Embed Size (px)
Citation preview

Les déficits constitutionnels en FVII
Muriel Giansily-BlaizotDépartement d’hématologie biologique
CHU de Montpellier
Le 30 janvier 2015, Lyon

Les déficits constitutionnels en FVII
Définition et généralités

Définition
� En théorie: FVII:C <70% (N: 70-140%)� En pratique: FVII:C <30%
� Suspecté devant l’association�Allongement du TQ�TCA normal
� Bien différencier les déficits constitutionnels ou héréditaires et les déficits acquis.
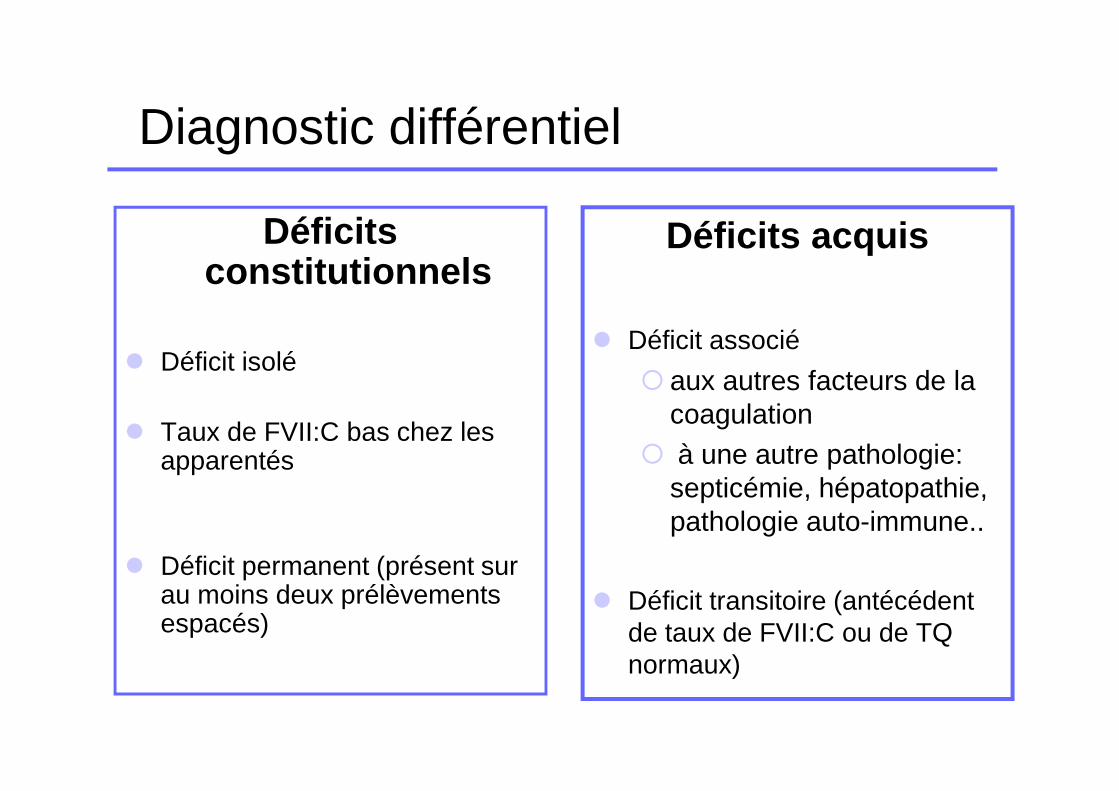
Diagnostic différentiel
Déficits constitutionnels
� Déficit isolé
� Taux de FVII:C bas chez les apparentés
� Déficit permanent (présent sur au moins deux prélèvements espacés)
Déficits acquis
� Déficit associé
� aux autres facteurs de la coagulation
� à une autre pathologie: septicémie, hépatopathie, pathologie auto-immune..
� Déficit transitoire (antécédent de taux de FVII:C ou de TQ normaux)

Exemples de déficits acquis en FVII
� En raison de sa ½ vie très courte, le FVII est le premier à diminuer en cas de :� Hypovitaminose K� Insuffisance hépatique débutante
� Grandes infections (septicémies…) la diminution du taux de FVII:C est transitoire.
� Rares cas d’auto-anticorps anti-FVII (contemporain d’une néoplasie, d’une infection VIH, isolé)

Les déficits constitutionnels en FVII
Formes cliniques
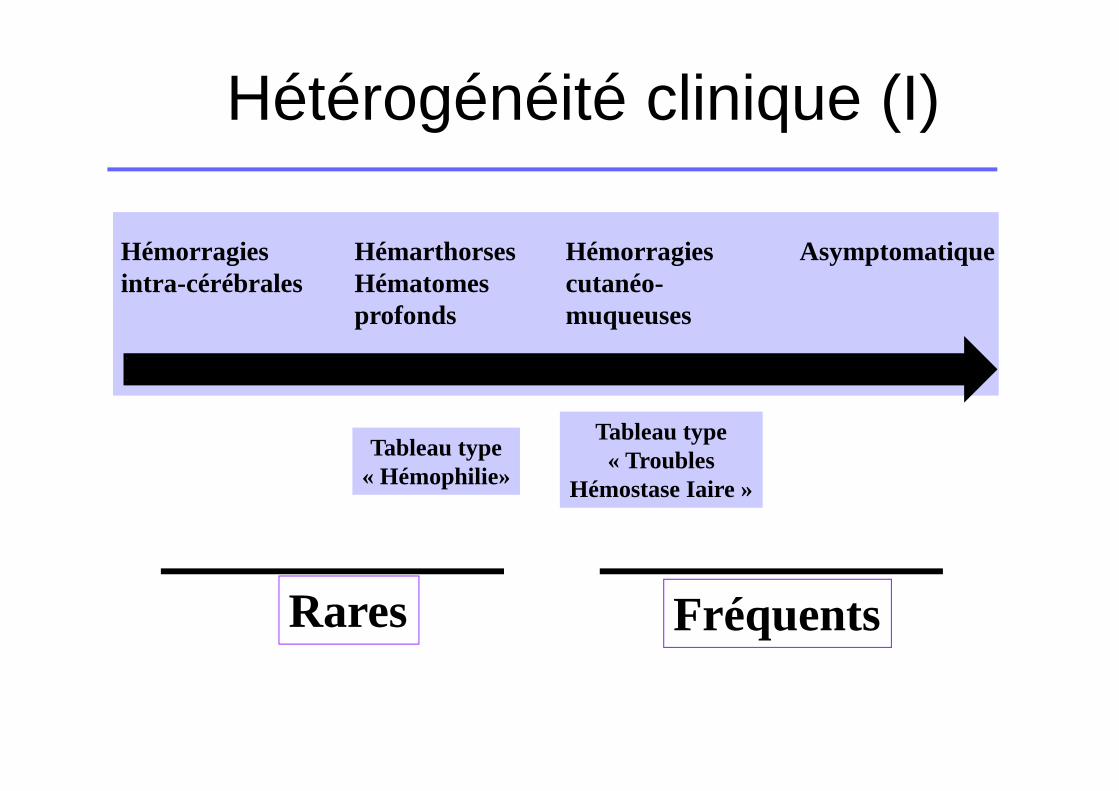
Hémorragies intra-cérébrales
HémarthorsesHématomes profonds
Hémorragies cutanéo-muqueuses
Asymptomatique
Tableau type« Hémophilie»
Tableau type« Troubles
Hémostase Iaire »
Rares Fréquents
Hétérogénéité clinique (I)

�Symptomatiques� Hémorragies intracérébrales (10-15%)� Hémarthroses et hématomes (20-25%)� Hémorragies cutanéo-muqueuses (65%)
�Asymptomatiques (sous- estimé)
Hétérogénéité clinique (II)

Hémorragies intra-cérébrales
� Rares (5 cas recensés en France)
� Gravissimes le plus souvent létales +++
� Dans les premières semaines de vie (3-5j à 3-4 mois)� Signes avant-coureurs: épistaxis, saignement à la chute du
cordon, ecchymoses faciles.� Hypertension intra-crânienne brutale sans traumatisme
déclenchant le plus souvent.
� Récidives fréquentes en l’absence de traitement.

Tableau « de type hémophilie »
�Rares (9 cas recensés en France)
�Hémarthoses et hématomes identiques à ceux de l’hémophilie�Spontanée ou post traumatique�Délabrement articulaire fréquent
� Traitement au long cours

Tableaux de type « troubles de l’hémostase primaire »
� Fréquent, 60% selon les études
� Hémorragies cutaneo-muqueuses� Epistaxis, gingivorragies, ecchymoses faciles, � ménorrhagies etc…
� Complications hémorragiques post-chirurgicales possibles mais non obligatoires.
� Pas de traitement au long cours et indication du traitement en préopératoire difficile à poser

Tableau « asymptomatique »
� Très fréquent (difficile à chiffrer car sous-estimé)
� Complications hémorragiques post-chirurgicales possibles mais non obligatoires.
� Pose un problème majeur d’indication du traitement substitutif en préopératoire.
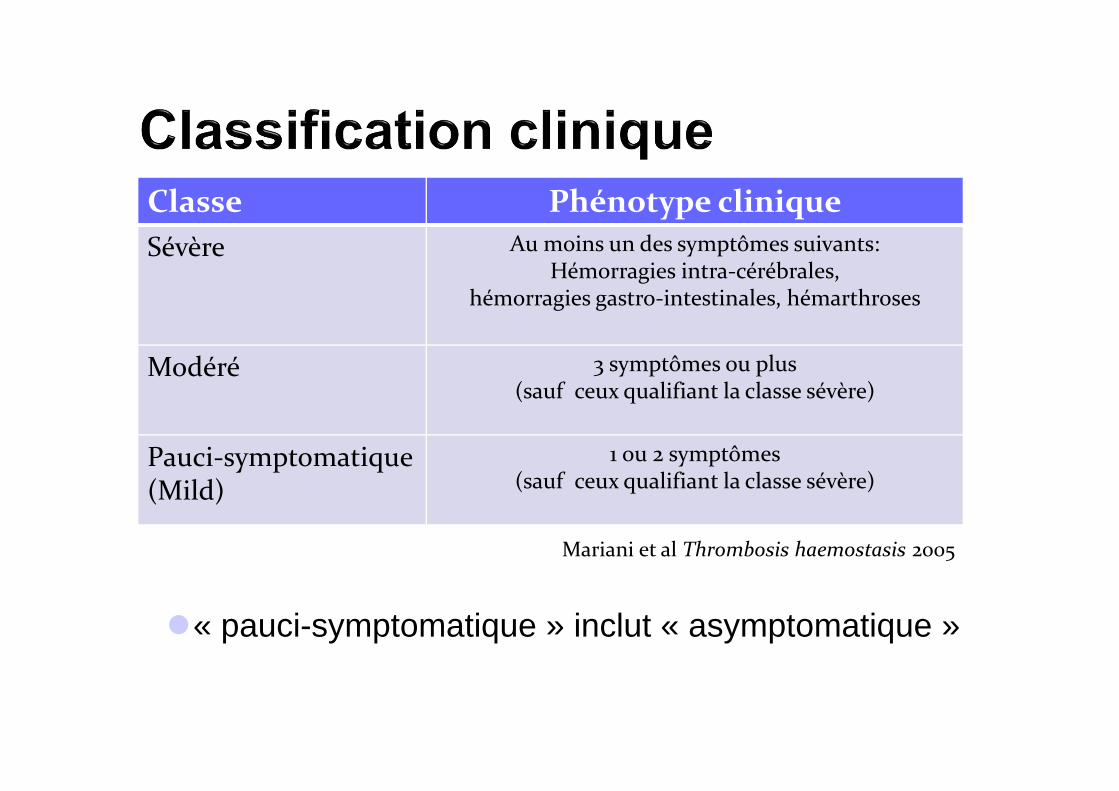
�« pauci-symptomatique » inclut « asymptomatique »
Classe Phénotype clinique
Sévère Au moins un des symptômes suivants: Hémorragies intra-cérébrales,
hémorragies gastro-intestinales, hémarthroses
Modéré 3 symptômes ou plus (sauf ceux qualifiant la classe sévère)
Pauci-symptomatique (Mild)
1 ou 2 symptômes (sauf ceux qualifiant la classe sévère)
Mariani et al Thrombosis haemostasis 2005

Les déficits constitutionnels en FVII
Physiopathologie

Le facteur VII de la coagulation
Un facteur clé aux multiples interactions
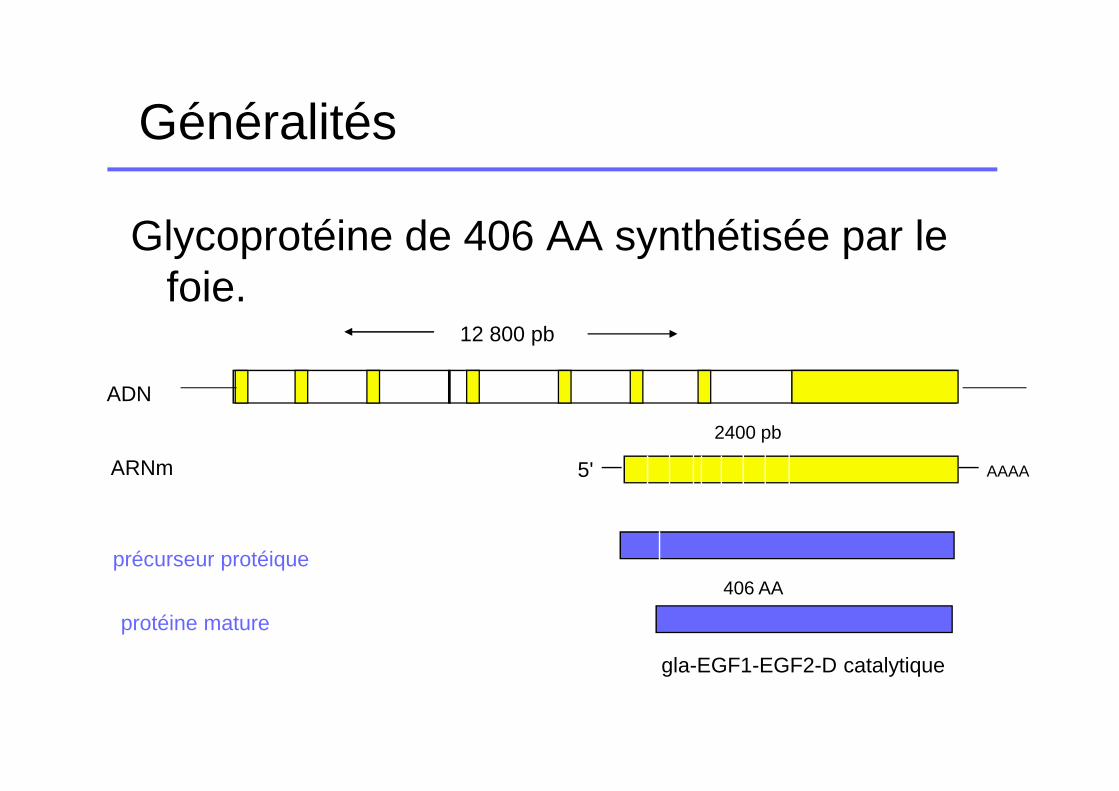
Généralités
Glycoprotéine de 406 AA synthétisée par le foie.
ADN
ARNm
précurseur protéique
protéine mature
12 800 pb
2400 pb
406 AA
gla-EGF1-EGF2-D catalytique
5' AAAA
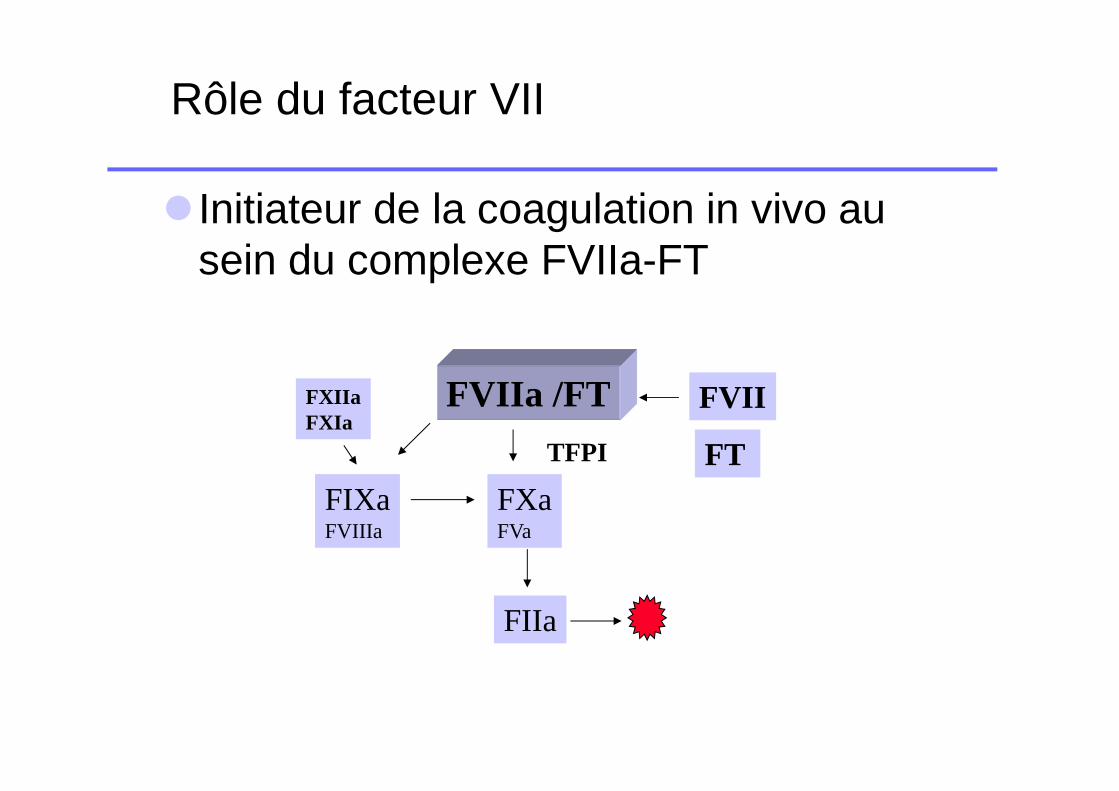
Rôle du facteur VII
� Initiateur de la coagulation in vivo au sein du complexe FVIIa-FT
FVIIa /FT
FXaFVa
FIXaFVIIIa
FIIa
FXIIaFXIa
FVII
FTTFPI
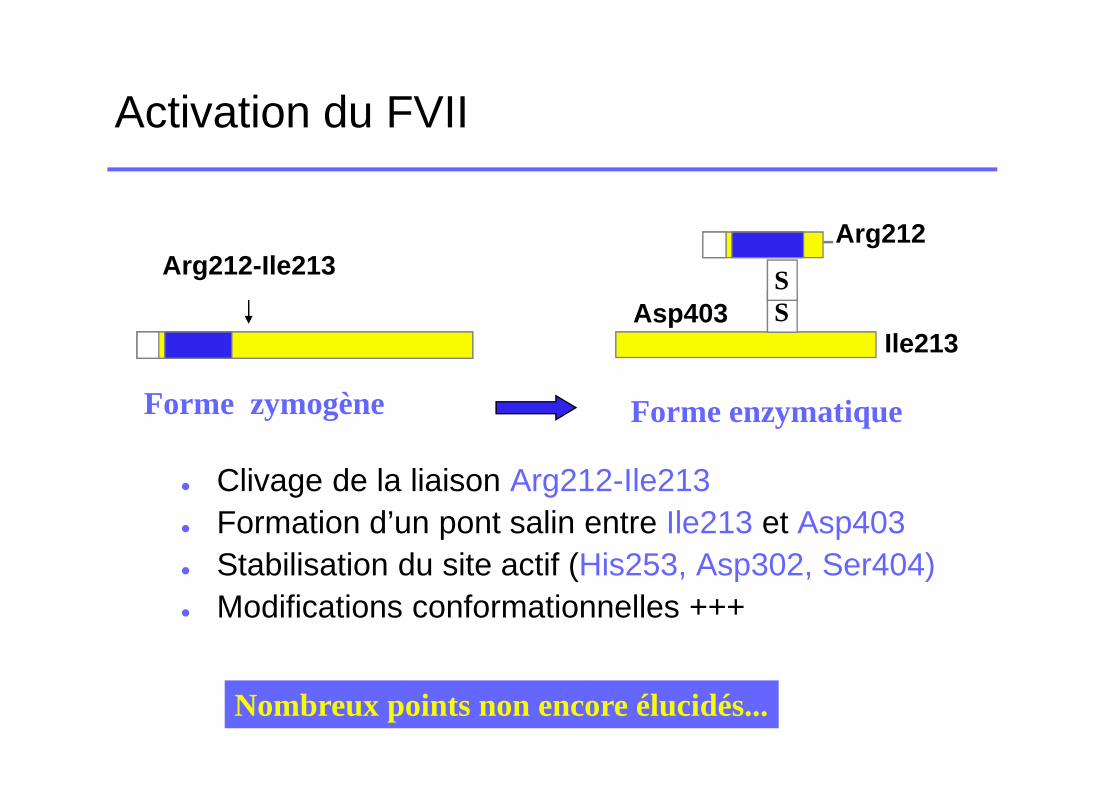
Activation du FVII
� Clivage de la liaison Arg212-Ile213� Formation d’un pont salin entre Ile213 et Asp403� Stabilisation du site actif (His253, Asp302, Ser404)� Modifications conformationnelles +++
Forme enzymatique
SS
Arg212
Asp403Ile213
Forme zymogène
Arg212-Ile213
Nombreux points non encore élucidés...
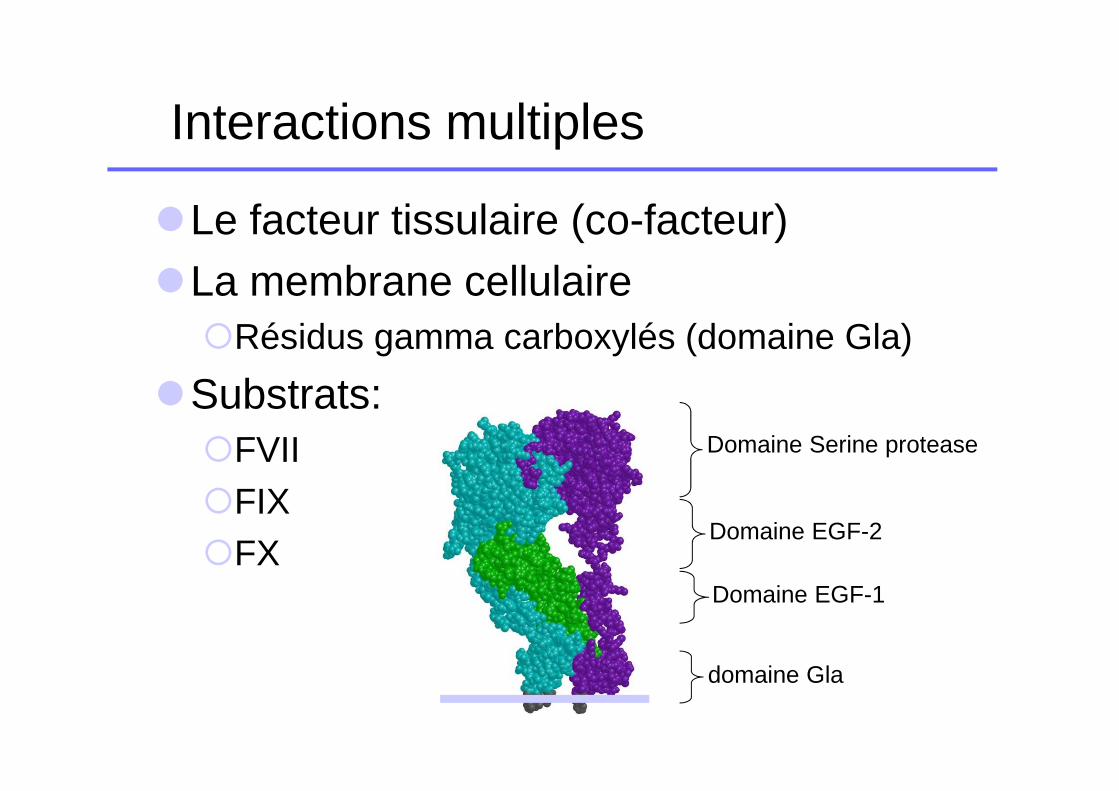
Interactions multiples
�Le facteur tissulaire (co-facteur)�La membrane cellulaire
�Résidus gamma carboxylés (domaine Gla)
�Substrats:�FVII�FIX�FX
Domaine Serine protease
Domaine EGF-2
Domaine EGF-1
domaine Gla
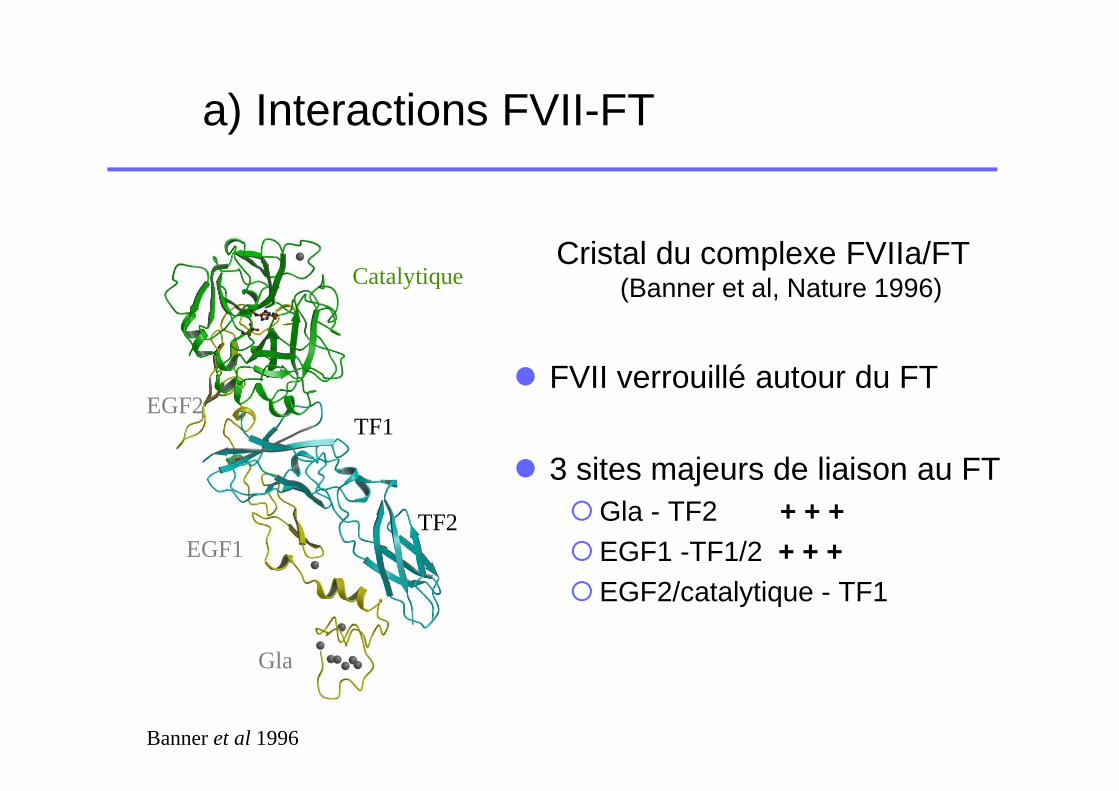
a) Interactions FVII-FT
Cristal du complexe FVIIa/FT (Banner et al, Nature 1996)
� FVII verrouillé autour du FT
� 3 sites majeurs de liaison au FT�Gla - TF2 + + +�EGF1 -TF1/2 + + +�EGF2/catalytique - TF1
Banner et al 1996
TF1
TF2
Gla
EGF1
EGF2
Catalytique

b) Interactions avec les membranes cellulaires
�Mb des plaquettes, cellules endothéliales
�Liaison aux phospholipides mb par les résidus « Gla » (ions calcium)
�Acide glutamique (Glu) gamma carboxylés (Gla) (réaction enzymatique dépendante de la vit K)
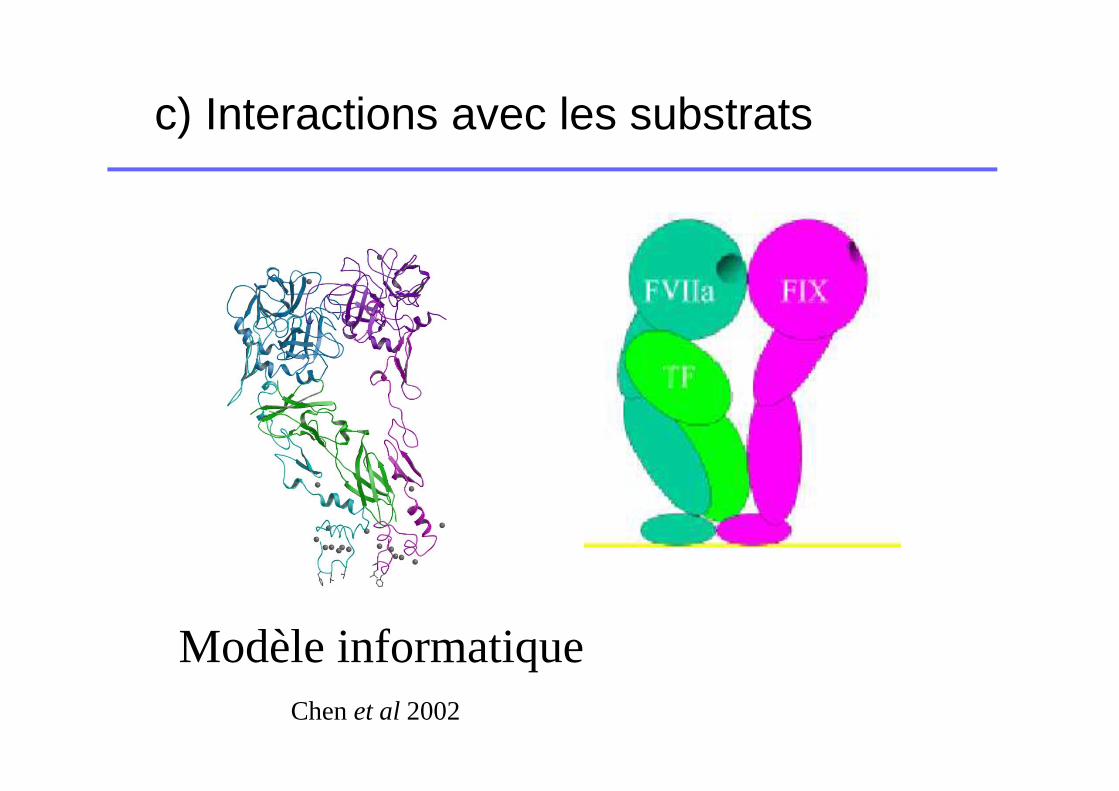
c) Interactions avec les substrats
Modèle informatiqueChen et al 2002

Le facteur VII de la coagulation
Est-il indispensable à la vie?

Souris knock out F7-/- (Rosen et al 1997)
� Souris knock out F7-/- (Rosen et al 1997) �Développement embryonnaire en apparence sans anomalie
(et pourtant transfert maternel de FVII non détectable).
�Décèdent rapidement d’hémorragies dans la première semaine après la naissance:� 70% à 24h (hémorragies intra abdominales)� Les autres à une semaine (hémorragies intra-cérébrales)
� Souris knock out F7+/- (Rosen et al 1997)�Environ 10% de FVII par rapport aux souris adultes +/+.�Ne présentent aucun signe hémorragique

Déficit en FVII: Anomalies du gène F7
Mutations ponctuelles
� Les plus fréquentes� À l’origine de déficits
très hétérogènes en FVII
Grands réarrangements du gène F7
� Rares� Parfois associés à des
anomalies du gène F10
Gene F7 14.8 kB Gène F10 26.7 kB
Chromosome 13

Anomalies du gène F7
� Mutations ponctuelles (tous types)
� Rares grands réarrangements:�Délétion gène complet (+/- avec le F10)� insertion/délétion etc…
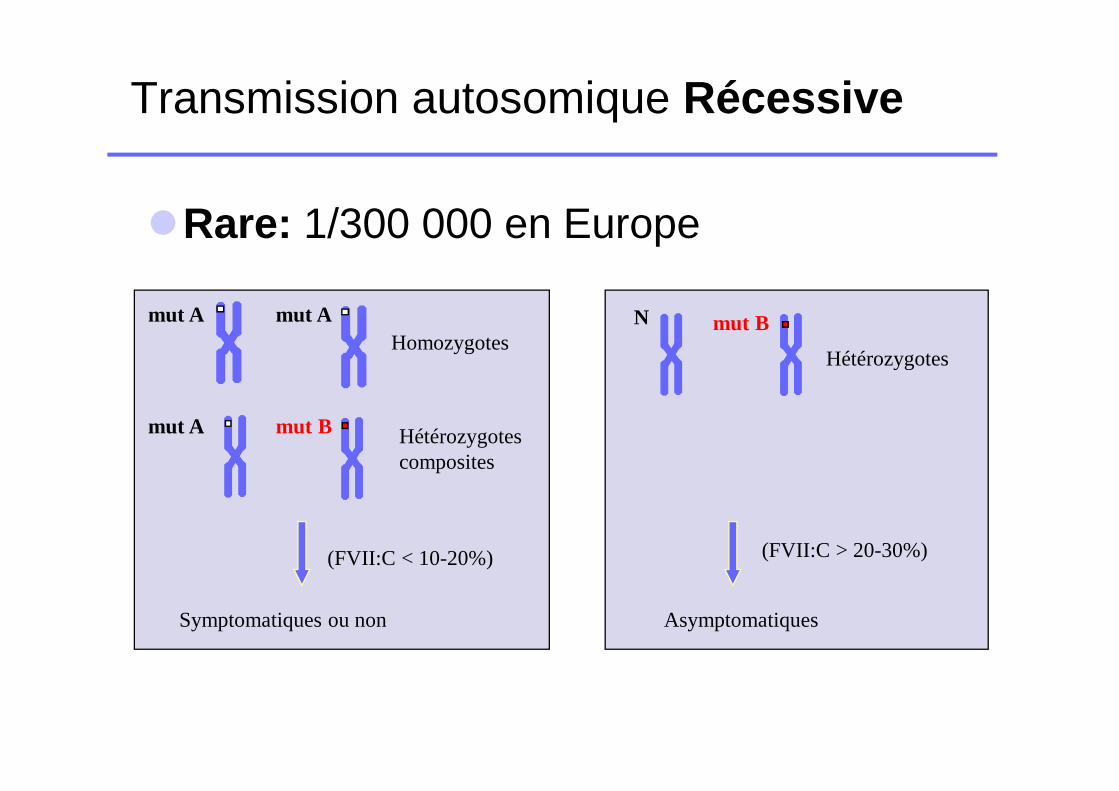
Transmission autosomique Récessive
�Rare: 1/300 000 en Europe
Hétérozygotes composites
mut A mut A
Symptomatiques ou non
Homozygotes
(FVII:C < 10-20%)
mut A mut B
mut BN
Hétérozygotes
(FVII:C > 20-30%)
Asymptomatiques

Les déficits constitutionnels en FVII
Diagnostic biologique

Diagnostic positif
�Dosage spécifique du FVII:C�Pose le diagnostic�À contrôler sur deux prélèvements espacés
�Dosage du FVII: Ag�Classe le déficit en quantitatif ou qualitatif
(intéressant pour le biologiste moléculaire)
�Pas d’intérêt clinique pour le patient.

Dosage du FVII:C
�Pas de consensus sur la thromboplastine
utilisée
�Origine humaine (recombinante ou extraction)
et origine animale
�Certains variants FVII donnent des valeurs de
FVII:C différentes selon la thromboplastine
utilisée

Thromboplastines, exemples
�Humaines�Recombinantes, exemples
� Gamme RecombiPlastin (IL)�Innovin (Dade Behring)
�Extraction placentaire, exemple�Thromborel S (Dade Behring)
�Animales�Lapin, exemple
�Gamme Neoplastin (Stago)
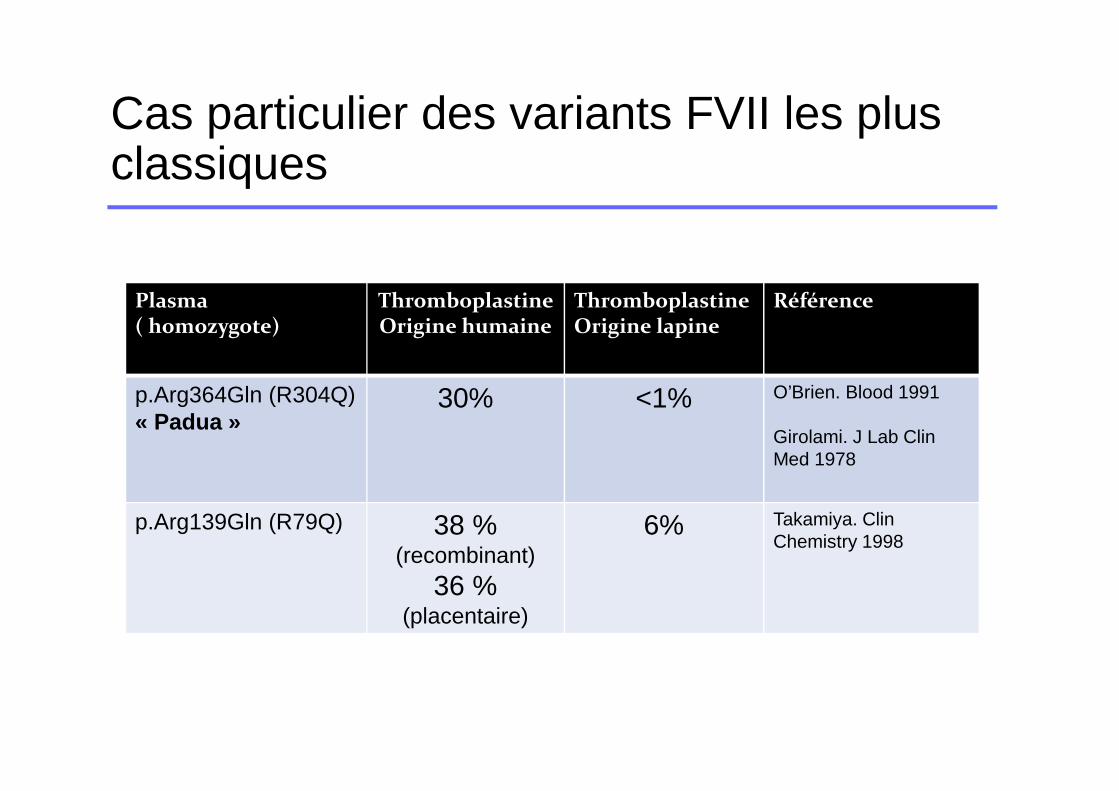
Plasma( homozygote)
Thromboplastine Origine humaine
ThromboplastineOrigine lapine
Référence
p.Arg364Gln (R304Q) « Padua »
30% <1% O’Brien. Blood 1991
Girolami. J Lab Clin Med 1978
p.Arg139Gln (R79Q) 38 % (recombinant)
36 % (placentaire)
6% Takamiya. Clin Chemistry 1998
Cas particulier des variants FVII les plus classiques
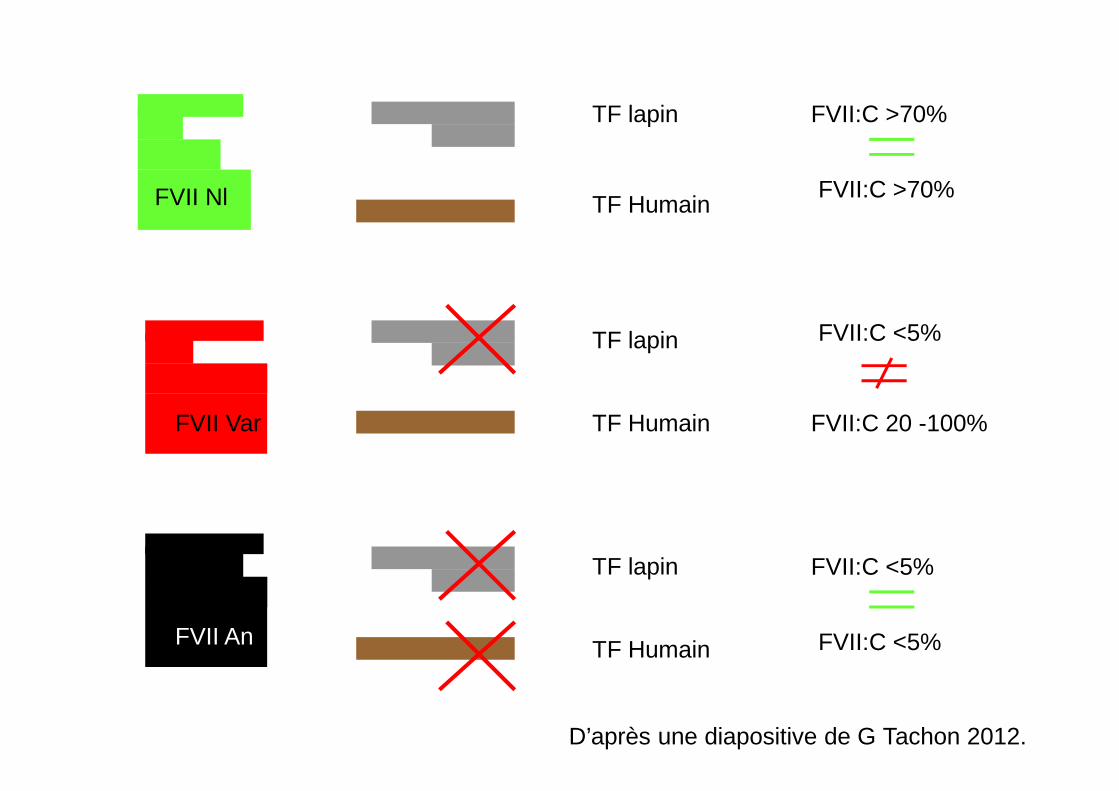
D’après une diapositive de G Tachon 2012.
FVII An
TF lapin
TF Humain
FVII:C <5%
FVII:C <5%
FVII Nl
TF lapin
TF Humain
FVII:C >70%
FVII:C >70%
FVII Var
TF lapin
TF Humain
FVII:C <5%
FVII:C 20 -100%

Dosage du FVII:C
Dans l’idéal, contrôler le taux de FVII:C avec une
thromboplastine humaine (recombinante).
� Les taux de FVII :C sont mieux corrélés avec les
phénotypes cliniques
�Variants FVII Padua et 139 le plus souvent
asymptomatiques et FVII:C plus élevés avec une
thromboplastine recombinante humaine…

Dosage du FVII:Ag (Kit Elisa)
� Pas d’utilité clinique� Le FVIIag n’est pas un facteur prédictif du risque hémorragique
� Utile au biologiste moléculaire (intérêt diagnostic )� Pour une bonne corrélation entre le génotype et le type de FVII
(quantitatif ou qualitatif)
� Utile pour la compréhension des mécanismes
mutationnel (intérêt scientifique)

Définitions et mécanismes
� Quantitatif� Diminution en quantité
d’un facteur qualitativement normal
� Mécanismes possibles?�Défaut de synthèse �Défaut de sécrétion�…
� Qualitatif� Synthèse en quantité
normale d’un facteur qualitativement anormal
� Mécanismes possibles?�Altération d’une fonction
(site catalytique, liaison aux autres facteurs, liaison aux phospholipides…)
Mutation dépendant…

Relation entre types de mutations et types de déficit
� Chaque mutation va avoir un impact différent sur la protéine codée
� Mutations qui empêchent la synthèse du FVII (associées à un déficit …….)
� Mutations qui ne gènent pas la synthèse mais qui rendent le FVII non fonctionnel (associées à un déficit …….)

“Catalogue des mutations F7”
� De nombreuses mutations sont classées en:�Associées à un déficit quantitatif�Associées à un déficit qualitatif
� “Facile” pour les homozygotes mais “plus compliqué” pour les hétérozygotes composites” (deux mutations différentes)

Les déficits constitutionnels en FVII
Diagnostic moléculaire

Diagnostic moléculaire gène F7
� Quelles stratégies d’étude?�Séquençage direct « à l’aveugle »? �Technique ciblée sur un type de mutation précis? �Méthodes de dosage génique?
� Elle dépend de plusieurs paramètres:�Mutation récurrente connue?�Taille du gène?�Type de mutations les plus fréquemment
recherchées?

Diagnostic moléculaire gène F7
� Dépend de plusieurs paramètres:
�Mutation récurrente connue? NON�Taille du gène? Petite�Type de mutations les plus fréquemment
recherchées? Mutations ponctuelles
Séquençage direct en première intention
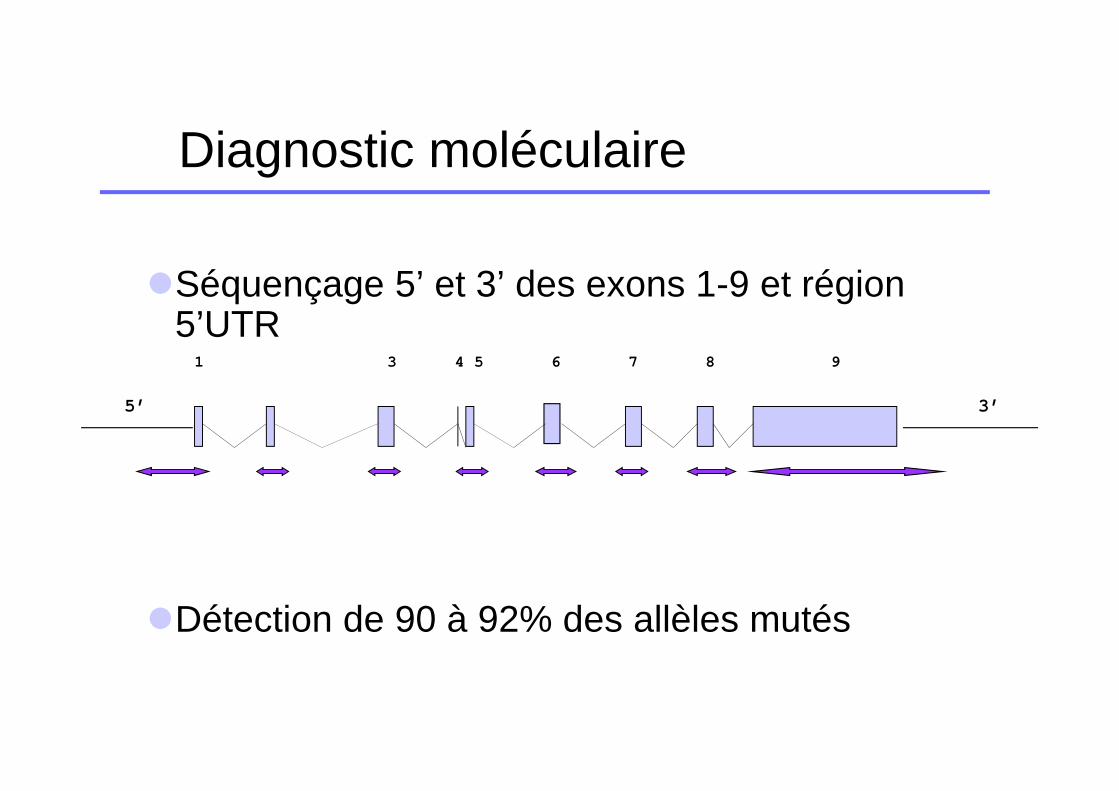
Diagnostic moléculaire
�Séquençage 5’ et 3’ des exons 1-9 et région 5’UTR
�Détection de 90 à 92% des allèles mutés
1 3 4 5 6 7 8 9
5’ 3’

Techniques de dosage génique
�Real time quantitative PCR�Semi-quantitative multiplex fluorescent-PCR
(SQF-PCR)�Multiplex ligation-dependant probe amplification
(MLPA)�…
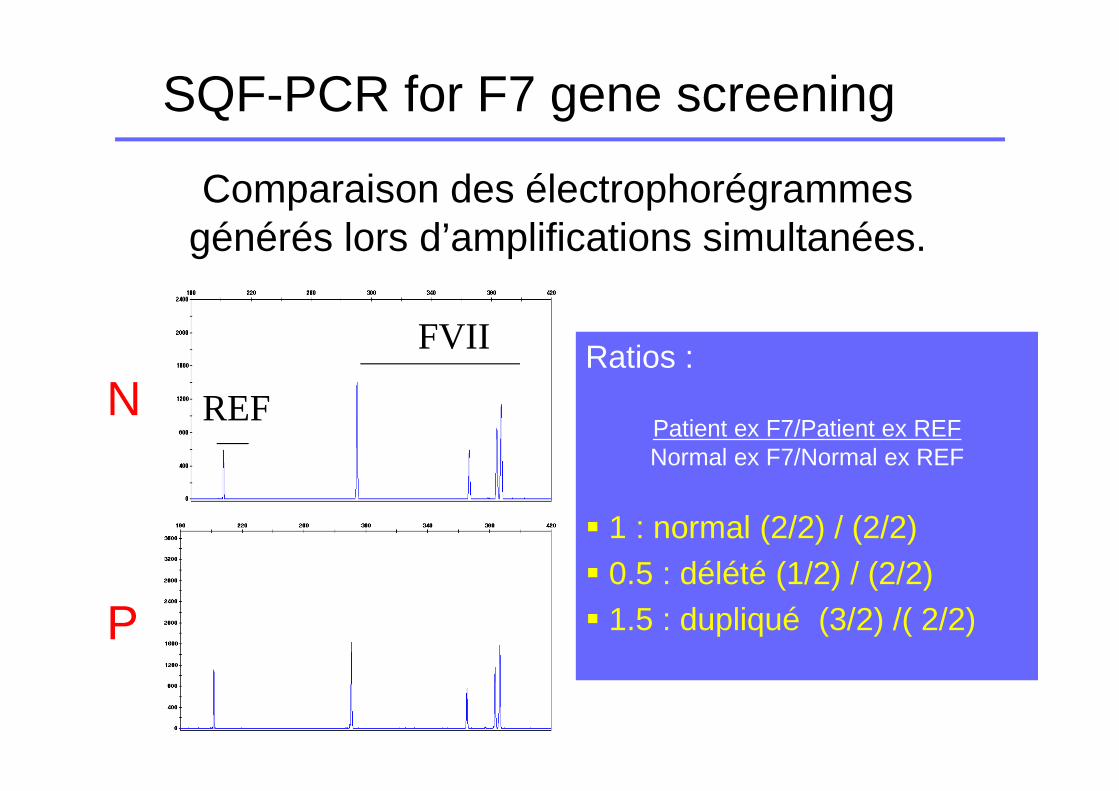
SQF-PCR for F7 gene screening
Comparaison des électrophorégrammes générés lors d’amplifications simultanées.
Ratios :
Patient ex F7/Patient ex REFNormal ex F7/Normal ex REF
� 1 : normal (2/2) / (2/2)� 0.5 : délété (1/2) / (2/2)� 1.5 : dupliqué (3/2) /( 2/2)P
NFVII
REF
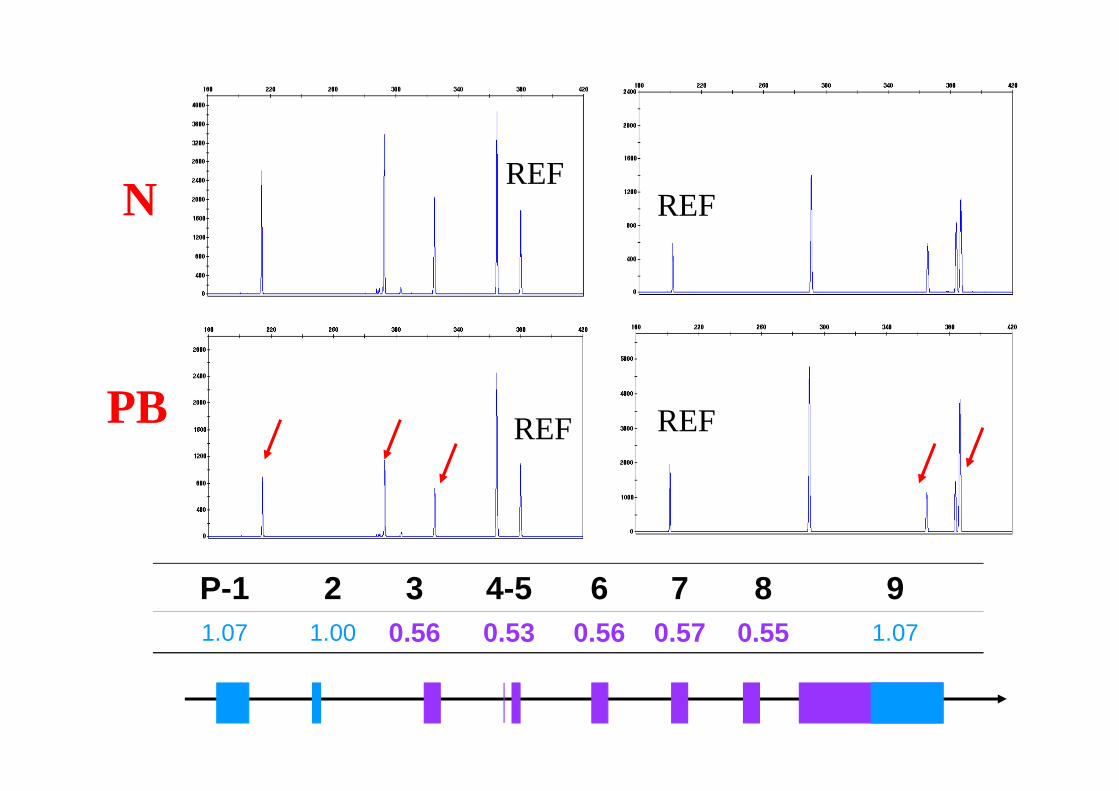
PB
N
P-1 2 3 4-5 6 7 8 91.07 1.00 0.56 0.53 0.56 0.57 0.55 1.07
REF
REF REF
REF

Les déficits constitutionnels en FVII
Correlations génotype -phénotype

Mécanismes mutationnels
� Conséquences phénotypiques des mutations du gène F7 très variables selon le type de mutation
� Mutations ponctuelles�exoniques F7� introniques F7�des régions régulatrices 5’
� Mutations par grands réarrangements du gène

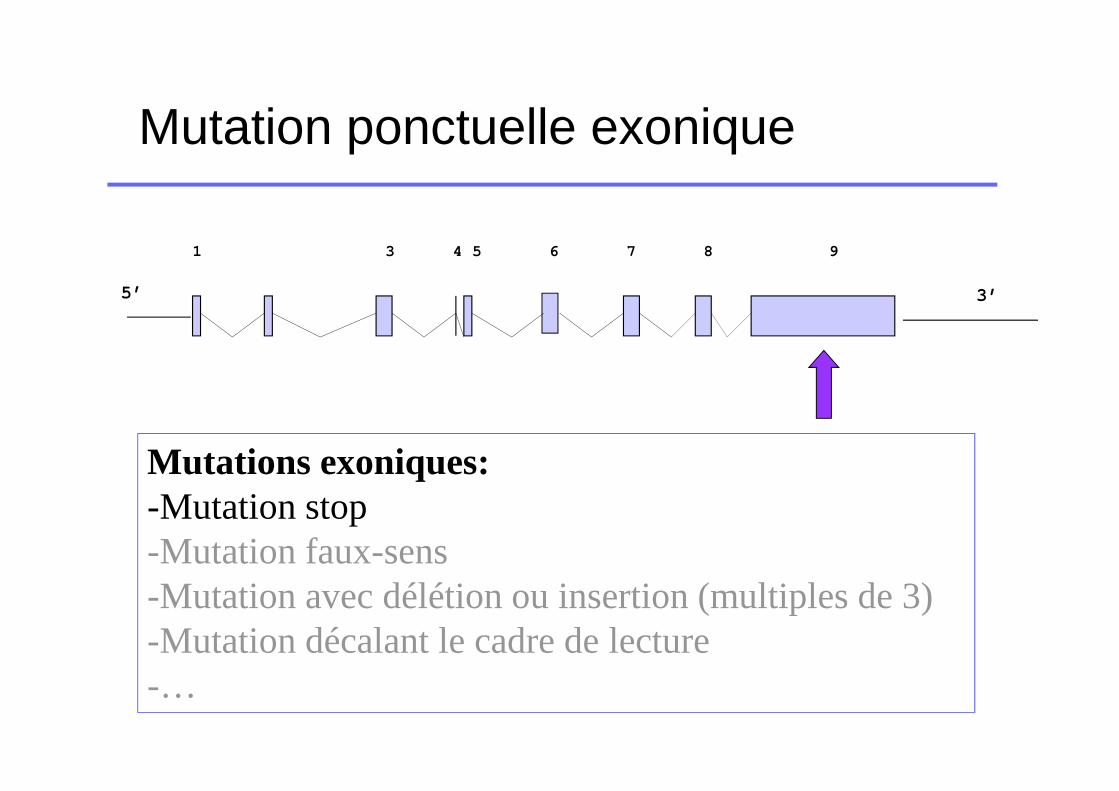
Mutation ponctuelle exonique
5’
1 3 4 5 6 7 8 9
3’
Mutations exoniques:-Mutation stop-Mutation faux-sens-Mutation avec délétion ou insertion (multiples de 3)-Mutation décalant le cadre de lecture-…
Mutation ponctuelle exonique
5’
1 3 4 5 6 7 8 9
3’
Mutations exoniques:-Mutation stop-Mutation faux-sens-Mutation avec délétion ou insertion (multiples de 3)-Mutation décalant le cadre de lecture-…
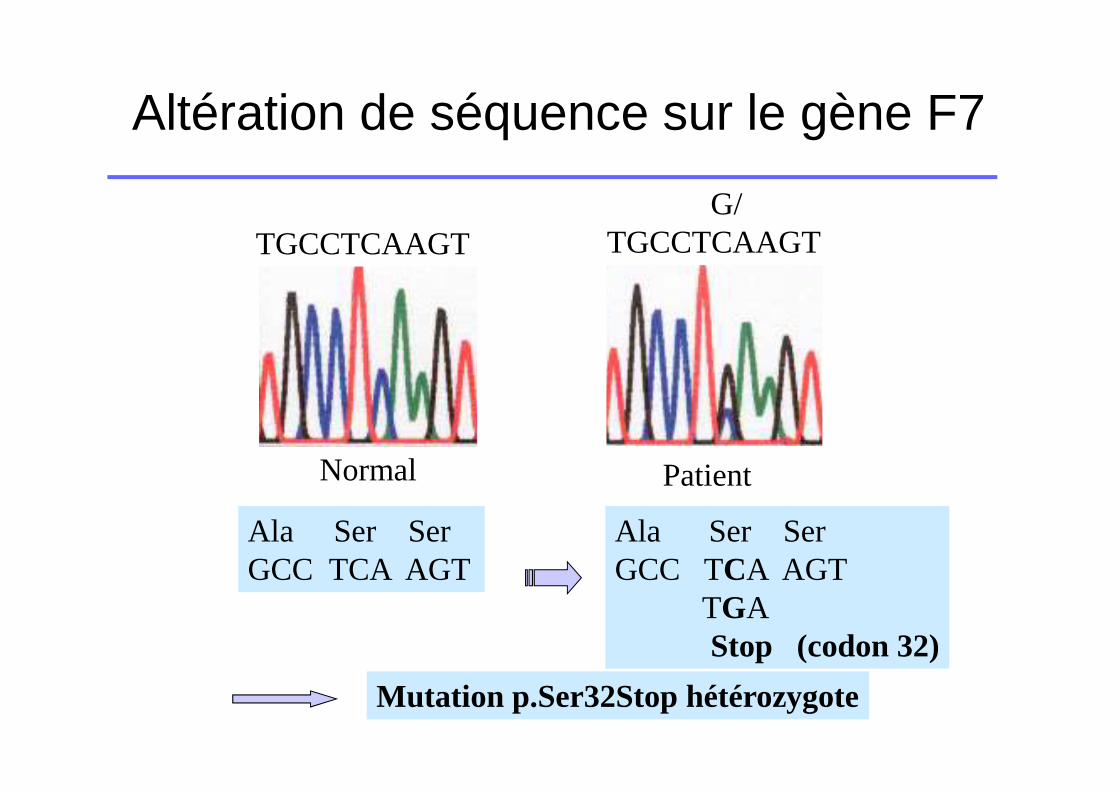
Altération de séquence sur le gène F7
TGCCTCAAGTG/
TGCCTCAAGT
Normal Patient
Ala Ser SerGCC TCA AGT
Ala Ser SerGCC TCA AGT
TGAStop (codon 32)
Mutation p.Ser32Stop hétérozygote
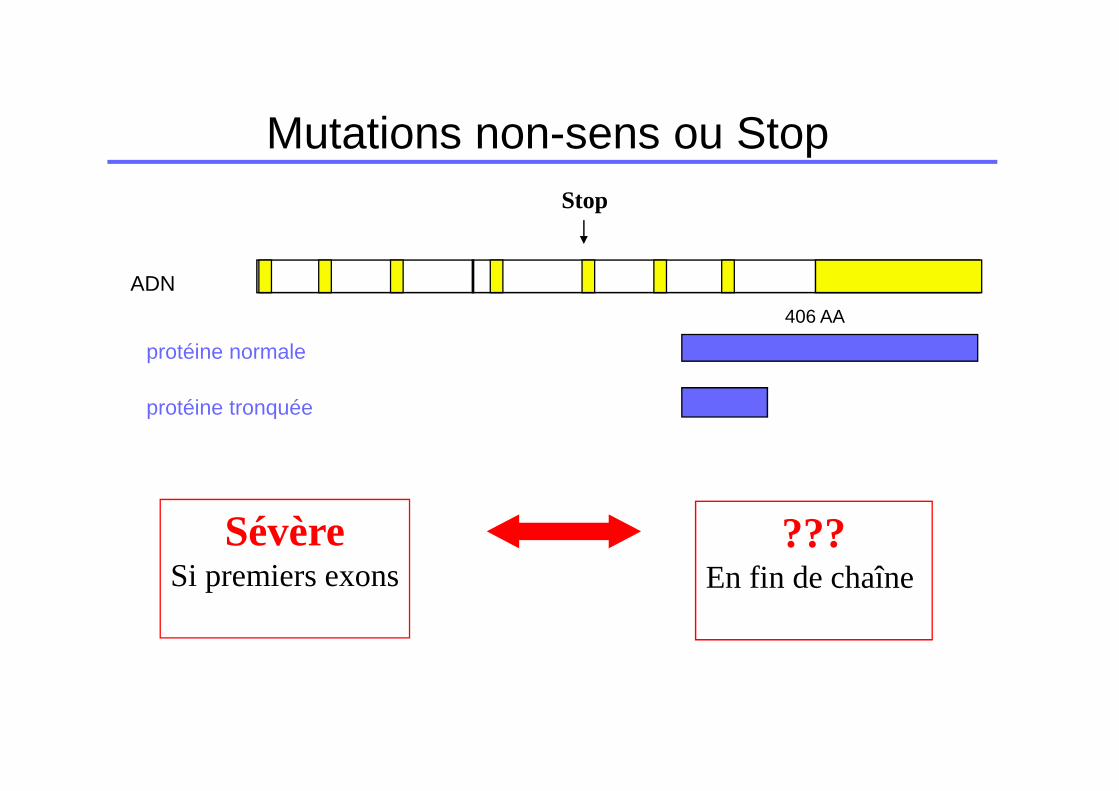
Mutations non-sens ou Stop
ADN
Stop
protéine tronquée
protéine normale
406 AA
SévèreSi premiers exons
???En fin de chaîne
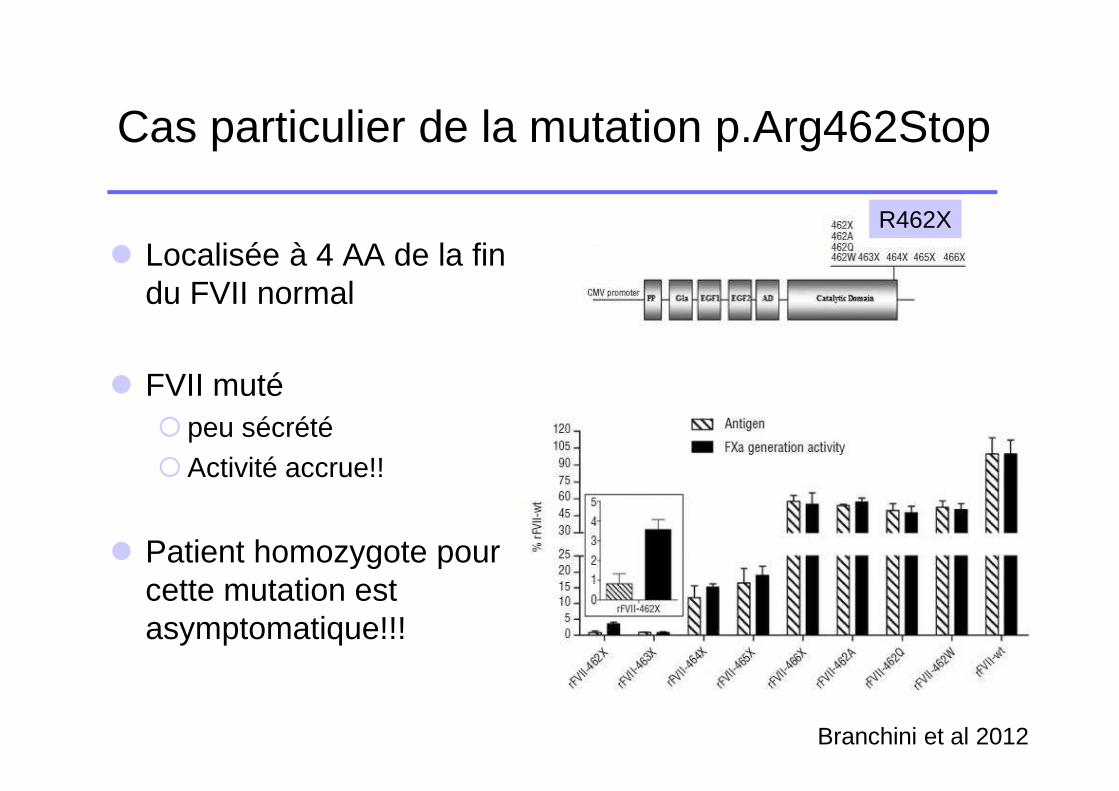
Cas particulier de la mutation p.Arg462Stop
� Localisée à 4 AA de la fin du FVII normal
� FVII muté � peu sécrété�Activité accrue!!
� Patient homozygote pour cette mutation est asymptomatique!!!
R462X
Branchini et al 2012
Mutation ponctuelle exonique
5’
1 3 4 5 6 7 8 9
3’
Mutations exoniques:-Mutation stop-Mutation faux-sens-Mutation avec délétion ou insertion (multiples de 3)-Mutation décalant le cadre de lecture-…
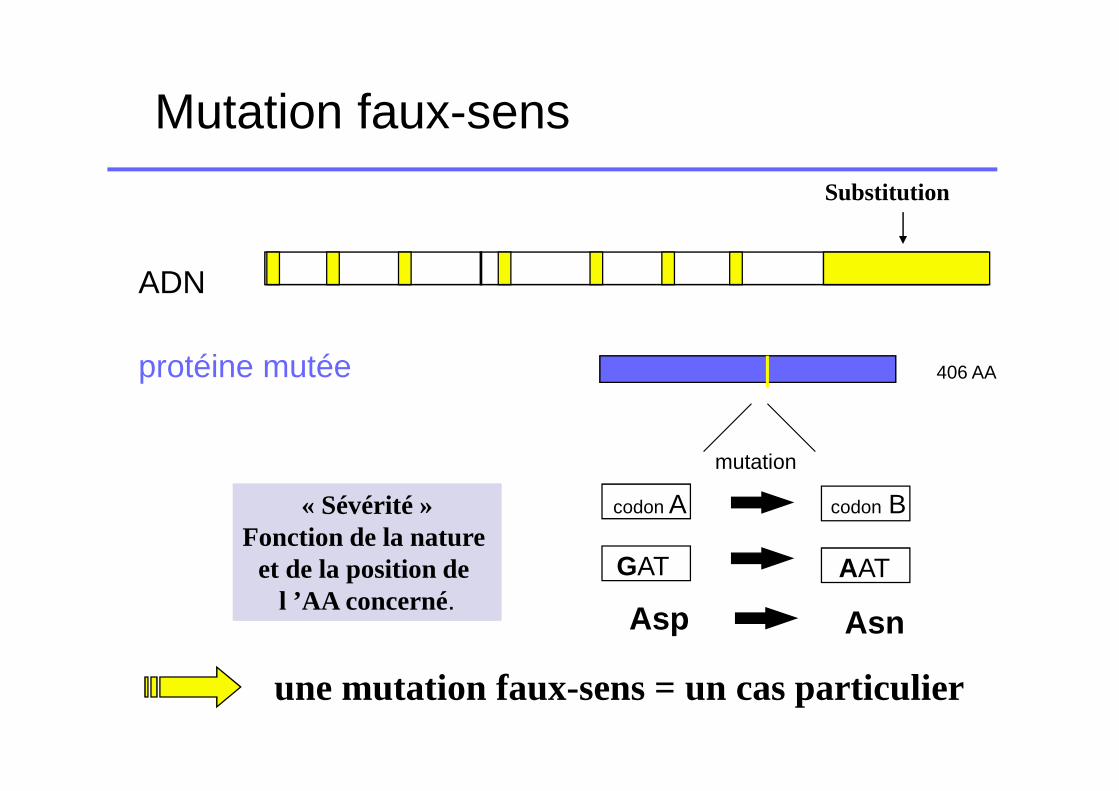
Mutation faux-sens
ADN
Substitution
protéine mutée 406 AA
codon A codon B
GAT AAT
Asp Asn
mutation
« Sévérité »Fonction de la nature
et de la position de l ’AA concerné.
une mutation faux-sens = un cas particulier

Mutation faux-sens
� « Destructuration » de la molécule FVII:déficit quantitatif
� Altération d’une fonction FVII �Liaison TF�Liaison substrat (FIX, FX)�Liaison phospholipides membranaires� fonction catalytique (site, activation etc..)
déficit qualitatif
� Combinaison des deuxdéficit à la fois quantitatif et qualitatif

Exemples de « destructuration de la molécule »
� Perte ou création de ponts disulfures� Encombrement stérique � Concerne souvent des AA enfouis
� Difficile à prédire sur la simple séquence en AA� argument bio info: remplacement hétérogène
(résidu hydrophobe enfoui par un résidu chargé et vice-versa), calcul de stabilisation/déstabilisation de mutations
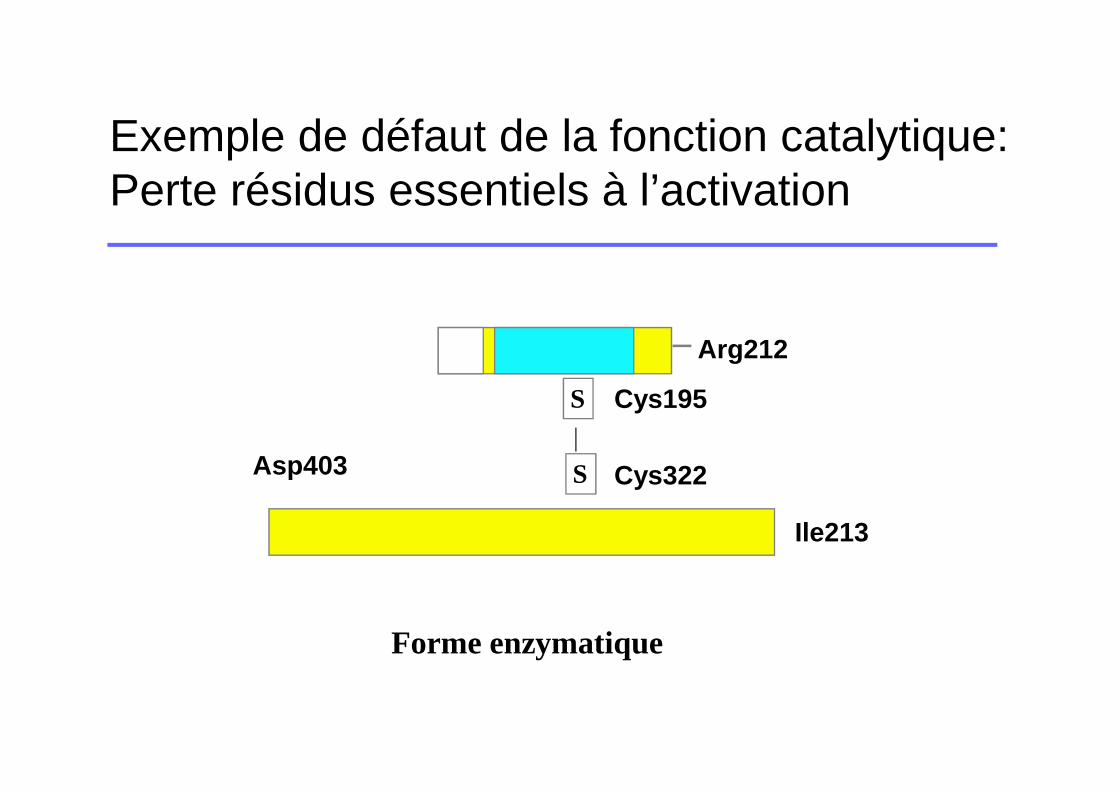
Exemple de défaut de la fonction catalytique: Perte résidus essentiels à l’activation
Forme enzymatique
S
S
Arg212
Asp403
Ile213
Cys195
Cys322
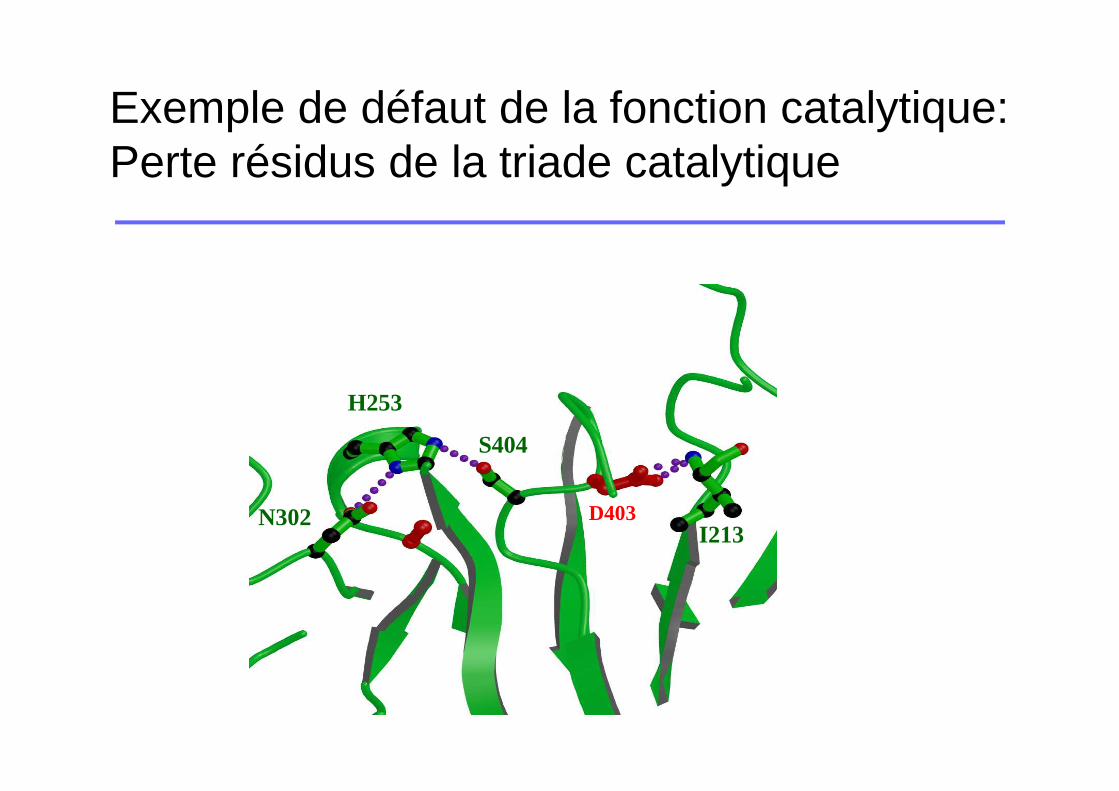
Exemple de défaut de la fonction catalytique: Perte résidus de la triade catalytique
H253
S404
I213D403N302

Mutations altérant la fonction d’activation ou la fonction catalytique
�Pas de FVII activé fonctionnel
�Déficits quantitatif ou qualitatif selon les mutations
�Tableaux cliniques catastrophiques chez les homozygotes
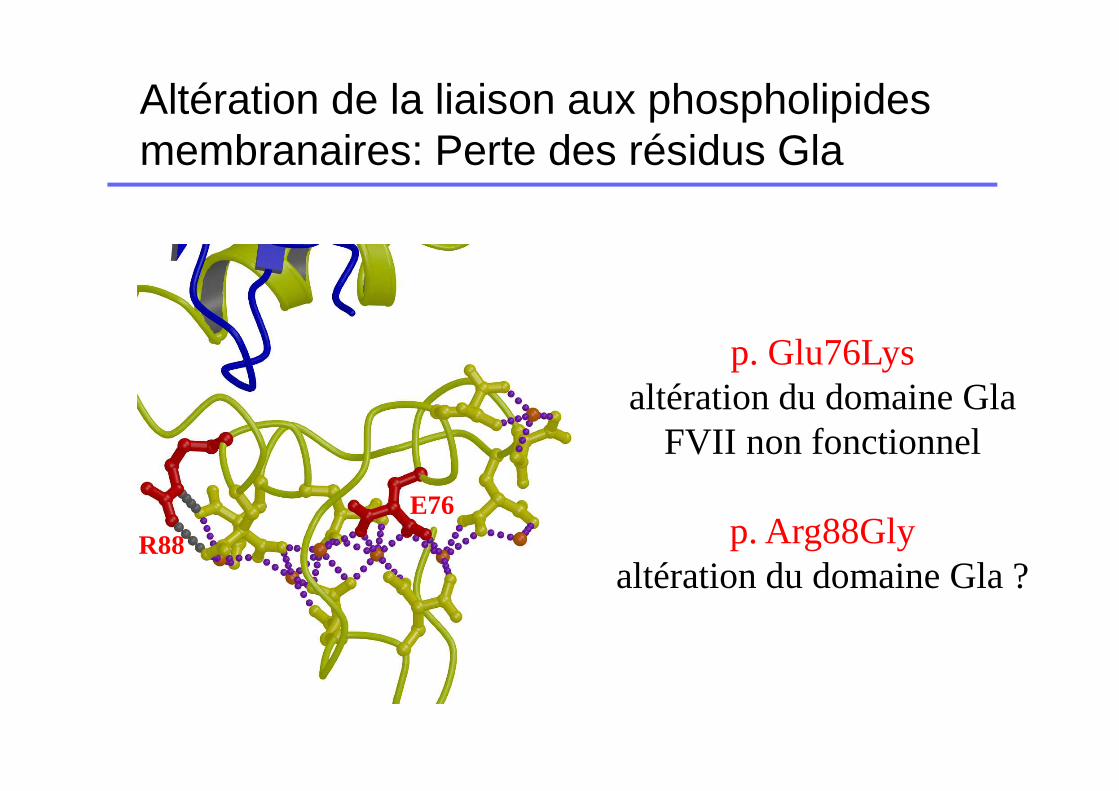
Altération de la liaison aux phospholipides membranaires: Perte des résidus Gla
p. Glu76Lysaltération du domaine Gla
FVII non fonctionnel
p. Arg88Glyaltération du domaine Gla ?
E76
R88
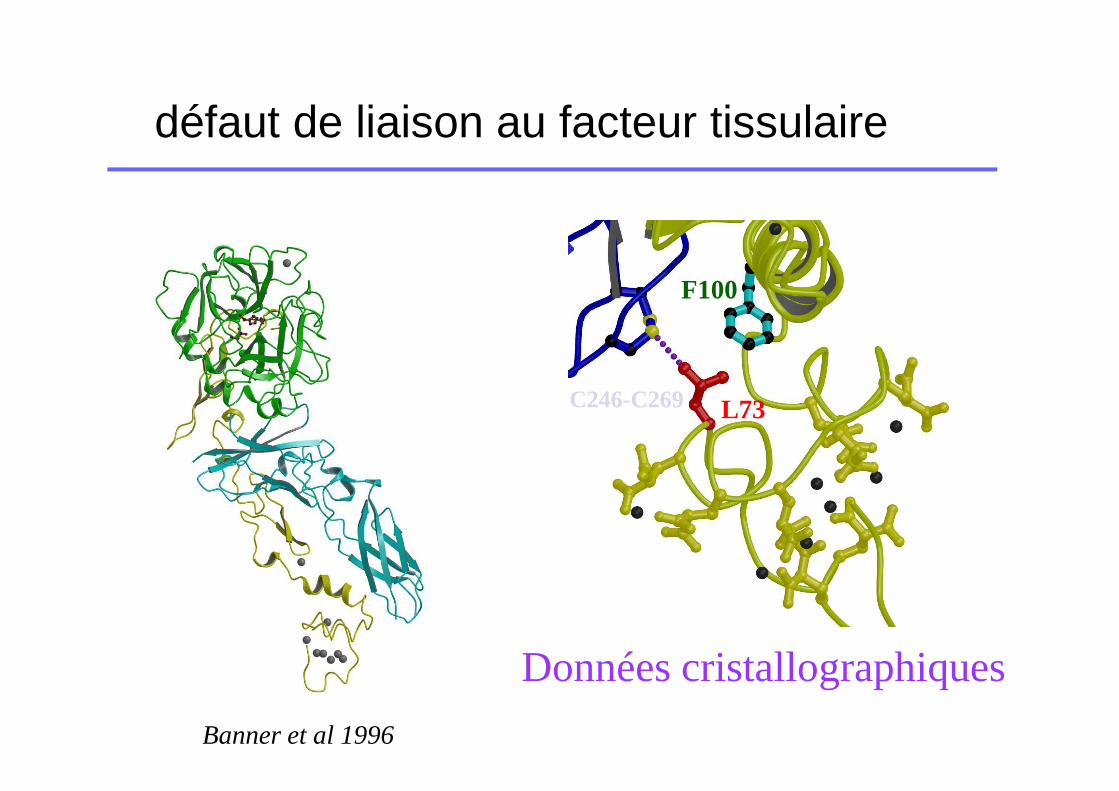
défaut de liaison au facteur tissulaire
L73C246-C269
F100
Données cristallographiques
Banner et al 1996

Mutations altérant la liaison FT
� Souvent déficits de type qualitatifs
� Moins sévères que les précédentes (nombreux point d’ancrage sur le FT?? Suppléance mutuelle??)
� Cas particulier des variants FVII dont le dosage varie en fonction de la thromboplastine (séquences TF différentes entre l’homosapiens et le lapin).
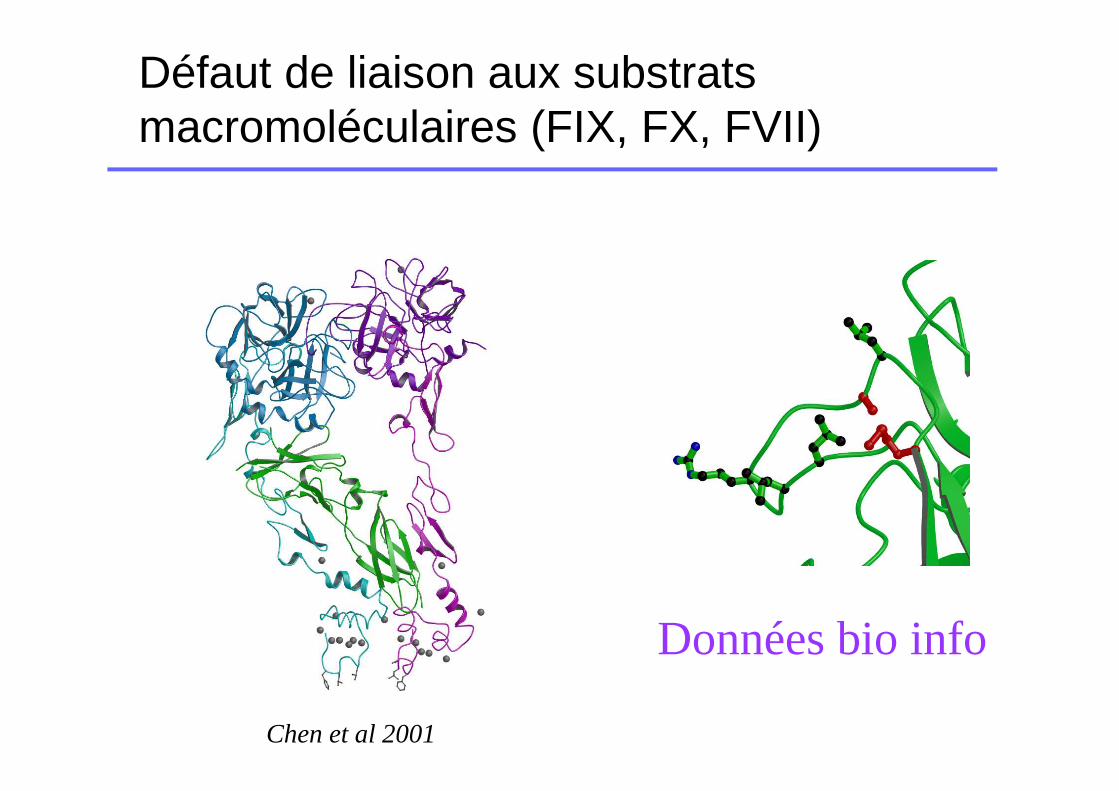
Défaut de liaison aux substrats macromoléculaires (FIX, FX, FVII)
Données bio info
Chen et al 2001

Exemple p.Met358Ile
� Mutation sur une boucle exposée au solvent en lien avec le FIX
� Le FVII est sécrété en quantité normale (FVII:Ag normal)
� Le FVII muté reconnait mal le substrat (FVII:C diminué)
� Déficit qualitatif pur si homozygotie
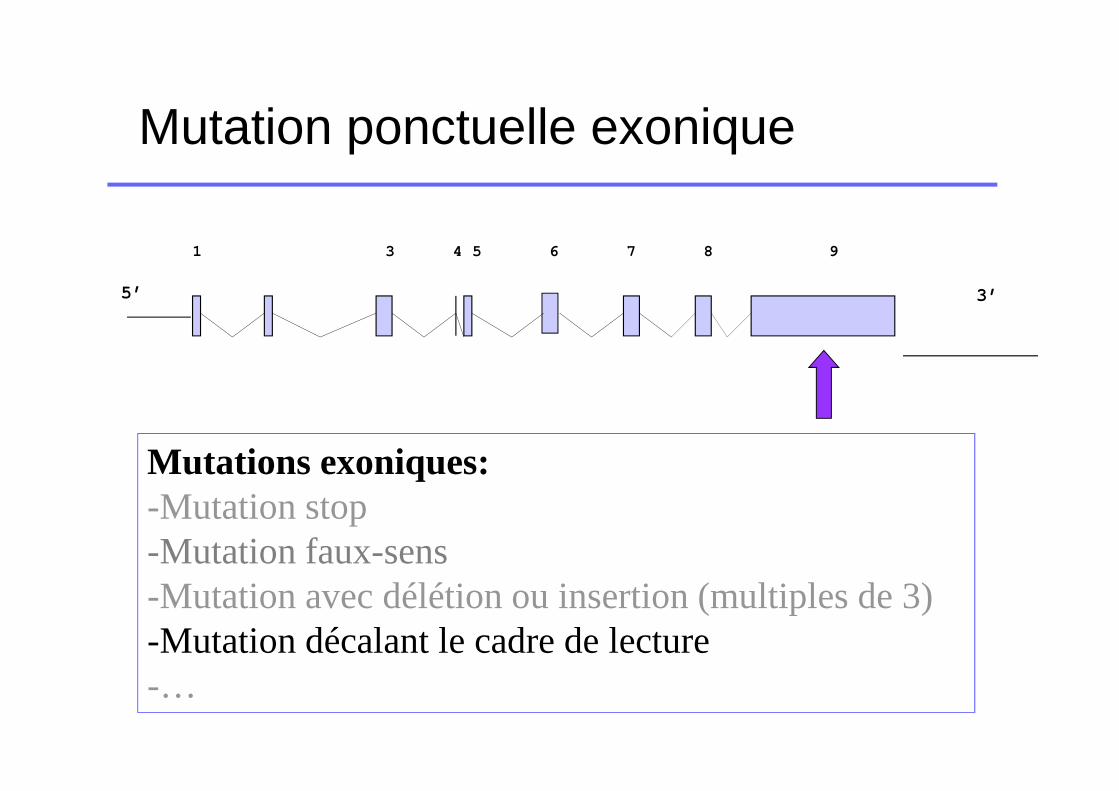
Mutation ponctuelle exonique
5’
1 3 4 5 6 7 8 9
3’
Mutations exoniques:-Mutation stop-Mutation faux-sens-Mutation avec délétion ou insertion (multiples de 3)-Mutation décalant le cadre de lecture-…
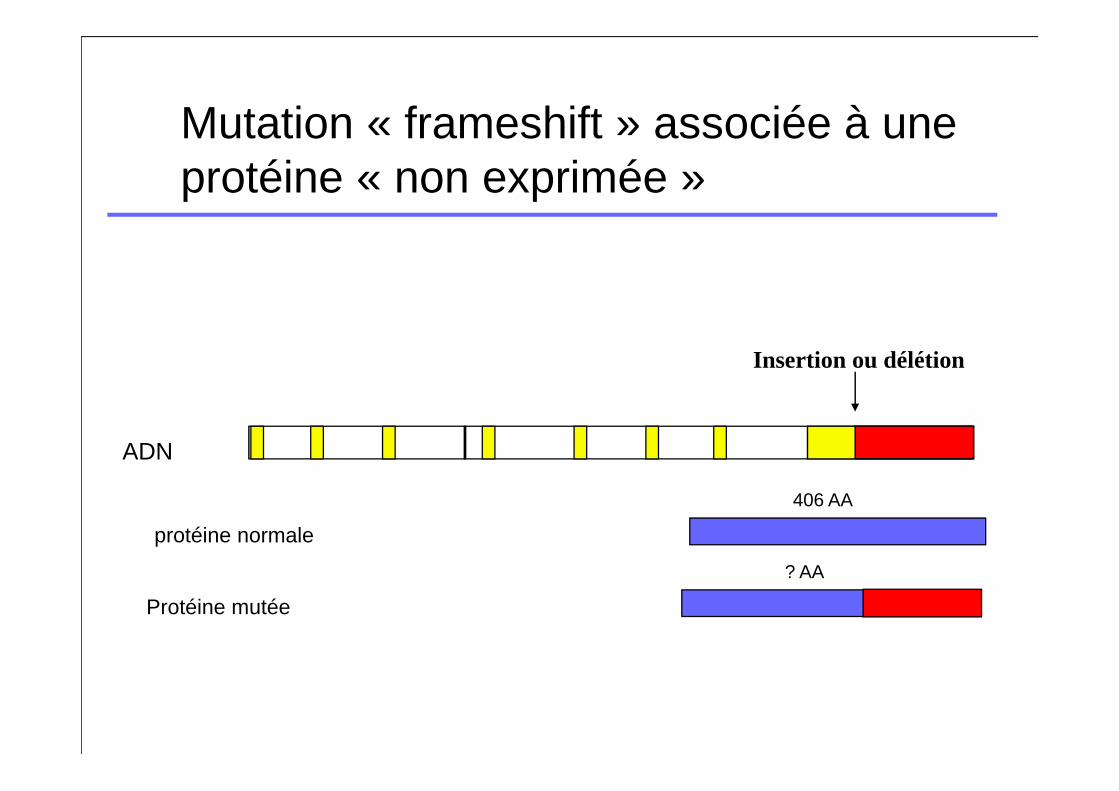
Mutation « frameshift » associée à une protéine « non exprimée »
Insertion ou délétion
ADN
protéine normale
406 AA
Protéine mutée
? AA
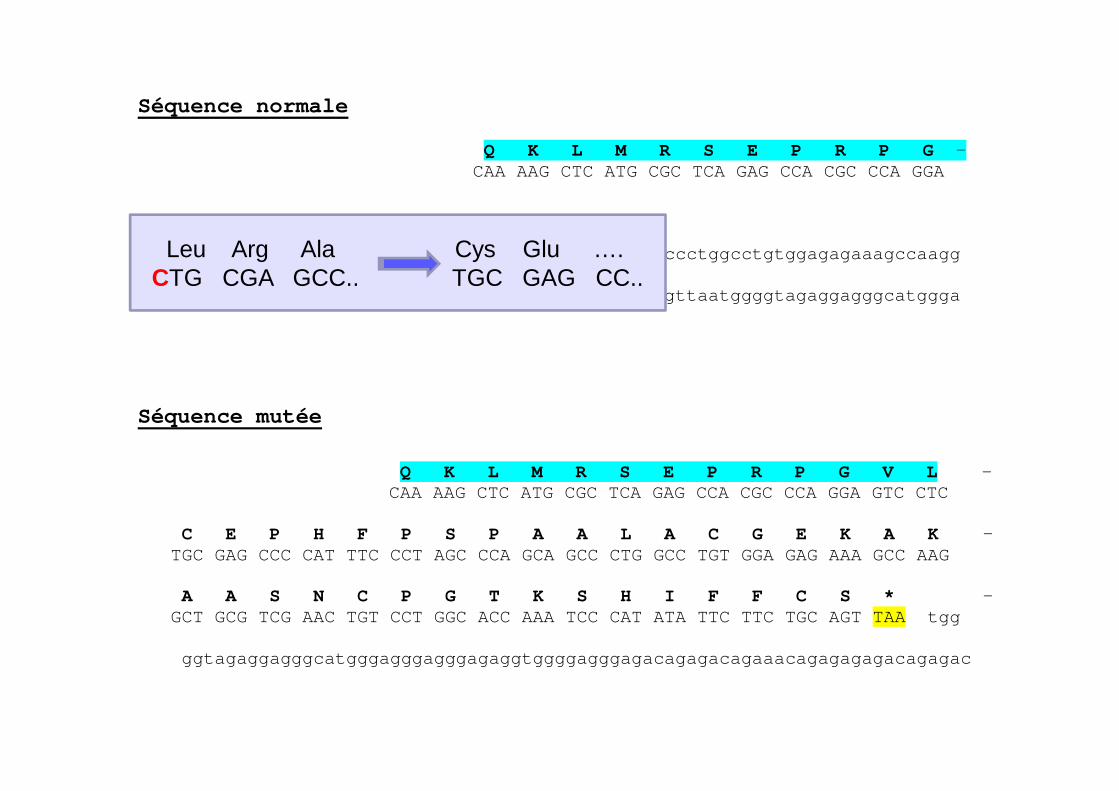
Séquence normale Q K L M R S E P R P G - CAA AAG CTC ATG CGC TCA GAG CCA CGC CCA GGA V L L R A P F P * GTC CTC CTG CGA GCC CCA TTT CCC TAG cccagcagccc tggcctgtggagagaaagccaagg ctgcgtcgaactgtcctggcaccaaatcccatatattcttctgcagtt aatggggtagaggagggcatggga Séquence mutée
Q K L M R S E P R P G V L - CAA AAG CTC ATG CGC TCA GAG CCA CGC CCA GGA G TC CTC C E P H F P S P A A L A C G E K A K - TGC GAG CCC CAT TTC CCT AGC CCA GCA GCC CTG GCC TGT GGA GAG AAA GCC AAG A A S N C P G T K S H I F F C S * - GCT GCG TCG AAC TGT CCT GGC ACC AAA TCC CAT ATA TTC TTC TGC AGT TAA tgg ggtagaggagggcatgggagggagggagaggtggggagggagacaga gacagaaacagagagagacagagac
Leu Arg Ala Cys Glu …. CTG CGA GCC.. TGC GAG CC..

Mutation décalant les cadres de lecture
� Mutation survenant dans les premiers exons� Classiquement pas de protéine fonctionnelle produite�FVII:C et FVII:Ag effondrés chez les homozygotes�Tableaux cliniques catastrophiques chez les
homozygotes
� Mutation survenant en fin de chaîne�Plus variable mais reste globalement très sévère�Déficit quantitatif chez les homozygotes
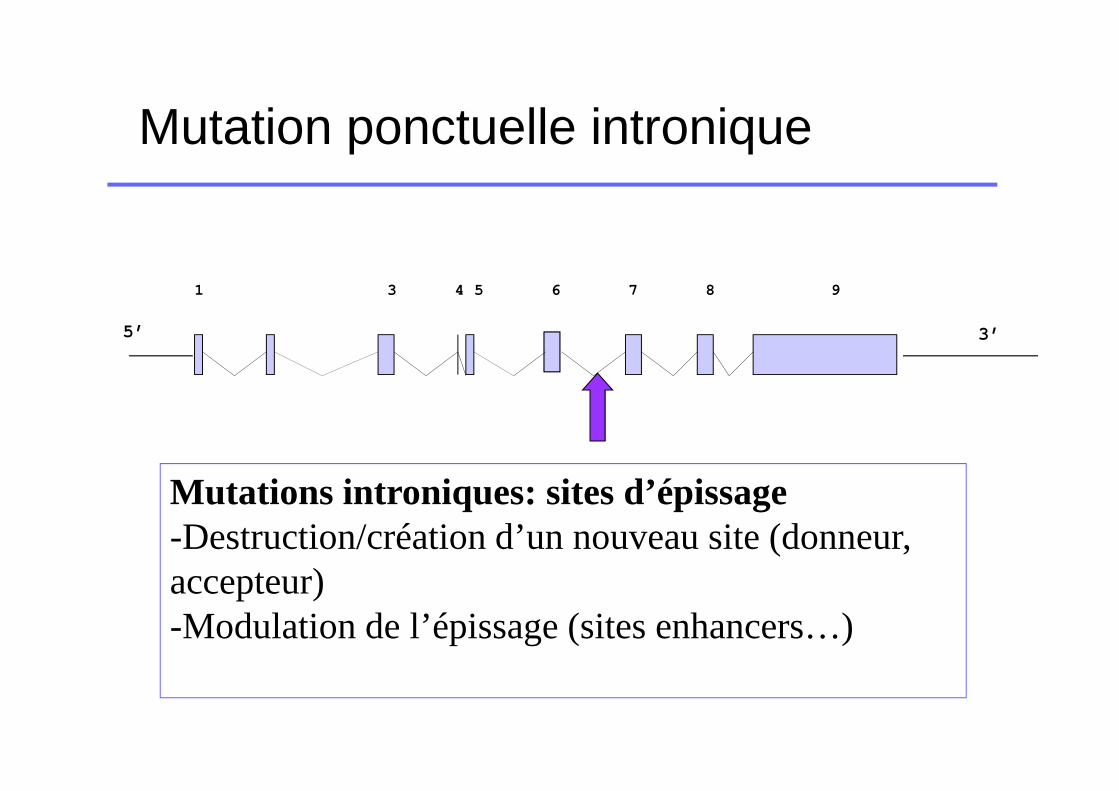
Mutation ponctuelle intronique
5’
1 3 4 5 6 7 8 9
3’
Mutations introniques: sites d’épissage-Destruction/création d’un nouveau site (donneur, accepteur)-Modulation de l’épissage (sites enhancers…)
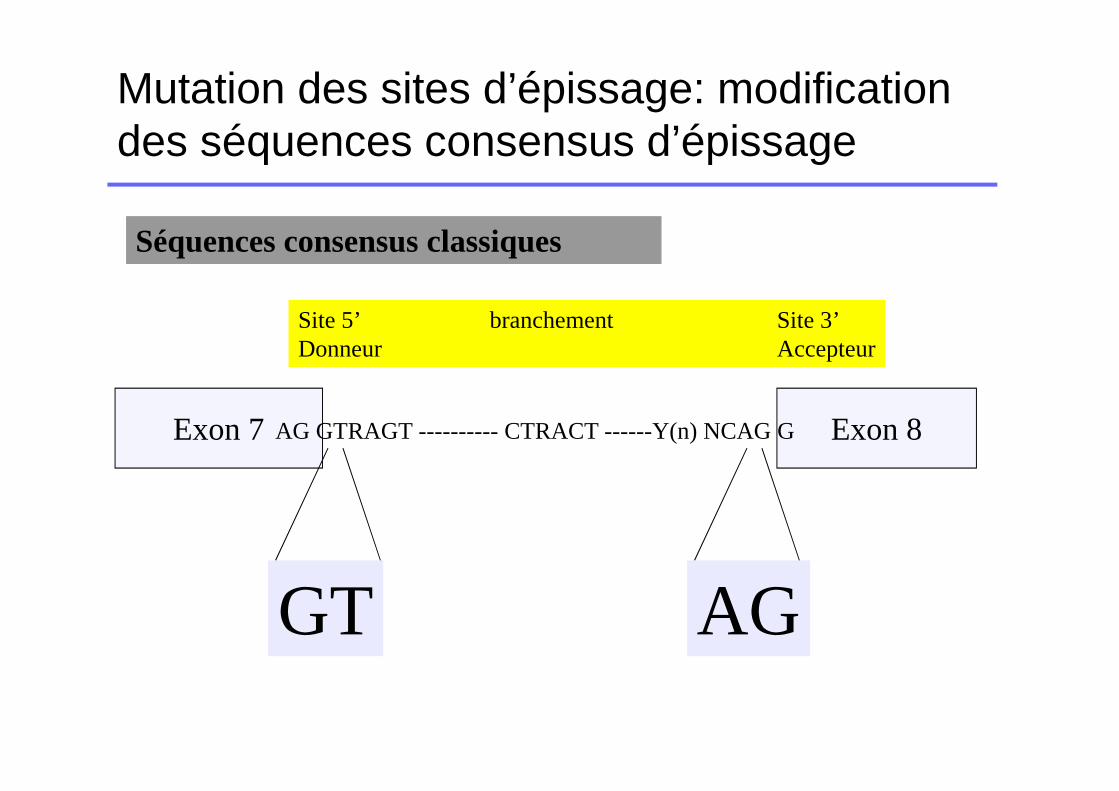
Mutation des sites d’épissage: modification des séquences consensus d’épissage
Exon 7 Exon 8AG GTRAGT ---------- CTRACT ------Y(n) NCAG G
Site 5’ branchement Site 3’Donneur Accepteur
Séquences consensus classiques
GT AG

ttcttt agC ACT AAG TCA GGA GAT TGG AAA AGC AAA TGC TTT 84 9 Y T T D T E C D L T D E I V K D V K 68 TAC ACA ACA GAC ACA GAG TGT GAC CTC ACC GAC GAG ATT GTG AAG GAT GTG AAG 903 Q T Y L A R V F S Y P A G N V E S T 86 CAG ACG TAC TTG GCA CGG GTC TTC TCC TAC CCG GCA GGG AAT GTG GAG AGC ACC 957 G S A G E P L Y E N S P E F T P Y L 104 GGT TCT GCT GGG GAG CCT CTG TAT GAG AAC TCC CCA GAG TTC ACA CCT TAC CTG 1011 E 105 GAG A gt aagtggcttgggctgtaataccgttcattcttgttagaaacgtctgaacat tctcgtgatcttg 1081 tgcctttaggggctacaaaattaaaaatatttattctttttttctcag aaactggtatgtaaggccgccacg 1153 cctgcttctttcagggagctggttctatgcacatgttttatatgagag ataattaagttgtcaattgtgata 1225 T N 107 acaaaacaggatttgactttgtacagaattctttggttccaaccaagc tcatttcctttgtttc agCA AAC 1296 L G Q P T I Q S F E Q V G T K V N V 125 CTC GGA CAG CCA ACA ATT CAG AGT TTT GAA CAG GTG GGA ACA AAA GTG AAT GTG 1350 T V E D E R T L V R R N N T F L S L 143 ACC GTA GAA GAT GAA CGG ACT TTA GTC AGA AGG AAC AAC ACT TTC CTA AGC CTC 1404 R D V F G K D L I Y T L Y Y W K S S 161 CGG GAT GTT TTT GGC AAG GAC TTA ATT TAT ACA CTT TAT TAT TGG AAA TCT TCA 1458 S S G K 165 AGT TCA GGA AAG gt gagcattttttaatttgtttttatgacctgttttaaattgtgaatacttgg gt
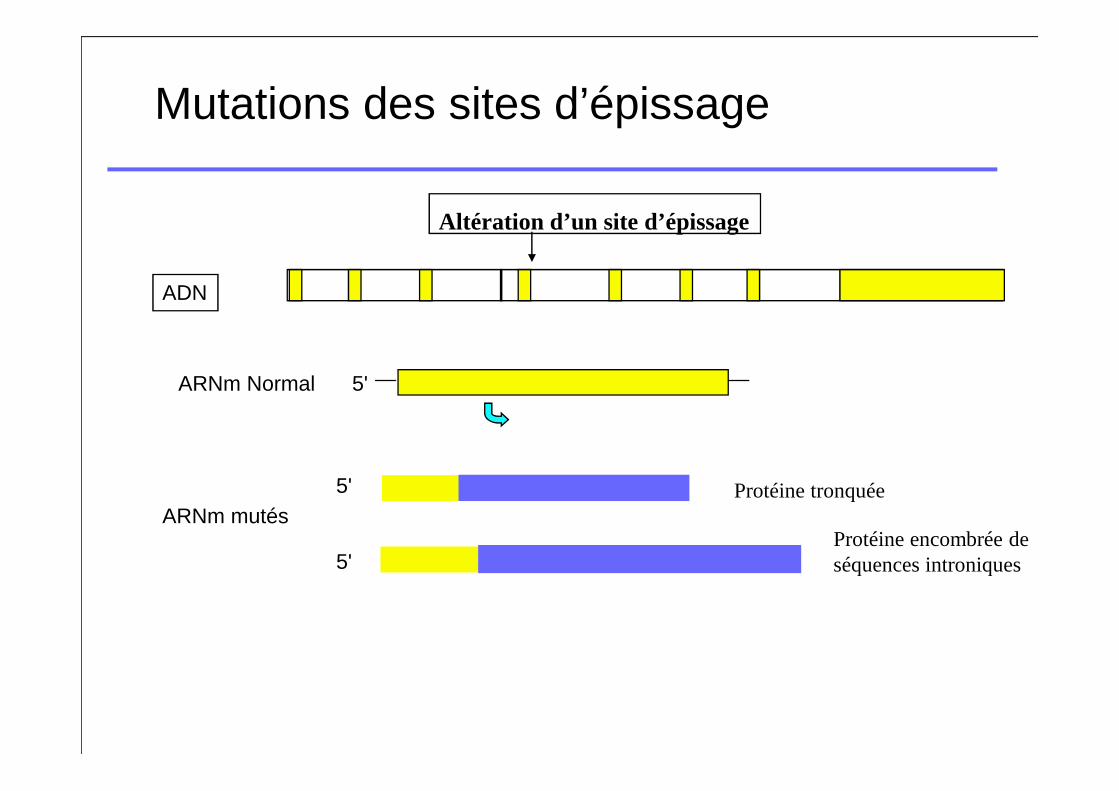
Mutations des sites d’épissage
ADN
Altération d’un site d’épissage
ARNm Normal 5'
ARNm mutés5'
5'
Protéine tronquée
Protéine encombrée de séquences introniques

Mutation des sites d’épissage
� Mutation des sites consensus « AG » ou « GT »� Classiquement pas de protéine produite�FVII:C et FVII:Ag effondrés chez les homozygotes�Tableaux cliniques catastrophiques chez les
homozygotes
� Autre type�Très variables car on a une compétition entre le site
sauvage et le muté: on a souvent un peu de FVII normal résiduel (tableaux cliniques moins graves)
Mutation ponctuelle des zones régulatrices
5’
1 3 4 5 6 7 8 9
3’
-Mutation des sites de transcription-…
-Conséquences phénotypiques très variables
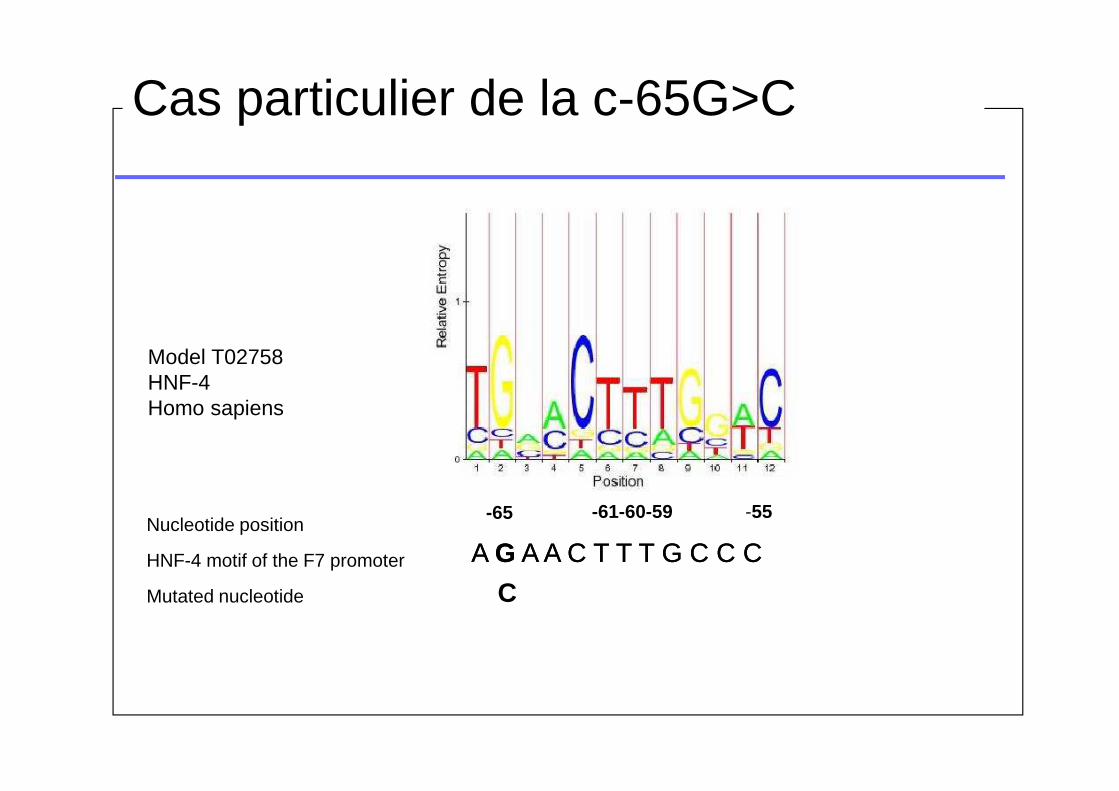
Model T02758HNF-4Homo sapiens
Nucleotide position
HNF-4 motif of the F7 promoter
Mutated nucleotide C
A G A A C T T T G C C CA G A A C T T T G C C C
-65 -61-60-59 -55
Cas particulier de la c-65G>C
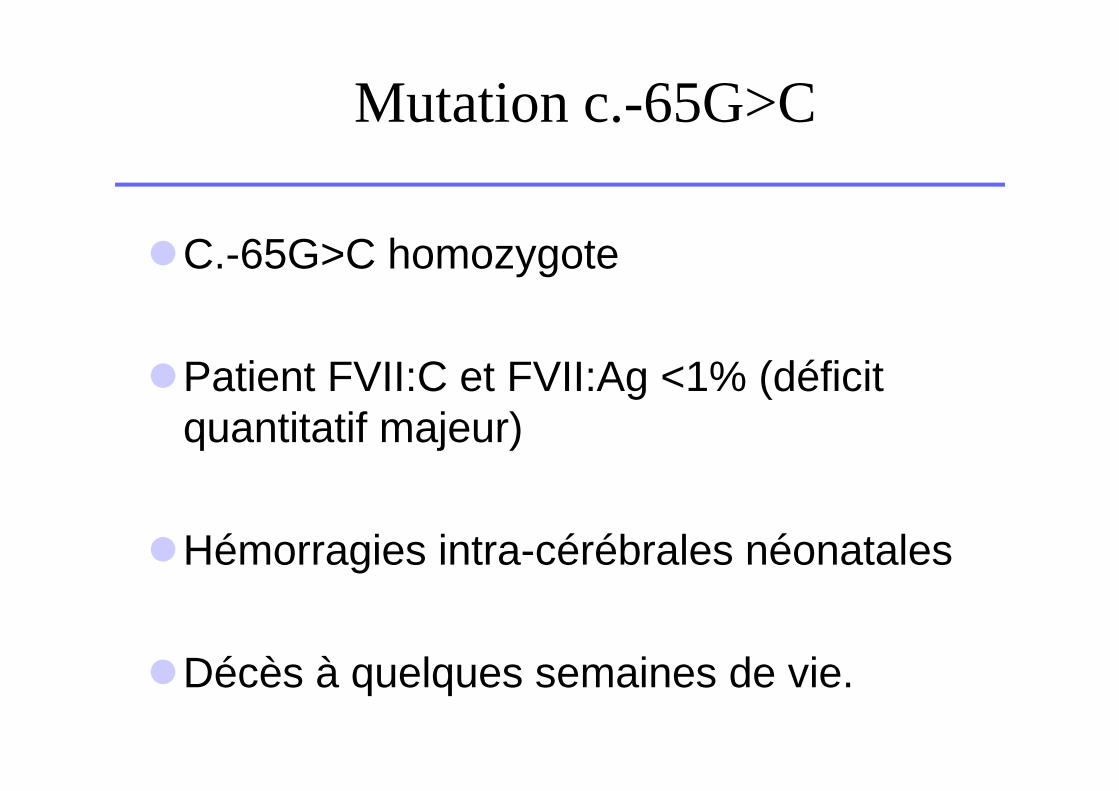
Mutation c.-65G>C
�C.-65G>C homozygote
�Patient FVII:C et FVII:Ag <1% (déficit quantitatif majeur)
�Hémorragies intra-cérébrales néonatales
�Décès à quelques semaines de vie.
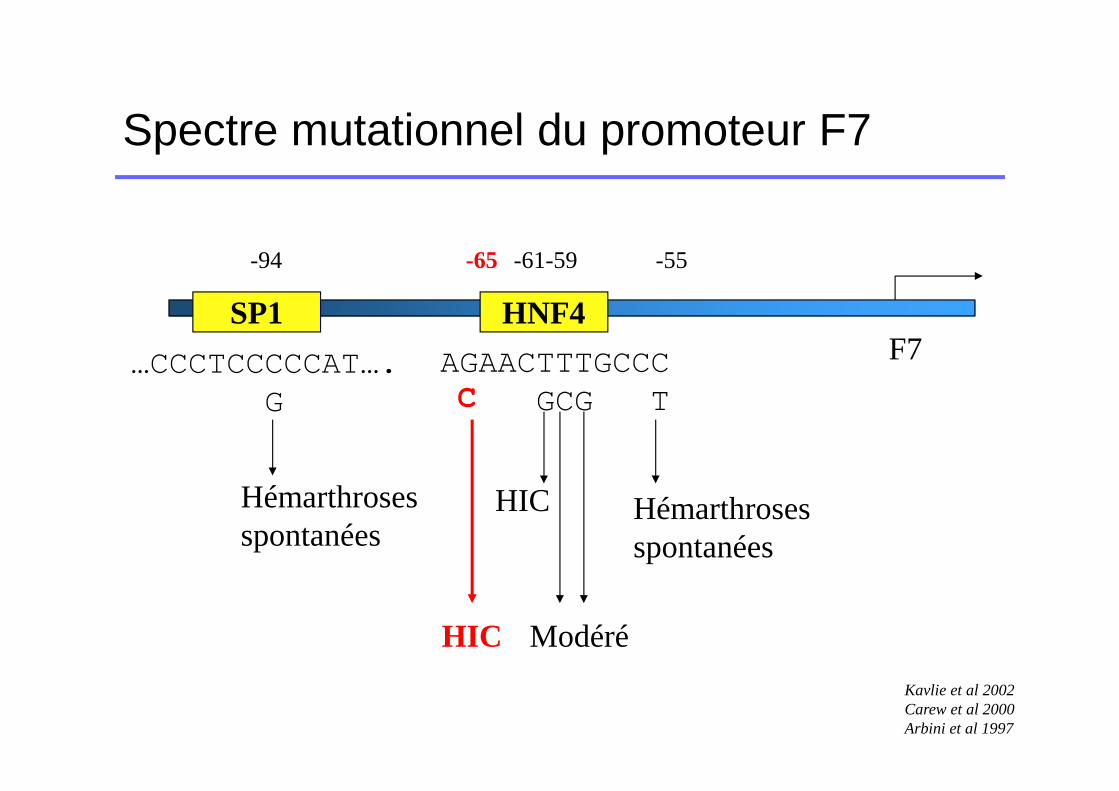
Spectre mutationnel du promoteur F7
HNF4SP1F7AGAACTTTGCCC
GCG T
HIC Hémarthrosesspontanées
Modéré
…CCCTCCCCCAT….G
Hémarthrosesspontanées
-94 -61-59 -55
Kavlie et al 2002Carew et al 2000Arbini et al 1997
-65
HIC
C

�Classe Sévère �pas de protéine produite
�Mutations stop, frameshift etc…�Grandes délétions
�protéine avec une fonction abolie�Perte des résidus essentiels (triade catalytique par exemple)
�Classe Modérée à Neutre� protéine anormale avec une fonction résiduelle
�La plupart des mutations faux-sens (altération liaison FT par exemple)
� protéine résiduelle normale.Collaboration, JL Pellequer et W Chen,
CEA site de Marcoules
Classification moléculaire

SévèresModéréespauci-symptomatiques
�Mise en jeu du pronostic fonctionnel voire vital
�FVII:C<5%�Mutations du gène F7
spécifiques (les deux mutations: classe sévère)
�15 à 20 cas en France
�Syndrome hémorragique moins marqué voire absent
�FVII:C 1-30%�Grand spectre mutationnel
(au moins une mutation classe modérée à neutre)
�300 cas en France (sous-estimé)
Lien entre les Classifications??

Hétérogénéité moléculaire
�Ce n’est déjà pas simple quand on raisonne avec un seul allèle (cas de l’hémophilie A ou B) ou avec des patients homozygotes
�Cela se complique énormément si:�on a deux mutations différentes�Il existe des polymorphismes qui peuvent être
associé avec l’une ou l’autre des mutations….

Hétérogénéité moléculaire
�Plus de 200 mutations ponctuelles sur le gène F7.
�Aucune mutation prépondérante
� Majorité de mutations faux-sens
� Nombreux hétérozygotes composites (multiplication des combinaisons génotypiques)
Hétérozygotes composites
mut A mut B
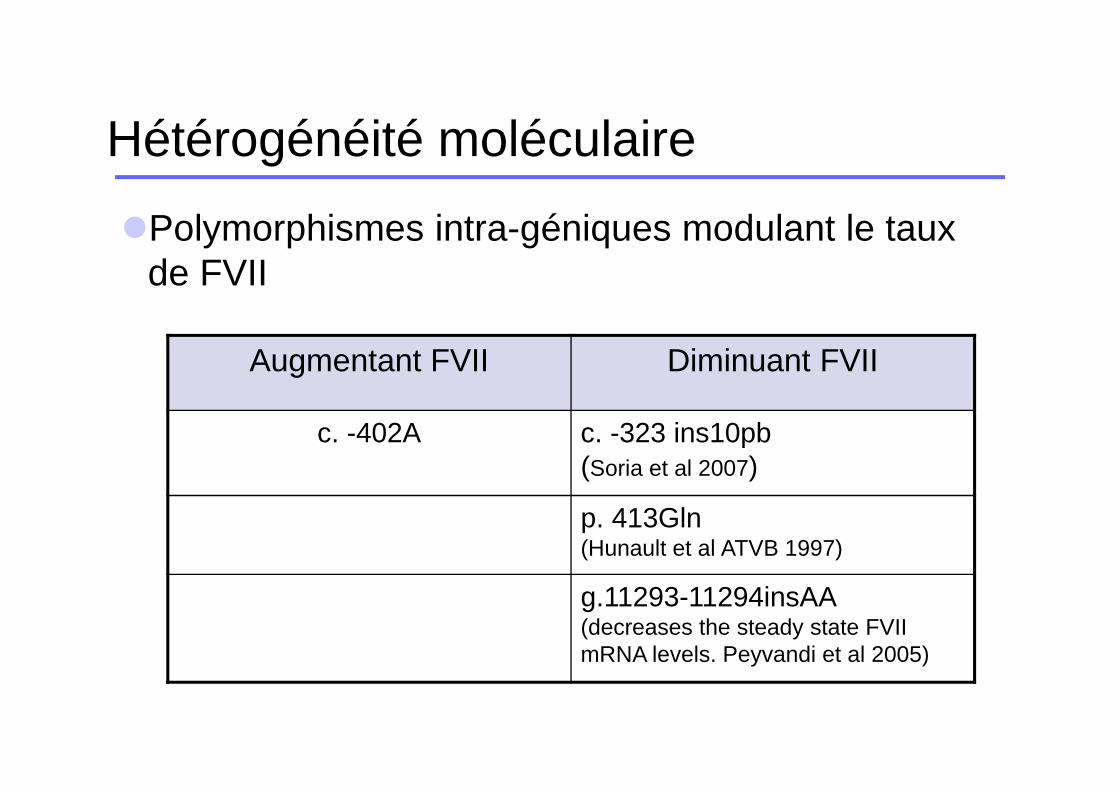
�Polymorphismes intra-géniques modulant le taux de FVII
Augmentant FVII Diminuant FVII
c. -402A c. -323 ins10pb(Soria et al 2007)
p. 413Gln(Hunault et al ATVB 1997)
g.11293-11294insAA(decreases the steady state FVII mRNA levels. Peyvandi et al 2005)
Hétérogénéité moléculaire
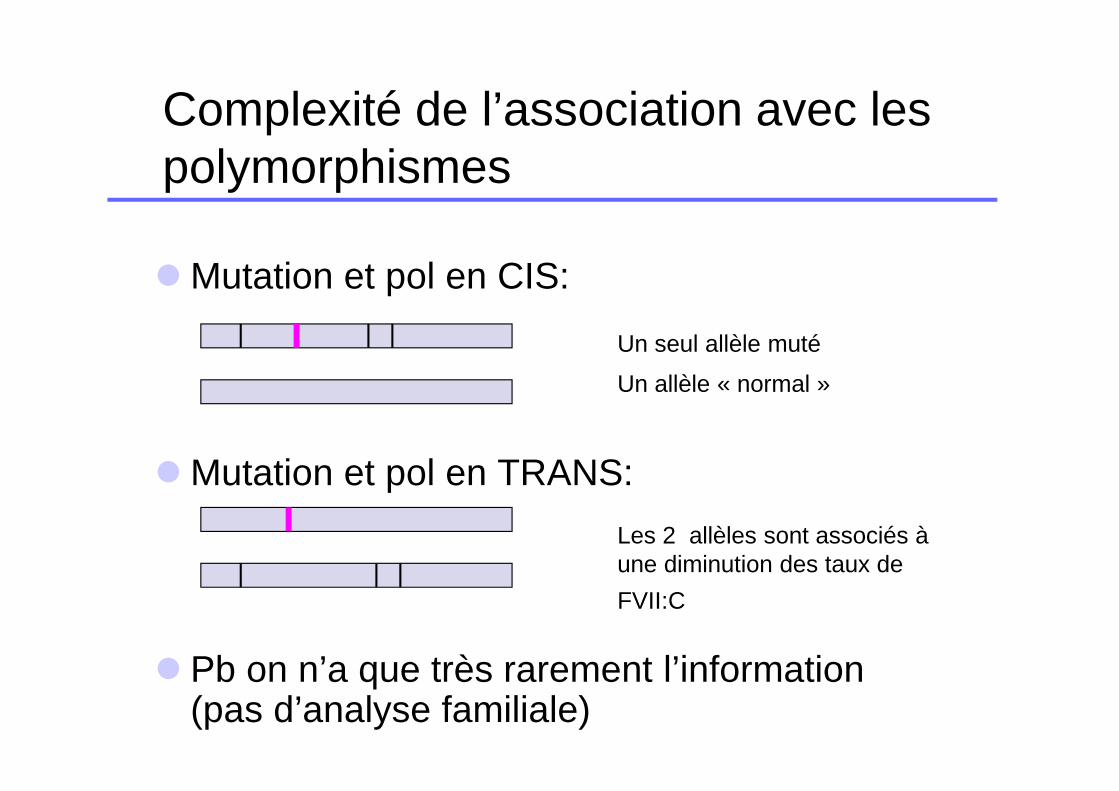
Complexité de l’association avec les polymorphismes
� Mutation et pol en CIS:
� Mutation et pol en TRANS:
� Pb on n’a que très rarement l’information (pas d’analyse familiale)
Un seul allèle muté
Un allèle « normal »
Les 2 allèles sont associés à une diminution des taux de
FVII:C

Exemples d’interprétations
Exemples où le FVII:Ag nous a permis d’éviter des erreurs….

Cas n°1
� Jeune patient de 25 ans �FVII:C= 8% �FVII:Ag= 12%
�Homozygote pour la mutation p.Ala304Val�Cette mutation est connue pour altérer la sécrétion
du FVII
� Validez vous ce bilan?

Cas n°2
� Patiente de 42 ans �FVII:C= 5%�FVII:Ag= 89%
�Homozygote pour la mutation p.Ala354Val�Cette mutation est connue pour altérer la liaison du
FVII au substrat (FIX ou FX) mais ne gène pas la sécrétion du FVII
� Validez vous ce bilan?

Cas n°3
�Patient de 2 ans sous prophylaxie pour des hémorragies intra-cérébrales�FVII:C <1% �FVII:Ag <1%
�Homozygote pour la mutation p.Ser112Stop
�Validez vous ce bilan?

Cas n°4
� Patient de 65 ans �FVII:C= 10%�FVII:Ag= 51%
�Hétérozygote composite� pour la mutation p.Ala304Val (connue pour altérer la
sécrétion du FVII)�Pour la mutation p.Met358Ile (connue pour altérer la
liaison du FVII au substrat mais ne gène pas la sécrétion du FVII)
� Validez vous ce bilan?
p.Ala304Val p.Met358Ile

Cas n°5
� Patiente de 50 ans �FVII:C= 4%�FVII:Ag= 35%
�Homozygote pour la mutation p.Arg88Gly (connue pour “dé-structurer” le domaine GLA c’est à dire donner une protéine instable qui se lie mal à la membrane phospholipidique)
� Validez vous ce bilan?
Mb

Cas n°6
� Patiente de 51 ans �FVII:C <5%�FVII:Ag= 45%
�Homozygote pour la mutation p.Arg364Gln�Cette mutation est connue pour altérer la liaison du
FVII au TF mais ne gène pas du tout la sécrétion du FVII
� Validez vous ce bilan?

Réponse non
� Avec ce génotype, nous aurions dû avoir:� FVII: C <5% (attention à la thromboplastine utilisée)� FVII: Ag entre 80 et 100%
� Chez cette patiente:� le taux de FVII:Ag à 45% est trop bas � La mutation p.Arg364Gln (facteur VII Padua) est un variant connu
pour avoir des discordances de dosage du FVII:C selon la thromboplastine. Quelle est la thromboplastine utilisée ici???
� Quelles sont les hypothèses?� Que faites vous?

Réponse non
� Le taux de FVII:Ag à 45% est trop bas� Erreur de dosage? On confirme sur l’aliquot
� La thromboplastine utilisée est d’origine lapine� Nous n’avions pas de plasma, nous demandons au prescripteur
s’il peut contrôler le FVII:C avec une thromboplastine d’originehumaine
� FVII:C= 9% avec Inovin® � Or, un homozygote p.Arg364Gln doit avoirdes taux de FVII:C en
thromboplastine humaine aux environs de 30%
Quelles sont vos hypothèses?
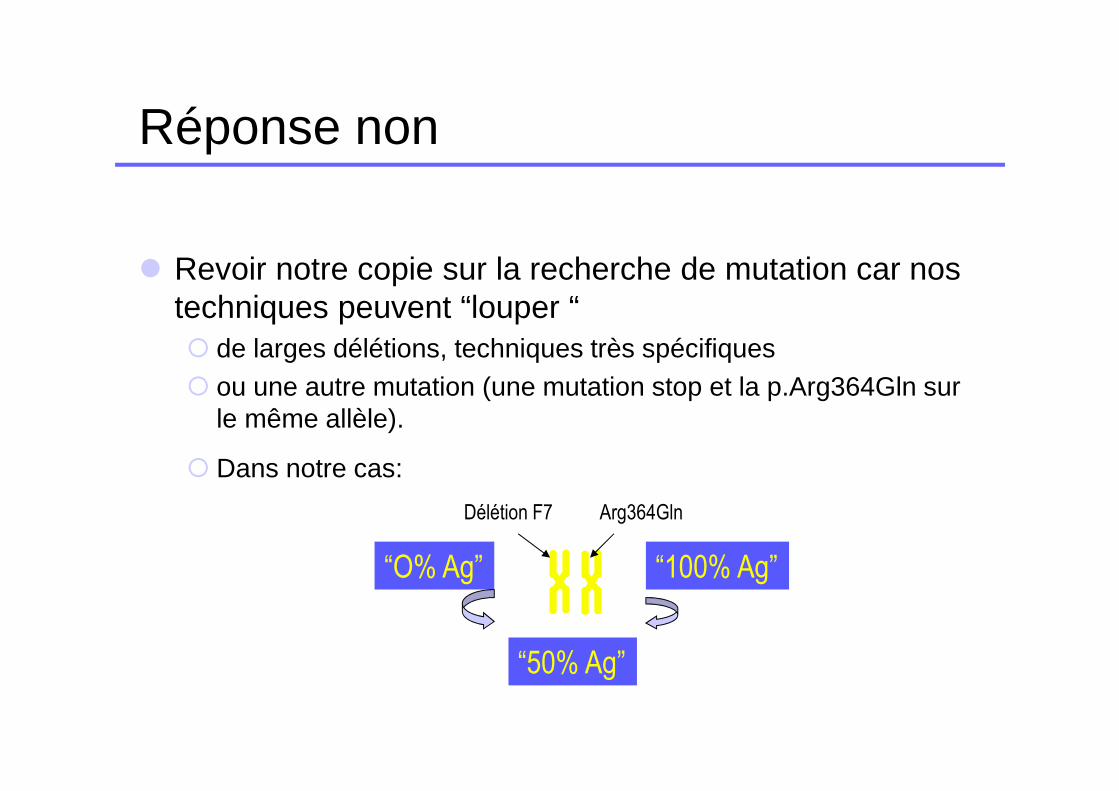
Réponse non
� Revoir notre copie sur la recherche de mutation car nos techniques peuvent “louper “ � de larges délétions, techniques très spécifiques � ou une autre mutation (une mutation stop et la p.Arg364Gln sur
le même allèle).
� Dans notre cas:
Délétion F7 Arg364Gln
“O% Ag” “100% Ag”
“50% Ag”
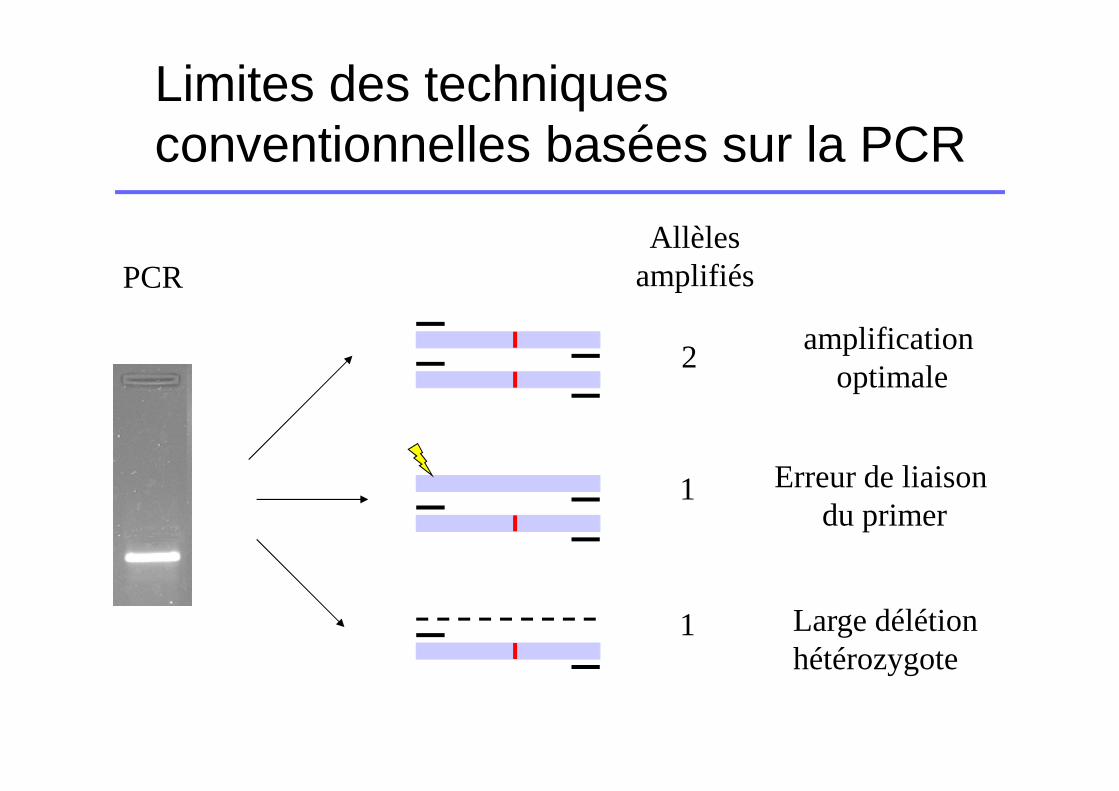
Limites des techniques conventionnelles basées sur la PCR
Allèlesamplifiés
2
1
1
PCR
Erreur de liaison du primer
Large délétion hétérozygote
amplification optimale
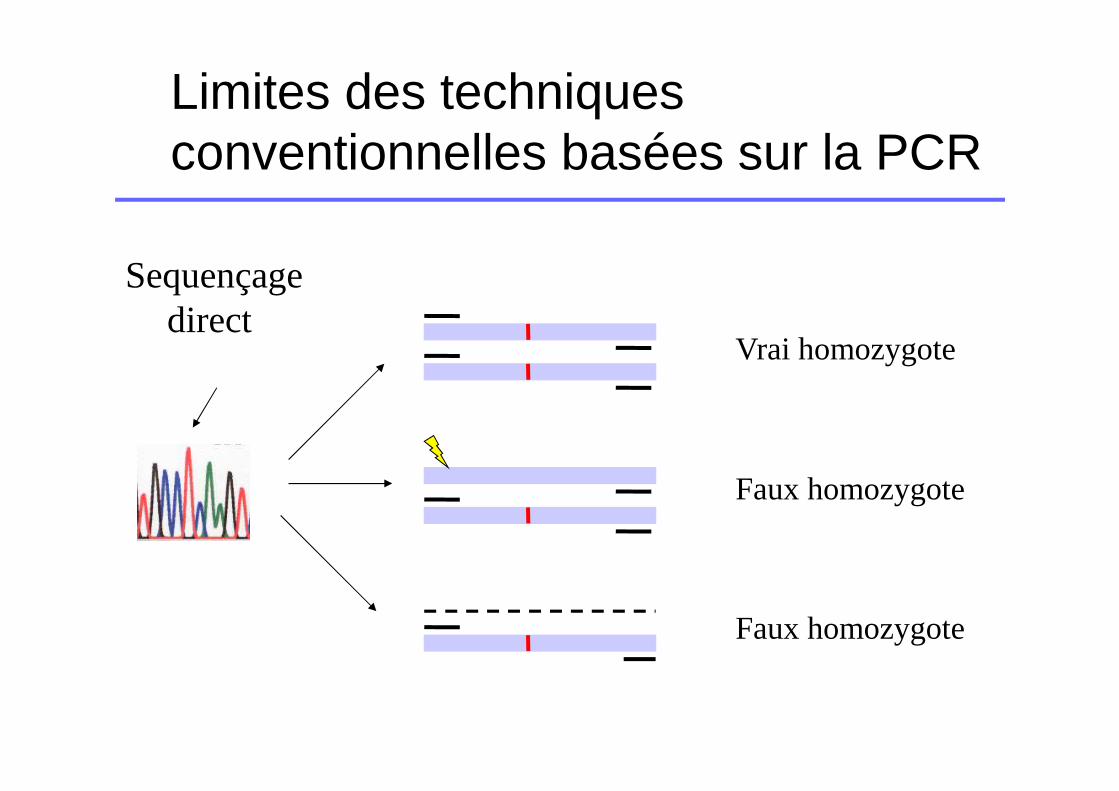
Limites des techniques conventionnelles basées sur la PCR
Sequençagedirect
Vrai homozygote
Faux homozygote
Faux homozygote

Cas n°7
� Patiente de 15 ans � FVII:C = 4% en thromboplastine de lapin� FVII:C = 70% en thromboplastine humaine� FVII:Ag = 78%
�Hétérozygote composite p.Arg139Gln/p.Arg337His
� La mutation p.Arg139Gln est connue pour altérer la liaison du FVII au TF et montrer des différences de taux de FVII:C selon la thromboplastine mais ne gène pas la sécrétion du FVII
� La mutation p.Arg337His n’est pas connue.
� Validez vous ce bilan? Si oui, qu’en pensez-vous?
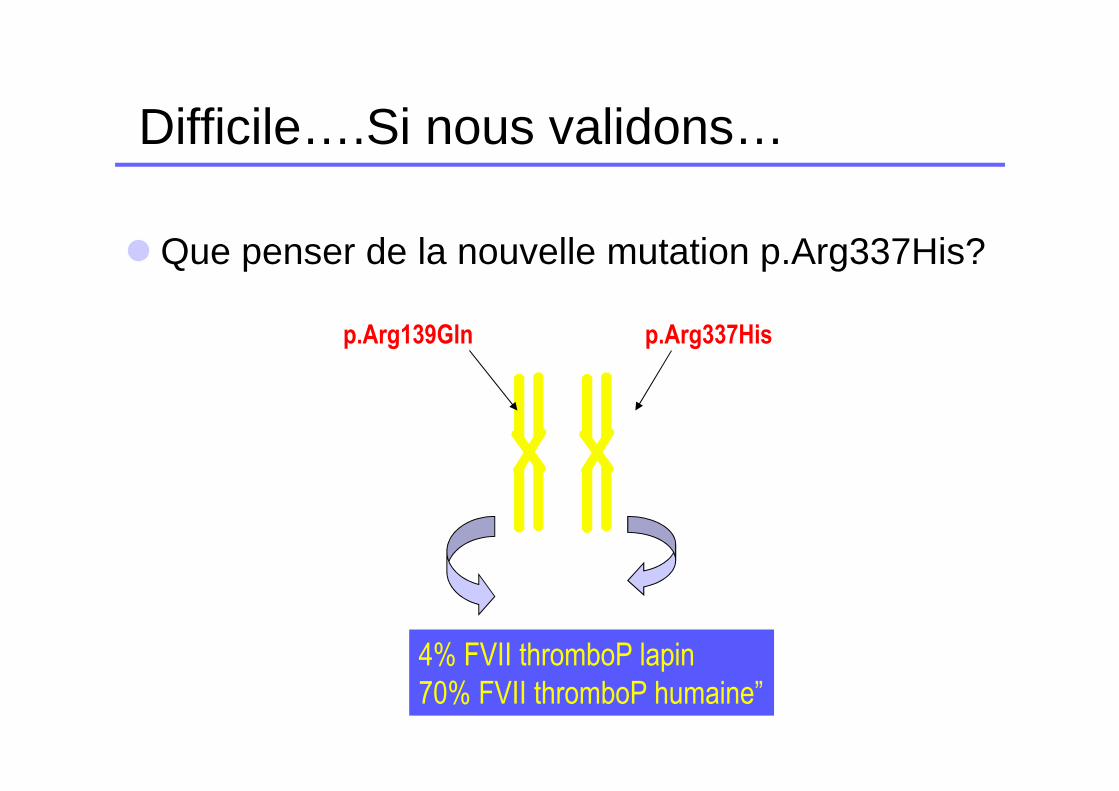
Difficile….Si nous validons…
� Que penser de la nouvelle mutation p.Arg337His?
p.Arg139Gln p.Arg337His
4% FVII thromboP lapin
70% FVII thromboP humaine”
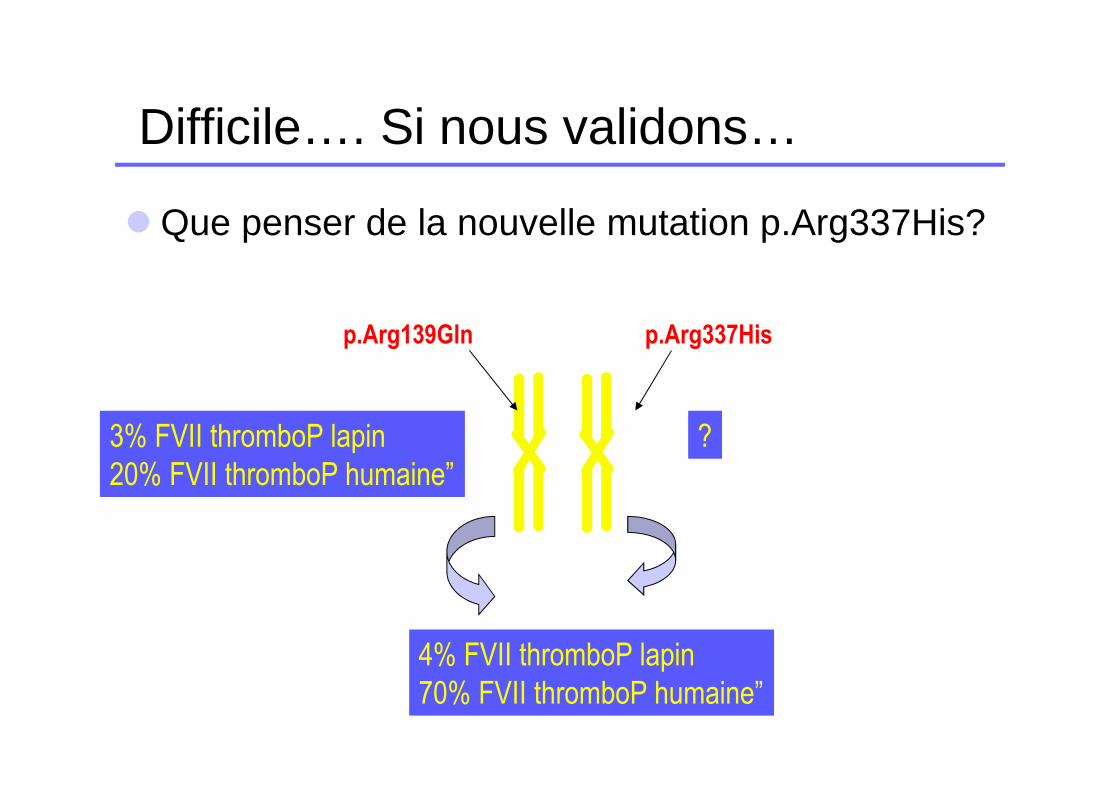
Difficile…. Si nous validons…
� Que penser de la nouvelle mutation p.Arg337His?
p.Arg139Gln p.Arg337His
3% FVII thromboP lapin
20% FVII thromboP humaine”
?
4% FVII thromboP lapin
70% FVII thromboP humaine”
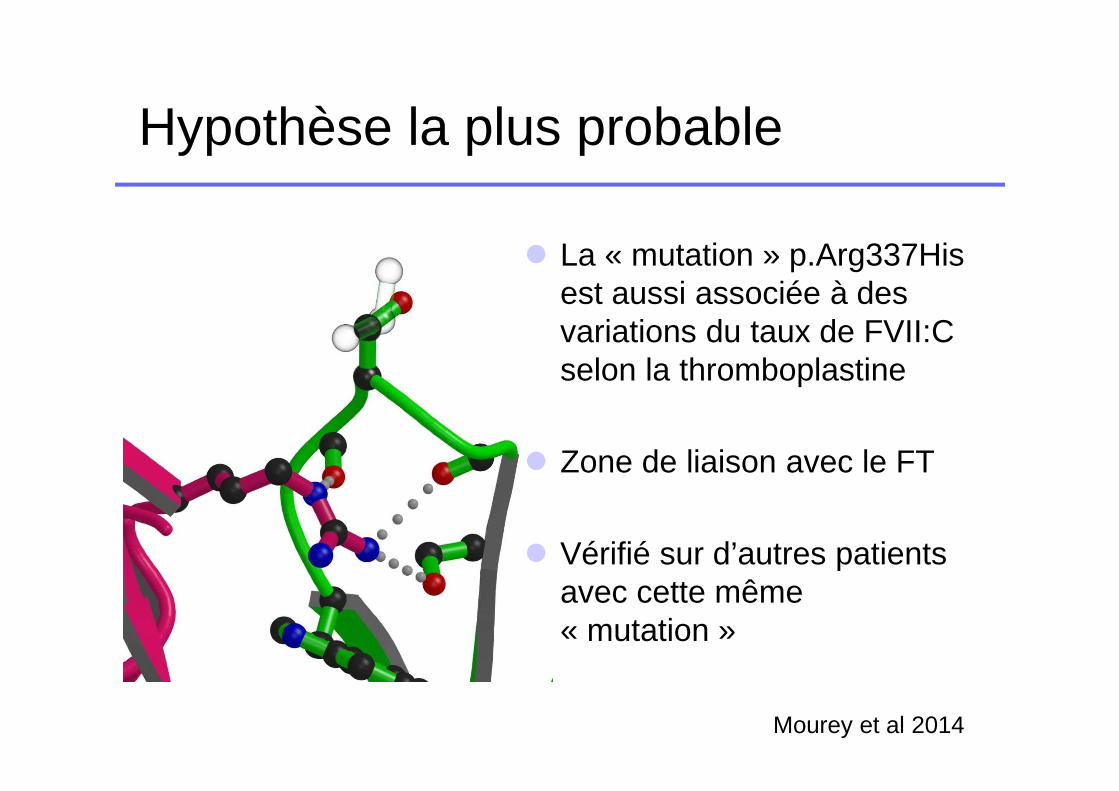
Hypothèse la plus probable
� La « mutation » p.Arg337His est aussi associée à des variations du taux de FVII:C selon la thromboplastine
� Zone de liaison avec le FT
� Vérifié sur d’autres patients avec cette même « mutation »
Mourey et al 2014

Synthèse
�Valeurs valides de FVII:C de FVII:Ag�Concordant avec le génotype
�Concordant avec le phénotype clinique???

Les paramètres biologiques sont-ils prédictifs du risque hémorragique chez les patients déficitaires
en FVII?
Question

Pas vraiment ….Il n’existe pas de test plasmatique sensible et spécifique permettant de prédire la sévérité hémorragique des patients et leur risque opératoire.
FVII:Ag et TFPI : NON
FVII:C et FVIIa :« piètres discriminants »
Mariani et al 2004Giansily-Blaizot et al 2004
� Forts recoupements entre les groupes symptomatiques et asymptomatiques
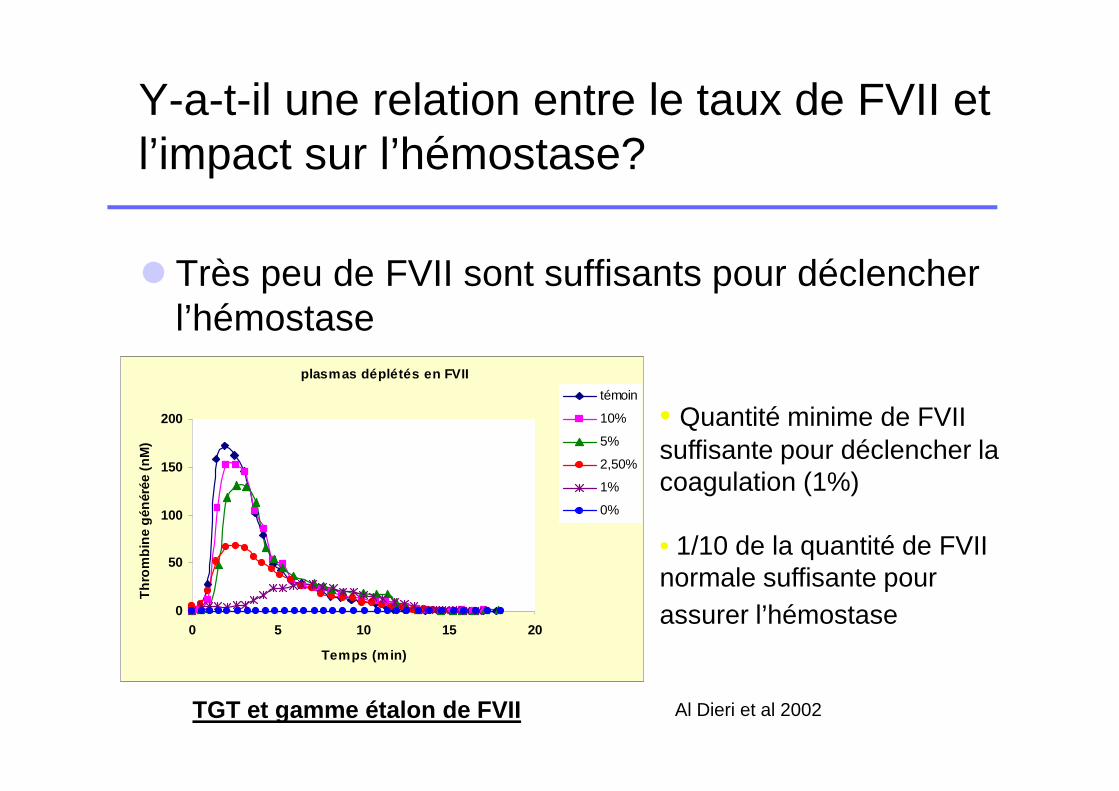
Y-a-t-il une relation entre le taux de FVII et l’impact sur l’hémostase?
� Très peu de FVII sont suffisants pour déclencher l’hémostase
plasmas déplétés en FVII
0
50
100
150
200
0 5 10 15 20
Temps (min)
Thr
ombi
ne g
énér
ée (
nM)
témoin
10%
5%
2,50%
1%
0%
• Quantité minime de FVII suffisante pour déclencher la coagulation (1%)
• 1/10 de la quantité de FVII normale suffisante pour assurer l’hémostase
Al Dieri et al 2002TGT et gamme étalon de FVII
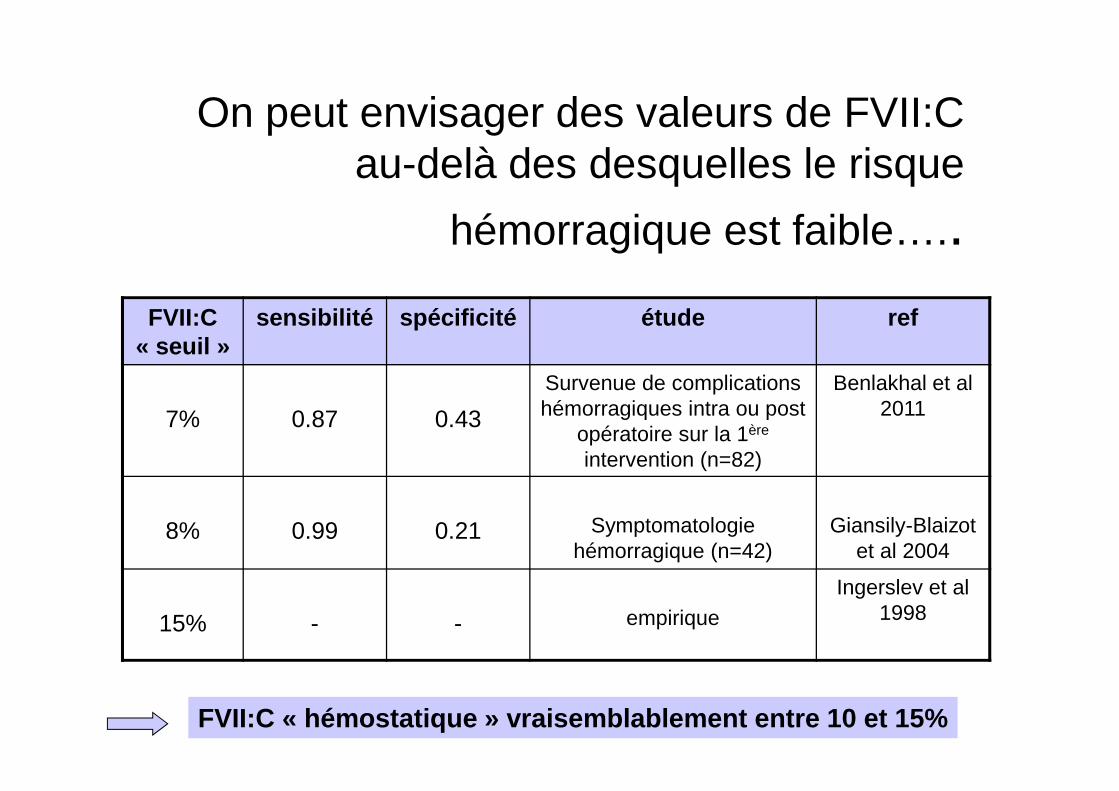
On peut envisager des valeurs de FVII:C au-delà des desquelles le risque
hémorragique est faible…..FVII:C
« seuil »sensibilité spécificité étude ref
7% 0.87 0.43
Survenue de complications hémorragiques intra ou post
opératoire sur la 1ère
intervention (n=82)
Benlakhal et al 2011
8% 0.99 0.21 Symptomatologie hémorragique (n=42)
Giansily-Blaizot et al 2004
15% - - empiriqueIngerslev et al
1998
FVII:C « hémostatique » vraisemblablement entre 10 et 15%
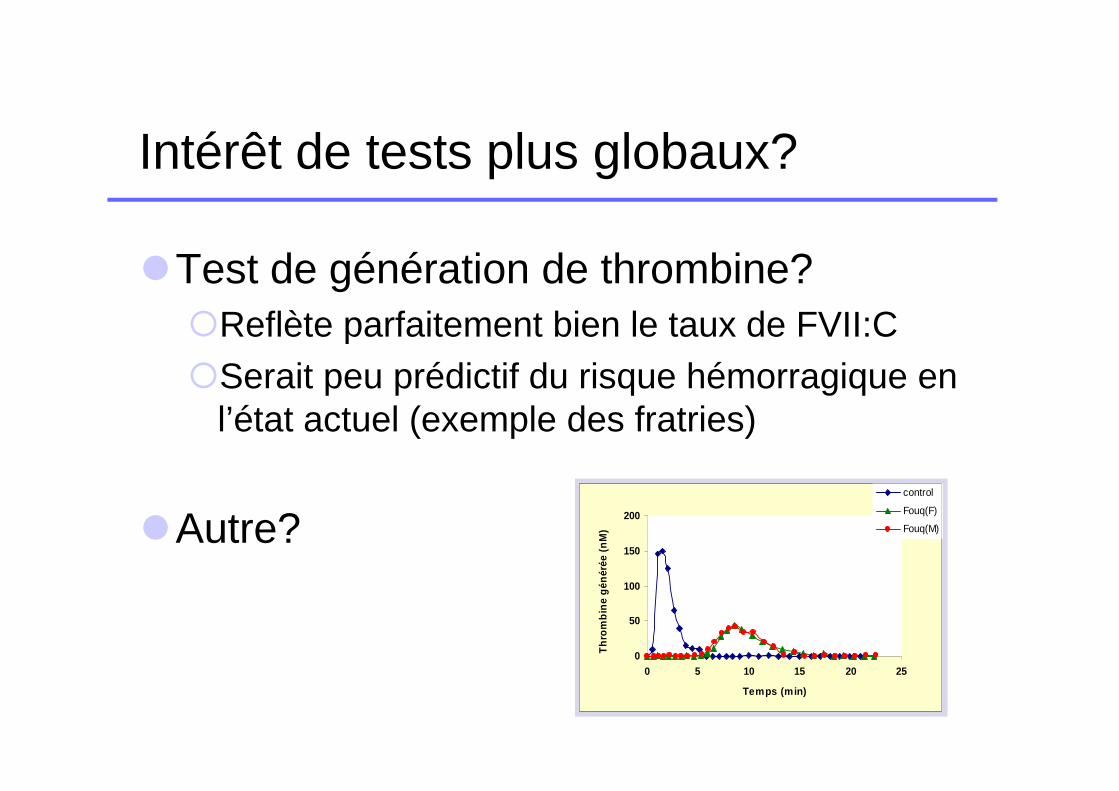
Intérêt de tests plus globaux?
�Test de génération de thrombine?�Reflète parfaitement bien le taux de FVII:C�Serait peu prédictif du risque hémorragique en
l’état actuel (exemple des fratries)
�Autre?
0
50
100
150
200
0 5 10 15 20 25
Temps (min)
Thr
ombi
ne g
énér
ée (
nM)
control
Fouq(F)
Fouq(M)

Les déficits constitutionnels en FVII
Quelques mots du traitement
Molécules disponibles et surveillance

Traitement
• FVII activé recombinant (rFVIIa) : AMM• 15 à 30 µg/kg toutes les 4 à 6 heures jusqu’à obtention de l’hémostase
(Doses et fréquence à adapter au cas par cas)
• Contre-indications : hypersensibilité connue au principe actif ou aux excipients• Mises en garde : toutes conditions pathologiques où le facteur tissulaire peut être
libéré de façon plus importante que la normale
• Complexe prothrombinique humain (PPSB) : AMM • en seconde intention lorsque « aucun facteur de coagulation spécifique de haute
pureté n’est disponible »
• FVII plasmatique (pdFVII) : en ATU • si contre indication aux rFVIIa et PPSB.

Surveillance et adaptation du Trt
� Pour le rFVIIa, il n’existe pas de paramètre biologique pour suivre le trt:�FVII:C donne des valeurs ininterprétables (> 400%!!)� Intérêt du TGT (à suivre)
� Pour le FVII plasmatique: on peut doser�FVII:C
� Penser à vérifier l’absence d’anticorps à distance d’une injection de FVII

Conclusion
� Grande hétérogénéité des déficits
� Progrès faits dans les corrélations génotypes phénotypes cliniques
� Pas de tests prédictifs du risque hémorragique mais des pistes
� Importance de l’apport des registres pour ces maladies rares.





























