Embed Size (px)
Citation preview

Avancées dans les dystrophie s musculaire s de Duchenne et de Becker
> Myopathie de Duchenne (DMD) > Myopathie de Duchenne de Boulogne > Duchenne muscular dystrophy > Myopathie de Becker (BMD) > Becker muscular dystrophy
Ce document présente l'état actuel des connaissances scientifiques sur les dystrophies musculaires de Duchenne et de Becker, mis à jour à l'occasion des Journées des Familles 2013 de l'AFM-Téléthon. Il est téléchargeable sur le site internet de l'AFM-Téléthon : WEB www.afm-telethon.fr . Pour en savoir plus sur les dystrophies musculaires de Duchenne et de Becker, vous pouvez consulter le Zoom sur… la dystrophie musculaire de Duchenne, le Zoom sur… la dystrophie musculaire de Becker et les Repères Savoir et Comprendre qui traitent de sujets scientifiques, médicaux, psychologiques et sociaux. Destinés aux personnes atteintes de maladies neuromusculaires et à leurs familles, ils sont disponibles sur le site internet de l'AFM-Téléthon et auprès du Service régional de votre région. Ces documents ne peuvent en aucun cas se substituer à l'avis d'un médecin, même s'ils peuvent vous faciliter le dialogue avec l'équipe soignante.
JUIN 2013
AVANCÉES DE LA RECHERCHE

Avancées dans les dystrophies musculaires de Duchenne et de Becker Juin 2013
2/40 AFM-Téléthon>Myoinfo
SOMMAIRE Faits marquants dans les dystrophies musculaires de Duchenne et de Becker ......................................................... 3
Faits marquants dans la dystrophie musculaire de Duchenne4
Faits marquants dans la dystrophie musculaire de Becker .... 6
Que sont les dystrophies musculaires de Duchenne et de Becker ? ................................................................................ 7
Les dystrophies musculaires de Duchenne et de Becker sont dues à des anomalies de la dystrophine ................................ 7
Les anomalies de la dystrophine conduisent à un manque de force ...... 10
La faiblesse musculaire favorise l'apparition de déformations orthopédiques ............................................................................... 11
Différents mécanismes en jeu dans les dystrophies musculaires de Duchenne et de Becker ............................... 11
L'hypothèse mécanique .................................................................. 11
L'hypothèse calcique ...................................................................... 11
L'hypothèse vasculaire ................................................................... 12
L'hypothèse inflammatoire .............................................................. 12
Des bases de données pour mieux connaître les dystrophies musculaires de Duchenne et de Becker. .............................. 12
Une base de données pour mieux connaitre la dystrophine ................. 13
Les pistes thérapeutiques dans les dystrophies musculaires de Duchenne et de Becker ................................................... 13
De nombreuses pistes complémentaires ........................................... 13
Apporter des molécules thérapeutiques au cœur du noyau .................. 13
Les vecteurs viraux ........................................................................ 14
Les vecteurs non-viraux .................................................................. 14
Remplacer le gène altéré par un gène thérapeutique : la thérapie génique ........................................................................................ 14
Produire une mini-dystrophine chez l'homme ..................................... 15
Transférer le gène SERCA2 pour améliorer la fonction cardiaque ........... 15
Réparer le gène altéré à l'aide des méganucléases ............................. 16
Translecture de codon stop grâce à l'ataluren (PTC124) ..................... 17
Réparer l’ARN de la dystrophine : le trans-épissage ........................... 18
Remplacer la protéine : la piste de l’utrophine ................................... 20
Le SMT C1100 ............................................................................... 21
Le nabumétone .............................................................................. 21
Remplacer les cellules musculaires déficientes par des cellules souches : la thérapie cellulaire ....................................................................... 21
Réduire la fibrose .......................................................................... 24
Augmenter la masse musculaire ...................................................... 25
Prévenir l'atteinte de la fonction cardiaque ........................................ 26
Les bêta-bloquants et les inhibiteurs de l’enzyme de conversion de l’angiotensine ................................................................................ 26
Le sildénafil ................................................................................... 26
Le sildénafil et le tadalafil ................................................................ 26
L’éplérénone ................................................................................. 27
Les pistes thérapeutiques dans la dystrophie musculaire de Duchenne ............................................................................ 27
Corriger l'ARN pré-messager par le saut d’exon ................................. 28
Le drisapersen (ou GSK2402968) ..................................................... 31
Le PRO044 .................................................................................... 32
Le PRO045 .................................................................................... 32
Le PRO052, le PRO053 et le PRO055 ................................................. 33
L’éteplirsen (ou AVI-4658) .............................................................. 33
L’AAV-U7 ...................................................................................... 34
Les tricyclo-ADN, de nouveaux oligonucléotides antisens synthétiques prometteurs .................................................................................. 35

Avancées dans les dystrophies musculaires de Duchenne et de Becker Juin 2013
AFM-Téléthon>Myoinfo 3/40
Réduire l’inflammation ................................................................... 35
La prednisone ................................................................................ 36
L’IGF-1 ......................................................................................... 36
Diminuer le stress oxydatif ............................................................. 37
Améliorer la force musculaire ......................................................... 37
Stabiliser le canal calcium RyR1 ........................................................ 37
Les inhibiteurs de l'enzyme de conversion .......................................... 38
Les pistes thérapeutiques dans la dystrophie musculaire de Becker ................................................................................. 38
Les anti-myostatines ....................................................................... 38
Le tadalafil .................................................................................... 39
Le sildénafil ................................................................................... 39
* * *
Faits marquants dans les dystrophies musculaires de Duchenne et de Becker > Les bases de données UMD-DMD France et "eDystrophin " recueillent des données sur la dystrophine (gène et protéine) pour établir des relations entre l’anomalie génétique, les anomalies de la dystrophine et les signes cliniques de la maladie, identifier les patients pour la recherche clinique… > Thérapie génique ● Preuve de concept de l’efficacité d’une thérapie génique avec un gène de microdystrophine dans un modèle de chien atteint de DMD. > Translecture des codons stop ● Essais de phase III de l’ataluren en ouvert en cours aux États-Unis Recrutement en cours aux États-Unis de 122 personnes atteintes de DMD et BMD ayant participé à une étude précédente testant l'ataluren et de 96 personnes en Europe (dont la France), Australie, Israël et Canada. ● Essai international (y compris en France) de phase III de l’ataluren en double aveugle contre placebo chez 220 personnes atteintes de DMD et BMD en cours Recrutement en cours. > Augmenter la production d’utrophine ● Essai de phase I d’une nouvelle formulation de SMT-C110 chez 48 volontaires sains en cours d’analyse aux États-Unis Recrutement terminé. Données en cours d’analyse. > Thérapie cellulaire ● Preuve de concept d’une thérapie cellulaire par des cellules progénitrices du muscle chez une souris modèle. ● Essai de phase I/II de thérapie cellulaire par greffe de cellules souches mésenchymateuses du cordon ombilical humain en cours en Chine et en Inde Recrutement en cours de 15 personnes atteintes de DMD en Chine et de 30 personnes atteintes de DMD en Inde. > Réduire la fibrose

Avancées dans les dystrophies musculaires de Duchenne et de Becker Juin 2013
4/40 AFM-Téléthon>Myoinfo
● Le HT-100, un dérivé de l’halofuginone, a reçu la désignation de médicament orphelin pour le traitement de la DMD. > Augmenter la masse musculaire ● Essai du DT-200 en préparation chez des volontaires sains Le DT-200 est une molécule qui se lie aux récepteurs des androgènes et qui augmente la taille et la force musculaires. > Prévenir l’atteinte cardiaque ● Essai de phase II du périndopril et d’un bêtabloquant chez 60 enfants atteints de DMD en cours Recrutement en cours. ● Essai du sildénafil puis du tadalafil chez 12 personnes atteintes de DMD et de BMD, marchants, en cours aux États-Unis Recrutement en cours. ● Essai du sildénafil et du tadalafil chez 30 personnes atteintes de DMD et de BMD, marchants en cours aux États-Unis Recrutement en cours. ● Essai de l’éplerenone chez 40 personnes atteintes de DMD avec une atteinte cardiaque sans dysfonction du ventricule gauche en cours aux États-Unis Recrutement en cours.
Faits marquants dans la dystrophie musculaire de Duchenne

Avancées dans les dystrophies musculaires de Duchenne et de Becker Juin 2013
AFM-Téléthon>Myoinfo 5/40
> Saut de l’exon 51 ● Essai international de phase III du drisapersen en cours chez 180 personnes atteintes de DMD capables de marcher Recrutement terminé. ● Essai de phase II du drisapersen chez 54 personnes atteintes de DMD et capables de marcher (Europe et Australie) : Recrutement terminé. Données en cours d’analyse. Une phase d’extension s’adressant aux personnes ayant participé à l’un ou l’autre de ces deux essais est en cours. ● Essai de phase I du drisapersen chez 32 personnes atteintes de DMD et incapables de marcher (France et États-Unis) : Recrutement terminé. Données en cours d’analyse. ● Essai de phase II de l’éteplirsen (AVI-4658) chez 12 personnes atteintes de DMD : Recrutement terminé. Données en cours d’analyse. >Saut de l'exon 44 ● Essai de phase I/II de PRO044 chez 18 personnes atteintes de DMD en Belgique, Italie, Hollande et Suède Recrutement terminé. Phase clinique en cours. > Saut de l’exon 45 ● Essai de phase I/II de PRO045 chez 45 personnes atteintes de DMD en cours (Europe dont la France) Recrutement en cours. > Sauts des exons 52, 53 et 55 ● Etude préclinique de l'AAV-U7 (saut de l’exon 53) sur "corps entier" de chiens GRMD en cours ● Etudes précliniques de PRO052, PRO053 et PRO055 Obtention de la désignation de médicaments orphelins. > Les tricyclo-ADN L’injection de tricyclo-ADN dans des souris modèles de DMD restaure l’expression de la dystrophine et améliore la fonction musculaire > Réduire l’inflammation ● Essai international comparant 3 protocoles de traitement par corticoïdes chez 300 personnes atteintes de DMD en cours Recrutement en cours. ● Essai comparant l'efficacité de l’IGF-1 à celle des corticoïdes chez 40 personnes atteintes de DMD Recrutement terminé. Phase clinique en cours. > Diminuer le stress oxydatif ● Essai de phase II de l’idébénone (essai DELOS) chez 240 personnes atteintes de DMD - Recrutement terminé. Phase clinique en cours. > Améliorer la force musculaire ● Essai du périndopril chez 40 enfants atteints de DMD en France Recrutement terminé. Données en cours d'analyse.

Avancées dans les dystrophies musculaires de Duchenne et de Becker Juin 2013
6/40 AFM-Téléthon>Myoinfo
Faits marquants dans la dystrophie musculaire de Becker > Les anti-myostatines ● Essai de phase I d'un transfert du gène (thérapie génique) de la follistatine chez 15 personnes atteintes de BMD en cours aux États-Unis Recrutement en cours. > Améliorer l'irrigation sanguine du muscle ● Essai du tadalafil chez 48 hommes atteints de BMD en cours aux États-Unis Recrutement en cours. ● Essai du sildénafil chez 17 hommes atteints de BMD au Danemark Phase clinique terminé. Données en cours d'analyse.

Avancées dans les dystrophies musculaires de Duchenne et de Becker Juin 2013
AFM-Téléthon>Myoinfo 7/40
Que sont les dystrophies musculaires de Duchenne et de Becker ? Les dystrophies musculaires de Duchenne (DMD) et de Becker (BMD) sont des maladies rares, qui touchent l'ensemble des muscles de l'organisme (muscles squelettiques, muscle cardiaque et muscles lisses) : ce sont des myopathies. La maladie de Duchenne est liée à des mutations dans le gène DMD sur le chromosome X, entraînant l'absence de la protéine dystrophine. La maladie de Becker est liée à des mutations dans le gène DMD sur le chromosome X, entraînant un déficit partiel en protéine dystrophine et/ou une production d'une dystrophine de taille anormale. Dans les 2 cas, seuls les hommes sont atteints ; les femmes qui ont un chromosome X porteur d'une anomalie dans le gène DMD ne présentent aucune gêne, sauf exception, mais ce chromosome X avec l'anomalie peut se transmettre à leur descendance. La prise en charge médicale est, pour l’instant, symptomatique. Elle vise essentiellement à prévenir les complications, notamment orthopédiques, cardiaques et respiratoires, et à améliorer le confort de vie des personnes atteintes de dystrophies musculaires de Duchenne ou de Becker. Les corticostéroïdes sont de plus en plus largement prescrits pour leur capacité à ralentir l’évolution de la maladie.
Les dystrophies musculaires de Duchenne et de Becke r sont dues à des anomalies de la dystrophine Les dystrophies musculaires de Duchenne et de Becker sont dues à une modification de l'ADN : ce sont des maladies génétiques. Ces anomalies de l'ADN touchent le gène DMD. Le gène DMD est localisé sur le chromosome X. Il a été identifié en 1986. La protéine qu'il code, la dystrophine, a été découverte en 1987. Le gène de la dystrophine ou gène DMD est le plus grand gène humain connu. Il occupe à lui seul à peu près 1% de la longueur totale du chromosome X. Sa très grande taille le rend statistiquement plus vulnérable que d’autres gènes humains à la survenue spontanée de petites ou grandes modifications. La dystrophine est une protéine spécifique des fibres musculaires. Allongée en bâton, elle est localisée sous la membrane cellulaire de la fibre musculaire. Elle est associée à des protéines qui forment ensemble un complexe reliant l'extérieur (matrice extracellulaire) et l'intérieur de la fibre musculaire, à travers la membrane cellulaire. On nomme ce complexe de protéines associées à la dystrophine, le complexe DAP pour "dystrophin associated proteins". Ce dispositif constitue un lien mécanique essentiel entre l'intérieur et l'extérieur de la cellule musculaire.
Une maladie est dite rare quand elle touche moins d'une personne sur 2 000. Les maladies rares font l'objet d'une politique de santé publique commune dans les domaines de la recherche, de l'information et de la prise en charge.
Les maladies d'origine génétique sont des maladies dues à des anomalies de l'ADN, c'est-à-dire de l'information qui détermine le fonctionnement biologique de notre organisme. Cette information est présente dans nos cellules sous forme de chromosomes. Nous l'héritons de nos parents et nos enfants héritent de la nôtre. C'est pourquoi les maladies génétiques sont souvent familiales, c'est-à-dire qu'il peut y avoir plusieurs membres d'une même famille atteints par la maladie génétique.
Avancées dans les dystrophies musculaires de Duchenne et de Becker Juin 2013
8/40 AFM-Téléthon>Myoinfo
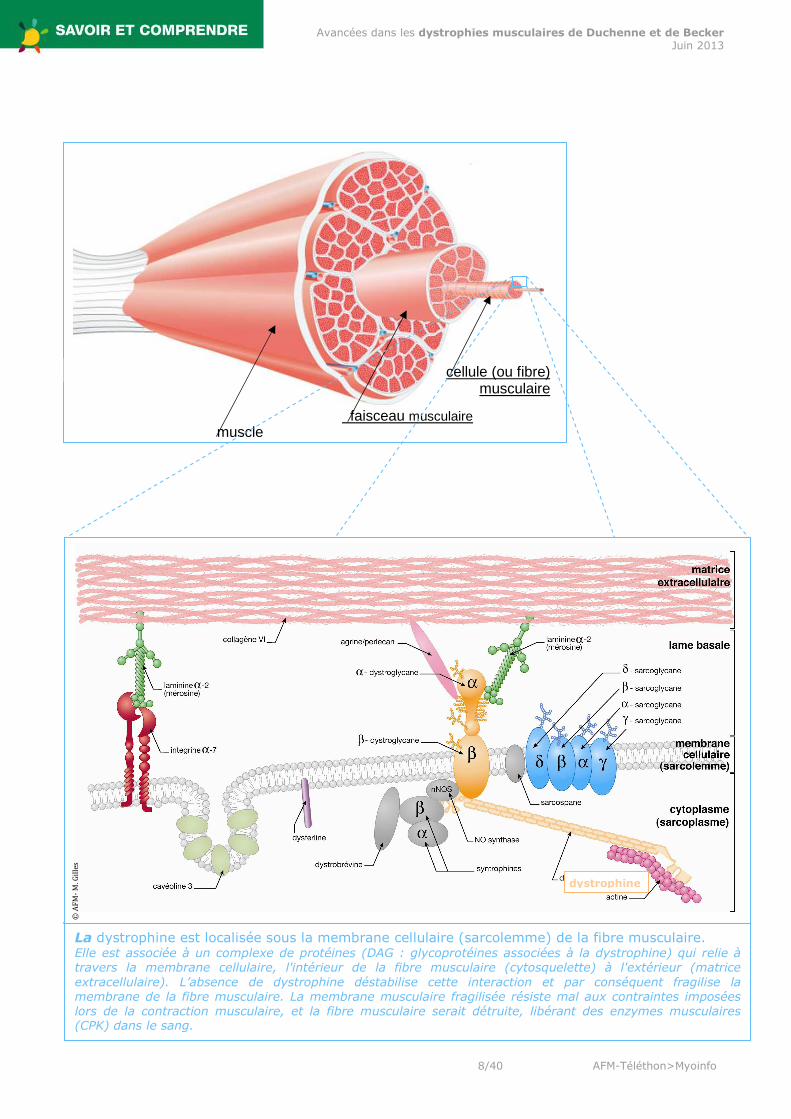
muscle faisceau musculaire
cellule (ou fibre) musculaire
La dystrophine est localisée sous la membrane cellulaire (sarcolemme) de la fibre musculaire. Elle est associée à un complexe de protéines (DAG : glycoprotéines associées à la dystrophine) qui relie à travers la membrane cellulaire, l'intérieur de la fibre musculaire (cytosquelette) à l'extérieur (matrice extracellulaire). L’absence de dystrophine déstabilise cette interaction et par conséquent fragilise la membrane de la fibre musculaire. La membrane musculaire fragilisée résiste mal aux contraintes imposées lors de la contraction musculaire, et la fibre musculaire serait détruite, libérant des enzymes musculaires (CPK) dans le sang.
© A
FM
- M
. G
ille
s
dystrophine
Avancées dans les dystrophies musculaires de Duchenne et de Becker Juin 2013
AFM-Téléthon>Myoinfo 9/40
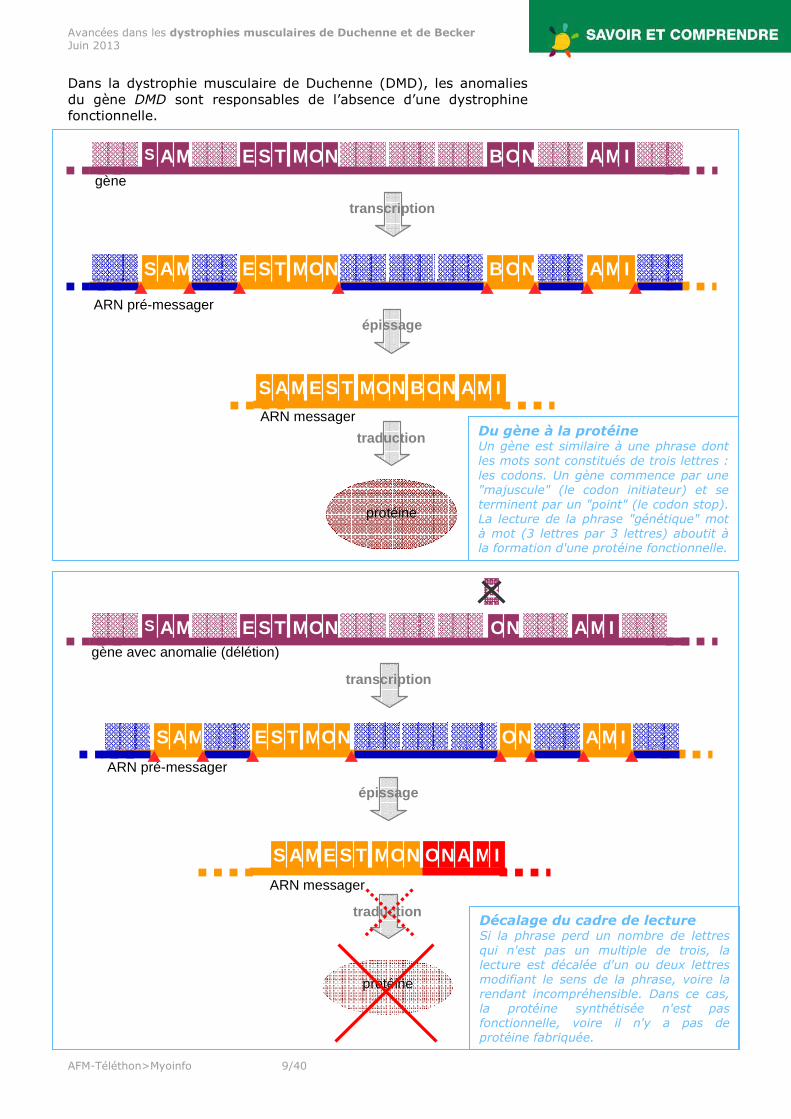
Dans la dystrophie musculaire de Duchenne (DMD), les anomalies du gène DMD sont responsables de l’absence d’une dystrophine fonctionnelle.
ONM S A M E T S I MAON
MOS A M E T S I MAN ON
épissage
transcription
traduction
protéine
ARN pré-messager
ARN messager
ONM S A M E T S I MAONgène avec anomalie (délétion)
B
Décalage du cadre de lecture Si la phrase perd un nombre de lettres qui n'est pas un multiple de trois, la lecture est décalée d'un ou deux lettres modifiant le sens de la phrase, voire la rendant incompréhensible. Dans ce cas, la protéine synthétisée n'est pas fonctionnelle, voire il n'y a pas de protéine fabriquée.
ONM S A M E T S I MAB ON
ONM S A M E T S I MAB ON
ONMS A ME T S I MAB ON
protéine
gène
ARN pré-messager
ARN messager
transcription
épissage
traduction
Du gène à la protéine Un gène est similaire à une phrase dont les mots sont constitués de trois lettres : les codons. Un gène commence par une "majuscule" (le codon initiateur) et se terminent par un "point" (le codon stop). La lecture de la phrase "génétique" mot à mot (3 lettres par 3 lettres) aboutit à la formation d'une protéine fonctionnelle.

Avancées dans les dystrophies musculaires de Duchenne et de Becker Juin 2013
10/40 AFM-Téléthon>Myoinfo
Dans la dystrophie musculaire de Becker (BMD), les anomalies du gène DMD respectent le cadre de lecture (un nombre de bases multiple de trois est maintenu) et sont responsables d'une réduction de la taille de la dystrophine produite. Les anomalies de la dystrophine conduisent à un man que de force L'absence de dystrophine (pour la DMD), son déficit partiel et/ou la réduction de sa taille (pour la BMD) rendent le complexe DAP instable et l'enveloppe des fibres musculaires, la membrane des cellules, plus fragile. Les cellules musculaires ont alors tendance à s'abîmer quand elles se contractent. Dans les 2 maladies, la fragilité des cellules ou fibres musculaires aboutit à leur destruction (on parle de dégénérescence) entraînant une nécrose du tissu musculaire. Cette nécrose s'accompagne de mécanismes de régénération, grâce à des cellules souches adultes spécialisées - les cellules satellites - présentes dans le muscle, qui visent à réparer le tissu musculaire atteint. Au fil du temps, les cycles de nécrose-régénération se succèdent. Dans un premier temps, les capacités de régénération compensent la dégénérescence et il n'y a pas ou peu de manifestations de la maladie. Secondairement, les mécanismes de régénération sont dépassés et la dégénérescence l'emporte entraînant l'apparition d'un manque de force musculaire. Petit à petit, les fibres musculaires sont remplacées par du tissu conjonctif (fibrose) et du tissu graisseux.
Les cellules souches possèdent à la fois la capacité
de se multiplier à l’identique pour produire de nouvelles
cellules souches (auto-renouvellement) et celle de
donner naissance, dans des conditions déterminées, à des cellules différenciées (cellules
sanguines, cellules du foie, cellules musculaires...).
La nécrose cellulaire est une mort accidentelle des cellules, due à des facteurs extérieurs
(manque d'oxygène, intoxication, maladie...). Si la
cellule est trop endommagée, elle se nécrose, libérant son contenu et provoquant une inflammation et des lésions
des tissus alentours.
S A M E T S I M A
S A M E T S I M A
épissage
transcription
S A M E T S I M A
O N B O N
délétion respectant le cadre de lecture
Des anomalies qui respectent le cadre de lecture du message génétique. Chez la majorité des personnes atteintes de dystrophie musculaire de Becker, les anomalies dans le gène DMD correspondent à une perte d'un nombre de lettres multiple de trois. Par exemple, l'information "Samestmonbonami." lue codons par codons donne la phrase "Sam est mon bon ami.". S'il y a une absence (délétion) des lettres "MON", "BON" et de l'intervalle non codant dont la taille respecte un multiple de 3 lettres, la phrase donne "Sam est ami". Cette phrase est incomplète mais garde un sens, comme la dystrophine, qui est raccourcie et peut conserver sa fonction dans la dystrophie musculaire de Becker.

Avancées dans les dystrophies musculaires de Duchenne et de Becker Juin 2013
AFM-Téléthon>Myoinfo 11/40
La faiblesse musculaire favorise l'apparition de déformations orthopédiques Le manque de force musculaire qui résulte du processus dystrophique rend les mouvements moins amples et moins nombreux. Certains muscles s'affaiblissent plus vite ou plus que d'autres, entrainant un déséquilibre de force. L'utilisation d'autres muscles pour pallier la faiblesse de certains groupes musculaires modifie la gestuelle. En l'absence de prise en charge, le manque de mouvements et/ou les postures de compensation et/ou le déséquilibre de force entre différents muscles favorisent le raccourcissement de certains muscles et de leurs tendons (rétractions musculo-tendineuses) ainsi que l'enraidissement progressif de certaines articulations et de leurs ligaments. Les médecins parlent de déformations orthopédiques. En effet, avec ou sans maladie musculaire, un muscle immobilisé à cause d’une maladie (comme une myopathie) ou d’un traumatisme (nécessitant une contention par plâtre par exemple) a tendance à perdre du volume et de la force (il s'atrophie), à devenir fibreux (il perd son élasticité) et à se rétracter (il se raccourcit).
Différents mécanismes en jeu dans les dystrophies musculaires de Duchenne et de Becker Pour mieux comprendre ces deux maladies, les chercheurs tentent d'identifier précisément les mécanismes par lesquels l'absence (DMD), le déficit partiel et/ou la taille réduite de la dystrophine (BMD) provoquent la dégénérescence des fibres musculaires. Plusieurs hypothèses sur les mécanismes en cause ont été émises. Il est probable que plusieurs mécanismes différents rentrent en jeu et qu'ils interagissent entre eux pour aboutir finalement à la dégénérescence des fibres musculaires. Pour pouvoir comprendre ces mécanismes, les chercheurs étudient des modèles animaux qui reproduisent la myopathie de Duchenne. Les principaux modèles animaux de la myopathie de Duchenne sont : la souris mdx, la souris dKO (qui n’exprime ni dystrophine, ni utrophine), le chien GRMD, le chien CXMD, le ver C. elegans dystrophique, le poisson zèbre dystrophique… L'hypothèse mécanique Un défaut de dystrophine entraîne une rupture du lien entre l'intérieur et l'extérieur de la fibre musculaire, provoquant une fragilisation de la membrane de la cellule musculaire et sa rupture au moment des contractions musculaires. L'hypothèse calcique Le calcium joue un rôle important dans le fonctionnement du muscle, en particulier lors de la contraction musculaire. Il est aussi au centre de très nombreux processus cellulaires et une modification des quantités de calcium dans la cellule peut avoir des effets très néfastes. Dans les dystrophies musculaires de Duchenne et de Becker, la membrane de la fibre musculaire est davantage perméable et permet notamment au calcium d’entrer. L'augmentation de l'entrée de calcium dans la fibre musculaire entraîne une augmentation de la concentration intracellulaire de calcium, qui induit l'activation d'enzymes qui dégradent des protéines, les protéases. Ces protéases amplifient une cascade de réactions aboutissant à la destruction de la cellule musculaire.
Un modèle animal est un animal qui reproduit les caractéristiques de la maladie (à la fois sur le plan génétique et sur le plan clinique) permettant l'étude des mécanismes de la maladie ou l'essai de traitements.
Une enzyme est une protéine qui permet, facilite ou accélère spécifiquement telle ou telle réaction chimique dans notre organisme (digestion cellulaire, synthèse de protéines, réplication d'ADN...).

Avancées dans les dystrophies musculaires de Duchenne et de Becker Juin 2013
12/40 AFM-Téléthon>Myoinfo
L'hypothèse vasculaire Parmi les différentes protéines qui participent au complexe des protéines associées à la dystrophine (DAP), on trouve l'enzyme NOS ou "NO synthase" musculaire. Cette enzyme permet la synthèse du monoxyde d'azote (NO) dans les cellules musculaires. Le monoxyde d'azote entraîne une dilatation locale des vaisseaux sanguins. Cette vasodilatation, en améliorant l'irrigation sanguine du muscle, augmente l'apport d'oxygène et de nutriments nécessaires au muscle lors d'un effort. Dans les dystrophies musculaires de Duchenne et de Becker, le complexe DAP est déstabilisé et la NOS est délocalisée. Le déficit en NOS pourrait aggraver l'insuffisance d'irrigation sanguine (ischémie) du muscle et donc la fatigue musculaire. Ce même mécanisme pourrait également participer à l'inflammation du tissu musculaire observée dans la DMD. L'hypothèse inflammatoire Dans la dystrophie musculaire de Duchenne (DMD), il existe une inflammation du tissu musculaire observée à l'examen au microscope de la biopsie musculaire. Celle-ci pourrait contribuer à la dégénérescence des fibres musculaires. Un argument en faveur de cette hypothèse est l’effet bénéfique des corticoïdes (anti-inflammatoires) dans la DMD.
Des bases de données pour mieux connaître les dystrophies musculaires de Duchenne et de Becker. Le développement d’un registre de patients permet d’effectuer un recensement exhaustif des personnes atteintes d'une maladie et de préciser l’histoire naturelle de celle-ci. La détermination de l’histoire naturelle d'une maladie est un pré-requis important avant la mise en place de traitements ou d'essais cliniques. La base de données UMD-DMD France, soutenue par l’AFM-Téléthon et impliquant l’ensemble des laboratoires français de diagnostic moléculaire et des Centres de Référence des maladies neuromusculaires, va permettre de mieux comprendre les mutations du gène DMD, mieux connaître l’évolution à long terme de la maladie, d'établir des corrélations génotype/phénotype et d’identifier les patients pour des essais cliniques ou des recherches scientifiques. La base de données UMD-DMD France en pratique - Un formulaire de consentement doit être impérativement co-signé par la personne porteuse d’une anomalie dans le gène DMD et par le médecin spécialiste qui la suit. - .Le formulaire de consentement est disponible sur le site : WEB www.umd.be/DMD/ - Toutes les données, médicales, cliniques et génétiques recueillies sont strictement confidentielles et rendues anonymes. - Les patients peuvent à tout moment accéder, rectifier ou demander le retrait de leurs données de la banque - Pour plus d’informations sur la base UMD-DMD France : WEB www.umd.be/DMD/
Les études de corrélations génotype/phénotype
recherchent l'existence de liens entre les caractéristiques génétiques, le génotype, et les caractéristiques s'exprimant de façon apparente, le phénotype
(taille, couleur et forme des yeux, couleur des cheveux,
manifestation d'une maladie...). On peut ainsi identifier une
relation plus ou moins étroite entre la présence d'une
anomalie génétique de tel ou tel type et celle de telles ou telles manifestations d'une
maladie génétique.
Ce que les médecins appellent l'histoire naturelle d'une
maladie est la description des différentes manifestations
d'une maladie et de leur évolution au cours du temps
en l'absence de tout traitement (médicaments, kinésithérapie,
chirurgie…).

Avancées dans les dystrophies musculaires de Duchenne et de Becker Juin 2013
AFM-Téléthon>Myoinfo 13/40
Une base de données pour mieux connaitre la dystrop hine La base de données "eDystrophin", soutenue par l’AFM-Téléthon, recense et décrit les anomalies de la protéine dystrophine en fonction des anomalies génétiques en cause. Grâce à un logiciel, la structure tridimensionnelle de la protéine peut être analysée qu’elle soit entière ou anormale. Cela renseigne sur la forme de la protéine mutée, si elle conserve sa structure filamenteuse, ses sites d’interaction avec d’autres protéines, ses propriétés... Cet outil permet d’établir des relations entre l'anomalie génétique (génotype), la forme de la dystrophine anormale et les signes cliniques de la maladie (phénotype). Il peut servir ainsi à mieux concevoir les stratégies de saut d’exon, par exemple. La base de données eDystrophin - En juillet 2012, cette base de données répertoriait 209 mutations dans le cadre de lecture, provenant de 945 patients principalement atteints de BMD. WEB http://edystrophin.genouest.org/
Les pistes thérapeutiques dans les dystrophies musculaires de Duchenne et de Becker Comme les dystrophies musculaires de Becker (BMD) et de Duchenne (DMD) sont dues à l'altération du même gène DMD, de nombreuses approches thérapeutiques développées pour la myopathie de Duchenne sont ou seront applicables ou transposables à la dystrophie musculaire de Becker, notamment la thérapie génique, la thérapie cellulaire et certaines approches médicamenteuses. De nombreuses pistes complémentaires Pour réussir à traiter les dystrophies musculaires de Duchenne et de Becker, les chercheurs ont développé des pistes thérapeutiques complémentaires ciblant ces maladies à différents niveaux, de leur origine génétique à leurs conséquences cliniques : - pallier l'altération du gène : la thérapie génique ; - corriger le gène : les méganucléases ; - modifier l'anomalie au niveau de l'ARN messager : la translecture de codons Stop prématurés, le transépissage ; - remplacer la protéine déficitaire : l'utrophine ; - apporter de nouvelles cellules musculaires : la thérapie cellulaire ; - réduire la destruction des fibres musculaires : les anti-protéases, les facteurs de croissance, les inhibiteurs de canaux calciques, les donneurs de NO,… - réduire la fibrose : les anti-fibrotiques - compenser la perte de masse musculaire : les anti-myostatines ; - améliorer la force musculaire : stabiliser RyR1, les inhibiteurs de l'enzyme de conversion, - protéger la fonction cardiaque : les médicaments de l'insuffisance cardiaque. Apporter des molécules thérapeutiques au cœur du no yau Les chercheurs ont développé différents types de vecteurs et de techniques permettant l’administration d’un gène thérapeutique ou d’un oligonucléotide antisens jusqu'au noyau des cellules malades : les vecteurs viraux (virus dont les éléments qui rendent malade sont remplacés par le gène thérapeutique), les vecteurs non viraux (plasmide, vecteurs lipidiques,…) et des techniques "physiques"
Un oligonucléotide antisens est un fragment d'ARN, généralement synthétisé en laboratoire qui se lie spécifiquement à un ARN messager naturel (la séquence de l’oligonucléotide antisens est complémentaire de celle de l'ARN messager). Il peut ainsi modifier l’ARN messager (saut ou incorporation d’exon(s) en intervenant à l'étape de sa maturation (l’épissage) .

Avancées dans les dystrophies musculaires de Duchenne et de Becker Juin 2013
14/40 AFM-Téléthon>Myoinfo
(électroporation, biolistique...). Chaque méthode présente des avantages et des inconvénients. Les vecteurs viraux Les vecteurs viraux les plus utilisés pour cibler le muscle squelettique in vivo sont les AAV (virus associés aux adénovirus). Ils présentent cependant l'inconvénient de ne pouvoir incorporer que des gènes de petite taille contrairement aux vecteurs non-viraux. Le gène de la dystrophine humaine étant très grand, les chercheurs "fabriquent" des mini-gènes de dystrophine qui codent une micro- ou une mini-dystrophine, c'est-à-dire des dystrophines plus courtes et fonctionnelles. Les oligonucléotides antisens peuvent également être inclus dans un minigène codant pour un petit ARN (U1 ou U7). Dans l’approche vectorisée, un AAV transporte au sein de la cellule ce mini-gène (U1 ou U7) qui comprend l’oligonucléotide antisens. Le vecteur viral permet d’atteindre le noyau des cellules de façon très efficace et ainsi la production du mini-gène. On parle d’oligonucléotides antisens vectorisés, par opposition aux oligonucléotides antisens libres, injectés tels quels. Un des problèmes posé par les AAV est celui de la réaction du système de défense immunitaire de l'organisme qu'ils peuvent provoquer. Pour contourner cette difficulté, les chercheurs utilisent des traitements qui modulent l'activité du système immunitaire (immunosuppresseurs) pour modérer, voire éteindre complètement, la réaction immunitaire de rejet contre le vecteur viral et le produit du gène thérapeutique. Les vecteurs non-viraux Une autre solution consiste à transporter le gène thérapeutique dans des vecteurs non viraux. Plusieurs pistes sont explorées, avec des vecteurs synthétiques (plasmide, vecteurs lipidiques…) ou des chromosomes artificiels humains. Ces vecteurs présentent les avantages de pouvoir contenir des gènes de grande taille et de provoquer une réaction immunitaire moindre. En revanche, le transfert de gène est moins efficace qu’avec les AAV, par exemple. Remplacer le gène altéré par un gène thérapeutique : la thérapie génique La thérapie génique consiste à remplacer le gène défectueux ou manquant par un gène thérapeutique. Dans les dystrophies musculaires de Duchenne et de Becker, il s'agit de remplacer le gène DMD altéré par un gène de dystrophine sans anomalie. Les études menées chez la souris mdx ont montré que les AAV transportant un gène de micro- ou de mini-dystrophine restaurent l'expression de la micro-ou mini-dystrophine en quantité quasi-normale et ce dans de nombreux muscles squelettiques et dans le muscle cardiaque. La force des muscles traités est souvent améliorée ainsi que l'espérance de vie des souris. Très récemment, des travaux ont rapporté la preuve de concept dans un modèle de chien de l’efficacité d’une thérapie génique avec un gène de micro-dystrophine. L’administration d’un AAV9 transportant une micro-dystrophine dans un muscle de la patte antérieure de chiens dystrophiques a entrainé une expression nette
Le virus adéno-associé (AAV pour adeno-associated virus) est un petit virus à ADN, qui
peut infecter l'être humain. Toutefois, il ne provoque pas
de maladie et n'entraine qu'une réponse immunitaire de
défense modérée Il est utilisé en génie génétique comme
vecteur pour la thérapie génique.

Avancées dans les dystrophies musculaires de Duchenne et de Becker Juin 2013
AFM-Téléthon>Myoinfo 15/40
de microdystrophine, une restauration du complexe de glycoprotéines associées à la dystrophine (complexe DAP) ainsi qu’une réduction des lésions tissulaires dans les muscles des chiens dystrophiques traités. Produire une mini-dystrophine chez l'homme En 2010, les résultats d’un essai de thérapie génique visant à transporter un gène de dystrophine raccourcie et fonctionnelle (mini-dystrophine) ont montré des fibres musculaires exprimant la mini-dystrophine chez 2 des 6 patients traités. Les réponses immunitaires spécifiques à la mini-dystrophine obtenues ont été hétérogènes (aucune réponse immunitaire chez 2 individus, une réponse immunitaire retardée chez 2 autres et une réponse détectée avant même que le traitement ne débute chez les 2 derniers,…). L’existence d’une réaction immunitaire provoquée par la dystrophine transférée mais aussi par la dystrophine présente avant le début du traitement, doit être mieux comprise et mieux gérée pour développer de nouvelles thérapies. Dans des conditions similaires, l’injection intramusculaire de plasmide portant la dystrophine entière n’engendrait pas de réponse immunitaire chez les 9 patients Duchenne et Becker qui ont participé à cette étude en 2004. Une équipe américaine a mis au point un nouveau vecteur viral, le vecteur AAV2.5, comportant un mélange de structure des vecteurs AAV2 et AAV1. Avec ce vecteur, le transfert de gène dans les muscles squelettiques est plus efficace et la réponse immunitaire est plus faible. En 2011, un essai clinique de phase I, randomisé, contrôlé, en double aveugle a évalué l’injection du vecteur AAV2.5 transportant la mini-dystrophine dans le bras (biceps) de 6 patients atteints de DMD. Dans l’autre bras, un contrôle salin ou un vecteur AAV2.5 vide (sans gène de mini-dystrophine) a été injecté. Le vecteur AAV2.5 a été détecté chez tous les patients. Il n’y a pas eu de différence concernant la réponse immunitaire dirigée contre le vecteur ou le placebo et le vecteur AAV2.5 a été bien toléré. Transférer le gène SERCA2 pour améliorer la fonction cardiaque Une autre piste thérapeutique en cours d’étude consiste à faire surexprimer une pompe à calcium, SERCA2a. Lors de la relaxation du muscle cardiaque, le calcium est recapté du cytoplasme dans le réticulum sarcoplasmique par SERCA2a. Dans les DMD et BMD, comme dans l’insuffisance cardiaque, l’expression de SERCA2a est fortement diminuée entrainant une accumulation du calcium dans le cytoplasme. L’injection unique d’un AAV9 transportant le gène SERCA2a dans la veine de la queue de souris mdx âgées de 12 mois a montré que, 8 mois plus tard, les cellules cardiaques des souris traitées exprimaient toutes l’AAV9 injecté. La fonction cardiaque a été significativement améliorée chez ces souris traitées. Cette approche est actuellement étudiée dans l’insuffisance cardiaque dans le cadre d’un essai de phase I/II chez l'homme (pas atteints de DMD ou de BMD).
Lors d'un essai clinique de phase I , un médicament dont l'intérêt thérapeutique a été montré sur des modèles animaux et/ou cellulaires est administré pour la 1ère fois à un petit groupe de volontaires sains, plus rarement à des malades, afin d'évaluer leur tolérance à la substance selon la dose administrée. >> Essais cliniques et maladies neuromusculaires, Repères Savoir & Comprendre, AFM, Juillet 2010.
Le réticulum sarcoplasmique est un réseau complexe de cavités à l'intérieur de la cellule musculaire, constituant un compartiment cellulaire dans lequel le calcium nécessaire à la contraction musculaire est mis en réserve.
Le cytoplasme est la substance à l'intérieur des cellules, dans laquelle baignent les différents éléments cellulaires : l'ossature de la cellule (cytosquelette), le noyau, les structures spécialisées assurant les fonctions de la cellule (organites), des réserves (inclusions) et des éléments libres (protéines, nutriments...).
Dans un essai randomisé , les participants sont répartis par tirage au sort dans les différents groupes.
Un essai contrôlé est un essai qui compare l'efficacité de la substance testée à celle d'un placebo ou d'une substance active connue : une partie des participants prend un placebo ou une autre substance active et constitue un groupe "contrôle".
Un essai en ouvert est un essai thérapeutique dans lequel les médecins et les participants ont connaissance du traitement prescrit.

Avancées dans les dystrophies musculaires de Duchenne et de Becker Juin 2013
16/40 AFM-Téléthon>Myoinfo
Réparer le gène altéré à l'aide des méganucléases Les méganucléases sont des ciseaux moléculaires qui coupent l'ADN. Ces enzymes sont capables de couper une portion de gène avec une très grande précision. Une portion saine peut être substituée à la portion anormale et le gène être ainsi physiquement réparé. Développée par la société de biotechnologie Cellectis, cette "chirurgie du génome" par méganucléases s'appuie sur les systèmes de réparation de l'ADN naturels de la cellule. L'AFM-Téléthon et Cellectis ont lancé le 24 juillet 2008, un programme sur 5 ans pour développer des méganucléases spécifiques de 7 gènes, dont le gène de la dystrophine. La première méganucléase dirigée sur le gène de la dystrophine a été livrée par Cellectis en octobre 2009. Ces molécules sont en cours de validation sur des cultures de cellules musculaires humaines ou dans des modèles animaux, préalables nécessaires à leur utilisation dans d'éventuelles applications chez l'homme.
La thérapie génique consiste à administrer un gène thérapeutique dans des cellules où le gène est défectueux ou manquant. Les chercheurs ont développé différents types de vecteurs et de techniques permettant le transfert de ces gènes dans l’organisme : vecteurs viraux (virus dont les éléments pathogènes sont remplacés par le gène-médicament), vecteurs synthétiques (composés chimiques se liant au gène thérapeutique) et techniques "physiques" (électroporation, biolistique...). Les vecteurs viraux les plus utilisés in vivo, sont les AAV (associated adeno virus).
© A
FM
- D. H
oa
ra
u

Avancées dans les dystrophies musculaires de Duchenne et de Becker Juin 2013
AFM-Téléthon>Myoinfo 17/40
Translecture de codon stop grâce à l'ataluren (PTC1 24) On estime que 10 à 15% des personnes atteintes de dystrophie musculaire de Duchenne (DMD) ou de Becker (BMD) présentent des anomalies génétiques de type "non-sens". Ce type d'anomalie conduit à la formation d’un "codon stop" prématuré dans l’ARN messager qui met ainsi fin à la synthèse de la dystrophine. Cela aboutit à la production d'une dystrophine plus courte que la dystrophine normale et qui n'est pas fonctionnelle. L'ataluren (ou PTC124) est une molécule qui passe outre les codons stop prématurés des gènes (translecture). Il est utilisé dans les dystrophies musculaires de Duchenne et de Becker pour restaurer la production d'une dystrophine fonctionnelle. Un essai de phase III, en ouvert, est actuellement en cours de recrutement de 122 patients atteints de DMD ou de BMD aux États-Unis. Il concerne les personnes qui ont participé à l’essai de phase II qui avait montré que la dose de 40 mg/kg était plus efficace que la dose de 80mg/kg. Les participants recevront, pendant 9 mois, un traitement par ataluren à la dose de 40 mg/kg/j en trois prises.
Au cours d'un essai clinique de phase III , un médicament, pour lequel on a déterminé lors d'essais antérieurs l'innocuité et le dosage optimum (essais de phase I et II), est administré à un grand groupe de malades, sur une longue durée, dans le but d'évaluer son efficacité thérapeutique en la comparant à celle d'un traitement de référence ou un placebo. Il permet aussi de mettre en évidence les interactions indésirables et les effets secondaires du traitement à moyen terme. Au terme de cet essai, le médicament peut obtenir une autorisation de mise sur le marché. >> Essais cliniques et maladies neuromusculaires, Repères Savoir & Comprendre, AFM, Juillet 2010.

Avancées dans les dystrophies musculaires de Duchenne et de Becker Juin 2013
18/40 AFM-Téléthon>Myoinfo
En Europe, Australie, Israël et Canada, une phase d’extension a débuté. Dans cet essai de phase III en ouvert, 96 patients qui ont déjà reçu de l’ataluren au cours d’un essai précédent seront inclus. La tolérance et l’innocuité du traitement par 40 mg/kg/j seront évaluées pendant 48 semaines. Essai en cours de recrutement • Essai de phase III en ouvert du traitement oral de l’ataluren : 3 fois par jour (10 mg/kg le matin, 10 mg/kg le midi et 20 mg/kg le soir) pendant 48 semaines. - Recrutement en cours de 96 personnes atteintes de DMD ou de BMD ayant déjà reçu de l'ataluren lors d'un essai précédent. En décembre 2012, la société PTC Therapeutics, qui développe l’ataluren, a annoncé dans un communiqué de presse qu’elle avait fait une demande d’approbation conditionnelle pour l’ataluren auprès de l'Agence Européenne des Médicaments (EMA). Une approbation conditionnelle est attribuée aux candidats-médicaments ayant un ratio bénéfice/risque positif, mais nécessitant des études complémentaires pour confirmer ces données. Elle permet notamment aux candidats-médicaments une mise à disposition sur le marché, pendant que des études complémentaires continuent. En parallèle à cette demande, PTC Therapeutics a débuté un nouvel essai international (y compris en France) de phase III, randomisé, en double aveugle, contre placebo de l’ataluren. Cet essai vise à déterminer l’innocuité et la tolérance de l’ataluren à la dose de 40 mg/kg par jour pendant 48 semaines chez 220 personnes atteintes de DMD ou de BMD. A l’heure actuelle, le recrutement n’a commencé qu’aux États-Unis. Essai en cours de recrutement • Essai international de phase III, randomisé, en double aveugle, contrôlé de l’ataluren en 3 prises orales par jour (10 mg/kg le matin, 10 mg/kg le midi et 20 mg/kg le soir). - Nombre de participants prévu : 220 personnes atteintes de DMD ou de BMD. Réparer l’ARN de la dystrophine : le trans-épissage Le trans-épissage est une technologie de réparation de l’ARN qui permet de remplacer une séquence défectueuse par une séquence thérapeutique. Les chercheurs forcent la machinerie d’épissage de la cellule à lier la séquence thérapeutique portée par une molécule dite "de trans-épissage" à l’ARN de la dystrophine anormal produit par la cellule. Ainsi, la dystrophine produite à partir de l’ARN réparé par trans-épissage est complète et donc entièrement fonctionnelle. L’intérêt majeur du trans-épissage est que cette stratégie thérapeutique au niveau de l’ARN peut réparer tous les types d’anomalie génétique : les mutations ponctuelles, les délétions et les duplications, et même les défauts portés par des exons indispensables à la fonction de la protéine.
L'épissage est une étape de la fabrication des protéines.
Dans la première étape, la transcription, le message du gène est "transcrit" en ARN
messager (un peu comme une photocopie de la région d'ADN
qui porte le gène). Dans la seconde étape,
l'épissage, l'ARN messager est "épissé" : certaines parties
(les séquences non codantes ou "introns")sont coupées et les morceaux restants sont
réunis en un seul brin d'ARN messager mature (qui ne
comprends que les séquences codantes ou "exons") qui ne
contient que les informations nécessaires pour guider la
synthèse de la protéine.
Avancées dans les dystrophies musculaires de Duchenne et de Becker Juin 2013
AFM-Téléthon>Myoinfo 19/40
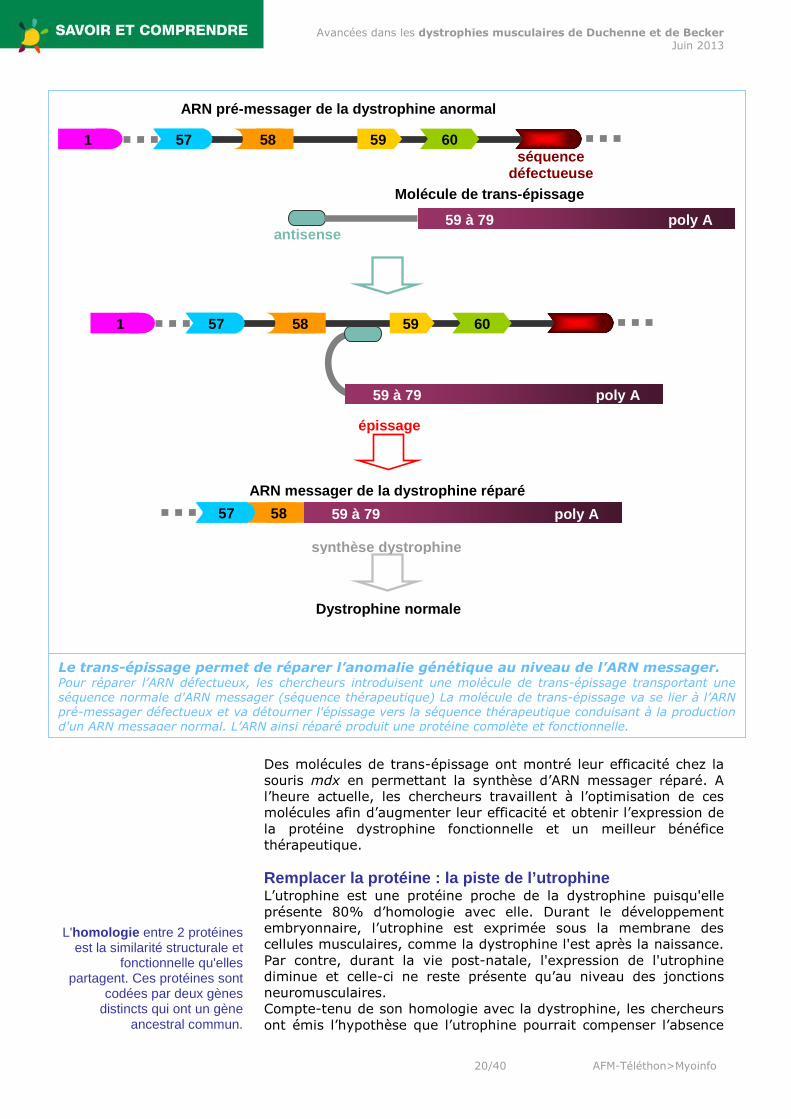
De plus, une même molécule de trans-épissage pouvant réparer des anomalies localisées à différents endroits du gène de la dystrophine, le développement d’un tel médicament s’adressera à un plus grand nombre de patients.
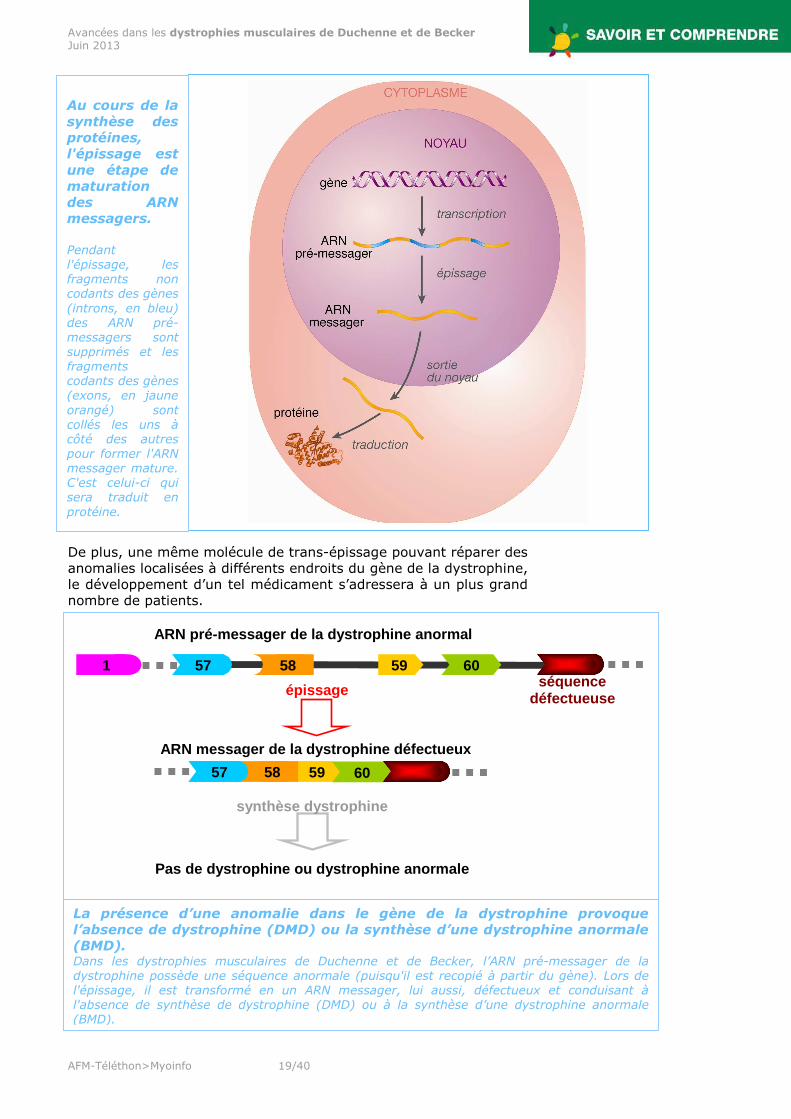
Au cours de la synthèse des protéines, l'épissage est une étape de maturation des ARN messagers. Pendant l'épissage, les fragments non codants des gènes (introns, en bleu) des ARN pré-messagers sont supprimés et les fragments codants des gènes (exons, en jaune orangé) sont collés les uns à côté des autres pour former l'ARN messager mature. C'est celui-ci qui sera traduit en protéine.
séquence défectueuse
59 60 58 57
ARN pré-messager de la dystrophine anormal
ARN messager de la dystrophine défectueux
épissage
59 60 58 57 1
synthèse dystrophine
Pas de dystrophine ou dystrophine anormale
La présence d’une anomalie dans le gène de la dystrophine provoque l’absence de dystrophine (DMD) ou la synthèse d’une dystrophine anormale (BMD). Dans les dystrophies musculaires de Duchenne et de Becker, l’ARN pré-messager de la dystrophine possède une séquence anormale (puisqu'il est recopié à partir du gène). Lors de l'épissage, il est transformé en un ARN messager, lui aussi, défectueux et conduisant à l'absence de synthèse de dystrophine (DMD) ou à la synthèse d’une dystrophine anormale (BMD).
Avancées dans les dystrophies musculaires de Duchenne et de Becker Juin 2013
20/40 AFM-Téléthon>Myoinfo
Des molécules de trans-épissage ont montré leur efficacité chez la souris mdx en permettant la synthèse d’ARN messager réparé. A l’heure actuelle, les chercheurs travaillent à l’optimisation de ces molécules afin d’augmenter leur efficacité et obtenir l’expression de la protéine dystrophine fonctionnelle et un meilleur bénéfice thérapeutique. Remplacer la protéine : la piste de l’utrophine L’utrophine est une protéine proche de la dystrophine puisqu'elle présente 80% d’homologie avec elle. Durant le développement embryonnaire, l’utrophine est exprimée sous la membrane des cellules musculaires, comme la dystrophine l'est après la naissance. Par contre, durant la vie post-natale, l'expression de l'utrophine diminue et celle-ci ne reste présente qu’au niveau des jonctions neuromusculaires. Compte-tenu de son homologie avec la dystrophine, les chercheurs ont émis l’hypothèse que l’utrophine pourrait compenser l’absence
L'homologie entre 2 protéines est la similarité structurale et
fonctionnelle qu'elles partagent. Ces protéines sont
codées par deux gènes distincts qui ont un gène
ancestral commun.
Le trans-épissage permet de réparer l’anomalie génétique au niveau de l’ARN messager. Pour réparer l’ARN défectueux, les chercheurs introduisent une molécule de trans-épissage transportant une séquence normale d'ARN messager (séquence thérapeutique) La molécule de trans-épissage va se lier à l’ARN pré-messager défectueux et va détourner l'épissage vers la séquence thérapeutique conduisant à la production d'un ARN messager normal. L’ARN ainsi réparé produit une protéine complète et fonctionnelle.
59 60 58 57 1
59 à 79 poly A
59 à 79 poly A antisense
ARN pré-messager de la dystrophine anormal
ARN messager de la dystrophine réparé
séquence défectueuse
59 60 58 57 1
synthèse dystrophine
Dystrophine normale
Molécule de trans-épissage
58 57 59 à 79 poly A
épissage

Avancées dans les dystrophies musculaires de Duchenne et de Becker Juin 2013
AFM-Téléthon>Myoinfo 21/40
de dystrophine dans la DMD ou le déficit partiel en dystrophine dans la BMD. De nombreuses études ont été menées pour augmenter l'expression (surexpression) de l’utrophine chez la souris mdx. Les chercheurs ont utilisé différentes approches : souris mdx transgéniques surexprimant génétiquement le gène de l’utrophine, approche pharmacologique (travaux pionniers sur l’effet du butyrate d’arginine), thérapie génique. Les résultats ont montré que l’utrophine, lorsqu'elle est fortement surexprimée dès le stade embryonnaire, peut remplacer efficacement et fonctionnellement la dystrophine. Par contre, les surexpressions stimulées après la naissance - chez des souris mdx adultes - ont donné des résultats moins convaincants. Le SMT C1100 Le SMT C1100 est une molécule conçue par la société Summit PLC pour augmenter la production d’utrophine dans le muscle. Les résultats d’un premier essai de phase I du SMT C1100 chez des volontaires sains ont montré que la molécule a été bien tolérée. Cependant, elle présentait une faible activité thérapeutique liée à une composition de la molécule (formulation) inadaptée (seule une très faible quantité de produit a atteint la circulation sanguine). Une nouvelle formulation de SMT C1100, conçue pour être mieux absorbée et atteindre plus facilement les muscles, a été développée. Elle a été évaluée, à des doses croissantes de 50 mg/kg à 400 mg/kg, lors d’un essai clinique de phase I, en double aveugle contre placebo, chez 48 volontaires sains. Des résultats préliminaires de cet essai ont montré qu’à la dose de 400 mg/kg, le taux sanguin de SMT C1100 chez les volontaires sains atteint celui qui est requis pour obtenir une augmentation de production d’utrophine de 50 %, in vitro, dans des cellules de patients atteints de DMD en culture. L’analyse des résultats de l’essai de la nouvelle formulation se poursuit actuellement. En mars 2013, la société Summit PLC a annoncé qu’elle comptait planifier au second semestre 2013 un essai clinique pour évaluer le candidat médicament SMT C1100 dans la dystrophie musculaire de Duchenne. Le SMT C1100 a obtenu le statut de médicament orphelin en Europe et aux États-Unis. Essai en cours d’analyse • Essai de phase I pour évaluer la tolérance et le devenir de la nouvelle formulation de SMT C1100 dans l’organisme (pharmacocinétique) chez des volontaires sains. • Les résultats préliminaires ont mis en évidence une augmentation des taux sanguins de SMT C1100. Le nabumétone En 2011, une équipe américaine a mis en évidence une nouvelle molécule, le nabumetone, qui augmente l’expression de l’utrophine dans des cultures de cellules et dont l’innocuité et le devenir de la molécule dans l’organisme (c'est-à-dire la pharmacocinétique) sont déjà connus chez l’homme. Remplacer les cellules musculaires déficientes par des cellules souches : la thérapie cellulaire Contrairement aux méthodes pharmacologiques basées sur l'emploi de molécules chimiques, la thérapie cellulaire est fondée sur l'utilisation de cellules vivantes. L'objectif est de remplacer des
Les cellules souches possèdent à la fois la capacité de se multiplier à l’identique pour produire de nouvelles cellules souches (auto-renouvellement) et celle de donner naissance, dans des conditions déterminées, à des cellules différenciées (cellules sanguines, cellules du foie, cellules musculaires...).

Avancées dans les dystrophies musculaires de Duchenne et de Becker Juin 2013
22/40 AFM-Téléthon>Myoinfo
cellules déficientes ou anormales par des cellules souches. Cette technique consiste à prélever des cellules soit chez le patient à traiter (autogreffe), soit chez un donneur (allogreffe). Ces cellules sont ensuite purifiées, modifiées, puis multipliées en laboratoire. Elles sont alors injectées dans l'organe à traiter de la personne malade. Différents types de cellules sont actuellement utilisés par les chercheurs dans le but de remplacer les cellules musculaires qui ont dégénéré dans la dystrophie musculaire de Duchenne (DMD) ou celle de Becker (BMD) : myoblastes, cellules souches sanguines spécifiques (CD133+), mésangioblastes, cellules souches dérivées du muscle (MDSC, MuStem), cellules stromales de la moelle osseuse, cellules souches adipeuses (hMADS), cellules progénitrices du muscle, cellules souches mésenchymateuses de sang de cordon ombilical... Il s'agit généralement de cellules souches adultes. Cependant, les cellules souches adultes ont des capacités d'auto-renouvellement et de différenciation inférieures aux cellules souches embryonnaires. L'utilisation de cellules souches embryonnaires est séduisante mais est encore au stade de la recherche fondamentale
La thérapie cellulaire est fondée sur l'utilisation de cellules souches vivantes. Cette technique consiste à prélever ces cellules soit chez le patient à traiter (autogreffe), soit chez un donneur (allogreffe), à les purifier, les multiplier et éventuellement les modifier. Ces cellules seront ensuite réimplantées chez le malade pour remplacer des cellules déficientes ou disparues, du fait de la maladie.
Prélèvement de cellules souches (adultes ou
embryonnaires )
Culture de cellules en laboratoire (purification,
prolifération, modification...)
DONNEUR
PATIENT
A TRAITER
PATIENT TRAITÉ
Allogreffe Autogreffe de cellules modifiées
PATIENT TRAITÉ
+
Traitement immuno-
supresseur à vie

Avancées dans les dystrophies musculaires de Duchenne et de Becker Juin 2013
AFM-Téléthon>Myoinfo 23/40
(sur des modèles cellulaires et animaux) pour des raisons de bioéthique et de techniques. Les myoblastes sont les cellules précurseurs du muscle. En cas de lésion musculaire, elles se différencient en fibres musculaires pour réparer le muscle lésé. Les myoblastes peuvent être obtenus de deux façons : - soit ils sont prélevés dans le muscle d’un donneur sain puis transplantés chez le patient (allogreffe) qui devra alors être traité par immunosuppresseurs ; - soit les myoblastes sont prélevés chez le patient, corrigés génétiquement in vitro puis retransplantés chez le patient (autogreffe). Des cellules souches dérivées du muscle, les cellules MuStem, peuvent aussi être transférer par thérapie cellulaire en vue de régénérer le muscle. En 2011, une équipe française, soutenue par l’AFM-Téléthon, a isolé des cellules MuStem à partir de muscle squelettiques de chien en bonne santé. L’injection de ces cellules à des chiens GRMD, modèle de la DMD, a permis d’améliorer la régénération de fibres musculaires, la restauration de l’expression de dystrophine et une stabilisation de l’état clinique des chiens transplantés. Ces cellules ont également été obtenues à partir de biopsies musculaires humaines. Des cellules souches sanguines (cellules hématopoïétiques) spécifiques (CD133+) peuvent dans des conditions précises, se transformer en cellules musculaires. Le projet MyoAmp, financé par l’Union Européenne, a été lancé en janvier 2007 pour une durée de 3 ans. Son objectif était de mettre au point un protocole de thérapie cellulaire associé à un saut d’exon dans la DMD. Il rassemblait des biologistes cellulaires, des cliniciens et des entreprises de biotechnologies de cinq pays européens (France, Italie, Suède, Allemagne et Angleterre). Quatre associations de malades y étaient associées (Association Française contre les Myopathies, Duchenne Parent Project, France et Royaume-Uni, Association Monégasque Contre les Myopathies). Un communiqué publié en avril 2011 a annoncé que MyoAmp avait réussi à définir les conditions nécessaires pour isoler et amplifier des cellules souches sanguines humaines sans danger et de façon reproductible. Celles-ci pourront être utilisées pour des essais cliniques dans des conditions de BMP pour Bonnes Pratiques de Fabrication (ou Good manufacturing practices, GMP). Ce succès est notamment dû à la relation étroite qui a existé entre les associations de patients, les cliniciens et les scientifiques. Les mésangioblastes sont des cellules souches des parois des vaisseaux sanguins. Un essai italien de phase I/II de thérapie cellulaire par greffe de mésangioblastes est en cours chez des personnes atteintes de DMD. L’essai vise à évaluer l’innocuité de ce protocole et l'évolution de la fonction musculaire. Trois premiers patients ont été inclus à l’essai. Si la tolérance est bonne, 3 patients supplémentaires seront inclus. Essai en cours en Italie • Essai de phase I/II, contrôlé évaluant la faisabilité et l'innocuité de l'injection intra-artérielle de mésangioblastes de donneur compatible chez 6 garçons atteints de DMD âgés de 6 à 14 ans. - Suivi prévu pendant 3 ans.
Un essai de phase I/II vise à démontrer la faisabilité, la tolérance et si possible l'efficacité d'un traitement en cours d'évaluation chez l'homme. >> Essais cliniques et maladies neuromusculaires, Repères Savoir & Comprendre, AFM, Juillet 2010.

Avancées dans les dystrophies musculaires de Duchenne et de Becker Juin 2013
24/40 AFM-Téléthon>Myoinfo
Les cellules stromales de la moelle osseuse fournissent un support structural et fonctionnel aux cellules souches du sang (cellules hématopoïétiques). Dans des conditions particulières de culture, ces cellules stromales de la moelle osseuse sont capables de se différencier en cellules du muscle squelettique. Des modèles de souris ou de chiens atteints de DMD ayant reçu une injection de cellules souches mésenchymateuses humaines - à l’origine de la moelle osseuse mais aussi de muscle – ont exprimé la dystrophine humaine dans les muscles. Les cellules progénitrices du muscle peuvent être dérivées de cellules souches embryonnaires ou de cellules souches pluripotentes induites (iPS) pour se différencier en cellules musculaires. En 2012, une étude a apporté la preuve de concept de l’efficacité d’une thérapie cellulaire par les cellules progénitrices du muscle chez une souris modèle. Cette stratégie a entrainé une régénération in vivo du muscle squelettique (avec expression de dystrophine) s’accompagnant d’une récupération de force musculaire chez ces souris. Les cellules souches mésenchymateuses du cordon ombilical humain (hUC-MSCs) peuvent se différencier en de multiples types de cellules. Prélevées dans le sang du cordon après l’accouchement (sans effet néfaste pour la mère ou pour son nouveau né), ces jeunes cellules sont encore immatures (non spécialisées). Cela leur donne un avantage tant au niveau de leur immunologie (en augmentant la probabilité de compatibilité génétique entre deux individus) que d’un risque moindre de rejet du greffon lors d’une thérapie cellulaire. En Chine, un essai de phase I/II en ouvert est en cours de recrutement de 15 personnes atteintes de DMD. Cet essai a pour but d’évaluer l’innocuité et l’efficacité d’une transplantation de cellules souches mésenchymateuses du cordon ombilical humain dans la DMD. En Inde, un autre essai de phase I/II en ouvert est en cours de recrutement de 30 personnes atteintes de DMD. Dans la même lignée, cet essai vise aussi à évaluer l’innocuité et l’efficacité d’une greffe de cellules souches mésenchymateuses du cordon ombilical humain chez des personnes atteintes de DMD. Essais en cours de recrutement en Chine et en Inde • Essai de phase I/II en ouvert évaluant l'innocuité et l’efficacité de la transplantation de cellules hUC-MSCs - Recrutement en cours de 15 garçons atteints de DMD, âgés de 5 à 12 ans, en Chine et de 30 garçons atteints de DMD, âgés de 5 à 12 ans, en Inde. Réduire la fibrose Dans les myopathies de Duchenne ou de Becker, la dégénérescence musculaire conduit au développement de fibrose, c’est-à-dire que, petit-à-petit, les fibres musculaires sont remplacées par du tissu fibreux. A partir de souris mdx, qui possèdent un gène de la dystrophine muté mais qui présentent des manifestations de la maladie moins sévères que celles de l’homme, une équipe française, soutenue par l’AFM-téléthon, a mis au point un nouveau modèle de souris avec fibrose, qu’elle a appelé fib-mdx. Ce nouveau modèle permettra d'étudier et d'analyser les mécanismes qui président au
Une cellule progénitrice est la progéniture précoce d'une
cellule souche. Contrairement à une cellule souche, la cellule
progénitrice peut uniquement se multiplier, se différencier
mais ne peut plus se renouveler.

Avancées dans les dystrophies musculaires de Duchenne et de Becker Juin 2013
AFM-Téléthon>Myoinfo 25/40
développement de la fibrose et de trouver les moyens éventuels pour y remédier. L’halofuginone L’halofuginone est un composé dérivé d’une herbe chinoise qui inhibe la fibrose et l'inflammation. Administrée à un modèle de souris de la DMD, elle diminue la fibrose, favorise la multiplication des cellules musculaires et améliore les fonctions cardiaque et respiratoire des souris traitées. Le HT-100, un dérivé de l’halofuginone, est un candidat-médicament développé par la société DART Therapeutics (en association avec HALO Therapeutics) pour réduire la fibrose et favoriser la régénération du muscle. En avril 2012, le HT-100 a reçu par la Commission Européenne la désignation de médicament orphelin pour le traitement de la DMD. Augmenter la masse musculaire Dans les dystrophies musculaires de Duchenne (DMD) et de Becker (BMD), la dégénérescence des fibres musculaires entraîne une perte de masse musculaire. En augmentant la masse musculaire, les chercheurs espèrent améliorer la force musculaire des personnes atteintes de DMD ou de BMD. L’ACE-031 La myostatine est un inhibiteur de la croissance musculaire, existant à l'état naturel dans l'organisme (endogène). Les chercheurs essaient de favoriser la croissance musculaire en bloquant l'action de la myostatine. La suppression de la myostatine chez la souris mdx atténue la dystrophie musculaire. Un essai clinique de phase II en double aveugle, visant à évaluer l'innocuité, la tolérance et le devenir dans l'organisme (pharmacocinétique) de différentes doses d'ACE-031, un inhibiteur de la myostatine, chez 88 patients atteints de DMD ou de BMD et traités par corticothérapie a démarré en 2010. Puis, une extension de cette phase II pendant 12 semaines supplémentaires a été mise en place chez 76 de ces participants. Fin avril 2011, les sociétés Acceleron Pharma et Shire (associées sur les études ACE-031 depuis 2010) ont annoncé l’interruption de cette étude suite à la survenue d’effets secondaires chez certains participants En mai 2013, un communiqué de presse a annoncé que les sociétés Acceleron Pharma et Shire avait fini leur collaboration et ne redemareraient pas le développement de ce programme. Le DT-200 La société DART Therapeutics développe une molécule de la famille des « modulateurs sélectifs des récepteurs aux androgènes » (dits SARMs), le DT-200, qui a montré des effets positifs au cours d’études précliniques dans la DMD (augmentation de la taille et de la force musculaires). En mars 2013, la société a annoncé le démarrage d'’un essai clinique au second semestre 2013 pour évaluer les effets de DT-200 sur la force et la taille du muscle dans la DMD. Cet essai clinique concerne des volontaires sains (phase I). Selon les résultats de cet essai de phase I, le DT-200 sera testé chez des personnes atteintes de DMD. Si l' efficacité du DT-200 était démontrée, il pourrait, faire partie du cocktail thérapeutique utilisable dans la DMD.
Un médicament orphelin est un médicament développé pour le traitement d’une maladie dite orpheline, c’est-à-dire une maladie rare. Pour les entreprises pharmaceutiques, le coût de mise sur le marché d’un produit préconisé dans une maladie rare ne serait pas couvert par les ventes attendues sur ce marché "restreint" du fait du peu de personnes concernées. C'est pourquoi, sous la pression des associations de maladies rares, une politique incitative économique a été mise en place pour encourager les entreprises pharmaceutiques à développer et à commercialiser des médicaments orphelins à destination des patients atteints de maladies rares et laissées pour compte. WEB www.eurordis.org/fr > Médicaments orphelins

Avancées dans les dystrophies musculaires de Duchenne et de Becker Juin 2013
26/40 AFM-Téléthon>Myoinfo
Prévenir l'atteinte de la fonction cardiaque Les bêta-bloquants et les inhibiteurs de l’enzyme d e conversion de l’angiotensine L'utilisation des inhibiteurs de l'enzyme de conversion de l'angiotensine (IEC), traitement classique de l'insuffisance cardiaque, a montré son intérêt pour retarder les effets de l'atteinte cardiaque chez les garçons atteints de dystrophies musculaires de Duchenne à partir de l'âge de 10 ans. Les bêta-bloquants sont des médicaments dont l’efficacité est prouvée dans le traitement de l'insuffisance cardiaque avérée. Un essai clinique de phase III ayant pour objectif d'évaluer l'efficacité des bêta-bloquants en association avec les inhibiteurs de l'enzyme de conversion de l'angiotensine (IEC) sur l’apparition et la progression de l'atteinte cardiaque dans la DMD est en cours de recrutement. Les garçons, âgés de 10 à 15 ans avec une fraction d’éjection cardiaque supérieure ou égale à 50%, sont suivis pendant 5 ans. Trois mesures de la fraction d'éjection systolique seront réalisées : une au début de l'étude, une au bout de 3 ans et une au bout des 5 ans. Essai en cours de recrutement • Essai français, multicentrique, de phase III du traitement par bêta-bloquant en association avec des inhibiteurs de l'enzyme de conversion (IEC) - Recrutement en cours de 60 garçons atteints de DMD, âgés de 10 à 15 ans, avec une fraction d’éjection cardiaque supérieure ou égale à 50% - Investigateur principal : Pr H.M. Bécane (Institut de Myologie, Paris, France) Une étude en ouvert commencée en décembre 2008 en Italie (Rome), est en cours pour comparer l'effet sur la prévention de l'atteinte cardiaque d'un traitement par un bêta-bloquant, d'une part, et par un inhibiteur de l'enzyme de conversion, d'autre part, chez des personnes atteintes de DMD ou de BMD, âgés de 2 à 45 ans. Les premiers résultats ne sont pas prévus avant fin 2012. Le sildénafil Une étude préclinique, publiée en octobre 2010, a montré des effets cardioprotecteurs, chez des souris mdx âgées, du traitement oral par sildénafil, une molécule qui favorise la relaxation des vaisseaux (vasodilatation). Le traitement entraine une protection à long terme et un rétablissement rapide de la fonction cardiaque. Même lorsqu’il est débuté après le développement de l’atteinte cardiaque, les symptômes sont rapidement améliorés. Un essai de phase II, randomisé, en double aveugle du sildénafil chez 30 patients atteints de DMD et de BMD, vise à évaluer les effets du sildénafil sur la fonction cardiaque des patients lors d’un traitement de 12 mois pour le premier groupe, ou de 6 mois par placebo puis 6 mois par le sildénafil pour le second groupe. Cet essai qui était en cours, aux États-Unis, vient d’être suspendu. Essai suspendu aux États-Unis • Essai de phase II, en double aveugle du sildénafil : 20 mg de sildénafil 3 fois par jour. Le sildénafil et le tadalafil Comme le sildénafil, le tadalafil est une molécule qui favorise la vasodilatation. Des études ont montré que le tadalafil et le sildenafil

Avancées dans les dystrophies musculaires de Duchenne et de Becker Juin 2013
AFM-Téléthon>Myoinfo 27/40
améliorent le débit sanguin dans des modèles de souris atteintes de dystrophie musculaire. Deux essais cliniques avec le sildénafil et le tadalafil sont en cours aux États-Unis. Un essai de phase I, en ouvert est en cours de recrutement de 12 garçons atteints de DMD, âgés de 7 à 15 ans, encore marchants. Cet essai a pour objectif de tester l'innocuité et l'effet de 2 doses de sildénafil puis de tadalafil sur un temps court. Un essai de phase II/III, croisé et randomisé, en ouvert, a pour but d'évaluer l'efficacité et l'innocuité du sildénafil et du tadalafil sur le débit sanguin musculaire durant l'exercice. Trente garçons atteints de DMD, âgés de 7 à 15 ans, encore marchants, sont en cours de recrutement pour cet essai. Ils recevront des doses croissantes soit de sildénafil, soit de tadalafil pendant 2 semaines. Essais en cours de recrutement aux États-Unis • Essai de phase I, en ouvert de 2 doses de sildénafil puis de tadalafil - En cours de recrutement de 12 personnes atteintes de DMD et capables de marcher. • Essai de phase II/III, croisé et randomisé, en ouvert du sildénafil et du tadalafil sur le débit sanguin musculaire durant l'exercice chez 30 personnes atteintes de DMD et capables de marcher. - Recrutement des participants en cours. L’éplérénone L'aldostérone est une hormone naturelle qui joue un rôle dans le maintien d'une bonne pression artérielle. Les anti-aldostérone tels que l'éplérénone bloquent son action en se fixant sur les récepteurs de l'aldostérone . Des études réalisées dans des souris modèles de la DMD ont montré que des molécules anti-aldostérone, indiquées dans le traitement de l’insuffisance cardiaque avérée, préservent les fonctions musculaire et cardiaque des souris. Un essai randomisé, en double aveugle, contre placebo, évalue l'efficacité de 25 mg/jour d'éplérénone pendant un an sur la fonction musculaire cardiaque ainsi que sur la fibrose du muscle cardiaque, chez 40 garçons âgés d'au moins 7 ans, atteints de DMD avec un muscle cardiaque atteint mais sans dysfonction ventriculaire gauche. La fonction cardiaque et la fibrose sont évaluées grâce à l'IRM. Essais en cours de recrutement aux États-Unis • Essai randomisé, en double aveugle, contre placebo : 25 mg/jour d'éplerenone - En cours de recrutement de 40 personnes atteintes de DMD avec une atteinte cardiaque mais sans dysfonction du ventricule gauche.
Les pistes thérapeutiques dans la dystrophie musculaire de Duchenne Si les progrès dans la recherche de pistes thérapeutiques concernent la dystrophie musculaire de Duchenne et la dystrophie musculaire de Becker, certaines pistes thérapeutiques comme le saut d'exon s’appliquent plus particulièrement à la dystrophie musculaire de Duchenne.
Avancées dans les dystrophies musculaires de Duchenne et de Becker Juin 2013
28/40 AFM-Téléthon>Myoinfo
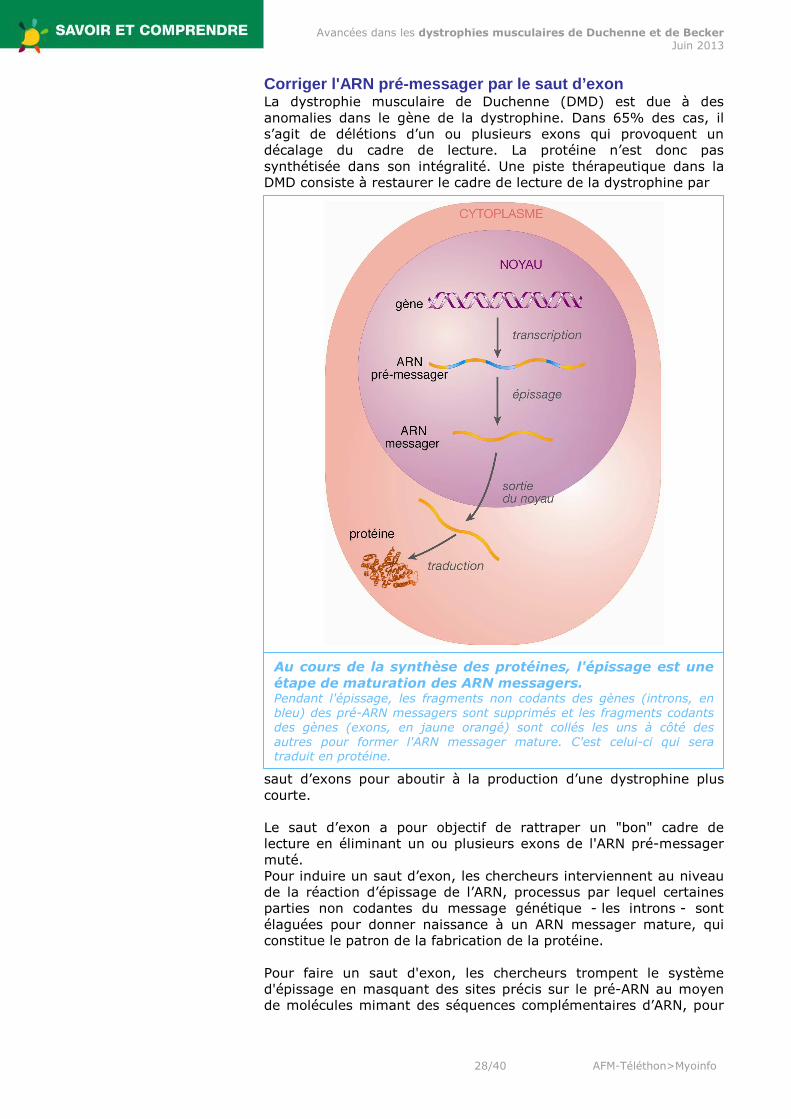
Corriger l'ARN pré-messager par le saut d’exon La dystrophie musculaire de Duchenne (DMD) est due à des anomalies dans le gène de la dystrophine. Dans 65% des cas, il s’agit de délétions d’un ou plusieurs exons qui provoquent un décalage du cadre de lecture. La protéine n’est donc pas synthétisée dans son intégralité. Une piste thérapeutique dans la DMD consiste à restaurer le cadre de lecture de la dystrophine par
saut d’exons pour aboutir à la production d’une dystrophine plus courte. Le saut d’exon a pour objectif de rattraper un "bon" cadre de lecture en éliminant un ou plusieurs exons de l'ARN pré-messager muté. Pour induire un saut d’exon, les chercheurs interviennent au niveau de la réaction d’épissage de l’ARN, processus par lequel certaines parties non codantes du message génétique - les introns - sont élaguées pour donner naissance à un ARN messager mature, qui constitue le patron de la fabrication de la protéine. Pour faire un saut d'exon, les chercheurs trompent le système d'épissage en masquant des sites précis sur le pré-ARN au moyen de molécules mimant des séquences complémentaires d’ARN, pour
Au cours de la synthèse des protéines, l'épissage est une étape de maturation des ARN messagers. Pendant l'épissage, les fragments non codants des gènes (introns, en bleu) des pré-ARN messagers sont supprimés et les fragments codants des gènes (exons, en jaune orangé) sont collés les uns à côté des autres pour former l'ARN messager mature. C'est celui-ci qui sera traduit en protéine.
Avancées dans les dystrophies musculaires de Duchenne et de Becker Juin 2013
AFM-Téléthon>Myoinfo 29/40
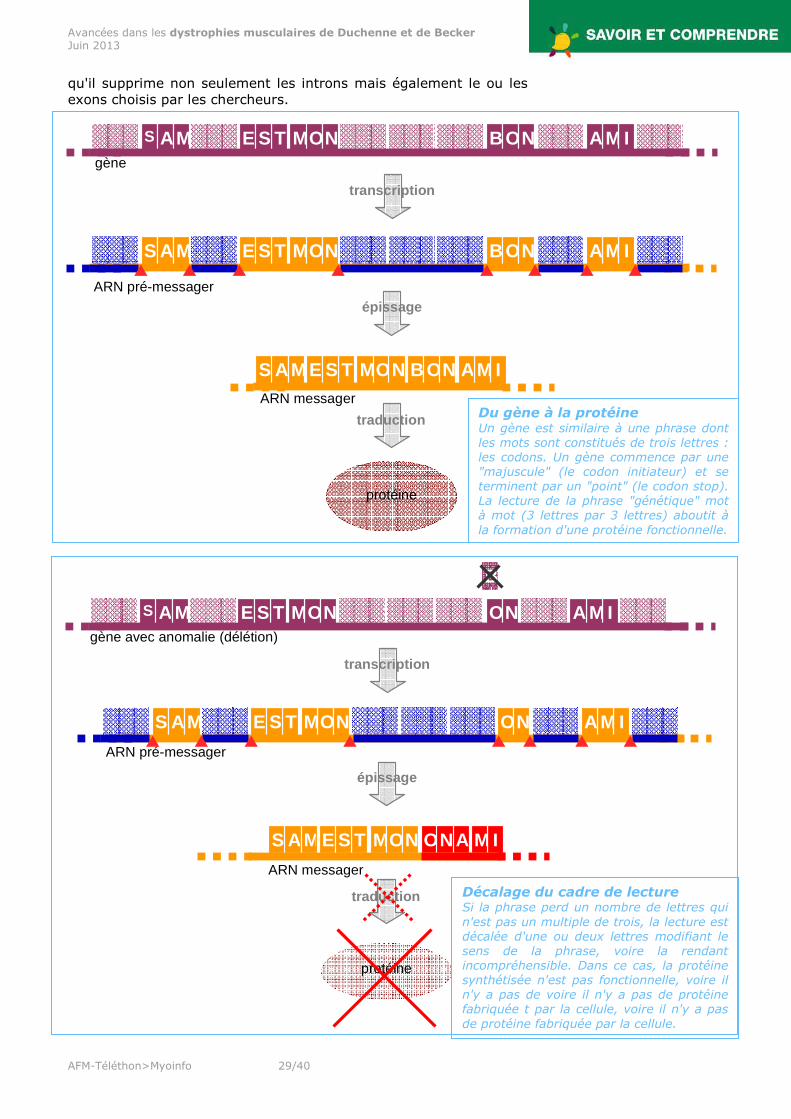
qu'il supprime non seulement les introns mais également le ou les exons choisis par les chercheurs.
ONM S A M E T S I MAB ON
ONM S A M E T S I MAB ON
ONMS A M E T S I MAB ON
protéine
gène
ARN pré-messager
ARN messager
transcription
épissage
traduction
Du gène à la protéine Un gène est similaire à une phrase dont les mots sont constitués de trois lettres : les codons. Un gène commence par une "majuscule" (le codon initiateur) et se terminent par un "point" (le codon stop). La lecture de la phrase "génétique" mot à mot (3 lettres par 3 lettres) aboutit à la formation d'une protéine fonctionnelle.
ONM S A M E T S I MAON
MOS A M E T S I MAN ON
épissage
transcription
traduction
protéine
ARN pré-messager
ARN messager
ONM S A M E T S I MAONgène avec anomalie (délétion)
B
Décalage du cadre de lecture Si la phrase perd un nombre de lettres qui n'est pas un multiple de trois, la lecture est décalée d'une ou deux lettres modifiant le sens de la phrase, voire la rendant incompréhensible. Dans ce cas, la protéine synthétisée n'est pas fonctionnelle, voire il n'y a pas de voire il n'y a pas de protéine fabriquée t par la cellule, voire il n'y a pas de protéine fabriquée par la cellule.
Avancées dans les dystrophies musculaires de Duchenne et de Becker Juin 2013
30/40 AFM-Téléthon>Myoinfo
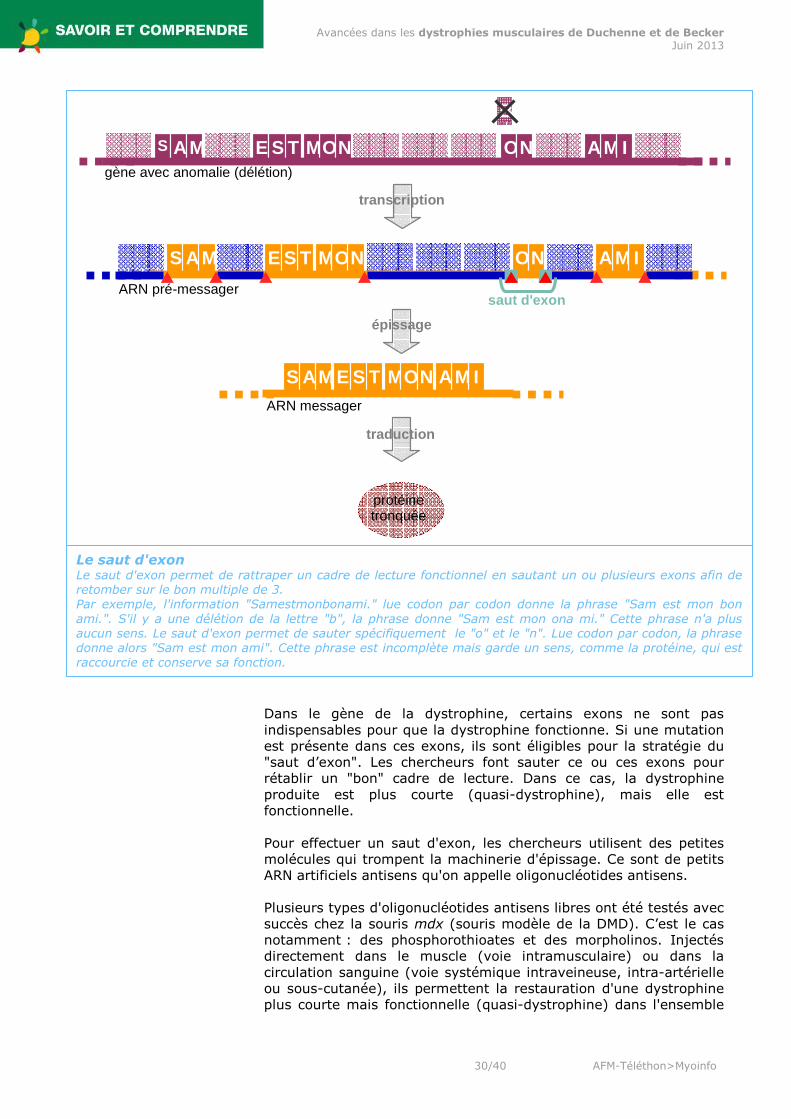
Dans le gène de la dystrophine, certains exons ne sont pas indispensables pour que la dystrophine fonctionne. Si une mutation est présente dans ces exons, ils sont éligibles pour la stratégie du "saut d’exon". Les chercheurs font sauter ce ou ces exons pour rétablir un "bon" cadre de lecture. Dans ce cas, la dystrophine produite est plus courte (quasi-dystrophine), mais elle est fonctionnelle. Pour effectuer un saut d'exon, les chercheurs utilisent des petites molécules qui trompent la machinerie d'épissage. Ce sont de petits ARN artificiels antisens qu'on appelle oligonucléotides antisens. Plusieurs types d'oligonucléotides antisens libres ont été testés avec succès chez la souris mdx (souris modèle de la DMD). C’est le cas notamment : des phosphorothioates et des morpholinos. Injectés directement dans le muscle (voie intramusculaire) ou dans la circulation sanguine (voie systémique intraveineuse, intra-artérielle ou sous-cutanée), ils permettent la restauration d'une dystrophine plus courte mais fonctionnelle (quasi-dystrophine) dans l'ensemble
ONM S A M E T S I MAON
MOS A M E T S I MAN
épissage
transcription
traduction
protéine tronquée
ARN pré-messager
ARN messager
ONM S A M E T S I MAONgène avec anomalie (délétion)
B
saut d'exon
Le saut d'exon Le saut d'exon permet de rattraper un cadre de lecture fonctionnel en sautant un ou plusieurs exons afin de retomber sur le bon multiple de 3. Par exemple, l'information "Samestmonbonami." lue codon par codon donne la phrase "Sam est mon bon ami.". S'il y a une délétion de la lettre "b", la phrase donne "Sam est mon ona mi." Cette phrase n'a plus aucun sens. Le saut d'exon permet de sauter spécifiquement le "o" et le "n". Lue codon par codon, la phrase donne alors "Sam est mon ami". Cette phrase est incomplète mais garde un sens, comme la protéine, qui est raccourcie et conserve sa fonction.

Avancées dans les dystrophies musculaires de Duchenne et de Becker Juin 2013
AFM-Téléthon>Myoinfo 31/40
des muscles squelettiques et entraînent une amélioration de la force musculaire chez la souris. Les morpholinos ont également été testés chez les chiens CXMDj (modèles canins de la DMD) et ont permis d'améliorer leurs performances motrices. Le drisapersen (ou GSK2402968) Le drisapersen (anciennement GSK2402968 ou PRO051) est un oligonucléotide antisens libre (phosphorothioate) développé par la société de biotechnologie Prosensa en collaboration avec les laboratoires GlaxoSmithKline, qui vise le saut de l’exon 51. Deux essais contrôlés en double aveugle sont actuellement en cours chez des personnes atteintes de DMD et ambulantes : - un essai multicentrique international de phase III pour évaluer l'efficacité et la tolérance de 6 mg/kg par semaine de drisapersen ; - un essai européen et australien de phase II pour évaluer l’innocuité et la tolérance de 6 mg/kg de drisapersen, une ou deux fois par semaine. Essais en cours • Essai de phase III multicentrique international, contrôlé, en double aveugle, du traitement par drisapersen :6 mg/kg une fois par semaine en sous-cutané. - Recrutement terminé de 180 personnes atteintes de DMD et capables de marcher. - Fin estimée de l’essai pour juillet 2013. - Investigateur principal en France : Pr I. Desguerre (Hôpital Necker, Paris). • Essai de phase II européen et australien, contrôlé, en double aveugle, du traitement par drisapersen : 6 mg/kg une ou deux fois par semaine en sous-cutané. - Recrutement terminé des 54 personnes atteintes de DMD et capables de marcher. - Essai en cours d’analyse. - Investigateur principal en France : Pr T. Voit (Institut de Myologie, Paris). Une phase d'extension en ouvert s'adressant aux personnes ayant terminé les essais de phase II ou III dans lesquels elles étaient incluses, permet de poursuivre l’évaluation de la tolérance et de l'efficacité de 6 mg/kg de drisapersen sur le long terme (minimum 2 ans). Les personnes qui ne souhaitent pas poursuivre l'essai en ouvert, ou qui ont présenté des effets secondaires trop importants, peuvent continuer à être suivies dans le cadre de l'essai en ouvert, sur la base de l'évolution naturelle de la maladie, sans prendre de traitement. Extension en cours des essais de phases II et III • Extension en ouvert ne concernant que les patients ayant participé aux essais en double aveugle de phase II et de phase III : 6 mg/kg par semaine, sur un minimum de 2 ans. - Fin estimée de l’essai pour la fin de l’année 2014. Pour les personnes atteintes de DMD et ne marchant plus, un essai de phase I, contrôlé, en double aveugle, a évalué, en France et aux États-Unis, l’innocuité, la tolérance et l’évolution du produit dans le sang au fil du temps (pharmacocinétique) d’une injection sous-cutanée de 3 doses de drisapersen (3 mg/kg, 6 mg/kg, 9 mg/kg).

Avancées dans les dystrophies musculaires de Duchenne et de Becker Juin 2013
32/40 AFM-Téléthon>Myoinfo
Les doses ont été évaluées l'une après l'autre après analyse des données de sécurité de la dose précédente. Des premiers résultats de cet essai ont été annoncés en novembre 2012 par la société GSK. Ils n'ont pas encore été publiés dans une revue scientifique. Le taux sanguin de drisapersen retrouvé chez les patients traités est proportionnel aux doses injectées de 3 ou 6 mg/kg mais pas à la dose de 9 mg/kg. L’effet indésirable le plus fréquemment rapporté est une décoloration et une induration de la zone d’injection chez les 12 patients qui ont reçu 3 ou 6 mg/kg de produit. Cet effet indésirable a été également retrouvé chez 2 des 5 patients ayant reçu le placebo. Quant aux 3 patients qui ont été traités avec 9 mg/kg de drisapersen, ils ont présenté une inflammation du site d’injection et de la fièvre. La société GSK a précisé que, même si des effets indésirables ont été constatés, il n’y a pas eu d’effet indésirable grave. Essai en cours d’analyse en France et aux États-Uni s • Essai de phase I, contrôlé, en double aveugle, d'une injection sous-cutanée unique de drisapersen :3 mg/kg, 6 mg/kg, 9 mg/kg chez des garçons atteints de DMD non marchant. - Essai en cours d’analyse. - Investigateur principal en France : Pr T. Voit (Institut de Myologie, Paris). En février 2013, la société GSK a annoncé qu’un petit pourcentage des garçons sous drisapersen a présenté des effets secondaires réversibles à l’arrêt du traitement (diminution du nombre de plaquettes et globules blancs dans le sang et excès de protéines dans les urines). La société GSK a rappelé que 96 % des garçons inclus n’ont pas manifesté d’effets secondaires et continuent de recevoir le candidat médicament : 117 garçons participant à l’essai de phase III ont déjà reçu le drisapersen pendant 24 semaines tandis que 41 autres l’ont reçu pendant 48 semaines, sans effets secondaires de ce type. Si ces effets secondaires du drisapersen doivent être surveillés sur le long terme, la société GSK en poursuit le développement clinique.
Le PRO044 Une autre molécule développée par la société Prosensa, le PRO044, un oligonucléotide antisens libre (phosphorothioates) visant le saut de l’exon 44, fait actuellement l’objet d’un essai de phase I/II. L’objectif de cet essai est d'évaluer chez 18 patients atteints de dystrophie musculaire de Duchenne l’innocuité du PRO044 administré par voie sous-cutanée pendant cinq semaines, avec d’une période d’observation de 13 semaines. Essai en cours en Belgique, Italie, Hollande et Suè de • Essai de phase I/II en ouvert d'une injection sous-cutanée de PRO044 à 0,5 mg/kg, 1,5 mg/kg, 5 mg/kg, 8 mg/kg, 10 mg/kg ou 12 mg/kg une fois par semaine pendant 5 semaines chez 18 personnes atteintes de DMD. - Recrutement terminé. Les données sont en cours d'analyse. - Investigateur principal : Dr N. Goemans (Louvain, Belgique) Le PRO045 Les laboratoires Prosensa et GSK travaillent sur un programme de développement pré-clinique et clinique du saut de l’exon 45, le PRO045. Le PRO045 a obtenu le statut de médicament orphelin.

Avancées dans les dystrophies musculaires de Duchenne et de Becker Juin 2013
AFM-Téléthon>Myoinfo 33/40
Un essai international de phase I/II en ouvert de PRO045 a démarré début 2013. Il vise à évaluer l’innocuité, la tolérance, l’efficacité et le devenir dans l'organisme (pharmacocinétique) de doses croissantes de PRO045 chez 45 garçons, âgés de 5 à 18 ans et marchants. Cet essai se déroulera en France, en Belgique, en Italie, aux Pays Bas et au Royaume-Uni. Le recrutement des participants n'a démarré qu'en Belgique. Essai en cours • Essai de phase I/II en ouvert d'une injection unique de PRO045 pendant 48 semaines à la dose de 0,15 mg/kg, 1 mg/kg, 3 mg/kg, 6 mg/kg ou 9 mg/kg chez des garçons atteints de DMD marchants. - Recrutement en cours en Belgique. - Investigateur principal en France : Pr T. Voit (Institut de Myologie, Paris). Le PRO052, le PRO053 et le PRO055 Les oligonucléotides PRO052, PRO053 et le PRO055 qui visent respectivement au saut de l’exon 52, 53 et 55 sont en développement préclinique. Ces trois oligonucléotides antisens ont été récemment désignés " médicaments orphelins " en Europe par l’Agence Européenne de Médecine (EMA). L’éteplirsen (ou AVI-4658) L’éteplirsen (ou AVI-4658 ) est un oligonucléotide antisens libre de type morpholino, visant le saut de l’exon 51 de la dystrophine, développé par la société Sarepta Therapeutics (anciennement appelée AVI BioPharma). Un essai de phase I/II, réalisé en ouvert au Royaume-Uni, a évalué la tolérance et l’efficacité de doses croissantes d’éteplirsen. Dix-neuf garçons atteints de DMD, âgés de 5 à 15 ans, ont été traités en intraveineux une fois par semaine à 6 doses différentes (0,5 mg/kg, 1 mg/kg, 2 mg/kg, 4 mg/kg, 10 mg/kg et 20 mg/kg) pendant 12 semaines, avec une phase de suivi de 14 semaines après la fin de leur traitement. Les résultats publiés en 2011 ont été très encourageants : le traitement a été bien toléré et les analyses ont montré chez 7 participants, une nouvelle production de dystrophine significative et dépendante de la dose injectée. Les 3 personnes avec les meilleures réponses ont présenté jusqu’à 15%, 21% et 55% de fibres exprimant la dystrophine après traitement. Par ailleurs, les chercheurs ont montré que cette dystrophine ainsi produite fonctionne correctement dans le muscle et qu’elle n’entraine pas de réponse immunitaire. Suite à ces résultats encourageants, un essai de phase II a démarré en juin 2012 aux États-Unis avec pour investigateur principal J. Mendell (Columbus, États-Unis). L’objectif de cet essai consiste à évaluer pendant 24 semaines la tolérance, l’innocuité, le devenir du produit dans l'organisme (pharmacocinétique) et l’efficacité de 30 et 50 mg/kg d’eteplirsen chez 12 garçons atteints de DMD, âgés de 7 à 13 ans. Ainsi, pendant 24 semaines, 4 enfants ont reçu du placebo, 4 autres ont reçu 30 mg/kg/semaine d'éteplirsen et 4 autres encore, de l'éteplirsen à la dose de 50 mg/kg/semaine. En avril 2012, des résultats préliminaires ont montré qu'après 24 semaines de traitement à la dose de 30 mg/kg d’éteplirsen, la production de dystrophine est significativement augmentée par rapport à celle des garçons sous placebo. En revanche, il n’y a pas
Un médicament orphelin est un médicament développé pour le traitement d’une maladie dite orpheline, c’est-à-dire une maladie rare. Pour les entreprises pharmaceutiques, le coût de mise sur le marché d’un produit préconisé dans une maladie rare ne serait pas couvert par les ventes attendues sur ce marché "restreint" du fait du peu de personnes concernées. C'est pourquoi, sous la pression des associations de maladies rares, une politique incitative économique a été mise en place pour encourager les entreprises pharmaceutiques à développer et à commercialiser des médicaments orphelins à destination des patients atteints de maladies rares et laissées pour compte. WEB www.eurordis.org/fr > Médicaments orphelins

Avancées dans les dystrophies musculaires de Duchenne et de Becker Juin 2013
34/40 AFM-Téléthon>Myoinfo
eu de production de dystrophine pour les patients traités par 50 mg/kg pendant 12 semaines. Dans les 2 cas, le traitement a été bien toléré. La fonction musculaire n’a pas été améliorée mais est restée stable, même chez ceux qui ont reçu le placebo. Dans une phase d’extension en ouvert de cet essai de phase II, tous les garçons ont été mis sous éteplirsen : 6 reçoivent la dose de 30mg/kg/semaine, les 6 autres reçoivent 50 mg/kg/semaine. Cette phase d’extension a pour but d'évaluer l'efficacité, l'innocuité et la tolérance sur le long terme (80 semaines) des deux doses d'éteplirsen testées. La société pharmaceutique Sarepta Therapeutics a publié un communiqué de presse faisant le point sur cet essai après 48, 62 et 74 semaines de traitement. Les résultats obtenus à 74 semaines confirment les précédents, à savoir une bonne tolérance du produit, sans effet secondaire grave. En termes d’impact sur la perte de la marche, les garçons qui ont été traités dès le début de l’essai avec l’éteplirsen (6 garçons) stabilisent leur capacité à la marche puisqu’ils ne perdent en moyenne que 2,2 mètres de marche au test de 6 minutes, tandis que ceux qui ont reçu le placebo pendant 24 semaines puis l’éteplirsen les semaines suivantes (4 garçons), ont perdu en moyenne 64,6 mètres à ce test. Ces résultats encourageants doivent cependant être considérés avec prudence tant qu'ils ne sont pas complètement analysés et publiés dans une revue scientifique. De plus, compte tenu, en particulier, du petit nombre de participants à ce premier essai, l’intérêt de l’éteplirsen dans le traitement de la dystrophie musculaire de Duchenne devra être confirmé sur un plus grand nombre de patients. Essai en cours d’analyse aux États-Unis • Essai de phase II, contrôlé, randomisé, en double aveugle d'une injection intraveineuse pendant 24 semaines d’éteplirsen : un groupe (4 garçons) a reçu 30 mg/kg/semaine, un groupe (4 garçons) a reçu 50mg/kg/semaine et un groupe (4 garçons) a reçu le placebo,. - Essai en cours d'analyse. - Investigateur principal : Pr J. Mendell (Columbus, États-Unis) • Extension en ouvert de l’essai de phase II pendant 80 semaines : un groupe de 6 garçons reçoit la dose de 30mg/kg/semaine et un groupe de 6 garçons, la dose de 50 mg/kg/semaine. Fin de la phase clinique estimée pour fin 2013 L’AAV-U7 Le Généthon (Évry) et l’Institut de myologie (Paris) ont testé le saut d'exon avec des séquences antisens insérées dans le petit ARN nucléaire, snARN-U7, vectorisé dans un vecteur AAV, chez la souris mdx. Le traitement a restauré l'expression d'une quasi-dystrophine dans l'ensemble des muscles squelettiques et a entraîné une amélioration fonctionnelle. En 2012, une étude a montré, dans un modèle de souris sévère de DMD, n’exprimant ni la dystrophine, ni l’utrophine (souris dKO), qu’une injection intraveineuse d’AAV9-U7 rétablissait l’expression de la dystrophine à un niveau proche de la normale, dans tous les muscles y compris le cœur. La fonction musculaire a également été améliorée et la survie de ces souris modèle, allongée. Une étude a couplé chez la souris mdx deux pistes thérapeutiques, le saut d’exons avec un AAV-U7 et l’inhibition de l'action de la myostatine, un inhibiteur de la croissance musculaire qui existe à

Avancées dans les dystrophies musculaires de Duchenne et de Becker Juin 2013
AFM-Téléthon>Myoinfo 35/40
l'état naturel dans l'organisme. Lorsque l’AAV-U7 est administré seul, l’expression de la dystrophine est restaurée et la fonction musculaire améliorée. Lors de l’administration de l'inhibiteur de la myostatine seul, le poids de la souris, sa masse musculaire et la taille des fibres musculaires augmentent sans effet sur la force musculaire. Les effets de chacune des thérapeutiques administrées conjointement à la souris mdx s’additionnent sans effet synergique sur la fonction musculaire. Chez le chien GRMD, les chercheurs ont rétabli l'expression d'une quasi-dystrophine dans une patte d'un chien malade en utilisant un AAV-U7. Actuellement, leur travail consiste à essayer de traiter le corps entier de l'animal. L’expression à long terme de la dystrophine dans les cellules cardiaques après injection d’AAV6-snARN U7 dans le cœur de chien GRMD est restaurée au 13e mois. Récemment, d’autres travaux, chez le chien GRMD, soutenus par l’AFM-Téléthon, ont montré l’efficacité du saut d’exon par des snARN U7 optimisés transportés par un AAV1 avec restauration de l'expression de la dystrophine dans les fibres musculaires et restauration partielle de la force musculaire… Le suivi à 5 ans a permis de mieux comprendre la réponse immunitaire. Il a également mis en évidence une diminution progressive du nombre de fibres musculaires corrigées, suggérant la nécessité d’un traitement récurent pour maintenir les effets d’une telle procédure. Les tricyclo-ADN, de nouveaux oligonucléotides anti sens synthétiques prometteurs Depuis plusieurs années, l’équipe de C. Leumann (Suisse) travaille sur la synthèse d’oligonucléotides antisens plus performants dans le but d’améliorer leur affinité avec l’ARN, leur résistance aux enzymes nucléaires et leur distribution dans l’organisme. Ils ont découvert que les oligonucléotides antisens tricyclo-ADN, une forme d’oligonucléotide chimiquement modifiée, présentent de meilleures propriétés de distribution dans l’organisme et de liaison à l’ARN et sont capables de faciliter le saut d’exon. Les équipes de C. Leumann et L. Garcia se sont associées pour étudier le saut d’exons par des tricyclo-ADN, dans le gène de la dystrophine. Les premières expériences d’injection de tricyclo-ADN dans les muscles de souris mdx ont montré une forte restauration de la dystrophine 3 semaines plus tard. Des expériences complémentaires récentes d’injection systémique de tricyclo-ADN ont montré une restauration de la dystrophine aussi bien dans les muscles que dans le cœur de souris mdx et la détection de saut d’exon dans leur cerveau. Dans le modèle plus sévère des souris dKO, sans dystrophine ni utrophine, la fonction musculaire et la dystrophie ont été significativement améliorées. Réduire l’inflammation Dans la dystrophie musculaire de Duchenne (DMD), il existe une inflammation des muscles qui contribue à la nécrose des fibres musculaires. Pour contrecarrer ce phénomène, les chercheurs testent l’utilisation de différents immunosuppresseurs et anti-inflammatoires dans des modèles animaux de DMD, en particulier la souris mdx. Les études montrent, généralement, une diminution de la nécrose et une régénération des fibres musculaires. Les résultats restent mitigés sur la force musculaire des souris.

Avancées dans les dystrophies musculaires de Duchenne et de Becker Juin 2013
36/40 AFM-Téléthon>Myoinfo
Chez l'homme, parmi les différents types d’immunosuppresseurs et d'anti-inflammatoires testés, les corticoïdes sont les plus efficaces. L'analyse de toutes les études ayant testé l'efficacité des corticoïdes dans la dystrophie musculaire de Duchenne par une revue Cochrane (2005 et 2008) a conclu "qu’un traitement à base de corticostéroïdes dans la myopathie de Duchenne améliore la force et la fonction musculaires à court et à moyen-terme". D'autre part, il semble que l'efficacité des corticoïdes est d'autant plus importante que le traitement est donné tôt dans la maladie. Chez les adultes, il est nécessaire de faire des études supplémentaires pour confirmer les effets positifs en particulier sur la fonction respiratoire. Mais ils sont d’ores et déjà largement préconisés. La prednisone En 2011, un essai en double aveugle, randomisé, a comparé, dans la DMD, l’efficacité et l’innocuité d’un traitement par prednisone quotidiennement ou uniquement le week-end. Pendant 12 mois, 64 garçons atteints de DMD âgés de 4 à 10 ans ont été traités : 32 patients par 0,75 mg/kg par jour de prednisone, excepté le weekend où ils ont reçu un placebo, et 32 par 5 mg/kg par jour de prednisone le weekend et ont reçu un placebo les autres jours de la semaine. Les traitements, aussi bien tolérés dans les 2 groupes, ont été aussi efficaces sur la force musculaire dans les 2 groupes. Ces traitements, à 2 posologies différentes, offrent ainsi aux cliniciens la possibilité d’avoir plusieurs options thérapeutiques. Une autre étude a montré cette même année, que la prise de prednisone n’empêchait pas de participer à un essai clinique avec oligonucléotides antisens. Aucune interférence entre un traitement par oligonucléotide antisens et la prise de prednisone chez la souris mdx ou dans des cellules musculaires de patients atteints de DMD n’a été observée. La prise de prednisone pourrait même améliorer l’effet des oligonucléotides antisens chez la souris mdx. Un essai international de phase III, randomisé, contrôlé, en double aveugle vient de débuter. Il a pour but de comparer 3 protocoles de traitement par des corticostéroïdes, pendant 60 mois, chez 300 garçons âgés de 4 à 7 ans : - 0,75 mg/kg/jour de prednisone, - 0,75 mg/kg/jour pendant 10 jours en alternance avec 10 jours sans traitement, - 0,9 mg/kg/jour de deflazacort. Ces différents traitements permettront de voir si la prise quotidienne de corticostéroïdes offre plus de bénéfices que la prise en alternance et si le déflazacort est mieux toléré que la prednisone. Essai en cours • Essai international de phase III, contrôlé, randomisé, en double aveugle de 3 protocoles de traitement par des corticostéroïdes chez 300 personnes atteintes de DMD âgés de 7 ans. - Recrutement en cours - Investigateur principal : Pr K. Bushby (Royaume-Uni) L’IGF-1 L’IGF-1 (pour Insulin Growth Factor I) est un facteur de croissance qui stimule la multiplication et la différenciation des cellules
Une revue Cochrane a pour but d'identifier quelles
pratiques de soins sont efficaces, celles qui ne
marchent pas et celles qui éventuellement sont néfastes.
Elle repose sur une compilation et une analyse exhaustive de la littérature
médicale et scientifique sur un sujet donné. Le processus suit une méthodologie rigoureuse :
recensement des études publiées, sélection de celles
qui sont méthodologiquement recevables, analyse de leurs
données combinées (méta-analyse). Le résultat de cette
méta-analyse fait généralement autorité.

Avancées dans les dystrophies musculaires de Duchenne et de Becker Juin 2013
AFM-Téléthon>Myoinfo 37/40
musculaires. L’IGF-1 pourrait, comme les corticoïdes, améliorer la fonction motrice, avec des effets secondaires moins importants. Pour évaluer les effets de l’IGF-1 par rapport aux corticoïdes sur la fonction musculaire dans la DMD, un essai de phase I/II randomisé, en simple aveugle est en cours aux États-Unis. Quarante garçons atteints de DMD et âgés de plus de 5 ans ont été inclus dans cet essai ; le groupe contrôle recevra uniquement le traitement standard de corticoïdes, l’autre groupe recevra également de l’IGF-1. Essai en cours aux États-Unis • Essai de phase I/II, randomisé, en simple aveugle de l’IGF-1 chez 40 personnes atteintes de DMD, âgés de plus de 5 ans. - Recrutement terminé. Diminuer le stress oxydatif Le stress oxydatif pouvant également jouer un rôle dans la dégénérescence des cellules musculaires, la société suisse Santhera, a développé un médicament, l’idébénone. Cette molécule, très proche du coenzyme Q10, est connue pour ses vertus anti-oxydantes. Un essai de phase II, appelé DELPHI et mené en Suisse par la Société Santhera, chez 21 patients atteints de dystrophie musculaire de Duchenne (DMD) avec une insuffisance cardiaque a montré que l'idébénone est bien tolérée. L'idébénone a amélioré significativement la fonction respiratoire des patients traités et avait tendance à exercer un effet cardioprotecteur. Une étude d’extension de cet essai (essai DELPHI-E) a été réalisée pour observer ses effets bénéfiques à plus long terme. Les résultats de cette étude d’extension n’ont pas encore été publiés. Une analyse complémentaire des résultats de l’essai DELPHI publiée récemment a mis en évidence un impact négatif du traitement par corticoïdes sur les effets de l’idébénone. L’effet bénéfique de l’idébénone a été significativement plus élevé chez les participants de l’essai DELPHI ne prenant pas de corticoïdes. Ces résultats ont été pris en compte dans le design de l’essai DELOS, un essai de phase III, toujours en cours, dont l’objectif est d’évaluer l'efficacité, l'innocuité et la tolérance de 900 mg/j d'idébénone pendant un an chez 240 garçons atteints de DMD, âgés de 10 à 18 ans. Essai en cours • Essai de phase III, multicentrique international, contrôlé, en double aveugle de 900 mg/kg par jour d'idébénone par voie orale. - Recrutement terminé de 240 personnes atteintes de DMD, âgées de 10 à 18 ans - Fin estimée en fin d’année 2013. - Investigateur principal en France : Pr T. Voit (Institut de Myologie, Paris) Améliorer la force musculaire Stabiliser le canal calcium RyR1 Dans la dystrophie musculaire de Duchenne (DMD), la perte de dystrophine entraine une désorganisation du complexe de protéines associées à la dystrophine (complexe DAP). Cette désorganisation perturbe les flux de calcium provoquant une augmentation de la concentration en calcium dans les cellules qui conduit à la mort cellulaire.
Le stress oxydatif correspond à une situation où la cellule ne contrôle plus la présence excessive de molécules toxiques issues de la respiration cellulaire : les radicaux libres. En excès, ces radicaux libres peuvent endommager les cellules et l’ADN.

Avancées dans les dystrophies musculaires de Duchenne et de Becker Juin 2013
38/40 AFM-Téléthon>Myoinfo
Dans la souris modèle mdx, des défauts fonctionnels et structuraux du récepteur de la ryanodyne (RyR1), un canal calcique, localisé dans le réticulum sarcoplasmique, ont été identifiés. RyR1 intervient dans la contraction musculaire en régulant les mouvements du calcium à l’intérieur de la fibre musculaire. Depuis quelques années, l’intérêt se porte sur la molécule S107, un stabilisateur de canal calcium qui interagit avec RyR1. Dans les souris mdx, S107 empêche l’augmentation anormale de calcium, réduit l’atteinte musculaire et améliore la fonction motrice. A ce jour, la société ARMGO Pharma explore les bénéfices de cette molécule dans le modèle mdx, mais aussi pour d’autres myopathies. Une équipe française dirigée par F. Pietri-Rouxel (Institut de Myologie, Paris), soutenue par l’AFM, a montré en 2012 que l’anomalie de la fonction de RyR1 est présente chez les patients atteints de dystrophies musculaires de Duchenne et Becker. Ces résultats confirment que les DMD et BMD devraient pouvoir bénéficier du développement d’approches pharmacologiques destinées à améliorer la stabilité du RyR1. Les inhibiteurs de l'enzyme de conversion Les inhibiteurs de l'enzyme de conversion de l'angiotensine (IEC) ont fait la preuve de leur action bénéfique sur la fonction cardiaque dans la DMD. Chez la souris mdx, l'inhibition de l'angiotensine au niveau musculaire a permis de maintenir les capacités de régénération du muscle squelettique. Un essai en double aveugle contre placebo du perindopril (un IEC) chez 40 enfants de 3 à 7 ans atteints de DMD a évalué leurs effets sur la fonction musculaire au bout de 2 ans de traitement. L'évaluation porte sur les capacités de marche, sur la force musculaire de certains muscles, la fonction respiratoire, ainsi que la prévention de l'apparition ou de la progression de l'atteinte cardiaque. Des IRM cardiaques et musculaires (notamment pour quantifier la fibrose) sont réalisées dans certains centres. L’essai est terminé et en cours d’analyse. Essai en cours d’analyse • Essai français contrôlé, en double aveugle du traitement par perindopril : 0,150 mg/kg par jour - Essai en cours d’analyse. - Investigateur principal : Dr I. Desguerre (Centre de Référence Neuromusculaire Garches-Necker-Mondor-Hendaye, Paris, France)
Les pistes thérapeutiques dans la dystrophie musculaire de Becker Si les progrès de la recherche concernant les pistes thérapeutiques dans la dystrophie musculaire de Duchenne peuvent faire avancer la recherche dans la dystrophie musculaire de Becker, certaines pistes thérapeutiques sont plus particulièrement explorées dans la dystrophie musculaire de Becker. Les anti-myostatines Pour augmenter la taille et la force des fibres musculaires, en inhibant la voie de la myostatine (un inhibiteur de la croissance musculaire qui existe à l'état naturel dans l'organisme), une piste thérapeutique consiste à administrer par thérapie génique le gène de la follistatine, un inhibiteur naturel de la myostatine.

Avancées dans les dystrophies musculaires de Duchenne et de Becker Juin 2013
AFM-Téléthon>Myoinfo 39/40
Un essai de phase I en ouvert coordonné par J. Mendell (Colombus, États-Unis) est en cours aux États-Unis pour évaluer l’innocuité de l’injection du gène de la follistatine à l’aide d’un AAV dans la cuisse ou la jambe de 15 patients atteints de BMD. La taille des fibres musculaires sera observée par biopsie 180 jours après l’administration. La fin de l’essai est prévue pour fin 2015. Essai en cours aux États-Unis • Essai de phase I en ouvert du transfert du gène de la follistatine - Recrutement en cours de 15 personnes atteintes de BMD - Fin estimée de l’essai pour la fin de l’année 2015. - Investigateur principal : J. Mendell (Colombus, États-Unis) Le tadalafil Au commencement d’un exercice physique, se produit un réflexe de contraction des vaisseaux sanguins musculaires. Si l’effort se prolonge, le muscle sécrète de l’oxyde nitrique (NO) qui entraine une dilatation des vaisseaux sanguins musculaires. Cette dilatation des vaisseaux permet un plus grand débit sanguin dans le muscle apportant une plus grande quantité d’oxygène et de nutriments aux muscles dont les besoins sont augmentés par l'exercice. Dans les myopathies de Duchenne (DMD) et de Becker (BMD), la NO synthase (NOS), qui fabrique le NO et qui est habituellement fixée à la dystrophine, est délocalisée, elle flotte dans le cytoplasme. Cette délocalisation entraine un manque de NO : la dilatation des vaisseaux sanguins lors d’exercice prolongé est de ce fait diminuée. Le tadalafil est un inhibiteur de la phosphodiestérase 5 (PDE5) qui prolonge les effets du NO et favorise la dilatation des vaisseaux. Une étude réalisée aux États-Unis a évalué les effets de la prise d’une dose unique de tadalafil sur le débit sanguin musculaire d'effort chez 10 hommes atteints de myopathie de Becker, âgés de 18 à 55 ans, ambulants et sans problème cardiaque. Les résultats ont montré une restauration complète du débit sanguin chez les patients atteints de BMD ayant pris du tadalafil. Malgré ces résultats encourageants, des études à plus grande échelle sont nécessaires avant de recommander ce type de traitement dans la dystrophie musculaire de Becker. Les auteurs ont reçu un financement du NIH pour poursuivre leurs recherches dans le cadre d’une étude multicentrique sur un plus grand nombre de personnes atteintes de dystrophie musculaire de Becker et pendant une plus longue période. Un essai de phase IV est d’ailleurs en cours aux États-Unis pour évaluer l’efficacité du tadalafil sur le débit sanguin musculaire au cours de l’exercice chez 48 hommes atteints de BMD. Essai en cours aux États-Unis • Essai de phase IV, en double aveugle, contre placebo du tadalafil sur le débit sanguin musculaire au cours de l'exercice : administré avant l'effort (contraction des muscles de l'avant-bras), en deux fois, une première dose de 10 mg la veille puis une seconde de 20 mg le jour même chez 48 hommes atteints de dystrophie musculaire de Becker et âgés de 18 à 55 ans. - Recrutement en cours . Le sildénafil Comme le tadalafil, le sildénafil est une molécule aux propriétés vasodilatatrices. Un essai de phase II, en double aveugle, contre placebo, évalue l'efficacité de 60 mg/jour (3 x 20 mg/j) de sildénafil, pendant 4

Avancées dans les dystrophies musculaires de Duchenne et de Becker Juin 2013
40/40 AFM-Téléthon>Myoinfo
semaines, sur les fonctions musculaire, cardiaque et cognitives, chez 17 patients atteints de BMD. Essai en cours d’analyse au Danemark • Essai de phase II, en double aveugle, contre placebo du sildénafil sur les fonctions musculaire, cardiaque et cognitives chez 17 hommes atteints de dystrophie musculaire de Becker et âgés de 18 minimum. - Phase clinique terminée. Données en cours d’analyse. >> Tout au long de l'année, suivez l'actualité de la recherche dans les maladies neuromusculaires sur WEB www.afm-telethon.fr > Actualités > Toute l'actualité.





























