Embed Size (px)
Citation preview

CORTES José Année 2000/2001
Lycée Technique MARIE CURIE 16 Bd Jeanne d'Arc 13005 MARSEILLE
Rapport de stage
Deuxième année de BTS Biotechnologie
Etude de deux polymorphismes du gène du TAFI Optimisation d’une PCR Multiplex
Maître de stage: Mme Mireille HENRY Laboratoire d'Hématologie
EPI 99-36 Faculté de Médecine La Timone
27 Bd. Jean Moulin 13385 Marseille Cedex 5.

Remerciements
Je tiens à remercier Mme Irène JUHAN-VAGUE et tout le personnel de l’EPI 99-36
de m’avoir accueilli durant mes stages de première et deuxième année de BTS.
Je remercie tout particulièrement Mme Mireille HENRY de m’avoir attendu pour la
mise au point du multiplex de l’exon IX et de l’exon VI, pour sa patience, pour le temps
qu’elle a su me consacrer tout au long de ces deux années et pour son aide précieuse dans la
rédaction de ce rapport.

SOMMAIRE
Numéro de page
PARTIE ECONOMIQUE I. Institut National de la Santé et de la Recherche Médicale (INSERM). 1 II. Historique du laboratoire. 2 III. Organigramme du personnel de laboratoire. 2 IV. Financement du laboratoire. 2 V. Hygiène et sécurité. 4 5.1. Les trois salles de manipulation du laboratoire. 4 5.1.1. La salle de culture cellulaire. 4 5.1.2. La salle de biologie moléculaire. 5 5.1.3. La salle de lecture UV. 5 5.2. Traitement des réactifs et du matériel avant et après utilisation. 5 PARTIE SCIENTIFIQUE I. Introduction. 7 1.1. Généralités sur le TAFI. 7 1.2. Rôle du TAFI dans le processus de fibrinolyse. 8 1.3. Le gène du TAFI. 9 1.4. Etude et objectif de l’équipe. 9 1.5. Ma participation à ce travail. 10 II. De l’échantillon sanguin à la PCR. 11
2.1. Extraction de l’ADN. 11 2.2. La PCR. 11
2.2.1. Introduction. 11 2.2.2. Le choix des amorces. 12 2.2.3. Les trois étapes de la PCR. 12 2.2.3.1. La dénaturation de l’ADN génomique double brin. 12 2.2.3.2. L’hybridation des amorces. 12 2.2.3.3. La synthèse de l’ADN (l’élongation. 13 2.2.4. Le nombre de cycles. 13 2.2.5. La Taq DNA polymérase. 14
III. Les polymorphismes analysés. 14
3.1. Le C+1058 T. 14 3.2. Le A+661 G. 17
3.3. Le multiplex C+1058 T et A+661 G. 19

IV. Optimisation du Multiplex. 21 V. Conclusions. 25
5.1. Scientifique. 25 5.2. Personnelle. 26 ANNEXES Annexe 1 : Gène du TAFI. Annexe 2 : La technique SSCP (Single Strand Conformation polymorphism). Annexe 3 : Extraction de l’ADN, méthode au NaCl. Annexe 4 : Extraction de l’ADN, méthode au phénol/chloroforme. Annexe 5 : La PCR. Annexe 6 : Préparation des réactifs pour l’extraction des DNA. Annexe 7 : Bibliographie.

Liste des abréviations Arg : Arginine. BET : Bromure d'éthidium. CPU : Carboxypeptidase unstable. dNTP : Désoxynucléotide triphosphate. INSERM : Institut National de la Santé Et de la Recherche Médicale. kb : Kilobase ou kilopaire de base (1kb = 1000pb). Lys : Lysine. MW : Molecular Weight. NBT- BCIP : Nitro Blue Tetrazolium - 5-Bromo-4-Chloro-3-Indolyl Phosphate. PAI-l: Plasminogen Activator Inhibitor de type 1. pb : paire de base. PCR : Polymerisation Chain Reaction. qsp : quantité suffisante pour… SDS : Sodium Dodecyl Sulfate. SSCP : Single Strand Conformation Polymorphism. TAFI : Thrombin Activatable Fibrinolysis Inhibitor. TAFIa : TAFI activé. t-PA : tissue Plasminogen Activator. T°opt : Température optimale. Tp : Tampon.

PARTIE
ECONOMIQUE

PRESENTATION DU LABORATOIRE D’ACCUEIL
Le laboratoire EPI 99-36, où j’ai effectué mon stage, est dirigé par le Professeur Irène
JUHAN-VAGUE et a pour intitulé Fibrinolyse et pathologie vasculaire .
I. Institut National de la Santé et de la Recherche Médicale (INSERM) :
C'est un organisme public de recherche qui développe son activité dans la biologie, la
médecine et la santé. Il est placé sous la tutelle des ministres chargés de la recherche et de la
santé. Sa finalité est de contribuer au développement des connaissances scientifiques en
biologie humaine et de participer aux progrès diagnostiques, thérapeutiques et préventifs.
Il bénéficie d’un budget de 3 milliards de FF (un peu plus de 457 millions d’€).
En 2001, l'INSERM possède, en France :
ü Un réseau de 326 laboratoires et 61 équipes de recherche, implantés, dans leur quasi-
totalité, au sein des hôpitaux et dans les universités.
ü Plus de 60 instituts fédératifs de recherche.
ü 21 contrats de recherche INSERM.
ü 17 centres d'investigations cliniques avec les hôpitaux.
ü Une communauté scientifique, médicale et technique de 10 000 personnes,
dont 2971 chercheurs et 3240 ingénieurs, techniciens et administratifs, salariés de
l’INSERM, 727 médecins-hospitaliers, environ 1874 hospitalo-universitaires et près de
1732 boursiers, 769 chercheurs, boursiers et étudiants étrangers.
Il y a 266 partenaires industriels de l’INSERM dont 48 % provenant de France, 22% d’Europe
et 30% d’autres pays (majoritairement des Etats-Unis). Près de 98% des laboratoires INSERM
coopèrent avec l’étranger.

Les principaux axes de recherche dans la région PACA :
ü L'immunologie et la génétique.
ü La génétique moléculaire et la biologie du développement.
ü La neurobiologie.
ü La nutrition.
II. Historique du laboratoire :
Il fut créé en octobre 1993 après la demande d’un Contrat Jeune Formation (CJF). Il fut
accepté par les instances scientifiques de l'INSERM pour une durée légale de 3 ans sous le
titre de "Fibrinolyse et Pathologie Vasculaire". Le professeur Irène JUHAN-VAGUE et son
équipe demandèrent ensuite un Contrat de Recherche INSERM (CRI) qui fut accepté en 1997
pour une période de 5 ans sur la même thématique. En janvier 1999, le laboratoire fut
transformé en Equipe Propre INSERM (EPI).
III. Organigramme du personnel du laboratoire.
Voir organigramme page suivante. IV. Financement du laboratoire.
Les origines du financement du laboratoire sont diverses. Le laboratoire est financé par
l’I.N.S.E.R.M., par l'Université, par un programme hospitalier de recherche (PHRC) et par
des contrats avec des entreprises.
Pour l'année 1999, le budget total alloué à l'EPI 99-36 est de 885 000 FF (˜ 135000 €) .
- I.N.S.E.R.M 500 000 FF (˜ 76300 €)
- Contrats divers INSERM 135 000 FF (˜ 25600 €)
- Université 100 000 FF (˜ 15300 €)
- Contrats divers Industrie 100 000 FF (˜ 15300 €)
- PHRC 50 000 FF (˜ 7650 €)
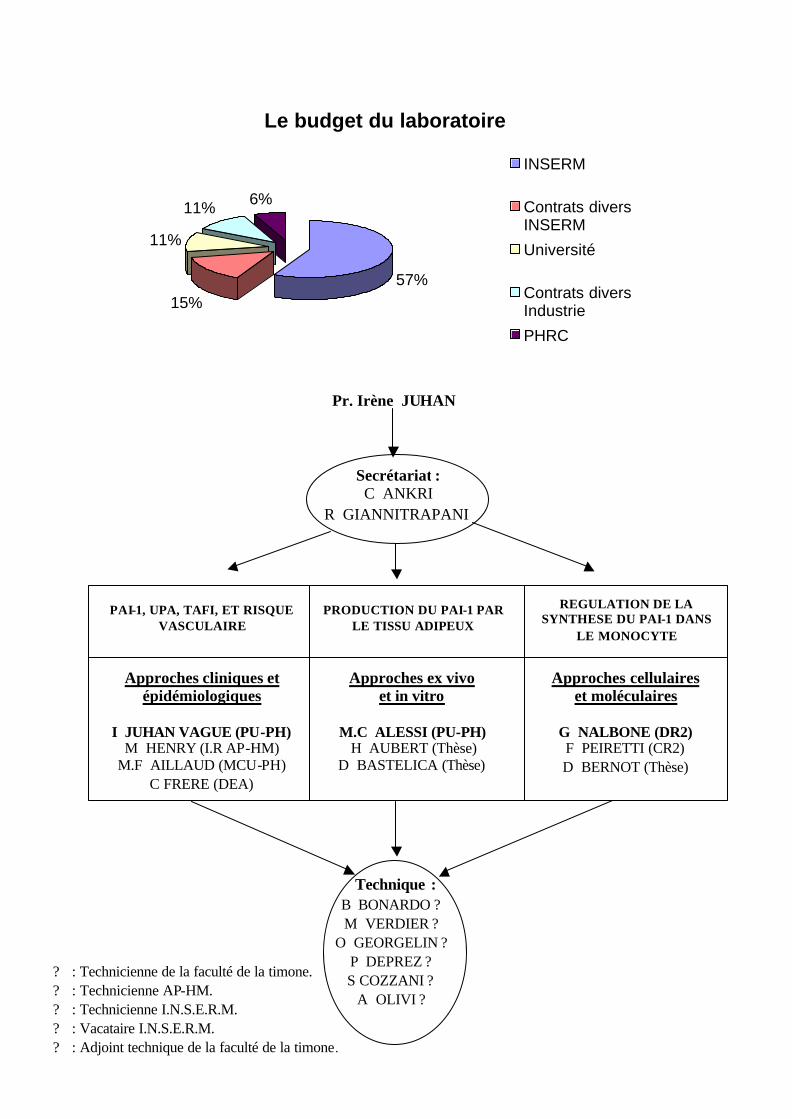
Le budget du laboratoire
57%15%
11%
11% 6%
INSERM
Contrats diversINSERM
Université
Contrats diversIndustrie
PHRC
Technique : B BONARDO ? M VERDIER ?
O GEORGELIN ? P DEPREZ ? S COZZANI ?
A OLIVI ?
? : Technicienne de la faculté de la timone. ? : Technicienne AP-HM. ? : Technicienne I.N.S.E.R.M. ? : Vacataire I.N.S.E.R.M. ? : Adjoint technique de la faculté de la timone.
Pr. Irène JUHAN
Secrétariat : C ANKRI
R GIANNITRAPANI
PAI-1, UPA, TAFI, ET RISQUE VASCULAIRE
PRODUCTION DU PAI-1 PAR LE TISSU ADIPEUX
REGULATION DE LA SYNTHESE DU PAI-1 DANS
LE MONOCYTE
Approches cliniques et épidémiologiques
I JUHAN VAGUE (PU-PH)
M HENRY (I.R AP-HM) M.F AILLAUD (MCU-PH)
C FRERE (DEA)
Approches ex vivo et in vitro
M.C ALESSI (PU-PH)
H AUBERT (Thèse) D BASTELICA (Thèse)
Approches cellulaires et moléculaires
G NALBONE (DR2) F PEIRETTI (CR2) D BERNOT (Thèse)

Légende :
MCU-PH : Maître de Conférence des Universités - Praticien Hospitalier
PU-PH : Professeur des Universités - Praticien Hospitalier
IR : Ingénieur de Recherche
DR : Directeur de Recherche
CR : Chargé de Recherche
DEA : Diplôme d’Etudes Approfondies
V. Hygiène et sécurité :
Dans un laboratoire qui manipule des produits biologiques humains, animaux ou
microbiologiques, le port de la blouse, des gants et la connaissance des règles de sécurité à
adopter pour l'utilisation de tel ou tel réactif est obligatoire. C'est pour cela que le comité
d'hygiène et sécurité de l'INSERM met en place pour les nouveaux employés un stage de
formation de 3 jours au cours duquel toutes ces notions d’hygiène et de sécurité sont
enseignées.
5.1. Les 3 salles de manipulation du laboratoire :
5.1.1. La salle de culture cellulaire :
La salle de culture cellulaire est une pièce en surpression, séparée de l'extérieur par
un sas également en surpression. Les manipulations sur les cellules, les échantillons
sanguins, les prélèvements d'origine humaine ou animale (graisses, cordons ombilicaux)
ou les bactéries transformées par des plasmides doivent être réalisées avec des gants en
latex stériles, sous hotte à flux laminaire en respectant strictement les règles de sécurité.
L'utilisation des gants en latex stériles et de la hotte à flux laminaire permet d'éviter la
contamination des échantillons par l'expérimentateur ou inversement.

5.1.2. La salle de biologie moléculaire :
Les manipulations de biologie moléculaire demandent de nombreuses précautions
afin d’éviter les contaminations inter-échantillons comme : le changement fréquent de
gants, des pointes et des pipettes stériles entre chaque manipulation. Et des pipetages
précis et délicats pour ne pas contaminer les pipettes.
5.1.3. La salle de lecture UV :
Cette salle est réservée à la lecture des gels d'agarose contenant du bromure
d'éthidium (BET) ; pour cela elle possède une lampe UV et une caméra reliée à un
ordinateur muni d'une imprimante photographique.
Le BET est une substance mutagène (ses molécules vont s'intercaler de façon
réversible entre les bases de l'ADN). Sa manipulation nécessite le port de gants. Les
rayons UV produisent sur la peau et la rétine des brûlures. Leur manipulation nécessite
donc une vitre protectrice et le port de lunettes spéciales.
5.2. Traitement des réactifs et du matériel avant et après leur utilisation.
Les matériels et réactifs utilisés pour les manipulations sur les produits biologiques
doivent être stériles. Cette stérilisation est réalisée dans un four Pasteur pour le matériel en
verre ou en Pyrex et dans un autoclave pour les réactifs et le matériel en plastique.
Les déchets obtenus lors des manipulations sont traités:
• Les déchets solides sont jetés dans une poubelle spéciale qui sera incinérée.
• Les déchets biologiques liquides sont recueillis dans un récipient contenant de l’eau de
Javel (pour détruire les contaminants).
• Les déchets chimiques (comme le BET) sont recueillis dans un récipient qui est spécifique
à chacun des déchets.

Les récipients contenant les déchets biologiques liquides et les déchets chimiques sont
récupérés par des organismes agréés pour le traitement des déchets.
Mr OLIVI André s'occupe de la stérilisation et de l’élimination des déchets.

PARTIE
SCIENTIFIQUE

I. INTRODUCTION
1.1. Généralités sur le TAFI.
Le TAFI (Thrombin Activatable Fibrinolysis Inhibitor) a été décrit récemment. Il est
également nommé procarboxypeptidase instable (PCPU), ou procarboxypeptidase B, la
présence de résidus glycosylés le différenciant des carboxypeptidases B tissulaires. Il s’agit
d’une glycoprotéine monocaténaire de 401 acides aminés et de poids moléculaire 60 kDa. Il
est synthétisé par le foie sous la forme d’un zymogène et sécrété dans la circulation ; sa
concentration plasmatique est de 2 à 5 µg/ml, soit 30 à 80 nM, avec cependant de grandes
variations interindividuelles. Sa voie d’élimination n’est pas connue.
L’activation du TAFI nécessite son clivage au niveau de l’arginine 92 par des enzymes
telles que la trypsine, la thrombine et la plasmine. Cette protéolyse conduit à la génération de
plusieurs fragments dont un de 35 kDa (TAFI activé ou TAFIa), support de l’activité
enzymatique de la molécule. Les concentrations de thrombine nécessaires à l’activation du
TAFI sont très élevées (≅ 500 nM) ; cette réaction est fortement accélérée (x1260) quand la
thrombine est engagée dans le complexe thrombine-thrombomoduline.
Le TAFIa est une carboxypeptidase basique dont la fonction est d’éliminer, sur la
fibrine partiellement dégradée, les résidus arginine et lysine en position carboxyterminale. Ces
résidus sont indispensables à l’assemblage des constituants du système fibrinolytique à la
surface de la fibrine. Ce phénomène entraîne la diminution de la fixation du plasminogène à la
fibrine, la réduction de l’activation du plasminogène par le t-PA et de la conversion du Glu-
plasminogène en Lys-plasminogène. La fibrine en cours de dégradation a un rôle de cofacteur
dans ces deux dernières fonctions.

1.2. Rôle du TAFI dans le processus fibrinolytique
Le TAFI semble ainsi avoir un rôle majeur dans la régulation de la balance entre
coagulation et fibrinolyse.
Le rôle du TAFI en pathologie n’est pas connu. Cependant, l’ensemble des données
concernant ses propriétés suggère que des modifications de son taux plasmatique puissent être
impliquées dans des syndromes hémorragiques ou thrombotiques.
L’enrichissement en TAFIa d’un système purifié ou d’un plasma déplété en TAFIa,
augmente le temps de lyse du caillot d’une manière dose dépendante. Une activation exagérée
du TAFI pourrait ainsi constituer un facteur de risque thrombotique. Le TAFIa serait
également impliqué dans l’activité profibrinolytique attribuée à la protéine C activée.
Dans une population de volontaires sains, il a été observé une grande dispersion des
temps de lyse du caillot, corrélée positivement à la concentration plasmatique de TAFI
X Xa, Va V
Ca²+PL
Prothrombine (II) Thrombine (IIa)
Fibrinogène Fibrine monomère
Fibrine (polymérisée) insoluble
XIII
XIIIa
Fibrine lysée
Plasmine Plasminogène
TTAAFFIIaa
y

antigène. Cette grande variabilité interindividuelle des taux de TAFI est faiblement influencée
par les facteurs environnementaux, laissant supposer leur déterminisme génétique.
1.3. Le gène du TAFI
Il est porté par le chromosome 13, en position q14.11. Ce gène de 48 kb est composé
de 11 exons et de 10 introns (èAnnexe 1). Le gène est totalement séquencé (via le
programme génome humain).
La région promotrice comporte 2700 nucléotides. Elle présente de multiples sites
d’initiation de la transcription ( 9 sites majeurs et 2 utilisés moins fréquemment). On note,
comme pour les gènes codant pour certains facteurs de la coagulation vitamine K dépendants
(facteurs VII, IX, X, protéine C), l’absence de TATA box . Les nucléotides –141 à –73
semblent essentiels pour l’expression du gène du TAFI dans les cellules hépatiques.
La région 3’ non traduite contient 3 sites de polyadénylation différents. Les 3 ARN
messagers ont été retrouvés.
Deux polymorphismes ont été identifiés dans la région codante du gène par Zhao et al : une
mutation silencieuse C→T sur le nucléotide 678 (exon VII), et une substitution A→G en
position 505 (exon V) entraînant le remplacement d’une thréonine par une alanine en position
147 de la protéine. Ce dernier polymorphisme est fréquent au sein des populations étudiées
(fréquence Ala de la vaiation Ala 147 Thr = 0,76).
1.4. Etude et objectif de l’équipe
L’équipe dans laquelle j’ai été intégré, à analyser la région promotrice et la région 3’non
traduite du gène du TAFI par la technique SSCP (Single Strand Conformation
Polymorphism) (èannexe 2) à partir de l’ADN de vingt individus avec un taux élevé de

TAFI et de vingt individus ayant un taux bas. Les résultats concernant les régions
promotrice et 3’ non traduite sont publiés (travaux acceptés pour publication dans Blood).
Ainsi neuf nouveaux polymorphismes génétiques ont été trouvés : cinq mutations dans
la région promotrice, deux dans la région codante et deux dans la région 3’ non traduite.
Les études génotypiques des populations de sujets sains ont permis de conclure au fort
déterminisme génétique du taux plasmatique de TAFI (relation génotype /phénotype). Les
polymorphismes G+505 A et C+1542 G expliquent 61,6% de la variabilité de ce taux. Plus
précisément, l’allèle G du polymorphisme G+505 A est associé à des taux bas de TAFI
plasmatique, tandis que l’allèle C du polymorphisme C+1542 G est lié à des taux élevés.
La relation génotype /maladie n’a pas encore été établie.
Actuellement, la mise au point des techniques de génotypage, et l’analyse génotypique
d’une population à risque élevé de thrombose ou ayant développé un infarctus du
myocarde, et d’une population témoin appariée en âge et en sexe devrait permettre
d’établir le lien génotype /maladie. Des études concernant le poids génétique des
polymorphismes sont réalisées dans des familles nucléaires (deux parents et deux à trois
enfants) ou à partir d’ADN d’autres races (Noirs de Côte d’Ivoire). L’étude de la région
codante est en cours.
1.5. Ma participation à ce travail
Mon travail a consisté en la mise au point de l’analyse conjointe de deux mutatio ns
récemment mise en évidence : une situé au niveau de l’exon VI, il s’agit d’une mutation
silencieuse G+663 A (en position 663 de la séquence codante), et l’autre située au début
de l’exon IX correspondant à une variation CèT en position 1040 de la séquence codante,
entraînant une variation Thréonine è Isoleucine de l’acide aminé 325. La discrimination

allélique est réalisée grâce à la mise au point d’une technique de PCR spécifique d’allèle
en multiplex. La finalité étant de pouvoir réaliser conjointement deux génotypages.
II. De l’échantillon sanguin à la PCR.
2.1. Extraction de l’ADN.
Pour amplifier une séquence d’acide nucléique, il faut tout d’abord extraire l’ADN de la
cellule et le purifier tout en éliminant les inhibiteurs des polymérases, notamment
l’hémoglobine.
Deux protocoles d’extraction sont utilisés au laboratoire : nous partons d’un
prélèvement sanguin anticoagulé par l’EDTA. Après élimination des globules rouges et de
l’hémoglobine, le culot de globules blancs est lysé par du SDS les protéines digérées par la
protéinase K puis précipitées, soit grâce à une forte concentration en NaCl (èannexe 3), soit
par une extraction par le phénol et le chloroforme (èannexe 4). L’ADN génomique est alors
précipité par de l’éthanol, récupéré par centrifugation, séché et solubilisé soit dans de l’eau
distillée stérile soit dans du tampon (TE) qui contient de l’EDTA (inhibiteur des nucléases).
L’ADN obtenu avec extraction au phénol/chloroforme est de meilleure qualité car mieux
débarrassé des protéines contaminantes, sa conservation sera meilleure.
Les extraits d’ADN sont mesurés à 260 nm et 280 nm pour vérifier leur pureté, puis
sont ajustés à 250µg/ml. Ils sont dilués au 1/10e pour les PCR ultérieures.
2.2. La PCR.
2.2.1. Introduction.
La PCR (Polymerase Chain Reaction) est une méthode d’amplification génique
apparue en octobre 1985 grâce à Kary Mullis.

Elle consiste à multiplier une petite séquence d’acide nucléique (ADN ou ADNc),
par action d’une ADN polymérase thermostable, isolée d’une bactérie thermophile :
Thermus aqua ticus.
2.2.2. Le choix des amorces.
La taille des amorces est généralement comprise entre 15 et 30 nucléotides. Un
pourcentage de G+C de l’ordre de 40 à 60% est nécessaire.
Lors de la conception des amorces, il faut éviter les séquences palindromiques, qui
induiraient une structure secondaire interne (un repliement dans l’espace), et les
appariements entre paires d’amorces. Ces dernières doivent avoir une température de
fusion (T°m) approximativement identique.
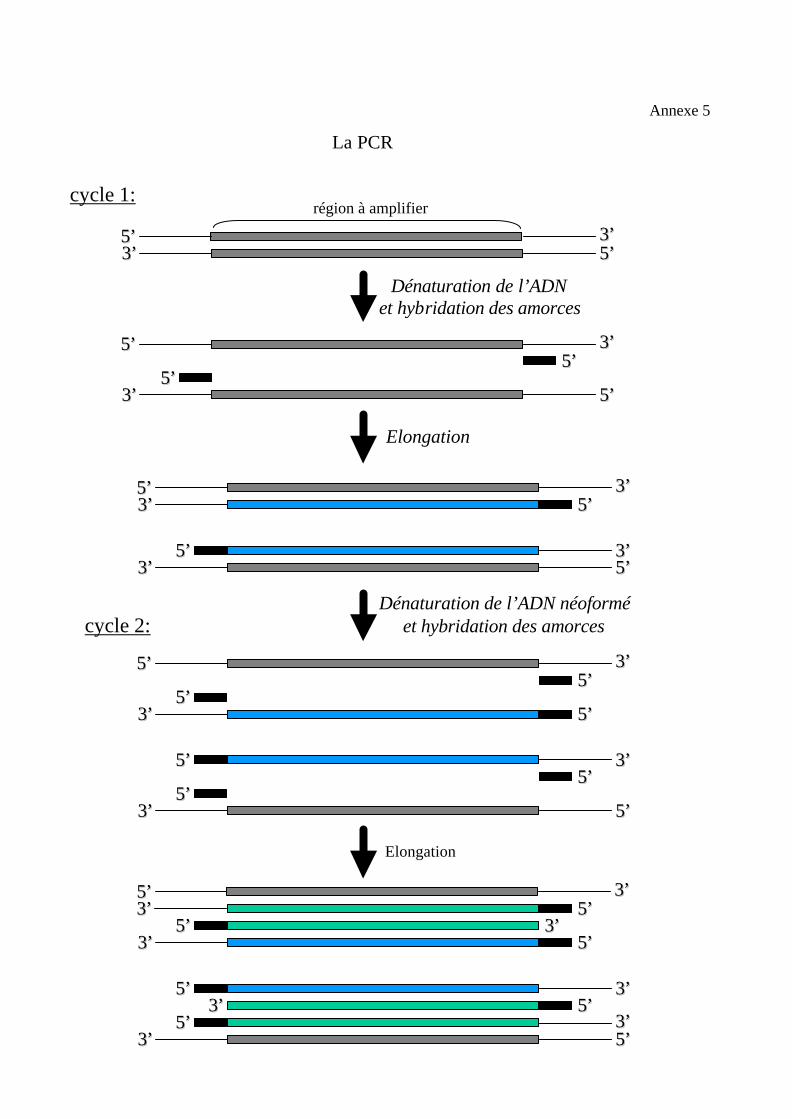
2.2.3. Les 3 étapes de la PCR. (èannexe 5)
2.2.3.1. La dénaturation de l’ADN génomique double brin.
Un chauffage à 94°C pendant 15 secondes à 2 minutes permet la séparation
des deux brins d’ADN, de façon à servir de matrice pour les amorces et la polymérase.
Une dénaturation incomplète, de l’ADN ou des produits de PCR, diminue le
rendement. Une dénaturation excessive entraîne la dégradation de l’enzyme.
2.2.3.2. L’hybridation des amorces.
À une température comprise entre de 50 et 60°C, les amorces se fixent sur
l’ADN cible, par complémentarité, permettant ainsi la synthèse d’ADN par la
polymérase.
Cette hybridation a une durée de 30 à 60 secondes.
Une bonne température d’hybridation est de 5 à 10°C au dessus de la température de
fusion théoriquement calculée des amorces ; en fait, la température d’hybridation des

amorces est fixée empiriquement pour chaque couple d’amorces en fonction de la
spécificité du produit de la réaction et de son rendement.
Une augmentation de température diminue les appariements non spécifiques des
amorces et augmente la spécificité des séquences amplifiées. Mais si la température
est trop élevée, les amorces ne pourront plus s’hybrider et l’ADN ne sera plus
amplifié.
2.2.3.3. La synthèse d’ADN (l’élongation).
La synthèse des nouveaux brins d’ADN débute lorsque la température
optimale (T°opt) de l’enzyme est atteinte (T° d’élongation). Pour la plupart des ADN
polymérases utilisées en PCR, la T° d’élongation est de 72°C.
L’élongation nécessite la présence de désoxynucléotides triphosphates (dNTP) et
d’amorces fixées en amont de la séquence à amplifier.
Cette étape clôture le cycle d’une PCR.
De retour à 94°C, un autre cycle commence.
La durée de l’élongation dépend de la longueur de la séquence à amplifier. On
considère que 1 minute à 72°C suffit pour l’élongation d’1kb d’ADN. La
concentration en dNTP étant le facteur limitant.
2.2.4. Le nombre de cycles.
En théorie, la quantité d’ADN obtenue par PCR est de 2n, où n représente le
nombre de cycle.
30 cycles suffisent généralement pour obtenir une concentration d’ADN permettant sa
visualisation sur gel d’agarose, son clonage et son séquençage.
Toutes les PCR que j’ai effectuées dans ce laboratoire comportaient 40 cycles.

2.2.5. La Taq DNA polymérase.
Nous utilisons une BioTaq polymérase (Quantum Appligène) délétée sur sa partie
N-terminale lui permettant de résister aux hautes températures et évitant l’action
exonucléasique de l’enzyme.
La technique de PCR est utilisée non seulement pour amplifier un fragment
d’ADN, mais également pour les techniques de génotypage.
La BioTaq est commercialisée à 5U/µl et la composition du tampon réactionnel
est : Tris HCl 20mM ; pH (25°C) 9,1 ; Sulfate d’ammonium 16mM ; MgCl2 3,5mM ;
Sérum Albumine Bovine 0,15mg/ml ; Température 72°C.
III. Les polymorphismes analysés.
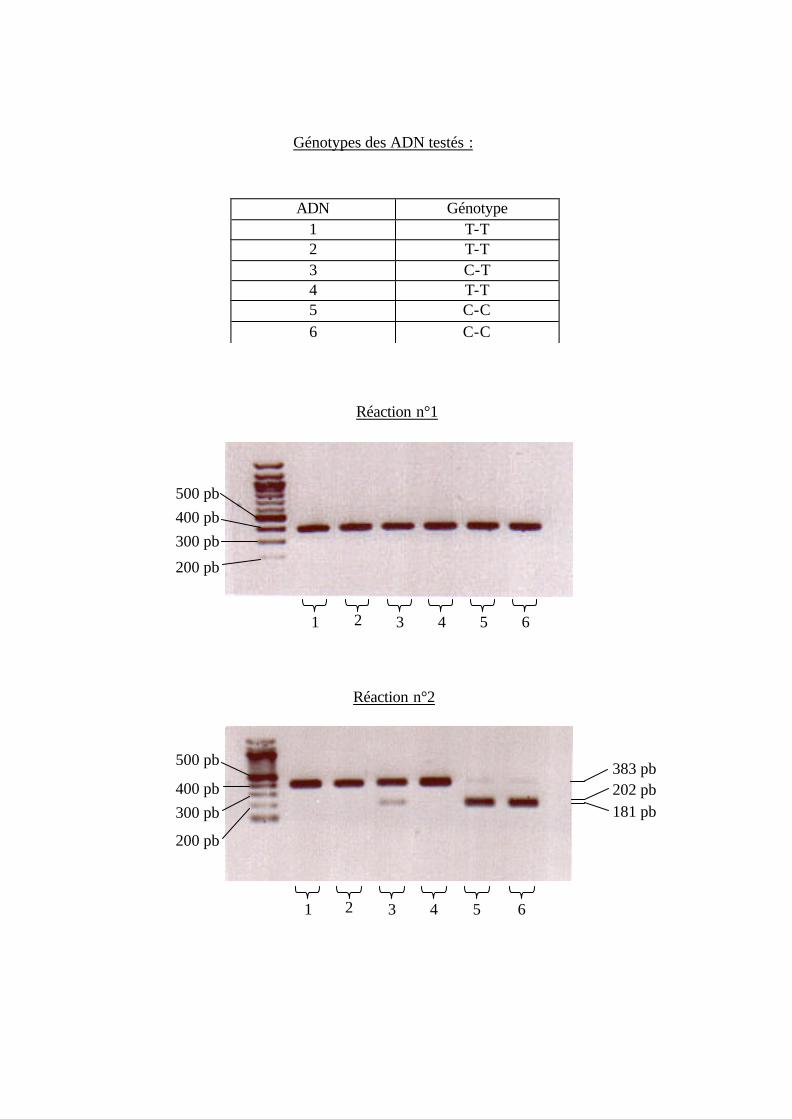
3.1. Le C+1040 T.
L’exon IX du TAFI est de petite taille (88 nucléotides). Pour réaliser l’analyse
génotypique de cette variation CèT sur gel d’agarose à 2%, il faut amplifier une plus
grande région de l’ADN de façon à obtenir des fragments d’ADN facile à analyser sur gel
d’agarose. Pour cela, il a fallu choisir un couple d’amorces sur les introns situés de part et
d’autre de l’exon grâce à un logiciel (Primer Express). La séquence des introns flanquants
l’exon IX a été recherchée dans les banques de données*.
La mise au point de la technique est réalisée à partir de six ADN génomiques
séquencés et de génotype connu pour la région cible, par la technique de PCR (4µl dNTP,
2,5µl Tp10X, 0,25µl amorce ss et as, 18µl H2O, 0,075µl Taq polymérase, 3µl ADN, 57°C)
et les fragments ss-as obtenus ont la taille attendue (èréaction n°1).
L’analyse des cartes de restriction des deux variants de l’exon IX (séquence C ou T)
révèle la présence d’un site de restriction supplémentaire pour l’enzyme Spe I en présence
de l’allèle C. L’utilisation de cette particularité pourrait s’avérer suffisante pour le
* http://www.ncbi.nlm.nih.gov/blast/blast.cgi

génotypage si l’enzyme Spe I n’était pas aussi chère (1730 FF les 1500 unités). Donc la
mise au point d’une technique alternative est nécessaire.
Les produits de PCR sont digérés par l’enzyme Spe I ((5µl produit de PCR, 1,5µl
Tp10X, 10 unités Spe I, 7,5µl H2O, 37°C, 90 minutes) (èréaction n°2)). Seuls les ADN de
séquence C ont été digérés par l’enzyme de restriction Spe I. Donc le fragment amplifié
correspond bien au fragment attendu, aussi bien en taille qu’en profil de restriction (donc la
variation CèT est bien présente dans le fragment amplifié).
Ce nouveau couple d’amorce peut être utilisé pour le génotypage de ce
polymorphisme.
La PCR de l’exon IX étant au point, nous avons essayé de réaliser une PCR spécifique
d’allèle permettant le génotypage et sa lecture sur gel d’agarose 2%. Cette technique
repose sur deux PCR, chacune d’elle correspondant à l’analyse de l’un ou l’autre allèle. Le
mélange réactionnel contient les amorces sens et antisens et l’amorce spécifique sens de
l’allèle C ou T plus : les dNTP, le tampon de l’enzyme, l’ADN polymérase et l’ADN
génomique.
Les conditions expérimentales seront établies, afin de favoriser l’hybridation de
l’amorce spécifique d’allèle, quand sa séquence complémentaire est présente. En l’absence
d’hybridation de l’amorce allèle spécifique sur l’ADN matrice, de part l’absence de
l’allèle, les fragments correspondant à l’hybridation des sondes sens et antisens pourront
être visualisé sur gel d’agarose plus BET.
La technique PCR allèle spécifique se déroule comme une PCR classique à ceci
près que l’on rajoute, au milieu réactionnel, une amorce spécifique d’allèle et que l’on
cherche à favoriser son hybridation.
La théorie de la réaction est schématisée ci-après :
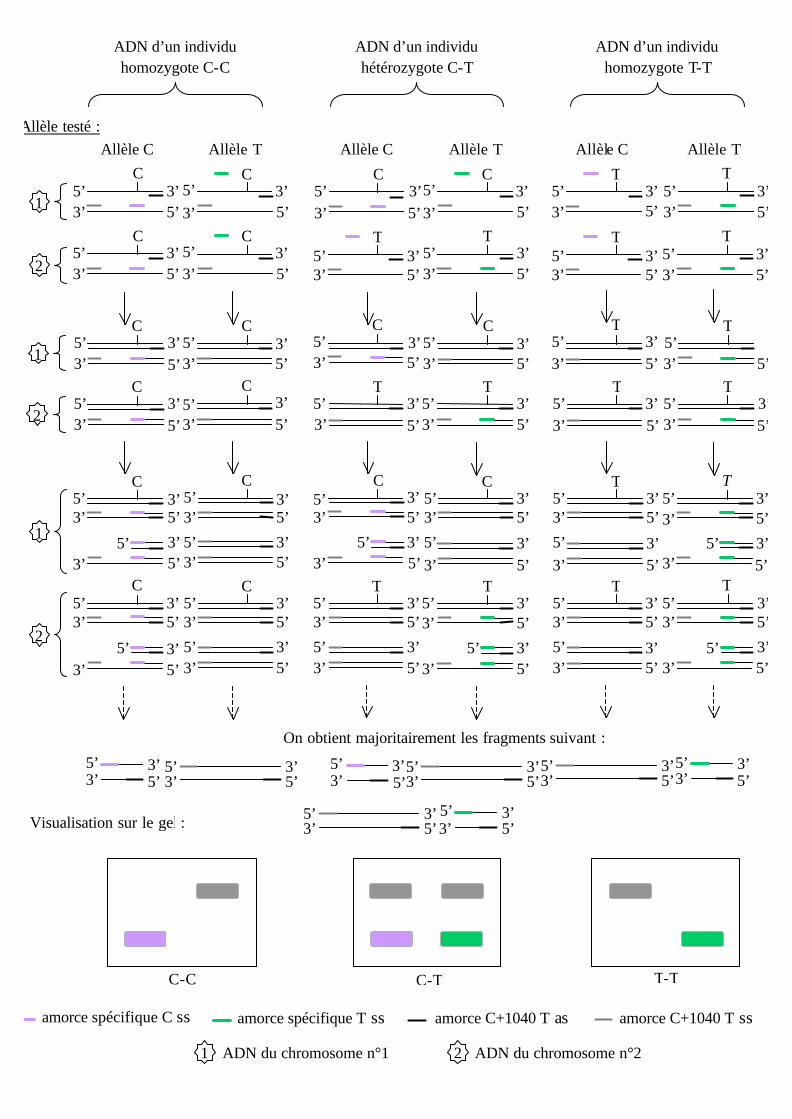
amorce spécifique C ss amorce spécifique T ss amorce C+1040 T as amorce C+1040 T ss
Visualisation sur le gel :
2
On obtient majoritairement les fragments suivant :
Allèle testé : Allèle C Allèle T
C
5’ 3’5’ 3’ 5’ 3’
5’ 3’
C
3’
5’C
3’
5’ 5’
3’ 5’
3’
5’ 3’ 5’ 3’
5’ 3’ 5’ 3’
C
C 5’ 3’
5’ 3’ 5’
C
3’ 3’ 5’
C
5’ 3’5’ 3’ 5’ 3’
5’ 3’
C
C 5’ 3’
5’ 3’ 5’
C
3’ 3’ 5’
5’
3’ 5’
3’ 3’
5’C
3’
5’
5’ 3’ 5’ 3’
5’ 3’ 5’ 3’
C
5’3’5’ 3’
5’ 3’
T T
3’ 5’
T 5’ 3’
5’ 3’ 5’
T
3’ 5’
5’3’
3’5’T
3’ 5’
3’5’
5’ 3’ 5’ 3’
5’ 3’ 5’ 3’
T
5’ 3’ 5’ 3’
T T
5’3’ 5’ 3’
T 5’ 3’
5’ 3’5’
T
3’ 3’5’
5’3’
3’5’T
3’ 5’
3’5’
5’ 3’ 5’ 3’
5’ 3’ 5’ 3’
T
C
5’ 3’ 5’ 3’
5’
3’ T
5’
3’ 5’ 3’
5’3’
5’ 3’
3’ 5’ C
3’ 5’
3’ 5’
5’ 3’ T
5’ 3’
5’ 3’ C
5’ 3’
5’ 3’ 5’ 3’
T T
5’ 3’ 5’ 3’
5’ 3’ 5’ 3’
C
T 5’ 3’
5’ 3’
C5’ 3’
5’ 3’
5’ 3’ 5’ 3’
5’ 3’ 5’ 3’
T
5’ 3’ 5’ 3’
5’ 3’ 5’ 3’
C
Allèle C Allèle C Allèle T Allèle T
ADN d’un individu homozygote C-C
ADN d’un individu homozygote T-T
ADN d’un individu hétérozygote C-T
5’ 5’ 3’
3’ 5’
5’ 3’ 3’ 5’
5’ 3’
3’ 5’
5’ 3’ 3’
5’5’ 3’
3’
5’ 5’ 3’
3’
5’ 5’ 3’ 3’
5’ 5’ 3’ 3’
1
1
1
2
2
1 ADN du chromosome n°1 2 ADN du chromosome n°2
C-C T-T C-T

Après analyse des résultats, nous avons essayé de réaliser une PCR allèle spécifique à
57°C sur les six ADN de génotype connu pour estimer la qualité du génotypage.
Mode opératoire :
dNTP 4µl Tp10X 2,5µl
Amorce ss 0,25µl Amorce as 0,25µl
Amorce spécifique C/T 0,25µl H2O 17,75µl
Taq polymérase 0,075µl ADN 3µl
Conclusion : La réaction se déroule parfaitement à 57°C puisque les génotypes attendus (1, 2
et 4 : T-T ; 3 : C-T ; 5 et 6 : C-C) sont retrouvés et lus très aisément et sans ambiguïté.
3.2. Le G+663 A.
L’exon VI est amplifié entièrement, en utilisant le couple d’amorce utilisé pour le
criblage de la région codante. Ces amorces sont localisées dans les parties chevauchantes
exon- intron.
Le polymorphisme est identifié et localisé par séquençage. La carte de restriction de
chaque allèle est identique, donc il est impossible de les différencier par méthode
enzymatique. Il nous a fallu mettre au point la PCR allèle spécifique pour pouvoir
génotyper ce polymorphisme, dont la théorie est schématisée ci-dessous.
Deux PCR sont réalisées en parallèle, chacune des PCR contenant l’amorce spécifique
de l’un des deux allèles.
Résultat :
100 pb 200 pb 300 pb 400 pb 500 pb
1 2 6 5 4 3
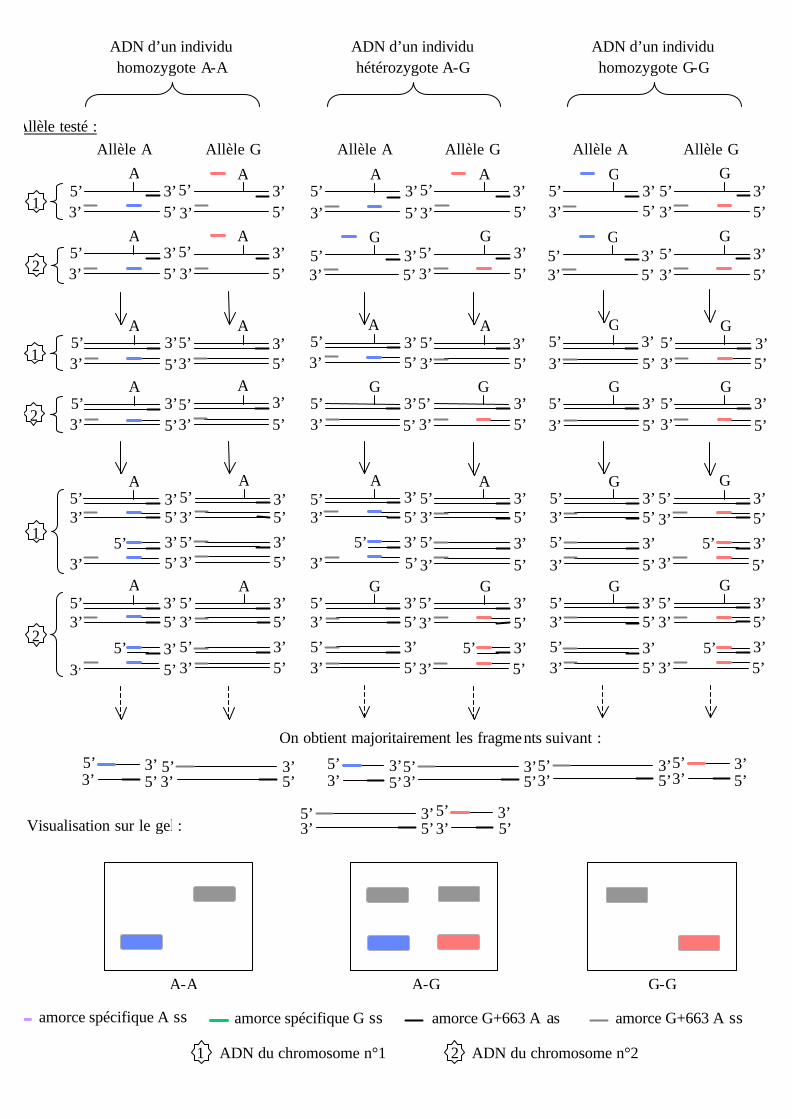
amorce spécifique A ss amorce spécifique G ss amorce G+663 A as amorce G+663 A ss
Visualisation sur le gel :
2
Allèle testé : Allèle A Allèle G
A
5’ 3’ 5’ 3’ 5’ 3’
5’ 3’
A
3’
5’A
3’
5’ 5’
3’ 5’
3’
5’ 3’ 5’ 3’
5’ 3’ 5’ 3’
A
A 5’ 3’
5’ 3’ 5’
A
3’ 3’ 5’
A
5’ 3’ 5’ 3’ 5’ 3’
5’ 3’
A
A 5’ 3’
5’ 3’ 5’
AA
3’ 3’ 5’
5’
3’ 5’
3’ 3’
5’A
3’
5’
5’ 3’ 5’ 3’
5’ 3’ 5’ 3’
A
5’ 3’ 5’ 3’
5’ 3’
G G
3’ 5’
G5’ 3’
5’ 3’5’
G
3’ 3’5’
5’ 3’
3’ 5’ G
3’ 5’
3’ 5’
5’ 3’ 5’ 3’
5’ 3’ 5’ 3’
G
5’ 3’ 5’ 3’
G G
5’ 3’ 5’ 3’
G5’ 3’
5’ 3’ 5’
G
3’ 3’ 5’
5’ 3’
3’ 5’ G
3’ 5’
3’ 5’
5’ 3’ 5’ 3’
5’ 3’ 5’ 3’
G
A
5’ 3’ 5’ 3’
5’
3’ G
5’
3’ 5’ 3’
5’3’
5’ 3’
3’ 5’A
3’ 5’
3’ 5’
5’ 3’ G
5’ 3’
5’ 3’ A
5’ 3’
5’ 3’ 5’ 3’
G G
5’ 3’ 5’ 3’
5’ 3’ 5’ 3’
A
G 5’ 3’
5’ 3’
A5’ 3’
5’ 3’
5’ 3’ 5’ 3’
5’ 3’ 5’ 3’
G
5’ 3’ 5’ 3’
5’ 3’ 5’ 3’
A
Allèle A Allèle A Allèle G Allèle G
ADN d’un individu homozygote A-A
ADN d’un individu homozygote G-G
ADN d’un individu hétérozygote A-G
On obtient majoritairement les fragments suivant :
5’5’ 3’
3’ 5’
5’ 3’ 3’ 5’
5’ 3’
3’ 5’
5’ 3’ 3’
5’ 5’3’
3’
5’ 5’ 3’
3’
5’ 5’ 3’ 3’
5’ 5’ 3’ 3’
1
1
1
2
2
1 ADN du chromosome n°1 2 ADN du chromosome n°2
A-A A-G G-G

On a réalisé un premier essai à 56°C dans les conditions opératoir es suivantes : 4µl
dNTP, 2,5µl Tp10X, 0,25µl amorces sens, antisens et spécifique d’allèle A ou G, 17,75µl
H2O, 0,075µl Taq polymérase, 3µl ADN génomique et 40 cycles. Mais la réaction est peu
spécifique donc un deuxième essai est réalisé sur des ADN de séquence et de génotype
connu à 59°C dans les même conditions. Les génotypes sont corrects et la lecture est facile
et sans ambiguïté. Ces conditions seront donc appliquées pour le génotypage de cette
variation.
Ces deux techniques de génotypage étant au point, nous avons tenté de les réunir en
une seule manipulation nommée “multiplex”.
3.3. Le multiplex : C+1040 T et G+663 A.
Le multiplex est envisageable car la position des amorces a été choisie afin que la
taille des bandes amplifiées soit analysable sur le même gel d’agarose. La taille des
fragments du génotypage de l’exon IX sont supérieures à celles de l’exon VI. Ainsi, sur le
gel d’agarose à 2%, on lira le génotype de l’exon IX avant celui de l’exon VI. On ajoute
donc au milieu réactionnel une amorce sens, une amorce antisens ainsi qu’une amorce
spécifique de l’allèle C ou T de séquence sens pour l’exon IX, et idem pour l’exon VI avec
une amorce sens spécifique de l’allèle A ou G.
Séquences des amorces et tailles des fragments amplifiés (en pb) :
C+1040 T SS : CCTTATTTATTTGCCTTTTA AS : CTATTATGCCTTCTGTTCCTA
383 C : TCGTGCTATTGAGAAAAC T : TCGTGCTATTGAGAAAAT
202
G+663 A SS : ATACTTGGTTTAATTAGCAG AS : TTCTTTGCCTTTTCTCCTAC
145 A : GATTTCTATGTTATGCCA G : GATTTCTATGTTATGCCG
60
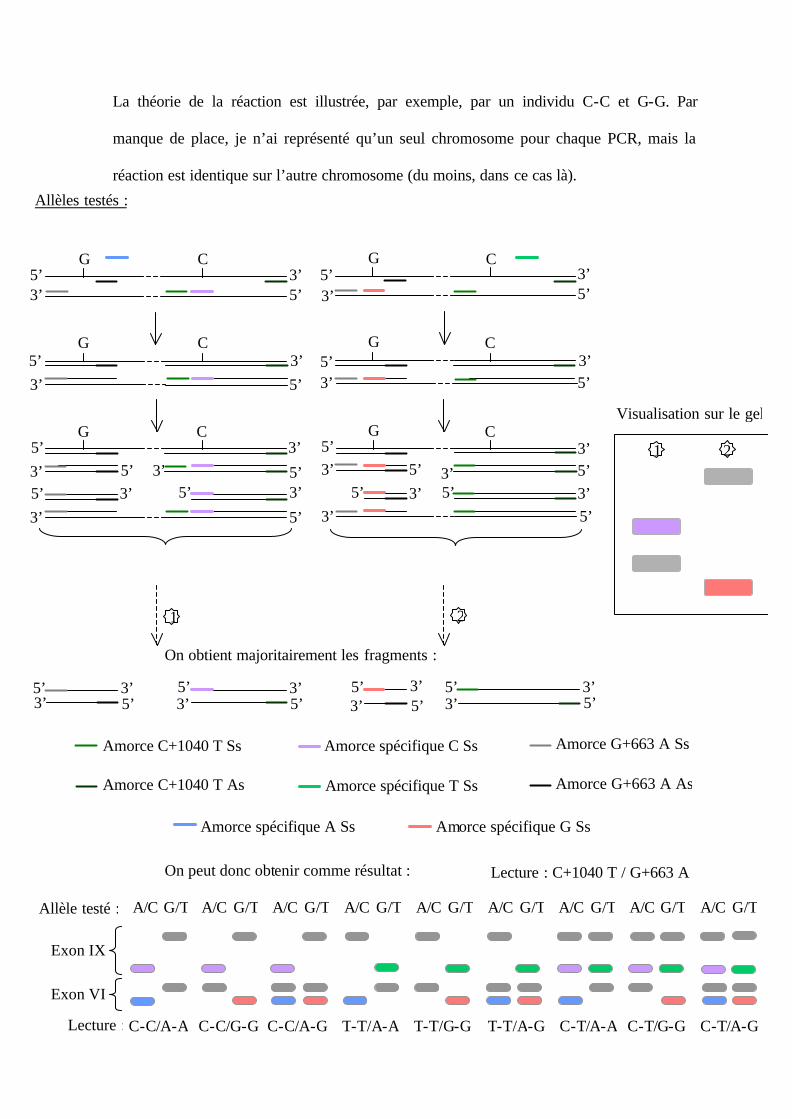
La théorie de la réaction est illustrée, par exemple, par un individu C-C et G-G. Par
manque de place, je n’ai représenté qu’un seul chromosome pour chaque PCR, mais la
réaction est identique sur l’autre chromosome (du moins, dans ce cas là).
On obtient majoritairement les fragments :
21
5’ 3’ 3’ 5’ 3’ 3’ 3’ 3’ 3’ 3’ 5’ 5’
5’ 5’ 5’ 5’
3’ 5’
3’
3’
5’
5’
3’
5’ 3’
5’
3’ 5’
3’
5’
C
C
C
3’ 5’
G
G
G
3’ 5’ 3’
5’
5’ 3’
5’ 3’
5’ 3’
3’
C
C
C
5’ 3’
5’
3’ 5’
3’ 5’ 3’
G
G
G
5’ 5’
5’ 3’
3’
Allèles testés :
21
Visualisation sur le gel
Amorce C+1040 T Ss
Amorce C+1040 T As
Amorce G+663 A Ss
Amorce G+663 A As
Amorce spécifique C Ss
Amorce spécifique T Ss
Amorce spécifique A Ss Amorce spécifique G Ss
A/C G/T A/C G/T A/C G/T A/C G/T A/C G/T A/C G/T A/C G/T A/C G/T A/C G/T
C-C/A-A C-C/G-G C-C/A-G T-T/A-A T-T/G-G T-T/A-G C-T/A-A C-T/G-G C-T/A-G Lecture :
Allèle testé :
On peut donc obtenir comme résultat : Lecture : C+1040 T / G+663 A
Exon IX
Exon VI

En pratique, il faut veiller à ce qu’il n’y ait pas compétition entre les deux PCR allèles
spécifiques. Pour cela, il faut jouer sur la température d’hybridation des amorces et la
concentration rela tive de chacune d’elles ; car la PCR la plus efficace va tirer la réaction et
s’amplifier majoritairement.
IV. Optimisation du multiplex.
Nous avons essayé d’améliorer la spécificité de ces réactions en faisant varier différents
facteurs.
Les différents essais et leur conclusion.
Nous avons utilisé pour cela 6 ADN dont le génotype a été déterminé par séquençage
direct
Ø La PCR allèle spécifique de l’exon VI nécessite une température d’hybridation de 59°C, et
celle de l’exon IX est de 57°C. Nous avons donc réalisé un essai de multiplex à une
température moyenne : 58°C.
Le marqueur de taille utilisé est un MW ladder 100 bp : fragment de 100 à 1000 pb.
Essai Référence C+1040 T G+663 A
1 209 T-T A-A
2 344 T-T A-A
3 217 C-T A-G
4 441 T-T A-G
5 127 C-C G-G
6 360 C-C G-G
7 témoin sans DNA
des produits de PCR :
ADN Génotype 1 T-T 2 T-T 3 C-T 4 T-T 5 C-C 6 C-C
Génotypes des ADN testés :
200 pb
300 pb 400 pb 500 pb
Réaction n°1
1 2
3 4 5 6
200 pb
300 pb 400 pb
500 pb
Réaction n°2
1 2
3 4 5 6
202 pb 181 pb
383 pb
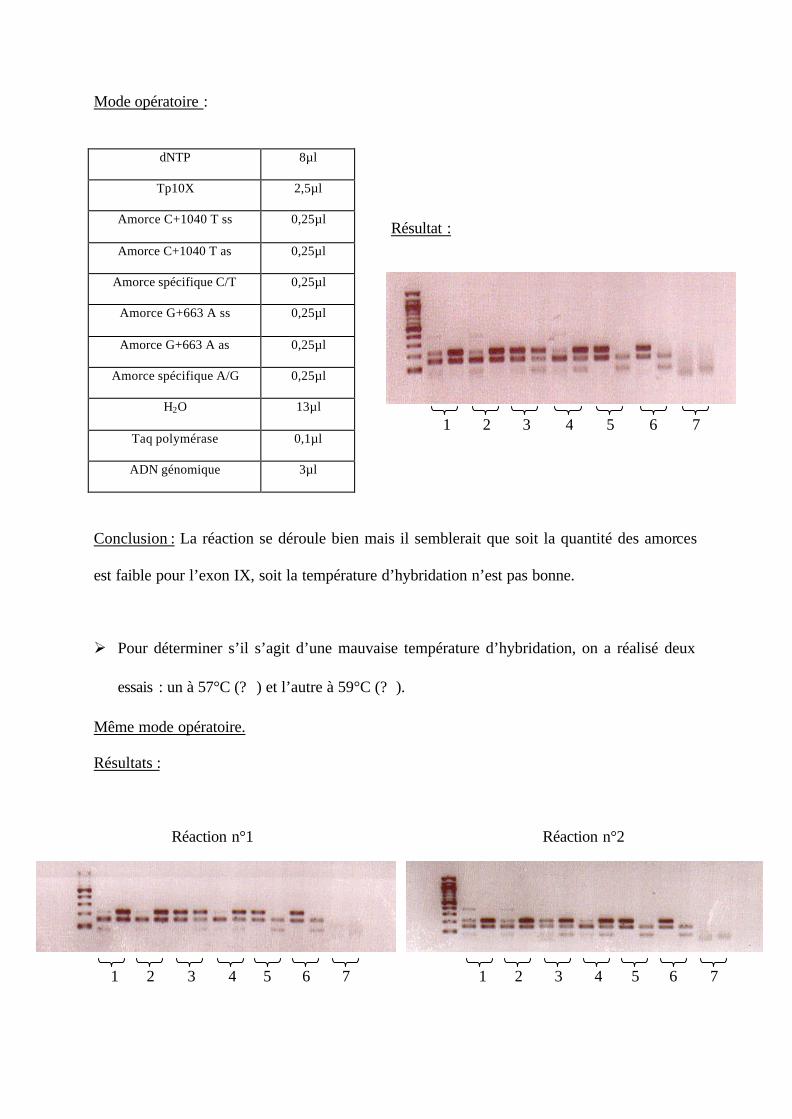
Mode opératoire :
dNTP 8µl
Tp10X 2,5µl
Amorce C+1040 T ss 0,25µl
Amorce C+1040 T as 0,25µl
Amorce spécifique C/T 0,25µl
Amorce G+663 A ss 0,25µl
Amorce G+663 A as 0,25µl
Amorce spécifique A/G 0,25µl
H2O 13µl
Taq polymérase 0,1µl
ADN génomique 3µl
Conclusion : La réaction se déroule bien mais il semblerait que soit la quantité des amorces
est faible pour l’exon IX, soit la température d’hybridation n’est pas bonne.
Ø Pour déterminer s’il s’agit d’une mauvaise température d’hybridation, on a réalisé deux
essais : un à 57°C (? ) et l’autre à 59°C (? ).
Même mode opératoire.
Résultats :
Résultat :
1 2 3 4 5 6 7
Réaction n°1 Réaction n°2
1 2 3 4 5 6 7 1 2 3 4 5 6 7

Conclusion : Malgré l’absence du fragment sens-antisens, à 57°C, l’exon IX est facilement
lisible contrairement à l’exon VI (sauf pour les génotypes n°5 et n°6). La réaction est peu
spécifique à 59°C (sauf pour les génotypes n°5 et n°6). Nous allons changer de température
pour favoriser les fragments sens-antisens de l’exon IX.
Ø Pour essayer de rétablir la balance entre les fragments sens-antisens des deux
génotypages, on a augmenté la quantité des amorces ss, as et spécifique de l’exon IX et de
l’amorce spécifique de l’exon VI à deux températures différentes : 56°C (? ) et 58°C (? ).
Mode opératoire :
DNTP 8µL Amorce G+663 A ss 0,25µl
Tp10X 2,5µl Amorce G+663 A as 0,25µl
Amorce C+1040 T ss 0,5µl Amorce spécifique A/G 0,5µl
Amorce C+1040 T as 0,5µl ADN génomique 5µl
Amorce spécifique C/T 0,5µl Taq polymérase 0,1µl
H2O 7µl
Résultats : (absence de témoins).
Réaction n°2 Réaction n°1
1 2 3 4 5 6 1 2 3 4 5 6

Conclusion : Le génotypage de l’exon IX, à 56°C, est trop favorisé par rapport au génotypage
de l’exon VI. A 58°C, malgré la présence homogène du fragment sens-antisens de l’exon VI,
les deux génotypes sont correctement lisibles. Néanmoins, la bande spécifique-antisens de
l’exon VI est trop faible.
Ø Pour rendre prioritaire le fragment spécifique-antisens de l’exon VI face au fragment sens-
antisens, on a diminué la quantité de moitié la quantité d’amorce G+663 A sens. La
réaction est testée à une température de 57°C.
Mode opératoire :
dNTP 8µl
Tp10X 2,5µl
Amorce C+1040 T ss 0,5µl
Amorce C+1040 T as 0,5µl
Amorce spécifique C/T 0,5µl
Amorce G+663 A ss 0,125µl
Amorce G+663 A as 0,25µl
Amorce spécifique A/G 0,5µl
H2O 7µl
Taq polymérase 0,1µl
ADN génomique 5µl
Conclusion : Malgré l’absence totale des fragments sens-antisens du génotypage de l’exon IX,
la réaction et la lecture sont très satisfaisante.
Résultat :
1 2 3 4 5 6 7
*
* : Quantité insuffisante d’ADN.

Ø Pour valider ces conditions opératoires, on a réalisé ce multiplex sur 47 ADN génotypés,
pour les exon VI et exon IX, en simplex.
Les résultats sont identiques dans les deux cas.
La reproductibilité et la stabilité de la réaction, à 57°C et dans ces conditions opératoires,
nous permettent de valider cette technique.
Voici un exemple de quelques résultats obtenus :
V. Conclusions.
5.1. Scientifique.
Je vais conclure ce rapport sur une étude épidémiologique réalisé en utilisant cette
technique. Nous avons analysé une population marseillaise de 85 patients atteints
d’infarctus et de 121 sujets témoins appariés en âge et en sexe. Les résultats sont les
suivants : la fréquence de l’allèle A du polymorphisme G+663 A est de 0,25 pour les
patients et de 0,27 pour les témoins, et la fréquence de l’allèle T du polymorphisme
C+1040 T est de 0,14 pour les patients et de 0,24 pour les témoins.
La différence cas /témoins a un faible écart-type (p=0,04), donc les résultats obtenus
sont significatifs.
C-T C-C C-T C-C C-T C-C C-T C-C
A-G G-G A-G G-G G-G G-G G-G G-G
Exon IX :
Exon VI :

Donc le polymorphisme de l’exon IX semble être corrélé avec la pathologie :
l’infarctus du myocarde. Cette étude doit être complétée car le nombre d’individus inclus
dans le protocole est trop faible.
L’exon VI est en fort déséquilibre de liaison avec les autres polymorphismes de la
région promotrice et de la région 3’ non traduite, contrairement à l’exon IX qui est
faiblement lié.
Nous pouvons également envisager que le changement d’un acide aminé,
correspondant à la variation de séquence nucléotidique de l’exon IX du TAFI, peut
modifier la fonction de la protéine et augmenter ainsi le risque d’apparition d’un infarctus
du myocarde.
Ces deux polymorphismes sont liés aux variations du taux plasmatique du TAFI. La
variation de l’exon IX explique 29% de la variation du taux alors que, l’exon VI en
explique 23%.
Cette liaison paraît faible mais elle est très forte par rapport aux résultats trouvés avec
d’autres molécules. Pour le PAI-1 par exemple (un autre inhibiteur de la fibrinolyse), la
génétique explique seulement 2% de la variation du taux plasmatique.
5.2. Personnelle.
Ce stage m’a permis de comprendre que la recherche consiste le plus souvent à
optimiser des méthodes pour chacune des étapes du travail que l’on réalise.
Il m’a fait découvrir un aspect très intéressant de la recherche en biologie moléculaire :
l’aspect ethnologique.
J’ai pris conscience de l’importance des outils informatiques et notamment de
l’Internet dans la démarche scientifique.

L’ambiance sympathique, l’autonomie et la confiance qui m’a été accordé, m’ont
donné le goût du travail d’équipe, une idée concrète du travail de technicien de laboratoire
et me font regretter la durée trop courte de ce stage.

ANNEXES
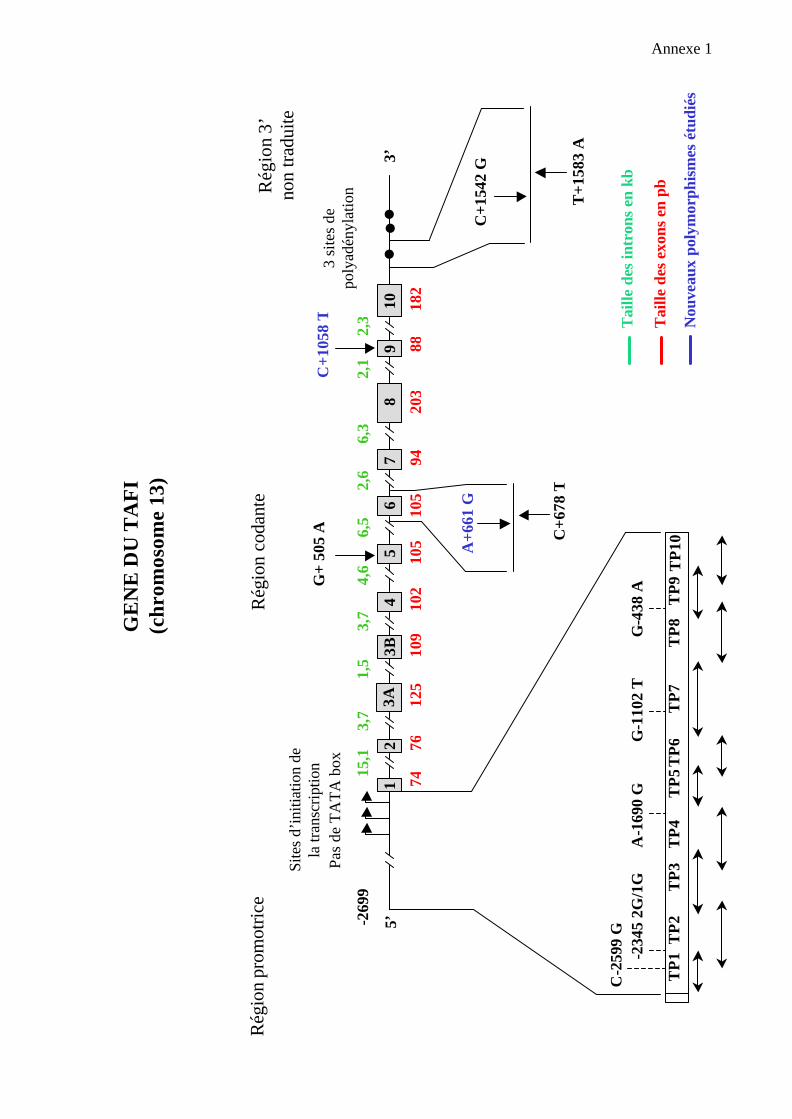
Annexe 1
3 si
tes
de
poly
adén
ylat
ion
Site
s d’
initi
atio
n de
la
tran
scrip
tion
Pas
de T
AT
A b
ox
G+
505
A
GE
NE
DU
TA
FI
(chr
omos
ome
13)
Rég
ion
prom
otric
e R
égio
n co
dant
eR
égio
n 3’
no
n tra
duite
C+1
542
G
T+1
583
A
TP
1T
P2
TP
3T
P4
TP
5T
P6
TP
7T
P8
TP
9
C-2
599
G
-234
5 2G
/1G
A
-169
0 G
G
-110
2 T
G
-438
A T
P10
lll
3’
-269
9
7488
7612
5 20
310
910
210
510
594
182
15,1
3,7
2,3
1,5
4,6
3,7
2,6
6,5
6,3
2,1
5’
A+6
61 G
C+6
78 T
C+1
058
T Tai
lle d
es in
tron
s en
kb
Tai
lle d
es e
xons
en
pb
Nou
veau
x po
lym
orph
ism
es é
tudi
és
2
3A
3B
4
5 6
7
8
9
1
1
0

La technique SSCP.
(Single Strand Conformation Polymorphism)
Cette technique permet l’analyse des ADN simples brins par migration
électrophorétique en gel de polyacrylamide non dénaturant. Les ADN simples brins migrent
en fonction de leur structure tertiaire, et donc de leur séquence nucléotidique.
Le changement d’une seule base se traduit par une différence de mobilité entre brin muté et
brin sauvage.
Une fraction du produit de PCR est dénaturée par chauffage puis déposée sur un gel
d’acrylamide non dénaturant à 6% ou 10% contenant ou non 10% de glycérol à température
ambiante.
Une différence dans les profils de migration, révèle l’existence d’une variation de séquence.
Dans ce laboratoire, il a été utilisé un marquage dit “froid”. Les fragments d’ADN sont
marqués par incorporation de dUTP biotine en cours de PCR.
Après transfert sur membrane, l’ADN est révélé grâce à la réaction streptavidine-NBT-BCIP.
L’existence des variations de séquence devra être confirmée par séquençage.
Annexe 2
Hétérozygote
Homozygote Homozygote
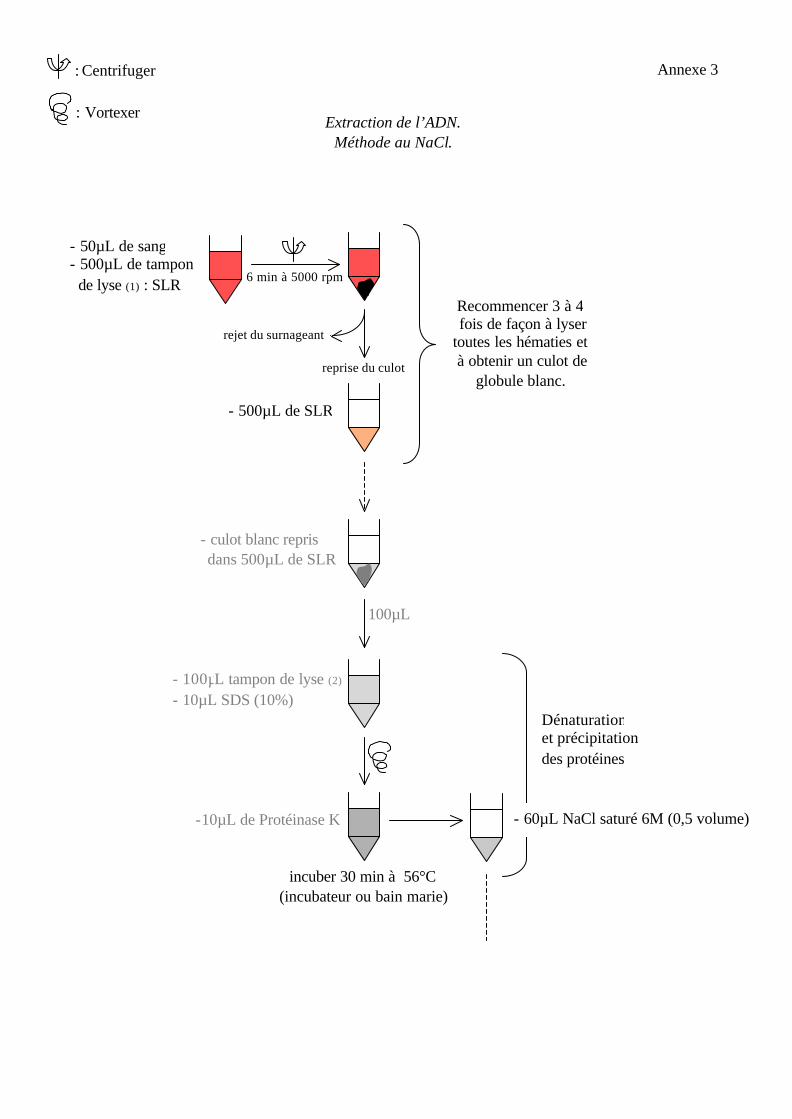
Extraction de l’ADN. Méthode au NaCl.
- 50µL de sang - 500µL de tampon de lyse (1) : SLR 6 min à 5000 rpm
reprise du culot
rejet du surnageant
- 500µL de SLR
Recommencer 3 à 4 fois de façon à lyser toutes les hématies et à obtenir un culot de
globule blanc.
- culot blanc repris dans 500µL de SLR
100µL
- 100µL tampon de lyse (2)
- 10µL SDS (10%)
-10µL de Protéinase K
incuber 30 min à 56°C (incubateur ou bain marie)
- 60µL NaCl saturé 6M (0,5 volume)
Dénaturation et précipitation des protéines
Annexe 3 : Centrifuger
: Vortexer
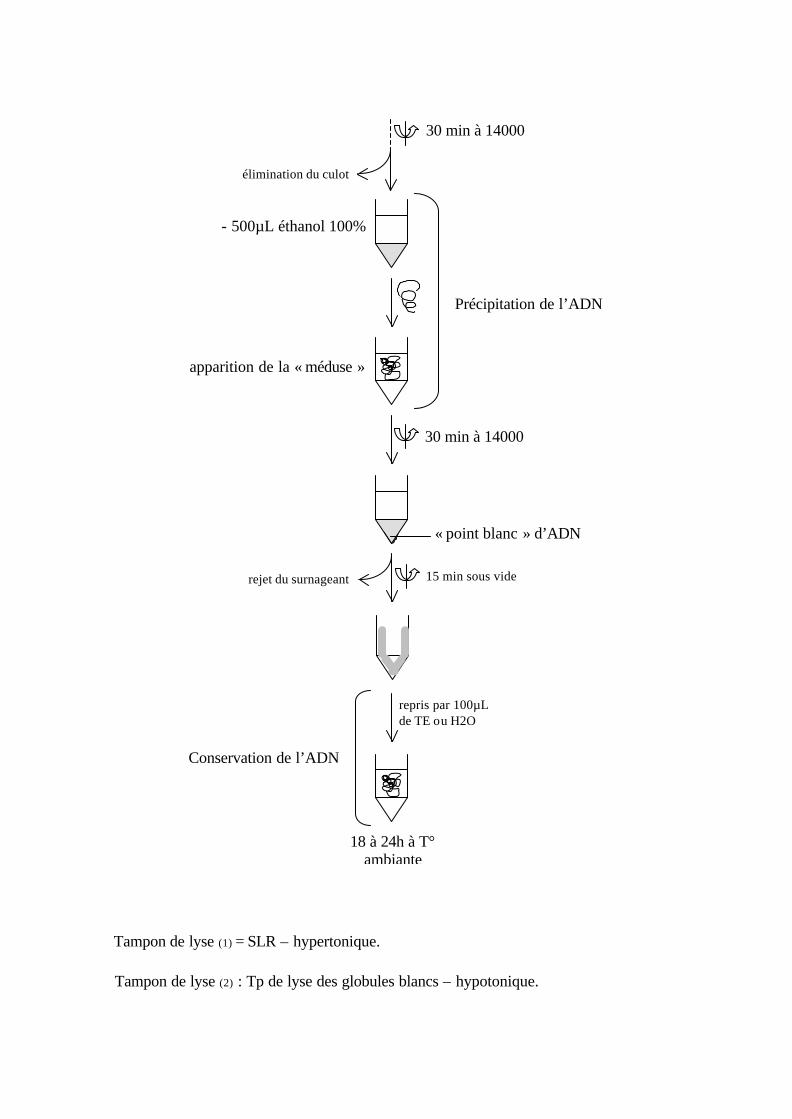
Tampon de lyse (1) = SLR – hypertonique.
30 min à 14000 rpm
élimination du culot
- 500µL éthanol 100%
apparition de la « méduse »
30 min à 14000
Précipitation de l’ADN
« point blanc » d’ADN
rejet du surnageant 15 min sous vide
repris par 100µL de TE ou H2O
18 à 24h à T° ambiante
Conservation de l’ADN
Tampon de lyse (2) : Tp de lyse des globules blancs – hypotonique.
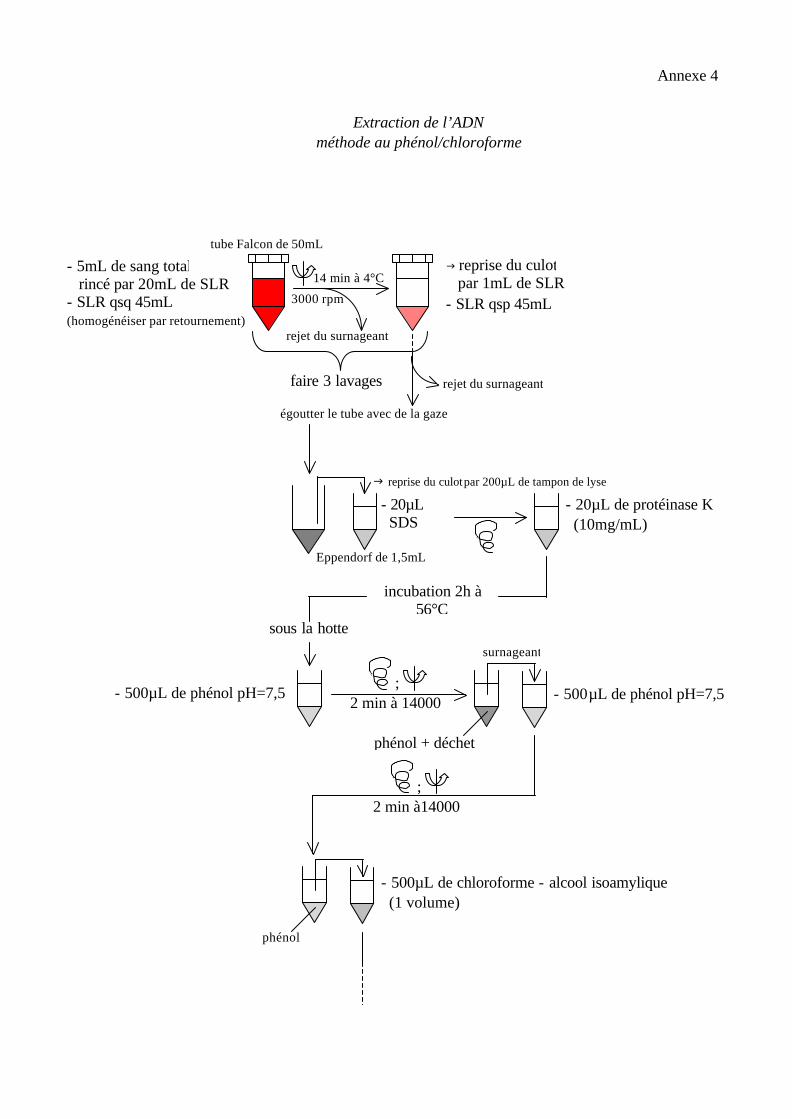
Extraction de l’ADN méthode au phénol/chloroforme
- 5mL de sang total rincé par 20mL de SLR - SLR qsq 45mL (homogénéiser par retournement)
14 min à 4°C
3000 rpm
rejet du surnageant (1)
g reprise du culot par 1mL de SLR - SLR qsp 45mL
faire 3 lavages rejet du surnageant
égoutter le tube avec de la gaze
g reprise du culot par 200µL de tampon de lyse
tube Falcon de 50mL
Eppendorf de 1,5mL
- 20µL SDS 10%
- 20µL de protéinase K (10mg/mL)
incubation 2h à 56°C
sous la hotte
; - 500µL de phénol pH=7,5
2 min à 14000
surnageant
- 500µL de phénol pH=7,5
; 2 min à14000
phénol + déchet
phénol
- 500µL de chloroforme - alcool isoamylique (1 volume)
Annexe 4
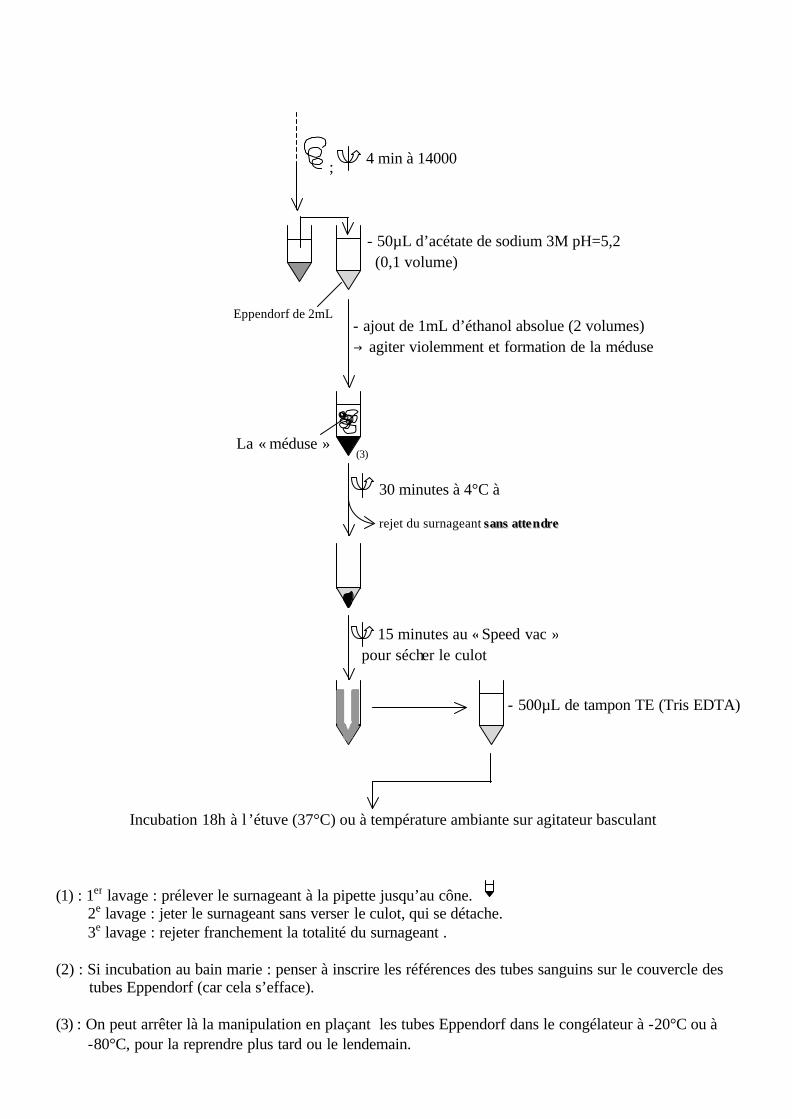
(1) : 1er lavage : prélever le surnageant à la pipette jusqu’au cône. 2e lavage : jeter le surnageant sans verser le culot, qui se détache. 3e lavage : rejeter franchement la totalité du surnageant . (2) : Si incubation au bain marie : penser à inscrire les références des tubes sanguins sur le couvercle des tubes Eppendorf (car cela s’efface). (3) : On peut arrêter là la manipulation en plaçant les tubes Eppendorf dans le congélateur à -20°C ou à -80°C, pour la reprendre plus tard ou le lendemain.
- 500µL de tampon TE (Tris EDTA)
; 4 min à 14000
- 50µL d’acétate de sodium 3M pH=5,2 (0,1 volume)
Eppendorf de 2mL - ajout de 1mL d’éthanol absolue (2 volumes) g agiter violemment et formation de la méduse
La « méduse » (3)
rejet du surnageant ss aannss aattttee nn ddrree
30 minutes à 4°C à
15 minutes au « Speed vac » pour sécher le culot
Incubation 18h à l ’étuve (37°C) ou à température ambiante sur agitateur basculant
33’’
55’’
La PCR
cycle 1:
cycle 2:
33’’ 55’’55’’33’’
région à amplifier
33’’ 55’’55’’
55’’33’’55’’
55’’
55’’33’’
55’’
55’’
33’’
33’’
Dénaturation de l’ADN et hybridation des amorces
Elongation
Dénaturation de l’ADN néoforméet hybridation des amorces
33’’ 55’’
33’’
55’’
55’’33’’
55’’ 33’’55’’
55’’
55’’
Elongation
33’’ 55’’
33’’
55’’33’’
55’’ 33’’55’’
55’’
55’’ 33’’
33’’ 33’’
33’’
Annexe 5
55’’
55’’

PREPARATION DES REACTIFS POUR L’EXTRACTION DES DNA.
1. TRIS-HCI 1M pH 7,5 (MW 121,1 1)
Peser 60,56 g dissoudre dans 900 ml de H2O ultrapure stérile, ajuster le p H à 7,5 avec HCI fumant et compléter à 100 ml. (stérilisation par autoclavage) conservation à + 4°C
2. MgCl 2-6H 2O 2M (MW 203,30) Peser 81,32 g et dissoudre dans 200 ml de H2O ultrapure stérile. (stérilisation par filtration)
3. NaCI 4 M: Sodium Chloride Sigma 1 M = 58,44 soit 233,76 g/1 soit 1 1,688g/50cc 4. EDTA 0,5M Ph 8 (MW 372,3)
Peser 93,05 g, dissoudre dans 250 ml de H2O ultrapure stérile puis ajuster le pH à 8 avec de la soude puis compléter avec H2O qsp 500 ml. (stérilisation par la chaleur) se conserve à + 4'C.
5. S L R(1) TRIS-HCI pH 7,5 1M 10ml 10mM MgCl2-6H2O 2M 2,5ml 5mM NaCl 4M 2,5ml 10mM H2O qsp 1l
(filtrer sur 0,45µ et conserver à + 4°C)
6. TAMPON DE LYSE DES GLOBULES BLANCS(2) TRIS-HCI pH 7,5 1M 500µl 10mM NaCl 5M 5ml 400mM EDTA pH 8 0,5M 200µl 2mM H2O qsp 50ml (filtrer sur 0,2µ et conserver à +4°C)
7. SDS 10% Laury] sulfate de sigma Peser 5 g de sodium dodecyl sulfate (produit désagréable à inhaler car très agressif pour les muqueuses, prendre garde de ne pas le respirer) compléter à 50ml exactement (dans une éprouvette) avec de l'eau distillée stérile. Puis stériliser. Se conserve à température ambiante.
8 . Protéinase K Cette enzyme, reçue en général à l'état lyophilisé, est régénérée avec de l'eau distillée stérile afin d'avoir une concentration finale de 10 mg/ml. Quand la solubilisation est complète le produit est aliquoté à raison de 500µl par tube et conservé à –20°C jusqu'à utilisation. Les tubes décongelés peuvent être recongelés.
9. NaCl saturé (6M) Peser la quantité nécessaire de NaCl pour obtenir une solution 6M (soit : poids moléculaire du NaCl x6x par le volume en litre que l'on veut préparer), ajouter une quantité d'eau distillée inférieure au volume final que l'on veut obtenir, mettre en agitation et chauffer afin de faciliter la solubilisation. Elle ne sera jamais complétée. Transvaser dans une éprouvette.afin d'ajuster le volume d'eau distillée. Stériliser. Pour que les conditions de salinité soit bonnes et que la déprotéinisation soit effective, du NACl non solubilisé doit rester au fond du flacon.
NACl : sodium chloride sigma ex : 1M = 58,44 g è 87,66 g/250 cc
La solution saturée se conserve à température ambiante.
Annexe 6

10. ETHANOL 100 (ABSOLU) (conserver à température ambiante)
11. TRIS-EDTA (TE)
TRIS-HCI p H 7,5 1M 1 ml 10mM EDTA p H 8 0,5M 200 µl 1mM H2O qsp 100 ml (stérilisation par la chaleur et conserver à + 4°C)
12. Phénol saturé en Tris PH 7,5
Nous recevons le phénol non saturé en tris et non équilibré en pH. Il faut donc ajouter dans le flacon de phénol la quantité de Tris nécessaire (fourni par le fabricant au moins 12 heures à l'avance, laisser le flacon à température ambiante en l'agitant de temps en temps. (Attention ce réactif est à la fois très toxique et corrosif. Il faut absolument le manipuler sous une sorbone en fonctionnement, vitre de protection baissée, renfermer les déchets dans un flacon étanche. Si l'on reçoit une goutte sur la peau, la brûlure est vive, il faut se rincer à l'eau très rapidement.). Se servir de la phase inférieure pour l'extraction. A conserver au réfrigérateur.
13. Chloroforme-Alcool iso-amylique 29/1
Mélanger 29 volumes de chloroforme (qualité biologie moléculaire) avec 1 volume d'alcool iso-amylique. A conserver au réfrigérateur.
14. Acétate de sodium 3M pH 5,2 Peser la quantité nécessaire d'acétate de sodium pour avoir une solution 3M ajouter très peu d'eau puis ajuster le pH à 5,2 avec de l'acide acétique concentré. (Attention il faut beaucoup d'acide et peu d'eau). Ajuster le volume dans une éprouvette avec de l'eau distillée stérile et stériliser. (Ne pas préparer plus de 50 ml de réactif car le volume utilisé est très faible et la pollution facile).
Pour acétate de sodium : 1 M + 82,03 Peser 12,3 g à dissoudre dans 30 cc H2O ultrapure stérile puis ajuster à pH 5,2 avec acide acétique et compléter à 50 cc. Stériliser.
Annexe 7

Bibliographie.
Henry M, Aubert H, Morange P.E, Nanni I, Alessi M.C, Tiret L, Juhan-Vague I. Identification of
polymorphisms in the promoter and the 3’ region of the TAFI gene : Evidence that plasma TAFI
antigen levels are strongly genetically controlled. Blood. 2001.





























