Embed Size (px)
Citation preview

Journal of Molecular Structure (Theochem), 138 (1986) 271-281 Elsevier Science Publishers B.V., Amsterdam -Printed in The Netherlands
ETUDE CONFORMATIONNELLE DE LA PROSTAGLANDINE El: RECHERCHE DES CONFORMATIONS DE BASSES ENERGIES AU MOYEN DE LA MECANIQUE MOLECULAIRE”
G. ROBINET, J. DEVILLERS et A. LATTES
lJA au CNRS N” 470, Universite Paul Sabatier, 118, Route de Narbonne, 31062 Toulouse Cedex (France)
M. BARTHELAT
Laboratoire des Heterocycles du Phosphore et de I’Azote, 118, Route de Narbonne, 31062 Toulouse Cedex (France)
(Recu le 15 septembre 1985; en forme finale le 15 decembre 1985)
*Ce travail est dkdi4 ii la mlmoire du Professeur F. G. Mathis.
ABSTRACT
(A conformational study of the prostaglandin El: investigation of the least-energy con- formations by using molecular mechanics.)
A study by molecular mechanics, using the MM2 program, shows that for the prosta- glandin El there are several conformations whose energies are lower than those of the two forms existing in the solid state. These last two appear to be rotamers slightly dis- torted by the packing forces. The formation of an hydrogen-bond network compensates the excess of energy of these last two forms. The pentagonal ring of the molecule is not submitted to a conformational equilibrium. The two types of least-energy conformations found in the calculation thus show the same ring geometry as is found in the crystal. However, they differ essentially in the orientation of the unsaturated chain, but they nevertheless remain almost planar due to attractive forces between the two chains.
RBSUMB
Une etude par la mCanique moleculaire, au moyen du programme MM2, montre que pour la prostaglandine El il existe de nombreuses conformations d’bnergies inferieures 2 celles des deux formes pri%entes dans le cristal. Ces dernieres apparaissent comme des rotameres legerement deform& par le “packing” et dont l’excks d’Bnergie est cornpen& par 1’Btablissement du r&au de liaisons hydrogene. La partie cyclique de la molbule n’est pas le siege d’un Bquilibre conformationnel et les deux types de conformations les plus basses en Bnergie trouvees presentent une forme de cycle identique B celle pr&ente dans le cristal. En revanche elles se distinguent essentiellement par l’orientation de la chaine insaturee. Toutefois, en raison des forces d’attraction entre les deux chaines, ces formes gardent une quasi-plan&t&
Les prostaglandines sont des molecules qui ont une t&s large gamme d’activites biologiques [ 1, 21 dont certaines se rapprochent de celles devo- lues parfois aux hormones steroi’diques. Les analogies structurales entre ces deux types de composes sont peu Qvidentes mais, comme le montre la Fig. 1,
0166-1280/86/$03.50 0 1986 Elsevier Science Publishers B.V.

212
OH
PGE I
HO Oertrone
OH
C -OH
Ecdysone
Fig. 1. Analogie possible de conformation de la prostaglandine El avec certains sthols.
on peut imaginer une analogie conformationnelle entre une prostaglandine et certains sterols. C’est pourquoi il nous a semble interessant de rechercher, au moyen de la mkcanique moleculaire et sur l’exemple de la prostaglandine El, la possibilite d’existence, independante de l’environnement, d’etats confor- mationnels de cette molecule qui miment les st&ols.
Les etudes physicochimiques et les approches par le calcul [3-U] ont deja apporte des renseignements precieux, mais en ce qui concerne les etudes theoriques, les methodes utilisees jusqu’a ce jour ont 6th simplifiees dans une mesure telle (par exemple en negligeant l’energie de torsion [18] ) qu’une nouvelle approche nous a paru justifiee. Ce travail a done Cte la recherche des conformations de basses energies de la PGEl. Nous avons choisi la mdthode de Westheimer, Hendrickson et Wiberg [ 191 telle qu’elle est mise en oeuvre dans les programmes d’Allinger et ~011. [20]. Le champ de force utilise est celui inclus dans le programme MM2 [ 211.
MhTHODOLOGIE
La prostaglandine El, molecule en C20, possede un grand nombre de degres de liberte de rotation autour des liaisons C-C qui, combine aux defor- mations eventuelles du cycle a cinq cha’inons, laisse prevoir un nombre con- siderable de conformations. Ces conformations sont separees par des bar-r&es Bnergetiques qui ne peuvent &re franchies dans le processus d’optimisation du type “methode de plus grande pent&’ [ 191.
A defaut de pouvoir realiser une exploration complete de l’hypersurface:
Energie de contrainte = f (parametres geometriques)
il est important de se fixer des conformations de depart suffisamment nombreuses et judicieusement choisies afin d’eviter, autant que faire se peut, de considerer un minimum relatif comme Ctant le minimum absolu.
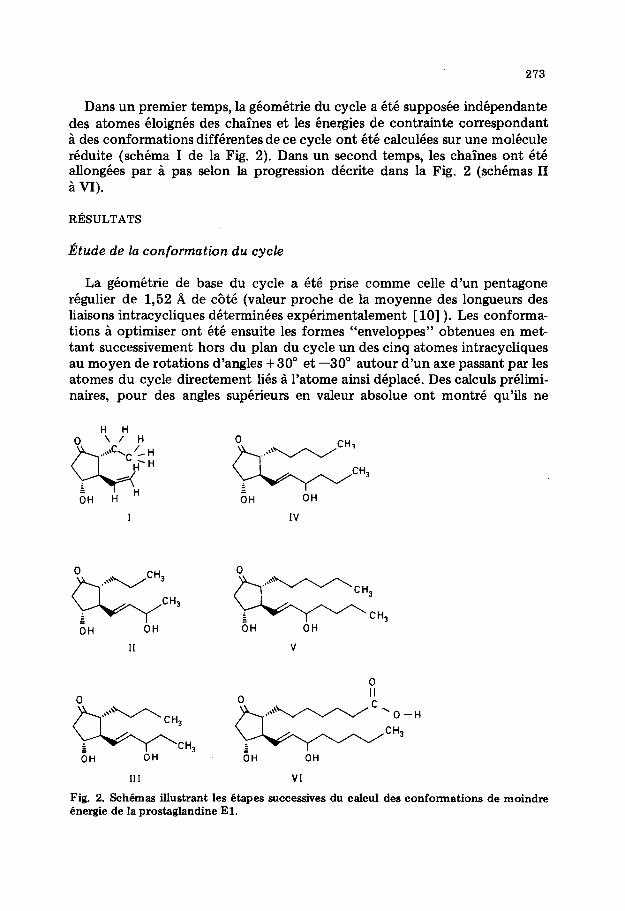
273
Dans un premier temps, la geometric du cycle a et6 suppoke independante des atomes eloignes des chaines et les energies de contrainte correspondant i des conformations differentes de ce cycle ont et6 calculees sur une molecule reduite (schema I de la Fig. 2). Dans un second temps, les chaines ont QtC allongees par a pas selon la progression d&rite dans la Fig. 2 (schemas II 1 VI).
RfiSULTATS
lhude de la conformation du cycle
La gdometrie de base du cycle a et6 prise comme celle d’un pentagone regulier de 1,52 R de cotd (valeur proche de la moyenne des longueurs des liaisons intracycliques d&erminees experimentalement [lo] ). Les conforma- tions 1 optimiser ont et6 ensuite les formes “enveloppes” obtenues en met- tant successivement hors du plan du cycle un des cinq atomes intracycliques au moyen de rotations d’angles + 30” et -30” autour d’un axe passant par les atomes du cycle directement lies a l’atome ainsi deplace. Des calculs prelimi- naires, pour des angles superieurs en valeur absolue ont montre qu’ils ne
H H 0 \/ H ECH,
EH OH
IV
KC., eLH3
OH OH OH
II V
GLH3 @&--:. OH OH OH
111 VI
Fig. 2. Sch6mas illustrant les &apes successives du celcul des conformations de moindre Bnergie de la prostaglandine El.
274
menaient jamais 1 des conformations supplementaires differentes. 11 faut noter ici que les optimisations de l’energie de contrainte pour dix geometries “demi-chaises” n’ont pas conduit i des conformations autres que celles trouvees 1 partie des formes “enveloppes”.
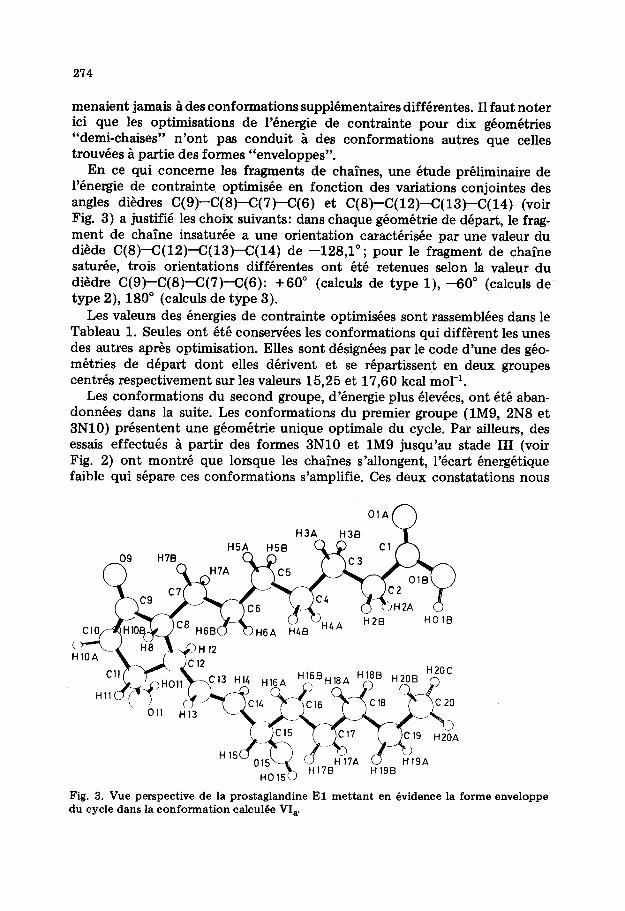
En ce qui concerne les fragments de chaines, une etude preliminaire de l’energie de contrainte. optimisee en fonction des variations conjointes des angles diedres C( 9)-C( 8)-C( 7)--C(6) et C( 8)-C( 12)-C( 13)-C( 14) (voir Fig. 3) a justifie les choix suivants: dans chaque geometric de depart, le frag- ment de chaine insaturee a une orientation caracterisee par une valeur du diede C(S)-C(l2)<(13)-C(14) de -128,l”; pour le fragment de chaine saturee, trois orientations differentes ont ete retenues selon la valeur du diedre C(9)-C(8)<(7)-C(6): +60” (calculs de type l), -60” (calculs de type 2), 180” (calculs de type 3).
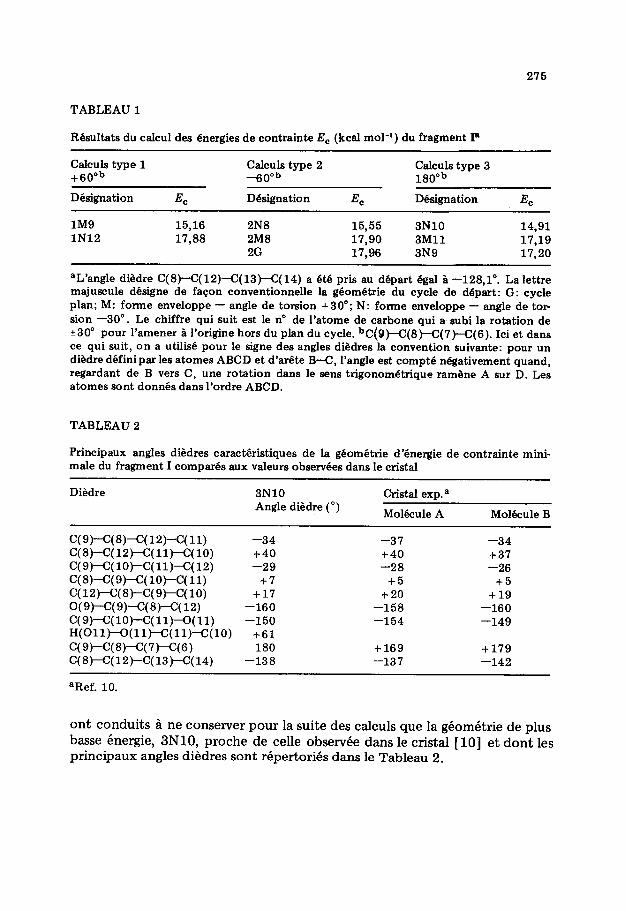
Les valeurs des energies de contrainte optimisees sont rassemblees dans le Tableau 1. Seules ont 6th conservees les conformations qui different les unes des autres apres optimisation. Elles sont designees par le code d’une des g&o- metries de depart dont elles d&vent et se repartissent en deux groupes cent& respectivement sur les valeurs 1525 et 17,60 kcal mol-‘.
Les conformations du second groupe, d’energie plus klevees, ont et& aban- don&es dans la suite. Les conformations du premier groupe (lM9, 2N8 et 3NlO) presentent une geomdtrie unique optimale du cycle. Par ailleurs, des essais effect&s B partir des formes 3NlO et lM9 jusqu’au stade III (voir Fig. 2) ont montre que lorsque les chaines s’allongent, l’ecart Cnergetique faible qui &pare ces conformations s’amplifie. Ces deux constatations nous
Fig. 3. Vue perspective de la prostaglandine El mettant en dvidence la forme enveloppe du cycle dans la conformation calcul6e VI,
275
TABLEAU 1
Resultats du caIcu1 des energies de contrainte E, (kcai mol-‘) du fragment I*
Calculs type 1 +60°b
CaIculs type 2 -60nb
CaIculs type 3 180°b
Designation E, Designation Ec Designation EC
lM9 15,16 2N8 15,55 3NlO 14,91 lN12 17,88 2M8 17,90 3Mll 17,19
2G 17,96 3N9 17,20
aL’angle diedre C( B)-C( 12)-C( 13~C( 14) a et6 pris au depart Bgai B -128,l”. La lettre majuscule designe de facon conventionnelle la geomdtrie du cycle de depart: G: cycle plan; M: forme enveloppe - angle de torsion t30'; N: forme enveloppe - angle de tor sion -30”. Le chiffre qui suit est le no de l’atome de carbone qui a subi la rotation de +30° pour l’amener a l’origine hors du pian du cycle. bC(9)-C(8)-C(7)-C(6). Ici et dans ce qui suit, on a utilisi! pour le signe des angles diedres Ia convention suivante: pour un diedre definipar les atomes ABCD et d’arete B-C, l’angle est compte negativement quand, regardant de B vers C, une rotation dans le sens trigonometrique ramene A sur D. Les atomes sont donnh dans l’ordre ABCD.
TABLEAU 2
Principaux angles diedres caractdristiques de la geomgtrie d’bnergie de contrainte mini- male du fragment I compares aux valeurs observees dans le cristal
Diedre 3NlO CristaI exp. a Angle diedre (“)
Molbule A Molecule B
C(9)-C(B)-C(12)-C(ll) -34 -37 -34 C(B)-C(12)-C(ll)-C(10) t40 t40 t37 c(9)-C(1o)-c(11)-C(12) -29 -28 -26 C(B)-C(9)-C(lO)-C(ll) t7 t5 t5 C(12)-C(S)-C(9)-C(l0) t17 +20 t19 O(9)-C(9)-C(B)-C(12) -160 -158 -160 c(9)-C(10)-c(11)--0(11) -150 -154 -149 H(Oll)--0(11jC(11)-C(10) t61 C(9)-C(B)-C(Sr)-C(S) 180 t169 t 179 C(B)-C(12)-C(13)-C(14) -138 -137 -142
aRef. 10.
ont conduits 1 ne conserver pour la suite des calculs que la gCom&rie de plus basse hnergie, 3N10, proche de celle observde dans le cristal [lo] et dont les principaux angles diedres sont rkpertorih dans le Tableau 2.

276
Allongement des chaines
L’allongement simultane des deux chaines a btQ mene conformement h la stategie definie plus haut et en suivant les &apes d&rites dans la Fig. 2.
Chaque fragment est obtenu a partir du precedent, par substitution aux extremites des deux chaines, en faisant les combinaisons de toutes les orien- tations possibles et l’energie est optimisee dans chaque cas. 11 en resulte, pour chacun des fragments II a V, neuf geometries de depart qui ont men& apres optimisation, B neuf conformations distinctes a la fois par l’energie et la gee- metric. Pour la molecule complete VI, le nombre de geometries de depart est double 1 cause des deux orientations possibles du groupement -C(O)OH. Dans le passage d’un fragment a un autre, la geometric de base retenue pour la substitution a Qte celle de plus basse dnergie resultant de l’optimisation precedente. Les resultats sont rassembles dans le Tableau 3.
DISCUSSION
Les dix-huit conformations ainsi obtenues dont l’energie de contrainte s’etage entre 24,00 et 26,78 kcal mol-’ devraient stre comparees aux deux conformations experimentales A et B determinees par Spek [lo] ; ici et dans la suite, on designe par A et B les deux conform&es de la PGEl tels qu’ils sont dans le cristal. Cependant l’energie de contrainte calculee pour ces con- formations experimentales serait artificiellement augment&e par l’imprecision sur les longueurs et les angles de liaison notamment. A celi s’ajoute l’effet des forces de Van der Waals dues a l’empilement. C’est pourquoi il nous a paru plus interessant de comparer nos r&ultats avec les formes relaxees de
TABLEAU 3
Resultats des minimisations pour les edifices I 1 VI (voir Fig. 2).
Edifice E de contrainte E de contrainte la plus basse apres immediatement minimisation* superieure*
E de contrainte maximale apres minimisation*
IIh 17,66 17,91 20,32 IIIh 18,58 l&69 20,27
Fb 19,29 19,89 19,76 20,30 22,34 21,94 VIC
: 24,00 24,38 25,72 25.15 25,51 26,78
*En kcal mol-‘. bPour les structures II B V les 9 gbom&tries de depart menent B 9 confor mations distinctes par l’bnergie et la geomgtrie. Seules ont et& repertoriees pour chaque structure, l’energie la plus basse, celle immddiatement superieure et la plus haute. CPour la molecule complete VI on a 9 structures a caracterisees par un angle diedre C(3)-C(2)- C( l)-0( 1A) voisin de 0” et 9 structures b d’angle diedre C(3 )-C( 2)-C( l)-C( 1A) voisin de 180”.

277
A et B, calculees par le mQme processus d’optimisation. Les conformations ainsi obtenues et que nous noterons A,,t et Bopt different entre elles par l’energie de contrainte (25,04 et 25,57 kcal mol-‘, respectivement) et essen- tiellement par l’orientation du groupe -C(O)OH. La comparaison de A, avec A et de B, avec B montre des differences de longueurs de liaison de 3.10v2 A en moyenne et des differences d’angles de liaisons de 3” en moyenne. En ce qui conceme la conformation du cycle, le Tableau 4 montre aussi que les formes optimisees restent tres proches des formes A et B. En revanche la modification plus importante de certains angles diedres des chaines (Tableau 5) peut s’interpreter, pour la molecule B, comme une consequence de la suppression des liaisons hydrogene intermoleculaires entre les atomes H(OlB) et O(lA) de la molecule de type B avec respectivement les atomes O(9) et H(Ol1) d’une molecule de type A (rotation du groupement C(O)OH de la molecule B de 45”) voir ref. 10). Des effets de ce type ont etd ainsi expliques dans les amides et les peptides [ 221.
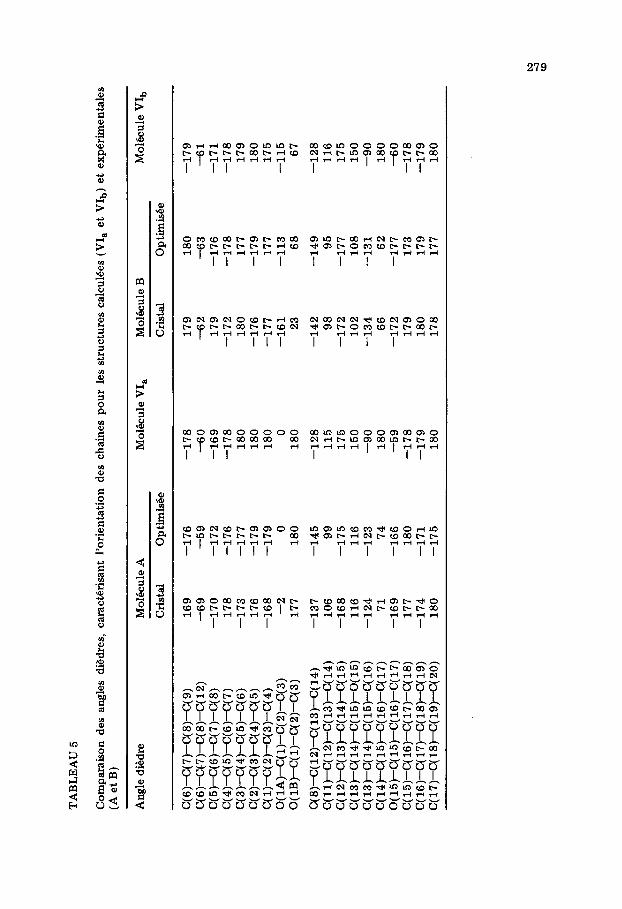
Parmi les dix-huit conformations obtenues par le calcul, trois d’entre elles, ayant la miZme orientation de la fonction acide que A, ont une Cnergie de contrainte inferieure a celle de A optimide: 24,00 (conformation notee VI,), 24,38 et 24,59 kcal mol- l. De mi$me trois conformations ayant la meme orientation du groupe -C(O)OH que B ont une energie de contrainte inferieure a celle de Bopt: 25,15 (conformation notee VIb), 25,51 et 25,55 kcal mol-‘. La comparaison entre VI, et Aopt d’une part et VI, et B opt d’autre part montre que les longueurs et angles de liaisons restent tres proches (Ccarts moyens: 10v3 8, 1”). En ce qui conceme les angles dibdres (voir Tableaux 4 et 5) la forme experimentale du cycle se trouve confirm&e comme &ant celle de plus basse Cnergie: c’est une enveloppe dont la pointe du rabat est constituee par l’atome C(12) comme le montre la Fig. 3. Les differences les plus importantes concement les valeurs des dikdres dont l’ari?te Porte l’atome C(15). L’ecart atteint 106” (espece A) ou 118” (espece B) pour le diedre C( 14)-C( 15)-C( 16)-C( 17)-; c’est done surtout au niveau de l’orientation de la chaine insaturke que les conformations calculees VI, et VI,, different des formes Aopt et Bopt et des geometries cristallines. Un effet comparable a ete mis en evidence par Kothekar, par une methode de calcul differente pour la PGBl [17]. Cet &art est tres inferieur pour les diedres aux extremites des chafnes. En consequence, si la distance entre deux atomes de carbone situ& aux extremites de chacune des chaines est h peu p&s la meme dans VI, et Aopt d’une part et VI,, et Bopt d’autre part, les distances C(6). . C(14), C(4) . . . C(16) et C(2). . . C(18) sont tres dimi- n&es dans les conformations VI, et VI,, (voir Tableau 6). On remarquera que les atomes concern& sont ceux qui par etablissement de liaisons forme- raient les cycles hexagonaux accoles dans une structure steroi’dique. Les conformations calculees VI, et VI,, sont done plus aptes a mimer les sterols que les conformations trouvees 1 l’dtat solide, mEme optimides.
Pour la structure calculee VI,, les cinq cent quatre vingt huit interactions attractives entre atomes appartenant a des chaines differentes apportent une
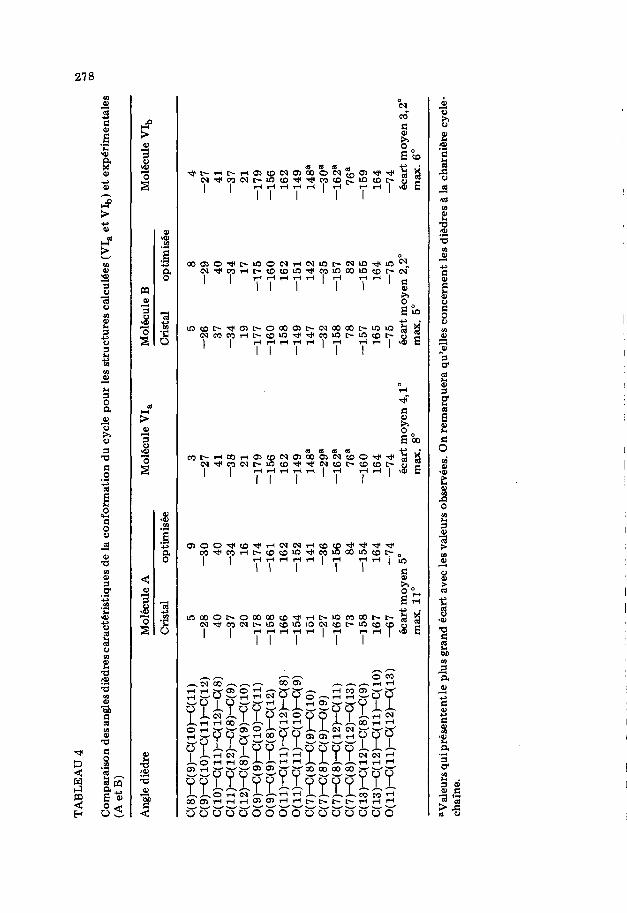
TA
BL
EA
U
4 N
-l
m
C
ompa
rais
on
desa
ngl
esdi
edre
scar
acte
rist
iqu
es
de l
a co
nfo
rmat
ion
du
cyc
le
pou
r le
s st
ruct
ure
s ca
lcu
lees
(V
I,
et V
Ib)
et e
xper
imen
tale
s (A
et
B)
An
gle
dik
dre
Mol
Qu
le
A
Mol
iku
le
VI,
M
olC
ule
B
M
olk
ule
V
I,
Cri
stal
op
tim
isee
C
rist
al
opti
mis
ke
C( 8
)-C
( 9)
-C(
lo)-
C(
11)
c(9)
-c(1
o)-c
(11)
-c(1
2)
C( l
o)-C
( ll
)--C
(12)
-C(
8)
C(H
)-C
(12)
-C(8
)-C
(9)
C( 1
2)-C
( 8)
-C(
9)-C
( 10
) O
( 9)-
C(
9)-C
( lO
)-C
( 11
) 0(
9)+
J(9)
~(8
)--c
(l2)
O
(ll)
-C(l
l)--
C(1
2)-C
(8)
o(ll
)-c(
11)-
c(lo
)-c(
9)
C(7
)--c
(8)-
-c(9
)--c
(lO
) C
(7)+
(8W
(9)+
(9)
C(7
)-C
(8)-
C(1
2)-C
(11)
C
( 7)-
C(
8)-C
( 12
)-C
( 13
) C
(13)
-C(1
2)-C
(8)-
C(9
) C
(13)
-C(1
2)-C
(ll)
-C(1
0)
O(l
l)-C
(ll)
-C(l
2)-C
(13)
5 9
3 5
8 4
-28
-30
-27
-26
-29
-27
40
40
41
37
40
41
-37
-34
-38
-34
-34
-37
20
16
21
19
17
21
-178
-1
74
-179
-1
77
-175
-1
79
-158
-1
61
-156
-1
60
-160
-1
56
166
162
162
158
162
162
-154
-1
52
-149
-1
49
-151
-1
49
151
141
148’
14
7 14
2 14
8a
-27
-36
-2ga
-3
2 -3
5 -3
0a
-165
-1
56
-162
’ -1
58
-157
-1
62a
73
84
76a
78
82
76a
-158
-1
54
-160
-1
57
-155
-1
59
167
164
164
165
164
164
-67
-74
-74
-75
-75
-74
&ar
t m
oyen
5”
&
zart
moy
en
4,l”
G
cart
moy
en
2,2”
&
art
moy
en
3,2”
m
ax.
11”
max
. 8”
m
ax.
5”
max
. 6”
aVal
eurs
qu
i pr
esen
ten
t le
plu
s gr
and
&ar
t av
ec l
es v
aleu
rs o
bser
vees
. O
n r
emar
quer
a qu
’ell
es
con
cern
ent
les
dibd
res
a la
ch
arn
iere
cy
cle-
ch
afn
e.
._
_ -
TA
BL
EA
U
5
Com
par
aiso
n
des
an
gles
d
ied
res,
ca
ract
eris
ant
l’or
ien
tati
on
des
ch
ain
es
pou
r le
s st
ruct
ure
s ca
lcu
lees
(V
I,
et V
Ib)
et e
xper
imen
tale
s (A
et
B)
An
gle
die
dre
Ct6
)-C
(7)-
C(8
)-C
(9)
C( 6
)-C
(-7)
--c(
8)
-C(
12)
C(5
)+?6
)-c(
7)-C
(8)
C(4
)-C
(5)-
C(6
)+X
7)
C(3
)_C
(4)-
C(5
)-C
(6)
C(2
)--C
(3)-
C(4
)+(5
) C
(l)-
C(2
)--C
(3)-
C(4
) 0(
lA
)-C
( 1)
-C(
2)-C
( 3)
O
( IS
)-C
( 1)
-C(
2)-C
(3)
C( 8
)-C
( 12
)-C
( 13
)-C
( 14
) C
(ll)
-C(1
2)-C
(13)
-C(1
4)
C( 1
2~C
( 13
)-C
(14)
-C(
15)
C( 1
3)-C
( 14
)-C
( 15
)-0(
15
) C
(13)
-C(1
4)-C
(15)
+(1
6)
C( 1
4)-C
( 15
)-C
( 16
)-C
( 17
) 0(
15)-
C(
15)-
C(1
6)-C
( 17
) C
( 15)
-C(
16)-
C(
17)-
C(
18)
C( 1
6~C
( 17
)-C
( 18
)-C
( 19
) C
( 17)
-C(
18)-
C(1
9)-C
( 20
)
Mol
ecu
le A
cris
tal
Op
tim
is6e
169
-176
-6
9 -5
9 -1
70
-172
17
8 -1
76
-173
-1
77
176
-179
-1
68
-179
-2
0
177
180
-137
-1
45
106
99
-168
-1
75
116
116
-124
-1
23
71
74
-169
-1
66
177
180
-174
-1
71
180
-175
Mol
bu
le
VI,
-178
-6
0 -1
69
-178
18
0 18
0 18
0 0 18
0
-128
11
5 17
5 15
0 -9
0 180
-59
-178
-1
79
180
Mol
bu
le
B
Mol
bu
le
VI,
Cri
stai
O
pti
mis
~e
179
180
-179
-6
2 -6
3 -6
1 17
9 -1
76
-171
-1
72
-178
-1
78
180
177
179
-176
-1
79
180
-177
17
7 17
5 -1
61
-113
-1
15
23
68
67
-142
-1
49
-128
98
95
11
6 -1
72
-177
17
5 10
2 10
8 15
0 -1
34
-131
-9
0 66
62
18
0 -1
72
-177
-6
0 17
9 17
3 -1
78
180
179
-179
17
8 17
7 18
0
280
TABLEAU 6
Distances (A) entre atomes de carbone appartenant aux 2 chaines tels qu’une liaison entre ces atomes entrainerait la formation de cycles B 6 chainons accol&
Conformation C(6)-C(l4) C(4)-C(16) C( 2)-C( 18)
A opt 3,93 5,58 5,04 B opt 4,Ol 5,80 5,44 VI, 3,55 3,87 4,Ol v1b 3,57 3,92 4,16
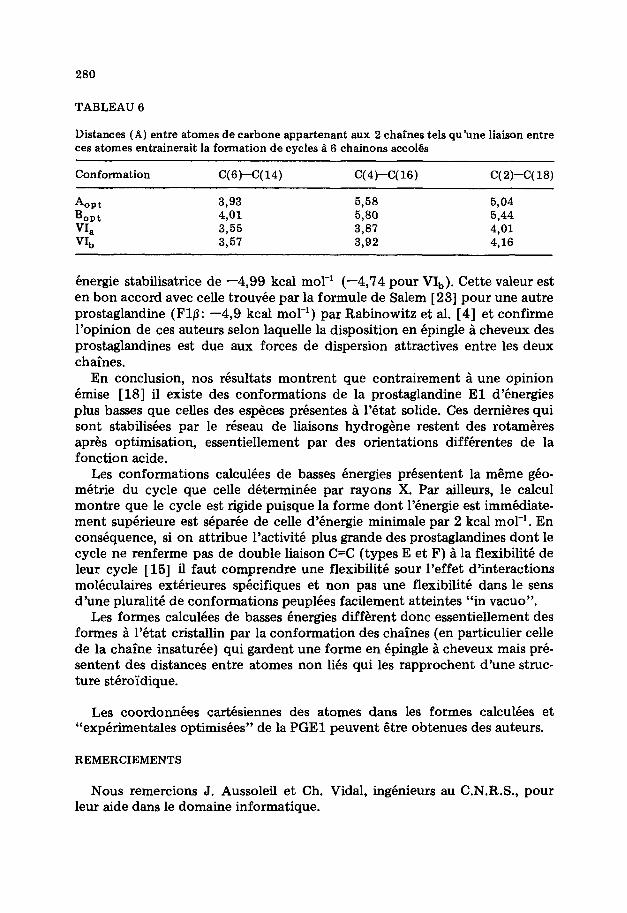
Cnergie stabilisatrice de -4,SS kcal mol-’ (-4,74 pour VI,). Cette valeur est en bon accord avec celle trouvee par la formule de Salem [ 231 pour une autre prostaglandine (Flp: -4,s kcal mol-‘) par Rabinowitz et al. [4] et confirme l’opinion de ces auteurs selon laquelle la disposition en epingle a cheveux des prostaglandines est due aux forces de dispersion attractives entre les deux chaines.
En conclusion, nos resultats montrent que contrairement a une opinion 6mise [18] il existe des conformations de la prostaglandine El d’energies plus basses que celles des especes presentes a l’etat solide. Ces dernieres qui sont stabilisees par le reseau de liaisons hydrogene restent des rotameres apres optimisation, essentiellement par des orientations differentes de la fonction acide.
Les conformations calculees de basses energies presentent la m6me geo- metric du cycle que celle determinCe par rayons X. Par ailleurs, le calcul montre que le cycle est rigide puisque la forme dont l’energie est immediate- ment superieure est sepame de celle d’energie minimale par 2 kcal mol-’ . En consequence, si on attribue l’activite plus grande des prostaglandines dont le cycle ne renferme pas de double liaison C=C (types E et F) a la flexibilite de leur cycle [ 151 il faut comprendre une flexibilite sour l’effet d’interactions moleculaires exterieures specifiques et non pas une flexibilite dans le sens d’une pluralite de conformations peuplees facilement atteintes “in vacua”.
Les formes calculees de basses energies different done essentiellement des formes a l’etat cristallin par la conformation des chafnes (en particulier celle de la chaine insaturee) qui gardent une forme en Bpingle a cheveux mais pre- sentent des distances entre atomes non lies qui les rapprochent d’une struc- ture steroi’dique.
Les coordonnees cartesiennes des atomes dans les formes calculees et “experimentales optimisees” de la PGEl peuvent Btre obtenues des auteurs.
REMERCIEMENTS
Nous remercions J. Aussoleil et Ch. Vidal, ingenieurs au C.N.R.S., pour leur aide dans le domaine informatique.

281
BIBLIOGRAPHIE
1 P. Crabbe, Chem. Brit., 11 (1975) 132. 2 S. Bergstrom, Angew. Chem. Int. Ed., 22 (1983) 858. 3 0. Korver, Rec. Trav. Chim. Pays-Bas, 88 (1969) 1070. 4 I. Rabinowitz, P. Ramwell et P. Davison, Nature New Biol., 233 (1971) 88. 5 J. R. Hoyland et L. B. Kier, J. Med. Chem., 15 (1972) 84. 6 G. F. Cooper et J. Fried, Proc. Nat. Acad. Sci. USA, 70 (1973) 1579. 7 J. W. Edmonds et W. L. Duax, Prostaglandins, 5 (1974) 275. 8 A. Murakami et Y. Akahori, Chem. Pharm. Bull., 22 (1974) 1133. 9 G. T. DeTitta, Science, 191(1975) 1271.
10 A. L. Spek, Acta Crystallogr., Sect. B: 33 (1977) 816. 11 G. T. DeTitta, D. A. Langs et J. W. Edmonds, Biochemistry, 18 (1979) 3387. 12A. Murakami et Y. Akahori, Chem. Pharm. Bull, 27 (1979) 548. 13 G. T. DeTitta, D. A. Langs, J. W. Edmonds et W. L. Duax, Acta Crystallogr., Sect. B:
36 (1980) 638. 14 S. Grigoras, I. Rusu, E. Pausescu, A. Medesan et S. Maldoveanu, Int. J. Quantum
Chem., 18 (1980) 501. 15 V. Kothekar et S. Dutta, Int. J. Quantum Chem., 18 (1980) 891. 16 V. Kothekar, Int. J. Quantum Chem., 20 (1981) 167. 17 V. Kothekar, Int. J. Quantum Chem. Suppl., 9 (1982) 281. 18 V. Kothekar, J. Theor. Biol., 94 (1982) 943. 19 (a) F. H. Westheimer, in M. S. Newman (Ed.), Steric Effects in Organic Chemistry,
Wiley, New York, 1956, p. 523. (b) J. B. Hendrickson, J. Am. Chem. Sot., 83 (1961) 4537; 84 (1962) 3355; 86 (1964) 4854. (c) K. B. Wiberg, J. Am. Chem. Sot., 87 (1965) 1070.
20 U. Burkert et N. L. Allinger, Molecular Mechanics, A.C.S., Monograph 177, American Chemical Society, Washington DC, 1982, p. 17.
21 N. L. Allinger, J. Am. Chem. Sot., 99 (1977) 8127; N. L. AIIinger and Y. H. Yuh, Q.C.P.E., 13 (1981) 395.
22 J. CaiIIet, P. Claverie et B. Pullman, Theor. Chim. Acta, 47 (1978) 17. 23 L. Salem, J. Chem. Phys., 37 (1962) 2100.





























