Embed Size (px)
Citation preview

LABEL DE CHIMIE THEORIQUEILE DE France – NORD
Edition 2018
Coordination : P Reinhardt (LCT, UPMC), I Demachy (LCP, Orsay)
Les enseignements se dérouleront du 18 janvier au 2 février :
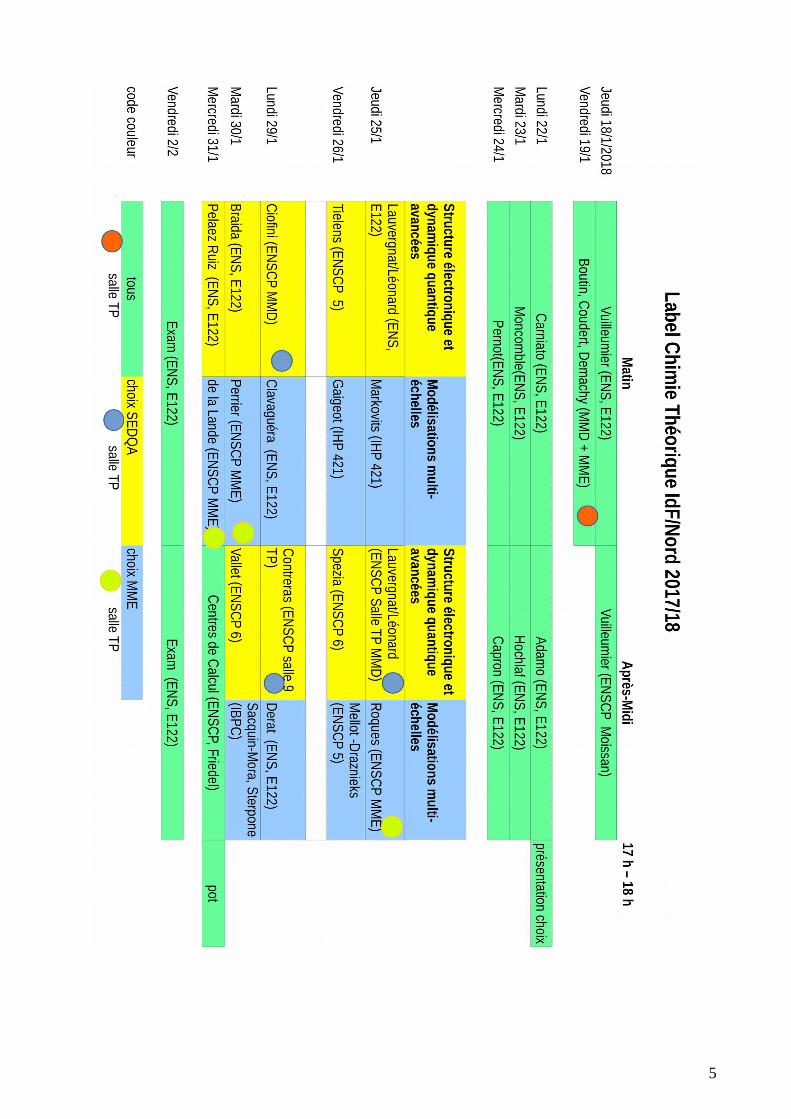
18/01 au 24/01 : séquence introductive commune à tous25/01 au 31/01 : séquence d’approfondissement au choix entre
Structure électronique et dynamique quantique avancées Modélisations multi-échelles
Vendredi 2 février : examens écrits 10h-12h et 14h-16h
Les lieux des cours et travaux pratiques sont situés sur le campus Curie de l’Université de Paris et à l’ENS Paris, à savoir
L’institut Henri Poincaré (IHP)
L’Ecole National Supérieur de Chimie de Paris, Chimie ParisTech
Salles de TP, ENSCP, Chimie ParisTech
L’ENS Paris, 24 rue l'Homond, salle E122
L’Institut de Biologie Physico-chimique (IBPC)
Equipe d’enseignants Benoît Braïda, Alexis Markovits, Julia Contreras, Riccardo Spezia : LCT,
UPMC Frédérik Tielens, Université de Bruxelles Etienne Derat : IPCM, UPMC Rodolphe Vuilleumier, Anne Boutin : Laboratoire PASTEUR, ENS Paris François-Xavier Coudert, Carlo Adamo, Ilaria Ciofini : Chimie Paris Tech Stéphane Carniato, Nathalie Capron : LCPMR, UPMC Majdi Hochlaf, Céline Léonard : MSME, Marne-la-Vallée Aurélien Moncomble, Valérie Vallet, Daniel Pelaez Ruiz : LASIR et PhLAM,
Université de Lille 1 Pascal Pernot, David Lauvergnat, Isabelle Demachy, Carine Clavaguéra,
Aurélien de la Lande : LCP, Paris-Sud Jérôme Roques : IPNO, Paris-Sud Caroline Mellot-Drasnieks : Collège de France Sophie Sacquin-Mora, Fabio Sterpone : IBPC, CNRS Marie-Pierre Gaigeot : LAMBE, Evry Val d’Essonne
1
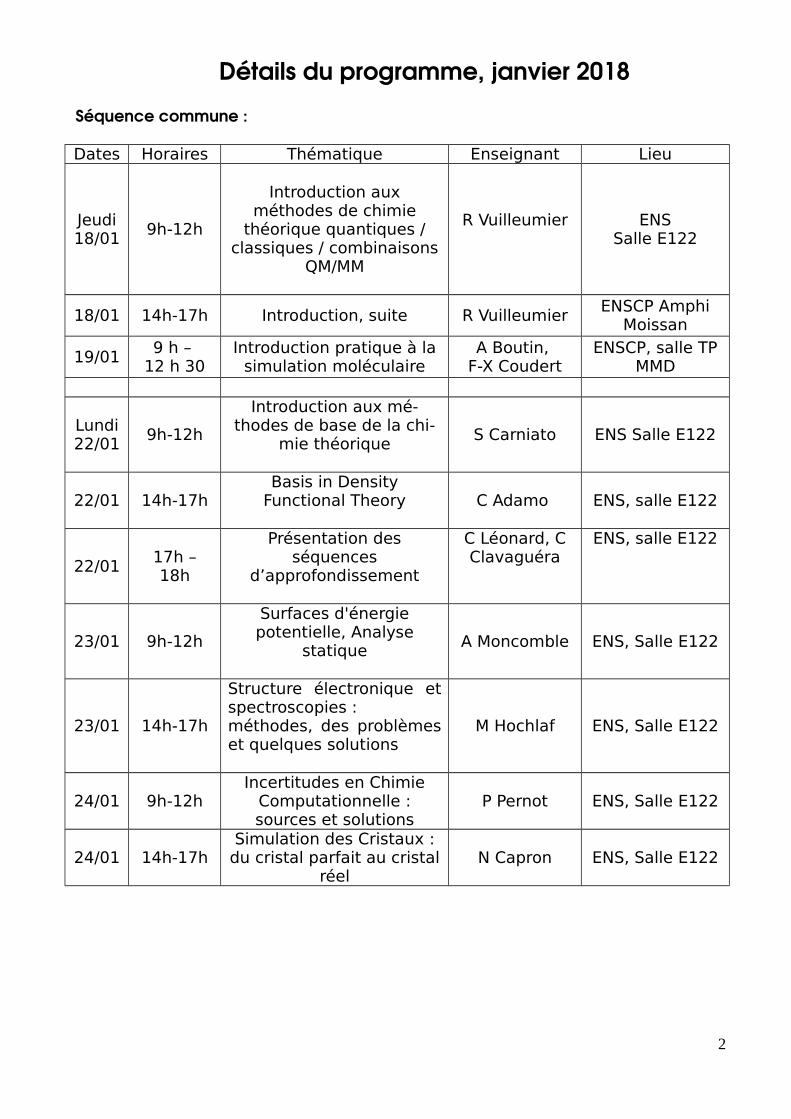
Détails du programme, janvier 2018
Séquence commune :
Dates Horaires Thématique Enseignant Lieu
Jeudi18/01
9h-12h
Introduction auxméthodes de chimie
théorique quantiques /classiques / combinaisons
QM/MM
R Vuilleumier ENSSalle E122
18/01 14h-17h Introduction, suite R VuilleumierENSCP Amphi
Moissan
19/019 h –
12 h 30Introduction pratique à la
simulation moléculaireA Boutin,
F-X CoudertENSCP, salle TP
MMD
Lundi22/01
9h-12h
Introduction aux mé-thodes de base de la chi-
mie théoriqueS Carniato ENS Salle E122
22/01 14h-17hBasis in Density
Functional Theory C Adamo ENS, salle E122
22/0117h –18h
Présentation desséquences
d’approfondissement
C Léonard, CClavaguéra
ENS, salle E122
23/01 9h-12h
Surfaces d'énergiepotentielle, Analyse
statiqueA Moncomble ENS, Salle E122
23/01 14h-17h
Structure électronique etspectroscopies :méthodes, des problèmeset quelques solutions
M Hochlaf ENS, Salle E122
24/01 9h-12hIncertitudes en Chimie
Computationnelle :sources et solutions
P Pernot ENS, Salle E122
24/01 14h-17hSimulation des Cristaux :du cristal parfait au cristal
réelN Capron ENS, Salle E122
2
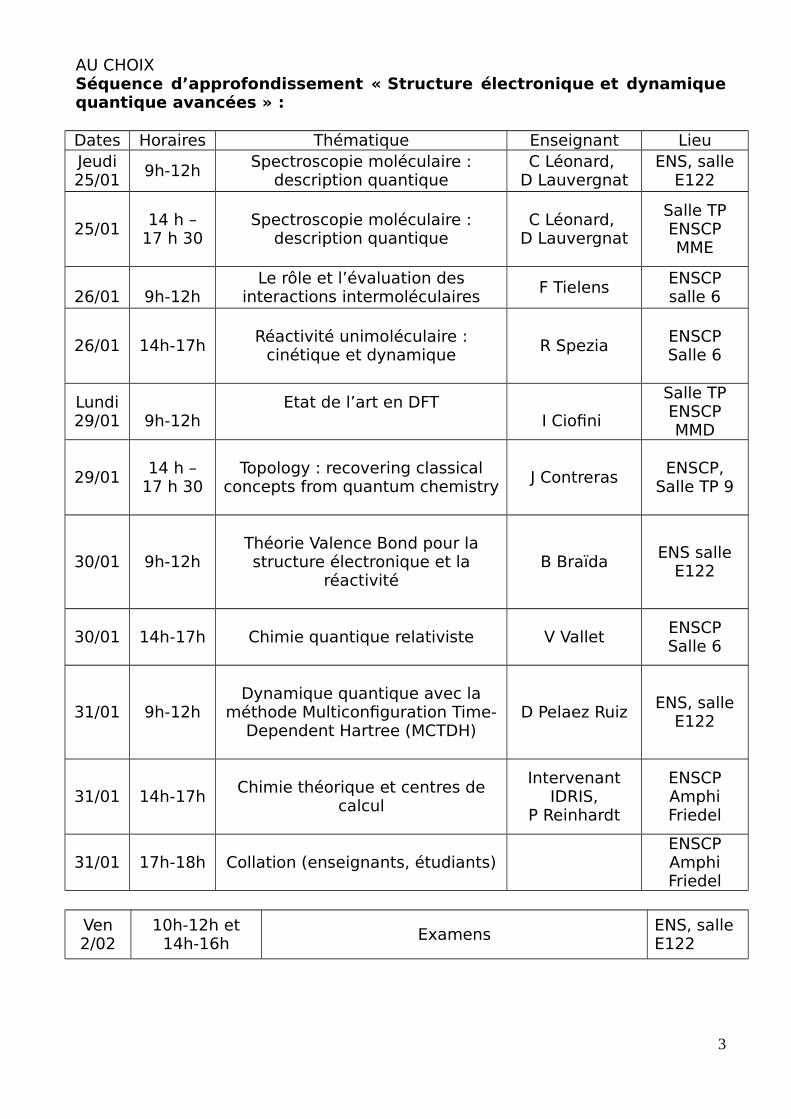
AU CHOIXSéquence d’approfondissement « Structure électronique et dynamiquequantique avancées » :
Dates Horaires Thématique Enseignant LieuJeudi25/01
9h-12hSpectroscopie moléculaire :
description quantiqueC Léonard,
D LauvergnatENS, salle
E122
25/0114 h –
17 h 30Spectroscopie moléculaire :
description quantiqueC Léonard,
D Lauvergnat
Salle TPENSCPMME
26/01 9h-12hLe rôle et l’évaluation des
interactions intermoléculairesF Tielens
ENSCPsalle 6
26/01 14h-17hRéactivité unimoléculaire :
cinétique et dynamiqueR Spezia
ENSCPSalle 6
Lundi29/01 9h-12h
Etat de l’art en DFTI Ciofini
Salle TPENSCPMMD
29/0114 h –
17 h 30Topology : recovering classical
concepts from quantum chemistryJ Contreras
ENSCP,Salle TP 9
30/01 9h-12hThéorie Valence Bond pour lastructure électronique et la
réactivitéB Braïda
ENS salleE122
30/01 14h-17h Chimie quantique relativiste V ValletENSCPSalle 6
31/01 9h-12hDynamique quantique avec la
méthode Multiconfiguration Time-Dependent Hartree (MCTDH)
D Pelaez RuizENS, salle
E122
31/01 14h-17hChimie théorique et centres de
calcul
IntervenantIDRIS,
P Reinhardt
ENSCPAmphiFriedel
31/01 17h-18h Collation (enseignants, étudiants)ENSCPAmphiFriedel
Ven2/02
10h-12h et14h-16h
ExamensENS, salle E122
3
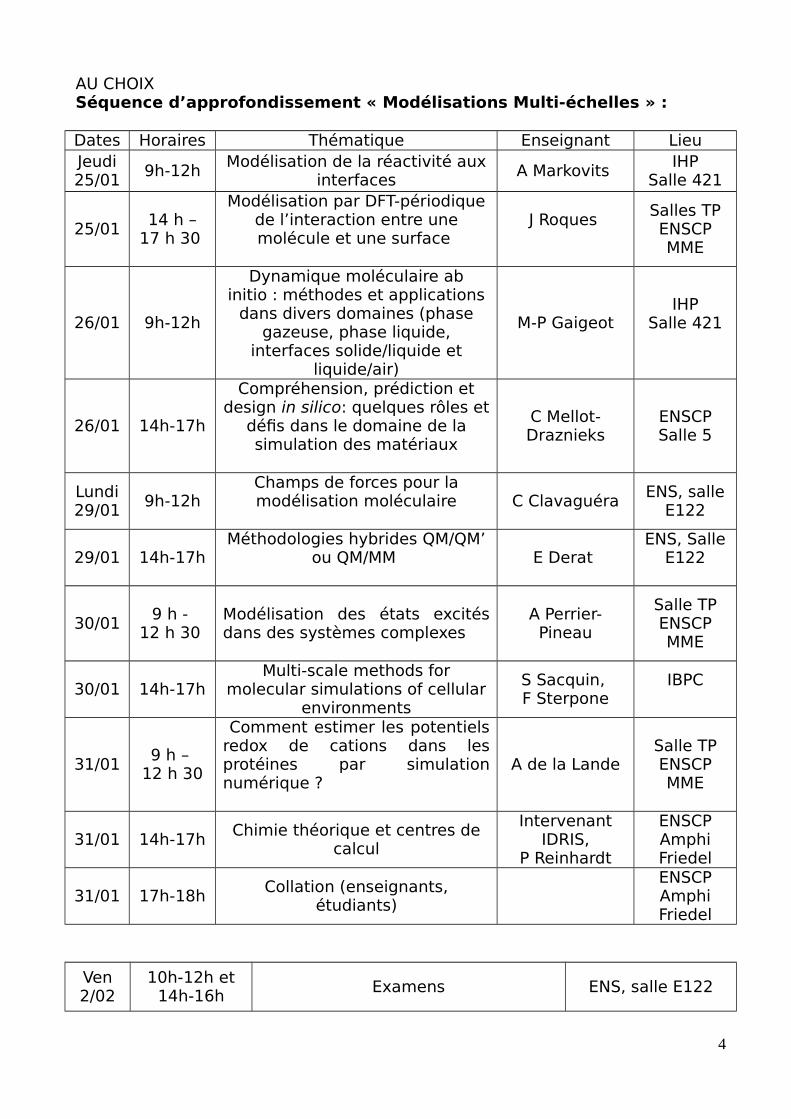
AU CHOIXSéquence d’approfondissement « Modélisations Multi-échelles » :
Dates Horaires Thématique Enseignant LieuJeudi25/01
9h-12hModélisation de la réactivité aux
interfacesA Markovits
IHPSalle 421
25/0114 h –
17 h 30
Modélisation par DFT-périodiquede l’interaction entre unemolécule et une surface
J Roques Salles TPENSCPMME
26/01 9h-12h
Dynamique moléculaire abinitio : méthodes et applications
dans divers domaines (phasegazeuse, phase liquide,
interfaces solide/liquide etliquide/air)
M-P GaigeotIHP
Salle 421
26/01 14h-17h
Compréhension, prédiction etdesign in silico: quelques rôles et
défis dans le domaine de lasimulation des matériaux
C Mellot-Draznieks
ENSCPSalle 5
Lundi29/01
9h-12hChamps de forces pour lamodélisation moléculaire C Clavaguéra
ENS, salleE122
29/01 14h-17hMéthodologies hybrides QM/QM’
ou QM/MM E Derat ENS, Salle
E122
30/019 h -
12 h 30 Modélisation des états excitésdans des systèmes complexes
A Perrier-Pineau
Salle TPENSCPMME
30/01 14h-17hMulti-scale methods for
molecular simulations of cellularenvironments
S Sacquin, F Sterpone
IBPC
31/019 h –
12 h 30
Comment estimer les potentielsredox de cations dans lesprotéines par simulationnumérique ?
A de la LandeSalle TPENSCPMME
31/01 14h-17hChimie théorique et centres de
calcul
IntervenantIDRIS,
P Reinhardt
ENSCPAmphiFriedel
31/01 17h-18hCollation (enseignants,
étudiants)
ENSCPAmphiFriedel
Ven2/02
10h-12h et14h-16h
Examens ENS, salle E122
4
5

Introduction aux méthodes de chimie théoriquequantiques / classiques / combinaisons QM/MM
Enseignant : Vuilleumier Rodolphe, professeur, Ecole Normale Supérieure
Objectifs de l’enseignement:L’objectif de cet enseignement est d’offrir une introduction et un cadre commun pourl’ensemble des cours proposés au cours du Label. Après un rappel historique du calculatomique et moléculaire, nous montrerons la nécessité d’employer des approximationspour la description des systèmes chimiques. Partant de l’approximation deSchrödinger, nous discuterons les différents objets auxquels s’intéressent lessimulations et les grandes classes de calculs atomiques et moléculaires. Ceci nouspermettra de faire un tour des différents enseignements proposés pendant ces deuxsemaines de Label en discutant d’une part des approximations faites, de leurjustification et des problèmes physico-chimiques que ces méthodes permettent detraiter.
Plan succinct :1) Introduction – Historique du calcul atomique et moléculaire2) Approximation de Born-Oppenheimer3) Méthode d’interaction de configuration, méthode Hartree-Fock4) Simulations de dynamique moléculaire5) Simulations Monte-Carlo6) Extensions, méthodes mixtes QM/MM7) Présentation des différents enseignements des deux semaines
Prérequis : Mécanique quantique niveau licence, thermodynamique
Lectures complémentaires conseillées : D. Frenkel and B. Smit « Understanding molecular simulations : from algorithms toapplications » (Academic Press, 2002)M. Tuckerman « Statistical Mechanics: Theory and Molecular Simulation » (OxfordGraduate Texts, 2010)
Introduction pratique à la simulation moléculaireEnseignants : Boutin, Anne, Directrice de recherche CNRS, professeure associéeENS, CNRS / ENS, François Xavier Coudert, Chargé de recherche CNRS, ChimieParis Tech. Isabelle Demachy, LCP, Paris-Sud
Objectifs de l’enseignement :Sous la forme de travaux pratiques, cet enseignement vise à donner aux étudiantsune expérience directe et concrète de la simulation moléculaire. À l’aide d’un codetrès simple de simulation moléculaire (dynamique moléculaire et Monte Carlo) sur desatomes de Lennard-Jones, on mettra en évidence par une démarche d’investigation lesnotions clefs de la simulation moléculaire et de la thermodynamique statique :ensembles thermodynamiques, équilibre et convergence, ergodicité, observables enphase condensées, etc.
Plan succinct : Introduction, structure d’un code de simulation moléculaire Premières simulations : un cristal d’argon Analyse de résultats : notions d’observables, d’équilibre, de convergence Application pratique à l’études des transitions de phase
Prérequis :Forces intermoléculaires, bases de thermodynamique statistiqueLectures complémentaires conseillées : Logiciel AtomTouch sur iOS / Android
6

Introduction aux méthodes de base de la chimie théorique :
application aux processus d’excitation/ionisation en couches internesEnseignant : Stéphane Carniato, Professeur, Université Pierre et Marie Curie
Objectifs de l’enseignement :L’étude des processus complexes survenant lors (relaxation électronique, ionisationsmultiples), ou à l’issue (dynamique nucléaire, émission X, diffusion Raman de rayonsX, effet Auger) de l’excitation/ionisation en couches internes de systèmes atomiques,moléculaires, ou solides a connu un essor considérable ces dernières années avecl’apparition de nouvelles sources de lumière (sources synchrotron 4ème génération,XFEL). Lors du processus d’excitation/ionisation impliquant les couches internes lastructure électronique de l’état excité est très différente de l’état fondamental. Ladescription précise des effets de relaxation et de corrélation électronique requiertalors un soutien théorique indispensable (énergie, couplage spin-‐orbite, surface depotentiel d’états excités, etc…). L’objectif du cours est une introduction aux méthodesde chimie théorique (Hartree-‐Fock, Post-‐HF) avec en toile de fond, leurs applicationspour la simulation des spectres de photoélectrons et de photoabsorption dans ledomaine des rayons X. Après une présentation des équations « de base » Hartree-‐Fock nous verrons comment il est possible de déterminer et calculer dans de cassimples les énergies d’ionisation et simuler les processus d’excitation multiples (Ex:processus shake-‐up). Cette présentation sera agrémentée de plusieurs exemplessimples utilisant les équations établies au fil du cours.
Plan succinct :A-‐Méthode Hartree-‐Fock (HF)
1-‐Equations HF, principe SCF et méthode d’orthogonalisation de Löwdin → Cas de H2
2-‐Applications: énergie de liaison des niveaux de coeura-‐Modèle des orbitales gelées (Approximation de Koopmans)b-‐Traitement de Relaxation électroniquec-‐Exemples
B-‐Corrélation électronique: Approches Post-‐HF (MP, CI)1-‐Approche Perturbative: Møller-‐Plesset (MP2)2-‐Approche Variationnelle: Interaction de configurations (CI)3-‐Rôle du spin (génération des fonctions propres (S2, Sz) de spin → Retour sur le cas de H2
4-‐Applications: Spectroscopies des couches internes (XPS, NEXAFS, processus K-‐2V)
Pré-‐requis : Goût pour la simulation et la programmation Manipulation des matrices : produit de matrices, matrice adjointe, unitaire, diagonalisation, vecteurs propres, valeurs propres. Base de mécanique quantique (Ex : matrices de Pauli, opérateurs, moments de transition, déterminants de Slater, équation de Schrödinger dépendante et indépendante du temps) Notions sur la photoémission (XPS, ESCA) et photoabsorption (NEXAFS)
Lectures complémentaires conseillées : Modern quantum chemistry : Introduction to advanced electronic structure theory : A. Szabo & N. S. Ostlund. NEXAFS spectroscopy, J. Stöhr, Springer series in surface sciences 25.
7

Basis in Density Functional Theory Enseignant : Carlo Adamo, Professeur, Chimie Paris Tech
Objectifs de l’enseignement :During the course the basic concepts of Density Functional Theory will be given.Starting from the first models seeking for an expression of the total energy as afunction of the energy only, the basic theorems of Density Functional Theory will bederived. The Kohn Sham method will be described together with the most commonlyused classes of exchange and correlation functionals. The accuracy of these modernDFT based approaches will be critically discussed. This course is completed by a practical aimed at exemplifying the use of DFT for thecalculation of different properties of molecular systems in solution and/or complexenvironments. The students will choose between the calculation of IR/Raman spectra,UV-Vis spectra or EPR and NMR parameters of simple molecular compounds.
Plan succinct :- DFT before DFT : from Thomas-Fermi model to Slater X- The Hohenberg Kohn theorems- The constrained-search formulation- The Kohn Sham method- The functionals : from LDA to double hybrids- Examples of limits and drawback in DFT- How to build a new functional
Prérequis :Basis in quantum chemistry
Lectures complémentaires conseillées : Parr & Yang “Density-Functional Theory of Atoms and Molecules” Oxford Science Publications
Surfaces d'énergie potentielle – Analyse statiqueEnseignant : Aurélien Moncomble, maître de conférences, Université de Lille
Objectifs de l’enseignement :Préciser l'intérêt pour la détermination de points stationnaires sur une surfacesFormaliser les problèmes d'optimisationPrésenter les grandes catégories d'algorithmes de recherche de minimaVoir quelques paramètres à contrôler dans un cas pratiqueAborder des notions sur la localisation d'autres points particuliersDétailler quelques algorithmes et leur implémentation
Plan succinct :1. Points stationnaires sur une surface d'énergie potentielle2. Algorithmes de recherches de minima3. Étude vibrationnelle sur un point stationnaire4. Recherche de chemins réactionnels5. Ouverture : points de croisement et intersections coniques
Prérequis :Notion de surface d'énergie potentielleBases d'analyse des fonctions de plusieurs variables (gradient, hessienne)Lectures complémentaires conseillées : http://www.mat.univie.ac.at/~neum/glopt.html (complexe mais très complet)http://apps.nrbook.com/empanel/index.html (Numerical Recipes, chap. 10)
8

Structure électronique et spectroscopies : méthodes, desproblèmes et quelques solutionsEnseignant : Majdi Hochlaf, Professeur, Université de Marne la Vallée
Objectifs de l’enseignement : Présentation des méthodologies des calculs électroniques pour générer des sur-
faces d’énergie potentielle telles que les méthodes de calcul ab-initio (MCSCF,MR-CI, CCSD(T)).
Description des états électroniques excités. Déterminations des propriétés (couplages, vibroniques, spin-orbite, …). Type de coordonnées internes et leur utilisation (e.g. coordonnées internes, de
Jacobi, …) Illustrations à travers d’exemples : états fondamental et excités de molécules
diatomiques et polyatomiques ayant un intérêt biologique, atmosphérique, as-trochimique et environnemental.
Plan succinct :Calculs électroniques :
bases atomiques : Slater, Gaussiennes, Pseudo potentiels ; Contraction Corrélation électronique : Méthodes mono et multi configurationelles
Propriétés : moment dipolaire, moment de transition, couplages spin-orbite, couplageadiabatique, …Problème nucléaire : Type de coordonnées, approche perturbative, approchevariationnelleComparaison des résultats théoriques aux résultats expérimentaux :affirmation/infirmation/addendaUtilisation des surfaces d’énergie potentielle pour la spectroscopie, les collisionsréactives et non-réactives.
Prérequis :Cours de S. Carniato sur les méthodes de calculs électroniques
Lectures complémentaires conseillées : J. M. Hollas. Spectroscopie. Dunod.Paris, 2003.Molecular Quantum Mechanics, P. Atkins and R. Friedman, Oxford University PressStructure électronique des molécules, Y. Jean et F. Volatron, Dunod Introduction to Computational, F. Jensen, WileyMolecular modeling, Principles and applications, A. R. Leach, Pearson Education EMA
Simulation des Cristaux : du cristal parfait au cristal réelEnseignant : Nathalie Capron, maître de conférences, Université Pierre et Marie Curie
Objectifs de l’enseignement :Après une rapide description (sur une base de documents dont les étudiants pourrontse servir seuls) des cristaux à travers leur appellation en terme de groupes d’espace,le cours tient à présenter les généralités de la simulation numérique, puis lesspécificités particulières aux solides (quels objets étudier et de quelle manière), grâcenotamment à des confrontations (ondes planes versus orbitales localisées, DFT versusHF, espace réciproque versus espace réel). Des exemples concrets (défauts dans TiO2,joints de grain dans HfO2) seront discutés.
Plan succinct :1) Rappels de Cristallographie – Energie des Cristaux2) Description de l’espace réciproque (définitions, cellule de Wigner-Seitz et zones deBrillouin, lien espace réel – espace réciproque, transformée de Fourier)
9

3) Simulation numérique : généralités et spécificités pour les systèmes cristallins,principaux paramètres à vérifier pour une bonne simulation4) Description des solides réels : types de défauts -0 D, 1 D, 2 D, 3D – et concentrationde défauts dans les modèles envisageables5) Problèmes / Avantages posés par les structures périodiques6) Codes existant (Crystal, Siesta, Wien2k, VASP, …)
Prérequis : notions mathématiques (espaces vectoriels, séries de Fourier,…), notionsde cristallographie géométrique, notions de chimie quantique
Lectures complémentaires conseillées : figureront sur le document de cours
Incertitudes en Chimie Computationnelle : sourceset solutions.Enseignant : Pascal Pernot, Directeur de recherche CNRS, Université Paris-Sud
Objectifs de l’enseignement :Présenter les principales sources d'erreur dans divers domaines de la chimiecomputationnelle pour arriver à une définition opérationnelle de l'incertitude associéeà un résultat de calcul. Les méthodes statistiques standard pour la gestion desincertitudes sont présentées et appliquées sur plusieurs exemples de la littératurerécente.
Plan succinct :1. Le concept de Mesure Virtuelle2. Les sources d'erreur en chimie computationnelle - erreurs aléatoires vs.
systématiques3. Modélisation statistique et propagation des incertitudes4. Etudes de cas
Prérequis : statistique descriptive élémentaire (moyenne, variance, covariance,
histogramme...) notions de probabilités (variable aléatoire, loi de distribution...)
Lectures complémentaires conseillées : J. Tellinghuisen. Statistical error propagation. J. Phys. Chem. A 105:3917–3921,
2001. K. K. Irikura, R. D. Johnson, and K. R. N. Uncertainty associated with virtual
measurements from computational quantum chemistry models. Metrologia41:369–375, 2004.
10

SEQUENCE D’APPROFONDISSEMENT
Structure électronique et dynamique quantiqueavancées
11

Spectroscopie moléculaire : description quantiqueEnseignants : Céline Léonard, professeur, Université Paris-Est Marne la ValléeDavid Lauvergnat, directeur de recherche, CNRS-Université Paris-Sud
Objectifs de l’enseignement :Le but de ce cours est de montrer les différentes étapes pour simuler un spectrevibrationnel à partir de la résolution de l’équation de Schrödinger liée àl’approximation de Born-Oppenheimer. Le point de départ est la détermination de lasurface d’énergie potentielle, puis il faut trouver les coordonnées ‘vibrationnelles’ lesmieux adaptées. Pour finir il faut résoudre l’équation de Schrödinger pour lesmouvements internes des noyaux.
Plan succinct :I. Introduction : spectroscopie vibrationnelle, pourquoi dépasser le modèle har-
monique ?II. Détermination d’une surface d’énergie potentielle
- Etude de la structure électronique de façon à bien choisir la méthode ab-initio(méthodes mono-configurationnelles ou pas, espaces actifs : diagramme d’OM, voiesde dissociation ; symétrie : étude de la symétrie des points remarquables de lasurface, règles de corrélation, diagramme de Walsh) - Choix des coordonnées pour la description du mouvement des noyaux- Détermination de la fonction analytique représentant la surface
III. Détermination d’un spectre vibrationnelL'une des difficultés en dynamiques quantique est de trouver une représentationcompacte des fonctions d'onde pour rendre les calculs des spectres vibrationnelsfaisables. Plusieurs aspects permettront d'illustrer le contournement de cettedifficulté :- Influence des coordonnées et la conséquence sur l'opérateur énergie cinétique- Modèles physiques : est-il utile de traiter tous les degrés de liberté ?- Comment éviter une croissance exponentielle de la taille de la base ?
Prérequis : Cours de liaison chimique, cours de la semaine 1, modèle de l’oscillateurharmonique
Lectures complémentaires conseillées : - Molecular vibrations, Wilson, Decius and Croos, Dover- Léonard C., Carbonnière Ph., Boudon V., Gabard T., Talbi D., La modélisation desvibrations des molécules : enjeux et applications, Edition spéciale de l’ActualitéChimique, vol. 382-383, Fev.-Mars (2014).
12

Le rôle et l’évaluation des interactions intermoléculairesdans la structure et les propriétés physico-chimique desystèmes d’origine biominérales
Enseignant : Frederik Tielens, professeur, Université de Bruxelles
Objectifs de l’enseignement :La DFT a provoqué depuis les années 90 une révolution dans les possibilités à décrirela structure électronique des solides. L’application de cette théorie en combinaisonavec les ressources de calculs toujours plus puissantes, permet aujourd’hui de calculerdes systèmes de plus en plus complexes. Parmi ces systèmes complexes on retrouvele solide bio minéral. Ces minéraux ayant des interfaces et des structures bulk les pluscomplexes existant. Des systèmes solides en interaction avec une phase liquide,combinant tous types d’interactions intermoléculaires. La modélisation de systèmesd’origine bio minérales ont un double challenge : 1 proposer un modèle structuraleréaliste et calculable et 2. Proposer une méthode ou stratégie de calcul permettant ladescription des interactions intermoléculaires les plus adaptées pour ces systèmes.
Plan succinct :Interactions classiques (électrostatique, induction, dispersion)Choix de modèle de géométrie – défauts de la DFT –Rappels méthodologiques, DFT et mécanique moléculaire.Applications : La silice amorphe, l’oxalate de calcium, l’hydroxyapatite et le carbonatede calcium
Prérequis :Introduction à la chimie quantiqueIntroduction à la chimie des matériaux
Réactivité unimoléculaire : cinétique et dynamiqueEnseignants : Riccardo Spézia, chargé de recherche, CNRS – Université Pierre et MarieCurie
Objectifs de l’enseignement : L’objectif du cours est de donner les basesthéoriques de la réactivité chimique ; en particulier la réactivité uni-moléculaire seraprise comme exemple en termes de fragmentation et d’isomérisation (les mêmesconcepts sont applicables à la réactivité bi-moléculaire). Nous traiterons d’abord lathéories statistique RRKM qui donne accès de façon relativement simple aux constantcinétiques à partir des connaissances des minima et points de selle. Ensuite, nousmontrerons que certains effets dynamiques peuvent être importants et que ladynamique directe peut donner des informations sur ces comportements non-statistiques.
Plan succinct :Nous commencerons par l’exposition de la théorie RRKM et donc de la théorie de l’étatde transition et le concept de relaxation interne vibrationnelle (IVR). En particuliernous montrerons comme il est possible de dériver la formule qui donne la valeur de laconstante cinétique de réaction micro-canonique. A partir de l’équation de base, nousmontrerons ensuite comme inclure les effets rotationnelle et d’effet tunnel. Ladétermination des états de transitions dites « loose » sera aussi montré. Ensuite, nousdécrirons les possibles sources qui donnent lieu aux réactivités non-RRKM. Enfin, nousmontrerons comme la dynamique directe peut mettre en évidence certains effets non-statistiques : fragmentation directe (non-IVR) et bifurcations. Dans se cadre nousdonnerons les recettes élémentaires pour faire des dynamiques de collision qui sont àla base de certaines processus de réactivité uni-moléculaire. Cette approche sera
13

mise dans le contexte de la modélisation théorique de la spectrométrie de masse.
Prérequis : Concept de surface d’énergie potentielle, localisation et caractérisationdes minima et points de selle. Bases de thermodynamique statistique (espace desphases, fonction de partition). Bases de dynamique moléculaire Born-Oppenheimer.Energies moléculaires rotationnelle et vibrationnelle quantiques (niveaux quantiques).
Lectures complémentaires conseillées : T.Baer and W.L.Hase. UnimolecularReaction Dynamics. Theory and Experiments. Oxford University Press, 1996.
Etat de l’art en DFTEnseignant : Ilaria Ciofini, Directrice de recherche, CNRS-Chimie Paris Tech
Application et exploration de méthodes de DFT par la pratique.
Topology : recovering classical concepts from quantumchemistry Enseignante : Julia Contreras-Garcia, chargée de Recherche, CNRS – Université Pierreet Marie Curie
Objectifs de l’enseignement :Theoretical chemistry usually gives a cold and distant approach to chemical bond.However new visual tools are currently being developed which enable an intuitivevisualization and quantification of bonds of different types. Students will be able to visualize covalent and non-covalent interactions and theirchange along a reaction. During the course we will review topological tools for thevisualization of covalent (ELF [1]) and non covalent (NCI [1]) interactions that will allowthem to understand the theoretical meaning of chemical bonding while retaining itsrelationship to physical concepts, such as the electron density.
Plan succinct :1. Overview of theoretical and experimental approaches to determine chemical
bonds2. Topological concepts3. Chemical functions: Electron density, ELF and NCI. 4. Chemical bond change & reactivity
These concepts will be settled through a Practical class (computer room). Each pair ofstudents will be given a text book example (e.g. ethanol, ethane, water, branchedoctane). They will have to present their results to the class and describe the bond typeencountered.
Prérequis :Quantum chemistry
Théorie Valence Bond pour la structure électronique et laréactivitéEnseignant : Benoît Braïda, maître de conférences, Université Pierre et Marie Curie
Objectifs de l’enseignement et plan succinct :
14

La théorie Valence Bond (VB), formulée au tournant des années 1930, a depuis évoluéen parallèle de la théorie des orbitales moléculaires (OM), des développements baséssur la théorie VB étant notamment à l’origine de deux prix Nobel. La théorie VB al’intérêt d’offrir une connexion directe entre mécanique quantique et concepts usuelsde chimie moléculaires (structures de Lewis, résonance, langage des flèches enréactivité,…), et bien que peu enseignée elle est ainsi un outil de choix en matièred’étude de problèmes de structure électronique comme de réactivité. Le premier coursaura pour but de brièvement poser les bases de la théorie VB, et d’illustrer son intérêten matière d’étude de la réactivité chimique avec quelques applications récentesmarquantes.
Prérequis : Cours de 1ère semaine de Stéphane Carniato
Lectures complémentaires conseillées : Pour aller plus loin :https://wiki.lct.jussieu.fr/workshop/index.php/Basic_literature_on_VB_theory
Chimie quantique relativisteEnseignant : Valérie Vallet, directrice de Recherche, CNRS – Université de Lille
Objectifs de l’enseignement : Comprendre les bases de la mécanique quantique relativiste. Apprécier l’importance des effets relativistes sur la structure électronique et les
propriétés de systèmes contenant des atomes lourds Connaitre les différents Hamiltoniens relativistes et leurs différentes approxima-
tions : 1/ Hamiltonien de Dirac-Coulomb Breit, 2/ Hamiltoniens à 2-composantes, 1-composantes ; 3/ bases atomiques relativistes, et concepts de potentiels effectifsde cœur relativistes
Plan succinct :Description générique des effets relativistesEquation de Dirac pour un système mono et multiélectroniqueApproximations de l’équation de DiracConcept de potentiels effectifs de cœur Exemples démontrant l’importance des effets relativistes
Prérequis : Mécanique quantique, et chimie quantique : notion de bases atomiques, LCAO, or-
bitales, structure de bande, couplage angulaires Notion de relativité restreinte
Lectures complémentaires conseillées : Relativistic Quantum Chemistry: The Fundamental Theory of Molecular Science,Markus Reiher, Alexander Wolf, Wiley 2009Introduction to Relativistic Quantum Chemistry, Kenneth G. Dyall and Knut Faegri, Jr.,Oxford University Press, 2007.
Dynamique quantique avec la méthode Multiconfiguration
15

Time-Dependent Hartree (MCTDHEnseignant : Daniel Pelaez Ruiz, maître de conférences, Université de Lille 1
Objectifs de l’enseignement :• Identifier des systèmes ayant besoin d’une description quantique. • Se familiariser avec la méthodologie de dynamique quantique. • Présenter la méthodologie de dynamique quantique MCTDH. • Présenter le Modèle de Couplage Vibronique dans le cadre MCTDH.
Plan succinct :On se propose de présenter d’une manière globale la méthodologie de dynamiquequantique MCTDH appliquée à des systèmes moléculaires de grande taille. Plusprécisément, on va se focaliser sur la description des processus photo-induits.
Prérequis :Connaissance de base de structure électronique (approximation de Born-Oppenheimeret Surfaces d’Énergie Potentielle). Connaissance de base du concept de paquetd’ondes.
Lectures complémentaires conseillées : - Revue MCTDH [M. H. Beck, A. Jäckle, G. A. Worth et H.-D. Meyer, The
multiconfiguration time-dependent Hartree method: A highly efficient algorithm forpropagating wavepackets, Physics Reports 324, 1 (2000)].
- Revue sur calcul d’états vibrationnels [J. M. Bowman, T. Carrington, et H.-D. MeyerVariational Quantum Approaches for Computing Vibrational Energies of PolyatomicMolecules, Molecular Physics 106, 2145-2182 (2008)].
16

SEQUENCE D’APPROFONDISSEMENT
MODELISATIONS MULTI-ECHELLES
17

Modélisation de la réactivité aux interfacesEnseignant : Alexis Markovits, professeur, Université Pierre et Marie Curie
Objectifs de l’enseignement :Comprendre la structure et la réactivé du solide par le raisonnement du chimiste.
Plan succinct :I). Quelques outils du chimiste pour décrire le solideII) Quelques exemples de modélisation à la surface du solide
Prérequis :A peine un peu de Chimie orbitalaire.
Lectures complémentaires conseillées : Solids and Surfaces: A Chemist's View of Bonding in Extended Structures par MonsieurRoald Hoffmann
Modélisation par DFT-périodique de l’interaction entre unemolécule et une surface. Enseignant : Jérôme Roques, maître de conférences, Université Paris-Sud
Objectifs de l’enseignement :Illustrer, au travers d’un exemple, la démarche à suivre pour modéliser l’interactionentre une molécule et une surface en utilisant une approche périodique couplée à uneméthode statique de calcul utilisant la théorie de la fonctionnelle de la densité.
Plan succinct :1) Modélisation d’un cristal.2) Modélisation d’une surface.3) Modélisation d’une molécule dans le vide.4) Etude de l’interaction d’une molécule avec un modèle de surface.
Pré-requis :Cours d’Alexis Markovits sur la ’’Modélisation de la réactivité aux interfaces’’
Lectures complémentaires conseillées :http://cms.mpi.univie.ac.at/vasp/vasp/vasp.html
Dynamique moléculaire ab initio : méthodes et applicationsdans divers domaines (phase gazeuse, phase liquide,interfaces solide/liquide et liquide/air)Enseignant : Marie-Pierre Gaigeot, professeur, Université d’Evry Val d’Essonne
Objectifs de l’enseignement :Dans cet enseignement, nous présentons les principes des dynamiques moléculairesab initio, principalement dans la représentation de la fonctionnelle de la densité (DFT),et montrons de nombreuses applications avec un but précis : montrer que cetteméthode est actuellement appliquée de façon « quasi-routinière » pour traiter dessystèmes moléculaires et des assemblages moléculaires de complexités diverses.Nous voyons des exemples de molécules et clusters en phase gazeuse, des moléculesimmergées dans des liquides, des systèmes inhomogènes complexes comme lesinterfaces solide/liquide et liquide/air. Nous voyons également comment uneobservable comme la spectroscopie vibrationnelle peut être modélisée dans cesdifférents états de la matière, comme fil conducteur.
18

Plan succinct :- méthodes- applications en phase gazeuse- applications en liquides- applications pour des interfaces solide/liquide, liquide/air
Prérequis :Notions de dynamique moléculaire, notions de représentation électronique DFT
Lectures complémentaires conseillées : Ab initio molecular dynamics, Basic theory and advanced methods, D. Marx, J. Hutter,Cambridge University Press, 2012
Compréhension, prédiction et design in silico : quelquesrôles et défis dans le domaine de la simulation desmatériaux Enseignant : Caroline Mellot-Draznieks, directrice de recherche, CNRS-Collègede France
Objectifs de l’enseignement :Les matériaux à charpente hybride ou MOFs (Metal Organic Frameworks) constituentune famille fascinante de solides cristallisés, tant par leur structure cristalline poreuseque par leur versatilité chimie en termes de fonctionnalisation, qui ouvrent la voie à unlarge champ d’applications (catalyse, séparation de gaz, photochimie, sensing, drugdelivery…). Nous nous attarderons sur quelques exemples choisis de matériaux decette famille pour aborder leur simulation en termes de structures cristallines,propriétés à l’interface, propriété électronique, polymorphisme. Nous nous attacheronsà illustrer l’adéquation entre la méthode de simulation choisie (ex : champ deforce versus DFT, dynamique versus optimisation de géométrie) et la question poséeau simulateur, celle-ci relevant de la compréhension d’une propriété connue, de saprédiction voire même du design, ie lorsqu’on anticipe l’existence d’un nouveaumatériau doté d’une propriété « cible ».
Plan succinct : Simulations en rapport avec la caractérisation expérimentale du so-
lide i) pour assister la résolution structurale (DRX de laboratoire ou synchro-tron) ii) pour anticiper un polymorphisme iii) ou aider à l’interprétation despectres RMN du solide
Impact de la structure électronique du solide sur ses propriétés : i)cas de solides « flexibles » sous stimulus (adsorption de gaz, température) ii)impact du linker organique constitutif du MOF sur sa stabilité.
Comment utiliser les simulations pour aller vers de nouveaux so-lides fonctionnels ? Quelques exemples de design in silico de nouveauxsolides suivis … de leur synthèse !
Prérequis :Méthodes de mécanique moléculaire, champ de force, DFT, diffraction, structurecristalline
19

Champs de forces pour la modélisation moléculaireEnseignant : Carine Clavaguéra, chercheur CNRS – Université Paris-Sud
Objectifs de l’enseignement :Comprendre la construction d’un champ de forces. Connaître les différents types dechamps de forces existants. Donner l’état de l’art et les développements récents.Connaître les domaines d’application et les limitations des champs de forces.Illustration par différents exemples.
Plan succinct :- panorama général des champs de forces pour la modélisation moléculaire- modèles mathématiques et paramétrisation- champs de forces polarisables- champs de forces réactifs- applications
Prérequis :Cours d’introduction aux méthodes de chimie théorique de la première semaine dulabel de chimie théorique
Lectures complémentaires conseillées : Molecular modelling : principles and applications, A. R. Leach, Addison WesleyLongman, 2001 (2ème édition)
Méthodologies hybrides QM/QM’ ou QM/MM
Enseignant : Etienne Derat, maître de conférences, Université Pierre et MarieCurie
Objectifs de l’enseignement :Cet enseignement se donne pour objectif de décrire les méthodes permettant desimuler avec une composante en mécanique quantique les systèmes moléculairescomplexes. Pour décrire la réactivité, il est nécessaire de traiter le système parmécanique quantique. Ceci pose un problème pour les systèmes de grande taille et/oude grande complexité. Une idée simple mais parfois complexe à mettre en œuvreconsiste à séparer le système en plusieurs parties. Ce cours montrera les concepts lesplus fréquemment utilisés dans ce domaine et se terminera par une présentationpratique de calcul QM/MM.
Plan succinct :Historique du concept de partition en modélisation ; Méthodes additives etsoustractives ; Méthodes de coupures entre les différentes zones ; Traitement del’interaction entre les différents niveaux de calcul ; Présentation des logicielsstandards permettant d’effectuer un calcul QM/QM’ ou QM/MM ;
Pré-requis :Connaissance des méthodes de modélisation classiques et quantiques
Lectures complémentaires conseillées : (1) Lin, H.; Truhlar, D. G. Theoretical Chemistry Accounts. 2006, 185–199.(2) Senn, H. M.; Thiel, W. Angewandte Chemie International Edition. 2009, 1198–
1229.(3) Chung, L. W.; Sameera, W. M. C.; Ramozzi, R.; Page, A. J.; Hatanaka, M.;
Petrova, G. P.; Harris, T. V.; Li, X.; Ke, Z.; Liu, F.; Li, H.-B.; Ding, L.; Morokuma,K. Chemical Reviews., 2015, 5678-5796.
20

21

Modélisation des états excités dans les systèmes complexesEnseignant : Aurélie Perrier-Pineau, maître de conférences, Université ParisDiderot – Chimie Paris Tech
Objectifs de l’enseignement :Au cours de cette séance de travaux pratiques, nous nous intéresserons à lamodélisation des états excités au sein de molécules dites « complexes », présentantun intérêt pour les nanosciences. Nous nous intéresserons en particulier aux nano-interrupteurs organiques (molécules “photoswitch”) et aux moléculesphotosensibilisatrices utilisées dans les cellules solaires.Pour ces systèmes de “grande” taille, la Théorie de la Fonctionnelle de la DensitéDépendante du Temps (TD-DFT) constitue l'outil de choix pour atteindre unedescription de leurs propriétés optiques et une analyse de leur photoréactivité. Dansce cadre, nous analyserons les fichiers de sortie obtenus à l'aide du code Gaussian 09.Nous analyserons en particulier les spectres d'absorption, d'émission et les éventuelstransferts de charge et/ou d'électrons obtenus au sein de ces systèmes aprèsphotoexcitation.
Plan succinct :I. Prise en main : Etude du Spectre d'absorption du formaldéhyde II. Etude des états excités d'un photoswitch organique : molécule modèle dedithiényléthène III. Etude d'un composé organique de type « Push-Pull »IV. Etude approfondie du transfert de charge
Prérequis :DFT, modélisation des états excités (méthode post-Hartree Fock et TD-DFT)
Lectures complémentaires conseillées : Essentials of Computational Chemistry, Theories and Models, Christopher J. Cramer,Wiley
Multi-scale methods for molecular simulations of cellularenvironments Enseignants : Sophie Sacquin Mora, chargée de recherche, CNRS, FabioSterpone, chargé de recherche, Institut de biologie physico-chimique
Objectifs de l’enseignement :In this lesson the modeling of macromolecules in the cellular environment will beintroduced. As a first step the key effects of macromolecular crowding on proteinmobility and stability will be exposed. We will then pause on the necessarysimplifications required to model macromolecules under the crowded cellularconditions, namely the definition of water free coarse-grained model. As final and mainingredient we will explain how hydrodynamic interactions can be introduced inmolecular simulations in order to explicitly account for solvent mediated interactionsand correlations.
Plan succinct :1. Stability, mobility under crowding: excluded volume effect, specific electrostatic
interactions.2. Toward simulations of cellular environments: Water-free coarse-grained models.3. How to account for solvent mediated interactions: The Brownian Dynamics, and
Lattice-Boltzmann Molecular Dynamics.
Prérequis :
22

Basic statistical-mechanics, Thermodynamics
Lectures complémentaires conseillées : T. Frembgen-Kesner and A. H. Elcock , « Computer Simulations of the BacterialCytoplasm », Biophys. Rev. 2013, 5, 109-119.S B Zimmerman, and A P Minton, « Macromolecular Crowding: Biochemical, Biophysi-cal, and Physiological Consequences », 1993, 22, 27-65.
Comment estimer les potentiels redox de cations dans lesprotéines par simulation numérique ?Enseignant : Aurélien De la Lande, Chargé de recherche, CNRS-Université Paris-Sud
Objectifs de l’enseignement :L'objectif de ce TP sur ordinateur est d'illustrer comment les simulations numériquespermettent de calculer des propriétés macroscopiques tels que les potentiels redoxdans des systèmes moléculaires complexes. En prenant l'exemple d'une globine(petite protéine contenant un hème), nous verrons comment relier les énergiesélectroniques calculées par mécanique moléculaire et par chimie quantique auxpotentiels redox et les comparer aux valeurs expérimentales. Nous verrons l'influence des durées de simulations de dynamiques moléculaires sur lafiabilité des résultats. Par ailleurs nous verrons dans quelles mesures différents typesde champs de forces ou des approches QM/MM peuvent fournir des valeurs plus oumoins proches des données expérimentales.
Plan succinct :- Partie théorique : calcul d'une enthalpie libre de réduction grâce à la théorie de laréponse linaire. - Partie pratique : analyse des trajectoires de dynamique moléculaire, calcul du bilanthermodynamique d'oxydation de l'hème dans la globine. Effet de la longueur desimulation. Analyse des contributions des résidus de la protéine et de l'eau sur le bilanthermodynamique de la réaction. Comparaison des résultats obtenus avec un champde force polarisable pour l'eau.Prérequis :-Notions de thermodynamique statistiques et d'électrochimie : potentiels redox,enthalpie libre d'oxydation/réduction, fonctions de partition.-Notions d'électrostatiques : loi de Coulomb, induction électronique (dipoles induits,interactions charge/dipoles)-Notions de champs de force, approches QM/MM
Lectures complémentaires conseillées : Texte de remise du prix Nobel de Marcus : http://www.nobelprize.org/nobel_prizes/chemistry/laureates/1992/marcus-lecture.htmlTechniques de simulation des potentiels redox dans les protéines à hème :J. Blumberger. Phys. Chem. Chem. Phys. 2008, 10, 5651
23





























