Embed Size (px)
Citation preview

UNIVERSITE TOULOUSE III – PAUL SABATIER
U.F.R sciences de la vie et de la terre
THESE
Pour obtenir le grade de
DOCTEUR DE L’UNIVERSITE TOULOUSE III
Discipline : Innovation pharmacologique
Présentée et soutenue par
Sandra DE BARROS
Le 29 octobre 2007
Les métalloprotéases matricielles 2 et 9 et la différenciation des
cellules progénitrices du tissu adipeux humain
JURY
Professeur Marie-Christine RIO Rapporteur
Docteur Jean-Philippe BASTARD Rapporteur
Docteur Annie QUIGNARD-BOULANGE Examinateur
Professeur Philippe VALET Président
Docteur Jean GALITZKY Directeur de thèse
1

A mes parents
A Christophe, Carole et Adrien,
A Laurent.
2

Remerciements
En préambule à ce travail, je tiens à exprimer toute ma reconnaissance et mes sincères
remerciements à tous ceux qui ont participé de près ou de loin à ce travail.
Je tiens à remercier le Professeur Marie-Christine RIO et le Docteur Jean-Philippe
BASTARD d’avoir spontanément accepté d’évaluer ce travail en tant que rapporteur.
J’exprime également ma gratitude au Professeur Philippe VALET et au Docteur Annie
QUIGNARD-BOULANGE pour avoir si gentiment acceptés de participer à ce jury de thèse.
Les recherches qui ont fait l’objet de cette thèse ont été effectuées dans l’unité
INSERM U858 équipe 1, encadrées par les Docteurs Jean GALITZKY, Virginie
BOURLIER, Coralie SENGENES et Anne BOULOUMIE. Je leur suis très profondément
reconnaissante de m’avoir apporté leur soutien, leur confiance et leur disponibilité au cours
de ces trois années de thèse.
Je tiens tout particulièrement à remercier Max LAFONTAN pour ses précieux
conseils.
Je dis un grand merci à tous les membres (ou ex-membres) de l’équipe des allées et
particulièrement à Karine, Alec et Marie-Adeline pour toute l’aide qu’elles ont pu
m’apporter.
J’adresse mes remerciements à l’ensemble du personnel de l’unité INSERM U858 et
plus particulièrement à l’ex-unité INSERM U586 dirigé par le professeur Dominique
LANGIN, pour les nombreux services rendus. Une petite attention particulière pour Danièle,
Carine et Corinne régulièrement « nounous » des plaques taqman, ainsi que Pascale et Lydia
pour toutes les questions administratives que j’ai pu rencontrer… Je tiens également à
remercier les personnes du Laboratoire de Pharmacologie Médicale et Clinique, ainsi que
celle du Centre de Pharmacovigilance, deux services dirigés par le Professeur Jean-Louis
MONTASTRUC que je remercie.
Enfin, je ne saurai clore cette partie sans adresser mes sincères remerciements à ma
famille. J’adresse un grand merci à mes parents, à mon frère, à Carole et à Adrien. Je
remercie également Laurent pour sa présence, sa patience et son soutien quotidien.
3

Table des matières
Table des matières 4
Liste des figures 7
Liste des tableaux 9
Principales abréviations utilisées 10
Avant propos 12
Introduction 14
I- Le tissu adipeux 14
A) Les fonctions du tissu adipeux 14
A.1) Les fonctions métaboliques du tissu adipeux 15
A.1.1) Le stockage des lipides : la lipogenèse et la synthèse des
triglycérides 15
A.1.2) La mobilisation des lipides : la lipolyse 16
A.2) Les fonctions sécrétoires du tissu adipeux 17
B) Le développement normal du tissu adipeux 18
B.1) Le processus de détermination adipocytaire 19
B.2) Le processus de différenciation adipocytaire 22
B.2.1) Les étapes du processus de différenciation adipocytaire 22
B.2.2) Contrôle transcriptionnel de la différenciation adipocytaire 23
B.2.2.1) Les PPARs 23
B.2.2.2) Les C/EBPs 24
B.2.2.3) Le facteur SREBP-1 24
B.2.2.4) Les autres facteurs de transcription 25
B.2.3) Régulation hormonale et intracellulaire de la
différenciation adipocytaire 25
B.3) La vascularisation du tissu adipeux 26
C) Le développement excessif du tissu adipeux : l’obésité 28
C.1) Les modifications cellulaires du tissu adipeux 29
C.1.1) Les adipocytes et les cellules progénitrices/souches 29
C.1.2) Les cellules endothéliales 29
C.1.3) Les macrophages 30
4

C.2) Les modifications des fonctions métaboliques du tissu adipeux 31
C.3) Les modifications des fonctions sécrétoires du tissu adipeux 32
D) La régression du tissu adipeux : la lipoatrophie 34
D.1) Les modifications cellulaires du tissu adipeux 35
D.1.1) Les adipocytes 35
D.1.2) Les cellules progénitrices/souches 37
D.1.3) Les cellules endothéliales 39
D.1.4) Les macrophages 40
D.2) Les modifications des fonctions métaboliques du tissu adipeux 40
D.3) Les modifications des fonctions sécrétoires du tissu adipeux 41
II- Les métalloprotéases matricielles 43
A) Classification et structure 43
B) Les régulations des MMPs 45
B.1) Les Régulations transcriptionnelles et post-transcriptionnelles 45
B.2) Les régulations de la sécrétion 46
B.3) Les processus de maturation des MMPs 46
B.4) Les processus d’inhibition des MMPs 48
C) Mode d’action des MMPs 49
D) Les fonctions biologiques et pathologiques des MMPs 50
III- Les métalloprotéases matricielles dans le tissu adipeux 54
A) La régulation de l’adipogenèse 54
B) La régulation de l’angiogenèse 55
C) La régulation de l’infiltration macrophagique 55
D) Les régulateurs des MMPs dans le tissu adipeux 56
Objectifs 58
Résultats 59
I- Implication des MMPs dans la différenciation adipocytaire des
cellules de la fraction stroma-vasculaire du tissu adipeux humain 59
A) Influence d’un inhibiteur des MMPs et des inhibiteurs de la protéase du VIH sur
5

la différenciation adipocytaire 59
B) Influence d’un anticorps neutralisant dirigé contre MMP-9 sur la
différenciation adipocytaire 61
C) Détermination des mécanismes impliqués dans la chute de l’expression de
MMP-9 par les inhibiteurs de la protéase du VIH 62
D) Études préliminaires et perspectives sur l’étude des cibles moléculaires de
MMP- 9 65
II- Implication des MMPs dans la différenciation des cellules CD34+/CD31-
isolées de la fraction stroma-vasculaire du tissu adipeux humain 68
A) Influence d’un inhibiteur des MMPs sur la différenciation adipocytaire
des cellules CD34+/CD31- 69
B) Mise au point d’une condition de culture permettant simultanément les
capacités adipogéniques et angiogéniques des cellules CD34+/CD31- 70
B.1) Etude de l’accumulation intracellulaire des triglycérides et de la
formation d’un réseau cellulaire positif pour le CD31 par les cellules
CD34+/CD31- cultivées dans un milieu mixte 70
B.2) Etude de l’expression en ARNm des marqueurs adipocytaires et
endothéliaux par les cellules CD34+/CD31- cultivées dans un milieu
mixte 72
B.3) Etude cinétique sur l’évolution des cellules CD34+/CD31- au cours de la
culture en milieu mixte. 73
C) Implication des contacts cellule-cellule dans les capacités adipogéniques et
angiogéniques des cellules CD34+/CD31- 76
C.1) Influence de la densité cellulaire sur les cellules CD34+/CD31- cultivées
en milieu mixte 76
C.2) Influence de la communication cellulaire via les jonctions gaps sur les
cellules CD34+/CD31- cultivées en milieu mixte 77
Conclusions et perspectives 80
Références bibliographiques 83
Annexes 103
6

Liste des figures
Figure 1 : Le tissu adipeux
Figure 2 : Lipogenèse et synthèse des triglycérides dans l’adipocyte
Figure 3 : Contrôle de la lipolyse dans l’adipocyte humain
Figure 4 : Les sécrétions du tissu adipeux
Figure 5 : Détermination et différenciation adipocytaires
Figure 6: Evolution de la matrice extracellulaire et de la forme cellulaire au cours de la
différenciation adipocytaire
Figure 7 : Progression des facteurs de transcription au cours de l’adipogènese dans les
préadipocytes de la lignée 3T3-L1
Figure 8 : Balance entre les facteurs de transcription pro- et anti-adipogéniques
Figure 9 : Comparaison entre les modifications du tissu adipeux lors d’une obésité ou d’une
lipoatrophie
Figure 10 : Les métalloprotéases matricielles
Figure 11 : Régulation de l’activité des métalloprotéases matricielles
Figure 12 : Eléments de liaison aux facteurs de transcription sur les promoteurs des
métalloprotéases matricielles
Figure 13 : Mode d’action des métalloprotéases matricielles
Figure 14 : Effet d’un anticorps neutralisant pour la MMP-9 sur la différenciation
adipocytaire des cellules de la FSV du tissu adipeux humain
Figure 15 : Hypothèse du mode d’action des IPs-VIH sur l’expression génique de MMP-9
Figure 16 : Influence d’un substrat matriciel sur la différenciation adipocytaire des cellules
de la FSV du TA humain
Figure 17 : Technique d’isolement des différentes populations de la FSV du tissu adipeux
humain
Figure 18 : Sécrétion des MMP-2 et -9 au cours de la différenciation adipocytaire des
cellules progénitrices CD34+/CD31- isolées
Figure 19 : Influence du batimastat sur la différenciation adipocytaire des cellules
progénitrices CD34+/CD31-
Figure 20 : Détermination de l’accumulation des TGs et de la formation d’un réseau
cellulaire positif pour le CD31 des cellules CD34+/CD31- cultivées en milieu adipogénique,
endothélial ou mixte
7

Figure 21 : Détermination de l’expression en ARNm des marqueurs adipocytaires et
endothéliaux des cellules CD34+/CD31- cultivées en milieu adipogénique, endothélial ou
mixte
Figure 22 : Evolution du contenu en ADN, TGs et du réseau cellulaire positif pour le CD31
des cellules CD34+/CD31- cultivées en milieu mixte
Figure 23 : Evolution de l’expression en ARNm des marqueurs adipocytaires et endothéliaux
des cellules CD34+/CD31- cultivées en milieu mixte
Figure 24 : Influence de la présence de cellules endothéliales matures sur la formation du
réseau cellulaire positif pour le CD31 des cellules CD34+/CD31- cultivées en milieu mixte
Figure 25 : Influence de la densité cellulaire sur les capacités adipogéniques et
angiogéniques des cellules CD34+/CD31- cultivées en milieu mixte
Figure 26 : Evolution de l’expression en ARNm des connexines 43 et 45 des cellules
CD34+/CD31- cultivées en milieu mixte
Figure 27 : Effet d’un inhibiteur des jonctions gaps sur les capacités adipogéniques et
angiogéniques des cellules CD34+/CD31- cultivées en milieu mixte
8

Liste des tableaux
Tableau 1: Les différents modèles de culture cellulaire utilisés pour l’étude de l’adipogenèse
Tableau 2: Facteurs extracellulaires et molécules de signalisation régulant l’adipogenèse
Tableau 3 : Classification des adultes maigres, normaux, en surpoids et obèses selon l’IMC
Tableau 4: Sécrétions du tissu adipeux modulées ou non au cours de l’obésité
Tableau 5: Classification des différentes lipoatrophies humaines
Tableau 6: Impact des IPs-VIH et des INRTs sur les sécrétions adipocytaires in vitro et in
vivo
Tableau 7: Classification des métalloprotéases matricielles
Tableau 8 : Les activateurs des MMPs
Tableau 9: Phénotypes des souris déficientes pour les MMPs
Tableau 10: Les inhibiteurs des MMPs dans le cancer
Tableau 11: Composition des milieux adipogénique, endothélial et mixte utilisés
Tableau 12: Détermination de l’expression en ARNm des marqueurs adipocytaires et
endothéliaux par des adipocytes matures, des cellules progénitrices CD34+/CD31- et des
cellules endothéliales capillaires CD34+/CD31+
Tableau 13: Expression en ARNm des connexines 43, 45, 40 et 37 dans des adipocytes
matures, des cellules progénitrices CD34+/CD31- et des cellules endothéliales capillaires
CD34+/CD31+
9

Principales abréviations utilisées
ACC : Acetyl Co-A carboxylase
ADAM: A Disintegrine And Metalloprotease domain
AGA: 18α-glycyrrhetinic acid
AGNE: acide gras non estérifié
AP-1: Activator protein-1
aP2 : fatty acid binding protein
ATGL : adipocyte triacylglycerol lipase
bFGF : basic fibroblast growth factor
bHLH : basic helix-loop helix
BMP : Bone morphogenetic protein
CD : Cluster de différenciation
C/EBP : CCAAT/enhancer binding protein
CHOP-10 : C/EBP homologous protein 10
CRABP-1 : Cytoplasmic retinoic acid binding protein-1
ECBM : Endothelial cell basal medium
ECGM : Endothelial cell growth medium
FAS : Fatty acid synthase
FSV: Fraction stroma-vasculaire
GLUT : Glucose transporteur
GZA : glycyrrhizic acid
IDV : Indinavir
IGF : Insulin growth factor
IκB: Inhibitor NF-κB
IL : Interleukine
IMC : Indice de masse corporelle
INRT : Inhibiteur nucléosidique de la réverse transcriptase
INNRT: Inhibiteur non nucléosidique de la réverse transcriptase
IPs-VIH : Inhibiteur de la protéase du VIH
LHS : Lipase hormono-sensible
LPL : Lipoprotéine lipase
MCP-1 : Monocyte chemoattractant proteine 1
10

MEC : Matrice extracellulaire
MMP : Métalloprotéase matricielle
MT-MMP : Métalloprotéase matricielle de type membranaire
NF-κB : Nuclear factor kappa B
NFV : Nelfinavir
PAI-1 : Plasminogen activator inhibitor 1
PDGF : Platelet-derived growth factor
PKA : Protein kinase A
RT-PCR :Real-time polymerase chain reaction
RTV : Ritonavir
RXR : Récepteur de l’acide 9-cis-rétinoique
SDF-1 : Stromal cell-derived factor-1
SQV : Saquinavir
SREBP: Sterol regulatory element binding protein
TA : Tissu Adipeux
TCF/LEF : T-cell factor/lymphoid enhancer factor
TIMP : Tissue inhibitor of matrix metalloproteinases
TGs : Triglycérides
TNF-α : Tumor necrosis factor α
VEGF : Vascular endothelial growth factor
VIH : Virus de l’immunodéficience humaine
VLDL : Very low density lipoprotein
vWF : von Willebrand Factor
11

Avant-propos
Le tissu adipeux (TA) adulte peut subir des processus de développement excessif
(surpoids, obésité) mais aussi des processus de régression (lipoatrophie). Ces deux
phénomènes entraînent des altérations de la composition cellulaire et des fonctions du TA. Il
est désormais reconnu qu’un excès ou une perte de masse grasse constitue un facteur
aggravant de nombreuses pathologies telles que le diabète de type 2 et les désordres
cardiovasculaires.
Le surpoids touche, aujourd’hui, près d’un milliard de personnes à l’échelle de la
planète, soit un sixième de la population mondiale. Parmi elles, 300 millions sont obèses et
l’obésité infantile est en plein essor. Les causes de l’obésité sont multiples et dépendent aussi
bien de facteurs génétiques qu’environnementaux. A l’heure actuelle, quelques médicaments
avec une indication pour l’obésité (orlistat, sibutramine) ont été développés mais leur
efficacité reste insuffisante.
Les syndromes de lipodystrophies étaient des événements rares (moins de 1 cas sur
100000) et de causes principalement génétiques. Ces dix dernières années sont apparues des
formes de lipodystrophies liées aux traitements antirétroviraux de l’infection par le virus de
l’immunodéficience humaine (VIH). En effet, si l’utilisation des inhibiteurs de la protéase du
VIH (IPs-VIH) et des inhibiteurs nucléosidiques de la reverse transcriptase (INRTs) ont
permis de diminuer significativement la morbidité et la mortalité des patients infectés par le
VIH, plus de 50% des patients traités ont développé un syndrome lipodystrophique se
caractérisant par une redistribution des dépôts adipeux et des complications métaboliques. Le
signe clinique majeur du syndrome lipodystrophique est une lipoatrophie au niveau de la face
et des membres associée ou non à une accumulation du TA au niveau des régions dorso-
cervicales et abdominales. Quelques approches thérapeutiques pour le traitement des
lipoatrophies ont été développées (thiazolidinedione, metformine) mais elles agissent plus sur
les complications métaboliques associées que sur l’altération du TA elle-même.
Ainsi, une meilleure connaissance des mécanismes impliqués dans l’extension et la
régression de la masse grasse est maintenant nécessaire afin d’identifier de nouvelles cibles
thérapeutiques. Dans ce chapitre d’introduction, nous allons dans une première partie nous
intéresser au TA, à ses fonctions et à son développement normal. Nous aborderons aussi les
modifications tant cellulaires que fonctionnelles que le TA subit lors d’une obésité ou d’une
lipoatrophie. Puis dans une deuxième partie, nous nous intéresserons aux principaux acteurs
12

impliqués dans la régulation des phénomènes de remodelage tissulaire, les métalloprotéases
matricielles ou MMPs. Leur classification, leur structure ainsi que la régulation de leur
activité seront présentées. Enfin, leur mode d’action et les fonctions biologiques auxquelles
ces protéases sont associées seront décrits. Enfin nous aborderons le rôle potentiel de ces
enzymes dans la plasticité du TA.
Nos travaux de recherche portant sur la caractérisation (i) du rôle de la MMP-9 dans
les processus de différenciation adipocytaire et (ii) des interactions entre les inhibiteurs de
protéases utilisées lors de la thérapie du VIH avec les voies dépendantes de MMP-9, seront
ensuite exposés. Enfin, une dernière partie de notre travail portant sur la caractérisation des
cellules progénitrices du TA humain en condition de culture permettant d’exprimer leurs
capacités angiogéniques et adipogéniques sera décrite.
13

Introduction
I. Le tissu adipeux
Le tissu adipeux (TA) est doué d’une importante plasticité. Il est en effet capable de
croître ou d’involuer de manière spectaculaire. Avant de nous intéresser au développement
excessif du TA ainsi qu’à sa capacité de régression, un aperçu de la fonction propre du TA
nous semble nécessaire.
A) Les fonctions du tissu adipeux
Deux grands types de TA sont présents chez les mammifères : le TA blanc et le TA
brun. Ces deux tissus ont des propriétés biochimiques et fonctionnelles distinctes. Le TA
brun se retrouve surtout chez les rongeurs et, chez l’homme, il est initialement présent chez le
nouveau-né et joue un rôle clé dans la gestion de la thermogenèse (libération d’énergie sous
forme de chaleur). Le TA blanc, dont nous parlerons exclusivement dans la suite de ce
travail, constitue la principale réserve d’énergie de l’organisme. Il comprend différents dépôts
anatomiques localisés dans les territoires sous-cutanés et viscéraux. Cette notion de
localisation du TA est essentielle puisque selon que le TA soit sous-cutané ou viscéral, son
organisation cellulaire va varier ainsi que son activité métabolique et sécrétoire (81, 136,
270).
Au cours de ces dernières années, les recherches sur le TA ont permis de faire
émerger les concepts et rôles clés du TA dans le contrôle des flux métaboliques ainsi que la
notion de TA endocrine. Ainsi, le TA n’est plus décrit comme un simple tissu de soutien et de
protection. Les adipocytes sont le type cellulaire majeur du TA. Ils assurent la fonction
métabolique du tissu (stockage et dégradation des triglycérides (TGs)) mais sont également
considérés comme une source cellulaire des sécrétions du TA. L’adipocyte est caractérisé par
une morphologie polygonale et une grande taille avec un diamètre moyen de 70µm chez le
sujet normo-pondéré. La quasi-totalité de la cellule est occupée par une unique vacuole
lipidique et le cytoplasme et les organites constituent un fin liseré périphérique (figure 1A).
Ces caractéristiques ainsi que sa flottaison in vitro permettent de le distinguer et de l’isoler
facilement de la fraction non adipocytaire du TA, la fraction stroma-vasculaire (FSV),
14
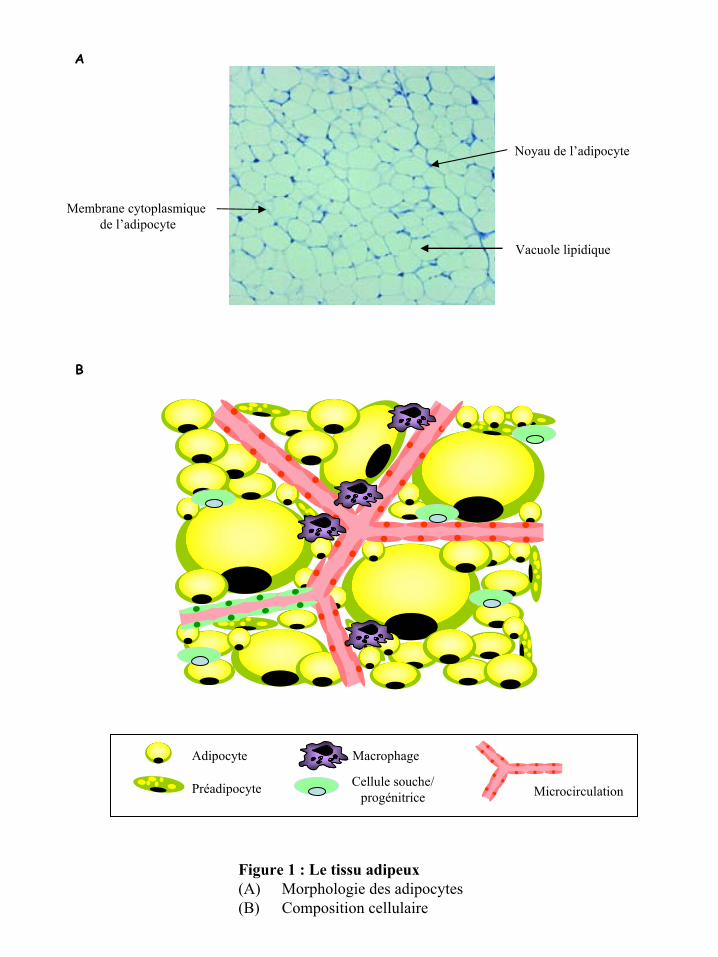
Figure 1 : Le tissu adipeux(A)
Morphologie des adipocytes(B)
Composition cellulaire
B
A
Microcirculation
Adipocyte
Préadipocyte
Macrophage
Cellule
souche/progénitrice
Vacuole lipidique
Noyau de l’adipocyte
Membrane cytoplasmique de l’adipocyte

contenant principalement des cellules progénitrices/souches, des cellules vasculaires et des
cellules immuno-inflammatoires (figure 1B).
A.1) Les fonctions métaboliques du tissu adipeux
Brièvement, nous aborderons dans les paragraphes suivants les mécanismes et les
voies de régulation des principales fonctions métaboliques adipocytaires : le stockage des
lipides (lipogenèse et synthèse de TGs) et leur dégradation (la lipolyse).
A.1.1) Le stockage des lipides : la lipogenèse et la synthèse des triglycérides
Le stockage des lipides dans l’adipocyte se fait par 2 voies (figure 2). La première
voie correspond à la capture directe des TGs associés aux chylomicrons et aux lipoprotéines
de très faible densité (ou VLDL pour « very low density lipoprotein ») circulants, provenant
de l’alimentation ou de la lipogenèse hépatique. La LPL (lipoprotéine lipase), produite et
sécrétée par les adipocytes, est ancrée par des protéoglycanes à héparane sulfate à la surface
endothéliale (87). En coopération avec GPIHBP1 (glycosylphosphatidylinositol-anchored
high density lipoprotein-binding protein 1) récemment décrite comme étant exprimée à la
surface endothéliale et comme jouant le rôle de récepteur potentiel aux chylomicrons et
VLDL, la LPL hydrolyse les lipoprotéines (11, 297). Les acides gras non estérifiés issus de
ce clivage sont ensuite recaptés par l’adipocyte, qui les transforme alors en acyl-Co enzyme
A (Acyl-CoA). Ils seront ré-estérifiés en TGs en présence de glycérol. La deuxième voie
correspond à la lipogenèse de novo. Le terme de lipogenèse proprement dit désigne la
néosynthèse d’acides gras à partir du glucose. Après l’entrée du glucose dans l’adipocyte,
grâce à des transporteurs spécifiques (GLUT-1 et 4), le glucose est dégradé en pyruvate par le
processus de la glycolyse. A partir du pyruvate, l’acétyl-CoA carboxylase (ACC) et la
synthase des acides gras (FAS, « fatty acid synthase ») interviennent de façon successive
pour catalyser la formation des acides gras à longue chaîne saturée. L’action des différentes
désaturases permet de synthétiser des acides gras plus ou moins saturés. Comme
précédemment, ces acides gras sont ensuite ré-estérifiés pour donner les TGs. Il est nécessaire
de préciser que chez les rongeurs, la lipogenèse est réalisée dans le foie et le TA. Par contre,
chez l’homme, la lipogenèse est majoritairement hépatique et elle n’est qu’accessoire dans le
TA (145), excepté suite à un régime alimentaire riche en glucides (105). De nombreux
facteurs endocrines et paracrines tels que les catécholamines, l’hormone de croissance et la
15
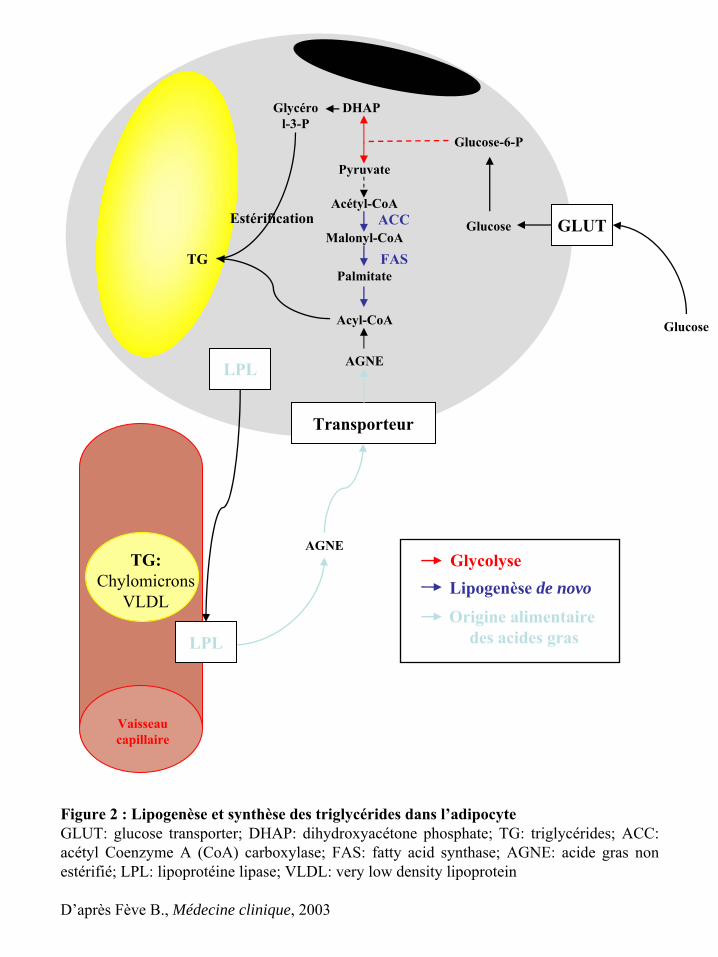
Figure 2 : Lipogenèse et synthèse des triglycérides dans l’adipocyteGLUT: glucose transporter; DHAP: dihydroxyacétone
phosphate; TG: triglycérides; ACC: acétyl
Coenzyme A (CoA) carboxylase; FAS: fatty
acid
synthase; AGNE: acide gras non estérifié; LPL: lipoprotéine lipase; VLDL: very
low
density
lipoprotein
D’après Fève B., Médecine clinique, 2003
Glucose
GLUTGlucose
TG
Glucose-6-P
Transporteur
AGNE
AGNE
TG:Chylomicrons
VLDL
LPL
LPL
Vaisseaucapillaire
Acétyl-CoA
Palmitate
Acyl-CoA
Pyruvate
DHAPGlycéro
l-3-P
Malonyl-CoAEstérification ACC
FAS
Lipogenèse de novoGlycolyse
Origine alimentaire des acides gras

leptine sont capables de contrôler le stockage des TGs mais le facteur majeur est l’insuline
qui stimule cette voie en augmentant le captage du glucose via le recrutement du transporteur
GLUT-4 à la membrane, et en activant les enzymes glycolytiques et lipogéniques (120). De
façon intéressante, les effets inhibiteurs de la leptine et de l’hormone de croissance sur le
stockage des TGs passent par l’inhibition des effets stimulateurs de l’insuline.
A.1.2) La mobilisation des lipides : la lipolyse
La lipolyse assure la dégradation des TGs contenus dans les vacuoles lipidiques de
l’adipocyte, en glycérol et acides gras non estérifiés. Plusieurs lipases interviennent de façon
séquentielle dans cette hydrolyse des réserves lipidiques (figure 3). La lipase hormono-
sensible (LHS) clive les TGs en diglycérides puis les diglycérides en monoglycérides, et
constitue l’enzyme limitante du processus lipolytique. La lipase des monoglycérides (LMG)
dégrade ensuite les monoglycérides en glycérol et acides gras non estérifiés. Cependant, la
mise en évidence d’une activité lipolytique diminuée mais toujours présente chez des souris
invalidées pour le gène de la LHS a laissé supposer l’existence d’au moins une autre lipase
capable d’hydrolyser les TGs dans les adipocytes (196). Trois laboratoires différents ont
identifié une nouvelle lipase dénommée « adipocyte triacyglycerol lipase » (ATGL),
isoforme ζ d’une phospholipase indépendante du calcium (iPLA2ζ) ou desnutrine (113, 277,
303). L’ATGL jouerait un rôle clé dans la lipolyse basale, mais l’hydrolyse des TGs et des
diglycérides par la LHS demeurerait l’étape limitante dans la lipolyse stimulée par les
principaux agents lipolytiques chez l’homme, les catécholamines et le peptide atrial
natriuretique (142). Les mécanismes susceptibles de réguler l’activité de la LHS sont bien
connus et sont présentés dans la figure 3 (135). L’activité de cette lipase dépend
spécifiquement de sa phosphorylation réversible par une kinase dépendante de l’AMPc
(PKA) (100) ou du GMPc (PKG) (240). La phosphorylation de la LHS active l’enzyme, en
démasquant le site catalytique. Par ailleurs, cette phosphorylation permet une redistribution
de la lipase du cytoplasme vers la vacuole lipidique. Les périlipines, protéines abondamment
exprimées à la surface des gouttelettes lipidiques, interviennent dans la localisation et la
stabilisation de la LHS à la surface des vacuoles. La phosphorylation des périlipines par la
PKA et la PKG réduit leur ancrage à la gouttelette lipidique et favorise ainsi l’accès de la
LHS à ses substrats lipidiques.
16
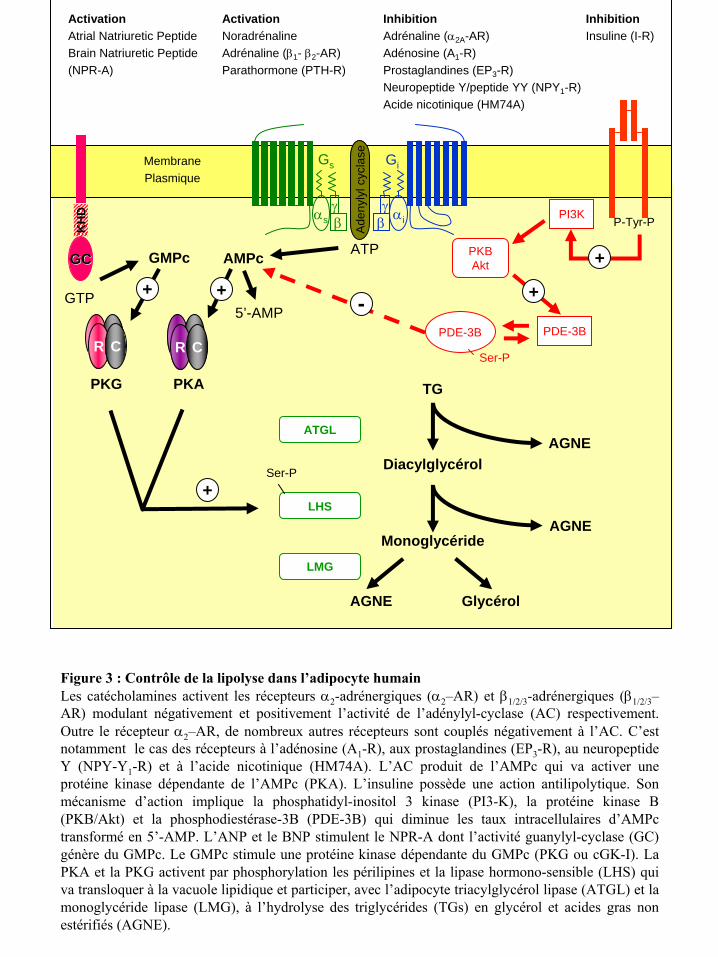
Figure 3 : Contrôle de la lipolyse dans l’adipocyte humainLes catécholamines activent les récepteurs α2
-adrénergiques (α2
–AR) et β1/2/3
-adrénergiques (β1/2/3
–
AR) modulant négativement et positivement l’activité de l’adénylyl-cyclase
(AC) respectivement. Outre le récepteur α2
–AR, de nombreux autres récepteurs sont couplés négativement à l’AC. C’est notamment le cas des récepteurs à l’adénosine (A1
-R), aux prostaglandines (EP3
-R), au neuropeptide Y (NPY-Y1
-R) et à l’acide nicotinique (HM74A). L’AC produit de l’AMPc
qui va activer une protéine kinase dépendante de l’AMPc
(PKA). L’insuline possède une action antilipolytique. Son mécanisme d’action implique la phosphatidyl-inositol
3 kinase (PI3-K), la protéine kinase B (PKB/Akt) et la phosphodiestérase-3B (PDE-3B) qui diminue les taux intracellulaires d’AMPc
transformé
en 5’-AMP. L’ANP et le BNP stimulent le NPR-A dont l’activité guanylyl-cyclase
(GC) génère du GMPc. Le GMPc
stimule une protéine kinase dépendante du GMPc
(PKG ou cGK-I). La PKA et la PKG activent par phosphorylation les périlipines
et la lipase hormono-sensible
(LHS) qui va transloquer à la vacuole lipidique et participer, avec l’adipocyte triacylglycérol
lipase (ATGL) et la monoglycéride
lipase (LMG), à l’hydrolyse des triglycérides (TGs) en glycérol et acides gras non estérifiés (AGNE).
Ade
nyly
lcyc
lase
αsγβ αi
γβ
Gs Gi
InhibitionAdrénaline (α2A -AR)Adénosine (A1 -R)Prostaglandines (EP3 -R)Neuropeptide Y/peptide YY (NPY1 -R)Acide nicotinique (HM74A)
ActivationNoradrénalineAdrénaline (β1 - β2 -AR)Parathormone (PTH-R)
ATPAMPc
PKA
R CR C
InhibitionInsuline (I-R)
GCGC
KH
D
GTP
GMPc
ActivationAtrial Natriuretic PeptideBrain Natriuretic Peptide(NPR-A)
P-Tyr-PPI3K
PKBAkt
PDE-3BPDE-3B
Ser-P
-
+
+
PKG
R CR C
5’-AMP
LHS
Ser-P
MembranePlasmique
+ +
+
TG
Diacylglycérol
Monoglycéride
AGNE Glycérol
AGNE
AGNEATGL
LMG

A.2) Les fonctions sécrétoires du tissu adipeux
D’un point de vue quantitatif, les acides gras libres, issus de la lipolyse et libérés dans
la circulation sanguine, représentent le principal produit de sécrétion du TA (265). Cependant
le TA est également capable de synthétiser et de sécréter une multitude de facteurs qui
agissent soit localement de façon autocrine et/ou paracrine, soit parfois de façon endocrine.
On désigne sous le terme « adipokines », l’ensemble des facteurs bioactifs produits et
sécrétés par le TA. Historiquement, la LPL est la première adipokine découverte (49).
Toutefois, c’est la découverte de la leptine, hormone synthétisée par le TA et intervenant
dans le contrôle de la prise alimentaire, qui a permis au TA d’acquérir le statut d’organe
endocrine (302). Suite à cette découverte, d’autres facteurs produits et sécrétés par le TA ont
été décrits, on en compte actuellement plus d’une cinquantaine qui diffèrent tant au niveau de
leur structure que de leur fonction : des hormones (stéroïdes et glucocorticoïdes), des
cytokines (TNFα, IL-6, IL-8), des facteurs de croissance (TGF-β), des protéases (MMP-2 et
9) et des anti-protéases (TIMP, PAI-1) (81). Plusieurs fonctions physiologiques leur sont
associées, du contrôle du métabolisme lipidique, à l’angiogenèse et l’inflammation (figure 4).
Excepté la leptine et l’adiponectine considérées comme spécifiques des adipocytes, les
adipokines apparaissent comme étant également exprimées et produites par les cellules de la
fraction non adipocytaire du TA, i.e la FSV (72). Des différences dans l’activité de sécrétion
mais également dans le type d’adipokines ont été décrites dans les TA humains sous-cutanés
et viscéraux (72, 136).
En résumé, l’adipocyte présente deux fonctions principales, le stockage des
lipides selon le processus de lipogenèse ainsi que celui de la synthèse des TGs, et la
mobilisation des lipides via le processus de lipolyse. Ces activités métaboliques sont
étroitement régulées par des signaux neuro-humoraux. De plus, grâce à une capacité
sécrétoire aussi multiple que variée, le TA joue un rôle endocrine permettant d’assurer
une communication avec de nombreux organes et tissus mais aussi un rôle paracrine et
autocrine permettant une communication locale entre les différents types cellulaires qui
le composent.
Voyons à présent les mécanismes impliqués dans le développement normal de la masse
grasse. Nous nous intéresserons ensuite aux modifications cellulaires du TA et de ses
principales fonctions au cours d’un développement excessif ou d’une régression.
17
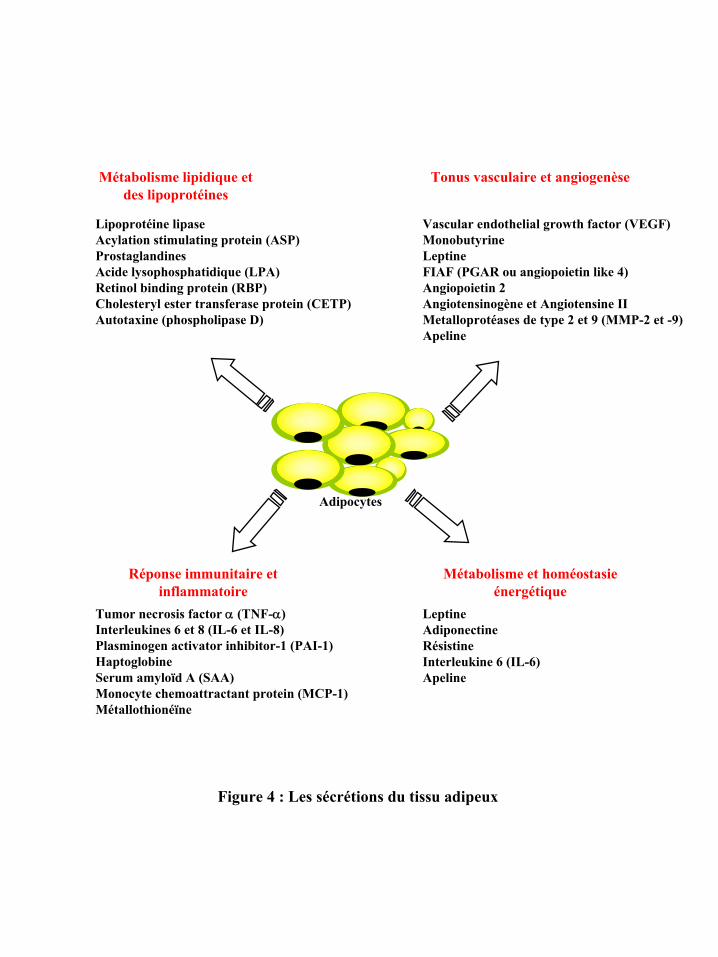
Figure 4 : Les sécrétions du tissu adipeux
Adipocytes
Métabolisme lipidique et des lipoprotéines
Lipoprotéine lipaseAcylation stimulating
protein
(ASP)Prostaglandines Acide lysophosphatidique
(LPA)Retinol
binding
protein
(RBP)Cholesteryl
ester transferase
protein
(CETP)Autotaxine
(phospholipase
D)
Tonus vasculaire et angiogenèse
Vascular
endothelial
growth
factor
(VEGF)MonobutyrineLeptineFIAF (PGAR ou angiopoietin
like
4)Angiopoietin
2Angiotensinogène
et Angiotensine IIMetalloprotéases
de type 2 et 9 (MMP-2 et
-9)Apeline
Réponse immunitaire et inflammatoire
Tumor
necrosis
factor
α
(TNF-α)Interleukines 6 et 8 (IL-6 et
IL-8)Plasminogen
activator
inhibitor-1 (PAI-1)HaptoglobineSerum
amyloïd
A (SAA)Monocyte chemoattractant
protein
(MCP-1)Métallothionéïne
Métabolisme et homéostasie énergétique
LeptineAdiponectineRésistineInterleukine 6 (IL-6)Apeline
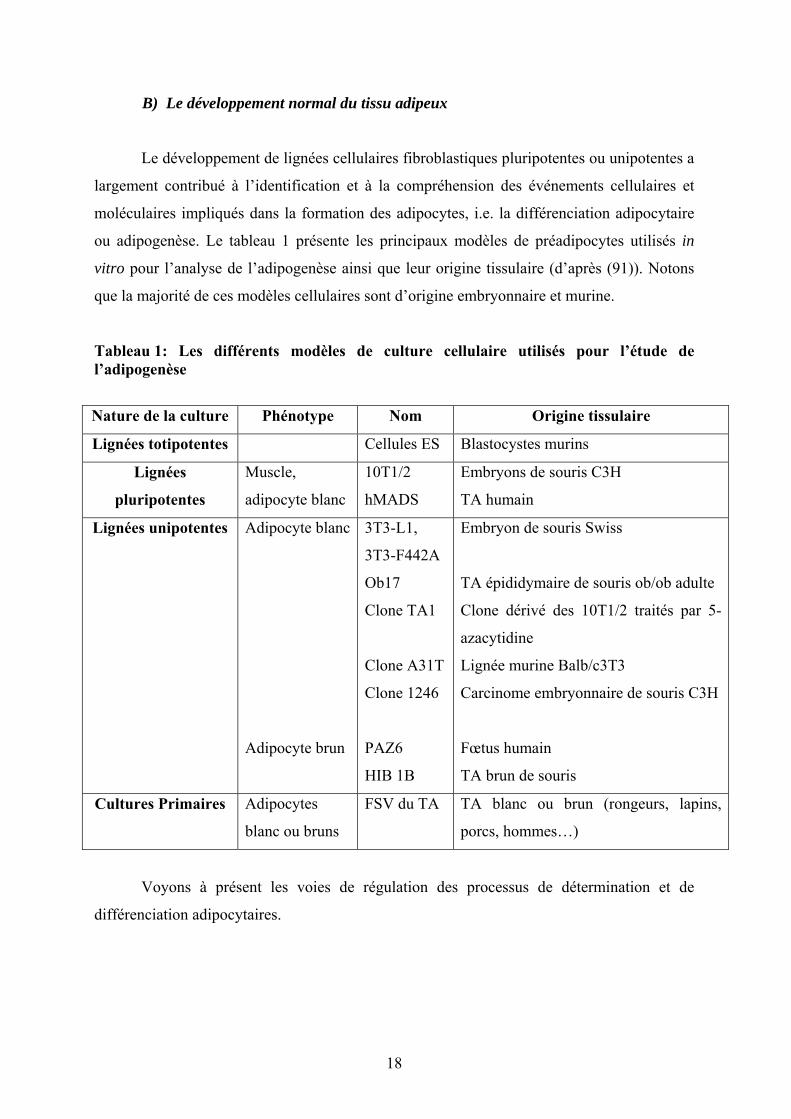
B) Le développement normal du tissu adipeux
Le développement de lignées cellulaires fibroblastiques pluripotentes ou unipotentes a
largement contribué à l’identification et à la compréhension des événements cellulaires et
moléculaires impliqués dans la formation des adipocytes, i.e. la différenciation adipocytaire
ou adipogenèse. Le tableau 1 présente les principaux modèles de préadipocytes utilisés in
vitro pour l’analyse de l’adipogenèse ainsi que leur origine tissulaire (d’après (91)). Notons
que la majorité de ces modèles cellulaires sont d’origine embryonnaire et murine.
Tableau 1: Les différents modèles de culture cellulaire utilisés pour l’étude de l’adipogenèse
Nature de la culture Phénotype Nom Origine tissulaire
Lignées totipotentes Cellules ES Blastocystes murins
Lignées
pluripotentes
Muscle,
adipocyte blanc
10T1/2
hMADS
Embryons de souris C3H
TA humain
Lignées unipotentes Adipocyte blanc
Adipocyte brun
3T3-L1,
3T3-F442A
Ob17
Clone TA1
Clone A31T
Clone 1246
PAZ6
HIB 1B
Embryon de souris Swiss
TA épididymaire de souris ob/ob adulte
Clone dérivé des 10T1/2 traités par 5-
azacytidine
Lignée murine Balb/c3T3
Carcinome embryonnaire de souris C3H
Fœtus humain
TA brun de souris
Cultures Primaires Adipocytes
blanc ou bruns
FSV du TA TA blanc ou brun (rongeurs, lapins,
porcs, hommes…)
Voyons à présent les voies de régulation des processus de détermination et de
différenciation adipocytaires.
18

B.1) Le processus de détermination adipocytaire
Comme l’indique la figure 5, la détermination adipocytaire correspond à l’orientation
d’une cellule précurseur, supposée d’origine mésenchymateuse, vers la lignée adipocytaire.
Bien que les mécanismes impliqués dans ce processus soient encore peu connus, certains
facteurs susceptibles d’orienter le précurseur mésenchymateux vers le lignage adipeux ont été
identifiés (pour revue (79, 226)). C’est notamment le cas des voies de signalisation de delta
fos B et de l’acide rétinoïque mais aussi de Wnt, une glycoprotéine sécrétée impliquée dans
la croissance et le devenir cellulaire et des BMPs (bone morphogenic protein) appartenant à
la superfamille du TGF-β et impliqués dans le devenir des cellules mésenchymateuses. Le
facteur delta fos B inhibe l’adipogenèse en favorisant la différenciation ostéogénique (234).
L’activation de la voie Wnt inhibe également la différenciation adipocytaire en favorisant
l’orientation myogénique (230). Enfin, une exposition des cellules mésenchymateuses
multipotentes CH310T1/2 à du BMP-4 engage les cellules vers un lignage adipogénique
(261). L’influence du BMP-2 sur ces cellules CH310T1/2 semble en revanche plus complexe
puisqu’à faibles concentrations il stimule l’engagement vers le lignage adipogénique alors
qu’à de fortes concentrations il stimule l’engagement vers le lignage ostéogénique (283).
Il est à noter que l’origine des précurseurs des adipocytes n’est pas claire. Chez
l’embryon, l’apparition d’adipocytes est concomitante à l’apparition du système vasculaire
(52) et la culture de cellules souches embryonnaires murines en présence d’acide rétinoïque
promeut la différenciation adipocytaire (58, 207). Chez l’adulte, des cellules
mésenchymateuses de la moelle osseuse caractérisées par leur capacité d’adhésion sur
plastique, leur capacité de prolifération et leur expression de différents marqueurs de surface
tels que le CD105 et l’absence du marqueur des cellules souches hématopoïétiques CD34,
sont capables in vitro de s’orienter vers des voies adipogéniques, chondrogéniques et
ostéogéniques (210). De plus, des analyses clonales ont permis de montrer qu’une même
cellule conservait ces capacités multiples de détermination (210). Récemment, une étude
réalisée chez des souris soumises à un régime gras a permis de montrer par l’utilisation
d’approches de repopulation des cellules de la moelle osseuse par des cellules souches qui
expriment une protéine fluorescente verte (GFP) que certains adipocytes des dépôts profonds
pourraient être originaires de cellules de la moelle osseuse (54). De plus, des cellules
sanguines circulantes ont été montrées comme pouvant exprimer une capacité de
différenciation adipocytaire, suggérant que des cellules de la moelle osseuse peuvent, sous
des stimuli à préciser, être mobilisées dans la circulation sanguine et être recrutées dans les
19
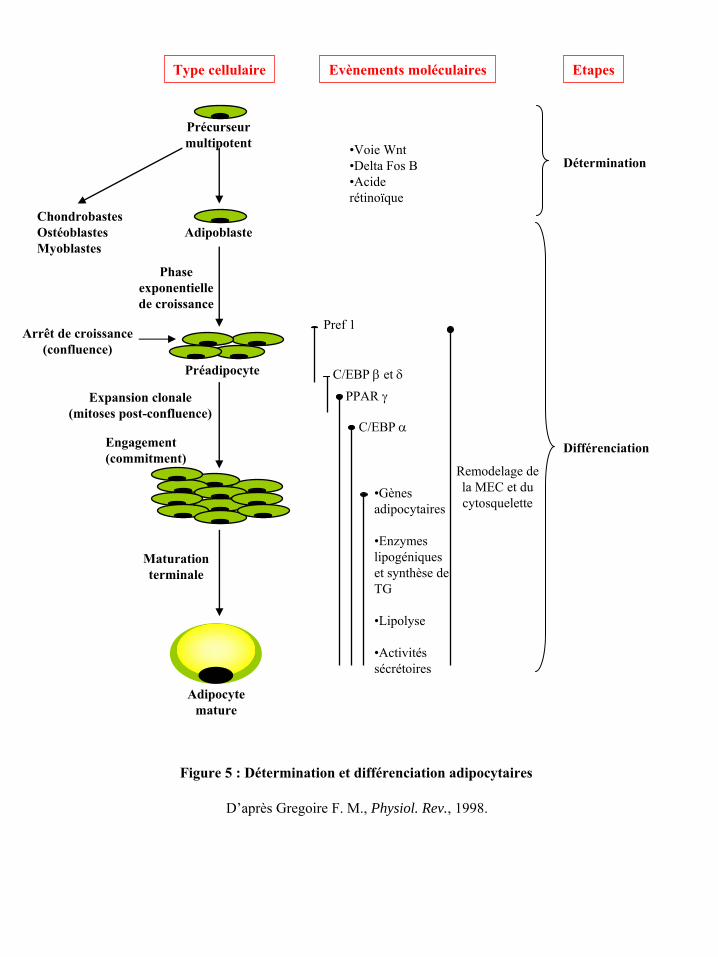
Figure 5 : Détermination et différenciation adipocytaires
D’après Gregoire
F. M., Physiol. Rev., 1998.
Type cellulaire Evènements moléculaires Etapes
Précurseur multipotent
AdipoblasteChondrobastesOstéoblastes Myoblastes
Détermination•Voie Wnt•Delta Fos B•Acide rétinoïque
Différenciation
Phase exponentielle de croissance
Préadipocyte
Arrêt de croissance (confluence)
Remodelage de la MEC et du cytosquelette
Pref
1
C/EBP β
et δPPAR γ
C/EBP α
•Gènes adipocytaires
•Enzymes lipogéniques et synthèse de TG
•Lipolyse
•Activités sécrétoires
Adipocyte mature
Maturation terminale
Expansion clonale (mitoses post-confluence)
Engagement(commitment)

dépôts adipeux (101). Le TA adulte contient également au sein de la FSV des cellules
résidentes capables d’exprimer des capacités multiples de différenciation (304). De plus,
diverses approches in vivo ont montré la capacité réparatrice de ces cellules dans des modèles
animaux d’ischémie vasculaire et cardiaque ou de dommages osseux et musculaires. La revue
qui suit : « le TA un donneur de cellules souches » présente ces données.
20

Cah. Nutr. Diét., 42, 2, 2007
73
biologie générale
biologie générale
LE TISSU ADIPEUX : UN DONNEUR DE CELLULES SOUCHES ?*
Anne BOULOUMIÉ, Sandra DE BARROS, Marie MAUMUS, Jean GALITZKY, Coralie SENGENES
Les cellules souches sont caractérisées par leur capacité d’autorenouvelle-ment et leur capacité à produire des cellules différenciées fonctionnelles.Différents types de cellules souches sont définis en fonction de leur ori-gine et de leur devenir : (1) les cellules souches embryonnaires totipoten-tes capables de former toutes les couches embryonnaires ainsi que lesannexes extra-embryonnaires qui supportent la croissance de l’embryon,(2) les cellules souches embryonnaires pluripotentes qui donnent nais-sance au mésoderme, endoderme et ectoderme, (3) les cellules souchesmultipotentes, capables de générer plusieurs types cellulaires, (4) lescellules progénitrices ou précurseurs qui présentent une capacité limitéed’autorenouvellement et qui se différencient en un type cellulaire défini[1].
Des progrès récents dans l’isolement et la caractérisa-tion des cellules souches/progénitrices ont conduit à lamise en évidence de cellules présentant certaines pro-priétés de cellules souches/progénitrices dans divers tissusadultes, tels que le cœur, le rein, le cerveau, le muscleet également le tissu adipeux. Ces cellules constitue-raient un réservoir de cellules réparatrices prêtes à êtremobilisées et à se différencier en réponse à une isché-mie et/ou à un dommage. Ces cellules présentent unintérêt croissant en médecine réparative et régénérativeavec un rôle thérapeutique potentiel dans une grandevariété d’applications cliniques. Cependant, la présencede cellules souches/progénitrices dans les tissus adultesa conduit à de nombreuses questions et controversesquant à leurs identités et potentialités réelles de diffé-renciation [2].
Chez l’adulte, la moelle osseuse est considérée comme letissu réservoir de cellules souches [3]. Deux familles au moinsde cellules souches/progénitrices qui représentent une frac-tion mineure de la population totale de la moelle (0,001 à0,01 % des cellules nuclées), ont été identifiées : les cellulessouches hématopoiétiques qui comprennent des cellulesmultipotentes capables de reconstituer l’ensemble deslignées hématopoiétiques, des cellules progénitrices mono-potentes donnant naissance à un lignage, et les cellules sou-ches mésenchymateuses qui présentent la capacité de sedifférencier en cellules des tissus connectifs, dont les os, letissu adipeux et le cartilage. Une population de cellulesimmatures capables de prolifération sans sénescence etcapables de s’orienter vers des lignages distincts, i.e. endo-thélial, endodermal et neural
in vitro
et
in vivo
, a été égale-ment décrite dans la moelle adulte et définie comme « cellulessouches adultes multipotentes » [4]. Cependant, leur exis-tence est soumise à de fortes controverses. Finalement, descellules endothéliales progénitrices sont également présentesdans la moelle osseuse. Ces cellules contribueraient à la néo-vascularisation de tissus ischémiques après leur mobilisationet leur recrutement vers les sites ischémiques [5].En plus de la moelle osseuse, d’autres sources de cellulessouches/progénitrices ont été décrites chez l’adulte, telsque la peau [6], le muscle [7], le cerveau [8] et le tissuadipeux humain [9].
Inserm, Équipe Avenir, INSERM U858, Institut de médecine moléculaire de Rangueil, Toulouse ; Université Paul Sabatier, Institut Louis Bugnard IFR31, Toulouse.
Correspondance : Anne Bouloumié, INSERM équipe AVENIR, Faculté de méde-cine, 37, allées Jules Guesde, 31000 Toulouse.Email : [email protected]
* Conférence donnée dans le cadre de la 47e Journée Annuelle de Nutrition et de Diététique, le vendredi 26 janvier 2007.

74
Cah. Nutr. Diét., 42, 2, 2007
biologie générale
Caractéristiques
in vitro
des cellules souches/progénitrices du tissu adipeux humain
La présence de cellules progénitrices dans le tissu adipeuxhumain est reconnue depuis les travaux de Bjorntorp surl’évolution de la cellularité de la masse adipeuse avec ledéveloppement de l’obésité.
Les préadipocytes
En effet, ses travaux ont montré clairement que l’obésitéchez l’homme se caractérise par une phase d’hypertro-phie adipocytaire suivie d’une phase d’hyperplasie adi-pocytaire [10, 11]. Les adipocytes étant des cellulesmatures différenciées, incapables de proliférer. L’aug-mentation du nombre d’adipocytes dans le tissu adipeux(TA) de patients obèses est expliquée par la différencia-tion de cellules progénitrices présentes dans la fractionstroma-vasculaire (FSV), i.e. la fraction non-adipocytaire,du TA [12]. En effet, l’isolement de la FSV de TA humainet sa mise en culture
in vitro
en condition adipogénique[13, 14], i.e. en présence de facteurs favorisant la diffé-renciation adipocytaire tels que l’insuline, la triodothryro-nine et le cortisol, conduit à la formation de cellules
invitro
qui expriment des transcripts spécifiques des adipo-cytes. De plus, ces cellules différenciées présentent desactivités fonctionnelles métaboliques, i.e. lipolyse et lipo-génèse, et sécrétoires (production de leptine) des adipo-cytes matures. Des expériences chez les rongeurs ontpermis de plus de montrer que ces cellules forment
invitro
des adipocytes matures fonctionnels au site de leurinjection [15, 16]. Ces résultats démontrent la présence,au sein du TA, de cellules immatures capables de se dif-férencier
in vitro
et
in vivo
en adipocytes fonctionnels.
Différenciation
in vitro
des cellules de la FSV du TA humain
Outre leur capacité de différenciation en adipocyte
invitro
, de nombreux laboratoires ont, par la suite, montréque les cellules de la FSV humaine pouvaient acquérirselon les conditions de culture
in vitro
des marqueursbiochimiques caractéristiques de lignages cellulaires variés[17] : lignage ostéogénique [18], chondrogénique [19,20] et myogénique [21, 22] mais également l’expressionde marqueurs du lignage neuronal [21, 23], endothélial[24-26], épithélial [27-29] et même cardiaque [30]. Deplus, des approches « clonales », c’est-à-dire d’analysedes capacités de différenciation des cellules dérivantd’une cellule unique après multiples passages, ont montréque plusieurs clones cellulaires pouvaient se différencieren plusieurs lignages [22, 23], suggérant la présence decellules de type « cellules souches adultes multipotentes »et non pas seulement un mélange de population de cel-lules progénitrices unipotentes. Ces résultats sont enaccord avec les résultats obtenus par le groupe de Chris-tian Dani [31] montrant dans la FSV isolée à partir deTA de jeunes donneurs, la présence de cellules plusimmatures et multipotentes (hMADS ou Human Multipo-tent Adipose Derived Stem Cell). Des expériences paral-lèles chez les rongeurs mettaient également en évidenceune plasticité importante des cellules de la FSV des TAsmurins [32-34].
Limites des approches
in vitro
sur les cellules de la FSV du TA humain
La majorité des approches réalisées
in vitro
utilisentdes phases d’expansion cellulaire en présence de fortesconcentrations de sérum, conditions associées à des alté-rations phénotypiques des cellules [35]. En effet, alors queles cellules natives, i.e. directement isolées, de la FSVhumaine expriment des marqueurs membranaires de typeCD34, marqueur des cellules souches hématopoiétiques,et sont dépourvues des marqueurs des cellules souchesmésenchymateuses Stro-1 et CD105, les cellules placéesen culture expriment un profil inverse, i.e. perte du CD34et gain de CD105 et Stro-1 [25, 36-38]. De plus, peu oupas d’analyses fonctionnelles ont été associées aux étudesrelatives aux potentialités de différenciation des cellules.Ces analyses sont cependant nécessaires pour statuerclairement quant à la capacité réelle de différenciation encellules fonctionnelles. De plus, certains résultats obtenuschez les rongeurs n’ont pas été confirmés chez l’hommesuggérant que les capacités de différenciation des cellulesde la FSV humaine sont plus restreintes que chez le rongeur.Enfin, la FSV du TA n’est pas constituée d’une populationde cellules homogènes mais contient, comme son nom l’indi-que, des populations cellulaires de la composante vascu-laire [25] et de la composante stromale, i.e. macrophageset lymphocytes infiltrés [39].
Isolement des cellules progénitrices/souches natives de la FSV du TA humain
Afin d’isoler et de caractériser la population de cellulesprogénitrices présentes dans la FSV du TA humain, notregroupe a développé une approche d’immunosélection/déplétion permettant d’isoler les différentes populationscellulaires. Nous avons ainsi démontré que les cellules« natives » positives pour le CD34 et négatives pour lemarqueur endothélial CD31 sont la seule population cel-lulaire parmi les cellules endothéliales capillaires (CD34+/CD31+), les macrophages (CD34–/CD14+) et les cellulesCD34–/CD31–/CD14–, à présenter la capacité de se dif-férencier en adipocytes
in vitro
[38]. De plus, une partiede cette même population cellulaire possède, en milieu deculture endothélial, la capacité d’exprimer des marqueursendothéliaux et de s’organiser en structures semblables àdes capillaires [25]. Ces résultats montrent que la fractioncellulaire CD34+/CD31– native, i.e. n’ayant subi aucuneétape d’expansion
in vitro
, de la FSV du TA humain pré-sente des propriétés de cellules progénitrices capables dedonner naissance à des cellules qui expriment des mar-queurs phénotypiques des adipocytes et des cellules endo-théliales. Il reste à déterminer si cette population cellulairenative est constituée d’un mélange de cellules progénitricesunipotentes ou de cellules souches multipotentes.
Caractéristiques
in vivo
des cellules souches/progénitrices du TA humain
Les approches
in vitro
ont donc apporté des observationsintéressantes sur la plasticité potentielle des cellules dérivéesde la FSV du TA humain, justifiant la mise en place d’appro-ches
in vivo
d’injection de cellules humaines dans desmodèles animaux présentant des ischémies/dommages

Cah. Nutr. Diét., 42, 2, 2007
75
biologie générale
tissulaires afin de suivre la potentialité réparatrice de cescellules. Les mécanismes responsables des interactionsentre cellules implantées et cellules hôtes restent à êtrecaractérisés. Cependant, des travaux récents apportent desdonnées intéressantes concernant la réponse de l’hôteainsi que le comportement des cellules injectées en fonctiondes environnements locaux distincts.
Réponse immune de l’hôte
Les cellules dérivées de la FSV des TAs humains semblentprésenter une nature non-immunogénique [40]. En effet,
in vitro
les cellules ne provoquent pas d’alloréactivité delymphocytes incompatibles mais apparaissent de plusexercer des propriétés immunosuppréssives [40, 41]. Deplus,
in vivo
, l’implantation des cellules humaines chezdes souris immunocompétentes n’induit pas d’accumulationde lymphocytes au site d’implantation [31]. Les processusexacts impliqués nécessitent une caractérisation plus appro-fondie. Cependant, ces observations pourraient avoir unimpact important en termes de thérapie allogénique ou pourle transfert de cellules dans un organisme hôte dérivéesd’un autre donneur [42].
Mécanismes d’adressage des cellules
Plusieurs types de modes d’administration des cellules ontété utilisés de l’injection intraveineuse à l’application directeau site endommagé. Quand les cellules sont injectées parvoie systémique chez la souris, les cellules s’infiltrent enconditions basales dans différents tissus [43] dont lespoumons, le foie, le cœur, le thymus et le cerveau [44].Toutefois, en présence de dommages tissulaires, une infil-tration plus élevée est observée au niveau du site endom-magé. Par exemple, une hépatectomie partielle chez lasouris conduit à l’augmentation de l’infiltration des cellulesdérivées de la FSV dans le foie [44]. Les mécanismes quiguident les cellules aux sites endommagés ne sont pasconnus. Nous avons initié des expériences sur la capacitéde migration des cellules CD34+/CD31–. Nos résultatsmontrent que les cellules migrent fortement en réponseà un gradient de facteurs produits par les cellules endo-théliales. Des recherches complémentaires sont nécessairespour déterminer quels facteurs, tels que des chimiokinesou des signaux relatifs à l’état hypoxique, sont responsablesde l’attraction des cellules souches/progénitrices de laFSV des TAs humains.
Devenir
in vivo
Le principe de la thérapie cellulaire est que les cellules sou-ches/progénitrices vont au niveau du site endommagé sedifférencier sous l’influence de facteurs locaux en cellulesd’un phénotype approprié. Cependant la différenciation« réelle » des cellules souches/progénitrices
in vivo
estsoumise à controverse et plusieurs processus alternes ontété suggérés comme participant aux activités réparatricesdes cellules telles que la fusion cellulaire aboutissant à lareprogrammation nucléaire et la production par les cellulessouches/progénitrices de facteurs trophiques et « répara-teurs » agissant de manière paracrine sur les cellules hôtes.La capacité réparatrice des cellules souches/progénitricesde la FSV du TA humain a été clairement mise en évidencedans différents modèles chez la souris. Dans un modèle desouris immunotolérante avec une ischémie de la patte arrièreprovoquée par ligature de l’artère fémorale, l’injection decellules de la FSV brute [24, 45, 46] ou des cellules
CD34+/CD31– [25] conduit à une amélioration de lanéovasularisation du membre ischémique. Toutefois, il resteà déterminer si cette néovascularisation est due à uneincorportion des cellules injectées dans la vasculature dela souris et à leur différenciation en cellules endothélialesou à la production locale par les cellules injectées de fac-teurs proangiogéniques [46, 47]. Des activités réparatricesde dommages osseux ont été également rapportées dansdifférents modèles murins [48, 49]. Enfin, l’injection descellules souches multipotentes chez des souris « mdx »,modèle de myopathie de Duchenne, est associée à uneexpression importante et prolongée de la dystrophinehumaine, protéine nécessaire à l’intégrité de la fibre muscu-laire [31]. Des expériences en parallèle sur les cellules dela FSV murine montrent également des capacités réparatricesmultiples [50-52].
Limites et questions
Un nombre important d’observations suggère que le proces-sus néoplastique impliquerait des cellules souches [53].Une étude récente montre le risque de transformation descellules de la FSV. En effet, la culture à long terme descellules de la FSV du TA humain adulte a été associée àl’immortalisation et la transformation spontanée des cellules,conduisant
in vivo
à la formation de tumeur après injec-tion des cellules chez la souris athymique [54]. Des étudescomplètes sont donc nécessaires pour établir clairement lerisque associé à l’expansion
in vitro
des cellules de la FSVhumaine.Le déclin du potentiel régénératif est une signature duvieillissement et pourrait être dû à des changements relatifsà l’âge des cellules souches spécifiques des tissus. Des étudesrécentes chez la souris montrent que le déclin de l’activitédes cellules progénitrices lié au vieillissement peut êtreréversible suite à une exposition à un environnement« jeune » alors qu’à l’inverse l’environnement d’un animalâgé empêche la régénération tissulaire stimulée par descellules progénitrices issues d’animaux « jeunes » [55]. Lesétudes de l’équipe de C. Dani montrant la présence decellules souches multipotentes dans la FSV ont été réali-sées chez des patients jeunes [31]. Il serait maintenantintéressant de déterminer l’impact de l’âge sur les cellulessouches/progénitrices du tissu adipeux tant au niveau de leurpotentialité intrinsèque que sur leur interaction avec l’envi-ronnement.Finalement, la présence de ces cellules dans la FSV posela question de leur origine et de leur spécificité. Récem-ment, Hong
et al.
[56] a décrit que les fibrocytes circulantCD45+ étaient capables en culture d’accumuler des trigly-cérides. Les études de Crossno
et al.
[57] réalisées chezla souris en transplantant des cellules de la moelle osseusede souris transgéniques qui expriment la protéine fluores-cente verte (GFP) à des souris sauvages soumises à unrégime riche en graisse, montrent l’apparition d’adipocytesGFP positifs dans les tissus adipeux. Ces deux études sug-gèrent que certaines cellules progénitrices du tissu adipeuxpourraient avoir une origine sanguine/moelle osseuse.Nos expériences montrent que les cellules CD34+/CD31–circulantes sanguines ne présentent cependant pas lesmêmes potentialités que les cellules CD34+/CD31– dutissu adipeux humain, i.e. production de colonies de cellulessanguines mais absence de différenciation adipocytaire[38], ce qui suggère que si ces cellules ont la même ori-

76
Cah. Nutr. Diét., 42, 2, 2007
biologie générale
gine, le micro-environnement dans lequel elles résidentdéfinit leur potentialité et leur spécificité. Des étudescomplémentaires sont donc nécessaires pour mieux définirl’influence du micro-environnement sur la potentialité descellules souches/progénitrices.
Conclusion
De nombreuses données obtenues
in vitro
et
in vivo
montrent la présence au sein des TAs humains de popu-lations cellulaires présentant des capacités similaires auxcellules progénitrices/souches. La capacité multiple de dif-férenciation reste soumise à controverse et nécessite desrecherches supplémentaires pour statuer clairement. Entreautres, des méthodes standardisées et validées d’isolement,de caractérisation, de sélection, de purification, de culture,de stockage et d’injection des cellules à partir du TA humaindoivent être développées. Les cellules souches/progénitricesdu TA humain pourraient, de part leur accessibilité et leurnombre au sein de la FSV, devenir un modèle cellulaireintéressant pour des approches de thérapie cellulaire pourla régénération et la réparation tissulaire [58, 59]. Toute-fois, de nombreux challenges scientifiques restent encoreà être réalisés avant d’envisager de telles approches. Lesmécanismes d’infiltration et d’adressage des cellules doiventêtre mieux caractérisés ainsi que le rôle relatif de la diffé-renciation, fusion et production de facteurs paracrines dansles effets réparateurs de ces cellules.
Résumé
De nombreux laboratoires ont montré que les cellules issuesde la fraction stroma-vasculaire des tissus adipeux humainspouvaient exprimer
in vitro
selon les conditions de culturedes marqueurs biochimiques de lignages cellulaires variés :adipogénique, ostéogénique, chondrogénique, myogénique,endothélial, neuronal et épithélial. De plus, diverses appro-ches
in vivo
ont montré la capacité réparatrice des cellulesde la fraction stroma-vasculaire du tissu adipeux humaindans des modèles animaux d’ischémie vasculaire et cardiaqueou de dommages osseux et musculaires. Ces résultats sug-gèrent fortement que le tissu adipeux humain contient descellules qui présentent des propriétés similaires à cellesdes cellules souches ou progénitrices. Toutefois, la natureexacte de ces cellules, leur potentialité réelle de différen-ciation
in vivo
ainsi que les mécanismes impliqués dansleur capacité de réparation et de régénération restent à êtrecaractérisés pour envisager leur utilisation dans des appro-ches de régénération et réparation tissulaire.
Mots-clés :
Cellule progénitrice – Préadipocyte – Cellulesendothéliales – Réparation – Régénération.
Abstract
Several laboratories have shown that the cells from thestroma-vascular fraction of the human adipose tissueexpress, depending on cell culture conditions, bioche-mical markers of multiple cell lineages including adipo-genic, osteogenic, chondrogenic, myogenic, endothelial,neuronal and epithelial cell lineage. Furthermore, various
in vivo approaches revealed the ability of the cells deri-ved from the stroma-vacsular fraction of adipose tissueto repair ischemic or damaged tissues. Altogether thesedata strongly suggest that the stroma-vascular fractionof the human adipose tissue contains cells that exhibitproperties like stem/progenitor cells. However, the exactnature of the cells, their potentiality to lead to diffe-rentiated cells in vivo as well as the mechanisms involvedin their repair capability remain to be characterized toconsider their use in regenerative and reparative medi-cine.
Key-words:
Progenitor – Preadipocyte – Endothelial cell– Repair – Regeneration.
Bibliographie
[1] Lakshmipathy U., Verfaillie C. – Stem cell plasticity.
BloodRev.
, 2005,
19
, 29-38.[2] Pessina A., Gribaldo L. – The key role of adult stem cells:
therapeutic perspectives.
Curr. Med. Res. Opin.
, 2006,
22
,2287-2300.
[3] Pittenger M.F., Mackay A.M., Beck S.C.
et al
. – Multili-neage potential of adult human mesenchymal stem cells.
Science
, 1999,
284
, 143-147.[4] Jiang Y., Jahagirdar B.N., Reinhardt R.L.
et al.
– Pluri-potency of mesenchymal stem cells derived from adultmarrow.
Nature
, 2002,
418
, 41-49.[5] Asahara T., Murohara T., Sullivan A.
et al.
– Isolation ofputative progenitor endothelial cells for angiogenesis.
Science
, 1997,
275
, 964-967.[6] Toma J.G., Akhavan M., Fernandes K.J.
et al.
– Isolationof multipotent adult stem cells from the dermis of mam-malian skin.
Nat. Cell. Biol.
, 2001,
3
, 778-784.[7] Jackson K.A., Mi T., Goodell M.A. – Hematopoietic
potential of stem cells isolated from murine skeletal muscle.Proc.
Natl. Acad. Sci. USA
, 1999,
96
, 14482-14486.[8] Bjornson C.R., Rietze R.L., Reynolds B.A., Magli M.C.,
Vescovi A.L. – Turning brain into blood: a hematopoieticfate adopted by adult neural stem cells in vivo.
Science
,1999,
283
, 534-537.[9] Zuk P.A., Zhu M., Mizuno H.
et al.
– Multilineage cells fromhuman adipose tissue: implications for cell-based thera-pies.
Tissue Eng.
, 2001,
7
, 211-228.[10] Bjorntorp P., Gustafson A., Persson B. – Adipose tissue
fat cell size and number in relation to metabolism in endo-genous hypertriglyceridemia.
Acta Med. Scand.
, 1971,
190
, 363-367.[11] Sjostrom L., Smith U., Krotkiewski M., Bjorntorp P. – Cel-
lularity in different regions of adipose tissue in young menand women.
Metabolism
, 1972,
21
, 1143-1153.[12] Bjorntorp P., Karlsson M., Pertoft H., Pettersson P., Sjos-
trom L., Smith U. – Isolation and characterization of cellsfrom rat adipose tissue developing into adipocytes.
J. LipidRes.
, 1978,
19
, 316-324.[13] Deslex S., Negrel R., Vannier C., Étienne J., Ailhaud G.
– Differentiation of human adipocyte precursors in a che-mically defined serum-free medium.
Int. J. Obes.
, 1987,
11
, 19-27.[14] Hauner H., Entenmann G., Wabitsch M.
et al.
– Promo-ting effect of glucocorticoids on the differentiation ofhuman adipocyte precursor cells cultured in a chemicallydefined medium.
J. Clin. Invest.
, 1989,
84
, 1663-1670.[15] Vannier C., Gaillard D., Grimaldi P.
et al
. – Adiposeconversion of ob17 cells and hormone-related events.
Int.J. Obes.
, 1985,
9
(Suppl. 1), 41-53.[16] Choi Y.S., Cha S.M., Lee Y.Y., Kwon S.W., Park C.J.,
Kim M. – Adipogenic differentiation of adipose tissue

Cah. Nutr. Diét., 42, 2, 2007
77
biologie générale
derived adult stem cells in nude mouse.
Biochem. Bio-phys. Res. Commun.
, 2006,
345
, 631-637.[17] Gimble J., Guilak F. – Adipose-derived adult stem cells:
isolation, characterization, and differentiation potential.
Cytotherapy
, 2003, 5, 362-369.[18] Halvorsen Y.D., Franklin D., Bond A.L. et al. – Extracel-
lular matrix mineralization and osteoblast gene expressionby human adipose tissue-derived stromal cells. Tissue Eng.,2001, 7, 729-741.
[19] Erickson G.R., Gimble J.M., Franklin D.M., Rice H.E.,Awad H., Guilak F. – Chondrogenic potential of adiposetissue-derived stromal cells in vitro and in vivo. Biochem.Biophys. Res. Commun., 2002, 290, 763-769.
[20] Estes B.T., Wu A.W., Guilak F. – Potent induction ofchondrocytic differentiation of human adipose-derivedadult stem cells by bone morphogenetic protein 6. Arth-ritis Rheum., 2006, 54, 1222-1232.
[21] Zuk P.A., Zhu M., Ashjian P. et al. – Human adipose tis-sue is a source of multipotent stem cells. Mol. Biol. Cell.,2002, 13, 4279-4295.
[22] Rodriguez L.V., Alfonso Z., Zhang R., Leung J., Wu B.,Ignarro L.J. – Clonogenic multipotent stem cells inhuman adipose tissue differentiate into functional smoothmuscle cells. Proc. Natl. Acad. Sci. USA, 2006, 103,12167-12172.
[23] Guilak F., Lott K.E., Awad H.A. et al. – Clonal analysisof the differentiation potential of human adipose-derivedadult stem cells. J. Cell. Physiol., 2006, 206, 229-237.
[24] Planat-Benard V., Silvestre J.S., Cousin B. et al. – Plas-ticity of human adipose lineage cells toward endothelialcells: physiological and therapeutic perspectives. Circula-tion, 2004, 109, 656-663.
[25] Miranville A., Heeschen C., Sengenes C., Curat C.A.,Busse R., Bouloumie A. – Improvement of postnatal neo-vascularization by human adipose tissue-derived stemcells. Circulation, 2004, 110, 349-355.
[26] Martinez-Estrada O.M., Munoz-Santos Y., Julve J.,Reina M., Vilaro S. – Human adipose tissue as a sourceof Flk-1+ cells: new method of differentiation and expan-sion. Cardiovasc. Res., 2005, 65, 328-333.
[27] Brzoska M., Geiger H., Gauer S., Baer P. – Epithelial dif-ferentiation of human adipose tissue-derived adult stem cells.Biochem. Biophys. Res. Commun., 2005, 330, 142-150.
[28] Talens-Visconti R., Bonora A., Jover R. et al. – Hepato-genic differentiation of human mesenchymal stem cellsfrom adipose tissue in comparison with bone marrowmesenchymal stem cells. World J. Gastroenterol., 2006,12, 5834-5845.
[29] Timper K., Seboek D., Eberhardt M. et al. – Human adi-pose tissue-derived mesenchymal stem cells differentiateinto insulin, somatostatin, and glucagon expressing cells.Biochem. Biophys. Res. Commun., 2006, 341, 1135-1140.
[30] Gaustad K.G., Boquest A.C., Anderson B.E., Gerdes A.M.,Collas P. – Differentiation of human adipose tissue stemcells using extracts of rat cardiomyocytes. Biochem. Bio-phys. Res. Commun., 2004, 314, 420-427.
[31] Rodriguez A.M., Pisani D., Dechesne C.A. et al. – Trans-plantation of a multipotent cell population from humanadipose tissue induces dystrophin expression in the immu-nocompetent mdx mouse. J. Exp. Med., 2005, 201,1397-1405.
[32] Planat-Benard V., Menard C., Andre M. et al. – Sponta-neous cardiomyocyte differentiation from adipose tissuestroma cells. Circ. Res., 2004, 94, 223-229.
[33] Cousin B., Andre M., Arnaud E., Penicaud L., Casteilla L.– Reconstitution of lethally irradiated mice by cells isolatedfrom adipose tissue. Biochem. Biophys. Res. Commun.,2003, 301, 1016-1022.
[34] Charriere G., Cousin B., Arnaud E. et al. – Preadipocyteconversion to macrophage. Evidence of plasticity. J. Biol.Chem., 2003, 278, 9850-9855.
[35] Javazon E.H., Beggs K.J., Flake A.W. – Mesenchymalstem cells: paradoxes of passaging. Exp. Hematol.,2004, 32, 414-425.
[36] McIntosh K., Zvonic S., Garrett S. et al. – The immuno-genicity of human adipose-derived cells: temporal chan-ges in vitro. Stem Cells, 2006, 24, 1246-1253.
[37] Mitchell J.B., McIntosh K., Zvonic S. et al. – Immunophe-notype of human adipose-derived cells: temporal changesin stromal-associated and stem cell-associated markers.Stem Cells, 2006, 24, 376-385.
[38] Sengenes C., Lolmede K., Zakaroff-Girard A., Busse R.,Bouloumie A. – Preadipocytes in the human subcutaneousadipose tissue display distinct features from the adultmesenchymal and hematopoietic stem cells. J. Cell. Phy-siol., 2005, 205, 114-122.
[39] Bouloumie A., Curat C.A., Sengenes C., Lolmede K.,Miranville A., Busse R. – Role of macrophage tissue infil-tration in metabolic diseases. Curr. Opin. Clin. Nutr.Metab. Care, 2005, 8, 347-354.
[40] Yanez R., Lamana M.L., Garcia-Castro J., Colmenero I.,Ramirez M., Bueren J.A. – Adipose tissue-derived mesen-chymal stem cells have in vivo immunosuppressive pro-perties applicable for the control of the graft-versus-hostdisease. Stem Cells, 2006, 24, 2582-2591.
[41] Puissant B., Barreau C., Bourin P. et al. – Immunomo-dulatory effect of human adipose tissue-derived adult stemcells: comparison with bone marrow mesenchymal stemcells. Br. J. Haematol., 2005, 129, 118-129.
[42] Niemeyer P., Kornacker M., Mehlhorn A. et al. – Compa-rison of Immunological Properties of Bone Marrow Stro-mal Cells and Adipose Tissue-Derived Stem Cells Beforeand After Osteogenic Differentiation in Vitro. Tissue Eng.,2007, 13, 111-121.
[43] Meyerrose T.E., De Ugarte D.A., Hofling A.A. et al. – Invivo Distribution of Human Adipose-Derived MSC. StemCells, 2007, 25, 220-227.
[44] Kim D.H., Je C.M., Sin J.Y., Jung J.S. – Effect of partialhepatectomy on in vivo engraftment after intravenousadministration of human adipose tissue stromal cells inmouse. Microsurgery, 2003, 23, 424-431.
[45] Moon M.H., Kim S.Y., Kim Y.J. et al. – Human adiposetissue-derived mesenchymal stem cells improve postnatalneovascularization in a mouse model of hindlimb ische-mia. Cell. Physiol. Biochem., 2006, 17, 279-290.
[46] Rehman J., Traktuev D., Li J. et al. – Secretion of angio-genic and antiapoptotic factors by human adipose stromalcells. Circulation, 2004, 109, 1292-1298.
[47] Sumi M., Sata M., Toya N., Yanaga K., Ohki T., Nagai R.– Transplantation of adipose stromal cells, but not matureadipocytes, augments ischemia-induced angiogenesis.Life Sci., 2007, 80, 559-565.
[48] Hattori H., Masuoka K., Sato M. et al. – Bone formationusing human adipose tissue-derived stromal cells and abiodegradable scaffold. J. Biomed. Mater. Res. B. Appl.Biomater., 2006, 76, 230-239.
[49] Peterson B., Zhang J., Iglesias R. et al. – Healing of cri-tically sized femoral defects, using genetically modifiedmesenchymal stem cells from human adipose tissue. Tis-sue Eng., 2005, 11, 120-129.
[50] Miyahara Y., Nagaya N., Kataoka M. et al. – Monolayeredmesenchymal stem cells repair scarred myocardium aftermyocardial infarction. Nat. Med., 2006, 12, 459-465.
[51] Bacou F., el Andalousi R.B., Daussin P.A. et al. – Trans-plantation of adipose tissue-derived stromal cells increasesmass and functional capacity of damaged skeletal muscle.Cell Transplant., 2004, 13, 103-111.
[52] Conejero J.A., Lee J.A., Parrett B.M. et al. – Repair ofpalatal bone defects using osteogenically differentiated fat-

78 Cah. Nutr. Diét., 42, 2, 2007
biologie générale
derived stem cells. Plast. Reconstr. Surg., 2006, 117,857-863.
[53] Marx J. – Cancer research. Mutant stem cells may seedcancer. Science, 2003, 301, 1308-1310.
[54] Rubio D., Garcia-Castro J., Martin M.C. et al. – Sponta-neous human adult stem cell transformation. Cancer Res.,2005, 65, 3035-3039.
[55] Conboy I.M., Conboy M.J., Wagers A.J., Girma E.R,Weissman I.L., Rando TA. – Rejuvenation of aged proge-nitor cells by exposure to a young systemic environment.Nature, 2005, 433, 760-764.
[56] Hong K.M., Burdick M.D., Phillips R.J., Heber D., Strie-ter R.M. – Characterization of human fibrocytes as circu-
lating adipocyte progenitors and the formation of humanadipose tissue in SCID mice. Faseb, 2005, 19, 2029-2031.
[57] Crossno J.T., Majka S.M., Grazia T., Gill R.G., Klemm D.J.– Rosiglitazone promotes development of a novel adipocytepopulation from bone marrow-derived circulating proge-nitor cells. J. Clin. Invest., 2006, 116, 3220-3228.
[58] Rodriguez A.M., Elabd C., Amri E.Z., Ailhaud., Dani C.– The human adipose tissue is a source of multipotentstem cells. Biochimie, 2005, 87, 125-128.
[59] Casteilla L., Planat-Benard V., Cousin B. et al. – Plasticityof adipose tissue: a promising therapeutic avenue in thetreatment of cardiovascular and blood diseases? Arch.Mal. Cœur Vaiss., 2005, 98, 922-926.

Afin d’isoler et de caractériser la population de cellules progénitrices présentes dans
la FSV, notre groupe a développé une technique d’isolement des différentes populations
cellulaires de la FSV qui se fonde sur l’utilisation de nanoparticules magnétiques couplées à
des anticorps dirigés spécifiquement contre les marqueurs de surface CD34 (marqueur des
cellules souches hématopoïétiques et des cellules endothéliales capillaires), CD31 (marqueur
des cellules endothéliales et de la lignée hématopoïétique) et CD14 (marqueur des
monocytes/macrophages). Nous avons ainsi démontré que les cellules positives pour le CD34
et négatives pour le CD31 (CD34+/CD31-) sont la seule population cellulaire présente dans
la FSV capable de présenter la capacité de différenciation adipocytaire en condition
adipogénique (241). De plus, notre groupe a récemment mis en évidence que cette population
cellulaire CD34+/CD31- possède, en milieu endothélial, la capacité d’exprimer des
marqueurs endothéliaux et de s’organiser en structures semblables à des capillaires (173). Ces
résultats indiquent que la fraction cellulaire CD34+/CD31- de la FSV du TA humain, n’ayant
subi aucune étape d’extension in vitro, présente des propriétés de cellules progénitrices
capables de donner naissance à des cellules qui expriment des marqueurs phénotypiques des
adipocytes et des cellules endothéliales. Il reste cependant à déterminer si cette population
cellulaire est constituée d’un mélange de cellules progénitrices unipotentes ou de cellules
souches multipotentes. D’autre part et de façon inattendue, les cellules CD34+/CD31-
présentent des caractéristiques différentes des cellules souches mésenchymateuses et
hématopoïétiques puisqu’elles ne sont capables ni d’exprimer le marqueur CD105, propre
aux cellules souches mésenchymateuses, ni douées d’activité hématopoïétique, en conditions
de culture adaptées (241). Ainsi, l’origine mésenchymateuse des précurseurs adipocytaires
communément admise jusqu’ici est remise en question. Toutefois, il est important de noter
que la quantité de cellules souches mésenchymateuses dans la moelle osseuse est très faible
et que leur isolement requiert des phases d’expansion in vitro (210). Ainsi, aucune étude n’a
pu pour l’instant être réalisée sur des cellules souches mésenchymateuses fraîchement
isolées. L’expression de nombreux marqueurs de surface est modulée par la culture et l’état
de prolifération des cellules (112). Ainsi, l’expression du marqueur CD34 est connue pour
être diminuée alors que celle de CD105 est augmentée par l’état de prolifération cellulaire
(170, 173, 174, 241). On peut donc penser que l’expression du CD34 des cellules
progénitrices CD34+/CD31- du TA et l’absence du marqueur CD105 est relative au fait que
ces cellules n’ont pas subi de phases d’expansion. Il est à noter que certaines équipes
suggèrent que les précurseurs adipocytaires pourraient être originaires d’une
dédifférenciation des adipocytes matures (5, 186, 211). Cependant, peu de données sont en
21

accord avec cette hypothèse et l’on peut supposer que cette dédifférenciation apparente des
adipocytes pourrait être due à une contamination des adipocytes matures par des cellules
progénitrices. En effet, les études de Miyazaki et al. montrent qu’en condition de culture
flottantes (c’est-à-dire quand la surface cellulaire adhérente se trouve au-dessus des cellules),
des progéniteurs adipocytaires, fixés sur les adipocytes, sont capables de migrer sur la surface
de culture et de se différencier en adipocytes (175).
B.2) Le processus de différentiation adipocytaire
B.2.1) Les étapes du processus de différenciation adipocytaire
La différenciation adipocytaire proprement dite ou adipogenèse représente le passage
du préadipocyte à l’adipocyte mature. Après une phase de croissance exponentielle, le
précurseur déterminé vers la lignée adipocytaire ou adipoblaste marque un arrêt de croissance
à confluence (figure 5). Ces cellules deviennent alors des préadipocytes et s’engagent dans
l’adipogenèse proprement dite. Tout au long du processus d’adipogenèse vont se succéder
différents événements morphologiques, biochimiques et moléculaires, que l’on regroupe en
deux catégories, les événements précoces et tardifs.
Les événements précoces concernent en particulier le remodelage de la matrice
extracellulaire (MEC) et du cytosquelette (figure 6). Au cours de la différenciation
adipocytaire, la composition de la MEC est fortement modifiée. D’une structure riche en
fibronectine, sa composition va évoluer vers une structure de type lame basale, composée
essentiellement de laminine, collagène IV et d’entactine (151). En effet, une diminution de la
production du collagène fibrillaire de type I et III et de la fibronectine et une augmentation de
la production de la laminine, des protéoglycans, de l’entactine et du collagène de type IV ont
été observés au cours de la différenciation des préadipocytes d’origine murine et bovine (132,
133, 184). Des cultures de préadipocytes murins, porcins et humains réalisées sur différents
supports matriciels ont souligné la variation d’adipogenèse selon le type de matrice utilisé
suggérant le rôle essentiel de la composition et de l’organisation de la MEC dans la
différenciation adipocytaire (95, 98, 133). Ainsi, l’ensemble de ces données suggère
l’importance du remodelage de la MEC durant le processus adipogénique. Grâce à un
remodelage matriciel, les tensions avec le cytosquelette sont modifiées. Des changements
dans la forme de la cellule sont possibles par une réorganisation du réseau intracellulaire
d’actine (252). L’ensemble de ces remodelages matriciels, du cytosquelette et de la forme de
22

Figure 6 : Evolution de la matrice extracellulaire et de la forme cellulaire au cours de la différenciation adipocytaire
D’après Lilla
J, Am J Path, 2002
FibronectineCollagène I, III
Collagène IV et Entactine
Structure fibroblastique des préadipocytes
Confluence et engagement des préadipocytes
Remodelage matriciel via des protéases
Remodelage du cytosqueletteet transcription de gènes
adipogéniques
(C/EBP et PPARγ)
Synthèse de la MEC par les préadipocytes
au coursde la différenciation
Adipocytes matures
C/EBPPPARγ
Synthèse d’aP2, LPL, FAS …

la cellule permet alors aux préadipocytes de subir une étape d’expansion clonale liée à une
série de réplications de l’ADN ou mitoses dites post-confluentes.
A la suite de ces événements précoces, les préadipocytes sont dits « engagés dans la
différenciation adipocytaire ». En anglais, on parle encore de « commitment » des
préadipocytes. Enfin, une étape de maturation terminale permet d’obtenir les adipocytes
matures. Cette maturation terminale est marquée par l’acquisition des propriétés
métaboliques et sécrétoires des adipocytes matures (91).
B.2.2) Contrôle transcriptionnel de la différenciation adipocytaire
Les changements morphologiques majeurs qui accompagnent la différenciation
adipocytaire, tels que l’arrondissement des cellules et l’accumulation progressive de vacuoles
lipidiques dans le cytoplasme, sont la conséquence de variation d’expression d’un très grand
nombre de gènes. Cette reprogrammation génétique profonde est sous le contrôle de facteurs
de transcription pro- ou anti-adipogéniques dont les principaux sont les facteurs PPARs
(Peroxisome proliferator-activated receptors), les C/EBPs (CCAAT/enhancer binding
proteins) et le facteur ADD1/SREBP-1c (Adipocyte determination and differentiation factor
1/sterol regulatory element binding protein-1c) (pour revue (74, 226).
B.2.2.1) Les PPARs
Les facteurs de transcription PPARs appartiennent à la superfamille des récepteurs
nucléaires. Une fois activés par la liaison à leur ligand, ils forment des hétérodimères avec les
récepteurs de l’acide 9-cis-rétinoïque (RXR) qui reconnaissent spécifiquement des éléments
de réponse PPRE (« PPAR Responsive Element ») situés dans les régions promotrices de
gènes cibles. Chez les mammifères, trois isoformes de PPARs α, β (ou δ ou NUC-1 (nuclease
abnormal-1)), et γ ont été décrites, chacun codé par un gène différent et caractérisés par une
distribution tissulaire, des ligands, des co-facteurs et des fonctions qui leur sont propres. De
multiples études démontrent que PPARγ, et plus particulièrement PPARγ2, est
préférentiellement exprimée dans les adipocytes et joue un rôle majeur dans le contrôle
transcriptionnel de la maturation terminale de l’adipocyte. En effet, il transactive de
nombreux gènes marqueurs de la différenciation adipocytaire, tels que celui de la LPL ou
celui codant pour la protéine de transport aux acides gras (aP2) (228). Plusieurs ligands
capables d’activer le PPARγ majorent le taux de conversion adipocytaire et l’amplitude de la
23

différenciation. C’est notamment le cas des thiazolidinediones qui sont utilisées dans le
traitement du diabète et de l’insulino-résistance. De plus, la 15-déoxy-δ prostanglandine et
l’acide lysophosphatidique (LPA) sont décrits comme des activateurs du facteur de
transcription (23, 171). Toutefois leur affinité pour le PPARγ est beaucoup plus faible que
celle habituellement décrite pour les récepteurs nucléaires avec leurs ligands physiologiques.
B.2.2.2) Les C/EBPs
Les facteurs C/EBPs appartiennent à la famille des facteurs de transcription à motifs
basiques et glissières de leucines (bHLH, « basic Helix Loop Helix »). Le domaine basique
permet à ces facteurs de se lier à l’ADN alors que le motif en glissières de leucines leur
permet de s’homo- ou de s’hétéro-dimériser. Trois isoformes de C/EBPs, α, β et γ, possèdent
un rôle inducteur dans l’adipogenèse (227, 228). Les C/EBPs β et γ sont induits de façon très
précoce et transitoire au cours de l’adipogenèse, et jouent un rôle clé dans l’enclenchement de
la maturation terminale. L’induction de C/EBPα se fait plus tardivement et permet de
transactiver différents gènes marqueurs de la différenciation adipocytaire tels qu’aP2, PPARγ
et la leptine mais également d’induire sa propre expression (227, 228). Récemment, des
travaux ont montré que des souris déficientes pour CHOP-10 (CCAAT/enhancer binding
protein (C/EBP) homologous protein), un facteur de transcription anti-adipogénique
appartenant à la famille des C/EBPs présentent une adiposité importante mais aucun désordre
métabolique associé (6). Cet effet semble impliquer les facteurs C/EBPs.
Les nombreuses données expérimentales obtenues depuis ces dernières années et sur
les lignées murines principalement, ont permis de proposer un modèle de progression des ces
facteurs de transcription au cours de l’adipogenèse (figure 7) (7).
B.2.2.3) Le facteur SREBP-1
Le facteur ADD1/SREBP-1 fait partie de la famille des facteurs à motifs hélice-
boucle-hélice (bHLH-LZ pour basic helix-loop-helix-leucine zipper). Le domaine HLH
permet à ces facteurs de former des homo- ou hétero-dimères avec des protéines de la même
famille. Après dimérisation, le complexe se lie par le domaine basique à des séquences
spécifiques dites E-box, situées au niveau des promoteurs de gènes cibles. De nombreuses
études ont montré l’implication de SREBP-1 au niveau hépatique et adipocytaire dans la
24
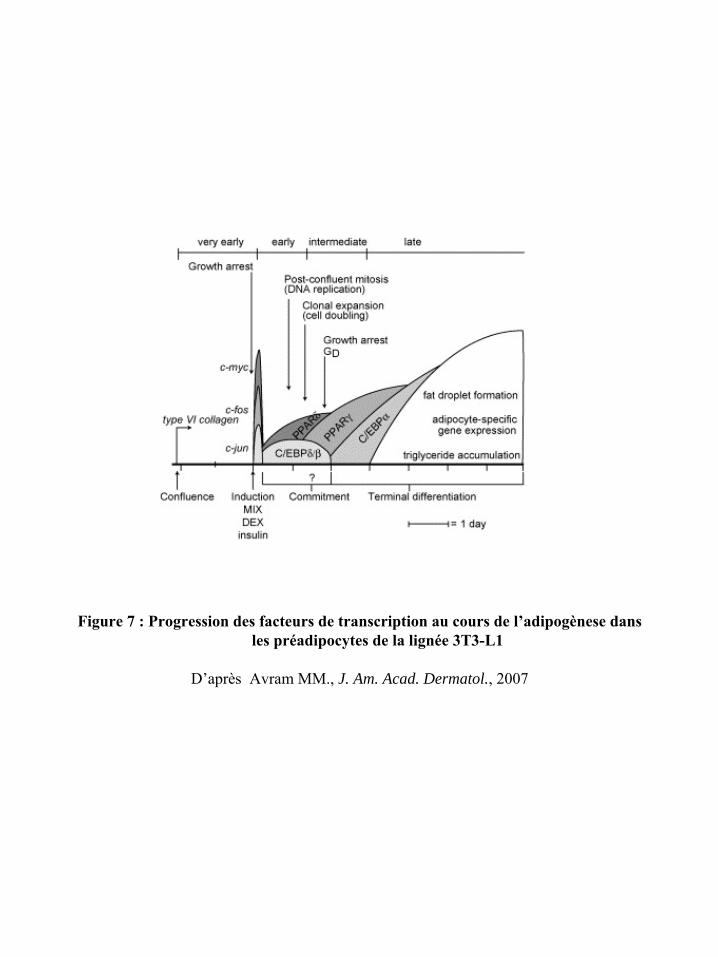
Figure 7 : Progression des facteurs de transcription au cours de l’adipogènese
dans les préadipocytes
de la lignée 3T3-L1
D’après Avram
MM., J. Am. Acad. Dermatol., 2007

régulation de nombreux gènes impliqués dans le métabolisme du cholestérol, des TG et des
acides gras (65, 103). Au niveau du TA, il favorise l’adipogenèse en induisant l’expression de
PPARγ (73, 122).
B.2.2.4) Les autres facteurs de transcription
En dehors de ces intervenants essentiels dans l’adipogenèse, d’autres facteurs
transcriptionnels aux fonctions pro- ou anti-adipogéniques constituent potentiellement des
régulateurs importants de la différenciation adipocytaire. Ces différents facteurs sont
identifiés et présentés dans la figure 8 (d’après revue (78)). Notons que les facteurs de
transcription Kruppel-like factor 15 et 5 (KLF15 et 5), appartenant à la famille des facteurs à
doigts de zinc, sont décrits comme étant pro-adipogéniques alors que KLF2 est anti-
adipogénique (177, 194, 292). Le facteur Krox 20 apparaît comme le facteur le plus précoce
induit directement par les signaux hormonaux (39). De manière intéressante, le facteur de
transcription CREB (cAMP-response-element binding protein) a été impliqué dans
l’accumulation des TGs dans des préadipocytes murins soumis à une dysfonction
mitochondriale par un traitement avec l’antimycin A (272).
B.2.3) Régulation hormonale et intracellulaire de la différenciation adipocytaire
Le processus d’adipogenèse que nous venons d’aborder est hautement contrôlé par de
nombreux facteurs de nature très variée: hormones, cytokines, nutriments et molécules de
signalisation cellulaire. Les principaux facteurs connus sont présentés dans le tableau 2 et
classés selon leur effet pro- ou anti-adipogénique.
25
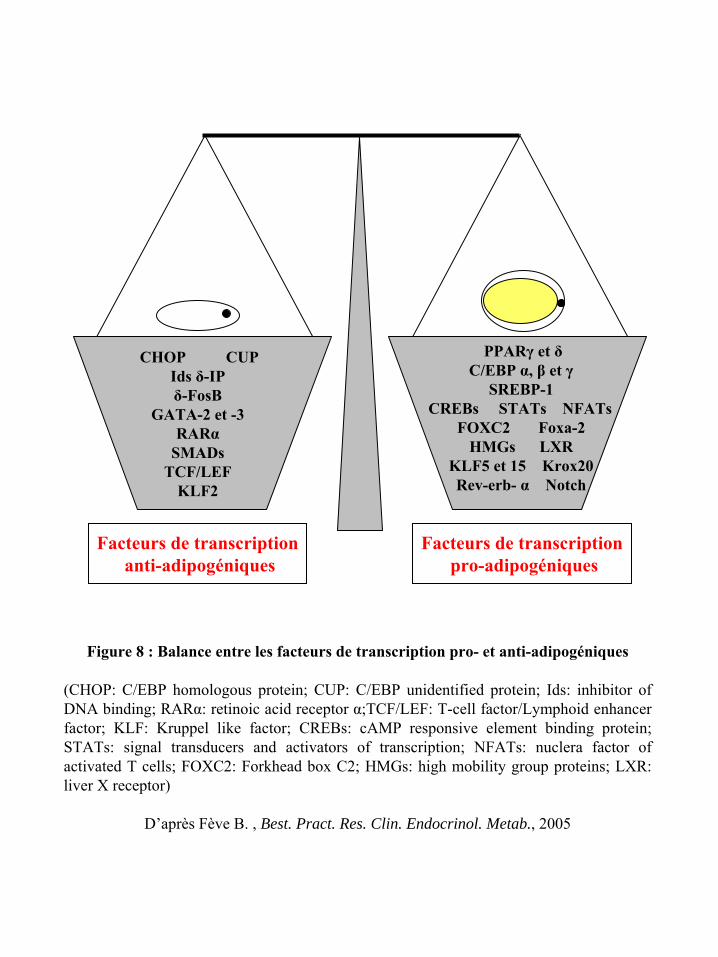
CHOP CUPIds
δ-IPδ-FosB
GATA-2 et -3RARα
SMADsTCF/LEF
KLF2
PPARγ
et δC/EBP α, β
et γSREBP-1
CREBs
STATs
NFATsFOXC2 Foxa-2
HMGs
LXRKLF5 et 15 Krox20Rev-erb-
α
Notch
Facteurs de transcriptionpro-adipogéniques
Facteurs de transcriptionanti-adipogéniques
Figure 8 : Balance entre les facteurs de transcription pro-
et anti-adipogéniques
(CHOP: C/EBP homologous
protein; CUP: C/EBP unidentified
protein; Ids: inhibitor
of
DNA binding; RARα: retinoic
acid
receptor
α;TCF/LEF: T-cell
factor/Lymphoid
enhancer
factor; KLF: Kruppel
like
factor; CREBs: cAMP
responsive element
binding
protein;
STATs: signal transducers
and
activators
of
transcription; NFATs: nuclera
factor
of
activated
T cells; FOXC2: Forkhead
box C2; HMGs: high
mobility
group proteins; LXR: liver
X receptor)
D’après Fève B. , Best. Pract. Res. Clin. Endocrinol. Metab., 2005
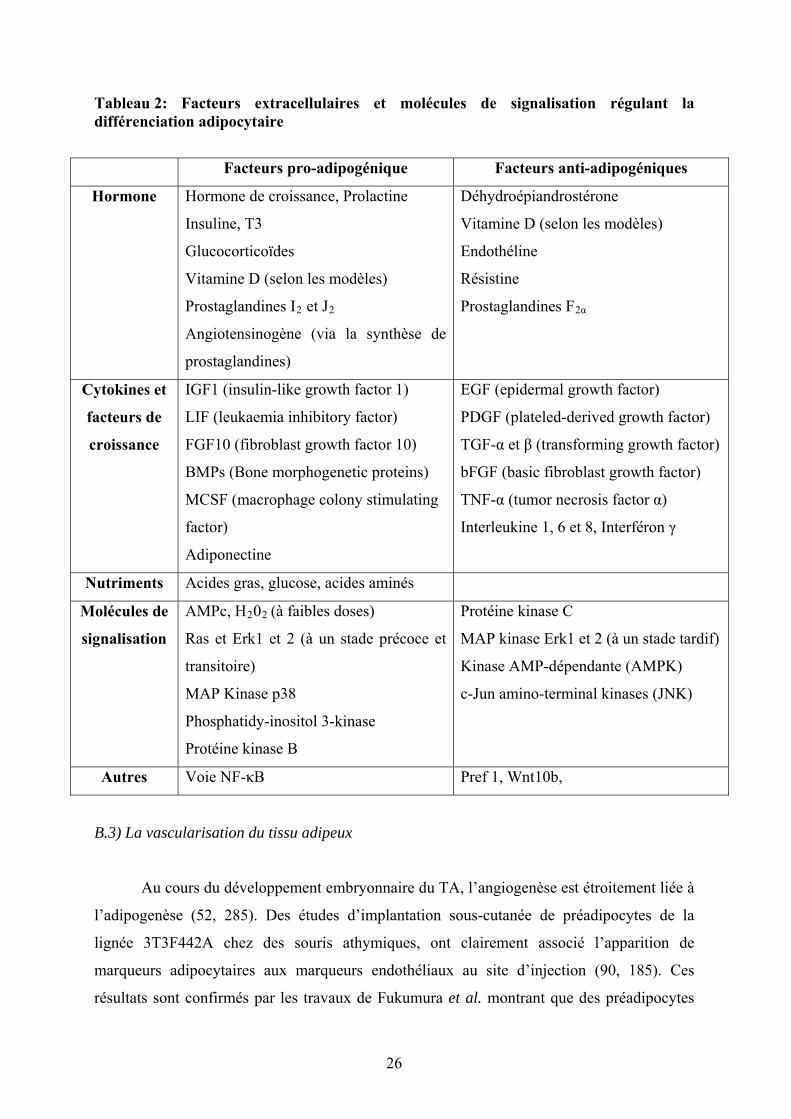
Tableau 2: Facteurs extracellulaires et molécules de signalisation régulant la différenciation adipocytaire
Facteurs pro-adipogénique Facteurs anti-adipogéniques
Hormone Hormone de croissance, Prolactine
Insuline, T3
Glucocorticoïdes
Vitamine D (selon les modèles)
Prostaglandines I2 et J2
Angiotensinogène (via la synthèse de
prostaglandines)
Déhydroépiandrostérone
Vitamine D (selon les modèles)
Endothéline
Résistine
Prostaglandines F2α
Cytokines et
facteurs de
croissance
IGF1 (insulin-like growth factor 1)
LIF (leukaemia inhibitory factor)
FGF10 (fibroblast growth factor 10)
BMPs (Bone morphogenetic proteins)
MCSF (macrophage colony stimulating
factor)
Adiponectine
EGF (epidermal growth factor)
PDGF (plateled-derived growth factor)
TGF-α et β (transforming growth factor)
bFGF (basic fibroblast growth factor)
TNF-α (tumor necrosis factor α)
Interleukine 1, 6 et 8, Interféron γ
Nutriments Acides gras, glucose, acides aminés
Molécules de
signalisation
AMPc, H202 (à faibles doses)
Ras et Erk1 et 2 (à un stade précoce et
transitoire)
MAP Kinase p38
Phosphatidy-inositol 3-kinase
Protéine kinase B
Protéine kinase C
MAP kinase Erk1 et 2 (à un stade tardif)
Kinase AMP-dépendante (AMPK)
c-Jun amino-terminal kinases (JNK)
Autres Voie NF-κB Pref 1, Wnt10b,
B.3) La vascularisation du tissu adipeux
Au cours du développement embryonnaire du TA, l’angiogenèse est étroitement liée à
l’adipogenèse (52, 285). Des études d’implantation sous-cutanée de préadipocytes de la
lignée 3T3F442A chez des souris athymiques, ont clairement associé l’apparition de
marqueurs adipocytaires aux marqueurs endothéliaux au site d’injection (90, 185). Ces
résultats sont confirmés par les travaux de Fukumura et al. montrant que des préadipocytes
26

transfectés par un dominant négatif de PPARγ empêchant l’adipogenèse, inhibe
l’angiogenèse au site d’implantation. En parallèle, l’inhibition de l’angiogenèse par un
traitement avec un anticorps neutralisant anti-VEGF-R2, inhibe l’adipogenèse (83). Ainsi,
adipogenèse et angiogenèse sont donc deux phénomènes étroitement liés.
Le rôle de l’endothélium dans le développement du TA est reconnu depuis longtemps
(285). Cependant, il a été négligé par le fait que le TA était généralement considéré comme
peu vascularisé. Cette observation est essentiellement liée à la difficulté d’accès aux
capillaires du TA, du fait de la grande taille des adipocytes matures et de leur organisation en
couches multiples. En effet, l’analyse du TA après une période de jeûne chez l’animal qui
s’accompagne d’une diminution du diamètre des adipocytes a permis de mettre en évidence
l’ensemble du réseau vasculaire de ce tissu (225). Ainsi, en évaluant la densité capillaire du
TA ou en marquant les érythrocytes au chrome 51, des études ont montré que le réseau
vasculaire du TA est aussi important que celui du muscle squelettique et que chaque
adipocyte présente des contacts étroits avec au moins un capillaire sanguin (94, 107).
L’importance de l’endothélium pourrait en partie s’expliquer par le rôle clé de la surface
endothéliale dans le contrôle de la lipogenèse adipocytaire via la LPL, enzyme sécrétée par
les adipocytes et active à la surface des cellules endothéliales. En effet, les cellules
endothéliales semblent jouer un rôle direct dans les phénomènes de trafic intercellulaire de la
LPL par la sécrétion d’un facteur qui, sous stimulation de l’insuline, libèrerait la LPL des
protéoglycans à héparane sulfate adipocytaires (126).
En résumé, les lignées cellulaires murines ont permis une étude approfondie des
processus de détermination et de différenciation adipocytaires. Le processus de
détermination adipocytaire est cependant peu connu contrairement à la différenciation
adipocytaire qui a suscité de nombreuses études. Par ailleurs, la mise en évidence
récente de cellules souches multipotentes au sein de la FSV permet au TA d’acquérir un
nouveau statut de « source d’approvisionnement de cellules souches/progénitrices »
pour d’éventuelles thérapies cellulaires. Cependant, de nombreux points restent encore
à élucider, notamment déterminer si la FSV du TA renferme un pool de cellules souches
aux potentialités variées ou bien une seule et unique cellule multipotente. De même,
malgré des similitudes, il est nécessaire de déterminer si les cellules
souches/progénitrices issues du TA adulte sont identiques aux préadipocytes d’origine
embryonnaire et si des différences inter-espèces sont observées (hommes vs rongeurs).
27

C) Le développement excessif du tissu adipeux : l’obésité
L’indice de masse corporelle (IMC), rapport du poids sur la taille au carré, permet
d’évaluer la corpulence d’un individu adulte. Le tableau 3 présente la classification des
adultes de poids normal, en surpoids, obèse ou maigre (sous-poids).
Tableau 3: Classification des adultes maigres, normaux, en surpoids et obèses selon l’IMC
Classification IMC (kg/m2)
Sous-poids
Maigreur sévère
Maigreur modérée
Maigreur légère
< 18,50
< 16,00
16,00 - 16,99
17,00 - 18,49
Poids normal 18,50 - 24,99
Surpoids
Pré-obèse
≥ 25,00
25,00 - 29,99
Obésité
Obésité de classe I
Obésité de classe II
Obésité de classe III
≥ 30,00
30,00 - 34,99
35,00 - 39,99
≥ 40,00 Classification définie par l’organisation mondiale de la Santé D’après WHO expert consultation, The Lancet, 2004,.(48)
L’obésité correspond à une surcharge pondérale résultant d’un développement
excessif du TA. Les potentialités d’extension du TA sont uniques. En effet, la masse grasse
peut-être multipliée par quatre chez un individu obèse et peut représenter jusqu'à 60 à 70% de
sa masse corporelle.
De nombreux événements cellulaires et biochimiques sont responsables de la
croissance excessive du TA adulte. Dans les paragraphes suivants, nous allons voir les
modifications du TA lors de l’obésité, aussi bien dans sa composition cellulaire que dans ses
fonctions métaboliques et sécrétoires.
28

C.1) Les modifications cellulaires du tissu adipeux
C.1.1) Les adipocytes et les cellules souches/progénitrices
Les changements dans la masse du TA sont associés à des modifications de la taille et
du nombre des adipocytes (214). L’augmentation du volume des adipocytes correspond à une
hypertrophie adipocytaire. Le diamètre moyen d’un adipocyte peut aller jusqu’à 120µm chez
le sujet obèse. Selon l’hypothèse dite de la « taille critique », la cellule adipeuse se chargerait
en TGs jusqu'à atteindre une taille critique au-delà de laquelle une phase d’hyperplasie
apparente apparaîtrait au sein du tissu. Ces nouveaux adipocytes proviendraient de la
prolifération et/ou du recrutement de cellules progénitrices associés à leur différenciation. Les
études de Crossno et al montrent que des souris soumises à un régime gras sont capables de
recruter des cellules souches de la moelle osseuse dans le TA afin de former de nouveaux
adipocytes (54), cependant les mécanismes impliqués ne sont pas caractérisés. Ainsi deux
phases adipocytaires auraient lieu dans le TA une phase d’hypertrophie cellulaire suivie dans
les cas d’obésité sévère d’une hyperplasie adipocytaire (15, 249). Les mécanismes
responsables de l’hypertrophie adipocytaire s’expliquent par des altérations des activités
métaboliques (lipogenèse et lipolyse) des adipocytes. Nous le verrons dans le chapitre
suivant. L’augmentation du nombre d’adipocytes est moins bien définie. Récemment, les
travaux de Tchoukalova et al. indiquent que la proportion des préadipocytes de la FSV,
positifs pour le marqueur aP2 et négatifs pour le CD68 (un marqueur des macrophages), est
diminuée avec l’obésité (262). Ces travaux peuvent être associés à ceux de van Harmelen
indiquant que la capacité de différenciation adipocytaire des cellules de la FSV est
négativement corrélée avec l’IMC (271).
C.1.2) Les cellules endothéliales
Des travaux de notre groupe indiquent que l’évolution du réseau vasculaire est
parallèle à celui de la masse adipeuse suggérant que le développement du TA est associé à
une néovascularisation. En effet, le pourcentage de cellules endothéliales des capillaires
sanguins, caractérisées par la double expression CD34 et CD31, au sein de la FSV, reste
constant avec l’augmentation de l’IMC (173). La dépendance du TA vis-à-vis de son réseau
vasculaire au cours de l’obésité a été mise en évidence grâce à de récents travaux montrant
que des facteurs anti-angiogéniques limitent le développement de la masse grasse dans
29

différents modèles de souris obèses (19, 233). De plus, l’apoptose sélective des cellules
endothéliales, in vivo par l’utilisation d’un phage contenant un peptide pro-apoptotique
associé à la prohibitine, une protéine membranaire de surface, entraîne une réversion de
l’obésité chez la souris (129). Ainsi, le réseau vasculaire joue un rôle déterminant dans la
croissance mais également dans le maintien de la masse grasse. Les mécanismes impliqués
dans l’extension du réseau vasculaire lors du développement du TA ne sont pas connus.
Cependant, de nombreuses évidences montrent que les adipocytes sécrètent de nombreux
facteurs angiogéniques (16, 17, 154, 301) et que cette production est augmentée avec l’état
d’obésité (247). De plus, on peut supposer que les cellules CD34+/CD31- pourraient
également via des processus de vasculogenèse, i.e. différenciation en cellules endothéliales,
participer à l’augmentation du résseau vasculaire du TA. Récemment, une étude réalisée sur
un modèle de souris obèse db/db a évalué simultanément l’angiogenèse et l’adipogenèse et a
montré une étroite relation spatiale et temporelle entre la formation de nouveaux vaisseaux
sanguins à partir de vaisseaux préexistants et le processus de différenciation adipocytaire
(189).
C.1.3) Les macrophages
La présence de macrophages au sein du tissu adipeux a été mise en évidence depuis
longtemps par les histologistes (263) mais n’a suscité que très peu d’intérêt jusqu'à ces
dernières années. Des observations récentes suggèrent l’implication de cette population
cellulaire dans l’installation d’un état pro-inflammatoire, au sein du TA, lors de son
développement excessif. En effet dans différents modèles animaux d’obésités, une
accumulation de macrophages a été observée lors du développement de la masse grasse (286,
293). De plus, notre groupe a montré que le pourcentage de macrophages au sein de la SVF
est corrélé de façon positive avec l’IMC dans les tissus adipeux sous-cutanés (55) et
viscéraux (56) humains. Inversement, l’équipe de K. Clément a montré que la quantité de
macrophages infiltrés dans le TA est diminuée après une perte de poids chez des sujets
obèses (24, 46).
L’origine de cette augmentation de la population macrophagique au sein du TA des
sujets obèses n’est pas bien comprise. De par les similitudes d’expression génique entre
adipocytes et macrophages et l’observation des capacités de phagocytose décrites dans le
préadipocyte humain, il a été suggéré que les macrophages puissent provenir de la
différenciation des préadipocytes (235). Cependant notre équipe n’a pu mettre en évidence
30

d’activité hématopoïétique à partir de précurseurs adipocytaires humains (241). Des
expériences de transplantation de moelle osseuse chez la souris démontrent que 80% des
macrophages du TA sont d’origine médullaire, suggérant ainsi que les macrophages du TA
sont issus de l’infiltration de monocytes sanguins circulants (286). De plus, notre groupe a
montré que les sécrétions adipocytaires stimulent l’activation endothéliale conduisant à une
augmentation de l’adhérence et de la transmigration des monocytes sanguins à travers
l’endothélium du TA humain (55). En conditions normales, la fonction majeure des
macrophages résidents au sein d’un tissu est d’éliminer le matériel étranger et les cellules
apoptotiques, processus essentiel dans le maintien de l’homéostasie tissulaire. Le rôle des
macrophages au niveau du TA lui-même reste à être clairement défini. Toutefois, des travaux
récents chez la souris indiquent que l’infiltration des macrophages due à une obésité induite
par un régime hypercalorique semble être plus liée à l’insulino-résistance associée à l’obésité
qu’à l’augmentation de la masse grasse (155, 192, 286).
C.2) Les modifications des fonctions métaboliques du tissu adipeux
Au cours de l’obésité, une augmentation de la lipogenèse semble évidente. Quelques
études ont rapporté que l’activité de la LPL était associée à la taille des adipocytes (66, 75).
Un excès de masse grasse s’accompagne également d’une altération de la fonction lipolytique
du TA au repos et au cours de l’exercice physique (102). Plusieurs travaux in vitro menés au
laboratoire ont évalué l’effet de l’obésité et du surpoids sur la régulation adrénergique de la
lipolyse. Le nombre de récepteurs α2A-adrénergiques a été corrélé positivement avec la taille
des adipocytes. Ainsi, les récepteurs α2A-adrénergiques sont fortement exprimés dans le TA
sous-cutané où les adipocytes sont hypertrophiés, et bien plus dans le tissu fémoral
qu’abdominal suggérant une faible activité lipolytique de l’adrénaline dans le TA abdominal
voire antilipolytique dans le TA fémoral chez la femme (13, 166). Chez le sujet obèse, les
réponses lipolytiques mesurées in situ dans le TA sous-cutané, sont très diminuées par
rapport à des sujets de poids normal au cours d’un exercice physique. Ceci s’explique par une
activation soutenue des récepteurs α2A-adrénergiques, surnuméraires dans les adipocytes
hypertrophiés de l’obèse (255). Par ailleurs, de nombreux travaux mentionnent une perte de
sensibilité du TA sous-cutané aux catécholamines au repos, ce qui entraîne une diminution
des réponses β-adrénergiques. Cette résistance aux catécholamines dans l’adipocyte d’obèse
pourrait provenir d’une diminution de la fonction β2-adrénergique (70), d’une diminution de
l’activité et de l’expression de la LHS (143), ou encore de l’expression majoritaire d’une
31
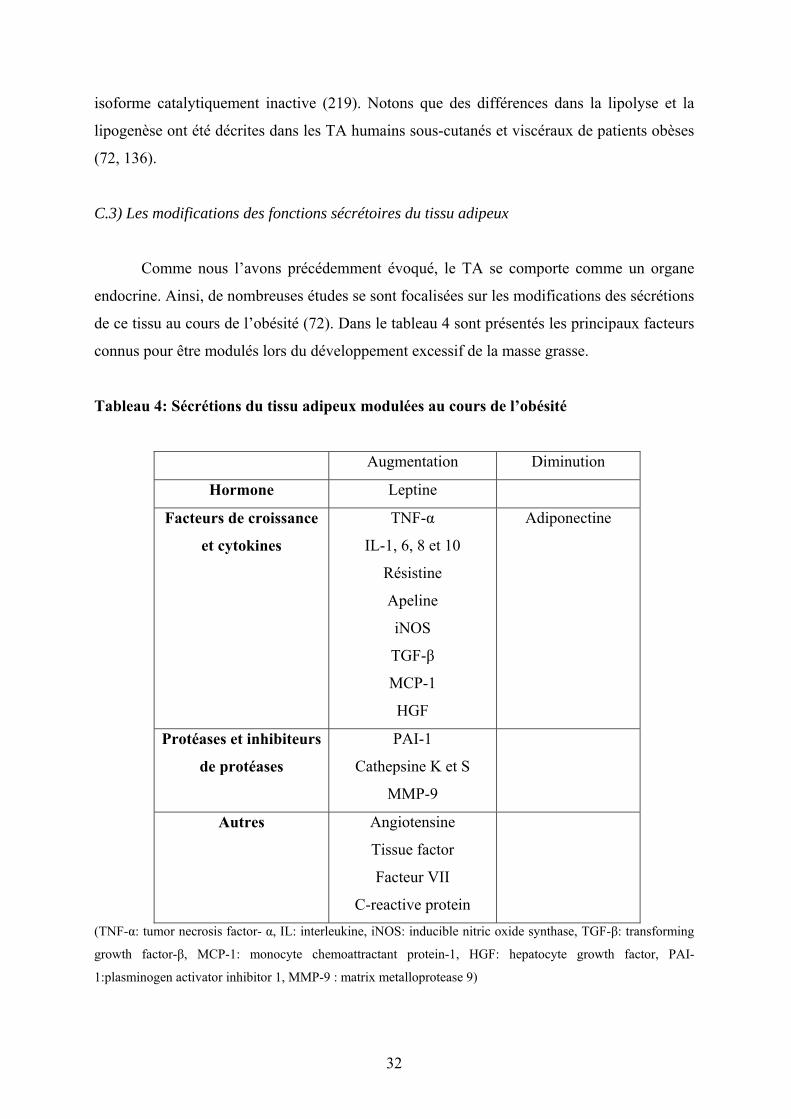
isoforme catalytiquement inactive (219). Notons que des différences dans la lipolyse et la
lipogenèse ont été décrites dans les TA humains sous-cutanés et viscéraux de patients obèses
(72, 136).
C.3) Les modifications des fonctions sécrétoires du tissu adipeux
Comme nous l’avons précédemment évoqué, le TA se comporte comme un organe
endocrine. Ainsi, de nombreuses études se sont focalisées sur les modifications des sécrétions
de ce tissu au cours de l’obésité (72). Dans le tableau 4 sont présentés les principaux facteurs
connus pour être modulés lors du développement excessif de la masse grasse.
Tableau 4: Sécrétions du tissu adipeux modulées au cours de l’obésité
Augmentation Diminution
Hormone Leptine
Facteurs de croissance
et cytokines
TNF-α
IL-1, 6, 8 et 10
Résistine
Apeline
iNOS
TGF-β
MCP-1
HGF
Adiponectine
Protéases et inhibiteurs
de protéases
PAI-1
Cathepsine K et S
MMP-9
Autres Angiotensine
Tissue factor
Facteur VII
C-reactive protein
(TNF-α: tumor necrosis factor- α, IL: interleukine, iNOS: inducible nitric oxide synthase, TGF-β: transforming
growth factor-β, MCP-1: monocyte chemoattractant protein-1, HGF: hepatocyte growth factor, PAI-
1:plasminogen activator inhibitor 1, MMP-9 : matrix metalloprotease 9)
32

En résume, l’obésité chez l’homme s’accompagne d’un remodelage important de la
masse grasse associé à des modulations des fonctions métaboliques et sécrétoires du TA.
Les mécanismes responsables de ces changements ne sont pas totalement définis mais
ces altérations semblent être impliquées directement dans la genèse des pathologies
associées à l’obèsité. Ainsi l’augmentation de la population macrophagique
inflammatoire et les changements dans la production d’adipokines dont principalement
la leptine et l’adiponectine sont deux éléments clés dans le développement de l’insulino-
résistance et de la dysfonction endothéliale, évènements précoces impliqués dans le
diabète de type II et les pathologies cardiovasculaires.
33

D) La régression du tissu adipeux : la lipoatrophie
En plus de ces capacités de croissance, le TA est également capable de régression. On
parle de lipoatrophie ou lipodystrophie. La classification des syndromes de lipoatrophies est
indiquée dans le tableau ci-dessous.
Tableau 5: Classification des différentes lipoatrophies humaines
D’après Reitman et al, TEM, 2000 et Garg et al, Clinical Cornestone, 2006
Classification des lipoatrophies humaines
1) Génétiques
- Lipodystrophie congénitale généralisée (Syndrome de Berardinelli-Seip)
Type 1 : mutations d’une acyltransférase codée par AGPAT2
Type 2 : mutations de la séipine
- Lipodystrophie familiale partielle
Type Dunnigan : mutations de la lamine A/C
Associée à PPARγ : mutations de PPARγ
Associée à Akt2/PKB : mutations de Akt/PKB
- Dysplasie mandibuloacrale
Type A, partielle, mutations de la lamine A/C
Type B, généralisée, mutations de ZMPSTE24 (une métalloprotéase à zinc)
- Syndromes progéroïdes (Syndrome de Werner, syndrome Cockayne)
- Syndrome de déficience en carbohydrate glycoprotéine
- Syndrome SHORT (Short stature, Hyperextensibility, Hernia, Ocular depression, Rieger
anormaly and Teething delay)
2) Présumées inflammatoires ou autoimmunes
- Lipoatrophie acquise généralisée (Syndrome de Lawrence)
- Lipoatrophie acquise partielle (Syndrome de Barraquer-Simons)
- Lipoatrophie acquise associée à des dermatomyosites
- Lipodystrophie associée à une hypocomplémentémie
3) Acquises, de causes inconnues
- Lipodystrophie associée aux traitements anti-VIH
34

Dans certains syndromes de lipoatrophie, l’absence localisée au niveau de la face et
des membres ou généralisée de TA est associée à une accumulation du TA au niveau des
régions abdominales, dorso-cervicales et mammaires. On parle dans ce cas de
lipodystrophies. Ces syndromes peuvent avoir des causes génétiques, immunes ou associées
aux traitements thérapeutiques lors d’infection en particulier pour le VIH (pour revue (25,
222). Paradoxalement elles sont, comme l’obésité, associées à des complications
métaboliques telles que l’hypertriglycéridémie, l’insulino-résistance, la stéatose hépatique et
les pathologies cardio-vasculaires.
Les syndromes lipoatrophiques étaient très rares (moins de 1 cas pour 100 000)
jusqu’à ces dix dernières années où sont apparues les formes liées aux traitements
antirétroviraux de l’infection par le VIH. Ce sont les inhibiteurs de la protéase du VIH (IPs-
VIH) qui ont été initalement associés à l’émergence de ce syndrome (31, 33, 34). Cependant,
des travaux démontrent que des patients séropositifs recevant des inhibiteurs nucléosidiques
de la reverse transcriptase du VIH en particulier la stavudine et la zidovudine (INRTs) et
naïfs pour les IPs-VIH développent également une lipoatrophie périphérique (20, 32, 157).
Par ailleurs, il a été rapporté que ces traitements pourraient agir en synergie entraînant une
atrophie profonde du TA (158, 269).
Au cours de ce paragraphe, nous allons faire le point sur les modifications cellulaires,
métaboliques et sécrétoires du TA lors d’une régression importante. Nous nous appuierons
essentiellement sur les données des lipoatrophies associées aux traitements anti-VIH qui sont
actuellement les lipoatrophies les plus fréquentes et les plus étudiées (276).
D.1) Les modifications cellulaires du tissu adipeux
Les études réalisées sur le TA sous-cutané des patients traités par les IPs-VIH et les
INRTs montrent une atteinte profonde de la morphologie du TA (62, 110, 152). Les effets
décrits des IP-VIH et des INRTs sur les différents types cellulaires qui composent le TA vont
être abordé dans les paragraphes suivants.
D.1.1) Les adipocytes
Une perte de poids est forcément associée à une diminution de la taille et/ou du
nombre des adipocytes. Si l’existence d’une différenciation adipocytaire après les phases de
développement normal est maintenant connue, la possibilité d’une perte des adipocytes est
35

moins bien documentée. Les études sur l’apoptose dans le TA sont peu abondantes (250).
Cependant, il est admis que l’apoptose peut se produire dans le TA et notamment dans les
adipocytes matures. Un certain nombre de facteurs ont été rapportés pour entraîner l’apoptose
des adipocytes in vitro, notamment des cytokines inflammatoires telles que le TNFα (213),
des flavonoïdes et des acides gras polyinsaturés (121, 188).
Une augmentation de l’apoptose des adipocytes a été décrite chez des patients infectés
par le VIH et ayant développé une lipoatrophie du TA suite aux traitements antirétroviraux
(62, 63, 110). Il est admis que les traitements antirétroviraux influent sur ce processus
apoptotique. En effet, il a été rapporté que le nelfinavir (NFV), un IP-VIH, favorise
l’apoptose des préadipocytes différenciés murins des lignées 3T3-L1 et 3T3F442A (12, 64).
Notons que les mécanismes directement mis en jeu ne sont pas encore définis. Par ailleurs,
ces effets apoptotiques ne sont pas retrouvés dans les préadipocytes murins non différenciés
(64). Le NFV a également été rapporté pour induire la nécrose des préadipocytes différenciés
de la lignée 3T3F442A via une augmentation des espèces réactives de l’oxygène (278)
associée à un stress du réticulum endoplasmique (264).
Concernant les INRTs, une étude indique que la stavudine (un INRT) entraîne une
apoptose des préadipocytes différenciés murins de la lignée 3T3F442A (28). De plus, des
patients traités avec la stavudine ont une augmentation de l’apoptose dans le TA mise en
évidence par la technique TUNEL (terminal deoxynucleotidyl transferase dUTP digoxigenin
nick-end labeling) (168). Il semblerait que l’apoptose adipocytaire induite par les INRTs soit
due à la genèse d’une toxicité mitochondriale. En effet, le TA des patients infectés par le VIH
et traités avec les INRTs, dont principalement la stavudine, présente des dysfonctionnements
mitochondriaux tels qu’une baisse des taux d’ADN mitochondrial (ADNmt) et des anomalies
de structure des mitochondries (169, 281). De plus, le remplacement de la stavudine par
d’autres INRTs tels que l’abacavir ou la zidovudine améliore les taux d’ADNmt et prévient
l’apoptose (168). Ces phénomènes peuvent s’expliquer par le fait que les INRTs pourraient
inhiber, outre la réverse transcriptase du VIH, d’autres ADN polymérases et notamment,
l’ADN polymérase mitochondriale γ. Cette polymérase est impliquée dans la réplication de
l’ADNmt et son inhibition conduit à une déplétion des taux d’ADNmt et/ou à des altérations
de la structure génomique de l’ADNmt (245) provoquant ainsi une augmentation des espéces
réactives de l’oxygène et l’apoptose cellulaire. L’apoptose des adipocytes joue donc un rôle
important dans l’apparition du syndrome de lipoatrophie associée aux traitements
antirétroviraux. Les travaux de Pajvani et al. ont permis de montrer par l’utilisation de souris
transgéniques surexprimant de manière conditionnelle la caspase 8, une enzyme pro-
36

apoptotique, spécifiquement dans les adipocytes, l’expression du transgène étant sous le
contrôle du promoteur d’aP2, que le déclenchement de l’apotose adipocytaire par activation
de la caspase 8 s’accompagnait d’un phénotype lipoatrophique. Ces travaux démontrent ainsi
que l’induction et le maintien d’une apoptose adipocytaire pourraient contribuer à la genèse
des lipoatrophies (201, 266)
D.1.2) Les cellules progénitrices/ souches
Outre les effets des composés antirétroviraux sur l’apoptose adipocytaire, de
nombreux travaux montrent que ces composés altèrent également le processus de
différenciation adipocytaire. De nombreuses études ont évalué l’effet des IPs-VIH tels que le
saquinavir (SQV), le nelfinavir (NFV) et l’indinavir (IDV) sur la différenciation des
préadipocytes murins des lignées 3T3-L1 et 3T3F442A et ont montré que ces inhibiteurs de
protéases limitent leur différenciation adipocytaire (64, 144, 176, 300). Chez l’homme, il
existe peu de données concernant les effets des IPs-VIH sur la différenciation adipocytaire.
Deux études réalisées sur des cellules de la FSV du TA humain d’origine mammaire et des
cellules souches mésenchymateuses humaines confirment les effets anti-adipogéniques de
l’IDV et du SQV (109, 288). A l’inverse, les effets des INRTs sur la différenciation des
préadipocytes murins ne sont pas clairs. Certaines études rapportent que ces inhibiteurs
entraînent une diminution de l’adipogenèse (224) et d’autres n’observent aucun effet (28,
198). Récemment un inhibiteur non nucléosidique de la reverse transcriptase du VIH
(INNRT), l’efavirenz, a également fait l’objet d’une étude in vitro. Il a été observé que cet
INNRT inhibe la différenciation des préadipocytes murins de la lignée 3T3-L1 et humains
(68) suggérant que la troisième classe de médicaments utilisée pour le traitement du VIH
contribue à l’altération de la masse grasse. In vivo, sur des biopsies de TA sous-cutané
provenant de patients infectés par le VIH et ayant développé une atrophie adipocytaire, suite
au traitement antirétroviral, des altérations des taux de CEBP/α, de PPARγ et de SREBP-1
ont également été décrites (10). Ces résultats suggèrent que les IPs-VIH altèrent les processus
de différenciation adipocytaire dans les étapes précoces. Cependant, les cibles exactes de ces
composés ne sont pas encore totalement définies.
L’effet anti-adipogénique des IPs-VIH pourrait tout d’abord s’expliquer par des
interactions avec la voie de signalisation de l’acide rétinoïque. En effet, la région catalytique
de la protéase du VIH qui lie les IPs-VIH a environ 60% d’homologie avec CRABP-1
(cytoplasmic retinoic acid binding protein-1), un régulateur intracellulaire des taux d’acide
37

rétinoïque. Ainsi, les IPs-VIH peuvent inhiber la fonction de CRABP-1 et limiter le processus
d’adipogenèse qui nécessite l’expression des gènes dépendants de l’acide rétinoïque (34). De
plus, les IPs-VIH ont été également décrits pour être des inhibiteurs du cytochrome P450 3A,
l’unique enzyme connue pour convertir l’acide rétinoïque en acide cis-9-rétinoïque, le ligand
du RXR (Retinoïd X Receptor) qui par la suite s’hétérodimérise avec PPARγ (34).
Cependant, il faut rester prudent quant à ces données puisque des études ont rapporté que des
altérations du facteur de transcription PPARγ n’expliquent pas la diminution, induite par les
IPs-VIH, de la différenciation des préadipocytes murins et humains (144, 288, 300).
Des interactions entre SREBP-1 et les IPs-VIH ont également été décrites. Ainsi, le
traitement des préadipocytes de la lignée 3T3F442A par l’IDV et le NFV s’accompagne
d’une séquestration du facteur de transcription SREBP-1 au niveau de l’enveloppe nucléaire
des préadipocytes contrairement aux préadipocytes contrôles qui accumulent SREBP-1 au
niveau nucléaire (30). Le mécanisme responsable de cette séquestration de SREBP-1
consécutive à un traitement par les IPs-VIH n’est pas totalement élucidé. Cependant, certains
travaux suggèrent un rôle clé de la lamine A, protéine majeure de l’enveloppe nucléaire. Le
facteur SREBP-1 est synthétisé sous forme d’un précurseur inactif ancré au niveau du
réticulum endoplasmique ou de l’enveloppe nucléaire puis maturé par clivage protéolytique.
La forme mature active de SREBP-1 peut ensuite être transférée dans le noyau et activer
directement la transcription génique (21, 22, 284). Une interaction entre SREBP-1 et la
lamine A/C a déjà été décrite (153). La lamine A est synthétisée sous forme de prélamine A
et sa maturation est assurée par clivage à l’aide d’une métalloprotéinase, FACE chez
l’homme et Zmpste24 chez la souris. Les souris déficientes pour Zmpste24 présentent une
lipoatrophie associée à une accumulation de prélamine A au niveau de l’enveloppe nucléaire
(206). Le rôle clé de la lamine A et de la régulation de l’interaction lamineA/SREBP-1 dans
le contrôle de la différenciation adipocytaire est de plus souligné par les syndromes majeurs
de lipoatrophies génétiques qui sont dus à des mutations de la lamine A. Enfin, récemment il
a été montré que les IPs-VIH provoquaient une accumulation de prélamine A dans des
fibroblastes humains, suggérant que les IPs-VIH pourraient interagir avec FACE pour inhiber
la maturation de la lamine et empêcher l’accès du noyau au facteur adipogénique SREBP-1
(27, 47).
Enfin, une interaction des IPs-VIH avec le protéasome qui constitue la principale
machinerie de dégradation protéique chez les eucaryotes (51) a été envisagée. Des études ont
rapporté que les IPs-VIH sont des inhibiteurs de l’activité protéasomique de certaines cellules
cancéreuses (239) mais aussi de préadipocytes murins de la lignée 3T3-L1 (202). Or, les
38

inhibiteurs spécifiques du protéasome tels que la lactacystine inhibent la différenciation des
myoblastes (124), des ostéoclastes (299) mais aussi la différenciation des préadipocytes de la
lignée 3T3-L1 (212). Nous verrons dans le chapitre Résultats les données que nous avons
obtenues sur les interactions entre les IPs-VIH et le protéasome dans les préadipocytes
humains.
A ce jour, les mécanismes moléculaires des INRTs responsables de la baisse de
l’adipogenèse observée dans les travaux de Roche et al. sont inconnus. Etant donné l’action
des INRTs sur la fonction mitochondriale, il est tentant de spéculer que les INRTs peuvent
diminuer l’adipogenèse via une augmentation des espèces réactives de l’oxygène. En effet,
les espèces réactives de l’oxygène diminuent la prolifération et la différenciation adipocytaire
des lignées murines (35, 36).
Ainsi, l’ensemble de ces données suggère que la lipoatrophie associée au traitement
antirétroviral semble également être due à un défaut du processus de différenciation
adipocytaire. D’ailleurs, une étude récente implique majoritairement une altération de
l’adipogenèse plutôt qu’une induction de l’apoptose des adipocytes dans la lipoatrophie de
patients infectés par le VIH et sous tri-thérapie (181).
D.1.3) Les cellules endothéliales
Chez les patients infectés par le VIH et sous traitement antirétroviral, peu de données
concernent les cellules endothéliales. On peut noter que l’infection au VIH est associée à
l’activation de cellules endothéliales, observée par une augmentation plasmatique des
marqueurs cellulaires de surface tels que VCAM (Vascular Cell Adhesion Molecule), E-
selectin et ICAM1 (Intercellular Adhesion Molecule) chez des patients séropositifs (40). Par
ailleurs, les traitements antirétroviraux entraînent des altérations de la fonction endothéliale
chez des patients sains (244) et des dommages de l’ADNmt dans les cellules endothéliales
humaines (114). Enfin, il a été observé, comme pour les adipocytes, une apoptose des cellules
endothéliales chez des patients infectés par le VIH et sous traitement antirétroviral (62).
Cependant, une augmentation de la densité des vaisseaux du TA atrophié de patients infectés
par le VIH et traités a également été observée (110) et on peut se demander si cette
augmentation apparente n’est pas relative en fait à la diminution du nombre et du volume
adipocytaire. Toutefois, il récemment a été observé que les INRTs favorisent l’angiogenèse
des cellules endothéliales humaines in vitro (61). A ce jour, il est difficile de conclure sur
39

l’effet de la régression du TA au niveau des cellules endothéliales. D’autres investigations
sont nécessaires.
D.1.4) Les macrophages
Dans le TA sous-cutané de patients infectés par le VIH et sous tri-thérapie est
observée une infiltration macrophagique (10, 62, 110, 190) similaire à celle observée dans le
TA de patients obèses (24, 286). Cette accumulation de macrophages pourrait être due à
l’apoptose des adipocytes. En effet, les travaux de Cinti et al. indiquent la présence de
macrophages dans les TA de souris et d’humains obèses au niveau des adipocytes nécrosés
(45). De manière identique, dans le modèle de souris lipoatrophiques surexprimant la caspase
8 dans le TA, il a été rapporté une augmentation du nombre de macrophages à proximité des
adipocytes en cours d’apoptose (201).
D.2) Les modifications des fonctions métaboliques du tissu adipeux
Les traitements antirétroviraux affectent également les deux grandes fonctions
métaboliques du TA. Ainsi, des études ont mis en évidence que les IPs-VIH diminuent la
lipogenèse des préadipocytes murins différenciés (64, 137, 144). Ces effets semblent être dus
à une diminution de l’activité de la LPL et de la synthèse de novo de lipides (218). Etant
donné que les IPs-VIH ont la capacité d’interagir avec le transporteur du glucose GLUT-4, il
a été suggéré que les effets des IPs-VIH sur la synthèse de novo de lipides passe par une
inhibition de l’activité de GLUT-4 et du captage du glucose (99, 179, 180). Récemment, une
inhibition de la lipogenèse a également été observée sur des préadipocytes murins
différenciés et traités avec des INRTs mais aussi avec l’efavirenz, un INNRT (68, 137).
La lipolyse est également touchée par les traitements antirétroviraux puisqu’ils sont
associés à une augmentation des taux circulants d’AGNE (93). L’activation de la lipolyse
semble être due à une combinaison complexe entre l’infection au VIH et les traitements
associés. En effet, une augmentation de la lipolyse a été rapportée chez des patients infectés
par le VIH et ce indépendamment du type de thérapie (268). In vitro, de nombreuses études
ont mis en évidence que les IPs-VIH stimulent la lipolyse des préadipocytes différenciés
murins (111, 144, 218, 231). De plus, une interruption de la prise de ces inhibiteurs diminue
l’activation de la lipolyse et les taux d’AGNE chez les patients (268). Récemment, une étude
40
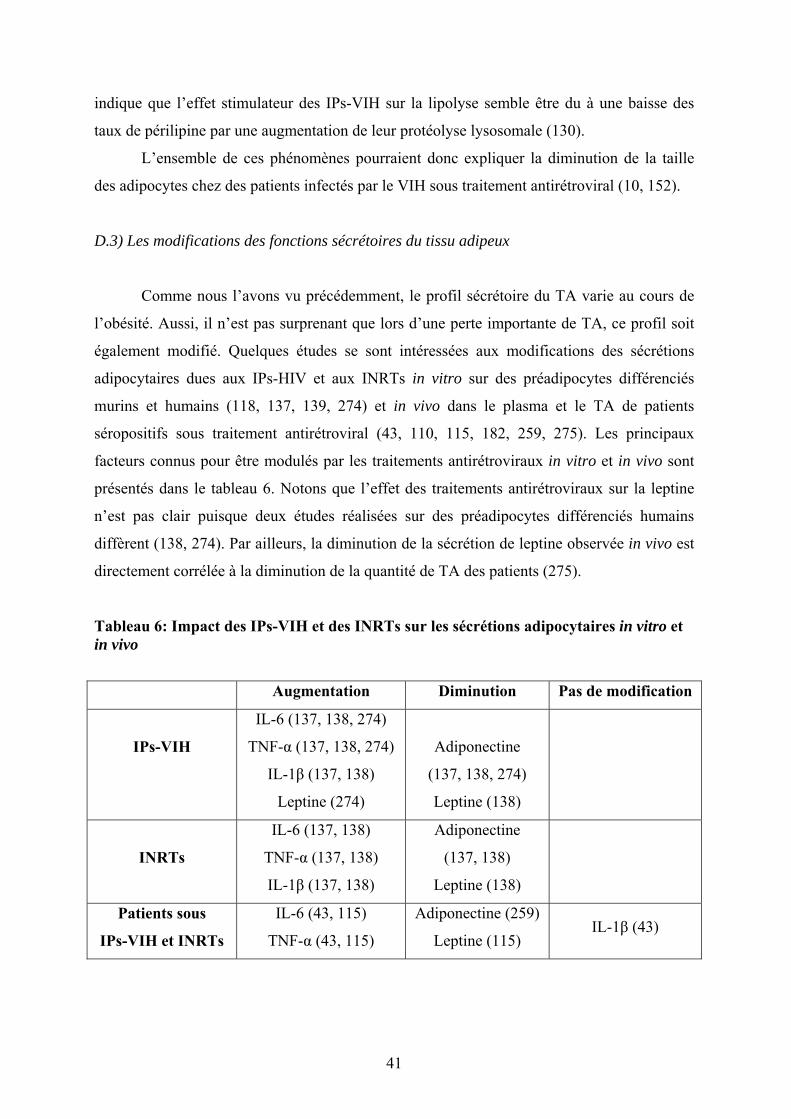
indique que l’effet stimulateur des IPs-VIH sur la lipolyse semble être du à une baisse des
taux de périlipine par une augmentation de leur protéolyse lysosomale (130).
L’ensemble de ces phénomènes pourraient donc expliquer la diminution de la taille
des adipocytes chez des patients infectés par le VIH sous traitement antirétroviral (10, 152).
D.3) Les modifications des fonctions sécrétoires du tissu adipeux
Comme nous l’avons vu précédemment, le profil sécrétoire du TA varie au cours de
l’obésité. Aussi, il n’est pas surprenant que lors d’une perte importante de TA, ce profil soit
également modifié. Quelques études se sont intéressées aux modifications des sécrétions
adipocytaires dues aux IPs-HIV et aux INRTs in vitro sur des préadipocytes différenciés
murins et humains (118, 137, 139, 274) et in vivo dans le plasma et le TA de patients
séropositifs sous traitement antirétroviral (43, 110, 115, 182, 259, 275). Les principaux
facteurs connus pour être modulés par les traitements antirétroviraux in vitro et in vivo sont
présentés dans le tableau 6. Notons que l’effet des traitements antirétroviraux sur la leptine
n’est pas clair puisque deux études réalisées sur des préadipocytes différenciés humains
diffèrent (138, 274). Par ailleurs, la diminution de la sécrétion de leptine observée in vivo est
directement corrélée à la diminution de la quantité de TA des patients (275).
Tableau 6: Impact des IPs-VIH et des INRTs sur les sécrétions adipocytaires in vitro et in vivo
Augmentation Diminution Pas de modification
IPs-VIH
IL-6 (137, 138, 274)
TNF-α (137, 138, 274)
IL-1β (137, 138)
Leptine (274)
Adiponectine
(137, 138, 274)
Leptine (138)
INRTs
IL-6 (137, 138)
TNF-α (137, 138)
IL-1β (137, 138)
Adiponectine
(137, 138)
Leptine (138)
Patients sous
IPs-VIH et INRTs
IL-6 (43, 115)
TNF-α (43, 115)
Adiponectine (259)
Leptine (115) IL-1β (43)
41

Les modifications importantes des sécrétions adipocytaires dues aux traitements
antirétroviraux in vitro et in vivo pourraient contribuer à l’atrophie du TA et aux
complications métaboliques qui lui sont associées. En effet, ces sécrétions peuvent altérer les
principales fonctions métaboliques des adipocytes ainsi que leur survie cellulaire. De plus,
certaines d’entre elles peuvent également moduler l’adipogenèse telles que le TNF-α et l’IL-6
(pour revue (89)).
Pour conclure, les lipoatrophies résultent donc d’une profonde altération de la
composition cellulaire et des grandes fonctions métaboliques et sécrétoires du TA. Suite
à un défaut du maintien de l’homéostasie énergétique lors d’une perte de TA, ces
phénomènes sont associés à des complications métaboliques similaires à celles
rapportées lors d’un excès de TA. A l’heure actuelle, quelques approches
thérapeutiques pour le traitement des lipoatrophies ont été développées
(thiazolidinedione, metformine) mais elles agissent plus sur les complications
métaboliques associées que sur l’altération du TA elle-même. Ainsi, une meilleure
connaissance des mécanismes impliqués dans la régression de la masse grasse est
nécessaire afin d’identifier de nouvelles cibles thérapeutiques.
En conclusion de cette première partie, le TA subit donc un remodelage important
lors d’un développement excessif (obésité) ou d’une régression (lipoatrophie) (pour
illustration figure 9). Ainsi, la modification de la composition cellulaire du TA et les
altérations des fonctions métaboliques et sécrétoires de ce tissu pourraient jouer un rôle clé
dans les pathologies associées à l’obésité et aux lipoatrophies telles que le diabète de type II
et les pathologies cardiovasculaires. Une meilleure compréhension des mécanismes
impliqués dans la perte et le gain de poids ainsi que la découverte de nouvelles cibles
thérapeutiques sont donc nécessaires.
Il est évident que la plasticité du TA est possible grâce à fort remodelage tissulaire, au
niveau cellulaire mais aussi au niveau matriciel. De nombreuses protéases ont été décrites
pour être impliquées dans la régulation du remodelage tissulaire. Toutefois, les MMPs sont
les acteurs majeurs du remodelage matriciel. Dans le chapitre suivant, nous aborderons la
classification, la structure ainsi que la régulation de l’activité des MMPs. Puis, les modes
d’action et les fonctions biologiques auxquels les MMPs sont associées seront présentés.
Enfin nous aborderons le rôle potentiel de ces enzymes dans la plasticité du TA.
42
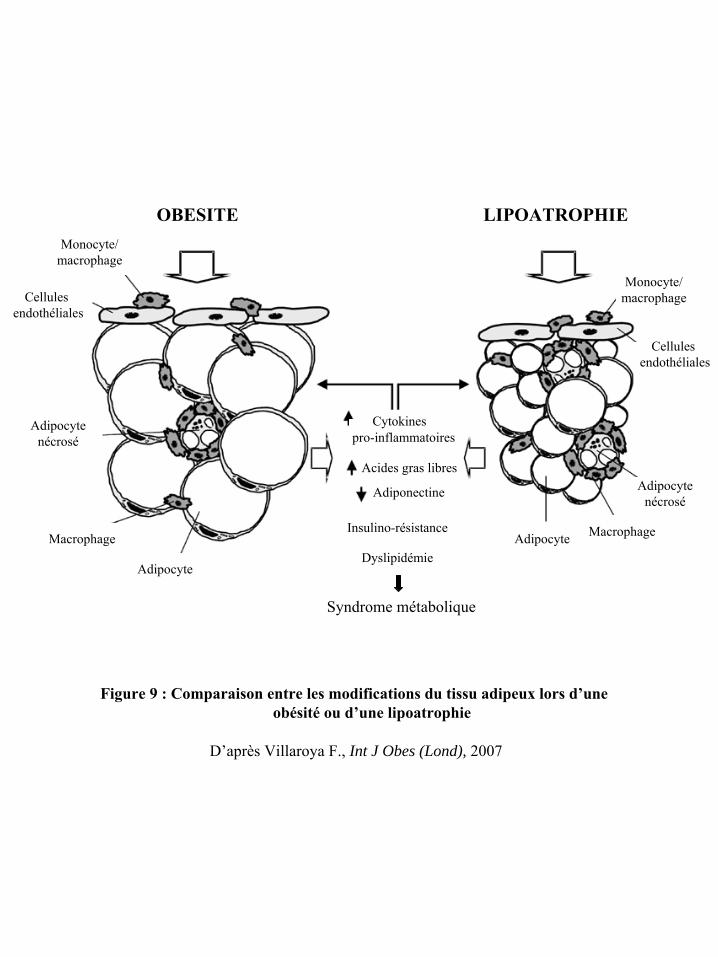
Figure 9 : Comparaison entre les modifications du tissu adipeux lors d’une obésité
ou d’une lipoatrophie
D’après Villaroya
F., Int J Obes (Lond), 2007
Monocyte/macrophage
Adipocyte
Macrophage
Adipocytenécrosé
Cellules endothéliales
OBESITE LIPOATROPHIE
Monocyte/macrophage
Adipocyte Macrophage
Adipocytenécrosé
Cellules endothéliales
Acides gras libres
Adiponectine
Insulino-résistance
Dyslipidémie
Syndrome métabolique
pro-inflammatoiresCytokines

II. Les métalloprotéases matricielles
Les MMPs font partie de la grande famille des metzincines. Elles sont impliquées
dans tous les processus nécessaires au développement et jouent un rôle majeur dans le
remodelage tissulaire via la dégradation de la MEC. Ainsi, elles sont associées à différentes
pathologies telles que l’arthrite rhumatoïde, les maladies cardiovasculaires et le
développement tumoral. Au cours de ce chapitre, nous allons nous intéresser aux MMPs en
général, à leur régulation, leur mode d’action et aux principales fonctions biologiques qui
leurs sont associées. Enfin, nous aborderons leur rôle dans le TA.
A) Classification et structure
Les MMPs constituent une famille d’endopeptidases à doigt de zinc comprenant, à ce
jour, au moins 24 membres dénommés soit par un nom descriptif, soit par un numéro. En
accord avec la spécificité de leur substrat et de leur structure, les membres de la famille des
MMPs ont été classés en au moins 5 familles distinctes: les collagénases, les gélatinases, les
stromélysines, les matrilysines et les MMPs de type membranaires (MT-MMPs) (Tableau 7).
Malgré une certaine spécificité de substrats, les spectres protéolytiques des différentes MMPs
sont assez larges et se recouvrent permettant ainsi la dégradation de l’ensemble des
composants de la MEC. Dans le tableau 7 se trouvent les différents substrats matriciels des
MMPs répertoriés grâce à des études in vitro (37, 254). Il est important de préciser qu’à ce
jour, tous les substrats des différentes MMPs ne sont pas encore connus et restent à être
déterminés. C’est notamment le cas des MMP-11 et -18.
La structure primaire des MMPs contient au minimum trois domaines: un pré-
domaine, un pro-domaine et un domaine catalytique proprement dit. Le pré-domaine, au
niveau N-terminal, correspond à un peptide signal. Il est nécessaire à la sécrétion des MMPs
puis est rapidement dégradé. Le pro-domaine maintient l’activité enzymatique sous forme
latente et est classiquement constitué d’une séquence peptidique comprenant un résidu
cystéine qui interagit avec l’atome de zinc au niveau du domaine catalytique (figure 10). Les
MMPs sont donc sécrétées sous forme latente ou zymogène. Pour être actives, le pro-
domaine doit être éliminé ou modifié par clivage protéolytique, libérant ainsi l’atome de zinc
du résidu cystéine et entraînant l’activation du domaine catalytique qui peut alors se lier à son
substrat. La plupart des MMPs possèdent en plus un domaine charnière riche en proline et un
domaine C-terminal contenant une séquence homologue à de l’hemopexine ou de la
43
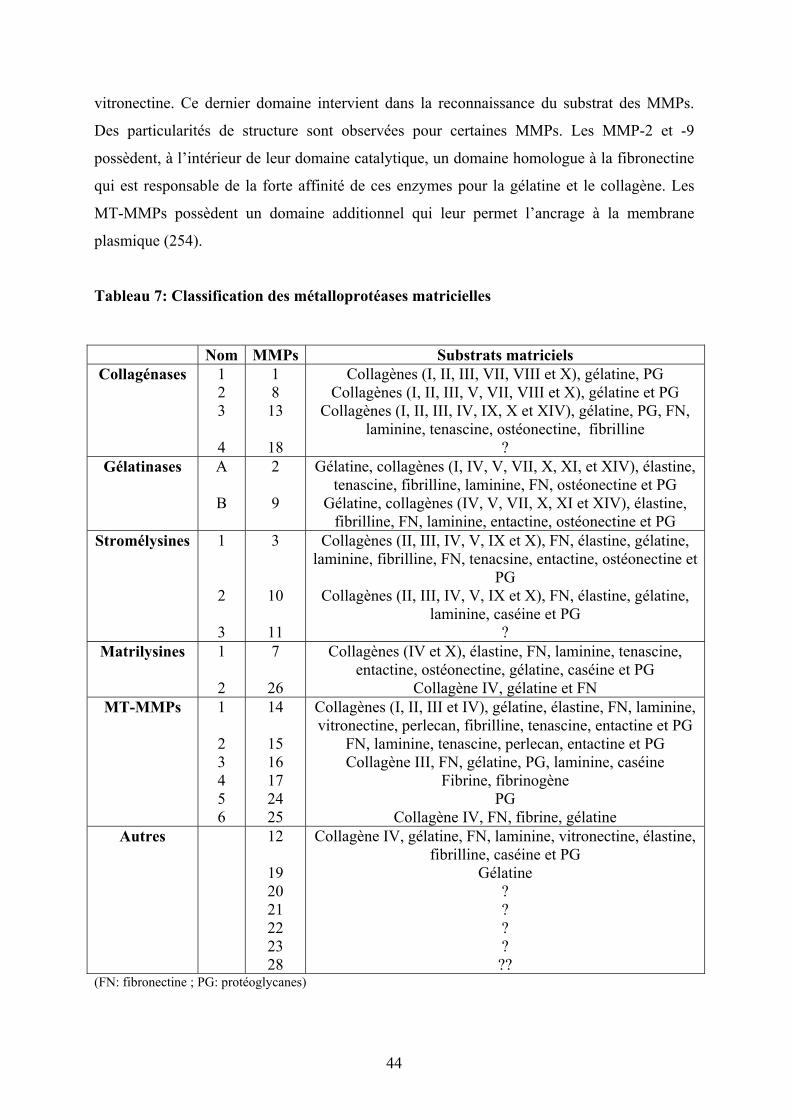
vitronectine. Ce dernier domaine intervient dans la reconnaissance du substrat des MMPs.
Des particularités de structure sont observées pour certaines MMPs. Les MMP-2 et -9
possèdent, à l’intérieur de leur domaine catalytique, un domaine homologue à la fibronectine
qui est responsable de la forte affinité de ces enzymes pour la gélatine et le collagène. Les
MT-MMPs possèdent un domaine additionnel qui leur permet l’ancrage à la membrane
plasmique (254).
Tableau 7: Classification des métalloprotéases matricielles
(FN: fibronectine ; PG: protéoglycanes)
Nom MMPs Substrats matriciels Collagénases
1 2 3 4
1 8 13
18
Collagènes (I, II, III, VII, VIII et X), gélatine, PG Collagènes (I, II, III, V, VII, VIII et X), gélatine et PG
Collagènes (I, II, III, IV, IX, X et XIV), gélatine, PG, FN, laminine, tenascine, ostéonectine, fibrilline
? Gélatinases
A
B
2 9
Gélatine, collagènes (I, IV, V, VII, X, XI, et XIV), élastine, tenascine, fibrilline, laminine, FN, ostéonectine et PG
Gélatine, collagènes (IV, V, VII, X, XI et XIV), élastine, fibrilline, FN, laminine, entactine, ostéonectine et PG
Stromélysines
1 2 3
3
10
11
Collagènes (II, III, IV, V, IX et X), FN, élastine, gélatine, laminine, fibrilline, FN, tenacsine, entactine, ostéonectine et
PG Collagènes (II, III, IV, V, IX et X), FN, élastine, gélatine,
laminine, caséine et PG ?
Matrilysines
1 2
7
26
Collagènes (IV et X), élastine, FN, laminine, tenascine, entactine, ostéonectine, gélatine, caséine et PG
Collagène IV, gélatine et FN MT-MMPs
1 2 3 4 5 6
14
15 16 17 24 25
Collagènes (I, II, III et IV), gélatine, élastine, FN, laminine, vitronectine, perlecan, fibrilline, tenascine, entactine et PG
FN, laminine, tenascine, perlecan, entactine et PG Collagène III, FN, gélatine, PG, laminine, caséine
Fibrine, fibrinogène PG
Collagène IV, FN, fibrine, gélatine Autres 12
19 20 21 22 23 28
Collagène IV, gélatine, FN, laminine, vitronectine, élastine, fibrilline, caséine et PG
Gélatine ? ? ? ? ??
44
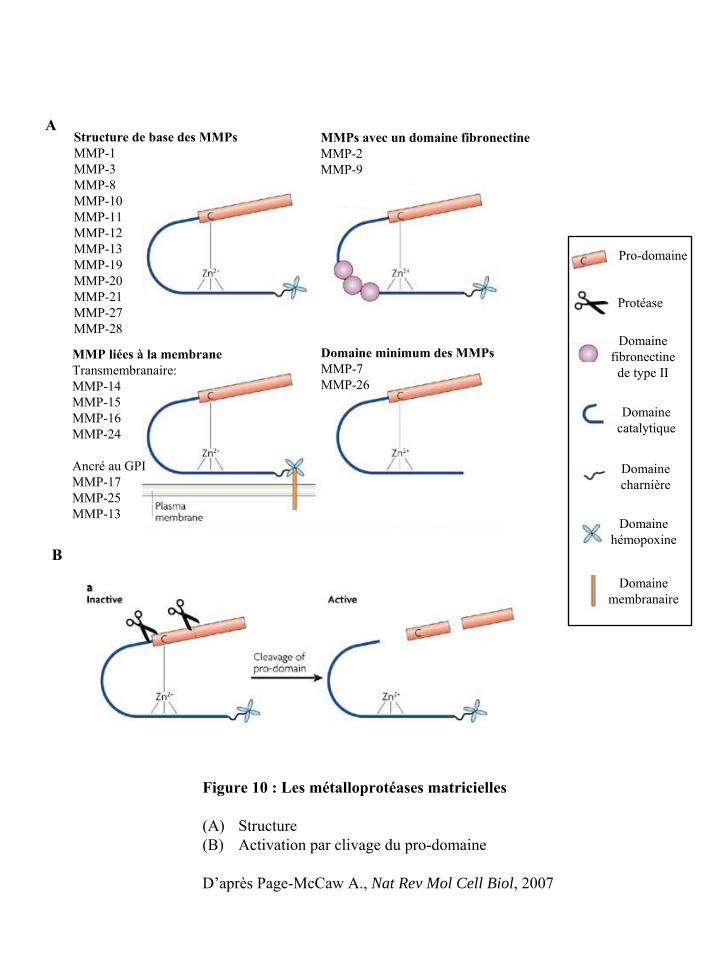
Figure 10 : Les métalloprotéases
matricielles
(A)
Structure(B)
Activation par clivage du pro-domaine
D’après Page-McCaw
A., Nat Rev Mol Cell Biol, 2007
Structure de base des MMPsMMP-1MMP-3MMP-8MMP-10MMP-11MMP-12MMP-13MMP-19MMP-20MMP-21MMP-27MMP-28
MMPs
avec un domaine fibronectineMMP-2MMP-9
MMP liées à
la membraneTransmembranaire:MMP-14MMP-15MMP-16MMP-24
Ancré
au GPIMMP-17MMP-25MMP-13
Domaine minimum des MMPsMMP-7MMP-26
A
B
Pro-domaine
Protéase
Domainemembranaire
Domainecatalytique
Domainehémopoxine
Domainecharnière
Domaine fibronectine
de type II

B) Les régulations des MMPs
Pour éviter une protéolyse excessive et des dommages tissulaires, l’activité des MMPs
est régulée par de multiples contrôles aussi bien au niveau de leur transcription, traduction et
sécrétion que de leur localisation, activation et inhibition (figure 11 et pour revue (254)).
Nous aborderons dans ce paragraphe les principales régulations des MMPs.
B.1) Les régulations transcriptionnelles et post-transcriptionnelles
L’expression des MMPs est normalement faible dans les tissus et est induite lorsqu’un
remodelage tissulaire est requis. Une large variété de facteurs solubles incluant des facteurs
de croissance et des cytokines tels que les interleukines 1α et β, les interférons α, β, et γ,
l’EGF, le FGF, le PDGF, le TGF-β et le TNF-α influent sur l’expression des MMPs. Il en est
de même pour les protéines de la MEC notamment les collagènes (290). De plus, des facteurs
non solubles tels que les contacts cellule-cellule et cellule-MEC ainsi que la forme de la
cellule modulent l’expression des MMPs (183, 254, 290). En effet, il a été observé que des
altérations morphologiques de divers types cellulaires entraînent une augmentation de
l’expression des collagénases (2) ainsi que l’activation de certaines intégrines (106).
L’étude des promoteurs des différentes MMPs a mis en évidence la présence de
plusieurs éléments de liaison pour divers facteurs de transcription tels qu’AP1 (activator
protein-1), PEA3 (polyoma enhancer A binding protein-3), NF-κB (nuclear factor-κB) et la
β-catenin (figure 12) (289, 295). Il est connu qu’AP1 lie les facteurs de transcription c-fos et
c-jun et que les sites PEA3 lient ceux de la famille ETS (E26 transformation specific). Le
facteur de transcription NF-κB est connu pour être impliqué dans les processus
d’inflammation et la β-caténine répond à la voie de signalisation de Wnt (187, 294). Comme
on peut l’observer dans la figure 12, plusieurs promoteurs des MMPs sont similaires en terme
de composition en éléments de liaison aux facteurs de transcription suggérant que plusieurs
MMPs sont co-régulées dans leur expression. Cependant et de manière surprenante, les
promoteurs des MMPs appartenant à la même famille telles que les MMP-2/MMP-9
(gélatinases) et les MMP-1/MMP-8 (collagénases) n’ont aucune similitude. Des groupes de
promoteurs de MMPs ont pu ainsi être formés en fonction de leur composition en élément de
liaison (figure 12 et pour revue (37). Il a ainsi été observé que l’expression des MMP-2, 14 et
28 est majoritairement « constitutive » avec tout de même une induction possible par certains
facteurs de croissance ou cytokines (37).
45
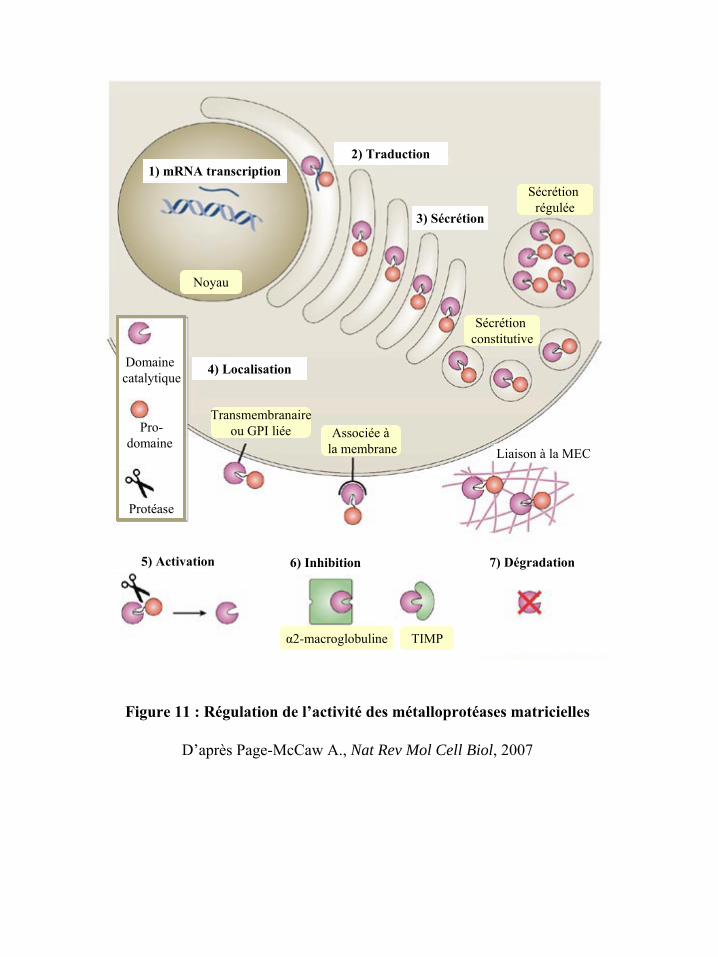
Figure 11 : Régulation de l’activité
des métalloprotéases
matricielles
D’après Page-McCaw
A., Nat Rev Mol Cell Biol, 2007
1) mRNA
transcription2) Traduction
3) Sécrétion
4) Localisation
5) Activation 6) Inhibition 7) Dégradation
Domaine catalytique
Pro-domaine
Protéase
Liaison à
la MEC
Sécrétion régulée
Noyau
Sécrétion constitutive
Transmembranaireou GPI liée Associée à
la membrane
α2-macroglobuline TIMP
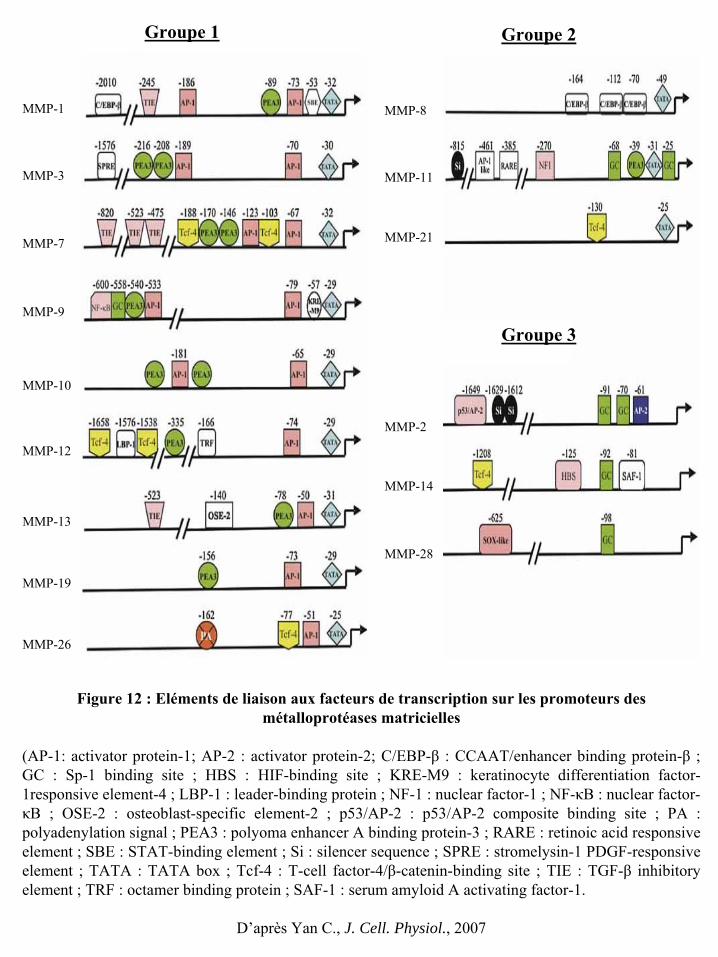
Figure 12 :
Eléments de liaison aux facteurs de transcription sur les promoteurs des métalloprotéases
matricielles
(AP-1: activator
protein-1; AP-2 : activator
protein-2; C/EBP-β
: CCAAT/enhancer
binding
protein-β
; GC : Sp-1 binding
site ; HBS : HIF-binding
site ; KRE-M9 : keratinocyte
differentiation
factor-
1responsive element-4 ; LBP-1 : leader-binding
protein
; NF-1 : nuclear
factor-1 ; NF-κB : nuclear
factor-
κB ; OSE-2 : osteoblast-specific
element-2 ; p53/AP-2 : p53/AP-2 composite binding
site ; PA : polyadenylation
signal ; PEA3 : polyoma
enhancer
A binding
protein-3 ; RARE : retinoic
acid
responsive element
; SBE : STAT-binding
element
; Si : silencer
sequence
; SPRE : stromelysin-1 PDGF-responsive
element
; TATA : TATA box ; Tcf-4 : T-cell
factor-4/β-catenin-binding
site ; TIE : TGF-β
inhibitory element
; TRF : octamer
binding
protein
; SAF-1 : serum
amyloid
A activating
factor-1.
D’après Yan
C., J. Cell. Physiol., 2007
MMP-8
MMP-11
MMP-21
MMP-2
MMP-14
MMP-28
Groupe 2
Groupe 3
MMP-1
MMP-3
MMP-13
MMP-7
MMP-9
MMP-10
MMP-12
MMP-19
MMP-26
Groupe 1

Il est important de signaler que la régulation transcriptionnelle des MMPs ne dépend
pas uniquement de la présence de site de liaison au niveau de leur promoteur. En effet, des
polymorphismes du promoteur et des régulations épigénétiques (i.e, la méthylation de l’ADN
ou des altérations de la structure de la chromatine) jouent également un rôle dans l’expression
des MMPs. Enfin des régulations post-transcriptionnelles des MMPs sont possibles et
correspondent le plus souvent à des processus de stabilisation ou de dégradation des transcrits
(pour revue (295)).
B.2) Les régulations de la sécrétion
Les MMPs sont sécrétées par de nombreux types cellulaires notamment les
fibroblastes, neutrophiles, macrophages, cellules épithéliales et endothéliales (290). Certains
types cellulaires tels que les macrophages et les neutrophiles, qui ont la capacité de stocker
les MMPs dans des granules intracytoplasmiques, régulent la sécrétion des MMPs par les
actions de la plasmine ou de la thrombine sur le processus de libération des granules (220).
B.3) Les processus de maturation des MMPs
Comme il a été précisé un peu plus tôt, l’activation des MMPs dépend de
l’élimination ou de la modification de leur pro-domaine par clivage protéolytique. Différents
types d’activation des MMPs ont été décrits.
En général, l’activation des MMPs se produit à l’extérieur de la cellule. Le tableau 8
présente les principaux activateurs des MMPs (pour revue (37)). La plasmine issue de la
dégradation du plasminogène est l’un des activateurs physiologiques majeur des MMPs et de
ce fait les activateurs de sa production tels que les activateurs du plasminogène de type
urokinase (uPA) ou de type tissulaire (tPA) le sont également (26). Les MMPs elles-mêmes
sont capables d’activer d’autres MMPs. De plus, il a été rapporté que d’autres protéases telles
que les kallikréines, les cathepsines et les chymases permettaient la maturation des MMPs
(67). Les espèces réactives de l’oxygène ont aussi été impliquées dans l’activation des MMPs
(217, 287).
Les travaux de Pei et Weiss ont démontré pour la première fois que la pro-MMP11
(ou pro-stromélysine 3) était activée à l’intérieur de la cellule avant sa sécrétion. Cette
activation se fait par les furines, une famille de sérine protéases (204). Plus tard, il a été
montré que cette activation par les furines concerne les MMPs contenant une séquence
46
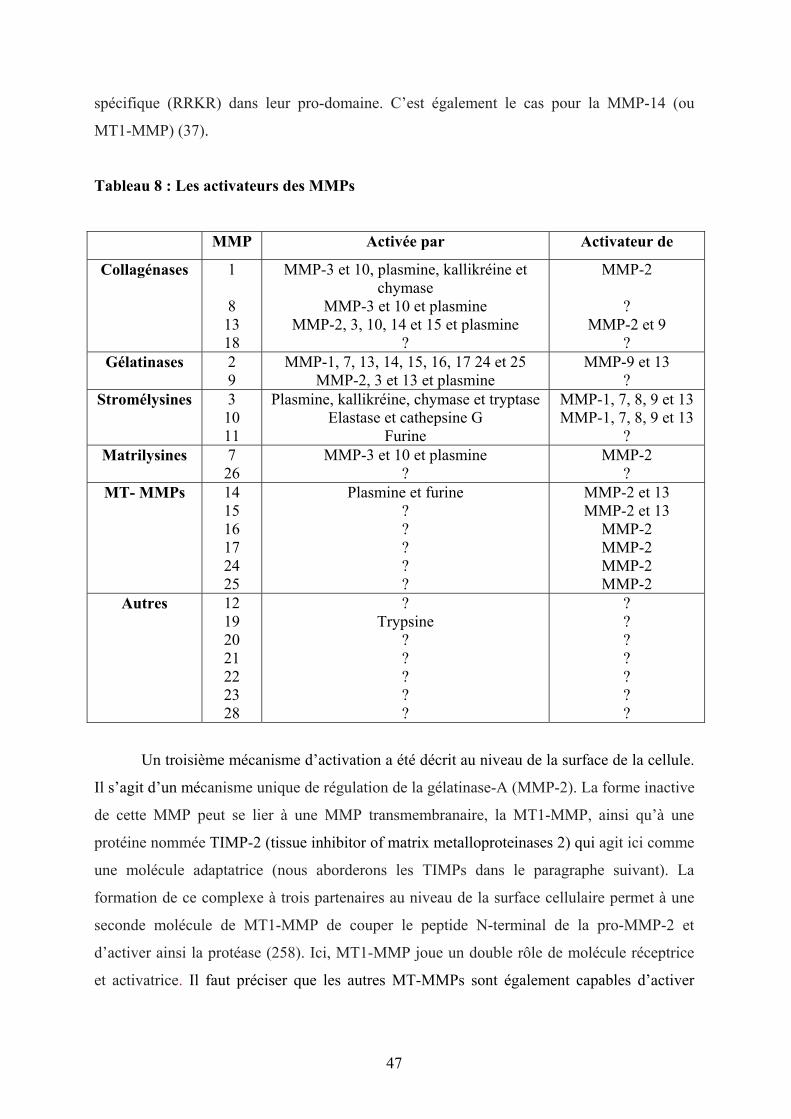
spécifique (RRKR) dans leur pro-domaine. C’est également le cas pour la MMP-14 (ou
MT1-MMP) (37).
Tableau 8 : Les activateurs des MMPs
MMP Activée par Activateur de
Collagénases
1
8 13 18
MMP-3 et 10, plasmine, kallikréine et chymase
MMP-3 et 10 et plasmine MMP-2, 3, 10, 14 et 15 et plasmine
?
MMP-2 ?
MMP-2 et 9 ?
Gélatinases
2 9
MMP-1, 7, 13, 14, 15, 16, 17 24 et 25 MMP-2, 3 et 13 et plasmine
MMP-9 et 13 ?
Stromélysines
3 10 11
Plasmine, kallikréine, chymase et tryptase Elastase et cathepsine G
Furine
MMP-1, 7, 8, 9 et 13 MMP-1, 7, 8, 9 et 13
? Matrilysines
7 26
MMP-3 et 10 et plasmine ?
MMP-2 ?
MT- MMPs
14 15 16 17 24 25
Plasmine et furine ? ? ? ? ?
MMP-2 et 13 MMP-2 et 13
MMP-2 MMP-2 MMP-2 MMP-2
Autres 12 19 20 21 22 23 28
? Trypsine
? ? ? ? ?
? ? ? ? ? ? ?
Un troisième mécanisme d’activation a été décrit au niveau de la surface de la cellule.
Il s’agit d’un mécanisme unique de régulation de la gélatinase-A (MMP-2). La forme inactive
de cette MMP peut se lier à une MMP transmembranaire, la MT1-MMP, ainsi qu’à une
protéine nommée TIMP-2 (tissue inhibitor of matrix metalloproteinases 2) qui agit ici comme
une molécule adaptatrice (nous aborderons les TIMPs dans le paragraphe suivant). La
formation de ce complexe à trois partenaires au niveau de la surface cellulaire permet à une
seconde molécule de MT1-MMP de couper le peptide N-terminal de la pro-MMP-2 et
d’activer ainsi la protéase (258). Ici, MT1-MMP joue un double rôle de molécule réceptrice
et activatrice. Il faut préciser que les autres MT-MMPs sont également capables d’activer
47

MMP-2 même si la protéolyse par la MT1-MMP reste prédominante (37). En revanche, les
activateurs physiologiques de nombreuses MMPs sont encore inconnus (tableau 8).
Une fois leur pro-domaine clivé, les MMPs sont protéolytiquement actives mais très
rapidement elles peuvent être inactivées par liaison à des protéines inhibitrices. Ces protéines
vont se fixer directement au niveau du domaine catalytique empêchant l’accès au substrat.
Dans le paragraphe qui suit nous nous intéresserons aux différents inhibiteurs endogènes des
MMPs.
B.4) Les processus d’inhibition des MMPs
In vivo, l’activité des MMPs est contrôlée par les TIMPs (tissue inhibitors of matrix
metalloproteinases). A ce jour, quatre TIMPs différents ont été identifiés (TIMP-1 à -4). Ce
sont des protéines de petites tailles (20 à 29 kDa), sécrétées mais qui peuvent être retrouvées
à la surface cellulaire associées à des protéines liées à la membrane plasmique ou séquestrées
au niveau de la MEC. C’est le cas de TIMP-3 qui peut interagir avec des protéoglycanes
(298). Les TIMPs sont exprimés par de nombreux types cellulaires incluant les fibroblastes,
les cellules épithéliales et endothéliales, les ostéoblastes, les chondroblastes, les cellules du
muscle squelettiques ainsi que des cellules cancéreuses. Tandis que l’expression de TIMP-2
est constitutive, celle des TIMP-1, 3 et 4 est inductible. Avec un certain degré de spécificité,
les TIMPs sont capables d’inhiber l’activité des MMPs. Ils forment un complexe
équimoléculaire avec les formes actives des MMPs où le domaine amino-terminal des TIMPs
se lie au domaine catalytique des MMPs et, de ce fait, bloque l’accès à la poche contenant
l’atome de zinc. Cette inhibition est réversible. Les TIMPs ont aussi la capacité d’inhiber
d’autres protéases telles que les ADAMs (A Disintegrine And Metalloprotease domain)
(141). La régulation des MMPs par les TIMPs est un facteur majeur dans le contrôle de la
dégradation de la MEC. Elles jouent également un rôle dans le contrôle de nombreux
processus cellulaires tels que la prolifération mais aussi l’apoptose et des événements
physiologiques tels que l’angiogenèse et pathophysiologiques tels que le développement
tumoral (41). Toutefois, de plus en plus d’évidences indiquent que certaines de ces fonctions
semblent être complètement indépendantes des MMPs. Par exemple, récemment, il a été
rapporté que TIMP-2 et 3 pouvaient lier l’intégrine α3 β1 et entraîner une cascade de
signalisation négative conduisant à un arrêt du cycle cellulaire des cellules endothéliales
(243). De la même manière, TIMP-3 est capable d’interagir avec le récepteur 2 du VEGF
empêchant ainsi l’accès à son substrat et entraînant une inhibition de l’angiogenèse (216).
48

Ces effets étant retrouvés quand les TIMPs sont mutés au niveau de leur domaine nécessaire
pour l’inhibition des MMPs, il est donc fortement suggéré que les TIMPs pourraient exercer
des fonctions indépendamment de leurs effets sur les MMPs.
L’α2-macroglobuline, une protéine de 722 KDa, peut également inhiber l’activité des
MMPs (251). Trouvée en abondance dans le plasma, elle serait, de ce fait, l’inhibiteur
endogène majeur des MMPs dans les liquides physiologiques alors que les TIMPs agiraient
plus localement dans l’espace extra- et péri-cellulaire.
D’autres protéines ont été rapportées pour inhiber certaines MMPs. C’est le cas de la
protéine RECK (Reversion-inducing cysteine-rich protein with kazal motifs), une
glycoprotéine GPI-ancrée qui inhibe les MMP-2, -9 et -14, et bloque l’angiogenèse (193).
Une déficience de cette protéine chez des souris est létale suggérant des fonctions
importantes de RECK dans le développement. Enfin, un certain nombre de protéines
contenant des séquences similaires au TIMPs peuvent inhiber les MMPs. Ce sont les « TIMP-
like molecules » qui incluent notamment les neltrines, les protéines frizzled, la PCPE (type 1
collagen C-proteinase enhancer protein) et le TFPI-2 (tissue factor pathway inhibitor 2) (pour
revue (8)).
Ainsi, l’activité des MMPs dans l’espace extracellulaire dépend de l’équilibre entre
leur inhibition et leur activation. C’est cet équilibre qui régule le renouvellement de la MEC
et le remodelage tissulaire durant les processus de développement normal ou pathologique.
Voyons à présent les différents modes d’action des MMPs.
C) Mode d’action des MMPs
La MEC est un réseau dynamique de protéines incluant du collagène, de la
fibronectine, de la laminine et des protéoglycanes qui forme une barrière structurale. La
dégradation de la MEC a été longtemps considérée comme un simple remodelage de cette
barrière structurale. Depuis, des études ont montré que la MEC n’était pas juste une structure
passive. Elle peut influencer le comportement des cellules et notamment leur détermination et
leur différenciation (257). Jusqu'à récemment, on pensait que les MMPs participaient à ces
processus via leurs effets sur la MEC mais il est maintenant clair que les MMPs ont d’autres
mécanismes d’action (figure 13 et pour revue (178)). Il a ainsi été rapporté que la dégradation
des composants de la MEC par les MMPs peut générer des fragments matriciels
49
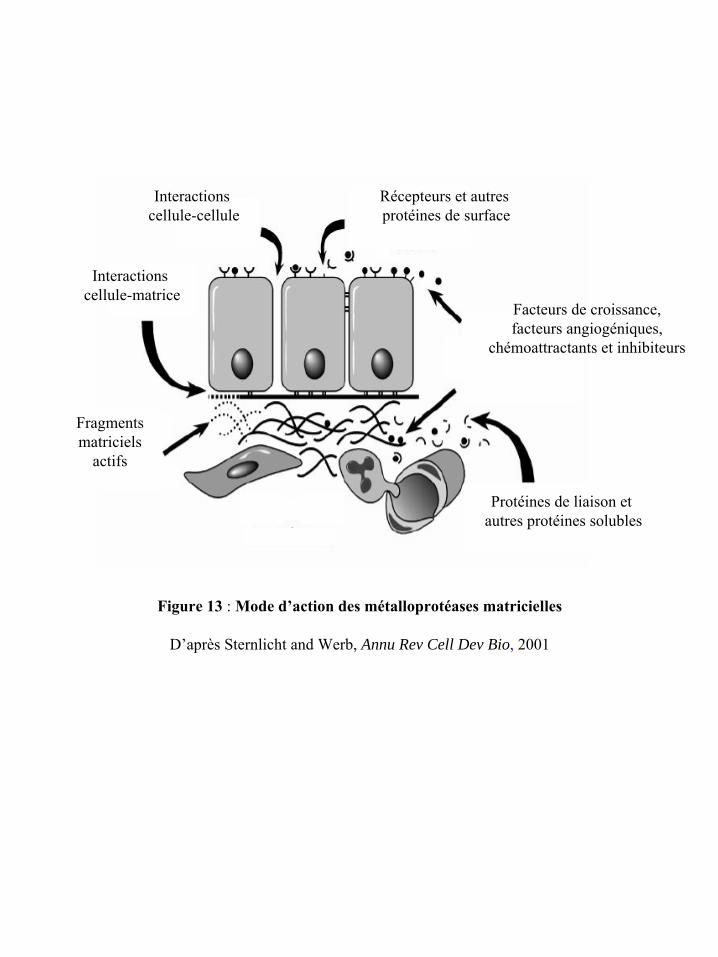
Interactions cellule-cellule
Interactions cellule-matrice
Récepteurs et autres protéines de surface
Protéines de liaison et autres protéines solubles
Facteurs de croissance,facteurs angiogéniques,
chémoattractants
et inhibiteurs
Fragments matriciels
actifs
Figure 13 : Mode d’action des métalloprotéases
matricielles
D’après Sternlicht
and
Werb, Annu Rev Cell Dev Bio,
2001

biologiquement actifs tels que l’endostatine (fragment du collagène XVIII) (77),
l’angiostatine (fragment du plasminogène) (195), des fragments de laminine et certains
peptides dérivés de la fibronectine (178). La MEC est également un réservoir de facteurs de
croissance et de cytokines qui sont libérés lors de la dégradation matricielle. C’est notamment
le cas du TGF-β lors de la dégradation de la décorine par les MMP-2, -3 ou -7 (108). En plus
de leur capacité à dégrader l’ensemble des composants de la MEC, les MMPs peuvent
également cliver de nombreuses protéines cellulaires de surface, péri-cellulaires ou
circulantes. Ainsi, les MMPs peuvent activer la forme pro-TGF-β et pro-TNF-α ou cliver les
IGFBPs (insulin-like growth factor binding protein) et par conséquent augmenter la
biodisponibilité de l’IGF (80). De plus, le changement de l’environnement cellulaire peut
indirectement moduler des voies dépendantes des protéines d’ancrages telles que celles des
intégrines. Enfin des études récentes suggèrent que certaines MMPs (MMP-2, -3, -11 et -14)
pourraient avoir également des cibles intracellulaires telles que PARP (poly ADP-ribose
polymérase, une enzyme de réparation à l’ADN (88, 134, 156, 248). Ces nouvelles données
rendent le champ d’action des MMPs encore plus large qu’il ne l’était.
D) Les fonctions biologiques et pathologiques des MMPs
Comme nous l’avons déjà évoqué, la MEC joue un rôle prépondérant dans le
comportement cellulaire (257). Il n’est donc pas surprenant que les principaux acteurs de son
remodelage aient un rôle tout aussi important.
Au niveau cellulaire, ces enzymes jouent un rôle majeur dans la migration cellulaire,
la prolifération, l’apoptose et la différenciation de nombreux types cellulaires notamment les
neurones, les chondrocytes, les ostéoblastes et les adipocytes (dont nous parlerons un peu
plus tard) (pour revue (162)). Récemment, des études ont également suggéré un rôle des
MMPs dans le devenir des cellules souches mésenchymateuses (159). En effet des travaux
indiquent que la composition et la rigidité de la MEC modulent l’orientation des cellules
souches mésenchymateuses vers différents lignages (69) par des modifications des adhésions
focales. De par leurs fonctions au niveau cellulaire, les MMPs régulent plusieurs processus
du développement incluant la morphogenèse, l’angiogenèse, la cicatrisation, la menstruation
et le développement mammaire et osseux (pour revue (280). Des approches de transgenèse
ont permis d’étudier les fonctions biologiques des MMPs dans le développement. A ce jour,
16 modèles de souris déficientes pour les MMPs ont été développés. Toutes les souris
déficientes pour les MMPs sont viables avec cependant des différences notoires sur les
50

phénotypes des animaux qui sont présentés dans le tableau 9 (199). Le fait que ces souris
soient viables, malgré l’importance des MMPs dans le développement, suggère de forts
mécanismes de compensation vis-à-vis d’autres MMPs.
Tableau 9: Phénotypes des souris déficientes pour les MMPs
Géne invalidé
Phénotype des animaux
MMP-2 Diminution de la taille corporelle, réduction de la néovascularisation, diminution de l’invasion primaire du conduit de la glande mammaire ; réduction du développement pulmonaire
MMP-3 Altération de la structure des jonctions neuromusculaires, altération de la morphogenèse de la glande mammaire
MMP-7 Défaut de l’immunité innée, diminution de la re-épithélialisation après une blessure du poumon
MMP-8 Augmentation des tumeurs de la peau, résistance aux hépatites létales induites par le TNFα
MMP-9 Défauts du développement osseux, défaut de la re-myélinisation neuronale après une blessure nerveuse, retard de la cicatrisation des fractures osseuses, altération du remodelage vasculaire, altération de l’angiogenèse
MMP-10 Augmentation de l’inflammation et de la mortalité en réponse à une infection ou une blessure
MMP-11 Retard de la tumorigenèse mammaire MMP-12 Diminution de la récupération d’une compression de la moelle épinière,
augmentation de l’angiogenèse due à une diminution de l’angiostatine MMP-13 Défaut du remodelage osseux, réduction des fibroses hépatiques, augmentation
de l’accumulation du collagène dans les plaques d’athérosclérose MMP-14 Défaut du remodelage squelettique, défaut de l’angiogenèse, inhibition de
l’éruption dentaire et de l’élongation des racines, défauts au niveau des poumons et des glandes submandibullaires
MMP-19 Obésité MMP-20 Défaut de l’émail dentaire MMP-23 Pas de phénotype rapporté MMP-24 Réponse anormale à une blessure du nerf sciatique MMP-28 Augmentation de la réponse inflammatoire D’après Page-McCaw A., Nat Rev Mol Cell Biol, 2007
En plus de ces données sur les principales fonctions des MMPs, il est important de
préciser la complexité des MMPs, en terme d’activité biologique. En effet, certaines d’entre
elles sont paradoxales, puisqu’une MMP peut avoir des effets diamétralement opposés. Par
exemple, il est connu que les MMP-2 et -9 favorisent l’angiogenèse mais des effets anti-
angiogéniques leurs sont également attribués (pour revue (232)). Il en est de même pour les
51

processus apoptotiques puisque les MMP-2, -3, -7 et -9 peuvent être pro- et anti-apoptotiques
(pour revue (160)).
Du fait de leurs fonctions biologiques, les MMPs ont été associées à diverses
pathologies. Ce sont des modulateurs importants de l’inflammation (pour revue (203)) et elles
sont fortement augmentées dans toutes les pathologies associées à une inflammation
notamment l’arthrite rhumatoïde. A ce jour, un inhibiteur des MMPs, le périostat, est utilisé
pour le traitement de la parodontite, une inflammation des tissus de soutien qui relient la dent
au maxillaire.
Des dérégulations des MMPs ont également été impliquées dans diverses pathologies
cardiovasculaires telles que l’athérosclérose et l’infarctus du myocarde (85, 253).
Enfin, l’importance des MMPs dans la progression du cancer n’est plus à démontrer
(60). Leur expression est fortement augmentée dans de nombreux cancers. Elles interviennent
dans toutes les phases du cancer c’est-à-dire de la croissance tumorale, l’invasion,
l’extravasation dans des sites plus éloignés ainsi que dans l’effet angiogénique requis pour
nourrir la formation de la nouvelle tumeur. Ainsi, les MMPs sont devenues la première cible
pour les programmes de découverte de nouveaux médicaments à but anti-cancéreux. De
nombreux inhibiteurs des MMPs ont été développés (tableau 10). Malgré un début très
prometteur, la survenue d’effets indésirables au cours des phases de développement clinique
et le peu de bénéfice par rapport aux effets indésirables a conduit à l’arrêt de nombreux
d’entre eux. A l’heure actuelle, d’autres inhibiteurs plus spécifiques sont en cours de
développement (pour revue (161).
52
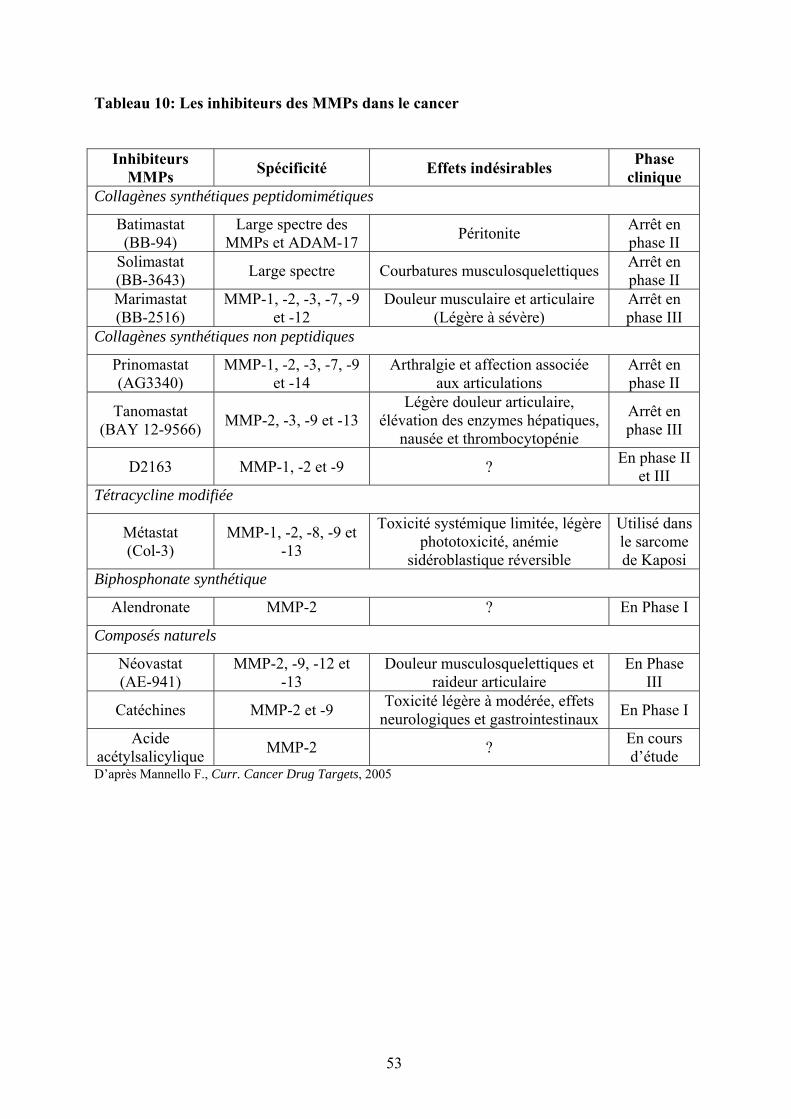
Tableau 10: Les inhibiteurs des MMPs dans le cancer
Inhibiteurs MMPs Spécificité Effets indésirables Phase
clinique Collagènes synthétiques peptidomimétiques
Batimastat (BB-94)
Large spectre des MMPs et ADAM-17 Péritonite Arrêt en
phase II Solimastat (BB-3643) Large spectre Courbatures musculosquelettiques Arrêt en
phase II Marimastat (BB-2516)
MMP-1, -2, -3, -7, -9 et -12
Douleur musculaire et articulaire (Légère à sévère)
Arrêt en phase III
Collagènes synthétiques non peptidiques
Prinomastat (AG3340)
MMP-1, -2, -3, -7, -9 et -14
Arthralgie et affection associée aux articulations
Arrêt en phase II
Tanomastat (BAY 12-9566) MMP-2, -3, -9 et -13
Légère douleur articulaire, élévation des enzymes hépatiques,
nausée et thrombocytopénie
Arrêt en phase III
D2163 MMP-1, -2 et -9 ? En phase II et III
Tétracycline modifiée
Métastat (Col-3)
MMP-1, -2, -8, -9 et -13
Toxicité systémique limitée, légère phototoxicité, anémie
sidéroblastique réversible
Utilisé dans le sarcome de Kaposi
Biphosphonate synthétique
Alendronate MMP-2 ? En Phase I
Composés naturels
Néovastat (AE-941)
MMP-2, -9, -12 et -13
Douleur musculosquelettiques et raideur articulaire
En Phase III
Catéchines MMP-2 et -9 Toxicité légère à modérée, effets neurologiques et gastrointestinaux En Phase I
Acide acétylsalicylique MMP-2 ? En cours
d’étude D’après Mannello F., Curr. Cancer Drug Targets, 2005
53

III. Les métalloporotéases matricielles dans le tissu adipeux
L’intérêt des MMPs dans le remodelage du TA est très récent et la majorité des études
ont été effectuées chez le rongeur. In vivo, dans des modèles de souris génétiquement obèses
(ob/ob et db/db) ou rendues obèses à l’aide d’un régime hyperlipidique, des variations
d’expression de différentes MMPs dans le TA ont été observées. On peut noter une
augmentation des MMP-2, -3, -11, -12, -13, -14, -19 et une diminution des MMP-7, -9, -16 et
-24 (38, 149, 164). De plus, un traitement au galardin, un inhibiteur des MMPs à large
spectre, limite le développement du TA de ces animaux (148). Ces premières données
suggèrent un rôle primordial du remodelage matriciel dans le développement de la masse
grasse.
A) La régulation de l’adipogenèse
In vitro, des travaux menés par notre équipe et celle de Croissandeau, indiquent que
des préadipocytes murins des lignées 3T3F442A (17) et 3T3L1 (53) produisent au cours de
leur différenciation adipocytaire deux MMPs appartenant à la famille des gélatinases : MMP-
2 et -9. Par ailleurs, l’inhibition de la différenciation adipocytaire est également observée
dans ces préadipocytes murins traités par des inhibiteurs des MMPs à large spectre
(batimastat et illomastat) ou des anticorps neutralisants dirigés contre les MMP-2 ou -9
suggérant un rôle majeur de ces deux MMPs dans l’adipogenèse chez les rongeurs (17, 53).
Récemment, la MT1-MMP a été identifiée comme importante dans le développement
de la masse grasse. En effet, les travaux de Chun et al. montrent que des souris déficientes
pour la MT1-MMP présentent un phénotype lipodystrophique (44). In vitro et de manière
surprenante, les préadipocytes issus de ces souris sont doués d’une adipogenèse normale dans
un modèle de culture classique en 2 dimensions. Cependant, lorsque ces préadipocytes sont
cultivés sur un support en 3 dimensions, leur adipogenèse est inhibée. Les auteurs démontrent
qu’in vivo les précurseurs adipocytaires sont entourés d’un riche réseau de fibres de collagène
de type 1. Ce réseau est profondément modifié notamment par la MT1-MMP en début de
différenciation. Ce remodelage modifie les contraintes physiques que fait subir la matrice de
collagène au cytosquelette des préadipocytes et induit des cascades de signalisation pour la
poursuite de la différenciation adipocytaire. En l’absence de MT1-MMP, le réseau de
collagène reste très dense, les contraintes du cytosquelette ne sont pas modifiées et le
54

programme de différenciation ne peut pas continuer. La MT1-MMP serait un facteur
adipogénique qui permet la croissance de l’organe adipeux en 3 dimensions.
De façon intéressante, trois autres MMPs, MMP-3 et -11 appartenant à la famille des
stromélysines (Stro1 et Stro-3 respectivement) et MMP-19, ont été décrites comme ayant des
effets inhibiteurs sur le développement du TA et sur l’adipogenèse. In vivo, des souris
transgéniques mutées pour la MMP-3 ont une adipogenèse au niveau de la glande mammaire
accélérée et une hypertrophie adipocytaire (3) et des souris déficientes pour les MMP-3, -11
et -19 soumises à un régime gras développent plus de TA que des souris contrôles (150, 163,
205). In vitro, Alexander et al. rapportent que la MMP-3 augmente au cours de la
différenciation des préadipocytes murins de la lignée 3T3 L1 et qu’un traitement avec des
inhibiteurs des MMPs (GM6001 et TIMP-1) accélère l’accumulation des lipides (3). Ces
données sont à l’opposé de celles obtenues par l’équipe de Croissandeau sur le même type
cellulaire. La spécificité et la sélectivité des inhibiteurs des MMPs utilisés pourraient
expliquer ces différences.
Enfin, en plus de leur rôle majeur dans la différenciation adipocytaire, il a récemment
été suggéré une implication des MMPs dans la détermination des cellules précurseurs vers le
lignage adipocytaire. En effet, les travaux d’Otto et al, indiquent qu’un traitement au BMP-4
entraîne une diminution des MMP-3 et -13 associée à l’engagement des cellules C3H10T1/2
vers le lignage adipogénique (197).
B) La régulation de l’angiogenèse
A ce jour, il existe peu de données concernant les MMPs et la régulation de
l’angiogenèse dans le TA. Toutefois, il a récemment été rapporté in vivo chez des rongeurs
que des macrophages positifs pour LYVE-1 (lymphatic vessel endothelial hyaluronan
receptor 1) et originaire de la moelle osseuse, induisent une angiogenèse notamment à travers
la sécrétion des MMP-9, -12 et -7 (42).
C) La régulation de l’infiltration macrophagique
La MMP-12 est requise pour la dégradation de la MEC par les macrophages et
l’invasion tissulaire (246). De plus, cette MMP est fortement augmentée chez des souris
obèses (38, 164). Les travaux de Huber et al. montrent que la MMP-12 est exprimée et
produite par les macrophages et les adipocytes matures suggérant que ces deux types
55

cellulaires contribuent à la production de MMP-12 dans le TA de sujets obèses. Ainsi, les
auteurs suggèrent que la production de MMP-12 pourrait favoriser l’infiltration
macrophagique mais aussi faciliter la croissance des adipocytes durant le développement de
la masse grasse (104). Toutefois cette hypothèse reste encore à démontrer. Récemment, les
travaux de Cho et al. montrent qu’un traitement avec un inhibiteur de l’activité des MMPs,
l’acide zoledronique, diminue le recrutement et l’infiltration des macrophages positifs pour
LYVE-1. Ces macrophages exprimant principalement les MMP-7, -9 et -12, l’implication de
l’activité de ces MMPs dans l’infiltration macrophagique est suggérée (42).
Chez l’homme, les études concernant les MMPs dans le TA sont peu nombreuses.
Notre équipe a rapporté que des préadipocytes humains en culture primaire produisent et
sécrètent les MMP-2 et -9 au cours de leur différenciation. Sachant qu’il existe de grandes
différences entre les modèles humains et murins, la confirmation de l’implication des MMPs
dans l’adipogenèse chez l’homme est nécessaire et a fait l’objet d’une partie de notre étude.
De plus, de manière intéressante, une augmentation de la MMP-9 plasmatique a été associée
à l’obésité (86) et sa diminution à une perte de poids (140) suggérant un rôle clé de cette
protéase dans le remodelage du TA humain.
D) Les régulateurs des MMPs dans le tissu adipeux
Des travaux ont impliqués TIMP-1 et -3 dans le développement de la masse grasse
chez la souris. Les travaux de Fata et al. sur l’involution de la glande mammaire dans des
souris déficientes pour TIMP-3 montrent une reconstitution adipocytaire accélérée durant le
remplacement des cellules épithéliales de la glande mammaire par la formation du TA (76).
Toutefois, un effet de TIMP-3 sur les autres dépôts de TA de ces souris n’est pas décrit. Les
travaux de Lijnen et al. indiquent que des souris déficientes pour TIMP-1 sont protégées
d’une obésité induite par un régime gras (147). De plus, une augmentation de l’adipogenèse
du TA mammaire est observée in vivo chez des souris surexprimant TIMP-1 et l’addition
d’une protéine recombinante TIMP-1 accélère la différenciation des préadipocytes de la
lignée 3T3L1 in vitro (3). Toutefois, les travaux de Demeulemeester et al. ne sont pas en
accord avec ces résultats et indiquent qu’une surexpression de TIMP-1 par un adénovirus
n’affecte pas le développement du TA des souris, in vivo et in vitro, et n’affecte pas
l’adipogenèse des préadipocytes de la lignée 3T3F442A (59). Concernant l’expression et la
sécrétion de TIMP-1, une augmentation de l’expression de TIMP-1 chez des souris
56

génétiquement obèses ou soumises à un régime gras a été décrite (38, 164) et au cours de
l’adipogenèse in vitro les résultats ne sont pas très clairs puisque des études indiquent qu’au
cours de la différenciation des préadipocytes de la lignée 3T3F442A, l’expression de TIMP-1
augmente (164), et d’autres, notamment notre groupe, que son expression et sa sécrétion
diminuent (17, 116). Chez l’homme, les taux plasmatiques de TIMP-1 sont corrélés avec le
pourcentage de la masse grasse mais pas à l’IMC ni au tour de taille (131). Des travaux
complémentaires seraient nécessaires pour mieux comprendre le rôle précis des TIMPs dans
le TA, notamment chez l’homme, et si leurs effets sont directement liés aux MMPs ou
indépendants.
En plus des TIMPs, d’autres régulateurs des MMPs tels que le système
plasminogène/plasmine jouent un rôle majeur dans le remodelage du TA. Toutefois, ce
dernier a déjà fait l’objet de nombreuses études et ne sera pas abordé ici.
Pour conclure, les MMPs sont nécessaires à tous les processus du développement
de l’organisme. Elles constituent une grande famille de protéases dont l’activité est
régulée à de multiples niveaux. Il est clairement admis que les substrats des MMPs
peuvent être des composants matriciels ou des facteurs solubles cellulaires rendant ainsi
le champ d’action des MMPs très vaste. Elles sont considérées comme des protéases
majeures dans le remodelage tissulaire. Cependant, quelques études seulement se sont
intéressées à l’implication des MMPs dans le remodelage du TA et aucune étude n’a
encore démontré un rôle de ces protéases dans le TA humain.
57

Objectifs
Comme nous l’avons déjà évoqué, le TA est soumis à des processus de
développement excessif (obésité) mais aussi de régression (lipoatrophie). Il est admis que
cette plasticité du TA n’est possible que grâce à un remodelage cellulaire et matriciel. Les
MMPs, comme nous venons de le voir, sont des acteurs majeurs du remodelage tissulaire.
Quelques études ont montré l’importance des MMPs dans la différenciation adipocytaire des
préadipocytes de lignées murines. De plus, les inhibiteurs de la protéase du VIH, décrits
comme associés à l’émergence du syndrome de lipodystrophie, sont également associés à une
inhibition de l’adipogenèse chez les rongeurs. Les effets anti-adipogéniques observés avec les
inhibiteurs de protéases matricielles et de protéases du VIH nous ont conduit à émettre
l’hypothèse d’un lien entre les IPs-VIH et les MMPs. L’influence de ces protéases dans
l’adipogenèse chez l’homme reste cependant à être déterminée.
Dans la première partie de ce travail de thèse, nous nous sommes intéressés à
l’implication des MMPs dans la différenciation des cellules issues de la fraction stroma-
vasculaire (FSV) du TA humain en culture primaire. L’effet des IPs-VIH sur la
différenciation de ces cellules en culture primaire a également été étudié ainsi qu’un lien
potentiel entre les IPs-VIH et les MMPs. Nous avons ainsi observé que les IPs-VIH affectent
l’expression génique de la MMP-9. Peu de données étant connues quant à la régulation
génique de la MMP-9 et aux cibles potentielles des IPs-VIH, les mécanismes moléculaires
responsables de la diminution de l’expression de MMP-9 par les IPs-VIH ont été déterminés.
Dans une deuxième partie de ce travail, sachant que la FSV est formée de plusieurs
populations cellulaires telles que les cellules progénitrices aux capacités adipogéniques et
angiogéniques (241), les cellules endothéliales capillaires et les cellules immuno-
inflammatoires, nous avons voulu confirmer l’influence de MMP-9 sur l’adipogénèse des
cellules progénitrices isolées. Puis, afin de déterminer les mécanismes qui contrôlent le
devenir des cellules progénitrices, une condition de culture permettant l’étude simultanée de
leurs capacités adipogéniques et angiogéniques a été mise au point.
58

Résultats
I) Implication des MMPs dans la différentiation adipocytaire des cellules de la
fraction stroma-vasculaire du tissu adipeux humain
Un rôle primordial des MMPs et du remodelage matriciel dans le développement du
TA est maintenant suggéré (38, 148, 149, 164). En effet, des travaux menés par notre équipe et
celle de Croissandeau, indiquent que des préadipocytes murins produisent les MMP-2 et -9
(17, 53). De plus, un traitement par des inhibiteurs des MMPs à large spectre (batimastat et
illomastat) ou des anticorps neutralisants dirigés contre les MMP-2 ou -9 limitent
l’adipogenèse suggérant un rôle majeur de ces 2 MMPs dans l’adipogenèse des préadipocytes
murins (17, 53). Toutefois, chez l’humain, les données concernant les MMPs dans le TA sont
peu nombreuses. Des travaux menés par notre équipe ont mis en évidence que des cellules
issues de la FSV du TA humain en culture primaire sécrètent les MMP-2 et -9 au cours de leur
différenciation adipocytaire.
Dans la publication 1, nous avons donc cherché à déterminer l’implication de ces
MMPs dans l’adipogenèse chez l’homme. En parallèle, nous avons également voulu confirmer
l’effet anti-adipogénique des IPs-VIH chez l’homme. En effet, si de nombreuses études ont
mis en évidence un effet inhibiteur des IP-VIHs sur la différenciation de préadipocytes murins
(64, 144, 176, 300), deux études seulement ont été réalisées sur des cellules souches
mésenchymateuses humaines et sur des préadipocytes humains d’origine mammaire (109,
288). Enfin, un lien potentiel entre les MMPs et les IPs-VIH a été recherché.
A) Influence d’un inhibiteur des MMPs et des inhibiteurs de la protéase du VIH
sur la différenciation adipocytaire
Publication 1 : Protease inhibitor treatments reveal specific involvement of matrix
metalloproteinase-9 in human preadipocyte differentiation.
V. Bourlier, A. Zakaroff-Girard, S. De Barros, C. Pizzacalla, V. Durand de Saint Front, M.
Lafontan, A. Bouloumié and J. Galitzky. J Pharmacol Exp Ther, 312:1272-1279, 2005
59

Protease Inhibitor Treatments Reveal Specific Involvement ofMatrix Metalloproteinase-9 in Human Adipocyte Differentiation
Virginie Bourlier, Alexia Zakaroff-Girard, Sandra De Barros, Christophe Pizzacalla,Veronique Durand de Saint Front, Max Lafontan, Anne Bouloumie, and Jean GalitzkyUnite de Recherche sur les Obesites, Institut National de la Sante et de la Recherche Medicale, Institut Louis Bugnard, HopitalRangueil, Universite Paul Sabatier, Toulouse, France (V.B., A.Z.-G., S.D.B., C.P., V.D.d.S.F., M.L., J.G.); and Institut furKardiovaskulare Physiologie, Frankfurt/Main, Germany (A.B.)
Received September 6, 2004; accepted November 9, 2004
ABSTRACTWe previously showed that human and murine 3T3-F442Apreadipocytes produced and released matrix metalloprotein-ases (MMPs) 2 and 9 and that a treatment by MMP inhibitorsresulted in the blockade of murine fat cell adipose conversion.In parallel, investigators reported that other protease inhibitors,the human immunodeficiency virus (HIV) protease inhibitors(PIs) involved in lipodystrophy in humans, also reduced theadipocyte differentiation process of several murine cell lines.The present work was performed to define the effects of MMPinhibitors and HIV-PIs on the human adipocyte differentiationprocess, to clarify the involvement of MMPs in the control ofhuman adipogenesis, and to determine whether HIV-PIs inter-act with MMPs in the control of this process. The effect of twoMMP inhibitor and four HIV-PI treatments on the differentiation
of primary culture human preadipocytes, as well as the putativerelationships between HIV-PIs and MMP-2 and -9 expression,release, or activity were investigated. We showed that MMPinhibitors and HIV-PIs reduced the human adipocyte differen-tiation process as assessed by the decrease of cell proteinand/or triglyceride contents and expression of fatty acid bind-ing protein and hormone-sensitive lipase, two adipocyte mark-ers. Unlike MMP inhibitors, HIV-PIs were devoid of any effectper se on recombinant MMP-2 and 9 activities but reduced theexpression and release of MMP-9 by human preadipocytes.Thus, the present study indicates that the modulation of theextracellular matrix components through the production and/oractivity of MMPs, and, more precisely, MMP-9 might be a keyfactor in the regulation of human adipose tissue development.
The mechanisms responsible for the growth of adiposetissue (AT) are still not well defined. Adipocyte hypertrophyand hyperplasia, due to the recruitment and differentiationof adipocyte precursor cells, or preadipocytes, into adipocytesare major cellular events in the development of the fat mass.Together with the hypertrophy and the hyperplasia events,recent investigations have clearly demonstrated that angio-genesis and extracellular matrix (ECM) remodeling are re-quired for coordinated growth of the fat depot (Lijnen et al.,2002; Rupnick et al., 2002). Among the enzymes involved inthe degradation of the ECM components, the matrix metal-loproteinase (MMP) family is considered to play a major role(Sternlicht and Werb, 2001). In a previous report, we dem-onstrated that mature human adipocytes and preadipocytes
produce and release two members of the MMP family,MMP-2 and -9, the expression and activity of which wereshown to be dependent on the adipocyte differentiation pro-cess (Bouloumie et al., 2001). Interestingly, we also reportedthat treatment of the murine 3T3-F442A preadipocytes,which also produced MMP-2 and -9 during adipose conver-sion, with MMP inhibitors decreased the rate of adipocytedifferentiation, suggesting that MMP activities are requiredfor adipogenesis in rodents. However, no data are availableconcerning the potential role of MMPs in the regulation of ATdevelopment in humans.
A lipodystrophy syndrome, characterized by body fat redis-tribution, hyperlipidemia, and insulin resistance, has beenassociated with the recent use of protease inhibitors (PIs) inthe therapy of AIDS as consequence of human immunodefi-ciency virus (HIV) infection (Carr et al., 1998). The mostprominent clinical sign of HIV-PI-associated lipodystrophy isa loss of subcutaneous fat (lipoatrophy) in the face and theextremities that can be accompanied, in some cases, by fat
This work was supported by Grants 01 128 and 02 197 from the AgenceNationale de Recherche sur le SIDA.
Article, publication date, and citation information can be found athttp://jpet.aspetjournals.org.
doi:10.1124/jpet.104.077263.
ABBREVIATIONS: AT, adipose tissue; ECM, extracellular matrix; MMP, matrix metalloproteinase; PI, protease inhibitor; HIV, human immunode-ficiency virus; IDV, indinavir; RTV, ritonavir; SQV, saquinavir; NFV, nelfinavir; RT-PCR, reverse transcriptase-polymerase chain reaction; HSL,hormone-sensitive lipase; aP2, fatty acid binding protein; NF-�B, nuclear factor-�B; IL, interleukin.
0022-3565/05/3123-1272–1279$20.00THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS Vol. 312, No. 3Copyright © 2005 by The American Society for Pharmacology and Experimental Therapeutics 77263/1194036JPET 312:1272–1279, 2005 Printed in U.S.A.
1272

accumulation in the neck, back, and visceral depots, suggest-ing that it most likely involves HIV-PI-mediated dysregula-tion of the balance between the development and the regres-sion of the AT depending on the anatomical location of the fatdeposits. Numerous studies have examined the effect of HIV-PIs on adipocyte differentiation of murine cell lines and evi-denced that they are potent inhibitors of adipogenesis in vitro(Zhang et al., 1999; Vernochet et al., 2003). Further analysisof the putative targets of HIV-PIs has focused on the tran-scription factors that regulate the expression of adipocyte-specific markers (Dowell et al., 2000; Caron et al., 2001).
The present work was performed to define the effects ofMMP inhibitors and HIV-PIs on the differentiation of prea-dipocytes isolated from the stroma vascular fraction of hu-man AT. It was also expected to clarify the involvement ofMMPs in the control of the adipocyte differentiation processin human AT and to determine whether HIV-PIs interactwith MMPs in the control of this process. We evidenced thatthe broad-spectrum MMP inhibitor batimastat strongly in-hibited human adipocyte differentiation. Moreover, HIV-PIssuch as indinavir (IDV), ritonavir (RTV), saquinavir (SQV),and nelfinavir (NFV) also lead to a strong reduction of thehuman adipocyte differentiation process by a mechanismthat may involve their inhibitory effect on MMP-9 expressionand release by treated preadipocytes. Finally, we showedthat MMP-9 inhibitor also reduced the differentiation of hu-man preadipocytes, suggesting that MMP-9 is specificallyinvolved in the batimastat-mediated inhibition of humanadipogenesis. These data suggest that the modulation of theECM components through the production and/or activity ofMMPs and, more precisely, MMP-9 might be a key regulatorof human AT development.
Materials and MethodsCell Culture and Treatment. Chemicals were from Sigma-Al-
drich (Saint-Quentin Fallavier, France), and cell culture reagentswere from Invitrogen (Cergy Pontoise, France), Roche Diagnostics(Meylan, France), or Cambrex Bio Science (Verviers, Belgium).
Human subcutaneous abdominal white AT was obtained frommoderately overweight women undergoing plastic surgery (mean age39 � 2 years, mean body mass index 26.5 � 0.8 kg/m2). The isolationof human AT-derived stromal cells and the culture of stromal prea-dipocytes (i.e., fat cell precursors) differentiated into adipocytes wereperformed as previously described (10). Briefly, sterile AT was cutinto small pieces and digested under agitation with collagenase (300U/ml) for 1 h 30 min at 37°C. After centrifugation, washing, andfiltration steps, the stromal cells were suspended in Dulbecco’s Mod-ified Eagle’s medium F-12 supplemented with 10% fetal calf serumand plated at 60,000 cells/cm2. After 24 h, the medium was changedfor medium consisting of Dulbecco’s Modified Eagle’s medium F-12supplemented with 33 �M biotin, 17 �M pantothenate, and 50 �g/mlgentamycin (basal medium) in the presence of 66 nM insulin, 1 nMtriiodothyronine, 100 nM cortisol, 10 �g/ml human transferrin (adi-pogenic medium), and, for the first 3 days, 1 �g/ml ciglitazone. Afterthe 3-day priming period, the cells were cultured in the adipogenicmedium supplemented with 1) the MMP inhibitor batimastat [BB94or [4-(N-hydroxyamino)-2R-isobutyl-3S-(thienylthiomethyl)-succi-nyl]-L-phenylalanine-N-methylamide, kindly provided by BritishBiotech, Oxford, UK] or MMP-9 inhibitor [MMP-9/MMP-13 inhibitorII or N-hydroxy-1-(4-methoxyphenyl)sulfonyl-4-benzyloxycarbon-ylpiperazine-2-carboxamide from Calbiochem, VWR International,Strasbourg, France]; or 2) HIV-PIs IDV (kindly provided by Merck,Whitehouse Station, NJ), SQV (kindly provided by Roche, WelwynGarden City, UK), RTV (kindly provided by Abbott Diagnostics,
Abbott Park, IL), or NFV (kindly provided by Roche Diagnostics) for10 days. Batimastat was dissolved in ethanol, whereas HIV-PIs andMMP-9 inhibitor were dissolved in pure dimethylsulfoxide. Controlcells were treated using an appropriate concentration of each vehicle.Medium was replaced every 2 days. Before the experiments, the cellswere placed in basal medium with or without 0.1% bovine serumalbumin (MMP inhibitors versus HIV-PIs, respectively) overnight,supplemented with the various treatments.
The medium was then collected and used in zymography analysis.Cell triglyceride and total protein contents were determined usingkits from Sigma-Aldrich and Bio-Rad S.A (Marnes-la-Coquette,France), respectively.
Gelatin Zymography. Proteins with gelatinolytic activity wereidentified by electrophoresis in the presence of SDS in 8% polyacryl-amide gels containing 1 mg/ml gelatin. Briefly, culture mediumbatches (20 �l) were directly loaded onto gels; after electrophoresis,proteins were renatured by exchanging SDS with 2.5% Triton X-100(20-min incubation repeated twice). The gels were then incubated for16 h at 37°C in 50 mM Tris-HCl, pH 8.8, 5 mM CaCl2, and 0.02%NaN3 and stained with Coomassie Blue. The presence of gelatino-lytic activity in the culture medium batches was visualized as un-colored areas on an otherwise blue gel. Migration of proteins wascompared with that of prestained molecular weight markers. Thegels were scanned by an imaging densitometer and quantified usingthe NIH image program (developed at the U.S. National Institutes ofHealth).
Fluorometric Activity Assay on Recombinant MMP-2 and-9. In vitro assays were performed on 96-well plates. Human recom-binant activated MMP-2 (2 nM) or MMP-9 (4 nM) (Calbiochem) werepreincubated in the presence or absence of increasing concentrationsof batimastat (from 0.01 nM to 0.01 �M) or HIV-PIs (from 1 nM to100 �M) for 30 min at 37°C. Twenty micromolar of the fluorogenicsubstrate Mca-Pro-Leu-Gly-Leu-Dnp-Ala-Arg (Bachem, Bubendorf,Switzerland) were then added to each well, and fluorescence (exci-tation, 325 nm; emission, 390 nm) was recorded every 2 min for 60min. MMP activity was evaluated by the maximum rate per minuteon five separate measurements in each well.
Real-Time Reverse Transcriptase-Polymerase Chain Reac-tion (RT-PCR). Changes in mRNA levels from specific genes werequantified by real-time RT-PCR. Total RNAs were extracted usingQiagen Rneasy Mini Kit according to the manufacturer’s instruc-tions, and RNA concentrations were determined using a fluorometricassay (Ribogreen; Molecular Probes, Eugene, OR). RNA (0.5 �g) wasreverse-transcribed using the ThermoScript RT system (Invitrogen)according to the manufacturer’s instructions. Reverse transcriptionwas also performed without Thermoscript enzyme on RNA samplesto check for any genomic DNA contamination. PCR primers weredesigned using Primer Express software according to the recommen-dations of Applied Biosystems (Foster City, CA). The forward andreverse primer sequences for fatty acid binding protein (aP2), hor-mone-sensitive lipase (HSL), and MMP-9, respectively, were as fol-lows: aP2, GCATGGCCAAACCTAACATGA (forward) and CCTGG-CCCAGTATGAAGGAAA (reverse); HSL, GTGCAAAGACGGAGGA-CCACTCCA (forward) and GACGTCTGGAGTTTCCCCTCAG (re-verse); and MMP-9, CCCTGGAGACCTGAGAACCA (forward) andCCACCCGAGTGTAACCATAGC (reverse).
Each amplification reaction was performed with 15 ng of cDNAsample in duplicate in 96-well optical reaction plates with a Gene-Amp 5700 sequence detection system. The PCR mixture containedforward and reverse primer mix (final concentration, 900 nM forHSL or MMP-9 and 300 nM for aP2) and SYBR Green PCR MasterMix. For ribosomal RNA control (18S rRNA), a mixture containingprimers and fluorogenic probe mix, TaqMan Universal PCR MasterMix (Applied Biosystems) was used. All reactions were performedunder the same conditions: 50°C for 2 min and 95°C for 10 min,followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. Resultswere analyzed with the GeneAmp 5700 software, and all values werenormalized to the levels of 18S rRNA.
MMP-9 Involvement in Human Adipocyte Differentiation 1273
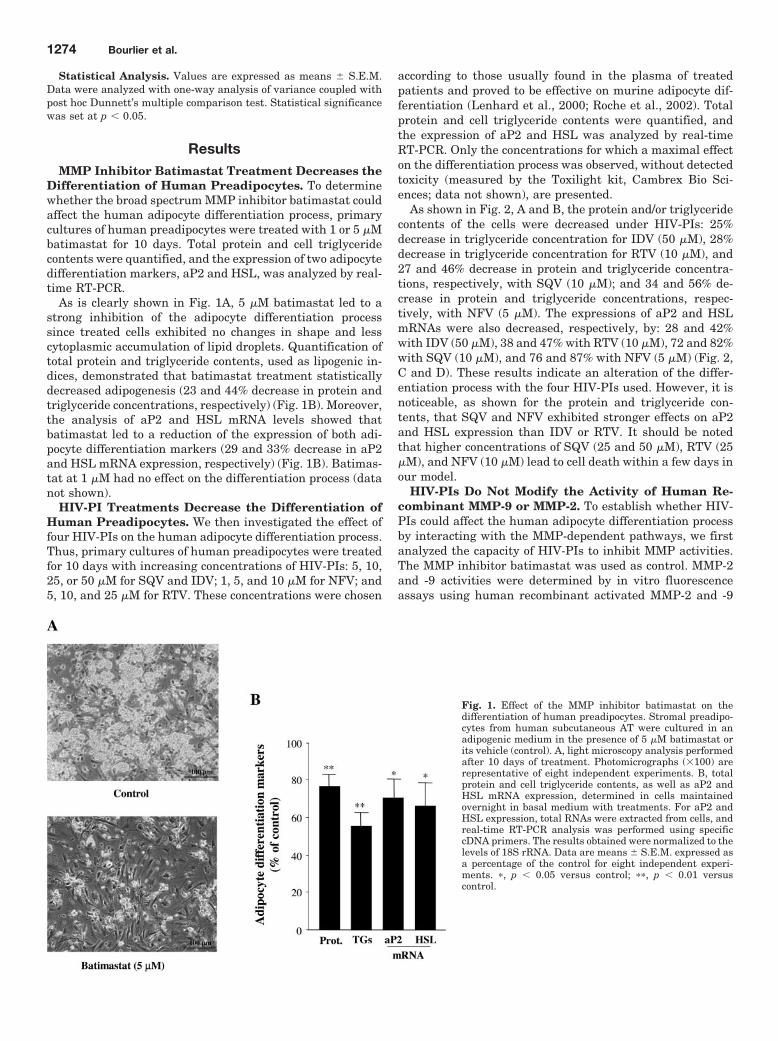
Statistical Analysis. Values are expressed as means � S.E.M.Data were analyzed with one-way analysis of variance coupled withpost hoc Dunnett’s multiple comparison test. Statistical significancewas set at p � 0.05.
ResultsMMP Inhibitor Batimastat Treatment Decreases the
Differentiation of Human Preadipocytes. To determinewhether the broad spectrum MMP inhibitor batimastat couldaffect the human adipocyte differentiation process, primarycultures of human preadipocytes were treated with 1 or 5 �Mbatimastat for 10 days. Total protein and cell triglyceridecontents were quantified, and the expression of two adipocytedifferentiation markers, aP2 and HSL, was analyzed by real-time RT-PCR.
As is clearly shown in Fig. 1A, 5 �M batimastat led to astrong inhibition of the adipocyte differentiation processsince treated cells exhibited no changes in shape and lesscytoplasmic accumulation of lipid droplets. Quantification oftotal protein and triglyceride contents, used as lipogenic in-dices, demonstrated that batimastat treatment statisticallydecreased adipogenesis (23 and 44% decrease in protein andtriglyceride concentrations, respectively) (Fig. 1B). Moreover,the analysis of aP2 and HSL mRNA levels showed thatbatimastat led to a reduction of the expression of both adi-pocyte differentiation markers (29 and 33% decrease in aP2and HSL mRNA expression, respectively) (Fig. 1B). Batimas-tat at 1 �M had no effect on the differentiation process (datanot shown).
HIV-PI Treatments Decrease the Differentiation ofHuman Preadipocytes. We then investigated the effect offour HIV-PIs on the human adipocyte differentiation process.Thus, primary cultures of human preadipocytes were treatedfor 10 days with increasing concentrations of HIV-PIs: 5, 10,25, or 50 �M for SQV and IDV; 1, 5, and 10 �M for NFV; and5, 10, and 25 �M for RTV. These concentrations were chosen
according to those usually found in the plasma of treatedpatients and proved to be effective on murine adipocyte dif-ferentiation (Lenhard et al., 2000; Roche et al., 2002). Totalprotein and cell triglyceride contents were quantified, andthe expression of aP2 and HSL was analyzed by real-timeRT-PCR. Only the concentrations for which a maximal effecton the differentiation process was observed, without detectedtoxicity (measured by the Toxilight kit, Cambrex Bio Sci-ences; data not shown), are presented.
As shown in Fig. 2, A and B, the protein and/or triglyceridecontents of the cells were decreased under HIV-PIs: 25%decrease in triglyceride concentration for IDV (50 �M), 28%decrease in triglyceride concentration for RTV (10 �M), and27 and 46% decrease in protein and triglyceride concentra-tions, respectively, with SQV (10 �M); and 34 and 56% de-crease in protein and triglyceride concentrations, respec-tively, with NFV (5 �M). The expressions of aP2 and HSLmRNAs were also decreased, respectively, by: 28 and 42%with IDV (50 �M), 38 and 47% with RTV (10 �M), 72 and 82%with SQV (10 �M), and 76 and 87% with NFV (5 �M) (Fig. 2,C and D). These results indicate an alteration of the differ-entiation process with the four HIV-PIs used. However, it isnoticeable, as shown for the protein and triglyceride con-tents, that SQV and NFV exhibited stronger effects on aP2and HSL expression than IDV or RTV. It should be notedthat higher concentrations of SQV (25 and 50 �M), RTV (25�M), and NFV (10 �M) lead to cell death within a few days inour model.
HIV-PIs Do Not Modify the Activity of Human Re-combinant MMP-9 or MMP-2. To establish whether HIV-PIs could affect the human adipocyte differentiation processby interacting with the MMP-dependent pathways, we firstanalyzed the capacity of HIV-PIs to inhibit MMP activities.The MMP inhibitor batimastat was used as control. MMP-2and -9 activities were determined by in vitro fluorescenceassays using human recombinant activated MMP-2 and -9
Fig. 1. Effect of the MMP inhibitor batimastat on thedifferentiation of human preadipocytes. Stromal preadipo-cytes from human subcutaneous AT were cultured in anadipogenic medium in the presence of 5 �M batimastat orits vehicle (control). A, light microscopy analysis performedafter 10 days of treatment. Photomicrographs (�100) arerepresentative of eight independent experiments. B, totalprotein and cell triglyceride contents, as well as aP2 andHSL mRNA expression, determined in cells maintainedovernight in basal medium with treatments. For aP2 andHSL expression, total RNAs were extracted from cells, andreal-time RT-PCR analysis was performed using specificcDNA primers. The results obtained were normalized to thelevels of 18S rRNA. Data are means � S.E.M. expressed asa percentage of the control for eight independent experi-ments. �, p � 0.05 versus control; ��, p � 0.01 versuscontrol.
1274 Bourlier et al.
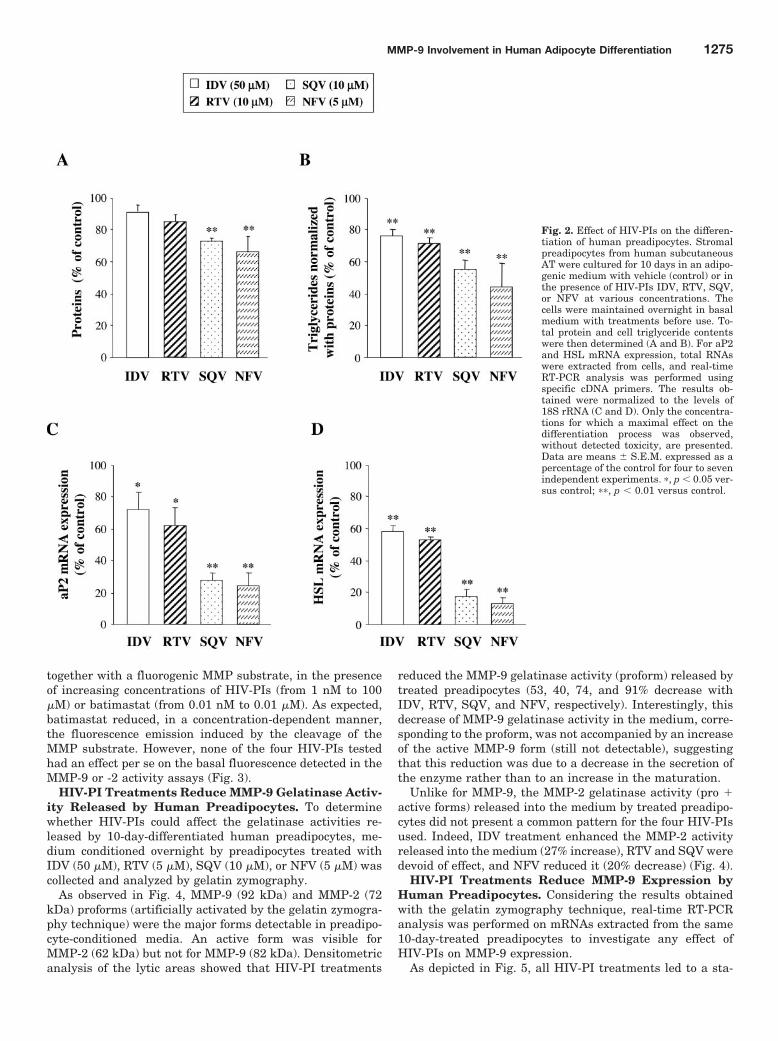
together with a fluorogenic MMP substrate, in the presenceof increasing concentrations of HIV-PIs (from 1 nM to 100�M) or batimastat (from 0.01 nM to 0.01 �M). As expected,batimastat reduced, in a concentration-dependent manner,the fluorescence emission induced by the cleavage of theMMP substrate. However, none of the four HIV-PIs testedhad an effect per se on the basal fluorescence detected in theMMP-9 or -2 activity assays (Fig. 3).
HIV-PI Treatments Reduce MMP-9 Gelatinase Activ-ity Released by Human Preadipocytes. To determinewhether HIV-PIs could affect the gelatinase activities re-leased by 10-day-differentiated human preadipocytes, me-dium conditioned overnight by preadipocytes treated withIDV (50 �M), RTV (5 �M), SQV (10 �M), or NFV (5 �M) wascollected and analyzed by gelatin zymography.
As observed in Fig. 4, MMP-9 (92 kDa) and MMP-2 (72kDa) proforms (artificially activated by the gelatin zymogra-phy technique) were the major forms detectable in preadipo-cyte-conditioned media. An active form was visible forMMP-2 (62 kDa) but not for MMP-9 (82 kDa). Densitometricanalysis of the lytic areas showed that HIV-PI treatments
reduced the MMP-9 gelatinase activity (proform) released bytreated preadipocytes (53, 40, 74, and 91% decrease withIDV, RTV, SQV, and NFV, respectively). Interestingly, thisdecrease of MMP-9 gelatinase activity in the medium, corre-sponding to the proform, was not accompanied by an increaseof the active MMP-9 form (still not detectable), suggestingthat this reduction was due to a decrease in the secretion ofthe enzyme rather than to an increase in the maturation.
Unlike for MMP-9, the MMP-2 gelatinase activity (pro �active forms) released into the medium by treated preadipo-cytes did not present a common pattern for the four HIV-PIsused. Indeed, IDV treatment enhanced the MMP-2 activityreleased into the medium (27% increase), RTV and SQV weredevoid of effect, and NFV reduced it (20% decrease) (Fig. 4).
HIV-PI Treatments Reduce MMP-9 Expression byHuman Preadipocytes. Considering the results obtainedwith the gelatin zymography technique, real-time RT-PCRanalysis was performed on mRNAs extracted from the same10-day-treated preadipocytes to investigate any effect ofHIV-PIs on MMP-9 expression.
As depicted in Fig. 5, all HIV-PI treatments led to a sta-
Fig. 2. Effect of HIV-PIs on the differen-tiation of human preadipocytes. Stromalpreadipocytes from human subcutaneousAT were cultured for 10 days in an adipo-genic medium with vehicle (control) or inthe presence of HIV-PIs IDV, RTV, SQV,or NFV at various concentrations. Thecells were maintained overnight in basalmedium with treatments before use. To-tal protein and cell triglyceride contentswere then determined (A and B). For aP2and HSL mRNA expression, total RNAswere extracted from cells, and real-timeRT-PCR analysis was performed usingspecific cDNA primers. The results ob-tained were normalized to the levels of18S rRNA (C and D). Only the concentra-tions for which a maximal effect on thedifferentiation process was observed,without detected toxicity, are presented.Data are means � S.E.M. expressed as apercentage of the control for four to sevenindependent experiments. �, p � 0.05 ver-sus control; ��, p � 0.01 versus control.
MMP-9 Involvement in Human Adipocyte Differentiation 1275
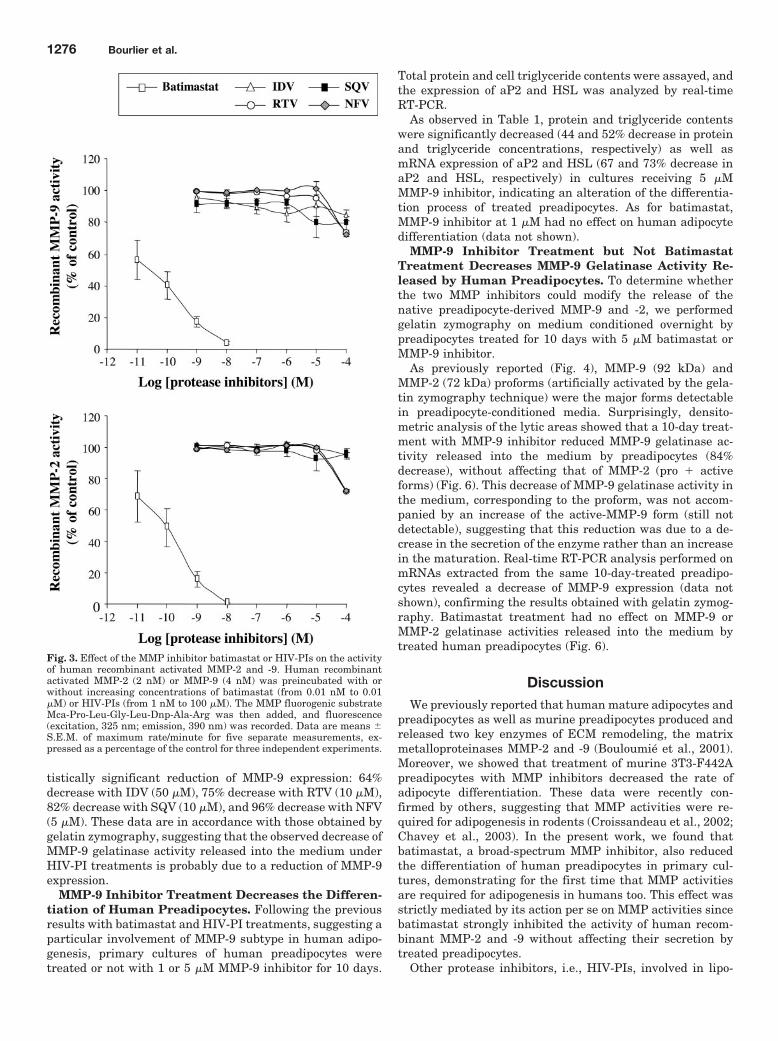
tistically significant reduction of MMP-9 expression: 64%decrease with IDV (50 �M), 75% decrease with RTV (10 �M),82% decrease with SQV (10 �M), and 96% decrease with NFV(5 �M). These data are in accordance with those obtained bygelatin zymography, suggesting that the observed decrease ofMMP-9 gelatinase activity released into the medium underHIV-PI treatments is probably due to a reduction of MMP-9expression.
MMP-9 Inhibitor Treatment Decreases the Differen-tiation of Human Preadipocytes. Following the previousresults with batimastat and HIV-PI treatments, suggesting aparticular involvement of MMP-9 subtype in human adipo-genesis, primary cultures of human preadipocytes weretreated or not with 1 or 5 �M MMP-9 inhibitor for 10 days.
Total protein and cell triglyceride contents were assayed, andthe expression of aP2 and HSL was analyzed by real-timeRT-PCR.
As observed in Table 1, protein and triglyceride contentswere significantly decreased (44 and 52% decrease in proteinand triglyceride concentrations, respectively) as well asmRNA expression of aP2 and HSL (67 and 73% decrease inaP2 and HSL, respectively) in cultures receiving 5 �MMMP-9 inhibitor, indicating an alteration of the differentia-tion process of treated preadipocytes. As for batimastat,MMP-9 inhibitor at 1 �M had no effect on human adipocytedifferentiation (data not shown).
MMP-9 Inhibitor Treatment but Not BatimastatTreatment Decreases MMP-9 Gelatinase Activity Re-leased by Human Preadipocytes. To determine whetherthe two MMP inhibitors could modify the release of thenative preadipocyte-derived MMP-9 and -2, we performedgelatin zymography on medium conditioned overnight bypreadipocytes treated for 10 days with 5 �M batimastat orMMP-9 inhibitor.
As previously reported (Fig. 4), MMP-9 (92 kDa) andMMP-2 (72 kDa) proforms (artificially activated by the gela-tin zymography technique) were the major forms detectablein preadipocyte-conditioned media. Surprisingly, densito-metric analysis of the lytic areas showed that a 10-day treat-ment with MMP-9 inhibitor reduced MMP-9 gelatinase ac-tivity released into the medium by preadipocytes (84%decrease), without affecting that of MMP-2 (pro � activeforms) (Fig. 6). This decrease of MMP-9 gelatinase activity inthe medium, corresponding to the proform, was not accom-panied by an increase of the active-MMP-9 form (still notdetectable), suggesting that this reduction was due to a de-crease in the secretion of the enzyme rather than an increasein the maturation. Real-time RT-PCR analysis performed onmRNAs extracted from the same 10-day-treated preadipo-cytes revealed a decrease of MMP-9 expression (data notshown), confirming the results obtained with gelatin zymog-raphy. Batimastat treatment had no effect on MMP-9 orMMP-2 gelatinase activities released into the medium bytreated human preadipocytes (Fig. 6).
DiscussionWe previously reported that human mature adipocytes and
preadipocytes as well as murine preadipocytes produced andreleased two key enzymes of ECM remodeling, the matrixmetalloproteinases MMP-2 and -9 (Bouloumie et al., 2001).Moreover, we showed that treatment of murine 3T3-F442Apreadipocytes with MMP inhibitors decreased the rate ofadipocyte differentiation. These data were recently con-firmed by others, suggesting that MMP activities were re-quired for adipogenesis in rodents (Croissandeau et al., 2002;Chavey et al., 2003). In the present work, we found thatbatimastat, a broad-spectrum MMP inhibitor, also reducedthe differentiation of human preadipocytes in primary cul-tures, demonstrating for the first time that MMP activitiesare required for adipogenesis in humans too. This effect wasstrictly mediated by its action per se on MMP activities sincebatimastat strongly inhibited the activity of human recom-binant MMP-2 and -9 without affecting their secretion bytreated preadipocytes.
Other protease inhibitors, i.e., HIV-PIs, involved in lipo-
Fig. 3. Effect of the MMP inhibitor batimastat or HIV-PIs on the activityof human recombinant activated MMP-2 and -9. Human recombinantactivated MMP-2 (2 nM) or MMP-9 (4 nM) was preincubated with orwithout increasing concentrations of batimastat (from 0.01 nM to 0.01�M) or HIV-PIs (from 1 nM to 100 �M). The MMP fluorogenic substrateMca-Pro-Leu-Gly-Leu-Dnp-Ala-Arg was then added, and fluorescence(excitation, 325 nm; emission, 390 nm) was recorded. Data are means �S.E.M. of maximum rate/minute for five separate measurements, ex-pressed as a percentage of the control for three independent experiments.
1276 Bourlier et al.
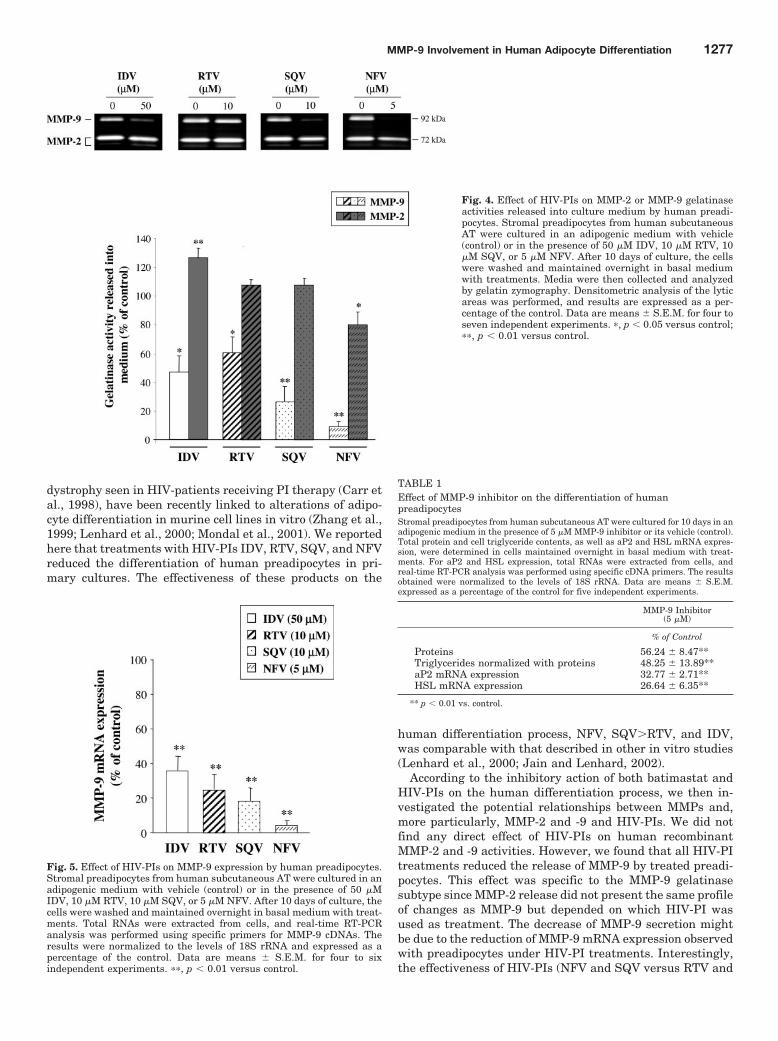
dystrophy seen in HIV-patients receiving PI therapy (Carr etal., 1998), have been recently linked to alterations of adipo-cyte differentiation in murine cell lines in vitro (Zhang et al.,1999; Lenhard et al., 2000; Mondal et al., 2001). We reportedhere that treatments with HIV-PIs IDV, RTV, SQV, and NFVreduced the differentiation of human preadipocytes in pri-mary cultures. The effectiveness of these products on the
human differentiation process, NFV, SQV�RTV, and IDV,was comparable with that described in other in vitro studies(Lenhard et al., 2000; Jain and Lenhard, 2002).
According to the inhibitory action of both batimastat andHIV-PIs on the human differentiation process, we then in-vestigated the potential relationships between MMPs and,more particularly, MMP-2 and -9 and HIV-PIs. We did notfind any direct effect of HIV-PIs on human recombinantMMP-2 and -9 activities. However, we found that all HIV-PItreatments reduced the release of MMP-9 by treated preadi-pocytes. This effect was specific to the MMP-9 gelatinasesubtype since MMP-2 release did not present the same profileof changes as MMP-9 but depended on which HIV-PI wasused as treatment. The decrease of MMP-9 secretion mightbe due to the reduction of MMP-9 mRNA expression observedwith preadipocytes under HIV-PI treatments. Interestingly,the effectiveness of HIV-PIs (NFV and SQV versus RTV and
Fig. 5. Effect of HIV-PIs on MMP-9 expression by human preadipocytes.Stromal preadipocytes from human subcutaneous AT were cultured in anadipogenic medium with vehicle (control) or in the presence of 50 �MIDV, 10 �M RTV, 10 �M SQV, or 5 �M NFV. After 10 days of culture, thecells were washed and maintained overnight in basal medium with treat-ments. Total RNAs were extracted from cells, and real-time RT-PCRanalysis was performed using specific primers for MMP-9 cDNAs. Theresults were normalized to the levels of 18S rRNA and expressed as apercentage of the control. Data are means � S.E.M. for four to sixindependent experiments. ��, p � 0.01 versus control.
Fig. 4. Effect of HIV-PIs on MMP-2 or MMP-9 gelatinaseactivities released into culture medium by human preadi-pocytes. Stromal preadipocytes from human subcutaneousAT were cultured in an adipogenic medium with vehicle(control) or in the presence of 50 �M IDV, 10 �M RTV, 10�M SQV, or 5 �M NFV. After 10 days of culture, the cellswere washed and maintained overnight in basal mediumwith treatments. Media were then collected and analyzedby gelatin zymography. Densitometric analysis of the lyticareas was performed, and results are expressed as a per-centage of the control. Data are means � S.E.M. for four toseven independent experiments. �, p � 0.05 versus control;��, p � 0.01 versus control.
TABLE 1Effect of MMP-9 inhibitor on the differentiation of humanpreadipocytesStromal preadipocytes from human subcutaneous AT were cultured for 10 days in anadipogenic medium in the presence of 5 �M MMP-9 inhibitor or its vehicle (control).Total protein and cell triglyceride contents, as well as aP2 and HSL mRNA expres-sion, were determined in cells maintained overnight in basal medium with treat-ments. For aP2 and HSL expression, total RNAs were extracted from cells, andreal-time RT-PCR analysis was performed using specific cDNA primers. The resultsobtained were normalized to the levels of 18S rRNA. Data are means � S.E.M.expressed as a percentage of the control for five independent experiments.
MMP-9 Inhibitor(5 �M)
% of Control
Proteins 56.24 � 8.47**Triglycerides normalized with proteins 48.25 � 13.89**aP2 mRNA expression 32.77 � 2.71**HSL mRNA expression 26.64 � 6.35**
** p � 0.01 vs. control.
MMP-9 Involvement in Human Adipocyte Differentiation 1277
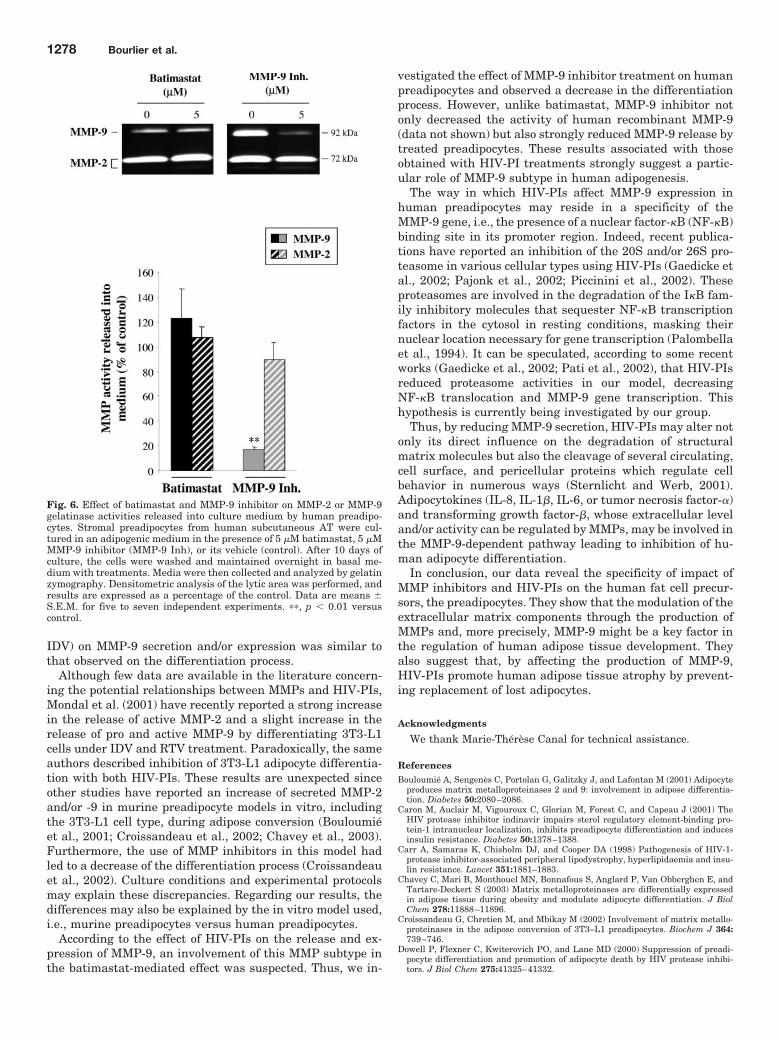
IDV) on MMP-9 secretion and/or expression was similar tothat observed on the differentiation process.
Although few data are available in the literature concern-ing the potential relationships between MMPs and HIV-PIs,Mondal et al. (2001) have recently reported a strong increasein the release of active MMP-2 and a slight increase in therelease of pro and active MMP-9 by differentiating 3T3-L1cells under IDV and RTV treatment. Paradoxically, the sameauthors described inhibition of 3T3-L1 adipocyte differentia-tion with both HIV-PIs. These results are unexpected sinceother studies have reported an increase of secreted MMP-2and/or -9 in murine preadipocyte models in vitro, includingthe 3T3-L1 cell type, during adipose conversion (Bouloumieet al., 2001; Croissandeau et al., 2002; Chavey et al., 2003).Furthermore, the use of MMP inhibitors in this model hadled to a decrease of the differentiation process (Croissandeauet al., 2002). Culture conditions and experimental protocolsmay explain these discrepancies. Regarding our results, thedifferences may also be explained by the in vitro model used,i.e., murine preadipocytes versus human preadipocytes.
According to the effect of HIV-PIs on the release and ex-pression of MMP-9, an involvement of this MMP subtype inthe batimastat-mediated effect was suspected. Thus, we in-
vestigated the effect of MMP-9 inhibitor treatment on humanpreadipocytes and observed a decrease in the differentiationprocess. However, unlike batimastat, MMP-9 inhibitor notonly decreased the activity of human recombinant MMP-9(data not shown) but also strongly reduced MMP-9 release bytreated preadipocytes. These results associated with thoseobtained with HIV-PI treatments strongly suggest a partic-ular role of MMP-9 subtype in human adipogenesis.
The way in which HIV-PIs affect MMP-9 expression inhuman preadipocytes may reside in a specificity of theMMP-9 gene, i.e., the presence of a nuclear factor-�B (NF-�B)binding site in its promoter region. Indeed, recent publica-tions have reported an inhibition of the 20S and/or 26S pro-teasome in various cellular types using HIV-PIs (Gaedicke etal., 2002; Pajonk et al., 2002; Piccinini et al., 2002). Theseproteasomes are involved in the degradation of the I�B fam-ily inhibitory molecules that sequester NF-�B transcriptionfactors in the cytosol in resting conditions, masking theirnuclear location necessary for gene transcription (Palombellaet al., 1994). It can be speculated, according to some recentworks (Gaedicke et al., 2002; Pati et al., 2002), that HIV-PIsreduced proteasome activities in our model, decreasingNF-�B translocation and MMP-9 gene transcription. Thishypothesis is currently being investigated by our group.
Thus, by reducing MMP-9 secretion, HIV-PIs may alter notonly its direct influence on the degradation of structuralmatrix molecules but also the cleavage of several circulating,cell surface, and pericellular proteins which regulate cellbehavior in numerous ways (Sternlicht and Werb, 2001).Adipocytokines (IL-8, IL-1�, IL-6, or tumor necrosis factor-�)and transforming growth factor-�, whose extracellular leveland/or activity can be regulated by MMPs, may be involved inthe MMP-9-dependent pathway leading to inhibition of hu-man adipocyte differentiation.
In conclusion, our data reveal the specificity of impact ofMMP inhibitors and HIV-PIs on the human fat cell precur-sors, the preadipocytes. They show that the modulation of theextracellular matrix components through the production ofMMPs and, more precisely, MMP-9 might be a key factor inthe regulation of human adipose tissue development. Theyalso suggest that, by affecting the production of MMP-9,HIV-PIs promote human adipose tissue atrophy by prevent-ing replacement of lost adipocytes.
Acknowledgments
We thank Marie-Therese Canal for technical assistance.
ReferencesBouloumie A, Sengenes C, Portolan G, Galitzky J, and Lafontan M (2001) Adipocyte
produces matrix metalloproteinases 2 and 9: involvement in adipose differentia-tion. Diabetes 50:2080–2086.
Caron M, Auclair M, Vigouroux C, Glorian M, Forest C, and Capeau J (2001) TheHIV protease inhibitor indinavir impairs sterol regulatory element-binding pro-tein-1 intranuclear localization, inhibits preadipocyte differentiation and inducesinsulin resistance. Diabetes 50:1378–1388.
Carr A, Samaras K, Chisholm DJ, and Cooper DA (1998) Pathogenesis of HIV-1-protease inhibitor-associated peripheral lipodystrophy, hyperlipidaemia and insu-lin resistance. Lancet 351:1881–1883.
Chavey C, Mari B, Monthouel MN, Bonnafous S, Anglard P, Van Obberghen E, andTartare-Deckert S (2003) Matrix metalloproteinases are differentially expressedin adipose tissue during obesity and modulate adipocyte differentiation. J BiolChem 278:11888–11896.
Croissandeau G, Chretien M, and Mbikay M (2002) Involvement of matrix metallo-proteinases in the adipose conversion of 3T3–L1 preadipocytes. Biochem J 364:739–746.
Dowell P, Flexner C, Kwiterovich PO, and Lane MD (2000) Suppression of preadi-pocyte differentiation and promotion of adipocyte death by HIV protease inhibi-tors. J Biol Chem 275:41325–41332.
Fig. 6. Effect of batimastat and MMP-9 inhibitor on MMP-2 or MMP-9gelatinase activities released into culture medium by human preadipo-cytes. Stromal preadipocytes from human subcutaneous AT were cul-tured in an adipogenic medium in the presence of 5 �M batimastat, 5 �MMMP-9 inhibitor (MMP-9 Inh), or its vehicle (control). After 10 days ofculture, the cells were washed and maintained overnight in basal me-dium with treatments. Media were then collected and analyzed by gelatinzymography. Densitometric analysis of the lytic area was performed, andresults are expressed as a percentage of the control. Data are means �S.E.M. for five to seven independent experiments. ��, p � 0.01 versuscontrol.
1278 Bourlier et al.

Gaedicke S, Firat-Geier E, Constantiniu O, Lucchiari-Hartz M, Freudenberg M,Galanos C, and Niedermann G (2002) Antitumor effect of the human immunode-ficiency virus protease inhibitor ritonavir: induction of tumor-cell apoptosis asso-ciated with perturbation of proteasomal proteolysis. Cancer Res 62:6901–6908.
Jain RG and Lenhard JM (2002) Select HIV protease inhibitors alter bone and fatmetabolism ex vivo. J Biol Chem 277:19247–19250.
Lenhard JM, Furfine ES, Jain RG, Ittoop O, Orband-Miller LA, Blanchard SG,Paulik MA, and Weiel JE (2000) HIV protease inhibitors block adipogenesis andincrease lipolysis in vitro. Antiviral Res 47:121–129.
Lijnen HR, Maquoi E, Hansen LB, Van Hoef B, Frederix L, and Collen D (2002)Matrix metalloproteinase inhibition impairs adipose tissue development in mice.Arterioscler Thromb Vasc Biol 22:374–379.
Mondal D, Larussa VF, and Agrawal KC (2001) Synergistic antiadipogenic effects ofHIV type 1 protease inhibitors with tumor necrosis factor alpha: suppression ofextracellular insulin action mediated by extracellular matrix-degrading proteasesAIDS Res Hum Retroviruses 17:1569–1584.
Pajonk F, Himmelsbach J, Riess K, Sommer A, and McBride WH (2002) The humanimmunodeficiency virus (HIV)-1 protease inhibitor saquinavir inhibits proteasomefunction and causes apoptosis and radiosensitization in non-HIV-associated hu-man cancer cells. Cancer Res 62:5230–5235.
Palombella VJ, Rando OJ, Goldberg AL, and Maniatis T (1994) The ubiquitin-proteasome pathway is required for processing the NF-kappa B1 precursor proteinand the activation of NF-kappa B. Cell 78:773–785.
Pati S, Pelser CB, Dufraine J, Bryant JL, Reitz MS Jr, and Weichold FF (2002)
Antitumorigenic effects of HIV protease inhibitor ritonavir: inhibition of Kaposisarcoma. Blood 99:3771–3779.
Piccinini M, Rinaudo MT, Chiapello N, Ricotti E, Baldovino S, Mostert M, and TovoPA (2002) The human 26S proteasome is a target of antiretroviral agents. AIDS16:693–700.
Roche R, Poizont-Martin I, Martin-El Yazidi C, Compe E, Gastaut JA, Torresani J,and Planells R (2002) Effects of antiretroviral drug combinations on the differen-tiation of adipocytes. AIDS 16:13–20.
Rupnick MA, Panigrahy D, Zhang CY, Dallabrida SM, Lowell BB, Langer R, andFolkman MJ (2002) Adipose tissue mass can be regulated through the vasculature.Proc Natl Acad Sci USA 99:10730–10735.
Sternlicht MD and Werb Z (2001) How matrix metalloproteinases regulate cellbehavior. Annu Rev Cell Dev Biol 17:463–516.
Vernochet C, Azoulay S, Duval D, Guedj R, Ailhaud G, and Dani C (2003) Differen-tial effect of HIV protease inhibitors on adipogenesis: intracellular ritonavir is notsufficient to inhibit differentiation. AIDS 17:2177–2180.
Zhang B, MacNaul K, Szalkowski D, Li Z, Berger J, and Moller DE (1999) Inhibitionof adipocyte differentiation by HIV protease inhibitors. J Clin Endocrinol Metab84:4274–4277.
Address correspondence to: Dr. Virginie Bourlier, Institut National de laSante et de la Recherche Medicale U586, 37 Allees Jules Guesde, 31073Toulouse, France. E-mail: [email protected]
MMP-9 Involvement in Human Adipocyte Differentiation 1279

Discussion de la publication 1
Nous avons travaillé sur des cultures primaires de cellules de la FSV du TA humain,
issues de dermolipectomies abdominales. Nos résultats montrent que les cellules de la FSV
cultivées dans un milieu adipogénique et traitées pendant 10 jours avec un inhibiteur des
MMPs à large spectre, le batimastat, ont une adipogenèse altérée. En effet, il a été constaté
que les cellules traitées ont un contenu intracellulaire en TGs diminué par rapport aux cellules
contrôles. Il en est de même pour l’expression en ARNm de deux marqueurs adipocytaires,
aP2 et la LHS. Ces données indiquent que le batimastat limite la différenciation adipocytaire
des cellules de la FSV du TA humain suggérant un rôle majeur des MMPs dans l’adipogenèse
chez l’homme.
En parallèle, nous avons déterminé l’effet de quatre inhibiteurs de la protéase du VIH:
l’indinavir (IDV), le ritonavir (RTV), le saquinavir (SQV) et le nelfinavir (NFV) sur la
différenciation des cellules de la FSV du TA humain. Nous avons observé que les quatre IPs-
VIH utilisés limitent la différenciation adipocytaire des cellules de la FSV en culture
primaire. En accord avec nos résultats, une étude récente indiquent que le RTV bloque
l’adipogenèse des préadipocytes humains de type SGBS (Syndrome de Simpson-Golabi-
Behmel) (92, 123). Il faut préciser que dans notre étude en accord avec des résultats décrits
dans d’autres études in vitro sur des préadipocytes murins que le SQV et le NFV montrent
des effets plus marqués sur la différenciation adipocytaire que l’IDV et le RTV (109, 144).
Ainsi, le syndrome de lipoatrophie associé aux IPs-VIH pourrait donc être relatif aux effets
apoptotiques observés sur les adipocytes matures (12, 64) mais également aux effets négatifs
sur l’adipogenèse.
Les effets anti-différenciant obtenus avec les inhibiteurs de protéases matricielles tels
que le batimastat et les inhibiteurs de la protéase du VIH, nous ont conduit à émettre
l’hypothèse d’un lien entre les MMPs et les IPs-VIH. Afin de déterminer si les IPs-VIH
affectent la différenciation adipocytaire via une voie dépendante des MMPs, nous avons
étudié leur capacité à inhiber directement l’activité des MMP-2 et -9 par une approche in
vitro en utilisant un substrat synthétique des MMPs qui libère un peptide fluorescent après
l’action des MMP-2 et -9 recombinantes en présence des IPs-VIH ou d’inhibiteurs des
MMPs. Aucun effet direct des IPs-VIH sur l’activité des protéines humaines recombinantes
MMP-2 et -9 n’a été observé contrairement au batimastat. Cependant et de manière
surprenante, nous avons observé que les quatre IPs-VIH diminuent spécifiquement l’activité
gélatinase de MMP-9 relarguée dans le milieu de culture alors que l’activité de MMP-2 reste
60

maintenue. Cette diminution de la sécrétion de MMP-9 par les IPs-VIH est associée à une
diminution des taux de transcrits pour MMP-9 suggérant que l’effet anti-adipogénique des
IPs-VIH est associé à l’inhibition spécifique de MMP-9 via un effet transcriptionnel sur son
expression. Le rôle spécifique de MMP-9 dans l’adipogenèse a été confirmé par un inhibiteur
dirigé contre MMP-9 (MMP-9/MMP-13 inhibitor II). Cet inhibiteur diminue la
différenciation des cellules de la FSV du TA humain et réduit l’activité mais également
l’expression de MMP-9. De plus, l’effet d’un anticorps neutralisant dirigé contre la MMP-9
dans la différenciation adipocytaire des cellules de la FSV du TA humain a fait l’objet d’une
étude complémentaire.
B) Influence d’un anticorps neutralisant dirigé contre la MMP-9 sur la
différenciation adipocytaire
Comme pour les précédents traitements décrits dans la publication 1, les cellules de la
FSV ont été cultivées en milieu adipogénique et traitées avec un anticorps neutralisant dirigé
spécifiquement contre la MMP-9 (Chemicon). Comme le montre la figure 14, après 10 jours
de traitement, le contenu intracellulaire en TGs est fortement diminué ainsi que l’expression
en ARNm des marqueurs adipocytaire aP2 et LHS. Ces résultats indiquent donc que l’activité
de MMP-9 est spécifiquement requise pour le bon déroulement de la différenciation
adipocytaire chez l’homme et suggèrent que les effets anti-adipogéniques des IPs-VIH
pourraient être médiés par la chute de l’expression de MMP-9.
Comme nous l’avons évoqué dans le chapitre d’introduction, les mécanismes
d’actions des IPs-VIH sur l’adipogenèse sont encore peu connus. Des travaux ont rapporté
que les IPs-VIH peuvent altérer la maturation et la stabilité de la lamine A/C, perturbant ainsi
la localisation cellulaire de SREBP-1 et l’adipogenèse de préadipocytes murins (29). Il a
également été décrit que les IPs-VIH pouvaient inhiber l’activité du protéasome de certaines
cellules cancéreuses (4, 239) et plus récemment celle des préadipocytes murins de la lignée
3T3-L1 (202). En revanche, l’effet des IPs-VIH sur l’activité du protéasome des
préadipocytes humains reste à déterminer et a fait l’objet de la suite de ce travail. L’activité
du protéasome a de plus été associée à de nombreuses fonctions cellulaires telles que la
progression dans le cycle cellulaire, l’apoptose et la régulation de l’expression de nombreux
gènes notamment en modulant l’activité du facteur de transcription NF-κB (1, 125). Quelques
données parcellaires montrent que l’expression génique de MMP-9, contrairement à celle de
61
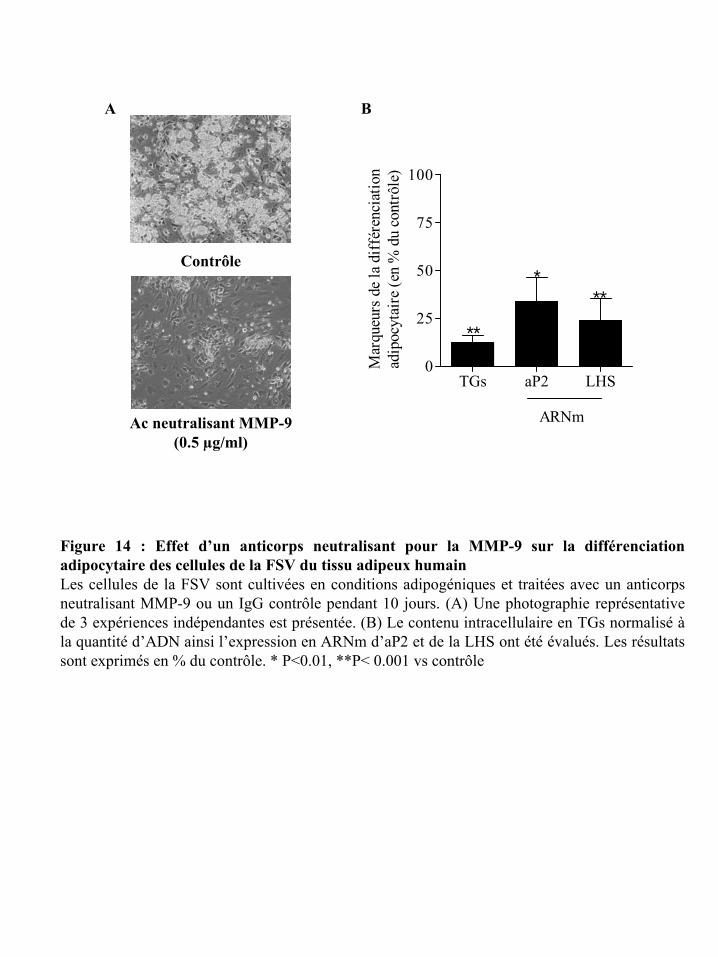
TGs aP2 LHS0
25
50
75
100
**
***
ARNm
Mar
queu
rs d
e la
dif
fére
ncia
tion
adip
ocyt
aire
(en
% d
u co
ntrô
le)
Contrôle
Ac
neutralisant MMP-9(0.5 µg/ml)
A B
Figure 14 : Effet d’un anticorps neutralisant pour la MMP-9 sur la différenciation adipocytaire
des cellules de la FSV du tissu adipeux humainLes cellules de la FSV sont cultivées en conditions adipogéniques
et traitées avec un anticorps neutralisant MMP-9 ou un IgG
contrôle pendant 10 jours. (A) Une photographie représentative de 3 expériences indépendantes est présentée. (B) Le contenu intracellulaire en TGs
normalisé
à
la quantité
d’ADN ainsi l’expression en ARNm
d’aP2 et de la LHS ont été
évalués. Les résultats sont exprimés en % du contrôle. * P<0.01, **P< 0.001 vs contrôle

MMP-2, peut-être régulée par NF-κB. En effet, au niveau du promoteur de MMP-9, un site de
liaison pour ce facteur de transcription a été décrit (236, 256). Ainsi, en regroupant
l’ensemble de ces données, nous avons émis l’hypothèse que les IPs-VIH diminuent l’activité
protéasomique des préadipocytes humains empêchant ainsi la dégradation d’IκB, l’inhibiteur
de NF-κB, qui interagit avec NF-κB et le séquestre dans le cytoplasme. Par conséquent, NF-
κB ne peut pas être transféré dans le noyau et entraîner la transcription de gènes notamment
celui de MMP-9. Pour plus de clarté, cette hypothèse est schématisée dans la figure 15.
C) Détermination des mécanismes impliqués dans la chute de l’expression de
MMP-9 par les inhibiteurs de la protéase du VIH
Publication 2 : Inhibition of human preadipocyte proteosomal activity, by HIV protease
inhibitor or specific inhibitor lacatacystin, leads to a defect in adipogenesis which involves
matrix metalloproteinase-9. S. De Barros, A. Zakaroff-Girard, M. Lafontan, J. Galitzky and
V. Bourlier J Pharmacol Exp Ther, 320:291-299, 2007
62
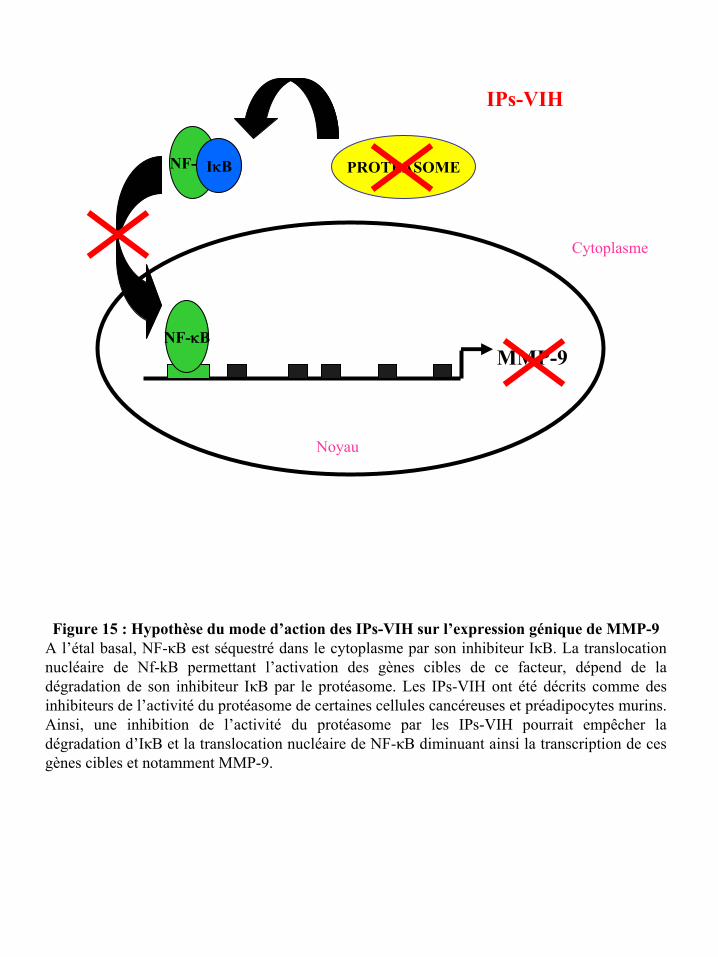
MMP-9NF-κB
NF-κBIκB
Noyau
Cytoplasme
PROTEASOME
IPs-VIH
Figure 15 : Hypothèse du mode d’action des IPs-VIH
sur l’expression génique de MMP-9A l’étal basal, NF-κB est séquestré
dans le cytoplasme par son inhibiteur IκB. La translocation nucléaire de Nf-kB
permettant l’activation des gènes cibles de ce facteur, dépend de la
dégradation de son inhibiteur IκB par le protéasome. Les IPs-VIH
ont été
décrits comme des inhibiteurs de l’activité
du protéasome
de certaines cellules cancéreuses et préadipocytes
murins. Ainsi, une inhibition de l’activité
du protéasome
par les IPs-VIH
pourrait empêcher la
dégradation d’IκB et la translocation nucléaire de NF-κB diminuant ainsi la transcription de ces gènes cibles et notamment MMP-9.

Inhibition of Human Preadipocyte Proteasomal Activity byHIV Protease Inhibitors or Specific Inhibitor LactacystinLeads to a Defect in Adipogenesis, Which Involves MatrixMetalloproteinase-9
Sandra De Barros, Alexia Zakaroff-Girard, Max Lafontan, Jean Galitzky,and Virginie BourlierUnite de Recherche sur les Obesites, Institut National de la Sante et de la Recherche Medical Unite 586, Institut LouisBugnard, Hopital Rangueil, Universite Paul Sabatier, Toulouse, France
Received August 2, 2006; accepted October 10, 2006
ABSTRACTIn a previous publication, we reported that human immunode-ficiency virus (HIV) protease inhibitors (PIs) inhibited the differ-entiation of human preadipocytes in primary culture, reducingthe expression and secretion of matrix metalloproteinase 9(MMP-9). The present work was performed to clarify this mech-anism. Interestingly, HIV-PIs have been reported to be inhibi-tors of the proteasome complex, which is known to regulatenuclear factor (NF)-�B activation and transcription of its targetgenes, among them MMP-9. We thus investigated the potentialinvolvement of the proteasome in the antiadipogenic effects ofHIV-PIs. The effect of four HIV-PIs was tested on preadipocyteproteasomal activity, and chronic treatment with the specificproteasome inhibitor lactacystin was performed to evaluatealterations of adipogenesis and MMP-9 expression/secretion.Finally, modifications of the NF-�B pathway induced by eitherHIV-PIs or lactacystin were studied. We demonstrated that
preadipocyte proteasomal activity was decreased by severalHIV-PIs and that chronic treatment with lactacystin mimickedthe effects of HIV-PIs by reducing adipogenesis and MMP-9expression/secretion. Furthermore, we observed an intracellu-lar accumulation of the NF-�B inhibitor, I�B�, with chronictreatment with HIV-PIs or lactacystin as well as a decrease inMMP-9 expression induced by acute tumor necrosis factor-�stimulation. These results indicate that inhibition of the protea-some by specific (lactacystin) or nonspecific (HIV-PIs) inhibitorsleads to a reduction of human adipogenesis, and they thereforeimplicate deregulation of the NF-�B pathway and the relateddecrease of the key adipogenic factor, MMP-9. This study addssignificantly to recent reports that have linked HIV-PI-relatedlipodystrophic syndrome with altered proteasome function, en-doplasmic reticulum stress, and metabolic disorders.
Proteasomes are highly conserved multimeric peptidasespresent in all eukaryotic cells. Whereas various forms ofproteasomes exist, the 20S proteasome (multicatalytic core ofall proteasomes) and the 26S proteasome (20S core cappedwith two 19S regulatory units) are major proteasomes. Pro-teasomes were initially thought to be just recyclers of dam-aged or misfolded proteins, but over the last decade theactivity of this enzymatic complex has been found to be of
critical importance for many cellular functions. Cell-cycleprogression, oncogenesis, apoptosis, regulation of gene ex-pression, and inflammation or immune surveillance are allphysiological functions regulated by the proteasome pathway(Kisselev and Goldberg, 2001). Among the substrates of theproteasome, cyclins (e.g., cyclin B1), cyclin-dependent kinaseinhibitors (e.g., p21 and p27), tumor suppressors (e.g., p53),and inhibitors (I�B) or precursors (e.g., p105) of the tran-scription factor NF-�B are well established (Kisselev andGoldberg, 2001; Adams, 2004). Accordingly, the proteasomecomplex has emerged as an attractive target for cancer ther-apy, and numerous proteasome inhibitors have been devel-oped (Adams, 2004; Voorhees and Orlowski, 2006).
This work was supported by Grants 01 128 and 02 197 from the AgenceNationale de Recherche sur le SIDA et les hepatites virales.
Article, publication date, and citation information can be found athttp://jpet.aspetjournals.org.
doi:10.1124/jpet.106.111849.
ABBREVIATIONS: I�B, inhibitor of nuclear factor-�B; NF-�B, nuclear factor-�B; HIV, human immunodeficiency virus; PI, protease inhibitor; AT,adipose tissue; MMP, matrix metalloproteinase; SQV, saquinavir; NFV, nelfinavir; TNF, tumor necrosis factor; Bay 11-7802, (E)-3-[(4-methylphe-nyl)-sulfonyl]-2-propenenitrile; IDV, indinavir; RTV, ritonavir; RFU, relative fluorescence units; TBST, Tris-buffered saline/Tween 20; RT, reversetranscriptase; PCR, polymerase chain reaction; aP2, fatty acid binding protein; HSL, hormone-sensitive lipase; ANOVA, analysis of variance.MG-132, carbobenzoxy-L-leucyl-L-leucyl-L-leucinal; TG, triglyceride.
0022-3565/07/3201-291–299$20.00THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS Vol. 320, No. 1Copyright © 2007 by The American Society for Pharmacology and Experimental Therapeutics 111849/3162833JPET 320:291–299, 2007 Printed in U.S.A.
291

Recently, human immunodeficiency virus (HIV) proteaseinhibitors (PIs), central components of highly active antiret-roviral therapy, have been reported by some authors to di-rectly inhibit the activities of murine and human 20S and/or26S proteasomes in purified fractions or cellular extracts(Andre et al., 1998; Gaedicke et al., 2002; Pajonk et al., 2002;Piccinini et al., 2002). Indeed, several cleavage sites used bythe HIV protease, which were once thought to be unique anddistinct from those of mammalian proteases (predicting in-hibitor selectivity), were found to be similar to those recog-nized by the 20S proteasome (Schmidtke et al., 1999). Al-though HIV-PIs have been successfully used in clinicaltherapy of HIV infection, they were also rapidly associatedwith the emergence of a lipodystrophic syndrome observed inpatients treated with highly active antiretroviral therapy,which includes dyslipidemia, insulin resistance, central adi-posity, and peripheral lipoatrophy. It is now clear that themechanism associated with PI-induced metabolic abnormal-ities is multifactorial (for reviews, see Hui, 2003; Rudich etal., 2005) but whether the action of HIV-PIs on proteasomalactivity is related to their side effects has been poorly inves-tigated to date.
We previously reported, in accordance with in vivo studiesperformed on adipose tissue biopsies from HIV-infected lipo-dystrophic patients (Bastard et al., 2002; Lloreta et al., 2002),that PIs reduced the differentiation of human preadipocytes(i.e., adipocyte precursors) in primary culture isolated fromsubcutaneous abdominal adipose tissue (AT) (Bourlier et al.,2005). This effect was attributed to an indirect action ofHIV-PIs on the expression and secretion of the matrix met-alloproteinase-9 (MMP-9), an enzyme implicated in extracel-lular matrix remodeling and significantly involved in thehuman adipogenic process in vitro (Bourlier et al., 2005).Because the MMP-9 promoter contains an NF-�B site (Satoand Seiki, 1993; St-Pierre et al., 2004) and because protea-somal activity is involved in the activation of NF-�B path-way, we investigated the potential role of the proteasome inthe antiadipogenic effect of HIV-PIs. We demonstrate herethat HIV-PIs, particularly saquinavir (SQV) and nelfinavir(NFV), were direct inhibitors of the human preadipocyte 20Sand 26S proteasomes. We found that chronic lactacystin (aspecific proteasome inhibitor) treatment mimicked the ef-fects of HIV-PIs by decreasing the differentiation process andthe expression and secretion of MMP-9. Chronic treatmentwith either HIV-PIs (SQV or NFV) or lactacystin led to theaccumulation of the I�B� protein, the inhibitor of the tran-scription factor NF-�B and a substrate of the proteasome.Finally, acute treatment with TNF-� (activator of NF-�B)induced a potent increase in MMP-9 expression, which wassignificantly reduced by SQV, lactacystin, or (E)-3-[(4-meth-ylphenyl)-sulfonyl]-2-propenenitrile (Bay 11-7082) (inhibitorof NF-�B activation) cotreatments. Overall, our data indicatethat HIV-PIs, by affecting proteasomal activity, alter theNF-�B pathway and implicate the reduction of MMP-9 activ-ity in their antiadipogenic effect.
Materials and MethodsCell Culture and Treatment. Chemicals were from Sigma-
Aldrich (Saint-Quentin Fallavier, France). Cell culture reagentswere either from Life Technologies (Cergy Pontoise, France), RocheDiagnostics (Meylan, France), or Cambrex Biosciences (Verviers,Belgium).
Human subcutaneous abdominal white AT was obtained frommoderately overweight women undergoing plastic surgery (mean age46 � 2 years and mean body mass index 27.5 � 0.7 kg/m2). Theisolation of human AT-derived stromal cells and the culture of stro-mal preadipocytes differentiated into adipocytes were performed asdescribed previously (Bouloumie et al., 2001). In brief, sterile AT wascut into small pieces and digested under agitation with collagenase(2 mg/ml) (Serva; Coger, Paris, France) for �1 h at 37°C. Aftercentrifugation, washing, and filtration steps, the stromal cells weresuspended in Dulbecco’s modified Eagle’s medium F-12 supple-mented with 10% fetal bovine serum and plated at 60,000 cells/cm2.After 24 h, the medium was replaced by a medium consisting ofDulbecco’s modified Eagle’s medium F-12 supplemented with 33 �Mbiotin, 17 �M pantothenate, and 50 �g/ml gentamicin (basal me-dium) in the presence of 66 nM insulin, 1 nM triiodothyronine, 100nM cortisol, and 10 �g/ml human transferrin (adipogenic medium)and 1 �g/ml ciglitazone for the first 3 days. After the 3-day primingperiod, the cells were used for experiments or cultured in the adipo-genic medium supplemented with 1) the proteasome inhibitor lacta-cystin or 2) HIV-PIs IDV (kindly provided by Merck Laboratories,Rahway, NJ) or SQV (kindly provided by Roche, Welwyn GardenCity, UK), RTV (kindly provided by Abbott Laboratories, Chicago,IL), or NFV (kindly provided by Roche Diagnostics, Mannheim,France) for 10 days. Lactacystin and HIV-PIs were dissolved in puredimethyl sulfoxide. Control cells were treated using an appropriateconcentration of vehicle (�0.25%) after verification that dimethylsulfoxide was without effect on the various parameters analyzed (i.e.,lipogenic index, adipogenic gene, or MMP secretion). Medium wasreplaced every 2 days. Before some experiments, cells were placedovernight in basal medium supplemented with the various treat-ments.
The medium was then collected and used in zymography analysis.Cellular triglyceride and DNA contents were determined using kitsfrom Sigma-Aldrich and Molecular Probes (PicoGreen; Interchim,Montlucon, France), respectively, according to the manufacturer’sinstructions. DNA content was used to normalize data (triglyceridecontent and MMP secretion) and to evaluate any cytotoxic effect oftreatments.
Gelatin Zymography. Proteins with gelatinolytic activity (in-cluding the gelatinase A “MMP-2” and the gelatinase B “MMP-9”)were identified by electrophoresis in the presence of SDS in 8%polyacrylamide gels containing 1 mg/ml gelatin. In brief, culturemedium aliquots (20 �l) were directly loaded onto gels, and, afterelectrophoresis, proteins were renatured by exchanging SDS with2.5% Triton X-100 (20-min incubation repeated twice). The gels werethen incubated for 16 h at 37°C in 50 mM Tris-HCl (pH 8.8), 5 mMCaCl2, and 0.02% NaN3 and stained with Coomassie Blue. Thepresence of gelatinolytic activity in the culture medium aliquots wasvisualized as clear areas on an otherwise blue gel. Migration ofproteins was compared with that of prestained molecular weightmarkers. The gels were scanned by an imaging densitometer andquantified using the NIH Image program (developed at the NationalInstitutes of Health, Bethesda, MD).
Fluorimetric Proteasome Activity Assay. Chymotrypsin-likeproteasome activity was performed on 96-well plates using the spe-cific fluorogenic substrate N-succinyl-Leu-Leu-Val-Tyr7-amido-4-methylcoumarin (Sigma). In brief, cells contained in one well werescraped in 35 �l of lysis buffer containing 25 mM Tris-HCl (pH 7.5),0.1% Nonidet P-40, 10% glycerol, 5 mM MgCl2, 1 mM ATP, and 10mM KCl. Protein concentration was determined using a Bio-Rad kit(Bio-Rad, Marnes-la Coquette, France), and 10 �g were incubatedalone or with increasing concentrations of lactacystin (0.01–10 �M)or HIV-PIs (1, 10, and 50 �M) for 30 min at 37°C in 20 mM Tris-HCl(pH 8), 0.5 mM EDTA, and 0.035% SDS to assess 20S proteasomeactivity or in 20 mM Tris-HCl (pH 8), 1 mM ATP, and 2 mM MgCl2to assess 26S proteasome activity, according to the method of Rock etal. (1994). Then, fluorogenic substrate was added to a final concen-tration of 50 �M, and fluorescence was monitored at �excitation 360
292 De Barros et al.

nm/�emission 460 nm every 5 min over a 2-h period using a FluoroscanAscent FL (Labsystems, Cergy Pontoise, France). Chymotrypsin-likeproteasome activities of 20S and 26S were evaluated by the maximalrate (relative fluorescence units per minute) on seven separate mea-surements in each well.
Western Blot. For Western blot analysis of I�B� and I�B�, cellscontained in two wells were scraped in 70 �l of lysis buffer containing20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM CaCl2, 1 mM MgCl2,1 mM Na3VO4, 1% Nonidet P-40, 10% glycerol, and a mix of proteaseinhibitors (Complete; Roche Diagnostics). The protein concentrationwas determined, and 35 to 50 �g of protein were loaded and sepa-rated by electrophoresis on a 12% SDS-polyacrylamide gel underdenaturing conditions. After transfer to polyvinylidene difluoridemembranes (Immobilon; Millipore, Bedford, MA) and Ponceau stain-ing to verify equal loading of the lanes, membranes were blocked for1 h with TBST [50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 0.1% Tween20] containing 5% nonfat milk, following by incubation overnight at4°C with primary antibody against I�B� (ab7545) or I�B� (ab7547)(Abcam, Cambridge, UK) at a dilution of 1:1000 in TBST containing1% nonfat milk. Then, membranes were washed with TBST andincubated for 1 h with a peroxidase-conjugated secondary antibody(Pierce Chemical, Brebieres, France) at a dilution of 1:50,000 inTBST containing 1% nonfat milk and washed again. The immuno-complexes were detected using a chemiluminescence reagent kit(SuperSignal West Dura Extended Duration Substrate; PierceChemical, Rockford, IL). Human Daudi cell lysate (Imgenex, SanDiego, CA) was used as positive control.
For Western blot analysis of NF-�B p65, cells contained in 12 wellswere washed twice with cold phosphate-buffered saline and preparedusing NE-PER Nuclear and Cytoplasmic Extraction Reagents kit(Pierce) according to the manufacturer’s instructions. Protein con-centrations in nuclear and cytoplasmic extracts were determined asdescribed above: 20 to 45 �g of proteins were loaded and separated byelectrophoresis on a 12% SDS-polyacrylamide gel electrophoresisunder denaturing conditions. After transfer to polyvinylidene diflu-oride membranes and Ponceau staining to verify equal loading of thelanes, a WesternBreeze Chemiluminescent Western Blot Immuno-detection Kit (Invitrogen, Cergy Pontoise, France) was used for sig-nal detection. Briefly and according to the manufacturer’s instruc-tions, membranes were blocked, washed, and then incubated with aprimary antibody against NF-�B p65 N-terminal (Santa Cruz Bio-technology, Le Perray-en-Yvelines, France) at a dilution of 1:1000.After washing, membranes were incubated with an alkaline phos-phatase-conjugated secondary antibody provided with the kit, andimmunocomplexes were detected after the addition of chemilumines-cent substrate. Human nuclear extract from A-431 cells (Santa CruzBiotechnology) was used as positive control.
The autoradiographs were scanned by an imaging densitometerand quantified using the NIH Image program. To normalize results,the densitometric value of each band of interest (i.e., I�B�, I�B�, andNF-�B p65) was reported to the densitometric value of total loadedproteins in the corresponding lane, obtained after scanning andquantification of the Ponceau staining (NIH Image program).
Real-Time RT-PCR. Changes in mRNA levels from specificgenes were quantified by real-time RT-PCR. Total RNAs were ex-tracted using a QIAGEN RNeasy Mini Kit according to the manu-facturer’s instructions, and RNA concentrations were determinedusing a Molecular Probes fluorimetric assay (RiboGreen; Interchim).RNA (0.5 �g) was reverse-transcribed using the Superscript II sys-tem (Invitrogen) according to the manufacturer’s instructions. Re-verse transcription was also performed without Thermoscript en-zyme on RNA samples to check for any genomic DNA contamination.PCR primers were designed using Primer Express software accord-ing to the recommendations of Applied Biosystems (Courtaboeuf,France). The forward and reverse primer sequences for fatty acidbinding protein (aP2), hormone-sensitive lipase (HSL), MMP-9,MMP-2, and I�B� were as follows (5�–3�): aP2, GCATGGCCAAAC-CTAACATGA (forward) and CCTGGCCCAGTATGAAGGAAA (re-
verse); HSL, GTGCAAAGACGGAGGACCACTCCA (forward) andGACGTCTCGGAGTTTCCCCTCAG; MMP-9, CCCTGGAGACCTG-AGAACCA (forward) and CCACCCGAGTGTAACCATAGC (re-verse); MMP-2 CACCCATTTACACCTACACCAAGA (forward) andAGAGCTCCTGAATGCCCTTGA (reverse), and I�B� TACGACGAC-ATTGTGGTTCACA (forward) and GGGTCGTCAGGAAGAGGTTTT(reverse).
Each amplification reaction was performed with 15 ng of cDNAsample in duplicate in 96-well optical reaction plates with a Gene-Amp 7500 sequence detection system. The PCR mixture containedforward and reverse primer mix (final concentration: 900 nM forHSL, MMP-9, or MMP-2 and 300 nM for aP2 or I�B�) and SYBRGreen PCR Master Mix. For ribosomal RNA control (18S rRNA), amixture containing primers and fluorogenic probe mix, TaqMan Uni-versal PCR Master Mix (Applied Biosystems) was used. All reactionswere performed under the same conditions: 50°C for 2 min and 95°Cfor 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min.Results were analyzed with the GeneAmp 7500 software, and allvalues were normalized to the levels of 18S rRNA. An interindividualvariability of 10.46% within the control values was observed for allPCR data (aP2, HSL, MMP-2, MMP-9, and I�B�).
Statistical Analysis. Values are expressed as means � S.E.M.from n independent experiments. Statistical analysis was performedusing Student’s t test for paired data or one-way analysis of variance(ANOVA) coupled with post hoc Dunnett’s multiple-comparison test(GraphPad Prism 4; GraphPad Software Inc., San Diego, CA). Val-ues of p � 0.05 were considered statistically significant.
ResultsHIV-PIs or Lactacystin Reduced Proteasome Activ-
ity from Human Preadipocyte Homogenates. To vali-date our hypothesis, we first investigated the effect of HIV-PIs on proteasome activity of human preadipocytes. After the3-day priming period, undifferentiated preadipocytes werescraped into specific lysis buffer, and cellular homogenateswere obtained. Then, homogenates were pretreated (30 min,37°C) with increasing concentrations of the proteasome in-hibitor lactacystin (from 0.01 to 10 �M) or HIV-PIs (1, 10,and 50 �M), and 20S and 26S chymotrypsin-like proteasomeactivities from these lysates were determined by in vitrofluorescence assays with the specific fluorogenic substrateN-succinyl-Leu-Leu-Val-Tyr7-amido-4-methylcoumarin.
As expected, lactacystin reduced, in a concentration-depen-dent manner, the fluorescence emission induced by the cleav-age of the substrate by the 20S and 26S proteasome (controlmaximal rate: 1.62 � 0.84 and 1.13 � 0.21 RFU/min, respec-tively) (Fig. 1, top and bottom). Similar results were obtainedwith another known proteasome inhibitor, carbobenzoxy-L-leucyl-L-leucyl-L-leucinal (MG-132) (data not shown). Asshown on the top of Fig. 1, HIV-PIs, except IDV, decreasedchymotrypsin-like activity of the 20S proteasome in a con-centration-dependent manner with the following rank of ef-ficiency: SQV � NFV � RTV. Higher concentrations of HIV-PIs were necessary to reduce the 26S chymotrypsin-likeproteasome activity in the cellular extracts (Fig. 1, bottom).
Chronic Lactacystin Treatment Decreased the Dif-ferentiation of Human Preadipocytes. To determinewhether the inhibition of the proteasome per se could affectthe human adipocyte differentiation process, primary cul-tures of human preadipocytes were placed in an adipogenicmedium in the presence of 0.1 or 0.5 �M lactacystin for 10days. Cellular triglyceride was quantified, and the expres-sion of two adipocyte differentiation markers, aP2 and HSL,was analyzed by real-time RT-PCR.
Proteasome Inhibition Reduces Human Adipogenesis 293
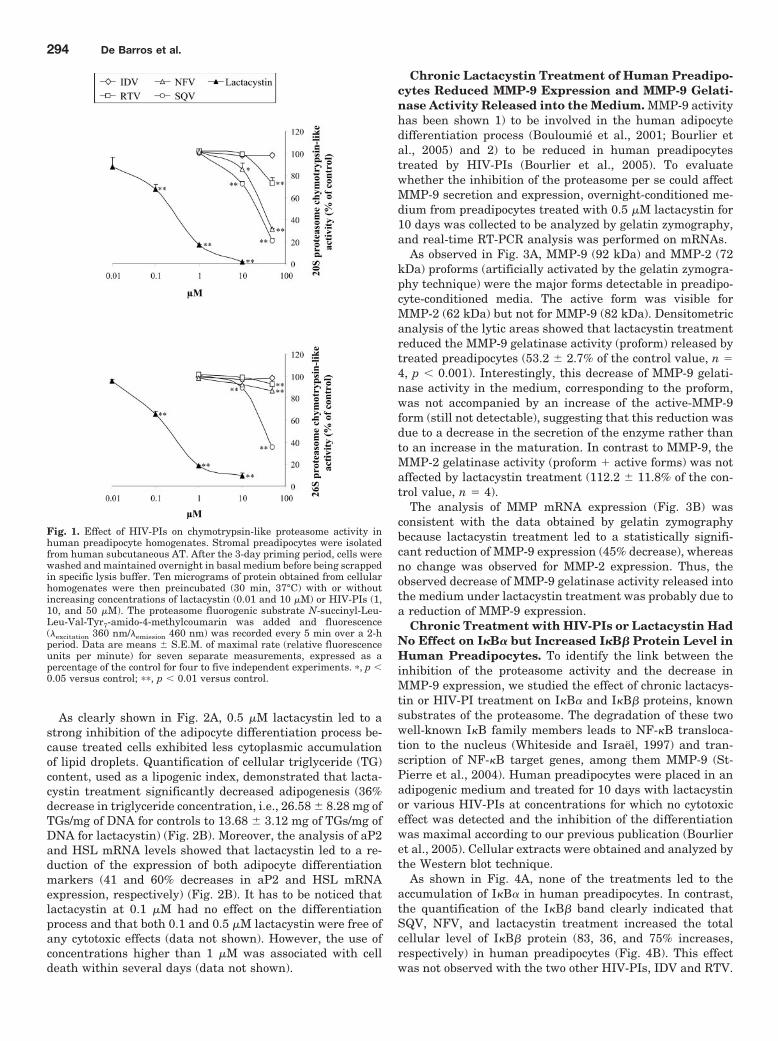
As clearly shown in Fig. 2A, 0.5 �M lactacystin led to astrong inhibition of the adipocyte differentiation process be-cause treated cells exhibited less cytoplasmic accumulationof lipid droplets. Quantification of cellular triglyceride (TG)content, used as a lipogenic index, demonstrated that lacta-cystin treatment significantly decreased adipogenesis (36%decrease in triglyceride concentration, i.e., 26.58 � 8.28 mg ofTGs/mg of DNA for controls to 13.68 � 3.12 mg of TGs/mg ofDNA for lactacystin) (Fig. 2B). Moreover, the analysis of aP2and HSL mRNA levels showed that lactacystin led to a re-duction of the expression of both adipocyte differentiationmarkers (41 and 60% decreases in aP2 and HSL mRNAexpression, respectively) (Fig. 2B). It has to be noticed thatlactacystin at 0.1 �M had no effect on the differentiationprocess and that both 0.1 and 0.5 �M lactacystin were free ofany cytotoxic effects (data not shown). However, the use ofconcentrations higher than 1 �M was associated with celldeath within several days (data not shown).
Chronic Lactacystin Treatment of Human Preadipo-cytes Reduced MMP-9 Expression and MMP-9 Gelati-nase Activity Released into the Medium. MMP-9 activityhas been shown 1) to be involved in the human adipocytedifferentiation process (Bouloumie et al., 2001; Bourlier etal., 2005) and 2) to be reduced in human preadipocytestreated by HIV-PIs (Bourlier et al., 2005). To evaluatewhether the inhibition of the proteasome per se could affectMMP-9 secretion and expression, overnight-conditioned me-dium from preadipocytes treated with 0.5 �M lactacystin for10 days was collected to be analyzed by gelatin zymography,and real-time RT-PCR analysis was performed on mRNAs.
As observed in Fig. 3A, MMP-9 (92 kDa) and MMP-2 (72kDa) proforms (artificially activated by the gelatin zymogra-phy technique) were the major forms detectable in preadipo-cyte-conditioned media. The active form was visible forMMP-2 (62 kDa) but not for MMP-9 (82 kDa). Densitometricanalysis of the lytic areas showed that lactacystin treatmentreduced the MMP-9 gelatinase activity (proform) released bytreated preadipocytes (53.2 � 2.7% of the control value, n �4, p � 0.001). Interestingly, this decrease of MMP-9 gelati-nase activity in the medium, corresponding to the proform,was not accompanied by an increase of the active-MMP-9form (still not detectable), suggesting that this reduction wasdue to a decrease in the secretion of the enzyme rather thanto an increase in the maturation. In contrast to MMP-9, theMMP-2 gelatinase activity (proform � active forms) was notaffected by lactacystin treatment (112.2 � 11.8% of the con-trol value, n � 4).
The analysis of MMP mRNA expression (Fig. 3B) wasconsistent with the data obtained by gelatin zymographybecause lactacystin treatment led to a statistically signifi-cant reduction of MMP-9 expression (45% decrease), whereasno change was observed for MMP-2 expression. Thus, theobserved decrease of MMP-9 gelatinase activity released intothe medium under lactacystin treatment was probably due toa reduction of MMP-9 expression.
Chronic Treatment with HIV-PIs or Lactacystin HadNo Effect on I�B� but Increased I�B� Protein Level inHuman Preadipocytes. To identify the link between theinhibition of the proteasome activity and the decrease inMMP-9 expression, we studied the effect of chronic lactacys-tin or HIV-PI treatment on I�B� and I�B� proteins, knownsubstrates of the proteasome. The degradation of these twowell-known I�B family members leads to NF-�B transloca-tion to the nucleus (Whiteside and Israel, 1997) and tran-scription of NF-�B target genes, among them MMP-9 (St-Pierre et al., 2004). Human preadipocytes were placed in anadipogenic medium and treated for 10 days with lactacystinor various HIV-PIs at concentrations for which no cytotoxiceffect was detected and the inhibition of the differentiationwas maximal according to our previous publication (Bourlieret al., 2005). Cellular extracts were obtained and analyzed bythe Western blot technique.
As shown in Fig. 4A, none of the treatments led to theaccumulation of I�B� in human preadipocytes. In contrast,the quantification of the I�B� band clearly indicated thatSQV, NFV, and lactacystin treatment increased the totalcellular level of I�B� protein (83, 36, and 75% increases,respectively) in human preadipocytes (Fig. 4B). This effectwas not observed with the two other HIV-PIs, IDV and RTV.
Fig. 1. Effect of HIV-PIs on chymotrypsin-like proteasome activity inhuman preadipocyte homogenates. Stromal preadipocytes were isolatedfrom human subcutaneous AT. After the 3-day priming period, cells werewashed and maintained overnight in basal medium before being scrappedin specific lysis buffer. Ten micrograms of protein obtained from cellularhomogenates were then preincubated (30 min, 37°C) with or withoutincreasing concentrations of lactacystin (0.01 and 10 �M) or HIV-PIs (1,10, and 50 �M). The proteasome fluorogenic substrate N-succinyl-Leu-Leu-Val-Tyr7-amido-4-methylcoumarin was added and fluorescence(�excitation 360 nm/�emission 460 nm) was recorded every 5 min over a 2-hperiod. Data are means � S.E.M. of maximal rate (relative fluorescenceunits per minute) for seven separate measurements, expressed as apercentage of the control for four to five independent experiments. �, p �0.05 versus control; ��, p � 0.01 versus control.
294 De Barros et al.
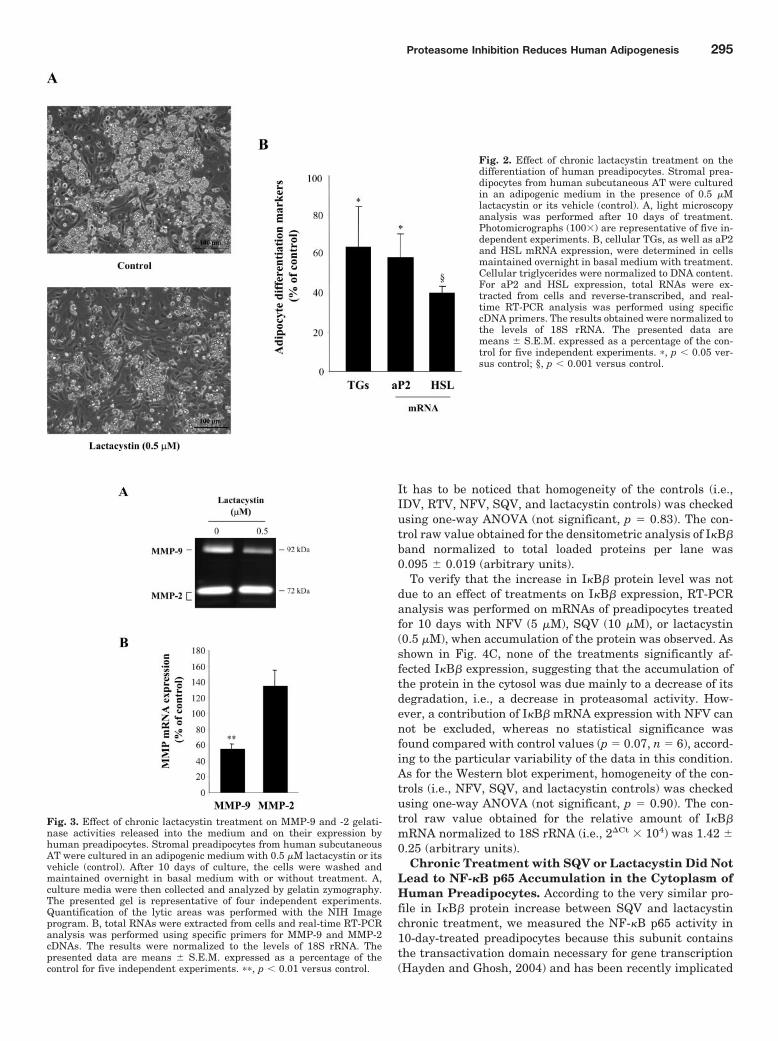
It has to be noticed that homogeneity of the controls (i.e.,IDV, RTV, NFV, SQV, and lactacystin controls) was checkedusing one-way ANOVA (not significant, p � 0.83). The con-trol raw value obtained for the densitometric analysis of I�B�band normalized to total loaded proteins per lane was0.095 � 0.019 (arbitrary units).
To verify that the increase in I�B� protein level was notdue to an effect of treatments on I�B� expression, RT-PCRanalysis was performed on mRNAs of preadipocytes treatedfor 10 days with NFV (5 �M), SQV (10 �M), or lactacystin(0.5 �M), when accumulation of the protein was observed. Asshown in Fig. 4C, none of the treatments significantly af-fected I�B� expression, suggesting that the accumulation ofthe protein in the cytosol was due mainly to a decrease of itsdegradation, i.e., a decrease in proteasomal activity. How-ever, a contribution of I�B� mRNA expression with NFV cannot be excluded, whereas no statistical significance wasfound compared with control values (p � 0.07, n � 6), accord-ing to the particular variability of the data in this condition.As for the Western blot experiment, homogeneity of the con-trols (i.e., NFV, SQV, and lactacystin controls) was checkedusing one-way ANOVA (not significant, p � 0.90). The con-trol raw value obtained for the relative amount of I�B�mRNA normalized to 18S rRNA (i.e., 2�Ct 104) was 1.42 �0.25 (arbitrary units).
Chronic Treatment with SQV or Lactacystin Did NotLead to NF-�B p65 Accumulation in the Cytoplasm ofHuman Preadipocytes. According to the very similar pro-file in I�B� protein increase between SQV and lactacystinchronic treatment, we measured the NF-�B p65 activity in10-day-treated preadipocytes because this subunit containsthe transactivation domain necessary for gene transcription(Hayden and Ghosh, 2004) and has been recently implicated
Fig. 3. Effect of chronic lactacystin treatment on MMP-9 and -2 gelati-nase activities released into the medium and on their expression byhuman preadipocytes. Stromal preadipocytes from human subcutaneousAT were cultured in an adipogenic medium with 0.5 �M lactacystin or itsvehicle (control). After 10 days of culture, the cells were washed andmaintained overnight in basal medium with or without treatment. A,culture media were then collected and analyzed by gelatin zymography.The presented gel is representative of four independent experiments.Quantification of the lytic areas was performed with the NIH Imageprogram. B, total RNAs were extracted from cells and real-time RT-PCRanalysis was performed using specific primers for MMP-9 and MMP-2cDNAs. The results were normalized to the levels of 18S rRNA. Thepresented data are means � S.E.M. expressed as a percentage of thecontrol for five independent experiments. ��, p � 0.01 versus control.
Fig. 2. Effect of chronic lactacystin treatment on thedifferentiation of human preadipocytes. Stromal prea-dipocytes from human subcutaneous AT were culturedin an adipogenic medium in the presence of 0.5 �Mlactacystin or its vehicle (control). A, light microscopyanalysis was performed after 10 days of treatment.Photomicrographs (100) are representative of five in-dependent experiments. B, cellular TGs, as well as aP2and HSL mRNA expression, were determined in cellsmaintained overnight in basal medium with treatment.Cellular triglycerides were normalized to DNA content.For aP2 and HSL expression, total RNAs were ex-tracted from cells and reverse-transcribed, and real-time RT-PCR analysis was performed using specificcDNA primers. The results obtained were normalized tothe levels of 18S rRNA. The presented data aremeans � S.E.M. expressed as a percentage of the con-trol for five independent experiments. �, p � 0.05 ver-sus control; §, p � 0.001 versus control.
Proteasome Inhibition Reduces Human Adipogenesis 295
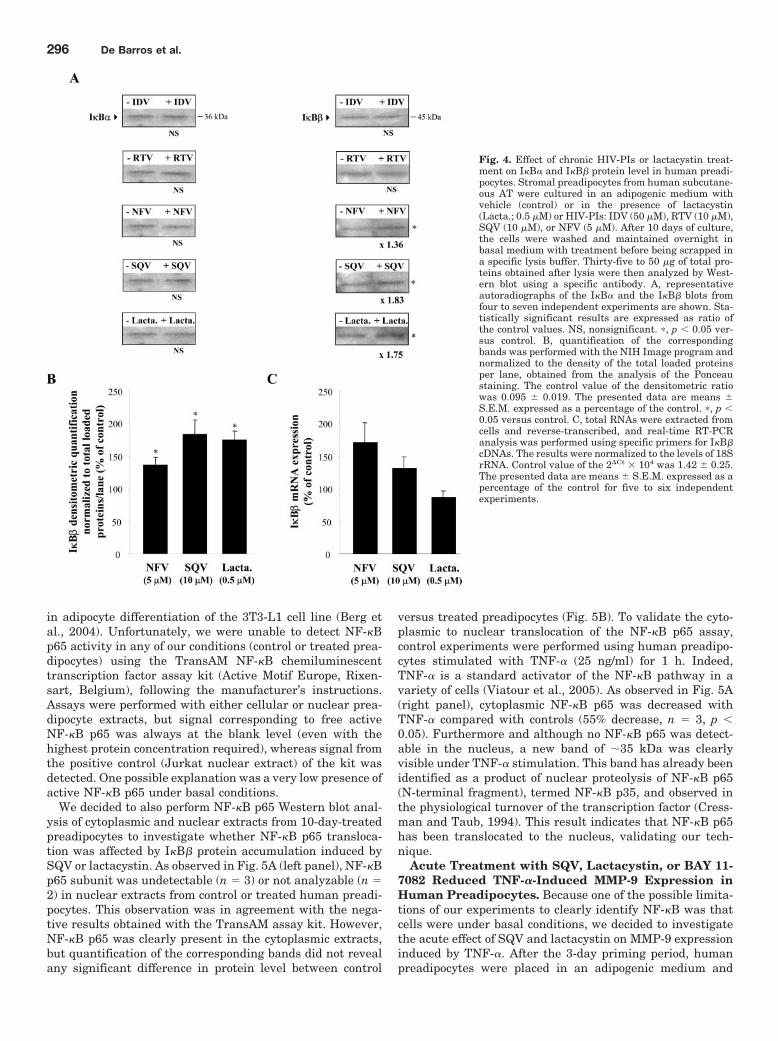
in adipocyte differentiation of the 3T3-L1 cell line (Berg etal., 2004). Unfortunately, we were unable to detect NF-�Bp65 activity in any of our conditions (control or treated prea-dipocytes) using the TransAM NF-�B chemiluminescenttranscription factor assay kit (Active Motif Europe, Rixen-sart, Belgium), following the manufacturer’s instructions.Assays were performed with either cellular or nuclear prea-dipocyte extracts, but signal corresponding to free activeNF-�B p65 was always at the blank level (even with thehighest protein concentration required), whereas signal fromthe positive control (Jurkat nuclear extract) of the kit wasdetected. One possible explanation was a very low presence ofactive NF-�B p65 under basal conditions.
We decided to also perform NF-�B p65 Western blot anal-ysis of cytoplasmic and nuclear extracts from 10-day-treatedpreadipocytes to investigate whether NF-�B p65 transloca-tion was affected by I�B� protein accumulation induced bySQV or lactacystin. As observed in Fig. 5A (left panel), NF-�Bp65 subunit was undetectable (n � 3) or not analyzable (n �2) in nuclear extracts from control or treated human preadi-pocytes. This observation was in agreement with the nega-tive results obtained with the TransAM assay kit. However,NF-�B p65 was clearly present in the cytoplasmic extracts,but quantification of the corresponding bands did not revealany significant difference in protein level between control
versus treated preadipocytes (Fig. 5B). To validate the cyto-plasmic to nuclear translocation of the NF-�B p65 assay,control experiments were performed using human preadipo-cytes stimulated with TNF-� (25 ng/ml) for 1 h. Indeed,TNF-� is a standard activator of the NF-�B pathway in avariety of cells (Viatour et al., 2005). As observed in Fig. 5A(right panel), cytoplasmic NF-�B p65 was decreased withTNF-� compared with controls (55% decrease, n � 3, p �0.05). Furthermore and although no NF-�B p65 was detect-able in the nucleus, a new band of �35 kDa was clearlyvisible under TNF-� stimulation. This band has already beenidentified as a product of nuclear proteolysis of NF-�B p65(N-terminal fragment), termed NF-�B p35, and observed inthe physiological turnover of the transcription factor (Cress-man and Taub, 1994). This result indicates that NF-�B p65has been translocated to the nucleus, validating our tech-nique.
Acute Treatment with SQV, Lactacystin, or BAY 11-7082 Reduced TNF-�-Induced MMP-9 Expression inHuman Preadipocytes. Because one of the possible limita-tions of our experiments to clearly identify NF-�B was thatcells were under basal conditions, we decided to investigatethe acute effect of SQV and lactacystin on MMP-9 expressioninduced by TNF-�. After the 3-day priming period, humanpreadipocytes were placed in an adipogenic medium and
Fig. 4. Effect of chronic HIV-PIs or lactacystin treat-ment on I�B� and I�B� protein level in human preadi-pocytes. Stromal preadipocytes from human subcutane-ous AT were cultured in an adipogenic medium withvehicle (control) or in the presence of lactacystin(Lacta.; 0.5 �M) or HIV-PIs: IDV (50 �M), RTV (10 �M),SQV (10 �M), or NFV (5 �M). After 10 days of culture,the cells were washed and maintained overnight inbasal medium with treatment before being scrapped ina specific lysis buffer. Thirty-five to 50 �g of total pro-teins obtained after lysis were then analyzed by West-ern blot using a specific antibody. A, representativeautoradiographs of the I�B� and the I�B� blots fromfour to seven independent experiments are shown. Sta-tistically significant results are expressed as ratio ofthe control values. NS, nonsignificant. �, p � 0.05 ver-sus control. B, quantification of the correspondingbands was performed with the NIH Image program andnormalized to the density of the total loaded proteinsper lane, obtained from the analysis of the Ponceaustaining. The control value of the densitometric ratiowas 0.095 � 0.019. The presented data are means �S.E.M. expressed as a percentage of the control. �, p �0.05 versus control. C, total RNAs were extracted fromcells and reverse-transcribed, and real-time RT-PCRanalysis was performed using specific primers for I�B�cDNAs. The results were normalized to the levels of 18SrRNA. Control value of the 2�Ct 104 was 1.42 � 0.25.The presented data are means � S.E.M. expressed as apercentage of the control for five to six independentexperiments.
296 De Barros et al.
treated for 24 h with TNF-� (25 ng/ml) alone or in cotreat-ment with SQV (10 �M), lactacystin (0.5 �M), or BAY 11-7082 (1 �M), an inhibitor of I�B phosphorylation, which isinvolved in the NF-�B activation (Pierce et al., 1997). MMP-9expression was analyzed by real-time RT-PCR.
As shown in Fig. 6, TNF-� treatment strongly enhancedMMP-9 expression (10 times) in human preadipocytes. Asexpected, cotreatment with BAY 11-7082, the inhibitor of theNF-�B pathway, reduced the MMP-9 expression induced byTNF-� (27% decrease). Similar results were obtained withSQV or lactacystin cotreatment, because MMP-9 expressionwas decreased by 48 and 22%, respectively, implicatingNF-�B as the link between proteasome activity and MMP-9expression in our experiments. It should be noted that simi-lar data were obtained when SQV was replaced by NFV (5�M) or when stimulation was performed with interleukin-1�(20 ng/ml) in place of TNF-� (data not shown). Comparedwith TNF-�, which has been shown to involve preferentiallyI�B�, interleukin-1� is known to induce both I�B� and I�B�degradation when activating the NF-�B pathway (Thompsonet al., 1995).
DiscussionWe previously reported that HIV-PIs reduced the differen-
tiation of human preadipocytes in primary culture. This ef-fect was attributed to the HIV-PI-induced decrease of MMP-9expression and secretion since we showed that blocking ofMMP-9 activity by pharmacological agents led to the inhibi-tion of human adipogenesis (Bourlier et al., 2005). In thepresent work, we report that HIV-PIs decreased the activityof the two major proteasome complexes, the 20S and the 26Sproteasomes, in human preadipocytes. Interestingly, weshowed that lactacystin, a proteasome inhibitor, mimickedthe effects of HIV-PIs in reducing both human adipogenesisand MMP-9 expression and secretion. Finally, some of ourresults suggested that perturbations of the NF-�B pathwayare the link between the inhibition of proteasomal activityand the decrease in MMP-9 expression. Indeed, even if wewere unable to detect NF-�B p65 signal changes under basalconditions, we did observe that chronic treatments (10 days)with HIV-PIs (SQV and NFV) or lactacystin induced cellularI�B� accumulation. Furthermore, SQV or lactacystin re-duced the expression of MMP-9 induced by acute treatment(24 h) with TNF-�, a classic activator of NF-�B.
The inhibition of proteasomal activity and, more precisely,proteasome chymotrypsin-like activity by HIV-PIs was ini-tially reported few years ago by Andre et al., (1998) andlinked to a decrease of antigen presentation and T-cell re-sponse in infected mice. Here we show, for the first time, thatproteasomes from human adipocyte precursors, which aremajor cell targets of HIV-PIs involved in the lipodystrophicsyndrome, are also directly inhibited by several HIV-PIs attherapeutic concentrations. These data agree with recentobservations made with the murine the 3T3-L1 cell linemodel of preadipocytes (Parker et al., 2005). As have otherinvestigators, we found that the 20S proteasome was more
Fig. 5. Effect of chronic SQV or lactacystin (Lacta.) treatment on cyto-plasmic and nuclear NF-�B p65 protein level in human preadipocytes.Stromal preadipocytes from human subcutaneous AT were cultured in anadipogenic medium with vehicle (control) or in the presence of lactacystin(0.5 �M) or SQV (10 �M). After 10 days of culture, the cells were washedand maintained overnight in basal medium with treatment before beingscrapped and prepared using NE-PER nuclear and cytoplasmic extrac-tion reagents kit. Twenty to 45 �g of proteins from cytoplasmic (Cyto.) ornuclear (Nucl.) extracts were then analyzed by Western blot using aspecific anti-NF-�B p65 subunit antibody. A representative autoradio-graph of five independent experiments is shown (A, left panel). Quanti-fication of the corresponding bands in the cytoplasmic extracts was per-formed with the NIH Image program and normalized to the density of thetotal loaded proteins per lane, obtained from analysis of Ponceau staining(B).The data presented are means � S.E.M. expressed as a percentage ofthe control. N/A, not analyzable. To validate the technique, human prea-dipocytes were acutely stimulated with TNF-� (25 ng/ml, 1 h), and West-ern blot analysis was performed. A representative autoradiograph of 3independent experiments is shown (A, right).
Fig. 6. Effect of acute SQV or lactacystin treatment on TNF-�-inducedMMP-9 expression in human preadipocytes. Stromal preadipocytes wereisolated from human subcutaneous AT. After the 3-day priming period,cells were placed in an adipogenic medium for 24 h, with or withoutTNF-� (25 ng/ml) alone or in cotreatment with lactacystin (0.5 �M), SQV(10 �M), or BAY 11-7082 (1 �M), an inhibitor of NF-�B activation. TotalRNAs were then extracted from cells and reverse-transcribed, and real-time RT-PCR analysis was performed using specific primers for MMP-9cDNAs. The results were normalized to the levels of 18S rRNA. Thepresented data are means � S.E.M. expressed as a percentage of theTNF-� effect alone for three to four independent experiments. �, p � 0.05versus TNF-� alone; ��, p � 0.01 versus TNF-� alone.
Proteasome Inhibition Reduces Human Adipogenesis 297

sensitive to HIV-PIs than the 26S proteasome (Gaedicke etal., 2002; Pajonk et al., 2002). One reason may be the differ-ent tertiary structure between the 20S and 26S proteasomes,leading to better accessibility of the inhibitors to the pro-teolytic sites of the 20S proteasome (Piccinini et al., 2005,Voorhees and Orlowski, 2006). We found that all HIV-PIs didnot have the same potency in inhibiting the chymotrypsin-like proteasome activity in our model. Indeed SQV and NFVwere the most effective, followed by RTV, whereas IDV waswithout effect. Similar observations were made in the rarepublications comparing the effect of HIV-PIs on human pro-teasomal activity measured on purified proteasome fromerythrocytes (Piccinini et al., 2002, 2005; Parker et al., 2005).It has to be noted that even if all HIV-PIs were weakerinhibitors of proteasomal activity than the specific lactacys-tin inhibitors in our in vitro assay, SQV and NFV (not RTVnor IDV), can accumulate greatly into cells (20 to �80 times)(Jones et al., 2001; Janneh et al., 2003). Thus, it can beexpected that intracellular concentrations of these HIV-PIsare much higher that the ones tested here.
Interestingly, SQV and NFV were the HIV-PIs with themajor effect on the differentiation of human preadipocytes inour previous work (Bourlier et al., 2005). We thus investi-gated whether a blockade of proteasomal activity can affecthuman adipogenesis. We showed that chronic lactacystintreatment of preadipocytes in an adipogenic medium led to adecrease of the various differentiation markers studied (tri-glyceride content and aP2 and HSL expression). Althoughsome authors have already implicated the proteasome inadipogenesis, their observations were on a different model,the murine 3T3-L1 cell line, using more or less specific pro-teasome inhibitors (calpain/proteasome), and contradictoryresults were obtained (Patel and Lane, 1999; Nguyen et al.,2000; Prince et al., 2002). As expected and in association withthe reduction of adipogenesis, we found that lactacystintreatment, as that with HIV-PIs, selectively decreasedMMP-9 expression and activity released into the culturemedium by preadipocytes. The reduction of MMP-9 activityper se may be responsible for the antiadipogenic effect oflactacystin since we have previously demonstrated thattreatment of human preadipocytes with pharmacological in-hibitors of MMP-9 activity decreased their differentiation(Bourlier et al., 2005). However we cannot exclude any otheradditional phenomenon implicated in adipogenesis thatmight be disturbed by proteasome inhibitor treatment.
Reduction of MMP-9 secretion with proteasome inhibitorshas previously been reported for distinct cellular types invitro involving NF-�B pathway perturbations (Ikebe et al.,1998; Kolev et al., 2003; Lu and Wahl, 2005). We were notable to find any modification in the activity and repartition ofNF-�B p65 within the cytosol and the nucleus in associationwith chronic SQV or lactacystin treatment in our model,using two different techniques (enzyme-linked immunosor-bent assay-based TransAM NF-�B assay kit and Westernblot). This may be partly explained by the fact that in theresting conditions that we studied, the part of active NF-�Bp65 was too weak to be detected. This hypothesis is in agree-ment with one of the rare published reports on the subjectshowing that NF-�B p65 is mostly cytosolic in unstimulatedhuman preadipocytes (Chung et al., 2005). Nevertheless, weshowed that the same chronic treatment with NFV, SQV, orlactacystin (but not with IDV nor RTV) led to intracellular
accumulation of I�B� protein. In contrast, none of the HIV-PIs or lactacystin was effective in modifying the I�B� proteinlevel. These results agree with the presumed roles of thesetwo members of the same family because 1) I�B� has beencorrelated to transient activation of NF-�B and I�B� to per-sistent activation that yields a more permanent change(Thompson et al., 1995) and 2) I�B�, but not I�B�, has beenshown to function as a classic cytoplasmic inhibitor of NF-�Bdimers in resting cells (Malek et al., 2001). Because bothNF-�B inhibitors have been shown to be substrates of theproteasome (Weil et al., 1997) and because no parallel in-crease in I�B� mRNA expression was observed under ourconditions, the increase in I�B� protein most probably re-flects the reduction of its degradation resulting from protea-somal activity inhibition induced by the treatments. Further-more, a different set of experiments conducted with theclassic NF-�B pathway activator TNF-� (Viatour et al., 2005)showed that MMP-9 expression was up-regulated within 24 hof treatment in our model, but also that this up-regulationcould be reduced either by SQV, lactacystin, or the NF-�Binhibitor, Bay 11-7082. Overall, according to these data onresting and TNF-�-stimulated human preadipocytes, we caneasily imagine that the decrease in MMP-9 expression ob-served with lactacystin and some HIV-PIs (SQV and NFV)arose in part from perturbations in the NF-�B pathway.
To our knowledge, this is the first investigation showingthat HIV-PIs are potent inhibitors of human preadipocyteproteasomal activity and that chronic low-level proteasomeinhibition reduces human adipogenesis. Disorders of theNF-�B pathway, resulting from proteasome inhibition maypartly explain the decrease in MMP-9 expression and secre-tion potentially responsible for adipogenesis reduction (Bour-lier et al., 2005) and observed with both lactacystin andHIV-PI treatment. Our in vitro results suggest that HIV-PI-induced perturbation of preadipocyte to adipocyte balancenoted in clinical studies (Bastard et al., 2002; Lloreta et al.,2002) may also target preadipocyte proteasome function. In-terestingly, a role for the proteasome in more metabolic as-pects of the HIV-related lipodystrophic syndrome has previ-ously been reported. Indeed, apolipoprotein B and sterolregulatory element binding protein degradation have beenshown to be altered by HIV-PI-related proteasome inhibitionand involved in lipid metabolism disorders (Liang et al.,2001; Hui, 2003). More recently, Parker et al. (2005) haslinked the reduction of proteasomal activity induced by HIV-PIs and the dyslipidemia to endoplasmic reticulum stress.Finally, attention is now being focused on HIV-PI-inducedproteasomal alterations and disturbances of glucose metab-olism because proteasome dysfunction and/or endoplasmicreticulum stress has been closely related to insulin resistance(Ozcan et al., 2004; Rome et al., 2004). All of this informationwill help to better understand the etiology of the lipodystro-phic syndrome but may also be of great interest for the recentclinical use of proteasome inhibitors in cancer therapy (i.e.,bortezomib treatment for multiple myeloma) and possibleeffects on adipose tissue development.
Acknowledgments
We thank Drs. Benarous, Belhaouri, and Jougla for help in ob-taining the adipose tissue samples. We also thank Dr. Sorisky forcritical review of this article.
298 De Barros et al.

ReferencesAdams J (2004) The development of proteasome inhibitors as cancer drugs. Cancer
Cell 5:417–421.Andre P, Groettrup M, Klenerman P, Giuli R, Booth BL Jr, Cerundolo V, Bonneville
M, Jotereau F, Zinkernagel RM, and Lotteau V (1998) An inhibitor of HIV-1protease modulates proteasome activity, antigen presentation, and T cell re-sponses. Proc Natl Acad Sci USA 95:13120–13124.
Bastard JP, Caron M, Vidal H, Jan V, Auclair M, Vigouroux C, Luboinski J, LavilleM, Maachi M, Girard PM, et al. (2002) Association between altered expression ofadipogenic factor SREBP1 in lipoatrophic adipose tissue from HIV-1-infectedpatients and abnormal adipocyte differentiation and insulin resistance. Lancet359:1026–1031.
Berg AH, Lin Y, Lisanti MP, and Scherer PE (2004) Adipocyte differentiationinduces dynamic changes in NF-�B expression and activity. Am J Physiol 287:E1178–E1188.
Bouloumie A, Sengenes C, Portolan G, Galitzky J, and Lafontan M (2001) Adipocyteproduces matrix metalloproteinases 2 and 9: involvement in adipose differentia-tion. Diabetes 50:2080–2086.
Bourlier V, Zakaroff-Girard A, De Barros S, Pizzacalla C, Durand de Saint Front V,Lafontan M, Bouloumie A, and Galitzky J (2005) Protease inhibitor treatmentsreveal specific involvement of matrix metalloproteinase-9 in human adipocytedifferentiation. J Pharmacol Exp Ther 312:1272–1279.
Chung S, Brown JM, Provo JN, Hopkins R, and McIntosh MK (2005) Conjugatedlinoleic acid promotes human adipocyte insulin resistance through NF�B-dependent cytokine production. J Biol Chem 280:38445–38456.
Cressman DE and Taub R (1994) Physiologic turnover of nuclear factor �B bynuclear proteolysis. J Biol Chem 269:26594–26597.
Gaedicke S, Firat-Geier E, Constantiniu O, Lucchiari-Hartz M, Freudenberg M,Galanos C, and Niedermann G (2002) Antitumor effect of the human immunode-ficiency virus protease inhibitor ritonavir: induction of tumor-cell apoptosis asso-ciated with perturbation of proteasomal proteolysis. Cancer Res 62:6901–6908.
Hayden MS and Ghosh S (2004) Signaling to NF-�B. Genes Dev 18:2195–2224.Hui DY (2003) Effects of HIV protease inhibitor therapy on lipid metabolism. Prog
Lipid Res 42:81–92.Ikebe T, Takeuchi H, Jimi E, Beppu M, Shinohara M, and Shirasuna K (1998)
Involvement of proteasomes in migration and matrix metalloproteinase-9 produc-tion of oral squamous cell carcinoma. Int J Cancer 77:578–585.
Janneh O, Hoggard PG, Tjia JF, Jones SP, Khoo SH, Maher B, Back DJ, andPirmohamed M (2003) Intracellular disposition and metabolic effects of zidovu-dine, stavudine and four protease inhibitors in cultured adipocytes. Antiviral Ther8:417–426.
Jones K, Hoggard PG, Sales SD, Khoo S, Davey R, and Back DJ (2001) Differencesin the intracellular accumulation of HIV protease inhibitors in vitro and the effectof active transport. AIDS 15:675–681.
Kisselev AF and Goldberg AL (2001) Proteasome inhibitors: from research tools todrug candidates. Chem Biol 8:739–758.
Kolev K, Skopal J, Simon L, Csonka E, Machovich R, and Nagy Z (2003) Matrixmetalloproteinase-9 expression in post-hypoxic human brain capillary endothelialcells: H2O2 as a trigger and NF-�B as a signal transducer. Thromb Haemost90:528–537.
Liang JS, Distler O, Cooper DA, Jamil H, Deckelbaum RJ, Ginsberg HN, and SturleySL (2001) HIV protease inhibitors protect apolipoprotein B from degradation bythe proteasome: a potential mechanism for protease inhibitor-induced hyperlipid-emia. Nat Med 7:1327–1331.
Lloreta J, Domingo P, Pujol RM, Arroyo JA, Baixeras N, Matias-Guiu X, GilaberteM, Sambeat MA, and Serrano S (2002) Ultrastructural features of highly activeantiretroviral therapy-associated partial lipodystrophy. Virchows Arch Medicine441:599–604.
Lu Y and Wahl LM (2005) Production of matrix metalloproteinase-9 by activatedhuman monocytes involves a phosphatidylinositol-3 kinase/Akt/IKK�/NF-�B path-way. J Leukoc Biol 78:259–265.
Malek S, Chen Y, Huxford T, and Ghosh G (2001) I�B�, but not I�B�, functions asa classical cytoplasmic inhibitor of NF-�B dimers by masking both NF-�B nuclearlocalization sequences in resting cells. J Biol Chem 276:45225–45235.
Nguyen AT, Gagnon A, Angel JB, and Sorisky A (2000) Ritonavir increases the levelof active ADD-1/SREBP-1 protein during adipogenesis. AIDS 14:2467–2473.
Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, GorgunC, Glimcher LH, and Hotamisligil GS (2004) Endoplasmic reticulum stress linksobesity, insulin action, and type 2 diabetes. Science (Wash DC) 306:457–461.
Pajonk F, Himmelsbach J, Riess K, Sommer A, and McBride WH (2002) The humanimmunodeficiency virus (HIV)-1 protease inhibitor saquinavir inhibits proteasomefunction and causes apoptosis and radiosensitization in non-HIV-associated hu-man cancer cells. Cancer Res 62:5230–5235.
Parker RA, Flint OP, Mulvey R, Elosua C, Wang F, Fenderson W, Wang S, Yang WP,and Noor MA (2005) Endoplasmic reticulum stress links dyslipidemia to inhibitionof proteasome activity and glucose transport by HIV protease inhibitors. MolPharmacol 67:1909–1919.
Patel YM and Lane MD (1999) Role of calpain in adipocyte differentiation. Proc NatlAcad Sci USA 96:1279–1284.
Piccinini M, Rinaudo MT, Anselmino A, Buccinna B, Ramondetti C, Dematteis A,Ricotti E, Palmisano L, Mostert M, and Tovo PA (2005) The HIV protease inhib-itors nelfinavir and saquinavir, but not a variety of HIV reverse transcriptaseinhibitors, adversely affect human proteasome function. Antivir Ther 10:215–223.
Piccinini M, Rinaudo MT, Chiapello N, Ricotti E, Baldovino S, Mostert M, and TovoPA (2002) The human 26S proteasome is a target of antiretroviral agents. AIDS16:693–700.
Pierce JW, Schoenleber R, Jesmok G, Best J, Moore SA, Collins T, and Gerritsen ME(1997) Novel inhibitors of cytokine-induced I�B� phosphorylation and endothelialcell adhesion molecule expression show anti-inflammatory effects in vivo. J BiolChem 272:21096–21103.
Prince AM, May JS, Burton GR, Lyle RE, and McGehee RE Jr (2002) Proteasomaldegradation of retinoblastoma-related p130 during adipocyte differentiation. Bio-chem Biophys Res Commun 290:1066–1071.
Rock KL, Gramm C, Rothstein L, Clark K, Stein R, Dick L, Hwang D, and GoldbergAL (1994) Inhibitors of the proteasome block the degradation of most cell proteinsand the generation of peptides presented on MHC class I molecules. Cell 78:761–771.
Rome S, Meugnier E, and Vidal H (2004) The ubiquitin-proteasome pathway is a newpartner for the control of insulin signaling. Curr Opin Clin Nutr Metab Care7:249–254.
Rudich A, Ben-Romano R, Etzion S, and Bashan N (2005) Cellular mechanisms ofinsulin resistance, lipodystrophy and atherosclerosis induced by HIV proteaseinhibitors. Acta Physiol Scand 183:75–88.
St-Pierre Y, Couillard J, and Van Themsche C (2004) Regulation of MMP-9 geneexpression for the development of novel molecular targets against cancer andinflammatory diseases. Expert Opin Ther Targets 8:473–489.
Sato H and Seiki M (1993) Regulatory mechanism of 92 kDa type IV collagenase geneexpression which is associated with invasiveness of tumor cells. Oncogene 8:395–405.
Schmidtke G, Holzhutter HG, Bogyo M, Kairies N, Groll M, de Giuli R, Emch S, andGroettrup M (1999) How an inhibitor of the HIV-I protease modulates proteasomeactivity. J Biol Chem 274:35734–35740.
Thompson JE, Phillips RJ, Erdjument-Bromage H, Tempst P, and Ghosh S (1995)I�B-� regulates the persistent response in a biphasic activation of NF-�B. Cell80:573–582.
Viatour P, Merville MP, Bours V, and Chariot A (2005) Phosphorylation of NF-�Band I�B proteins: implications in cancer and inflammation. Trends Biochem Sci30:43–52.
Voorhees PM and Orlowski RZ (2006) The proteasome and proteasome inhibitors incancer therapy. Annu Rev Pharmacol Toxicol 46:189–213.
Weil R, Laurent-Winter C, and Israel A (1997) Regulation of I�B� degradation:similarities to and differences from I�B�. J Biol Chem 272:9942–9949.
Whiteside ST and Israel A (1997) I�B proteins: structure, function and regulation.Semin Cancer Biol 8:75–82.
Address correspondence to: Dr. Virginie Bourlier, INSERM U586, 37Allees Jules Guesde, 31000 Toulouse, France. E-mail: [email protected]
Proteasome Inhibition Reduces Human Adipogenesis 299

Discussion de la publication 2
Dans cette étude, nous avons voulu déterminer les mécanismes impliqués dans la
diminution de l’expression de MMP-9 par les IPs-VIH. Nous avons tout d’abord évalué
l’effet des IPs-VIH sur l’activité du protéasome des cellules de la FSV du TA humain.
L’activité chymotrypsine des deux formes majeures du protéasome, le protéasome 20S qui
constitue la partie multicatalytique de tous les protéasomes et le protéasome 26S constitué du
protéasome 20S coiffé par deux sous-unités régulatrices 19S a été déterminée sur des extraits
protéiques de cellules de la FSV non différenciées. Nous avons observé que le SQV, le NFV
et le RTV diminuent l’activité du protéasome 20S et dans une moindre mesure celle du
protéasome 26S. Cette différence de sensibilité entre les activités des protéasomes 20S et le
26S a déjà été rapportée (84, 200) et pourrait s’expliquer par un problème d’accessibilité des
IPs-VIH aux sites catalytiques situés sur le protéasome 20S quand celui-ci est coiffé par les
sous-unités 19S c’est-à-dire sous une forme 26S (208, 279). Il est important de préciser que
l’inhibition de l’activité protéasomique observée par les IPs-VIH est très inférieure à celle
observée avec un inhibiteur spécifique du protéasome, la lactacystine. De plus, les
concentrations des IPs-VIH effectives sur l’inhibition du protéasome d’extraits protéiques des
cellules de la FSV sont plus élevées que celles requises pour l’inhibition de la différenciation
adipocytaire in vitro. Toutefois, les IPs-VIH tels que le SQV et le NFV ont été décrits comme
s’accumulant dans le cytoplasme cellulaire conduisant à une augmentation de la
concentration intracellulaire de 20 à 80 fois (111, 117, 273, 274).
Afin de déterminer si l’inhibition du protéasome affecte le processus de
différenciation adipocytaire, les cellules de la FSV du TA humain ont été traitées avec la
lactacystine. Après 10 jours de traitement, nous avons observé une diminution de
l’adipogenèse associée à une baisse de la sécrétion et de l’expression de MMP-9, sans atteinte
de MMP-2. Récemment, en accord avec nos résultats, les travaux de Li et al. ont montré que
la lactacystine inhibe également la différenciation des préadipocytes murins de la lignée 3T3-
L1 (146). L’ensemble de ces données suggère que le protéasome joue un rôle majeur dans
l’adipogenèse des cellules de la FSV ainsi que dans le contrôle de l’expression génique de
MMP-9. Il est tentant de spéculer que l’un des mécanismes impliqués dans l’effet anti-
adipogénique de la lactacystine est lié à une inhibition de l’activité de MMP-9. Toutefois,
d’autres voies peuvent être impliquées. Ainsi, il a été décrit que l’inhibition du protéasome
des préadipocytes murins entraîne une augmentation de CHOP-10, un facteur de transcription
anti-adipogénique (146). Les mécanismes impliqués dans cet effet sont encore inconnus.
63

Nous avons ensuite recherché des éventuelles perturbations de la voie NF-κB. Dans
un premier temps, nous nous sommes intéressés à IκB et avons observé, par des analyses de
western blot, une augmentation de la protéine IκBβ dans les préadipocytes humains traités
avec le SQV et le NFV pendant 10 jours suggérant une diminution de sa dégradation via le
protéasome. Dans un second temps, nous avons souhaité mettre en évidence un effet direct
des IPs-VIH sur le facteur de transcription NF-κB. Cependant, malgré plusieurs tentatives,
aucun effet direct des IPs-VIH et de la lactacystine sur l’activité et la localisation cellulaire
(cytoplasmique ou nucléaire) de NF-κB n’a pu être mis en évidence. Ces résultats pourraient
s’expliquer par le fait que nous avons travaillé dans des conditions basales c’est-à-dire non
stimulées. Nous avons donc stimulé les cellules de la FSV avec un activateur connu de la voie
NF-κB, le TNF-α. Comme attendu, suite à un traitement aigu avec le TNF- α, l’expression de
MMP-9 est fortement augmentée et lors d’un co-traitement avec la lactacystine et les IPs-VIH
tels que le SQV et le NFV, cette réponse est diminuée. Si l’on considère ces différents
résultats sur les préadipocytes humains en conditions basales et stimulées, on peut penser que
la chute de l’expression de MMP-9 induite par les IPs-VIH et la lactacystine est en partie due
à des perturbations de la voie NF-κB.
Nos résultats obtenus in vitro suggèrent donc que les IPs-VIH participent à l’atrophie
du TA en inhibant l’activité du protéasome des cellules de la FSV du TA humain. De manière
intéressante, l’inhibition du protéasome par les IPs-VIH a également été associée aux
complications métaboliques décrites dans les lipoatrophies induites par les IPs-VIH telles que
l’insulino-résistance et les désordres dans le métabolisme lipidique. Ainsi, compte tenu de
l’ensemble de ces données, il est nécessaire de rester vigilant quant aux effets indésirables
potentiels des inhibiteurs du protéasome dont l’un d’entre eux, le bortezomib est à ce jour
utilisé pour le traitement du cancer.
En résumé de cette première partie, nos résultats montrent que les MMPs et
particulièrement la MMP-9 est un acteur majeur dans la différenciation adipocytaire des
cellules de la FSV du TA humain. De plus, MMP-9 semble être une cible potentielle des IPs-
VIH. En effet, l’expression génique et la sécrétion de cette MMP est diminuée par les IPs-
VIH. Les mécanismes impliqués dans cet effet ont été déterminés et passent en partie par une
inhibition de l’activité protéasomique des cellules de la FSV par les IPs-VIH, entraînant des
perturbations de la voie NF-κB, facteur de transcription impliqué dans l’expression génique
de MMP-9. La MMP-9 joue donc un rôle majeur dans le processus de différenciation
adipocytaire chez l’homme.
64

En perspective à ce travail, les cibles moléculaires de MMP-9 responsables de l’effet
modulateur de la différenciation adipocytaire restent à définir. Nos études préliminaires ainsi
que les perspectives envisagées dans cet axe vont être brièvement présentées dans le
paragraphe qui suit.
D) Etudes préliminaires et perspectives sur l’étude des cibles moléculaires de
MMP-9
Comme nous l’avons évoqué l’effet des MMPs s’exerce via leurs actions sur les
composants de la MEC mais aussi via leurs actions sur des facteurs solubles activables par
ces MMPs et/ou des facteurs piégés dans la MEC.
Afin de déterminer si l’action de MMP-9 passe par la dégradation des composants
matriciels, nous avons d’abord évalué l’influence de différents supports matriciels tels que
des supports de collagène, fibronectine et gélatine, connus pour être des substrats de la MMP-
9 (57, 254), sur l’adipogenèse des cellules de la FSV du TA humain. Pour cela, des cellules
de la FSV du TA ont été ensemencées (60 000 cellules/cm2) sur ces différents supports et
mises en conditions adipogéniques. Après 10 jours de culture, le contenu intracellulaire en
TGs a été quantifié. Comme on peut l’observer dans la figure 16, aucun des supports
matriciels testés n’affecte l’accumulation lipidique des préadipocytes humains, un résultat ne
suggérant aucune altération de leur adipogenèse.
Ces données sont en accord avec d’autres travaux soulignant l’absence d’effet d’un
support de collagène (191) et de fibronectine (95) sur l’adipogenèse des cellules de la FSV de
TA humains et porcins, respectivement. A l’inverse, d’autres travaux ont rapporté un effet
anti-différenciant d’un support de fibronectine sur des préadipocytes d’origine murine et
humaine (98, 191, 260). Les différences observées dans ces études sont surprenantes et
pourraient s’expliquer par le modèle cellulaire et/ou les conditions de culture utilisées. On
note qu’un support de laminine décrit pour activer l’adipogenèse des préadipocytes porcins
(95) a également été testé mais ce support n’a pas permis l’adhérence des cellules de la FSV
du TA humain. Ainsi, ces résultats ne révélant aucun effet du support matriciel sur
l’adipogenèse, il est tentant de suggérer que l’activité de MMP-9 n’est pas due à la
dégradation de la MEC. Il faut cependant rester prudent car il est fort possible que l’action de
MMP-9 passe par la dégradation d’une MEC sécrétée par la cellule, elle-même, et non pas par
celle déposée au préalable sur un support. De plus, il est possible que le modèle de culture en
2 dimensions utilisé pour nos cellules ne permette pas d’observer un effet du support
65
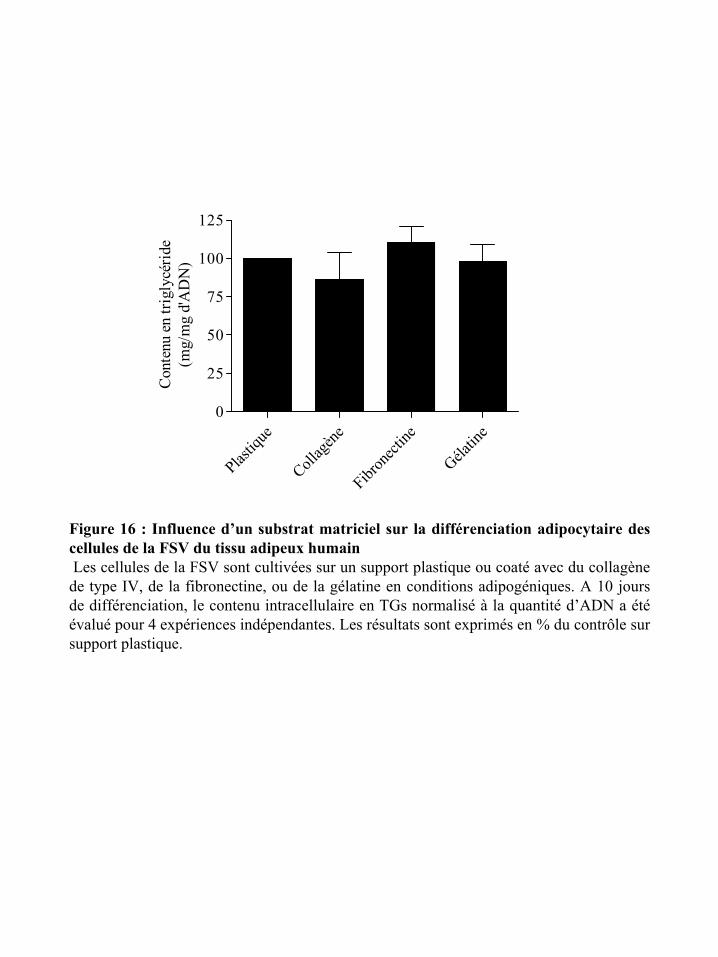
Plastiq
ue
Collag
ène
Fibron
ectine
Gélatin
e0
25
50
75
100
125
Con
tenu
en
trig
lycé
ride
(mg/
mg
d'A
DN
)
Figure 16 : Influence d’un substrat matriciel sur la différenciation adipocytaire
des cellules de la FSV du tissu adipeux humainLes cellules de la FSV sont cultivées sur un support plastique ou coaté
avec du collagène de type IV, de la fibronectine, ou de la gélatine en conditions adipogéniques. A 10 jours de différenciation, le contenu intracellulaire en TGs
normalisé
à
la quantité
d’ADN a été
évalué
pour 4 expériences indépendantes. Les résultats sont exprimés en % du contrôle sur support plastique.

matriciel. L’influence de différents supports matriciels dans un modèle de culture en 3
dimensions est à envisager. En effet, une étude récente montre que des effets de la MT1-
MMP ne sont révélés que sur un modèle de culture en 3 dimensions (44).
En parallèle, il serait très intéressant de caractériser les principaux composants
matriciels sécrétés par les cellules de la FSV du TA humain en cours de différenciation
traitées ou non avec un inhibiteur de MMP-9. Une telle étude permettrait non seulement de
déterminer l’évolution de la MEC au cours de l’adipogenèse chez l’homme mais aussi de
définir les effets de cette métalloprotéase sur les composants matriciels. De plus, MMP-9
étant connu pour générer des fragments matriciels biologiquement actifs tels que
l’endostatine (77) et certains peptides dérivés de la fibronectine (82), un traitement des
cellules de la FSV du TA humain par des fragments matriciels pourraient être envisagé. En
effet, les travaux de Kamiya et al. montrent qu’un fragment issu de la dégradation de la
fibronectine stimule la différenciation des préadipocytes de la lignée ST-13 (119).
Enfin, la recherche des cibles non matricielles susceptibles de moduler et réguler le
comportement cellulaire pourrait également être envisagée. De nombreuses protéines
cellulaires de surface, péri-cellulaires ou circulantes peuvent être clivées par les MMPs
régulant ainsi leur biodisponibilité. Cependant, peu de données existent chez l’homme. Il
nous semblerait donc intéressant de caractériser notamment la production des facteurs de
croissance et/ou cytokines par les cellules de la FSV du TA humain au cours de la
différenciation. Cette approche pourrait être réalisée grâce à l'utilisation de la technique des
immunoarrays dédiés aux « cytokines » qui permet d’identifier et de quantifier, dans un
milieu de culture ou dans un lysat cellulaire, des protéines différentes décrites comme étant
des cibles de MMP-9 dans d’autres modèles cellulaires et dont les effets sur la différenciation
cellulaire ont été également mis en évidence telles que les TGFs, TNFs, ILs, IGF-I, IGFBPs.
En parallèle, l’influence d’un traitement avec un inhibiteur de MMP-9 pourra être évaluée et
ce, afin de mettre en évidence les facteurs de croissance et/ou cytokines dont le taux
extracellulaire est modifié par la présence de MMP-9 soit par leur clivage direct soit par leur
libération de la MEC.
Pour conclure, l’étude des cibles moléculaires de MMP-9 constitue donc un axe de
recherche dont le développement pourrait permettre de mieux comprendre les effets de la
MMP-9. Cependant, les cellules de la FSV étant une population cellulaire hétérogène, on peut
se demander quels types cellulaires sont les cibles de MMP-9. Récemment, des travaux de
notre groupe ont permis d’isoler et de caractériser la population cellulaire contenant les
66

précurseurs adipocytaires (241). Dans la suite de ce travail, nous avons déterminé si l’effet de
la MMP-9 est retrouvé dans ces précurseurs adipocytaires spécifiquement isolés.
67

II- Implication des MMPs dans la différenciation des cellules progénitrices
CD34+/CD31- isolées de la fraction stroma-vasculaire du tissu adipeux humain
La caractérisation et l’isolement de la population cellulaire contenant les précurseurs
adipocytaires a été possible grâce au développement d’une technique d’isolement des
différentes populations cellulaires de la FSV qui se fonde sur l’utilisation de nanoparticules
magnétiques couplées à des anticorps dirigés spécifiquement contre les marqueurs de surface
CD34 (marqueur des cellules souches hématopoïétiques et des cellules endothéliales
capillaires), CD31 (marqueur des cellules endothéliales et de la lignée hématopoïétique) et
CD14 (marqueur des monocytes/macrophages) (figure 17). La fraction cellulaire
CD34+/CD31- est la seule à exprimer les marqueurs de différenciation spécifiques des
adipocytes et à acquérir les activités métaboliques adipocytaires, en réponse aux conditions
adipogéniques (241).
Notre groupe a également mis en évidence que ces cellules CD34+/CD31-, cultivées
en condition de culture endothéliales, changent de morphologie et d’organisation et
expriment des marqueurs spécifiques des cellules endothéliales tels que le CD31 et le facteur
de Von Willebrand (vWF). De plus, leur injection dans la patte ischémiée d’une souris
athymique entraîne une augmentation du flux sanguin et de la densité capillaire au niveau du
membre ischémié (173). D’autres laboratoires ont rapporté des capacités pro-angiogéniques
similaires des cellules issues de la FSV du TA (211, 221, 291). Par conséquent, la fraction
cellulaire CD34+/CD31- pourrait constituer un pool local de cellules progénitrices/souches
qui serait impliqué dans la formation non seulement des nouveaux adipocytes mais aussi des
capillaires accompagnant le développement de la masse grasse.
Les mécanismes impliqués dans le contrôle du devenir des cellules progénitrices
CD34+/CD31- restent à déterminer. Il a été rapporté que le microenvironnement pouvait
réguler le devenir des cellules souches hématopoïétiques et mésenchymateuses (127, 229). En
effet, de nombreux facteurs solubles (cytokines et facteurs de croissance) ainsi que la
composition et l’élasticité de la MEC contrôlent le devenir des cellules souches (14, 69, 71,
172). De plus, une récente publication indique que l’engagement des cellules souches
mésenchymateuses vers le lignage adipogénique et ostéogénique est régulé par la densité
cellulaire d’ensemencement qui altère la forme de la cellule et module les interactions
cellulaires (167).
Dans la deuxième partie de ce travail de thèse, nous avons d’abord vérifié et confirmé
l’implication des MMPs dans la différenciation adipocytaire des cellules CD34+/CD31-
68
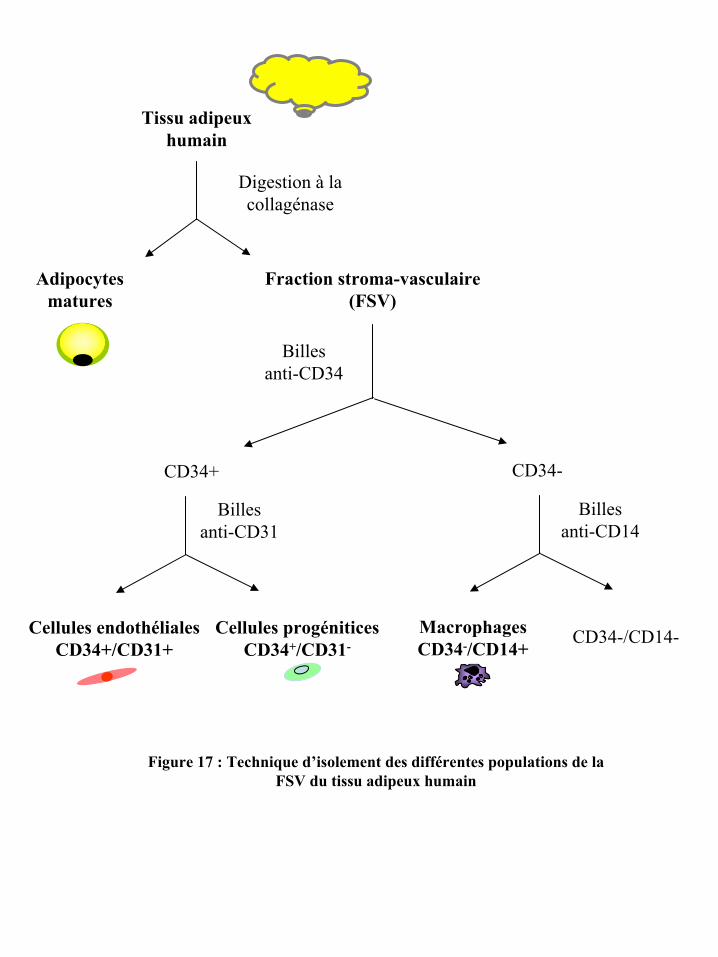
Fraction stroma-vasculaire(FSV)
CD34-
Cellules progénitices CD34+/CD31-
CD34+
Billes anti-CD34
Billesanti-CD14
Digestion à
la collagénase
Adipocytes matures
Billesanti-CD31
Macrophages CD34-/CD14+
CD34-/CD14-
Figure 17 : Technique d’isolement des différentes populations de la FSV du tissu adipeux humain
Tissu adipeux humain
Cellules endothéliales CD34+/CD31+

spécifiquement isolées. Suite à cet ensemble de résultats sur les cellules CD34+/CD31-, nous
nous sommes ensuite demandés si l’effet anti-adipogénique observé avec des inhibiteurs de
l’activité et/ou de l’expression des MMPs, particulièrement MMP-9, modulait également les
autres capacités de ces cellules dont la capacité angiogénique. Ainsi, afin d’étudier les
mécanismes qui contrôlent le devenir des cellules CD34+/CD31- nous avons mis au point
une condition de culture permettant l’étude simultanée de l’adipogenèse et de l’angiogenèse.
A) Influence d’un inhibiteur des MMPs sur la différenciation adipocytaire des
cellules CD34+/CD31-
Nous avons isolé puis cultivé les cellules CD34+/CD31- dans des conditions
adipogéniques similaires à celles décrites dans la première partie de ce travail. L’activité
gélatinase des MMP-2 et -9 relarguée dans le milieu de culture au cours de la différenciation
a tout d’abord été évaluée par la technique de zymographie à la gélatine comme décrite dans
les précédentes publications. Comme le montre la figure 18, la sécrétion des MMP-2 et -9 par
les cellules CD34+/CD31- augmente au cours de la différenciation adipocytaire des cellules
CD34+/CD31- isolées. Ces données indiquent que la production de MMP-2 et -9 est
strictement modulée par la différenciation adipocytaire et ne peut être attribuée à la
modulation de la présence ou de l’état d’activation des autres types cellulaires (macrophages,
cellules endothéliales) de la FSV.
L’implication des MMPs dans la différenciation adipocytaire des cellules
CD34+/CD31- a ensuite été déterminée. Pour cela et de manière similaire à la FSV, les
cellules CD34+/CD31- ont été traitées avec un inhibiteur des MMPs à large spectre, le
batimastat. Après 10 jours de traitement, une diminution de l’accumulation des gouttelettes
lipidiques a été observée et confirmée par le dosage du contenu intracellulaire en TGs (figure
19). Ces données sont en accord avec celles obtenues sur les cellules de la FSV. Elles
suggèrent un rôle majeur des MMPs dans le processus de différenciation adipocytaire des
cellules progénitrices CD34+/CD31- isolées. Cependant, une étude complète serait nécessaire
afin de préciser les effets du batimastat sur l’expression de gènes marqueurs de la
différenciation adipocytaire (gènes précoces tels qu’aP2 et PPARγ et gènes tardifs tels que la
LHS et FAS).
69

StandardJ0 J10MMP-9
MMP-2
Figure 18 : Sécrétion des MMP-2 et -9 au cours de la différenciation adipocytaire
des cellules progénitrices
CD34+/CD31-
isoléesAprès avoir été
isolées de la FSV, les cellules CD34+/CD31-
sont cultivées en conditions adipogéniques. L’activité
gélatinase
des MMP-2 et -9 relarguée
dans le milieu de culture à
0 (J0) et 10 (J10) jours de différenciation est évaluée par zymographie
à
la gélatine. L’utilisation d’un standard MMPs
nous a permis de détecter les bandes spécifiques de MMP-2 et -9 actives. Le gel présenté
est représentatif de 2 expériences indépendantes.
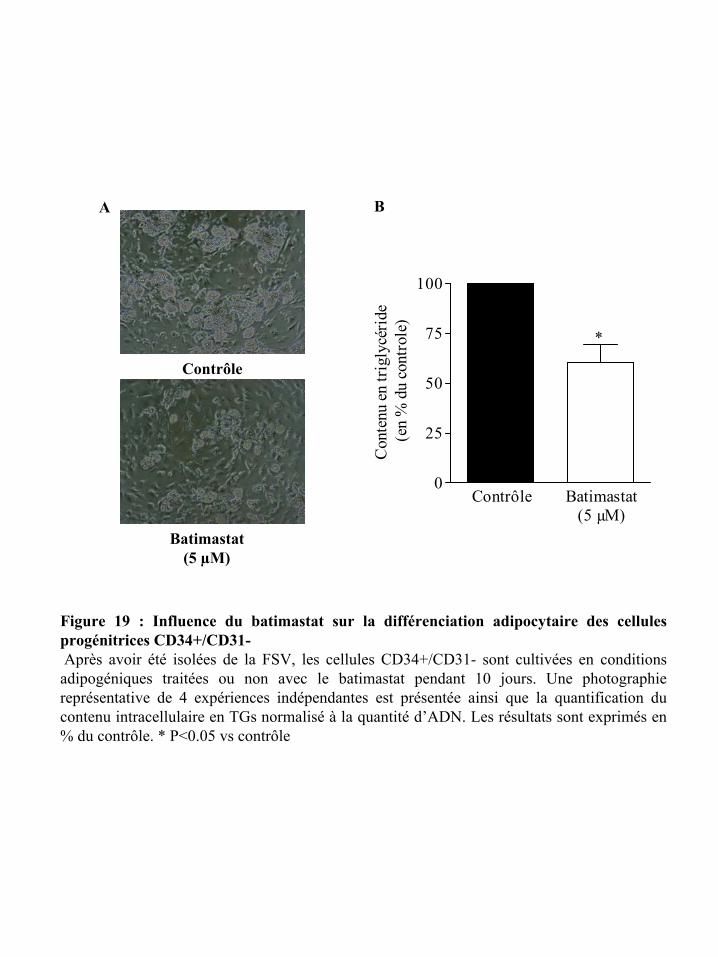
0
25
50
75
100
*
Contrôle Batimastat (5 µM)
Con
tenu
en
trig
lycé
ride
(en
% d
u co
ntro
le)
Contrôle
Batimastat(5 µM)
A B
Figure 19 : Influence du batimastat
sur la différenciation adipocytaire
des cellules progénitrices
CD34+/CD31-Après avoir été
isolées de la FSV, les cellules CD34+/CD31-
sont cultivées en conditions adipogéniques
traitées ou non avec le batimastat
pendant 10 jours. Une photographie représentative de 4 expériences indépendantes est présentée ainsi que la quantification du contenu intracellulaire en TGs
normalisé
à
la quantité
d’ADN. Les résultats sont exprimés en % du contrôle. * P<0.05 vs contrôle

Des travaux de notre groupe ayant démontré que les cellules progénitrices
CD34+/CD31- sont également capables d’angiogenèse dans des conditions de culture
endothéliales (173), il nous a semblé intéressant de déterminer si l’effet anti-adipogénique
observé avec le batimastat pouvait correspondre à une réorientation des cellules
CD34+/CD31- vers le lignage endothélial et/ou à une simple inhibition de la différenciation
adipocytaire. Ainsi, afin de déterminer les mécanismes impliqués dans le devenir des cellules
progénitrices CD34+/CD31-, nous avons mis au point une condition de culture permettant
l’étude simultanée des capacités adipogéniques et angiogéniques.
B) Mise au point d’une condition de culture permettant simultanément les
capacités adipogéniques et angiogéniques des cellules CD34+/CD31-
B.1) Etude de l’accumulation intracellulaire des triglycérides et de la formation d’un réseau
cellulaire positif pour le CD31 par les cellules CD34+/CD31- cultivées dans un milieu mixte
Des cellules progénitrices CD34+/CD31- ont été ensemencées (120 000 cellules/cm2)
dans un milieu de type ECBM (endothelial cell basal medium) supplémenté avec 10% de
sérum de veau foetal et dans des puits coatés avec de la fibronectine (5 µg/cm2) afin de
favoriser l’angiogenèse. Après 24 heures, les cellules ont été mises en présence d’un milieu
adipogénique, endothélial ou d’un milieu mixte correspondant à une combinaison des milieux
adipogéniques et endothéliaux. La composition de ces trois milieux est présentée dans le
tableau 11. Notons que dans cette étude, étant donné les effets apoptotiques de la ciglitazone
sur les cellules endothéliales (165), cet agoniste PPARγ a été remplacé par la rosiglitazone
décrite à l’inverse comme pro-angiogénique (209, 282).
Après 10 jours de culture, les cellules CD34+/CD31- cultivées en milieu
adipogénique et mixte montrent un phénotype adipocytaire caractérisé par l’accumulation de
gouttelettes lipidiques (figure 20A). Comme on peut le voir sur la figure 20B, une forte
augmentation du contenu intracellulaire en TG est observée au cours de la différenciation des
cellules CD34+/CD31- cultivées en milieu adipogénique et mixte. Cependant, aucune
différence dans le contenu intracellulaire en TG n’est observée si l’on compare le milieu
adipogénique versus le milieu mixte. En effet, le pourcentage de cellules différenciées en
adipocytes est similaire dans les milieux adipogénique et mixte (66.4 ± 4.4 % et 70.1 ± 1.5 %,
respectivement). En revanche, après 10 jours de culture en milieu endothélial, les cellules
CD34+/CD31- ne stockent pas de lipides.
70
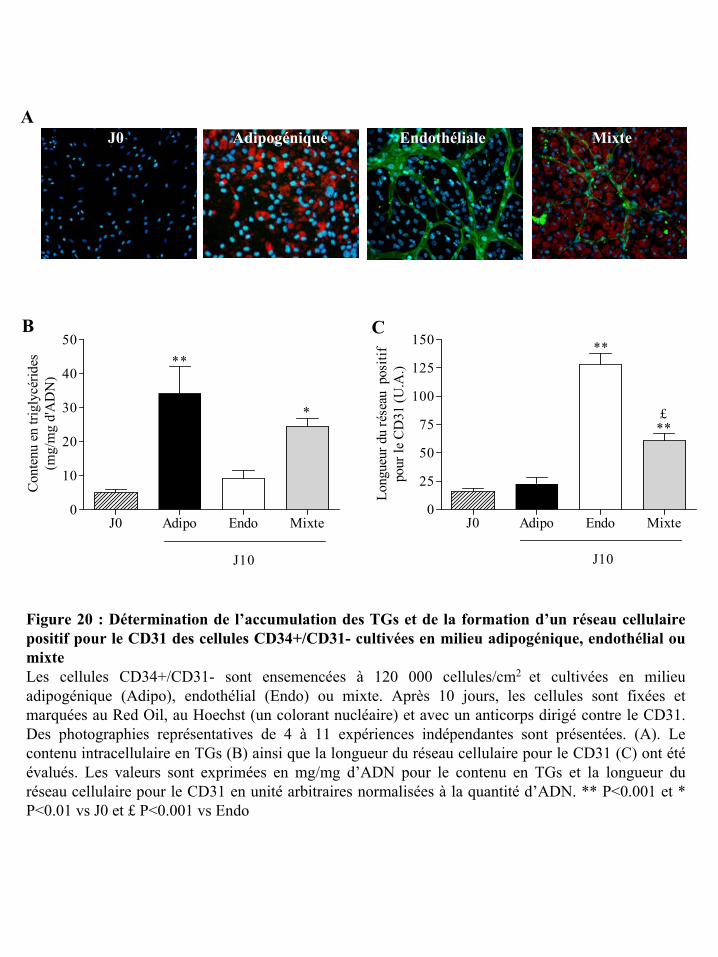
J0 Adipo Endo Mixte0
25
50
75
100
125
150 **
**
J10
£
Long
ueur
du
rése
au p
ositi
fpo
ur le
CD
31 (U
.A.)
J0 Adipo Endo Mixte0
10
20
30
40
50**
*
J10
Con
tenu
en
trigl
ycér
ides
(mg/
mg
d'A
DN
)
B
A
C
MixteEndothélialeAdipogéniqueJ0
Figure 20
: Détermination de l’accumulation des TGs
et de la formation d’un réseau cellulaire positif pour le CD31 des cellules CD34+/CD31-
cultivées en milieu adipogénique, endothélial ou mixte Les cellules CD34+/CD31-
sont ensemencées à
120
000 cellules/cm2 et cultivées en milieu adipogénique
(Adipo), endothélial (Endo) ou mixte. Après 10 jours, les cellules sont fixées et marquées au Red
Oil, au Hoechst
(un colorant nucléaire) et avec un anticorps dirigé
contre le CD31. Des photographies représentatives de 4 à
11 expériences indépendantes sont présentées. (A). Le contenu intracellulaire en TGs
(B) ainsi que la longueur du réseau cellulaire pour le CD31 (C) ont été
évalués. Les valeurs sont exprimées en mg/mg
d’ADN pour le contenu en TGs
et la longueur du réseau cellulaire pour le CD31 en unité
arbitraires normalisées à
la quantité
d’ADN. ** P<0.001 et * P<0.01 vs J0 et £
P<0.001 vs Endo

Tableau 11: Composition des milieux adipogénique, endothélial et mixte utilisés
Milieu adipogénique Milieu endothélial Milieu mixte
ECBM
Insuline (66 nmol/L)
T3 (1 nmol/L)
Cortisol (100 nmol/L)
Transferrine (10 µg/mL)
Rosiglitazone (3 µmol/L)
ECGM
VEGF (0.5 ng/mL)
IGF-1 (20 ng/mL)
ECGM
Insuline (66 nmol/L)
T3 (1 nmol/L)
Transferrine (10 µg/mL)
Rosiglitazone (3 µmol/L)
VEGF (0.5 ng/mL)
IGF-1 (20 ng/mL) (ECBM: endothelial cell basal medium, ECGM: endothelial cell growth medium constitué d’un milieu de type ECBM supplémenté avec des facteurs de croissance tels que l’EGF (0.1 ng/ml), le bFGF (1 ng/ml) et l’hydrocortisone (1 µg/ml)) et 2 % de sérum de veau foetal)
En parallèle, des analyses d’immnunohistochimie ont permis d’évaluer l’apparition
d’un marquage spécifique pour le CD31, un marqueur des cellules endothéliales (pour les
méthodes voir (173)). Comme on peut l’observer dans les figures 20A et C, la culture des
cellules CD34+/CD31- en milieu endothélial et mixte entraîne la formation d’un réseau
cellulaire positif pour le CD31. Cependant, en milieu mixte, la longueur du réseau cellulaire
positif pour le CD31 est plus faible qu’en milieu endothélial suggérant une perte du potentiel
angiogénique en milieu mixte. En effet, le pourcentage de cellules positives pour le CD31 est
plus faible en milieu mixte comparé au milieu endothélial (14.4 ± 1.1 % et 19.1 ± 2.4 % en
milieu mixte et endothélial, respectivement, P<0.05, milieu endothélial vs mixte). En
revanche, la culture des cellules CD34+/CD31- en milieu adipogénique n’entraîne pas la
formation d’un réseau positif pour le CD31. Il est important de préciser que le milieu mixte
comme le milieu endothélial est constitué d’un milieu de base de type ECGM qui contient
des facteurs de croissance et 2% de sérum de veau foetal. Ainsi, les cellules CD34+/CD31-
montrent une prolifération en milieu mixte et endothélial mais pas en milieu adipogénique.
Cette donnée est mise en évidence dans les photographies présentées dans la figure 20A
grâce au marquage des noyaux par le Hoechst, un colorant nucléaire, et confirmée par la
quantification du contenu cellulaire en ADN. En effet, au cours de la culture, une forte
augmentation (3,5 fois) du contenu en ADN dans les milieux endothélial (données
personnelles) et mixte (figure 22A) est observée. Toutefois, notons qu’aucune différence dans
le contenu en TGs, lorsqu’il est normalisé à la quantité d’ADN, n’a été observé lors de la
culture des cellules CD34+/CD31- dans un milieu adipogénique constitué d’une base ECBM
71

ou d’une base ECGM, suggérant que la prolifération de ces cellules dans le milieu ECGM ne
modifie pas le niveau de différenciation adipocytaire.
L’ensemble de ces résultats montre que les cellules CD34+/CD31- cultivées en milieu
mixte peuvent à la fois accumuler des lipides et former un réseau cellulaire positif pour le
CD31.
B.2) Etude de l’expression en ARNm des marqueurs adipocytaires et endothéliaux par les
cellules CD34+/CD31- cultivées dans un milieu mixte
Afin de confirmer les capacités adipogéniques et angiogéniques des cellules
CD34+/CD31- en milieu mixte, nous avons voulu évaluer l’expression en ARNm des
marqueurs adipocytaires et endothéliaux par des analyses de RT-PCR. L’expression de ces
marqueurs a été tout d’abord déterminée dans des cellules CD34+/CD31-, des adipocytes
matures et des cellules endothéliales capillaires CD34+/CD31+ fraîchement isolées. Les
résultats présentés dans le tableau 12, montrent comme attendu que les transcrits pour les
marqueurs adipocytaires sont détectés majoritairement dans les adipocytes matures et les
transcrits pour les marqueurs endothéliaux sont détectés majoritairement dans les cellules
endothéliales CD34+/CD31+. On peut cependant noter l’expression non négligeable d’aP2 et
de PPARγ dans les cellules endothéliales capillaires CD34+/CD31+. Concernant PPARγ, ces
résultats sont en accord avec d’autres observations (18). En revanche, aucune autre étude ne
s’est intéressée à l’expression d’aP2 dans les cellules endothéliales. L’ensemble de ces
résultats suggèrent donc que les cellules CD34+/CD31- fraîchement isolées expriment peu les
marqueurs adipocytaires et endothéliaux comparés aux adipocytes matures et aux cellules
endothéliales CD34+/CD31+, respectivement.
L’expression de ces différents marqueurs adipocytaires et endothéliaux dans les
cellules CD34+/CD31- cultivées en milieu adipogénique, endothélial et mixte pendant 10
jours a ensuite été déterminée. Comme le montre la figure 21A, en milieu mixte, une
augmentation significative des taux de transcrits codant pour les marqueurs adipocytaires,
excepté PPARγ, est observée par rapport au milieu endothélial. De plus, une augmentation en
ARNm de tous les marqueurs endothéliaux est observée dans les cellules CD34+/CD31- en
milieu mixte par rapport au milieu adipogénique (figure 21B). L’ensemble de ces résultats
montre que la culture des cellules CD34+/CD31- en milieu mixte pendant 10 jours induit
l’expression des ARNm codants pour des marqueurs adipocytaires d’une part et des
marqueurs endothéliaux d’autre part.
72
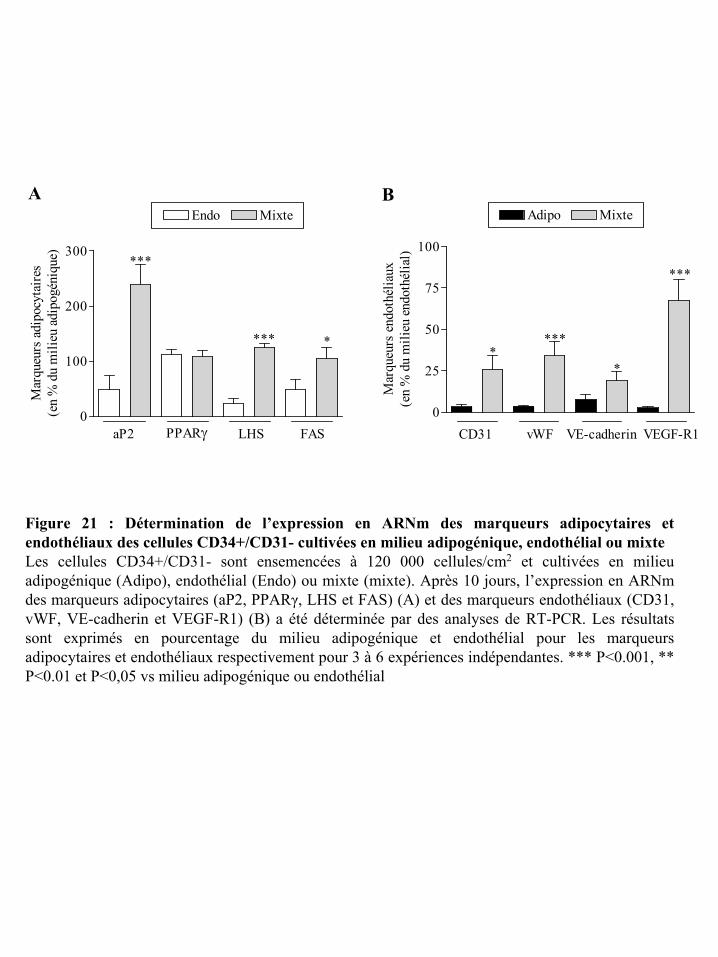
A B
0
100
200
300
FASLHSPPARγaP2
***
*** *
Endo Mixte
Mar
queu
rs a
dipo
cyta
ires
(en
% d
u m
ilieu
adi
pogé
niqu
e)
0
25
50
75
100
CD31 vWF VE-cadherin VEGF-R1
****
*
***
Adipo Mixte
Mar
queu
rs e
ndot
hélia
ux(e
n %
du
mili
eu e
ndot
hélia
l)
Figure 21 : Détermination de l’expression en ARNm
des marqueurs adipocytaires
et endothéliaux des cellules CD34+/CD31-
cultivées en milieu adipogénique, endothélial ou mixteLes cellules CD34+/CD31-
sont ensemencées à
120
000 cellules/cm2
et cultivées en milieu adipogénique
(Adipo), endothélial (Endo) ou mixte (mixte). Après 10 jours, l’expression en ARNm
des marqueurs adipocytaires
(aP2, PPARγ, LHS et FAS) (A) et
des marqueurs endothéliaux (CD31, vWF, VE-cadherin
et VEGF-R1) (B) a été
déterminée par des analyses de RT-PCR. Les résultats sont exprimés en pourcentage du milieu adipogénique
et endothélial pour les marqueurs adipocytaires
et endothéliaux respectivement pour 3 à
6 expériences indépendantes. *** P<0.001, ** P<0.01 et P<0,05 vs milieu adipogénique
ou endothélial
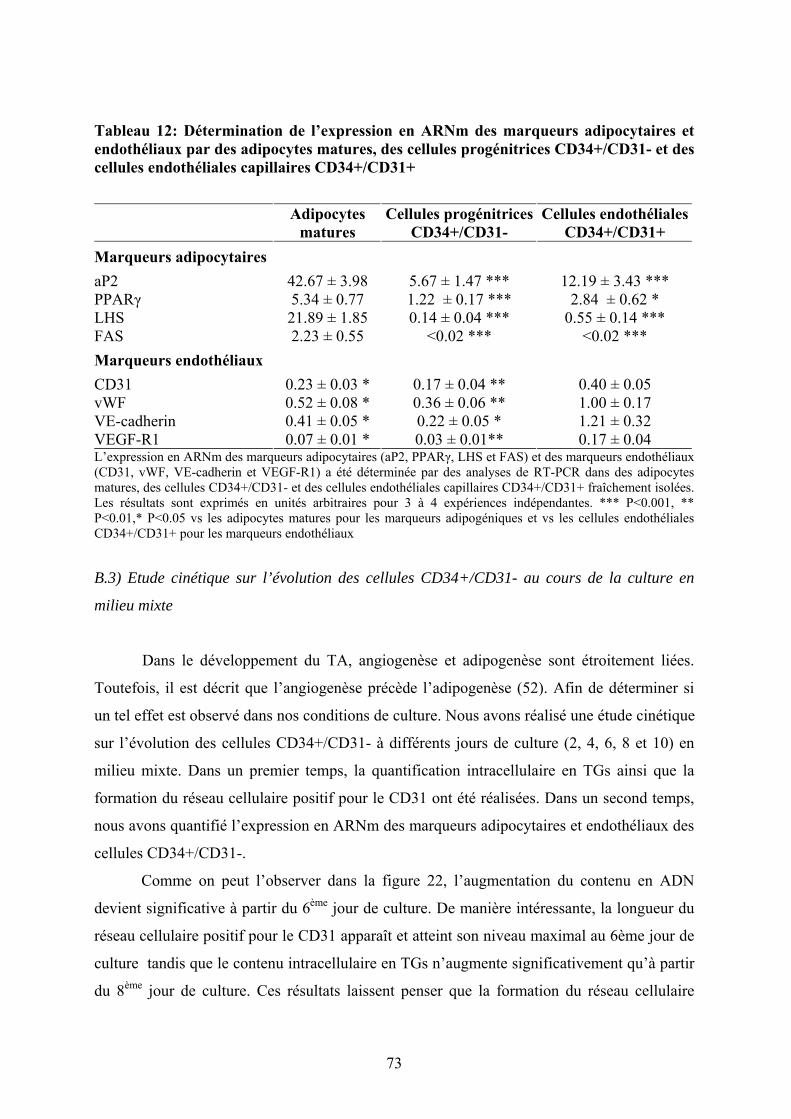
Tableau 12: Détermination de l’expression en ARNm des marqueurs adipocytaires et endothéliaux par des adipocytes matures, des cellules progénitrices CD34+/CD31- et des cellules endothéliales capillaires CD34+/CD31+
Adipocytes matures
Cellules progénitricesCD34+/CD31-
Cellules endothéliales CD34+/CD31+
Marqueurs adipocytaires aP2 42.67 ± 3.98 5.67 ± 1.47 *** 12.19 ± 3.43 *** PPARγ 5.34 ± 0.77 1.22 ± 0.17 *** 2.84 ± 0.62 * LHS 21.89 ± 1.85 0.14 ± 0.04 *** 0.55 ± 0.14 *** FAS 2.23 ± 0.55 <0.02 *** <0.02 *** Marqueurs endothéliaux CD31 0.23 ± 0.03 * 0.17 ± 0.04 ** 0.40 ± 0.05 vWF 0.52 ± 0.08 * 0.36 ± 0.06 ** 1.00 ± 0.17 VE-cadherin 0.41 ± 0.05 * 0.22 ± 0.05 * 1.21 ± 0.32 VEGF-R1 0.07 ± 0.01 * 0.03 ± 0.01** 0.17 ± 0.04 L’expression en ARNm des marqueurs adipocytaires (aP2, PPARγ, LHS et FAS) et des marqueurs endothéliaux (CD31, vWF, VE-cadherin et VEGF-R1) a été déterminée par des analyses de RT-PCR dans des adipocytes matures, des cellules CD34+/CD31- et des cellules endothéliales capillaires CD34+/CD31+ fraîchement isolées. Les résultats sont exprimés en unités arbitraires pour 3 à 4 expériences indépendantes. *** P<0.001, ** P<0.01,* P<0.05 vs les adipocytes matures pour les marqueurs adipogéniques et vs les cellules endothéliales CD34+/CD31+ pour les marqueurs endothéliaux
B.3) Etude cinétique sur l’évolution des cellules CD34+/CD31- au cours de la culture en
milieu mixte
Dans le développement du TA, angiogenèse et adipogenèse sont étroitement liées.
Toutefois, il est décrit que l’angiogenèse précède l’adipogenèse (52). Afin de déterminer si
un tel effet est observé dans nos conditions de culture. Nous avons réalisé une étude cinétique
sur l’évolution des cellules CD34+/CD31- à différents jours de culture (2, 4, 6, 8 et 10) en
milieu mixte. Dans un premier temps, la quantification intracellulaire en TGs ainsi que la
formation du réseau cellulaire positif pour le CD31 ont été réalisées. Dans un second temps,
nous avons quantifié l’expression en ARNm des marqueurs adipocytaires et endothéliaux des
cellules CD34+/CD31-.
Comme on peut l’observer dans la figure 22, l’augmentation du contenu en ADN
devient significative à partir du 6ème jour de culture. De manière intéressante, la longueur du
réseau cellulaire positif pour le CD31 apparaît et atteint son niveau maximal au 6ème jour de
culture tandis que le contenu intracellulaire en TGs n’augmente significativement qu’à partir
du 8ème jour de culture. Ces résultats laissent penser que la formation du réseau cellulaire
73
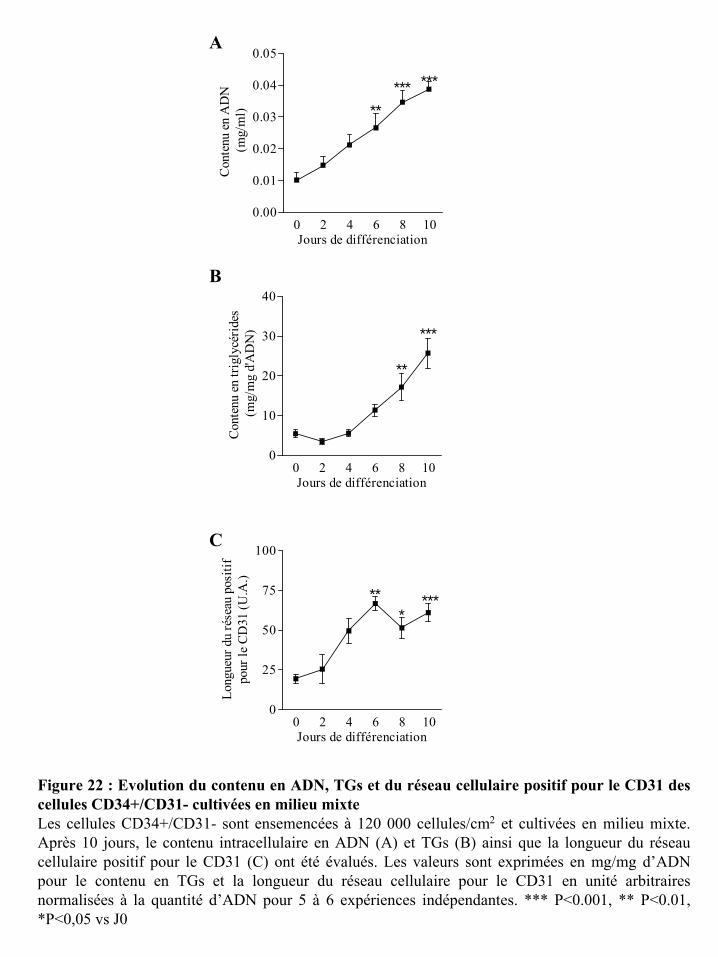
C
B
A
0 2 4 6 8 100.00
0.01
0.02
0.03
0.04
0.05
***
*****
Jours de différenciationC
onte
nu e
n A
DN
(mg/
ml)
0 2 4 6 8 100
25
50
75
100
***
***
Jours de différenciation
Long
ueur
du
rése
au p
ositi
fpo
ur le
CD
31 (U
.A.)
0 2 4 6 8 100
10
20
30
40
**
***
Jours de différenciation
Con
tenu
en
trig
lycé
ride
s (m
g/m
g d'
AD
N)
Figure 22
: Evolution du contenu en ADN, TGs
et du réseau cellulaire positif pour le CD31 des cellules CD34+/CD31-
cultivées en milieu mixteLes cellules CD34+/CD31-
sont ensemencées à
120
000 cellules/cm2
et cultivées en milieu mixte. Après 10 jours, le contenu intracellulaire en ADN (A) et TGs
(B) ainsi que la longueur du réseau cellulaire positif pour le CD31 (C) ont été
évalués. Les valeurs sont exprimées en mg/mg
d’ADN pour le contenu en TGs
et la longueur du réseau cellulaire pour le CD31 en unité
arbitraires normalisées à
la quantité
d’ADN pour 5 à
6 expériences indépendantes. *** P<0.001, ** P<0.01, *P<0,05 vs J0

positif pour le CD31 précède l’accumulation des TGs. Cependant, il est difficile de comparer
l’apparition d’un marqueur avec la mise en place d’une activité métabolique.
L’étude de l’expression en ARNm des marqueurs adipocytaires et endothéliaux, au
cours de la culture des cellules CD34+/CD31- en milieu mixte nous a permis de compléter
ces résultats. En effet, on note que l’expression d’aP2 et PPARγ, des marqueurs précoces du
processus adipogénique, apparaît et atteint son maximum dès le 4ème jour tandis que
l’expression de FAS, connu pour être un marqueur de l’activité métabolique, apparaît dès le
4ème jour mais atteint son maximum au 8ème jour de culture (figure 23). Concernant les
marqueurs endothéliaux, nous avons constaté que l’expression du CD31 augmente
significativement dès le 4ème jour de culture et une tendance similaire pour le facteur vWF
semble être observée même si aucune différence significative n’a pu être mise en évidence du
fait d’une forte variabilité interindividuelle. Ces résultats semblent indiquer qu’au cours de la
culture l’expression des marqueurs endothéliaux étudiés atteint un niveau maximal plus
rapidement que l’expression de FAS suggérant que les capacités angiogéniques apparaissent
plus précocement que les capacités adipogéniques. Toutefois, il n’est pas exclu que
l’accumulation des TGs nécessite plus de temps que l’organisation d’un réseau cellulaire
positif pour le CD31. Il est important de préciser que les marqueurs endothéliaux utilisés pour
ce travail sont décrits dans la littérature comme des marqueurs détectés spécifiquement dans
les cellules endothéliales matures (223). Leur expression en fonction de la différenciation
endothéliale n’est pas connue et à ce jour il n’est pas décrit de marqueurs géniques
spécifiques de la différenciation des cellules endothéliales. Par ailleurs, il faut également
rester prudent quant à la spécificité des marqueurs de la différenciation adipocytaire. En effet,
certains sont également détectés dans les cellules endothéliales matures notamment PPARγ et
aP2.
Pour conclure, l’ensemble de ces résultats indique que les cellules CD34+/CD31-
cultivées en milieu mixte pendant 10 jours expriment à la fois des marqueurs adipocytaires et
endothéliaux. Cette condition de culture constitue donc un nouvel outil pour l’étude des
mécanismes contrôlant le devenir des cellules progénitrices CD34+/CD31-.
74
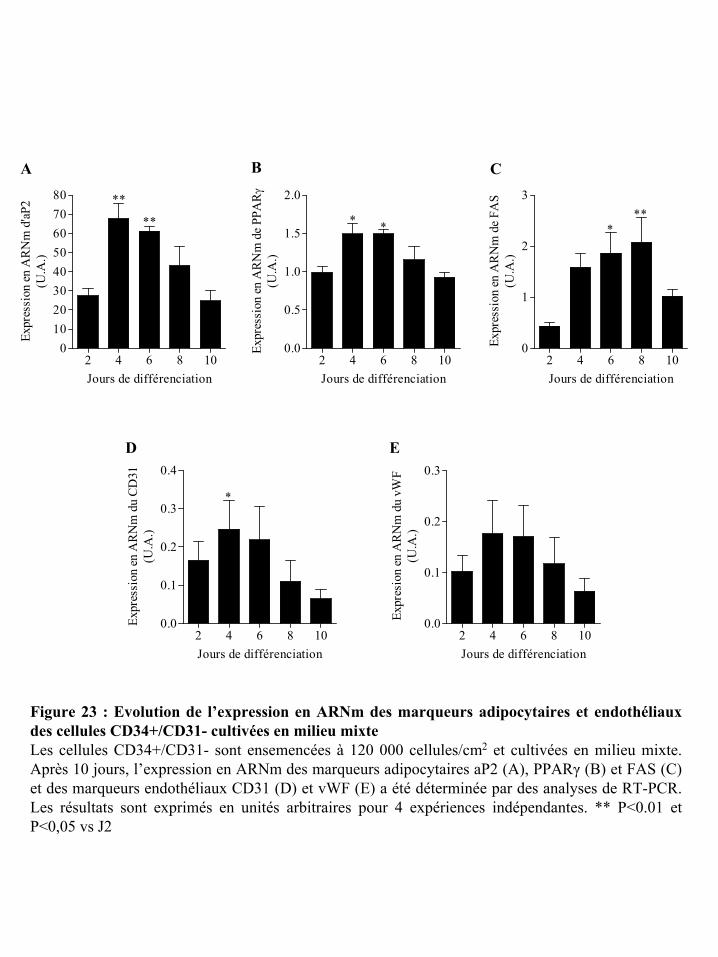
2 4 6 8 100.0
0.5
1.0
1.5
2.0
* *
Jours de différenciation
Expr
essi
on e
n A
RN
m d
e PP
ARγ
(U.A
.)
2 4 6 8 100
1020304050607080
*
**
**
Jours de différenciation
Expr
essi
on e
n A
RN
m d
'aP2
(U.A
.)
2 4 6 8 100
1
2
3
***
Jours de différenciation
Expr
essi
on e
n A
RN
m d
e FA
S(U
.A.)
2 4 6 8 100.0
0.1
0.2
0.3
Jours de différenciation
Expr
esio
n en
AR
Nm
du
vWF
(U.A
.)
2 4 6 8 100.0
0.1
0.2
0.3
0.4
*
Jours de différenciation
Expr
essi
on e
n A
RN
m d
u C
D31
(U.A
.)
A CB
ED
Figure 23
: Evolution de l’expression en ARNm
des marqueurs adipocytaires
et endothéliaux des cellules CD34+/CD31-
cultivées en milieu mixteLes cellules CD34+/CD31-
sont ensemencées à
120
000 cellules/cm2
et cultivées en milieu mixte. Après 10 jours, l’expression en ARNm
des marqueurs adipocytaires
aP2 (A), PPARγ
(B) et FAS (C) et
des marqueurs endothéliaux CD31 (D) et vWF
(E) a été
déterminée par des analyses de RT-PCR. Les résultats sont exprimés en unités arbitraires pour 4 expériences indépendantes. ** P<0.01 et P<0,05 vs J2

Toutefois, nous nous sommes demandés si l’effet angiogénique observé par les
cellules CD34+/CD31- n’est pas du à une contamination par des cellules endothéliales
matures CD34+/CD31+. Des analyses de cytométrie de flux ont été effectuées pour
déterminer la pureté de la fraction CD34+/CD31-. Il a été observé que 92.9 ± 0.8 % des
cellules retrouvées dans cette fraction sont CD34+/CD31- (données personnelles). Ainsi, la
possibilité d’une contamination par des cellules endothéliales est faible. Cependant, afin de
déterminer l’influence de la présence des cellules endothéliales capillaires matures sur les
cellules CD34+/CD31- en milieu mixte, des expériences de coculture (CD34+/CD31- et
CD34+/CD31+) ont été réalisées. Les cellules CD34+/CD31- ont été ensemencées dans un
milieu ECBM/10% sérum de veau fœtal à 120000 cellules/puits en présence ou non de
cellules endothéliales capillaires CD34+/CD31+ (5000 cellules/puits). Après 24 heures (J0) et
10 jours de culture en milieu mixte (J10), le marquage positif pour le CD31 a été quantifié.
Les résultats présentés à la figure 24, montrent qu’après 24 heures, le nombre de cellules
positives pour le CD31 lors de la coculture est augmenté (P<0,05 contrôle vs coculture) du à
la présence de cellules endothéliales capillaires matures CD34+/CD31+. Cependant, après 10
jours de culture, aucune différence dans la longueur du réseau cellulaire pour le CD31 n’est
observée entre les cellules CD34+/CD31- seules ou en coculture. Ce résultat indiquent que
les cellules endothéliales matures quiescentes CD34+/CD31+ cultivées en milieu mixte ne
s’organisent pas en réseau à la différence des cellules CD34+/CD31-. L’ensemble de ces
données suggère donc que l’effet angiogénique observé est du à un potentiel angiogénique
des cellules CD34+/CD31- plutôt qu’à une contamination par des cellules endothéliales
CD34+/CD31+.
La mise au point de cette condition de culture permettant de révéler simultanément les
capacités adipogéniques et angiogéniques des cellules CD34+/CD31- nous a permis d’initier
des études sur les mécanismes potentiels impliqués dans la régulation de l’expression de cette
double fonctionnalité des cellules CD34+/CD31-. Trois axes ont été envisagés : l’influence
des contacts cellule-cellule, l’influence de la MEC et de son remodelage notamment via les
MMPs et l’influence des facteurs paracrines. Les résultats préliminaires que nous avons
obtenus sont présentés dans le paragraphe suivant.
75
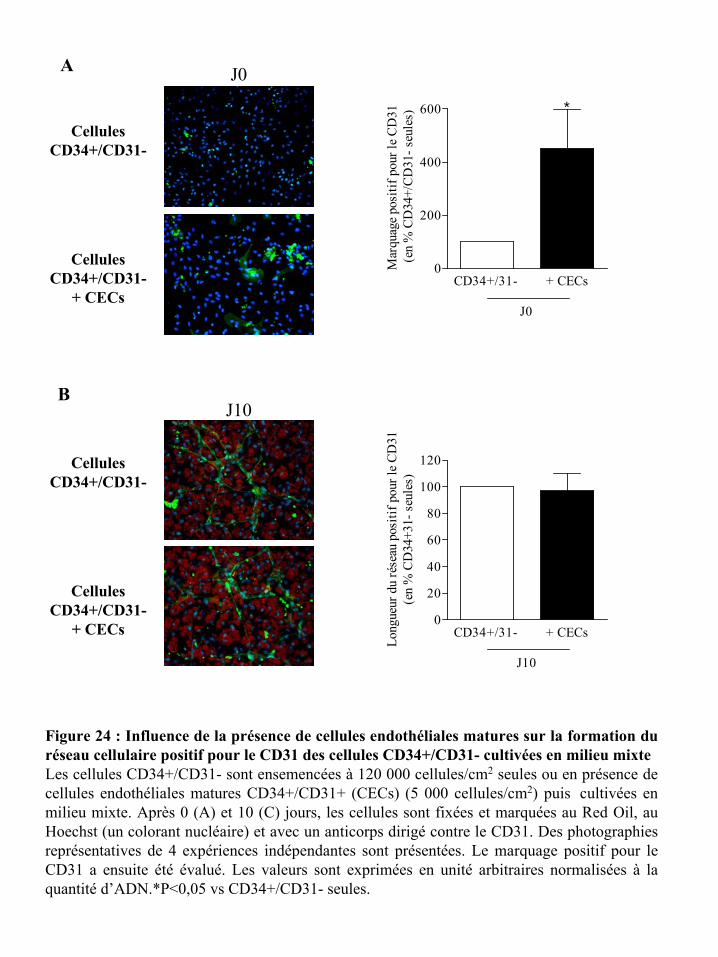
Figure 24
: Influence de la présence de cellules endothéliales matures sur la formation du réseau cellulaire positif pour le CD31 des cellules CD34+/CD31-
cultivées en milieu mixteLes cellules CD34+/CD31-
sont ensemencées à
120
000 cellules/cm2
seules ou en présence de cellules endothéliales matures CD34+/CD31+ (CECs) (5 000 cellules/cm2) puis
cultivées en milieu mixte. Après 0 (A) et 10 (C) jours, les cellules sont fixées et marquées au Red
Oil, au Hoechst
(un colorant nucléaire) et avec un anticorps dirigé
contre le CD31. Des photographies représentatives de 4 expériences indépendantes sont présentées. Le marquage positif pour le CD31 a ensuite été
évalué. Les valeurs sont exprimées en unité
arbitraires normalisées à
la quantité
d’ADN.*P<0,05 vs CD34+/CD31-
seules.
CD34+/31- + CECs0
200
400
600 *
Mar
quag
e pos
itif p
our l
e C
D31
(en
% C
D34
+/C
D31
- seu
les)
J0
J0
J10
A
B
Cellules CD34+/CD31-
Cellules CD34+/CD31-
+ CECs
Cellules CD34+/CD31-
Cellules CD34+/CD31-
+ CECs CD34+/31- + CECs0
20
40
60
80
100
120
J10
Long
ueur
du
rése
au p
ositi
f pou
r le
CD
31(e
n %
CD
34+3
1- s
eule
s)

C) Implication des contacts cellule-cellule dans les capacités adipogéniques et
angiogéniques des cellules CD34+/CD31-
C.1) Influence de la densité cellulaire sur les cellules CD34+/CD31- cultivées en milieu mixte
Etant donné que les cellules CD34+/CD31- ont une capacité de prolifération
importante en milieu mixte, la densité cellulaire augmente avec le temps. L’influence des
contacts cellule-cellule sur le devenir des cellules progénitrices CD34+/CD31- a donc été
déterminée en modulant la densité cellulaire d’ensemencement. Pour cela, les cellules ont été
ensemencées à trois densités différentes (2 000, 20 000 ou 120 000 cellules/cm2) et cultivées
pendant 10 jours en milieu mixte. Comme on peut le voir dans la figure 25A, quelle que soit
la densité cellulaire d’ensemencement, la quantité cellulaire d’ADN augmente au cours de la
culture et à 10 jours de culture cette quantité d’ADN est identique pour les trois densités. Une
augmentation du contenu intracellulaire en TGs est également observée quelque soit la
densité cellulaire (figure 25B). Cependant, le contenu intracellulaire en TGs est plus faible si
l’on compare la densité d’ensemencement la plus basse (2000 cellules/cm2) et la plus élevée
(120000 cellules/cm2) suggérant un retard de l’adipogenèse lorsque les cellules sont
ensemencées à faible densité. Concernant, la formation du réseau cellulaire positif pour le
CD31, il n’est observé qu’à la plus forte densité (figure 25C) suggérant que les contacts
cellule-cellule initiaux sont nécessaires à l’expression des capacités angiogéniques des
cellules CD34+/CD31-. L’ensemble de ces données suggère donc que la densité cellulaire
d’ensemencement peut moduler le devenir des cellules CD34+/CD31-.
L’influence de la densité cellulaire sur le devenir cellulaire est connue puisque des
travaux ont déjà suggéré que la densité cellulaire influence la différenciation des cellules
souches mésenchymateuses (210). Cependant, ce sont les travaux de Mc Beath et al. qui ont
clairement montré que la densité cellulaire d’ensemencement module l’engagement des
cellules souches mésenchymateuses vers le lignage adipogénique et ostéogénique (167). Ces
effets relatifs à la densité cellulaire peuvent être dus à des modulations de la forme de la
cellule et des tensions du cytosquelette comme il a déjà été suggéré (167) mais également à la
modulation des contacts et des communications entre les cellules.
De nombreuses études ont montré l’importance des jonctions gaps dans le contrôle
des processus de myogenèse (9, 215), d’ostéogenèse (237) mais aussi d’adipogenèse (238).
Récemment, une étude indique que la communication par les jonctions gaps est nécessaire à
l’expansion clonale durant la différenciation des préadipocytes de la lignée 3T3-L1 (296).
76
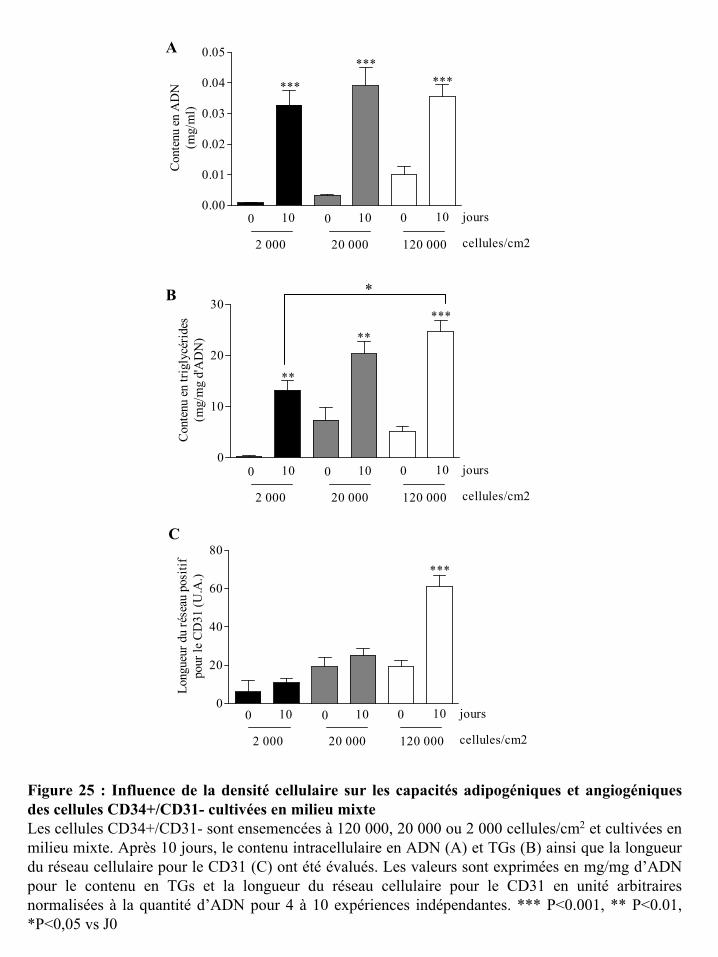
B
A
0.00
0.01
0.02
0.03
0.04
0.05
0 10 0 10 0 10
2 000
jours
20 000 120 000 cellules/cm2
***
******
Con
tenu
en
AD
N(m
g/m
l)
0
10
20
30
0 10 0 10 0 10
2 000
jours
20 000 120 000 cellules/cm2
**
***
**
Con
tenu
en
trigl
ycér
ides
(mg/
mg
d'AD
N)
0
20
40
60
80
0 10 0 10 0 10
2 000
jours
20 000 120 000 cellules/cm2
***
Long
ueur
du
rése
au p
ositi
fpo
ur le
CD
31 (U
.A.)
C
*
Figure 25 : Influence de la densité
cellulaire sur les capacités adipogéniques
et angiogéniques
des cellules CD34+/CD31-
cultivées en milieu mixteLes cellules CD34+/CD31-
sont ensemencées à
120
000, 20
000 ou 2
000 cellules/cm2
et cultivées en milieu mixte. Après 10 jours, le contenu intracellulaire en ADN (A) et TGs
(B) ainsi que la longueur du réseau cellulaire pour le CD31 (C) ont été
évalués. Les valeurs sont exprimées en mg/mg
d’ADN pour le contenu en TGs
et la longueur du réseau cellulaire pour le CD31 en unité
arbitraires normalisées à
la quantité
d’ADN pour 4 à
10 expériences indépendantes. *** P<0.001, ** P<0.01, *P<0,05 vs J0

Dans la suite de ce travail, l’implication de la communication par les jonctions gaps dans les
capacités angiogéniques et adipogéniques des cellules CD34+/CD31- a été évaluée.
C.2) Influence de la communication cellulaire via les jonctions gaps sur les cellules
CD34+/CD31- cultivées en milieu mixte
Les jonctions gaps forment un canal intercellulaire essentiellement constitué d’un
assemblage de connexines. Nous avons tout d’abord évalué l’expression en ARNm des
connexines 43, 45, 40 et 37 dans des adipocytes matures, des cellules progénitrices
CD34+/CD31- et des cellules endothéliales capillaires CD34+/CD31+ fraîchement isolées.
Comme on peut le voir dans le tableau 13, toutes les connexines, excepté la connexine 37,
sont détectées dans les trois types cellulaires testés. Le niveau d’ARNm de la connexin 40 est
cependant très faible et on note que les cellules endothéliales capillaires expriment des
niveaux plus élevés de connexine 43 que les adipocytes matures.
L’expression en ARNm des connexines 43 et 45 a ensuite été évaluée au cours de la
culture des cellules progénitrices CD34+/CD31- en milieu mixte. Les résultats sont présentés
dans la figure 26 et indiquent clairement que l’expression en ARNm de ces connexines
diminue au cours de la culture. Ces résultats sont en accord avec les travaux d’Umezawa et
al. montrant que l’expression de la connexine 43 est diminué au cours de la différenciation
adipocytaire des cellules stromales de la moelle osseuse H-1/A d’origine murine (267).
Tableau 13: Expression en ARNm des connexines 43, 45, 40 et 37 dans des adipocytes matures, des cellules progénitrices CD34+/CD31- et des cellules endothéliales capillaires CD34+/CD31+
Types de connexines
Adipocytes matures
Cellules progénitricesCD34+/CD31-
Cellules endothéliales CD34+/CD31+
Connexine 43 0.20 ± 0.06 0.51 ± 0.06 0.85 ± 0.21 *
Connexine 45 0.05 ± 0.01 0.14 ± 0.06 0.18 ± 0.08
Connexine 40 <0.02 <0.02 0.04 ± 0.01
Connexine 37 Non détectable Non détectable Non détectable L’expression en ARNm des connexines 43, 45, 40 et 37 a été déterminée par des analyses de RT-PCR dans des adipocytes matures, des cellules CD34+/CD31- et des cellules endothéliales CD34+/CD31+ fraîchement isolées. Les résultats sont exprimés en unités arbitraires pour 3 expériences indépendantes. * P<0.05 vs les adipocytes matures
77
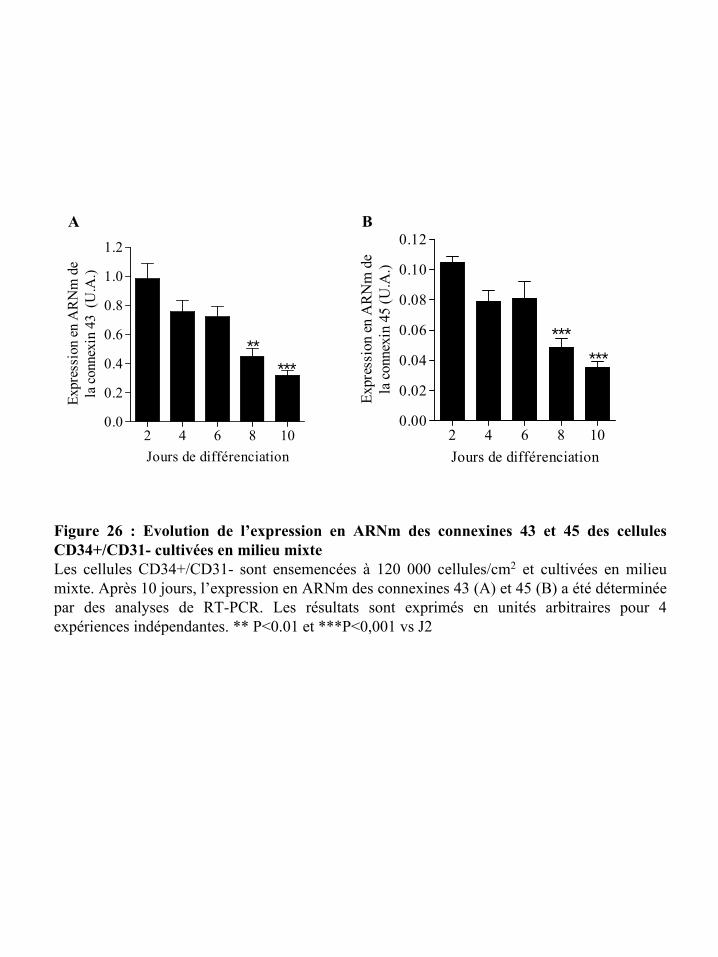
2 4 6 8 100.0
0.2
0.4
0.6
0.8
1.0
1.2
*****
Jours de différenciation
Expr
essi
on e
n A
RN
m d
ela
con
nexi
n 43
(U
.A.)
2 4 6 8 100.00
0.02
0.04
0.06
0.08
0.10
0.12
******
Jours de différenciation
Expr
essi
on e
n A
RN
m d
ela
con
nexi
n 45
(U.A
.)
Figure 26 : Evolution de l’expression en ARNm
des connexines
43 et 45 des cellules CD34+/CD31-
cultivées en milieu mixteLes cellules CD34+/CD31-
sont ensemencées à
120
000 cellules/cm2
et cultivées en milieu mixte. Après 10 jours, l’expression en ARNm
des connexines
43 (A) et 45 (B) a été
déterminée par des analyses de RT-PCR. Les résultats sont exprimés en unités arbitraires pour 4 expériences indépendantes. ** P<0.01 et ***P<0,001 vs J2
BA

Le rôle direct de la communication par les jonctions gaps sur les capacités
adipogéniques et angiogéniques des cellules CD34+/CD31- a été déterminé grâce à
l’utilisation d’un inhibiteur des jonctions gaps, le 18α-glycyrrhetinic acide (AGA). Les
cellules ont été traitées pendant 10 jours avec 5 et 10 µM d’AGA ou de GZA (glycyrrhizic
acide) un analogue peptidique de l’AGA sans effet sur les jonctions gaps. Comme on peut le
voir dans la figure 27, le GZA n’a aucun effet significatif ni sur le contenu intracellulaire en
TGs ni sur l’apparition du réseau cellulaire positif pour le CD31. En revanche, l’AGA à 10
µM diminue de façon significative le contenu intracellulaire en TGs mais n’affecte pas
significativement le réseau CD31 même si une tendance à l’augmentation du réseau est
observée. Il est important de noter qu’à une concentration de 10 µM, l’AGA n’a pas effet sur
le contenu cellulaire en ADN (données personnelles). Cependant, l’utilisation de cet
inhibiteur à des concentrations supérieures à 30 µM est associée à une mort cellulaire au bout
de quelques jours de culture.
Ces résultats indiquent que la communication cellulaire via les jonctions gaps est
requise pour l’adipogenèse des cellules CD34+/CD31- en accord avec les travaux de
Yanagiya et al. sur les préadipocytes murins (296). Toutefois, l’effet observé sur les cellules
CD34+/CD31- ne semble pas être du à une altération de l’expansion clonale comme c’est le
cas pour les préadipocytes murins décrit par les résultats de Yanigaya et al. car aucune
différence du contenu cellulaire en ADN dans les cellules contrôles et traitées n’a été
observée (données personnelles). Une étude plus complète est toutefois nécessaire afin de
préciser les effets d’un inhibiteur des jonctions gaps sur l’expression en ARNm des
marqueurs de la différenciation adipocytaire et de déterminer à quel stade de l’adipogenèse
les jonctions gaps sont requises.
En résumé de cette deuxième partie, nous avons mis au point une condition de
culture permettant simultanément l’étude des capacités angiogéniques et adipogéniques des
cellules CD34+/CD31-. Ce nouvel outil va permettre l’étude des mécanismes qui contrôlent
le devenir des cellules CD34+/CD31- et notamment l’influence du microenvironnement. Des
premiers résultats indiquent que les contacts cellule-cellule ainsi que la communication via
les jonctions gaps peuvent moduler le devenir des cellules CD34+/CD31-.
78
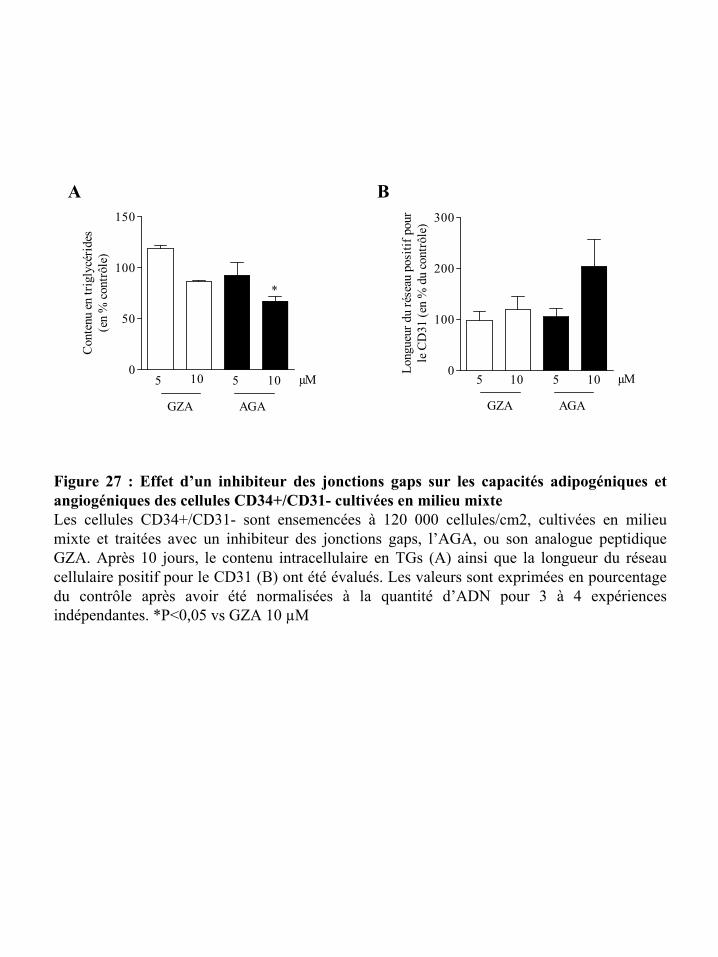
0
50
100
150
*
5 10 105 µM
GZA AGA
Con
tenu
en
trig
lycé
ride
s(e
n %
con
trôl
e)
0
100
200
300
5 10 105 µM
GZA AGALo
ngue
ur d
u ré
seau
pos
itif p
our
le C
D31
(en
% d
u co
ntrô
le)
BA
Figure
27 : Effet d’un inhibiteur des jonctions gaps sur les capacités adipogéniques
et angiogéniques
des cellules CD34+/CD31-
cultivées en milieu mixteLes cellules CD34+/CD31-
sont ensemencées à
120
000 cellules/cm2, cultivées en milieu mixte et traitées avec un inhibiteur des jonctions gaps, l’AGA, ou son analogue peptidique GZA. Après 10 jours, le contenu intracellulaire en TGs
(A) ainsi que la longueur du réseau cellulaire positif pour le CD31 (B) ont été
évalués. Les valeurs sont exprimées en pourcentage du contrôle après avoir été
normalisées à
la quantité
d’ADN pour 3 à
4 expériences indépendantes. *P<0,05 vs GZA 10 µM

En perspective à ce travail, l’influence des MMPs sur le devenir des cellules
CD34+/CD31- cultivées en milieu mixte est envisagé. En effet, nos résultats indiquent que
les cellules CD34+/CD31- isolées et cultivées en milieu adipogénique sécrètent les MMP-2 et
-9 et une inhibition de leur activité par le batimastat limite l’adipogenèse de ces cellules. Il
serait maintenant indispensable d’étudier les effets d’une inhibition des MMPs sur les
capacités adipogéniques et angiogéniques des cellules CD34+/CD31-. L’influence directe de
la MEC et notamment de ses composants matriciels tels que la fibronectine, le collagène, la
gélatine sur les capacités adipogéniques et angiogéniques des cellules CD34+/CD31-
cultivées en milieu mixte pourrait ensuite être déterminée. Ces études permettraient de définir
le rôle de la matrice et de sa dégradation dans le devenir des cellules progénitrices du TA
humain. Enfin, un troisième axe serait également intéressant et concerne l’influence de
facteurs paracrines sur le devenir de ces cellules. En effet, ces cellules contribuent
certainement à la croissance du TA en fournissant adipocytes et cellules endothéliales
nécessaires à son développement. Comme nous l’avons décrit dans l’introduction, le TA est
une source de nombreux facteurs ou adipokines, elles-mêmes originaires des différentes
populations cellulaires qui composent le TA. Ces sécrétions sont modulées au cours des
phases de croissance des dépôts adipeux. On peut donc se demander si ces adipokines
exercent un rôle dans le contrôle de la différenciation des cellules progénitrices. Ainsi,
l’influence des sécrétions par les autres types cellulaires du TA environnant les cellules
CD34+/CD31- tels que les macrophages, les cellules endothéliales, et les adipocytes matures
doit être envisagée par l’utilisation de milieu conditionnés en base ECBM (condition de
culture que nous avons vérifié comme étant favorable à la survie des cellules endothéliales,
macrophages et adipocytes matures isolés du TA humain) sur les cellules CD34+/CD31- en
milieu mixte.
79

Conclusions et perspectives
Ce travail de thèse a eu pour objectif de déterminer l’implication des MMPs dans la
différenciation des précurseurs adipocytaires humains. Nous avons démontré un rôle majeur
de l’activité des MMPs et plus particulièrement MMP-9 dans la différenciation adipocytaire
des cellules de la FSV du TA humain. De plus, MMP-9 pourrait être une cible de l’effet
inhibiteur des IPs-VIH sur la différenciation adipocytaire des cellules de la FSV du TA
humain. En effet, nous avons mis en évidence que les IPs-VIH diminuent l’expression et la
sécrétion de MMP-9. Les mécanismes impliqués dans cet effet passent en partie par une
inhibition de l’activité protéasomique des cellules de la FSV par les IPs-VIH, entraînant des
perturbations de la voie NF-κB, facteur de transcription impliqué dans l’expression génique
de MMP-9. L’étude des cibles moléculaires de MMP-9 impliquées dans les effets de cette
protéase sur la différenciation adipocytaire, telles que la dégradation des composants de la
MEC puisque celle-ci évolue au cours de la différenciation adipocytaire et/ou la
biodisponibilité de facteurs solubles qui modulent l’adipogenèse doit maintenant être
envisagé. De même les cibles cellulaires de MMP-9 au sein du TA doivent être mieux
définies.
L’ensemble de nos travaux a montré que la fraction cellulaire CD34+/CD31- contient
un pool de cellules progénitrices présentant une potentialité adipocytaire et endothéliale. A
présent, une nouvelle condition de culture permet simultanément l’étude des capacités
adipogéniques et angiogéniques de ces cellules. Cependant, il reste à déterminer si cette
population cellulaire CD34+/CD31- est constituée d’un mélange de cellules progénitrices
unipotentes ou de cellules bi ou multipotentes. Notons que, la différenciation in vitro en
adipocyte n’atteint jamais le pourcentage total de cellules, même si elle est nettement
supérieure aux cellules exprimant des marqueurs endothéliaux tels que le CD31. Cette
observation semble suggérer que les cellules CD34+/CD31- constituent une population
cellulaire hétérogène. Une expérience d’analyse clonale pourrait permettre de déterminer
l’homogénéité ou l’hétérogénéité de cette fraction cellulaire. Cependant ce type d’analyse est
basé sur l’évaluation des capacités angiogéniques et adipogéniques de cellules dérivées après
prolifération d’une cellule initiale. Or, l’effet de la prolifération sur le devenir cellulaire est à
définir étant donné que jusqu'à présent nos études ont été réalisées sur des cellules en culture
primaire non passagées. Nos résultats préliminaires sur les cellules CD34+/CD31- passagées
80

montrent une diminution des capacités angiogéniques alors que leur capacité adipogénique
est maintenue (données personnelles).
La présence de cette population cellulaire CD34+/CD31- au sein du TA humain pose
la question de leur provenance et de leur mobilisation potentielle. En effet, ces cellules
peuvent être résidentes et capable d’autorenouvellement, et/ou elles peuvent être recrutées à
partir des cellules progénitrices circulantes issues de la moelle osseuse comme l’ont suggéré
les travaux de Hong et al. (101). Différents types de signaux tels que le SDF-1 (stromal
derived factor-1) mais également MMP-9 ont été montrés comme participant au recrutement
des progéniteurs CD34+ avec un potentiel hématopoïétique et/ou hépatique dans le foie des
souris NOD/SCID après un dommage hépatique (128). On peut donc poser l’hypothèse d’un
rôle potentiel de MMP-9 sur le recrutement de cellules progénitrices circulantes au sein du
TA.
Une mobilisation potentielle des cellules CD34+/CD31- du TA est-elle possible ? Ces
cellules sont-elles capables de migrer et de quitter le TA ? Le phénomène de mobilisation a
été très étudié pour les cellules souches hématopoïétiques et consiste en leur libération depuis
la moelle osseuse jusqu'à la circulation sanguine. Cette mobilisation est stimulée par de
nombreux facteurs tels que le SDF-1 et le G-CSF (granulocyte colony-stimulating factor). De
manière intéressante, ces facteurs entraînent la production de MMPs et notamment MMP-9
dans les cellules hématopoïétiques (50, 128). De plus, l’administration de ces composés à des
souris déficientes pour MMP-9 ne permet pas la mobilisation cellulaire (96). Les mécanismes
impliqués dans la migration des cellules souches vers la circulation sanguine passe par
l’augmentation des quantités solubles de kit ligand, une glycoprotéine transmembranaire,
induite par MMP-9 (96) qui permet non seulement la mobilisation des cellules progénitrices
dans la circulation mais aussi la prolifération et la migration des mastocytes dans le tissu
(97). Récemment notre groupe a montré que les cellules CD34+/CD31- migrent en réponse à
des milieux conditionnés par des cellules endothéliales capillaires CD34+/CD31+ du TA
humain. Cet effet est inhibé par la toxine pertussique (un inhibiteur de la protéine Gi) et par
un anticorps dirigé contre le CXCR4, le récepteur du SDF-1. De plus, nous avons mis en
évidence que les cellules endothéliales CD34+/CD31+ produisent et sécrètent du SDF-1 et
que les sécrétions des cellules endothéliales CD34+/CD31+ permettent l’expression des
capacités angiogéniques des cellules CD34+/CD31- (242). L’ensemble de ces résultats est
présenté en annexe dans la publication 3. Le rôle de MMP-9 dans la migration des cellules
CD34+/CD31- est à envisager d’autant plus que les expériences de migration des cellules
81

CD34+/CD31- présentés dans ce travail ont été réalisés sur un support de gélatine (données
personnelles).
Enfin, l’implication de MMP-9 in vivo reste à être définie. Afin de déterminer si les
effets de MMP-9 observés in vitro ont une signification physiologique ou physiopathologique
lors d’un développement ou d’une régression de TA, la caractérisation du TA et de ses
capacités d’extension dans un modèle original de souris invalidées pour le gène de MMP-9
(souris KO MMP-9) sont à envisager. Nos premiers résultats, obtenus sur les souris KO
MMP-9, sous tendent l’implication de cette métalloprotéase dans le développement du TA.
En effet, en conditions nutritionnelles standard, l’absence de MMP-9 est associée à une
diminution de 37% du TA sous cutané et de 27% du TA intra-abdominal (n=6), sans
variation de la masse corporelle totale par rapport aux souris contrôles. De plus, la quantité de
triglycérides stockés dans ces tissus est deux fois plus faible chez les souris KO MMP-9.
Suite à ces résultats, il nous apparaît nécessaire de mieux caractériser ce modèle. Pour cela,
dans un premier temps nous envisageons l’analyse du TA des souris KO MMP-9 aussi bien
dans sa structure que dans sa composition cellulaire et matricielle. En effet, il est important
de définir si la différence de masse adipeuse entre les souris KO MMP-9 et les souris
contrôles est due à une diminution du nombre et/ou de la taille des adipocytes matures. Des
éventuelles modifications de la vasculature, de la présence de macrophages et de la MEC
dans les TA des souris KO MMP-9 et contrôles seront recherchées. De la même manière, il
serait intéressant de définir la composition cellulaire de la FSV isolée des souris KO MMP-9
et contrôles. Enfin, l’isolement des différentes populations cellulaires de la FSV issues des
souris KO MMP-9 et contrôles, par l’utilisation de nanoparticules magnétiques couplées à
des anticorps d’intérêt ainsi que l’évaluation des capacités adipogéniques et angiogéniques in
vitro sur les cellules progénitrices ainsi obtenues pourraient être envisagé. Dans un second
temps, une cinétique de développement et/ou de régression du TA des souris KO MMP-9
pourrait être envisagée. L’étude des différents types cellulaires des TA et des composants
matriciels de ces souris devenues obèses ou maigres pourra être réalisée, comme
précédemment décrit .
Pour conclure, ce travail démontre l’importance des MMPs dans le processus de
différenciation adipocytaire des cellules progénitrices du TA humain. Les MMPs pourraient
ainsi contribuer au développement de la masse grasse. Bien que leur rôle exact reste à
déterminer les MMPs pourraient devenir de nouvelles cibles d’intérêt dans le développement
de traitements efficaces pour l’obésité et la lipoatrophie.
82

Références bibliographiques 1. Adams, J. 2004. The development of proteasome inhibitors as anticancer drugs.
Cancer Cell 5:417-21. 2. Aggeler, J., S. M. Frisch, and Z. Werb. 1984. Changes in cell shape correlate with
collagenase gene expression in rabbit synovial fibroblasts. J Cell Biol 98:1662-71. 3. Alexander, C. M., S. Selvarajan, J. Mudgett, and Z. Werb. 2001. Stromelysin-1
regulates adipogenesis during mammary gland involution. J Cell Biol 152:693-703. 4. Andre, P., M. Groettrup, P. Klenerman, R. de Giuli, B. L. Booth, Jr., V.
Cerundolo, M. Bonneville, F. Jotereau, R. M. Zinkernagel, and V. Lotteau. 1998. An inhibitor of HIV-1 protease modulates proteasome activity, antigen presentation, and T cell responses. Proc Natl Acad Sci U S A 95:13120-4.
5. Aoki, S., S. Toda, T. Sakemi, and H. Sugihara. 2003. Coculture of endothelial cells and mature adipocytes actively promotes immature preadipocyte development in vitro. Cell Struct Funct 28:55-60.
6. Ariyama, Y., H. Shimizu, T. Satoh, T. Tsuchiya, S. Okada, S. Oyadomari, and M. Mori. 2007. Chop-deficient mice showed increased adiposity but no glucose intolerance. Obesity (Silver Spring) 15:1647-56.
7. Avram, M. M., A. S. Avram, and W. D. James. 2007. Subcutaneous fat in normal and diseased states 3. Adipogenesis: from stem cell to fat cell. J Am Acad Dermatol 56:472-92.
8. Baker, A. H., D. R. Edwards, and G. Murphy. 2002. Metalloproteinase inhibitors: biological actions and therapeutic opportunities. J Cell Sci 115:3719-27.
9. Balogh, S., C. C. Naus, and P. A. Merrifield. 1993. Expression of gap junctions in cultured rat L6 cells during myogenesis. Dev Biol 155:351-60.
10. Bastard, J. P., M. Caron, H. Vidal, V. Jan, M. Auclair, C. Vigouroux, J. Luboinski, M. Laville, M. Maachi, P. M. Girard, W. Rozenbaum, P. Levan, and J. Capeau. 2002. Association between altered expression of adipogenic factor SREBP1 in lipoatrophic adipose tissue from HIV-1-infected patients and abnormal adipocyte differentiation and insulin resistance. Lancet 359:1026-31.
11. Beigneux, A. P., B. S. Davies, P. Gin, M. M. Weinstein, E. Farber, X. Qiao, F. Peale, S. Bunting, R. L. Walzem, J. S. Wong, W. S. Blaner, Z. M. Ding, K. Melford, N. Wongsiriroj, X. Shu, F. de Sauvage, R. O. Ryan, L. G. Fong, A. Bensadoun, and S. G. Young. 2007. Glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1 plays a critical role in the lipolytic processing of chylomicrons. Cell Metab 5:279-91.
12. Ben-Romano, R., A. Rudich, S. Etzion, R. Potashnik, E. Kagan, U. Greenbaum, and N. Bashan. 2006. Nelfinavir induces adipocyte insulin resistance through the induction of oxidative stress: differential protective effect of antioxidant agents. Antivir Ther 11:1051-60.
13. Berlan, M., and M. Lafontan. 1985. Evidence that epinephrine acts preferentially as an antilipolytic agent in abdominal human subcutaneous fat cells: assessment by analysis of beta and alpha 2 adrenoceptor properties. Eur J Clin Invest 15:341-8.
14. Bi, Y., C. H. Stuelten, T. Kilts, S. Wadhwa, R. V. Iozzo, P. G. Robey, X. D. Chen, and M. F. Young. 2005. Extracellular matrix proteoglycans control the fate of bone marrow stromal cells. J Biol Chem 280:30481-9.
15. Bjorntorp, P., A. Gustafson, and B. Persson. 1971. Adipose tissue fat cell size and number in relation to metabolism in endogenous hypertriglyceridemia. Acta Med Scand 190:363-7.
83

16. Bouloumie, A., H. C. Drexler, M. Lafontan, and R. Busse. 1998. Leptin, the product of Ob gene, promotes angiogenesis. Circ Res 83:1059-66.
17. Bouloumie, A., C. Sengenes, G. Portolan, J. Galitzky, and M. Lafontan. 2001. Adipocyte produces matrix metalloproteinases 2 and 9: involvement in adipose differentiation. Diabetes 50:2080-6.
18. Braissant, O., F. Foufelle, C. Scotto, M. Dauca, and W. Wahli. 1996. Differential expression of peroxisome proliferator-activated receptors (PPARs): tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology 137:354-66.
19. Brakenhielm, E., R. Cao, B. Gao, B. Angelin, B. Cannon, P. Parini, and Y. Cao. 2004. Angiogenesis inhibitor, TNP-470, prevents diet-induced and genetic obesity in mice. Circ Res 94:1579-88.
20. Brinkman, K., J. A. Smeitink, J. A. Romijn, and P. Reiss. 1999. Mitochondrial toxicity induced by nucleoside-analogue reverse-transcriptase inhibitors is a key factor in the pathogenesis of antiretroviral-therapy-related lipodystrophy. Lancet 354:1112-5.
21. Brown, M. S., and J. L. Goldstein. 1999. A proteolytic pathway that controls the cholesterol content of membranes, cells, and blood. Proc Natl Acad Sci U S A 96:11041-8.
22. Brown, M. S., and J. L. Goldstein. 1997. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 89:331-40.
23. Brun, R. P., P. Tontonoz, B. M. Forman, R. Ellis, J. Chen, R. M. Evans, and B. M. Spiegelman. 1996. Differential activation of adipogenesis by multiple PPAR isoforms. Genes Dev 10:974-84.
24. Cancello, R., C. Henegar, N. Viguerie, S. Taleb, C. Poitou, C. Rouault, M. Coupaye, V. Pelloux, D. Hugol, J. L. Bouillot, A. Bouloumie, G. Barbatelli, S. Cinti, P. A. Svensson, G. S. Barsh, J. D. Zucker, A. Basdevant, D. Langin, and K. Clement. 2005. Reduction of macrophage infiltration and chemoattractant gene expression changes in white adipose tissue of morbidly obese subjects after surgery-induced weight loss. Diabetes 54:2277-86.
25. Capeau, J., J. Magre, O. Lascols, M. Caron, V. Bereziat, C. Vigouroux, and J. P. Bastard. 2005. Diseases of adipose tissue: genetic and acquired lipodystrophies. Biochem Soc Trans 33:1073-7.
26. Carmeliet, P., L. Moons, R. Lijnen, M. Baes, V. Lemaitre, P. Tipping, A. Drew, Y. Eeckhout, S. Shapiro, F. Lupu, and D. Collen. 1997. Urokinase-generated plasmin activates matrix metalloproteinases during aneurysm formation. Nat Genet 17:439-44.
27. Caron, M., M. Auclair, B. Donadille, V. Bereziat, B. Guerci, M. Laville, H. Narbonne, C. Bodemer, O. Lascols, J. Capeau, and C. Vigouroux. 2007. Human lipodystrophies linked to mutations in A-type lamins and to HIV protease inhibitor therapy are both associated with prelamin A accumulation, oxidative stress and premature cellular senescence. Cell Death Differ.
28. Caron, M., M. Auclair, C. Lagathu, A. Lombes, U. A. Walker, M. Kornprobst, and J. Capeau. 2004. The HIV-1 nucleoside reverse transcriptase inhibitors stavudine and zidovudine alter adipocyte functions in vitro. Aids 18:2127-36.
29. Caron, M., M. Auclair, H. Sterlingot, M. Kornprobst, and J. Capeau. 2003. Some HIV protease inhibitors alter lamin A/C maturation and stability, SREBP-1 nuclear localization and adipocyte differentiation. Aids 17:2437-44.
30. Caron, M., M. Auclair, C. Vigouroux, M. Glorian, C. Forest, and J. Capeau. 2001. The HIV protease inhibitor indinavir impairs sterol regulatory element-binding
84

protein-1 intranuclear localization, inhibits preadipocyte differentiation, and induces insulin resistance. Diabetes 50:1378-88.
31. Carr, A. 2000. HIV protease inhibitor-related lipodystrophy syndrome. Clin Infect Dis 30 Suppl 2:S135-42.
32. Carr, A., J. Miller, M. Law, and D. A. Cooper. 2000. A syndrome of lipoatrophy, lactic acidaemia and liver dysfunction associated with HIV nucleoside analogue therapy: contribution to protease inhibitor-related lipodystrophy syndrome. Aids 14:F25-32.
33. Carr, A., K. Samaras, S. Burton, M. Law, J. Freund, D. J. Chisholm, and D. A. Cooper. 1998. A syndrome of peripheral lipodystrophy, hyperlipidaemia and insulin resistance in patients receiving HIV protease inhibitors. Aids 12:F51-8.
34. Carr, A., K. Samaras, D. J. Chisholm, and D. A. Cooper. 1998. Pathogenesis of HIV-1-protease inhibitor-associated peripheral lipodystrophy, hyperlipidaemia, and insulin resistance. Lancet 351:1881-3.
35. Carriere, A., M. C. Carmona, Y. Fernandez, M. Rigoulet, R. H. Wenger, L. Penicaud, and L. Casteilla. 2004. Mitochondrial reactive oxygen species control the transcription factor CHOP-10/GADD153 and adipocyte differentiation: a mechanism for hypoxia-dependent effect. J Biol Chem 279:40462-9.
36. Carriere, A., Y. Fernandez, M. Rigoulet, L. Penicaud, and L. Casteilla. 2003. Inhibition of preadipocyte proliferation by mitochondrial reactive oxygen species. FEBS Lett 550:163-7.
37. Chakraborti, S., M. Mandal, S. Das, A. Mandal, and T. Chakraborti. 2003. Regulation of matrix metalloproteinases: an overview. Mol Cell Biochem 253:269-85.
38. Chavey, C., B. Mari, M. N. Monthouel, S. Bonnafous, P. Anglard, E. Van Obberghen, and S. Tartare-Deckert. 2003. Matrix metalloproteinases are differentially expressed in adipose tissue during obesity and modulate adipocyte differentiation. J Biol Chem 278:11888-96.
39. Chen, Z., J. I. Torrens, A. Anand, B. M. Spiegelman, and J. M. Friedman. 2005. Krox20 stimulates adipogenesis via C/EBPbeta-dependent and -independent mechanisms. Cell Metab 1:93-106.
40. Chi, D., J. Henry, J. Kelley, R. Thorpe, J. K. Smith, and G. Krishnaswamy. 2000. The effects of HIV infection on endothelial function. Endothelium 7:223-42.
41. Chirco, R., X. W. Liu, K. K. Jung, and H. R. Kim. 2006. Novel functions of TIMPs in cell signaling. Cancer Metastasis Rev 25:99-113.
42. Cho, C. H., Y. J. Koh, J. Han, H. K. Sung, H. Jong Lee, T. Morisada, R. A. Schwendener, R. A. Brekken, G. Kang, Y. Oike, T. S. Choi, T. Suda, O. J. Yoo, and G. Y. Koh. 2007. Angiogenic role of LYVE-1-positive macrophages in adipose tissue. Circ Res 100:e47-57.
43. Christeff, N., J. C. Melchior, P. de Truchis, C. Perronne, and M. L. Gougeon. 2002. Increased serum interferon alpha in HIV-1 associated lipodystrophy syndrome. Eur J Clin Invest 32:43-50.
44. Chun, T. H., K. B. Hotary, F. Sabeh, A. R. Saltiel, E. D. Allen, and S. J. Weiss. 2006. A pericellular collagenase directs the 3-dimensional development of white adipose tissue. Cell 125:577-91.
45. Cinti, S., G. Mitchell, G. Barbatelli, I. Murano, E. Ceresi, E. Faloia, S. Wang, M. Fortier, A. S. Greenberg, and M. S. Obin. 2005. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J Lipid Res 46:2347-55.
85

46. Clement, K., N. Viguerie, C. Poitou, C. Carette, V. Pelloux, C. A. Curat, A. Sicard, S. Rome, A. Benis, J. D. Zucker, H. Vidal, M. Laville, G. S. Barsh, A. Basdevant, V. Stich, R. Cancello, and D. Langin. 2004. Weight loss regulates inflammation-related genes in white adipose tissue of obese subjects. Faseb J 18:1657-69.
47. Coffinier, C., S. E. Hudon, E. A. Farber, S. Y. Chang, C. A. Hrycyna, S. G. Young, and L. G. Fong. 2007. From the Cover: HIV protease inhibitors block the zinc metalloproteinase ZMPSTE24 and lead to an accumulation of prelamin A in cells. Proc Natl Acad Sci U S A 104:13432-7.
48. consultation, W. e. 2004. Appropriate body-mass index for Asian populations and its implications for policy and intervention strategies. The Lancet:157-163.
49. Cook, K. S., D. L. Groves, H. Y. Min, and B. M. Spiegelman. 1985. A developmentally regulated mRNA from 3T3 adipocytes encodes a novel serine protease homologue. Proc Natl Acad Sci U S A 82:6480-4.
50. Cottler-Fox, M. H., T. Lapidot, I. Petit, O. Kollet, J. F. DiPersio, D. Link, and S. Devine. 2003. Stem cell mobilization. Hematology Am Soc Hematol Educ Program:419-37.
51. Coux, O., K. Tanaka, and A. L. Goldberg. 1996. Structure and functions of the 20S and 26S proteasomes. Annu Rev Biochem 65:801-47.
52. Crandall, D. L., G. J. Hausman, and J. G. Kral. 1997. A review of the microcirculation of adipose tissue: anatomic, metabolic, and angiogenic perspectives. Microcirculation 4:211-32.
53. Croissandeau, G., M. Chretien, and M. Mbikay. 2002. Involvement of matrix metalloproteinases in the adipose conversion of 3T3-L1 preadipocytes. Biochem J 364:739-46.
54. Crossno, J. T., Jr., S. M. Majka, T. Grazia, R. G. Gill, and D. J. Klemm. 2006. Rosiglitazone promotes development of a novel adipocyte population from bone marrow-derived circulating progenitor cells. J Clin Invest 116:3220-8.
55. Curat, C. A., A. Miranville, C. Sengenes, M. Diehl, C. Tonus, R. Busse, and A. Bouloumie. 2004. From blood monocytes to adipose tissue-resident macrophages: induction of diapedesis by human mature adipocytes. Diabetes 53:1285-92.
56. Curat, C. A., V. Wegner, C. Sengenes, A. Miranville, C. Tonus, R. Busse, and A. Bouloumie. 2006. Macrophages in human visceral adipose tissue: increased accumulation in obesity and a source of resistin and visfatin. Diabetologia 49:744-7.
57. Dang, D., Y. Yang, X. Li, A. Atakilit, J. Regezi, D. Eisele, D. Ellis, and D. M. Ramos. 2004. Matrix metalloproteinases and TGFbeta1 modulate oral tumor cell matrix. Biochem Biophys Res Commun 316:937-42.
58. Dani, C., A. G. Smith, S. Dessolin, P. Leroy, L. Staccini, P. Villageois, C. Darimont, and G. Ailhaud. 1997. Differentiation of embryonic stem cells into adipocytes in vitro. J Cell Sci 110 ( Pt 11):1279-85.
59. Demeulemeester, D., I. Scroyen, G. Voros, J. Snoeys, B. De Geest, D. Collen, and H. R. Lijnen. 2006. Overexpression of tissue inhibitor of matrix metalloproteinases-1 (TIMP-1) in mice does not affect adipogenesis or adipose tissue development. Thromb Haemost 95:1019-24.
60. Deryugina, E. I., and J. P. Quigley. 2006. Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev 25:9-34.
61. Di Simone, N., M. De Santis, E. Tamburrini, F. Di Nicuolo, M. B. Lucia, P. Riccardi, S. D'Ippolito, R. Cauda, and A. Caruso. 2007. Effects of antiretroviral therapy on tube-like network formation of human endothelial cells. Biol Pharm Bull 30:982-4.
86

62. Domingo, P., X. Matias-Guiu, R. M. Pujol, E. Francia, E. Lagarda, M. A. Sambeat, and G. Vazquez. 1999. Subcutaneous adipocyte apoptosis in HIV-1 protease inhibitor-associated lipodystrophy. Aids 13:2261-7.
63. Domingo, P., F. Vidal, J. C. Domingo, S. Veloso, M. A. Sambeat, F. Torres, J. J. Sirvent, J. Vendrell, X. Matias-Guiu, and C. Richart. 2005. Tumour necrosis factor alpha in fat redistribution syndromes associated with combination antiretroviral therapy in HIV-1-infected patients: potential role in subcutaneous adipocyte apoptosis. Eur J Clin Invest 35:771-80.
64. Dowell, P., C. Flexner, P. O. Kwiterovich, and M. D. Lane. 2000. Suppression of preadipocyte differentiation and promotion of adipocyte death by HIV protease inhibitors. J Biol Chem 275:41325-32.
65. Eberle, D., B. Hegarty, P. Bossard, P. Ferre, and F. Foufelle. 2004. SREBP transcription factors: master regulators of lipid homeostasis. Biochimie 86:839-48.
66. Edens, N. K., S. K. Fried, J. G. Kral, J. Hirsch, and R. L. Leibel. 1993. In vitro lipid synthesis in human adipose tissue from three abdominal sites. Am J Physiol 265:E374-9.
67. Eeckhout, Y., and G. Vaes. 1977. Further studies on the activation of procollagenase, the latent precursor of bone collagenase. Effects of lysosomal cathepsin B, plasmin and kallikrein, and spontaneous activation. Biochem J 166:21-31.
68. El Hadri, K., M. Glorian, C. Monsempes, M. N. Dieudonne, R. Pecquery, Y. Giudicelli, M. Andreani, I. Dugail, and B. Feve. 2004. In vitro suppression of the lipogenic pathway by the nonnucleoside reverse transcriptase inhibitor efavirenz in 3T3 and human preadipocytes or adipocytes. J Biol Chem 279:15130-41.
69. Engler, A. J., S. Sen, H. L. Sweeney, and D. E. Discher. 2006. Matrix elasticity directs stem cell lineage specification. Cell 126:677-89.
70. Enoksson, S., M. Talbot, F. Rife, W. V. Tamborlane, R. S. Sherwin, and S. Caprio. 2000. Impaired in vivo stimulation of lipolysis in adipose tissue by selective beta2-adrenergic agonist in obese adolescent girls. Diabetes 49:2149-53.
71. Even-Ram, S., V. Artym, and K. M. Yamada. 2006. Matrix control of stem cell fate. Cell 126:645-7.
72. Fain, J. N., A. K. Madan, M. L. Hiler, P. Cheema, and S. W. Bahouth. 2004. Comparison of the release of adipokines by adipose tissue, adipose tissue matrix, and adipocytes from visceral and subcutaneous abdominal adipose tissues of obese humans. Endocrinology 145:2273-82.
73. Fajas, L., K. Schoonjans, L. Gelman, J. B. Kim, J. Najib, G. Martin, J. C. Fruchart, M. Briggs, B. M. Spiegelman, and J. Auwerx. 1999. Regulation of peroxisome proliferator-activated receptor gamma expression by adipocyte differentiation and determination factor 1/sterol regulatory element binding protein 1: implications for adipocyte differentiation and metabolism. Mol Cell Biol 19:5495-503.
74. Farmer, S. R. 2006. Transcriptional control of adipocyte formation. Cell Metab 4:263-73.
75. Farnier, C., S. Krief, M. Blache, F. Diot-Dupuy, G. Mory, P. Ferre, and R. Bazin. 2003. Adipocyte functions are modulated by cell size change: potential involvement of an integrin/ERK signalling pathway. Int J Obes Relat Metab Disord 27:1178-86.
76. Fata, J. E., K. J. Leco, E. B. Voura, H. Y. Yu, P. Waterhouse, G. Murphy, R. A. Moorehead, and R. Khokha. 2001. Accelerated apoptosis in the Timp-3-deficient mammary gland. J Clin Invest 108:831-41.
87

77. Ferreras, M., U. Felbor, T. Lenhard, B. R. Olsen, and J. Delaisse. 2000. Generation and degradation of human endostatin proteins by various proteinases. FEBS Lett 486:247-51.
78. Feve, B. 2005. Adipogenesis: cellular and molecular aspects. Best Pract Res Clin Endocrinol Metab 19:483-99.
79. Fève, B. 2003. Physiologie et différenciation du tissu adipeux. Médecine clinique endocrinologie et diabète 8:34-43.
80. Fowlkes, J. L., K. M. Thrailkill, D. M. Serra, K. Suzuki, and H. Nagase. 1995. Matrix metalloproteinases as insulin-like growth factor binding protein-degrading proteinases. Prog Growth Factor Res 6:255-63.
81. Frayn, K. N., F. Karpe, B. A. Fielding, I. A. Macdonald, and S. W. Coppack. 2003. Integrative physiology of human adipose tissue. Int J Obes Relat Metab Disord 27:875-88.
82. Fukai, F., M. Ohtaki, N. Fujii, H. Yajima, T. Ishii, Y. Nishizawa, K. Miyazaki, and T. Katayama. 1995. Release of biological activities from quiescent fibronectin by a conformational change and limited proteolysis by matrix metalloproteinases. Biochemistry 34:11453-9.
83. Fukumura, D., A. Ushiyama, D. G. Duda, L. Xu, J. Tam, V. Krishna, K. Chatterjee, I. Garkavtsev, and R. K. Jain. 2003. Paracrine regulation of angiogenesis and adipocyte differentiation during in vivo adipogenesis. Circ Res 93:e88-97.
84. Gaedicke, S., E. Firat-Geier, O. Constantiniu, M. Lucchiari-Hartz, M. Freudenberg, C. Galanos, and G. Niedermann. 2002. Antitumor effect of the human immunodeficiency virus protease inhibitor ritonavir: induction of tumor-cell apoptosis associated with perturbation of proteasomal proteolysis. Cancer Res 62:6901-8.
85. Galis, Z. S., and J. J. Khatri. 2002. Matrix metalloproteinases in vascular remodeling and atherogenesis: the good, the bad, and the ugly. Circ Res 90:251-62.
86. Ghanim, H., A. Aljada, D. Hofmeyer, T. Syed, P. Mohanty, and P. Dandona. 2004. Circulating mononuclear cells in the obese are in a proinflammatory state. Circulation 110:1564-71.
87. Goldberg, I. J. 1996. Lipoprotein lipase and lipolysis: central roles in lipoprotein metabolism and atherogenesis. J Lipid Res 37:693-707.
88. Golubkov, V. S., S. Boyd, A. Y. Savinov, A. V. Chekanov, A. L. Osterman, A. Remacle, D. V. Rozanov, S. J. Doxsey, and A. Y. Strongin. 2005. Membrane type-1 matrix metalloproteinase (MT1-MMP) exhibits an important intracellular cleavage function and causes chromosome instability. J Biol Chem 280:25079-86.
89. Gougeon, M. L., L. Penicaud, B. Fromenty, P. Leclercq, J. P. Viard, and J. Capeau. 2004. Adipocytes targets and actors in the pathogenesis of HIV-associated lipodystrophy and metabolic alterations. Antivir Ther 9:161-77.
90. Green, H., and O. Kehinde. 1979. Formation of normally differentiated subcutaneous fat pads by an established preadipose cell line. J Cell Physiol 101:169-71.
91. Gregoire, F. M., C. M. Smas, and H. S. Sul. 1998. Understanding adipocyte differentiation. Physiol Rev 78:783-809.
92. Grigem, S., P. Fischer-Posovszky, K. M. Debatin, E. Loizon, H. Vidal, and M. Wabitsch. 2005. The effect of the HIV protease inhibitor ritonavir on proliferation, differentiation, lipogenesis, gene expression and apoptosis of human preadipocytes and adipocytes. Horm Metab Res 37:602-9.
88

93. Hadigan, C., S. Borgonha, J. Rabe, V. Young, and S. Grinspoon. 2002. Increased rates of lipolysis among human immunodeficiency virus-infected men receiving highly active antiretroviral therapy. Metabolism 51:1143-7.
94. Hausberger, F. X., and M. M. Widelitz. 1963. Distribution of labeled erythrocytes in adipose tissue and muscle in the rat. Am J Physiol 204:649-52.
95. Hausman, G. J., J. T. Wright, and R. L. Richardson. 1996. The influence of extracellular matrix substrata on preadipocyte development in serum-free cultures of stromal-vascular cells. J Anim Sci 74:2117-28.
96. Heissig, B., K. Hattori, S. Dias, M. Friedrich, B. Ferris, N. R. Hackett, R. G. Crystal, P. Besmer, D. Lyden, M. A. Moore, Z. Werb, and S. Rafii. 2002. Recruitment of stem and progenitor cells from the bone marrow niche requires MMP-9 mediated release of kit-ligand. Cell 109:625-37.
97. Heissig, B., S. Rafii, H. Akiyama, Y. Ohki, Y. Sato, T. Rafael, Z. Zhu, D. J. Hicklin, K. Okumura, H. Ogawa, Z. Werb, and K. Hattori. 2005. Low-dose irradiation promotes tissue revascularization through VEGF release from mast cells and MMP-9-mediated progenitor cell mobilization. J Exp Med 202:739-50.
98. Hemmrich, K., D. von Heimburg, K. Cierpka, S. Haydarlioglu, and N. Pallua. 2005. Optimization of the differentiation of human preadipocytes in vitro. Differentiation 73:28-35.
99. Hertel, J., H. Struthers, C. B. Horj, and P. W. Hruz. 2004. A structural basis for the acute effects of HIV protease inhibitors on GLUT4 intrinsic activity. J Biol Chem 279:55147-52.
100. Holm, C., T. Osterlund, H. Laurell, and J. A. Contreras. 2000. Molecular mechanisms regulating hormone-sensitive lipase and lipolysis. Annu Rev Nutr 20:365-93.
101. Hong, K. M., M. D. Burdick, R. J. Phillips, D. Heber, and R. M. Strieter. 2005. Characterization of human fibrocytes as circulating adipocyte progenitors and the formation of human adipose tissue in SCID mice. Faseb J 19:2029-31.
102. Horowitz, J. F. 2001. Regulation of lipid mobilization and oxidation during exercise in obesity. Exerc Sport Sci Rev 29:42-6.
103. Horton, J. D., J. L. Goldstein, and M. S. Brown. 2002. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest 109:1125-31.
104. Huber, J., M. Loffler, M. Bilban, M. Reimers, A. Kadl, J. Todoric, M. Zeyda, R. Geyeregger, M. Schreiner, T. Weichhart, N. Leitinger, W. Waldhausl, and T. M. Stulnig. 2007. Prevention of high-fat diet-induced adipose tissue remodeling in obese diabetic mice by n-3 polyunsaturated fatty acids. Int J Obes (Lond) 31:1004-13.
105. Hudgins, L. C., A. Baday, M. K. Hellerstein, T. S. Parker, D. M. Levine, C. E. Seidman, R. A. Neese, J. D. Tremaroli, and J. Hirsch. 2007. The effect of dietary carbohydrate on genes for fatty acid synthase and inflammatory cytokines in adipose tissues from lean and obese subjects. J Nutr Biochem.
106. Huhtala, P., M. J. Humphries, J. B. McCarthy, P. M. Tremble, Z. Werb, and C. H. Damsky. 1995. Cooperative signaling by alpha 5 beta 1 and alpha 4 beta 1 integrins regulates metalloproteinase gene expression in fibroblasts adhering to fibronectin. J Cell Biol 129:867-79.
107. IG, G., and S. MA. 1945. Blood vessels in fat tissue: relation to problems of gas exchange. J exp Med 81:219-232.
108. Imai, K., A. Hiramatsu, D. Fukushima, M. D. Pierschbacher, and Y. Okada. 1997. Degradation of decorin by matrix metalloproteinases: identification of the
89

cleavage sites, kinetic analyses and transforming growth factor-beta1 release. Biochem J 322 ( Pt 3):809-14.
109. Jain, R. G., and J. M. Lenhard. 2002. Select HIV protease inhibitors alter bone and fat metabolism ex vivo. J Biol Chem 277:19247-50.
110. Jan, V., P. Cervera, M. Maachi, M. Baudrimont, M. Kim, H. Vidal, P. M. Girard, P. Levan, W. Rozenbaum, A. Lombes, J. Capeau, and J. P. Bastard. 2004. Altered fat differentiation and adipocytokine expression are inter-related and linked to morphological changes and insulin resistance in HIV-1-infected lipodystrophic patients. Antivir Ther 9:555-64.
111. Janneh, O., P. G. Hoggard, J. F. Tjia, S. P. Jones, S. H. Khoo, B. Maher, D. J. Back, and M. Pirmohamed. 2003. Intracellular disposition and metabolic effects of zidovudine, stavudine and four protease inhibitors in cultured adipocytes. Antivir Ther 8:417-26.
112. Javazon, E. H., K. J. Beggs, and A. W. Flake. 2004. Mesenchymal stem cells: paradoxes of passaging. Exp Hematol 32:414-25.
113. Jenkins, C. M., D. J. Mancuso, W. Yan, H. F. Sims, B. Gibson, and R. W. Gross. 2004. Identification, cloning, expression, and purification of three novel human calcium-independent phospholipase A2 family members possessing triacylglycerol lipase and acylglycerol transacylase activities. J Biol Chem 279:48968-75.
114. Jiang, B., V. Y. Hebert, Y. Li, J. M. Mathis, J. S. Alexander, and T. R. Dugas. 2007. HIV antiretroviral drug combination induces endothelial mitochondrial dysfunction and reactive oxygen species production, but not apoptosis. Toxicol Appl Pharmacol.
115. Johnson, J. A., J. B. Albu, E. S. Engelson, S. K. Fried, Y. Inada, G. Ionescu, and D. P. Kotler. 2004. Increased systemic and adipose tissue cytokines in patients with HIV-associated lipodystrophy. Am J Physiol Endocrinol Metab 286:E261-71.
116. Johnson, M. D., H. R. Kim, L. Chesler, G. Tsao-Wu, N. Bouck, and P. J. Polverini. 1994. Inhibition of angiogenesis by tissue inhibitor of metalloproteinase. J Cell Physiol 160:194-202.
117. Jones, K., P. G. Hoggard, S. D. Sales, S. Khoo, R. Davey, and D. J. Back. 2001. Differences in the intracellular accumulation of HIV protease inhibitors in vitro and the effect of active transport. Aids 15:675-81.
118. Jones, S. P., O. Janneh, D. J. Back, and M. Pirmohamed. 2005. Altered adipokine response in murine 3T3-F442A adipocytes treated with protease inhibitors and nucleoside reverse transcriptase inhibitors. Antivir Ther 10:207-13.
119. Kamiya, S., R. Kato, M. Wakabayashi, T. Tohyama, I. Enami, M. Ueki, H. Yajima, T. Ishii, H. Nakamura, T. Katayama, J. Takagi, and F. Fukai. 2002. Fibronectin peptides derived from two distinct regions stimulate adipocyte differentiation by preventing fibronectin matrix assembly. Biochemistry 41:3270-7.
120. Kersten, S. 2001. Mechanisms of nutritional and hormonal regulation of lipogenesis. EMBO Rep 2:282-6.
121. Kim, H. K., M. Della-Fera, J. Lin, and C. A. Baile. 2006. Docosahexaenoic acid inhibits adipocyte differentiation and induces apoptosis in 3T3-L1 preadipocytes. J Nutr 136:2965-9.
122. Kim, J. B., P. Sarraf, M. Wright, K. M. Yao, E. Mueller, G. Solanes, B. B. Lowell, and B. M. Spiegelman. 1998. Nutritional and insulin regulation of fatty acid synthetase and leptin gene expression through ADD1/SREBP1. J Clin Invest 101:1-9.
123. Kim, R. J., C. G. Wilson, M. Wabitsch, M. A. Lazar, and C. M. Steppan. 2006. HIV protease inhibitor-specific alterations in human adipocyte differentiation and metabolism. Obesity (Silver Spring) 14:994-1002.
90

124. Kim, S. S., S. Rhee, K. H. Lee, J. H. Kim, H. S. Kim, M. S. Kang, and C. H. Chung. 1998. Inhibitors of the proteasome block the myogenic differentiation of rat L6 myoblasts. FEBS Lett 433:47-50.
125. Kisselev, A. F., and A. L. Goldberg. 2001. Proteasome inhibitors: from research tools to drug candidates. Chem Biol 8:739-58.
126. Knutson, V. P. 2000. The release of lipoprotein lipase from 3T3-L1 adipocytes is regulated by microvessel endothelial cells in an insulin-dependent manner. Endocrinology 141:693-701.
127. Kolf, C. M., E. Cho, and R. S. Tuan. 2007. Mesenchymal stromal cells. Biology of adult mesenchymal stem cells: regulation of niche, self-renewal and differentiation. Arthritis Res Ther 9:204.
128. Kollet, O., S. Shivtiel, Y. Q. Chen, J. Suriawinata, S. N. Thung, M. D. Dabeva, J. Kahn, A. Spiegel, A. Dar, S. Samira, P. Goichberg, A. Kalinkovich, F. Arenzana-Seisdedos, A. Nagler, I. Hardan, M. Revel, D. A. Shafritz, and T. Lapidot. 2003. HGF, SDF-1, and MMP-9 are involved in stress-induced human CD34+ stem cell recruitment to the liver. J Clin Invest 112:160-9.
129. Kolonin, M. G., P. K. Saha, L. Chan, R. Pasqualini, and W. Arap. 2004. Reversal of obesity by targeted ablation of adipose tissue. Nat Med 10:625-32.
130. Kovsan, J., R. Ben-Romano, S. C. Souza, A. S. Greenberg, and A. Rudich. 2007. Regulation of adipocyte lipolysis by degradation of the perilipin protein: nelfinavir enhances lysosome-mediated perilipin proteolysis. J Biol Chem 282:21704-11.
131. Kralisch, S., M. Bluher, A. Tonjes, U. Lossner, R. Paschke, M. Stumvoll, and M. Fasshauer. 2007. Tissue inhibitor of metalloproteinase-1 predicts adiposity in humans. Eur J Endocrinol 156:257-61.
132. Kubo, Y., S. Kaidzu, I. Nakajima, K. Takenouchi, and F. Nakamura. 2000. Organization of extracellular matrix components during differentiation of adipocytes in long-term culture. In Vitro Cell Dev Biol Anim 36:38-44.
133. Kuri-Harcuch, W., C. Arguello, and M. Marsch-Moreno. 1984. Extracellular matrix production by mouse 3T3-F442A cells during adipose differentiation in culture. Differentiation 28:173-8.
134. Kwan, J. A., C. J. Schulze, W. Wang, H. Leon, M. Sariahmetoglu, M. Sung, J. Sawicka, D. E. Sims, G. Sawicki, and R. Schulz. 2004. Matrix metalloproteinase-2 (MMP-2) is present in the nucleus of cardiac myocytes and is capable of cleaving poly (ADP-ribose) polymerase (PARP) in vitro. Faseb J 18:690-2.
135. Lafontan, M. 2005. Fat cells: afferent and efferent messages define new approaches to treat obesity. Annu Rev Pharmacol Toxicol 45:119-46.
136. Lafontan, M., and M. Berlan. 2003. Do regional differences in adipocyte biology provide new pathophysiological insights? Trends Pharmacol Sci 24:276-83.
137. Lagathu, C., J. P. Bastard, M. Auclair, M. Maachi, M. Kornprobst, J. Capeau, and M. Caron. 2004. Antiretroviral drugs with adverse effects on adipocyte lipid metabolism and survival alter the expression and secretion of proinflammatory cytokines and adiponectin in vitro. Antivir Ther 9:911-20.
138. Lagathu, C., B. Eustace, M. Prot, D. Frantz, Y. Gu, J. P. Bastard, M. Maachi, S. Azoulay, M. Briggs, M. Caron, and J. Capeau. 2007. Some HIV antiretrovirals increase oxidative stress and alter chemokine, cytokine or adiponectin production in human adipocytes and macrophages. Antivir Ther 12:489-500.
139. Lagathu, C., M. Kim, M. Maachi, C. Vigouroux, P. Cervera, J. Capeau, M. Caron, and J. P. Bastard. 2005. HIV antiretroviral treatment alters adipokine expression and insulin sensitivity of adipose tissue in vitro and in vivo. Biochimie 87:65-71.
91

140. Laimer, M., S. Kaser, M. Kranebitter, A. Sandhofer, G. Muhlmann, H. Schwelberger, H. Weiss, J. R. Patsch, and C. F. Ebenbichler. 2005. Effect of pronounced weight loss on the nontraditional cardiovascular risk marker matrix metalloproteinase-9 in middle-aged morbidly obese women. Int J Obes (Lond) 29:498-501.
141. Lambert, E., E. Dasse, B. Haye, and E. Petitfrere. 2004. TIMPs as multifacial proteins. Crit Rev Oncol Hematol 49:187-98.
142. Langin, D., A. Dicker, G. Tavernier, J. Hoffstedt, A. Mairal, M. Ryden, E. Arner, A. Sicard, C. M. Jenkins, N. Viguerie, V. van Harmelen, R. W. Gross, C. Holm, and P. Arner. 2005. Adipocyte lipases and defect of lipolysis in human obesity. Diabetes 54:3190-7.
143. Large, V., S. Reynisdottir, D. Langin, K. Fredby, M. Klannemark, C. Holm, and P. Arner. 1999. Decreased expression and function of adipocyte hormone-sensitive lipase in subcutaneous fat cells of obese subjects. J Lipid Res 40:2059-66.
144. Lenhard, J. M., E. S. Furfine, R. G. Jain, O. Ittoop, L. A. Orband-Miller, S. G. Blanchard, M. A. Paulik, and J. E. Weiel. 2000. HIV protease inhibitors block adipogenesis and increase lipolysis in vitro. Antiviral Res 47:121-9.
145. Letexier, D., C. Pinteur, V. Large, V. Frering, and M. Beylot. 2003. Comparison of the expression and activity of the lipogenic pathway in human and rat adipose tissue. J Lipid Res 44:2127-34.
146. Li, X., H. Y. Huang, J. G. Chen, L. Jiang, H. L. Liu, D. G. Liu, T. J. Song, Q. He, C. G. Ma, D. Ma, H. Y. Song, and Q. Q. Tang. 2006. Lactacystin inhibits 3T3-L1 adipocyte differentiation through induction of CHOP-10 expression. Biochem Biophys Res Commun 350:1-6.
147. Lijnen, H. R., D. Demeulemeester, B. Van Hoef, D. Collen, and E. Maquoi. 2003. Deficiency of tissue inhibitor of matrix metalloproteinase-1 (TIMP-1) impairs nutritionally induced obesity in mice. Thromb Haemost 89:249-55.
148. Lijnen, H. R., E. Maquoi, L. B. Hansen, B. Van Hoef, L. Frederix, and D. Collen. 2002. Matrix metalloproteinase inhibition impairs adipose tissue development in mice. Arterioscler Thromb Vasc Biol 22:374-9.
149. Lijnen, H. R., E. Maquoi, P. Holvoet, A. Mertens, F. Lupu, P. Morange, M. C. Alessi, and I. Juhan-Vague. 2001. Adipose tissue expression of gelatinases in mouse models of obesity. Thromb Haemost 85:1111-6.
150. Lijnen, H. R., H. B. Van, L. Frederix, M. C. Rio, and D. Collen. 2002. Adipocyte hypertrophy in stromelysin-3 deficient mice with nutritionally induced obesity. Thromb Haemost 87:530-5.
151. Lilla, J., D. Stickens, and Z. Werb. 2002. Metalloproteases and adipogenesis: a weighty subject. Am J Pathol 160:1551-4.
152. Lloreta, J., P. Domingo, R. M. Pujol, J. A. Arroyo, N. Baixeras, X. Matias-Guiu, M. Gilaberte, M. A. Sambeat, and S. Serrano. 2002. Ultrastructural features of highly active antiretroviral therapy-associated partial lipodystrophy. Virchows Arch 441:599-604.
153. Lloyd, D. J., R. C. Trembath, and S. Shackleton. 2002. A novel interaction between lamin A and SREBP1: implications for partial lipodystrophy and other laminopathies. Hum Mol Genet 11:769-77.
154. Lolmede, K., V. Durand de Saint Front, J. Galitzky, M. Lafontan, and A. Bouloumie. 2003. Effects of hypoxia on the expression of proangiogenic factors in differentiated 3T3-F442A adipocytes. Int J Obes Relat Metab Disord 27:1187-95.
155. Lumeng, C. N., J. L. Bodzin, and A. R. Saltiel. 2007. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest 117:175-84.
92

156. Luo, D., B. Mari, I. Stoll, and P. Anglard. 2002. Alternative splicing and promoter usage generates an intracellular stromelysin 3 isoform directly translated as an active matrix metalloproteinase. J Biol Chem 277:25527-36.
157. Madge, S., S. Kinloch-de-Loes, D. Mercey, M. A. Johnson, and I. V. Weller. 1999. Lipodystrophy in patients naive to HIV protease inhibitors. Aids 13:735-7.
158. Mallal, S. A., M. John, C. B. Moore, I. R. James, and E. J. McKinnon. 2000. Contribution of nucleoside analogue reverse transcriptase inhibitors to subcutaneous fat wasting in patients with HIV infection. Aids 14:1309-16.
159. Mannello, F. 2006. Multipotent mesenchymal stromal cell recruitment, migration, and differentiation: what have matrix metalloproteinases got to do with it? Stem Cells 24:1904-7.
160. Mannello, F., F. Luchetti, E. Falcieri, and S. Papa. 2005. Multiple roles of matrix metalloproteinases during apoptosis. Apoptosis 10:19-24.
161. Mannello, F., G. Tonti, and S. Papa. 2005. Matrix metalloproteinase inhibitors as anticancer therapeutics. Curr Cancer Drug Targets 5:285-98.
162. Mannello, F., G. A. Tonti, G. P. Bagnara, and S. Papa. 2006. Role and function of matrix metalloproteinases in the differentiation and biological characterization of mesenchymal stem cells. Stem Cells 24:475-81.
163. Maquoi, E., D. Demeulemeester, G. Voros, D. Collen, and H. R. Lijnen. 2003. Enhanced nutritionally induced adipose tissue development in mice with stromelysin-1 gene inactivation. Thromb Haemost 89:696-704.
164. Maquoi, E., C. Munaut, A. Colige, D. Collen, and H. R. Lijnen. 2002. Modulation of adipose tissue expression of murine matrix metalloproteinases and their tissue inhibitors with obesity. Diabetes 51:1093-101.
165. Margeli, A., G. Kouraklis, and S. Theocharis. 2003. Peroxisome proliferator activated receptor-gamma (PPAR-gamma) ligands and angiogenesis. Angiogenesis 6:165-9.
166. Mauriege, P., J. Galitzky, M. Berlan, and M. Lafontan. 1987. Heterogeneous distribution of beta and alpha-2 adrenoceptor binding sites in human fat cells from various fat deposits: functional consequences. Eur J Clin Invest 17:156-65.
167. McBeath, R., D. M. Pirone, C. M. Nelson, K. Bhadriraju, and C. S. Chen. 2004. Cell shape, cytoskeletal tension, and RhoA regulate stem cell lineage commitment. Dev Cell 6:483-95.
168. McComsey, G. A., D. M. Paulsen, J. T. Lonergan, S. M. Hessenthaler, C. L. Hoppel, V. C. Williams, R. L. Fisher, C. L. Cherry, C. White-Owen, K. A. Thompson, S. T. Ross, J. E. Hernandez, and L. L. Ross. 2005. Improvements in lipoatrophy, mitochondrial DNA levels and fat apoptosis after replacing stavudine with abacavir or zidovudine. Aids 19:15-23.
169. McComsey, G. A., and U. A. Walker. 2004. Role of mitochondria in HIV lipoatrophy: insight into pathogenesis and potential therapies. Mitochondrion 4:111-8.
170. McIntosh, K., S. Zvonic, S. Garrett, J. B. Mitchell, Z. E. Floyd, L. Hammill, A. Kloster, Y. Di Halvorsen, J. P. Ting, R. W. Storms, B. Goh, G. Kilroy, X. Wu, and J. M. Gimble. 2006. The immunogenicity of human adipose-derived cells: temporal changes in vitro. Stem Cells 24:1246-53.
171. McIntyre, T. M., A. V. Pontsler, A. R. Silva, A. St Hilaire, Y. Xu, J. C. Hinshaw, G. A. Zimmerman, K. Hama, J. Aoki, H. Arai, and G. D. Prestwich. 2003. Identification of an intracellular receptor for lysophosphatidic acid (LPA): LPA is a transcellular PPARgamma agonist. Proc Natl Acad Sci U S A 100:131-6.
172. Metallo, C. M., J. C. Mohr, C. J. Detzel, J. J. Pablo, B. J. Wie, and S. P. Palecek. 2007. Engineering the stem cell microenvironment. Biotechnol Prog 23:18-23.
93

173. Miranville, A., C. Heeschen, C. Sengenes, C. A. Curat, R. Busse, and A. Bouloumie. 2004. Improvement of postnatal neovascularization by human adipose tissue-derived stem cells. Circulation 110:349-55.
174. Mitchell, J. B., K. McIntosh, S. Zvonic, S. Garrett, Z. E. Floyd, A. Kloster, Y. Di Halvorsen, R. W. Storms, B. Goh, G. Kilroy, X. Wu, and J. M. Gimble. 2006. Immunophenotype of human adipose-derived cells: temporal changes in stromal-associated and stem cell-associated markers. Stem Cells 24:376-85.
175. Miyazaki, T., Y. Kitagawa, K. Toriyama, M. Kobori, and S. Torii. 2005. Isolation of two human fibroblastic cell populations with multiple but distinct potential of mesenchymal differentiation by ceiling culture of mature fat cells from subcutaneous adipose tissue. Differentiation 73:69-78.
176. Mondal, D., V. F. Larussa, and K. C. Agrawal. 2001. Synergistic antiadipogenic effects of HIV type 1 protease inhibitors with tumor necrosis factor alpha: suppression of extracellular insulin action mediated by extracellular matrix-degrading proteases. AIDS Res Hum Retroviruses 17:1569-84.
177. Mori, T., H. Sakaue, H. Iguchi, H. Gomi, Y. Okada, Y. Takashima, K. Nakamura, T. Nakamura, T. Yamauchi, N. Kubota, T. Kadowaki, Y. Matsuki, W. Ogawa, R. Hiramatsu, and M. Kasuga. 2005. Role of Kruppel-like factor 15 (KLF15) in transcriptional regulation of adipogenesis. J Biol Chem 280:12867-75.
178. Mott, J. D., and Z. Werb. 2004. Regulation of matrix biology by matrix metalloproteinases. Curr Opin Cell Biol 16:558-64.
179. Murata, H., P. W. Hruz, and M. Mueckler. 2002. Indinavir inhibits the glucose transporter isoform Glut4 at physiologic concentrations. Aids 16:859-63.
180. Murata, H., P. W. Hruz, and M. Mueckler. 2002. Investigating the cellular targets of HIV protease inhibitors: implications for metabolic disorders and improvements in drug therapy. Curr Drug Targets Infect Disord 2:1-8.
181. Mynarcik, D., L. X. Wei, E. Komaroff, R. Ferris, M. McNurlan, and M. Gelato. 2005. Chronic loss of subcutaneous adipose tissue in HIV-associated lipodystrophy may not be associated with accelerated apoptosis. J Acquir Immune Defic Syndr 38:367-71.
182. Mynarcik, D. C., T. Combs, M. A. McNurlan, P. E. Scherer, E. Komaroff, and M. C. Gelato. 2002. Adiponectin and leptin levels in HIV-infected subjects with insulin resistance and body fat redistribution. J Acquir Immune Defic Syndr 31:514-20.
183. Nagase, H., and J. F. Woessner, Jr. 1999. Matrix metalloproteinases. J Biol Chem 274:21491-4.
184. Nakajima, I., T. Yamaguchi, K. Ozutsumi, and H. Aso. 1998. Adipose tissue extracellular matrix: newly organized by adipocytes during differentiation. Differentiation 63:193-200.
185. Neels, J. G., T. Thinnes, and D. J. Loskutoff. 2004. Angiogenesis in an in vivo model of adipose tissue development. Faseb J 18:983-5.
186. Negrel, R., P. Grimaldi, C. Forest, and G. Ailhaud. 1985. Establishment and characterization of fibroblast-like cell lines derived from adipocytes with the capacity to redifferentiate into adipocyte-like cells. Methods Enzymol 109:377-85.
187. Nelson, W. J., and R. Nusse. 2004. Convergence of Wnt, beta-catenin, and cadherin pathways. Science 303:1483-7.
188. Nelson-Dooley, C., M. A. Della-Fera, M. Hamrick, and C. A. Baile. 2005. Novel treatments for obesity and osteoporosis: targeting apoptotic pathways in adipocytes. Curr Med Chem 12:2215-25.
94

189. Nishimura, S., I. Manabe, M. Nagasaki, Y. Hosoya, H. Yamashita, H. Fujita, M. Ohsugi, K. Tobe, T. Kadowaki, R. Nagai, and S. Sugiura. 2007. Adipogenesis in obesity requires close interplay between differentiating adipocytes, stromal cells, and blood vessels. Diabetes 56:1517-26.
190. Nolan, D., E. Hammond, I. James, E. McKinnon, and S. Mallal. 2003. Contribution of nucleoside-analogue reverse transcriptase inhibitor therapy to lipoatrophy from the population to the cellular level. Antivir Ther 8:617-26.
191. O'Connor, K. C., H. Song, N. Rosenzweig, and D. A. Jansen. 2003. Extracellular matrix substrata alter adipocyte yield and lipogenesis in primary cultures of stromal-vascular cells from human adipose. Biotechnol Lett 25:1967-72.
192. Odegaard, J. I., R. R. Ricardo-Gonzalez, M. H. Goforth, C. R. Morel, V. Subramanian, L. Mukundan, A. R. Eagle, D. Vats, F. Brombacher, A. W. Ferrante, and A. Chawla. 2007. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature 447:1116-20.
193. Oh, J., R. Takahashi, S. Kondo, A. Mizoguchi, E. Adachi, R. M. Sasahara, S. Nishimura, Y. Imamura, H. Kitayama, D. B. Alexander, C. Ide, T. P. Horan, T. Arakawa, H. Yoshida, S. Nishikawa, Y. Itoh, M. Seiki, S. Itohara, C. Takahashi, and M. Noda. 2001. The membrane-anchored MMP inhibitor RECK is a key regulator of extracellular matrix integrity and angiogenesis. Cell 107:789-800.
194. Oishi, Y., I. Manabe, K. Tobe, K. Tsushima, T. Shindo, K. Fujiu, G. Nishimura, K. Maemura, T. Yamauchi, N. Kubota, R. Suzuki, T. Kitamura, S. Akira, T. Kadowaki, and R. Nagai. 2005. Kruppel-like transcription factor KLF5 is a key regulator of adipocyte differentiation. Cell Metab 1:27-39.
195. O'Reilly, M. S., L. Holmgren, Y. Shing, C. Chen, R. A. Rosenthal, M. Moses, W. S. Lane, Y. Cao, E. H. Sage, and J. Folkman. 1994. Angiostatin: a novel angiogenesis inhibitor that mediates the suppression of metastases by a Lewis lung carcinoma. Cell 79:315-28.
196. Osuga, J., S. Ishibashi, T. Oka, H. Yagyu, R. Tozawa, A. Fujimoto, F. Shionoiri, N. Yahagi, F. B. Kraemer, O. Tsutsumi, and N. Yamada. 2000. Targeted disruption of hormone-sensitive lipase results in male sterility and adipocyte hypertrophy, but not in obesity. Proc Natl Acad Sci U S A 97:787-92.
197. Otto, T. C., R. R. Bowers, and M. D. Lane. 2007. BMP-4 treatment of C3H10T1/2 stem cells blocks expression of MMP-3 and MMP-13. Biochem Biophys Res Commun 353:1097-104.
198. Pacenti, M., L. Barzon, F. Favaretto, K. Fincati, S. Romano, G. Milan, R. Vettor, and G. Palu. 2006. Microarray analysis during adipogenesis identifies new genes altered by antiretroviral drugs. Aids 20:1691-705.
199. Page-McCaw, A., A. J. Ewald, and Z. Werb. 2007. Matrix metalloproteinases and the regulation of tissue remodelling. Nat Rev Mol Cell Biol 8:221-33.
200. Pajonk, F., J. Himmelsbach, K. Riess, A. Sommer, and W. H. McBride. 2002. The human immunodeficiency virus (HIV)-1 protease inhibitor saquinavir inhibits proteasome function and causes apoptosis and radiosensitization in non-HIV-associated human cancer cells. Cancer Res 62:5230-5.
201. Pajvani, U. B., M. E. Trujillo, T. P. Combs, P. Iyengar, L. Jelicks, K. A. Roth, R. N. Kitsis, and P. E. Scherer. 2005. Fat apoptosis through targeted activation of caspase 8: a new mouse model of inducible and reversible lipoatrophy. Nat Med 11:797-803.
202. Parker, R. A., O. P. Flint, R. Mulvey, C. Elosua, F. Wang, W. Fenderson, S. Wang, W. P. Yang, and M. A. Noor. 2005. Endoplasmic reticulum stress links
95

dyslipidemia to inhibition of proteasome activity and glucose transport by HIV protease inhibitors. Mol Pharmacol 67:1909-19.
203. Parks, W. C., C. L. Wilson, and Y. S. Lopez-Boado. 2004. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat Rev Immunol 4:617-29.
204. Pei, D., and S. J. Weiss. 1995. Furin-dependent intracellular activation of the human stromelysin-3 zymogen. Nature 375:244-7.
205. Pendas, A. M., A. R. Folgueras, E. Llano, J. Caterina, F. Frerard, F. Rodriguez, A. Astudillo, A. Noel, H. Birkedal-Hansen, and C. Lopez-Otin. 2004. Diet-induced obesity and reduced skin cancer susceptibility in matrix metalloproteinase 19-deficient mice. Mol Cell Biol 24:5304-13.
206. Pendas, A. M., Z. Zhou, J. Cadinanos, J. M. Freije, J. Wang, K. Hultenby, A. Astudillo, A. Wernerson, F. Rodriguez, K. Tryggvason, and C. Lopez-Otin. 2002. Defective prelamin A processing and muscular and adipocyte alterations in Zmpste24 metalloproteinase-deficient mice. Nat Genet 31:94-9.
207. Phillips, B. W., C. Vernochet, and C. Dani. 2003. Differentiation of embryonic stem cells for pharmacological studies on adipose cells. Pharmacol Res 47:263-8.
208. Piccinini, M., M. T. Rinaudo, A. Anselmino, B. Buccinna, C. Ramondetti, A. Dematteis, E. Ricotti, L. Palmisano, M. Mostert, and P. A. Tovo. 2005. The HIV protease inhibitors nelfinavir and saquinavir, but not a variety of HIV reverse transcriptase inhibitors, adversely affect human proteasome function. Antivir Ther 10:215-23.
209. Pistrosch, F., K. Herbrig, U. Oelschlaegel, S. Richter, J. Passauer, S. Fischer, and P. Gross. 2005. PPARgamma-agonist rosiglitazone increases number and migratory activity of cultured endothelial progenitor cells. Atherosclerosis 183:163-7.
210. Pittenger, M. F., A. M. Mackay, S. C. Beck, R. K. Jaiswal, R. Douglas, J. D. Mosca, M. A. Moorman, D. W. Simonetti, S. Craig, and D. R. Marshak. 1999. Multilineage potential of adult human mesenchymal stem cells. Science 284:143-7.
211. Planat-Benard, V., J. S. Silvestre, B. Cousin, M. Andre, M. Nibbelink, R. Tamarat, M. Clergue, C. Manneville, C. Saillan-Barreau, M. Duriez, A. Tedgui, B. Levy, L. Penicaud, and L. Casteilla. 2004. Plasticity of human adipose lineage cells toward endothelial cells: physiological and therapeutic perspectives. Circulation 109:656-63.
212. Prince, A. M., J. S. May, G. R. Burton, R. E. Lyle, and R. E. McGehee, Jr. 2002. Proteasomal degradation of retinoblastoma-related p130 during adipocyte differentiation. Biochem Biophys Res Commun 290:1066-71.
213. Prins, J. B., C. U. Niesler, C. M. Winterford, N. A. Bright, K. Siddle, S. O'Rahilly, N. I. Walker, and D. P. Cameron. 1997. Tumor necrosis factor-alpha induces apoptosis of human adipose cells. Diabetes 46:1939-44.
214. Prins, J. B., and S. O'Rahilly. 1997. Regulation of adipose cell number in man. Clin Sci (Lond) 92:3-11.
215. Proulx, A., P. A. Merrifield, and C. C. Naus. 1997. Blocking gap junctional intercellular communication in myoblasts inhibits myogenin and MRF4 expression. Dev Genet 20:133-44.
216. Qi, J. H., Q. Ebrahem, N. Moore, G. Murphy, L. Claesson-Welsh, M. Bond, A. Baker, and B. Anand-Apte. 2003. A novel function for tissue inhibitor of metalloproteinases-3 (TIMP3): inhibition of angiogenesis by blockage of VEGF binding to VEGF receptor-2. Nat Med 9:407-15.
217. Rajagopalan, S., X. P. Meng, S. Ramasamy, D. G. Harrison, and Z. S. Galis. 1996. Reactive oxygen species produced by macrophage-derived foam cells regulate
96

the activity of vascular matrix metalloproteinases in vitro. Implications for atherosclerotic plaque stability. J Clin Invest 98:2572-9.
218. Ranganathan, S., and P. A. Kern. 2002. The HIV protease inhibitor saquinavir impairs lipid metabolism and glucose transport in cultured adipocytes. J Endocrinol 172:155-62.
219. Ray, H., M. Beylot, P. Arner, D. Larrouy, D. Langin, C. Holm, and V. Large. 2003. The presence of a catalytically inactive form of hormone-sensitive lipase is associated with decreased lipolysis in abdominal subcutaneous adipose tissue of obese subjects. Diabetes 52:1417-22.
220. Raza, S. L., L. C. Nehring, S. D. Shapiro, and L. A. Cornelius. 2000. Proteinase-activated receptor-1 regulation of macrophage elastase (MMP-12) secretion by serine proteinases. J Biol Chem 275:41243-50.
221. Rehman, J., D. Traktuev, J. Li, S. Merfeld-Clauss, C. J. Temm-Grove, J. E. Bovenkerk, C. L. Pell, B. H. Johnstone, R. V. Considine, and K. L. March. 2004. Secretion of angiogenic and antiapoptotic factors by human adipose stromal cells. Circulation 109:1292-8.
222. Reitman, M. L., E. Arioglu, O. Gavrilova, and S. I. Taylor. 2000. Lipoatrophy revisited. Trends Endocrinol Metab 11:410-6.
223. Reyes, M., A. Dudek, B. Jahagirdar, L. Koodie, P. H. Marker, and C. M. Verfaillie. 2002. Origin of endothelial progenitors in human postnatal bone marrow. J Clin Invest 109:337-46.
224. Roche, R., I. Poizot-Martin, C. M. Yazidi, E. Compe, J. A. Gastaut, J. Torresani, and R. Planells. 2002. Effects of antiretroviral drug combinations on the differentiation of adipocytes. Aids 16:13-20.
225. Rosell, S., and E. Belfrage. 1979. Blood circulation in adipose tissue. Physiol Rev 59:1078-1104.
226. Rosen, E. D., and O. A. MacDougald. 2006. Adipocyte differentiation from the inside out. Nat Rev Mol Cell Biol 7:885-96.
227. Rosen, E. D., and B. M. Spiegelman. 2000. Molecular regulation of adipogenesis. Annu Rev Cell Dev Biol 16:145-71.
228. Rosen, E. D., C. J. Walkey, P. Puigserver, and B. M. Spiegelman. 2000. Transcriptional regulation of adipogenesis. Genes Dev 14:1293-307.
229. Ross, J., and L. Li. 2006. Recent advances in understanding extrinsic control of hematopoietic stem cell fate. Curr Opin Hematol 13:237-42.
230. Ross, S. E., N. Hemati, K. A. Longo, C. N. Bennett, P. C. Lucas, R. L. Erickson, and O. A. MacDougald. 2000. Inhibition of adipogenesis by Wnt signaling. Science 289:950-3.
231. Rudich, A., S. Vanounou, K. Riesenberg, M. Porat, A. Tirosh, I. Harman-Boehm, A. S. Greenberg, F. Schlaeffer, and N. Bashan. 2001. The HIV protease inhibitor nelfinavir induces insulin resistance and increases basal lipolysis in 3T3-L1 adipocytes. Diabetes 50:1425-31.
232. Rundhaug, J. E. 2005. Matrix metalloproteinases and angiogenesis. J Cell Mol Med 9:267-85.
233. Rupnick, M. A., D. Panigrahy, C. Y. Zhang, S. M. Dallabrida, B. B. Lowell, R. Langer, and M. J. Folkman. 2002. Adipose tissue mass can be regulated through the vasculature. Proc Natl Acad Sci U S A 99:10730-5.
234. Sabatakos, G., N. A. Sims, J. Chen, K. Aoki, M. B. Kelz, M. Amling, Y. Bouali, K. Mukhopadhyay, K. Ford, E. J. Nestler, and R. Baron. 2000. Overexpression of DeltaFosB transcription factor(s) increases bone formation and inhibits adipogenesis. Nat Med 6:985-90.
97

235. Saillan-Barreau, C., B. Cousin, M. Andre, P. Villena, L. Casteilla, and L. Penicaud. 2003. Human adipose cells as candidates in defense and tissue remodeling phenomena. Biochem Biophys Res Commun 309:502-5.
236. Sato, H., and M. Seiki. 1993. Regulatory mechanism of 92 kDa type IV collagenase gene expression which is associated with invasiveness of tumor cells. Oncogene 8:395-405.
237. Schiller, P. C., G. D'Ippolito, W. Balkan, B. A. Roos, and G. A. Howard. 2001. Gap-junctional communication is required for the maturation process of osteoblastic cells in culture. Bone 28:362-9.
238. Schiller, P. C., G. D'Ippolito, R. Brambilla, B. A. Roos, and G. A. Howard. 2001. Inhibition of gap-junctional communication induces the trans-differentiation of osteoblasts to an adipocytic phenotype in vitro. J Biol Chem 276:14133-8.
239. Schmidtke, G., H. G. Holzhutter, M. Bogyo, N. Kairies, M. Groll, R. de Giuli, S. Emch, and M. Groettrup. 1999. How an inhibitor of the HIV-I protease modulates proteasome activity. J Biol Chem 274:35734-40.
240. Sengenes, C., A. Bouloumie, H. Hauner, M. Berlan, R. Busse, M. Lafontan, and J. Galitzky. 2003. Involvement of a cGMP-dependent pathway in the natriuretic peptide-mediated hormone-sensitive lipase phosphorylation in human adipocytes. J Biol Chem 278:48617-26.
241. Sengenes, C., K. Lolmede, A. Zakaroff-Girard, R. Busse, and A. Bouloumie. 2005. Preadipocytes in the human subcutaneous adipose tissue display distinct features from the adult mesenchymal and hematopoietic stem cells. J Cell Physiol 205:114-22.
242. Sengenes, C., A. Miranville, M. Maumus, S. de Barros, R. Busse, and A. Bouloumie. 2007. Chemotaxis and Differentiation of Human Adipose Tissue CD34+/CD31- Progenitor Cells: Role of SDF-1 Released by Adipose Tissue Capillary Endothelial Cells. Stem Cells.
243. Seo, D. W., H. Li, L. Guedez, P. T. Wingfield, T. Diaz, R. Salloum, B. Y. Wei, and W. G. Stetler-Stevenson. 2003. TIMP-2 mediated inhibition of angiogenesis: an MMP-independent mechanism. Cell 114:171-80.
244. Shankar, S. S., and M. P. Dube. 2004. Clinical aspects of endothelial dysfunction associated with human immunodeficiency virus infection and antiretroviral agents. Cardiovasc Toxicol 4:261-9.
245. Shikuma, C. M., N. Hu, C. Milne, F. Yost, C. Waslien, S. Shimizu, and B. Shiramizu. 2001. Mitochondrial DNA decrease in subcutaneous adipose tissue of HIV-infected individuals with peripheral lipoatrophy. Aids 15:1801-9.
246. Shipley, J. M., R. L. Wesselschmidt, D. K. Kobayashi, T. J. Ley, and S. D. Shapiro. 1996. Metalloelastase is required for macrophage-mediated proteolysis and matrix invasion in mice. Proc Natl Acad Sci U S A 93:3942-6.
247. Silha, J. V., M. Krsek, P. Sucharda, and L. J. Murphy. 2005. Angiogenic factors are elevated in overweight and obese individuals. Int J Obes (Lond) 29:1308-14.
248. Si-Tayeb, K., A. Monvoisin, C. Mazzocco, S. Lepreux, M. Decossas, G. Cubel, D. Taras, J. F. Blanc, D. R. Robinson, and J. Rosenbaum. 2006. Matrix metalloproteinase 3 is present in the cell nucleus and is involved in apoptosis. Am J Pathol 169:1390-401.
249. Sjostrom, L., U. Smith, M. Krotkiewski, and P. Bjorntorp. 1972. Cellularity in different regions of adipose tissue in young men and women. Metabolism 21:1143-53.
250. Sorisky, A., R. Magun, and A. M. Gagnon. 2000. Adipose cell apoptosis: death in the energy depot. Int J Obes Relat Metab Disord 24 Suppl 4:S3-7.
98

251. Sottrup-Jensen, L., and H. Birkedal-Hansen. 1989. Human fibroblast collagenase-alpha-macroglobulin interactions. Localization of cleavage sites in the bait regions of five mammalian alpha-macroglobulins. J Biol Chem 264:393-401.
252. Spiegelman, B. M., and S. R. Farmer. 1982. Decreases in tubulin and actin gene expression prior to morphological differentiation of 3T3 adipocytes. Cell 29:53-60.
253. Spinale, F. G. 2002. Matrix metalloproteinases: regulation and dysregulation in the failing heart. Circ Res 90:520-30.
254. Sternlicht, M. D., and Z. Werb. 2001. How matrix metalloproteinases regulate cell behavior. Annu Rev Cell Dev Biol 17:463-516.
255. Stich, V., I. De Glisezinski, F. Crampes, J. Hejnova, J. M. Cottet-Emard, J. Galitzky, M. Lafontan, D. Riviere, and M. Berlan. 2000. Activation of alpha(2)-adrenergic receptors impairs exercise-induced lipolysis in SCAT of obese subjects. Am J Physiol Regul Integr Comp Physiol 279:R499-504.
256. St-Pierre, Y., J. Couillard, and C. Van Themsche. 2004. Regulation of MMP-9 gene expression for the development of novel molecular targets against cancer and inflammatory diseases. Expert Opin Ther Targets 8:473-89.
257. Streuli, C. 1999. Extracellular matrix remodelling and cellular differentiation. Curr Opin Cell Biol 11:634-40.
258. Strongin, A. Y., I. Collier, G. Bannikov, B. L. Marmer, G. A. Grant, and G. I. Goldberg. 1995. Mechanism of cell surface activation of 72-kDa type IV collagenase. Isolation of the activated form of the membrane metalloprotease. J Biol Chem 270:5331-8.
259. Sutinen, J., E. Korsheninnikova, T. Funahashi, Y. Matsuzawa, T. Nyman, and H. Yki-Jarvinen. 2003. Circulating concentration of adiponectin and its expression in subcutaneous adipose tissue in patients with highly active antiretroviral therapy-associated lipodystrophy. J Clin Endocrinol Metab 88:1907-10.
260. Taleb, S., R. Cancello, K. Clement, and D. Lacasa. 2006. Cathepsin s promotes human preadipocyte differentiation: possible involvement of fibronectin degradation. Endocrinology 147:4950-9.
261. Tang, Q. Q., T. C. Otto, and M. D. Lane. 2004. Commitment of C3H10T1/2 pluripotent stem cells to the adipocyte lineage. Proc Natl Acad Sci U S A 101:9607-11.
262. Tchoukalova, Y., C. Koutsari, and M. Jensen. 2007. Committed subcutaneous preadipocytes are reduced in human obesity. Diabetologia 50:151-7.
263. Tedeschi. 1965. Pathological anatomy of adipose tissue.:141-68. 264. Tran, H., S. Robinson, I. Mikhailenko, and D. K. Strickland. 2003. Modulation of
the LDL receptor and LRP levels by HIV protease inhibitors. J Lipid Res 44:1859-69. 265. Trayhurn, P., and J. H. Beattie. 2001. Physiological role of adipose tissue: white
adipose tissue as an endocrine and secretory organ. Proc Nutr Soc 60:329-39. 266. Trujillo, M. E., U. B. Pajvani, and P. E. Scherer. 2005. Apoptosis through targeted
activation of caspase 8 ("ATTAC-mice"): novel mouse models of inducible and reversible tissue ablation. Cell Cycle 4:1141-5.
267. Umezawa, A., and J. Hata. 1992. Expression of gap-junctional protein (connexin 43 or alpha 1 gap junction) is down-regulated at the transcriptional level during adipocyte differentiation of H-1/A marrow stromal cells. Cell Struct Funct 17:177-84.
268. van der Valk, M., G. Allick, G. J. Weverling, J. A. Romijn, M. T. Ackermans, J. M. Lange, B. L. van Eck-Smit, C. van Kuijk, E. Endert, H. P. Sauerwein, and P. Reiss. 2004. Markedly diminished lipolysis and partial restoration of glucose metabolism, without changes in fat distribution after extended discontinuation of
99

protease inhibitors in severe lipodystrophic human immunodeficient virus-1-infected patients. J Clin Endocrinol Metab 89:3554-60.
269. van der Valk, M., E. H. Gisolf, P. Reiss, F. W. Wit, A. Japour, G. J. Weverling, and S. A. Danner. 2001. Increased risk of lipodystrophy when nucleoside analogue reverse transcriptase inhibitors are included with protease inhibitors in the treatment of HIV-1 infection. Aids 15:847-55.
270. Van Harmelen, V., K. Rohrig, and H. Hauner. 2004. Comparison of proliferation and differentiation capacity of human adipocyte precursor cells from the omental and subcutaneous adipose tissue depot of obese subjects. Metabolism 53:632-7.
271. van Harmelen, V., T. Skurk, K. Rohrig, Y. M. Lee, M. Halbleib, I. Aprath-Husmann, and H. Hauner. 2003. Effect of BMI and age on adipose tissue cellularity and differentiation capacity in women. Int J Obes Relat Metab Disord 27:889-95.
272. Vankoningsloo, S., A. De Pauw, A. Houbion, S. Tejerina, C. Demazy, F. de Longueville, V. Bertholet, P. Renard, J. Remacle, P. Holvoet, M. Raes, and T. Arnould. 2006. CREB activation induced by mitochondrial dysfunction triggers triglyceride accumulation in 3T3-L1 preadipocytes. J Cell Sci 119:1266-82.
273. Vernochet, C., S. Azoulay, D. Duval, R. Guedj, G. Ailhaud, and C. Dani. 2003. Differential effect of HIV protease inhibitors on adipogenesis: intracellular ritonavir is not sufficient to inhibit differentiation. Aids 17:2177-80.
274. Vernochet, C., S. Azoulay, D. Duval, R. Guedj, F. Cottrez, H. Vidal, G. Ailhaud, and C. Dani. 2005. Human immunodeficiency virus protease inhibitors accumulate into cultured human adipocytes and alter expression of adipocytokines. J Biol Chem 280:2238-43.
275. Vigouroux, C., M. Maachi, T. H. Nguyen, C. Coussieu, S. Gharakhanian, T. Funahashi, Y. Matsuzawa, I. Shimomura, W. Rozenbaum, J. Capeau, and J. P. Bastard. 2003. Serum adipocytokines are related to lipodystrophy and metabolic disorders in HIV-infected men under antiretroviral therapy. Aids 17:1503-11.
276. Villarroya, F., P. Domingo, and M. Giralt. 2007. Lipodystrophy in HIV 1-infected patients: lessons for obesity research. Int J Obes (Lond).
277. Villena, J. A., S. Roy, E. Sarkadi-Nagy, K. H. Kim, and H. S. Sul. 2004. Desnutrin, an adipocyte gene encoding a novel patatin domain-containing protein, is induced by fasting and glucocorticoids: ectopic expression of desnutrin increases triglyceride hydrolysis. J Biol Chem 279:47066-75.
278. Vincent, S., F. Tourniaire, C. M. El Yazidi, E. Compe, O. Manches, R. Plannels, and R. Roche. 2004. Nelfinavir induces necrosis of 3T3F44-2A adipocytes by oxidative stress. J Acquir Immune Defic Syndr 37:1556-62.
279. Voorhees, P. M., and R. Z. Orlowski. 2006. The proteasome and proteasome inhibitors in cancer therapy. Annu Rev Pharmacol Toxicol 46:189-213.
280. Vu, T. H., and Z. Werb. 2000. Matrix metalloproteinases: effectors of development and normal physiology. Genes Dev 14:2123-33.
281. Walker, U. A., M. Bickel, S. I. Lutke Volksbeck, U. P. Ketelsen, H. Schofer, B. Setzer, N. Venhoff, V. Rickerts, and S. Staszewski. 2002. Evidence of nucleoside analogue reverse transcriptase inhibitor--associated genetic and structural defects of mitochondria in adipose tissue of HIV-infected patients. J Acquir Immune Defic Syndr 29:117-21.
282. Wang, C. H., N. Ciliberti, S. H. Li, P. E. Szmitko, R. D. Weisel, P. W. Fedak, M. Al-Omran, W. J. Cherng, R. K. Li, W. L. Stanford, and S. Verma. 2004. Rosiglitazone facilitates angiogenic progenitor cell differentiation toward endothelial lineage: a new paradigm in glitazone pleiotropy. Circulation 109:1392-400.
100

283. Wang, E. A., D. I. Israel, S. Kelly, and D. P. Luxenberg. 1993. Bone morphogenetic protein-2 causes commitment and differentiation in C3H10T1/2 and 3T3 cells. Growth Factors 9:57-71.
284. Wang, X., R. Sato, M. S. Brown, X. Hua, and J. L. Goldstein. 1994. SREBP-1, a membrane-bound transcription factor released by sterol-regulated proteolysis. Cell 77:53-62.
285. Wassermann, F. 1965. The development of adipose tissue. Renold A, Cahill G (eds). 286. Weisberg, S. P., D. McCann, M. Desai, M. Rosenbaum, R. L. Leibel, and A. W.
Ferrante, Jr. 2003. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 112:1796-808.
287. Weiss, S. J., G. Peppin, X. Ortiz, C. Ragsdale, and S. T. Test. 1985. Oxidative autoactivation of latent collagenase by human neutrophils. Science 227:747-9.
288. Wentworth, J. M., T. P. Burris, and V. K. Chatterjee. 2000. HIV protease inhibitors block human preadipocyte differentiation, but not via the PPARgamma/RXR heterodimer. J Endocrinol 164:R7-R10.
289. Westermarck, J., and V. M. Kahari. 1999. Regulation of matrix metalloproteinase expression in tumor invasion. Faseb J 13:781-92.
290. Woessner, J. F., Jr. 1991. Matrix metalloproteinases and their inhibitors in connective tissue remodeling. Faseb J 5:2145-54.
291. Wosnitza, M., K. Hemmrich, A. Groger, S. Graber, and N. Pallua. 2007. Plasticity of human adipose stem cells to perform adipogenic and endothelial differentiation. Differentiation 75:12-23.
292. Wu, J., S. V. Srinivasan, J. C. Neumann, and J. B. Lingrel. 2005. The KLF2 transcription factor does not affect the formation of preadipocytes but inhibits their differentiation into adipocytes. Biochemistry 44:11098-105.
293. Xu, H., G. T. Barnes, Q. Yang, G. Tan, D. Yang, C. J. Chou, J. Sole, A. Nichols, J. S. Ross, L. A. Tartaglia, and H. Chen. 2003. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest 112:1821-30.
294. Yamamoto, Y., and R. B. Gaynor. 2001. Therapeutic potential of inhibition of the NF-kappaB pathway in the treatment of inflammation and cancer. J Clin Invest 107:135-42.
295. Yan, C., and D. D. Boyd. 2007. Regulation of matrix metalloproteinase gene expression. J Cell Physiol 211:19-26.
296. Yanagiya, T., A. Tanabe, and K. Hotta. 2007. Gap-junctional communication is required for mitotic clonal expansion during adipogenesis. Obesity (Silver Spring) 15:572-82.
297. Young, S. G., B. S. Davies, L. G. Fong, P. Gin, M. M. Weinstein, A. Bensadoun, and A. P. Beigneux. 2007. GPIHBP1: an endothelial cell molecule important for the lipolytic processing of chylomicrons. Curr Opin Lipidol 18:389-96.
298. Yu, W. H., S. Yu, Q. Meng, K. Brew, and J. F. Woessner, Jr. 2000. TIMP-3 binds to sulfated glycosaminoglycans of the extracellular matrix. J Biol Chem 275:31226-32.
299. Zavrski, I., H. Krebbel, B. Wildemann, U. Heider, M. Kaiser, K. Possinger, and O. Sezer. 2005. Proteasome inhibitors abrogate osteoclast differentiation and osteoclast function. Biochem Biophys Res Commun 333:200-5.
300. Zhang, B., K. MacNaul, D. Szalkowski, Z. Li, J. Berger, and D. E. Moller. 1999. Inhibition of adipocyte differentiation by HIV protease inhibitors. J Clin Endocrinol Metab 84:4274-7.
101

301. Zhang, Q. X., C. J. Magovern, C. A. Mack, K. T. Budenbender, W. Ko, and T. K. Rosengart. 1997. Vascular endothelial growth factor is the major angiogenic factor in omentum: mechanism of the omentum-mediated angiogenesis. J Surg Res 67:147-54.
302. Zhang, Y., R. Proenca, M. Maffei, M. Barone, L. Leopold, and J. M. Friedman. 1994. Positional cloning of the mouse obese gene and its human homologue. Nature 372:425-32.
303. Zimmermann, R., J. G. Strauss, G. Haemmerle, G. Schoiswohl, R. Birner-Gruenberger, M. Riederer, A. Lass, G. Neuberger, F. Eisenhaber, A. Hermetter, and R. Zechner. 2004. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science 306:1383-6.
304. Zuk, P. A., M. Zhu, P. Ashjian, D. A. De Ugarte, J. I. Huang, H. Mizuno, Z. C. Alfonso, J. K. Fraser, P. Benhaim, and M. H. Hedrick. 2002. Human adipose tissue is a source of multipotent stem cells. Mol Biol Cell 13:4279-95.
102

103
Annexes
Publication 3 :
Chemotaxis and Differentiation of Human Adipose Tissue CD34+/CD31- Progenitor
Cells: Role of SDF-1 Released by Adipose Tissue Capillary Endothelial Cells
Coralie Sengenès, Alexandra Miranville, Marie Maumus, Sandra de Barros, Rudi Busse and
Anne Bouloumié. Stem Cells, 2007

Chemotaxis and Differentiation of Human Adipose Tissue CD34�/CD31� Progenitor Cells: Role of Stromal Derived Factor-1 Releasedby Adipose Tissue Capillary Endothelial Cells
CORALIE SENGENÈS,a ALEXANDRA MIRANVILLE,b MARIE MAUMUS,a SANDRA DE BARROS,a RUDI BUSSE,b
ANNE BOULOUMIÉa
aInstitut National de la Sante et de la Recherche Medicale, U858, AVENIR Team, I2MR, Paul Sabatier University,IFR31, Toulouse, France; bInstitute of Cardiovascular Physiology, Johann Wolfgang Goethe University,Frankfurt am Main, Germany
Key Words. Human CD34� cells • Stromal derived factor-1 • Endothelial differentiation • Chemotaxis • CXCR-4 • MatrigelMicrovasculature • Progenitor cells
ABSTRACT
The native CD34�/CD31� cell population present in thestroma-vascular fraction of human adipose tissue (hAT)displays progenitor cell properties since they exhibit adipo-cyte- and endothelial cell-like phenotypes under appropriatestimuli. To analyze the signals within hAT regulating theirphenotypes, the influence of hAT-derived capillary endothe-lial cells (CECs) was studied on the chemotaxis and differ-entiation of the hAT-CD34�/CD31� cells. Conditioned me-dium from hAT-CECs led to a strong chemotaxis of thehAT-CD34�/CD31� cells that was inhibited with pretreat-ments with pertussis toxin, CXCR-4 antagonist, or neutral-izing antibodies. Furthermore, hAT-CECs produced andsecreted the CXCR-4 ligand, that is, the stromal derivedfactor-1 (SDF-1). Finally, hAT-CECs induced the differen-
tiation of hAT-CD34�/CD31� cells toward an endothelialcell (EC) phenotype. Indeed, hAT-CECs and -CD34�/CD31� cell coculture stimulated in a two-dimensional sys-tem the expression of the EC CD31 marker by the hAT-progenitor cells and, in a three-dimensional approach, theformation of capillary-like structures via a SDF-1/CXCR-4dependent pathway. Thus, the migration and differentiationof hAT progenitor cells are modulated by hAT-CEC-derivedfactors. SDF-1, which is secreted by hAT-derived CECs, andits receptor CXCR-4, expressed by hAT-derived progenitorcells, may promote chemotaxis and differentiation of hAT-derived progenitor cells and thus contribute to the forma-tion of the vascular network during the development of hAT.STEM CELLS 2007;25:2269–2276
Disclosure of potential conflicts of interest is found at the end of this article.
INTRODUCTION
The excessive human adipose tissue (hAT) development isassociated with adipocyte hypertrophy and hyperplasia [1, 2] aswell as an extension of the capillary network [3–5]. The pro-cesses involved in the increased number of mature adipocytes aswell as the neovascularization within the fat mass are still to beclearly defined. We have described a cell population, present inthe native stroma-vascular fraction (SVF) of hAT, characterizedas positive for the hematopoietic stem cell marker CD34 andnegative for the endothelial cell marker CD31. The CD34�/CD31� cell population was the only cell population of the SVFunder adipogenic culture conditions among the capillary endo-thelial cells (CECs), the macrophages, and the cells negative forthe CD34, CD31, and CD14 markers able to express adipocyte-specific markers and metabolic lipolytic and lipogenic activities.Furthermore, under endothelial cell culture conditions, theCD34�/CD31� cells changed their morphology and organiza-tion and expressed endothelial cell (EC)-specific markers suchas CD31 and von Willebrand factor [5]. In vivo, their injectionled to an increase in blood flow and capillary density within the
ischemic muscle in the athymic mouse model of hindlimbischemia, whereas injection with native hAT-derived CD34�
cells exerted no effects [5]. Other laboratories have reportedsimilar proangiogenic abilities of the whole adipose tissue (AT)adherent SVF cells [6, 7]. The further characterization of thenative CD34�/CD31� cells has shown that they did not expressthe classic mesenchymal stem cell markers (CD105 and Stro-1)and displayed different features from the adult hematopoieticstem cells since they did not form blood colony in hematopoieticassays [8]. Thus, the native CD34�/CD31� cell population ofhAT can be considered a local pool of adipocyte as well asendothelial progenitor cells that might play a role in the forma-tion of adipocytes but also of capillaries accompanying thedevelopment of the fat mass. Since the microenvironment, thatis, soluble factors such as the stromal derived factor-1 (SDF-1)[9–11], cellular, and extracellular matrix components have beenshown to regulate the biology of hematopoietic and mesenchy-mal stem cells [12–14], the present study was undertaken tocharacterize the effects of signals arising from hAT microenvi-ronment on the chemotaxis and differentiation of the hATprogenitor cells. Our data delineate the concept that hAT-de-rived CECs induce the chemotaxis and support the endothelial
Correspondence: Coralie Sengenes, Ph.D., Team 1, “Vascular network, progenitor cells and immune cells from adipose tissue,” InstitutNational de la Sante et de la Recherche Medicale U858/I2MR, BP84225, 31432 Toulouse Cedex 4, France. Telephone: �33 5 6114 5975;Fax: �33 5 6125 5116; e-mail: [email protected] Received March 14, 2007; accepted for publication May 17,2007; first published online in STEM CELLS EXPRESS May 24, 2007. ©AlphaMed Press 1066-5099/2007/$30.00/0 doi: 10.1634/stemcells.2007-0180
TISSUE-SPECIFIC STEM CELLS
STEM CELLS 2007;25:2269–2276 www.StemCells.com
at Inserm D
isc Doc on Septem
ber 7, 2007 w
ww
.StemC
ells.comD
ownloaded from

differentiation potential of the hAT-derived progenitor cells viathe SDF-1/CXCR-4 axis within hAT.
MATERIALS AND METHODS
Isolation of the SVF from hATHuman AT was obtained from individuals undergoing plastic sur-gery by lipoaspiration. The study was approved by the ethicalcommittees of Toulouse and Frankfurt am Main hospital universi-ties. Subcutaneous AT was obtained from a total of 38 individuals(mean body mass index: 26.07 � 1.01 kg/m2). The AT was digestedusing collagenase (300 U/ml in phosphate-buffered saline [PBS],2% bovine serum albumin [BSA], pH 7.4) for 30 minutes underconstant shaking. Following removal of the floating mature adipo-cytes, the lower layer containing the SVF was centrifuged (300g, 10minutes) and the pellet resuspended in erythrocyte lysis buffer (155mmol/l NH4Cl; 5.7 mmol/l K2HPO4; 0.1 mmol/l EDTA, pH 7.3) for10 minutes. After successive filtrations through 100-, 70-, and40-�m sieves, the cells were resuspended in PBS/2% fetal calfserum (FCS).
Isolation of the CD34�/CD31� Progenitor Cells andthe CD34�/CD31� CECs from the SVF of hATCD34� cells were isolated from the SVF using CD34-coupledmagnetic microbeads as previously described [5]. Briefly, the SVFwas incubated (room temperature, 15 minutes) with the StemCellTechnologies (St. Katharinen, Germany, http://www.stemcell.com)positive selection cocktail. Following the addition of magneticnanoparticles, cells were recovered by successive magnetic sortingsteps. The CD34� cells were suspended in PBS/0.1% BSA. TheCD34� population was depleted from the CD31� cells using DynalBiotech (Hamburg, Germany, http://www.dynalbiotech.com) CD31-coupled magnetic microbeads (50 �l/ml). After incubation (4°C, 20minutes), the cell suspension containing the beads, suspended in 10ml of PBS/0.1% BSA, was exposed to the magnet for 1 minute. Themagnetic bead-free fraction, the CD34�/CD31� cells (hAT-derivedprogenitor cells), was collected, centrifuged (250g, 10 minutes), andsuspended in the culture medium and the CD34�/CD31� cellfraction (hAT-derived CECs) was removed from the magnet andsuspended in endothelial cell culture medium. The CD34�/CD31�
cell content of the selected fraction was 92.2% � 1.2%.
Cell CultureThe hAT-derived CECs were cultured in Endothelial Cell GrowthMedium MV (ECGM-MV) (PromoCell, Heidelberg, Germany,http://www.promocell.com) on human fibronectin coated culturedishes. Human AT-derived CECs were used until passage 2 [5, 15].The human AT-derived progenitor cells were cultured in Endothe-lial Cell Basal Medium (ECBM, which contains no growth factors)(PromoCell) supplemented with 10% FCS. Progenitor cells wereused until passage 1 [5]. Human umbilical vein endothelial cells(HUVECs) were isolated [16] and cultured until passage 8.
Preparation of Conditioned Media and Enzyme-Linked Immunosorbent AssayHuman AT-derived CECs, hAT-derived progenitor cells, hAT-res-ident macrophages (CD14�/CD31�) that were isolated as previ-ously described [15], and HUVECs were incubated with an equalamount of basal ECBM supplemented with 0.1% BSA for 24 hours.Mature adipocytes (400,000 cells) were included in fibrin gels (1.5mg fibrinogen/ml ECBM supplemented with 25 units/ml �-throm-bin) and cultured in ECBM supplemented with 0.1% BSA for 24hours. Control gels were prepared without adipocytes. The condi-tioned media were collected, centrifuged (1,000g, 10 minutes, 4°C),and stored at �70°C until use. Release of SDF-1 was measured inconditioned media by enzyme-linked immunosorbent assay accord-ing to the instructions of the manufacturer (RayBiotech Inc.,Norcross, GA, http://www.raybiotech.com). Note that the SDF-1levels were measured in conditioned media from P0, P1, and P2
human AT-derived CECs and no statistical significant differenceswere found (not shown).
Transmigration AssayTransmigration assays of hAT-derived progenitor cells were per-formed using 8-�m pore HTS FluoroBlock inserts (Falcon; BDBiosciences, San Diego, http://www.bdbiosciences.com) coatedwith bovine gelatin (0.15 mg/cm2). The progenitor cells wereloaded at 50,000 cells per insert (upper chamber) and conditionedmedia, SDF-1 in the presence or not of AMD3100 (Sigma-Aldrich,St. Louis, http://www.sigmaaldrich.com) or CXCR-4 neutralizingantibody (clone 12G5; R&D Systems Inc., Minneapolis, http://www.rndsystems.com), were added to the lower chamber. The cellswere allowed to migrate into the lower chamber for 20–24 hours.After the assays, the inserts were fixed (4% paraformaldehyde) andthe nuclei were stained with 4,6-diamidino-2-phenylindole. Forblocking experiments, the progenitor cells were preincubated withthe indicated inhibitors at 37°C for 1 hour. The cell transmigrationwas quantified by counting the number of nuclei using a computer-assisted microscope (Nikon, Dusseldorf, Germany, http://www.nikon.com) at three distinct positions.
RNA Extraction and Real-Time QuantitativePolymerase Chain Reaction AssayTotal RNAs were extracted from human mature adipocytes, hAT-CD34�/CD31� cells, hAT-macrophages, and hAT-CD34�/CD31�
CECs using the RNeasy kit (Qiagen, Hilden, Germany, http://www1.qiagen.com). RNA concentrations were determined using afluorometric assay (RiboGreen). RNA (0.5 �g) was reverse-tran-scribed using the SuperScript III RNase H RT system (Invitrogen,Carlsbad, CA, http://www.invitrogen.com) according to the manu-facturer’s instructions (random hexamers and deoxynucleoside-5�-triphosphates were from Life Technologies, Rockville, MD, http://www.lifetech.com). Reverse transcription was also performedwithout superscript enzyme on RNA samples to ensure the absenceof contaminating genomic DNA. Primers for human SDF-1(CXCL12), hypoxia inducible factor-1� (HIF-1�), and CXCR-4were provided by Applied Biosystems (Assay-On-Demand:Hs00171022_m1, Hs00153153_m1, and Hs00607978_m1, respec-tively; Foster City, CA, http://www.appliedbiosystems.com). Allamplification reactions were performed in duplicate from 20 ngcDNA using the Mx4000 Multiplex Quantitative PCR System(Stratagene, La Jolla, CA, http://www.stratagene.com) using thefollowing conditions: 50°C for 2 minutes and 95°C for 10 minutesfollowed by 40 cycles at 95°C for 15 seconds and 60°C for 1minute. Results were analyzed with Stratagene MX4000 software,and all values were normalized to the levels of the ribosomal RNAs.
Coculture Between hAT-Derived CECs andhAT-Derived CD34�/CD31� Progenitor CellsHuman AT-derived CECs plated at 60,000 cells per cm2 werecultured in ECGM-MV on human fibronectin-coated 48-well cellculture plates until confluence. Then, hAT-derived progenitor cellswere labeled with PHK26 according to the instructions of themanufacturer (Sigma-Aldrich) and 2,000–3,000 cells per well wereplaced in each well in ECBM/2% SVF. After 10 days of coculture,the cocultured cells were fixed and stained with the indicatedantibodies.
Three-Dimensional Coculture ModelHuman AT-derived CECs plated at 60,000 cells per cm2 werecultured in ECGM-MV on human fibronectin-coated 48-well cellculture plates. Once confluent, the hAT-CECs were covered with200 �l of unpolymerized BD Matrigel Matrix Growth Factor Re-duced (BD Biosciences) and incubated at 37°C for 30–45 minutes.Human AT-derived progenitor cells were harvested with trypsin andlabeled with PHK26 according to the instructions of the manufac-turer. The progenitor cells were resuspended in ECBM/0.1% BSAand 2,000–3,000 cells per well were loaded onto the matrigel-coated wells (200 �l of cell suspension per well). After 24 hours of“coculture,” progenitor cell tube formation (red fluorescent net-
2270 Adipose Tissue Stem and Endothelial Cell Crosstalk
at Inserm D
isc Doc on Septem
ber 7, 2007 w
ww
.StemC
ells.comD
ownloaded from
work) was assessed with a computer-assisted microscope (Nikon).Microphotographs of the center of each well were taken, and tubeformation was quantified by measuring the total tube length usingLucia image software (Nikon, Tokyo, http://www.nikon.com). Tubeformation of progenitor cells alone was used as a control. Forblocking experiments, the progenitor cells were preincubated withthe indicated inhibitors at 37°C for 1 hour.
ImmunocytochemistryCells were fixed with 4% paraformaldehyde and immunostainedwith anti-human CXCR-4 monoclonal antibody (Abcam, Cam-bridge, U.K., http://www.abcam.com) (1:50) or platelet endothelialcell adhesion molecule-1 (CD31) monoclonal antibody (Dako,Glostrup, Denmark, http://www.dako.com) (1:10) followed bystaining with fluorescently labeled secondary antibody (1:200) (In-vitrogen).
Statistical AnalysisData are expressed as mean � SEM from at least three independentexperiments. Statistical analysis was performed by Student’s t testor one-way analysis of variance followed by a Bonferroni post hoctest when appropriate. Differences were considered significantwhen p � .05.
RESULTS
AT-Derived CECs Induce the Chemotaxis of theCD34�/CD31� CellsThe effect of hAT-CECs on the migration of hAT-derived CD34�/CD31� cells was assessed in modified Boyden chamber assaysconsisting of two cell chambers separated by a polycarbonate filter(8-�m pores). The lower chamber was filled up with control mediaor 24 hour-conditioned media from confluent hAT-CECs, matureadipocytes, or confluent HUVECs, and the cell suspension ofhAT-derived CD34�/CD31� cells was placed in the upper cham-ber. Cells that migrated across the filter and attached to the underpart of the filter after 24 hours were counted after nuclei staining.As depicted in Figure 1A, conditioned media from mature adipo-cytes or from HUVECs did not induce any cell migration. Con-versely, conditioned media from hAT-CECs led to the robustmigration of the CD34�/CD31� cells (5.7 � 0.7-fold increase, n �21, p � .001). Human AT-CEC conditioned media failed to inducethe migration of hAT-derived CD34�/CD31� cells after proteindenaturation of the conditioned media (Fig. 1B). Moreover, thestimulatory effect of conditioned media derived from hAT-CECswas lost when the conditioned media were applied in the upperchamber together with the CD34�/CD31� cells. Taken together,the results showed that secreted proteins from hAT-CECs stimu-lated the directed chemotactic migration of the hAT-derivedCD34�/CD31� (Fig. 1B). In addition, preincubation of hAT-de-rived progenitor cells with increasing concentrations of pertussistoxin (PTX) (200 and 400 ng/ml) led to a concentration-dependentinhibition of the hAT-CEC-mediated migration (76% � 9% ofinhibition for 400 ng/ml of PTX, p � .001, n � 4) (Fig. 1B).
The CXCR-4/SDF-1 Axis Is Involved in theChemotaxis of the CD34�/CD31� Cells Mediated byAT-Derived CECsTo further identify the nature of the factor(s) involved in suchan effect, the CD34�/CD31� cells were preincubated with aCXCR-4 antagonist (AMD3100, 10 �g/ml) or with neutralizinganti-CXCR-4 antibody (10 �g/ml), and the effect of hAT-CECconditioned media was then assessed. As shown in Figure 2, the
Figure 1. Human adipose tissue (AT)-derived CECs induce the che-motaxis of hAT-derived CD34�/CD31� progenitor cells. (A): Trans-well migration of human AT-derived progenitor cells in the presence ofcontrol medium (Control) or conditioned media from adipocytes (n �14), hAT-CECs (n � 21), and HUVECs (n � 3). � p � .001 whencompared with control migration. (B): Transwell migration of hAT-derived progenitor cells in response to hAT-CEC conditioned mediumin the lower chamber (hAT-CEC conditioned medium), in the upperchamber (upper chamber hAT-CECs) (n � 3), after 1-hour preincuba-tion with increasing concentrations of pertussis toxin (n � 4) or afterprotein denaturated hAT-CEC-conditioned medium (heated AT-CECs)(n � 4). � p � .01, �� p � .001 when compared with migration inducedby hAT-CEC-conditioned medium. Abbreviations: CECs, capillary en-dothelial cells; hAT, human adipose tissue; HUVECS, human umbilicalvein endothelial cells; PTX, pertussis toxin.
Figure 2. The stromal derived factor-1/CXCR-4 axis is involved in thehAT-derived progenitor cell chemotaxis toward hAT-CEC conditionedmedia. Transwell migration of hAT-derived progenitor cells in thepresence of hAT-derived CEC-conditioned media after 1 hour of pre-incubation with 10 �g/ml of the CXCR-4 antagonist AMD3100(�AMD3100) (n � 6) or with 10 �g/ml of an anti-CXCR-4 neutralizingantibody (�Anti-CXCR-4) (n � 6). � p � .01 when compared withmigration induced by hAT-CEC-conditioned media. Abbreviations:CECs, capillary endothelial cells; hAT, human adipose tissue.
2271Sengenes, Miranville, Maumus et al.
www.StemCells.com
at Inserm D
isc Doc on Septem
ber 7, 2007 w
ww
.StemC
ells.comD
ownloaded from
hAT-CEC-mediated chemotaxis was suppressed when the pro-genitor cells were incubated with AMD3100 or with anti-
CXCR-4 neutralizing antibody (77% � 11% and 96% � 4% ofinhibition, respectively, p � .01, n � 6). Moreover, as depictedin Figure 3A, increasing concentrations of recombinant SDF-1�or -1�, ligands for the CXCR-4, induced a concentration-depen-dent chemotaxis of hAT-derived progenitor cells that was sig-nificantly inhibited when the progenitor cells were preincubatedwith AMD3100 (58% � 7% of inhibition, p � .01, n � 3) orneutralizing anti-CXCR-4 antibody (72% � 3% of inhibition,p � .01, n � 3) (Fig. 3B). Of note, the chemotaxis of theCD34�/CD31� cells toward granulocyte monocyte colonystimulating factor (20–100 ng/ml), stem cell factor (20–100ng/ml), monocyte chemoattractant protein 1 (1–20 ng/ml), andinterleukin 8 (20 ng/ml) was also evaluated, and none of thechemokines induced a statistically significant migration (datanot shown).
CXCR-4 Is Expressed on CD34�/CD31� Cells andSDF-1 Is Produced by hAT-Derived CECsThe expression of CXCR-4 in hAT-derived CD34�/CD31�
cells was then investigated by real-time polymerase chain reac-tion (PCR) analysis. The results showed that transcripts forCXCR-4 were present in hAT-derived CD34�/CD31� cells(Fig. 4B). Moreover, as presented in Figure 4A, the CXCR-4protein expression was detected by immunocytochemistry onhAT-CD34�/CD31� cells. Additionally, there was a positivecorrelation between the level of transcripts encoding forCXCR-4 in hAT progenitor cells and the one of HIF-1� in thecorresponding hAT-CECs (Fig. 4B, p � .04, n � 14). Inparallel, SDF-1 mRNA expression was assessed in hAT-derivedCECs and compared with the one of HUVECs and other cellsfrom the SVF such as hAT-macrophages. As depicted in Figure4C, the SDF-1 mRNA was mainly expressed by hAT-derivedCECs and was below Ct values of 40 in HUVECs. Consistentwith the mRNA expression, SDF-1 release was significantlyhigher in hAT-derived CECs as compared with other cell frac-tions present in AT-derived SVF such as macrophages (412.2 �92.9 pg/ml vs. 34.4 � 18.1 pg/ml, respectively, p � .001, n �3–9) (Fig. 4D). The production of SDF-1 by HUVECs was 42
Figure 3. Functionality of the CXCR-4/SDF-1 axis in human adiposetissue. (A): Human adipose tissue (AT)-derived progenitor cells wereexposed to increasing concentrations of SDF-1� or SDF-1� (10–100ng/ml), and transwell migration was evaluated after 24 hours (n � 3–8);� p � .01 and �� p � .001 compared with control. (B): HumanAT-derived progenitor cells were preincubated (1 hour) with 10 �g/mlCXCR-4 antagonist AMD3100 (n � 3) or with 10 �g/ml anti-CXCR-4neutralizing antibody (n � 3). Transwell migration toward 100 ng/mlSDF-1� was then evaluated after 24 hours. $ p � .001 compared withcontrol, � p � .01 and �� p � .001 compared with SDF-1� inducedmigration. Abbreviation: SDF-1, stromal derived factor-1.
Figure 4. Expression of SDF-1 andCXCR-4 in hAT. (A): Upper panel: hAT-derived CD34�/CD31� progenitor cellswere fixed and stained with antibodies di-rected against CXCR-4 receptor followedby fluorescein isothiocyanate (FITC)-con-jugated secondary antibodies. Lower panel:hAT-CD34�/CD31� cells were stainedwith FITC-conjugated secondary antibodies.Representative fluorescence microscopyanalysis is shown from n � 3. (B): Correla-tion between mRNA levels of CXCR-4 inhAT-progenitor cells and HIF-1 levels inhAT-CECs quantified by real-time polymer-ase chain reaction (PCR) experiments (p �.04, n � 14). (C): SDF-1 mRNA expressionin hAT-macrophages (n � 3), HUVECs(n � 4), and hAT-CECs (n � 6) quantifiedby real-time PCR experiments. (D): En-zyme-linked immunosorbent assay of 24-hour conditioned media from hAT-macro-phages (n � 3), HUVECs (n � 4), andhAT-CECs (n � 9). � p � .05 versus mac-rophage SDF-1 secretion. Abbreviations:A.U., arbitrary units; CECs, capillary endo-thelial cells; hAT, human adipose tissue;HIF-1, hypoxia inducible factor-1; HUVECS,human umbilical vein endothelial cells;SDF-1, stromal derived factor-1.
2272 Adipose Tissue Stem and Endothelial Cell Crosstalk
at Inserm D
isc Doc on Septem
ber 7, 2007 w
ww
.StemC
ells.comD
ownloaded from

times lower than the one of hAT-CECs (9.84 � 3.81 pg/ml,n � 4).
AT-Derived CECs Support the Differentiation ofAT-Derived Progenitor Cells Toward an EndothelialCell PhenotypeCoculture experiments of hAT-derived CECs and CD34�/CD31� cells were performed. Human AT-derived CD34�/CD31� cells were labeled with PKH26 and seeded onto con-fluent hAT-derived CECs (Fig. 5A). After 10 days of coculture,CD31 expression in the formal CD31 negative AT-derivedprogenitor cells (Fig. 5B) was analyzed. hAT-derived CECsformed a dense CD31 positive network into which PKH26positive AT-derived progenitor cells incorporated and expressedthe CD31 marker (Fig. 5C). To further analyze the effect of thehAT-derived CECs on the differentiation of the CD34�/CD31�
cells, a three-dimensional coculture model was developed.PKH26 labeled CD34�/CD31� cells were seeded on the top of
a growth factor-reduced matrigel under which confluent hAT-derived CECs were present or not. As shown in Figure 6A,CD34�/CD31� cells alone did neither migrate nor form capil-lary-like structures. Interestingly, the presence of hAT-derivedECs under the matrigel dramatically promoted the organizationof hAT-CD34�/CD31� cells into branched structures andpseudotubes (360% � 48% of the total length obtained withhAT-CD34�/CD31� cells alone, p � .01, n � 7) (Fig. 6B).Given the chemotactic effect mediated by the CXCR-4/SDF-1axis, we studied the involvement of this system in the hAT-CEC-dependent tube formation by using a neutralizing anti-CXCR-4 antibody, the CXCR-4 antagonist AMD3100, or per-tussis toxin (not shown). As depicted in Figure 6C and 6D, thetube formation induced by hAT-derived CECs was significantlyinhibited in a concentration-dependent manner by the inhibitors(24% � 12% and 67% � 5% of inhibition with 10 �g/ml and100 �g/ml AMD3100, respectively; 41% � 15% and 93% �2% of inhibition for 10 �g/ml and 20 �g/ml neutralizing anti-CXCR-4 antibody, respectively; 85% � 0.5% of inhibition for200 ng/ml pertussis toxin, n � 4–7).
DISCUSSION
Although reports show that adipogenesis and angiogenesis aretightly correlated during the growth of fat mass [3, 17–20], littleis known about the mechanisms that regulate the extension of itsvasculature. In the present study we report that hAT-derivedCECs induce the chemotaxis of hAT-derived progenitor cells.Moreover, we demonstrate that the presence of hAT-derivedCECs promotes the differentiation and organization of hAT-derived hAT-CD34�/CD31� progenitor cells into capillary-likestructures via a SDF-1/CXCR-4-dependent pathway.
In the last years, several reports have contributed to theevidence that the SVF cells are phenotypically heterogeneousand can express under specific culture conditions adipogenic,osteogenic, chondrogenic, and myogenic lineage markers butalso endothelial, epithelial, and neural markers [5, 8, 21–23].We have previously shown that native immunoselected CD34�/CD31� cells present in the hAT-derived SVF display angio-genic and adipogenic potentials. Indeed, this cell population isable to differentiate into adipocyte-like cells [8] and into endo-thelial-like cells in vitro and in vivo to promote the neovascu-larization of ischemic muscle in mice [5]. Similar results werealso described using adherent hAT-SVF [6, 7]. The mechanismsunderlying such proangiogenic effects are still to be defined.Although the hAT-derived progenitor cells were shown to inte-grate into vessels in vivo [5, 6], they could also contribute toneovascularization through the production of proangiogenic fac-tors [7, 24]. Nevertheless, such a population of progenitor cellsmay participate locally to the extension of the blood capillarynetwork known to be associated with fat mass development. Thepresent study was undertaken to analyze the potential crosstalkwithin the hAT between hAT-CD34�/CD31� progenitor cellsand CECs. Indeed, the EC barrier has already been shown tointeract with progenitor/stem cells [25–27]. Human AT-derivedCECs specifically stimulated the migration of hAT-derivedCD34�/CD31� progenitor cells through the release of solubleproteins. Human AT-derived CECs induced a chemotactic re-sponse since no random motility was observed when the cellswere incubated in the same compartment with endothelial cell-derived conditioned media. Moreover, since hAT-derivedCD34�/CD31� progenitor cell chemotaxis was inhibited in thepresence of pertussis toxin, which specifically prevents activa-tion of Gi proteins coupled to chemokine receptors, the involve-ment of a chemokine ligand/receptor interaction was strongly
Figure 5. Human AT-derived progenitor cells incorporate into thenetwork formed by hAT-CECs and express platelet endothelial celladhesion molecule-1 (PECAM-1). (A): Confluent hAT-CECs werefixed and stained with antibodies directed against PECAM-1 followedby fluorescein isothiocyanate (FITC)-conjugated secondary antibodies.(B): Confluent PKH26 hAT-CD34�/CD31� progenitor cells werefixed and stained with antibodies against PECAM-1 followed by FITC-conjugated secondary antibodies. (C): Confluent hAT-CECs were cul-tured in the presence of PKH26-labeled hAT-derived progenitor cellsfor 10 days of coculture in endothelial basal medium/2% fetal calfserum. Cells were fixed and stained with antibodies directed againstPECAM-1 followed by FITC-conjugated secondary antibodies. Repre-sentative fluorescence microscopy analysis is shown from n � 3 inde-pendent experiments. Abbreviations: CECs, capillary endothelial cells;DAPI, 4,6-diamidino-2-phenylindole; hAT, human adipose tissue.
2273Sengenes, Miranville, Maumus et al.
www.StemCells.com
at Inserm D
isc Doc on Septem
ber 7, 2007 w
ww
.StemC
ells.comD
ownloaded from
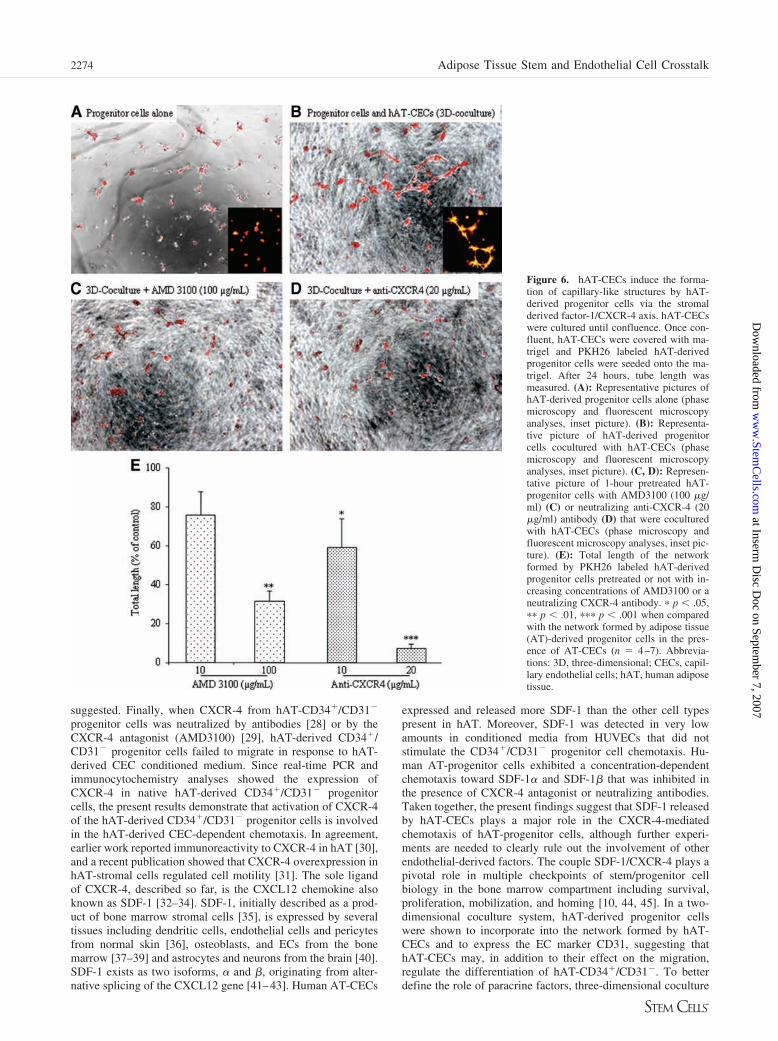
suggested. Finally, when CXCR-4 from hAT-CD34�/CD31�
progenitor cells was neutralized by antibodies [28] or by theCXCR-4 antagonist (AMD3100) [29], hAT-derived CD34�/CD31� progenitor cells failed to migrate in response to hAT-derived CEC conditioned medium. Since real-time PCR andimmunocytochemistry analyses showed the expression ofCXCR-4 in native hAT-derived CD34�/CD31� progenitorcells, the present results demonstrate that activation of CXCR-4of the hAT-derived CD34�/CD31� progenitor cells is involvedin the hAT-derived CEC-dependent chemotaxis. In agreement,earlier work reported immunoreactivity to CXCR-4 in hAT [30],and a recent publication showed that CXCR-4 overexpression inhAT-stromal cells regulated cell motility [31]. The sole ligandof CXCR-4, described so far, is the CXCL12 chemokine alsoknown as SDF-1 [32–34]. SDF-1, initially described as a prod-uct of bone marrow stromal cells [35], is expressed by severaltissues including dendritic cells, endothelial cells and pericytesfrom normal skin [36], osteoblasts, and ECs from the bonemarrow [37–39] and astrocytes and neurons from the brain [40].SDF-1 exists as two isoforms, � and �, originating from alter-native splicing of the CXCL12 gene [41–43]. Human AT-CECs
expressed and released more SDF-1 than the other cell typespresent in hAT. Moreover, SDF-1 was detected in very lowamounts in conditioned media from HUVECs that did notstimulate the CD34�/CD31� progenitor cell chemotaxis. Hu-man AT-progenitor cells exhibited a concentration-dependentchemotaxis toward SDF-1� and SDF-1� that was inhibited inthe presence of CXCR-4 antagonist or neutralizing antibodies.Taken together, the present findings suggest that SDF-1 releasedby hAT-CECs plays a major role in the CXCR-4-mediatedchemotaxis of hAT-progenitor cells, although further experi-ments are needed to clearly rule out the involvement of otherendothelial-derived factors. The couple SDF-1/CXCR-4 plays apivotal role in multiple checkpoints of stem/progenitor cellbiology in the bone marrow compartment including survival,proliferation, mobilization, and homing [10, 44, 45]. In a two-dimensional coculture system, hAT-derived progenitor cellswere shown to incorporate into the network formed by hAT-CECs and to express the EC marker CD31, suggesting thathAT-CECs may, in addition to their effect on the migration,regulate the differentiation of hAT-CD34�/CD31�. To betterdefine the role of paracrine factors, three-dimensional coculture
Figure 6. hAT-CECs induce the forma-tion of capillary-like structures by hAT-derived progenitor cells via the stromalderived factor-1/CXCR-4 axis. hAT-CECswere cultured until confluence. Once con-fluent, hAT-CECs were covered with ma-trigel and PKH26 labeled hAT-derivedprogenitor cells were seeded onto the ma-trigel. After 24 hours, tube length wasmeasured. (A): Representative pictures ofhAT-derived progenitor cells alone (phasemicroscopy and fluorescent microscopyanalyses, inset picture). (B): Representa-tive picture of hAT-derived progenitorcells cocultured with hAT-CECs (phasemicroscopy and fluorescent microscopyanalyses, inset picture). (C, D): Represen-tative picture of 1-hour pretreated hAT-progenitor cells with AMD3100 (100 �g/ml) (C) or neutralizing anti-CXCR-4 (20�g/ml) antibody (D) that were coculturedwith hAT-CECs (phase microscopy andfluorescent microscopy analyses, inset pic-ture). (E): Total length of the networkformed by PKH26 labeled hAT-derivedprogenitor cells pretreated or not with in-creasing concentrations of AMD3100 or aneutralizing CXCR-4 antibody. � p � .05,�� p � .01, ��� p � .001 when comparedwith the network formed by adipose tissue(AT)-derived progenitor cells in the pres-ence of AT-CECs (n � 4–7). Abbrevia-tions: 3D, three-dimensional; CECs, capil-lary endothelial cells; hAT, human adiposetissue.
2274 Adipose Tissue Stem and Endothelial Cell Crosstalk
at Inserm D
isc Doc on Septem
ber 7, 2007 w
ww
.StemC
ells.comD
ownloaded from

experiments were performed. The results clearly showed thathAT-derived progenitor cells migrate and assemble into capil-lary-like structures only in the presence of hAT-CECs. More-over, when CXCR-4 either was antagonized or neutralized,hAT-progenitor cells failed to appropriately migrate and assem-ble into tubular structures on matrigel substrate.
Taken together, the present results show that hAT-derivedCECs, through their production of paracrine factors such asSDF-1, modulate the migration but also the differentiation andorganization of the hAT-derived CD34�/CD31� progenitorcells through the activation of CXCR-4. CXCR-4 mRNA levelsare known to be upregulated by hypoxia through HIF-1 activa-tion via the hypoxia responsive element in the 5� region of theCXCR4 gene [46, 47]. In the murine AT, hypoxic areas havebeen shown to be associated with obesity [48]. Interestingly, themRNA levels of CXCR-4 from the CD34�/CD31� were foundto be positively correlated to HIF-1� transcripts of the CECs. Arecent publication has reported that the recruitment of CXCR-4positive progenitor cells to regenerating foci or tissues wasmediated by hypoxic gradients via HIF-1 induced expression ofCXCR-4/SDF-1 axis [28]. Moreover, SDF-1 expression in isch-emic tissue has been shown to primarily localize in ECs [36]. Itis thus tempting to speculate that hypoxia within the hAT maypromote the migration of resident hAT-derived CD34�/CD31�
progenitor cells at active sites of neovascularization in the fatmass.
All together, the present results show that the migration anddifferentiation of hAT-derived progenitor cells are modulatedby factors originating from hAT-derived CECs. The SDF-1/CXCR-4 axis appears to play a key role. Such an effect may beinvolved in the recruitment of hAT-derived progenitor cells tofoci where SDF-1 is highly expressed such as neovasculariza-tion sites to promote the extension of the capillary network thatoccurs during the growth of the fat mass.
ACKNOWLEDGMENTS
We are grateful to Valerie Wegner and Pauline Decaunes fortheir excellent technical assistance. We also thank Dr. MarieSanson for her special help with HUVECs. This work wassupported by grants from AVENIR INSERM, Alexander vonHumboldt Foundation, and Sofja Kovalevskaja Price (HumboldtFoundation and the German Federal Ministry of Education andResearch).
DISCLOSURE OF POTENTIAL CONFLICTS
OF INTEREST
The authors indicate no potential conflicts of interest.
REFERENCES
1 Hirsch J, Batchelor B. Adipose tissue cellularity in human obesity. ClinEndocrinol Metab 1976;5:299–311.
2 Bjorntorp P, Sjostrom L. Adipose tissue cellularity. Int J Obes 1979;3:181–187.
3 Crandall DL, Hausman GJ, Kral JG. A review of the microcirculation ofadipose tissue: Anatomic, metabolic, and angiogenic perspectives. Mi-crocirculation 1997;4:211–232.
4 Bouloumie A, Lolmede K, Sengenes C et al. Angiogenesis in adiposetissue. Ann Endocrinol (Paris) 2002;63:91–95.
5 Miranville A, Heeschen C, Sengenes C et al. Improvement of postnatalneovascularization by human adipose tissue-derived stem cells. Circula-tion 2004;110:349–355.
6 Planat-Benard V, Silvestre JS, Cousin B et al. Plasticity of humanadipose lineage cells toward endothelial cells: Physiological and thera-peutic perspectives. Circulation 2004;109:656–663.
7 Rehman J, Traktuev D, Li J et al. Secretion of angiogenic and antiapop-totic factors by human adipose stromal cells. Circulation 2004;109:1292–1298.
8 Sengenes C, Lolmede K, Zakaroff-Girard A et al. Preadipocytes in thehuman subcutaneous adipose tissue display distinct features from theadult mesenchymal and hematopoietic stem cells. J Cell Physiol 2005;205:114–122.
9 Aiuti A, Webb IJ, Bleul C et al. The chemokine SDF-1 is a chemoat-tractant for human CD34� hematopoietic progenitor cells and providesa new mechanism to explain the mobilization of CD34� progenitors toperipheral blood. J Exp Med 1997;185:111–120.
10 Lapidot T, Kollet O. The essential roles of the chemokine SDF-1 and itsreceptor CXCR4 in human stem cell homing and repopulation of trans-planted immune-deficient NOD/SCID and NOD/SCID/B2m(null) mice.Leukemia 2002;16:1992–2003.
11 Son BR, Marquez-Curtis LA, Kucia M et al. Migration of bone marrowand cord blood mesenchymal stem cells in vitro is regulated by stromal-derived factor-1-CXCR4 and hepatocyte growth factor-c-met axes andinvolves matrix metalloproteinases. STEM CELLS 2006;24:1254–1264.
12 Fuchs E, Tumbar T, Guasch G. Socializing with the neighbors: Stemcells and their niche. Cell 2004;116:769–778.
13 Shen Q, Goderie SK, Jin L et al. Endothelial cells stimulate self-renewaland expand neurogenesis of neural stem cells. Science 2004;304:1338–1340.
14 Kaplan RN, Psaila B, Lyden D. Niche-to-niche migration of bone-marrow-derived cells. Trends Mol Med 2007;13:72–81.
15 Curat CA, Miranville A, Sengenes C et al. From blood monocytes toadipose tissue-resident macrophages: Induction of diapedesis by humanmature adipocytes. Diabetes 2004;53:1285–1292.
16 Busse R, Lamontagne D. Endothelium-derived bradykinin is responsiblefor the increase in calcium produced by angiotensin-converting enzymeinhibitors in human endothelial cells. Naunyn Schmiedebergs Arch Phar-macol 1991;344:126–129.
17 Zhang QX, Magovern CJ, Mack CA et al. Vascular endothelial growthfactor is the major angiogenic factor in omentum: Mechanism of theomentum-mediated angiogenesis. J Surg Res 1997;67:147–154.
18 Fukumura D, Ushiyama A, Duda DG et al. Paracrine regulation ofangiogenesis and adipocyte differentiation during in vivo adipogenesis.Circ Res 2003;93:e88–97.
19 Brakenhielm E, Cao R, Gao B et al. Angiogenesis inhibitor, TNP-470,prevents diet-induced and genetic obesity in mice. Circ Res 2004;94:1579–1588.
20 Neels JG, Thinnes T, Loskutoff DJ. Angiogenesis in an in vivo model ofadipose tissue development. FASEB J 2004;18:983–985.
21 Zuk PA, Zhu M, Mizuno H et al. Multilineage cells from human adiposetissue: implications for cell-based therapies. Tissue Eng 2001;7:211–228.
22 De Ugarte DA, Alfonso Z, Zuk PA et al. Differential expression of stemcell mobilization-associated molecules on multi-lineage cells from adi-pose tissue and bone marrow. Immunol Lett 2003;89:267–270.
23 Rodriguez AM, Elabd C, Amri EZ et al. The human adipose tissue is asource of multipotent stem cells. Biochimie 2005;87:125–128.
24 Nakagami H, Maeda K, Morishita R et al. Novel autologous cell therapyin ischemic limb disease through growth factor secretion by culturedadipose tissue-derived stromal cells. Arterioscler Thromb Vasc Biol2005;25:2542–2547.
25 Papayannopoulou T. Bone marrow homing: The players, the playfield,and their evolving roles. Curr Opin Hematol 2003;10:214–219.
26 Lapidot T, Dar A, Kollet O. How do stem cells find their way home?Blood 2005;106:1901–1910.
27 Schmidt A, Ladage D, Steingen C et al. Mesenchymal stem cells trans-migrate over the endothelial barrier. Eur J Cell Biol 2006;85:1179–1188.
28 Ceradini DJ, Kulkarni AR, Callaghan MJ et al. Progenitor cell traffickingis regulated by hypoxic gradients through HIF-1 induction of SDF-1. NatMed 2004;10:858–864.
29 De Clercq E. Inhibition of HIV infection by bicyclams, highly potent andspecific CXCR4 antagonists. Mol Pharmacol 2000;57:833–839.
30 Hazan U, Romero IA, Cancello R et al. Human adipose cells expressCD4, CXCR4, and CCR5 [corrected] receptors: A new target cell typefor the immunodeficiency virus-1? FASEB J 2002;16:1254–1256.
31 Cho HH, Kyoung KM, Seo MJ et al. Overexpression of CXCR4 in-creases migration and proliferation of human adipose tissue stromal cells.Stem Cells Dev 2006;15:853–864.
32 Kucia M, Reca R, Miekus K et al. Trafficking of normal stem cells andmetastasis of cancer stem cells involve similar mechanisms: Pivotal roleof the SDF-1-CXCR4 axis. STEM CELLS 2005;23:879–894.
33 Stellos K, Gawaz M. Platelets and stromal cell-derived factor-1 inprogenitor cell recruitment. Semin Thromb Hemost 2007;33:159–164.
2275Sengenes, Miranville, Maumus et al.
www.StemCells.com
at Inserm D
isc Doc on Septem
ber 7, 2007 w
ww
.StemC
ells.comD
ownloaded from

34 Busillo JM, Benovic JL. Regulation of CXCR4 signaling. BiochimBiophys Acta 2007;1768:952–963.
35 Bleul CC, Farzan M, Choe H et al. The lymphocyte chemoattractantSDF-1 is a ligand for LESTR/fusin and blocks HIV-1 entry. Nature1996;382:829–833.
36 Salvucci O, Yao L, Villalba S et al. Regulation of endothelial cellbranching morphogenesis by endogenous chemokine stromal-derivedfactor-1. Blood 2002;99:2703–2711.
37 Imai K, Kobayashi M, Wang J et al. Selective secretion of chemoattrac-tants for haemopoietic progenitor cells by bone marrow endothelial cells:A possible role in homing of haemopoietic progenitor cells to bonemarrow. Br J Haematol 1999;106:905–911.
38 Ponomaryov T, Peled A, Petit I et al. Induction of the chemokinestromal-derived factor-1 following DNA damage improves human stemcell function. J Clin Invest 2000;106:1331–1339.
39 Yun HJ, Jo DY. Production of stromal cell-derived factor-1 (SDF-1) andexpression of CXCR4 in human bone marrow endothelial cells. J KoreanMed Sci 2003;18:679–685.
40 Tanabe S, Heesen M, Yoshizawa I et al. Functional expression of theCXC-chemokine receptor-4/fusin on mouse microglial cells and astro-cytes. J Immunol 1997;159:905–911.
41 Tashiro K, Tada H, Heilker R et al. Signal sequence trap: A cloning
strategy for secreted proteins and type I membrane proteins. Science1993;261:600–603.
42 Shirozu M, Nakano T, Inazawa J et al. Structure and chromosomallocalization of the human stromal cell-derived factor 1 (SDF1) gene.Genomics 1995;28:495–500.
43 De La Luz Sierra M, Yang F, Narazaki M et al. Differential processingof stromal-derived factor-1alpha and stromal-derived factor-1beta ex-plains functional diversity. Blood 2004;103:2452–2459.
44 Lapidot T, Petit I. Current understanding of stem cell mobilization: Theroles of chemokines, proteolytic enzymes, adhesion molecules, cyto-kines, and stromal cells. Exp Hematol 2002;30:973–981.
45 Aicher A, Zeiher AM, Dimmeler S. Mobilizing endothelial progenitorcells. Hypertension 2005;45:321–325.
46 Staller P, Sulitkova J, Lisztwan J et al. Chemokine receptor CXCR4downregulated by von Hippel-Lindau tumour suppressor pVHL. Nature2003;425:307–311.
47 Zagzag D, Lukyanov Y, Lan L et al. Hypoxia-inducible factor 1 andVEGF upregulate CXCR4 in glioblastoma: Implications for angiogenesisand glioma cell invasion. Lab Invest 2006;86:1221–1232.
48 Hosogai N, Fukuhara A, Oshima K et al. Adipose tissue hypoxia inobesity and its impact on adipocytokine dysregulation. Diabetes 2007;56:901–911.
2276 Adipose Tissue Stem and Endothelial Cell Crosstalk
at Inserm D
isc Doc on Septem
ber 7, 2007 w
ww
.StemC
ells.comD
ownloaded from

Titre et résumé en anglais
Matrix metalloproteases 2 and 9 and the differentiation of human adipose
tissue progenitor cells
Human adipose tissue (AT) has long attracted attention because of its capacity for
excessive development and regression. Cellular and matrix remodelling can explain human
AT plasticity. The matrix metalloproteinase (MMP) family is considered to play a major role
in tissue remodelling. In a previous report, we demonstrated that the stroma-vascular fraction
(SVF) of human AT produce and release MMP-2 and -9 during adipocyte differentiation in
vitro. In this work, we evidenced that decrease of MMP-9 activity by batimastat and
expression by HIV protease inhibitors decrease adipocyte differentiation of SVF cells. We
showed that the way in which HIV protease inhibitor affect MMP-9 expression involve
perturbations of proteasome and transcription factor NF-κB activities. Since the SVF of
human AT represents a heterogeneous cell population containing capillary endothelial cells,
inflammatory cells and progenitor cells that are able, under appropriate culture conditions, to
express adipogenic and angiogenic abilities, we would like to determine MMP-9 involvement
in the fate of progenitor cells. We defined a new culture condition in which progenitor cells
express simultaneously adipogenic and angiogenic abilities. These data strongly suggests that
MMP-9 modulate the differentiation of human progenitor cells and contribute to human AT
remodelling and thus in this excessive development and/or regression.
104

105
Auteur : Sandra DE BARROS
Titre : Les métalloprotéases matricielles 2 et 9 et la différenciation des cellules progénitrices
du tissu adipeux humain
Directeur de thèse : Dr Jean GALITZKY
Lieu et date de soutenance : Toulouse, le 29 octobre 2007 Résumé : Le tissu adipeux (TA) peut se développer de manière excessive (obésité) mais
aussi régresser (lipoatrophie). Cette plasticité peut s’expliquer par un remodelage cellulaire et
matriciel. Les métalloprotéases matricielles (MMPs) sont des acteurs majeurs du remodelage
tissulaire. Des travaux précédents menés au sein de l’équipe ont montré que les cellules de la
fraction stroma vasculaire (FSV) du TA humain sécrètent les MMP-2 et -9 au cours de la
différenciation adipocytaire in vitro. Durant ce travail, nous avons observé que l’inhibition de
l’activité de MMP-9 par le batimastat et de son expression par les inhibiteurs de la protéase
du VIH limite la différenciation adipocytaire des cellules de la FSV. L’étude des mécanismes
moléculaires responsables de la diminution de l’expression de MMP-9 par les inhibiteurs de
la protéase du VIH a montré que l’expression de MMP-9 est régulée par une voie impliquant
l’activité du protéasome et le facteur de transcription NF-κB. Sachant que la FSV est une
population cellulaire hétérogène contenant des cellules endothéliales, des cellules immuno-
inflammatoires et des cellules progénitrices aux capacités adipogéniques et angiogéniques,
nous avons souhaité déterminer l’implication de MMP-9 dans le devenir des cellules
progénitrices. Pour cela, une condition de culture permettant simultanément l’expression des
capacités adipogéniques et angiogéniques des cellules progénitrices a été mise au point.
L’ensemble de ce travail indique que MMP-9 influence la différenciation des cellules
progénitrices du TA humain et contribuent au remodelage du TA lors de son développement
et/ou de sa régression. Mots clés : tissu adipeux, MMP, adipogenèse, cellules progénitrices Discipline : Innovation pharmacologique Intitulé et adresse du laboratoire :
Equipe 1/Avenir - Réseau vasculaire, cellules progénitrices et cellules immnunitaires du tissu
adipeux- INSERM U858/I2MR, 37 Allées Jules Guesde, 31000 Toulouse





























