Embed Size (px)
Citation preview

Hémoglobinopathies
Jacques Elion
Module de Médecine MoléculaireAnnée universitaire 2012-13
Faculté de MédecineInstitut national de la santé et de la recherche médicale

Hemoglobinopathies :
• pathologie monogénique la plus commune au monde
• transmission récessive(rare cas transmission dominante)
• Hétérogénité clinique : du porteurasymptomatique à la mort foetale in utéro
Tokyo 99
InsermUMR 665

Globin Gene Organization

Expression of Different Globins

Classification:Abnormal hemoglobins: structural abnormalitiesThalassemias: synthesis defects
the difference is not in geneticsPrimary defects are similar (point mutations, deletions…)Some mutations give rise to a mixed defect
the difference is in pathophysiology:The protein can be functionally normal/abnormalThe protein is quantitatively deficient/absent
Hb A = 2 2
InsermUMR 665

1.the thalassemias
decreased synthesis of the or globin chains
- and -thalassemias
- two types (i.e. ° or +-thal)
InsermUMR 665

1.the thalassemias
- -thalassemias - -thalassemias - thalassemias with an abnormal Hb
InsermUMR 665

Pathophysiology of Pathophysiology of --thalassemiathalassemia
/ chain disequilibrium
Excess of -chains
Inclusion bodies,ineffective erythropoiesis, hemolysis
Anemia
Proteolysis
Persistence of fetal hemoglobin- cell selection- absolute increase of HbF ?
Bone Iron Jaundice Infectiondisease loading
InsermU665

CCTCACCC
CCACACCC
CCAATATAAA
-106 -91 -76 -31
ATG
NullNullMutationsMutations
(FS,NS)
CapCapSiteSite
Initiation Initiation CodonCodon
CleavageCleavagemutationsmutations
AATAAA
Stop Stop CodonCodon
SpliceSpliceMutationsMutations
Mutations Mutations PromotorPromotor
--thalassemias: thalassemias: point mutations are the most common causepoint mutations are the most common cause

Cao A, Moi P.
Regulation of the globin genes.Pediatr Res. 2002 Apr;51(4):415-21.
Rarely, -thalassemias result from deletions
- of the structural genes
- of major regulatory regions

-thalassaemia major:Cooley’s anemiaa life-threatening transfusion-dependent condition
more than 200 -thal mutations described to date
InsermU665

-thalassaemia intermedia:a mild transfusion-independent condition mayresult from mild mutations
D.J. Weatherall, Nature Reviews Genetics, 2001, 2/245-255 InsermU665

Allelic combination* participates to clinical heterogeneity*compound heterozygozity
D.J. Weatherall, Nature Reviews Genetics, 2001, 2/245-255
Mild -thalalleles are indicated in bold
InsermU665

1.the thalassemias
- -thalassemias - -thalassemias - thalassemias with an abnormal Hb
InsermUMR 665

--thalassemiasthalassemias
FunctionalFunctional--genesgenes
44
33
22
22
11
00
normalnormal
HeterozygousHeterozygous ++ --thalthal
HomozygousHomozygous ++--thalthal
HeterozygousHeterozygous 00--thalthal
Hb H Hb H diseasedisease
HydropsHydrops fetalisfetaliswithwith Hb BartHb Bart’’ss
Most commonly results from deletions InsermU665

Hydrops Fetalis

Chui DH, Fucharoen S, Chan V.
Blood. 2003 Feb 1;101(3):791-800.
In most cases, -thalassemias result from large deletions
+
+
0
0
0
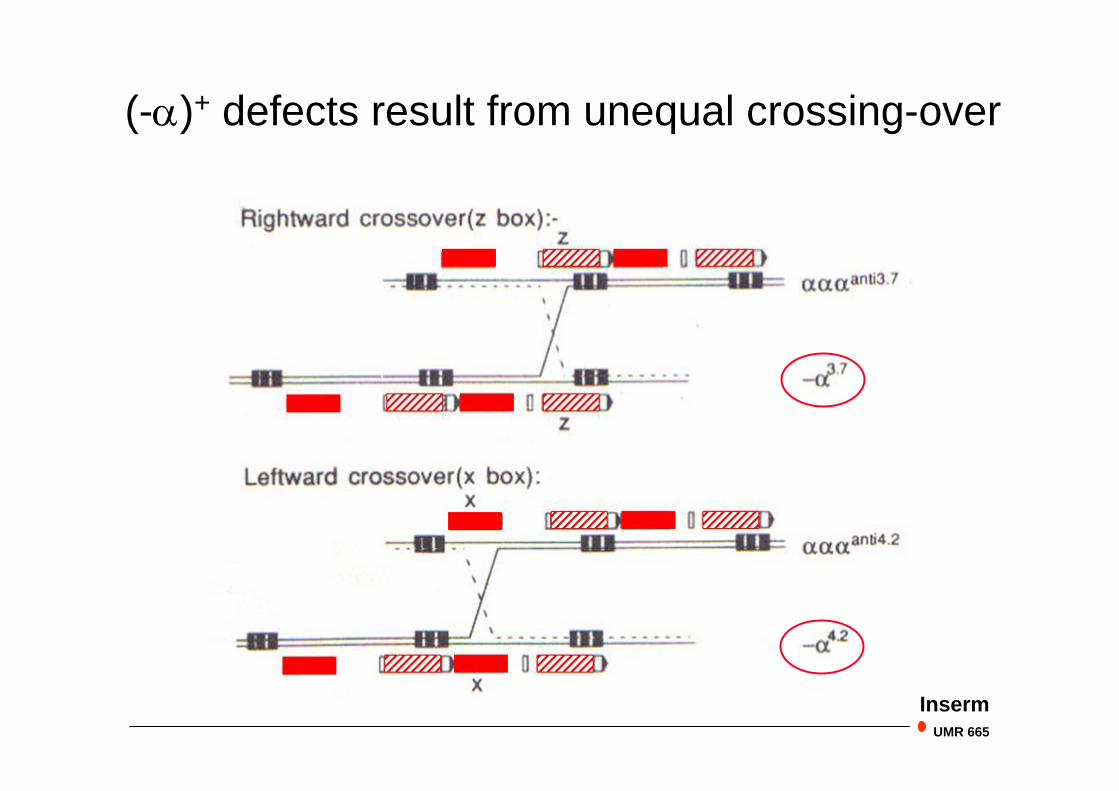
(-)+ defects result from unequal crossing-over
InsermUMR 665

Geographical distribution of -thalassemia
D.J. Weatherall, Nature Reviews Genetics, 2001, 2/245-255Inserm
U665

3. abnormal hemoglobins
- classification - common Hb variants - sickle cell disease
InsermUMR 665

Central cavity:2,3-DPG bindingabnormal affinity
11 contacts:unstable Hbs
2
1
2
12 contacts:abnormal affinity
Heme pocket:Hb Mabnormal affinityunstable Hbs
Surface:No functional consequencesEXCEPT Hb S
Classification of hemoglobin variants
1

Haemoglobin S = Sickle Cell Disease
Other frequent haemoglobin variants such as HbC or HbE in the homozygous state result in mild haemolyticanaemia or are asymptomatic
BUT
compound heterozygosities such as- S/C- E/-thal
produce moderate to severe phenotypes
OBVIOUSLY such associations are most oftenencountered in populations in which the two types of defect coexist
Frequent Haemoglobin variants

Original geographic distribution of HbS
Hb S
Malaria
InsermUMR 665
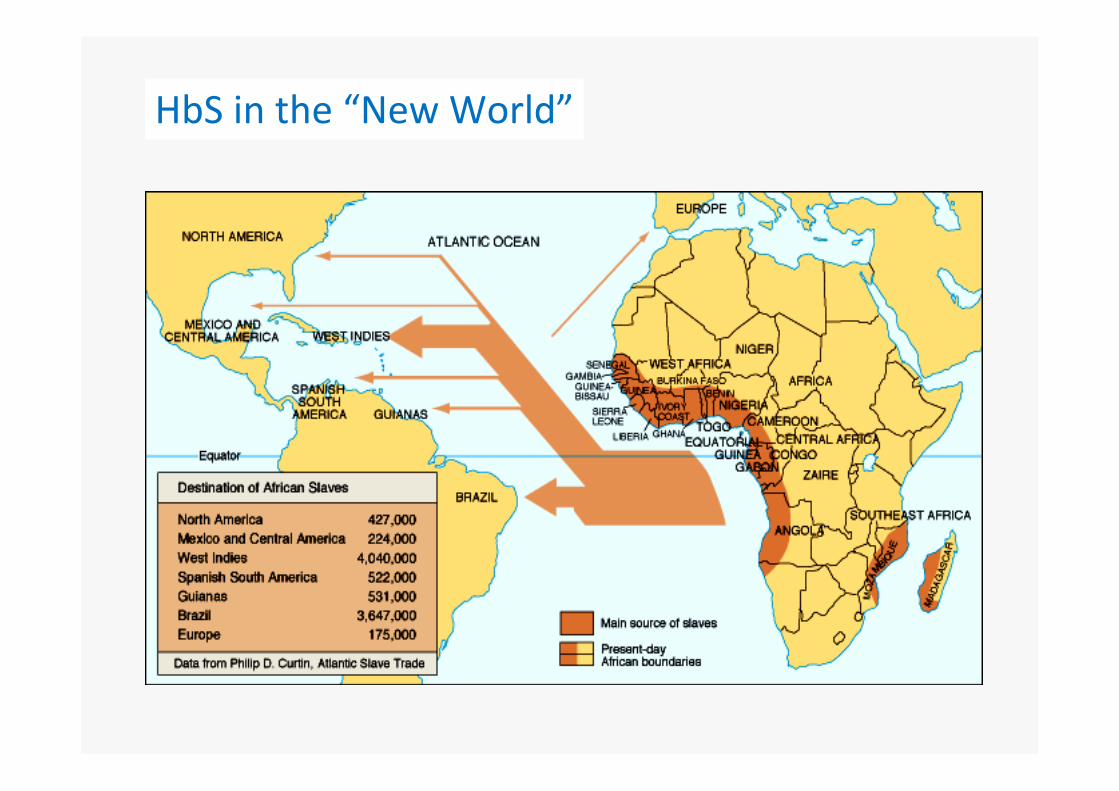
HbS in the “New World”

Original geographic distribution of frequenthaemoglobin variants
HbC
HbE
InsermUMR 665

Geographic distribution of haemoglobin E
gene frequency
<.02
.02-.10
> .10
2nd most commonvariant after HbS
InsermUMR 665

Haemoglobin E
HbE heterozygotes are asymptomatic- with normal Hb, but microcytosis (average MCV 73 fl)- HbE is expressed at 30-35%
HbE homozygotes are also asymptomatic- Hb is normal or slightly decreased- microcytosis (average MCV 67 fl)
HbE/-thal compound heterozygotes present withan often severe thalassemic syndrome
HbE results from a mixed defect which associates a Hb variant and a -thalassaemia determinant

Haemoglobin E defect
HBB:c.G79AHBB p.Glu26Lys
normal splicing
alternative splicing
part of the mRNA precursor is abnormally spliced, resulting in a +-thal allele
InsermUMR 665

Geographic distribution of haemoglobin C
gene frequency
.01
.02
.04
.08
.10
3rd mostcommon variant
InsermUMR 665

InsermUMR 665

5 ' 3 'LCR G A
Chromosome 11
S Glu
chaîne de globine S
l
polymérisation
GTG codon 6Gène SA
Val
oxy HbS desoxy HbS
globule rouge
Falciformationdéformationrigidificationfragilisation
Vaso-occlusion
Anémiehémolytique Inserm
UMR 763
HbS
Hb S : Drépanocytose
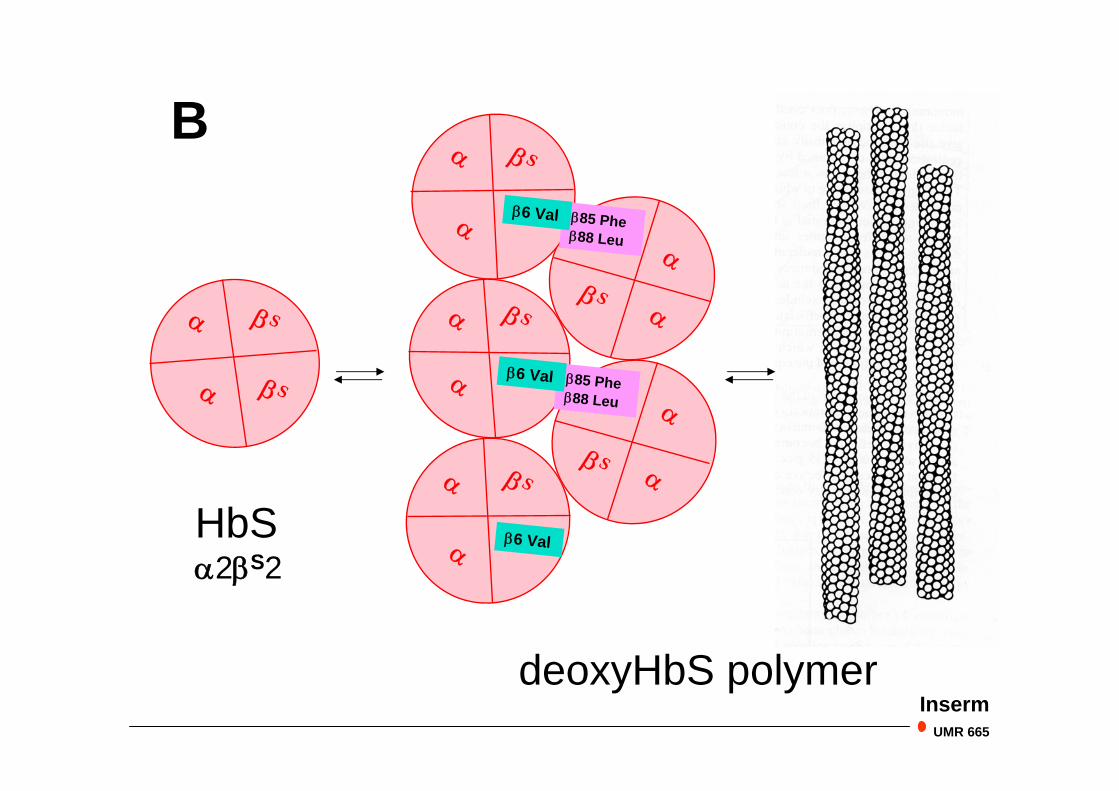
HbS2S2
deoxyHbS polymer
S
S
S
S
S
S
S
85 Phe88 Leu
6 Val
6 Val
85 Phe88 Leu
6 Val
B
InsermUMR 665

InsermU665

Globules rouges normaux et falciformés
InsermUMR 665

H2O2
-O2
Hb S
O2
protéinebande 3
spectrine
ankirine
corps de Heinz
Libération devésicules
*OH
IgG
OH
Fe
protéinebande 3
OH
hémichromes
Fe+3
Microenvironnementoxydant
Distorsion
polymères
Ca+2
Canal GardosK+
K+Cl-
CotransportK-Cl
Activation des transport ioniques et déshydratation Inserm
UMR 665
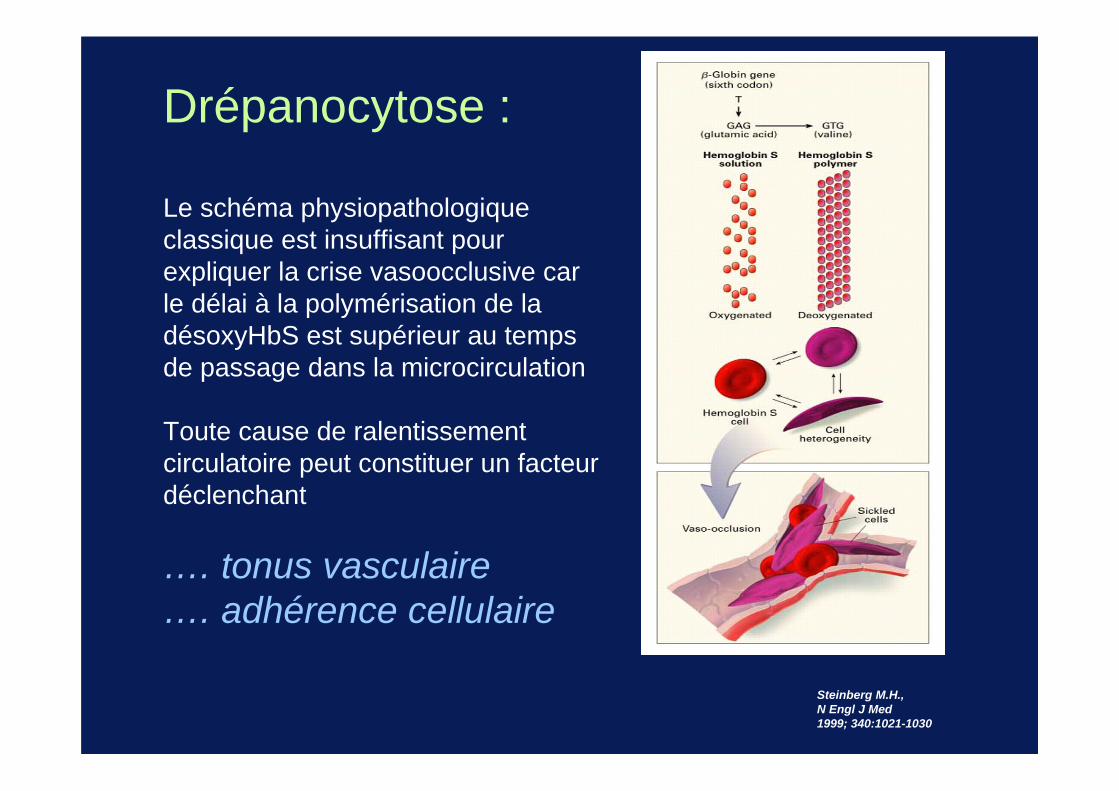
Steinberg M.H., N Engl J Med 1999; 340:1021-1030
Drépanocytose :
Le schéma physiopathologique classique est insuffisant pour expliquer la crise vasoocclusive car le délai à la polymérisation de la désoxyHbS est supérieur au temps de passage dans la microcirculation
Toute cause de ralentissement circulatoire peut constituer un facteur déclenchant
…. tonus vasculaire…. adhérence cellulaire

Drépanocytose
InsermUMR 665
-nouvelles données physiopathologiques :
- adhérence des GR à l’endothélium
- activation cellulaire et rôle de l’hypoxie
- environnement pro-inflammatoire
- dysfonctionnement vasculaire
- un seul médicament actif : hydroxyurée (1995)
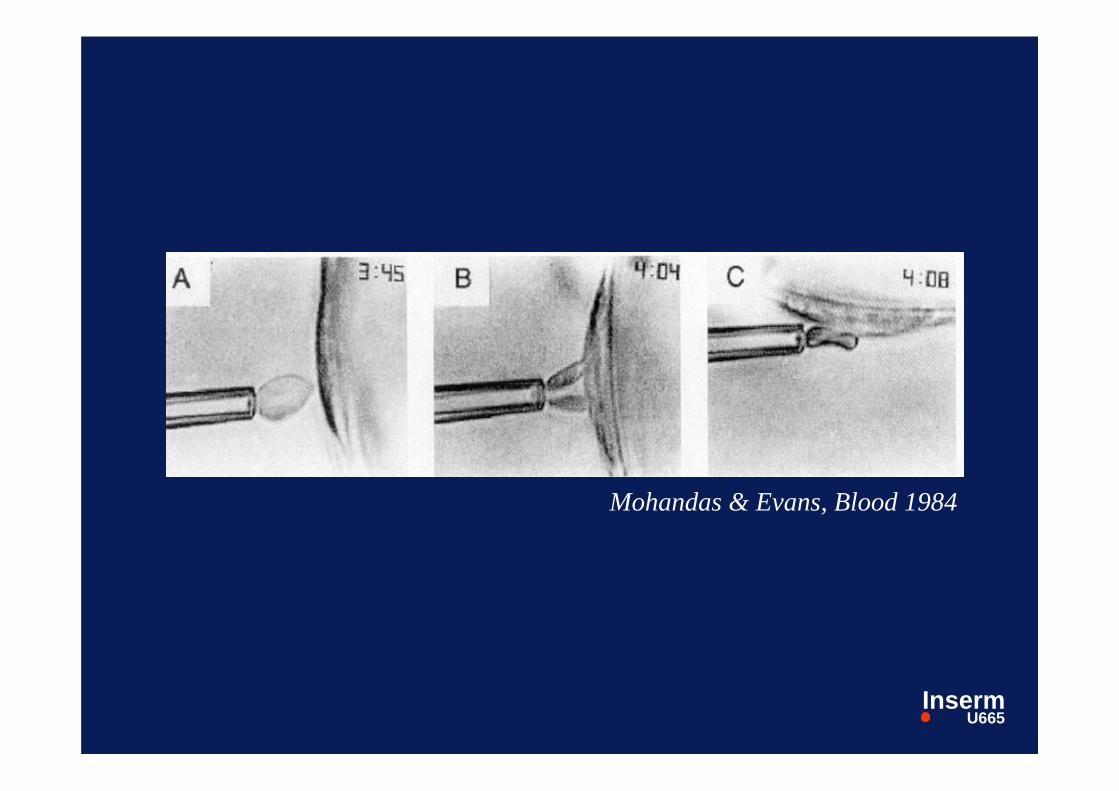
Mohandas & Evans, Blood 1984
InsermU665

Adhérence des GR drépanocytairesà l’endothélium :
Trois questions:
- quel territoire vasculaire ?- quels globules rouges ?- quels mécanismes moléculaires ?
InsermU665
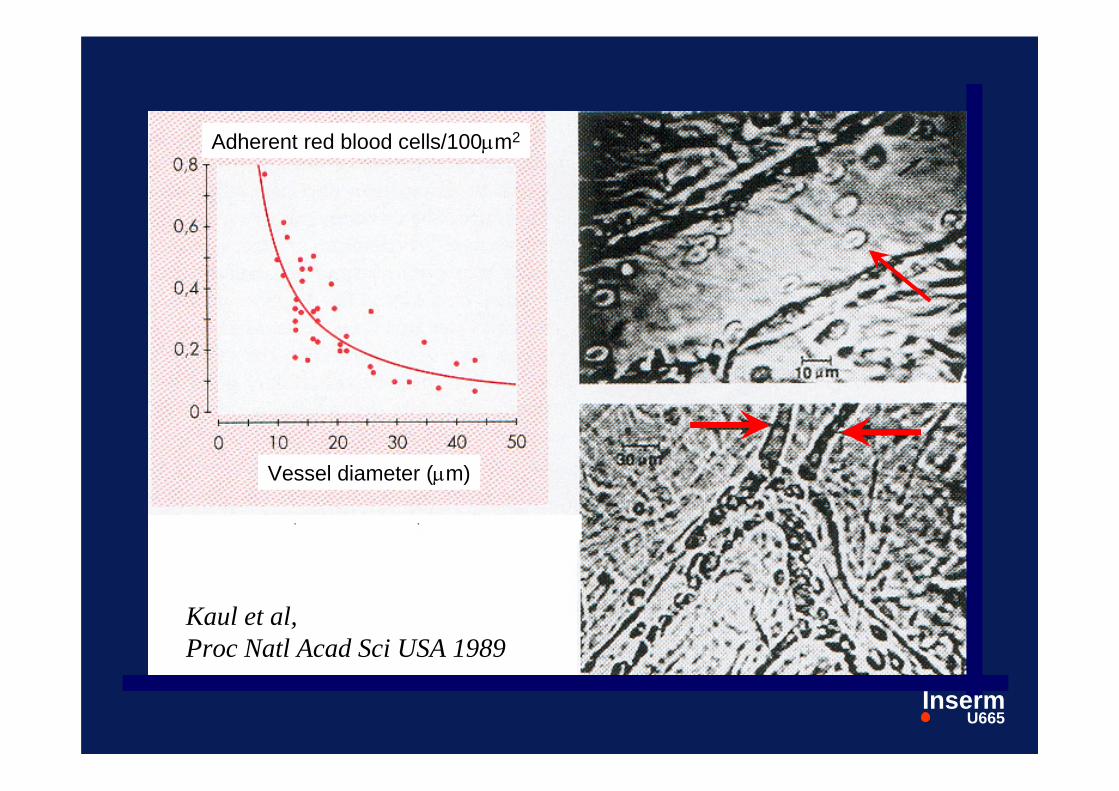
Kaul et al,Proc Natl Acad Sci USA 1989
Vessel diameter (m)
Adherent red blood cells/100m2
InsermU665

Adhesion des GR drépanocytairesà l’endothélium :
Trois questions:
- quel territoire vasculaire ?
veinules post-capillaires- quels globules rouges ?- quels mécanismes moléculaire ?
InsermU665

Adhesion des GR drépanocytairesà l’endothélium :
Trois questions:
- quel territoire vasculaire ?- quels globules rouges ?
Gr jeunes : "réticulocytes de stress"(cellules vieillies)
- quels mécanismes moléculaires ?Inserm
U665

Les réticulocytes de stress expriment des protéinesd’adhérence normalement présentes sur les précurseursérythroïdes dans la moelle
Elion & LabieHématologie 1998
InsermUMR 665

Matrice sous-endothéliale
IgG
FcR
GR vieilli
laminine
VLA4VCAM1
Reticulocyte de stress
CD36
CD36
TSP
GP1b/2b3a-like
HMW vWF
GP1b-likecellules endothéliales
BCAM/Lu
Erythropoièse accrue
Oxydationdéshydratation
Activation endothéliale
plaquettes
Flux sanguin
Les acteurs de l’adhérence cellulaire dans la drépanocytose
InsermUMR 665

Les acteurs de l’adhésion cellulaire dans la drépanocytose
- réticulocytes de stress +++- GR matures- GR vieillis
- cellules endothéliales activées- structures sous-endothéliales
InsermU665

In Sickle Cell Disease
abnormal cell adhesion to the endothelium plays a major in targetting vaso-occlusion
from JP Cartron SCADHESION project C Le Van Kim, PL Tharaux, J Elion
….but so does the vascular toneInserm
UMR 665

Tonus vasculaire
-
Deux médiateurs produits par l ’endothélium sont élevés dans la drépanocytose :
Endothéline-1 vasoconstricteur
NO vasodilatateurmais l’hyperproduction de NO est «futile»car destruction du NO par l’hémolyse
vasoconstrictionInserm
UMR 665

blood
subendothelium
Interactions between the endothelium and circulatingblood cells play a central role in vascular biology
EC
RBCs
EC
InsermU665

Barrier between blood and the subendothelial structuresCrucial role in controlling
vascular tonethrombogenesisvascular remodeling
Numerous secretory propertiesanti- / pro-coagulant proteinsvasodilators (NO) / vasoconstrictors (Et-1)growth factors
Expressing membrane receptorscell adhesion
Activated by various stimulihypoxiainflammatory cytokines
The vascular endothelium
InsermUMR 665

Barrier between blood and the subendothelial structuresCrucial role in controlling
vascular tonethrombogenesisvascular remodeling
Numerous secretory propertiesanti- / pro-coagulant proteinsvasodilators (NO) / vasoconstrictors (Et-1)growth factors
Expressing membrane receptorscell adhesion
Activated by various stimulihypoxiainflammatory cytokines
The vascular endothelium
InsermUMR 665

Endothelium-derived nitric oxyde (NO)is a major actor of vascular biology
- vasodilation- inhibition of platelet activation- endothelial expression of adhesion molecules
NO activates the soluble guanylate cyclase (sGC) in smoothmuscle cells vasodilation
Haemoglobin is the most potent NO scavengerparadox at NO working as a diffusible effector and
its proximity to large amounts of Hb in the RBC
InsermUMR 665

but physiologically, physical and dynamical barriersprevent NO destruction by Hb
NO release in hypoxic tissues InsermUMR 665

Gladwin et al. Nature Medicine, 2003
but physiologically, physical and dynamical barriersprevent NO destruction by Hband rather Hb in the RBC acts as a NO transporter
NO release in hypoxic tissues

Gladwin et al. Nature Medicine, 2003
Nitrites may act as a second important NO transporter
Paracrine vasodilation
InsermUMR 665

Haemolytic anaemiasNO reacts 1,000 times more rapidly with free Hb than with RBCs
NO scavenging is complete
Common vascular complicationsPulmonary hypertension, cutaneaous ulceration, acute and chronicrenal failure
InsermUMR 665
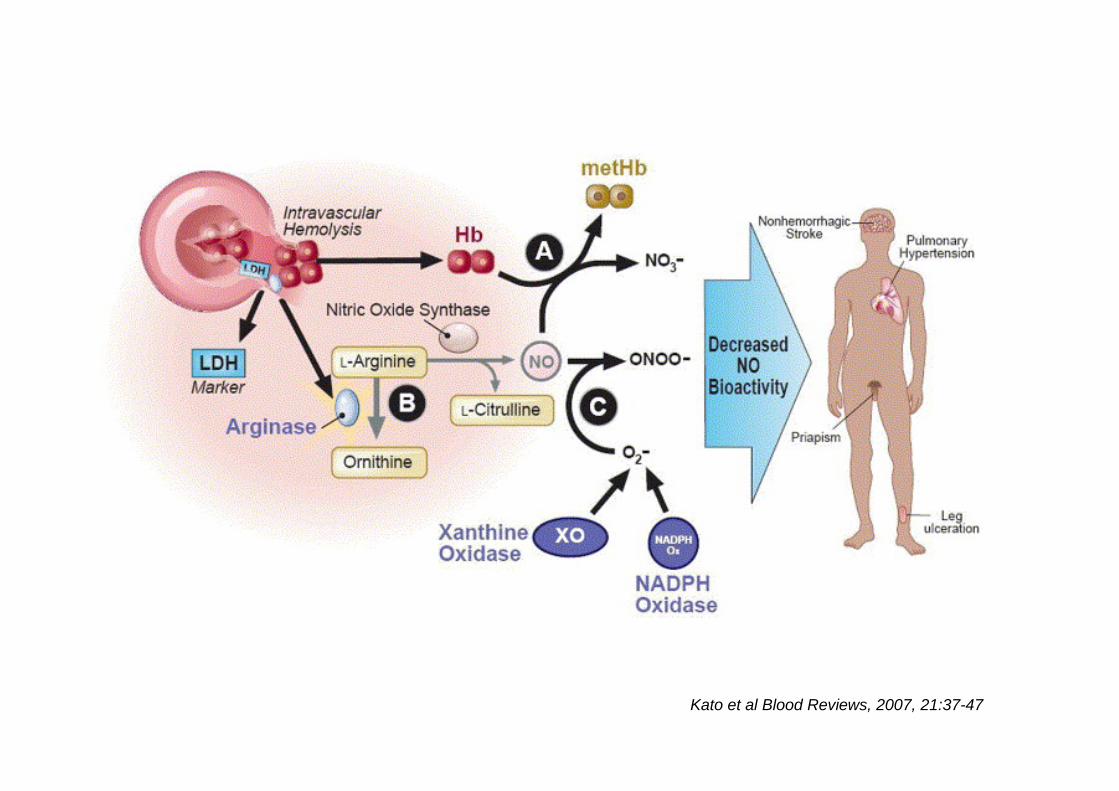
Kato et al Blood Reviews, 2007, 21:37-47
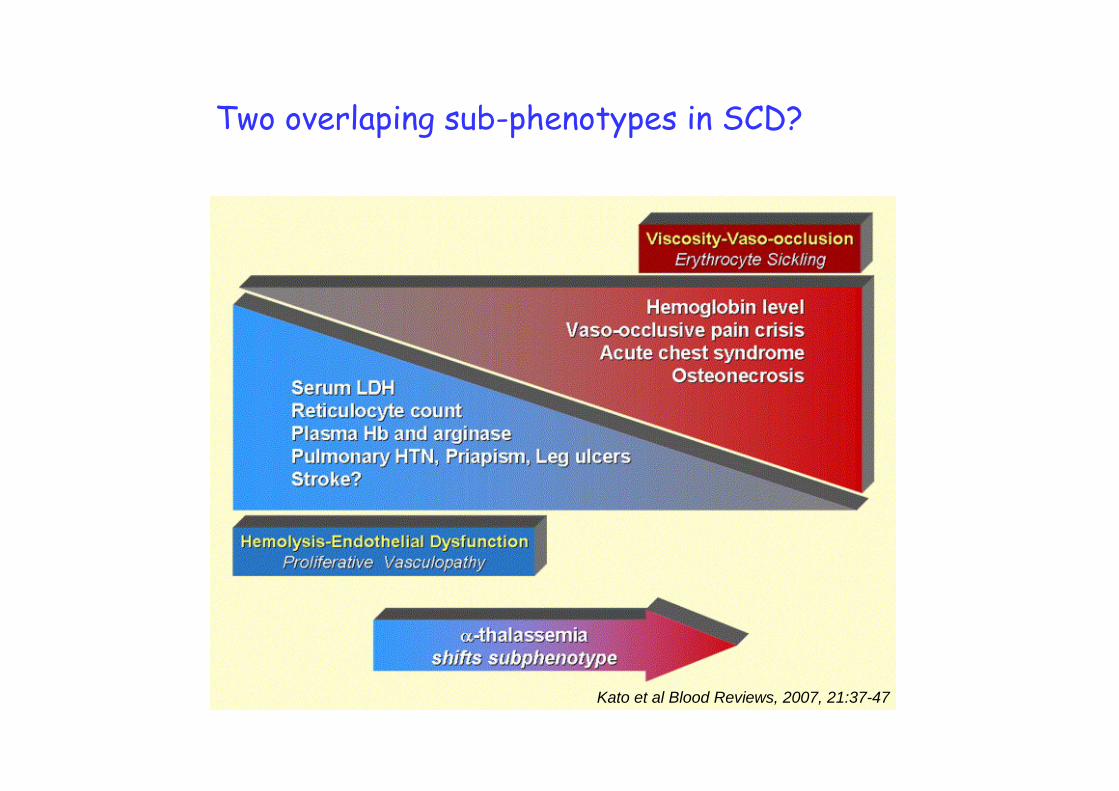
Kato et al Blood Reviews, 2007, 21:37-47
Two overlaping sub-phenotypes in SCD?

Therapeutic implications
NO production
NO destruction
NO response
Kyle Mack and Kato Int J Biochem Cell Biol, 2006, 38:1237-43

4. Genetic modifyers
- in sickle cell disease - in -thalassemia
InsermUMR 665

SICKLE CELL ANEMIA
Alleviating genetic factors
• -thalassemia• high output of fetal hemoglobin (HbF)• lowered expression of the S gène (India)
InsermU665

- SCD onset parallels extinction of HbF expression during development
- High HbF levels are statistically associated with a milderclinical expression of the disease
- HbF prevents growth of the deoxyHbS polymer
FetalFetal hemoglobinhemoglobin and SCDand SCD
rationale for the treatment of severe forms of SCD by hydroxyurea
InsermU665

Propensity for maintaining high HbF levels is under genetic control
- at the cellular level (F cell number) - at the molecular level (HbF/F cell)
polygenic modulationdeterminants within the -Gb cluster (HPFH is rare)polymorphisms = RFLPs, microsatellites
FetalFetal hemoglobinhemoglobin
InsermU665

3. Genetic modifyers
- in sickle cell disease - in -thalassemia
InsermUMR 665

Excess of -globin chainsis the pathophysiological hallmark of -thalassaemias
Secondary modifiers -chain excess mild phenotypes
- -thalassaemia- increased fetal -chain production (high HbF)
linked to the -cluster (chr 11)unlinked (chr X: FCP locus?, chr 6: HBS1L-MYB,
chr 2: BCL11A, chr 19: KLF1)
-chain excess severe phenotype- -globin gene expansion
Inserm
U665

























