Embed Size (px)
Citation preview

A07 1IFT3295 - F. Major
Structure des protéines
• Importance de la structure des protéines• Structure primaire : la séquence d’acides aminés• Structure secondaire : structure tridimensionnelle locale• Structure tertiaire : structure tridimensionnelle globale• Structure quaternaire : associations de plusieurs chaînes
peptidiques• Conclusion

A07 2IFT3295 - F. Major
Importance de la structure des protéines
• Une importante fraction de la structure et de lafonction de la cellule implique les protéines.
• La structure d’une protéine lui confère sa fonction.

A07 3IFT3295 - F. Major
Structure primaire : la séquence d’acides aminés
• Les protéines sont des polymères linéaires(séquence) d’acides aminés.– Proposé par Fischer et Hofmeister en 1902.
• La séquence d’une protéine détermine sa structuretridimensionnelle.
– Anfinsen CB, Haber E, Sela M, White FH, the kinetics of formation of native ribonucleaseduring oxydation of the reduced polypeptide chain, PNAS 47 (1961) 1309-1314.
• On appelle peptide, une protéine de massemoléculaire de moins de 1000 daltons (g/mol).

A07 4IFT3295 - F. Major
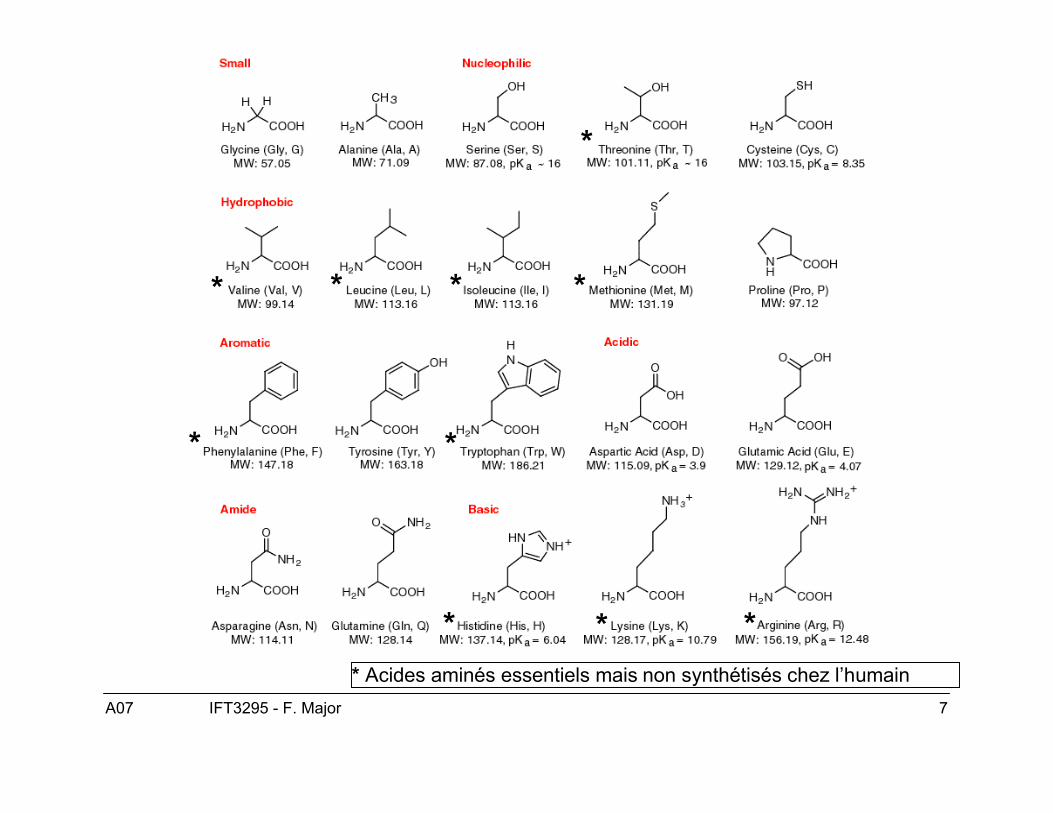
Acides aminés
Petites molécules composés d’un groupe amide(NH2), un carboxyle (COOH) et d’un hydrogèneattaché au carbone central (α). Chaque acide aminépossède également une chaîne latérale (groupe R)attaché lui aussi au Cα. Ce dernier est spécifique autype d’acide aminé et qui lui confère ses propriétéschimiques.

A07 5IFT3295 - F. Major
Groupes d’un acide aminé
Chaîne latérale
Groupe acideou carboxyleGroupe
amide
R : un des 20 acides aminés
A07 6IFT3295 - F. Major
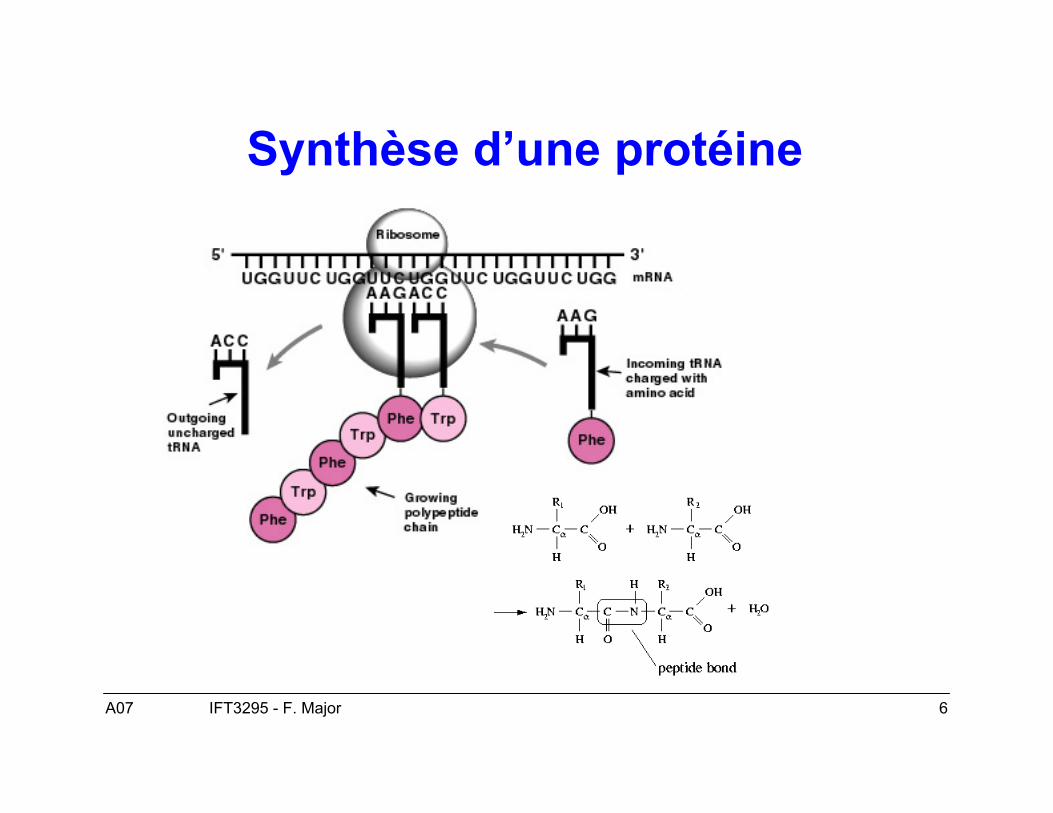
Synthèse d’une protéine
A07 7IFT3295 - F. Major
* * *
* *
*
*
** ** Acides aminés essentiels mais non synthétisés chez l’humain

A07 8IFT3295 - F. Major
Alimentation et acides aminés
Le lait et les blancs d’oeufs sont des protéinescomplètes, ils contiennent 8 acides aminés essentielsnon synthétisés par l’humain.
A07 9IFT3295 - F. Major
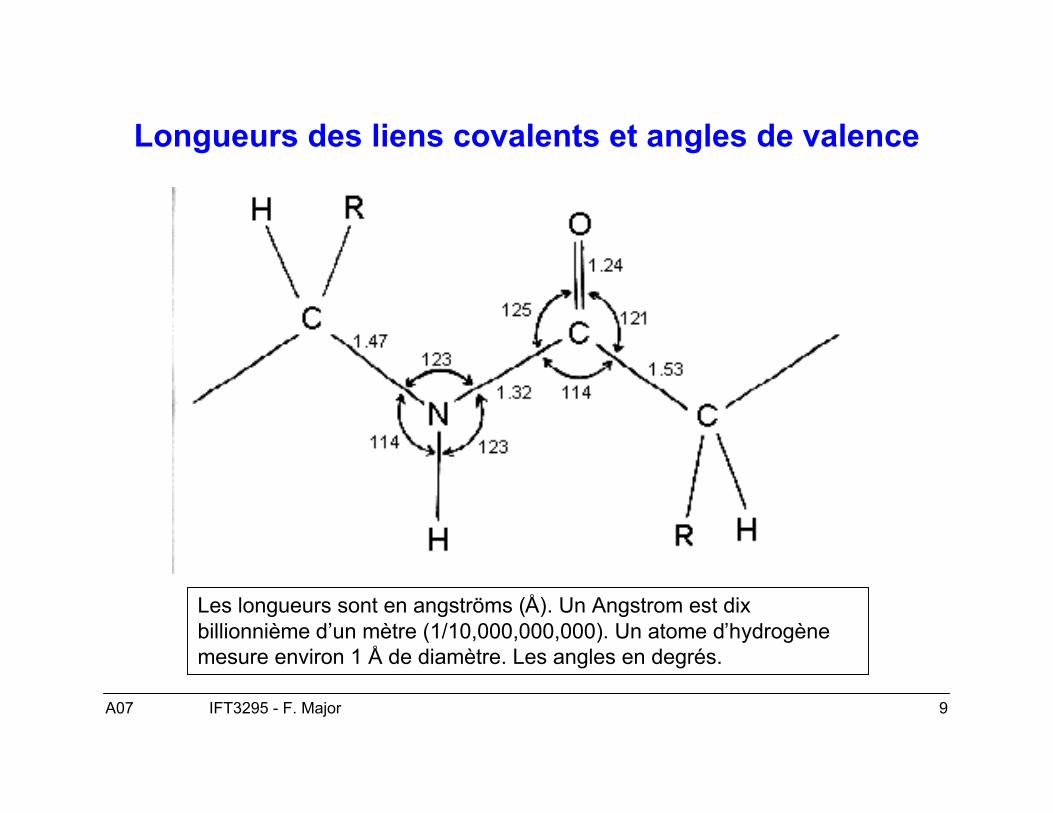
Longueurs des liens covalents et angles de valence
Les longueurs sont en angströms (Å). Un Angstrom est dixbillionnième d’un mètre (1/10,000,000,000). Un atome d’hydrogènemesure environ 1 Å de diamètre. Les angles en degrés.
A07 10IFT3295 - F. Major
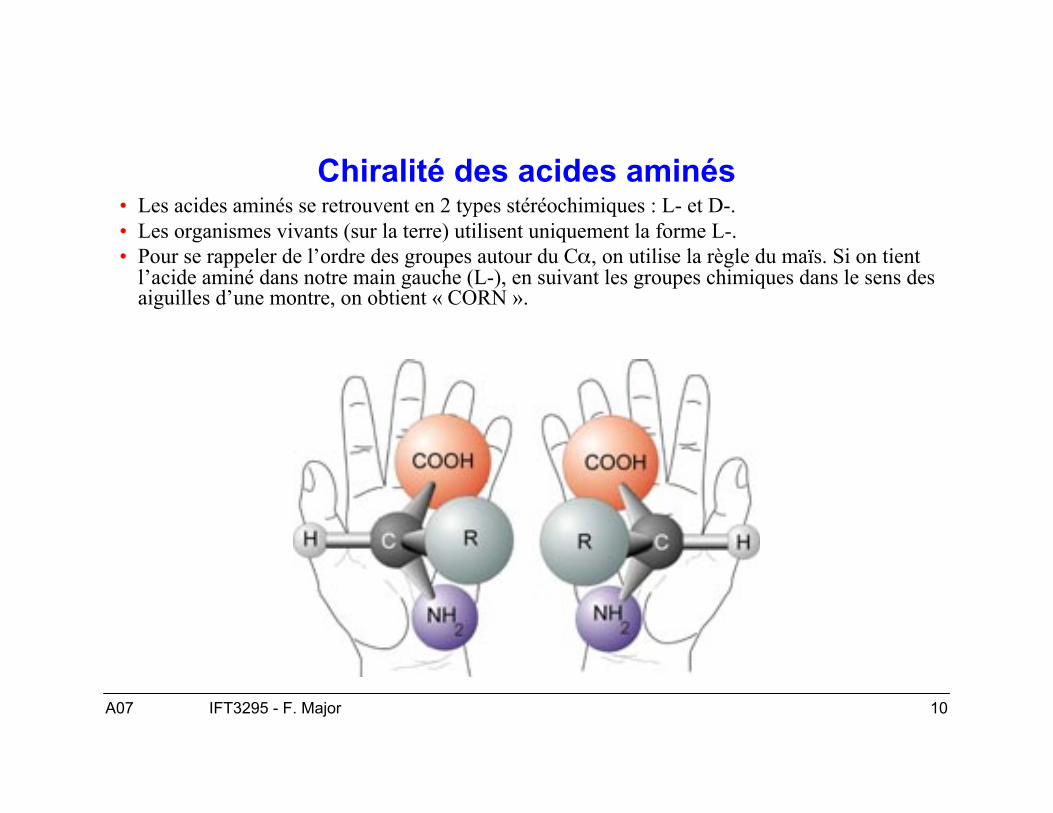
Chiralité des acides aminés• Les acides aminés se retrouvent en 2 types stéréochimiques : L- et D-.• Les organismes vivants (sur la terre) utilisent uniquement la forme L-.• Pour se rappeler de l’ordre des groupes autour du Cα, on utilise la règle du maïs. Si on tient
l’acide aminé dans notre main gauche (L-), en suivant les groupes chimiques dans le sens desaiguilles d’une montre, on obtient « CORN ».
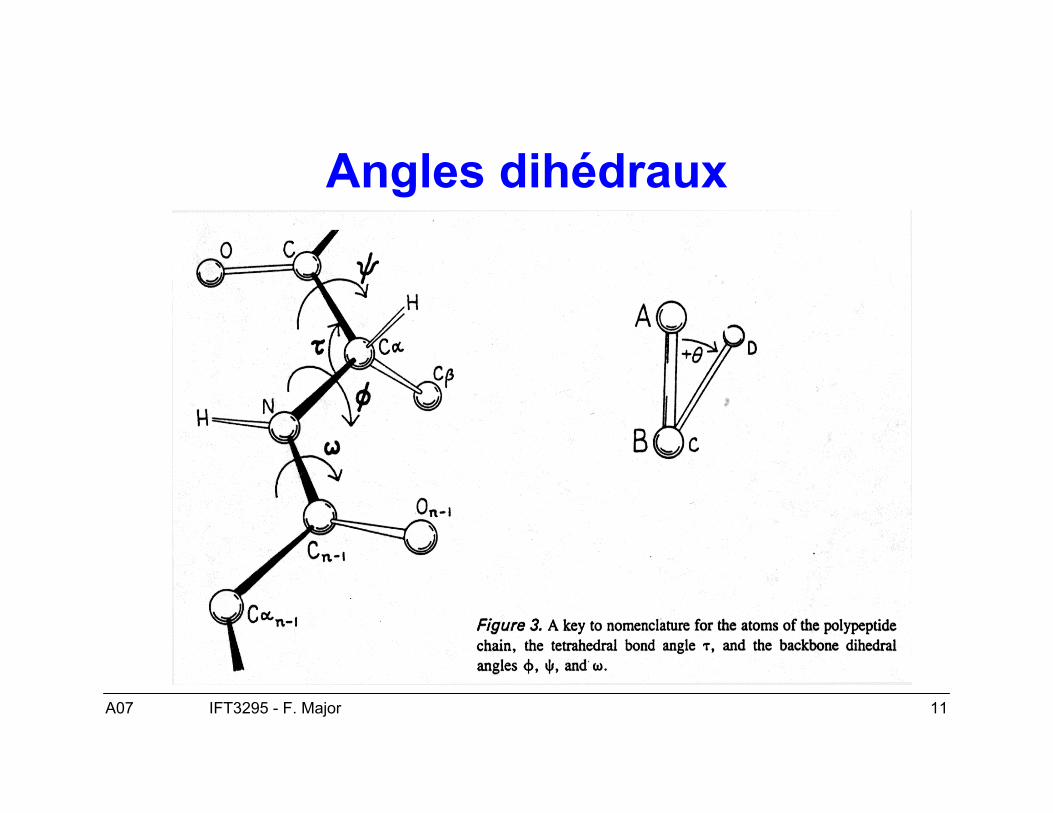
A07 11IFT3295 - F. Major
Angles dihédraux

A07 12IFT3295 - F. Major
Structure secondaire : structuretridimensionnelle locale
• Deux conformations dominantes :– Hélices α– Feuillets β
• Les angles ϕ et ψ déterminent la conformationlocale de la chaîne principale des acides aminés.
• Les ponts hydrogènes stabilisent les éléments destructure secondaire.
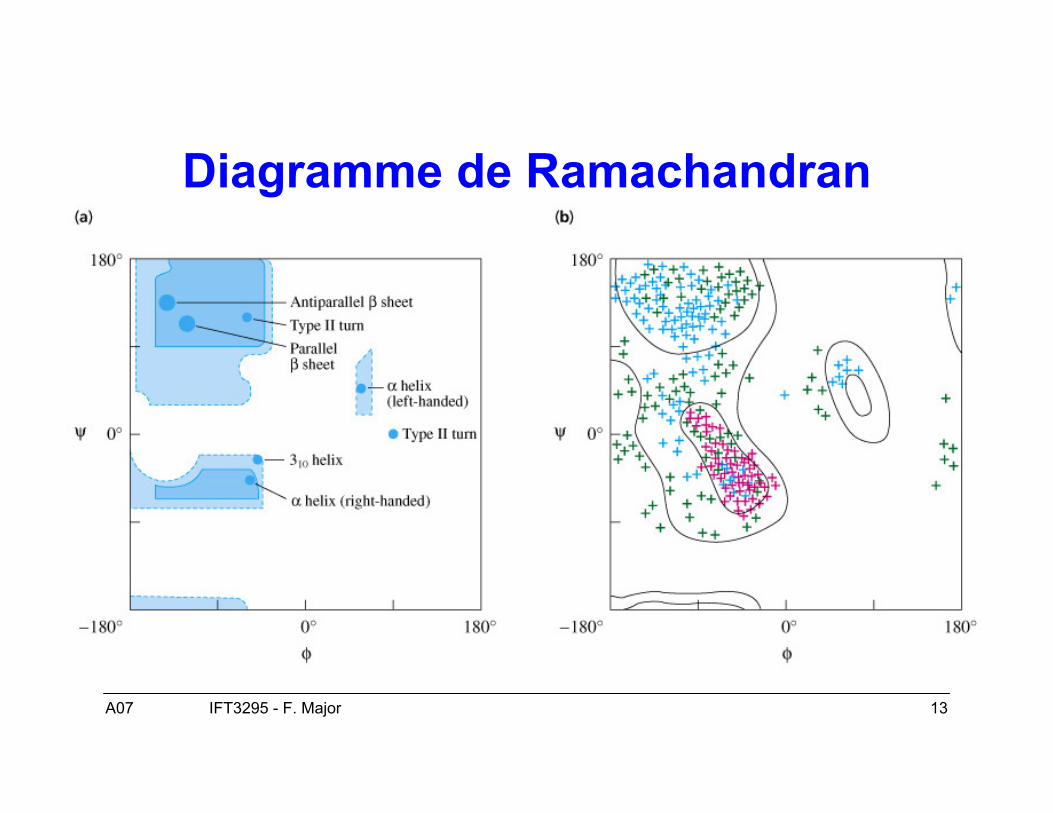
A07 13IFT3295 - F. Major
Diagramme de Ramachandran
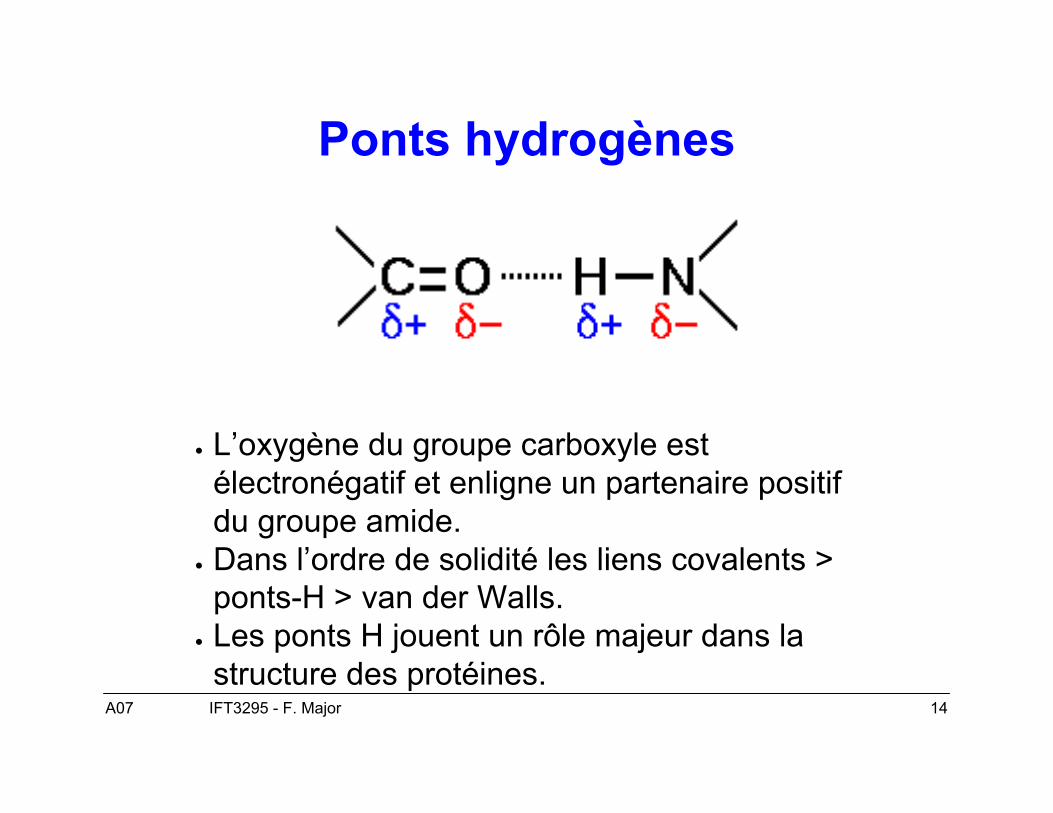
A07 14IFT3295 - F. Major
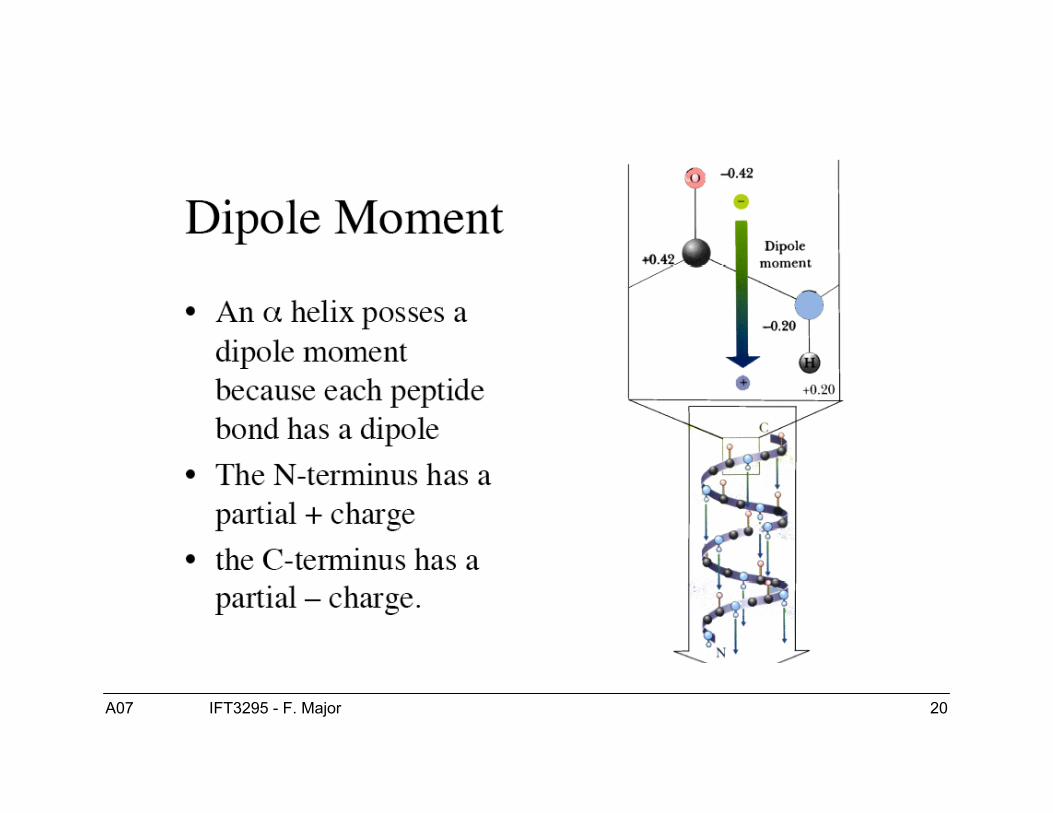
Ponts hydrogènes
● L’oxygène du groupe carboxyle estélectronégatif et enligne un partenaire positifdu groupe amide.
● Dans l’ordre de solidité les liens covalents >ponts-H > van der Walls.
● Les ponts H jouent un rôle majeur dans lastructure des protéines.

A07 15IFT3295 - F. Major
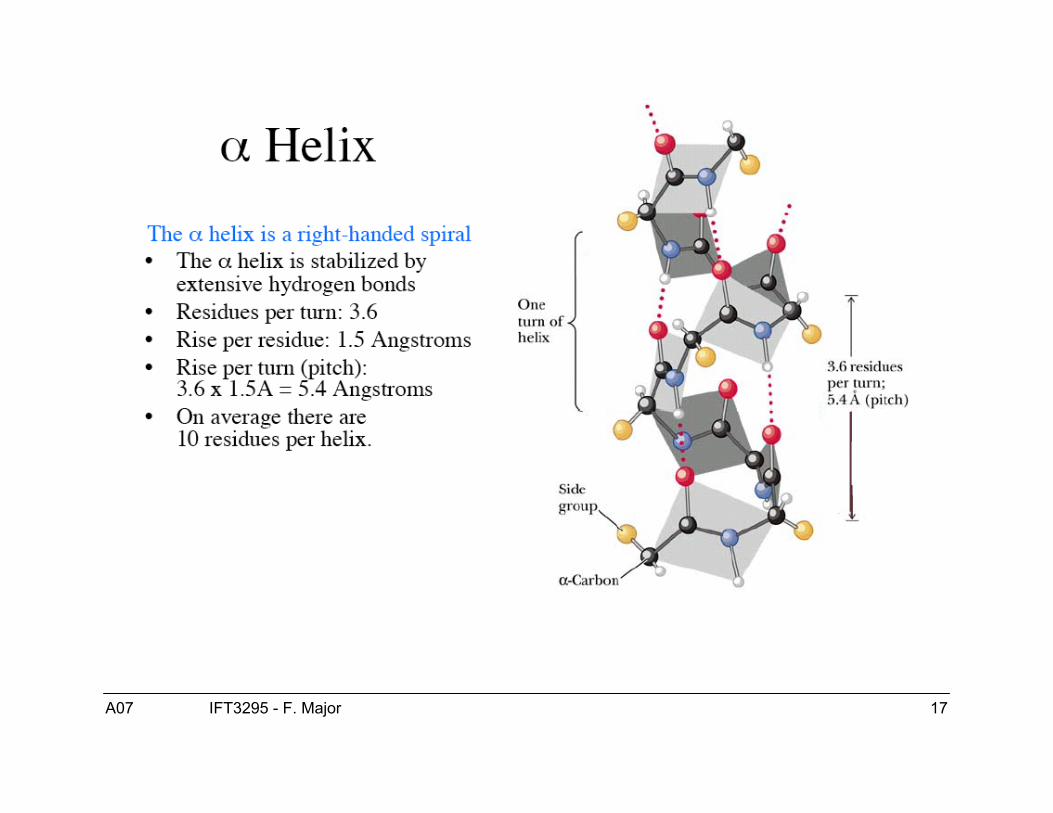
Hélice αPrédite par Pauling et ses collaborateurs en 1951.
Confirmée expérimentalement + de 10 ans + tard…
L. Pauling, R. B. Corey, H. R. Branson, The structure of proteins: Two hydrogen-bondedhelical configurations of the polypeptide chain, PNAS (USA) 37 (1951) 205-211.
C. C. Blake, D. F. Koenig, G. A. Mair, A. C. North, D. C. Phillips, V. R. Sarma,Structure of hen egg-white lysozyme. a three-dimensional fourier synthesis at 2angstrom resolution, Nature 206 (1965) 757-761.
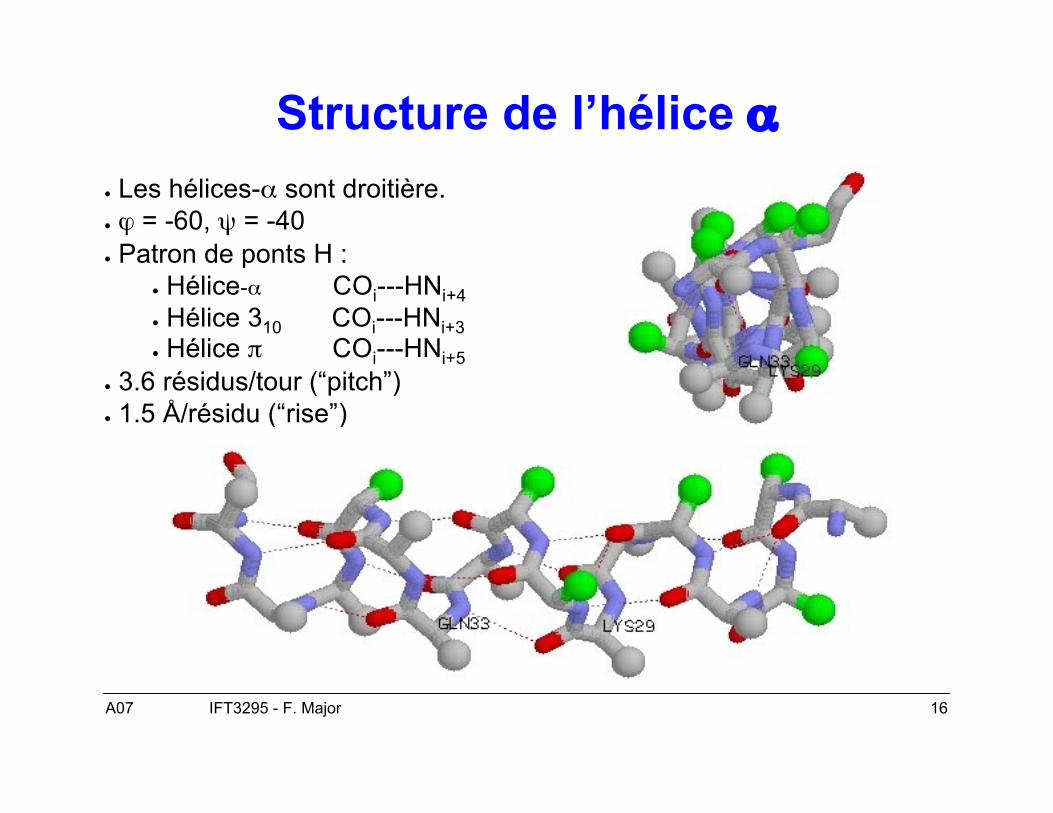
A07 16IFT3295 - F. Major
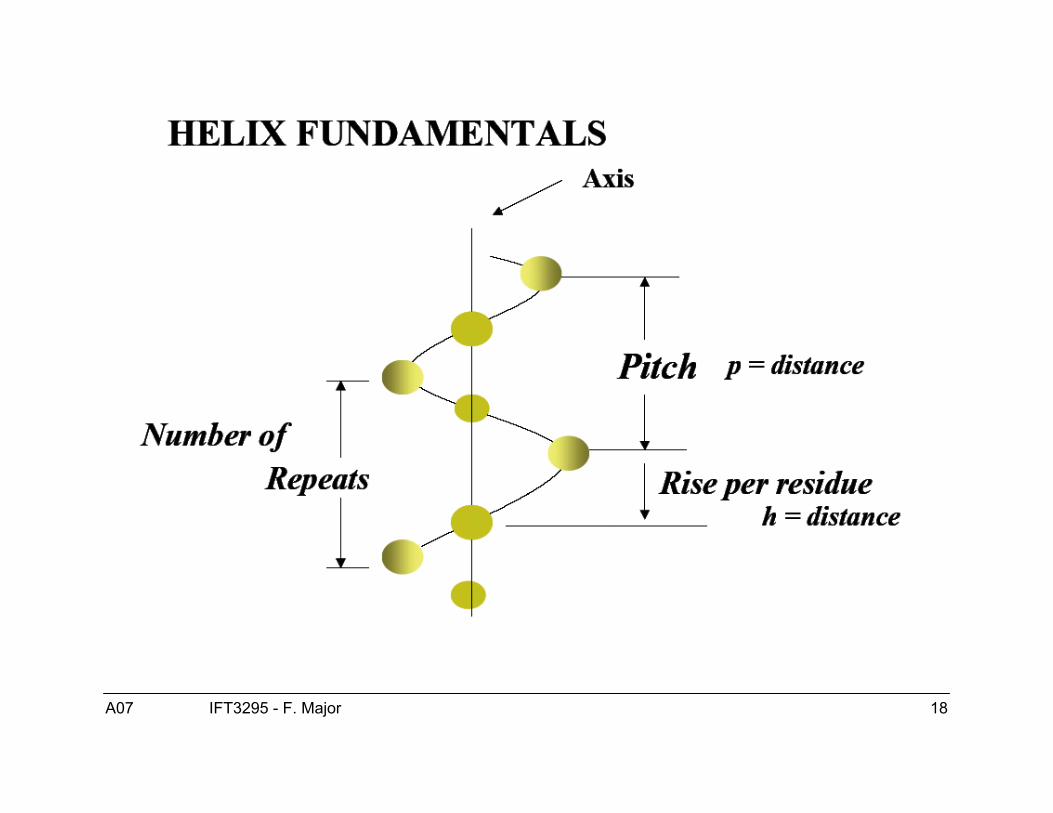
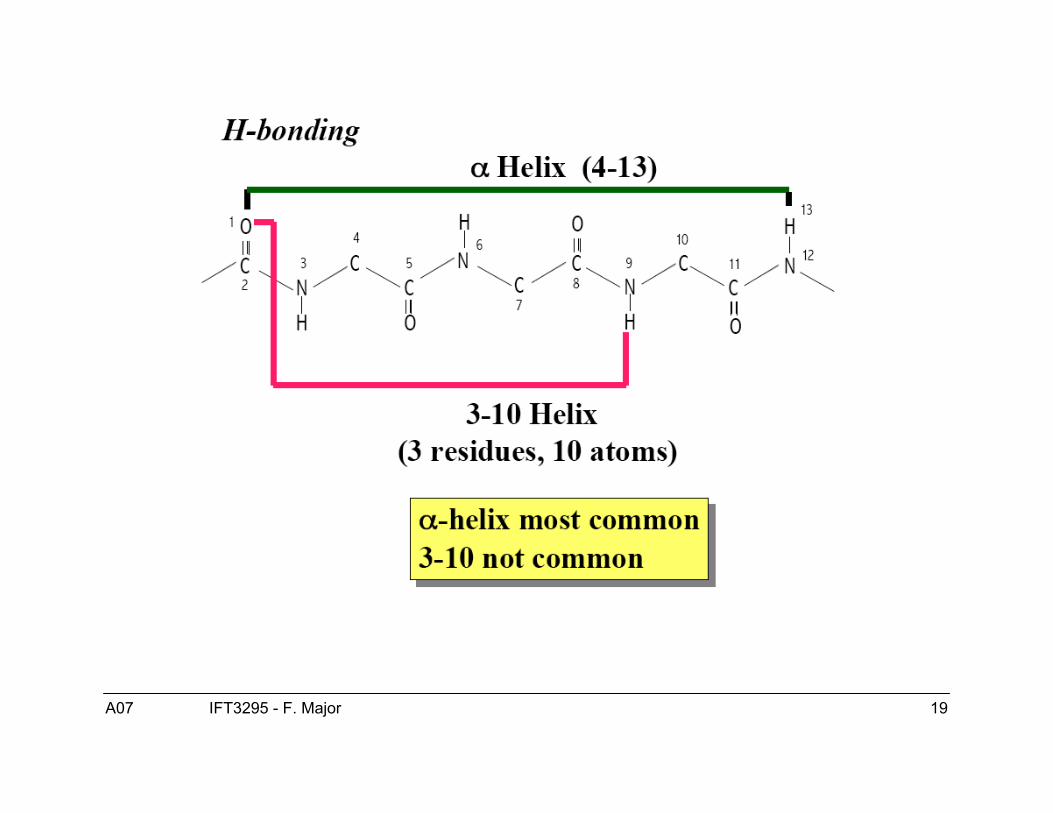
Structure de l’hélice α● Les hélices-α sont droitière.● ϕ = -60, ψ = -40● Patron de ponts H :
● Hélice-α COi---HNi+4● Hélice 310 COi---HNi+3● Hélice π COi---HNi+5
● 3.6 résidus/tour (“pitch”)● 1.5 Å/résidu (“rise”)
A07 17IFT3295 - F. Major
A07 18IFT3295 - F. Major
A07 19IFT3295 - F. Major
A07 20IFT3295 - F. Major

A07 21IFT3295 - F. Major
Code PDB 1A7M

A07 22IFT3295 - F. Major
Feuillets βPrédits par Pauling et ses collaborateurs en 1951.
L. Pauling, R. B. Corey, The pleated sheet, a new layer configuration ofpolypeptide chains, PNAS (USA) 37 (1951) 251-256.
Formés et stabilisés par des ponts-H entre des brinspolypeptidiques adjacents, les brins-β.
Il y a n! façons d’agencer n brins et puisque nous avons 2possibilités d’orientation pour chaque brin, on a n! x 2n
arrangements possibles.
Par contre, plusieurs arrangements sont symétriques, en fait on a 2axes de symétrie, alors on aura ¼ * n! * 2n = n! * 2(n - 2)
arrangements de feuillets pour n brins.n M2 23 124 965 960
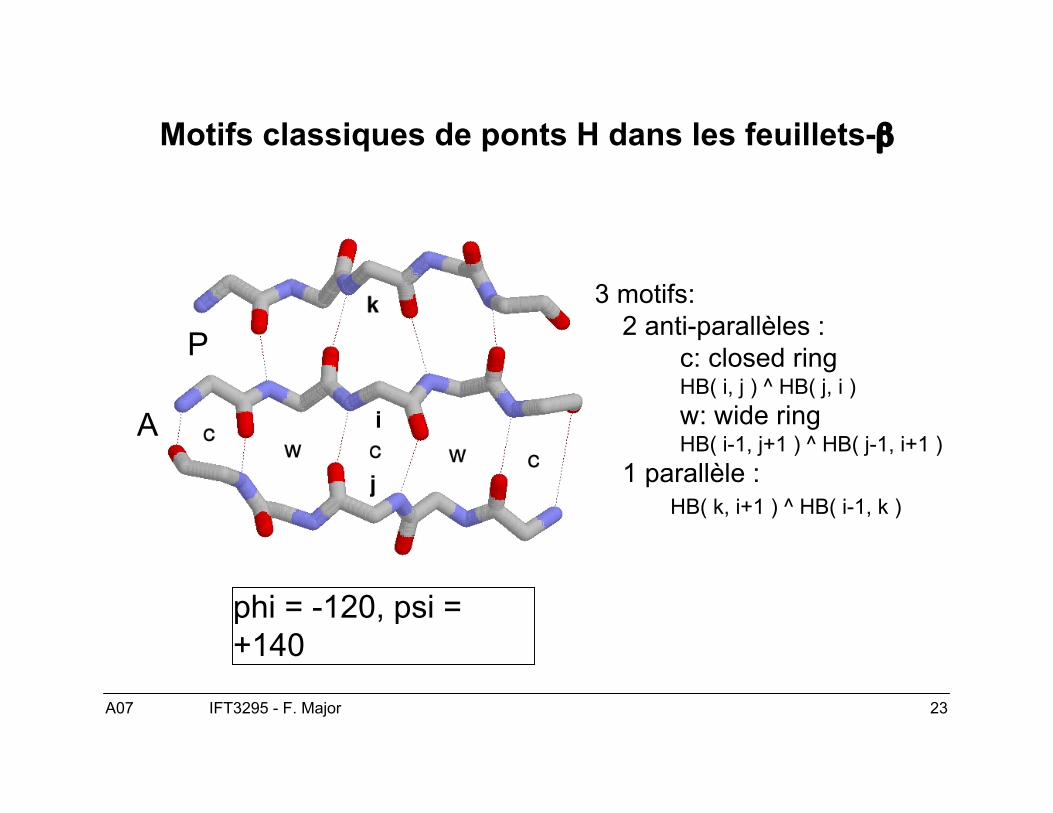
A07 23IFT3295 - F. Major
Motifs classiques de ponts H dans les feuillets-β
3 motifs:2 anti-parallèles :
c: closed ringHB( i, j ) ^ HB( j, i )w: wide ringHB( i-1, j+1 ) ^ HB( j-1, i+1 )
1 parallèle :HB( k, i+1 ) ^ HB( i-1, k )
P
A
phi = -120, psi =+140
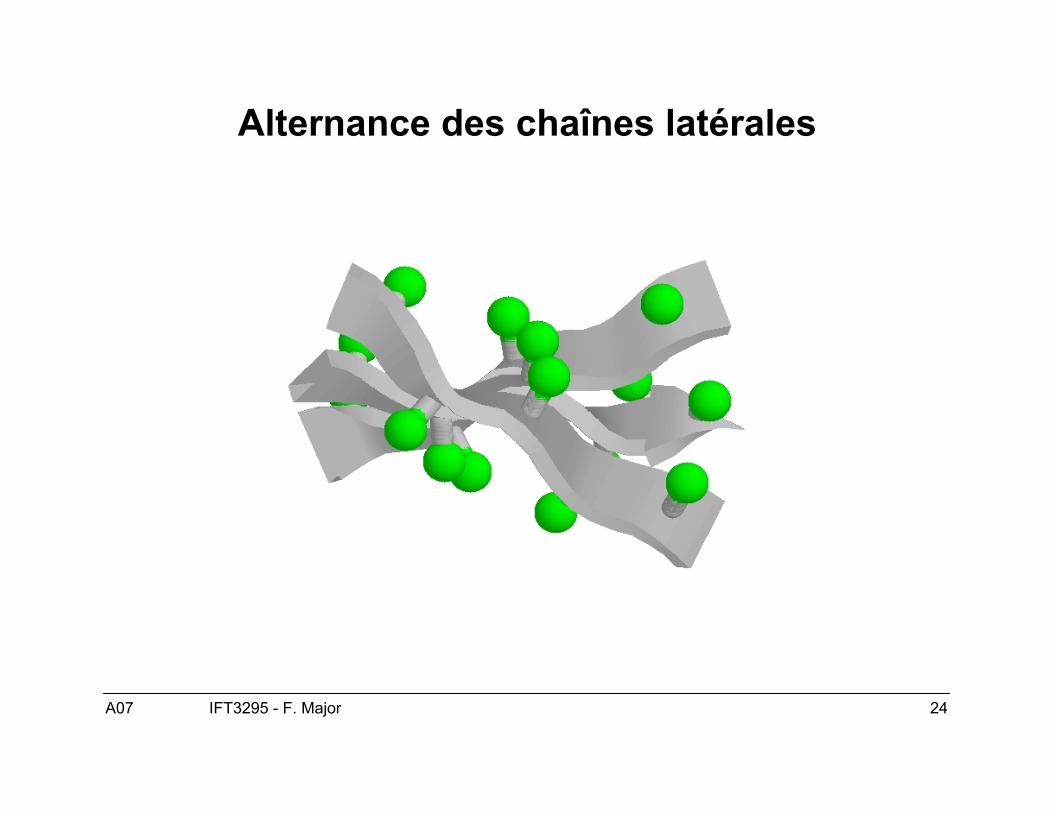
A07 24IFT3295 - F. Major
Alternance des chaînes latérales
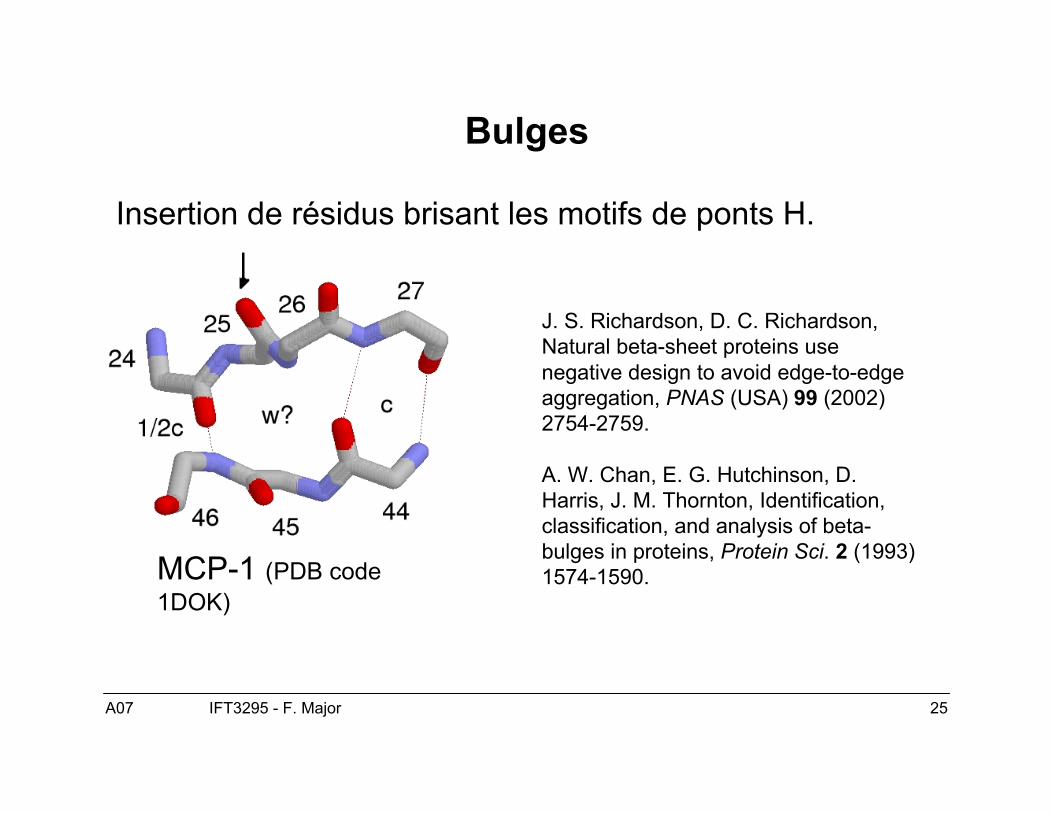
A07 25IFT3295 - F. Major
Bulges
Insertion de résidus brisant les motifs de ponts H.
J. S. Richardson, D. C. Richardson,Natural beta-sheet proteins usenegative design to avoid edge-to-edgeaggregation, PNAS (USA) 99 (2002)2754-2759.
A. W. Chan, E. G. Hutchinson, D.Harris, J. M. Thornton, Identification,classification, and analysis of beta-bulges in proteins, Protein Sci. 2 (1993)1574-1590.MCP-1 (PDB code
1DOK)

A07 26IFT3295 - F. Major
Structure tertiaire : structuretridimensionnelle globale
• Interactions du squelette responsables de lagénération de la structure secondaire.
• Interactions des chaînes latérales responsables dela structure finale.
• Toute protéine possède un structure tertiairedistinctive qui lui confère sa fonction.
• La structure d’une protéine est maintenue à traversl’évolution malgré les mutations qu’elle peutsubir.

A07 27IFT3295 - F. Major
« Fold » d’une protéine• On réfère souvent à la structure tridimensionnelle finale d’une protéine
comme de son « fold ».• Le processus par lequel la protéine atteint sa structure finale s’appelle
« protein folding » (repliement des protéines).• La structure primaire (séquence) contient toute l’information nécessaire
pour enclencher le processus de « folding » .• L’espace des séquences possédant une structure ordonnée est petit par
rapport à l’espace des séquences (séquence aléatoire ne possède pas destructure organisée).
• Malgré la nature déterministe du processus de « folding », il est toujoursimpossible de prédire la structure finale d’une protéine à partir de saséquence.

A07 28IFT3295 - F. Major
Domaines et motifs• On reconnaît des domaines et des motifs à travers les « fold » de protéines.• Les domaines sont des sections compactes qui représentent structuralement
(et fonctionnellement) des régions indépendantes.• Un domaine est un morceau qui maintiendrait sa structure même s’il était
séparé de la protéine.• Les motifs (qu’on appelle aussi super structures secondaires) sont de petites
sous structures qui ne sont pas structuralement indépendantes et quiconsiste généralement de quelques éléments de structures secondaires(morceaux de structures secondaires).
• Les motifs sont souvent associés à des fonctions et représentent des unitésminimales fonctionnelles (plusieurs motifs peuvent se combiner endomaines spécifiques).

A07 29IFT3295 - F. Major
Interactions tertiaires
• Les interactions intramoléculaires, comme dans lecas des éléments de structure secondaire, sont trèsimportantes pour stabiliser la structure tertiaire.
• À cause de la diversité des 20 acides aminés,plusieurs types d’interactions sont possibles.
• Les protéines existent dans l’eau et les interactionsintermoléculaires avec les molécules d’eau(solvant) sont aussi à considérer dans le « fold »final.

A07 30IFT3295 - F. Major
Noyau hydrophobique• Les résidus possédant des chaînes latérales hydrophobiques ont tendance à se
retrouver à l’intérieur du « fold » de la protéine alors que les résidus chargés etpolaires se retrouvent à la surface pour interagir avec les molécules d’eaupolaires et les ions chargés.
• Comme le squelette des résidus hydrophobiques est polaire, les groupespolaires du squelette doivent être neutralisés pour participer au noyauhydrophobique, ce qui est le cas pour le squelette dans les éléments destructure secondaire où la neutralisation de ces groupes vient de la formationdes ponts H et d’où la participation importante des éléments de structuresecondaire dans les noyaux hydrophobiques des protéines.
• C’est aussi le cas des résidus polaires libres de structure secondaire quipeuvent participer dans le noyau hydrophobique si leurs groupes polairesforment des ponts H avec d’autres résidus polaires libres ou avec desmolécules d’eau.
• Même phénomène pour les résidus chargés pouvant se lier avec d’autresrésidus de charges opposées, définit par Stearn en 1939, et connu sous le nomde paire ionique.
• On observe même la formation de liaisons covalentes de type ponts salinsentre deux groupes thiols (-SH) de deux cystéines, le seul acide aminé pouvantformer de telles liaisons. Ce type d’interaction stabilise fortement le « fold »d’une protéine.

A07 31IFT3295 - F. Major
Modifications
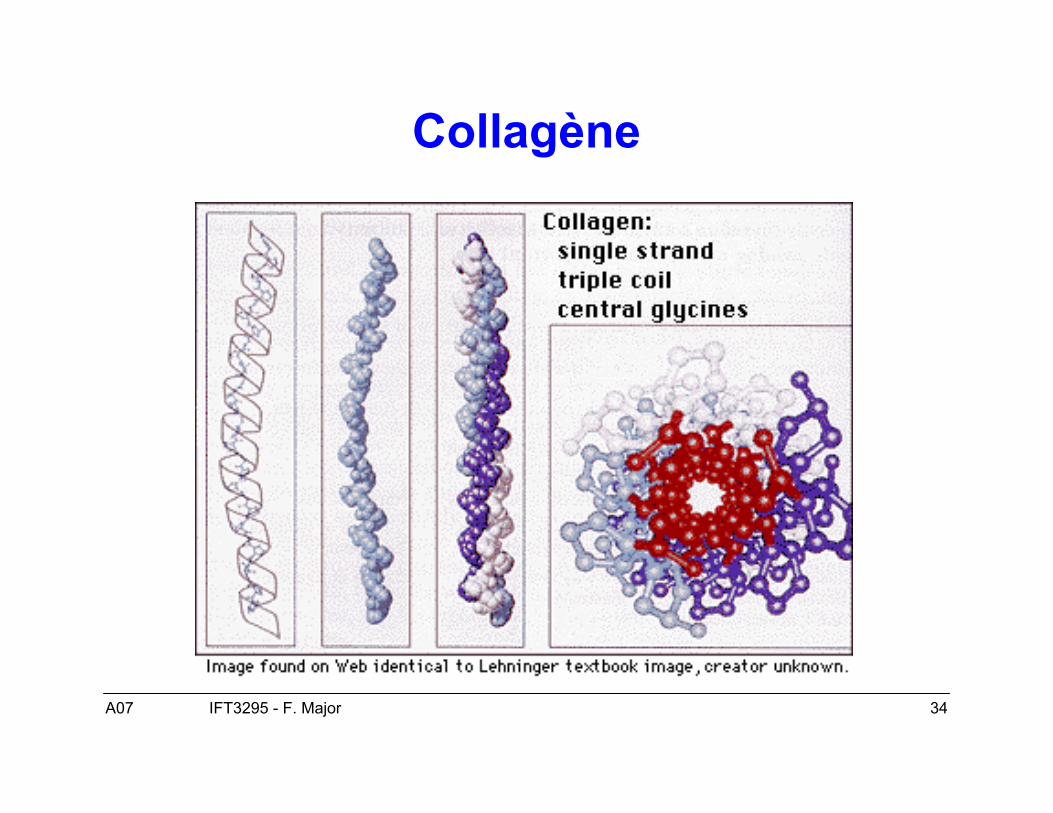
• Les interactions entre les protéines et des molécules non polypeptidiquespeuvent modifier la structure et la fonction des protéines (cf. fibres decollagène fait usage d’une hydroxyproline pour stabiliser sa structure).
• Les hydrates de carbone et les lipides peuvent former des liaisons covalentesavec certains résidus des protéines (cf. hydrates de carbone sont associés à lalocalisation intracellulaire alors que les lipides jouent un rôle important pourles protéines membranaires et trans-membranaires).
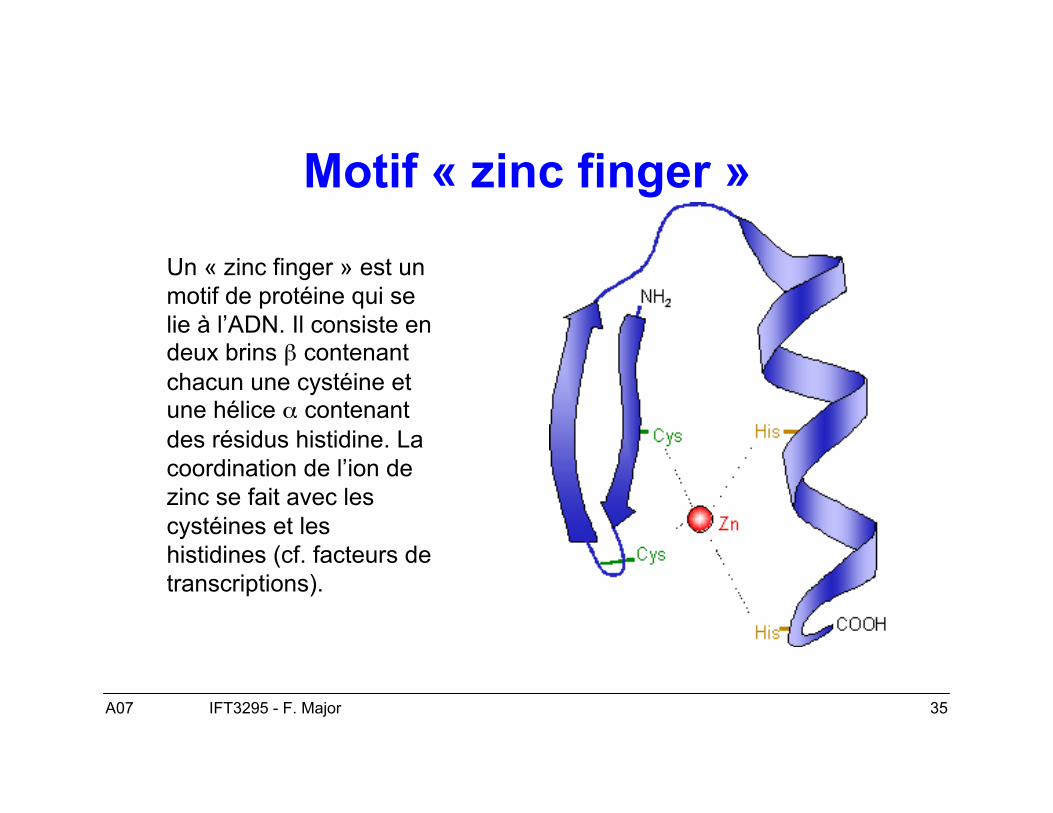
• Finalement, les protéines s’associent à des ions métalliques, comme lesenzymes qui les utilisent comme co-facteurs et d’autres en ont besoin pour leurstructure, comme le motif de « zinc-finger » impliqué dans des interactionsentre protéines et ADN qui nécessite une interaction entre des ions de zincavec des cystéines ou des cystéines et des histidines.
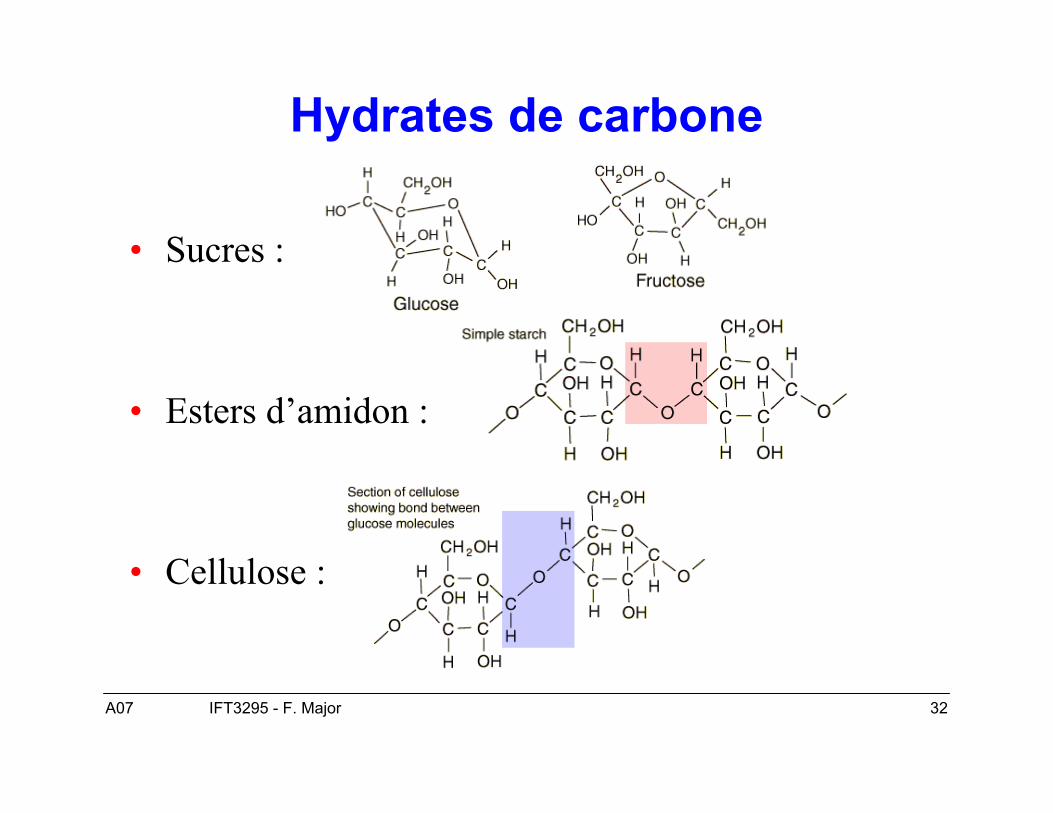
A07 32IFT3295 - F. Major
Hydrates de carbone
• Sucres :
• Esters d’amidon :
• Cellulose :
A07 33IFT3295 - F. Major
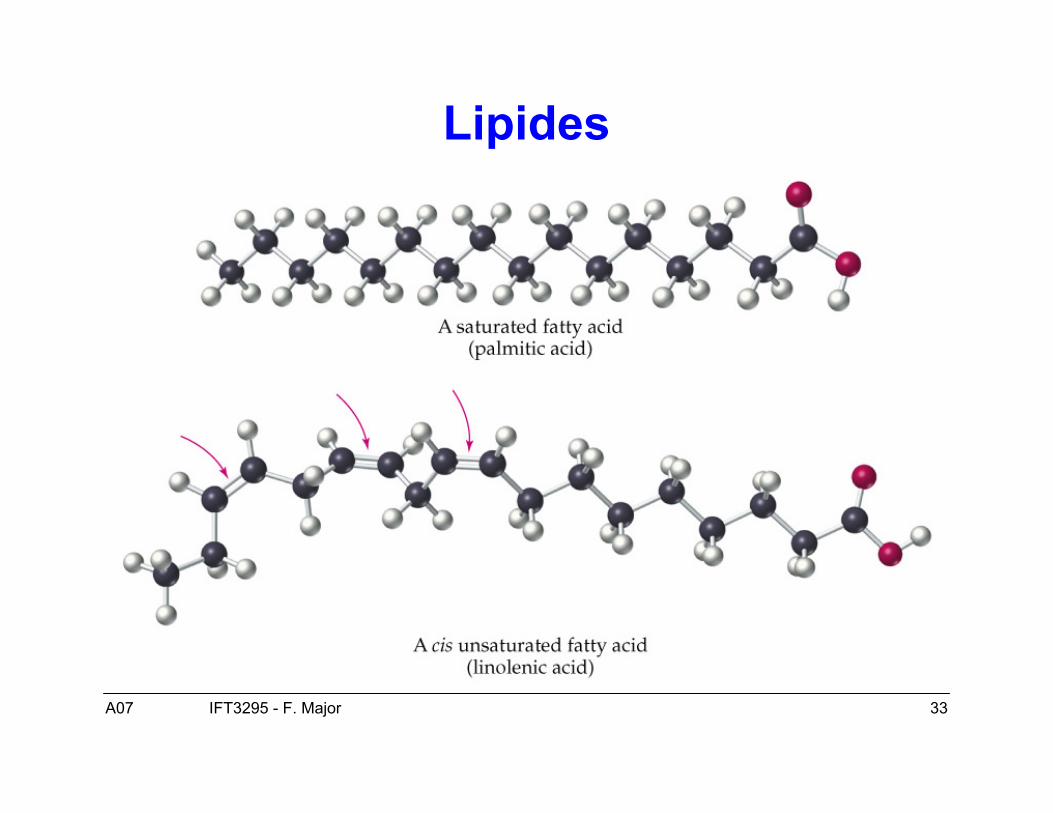
Lipides
A07 34IFT3295 - F. Major
Collagène
A07 35IFT3295 - F. Major
Motif « zinc finger »
Un « zinc finger » est unmotif de protéine qui selie à l’ADN. Il consiste endeux brins β contenantchacun une cystéine etune hélice α contenantdes résidus histidine. Lacoordination de l’ion dezinc se fait avec lescystéines et leshistidines (cf. facteurs detranscriptions).

A07 36IFT3295 - F. Major
« Zinc fingers » liés à l’ADN

A07 37IFT3295 - F. Major
Espace de repliement et évolution
• L’espace de repliement est limité.• On croit actuellement qu’il y aurait environ 1000 « fold » principaux dans la nature.• Deux forces qui ont limité la diversité :
– Évolution divergente. Existence d’un petit nombre de « fold » ancestraux. Fonctions de baseassurées par un certains nombre de « fold » qui ont ensuite évolués lentement à travers l’espacedes mutations et de la diversification des fonctions. On dit que la structure dicte la fonctionmais il y a des cas de « fold » et séquences similaires qui ont des fonctions différentes. Ladifférence existe dans un petit nombre de résidus clés. La non observation de certains « fold »s’explique par le fait que ces derniers n’ont jamais été requis ou développés par les organismes.
– Évolution convergente. Le nombre de « fold » est petit parce qu’il y a des conformationsbiophysiquement favorables et qu’ils ont été créés à répétition de manière indépendante.Certains « fold » similaires n’ont pas de séquences similaires et ne partagent pas d’ancêtrecommun. La non observation de certains « fold » s’explique par le fait que ces derniers sontstructuralement défavorisés.
– Les deux types d’évolution ne sont pas nécessairement exlusifs.

A07 38IFT3295 - F. Major
Classification biochimique des « folds »
• On regroupe les protéines en trois catégories selonleurs propriétés biochimiques : globulaires,membranaires et fibreuses.

A07 39IFT3295 - F. Major
Protéines globulaires
• Existent dans l’état aqueux et se replient demanière compacte avec un noyau hydrophobiqueet une surface polaire.– Bien représentées dans la PDB parce qu’elles sont plus
facile à cristalliser simplifiant la détermination de leurstructure. Les 2 premières structures cristallines étaientglobulaires : myoglobine et hémoglobine.

A07 40IFT3295 - F. Major
Myoglobine (1A6M)

A07 41IFT3295 - F. Major
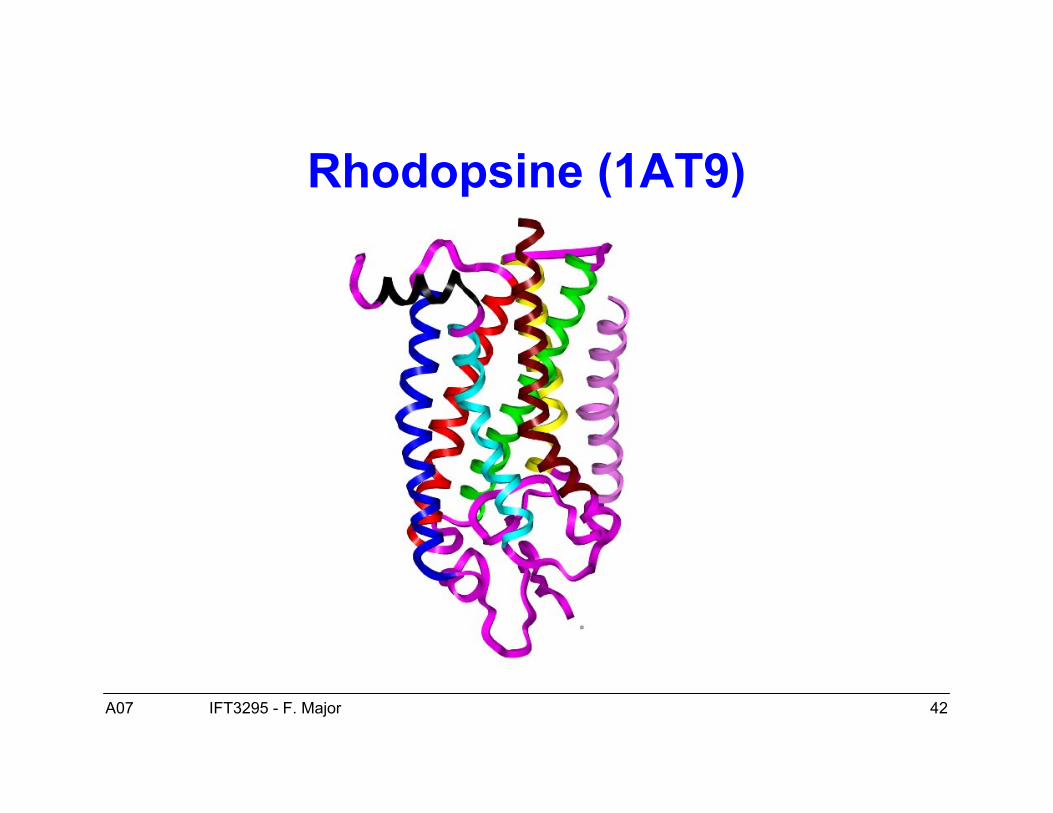
Protéines membranaires
• Plusieurs propriétés communes avec les protéines globulaires.• Cependant :
– Elles existent dans la membrane de la cellule, un milieu très différent dumilieu aqueux des protéines globulaires, un milieu plutôt hydrophobique.Cela implique que la surface de ces protéines est hydrophobique (nonpolaire).
– Elles sont plus difficile à cristalliser, elles sont donc moins bienreprésentées dans la PDB et moins connues par conséquence. Cellesconnues suggèrent un mécanisme de repliement similaire à celui desprotéines globulaires avec les mêmes éléments de structure secondaire.
• Un exemple typique est la rhodopsine (1AT9).
A07 42IFT3295 - F. Major
Rhodopsine (1AT9)

A07 43IFT3295 - F. Major
Protéines fibreuses
• Diffèrent des deux autres types. Elles sont constituées derépétitions d’acides aminés qui forment de simples etlongues fibres.
• Elles jouent un rôle structurale dans les organismes(forment les tissus).
• Consistent souvent en un seul type de structure secondairerépété dans de très longues séquences ou sont simplementatypiques et sans structure particulière.
• Cas typique les fibres de collagène (1QSU).
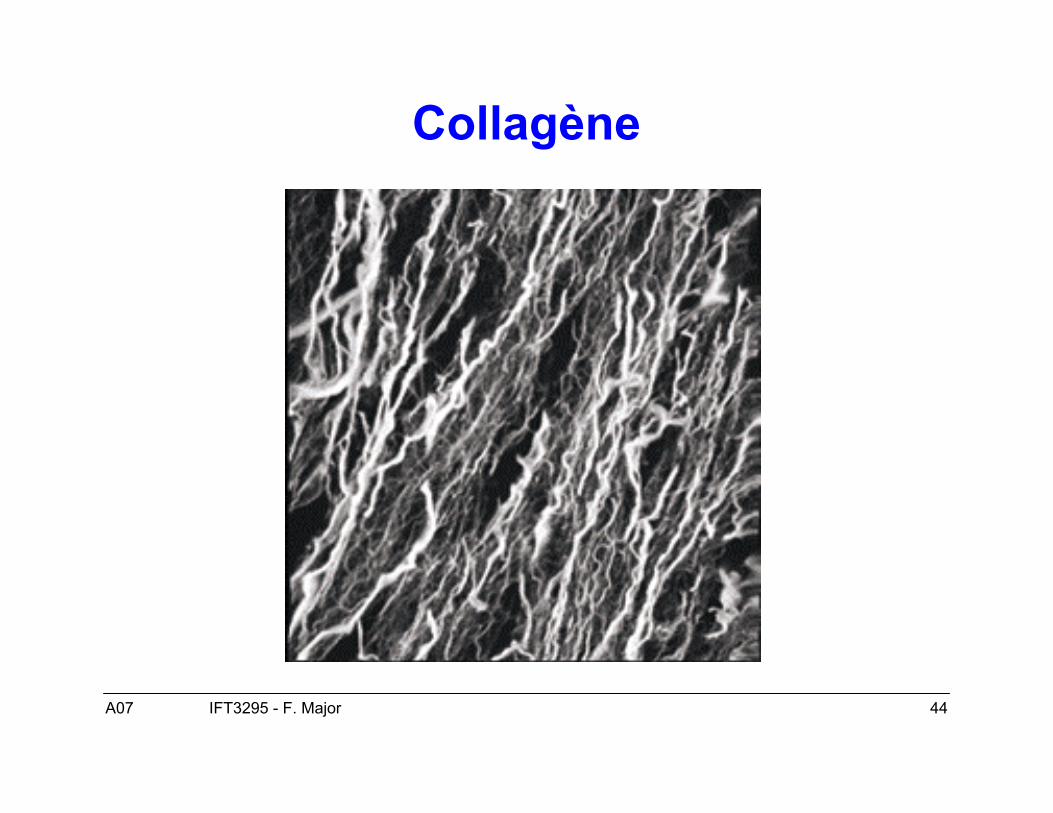
A07 44IFT3295 - F. Major
Collagène

A07 45IFT3295 - F. Major
Classification structurale des « folds »
• Basée sur les éléments de structure secondaire, ona 4 groupes :– Tout α– Tout β– α / β (brins β reliés par des hélices α)– α + β (mélanges α et β non alternés)– 2 classifications à voir plus tard dans le cours : SCOP et
CATH.

A07 46IFT3295 - F. Major
Représentation graphiques des « folds »
• Diagramme topologique simple.• Diagramme caricaturale.• Diagramme TOPS.
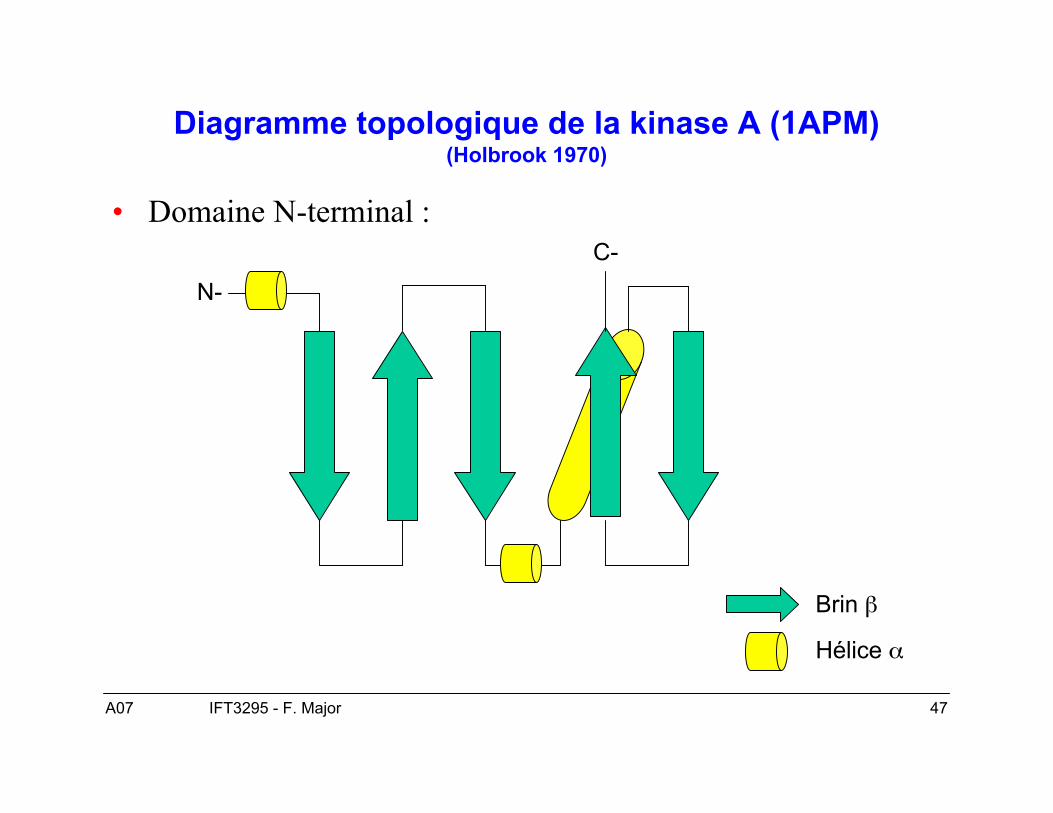
A07 47IFT3295 - F. Major
Diagramme topologique de la kinase A (1APM)(Holbrook 1970)
• Domaine N-terminal :
N-
C-
Brin β
Hélice α
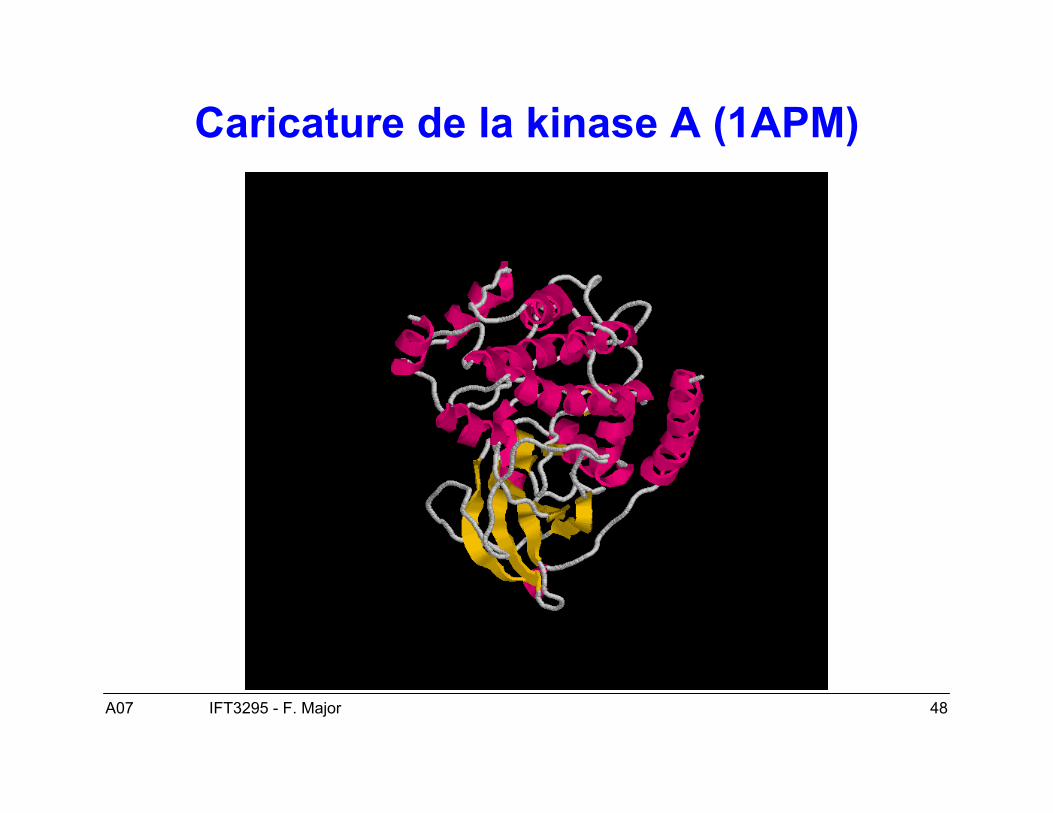
A07 48IFT3295 - F. Major
Caricature de la kinase A (1APM)
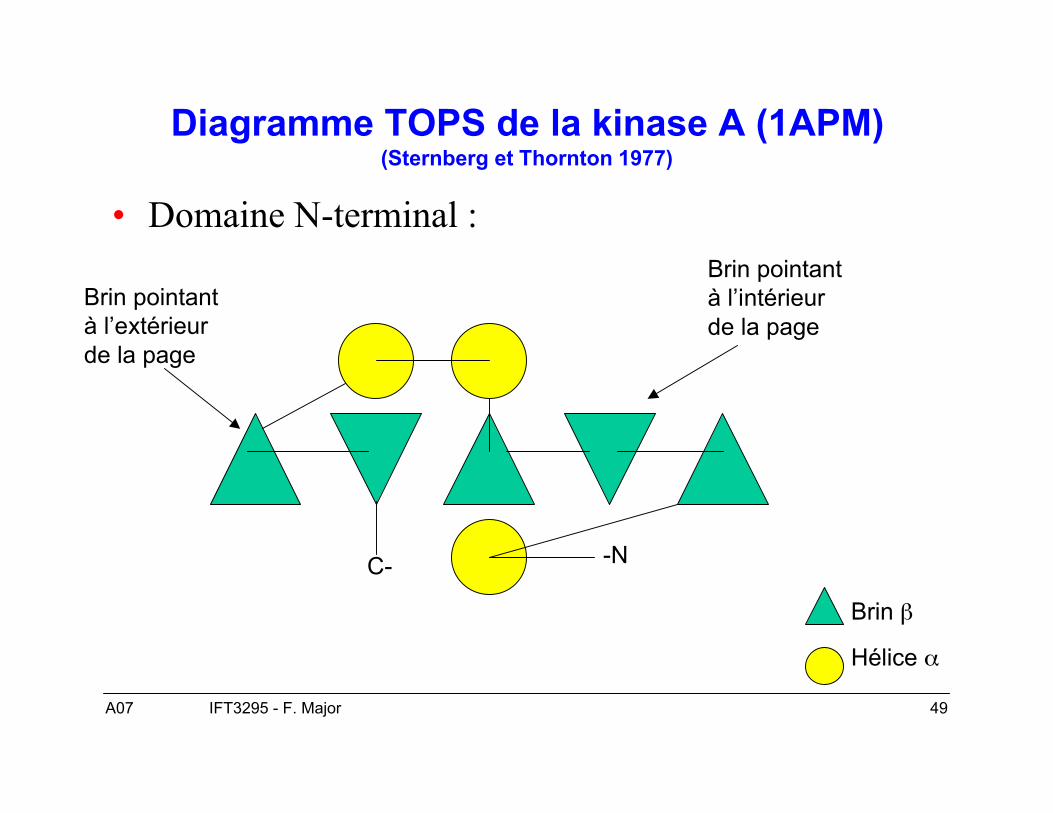
A07 49IFT3295 - F. Major
Diagramme TOPS de la kinase A (1APM)(Sternberg et Thornton 1977)
• Domaine N-terminal :
-NC-
Brin β
Hélice α
Brin pointantà l’extérieurde la page
Brin pointantà l’intérieurde la page

A07 50IFT3295 - F. Major
Structure quaternaire :associations de plusieurs chaînes peptidiques
• La structure tertiaire décrit l’organisation structurale d’une chaînepolypeptidique mais plusieurs protéines fonctionnent en mode multimère (soitplusieurs copies d’un monomère).
• Ces protéines possèdent une structure quaternaire.• Celles qui sont composées de la même sous unité sont dites homomériques et
celles dont les sous unités diffèrent sont dires hétéromérique.• Les interactions multimériques sont constituées des mêmes types que dans les
noyaux des protéines globulaires, soit des chaînes latérales non polaires, desgroupes pouvant former des ponts H ou des ponts disulfures.
• Trois des avantages d’avoir des structures quaternaires sont :– Coopérativité (hémoglobine)– Co-localisation de fonction (tryptophane synthétase)– Combinaisons de sous unités (immunoglobuline)
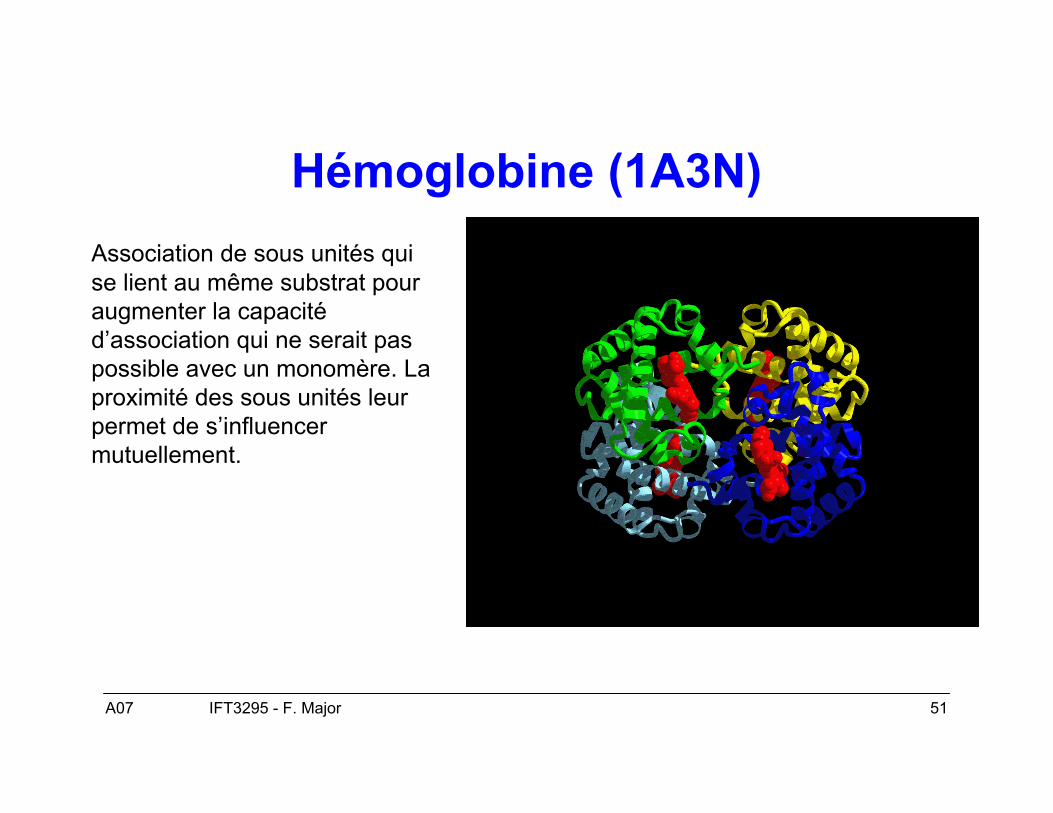
A07 51IFT3295 - F. Major
Hémoglobine (1A3N)Association de sous unités quise lient au même substrat pouraugmenter la capacitéd’association qui ne serait paspossible avec un monomère. Laproximité des sous unités leurpermet de s’influencermutuellement.
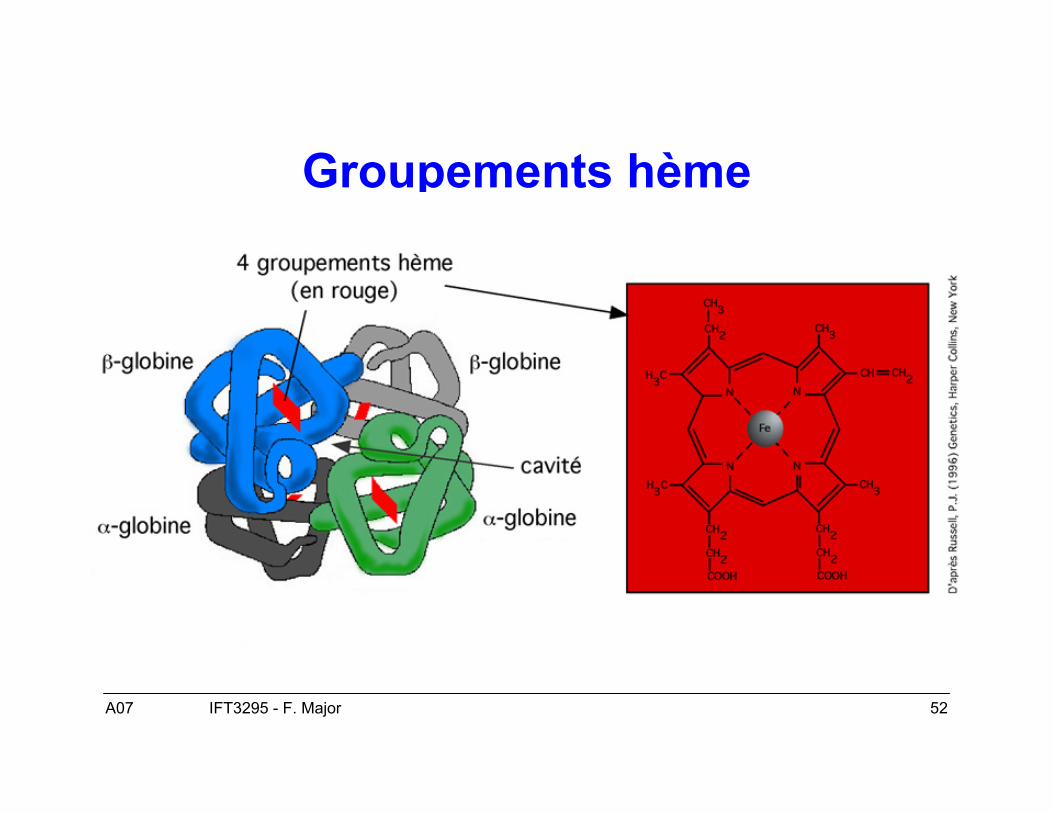
A07 52IFT3295 - F. Major
Groupements hème
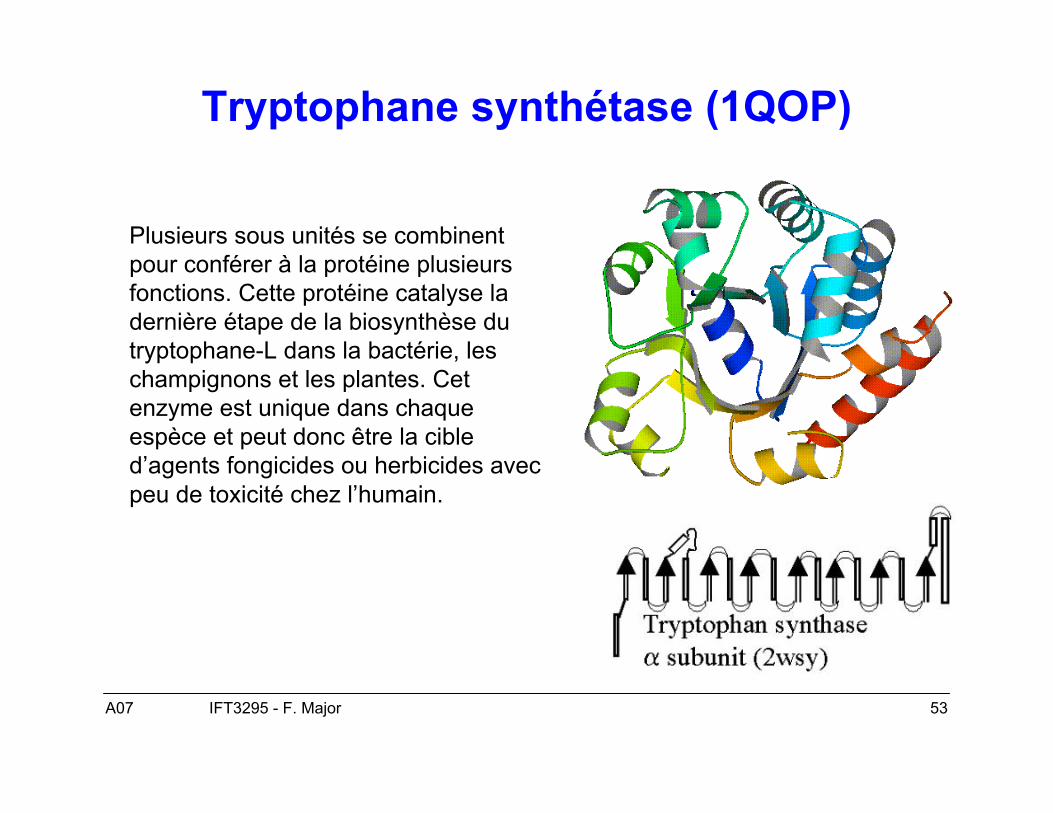
A07 53IFT3295 - F. Major
Tryptophane synthétase (1QOP)
Plusieurs sous unités se combinentpour conférer à la protéine plusieursfonctions. Cette protéine catalyse ladernière étape de la biosynthèse dutryptophane-L dans la bactérie, leschampignons et les plantes. Cetenzyme est unique dans chaqueespèce et peut donc être la cibled’agents fongicides ou herbicides avecpeu de toxicité chez l’humain.
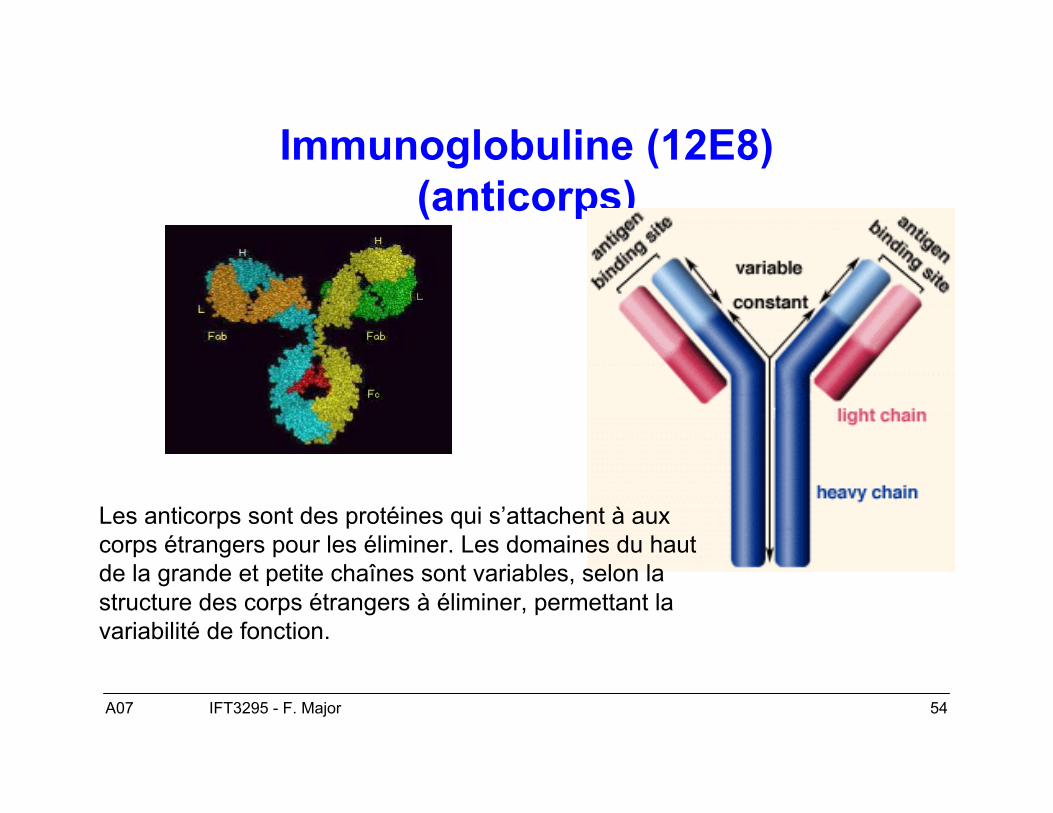
A07 54IFT3295 - F. Major
Immunoglobuline (12E8)(anticorps)
Les anticorps sont des protéines qui s’attachent à auxcorps étrangers pour les éliminer. Les domaines du hautde la grande et petite chaînes sont variables, selon lastructure des corps étrangers à éliminer, permettant lavariabilité de fonction.

A07 55IFT3295 - F. Major
Conclusion
• Les bases structurales des protéines ont été établies dansles années 70.
• On possède maintenant les outils pour les visualiser et lesanalyser mais ces outils pourraient être améliorés.
• On ne sait toujours pas comment se replie une protéinedans l’espace tridimensionnelle et à partir de sa séquence.
• Dans les années qui viennent, on devrait pouvoir mieuxcomprendre les relations qui existent entre la séquence, lastructure et la fonction des protéines.





























