Embed Size (px)
Citation preview

RAPPORT DE STAGE
LEA-DB : Base de données et site Web
dédiés à l'étude des LEA
Avril à août 2007
Responsables de stage :J.-M. Richer, LERIA, AngersE. Jaspard, UMR PMS 1191, AngersG. Hunault, HIFI UPRES 3859, IFR 132, Angers
Laurent CHATELAINMaster Pro BioInformatique

Université de Nantes Rapport de stage
Remerciements
Je remercie tout d'abord J-M. RICHER, maître de stage et membre du Laboratoire d'Etude et
de Recherche en Informatique d'Angers, qui m'a apporté l'encadrement nécessaire à la réalisation du
projet.
Mes remerciements sont adressés en particulier à E. JASPARD, également maître de stage,
et G. HUNAULT pour leur disponibilité quasi permanente et pour leur précieux soutien.
Pour finir, je remercie toute l'équipe du département informatique de la Faculté d'Angers
pour leur accueil, et pour les bonnes conditions de travail dans lesquelles j'ai pu effectuer mon
stage.
Master Pro BioInformatique Laurent CHATELAINPage 1

Université de Nantes Rapport de stage
Table des matières
Introduction ......................................................................................................................................... 41 Contexte général................................................................................................................................5
1.1 Introduction................................................................................................................................51.2 Positionnement vis-à-vis des enjeux socio-économiques...............................................................6
1.3 Objectifs de l'unité UMR PMS.................................................................................................. 62 Présentation des LEA........................................................................................................................ 7
2.1 Localisation et expression..........................................................................................................72.2 Caractéristiques structurales...................................................................................................... 72.3 Cas des déhydrines...................................................................................................................11
3 LEA-DB : aspect base de données.................................................................................................. 143.1 La base de données.................................................................................................................. 14
3.1.1 Modélisation.....................................................................................................................143.1.2 Structure détaillée............................................................................................................ 15
3.2 Récupération des données .......................................................................................................193.2.1 Principe............................................................................................................................ 193.2.2 Sources de données.......................................................................................................... 213.2.3 Récupération des données................................................................................................ 223.3 Extraction des données et remplissage de la base : programmation Perl............................223.3.1 Analyse des fichiers à parcourir.......................................................................................223.3.2 Le programme.................................................................................................................. 24
4 LEA-DB : aspect site Web.............................................................................................................. 274.1 Généralités............................................................................................................................... 274.2 Organisation du site et organisation d'une page type...............................................................294.3 Fonctionnalités de LEA-DB.................................................................................................... 30
4.3.1 Consultation .................................................................................................................... 314.3.2 Recherche de données dans la base..................................................................................334.3.3 Les outils ......................................................................................................................... 344.3.4 Comparaison des résultats avec d'autres outils sur Internet.............................................364.3.5 Autres pages .................................................................................................................... 38
5 Perspectives de recherches et améliorations possibles.................................................................... 40Conclusion..........................................................................................................................................43Bibliographie...................................................................................................................................... 44ANNEXES......................................................................................................................................... 44
Master Pro BioInformatique Laurent CHATELAINPage 2

Université de Nantes Rapport de stage
Introduction
Découvertes chez les plantes, les protéines LEA (« Late Embryogenesis Abundant
proteins ») recouvrent un grand nombre de protéines très variées et sont principalement associées à
la tolérance au stress hydrique résultant de la dessiccation ou d'un choc thermique dû au froid. De
nombreuses fonctions ont été associées aux LEA, mais leur rôle précis n'est pas encore connu. Ces
protéines assurant des rôles aussi importants que la protection des structures cellulaires, il semble
judicieux de tenter de mieux les comprendre et de mieux les caractériser.
L'objectif de ce stage est de créer et d’exploiter une base de données contenant des informations
relatives à ces protéines, dans le but de mettre en lumière des données relatives à la relation
structure-fonction de ces protéines, et de fournir une base exploitable directement par les biologistes
travaillant sur ce domaine.
La première phase a consisté à s'approprier les notions fondamentales au sujet des LEA de
manière à bien saisir les enjeux mais aussi les difficultés du sujet à traiter.
Le stage s'organise ensuite en deux grandes étapes du point de vue informatique : la création d'une
base de données en elle-même, puis le développement de l'interface Web qui permet de l'exploiter.
Le remplissage de cette base a constitué une partie importante du projet, tant sur le plan de la
réflexion quant aux données à conserver en vue des études à mener, que de la façon de récupérer
ces informations.
Nous fournirons en fin de rapport quelques propositions pour enrichir et faire évoluer la base
de données et son interface, ainsi que les premières pistes de recherches que les biologistes auront la
possibilité d'emprunter pour l'analyse des données de la base.
Master Pro BioInformatique Laurent CHATELAINPage 3

Université de Nantes Rapport de stage
1 Contexte général
Ce projet de base de données relative aux LEA est issu de la collaboration entre Jean-Michel
RICHER du laboratoire LERIA (Laboratoire d'Etude et de Recherche en Informatique d'Angers),
qui participe également au projet BIL (BioInformatique Ligérienne), Emmanuel JASPARD de
l'UMR PMS (Physiologie Moléculaire des Semences) et Gilles HUNAULT du laboratoire HIFI
(Hémodynamique et Fibrose Hépatique). L'UMR PMS travaille depuis de nombreuses années sur
ces protéines et a notamment découvert la première LEA de la matrice mitochondriale PsLEAm
(Grelet et al.,2005; Tolleter et al,, 2007).
1.1 Introduction
L'UMR 1191 Physiologie Moléculaire des Semences (UMR PMS) a été constituée le 1er
janvier 2000 entre l'Université d'Angers, l'Institut National d'Horticulture (INH) et l'INRA
(département de Biologie Végétale).
L'objectif de cette structuration était de rassembler des équipes de recherche déjà engagées dans des
travaux sur les semences, afin de conforter, sur le plan de la recherche fondamentale, le pôle
semence bien établi en Anjou par la présence de nombreux industriels et instituts techniques.
Au sein du dispositif INRA, l'UMR se distingue par sa forte proportion d'enseignants-chercheurs
parmi l'effectif chercheur (11/13). L'UMR a été favorablement renouvelée en 2007 avec comme
recommandations de recentrer les travaux sur un nombre réduit d'espèces et de développer des
approches de validation fonctionnelle des gènes.
1.2 Positionnement vis-à-vis des enjeux socio-économiques
La production française de semences atteint environ 1,3 millions de tonnes, plaçant ainsi
notre pays au premier rang européen et au deuxième rang mondial après les États-Unis. Réalisant un
chiffre d'affaires de 1800 M€, la France a exporté pour plus de 400 M€ lors de la campagne 2004-
2005 (sources GNIS, Groupement National Interprofessionnel des Semences et plants).
Master Pro BioInformatique Laurent CHATELAINPage 4

Université de Nantes Rapport de stage
La région des Pays de la Loire, assurant en moyenne 10% de la production, est une zone
traditionnelle et historique de multiplication des semences. Outre les producteurs, elle regroupe
également tous les acteurs de la filière publics et privés.
La qualité des semences représente un enjeu stratégique et économique important. En effet,
le marché très compétitif oblige les producteurs de semences à garantir la qualité de leur produit
envers leurs clients. En terme de qualité physiologique, il s'agit d'assurer que les semences vendues
soient vigoureuses, lèvent rapidement, de manière homogène et tolèrent la conservation prolongée.
1.3 Objectifs de l'unité UMR PMS
L'objectif des recherches de cette unité est d'acquérir une meilleure compréhension de la
biologie des semences au cours de la germination et de la levée, en vue d'en assurer la qualité
physiologique.
Pour ce qui nous intéresse plus particulièrement dans le cadre de ce stage, l'équipe d'O. Leprince a
pour objectif général de mieux comprendre les mécanismes protecteurs et de régulation impliqués
dans la tolérance à la dessiccation ainsi que les facteurs moléculaires impliqués dans la conservation
des semences.
La tolérance à la dessiccation (TD) correspond à la propriété de résister sans dommage à la
perte presque totale de l'eau cellulaire. Chez les semences orthodoxes, elle est acquise pendant la
phase de remplissage des graines et perdue au cours de la germination. Il s'agit d'un phénomène
multifactoriel qui repose sur au moins deux composantes :
1) la synthèse de molécules protectrices tels que les sucres non réducteurs et les protéines de
stress LEA et HSP (Heat Shock Protein) (Hoekstra et al., 2001)
2) la répression coordonnée du métabolisme et l'élimination de réactions néfastes engendrées
par des dysfonctionnements métaboliques pendant la dessiccation (Leprince et al., 2000). En
termes de régulation, on sait que l'ABA (Acide Abscissique) joue un rôle majeur mais les
voies de signalisation sont loin d'être connues.
Master Pro BioInformatique Laurent CHATELAINPage 5

Université de Nantes Rapport de stage
2 Présentation des LEA
Les LEA sont des protéines en majorité très hydrophiles qui semblent impliquées dans la
protection des structures cellulaires, ce qui en fait des protéines potentiellement très importantes
dans la vie des cellules. Elles sont très nombreuses et diverses et forment un très vaste ensemble de
familles de protéines.
2.1 Localisation et expression
Une des premières caractéristiques de ces protéines est qu'elles sont ubiquitaires : on les
retrouve dans de nombreux compartiments cellulaires comme le noyau, le cytoplasme, le reticulum
endoplasmique et elles ont été récemment découvertes au niveau de la mitochondrie.
Leur importance se manifeste également dans la diversité des espèces chez lesquelles on les a
identifiées (plantes, bactéries, nématodes). Cependant les LEA n'ont toujours pas été mises en
évidence chez l'homme, d'où l'intérêt et les interrogations qu'elles suscitent : sont-elles présentes
chez l'homme et quelle est leur fonction ?
Chez les plantes, elles sont fortement exprimées pendant la maturation des graines ainsi que dans
les tissus végétatifs en réponse à différents stress : hydrique (déshydratation), thermique (froid),
salin.
2.2 Caractéristiques structurales
Ainsi les LEA sont connues depuis près de 30 ans (Dure et al., 1981) mais leur fonction
précise demeure inconnue (vs. une activité enzymatique par exemple). La ou les fonctions des LEA
ne sont donc pas caractérisées même si des études ont réussi à dégager quelques grands rôles
comme la résistance à la déshydratation.
Certaines caractéristiques structurales ont cependant été mises en évidence.
Tout d'abord ces protéines sont très hydrophiles de par leur forte teneur en acides aminés
hydrophiles. Les LEA du groupe 2 joueraient ainsi un rôle de surfactant et préviendraient
l'agrégation des macromolécules, participant de cette manière au maintien de leur intégrité.
Master Pro BioInformatique Laurent CHATELAINPage 6

Université de Nantes Rapport de stage
Des travaux récents chez Arabidopsis thaliana utilisant des mutants KO du gène ATEM6
(un gène codant une LEA) ont montré que les graines présentaient une déshydratation et une
maturation précoce (Manfree, Lanni 2006). L'une des fonctions liée à cette caractéristique
structurale serait donc de réguler la perte en eau en intervenant pendant la maturation de l'embryon.
Ensuite, une caractéristique tout à fait remarquable de ces protéines est qu'elles semblent
échapper au dogme reliant la structure d'une protéine à sa fonction : en effet elles sont non
structurées dans leur forme native, et elles s'enrichissent en structures secondaires dans des
conditions dénaturantes.
A titre d'exemple, la structure secondaire montre que MtEm6 et PM25 (2 LEA) sont non structurées
à l'état hydraté avec un pourcentage en hélice α et feuillet β de 10 et 15%. Un séchage lent induit
une augmentation significative de la teneur en hélice α (Em6, 60%; PM25, 56%). Toutefois chez
PM25 il induit également une augmentation de la proportion en feuillet β (25%). Cette structure en
feuillet β pourrait être liée à une fonction spécifique à l'état sec.
Enfin, certaines LEA possèdent des motifs répétés qui ont permis de classer ces protéines
comme nous allons le voir.
Les LEA sont étudiées depuis longtemps puisque des équipes comme celles de Dure s’y
intéressaient déjà dans les années 1980 (classification en 1989), puis Wise et Tunnacliffe par la
suite. Malgré cela elles restent mal connues.
Mais ces équipes, même si cela reste complexe, ont trouvé des critères de classification. Tout
d'abord des critères basés sur les séquences protéiques.
Ainsi, sur la base de similarités de séquences, elles ont été divisées en plusieurs groupes:
Tableau 1 : Groupes de LEA et motifs associés (Tunnacliffe & Wise, 2007)
Groupe Motif Référence Pfam1 CGQTRREQLGEEGYSQMRGK Cuming (1999) PF004772a Segment « Y » = DEYGNP Close (1997) PF002572b Segment « S » = Sn Close (1997) PF002572c Segment « K » = EKKGIMDKIKEKLPG Close (1997) PF002573 TAQAAKEKAXE Cuming (1999) PF029874 PF03760
Master Pro BioInformatique Laurent CHATELAINPage 7

Université de Nantes Rapport de stage
On remarque aussi que cette classification associe chaque groupe à une famille de protéines
(Pfam) (Bateman et al., 2004). La base de données Pfam contient des informations sur les domaines
et les familles protéiques.
A remarquer également que dans la classification de Wise & Tunnacliffe de 2007, les groupes 5 à 9
ont disparu par rapport à la classification de 2003, et que les protéines de ces groupes ont donc été
redistribuées dans les 4 groupes restants.
Il faut aussi noter que la faible complexité de ces protéines rend difficiles les alignements. En effet
les répétitions de motifs diminuent la variabilité des acides aminés entre les différentes protéines à
comparer, ce qui complique la discrimination de celles-ci, car moins de critères les distinguent.
L'absence de structure 3D disponible rend impossible les études d'homologies de structures, à moins
de se baser sur des prédictions que l'on peut obtenir à l'aide de logiciels.
Les analyses POPPS (Proteins or Oligonucleodides Probability Profiles) des séquences
constituent également des critères de classification. Cela consiste en la mesure des fréquences de
chaque acide aminé dans les protéines de chaque groupe. Il a été observé que certains mono/di ou
tri-peptides étaient sur ou sous représentés dans un groupe donné, et mieux, que certaines de ces sur
ou sous abondances peuvent servir de facteur discriminant pour classer une protéine dans un groupe
ou dans un autre (Tunnacliffe, Wise, 2007).
Ces analyses POPPs ont conduit à la définition de SuperFamilles (SF) de LEA, avec une ou
plusieurs superfamilles comprenant chacun des groupes initiaux. Ces superfamilles sont donc le
reflet de la structure des groupes de LEA.
Tableau 2 : Résultats des analyses POPPs
Groupe SF Eléments discriminants1a 4 +E, +G, +EG, +GE, +GG, +KG, +QE, +RK, +GGE, +KGG
1b 6 +E, +G, +DE, +EG, +ES, +GG, +GQ, +RE, +RK, +ARE, +DES, +REG
2a 1 +G, -L, +EK, +GG, +GT, +EKL, +IKE, +KEK, +KIK, +KKG, +KLP, +LPG
2b 10 -F, +G, -I, +AG, +EK, +GG, +GQ, +KE, +SS, +EKL, +GAG, +IKE, +KEK, +KLP, +LPG, +SSS
2c 3 -F, -I, -L, -R, -W, +DK, +EK, +KK, +KL, +LP, +TH, +EKK, +KEK, +KLP, +LPG
3a 2 +A, -C, +E, -F, -I, +K, -L, -P, +AE, +AK, +EK, +ET, +GE, +GK, +KE, +AAE, +AKD, +EKA
3b 5 +A, -I, +K, -L, -P, +Q, +T, -V, +AA, +AQ, +EK, +KE, +KT, +QA, +QQ, +QS, +QT, +TQ, +AAK, +AQA, +EKT, +QAA, +TQQ
6 7 +A, -F, -L, +AA, +AE, +MQ, +QS, +VA, +AAA, +GVA, +QSA, +SAA
Master Pro BioInformatique Laurent CHATELAINPage 8

Université de Nantes Rapport de stage
Puis sur la base de cette classification, obtenue grâce aux différents critères que nous venons
de voir, ces équipes sont parvenues à associer des mots-clés à chacun des groupes de leur
classification, ce qui se révèle être d'un grand intérêt pour les analyses syntaxiques puis sémantiques
de la fonction des LEA que l'on envisage par la suite.
Tableau 3 : Mots-clés associés aux superfamilles de LEA, et donc aux groupes de LEA
Groupe SF Mots-clés
1 4 histone H4, chromosomal protein, nuclear protein, DNA binding
1 6 dsRNA binding, DNA gyrase, breakage, CLP, ATP binding
2 1 Break, ATP binding, protein biosynthesis, topoisomerase, repair
2 3 Coiled, coil, nuclear protein, caldesmon, histone H1, chaperone, tropomyosin filament, break, DNA topoisomerase
2 8 DNA topoisomerase, nuclear protein, HMG box, coiled coil
2 9 Transcriptional inhibition, glycosyl hydrolase, nuclear protein,
2 10 nuclear protein, DNA binding, transcription regulation, intermediate filament, keratin, chaperone, homeobox, coiled coil, HMG box domain, cytoskeleton
3 2 Chaperone, coiled coil, tropomyosin, stress, filament, phosphorylation, caldesmon elongation factor, neurofilament, actin binding, cytoskeleton, rotamase
3 5 Coiled coil, histone H1, filament, nuclear protein, neurofilament, flagella, HAMP domain, synuclein, DNA binding, hsp70
6 7 Groel protein, nuclear protein, histone H1, chaperonin, DNA binding, HAMP domain, synuclein, transcription regulation
Il faut ajouter à ces critères des règles basées sur les profils d'hydrophobicité des protéines, qui
permettent de classer les LEA dans des sous-groupes (Tunnacliffe, Wise, 2007).
Tableau 4 : Règles de classification des LEA
Groupe Règles2a H < = 0.15 et aromatiques > = 0.077 et min_hyph < = -1.97et chargés < = 0.42
2b L > = 0.23 et H < = 0.3 et ave_hyph > = -1.233 et ave_hyph < = -0.978
2c aromatiques > = 0.077 and min_hyph < = -2.743 et chargés >= 0.4
3 H > = 0.34
1 E > = 0.02 et ave_hyph < = -1.241
lea5 max_hyph > = 1.0 et ave_hyph < = -0.3
lea14 aliphatiques> = 0.25
6 H > = 0.25 et max_hyph > = 0.5
4 Autres
Les règles doivent être appliquées de haut en bas (si ... sinon).
Master Pro BioInformatique Laurent CHATELAINPage 9

Université de Nantes Rapport de stage
2.3 Cas des déhydrines
Malgré la complexité de leur analyse, certaines LEA ont été plus particulièrement étudiées,
il s'agit des LEA du groupe 2, appelées aussi déhydrines, et qui sont donc aujourd'hui les mieux
connues.
Il a ainsi été défini des fonctions plus précises pour cette classe, en fonction de la présence
de certains patterns. Un motif est généralement un segment court, continu et non ambigu d'une
séquence alors qu'un pattern a une structure plus complexe. Il est souvent composé de différents
motifs qui peuvent être plus ou moins éloignés les uns des autres et sa définition peut comporter des
exclusions ou des associations de motifs.
L’alignement multiple de plusieurs séquences dotées de la même fonction est utilisé pour définir un
motif consensus (ensemble de positions conservées) qui peut alors servir de " signature "
fonctionnelle : toute nouvelle séquence dans laquelle ce motif est détecté est alors réputée
correspondre à une fonction similaire.
Les motifs K (EKKGIMDKIKEKLPG) sont souvent retrouvés dans la partie C-terminale
des protéines et peuvent être répétés de 1 à 11 fois. Des prédictions de structure indiquent une
propension à former des hélices α amphipathiques de classe A (Close, 1996, 1997; Campbell and
Close, 1997; Cuming, 1999).
Les motifs Y (DEYGNP) sont plutôt situés en partie N-terminale des protéines et concernent
une grande partie des LEA du groupe 2.
Les motifs S (Sn) peuvent subir des phosphorylations et seraient ainsi associés à la
localisation nucléaire (Goday et al., 1994; Godoy et al., 1994) .
Les motifs Φ correspondent à des enchaînements d'acides aminés polaires riches en
glycines. Il leur a été attribué un rôle dans les interactions avec les surfaces hydrophobes des
macromolécules cytoplasmiques ou nucléaires, empêchant ainsi leur agrégation (Campbell and
Close, 1997).
Prenons par exemple une déhydrine qui présenterait un motif Y, puis un motif S puis n motifs K :
les auteurs ont réussi à déterminer que cette protéine serait plutôt associée à la fixation aux
phospholipides des membranes des vésicules lipidiques.
Master Pro BioInformatique Laurent CHATELAINPage 10

Université de Nantes Rapport de stage
Tableau 5 : Association de patterns à des fonctions plus précises des déhydrines
YSKn KnS Skn, Kn, KnS- Fixation aux phospholipides des vésicules lipidiques
- Fixation aux métaux- Protection contre la peroxydation par radicaux hydroxyles- Cryoprotection
- Processus d'acclimatation au froid
Intérêts de la base
Ainsi on voit bien l'importance de ces protéines et donc les nombreux intérêts d’une base de
données pour les LEA.
1. Il n’existe pas de base de données dédiée aux LEA.
2. Son contenu a été mûrement réfléchi par un biochimiste, un informaticien et un statisticien. Il
constitue un recueil de données spécialisées sur cette vaste famille de protéines.
3. La base de données contient plusieurs milliers de séquences. Ce nombre est beaucoup plus
important que ceux sur lesquelles les études bioinformatiques antérieures reposaient.
4. Aucune structure de LEA n’est connue.
5. Par ailleurs la définition de leur fonction est floue, imprécise. Elle est souvent reliée à des
phénomènes physiologiques amples qui mettent en jeu un très grand nombre de protéines
diverses, dont les LEA.
6. En conséquence, la relation structure – fonction des LEA (groupe par groupe) est inconnue.
7. Des données expérimentales sont donc absolument nécessaires. Mais elles n’ont trait qu’à une
seule protéine LEA. Ce sont des données individuelles, pas forcément extrapolables à
l’ensemble des LEA ou à des sous-ensembles.
Master Pro BioInformatique Laurent CHATELAINPage 11

Université de Nantes Rapport de stage
8. De ce fait, l’analyse prospective par des outils bioinformatiques s’appuyant sur la base
de données LEA est une approche complémentaire aux données expérimentales.
9. Et ce d’autant plus, qu’à contrario, la fouille de données permet de traiter l’ensemble des
protéines afin de les classer, de les différencier : d’en obtenir une sorte de « carte d’identité »,
groupe par groupe.
10. Elle permet aussi d’inclure des protéines considérées comme non LEA et inversement
d’exclure des protéines « réputées » LEA qui n’en sont pas.
11. La structure de la base de données permet un grand nombre d’analyses dans le but d’étudier
la relation structure – fonction des LEA :
- analyse syntaxique et sémantique des mots-clés liés à la fonction, à la localisation
cellulaire, au stade de développement
- recherche de motifs consensus, de signatures des LEA groupe par groupe
- recherche de domaines protéiques
- caractérisation physico-chimique : masse molaire, point isoélectrique, composition en acides
aminés, profils d’hydrophobicité, propension à être nativement non repliée, …
Master Pro BioInformatique Laurent CHATELAINPage 12

Université de Nantes Rapport de stage
3 LEA-DB : aspect base de données
3.1 La base de données
Pour cette partie du projet, nous avons utilisé MySQL pour le développement de la base.
3.1.1 Modélisation
La première grande étape du projet a consisté en la définition de la base. Cette phase est
primordiale pour l'étude ultérieure des LEA et requiert donc toutes les attentions. En effet, on
souhaite remplir la base avec des données relatives aux LEA sélectionnées grâce à la requête
précise que nous verrons plus loin (section 3.2.1). La difficulté réside dans le choix de champs qui
devront se révéler pertinents lors des fouilles de données qui suivront. Nous nous sommes donc
réunis à plusieurs reprises pour décider des données qu'il nous semblait judicieux de conserver pour
chaque protéine. De ces entretiens est apparue la structure de la base.
Il est à noter que les informations sur les protéines sont extraites de fichiers « GenPept »,
format de fichier spécifique au NCBI (National Center for Biotechnology Information,
http://www.ncbi.nlm.nih.gov/), contenant de nombreuses données mais dont seulement une partie
nous intéresse.
Le NCBI est une ressource nationale américaine pour l'information biologique moléculaire. Il
développe des bases de données publiques telles que GenBank, dbSNP ou RefSeq, conduit des
recherches dans la biologie informatique, développe des logiciels pour analyser des données de
génome et fournir des informations biomédicales. Il est en quelque sorte le pendant américain de
l'Institut européen de bioinformatique (EBI).
La base est constituée de 5 tables, dont 3 tables contenant les informations sur les protéines
sont reliées entre elles par l'identifiant de la protéine. Les deux autres tables sont indépendantes et
sont destinées à stocker les mots-clés utiles aux recherches d'une part, et les données de connexion
des différents utilisateurs privilégiés d'autre part.
Master Pro BioInformatique Laurent CHATELAINPage 13

Université de Nantes Rapport de stage
3.1.2 Structure détaillée
Nous avons d'abord la table 'proteins' contenant les différentes informations sur les
protéines, la table 'fastas' contenant les séquences des protéines au format Fasta, la table 'notes'
contenant des informations biologiques, la table 'keywords' contenant les mots clés destinés aux
recherches qui seront développées dans la section 4.3.2, et la table 'membres' qui permet de gérer
des types d'utilisateurs possédant chacun des droits différents sur la base.
Les séquences Fasta, qui sont elles aussi des données relatives aux protéines ont été volontairement
séparées dans une autre table (fastas) pour des raisons de performances. En effet, les séquences
peuvent constituer des données assez lourdes (jusqu'à 800 acides aminés pour les LEA les plus
longues), or la plupart des requêtes ne faisaient pas appel aux séquences, d'où l'idée de les mettre à
part pour éviter de les manipuler inutilement.
Commençons par la table 'proteins'. Cette table contient 20 champs que nous allons
détailler.
Tout d'abord le champ 'accession' : il s'agit d'un code unique attribué à chaque publication
de protéine dans la base de données du NCBI (Genbank) qui constitue donc une clé intéressante
pour l'identification des protéines stockées. Ce code consiste en une chaîne de caractères et est donc
stockée sous un type varchar(15). Dans un fichier « GenPept » il s'agit de la première donnée de la
ligne 'LOCUS'. Ce champ sert de clé primaire.
Le nom de la protéine (champ 'name') est également stocké sous un type varchar(50). Dans le
fichier « GenPept » il s'agit de la première information retrouvée sur la ligne 'DEFINITION', c'est à
dire ce qui se trouve avant le '['.
La longueur de la séquence en acides aminés de la protéine (champ 'length'), est stockée sous un
type int(5). On retrouve cette information en deuxième position de la ligne 'LOCUS' du fichier
« GenPept ».
L'organisme (champ 'organism') dont est issue la LEA est stocké sous un type varchar(50). Il s'agit
de l'information portée sur la ligne 'SOURCE / ORGANISM' du fichier « GenPept ».
Le règne (champ 'reign') dont elle est issue est stocké sous un type varchar(50). Il s'agit de
l'information portée sur la deuxième ligne de 'SOURCE / ORGANISM' du fichier « GenPept »
après le premier ';'.
Master Pro BioInformatique Laurent CHATELAINPage 14

Université de Nantes Rapport de stage
Le champ 'uniprot' contient l'identifiant Uniprot de la protéine lorsqu'il est présent dans le fichier
« GenPept ». On le retrouve sur la ligne 'DBSOURCE / accession' et est stocké sous un type
varchar(15).
Le champ 'authors' contient les noms des auteurs, le champ 'title' contient le titre de l'article et le
champ 'journal' contient les références du journal lié à la publication de la séquence, chacun étant
stocké sous un type varchar(300). Si plusieurs références sont mentionnées, seule la première est
conservée comme nous le verrons dans la section 3.3.2. On récupère donc les lignes 'AUTHORS',
'TITLE' et 'JOURNAL' correspondants à la REFERENCE numéro 1 du fichier « GenPept ».
Notons que le champ 'title' contient beaucoup de mots-clés qui nous serviront pour l’analyse des
données de la base.
Le champ 'function' contient la fonction qui est associée à la protéine, et est stocké sous un type
varchar(200). On retrouve ces informations sur la ligne 'FEATURES / Protein / function=', situées
entre guillemets.
Le champ 'tissue_type' contient le tissu dans lequel la protéine a été identifiée (graine, feuille,
racine...) et est stocké sous un type varchar(50).On retrouve ces informations sur la ligne
'FEATURES / source / tissue_type=', situées entre guillemets.
Le champ 'dev_stage' correspond au stade de développement où cette protéine a été identifiée , et
est stocké sous un type varchar(30). On retrouve cette information sur la ligne 'FEATURES /
SOURCE / dev_stage=' entre des guillemets.
Les 3 champs suivants contiennent les différentes appellations d’une même protéine, et chacun est
stocké sous un type varchar(100). On retrouve ces informations à différents endroits du fichier
GenPept : le champ 'product' contient les informations entre les guillemets de la ligne 'FEATURES
/ Protein / product=', le champ 'protein_name' celles de la ligne 'FEATURES / Protein / name=' (et
séparées par un ';' si elles sont plusieurs). Enfin le champ 'gene' contient les données situées entre
les guillemets de la ligne 'FEATURES / CDS /gene='.
Les 3 champs suivants concernent les données relatives à la fonction supposée de la protéine et
chacun est stocké sous un type varchar(300). On récupère pour cela les informations situées entre
les guillemets des lignes 'FEATURES / Region / region_name=' pour le champ 'region_name',
Master Pro BioInformatique Laurent CHATELAINPage 15

Université de Nantes Rapport de stage
'FEATURES / Region / note=' avant le ';' pour le champ 'region_note', et 'FEATURES / CDS /
note=' pour le champ 'putative_function'.
Le champ 'pfam' indique la présence de motifs protéiques conservés, et est stocké sous un type
varchar(30). Cette donnée est récupérée dans la ligne 'FEATURES / Region / note=' après le ';'.
Le champ 'cdd' constitue un lien vers la base NCBI des domaines protéiques conservés, et est
stocké sous un type varchar(30). Il s'agit de récupérer le lien situé sur la ligne 'FEATURES / Region
/ db_xref='.
Les protéines contiennent souvent plusieurs modules ou domaines, chacun possédant une origine et
une fonction évolutives différentes. Le CDD (NCBI's Conserved Domain Database) est une
collection d'alignements multiples de séquences de domaines ancestraux et de protéines. Un outil
disponible sur le site permet d'identifier le domaine présent dans la protéine passée en paramètre, et
c'est la référence du domaine qui est stockée dans ce champ.
Le champ 'date' contient la date de publication de l'article, et est stocké sous un type int(5) car on ne
retient que l'année. Cette donnée est située en cinquième position de la ligne 'LOCUS'.
Il est important de noter que tous les champs ne sont pas obligatoirement remplis pour toutes les
protéines, et dans ce cas les champs non renseignés sont remplis avec « N/A » (pour : « Non
Available ») dans la base.
La table 'fastas' contient bien sûr le numéro d'accession de la protéine comme clé de la table,
ainsi que sa séquence. Le parti a été pris de séparer cette donnée des autres informations concernant
la protéine, car il s'agit de données plus volumineuses et qui ne seront pas utilisées à chaque étape
de l’analyse des données de la base.
Le champ 'fasta' contient la séquence protéique au format Fasta, et est donc stocké sous un type
varchar(4000). On retrouve cette information sur la ligne 'ORIGIN' du fichier « GenPept ».
La table 'notes' contient également le numéro d'accession comme clé de la table, ainsi que 2
autres champs d'informations et 2 champs relatifs aux travaux de Wise de 2003.
Le champ 'remarks' contient diverses informations portées par le biologiste, et est stocké sous un
Master Pro BioInformatique Laurent CHATELAINPage 16

Université de Nantes Rapport de stage
type varchar(200).
Les champs 'our_classif', 'wise_group', 'wise_pfam' indiquent, le cas échéant, respectivement la
classe de LEA à laquelle appartient une protéine selon nos critères, sa classification selon Wise et la
classification Pfam que Wise lui a associée. Ils sont stockés sous des types varchar(50), varchar(15)
et varchar(15).
La table 'keywords' ne contient que le champ 'keyword' qui en constitue la clé. Il est stocké
sous un type varchar(30) et sert pour la recherche des LEA par mot clé.
La table 'membres' contient le 'login' comme clé (varchar(15)) ainsi que le champ 'mdp'
(varchar(15)) pour « mot de passe », qui servent à la validation de la connexion de l'utilisateur à la
base, lui autorisant ainsi l'accès à des fonctionnalités supplémentaires qui seront détaillées dans la
section 4.2.5.
3.2 Récupération des données
3.2.1 Principe
Comme nous l'avons vu, les LEA ne sont pas bien caractérisées, et les analyses
bioinformatiques menées par Wise et Tunnacliffe portaient seulement sur un petit échantillon de
protéines (environ 150). Ici, l'objectif est d'étendre cette étude à une population beaucoup plus large
pour tenter d'élargir les règles déjà établies, et établir la relation structure-fonction de ces LEA.
Mais comment travailler sur les LEA alors que l'on n'en connaît pas de définition précise ?
D'où l'importance d'une requête aussi efficace et pertinente que possible pour récupérer le
maximum de LEA, tout en réalisant un tri sur les mots-clés qui ne donnaient pas de réponse
satisfaisante, c'est à dire des protéines n'ayant a priori pas de lien avec les LEA et qu'il ne semble
donc pas intéressant d'intégrer à l'étude envisagée.
Cette requête est construite en 2 étapes : la première consiste à chercher toutes les protéines
associées aux mots-clés que l'on peut eux-mêmes associer aux LEA (comme dehydrin OR glycine
rich OR unstructured), et ceci est réalisé grâce à l'opérateur 'or'.
Master Pro BioInformatique Laurent CHATELAINPage 17

Université de Nantes Rapport de stage
Requête de départ :
late embryogenesis abundant OR Lea OR dehydrin OR glycine rich OR unstructured OR ion sequestration OR dehydration OR dehydration stress OR dehydration-stress OR dehydration induced OR dehydration-induced OR dehydration shock OR desiccation OR desiccation stress OR desiccation-stress OR desiccation induced OR desiccation shock OR cold stress OR cold-stress OR cold induced OR cold-induced OR cold shock OR cold-shock OR water stress OR water-stress OR water induced OR water shock OR salt stress OR salt-stress OR salt induced OR salt-induced OR salt shock OR salt-shock OR abcisic acid OR abcisic acid induced OR abcisic acid-induced OR ABA OR ABA induced OR ABA-induced
Cette requête a renvoyé 55358 hits le 23/07/07 contre 53397 le 06/06/07 : on voit l'inflation
galopante de protéines 'estampillées' LEA, et donc l'intérêt de notre étude, mais aussi la nécessité de
filtrer toutes ces réponses.
C'est l'objet de la seconde étape, qui vise à éliminer le plus possible de réponses 'inadéquates' grâce
à l'opérateur 'not'. L'exemple le plus frappant concerne les protéines qui sont retournées uniquement
à cause de leur auteur dont le nom se trouve être 'Lea'.
Suite de la requête :
NOT enolase NOT hsp NOT hsps NOT kinase NOT dehydrogenase NOT oxidase NOT oxygenase NOT dioxygenase NOT reductase NOT F0 NOT aldolase NOT chitinase NOT dehydrase NOT hydrolase NOT hydrolyase NOT dehydratase NOT dehydrolase NOT dehydroquinase NOT phosphatase NOT amylase NOT beta-lactamase NOT glucuronosyltransferase NOT polygalacturonase NOT protease NOT proteinase NOT fucosyltransferase NOT sialyltransferase NOT acyltransferase NOT transferase NOT ribonuclease NOT thioredoxin NOT phospholipase NOT aminotransferase NOT torsin NOT casein NOT fasciclin NOT hemolin NOT immunoglobulin NOT noxin NOT aconitase NOT endochitinases NOT polymerase NOT crotonase NOT alpha-enolase NOT caltractin NOT kinase NOT chondrocalcin NOT K-casein NOT casein
Puis : NOT patent NOT antigen NOT lethal NOT strictosidine NOT oleosin NOT capsid NOT echovirus NOT ribulose NOT chlorophyll NOT cytochrome NOT photosystem NOT OLIGOSACCHARIDE NOT microglobulin NOT pyruvate NOT glycogen NOT glycosyl NOT prenyl NOT sporulation NOT needle NOT wall NOT betaine NOT salivaricin NOT cadherin NOT plastid-targeted NOT Shikimate NOT proline-rich NOT nicotinic NOT Chitin NOT phenylethanolamine NOT annexin NOT sarcoma NOT ankyrin NOT oocytes NOT S-adenosylmethionine NOT O-succinylbenzoate NOT cancer NOT channel NOT Disulfide NOT Luminescent NOT fluorescent NOT allergen NOT hardening NOT enhancer NOT Alzheimer NOT Glucagon NOT Capsid NOT neuroprotective
Puis : NOT leak NOT AroA NOT pMA1951 NOT CG5481 NOT LD06565p NOT Cd55 NOT CSCN1 NOT Bbcrasp NOT lti NOT BAA23547 NOT NP_563710 NOT NP_172994 NOT pipd NOT ipad NOT egf NOT PsbA NOT elip NOT rd22 NOT AtPIP5K1 NOT RiP-15 NOT FabZ NOT EF1 NOT SNF4 NOT bZIP NOT HIV-1 NOT fxuA NOT CzcD NOT BLT14 NOT TIP1 NOT F1 NOT Tim17 NOT PIP1 NOT TAZ1 NOT AZF3 NOT CSDE1 NOT SPBc2 NOT Csda
Master Pro BioInformatique Laurent CHATELAINPage 18

Université de Nantes Rapport de stage
Puis : NOT lea[Author] NOT Reshef[Author]
Puis : NOT fragment NOT peptide NOT partial
Puis : AND 80:800[Sequence Length]
Cependant il faut rester vigilant dans cette étape de tri, car on est tenté d'écarter des termes
comme 'transcription factor', qui ramènent énormément de réponses, pour simplifier l'analyse
ultérieure. Mais des auteurs attribuent aux LEA des propriétés de liaison à l'ADN , tout comme les
facteurs de transcription le font, et exclure un terme comme celui-ci risquerait donc d'ignorer des
protéines potentiellement intéressantes pour notre étude. Il ne faut donc pas être trop stringeant
pour ne pas perdre des informations potentiellement importantes. C'est là que réside toute la
difficulté de ce travail de sélection, mais cette méthode d’élimination grâce à des termes choisis
avec soin, nous permet d'obtenir 32000 réponses.
De plus, sur ces données ainsi récupérées, nous en avons parcouru beaucoup et elles semblaient tout
à fait pertinentes et adaptées aux analyses envisagées.
Il faut noter que le nombre élevé de retours est dû à la requête qui n'est que partielle, et vouée à
s'affiner en fonction des résultats qui seront obtenus lors des fouilles de données.
3.2.2 Sources de données
Il existe plusieurs bases de données protéiques que l'on peut interroger pour trouver des
informations. Plusieurs possibilités s'offraient donc à nous pour récupérer les fichiers de données
sur les protéines issues de la requête.
Tout d'abord, la base Swissprot (http://expasy.org/sprot/) qui contient de nombreuses
protéines, et pour chacune d'entre elles beaucoup d'informations intéressantes pour l'étude
envisagée. Cette base présente également l'intérêt d'attribuer à chaque protéine répertoriée un
identifiant unique (id Uniprot), dont l'avantage est qu'il est reconnu par de nombreux outils
bioinformatiques sur Internet.
Cependant, cette source de données n'a pas été retenue compte tenu de la faible performance des
requêtes possibles. En effet, les requêtes sur ce site n'autorisent pas l'utilisation d'opérateurs
booléens (ou bien 3 seulement dans les recherches avancées), et ne permettent donc pas un filtre
assez efficace pour notre étude.
Master Pro BioInformatique Laurent CHATELAINPage 19

Université de Nantes Rapport de stage
Une autre base protéique très connue est la PDB (Protein Data Bank,
http://www.rcsb.org/pdb/home/home.do), mais cette base qui gère des données sur des structures de
protéines ne présente pas d'intérêt pour nous, puisqu'on sait qu'on ne dispose pas de structure de
LEA à ce jour.
Enfin, le NCBI présente un grand intérêt pour nous puisqu'il permet d'effectuer des requêtes
assez complètes, ce qui assure un bon filtrage des protéines qui seront retenues. C'est donc cette
base que nous avons retenue pour récupérer les fichiers de données. Elle propose aussi un format de
fichier intéressant car contenant de nombreuses informations : le format « GenPept ».
Ce format présente cependant des limites puisque les règles de formatage ne semblent pas définies
de manière stricte, ou bien ne sont pas respectées scrupuleusement par les équipes publiant leurs
travaux.
Cette base de données présente aussi l'inconvénient d'identifier les protéines grâce à un numéro
interne, et qui n'est donc pas reconnu par d'autres outils sur Internet, ou du moins pas pour tous les
identifiants. Mais l'important à cette étape du projet reste essentiellement de récupérer des données
protéiques adaptées, d'où l'intérêt de ce site.
3.2.3 Récupération des données
Pour récupérer les fichiers on envoie la requête dans la partie protéique de la banque de
données du NCBI, afin de récupérer les fichiers « GenPept ».
Notons plusieurs choses :
tout d'abord, le fichier récupéré doit être enregistré avec l'extension '.html' car le programme
d'extraction des informations utilise les balises inhérentes à ce langage.
Ensuite, le NCBI ne permet pas d'obtenir des fichiers « GenPept » de plus de 500 protéines, d'où
une première tentative d'automatisation de la récupération des résultats.
Mais cette tentative n'a pour l'instant pas abouti à cause d'un problème de 'cookies', ce qui rend
assez fastidieuse la récupération des fichiers qui serviront à l'alimentation de la base puisqu'elle
nécessite de rapatrier manuellement les fichiers de protéines par paquets de 500.
Enfin, les fichiers « GenPept » doivent pour l'instant être placés dans le répertoire contenant le code
source pour être utilisables pour remplir la base.
Master Pro BioInformatique Laurent CHATELAINPage 20

Université de Nantes Rapport de stage
3.3 Extraction des données et remplissage de la base : programmation Perl
L'essentiel de cette partie du projet concerne l'extraction de certaines données choisies parmi
les nombreuses informations contenues dans les fichiers « GenPept » récupérés. Nous utilisons pour
cela le langage Perl car il est très efficace et pratique dans le parcours et l'analyse de chaînes de
caractères.
3.3.1 Analyse des fichiers à parcourir
Ce sont des fichiers formatés contenant les informations sur chaque protéine, dont seulement
certaines nous intéressent.
Même si ce format de fichier permet de retrouver certaines données de façon assez régulière comme
le numéro d'accession ou la taille, d'autres en revanche comme la fonction associée ne sont pas
présentes systématiquement, ou bien apparaissent sous diverses formes. Ceci constitue donc la
principale difficulté de l'extraction des données.
La résolution de ce problème réside dans une analyse détaillée de la structure d'un grand nombre de
fichiers « GenPept », de manière à faire émerger leur organisation 'moyenne'. Celle qui s'en dégage
est la suivante :
<pre class="genbank"> début d'une protéineLOCUS accession, length, dateDEFINITION nameACCESSIONVERSIONDBSOURCEKEYWORDSSOURCE uniprot
ORGANISM organism, reignREFERENCE
AUTHORS ou CONSRTM authorsTITLE titleJOURNAL journalPUBMED ou REMARK ou <a
FEATURESsource /organism
(/strain)
Master Pro BioInformatique Laurent CHATELAINPage 21

Université de Nantes Rapport de stage
(/dev_stage) dev_stage/db_xref/tissue_type tissue_type
(>gene< /gene/locus_tag/note)
>Protein< /name protein_name/product product(/gene)(/locus_tag)/product/function function
(>sig_peptide< /product /calculated_mol_wt)(>transit_peptide<) (>mat_peptide<)(>Region> (/gene)
(/locus_tag)/region_name region_name(/experiment)/note region_note, pfam(/FTId)/db_xref) cdd
(>Sec_Str<) (>Bond<)>CDS< /gene gene
(/locus_tag)/coded_by(/inference)(/note) putative_function(/transl_table)(/db_xref)(/codon_start)
ORIGIN fasta// fin de la séquence fasta</pre> fin de la protéine
3.3.2 Le programme
Le programme exécutant cette extraction est 'recup.pl'. Il se compose de 6 parties :
-quelques fonctions :
La fonction 'sansApostrophe($chaine)' permet d'échapper, au sens informatique du terme,
toutes les apostrophes de la chaîne qui lui est fournie grâce à une expression régulière. Ceci est
nécessaire à cause de certains noms anglo-saxons (O'Brien par exemple) qui posent des problèmes
de syntaxe lors de la construction des requêtes SQL.
La fonction 'init()' permet d'initialiser chaque champ à « N/A » (pour : « Not Available »)
par défaut.
La fonction 'sansEspaceDebutEtFin($chaine)' permet d'enlever les espaces de début et fin de
chaîne du paramètre passé.
Master Pro BioInformatique Laurent CHATELAINPage 22

Université de Nantes Rapport de stage
La fonction 'mot($chaine,$nombre)' tronque la chaîne en autant de morceaux qu'elle
rencontre d'espace, et stocke chacun dans une case d'un tableau '@mots'. Le $nombre sert à
récupérer le mot situé dans la case d'indice $nombre.
-ouverture du fichier GenPept :
On vérifie ici la syntaxe de la commande entrée par l'utilisateur visant à exécuter le programme qui
doit être de la forme : perl recup.pl nom_de_fichier.html
-stockage du fichier dans une variable :
On stocke le fichier dans une chaîne de caractères pour lui appliquer des expressions régulières.
-extraction des informations :
On utilise pour cela de nombreuses expressions régulières.
Tout d'abord il s'agit de bien découper le fichier rapatrié protéine par protéine, et nous allons voir
pourquoi.
Sur un fichier bien formaté, il aurait été aisé d'extraire toutes les données avec une seule et
même expression appliquée à la chaîne entière, ce qui aurait permis un meilleur traitement.
Mais la possible absence de certaines informations pour certaines protéines génère des erreurs. En
effet, l'expression régulière est construite pour récupérer des données situées entre deux 'balises';
ainsi lorsqu'une donnée n'est pas présente pour une protéine, l'expression extrait tout ce qui est
compris entre la première balise correspondant à une première protéine, et la seconde pouvant
correspondre à une autre protéine située plus loin dans le fichier, d'où une incohérence entre le
numéro de la protéine et ses informations propres.
Master Pro BioInformatique Laurent CHATELAINPage 23
LEA 2
Homo sapiens
Université de Nantes Rapport de stage
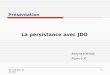
Sur cet exemple, si on ne découpe pas la chaîne en protéines (entre les balises '<pre
class="genbank">' et '</pre>'), le programme cherche la balise attendue jusqu'à la trouver (ici la
balise 'SOURCE').
<pre class="genbank"> début d'une protéine
</pre>
<pre class="genbank"> début d'une protéine
</pre>
On se préserve de cet écueil en découpant la chaîne en protéines successives c'est à dire entre les
balises '<pre class="genbank">' et '</pre>'.
On applique ensuite sur chacune d'entre elles des expressions régulières visant à ne sélectionner que
les informations d'intérêt. Ceci est relativement simple pour des champs comme « accession »,
« length », « date », « organism » et « fasta » qui sont remplis pour chaque protéine.
Cela devient plus difficile lorsqu'on veut obtenir le champ « reign », car les informations qui nous
intéressent ne se situent pas toujours au même endroit dans la rubrique du fichier. Il n'existe
cependant que deux cas différents (du moins sur les fichiers rencontrés et étudiés) qu'il suffit
d'envisager pour solutionner ce problème.
Les difficultés se font sentir lorsque des données ne figurent pas pour certaines protéines ou
alors sous diverses formes comme c'est le cas pour « authors » (ou consrtm), « title », « journal »
ainsi que pour de nombreuses sous-rubrique de « features ». On stocke donc les informations
relatives à « authors », « title » et « journal » dans une variable, à laquelle on applique une
expression pour en extraire les différentes sous-expressions.
Master Pro BioInformatique Laurent CHATELAINPage 24
ACCESSION
SOURCE
NAME
Protéine 1
LEA 1
ACCESSION
SOURCE
NAME
Protéine 2
accession = Protéine 1
name = LEA 2
source = Homo sapiens

Université de Nantes Rapport de stage
On fait de même pour les données situées dans la rubrique « features », mais la complexité est telle
qu'on est obligé de parcourir la variable avec autant d'expressions qu'il y a de champs à retenir, à
savoir 11.
-construction des requêtes pour chaque protéine :
Pour chaque protéine l'information étant stockée dans une variable qui porte le nom du champ, il est
aisé de construire la requête d'insertion dans la base. On écrit chaque requête dans le fichier
'sortie_qry.sql'.
-envoi de la commande d'exécution des requêtes :
Le programme recup.pl envoie enfin une commande système pour exécuter les requêtes inscrites
dans le fichier de sortie SQL. Cette commande est construite de la manière suivante :
mysql -u user -p -h host database < sortie_qry.sql
Nous avons décidé de ne garder que le contenu de la première occurrence d'une rubrique. En
effet, il arrive que la protéine en question soit citée par exemple dans plusieurs articles, ce qui
signifie plusieurs auteurs, titres et journaux. Cette décision arbitraire vient de l'observation que
ceux-ci n'apparaissent pas dans un ordre constant (ni chronologique ni alphabétique), ce qui
souligne d'ailleurs une fois de plus que le remplissage de ce genre de fichiers ne suit pas toujours
certaines normes d'où la nécessité de curation, même si des améliorations ont été constatées sur les
fichiers plus récents, notamment grâce aux différentes ontologies qui ont été mises en place.
Il faut noter que pour l'instant la base est alimentée avec 1048 protéines. Ces protéines sont
issues de 7 fichiers « html » de 200 protéines chacun, et la base devrait donc en contenir 1400. Le
formatage inconstant de ce genre de fichiers induit cette efficacité du programme de récupération
des données autour de 75%. Mais cette efficacité varie grandement entre les fichiers récupérés,
allant de 3 à 100% selon les fichiers rapatriés. En effet, même si les expressions régulières élaborées
pour extraire les informations envisagent de nombreux cas, elles ne sont pas encore exhaustives
compte tenu de la variabilité importante de l'organisation des fichiers « GenPept ».
Master Pro BioInformatique Laurent CHATELAINPage 25

Université de Nantes Rapport de stage
4 LEA-DB : aspect site Web
4.1 Généralités
LEA-DB est aussi l'interface Web qui permet d'accéder aux données de la base, ainsi qu'à
des fonctionnalités comme des outils applicables aux protéines. Elle a été conçue pour rendre la
navigation la plus simple possible, bien qu'étant assez sophistiquée et proposant différentes options
comme nous allons le voir.
Pour cette partie du projet, la programmation a été effectuée en PHP pour l'interface, avec
l'utilisation de feuilles de styles CSS, ainsi que des contrôles de réponse en Javascript.
Tout d'abord, la mise en forme du site a été réalisée en CSS (Cascading Style Sheet), ce qui
m'a permis d'aborder cet aspect intéressant de la mise en forme Internet, et présente divers
avantages.
En effet, la structure du document et la présentation peuvent être gérées dans des fichiers
séparés. De plus, le code HTML est considérablement réduit en taille et en complexité puisqu'il
contient des balises de présentation dont le contenu est diminué.
Enfin, les documents (pages « html ») faisant référence à une même feuille de styles, cela permet
une modification souple et rapide du site dans son ensemble et une présentation homogène de
l'ensemble des pages.
Toutes les pages du site sont organisées de la même façon : un bandeau de haut de page, un
menu à gauche pour accéder aux fonctionnalités du site, une zone d'affichage principale et enfin un
bas de page.
Master Pro BioInformatique Laurent CHATELAINPage 26

Université de Nantes Rapport de stage
Il est important de noter que la grande majorité des pages est validée XHTML 1.0
Transitional.
Ceci a entre autre intérêt de faciliter le travail d'indexation et d'analyse des pages, car il est
nécessaire de proposer des documents que les moteurs de recherche puissent reconnaître
facilement. XML est le parfait exemple d'un format conçu pour être à la fois lisible par l'oeil
humain et par la machine. XHTML tente d'offrir cette rigueur aux pages Web elles-mêmes, pour à
terme rendre le Web plus facile à traiter et donc plus accessible à tous.
Les tests de validité ont été réalisés grâce à un module de Firefox 2 : HTML Validator
(https://addons.mozilla.org/fr/firefox/addon/249), ainsi que le validateur du W3C
(http://validator.w3.org/).
Il se peut que certaines pages (de nouvelles pages, générées en fonction du contenu de la base) ne
soient pas valides, ceci étant principalement dû à l'affichage des données extraites par le programme
Perl. En effet, tous les cas ne sont pas envisageables pour récupérer les informations d'un fichier
« GenPept », et l'extraction pourra peut-être parfois générer des balises dans le code HTML, ce qui
provoquerait des erreurs lors de la validation, mais qui n'altèrerait en rien le bon fonctionnement du
site.
Master Pro BioInformatique Laurent CHATELAINPage 27

Université de Nantes Rapport de stage
4.2 Organisation du site et organisation d'une page type
La page d'accueil du site est 'index.php'. Elle est reliée à toutes les pages par le bouton
'Home' du menu de la zone de gauche. La structure du site et l'imbrication des programmes
apparaissent en annexe sur le schéma du même nom page 48.
A un traitement correspond une page : par exemple la fonctionnalité qui permet d'ajouter une
protéine se situe sur la page 'ajout.php', et le traitement (requête SQL) est exécuté par la page
'ajout_exe.php'.
Le code source a été organisé en quelques autres fichiers destinés à alléger l'ensemble ainsi
que dans un souci de modularité, et contenant des fragments de programme redondants. Tout
d'abord.
Le fichier 'funct.js' contient la seule fonction Javascript du site, pour bien isoler ce langage et ainsi
simplifier d’éventuelles modifications du code.
Le fichier 'fonction.php' contient les fonctions d'affichage, de navigation et de connexion à la base.
Enfin, le fichier 'fonction_prot' contient les fonctions relatives aux protéines, comme le calcul du
poids moléculaire, du point isoélectrique, ou encore de la composition en acides aminés.
Ensuite, dans un souci de simplification du code, quelques fonctions ont été écrites.
Tout d’abord le fichier 'style.css' qui contient le code gérant la mise en forme des pages, qu'on a
voulue externe pour ces mêmes raisons de modularité et de réduction du code.
L'entête de la majorité des pages étant le même, il a été mis dans une fonction 'doctype_head' qui
reprend le doctype du document (ligne de déclaration du type de document, qui indique au
navigateur dans quel type de HTML la page a été écrite) ainsi que le 'head' (code compris entre les
balises <head> et </head> de la mise en page HTML), et prend en paramètre un booléen indiquant
l'utilisation ou non de la fonction de validation Javascript.
Basée sur le même principe on retrouve la fonction 'header' d'affichage du bandeau supérieur, et la
fonction 'affiche_menu' d'affichage du menu latéral.
Master Pro BioInformatique Laurent CHATELAINPage 28

Université de Nantes Rapport de stage
<?php session_start(); // PAGE INDEX.PHPinclude("fonction.php");connexion(); //fonction.php, sélection et connexion à la base
//AFFICHAGE DU NOMBRE DE PROTEINES DANS LA BASE$requete="SELECT DISTINCT count(*) FROM proteins";$ressource=mysql_query($requete) or die ("Erreur SQL ".mysql_error()); $nb_tot=mysql_fetch_row($ressource);
doctype_head(0); //fonction.php, 1 pour utiliser la fonction de confirmation javascriptaffich_header(); //fonction.phpaffich_menu($nb_tot[0],1);//fonction.php, 1 pour afficher le nombre de protéines dans la base
?>
<div id="main"> <br/>
<?php
echo "<div class='texte'>";echo "The LEA database is dedicated to the «<b>Late Embryogenesis Abundant</b>» proteins.<br/><br/>
LEA proteins have been discovered in 1981. Although, they are almost associated with abiotic stress tolerance (particularly dehydration and cold stress), their actual function remains unkwown.<br/><br/>
The LEA database provides usefull data about the different sub-families (or groups) of LEA proteins for the analysis of their structure - function relationships. (<a href='references.php'>More...</a>)<br/><br/>";
echo "</div>";
?>
<br/><br/>
</div><div id="footer"> <i>© 2007 L.Chatelain</i> </div></body></html>
Master Pro BioInformatique Laurent CHATELAINPage 29

Université de Nantes Rapport de stage
4.3 Fonctionnalités de LEA-DB
LEA-DB est destinée à manipuler et analyser les informations contenues dans sa base, et
possède par conséquent diverses fonctionnalités que nous exposons ci-dessous.
4.3.1 Consultation
Il s'agit de concevoir une interface qui se veut conviviale, simple dans son utilisation pour
parcourir les informations que la base contient. Ceci est possible en cliquant sur le bouton
« consult » du menu qui donne accès à l'ensemble de protéines contenues dans la base.
L'utilisateur peut choisir le nombre de résultats qui seront affichés par page (20 à 200).
Possibilité lui est également donnée de choisir le mode d'affichage : en mode résumé
(« summary »), on affiche seulement le numéro d'accession, le nom, l'organisme et la fonction
associée alors qu'en mode détaillé (« details ») toutes les informations « publiques » de la protéine
apparaissent.
Ces informations « publiques » sont toutes les données de la protéines qui sont accessibles à
toute personne se rendant sur cette page, par opposition aux données réservées aux utilisateurs
privilégiés qui se sont connectés à la base grâce à un login et un mot de passe fournis par
l'administrateur (nous verrons comment devenir un utilisateur privilégié dans la section 4.2.5). Il
s'agit essentiellement du champ « remarks », édité par le biologiste, qui peut contenir des notes ou
conclusions sur une protéine.
L'utilisateur peut donc consulter les pages de données au moyen de boutons de navigation
standards. Néanmoins, la difficulté de cette navigation réside dans le fait que le nombre de résultats
par page n'étant pas fixe (puisque le choix est laissé à l'utilisateur), la requête d'affichage peut
varier. Pour résoudre ce problème il faut donc garder en mémoire le numéro de la première protéine
affichée dans la page, en plus du nombre désiré de résultats par page.
Master Pro BioInformatique Laurent CHATELAINPage 30

Université de Nantes Rapport de stage
Pour chaque protéine a été prévue une case à cocher, ce qui permet de sélectionner des
protéines lors du parcours de la base en vue de les étudier par la suite. On valide cette sélection en
cliquant sur le bouton « validate » ou tout autre bouton de cette barre de navigation, excepté « Reset
selection ». La validation de la sélection passe donc par l'envoi de ce formulaire (les cases à cocher)
à la page qui enregistre cette sélection, et ceci grâce à une fonction écrite en PHP.
Un bouton permet de sélectionner toutes les protéines apparaissant sur la page, de même qu'on peut
effacer la sélection pour choisir d'autres protéines.
Le mode d'affichage par défaut est le mode résumé, ce qui permet de réduire la taille des
pages de consultation. Mais lorsqu'au moins une protéine est sélectionnée, on peut afficher le mode
détaillé de la sélection grâce au bouton « Display selected proteins ». Ceci présente l'avantage de
pouvoir consulter les informations plus précises sur les protéines sélectionnées.
De même, on peut afficher les séquences au format Fasta des protéines de la sélection dans
une zone de texte grâce au bouton « Display sequences ». Ceci donne la possibilité de les
sélectionner et de les copier facilement, avant de les soumettre à un outil d'alignement comme Blast
ou ClustalW par exemple.
Master Pro BioInformatique Laurent CHATELAINPage 31

Université de Nantes Rapport de stage
4.3.2 Recherche de données dans la base
L'un des objectifs de ce projet est de confronter les caractéristiques des protéines qu'on aura
pu sélectionner en vu d'établir des règles sur les LEA.
Il peut donc être intéressant pour l'utilisateur d'effectuer des recherches selon certains critères, ce
que permet le bouton « Search » du menu.
On accède à une page qui propose de nombreux critères de recherche.
On peut tout d'abord rechercher une protéine grâce à son numéro d'accession. Il s'agit uniquement
ici de vérifier qu'une protéine est présente dans la base, car cette recherche ne peut renvoyer qu'un
résultat puisque basée sur un identifiant.
On peut ensuite effectuer une recherche grâce à un mot-clé contenu dans la table
« keywords », qui est d'ailleurs amenée à évoluer. En effet en avançant dans le travail sur les LEA,
certains mots-clés jusqu'alors absents de la table se révèleront certainement pertinents, et il sera
alors judicieux de les y inclure.
Ces recherches permettent d'isoler les protéines correspondant à un 'thème' précis, ce qui nous
rapproche des analyses syntaxiques puis sémantiques qui permettront peut-être de dégager des
fonctions plus précises pour les LEA.
Les requêtes associées portent sur les champs « name », « function », « product »,
« region_name », « region_note », « putative_function », qui ont été conservés car ils constituent
de véritables réservoirs de mots-clés dont l'analyse est susceptible de révéler des informations
importantes quant à la relation structure-fonction des LEA.
On peut également rechercher des protéines en fonction de l'organisme chez lequel on les a
découvertes. La liste de sélection est générée automatiquement avec les organismes retrouvés dans
la base. Il faut préciser que seuls le genre et l'espèce apparaissent car ce critère de recherche est
essentiellement destiné à discriminer les règnes animal et végétal. En effet, la première application
de la base LEA-DB porte sur des LEA d'origine végétale (PsLEAm et MtPM25 par exemple)
étudiées au sein de l'UMR PMS. Ceci a donc nécessité un traitement supplémentaire de la chaîne
obtenue pour ne conserver que les 2 premiers mots.
Cette étape est réalisée dans le programme d'extraction lui-même pour éviter de fournir des données
qui seraient à retransformer, grâce à la fonction 'mots' vue précédemment.
On peut encore rechercher les protéines selon leur taille, classée par tranches de 100 acides
aminés, par groupe de la classification de Wise lorsqu'il est disponible, ou par numéro d'accession
Pfam (Famille protéique), ou par numéro d'accession CDD (Domaines conservés) ou bien encore
Master Pro BioInformatique Laurent CHATELAINPage 32

Université de Nantes Rapport de stage
par date. Tous ces champs sont générés automatiquement en fonction du contenu de la base.
On sait que certains motifs servent à classer les LEA dans un groupe plutôt qu'un autre. Il est
donc intéressant d'utiliser la recherche par motif pour interroger la base. On peut par ce biais espérer
trouver les protéines appartenant à un groupe, et, ce pool de protéines étant nettement plus
important que lors des précédentes études, tenter de préciser les fonction des protéines de ce groupe.
Avec cet outil, la recherche s'effectue grâce à une expression régulière écrite en PHP qui cherche le
motif exact entré par l'utilisateur. En effet, les motifs (Y, S, K) mis en évidence pour chaque groupe
de LEA indiqués dans le tableau de la section 2.2 sont des séquences retrouvées telles quelles, et ne
montrent pas les quelques acides aminés qui peuvent être plus ou moins conservés de part et d'autre.
Il faut noter que toutes ces recherches sont effectuées sur l'ensemble des protéines de la base.
Mais l'aspect le plus intéressant de cet outil est qu'il autorise des recherches multicritères.
On peut en effet rechercher les protéines qui vérifient par exemple une certaine longueur et qui
possèdent un certain domaine conservé et présentant tel motif. La force de cet outil est de permettre
une recherche fine et aussi précise que l'idée du biologiste qui interroge la base.
La conception de cet outil a posé des difficultés dans l'automatisation de la construction des
requêtes, qui devaient être validées en SQL quelque soit le nombre de paramètres demandés pour la
recherche. De plus, tous les critères n'ont pas forcément la même syntaxe (motif, longueur, mot-
clé...), ce qui augmente la complexité de l'automatisation.
Concernant la recherche par mot-clé, on peut envisager d'effectuer des requêtes sur la base
avec plusieurs mots-clés. Des recherches portant sur plusieurs mots-clés reliés entre eux par des
opérateurs booléens pourraient se révéler fort riches en informations, en affinant encore une fois la
requête déjà complexe ayant servi à remplir la base.
4.3.3 Les outils
Pour exploiter les données de la base, le biologiste a besoin d'outils qui vont lui donner la
possibilité de « faire parler » toutes ces données, de donner du sens à ces informations.
Pour l'instant LEA-DB est enrichie de 4 outils, mais l'architecture du site permet d'en ajouter
aisément.
Master Pro BioInformatique Laurent CHATELAINPage 33

Université de Nantes Rapport de stage
Il est important de noter que ceux-ci ne s'appliquent qu'aux protéines de la sélection.
Le premier outil est une recherche de motifs : au moyen d'une expression régulière, le
programme recherche le motif passé en paramètre par l'utilisateur, et ce uniquement dans les
séquences des protéines précédemment choisies.
Le deuxième outil concerne la composition des protéines en acides aminés. En effet, comme
nous l'avons vu, les analyses POPPs ont révélé que certaines sur ou sous-représentations d'acides
aminés étaient caractéristiques d'un groupe donné de LEA. D'où la fonction qui calcule le
pourcentage de chaque acide aminé pour chaque protéine de la sélection.
Pour rendre la lecture plus facile et surtout plus parlante pour le biologiste, les acides aminés on été
classés en fonction de leurs propriétés physico-chimiques :
G : GlycineD : Aspartate = acidesE : Glutamate R : ArginineK : Lysine = basiquesA : AlanineL : Leucine = aliphatiquesI : IsoleucineV : ValineS : Sérine = hydroxylésT : ThréonineC : Cystéine = soufrésM : MéthionineF: PhénylalanineW : Tryptophane = aromatiquesY : TyrosineN : Asparagine = amidesQ : GlutamineP : Proline = imineH : Histidine
L'outil suivant consiste à calculer la masse molaire théorique de chaque protéine de la
sélection. Il s'agit pour cela de sommer les masses de chaque acide aminé constituant la protéine.
Mais il ne faut pas oublier de retirer la masse de chaque molécule d'eau éliminée lors de chaque
liaison peptidique entre les acides aminés. En effet, lors de la liaison entre deux acides aminés, la
réaction qui se produit entre le COOH d'un acide aminé et le NH2 de l'acide aminé suivant libère une
molécule d'eau qu'il faut donc retrancher à cette masse totale. Pour une protéine de n acides aminés
il existe donc (n-1) liaisons peptidiques:
masse protéine (n acides aminés) = ∑1 n (masse acide aminé) – (n-1)*(masse molécule d'eau)
Master Pro BioInformatique Laurent CHATELAINPage 34

Université de Nantes Rapport de stage
Mais l'outil le plus intéressant d'un point de vue personnel reste sans conteste le calcul du
point isoélectrique des protéines. En effet, cette partie du projet a fait appel à mes connaissances en
biochimie, que l'outil informatique a permis d'automatiser. Le point isoélectrique ou pH
isoélectrique (pI) est le pH où une molécule (ici une protéine ou un peptide) est sous sa forme
zwitterionique ou ion mixte, sa charge globale étant alors nulle.
L'algorithme consiste donc à parcourir les pH de 0 à 14 en recherchant celui pour lequel la
charge globale de la protéine est minimale. A un pH donné, chaque acide aminé possède une charge
globale particulière qui est la somme de toutes les charges portées par ses groupements ionisables
situés sur les chaînes latérales des acides aminés. Ainsi, en parcourant les pH avec un pas
suffisamment petit (0,01 unité ici), on trouve le pH pour lequel la charge est la plus proche de 0,
c'est à dire le pI.
On tient aussi compte des groupements NH2 et COOH terminaux dans ce calcul.
Le calcul du pI ne concerne donc qu'une partie des 20 acides aminés : D, E, C, Y, H, K, et R.
Notons que les valeurs de masses molaires comme celles des pK prises pour réaliser ces
calculs sont issues des tables de Wikipedia (http://fr.wikipedia.org/wiki/Acide_amin%C3%A9), car
elles semblaient être des valeurs moyennes entre les différentes autres tables consultées (EMBOSS,
DTASelect, Solomon).
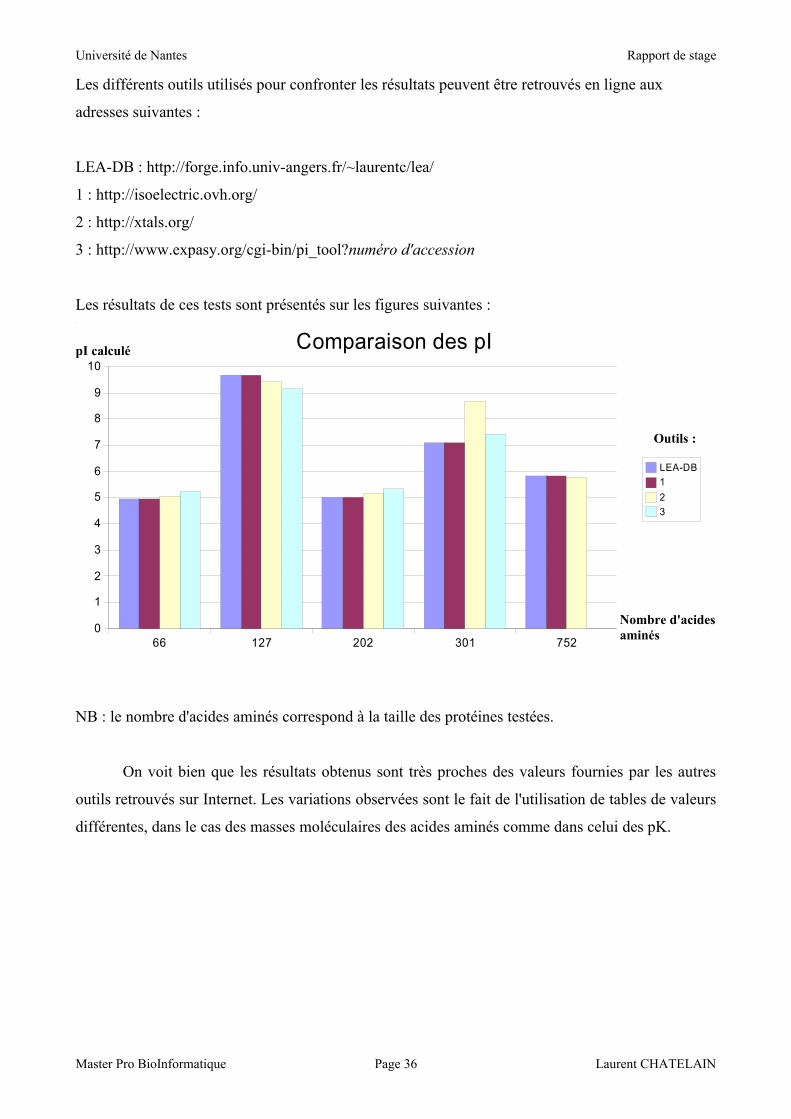
4.3.4 Comparaison des résultats avec d'autres outils sur Internet
Les outils de calcul de masse molaire et de pI ont ensuite fait l'objet de tests pour vérifier
que les algorithmes mis en place donnent des résultats satisfaisants.
Dès les premiers tests, les pI calculés par le programme étaient proches de ceux estimés par d'autres
outils sur Internet.
En revanche, en ce qui concerne les masses molaires, le programme annonçait toujours des
masses inférieures. Cette erreur me fit d'abord chercher du côté de l'algorithme, jusqu'à ce que je
pense au format même des données. En effet, il restait à la fin du traitement de la chaîne Fasta les
retours chariots, qui étaient comptés comme autant d'acides aminés, et qui de ce fait
correspondaient à autant de masses de molécules d'eau retranchées en trop. Depuis que ce problème
est résolu, les résultats sont proches des valeurs estimées par les autres outils.
Master Pro BioInformatique Laurent CHATELAINPage 35
Université de Nantes Rapport de stage
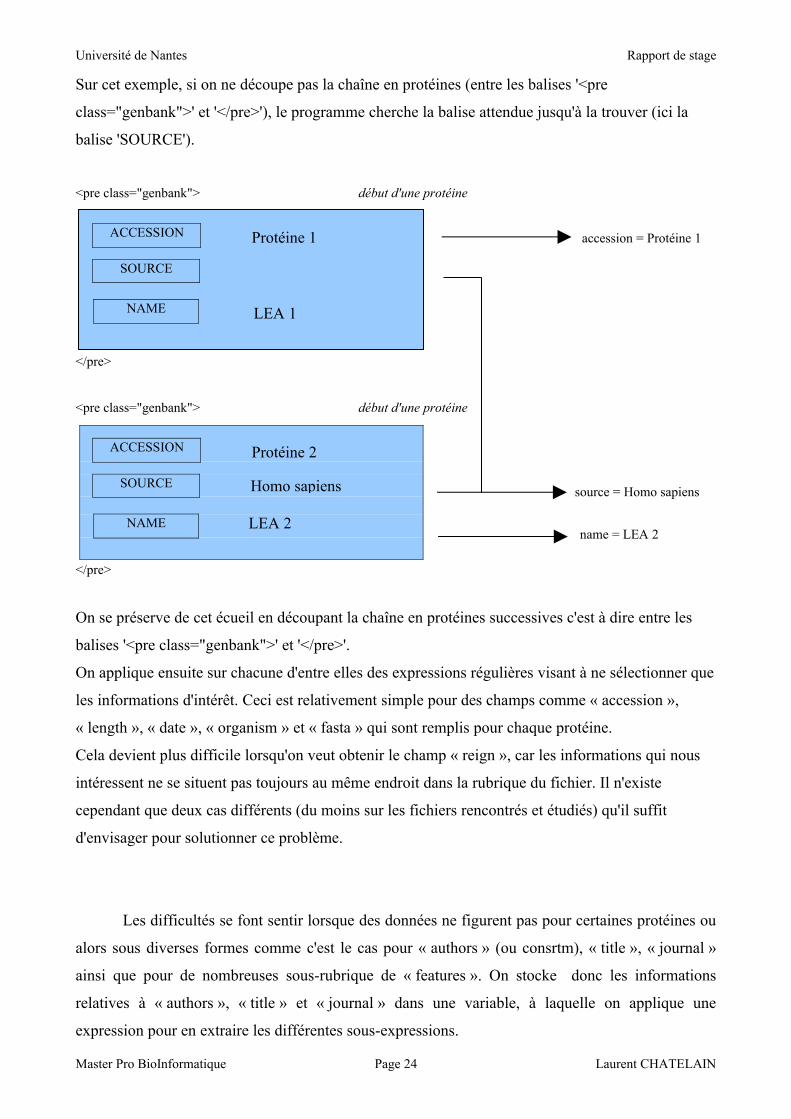
Les différents outils utilisés pour confronter les résultats peuvent être retrouvés en ligne aux
adresses suivantes :
LEA-DB : http://forge.info.univ-angers.fr/~laurentc/lea/
1 : http://isoelectric.ovh.org/
2 : http://xtals.org/
3 : http://www.expasy.org/cgi-bin/pi_tool?numéro d'accession
Les résultats de ces tests sont présentés sur les figures suivantes :
66 127 202 301 7520
1
2
3
4
5
6
7
8
9
10
Comparaison des pI
LEA-DB123
NB : le nombre d'acides aminés correspond à la taille des protéines testées.
On voit bien que les résultats obtenus sont très proches des valeurs fournies par les autres
outils retrouvés sur Internet. Les variations observées sont le fait de l'utilisation de tables de valeurs
différentes, dans le cas des masses moléculaires des acides aminés comme dans celui des pK.
Master Pro BioInformatique Laurent CHATELAINPage 36
Nombre d'acides aminés
pI calculé
Outils :
Université de Nantes Rapport de stage
66 127 202 301 7520
10000
20000
30000
40000
50000
60000
70000
80000
90000
Comparaison des masses molaire théoriques
LEA-DB123
4.3.5 Autres pages
Le site LEA-DB possède une page d'aide quant à l'utilisation des fonctionnalités proposées,
ainsi qu'une page pour contacter les membres de l'équipe ayant réalisé ce projet.
La page 'About LEA' propose un bref historique de la recherche sur les LEA, ainsi que la
bibliographie qui a été utilisée et des liens vers les articles en ligne.
Une fonctionnalité intéressante de ce site est de proposer un accès sécurisé à certaines
informations de la base. En effet, toutes les manipulations de données présentées jusqu'ici sont
accessibles à tous les utilisateurs, mais le site en prévoit certaines dont l'accès est restreint.
L'accès aux pages sensibles (erase.php, modif.php, suppress.php ...) qui permettent d'intervenir
directement sur les données contenues dans la base est donc soumis à la vérification des droits de
l'utilisateur.
Grâce au menu 'connect', l'utilisateur peut s'identifier par un login et mot de passe, et ainsi
utiliser d'autres fonctions réservées aux utilisateurs privilégiés. Ces utilisateurs privilégiés sont
gérés au niveau de la base de données, et pas au niveau du site.
L'utilisateur une fois connecté sous son compte peut ajouter une protéine en remplissant les champs
manuellement ou ajouter plusieurs protéines automatiquement, comme nous allons le voir.
Master Pro BioInformatique Laurent CHATELAINPage 37
Outils :
Nombre d'acides aminés
Masse calculée (g/mol)

Université de Nantes Rapport de stage
Il peut également modifier ou supprimer une protéine manuellement en navigant sur les
pages de consultation. Dans le cas de la modification, il est renvoyé vers une page contenant un
formulaire pré-rempli avec les informations de la protéine qu'il souhaite modifier. Nous avons pris
la décision de ne permettre la modification que des champs qui lui sont réservés ('remarks' et
'our_classif'), afin d'éviter de modifier des champs qui ne sont supposés être modifiés.
En ce qui concerne la suppression d'une protéine, une demande de confirmation écrite en Javascript
apparaît. Cette demande de confirmation assure une sécurité supplémentaire au biologiste. Elle a été
programmée en Javascript malgré notre décision d'éviter ce langage, car pour une validation aussi
simple, les interpréteurs donneront le même résultat.
Enfin, les dernières fonctionnalités réservées sont celles de remplissage et de vidage de la
base. La fonction "feed" dirige vers une page demandant le fichier html qui doit être utilisé pour
alimenter la base, puis lance le programme Perl d'extraction des données sur ce fichier.
La fonction 'erase' permet de vider les tables d'informations sur les protéines ('proteins' et
'fastas'), mais pas les autres, de manière à conserver une trace du travail effectué par le biologiste
(connecté en tant qu'utilisateur privilégié) (annotation du champ 'remarks' de la table 'notes').
Ces fonctions seront très utiles au biologiste. En effet il s'agit d'une base dédiée à la recherche, dont
la vocation est de manipuler des données sur des protéines, mais dont on sait d'une part que toutes
ne sont pas des LEA, et d'autre part qu'il manque certainement des LEA dans la base. Ainsi,
l'objectif est de travailler sur les données et de pouvoir vider puis ré-alimenter la base avec des
protéines issues d'une nouvelle requête qui nous semblera plus adaptée.
Pour l'instant ces informations de connexion sont fournies par l'administrateur à un
utilisateur intéressé, suite à un mail de contact par exemple (sur la page 'contiguïtés') mais on peut
envisager une inscription en ligne 'autonome', avec génération d'un login unique et d'un mot de
passe.
De plus, il existe à l'heure actuelle deux types d'utilisateurs. Mais l'accès a été prévu dans la
perspective d'en ajouter facilement de nouveaux. En effet, actuellement l'accès aux diverses
fonctions ou informations réservées est permis aux utilisateurs dont le login et mot de passe font
passer la variable de session 'droits' à 1.
On peut donc envisager de rajouter un champ 'droits' supplémentaire dans la table 'membres', qui
comporterait un entier. Ce nombre déterminerait le type d'utilisateur et donc les fonctionnalités
auxquelles il peut accéder. Il suffira ensuite de vérifier le champ 'droits' de l'utilisateur avant de
proposer ou non une fonctionnalité.
Master Pro BioInformatique Laurent CHATELAINPage 38

Université de Nantes Rapport de stage
5 Perspectives de recherches et améliorations possibles
L'objectif de l'UMR PMS est l'étude de la relation structure-fonction des LEA. Pour cela,
plusieurs extensions sont envisagées.
La première serait d'installer un outil d'alignements multiples en local : Multalin
(http://bioinfo.genopole-toulouse.prd.fr/multalin/multalin.html). Il s'agit d'un programme libre de
droits, efficace et dont l'interface est conviviale. L'intérêt d'un tel outil repose sur la possible mise
en évidence de motifs conservés, et ceci simplement grâce à l'option 'align' prévue sur les protéines
qui auront été préalablement sélectionnées.
A la différence de l'outil de recherche de motifs connus déjà proposé, cela permettrait de mettre en
évidence des motifs communs des protéines qu'on suppose avoir la même fonction. On cherche
donc ici des motifs inconnus qui seraient spécifiquement responsables d'une fonction protéique.
On peut ensuite envisager de rechercher des motifs connus. En effet, on prête à certaines
LEA des propriétés d'interaction avec l'ADN, ce qui nous suggère de rechercher des motifs
caractéristiques de facteurs de transcriptions. En première approche, l’outil de recherche de motifs
intégré au site semble tout à fait adapté.
De même on leur attribue un rôle protecteur de type HSP, ce qui nous indique qu'il serait intéressant
de rechercher des motifs de fixation de l'ATP, eux-mêmes présents sur ce type de protéines.
Effectuer des analyses syntaxiques puis sémantiques pourrait se révéler riche en
informations, en particulier en ce qui concerne la mise en évidence des fonctions assurées par les
LEA. Il est en effet déjà possible de réaliser des recherches syntaxiques basées sur des mots-clés, et
comme on l'a vu cela va permettre d'isoler des protéines associées à un 'thème' particulier.
Mais l'objectif est d'aller plus loin et de permettre des recherches sémantiques, de façon à
tirer du sens de ces données contenues dans la base. Pour cela il faut associer à chaque mot-clé les
différentes syntaxes que l'on est susceptible de rencontrer et qui possèdent le même sens.
Par exemple, pour les LEA du groupe 3 on peut retrouver 'group 3', 'LEA 3', LEA-3', et il s'agit
donc d'indiquer au programme de rechercher aussi ces termes lorsqu'on s'intéresse aux LEA de ce
groupe.
Master Pro BioInformatique Laurent CHATELAINPage 39

Université de Nantes Rapport de stage
Cela consiste en une sorte de lemmatisation : les mots (lemmes) d'une langue utilisent plusieurs
formes en fonction de leur genre (masculin ou féminin), leur nombre (un ou plusieurs), leur
personne (moi, toi, eux, ...), leur mode (indicatif, impératif, ...) donnant ainsi naissance à plusieurs
formes pour un même lemme.
La lemmatisation d'une forme d'un mot consiste à en prendre sa forme canonique, c'est à dire
l'infinitif pour un verbe, le masculin singulier pour les autres mots.
Ensuite, il serait donc possible de faire ressortir du sens de ces analyses. En effet une recherche du
type « DNA + binding + growth + factor + cell + division », prenant en plus compte les mots
dérivés grâce à la lemmatisation, indiquerait un rôle précis dans le cycle cellulaire au niveau du
noyau.
Parallèlement on peut effectuer des analyses statistiques sur la longueur des chaînes, la
composition en acides aminés, les propriétés physico-chimiques (profils d'hydrophobicité...). Ceci
est en partie permis par les outils intégrés à LEA-DB. Ces analyses permettraient de confronter les
résultats obtenus avec les règles déjà établies pour classer les LEA, et peut-être de faire émerger
certaines caractéristiques d'un groupe de protéines.
On peut enfin envisager d'utiliser la modélisation moléculaire. En effet, on pourrait utiliser
des logiciels libres de droit comme PyMol, Modeller pour prédire les structures des protéines.
Toutes les protéines testées seraient modélisées par le même logiciel, et on peut donc penser que les
résultats seraient homogènes. Cependant, un obstacle important demeure, car aucune vérification de
ces prédictions ne sera possible puisqu'on ne dispose d'aucune donnée sur la structure
tridimensionnelle des LEA.
En ce qui concerne l'aspect programmation du site, on pourrait envisager de faire évoluer
LEA-DB en programmation orientée objet car PHP le permet, mais cela ne nous semblait pas
adapté au début du projet. En revanche, la stratégie d’analyse des LEA s'est précisée au fil des
discussions et des réflexions. Maintenant que le plan se dessine et que le projet est plus avancé, il
semble que l'approche objet serait une bonne façon d'améliorer et de structurer le code.
On imagine très bien des objets de classe 'protéine', dont l'attribut 'accession 'serait ainsi
indissociable, et qui comporteraient des fonctions spécifiques comme les différents calculs effectués
sur les chaînes d'acides aminés, ce qui garantirait un code plus propre.
Master Pro BioInformatique Laurent CHATELAINPage 40

Université de Nantes Rapport de stage
Une autre amélioration possible serait de permettre à l'utilisateur de saisir une expression
régulière pour la recherche de motifs, ce qui mettrait en évidence des motifs dérivés de motifs
consensus qui ne l'auraient pas été par l'outil tel qu'il est proposé actuellement.
Master Pro BioInformatique Laurent CHATELAINPage 41

Université de Nantes Rapport de stage
Conclusion
Ces cinq mois de stage se sont révélés très positifs. Ce stage de fin de Master m'a apporté
plusieurs ouvertures.
Tout d'abord, j'ai pu appliquer mes connaissances sur les bases de données acquises par les
cours d'analyse des systèmes d'information et de SQL (Structure Query Language), mais également
en technologies Internet (PHP, XHTML, CSS) et en Perl.
Je suis également très satisfait de ce stage car j'ai trouvé vraiment enrichissant de travailler
à l'interface entre les biologistes et les informaticiens. En effet ce stage pratique m'a permis
d'apprendre beaucoup de choses en informatique, mais fut aussi l'occasion de nombreuses réflexions
sur l'aspect biologique du projet, et sur les possibilités qui s'offraient à nous pour le mener à bien.
Finalement, ce stage m'a permis d'affiner mes orientations professionnelles.
Concernant mes aspirations professionnelles, j'aimerais me tourner vers ce type de projets basés sur
la fouille de données, pour essayer de dégager du sens des informations biologiques disponibles sur
une problématique donnée.
Master Pro BioInformatique Laurent CHATELAINPage 42

Université de Nantes Rapport de stage
Bibliographie
Tolleter, D , Jaquinod M, Mangavel C, Passirani C, Saulnier P, Manon S, Teyssier E, Payet N,
Avelange-Macherel MH, Macherel D. . (2007) "Structure and Function of a Mitochondrial Late
Embryogenesis Abundant Protein Are Revealed by Desiccation" Plant Cell (May 25)
Tunnacliffe, A. & Wise, M.J. (2007) "The continuing conundrum of the LEA proteins"
Naturwissenschaften (May 4)
Abba', S. et al. (2006) "A dehydration-inducible gene in the truffle Tuber borchii identifies a novel
group of dehydrins" BMC Genomics 7, 39
Grelet, J. et al. (2005) "Identification in pea seed mitochondria of a late-embryogenesis abundant
protein able to protect enzymes from drying" Plant Physiol. 137, 157 - 167
Wise, M.J. (2003) "LEAping to conclusions: a computational reanalysis of late embryogenesis
abundant proteins and their possible roles" BMC Bioinformatics 4, 52
Wise M.J., Tunnacliffe A. (2004) POPP the question: What do LEA proteins do? Trends Plant Sci
9:13-17
Boudet J, Buitink J, Hoekstra FA, Rogniaux H, Larré C, Satour P, Leprince O (2006)
Comparative analysis of the heat stable proteome of radicles of Medicago truncatula seeds during
germination identifies late embryogenesis abundant proteins associated with desiccation tolerance.
Plant Physiol 140:1418,1436
Sites Internet :
http://fr.selfhtml.org/ (HTML, PHP, CSS, Perl)
http://www.info.univ-angers.fr/~gh/tuteurs/tutperl.htm (Perl)
http://css.alsacreations.com/ (CSS)
Master Pro BioInformatique Laurent CHATELAINPage 43
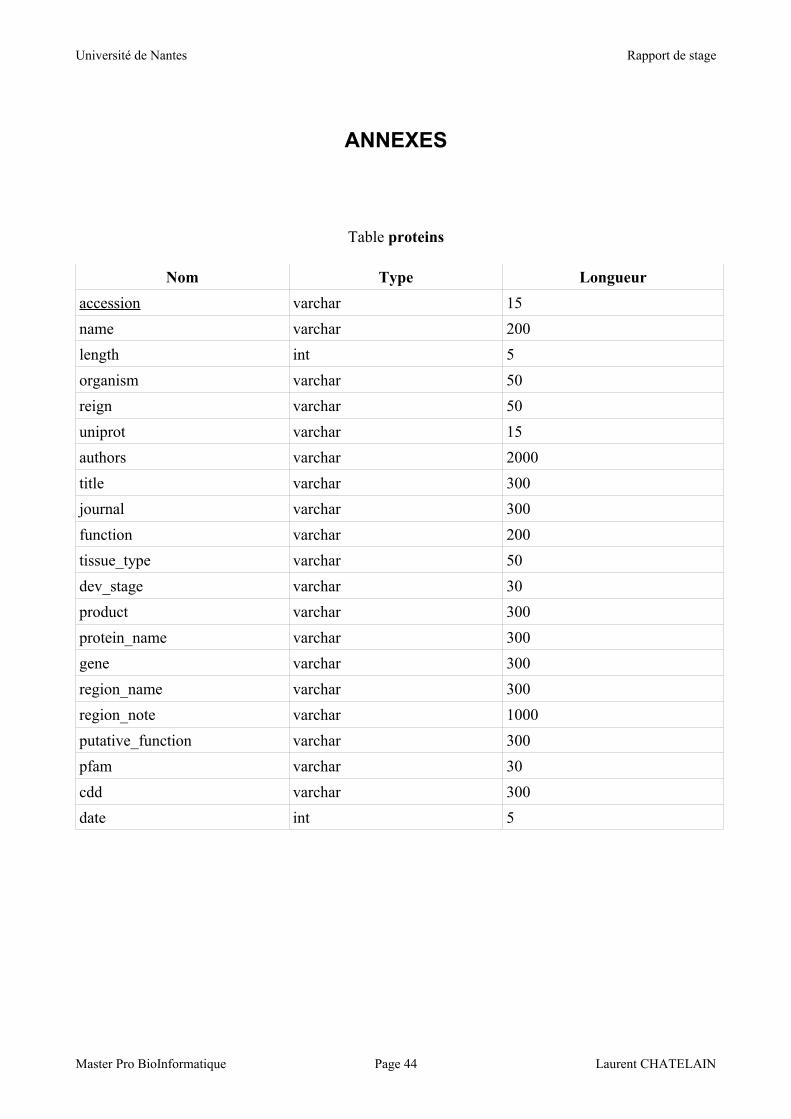
Université de Nantes Rapport de stage
ANNEXES
Table proteins
Nom Type Longueuraccession varchar 15name varchar 200length int 5organism varchar 50reign varchar 50uniprot varchar 15authors varchar 2000title varchar 300journal varchar 300function varchar 200tissue_type varchar 50dev_stage varchar 30product varchar 300protein_name varchar 300gene varchar 300region_name varchar 300region_note varchar 1000putative_function varchar 300pfam varchar 30cdd varchar 300date int 5
Master Pro BioInformatique Laurent CHATELAINPage 44
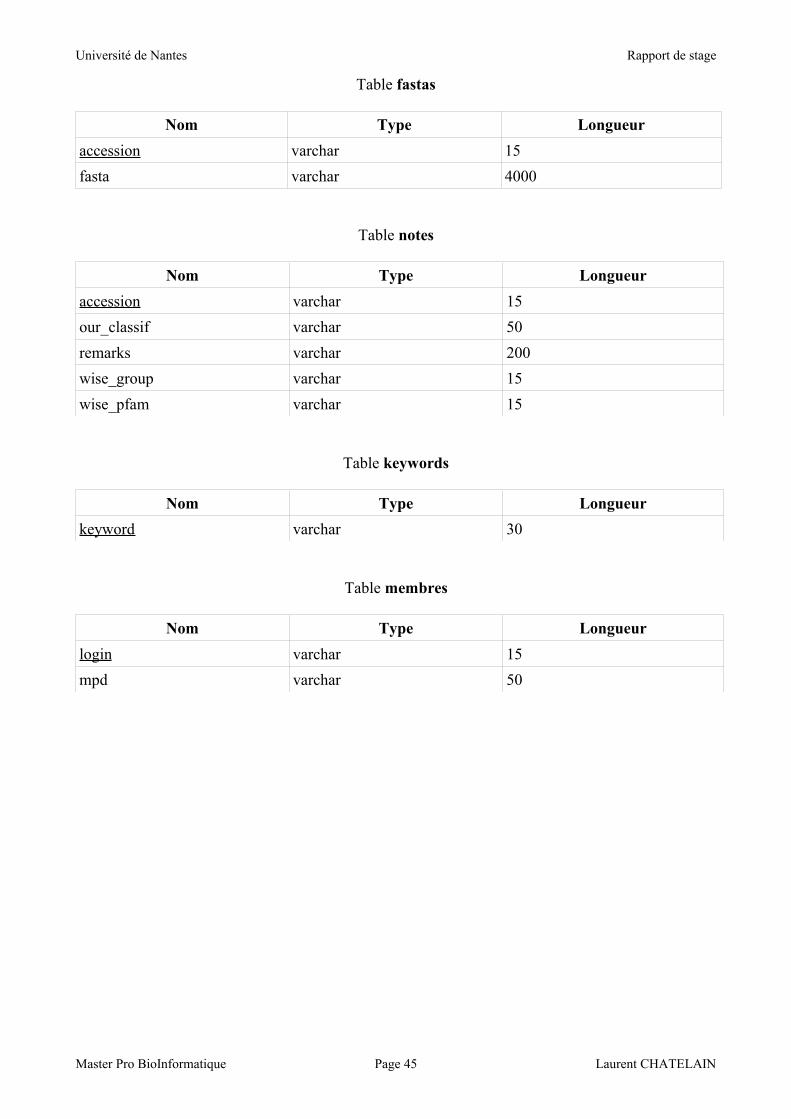
Université de Nantes Rapport de stage
Table fastas
Nom Type Longueuraccession varchar 15fasta varchar 4000
Table notes
Nom Type Longueuraccession varchar 15our_classif varchar 50remarks varchar 200wise_group varchar 15wise_pfam varchar 15
Table keywords
Nom Type Longueurkeyword varchar 30
Table membres
Nom Type Longueurlogin varchar 15mpd varchar 50
Master Pro BioInformatique Laurent CHATELAINPage 45
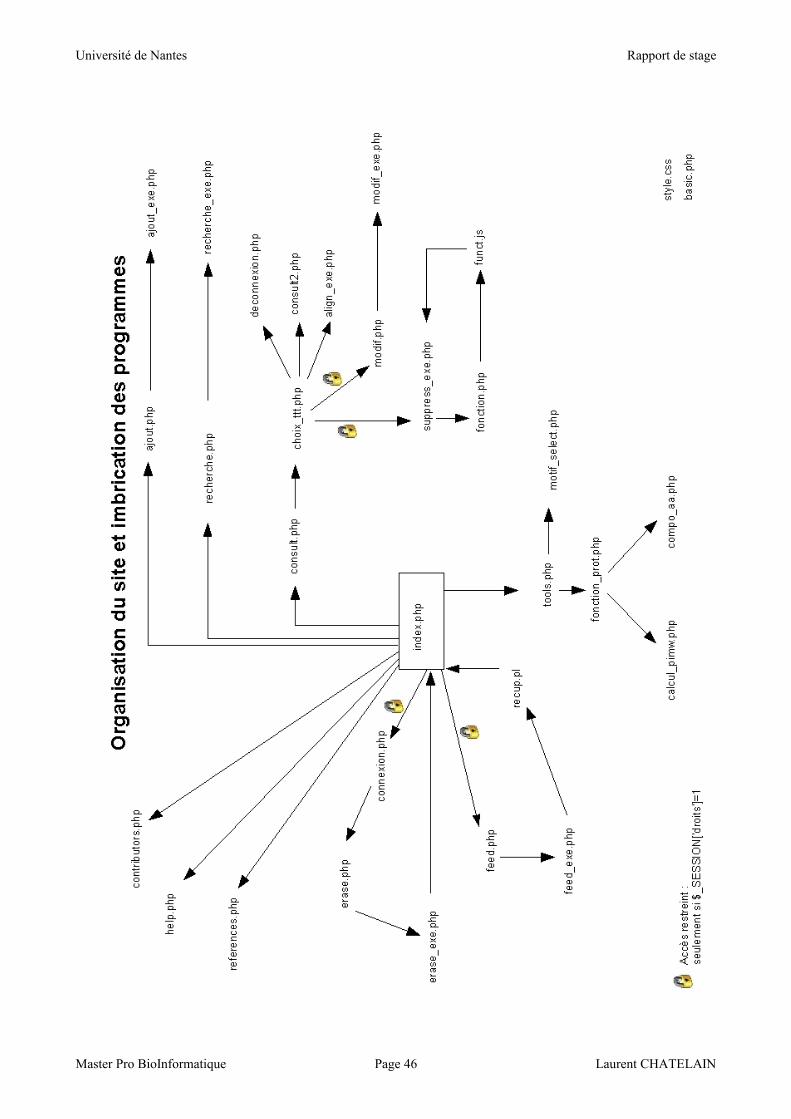
Université de Nantes Rapport de stage
Master Pro BioInformatique Laurent CHATELAINPage 46
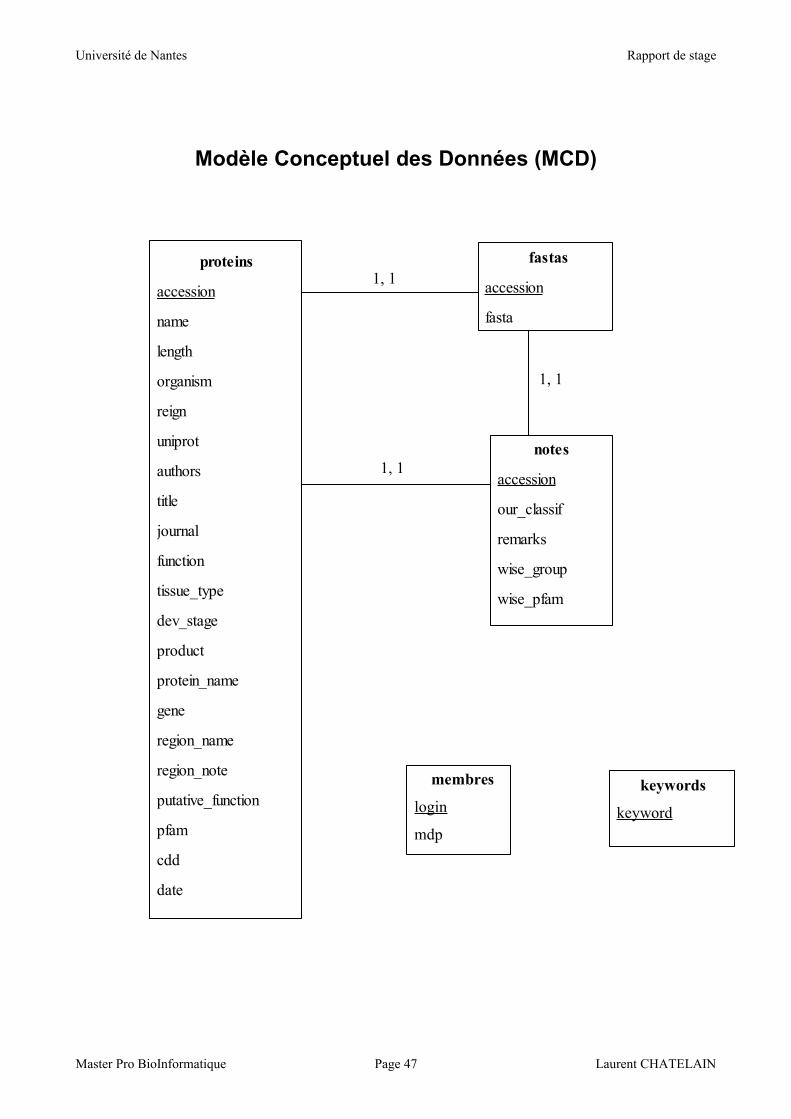
Université de Nantes Rapport de stage
Modèle Conceptuel des Données (MCD)
Master Pro BioInformatique Laurent CHATELAINPage 47
proteins
accession
name
length
organism
reign
uniprot
authors
title
journal
function
tissue_type
dev_stage
product
protein_name
gene
region_name
region_note
putative_function
pfam
cdd
date
fastas
accession
fasta
notes
accession
our_classif
remarks
wise_group
wise_pfam
membres
login
mdp
keywords
keyword
1, 1
1, 1
1, 1