Embed Size (px)
Citation preview

Viewpoints: Chemists on Chemistry
Chemistry of the Heaviest Elements—One Atom at a Time
Darleane C. Hoffman and Diana M. Lee
Viewpoints: Chemists on Chemistry is supported by a grant from The Camille and Henry Dreyfus Foundation, Inc.
Other Material Related toNuclear Chemistry in This Issue
Chemistry of the Heaviest Elements— 332One Atom at a Time
Outline
Historical Perspective
Theory
Experimental TechniquesProduction of Heavy Element Isotopes
for Chemical StudiesDetection and AnalysisChemical Separation MethodsNew Instrumentation
Studies of Chemical PropertiesEarliest StudiesRenaissance and New Results
FutureMore “In-Depth” Chemical StudiesChemistry beyond Seaborgium?
93
Np
In keeping with the goal of the Viewpoints series of the Journalof Chemical Education, this article gives a 75-year perspective ofthe chemistry of the heaviest elements, including a 50-year retrospec-tive view of past developments, a summary of current researchachievements and applications, and some predictions about excit-ing, new developments that might be envisioned within the next 25years. A historical perspective of the importance of chemical separa-tions in the discoveries of the transuranium elements from neptunium(Z = 93) through mendelevium (Z = 101) is given. The developmentof techniques for studying the chemical properties of mendeleviumand still heavier elements on the basis of measuring the radioactive
decay of a single atom (“atom-at-a-time” chemistry) and combiningthe results of many separate experiments is reviewed. The influenceof relativistic effects (expected to increase as Z2) on chemical prop-erties is discussed. The results from recent atom-at-a-time studies ofthe chemistry of the heaviest elements through seaborgium (Z = 106)are summarized and show that their properties cannot be readilypredicted based on simple extrapolation from the properties of theirlighter homologues in the periodic table. The prospects for extend-ing chemical studies to still heavier elements than seaborgium areconsidered and appear promising.
1
H
3
Li
4
2
Be
11 12
Na Mg
19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36
37
K Ca Sc Ti V Cr Mn Fe Co Ni Cu Zn Ga Ge As Se Br Kr
He
5
B
6
C
7
N
8
O
9
F
10
Ne
13
Al
14
Si
15
P
16
S
17
Cl
18
Ar
Rb
38
Sr
39
Y
40
Zr
41
Nb
42
Mo
43
Tc
44
Ru
45
Rh
46
Pd
47
Ag
48
Cd
49
In
50
Sn
51
Sb
52
Te
53
I
54
Xe
55
Cs
56
Ba
57-71
89-103
La*
58
72
Hf
73 74
W
75
Re Os
77
I r
78
Pt
79
Au
80
Hg
81
Tl
82
Pb
83
Bi
84
Po
85
At
86
Rn
87
Fr
88
Ra Ac*
104
Rf
105
Ha
107 108 109
Bh Hs Mt
Ce 59
Pr
60
Nd
61
Pm
62
Sm
63
Eu
64
Gd
65
Tb
66
Dy
67
Ho
68
Er
69
Tm
70
Yb
71
Lu
90
Th91
Pa 92
U93
Np 95
Am
96
Cm
97
Bk
98
Cf
99
Es
100
Fm
101
Md
102
No
103
Lr
(113) (114) (115) (116) (117) (118)
Lanthanides
Act inides
106
Ta
Sg
76
94
Pu
110 111 112
1
2
3 4 5 6 7 8 9 10 11 12
13 14 15 16 17
18Group
57
La
89
Ac
Nucleogenesis! A Game with Natural Rules 349for Teaching Nuclear Synthesis and DecayDonald J. Olbris and Judith Herzfeld
Before There Was Chemistry: The Origin 353of the Elements as an Introduction to ChemistryNeil Glickstein
Periodic Tables of Elemental Abundance 356Steven I. Dutch
A Different Approach to a 3-D Periodic System 359Including Stable Isotopes Alexandru T. Balaban
JChemEd.chem.wisc.edu • Vol. 76 No. 3 March 1999 • Journal of Chemical Education 331
WE. O. Lawrence Berkeley National Laboratory has madeavailable videos, still images, and excerpts from inter-views with nuclear chemists who have discovered andstudied the heaviest elements. These materials can befound on JCE Online at http://JChemEd.chem.wisc.edu/Journal/issues/1999/Mar/abs331.html.

Viewpoints: Chemists on Chemistry
332 Journal of Chemical Education • Vol. 76 No. 3 March 1999 • JChemEd.chem.wisc.edu
Historical Perspective
Ever since ancient times, humans have been fascinatedwith trying to understand the composition of the worldaround them. The Greek philosophers considered the problem“theoretically”, and as early as the 4th century B.C.E., Aristotleproposed that all matter consisted of varying proportions ofthe four “elements”—air, earth, fire, and water. Within thenext few hundred years humans isolated and used a numberof elements such as gold, silver, and tin, which were foundrelatively pure in nature, and removed others such as sulfur,lead, and mercury from their ores. In medieval times, alchemistsisolated and discovered additional elements and dreamed ofturning lead into gold using secret formulas and incantations,but to no avail. With the development of experimental scienceand the scientific method in the 18th century, the pace ofdiscovery of new elements accelerated rapidly. But uranium,discovered in 1789 in pitchblende from Saxony, Germany,by Martin Klaproth, was to remain the heaviest knownchemical element for more than 150 years.
Beginning in the mid-1930s, the new breed of nuclearscientists, including both chemists and physicists, becameintrigued with the possibility of synthesizing new “artificial”elements not found in nature. The ancient alchemists’ dreamof transmutation was finally realized in 1937 when the firstman-made element, technetium (Z = 43), was synthesized byC. Perrier and E. Segrè (1). At Berkeley, in experiments designedto investigate the newly discovered (1939) phenomenon of
nuclear fission, E. M. McMillan and P. H. Abelson in 1940chemically isolated and identified the new element neptunium(Z = 93) in the products of neutron irradiation of uranium (2).Shortly thereafter, in December 1940, G. T. Seaborg, E. M.McMillan, J. W. Kennedy, and A. C. Wahl (3) identified anisotope of plutonium, and in February 1941 the first chemi-cal separation of plutonium was performed by Seaborg’s firstgraduate student, Art Wahl (4). Although these experiments wereconducted as academic research and without governmentalfinancial support, the discoverers voluntarily withheld publica-tion until 1946 because of wartime security concerns aboutthe fissionability of plutonium.
By 1961 the elements through lawrencium (Z = 103) hadbeen discovered, thus completing the actinide series. Sincethen, nine transactinide elements have been produced and iden-tified, so the elements through 112 are now known. Figure 1shows the 1998 periodic table; the 24 elements discovered since1936 are shaded. Table 1 gives the names and symbols forthe transfermium elements approved by the InternationalUnion of Pure and Applied Chemistry (IUPAC) in August1997, ending a long period of dissent and confusion. It alsoended use of the unwieldy “provisionary” names unnilquadium,unnilpentium, unnilhexium, etc., which had begun to appearon periodic tables in our classrooms, though they were neverused by researchers in the field! IUPAC is now consideringclaims to priority of discovery of elements 110, 111, and 112and will request suggestions from the discoverers for names forthese elements. To avoid confusion, we shall continue to usehahnium (Ha) for element 105 in this article because hahniumwas used in all our previous publications on the chemistry of
Chemistry of the Heaviest Elements—One Atom at a Time
Darleane C. Hoffman and Diana M. LeeDepartment of Chemistry, University of California, Berkeley, and Nuclear Science Division, E. O. Lawrence BerkeleyNational Laboratory, Berkeley, CA 94720*
1
H
3
Li
4
2
Be
11 12
Na Mg
19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36
37
K Ca Sc Ti V Cr Mn Fe Co Ni Cu Zn Ga Ge As Se Br Kr
He
5
B
6
C
7
N
8
O
9
F
10
Ne
13
Al
14
Si
15
P
16
S
17
Cl
18
Ar
Rb
38
Sr
39
Y
40
Zr
41
Nb
42
Mo
43
Tc
44
Ru
45
Rh
46
Pd
47
Ag
48
Cd
49
In
50
Sn
51
Sb
52
Te
53
I
54
Xe
55
Cs
56
Ba
57
La
58
72
Hf
73 74
W
75
Re Os
77
I r
78
Pt
79
Au
80
Hg
81
Tl
82
Pb
83
Bi
84
Po
85
At
86
Rn
87
Fr
88
Ra
89
Ac
104
Rf
105
Ha
107 108 109
Bh Hs Mt
Ce
59
Pr
60
Nd
61
Pm
62
Sm
63
Eu
64
Gd
65
Tb
66
Dy
67
Ho
68
Er
69
Tm
70
Yb
71
Lu
90
Th
91
Pa
92
U
93
Np
95
Am
96
Cm
97
Bk
98
Cf
99
Es
100
Fm
101
Md
102
No
103
Lr
(113) (114) (115) (116) (117) (118)
Lanthanides
Act inides
106
Ta
Sg
76
94
Pu
110 111 112
1
2
3 4 5 6 7 8 9 10 11 12
13 14 15 16 17
18Group
Figure 1. Periodic table from 1998,showing the 112 known elements.Elements discovered after 1936 arein blue; undiscovered elements arein parentheses.
*http://gateway.lbl.gov and http://bgsmc01.lbl.gov.
Chemistry of the Heaviest Elements—One Atom at a Time
JChemEd.chem.wisc.edu • Vol. 76 No. 3 March 1999 • Journal of Chemical Education 333
94
Pu
element 105, having been approved by the American ChemicalSociety in 1994 before the IUPAC approval of the compromisenames shown in Table 1.
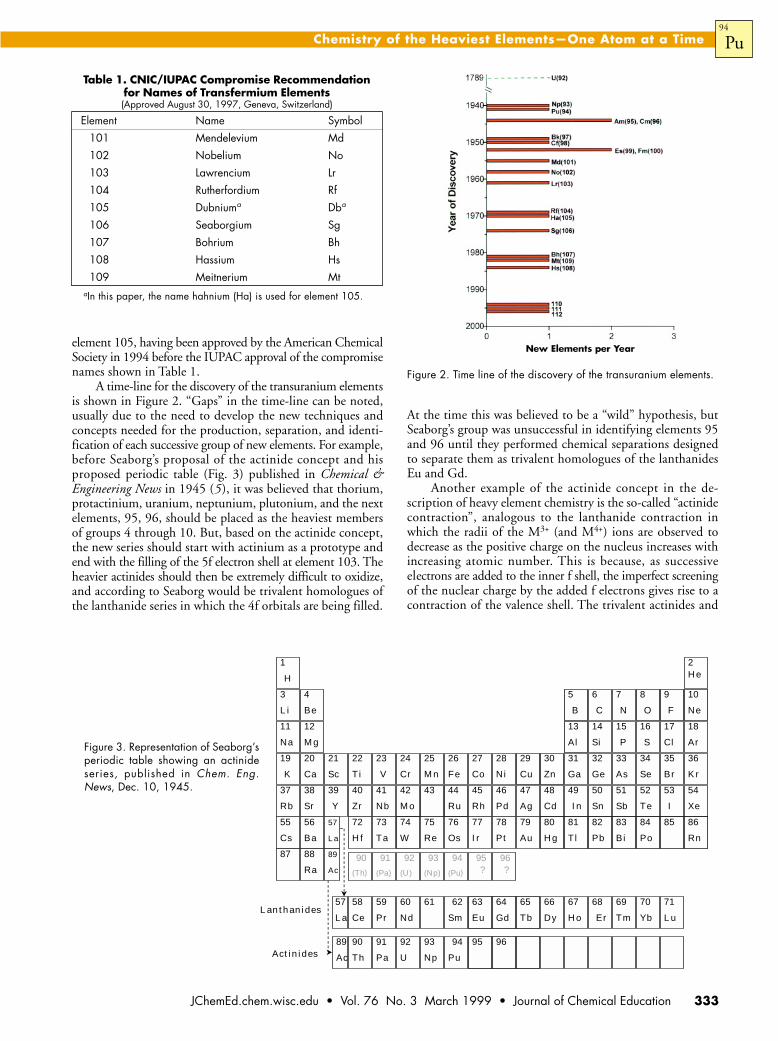
A time-line for the discovery of the transuranium elementsis shown in Figure 2. “Gaps” in the time-line can be noted,usually due to the need to develop the new techniques andconcepts needed for the production, separation, and identi-fication of each successive group of new elements. For example,before Seaborg’s proposal of the actinide concept and hisproposed periodic table (Fig. 3) published in Chemical &Engineering News in 1945 (5), it was believed that thorium,protactinium, uranium, neptunium, plutonium, and the nextelements, 95, 96, should be placed as the heaviest membersof groups 4 through 10. But, based on the actinide concept,the new series should start with actinium as a prototype andend with the filling of the 5f electron shell at element 103. Theheavier actinides should then be extremely difficult to oxidize,and according to Seaborg would be trivalent homologues ofthe lanthanide series in which the 4f orbitals are being filled.
At the time this was believed to be a “wild” hypothesis, butSeaborg’s group was unsuccessful in identifying elements 95and 96 until they performed chemical separations designedto separate them as trivalent homologues of the lanthanidesEu and Gd.
Another example of the actinide concept in the de-scription of heavy element chemistry is the so-called “actinidecontraction”, analogous to the lanthanide contraction inwhich the radii of the M3+ (and M4+) ions are observed todecrease as the positive charge on the nucleus increases withincreasing atomic number. This is because, as successiveelectrons are added to the inner f shell, the imperfect screeningof the nuclear charge by the added f electrons gives rise to acontraction of the valence shell. The trivalent actinides and
noitadnemmoceResimorpmoCCAPUI/CINC.1elbaTstnemelEmuimrefsnarTfosemaNrof)dnalreztiwS,aveneG,7991,03tsuguAdevorppA(
tnemelE emaN lobmyS101 muiveledneM dM201 muileboN oN301 muicnerwaL rL401 muidrofrehtuR fR501 muinbuD a bD a
601 muigrobaeS gS701 muirhoB hB801 muissaH sH901 muirentieM tM
aIn this paper, the name hahnium (Ha) is used for element 105.
Figure 2. Time line of the discovery of the transuranium elements.
Figure 3. Representation of Seaborg’speriodic table showing an actinideseries, published in Chem. Eng.News, Dec. 10, 1945.
1
H
3
L i
4
2
Be
11 12
Na M g
19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36
37
K Ca Sc Ti V Cr M n Fe Co Ni Cu Zn Ga Ge As Se Br K r
H e
5
B
6
C
7
N
8
O
9
F
10
Ne
13
Al
14
Si
15
P
16
S
17
Cl
18
Ar
Rb
38
Sr
39
Y
40
Zr
41
Nb
42
M o
43 44
Ru
45
Rh
46
Pd
47
Ag
48
Cd
49
I n
50
Sn
51
Sb
52
Te
53
I
54
Xe
55
Cs
56
Ba
58
72
H f
73 74
W
75
Re Os
77
I r
78
Pt
79
Au
80
H g
81
Tl
82
Pb
83
Bi
84
Po
85 86
Rn
87 88
Ra
89
Ac
Ce
60
Nd
61 62 63
Eu
64
Gd
65
Tb
66
Dy
67
H o
68
Er
69
Tm
70
Yb
71
L u
90
(Th)
91 92 93 95
Ta
76
94 96
59
Pr
62
Sm
57
L a
90
Th
92
U
93 62 95 9691
Pa
94
Pu
89
Ac
57
L a
L an t han i des
Act i n i des Np
(Pa) (U) (Np) (Pu) ? ?

Viewpoints: Chemists on Chemistry
334 Journal of Chemical Education • Vol. 76 No. 3 March 1999 • JChemEd.chem.wisc.edu
lanthanides are generally eluted from a cation exchange columnin order of the radii of the hydrated ions, with the largesthydrated ions eluting first. Thus Lr is eluted first, Md next,etc., and similarly for the lanthanides—Lu, Yb being first andthe rest following in order. This concept was utilized to performseparations of the individual actinides by elution from cationexchange columns with a variety of complexing agents, oneof the best being ammonium α-hydroxyisobutyrate (HIB).In the 1955 Md discovery experiment, 1.6-h 256Md wasseparated and chemically identified (6 ) by its elution with HIBfrom a carefully calibrated cation-exchange resin column inthe predicted position for trivalent element 101.
By the end of 1970, the actinide concept had been furthervalidated by chemical studies of the properties of No, Lr, andRf. According to the actinide hypothesis it was expected thatNo2+ might exist by analogy to its homologue Yb, which can bereduced from 3+ to 2+ with strong reducing agents. However,it was among the early surprises that not only was the 2+ stateof No achievable, it was actually the most stable oxidation statein aqueous solution! Thus the earliest report of the discovery(7 ) of nobelium, based on separating it as a trivalent actinide,could not have been correct. The chemical separations usedwould have separated No3+ but not No2+, which would havebeen present under the conditions of the experiment.
First studies of the solution chemistry of Lr and Rf,performed in 1970 by Silva et al. (8, 9), showed that for Lrthe trivalent state was again the most stable. Experiments withRf showed that the 4+ state was the most stable in aqueoussolution. Its properties were similar to those of Zr4+ and Hf 4+
and different from those of Lr3+ and the other trivalent actinides.These experiments thus confirmed that Lr actually did completethe actinide series and that Rf was the first transactinide ele-ment and the first member of a new 6d transition series. Rf waspositively identified by measuring the radioactive α-decay andhalf-life of about a minute of the well-known isotope 261Rf.
Experiments conducted in 1980 by Hulet et al. showedthat, in elutions from anion exchange columns, the chloridecomplexes of Rf also behave like those of the tetravalent group4 elements rather than those of the trivalent actinides (10).Some gas-phase experiments were conducted on Rf and Ha;but the atomic number of the species whose radioactivity wasbeing measured was not definitively established, so the resultsare somewhat suspect. No studies of the chemical propertiesof Ha in aqueous solution were reported until 1988.
Recent experiments have shown that although elements 104and 105 generally seem to belong to groups 4 and 5, theyalso show unexpected deviations in chemical properties fromtrends based on extrapolation from their lighter homologuesin the periodic table. In fact, Rf and Ha have been found tobehave more like the pseudo-homologues Th(IV) and Pa(V)under some conditions. On the other hand, preliminary studiesof Sg show that it behaves as the group 6 elements Mo andW, and not as the pseudo-homologue, U(VI). Recently,periodic tables similar to the one shown in Figure 4 have beenproposed (11–13). The first five actinides are shown in a stair-step arrangement leading from Rf down to the trivalentactinides Am and Cm, to reflect the similarities in chemicalbehavior between the early actinides and the elements ingroups 4 to 8. This arrangement shows Th and Pa as pseudo-homologues of the group 4 and 5 elements and, to a lesserextent, shows U, Np, and Pu as pseudo-homologues of the group6, 7, and 8 elements. Assessment of the validity of such arepresentation must await the results of more detailed studiesof the chemical behavior of the transactinide elements.
As illustrated in Figure 2, the definition of “heaviestelements” changes with time as new, still heavier elementsare discovered. In discussing the atom-at-a-time chemistry ofthe heaviest elements, Md is a natural place to begin. It wasdiscovered in 1955 and is the last of the heavy elements to beidentified initially by direct chemical separation techniques,
1
H
3
Li
4
2
Be
11 12
Na Mg
19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36
37
K Ca Sc Ti V Cr Mn Fe Co Ni Cu Zn Ga Ge As Se Br Kr
He
5
B
6
C
7
N
8
O
9
F
10
Ne
13
Al
14
Si
15
P
16
S
17
Cl
18
Ar
Rb
38
Sr
39
Y
40
Zr
41
Nb
42
Mo
43
Tc
44
Ru
45
Rh
46
Pd
47
Ag
48
Cd
49
In
50
Sn
51
Sb
52
Te
53
I
54
Xe
55
Cs
56
Ba
57-71
89-103
La*
58
72
Hf
73 74
W
75
Re Os
77
I r
78
Pt
79
Au
80
Hg
81
Tl
82
Pb
83
Bi
84
Po
85
At
86
Rn
87
Fr
88
Ra Ac*
104
Rf
105
Ha
107 108 109
Bh Hs Mt
Ce 59
Pr
60
Nd
61
Pm
62
Sm
63
Eu
64
Gd
65
Tb
66
Dy
67
Ho
68
Er
69
Tm
70
Yb
71
Lu
90
Th91
Pa 92
U93
Np 95
Am
96
Cm
97
Bk
98
Cf
99
Es
100
Fm
101
Md
102
No
103
Lr
(113) (114) (115) (116) (117) (118)
Lanthanides
Act inides
106
Ta
Sg
76
94
Pu
110 111 112
1
2
3 4 5 6 7 8 9 10 11 12
13 14 15 16 17
18Group
57
La
89
Ac
Figure 4. Periodic table showingearly actinides resembling group4–9 elements to a decreasingextent.

Chemistry of the Heaviest Elements—One Atom at a Time
JChemEd.chem.wisc.edu • Vol. 76 No. 3 March 1999 • Journal of Chemical Education 335
95
Am
and it is the first of the transuranium elements to be identifiedusing “one-atom-at-a-time” chemistry (6 ). That is, it waschemically identified by performing many chemical separationsin which a total of only 7 atoms of 256Md (half-life about 30min) were detected on a one-atom-at-a-time basis by measur-ing the radioactive decay of single atoms. An additional 8atoms of its radioactive decay product, 256Fm, were also sepa-rated and measured. The 256Md was produced at the 60-inchcyclotron at Berkeley by helium-ion bombardment of onlyabout 109 atoms of the very radioactive target, 20-day 253Es.
The heavier elements from nobelium (Z = 102) through112 must be produced in irradiations with heavier projectilesthan helium ions and were identified first by physical techniques.Many controversies developed concerning the discoveries ofelements 102, 103, 104, and105 because of the very lowproduction rates, the half-livesof minutes or less, and the ne-cessity for developing new tech-niques other than chemicalseparations for positive iden-tification of atomic number.The currently known isotopesof Md through Sg are shownin Figure 5 (bottom). Atom-at-a-time chemical studieshave now been performed onall these elements; the firststudies on Sg were reportedby Schädel et al. in 1997 (12,13). The known isotopes of Sgthrough element 112 are shownin Figure 5 (top), which clearlyshows that the half-lives de-crease rapidly with increasingatomic number, and that thecandidates for chemical studiesbeyond Sg are relatively fewand will depend on the discov-ery of longer-lived isotopes.
The production and studyof the heaviest elements requiresspecial facilities and capabilitiesand presents unique challengesto the chemist. These includespecial facilities for preparationand use of radioactive targets;an accelerator that can furnishhigh-intensity beams of lightto heavy ions; provision forpreventing contamination ofthe accelerator should a radio-active target rupture; methodsfor rapidly and efficientlytransporting the desired prod-ucts from the hostile radiationenvironment associated with theproduction site in the accelera-tor beam line to a facility out-side the radiation field wherechemistry can be performed;and selective and efficient
chemical separations for measurement and identification ofisotopes whose half-lives are minutes or less and that havevery small production rates. A single atom must be detectedby measuring its known radioactive decay properties. Forexample, in the chemical studies of Rf, only one atom wasdetected in about every ten one-minute chemical separations,resulting in detection of several atoms per hour. In recentlyreported studies of the 7- and 21-s 265,266Sg isotopes, only15 atoms were detected during weeks of running time. Con-sequently, chemical separation procedures that come to equi-librium rapidly and are valid on an atom-at-a-time basis mustbe used. Personnel trained in techniques of handling radio-activity and in nuclear and radiochemistry are essential. Suchfacilities and capabilities exist at only a few laboratories in
152150 154 156 158 160148
250 251 252 253 254 255 256 257 258 259 260 2620.3ms 0.6s 2.3 s 1.7m
55s 3.1m 2.9 s 25 s 1.2ms 58 m 106ms 5 msSF α α,SF α α α,SF α SF α,EC SF SF
146
247 248 249 250 251 252 253 254 255 256 257 258 259 2603 s 7 s 24 s 50 s 4 m 2 m 30m 10m 27 m 1.3 h 5.5 h 57m 52 d 1.6 h 27.8 dα αEC,α α,EC EC,α EC,α EC,α EC EC EC,α EC,α EC,α EC SF SFα
6 m~
0.3sIT
253 254 255 256 257 258 259 260 261 262 263?
253 254 255 256 257 258 259 260 261 2621.3 sα
13 sα
22 sα,EC α,EC α,EC α,ECα
SF SF
SF
SF
α,SF
α,SF α,SF α,SF α,SF?
EC
SF,α SF,α? SF
26 s 0.65 s 3.9 s 6.1 s 3 m 39 m 3.6 h
0.05 ms 0.02 ms 1.4 s 7 ms 4.8 s 13 ms 3 s 20 ms 78 sSF
47ms
257 258 260 261 262 263
α,SF α,SF α,SF SF,αEC,α EC,α1.3 s 4.4 s 1.5 s 1.8 s 34 s 27 s
Md 101
258 259 260 261 263 265 2662.9msSF α,SF? α,SF α,SF? α,SF? α α
0.5 s 4 ms 0.2 s .9s .3s 7 s 21 s
Half-life Range
1m to 1h
1s to 1m
< 1s
1h to 1d
> 1d
N
No 102
Lr 103
Rf 104
Ha 105
Sg 106
2s
SF
Half-life Range
1s to 1m
10ms to 1s
152 154 156 158
N
160 162 164
258 259 260 261 263 265 2662.9msSF α,SF? α,SF α,SF? α,SF? α α
0.5 s 4 ms 0.2 s 7 s 21 s
261 262 264
α,SF?12 ms 0.44s
α
263 264 265 267
α α ,SF? α? 0.5ms 0.8
ms
2699 s1.7
msα
33ms
266
α
26870 ms3.4ms
α
267? 269 271
α α4 us
273
α0.17ms
1.1ms
56ms
α0.1ms
110
272
α1.5ms111
277112 0.24ms
α
Sg 106
Bh 107
Hs 108
Mt 109
<10ms
0.1s
?
8ms
0.1s
α
0.9s
0.3s
Figure 5. Chart of the known transfermium isotopes as of 1998. Top: Seaborgium through element112. Bottom: Mendelevium through seaborgium. Modes of decay are indicated as follows: α =alpha-particle (4He2+) decay; SF = spontaneous fission; EC = electron-capture decay.

Viewpoints: Chemists on Chemistry
336 Journal of Chemical Education • Vol. 76 No. 3 March 1999 • JChemEd.chem.wisc.edu
the world. These include the Lawrence Berkeley NationalLaboratory (LBNL)1; Gesellschaft für Schwerionenforschung(GSI) near Darmstadt, Germany; the Joint Institute forNuclear Research (JINR), Dubna, Russia; and the PaulScherrer Institute (PSI) in Villigen, Switzerland.
With all the difficulties and challenges involved, why shouldwe go to so much effort to study the chemical properties ofthe heaviest chemical elements? These investigations areamong the most fundamental in all of chemistry in that theyare the only means for obtaining the information required toplace the heaviest elements in the appropriate group or series inthe periodic table and to compare their properties with thoseof their proposed lighter homologues. It was recognized ratherearly that the increasingly strong “relativistic effects” (14–17 ),predicted to increase proportionally as Z 2, would lead to everlarger deviations from the periodicity of the chemical proper-ties of the heavier elements based on simple extrapolation ofthese properties from those of their lighter homologues. As theatomic number, and hence the positive charge on the nucleus,increases, there is a contraction of the s and p electronicorbitals. This can give rise to changes in the electronic orbitalconfigurations, even extending out to the valence electrons.Consequently, changes in ionic and atomic radii, ionizationpotentials, most stable oxidation states (redox potentials), andcomplexing ability are predicted. Experimental studies of theheaviest elements provide the unique opportunity to compareactual experimental results with theoretical predictions inorder to assess the magnitude of the influence of relativisticeffects on chemical properties at the very highest atomicnumbers, where the effects should be largest and coulddrastically change chemical properties.
Theory
Studies of the chemical properties of the heaviest elementsare extremely challenging for theorists as well as for experimen-talists. The Schrödinger equation is no longer applicable andfully relativistic calculations must be performed. Fundamentalpredictions of the properties of the elements through Z = 172,based on atomic relativistic calculations, were reported more than25 years ago (14–17 ) and indicated the deviations in atomicproperties that might be expected owing to relativistic effects.
For example, in 1975, Pitzer reported the striking conclusion,based on initial results from Hartree–Fock relativistic calcula-tions, that elements 112 and 114 as well as 118 (eka-Rn) mightbe volatile, relatively inert gases (18). Relativistic atomic cal-culations also led to predictions that the transactinides wouldbe members of a 6d transition series and show similarities tothe 4d and 5d transition series elements.
In a 1988 review of relativistic effects on structuralchemistry, P. Pyykkö summarized the primary relativisticeffects on atomic orbitals as (i) contraction of the radius andenergetic stabilization of s and p electronic orbitals; (ii) spin-orbit splitting of the l > 0 orbitals; (iii) a resulting increase inthe radii and energetic destabilization of the outer d and all forbitals (19). As mentioned earlier, these effects can give rise todrastic changes in the chemical properties of the heaviest ele-ments. The diagram shown in Figure 6 of the nonrelativisticvs relativistic Dirac–Fock calculations (20) of the valenceorbital eigenvalues for the group 6 elements, Mo, W, and Sg,illustrates the large differences due to relativistic effects.
In recent reviews, Pershina (21, 22) gives excellentsummaries of the development of and recent improvements invarious computational methods for making more accurate rela-tivistic atomic calculations of heavy-element atomic properties.However, she points out that although these atomic calculationsgive some general guidance for experimental research, theydo not predict the behavior of molecular species, especially un-der particular experimental conditions. This still more difficultand complex theoretical problem was undertaken by Pershinaand coworkers in conjunction with our initial internationalcollaboration of scientists from the USA, Germany, Switzerland,and Russia to perform experimental studies of the compoundsof the heaviest elements. Systematic theoretical studies of heavy-element compounds using relativistic quantum-chemicalcalculations were combined with fundamental physicochemicalconsiderations to make detailed predictions (23, 24 ) of thevolatilities of the halides and the partitioning of the trans-actinides and their lighter homologues between aqueous andorganic solvents for specific experimental conditions.
Pershina and Fricke (25) have recently completed a reviewof theoretical studies of the molecular properties of the trans-actinides. Except for a very few early publications, most ofthese studies were initiated in the 1990s, indicating the
Figure 6. Valence orbital eigenvaluesof group 6 metals.

Chemistry of the Heaviest Elements—One Atom at a Time
JChemEd.chem.wisc.edu • Vol. 76 No. 3 March 1999 • Journal of Chemical Education 337
96
Cm
increasing interest of theorists in this new fieldwhich was sparked by the recent acquisition of ex-perimental data on Rf, Ha, and Sg. Relativisticmolecular codes have successfully described theelectronic structure of compounds and predicted mo-lecular properties for the gas-phase compounds ofthe transactinides and their complexes in aqueous so-lutions. Especially promising are predictions of ac-tual equilibria of reactions. The agreement betweencalculations (24 ) and experiment (26 ) “confirmedthe necessity for doing relativistic molecular orbital calculationsand the unreliability of the straightforward extrapolations ofproperties within the chemical groups”—4, 5, 6 of the peri-odic table. Additional theoretical calculations and new meth-ods must be developed to treat the still more complex chem-istry expected for elements 107 (Bh) and 108 (Hs), which liketheir lighter group 7 and 8 homologues should exhibit a muchlarger variety of oxidation states, but with preferential stabil-ity of the lower ones.
Experimental Techniques
Production of Heavy Element Isotopes for ChemicalStudies
Although Md and No were initially produced and iden-tified in atom-at-a-time studies, they can now be producedin much larger—although still not weighable—quantities byirradiation of larger targets of radioactive isotopes such as253,254Es and 249Cf. But beginning with Lr, owing to the veryshort half-lives of the isotopes and/or low production rates, allchemical experiments have been conducted on a one-atom-at-a-time basis. Normal chemical analysis techniques are usuallynot applicable and the atoms must be detected by measuringthe radioactive decay of a single atom at a time.
Typically, the longest-lived known isotope of the elementis used for chemical studies, if it can be made with a “reason-able” production cross section2 and if the appropriate targetmaterial and projectiles are available. The isotopes used forthe first chemistry performed on each of the transfermiumelements, together with their half-lives, production reactions,and the year of the experiments, are given in Table 2. These areall “compound” nucleus reactions, which means that the in-coming projectile and the target atom completely fuse to forma highly excited nucleus, which then can de-excite in a varietyof ways—fission being one of the most probable for the heavyelements. The desired reactions constitute only a small fractionof the large variety of unwanted reactions that occur.
Cartoon of Compound Nucleus Reaction
As can be seen from Figure 5 (bottom), considerably longer-lived isotopes of some of these elements have now been synthe-sized. In the case of the transnobelium isotopes, the productioncross sections are nanobarns or less, while those for fission orother competing reactions may be barns or millibarns.
In some later studies, “transfer” reactions, in which only aportion of the projectile is transferred to the target nucleus and
fuses with it, were used to produce neutron-rich products nearthe atomic number of the target in high yield. For example, stud-ies of Lr chemistry utilized 259,260Lr produced by transfer of 5,6Befragments from 22Ne projectiles to the rare (currently availableonly in microgram quantities) target nucleus 276-d 254Es.
In current studies of chemical properties, 3-min 260Lr,75-s 261Rf, and 34-s 262Ha produced in reactions of 18Oprojectiles with 249Bk (320 d) and 248Cm (3.5 × 105 y) targetsare normally used. These reactions have cross sections of afew nanobarns.
Detection and AnalysisBeginning with Md, methods were devised for removing
the desired reaction products from the hostile irradiation sitein the accelerator, rather than removing the highly radioactiveand difficult to prepare targets and chemically dissolving andprocessing them to remove the desired isotope. Owing to themomentum imparted to them during the nuclear reaction, thedesired reaction products (along with many of the unwantedrecoiling reaction products) will recoil out of the relativelythin targets and can be collected in a variety of ways
In the earliest studies, they were deposited on a thin“catcher” foil placed directly behind the target in the produc-tion chamber of the accelerator. The foil can then be removedmanually or remotely shuttled to a detection system, withoutdisturbing the accelerator vacuum, and analyzed directly forits α and spontaneous fission (SF) activity with appropriateradiation detectors. Alternatively, the collector foil can beremoved and chemically processed. In any case, the valuabletarget is not destroyed, and considerable decontamination fromall of the activity remaining in the target itself is achieved.
Later, gas transport systems were developed and utilizedto rapidly and efficiently transport reaction products attachedto various aerosols (e.g., water vapor, NaCl, KCl, MoO3, and Cclusters) to collection foils or automated systems for chemistryor direct measurements of radioactivity. A schematic diagramof the target system and the helium (KCl aerosol) gas transportsystem used by our Heavy Element Nuclear and Radiochemis-try Group at the LBNL 88-inch cyclotron is shown in Figure 7.It can be used to transport reaction products to a manualcollection site as shown, or to automated chemical processingsystems, directly to γ- or X-ray spectrometer systems, or to ourrotating wheel system, the Merry Go-Around (MGA) (Fig. 8)for measurement of α and SF activity.
One of the major problems in the determination ofchemical properties is to make sure that the detection methodcan positively identify the activity being measured as belongingto the desired element. A technique that can provide thisproof and has been widely used in the identification of newheavy elements is measuring the known half-life and the en-ergies of the α-particles of the isotope being studied and thetime relationships between its α-particles and those emitted
601–101stnemelE,seidutSlacimehCtsriFnidesUsepotosI.2elbaT)efil-flaH(epotosI noitcaeR raeY ecnerefeR
652 :)h3.1(dM 352 sE + 4 eH → 652 n+dM 5591 (.lateosroihG 6 )552 )nim1.3(oN 442 uP + 61 O → 552 n5+oN 8691 (.lateylaM 72 )652 )s62(rL 942 fC + 11 B → 652 n4+rL 0791 (.lateavliS 8 )162 )s57(fR 842 mC + 81 O → 162 n5+fR 0791 (.lateavliS 9 )262 )s53(aH 942 kB + 81 O → 262 n5+aH 8891 (.latehcirogerG 82 )
662,562 )s12,s7(gS 842 mC + 22 O → 662,562 n4,n5+gS 7991 (.lateledähcS 21 )

Viewpoints: Chemists on Chemistry
338 Journal of Chemical Education • Vol. 76 No. 3 March 1999 • JChemEd.chem.wisc.edu
Figure 7. Schematic of target system and helium (aerosol)gas transport system to 4-position collection site used atthe 88-inch cyclotron by the Berkeley Heavy ElementNuclear and Radiochemistry Group.
Figure 8. Darleane Hoffman and Diana Lee at the LBNL 88-inch cyclotron with the Merry Go-around (MG) rotatingwheel system, which is outside the cyclotron irradiation siteand shielding. During an experiment, aerosols containingthe recoiling reaction products are transported about 10m from the target chamber to the MG via the helium gastransport system through a capillary (shown next toHoffman’s right hand) and deposited successively on thinpolypropylene disks in the 80 collection positions aroundthe periphery of the wheel. The wheel is then stepped atappropriate time intervals for collection of the desired ac-tivity and the disks are positioned between detectors above(shown) and below the wheel for measurement of α andSF activity.
Figure 9. Ken Gregorich and Darleane Hoffman demonstrat-ing the simple setup for the “manual” chemistry performed inthe first studies of the solution chemistry of Ha. The fume hoodis outside the cyclotron shielding and the Lucite enclosure con-taining the collection wheel, shown schematically in Fig. 7,can be seen in the corner.

Chemistry of the Heaviest Elements—One Atom at a Time
JChemEd.chem.wisc.edu • Vol. 76 No. 3 March 1999 • Journal of Chemical Education 339
97
Bk
from its known daughters and even granddaughters. Since theatomic number (Z ) and mass number (A) of the daughternuclides are known, the parent atomic number and mass num-ber will simply be that of the daughter plus an α-particle;that is, Z = 2, A = 4. This technique was used in the discoveryand identification of Sg (element 106) in 1974 by Ghiorsoet al. (29) and in the confirmation of that discovery byGregorich et al. (30) in 1994, using the production reaction249Cf +
18O → 263Sg + 4n. The α-decay sequence shown below
was used to positively identify 263Sg using this method, whichis usually referred to as the “α-α correlation technique”.
Diagram of 263Sg α -Decay Chain% of α or SF decay is noted in the box for each nuclide; energies of themajor α groups are given underneath. The most intense group is underlined.
Although the SF fragments can be measured with greatsensitivity, such measurements do not provide positive iden-tification of the element that fissioned because the processitself destroys the original nucleus. The nucleus divides intotwo large fission fragments, which then de-excite by emittingneutrons, photons, and β-particles until they reach a long-lived or stable nucleus.
Schematic of Spontaneous Fission Process
A wide variety of mass “splits” is possible, and the Z ofeach of the two original fission fragments must be determinedfor each different split in order to determine the Z of theatom being studied. It has so far not been possible to measurethe Z and A of the fission fragments to infer the Z and A ofthe fissioning nucleus from atom-at-a-time measurements.Half-life measurements are insufficient to provide positiveidentification because the SF half-lives of the various elementsoverlap and cover an extremely wide range of values from4.5 × 109 years for 238U to milliseconds and microsecondsfor many of the isotopes of the transfermium elements shownin Figure 5. In early measurements, SF activity was detectedfrom the “tracks” made by the fragments in mica or glass
plates, but specially designed solid-state silicon detectors arenow typically used to measure the energies and decay of bothSF and α-activities.
Chemical Separation MethodsThe chemical procedures used in atom-at-a-time studies
must be rapid enough to accomplish in times comparable tothe half-lives of the isotopes used in the studies. Furthermore,the chemistry must give the same results for only a few atomsas for tracer and macro amounts. This typically limits thetechniques to ion exchange and gas chromatography, solventextraction, and other procedures with fast kinetics in whichthe atoms rapidly undergo many identical chemical reactionsbetween two-phase systems in which equilibrium is attainedrapidly. Adloff and Guillaumont (31) have thoroughly discussedthe validity of conclusions about chemical behavior obtainedfrom very small numbers of atoms. They defined an equilib-rium constant for such reactions in terms of the probabilitiesof finding the species in one phase or the other and concludedthat it is valid to combine the results of many separate one-atom-at-a-time experiments in order to get statistically sig-nificant results (32, 33).
Both manual and automated systems have been used toperform atom-at-a-time chemical separations of the heaviestelements. In the manual separations, after deposition of theactivity-laden aerosol on one of the disks positioned on thecollection wheel shown in Figure 7, the wheel is rotated andthe disk is manually removed. Subsequently, rather standardchemical procedures, usually liquid–liquid extractions or ion-exchange column separations, are performed very rapidly with5–10-µL volumes of solutions. The photo of the ratherordinary looking chemical fume hood and equipment (Fig. 9)shows the Lucite housing containing the collection stationfor the aerosols in the corner. This hood is outside the cyclotronvault and shielding, some 10 m away from the radiation field ofthe production chamber and beam line. The simple equipmentshown was used in the first studies of the aqueous chemistryof Ha, which established that it behaved similarly to the penta-valent group 5 elements and that its most stable oxidationstate in aqueous solution was 5+. A similarly simple setup withthe addition of stoppered mini-test-tubes and a centrifugewas used in liquid–liquid extraction studies of Rf. The successof such “manual” studies depends on the dexterity, speed, andendurance of the graduate students and staff who perform them,rather than on expensive, computer-controlled automatedsystems. However, although the automated systems are notnecessarily faster, they usually give more reproducible resultsand are more appropriate for around-the-clock experimentslasting weeks at a time.
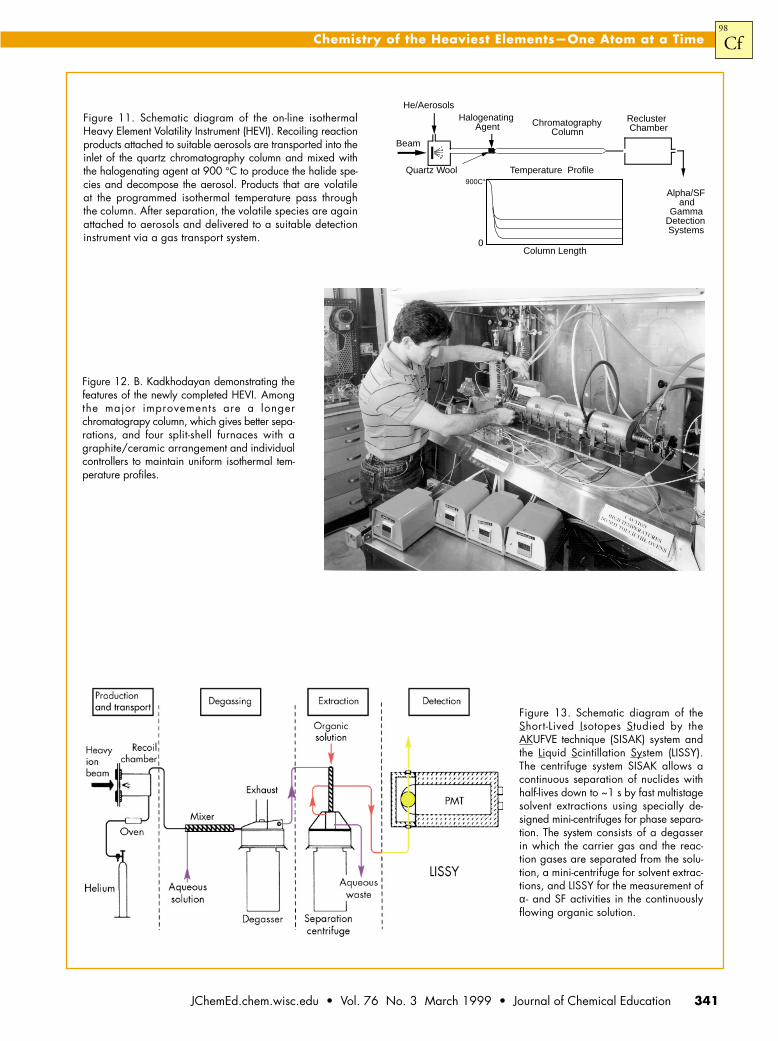
New InstrumentationA comprehensive review of the instrumentation for atom-
at-a-time chemistry of the heavy elements up to 1996 is givenin ref 34. Among the systems used in studies of the aqueouschemistry of the heaviest elements is the Automated RapidChemistry Apparatus (ARCA II, mini-ARCA) developed byscientists at the Institut für Kernchemie, Johannes Gutenberg-Universität Mainz and the GSI, Germany. Figure 10 illustratesthe complexity of the equipment compared to that for themanual experiments! ARCA II is used to perform rapid, repeated,high-pressure liquid chromatography column experiments onthe seconds time scale. The flow of various solutions is directed

Viewpoints: Chemists on Chemistry
340 Journal of Chemical Education • Vol. 76 No. 3 March 1999 • JChemEd.chem.wisc.edu
through small chromatography columns by microcomputercontrol of a series of pumps, valves, and mechanical sliders.It is equipped with two “magazines”, each containing 20 mini-chromatography columns (1.6 mm i.d. × 8 mm long). Theactivity-laden KCl aerosols from the gas transport system arecollected by pumping the carrier gas through a glass frit. After asuitable collection time, the KCl aerosol is dissolved in a smallvolume of solution and the flow is directed to one of thecolumns. The eluting solution is pumped through and thedesired fraction is collected on Ta or Pt plates held in positionson the hot-plate. A heat lamp and gas stream are also used tospeed evaporation, but it still takes approximately 35 s beforethe samples are dried and ready for a-spectroscopy measurements.
Both thermochromatographic and isothermal methodshave been used to study gas-phase properties. These areespecially suitable for studies of short-lived isotopes becausethe time-consuming step of evaporating a liquid sample isavoided. I. Zvara et al. (35, 36 ) at Dubna pioneered the useof the thermochromatographic method to study the halidesof Rf and Ha. Unfortunately, in these early studies only SFactivity was measured, so the identity of the element beingstudied is uncertain and the results cannot be considered de-finitive. The use of automated isothermal systems, such asthe On-Line Automated Gas Analyzer (OLGA) for studiesof the volatilities of the halides of the heaviest elements waspioneered by Gäggeler and his group (37 ) at the Paul ScherrerInstitute in Switzerland. In thermochromatographic systems,the chromatography column has a temperature gradient todeposit different species along the column according to theirvolatility. By contrast, in an isothermal system, the entire quartzcolumn after the high-temperature halogenation position iskept at a constant temperature. A series of temperatures isthen run and the amount of the given species passing throughthe column at each temperature is determined. An improvedisothermal system, the Heavy Element Volatility Instrument(HEVI), built at LBNL by B. Kadkhodayan et al. (38), gavebetter separations and more uniform isothermal temperatureprofiles. A schematic diagram of the HEVI isothermal gas-phase chemistry system is shown in Figure 11 and a photoof HEVI, just after its completion, is shown in Figure 12.Recently, in OLGA III (39), further improvements have been
made to reduce the time between production of the isotopeand final detection by miniaturizing and cooling the aerosolreclustering chamber so that isotopes with half-lives as shortas 1 to 4 s can be studied.
A schematic diagram (34) of the SISAK apparatus (40,41) is shown in Figure 13. This is a microcentrifuge systemfor performing liquid–liquid extractions on the seconds timescale, coupled to a continuously flowing liquid scintillationsystem (LISSY) for detection of α-α correlations. It has beenused in studies of the transactinides, but improvements insensitivity are needed to make measurements of nuclidesproduced with cross sections of nanobarns or less.
Studies of Chemical Properties
Earliest StudiesE. K. Hulet reviewed the results of studies of chemical
properties of the transeinsteinium elements as of 1983 (42).By the late 1970s chemical studies had established that themost stable oxidation states of the transfermium elements inaqueous solution were Md, 3+; No, 2+; Lr, 3+; and Rf , 4+; andthe redox potentials shown in Table 3 had been measured.The volatility of Md metal had been compared with that ofother actinide metals and because of its high volatility it wasbelieved to be a divalent metal. No experimental verificationof the electronic structure of Md had been attempted, but itwas calculated to be [Rn]5f137s2.
Although early gas-phase studies of the halides of Rf andHa (35, 36 ), suggested that they behaved like their lightergroup 4 and 5 homologues, only SF activity was detected,
Figure 10. Photo of microcomputer-controlled ARCA II(mini-ARCA). The two cartridges containing the 20 tinychromatographic columns can be seen above the Tefloncapillary, which delivers the eluting solution to Ta disksheld on the circular sample collector on the hot-plate. Aheat lamp to aid in drying the samples is just behind thecartridges; in front of it is an arrangement for blowingcool gas on the already dried disk before placing it in thesolid-state detector system.
slaitnetoPnoitcudeR.3elbaT
tnemelE(noitcaeRflaH E°/ )V a
III → II III →0 II →0)99(sE � 55.1 � 89.1 � 2.2
)001(mF � 51.1 � 59.1 � 73.2
)101(dM � 2.0 � 7.1 � 4.2
)201(oN 4.1 � 1.1 � 53.2
)301(rL (< � )44.0 � 60.2 —aRelative to the standard hydrogen electrode.
Chemistry of the Heaviest Elements—One Atom at a Time
JChemEd.chem.wisc.edu • Vol. 76 No. 3 March 1999 • Journal of Chemical Education 341
98
Cf
Beam
Temperature Profile
ChromatographyColumn
900C°
0Column Length
ReclusterChamber
Quartz Wool
Alpha/SFand
GammaDetectionSystems
He/AerosolsHalogenating
AgentFigure 11. Schematic diagram of the on-line isothermalHeavy Element Volatility Instrument (HEVI). Recoiling reactionproducts attached to suitable aerosols are transported into theinlet of the quartz chromatography column and mixed withthe halogenating agent at 900 °C to produce the halide spe-cies and decompose the aerosol. Products that are volatileat the programmed isothermal temperature pass throughthe column. After separation, the volatile species are againattached to aerosols and delivered to a suitable detectioninstrument via a gas transport system.
Figure 12. B. Kadkhodayan demonstrating thefeatures of the newly completed HEVI. Amongthe major improvements are a longerchromatograpy column, which gives better sepa-rations, and four split-shell furnaces with agraphite/ceramic arrangement and individualcontrollers to maintain uniform isothermal tem-perature profiles.
Figure 13. Schematic diagram of theShort-Lived Isotopes Studied by theAKUFVE technique (SISAK) system andthe Liquid Scintillation System (LISSY).The centrifuge system SISAK allows acontinuous separation of nuclides withhalf-lives down to ~1 s by fast multistagesolvent extractions using specially de-signed mini-centrifuges for phase separa-tion. The system consists of a degasserin which the carrier gas and the reac-tion gases are separated from the solu-tion, a mini-centrifuge for solvent extrac-tions, and LISSY for the measurement ofα - and SF activities in the continuouslyflowing organic solution.

Viewpoints: Chemists on Chemistry
342 Journal of Chemical Education • Vol. 76 No. 3 March 1999 • JChemEd.chem.wisc.edu
and therefore the identity of the species being measured couldnot be positively attributed to element 104 or 105. No studiesof the aqueous properties of Ha (Z = 105) had been per-formed at all. The longest-lived known isotope of Sg was263Sg, which had a half-life of only 0.9 s and no chemicalstudies had yet been attempted.
Renaissance and New ResultsAfter the pioneering studies of the chemical properties of
Md, No, Lr, and Rf came a rather long hiatus extending fromthe 1970s until the late 1980s, when there was a “renaissance”of interest in Lr and the transactinides. This was sparked inpart by predictions (summarized by Keller [43] in 1984) thatrelativistic effects might cause qualitative changes in electronicconfigurations such that the valence electron structure of Lrmight be 7s27p1/2—rather than 6d7s2, as would be expectedby analogy with the 5d6s2 valence configuration of Lr’s homo-logue Lu. Similarly, Rf and Ha might also be expected toexhibit stabilized 7p1/2 orbitals. These relativistic effects mightresult in chemical properties significantly different from thosepredicted by simple extrapolation of periodic table trends.Our Heavy Element Nuclear and Radiochemistry Group atLBL, composed largely of graduate students from the Chem-istry Department of the University of California, Berke-ley, set out to investigate these effects on the chemical prop-erties of the elements at the end of the actinide series andthe beginning of the transactinide elements. Descriptions ofa few representative recent studies follow.
Although 3-min 260Lr was discovered in 1971, it was notused for studies of Lr chemistry until 1987, when we used itin manual studies designed to deduce the ionic radius of Lr3+.The position of Lr3+ in elutions from cation-exchange resincolumns with ammonium HIB was compared with those oftrivalent lanthanides. The time from production of 260Lr tothe beginning of the measurements of its α decay was 5 to 6minutes. Although only seven α-events were detected in morethan 20 separations, we found that Lr3+ eluted in nearly the sameposition as Er3+, showing that they had similar ionic radii(44 ). Later, a long-lasting collaboration was initiated whenthe GSI/Mainz group brought ARCA to LBL to conduct moreexperiments on Lr chemistry. Our earlier result was corrobo-rated and its statistical significance was improved (45). Fromthe ARCA experiments, Lr3+ was inferred to have an ionicradius of 0.0881 nm. This result was unexpected because itgives a difference of only 0.0015 nm between Md3+ (0.0896nm) and Lr3+, which differ by 2 Z. This difference is muchsmaller than the 2 Z separation of 0.0021 nm between thehomologous lanthanide ions Tm3+ and Lu3+. Attempts to reduceLr3+ with V2+ and Cr2+ in dilute HCl were unsuccessful (46 ),and the resulting limit for the reduction potential of theLr3+/Lr1(2)+ couple was found to be more negative than �0.44 V.
In 1988, discovery of 39-min 261Lr and 3.6-h 262Lr byLawrence Livermore National Laboratory/LBL collaboratorspermitted longer chemical procedures to be performed (47 ).Attempts to reduce Lr3+ with Sm2+ and coprecipitate Lr1+ withRb1+ tetraphenylborate or chloroplatinate were unsuccessful.The Lr3+/ Lr1+ couple was estimated to be even more negativethan �1.56 V, making it unlikely that Lr1+ can exist in aque-ous solutions.
Since no experiments on Ha in aqueous solution had yetbeen performed, we also began experiments on Ha in 1987simply to determine its most stable oxidation state in aqueous
solution. We postulated that Ha might have a 7s26d7p2 valenceconfiguration, rather than 6d37s2 as expected by analogy withthe 5d36s2 valence configuration of its lighter homologue,Ta(V). It was conceivable that relativistic effects stabilized the7s2 electrons sufficiently that the most stable state of Ha in aque-ous solution might be 3+ rather than 5+—unlike its lightergroup 5 homologues, Nb and Ta, for which 5+ is most stable.Gregorich et al. used so-called “glass chemistry” to performthe first aqueous chemistry experiments on element 105, using35-s 262Ha (28). KCl aerosols were deposited on glass coverslips held on the collector wheel (Fig. 7) in the Lucite enclo-sure shown in Figure 9. The cover slip was removed andplaced on the hot-plate, where 3 µL of concentrated nitricacid was added to dissolve the KCl and then fumed to dry-ness. Nitric acid was added and fumed to dryness again; thecover slip was washed with dilute nitric acid, dried, and trans-ferred to a detector for measurement of its radioactivity. Theenergy and time distribution of the α-decay and the detec-tion of time-correlated pairs of α-particles from the decay of262Ha and its 4.3-s daughter, 258Lr, provided positive identi-fication of the Ha. From 801 such manual separations, taking51 s each, 26 α decays from 262Ha were positively identified.These results showed that, indeed, Ha sorbed on glass sur-faces after fuming with nitric acid, as did its group 5 homo-logues, Nb(V) and Ta(V). The tetravalent group 4 elements,Zr and Hf, and the trivalent actinides were tested under thesame conditions and did not sorb. These experiments con-firmed the group 5 character of Ha and indicated that itshould be placed in the periodic table as the heaviest knownmember of group 5.
Manual liquid–liquid extraction experiments were thenconducted in which anionic fluoride species of Ta(V) extractedinto methylisobutylketone from mixed nitric–hydrofluoricacid solutions; but surprisingly, Ha(V) remained in the aque-ous phase with its lighter homologue Nb(V), which doesnot extract. This was the first indication of differences in thebehavior of Ha from its heavier group 5 homologue Ta(V) andwas the impetus for a long series of experiments to elucidatethe chemical behavior of Ha.
Chromatographic separations of Ha using ARCA II werethen conducted at the 88-inch LBL cyclotron jointly by theGSI/Mainz, PSI/Bern, and LBL/Berkeley groups in 1988 and1990 (48). These showed that Ha was eluted promptly fromcation exchange columns with HIB together with pentavalentNb, Ta, and Pa, whereas trivalent and tetravalent ions wereretained. Again, this provided proof that pentavalent Ha isthe most stable oxidation state in aqueous solution. How-ever, Ha again exhibited non-Ta-like behavior in elutions ofpentavalent Ha, Nb, and Pa sorbed on columns composedof the liquid anion exchanger triisooctylamine on an inertsupport. Ha behaved more like the pseudo-homologue, Pa(V).Nearly 2200 collection and separation cycles on a one-minutetime scale were carried out with ARCA II! The results, showingextractability in the order Ta > Nb > Ha > Pa, were consistentwith calculations of complex formation, but calculations ofextractability gave the reverse order (23). The discrepancy wasattributed to multiply charged F
� or mixed F–Cl complexes(23). New experiments with single halide systems were sug-gested for easier comparison with theoretical predictions basedon considerations of hydrolysis vs complex formation, fromelectronic structures calculated with an improved relativisticcode and Born’s theory of metal-ion extraction (24). These
Chemistry of the Heaviest Elements—One Atom at a Time
JChemEd.chem.wisc.edu • Vol. 76 No. 3 March 1999 • Journal of Chemical Education 343
99
Es
calculations indicated the extraction order should be Pa >>Nb ≈ Ha > Ta. First experiments conducted at LBNL byPaulus et al. with a pure HCl system and anion exchangechromatographic separations using ARCA II confirm thesepredictions of an inversion of the trend in properties in goingfrom the 5d to the 6d elements (26 ). More experiments willbe performed with HBr and HF.
Our group at LBNL has performed manual studies tocompare the extraction into tributylphosphate (TBP),triisooctylamine (TIOA), and TTA from aqueous solutionsof 261Rf with its lighter homologues Zr and Hf, and thepseudo-homologues Th(IV) and Pu(IV). These experimentsshow that Rf generally behaves as a group 4 element, but un-der some conditions, its behavior is more like Pu(IV) orTh(IV) (49, 50).
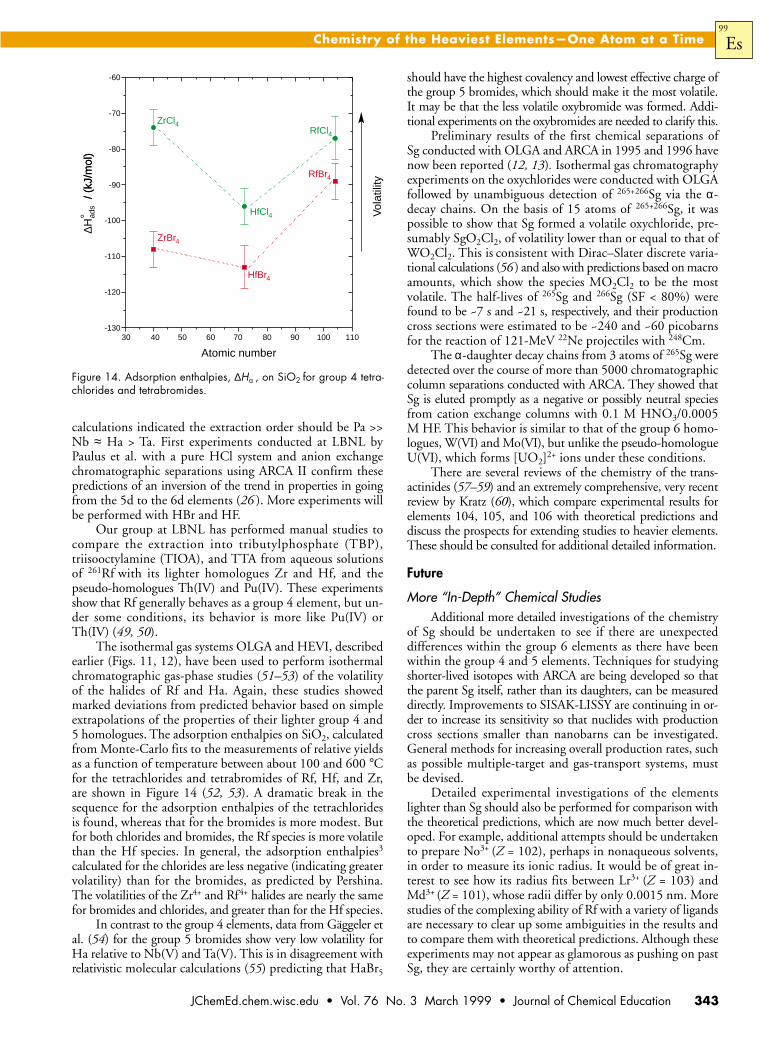
The isothermal gas systems OLGA and HEVI, describedearlier (Figs. 11, 12), have been used to perform isothermalchromatographic gas-phase studies (51–53) of the volatilityof the halides of Rf and Ha. Again, these studies showedmarked deviations from predicted behavior based on simpleextrapolations of the properties of their lighter group 4 and5 homologues. The adsorption enthalpies on SiO2, calculatedfrom Monte-Carlo fits to the measurements of relative yieldsas a function of temperature between about 100 and 600 °Cfor the tetrachlorides and tetrabromides of Rf, Hf, and Zr,are shown in Figure 14 (52, 53). A dramatic break in thesequence for the adsorption enthalpies of the tetrachloridesis found, whereas that for the bromides is more modest. Butfor both chlorides and bromides, the Rf species is more volatilethan the Hf species. In general, the adsorption enthalpies3
calculated for the chlorides are less negative (indicating greatervolatility) than for the bromides, as predicted by Pershina.The volatilities of the Zr4+ and Rf4+ halides are nearly the samefor bromides and chlorides, and greater than for the Hf species.
In contrast to the group 4 elements, data from Gäggeler etal. (54) for the group 5 bromides show very low volatility forHa relative to Nb(V) and Ta(V). This is in disagreement withrelativistic molecular calculations (55) predicting that HaBr5
should have the highest covalency and lowest effective charge ofthe group 5 bromides, which should make it the most volatile.It may be that the less volatile oxybromide was formed. Addi-tional experiments on the oxybromides are needed to clarify this.
Preliminary results of the first chemical separations ofSg conducted with OLGA and ARCA in 1995 and 1996 havenow been reported (12, 13). Isothermal gas chromatographyexperiments on the oxychlorides were conducted with OLGAfollowed by unambiguous detection of 265+266Sg via the α-decay chains. On the basis of 15 atoms of 265+266Sg, it waspossible to show that Sg formed a volatile oxychloride, pre-sumably SgO2Cl2, of volatility lower than or equal to that ofWO2Cl2. This is consistent with Dirac–Slater discrete varia-tional calculations (56 ) and also with predictions based on macroamounts, which show the species MO2Cl2 to be the mostvolatile. The half-lives of 265Sg and 266Sg (SF < 80%) werefound to be ~7 s and ~21 s, respectively, and their productioncross sections were estimated to be ~240 and ~60 picobarnsfor the reaction of 121-MeV 22Ne projectiles with 248Cm.
The α-daughter decay chains from 3 atoms of 265Sg weredetected over the course of more than 5000 chromatographiccolumn separations conducted with ARCA. They showed thatSg is eluted promptly as a negative or possibly neutral speciesfrom cation exchange columns with 0.1 M HNO3/0.0005M HF. This behavior is similar to that of the group 6 homo-logues, W(VI) and Mo(VI), but unlike the pseudo-homologueU(VI), which forms [UO2]2+ ions under these conditions.
There are several reviews of the chemistry of the trans-actinides (57–59) and an extremely comprehensive, very recentreview by Kratz (60), which compare experimental results forelements 104, 105, and 106 with theoretical predictions anddiscuss the prospects for extending studies to heavier elements.These should be consulted for additional detailed information.
Future
More “In-Depth” Chemical StudiesAdditional more detailed investigations of the chemistry
of Sg should be undertaken to see if there are unexpecteddifferences within the group 6 elements as there have beenwithin the group 4 and 5 elements. Techniques for studyingshorter-lived isotopes with ARCA are being developed so thatthe parent Sg itself, rather than its daughters, can be measureddirectly. Improvements to SISAK-LISSY are continuing in or-der to increase its sensitivity so that nuclides with productioncross sections smaller than nanobarns can be investigated.General methods for increasing overall production rates, suchas possible multiple-target and gas-transport systems, mustbe devised.
Detailed experimental investigations of the elementslighter than Sg should also be performed for comparison withthe theoretical predictions, which are now much better devel-oped. For example, additional attempts should be undertakento prepare No3+ (Z = 102), perhaps in nonaqueous solvents,in order to measure its ionic radius. It would be of great in-terest to see how its radius fits between Lr3+ (Z = 103) andMd3+ (Z = 101), whose radii differ by only 0.0015 nm. Morestudies of the complexing ability of Rf with a variety of ligandsare necessary to clear up some ambiguities in the results andto compare them with theoretical predictions. Although theseexperiments may not appear as glamorous as pushing on pastSg, they are certainly worthy of attention.
-60
-70
-80
-90
-100
-110
-120
-13030 40 50 60 70 80 90 100 110
∆H°
/ (k
J/m
ol)
/ (k
J/m
ol)
ads
ZrBr4
HfBr4
RfBr4
ZrCl4
HfCl4
RfCl4
Atomic number
Vol
atili
tyFigure 14. Adsorption enthalpies, ∆Ha , on SiO2 for group 4 tetra-chlorides and tetrabromides.
Viewpoints: Chemists on Chemistry
344 Journal of Chemical Education • Vol. 76 No. 3 March 1999 • JChemEd.chem.wisc.edu
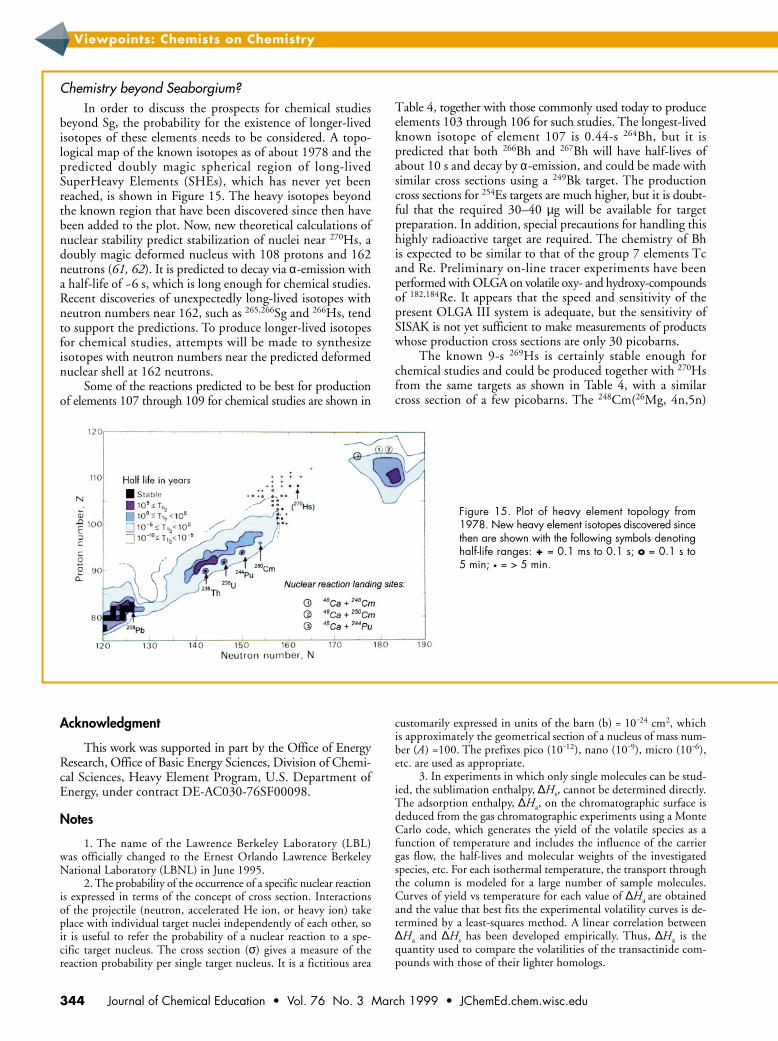
Chemistry beyond Seaborgium?In order to discuss the prospects for chemical studies
beyond Sg, the probability for the existence of longer-livedisotopes of these elements needs to be considered. A topo-logical map of the known isotopes as of about 1978 and thepredicted doubly magic spherical region of long-livedSuperHeavy Elements (SHEs), which has never yet beenreached, is shown in Figure 15. The heavy isotopes beyondthe known region that have been discovered since then havebeen added to the plot. Now, new theoretical calculations ofnuclear stability predict stabilization of nuclei near 270Hs, adoubly magic deformed nucleus with 108 protons and 162neutrons (61, 62). It is predicted to decay via α-emission witha half-life of ~6 s, which is long enough for chemical studies.Recent discoveries of unexpectedly long-lived isotopes withneutron numbers near 162, such as 265,266Sg and 266Hs, tendto support the predictions. To produce longer-lived isotopesfor chemical studies, attempts will be made to synthesizeisotopes with neutron numbers near the predicted deformednuclear shell at 162 neutrons.
Some of the reactions predicted to be best for productionof elements 107 through 109 for chemical studies are shown in
Table 4, together with those commonly used today to produceelements 103 through 106 for such studies. The longest-livedknown isotope of element 107 is 0.44-s 264Bh, but it ispredicted that both 266Bh and 267Bh will have half-lives ofabout 10 s and decay by α-emission, and could be made withsimilar cross sections using a 249Bk target. The productioncross sections for 254Es targets are much higher, but it is doubt-ful that the required 30–40 µg will be available for targetpreparation. In addition, special precautions for handling thishighly radioactive target are required. The chemistry of Bhis expected to be similar to that of the group 7 elements Tcand Re. Preliminary on-line tracer experiments have beenperformed with OLGA on volatile oxy- and hydroxy-compoundsof 182,184Re. It appears that the speed and sensitivity of thepresent OLGA III system is adequate, but the sensitivity ofSISAK is not yet sufficient to make measurements of productswhose production cross sections are only 30 picobarns.
The known 9-s 269Hs is certainly stable enough forchemical studies and could be produced together with 270Hsfrom the same targets as shown in Table 4, with a similarcross section of a few picobarns. The 248Cm(26Mg, 4n,5n)
Figure 15. Plot of heavy element topology from1978. New heavy element isotopes discovered sincethen are shown with the following symbols denotinghalf-life ranges: + = 0.1 ms to 0.1 s; o = 0.1 s to5 min; • = > 5 min.
Acknowledgment
This work was supported in part by the Office of EnergyResearch, Office of Basic Energy Sciences, Division of Chemi-cal Sciences, Heavy Element Program, U.S. Department ofEnergy, under contract DE-AC030-76SF00098.
Notes
1. The name of the Lawrence Berkeley Laboratory (LBL)was officially changed to the Ernest Orlando Lawrence BerkeleyNational Laboratory (LBNL) in June 1995.
2. The probability of the occurrence of a specific nuclear reactionis expressed in terms of the concept of cross section. Interactionsof the projectile (neutron, accelerated He ion, or heavy ion) takeplace with individual target nuclei independently of each other, soit is useful to refer the probability of a nuclear reaction to a spe-cific target nucleus. The cross section (σ) gives a measure of thereaction probability per single target nucleus. It is a fictitious area
customarily expressed in units of the barn (b) = 10�24 cm2, whichis approximately the geometrical section of a nucleus of mass num-ber (A) =100. The prefixes pico (10�12), nano (10�9), micro (10�6),etc. are used as appropriate.
3. In experiments in which only single molecules can be stud-ied, the sublimation enthalpy, ∆Hs, cannot be determined directly.The adsorption enthalpy, ∆Ha, on the chromatographic surface isdeduced from the gas chromatographic experiments using a MonteCarlo code, which generates the yield of the volatile species as afunction of temperature and includes the influence of the carriergas flow, the half-lives and molecular weights of the investigatedspecies, etc. For each isothermal temperature, the transport throughthe column is modeled for a large number of sample molecules.Curves of yield vs temperature for each value of ∆Ha are obtainedand the value that best fits the experimental volatility curves is de-termined by a least-squares method. A linear correlation between∆Ha and ∆Hs has been developed empirically. Thus, ∆Ha is thequantity used to compare the volatilities of the transactinide com-pounds with those of their lighter homologs.

Chemistry of the Heaviest Elements—One Atom at a Time
JChemEd.chem.wisc.edu • Vol. 76 No. 3 March 1999 • Journal of Chemical Education 345
100
Fm
270,269Hs reactions could also be used, but again, the crosssections are estimated to be only a few picobarns. Hassium isexpected to have a volatile tetroxide as do the group 8 elementsRu and Os.
The reaction 254Es (22Ne,4n) would be the best for pro-ducing 272Mt (~1 s) for chemical studies, as it has the largestestimated cross section; but again, 254Es is required. The crosssections for the other reactions shown are smaller and thehalf-lives of the products are shorter. The newly constructedBerkeley Gas-filled Separator (BGS) will be used to investi-gate the best production reactions and determine the half-lives and nuclear properties of Bh, Hs, and Mt isotopes be-fore chemical studies are attempted. However, to performchemical studies of Hs and Mt, significant increases in theproduction rates (perhaps by using multiple targets and gastransport systems) and in the efficiencies and speed of thevarious chemical separation techniques must be achieved.Another possibility under consideration is to use the BGS asa pre-separator to quickly separate the desired species from ahost of unwanted activities prior to chemical studies.
Smolanczuk recently calculated that 298114 should de-cay by α-emission with a half-life of only about 12 min, in-stead of the 109 y predicted 20 years ago (63). However, hepredicts half-lives as long as years for isotopes of element 110(50 y for 292110), months for 112, and minutes for 114 withneutron numbers near the spherical shell of 184 neutrons. Itis no longer expected that these spherical nuclei around thedoubly magic spherical nucleus will form an island in a sea ofinstability, but rather that they belong to an extended penin-sula of relatively long-lived nuclides (62). One of the mostexciting future experiments will be to look for the doubly de-formed lighter isotope 270Hs, to see if the theoretical predic-tions are correct. If so, a whole new vista of longer-lived iso-topes opens up before us. BGS might be used to separate and“stockpile” longer-lived isotopes on a collector plate for fu-ture chemical separations.
Some potential compound nucleus reactions with 48Caprojectiles and targets of the neutron-rich, very-long-livedisotopes 244Pu, 248Cm, and 250Cm are shown in Figure 15.
These might allow us to gain access to the doubly magicsuperheavy element (SHE) region. Discussions of some ofthese reactions as well as reactions with 26Mg and 36S projec-tiles are given in refs 64 and 65. Transfer and deeply inelas-tic reactions in which only a portion of the projectile is trans-ferred to the target, resulting in a nucleus with lower excita-tion energy, which is less likely to fission, are also being re-considered. Among the new techniques proposed for produc-tion of SHEs is the use of extremely neutron-rich, unstable(“radioactive”) beams (66 ), but increases in the intensitiesof these beams are still needed.
It appears likely that we will be able to study the chemistryof Bh in the near future. Then improvements in production rates,using different beams and targets, and increases in the efficiencyand speed of chemical separations should permit chemicalstudies of Hs and Mt within the next 20 years. Althoughit now appears probable that a host of isotopes of elements110 through 114 can exist that are sufficiently long-lived forchemical studies, imaginative new production reactions andingenious ways to study still smaller numbers of atoms mustbe devised in order to enter this new frontier region.
seidutSlacimehCrofsnoitcaeR.4elbaTZ noitcaeR σ bp/ t 2/1 s/ yaceD
301 942 (kB 81 ,O α )n3 062 rL 0009 081 α
401 842 (mC 81 )n5,O 162 fR 0005 57 α
501 942 (kB 81 )n5,O 262 aH 0006 43 α
601 842 (mC 22 )n5,eN 562 gS942 (fC 81 )n4,O 362 gS052 (fC 81 )n4,O 462 gS
052003002
529.0
2
ααα FS,
701 942 (kB 22 )n4,eN 762 hB452 (sE 61,81 ,O x )n 662-862 hB
03001~
0101~
αα
801 152 (fC 22 )n4,eN 962 sH252 (fC 22 )n4,eN 072 sH832 (U 63 )n4,S 072 sH
222
955
αα FS,α FS,
901 942 (kB 62 )n4,gM 172 tM252 (fC 32 )n4,aN 172 tM452 (sE 22,02 ,eN x )n 272-072 tM
3.01.0
1~
2.02.0
1~
ααα
General Reading
The Robert A. Welch Foundation, Fifty Years with Transuranium Ele-ments; Proceedings of Conference on Chemical ResearchXXXIV, October 22–23, 1990; Welch Foundation: Houston,TX, 1990.
The Robert A. Welch Foundation, Transactinide Elements; Proceed-ings of 41st Welch Conference on Chemical Research, Octo-ber 1997; Welch Foundation, Houston, TX, 1997.
Katz, J. J.; Seaborg, G. T.; Morss, L. Chemistry of the Actinide Ele-ments, 2nd ed.; Chapman and Hall: London, 1986.
Seaborg, G. T.; Loveland, W. D. The Elements Beyond Uranium;Wiley: New York, 1990.
Seaborg, G. T.; Loveland, W. In The New Chemistry; Hall, N., Ed.;Cambridge University Press: New York, in press; Chapter 1,The Elements Beyond Uranium.
Symposium on Transuranium Elements—A Half Century; Morss, L.R.; Fuger, J., Eds.; ACS Books; American Chemical Society:Washington, DC, 1992.
Hoffman, D. C. Chem. Eng. News 1994, 72 (May 2), 24–34.Seaborg, G. T.; Hobart, D. E. Summary of the Properties of the
Lanthanide and Actinide Elements; In IANCAS’ Frontiers inNuclear Chemistry; Sood, D. D.; Reddy, A. V. R.; Pujari, P. K.Eds.; Perfect Prints: Thane, India, 1996; pp 69–94.

Viewpoints: Chemists on Chemistry
346 Journal of Chemical Education • Vol. 76 No. 3 March 1999 • JChemEd.chem.wisc.edu
Literature Cited
l. Perrier, C.; Segrè, E. J. Chem. Phys. 1937, 5, 712.2. McMillan, E. M.; Abelson, P. H. Phys. Rev. 1940, 57, 1185.3. Seaborg, G. T.; McMillan, E. M.; Kennedy, J. W.; Wahl, A. C.
Phys. Rev. 1946, 69, 366.4. Seaborg, G. T.; Wahl, A. C.; Kennedy, J. W. Phys. Rev. 1946,
69, 367.5. Seaborg, G. T. Chem. Eng. News 1945, 23(Dec 10), 2190.6. Ghiorso, A.; Harvey, B. G.; Choppin, G. R.; Thompson, S. G.;
Seaborg, G. T. Phys. Rev. 1955, 98, 1518..7. Fields, P. R.; Friedman, A. M.; Milsted, J.; Atterling, H.; Forsling,
W.; Holm, L. W.; Astrom, B. Phys. Rev. 1957, 107, 1460.8. Silva, R. J.; Sikkeland, T.; Nurmia, M.; Ghiorso, A. Inorg. Nucl.
Chem. Lett. 1970, 6, 733.9. Silva, R.; Harris, J.; Nurmia, M.; Eskola, K.; Ghiorso, A. Inorg.
Nucl. Chem. Lett. 1970, 6, 871.10. Hulet, E. K.; Lougheed, R. W.; Wild, J. G.; Landrum, J. H.;
Nitschke, J. M.; Ghiorso, A. Inorg. Nucl. Chem. 1980, 42, 79.11. Herrmann, G. Nucl. Phys. News 1998, 8, 7.12. Schädel, M.; Brüchle, W.; Dressler, R.; Eichler, B.; Gäggeler, H.
W.; Günther, R.; Gregorich, K. E.; Hoffman, D. C.; Hübener,S.; Jost, D. T.; Kratz, J. V.; Paulus, W.; Schumann, D.; Timokhin,S.; Trautmann, N.; Türler, A.; Wirth G.; Yakuschev, A. Nature1997, 388, 55.
13. Schädel, M.; Brüchle, W.; Schausten, B.; Schimpf, E.; Jaeger, E.;Wirth, G.; Guenther, R.; Kratz, J. V.; Paulus, W.; Seibert, A.; Thoerle,P.; Trautmann, N.; Zauner, S.; Schumann, D.; Andrassy, M.; Misiak,R.; Gregorich, K. E.; Hoffman, D. C.; Lee, D. M.; Sylwester, E. R.;Nagame, Y.; Oura, Y. Radiochim. Acta 1997, 77, 149.
14. Fricke, B.; Greiner, W. Phys. Lett. 1969, 30B, 347.15. Fricke, B.; Waber, J. T. Actinides Rev. 1971, 1, 433.16. Mann, J. B. J. Chem. Phys. 1969, 51, 841.17. Desclaux, J.-P. At. Data Nucl. Data Tables 1973, 12, 311.18. Pitzer, K. S. J. Chem. Phys. 1975, 63, 1032.19. Pyykkö, P. Chem. Rev. 1988, 88, 563.20. Nash, C. E.; Bursten, B. E. URL: http://chemistry.ohio-state.edu/
~cnash/homepage.htm (accessed Jan 1999).21. Pershina, V. Chem. Rev. 1996, 96, 1977.22. Pershina, V. In Transactinide Elements; Proceedings of the 41st
Welch Conference on Chemical Research, October 1997; WelchFoundation: Houston, TX, 1997; pp 167–194.
23. Pershina, V.; Fricke, B.; Kratz, J. V.; Ionova, G. V. Radiochim.Acta 1994, 64, 37.
24. Pershina, V. Radiochim. Acta 1998, 80, 65, 75.25. Pershina, V.; Fricke, B. In Heavy Elements and Related New Phenom-
ena; Greiner, W.; Gupta, R. K., Eds.; World Scientific: Singapore, inpress (GSI-Preprint-98-26, April 1998).
26. Paulus, W.; Kratz, J. V.; Strub, E.; Zauner, S.; Brüchle, W.;Pershina, V.; Schädel, M.; Schausten, B.; Adams, J. L.; Gregorich,K. E.; Hoffman, D. C.; Laue, C.; Lee, D. M.; McGrath, C. A.;Shaughnessy, D. K.; Strellis, D. A.; Sylwester E. R. Proceedingsof International Conference on Actinides, Baden-Baden, Ger-many, Sep 1997; J. Alloys Comp. 1998, 271–273, 292.
27. Maly, J.; Sikkeland, T.; Silva, R.; Ghiorso, A. Science 1968, 160, 1114.28. Gregorich, K. E.; Henderson, R. A.; Lee, D. M.; Nurmia, M. J.;
Chasteler, R. M.; Hall, H. L.; Bennett, D. A.; Gannett, C. M.;Chadwick, R. B.; Leyba, J. D.; Hoffman, D. C.; Herrmann, G.Radiochim. Acta 1988, 43, 223.
29. Ghiorso, A.; Nitschke, J. M.; Alonso, J. R.; Alonso, C. T.; Nurmia,M.; Seaborg, G. T.; Hulet, E. K.; Lougheed, R. W. Phys. Rev. Lett.1974, 33, 1490.
30. Gregorich, K. E.; Lane, M. R.; Mohar, M. F.; Lee, D. M.; Kacher,C. D.; Sylwester, E. R.; Hoffman, D. C. Phys. Rev. Lett. 1994,72, 1423.
31. Adloff, J.-P.; Guillaumont, R. Fundamentals of Radiochemistry;CRC: Boca Raton, FL, 1993; pp 327–352.
32. Guillaumont, R.; Adloff, J.-P.; Peneloux, A. Radiochim. Acta 1989,46, 169.
33. Guillaumont, R.; Adloff, J.-P.; Peneloux, A.; Delamoye, P.Radiochim. Acta 1991, 54, 1.
34. Wierczinski, B.; Hoffman, D. C. In IANCAS’ Frontiers in NuclearChemistry; Sood, Reddy, Pujari, Eds.; Perfect Prints: Thane, In-dia, 1996; pp 171–191; LBL-37473 Preprint June 1995.
35. Zvara, I.; Chuburkov, Yu. T.; Tsaletka, R.; Shalaevskii, M. R. Sov.Radiochem. 1969, 11, 153, 161.
36. (a) Zvara, I.; Aikhler, V.; Belov, V. Z.; Zvarova, T. S.; Korotkin,Yu. S.; Shalaevskii, M. R.; Shchegolev, V. A.; Yussonnua, M. Sov.Radiochem. 1974, 16, 709. (b) Zvara, I.; Belov, V. Z.; Domanov,V. P.; Shalaevskii, M. R. Sov. Radiochem. 1976, 18, 328.
37. Gäggeler, H. W.; Jost, D. T.; Baltensperger, U.; Weber, A.; Kovacs,A.; Vermeulen, D.; Türler, A. Nucl. Instrum. Methods Phys. Res.1991, A309, 201.
38. Kadkhodayan, B.; Türler, A.; Gregorich, K. E.; Nurmia, M. J.;Lee, D. M.; Hoffman D. C. Nucl. Instrum. Methods Phys. Res.1991, A317, 254.
39. Eichler, B.; Türler, A.; Jost, D. T.; Gäggeler, H. W. Annual Re-port of Labor für Radio- and Umweltchemie; Paul ScherrerInstitut: Villigen, Switzerland, 1993; p 42.
40. Alstad, J.; Skarnemark, G.; Haberberger, F.; Herrmann, G.;Nähler, A.; Pense-Maskow, M.; Trautmann, N. J. Radioanal. Nucl.Chem. 1995, 189, 133.
41. Alstad, J.; Eberhardt, K.; Malmbeck, R.; Nähler, A.; Omtvedt,J. P.; Skarnemark, G.; Trautmann, N.; Wierczinski, B. In 4thInternational Conference on Nuclear and Radiochemistry, St.Malo, France, September 1996; Extended abstracts, IPN Orsay,1, A-P2.
42. Hulet, E. K. Radiochim. Acta 1983, 32, 7.43. Keller, O. L. Jr. Radiochim. Acta 1984, 37, 169.44. Hoffman, D. C.; Henderson, R. A.; Gregorich, K. E.; Bennett,
D. A.; Chasteler, R. M.; Gannett, C. M.; Hall, H. L.; Lee, D.M.; Nurmia, M. J.; Cai, S.; Agarwal, R.; Charlop, A. W.; Chu,Y. Y.; Silva, R. J. J. Radioanal. Nucl. Chem. 1988, 124, 135.
45. Brüchle, W.; Schädel, M.; Scherer, U. W.; Kratz, J. V.; Gregorich,K. E.; Lee, D.; Nurmia, M.; Chasteler, R.; Hall, H. L.; Henderson,R. A.; Hoffman, D. C. Inorg. Chim. Acta 1988, 146, 267.
46. Scherer, U. W.; Kratz, J. V.; Schädel, M.; Brüchle, W.; Gregorich,K. E.; Henderson, R. A.; Lee, D.; Nurmia, M.; Hoffman, D. C.Inorg. Chim. Acta 1988, 146, 249.
47. Lougheed, R. W.; Moody, K. J.; Dougan, R. J.; Wild, J. G.; Hulet,E. K.; Dupzyk, R. J.; Henderson, C. M.; Gannett, C. M.;Henderson, R. A.; Hoffman, D. C.; Lee, D. M.; Sümmerer, K.;Hahn, R. L. Nuclear Chemistry Division FY 87 Annual Report;Lawrence Livermore National Laboratory: Livermore, CA, 1987;UCAR 10062/87, 4-2.
48. Schädel, M.; Brüchle, W.; Jäger, E.; Schimpf, E. Radiochim. Acta1989, 48, 171.
49. Czerwinski, K. R.; Gregorich, K. E.; Hannink, N. J.; Kacher, C.D.; Kadkhodayan, B. A.; Kreek, S. A.; Lee, D. M.; Nurmia, M.J.; Türler, A.; Seaborg, G. T.; Hoffman, D. C. Radiochim. Acta1994, 64, 23; Radiochim. Acta 1994, 64, 29.
50. Kacher, C. D.; Gregorich, K. E.; Lee, D. M.; Watanabe, Y.;Kadkhodayan, B.; Yang, B. J.; Hsu, M.; Hoffman, D. C.; Bilewicz,A. Radiochim. Acta 1996, 75, 127; Radiochim. Acta 1996, 75, 135.
51. Türler, A.; Gäggeler, H. W.; Gregorich, K. E.; Barth, H.; Brüchle,W.; Czerwinski, K. R.; Gober, M. K.; Hannink, N. J.; Henderson,R. A.; Hoffman, D. C.; Jost, D. T.; Kacher, C. D.; Kadkhodayan,B.; Kovacs, J.; Kratz, J. V.; Kreek, S. A.; Lee, D. M.; Leyba, J.D.; Nurmia, M. J.; Schädel, M.; Scherer, U. W.; Schimpf, E.;Vermeulen, D.; Weber, A.; Zimmermann, H. P.; Zvara I. J.Radioanal. Nucl. Chem. 1992, 160, 327.
52. Kadkhodayan, B.; Türler, A.; Gregorich, K. E.; Baisden, P. A.;Czerwinski, K. R.; Eichler, E.; Gäggeler, H. W.; Hamilton, T. M.;Stoyer, N. J.; Jost, D. T.; Kacher, C. D.; Kovacs, A.; Kreek, S. A.;Lane, M. R.; Mohar, M. F.; Neu, M. P.; Sylwester, E. R.; Lee, D.M.; Nurmia, M. J.; Seaborg, G. T.; Hoffman, D. C. Radiochim.Acta 1996, 72, 169.
53. Sylwester, E. Ph.D. Thesis, Department of Chemistry, Univer-sity of California, Berkeley, CA, October 1998.
54. Gäggeler, H. Paul Scherrer Institut Report PSI-R-97-19, August1997; Proceedings, Actinides-97, Baden-Baden, Germany; J. AlloysCompd. in press.

Chemistry of the Heaviest Elements—One Atom at a Time
JChemEd.chem.wisc.edu • Vol. 76 No. 3 March 1999 • Journal of Chemical Education 347
101
Md
55. Pershina, V.; Sepp, W.-D.; Fricke, B.; Kolb, D.; Schädel, M.;Ionova, G. V. J. Chem. Phys. 1992, 97, 1116.
56. Pershina, V.; Fricke, B. J. Phys Chem. 1997, 100, 8748.57. Hoffman, D. C. Radiochim. Acta 1993, 61, 123; Radiochim. Acta
1996, 72, 1.58. Kratz, J. V. J. Alloys Compd. 1994, 213/214, 20.59. Schädel, M. Radiochim. Acta 1995, 70/71, 207.60. Kratz, J. V. Institut für Kernchemie, Johannes Gutenberg-
Universität Mainz; In Heavy Elements and Related New Phenomena;Greiner, W.; Gupta, R. K., Eds.; World Scientific: Singapore, inpress (Report IKMZ, March 1998).
61. Sobiczewski, A. Physics–Uspekhi 1996, 39, 885.
Diana M. LeeStaff ScientistNuclear Science DivisionLawrence Berkeley National LaboratoryM.S., Nuclear Chemistry, University of California, Berkeley, 1972B.S., Chemistry, University of California, Berkeley, 1961
Diana M. Lee has participated in research in nuclear andradiochemistry at the Lawrence Berkeley National Labora-tory since 1961. She developed charged-particle nuclear ac-tivation analysis techniques for sensitive analysis of a host ofelements throughout the periodic table, which were appliedin a variety of fundamental investigations. In recent years,she has been involved in investigations of spontaneous fis-sion properties, heavy element production mechanisms, andthe search for superheavy elements, and in development of“atom-at-a time” studies of the chemical and nuclear prop-erties of the heaviest elements.
Darleane C. HoffmanProfessor of the Graduate School, University of California atBerkeley, Faculty Sr. Scientist, Nuclear Science Division,Lawrence Berkeley National Laboratory & Charter Director,G. T. Seaborg Institute for Transactinium Science,Lawrence Livermore National LaboratoryPh.D., Physical (nuclear) Chemistry, 1951B.S., Chemistry, Iowa State University, Ames, Iowa, 1948
Darleane C. Hoffman is an internationally recognizedleader in the development of “atom-at-a-time” techniques forinvestigating the chemical and nuclear properties of the heavi-est elements and in the education of students in nuclear andradiochemistry. She came to Berkeley in 1984 as professorof chemistry and under her direction, 13 students have re-ceived Ph.D. degrees, three have received M.S. degrees, andthree more will receive degrees in 1999. During her tenureat Los Alamos National Laboratory from 1953 to 1984, sheand her coworkers discovered 244Pu in nature, per-formed pioneering studies of the spontaneous fis-sion process, and developed rapid radiochemicalseparations for short-lived fission products. In ad-dition, she has investigated the behavior and mi-gration of radionuclides in the environment. In1997, she received the President’s National Medalof Science for the discoveries of 244Pu in nature andsymmetric spontaneous fission of heavy nuclei, forpioneering studies of elements 104, 105, and 106,and for outstanding service to education of studentsin nuclear chemistry.
62. Sobiczewski, A. In Transactinide Elements; Proceedings of the 41stWelch Conference on Chemical Research, October 1997; WelchFoundation: Houston, TX, 1997; pp 368–384.
63. Smolanczuk, R. Phys. Rev. C 1997, 56, 812.64. Oganessian, Yu. Ts. In Transactinide Elements; Proceedings of the
41st Welch Conference on Chemical Research, October 1997;Welch Foundation: Houston, TX, 1997; pp 347–367.
65. Armbruster, P. In Transactinide Elements; Proceedings of the 41stWelch Conference on Chemical Research, October 1997; WelchFoundation: Houston, TX, 1997; pp 231–262.
66. Seaborg, G. T.; Loveland, W. In The New Chemistry; Hall, N., Ed.;Cambridge University Press: New York, in press; Chapter 1, p 18.
Chemistry of the Heaviest Elements—One Atom at a Time
Viewpoints: Chemists on Chemistry