Embed Size (px)
Citation preview

Synthkse totale des acides (+) et (-) nonactique a partir de carbohydrates1
ROBERT E. IRELAND ET JEAN-PAUL VEVERT Dioision of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, CA 91 125, U.S.A.
R e p le 17 juin 1980
ROBERT E. IRELAND et JEAN-PAUL VEVERT. Can. J. Chem. 59,572 (1981). La synthese des acides (-) et (+) nonactique 2a et 2b a ete realisee a partir respectivement de D-mannose (7) et de ~-gulono-y-
lactone (22). Elle fait intervenir comme Ctape cle le rearrangement [3,3]-sigmatropique de cetkne-acetals silyles IV qui permet le contrBle de la configuration du C-2 de l'acide nonactique. Ces cktene-acetals sont prepares a partir d'esters aliphatiques de glycals furanoiques 11, qui sont eux mkme synthetises en dix etapes a partir du carbohydrate precurseur. Les centres chiraux de ces glycals proviennent de la conservation durant cette suite de transformations des centres correspondants du monosaccharide de depart. Effectue avec ce genre de cetene-acetals le rearrangement de Claisen conduit a des composes comportant l'enchainement du type aldol desire. On tire egalement parti de la connaissance de la configuration absolue des carbones chiraux de l'acide nonactique pour determiner la conformation (chaise ou bateau?) de I'etat de transition du rearrangement [3,3]-sigmatropique.
ROBERT E. IRELAND and JEAN-PAUL VEVERT. Can. J. Chem. 59,572 (1981). The synthesis of (-) and (+) nonactic acids (2a) and (26) has been achieved starting from D-mannose (7) and D-gluono-y-lactone
(22) respectively. The key step in the synthesis is the [3,3]-sigmatropic rearrangement of the silylated ketene-acetals IV leading to control of the C-2 configuration of nonactic acid. The ketene-acetals were prepared from aliphatic esters of furanoid-glycals 11, which were prepared in ten steps from the carbohydrate precursor. The chiral sites of the glycals arise from the corresponding centres in the starting monosaccharide. T h ~ s type of ketene-acetal Claisen rearrangement leads to products containing the aldol portion required. At the same time knowledge of the absolute configuration of the chiral carbon atom of nonactic acid allows for the determination of the chair or boat form of the transition state of the [3,3]-sigmatropic rearrangement.
[Journal translation]
Introduction R I
La nonactine (1) macrotetrolide isole pour la premihe fois en 1955 par Prelog, Keller-Schierlein
,,\. o. ..KO et coll. ( 1 ) represente l'homologue le plus simple d'une famille de tetraesters macrocycliques a 32 rnernbres, connus sous le nom de nactines (2 ) et produits a partir de cultures de streptomyces (3) . R~ o L'interkt biologique suscite par ces composes %0 OI,2,,,,fi R > 1 0 resulte en partie de leurs proprietes anti- microbiennes (G+ , fungi) mais surtout de leur 0 R3 capacite a agir comme transporteurs d'ions a tra- 1 R I = R Z = R3 = R4 = CH
vers les membranes biologiques (4). Les nactines forment des complexes 1 : 1 avec de nombreux ions F3
CH3
des metaux alcalins et alcalino-terreux et plus par- ROOC G OH Rooc,2,,~Q,..XOH : H H ticulierement avec le potassium (5 ) qui se trouve CH3 C H ~ litteralement englobe par coordination des huit 2a R = H 2b R = H oxygenes des carbonyles et cycles tetrahy- 3a R = CH, 3b R = CH, drofuraniques a I'interieur du squelette carbone (6). Ces proprietes de transporteurs d'ions font CH3 CH3
entr'autre de ces composes de puissants inhibiteurs R O O C ~ ~ ~ R O O C ~ , + ~ ~ , , , , % ~ ~ H H
de la phosphorilation oxydative de I'ADP en ATP CH3 CH, (3) . La structure de la nonactine a ete elucidee par 4a R = H 4b R = H Prelog, Gerlach et coll. (7). Le macrocycle est 5a R = CH, s b R = CH, forme de quatre subunites d'acides nonactique (2) SCHEMA I relies entr'eux par des liaisons ester, avec alter- nance des Cnantiomeres (-) (2a) et (+) ( 2 b ) de telle h ~ d r o l ~ s e conduit 2 un melange racemique sorte qu'il presente une configuration *&so. Son d'acides nonactique dont la configuration absolue
des quatre carbones chiraux a Cte determinee (8). T I T Contribution NO. 6249. La synthese de la nonactine a Cte realisee par Ger-
0008-404218 11030572- 12$01.00/0 @ 1981 National Research Council of CanadaIConseil national de recherches du Canada
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
UN
IV O
F B
IRM
ING
HA
M o
n 11
/10/
14Fo
r pe
rson
al u
se o
nly.

IRELAND ET VEVERT 573
lach et coll. a partir de l'acide nonactique racemique (9) et par Schmidt et coll. (10) apartir de l'acide (-) nonactique (2a) et de derives des epi- meres (-) (4a) et (+) (4b) en position 8 (Schema I). La plupart des syntheses de l'acide nonactique pu- bliees a ce jour conduisent a un melange racemique (10-14).2 Elles font generalement intervenir dans l'etape cle pour contr6ler la configuration cis du tetrahydrofurane-2,5 disubstitue l'hydrogenation catalytique d'un cycle furanique. Seule celle rap- portee par Schmidt et coll. permet d'isoler l'acide (-) nonactique (2a) a partir d'un melange de quatre diastereoisomeres. Par inversion de Walden du C-8 des trois diastereoisomeres restants ces auteurs ont obtenus un melange duquel l'acide (+) nonactique (2b) a pu 2tre separe (10, 13). Cette synthese ne presente toutefois pas un tres haut degre de stereoselectivite. L'oxyde de S (-) propylene, non commercialement disponible, etant d'autre part utilise comme produit de depart, elle necessite la preparation de ce compose avec une grande purete optique a partir d'acide lactique (15).
En raison de l'inter2t grandissant porte aux sucres dans divers laboratoires dont le n6tre (16) en tant que produits de depart chiraux relativement peu onereux pour la synthkse de produits naturels comportant un nombre eleve de carbones as- symetriques, il nous est apparu interessant d'ela- borer une synthese des enantiomeres de l'acide nonactique a partir de cette classe de composes.
Rksultats et discussion Un derive du type V (Schema 2) peut btre con-
sidere cornme un precurseur interessant de l'acide n~nac t ique .~ La synthese d'un tel compose peut 2tre envisagee par formation de la liaison entre les atomes de carbone chiraux 2 et 3 des deux entites encerclees. De precedents travaux dans notre laboratoire ont demontre que la formation de telles liaisons carbone-carbone, pour l'obtention de derives du type aldol apparentes a V, pouvait 2tre realisee par un rearrangement de Claisen de cetenes-acetals silylks (17). Ceci permet de con- siderer l'alcool allylique heterocyclique I1 cornrne un intermediaire cle de cette synthese. I1 perrnet en effet d'obtenir le cetene-acetal silyle IV devant conduire apres le rearrangement [3,3]-sigrnatro- pique au precurseur desire. Alors que la configura- tion du carbone 3 de V dkcoule directement de celle du carbone 3 de IV, la configuration du carbone 2
Weir aussi P. A. Bartlett et K. K. Jernstedt, cite dans la ref. 2b.
3Pour des raisons de clarte nous n'avons represent6 que les precurseurs de I'enantiomkre (-).
resulte de la geometrie du cetene-acetal silyle et il devient donc important de contr6ler celle-ci lors de la formation de IV.
OTMS /
Des resultats anterieurs obtenus dans ce laboratoire font apparaitre que suivant le solvant ou la base employee, la deprotonation a -78°C de l'ester 6 (Schema 3) conduit a un melange de pro- portions variables des enolates Z et E (17, 18). Ainsi lorsque la deprotonation est effectuee par le diisopropylamidure de lithium (LDA) a -78°C dans le THF l'anion-enolate de type Z est forme preferentiellernent, tandis que la formation de l'anion-enolate de type E est favorisee quand cette deprotonation a lieu dans un melange de 23% d'hexamethylphosphotriamide (HMPA) dans le THF.
OLi R OLi
+'=( RCH,COOR' - r\ 6 R OR' OR'
Z E LDA:THF ZIE > 1 LDA:THF:23%HMPA ZIE < 1
Le rearrangement de Claisen des cetknes-acetals silyles tels que IV, obtenus par silylation du melange d'enolates correspondant, conduit a deux esters de silyle y,6 insatures diastereoisomeres V. I1 a ete montre qu'en serie acyclique en l'absence d'effets steriques particuliers ce rearrangement s'opkrait suivant un etat de transition du type chaise (18). La proportion des deux diastereoiso- meres forrnes ne depend donc que de la proportion relative des deux enolates E et Z et peut de ce fait 2tre prevue et orientee par un contr6le des condi- tions dans lesquelles la deprotonation est effectuee. Les resultats preliminaires obtenus en serie cy-
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
UN
IV O
F B
IRM
ING
HA
M o
n 11
/10/
14Fo
r pe
rson
al u
se o
nly.

574 CAN. J . CHEM. VOL. 59, 1981
clique semblaient indiquer le passage pour le rear- rangement [3,3]-sigmatropique par un inter- mediaire du type bateau. L'application de ce rear- rangement a la synthese de produits naturels de configuration connue tels que les deux enantio- meres de I'acide nonactique semblait donc devoir de plus fournir des renseignements plus precis quant a la nature de l'ktat de transition. Les resul- tats concernant la formation prCpondCrante de l'knolate Z dans le THF et celle prepondkrante de 1'Cnolate E en presence de HMPA semblant pouvoir 2tre transposks en serie cyclique, l'ob- tention d'une forte proportion de I'un des diastCrCoisomkres par rapport a l'autre devrait in- diquer le passage pratiquement exclusif pour le rearrangement de ce diast6rCoisomere majoritaire par un Ctat de transition unique, soit chaise, soit bateau. La comparaison ultkrieure de ce diastCrCoisomere avec le produit naturel, devrait indiquer par lequel de ces deux 6tats de transition le rearrangement a eu lieu. Par contre l'obtention des deux diastereoisomeres dans des proportions ten- dant vers 5050 devrait indiquer le passage competitif par ces deux Ctats de transition.
L'etape cle de la synthkse proposee devrait donc permettre d'obtenir de f a ~ o n specifique la configu- ration dCsiree des carbones 3 et 6 et sklectivement celle du carbone 2. Une methode generale de synthese de glycals optiquement actifs du type I1 a partir de sucres a ete preckdemment mise au point dans notre laboratoire, utilisant la reduction par le lithium dans l'amoniac des chlorures du type I (17b, c, 19).
Le D-(+)-mannose (7) (Schema 4) a etC choisi comme produit de depart pour la synthese de 1'Cnantiomere (-) ( k ) . I1 est en effet possible d'obtenir pour un prix modique des quantites im- portantes de I'isomere (+) de ce sucre qui possede la configuration desiree des carbones 4 et 3 appeles a devenir respectivement les carbones 2 et 3 de l'alcool allylique 11. Par une serie de transforma- tions connues, diacetonisation, benzylation, &pa- ration des deux anomkres et hydrolyse selective de l'un des acetonides, le melange d'anomeres a et P 7 conduit avec un rendement global de 76% a l'a-D- mannofuranoside de benzyle (8) (20). En prksence de dimethylaceta1 du dimCthylformamide celui-ci est transforme quantitativement en acetal du DMF (10) qui par chauffage dans l'anhydride acetique fournit I'olefine l l a (76,5% apres chromato- graphie) et 17,5% d'un compose 9 correspondant a une diacetylation des alcools en position 5 et 6 de 8 (21). La reduction par LiAlH, de ce diacktate per- met de regknbrer le diol 8 conduisant a un rende-
ment de 93% en olefine l l a sur la base du diol rkcupCr6.
Une modification de la mkthode preckdente, in- troduite par Hanessian et coll. consiste a trans- former 10 en iodure d'amonium par traitement par l'iodure de methyle, et a chauffer le sel obtenu dans le toluene (22). Contrairement a la methode originale, cette variante appliquee a 10 ne conduit pas a I'olefine dksiree mais a un iodo-formate resultant d'une substitution par l'ion iodure de l'oxygene en position 6 de 10. Cette reaction est probablement dGe au faible encombrement stkrique de cette position et devient la reaction attendue dans le cas des acetals des N,N-dimethylamides autres que ceux du DMF (28).
L'alcool 1k obtenu apres hydroboration oxyda- tive de l l a par le 9-BBN (963% apres chromato- graphie) est protege quantitativement sous forme de mCthoxym6thylether (MOM) (13a) par reaction de l'alcoolate de potassium correspondant avec l'ether chloromethylm~thylique dans le THF a 0°C. Une tentative de mise en place de ce groupe pro- tecteur suivant un mode operatoire consistant a faire reagir l'alcool et du dimCthoxymethane en presence de P,O, (23) n'a conduit qu'a des rende- ments mkdiocres (40% apres chromatographie) bien qu'employke avec succes dans notre laboratoire avec des substrats analogues. Le furanoside de benzyle (13a) traite par le lithium dans l'amoniac a -78°C permet d'isoler le lactol 14a avec un rendement de 79%. Celui-ci est trans- form6 en chlorure de mannofuranosyle (15a) par le CCl, et la PPh, dans le THF a reflux (91 3% apres distillation). La reduction par le lithium dans l'amoniac a -78°C de ce dernier conduit, comme prevu en skrie furanique, a un melange (10:l d'apres le spectre de 'H rmn du distillat) du glycal 17a (70%) et du derive 16a resultant d'une reduc- tion non suivie de fragmentation de 15a (17c, 19). Le rearrangement [3,3]-sigmatropique est effectuk sur le melange precedent, le glycal se dkcomposant partiellement sur gel de silice et Florisil.
Postulant le passage par un intermediaire du type bateau pour le rearrangement de Claisen, la configuration du carbone 2 de l'acide nonactique necessite la formation majoritaire de l'knolate de type Z. Celui-ci est obtenu par traitement par le diisopropylamidure de lithium dans le THF a -78°C du propionate resultant de I'estCrification du chlorure de propionyle par l'alcoolate (n-BuLi, THF, -78°C) correspondant de I'alcool allylique 17a du melange prkcedant. Le melange d'enolates est pikge a -78°C par le chlorure de trimethylsilyle (TMSCI) et le rearrangement s'opere durant le
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
UN
IV O
F B
IRM
ING
HA
M o
n 11
/10/
14Fo
r pe
rson
al u
se o
nly.

IRELAND ET VEVERT 575
rechauffement jusqu'a la temperature ambiante. Aprks hydrolyse des esters de trimkthylsilyle, le melange d'acides traite par le diazomethane con- duit aux deux esters y,6 insatures diastereoiso- meres lSa, qui presentent le mtme Rf en ccm et ne peuvent ttre separes a ce stade. L'hydrogination de la double liaison est realisee en presence de rhodium sur charbon. Toutefois une faible quan- tite de produits resultant d'hydrogenolyse et d'isomerisation de la double liaison etant egale- ment formes, l'emploi de platine sur charbon lui est prefere. Contrairement a leurs analogues insatures les deux esters diastereoisomkres 19a et 19'a peu- vent 2tre skpares par chromatographie sur gel de silice. L'examen du melange par cpv revkle qu'ils ont ete formes dans un rapport de 89: 11. Ce rapport eleve semble indiquer d'aprks le raisonnement pre- cedent le passage par un Ctat de transition unique pour le diastereoisomere majoritaire et cet Ctat de transition semble avoir ete du type bateau. Dans l'attente d'une confirmation univoque par com- paraison de l'acide nonactique synthetique avec le compose naturel, differentes constatations sem- blent soutenir cette hypothese. Apres separation le spectre de 'H rmn du diastCreoisomere majoritaire presente une resonance du methyle en position 2 a champ plus eleve (1,ll ppm) que le mkthyle corres- pondant du diaster~oisomkre minoritaire (1,20 ppm) et son Rf est d'autre part plus petit. Les differents derives et analogues de l'acide nonac- tique synthetises ont de mtme toujours prksente un Rf plus petit et une resonance du mkthyle en C-2 a champ plus eleve que les Cpimeres correspondants en C-2 (7b, 10a, 12). Les probabilites que le diastCreoisomere majoritaire presente comme prkvu la mtme configuration en C-2 que l'acide nonactique etaient donc Clevees et semblaient confirmer nos hypotheses. La synthkse a donc 6tC poursuivie avec ce diastCreoisomere, isole avec un rendement de 48,5% a partir de 17a.
La mtme suite de reactions avait ete initialement entreprise partir de 13'a, l'alcool primaire ttant protege sous forme d'ether de tert-butyldi- methylsilyle (TBDMS). Le rendement aprks le rearrangement de Claisen est toutefois tres faible (quelques %). Une explication ace fait pourrait ttre l'echange de l'ether de TBDMS avec l'alcoolate de lithium de 17'a, conduisant a des produits se decomposant durant l'hydrolyse. Un argument en faveur de cette explication dkcoule de l'observa- tion suivante. Lors de la formation du glycal 17'a on observe a cat6 des produits attendus un com- posC resultant d'un tel echange de l'ether de TBDMS. Voisine de 50% lorsque la reduction est
effectuee dans l'amoniac a -33°C sa formation peut cependant ttre ramenbe moins de 5% -78°C. 11 semble toutefois que dans le THF cet echange ne puisse 6tre evite.
L'alcool en position 8 de 20a est libere par chauffage de 19a dans l'acide chlorhydrique a 2% dans le methanol anhydre (96,5% apres chromato- graphie) et oxyde en aldehyde 21a avec un rende- ment de 90% (24). L'introduction du methyle en position 8 a etC effectuee par le dimethylcuprate de lithium a -20°C. I1 a ete montre que ce reactif, inerte vis-a-vis des cetones et esters carboxyliques, reagit avec les aldehydes (25). Les deux alcools diasterCoisomkres sont obtenus dans des propor- tions comparables. Le plus polaire devant corres- pondre a l'ester mkthylique de l'acide (-) nonac- tique (3a) (12) est isole avec un rendement de 39,5% et son Cpimere en C-8 (5a) avec un rendement de 45%. L'hydrolyse separee de ces deux Cpimeres par une solution aqueuse 2 N d'hydroxyde de potas- sium s'effectue en quelques minutes a temperature ambiante. Elle conduit l'acide (-) nonactique (2a) e ta son epimere en C-8 (4a) avec un rendement de 96% sans qu'aucun produit resultant d'une Cpimerisation en C-2 ou C-3 ne puisse 2tre detecte par ccm. Les caractkristiques analytiques et spec- trales de l'acide (-) nonactique et de son ester methylique sont en accord avec celles publiees a partir du produit resultant de l'hydrolyse de la di- nactine, et plus particulikrement en 'H rmn avec les deplacements chimiques et constantes de couplage du methyle en C-2 (3,7b). Comme prevu (12), des deux epimeres en C-8 le plus polaire presente les caracteristiques de l'acide nonactique. Le pouvoir rotatoire negatif obtenu pour 3a et 5a corrobore les deductions faites apres le rearrangement de Claisen, l'epimere en C-2 (S,R,S,S) de l'acide nonactique presentant un pouvoir rotatoire positif (7b).
L'enantiomkre (+) de l'acide nonactique (2b) a CtC synthetise suivant le mtme schema. Bien que commercialement disponible le L-(-)-mannose n'a pas etC choisi comme produit de depart en raison de son prix Cleve et la D-gulono-y-lactone (22) (Schema 5) presentant les configurations desirees des carbones 2, 3 et 4 lui est preferke. Apres con- version de 22 en diacetonide et reduction de la lactone par l'hydrure de diisobutyl aluminium (DIBAL) dans l'ether, les deux lactols anomeres obtenus sont proteges par benzylation et dpares par chromatographie. L'hydrolyse selective de l'un des acetonides de l'anomkre majoritaire p conduit au diol 24 avec un rendement global de 50%. Celui-ci est diaster6oisomkre du diol8, la configu-
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
UN
IV O
F B
IRM
ING
HA
M o
n 11
/10/
14Fo
r pe
rson
al u
se o
nly.

576 CAN. J. CHEM. VOL. 59, 1981
10 X', X2 = C /H
X 'N(CH,)Z 0 0
l l a Ih R = H 1% R = CH,OCH, 13'a R = TBDMS
1Q X = OH, R = MOM; 14'a X = OH, R = TBDMS 17a R = MOM 15a X = CI, R = MOM; 15'a X = CI, R = TBDMS 17'a R = TBDMS 16a X = H , R=MOM;16'a X = H , R = T B D M S
3a L h H3COOC * 2 . +
CH3 5a '. Q 19a X = CH20MOM; 19'a epirnere C-2 20a X = CH20H 21a X = CHO
(c) H,CCOCH3, H+; (d) NaH, PhCH2CI, DMF;(e) HCI conc., CH30H; (f) (CH,),NCH(OCH,),, CH2C12; (g) (H3CCO),0, 130°C; (h) 9-BBN; NaOH, H202; (i) KH, CICH,0CH3; (k) Li, NH,, -78°C; (1) Ph3P, CCI,; (rn) n-BuLi; n-C2HSCOCI; LDA, THF, -78°C; TMSCI; 25°C; H20 , OH-; CH2N2; (n) Hz, RhIC; (0 ) 2% HCI, CH30H; (p) (COCI),, DMSO; Et3N; (q) Li(CH3),Cu; (192 NKOH, H20 .
SCHEMA 4
ration du carbone 5 appele a perdre sa chiralite lors de l'etape suivante etant la mime pour ces deux composes. Sa conversion en olefine l l b enantio- mhe de l l a est effectuee suivant la sequence precedemment decrite. Par une serie de reactions identiques a celles utilisees pour obtenir l'knantio- mere (-), l'olefine l l b est transformke en aldehyde 21b. L'introduction du methyle en C-8 par reaction de 21a avec le dimethylcuprate de lithium ayant conduit precedemment a un melange des deux diastereoisomkres legerement plus riche en l'epi- mere non desire (4753 en produits isoles), nous avons envisage son incorporation possible par un autre reactif. La reaction de 21b avec un equivalent de bromure de methyl-magnesium dans l'ether a basse temperature conduit a des rendements en produits isoles apres chromatographie de 533% pour le (+) nonactate de methyle (36) et 4Wo pour son epimere en C-8 (Sb). Bien que tendant cette fois encore vers un rapport voisin de 1: 1 (57:43 en pro- duits isoles) l'utilisation du reactif de Grignard semble toutefois favoriser legerement la formation du diastereoisomere desire comme le montre l'examen du melange brut par ccm, et conduire a un rendement sensiblement superieur (933%). Ces resultats font apparaitre une stereoselectivite tres
faible, et sont en accord avec ceux recemment rap- portis par Still et Schneider relatifs a l'addition d'organometalliques sur des P-alkoxyaldehydes non substitues en u (29). L'hydrolyse comme precedemment de ces deux esters offre l'acide (+) (S ,S ,R ,R) nonactique et son epimere en C-8 en tout point equivalents au signe pres du pouvoir rotatoire, aux enantiomeres precedemment ob- tenus.
Conclusion La synthese des deux enantiomeres de l'acide
nonactique que nous venons de decrire montre une fois de plus l'intergt prksente par les sucres comme produits de depart chiraux relativement peu onereux. D'autre part elle souligne l'interit deja precedemment demontre (18) qui peut itre tire de l'utilisation du rearrangement [3,3]-sigmatropique de cetene-acetals silyles pour la formation de liaisons entre atomes de carbone dont la chiralite peut itre preselectionnee. Elle fait egalement ap- paraitre son application possible la formation de derives du type aldol, deja intensivement mise a profit lors de la synthese de l'acide lasalocide A (16). Elle presente en outre un haut degre de stereoselectivite, le seul centre non contr61e (C-8)
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
UN
IV O
F B
IRM
ING
HA
M o
n 11
/10/
14Fo
r pe
rson
al u
se o
nly.
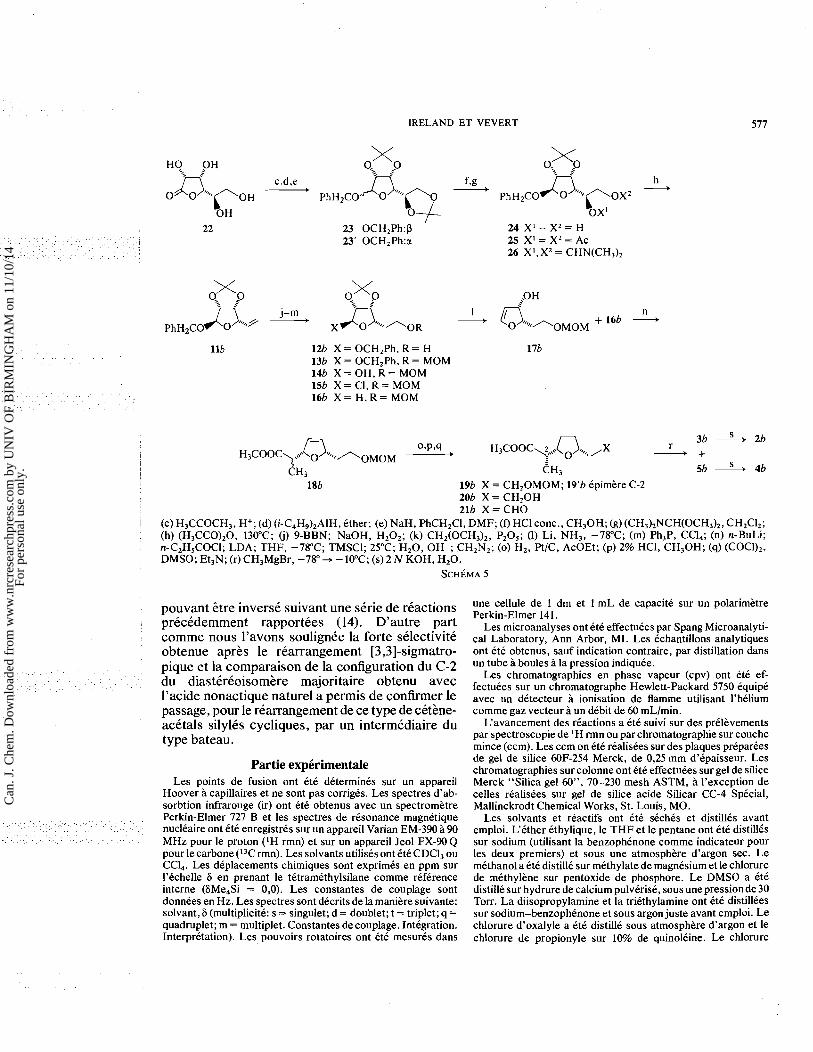
IRELAND ET VEVERT
llb 12b X = OCH,Ph. R = H 17b 13b X = 0CH2Ph, R = MOM 14b X = OH, R = MOM 15b X = CI, R = MOM 16b X = H , R = MOM
3b 2b 0.p.q H ~ c o o c ~ , ~ ~ ~ ~ ~ ~ ~ ~ ~ , ~ ~ ~ ~ ~ --f H ~ c o o c ~ , , , + Q , , , , , ,X - + r
I
I CH3 CH, 56 A 4b 18b 19b X = CH20MOM; 19'b epimere C-2
20b X = CH20H 21b X = CHO
1 (c) H,CCOCH3, H+; (d) (i-C4H9),A1H, ether; (e) NaH, PhCH2CI, DMF; (0 HCI conc., CH,OH; (g) (CH,),NCH(OCH,),, CH2ClZ; (h) (H,CCO),O, 130°C; (i) PBBN; NaOH, H202; (k) CH2(OCH3),, PZ05; (1) Li, NH,, -78°C; (m) Ph,P, CC14; (n) n-BuLi; n-C2HSCOCI; LDA; THF, -78°C; TMSCl; 25°C; H 2 0 , OH-; CH2N2; (0) H,, A/C, AcOEt; (p) 2% HCI, CH,OH; (q) (COCl),, DMSO; Et,N; (r) CH,MgBr, -78" + - 10°C; (s) 2 N KOH, H,O.
S C H ~ M A 5
pouvant stre inverse suivant une skrie de reactions precedemment rapportees (14). D'autre part comme nous l'avons soulignee la forte selectivite obtenue aprts le rearrangement [3,3]-sigmatro- pique et la comparaison de la configuration du C-2 du diastereoisomtre majoritaire obtenu avec l'acide nonactique nature1 a permis de confirmer le passage, pour le rearrangement de ce type de cettne- acetals silyles cycliques, par un intermediaire du type bateau.
Les points de fusion ont Cte ddtermines sur un appareil Hoover a capillaires et ne sont pas corrigis. Les spectres d'ab- sorbtion infrarouge (ir) ont etd obtenus avec un spectrometre Perkin-Elmer 727 B et les spectres de resonance magnetique nucleaire ont ete enregistris sur un appareil Varian EM-390 1 9 0 MHz pour le proton ('H rmn) et sur un appareil Jeol FX-90 Q pour le carbone ("C rmn). Les solvants utilises ont etd CDCl, ou CC14. Les deplacements chimiques sont exprimes en ppm sur I'dchelle 6 en prenant le tktramethylsilane comme reference interne (SMe4Si = 0,O). Les constantes de couplage sont donnkes en Hz. Les spectres sont decrits de la maniere suivante: solvant, 6 (multiplicite: s = singulet; d = doublet; t = triplet; q = quadruplet; m = multiplet. Constantes de couplage. Intdgration. Interprbtation). Les pouvoirs rotatoires ont i t6 mesurts dans
une cellule de 1 dm et 1 mL de capacite sur un polarimetre Perkin-Elmer 141.
Les microanalyses ont ete effectuees par Spang Microanalyti- cal Laboratory, Ann Arbor, MI. Les echantillons analytiques ont ete obtenus, sauf indication contraire, par distillation dans un tube boules a la pression indiquke.
Les chromatographies en phase vapeur (cpv) ont ete ef- fectuees sur un chromatographe Hewlett-Packard 5750 equip6 avec un detecteur a ionisation de flamme utilisant l'helium comme gaz vecteur a un debit de 60 mL/min.
L'avancement des reactions a ete suivi sur des prelevements par spectroscopie de 'H rmn ou par chromatographie sur couche mince (ccm). Les ccm on Cte realisees sur des plaques preparees de gel de silice 60F-254 Merck, de 0,25 mm d'kpaisseur. Les chromatographies sur colonne ont etC effectuees sur gel de silice Merck "Silica gel 60", 70-230 mesh ASTM, a l'exception de celles rkalisees sur gel de silice acide Silicar CC-4 Special, Mallinckrodt Chemical Works, St. Louis, MO.
Les solvants et rdactifs ont ete sechbs et distilles avant emploi. L'dther ethylique, le THF et le pentane ont CtC distilles sur sodium (utilisant la benzophenone comme indicateur pour les deux premiers) et sous une atmosphke d'argon sec. Le mdthanol a ete distill6 sur methylate de magnesium et le chlorure de methylene sur pentoxide de phosphore. Le DMSO a 6tC distille sur hydrure de calcium pulverise, sous une pression de 30 Tom. La diisopropylamine et la tridthylamine ont etC distillees sur sodium-benzophenone et sous argon juste avant emploi. Le chlorure d'oxalyle a etC distille sous atmosphere d'argon et le chlorure de propionyle sur 10% de quinoleine. Le chlorure
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
UN
IV O
F B
IRM
ING
HA
M o
n 11
/10/
14Fo
r pe
rson
al u
se o
nly.

578 CAN. J. CHEM. VOL. 59, 1981
d'amonium a Cte seche sur P205 a 80°C sous 1 Torr pendant deux jours.
Toutes les reactions ont ete realisees sous atmosphere d'ar- gon sec (a I'exception des hydrogenations et reductions dans I'amoniac) en legere surpression utilisant un systeme permettant alternativement d'evacuer et d'introduire le gaz inerte. Les reactions (hormis les hydrolyses finales) ont 6te effectuees avec du materiel (riacteurs, seringues, condenseurs, etc ...) preala- blement seche 12 h a 140°C et refroidi dans un dessiccateur sur sulfate de calcium anhydre. Les volumes inferieurs a 30 mL ont ete introduits a I'aide de seringues a travers des bouchons per- forables.
Par traitement habituel on entend: lavage (3 ou 4 fois) par une solution saturee en NaCl de la phase organique, sechage sur MgSO,, filtration, evaporation du solvant sous pression reduite et sechage du residu plusieurs heures sous une pression de I'ordre de Tom a temperature ambiante. Les distillations sur les quantites inferieures a 5 g ont kt6 r6alisCes dans un tube a boules par chauffage dans un four thermostate. Les points d'ebulition indiques se rapportent a ces distillations.
A une solution de 51,l g (164,7 mmol) debhenylm6thyl-2,3-0- (I-methvlethvlidbneba-D-mannofuranoside (8) (201 dans 600 m~ de ihlorire de ðylene, on ajoute 96 g'(0,8 mb~) de N,N- dimCthylformamide dimethylacetal fraichement redistill&. La solution est maintenue pendant 24 h sous atmosphere d'argon a temperature ambiante. Le solvant est evapor6 sous pression reduite a temperature inferieure 50°C. Le r6sidu conserve 12 h supplementaires sous un vide de 0,01 Tom est utilise sans autre purification au cours de l'etape suivante: lH rmn (CDCI,) 6: 1,32 et 1,45 (2% 2 x 3H, C(CH,),); 2,36(s, 6H, N(CH,),); 5,06(s, lH , PhCH20CHO-); 5,40 et 5,46 (2s (diastereoisomkres), lH , -0CHON:); 7,33 (s, SH, C6H5).
Phe'nylme'thyl 2,3-0-(1-me'thyle'fhylid2ne)- 5.6-dide'hydro- 5,6-didioxy-a-o-mannofuranoside (Ila)
Une solution du produit brut precedent dans 500 mL d'anhy- dride acetique est chauffke a 130°C pendant 1 heure. L'anhy- drique acetique est k a p o r e sous pression reduite et I'huile residuelle dissoute dans 500 mL d'ether. Apres lavage avec une solution B 5% de NaHCO, (4 x 75 mL) et traitement habituel on obtient une huile, qui chromatographike sur gel de silice (eluant: cyclohexanelAcOEt, 85: 15) fournit 34,8 g (76,5% a partir de 8; 96% sur la base du diol8 regenere) de l l a et 11,34 g (17,5%) de diacktate 9. La reduction de ce dernier par LiAIH, permet de regenerer 8 avec un rendement de 93%; Eb: 95-100°C (0,W Tom); R,: 0,4 (cyclohexane/AcOEt, 75:25); [aIDz5 +56,6" (c 1,14, CHCI,); ir (CHCI,): 1020, 1090, 1230, 1385, 1390, 3010, 3030,3090,3110~m-~; 'Hrmn (CDCI,)G: 1,30 et 1,45 (s, 2 x 3H, 2CH,); 5,08 (s, IH, -0CHO-); 5,20-5,53 (m, 2H, C=CH2); 5,8O-6,23 (m, IH, C=CH); 7,30 (S, 5H, C6H5). Anal. C ~ C . pour C,6H2,04: C 69,55, H 7,29; trouvee: C69,49, H 7,17.
Phe'nylme'thyl 2,3-041-me'thyle'thy1idkne)-5-de'oxy-a-D mannofuranoside (12a)
A 250mL (125 mmol) d'une solution 0,5 M de 9- borabicyclo[3.3.1]nonane (9-BBN) dans le THF4 agit6e magnetiquement sous atmosphere d'argon on ajoute a I'aide d'une ampoule B addition 32,5 g (1 17,6 mmol) de l l a en solution dans 130 mL de THF. La solution est maintenue 2 heures et demie a temperature ambiante. On ajoute successivement par I'ampoule a addition 75 mL d'ethanol, 50 mL d'une solution 6 M de NaOH et 25 mL d'une solution a 30% de H202 et chauffe le melange 1 heure et demie a 60°C. La phase aqueuse refroidie est
saturee en carbonate de potassium. La phase organique est skparee et traitke de manibre habituelle. Le residu chromato- graphie sur gel de silice (eluant: cyclohexanelAcOEt, 75:25 a 65:35) conduit a I'isolement de 33,4 g (96,5%) d'alcool12a sous forme d'huile; Eb: 120°C (0,05 Tom); R,: 0,50 (cyclo- hexanelAcOEt, 5050); [aIDz5 +97" (c 1,03, CHC13); ir (CHCI,): 1010,1085,1225,1385,1390,3560,3640cm-1; 1Hrmn(CDCI,)6: 1,30 et 1,43 (2s, 2 x 3H, C(CH,),); 3,60-3,90 (m, 2H, CCH20H); 4,52 (AB, JAB = 12 HZ, VAB = I3 HZ, 2H, 4 C H 2 P h ) ; 5,04 (s, IH,--0CHO-);7,32(s, 5H, C6H5);(CDC13 + D20)6: 3,72(t, J = 6 Hz, 2H, CCHzOH). Anal. calc. pour Cl6HZ2O5: C 65,29, H 7.53; trouvee: C 65.19. H7.66. . . .
Phe'nylme'thyl 2,3-0-(I-me'thyle'thylid2.ne)-5-de'oxy-6-0- me'thoxyme'thyl-a-o-mannofuranoside (13a)
Une suspension de KH a 23,6% dans I'huile5 (16,77 g; 99 mmol) est lavee avec 3 x 100 mL de pentane sous atmosphere d'argon. On ajoute 205 mL de THF, un crystal d'imidazole, refroidit la solution dans un bain de glace et additionne lente- ment sous agitation magnitique a I'aide d'une ampoule a addi- tion 24,2 g (82,2 mmol) de 12a en solution dans 205 mL de THF. A la fin de I'addition le melange est agite 15 min supplementaires a 0°C. On ajoute lentement a I'aide d'une seringue 9,43 mL (124 mmol) d'ether chloromethylm6thylique prealablement seche pendant 12 h sur chlorure de calcium, puis 2 h sur carbo- nate de potassium et filtr6. Le bain de glace est enleve. Le melange reactionnel maintenu a temperature ambiante pendant 7 h est alors hydrolyse avec 150 mL d'une solution saturee de bicarbonate de sodium et gard6 sous agitation durant la nuit. On ajoute 1 L d'ether et decante la phase organique qui est lavee avec 4 x 250 mL d'une solution saturee de bicarbonate de sodium, puis traitee de falon habituelle. On obtient 27,8g (100%) de 13a (une tache en ccm) qui est utilis6 sans autre purification au cours de 1'6tape suivante. Un echantillon analytique de 1 g est obtenu par chromatographie sur gel de silice (eluant: cyclohexanelAcOEt, 85: IS); Eb: 125-127°C (0,Ol Torr); R,: 0,38 (cyclohexane/AcOEt, 75:25); [aIDz5 +89,2" (c 0,95, CHCI,); ir (CHCI,): 1020, 1050, 1090, 1120, 1165, 1220, 1385, 1390,2960 cm-I; 'H rmn (CDCI,) 6: 1,30 et 1,43 (2s, 2 x 3H, C(CH,),); 2,O (d de t, J, = J2 = 6 Hz, 2H, -CH2CH20MOM); 3,33 (s, 3H, OCH,); 3,67 (t, J = 6,3 Hz, 2H, --CH20MOM); 4,03-4,30 (m, lH, --OCHCH2-); 4,61 (s large, 4H, -OCH20-- et -0CHCHO-); 4,53 (AB, JAB = 12 HZ, VAB = 17 HZ, 2H, -OCH2Ph); 5,03 (s, IH, -0CHO-); 7,31 (s, 5H, C6H5). Anal. calc. pour C,,HZ6O6: C 63,89, H 75'4; trouvee: C 63,93, H 7,68.
2,3-0-(1-Me'thyle'thylid2.ne)-5-de'oxy-6-O-me'thoxyme'thyl- (a et P)-~mannofuranose (14a)
A 1,2 L d'amoniac (redistill6 a partir d'une solution bleue de lithium dans I'amoniac) sous agitation -78°C on ajoute 1,25 g (180 mmol) de lithium en fil et aprbs 20 min, a I'aide d'une ampoule addition, une solution de 27,2 g (80,4 mmol) de 13a dans 40 mL de THF. Le bain refrigerant est enlev6 et la reaction maintenue sous reflux d'amoniac. Aprks 2 h, 10,7 g (0,2 mol) de NH4CI sec sont ajoutes par portions d'environ 0,5 g. On laisse alors lentement &vaporer I'amoniac sous un courant d'argon pendant que 500 mL d'ether anhydre sont ajoutks au milieu riactionnel. Lorsque I'amoniac est totalement Cvapore la sus- pension est filtree sur veme fritte et 1'6ther 6vaporb sous pres- sion reduite. Aprbs chromatographie sur gel de silice (kluant: cyclohexane/AcOEt, 60:40) on isole 15,7 g (79%) de lactol 14a. Le spectre de lH rmn de 14a montre la presence d'un seul anomere qui en solution chloroformique s'isomerise en un melange a et P; Eb: 105°C (0,02 Tom); Rf: 0,30 (cyclo-
40btenu chez Aldrich. 50btenu chez ALfa Products.
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
UN
IV O
F B
IRM
ING
HA
M o
n 11
/10/
14Fo
r pe
rson
al u
se o
nly.

IRELAND ET VEVERT
hexane/AcOEt, 50:50); ir (film): 1060, 1155, 1210, 1385, 1390, 2940, 3400 cm-I; IH rmn (CDCI,) 6: 1,31 et 1,43 (anomere majoritaire), 1,35 et 1,50 (anomere minoritaire) (2 x 2s, 2 x 3H, C(CH,),); 1,93 (d de t, J, = J, = 6 Hz, 2H,--CH,CH,OMOM); 3,35 (s, 3H, -OCH,); 3,65 (t, J = 6 HZ, 2H, -CH20MOM); 4,25 (d de t, J, = 3 Hz, J, = 6 Hz, lH, -OCHCH,-); 4,63 (s, 2H, -OCH,O-); 5,30 (s, "0,85H", -0CHO-anomere majoritaire). Anal. calc. pour C,,H2,06: C 53,22, H 8,12; trouvee: C 53,20, H 8,Ol.
Chlorure de 5-de'oxy-2,3-0-(l-me'thyle'thylid2ne)-6-0- me'thoxymPthyl-a-~mannofuranosyle (15a)
On chauffe a 90°C sous agitation magnetique et sous atmos- phere d'argon une solution de 993,l mg (4 mmol) de 14a, 2 mL (20 mmol) de tetrachlorure de carbone et 2,1 g (8 mmol) de triphenylphosphine dans 20 mL de THF. Le milieu rtactionnel homogene laisse rapidement precipiter de I'oxyde de triphenyl- phosphine. Aprts 3 h la solution refroidie est filtree sur verre fritte et le solvant evapore sous pression reduite. Le residu est triture avec du pentane sec, garde une demi-heure au refrigerateur, et I'oxyde de triphenylphosphine qui a cristallise est filtre. Cette dernikre operation est repetie (3 ou 4 fois) jusqu'a elimination complete de I'oxyde de triphenylphosphine. La distillation du produit brut dans un tube a boules permet d'obtenir 976mg (91,5%) de 15a; Eb: 90°C (0,02 Torr); [alDZ5 +94,4" (c 1.04, CHCI,); ir (film): 1050, 1090, 1160, 1215, 1385, 1390, 2965 cm-I; lH rmn (CDCI,) 6: 1,31 et 1,43 (2s, 2 x 3H, C(CH,),); 2,O (d de t, J, = J, = 6 Hz, 2H, -CH,CH,OMOM); 3,35 (s, 3H, -OCH3); 3.63 (t, J = 6 Hz, 2H, -CH,OMOM); 4,25-4,60(partie Xd'un AMX, lH,-OCHCH,-); 4,60 (s, 2H, -OCH,O-); 4,70 (partie M d'un AMX, JAM = 6 Hz, JMX = 3 Hz, lH, CICCCH); 4,93 (partie A d'un AMX, JAM = 6 Hz, lH, CICCH); 6,06 (s, lH, -0CHCI). Anal. calc. pour C, ,H,g05CI: C 49,54, H 7,18; trouvee: C 49,62, H 7.29.
Phe'nylm~rhyl2,3-0-(l-m~thylbrhylid>ne)-5-de'oxy-6 O-[dime'thyl-(I,I-dime'thyle'thyl)-silyl]-a-~-manno- furanoside (13'a)
Une solution de 5,88 g (20 mmol) de 12.0 dans le dimethylfor- mamide anhydre est agitee sous atmosphere d'argon en presence de 5,7 g (84 mmol) d'imidazole prealablement sublime et 6,33 g (42 mmol) de chlorure de tert-butyldimethylsilane. Apres 15 h a temperature ambiante 200 mLd'eau sont ajoutts au milieu reactionnel qui est extrait par 3 x 100 mL d'ether. Apres traitement habituel, I'huile obtenue est chromatographite sur gel de silice (eluant: cyclohexane/AcOEt, 75:25). On obtient 8,OS g (99%) d'kther silylt 13'a; Eb: 140°C (0.07 Torr); RI: 0,34 (cyclohexane/AcOEt, 75:25); [aIDZS +80,9" (c 0,93, CHCI,); ir (CHCI,): 840, 1015, 1095, 1230, 1265, 1385, 1390, 2875, 2950, 2970cm-I; 'H rmn (CDC1,)G: 0,09(s, 6H, Si(CH,),); 0,91 (s, 9H, I-C4Hg Si); 1,28 et 1,42 (2s, 2 x 3H, C(CH,),); 1,91 (d de t, J, = J, = 6 Hz, 2H, CH,CH,OTBDMS); 3.25 (t, J = 6,5 Hz, 2H, CHZOTBDMS); 4,53 (AB, JAB = 12 HZ, VAB = 18.6 HZ, 2H, OCH,Ar); 5,02 (s, lH, OCHOCH,Ar); 7,30 (s, SH, -C6H5). Anal. calc. pour C2,H3,Si05: C 64,67, H 838; trouvee: C 64,79, H 9,00.
2,3-0-(1-Me'thylPthylid2ne)-5-de'oxy-6-O-[dime'thyl- (1,l-dimbthyle'thy1)-silyll-a et p-mnannofuranose (14'a)
Obtenu a partir de 7,96 g (19,s mmol) de 13'a suivant le mode operatoire permettant d'obtenir 14a a partir de 13a. Apres chromatographie sur gel de silice on isole 5,35 g (86%) de 14'a; Eb: 97°C (0,02 Torr); RI: 0,45 (cyclohexane/AcOEt, 50:50); ir (CHCI,): 850, 1015, 1055, 1095, 1230, 1265, 1385, 1390, 2890, 2950, 2970, 3435, 3620 cm-I; 'H rmn (CDCI,) 6: 0,09 (s, 6H, Si(CH,),); 0,93 (s, 9H, r-C4HgSi); 1,30 et 1,43 (2s, 2 x 3H, C(CH,),); 1,93 (d de t, J, = J , = 6 Hz, 2H, CH,CH,OTBDMS); 3,73 (t, J = 6 Hz, 2H, CH,OTBDMS); 5,33 (s, lH, OCHOH).
Anal. calc. pour C15H3005Si: C 56,57, H9,49; trouvee: C 56,49, H 9,36.
Chlorure de 5-dboxy-2,3-0-(1-me'thyle'thylid2ne)-60- [dime'thyl-(l,l-dime'thyle'thyl)-silyl]-a-~mannofurano- syle (15'a)
Obtenu a partir de 647 mg (2,03 mmol) de 14'a suivant le mode operatoire permettant d'obtenir 15a a partir de 14a. Obtenu aprks distillation: 640 mg (94%) Eb: 81°C (0,02 Torr); [aID25 +82,7" (c 0,9, CHCI,); ir (CHCI,): 840, 1095, 1175, 1230, 1265, 1385, 1390, 2875, 2950, 2970 cm-I; lH rmn (CDCI,) 6: 0,09 (s, 6H, Si(CH,),); 0,93 (s, 9H, r-C4HgSi); 1,30 et 1,43 (2s. 2 x 3H, C(CH,),); 1,% (d de t, J, = J, = 6Hz, 2H, CH,CH,OTBDMS); 3,73 (t, J = 6 Hz, 2H, CH,OTBDMS); 4,30-4,53 (partie X d'un AMX, lH, OCHCH,); 4,73 (partie M d'un AMX, JAM = 6 HZ, JMx = 3 Hz, lH, CICCCH); 4,93 (partie A d'un AMX, JAM = 6 Hz, lH, CICCH); 6,10 (s, lH, CICHO). Anal. calc. pour C,5H2g04SiCI: C 53,47, H 8,68; trouvke: C 53,35, H 8,49.
2R-cis-2-(~-Me'thoxym~rhyl>noxye'rhyl)-2,3-dihydro-3- hydroxyfurane (17a)
A 80 mL d'amoniac (redistill6 i partir d'une solution bleue de lithium dans I'amoniac) sous agitation i -78°C on ajoute 110 mg (15,8 mmol; 5 eq.) de lithium en fil, et apres une demi-heure a I'aide d'une seringue une solution de 845 mg (3,17 mmol) de 15a dans 4 mL de THF. On rince avec 2 x 3 mL de THF et garde sous agitation a -78°C pendant une heure et demie. A la solution bleue est ajoute 1,34g (25 mmol) de NH4CI sec. L'amoniac est evapore sous courant d'argon et 100 mL d'kther et 2 g de MgSO, sont ajoutes au milieu reactionnel. Lorsque I'amoniac est evapore, la suspension est filtree sur verre fritte et I'ether evapore sous pression rBduite. L'huile residuelle est trituree avec un melange d'kther et de pentane (l:l), conservee 1 heure au refrigerateur et filtree. La distillation du produit brut conduit a I'obtention de 441 mg d'un melange de glycal 17a et de 1 ,5-di- deoxy-2,3-0-(1-methy1ethylidene)-6-0-m6hxym6thyl-~- mannofuranose (16a) dans un rapport 10:l ('H rmn) correspon- dant a un rendement de 70% en 17a. DifErentes tentatives de chromatographie du melange sur gel de silice ou Florisil ont conduit B une dCcomposition partielle du glycal; Eb du melange: 85°C (0,005 Torr); ir(fi1m): 1060,1115,1160,1606,3450cm-1; 'H rmn (CDCI,) 6: 1,90-2,40 (m, 2H, --CH,CH,OMOM); 3,35 (s, 3H, -OCH,); 3,67 (m, 2H, --CH,OMOM); 4,25 (d de t, J, = J, = 7 HZ, lH, C=COCH); 4,60 (s, 2H, -OCHZO-); 4,65-4,73 (m, lH, CHOH); 5,20 (d de d, J, = J2 = 5,4 Hz, lH, -OC=CH-); 6,56 (d, J = 5,4 Hz, lH, -OCH=C-).
[2R[2a(R* et S *),5a]]-2,EDihydro-5-(2-me'thoxy- me'thyl2noxye'thyl)-a-me'thyl-2-furanace'ae de me'thyle (184
A une solution de 441 mg du melange 10: 1 de 17a et 16a (2,22 mmol de glycal 17a) dans 10 mL de THF ?I -78°C sous atmos- phere d'argon, on ajoute sous agitation un crystal de phenan- troline et une solution 2,4 M de n-BuLi dans I'hexane4 jusqu'i apparition d'une coloration brunhre (0,96 mL; 2,3 mmol). Apres 5 min 0,2 mL (2.33 mmol) de chlorure de propionyle sont ajoutes. On enleve le bain rkfrigtrant, laisse remonter la temperature a - 10°C et refroidit a nouveau i -78°C. La solution est alors ajoutee B I'aide d'une aiguille i double entree B une solution 0,s N de 2,33 mmol de diisopropylamidure de lithium dans le THF, maintenue i -78°C sous agitation. Apres 15 minon ajoute 1,90 mL (1 1 mmol) du liquide surnageant obtenu aprks centrifugation d'un melange 3:l (vlv) de chlorure de tri- methylsilyle et de triethylamine, maintient le milieu reactionnel 15 min supplCmentaires a -78"C, enleve le bain refrigerant et garde B temperature ambiante pendant 20 h. La solution est alors hydrolysee a 0°C avec 50 mL d'une solution 0,s N de NaOH. Les solvants sont Bvapores sous pression riduite ?I temp6rature
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
UN
IV O
F B
IRM
ING
HA
M o
n 11
/10/
14Fo
r pe
rson
al u
se o
nly.

580 CAN. J. CHEM. VOL. 59, 1981
inferieure a 30"C, la phase aqueuse extraite par 3 x 50 rnL d'ether, acidifiee jusqu a pH 2,5, extraite par 7 x 100 rnL d'Cther et les phases CthCrees reunies traitees de f a ~ o n habituelle. Un exces d'une solution BthCree de diazornethane est alors ajoutC a I'huile obtenue, conduisant a 382 rng de 18a brut, qui est di- recternent hydrogen6 sans purification ulterieure. Un echantil- Ion analytique est obtenu par chromatographie sur gel de silice (Cluant: cyclohexanelAcOEt, 75:25); Eb: 80°C (0,05 Tom); Rf: 0,3 (cyclohexanelAcOEt, 60:40); ir (CHCI,): 1050, 1740 crn-I; 'H rrnn (CDCI,) 6: 1,11 (diast8reoisornere rnajoritaire) et 1,22 (diastCreoisornere rninoritaire) (2 x d, J = 7,2 Hz, 3H, -CH,); 1,65-1,96 (rn, 2H, -CH,CH,OMOM); 2,60 (d de q, J, = J, = 7,2 HZ, lH, -0,CCH); 3,33 (s, 3H, -OCH,); 3,63 (t, J = 6,5 HZ, 2H, -CH20MOM); 3,68 (s, 3H, -COOCH3); 4,58 (s, 2H, -OCH20-); 4,73-5,M (rn, 2H, HCOCH); 5,86 (m, 2H, -HC=CH-). Anal. calc. pour C12H2,0,: C 59,00, H 8,25; trouvee: C 58,76, H 8,26.
[2R[2a(R*),5a]] et [2R[2a(S*),5a]]-Te'trahydro-5- (2-rne'thoxyrne'thyltnoxye'thyl)-a-rne'thyl-2-furanace'tate de rne'thyle(l9a) et (l9'a)
L'ester brut 18a en solution dans 20 rnL de THE est hy- drogene dans un appareil de Pan en presence de 20mg de rhodium (5%) sur charbon pendant 4 h sous 4 atm. Apres filtra- tion du catalyseur et traiternent habitue1 un Bchantillon du pro- duit brut est analyse par cpv (SE30 10%; T: 115°C). L'avvarition de 2 pics se rec~uv~ant~~artiellement (tR: 60 et 63 &in) fait apparaitre que les 2 diastereoisorneres 19a et 19'a ont Cte forrnes dans une proportion de 89: 1 1. Le produit brut chromatographie sur gel de silice (eluant: cyclohexanelAcOEt, 85:15) conduit a I'isolernent de 265 rng de I'ester 19a (48,5% B partir du glycal 17a); Eb: 75°C (0,05 Tom); Rf: 0,26 (cyclohexanelAcOEt, 60:40); [aID2, - 13,6' (c 1,07, CHCI,); ir (film): 1050, 1120, 1160, 1205, 1742, 2970 crn-I; 'H rrnn (CDCI,) 6: 1,11 (d, J = 7,2 Hz, 3H,-CH,); 2,52(d deq, J, = J2 = 7,2Hz, 1H,-0,CCH); 3,33 (s, 3H, -OCH3); 3,58 (t, J = 6,5 HZ, 2H, -CH20MOM); 3,68 (s, 3H, -COOCH,); 3,78-4,13 (rn, 2H, -HCOCH-); 4,58 (s, 2H, -OCH,O-). Anal. calc. pour C,,H,,O,: C 58,52, H 9,00; trouvke: C 58,73, H 8,89.
On isole de mkrne 29,5ing (5,3% a partir du glycal 17a) de l'epimere 19'a; Rf: 0,3 (cyclohexanelAcOEt, 60:40); 'H rrnn (CDCI,) 6: 1,20 (d, J = 7,2 Hz, 3H, -CH,), 2,50 (d de q, J, = J, = 7 2 Hz, lH, -0,CCH); 3,33 (s, 3H, -OCH,); 3,58 (t, J = 6,5 Hz, 2H, -CH,OMOM); 3,65 (s, 3H, -COOCH,); 3,78-4,13 (rn, 2H, -HCOCH-); 4,58 (s, 2H, -OCH,O-).
[2R[2a(R*),5a]]-Te'trahydro-5-(2-hydroxykhy&x- rne'thyl-2-furanace'tate de rne'thyle (20a)
Une solution de 240 rng (0,97 rnmol) de 19a dans 50 mL de methanol anhydre contenant 2,1% d'acide chlorhydrique (prepare a partir de 1,7 rnL de chlorure d'acktyle et de 50 rnL de methanol) est chauffke a 45°C pendant 14 h sous atmosphere d'argon. Apres evaporation des solvants sous pression reduite, I'huile obtenue est chromatographike sur gel de silice (Cluant: cyclohexanelAcOEt, 65:35). On isole 190 mg (96,5%) d'ester- alcool20a; Eb: 83°C (0,M Tom); Rf: 0,21 (cyclohexanelAcOEt, 85:15); [aIDz5 -26,3O (C 1,06, CHCI,); ir (CHCI,): 1080, 1180 1210,1742,2875,3570 cm-I; IH rrnn (CDCI,) 6: 1,11 (d, J = 7,2 Hz, 3H, -CH,); 2,52 (d de q, J, = J, = 7,2 Hz, lH, -0,CCH-); 3,68 (s,3H, -COOCH,); 3,71 (t, J = 6,5 Hz, 2H, -CH,OH); 3,83-4,20 (rn, 2H, -HCOCH-). Anal. calc. pour C1,H,,0,: C 59,39, H 8,97; trouvke: C 59,21, H 8,97.
[2R[2a(R *),5a]]-Te'trahydro-5-(2-oxoe'thy1)-a- rne'thyl-2-furanace'tate de rne'thyle (21 a)
A une solution de 226 rng (0,155 rnL; 1,78 rnrnol) de chlorure
d'oxalyle dans 2 rnL de chlorure de rnkthylene agitee a -78°C sous atmosphere d'argon on ajoute 278 rng (0,252 rnL; 3,56 rnrnol) de DMSO et apres 5 rnin une solution de 180 rng (0,89 rnrnol) de 20n dans 8 rnL de THE. On rnaintient a cette temperature pendant 20 rnin, ajoute 900 rng (1,23 rnL; 8,9 rnrnol) de triethylarnine, enleve le bain refrigerant apres 5 rnin et garde sous agitation pendant une heure et dernie. Le melange reac- tionnel est alors hydrolysk et la phase aqueuse extraite par 3 x 30 rnL de chlorure de methylhe. Les phases organiques rCunies sont lavees avec respectivernent 2 x 20 rnL d'une solution a 2% d'HCI et 2 x 20 rnL d'une solution a 2% de bicarbonate de sodium, puis traitees de f a ~ o n habituelle. On isole apres chromatographie sur gel de silice (eluant: cyclohexanelAcOEt, 75:25), 160 rng (90%) d'aldehyde 21a; Eb: 70°C (0,03 Tom); Rf: 0,25 (cyclohexane/AcOEt, 5050); [aID2, -26,3O (c 1,M, CHCI,); ir (CHCI,): 1090, 1730 cm-I; 'H rrnn (CDCI,) 6: 1,11 (d, J = 7,2 Hz, 3H, -CH,); 3,68 (s, 3H, -COOCH,); 3,9-4,48 (rn, 2H, -HCOCH-); 9,82 (t, J = 1,3 Hz, lH, X H O ) .
[2R[2a(R*),5a(S*)]] et [2R[2a(R*),5a(R*)]]- Te'trahydro-5-(2-hydroxypropyl)-a-rne'thyl-2-furanace'tate de rne'thyle (3a) et (5a)
A une suspension de 162 rng (0,79rnrnol) du complexe (H,C),S.CuBr (26) dans 2 mL de pentane sous agitation a -20°C et sous atmosphere d'argon on ajoute goutte goutte 0,93 mL (1,5 rnmol) d'une solution 1,62 M de methyllithium dans ]'ether., Apres 20 rnin B cette temperature, une solution de 70 rng (0,35 rnmol) de 21a dans 3 rnL d'un melange 1: 1 (vlv) d'Cther et de pentane est ajoutC au milieu reactionnel qui est alors rnaintenu pendant 3 h une temperature comprise entre -20" et - 10°C. La suspension est versee dans lOmL d'une solution a pH 8 de NH3/NH4CI. La phase aqueuse est extraite par 3 x 20 rnL de CH2C12 et les phases organiques rCunies traitCes de f a ~ o n habituelle. Apres chrornatographie sur gel de silice (eluant: cyclohexanelAcOEt, 70:30) on isole 29,9 rng (39,5%) de 3a et 34,2 rng (45%) de 5a ainsi que 3 rng d'un melange de ces deux produits.
(2R,3R,6S,8S) Nonactate de rnCthyle (3a): Eb: 90°C (0,025 Tom); Rf: 0,13 (cyclohexanelAcOEt, 5050); [aIDzS -20,7" (c 0,925, CHCI,); ir (CHCI,): 1090, 1180, 1205, 1285, 1395, 1450, 1475,1740,3000,3535, crn-I; 'H rrnn(CDC1,)G: 1, l l (d, J = 7,2 Hz, 3H, -0,CCCH3); 1,18 (d, J = 6,3 Hz, 3H, HOCCH,); 2,50 (d de q, J, = J, = 7,2 Hz, 2H, -0,CCH et OH); 3,67 (s, 3H, -OCH,); 'H rmn (CC1,)G: 1,08(d, J = 7,2 Hz, 3H,-O,CCCH,); 1,12 (d, J = 6,3 Hz, 3H, HOCCH,); 2,43 (d de q, J, = J, = 7,2 Hz, lH, -0,CCH); 3,60 (s, 3H, -OCH3); I3C rrnn (CDCI,): 175,l; 81,O; 65,2; 51,7; 45.3; 42,9; 30,l; 28,8; 23,2; 13,5. Anal. calc. pour CI,H2,O4: C 61,09, H 9,32; trouvCe: C 61,18, H 9,46.
(2R,3R,6S,8R) Nonactate de rnkthyle (5a): Eb: 90°C (0,025 Tom); Rf: 0,17 (cyclohexanelAcOEt, 5050); [aIDz5 -33" (C 1,18, CHCI,), ir (CHCI,): identique a 3a; 'H rrnn (CDCI,) 6: 1,12 (d, J = 7,2 Hz, 3H, -0,CCCH3); 1,15 (d, J = 6,3 Hz, 3H, HOCCH,); 2,50 (d de q, J, = J, = 7,2 Hz, lH, -0,CCH); 3,67 (s, 3H, -OCH3); IH rrnn (CCI,) 6: 1,07 (d, J = 6,3 Hz, 3H, HOCCH,); 1,09 (d, J = 7,2 Hz, 3H, -O,CCCH,); 2,45 (d de q, J, = J, = 7,2Hz, lH, -0,CCH); 3,62 (s, 3H, -OCH,). Anal. calc. pour C,,H2,0,: C 61,09, H 9,32; trouvCe: C 60,94, H 9,20.
Acide [2R[2a(R*),5a(S*)]]-te'trahydro-5-(2-hydroxypropyl)-a- rne'thyl-2- furanacktique (2a)
A 19 rng (0,087 rnrnol) d'ester 3a on ajoute 0,5 rnL d'une solu- tion aqueuse 2 N de KOH et agite rnagnktiquernent 40 min ?I
temperature arnbiante. La solution est acidifike jusqu'8pH 1,5 et Cvaporte B sec a ternpkrature arnbiante sous pression rCduite. Le rksidu est triturC et liltre sur veme fritte avec 3 x 50 rnL de CHCI,. Apres evaporation du CHCI,, I'acide brut est
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
UN
IV O
F B
IRM
ING
HA
M o
n 11
/10/
14Fo
r pe
rson
al u
se o
nly.

IRELAND ET VEVERT
chromatographie sur gel de silice acide (eluant: cy- clohexanelAcOEt, 50:50). On receuille 17 mg (96%) d'acide (-) nonactique ( 2 ~ ) ; Eb: 130°C (0,003 Torr); R,: 0,l (cyclo- hexanelAcOEtlAcOH, 50:50:2). [aIDz5 -9,3"(c 1,0, CHCI,); ir (CHCI,): 1090, 1395, 1475, 1720, 2995, 3540 cm-I; 'H rmn (CDCI,) 8: 1,15 (d, J = 7,2 Hz, 3H, -O,CCCH,); 1,20 (d, J = 6,3 Hz, 3H, HOCCH,); 2,48 (d de q, J, = J, = 7,2 Hz, lH, -0,CCH); 6,39 (2H, OH); 'H rmn (CCI,) 6: 1,12 (d, J = 7,2 Hz,
, 3H, -02CCCH3); 1,14 (d, J = 6,3 HZ, 3H, HOCCH,); 2,43 (d de q, J, = J, = 7,2 Hz, lH, -0,CCH). Anal. calc. pour CloHl8O4: C 59,39, H 8,97; trouvee: C 59,23, H 8,98.
Acide [2R[2a(R*).5a(R*)]]-titrahydro-5- (2-hydroxypropy1)-a-rne'thyl-2-furanace'tique (4a)
On hydrolyse de la mCme f a ~ o n 11,5 mg (0,051 mmol) de 8-&pi nonactate de methyle (5a) et obtient apres chromatographie sur gel de silice acide 10 mg (93%) d'acide (-) 8-ipi nonactique (Q); R,: 0,12 (cyclohexanelAcOEtlAcOH, 50:50:2); [aIDZ5 --50,1° (C 1,0, CHCI,); ir (CHCI,): 1090,1395, 1435,1475,1770,2995,3350 cm-I; IHrmn (CDC1,)G: 1,12 (d, J = 7,2 Hz, 3H,-0ZCCCH3); 1,18 (d, J = 6,3 Hz, 3H, HOCCH,); 2,47 (d de q, J, = J, = 7,2 Hz, lH, -0,CCH); 5,88 (2H, OH); lH rmn (CCI,) 6: 1,09 (d, J = 7,2 HZ, 3H, -OZCCCH3); 1,14 (d, J = 6,3 HZ, 3H, HOCCH,); 2,42 (d de q, J, = J, = 7,2 Hz, lH, -0,CCH). Anal. calc. pour Cl0Hl8O4: C 59,39, H 8,97; trouvee: C 59,49, H 833.
PhPnylmithyl2,3:5,6-di-O-(l-mCthyle'thylid>ne)-(a et P)-D- gulofuranoside (23) et (23')
Obtenus a partir de 92g (0,353 mol) de 2,3:5,6-di-0-(1- m6thyl~thylid~ne)-(a et P)-D-gulofuranose (27) suivant le mode operatoire permettant d'isoler les mannofuranosides corres- pondants (20). La chromatographie du melange brut sur gel de silice (eluant: cyclohexane/AcOEt, 80:20) permet d'isoler 110 g d'anomtre p qui recristallisi5 dans I'Cther de petrole fournit 106,2 g (86%) de 23; F: 88-89°C (ether de petrole); Rf: 0,4 (cy- clohexanelAcOEt, 80:20); [alDz5 -64,Y' (c 1,03, acetone); ir (CHCI,): 855, 1015, 1090, 1160, 1230, 1380, 1390, 1465, 2960, 3000 cm-I; 'H rmn (CDCI,): 1,27; 1,40; 1,43 et 1,45 (4s. 12H, 2 C(CH3),); 4,60 (AB, JAB = 12 HZ, VAB = 20,8 HZ, 2H, -OCH2Ph); 5,15 (s, lH, OCHOCH,Ph); 7,31 (s, 5H, C6H5-). Anal. calc. pour Cl9H2,O6: C 65,13, H 7,48; trouvke: C 64,99, H 7,38.
On isole de mCme 6g (5%) d'a-glycoside 23': F: 8647°C (CH30H/H20); Rf: 0,17 (cyclohexanelAcOEt, 80:20); [aIDZ5 -63,4" (c 1,08, acetone); ir (CHCI,): 860,1030, 1080,1120,1175, 1230,1385,1390,1465,2960,3020 cm-I; 'Hrmn (CDC1,)G: 1,32; I ,38, 1,45 et 1,50 (4s, 12H, 2 C(CH3),);7,2-7,s (m, 5H, C6H5-). Anal. calc. pour Cl9H,,O,: C 65,13, H7,48; trouvCe: C65,06, H 7.39.
Phinylme'thyl 2.3-@(I-mithy1ithylidkne)-$-D gulofuranoside (24)
Obtenu par hydrolyse partielle de 105,8g (0,302 mol) de 23 suivant le mode operatoire permettant d'isoler I'a-man- nofuranoside 8 correspondant (20). Apres neutralisation le methanol est evapore sous pression reduite et le residu verse dans 2 L de CH,CI,. La phase organique est separCe et traitee de f a ~ o n habituelle. On obtient 92,7 g (99%) de 24. Un echantillon analytique est obtenu par recristallisatioa dans un melange AcOEtlether de petrole; F: 137-138°C; Rf: 0,43 (EtOAc); -%,3"(c 1,12, acetone); ir (CHCI,): 990,1015,1095,1175,1230, 1385,1390,2960,3030,3550,3620cm-I; 'H rmn (CDCI,) 6: 1,28 et 1,45 (2s, 6H, C(CH,),); 4,58 (AB, JAB = 12 HZ, vAB = 20,8 HZ, 2H, -OCH,Ph); 5,15 (s, lH, -OCHOCH,Ph); 7,31 (s, 5H, C6H5--). Anal. calc. pour C,,H,,O,: C 61,92, H 7,15; trouvCe: C 61,89, H 7,09.
Phe'nylmPthyl2,3-O-(l-mithyle'~hylid2ne)-5,6-(N, N- dimt!thylaminomithyl>ne)-0-~gulofuranoside (26)
MCme mode operatoire que pour le diastCreoisomere 10 a partir de 50,7 g (0,163 mol) de 24; 'H rmn (CDCI,) 8: 1,26 et 1,43 (2s, 6H, C(CH,),); 2,38 (s, 6H, -N(CH,),); 5,13 (s, IH, -OCHOCH2Ph); 5,43, (s, IH, (H3C)2NCH); 7,30 (s, 5H, C6H5).
Phinylme'thyl2,3-0-(l-mithyle'thylid2ne)-5,6-didihydr0-5,6- dide'oxy-P-~gulofuranoside (11 b)
MBme mode operatoire que pour I'enantiomere l l a ipartir du formamideacCtal26 brut. Obtenu: 34,15 g (755% apartir de24); F: 36,7-37,5"C (ether de petrole ?I -20°C); [aIDzs -54,4" (c 0,975, CHCI,). Anal. calc. pour C,6H2004: C 69,55, H 7,29; trouvee: C 69,50, H 7,19.
On isole de mCme 12,2 g (19%) de diacetate 25 qui est recycle en 24 apres reduction par LIAIH,.
Phe'nylme'thyl2,3-0-(I-mithyle'thylid>ne)-5-dioxy- P-D gulofuranoside (12b)
MCme mode operatoire que pour I'enantiomkre 12u, partir de 44g (0,16 mol) de I'olefine l lb . Obtenu: 44,4 g (95%); [aIDZS -99,0" (c l,O, CHCI,). Anal. calc. pour C,6H,20,: C 65,29, H 733; trouvee: C 65,15, H 7,47.
Le groupe protecteur MOM est plus efficacement introduit par reaction avec I'ether chloromCthylmethylique (cf. 13a) et le mode operatoire suivant (23). employe dans notre laboratoire avec succes sur des substrats etroitement apparentes a 12b, conduit avec ce compose a des rendements mediocres.
A une solution de 42 g (0,142 mol) de I'alcool 12b et 60 mL de CH,(OCH,), dans 600 mL de CH,CI, on ajoute sous agitation et sur une periode de 20 min 100 g d'un melange 1: 1 de P20S en poudre et de Celite. Apres une heure le milieu rkactionnel est verse dans 500 mL d'une solution a 10% de NaHCO, et extrait par 3 x 500mL d'ether. Les phases etherees reunies sont traitees de f a ~ o n habituelle. Le produit brut chromatographie sur gel de silice (eluant: cyclohexanelAcOEt, 90: 10) conduit a I'isolement de 19,3g (40%) de 13b; [aIDz5 -88,5" (c 1,075, CHCI,). Anal. calc. pour C,,H,,O,: C 63,89, H 7,74; trouvee: C 64.04, H 7,77.
2,3-0-(l-Me'thylithylid>ne)-5-de'oxy-6-O-me'thoxym~thyl- (a et P)-~gulofuranose (14b)
MCme mode operatoire que pour I'enantiomkre 1Q a partir de 18,45 g(54 mmol) de 13b. Obtenu: 11,l g(82%). Anal. calc. pour C11HZ006: C 53,22, H 8,12; trouvee: C 53,24, H 8,10.
Chlorure de 5-de'oxy-2,3-0-(I-mithyltthylid>ne)-6-0- mithoxymithyl-0-~gulofuranosyle (15 b)
M2me mode operatoire que pour le chlorure enantiomere 15a a partir de 1,97 g (7,93 mmol) de lactol 14b. Obtenu: 1,82g (86%); [ahz5 -92,s" (c 0.905, CHCI,). Anal. calc. pour C,,H1905CI: C 4934, H 7,18; trouvte: C 49,37, H 7,06.
2S-cis-2-(~-Me'thoxymirhyl>nox~thyl)-2,3-dihydr0-3- hydroxyfurane (17b)
Mbme mode operatoire que pour I'enantiomere 17a a partir de 1,8 g (6,75 mmol) du chlorure 15b. Obtenu: 1,095 g d'un melange 10:l ('H rmn) de glycal 17b et de 16b, correspondant un rendement de 82% en glycal17b.
[2S[2a(R* et S*),5~]]-2,5-Dihydro-5-(2-mPrhoxy- mithyl>noxyithyl)-a-me'thyl-2-furanacitae de me'rhyle (18b)
MCme mode operatoire que pour les enantiomkres respectifs
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
UN
IV O
F B
IRM
ING
HA
M o
n 11
/10/
14Fo
r pe
rson
al u
se o
nly.

582 CAN. J . CHEM. VOL. 59, 1981
correspondants 18a a partir de 1,09g du melange 10:l precedent, correspondant i 5,5 mmol de glycal17b.
[2S[2a(R*),Sa]] et [2S[2a(S *),5a]]-Te'trahydro-5- (2-m~thoxym~thyltnoxy~thyl)-a-me'thyl-2-furanace'tate de mithyle (19b) et(l9'b)
Obtenu par hydrog6nation i pression atmosphkrique d'une solution de I'ester insature brut precedent 18b dans 20 mL d'AcOEt en pr6sence de 100 mg de Pt (5%) sur charbon pendant une demi-heure. Apres filtration du catalyseur et traitement habitue1 de la phase organique, le produit brut est analyse par cpv (SE30 10%; T: 118°C). Le m8me chromatogramme que pour les Cnantiombres correspondants 19a et 19'a est obtenu, indi- quant que la formation des diast6reoisombres 19b et 19'b s'est effectuee dans une proportion de 86: 14. La chromatographie sur gel de silice (6luant: cyclohexane/AcOEt, 85:15) du produit brut conduit B I'isolement de 596 mg de I'ester 19b (44% a partir du glycal 17b); [alDZs +13,8" (c 0,995, CHCI,). Anal. calc. pour C,,H,,OS: C 58,52, H 9,00; trouvee: C 58,67, H 9,03.
On isole de mCme 66,3 mg (49% B partir du glycal 17b) de I'ester epimkre en C-2, 19'b; [aIDz5 -33,8" (c 1,005, CHCI,). Anal. calc. pour C,,H,,Os: C 58,52, H 9,00; trouvke: C 58,27, H 8,90.
[2S[2a(R*),5a]]-Te'trahydro-5-(2-hydroxye'thyl)-a-me'thyl- 2-furanace'tate de me'thyle (20b)
MBme mode operatoire que pour I'alcool enantiomere 19a a partir de 230 mg (0,93 mmol) de 19b. Obtenu: 179,5 mg (95%); [alDZ5 +26,0° (c 1,01, CHCI,). Anal. calc. pour C10H1804: C 59,39, H 8,97; trouvee: C 59,47, H 8,86.
[2S[2a(R*),5a]]-Te'trahydro-5-(2-oxoe'thyl)-a-me'thyl- 2-furanace'tate de me'thyle (21 b)
MCme mode operatoire que pour I'aldehyde enantiomere 21a B partir de 160 mg (0,79 mmol) de I'alcool 206. Obtenu: 144 mg (91%); [aIDz5 +28,Y(c 1,22, CHCI,). Anal. calc. pourC1,H,,0,: C 59,99, H 8.05; trouv6e: C 60,03, H 8,02.
[2S[2a(R*),5a(S*)]] et [2S[2a(R*),5a(R*)]]-Te'trahydro-5- (2-hydroxypropy1)-a-me'thyl-2-furanace'te de me'thyle (3 b) et (5 b)
A une solution de 130 mg (0,65 mmol) d'aldehyde 21b dans 5 mL d'ether anhydre agitke sous atmosphere d'argon i -78°C on ajoute a I'aide d'une seringue 5,5 mL (0,66 mmol) d'une solution 0,12 M de CH,MgBr dans I'ether, preparee par dilution d'une solution commerciale 3 M dans ]'ether., Le milieu reac- tionnel est maintenu une demi-heure i -78"C, puis une heure et demie a une temperature comprise entre -60 et -50°C. On laisse finalement remonter la temperature jusqu'a - 1O0C, hy- drolyse par une solution saturee de NH4C1 et extrait la phase aqueuse avec 4 x 50 mL d'6ther. Les phases ether6es r6unies sont trait6es de f a ~ o n habituelle. La chromatographie du produit brut sur gel de silice (6luant: cyclohexane/AcOEt, 70:30) con- duit B I'isolement de 13 mg d'aldehyde 21b n'ayant pas rkagi, 67,7 mg d'alcool 3b (533% calculC a partir de l'aldehyde re- couvre) et 51 mg d'alcool Cpimere 56 (40% calcule i partir de I'aldehyde recouvr6).
(2S,3S,6R,8R) Nonactate de methyle (3b): [aIDZ5 +22,1° (c 0.7, CHCI,). Anal. calc. pour C,,H,,O,: C 61,09, H 9,32; trouvee: C 61,13, H 9,33.
(2S,3S,6R,8S) Nonactate de mkthyle (5b): [aIDZS +32,9' (C 1,07, CHCI,). Anal. calc. pour C,,H,,O,: C 61,09, H 9,32; trouvCe: C 61,27, H 9,39.
partir de 19 mg (0,087 mmol) de (+) nonactate de methyle (3b). Obtenu: 16,9mg (95%) d'acide (+) nonactique (2b); [alDZS +10,3" (c 1,69, CHCI,). Anal. calc. pour CloH1,O4: C 59,39, H 8,97; trouvee: C 59,55, H 9.09.
Acide [2S[2a(R*),5a(R*)]]-Te'trahydro-5-(2-hydroxypropyl)-a- me'thyl-2-furanace'tique (4b)
MBme mode operatoire que pour l'acide (-) 8-Ppi nonactique (4a) B partir de 25,3 mg (0,117 mmol) de (+) 8-ipi nonactate de m6thyle (5b). Obtenu: 22,6 mg (95,5%) d'acide (+) 8-Ppi nonac- tique (4b); [alDZs +51,3" (c 1,01, CHCI,). Anal. calc. pour CloH,,04: C 59,39, H 8,97; trouvee: C 59,47, H 8,91.
Remerciements Les auteurs tiennent a remercier 1'United States
National Science Foundation pour l'attribution d'une bourse (No CHE 7821066) qui a permis la rkalisation de ce travail.
1. R. CORBAZ, L. ETTLINGER, E. GAUMANN, W. KELLER- SCHIERLEIN, F. KRADOFFER, L. NEIPP, V. -LOG et H. ZAHNER. Helv. Chim. Acta, 38, 1445 (1955).
2. (a) K. C. NICOLAOU. Tetrahedron, 33,683 (1977); (b) P. A. BARTLETT. Tetrahedron, 36,2 (1980).
3. W. KELLER-SCHIERLEIN et H. GERLACH. Fortschr. Chem. Org. Naturst. 26, 161 (1968).
4. (a) L. A. P. PIODA, H. A. WACHTER, R. E. DOHNER et W. SIMON. Helv. Chim. Acta, 50, 1373 (1967); (b) H. K. WIPF, L. A. P. PIODA, Z. STEFARAC et W. SIMON. Helv. Chim. Acta, 51,377, (1968). J. H. PRESTEGARD et S. I. CHAN. J. Am. Chem. Soc. 92, 4440 (1970). M. DOBLER, J. D. DuNITzet B. T. KILBOURN. Helv. Chim. Acta, 52,2573 (1969). (a) J. DOMINGUEZ, J. D. DUNITZ, H. GERLACH et V. PFE- LOG. Helv. Chim. Acta, 45, 129 (1962); (b) J. BECK, H. GERLACH, V. PFELOG et W. VOSER. Helv. Chim. Acta, 45, 621 (1962). H. GERLACH et V. PRELOG. Justus Liebigs Ann. Chem. 669, 121 (1963). H. GERLACH, K. OERTLE, A. THALMANN et S. SERVI. Helv. Chim. Acta, 58,2836 (1975). (a) U. SCHMIDT, J. GOMBOS, E. HASLINGER et H. ZAK. Chem. Ber. 109, 2628 (1976); (b) J. GOMBOS, E. HAS- LINGER, A. NIKIFOROV, H. ZAK et U. SCHMIDT. Monatsh. Chem. 106, 1043 (1975). G. BECK et E. HENSELEIT. Chem. Ber. 104,21(1971). H. GERLACH et H. WETTER. Helv. Chim. Acta, 57, 2306 (1974). (a) H. ZAK et U. SCHMIDT. Angew. Chem. Int. Ed. Engl. 14,432 (1975); (b) J. GOMBOS, E. HASLINGER, H. ZAK et U. SCHMIDT. Monatsh. Chem. 106,219 (1975); (c) J. GOMBOS, E. HASLINGER, H. ZAK et U. SCHMIDT. Tetrahedron Lett. 3391 (1975). M. J. ARGO, M. H. TRAMMELL et J. D. WHITE. J. Org. Chem. 41,2075 (1976). J. GOMBOS, E. HAS LINGER^^ U. SCHMIDT. Chem. Ber. 109, 2645 (1976). R. E. IRELAND, S. THAISRIVONGS et C. S. WILCOX. J. Am. Chem. Soc. 102, 1155 (1980). (a) R. E. IRELAND et C. S. WILCOX. Tetrahedron Lett. 2839 (1977); (b) R. E. IRELAND. C. S. WILCOX. S. THAISRIVONGS
Acide [2S[2a(R*),5a(S*)]]-Te'trahydro-5-(2-hydroxypropyl)-a- i t N: VANIER. Can. J. dhem. 57, 1743 (1979); (c) R. E. me'thyl-2-furanace'tique (2 b) IRELAND, S. THAISRIVONGS, N. VANIER et C. S. WILCOX.
MBme mode operatoire que pour I'acide (-) nonactique (2u) a J. Org. Chem. 45,48 (1980).
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
UN
IV O
F B
IRM
ING
HA
M o
n 11
/10/
14Fo
r pe
rson
al u
se o
nly.

IRELAND ET VEVERT 583
18. R. E. IRELAND, R. H. MUELLER et A. K. WILLARD. J. Am. Chem. Soc. 98,2868 (1976).
19. R. E. IRELAND, C. S. WILCOX et S. THAISRIVONGS. J. Org. Chem. 43,786 (1978).
20. J. S. BRIMACOMBE, F. HUNEDY et L. C. N. TUCKER. J. Chem. Soc. C, 1381 (1968).
21. F. W. EASTWOOD, K. J. HARRINGTON, J. S. JOSAN et J . L. PURA. Tetrahedron Lett. 5223 (1970).
22. S. HANESSIAN, A. BARGIOTTI et M. LARUE. Tetrahedron Lett. 737 (1978).
23. K. FUJI, S. NAKANO et F. FUJITE. Synthesis, 276 (1975). 24. A. J . MANCUSO, S. L. HUANG et D. SWERN. J. Org. Chem.
43,2480 (1978); K. OMURA et D. SWERN. Tetrahedron, 34, 1651 (1978).
25. E. BARREIRO, J. L. LUCHE, J. ZWEIG et P. CRABBE. Tet- rahedron Lett. 2353 (1975).
26. H. 0. HOUSE, CHIA-YEH CHU, J. M. WILKINS et M. J. UMEN. J. Org. Chem. 40,1460 (1975).
27. L. M. LERNER, B. D. KOHN et P. KOHN. J. Org. Chem. 33, 1780 (1968).
28. T. P. CULBERTSON. J. Org. Chem. 38,3624 (1973). 29. W. C. STILL et J. A. SCHNEIDER. Tetrahedron Lett. 1035
(1980).
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
UN
IV O
F B
IRM
ING
HA
M o
n 11
/10/
14Fo
r pe
rson
al u
se o
nly.





























