet de contourner cette enzyme et d’apporter la glucosamine aux cellules
de manière artificielle. Il serait à ce stade important de vérifier la spécificité et la pertinence
physiologique de nos résultats en déterminant notamment s’ils sont reproductibles par un traitement
avec une concentration élevée de glucose et reversés par l’utilisation d’un inhibiteur de la GFAT
comme l’azaserine. Ceci permettrait de savoir si la modification du métabolisme des gangliosides
observée est effectivement dépendante de la GFAT et d’une activation de la voie des hexosamines.
Nous avons montré lors de ce travail, dans deux modèles cellulaires différents, que les AGE et la
glucosamine sont capables d’affecter le profil en gangliosides en agissant sur les activités de
leurs enzymes de biosynthèse. La modification du métabolisme des gangliosides pourrait
constituer un nouveau mécanisme d’action potentiel des AGE et de la glucosamine. Les
mécanismes selon lesquels ces produits régulent les activités des enzymes de biosynthèse des
gangliosides n’a pas été explorée. Il s’agirait par exemple de déterminer si la modification du
métabolisme des gangliosides fait partie de voies déjà décrites. La régulation des enzymes de
biosynthèse des gangliosides par la glucosamine met-elle en jeu une O-glycosylation de facteurs de
transcription ou des enzymes elle-mêmes, ou bien se fait-elle par apport de substrats et action de
masse ? La régulation par les AGE passe-t-elle par RAGE ? Dans les cellules mésangiales, les AGE
et la glucosamine induisent des modifications des gangliosides similaires : une augmentation des
gangliosides de la série-a et une diminution du GD3. Il serait intéressant de déterminer comment
ces deux voies biochimiques activées lors du diabète, peuvent converger vers le métabolisme des
gangliosides, notamment si les AGE et la glucosamine agissent parallèlement et indépendamment
ou si au contraire les deux voies se chevauchent et empruntent les mêmes mécanismes. Les AGE et
la glucosamine peuvent activer différents facteurs de transcription, respectivement par suite d’une
interaction avec RAGE et d’une O-glycosylation, notamment NF-κB ou Sp1 (Du X.L. et al., 2000;
Goldberg H.J. et al., 2002; Lander H.M. et al., 1997), facteurs qui réguleraient la transcription de
différents gènes d’enzymes de biosynthèse des gangliosides (Furukawa K. et al., 1996; Zeng G. et
al., 1998; Zeng G. et al., 2003). Il est donc possible que la régulation du métabolisme des
170

gangliosides se déroule finalement à un niveau transcriptionnel. La mise en évidence de
mécanismes d’action communs entre la voie de la glycation et celle des hexosamines permettrait
d’envisager des actions thérapeutiques communes pour contrer les deux voies à la fois.
Un bon nombre des effets cellulaires des AGE semble être médié par une interaction avec des
récepteurs membranaires spécifiques. Les récepteurs aux AGE (R1, R2, R3) sont plutôt décrits
comme des voies de captation et d’élimination des AGE (Iacobini C. et al., 2003; Lu C. et al., 2004)
alors que le RAGE est avancé comme la première étape d’une voie de signalisation menant à des
altérations cellulaires variées. La présence de RAGE a été détectée dans les péricytes (Yonekura H.
et al., 2003) et les cellules mésangiales (Vlassara H., 2001). Le blocage de l’interaction entre les
AGE et RAGE dans nos modèles cellulaires, par l’utilisation d’anticorps spécifiques ou l’inhibition
de l’expression du récepteur par des olgonucléotides anti-sens ou des SiRNA par exemple
permettrait de déterminer si l’inhibition de prolifération et la modification du métabolisme des
gangliosides causées par les AGE sont dépendantes de RAGE. Par ailleurs, le stress oxydant est une
caractéristique générale de l’état diabétique, qui pourrait être un point commun entre toutes les
voies biochimiques menant aux microcomplications. Les AGE ont été décrits comme générateurs
de stress oxydant, notamment dans les péricytes rétiniens (Denis U. et al., 2002). Il serait intéressant
de vérifier par l’utilisation d’anti-oxydant par exemple, si le stress oxydant joue un rôle dans l’effet
anti-prolifératif des AGE et la perturbation du métabolisme des gangliosides dans les péricytes et
les cellules mésangiales.
Le métabolisme et le profil en gangliosides sont donc modifiés dans les péricytes et les cellules
mésangiales, en réponse aux AGE comme à la glucosamine. L’ensemble des résultats obtenus dans
cette étude suggère que les gangliosides pourraient être des médiateurs de l’effet anti-
prolifératif des AGE et de la glucosamine sur les péricytes et les cellules mésangiales mais les
mécanismes par lesquels les gangliosides affectent la croissance cellulaire n’ont pas été totalement
élucidés. Certaines modifications sont communes aux deux types cellulaires ou aux deux
traitements alors que d’autres sont spécifiques (Figure 64). C’est le cas par exemple pour le GD3
dont le taux diminue sauf dans les péricytes traités avec la glucosamine où il est augmenté. La
signification de ces différences n’est pas claire et nous ne savons pas précisément quel(s)
ganglioside(s) est (sont) responsable(s) de l’effet observé. Les effets de la modification du profil en
gangliosides doivent-ils être attribués à un ganglioside en particulier, à une série ou bien à la
perturbation de l’équilibre entre les différentes séries ? Par quels mécanismes les gangliosides
agissent-ils sur la prolifération cellulaire ? Il est envisageable que tel ou tel ganglioside augmenté en
réponse au traitement agisse selon un mécanisme propre. Nous avons montré que le GM2 et le GM1
sont capables de bloquer le cycle cellulaire des cellules mésangiales et de causer une hypertrophie
via l’augmentation d’une protéine inhibitrice du cycle cellulaire, p21Waf1/Cip1. Par ailleurs, le GD3 a
171

été décrit dans la littérature pour induire l’apoptose alors qu’un grand nombre de gangliosides de la
série-a ont été impliqués dans une interaction avec les récepteurs aux facteurs de croissance. Les
différents gangliosides augmentés en réponse aux traitements pourraient agir de concours pour
inhiber la prolifération cellulaire. Il serait dans un premier temps intéressant de définir le
mécanisme selon lequel la prolifération des péricytes et des cellules mésangiales est inhibée en
réponse aux AGE ainsi qu’à la glucosamine : apoptose, nécrose, ralentissement ou blocage du cycle
cellulaire… afin d’éclaircir le mode d’action des gangliosides.
↑ GM3, GM2, GM1
↓ GD3
↑ GM3 synthase
↓ GD3 synthase
↑ GM3, GM2, GM1
↓GD3
↑ GM3 synthase
↓ GD3 synthase
↓ GM3
↑ GM1, GD3
↑ GM3 synthase
↑↑ GM2, GM1
↓ GD3
↓ GM3 synthase
Glu
cosa
min
eAG
E
Péricytes Cellules mésangiales
↓ prolifération
Souris db/db
↑GM3
↑ GM3 synthase
↓ GD3 synthase
↑ GM3, GM2, GM1
↓ GD3
↑ GM3 synthase
↓ GD3 synthase
↑ GM3, GM2, GM1
↓GD3
↑ GM3 synthase
↓ GD3 synthase
↓ GM3
↑ GM1, GD3
↑ GM3 synthase
↑↑ GM2, GM1
↓ GD3
↓ GM3 synthase
Glu
cosa
min
eAG
E
Péricytes Cellules mésangiales
↓ prolifération↓ prolifération
Souris db/db
↑GM3
↑ GM3 synthase
↓ GD3 synthase
Figure 64 : Bilan des modifications du métabolisme des gangliosides dans les péricytes et les cellules mésangiales en
réponse aux AGE et à la glucosamine, et dans les souris db/db.
Cependant, dans les deux types cellulaires en réponse aux deux traitements, les modifications des
gangliosides semblent aller dans le sens d’une augmentation du flux vers les gangliosides de la
série-a : augmentation des gangliosides de la série-a et de l’activité GM3 synthase dans les péricytes
et les cellules mésangiales en réponse aux AGE, augmentation du GM1 et de l’activité GM3
synthase dans les péricytes et augmentation des gangliosides de la série-a dans les cellules
mésangiales traitées avec la glucosamine. De plus, une augmentation de l’activité GM3 synthase et
des taux de GM3 dans les cortex rénaux de souris db/db ont été mesurées (Figure 64). Les
gangliosides de la série-a et la GM3 synthase, enzyme limitante de leur biosynthèse, pourraient
constituer un dénominateur commun aux altérations de prolifération causées par les AGE et
la glucosamine dans les péricytes et les cellules mésangiales et par extension, commun à la
172

rétinopathie et à la néphropathie diabétiques. Là encore, il est nécessaire de vérifier la
pertinence physipathologique des observations faites en étudiant par exemple le métabolisme des
gangliosides au niveau des structures microvasculaires rétiniennes et rénales de patients diabétiques
notamment. La découverte de mécanismes communs aux deux microcomplications est
particulièrement attrayante car elle permettrait d’envisager des traitements efficaces sur les deux
microcomplications à la fois. L'inhibition de l'activité de la GM3 synthase et donc de l'augmentation
des gangliosides de la série-a pourrait permettre de limiter la chute de prolifération et/ou
l'hypertrophie des péricytes et des cellules mésangiales causées par l'environnement diabétique. La
GM3 synthase représenterait ainsi une cible thérapeutique commune contre les deux
microcomplications. L'intérêt des gangliosides, notamment de la série-a, et de la GM3 synthase
dans le cadre plus global du diabète est d’ailleurs vivement renforcé par de récents travaux
suggérant leur rôle dans les mécanismes de l'insulino-résistance (Sasaki A. et al., 2003; Tagami S.
et al., 2002; Yamashita T. et al., 2003).
173


REFERENCES BIBLIOGRAPHIQUES
174


The effect of intensive treatment of diabetes on the development and progression of long-term
complications in insulin-dependent diabetes mellitus. The Diabetes Control and Complications Trial
Research Group. N.Engl.J Med., 1993, Vol. 329, n°. 14, pp. 977-986. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group. Lancet, 1998a, Vol. 352, n°. 9131, pp. 837-853.
Tight blood pressure control reduces risks of type 2 diabetes and is cost effective. BMJ, 1998b, Vol. 317, n°. 7160, pp. B.
AKIMOTO Y., KREPPEL L. K., HIRANO H., HART G. W. Increased O-GlcNAc transferase in pancreas of rats with streptozotocin-induced diabetes. Diabetologia, 2000, Vol. 43, n°. 10, pp. 1239-1247.
AL DOUAHJI M., BRUGAROLAS J., BROWN P. A., STEHMAN-BREEN C. O., ALPERS C. E., SHANKLAND S. J. The cyclin kinase inhibitor p21WAF1/CIP1 is required for glomerular hypertrophy in experimental diabetic nephropathy. Kidney Int., 1999, Vol. 56, n°. 5, pp. 1691-1699.
ALESSANDRI G., DE CRISTAN G., ZICHE M., CAPPA A. P., GULLINO P. M. Growth and motility of microvascular endothelium are modulated by the relative concentration of gangliosides in the medium. J Cell Physiol, 1992, Vol. 151, n°. 1, pp. 23-28.
AOKI H., SATOH M., MITSUZUKA K., ITO A., SAITO S., FUNATO T., ENDOH M., TAKAHASHI T., ARAI Y. Inhibition of motility and invasiveness of renal cell carcinoma induced by short interfering RNA transfection of beta 1,4GalNAc transferase. FEBS Lett., 2004, Vol. 567, n°. 2-3, pp. 203-208.
ARAKI N., HIGASHI T., MORI T., SHIBAYAMA R., KAWABE Y., KODAMA T., TAKAHASHI K., SHICHIRI M., HORIUCHI S. Macrophage scavenger receptor mediates the endocytic uptake and degradation of advanced glycation end products of the Maillard reaction. Eur.J Biochem., 1995, Vol. 230, n°. 2, pp. 408-415.
ARDAIL D., POPA I., BODENNEC J., LOUISOT P., SCHMITT D., PORTOUKALIAN J. The mitochondria-associated endoplasmic-reticulum subcompartment (MAM fraction) of rat liver contains highly active sphingolipid-specific glycosyltransferases. Biochem.J, 2003, Vol. 371, n°. Pt 3, pp. 1013-1019.
ASO Y., INUKAI T., TAYAMA K., TAKEMURA Y. Serum concentrations of advanced glycation endproducts are associated with the development of atherosclerosis as well as diabetic microangiopathy in patients with type 2 diabetes. Acta Diabetol., 2000, Vol. 37, n°. 2, pp. 87-92.
ATTAWIA M. A., NAYAK R. C. Circulating antipericyte autoantibodies in diabetic retinopathy. Retina, 1999, Vol. 19, n°. 5, pp. 390-400.
BELTRAMO E., BUTTIGLIERI S., POMERO F., ALLIONE A., D'ALU F., PONTE E., PORTA M. A study of capillary pericyte viability on extracellular matrix produced by endothelial cells in high glucose. Diabetologia, 2003, Vol. 46, n°. 3, pp. 409-415.
BELTRAMO E., POMERO F., ALLIONE A., D'ALU F., PONTE E., PORTA M. Pericyte adhesion is impaired on extracellular matrix produced by endothelial cells in high hexose concentrations. Diabetologia, 2002, Vol. 45, n°. 3, pp. 416-419.
175

BHUNIA A. K., SCHWARZMANN G., CHATTERJEE S. GD3 recruits reactive oxygen species to induce cell proliferation and apoptosis in human aortic smooth muscle cells. J Biol.Chem., 2002, Vol. 277, n°. 19, pp. 16396-16402.
BIEBERICH E., FREISCHUTZ B., LIOUR S. S., YU R. K. Regulation of ganglioside metabolism by phosphorylation and dephosphorylation. J Neurochem., 1998, Vol. 71, n°. 3, pp. 972-979.
BIEBERICH E., TENCOMNAO T., KAPITONOV D., YU R. K. Effect of N-glycosylation on turnover and subcellular distribution of N-acetylgalactosaminyltransferase I and sialyltransferase II in neuroblastoma cells. J Neurochem., 2000, Vol. 74, n°. 6, pp. 2359-2364.
BJORN S. F., BANGSTAD H. J., HANSSEN K. F., NYBERG G., WALKER J. D., VIBERTI G. C., OSTERBY R. Glomerular epithelial foot processes and filtration slits in IDDM patients. Diabetologia, 1995, Vol. 38, n°. 10, pp. 1197-1204.
BOLOT G., DAVID M. J., KASAMA T., TAKI T., HANDA S., RICHARD M., PIGNAT J. C., THOMAS L., PORTOUKALIAN J. Occurrence of monosialosyl pentahexaosylceramide GalNAc-GM1 as specific tumor-associated ganglioside of human head and neck squamous cell carcinomas. Cancer Lett., 1999, Vol. 135, n°. 2, pp. 159-164.
BOLOT G., DAVID M. J., TAKI T., HANDA S., KASAMA T., RICHARD M., PIGNAT J. C., THOMAS L., PORTOUKALIAN J. Analysis of glycosphingolipids of human head and neck carcinomas with comparison to normal tissue. Biochem.Mol.Biol.Int, 1998, Vol. 46, n°. 1, pp. 125-135.
BOYD-WHITE J., WILLIAMS J. C., JR. Effect of cross-linking on matrix permeability. A model for AGE-modified basement membranes. Diabetes, 1996, Vol. 45, n°. 3, pp. 348-353.
BREMER E. G., HAKOMORI S. GM3 ganglioside induces hamster fibroblast growth inhibition in chemically-defined medium: ganglioside may regulate growth factor receptor function. Biochem.Biophys.Res Commun., 1982, Vol. 106, n°. 3, pp. 711-718.
BREMER E. G., HAKOMORI S., BOWEN-POPE D. F., RAINES E., ROSS R. Ganglioside-mediated modulation of cell growth, growth factor binding, and receptor phosphorylation. J.Biol.Chem., 1984, Vol. 259, n°. 11, pp. 6818-6825.
BREMER E. G., SCHLESSINGER J., HAKOMORI S. Ganglioside-mediated modulation of cell growth. Specific effects of GM3 on tyrosine phosphorylation of the epidermal growth factor receptor. J.Biol.Chem., 1986, Vol. 261, n°. 5, pp. 2434-2440.
BUCALA R., MAKITA Z., KOSCHINSKY T., CERAMI A., VLASSARA H. Lipid advanced glycosylation: pathway for lipid oxidation in vivo. Proc.Natl.Acad.Sci.U.S.A, 1993, Vol. 90, n°. 14, pp. 6434-6438.
CARUBIA J. M., YU R. K., MACALA L. J., KIRKWOOD J. M., VARGA J. M. Gangliosides of normal and neoplastic human melanocytes. Biochem.Biophys.Res Commun., 1984, Vol. 120, n°. 2, pp. 500-504.
CERIELLO A., BORTOLOTTI N., FALLETI E., TABOGA C., TONUTTI L., CRESCENTINI A., MOTZ E., LIZZIO S., RUSSO A., BARTOLI E. Total radical-trapping antioxidant parameter in NIDDM patients. Diabetes Care, 1997, Vol. 20, n°. 2, pp. 194-197.
176

CHAN K. F., Ganglioside-modulated protein phosphorylation. Partial purification and characterization of a ganglioside-inhibited protein kinase in brain. J Biol.Chem., 1988, Vol. 263, n°. 1, pp. 568-574.
CHEN B. H., JIANG D. Y., TANG L. S. [Advanced glycation end products induce apoptosis and expression of apoptotic genes in cultured bovine retinal capillary pericytes]. Zhonghua Yan.Ke.Za Zhi., 2003, Vol. 39, n°. 4, pp. 224-227.
CHEN G., LIU P., THURMOND D. C., ELMENDORF J. S. Glucosamine-induced insulin resistance is coupled to O-linked glycosylation of Munc18c. FEBS Lett., 2003, Vol. 534, n°. 1-3, pp. 54-60.
CHIBBER R., MOLINATTI P. A., ROSATTO N., LAMBOURNE B., KOHNER E. M. Toxic action of advanced glycation end products on cultured retinal capillary pericytes and endothelial cells: relevance to diabetic retinopathy. Diabetologia, 1997, Vol. 40, n°. 2, pp. 156-164.
CHUNG T. W., CHOI H. J., LEE Y. C., KIM C. H. Molecular mechanism for transcriptional activation of ganglioside GM3 synthase and its function in differentiation of HL-60 cells. Glycobiology, 2004.
COHEN-FORTERRE L., ANDRE J., MOZERE G., PEYROUX J., STERNBERG M. Kidney sialidase and sialyltransferase activities in spontaneously and experimentally diabetic rats. Influence of insulin and sorbinil treatments. Biochem.Pharmacol., 1990, Vol. 40, n°. 3, pp. 507-513.
COLELL A., GARCIA-RUIZ C., ROMAN J., BALLESTA A., FERNANDEZ-CHECA J. C. Ganglioside GD3 enhances apoptosis by suppressing the nuclear factor-kappa B-dependent survival pathway. FASEB J, 2001, Vol. 15, n°. 6, pp. 1068-1070.
COMER F. I., HART G. W. O-Glycosylation of nuclear and cytosolic proteins. Dynamic interplay between O-GlcNAc and O-phosphate. J Biol.Chem., 2000, Vol. 275, n°. 38, pp. 29179-29182.
COMPTON S. J., JONES C. G. Mechanism of dye response and interference in the Bradford protein assay. Anal.Biochem., 1985, Vol. 151, n°. 2, pp. 369-374.
CRESPO P. M., ZURITA A. R., DANIOTTI J. L. Effect of gangliosides on the distribution of a glycosylphosphatidylinositol-anchored protein in plasma membrane from Chinese hamster ovary-K1 cells. J Biol.Chem., 2002, Vol. 277, n°. 47, pp. 44731-44739.
CROCKER P. R., KELM S., DUBOIS C., MARTIN B., MCWILLIAM A. S., SHOTTON D. M., PAULSON J. C., GORDON S. Purification and properties of sialoadhesin, a sialic acid-binding receptor of murine tissue macrophages. EMBO J, 1991, Vol. 10, n°. 7, pp. 1661-1669.
CROOK M. A., PICKUP J. C., LUMB P. J., GIORGINO F., WEBB D. J., FULLER J. H., GEORGINO F. Relationship between plasma sialic acid concentration and microvascular and macrovascular complications in type 1 diabetes: the EURODIAB Complications Study. Diabetes Care, 2001, Vol. 24, n°. 2, pp. 316-322.
DALLA V. M., MASIERO A., ROITER A. M., SALLER A., CREPALDI G., FIORETTO P. Is podocyte injury relevant in diabetic nephropathy? Studies in patients with type 2 diabetes. Diabetes, 2003, Vol. 52, n°. 4, pp. 1031-1035.
DANZE P. M., TARJOMAN A., ROUSSEAUX J., FOSSATI P., DAUTREVAUX M. Evidence for an increased glycation of IgG in diabetic patients. Clin.Chim.Acta, 1987, Vol. 166, n°. 2-3, pp. 143-153.
177

DE CRISTAN G., MORBIDELLI L., ALESSANDRI G., ZICHE M., CAPPA A. P., GULLINO P. M. Synergism between gangliosides and basic fibroblastic growth factor in favouring survival, growth, and motility of capillary endothelium. J Cell Physiol, 1990, Vol. 144, n°. 3, pp. 505-510.
DE MARIA R., LENTI L., MALISAN F., D'AGOSTINO F., TOMASSINI B., ZEUNER A., RIPPO M. R., TESTI R. Requirement for GD3 ganglioside in. Science, 1997, Vol. 277, n°. 5332, pp. 1652-1655.
DEGENHARDT T. P., THORPE S. R., BAYNES J. W. Chemical modification of proteins by methylglyoxal. Cell Mol.Biol.(Noisy.-le-grand), 1998, Vol. 44, n°. 7, pp. 1139-1145.
DEGROOTE S., WOLTHOORN J., VAN MEER G. The cell biology of glycosphingolipids. Semin.Cell Dev.Biol., 2004, Vol. 15, n°. 4, pp. 375-387.
DELORME C., BRUSSOW H., SIDOTI J., ROCHE N., KARLSSON K. A., NEESER J. R., TENEBERG S. Glycosphingolipid binding specificities of rotavirus: identification of a sialic acid-binding epitope. J Virol., 2001, Vol. 75, n°. 5, pp. 2276-2287.
DENIS U., LECOMTE M., PAGET C., RUGGIERO D., WIERNSPERGER N., LAGARDE M. Advanced glycation end-products induce apoptosis of bovine retinal pericytes in culture: involvement of diacylglycerol/ceramide production and oxidative stress induction. Free Radic.Biol.Med., 2002, Vol. 33, n°. 2, pp. 236-247.
DOI T., VLASSARA H., KIRSTEIN M., YAMADA Y., STRIKER G. E., STRIKER L. J. Receptor-specific increase in extracellular matrix production in mouse mesangial cells by advanced glycosylation end products is mediated via platelet-derived growth factor. Proc.Natl.Acad.Sci.U.S.A, 1992, Vol. 89, n°. 7, pp. 2873-2877.
DOTTA F., GIANANI R., PREVITI M., LENTI L., DIONISI S., D'ERME M., EISENBARTH G. S., DI MARIO U. Autoimmunity to the GM2-1 islet ganglioside before and at the onset of type I diabetes. Diabetes, 1996, Vol. 45, n°. 9, pp. 1193-1196.
DOWLER J.G.F.HAMILTON A.M.P. Clinical features of diabetic eye disease. In : Textbook of diabetes. second edition. London : edited by J. C. Pickup and williams G., 1997, pp. 46.1-46.15.
DREYFUS H., GUEROLD B., FREYSZ L., HICKS D. Successive isolation and separation of the major lipid fractions including gangliosides from single biological samples. Anal.Biochem., 1997, Vol. 249, n°. 1, pp. 67-78.
DU X. L., EDELSTEIN D., ROSSETTI L., FANTUS I. G., GOLDBERG H., ZIYADEH F., WU J., BROWNLEE M. Hyperglycemia-induced mitochondrial superoxide overproduction activates the hexosamine pathway and induces plasminogen activator inhibitor-1 expression by increasing Sp1 glycosylation. Proc.Natl.Acad.Sci.U.S.A, 2000, Vol. 97, n°. 22, pp. 12222-12226.
ENGE M., BJARNEGARD M., GERHARDT H., GUSTAFSSON E., KALEN M., ASKER N., HAMMES H. P., SHANI M., FASSLER R., BETSHOLTZ C. Endothelium-specific platelet-derived growth factor-B ablation mimics diabetic retinopathy. EMBO J, 2002, Vol. 21, n°. 16, pp. 4307-4316.
FAN Y. P., WEISS R. H. Exogenous attenuation of p21(Waf1/Cip1) decreases mesangial cell hypertrophy as a result of hyperglycemia and IGF-1. J.Am.Soc.Nephrol., 2004, Vol. 15, n°. 3, pp. 575-584.
178

FISHMAN P. H., BRADY R. O. Biosynthesis and function of gangliosides. Science, 1976, Vol. 194, n°. 4268, pp. 906-915.
FONG D. S., AIELLO L., GARDNER T. W., KING G. L., BLANKENSHIP G., CAVALLERANO J. D., FERRIS F. L., III, KLEIN R. Diabetic retinopathy. Diabetes Care, 2003, Vol. 26, n°. 1, pp. 226-229.
FORBES J. M., SOULIS T., THALLAS V., PANAGIOTOPOULOS S., LONG D. M., VASAN S., WAGLE D., JERUMS G., COOPER M. E. Renoprotective effects of a novel inhibitor of advanced glycation. Diabetologia, 2001, Vol. 44, n°. 1, pp. 108-114.
FORBES J. M., THALLAS V., THOMAS M. C., FOUNDS H. W., BURNS W. C., JERUMS G., COOPER M. E. The breakdown of preexisting advanced glycation end products is associated with reduced renal fibrosis in experimental diabetes. FASEB J, 2003, Vol. 17, n°. 12, pp. 1762-1764.
FORRESTER J.V.KNOTT R.M. Pathogenesis of diabetic retinopathy and cataract. In : Textbook of diabetes. second edition. London : edited by J. C. Pickup and williams G., 1997, pp. 45.1-45.19.
FURUKAWA K., SOEJIMA H., NIIKAWA N., SHIKU H. Genomic organization and chromosomal assignment of the human beta1, 4-N-acetylgalactosaminyltransferase gene. Identification of multiple transcription units. J Biol.Chem., 1996, Vol. 271, n°. 34, pp. 20836-20844.
GARCIA-RUIZ C., COLELL A., MORALES A., CALVO M., ENRICH C., FERNANDEZ-CHECA J. C. Trafficking of ganglioside GD3 to mitochondria by tumor necrosis factor-alpha. J Biol.Chem., 2002, Vol. 277, n°. 39, pp. 36443-36448.
GARCIA-RUIZ C., COLELL A., PARIS R., FERNANDEZ-CHECA J. C. Direct interaction of GD3 ganglioside with mitochondria generates reactive oxygen species followed by mitochondrial permeability transition, cytochrome c release, and caspase activation. FASEB J, 2000, Vol. 14, n°. 7, pp. 847-858.
GARDINER T. A., ANDERSON H. R., STITT A. W. Inhibition of advanced glycation end-products protects against retinal capillary basement membrane expansion during long-term diabetes. J Pathol., 2003, Vol. 201, n°. 2, pp. 328-333.
GAROFALO T., MISASI R., MATTEI V., GIAMMARIOLI A. M., MALORNI W., PONTIERI G. M., PAVAN A., SORICE M. Association of the death-inducing signaling complex with microdomains after triggering through CD95/Fas. Evidence for caspase-8-ganglioside interaction in T cells. J Biol.Chem., 2003, Vol. 278, n°. 10, pp. 8309-8315.
GEOFFROY K., WIERNSPERGER N., LAGARDE M., EL BAWAB S. Bimodal effect of advanced glycation end products on mesangial cell proliferation is mediated by neutral ceramidase regulation and endogenous sphingolipids. J.Biol.Chem., 2004, Vol. 279, n°. 33, pp. 34343-34352.
GILLARD B. K., THOMAS J. W., NELL L. J., MARCUS D. M. Antibodies against ganglioside GT3 in the sera of patients with type I diabetes mellitus. J Immunol., 1989, Vol. 142, n°. 11, pp. 3826-3832.
GIRAUDO C. G., DANIOTTI J. L., MACCIONI H. J. Physical and functional association of glycolipid N-acetyl-galactosaminyl and galactosyl transferases in the Golgi apparatus. Proc.Natl.Acad.Sci.U.S.A, 2001, Vol. 98, n°. 4, pp. 1625-1630.
179

GOLDBERG H. J., WHITESIDE C. I., FANTUS I. G. The hexosamine pathway regulates the plasminogen activator inhibitor-1 gene promoter and Sp1 transcriptional activation through protein kinase C-beta I and -delta. J.Biol.Chem., 2002, Vol. 277, n°. 37, pp. 33833-33841.
GOLDBERG H. J., CAI W., PEPPA M., DARDAINE V., BALIGA B.S., URIBARRI J., VLASSARA H. Advanced glycoxidation end products in commonly consumed foods. J Am Diet Assoc., 2004, Vol. 104, n°. 8, pp. 1287-1291.
GOUNI-BERTHOLD I., SEUL C., KO Y., HESCHELER J., SACHINIDIS A. Gangliosides GM1 and GM2 induce vascular smooth muscle cell proliferation via extracellular signal-regulated kinase 1/2 pathway. Hypertension, 2001, Vol. 38, n°. 5, pp. 1030-1037.
GRIFFIN S. V., PICHLER R., WADA T., VAUGHAN M., DURVASULA R., SHANKLAND S. J. The role of cell cycle proteins in Glomerular disease. Semin.Nephrol., 2003, Vol. 23, n°. 6, pp. 569-582.
GRIFFITH L. S., SCHMITZ B. O-linked N-acetylglucosamine levels in cerebellar neurons respond reciprocally to pertubations of phosphorylation. Eur.J Biochem., 1999, Vol. 262, n°. 3, pp. 824-831.
GU X., PREUSS U., GU T., YU R. K. Regulation of sialyltransferase activities by phosphorylation and dephosphorylation. J Neurochem., 1995, Vol. 64, n°. 5, pp. 2295-2302.
HAKOMORI S., IGARASHI Y. Functional role of glycosphingolipids in cell recognition and signaling. J Biochem.(Tokyo), 1995, Vol. 118, n°. 6, pp. 1091-1103.
HAMMES H. P., ALT A., NIWA T., CLAUSEN J. T., BRETZEL R. G., BROWNLEE M., SCHLEICHER E. D. Differential accumulation of advanced glycation end products in the course of diabetic retinopathy. Diabetologia, 1999, Vol. 42, n°. 6, pp. 728-736.
HAMMES H. P., LIN J., RENNER O., SHANI M., LUNDQVIST A., BETSHOLTZ C., BROWNLEE M., DEUTSCH U. Pericytes and the pathogenesis of diabetic retinopathy. Diabetes, 2002, Vol. 51, n°. 10, pp. 3107-3112.
HAMMES H. P., MARTIN S., FEDERLIN K., GEISEN K., BROWNLEE M. Aminoguanidine treatment inhibits the development of experimental diabetic retinopathy. Proc.Natl.Acad.Sci.U.S.A, 1991, Vol. 88, n°. 24, pp. 11555-11558.
HAN I., KUDLOW J. E. Reduced O glycosylation of Sp1 is associated with increased proteasome susceptibility. Mol.Cell Biol., 1997, Vol. 17, n°. 5, pp. 2550-2558.
HANAI N., DOHI T., NORES G. A., HAKOMORI S. A novel ganglioside, de-N-acetyl-GM3 (II3NeuNH2LacCer), acting as a strong promoter for epidermal growth factor receptor kinase and as a stimulator for cell growth. J Biol.Chem., 1988, Vol. 263, n°. 13, pp. 6296-6301.
HARAGUCHI M., YAMASHIRO S., FURUKAWA K., TAKAMIYA K., SHIKU H., FURUKAWA K. The effects of the site-directed removal of N-glycosylation sites from beta-1,4-N-acetylgalactosaminyltransferase on its function. Biochem.J, 1995, Vol. 312 ( Pt 1), pp. 273-280.
HATTORI M., HORIUCHI R., HOSAKA K., HAYASHI H., KOJIMA I. Sialyllactose-mediated cell interaction during granulosa cell differentiation. Identification of its binding proteins. J Biol.Chem., 1995, Vol. 270, n°. 14, pp. 7858-7863.
180

HE C. J., KOSCHINSKY T., BUENTING C., VLASSARA H. Presence of diabetic complications in type 1 diabetic patients correlates with low expression of mononuclear cell AGE-receptor-1 and elevated serum AGE. Mol.Med., 2001, Vol. 7, n°. 3, pp. 159-168.
HE C. J., ZHENG F., STITT A., STRIKER L., HATTORI M., VLASSARA H. Differential expression of renal AGE-receptor genes in NOD mice: possible role in nonobese diabetic renal disease. Kidney Int, 2000, Vol. 58, n°. 5, pp. 1931-1940.
HEATH H., PATERSON R. A., HART J. C. Changes in the hydroxyproline, hexosamine and sialic acid of the diabetic human and beta, beta'-iminodipropionitrile-treated rat retinal vascular systems. Diabetologia, 1967, Vol. 3, n°. 6, pp. 515-518.
HEBERT L. F., JR., DANIELS M. C., ZHOU J., CROOK E. D., TURNER R. L., SIMMONS S. T., NEIDIGH J. L., ZHU J. S., BARON A. D., MCCLAIN D. A. Overexpression of glutamine:fructose-6-phosphate amidotransferase in transgenic mice leads to insulin resistance. J Clin.Invest, 1996, Vol. 98, n°. 4, pp. 930-936.
HORIE K., MIYATA T., MAEDA K., MIYATA S., SUGIYAMA S., SAKAI H., VAN YPERSOLE D. S., MONNIER V. M., WITZTUM J. L., KUROKAWA K. Immunohistochemical colocalization of glycoxidation products and lipid peroxidation products in diabetic renal glomerular lesions. Implication for glycoxidative stress in the pathogenesis of diabetic nephropathy. J Clin.Invest, 1997, Vol. 100, n°. 12, pp. 2995-3004.
HOSHI S., SHU Y., YOSHIDA F., INAGAKI T., SONODA J., WATANABE T., NOMOTO K., NAGATA M. Podocyte injury promotes progressive nephropathy in zucker diabetic fatty rats. Lab Invest, 2002, Vol. 82, n°. 1, pp. 25-35.
HYNDS D. L., SUMMERS M., VAN BROCKLYN J., O'DORISIO M. S., YATES A. J. Gangliosides inhibit platelet-derived growth factor-stimulated growth, receptor phosphorylation, and dimerization in neuroblastoma SH-SY5Y cells. J.Neurochem., 1995, Vol. 65, n°. 5, pp. 2251-2258.
IACOBINI C., AMADIO L., ODDI G., RICCI C., BARSOTTI P., MISSORI S., SORCINI M., DI MARIO U., PRICCI F., PUGLIESE G. Role of galectin-3 in diabetic nephropathy. J Am Soc Nephrol, 2003, Vol. 14, n°. 8 Suppl 3, pp. S264-S270.
IACOBINI C., MENINI S., ODDI G., RICCI C., AMADIO L., PRICCI F., OLIVIERI A., SORCINI M., DI MARIO U., PESCE C., PUGLIESE G. Galectin-3/AGE-receptor 3 knockout mice show accelerated AGE-induced glomerular injury: evidence for a protective role of galectin-3 as an AGE receptor. FASEB J, 2004, Vol. 18, n°. 14, pp. 1773-1775.
IBER H., VAN ECHTEN G., KLEIN R. A., SANDHOFF K. pH-dependent changes of ganglioside biosynthesis in neuronal cell culture. Eur.J Cell Biol., 1990, Vol. 52, n°. 2, pp. 236-240.
INOKUCHI J. I., UEMURA S., KABAYAMA K., IGARASHI Y. Glycosphingolipid deficiency affects functional microdomain formation in Lewis lung carcinoma cells. Glycoconj.J, 2000, Vol. 17, n°. 3 -4, pp. 239-245.
JACKSON S. P., TJIAN R. O-glycosylation of eukaryotic transcription factors: implications for mechanisms of transcriptional regulation. Cell, 1988, Vol. 55, n°. 1, pp. 125-133.
KADOR P. F., AKAGI Y., TAKAHASHI Y., IKEBE H., WYMAN M., KINOSHITA J. H. Prevention of retinal vessel changes associated with diabetic retinopathy in galactose-fed dogs by aldose reductase inhibitors. Arch.Ophthalmol., 1990, Vol. 108, n°. 9, pp. 1301-1309.
181

KALFA T. A., GERRITSEN M. E., CARLSON E. C., BINSTOCK A. J., TSILIBARY E. C. Altered proliferation of retinal microvascular cells on glycated matrix. Invest Ophthalmol.Vis.Sci., 1995, Vol. 36, n°. 12, pp. 2358-2367.
KANDA T., ARIGA T., YAMAWAKI M., YU R. K. GM3 regulates protein kinase systems in cultured brain microvascular endothelial cells. J Neurochem., 1993, Vol. 61, n°. 5, pp. 1969-1972.
KANETO H., XU G., SONG K. H., SUZUMA K., BONNER-WEIR S., SHARMA A., WEIR G. C. Activation of the hexosamine pathway leads to deterioration of pancreatic beta-cell function through the induction of oxidative stress. J Biol.Chem., 2001, Vol. 276, n°. 33, pp. 31099-31104.
KARACHALIAS N., BABAEI-JADIDI R., AHMED N., THORNALLEY P. J. Accumulation of fructosyl-lysine and advanced glycation end products in the kidney, retina and peripheral nerve of streptozotocin-induced diabetic rats. Biochem.Soc Trans., 2003, Vol. 31, n°. Pt 6, pp. 1423-1425.
KATOH N., Inhibition by gangliosides GM3, GD3 and GT1b of substrate phosphorylation by protein kinase C in bovine mammary gland and its reversal by phosphatidylserine. Life Sci., 1995, Vol. 56, n°. 3, pp. 157-162.
KELLY D. J., GILBERT R. E., COX A. J., SOULIS T., JERUMS G., COOPER M. E. Aminoguanidine ameliorates overexpression of prosclerotic growth factors and collagen deposition in experimental diabetic nephropathy. J Am Soc Nephrol, 2001, Vol. 12, n°. 10, pp. 2098-2107.
KIM J., SON J. W., LEE J. A., OH Y. S., SHINN S. H. Methylglyoxal induces apoptosis mediated by reactive oxygen species in bovine retinal pericytes. J Korean Med.Sci., 2004, Vol. 19, n°. 1, pp. 95-100.
KING G. L., BROWNLEE M. The cellular and molecular mechanisms of diabetic complications. Endocrinol Metab Clin.North Am, 1996, Vol. 25, n°. 2, pp. 255-270.
KOHNER E.M., The Lesions and Natural History of Diabetic Retinopathy. In : Chronic complications of diabetes. London : edited by J. C. Pickup and williams G., 1994, pp. 63-76.
KOHNER E.M., PORTA M., HYER S.L. The Pathogenesis of Diabetic Retinopathy and Cataract. In : Chronic complications of diabetes. London : edited by J. C. Pickup and williams G., 1994, pp. 52-62.
KOHNER E. M., PATEL V., RASSAM S. M. Role of blood flow and impaired autoregulation in the pathogenesis of diabetic retinopathy. Diabetes, 1995, Vol. 44, n°. 6, pp. 603-607.
KOJIMA N., HAKOMORI S. Cell adhesion, spreading, and motility of GM3-expressing cells based on glycolipid-glycolipid interaction. J Biol.Chem., 1991, Vol. 266, n°. 26, pp. 17552-17558.
KOJIMA N., KUROSAWA N., NISHI T., HANAI N., TSUJI S. Induction of cholinergic differentiation with neurite sprouting by de novo biosynthesis and expression of GD3 and b-series gangliosides in Neuro2a cells. J Biol.Chem., 1994, Vol. 269, n°. 48, pp. 30451-30456.
KOLM-LITTY V., SAUER U., NERLICH A., LEHMANN R., SCHLEICHER E. D. High glucose-induced transforming growth factor beta1 production is mediated by the hexosamine pathway in porcine glomerular mesangial cells. J.Clin.Invest, 1998, Vol. 101, n°. 1, pp. 160-169.
KOLTER T., PROIA R. L., SANDHOFF K. Combinatorial ganglioside biosynthesis. J Biol.Chem., 2002, Vol. 277, n°. 29, pp. 25859-25862.
182

KOOCHEKPOUR S., MERZAK A., PILKINGTON G. J. Vascular endothelial growth factor production is stimulated by gangliosides and TGF-beta isoforms in human glioma cells in vitro. Cancer Lett., 1996, Vol. 102, n°. 1-2, pp. 209-215.
KOPITZ J., VON REITZENSTEIN C., BURCHERT M., CANTZ M., GABIUS H. J. Galectin-1 is a major receptor for ganglioside GM1, a product of the growth-controlling activity of a cell surface ganglioside sialidase, on human neuroblastoma cells in culture. J Biol.Chem., 1998, Vol. 273, n°. 18, pp. 11205-11211.
KOTANI M., HOSOYA H., KUBO H., ITOH K., SAKURABA H., KUSUBATA M., INAGAKI M., YAZAKI S., SUZUKI Y., TAI T. Evidence for direct binding of intracellularly distributed ganglioside GM2 to isolated vimentin intermediate filaments in normal and Tay-Sachs disease human fibroblasts. Cell Struct.Funct., 1994, Vol. 19, n°. 2, pp. 81-87.
KREUTTER D., KIM J. Y., GOLDENRING J. R., RASMUSSEN H., UKOMADU C., DELORENZO R. J., YU R. K. Regulation of protein kinase C activity by gangliosides. J Biol.Chem., 1987, Vol. 262, n°. 4, pp. 1633-1637.
KRISTAL B. S., BROWN A. M. Apoptogenic ganglioside GD3 directly induces the mitochondrial permeability transition. J Biol.Chem., 1999, Vol. 274, n°. 33, pp. 23169-23175.
KROWLESKI A.S.WARRAM J.H. Clinical features and epidemiology of diabetic nephropathy. In : Textbook of diabetes. second edition. London : edited by J. C. Pickup and williams G., 1997, pp. 53.1-53.13.
KUAN C. J., AL DOUAHJI M., SHANKLAND S. J. The cyclin kinase inhibitor p21WAF1, CIP1 is increased in experimental diabetic nephropathy: potential role in glomerular hypertrophy. J.Am.Soc.Nephrol., 1998, Vol. 9, n°. 6, pp. 986-993.
KWAK D. H., RHO Y. I., KWON O. D., AHAN S. H., SONG J. H., CHOO Y. K., KIM S. J., CHOI B. K., JUNG K. Y. Decreases of ganglioside GM3 in streptozotocin-induced diabetic glomeruli of rats. Life Sci., 2003, Vol. 72, n°. 17, pp. 1997-2006.
LAN H. Y., Tubular epithelial-myofibroblast transdifferentiation mechanisms in proximal tubule cells. Curr.Opin.Nephrol Hypertens., 2003, Vol. 12, n°. 1, pp. 25-29.
LANDER H. M., TAURAS J. M., OGISTE J. S., HORI O., MOSS R. A., SCHMIDT A. M. Activation of the receptor for advanced glycation end products triggers a p21(ras)-dependent mitogen-activated protein kinase pathway regulated by oxidant stress. J Biol.Chem., 1997, Vol. 272, n°. 28, pp. 17810-17814.
LECOMTE M., PAGET C., RUGGIERO D., WIERNSPERGER N., LAGARDE M. Docosahexaenoic acid is a major n-3 polyunsaturated fatty acid in bovine retinal microvessels. J Neurochem., 1996, Vol. 66, n°. 5, pp. 2160-2167.
LI R., GAGE D., MCKALLIP R., LADISCH S. Structural characterization and in vivo immunosuppressive activity of neuroblastoma GD2. Glycoconj.J, 1996, Vol. 13, n°. 3, pp. 385-389.
LI W., YANOFF M., JIAN B., HE Z. Altered mRNA levels of antioxidant enzymes in pre-apoptotic pericytes from human diabetic retinas. Cell Mol.Biol.(Noisy.-le-grand), 1999, Vol. 45, n°. 1, pp. 59-66.
183

LINDAHL P., JOHANSSON B. R., LEVEEN P., BETSHOLTZ C. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science, 1997, Vol. 277, n°. 5323, pp. 242-245.
LISANTI M. P., SCHERER P. E., VIDUGIRIENE J., TANG Z., HERMANOWSKI-VOSATKA A., TU Y. H., COOK R. F., SARGIACOMO M. Characterization of caveolin-rich membrane domains isolated from an endothelial-rich source: implications for human disease. J Cell Biol., 1994, Vol. 126, n°. 1, pp. 111-126.
LIU Y., LI R., LADISCH S. Exogenous ganglioside GD1a enhances epidermal growth factor receptor binding and dimerization. J Biol.Chem., 2004, Vol. 279, n°. 35, pp. 36481-36489.
LLOYD K. O., FURUKAWA K. Biosynthesis and functions of gangliosides: recent advances. Glycoconj.J, 1998, Vol. 15, n°. 7, pp. 627-636.
LORENZI M., MONTISANO D. F., TOLEDO S., BARRIEUX A. High glucose induces DNA damage in cultured human endothelial cells. J Clin.Invest, 1986, Vol. 77, n°. 1, pp. 322-325.
LU C., HE J. C., CAI W., LIU H., ZHU L., VLASSARA H. Advanced glycation endproduct (AGE) receptor 1 is a negative regulator of the inflammatory response to AGE in mesangial cells. Proc.Natl.Acad.Sci.U.S.A, 2004, Vol. 101, n°. 32, pp. 11767-11772.
MAKINO H., SUGIYAMA H., KASHIHARA N. Apoptosis and extracellular matrix-cell interactions in kidney disease. Kidney Int Suppl, 2000, Vol. 77, pp. S67-S75.
MALISAN F., TESTI R. Lipid signaling in CD95-mediated apoptosis. FEBS Lett., 1999, Vol. 452, n°. 1-2, pp. 100-103.
MARSHALL S., The hexosamine signaling pathway: a new road to drug discovery. Current Opinion in Endocrinology and Diabetes, 2002, Vol. 9, pp. 160-167.
MARSHALL S., BACOTE V., TRAXINGER R. R. Discovery of a metabolic pathway mediating glucose-induced desensitization of the glucose transport system. Role of hexosamine biosynthesis in the induction of insulin resistance. J Biol.Chem., 1991, Vol. 266, n°. 8, pp. 4706-4712.
MARSHALL S., NADEAU O., YAMASAKI K. Dynamic actions of glucose and glucosamine on hexosamine biosynthesis in isolated adipocytes: Differential effects on glucosamine-6-phosphate, UDP-N-acetylglucosamine, and ATP levels. J.Biol.Chem., 2004.
MARTINA J. A., DANIOTTI J. L., MACCIONI H. J. Influence of N-glycosylation and N-glycan trimming on the activity and intracellular traffic of GD3 synthase. J Biol.Chem., 1998, Vol. 273, n°. 6, pp. 3725-3731.
MAUER S. M., STEFFES M. W., AZAR S., BROWN D. M. Effects of sorbinil on glomerular structure and function in long-term-diabetic rats. Diabetes, 1989, Vol. 38, n°. 7, pp. 839-846.
MAZLEN R. G., MUELLENBERG C. G., O'BRIEN P. J. L-Glutamine D-fructose 6-phosphate amidotransferase from bovine retina. Exp.Eye Res., 1970, Vol. 9, n°. 1, pp. 1-11.
MCALARNEY T., APOSTOLSKI S., LEDERMAN S., LATOV N. Characteristics of HIV-1 gp120 glycoprotein binding to glycolipids. J Neurosci.Res, 1994, Vol. 37, n°. 4, pp. 453-460.
184

MCLELLAN A. C., THORNALLEY P. J., BENN J., SONKSEN P. H. Glyoxalase system in clinical diabetes mellitus and correlation with diabetic complications. Clin.Sci.(Lond), 1994, Vol. 87, n°. 1, pp. 21-29.
MENE P., SIMONSON M.S., DUNN M.J. Physiology of the Mesangial Cell. Physiol Rev., 1989, Vol. 69, n°. 4, pp. 1347-1424.
MERZAK A., KOOCHEKPOUR S., PILKINGTON G. J. Adhesion of human glioma cell lines to fibronectin, laminin, vitronectin and collagen I is modulated by gangliosides in vitro. Cell Adhes.Commun., 1995, Vol. 3, n°. 1, pp. 27-43.
MEUILLET E. J., KROES R., YAMAMOTO H., WARNER T. G., FERRARI J., MANIA-FARNELL B., GEORGE D., REBBAA A., MOSKAL J. R., BREMER E. G. Sialidase gene transfection enhances epidermal growth factor receptor activity in an epidermoid carcinoma cell line, A431. Cancer Res., 1999, Vol. 59, n°. 1, pp. 234-240.
MEUILLET E. J., MANIA-FARNELL B., GEORGE D., INOKUCHI J. I., BREMER E. G. Modulation of EGF receptor activity by changes in the GM3 content in a human epidermoid carcinoma cell line, A431. Exp.Cell Res, 2000, Vol. 256, n°. 1, pp. 74-82.
MILJAN E. A., MEUILLET E. J., MANIA-FARNELL B., GEORGE D., YAMAMOTO H., SIMON H. G., BREMER E. G. Interaction of the extracellular domain of the epidermal growth factor receptor with gangliosides. J Biol.Chem., 2002, Vol. 277, n°. 12, pp. 10108-10113.
MILLER J. A., GRAVALLESE E., BUNN H. F. Nonenzymatic glycosylation of erythrocyte membrane proteins. Relevance to diabetes. J Clin.Invest, 1980, Vol. 65, n°. 4, pp. 896-901.
MIRKIN B. L., CLARK S. H., ZHANG C. Inhibition of human neuroblastoma cell proliferation and EGF receptor phosphorylation by gangliosides GM1, GM3, GD1A and GT1B. Cell Prolif., 2002, Vol. 35, n°. 2, pp. 105-115.
MITSUDA T., FURUKAWA K., FUKUMOTO S., MIYAZAKI H., URANO T., FURUKAWA K. Overexpression of ganglioside GM1 results in the dispersion of platelet-derived growth factor receptor from glycolipid-enriched microdomains and in the suppression of cell growth signals. J.Biol.Chem., 2002, Vol. 277, n°. 13, pp. 11239-11246.
MIZUTANI M., KERN T. S., LORENZI M. Accelerated death of retinal microvascular cells in human and experimental diabetic retinopathy. J Clin.Invest, 1996, Vol. 97, n°. 12, pp. 2883-2890.
MOON S.K., KIM H.M., LEE Y.C., KIM C.H. Disialoganglioside (GD3) synthase gene expression suppresses vascular smooth muscle cell responses via the inhibition of ERK1/2 phosphorylation, cell cycle progression, and matrix metalloproteinase-9 expression. J. Biol. Chem., 2004, Vol. 279, n°. 32, pp. 33063-33070
MULLARKEY C.J.BROWNLEE M. Biochemical Basis of Microvascular Disease. In : Chronic complications of diabetes. London : edited by J. C. Pickup and williams G., 1994, pp. 20-33.
MUNDEL P., KRIZ W. Structure and function of podocytes: an update. Anat.Embryol.(Berl), 1995, Vol. 192, n°. 5, pp. 385-397.
MUTOH T., TOKUDA A., MIYADAI T., HAMAGUCHI M., FUJIKI N. Ganglioside GM1 binds to the Trk protein and regulates receptor function. Proc.Natl.Acad.Sci.U.S.A, 1995, Vol. 92, n°. 11, pp. 5087-5091.
185

NAKANO J., YASUI H., LLOYD K. O., MUTO M. Biologic roles of gangliosides G(M3) and G(D3) in the attachment of human melanoma cells to extracellular matrix proteins. J Investig.Dermatol.Symp.Proc., 1999, Vol. 4, n°. 2, pp. 173-176.
NAKATSUJI Y., MILLER R. H. Selective cell-cycle arrest and induction of apoptosis in proliferating neural cells by ganglioside GM3. Exp.Neurol., 2001, Vol. 168, n°. 2, pp. 290-299.
NATALIZIO A., RUGGIERO D., LECOMTE M., LAGARDE M., WIERNSPERGER N. Glycosphingolipid changes induced by advanced glycation end-products. Biochem.Biophys.Res.Commun., 2001, Vol. 281, n°. 1, pp. 78-83.
NAYAK R. C., AGARDH C. D., KWOK M. G., STJERNQUIST H., FARTHING-NAYAK P. J., AGARDH E. Circulating anti-pericyte autoantibodies are present in Type 2 diabetic patients and are associated with non-proliferative retinopathy. Diabetologia, 2003, Vol. 46, n°. 4, pp. 511-513.
NAYAK R. C., BERMAN A. B., GEORGE K. L., EISENBARTH G. S., KING G. L. A monoclonal antibody (3G5)-defined ganglioside antigen is expressed on the cell surface of microvascular pericytes. J Exp.Med., 1988, Vol. 167, n°. 3, pp. 1003-1015.
NERLICH A. G., SAUER U., KOLM-LITTY V., WAGNER E., KOCH M., SCHLEICHER E. D. Expression of glutamine:fructose-6-phosphate amidotransferase in human tissues: evidence for high variability and distinct regulation in diabetes. Diabetes, 1998, Vol. 47, n°. 2, pp. 170-178.
NICHOLAS S. B., Advances in pathogenetic mechanisms of diabetic nephropathy. Cell Mol.Biol.(Noisy.-le-grand), 2003, Vol. 49, n°. 8, pp. 1319-1325.
NISHIKAWA T., EDELSTEIN D., DU X. L., YAMAGISHI S., MATSUMURA T., KANEDA Y., YOREK M. A., BEEBE D., OATES P. J., HAMMES H. P., GIARDINO I., BROWNLEE M. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature, 2000, Vol. 404, n°. 6779, pp. 787-790.
NIWA T., KATSUZAKI T., MIYAZAKI S., MIYAZAKI T., ISHIZAKI Y., HAYASE F., TATEMICHI N., TAKEI Y. Immunohistochemical detection of imidazolone, a novel advanced glycation end product, in kidneys and aortas of diabetic patients. J Clin.Invest, 1997, Vol. 99, n°. 6, pp. 1272-1280.
NOJIRI H., STROUD M., HAKOMORI S. A specific type of ganglioside as a modulator of insulin-dependent cell growth and insulin receptor tyrosine kinase activity. Possible association of ganglioside-induced inhibition of insulin receptor function and monocytic differentiation induction in HL-60 cells. J Biol.Chem., 1991, Vol. 266, n°. 7, pp. 4531-4537.
NOJIRI H., TAKAKU F., TERUI Y., MIURA Y., SAITO M. Ganglioside GM3: an acidic membrane component that increases during macrophage-like cell differentiation can induce monocytic differentiation of human myeloid and monocytoid leukemic cell lines HL-60 and U937. Proc.Natl.Acad.Sci.U.S.A, 1986, Vol. 83, n°. 3, pp. 782-786.
NOJIRI H., YAMANA H., SHIROUZU G., SUZUKI T., ISONO H. Glycotherapy for cancer: remodeling of ganglioside pattern as an effective approach for cancer therapy. Cancer Detect.Prev., 2002, Vol. 26, n°. 2, pp. 114-120.
NOMURA M., YAMAGISHI S., HARADA S., HAYASHI Y., YAMASHIMA T., YAMASHITA J., YAMAMOTO H. Possible participation of autocrine and paracrine vascular endothelial growth factors in hypoxia-induced proliferation of endothelial cells and pericytes. J Biol.Chem., 1995, Vol. 270, n°. 47, pp. 28316-28324.
186

NORES G. A., CAPUTTO R. Inhibition of the UDP-N-acetylgalactosamine: GM3, N-acetylgalactosaminyl transferase by gangliosides. J Neurochem., 1984, Vol. 42, n°. 5, pp. 1205-1211.
OBINECHE E. N., MENSAH-BROWN E., CHANDRANATH S. I., AHMED I., NASEER O., ADEM A. Morphological changes in the rat kidney following long-term diabetes. Arch.Physiol Biochem., 2001, Vol. 109, n°. 3, pp. 241-245.
OHGAMI N., NAGAI R., IKEMOTO M., ARAI H., KUNIYASU A., HORIUCHI S., NAKAYAMA H. Cd36, a member of the class b scavenger receptor family, as a receptor for advanced glycation end products. J Biol.Chem., 2001, Vol. 276, n°. 5, pp. 3195-3202.
OLDFIELD M. D., BACH L. A., FORBES J. M., NIKOLIC-PATERSON D., MCROBERT A., THALLAS V., ATKINS R. C., OSICKA T., JERUMS G., COOPER M. E. Advanced glycation end products cause epithelial-myofibroblast transdifferentiation via the receptor for advanced glycation end products (RAGE). J Clin.Invest, 2001, Vol. 108, n°. 12, pp. 1853-1863.
ORLIDGE A., D'AMORE P. A. Inhibition of capillary endothelial cell growth by pericytes and smooth muscle cells. J Cell Biol., 1987, Vol. 105, n°. 3, pp. 1455-1462.
PAGTALUNAN M. E., MILLER P. L., JUMPING-EAGLE S., NELSON R. G., MYERS B. D., RENNKE H. G., COPLON N. S., SUN L., MEYER T. W. Podocyte loss and progressive glomerular injury in type II diabetes. J Clin.Invest, 1997, Vol. 99, n°. 2, pp. 342-348.
PANZETTA P., ALLENDE M. L. Ganglioside expression during differentiation of chick retinal cells in vitro. Neurochem.Res, 2000, Vol. 25, n°. 1, pp. 163-169.
PEGUET-NAVARRO J., SPORTOUCH M., POPA I., BERTHIER O., SCHMITT D., PORTOUKALIAN J. Gangliosides from human melanoma tumors impair dendritic cell differentiation from monocytes and induce their apoptosis. J Immunol., 2003, Vol. 170, n°. 7, pp. 3488-3494.
PODESTA F., ROMEO G., LIU W. H., KRAJEWSKI S., REED J. C., GERHARDINGER C., LORENZI M. Bax is increased in the retina of diabetic subjects and is associated with pericyte apoptosis in vivo and in vitro. Am J Pathol., 2000, Vol. 156, n°. 3, pp. 1025-1032.
PORTERO-OTIN M., NAGARAJ R. H., MONNIER V. M. Chromatographic evidence for pyrraline formation during protein glycation in vitro and in vivo. Biochim.Biophys.Acta, 1995, Vol. 1247, n°. 1, pp. 74-80.
PUGLIESE G., TILTON R. G., SPEEDY A., CHANG K., SANTARELLI E., PROVINCE M. A., EADES D., SHERMAN W. R., WILLIAMSON J. R. Effects of very mild versus overt diabetes on vascular haemodynamics and barrier function in rats. Diabetologia, 1989, Vol. 32, n°. 12, pp. 845-857.
RADOFF S., VLASSARA H., CERAMI A. Characterization of a solubilized cell surface binding protein on macrophages specific for proteins modified nonenzymatically by advanced glycosylated end products. Arch.Biochem.Biophys., 1988, Vol. 263, n°. 2, pp. 418-423.
RELLIER N., RUGGIERO D., LECOMTE M., LAGARDE M., WIERNSPERGER N. Advanced glycation end products induce specific glycoprotein alterations in retinal microvascular cells. Biochem.Biophys.Res Commun., 1997, Vol. 235, n°. 2, pp. 281-285.
187

RELLIER N., RUGGIERO-LOPEZ D., LECOMTE M., LAGARDE M., WIERNSPERGER N. In vitro and in vivo alterations of enzymatic glycosylation in diabetes. Life Sci., 1999, Vol. 64, n°. 17, pp. 1571-1583.
RHO Y. I., KWAK D. H., LEE S., AHN S. H., BAEK S. H., SONG J. H., CHOO Y. K., CHOI B. K., JUNG K. Y. Mechanism for the negative regulation of cell proliferation by ganglioside GM3 in high glucose-treated glomerular mesangial cells. Life Sci., 2004, Vol. 75, n°. 1, pp. 51-62.
RIPPO M. R., MALISAN F., RAVAGNAN L., TOMASSINI B., CONDO I., COSTANTINI P., SUSIN S. A., RUFINI A., TODARO M., KROEMER G., TESTI R. GD3 ganglioside directly targets mitochondria in a bcl-2-controlled fashion. FASEB J, 2000, Vol. 14, n°. 13, pp. 2047-2054.
ROMEO G., LIU W. H., ASNAGHI V., KERN T. S., LORENZI M. Activation of nuclear factor-kappaB induced by diabetes and high glucose regulates a proapoptotic program in retinal pericytes. Diabetes, 2002, Vol. 51, n°. 7, pp. 2241-2248.
ROSEMAN S., The synthesis of complex carbohydrates by multiglycosyltransferase systems and their potential function in intercellular adhesion. Chem.Phys.Lipids, 1970, Vol. 5, n°. 1, pp. 270-297.
RUAN S., RAJ B. K., LLOYD K. O. Relationship of glycosyltransferases and mRNA levels to ganglioside expression in neuroblastoma and melanoma cells. J Neurochem., 1999, Vol. 72, n°. 2, pp. 514-521.
RUGGIERO D., LECOMTE M., MICHOUD E., LAGARDE M., WIERNSPERGER N. Involvement of cell-cell interactions in the pathogenesis of diabetic retinopathy. Diabetes Metab, 1997, Vol. 23, n°. 1, pp. 30-42.
RUGGIERO-LOPEZ D., RELLIER N., LECOMTE M., LAGARDE M., WIERNSPERGER N. Growth modulation of retinal microvascular cells by early and advanced glycation products. Diabetes Res Clin.Pract., 1997, Vol. 34, n°. 3, pp. 135-142.
RUSNATI M., URBINATI C., TANGHETTI E., DELL'ERA P., LORTAT-JACOB H., PRESTA M. Cell membrane GM1 ganglioside is a functional coreceptor for fibroblast growth factor 2. Proc.Natl.Acad.Sci.U.S.A, 2002, Vol. 99, n°. 7, pp. 4367-4372.
SACHINIDIS A., KRAUS R., SEUL C., MEYER ZU BRICKWEDDE M. K., SCHULTE K., KO Y., HOPPE J., VETTER H. Gangliosides GM1, GM2 and GM3 inhibit the platelet-derived growth factor-induced signalling transduction pathway in vascular smooth muscle cells by different mechanisms. Eur.J Cell Biol., 1996, Vol. 71, n°. 1, pp. 79-88.
SAITO M., ITO M., SUGIYAMA K. A specific loss of C-series gangliosides in pancreas of streptozotocin-induced diabetic rats. Life Sci., 1999, Vol. 64, n°. 20, pp. 1803-1810.
SAITO M., SUGIYAMA K. Gangliosides of rat eye lens: a severe reduction in the content of C-series gangliosides following streptozotocin treatment. Life Sci., 2000, Vol. 67, n°. 15, pp. 1891-1899.
SAKATA N., UESUGI N., TAKEBAYASHI S., NAGAI R., JONO T., HORIUCHI S., TAKEYA M., ITABE H., TAKANO T., MYINT T., TANIGUCHI N. Glycoxidation and lipid peroxidation of low-density lipoprotein can synergistically enhance atherogenesis. Cardiovasc.Res, 2001, Vol. 49, n°. 2, pp. 466-475.
188

SAKURAI S., YONEKURA H., YAMAMOTO Y., WATANABE T., TANAKA N., LI H., RAHMAN A. K., MYINT K. M., KIM C. H., YAMAMOTO H. The AGE-RAGE system and diabetic nephropathy. J Am Soc Nephrol, 2003, Vol. 14, n°. 8 Suppl 3, pp. S259-S263.
SANCHEZ S. S., ABREGU A. V., AYBAR M. J., SANCHEZ RIERA A. N. Changes in liver gangliosides in streptozotocin-induced diabetic rats. Cell Biol.Int, 2000, Vol. 24, n°. 12, pp. 897-904.
SAQR H. E., WALTERS J. D., GUAN Z., STOKES B. T., YATES A. J. Gangliosides inhibit PDGF-induced signal transduction events in U-1242 MG human glioma cells. Neurochem.Res, 1995, Vol. 20, n°. 11, pp. 1389-1395.
SASAKI A., HATA K., SUZUKI S., SAWADA M., WADA T., YAMAGUCHI K., OBINATA M., TATENO H., SUZUKI H., MIYAGI T. Overexpression of plasma membrane-associated sialidase attenuates insulin signaling in transgenic mice. J Biol.Chem., 2003, Vol. 278, n°. 30, pp. 27896-27902.
SCHEIDELER M. A., DAWSON G. Direct demonstration of the activation of UDP-N-acetylgalactosamine: [GM3]N-acetylgalactosaminyltransferase by cyclic AMP. J Neurochem., 1986, Vol. 46, n°. 5, pp. 1639-1643.
SCHLEICHER E. D., WEIGERT C. Role of the hexosamine biosynthetic pathway in diabetic nephropathy. Kidney Int.Suppl, 2000, Vol. 77, pp. S13-S18.
SCHNAAR R. L., COLLINS B. E., WRIGHT L. P., KISO M., TROPAK M. B., RODER J. C., CROCKER P. R. Myelin-associated glycoprotein binding to gangliosides. Structural specificity and functional implications. Ann.N.Y.Acad.Sci., 1998, Vol. 845, pp. 92-105.
SCHNAAR R. L., NEEDHAM L. K. Thin-layer chromatography of glycosphingolipids. Methods Enzymol., 1994, Vol. 230, pp. 371-389.
SENN H. J., ORTH M., FITZKE E., WIELAND H., GEROK W. Gangliosides in normal human serum. Concentration, pattern and transport by lipoproteins. Eur.J Biochem., 1989, Vol. 181, n°. 3, pp. 657-662.
SERRANO M. A., REGLERO A., CABEZAS J. A., GARCIA DIEZ L. C., CORRALES J. J., DE CASTRO S., MIRALLES J. M. Serum glycosidases in diabetes mellitus in relation to the retinopathy and to the length of the disease. Clin.Chim.Acta, 1983, Vol. 132, n°. 1, pp. 23-27.
SHAFI R., IYER S. P., ELLIES L. G., O'DONNELL N., MAREK K. W., CHUI D., HART G. W., MARTH J. D. The O-GlcNAc transferase gene resides on the X chromosome and is essential for embryonic stem cell viability and mouse ontogeny. Proc.Natl.Acad.Sci.U.S.A, 2000, Vol. 97, n°. 11, pp. 5735-5739.
SHAMSI F. A., PARTAL A., SADY C., GLOMB M. A., NAGARAJ R. H. Immunological evidence for methylglyoxal-derived modifications in vivo. Determination of antigenic epitopes. J.Biol.Chem., 1998, Vol. 273, n°. 12, pp. 6928-6936.
SHARMA K., JIN Y., GUO J., ZIYADEH F. N. Neutralization of TGF-beta by anti-TGF-beta antibody attenuates kidney hypertrophy and the enhanced extracellular matrix gene expression in STZ-induced diabetic mice. Diabetes, 1996, Vol. 45, n°. 4, pp. 522-530.
SHEETZ M. J., KING G. L. Molecular understanding of hyperglycemia's adverse effects for diabetic complications. JAMA, 2002, Vol. 288, n°. 20, pp. 2579-2588.
189

SHIKATA K., MAKINO H., SUGIMOTO H., KUSHIRO M., OTA K., AKIYAMA K., ARAKI N., HORIUCHI S., OTA Z. Localization of advanced glycation endproducts in the kidney of experimental diabetic rats. J Diabetes Complications, 1995, Vol. 9, n°. 4, pp. 269-271.
SIMON B. M., MALISAN F., TESTI R., NICOTERA P., LEIST M. Disialoganglioside GD3 is released by microglia and induces oligodendrocyte apoptosis. Cell Death.Differ., 2002, Vol. 9, n°. 7, pp. 758-767.
SIMPSON L. L., RAPPORT M. M. The binding of botulinum toxin to membrane lipids: phospholipids and proteolipid. J Neurochem., 1971, Vol. 18, n°. 9, pp. 1761-1767.
SINGH L. P., ANDY J., ANYAMALE V., GREENE K., ALEXANDER M., CROOK E. D. Hexosamine-induced fibronectin protein synthesis in mesangial cells is associated with increases in cAMP responsive element binding (CREB) phosphorylation and nuclear CREB: the involvement of protein kinases A and C. Diabetes, 2001, Vol. 50, n°. 10, pp. 2355-2362.
SINGH L. P., CROOK E. D. Hexosamine regulation of glucose-mediated laminin synthesis in mesangial cells involves protein kinases A and C. Am J Physiol Renal Physiol, 2000, Vol. 279, n°. 4, pp. F646-F654.
SINGH L. P., GREEN K., ALEXANDER M., BASSLY S., CROOK E. D. Hexosamines and TGF-beta1 use similar signaling pathways to mediate matrix protein synthesis in mesangial cells. Am J Physiol Renal Physiol, 2004, Vol. 286, n°. 2, pp. F409-F416.
SINGH R., BARDEN A., MORI T., BEILIN L. Advanced glycation end-products: a review. Diabetologia, 2001, Vol. 44, n°. 2, pp. 129-146.
SLEVIN M., KUMAR S., HE X., GAFFNEY J. Physiological concentrations of gangliosides GM1, GM2 and GM3 differentially modify basic-fibroblast-growth-factor-induced mitogenesis and the associated signalling pathway in endothelial cells. Int J Cancer, 1999, Vol. 82, n°. 3, pp. 412-423.
SOULIS T., THALLAS V., YOUSSEF S., GILBERT R. E., MCWILLIAM B. G., MURRAY-MCINTOSH R. P., COOPER M. E. Advanced glycation end products and their receptors co-localise in rat organs susceptible to diabetic microvascular injury. Diabetologia, 1997, Vol. 40, n°. 6, pp. 619-628.
STEFFES M. W., SCHMIDT D., MCCRERY R., BASGEN J. M. Glomerular cell number in normal subjects and in type 1 diabetic patients. Kidney Int, 2001, Vol. 59, n°. 6, pp. 2104-2113.
STITT A. W., The role of advanced glycation in the pathogenesis of diabetic retinopathy. Exp.Mol.Pathol., 2003, Vol. 75, n°. 1, pp. 95-108.
STITT A. W., BHADURI T., MCMULLEN C. B., GARDINER T. A., ARCHER D. B. Advanced glycation end products induce blood-retinal barrier dysfunction in normoglycemic rats. Mol.Cell Biol.Res Commun., 2000a, Vol. 3, n°. 6, pp. 380-388.
STITT A. W., BURKE G. A., CHEN F., MCMULLEN C. B., VLASSARA H. Advanced glycation end-product receptor interactions on microvascular cells occur within caveolin-rich membrane domains. FASEB J, 2000b, Vol. 14, n°. 15, pp. 2390-2392.
STITT A. W., HE C., VLASSARA H. Characterization of the advanced glycation end-product receptor complex in human vascular endothelial cells. Biochem.Biophys.Res Commun., 1999, Vol. 256, n°. 3, pp. 549-556.
190

STITT A. W., HUGHES S. J., CANNING P., LYNCH O., COX O., FRIZZELL N., THORPE S. R., COTTER T. G., CURTIS T. M., GARDINER T. A. Substrates modified by advanced glycation end-products cause dysfunction and death in retinal pericytes by reducing survival signals mediated by platelet-derived growth factor. Diabetologia, 2004, Vol. 47, n°. 10, pp. 1735-1746.
STITT A. W., LI Y. M., GARDINER T. A., BUCALA R., ARCHER D. B., VLASSARA H. Advanced glycation end products (AGEs) co-localize with AGE receptors in the retinal vasculature of diabetic and of AGE-infused rats. Am J Pathol., 1997, Vol. 150, n°. 2, pp. 523-531.
SUZUKI Y., Gangliosides as influenza virus receptors. Variation of influenza viruses and their recognition of the receptor sialo-sugar chains. Prog.Lipid Res, 1994, Vol. 33, n°. 4, pp. 429-457.
SVENNERHOLM L., Quantitative estimation of sialic acids. II. A colorimetric resorcinol-hydrochloric acid method. Biochim.Biophys.Acta, 1957, Vol. 24, n°. 3, pp. 604-611.
TAGAMI S., INOKUCHI J. J., KABAYAMA K., YOSHIMURA H., KITAMURA F., UEMURA S., OGAWA C., ISHII A., SAITO M., OHTSUKA Y., SAKAUE S., IGARASHI Y. Ganglioside GM3 participates in the pathological conditions of insulin resistance. J.Biol.Chem., 2002, Vol. 277, n°. 5, pp. 3085-3092.
TAKAMIYA K., YAMAMOTO A., FURUKAWA K., YAMASHIRO S., SHIN M., OKADA M., FUKUMOTO S., HARAGUCHI M., TAKEDA N., FUJIMURA K., SAKAE M., KISHIKAWA M., SHIKU H., FURUKAWA K., AIZAWA S. Mice with disrupted GM2/GD2 synthase gene lack complex gangliosides but exhibit only subtle defects in their nervous system. Proc.Natl.Acad.Sci.U.S.A, 1996, Vol. 93, n°. 20, pp. 10662-10667.
TANG J., NEIDIGH J. L., COOKSEY R. C., MCCLAIN D. A. Transgenic mice with increased hexosamine flux specifically targeted to beta-cells exhibit hyperinsulinemia and peripheral insulin resistance. Diabetes, 2000, Vol. 49, n°. 9, pp. 1492-1499.
THOMAS C. P., BURONFOSSE A., PORTOUKALIAN J., FERTIL B. The gangliosides as a possible molecular coupling factor between the proportion of radiosensitive cells in vitro and the metastatic potential in vivo within a human melanoma cell line. Br.J Cancer, 1997, Vol. 75, n°. 5, pp. 639-649.
TIBERTI C., DOTTA F., ANASTASI E., TORRESI P., MULTARI G., VECCI E., ANDREANI D., DI MARIO U. Anti-ganglioside antibodies in new onset type 1 diabetic patients and high risk subjects. Autoimmunity, 1995, Vol. 22, n°. 1, pp. 43-48.
TIEMEYER M., YASUDA Y., SCHNAAR R. L. Ganglioside-specific binding protein on rat brain membranes. J Biol.Chem., 1989, Vol. 264, n°. 3, pp. 1671-1681.
TOMASSINI B., TESTI R. Mitochondria as sensors of sphingolipids. Biochimie, 2002, Vol. 84, n°. 2-3, pp. 123-129.
TOMINO Y., INOUE W., WATANABE S., YAGAME M., EGUCHI K., NOMOTO Y., SAKAI H. Detection of glomerular sialic acids in patients with diabetic nephropathy. Am J Nephrol, 1988, Vol. 8, n°. 1, pp. 21-26.
TOMINO Y., YAGAME M., INOUE W., WATANABE S., KANESHIGE H., NOMOTO Y., SAKAI H., TAKEDA M. Double immunofluorescence studies of glomerular sialic acids in patients with diabetic nephropathy. J Diabet.Complications, 1987, Vol. 1, n°. 2, pp. 61-64.
191

TREINS C., GIORGETTI-PERALDI S., MURDACA J., VAN OBBERGHEN E. Regulation of vascular endothelial growth factor expression by advanced glycation end products. J Biol.Chem., 2001, Vol. 276, n°. 47, pp. 43836-43841.
TREVISAN R., BARNES D., VIBERTI G.C. Pathogenesis of diabetic nephropathy. In : Textbook of diabetes. second edition. London : edited by J. C. Pickup and williams G., 1997, pp. 52.1-52.21.
TSILIBARY E. C., Microvascular basement membranes in diabetes mellitus. J Pathol., 2003, Vol. 200, n°. 4, pp. 537-546.
TSUBOI N., UTSUNOMIYA Y., KAWAMURA T., KAWANO T., HOSOYA T., OHNO T., YAMADA H. Ganglioside as an endogenous growth suppressor for glomerular mesangial cells. Kidney Int., 2001, Vol. 60, n°. 4, pp. 1378-1385.
VAN BROCKLYN J., BREMER E. G., YATES A. J. Gangliosides inhibit platelet-derived growth factor-stimulated receptor dimerization in human glioma U-1242MG and Swiss 3T3 cells. J.Neurochem., 1993, Vol. 61, n°. 1, pp. 371-374.
VAN ECHTEN G., SANDHOFF K. Ganglioside metabolism. Enzymology, Topology, and regulation. J Biol.Chem., 1993, Vol. 268, n°. 8, pp. 5341-5344.
VAN HEYNINGEN W. E., MILLER P. A. The fixation of tetanus toxin by ganglioside. J Gen.Microbiol., 1961, Vol. 24, pp. 107-119.
VLASSARA H., The AGE-receptor in the pathogenesis of diabetic complications. Diabetes Metab Res Rev., 2001, Vol. 17, n°. 6, pp. 436-443.
VLASSARA H., STRIKER L. J., TEICHBERG S., FUH H., LI Y. M., STEFFES M. Advanced glycation end products induce glomerular sclerosis and albuminuria in normal rats. Proc.Natl.Acad.Sci.U.S.A, 1994, Vol. 91, n°. 24, pp. 11704-11708.
VOSSELLER K., WELLS L., LANE M. D., HART G. W. Elevated nucleocytoplasmic glycosylation by O-GlcNAc results in insulin resistance associated with defects in Akt activation in 3T3-L1 adipocytes. Proc.Natl.Acad.Sci.U.S.A, 2002, Vol. 99, n°. 8, pp. 5313-5318.
WALKER J. D.VIBERTI G.C. Aetiology and Pathogenesis of Diabetic Nephropathy: Clues from Early Functional Abnormalities. In : Chronic complications of diabetes. London : edited by J. C. Pickup and williams G., 1994, pp. 146-161.
WANG X. Q., SUN P., O'GORMAN M., TAI T., PALLER A. S. Epidermal growth factor receptor glycosylation is required for ganglioside GM3 binding and GM3-mediated suppression [correction of suppresion] of activation. Glycobiology, 2001a, Vol. 11, n°. 7, pp. 515-522.
WANG X. Q., SUN P., PALLER A. S. Inhibition of integrin-linked kinase/protein kinase B/Akt signaling: mechanism for ganglioside-induced apoptosis. J Biol.Chem., 2001b, Vol. 276, n°. 48, pp. 44504-44511.
WANG X. Q., SUN P., PALLER A. S. Ganglioside induces caveolin-1 redistribution and interaction with the epidermal growth factor receptor. J Biol.Chem., 2002, Vol. 277, n°. 49, pp. 47028-47034.
192

WATANABE R., WU K., PAUL P., MARKS D. L., KOBAYASHI T., PITTELKOW M. R., PAGANO R. E. Up-regulation of glucosylceramide synthase expression and activity during human keratinocyte differentiation. J Biol.Chem., 1998, Vol. 273, n°. 16, pp. 9651-9655.
WAUTIER J. L., GUILLAUSSEAU P. J. Advanced glycation end products, their receptors and diabetic angiopathy. Diabetes Metab, 2001, Vol. 27, n°. 5 Pt 1, pp. 535-542.
WAUTIER J. L., WAUTIER M. P., SCHMIDT A. M., ANDERSON G. M., HORI O., ZOUKOURIAN C., CAPRON L., CHAPPEY O., YAN S. D., BRETT J., . Advanced glycation end products (AGEs) on the surface of diabetic erythrocytes bind to the vessel wall via a specific receptor inducing oxidant stress in the vasculature: a link between surface-associated AGEs and diabetic complications. Proc.Natl.Acad.Sci.U.S.A, 1994, Vol. 91, n°. 16, pp. 7742-7746.
WAUTIER J. L., ZOUKOURIAN C., CHAPPEY O., WAUTIER M. P., GUILLAUSSEAU P. J., CAO R., HORI O., STERN D., SCHMIDT A. M. Receptor-mediated endothelial cell dysfunction in diabetic vasculopathy. Soluble receptor for advanced glycation end products blocks hyperpermeability in diabetic rats. J Clin.Invest, 1996, Vol. 97, n°. 1, pp. 238-243.
WAUTIER M. P., CHAPPEY O., CORDA S., STERN D. M., SCHMIDT A. M., WAUTIER J. L. Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am J Physiol Endocrinol Metab, 2001, Vol. 280, n°. 5, pp. E685-E694.
WEIGERT C., FRIESS U., BRODBECK K., HARING H. U., SCHLEICHER E. D. Glutamine:fructose-6-phosphate aminotransferase enzyme activity is necessary for the induction of TGF-beta1 and fibronectin expression in mesangial cells. Diabetologia, 2003, Vol. 46, n°. 6, pp. 852-855.
WEIS F. M., DAVIS R. J. Regulation of epidermal growth factor receptor signal transduction. Role of gangliosides. J.Biol.Chem., 1990, Vol. 265, n°. 20, pp. 12059-12066.
WELLS L., VOSSELLER K., HART G. W. Glycosylation of nucleocytoplasmic proteins: signal transduction and O-GlcNAc. Science, 2001, Vol. 291, n°. 5512, pp. 2376-2378.
WENDT T., TANJI N., GUO J., HUDSON B. I., BIERHAUS A., RAMASAMY R., ARNOLD B., NAWROTH P. P., YAN S. F., D'AGATI V., SCHMIDT A. M. Glucose, glycation, and RAGE: implications for amplification of cellular dysfunction in diabetic nephropathy. J Am Soc Nephrol, 2003, Vol. 14, n°. 5, pp. 1383-1395.
WESTWOOD M. E., MCLELLAN A. C., THORNALLEY P. J. Receptor-mediated endocytic uptake of methylglyoxal-modified serum albumin. Competition with advanced glycation end product-modified serum albumin at the advanced glycation end product receptor. J Biol.Chem., 1994, Vol. 269, n°. 51, pp. 32293-32298.
WOLF G., Cell cycle regulation in diabetic nephropathy. Kidney Int.Suppl, 2000, Vol. 77, pp. S59-S66.
WOLF G., SCHROEDER R., THAISS F., ZIYADEH F. N., HELMCHEN U., STAHL R. A. Glomerular expression of p27Kip1 in diabetic db/db mouse: role of hyperglycemia. Kidney Int., 1998, Vol. 53, n°. 4, pp. 869-879.
WOLF G., SHARMA K., CHEN Y., ERICKSEN M., ZIYADEH F. N. High glucose-induced proliferation in mesangial cells is reversed by autocrine TGF-beta. Kidney Int, 1992, Vol. 42, n°. 3, pp. 647-656.
193

WOLF G., ZIYADEH F. N. Molecular mechanisms of diabetic renal hypertrophy. Kidney Int, 1999, Vol. 56, n°. 2, pp. 393-405.
YAMADA K. M., CRITCHLEY D. R., FISHMAN P. H., MOSS J. Exogenous gangliosides enhance the interaction of fibronectin with ganglioside-deficient cells. Exp.Cell Res, 1983, Vol. 143, n°. 2, pp. 295-302.
YAMAGISHI S., AMANO S., INAGAKI Y., OKAMOTO T., KOGA K., SASAKI N., YAMAMOTO H., TAKEUCHI M., MAKITA Z. Advanced glycation end products-induced apoptosis and overexpression of vascular endothelial growth factor in bovine retinal pericytes. Biochem.Biophys.Res.Commun., 2002a, Vol. 290, n°. 3, pp. 973-978.
YAMAGISHI S., HSU C. C., TANIGUCHI M., HARADA S., YAMAMOTO Y., OHSAWA K., KOBAYASHI K., YAMAMOTO H. Receptor-mediated toxicity to pericytes of advanced glycosylation end products: a possible mechanism of pericyte loss in diabetic microangiopathy. Biochem.Biophys.Res Commun., 1995, Vol. 213, n°. 2, pp. 681-687.
YAMAGISHI S., INAGAKI Y., OKAMOTO T., AMANO S., KOGA K., TAKEUCHI M., MAKITA Z. Advanced glycation end product-induced apoptosis and overexpression of vascular endothelial growth factor and monocyte chemoattractant protein-1 in human-cultured mesangial cells. J.Biol.Chem., 2002b, Vol. 277, n°. 23, pp. 20309-20315.
YAMAGISHI S., KOBAYASHI K., YAMAMOTO H. Vascular pericytes not only regulate growth, but also preserve prostacyclin-producing ability and protect against lipid peroxide-induced injury of co-cultured endothelial cells. Biochem.Biophys.Res Commun., 1993, Vol. 190, n°. 2, pp. 418-425.
YAMAMOTO Y., KATO I., DOI T., YONEKURA H., OHASHI S., TAKEUCHI M., WATANABE T., YAMAGISHI S., SAKURAI S., TAKASAWA S., OKAMOTO H., YAMAMOTO H. Development and prevention of advanced diabetic nephropathy in RAGE-overexpressing mice. J Clin.Invest, 2001, Vol. 108, n°. 2, pp. 261-268.
YAMASHITA T., HASHIRAMOTO A., HALUZIK M., MIZUKAMI H., BECK S., NORTON A., KONO M., TSUJI S., DANIOTTI J. L., WERTH N., SANDHOFF R., SANDHOFF K., PROIA R. L. Enhanced insulin sensitivity in mice lacking ganglioside GM3. Proc.Natl.Acad.Sci.U.S.A, 2003, Vol. 100, n°. 6, pp. 3445-3449.
YAMASHITA T., WADA R., SASAKI T., DENG C., BIERFREUND U., SANDHOFF K., PROIA R. L. A vital role for glycosphingolipid synthesis during development and differentiation. Proc.Natl.Acad.Sci.U.S.A, 1999, Vol. 96, n°. 16, pp. 9142-9147.
YANG C. W., VLASSARA H., PETEN E. P., HE C. J., STRIKER G. E., STRIKER L. J. Advanced glycation end products up-regulate gene expression found in diabetic glomerular disease. Proc.Natl.Acad.Sci.U.S.A, 1994, Vol. 91, n°. 20, pp. 9436-9440.
YANG X., SU K., ROOS M. D., CHANG Q., PATERSON A. J., KUDLOW J. E. O-linkage of N-acetylglucosamine to Sp1 activation domain inhibits its transcriptional capability. Proc.Natl.Acad.Sci.U.S.A, 2001, Vol. 98, n°. 12, pp. 6611-6616.
YATES A. J., SAQR H. E., VAN BROCKLYN J. Ganglioside modulation of the PDGF receptor. A model for ganglioside functions. J.Neurooncol., 1995, Vol. 24, n°. 1, pp. 65-73.
194

YEH C. H., STURGIS L., HAIDACHER J., ZHANG X. N., SHERWOOD S. J., BJERCKE R. J., JUHASZ O., CROW M. T., TILTON R. G., DENNER L. Requirement for p38 and p44/p42 mitogen-activated protein kinases in RAGE-mediated nuclear factor-kappaB transcriptional activation and cytokine secretion. Diabetes, 2001, Vol. 50, n°. 6, pp. 1495-1504.
YONEKURA H., YAMAMOTO Y., SAKURAI S., PETROVA R. G., ABEDIN M. J., LI H., YASUI K., TAKEUCHI M., MAKITA Z., TAKASAWA S., OKAMOTO H., WATANABE T., YAMAMOTO H. Novel splice variants of the receptor for advanced glycation end-products expressed in human vascular endothelial cells and pericytes, and their putative roles in diabetes-induced vascular injury. Biochem.J., 2003, Vol. 370, n°. Pt 3, pp. 1097-1109.
YOSHINO H., ARIGA T., LATOV N., MIYATAKE T., KUSHI Y., KASAMA T., HANDA S., YU R. K. Fucosyl-GM1 in human sensory nervous tissue is a target antigen in patients with autoimmune neuropathies. J Neurochem., 1993, Vol. 61, n°. 2, pp. 658-663.
YUSUF H. K., SCHWARZMANN G., POHLENTZ G., SANDHOFF K. Oligosialogangliosides inhibit GM2- and GD3-synthesis in isolated Golgi vesicles from rat liver. Biol.Chem.Hoppe Seyler, 1987, Vol. 368, n°. 5, pp. 455-462.
ZADOR I. Z., DESHMUKH G. D., KUNKEL R., JOHNSON K., RADIN N. S., SHAYMAN J. A. A role for glycosphingolipid accumulation in the renal hypertrophy of streptozotocin-induced diabetes mellitus. J Clin.Invest, 1993, Vol. 91, n°. 3, pp. 797-803.
ZATZ R., BRENNER B. M. Pathogenesis of diabetic microangiopathy. The hemodynamic view. Am J Med., 1986a, Vol. 80, n°. 3, pp. 443-453.
ZATZ R., DUNN B. R., MEYER T. W., ANDERSON S., RENNKE H. G., BRENNER B. M. Prevention of diabetic glomerulopathy by pharmacological amelioration of glomerular capillary hypertension. J Clin.Invest, 1986b, Vol. 77, n°. 6, pp. 1925-1930.
ZEBDA N., PEDRON S., REBBAA A., PORTOUKALIAN J., BERTHIER-VERGNES O. Deficiency of ganglioside biosynthesis in metastatic human melanoma cells: relevance of CMP-NeuAc:LacCer alpha 2-3 sialyltransferase (GM3 synthase). FEBS Lett., 1995, Vol. 362, n°. 2, pp. 161-164.
ZENG G., ARIGA T., GU X. B., YU R. K. Regulation of glycolipid synthesis in HL-60 cells by antisense oligodeoxynucleotides to glycosyltransferase sequences: effect on cellular differentiation. Proc.Natl.Acad.Sci.U.S.A, 1995, Vol. 92, n°. 19, pp. 8670-8674.
ZENG G., GAO L., XIA T., TENCOMNAO T., YU R. K. Characterization of the 5'-flanking fragment of the human GM3-synthase gene. Biochim.Biophys.Acta, 2003, Vol. 1625, n°. 1, pp. 30-35.
ZENG G., GAO L., YU R. K. Isolation and functional analysis of the promoter of the rat CMP-NeuAc:GM3 alpha2,8 sialyltransferase gene 1. Biochim.Biophys.Acta, 1998, Vol. 1397, n°. 2, pp. 126-130.
ZHENG M., FANG H., TSURUOKA T., TSUJI T., SASAKI T., HAKOMORI S. Regulatory role of GM3 ganglioside in alpha 5 beta 1 integrin receptor for fibronectin-mediated adhesion of FUA169 cells. J Biol.Chem., 1993, Vol. 268, n°. 3, pp. 2217-2222.
ZICHE M., ALESSANDRI G., GULLINO P. M. Gangliosides promote the angiogenic response. Lab Invest, 1989, Vol. 61, n°. 6, pp. 629-634.
195

ZICHE M., MORBIDELLI L., ALESSANDRI G., GULLINO P. M. Angiogenesis can be stimulated or repressed in vivo by a change in GM3:GD3 ganglioside ratio. Lab Invest, 1992, Vol. 67, n°. 6, pp. 711-715.
ZIYADEH F. N., COHEN M. P. Effects of glycated albumin on mesangial cells: evidence for a role in diabetic nephropathy. Mol.Cell Biochem., 1993, Vol. 125, n°. 1, pp. 19-25.
ZIYADEH F. N., SHARMA K., ERICKSEN M., WOLF G. Stimulation of collagen gene expression and protein synthesis in murine mesangial cells by high glucose is mediated by autocrine activation of transforming growth factor-beta. J Clin.Invest, 1994, Vol. 93, n°. 2, pp. 536-542.
196

PUBLICATIONS
197


a-series gangliosides mediate the effects of Advanced Glycation End-products on pericyte and mesangial cell proliferation A common mediator for retinal and renal microangiopathy ?
DIABETES, VOL. 54, JANUARY 2005
Running title: gangliosides as mediators of AGE effects
Elodie Masson, Lysiane Troncy, Daniel Ruggiero, Nicolas Wiernsperger, Michel Lagarde, and
Samer El Bawab
Diabetic Microangiopathy Research Unit, MERCK Santé-INSERM UMR 585, INSA-Lyon
Corresponding author: S. El Bawab, Diabetic Microangiopathy Research Unit, MERCK Santé-INSERM
U585, INSA-Lyon, Louis Pasteur Bldg, 69621 Villeurbanne Cedex, France.
Phone: (+33) 4 72 43 81 12
Fax: (+33) 4 72 43 81 13
E-mail: [email protected]

Abstract Advanced glycation end-products (AGE) are involved in the development of microvascular
complications, including alterations of retinal pericyte and renal mesangial cell growth occurring
during diabetic retinopathy (DR) and nephropathy (DN), respectively. As gangliosides are
implicated in the regulation of cell proliferation, we hypothesized that AGE could exert cellular
effects in part by modulating ganglioside levels. Results of the present study indicate that AGE
caused an inhibition of both bovine retinal pericyte (BRP) and rat renal mesangial cell (RMC)
proliferation, associated with an increase of a-series gangliosides consecutive to GM3 synthase
activity increase and GD3 synthase activity inhibition. Similar modifications were also found in
renal cortex of diabetic db/db mice as compared to controls. Treatment of BRP and RMC with
exogenous a-series gangliosides decreased proliferation and blockade of a-series gangliosides with
specific antibodies partially protected the two cell types from the AGE-induced proliferation
decrease. Further, inhibition of GM3 synthase using specific SiRNA partially reversed the AGE
effects on mesangial cell proliferation. These results suggest that a-series gangliosides are mediators
of the adverse AGE effects on BRP and RMC proliferation. They also raise the hypothesis of
common mechanisms involved in the development of DR and DN.
Abbreviations: AGE, advanced glycation end-products; BRP, bovine retinal pericytes; BSA, bovine
serum albumin; DMEM, dulbecco’s modified eagle’s medium; DN, diabetic nephropathy; DR,
diabetic retinopathy; HPTLC, high-performance-thin-layer chromatography; PBS, phosphate-
buffered saline; RAGE, receptor for advanced glycation end-products; RMC, rat mesangial cells.

Microangiopathy is a chronic complication of diabetes mellitus characterized by structural and
functional alterations of microvessels. Retina and kidney are two main targets of the pathology
leading to diabetic retinopathy (DR) and nephropathy (DN), respectively. Diabetic retinopathy is
the second cause of blindness in developed countries (1). Capillaries undergo progressive structural
alterations such as basement membrane thickening, specific loss of pericytes and subsequent
modifications of endothelial cell proliferation and function. Combined with ischemia, these
alterations damage microvessel walls and favor excessive capillary permeability, edema,
microaneurisms and hemorrage, thereby threatening vision (2). Nephropathy is considered a major
cause of mortality in diabetic patients (3). One of the main features of the pathology is glomerular
enlargement. This is the consequence of basement membrane thickening and expansion of the
mesangium due to hypertrophy of growth-arrested mesangial cells and accumulation of
extracellular matrix proteins. Combined with haemodynamic defects, these events trigger
glomerular sclerosis, impaired glomerular filtration rate and microalbuminuria resulting in severe
renal insufficiency (4).
On the other hand, although the pathogenic basis of DR and DN are not fully understood at cellular
and molecular levels, regulation of cell proliferation, cell-cell and cell-matrix interactions seem to
play important role. A number of biochemical hypotheses explaining the mechanisms involved in
the development of diabetic microvascular complications have been proposed, among others, the
formation of Advanced Glycation End-products (AGE) (reviewed in (5)). Reducing sugars such as
glucose react non-enzymatically with amino-groups of proteins, lipids and nucleic acids through a
series of reactions finally producing AGE. Glycation is glucose concentration-dependent and is thus
enhanced in diabetes. It occurs preferentially with long-lived proteins exposed to blood glucose
such as extracellular matrix or circulatory proteins, altering their structure and function. In addition,
AGE can bind membrane receptors to induce cellular responses via generation of oxidative stress
(6,7), activation of nuclear factor-κB (5,7), and expression of various genes such as pro-
inflammatory cytokines or adhesion molecules (8,9). All these modifications have important
biological effects that can explain many of the changes observed in diabetic microvascular
complications, including increase in vascular permeability, increase in extracellular matrix
production and stiffness, and alteration in cell-matrix interactions and cell growth (10). Indeed,
many in vivo and in vitro studies have pointed out the implication of AGE in the development of
diabetic retinopathy and nephropathy (10,11).
Gangliosides are glycosphingolipids concentrated in plasma membrane microdomains and
characterized by the presence of sialic acid in their structure. Successive sialylations of
lactosylceramide lead to monosialoganglioside GM3, disialoganglioside GD3, and
trisialoganglioside GT3. These gangliosides are then converted by the sequential actions of

glycosyltransferases and sialyltransferases into more complex gangliosides to form the a-, b-, and c-
series, respectively (12). Gangliosides are known to play major roles in cell-cell and cell-matrix
recognition via interactions with adhesion receptors such as integrins, matrix proteins, or with other
glycosphingolipids. Moreover, gangliosides, and in particular the a-series gangliosides, have been
implicated in the regulation of cell proliferation, through modulation of different growth factor
activities (13). As AGE are implicated in many perturbations of cell growth and viability occurring
during DR and DN, we hypothesized that gangliosides could participate in mediating their effects.
In the present study, we investigated the effect of AGE on the proliferation of two cell types, retinal
pericytes and renal mesangial cells, involved in DR and DN, respectively. Results indicated that
AGE cause an inhibition of proliferation of both cell types at least in part by accumulating the a-
series gangliosides. The observations suggest that the a-series gangliosides might represent a
common mechanism involved in cellular alterations occurring during the development of DR and
DN.

RESEARCH DESIGN AND METHODS
Cell isolation and culture. Bovine retinal pericytes (BRP) were obtained from microvessels of
bovine retinas as previously described (14). Briefly, microvessels fragments were isolated from
freshly dissected bovine retinas after Dounce homogenization and collagenase-dispase digestion.
Microvessels fragments were then trapped onto a 40 µm nylon filter and plated in 6 cm fibronectin-
coated dishes. Primary cultures were grown in dulbecco’s modified eagle’s medium (DMEM)
supplemented with 10% fetal bovine serum (Gibco, Invitrogen Corporation, N.Y., USA), 1%
glutamine and 1% penicillin/streptomycin (Sigma). Outgrowth of pericytes from the microvessels
occurred after about 48 h. BRP cultures were 100% pure as characterized on the basis of
morphological criteria and immunostaining as previously described (14). Cells were cultured up to
the second passage during which BRP were treated.
Rat renal mesangial cells (RMC) were obtained from glomeruli purified from young male Wistar
rat (Charles River, L’Arbresle, Fance). Briefly, cortex fragments were isolated from freshly
removed kidneys. Glomeruli were separated by differential sieving as previously described (15) and
plated in 6 cm fibronectin-coated dishes in DMEM supplemented with 20% fetal bovine serum, 1%
glutamine and 1% penicillin/streptomycin. Outgrowth of RMC from the glomeruli occurred after
about 3 weeks and cells were cultured in the same medium supplemented with 15% fetal bovine
serum and used between 5th and 15th passages. RMC were characterized by morphological criteria
and by immunostaining as previously described (15)
Isolation of mice renal cortex. Renal cortex fragments were isolated from kidneys of eleven weeks
diabetic db/db and control db/m mice (Charles River). Briefly, animals were anaesthetized, sacrified
and kidneys were removed. Kidney cortex fragments were then dissected and mechanically
homogenized with a Dounce homogenizer in 25 mM Hepes, 1 mM EDTA, 10 µl/ml proteases
inhibitors.
AGE preparation and treatment. AGE were prepared as previously described (16) by incubation
of fatty acid free and low endotoxin bovine serum albumin (7.2 mg/ml final concentration) (Sigma)
with 100 mM methylglyoxal (Sigma) at 37°C for 50 hours. Bovine serum albumin (BSA) in the
absence of methylglyoxal was incubated in the same conditions and used as a control preparation
(control BSA). AGE or control BSA (3 µM final concentrations) were added to the culture media
24 h after seeding. Each cell type was treated during one passage (about 7 days for BRP and 4 days
for RMC). Cultured medium was replaced by fresh medium every 2 days.

Measurement of cell growth. At the end of the treatment, cells were harvested with trypsin and
cell pellets were washed twice with ice-cold phosphate-buffered saline (PBS) (Sigma). For each
sample, an aliquot of cells was counted using a haemocytometer to determine the cell number.
Another aliquot was used to measure proteins according to Bradford.
Ganglioside analysis. For metabolic labelling of gangliosides, 0.2 µCi/ml or 1 µCi/ml for labelling
experiments with high specific activity (GM2 and GM1 measurement experiments) of [14C(U)]D-
Galactose (329.5 mCi/mmol) (PerkinElmer Life Sciences, Boston, MA) were added to the media
overnight. Cells were then collected by tripsynation and washed 3 times with PBS. Extraction of
gangliosides from cell pellets or cortex homogenates was performed as previously described by
Natalizio et al. (17). Briefly, total lipids were extracted with chloroform (C)/methanol (M) (1:1, v/v)
and submitted to partition with C/M/PBS 1 mM (10:10:7, v/v/v). The upper phases containing
gangliosides were next desalted on C18 silica gel column (Waters Corporation, Milford, MA) and
analyzed by HPTLC (Merck, Darmstadt, Germany). Plates were developed in C/M/0.2% CaCl2
(55:45:10, v/v/v) and gangliosides were visualized by autoradiography using phosphor screen and
Storm 820 (Molecular Dynamics, Amersham Pharmacia Biotech, Piscataway, USA) and by
resorcinol staining (resorcinol 0.3% (Sigma), 0.03% CuSO4, 30% HCl) with Image Master VDS-
CL (Amersham Pharmacia Biotech). Quantification was done with Image Quant (Molecular
Dynamics). As GT1b is absent in both BRP and RMC ganglioside profiles, it was added as internal
standard in the samples before lipid extraction.
Measurement of GM3 and GD3 synthase activities. At the end of treatment, cells were washed
with PBS, incubated for 20 min at 4°C in 50 µl lysis buffer (20 mM sodium cacodylate pH 6.6
(Sigma), 0.2% Triton X-100, 1 mM EDTA, 10 µl/ml proteases inhibitors, Calbiochem, La Jolla,
CA, USA) and then collected by scrapping. Four dishes of BRP (~2-3 × 106 cells) and 3 dishes of
RMC (~3-6 × 106 cells) were pooled. Cell lysates were centrifuged at 10,000g for 5 min, and
proteins in the supernatants were used to measure enzyme activity. Cortex homogenates were
centrifuged at 1000g for 2 min, and the postnuclear supernatant was used to measure enzyme
activities. Equal amounts of proteins from each sample (about 500 µg for cells and 200-400 µg for
cortex) were used to perform the assay. Samples were mixed with an equal volume of reaction
buffer containing at final concentrations: 0.1 mM lactosylceramide or GM3 (Matreya, Biovalley,
Marne la Vallée, France) for GM3 and GD3 synthase activities, respectively, and 4 µCi/ml [sialic-
4,5,6,7,8,9-14C]CMP-sialic acid (325.2 mCi/mmol) (PerkinElmer Life Sciences), 100 µM CMP-
sialic acid (Sigma), 10 mM MgCl2, 0.2% Triton X-100, 100 mM sodium cacodylate pH 6.6. After

shaking, the reaction mixtures were incubated at 37°C for 150 min. Reactions were stopped by
loading the samples on silica gel 60 columns (Merck) to separate excess substrates from products.
After washing the columns with water, gangliosides were eluted with C/M (1:1, v/v), finally
separated by TLC (Merck) and reaction products were revealed by autoradiography. Enzyme
activities were expressed as pmoles of product formed/hour/mg proteins.
Treatment with exogenous gangliosides. To evaluate the effect of exogenous gangliosides on
BRP and RMC proliferation, cells were cultured in 96 wells plates. Exogenous glycolipids GM3,
GM2, GM1, GD1a, glucosylceramide and lactosylceramide (Matreya) were added to the culture
complete medium at a final concentration of 50 µM in the form of complexes with BSA at a ratio of
1:1 in DMEM–10 mM Hepes, pH 7.4. At the end of the treatment, cells were washed twice with
PBS and lysed for 30 min at 37°C in 50 µl Ripa lysis buffer (PBS 10 mM, NP40 1% (Pierce, Perbio
Science, Brebières, France), sodium deoxycholate 0.5%, SDS 0.1%, 10 µl/ml proteases inhibitors).
Total proteins were finally measured using the BCA protein assay (Pierce) to assess cell
proliferation, as in our experiments cell number correlated with total protein concentrations (see
Fig. 1).
Treatment with anti-a-series ganglioside antibodies. In order to block the potential effects of a-
series gangliosides, cells were cultured in 96 wells plates and treated with 3 µM of AGE or control
BSA in the presence or absence of 50 µg/ml of polyclonal anti–GM2 (Calbiochem) or anti–GM1
(Matreya) antibodies. Rabbit IgG (Sigma) was used as a control. At the end of the treatment, cells
were washed twice with PBS, lysed in 50 µl Ripa lysis buffer and proteins were measured to assess
cell proliferation.
Transfection of RMC with GM3 synthase SiRNA. In order to block GM3 synthase activity,
RMC were grown in 6-wells plates to 30 % confluence and transfected with specific GM3 synthase
SiRNA designed against rat cDNA sequence (Ambion, Huntingdon, UK) using Oligofectamine
reagent (Invitrogen Corporation). The GM3 synthase antisense sequence used was
GGGUUAUUCUGAACAUGUUtt. In preliminary experiments, dose-response study was
performed by transfecting cells with increasing concentrations of SiRNA (0-800 nM). After 72 h of
transfection, GM3 synthase activity was measured on homogenates of transfected cells.
Proliferation was next assayed in SiRNA transfected cells. To this end, 24 h after transfection with
400 nM SiRNA, cells were treated with 3 µM of either control BSA or AGE (3 days). Cells were
then washed twice with PBS, lysed in Ripa lysis buffer and proteins were measured to assess cell
proliferation.

Statistical analysis. Data are expressed as means ± SEM and presented as percentage of controls.
In cell studies, the Wilcoxon signed-rank test was used to define the significance of the difference
between groups. For GM3 and GD3 synthase activity in mice experiments, the Student t-test was
used. p< 0.05 was considered as statistically significant.

RESULTS AGE inhibit pericyte and mesangial cell proliferation. To compare the effects of AGE on
pericyte and mesangial cell proliferation, cells were treated with either BSA or AGE 3 µM for 4-7
days. Cell counting revealed that AGE reduced pericyte and mesangial cell numbers by 33% and
40%, respectively (Fig. 1). Total proteins were also measured, found diminished, and correlated to
cell number. The results show similar adverse effects of AGE on both pericyte and mesangial cell
proliferation, and led us to investigate common mechanisms involved in the AGE response in these
two cell types.
AGE increase the a-series gangliosides in pericytes and mesangial cells. Previous results from
our laboratory suggested that AGE modulate gangliosides profile of retinal microvascular cells (17).
To this end, ganglioside patterns were analyzed in BRP and RMC in response to BSA control or
AGE. As shown in Figure 2A, ganglioside profile is cell type specific. Under control conditions,
main gangliosides in pericytes were GM3 (63% of all gangliosides detected) and GM1 (9%) of the
a-series gangliosides (Fig. 2B), and GD3 (28%) of the b-series gangliosides. Mesangial cell profile
differed by the very low amount of GD3 (5%), and in the a-series, by the presence of GD1a (20%),
with GM3 remaining the major ganglioside (75%).
An increase of the a-series gangliosides and a decrease of the b-series gangliosides were observed
in both AGE-treated cell types. In pericytes, the a-series gangliosides GM3 and GM1 were
increased by about 40% whereas the b-series ganglioside GD3 was decreased by 24% (Fig. 3A). In
mesangial cells, GM3 was increased by 33% whereas GD3 was decreased by 30%; GD1a levels
were not affected (Fig. 3B). Similar results were obtained by autoradiography analysis after
galactose labelling (data not shown). As the a-series GM2 in BRP, GM2 and GM1 in RMC were
hardly detectable by resorcinol staining, cells were labeled with [14C]D-Galactose at high specific
activity and gangliosides were then analyzed in control and AGE treated cells. Results revealed that
GM2 was also increased by 55% in BRP (Fig 3C) and that GM2 and GM1 were increased by 25-
35% in RMC (Fig 3D). These results indicate that AGE induce similar gangliosides pattern
modifications in both pericytes and mesangial cells. They also suggest that the increase of the a-
series gangliosides might be a common mechanism underlying the decrease of cell proliferation.
AGE modulate GM3 and GD3 synthase activities in pericytes and mesangial cells. To
investigate the mechanism responsible for the observed a-series ganglioside increase, GM3 and
GD3 synthase activities were measured in control and treated cells. Results presented in Figure 4A
show that AGE treatment increased GM3 synthase activity by about 80% in pericytes and 50% in

mesangial cells, most likely through increase of Vmax of the reaction (data not shown). In parallel,
AGE treatment decreased GD3 synthase activity by about 60% in both cell types (Fig. 4B). These
results suggest that AGE exert similar mechanisms in RMC and BRP leading to a-series
gangliosides accumulation.
A-series gangliosides inhibit pericyte and mesangial cell proliferation. Previous literature data
have described gangliosides as growth suppressors notably in mesangial cells (18) and implicated
the a-series gangliosides in inhibition of cell proliferation (18-23). Thus, we investigated whether
exogenous addition of the a-series gangliosides affects pericyte and mesangial cell proliferation.
Cells were treated during one passage with 50 µM gangliosides and, as a control, with the non-
sialylated precursors glucosylceramide and lactosylceramide. Figure 5A shows that GM2, GM1 and
GD1a inhibited most efficiently pericyte and mesangial cell proliferation by 15-30%. GM3 slightly
inhibited pericyte proliferation but had no significant effect on mesangial cells. Glucosylceramide
and lactosylceramide used as controls had no effect. Because GM2 and GM1 are increased in BRP
and RMC in response to AGE, and as they alter proliferation of both cell types, we hypothesized
that these a-series gangliosides might be common mediators of the AGE effect. Thus, cells were
treated with AGE in the presence of anti-GM1 and anti-GM2 antibodies and proliferation measured.
In the control BSA-treated cells, proliferation was not different in the presence or absence of anti-
gangliosides antibodies (data not shown), and addition of IgG as a control to the AGE treatment did
not affect the AGE response. Treatment of cells with AGE in the presence of anti-GM2 and anti-
GM1 antibodies partially prevented the proliferation decrease of pericytes and mesangial cells (Fig.
5B). Together these observations suggest that the a-series gangliosides, and in particular GM2 and
GM1, are common mediators of the AGE-induced inhibition of proliferation of pericytes and
mesangial cells.
GM3 synthase SiRNA partially protects against the AGE effects. To further support the role of
a-series gangliosides in mediating the AGE effects, RMC were transfected with GM3 synthase
SiRNA. Preliminary experiments showed that the designed SiRNA indeed inhibited GM3 synthase
activity in RMC (Fig. 6A). Next, proliferation was measured in the transfected and treated cells. As
shown in Figure 6B, the effects of AGE on RMC proliferation were partially reversed in cells
transfected with GM3 synthase SiRNA. These results clearly show the implication of gangliosides
in mediating the AGE effects. It must be indicated that the partial effects could be explained by: (i)
the AGE effects were less potent than in previous experiments as cells were treated at higher
confluency in order to improve transfection efficiency and (ii) GM3 synthase activity was inhibited
only by about 50%. In fact, SiRNA concentrations higher than 400nM were not used to avoid non-

specific and toxic effects.
GM3 and GD3 synthase activities, and GM3 levels are modified in renal cortex of diabetic
mice. To assess the effect of a diabetic environment on ganglioside biosynthetic pathway ex vivo,
GM3 and GD3 synthase activities were measured on homogenates of renal cortex of the diabetic
db/db mice model. Results presented in Figure 7A-B show that GM3 synthase activity was
increased by 80% whereas GD3 synthase activity was decreased by 50% in diabetic mice renal
cortex as compared to controls (db/m). Gangliosides were also analyzed and revealed an increase,
although not statistically significant, of GM3 levels in the renal cortex of diabetic db/db mice
(Figure 7C). Altogether these results support the pathophysiological significance of the results
obtained in RMC and BRP treated with AGE.

DISCUSSION
Numerous studies have pointed out the crucial role of pericytes and mesangial cells in the
development of diabetic retinopathy (DR) and nephropathy (DN), respectively. Indeed, retinal
pericytes loss is one of the earliest hallmarks of DR, and growth-arrest, hypertrophy, and loss of
renal mesangial cells are typical features of DN. Thus, regulation of these two cell type proliferation
appears to be a pivotal pathophysiological process in microvascular complications. AGE, glycated
protein derivatives that notably accumulate in kidneys (24) and retina (25) of diabetic patients, have
been widely involved in the development of microvascular complications (5). In the present study,
we investigated the effect of AGE on BRP and RMC proliferation. Results showed that AGE
treatment indeed inhibited both cell types proliferation. These findings are consistent with previous
results obtained with other types of AGE in pericytes (26,27) and mesangial cells (28,29), thus
highlighting their pathophysiological relevance.
The effects of AGE have been extensively studied. However, mechanisms leading to alteration of
cell function and viability remain incompletely understood. On the other hand, although
gangliosides play a major role in cell growth and apoptosis, their potential involvement in diabetic
microvascular complications has been poorly studied. In the present work, we show that treatment
of BRP and RMC with AGE resulted in an increase of the a-series gangliosides concomitant with a
decrease of the b-series in both cell types. This increase could be explained by the observed
concomitant activation of GM3 synthase and inhibition of GD3 synthase activities, suggesting that
AGE regulate activity of these enzymes through yet undefined mechanisms.
On the other hand, evidence that the a-series gangliosides are likely to be responsible for the
observed proliferation decrease has been provided in the present study. Exogenous a-series
gangliosides application, particularly GM2 and GM1, inhibited BRP and RMC proliferation.
Moreover, treatment of cells with AGE in the presence of anti-GM1 or anti-GM2 partially protected
BRP and RMC against the proliferation decrease caused by AGE. Finally, inhibition of GM3
synthase using specific SiRNA partially reversed the AGE effects on RMC proliferation. Together,
these results suggest that the a-series gangliosides, and in particular GM2 and GM1, are involved in
mediating the anti-proliferative effect of AGE on BRP and RMC. The partial protection observed
using anti-GM2 and anti-GM1 antibodies or SiRNA approach might suggest that other pathways
are also involved in mediating the AGE effect. For example, a cascade involving oxidative stress
and ceramide accumulation has been proposed to induce BRP apoptosis in response to AGE (16).
The role of the a-series gangliosides is further supported by numerous studies showing that a-series
gangliosides decrease cell proliferation by modulating growth factor receptors signaling. For
example, a-series gangliosides (GM3, GM2, GM1, GD1a) have been described to inhibit epidermal

growth factor and platelet-derived growth factor receptor phosphorylation and signaling in different
cell types (19-22,30-32).
The potential role of gangliosides in diabetic microvascular complications is also supported by ex
vivo observations. Indeed, the present results show that GM3 and GD3 synthase activities are
increased and decreased, respectively, and that GM3 tends to accumulate in renal cortex of diabetic
db/db mice. Moreover, GM3 has been shown to accumulate in kidney of streptozotocin-induced
diabetic rats, and kidney hypertrophy was inhibited when ganglioside biosynthesis was blocked,
suggesting a crucial role of gangliosides in diabetes-induced renal hypertrophy (33).
On the other hand, recent studies have suggested the involvement of GM3 synthase and a-series
gangliosides in insulin resistance. Thus, GM3 synthase activity and its mRNA have been shown to
be increased in adipocytes treated with low concentrations of tumor necrosis factor α, and GM3
synthase mRNA levels were found to be increased in adipose tissues of Zucker fa/fa rats (34).
Furthermore, GM3 synthase knock-out mice showed increased sensitivity to insulin by enhanced
insulin receptor phosphorylation and mice were protected against high fat diet-induced insulin
resistance (35). Finally, mice overexpressing the human sialidase NEU3 showed reduced insulin-
stimulated phosphorylation of the insulin receptor and the insulin receptor substrate, suggesting that
GM2 and GM1, the possible sialidase products in transgenic tissues, attenuate insulin signaling
(36).
In conclusion, our data suggest that the a-series gangliosides, and in particular GM2 and GM1, are
mediators of the adverse AGE effects on pericyte and mesangial cell proliferation. Further,
gangliosides pathway appears to be a new and common AGE mechanism of action in both BRP and
RMC. These data also raise GM3 synthase and a-series gangliosides as potential targets for the
treatment of both DR and DN.

Reference List
1. Fong DS, Aiello L, Gardner TW, King GL, Blankenship G, Cavallerano JD, Ferris FL, III,
Klein R: Diabetic retinopathy. Diabetes Care 26:226-229, 2003
2. Forrester J.V., Knott R.M.: Pathogenesis of diabetic retinopathy and cataract. In Textbook of diabetes. Pickup J., Williams G, Eds. Oxford, 1997, p. 45.1-45.19
3. Krolewski AS, Warram JH: Clinical features and epidemiology of diabetic nephropathy. In Textbook of diabetes. Pickup J, Williams G, Eds. Oxford, 1997, p. 53.1-53.13
4. Wolf G: Cell cycle regulation in diabetic nephropathy. Kidney Int.Suppl 77:S59-S66, 2000
5. Singh R, Barden A, Mori T, Beilin L: Advanced glycation end-products: a review. Diabetologia 44:129-146, 2001
6. Lal MA, Brismar H, Eklof AC, Aperia A: Role of oxidative stress in advanced glycation end product-induced mesangial cell activation. Kidney Int. 61:2006-2014, 2002
7. Schmidt AM, Hori O, Brett J, Yan SD, Wautier JL, Stern D: Cellular receptors for advanced glycation end products. Implications for induction of oxidant stress and cellular dysfunction in the pathogenesis of vascular lesions. Arterioscler.Thromb. 14:1521-1528, 1994
8. Hofmann MA, Drury S, Fu C, Qu W, Taguchi A, Lu Y, Avila C, Kambham N, Bierhaus A, Nawroth P, Neurath MF, Slattery T, Beach D, McClary J, Nagashima M, Morser J, Stern D, Schmidt AM: RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell 97:889-901, 1999
9. Schmidt AM, Hori O, Chen JX, Li JF, Crandall J, Zhang J, Cao R, Yan SD, Brett J, Stern D: Advanced glycation endproducts interacting with their endothelial receptor induce expression of vascular cell adhesion molecule-1 (VCAM-1) in cultured human endothelial cells and in mice. A potential mechanism for the accelerated vasculopathy of diabetes. J.Clin.Invest 96:1395-1403, 1995
10. Stitt AW: The role of advanced glycation in the pathogenesis of diabetic retinopathy. Exp.Mol.Pathol. 75:95-108, 2003
11. Wautier JL, Guillausseau PJ: Advanced glycation end products, their receptors and diabetic angiopathy. Diabetes Metab 27:535-542, 2001
12. van Echten G, Sandhoff K: Ganglioside metabolism. Enzymology, Topology, and regulation. J.Biol.Chem. 268:5341-5344, 1993

13. Hakomori S: Bifunctional role of glycosphingolipids. Modulators for transmembrane signaling and mediators for cellular interactions. J.Biol.Chem. 265:18713-18716, 1990
14. Lecomte M, Paget C, Ruggiero D, Wiernsperger N, Lagarde M: Docosahexaenoic acid is a major n-3 polyunsaturated fatty acid in bovine retinal microvessels. J.Neurochem. 66:2160-2167, 1996
15. Geoffroy K, Wiernsperger N, Lagarde M, El Bawab S: Bimodal effect of advanced glycation end products on mesangial cell proliferation is mediated by neutral ceramidase regulation and endogenous sphingolipids. J.Biol.Chem. 279:34343-34352, 2004
16. Denis U, Lecomte M, Paget C, Ruggiero D, Wiernsperger N, Lagarde M: Advanced glycation end-products induce apoptosis of bovine retinal pericytes in culture: involvement of diacylglycerol/ceramide production and oxidative stress induction. Free Radic.Biol.Med. 33:236-247, 2002
17. Natalizio A, Ruggiero D, Lecomte M, Lagarde M, Wiernsperger N: Glycosphingolipid changes induced by advanced glycation end-products. Biochem.Biophys.Res.Commun. 281:78-83, 2001
18. Tsuboi N, Utsunomiya Y, Kawamura T, Kawano T, Hosoya T, Ohno T, Yamada H: Ganglioside as an endogenous growth suppressor for glomerular mesangial cells. Kidney Int. 60:1378-1385, 2001
19. Bremer EG, Hakomori S, Bowen-Pope DF, Raines E, Ross R: Ganglioside-mediated modulation of cell growth, growth factor binding, and receptor phosphorylation. J.Biol.Chem. 259:6818-6825, 1984
20. Bremer EG, Schlessinger J, Hakomori S: Ganglioside-mediated modulation of cell growth. Specific effects of GM3 on tyrosine phosphorylation of the epidermal growth factor receptor. J.Biol.Chem. 261:2434-2440, 1986
21. Hynds DL, Summers M, Van Brocklyn J, O'Dorisio MS, Yates AJ: Gangliosides inhibit platelet-derived growth factor-stimulated growth, receptor phosphorylation, and dimerization in neuroblastoma SH-SY5Y cells. J.Neurochem. 65:2251-2258, 1995
22. Mirkin BL, Clark SH, Zhang C: Inhibition of human neuroblastoma cell proliferation and EGF receptor phosphorylation by gangliosides GM1, GM3, GD1A and GT1B. Cell Prolif. 35:105-115, 2002
23. Slevin M, Kumar S, He X, Gaffney J: Physiological concentrations of gangliosides GM1, GM2 and GM3 differentially modify basic-fibroblast-growth-factor-induced mitogenesis and the associated signalling pathway in endothelial cells. Int.J.Cancer 82:412-423, 1999

24. Tanji N, Markowitz GS, Fu C, Kislinger T, Taguchi A, Pischetsrieder M, Stern D, Schmidt AM, D'Agati VD: Expression of advanced glycation end products and their cellular receptor RAGE in diabetic nephropathy and nondiabetic renal disease. J.Am.Soc.Nephrol. 11:1656-1666, 2000
25. Hammes HP, Alt A, Niwa T, Clausen JT, Bretzel RG, Brownlee M, Schleicher ED: Differential accumulation of advanced glycation end products in the course of diabetic retinopathy. Diabetologia 42:728-736, 1999
26. Chibber R, Molinatti PA, Rosatto N, Lambourne B, Kohner EM: Toxic action of advanced glycation end products on cultured retinal capillary pericytes and endothelial cells: relevance to diabetic retinopathy. Diabetologia 40:156-164, 1997
27. Yamagishi S, Amano S, Inagaki Y, Okamoto T, Koga K, Sasaki N, Yamamoto H, Takeuchi M, Makita Z: Advanced glycation end products-induced apoptosis and overexpression of vascular endothelial growth factor in bovine retinal pericytes. Biochem.Biophys.Res.Commun. 290:973-978, 2002
28. Yamagishi S, Inagaki Y, Okamoto T, Amano S, Koga K, Takeuchi M, Makita Z: Advanced glycation end product-induced apoptosis and overexpression of vascular endothelial growth factor and monocyte chemoattractant protein-1 in human-cultured mesangial cells. J.Biol.Chem. 277:20309-20315, 2002
29. Ziyadeh FN, Cohen MP: Effects of glycated albumin on mesangial cells: evidence for a role in diabetic nephropathy. Mol.Cell Biochem. 125:19-25, 1993
30. Meuillet EJ, Kroes R, Yamamoto H, Warner TG, Ferrari J, Mania-Farnell B, George D, Rebbaa A, Moskal JR, Bremer EG: Sialidase gene transfection enhances epidermal growth factor receptor activity in an epidermoid carcinoma cell line, A431. Cancer Res. 59:234-240, 1999
31. Mitsuda T, Furukawa K, Fukumoto S, Miyazaki H, Urano T, Furukawa K: Overexpression of ganglioside GM1 results in the dispersion of platelet-derived growth factor receptor from glycolipid-enriched microdomains and in the suppression of cell growth signals. J.Biol.Chem. 277:11239-11246, 2002
32. Yates AJ, Saqr HE, Van Brocklyn J: Ganglioside modulation of the PDGF receptor. A model for ganglioside functions. J.Neurooncol. 24:65-73, 1995
33. Zador IZ, Deshmukh GD, Kunkel R, Johnson K, Radin NS, Shayman JA: A role for glycosphingolipid accumulation in the renal hypertrophy of streptozotocin-induced diabetes mellitus. J.Clin.Invest 91:797-803, 1993

34. Tagami S, Inokuchi JJ, Kabayama K, Yoshimura H, Kitamura F, Uemura S, Ogawa C, Ishii A, Saito M, Ohtsuka Y, Sakaue S, Igarashi Y: Ganglioside GM3 participates in the pathological conditions of insulin resistance. J.Biol.Chem. 277:3085-3092, 2002
35. Yamashita T, Hashiramoto A, Haluzik M, Mizukami H, Beck S, Norton A, Kono M, Tsuji S, Daniotti JL, Werth N, Sandhoff R, Sandhoff K, Proia RL: Enhanced insulin sensitivity in mice lacking ganglioside GM3. Proc.Natl.Acad.Sci.U.S.A 100:3445-3449, 2003
36. Sasaki A, Hata K, Suzuki S, Sawada M, Wada T, Yamaguchi K, Obinata M, Tateno H, Suzuki H, Miyagi T: Overexpression of plasma membrane-associated sialidase attenuates insulin signaling in transgenic mice. J.Biol.Chem. 278:27896-27902, 2003

Legends :
Fig. 1. AGE inhibit pericyte and renal mesangial cell proliferation. Cells were exposed to 3 µM BSA or AGE for 4 (RMC) and 7 (BRP) days. Cells were then trypsinized and counted using haemocytometer and total proteins were measured. Results are presented as percentage of control BSA and represent means ± SEM of 6 (BRP) and 9 (RMC) independent experiments each performed in duplicates. * p < 0.05 vs control BSA. Fig. 2. Gangliosides pattern of pericytes and mesangial cells. (A) Gangliosides were extracted, separated by HPTLC, then detected by resorcinol staining and quantified. GT1b, absent in both cell types, was used as internal standard. (B) Scheme representing gangliosides biosynthesis pathway, modified from (12). Cer, ceramide; Glccer, glucosylceramide; Laccer, lactosylceramide. Fig. 3. AGE modulate gangliosides pattern in pericytes and mesangial cells. Pericytes (A) or mesangial cells (B) were exposed to 3 µM BSA or AGE for 7 and 4 days, respectively. Gangliosides were analyzed by HPTLC and revealed by resorcinol spraying as described under Research designs and Methods. In control conditions, GM3, GM1 and GD3 quantities in BRP were 2.39 ± 0.19, 0.34 ± 0.03 and 1.05 ± 0.11 µg/mg proteins, respectively. GM3, GD3 and GD1a quantities in RMC were 4.03 ± 0.28, 0.27 ± 0.07 and 1.08 ± 0.11 µg/mg proteins, respectively. Results are expressed as percentage of control BSA and represent means ± SEM of 6 (BRP) and 9 (RMC) independent experiments each performed in duplicates. * p<0.05 vs control BSA. (C) and (D) Gangliosides were metabolically labelled with 1 µCi/ml [14C]Galactose and analyzed by autoradiography. Results are expressed as percentage of control BSA and represent means ± SEM of 3 independent experiments.
Fig. 4. AGE increase GM3 synthase activity and decrease GD3 synthase activity in pericytes and mesangial cells. Cells were treated with 3 µM BSA or AGE. GM3 (A) and GD3 (B) synthase activities were measured on cell homogenates as described under Research design and Methods. Control activities were 2.7 and 0.8 pmoles/hour/mg proteins for GM3 and GD3 synthase, respectively, in BRP; 5.1 and 0.7 pmoles/hour/mg proteins for GM3 and GD3 synthase, respectively, in RMC. Results are expressed as percentage of control BSA and represent means ± SEM of 4-5 independent experiments. * p<0.05 vs control BSA. Fig. 5. A-series gangliosides inhibit cell proliferation. (A) Pericytes or mesangial cells were treated with gangliosides-BSA complexes for one passage. (B) Cells were treated with 3 µM BSA or AGE in the presence or absence of 5µg per well of IgG, anti-GM2 or anti-GM1 polyclonal antibodies. At the end of the treatment, total proteins were measured. Results are expressed as percentage of control and represent means ± SEM of 5-6 independent experiments each performed in triplicates. * p<0.05 vs control (A) or AGE treated cells (B).
Fig. 6. Transfection with GM3 synthase SiRNA protects RMC.

(A) TLC separation of GM3 product obtained from GM3 synthase activity in homogenates of RMC. Mesangial cells were transfected with increasing concentrations of GM3 synthase SiRNA and GM3 synthase activity was measured after 72 h. Ctr, cells transfected with oligofectamine without SiRNA. (B) 24 h after transfection with 400 nM GM3 synthase SiRNA, cells were treated with 3 µM BSA control or AGE. At the end of treatment, total proteins were measured. Results are expressed as percentage of control BSA and represent means ± SEM of 6 independent experiments. * p<0.05 vs AGE treated control cells.
Fig. 7. GM3 and GD3 synthase activities, and GM3 levels are modulated in renal cortex of diabetic mice. GM3 (A) and GD3 (B) synthase activities were measured on homogenates of renal cortex from control (db/m) and db/db mice. Control activities were 0.6 and 21 pmoles/hour/mg proteins for GM3 and GD3 synthase, respectively. (C) GM3 levels of control (db/m) and db/db mice renal cortex are represented. In control mice, GM3 levels were 66 ± 9 ng/mg proteins. Results are expressed as percentage of control and represent means ± SEM of 4-6 animals. * p<0.05 vs control mice.
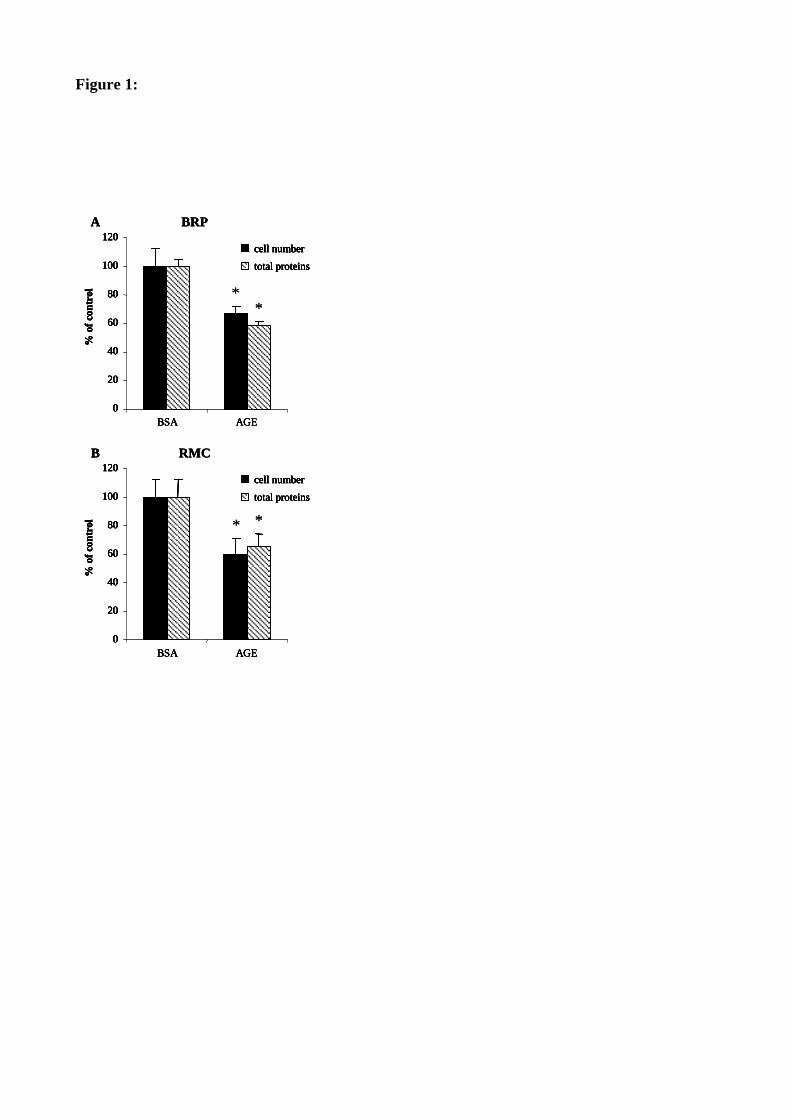
Figure 1:
0
20
40
60
80
100
120
% o
f con
trol
cell number
total proteins
A BRP
B RMC
0
20
40
60
80
100
120
% o
f con
trol
BSA AGE
BSA AGE
cell number
total proteins
**
* *
0
20
40
60
80
100
120
% o
f con
trol
cell number
total proteins
A BRP
B RMC
0
20
40
60
80
100
120
% o
f con
trol
BSA AGE
BSA AGE
cell number
total proteins
0
20
40
60
80
100
120
% o
f con
trol
0
20
40
60
80
100
120
0
20
40
60
80
100
120
% o
f con
trol
cell number
total proteins
cell numbercell number
total proteinstotal proteins
A BRP
B RMC
0
20
40
60
80
100
120
% o
f con
trol
0
20
40
60
80
100
120
0
20
40
60
80
100
120
% o
f con
trol
BSA AGEBSA AGE
BSA AGEBSA AGE
cell number
total proteins
cell numbercell number
total proteinstotal proteins
**
* *
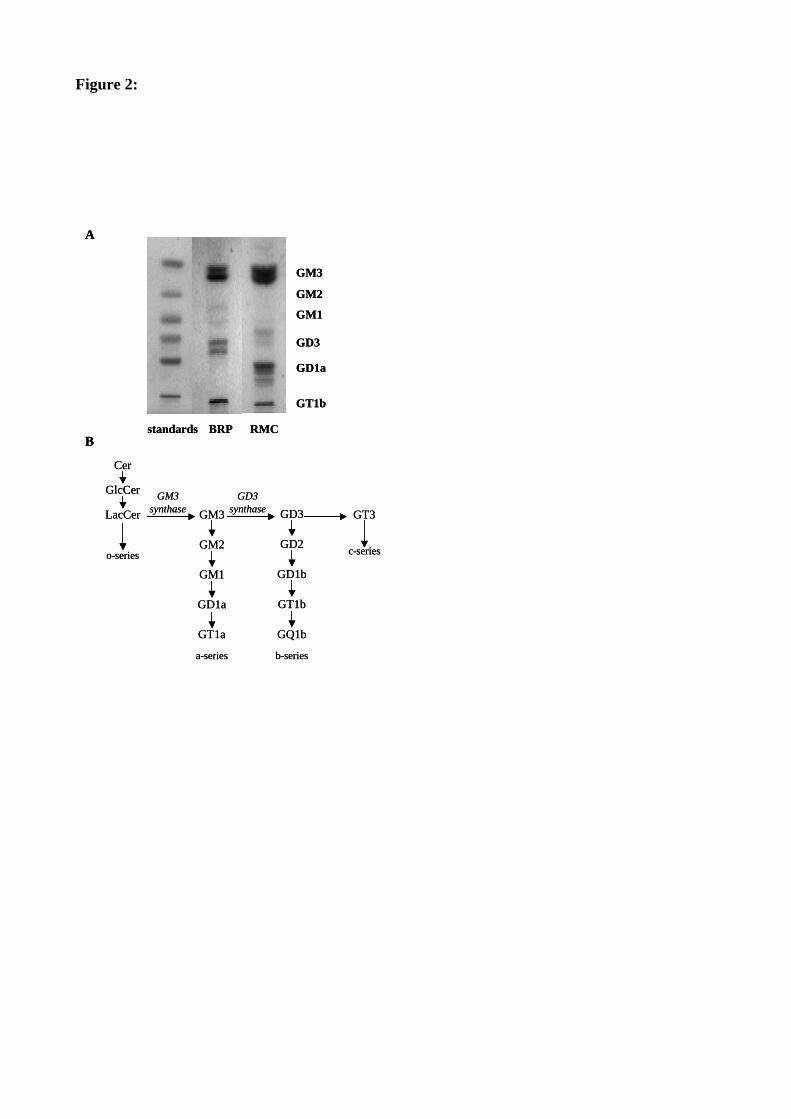
Figure 2:
GM3
GT1b
GD3
GM2
GM1
GD1a
BRP RMCstandards
A
Cer
GlcCer
LacCer
GT1a
GD2
GD1b
GT1b
GQ1b
GT3
a-series
GM3 synthase
c-series
b-series
o-series
GM3
GM1
GM2
GD3
GD1a
B
GD3 synthase
GM3
GT1b
GD3
GM2
GM1
GD1a
BRP RMCstandards
A
Cer
GlcCer
LacCer
GT1a
GD2
GD1b
GT1b
GQ1b
GT3
a-series
GM3 synthase
c-series
b-series
o-series
GM3
GM1
GM2
GD3
GD1a
B
GD3 synthase
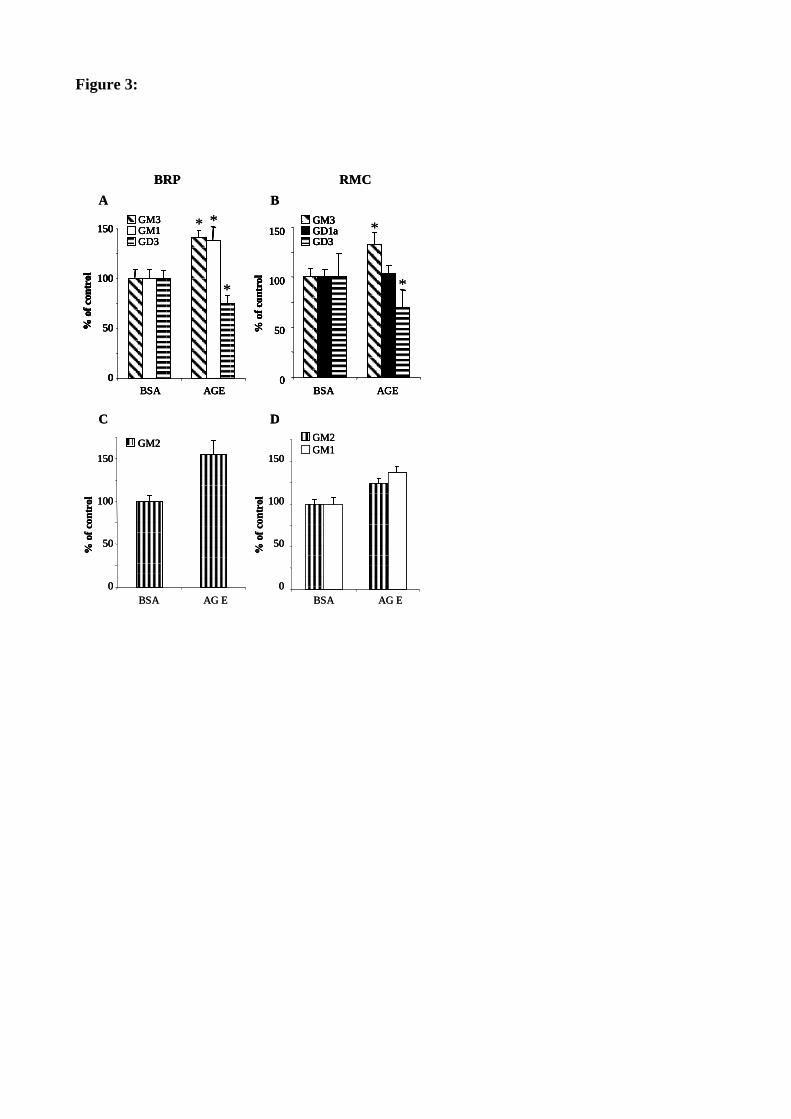
Figure 3:
BSA AGE0
50
100
150
% o
f con
trol
GM3GM1GD3
A B
C D
BRP RMC
GM3GD1aGD3
0
50
100
150
% o
f con
trol
BSA AGE
BSA AG E0
50
100
150
% o
f con
trol
GM2 GM2GM1
* *
*
*
*
BSA AG E0
50
100
150
% o
f con
trol
BSA AGE0
50
100
150
% o
f con
trol
GM3GM1GD3
BSA AGEBSA AGE0
50
100
150
% o
f con
trol
0
50
100
150
% o
f con
trol
0
50
100
150
% o
f con
trol
GM3GM1GD3
GM3GM1GD3
A B
C D
BRP RMC
GM3GD1aGD3
0
50
100
150
% o
f con
trol
BSA AGE
GM3GD1aGD3
GM3GM3GD1aGD1aGD3GD3
0
50
100
150
% o
f con
trol
0
50
100
150
% o
f con
trol
0
50
100
150
% o
f con
trol
BSA AGEBSA AGE
BSA AG E0
50
100
150
% o
f con
trol
0
50
100
150
% o
f con
trol
GM2GM2 GM2GM1GM2GM1
* *
*
*
*
BSA AG E0
50
100
150
% o
f con
trol
0
50
100
150
% o
f con
trol
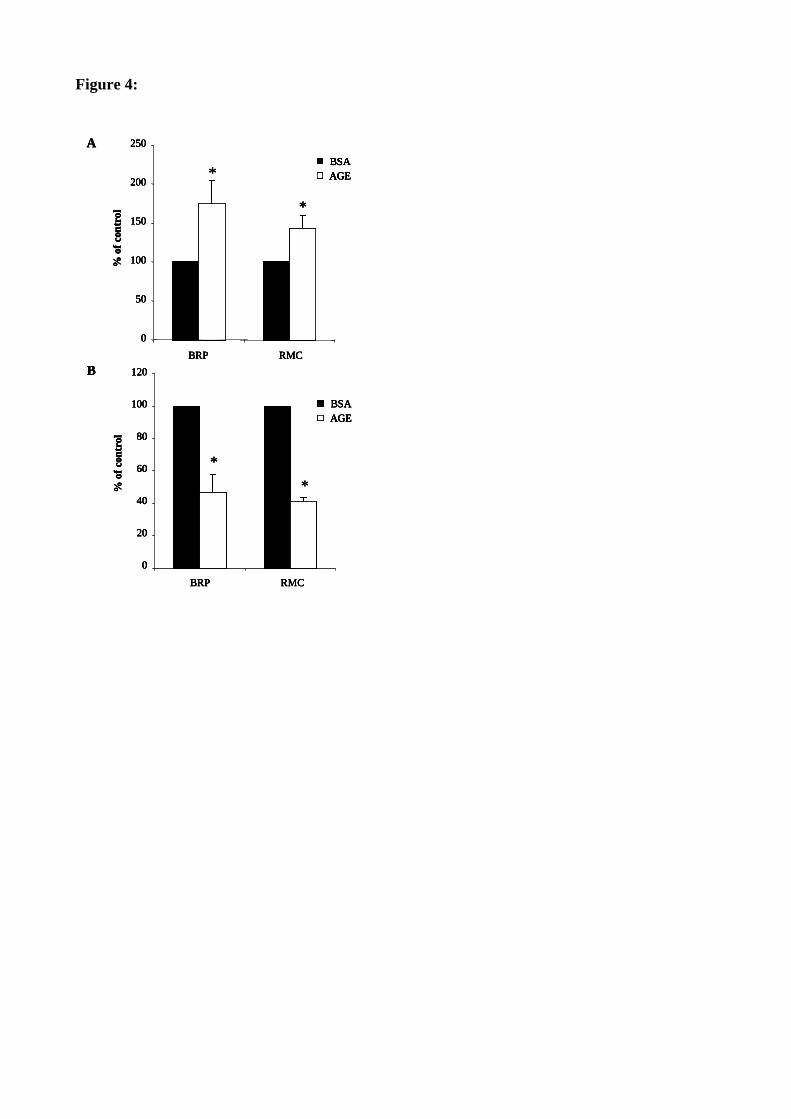
Figure 4:
0
50
100
150
200
250
BRP RMC
% o
f con
trol
BSAAGE
*
*
A
B
0
20
40
60
80
100
120
BRP RMC
% o
f con
trol
BSAAGE
**
0
50
100
150
200
250
BRP RMC
% o
f con
trol
BSAAGE
*
*
A
0
50
100
150
200
250
BRP RMC
% o
f con
trol
BSAAGE
*
*
0
50
100
150
200
250
BRP RMC
% o
f con
trol
BSAAGEBSAAGE
*
*
A
B
0
20
40
60
80
100
120
BRP RMC
% o
f con
trol
BSAAGE
**
B
0
20
40
60
80
100
120
BRP RMC
% o
f con
trol
BSAAGE
0
20
40
60
80
100
120
BRP RMC
% o
f con
trol
BSAAGEBSAAGE
**
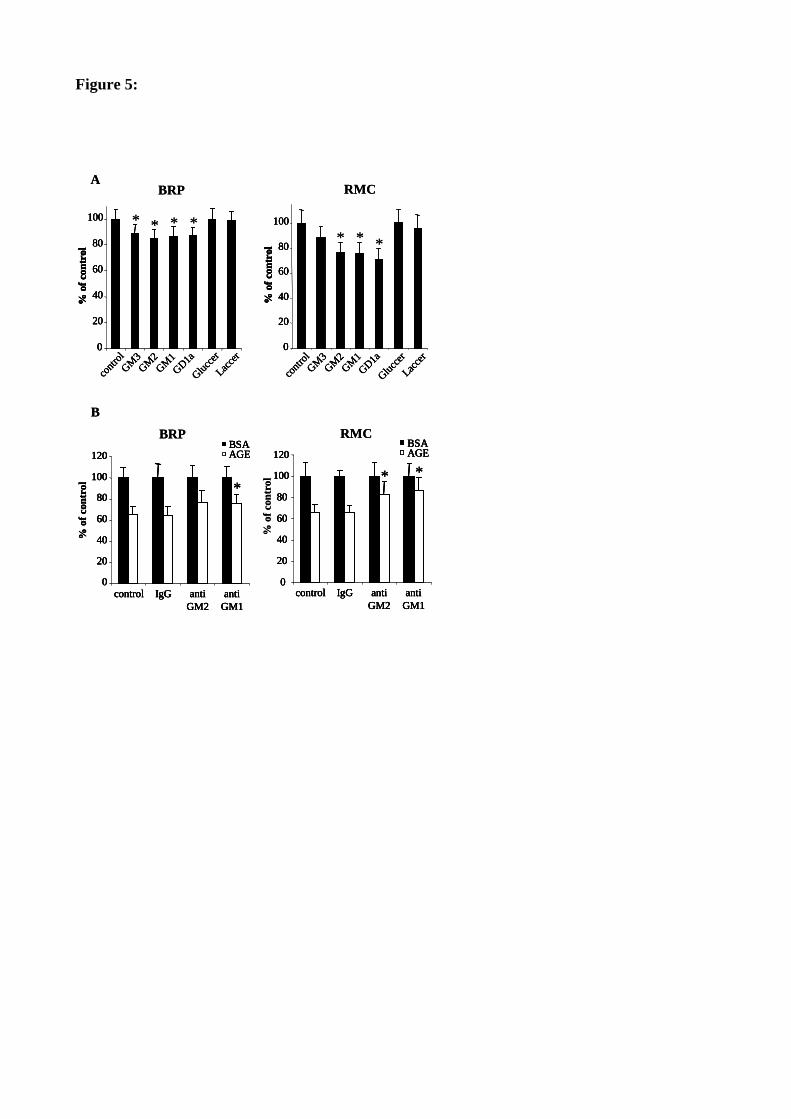
Figure 5:
A
B
BRP
contr
olGM3
GM2GM1
GD1a
Gluccer
Laccer
0
20
40
60
80
100
% o
f con
trol
* * * *
RMC
contr
olGM3
GM2GM1
GD1a
Gluccer
Laccer
0
20
40
60
80
100
% o
f con
trol
* * *
BRP
0
20
40
60
80
100
120
% o
f con
trol
BSAAGE
control anti GM2
anti GM1
IgG
*
RMC
% o
f con
trol
0
20
40
60
80
100
120
* *
BSAAGE
control anti GM2
anti GM1
IgG
A
B
BRP
contr
olGM3
GM2GM1
GD1a
Gluccer
Laccer
0
20
40
60
80
100
% o
f con
trol
contr
olGM3
GM2GM1
GD1a
Gluccer
Laccer
0
20
40
60
80
100
% o
f con
trol
0
20
40
60
80
100
% o
f con
trol
* * * *
RMC
contr
olGM3
GM2GM1
GD1a
Gluccer
Laccer
0
20
40
60
80
100
% o
f con
trol
contr
olGM3
GM2GM1
GD1a
Gluccer
Laccer
0
20
40
60
80
100
% o
f con
trol
0
20
40
60
80
100
% o
f con
trol
* * *
BRP
0
20
40
60
80
100
120
% o
f con
trol
BSAAGE
control anti GM2
anti GM1
IgG
*
0
20
40
60
80
100
120
0
20
40
60
80
100
120
% o
f con
trol
BSAAGEBSAAGE
control anti GM2
anti GM1
IgGcontrol anti GM2
anti GM1
IgG
*
RMC
% o
f con
trol
0
20
40
60
80
100
120
* *
BSAAGE
control anti GM2
anti GM1
IgG0
20
40
60
80
100
120
0
20
40
60
80
100
120
* *
BSAAGEBSAAGE
control anti GM2
anti GM1
IgGcontrol anti GM2
anti GM1
IgG
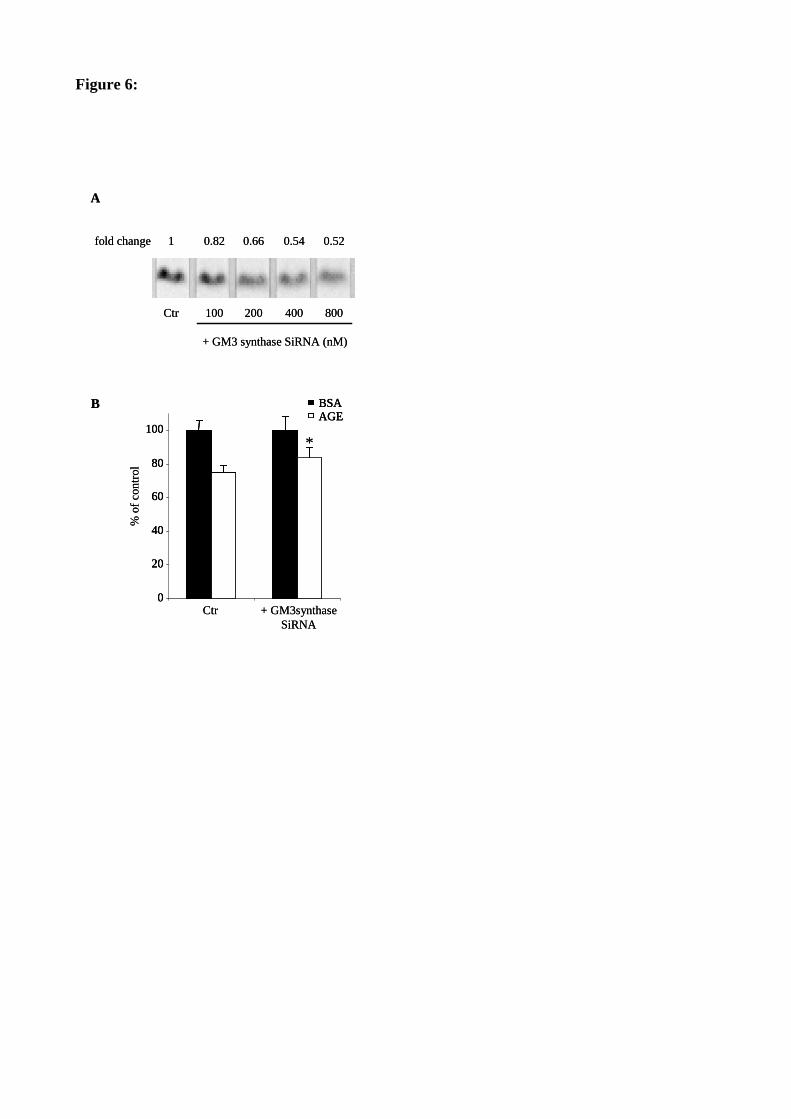
Figure 6:
A
B
Ctr + GM3synthaseSiRNA
*
% o
f con
trol
BSAAGE
0
20
40
60
80
100
+ GM3 synthase SiRNA (nM)
200 400 800Ctr 100
fold change 1 0.82 0.66 0.54 0.52
A
B
Ctr + GM3synthaseSiRNA
*
% o
f con
trol
BSAAGEBSAAGE
0
20
40
60
80
100
0
20
40
60
80
100
+ GM3 synthase SiRNA (nM)
200 400 800Ctr 100
fold change 1 0.82 0.66 0.54 0.52
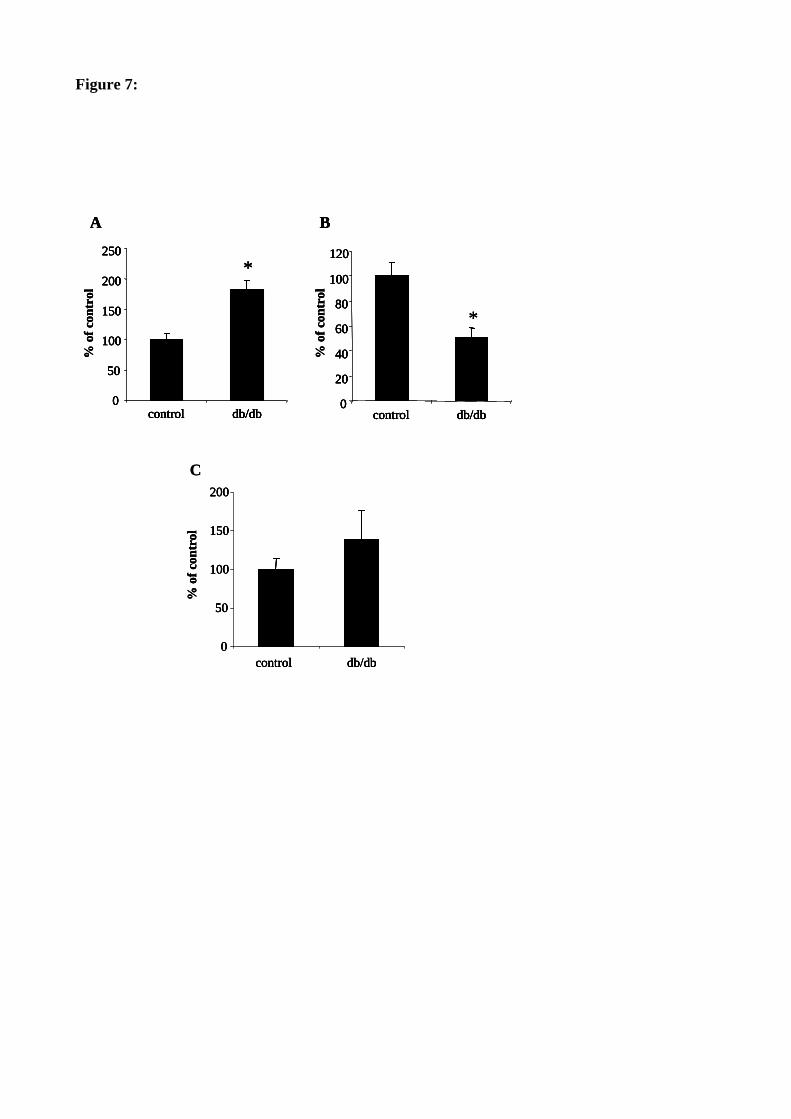
Figure 7:
control db/db0
50
100
150
200
250
% o
f con
trol
A
*%
of c
ontr
ol
0
20
40
60
80
100
120
control db/db
B
*
C
% o
f con
trol
control db/db0
50
100
150
200
control db/db0
50
100
150
200
250
% o
f con
trol
A
*%
of c
ontr
ol
0
20
40
60
80
100
120
control db/db
B
*
control db/db0
50
100
150
200
250
control db/dbcontrol db/db0
50
100
150
200
250
0
50
100
150
200
250
% o
f con
trol
A
*%
of c
ontr
ol
0
20
40
60
80
100
120
control db/db0
20
40
60
80
100
120
0
20
40
60
80
100
120
control db/dbcontrol db/db
B
*
C
% o
f con
trol
control db/db0
50
100
150
200
% o
f con
trol
control db/dbcontrol db/db0
50
100
150
200
0
50
100
150
200

Involvement of gangliosides in glucosamine-induced
proliferation decrease of retinal pericytes
GLYCOBIOLOGY ADVANCE ACCESS PUBLISHED DECEMBER 29, 2004
Elodie Masson, Nicolas Wiernsperger, Michel Lagarde, and Samer El Bawab
Diabetic Microangiopathy Research Unit, MERCK Santé-INSERM UMR 585, INSA-Lyon
Corresponding author: S. El Bawab, Diabetic Microangiopathy Research Unit, MERCK Santé-INSERM
U585, INSA-Lyon, Louis Pasteur Bldg, 69621 Villeurbanne Cedex, France.
Phone: (+33) 4 72 43 81 12
Fax: (+33) 4 72 43 81 13
E-mail: [email protected]
Keywords: diabetic retinopathy/gangliosides/hexosamine pathway/pericytes

Abstract The hexosamine pathway (HP) is a biochemical hypothesis recently proposed explaining cellular
alterations occurring during diabetic microvascular complications. Diabetic retinopathy is a
common microvascular complication of diabetes, and it is known that cell proliferation is severely
affected during the development of the disease. Particularly, early stages are characterized by death
of the retinal microvascular cells, pericytes. Gangliosides have often been described to regulate cell
growth, however very few studies focused on the potential role of gangliosides in diabetic
microvascular alterations. The aim of the present study was to investigate the effect of the HP
activation on pericyte proliferation and to determine the potential implication of gangliosides in this
process. Results indicate first that HP activation, mimicked by glucosamine treatment, decreased
pericyte proliferation. Second, glucosamine treatment induced a modification of gangliosides
pattern, particularly, GM1 and GD3 were significantly increased. Next, results showed that
exogenous addition of a-series gangliosides (GM3, GM2,GM1, GD1a) and b-series ganglioside
(GD3) caused a decrease of pericyte proliferation, whereas non-sialylated precursors
glucosylceramide and lactosylceramide were without effect. Further, when ganglioside biosynthesis
was blocked using PPMP, a glucosylceramide synthase inhibitor, the effects of glucosamine on
pericyte proliferation were partially reversed. Our results suggest that, in retinal pericytes,
gangliosides and particularly GM1 and GD3 that are increased in response to glucosamine, are
involved in the anti-proliferative effect of glucosamine. These observations also underlie the
potential involvement of gangliosides in a pathological context, such as diabetic microvascular
complications.

Introduction
Gangliosides are glycosphingolipids characterized by the presence of sialic acid in their
oligosaccharidic portion. After the initial glycosylation step of ceramide catalyzed by
glucosylceramide synthase, successive sialylations and glycosylations lead to a wide diversity of
gangliosides. They are concentrated in plasma membranes with a cell-specific pattern. They are
found in most tissues of the body and are particularly abundant in brain and nervous tissues (van
Echten et al., 1993). Gangliosides are known to exert a bifunctional role (Hakomori, 1990). On one
hand, they play major roles in cell-cell and cell-matrix recognition via interactions with adhesion
receptors such as integrins, matrix proteins (collagen and fibronectin), or with other
glycosphingolipids, giving them important role in morphogenesis and oncogenesis. In addition, a
number of gangliosides may act as receptors of bacteria, viruses or toxins. On the other hand,
gangliosides are known to regulate transmembrane signaling by modulating functional membrane
proteins. Thus, they have been described to regulate cell proliferation through modulation of growth
factor receptor-associated kinases such as EGF and PDGF receptors (Bremer et al., 1986; Mirkin et
al., 2002; Sachinidis et al., 1996; Van Brocklyn et al., 1993). The importance of gangliosides in
regulating cell proliferation is also supported by studies implicating them in cell cycle arrest
processes via cyclin-dependent kinase inhibitors (Nakatsuji et al., 2001) or in apoptosis through
mitochondrial events (Kristal et al., 1999; Rippo et al., 2000).
It is well documented that regulation of cell proliferation is severely affected in retinopathy, a main
microvascular complication of diabetes. Death of retinal pericytes, microvascular contractile cells,
is indeed one of the earliest retinal changes, induced by hyperglycemia. Pericyte loss and
subsequent modification of endothelial cell proliferation and function, combined with basement
membrane thickening, trigger structural and functional alterations of capillaries. These chronic
alterations establish edema, microaneurisms and hemorrage that at ultimate stages can lead to
blindness (Forrester J.V. et al., 1997).
Although the pathogenic basis of diabetic microangiopathy is not fully understood at cellular and
molecular levels, it is clear that regulations of cell proliferation play an important role. The
hexosamine pathway (HP), a novel glucose-sensing pathway, has been implicated in
hyperglycemia-induced diabetic vascular alterations (Marshall et al., 1991). After entering cells,
glucose is rapidly phosphorylated and converted to fructose-6-phosphate (F-6-P) which is mainly
catabolized through the glycolytic pathway. Nevertheless, under hyperglycemic conditions, a
greater part of F-6-P can be converted to glucosamine-6-phosphate by glutamine:F-6-P
amidotransferase (GFAT), the first and rate-limiting enzyme of the hexosamine biosynthetic

pathway. Glucosamine-6-phosphate then undergoes rapid conversion to various hexosamine
products, including UDP-N-acetylglucosamine (UDP-GlcNAc). Thus, the HP can be activated
under the hyperglycemic conditions of a diabetic state by increased availability of substrate (F-6-P)
and/or GFAT activation. HP exerts its effects via two described mechanisms. First, hexosamine
product biosynthesis provides glycosidic precursors for the synthesis of glycoproteins, glycolipids
and proteoglycans. These intermediates can especially be used in the endoplasmic reticulum to form
complex glycoproteins, secreted or inserted into the membrane where they can modulate
transmembrane signaling or extracellular interactions. Additionally, UDP-GlcNAc can be used as
precursor for CMP-sialic acid, a substrate of ganglioside biosynthesis, raising the possibility of a
connection between HP and the ganglioside pathway. Second, cytosolic or nuclear proteins can be
covalently modified by the addition of a single monosaccharide GlcNAc, a product of the HP. This
O-linked glycosylation occurs on key intracellular proteins playing roles in controlling gene
expression, cell growth and division, enzyme activity, or structural integrity of the cytoskeleton
(Marshall, 2002). Because the potential targets of these O-glycosylation modifications are
numerous, the HP may be involved in many pathological mechanisms.
A growing number of studies suggest a role of HP in glucose-induced insulin resistance (Hebert, Jr.
et al., 1996; Kaneto et al., 2001; Vosseller et al., 2002) and in diabetic nephropathy, particularly in
renal mesangial cell alterations (Kolm-Litty et al., 1998; Schleicher et al., 2000; Singh et al., 2001).
However very few studies, if any, have examined the involvement of HP in the development of
diabetic retinopathy. Moreover, although gangliosides play major role in cell proliferation, their
potential involvement in diabetic microvascular complications and particularly in retinal pericyte
growth perturbations has been poorly studied.
The aim of the present study was to investigate the effect of the HP activation on pericyte
proliferation, ganglioside profile, and potential link between the two events. Results indicated that
glucosamine modifies pericyte ganglioside pattern concomitantly to a decrease of cell proliferation.
Further, addition of exogenous gangliosides exerted anti-proliferative effects on pericytes,
suggesting that gangliosides are involved in mediating the adverse glucosamine effects on pericyte
proliferation.

Results
Glucosamine treatment inhibits pericyte proliferation
To evaluate the effect of HP on pericyte growth, cells were treated with glucosamine which
bypasses the rate limiting enzyme GFAT and mimics an exacerbation of the HP. Cells were
exposed to 5mM glucosamine for 1 to 7 days and proliferation was estimated by cell counting and
total proteins measurement. As shown in Figure 1, time-course experiments revealed that
glucosamine potently decreased pericyte proliferation starting at 1-2 days of treatment. Cell
counting revealed that glucosamine reduced pericyte number by 32% and 40% after 2 and 7 days of
treatment, respectively. A decrease of total proteins was also observed in glucosamine treated cells
and correlated to cell number (Fig. 1C). To investigate the effects of glucosamine on cell
proliferation, apoptosis was measured in cells treated for 7 days with 5mM glucosamine.
Measurement of caspase 3 activation and determination of DNA fragmentation using an
oligonucleosomes determination kit showed that glucosamine did not induce apoptosis of bovine
retinal pericytes (BRP), at least under our culture conditions (data not shown). These results suggest
that the growth inhibition observed in response to glucosamine might involve cell cycle events.
Glucosamine modulates ganglioside profile
Ganglioside pattern of bovine retinal pericytes was analyzed in control and cells treated with 5mM
glucosamine. Under control conditions, the main gangliosides were GM3 (60% of all gangliosides
detected), GD3 (28%) and GM1 (12%) (Fig. 2A). As shown in Figure 2, important modifications of
ganglioside profile were observed in response to glucosamine (Fig 2B-C). Thus, GM1 and GD3
masses were increased by 27 and 19%, and by 85 and 105%, after 2 and 7 days of treatment,
respectively. Conversely, GM3 mass was decreased by 21 and 32% after 2 and 7 days of treatment,
respectively. Similar results were obtained by autoradiography analysis after galactose labeling
(data not shown). These results suggest a possible connection between the hexosamine pathway and
the ganglioside biosynthesis pathway. As the glucosamine effects were maximal at 7 days,
subsequent experiments were performed at this time of treatment.
Glucosamine increases GM3 synthase activity
To investigate the mechanism responsible for the observed increase of GM1 and GD3 masses, GM3
synthase activity, a rate-limiting enzyme for the synthesis of gangliosides was measured in control
and treated cells. Results presented in Figure 3 show that glucosamine treatment increased GM3
synthase activity by about 160%. As GM3 is a precursor of complex gangliosides, its decrease

despite the augmentation of GM3 synthase activity suggests that GM3 was metabolized to GM1
and GD3 that are both increased in response to glucosamine.
Treatment with exogenous gangliosides inhibits pericyte proliferation
Since GM1 and GD3 were increased concomitantly to a decrease of cell proliferation in response to
glucosamine, we further explored the hypothesis that gangliosides are involved in mediating the
glucosamine effects. Thus, we investigated whether the addition of exogenous gangliosides affects
pericyte proliferation. Cells were treated during one passage (7 days) with 10, 50 or 100 µM
gangliosides and, as a control, with the non-sialylated precursors glucosylceramide and
lactosylceramide. Results presented in Figure 4 show that GM1 and GD3 inhibited pericyte
proliferation in a dose-dependent manner whereas glucosylceramide and lactosylceramide used as
controls showed no effects. Inhibition of cell proliferation reached 25% at ganglioside concentration
of 100 µM. The effect of a-series gangliosides that were not increased in response to glucosamine
was also tested. Results indicated that GM3, GM2, and GD1a also inhibited BRP proliferation (data
not shown), suggesting that the presence of sialic acid is required in the active structure leading to
inhibition of BRP proliferation. It should be indicated however, that treatment with exogenous
gangliosides resulted in rather modest effects as compared to the effects of glucosamine. These data
might be explained by the fact that only a small proportion of added gangliosides are inserted into
cell membranes. Additionally, exogenous gangliosides treatment may result in the generation of cell
membrane pool functionally different from the gangliosides pool formed endogenously in response
to glucosamine treatment.
PPMP protects against the inhibition of proliferation caused by glucosamine
Next, a complementary approach was used to further support the involvement of gangliosides in the
anti-proliferative effects of glucosamine on pericytes. PPMP, an inhibitor of glucosylceramide
synthase was used to block the increase of gangliosides caused by glucosamine. As shown in Figure
5, treatment of BRP with glucosamine in the presence of increasing concentrations of PPMP
partially protected cells against proliferation decrease, in a dose-dependent manner.
Altogether, these results support the hypothesis that GM1 and GD3 increase is involved, at least in
part, in the proliferation decrease of pericytes treated with glucosamine.

Discussion
A growing number of studies indicate that the hexosamine pathway may be causally involved in the
development of diabetic microvascular complications, particularly nephropathy. Many of the
pathological features of diabetic nephropathy are also common to diabetic retinopathy. Further, as it
is known that retina expresses active GFAT and that hexosamine content is increased in retinal
tissues in human and rats with diabetes (Heath et al., 1967; Mazlen et al., 1970), this raises the
hypothesis that the HP could possibly be implicated in diabetic retinopathy development and
pericyte loss. On the other hand, gangliosides are known to play a role in cell differentiation and
growth regulation. Altered ganglioside levels have been associated with oncogenic transformation
or inherited disease such as Farber’s disease (Farina et al., 2000; Manzi et al., 1990; Zeng et al.,
2000), although very little is known concerning the mechanism or the biological significance of
these changes. However, ganglioside changes and their involvement in diabetes where cell growth
is also altered have been poorly studied. In the present study, ganglioside modifications and their
consequences on proliferation of pericytes submitted to diabetic environment were investigated.
Glucosamine, that is avidly taken up by the glucose transporter and phosphorylated by hexokinase
when added to cells (Schleicher et al., 2000), was used to mimic the high glucose environment
effects that are mediated by the HP in our model. Pericytes were chosen as their growth is
particularly affected in diabetic microangiopathy. Results demonstrate that glucosamine treatment
induces modification of BRP ganglioside profile with an increase of GM1 and GD3 and a decrease
of GM3. Moreover, glucosamine increased GM3 synthase activity, which regulates GM3
biosynthesis, a precursor of complex gangliosides. Thus, by increasing GM3 synthase activity,
glucosamine could drive complex ganglioside biosynthesis. This hypothesis is supported by the
observed increase of GM1 and GD3. Glucosamine regulates GM3 synthase activity through yet
undefined mechanism. One possible explanation is through regulation of GM3 synthase gene
expression. In fact, human GM3 synthase promoter presents putative binding sites for SP1 (Zeng et
al., 2003), a transcription factor whose activity is known to be regulated by O-linked N-
acetylglucosamine modification (Goldberg et al., 2002). On the other hand, it should be also
considered that glucosamine can lead to increased ganglioside biosynthesis by providing excessive
substrates that increase the flux of ganglioside biosynthesis in glucosamine-treated pericytes.
Indeed, UDP-GlcNAc can be converted into CMP-sialic acid, a precursor of ganglioside
biosynthesis.
Next, results showed that glucosamine causes a decrease of pericyte proliferation. This proliferation
decrease is partially reproduced by the application of exogenous gangliosides including GM1 and
GD3, both increased in response to glucosamine. This observation suggests a role of gangliosides in

mediating glucosamine effects. Of note, previous studies have documented that only 20% of
exogenously added gangliosides are inserted into cell membranes (Bremer et al., 1984). This might
explain the less potent effects of exogenous gangliosides on decreasing pericyte proliferation as
compared to glucosamine. These results are in accordance with other studies showing that
gangliosides can decrease cell growth, by modulating growth factor receptor signaling. For
example, GM1 has been shown to inhibit epidermal growth factor and platelet-derived growth
factor receptor phosphorylation and signaling in different cell types (Bremer et al., 1986; Mirkin et
al., 2002; Sachinidis et al., 1996; Van Brocklyn et al., 1993). Moreover, GD3 has been described as
an intracellular mediator of apoptosis, particularly by inducing mitochondrial permeability
transition, cytochrome c release and caspase activation (Garcia-Ruiz et al., 2000; Kristal et al.,
1999; Rippo et al., 2000). A complementary approach using PPMP, an inhibitor of
glucosylceramide synthase, was used next. Addition of PPMP to glucosamine treatment partially
protected pericytes against the proliferation decrease, supporting a role of gangliosides in mediating
the glucosamine effects. These data also suggest that unknown additional pathways are also
involved in causing decrease of pericyte proliferation in response to glucosamine. As discussed
earlier, the hexosamine pathway can affect cell proliferation through several pathways and
mechanisms. Activation of the hexosamine pathway leads to O-GlcNAc modifications of proteins
such as transcription factors (Wells et al., 2001). For example it has been shown in bovine aortic
endothelial cells that the transcription factor SP1 is modified by O-GlcNAc in response to
hyperglycemia or elevated glucosamine (Du et al., 2000). Increased O-GlcNAc modification of SP1
increases SP1 transactivation and SP1-dependent expression of TGF-β1 and PAI-1 (Du et al.,
2000). Interestingly, SP1 has also been shown to regulate the expression of proteins involved in cell
cycle regulation such as p21waf1/cip1 (Gartel et al., 1999). As in our study, glucosamine did not inhibit
BRP proliferation through apoptosis, it would be of great interest to investigate the effect of
glucosamine on cell cycle proteins such as p21waf1/cip1 in these cells.
In conclusion, the present study indicates that glucosamine increases ganglioside biosynthesis in
pericytes and suggests that gangliosides are likely involved in the anti-proliferative effect of
glucosamine in this cell type. These observations also indicate that the ganglioside pattern can be
modified by a diabetic environment and extend the role of gangliosides into the field of diabetic
microvascular complications.

Materials and methods
Cell isolation and culture
Bovine retinal pericytes (BRP) were isolated from microvessels of bovine retinas as previously
described (Lecomte et al., 1996). Briefly, retinas were dissected under sterile conditions from
freshly enucleated bovine eyes obtained from a local slaughterhouse. Retinas (2 per culture dish)
were minced into small pieces and homogenized in a Dounce homogenizer in Hanks’ balanced salt
solution (HBSS) buffer (HBSS supplemented with 10 mM Hepes, pH 7.4, 1% antibiotics, 0.5%
bovine serum albumin (BSA), Sigma, Saint-Quentin Fallavier, France). The homogenates were then
digested at 37°C for 20 min in a solution of collagenase/dispase (Roche Diagnostics, Mannheim,
Germany). After digestion, microvessels fragments were trapped onto a 40 µm nylon filter and
plated in 6 cm fibronectin-coated dishes. Outgrowth of pericytes from the microvessels occurred
after about 48 h. Primary cultures were grown in dulbecco’s modified eagle’s medium (DMEM,
Sigma) supplemented with 10% fetal bovine serum (Gibco, Invitrogen Corporation, N.Y., USA),
1% glutamine and 1% penicillin/streptomycin (Sigma). Cells were passaged with trypsin-EDTA
(Sigma) (1:3) and cultured up to the second passage during which they were treated. Glucosamine
(Sigma) was added to the culture medium at 5 mM final concentration and culture medium was
replaced every two days. BRP were treated for 1 to 7 days.
Ganglioside analysis
At the end of the treatment period, pericytes (5-8 × 105 cells) were collected by trypsination and
washed twice with phosphate-buffered saline (PBS, Sigma). For metabolic labelling of
gangliosides, 0.2 µCi/ml of [14C(U)]D-Galactose (329.5 mCi/mmol) (PerkinElmer Life Sciences,
Boston, MA) was added to the media overnight before harvesting as galactose incorporates into all
gangliosides. Cell pellets were dispersed into 2 ml chloroform (C)/methanol (M) (1:1, v/v), mixed
thoroughly and extracted overnight at 4°C. After centrifugation, the residues were extracted twice
with 2 ml of the same solvent. The pooled extracts of total lipids were evaporated to dryness and
submitted to partition with C/M/PBS 1 mM (10:10:7, v/v/v). The upper phases containing
gangliosides were next desalted on C18 silica gel column (Waters Corporation, Milford, MA) and
analyzed by HPTLC (Merck, Darmstadt, Germany). Plates were developed in C/M/0.2% CaCl2
(55:45:10, v/v/v) and gangliosides were visualized by autoradiography using phosphor screen and
Storm 820 (Molecular Dynamics, Amersham Pharmacia Biotech, Piscataway, USA) and by
resorcinol staining (gangliosides-specific stain: resorcinol 0.3% (Sigma), 0.03% CuSO4, 30% HCl)
with Image Master VDS-CL (Amersham Pharmacia Biotech). Gangliosides were identified by

comigration with standards (Matreya, Biovalley, Marne la Vallée, France). Quantification was done
by densitometry analysis with Image Quant (Molecular Dynamics). As GT1b is absent in BRP
ganglioside profile, it was added as an internal standard in the samples before lipid extraction.
Results were adjusted to the total proteins of the sample and the extraction efficiency.
Measurement of GM3 synthase activity
At the end of treatment, cells were washed with PBS, incubated for 20 min at 4°C in 50 µl lysis
buffer (20 mM sodium cacodylate pH 6.6 (Sigma), 0.2% Triton X-100, 1 mM EDTA, 10 µl/ml
proteases inhibitors, Calbiochem, Merck KGaA, Darmstadt, Germany) and then collected by
scrapping. Four dishes of BRP (~2-3 × 106 cells) were pooled. Cell lysates were centrifuged at
10,000g for 5 min, and proteins in the supernatants were used to measure GM3 synthase activity.
Equal amounts of proteins from each sample (about 500 µg) were used to perform the assay.
Samples were mixed with an equal volume of reaction buffer containing at final concentrations: 0.1
mM lactosylceramide (Matreya), 4 µCi/ml [sialic-4,5,6,7,8,9-14C]CMP-sialic acid (325.2
mCi/mmol) (PerkinElmer Life Sciences), 100 µM CMP-sialic acid (Sigma), 10 mM MgCl2, 0.2%
Triton X-100, 100 mM sodium cacodylate pH 6.6. After shaking, the reaction mixtures were
incubated at 37°C for 150 min. Reactions were stopped by loading the samples on silica gel 60
columns (Merck) to separate excess of substrates from products. After washing the columns with
water, gangliosides were eluted with C/M (1:1, v/v) and the solvent dried under nitrogen.
Gangliosides were finally separated by TLC (Merck) and reaction products were revealed by
autoradiography. GM3 synthase activity was expressed as pmoles of GM3 produced per hour/mg
proteins.
Measurement of cell growth
After each treatment period, cells were harvested with trypsin and cell pellets were washed twice
with PBS. For each sample, an aliquot of cells was counted using a haemocytometer to determine
the cell number, another aliquot was used to measure proteins according to Bradford.
Treatment with exogenous gangliosides
To evaluate the effect of exogenous gangliosides on BRP proliferation, cells were cultured in 96
wells plates. Exogenous glycolipids GM1, GD3, glucosylceramide and lactosylceramide (Matreya)
were added to the culture complete medium at final concentrations of 10, 50 or 100 µM in the form
of complexes with BSA at a ratio of 1:1 in DMEM–10 mM Hepes, pH 7.4 to facilitate their

incorporation within the cell. At the end of treatment, cells were washed twice with PBS and lysed
for 30 min at 37°C in 50 µl Ripa lysis buffer (PBS 10 mM, NP40 1% (Pierce, Perbio Science,
Brebières, France), sodium deoxycholate 0.5%, SDS 0.1%, 10 µl/ml proteases inhibitors). Total
proteins were finally measured using the BCA protein assay (Pierce) to assess cell proliferation, as
in our experiments cell numbers correlated with total protein concentrations (see Fig. 1).
PPMP treatment
In order to inhibit ganglioside biosynthesis, DL-threo-1-Phenyl-2-palmitoylamino-3-morpholino-1-
propanol (PPMP, Sigma), an inhibitor of glucosylceramide synthase that catalyses the first step of
ganglioside biosynthesis, was used. BRP were cultured in 96 wells plates and treated with 5 mM
glucosamine in the presence of increasing concentrations of PPMP for 7 days. PPMP was not used
at concentrations higher than 1µM as it caused cellular morphological alterations and toxic effects.
At the end of treatment, cells were washed twice with PBS, lysed in Ripa and proteins were
measured to assess cell proliferation as described above.
Statistical analysis
Data are expressed as means ± SEM and presented as percentage of controls. The Wilcoxon signed-
rank test was used to define the significance of the difference between groups. p< 0.05 was
considered as statistically significant.

Abbreviations BRP, bovine retinal pericytes; DMEM, dulbecco’s modified eagle’s medium; DR, diabetic
retinopathy; Glccer, glucosylceramide; GlcN, glucosamine; Laccer, lactosylceramide; HBSS,
Hanks’ balanced salt solution; HP, hexosamine pathway; HPTLC, high-performance thin-layer
chromatography; PBS, phosphate-buffered saline; PPMP, DL-threo-1-Phenyl-2-palmitoylamino-3-
morpholino-1-propanol.

References
Bremer, E.G., Hakomori, S., Bowen-Pope, D.F., Raines, E., and Ross, R. (1984) Ganglioside-
mediated modulation of cell growth, growth factor binding, and receptor phosphorylation.
J.Biol.Chem., 259, 6818-6825.
Bremer, E.G., Schlessinger, J., and Hakomori, S. (1986) Ganglioside-mediated modulation of cell
growth. Specific effects of GM3 on tyrosine phosphorylation of the epidermal growth factor
receptor. J.Biol.Chem., 261, 2434-2440.
Du, X.L., Edelstein, D., Rossetti, L., Fantus, I.G., Goldberg, H., Ziyadeh, F., Wu, J., and Brownlee,
M. (2000) Hyperglycemia-induced mitochondrial superoxide overproduction activates the
hexosamine pathway and induces plasminogen activator inhibitor-1 expression by increasing Sp1
glycosylation. Proc.Natl.Acad.Sci.U.S.A, 97, 12222-12226.
Farina, F., Cappello, F., Todaro, M., Bucchieri, F., Peri, G., Zummo, G., and Stassi, G. (2000)
Involvement of caspase-3 and GD3 ganglioside in ceramide-induced apoptosis in Farber disease.
J.Histochem.Cytochem., 48, 57-62.
Forrester J.V. and Knott R.M. Pickup J. and Williams G. (1997) Pathogenesis of diabetic
retinopathy and cataract. Blackwell Science, 45.1-45.19.
Garcia-Ruiz, C., Colell, A., Paris, R., and Fernandez-Checa, J.C. (2000) Direct interaction of GD3
ganglioside with mitochondria generates reactive oxygen species followed by mitochondrial
permeability transition, cytochrome c release, and caspase activation. FASEB J., 14, 847-858.
Gartel, A.L. and Tyner, A.L. (1999) Transcriptional regulation of the p21((WAF1/CIP1)) gene.
Exp.Cell Res, 246, 280-289.
Goldberg, H.J., Whiteside, C.I., and Fantus, I.G. (2002) The hexosamine pathway regulates the
plasminogen activator inhibitor-1 gene promoter and Sp1 transcriptional activation through protein
kinase C-beta I and -delta. J.Biol.Chem., 277, 33833-33841.

Hakomori, S. (1990) Bifunctional role of glycosphingolipids. Modulators for transmembrane
signaling and mediators for cellular interactions. J.Biol.Chem., 265, 18713-18716.
Heath, H., Paterson, R.A., and Hart, J.C. (1967) Changes in the hydroxyproline, hexosamine and
sialic acid of the diabetic human and beta, beta'-iminodipropionitrile-treated rat retinal vascular
systems. Diabetologia, 3, 515-518.
Hebert, L.F., Jr., Daniels, M.C., Zhou, J., Crook, E.D., Turner, R.L., Simmons, S.T., Neidigh, J.L.,
Zhu, J.S., Baron, A.D., and McClain, D.A. (1996) Overexpression of glutamine:fructose-6-
phosphate amidotransferase in transgenic mice leads to insulin resistance. J.Clin.Invest, 98, 930-
936.
Kaneto, H., Xu, G., Song, K.H., Suzuma, K., Bonner-Weir, S., Sharma, A., and Weir, G.C. (2001)
Activation of the hexosamine pathway leads to deterioration of pancreatic beta-cell function
through the induction of oxidative stress. J.Biol.Chem., 276, 31099-31104.
Kolm-Litty, V., Sauer, U., Nerlich, A., Lehmann, R., and Schleicher, E.D. (1998) High glucose-
induced transforming growth factor beta1 production is mediated by the hexosamine pathway in
porcine glomerular mesangial cells. J.Clin.Invest, 101, 160-169.
Kristal, B.S. and Brown, A.M. (1999) Ganglioside GD3, the mitochondrial permeability transition,
and apoptosis. Ann.N.Y.Acad.Sci., 893, 321-324.
Lecomte, M., Paget, C., Ruggiero, D., Wiernsperger, N., and Lagarde, M. (1996) Docosahexaenoic
acid is a major n-3 polyunsaturated fatty acid in bovine retinal microvessels. J.Neurochem., 66,
2160-2167.
Manzi, A.E., Sjoberg, E.R., Diaz, S., and Varki, A. (1990) Biosynthesis and turnover of O-acetyl
and N-acetyl groups in the gangliosides of human melanoma cells. J.Biol.Chem., 265, 13091-
13103.
Marshall, S. (2002) The hexosamine signaling pathway: a new road to drug discovery. 9, 160-167.

Marshall, S., Bacote, V., and Traxinger, R.R. (1991) Discovery of a metabolic pathway mediating
glucose-induced desensitization of the glucose transport system. Role of hexosamine biosynthesis
in the induction of insulin resistance. J.Biol.Chem., 266, 4706-4712.
Mazlen, R.G., Muellenberg, C.G., and O'Brien, P.J. (1970) L-Glutamine D-fructose 6-phosphate
amidotransferase from bovine retina. Exp.Eye Res., 9, 1-11.
Mirkin, B.L., Clark, S.H., and Zhang, C. (2002) Inhibition of human neuroblastoma cell
proliferation and EGF receptor phosphorylation by gangliosides GM1, GM3, GD1A and GT1B.
Cell Prolif., 35, 105-115.
Nakatsuji, Y. and Miller, R.H. (2001) Selective cell-cycle arrest and induction of apoptosis in
proliferating neural cells by ganglioside GM3. Exp.Neurol., 168, 290-299.
Rippo, M.R., Malisan, F., Ravagnan, L., Tomassini, B., Condo, I., Costantini, P., Susin, S.A.,
Rufini, A., Todaro, M., Kroemer, G., and Testi, R. (2000) GD3 ganglioside as an intracellular
mediator of apoptosis. Eur.Cytokine Netw., 11, 487-488.
Sachinidis, A., Kraus, R., Seul, C., Meyer zu Brickwedde, M.K., Schulte, K., Ko, Y., Hoppe, J., and
Vetter, H. (1996) Gangliosides GM1, GM2 and GM3 inhibit the platelet-derived growth factor-
induced signalling transduction pathway in vascular smooth muscle cells by different mechanisms.
Eur.J.Cell Biol., 71, 79-88.
Schleicher, E.D. and Weigert, C. (2000) Role of the hexosamine biosynthetic pathway in diabetic
nephropathy. Kidney Int.Suppl, 77, S13-S18.
Singh, L.P., Andy, J., Anyamale, V., Greene, K., Alexander, M., and Crook, E.D. (2001)
Hexosamine-induced fibronectin protein synthesis in mesangial cells is associated with increases in
cAMP responsive element binding (CREB) phosphorylation and nuclear CREB: the involvement of
protein kinases A and C. Diabetes, 50, 2355-2362.
Van Brocklyn, J., Bremer, E.G., and Yates, A.J. (1993) Gangliosides inhibit platelet-derived growth
factor-stimulated receptor dimerization in human glioma U-1242MG and Swiss 3T3 cells.
J.Neurochem., 61, 371-374.

van Echten, G. and Sandhoff, K. (1993) Ganglioside metabolism. Enzymology, Topology, and
regulation. J.Biol.Chem., 268, 5341-5344.
Vosseller, K., Wells, L., Lane, M.D., and Hart, G.W. (2002) Elevated nucleocytoplasmic
glycosylation by O-GlcNAc results in insulin resistance associated with defects in Akt activation in
3T3-L1 adipocytes. Proc.Natl.Acad.Sci.U.S.A, 99, 5313-5318.
Wells, L., Vosseller, K., and Hart, G.W. (2001) Glycosylation of nucleocytoplasmic proteins: signal
transduction and O-GlcNAc. Science, 291, 2376-2378.
Zeng, G., Gao, L., Birkle, S., and Yu, R.K. (2000) Suppression of ganglioside GD3 expression in a
rat F-11 tumor cell line reduces tumor growth, angiogenesis, and vascular endothelial growth factor
production. Cancer Res., 60, 6670-6676.
Zeng, G., Gao, L., Xia, T., Tencomnao, T., and Yu, R.K. (2003) Characterization of the 5'-flanking
fragment of the human GM3-synthase gene. Biochim.Biophys.Acta, 1625, 30-35.

Legends:
Fig. 1. Glucosamine inhibits pericyte proliferation.
Pericytes were exposed to 5 mM glucosamine for a maximum of 7 days. Cells were periodically
trypsinized, counted and total proteins measured. Representative growth curves corresponding to
cell number expressed in 105 cells (A) and total proteins expressed in µg (B) are shown. (C) Cell
number counting and total protein measurement at 2 and 7 days are represented. Results are
expressed as percentage of control and represent means ± SEM of 3-7 independent experiments. * p
< 0.05 vs control. GlcN, glucosamine.
Fig. 2. Glucosamine modulates ganglioside pattern.
Pericytes were exposed to 5 mM glucosamine for 2 (B) or 7 (C) days, and then collected.
Gangliosides were analysed by HPTLC and resorcinol staining. Representative HPTLC of
gangliosides after 7 days treatment revealed by resorcinol (A) and densitometric quantification of 3-
7 independent experiments (B, C) are shown. Results are expressed as µg of gangliosides per mg of
proteins and represent means ± SEM. * p<0.05 vs control. Std, standard. The band on the origin
was unknown, therefore it was not quantified.
Fig. 3. Glucosamine increases GM3 synthase activity.
Pericytes were treated with 5 mM glucosamine for 7 days. GM3 synthase activity was measured on
cell homogenates as described in Materials and methods. (A) Autoradiography of a representative
TLC plate of GM3 synthase activity products is shown. (B) Control activity was 4.2 pmol/hour/mg
proteins. Results are expressed as percentage of control and represent means ± SEM of 8
independent experiments. * p<0.05 vs control.
Fig. 4. Exogenous gangliosides inhibit cell proliferation.
Pericytes were treated with increasing concentrations of GM1, GD3, glucosylceramide (Gluccer) or
lactosylceramide (Laccer)-BSA complexes for 7 days. At the end of treatment, cells were washed,
lysed and total proteins were measured. Results are expressed as percentage of control and represent
means ± SEM of 4 independent experiments each performed in triplicates. * p<0.05 vs control.
Fig. 5. PPMP protects against glucosamine effects.
Pericytes were treated with 5 mM glucosamine in the presence or absence of 0.1, 0.25, 0.5, or 1 µM

PPMP for 7 days. At the end of treatment, cells were washed, lysed, and total proteins measured.
Results are expressed as µg of proteins per well and represent means ± SEM of 6 independent
experiments each performed in triplicates. * p<0.05 vs glucosamine treated cells.
Fig. 1.
0
20
40
60
80
100
120
% o
f con
trol
cell numbertotal proteins
glucosamine (mM)
0 5
2 days 7 days
**
C
0
1
2
3
4
cell
num
ber (
105 )
control5 mM GlcN
0 2 4 6days
0
50
100
150
200
250
tota
l pro
tein
s (µg
)
0 2 4 6days
control5 mM GlcN
BA
0
20
40
60
80
100
120
% o
f con
trol
cell numbertotal proteins
glucosamine (mM)
0 5
2 days 7 days
**
0
20
40
60
80
100
120
% o
f con
trol
cell numbertotal proteins
glucosamine (mM)
0 5
2 days 7 days
**
C
0
1
2
3
4
cell
num
ber (
105 )
control5 mM GlcN
0 2 4 6days
0
1
2
3
4
cell
num
ber (
105 )
control5 mM GlcNcontrol5 mM GlcN
0 2 4 6days
0 2 4 6days
0
50
100
150
200
250
tota
l pro
tein
s (µg
)
0 2 4 6days
control5 mM GlcN
0
50
100
150
200
250
tota
l pro
tein
s (µg
)
0 2 4 6days
0 2 4 6days
control5 mM GlcNcontrol5 mM GlcN
BA

Fig. 2.
A
B
(internal std)
0 5glucosamine (mM)
std std
GM3
GM2GM1
GD3
GM3
originGT1b
C
µg g
angl
iosi
des
/ mg
prot
eins
0 5glucosamine (mM)
0
0.5
1.0
1.5
2.0
2.5
3.0 GM3GM1GD3
*GM3 GM1GD3
*
*
0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
µg g
angl
iosi
des
/ mg
prot
eins
0 5glucosamine (mM)
2 days 7 days
A
B
(internal std)
0 5glucosamine (mM)
std std
GM3
GM2GM1
GD3
GM3
originGT1b
C
µg g
angl
iosi
des
/ mg
prot
eins
0 5glucosamine (mM)
0
0.5
1.0
1.5
2.0
2.5
3.0 GM3GM1GD3
µg g
angl
iosi
des
/ mg
prot
eins
0 5glucosamine (mM)0 5
glucosamine (mM)
0
0.5
1.0
1.5
2.0
2.5
3.0 GM3GM1GD3
GM3GM1GD3
*GM3 GM1GD3
*
*
0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
µg g
angl
iosi
des
/ mg
prot
eins
0 5glucosamine (mM)
*GM3 GM1GD3
*
*
GM3 GM1GD3
GM3GM3 GM1GM1GD3GD3
*
*
0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
µg g
angl
iosi
des
/ mg
prot
eins
0 5glucosamine (mM)0 5
glucosamine (mM)
2 days 7 days
Fig. 3.
0
50
100
150
200
250
300
350
0 5glucosamine (mM)
*
% o
f con
trol
BA
origin
GM3
0 5glucosamine
(mM)
0
50
100
150
200
250
300
350
0 5glucosamine (mM)
*
% o
f con
trol
B
0
50
100
150
200
250
300
350
0 5glucosamine (mM)
*
% o
f con
trol
BA
origin
GM3
0 5glucosamine
(mM)
A
origin
GM3
0 5glucosamine
(mM)
origin
GM3
0 5glucosamine
(mM)
Fig. 4.
0
20
40
60
80
100
120
control GM1 GD3 Gluccer Laccer
% o
f con
trol
**
**
10µM50µM100µM
0
20
40
60
80
100
120
control GM1 GD3 Gluccer Laccer
% o
f con
trol
**
**
0
20
40
60
80
100
120
control GM1 GD3 Gluccer Laccer
% o
f con
trol
**
**
10µM50µM100µM
10µM50µM100µM
Fig. 5.
PPMP (µM)
Prot
eins
(µg)
0glucosamine 5mM
* * * *
0
1
2
3
4
5
6
7
8
0 1 0 0.1 0.25 0.5 1
PPMP (µM)
Prot
eins
(µg)
0glucosamine 5mM0glucosamine 5mMglucosamine 5mM
* * * *
0
1
2
3
4
5
6
7
8
0 1 0 0.1 0.25 0.5 10
1
2
3
4
5
6
7
8
0 1 0 0.1 0.25 0.5 1


Glucosamine induces cell-cycle arrest and hypertrophy of
mesangial cells: implication of gangliosides
BIOCHEMICAL JOURNAL IMMEDIATE PUBLICATION. Published on 17 Jan 2005.
Short title: glucosamine, gangliosides, and hypertrophy of mesangial cells Elodie Masson, Nicolas Wiernsperger, Michel Lagarde, and Samer El Bawab
Diabetic Microangiopathy Research Unit, MERCK-Santé/INSERM UMR 585, INSA-Lyon
Corresponding author: S. El Bawab, Diabetic Microangiopathy Research Unit, MERCK-Santé/INSERM
U585, INSA-Lyon, Louis Pasteur Bldg, 69621 Villeurbanne Cedex, France.
Phone: (+33) 4 72 43 81 12
Fax: (+33) 4 72 43 81 13
E-mail: [email protected]
Keywords : hexosamine pathway, diabetic nephropathy, hypertrophy, mesangial cells, gangliosides

SYNOPSIS
Alterations of proliferation and hypertrophy of renal mesangial cells are typical features of diabetic
nephropathy. The hexosamine pathway has been proposed as one biochemical hypothesis
explaining microvascular alterations of diabetic nephropathy, however, its involvement in the
regulation of mesangial cell growth or hypertrophy has been poorly studied. On the other hand,
although gangliosides are known to regulate cell proliferation, their potential role in mesangial cell
growth perturbations has hardly been explored. In the present study, we investigated the effects of
the hexosamine pathway activation, mimicked by glucosamine treatment, on mesangial cell growth
and hypertrophy, and the potential implication of gangliosides in these processes. Results indicate
that glucosamine induced hypertrophy of mesangial cells as measured by an increase of proteins to
cell ratio, and caused cell-cycle arrest through increased cyclin-dependent kinase inhibitor
p21Waf1/Cip1 expression. Further, glucosamine treatment resulted in a massive increase of
gangliosides GM2 and GM1 levels. Treatment of cells with exogenous GM2 and GM1 reproduced
0.5 mM glucosamine effects on p21Waf1/Cip1 expression, cell-cycle arrest and hypertrophy,
suggesting that gangliosides GM2 and GM1 are likely involved in mediating glucosamine effects.
These results document a new role of the hexosamine pathway in the regulation of mesangial cell
growth and hypertrophy. They also suggest a potential new mechanism of action of the hexosamine
pathway through modulation of ganglioside levels.

INTRODUCTION Diabetic nephropathy (DN) is the most frequent cause of end stage renal disease. DN affects
approximately 30% of patients after 20 years of diabetes, and exposes them to higher mortality. The
pathology is characterized by complex structural changes including renal hypertrophy, basement
membrane thickening, and progressive accumulation of extracellular matrix components. Combined
with haemodynamic disturbances, these changes lead to glomerulosclerosis, microalbuminuria and
finally to a decline in renal function [1]. Hypertrophy of mesangial cells and increased secretion of
matrix proteins contribute to glomerular enlargement, which is considered as one of the earliest
alterations of DN. Mesangial cells growth during the diabetic state consists in an early, transient and
limited proliferation, followed by growth-arrest and hypertrophy. For example, it has been shown in
kidneys of streptozotocin-induced diabetic animals, that mesangial cells arrest at the G1 phase and
undergo sustained hypertrophy associated with an increase in the synthesis and deposition of
extracellular matrix proteins [2]. The cyclin-cyclin dependent kinase complex inhibitors,
p21Waf1/Cip1 and p27Kip1, have been implicated in this blockade, as shown in mesangial cells treated
with high glucose concentrations [3,4]. Increased expression of p21Waf1/Cip1 and p27Kip1 has also
been shown in mesangial cells of streptozotocin-induced diabetic rats and in diabetic db/db mice
[3,5].
Numerous hypotheses have been proposed to explain glucose toxicity and diabetic microvascular
alterations. In 1991, Marshall et al. first reported on a new pathway mediating glucose effects, the
hexosamine pathway (HP) [6]. After entering cells, glucose is rapidly phosphorylated and converted
to fructose-6-phosphate (F-6-P), which is mainly catabolized through the glycolytic pathway. Under
hyperglycemic conditions however, a greater part of F-6-P can be converted into glucosamine-6-
phosphate by the action of glutamine: F-6-P amidotransferase (GFAT), the first and rate-limiting
enzyme of the hexosamine biosynthetic pathway. Glucosamine-6-phosphate is then metabolized
into various hexosamine products, including UDP-N-acetylglucosamine (UDP-GlcNAc). Thus, the
HP exerts its effects by providing glycosidic precursors for glycoproteins, glycolipids and
proteoglycans. Additionally, the HP could mediate its effects through a single O-linked
glycosylation. In fact, GlcNAc, a product of the HP, can covalently modify and affect activities of
proteins playing roles in the control of gene expression, cell growth and division, enzyme activity,
or structural integrity of the cytoskeleton [7,8].
The HP was first implicated in glucose-induced desensitization of the insulin response [6]. In vivo,
acute glucosamine infusion induces insulin-resistance in normoglycemic rats [9]. Moreover,
overexpression of GFAT in mouse skeletal muscle [10] or in β cells [11] has proven the role of the
HP in insulin-resistance in animals and highlighted the importance of this pathway. Recent data also

suggested a role of the HP in the development of diabetic microvascular complications such as
nephropathy. Thus, the HP has been shown to activate through a mechanism involving the
transcription factor Sp1 the promoter of the Plasminogen Activator Inhibitor-1 gene, a regulator of
extracellular matrix production [12,13]. Moreover, HP activation has been largely described to
induce TGFβ production and matrix protein synthesis in mesangial cells [14-17]. However, the
involvement of the HP in the regulation of mesangial cell growth or hypertrophy has been poorly
studied.
Gangliosides are glycosphingolipids characterized by the presence of sialic acid in their
oligosaccharidic part. They are concentrated in the plasma membrane with a cell-specific pattern,
and found in most tissues of the body, especially in brain and nervous tissues [18]. Gangliosides
play major roles in cell-cell and cell-matrix recognition via interactions with adhesion receptors
such as integrins, matrix proteins (collagen and fibronectin), or with other glycosphingolipids [19].
On the other hand, gangliosides are known to regulate transmembrane signalling by modulating
functional membrane proteins. Thus, they have been described to regulate proliferation of different
cell types through modulation of growth factor receptor-associated kinases such as EGF and PDGF
receptor [20-23]. In spite of these described effects of gangliosides in regulating cell proliferation,
their implication in mesangial cell growth perturbations occurring during DN, has not been studied
yet.
The aim of the present study was to investigate the effect of HP activation, mimicked by
glucosamine treatment, on mesangial cell growth and hypertrophy, and to determine the potential
role of gangliosides in mediating the glucosamine effects. Results show that 0.5 mM glucosamine
inhibited mesangial cell growth and induced hypertrophy by increasing p21Waf1/Cip1 expression and
arresting cells at the G0/G1 phase. Moreover, glucosamine treatment increased very significantly
GM2 and GM1 gangliosides, and treatment with exogenous GM2 and GM1 reproduced
glucosamine effects on mesangial cell hypertrophy, cell-cycle arrest at the G0/G1 phase and
p21Waf1/Cip1 expression. The results suggest that glucosamine induces cell-cycle arrest and
hypertrophy of mesangial cells, at least in part, through GM2 and GM1 ganglioside production.

EXPERIMENTAL
Materials Sources of materials were as follows: animals, Charles River, L’Arbresle, Fance; fetal bovine
serum, Gibco, Invitrogen Corporation, N.Y., USA; all other cell culture products, RNAse,
resorcinol, Sigma, Saint-Quentin Fallavier, France; D-[U-14C]galactose and D-[1-14C]glucosamine
hydrochloride, Amersham Pharmacia Biotech, Buckinghamshire, UK; HPTLC, Merck, Darmstadt,
Germany; C18 silica gel column, Waters Corporation, Milford, MA; gangliosides, Matreya,
Biovalley, Marne la Vallée, France; fluorescein-labelled cholera toxin B subunit, Molecular Probes,
Leiden, The Netherlands; NP40 1 %, Pierce, Perbio Science, Brebières, France; monoclonal
antibodies, BD Biosciences, Le Pont de Claix, France.
Methods Mesangial cell isolation and culture Renal mesangial cells (RMC) were obtained from glomeruli of young male Wistar rat kidneys
isolated by differential sieving, as previously described [24]. RMC were characterized by
morphological criteria (stellate, spindle-shaped when confluent) and by negative staining for Factor
VIII and cytokeratin and their prominent cytoskeletal staining for actin. Cells were cultured in
DMEM supplemented with 15 % fetal bovine serum, 1 % glutamine and 1 %
penicillin/streptomycin and used between passages 7 to 15. Under these conditions, cells reached
confluency after about 4 days, therefore, treatments were done for a maximum of 4 days.
Total proteins to cell count ratio RMC were treated with 0.5 mM or 5 mM glucosamine for 1 to 4 days. At the end of the treatment
period, cells were trypsinized and washed twice with ice-cold phosphate-buffered saline (PBS). For
each sample, an aliquot of cells was counted using a haemocytometer to determine the cell number.
Another aliquot was lysed and used to measure total proteins according to Bradford. The ratio of
total proteins to cell number expressed as micrograms per 105 cells was used as a hypertrophy
index.
Cell cycle analysis RMC were harvested with trypsin after different treatments, washed twice with PBS, fixed in 70 %
ethanol and stored at 4 °C until use. Before flow cytometry analysis, cells were washed in PBS,
centrifuged and cell pellets were resuspended in RNAse (1 mg/ml) for 20 min. Cells were then
stained for 15 min with propidium iodide in PBS (40 µg/ml final concentration) before analysis

with FACSCAN (Becton Dickinson, Franklin Lakes, N.J., USA) using CellQuest software (Becton
Dickinson).
Ganglioside analysis After 48 h of treatment, RMC (1-4 × 106 cells) were collected by trypsination and washed twice
with PBS. For metabolic labelling of gangliosides, 0.2 µCi/ml of D-[U-14C]galactose (303
mCi/mmol) were added to the media overnight before harvesting cells, as galactose is incorporated
into all gangliosides. Cell pellets were dispersed into 2 ml chloroform (C)/methanol (M) (1:1, v/v),
mixed thoroughly and extracted overnight at 4 °C. After centrifugation, the residues were extracted
twice with 2 ml of the same solvent. The pooled extracts of total lipids were evaporated to dryness
and submitted to partition with C/M/PBS 1 mM (10:10:7, v/v/v). The upper phases containing
gangliosides were next desalted on C18 silica gel column and analyzed by HPTLC. Plates were
developed in C/M/0.2 % CaCl2 (55:45:10, v/v/v) and gangliosides were visualized by
autoradiography using phosphor screen and Storm 820 (Molecular Dynamics, Amersham
Pharmacia Biotech, Piscataway, USA) and by resorcinol staining (gangliosides-specific stain:
resorcinol 0.3 % (Sigma), 0.03 % CuSO4, 30 % HCl) with Image Master VDS-CL (Amersham
Pharmacia Biotech). Gangliosides were identified by comigration with standards. GM2 and GM1
identities were confirmed after HPTLC separation using anti-GM2 antibody (generous gift from Dr.
J. Portoukalian, Lyon, France) and fluorescein-labelled choleratoxin B subunit that interact
specifically with GM1. Quantification was performed by densitometry analysis using Image Quant
(Molecular Dynamics). As GT1b is absent in RMC ganglioside profile, it was added as an internal
standard in the samples before lipid extraction. Results were then adjusted for extraction efficiency
and total proteins of the sample.
Radiolabelling with 14C glucosamine To investigate the hypothesis that glucosamine treatment can affect ganglioside profile by providing
substrates for the ganglioside biosynthesis, cells were labeled with 14C-glucosamine. Control cells
were incubated with 0.2 µCi/ml D-[1-14C]glucosamine for 48 h. Glucosamine-treated cells were
incubated with 2 mM glucosamine at a much higher specific activity of 0.5 mCi/ml D-[1-14C]glucosamine. After the incubation, cells were washed and the radioactivity associated to
gangliosides was then analyzed to test the ability of glucosamine to incorporate into ganglioside
structures.

Treatment with exogenous gangliosides Exogenous gangliosides GM2 and GM1 were added to the culture complete medium at a final
concentration of 60 µM. This concentration was used as it induces inhibition of RMC proliferation
close to the effects observed with 0.5 mM GlcN treatment. Gangliosides were added in the form of
complexes with BSA at a ratio of 1:1 in DMEM–10 mM Hepes, pH 7.4 to facilitate their
incorporation within the cell.
Western blot analysis At the end of the treatment period, cells were washed and collected in Ripa lysis buffer (PBS 10
mM, NP40 1 %, sodium deoxycholate 0.5 %, SDS 0.1 %, 10 µl/ml proteases inhibitors). To remove
insoluble materials, lysates were centrifuged at 10 000 g for 5 min. Samples (20 µg of lysates) were
then boiled for 5 min, loaded onto a 12 % SDS-polyacrylamide gel, electrophoresed, and transferred
to a PVDF membrane. The membranes were blocked with 5 % milk in PBS containing 0.05 %
Tween (PBST), and blotted with monoclonal antibodies (p21, 1 µg/ml; p27, 1:2000) in the blocking
solution. After extensive washing, the membranes were blotted with secondary anti-mouse antibody
at a dilution of 1:2000. The signal was visualized by enhanced chemiluminescence.
Statistical analysis Data are expressed as means ± SEM. The Wilcoxon signed-rank test was used to define the
significance of the difference between groups. p<0.05 was considered as statistically significant.

RESULTS
Glucosamine decreases mesangial cell proliferation and induces hypertrophy The effect of glucosamine on RMC proliferation and hypertrophy was investigated in this study.
Glucosamine was used to mimic HP activation because of its demonstrated ability to selectively
enter the hexosamine biosynthetic pathway and to bypass the rate-limiting enzyme GFAT [25]. To
this end, mesangial cells were treated with either 0.5 or 5 mM glucosamine, collected and cell
number and total proteins were measured. As shown in Figure 1, time-course experiments revealed
that glucosamine potently decreases proliferation. Glucosamine effect was evident starting from 48
h and, at the end of the treatment period, cell number in RMC treated with 0.5 mM and 5 mM
glucosamine represented only 41 % and 12 % of control, respectively (Figure 1A). This adverse
effect on cell proliferation was also measurable by a decrease of total proteins content (Figure 1B).
In addition, as the ratio of total protein content to cell number is a well-established measurement of
cellular hypertrophy, this parameter was used to determine whether the alteration of cell growth was
accompanied by cell hypertrophy. As shown in Figure 1C, glucosamine indeed induced an increase
of the proteins to cell number ratio. At the end of the treatment period, protein to cell number ratio
increased by 1.6- and 2-fold for cells treated with 0.5 mM and 5 mM glucosamine, respectively.
The increase of cell size, characteristic of cellular hypertrophy, was also obvious by microscopy, as
illustrated in Figure 1D. These observations indicate that glucosamine triggers a decrease of
proliferation as well as hypertrophy of mesangial cells. As these effects were evident at 0.5 mM
GlcN, and to avoid potential cytotoxicity with the relatively high concentration of 5 mM, the
following experiments were carried out using 0.5 mM GlcN. Moreover, as the glucosamine effects
were evident at 48 h, and to avoid contact inhibition at confluence (4 days), the subsequent
experiments were performed at this time of treatment.
Glucosamine arrests cell-cycle progression Results from previous experiments showed that glucosamine treatment blocks very early cell
proliferation and may thus contribute to cellular hypertrophy. Therefore, the mechanisms
responsible for cell growth inhibition in response to glucosamine were investigated next. Cell cycle
analysis by flow cytometry was performed on control and cells treated with either 0.5 mM or 5 mM
glucosamine. Results summarized in Table 1 indicate that 0.5 mM glucosamine increased the
proportion of cells in the G0/G1 phase of the cell cycle as compared to control cells. This suggests
that 0.5 mM glucosamine blocks the progression of RMC through the cell cycle by inhibiting the
G1/S transition phase, arresting cells at G0/G1 phase. Surprisingly, treatment of cells with higher
glucosamine concentration (5 mM) resulted in cell cycle blockade at the G2/M phase. To further

investigate this discrepancy, a dose-response study was performed. As shown in Fig. 2,
glucosamine concentrations lower than 1 mM arrested cells in G0/G1 phase, whereas higher
concentrations of glucosamine (2-5 mM) induced G2/M phase arrest. These observations suggest
that at high glucosamine concentrations (>1 mM), additional pathways are involved.
Glucosamine increases p21Waf1/Cip1 expression
P21Waf1/Cip1 and p27Kip1 are inhibitors of cyclin-cyclin dependent kinase complexes known to inhibit
the G1/S phase transition. Moreover, it has been shown that p21Waf1/Cip1 and p27Kip1 proteins are
involved in growth arrest at the G1/S phase transition and hypertrophy of RMC in response to high
glucose concentrations [2]. Therefore, the expression of p21Waf1/Cip1 and p27Kip1 proteins was
measured. As shown in Figure 3, p21Waf1/Cip1 expression in cells treated with glucosamine was about
3-fold higher than in control cells. These data are consistent with the cell cycle arrest shown by
flow cytometry analysis. In contrast however, p27Kip1 expression was decreased by glucosamine
treatment. These results suggest that unlike high ambient glucose effects on RMC, GlcN-induced
cell-cycle arrest is likely p21Waf1/Cip1-dependent.
Glucosamine increases GM2 and GM1 levels The hexosamine pathway has been implicated in the regulation of numerous enzyme and protein
activities and as a consequence may regulate several pathways. Moreover, because the HP provides
intermediates for the synthesis of glycoconjugates such as glycolipids, glucosamine treatment could
affect ganglioside pattern of mesangial cells. To investigate this possibility, ganglioside pattern was
analyzed by HPTLC in control and cells treated with either 0.5 mM or 5 mM glucosamine for 48 h.
Under control conditions, the main gangliosides observed in RMC were GM3 (70 % of all
gangliosides detected), GD1a (24 %) and GD3 (5 %). When cells were treated with glucosamine,
important modifications of the ganglioside profile were observed (Figure 4A). Thus, gangliosides
GM2 and GM1, hardly detectable in control cells, were strongly increased to reach about 1.5 and
0.4 µg/mg of proteins that corresponded to 20 % and 5 % of all gangliosides detected, respectively
(Figure 4B). Similar results were obtained by autoradiography analysis after galactose labelling
(data not shown). GM3 and GD1a levels were only slightly affected whereas GD3 was undetectable
in the glucosamine-treated cells. These results suggest a shift into a-series ganglioside biosynthesis
in response to glucosamine. To further investigate whether HP affects ganglioside biosynthesis,
control cells were labelled with exogenous radioactive glucosamine and gangliosides were extracted
and analyzed for radioactivity content. Results show that indeed glucosamine or its metabolites
incorporate into ganglioside biosynthesis pathway (Figure 4C). Next, cells were treated for 48 h

with 2 mM glucosamine at a specific activity of 0.5 mCi/ml. Ganglioside analysis revealed
radioactive GM3, GM2, GM1 and GD1a (Figure 4C). These results support the hypothesis of a
connection of the HP and ganglioside biosynthesis pathways.
GM2 and GM1 gangliosides induce mesangial cell hypertrophy Gangliosides are known to play a role in a variety of important cellular functions including
proliferation and differentiation. As GM2 and GM1 were massively increased concomitantly to
RMC hypertrophy in response to glucosamine, we further investigated the possibility that
gangliosides would be involved in mediating the glucosamine effects. Proteins to cell number ratio
was measured in control cells and in cells treated with exogenous gangliosides. Results presented in
Figure 5 show that exogenous addition of GM2 and GM1 decreases mesangial cell proliferation as
measured by cell number counting and increases proteins to cell number ratio by about 30 %,
suggesting that RMC undergo hypertrophy in response to GM2 and GM1 exposure. GM3, GD3,
GD1a and lactosylceramide, a non-sialylated precursor of gangliosides, were also tested. Whereas
GM3, GD3 and GD1a also induced mesangial cell hypertrophy similar to GM2 and GM1,
lactosylceramide had no effect (data not shown). These results suggest that sialic acid is part of the
active structure.
GM2 and GM1 gangliosides block cell-cycle progression by increasing p21Waf1/Cip1 expression As GM2 and GM1 induced RMC hypertrophy as observed for glucosamine, we next investigated
whether similar mechanisms were involved. First, cell-cycle of cells treated with exogenous GM2
and GM1 was analysed by flow cytometry. As shown in Table 1, gangliosides induced an
accumulation of cells in the G0/G1 phase, suggesting a blockade in cell-cycle progression at the
G1/S transition. Next, p21Waf1/Cip1 and p27Kip1 expression was measured. Results indicate that
p21Waf1/Cip1 expression tended to increase in response to GM2 and GM1 exposure whereas p27Kip1
expression was diminished (Figure 3). Altogether, these results suggest that glucosamine and
exogenous gangliosides (GM2 and GM1) inhibit RMC proliferation likely through similar
mechanisms, by increasing p21Waf1/Cip1 expression and blocking cells at the G0/G1 phase. They also
suggest that gangliosides GM2 and GM1 are, at least partly, mediators of the glucosamine effects.

DISCUSSION
Several lines of evidence indicate that the flux through the hexosamine pathway may be causally
involved in the development of diabetic nephropathy. Activation of the HP has been implicated in
signaling pathways that lead to matrix proteins overproduction [25], a major cause of glomerular
enlargement in the early stages of DN. For example, GlcN in particular has been shown to increase
the production of the known matrix-stimulating growth factor, TGFβ [26-28]. However, the
potential role of the HP in growth perturbation and hypertrophy of mesangial cells has been poorly
studied. Therefore, these hypotheses have been investigated in the present study.
Treatment of RMC with GlcN potently inhibited cell growth and induced hypertrophy as assessed
by the proteins to cell number ratio, in a time-dependent manner. Hypertrophy is the consequence
of increased protein content combined to absence of cell division. Thus, the effect of GlcN
treatment on cell-cycle progression of RMC was investigated next. Flow cytometry analyses
indicated that low concentration of GlcN (0.5 mM) arrested RMC cycle progression at the G0/G1
phase. On the other hand, 5 mM GlcN caused an accumulation of cells at the G2/M phase,
suggesting that high concentrations block mesangial cell-cycle at the G2/M transit. These
observations clearly show that low and high concentrations of GlcN exert different actions on
RMC. These differences could be explained when taking into account recent observations from
Marshall et al. in adipocytes [29]. Indeed, these authors showed that a high concentration of GlcN
(2 mM) causes massive accumulation of the hexosamine intermediate product GlcN-6-P that was
accompanied with ATP depletion. In contrast, these effects were not observed in response to low
concentrations of GlcN (0.25 mM) or to high glucose concentrations (20 mM). In RMC treated with
GlcN, ATP depletion or alternatively high GlcN concentrations through other unknown
mechanisms, could result ultimately to DNA damage leading to the arrest of cells in G2/M. Thus,
treatment of RMC with low GlcN concentration (0.5 mM) is likely to be more physiologically
relevant.
To gain better insight into the mechanisms responsible for cell-cycle arrest caused by GlcN, the
expression of cyclin-dependent kinase inhibitors (CDKI) p21Waf1/Cip1 and p27Kip1 was studied. Our
studies focused on p21Waf1/Cip1 and p27Kip1 as these proteins have been involved in G0/G1 arrest of
mesangial cells in different diabetic animal models as well as of cultured mesangial cells exposed to
high ambient glucose concentration [2]. Results indicated that p21Waf1/Cip1 expression was about 3-
fold increased by GlcN treatment as compared to control levels, suggesting that p21Waf1/Cip1 is
involved in the blockade of the cell-cycle progression observed in RMC exposed to GlcN. On the
contrary, p27Kip1 expression was found rather diminished in response to GlcN treatment. A

decreased or unchanged p27Kip1 expression has also been shown in glomeruli of diabetic animal
models [3,30] and in RMC treated with high glucose concentrations [3]. Thus, the effect of GlcN on
RMC cell-cycle arrest is most likely p21Waf1/Cip1-dependent. In addition, p21Waf1/Cip1 has been
described as a pivotal regulator of hypertrophy. Fan et al. showed that exogenous attenuation of
p21Waf1/Cip1 using antisense oligonucleotides, decreased hypertrophy of mesangial cells induced by
hyperglycemia and insulin growth factor-1 [31]. Moreover, in vivo, p21Waf1/Cip1 knockout diabetic
mice did not develop glomerular hypertrophy [32]. These observations suggest that the increased
p21Waf1/Cip1 expression may also be involved in the mechanisms leading to hypertrophy of RMC in
response to GlcN. Because glucosamine effects on RMC are similar to high ambient glucose
effects, one might speculate that the high glucose effects on RMC growth and hypertrophy are, at
least in part, mediated through a HP-induced p21Waf1/Cip1 pathway.
On the other hand, gangliosides are glycosphingolipids that have been involved in the regulation of
proliferation of various cell types. Further, it has been shown that when GlcN is added to cells, even
at low concentrations, marked elevation of UDP-GlcNAc levels is observed [7,29]. This glycosidic
precursor can be incorporated into ganglioside structures such as GM2 and GM1, after
isomerization to UDP-N-Acetyl-galactosamine. Therefore, we investigated whether ganglioside
pattern was affected in RMC submitted to GlcN treatment. Analyses revealed modification of
ganglioside pattern with a massive increase of gangliosides GM2 and GM1. These results suggest
that GlcN increases GM2 and GM1 levels by mass action by providing high amounts of precursors
such as UDP-GalNAc. Radiolabelling experiments with 14C glucosamine also supported a
connection of the HP and ganglioside biosynthesis pathways. However, other mechanisms
regulating ganglioside biosynthesis in response to GlcN should not be excluded. For example, GM2
synthase presents putative binding sites for Sp1, a transcription factor whose activity is up regulated
by GlcN [33].
Next, we assessed whether GM2 and GM1 were involved in the hypertrophy and cell-cycle arrest of
RMC in response to GlcN. Exogenous application of GM2 and GM1 resulted in an increase of the
proteins to cell number ratio. Further, cell-cycle analysis of RMC exposed to GM2 and GM1
showed an accumulation of cells at the G0/G1 phase that was associated to an increase of p21Waf1/Cip1
expression. Thus, exogenous GM2 or GM1 application reproduced the effects of 0.5 mM GlcN on
RMC hypertrophy and cell-cycle arrest. Few studies implicated gangliosides in cell-cycle arrest.
Nakatsuji et al. showed that ganglioside GM3 induces cell-cycle arrest and apoptosis of astrocytes
in part through CDKI p27Kip1 increased expression [34]. Our results further suggest that
gangliosides might also contribute to mechanisms leading to cell hypertrophy. However and as
gangliosides showed similar effects on p27Kip1 expression, results from the present study can not
rule out a role of p27Kip1 decrease in cell cycle arrest induced by glucosamine and gangliosides.

In conclusion, our results show that 0.5 mM GlcN induces cell-cycle arrest at the G0/G1 phase and
hypertrophy of RMC. Moreover, results suggest that gangliosides GM2 and GM1 are involved in
mediating GlcN effects in RMC. Observations from the present study raise new hypothesis into the
potential involvement of the HP in the development of diabetic nephropathy, and add on new
mechanism of action of the HP such as modulation of ganglioside levels in cells. Interestingly,
gangliosides have recently been implicated in insulin-resistance [35,36] and, as discussed earlier,
HP has been shown to induce insulin-resistance in vitro and in vivo. It would be of interest to
investigate whether gangliosides are involved in the HP-induced insulin-resistance.

ABBREVIATIONS
Abbreviations used: CDKI, cyclin-dependant kinase inhibitor; DMEM, dulbecco’s modified eagle’s
medium; DN, diabetic nephropathy; GFAT, glutamine: F-6-P amidotransferase; GlcN,
glucosamine; HBSS, Hanks’ balanced salt solution; HP, hexosamine pathway; HPTLC, high
performance thin layer chromatography; PBS, phosphate-buffered saline; RMC, renal mesangial
cell.

Reference List
1 Trevisan, R., Barnes, D. J., and Viberti, G. (1997) Pathogenesis of diabetic nephropathy. In
Textbook of diabetes (Pickup, J. and Williams, G., ed.), pp. 52.1-52.21, Blackwell Science,
Oxford
2 Wolf, G. (2000) Cell cycle regulation in diabetic nephropathy. Kidney Int.Suppl 77, S59-S66
3 Kuan, C. J., al Douahji, M., and Shankland, S. J. (1998) The cyclin kinase inhibitor
p21WAF1, CIP1 is increased in experimental diabetic nephropathy: potential role in
glomerular hypertrophy. J.Am.Soc.Nephrol. 9, 986-993
4 Wolf, G., Schroeder, R., Ziyadeh, F. N., Thaiss, F., Zahner, G., and Stahl, R. A. (1997) High
glucose stimulates expression of p27Kip1 in cultured mouse mesangial cells: relationship to
hypertrophy. Am.J.Physiol 273, F348-F356
5 Wolf, G., Schroeder, R., Thaiss, F., Ziyadeh, F. N., Helmchen, U., and Stahl, R. A. (1998)
Glomerular expression of p27Kip1 in diabetic db/db mouse: role of hyperglycemia. Kidney
Int. 53, 869-879
6 Marshall, S., Bacote, V., and Traxinger, R. R. (1991) Discovery of a metabolic pathway
mediating glucose-induced desensitization of the glucose transport system. Role of
hexosamine biosynthesis in the induction of insulin resistance. J.Biol.Chem. 266, 4706-4712
7 Marshall, S. (2002) The hexosamine signaling pathway: a new road to drug discovery. Current
Opinion in Endocrinology and Diabetes. 9, 160-167
8 Vosseller, K., Sakabe, K., Wells, L., and Hart, G. W. (2002) Diverse regulation of protein
function by O-GlcNAc: a nuclear and cytoplasmic carbohydrate post-translational
modification. Curr.Opin.Chem.Biol. 6, 851-857
9 Rossetti, L., Hawkins, M., Chen, W., Gindi, J., and Barzilai, N. (1995) In vivo glucosamine
infusion induces insulin resistance in normoglycemic but not in hyperglycemic conscious rats.
J.Clin.Invest 96, 132-140
10 Hebert, L. F., Jr., Daniels, M. C., Zhou, J., Crook, E. D., Turner, R. L., Simmons, S. T.,
Neidigh, J. L., Zhu, J. S., Baron, A. D., and McClain, D. A. (1996) Overexpression of

glutamine:fructose-6-phosphate amidotransferase in transgenic mice leads to insulin
resistance. J.Clin.Invest 98, 930-936
11 Tang, J., Neidigh, J. L., Cooksey, R. C., and McClain, D. A. (2000) Transgenic mice with
increased hexosamine flux specifically targeted to beta-cells exhibit hyperinsulinemia and
peripheral insulin resistance. Diabetes 49, 1492-1499
12 Goldberg, H. J., Scholey, J., and Fantus, I. G. (2000) Glucosamine activates the plasminogen
activator inhibitor 1 gene promoter through Sp1 DNA binding sites in glomerular mesangial
cells. Diabetes 49, 863-871
13 James, L. R., Fantus, I. G., Goldberg, H., Ly, H., and Scholey, J. W. (2000) Overexpression of
GFAT activates PAI-1 promoter in mesangial cells. Am.J.Physiol Renal Physiol 279, F718-
F727
14 Kolm-Litty, V., Sauer, U., Nerlich, A., Lehmann, R., and Schleicher, E. D. (1998) High
glucose-induced transforming growth factor beta1 production is mediated by the hexosamine
pathway in porcine glomerular mesangial cells. J.Clin.Invest 101, 160-169
15 Singh, L. P. and Crook, E. D. (2000) Hexosamine regulation of glucose-mediated laminin
synthesis in mesangial cells involves protein kinases A and C. Am.J.Physiol Renal Physiol
279, F646-F654
16 Singh, L. P., Alexander, M., Greene, K., and Crook, E. D. (2003) Overexpression of the
complementary DNA for human glutamine:fructose-6-phosphate amidotransferase in
mesangial cells enhances glucose-induced fibronectin synthesis and transcription factor cyclic
adenosine monophosphate-responsive element binding phosphorylation. J.Investig.Med. 51,
32-41
17 Weigert, C., Friess, U., Brodbeck, K., Haring, H. U., and Schleicher, E. D. (2003)
Glutamine:fructose-6-phosphate aminotransferase enzyme activity is necessary for the
induction of TGF-beta1 and fibronectin expression in mesangial cells. Diabetologia 46, 852-
855
18 van Echten, G. and Sandhoff, K. (1993) Ganglioside metabolism. Enzymology, Topology, and
regulation. J.Biol.Chem. 268, 5341-5344
19 Hakomori, S. (1990) Bifunctional role of glycosphingolipids. Modulators for transmembrane
signaling and mediators for cellular interactions. J.Biol.Chem. 265, 18713-18716

20 Bremer, E. G., Schlessinger, J., and Hakomori, S. (1986) Ganglioside-mediated modulation of
cell growth. Specific effects of GM3 on tyrosine phosphorylation of the epidermal growth
factor receptor. J.Biol.Chem. 261, 2434-2440
21 Mirkin, B. L., Clark, S. H., and Zhang, C. (2002) Inhibition of human neuroblastoma cell
proliferation and EGF receptor phosphorylation by gangliosides GM1, GM3, GD1A and
GT1B. Cell Prolif. 35, 105-115
22 Sachinidis, A., Kraus, R., Seul, C., Meyer zu Brickwedde, M. K., Schulte, K., Ko, Y., Hoppe,
J., and Vetter, H. (1996) Gangliosides GM1, GM2 and GM3 inhibit the platelet-derived
growth factor-induced signalling transduction pathway in vascular smooth muscle cells by
different mechanisms. Eur.J.Cell Biol. 71, 79-88
23 Van Brocklyn, J., Bremer, E. G., and Yates, A. J. (1993) Gangliosides inhibit platelet-derived
growth factor-stimulated receptor dimerization in human glioma U-1242MG and Swiss 3T3
cells. J.Neurochem. 61, 371-374
24 Geoffroy, K., Wiernsperger, N., Lagarde, M., and El Bawab, S. (2004) Bimodal effect of
advanced glycation end products on mesangial cell proliferation is mediated by neutral
ceramidase regulation and endogenous sphingolipids. J.Biol.Chem. 279, 34343-34352
25 Schleicher, E. D. and Weigert, C. (2000) Role of the hexosamine biosynthetic pathway in
diabetic nephropathy. Kidney Int.Suppl 77, S13-S18
26 Roos, M. D., Han, I. O., Paterson, A. J., and Kudlow, J. E. (1996) Role of glucosamine
synthesis in the stimulation of TGF-alpha gene transcription by glucose and EGF.
Am.J.Physiol 270, C803-C811
27 Weigert, C., Brodbeck, K., Lehmann, R., Haring, H. U., and Schleicher, E. D. (2001)
Overexpression of glutamine:fructose-6-phosphate-amidotransferase induces transforming
growth factor-beta1 synthesis in NIH-3T3 fibroblasts. FEBS Lett. 488, 95-99
28 Ziyadeh, F. N., Sharma, K., Ericksen, M., and Wolf, G. (1994) Stimulation of collagen gene
expression and protein synthesis in murine mesangial cells by high glucose is mediated by
autocrine activation of transforming growth factor-beta. J.Clin.Invest 93, 536-542
29 Marshall, S., Nadeau, O., and Yamasaki, K. (2004) Dynamic actions of glucose and
glucosamine on hexosamine biosynthesis in isolated adipocytes: differential effects on

glucosamine 6-phosphate, UDP-N-acetylglucosamine, and ATP levels. J Biol.Chem. 279,
35313-35319
30 Griffin, S. V., Pichler, R., Wada, T., Vaughan, M., Durvasula, R., and Shankland, S. J. (2003)
The role of cell cycle proteins in Glomerular disease. Semin.Nephrol. 23, 569-582
31 Fan, Y. P. and Weiss, R. H. (2004) Exogenous attenuation of p21(Waf1/Cip1) decreases
mesangial cell hypertrophy as a result of hyperglycemia and IGF-1. J.Am.Soc.Nephrol. 15,
575-584
32 al Douahji, M., Brugarolas, J., Brown, P. A., Stehman-Breen, C. O., Alpers, C. E., and
Shankland, S. J. (1999) The cyclin kinase inhibitor p21WAF1/CIP1 is required for glomerular
hypertrophy in experimental diabetic nephropathy. Kidney Int. 56, 1691-1699
33 Goldberg, H. J., Whiteside, C. I., and Fantus, I. G. (2002) The hexosamine pathway regulates
the plasminogen activator inhibitor-1 gene promoter and Sp1 transcriptional activation
through protein kinase C-beta I and -delta. J.Biol.Chem. 277, 33833-33841
34 Nakatsuji, Y. and Miller, R. H. (2001) Selective cell-cycle arrest and induction of apoptosis in
proliferating neural cells by ganglioside GM3. Exp.Neurol. 168, 290-299
35 Sasaki, A., Hata, K., Suzuki, S., Sawada, M., Wada, T., Yamaguchi, K., Obinata, M., Tateno,
H., Suzuki, H., and Miyagi, T. (2003) Overexpression of plasma membrane-associated
sialidase attenuates insulin signaling in transgenic mice. J.Biol.Chem. 278, 27896-27902
36 Tagami, S., Inokuchi, J. J., Kabayama, K., Yoshimura, H., Kitamura, F., Uemura, S., Ogawa,
C., Ishii, A., Saito, M., Ohtsuka, Y., Sakaue, S., and Igarashi, Y. (2002) Ganglioside GM3
participates in the pathological conditions of insulin resistance. J.Biol.Chem. 277, 3085-3092

FIGURE LEGENDS
Figure 1 Glucosamine decreases RMC growth and induces hypertrophy Mesangial cells were exposed to 0.5 mM or 5 mM glucosamine for 4 days. Every day, cells were collected, cell number was counted and total proteins were measured. Growth curves corresponding to cell number expressed in 105 cells (A) and total proteins expressed in µg (B) are shown. Total proteins to cell number ratio is presented in (C) expressed in µg proteins per 105 cells. Results represent means ± SEM of 4 independent experiments. (D) Microscopy phase images of cells under control conditions or cells treated with glucosamine. * p < 0.05 vs control.
Figure 2 Dose-dependent effects of glucosamine on cell-cycle arrest RMC were treated with increasing concentrations of GlcN for 48 hours. Cells were then collected and analysed by flow cytometry as described under Materials and Methods. Results are expressed as the percentage of cells in G0/G1, S, and G2/M.
Figure 3 Glucosamine, GM2 and GM1 gangliosides increase p21Waf1/Cip1 expression RMC were exposed to glucosamine (0.5 or 5 mM), GM2 or GM1 (60 µM) for 48 hours. Cells were then lysed and equal amounts of proteins immunobloted with monoclonal anti-p21Waf1/Cip1 (A) or anti-p27Kip1 (B)
antibodies. Membranes were immunobloted next with anti-β-actin to control for equal protein loading and
transfer. Representative immunoblots and quantification obtained by densitometric analysis of the bands are
shown. Results were corrected to β-actin signal and are expressed as fold change compared to control.
Results represent means ± SEM of 4 independent experiments.
Figure 4 Effect of glucosamine on ganglioside profile (A and B) RMC were treated with glucosamine (0.5 or 5 mM) for 48 hours and then collected. Gangliosides were extracted, purified and analysed by HPTLC and resorcinol staining. Representative HPTLC (A) and densitometric quantitation (B) are shown. Results are expressed as µg of gangliosides per mg of proteins and represent means ± SEM of 4 independent experiments each performed in duplicates. Under control
conditions, gangliosides quantities were 4.62 ± 0.37, 1.65 ± 0.11 and 0.30 ± 0.05 µg/mg proteins for GM3,
GD1a and GD3, respectively. ND, not detectable. * p < 0.05 vs control. (C) RMC were labelled for 48 h in the presence of 0.2 µCi/ml 14C glucosamine for control cells and 2 mM of glucosamine at a specific activity of 0.5 mCi/ml for treated cells. At the end of incubation, cells were washed extensively and gangliosides were extracted, purified, and separated by HPTLC as described under

Experimental procedures. Radioactivity associated to gangliosides was then analyzed by autoradiography of the HPTLC plates.
Figure 5 GM2 and GM1 gangliosides induce mesangial cells hypertrophy RMC were cultured in the presence of GM2 or GM1 (60 µM) for 48 hours. Control conditions were complete medium with BSA-10 mM Hepes pH 7.4. Cells were then harvested and counted using haemocytometer, and total proteins measured. Cell number expressed in 105 cells (A) and total proteins expressed in µg (B) are presented. Total proteins to cell number ratio, used to assess hypertrophy is expressed in µg proteins per 105 cells (C). Results represent means ± SEM of 4-6 independent experiments. * p < 0.05 vs control.
Table 1 Glucosamine, GM2 and GM1 gangliosides block cell-cycle of mesangial cells RMC were treated with glucosamine (0.5 or 5 mM) or cultured in the presence of GM2 or GM1 (60 µM) for 48 hours. Cells were then collected and analysed by flow cytometry as described under Experimental methods. Results are expressed as the percentage of cells in each phase of the cell-cycle G0/G1, S and G2/M. Results represent means ± SEM of 5 independent experiments.
Figure 1
C
A
B
0
20
40
60
80
100
0 1 2 3 4days
tota
l pro
tein
s / c
ell n
umbe
r controlGlcN 0.5 mMGlcN 5 mM
0
10
20
30
40
0 1 2 3 4 5days
cell
num
ber (
105 )
controlGlcN 0.5 mMGlcN 5 mM
0
200
400
600
800
1000
1200
tota
l pro
tein
s (µg
)
0 1 2 3 4 5days
controlGlcN 0.5 mMGlcN 5 mM
D
control
GlcN 0.5 mM
GlcN 5 mM
*** *
*
*
*C
A
B
0
20
40
60
80
100
0 1 2 3 4days
tota
l pro
tein
s / c
ell n
umbe
r controlGlcN 0.5 mMGlcN 5 mM
0
10
20
30
40
0 1 2 3 4 5days
cell
num
ber (
105 )
controlGlcN 0.5 mMGlcN 5 mM
0
200
400
600
800
1000
1200
tota
l pro
tein
s (µg
)
0 1 2 3 4 5days
controlGlcN 0.5 mMGlcN 5 mM
D
control
GlcN 0.5 mM
GlcN 5 mMC
A
B
0
20
40
60
80
100
0 1 2 3 4days
tota
l pro
tein
s / c
ell n
umbe
r controlGlcN 0.5 mMGlcN 5 mM
controlGlcN 0.5 mMGlcN 5 mM
0
10
20
30
40
0 1 2 3 4 5days
cell
num
ber (
105 )
controlGlcN 0.5 mMGlcN 5 mM
0
10
20
30
40
0 1 2 3 4 5days
0 1 2 3 4 5days
cell
num
ber (
105 )
controlGlcN 0.5 mMGlcN 5 mM
controlGlcN 0.5 mMGlcN 5 mM
0
200
400
600
800
1000
1200
tota
l pro
tein
s (µg
)
0 1 2 3 4 5days
0 1 2 3 4 5days
controlGlcN 0.5 mMGlcN 5 mM
controlGlcN 0.5 mMGlcN 5 mM
D
controlcontrol
GlcN 0.5 mMGlcN 0.5 mM
GlcN 5 mMGlcN 5 mM
*** *
*
*
*

Figure 2
0
50
60
70
80
90
100
0 0.5 1 2 5GlcN (mM)
G2/MSG0/G1
% o
f cel
ls
0
50
60
70
80
90
100
0 0.5 1 2 5GlcN (mM)
G2/MSG0/G1
G2/MSG0/G1
% o
f cel
ls
Figure 3
p27
GM2 GM1control 0.5 5GlcN (mM)
β-actin
B
0
0.2
0.4
0.6
0.8
1
1.2
1.4
fold
cha
nge
control 0.5 5 GM1 GM2GlcN (mM)
β-actin
p21
control 0.5 5 GM2 GM1GlcN (mM)
A
0
0.5
1
1.5
2
2.5
3
3.5
4
4.5
fold
cha
nge
control 0.5 5 GM1 GM2GlcN (mM)
p27
GM2 GM1control 0.5 5GlcN (mM)
β-actin
B
0
0.2
0.4
0.6
0.8
1
1.2
1.4
fold
cha
nge
control 0.5 5 GM1 GM2GlcN (mM)
0
0.2
0.4
0.6
0.8
1
1.2
1.4
fold
cha
nge
0
0.2
0.4
0.6
0.8
1
1.2
1.4
fold
cha
nge
control 0.5 5 GM1 GM2GlcN (mM)
control 0.5 5 GM1 GM2GlcN (mM)
β-actin
p21
control 0.5 5 GM2 GM1GlcN (mM)
A
0
0.5
1
1.5
2
2.5
3
3.5
4
4.5
fold
cha
nge
control 0.5 5 GM1 GM2GlcN (mM)
0
0.5
1
1.5
2
2.5
3
3.5
4
4.5
fold
cha
nge
control 0.5 5 GM1 GM2GlcN (mM)
control 0.5 5 GM1 GM2GlcN (mM)
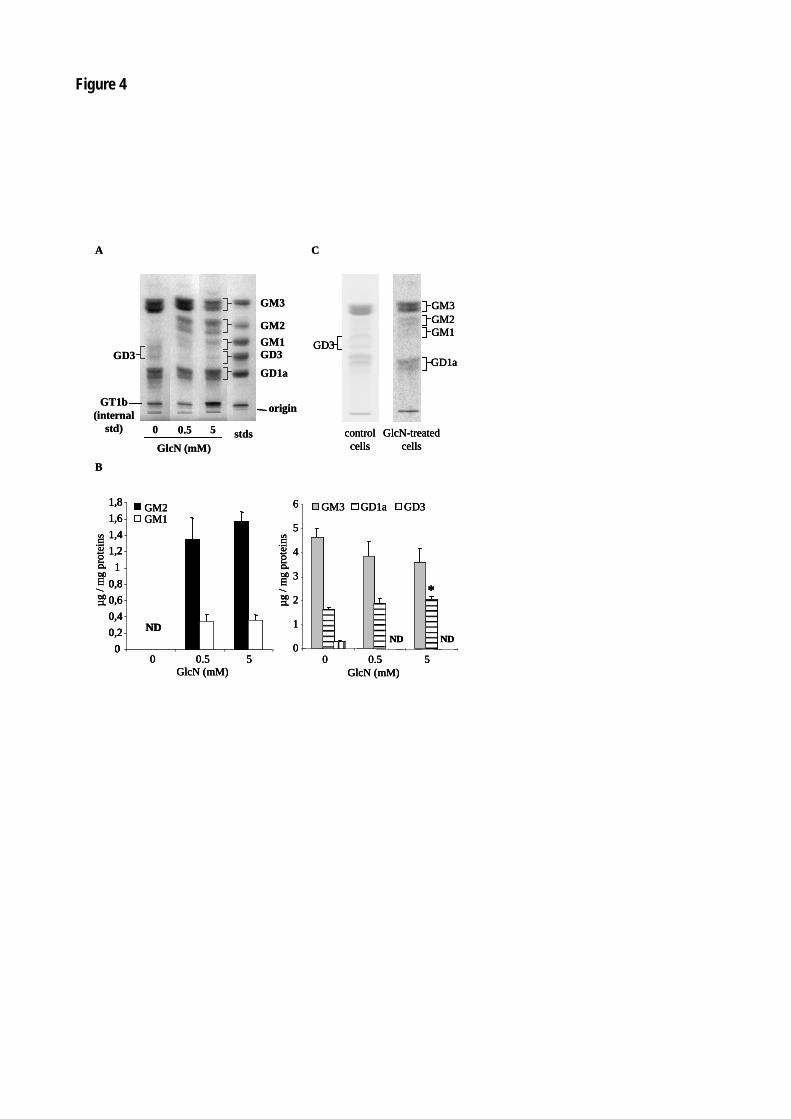
Figure 4
A
B
0 50.50
1
2
3
4
5
6
GlcN (mM)
µg /
mg
prot
eins
GM3 GD1a GD3
µg /
mg
prot
eins
ND
00,20,40,60,8
11,21,41,61,8 GM2
GM1
0 50.5GlcN (mM)
*
ND ND
GT1b (internal
std)
GlcN (mM)stds50 0.5
origin
GM1GM2
GM3
GD3
GD1aGD3
C
control cells
GlcN-treated cells
GM1GM2GM3
GD1aGD3
A
B
0 50.50
1
2
3
4
5
6
GlcN (mM)
µg /
mg
prot
eins
GM3 GD1a GD3
µg /
mg
prot
eins
ND
00,20,40,60,8
11,21,41,61,8 GM2
GM1
0 50.5GlcN (mM)
*
ND ND
0 50.50
1
2
3
4
5
6
GlcN (mM)
µg /
mg
prot
eins
GM3 GD1a GD3
µg /
mg
prot
eins
ND
00,20,40,60,8
11,21,41,61,8
00,20,40,60,8
11,21,41,61,8 GM2
GM1
0 50.5GlcN (mM)
*
ND ND
GT1b (internal
std)
GlcN (mM)stds50 0.5
origin
GM1GM2
GM3
GD3
GD1aGD3
C
control cells
GlcN-treated cells
GM1GM2GM3
GD1aGD3
control cells
GlcN-treated cells
GM1GM2GM3
GD1aGD3
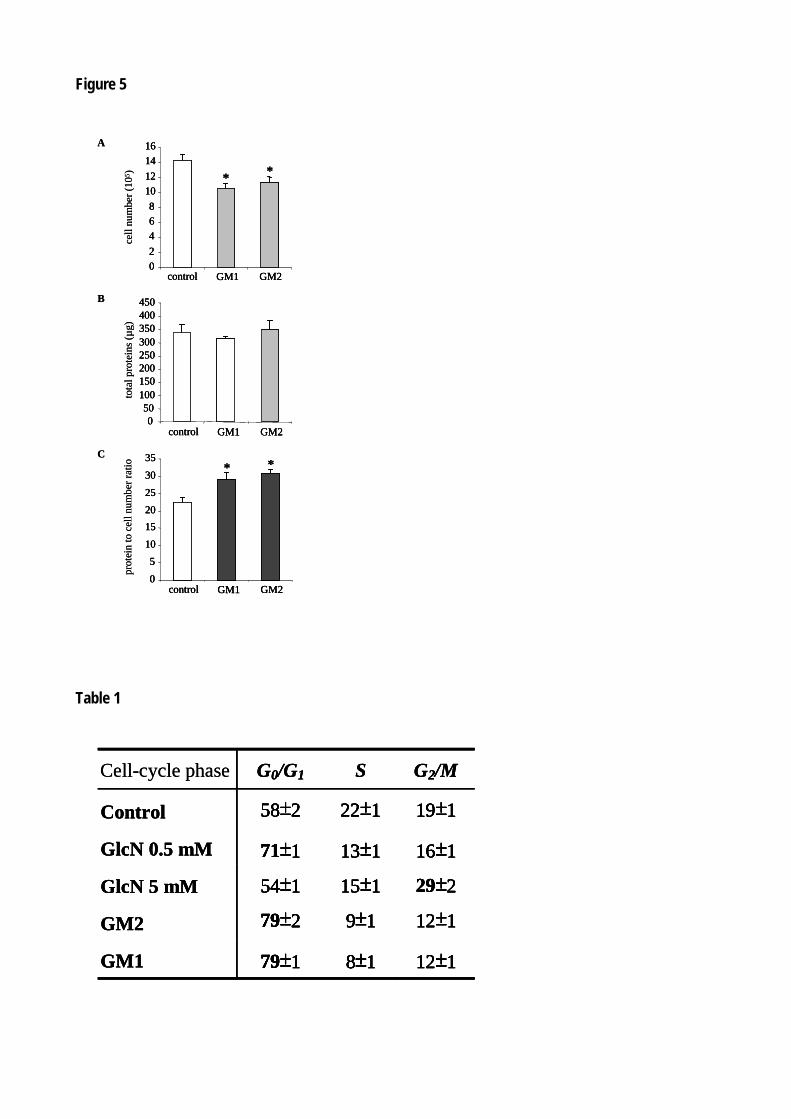
Figure 5
A
control GM1 GM2
cell
num
ber (
105 )
B
tota
l pro
tein
s (µg
)
control GM1 GM2
C
prot
ein
to c
ell n
umbe
r rat
io
control GM1 GM2
* *
* *
02468
10121416
050
100150200250300350400450
0
51015202530
35
A
control GM1 GM2 control GM1 GM2
cell
num
ber (
105 )
B
tota
l pro
tein
s (µg
)
control GM1 GM2 control GM1 GM2
C
prot
ein
to c
ell n
umbe
r rat
io
control GM1 GM2 control GM1 GM2
* *
* *
02468
10121416
02468
10121416
050
100150200250300350400450
050
100150200250300350400450
0
51015202530
35
0
51015202530
35
Table 1
G0/G1 S G2/M
Control ±58 2 22±1 19±1
GlcN 0.5 mM 71±1 13±1 16±1
GlcN 5 mM 54±1 15±1 29±2
GM2 79±2 9±1 ±12 1
GM1 79±1 8±1 12±1
Cell-cycle phase G0/G1 S G2/M
Control ±58 2 22±1 19±1
GlcN 0.5 mM 71±1 13±1 16±1
GlcN 5 mM 54±1 15±1 29±2
GM2 79±2 9±1 ±12 1
GM1 79±1 8±1 12±1
G0/G1 S G2/M
Control ±58 2 22±1 19±1±58 258 2 22±122±1 19±119±1
GlcN 0.5 mM 71±1 13±1 16±171±171±1 13±113±1 16±116±1
GlcN 5 mM 54±1 15±1 29±254±154±1 15±115±1 29±229±2
GM2 79±2 9±1 ±12 179±279±2 9±19±1 ±12 112 1
GM1 79±1 8±1 12±179±179±1 8±18±1 12±112±1
Cell-cycle phase