Embed Size (px)
Citation preview

UE1 Biochimie et biologie moléculaire
Mardi 10/10/2017 de 15h30 à 17h30
Pr Hélène Cavé
Ronéotypeuse : Sabah Tirrani
Ronéoficheur : Wissam Azbabay
UE1 Du gène à la protéine mutante
Exemple de l’oncogènese
(2ème partie)
Les notions tombables sont indiqués à la dernière page.
UE1 Ronéo 3 cours 5 1/18

I) Quelles mutations dans les tumeurs
1) Mutations de p53
2) Fonction de p53
II) Étude de de délétions et de perte d’hétérozygoties
1) CGH Array
2) Etude de perte d’hétérozygoties des marqueurs polymorphes
III) Mutations affectant le cycle cellulaire
1) Délétion de RB
2) Délétion du locus INK4/ARF
3) Convergence des altérations oncogéniques du cycle cellulaire
IV) Oncogènes et gênes suppresseurs de tumeurs
V) Une nouvelle technique de séquencage
VI) Extension des cibles de l’oncogénèse
VII) Le développement d’un cancer
VIII) Clonalité tumorale
IX) Comment la cellule tumorale est capable d’accumuler plusieurs altérations
de son ADN
UE1 Ronéo 3 Cours 5 2/18

I) Quelles mutations dans les tumeurs
Panorama des altérations somatiques dans les ATL
C’est un schéma globale des ATL , maintenant on le voit très souvent dans les rapports scientifiques, il
permet d‘étudier une tumeur.
Chaque colonne correspond à une tumeur et chaque ligne à un gène, en general on met une tumeur
dans la case correspondant a l’intersection de la colonne et du gene quand il est muté ou on peux
utilise un code couleur qui va nous permettre de mettre en évidence pas mal de chose comme les types
de mutations, on organise souvent les choses en fonctions des mutations les plus fréquentes comme les
mutations des kinases et on regarde aussi toute les tumeurs et les types de mutations. On va étudier un
gène qui est très répandu dans les tumeurs, la P53.
Mutations de P53
Les anomalies de P53 sont la conséquence d’un certain nombre d’altérations (comme celle de mdm2
ou HPV). p53 est un facteur de transcription dont le gène est directement altéré dans les tumeurs par
des mutations récurrentes qui touchent la partie codant le domaine de liaison à l’ADN.
Fonctions de P53.
p53 agit comme un verrou de
sécurité pour un grand nombre de
situations et est au cœur d’un réseau
extrêmement complexe d’interactions
biologiques, c‘est donc une molécule
importante, au centre de la cellule.
Elle reçoit des signaux divers et
entraine une réponse adaptée faisant
intervenir de façon concerté l’arrêt du
cycle cellulaire, la réparation de
l’ADN et l’apoptose.
Un signal mitogène anormal (signal
qui pousse le cycle cellulaire via BCR-
ABL ou RAS mutée), ou des dommages de l’ADN (UV, rayon X, génotoxiques) activent p53.
Ue1 Ronéo 3 Cours 5 3/18

P53 empêche cette prolifération incontrolée dû au signal mitogène anormal et va limiter les dommages
de l’ADN en jouant son rôle de facteur de transcription et stimuler ses cibles transcriptionnelles qui
sont notamment p21 (qui freine le cycle cellulaire), BAX (protéine proapoptotique), gadd 45 (favorise
la réparation de l’ADN), mdm2 (inhibiteur de p53 qui permet un rétrocontrole négatif).
P53 est donc extrêmement majeure, elle agit comme une soupape de sécurité dans des conditions qui
peuvent être endommageable pour la cellule, mais elle est inactivée dans de nombreuses tumeurs par
des mécanismes d’une grande variabilité : soit directement par la mutation de son gène, indirectement
via les protéines de l’HPV ou en augmentant mdm2 qui va donc augmenter son rétrocontrole négatif.
II) Etude de délétions et de perte d’hétérozygotie
1) La CGH Array
La délétion est une perte de matériel génétique.
Attention : distinguer la délétion (plusieurs centaines de nt, taille suffisamment importante pour que
l’on ne le voit pas par séquençage) et les petites délétions (mise en évidence par des techniques de
séquençage).
Perte d’un chromosome : Monosomie
Mutations ponctuelles : Indel
Les délétions sont bien connues dans les cancers car il existe depuis longtemps des outils pour les
mettre en évidence comme le Caryotype(grandes délétions, chromosome entier) et le CGH array.
Le CGH array est technique utilisée dans les laboratoires de diagnostic. On veut comparer un génome
tumoral avec un génome témoin. C’est une technique d’hybridation d’ADN tumoral sur des sondes.
C’est une hybridation sur puce, une puce est un support physique sur lesquels sont fixés au préalable
des sondes oligonucléotides qui sont sensé reconnaitre des parties du génome.
On compare l’ADN tumoral à un ADN témoin. On commence par marquer chacun des ADN par des
fluorochromes différents puis on les mélange à quantité égale (mélange équimolaire). Ce n’est
qu’ensuite qu’on les hybride. La densité de couverture du génome dépend de la puce qu’on va utiliser.
S’il y a autant d’ADN tumoral
que d’ADN témoin, on obtient un
mélange de couleurs : il y autant
de vert que de rouge, on obtient
donc du jaune. On repère ainsi sur
la lame la résultante de
fluorescence après un traitement
bio-informatique. Les puces sont
miniaturisées, de façon à ce qu’on
puisse hybrider un nombre très
important de sondes, on peut donc
comme ça tester tout le génome,
c’est une technique
pangénomique.
Ue1 Ronéo 3 Cours 5 4/18

Par exemple, ici, le résultat montre l’ensemble des chromosomes. En jaune, on a tout ce qui est normal
en terme quantitatif (autant d’ADN tumoral que d’ADN témoin. Au chromosome 9,on trouve plus de
rouge : proportionnellement, on a donc plus d’ADN tumoral que d’ADN témoin. Il y a donc une
duplication dans la tumeur. A l’inverse, à la suite du chromosome 9, on a un marquage vert, ce qui
signifie qu’on a moins d’ADN tumoral. Il y a donc une délétion sur la tumeur. Cette technique est
beaucoup utilisée en cancérologie car on peut comme ça tester le génome entier.
Cette technique a néanmoins un handicap : dans le cas d’une petite tumeur, lors du prélèvement, il y a
un risque que l’on prélève aussi du tissu sain, ce qui fait que l’on est parfois gêné car il y a un mélange
de tissu sain et de tissu tumoral donc l’ADN tumoral ne sera pas tout à fait pur car il y aura aussi de
l’ADN sain ce qui risque de fausser les résultats. Il faut donc parfois utiliser plusieurs techniques
complémentaires.
2) Etude de perte d’hétérozygotie ou LOH (abréviation anglaise): les marqueurs polymorphes
Dans de l’ADN normal on a deux gènes avec deux allèles chacun. Dans l’ADN tumorale on a vu que
l’on parfois des délétions et parfois cette délétion déborde et ne s’arrête pas au bord de ce gène, or on
va avoir potentiellement ici des marqueurs hétérozygotes, par exemple un allèle A1 et un allèle A2.
Par le fait du hasard, et si les marqueurs délétés sont polymorphes (=on peut distinguer les deux copies
du gène dans la population, et donc un individu donné a des chances d’être hétérozygote) cette
délétion peut s’accompagner de la perte d’un allèle et donc de la perte d’hétérozygotie de l’ADN
tumorale (par exemple on a perdu A1 et il ne reste qu’A2) . On peut dire en retour que la mise en
évidence de la perte d’hétérozygotie va permettre de localiser les gènes inactives dans les tumeurs.
a) Qu’est-ce qu’un marqueur génétique ?
C’est est une séquence d’ADN qui peut être identifiée par un test simple et dont on connait
précisément la localisation et donc le locus de ce marqueur qui est généralement un locus unique.
Quand on parle de marqueur polymorphe et bien là c’est la même définition mais en plus c’est un
marqueur dont la séquence génétique diffèrent entre les membres d’une espèce et donc entre les 2
allèles d’un individu (un allèle provenant du père et l’autre provenant de la mère). Dans la population
générale, on a un pourcentage élevé d’hétérozygotie.
Ces marqueurs sont de deux types : les microsatellites et les SNP.
• Les microsatellite
(découvert plus tôt que les
SNP) est une répétition en
tandem de un à six pdb. En
général on les symbolise en
mettant entre parenthèse la
répétition avec son nombre
en indice. La plupart des
polymorphes sont la
duplication en tandem de C. Cette séquence répétée est
structurellement très à
même d’introduire des
variations lorsqu’elle est
recopiée car c’est un peu
moins facile pour l’ADN
polymérase de le recopier,
on va donc très vite avoir un CA de plus ou de moins et c’est cela qui fait que ce marqueur va
être polymorphe.
UE1 Ronéo 3 Cours 5 5/18
Bien qu’il existe des systèmes de réparations, quelques-uns vont rester et on va les retrouver
dans la génération d’après. C’est une source de diversité dans la population car ce
polymorphisme est différent d’un individu a l’autre, d’un allèle a l’autre.
C’est assez simple à étudier en mettant en évidence la taille du polymorphisme. On est capable
d’amplifier la région d’ADN, puis on est capable de faire migrer l’amplicon. La taille de l’amplicon va
varier selon le nombre de répétitions. Comme il s’agit d’un polymorphisme de taille, on peut
distinguer plusieurs allèles.
Exemple : sur la diapo ci-dessus on a une effectué une PCR et une migration électrophorétique :
l’individu hétérozygote apparait avec 2 pics donc deux allèles différents A1 et A2 qui a plus de
répétitions.
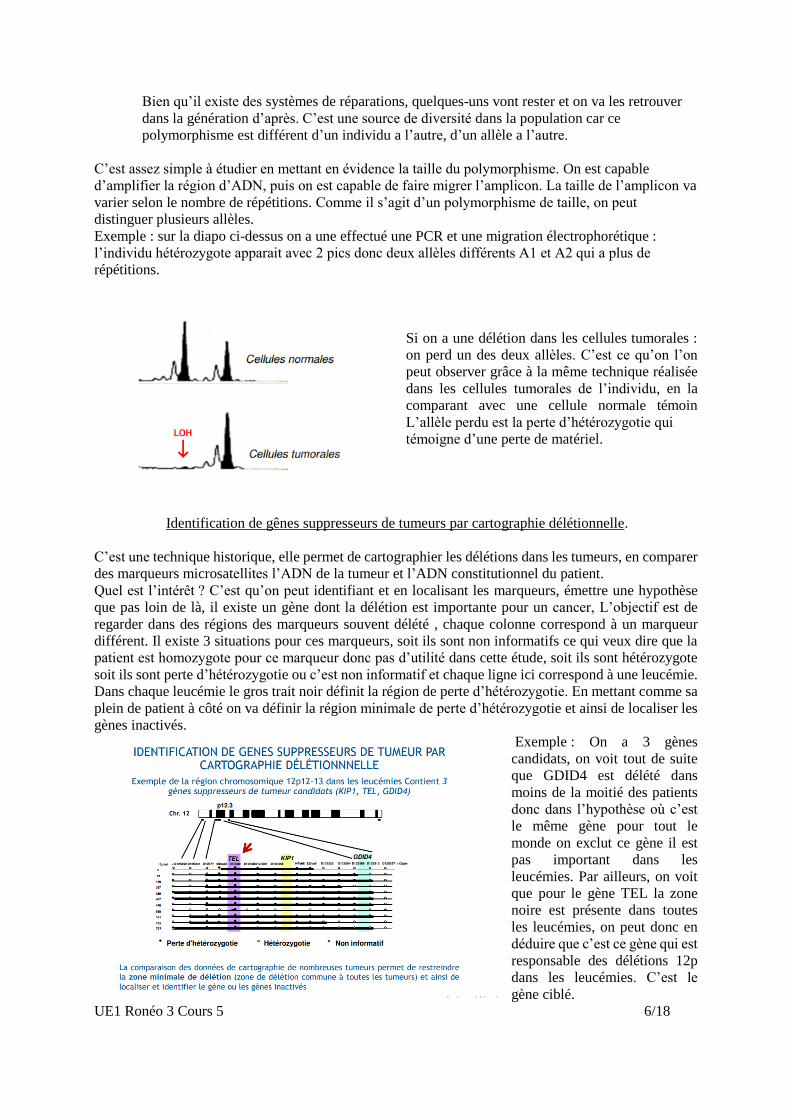
Si on a une délétion dans les cellules tumorales :
on perd un des deux allèles. C’est ce qu’on l’on
peut observer grâce à la même technique réalisée
dans les cellules tumorales de l’individu, en la
comparant avec une cellule normale témoin
L’allèle perdu est la perte d’hétérozygotie qui
témoigne d’une perte de matériel.
Identification de gênes suppresseurs de tumeurs par cartographie délétionnelle.
C’est une technique historique, elle permet de cartographier les délétions dans les tumeurs, en comparer
des marqueurs microsatellites l’ADN de la tumeur et l’ADN constitutionnel du patient.
Quel est l’intérêt ? C’est qu’on peut identifiant et en localisant les marqueurs, émettre une hypothèse
que pas loin de là, il existe un gène dont la délétion est importante pour un cancer, L’objectif est de
regarder dans des régions des marqueurs souvent délété , chaque colonne correspond à un marqueur
différent. Il existe 3 situations pour ces marqueurs, soit ils sont non informatifs ce qui veux dire que la
patient est homozygote pour ce marqueur donc pas d’utilité dans cette étude, soit ils sont hétérozygote
soit ils sont perte d’hétérozygotie ou c’est non informatif et chaque ligne ici correspond à une leucémie.
Dans chaque leucémie le gros trait noir définit la région de perte d’hétérozygotie. En mettant comme sa
plein de patient à côté on va définir la région minimale de perte d’hétérozygotie et ainsi de localiser les
gènes inactivés.
Exemple : On a 3 gènes
candidats, on voit tout de suite
que GDID4 est délété dans
moins de la moitié des patients
donc dans l’hypothèse où c’est
le même gène pour tout le
monde on exclut ce gène il est
pas important dans les
leucémies. Par ailleurs, on voit
que pour le gène TEL la zone
noire est présente dans toutes
les leucémies, on peut donc en
déduire que c’est ce gène qui est
responsable des délétions 12p
dans les leucémies. C’est le
gène ciblé.
UE1 Ronéo 3 Cours 5 6/18

• Les SNP correspond aux 2nd type de polymorphisme qui existe, ce sont des polymorphismes
nucléotidiques. Il s’agit de la variation d’un seul nucléotide à un emplacement spécifique du
génome (par exemple un G sur un allèle et un C sur l’autre). Ils sont présents dans plus d’1%
de la population. Les SNP sont beaucoup moins hétérozygotes et polymorphes que les autres
marqueurs car il n’existe que 4 possibilités, mais comme on en connait beaucoup (c’est la
forme la plus abondante de variation génétique), on compense par le nombre de marqueurs le
manque d’informativité de chacun. Ce sont devenus des outils de premier plan car ils ont
supplanté les microsatellites mais le principe reste le même car c’est la perte d’hétérozygotie
qui va nous permettre de localiser le problème. Pour les mettre en évidence, on peut utiliser le
séquençage Sanger.
Des techniques d’hybridation sur puce ont
aussi été développées, et elles sont beaucoup
plus adaptées à la mise en évidence des SNP,
ce sont les SNP-Array. Le principe de base est
similaire à la CGH Array : hybridation de
l’ADN marqué sur les puces, avec des sondes
qui vont reconnaitre chaque SNP.
Pour un patient donné, l’allèle c’est soit
hybidé sur le C auquel cas il est homozygote
G, soit le sur T auquel cas il est homozygote
A, soit sur les 2 auquel cas il est hétérozygote.
Cette analyse permet une étude
pangénomique, elle peut couvrir tout le
génome.
b) Etude des pertes d’hétérozygotie (LOH)
Ces pertes d’hétérozygoties vont révéler plusieurs types d’anomalie :
• Une haploinsuffisance : c’est à dire la perte d’un
allèle, parfois il suffira à créer une pathologie ou
un cancer mais c’est souvent insuffisant car il y
aura un système de compensation.
• Mutation de l’autre allèle : souvent on va
trouver en face un allèle portant une mutation
ponctuelle. On retrouve donc deux anomalies, un
allèle délété et un allèle muté.
• Perte d’hétérozygotie sans perte de matériel :
les 2 gènes sont présents, il n’existe pas de
délétion mais une duplication de l’allèle muté
donc il n’y a pas de variation du nombre de copies
(perte qualitative, mais non quantitative). C’est
une disomie uniparentale acquise (un individu hérité deux fois d’un chromosome d’un de
ses parents) C’est difficile à voir, la CGH Array ne le permet pas seul la SNP Array le permet.
c) Applications des études de SNP pangénomiques.
• Profilage des mutations somatiques dans les cancers : Les régions de déséquilibre allélique
désignent les délétions (ou amplifications) d’ADN dans les cancers
UE1 Ronéo 3 Cours 5 7/18

• Recherche de gènes de susceptibilité aux maladies (GWAS) ex : prédisposition aux cancers
→ Comparaison de fréquence allélique entre une population de patients et une population de
sujets contrôles sains
• Etudes pharmacogénomiques ex : comprendre le lien entre les polymorphismes
interindividuels et l’efficacité ou la toxicité des drogues = permet de vérifier pourquoi les
patients répondent ou non à leur traitement.
Toutes ces techniques sont très utilisés actuellement
III) Mutations affectant le cycle cellulaire.
Rappel : La cellule part en G1 avec 2n au
niveau de leurs chromosomes, on va
ensuite avoir une phase S dans lequel on
va dupliquer l’ADN pour avoir 4n. En fin
de G2 on passe en mitose ou on retourne
en 2n. Il existe 2 points de contrôle qui
permet d’éviter les bêtises, le 1er est en
G1/S qui va sonner le démarrage et
l’initiation de la réplication, le 2ème est
un contrôle de qualité pour contrôler si
l’étape très sensible de réplication de l’ADN c’est bien passée .
1) Délétion de RB (Rétinoblastoma)
C’est un gène dont les délétions ont été mises en évidence
de façon récurrente dans les cancers. Il a été identifié
comme causal dans les tumeurs du rétinoblastome. Il
code pour la protéine RB (pRB).
C’est un facteur du cycle cellulaire très important, elle va
intégrer un certain nombre de signaux intra et extra
cellulaires qui vont gouverner sa phosphorylation et c’est
ce qui va gouverner le passage ou pas en phase S donc
l’entrée en cycle cellulaire, Rb va enfaite séquestrer un
facteur de transcription qui est E2F : Quand RB n’est pas
phosphorylé E2F lui est lié et on reste en G1, si on a un signal mitogène, on va avoir une
phosphorylation de RB qui va se détacher de E2F qui va lui se fixer sur l’ADN et jouer son rôle de
facteur de transcription en agissant sur le gène de la Phase S donc par sa présence RB empêche que la
cellule entre en phase S de façon non contrôlé.
Dans les tumeurs on a une délétion de la région chromosomique 13 qui porte le gène de RB, on perd
donc la protéine qui n’est plus capable de séquestrer E2F qui va agir sur le cycle cellulaire de façon
positive et va provoquer une entrée incontrôlée dans la phase S, car elle ne répond plus à la
signalisation. La cellule est privée de gardien du cycle cellulaire.
2) Délétion du locus INK4/ARF (ou comment faire sauter 2 verrous d’un coup)
C’est un autre exemple qui va jouer sur le cycle cellulaire. Ce locus est très souvent muté dans les
leucémies et autres cancers. Il est spécial car il comprend en étroite proximité 3 gènes qui exercent un
rétrocontrôle négatif, mais pas forcément de la même manière.
UE1 Ronéo 3 Cours 5 8/18

On a :
• INK 4b qui code pour p15. Il inhibe les CDK (Kinases Cycline Dépendantes)
• INK4a qui code pour p16. Aussi un inhibiteur des CDK.
• ARF qui code pour p14 est un inhibiteur de mdm2 qui inhibe lui-même p53 qui freine le CC.
Ces gènes ont des promoteurs alternatifs : INK4a est synthétisé grâce aux exons E1alpha,E2,E3 et
ARF est synthétisé grâce aux exons E1bêta,E2,E3.
On réutilise donc du matériel génétique pour deux protéines différentes (ARF et INK4a). La
particularité c’est que selon que ce soit un promoteur ou l’autre E2 va être vu dans des cadres de
lecture différents.
Lorsqu’on commence à l’exon 1alpha ou l’exon 1beta, il y a un décalage du cadre de lecture. On a
donc deux cadres de lecture différents. On code ainsi deux protéines qui ont des fonctions totalement
différentes.
La proximité de ces 3 gènes codant pour les protéines freinant le cycle cellulaire fait que les délétions
sont un mode privilégié d’inhibition de ces molécules dans les cancers. C’est en effet très rentable :
avec une seule altération, on est capable d’éliminer 3 protéines. On a donc souvent des co-délétions de
ces trois gènes.
Ces protéines agissent enfaite aux 2 points de contrôle du
cycle cellulaire.
Au point de contrôle G1/S : p15 et P16 en inhibant les CDK
activent la protéine RB qui empêche E2F d’entrer en action.
Ainsi, en absence de P15 et p16, l’entrée dans le CC est
favorisée car RB est plus à même d’être phosphorylé et donc
inactive.
Au point de contrôle G2/M : ARF inhibe mdm2 et donc
active. P53 active p21 qui est un inhibiteur des CDK et donc
freine l’entrée dans le cycle cellulaire. En l’absence de ARF,
p53 n’est plus activée, donc l’entrée dans le cycle cellulaire
est favorisée.
Quand le génome délète les trois gènes, elle est capable de supprimer les deux points de contrôle ou de
les affaiblir, ce qui est favorable pour la progression tumorale.
UE1 Ronéo 3 Cours 5 9/18

3) Convergence des altérations oncogéniques du cycle cellulaire.
On peut observer que sur une même voie
biologique on peut avoir plusieurs façons de
l’affecter.
Soit par une mutation activatrice sur un gène
qui va réguler négativement le CC ou au
contraire par une mutation qui activer des
gènes qui vont coder pour des protéines qui
vont activer l’entrée dans le CC. A partir de
ce type d’aberrations dans les tumeurs on a
défini la notion d’oncogène et de gènes
suppresseurs de tumeur.
On a donc souvent une convergence des altérations oncogéniques. Elles sont multiples et
convergent vers un point précis, ici le cycle cellulaire.
IV) Notion d’oncogènes et de gènes suppresseurs de tumeurs Ce sont des notions défini sur la base des mutations qui vont les affecter.
1) Les oncogènes
Ils sont altérés dans les tumeurs de telle sorte qu’il ait gagné de la fonction, c’est une mutation gain de
fonction. Dans les cancers ils promeuvent la tumorigènese. Il suffit qu’une mutation soit présente pour
qu’il y ait une tumeur. Ces mutations sont dites dominantes au niveau cellulaire
2) Gènes suppresseurs de tumeurs.
Ils participent à supprimer les tumeurs par exemple les freins du cycle cellulaire. C’est une mutation
perte de fonction qui va pousser la cellule à devenir tumorale. Souvent dans cette situation, les deux
allèles doivent être mutés car en cas de mutation d’un seul allèle, on a certes une haploinsuffisance
mais il existe des systèmes de compensation qui protègeront la cellule. C’est donc une mutation
récessive au niveau cellulaire.
3) Conséquences des altérations génétiques.
• La surexpression inappropriée d’un
gène concerne un oncogène.
• Le gène de fusion : gènes touchés ni
oncogène ni gène suppresseur de tumeur, on va
en créer un nouveau qui sera un oncogène
• Inactivation par translocation : gène
suppresseur de tumeur
• Mutation ponctuelle : soit oncogène,
soit gène suppresseur de tumeur
• L’amplification concerne un oncogène
• Délétion, inactivation des deux allèles,
haploinsuffisance : gène suppresseur de tumeur
UE1 Ronéo 3 Cours 5 10/18

Plusieurs mécanismes pour inactiver la réponse P53 dans les cancers.
Petite synthèse de ce qu’on a vu : plusieurs
mécanismes vont pouvoir être utilisé dans
différents types d’oncogenèse ou parfois
chez un même patient parfois pour inactiver
la repose p53, On voit tous les chemins que
peux prendre la biologie d’une tumeur.
V) Une nouvelle technique de séquençage
Le séquençage de l’exome entier a beaucoup progressé ces dernières années.
La méthode de Sanger classique a une capacité limitée puisque que l’on partait d’une hypothèse on se
disait je vais aller voir dans ce gène là mais actuellement le séquençage nouvelle génération nous
permet de faire un séquençage de l’ensemble du génome et de l’exome (donc des régions codantes).
Il permet de s’affranchir d’hypothèse donc de débuter sans a priori sur ce que l’on cherche, ce qui
nous a permis de tomber sur des mutations de gènes qui n’étaient pas des gènes candidat initialement
et d’étendre la biologie des tumeurs.
On a maintenant une vue d’ensemble du génome de la tumeur et il y a la possibilité d’une «
Profondeur de lecture » importante (nombre élevé de lectures indépendantes de la séquence)
permettant la détection de mutations dans des sous-clones tumoraux.
Le fait de pouvoir accéder au séquençage du génome entier a permis d’avoir une image globale des
mutations dans les tumeurs.
Les cellules tumorales ont
accumulé plusieurs mutations de
l’ADN.
Une seule mutation ne suffit pas
car comme on le voit, le nombre
de mutations identifié dans les
cellules tumorales est
généralement supérieur à 1. On
peut même aller jusqu’à 500
mutations comme c’est le cas pour
le mélanome. Le taux de mutation
varie d’un type de tumeur à
l’autre. Mais on a également
trouvé des mutations dans des
gènes qu’on attendait pas en
tumorigenèse. Ces découvertes ont
étendu les cibles de l’oncogénèse.
UE1 Ronéo 3 Cours 5 11/18

VI) Extension des cibles de l’oncogenèse
Lorsque l’on a cherché les gènes de l’oncogenèse, on a trouvé tous les gènes du CC, mais également
certains du contrôle de l’expression des gènes, que l’on n’attendait pas forcément.
Les modifications épigénétiques touchent la transcription.
On peut les classer en 3 grands groupes :
• Modification post traductionnelle des histones (=protéine) qui vont jouer sur la compaction
de la chromatine
• Méthylation de l’ADN
• Remodelage de la chromatine : changement de l’ouverture de la chromatine
Ces modifications vont jouer sur la capacité que va avoir la cellule à transcrire les gènes.
a) Méthylation de l’ADN
C’est l’ajout réversible d’un CH3 sur une cytosine dans une séquence CG. On va obtenir une 5-méthyl
cytosine grâce à une enzyme : l’ADN méthyl transférase (DNMT).
Cette méthylation n’a pas lieu n’importe où, mais au niveau des séquences CG, qui ont une répartition
particulière. On les trouve sur les îlots CpG, très denses en séquences CG C’est une région qui fait
plus de 200 pdb et qui contient au moins 50% de CG.
Ils sont essentiellement présents dans la région promotrice des gènes et sont rares(1à2% du génome).
On a aussi des plages CpG : moins riches en CG, mais proche des promoteurs.
Cette méthylation est donc importante car elle
conditionne le niveau d’expression des gènes, dans
un sens de répression des gènes.
Dans les tumeurs, on trouve des anomalies de la
méthylation. Dans certains gènes, les îlots CpG ne
sont pas correctement méthylés, alors que dans
d’autres, ils sont trop méthylés, ce qui réprime la
transcription du gène. Certains gènes subissent donc
une extinction épigénétique par méthylation de l’ADN.
Au contraire, on peut avoir la déméthylation d’un oncogène. C’est souvent dû à une mutation de
l’enzyme DNMT.
b) Modification post-traductionnelle des histones
Il s’agit de modifier la queue des histones, l’extrémité N-ter des histones. Elles peuvent être acétylées
ou méthylées. Ces modifications influencent l’expression des gènes en modifiant la compaction de la
chromatine.
Pour mettre en œuvre ces modifications, plusieurs enzymes sont mis en cause, les méthyl transférases
ou les acétyl transférases.
Par exemple, l’EZH2 (Histone-lysine N-methyltransferase) va méthyler de façon spécifique des
histones et fait partie d’un complexe multiprotéique plus large : le complexe répresseur Polycomb 2.
Là encore, dans les cancers on trouve des mutations de ce gène, EZH2 est mutés dans un certain
nombre de tumeurs et on trouve aussi des mutations d’autres protéines du complexe. On va perdre la
marque methylé qui va être remplacés par une marque acetylé, Ce ne sont plus du tous les mêmes
facteurs de transcription qui vont reconnaitre ces zones qui lieu d’être réprimés ils vont être activés,
donc des zones entières vont s’ouvrir à la transcription. Il y a une dérégulation épigénétique au niveau
des cancers
UE1 Ronéo 3 Cours 5 12/18

Les systèmes cellulaires ciblés par les mutations convergent sur les voies multiples, complémentaires
et qui s’ajoutent entre elles pour acquérir un génotype cancéreux.
VII) Le développement d’un cancer
Cela nous montre que le développement d’un cancer passe par l’accumulation de plusieurs mutations
et altérations. Si on reprend l’exemple de RAS, si on a seulement une mutation de RAS mais que la
mort cellulaire fonctionne, cette mutation ne va pas être efficace, par contre s’il y a aussi une altération
de la mort cellulaire, là on aura une prolifération extrêmement active.
On voit donc bien qu’une mutation ne suffit pas, cela a notamment été mis en évidence dans un type
de tumeur, la tumeur colique. Lorsque l’on observe cette tumeur, on voit qu’en fonction des différents
territoires, on a des mutations différentes. Cette notion de multi-étapes est notamment bien décrit pour
le cancer colorectal avec polypose, il commence par une extension tumorale bénigne, en phase initiale,
la mutation du gène APC est suffisante pour
conclure qu’il existe un petit adénome,
ensuite on retrouve des adénomes malins
avec cette fois des mutations de RAS, enfin
au niveau du carcinome on retrouve une
diversité des mutations des cellules. Cela
nous a permis de décrire l’histoire de la
tumeur. (Donné à titre d’exemple, pas à
apprendre)
VIII) Clonalité tumorale
Cela nous amène à réfléchir sur l’origine de la tumeur, sur l’essence d’un clone tumoral. Les cellules
tumorales proviennent tous d’une même cellule et ces cellules vont acquérir les caractéristiques du
cancer à travers un processus de mutation et de sélection.
Cela nous permet de mieux comprendre l’évolution des espèces selon la théorie de Darwin : la cellule
mutée aura un avantage sélectif par rapport à ses voisines. L’environnement aussi joue un rôle car elle
fournit une pression de sélection et la cellule peut avoir plus de chances, grâce à sa mutation,
d’échapper à cette pression. Or si une cellule passe les barrières de sélection, elle va proliférer et
survivre et donc s’accumuler : on a alors le développement d’un clone cellulaire toutes issues de la
même cellule initiale. Mais, il doit y avoir accumulation au cours du temps de mutations somatiques
qui vont se déclarer dans une seule cellule : on a la formation d’un sous clone Du fait donc de
l’accumulation et de mutations secondaires, on a un matériel tumoral hétérogène.
Attention : les sous-clones coexistent et l’apparition d’un sous-clone ne fait pas disparaître le dernier.
UE1 Ronéo 3 Cours 5 13/18

Il existe deux modes de développement d’une tumeur, dépendant de la cellule créant le sous clone.
Le modèle de développement ramifié est plus rapide.
Quand un sous clone est dominant, on peut faire le diagnostic de la tumeur. On peux donc finalement
dire que la tumeur consiste en la coexistence de plein de maladies un peu différentes les unes des
autres dans une même entité. Comme la charge mutationnelle est différente entre les clones, on aura
certains clones plus résistants que d’autres aux traitements.
L’hétérogénéité d’une tumeur nous renseigne sur la séquence d’apparitions des mutations.
Exemple d’une tumeur rénale avec métastase pulmonaire.
Des chercheurs ont étudié deux biopsies de la tumeur obtenus à deux régions différentes du rein et une
biopsie de la métastase. Ils ont recherché les mutations en se
demandant si elles étaient spécifiques du tissu dans lequel on les
trouvait. On se rend compte que ces trois prélèvements n’ont pas les
mêmes types de mutations.
On peut ainsi reconstruire l’histoire de l’apparition de la mutation. On
commence par chercher la mutation initiatrice parmi les mutations
présentes dans tous les dérivés de la tumeur.
Ensuite, les mutations retrouvées uniquement dans la métastase sont
celles qui ont procuré aux cellules leurs capacités de migrations. Enfin,
les cellules restées dans la tumeur rénale ont des mutations différentes
selon l’endroit.
Il s’agit donc de deux manières différentes d’évoluer pour la tumeur. C’est plutôt l’effet du hasard qui
crée l’avantage sélectif qui permet la tumeur, il n’existe pas de prédestination de la tumeur. On a ainsi
la formation de l’architecture clonale d’une tumeur, qui hiérarchise les clones les uns par rapport aux
autres et qui nous renseigne sur la séquence d’apparition des mutations avant le diagnostic.
IX) Comment la cellule tumorale est-elle capable d’accumuler plusieurs
altérations de son ADN ? Les altérations de l’ADN surviennent en permanence au cours de la vie de la cellule à cause de
nombreuses raisons. Elles peuvent être dues à des erreurs de réplications de l’ADN, des agressions
endogènes ou exogènes, ou elles peuvent être juste des altérations spontanées.
UE1 Ronéo 3 Cours 5 14/18

Or il existe un certain nombre de protections et de système de réparations qui sont sensé pouvoir
réparer tout ça et nous assurer une santé de fer. Mais pourtant les cancers existent.
Pourquoi et qu’est ce qui fait que cet équilibre est rompu ? (pas à apprendre, elle dit que c’est juste
une discussion pour nous apprendre à avoir un esprit critique)
La 1ère hypothèse est que peut-être ce sont les modes de vie, les conduites à risque comme fumer ou
manger gras ou encore la pollution qui influencent sur cet équilibre. Beaucoup de personnes utilisent
ces données pour avoir un mode de vie qui protège du cancer, mais ce raisonnement est aussi très
culpabilisant.
Vogelstein et ses collègues ont posé une autre hypothèse : comme ces agressions sont le résultat du
fonctionnement normal des cellules, on aura des cancers dans les cellules qui se multiplient beaucoup.
Ils ont pris différents types de tumeurs et évalué le nombre de divisions cellulaires avec le risque de
cancer. C’est relativement corrélé.
Ils ont donc conclu que le cancer est associé au nombre de division du tissu qui le porte. Cela
n’explique que quelques cancers. Cela voudrait dire que l’on y est pour rien, le cancer est la résultante
d’anomalies endogènes et spontanées.
Cependant cette hypothèse a provoqué de nombreuses protestations notamment chez le corps médical :
faut-il arrêter de dire aux gens d’arrêter de fumer ? Puis une autre hypothèse a été avancée : Si le
nombre de cellules et de divisions cellulaires sont vraiment les principaux déterminants de
l’accumulation de mutations oncogéniques, le risque de cancer devrait augmenter avec la taille
corporelle et avec la durée de vie (reflets du nombre de cellules).
On a alors pris comme exemple les mammifères pour voir leur taux de cancer et si cette hypothèse
pouvait être validée. On a pris des nécropsies d’animaux décédés pour regarder la fréquence des
tumeurs, et le mettre en regard de données connues (taille et durée de vie des animaux). Le résultat est
que le cancer est indépendant de la taille et la durée de vie.
Néanmoins, on a observé que l’éléphant, très gros animal qui vit très longtemps, avait un taux de
cancer très bas, nettement inférieur à l’homme. On s’est alors demandé pourquoi il était protégé des
tumeurs. A-t-il un moyen de lutter contre les agressions ? Est-il capable plus que l’Homme de faire
face à des perturbations et altérations ? On a donc soumis à des agressions des lymphocytes
d’éléphants et d’homme par des radiations ionisantes, entraînant des cassures de l’ADN. La réponse va
être la réparation. Chez l’éléphant africain, il y a plus de lymphocytes qui entrent en apoptose que
l’homme: il a été mieux capable de se défendre contre les radiations ionisantes. Cela renvoie à la
capacité qu’a un organisme de se défendre face aux agressions.
Le génome de l’éléphant contient 40 copies de P53 : il a un très gros arsenal individuel or si on le
compare avec l’homme sain et ses deux copies de l’allèle de p53 et une personne atteinte du syndrome
de Li Fraumeni (ont perdu un allèle de P53 = prédisposition au cancer dès l’âge pédiatrique), on peut
clairement observer la corrélation entre le nombre de copies de p53 et la protection face aux cancers.
Nous ne sommes pas tous égaux devant le cancer.
On ne peut donc pas vraiment savoir pourquoi on fait un cancer et pourquoi on en fait pas car
beaucoup de facteurs entrent en compte, c’est encore un sujet d’hypothèse et de considérations.
Les signatures mutationnelles
Le taux et la nature des mutations varie énormément d’un type de cancer à l’autre. Les signatures
mutationnelles correspondent enfaite à des processus oncogéniques distincts.
UE1 Ronéo 3 Cours 5 15/18

Par exemple, les tumeurs
pédiatriques contiennent peu de
mutations car l’organisme n’a pas
eu le temps de les accumuler. Au
contraire, les organes en contact
avec l’extérieur, subissent plus de
mutations par les agressions
exogènes.
Cela nous permet d’en savoir plus sur la cause de ces cancers.
Par exemple, on sait que l’exposition à certains toxiques à l’origine du cancer du poumon va être à
l’origine de transversions G -> T suite aux hydrocarbones aromatiques.
Dans les mélanomes, on a aussi des mutations qui signent l’exposition aux UV : des transitions
CC->TT .
Le vieillissement quant à lui n’entraîne pas un type spécifique de mutation mais plutôt une
accumulation d’altérations spontanées, ce qui explique pourquoi on a pas les mêmes tumeurs chez
l’enfant et chez l’adulte.
Et on a aussi mis en évidence des signatures mutationnelles qui témoignent d’une instabilité du
génome, c’est globalement des défauts des systèmes de réparations.
Par exemple, les lymphocytes sont des cellules très sujets aux mutations, ils sont le site de la
recombinaison VDG qui génère des Immunoglobulines des récepteurs T par la cassure et la religation
de l’ADN. Les lymphocytes sont susceptibles d’avoir des anomalies « off target » c’est-à-dire sur
d’autres gènes que les Ig : cette anomalie est ainsi « signée » car on y retrouve les signes de la
religation, mécanisme très particulier.
Les différents points de contrôle.
• Contrôle de la stabilité chromosomique Point de contrôle G2>M (P53, …)
• Système de réparation des mésappariements : Intervient lorsque les mutations de l’ADN
résultent d’erreurs de la réplication (dérapage de l’ADN polymérase)
• Système de réparation NER (Nucleotide Excision Repair) : Système de réparation de
mutations induites par des carcinogènes environnementaux (UV, carcinogènes chimiques).
Tous ces points de contrôle peuvent être altérés lors d’un cancer.
Le système de réparation des mésappariements.
Il intervient lorsque les mutations de l’ADN résultent d’erreurs de la réplication (dérapage de l’ADN
polymérase) et il comprend les gènes hMSH2, hMLH1, hPMS1, hPMS2, hMSH6
Exemple : Le cancer du colon non polyposique est un cancer peut avoir des prédispositions génétiques
par des mutations constitutionnelles du SRM (MLH1 et MSH2). C’est généralement une maladie à
transmission autosomique dominante mais on peut aussi avoir des formes sporadiques dues soit à des
mutations ponctuelles soit à des extinctions épigénétiques de ces gènes, avec notamment MLH1 qui va
pouvoir avoir une hyperméthylation de son promoteur.
Les séquences répétées sont particulièrement touchées par les dérapages de l’ADN polymérase. Quand
on a un défaut d’un de ces gènes, on a un génotype bien particulier au niveau des séquences les plus
susceptibles de donner lieu à ces dérapages : ce sont les microsatellites.
UE1 Ronéo 3 Cours 5 16/18

Les microsatellites sont donc utilisés pour faire le profil des cancers du côlon lié à des défauts de
réparation des mésappariement : c’est l’instabilité des microsatellites.
On amplifie un panel de ces séquences grâce à la PCR, et lorsqu’au moins 5 ont cette allure-là, on
range le patient dans cette famille de cancers..
Extension de la notion de gènes suppresseurs de tumeurs.
Cette notion qu’on a vu dans le CC s’applique aussi aux gènes de contrôle des altérations de l’ADN,
au gènes de réparations c’est-à-dire que s’il y a une perte d’efficacité ils vont se conduire comme des
gènes suppresseurs de tumeurs, ils vont favoriser l’apparition de mutations dans d’autres gènes
suppresseurs de tumeurs. Ils vont agir en quelques sortes par ricochet sur d’autres gènes.
Les cellules tumorales partagent certaines propriétés.
Au-delà des premières caractéristiques
qu’on a évoqué comme le dérèglement
du CC et de l’apoptose, on voit des
caractéristiques dites émergentes comme
l’instabilité génomique et on verra au
cours prochain comment la
reprogrammation énergétique fait partie
des caractéristiques émergente des
cancers.
On sait maintenant détecter les mutations, le nouveau défi des années qui viennent est de maintenant
comprendre les conséquences fonctionnelles des mutations car même si il y a 500 mutations, à priori
toute ne vont pas être nécessaire car il y a un certain nombre de mutations dites conductrices, c’est à
dire qui vont conduire la sélection des tumeurs qui les portent et à coté il y a des mutations passagères
qui n’ont pas de rôle propre fonctionnelle dans les tumeurs.
Tout ça pourquoi ?
L’identification des oncogènes / gènes suppresseurs de tumeur et la compréhension de la structure des
tumeurs permet :
• de caractériser les propriétés des cellules tumorales
• d’améliorer le diagnostic, la classification et l’évaluation pronostique des cancers
• d’envisager des « thérapeutiques ciblées »
UE1 Ronéo 3 Cours 5 17/18

UE1 Ronéo 3 Cours 5 18/18





























