Embed Size (px)
Citation preview

Thermodynamique
des principes
à l'étude de systèmes industriels
Claude Rozé
Cours de troisième année de licence
Dernière révision : 8 novembre 2013


Felix qui potuit rerum cognoscere causas
(Heureux celui qui a pu pénétrer les causes secrètes des choses)
Virgile, Géorgiques, II, 489
et
Barbe-Rouge dans La galère d'Astérix


Ce document est le support du cours donné aux étudiants de troisième année de licence PMSI (Physique,Mécanique et Sciences de l'Ingénieur) de l'UFR des Sciences et Techniques de l'Université de Rouen, de parcoursMécanique et ingénierie et Gestion des Systèmes Industriels : Maîtrise de l'Energie. Il est diusé en espérant qu'ilsera utile, mais peut encore comporter des erreurs ou inexactitudes, qui seront corrigées progressivement.
Le but de ce cours est de donner une formation générale théorique sur la thermodynamique à partir d'uneformulation purement axiomatique, mais en essayant de retrouver les formulations "classiques". Comme toutexposé de ce type, il existe des ambiguïtés qu'il est très dicile de lever, sauf si on accepte de mathématisercomplètement l'exposé, ce qui est exclu pour les formations envisagées.


Table des matières
1 Principes généraux 11 Généralités . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 12 Le système thermodynamique à l'équilibre . . . . . . . . . . . . . . . . . . . . . . . . . . . 23 Transformation d'un système entre deux états d'équilibre . . . . . . . . . . . . . . . . . . 64 Cas particulier des systèmes ouverts en écoulement . . . . . . . . . . . . . . . . . . . . . . 12
2 Application des principes au uide homogène 211 Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 212 Relations entre coecients calorimétriques . . . . . . . . . . . . . . . . . . . . . . . . . . . 213 Fonctions thermodynamiques . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 244 Équations d'état . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 32
3 Le corps pur sous diérentes phases 431 Dénitions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 432 Description phénoménologique des changements d'état . . . . . . . . . . . . . . . . . . . . 433 L'équation de Van der Waals comme modèle pour le changement d'état . . . . . . . . . . 454 Relations thermodynamiques du changement d'état . . . . . . . . . . . . . . . . . . . . . . 505 Présentation des diagrammes thermodynamiques . . . . . . . . . . . . . . . . . . . . . . . 54
4 Mélange en l'absence de réaction chimique 571 Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 572 Potentiel chimique d'un gaz . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 583 Potentiel chimique en phase liquide . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 604 Équilibre liquide-vapeur d'une solution binaire . . . . . . . . . . . . . . . . . . . . . . . . 62
5 Thermodynamique des processus irréversibles 671 Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 672 Bilan local . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 673 L'hypothèse de l'équilibre thermodynamique local . . . . . . . . . . . . . . . . . . . . . . . 674 Thermodynamique hors équilibre en régime linéaire . . . . . . . . . . . . . . . . . . . . . . 705 Exemples . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 716 État stationnaire hors équilibre . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 73


Chapitre 1
Principes généraux
1 Généralités
1.1 Introduction
Le but de la thermodynamique est de décrire de façon générale les systèmes sur le plan énergétique,ainsi que leurs transformations. Le choix qui est fait dans ce cours est de travailler en trois étapes distinctes[1] :
a. La thermodynamique de l'équilibreIl s'agit de déterminer quelles sont les variables qui permettent de décrire l'état d'équilibre dusystème étudié et d'écrire toutes les relations existant entre ces variables.
b. La thermodynamique des transformations d'un équilibre à un autre équilibreOn considère un système à l'équilibre et on lui fait subir une transformation, soit par la mise enplace d'une nouvelle contrainte, soit en levant une contrainte. Le système va relaxer vers un nouvelétat d'équilibre. On peut alors eectuer des bilans sur certaines variables, qui ont été modiées parla transformation. Des lois immuables peuvent être observées et aboutissent aux postulats que sontle premier principe et le second principe.
c. La thermodynamique des processus irréversiblesLa thermodynamique issue du premier et du second principe est limitée à des bilans entre étatd'équilibre initial et état d'équilibre nal. Ces principes ne permettent pas d'avoir des informationssur le système en cours de transformation, sauf dans le cas idéal d'une transformation réversible. Onajoute alors l'hypothèse d'équilibre thermodynamique local, c'est à dire que, même si un systèmen'est pas à l'équilibre, on peut le diviser en sous-systèmes élémentaires, susamment petits pourqu'ils soient considérés à l'équilibre. Les relations décrites en (a) peuvent être écrites au niveau localet les deux principes décrits en (b) deviennent des équations de bilan local.Cette hypothèse ouvre la voie à la thermodynamique des processus irréversibles, permettant d'éta-blir certaines lois de dissipation (par exemple, la loi de Fourier pour la dissipation de l'énergiethermique) et d'expliquer les phénomènes couplés (thermocouple, etc.).
L'autre parti-pris de ce cours est de rester au niveau macroscopique, ce qui est une gageure en ther-modynamique. En eet, toute grandeur et notion macroscopique est le résultat d'un comportement d'en-semble d'un grand nombre d'éléments (les molécules, en général). L'approche la plus rigoureuse est doncla thermostatistique, qui permet de retrouver les grandeurs macroscopiques en faisant des statistiquesd'ensemble. Rester au niveau macroscopique (phénoménologique) ne permet pas toujours de dénir ri-goureusement certaines variables : comment dénir sans ambigüité l'énergie interne ou l'entropie ? Lapirouette consiste à ne pas les dénir, mais à les introduire par des postulats : c'est cet artice qui estutilisé dans les pages qui suivent. Les annexes de ce chapitre donneront malgré tout une interprétationmicroscopique de certaines notions introduites.
1.2 Système thermodynamique
Un système Σ est une portion d'espace contenant de la matière et séparé du reste de l'espace parune surface réelle ou ctive.
Un système thermodynamique est nécessairement macroscopique : les lois de la thermodynamiquesont la manifestation macroscopique de comportements microscopiques d'un grand nombre d'éléments

2 Principes généraux
microscopiques (molécules, atomes, etc.). L'ordre de grandeur du nombre d'éléments est la mole : une molede molécules contient NA = 6.02×1023 1 molécules et se comporte comme un système thermodynamique.
1.3 Grandeur extensive et grandeur intensive
1.3.1 Dénitions
Une grandeur G est extensive si sa valeur est proportionnelle à la quantité de matière. Une grandeurest intensive si elle n'en dépend pas.
Considérons une grandeur extensive G qui est fonction uniquement d'autres grandeurs extensivesXi : si le système est augmenté d'un facteur λ, les variables Xi sont elles aussi multipliées par λ. Gest également multiplié par λ. Donc :
λG(Xi) = G(λXi)
On dit alors que la fonction G(Xi) est homogène de degré 1 par rapport aux variables Xi.
1.3.2 Bilan d'une grandeur extensive
Soit G une grandeur extensive quelconque. On considère un système de volume V , limité par unesurface Σ, le bilan de la grandeur G sur un intervalle de temps ∆t s'écrit :
∆G = G(e) +G(i)
où∆G est la variation de la grandeur G pendant le temps∆t, G(e) est l'échange de G venant de l'extérieuret G(i) la quantité de G produite à l'intérieur.
Grandeur conservative : une grandeur G est conservative si sa production est nulle (par exemple, lamasse, la charge, en physique classique). G ne peut alors évoluer que par échange avec l'extérieur.
2 Le système thermodynamique à l'équilibre
2.1 Caractérisation d'un système à l'équilibre
On admet que l'on peut caractériser un système thermodynamique à l'équilibre par un ensemble devariables extensives Xi, i = 1 . . . n, auxquelles on ajoute obligatoirement une variable extensive Uappelée énergie interne.
Par exemple, si on considère un uide pur homogène, les variables permettant de le caractériser àl'équilibre sont :
le volume V , la quantité de matière, c'est à dire le nombre de molécules ou le nombre de moles N , l'énergie interne U .L'énergie est une grandeur qui varie dès qu'un système change d'état. Ce changement d'état peut être
(i) macroscopique (variation de vitesse et/ou de position) et l'énergie cinétique et l'énergie potentielledu système évolue, (ii) microscopique (variation de vitesse de position des molécules, transformationdes molécules par réaction chimique, etc.). L'énergie interne est la variable qui représente la somme desénergies des molécules contenues dans le système et de leurs interactions. Autrement, l'énergie internecomptabilise l'énergie cinétique microscopique des molécules (translation, vibration, rotation, etc.) et leurénergie potentielle (chimie, interactions électromagnétiques, etc.).
Les variables Xi, U ne sont pas encore susantes pour décrire parfaitement le système. On introduitune nouvelle variable extensive, appelée entropie, notée S. L'entropie est une grandeur proportionnelleau nombre de congurations microscopiques (positions et vitesses des molécules, états d'excitation, etc.)correspondant à une même conguration macroscopique, c'est à dire non discernables au niveau macro-scopique. Soit Ω le nombre de congurations microscopiques d'un système, la thermostatistique postuleque tous ces "micro-états" sont équiprobables et que S = k log Ω (voir annexe).
1. NA est le nombre d'Avogadro.

2 Le système thermodynamique à l'équilibre 3
2.2 Postulat pour un système à l'équilibre
Un système à l'équilibre thermodynamique est caractérisé par une fonction entre les variables exten-sives U , S et Xi, i = 1 . . . n : f(U, S,X1, . . . , Xn) = 0. On peut écrire également 2 S = fS(U,X1, . . . , Xn)ou U = fU (S,X1, . . . , Xn). On admet que les fonctions fS et fU possèdent les propriétés suivantes :
a. elles sont continues et au moins deux fois diérentiables,b. fS est strictement croissante par rapport à l'énergie interne U ,c. on admet que 3 :
S → 0 lorsque∂fU∂S
→ 0
Remarque : Au lieu d'utiliser fS et fU , on note plus simplement S = S(U,X1, . . . , Xn) et U = U(S,X1, . . . , Xn). Les variables extensives caractérisant un uide homogène sont : le volume V , le nombre de molesN . La fonction U = U(S, V,N) décrit donc complètement un uide à l'équilibre. Cette fonctionpeut être représentée par un diagramme thermodynamique.
2.3 Température et variables intensives
On considère un système caractérisé par U = U(S,X1, . . . , Xn). On dénit la température thermody-namique par :
T =∂U
∂S
Il s'agit d'une variable intensive. La température est nécessairement positive compte tenu que S est unefonction croissante de U .
On peut également dériver la fonction U par rapport à chacune des variables Xi. On dénit ainsi unnouvelle variable intensive :
fi =∂U
∂Xi
La diérentielle de l'énergie interne s'écrit donc :
dU = TdS +
n∑i=1
fidXi (1.1)
Il s'agit de la relation de Gibbs, où à chaque variable extensive Xi correspond une variable intensiveconjuguée fi.
Cas d'un uide homogène : considérons un système ouvert contenant un uide pur homogène, c'està dire déni par son énergie interne U = U(S, V,N). La température du uide est donnée par :
T =∂U
∂S
On appelle pression, la variable intensive notée p, dénie par :
−p = ∂U
∂V
On appelle potentiel chimique, la variable intensive, notée µ, dénie par :
µ =∂U
∂N
2. Cela est justié par le théorème des fonctions implicites :Soit f une fonction continue dérivable de R2 dans R. Soit (x, y) un point tel que f(x, y) = 0 et tel que la dérivée partiellede f, par rapport à la deuxième variable, ne soit pas nulle en (x, y). Il existe une fonction φ continue dérivable dénie surun voisinage de x tel que :
f(x, y) = 0 ⇐⇒ φ(x) = y
Dans notre cas, la fonction f dépend obligatoirement de U et S, quels que soient les autres paramètres Xi. On peut donctoujours trouver les fonctions fU et fS .
3. Il s'agit de l'énoncé de Planck du postulat de Nerst, appelé "Troisième principe de la thermodynamique".

4 Principes généraux
Le potentiel chimique s'interprète comme l'énergie interne propre des molécules composant le système : sides molécules entrent ou sortent du système, chacune d'entre elles emporte avec elle une énergie interneµ.
La relation de Gibbs ou forme diérentielle de la relation fondamentale U(S, V,N) devient :
dU = TdS − pdV + µdN
Si le uide n'est plus pur, mais est un mélange de m espèces chimiques diérentes, la relation de Gibbsdevient :
dU = TdS − pdV +m∑j=1
µjdNj
où µj est le potentiel chimique de l'espèce j.
2.4 Relation de Gibbs-Duhem
L'énergie interne est une fonction homogène de degré 1 par rapport à ses variables :
U(λS, λX1, . . . , λXn) = λU(S,X1, . . . , Xn)
Dérivons par rapport à λ :
∂U(λS, λXi)
∂(λS)
∂(λS)
∂λ+
n∑i=1
∂U(λS, λXi)
∂(λXi)
∂(λXi)
∂λ=U(S,Xi)
S∂U(λS, λXi)
∂(λS)+
n∑i=1
Xi∂U(λS, λXi)
∂(λXi)=U
Par simple changement de variable :
S∂U
∂S+
n∑i=1
Xi∂U
∂Xi= U
or∂U
∂S= T et
∂U
∂Xi= fi, on en déduit l'équation d'Euler :
U = TS +
n∑i=1
fiXi
Prenons la diérentielle de cette équation et soustrayons la relation de Gibbs. On obtient la relation deGibbs-Duhem :
SdT +n∑i=1
Xidfi = 0
Exemple : pour un uide homogène, l'équation d'Euler s'écrit : U = TS − PV + µN et la relation deGibbs-Duhem :
SdT +Ndµ− V dp = 0
Si le système est composé de molécules de m espèces chimiques diérentes, cette relation devient :
SdT − V dp+m∑i=1
Njdµj = 0
sachant que la relation de Gibbs devient dU = TdS − pdV +∑mj=1 µjdNj et que l'équation d'Euler est
U = TS − PV +∑mi=j µjNj .

2 Le système thermodynamique à l'équilibre 5
2.5 Équations d'état
Si la plupart des variables intensives et extensives sont directement accessibles à l'expérience, il n'enest pas de même de l'entropie. C'est la raison pour laquelle on va rechercher des relations équivalentes àla relation fondamentale U = U(S,X1, . . . , Xn) contenant la même information, mais ne contenant pasS. On propose donc la dénition suivante :
Une équation d'état est une relation entre les paramètres intensifs et extensifs de ce système, mais necontenant pas l'entropie S.
Si le système est décrit par n variables extensives, auxquelles on ajoute l'entropie S, alors la fonctionU = U(S,Xi) permet d'obtenir :
T =∂U
∂S= T (S,X1, . . . , Xn)
fi =∂U
∂Xi= fi(S,X1, . . . , Xn), i = 1, . . . , n
En éliminant S, on obtient n équations d'état :
fi = fi(T,X1, . . . , Xn), i = 1, . . . , n
Il y a exactement la même information dans ces n équations d'état, que dans la relation fondamentaleU = U(S,X1, . . . , Xn).
Cas d'un uide homogène : le uide homogène est décrit par les variables extensives V et N . Dansce cas la relation fondamentale est U = U(S, V,N). On en déduit les relations :
T = T (S, V,N) (1.2)
P = P (S, V,N) (1.3)
µ = µ(S, V,N) (1.4)
En éliminant S, on obtient deux équations d'état :
P = P (T, V,N) (1.5)
µ = µ(T, V,N) (1.6)
Ainsi, le uide peut être parfaitement décrit, soit par la relation U = U(S, V,N), soit par deux équationsd'état. Cependant, on n'utilise en général pas l'équation (1.6). On la remplace par la relation fondamentaledans laquelle on a substitué l'entropie par la température.
Dans la pratique, pour connaître les caractéristiques complètes d'un uide, on peut utiliser : la relation U = U(S, V,N), ou plutôt u = u(s, v) où u est l'énergie interne molaire, s l'entropiemolaire et v le volume molaire. Elle prend presque toujours la forme d'un diagramme expérimentaldu type de la gure 2.5). De plus, on n'utilise rarement la variable u, mais des variables dérivéestelle que l'enthalpie ou des variables facilement mesurables, telles que température et pression.
une équation d'état de type f(p, v, T ) = 0 (équation d'état du gaz parfait, de Van der Waals, etc.)et une équation donnant les variations d'une autre variable en fonction de la pression et de latempérature (souvent, la capacité calorique molaire du gaz à pression constante, en fonction de latempérature et de la pression cp = cp(p, T ) [18], qui sera dénie au chapitre suivant).
Exemple : considérons la relation fondamentale :
U = exp
[2S
3RN
]V −2/3N5/3 (1.7)
où U est l'énergie interne, S l'entropie, N le nombre de moles et V le volume. R est une constante.
T =∂U
∂S=
2
3NRexp
[2S
3NR
]V −2/3N5/3 (1.8)
p = −∂U∂V
=2
3exp
[2S
3NR
]V −5/3N5/3 (1.9)
µ =∂U
∂N= exp
[2S
3RN
]V −2/3
5
3N2/3 − 2S
3RN1/3
(1.10)

6 Principes généraux
4
u
s
v
v
vv1 2
3
Figure 1.1 Diagramme donnant l'énergie interne molaire u = u(s, v) en fonction de l'entropie molaires et du volume molaire v
L'expression de T donne :
exp
[2S
3RN
]=
3
2
V 2/3
N2/3RT
que l'on reporte dans l'expression de p et on obtient PV = NRT , c'est à dire l'équation d'état desgaz parfaits. L'équation (1.10) permettrait d'obtenir l'expression du potentiel chimique. Plutôt que deconsidérer cette expression comme une équation d'état, on élimine S entre (1.8) et (1.7) :
T =2
3NRU ou encore U =
3
2NRT, que l'on note U = NCvT
où Cv est la capacité calorique molaire à volume constant (qui sera dénie dans le cas général au chapitresuivant). Finalement, la relation fondamentale (1.8) caractérise un gaz parfait de capacité caloriquemolaire à volume constant Cv = 3/2R.
3 Transformation d'un système entre deux états d'équilibre
La partie précédente traitait uniquement de l'état d'équilibre d'un système thermodynamique. In-téressons nous maintenant à une transformation entre deux états d'équilibre. On peut alors écrire deséquations de bilan sur des grandeurs extensives, en particulier l'énergie interne et l'entropie.
3.1 Dénitions
On peut dénir un système en fonction des types d'échanges qu'il eectue avec l'extérieur. En parti-culier,
un système isolé n'échange ni matière, ni énergie avec l'extérieur, un système fermé n'échange pas de matière avec l'extérieur, mais uniquement de l'énergie, un système ouvert échange matière et énergie avec l'extérieur.Par convention, tous les échanges de chaleur, de matière et de travail mécanique sont représentés
par une valeur algébrique, positif si l'échange se fait de l'extérieur vers l'intérieur, négatif s'il se fait del'intérieur vers l'extérieur.
On peut être plus précis dans le cas d'un système uide. Un système est : isotherme s'il évolue à température constante, isochore s'il évolue à volume constant, isobare s'il évolue à pression constante.Un système ouvert est stationnaire lorsque les ux d'énergie et de masse qui traversent la surface
limitant ce système sont constants et que sa masse et son énergie restent constantes.

3 Transformation d'un système entre deux états d'équilibre 7
3.2 Conservation de l'énergie
3.2.1 Énergie totale
On appelle énergie totale d'un système la grandeur :
E = Ec + Ep + U
où Ec est l'énergie cinétique macroscopique du système, Ep l'énergie potentielle macroscopique et Ul'énergie interne.
3.2.2 Postulat sur la conservation de l'énergie
L'énergie totale d'un système est une grandeur conservative
On peut énoncer ce même postulat de la façon suivante :la variation d'énergie totale entre deux instants de toute transformation est égale à l'énergie échangée
avec l'extérieur :
∆E = E(e)
Remarque : la conservation de l'énergie est une des lois de conservation de la physique classique.Les autres sont : la conservation de la masse, la conservation de la quantité de mouvement (appelée loifondamentale de la dynamique en mécanique) et la conservation de la charge.
3.2.3 Forme classique du premier principe
Pour un système dont les énergies potentielles et cinétiques macroscopiques sont négligeables, on a :
∆U = E(e)
L'énergie peut être échangée, soit en modiant les variables macroscopiques du système (volume, par exemple) : cet échange estalors appelée travail 4,
soit sans variation de ces variables, par un transfert au niveau microscopique : il s'agit de la chaleur, soit en échangeant de la matière.Considérons un système fermé. Pour celui-ci, l'échange avec l'extérieur est uniquement un échange
d'énergie de type travail ou chaleur :
E(e) =W +Q
où W est le travail et Q est la chaleur. On en déduit l'énoncé classique du premier principe :
Énoncé : pour un système fermé, pour lequel on néglige l'énergie cinétique macroscopique etl'énergie potentielle macroscopique, subissant une transformation quelconque entre deux étatsd'équilibre, la variation d'énergie interne s'écrit :
∆U =W +Q
3.2.4 Quelques cas particuliers
Si le système est ouvert, il convient d'ajouter un terme correspondant à l'énergie emportée par lamatière échangée avec l'extérieur. On obtient une expression peu utilisée. En eet, dans la pratique,l'échange de matière avec l'extérieur se fait souvent par des tuyauteries dans lesquelles circulentdu uide à pression constante. Un énoncé du premier principe dans ce cas sera établi dans unparagraphe ultérieur.
4. Une interprétation microscopique de la diérence entre transfert d'énergie par travail et transfert d'énergie thermiquepeut être faite : voir annexe 4

8 Principes généraux
En mécanique, on néglige généralement les eets thermiques et la notion d'énergie interne n'apparaîtpas puisqu'aucune évolution de nature microscopique n'est envisagée. On retrouve alors le théorèmede l'énergie cinétique :
∆(Ec + Ep) =W
où Ep est l'énergie potentielle macroscopique etW le travail des forces ne dérivant pas d'un potentiel.Certaines de ces forces sont des forces dissipatives (forces de frottement par exemple) :
∆(Ec + Ep) =W +Wd
Ces forces correspondent à une conversion de travail en chaleur, qui peut être soit dissipée à l'exté-rieur du système (échange thermique Q), soit accroître la température du système (augmentationde l'énergie interne). On retrouve :
∆(Ec + Ep + U) =W +Q
La transformation est adiabatique s'il n'y a pas d'échange d'énergie thermique (pas de chaleur).On en déduit alors :
∆U =W
Si le système décrit un cycle, l'état d'équilibre nal est le même que l'état d'équilibre initial. Donc,au bout d'un cycle :
∆U = 0 =W +Q
Le travail échangé par le système dépend des forces exercées sur ce système. Ainsi, pour des forces de pression, le travail s'écrit : W = −
∫pext.dV (voir annexe 2).
pour des forces électriques, il s'écrit : W = −∫Φ.dq où Φ est la diérence de potentiel et dq la
quantité de charge. le travail des forces de tension supercielle s'écrivent :W =
∫σdA, où σ est la tension supercielle
et dA la variation d'aire de la surface. le travail des forces de gravité s'écrivent : W =
∫mgdz où m est la masse, g l'accélération de la
gravité et dz l'altitude. le travail d'un l élastique est : W =
∫T.dx, où T est la tension du l et dx son élongation.
le travail des forces magnétiques est : W =∫B.dM , où B est le champ magnétique et dM est la
magnétisation du matériau.On admet que le travail est toujours de la forme dW = f(e)i.dXi où f(e)i est la force exercée par lesystème extérieur et dXi la réponse du système.
3.3 Second principe
3.3.1 Postulat
Énoncé : La variation d'entropie d'un système entre un état initial d'équilibre et un état nald'équilibre d'une transformation quelconque s'écrit :
∆S = S(e) + S(i) (1.11)
où : S(e) est l'échange d'entropie,a. parce qu'il y a un transfert de matière,b. parce qu'il y a un transfert d'énergie sous forme de chaleur.
S(i) est la production d'entropie et est nécessairement positive ou nulle.
3.3.2 Conséquences
Pour un système fermé :∆S = S(e) + S(i)
avec S(i) ≥ 0 et S(e) ne dépend que du transfert d'énergie thermique Q.

3 Transformation d'un système entre deux états d'équilibre 9
On remarque que l'échange d'énergie sous forme de travail n'intervient pas dans le bilan entro-pique : le travail ne change pas l'entropie. En revanche, si on sait que l'échange d'énergie thermiqueproduit un ux d'entropie, on ne sait pas calculer son expression dans le cas général. On verradans un paragraphe ultérieur que lorsque cette échange se fait avec un système extérieur restant àtempérature constante, l'échange d'entropie est Q/T .
De même, on sait que S(i) ≤ 0, mais on ne sait pas calculer son expression en général. Pour un système fermé :
∆S = S(e) + S(i)
avec S(i) ≥ 0 et S(e) ne dépend que du transfert d'énergie thermique Q. Pour un système isolé, on a ∆S = S(i) > 0, où S(i) ≥ 0 est la production d'entropie au cours dela transformation. En particulier, considérons un système isolé auquel on enlève une contrainte.Le système évoluera vers un nouvel état d'équilibre, dont l'entropie est plus grande que celle del'état d'équilibre initial. Cet état d'équilibre est nécessairement un maximum, sinon l'évolution sepoursuivrait.En conséquence, l'entropie S(U,Xi) d'un système isolé est maximum par rapport à n'importe quellemodication d'une variable extensive.
Considérons deux systèmes homogènes (1) et (2) à l'équilibre, dénis respectivement par (U (1), X(1)i )
et (U (2), X(2)i ). Combinons les, sans qu'il y ait d'échange avec l'extérieur. Le système composite est
alors déni par les variables extensives (U (1)+U (2), X(1)i +X
(2)i ). En revanche, l'entropie du système
composite est nécessairement supérieure à l'entropie des systèmes pris séparément :
S(1)+(2) ≥ S(1) + S(2)
S(U (1) + U (2), X(1)i +X
(2)i ) ≥ S((U (1), X
(1)i ) + S(U (2), X
(2)i )
Ceci est vrai pour toute proportion λ :
S(λU (1) + (1− λ)U (2), λX(1)i + (1− λ)X
(2)i ) ≥ λS((U (1), X
(1)i ) + (1− λ)S(U (2), X
(2)i )
On dit que l'entropie est une fonction concave de ses arguments extensifs. Pour une transformation adiabatique :
∆S = S(i)
où S(i) ≥ 0 est la production d'entropie au cours de la transformation.
3.3.3 Transformation réversible et transformation irréversible
On appelle transformation réversible une transformation qui se fait de façon tellement lente quel'on peut considérer que le système est à tout instant de la transformation à l'équilibre.
Quelques propriétés et conséquences : La notion de réversibilité est évidemment une limite mathématique, non atteignable dans la réalité. Toute transformation qui n'est pas réversible est irréversible. En tout point d'une transformation réversible, il y a équilibre. Donc, conformément au paragraphe 2,la relation fondamentale U(S,Xi) (i = 1 . . . n) ou, de façon équivalente, les n équations d'état dusystème étudié sont dénies tout au long de la transformation. Si le système est un uide, les notionsde température, pression, etc. sont alors parfaitement dénies à tout instant et ces grandeurs sontuniformes dans tout le système. Les relations de Gibbs et de Gibbs-Duhem sont utilisables, ainsique l'équation d'Euler.En particulier, un système réversible non adiabatique mis en contact avec un système extérieur àla température T se met à tout instant à cette température T . De même, un système fermé parun piston évoluant de façon réversible a une pression interne p à tout instant égale à la pressionexercée sur le piston pext. Le travail élémentaire est donc dW = −p.dV , où p est la pression dans legaz. De façon générale, toute forme de travail élémentaire est de la forme dW = fi.dXi, où fi estla force extérieure exercée sur le système, égale à la réponse du système caractérisée par la variableintensive fi.

10 Principes généraux
Au cours d'une transformation réversible, la production d'entropie est nulle.Démonstration : considérons un système fermé évoluant de façon réversible. En tout point de latransformation, le système est à l'équilibre et on peut dénir une fonction U(t) = U(S(t), Xi(t))valable à tout instant t. Entre deux instants t et t+ dt, on peut écrire dU = TdS +
∑fidXi
5.D'autre part, entre ces deux mêmes instants, le premier principe donne : dU = dQ + dW . Or, letravail élémentaire est dW =
∑fidXi. En identiant, on obtient dS = dQ/T .
En introduisant cette formule dans l'expression du second principe, on en déduit que la productiond'entropie est nulle.
Pour une transformation irréversible, la fonction U(S,Xi) n'existe qu'à l'état initial et à l'état nal.Les notions de température, pression, etc. n'existent pas en dehors de ces instants.
Applications à l'étude de systèmes : dans le cas d'une transformation irréversible entre un étatd'équilibre initial t0 et un instant d'équilibre nal t1, la relation fondamentale ne peut être écrite qu'àl'instant initial et à l'instant nal : la température, la pression, etc. ne sont pas dénies au cours de latransformation. On essaiera donc d'obtenir les informations concernant cette transformation, en écrivantle premier et le second principe. Le nombre d'inconnues peut alors être supérieur au nombre d'équationsdisponibles. Une technique de calcul, classique en thermodynamique, consiste alors à utiliser la procéduresuivante.
On imagine une transformation réversible physiquement possible, partant du même état initial etarrivant au même état nal. On a alors :
• pour la transformation réversible, on peut calculer ∆U = W rev + Qrev et ∆S = Srev
(e) en intégrantla relation fondamentale,
• pour la transformation réelle, ∆U = Qirr +W irr et ∆S = Sirr
(e) + Sirr
(i).Par identication des ∆U et ∆S entre ces deux transformations, on obtient deux équations supplémen-taires.
3.3.4 Système en contact avec un thermostat
On appelle thermostat (ou réservoir de température ou source de chaleur) un système de grandedimension qui reste à l'équilibre à température constante quel que soit l'échange d'énergie thermiquequ'on eectue avec lui.
Pour un système échangeant de l'énergie thermique Q avec un thermostat à température T , le termed'échange d'entropie S(e) du second principe est Q/T .
Démonstration : Considérons un système fermé Σ en contact avec un thermostat R de température Tconstante. Σ échange de l'énergie sous forme thermique avec le thermostat et échange du travail W avecl'extérieur à R∪ Σ. L'ensemble R∪ Σ forme un système isolé, auquel on applique le premier principe :
∆U(R∪ Σ) = ∆U(Σ) + ∆U(R) =W
Appelons Q la chaleur échangée par Σ avec R :
∆U(R) = −Q
De même, l'ensemble R ∪ Σ formant un système isolé, le second principe donne :
∆S(R∪ Σ) = ∆S(Σ) + ∆S(R) ≥ 0
R est un thermostat et est donc en permanence à l'équilibre. Il est caractérisé par une fonction U = U(S).La relation de Gibbs se réduit à :
dU(R) = TdS(R) ou en intégrant ∆U(R) = T∆S(R)
Autrement dit :
∆S(R) = S(e)(R) =∆U(R)
T= −Q
T
Le second principe appliqué à Σ seul donne donc :
∆S(Σ) = S(e)(Σ) + S(i)(Σ)
5. Dans ce cas, la notation diérentielle représente une transformation innitésimale, tandis qu'au paragraphe 2, lesystème était à l'équilibre et on étudiait la fonction U = U(S,Xi) au sens mathématique du terme.

3 Transformation d'un système entre deux états d'équilibre 11
L'échange d'entropie ne dépend que de la chaleur et ne se fait qu'avec R :
∆S(Σ) = −S(e)(R) + S(i)(Σ) =Q
T+ S(i)(Σ)
On en déduit le second principe pour un système fermé échangeant de l'énergie thermique Q avec unthermostat à température T :
∆S =Q
T+ S(i)
avec S(i) ≥ 0.On étend ce résultat à un système en relation avec n thermostats à température Ti :
∆SR =
n∑i=1
QiTi
+ S(i)
avec S(i) ≥ 0.On peut étendre ce résultat à un système Σ échangeant de l'énergie thermique avec un système
extérieur R évoluant de façon réversible (c'est à dire dont la température T est xée à tout instant) :
∆SR =
∫dQ
T+ S(i)
avec S(i) ≥ 0.
3.3.5 Machine cyclique
Une machine cyclique ne peut fournir du travail (W < 0) à partir d'une seule source de chaleur. Ils'agit d'une conséquence du second principe 6.
Démonstration : le premier principe donne dans le cas d'une machine cyclique échangeant de l'énergiethermique avec une seule source de chaleur :
∆U = 0 =W +Q
avec Q > 0. Le bilan entropique s'écrit :
∆S = 0 =Q
T+ S(i)
Si Q > 0, cela implique que S(i) < 0, ce qui est contraire au second principe.
Considérons donc une machine cyclique produisant du travail (W < 0) en échangeant de l'énergiethermique avec une source chaude (C) à température Tc et une source froide (F ) à température Tf , telleque Tf < Tc. Appelons Qc l'énergie thermique échangée avec (C) et Qf l'énergie thermique échangéeavec (F ). Quel est le signe de Qc et Qf ?
premier cas : admettons Qc > 0 et Qf > 0. Dans ce cas, introduisons une troisième source detempérature T , telle que T > Tc et T > Tf . Considérons le système constitué du moteur et dessources (C) et (F ). Ce système eectue des cycles avec une seule source de chaleur, ce qui estimpossible.
second cas : admettons que Qc < 0 et Qf > 0. La source froide donne de l'énergie thermique et lasource chaude en reçoit. On peut à nouveau imaginer une troisième source de température T telleque Tc > T > Tf , qui recevrait Qc de la source (C) et rendrait Qf à la source (F ). On obtient ànouveau une machine cyclique monotherme, ce qui est impossible.
En conséquence, Qc > 0 et Qf < 0.
Considérons un tel cycle et calculons en le rendement. On appelle rendement (ou ecacité) le rapportentre le gain et la dépense. Dans notre cas, le rendement est déni par
η =−QcW
6. C'est l'énoncé du second principe proposé par William Thomson (ennobli en 1886 et devenu Lord Kelvin).

12 Principes généraux
Le premier principe s'écrit pour un cycle :
∆U = 0 =W +Qc +Qf ⇒ Qf = −W −Qc
Le bilan entropique s'écrit :
∆S = 0 =QcTc
+QfTf
+ S(i)
Sachant que S(i) ≥ 0 :QcTc
+−W −Qc
Tf≤ 0
On en déduit le rendement :
η =−WQc
≤ 1− TfTc
Le rendement maximum ηc = 1 − Tf/Tc s'appelle rendement de Carnot et s'obtient pour une transfor-mation réversible 7. Le rendement de tout moteur réel doit donc être comparé à cette valeur.
4 Cas particulier des systèmes ouverts en écoulement
Le premier principe pour un système ouvert s'écrit simplement en ajoutant un terme d'échange d'éner-gie interne dû au ux de matière échangée avec l'extérieur :
∆U =W +Q+ E(e)
De même, le second principe pour un système ouvert s'obtient en ajoutant un terme au ux d'entropieS(e), correspondant à l'entropie transportée par la matière échangée avec l'extérieur.
Dans la pratique, les systèmes ouverts sont souvent des machines possédant une ou des entrées deuide à pression connue et une ou des sorties de uide à pression connue : pompe, réacteur chimique, etc.Il est donc intéressant de chercher des expressions du premier et second principes adaptés aux systèmesouvert en écoulement.
4.1 Le premier principe
Considérons un système ouvert en écoulement. Il se compose d'une enceinte limitée par une surfaceΣ, possédant une entrée par laquelle du uide arrive à la pression pe et une sortie d'où le uide est extraità la pression ps 8.
Bilan de masse : appelons m(t) la masse contenue dans Σ à l'instant t. Pendant une durée dt, cettemasse va évoluer du fait qu'une quantité dme entre et qu'une quantité de matière dms sort de l'enceinte.Le bilan de masse entre t et t+ dt s'écrit simplement :
m(t+ dt)−m(t) = dme − dms (1.12)
On remarque que m(t) + dme = m(t + dt) + dms : le système constitué par la masse contenue dans Σau temps t et de la masse dme qui va entrer entre les instants t et t + dt contient la même quantité dematière que le système constitué par la masse contenue dans Σ au temps t + dt et de la masse dms quiest sortie entre les instants t et t+ dt. Il s'agit donc d'un système fermé que l'on note Σse.
Bilan d'énergie : appliquons le premier principe au système Σse. L'énergie totale contenue dans cesystème à l'instant t est :
E(t) + eedme
A l'instant t+ dt, ce même système contient l'énergie totale :
E(t+ dt) + esdms
7. Il s'agit de l'énoncé historique du second principe par Carnot (1796-1832).8. Le raisonnement qui suit est généralisable à des nombres quelconques n d'entrées et m de sorties.

4 Cas particulier des systèmes ouverts en écoulement 13
où E(t) désigne l'énergie totale contenu dans Σ à l'instant t, ee et es les énergies massiques respectivementdu uide entrant et du uide sortant. Le premier principe s'écrit :
dE + [edm]se = dQ+ dW (1.13)
où dQ désigne l'énergie thermique échangée par l'enceinte avec l'extérieur et dW le travail apporté auuide.
On peut diviser le travail dW en deux parties : le travail des forces de pression permettant de faire entrer et sortir le uide, agissant sur dme etdms ;
les travaux échangés à travers la surface Σ, qu'on notera dWu (le travail "utile").Le travail des forces de pression agissant sur le uide entrant est :
dWe = pedVe = pevedme
où dVe est le volume de uide entrant entre les instants t et t + dt et ve le volume massique du uideentrant. De même, le travail échangé pour faire sortir le uide est : psvsdms. Compte tenu de la conventionde signe habituelle, le travail des forces de pression reçu par le système est :
pevedme − psvsdms
Finalement, le bilan d'énergie (1.13) s'écrit :
dE + [(e+ pv)dm]se = dWu + dQ
où e représente l'énergie massique totale, composée de : l'énergie cinétique massique du uide : V2/2, où V est la vitesse du uide, l'énergie potentielle du uide, due aux champs de force extérieurs : ep, l'énergie interne massique u.
Le bilan énergétique s'explicite alors :
dE +
[(V2
2+ ep + u+ pv
)dm
]se
= dWu + dQ
On voit apparaître une grandeur énergétique h = u + pv, qui est adéquate pour décrire l'énergie enécoulement et qu'on appelle enthalpie massique.
En terme de puissance, on obtient :
dE
dt+
[m
(V2
2+ ep + h
)]se
= Wu + Qth
où m est le débit massique, Wu la puissance mécanique "utile", Qth la puissance thermique.
Régime stationnaire : si le système Σ ne comporte qu'une entrée et une sortie et est en régimestationnaire : dme = dms, dE = 0. Le premier principe s'écrit :
m
[(V2
2+ ep + h)
]se
= Wu + Qth
Quelques exemples : considérons un écoulement de liquide dans un tuyau de section quelconque, indéformable et adiaba-tique. L'énergie potentielle se réduit au potentiel de gravité. Dans ce cas, en écoulement stationnaire,[
m(V 2
2+ ep + h)
]se
= 0 (1.14)
m
[(V 2
2+ ep + h)
]se
= 0 (1.15)
V2
2+ ep + h = cste (1.16)
V2
2+ ep + u+ pv = cste (1.17)

14 Principes généraux
L'énergie interne d'un liquide ne dépend que de la température : u = u(T ). Or celle ci ne varie pasdans le tuyau. Ce terme est donc constant. On obtient donc :
V2
2+ gz + pv = cste (1.18)
V2
2g+ z +
p
ρg= cste (1.19)
On retrouve la loi de Bernoulli bien connue en mécanique des uides. cas d'une chaudière en régime stationnaire : la chaudière possède une entrée pour l'eau et une pourle mélange carburant/air. Elle possède une sortie pour l'eau chaude et une pour les gaz brûlés. Unechaudière idéale est adiabatique. En conséquence, le premier principe se ramène à :
m(eau) [hs(eau)− he(eau)] + m(gaz) [hs(gaz brûlés)− he(gaz frais)] = 0
L'énergie chimique de combustion est donc transférée sous forme de chaleur à l'eau. pour un réacteur d'avion :
m
[hs +
V2s
2− he
]= W = 0
où he est l'enthalpie du carburant et du comburant et hs l'enthalpie des gaz brûlés, Vs la vitessede sortie des gaz, la vitesse d'entrée du carburant étant négligeable. La vitesse de sortie induit unequantité de mouvement mVs par unité de temps, qui est la force de réaction (poussée).
pour une pompe en régime permanent agissant sur un liquide (masse volumique constante) :
mps − peρ
= W > 0
où ρ est la masse volumique du liquide.
4.2 Le second principe
On reprend les notations du paragraphe précédent. Pour un système échangeant de l'énergie thermiqueavec n thermostats à température Ti, le bilan entropique s'écrit :
dS
dt=
n∑i=1
QiTi
+ [ms]es + σ
où se est l'entropie massique en entrée, ss est l'entropie massique en sortie. σ > 0 est le taux de productiond'entropie et dQ la chaleur échangée avec l'extérieur à la température Ti.
Régime stationnaire : dans le cas d'un régime stationnaire : me = ms, dS = 0 et dans ce cas :
m [s]se =
n∑i=1
QiTi
+ σ

4 Cas particulier des systèmes ouverts en écoulement 15
Annexes
1. Forme diérentielle et diérentielle totale
On considère l'application suivante d'un espace de dimension n appelé E (l'espace des variables ther-modynamiques par exemple) dans Rn :
M(x1, . . . , xn) ∈ E 7→ (A1(x1, . . . , xn), A2(x1, . . . , xn), . . . , An(x1, . . . , xn))
On dénit la forme diérentielle dF par :
dF =n∑i=1
Ai(x1, . . . , xn)dxi
Soit C la courbe reliant le pointM1 au point M2 de E , alors la variation de f le long de cette courbe par :
IF (M1 →M2) =
∫CdF =
∫ M2
M1
dF
En général, IF dépend du chemin C reliant les points M1 et M2.Dans le cas où il existe une fonction F telle que :
∂F
∂xi= Ai(x1, . . . , xn)
on dit que dF est une diérentielle totale et alors l'intégrale :
IF (M1 →M2) = ∆F = F (M2)− F (M1)
ne dépend pas du chemin suivi pour aller de M1 à M2. Le critère permettant de savoir si une formediérentielle est une diérentielle totale est donné par les conditions de Cauchy :
∂2F
∂xi∂xj=
∂2F
∂xj∂xi
Dans certains cas, bien que la forme diérentielle df ne soit pas une diérentielle totale, il existe unefonction g(x1, . . . , xn), telle que g.df = dF est une diérentielle totale. On dit alors que g est un facteurintégrant.
Exemple : considérons la forme diérentielle
df = (x+ y) dx+ 2xy dy
et intégrons la entre le point M1 = (0, 1) et le point M2 = (1, 3) en suivant deux chemins (g. 4.2) : la droite C d'équation : y = 2x+ 1, la parabole C′ d'équation : y = 2x2 + 1On trouve : la droite C :
If (M1 →M2) =
∫C(x+ y) dx+ 2xy dy
or, le long de cette courbe, on a y = 2x+ 1 et dy = 2 dx, donc :
If (M1 →M2) =
∫ 1
x=0
(8x2 + 3x+ 5) dx
On obtient :
If (M1 →M2) =55
6

16 Principes généraux
0x
y
1
2
3
4
21
C’
C
Figure 1.2 Chemins d'intégration
la courbe C′ : le long de cette courbe, on a y = 2x2 + 1 et dy = 4x dx, donc :
If (M1 →M2) =
∫ 1
x=0
(16x4 + 10x2 + x+ 1) dx
On obtient :
If (M1 →M2) =241
30
En revanche, considérons la forme diérentielle :
dF = (2x+ y) dx+ x dy
On remarque qu'il existe une fonction F telle que :
∂F
∂x= 2x+ y
∂F
∂y= x
Il sut de choisir :F = x2 + xy
On vérie alors aisément que si on intègre la forme diérentielle dF entre M1 et M2 vaut :
IF (M1 →M2) = ∆F = F (M1)− F (M2) = 4
et que ce résultat est identique que l'on suive le chemin C ou le chemin C′.
Application à la thermodynamique : si on considère un système fermé constitué d'un uide carac-térisé par la température T et le volume V , alors la chaleur transférée à l'extérieur est dénie de façongénérale par la forme diérentielle :
dQ = CvdT + lvdp
où Cv et lv sont des coecients (dénis au chapitre suivant) qui sont tous deux des fonctions de T et V .Au cours de la transformation C de ce système entre M1 = (T1, V1) et M2 = (T2, V2), la chaleur échangées'obtient en intégrant dQ :
Q =
∫CdQ =
∫CCvdT + lvdV
Cette quantité d'énergie dépend du type de transformation eectuée : ce n'est pas une diérentielle totale.Considérons de même le travail élémentaire :
dW = −pdV

4 Cas particulier des systèmes ouverts en écoulement 17
Le travail échangé est :
W = −∫CpdV
Cette énergie dépend également du type de transformation eectuée.En revanche, considérons la somme :
dU = dQ+ dW = CvdT + (lv − p)dV
Le premier principe arme que cette forme diérentielle est une diérentielle totale, c'est à dire que :∫CdU = ∆U = U(M2)− U(M1)
Autrement dit, les variations de la fonction U ne dépend pas du chemin suivi. On dit que U est unefonction d'état, que l'on baptise "énergie interne".
Le second principe indique que la fonction 1/T est un facteur intégrant de la forme diérentielle dQ.Autrement dit, si on dénit :
dS =dQ
T
et alors : ∫CdS = ∆S = S(M2)− S(M1)
ne dépend pas du chemin suivi. On appelle la fonction d'état S "entropie".
2. Calcul du travail pour un uide
Considérons un système limité par une surface Σ dont la surface évolue au cours du temps. Les forcesdFext exercées sur cette surface eectuent un travail. Au cours d'un déplacement dr d'un élément desurface, le travail de ces forces est :
dW =
∫S
dFext.dr = −∫S
pext dS n.dr
où pext est le pression locale correspondant à la force dFext, dS orientée par la normale n dirigée versl'extérieur. Le produit dSn.dr correspond au volume dV balayé par la surface au cours de son déplacement.On obtient :
dW =
∫Σ
−pextdV
Il convient de tenir compte du fait que la pression externe pext n'est pas nécessairement uniforme surtoute la surface du système.
Si on a une transformation réversible, la pression interne est à tout instant en équilibre avec la pressionexterne imposée (équilibre mécanique). On obtient :
dW =
∫Σ
−pdV
3. Dénition microscopique de l'entropie
Considérons un système en équilibre d'énergie interne U et de volume V . A son état macroscopiquecorrespond un grand nombre d'états microscopiques : pour un gaz, un état microscopique est dénipar la position et la vitesse de toutes les molécules composant le gaz. Chacune de ces congurationsmicroscopiques a une probabilité pi d'apparaître. On dénit l'entropie par :
S(U) = −kB∑i∈Ω
pi log pi (1.20)
où kB est la constante de Boltzmann.

18 Principes généraux
Soit Ω le nombre total de congurations microscopiques accessibles, en admettant qu'elles ont toutesla même probabilité, on déduit :
pi =1
Ω
et la relation (1.20) devient :S(U) = kB log Ω
Ainsi l'entropie est d'autant plus grande, que le nombre de congurations microscopiques accessibles pourune même conguration macroscopique est grand.
Considérons maintenant un système pouvant se trouver dans des états d'énergie ϵi avec la probabilitépi (il n'y a plus équiprobabilité des congurations microscopiques). À l'équilibre, l'entropie est maximum.On recherche donc le maximum de S en utilisant la relation 1.20, sachant que les probabilités pi et lesénergies ϵi sont soumises aux contraintes :∑
i
pi = 1 et U =∑i
piϵi
Le calcul donne :
pi =exp [−ϵi/kBT ]
Z
avec Z =∑i exp [−ϵi/kBT ].
4. Interprétation microscopique du travail et de la chaleur
D'un point de vue microscopique, l'énergie interne d'un système est donnée par :
U =∑i
piϵi
où pi est la probabilité pour le système d'être dans l'état d'énergie ϵi. De la sorte, U peut changer àcause des changements de ϵi, c'est à dire des niveaux d'énergie accessibles au système, ou à cause deschangements de probabilités pi. Considérons une variation élémentaire d'énergie. Elle peut s'écrire :
dU =∑i
dpiϵi + pidϵi
On appelle alors le premier terme "chaleur" ou "transfert d'énergie thermique" et on l'identie à TdS,tandis que le second est appelé "travail" et identié à −pdV . L'entropie est associé aux changements depopulations des états d'énergie, alors que le travail est relié au changement des niveaux d'énergie desétats.
a) Le travail
Admettons que les énergies ϵj ne dépendent que du volume, alors :
dϵj =∂ϵj∂V
∣∣∣∣S
dV
où on dérive en gardant les probabilités pj constants, c'est à dire S constant. Dans ce cas,
∑j
pjdϵj =∑j
pj∂ϵj∂V
∣∣∣∣S
dV
Puisque pj est constant : ∑j
pjdϵj =∂
∂V
∑j
pjϵjdV =∂U
∂V
∣∣∣∣S
dV = −pdV

4 Cas particulier des systèmes ouverts en écoulement 19
b) La chaleur
On dénit l'entropie d'un ensemble de Gibbs par :
SG = k
N logN −∑j
nj log nj
Il s'agit de l'entropie d'un ensemble de N systèmes équivalents. Or on s'intéresse à l'entropie moyennedu système. On utilise N =
∑j nj , de sorte que :
SG = k
∑j
nj logN −∑j
nj log nj
(1.21)
= −k∑j
nj lognjN
(1.22)
L'entropie moyenne est donc SG divisé par N :
S = −k∑j
njN
lognjN
On identie nj/N comme étant pj , probabilité que le système soit dans l'état j. On obtient la formuleusuelle de l'entropie pour un système non isolé :
S = −k∑j
pj log pj
Utilisons cette expression pour relier S et chaleur. La diérentielle de S est :
dS = −k∑j
(dpj log pj + dpj)
Le second terme donne zéro, puisque la probabilité totale est constante. Nous avons donc :
dS = −k∑j
dpj log pj
Pour le terme en log pj , nous utilisons le facteur de Boltzmann :
pj =e−ϵjkT
Z⇔ log pj = −
( ϵjkT
+ Z)
De la sorte :dS = k
∑j
( ϵjkT
+ Z)dpj
Le second terme est nul puisque Z est une constante et que la somme des dpj est nulle. Donc seul lepremier terme contribue :
dS =1
T
∑j
ϵjdpj (1.23)
TdS =∑j
ϵjdpj (1.24)

20 Principes généraux

Chapitre 2
Application des principes au fluide
homogène
1 Introduction
Si le système thermodynamique étudié est constitué d'un uide homogène, les variables extensivesqui permettent de le décrire sont son volume V et le nombre de moles N , auxquelles on ajoute l'entropieS. Toute la connaissance que l'on peut avoir sur ce uide à l'équilibre est contenue dans la fonctionU = U(S, V,N), que l'on ne connaît pas en général. Pour des raisons pratiques, on utilise pour le décrireles variables pression p, volume V et T . Ces variables ne sont pas indépendantes, puisqu'elles sont reliéespar une équation d'état f(p, V, T ) = 0. Parmi ces trois variables, seules deux sont indépendantes.
2 Relations entre coecients calorimétriques
2.1 Dénition des coecients calorimétriques
Au cours d'une transformation élémentaire réversible, la quantité de chaleur dQ peut s'exprimer enfonction de deux des trois variables p, V et T . Suivant le couple de variables choisie, on peut alors écrireles formes diérentielles suivantes :
dQ(T, V ) = Cv(T, V ) dT + lv(T, V ) dV (2.1)
dQ(T, p) = Cp(T, p) dT + lp(T, p) dp (2.2)
dQ(V, p) = λv(V, p) dV + λp(V, p) dp (2.3)
qui dénissent les coecients calorimétriques : Cv (capacité calorique à volume constant), Cp (capacitécalorique à pression constante), lv (coecient de chaleur de détente isotherme), lp (coecient de chaleurde compression isotherme), λv (coecient de chaleur de compression isochore) et λp (coecient de chaleurde détente isobare). Ces coecients ne sont bien sûr pas indépendants. En eet, eectuons un changementde variables en remplaçant V par p :
dV =∂V
∂TdT +
∂V
∂pdp
En remplaçant dans la relation (2.1) :
dQ =
[Cv + lv
∂V
∂T
]dT +
[lv∂V
∂p
]dp
Par identication avec (2.2), on en déduit :
lv = (Cp − Cv)∂T
∂V
∣∣∣∣p
(2.4)
et lp = lv∂V
∂p
∣∣∣∣T
(2.5)

22 Application des principes au fluide homogène
Le même type de changement de variables donne les relations suivantes :
lp = −(Cp − Cv)∂T
∂p
∣∣∣∣V
, λp = Cv∂T
∂p
∣∣∣∣V
, λv = Cp∂T
∂V
∣∣∣∣p
Rapport des capacités caloriques ou des chaleurs spéciques : γ = Cp/Cv est appelé rapportdes chaleurs spéciques.
2.2 Relations de Clapeyron
2.2.1 Calcul de lv
Pour un système fermé contenant un uide homogène, on a dU = TdS − pdV , donc :
dU = dQ− pdV
dS =dQ
T
En utilisant (2.1) :
dU = CvdT + (lv − p)dV
dS = CvdT
T+ lv
dV
T
Les fonctions U et S sont des diérentielles totales : elles caractérisent complètement le système à l'équi-libre. On peut donc utiliser le critère de Cauchy (égalité des dérivées secondes 1) et on en déduit :
∂(lv − p)
∂T=∂Cv∂V
∂(lv/T )
∂T=∂(Cv/T )
∂V(2.6)
c'est à dire :
∂lv∂T
− ∂p
∂T=∂Cv∂V
1
T
∂lv∂T
− 1
T 2lv =
1
T
∂Cv∂V
⇒ ∂lv∂T
− 1
Tlv =
∂Cv∂V
On en déduit une des relations de Clapeyron :
lv = T∂p
∂T
∣∣∣∣V
(2.7)
2.2.2 Calcul de lp
On peut faire le même raisonnement en utilisant l'enthalpie :
dH = dU + pdV + V dp = dQ− pdV + pdV + V dp = dQ+ V dp
dS =dQ
T
1. Considérons une fonction de n variables f(x1, . . . , xn), la dérivée de cette fonction :
df =∂f
∂x1dx1 + . . .+
∂f
∂xndxn
On a l'égalité entre les dérivées secondes de f :∂2f
∂xi∂xj=
∂2f
∂xj∂xi

2 Relations entre coecients calorimétriques 23
En utilisant (2.2) :
dH = CpdT + (lp + V )dp
dS = CpdT
T+ lp
dP
T
En écrivant l'égalité des dérivées secondes, on en déduit :
∂(lp − V )
∂T=∂Cp∂p
∂(lp/T )
∂T=∂(Cp/T )
∂p(2.8)
On en déduit la seconde relation de Clapeyron :
lp = −T ∂V
∂T
∣∣∣∣p
2.3 Relations de Clausius
Compte tenu de (2.6) et (2.8), on en déduit également les relations de Clausius :
∂Cv(T, V )
∂V= T
∂2p
∂T 2et∂Cp(T, p)
∂p= −T ∂
2V
∂T 2
Les relations de Clausius permettent, connaissant Cp(T, p) à une pression p0 donnée de calculer parintégration sa valeur à toute pression. De même, connaissant Cv(T, V ) à un volume donné, on peut parintégration sur V calculer sa valeur à tout autre volume.
2.4 Relation de Mayer
En écrivant l'égalité entre lv calculé par les relations (2.4) et (2.7), on en déduit :
Cp − Cv = T∂p
∂T
∂V
∂T
On en déduit que si on connaît la fonction Cv(T, V ) ou la fonction Cp(T, p), on peut en déduire l'autrepar la relation de Mayer.
2.5 Applications
Finalement, si on connaît une équation d'état sous la forme f(p, V, T ) = 0 et une des capacités calori-ques à pression ou à volume constant, on peut calculer toutes les autres grandeurs thermodynamiques,en particulier l'énergie interne U , l'entropie S et l'enthalpie H.
2.5.1 Le gaz parfait
Considérons un gaz parfait déni par son équation d'état pV = RT (pour une mole de gaz) etCv =cste. Dans ce cas, on obtient :
relations de Clapeyron :lv = p et lp = −V
relation de Mayer :Cp − Cv = R
on en déduit l'expression de l'énergie interne :
U = CvT + cste
et celle de l'enthalpie :H = CpT + cste
L'expression de l'entropie est :
S = Cv log T +R log V + cste ou S = Cp log T −R log p+ cste

24 Application des principes au fluide homogène
Exercice : montrer qu'au cours d'une transformation adiabatique réversible d'un gaz parfait à capacitéscaloriques constantes, on a pV γ =constante, où γ est le rapport des capacités caloriques, p la pressionet V le volume.
2.5.2 Le gaz de Van der Waals
On considère un gaz de Van der Waals, c'est à dire déni par l'équation d'état (pour une mole) :(p+
a
V 2
)(V − b) = RT,
pour lequel on admet que Cv est constant. L'énergie interne s'écrit :
U = CvT − a
V
et l'entropie :S = Cv log T +R log(V − b) + cste
3 Fonctions thermodynamiques
3.1 Introduction
L'objectif de ce paragraphe est double : trouver des fonctions mettant en jeu des variables adéquates au transformations considérée et conte-nant la même information que la relation fondamentale U(S,Xi),
trouver des fonctions permettant de caractériser l'équilibre d'un système thermodynamique.
3.2 Notion de réservoir
Considérons un système caractérisé, entre autres, par la variable extensiveXi, dont la variable intensiveassociée est fi.
Dénition : un système R est un réservoir si, mis en contact avec un système Σ et échangeant avec luiuniquement une variable extensive X, toutes les transformations de Σ se font à f constante. Les variations∆X sont supposées négligeables devant la capacité en X du réservoir.
L'énergie fournie à Σ par le réservoir est :
∆U = −f.∆XR = f.∆XΣ
où ∆XR est la variation en X du réservoir et ∆XΣ celle du système Σ.
Dénition : on dit que R est une source de f .
Exemple : source de température (thermostat) → transformation isotherme, avec ∆U = T∆SR, source de pression → transformation isobare, avec ∆U = −P∆VR.
3.3 La transformée de Legendre
Soit f(x) une fonction convexe, c'est-à-dire donc la dérivée seconde est positive (f ′′(x) > 0). Posonsp = ∂f/∂x. On cherche une fonction g(p) de la variable p qui contienne la même information que dans f .
On procède de la façon suivante : pour chaque point x0, l'équation de la droite tangente à f(x0) enx = x0 s'écrit y(x) = f ′(x0)(x−x0)+ f(x0). Cette fonction tangente est unique, puisque la fonction f(x)étant convexe, sa dérivée f ′(x) est une fonction croissante monotone. Il existe donc pour chaque point x,une et une seule fonction droite tangente (D).
Cette tangente d'équation y(x) = f ′(x0)x− [f ′(x0)x0 + f(x0)] est parfaitement décrite si on connaîtles deux grandeurs f ′(x0) et −f ′(x0)x0+f(x0). Autrement dit, si on connaît p = f ′(x0), la fonction g(p) =

3 Fonctions thermodynamiques 25
f − px0 permet d'avoir toute l'information sur la fonction originale f . On appelle g(p) la transforméede Legendre de la fonction f .
Comme la fonction f ′ est monotone, il existe une seule valeur x telle que p = f ′(x). On a doncx = f ′−1(p). La fonction g s'écrit donc explicitement en fonction de p par :
g(p) = f(f ′−1(p))− pf ′−1(p)
Exemple : soit f(x) = x lnx. Sa dérivée est p(x) = f ′(x) = lnx + 1, ou encore x = ep−1. Calculonsg(x) = f(x) − p(x).x, c'est-à-dire g = x lnx − (lnx + 1)x = −x. On remplace x par son expression enfonction de p et la transformée de Legendre de f(x) = x lnx est g(p) = −ep−1.
Remarques : Si ∂2f/∂2x ≥ 0 (f est convexe) alors sa transformée de Legendre g est concave, c'est à dire∂2g/∂2p ≤ 0.En eet, g = f − px, donc x = −∂g/∂p. On dérive une seconde fois : ∂x/∂p = −∂2g/∂p2. D'autrepart :
∂p
∂x=
∂
∂x
(∂f
∂x
)=∂2f
∂x2
On a donc :∂2g
∂p2= − 1
∂2f/∂x2
La réciproque est évidemment vraie. g ne dépend que de la pente p. En eet, dg = df − pdx− xdp, or df = p.dx, donc dg = −x.dp. La transformée de Legendre est involutive : si g est la transformée de Legendre de f , la transforméede Legendre de g est f .
3.4 Application de la transformée de Legendre au système uide
On va chercher des fonctions, transformées de Legendre pour diérentes variables intensives en utilisantla relation fondamentale 2 U = U(S, V,N) valable pour un uide homogène. Elles auront une utilisationprivilégiée pour des transformations avec un réservoir maintenant une ou plusieurs variables intensivesconstantes.
Enthalpie
On dénit, par transformée de Legendre de la fonction U(S, V,N) par rapport à la variable p =−∂U/∂V , l'enthalpie :
H = U + pV
La forme diérentielle correspondante est : dH = TdS+V dp+µdN . Elle est adaptée aux transformationsd'un système fermé évoluant à pression constante. L'équation d'Euler se met sous la forme : H = TS+µNou dans le cas général : H = TS +
∑i fiXi.
Énergie libre
Si on eectue la transformée de Legendre de la fonction U par rapport à la variable T = ∂U/∂S, onobtient l'énergie libre 3 ou fonction de Helmholtz :
F = U − TS
La forme diérentielle correspondante est : dF = −SdT−pdV +µdN . Elle est adaptée aux transformationsd'un système fermé à température constante. L'équation d'Euler se met sous la forme : F = −pV + µNou dans le cas général : F = −pV +
∑i fiXi.
2. La transformation de Massieu est la transformation de Legendre appliquée à la relation fondamentale sous formeentropique : S = S(U, V,N). Elle est moins utilisée.
3. En anglais, Helmholtz free energy, notée A.

26 Application des principes au fluide homogène
Enthalpie libre
Si on eectue la double transformée de Legendre par rapport à p et à T , on obtient l'enthalpie libreou potentiel de Gibbs 4 :
G = U + pV − TS = H − TS
La forme diérentielle correspondante est : dG = −SdT+V dp+µdN . Elle est adaptée aux transformationseectuées par un système fermé à pression et température constantes.
On remarque que l'équation d'Euler donne : G = U + pV − TS = TS − pV +∑i µiNi + pV − TS =∑
i µiNi. Dans le cas où le système ne comprend qu'un seul composant, l'équation d'Euler se met sousla forme :
G = µN
L'enthalpie libre molaire s'identie au potentiel chimique.
Grand potentiel
Si on eectue la double transformée de Legendre par rapport à la température T et au potentielchimique µ, on obtient le grand potentiel :
J = U − TS − µN = F − µN
Sa forme diérentielle est : dJ = −SdT −Ndµ−pdV . L'équation d'Euler devient : J = −pV . On emploiecette fonction pour les systèmes couplés à un thermostat et à un réservoir de particules évoluant à volumeconstant. Cette fonction apparaît en thermodynamique statistique.
3.5 Relations de Maxwell
Pour la forme diérentielle de chaque fonction thermodynamique, il est possible d'écrire une identitéentre les dérivées secondes (relations de Cauchy). Par exemple, considérons la forme diérentielle :
dU = TdS − pdV + µdN
On peut écrire :∂T
∂V= − ∂p
∂S,∂T
∂N=∂µ
∂S,− ∂p
∂N=∂µ
∂V
De façon générale, si un système est décrit par les couples (fi, Xi), i = 1 à n, où fi est la variable intensiveconjuguée de la variable intensive Xi par rapport à l'énergie interne, ces relations prennent le nom derelations de Maxwell et ont la forme :
∂fi∂Xj
=∂fj∂Xi
∂Xi
∂Xj= −∂fj
∂fi
3.6 Relations d'Helmholtz
Première formulation
La forme diérentielle de l'énergie libre est dF = −PdV − SdT . Autrement dit,∂F
∂T= −S. En
remplaçant dans l'expression de dénition de l'énergie libre F = U − TS, on trouve :
F = U + T∂F
∂T
En divisant tous les termes par T 2, on obtient :
1
T
∂F
∂T− F
T 2= − U
T 2ou encore
∂(F/T )
∂T= − U
T 2(première relation de Helmholtz)
4. En anglais, Gibbs free energy.

3 Fonctions thermodynamiques 27
De même, de la forme diérentielle dG = V dP − SdT , on en déduit∂G
∂T= −S. En remplaçant dans
l'expression de dénition de l'enthalpie libre G = H − TS, on obtient :
G = H + T∂G
∂T
En divisant tous les termes par T 2, on trouve :
1
T
∂G
∂T− G
T 2= − H
T 2ou
∂(G/T )
∂V= −H
T 2(seconde relation de Helmholtz)
Seconde formulation
Considérons une transformation se déroulant à température et volume constants et menant de l'état1 à l'état 2. F1 et F2 sont respectivement les énergies libres de l'état initial et de l'état nal. Soit la mêmetransformation se déroulant à la température T + dT . Exprimons la variation de ∆F = F2 − F1 avec latempérature.
Les relations de Helmholtz permettent d'écrire :
F1 − U1
T=∂F1
∂TetF2 − U2
T=∂F2
∂T
On obtient :F2 − F1 − (U2 − U1)
T=∂F1
∂T− ∂F2
∂T=∂(F2 − F1)
∂T
Finalement :
∆F = ∆U + T∂∆F
∂T
De même, on peut démontrer :
∆G = ∆H + T∂∆G
∂T
3.7 Équation d'Helmholtz
On utilise la diérentielle de l'énergie dU = TdS − pdV :
dS =1
T(dU + pdV )
=1
T
[∂U
∂VdV +
∂U
∂TdT
]+p
TdV
=
[1
T
∂U
∂V+p
T
]dV +
1
T
∂U
∂TdT
La condition de Cauchy implique :
∂
∂T
[1
T
∂U
∂V+p
T
]=
∂
∂V
[1
T
∂U
∂T
]On en déduit la relation suivante :
∂U
∂V= T 2 ∂
∂T
[ pT
]Application : la relation trouvée au paragraphe précédent peut être intégrée :
U(S, V ) = U(S, V0) +
∫ V
V0
T 2 ∂
∂T
[ pT
]dV
Lorsque V0 tend vers l'inni, le gaz devient très dilué et son comportement s'approche de celui d'un gazparfait :
U(S, V ) = Ugp +
∫ V
∞T 2 ∂
∂T
[ pT
]dV

28 Application des principes au fluide homogène
La diérence U(S, V )− Ugp s'appelle énergie interne résiduelle 5.On peut facilement en déduire l'enthalpie résiduelle, l'entropie résiduelle, etc. (voir annexe 1). On
trouve :
H(S, p)−Hgp =
∫ V
∞T 2 ∂
∂T
[ pT
]dV + pV −RT
Connaissant la capacité calorique du gaz, lorsqu'il est très dilué, appelée capacité calorique du gazparfait Cp,gp(T ), on peut en déduire l'expression de l'enthalpie à toute pression et à toute température.On notera que le second terme intégral est calculable si on connaît l'équation d'état du gaz.
De la même façon, il est possible de calculer (annexe 1) l'entropie résiduelle :
S(T, p)− Sgp =
∫ V
∞
[∂p
∂T− R
V
]dV
et l'enthalpie libre résiduelle :
G(T, p)−Ggp =
∫ V
∞
[−p+ RT
V
]dV −RT log
pV
RT+ pV −RT
3.8 Équilibre et stabilité d'un système thermodynamique
L'équilibre et la stabilité sont dénis par les variations d'un potentiel par rapport aux variables Xi
susceptibles de varier. Si f est un potentiel, la position d'équilibre Xi,0 sera déterminée par∂f
∂Xi
∣∣∣∣Xi,0
= 0.
La stabilité dépendra alors du comportement de f au voisinage de ce point d'équilibre (g. 2.1) :(a) stabilité : le système retourne à la position d'équilibre quelle que soit la perturbation subie,(b) instabilité : sous l'action de toute perturbation, le système s'éloignera de la position d'équilibrepour en retrouver une autre,
(c) métastabilité : le système revient à la position d'équilibre si la perturbation n'est pas trop impor-tante. Si la perturbation est importante, il trouve un autre état d'équilibre.
(d) stabilité neutre : toute perturbation déplace la position d'équilibre du système.
(d) stabilité neutre
(a) stable (b) instable
(c) métastable
Figure 2.1 Diérents comportements vis à vis de la stabilité
3.8.1 Concavité de l'entropie
On a vu que l'entropie est une fonction concave. Autrement dit :
S(λU (1) + (1− λ)U (2), λX(1)i + (1− λ)X
(2)i ) ≥ λS((U (1), X
(1)i ) + (1− λ)S(U (2), X
(2)i )
En particulier, si λ = 1/2, X(1)i = Xi + δXi et X
(2)i = Xi − δXi, U (1) = U + δU et U (2) = U − δU , on
obtient :2S(U,Xi) ≥ S(U + δU,Xi + δXi) + S(U − δU,Xi − δXi)
5. Les grandeurs résiduelles sont appelées departure functions en anglais

3 Fonctions thermodynamiques 29
On peut développer les deux termes de droite :
S(U + δU,Xi + δXi)− S(U,Xi) =∂S
∂U∆U +
∂S
∂Xi∆Xi +
1
2
∂2S
∂U2∆U2 +
1
2
∂2S
∂X2i
∆X2i +
∂2S
∂U∂Xi∆U∆Xi
S(U − δU,Xi − δXi)− S(U,Xi) = − ∂S
∂U∆U − ∂S
∂Xi∆Xi +
1
2
∂2S
∂U2∆U2 +
1
2
∂2S
∂X2i
∆X2i +
∂2S
∂U∂Xi∆U∆Xi
On additionne et on obtient :
S(U + δU,Xi+ δXi)+S(U − δU,Xi− δXi)− 2S(U,Xi) =∂2S
∂U2∆U2+
∂2S
∂X2i
∆X2i +2
∂2S
∂U∂Xi∆U∆Xi ≤ 0
On en déduit :∂2S
∂U2≤ 0,
∂2S
∂X2i
≤ 0 et∂2S
∂U2
∂2S
∂X2i
−(
∂2S
∂U∂Xi
)2
≤ 0
3.8.2 Convexité de l'énergie interne
L'énergie interne est minimum à l'équilibre.
La démonstration mathématique est la suivante : on considère deux systèmes notés (1) et (2). Dé-nissons U (1) = U((S(1), X
(1)i ) et U (2) = U(S(2), X
(2)i ), telles que S(1) = S((U (1), X
(1)i ) et S(2) =
S(U (2), X(2)i ). Puisque S est concave,
S(λU (1) + (1− λ)U (2), λX(1)i + (1− λ)X
(2)i ) ≥ λS((U (1), X
(1)i ) + (1− λ)S(U (2), X
(2)i )
Or les fonctions U = U(S,Xi) et S = S(U,Xi) sont inverses l'une de l'autre. Autrement dit, U =U(S(W,Xi), Xi), pour tout W . Donc :
λU (1) + (1− λ)U (2) = U(S(λU (1) + (1− λ)U (2), λX(1)i + (1− λ)X
(2)i ) (2.9)
≥ U(λS(U (1), X(1)i ) + (1− λ)S(U (2), X
(2)i ), λX
(1)i + (1− λ)X
(2)i ) (2.10)
puisque ∂U/∂S = T > 0. On en déduit :
λU(S(1), X(1)i ) + (1− λ)U(S(2), X
(2)i ) ≥ U(λS(1) + (1− λ)S(2), λX
(1)i + (1− λ)X
(2)i )
Donc, U est convexe. On obtient :
∂2U
∂S2≤ 0,
∂2U
∂X2i
≤ 0 et∂2U
∂S2
∂2U
∂X2i
−(
∂2U
∂S∂Xi
)2
≤ 0
3.8.3 Conséquences : conditions de stabilité d'un système uide homogène
On considère un uide homogène déni par la fonction U = U(S, V ). On pose :
USS =∂2U
∂S2
UV V =∂2U
∂V 2
USV =∂2U
∂S∂V
Les conditions précédentes donnent :
∂T
∂S> 0,
∂p
∂V< 0 et
∂T
∂V.∂p
∂S− ∂T
∂S.∂p
∂V> 0
La dernière condition peut également se ré-écrire, compte tenu des relations de Maxwell :(∂T
∂V
)2
+∂T
∂S.∂p
∂V< 0
On en déduit les conséquences suivantes concernant les coecients calorimétriques :

30 Application des principes au fluide homogène
Le signe de Cv : la diérentielle de l'entropie s'écrit dS =CvTdT +
lvTdV . On en déduit USS =
∂T
∂S=
T
Cv> 0 c'est à dire Cv > 0.
Le signe de Cp : la diérentielle de l'entropie S(T, p) s'écrit également dS =CpTdT +
lpTdp. On en
déduit que :CpT
=∂S
∂T(2.11)
D'autre part :
dT =∂T
∂SdS +
∂T
∂VdV (2.12)
dp =∂p
∂SdS +
∂p
∂VdV (2.13)
D'autre part :
T =∂U
∂Set − p =
∂U
∂V
En remplaçant dans (2.12) et (2.13), on obtient :
dT = USSdS + USV dV
−dp = UV SdS + UV V dV
ou à pression constante :
dT = USSdS + USV dV
0 = UV SdS + UV V dV
En éliminant dV dans les équations ci-dessus :
dT =USSUV V − USV UV S
UV VdS
La relation (2.11) s'écrit alors :CpT
=UV V
USSUV V − USV UV SLes conditions de stabilité montrent que le numérateur et le dénominateur doivent être positifs, doncCp > 0
Le signe de Cp − Cv : comparons les coecients Cp et Cv :
1
T(Cp − Cv) =
UV VUSSUV V − USV UV S
− 1
USS
=U2SV
USS(USSUV V − USV UV S)
Cette expression est positive, donc Cp > CvCeci a une conséquence sur la relation de Mayer qui s'écrit :
Cp − Cv = T∂p
∂T
∂V
∂T
Sachant que :∂p
∂T
∂T
∂V
∂V
∂p= −1
Du fait que Cp > Cv, on en déduit l'inégalité :
∂p
∂V< 0
Ceci s'interprète de la façon suivante : le volume ne peut que diminuer lorsque la pression augmente.Toute autre situation est instable.

3 Fonctions thermodynamiques 31
3.8.4 Équilibre d'un système couplé à un thermostat
Considérons un système couplé à un thermostat à température T . L'ensemble comprenant le systèmeétudié Σ et le réservoir R est fermé et isolé.
A l'équilibre, la condition d'entropie maximum peut être appliquée à l'ensemble Σ ∪R, c'est à dire :dSΣ∪R = 0 et d2SΣ∪R < 0. Autrement dit :
dSΣ + dSR = 0, d2SΣ + d2SR < 0 (2.14)
La variation d'entropie du réservoir est dSR =dURT
= −dUΣ
T, puisque l'énergie interne de Σ ∪ R est
constante. Les conditions (2.14) donnent donc :
dS − dURT
= 0, d2S − d2URT
< 0
et puisque T est constant :
d(UΣ − TSΣ) = dFΣ = 0, d2(UΣ − TSΣ) = d2FΣ > 0
L'équilibre d'un système en contact thermique avec un thermostat est l'état qui minimise l'énergielibre. Autrement dit, l'énergie libre d'un système isotherme stable est une fonction concave.
3.8.5 Équilibre d'un système couplé à un réservoir de pression
Considérons un système couplé à un réservoir de pression à pression p. L'ensemble comprenant lesystème étudié Σ et le réservoir R est fermé et isolé. La position d'équilibre est celle qui maximisel'entropie à énergie constante ou celle qui minimise l'énergie à entropie constante.
Le minimum de l'énergie pour le système total Σ ∪ R s'écrit dUΣ∪R = 0 et d2UΣ∪R > 0. Autrementdit,
dUΣ + dUR = 0, d2(UΣ + UR) > 0
Le réservoir réalise un travail sur le système Σ : dUR = −p dVR = −dUΣ = p dVΣ. La condition deminimum donne donc :
dUΣ + pdVΣ = 0, d2UΣ + pd2VΣ > 0
Puisque p est constant :
d(UΣ + pVΣ) = dHΣ = 0, d2(UΣ + pVΣ) = d2HΣ > 0
L'enthalpie est minimum à l'équilibre, sous la contrainte que la pression soit égale à celle du réservoir.Autrement dit, l'enthalpie d'un système isobare stable est une fonction convexe.
3.8.6 Équilibre d'un système couplé à un réservoir de pression et à un thermostat
La condition de minimum de l'énergie à température et pression donnée se traduit par la conditionde minimum de l'énergie libre de Gibbs G = U − TS + pV à température et pression constante, égalesà celles du réservoir. Autrement dit, l'enthalpie libre d'un système isobare et isotherme stable est unefonction convexe.
3.9 Travail maximum récupérable
Pour une transformation isotherme
On considère un système Σ homogène fermé, évoluant à température constante T en échangeant lachaleur Q avec un thermostat R. Il peut aussi échanger du travail W avec l'extérieur à (Σ∪R). Puisquele système (Σ ∪R) n'échange que du travail avec l'extérieur, l'entropie ne peut que s'accroître et tendrevers son maximum. Le premier principe s'écrit :
∆U = ∆UΣ +∆UR =W

32 Application des principes au fluide homogène
avec ∆UR = −Q. Le second principe implique :
∆S = ∆SΣ +∆SR ≥ 0
avec ∆SR = −Q/T . On en déduit :
∆SΣ − Q
T≥ 0
∆SΣ +∆URT
≥ 0
∆SΣ +W −∆UΣ
T≥ 0
Finalement ∆UΣ − T∆SΣ ≤ W et donc ∆F ≤ W . Sachant que W est négatif, la valeur maximale dutravail fournie par un système fermé évoluant à T constant est donnée par |∆F | et sera obtenu pour unfonctionnement réversible.
Pour une transformation isotherme et isobare
On considère un système Σ homogène fermé, évoluant à température constante T et à pression pconstante en échangeant la chaleur Q et du travail de pression Wp = p∆V avec le thermostat-réservoirde pression R. Il peut aussi échanger du travail Wu avec l'extérieur à (Σ∪R). Puisque le système (Σ∪R)n'échange que du travail avec l'extérieur, l'entropie ne peut que s'accroître et tendre vers son maximum.Le premier principe s'écrit :
∆U = ∆UΣ +∆UR =Wu
avec ∆UR = −Q+ p∆V . Le second principe implique :
∆S = ∆SΣ +∆SR ≥ 0
avec ∆SR = −Q/T . On en déduit :
∆SΣ − Q
T≥ 0
∆SΣ − ∆UR − p∆V
T≥ 0
∆SΣ − Wu −∆UΣ − p∆V
T≥ 0
Finalement ∆UΣ+p∆V −T∆SΣ ≤Wu et donc ∆G ≤Wu. Sachant queW est négatif, la valeur maximaledu travail fournie par un système fermé évoluant à T et à p constants est donnée par |∆G| et sera obtenupour un fonctionnement réversible.
4 Équations d'état
4.1 Coecients de réponse ou coecients thermoélastiques
Ce sont des coecients que l'on mesure facilement et qui permettent de déduire une fonction d'étatdu uide f(p, V, T ) = 0. On dénit :
le coecient de compressibilité isotherme :
χT = − 1
V
(∂V
∂p
)T
= − 1
V
(∂2G
∂p2
)T
le coecient de dilatation isobare ou expansion thermique :
α =1
V
(∂V
∂T
)p
=1
V
(∂2G
∂T 2
)p

4 Équations d'état 33
le coecient de compression isochore :
β =1
p
(∂p
∂T
)V
le coecient de compressibilité adiabatique :
χS = − 1
V
(∂V
∂p
)S
= − 1
V
(∂2H
∂p2
)S
Remarque : ces coecients ne sont pas indépendants :
α = pβχT
Application :a. Montrer que pour un gaz parfait :
α =1
T, β =
1
T, χT =
1
p
b. Expérimentalement, on a déterminé l'évolution des coecients thermoélastiques αP et κT enfonction des variables thermodynamiques T et p (pour une mole de gaz) :
α =R
RT + bp, β =
1
Tet χT =
RT
p(RT + bp)
où R est la constante des gaz parfaits et b une constante caractéristique du gaz réel étudié. Endéduire l'équation d'état du gaz : p(V − b) = RT .
c. Montrer que pour un gaz de Van der Waals :
α =nR
V
[p− n2a
V 2+
2n3ab
V 3
] , β =nR
p(V − nb), χT =
Vnb
V
[p− n2a
V 2+
2n3ab
V 3
]d. Montrer que :
cp − cv =V Tβ2
χt
4.2 Diverses équations d'état
Dans les équations suivantes, les variables sont dénies comme suit : p pression, V volume molaire, T température.
4.2.1 Loi des gaz parfaits
La loi des gaz parfaits correspond à un ensemble de molécules sans interaction entre elles. Elle s'écrit :
pv = RT
avec R =8.31451 J/mol.K, appelée constante des gaz parfaits et v est le volume molaire.
4.2.2 L'équation de Van der Waals
L'équation d'état de Van der Waals peut être présentée comme une extension de la loi des gaz par-faits pV = nRT au cas où il existe des interactions à courte distance (attraction) et à longue distance(répulsion) entre molécules. La pression est liée à l'énergie cinétique : il s'agit de la densité de ux dequantité de mouvement des molécules. Dans le cas où il existe des interactions, il convient d'ajouter àl'énergie cinétique une énergie potentielle d'interaction. Celle-ci est proportionnelle à −(N/V )N , où Nest le nombre de molécules. En eet, chacune des N molécules interagit avec les autres dans une certaine

34 Application des principes au fluide homogène
sphère d'inuence de rayon r0. Le nombre de molécules dans cette sphère est proportionnelle à la densitéde molécules N/V .
La pression est proportionnelle à la densité volumique d'énergie, donc la pression totale dans le gazest :
p = pcin − a
(N
V
)2
c'est à dire que la pression "cinétique" s'écrit :
pcin = p+ a
(N
V
)2
D'autre part, on admet que les molécules ne peuvent se rapprocher en deça d'un certain rayon. Autrementdit, le volume accessible aux molécules doit être diminuée du volume que ces molécules occupent elles-mêmes :
Vf = V −Nb
où b est le co-volume ou volume exclus. En écrivant la loi des gaz parfaits avec pcin et Vf :
pcinVf = nRT,
on obtient l'équation d'état du gaz de Van der Waals par mole de gaz :
p =RT
v − b− a
v2
où v le volume molaire et T la température.
Calcul des fonctions thermodynamiques On admet dans la suite que la seconde équation d'étatest cv=constante.
L'énergie interne : la diérentielle de l'énergie interne molaire s'écrit du = cvdT+(lv−p)dv. Comptetenu de la relation de Clapeyron :
lv = T∂p
∂T=
RT
v − b= p+
a
v2
La diérentielle de l'énergie interne est donc du = cvdT + av2 dv, ce qui, une fois intégrée, donne :
u = cvT − a
v+K
où K est une constante. L'entropie : la diérentielle de l'entropie molaire est ds = cv
dTT + R
v−bdv, qui donne par intégration :
s = cv log T +R log(v − b) +K
où K est une constante. L'énergie libre : sachant que l'énergie libre est f = u− Ts, on en déduit :
f = −av−RT log(v − b) + cvT (1− log T ) +K
où K est une constante. L'enthalpie libre : sachant que l'enthalpie libre est g = h− Ts = u+ pv− Ts = f + pv, on obtient :
g =RTv
v − b− 2a
v−RT log(v − b) + cvT (1− log T ) +K
où K est une constante.Proposée en 1873, l'équation de Van der Waals est la première à approcher le comportement des gaz
réels. Cette équation permet de prédire l'apparition de liquide, mais elle reste éloignée de la réalité. Ellereste cependant intéressante sur le plan pédagogique.

4 Équations d'état 35
4.2.3 L'équation du viriel
pv
RT= 1 +
B
v+C
v2+D
v3+ . . .
Bien que n'étant pas l'équation d'état la plus pratique, elle est intéressante, parce qu'elle est directementissue de la thermodynamique statistique. Si on fait des hypothèses sur la forme des interactions intermo-léculaires, des expressions théoriques peuvent être développées pour chacun des coecients : B représenteles interactions de paires, C de triplets, etc.
4.2.4 Équation d'état de Redlich-Kwong
p =RT
v − b− a√
Tv (v + b)
avec :
a = 0.42748R2T 2.5
c
pcet b = 0.08664
RTcpc
(2.15)
où Tc et pc sont des constantes appelées respectivement température critique et pression critique, dont onverra la signication précise au chapitre suivant.
L'équation de Redlich-Kwong garde une forme relativement simple. Bien que supérieure à l'équationde Van der Waals, elle se comporte mal par rapport à la phase liquide et ne peut prédire avec précisionles équilibres liquide-vapeur. Cependant, elle peut être utilisée conjointement avec des corrélations pourla phase liquide.
Cette équation est tout à fait adéquate pour la phase gazeuse, lorsque le rapport entre la pression etla pression critique est inférieur à 1/2 du rapport de la température à la température critique.
4.2.5 L'équation de Redlich-Kwong-Soave
Il s'agit d'une équation dérivée de la précédente. Le terme a/√T de l'équation de Redlich-Kwong est
remplacé par la fonction α(ω, T ), de façon à mieux représenter la pression de vapeur des hydrocarbures :
p =RT
v − b− aα
v (v + b)
avec :
a = 0.42747× R2T 2c
pc, b = 0.08664× RTc
pc(2.16)
α =[1 +
(0.48508 + 1.55171ω − 0.15613ω2
) (1− T 0.5
r
)]2(2.17)
où Tr est la température réduite : Tr = T/Tc et ω est le facteur accentrique de l'espèce chimique.
4.2.6 L'équation de Peng-Robinson
p =RT
v − b− aα
v2 + 2bv − b2
où R est la constante des gaz parfaits et :
a = 0.45724× R2T 2c
pc, b = 0.07780× RTc
pc(2.18)
α =[1 +
(0.37464 + 1.54226ω − 0.26992ω2
) (1− T 0.5
r
)]2(2.19)
où Tr est la température réduite et ω est le facteur accentrique.L'équation de Peng-Robinson a été proposée en 1976 et a les propriétés suivantes : les paramètres sont en termes propriétés critiques et du facteur accentrique, le modèle permet d'obtenir une précision acceptable près du point critique (c'est à dire lorsqueT ≃ Tc et p ≃ pc), particulièrement pour les calculs de coecient de compressibilité et de massevolumique du liquide,

36 Application des principes au fluide homogène
les lois de mélanges n'emploie qu'un simple paramètre d'interaction binaire, indépendant de latempérature, de la pression et de la composition,
l'équation peut être appliquée pour tout calcul de propriétés des uides dans les processus concer-nant le gaz naturel.
Dans les grandes lignes, l'équation de Peng-Robinson a des performances similaires à l'équation deSoave, mais est généralement supérieure pour la prédiction des masses volumiques des espèces, en parti-culier, celles qui ne sont pas polaires.
4.2.7 L'équation BWRS
L'équation d'état de Benedict-Webb-Rubin-Starling (BWRS) est une corrélation générale utilisantles constantes (8 à 11) des composés purs pour mieux prédire les équilibres de la phase vapeur. Elleest construite pour la prédiction des propriétés des mélanges d'hydrocarbures avec N2, H2 et H2S. Lesrésultats des prédictions des équilibres vapeur-liquide pour le gaz naturel liquéé (LNG), les gaz naturelssynthétiques (SNG) et les gaz de pétrole liquéés (LPG) sont excellents. Les valeurs des paramètres pourdiérentes substances peuvent être trouvées dans la référence [19].

4 Équations d'état 37
Annexe 1
1. Grandeurs résiduelles
a. L'énergie interne
On a vu que :
U(S, V )− Ugp(S, V ) =
∫ V
∞T 2 ∂
∂T
[ pT
]dV
=
∫ V
∞
[T∂p
∂T− p
]dV
On peut introduire le facteur de compressibilité Z =PV
RT:
p =ZRT
V⇒ ∂p
∂T=ZR
V+RT
V
∂Z
∂T
L'énergie interne résiduelle se met ainsi sous la forme :
U − UgpRT
= T
∫ V
∞
∂Z
∂T
dV
V
L'enthalpie résiduelle se calcule par H = U + pV , soit :
H(S, V ) = Ugp +RT 2
∫ V
∞
∂Z
∂T
dV
V+ pV
= Ugp +RT 2
∫ V
∞
∂Z
∂T
dV
V+ ZRT
Or Hgp = Ugp + pV = Ugp +RT , on en déduit :
H −Hgp
RT= T
∫ V
∞
∂Z
∂T
dV
V+ (Z − 1)
b. Application à l'équation de Van der Waals
Considérons l'équation d'état :
p =RT
V − b− a
V 2⇒ pV
RT= Z =
V
V − b− a
V RT
On en déduit :∂Z
∂T=
a
V RT 2
L'intégrale devient : ∫ V
∞
a
V 2RT 2dV =
[− a
V RT 2
]V∞
= − a
V RT 2
L'enthalpie résiduelle est alors :
H −Hgp
RT= − a
V RT+ (Z − 1) =
V
V − b− 2a
V RT− 1 =
b
V − b− 2a
V RT
c. L'enthalpie résiduelle en fonction de la pression
On peut calculer ces mêmes grandeurs en fonction de la pression p : on utilise la diérentielle del'enthalpie dH = TdS + V dp :
dS =1
T(dH − V dp)
=1
T
[∂H
∂pdp+
∂H
∂TdT
]− V
Tdp
=
[1
T
∂H
∂p− V
T
]dp+
1
T
∂H
∂TdT

38 Application des principes au fluide homogène
La condition de Cauchy implique :
∂
∂T
[1
T
∂H
∂p− V
T
]=
∂
∂p
[1
T
∂H
∂T
]On en déduit la relation suivante :
∂H
∂p= V − T
∂V
∂T
Soit :
H −Hgp =
∫ p
0
[V − T
∂V
∂T
]dp
Si on l'écrit en fonction du facteur de compressibilité Z :
∂V
∂p=∂(ZRT/p)
∂T=ZR
p+RT
p
(∂Z
∂T
)soit :
V − T∂V
∂T=ZRT
p− T
[ZR
p+RT
p
(∂Z
∂T
)]= −RT 2
(∂Z
∂T
)Donc :
H −Hgp
RT= −T
∫ p
0
[1
p
(∂Z
∂T
)]dp
On peut également introduire les grandeurs réduites :
Tr =T
Tc, pr =
p
pc
où Tc et pc sont respectivement la température critique et la pression critique. On en déduit :
H =H −Hgp
RTc= −T 2
r
∫ pr
0
[1
pRpc
(∂Z
∂Tr
)]dpr
Cette grandeur est calculée sur le diagramme (4.2.7).
d. L'entropie résiduelle en fonction de la pression
L'entropie s'écrit :
S − Sgp =
∫ p
0
[∂S
∂p−(∂S
∂p
)gp
]dp
Or∂S
∂p= −∂V
∂Tet
(∂S
∂p
)gp
= −Rp
D'où :
S − Sgp =
∫ p
0
[R
p− ∂V
∂T
]dp
ou en fonction de Z :
S − Sgp =
∫ p
0
[(1− Z)R
p− RT
p
(∂z
∂T
)]dp
En termes de grandeurs réduites :
S =S − Sgp
R=H −Hgp
RTRTc+
∫ pr
0
(1− Z)
prdpr
Cette grandeur est calculée sur le diagramme (4.2.7).

4 Équations d'état 39
e. Application
Du propane subit une détente de p1 =5.21 MPa et T1 =407 K à p2 =4.26 Mpa et T2 =370 K. Calculerl'échange thermique qui en résulte par unité de masse de propane.
La température critique du propane est Tc = 370 K et sa pression critique est pc = 4.26 MPa. On endéduit les températures et pressions réduites :
Tr1 = T1/Tc = 1.1, Tr2 = T1/Tc = 1.1
pr1 = p1/pc = 1.2, pr2 = 1.0
La masse molaire du propane est M = 44 g/mol. La capacité thermique à pression constante du propaneest cp = 1.6794 kJ/(kg.K).
La diérence d'enthalpie est :
h2 − h1 = h2,gp − h1,gp + (h2 − h2,gp) + (h1 − h1,gp)
= cp(T2 − T1)−R
MTc(H2 −H1)
= 1.6794× (370− 407) + 0.1885× 370× (1.45− 2.5)
= −135.37 kJ/kg
L'erreur que l'on fait si on suppose que le propane est un gaz parfait est de :
RTc(H2 −H1)
h2 − h1=
73.23
135.37= 54%
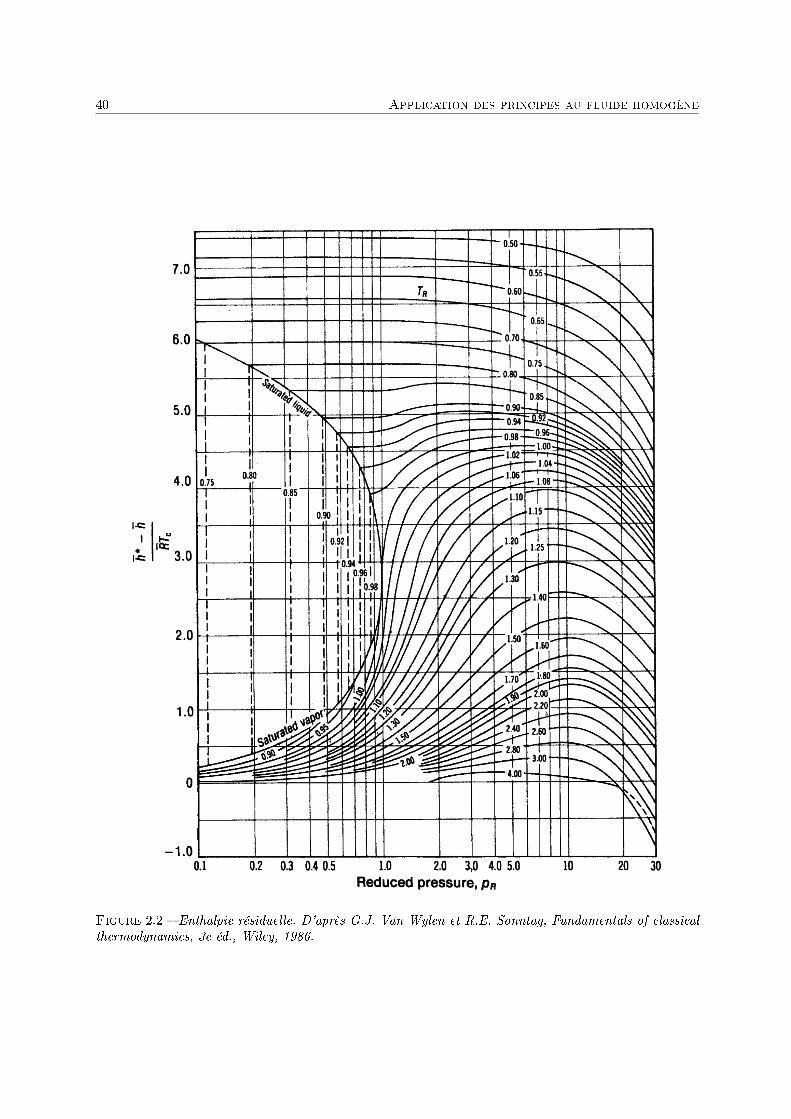
40 Application des principes au fluide homogène
Figure 2.2 Enthalpie résiduelle. D'après G.J. Van Wylen et R.E. Sonntag, Fundamentals of classicalthermodynamics, 3e éd., Wiley, 1986.
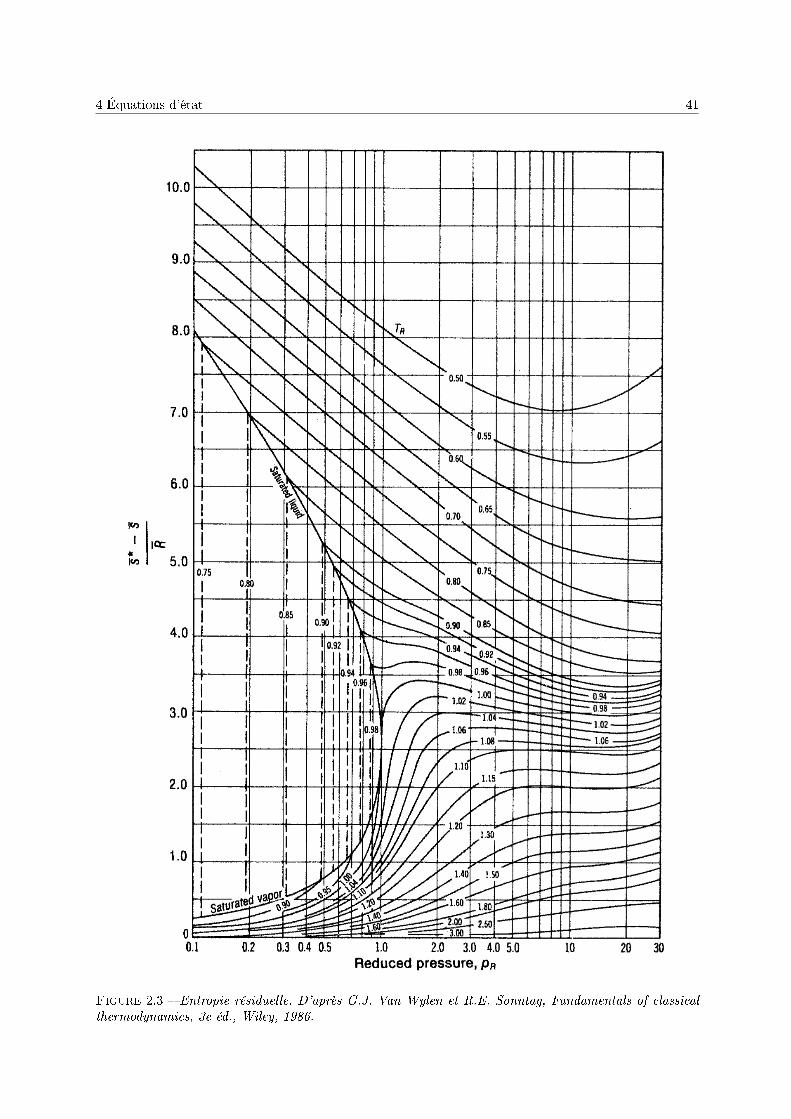
4 Équations d'état 41
Figure 2.3 Entropie résiduelle. D'après G.J. Van Wylen et R.E. Sonntag, Fundamentals of classicalthermodynamics, 3e éd., Wiley, 1986.

42 Application des principes au fluide homogène

Chapitre 3
Le corps pur sous différentes
phases
1 Dénitions
On appelle phase, toute partie homogène, possédant des propriétés macroscopiques particulières(indice de réfraction, dureté, viscosité, etc.), séparée des autres parties du système par une surface dénie.
On dit qu'il y a changement (ou transition) de phase, si en modiant continûment une contraintedu système, le système passe d'une phase à l'autre (de façon continue ou discontinue). Il y a alorsmodication des propriétés physiques.
2 Description phénoménologique des changements d'état
La matière peut se trouver habituellement sous trois phases : solide, liquide. Cependant, dans chacunede ces familles peuvent éventuellement exister plusieurs phases (surtout pour les solides). En plus de cesphases, peuvent exister des états intermédiaires : mésomorphe (combinant les propriétés du solide etdu liquide comme les cristaux liquides) et métastable (phase existante dans des conditions où elle estnormalement instable). On peut également considérer le plasma (mélange d'ions, atomes et d'électrons)comme une phase de la matière.
2.1 Les phases habituelles
On peut distinguer les phases solides, liquide et gaz par l'ordre sous lequel on trouve les moléculesqui les composent au niveau microscopique. On dénit pour cela la fonction de distribution radiale oufonction de corrélation de paires g(r) : à partir d'une molécule, on détermine le nombre de molécules quel'on rencontre en fonction de la distance à laquelle on s'éloigne (g. 3.1).
r
Figure 3.1 Dénition de la fonction de distribution radiale g(r)
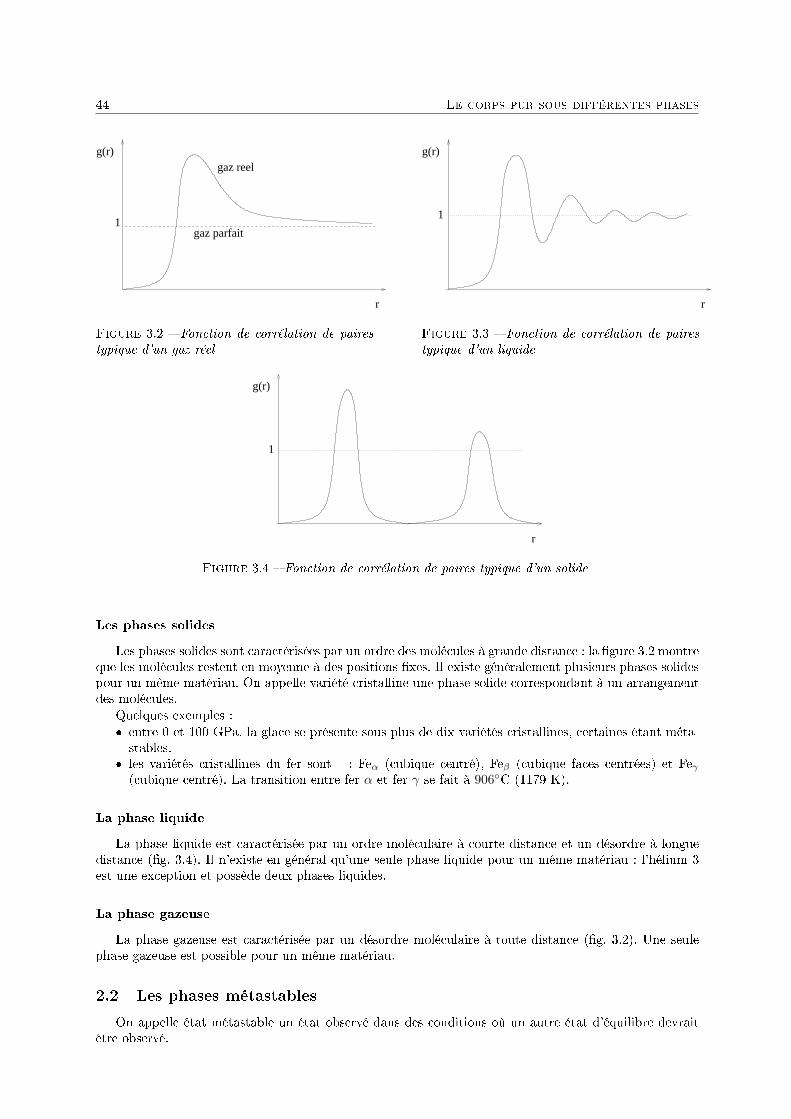
44 Le corps pur sous différentes phases
gaz parfait
g(r)
r
gaz reel
1
Figure 3.2 Fonction de corrélation de pairestypique d'un gaz réel
g(r)
r
1
Figure 3.3 Fonction de corrélation de pairestypique d'un liquide
1
g(r)
r
Figure 3.4 Fonction de corrélation de paires typique d'un solide
Les phases solides
Les phases solides sont caractérisées par un ordre des molécules à grande distance : la gure 3.2 montreque les molécules restent en moyenne à des positions xes. Il existe généralement plusieurs phases solidespour un même matériau. On appelle variété cristalline une phase solide correspondant à un arrangementdes molécules.
Quelques exemples : entre 0 et 100 GPa, la glace se présente sous plus de dix variétés cristallines, certaines étant méta-stables.
les variétés cristallines du fer sont : Feα (cubique centré), Feβ (cubique faces centrées) et Feγ(cubique centré). La transition entre fer α et fer γ se fait à 906C (1179 K).
La phase liquide
La phase liquide est caractérisée par un ordre moléculaire à courte distance et un désordre à longuedistance (g. 3.4). Il n'existe en général qu'une seule phase liquide pour un même matériau : l'hélium 3est une exception et possède deux phases liquides.
La phase gazeuse
La phase gazeuse est caractérisée par un désordre moléculaire à toute distance (g. 3.2). Une seulephase gazeuse est possible pour un même matériau.
2.2 Les phases métastables
On appelle état métastable un état observé dans des conditions où un autre état d'équilibre devraitêtre observé.

3 L'équation de Van der Waals comme modèle pour le changement d'état 45
Ainsi, partant d'un liquide, il est parfois possible de maintenir cet état en dessous de la températurede solidication de la substance : on a alors un liquide surfondu 1, correspondant à un état métastable.
Dans le cas de la silice, la transition vitreuse, correspondant à une solidication métastable du liquide,permet d'obtenir un verre. On a alors un état solide non organisé, c'est à dire non cristallin, qui necorrespond pas à un état thermodynamique d'équilibre.
2.3 Vocabulaire des changements de phase
La solidication correspond au passage de la phase liquide vers la phase solide. La fusion correspond au passage de la phase solide à la phase liquide. La vaporisation correspond au passage de la phase liquide vers la phase gazeuse. La liquéfaction est le passage de la phase gazeuse vers la phase liquide. La sublimation est le passage de la phase solide à la phase gazeuse. La condensation est le passage de la phase gazeuse à la phase solide.On distinguera deux cas de vaporisation : l'évaporation qui s'eectue à toute température, qui est un phénomène de surface, où le liquidepasse sous la forme gazeuse dans un autre gaz. Par exemple, quelque soit la température, l'eauliquide contenue dans un récipient va s'évaporer dans l'air. Ce problème ne concerne donc plus uncorps pur, mais un mélange (voir chapitre suivant).
l'ébullition qui est le passage du liquide à l'état gazeux dans l'ensemble de son volume, et non plusseulement à travers sa surface de séparation. Ce phénomène arrive à une température donnée pourune pression xée.
2.4 Le diagramme pV T
Sur un diagramme pV T (g. 3.5), on distingue : le liquide comprimé, le liquide saturé, la vapeur saturée, le mélange liquide-vapeur, la vapeur sur-chauée,
la température de saturation, la pression de saturation, courbe de saturation, courbe d'ébullition(apparition des premières bulles de gaz), courbe de rosée (apparition des premières gouttes deliquide),
la chaleur latente de fusion ou d'évaporation, le point critique : point au dessus duquel il n'y a plus de séparation liquide vapeur, le point triple : pression et température correspondant à la co-existance des trois phases (solide,liquide et vapeur). La proportion de solide, liquide et vapeur ne dépendent alors que du volume.
les courbes spinodales (g. 3.7) sont les positions des états métatables possibles.
Remarque : la zone de coexistence du liquide et de la vapeur est un domaine fermé, alors que la zone decoexistence liquide-solide est innie. Ceci s'explique par la notion d'ordre : la diérence d'ordonnancementdes molécules est quantitative entre le gaz et le liquide, alors qu'elle est de nature réellement diérenteentre le liquide et le gaz.
Exemple : le point triple de l'eau se situe en pT = 611 Pa et TT = 273, 16 K. L'eau peut alors setrouver simultanément dans l'état solide, liquide et gazeux.
3 L'équation de Van der Waals comme modèle pour le change-ment d'état
L'équation d'état de Van der Waals est l'équation la plus simple permettant de calculer l'équilibreliquide/vapeur d'un uide. Elle comprend deux paramètres a et b modélisant les interactions entre molé-cules. Son expression est :
(p+a
v2)(v − b) = RT
1. On peut refroidir de l'eau liquide en laboratoire aux alentours de -40°C, c'est à dire un peu au dessous de la températurede l'eau surfondue présente dans certains nuages de l'atmosphère [20].
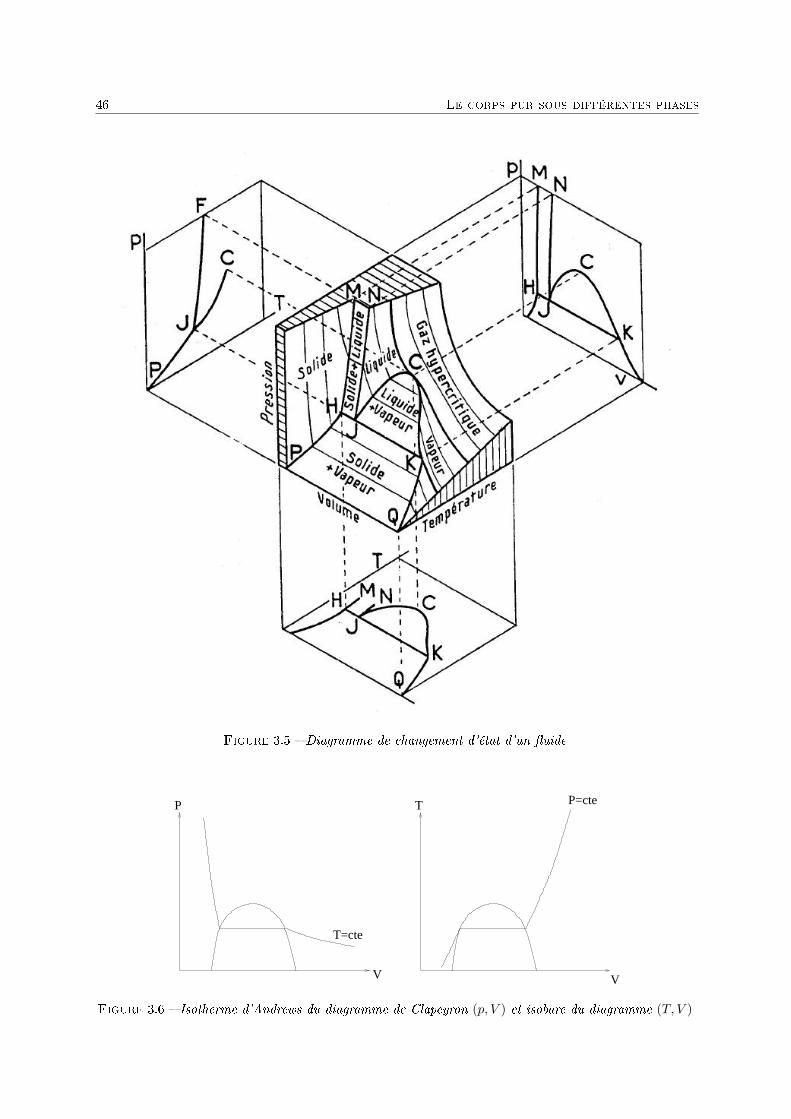
46 Le corps pur sous différentes phases
Figure 3.5 Diagramme de changement d'état d'un uide
V
T P=cteP
V
T=cte
Figure 3.6 Isotherme d'Andrews du diagramme de Clapeyron (p, V ) et isobare du diagramme (T, V )
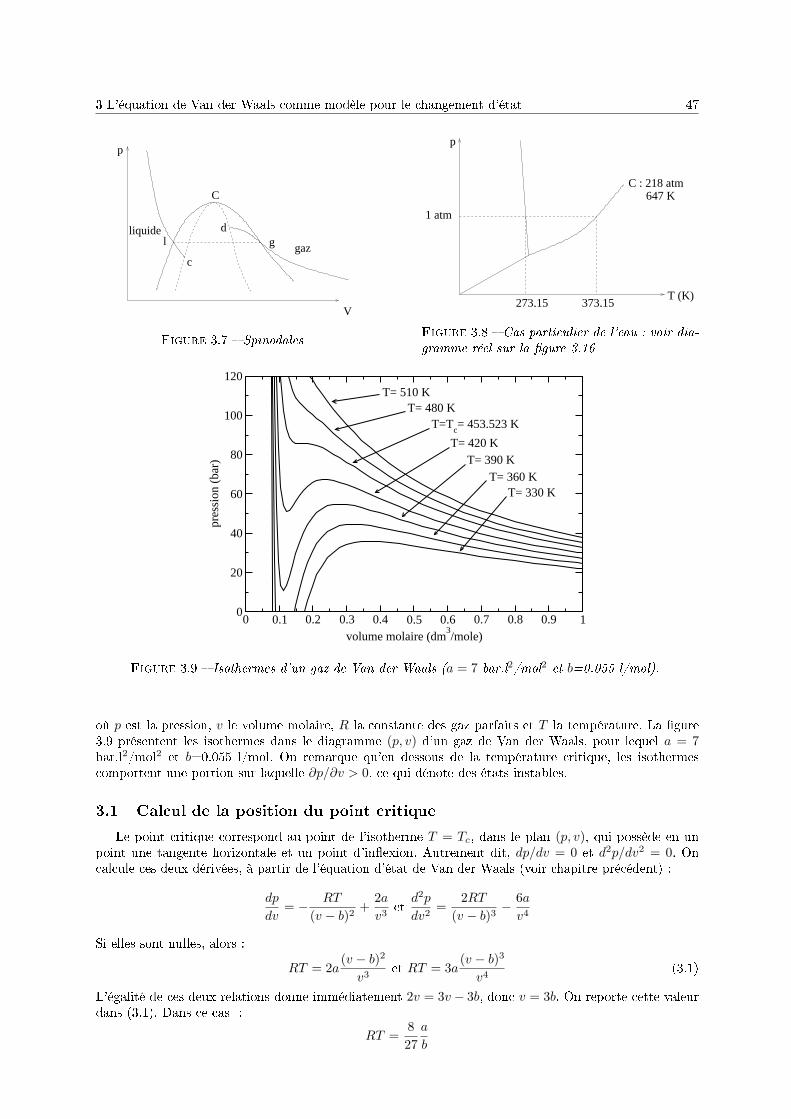
3 L'équation de Van der Waals comme modèle pour le changement d'état 47
gaz
p
V
C
l g
c
dliquide
Figure 3.7 Spinodales
p
273,15 373,15T (K)
1 atm
C : 218 atm 647 K
Figure 3.8 Cas particulier de l'eau : voir dia-gramme réel sur la gure 3.16
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1volume molaire (dm
3/mole)
0
20
40
60
80
100
120
pres
sion
(ba
r)
T= 330 KT= 360 K
T= 390 KT= 420 K
T=Tc= 453.523 K
T= 480 KT= 510 K
Figure 3.9 Isothermes d'un gaz de Van der Waals (a = 7 bar.l2/mol2 et b=0.055 l/mol).
où p est la pression, v le volume molaire, R la constante des gaz parfaits et T la température. La gure3.9 présentent les isothermes dans le diagramme (p, v) d'un gaz de Van der Waals, pour lequel a = 7bar.l2/mol2 et b=0.055 l/mol. On remarque qu'en dessous de la température critique, les isothermescomportent une portion sur laquelle ∂p/∂v > 0, ce qui dénote des états instables.
3.1 Calcul de la position du point critique
Le point critique correspond au point de l'isotherme T = Tc, dans le plan (p, v), qui possède en unpoint une tangente horizontale et un point d'inexion. Autrement dit, dp/dv = 0 et d2p/dv2 = 0. Oncalcule ces deux dérivées, à partir de l'équation d'état de Van der Waals (voir chapitre précédent) :
dp
dv= − RT
(v − b)2+
2a
v3etd2p
dv2=
2RT
(v − b)3− 6a
v4
Si elles sont nulles, alors :
RT = 2a(v − b)2
v3et RT = 3a
(v − b)3
v4(3.1)
L'égalité de ces deux relations donne immédiatement 2v = 3v − 3b, donc v = 3b. On reporte cette valeurdans (3.1). Dans ce cas :
RT =8
27
a
b

48 Le corps pur sous différentes phases
gaz Tc (K) pcvc/(RTc)Ne 44.8 0.305Ar 150.7 0.292Kr 209.9 0.290Xe 289.8 0.288N2 126.0 0.292O2 154.0 0.292CO 133.0 0.294CH4 190.3 0.289
Table 3.1 Validité de l'approximation de Van der Waals pour quelques gaz
On reporte RT et v dans l'équation de Van der Waals, pour obtenir la pression :
p =RT
v − b− a
v2=
a
27b2
On en déduit les coordonnées du point critique : Tc =8a
27bR, pc =
a
27b2, vc = 3b. Il existe une relation
entre Tc, vc et pc :pcvcRTc
=3
8= 0.375. Le respect de cette relation permet d'évaluer la pertinence du
modèle de gaz de Van der Waals pour des gaz réels (table 3.1).
Loi des états correspondants : dénissons les coordonnées réduites T ∗ = T/Tc, v∗ = v/vc etp∗ = p/pc. On obtient :
(p∗ +3
v∗2)(3v∗ − 1) = 8T ∗
3.1.1 La transition liquide-gaz
Lorsque T < Tc, l'isotherme comporte une partie instable. L'énergie libre est le potentiel thermody-namique pour une transformation à température constante. Pour le gaz de Van der Waals, son expressionvaut :
f(T, v) = CvT (1− log T ) +a
v−RT log(v − b) + u0 − Ts0
où f est l'énergie libre molaire, Cv est la chaleur massique à volume constant, u0 et s0 sont des constantes.Ceci peut s'écrire :
f(T, v) =a
v−RT log(v − b) +K(T )
où K(T ) est une constante dépendant de la température T . L'énergie libre est représentée sur la gure3.10, pour T = 390 K et pour les mêmes coecients a et b que pour la gure 3.9, simultanément àl'isotherme dans le diagramme (p, v). La courbe représentative de l'énergie libre présente deux pointsd'inexion A et B, dénis par :
∂2f
∂v2= 0
Sachant que ∂f/∂v = −p, ces points d'inexion correspondent à des dérivées nulles dans le plan (p, v).Entre les points d'inexion, l'énergie libre est concave, alors qu'elle est convexe à l'extérieur. Autrementdit, le système homogène est intrinsèquement instable entre les volumes molaires vA et vB . Il va chercherun nouvel état stable en minimisant son énergie libre. Pour cela, il se scinde en deux parties (deuxphases), une de volume molaire vC et l'autre de volume molaire vD, de sorte que l'énergie libre dumélange hétérogène est plus faible que l'énergie libre du mélange homogène (g. 3.11), avec un volumemolaire global identique :
f ′M = fD + (fC − fD)vM − vDvC − vD
< fM
Les points C et D sont les points de la courbe représentative de f , situés de part et d'autre de la portionconcave et ayant la même tangente. Les dérivées de l'énergie libre ∂f/∂v|C au point C et ∂f/∂v|D au
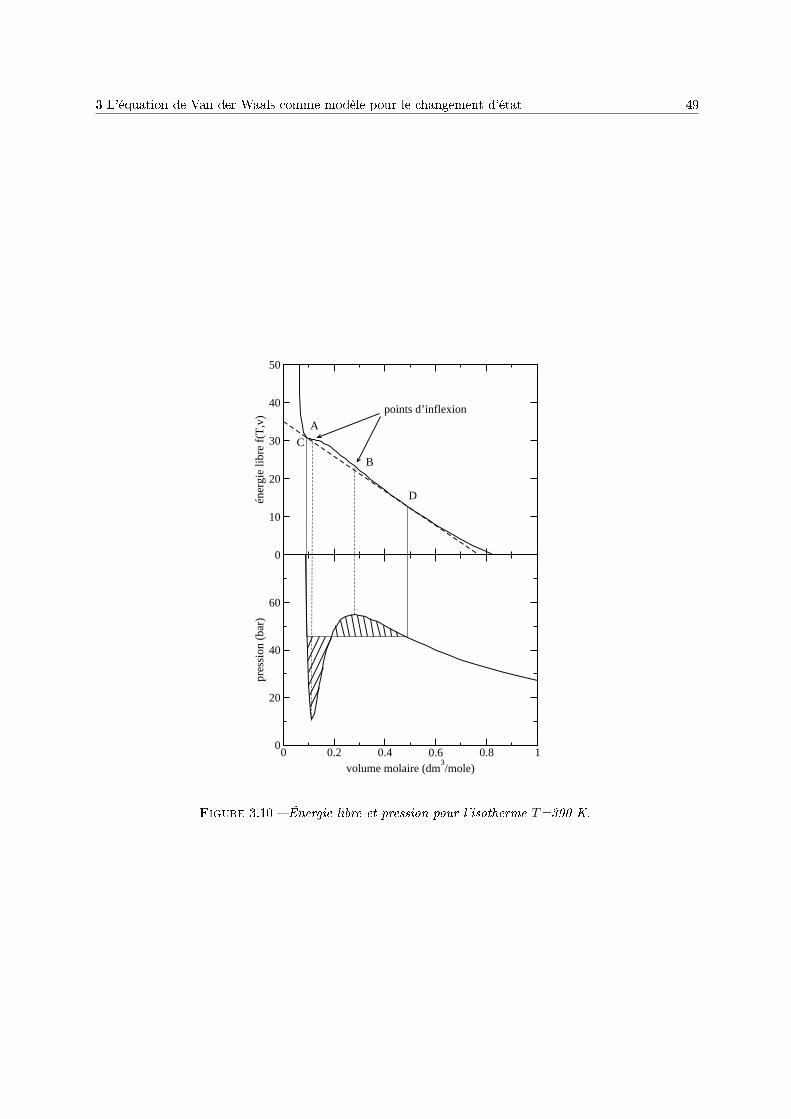
3 L'équation de Van der Waals comme modèle pour le changement d'état 49
0 0.2 0.4 0.6 0.8 1volume molaire (dm
3/mole)
0
20
40
60
pres
sion
(ba
r)
0
10
20
30
40
50
éner
gie
libre
f(T
,v) points d’inflexion
A
B
C
D
Figure 3.10 Énergie libre et pression pour l'isotherme T=390 K.

50 Le corps pur sous différentes phases
v
C
D
f(v)
f’
f
f
v v v
fCM
M
D
C M D
M’B
MA
Figure 3.11 Minimisation de l'énergie libre
point D sont dont identiques et égales à une pression p0. Les points C et D appartiennent à la mêmedroite de pente −p0 :
fC − fD = −p0(vC − vD) (3.2)
D'autre part, la diérentielle de l'énergie libre s'écrit, à température constante, df = −p dv, soit
fC − fD = −∫ vC
vD
pdv
En combinant ces deux équations, on obtient :∫ vD
vC
(p− p0)dv = 0
ce qui signie que la pression p0 est dénie par l'égalité des aires hachurées dans diagrammes (p, v) de lagure 3.10. On remarque également, l'équation (3.2) se met sous la forme :
fC + p0vC = fD + p0vD ou gC = gD
Autrement dit, il y égalité des enthalpies libres molaires ou potentiels chimiques entre les deux phases.Remarque : lorsque le point M se trouve entre C et A, l'énergie libre est localement convexe. Il s'agit
cependant d'un état métastable. En eet, sous l'action de perturbations, le système va encore se diviseren deux phases, de volumes molaires vC et vD.
Finalement, on observe : des états stables à gauche de C et à droite de D, des états métastables sur les portions CA et BD, des états instables sur la portion AB.
Le seul état stable sur toute la portion CD est un système comportant deux phases à la température T0et à la pression p0.
4 Relations thermodynamiques du changement d'état
4.1 Équilibre entre les phases d'un corps pur
Un uide est nécessairement décrit par les couples de variables intensives/extensives suivants : (T, S),(−p, V ) et (µ,N). A l'équilibre, on a donc :
T1 = T2 (3.3)
p1 = p2 (3.4)
µ1 = µ2 (3.5)

4 Relations thermodynamiques du changement d'état 51
Démonstration : soit deux corps en équilibre dans un système isolé. On a donc le premier principe :
U = U1 + U2 = cte (3.6)
V = V1 + V2 = cte (3.7)
N = N1 +N2 = cte (3.8)
Et comme il s'agit d'un état d'équilibre :
S maximal →(∂S
∂Xi
)Xk =i
= 0
Par conséquent, prenons X1 = U1 :
∂S
∂U1=∂S1
∂U1+∂S2
∂U1=∂S1
∂U1+∂S2
∂U2
∂U2
∂U1
or :∂U2
∂U1=
∂U
∂U1− ∂U1
∂U1= 0− 1 = −1
donc :∂S
∂U1=∂S1
∂U1− ∂S2
∂U2= 0
c'est à dire :
∂S1
∂U1=
∂S2
∂U2(3.9)
1
T1=
1
T2(3.10)
De même, pour X1 = V1, on obtient p1 = p2 et pour X1 = N1, µ1 = µ2.
4.2 L'équation de Clapeyron
On cherche une relation entre la chaleur latente de vaporisation Lv et les variations de la pression devapeur saturante ps = ps(T ).
Pour cela, considérons un corps pur dans un état d'équilibre sous les phases liquide (l) et vapeur (v)à une température T et une pression p xées, et possédant une masse constante. Il s'agit en fait d'unéquilibre dynamique, des particules pouvant passer d'une phase à l'autre. Le nombre de moles nl et nvdans chacune des phase varient en permanence, mais gardent la même valeur moyenne qui est la seulegrandeur macroscopique accessible. D'autre part, la somme nl + nv reste rigoureusement constante.
A l'équilibre, les enthalpies libres molaires (potentiels chimiques) de chaque phase, µl et µv sont égales,sinon l'ensemble des molécules passeraient dans la phase ayant le potentiel chimique le plus faible. Larelation :
µl(T, p) = µv(T, p)
fournit donc une relation entre T et p. Cette relation, que l'on note p = ps(T ), est une loi d'équilibre entreles deux phases. Écrivons l'égalité entre les potentiels chimiques en (T, P ) et en un état (T + dT, p+ dp)très voisin (g. 3.12) :
µl(T, p) = µv(T, p) et µl(T + dT, p+ dp) = µv(T + dT, p+ dp)
En soustrayant, on obtient :
dµv = µv(T + dT, p+ dp)− µv(T, p) = µl(T + dT, p+ dp)− µl(T, p) = dµl
c'est à dire : (∂µv∂T
)p
dT +
(∂µv∂p
)T
dp =
(∂µl∂T
)p
dT +
(∂µl∂p
)T
dp (3.11)
−svdT + vvdp = −sldT + vldp (3.12)
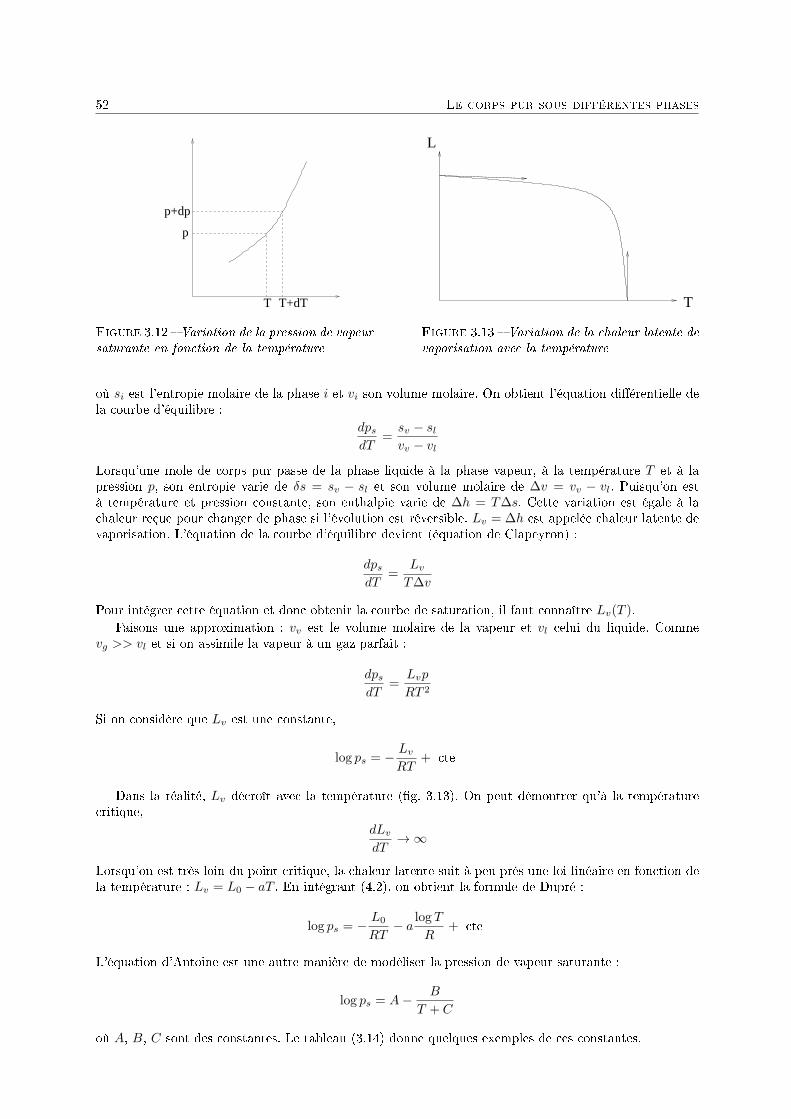
52 Le corps pur sous différentes phases
T T+dT
p
p+dp
Figure 3.12 Variation de la pression de vapeursaturante en fonction de la température
L
T
Figure 3.13 Variation de la chaleur latente devaporisation avec la température
où si est l'entropie molaire de la phase i et vi son volume molaire. On obtient l'équation diérentielle dela courbe d'équilibre :
dpsdT
=sv − slvv − vl
Lorsqu'une mole de corps pur passe de la phase liquide à la phase vapeur, à la température T et à lapression p, son entropie varie de δs = sv − sl et son volume molaire de ∆v = vv − vl. Puisqu'on està température et pression constante, son enthalpie varie de ∆h = T∆s. Cette variation est égale à lachaleur reçue pour changer de phase si l'évolution est réversible. Lv = ∆h est appelée chaleur latente devaporisation. L'équation de la courbe d'équilibre devient (équation de Clapeyron) :
dpsdT
=LvT∆v
Pour intégrer cette équation et donc obtenir la courbe de saturation, il faut connaître Lv(T ).Faisons une approximation : vv est le volume molaire de la vapeur et vl celui du liquide. Comme
vg >> vl et si on assimile la vapeur à un gaz parfait :
dpsdT
=Lvp
RT 2
Si on considère que Lv est une constante,
log ps = − LvRT
+ cte
Dans la réalité, Lv décroît avec la température (g. 3.13). On peut démontrer qu'à la températurecritique,
dLvdT
→ ∞
Lorsqu'on est très loin du point critique, la chaleur latente suit à peu près une loi linéaire en fonction dela température : Lv = L0 − aT . En intégrant (4.2), on obtient la formule de Dupré :
log ps = − L0
RT− a
log T
R+ cte
L'équation d'Antoine est une autre manière de modéliser la pression de vapeur saturante :
log ps = A− B
T + C
où A, B, C sont des constantes. Le tableau (3.14) donne quelques exemples de ces constantes.
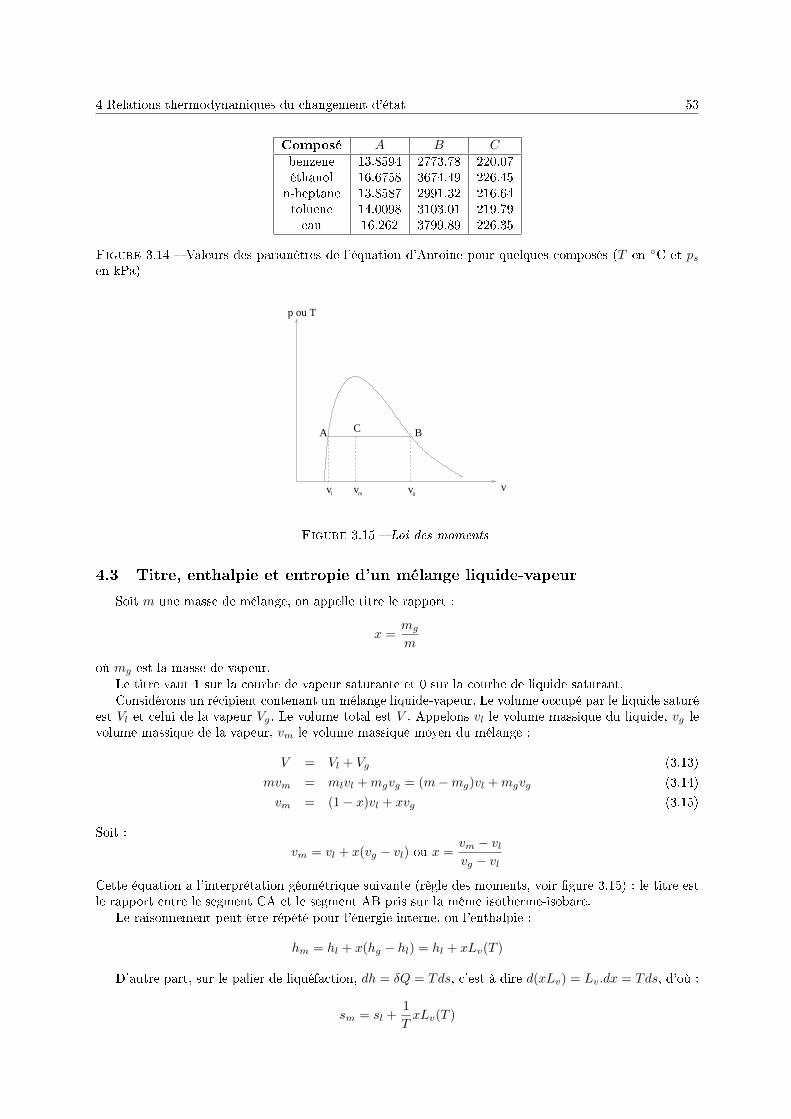
4 Relations thermodynamiques du changement d'état 53
Composé A B Cbenzene 13.8594 2773.78 220.07éthanol 16.6758 3674.49 226.45
n-heptane 13.8587 2991.32 216.64toluene 14.0098 3103.01 219.79eau 16.262 3799.89 226.35
Figure 3.14 Valeurs des paramètres de l'équation d'Antoine pour quelques composés (T en C et psen kPa)
v
p ou T
A BC
vvvl m g
Figure 3.15 Loi des moments
4.3 Titre, enthalpie et entropie d'un mélange liquide-vapeur
Soit m une masse de mélange, on appelle titre le rapport :
x =mg
m
où mg est la masse de vapeur.Le titre vaut 1 sur la courbe de vapeur saturante et 0 sur la courbe de liquide saturant.Considérons un récipient contenant un mélange liquide-vapeur. Le volume occupé par le liquide saturé
est Vl et celui de la vapeur Vg. Le volume total est V . Appelons vl le volume massique du liquide, vg levolume massique de la vapeur, vm le volume massique moyen du mélange :
V = Vl + Vg (3.13)
mvm = mlvl +mgvg = (m−mg)vl +mgvg (3.14)
vm = (1− x)vl + xvg (3.15)
Soit :
vm = vl + x(vg − vl) ou x =vm − vlvg − vl
Cette équation a l'interprétation géométrique suivante (règle des moments, voir gure 3.15) : le titre estle rapport entre le segment CA et le segment AB pris sur la même isotherme-isobare.
Le raisonnement peut être répété pour l'énergie interne, ou l'enthalpie :
hm = hl + x(hg − hl) = hl + xLv(T )
D'autre part, sur le palier de liquéfaction, dh = δQ = Tds, c'est à dire d(xLv) = Lv.dx = Tds, d'où :
sm = sl +1
TxLv(T )
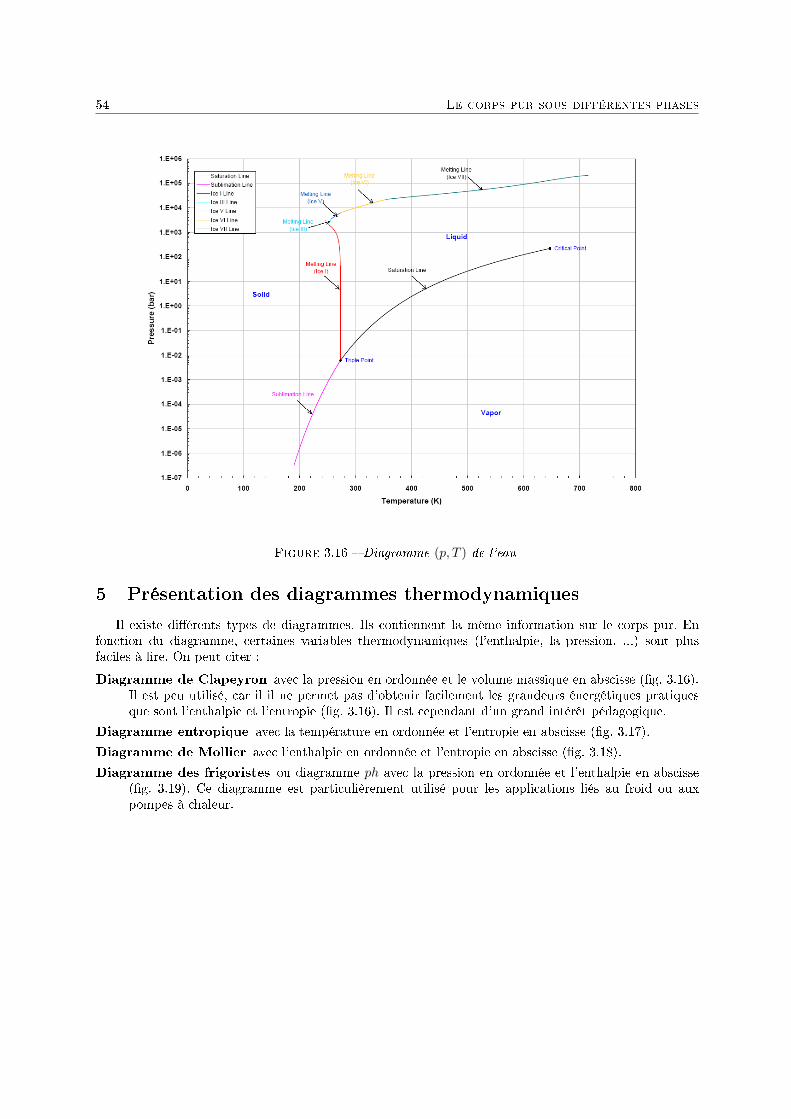
54 Le corps pur sous différentes phases
Figure 3.16 Diagramme (p, T ) de l'eau
5 Présentation des diagrammes thermodynamiques
Il existe diérents types de diagrammes. Ils contiennent la même information sur le corps pur. Enfonction du diagramme, certaines variables thermodynamiques (l'enthalpie, la pression, ...) sont plusfaciles à lire. On peut citer :
Diagramme de Clapeyron avec la pression en ordonnée et le volume massique en abscisse (g. 3.16).Il est peu utilisé, car il il ne permet pas d'obtenir facilement les grandeurs énergétiques pratiquesque sont l'enthalpie et l'entropie (g. 3.16). Il est cependant d'un grand intérêt pédagogique.
Diagramme entropique avec la température en ordonnée et l'entropie en abscisse (g. 3.17).
Diagramme de Mollier avec l'enthalpie en ordonnée et l'entropie en abscisse (g. 3.18).
Diagramme des frigoristes ou diagramme ph avec la pression en ordonnée et l'enthalpie en abscisse(g. 3.19). Ce diagramme est particulièrement utilisé pour les applications liés au froid ou auxpompes à chaleur.
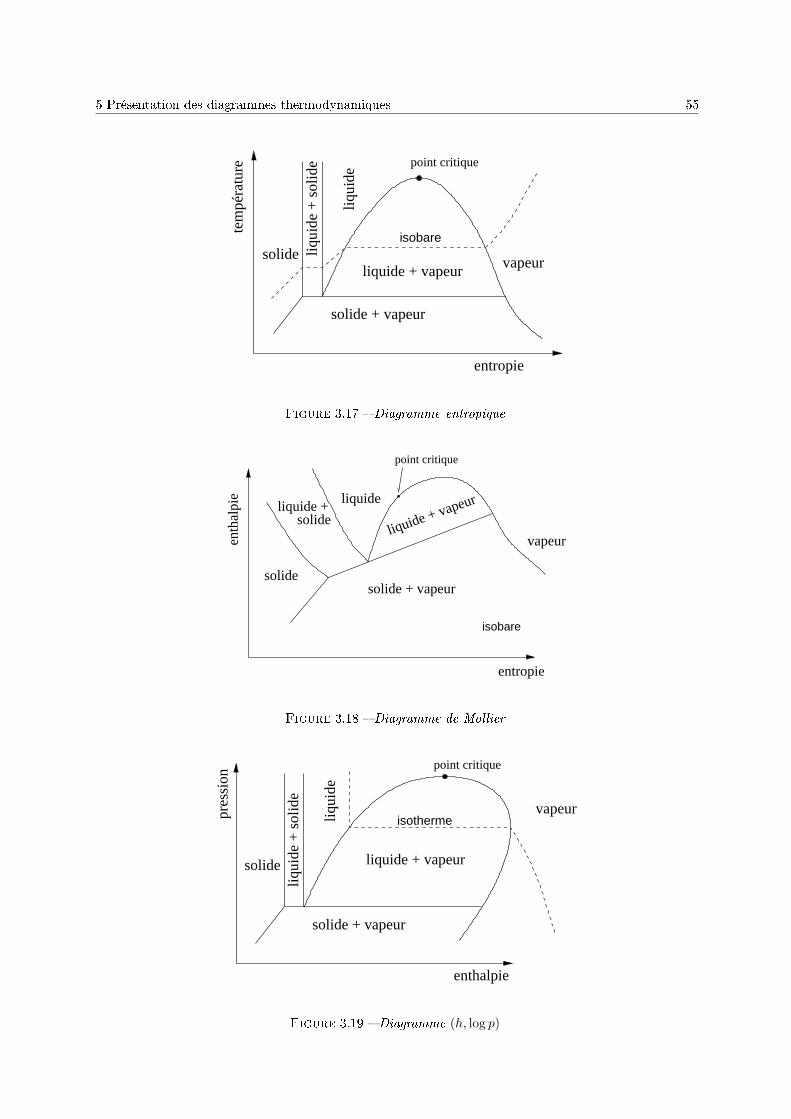
5 Présentation des diagrammes thermodynamiques 55
point critique
solide
solide + vapeur
liqui
de
liquide + vapeur
tem
péra
ture
entropie
liqui
de +
sol
ide
vapeur
isobare
Figure 3.17 Diagramme entropique
point critique
solide + vapeur
vapeur
solide
liquide + solide
liquide
liquide + vapeur
isobare
entropie
enth
alpi
e
Figure 3.18 Diagramme de Mollier
point critique
enthalpie
pres
sion
liquide + vapeursolide
solide + vapeur
liqui
de +
sol
ide
liqui
de
vapeurisotherme
Figure 3.19 Diagramme (h, log p)

56 Le corps pur sous différentes phases

Chapitre 4
Mélange en l'absence de réaction
chimique
1 Introduction
1.1 Variance d'un système
La variance d'un système est le nombre de variables indépendantes caractérisant ce système à l'équi-libre.
Soit φ le nombre de phases d'un système contenant c constituants. On peut utiliser les fractionsmolaires x(k)i pour représenter la composition de chaque phase, avec :
x(k)i =
n(k)i
n(k)avec n(k) =
c∑i=1
n(k)i
où n(k)i est le nombre de moles du constituants i sous la phase k et n(k) le nombre total de moles dans laphase k. Le système est alors décrit par c.φ fractions molaires. On ajoute la pression et la température,on obtient c.φ+ 2 variables.
Comptons le nombre de relations entre ces variables : pour chaque phase k, la somme des fractionsmolaires vaut 1 : ∑
i
x(k)i = 1
soit φ relations.Il y a égalité des potentiels chimiques, c'est à dire que pour chaque constituant :
µ(1)i = µ
(2)i = . . . = µ
(φ)i
soit φ− 1 relations par constituant, c'est à dire c(φ− 1) relations au total.Le nombre de variables indépendantes est donc :
ν = (c.φ+ 2)− φ− c(φ− 1)
D'où la règle des phases de Gibbs :
ν = c+ 2− φ
Application au corps pur sous plusieurs phases : si deux phases coexistent dans le système,la variance (nombre de degrés de liberté) vaut 1 et, par exemple, la seule variable T sut à décrirele système. La pression est donc une fonction de T . La région de coexistence des deux phases est parconséquent localisée dans un diagramme (P, T ) par une courbe d'équation : P = Peq(T ).
Si le corps apparaît sous trois phases, alors la variance tombe à zéro : il ne peut y avoir coexistencedes trois phases que pour une seule valeur de p, T et V , appelée point triple.

58 Mélange en l'absence de réaction chimique
1.2 Cas particulier de l'équilibre liquide/vapeur
Un mélange multi-constituants atteint un état d'équilibre liquide-vapeur quand le potentiel chimiquede chaque espèce i est le même dans les phases liquide et vapeur :
µi(liq) = µi(vap)
La résolution d'un problème d'équilibre entre phases passe, dans le cas général, par le calcul des potentielschimiques de tous les constituants dans les phases en présence.
2 Potentiel chimique d'un gaz
2.1 Le gaz parfait
Pour un gaz parfait, la diérentielle de l'enthalpie libre molaire s'écrit :
dg = dµ = −sdT + vdp
où s est l'entropie molaire et v le volume molaire. On a donc :
∂µ
∂p= v
Si le gaz est parfait, pv = RT , donc :∂µ
∂p=RT
p
On intègre et on obtient l'expression du potentiel chimique :
µ = µ0(T ) +RT logp
p0(4.1)
où µ0(T ) est le potentiel standard du gaz parfait à la pression de référence p0.
2.2 Mélange de gaz parfaits
On considère désormais un mélange de gaz parfaits Gi. Lorsque le mélange de gaz parfaits est lui-même un gaz parfait, on dit que le mélange est idéal. Cela signie qu'il n'y a aucune interaction entre lesmolécules des diérents gaz composant le mélange.
On dénit la pression partielle pi du gaz Gi par la pression qui existerait dans le volume si le gaz Giétait seul. Autrement dit,
pi =NiRT
V
où Ni est le nombre de moles de Gi.
Pression partielle d'un gaz parfait : dans le cas d'un mélange idéal de gaz parfaits, on a :
p =NRT
V=
∑iNiRT
V
On en déduit la loi de Dalton :
p =∑i
pi ou pi =NiNp = xip
où xi est la fraction molaire du gaz Gi.

2 Potentiel chimique d'un gaz 59
Potentiel chimique d'un gaz dans un mélange idéal de gaz parfaits : reprenons la formediérentielle de l'enthalpie libre, qui s'écrit dans ce cas :
dG =∑i
µidNi + V dp− SdT
La diérentielle dG est totale. On peut donc appliquer le critère de Cauchy et on obtient l'équationsuivante, qui est, par ailleurs, une des relations de Maxwell :
∂µi∂p
=∂V
∂Ni
Sachant que le mélange est un gaz parfait : pV =∑iNiRT , donc :
∂µi∂p
=RT
p
En multipliant chaque membre par 1/xi, on obtient :
∂µi∂pi
=RT
pi
Soit en intégrant :
µi(T, p) = µ0i (T ) +RT log
pip
Grandeurs de mélange : on considère n gaz parfaits Ai, pris à la température T et à la pression p,que l'on mélange et que l'on ramène à la température T et la pression p.
L'enthalpie libre de mélange est :
∆Gm =∑i
µini −∑i
µ0ini =
∑niRT log xi
Cette variation est négative. On remarque que ∆Gm/T ne dépend pas de la température. Autrement dit :
∂
∂T
(∆GmT
)= 0
−∆Hm
T 2= 0
Autrement dit, l'enthalpie de mélange est nulle : le mélange idéal se fait sans échange de chaleur. On a :
∆Gm = ∆Hm − T∆Sm
On en déduit l'entropie de mélange :
∆Sm = −(∂∆Gm∂T
)p,ni
= −R∑i
ni log xi
La variation de volume est :
∆Vm =
(∂∆Gm∂p
)T,ni
= 0
Le mélange idéal se fait sans variation de volume.
2.3 Gaz réels
Dans le cas des gaz réels, on choisit de conserver la même forme pour l'expression du potentielchimique, en écrivantl'une des relations suivantes :
µi = µ0i (T ) +RT log γi
pip0
(4.2)
µi = µ0i (T ) +RT log
fip0
(4.3)

60 Mélange en l'absence de réaction chimique
où fi est la fugacité 1 du constituant i et γi = fi/pi s'appelle coecient de fugacité. Ce coecient est telque :
limp→0
γi = 1
µ0i (T ) est le potentiel chimique du gaz lorsque la fugacité est égale à la pression de référence p0 (en
général, 1 bar).La fugacité mesure l'écart à l'idéalité du mélange :
µi = µ0i (T ) +RT
fip0
µideali = µ0i (T ) +RT
pip0
donc :
fi = pi exp
[µi − µideali
RT
]
Exercice : montrer que pour un gaz de Van der Waals, on a :
log f = logRT
V − b+
b
V − b− 2a
RTV
3 Potentiel chimique en phase liquide
3.1 Corps pur
On reprend la forme diérentielle de l'enthalpie libre dG = −SdT + V dp. On intègre à températurexée : G(T, P ) = G0(T ) +
∫ pp0V dp. Le volume est à peu près constant : V ≃ V0. Donc :
G(T, p) = G0(T ) + V0(p− p0) c'est à dire µ(T, p) = µ0(T ) + v0(p− p0)
où v0 est le volume molaire.
Remarques : on aurait pu à nouveau utiliser les relations de Maxwell. la variation du potentiel chimique avec la pression est faible : v0(p− p0) ≪ µ0(T )
3.2 Mélange idéal
Dans ce cas, il n'y a pas d'interaction entre les composants. Le potentiel chimique s'écrit alors, pourle composant i, de volume molaire vi, à la concentration xi (nombre de moles du composant divisé parle nombre total de moles) :
µi(x, T, p) = µi(corps pur, T, p) +RT log xi (4.4)
µi(x, T, p) = µ0i (corps pur, T, p0) + vi(p− p0) +RT log xi (4.5)
3.3 Mélange réel
On pose :µi(T, p) = µ0
i (T, p) + vi(p− p0) +RT log ai
où ai est l'activité : ai = γixi et γi le coecient d'activité, qui repond à la condition :
limx→1
γi = 1
Si dans le cas du mélange idéal de liquide, on a, comme dans le cas du mélange idéal de gaz, aucunevariation de volume et d'enthalpie, ceci n'est plus le cas dans le mélange réel. Par exemple, on mélange à
1. Le mot fugacité est lié à la tendance du composant de "fuir" la phase en question. L'unité de la fugacité est la mêmeque celle de la pression.
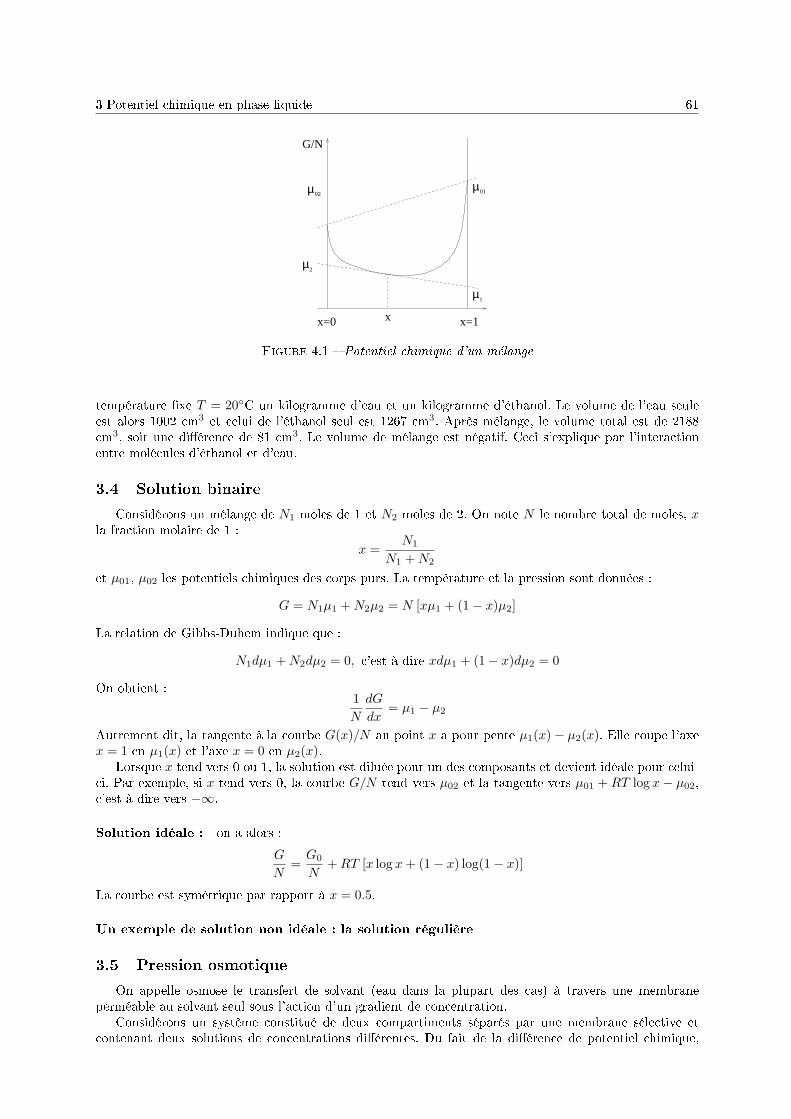
3 Potentiel chimique en phase liquide 61
G/N
x=0 x=1x
µ
µ
µµ
2
1
0102
Figure 4.1 Potentiel chimique d'un mélange
température xe T = 20C un kilogramme d'eau et un kilogramme d'éthanol. Le volume de l'eau seuleest alors 1002 cm3 et celui de l'éthanol seul est 1267 cm3. Après mélange, le volume total est de 2188cm3, soit une diérence de 81 cm3. Le volume de mélange est négatif. Ceci s'explique par l'interactionentre molécules d'éthanol et d'eau.
3.4 Solution binaire
Considérons un mélange de N1 moles de 1 et N2 moles de 2. On note N le nombre total de moles, xla fraction molaire de 1 :
x =N1
N1 +N2
et µ01, µ02 les potentiels chimiques des corps purs. La température et la pression sont données :
G = N1µ1 +N2µ2 = N [xµ1 + (1− x)µ2]
La relation de Gibbs-Duhem indique que :
N1dµ1 +N2dµ2 = 0, c'est à dire xdµ1 + (1− x)dµ2 = 0
On obtient :1
N
dG
dx= µ1 − µ2
Autrement dit, la tangente à la courbe G(x)/N au point x a pour pente µ1(x)− µ2(x). Elle coupe l'axex = 1 en µ1(x) et l'axe x = 0 en µ2(x).
Lorsque x tend vers 0 ou 1, la solution est diluée pour un des composants et devient idéale pour celui-ci. Par exemple, si x tend vers 0, la courbe G/N tend vers µ02 et la tangente vers µ01 +RT log x− µ02,c'est à dire vers −∞.
Solution idéale : on a alors :
G
N=G0
N+RT [x log x+ (1− x) log(1− x)]
La courbe est symétrique par rapport à x = 0.5.
Un exemple de solution non idéale : la solution régulière
3.5 Pression osmotique
On appelle osmose le transfert de solvant (eau dans la plupart des cas) à travers une membraneperméable au solvant seul sous l'action d'un gradient de concentration.
Considérons un système constitué de deux compartiments séparés par une membrane sélective etcontenant deux solutions de concentrations diérentes. Du fait de la diérence de potentiel chimique,

62 Mélange en l'absence de réaction chimique
membrane
∆p
eau pure
solution
Figure 4.2 Diérence de pression due à l'osmose
le phénomène d'osmose va se traduire par un ux d'eau dirigé de la solution diluée vers la solutionconcentrée.
Si l'on essaie d'empêcher ce ux d'eau en appliquant une pression sur la solution concentrée, la quantitéd'eau transférée par osmose va diminuer. Il arrivera un moment où la pression appliquée sera telle que leux d'eau va s'annuler. Cette pression d'équilibre est appelée pression osmotique.
Une augmentation de la pression au delà de la pression osmotique va se traduire par un ux d'eaudirigé en sens inverse du ux osmotique, c'est à dire de la solution concentrée vers la solution diluée :c'est le phénomène d'osmose inverse.
Calculons la pression osmotique (g. 3.5) : du côté de la solution où le soluté présente une concentrationx, le potentiel chimique de l'eau vaut :
µ1 = µ(x = 1, p0) +RT log(1− x) = µ0 +RT log(1− x)
Du côté du solvant pur, le potentiel chimique est :
µ2 = µ0 + v0(p− p0)
De part et d'autre de la membrane, les potentiels chimiques sont égaux. De la sorte,
v0(p− p0) = RT log(1− x) ≃ −RTx
On a donc :
∆p =RTx
v0
Application : le passage de certains uides dans les cellules vivantes à travers la membrane cellulaire, dans les reins, les produits métaboliques et autres impuretés contenus dans le sang sont éliminéspar osmose à travers une membrane semi-perméable vers l'urine,
la pression osmotique est le mécanisme responsable de la montée de l'eau dans les plantes. Lesfeuilles perdent de l'eau à travers la transpiration, donc la concentration de solutés dans leur uideest plus élevée. L'eau est poussée des racines à travers le tronc et les branches vers les feuilles parla pression osmotique. Dans le cas des grands arbres, la pression osmotique peut atteindre jusqu'à10 à 15 atm (ou 10 à 15 fois la pression atmosphérique) !
une application importante de la pression osmotique résultant de l'osmose est la détermination desmasses molaires de macromolécules.
Remarque : les propriétés d'une solution est aecté par le nombre totale de particules dissoutes dansla solution. Ainsi, le sel NaCl se séparant en deux ions dans l'eau aura un eet deux fois plus importantque du glucose à concentrations égales.
4 Équilibre liquide-vapeur d'une solution binaire
La pression et la température sont xées. La fraction molaire du composant 1 dans la phase liquideest notée x. La fraction molaire de ce même composant dans la phase gazeuse est notée y.

4 Équilibre liquide-vapeur d'une solution binaire 63
4.1 Mélange idéal
On fait les trois hypothèses suivantes : la vapeur est un mélange de gaz parfaits, le liquide est incompressible, la solution liquide est idéale.
4.1.1 Diagramme isotherme
Dans le cas du mélange idéal, la pression totale est la somme des pressions partielles p1 et p2. Il y aégalité des potentiels chimiques de chaque composant dans chacune des phases :
µ1(l) = µ1(g) et µ2(l) = µ2(g)
D'autre part, si l'un des composant est pur, la pression totale est égale à la pression de vapeur saturantede ce composant à la température T . Dans ce cas :
µ1 = µ1(x = 1, liq) +RT log x = µ1(p = ps1, gaz) +RT logp1ps1
Donc :
p1 = xps1 (4.6)
p2 = (1− x)ps2 (4.7)
Cette loi indiquant que la pression partielle d'un composé dans la vapeur est proportionnel à la compo-sition du liquide (loi de Raoult). On en déduit que la pression totale de la vapeur s'écrit :
p = xps1 + (1− x)ps2 (4.8)
Cherchons à faire apparaître la composition de la vapeur y dans cette expression. On a p1 = yp (loi deDalton). Donc en utilisant (4.6), on eectue le changement de variable x = yp/ps1 dans la relation (4.8).On obtient alors :
p =ps1ps2
ps1 + y(ps2 − ps1)(4.9)
La fonction p(x) est linéaire, tandis que la fonction p(y) est un arc d'hyperbole. Si on admet que ps1 > ps2,on obtient une courbe telle que celle de la gure 4.3. Il s'agit du diagramme isotherme. La partie supérieureest la zone de liquide homogène. La partie inférieure la zone de vapeur homogène et la zone entre les deuxcourbes représentent les états où les phases liquide et vapeur sont présentes simultanément. La courbesupérieur (droite) est appelée courbe d'ébullition et celle du bas courbe de rosée.
4.1.2 Diagramme isobare
Dans la pratique, la pression est imposée et on utilise plutôt un diagramme isobare. Sachant que ps1et ps2 sont fonction de la température, on obtient une courbe isobare du type de la gure 4.4, où Teiest la température d'ébullition du composant i pur. La phase vapeur homogène est alors dans la partiesupérieure et la phase liquide homogène dans la partie inférieure. Ainsi, si on chaue le mélange liquide,la température augmente jusqu'à la courbe d'ébullition où la première bulle de vapeur se forme. Puis latempérature continue d'augmenter jusqu'à disparition de la dernière goutte de liquide et on reste alorsen présence d'une seule phase vapeur.
Si on xe la température et la pression dans le domaine liquide+vapeur, on lit directement les com-positions de la phase vapeur sur la courbe de rosée et celle de la phase liquide sur la courbe d'ébullition.
Distillation : en chauant le mélange de composition x, on récupère une phase vapeur de compositiony > x, enrichi en composé le plus volatil. Si on condense cette phase et que l'on chaue à nouveau, onobtient un mélange de plus en plus riche en composé le plus volatil.
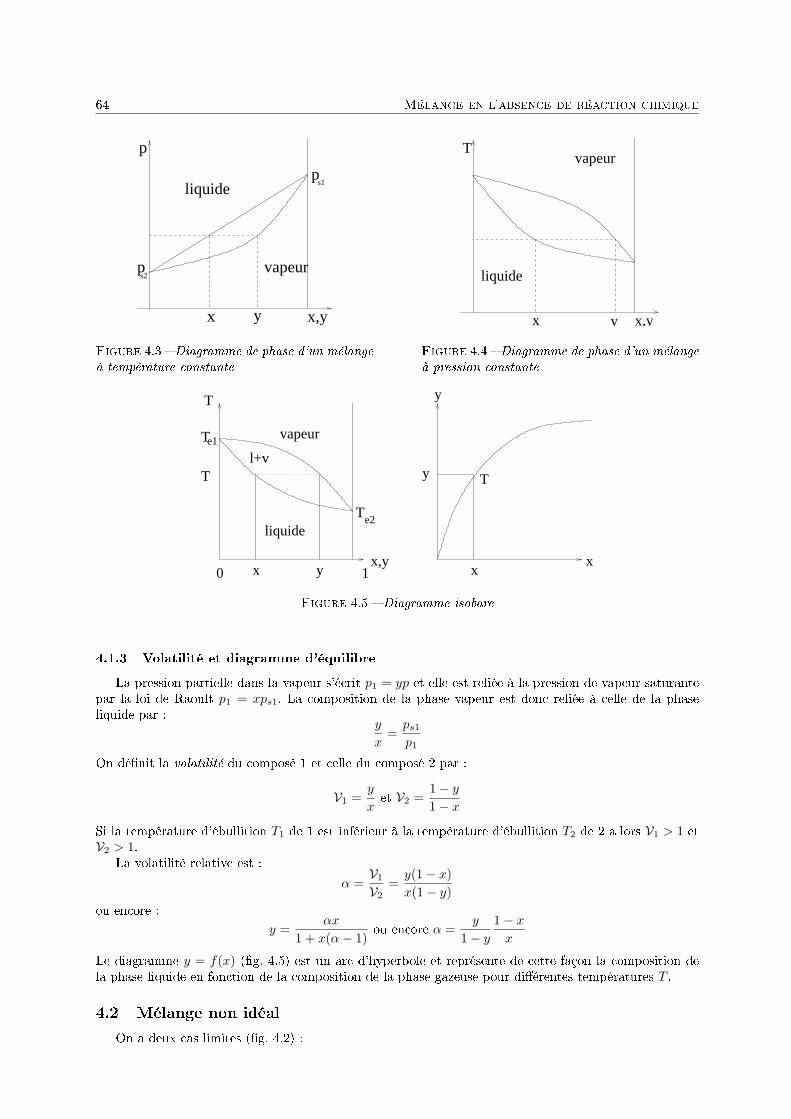
64 Mélange en l'absence de réaction chimique
s2
p
x,yx y
ps1
liquide
vapeurp
Figure 4.3 Diagramme de phase d'un mélangeà température constante
x,yx
T
liquide
vapeur
y
Figure 4.4 Diagramme de phase d'un mélangeà pression constante
T
T
0 1x y
vapeur
liquide
l+v
xx
y
y
x,y
T T
e2
Te1
Figure 4.5 Diagramme isobare
4.1.3 Volatilité et diagramme d'équilibre
La pression partielle dans la vapeur s'écrit p1 = yp et elle est reliée à la pression de vapeur saturantepar la loi de Raoult p1 = xps1. La composition de la phase vapeur est donc reliée à celle de la phaseliquide par :
y
x=ps1p1
On dénit la volatilité du composé 1 et celle du composé 2 par :
V1 =y
xet V2 =
1− y
1− x
Si la température d'ébullition T1 de 1 est inférieur à la température d'ébullition T2 de 2 a lors V1 > 1 etV2 > 1.
La volatilité relative est :
α =V1
V2=y(1− x)
x(1− y)
ou encore :
y =αx
1 + x(α− 1)ou encore α =
y
1− y
1− x
x
Le diagramme y = f(x) (g. 4.5) est un arc d'hyperbole et représente de cette façon la composition dela phase liquide en fonction de la composition de la phase gazeuse pour diérentes températures T .
4.2 Mélange non idéal
On a deux cas limites (g. 4.2) :
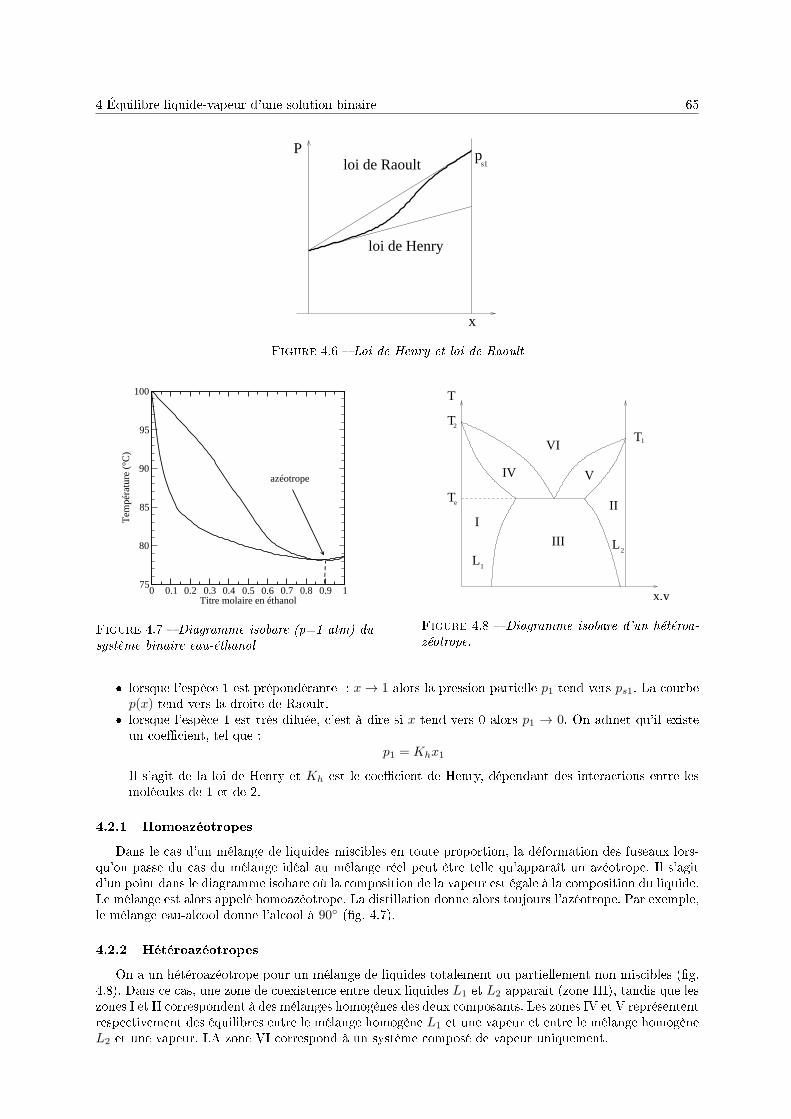
4 Équilibre liquide-vapeur d'une solution binaire 65
Ploi de Raoult
loi de Henry
x
ps1
Figure 4.6 Loi de Henry et loi de Raoult
0 0.1 0.2 0.3 0.40.5 0.6 0.7 0.8 0.9 1Titre molaire en éthanol
75
80
85
90
95
100
Tem
péra
ture
(°C
)
azéotrope
Figure 4.7 Diagramme isobare (p=1 atm) dusystème binaire eau-éthanol
x,y
T
TT
T
LL
I
III
II
IV V
VI
e
2
1
1
2
Figure 4.8 Diagramme isobare d'un hétéroa-zéotrope.
lorsque l'espèce 1 est prépondérante : x→ 1 alors la pression partielle p1 tend vers ps1. La courbep(x) tend vers la droite de Raoult.
lorsque l'espèce 1 est très diluée, c'est à dire si x tend vers 0 alors p1 → 0. On admet qu'il existeun coecient, tel que :
p1 = Khx1
Il s'agit de la loi de Henry et Kh est le coecient de Henry, dépendant des interactions entre lesmolécules de 1 et de 2.
4.2.1 Homoazéotropes
Dans le cas d'un mélange de liquides miscibles en toute proportion, la déformation des fuseaux lors-qu'on passe du cas du mélange idéal au mélange réel peut être telle qu'apparaît un azéotrope. Il s'agitd'un point dans le diagramme isobare où la composition de la vapeur est égale à la composition du liquide.Le mélange est alors appelé homoazéotrope. La distillation donne alors toujours l'azéotrope. Par exemple,le mélange eau-alcool donne l'alcool à 90 (g. 4.7).
4.2.2 Hétéroazéotropes
On a un hétéroazéotrope pour un mélange de liquides totalement ou partiellement non miscibles (g.4.8). Dans ce cas, une zone de coexistence entre deux liquides L1 et L2 apparaît (zone III), tandis que leszones I et II correspondent à des mélanges homogènes des deux composants. Les zones IV et V représententrespectivement des équilibres entre le mélange homogène L1 et une vapeur et entre le mélange homogèneL2 et une vapeur. LA zone VI correspond à un système composé de vapeur uniquement.
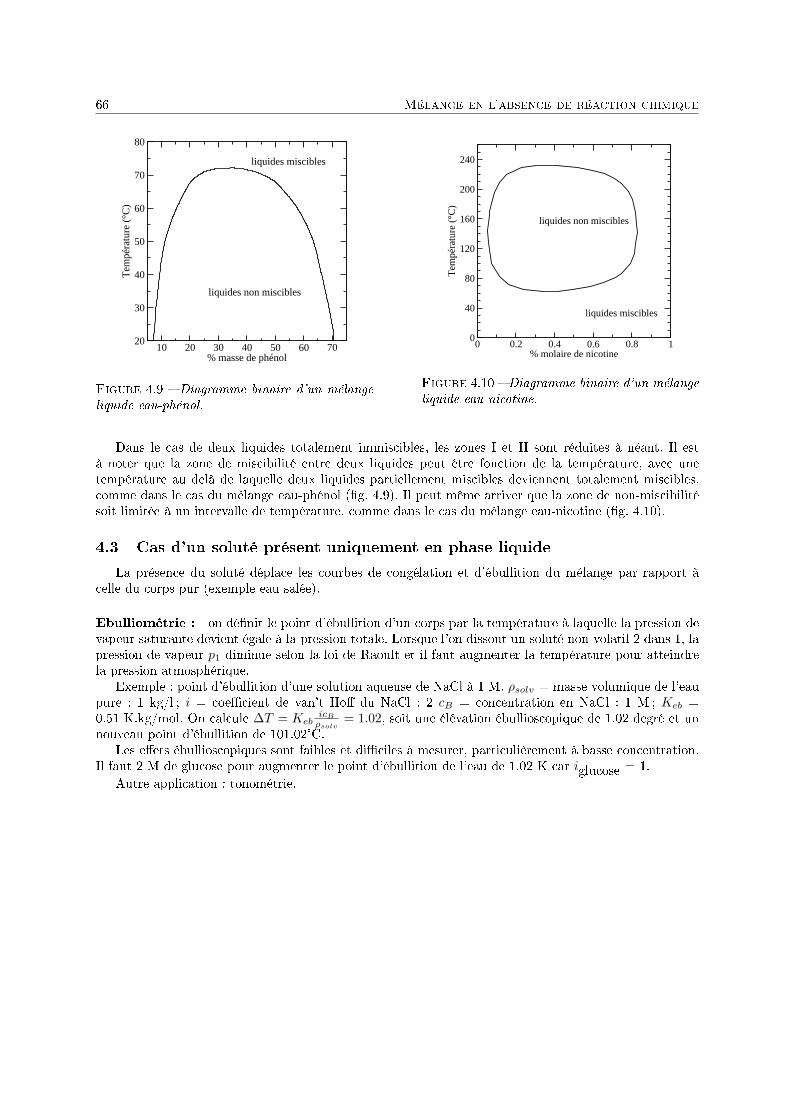
66 Mélange en l'absence de réaction chimique
10 20 30 40 50 60 70% masse de phénol
20
30
40
50
60
70
80
Tem
péra
ture
(°C
)
liquides non miscibles
liquides miscibles
Figure 4.9 Diagramme binaire d'un mélangeliquide eau-phénol.
0 0.2 0.4 0.6 0.8 1% molaire de nicotine
0
40
80
120
160
200
240
Tem
péra
ture
(°C
)
liquides non miscibles
liquides miscibles
Figure 4.10 Diagramme binaire d'un mélangeliquide eau-nicotine.
Dans le cas de deux liquides totalement immiscibles, les zones I et II sont réduites à néant. Il està noter que la zone de miscibilité entre deux liquides peut être fonction de la température, avec unetempérature au delà de laquelle deux liquides partiellement miscibles deviennent totalement miscibles,comme dans le cas du mélange eau-phénol (g. 4.9). Il peut même arriver que la zone de non-miscibilitésoit limitée à un intervalle de température, comme dans le cas du mélange eau-nicotine (g. 4.10).
4.3 Cas d'un soluté présent uniquement en phase liquide
La présence du soluté déplace les courbes de congélation et d'ébullition du mélange par rapport àcelle du corps pur (exemple eau salée).
Ebulliométrie : on dénit le point d'ébullition d'un corps par la température à laquelle la pression devapeur saturante devient égale à la pression totale. Lorsque l'on dissout un soluté non-volatil 2 dans 1, lapression de vapeur p1 diminue selon la loi de Raoult et il faut augmenter la température pour atteindrela pression atmosphérique.
Exemple : point d'ébullition d'une solution aqueuse de NaCl à 1 M. ρsolv = masse volumique de l'eaupure : 1 kg/l ; i = coecient de van't Ho du NaCl : 2 cB = concentration en NaCl : 1 M ; Keb =0.51 K.kg/mol. On calcule ∆T = Keb
icBρsolv
= 1.02, soit une élévation ébullioscopique de 1.02 degré et unnouveau point d'ébullition de 101.02°C.
Les eets ébullioscopiques sont faibles et diciles à mesurer, particulièrement à basse concentration.Il faut 2 M de glucose pour augmenter le point d'ébullition de l'eau de 1.02 K car iglucose = 1.
Autre application : tonométrie.

Chapitre 5
Thermodynamique des processus
irréversibles
1 Introduction
L'étude de systèmes réels impose un formalisme permettant de travailler dans des états hors équilibre.Pour cela, on va supposer que le système peut être divisé en éléments de volume susamment grandspour contenir une grand nombre de molécules (la thermodynamique phénoménologique est applicable) etsusamment petits pour que l'on puissent considérer qu'ils sont à l'équilibre.
2 Bilan local
Soit G une grandeur extensive quelconque. On rappelle que le bilan global de cette grandeur pour unvolume V immobile limité par une surface Σ s'écrit :
∆G = G(e) +G(i)
où G(e) représente l'échange à travers Σ et G(i) la production de G. On peut également faire un bilanlocal. Pour cela, dénissons la densité volumique de G par :
g =dG
dVc'est à dire G =
∫∫∫V
g dV
Si on considère un intervalle de temps dt, le bilan global s'écrit :
∂
∂t
∫∫∫V
g dV = −∫∫
Σ
φg.n dS +
∫∫∫V
p dV
où φg est le vecteur densité de ux de G, p est le taux de production de G et n la normale à Σ orientéevers l'extérieur. Or le théorème de Green permet de transformer une intégrale sur une surface en uneintégrale sur le volume contenu : ∫∫
Σ
φg.n dS =
∫∫∫V
−→∇.φgdV
On peut ainsi écrire l'équation locale :∂g
∂t+−→∇ .φg = p (5.1)
3 L'hypothèse de l'équilibre thermodynamique local
Il s'agit du premier postulat de la thermodynamique des phénomènes irréversibles.
Énoncé : à l'intérieur de chaque élément de volume du système étudié, il existe un état d'équilibrelocal pour lequel l'entropie volumique se présente sous la même fonction des variables macroscopiquesvolumiques que dans le cas de l'équilibre.

68 Thermodynamique des processus irréversibles
3.1 Relations de Gibbs et Gibbs-Duhem
La relation de Gibbs peut alors s'écrire en fonction des variables volumiques : considérons un systèmedéni par l'énergie interne U , l'entropie S, le volume V et le nombre de moles de chacun de ses composantsNk, k = 1 . . . n. Posons U = u.V , S = s.V et Nk = nk.V . La relation de Gibbs s'écrit pour un volume Voù température, pression et potentiel chimique sont uniformes :
dU = TdS − pdV +
n∑k=1
µkdNk (5.2)
On a alors dU = udV + V du, dS = sdV + V ds et dNk = nkdV + V dnk. De sorte que (5.2) peut s'écrire :
V
[du− Tds−
n∑k=1
µkdnk
]+ dV
[u− Ts+ p−
n∑k=1
µknk
]= 0
Or l'équation d'Euler par unité de volume permet d'écrire :
u = Ts− p+
n∑k=1
µkdnk (5.3)
On en déduit la relation de Gibbs en termes de variables volumiques :
du = Tds+
n∑k=1
µkdnk (5.4)
On en déduit la relation de Gibbs-Duhem en termes de variables volumiques :
sdT − dp+n∑k=1
nkdµk = 0 (5.5)
Ainsi, l'hypothèse de l'équilibre thermodynamique local impose que pour tout élément de volume quel'on suit dans son mouvement, les relations (5.3), (5.4) et (5.5) sont toujours valables.
3.2 Calcul de la production d'entropie
L'équation de bilan local introduite au paragraphe (2) peut être appliqué à la masse, à chaque compo-sant du système, à l'énergie totale et à l'entropie. Considérons un système constitué de diérentes espèceschimiques, sans réaction. Chaque espèce est caractérisée par sa fraction molaire xk = nk/
∑k nk = nk/n.
3.2.1 Conservation des espèces chimiques
Conformément à (5.1), écrivons la conservation de l'espèce k :
∂nk∂t
= −−→∇ .jk (5.6)
où nk est le nombre de moles de l'espèce k par unité de volume, jk = nkvk la densité de ux de l'espècek.
Il est pratique de considérer la vitesse d'un composant k (vitesse de convection) comme la somme dela vitesse du centre de gravité de l'élément de volume et d'une vitesse de diusion. La vitesse de centrede gravité (vitesse barycentrique) est :
v =
∑k nkvk∑k nk
=
∑k nkvkn
où n est le nombre total de moles par unité de volume, vk la vitesse de convection du composant k. Ondénit la densité de ux de matière par :
jn = nv

3 L'hypothèse de l'équilibre thermodynamique local 69
La vitesse de diusion v′k de l'espèce k est alors :
v′k = vk − v
La densité de ux molaire jk de l'espèce k peut s'écrire :
jk = nkvk = nk(v + v′k) = xk jn + j′k
où xk est la fraction molaire de l'espèce k dans le mélange. On remarque que la somme des densités deux de diusion est nulle :
∑k
j′k =∑k
jk − jn
[∑k
xk
]= jn − jn = 0
3.2.2 Conservation de la matière
En sommant les relation précédentes sur toutes les espèces chimiques, on obtient l'équation de conser-vation de la masse :
∂n
∂t= −
−→∇ .jn (5.7)
3.2.3 Conservation de l'énergie totale
On néglige l'énergie cinétique. De la sorte, le bilan d"'énergie se ramène à une bilan d'énergie interne(premier principe) :
∂u
∂t= −
−→∇ . je
où je est la densité de ux d'énergie interne.
3.2.4 Conservation de l'entropie
Le bilan entropique local s'écrit :∂s
∂t= −
−→∇ .js + σ (5.8)
où js est la densité de ux d'entropie et σ la production volumique d'entropie (σ ≥ 0).
3.2.5 Application de l'hypothèse de l'équilibre thermodynamique local
Utilisons désormais la relation de Gibbs Tds = du−∑k µkdnk, c'est à dire :
∂s
∂t=
1
T
∂u
∂t−∑k
µkT
∂nk∂t
En utilisant les lois de conservation :
∂s
∂t= − 1
T
−→∇ .je +∑k
µkT
−→∇ .jk
Sachant que−→∇ .(f j) = j.
−→∇f + f
−→∇ .j ou encore
−→∇.(f j)− j.
−→∇f = f
−→∇ .j, on en déduit :
∂s
∂t= −
−→∇.
[jeT
−∑k
µkTjk
](5.9)
+je.−→∇[1
T
]−∑k
jk.−→∇[µkT
](5.10)

70 Thermodynamique des processus irréversibles
En comparant avec la relation (5.8), on trouve :
js =1
T
[je −
∑k
µk jk
](5.11)
σ = je.−→∇ 1
T−∑k
jk.−→∇[µkT
](5.12)
En réalité, le potentiel chimique dépend de la pression, de la température et des nk : après calcul (voirannexe), on trouve :
σ = jq.−→∇(1
T
)− jv.
−→∇pT
−∑k
j′k.(−→∇µk)p,TT
(5.13)
où jv est la densité de ux de volume molaire :
jv = jn∑k
xkvk = jnv = nvv = v
où v = 1/n.
3.2.6 Présence de champs externes
La présence de champs externes et d'autres formes d'énergie peut être prise simplement en compte,en utilisant le concept de potentiel chimique généralisé :
µ∗k = µk(T, p, x) +Mk
(V2
2+ gz + . . .
)+ qkϕ+ . . .
= µk(T, p, x) +∑i
τikψi
où V2/2 représente l'énergie cinétique macroscopique, gz le potentiel de gravité, qk la charge électriqueet ϕ le potentiel électrique.
Considérons le cas où on a une seule forme de champ externe. On en déduit l'enthalpie généralisée :
h∗k = −T 2 ∂
∂T
(µ∗k
T
)p,x
= hk +∑k
τkψ
L'entropie molaire partielle reste inchangée. Pour la variation de la densité d'énergie totale, on pourraégalement écrire :
∂e
∂t=∂u∗
∂tavec u∗ = h∗ − p. Dans l'expression de la production d'entropie, de nouveaux termes apparaissent dus àla décomposition de l'expression
−→∇(µk/T ), du type :∑
k
τk jk.
−→∇ψT
= jψ.
−→∇ψT
en posant jψ =∑k τk jk.
En particulier, si on considère le transport de particules chargées, on dénit la densité de ux decourant électrique (en A/m2) et −−→∇ψ = E le champ électrique (en V/m).
4 Thermodynamique hors équilibre en régime linéaire
4.1 Postulats
Second postulat : la production d'entropie s'écrit :
σ =∑i
ji.Fi
où ji est le ux d'une grandeur extensive et Fi est une force thermodynamique.

5 Exemples 71
Troisième postulat : les ux et les forces thermodynamiques de même ordre tensoriel sont reliés(principe de Curie). On admet que dans le domaine du proche équilibre, la relation est linéaire :
ji =∑k
LikFk
où Lik est un tenseur constant, symétrique (relations d'Onsager).
Conséquences : la production d'entropie devient :
σ =∑j,k
LjkFjFk > 0
La matrice Ljk est donc dénie positive. En particulier, pour une matrice de dimension 2,
L11 > 0, L22 > 0, (L12 + L21)2 < 4L11L22
4.2 Application
Reprenons l'expression de la production d'entropie (5.19). Les relations phénoménologiques s'écrivent :
jq = Lqq−→∇(1
T
)+ Lqv
(−−→∇pT
)+∑k
Lqk−(
−→∇µk)p,TT
jv = Lvq−→∇(1
T
)+ Lvv
(−−→∇pT
)+∑k
Lvk−(
−→∇µk)p,TT
j′k = Lkq−→∇(1
T
)+ Lkv
(−−→∇pT
)+∑k
Lkk−(
−→∇µk)p,TT
ξ = LaaA
T
Si on néglige les couplages, on retrouve les lois classiques :
jq = Lqq−→∇(1
T
)= −λ
−→∇T (loi de Fourier)
jv = Lvv
(−−→∇pT
)= kD
−→∇p (loi de Darcy)
j′k = Lkk−(
−→∇µk)p,TT
= −DF−→∇ck (loi de Fick)
ξ = LaaA
T= k exp(α/T ) (loi d'Arrhénius)
5 Exemples
5.1 Conduction dans une barre
températureTA x
température TB
Figure 5.1 Conduction dans une barre
On considère une tige entourée d'un revêtement adiabatique, de longueur l de section Σ, de massevolumique ρ et de chaleur massique C (g. 5.1). Cette tige est initialement à la température T0. A

72 Thermodynamique des processus irréversibles
l'instant t = 0, ses extrémités A et B sont mises en contact avec deux sources de température TA et TBrespectivement.
Pour t > 0, la température T (x, t) est alors une fonction de x et du temps t.Considérons une portion de tige comprise entre x et x+dx et appliquons le premier principe : dU = δQ.
L'énergie interne massique est : u = CT+ constante, donc :
dU = ρΣCdTdx (5.14)
D'où :
ρΣdxCdT = [ΣJq(x, t)− ΣJq(x+ dx, t)] dt
= −∂Jq∂x
dxdtΣ
où Jq(x, t) est la densité de ux d'énergie thermique qui traverse la section de la tige à l'abscisse x. Onobtient :
ρC∂T
∂t= −∂Jq
∂x(5.15)
L'échange d'entropie de cette portion avec les portions voisines est uniquement associée à la chaleur.D'où :
dSe =dQ(x)
T (x)− dQ(x+ dx)
T (x+ dx)
= Σdt
[Jq(x)
T (x)− Jq(x+ dx)
T (x+ dx)
]= −Σdtdx
∂
∂x
[Jq(x)
T (x)
](5.16)
La variation totale d'entropie est alors dS = dSe + dSi entre t et t+ dt. Sachant que la diérentiellede l'énergie interne s'écrit : dU = TdS, en utilisant (5.14), on obtient :
dS = ρΣdxC
TdT (5.17)
On déduit la création d'entropie σs :
dSi = dS − dSe = Σdtdx
[ρC
T
∂T
∂t+
∂
∂x
(Jq(x)
T (x)
)]= σsΣdx
d'où :
σs = ρC
T
∂T
∂t+∂(Jq/T )
∂x
En utilisant (2.2), on trouve :
σs = − 1
T
∂Jq∂x
+1
T
∂Jq∂x
+ Jq∂(1/T )
∂xDonc :
σs = Jq∂(1/T )
∂xLa production est le produit du ux de chaleur et de la force thermodynamique :
F =−→∇(1
T
)Les relations d'Onsager permettent de déduire qu'il existe une constante L telle que :
Jq = L∂(1/T )
∂x
Il s'agit tout simplement de la loi de Fourier de diusion de la chaleur :
Jq = −λ−→∇T
avec λ =L
T 2.

6 État stationnaire hors équilibre 73
5.2 Eets thermoélectriques
Considérons un système constitué de deux ls de métaux diérents. On examine les eets thermoélec-triques où eet thermique et eet électrique sont couplés. Le ux de chaleur est appelé jq et le courantélectrique je. Les relations d'Onsager deviennent :
jq = Lqq−→∇(1
T
)+ Lqe
E
T
je = Leq−→∇(1
T
)+ Lee
E
T
Si les gradients ne se font que sur une seule dimension x :
jq = − 1
T 2Lqq
∂T
∂x+ Lqe
E
T
je = − 1
T 2Leq
∂T
∂x+ Lee
E
T
5.2.1 Eet Seebeck
Les jonctions entre les deux métaux sont maintenus à des températures diérentes. Aucun courantélectrique ne circule dans le circuit. Les relations d'Onsager donnent :
jq = − 1
T 2Lqq
∂T
∂x+ Lqe
E
T
0 = −Leq∂T
∂x+ LeeTE
L'intégration de la seconde équation permet d'obtenir une relation entre diérence de température etdiérence de potentiel. Si on admet que la diérence de température n'est pas trop importante, il existene température moyenne T , de sorte que
∫TEdx ≃ T
∫Edx = −T∆ϕ. Donc :
Leq = −LeeT(∆ϕ
∆T
)I=0
= Leeϵ
où la puissance thermoélectrique ϵ est de l'ordre de 10−5 V/K.
5.2.2 Eet Peltier
Les jonctions entre les deux métaux sont maintenus à une même température, pendant qu'un courantélectrique constant je circule dans le circuit. Les relations d'Onsager donnent :
jq = LqeE
T
je = LeeE
T
Le courant provoque donc un ux de chaleur jq. La seconde relation est la loi d'Ohm : Lee = T/r où r
est la résistance par unité de longueur. On pose Π =(jqje
)de sorte que : Lqe = ΠLee = ΠT/r.
Π est de l'ordre de 10−5 J/(A.s).
Remarque : la relation de réciprocité d'Onsager Lqe = Leq donne une relation entre eet Seebeck etl'eet Peltier : ϵ = Π/T .
6 État stationnaire hors équilibre
6.1 Théorème de Prigogine
Énoncé : si l'état stationnaire d'un système ouvert en régime permanent est susamment proche del'état d'équilibre (régime linéaire), la création d'entropie est minimale.

74 Thermodynamique des processus irréversibles
6.2 Conduction dans une barre
Reprenons l'exemple exposé plus haut. Calculons la production totale d'entropie dans la tige :
P = Σ
∫ l
0
σsdx
On dérive par rapport au temps :
∂P
∂t= Σ
∫ l
x=0
∂σs∂t
dx = Σ
∫ l
0
∂
∂t
[L
(∂1/T
∂x
)2]dx
= Σ
∫ l
0
2L∂(1/T )
∂x
∂2(1/T )
∂x∂tdx = 2Σ
∫ l
0
Jq∂2(1/T )
∂x∂tdx
On intègre par parties :
Jq → ∂Jq∂x
∂2(1/T )
∂x∂t→ ∂(1/T )
∂t
D'où :
∂P
∂t= 2Σ
[Jq∂(1/T )
∂t
]l0
− 2
∫ l
0
∂Jq∂x
∂(1/T )
∂tdx
= −2Σ
∫ l
0
(−ρC ∂T
∂t
).
(− 1
T 2
∂T
∂t
)dx
puisque T est constant en A et B.On obtient :
∂P
∂t= −2ΣρC
∫ l
0
1
T 2
(∂T
∂t
)2
dx
La production d'entropie est minimale si∂T
∂t= 0, c'est à dire à l'état stationnaire.

6 État stationnaire hors équilibre 75
6.2.1 Annexe
Dénissons le ux de chaleur par :
jq = T js −∑k
sk jk (5.18)
où sk est l'entropie molaire partielle du composant k et vaut sk =∂s
∂nk. La densité de ux d'énergie
devient d'après (5.11) :
je = T js +∑k
µk jk = jq +∑k
(µk + Tsk )jk
= jq +∑k
hk jk
En eet, G = H − TS, soit∑k µknk =
∑k hknk + T
∑k sknk, où hk est l'enthalpie molaire partielle.
La production d'entropie s'écrit :
σ = jq.−→∇ 1
T−∑k
jk.
[hk
−→∇ 1
T−−→∇ µkT
]+A
Tξ
= jq.−→∇ 1
T−∑k
jk.
[−hkT 2
−→∇T −
−→∇ µkT
]+A
Tξ
Sachant que hk = −T 2
(∂µk/T
∂T
)p,nk
, on en déduit :
σ = jq.−→∇ 1
T+A
Tξ −
∑k
jk.
[(∂µk/T
∂T
)p,nk
−→∇T −
−→∇ µkT
]
Or :
−→∇ µkT
=
(∂µk/T
∂T
)p,nk
−→∇T +
(∂µk/T
∂p
)T,nk
−→∇p+
∑i
(∂µk/T
∂ni
)p,T
−→∇ni
Donc : (∂µk/T
∂T
)p,nk
−→∇T −
−→∇ µkT
= −(∂µk/T
∂p
)T,nk
−→∇p−
∑i
(∂µk/T
∂ni
)p,T
−→∇ni
= − 1
T
[(∂µk∂p
)T,nk
−→∇p+
∑i
(∂µk∂ni
)p,T
−→∇ni
]
Sachant que ∂µk/∂p = vk, où vk est le volume molaire de l'espèce k :
(∂µk/T
∂T
)p,nk
−→∇T −−→∇ µkT
= − 1
T
[vk
−→∇p+∑i
(∂µk∂ni
)p,T
−→∇ni
]
on en déduit :
σ = jq.−→∇ 1
T+A
Tξ +
∑k
jk.vk
−→∇pT
+∑k
jkT.∑i
(∂µk∂ni
)T,p,nk
−→∇ni

76 Thermodynamique des processus irréversibles
On introduit les densités de ux de diusion (jk = xk jn + j′k) :
σ = jq.−→∇ 1
T+A
Tξ
+jn.∑k
[xkvk
−→∇pT
+xkT
∑i
(∂µk∂ni
)T,p,ni
−→∇ni
]
+∑k
j′k.
[vk
−→∇pT
+1
T
∑i
(∂µk∂ni
)T,p,ni
−→∇ni
]
= jq.−→∇ 1
T+A
Tξ
+jv.
−→∇pT
+ jn.1
T
∑k
xk(−→∇µk)T,p +
∑k
j′k1
T
∑i
(∂µk∂ni
)T,p,ni
−→∇ni
où jv est la densité de ux de volume molaire :
jv = jn∑k
xkvk = jnv = nvv = v
où v = 1/n.Par ailleurs, l'équation de Gibbs-Duhem (équation 5.5) montre que :∑
k
xk(−→∇µk)T,p = 0
La production d'entropie s'écrit nalement :
σ = jq.−→∇(1
T
)− jv.
−→∇pT
−∑k
j′k.(−→∇µk)p,TT
+A
Tξ (5.19)

Bibliographie
[1] L. Evans, Entropy and Partial Dierential Equations, Lecture Notes at UC Berkeley.
[2] L. Napolitano. Thermodynamique des systï¾½mes composites en ï¾½quilibre et hors ï¾½quilibre.Mï¾½moires de sciences physiques, fascicule 71. Gauthier-Villars, Paris, 1971.
[3] S.M. Blinder. Mathematical methods in elementary thermodynamics. Journal of Chemical Education,43(2), 85-92, 1996
[4] R. Balian. Du microscopique au macroscopique. Cours de physique statistique de l'ï¾½cole Polytech-nique. Ellipses, Paris, 1982
[5] M. Dubeck. Thermodynamique. Les irrï¾½versibilitï¾½s. Eska, Paris, 1993.
[6] P. Glansdor et I. Prigogine. Structure, stabilitï¾½ et uctuations. Masson, Paris, 1971.
[7] A. Marchand et A. Pacault. La thermodynamique mot ï¾½ mot. De Boeck-Wesmael, Bruxelles, 1995.
[8] M. Le Bellac et F. Mortessagne. Thermodynamique statistique. Equilibre et hors ï¾½quilibre. Dunod,Paris, 2001.
[9] J.P. Pï¾½rez. Thermodynamique. Fondements et applications. Masson, Paris, 1997.
[10] Y.A. Çengel et M.A. Boles. Thermodynamics. An engineering approach. McGraw Hill, New York,1998.
[11] D. Clodic, Y.S. Chang, A.M. Pougin. Evaluation de uides frigorigï¾½nes ï¾½ faible GWP pour lefroid domestique, le froid commercial, les transports frigoriques, la climatisation automobile. Rapportcommandï¾½ par le ministï¾½re de l'environnement. Centre ï¾½nergï¾½tique de l'Ecole des Mines deParis, Paris, 1999.
[12] N. Pottier. Physique statistique hors d'ï¾½quilibre : ï¾½quation de Boltzmann, rï¾½ponse linï¾½aire.Cours de DEA de Physique des Solides. Paris, 1999-2000.
[13] E.H. Lieb and J. Yngvason. The physics and mathematics of the second law of thermodynamics.Physics Reports, 310, 1-96 (1999).
[14] I. Prigogine, D. Kondepuli. Thermodynamique. Des moteurs thermiques aux structures dissipatives.O. Jacob, Paris, 1999.
[15] I. Prigogine, R. Defay. Thermodynamique Chimique. Desoer, Liï¾½ge, 1947.
[16] I. Prigogine. Introduction to thermodynamics of irreversible processes. John Wiley and Sons, NewYork, 1962.
[17] P. Infelta. Introductory thermodynamics, BrownWalker Press, 2004.
[18] B.E. Poling, J.M. Prausnitz, J.P. O'Connell. The properties of gases and liquids. McGraw Hill, 5thed., 2001.
[19] K.E. Starling, Fluid properties for light petroleum systems. Gulf publishing company, 1973.
[20] P. Taborek, Nucleation in emulsied supercooled water. Physical Review B, 32(9), 5902-5906, 1985.





























