Embed Size (px)
Citation preview

A
A
Cé
So
0h
nnales de pathologie (2014) 34, 124—129
Disponible en ligne sur
ScienceDirectwww.sciencedirect.com
RTICLE ORIGINAL
arcinomes sarcomatoïdes du poumon :tude rétrospective de 28 cas
arcomatoid carcinoma of the lung: Retrospective studyf 28 cases
Alia Zehania,∗, Aïda Ayadi-Kaddoura, Adel Marghli b,Hela Maamouria, Lamia Kassarc, Tarek Kilanib,Faouzi El Meznia
a Service d’anatomie et de cytologie pathologiques, hôpital Abderrahman Mami, Ariana,Tunisieb Service de chirurgie thoracique, hôpital Abderrahman Mami, Ariana, Tunisiec Service de dermatologie, hôpital Mahmoud Matri, Ariana, Tunisie
Accepté pour publication le 18 decembre 2013Disponible sur Internet le 19 mars 2014
MOTS CLÉSPoumon ;Carcinomesarcomatoïde ;Tumeur maligne ;Chirurgie ;Pronostic
RésuméIntroduction. — Le carcinome sarcomatoïde (CS) du poumon est une tumeur maligne rare,constitué d’un groupe hétérogène de tumeurs peu différenciées non à petites cellules quicomportent un contingent sarcomateux ou sarcomatoïde. La classification de l’OMS (2004)distingue dans ce groupe 5 sous-types histologiques qui représentent un continuum de diffé-renciation épithéliale et mésenchymateuse : le carcinome pléomorphe, le carcinome à cellulesgéantes, le carcinome à cellules fusiformes, le carcinosarcome et le blastome pulmonaire. Lediagnostic est anatomopathologique et nécessite un bon échantillonnage de la tumeur.Patients et méthodes. — Étude rétrospective de 28 CS du poumon diagnostiqués entre 1993 et2010 avec une analyse des aspects anatomocliniques.Résultats. — Notre série était constituée de 25 hommes et de 3 femmes avec un âge moyen de62,9 ans (48—75). La symptomatologie était dominée par des signes respiratoires. L’imageriemontrait une masse pulmonaire, envahissant la paroi et/ou la plèvre dans 5 cas. Le diagnosticétait dans tous les cas anatomopathologique. Ces 28 tumeurs se répartissaient en 19 carcinomespléomorphes (67,8 %), 4 carcinomes à cellules géantes (14,3 %), 1 carcinome à cellules fusi-formes (3,6 %) et 4 carcinosarcomes (14,3 %). Le traitement était chirurgical dans 27 cas. Lestraitements associés étaient une chimiothérapie néoadjuvante (3 cas) ou adjuvante (1 cas)et/ou une radiothérapie préopératoire (1 cas). Sept patients sont décédés. Les autres patientsétaient perdus de vue.Conclusion. — Les CS sont des tumeurs hautement agressives avec un potentiel métastatiqueimportant et un haut risque de récidive. Son pronostic est plus sombre que celui des autrescancers non à petites cellules.© 2013 Elsevier Masson SAS. Tous droits réservés.
∗ Auteur correspondant. Rue Mohamed Rached Béji, résidence Ichbilia n : 1, 2038 Ennasr 2, Tunis, Tunisie.Adresse e-mail : [email protected] (A. Zehani).
242-6498/$ — see front matter © 2013 Elsevier Masson SAS. Tous droits réservés.ttp://dx.doi.org/10.1016/j.annpat.2013.12.001

Carcinome sarcomatoïde du poumon 125
KEYWORDSLung;
Summaryd carted nIt repes arosarg of
wentre re
ulathe syulmocasehic
nty- or an 7 p
are
gnosi. All
Sarcomatoidcarcinoma;Malignant tumors;Surgery;Prognosis
Introduction. — Sarcomatoization as poorly differentiaor sarcoma-like elements.
differentiation. Five subtypgiant cell carcinoma, carcinand requires a good samplinPatients and methods. — Tbetween 1993 and 2010, weteristics.Results. — The patient pop62.9 years (48—75 years). Tging features showed a pdiagnosis was made in all
as below: into 19 pleomorpand 4 carcinosarcomas. Twewere neoadjuvant (3 cases)(5 cases). Deaths occurred iConclusion. — These tumorsrates of recurrence. Its pro© 2013 Elsevier Masson SAS
Introduction
Le carcinome sarcomatoïde du poumon est une tumeurmaligne rare, constitué d’un groupe hétérogène de tumeurspeu différenciées non à petites cellules qui comportent uncontingent sarcomateux ou sarcomatoïde, représentant aumoins 10 % de la prolifération [1]. La dernière classificationde l’OMS (2004) distingue dans ce groupe 5 sous-types histo-logiques qui représentent un continuum de différenciationépithéliale et mésenchymateuse : le carcinome pléomorphe,le carcinome à cellules géantes, le carcinome à cellules fusi-formes, le carcinosarcome et le blastome pulmonaire [2].Le diagnostic est anatomopathologique et nécessite un bonéchantillonnage de la tumeur. Ces tumeurs sont hautement
agressives avec un potentiel métastatique important et unhaut risque de récidive [3,4]. Le pronostic dépend essentiel-lement de la taille et de l’extension tumorale. Son pronosticest plus sombre que celui des autres cancers non à petitescellules, malgré une prise en charge pluridisciplinaire avecun taux de survie à 5 ans de 24 % [3].Cette série de 28 cas, associée à une revue de la lit-térature, a pour but de caractériser sur le plan clinique,morphologique et immunohistochimique ce type de tumeuret de dégager des implications thérapeutiques et pronos-tiques.
Patients et méthodes
Patients
Les dossiers de 28 cas de carcinomes sarcomatoïdes dupoumon diagnostiqués et opérés dans notre institutiondu 1er janvier 1993 au 31 décembre 2010 ont été revus àl’occasion de cette étude.
Méthodes
Tous les patients ont eu un examen clinique complet, uneradiographie du thorax, une fibroscopie bronchique et une
cinoma (SC) of the lung is defined by the World Health Organi-on-small cell carcinoma that contains a component of sarcomaresents an overall continuum of epithelial and mesenchymal
e recognized: pleomorphic carcinoma, spindle cell carcinoma,coma, and pulmonary blastoma. The diagnosis is pathological
the tumor.y-eight cases of primary sarcomatoid carcinoma, diagnosedviewed retrospectively, noting the clinicopathological charac-
ion consisted of 25 males and 3 females with mean age ofmptomatology was dominated by respiratory symptoms. Ima-nary mass invading pleura or thoracic wall in 5 cases. Thes on histological examination. These 28 tumors were dividedcarcinomas, 4 giant cell carcinomas, 1 spindle cell carcinomaseven tumors were treated surgically. Associated treatmentsdjuvant chemotherapy (1 case) and preoperative radiotherapyatients. Twenty-two patients were lost to follow up.frequently symptomatic, are locally advanced, and have highers is worse than that of other non-small cell lung cancer.rights reserved.
tomodensitométrie (TDM) thoracique. Dans le cadre du biland’extension, une TDM abdominalo-pelvienne et une TDMcérébrale ont été pratiquées dans tous les cas. Pour étu-dier le siège exact des tumeurs, nous nous sommes basés surles données de l’imagerie. La taille tumorale a été appré-ciée à l’examen macroscopique pour les tumeurs opéréeset sur l’imagerie pour le cas qui n’a pas été réséqué. Lastadification de la tumeur a été faite selon la classificationTNM 2009. Le diagnostic était dans tous les cas anatomo-pathologique, porté sur une pièce d’exérèse (27 cas) ou surune biopsie chirurgicale (1 cas). La classification de réfé-rence a été celle de l’OMS (2004). Les cas où le diagnosticde carcinome sarcomatoïde a été évoqué sur des biopsiesbronchiques ou transpariétales n’ont pas été inclus dans
cette étude. Tous nos prélèvements ont été fixés au formolpuis inclus en paraffine. Ils ont été coupés à 4 microns puiscolorés à l’hématoxyline-éosine. Les coupes histologiquesont été examinées au microscope optique conventionnel.Une étude immunohistochimique a été pratiquée dans tousles cas. Les coupes tissulaires de 3 à 4 microns d’épaisseuront été confectionnées et montées sur des lames en verresialinisées. Un panel d’anticorps a été utilisé associant :PS100, EMA, desmine, actine, vimentine, pancytokératinetype AE1-AE3 et TTF1. La technique immunohistochimiqueutilisée était celle de la streptavidine-biotine complexe.Nous avons procédé, au cours de cette étude, à uneanalyse détaillée de notre série en fonction de l’âge, dusexe, des circonstances de découverte, de la présentationradiologique, de l’aspect macroscopique et histologique, dutraitement et des différentes modalités évolutives.
Résultats
Épidémiologie et clinique
L’âge moyen de nos patients était de 62,9 ans avec desextrêmes allant de 48 à 75 ans et un pic de fréquence entre60 et 70 ans. Notre série était constituée de 25 hommes et
1 A. Zehani et al.
3dl9tpgamlsdDfitc
E
Ldbmddérptehhd(e(eée
T
LuCt(t
É
LcI4cteéccgcàcc
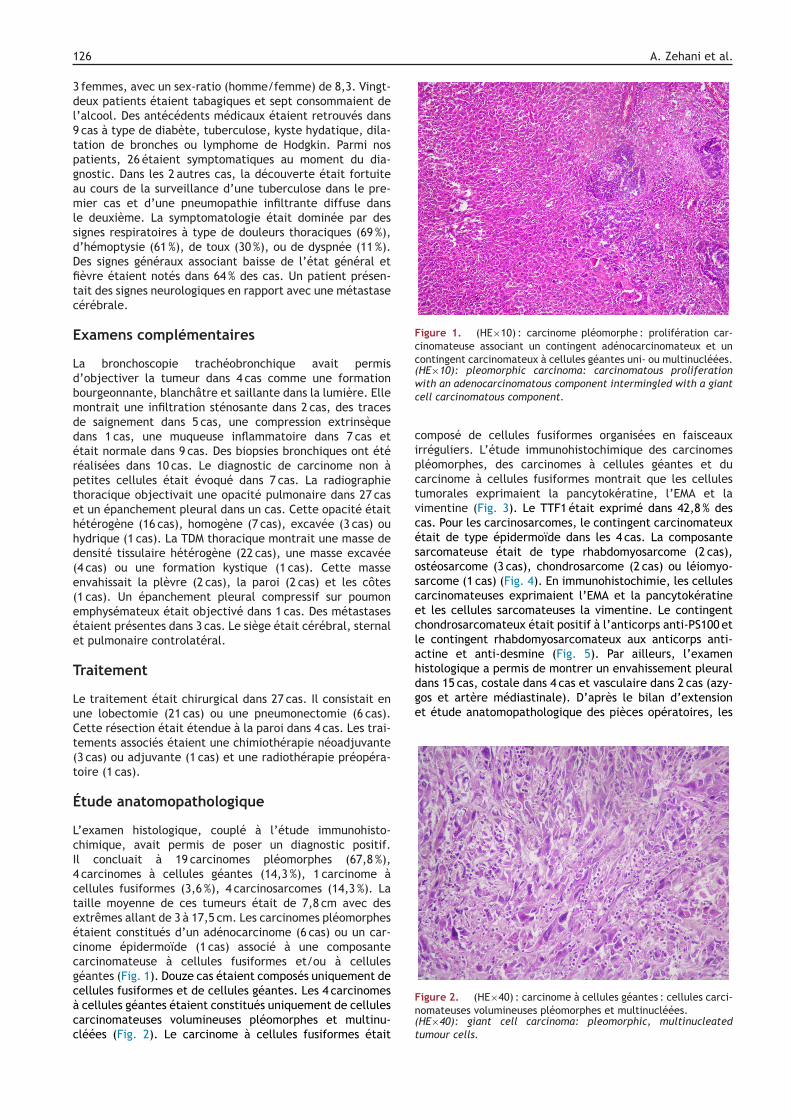
Fcc(HE×10): pleomorphic carcinoma: carcinomatous proliferationwith an adenocarcinomatous component intermingled with a giantcell carcinomatous component.
composé de cellules fusiformes organisées en faisceauxirréguliers. L’étude immunohistochimique des carcinomespléomorphes, des carcinomes à cellules géantes et ducarcinome à cellules fusiformes montrait que les cellulestumorales exprimaient la pancytokératine, l’EMA et lavimentine (Fig. 3). Le TTF1 était exprimé dans 42,8 % descas. Pour les carcinosarcomes, le contingent carcinomateuxétait de type épidermoïde dans les 4 cas. La composantesarcomateuse était de type rhabdomyosarcome (2 cas),ostéosarcome (3 cas), chondrosarcome (2 cas) ou léiomyo-sarcome (1 cas) (Fig. 4). En immunohistochimie, les cellulescarcinomateuses exprimaient l’EMA et la pancytokératineet les cellules sarcomateuses la vimentine. Le contingentchondrosarcomateux était positif à l’anticorps anti-PS100 etle contingent rhabdomyosarcomateux aux anticorps anti-actine et anti-desmine (Fig. 5). Par ailleurs, l’examenhistologique a permis de montrer un envahissement pleural
26
femmes, avec un sex-ratio (homme/femme) de 8,3. Vingt-eux patients étaient tabagiques et sept consommaient de’alcool. Des antécédents médicaux étaient retrouvés dans
cas à type de diabète, tuberculose, kyste hydatique, dila-ation de bronches ou lymphome de Hodgkin. Parmi nosatients, 26 étaient symptomatiques au moment du dia-nostic. Dans les 2 autres cas, la découverte était fortuiteu cours de la surveillance d’une tuberculose dans le pre-ier cas et d’une pneumopathie infiltrante diffuse dans
e deuxième. La symptomatologie était dominée par designes respiratoires à type de douleurs thoraciques (69 %),’hémoptysie (61 %), de toux (30 %), ou de dyspnée (11 %).es signes généraux associant baisse de l’état général etèvre étaient notés dans 64 % des cas. Un patient présen-ait des signes neurologiques en rapport avec une métastaseérébrale.
xamens complémentaires
a bronchoscopie trachéobronchique avait permis’objectiver la tumeur dans 4 cas comme une formationourgeonnante, blanchâtre et saillante dans la lumière. Elleontrait une infiltration sténosante dans 2 cas, des tracese saignement dans 5 cas, une compression extrinsèqueans 1 cas, une muqueuse inflammatoire dans 7 cas ettait normale dans 9 cas. Des biopsies bronchiques ont étééalisées dans 10 cas. Le diagnostic de carcinome non àetites cellules était évoqué dans 7 cas. La radiographiehoracique objectivait une opacité pulmonaire dans 27 cast un épanchement pleural dans un cas. Cette opacité étaitétérogène (16 cas), homogène (7 cas), excavée (3 cas) ouydrique (1 cas). La TDM thoracique montrait une masse deensité tissulaire hétérogène (22 cas), une masse excavée4 cas) ou une formation kystique (1 cas). Cette massenvahissait la plèvre (2 cas), la paroi (2 cas) et les côtes1 cas). Un épanchement pleural compressif sur poumonmphysémateux était objectivé dans 1 cas. Des métastasestaient présentes dans 3 cas. Le siège était cérébral, sternalt pulmonaire controlatéral.
raitement
e traitement était chirurgical dans 27 cas. Il consistait enne lobectomie (21 cas) ou une pneumonectomie (6 cas).ette résection était étendue à la paroi dans 4 cas. Les trai-ements associés étaient une chimiothérapie néoadjuvante3 cas) ou adjuvante (1 cas) et une radiothérapie préopéra-oire (1 cas).
tude anatomopathologique
’examen histologique, couplé à l’étude immunohisto-himique, avait permis de poser un diagnostic positif.l concluait à 19 carcinomes pléomorphes (67,8 %),
carcinomes à cellules géantes (14,3 %), 1 carcinome àellules fusiformes (3,6 %), 4 carcinosarcomes (14,3 %). Laaille moyenne de ces tumeurs était de 7,8 cm avec desxtrêmes allant de 3 à 17,5 cm. Les carcinomes pléomorphestaient constitués d’un adénocarcinome (6 cas) ou un car-inome épidermoïde (1 cas) associé à une composantearcinomateuse à cellules fusiformes et/ou à celluleséantes (Fig. 1). Douze cas étaient composés uniquement deellules fusiformes et de cellules géantes. Les 4 carcinomes
cellules géantes étaient constitués uniquement de cellulesarcinomateuses volumineuses pléomorphes et multinu-léées (Fig. 2). Le carcinome à cellules fusiformes était
dge
Fn(t
igure 1. (HE×10) : carcinome pléomorphe : prolifération car-inomateuse associant un contingent adénocarcinomateux et unontingent carcinomateux à cellules géantes uni- ou multinucléées.
ans 15 cas, costale dans 4 cas et vasculaire dans 2 cas (azy-os et artère médiastinale). D’après le bilan d’extensiont étude anatomopathologique des pièces opératoires, les
igure 2. (HE×40) : carcinome à cellules géantes : cellules carci-omateuses volumineuses pléomorphes et multinucléées.HE×40): giant cell carcinoma: pleomorphic, multinucleatedumour cells.
Carcinome sarcomatoïde du poumon
Figure 3. (IHC×40) : immunohistochimie (carcinomepléomorphe) : expression intense de la pancytokératine detype AE1/AE3 au niveau du contingent glandulaire et modérée auniveau du contingent fuso-cellulaire.(IHC×40): immunochemistry (pleomorphic carcinoma): intenseexpression of AE1/AE3 in the glandular component and moderateexpression in the spindle cell component.
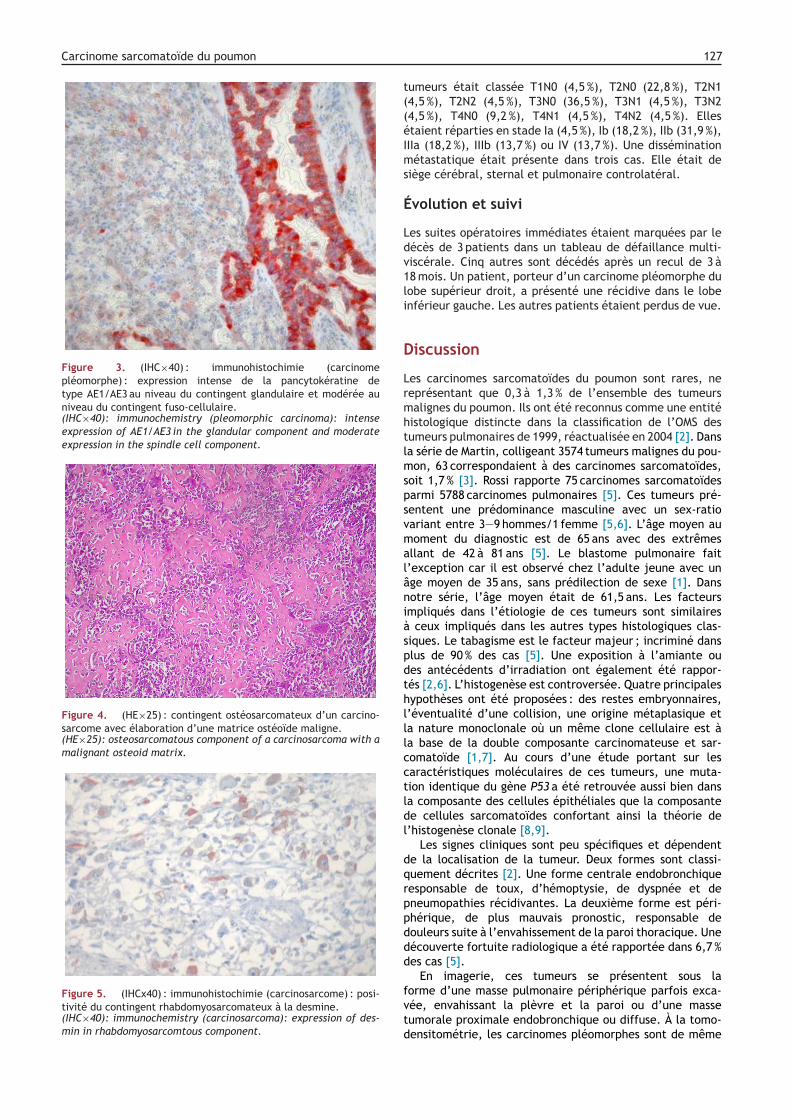
Figure 4. (HE×25) : contingent ostéosarcomateux d’un carcino-sarcome avec élaboration d’une matrice ostéoïde maligne.(HE×25): osteosarcomatous component of a carcinosarcoma with amalignant osteoid matrix.
Figure 5. (IHCx40) : immunohistochimie (carcinosarcome) : posi-tivité du contingent rhabdomyosarcomateux à la desmine.(IHC×40): immunochemistry (carcinosarcoma): expression of des-min in rhabdomyosarcomtous component.
127
tumeurs était classée T1N0 (4,5 %), T2N0 (22,8 %), T2N1(4,5 %), T2N2 (4,5 %), T3N0 (36,5 %), T3N1 (4,5 %), T3N2(4,5 %), T4N0 (9,2 %), T4N1 (4,5 %), T4N2 (4,5 %). Ellesétaient réparties en stade Ia (4,5 %), Ib (18,2 %), IIb (31,9 %),IIIa (18,2 %), IIIb (13,7 %) ou IV (13,7 %). Une disséminationmétastatique était présente dans trois cas. Elle était desiège cérébral, sternal et pulmonaire controlatéral.
Évolution et suivi
Les suites opératoires immédiates étaient marquées par ledécès de 3 patients dans un tableau de défaillance multi-viscérale. Cinq autres sont décédés après un recul de 3 à18 mois. Un patient, porteur d’un carcinome pléomorphe dulobe supérieur droit, a présenté une récidive dans le lobeinférieur gauche. Les autres patients étaient perdus de vue.
Discussion
Les carcinomes sarcomatoïdes du poumon sont rares, nereprésentant que 0,3 à 1,3 % de l’ensemble des tumeursmalignes du poumon. Ils ont été reconnus comme une entitéhistologique distincte dans la classification de l’OMS destumeurs pulmonaires de 1999, réactualisée en 2004 [2]. Dansla série de Martin, colligeant 3574 tumeurs malignes du pou-mon, 63 correspondaient à des carcinomes sarcomatoïdes,soit 1,7 % [3]. Rossi rapporte 75 carcinomes sarcomatoïdesparmi 5788 carcinomes pulmonaires [5]. Ces tumeurs pré-sentent une prédominance masculine avec un sex-ratiovariant entre 3—9 hommes/1 femme [5,6]. L’âge moyen aumoment du diagnostic est de 65 ans avec des extrêmesallant de 42 à 81 ans [5]. Le blastome pulmonaire faitl’exception car il est observé chez l’adulte jeune avec unâge moyen de 35 ans, sans prédilection de sexe [1]. Dansnotre série, l’âge moyen était de 61,5 ans. Les facteursimpliqués dans l’étiologie de ces tumeurs sont similairesà ceux impliqués dans les autres types histologiques clas-siques. Le tabagisme est le facteur majeur ; incriminé dansplus de 90 % des cas [5]. Une exposition à l’amiante oudes antécédents d’irradiation ont également été rappor-
tés [2,6]. L’histogenèse est controversée. Quatre principaleshypothèses ont été proposées : des restes embryonnaires,l’éventualité d’une collision, une origine métaplasique etla nature monoclonale où un même clone cellulaire est àla base de la double composante carcinomateuse et sar-comatoïde [1,7]. Au cours d’une étude portant sur lescaractéristiques moléculaires de ces tumeurs, une muta-tion identique du gène P53 a été retrouvée aussi bien dansla composante des cellules épithéliales que la composantede cellules sarcomatoïdes confortant ainsi la théorie del’histogenèse clonale [8,9].Les signes cliniques sont peu spécifiques et dépendentde la localisation de la tumeur. Deux formes sont classi-quement décrites [2]. Une forme centrale endobronchiqueresponsable de toux, d’hémoptysie, de dyspnée et depneumopathies récidivantes. La deuxième forme est péri-phérique, de plus mauvais pronostic, responsable dedouleurs suite à l’envahissement de la paroi thoracique. Unedécouverte fortuite radiologique a été rapportée dans 6,7 %des cas [5].
En imagerie, ces tumeurs se présentent sous laforme d’une masse pulmonaire périphérique parfois exca-vée, envahissant la plèvre et la paroi ou d’une massetumorale proximale endobronchique ou diffuse. À la tomo-densitométrie, les carcinomes pléomorphes sont de même

1
ds[eslipv
vgllltLLpedtcuàgufnLdp[qpcotààcisclRcdelCcLddsftclvtpsls
cLctmldllavpttadéddtdmrts[hpqeptlp
epclct
28
ensité que le muscle de voisinage et présentent un rehaus-ement périphérique après injection du produit de contraste10]. L’envahissement de la paroi thoracique ou de la plèvrest objectivé dans la moitié des cas. Les carcinosarcomese présentent comme une masse volumineuse, nécrosée,ocalement invasive avec un rehaussement hétérogène aprèsnjection du produit de contraste [11]. Pour les blastomesulmonaires, l’aspect est généralement celui d’une masseolumineuse, bien limitée, périphérique [12].
Le diagnostic de certitude est anatomopathologique,alidé par une étude immunohistochimique. Le dia-nostic sur des biopsies est difficile et incertain car’échantillonnage est insuffisant. Seul un examen complet dea pièce de résection permet le typage précis, en raison de’hétérogénéité histologique. Sur le plan macroscopique, lesumeurs périphériques sont bien limitées, jaune-grisâtres.es lobes supérieurs représentent le siège de prédilection.es lésions endobronchiques sont polypoïdes et souventédiculées, d’aspect blanc-grisâtre. La taille tumorale varientre 2 et 18 cm avec une taille moyenne de 7 cm. Des foyerse nécrose et d’hémorragie sont souvent observés [1]. His-ologiquement, le carcinome pléomorphe associe un adéno-arcinome (45 %), un carcinome à grandes cellules (25 %),n carcinome épidermoïde (8 %) ou un carcinoïde atypique
un contingent fait de cellules fusiformes et/ou de celluleséantes représentant au moins 10 % de la prolifération oun carcinome constitué uniquement de cellules géantes etusiformes (22 %) [5,13]. Les cellules fusiformes sont orga-isées en faisceaux, réalisant parfois un aspect storiforme.e stroma est fibreux ou myxoïde. Les cellules géantes sontiscohésives, uni- ou multinucléées, à cytoplasme éosino-hile dense, montrant souvent des images d’empéripolèse1,2,7,14]. Les emboles vasculaires et la nécrose sont fré-uents. Le carcinome à cellules fusiformes est un carcinomeeu différencié non à petites cellules extrêmement rare,omportant seulement un contingent de cellules fusiformesrganisées en faisceaux irréguliers. Un infiltrat inflamma-oire lympho-plasmocytaire peut être observé. Le carcinome
cellules géantes est un carcinome peu différencié non petites cellules comportant seulement un contingent deellules géantes pléomorphes, mono ou multinucléées. Des
mages d’empéripolèse et un infiltrat inflammatoire denseont fréquents. L’étude immunohistochimique montre uneoexpression des cellules tumorales aux marqueurs épithé-iaux et à la vimentine [15]. Dans la série rapportée parossi, la CK7 et le TTF1 sont exprimés dans le contingentarcinomateux dans respectivement 76 % et 58 % des cas,ans la composante sarcomatoïde dans 62 % et 43 % des cast dans les carcinomes constitués exclusivement de cel-ules géantes et/ou fusiformes dans 70 % et 55 % des cas. LaK20 est toujours négative. Le carcinosarcome associe deuxontingents tumoraux carcinomateux et sarcomateux [16].e contingent carcinomateux est le plus souvent de type épi-ermoïde (65 %) parfois de type adénocarcinome (25 %) oue type carcinome à grandes cellules (10 %). Le contingentarcomateux est constitué d’une prolifération de cellulesusiformes, avec par endroits une différentiation vers unissu spécifique hétérologue à type d’ostéosarcome, dehondrosarcome et/ou de rhabdomyosarcome. Cependant,a notion de carcinosarcome est très largement contro-ersée et pour un grand nombre d’experts en pathologiehoracique, ces tumeurs seraient des carcinomes dont lehénotype mime certains sarcomes ; une recherche exten-ive d’expression de cytokératine doit permettre de prouvera nature épithéliale de ces tumeurs. Par ailleurs, la pré-ence d’un stroma chondroïde ne fait pas d’un carcinome unldcu1Cdc
mgudr
tdtttdt[ed
A. Zehani et al.
arcinosarcome avec différenciation chondrosarcomateuse.’immunohistochimie montre la positivité du contingentarcinomateux à la CK. La composante chondrosarcoma-euse est positive à la PS 100 et la composante rhabdo-yosarcomateuse est positive aux marqueurs muscu-
aires. L’une des explications possibles de l’associationu carcinome épidermoïde avec le carcinosarcome et de’adénocarcinome avec le carcinome pléomorphe seraita tendance des carcinosarcomes à naître dans les voiesériennes principales et des carcinomes pléomorphes à sur-enir dans le poumon périphérique. Le blastome est lelus rare des carcinomes sarcomatoïdes ; défini comme uneumeur biphasique dont les 2 composantes mésenchyma-euse et épithéliale sont malignes et immatures ressemblantu poumon fœtal à 10—16 semaines de gestation, au stadee développement pseudo-glandulaire. La composantepithéliale ressemble à un adénocarcinome fœtal bienifférencié, réalisant souvent un aspect « endométrioïde »’étendue variable. Le stroma embryonnaire, de type blas-émateux, est fait de petites cellules ovales ou fusiformes,ispersées dans une substance myxoïde. Il peut renfer-er des foyers d’ostéosarcome, de chondrosarcome ou de
habdomyosarcome. En immunohistochimie, les cellules épi-héliales expriment intensément la CK et l’ACE. Les cellulestromales expriment la vimentine et l’actine muscle lisse1,2]. Ces tumeurs sont caractérisées par une extrêmeétérogénéité et une grande variabilité histologique ; ellesrêtent ainsi la confusion avec d’autres tumeurs tellesue les sarcomes en particulier l’histiocytofibrome malint le fibrosarcome, les carcinomes métastatiques ou desrocessus inflammatoires tels que la tumeur myofibroblas-ique inflammatoire [1,6,17]. Un échantillonnage large dea tumeur et le recours à une étude immunohistochimiqueermet de rectifier le diagnostic.
L’avancée de la compréhension des anomalies génétiquest moléculaires impliquées dans le carcinome broncho-ulmonaire a permis le développement de nouvelles molé-ules ciblant des protéines spécifiques du cancer. En effet,’utilisation de thérapies ciblées anti-EGFR dans les car-inomes à petites cellules apparaît comme une approchehérapeutique prometteuse. Cependant, la connaissance de
a biologie des CS est limitée et l’efficacité de ces thérapiesemeure inconnue. Dans la série rapportée par Lee et al.,olligeant 61 cas de carcinomes pléomorphes du poumon,ne mutation somatique de l’EGFR a été retrouvée dans9,6 %, une mutation du KRAS dans 9,8 % et une mutation de-KIT dans 4,9 % [18]. Italiano et al. suggèrent que la plupartes patients porteurs d’un CS ne peuvent pas bénéficier deette thérapie ciblée [19].Le traitement est chirurgical. Certains auteurs recom-andent d’opérer seulement les tumeurs sans métastases
anglionnaires [20]. Les traitements adjuvants sont parfoistilisés mais sans réelle efficacité [21]. Ils sont indiquésevant des tumeurs volumineuses envahissant la paroi tho-acique et avec des métastases ganglionnaires.
Le pronostic est sombre et l’évolution est plus péjora-ive que les autres carcinomes non micro-cellulaires [2]. Ilépend essentiellement de la taille et de l’extension de laumeur. D’autres facteurs pronostiques ont été rapportésels que les métastases ganglionnaires, les emboles lympha-iques et l’étendue de la nécrose [7]. Le caractère agressife ces tumeurs serait attribuable aux éléments sarcoma-eux qui permettent de développer une grande angiogenèse2]. La médiane de survie est de 31 mois pour un stade It de 9 mois pour un stade III. Le temps moyen de réci-ive est de 11 mois. Les métastases les plus fréquentes sont

[
[
[
[
[
[
tations different from ordinary nonsmall cell carcinoma. Lung
Carcinome sarcomatoïde du poumon
ganglionnaires, médiastinales, pulmonaires, cérébrales etcutanées [15,22]. Les décès surviennent dans 65,3 % aprèsun suivi variant entre 1 et 146 mois [5].
Déclaration d’intérêts
Les auteurs déclarent ne pas avoir de conflits d’intérêts enrelation avec cet article.
Références
[1] Franks TJ, Galvin JR. Sarcomatoid carcinoma of the lung. Histo-logic criteria and common lesions in the differential diagnosis.Arch Pathol Lab Med 2010;134:49—54.
[2] Corrin B, Chang YL, Rossi G, Koss MN, Geisinger K, Wick MR,et al. Sarcomatoid carcinoma. In: Travis WD, Brambilla E,
Müller-Hermelink HK, Harris CC, editors. World Health Orga-nisation Classification of Tumours. Pathology and Genetics ofTumours of the Lung, Pleura, Thymus and Heart. Lyon: IARCPress; 2004. p. 53—8.[3] Martin LW, Correa AM, Ordonez NG, Roth JA, Swisher SG, Vapor-ciyan AA, et al. Sarcomatoid carcinoma of the lung: a predictorof poor prognosis. Ann Thorac Surg 2007;84:973—80.
[4] Pelosi G, Sonzogni A, De Pas T, Galetta D, Veronesi G, SpaggiariL, et al. Review article: pulmonary sarcomatoid carcinomas: apractical overview. Int J Surg Pathol 2010;18:103—20.
[5] Rossi G, Cavazza A, Sturm N, et al. Pulmonary carcinomas withpleomorphic, sarcomatoid, or sarcomatous elements: a clini-copathologic and immunohistochemical study of 75 cases. AmJ Surg Pathol 2003;27:311—24.
[6] Nakajima M, Kasai T, Hashimoto H, et al. Sarcomatoid carci-noma of the lung: a clinicopathologic study of 37 cases. Cancer1999;86:608—16.
[7] Mochizuki T, Ishii G, Nagai K, Yoshida J, Nishimura M, MizunoT, et al. Pleomorphic carcinoma of the lung. Clinicopathologiccharacteristics of 70 cases. Am J Surg Pathol 2008;32:1727—35.
[8] Thompson L, Chang B, Barsky SH. Monoclonal origins ofmalignant mixed tumors (carcinosarcomas). Evidence for adivergent histogenesis. Am J Surg Pathol 1996;20:277—85.
[9] Holst VA, Finkelstein S, Colby TV, Myers JL, Yousem SA. p53 andK-ras mutational genotyping in pulmonary carcinosarcoma,spindle cell carcinoma, and pulmonary blastoma: implicationsfor histogenesis. Am J Surg Pathol 1997;21:801—11.
[
[
[
[
[
[
[
129
10] Kim TH, Kim SJ, Ryu YH, Lee HJ, Goo JM, et al. Pleomorphiccarcinoma of lung: comparison of CT features and pathologicfindings. Radiology 2004;232:554—9.
11] Sato S, Koike T, Yamato Y, Yoshiya K, Motono N, Takeshige M,et al. A case of rapidly growing pulmonary carcinosarcoma. IntJ Clin Oncol 2010;15:319—24.
12] Iwata T, Nishiyama N, Inoue K, Kawata Y, Izumi N, TsukiokaT, et al. Biphasic pulmonary blastoma: report of a case. AnnThorac Cardiovasc Surg 2007;13:40—3.
13] Rainosek DE, Ro JY, Ordonez NG, Kulaga AD, Ayala AG. Sarco-matoid carcinoma of the lung. A case with atypical carcinoidand rhabdomyosarcomatous components. Am J Clin Pathol1994;102:360—4.
14] Kikuchi R, Isowa N, Tokuyasu H, Kawasaki Y, Onuma H, MiuraH. Three cases of resected pleomorphic carcinoma. Ann ThoracCardiovasc Surg 2010;16:264—9.
15] Chang YL, Lee YC, Shih JY, Wu CT. Pulmonary pleomorphic(spindle) cell carcinoma: peculiar clinicopathologic manifes-
Cancer 2001;34:91—7.16] Yoshino N, Kubokura H, Yamauchi S, Ohaki Y, Koizumi K, Shimizu
K. A true pulmonary carcinosarcoma that required diagnosticdifferentiation from a pleomorphic adenoma: a case report.Ann Thorac Cardiovasc Surg 2009;15:42—5.
17] Wick MR, Ritter JH, Nappi O. Inflammatory sarcomatoid carci-noma of the lung: report of three cases and clinicopathologiccomparison with inflammatory pseudotumors in adult patients.Hum Pathol 1995;26:1014—21.
18] Italiano A, Cortot AB, Ilie M, Martel-Planche G, Fabas T, Pop D,et al. EGFR and KRAS status of primary sarcomatoid carcinomasof the lung: implications for anti-EGFR treatment of a rare lungmalignancy. Int J Cancer 2009;125:2479—82.
19] Lee S, Kim Y, Sun JM, La Choi Y, Kim JG, Shim YM, et al. Mole-cular profiles of EGFR, K-ras, c-met, and FGFR in pulmonarypleomorphic carcinoma, a rare lung malignancy. J Cancer ResClin Oncol 2011;137:1203—11.
20] Raveglia F, Mezzetti M, Panigalli T, Furia S, Giuliani L,Conforti S, et al. Personal experience in surgical manage-ment of pulmonary pleomorphic carcinoma. Ann Thorac Surg2005;78:1742—7.
21] Hountis P, Moraitis S, Dedeilias P, Ikonomidis P, DouzinasM. Sarcomatoid lung carcinomas: a case series. Cases J2009;2:7900—2.
22] Terada T. Sarcomatoid carcinoma of the lung presenting as acutaneous metastasis. J Cutan Pathol 2010;37:482—5.





























