Embed Size (px)
Citation preview

♥ Labbé Tyze et Soit Franc et Sage ♥12/02/2014UE 7 Tissu Sanguin, Hémostase primaire, Dr GUILLET BenoîtUn diaporama est disponible pour ceux qui le souhaitent mais nous l’avons entièrement inséré dans la frappe pour une meilleure compréhension et afin de concentrer le cours sur un seul support.
L’HÉMOSTASE PRIMAIRE
I- Les principaux acteurs
1-La paroi vasculaire :
- Les Cellules endothéliales - Le Sous-Endothélium : Tissu conjonctif avec fibres en particulier le collagène, des cellules telles que fibroblastes, fibres musculaires lisses (pour la vasoconstriction), macrophages … Le collagène sert surtout pour l'hémostase primaire, les cellules pour la coagulation plasmatique.
2-Le sang :
- Les Cellules sanguines : - Plaquettes : cellules centrales de l’hémostase primaire - Erythrocytes (GR) et leucocytes (GB)
- Le Plasma : Les 2 protéines de la coagulation les plus importantes pour l’hémostase primaire sont :
- Facteur Willebrand (grosse protéine)- Fibrinogène
II-Les étapes de l'hémostase primaireL'hémostase primaire est une phase vasculo-plaquettaire avec différentes étapes:
- La formation du clou plaquettaire - Vasoconstriction réflexe, immédiate et localisée (1er phénomène, mais très bref) : le
vaisseau lésé diminue son diamètre donc diminue la quantité de sang passant près de la brèche.
1/18

1-La formation du clou plaquettaire
Succession de 3 mécanismes plaquettaires : = Adhésion, Activation et Sécrétion puis Agrégation
a-L'Adhésion plaquettaire
On a une brèche vasculaire au niveau de laquelle le sous-endothélium est en contact avec le sang. L’adhésion des plaquettes au Sous-endothélium se fait grâce aux : - Glycoprotéines plaquettaires membranaires : GPIb-V-IX (permet la 1ère attache puis ) GPIa-IIa et GPVI - Facteur von Willebrand (vWF) - Collagène sous-endothélial
Schéma : On a une plaquette quiescente avec a la surface des glycoprotéines de surfaces ( GPIb-V-IX, GPIa-IIa, GP-VI ). La GPIIb-IIIa servira pour l’agrégation plaquettaire.Les granules denses alpha serviront pour la sécrétion.Le Facteur Willebrand est un multimère de haut poids moléculaire et qui circule sous forme globulaire.Le collagène qui est dans le sous endothélium. Le FW vient se collé sur le collagène et va s'étaler (changement de conformation). La plaquette elle circule en roulant, en rebondissant sur les cellules endothéliales, ici il n'y a plus de cellule endothéliale quand elle rebondit mais du collagène avec FW. Leurs Rc GPIb-V-IX se lie au FW, ceci entraîne un ralentissement de la plaquette, l’arrêt complet se fait grâce a GPIa-IIa et GPI-VI (Rc directs du collagène).Ces plaquettes immobilisées vont s'activer grâce à des signaux intracellulaires (notamment mobilisation du Ca).
2/18

b-L'Activation (et sécrétion) plaquettaire
L'adhésion plaquettaire entraîne l’Activation plaquettaire
L'activation plaquettaire comporte 4 éléments :
- Changement de forme = étalement avec émission de pseudopodes
- Sécrétion (excrétion) du contenu des granules denses et α-plaquettaires:
Il y a libération de plus de 1000 molécules différentes qui vont agir pour l’hémostase primaire (= pro-hémostase primaire) :- ADP qui va aller activer (recruter) les autres plaquettes circulantes - Sérotonine, Thromboxane A2 pour renforcer la vasoconstriction
- Modification conformationnelle de la GPIIb-IIIa à la surface plaquetaire
Le fait pour permettre l'agrégation plaquettaire
- Flip-Flop
C'est le retournement de la membrane cytoplasmique, face interne vers le milieu extra-cellulaire. Grâce a mécanisme enzymatique actif.Exposition externe de phospholipides anioniques = plate-forme fonctionnelle pour les protéines de la coagulation plasmatique.
Comme toute cellule de l’organisme (leucocyte, GB, neurone…) , après activation, les plaquettes vont modifier leur charge de surface et faire basculer les phospholipides anioniques (qui sont le + souvent sur le versant cytoplasmique) vers l’extérieur.On passe de cellules qui sont plutôt de type chargées positivement à des cellules très fortement négatives.
3/18

Schéma : La plaquette va s'étaler, changer de forme, elles vont adhérer et sécréter le contenu des granules denses et alpha. Cela entraine la formation d'un nuage d'ADP, Entrainant l'activation des plaquettes circulantes. Ainsi le caillot plaquettaire va grossir. Ensuite la plaquette va réaliser le flip-flop et la modification conformationnelle de la GPIIbIIIa.
c-L’agrégation plaquettaire
= Empilement des plaquettes recrutées et activées recouvrant la brèche vasculaire
=Sert de support essentiel à la coagulation plasmatique
Fait intervenir :- L’interaction des GPIIb-IIIa plaquettaires avec le fibrinogène plasmatique-Le vWF et la Fibronectine peuvent aussi se lier à la GPIIb-IIIa mais pas au niveau d'une veine, seulement au niveau des zones de bifurcations carotidiennes et coronariennes ( où il y a de fortes forces de cisaillement que l'on verra après )
- Formation de ponts :Plaquette – GPIIbIIIa ---- Fg ---- GPIIbIIIa - Plaquette
4/18

Schéma : On a formation d'un agrégat plaquettaire par liaison des plaquettes entre elles par l'intermédiaire de fibrinogène. Cet agrégat plaquettaire qui sécrète encore de l'ADP donc active encore d'autres plaquettes, va servir de support à la coagulation plasmatique.
2-La Vasoconstriction
-Immédiate après la brèche vasculaire mais se relache très vite.- Provoquée par 2 molécules sécrétées par les plaquettes activées :
- le Thromboxane A2 -la SérotonineEntraine la contraction des fibres musculaires lisses de la Média.Ephémère à Durée : 1 minute.
Conséquences (mécaniques)1) Atténuation des pertes hémorragiques (car on diminue le diamètre du vaisseau)2) Augmentation des forces de cisaillement locales (→ retentissement sur hémostase de manière importante)
III- L’HEMORRHEOLOGIE
Le flux sanguin : écoulement d’un fluide dans un cylindre creux rigide (modèle de flux newtonien).
Le sang circulant : circule sous forme de couches cylindriques concentriques glissant les unes sur les autres à une vitesse croissante, de la paroi vers le centre: Vcentre > Vparoi.
5/18

(2 couches glissants les unes sur les autres n’ont pas la même vitesse : plus on va vers le centre, plus le sang va vite.)
Dans un segment vasculaire rectiligne ( rare au niveau des vaisseaux ) : on aura un flux sanguin laminaire (couches concentriques parallèles).
« Appliquons ce modèle au sang »
Le flux a un front de vitesse parabolique laminaire (type Poiseuille)Il en résulte : - Une vitesse maximale et un cisaillement ( entre les couches concentriques ) minimal au centre du vaisseau - Inversement le long de la paroi (vitesse très ralenti et forces de cisaillement très augmentées).
Quand on fait une coupe longitudinale, on retrouve notre « front de vitesse » et on voit que le sang qui est au centre du vaisseau va plus vite que le sang qui longe la paroi vasculaire.
Forces de cisaillement : conséquences des mouvements relatifs de 2 couches de fluide contigües.
- Mesurées par le taux de cisaillement : G = DV / d (unité s-1)- DV : différence de vitesse entre 2 couches- d : distance entre les 2 couches
Plus la différence de vitesse entre 2 couches est grande, + G est élevée.
En se rapprochant de la paroi, le gradient de vitesse est très important du fait des forces de frottement (et autres) qui ralentissent la vitesse sur le bord, donc les forces de cisaillement seront très augmentées au voisinage de la paroi vasculaire.
Les taux de cisaillement augmentent avec : Diminution du diamètre vasculaire, car augmentation du différentielle de vitesse entre les couches (+ le vaisseau est petit, + ça va frotter)
6/18
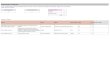
Augmentation du débit sanguin ( peu important dans le cadre de l'hémostase
G artériole >> G grosse veine.-Dans une artériole, on a un front de vitesse qui est très allongé, les gradients de vitesse sont très importants entre les couches, donc G importante.-Dans une grosse veine, le front de vitesse est beaucoup plus faible, une pression sanguine plus faible, un débit plus faible, des gradients de vitesse peu importants donc G plus faible
Donc il y a plus de cisaillement dans une artériole que dans une veine.
G artériole lésée >> G artériole normaleLa brèche provoque une vasoconstriction ce qui entraine donc une augmentation des différentielles de vitesses et donc des forces de cisaillement locales.
7/18

Exemple d’une artère coronarienne : le front de vitesse est très très étendu.Une plaque d’athérome forme un obstacle et ralenti de façon très importante le sang qui passe à son contact. Donc très brutalement, on a une augmentation des forces de cisaillement proche de la plaque d’athérome. Il y a donc augmentation du pouvoir d’hémostase primaire au contact d’une plaque d’athérome « il ne lui faut pas grand chose pour former un agrégat »
Si l’hémostase primaire se déclenche, il y a formation d’un caillot de plaquette + coagulation, donc obstruction du vaisseau ce qui provoquerait un IDM.
Les effets de l’augmentation des forces de cisaillement sur l’hémostase primaire :
1) Augmentation du nombre de collisions cellulaires On rappelle que dans un vaisseau : au centre il y a des grosses cellules (GR), en périphérie il y a des petites cellules (plaquettes) qui rebondissent.
Si les forces de cisaillement augmentent, les plaquettes rebondissent beaucoup plus souvent et beaucoup plus fortement sur les GB pour aller plus souvent et plus fortement au contact de la paroi vasculaire. Augmentation :
- Interactions plaquettes-paroi vasculaire ----> Adhésion plaquettaire augmente
- Interactions plaquettes-érythrocytes ------> Adhésion plaquettaire augmente
- Interactions plaquettes-plaquettes --------> Agrégation plaquettaire augmente
8/18

Ces chocs inter plaquettaires peuvent les activer.
2) Stimulation mécanique de la sécrétion plaquettaire --> Activation plaquettaire augmente (même celles à distance des forces de cisaillement élevées )
3) Modifications conformationnelles de certaines protéines :
Un cisaillement élevé peut provoquer à lui seul une modification de conformation de certaines protéines :
- vWF : de forme globulaire à forme étirée : Adhésion et agrégation plaquettaire augmentent
GPIb-V-IX : augmentation de l’affinité pour vWF : Adhésion et agrégation plaquettaire augmentent
Liaisons vWF / GPIIbIIIa : Adhésion et agrégation plaquettaire augmentent(Si G très élevé, sur bifurcations artérielles surtout athéromateuse.)
NB : L'influence des globules rouges (GR) : Les plaquettes rebondissent sur la paroi et les GR qui sont au centre des vaisseaux. S'il y a moins de GR, les plaquettes rebondissent moins. En cas d'anémie on peut avoir des problèmes d'hémostase.
IV. Exploration biologique de l'hémostase primaire :
A- Les principaux tests de première intention explorant l'hémostase primaire
1) Numération sanguine : Numération plaquettaires. (c'est la quantité => thrombopénie et pas la qualité, si mauvaise qualité thrombopathie, ex : pas de GP IIbIIIa) Taux d'hémoglobine et hématocrite. L'anémie est définie par le taux d'hémoglobine et quand elle est trop importante, elle entraine une anomalie de l'hémostase primaire responsable d'une aggravation du saignement.
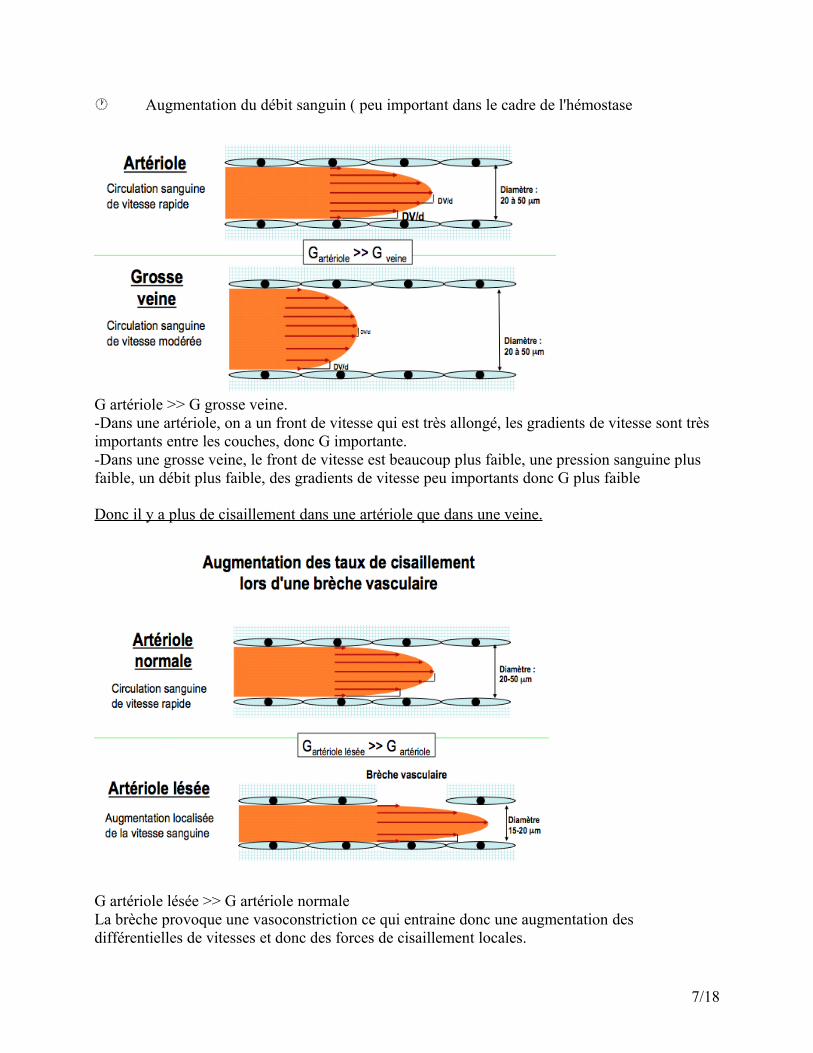
2) Temps de saignement : test globale de l'hémostase primaire(Ivy- incision ou Ivy-3 points)
9/18

3) Temps d'occlusion : (remplace de plus en plus le temps de saignement)Se fait sur une prise de sang.
4) Dosage des taux plasmatiques de : Fibrinogène vWD : Antigène→ quantité vWF : RCO (mesure de l'activité du facteur Willebrand ) → qualité
1) Numération sanguine
Sujet d'un prochain cours.
2) Le Temps de saignement (TS)
Principe du TS : Mesure in vivo du temps de saignement provoquée par l’incision de la couche superficielle de la peau (épiderme et derme superficiel)Les micro-vaisseaux ne nécessitent que l'hémostase primaire pour s'arrêter, le temps de saignement est donc spécifique de l'hémostase primaire et non de la coagulation.
Evaluation globale de l'hémostase primaire : Evaluation depuis la rupture de l'intégrité de la paroi vasculaire jusqu’au clou plaquettaire.
Les méthodes utilisées : Sous pression constante à 40mmHg par un brassard posé sur le 1/3 inférieur du bras. Temps de saignement sur la face antérieur de l'avant bras, à 5 cm du pli du coude Mesure du temps de saignement dès la brèche causée jusqu'à l'arrêt du saignement. (Attention à ne pas toucher la veine)
Deux méthodes principales de temps de saignement :
a) TS par Ivy-incisionIncision de 1mm de profondeur et 1 cm de longueurSang récupéré par un buvard sans toucher la brèche pour ne pas arracher le caillot, on laisse couler le sang sur le papier que l'on place 1 cm en dessous.Temps normal : 4 à 8 min (si déficit : 30 min environ )
b) TS par Ivy- 3 pointsPar une micro-lance calibrée : 3 ponctions séparées d'au moins 1 cmLe sang peut être récupéré par : Un buvard Un capillaire gradué avec mesure du volume de saignement.Valeurs normales :- Temps : 2 à 4 minutes.- Volume : < 120 μl
10/18
Temps de Saignement Temps de Saignement IvyIvy--IncisionIncision
Il existe une troisième méthode mais qui est fortement déconseillée, c'est la méthode de Duke, elle consiste à réaliser l'incision au niveau de l'oreille. Cette méthode est biaisé chez l'enfant par exemple car l'enfant à peur, il crie et augmente sa pression sanguine.
3) Le temps d'occlusion (ou « TS in vitro »)
- Un automate : PFA-100 TM (Dade-Behring)PFA : Platelet Function Analyser
Mesure de l'adhésion et de l'agrégation plaquettaire sur une membrane de collagène en présence d'agonistes (agents pro-agrégants)
Bonne détection de : Maladie de Willebrand. Thrombopathies par aspirine. Thrombopathies majeures.
11/18

Comment cela fonctionne :
Le PFA 100 simule in vitro l'hémostase primaire :
C'est un tube microcapillaire, la machine verse le sang total dans le tube à une extrémité avec des agents pro-agrégants plaquettaire → ADP ou épinéphrine, à une autre extrémité on trouve un détecteur optique reconnaissant un faisceau lumineux. Quand le sang passe, ça bloque le passage de la lumière et le détecteur ne reçoit rien.
A l’intérieure, une membrane recouverte de collagène avec au centre un orifice pour laisser passer le sang. (les plaquettes se fixent normalement sur le collagène lorsqu'elles sont activées)
Les plaquettes s'agrègent sur la membrane grâce aux deux activateurs plaquettaires que sont l'ADP et l'épinéphrine remplissant alors le trou de la membrane, le chronomètre s'arrête quand il y a occlusion de l’orifice par un caillot de plaquette et uniquement de plaquettes. La lumière passe donc et peut être reconnu par le détecteur optique.
/!/ Le temps d'occlusion n'explore pas la phase vasculaire !
Pathologies : -Scorbut, défaut de vitamine C donc défaut de synthèse de collagène, temps de saignement augmenté -Maladies héréditaire du collagène augmentant le temps de saignement.
4) Dosage du Facteur von Willebrand (vWF)
a) Dosage du vWD antigènePar méthode immunologiques
12/18

b) Dosage de l'activité cofacteur de la ristocétine (RCO)Etudie l'activité du vWFOn utilise ces particules de latex recouvertes de GPIb alpha que l'on met dans un tube avec le plasma du malade. (partie acellulaire mais contenant les protéines de la coagulation)On ajoute la RCO (ancien antibiotique) qui entraine une modification conformationnelle de la GPIb alpha favorisant la liaison GPIb alpha – vWF entre les microparticules de latex ce qui va provoquer une agglutination formant des agrégats( agglutinas de particules de latex) visibles à l'oeil nu alors qu'elles étaient auparavant translucide.
L'activité cofacteur de la RCO définit l'activité biologique du vWF
On rend le résultat en pourcentages de la normale exprimé en fonction de la population selon une courbe de Gauss. ( Biostats PACES <3 ) Normale : entre 50 à 150% (Si - de 50% = déficit en vWF / Si + de 150% = augmentation du vWF anormale )
Un peu de culture : La Ristocétine (RCO) est un ancien antibiotique des années 60 qui provoquait des thrombopénies et des saignements prolongés dû à la formation d'agrégats, Nowadays elle ne sert qu'à doser l'activité biologique du vWF.
c) Dosage du taux de Facteur VIII de la coagulationCar le vWF présente 2 fonctions biologiques principales :
1- Adhésion plaquettaire (hémostase primaire)2- Transport et protection du Facteur 8 plasmatique (coagulation)
Ce qui explique qu'un défaut du vWF entraine des défauts de la coagulation et de l'hémostase.
L'ensemble vWF: RCO + vWF : Ag + FVIII = Bilan Willebrand
Pour votre culture : Les gens du groupe O ont des taux qui ont tendance à être autour de 50%, c'est à dire de 30 à 60% à cause d'une diminution physiologique du vWF.
B- Les tests fonctionnels plaquettaires (à titre indicatif cela peut vous aider à comprendre mais pas vraiment à savoir)
Tests d'agrégation in vitro → Recherche de la thrombopathieAnalyse de la qualité de l'agrégation des plaquettes du patient en présence de différents activateurs. (=agonistes)
Agonistes les plus souvent utilisés : Acide arachidonique ADP
13/18

Collagène
Exemple : test d'agrégation à l'ADP.
La Densité optique est inversement proportionnelle à l’agrégation. T0 = on rajoute de l'ADP.La densité optique va diminuer → changement de forme puis 1ère vague d'agrégation : agrégation passive, se sont les plaquettes qui ont des récepteurs ADP qui vont dépendre donc directement de l'ADP exogène que l'on a mis, ces plaquettes sécrètent à leur tour de l'ADP = sécrétion autologue d'ADP d'origine endogène, apparition de la 2ème vague d'agrégation : agrégation active.
Pas de changement de forme : problème du cytosquelette.Anomalie de la première vague : anomalie du récepteur à l'ADP et de toute la chaine d'activation.Anomalie de la deuxième vague : anomalie de la sécrétion plaquettaire, difficulté de la sécrétion des granules alpha (assez fréquent dans la population).
V. Introduction à la pathologie :
A) Anomalies hémorrhéologiques :
1) Anomalies de la phase vasculaire (paroi vasculaire)
Exemples : Anomalies de synthèse du collagène :→ Pathologie acquise : scorbut (carence en vitamine C)→ Pathologie héréditaire : maladie d'Ehler-Danlos (anomalie héréditaire du collagène)
14/18

2) Anomalies sanguines
Ce sont les anémies, lorsque le taux d'Hb < 9g/dl.Diminution de l’interaction plaquettes – paroi vasculaire.
B) Anomalies de la phase plaquettaireDans cette partie il faut connaitre les exemples en gras de chaque anomalie, le reste ne fera qu'embellir votre copie.
1) Anomalies de l'adhésion (exemples)
- Anomalies du collagène (cf anomalie hémorrhéologiques)
- Maladie de Willebrand . Déficit héréditaire en facteur Willebrand. Autosomique Récessive
- Syndrome de Jean Bernard Soulier. Déficit en GP Ib/V/IX plaquettaires. Autosomique récessive
2) Anomalies de l'activation/sécrétion (exemples)
- Anomalies des récepteurs pro-activateurs Récepteur GP Ib/V/IX (cf ci dessus)
Récepteur de l'ADP (en particulier P2Y12)
- Déficit de sécrétion des granules Syndrome du pool vide : absence de granules alpha
Syndrome des plaquette grises : absence de granules denses.
- Anomalies du cytosquelette plaquettaire Syndrome de Wiskott-Aldrich : Anomalie de la protéine WASP impliquée dans la contractilité du cytosquelette Récessive liée à l'X.
- Anomalies des voies de signalisation intracellulaires Déficit de la cyclo-oxygénase (COX2)
3) Anomalies de l'agrégation plaquettaire (exemples)
- Maladie de Glanzmann Déficit du récepteur GP IibIIIa
15/18

Autosomique récessive La plus grave.
- Déficit en fibrinogène Hypo ou afibrinogénémie Autosomique récessive.
C) La maladie de Willebrand : IMPORTANT
Généralités :
Déficit en vWF : déficit le plus fréquent des facteurs de l'hémostase (1 à 2% de la population)Déficit qualitatif (fonctionnement de la protéine) ou quantitatif en vWFAnomalie du gène vWF (vWF) sur le chromosome 12. (uniquement)Entraine une répercussion sur la Transmission autosomique (habituellement dominante).
Rappels : les 2 fonctions du vWF :
- L’adhésion plaquettaire au site de lésion de la paroi vasculairePar liaison avec le collagène pariétal et GPIb alpha plaquettaire
- Le transport et la protection du Facteur VIII circulantConséquences : une diminution du taux de VWF => diminution proportionnelle du FVIII
Le bilan Willebrand :
- L’activité cofacteur de la ristocétine : VWF: RCO
Mesure l’activité plasmatique pro-adhésive plaquettaire du VWFLa ristocétine modifie conformation du VWF (en forme pro-adhésive)Mesure de la capacité du VWF à provoquer une agglutination de plaquettes ou de billes recouvertes de GPIb alpha. Résultats en % (N = 50-150%)
- Le taux de VWF antigène (« quantité ») : VWF:Ag Mesure le taux plasmatique quantitatif du VWF
- Le taux de facteur VIII activité : FVIII:CMesure le taux plasmatique d’activité pro-coagulante du FVIII
Autres tests utilisés pour le diagnostic de VWD afin d'affiner le diagnostic ( en retenir au moins un )
Test d’agrégation plaquettaire en présence de RCO Test de liaison VWF-FVIII Test de liaison VWF-Collagène …
16/18
Génotypage du gène vWF
Manifestations cliniques : IMPORTANT Hémorragies de type hémostase primaire, très typique : Ecchymoses (un saignement pas collecté : des bleus et ne fait pas mal ) différent de l'hématome gonflé et qui fait mal ( le sang est collecté) Hémorragies muqueuses en particulier épistaxis, méno-métrorragies, les saignements intestinaux ...
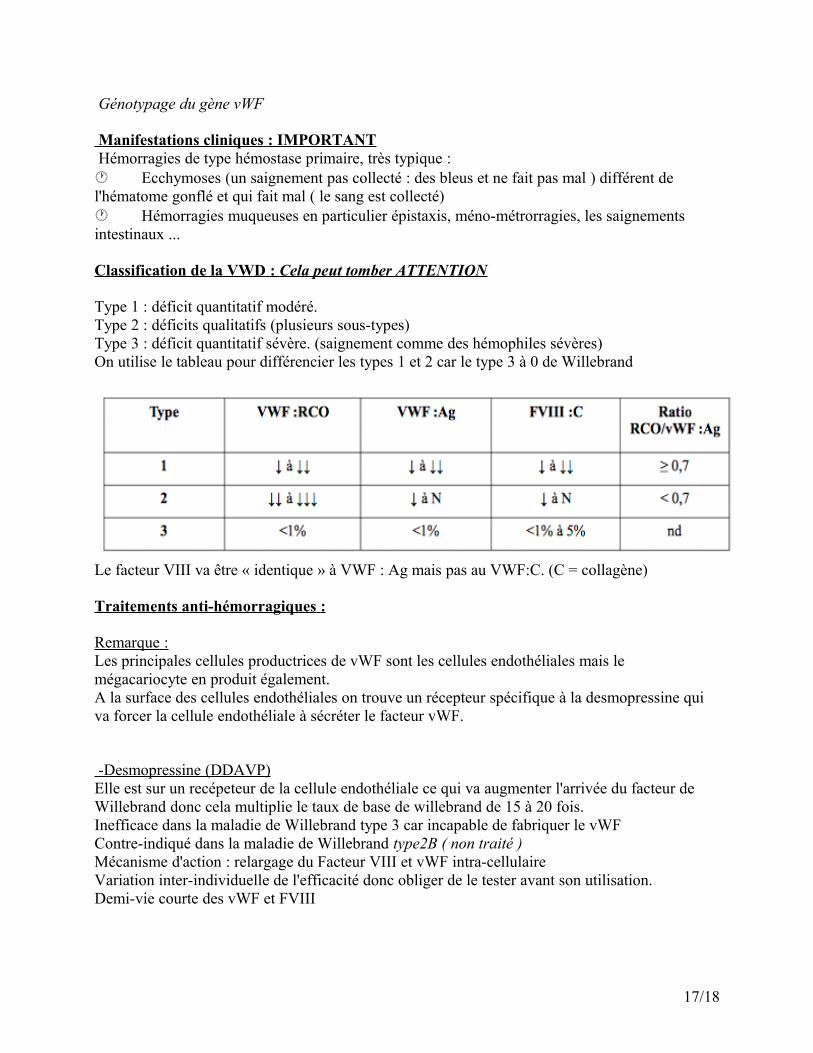
Classification de la VWD : Cela peut tomber ATTENTION
Type 1 : déficit quantitatif modéré.Type 2 : déficits qualitatifs (plusieurs sous-types)Type 3 : déficit quantitatif sévère. (saignement comme des hémophiles sévères)On utilise le tableau pour différencier les types 1 et 2 car le type 3 à 0 de Willebrand
Le facteur VIII va être « identique » à VWF : Ag mais pas au VWF:C. (C = collagène)
Traitements anti-hémorragiques :
Remarque : Les principales cellules productrices de vWF sont les cellules endothéliales mais le mégacariocyte en produit également.A la surface des cellules endothéliales on trouve un récepteur spécifique à la desmopressine qui va forcer la cellule endothéliale à sécréter le facteur vWF.
-Desmopressine (DDAVP)Elle est sur un recépeteur de la cellule endothéliale ce qui va augmenter l'arrivée du facteur de Willebrand donc cela multiplie le taux de base de willebrand de 15 à 20 fois.Inefficace dans la maladie de Willebrand type 3 car incapable de fabriquer le vWF Contre-indiqué dans la maladie de Willebrand type2B ( non traité )Mécanisme d'action : relargage du Facteur VIII et vWF intra-cellulaireVariation inter-individuelle de l'efficacité donc obliger de le tester avant son utilisation.Demi-vie courte des vWF et FVIII
17/18

- Concentrés de vWF : traitements substitutifs : D’origines plasmatiques donc issues de donneur de sang
Complications :
Risque de transmission virale ou ATNC (agent transmissible non conventionnel) quasi nul (surtout théoriques) Allo-anticorps anti-vWF décrit que pour la vWD type 3. (réaction car ne reconnaît pas le vWF) Immunisation contre le non soi car la maladie de type 3 ne connait pas le Willebrand donc cela entraine une inefficacité des injections.
18/18