Embed Size (px)
Citation preview

Myélofibrose
d o s s i e r
Correspondances en Onco-Hématologie - Vol. IX - n° 1 - janvier-février 2014 2323
Nouvelles anomalies moléculaires pronostiquesNew prognostic molecular abnormalitiesA. Murati*
RÉ
SU
MÉ
Su
mm
ar
y
» Depuis la découverte en 2005 de la mutation ponctuelle du gène JAK2 (JAK2 V617F) dans les syndromes myéloprolifératifs (SMP) non leucémie myéloïde chronique (LMC), de nombreuses mutations somatiques ont été mises en évidence dans les hémopathies myéloïdes ces dernières années, impliquant les gènes MPL, LNK, CBL, NF1, RAS, ASXL1, EZH2, SUZ12, TET2, DNMT3A, IDH1/2, SF3B1 et SRSF2. Il y a quelques semaines, la description des mutations de CALR a apporté une nouvelle pièce au puzzle moléculaire des SMP. L’étude de l’ensemble de ces mutations dans les myélofi broses a pour objectif d’identifi er de nouveaux marqueurs biologiques à visée diagnostique, pronostique et thérapeutique afi n d’élaborer une prise en charge ciblée des patients atteints de SMP non LMC.
Mots-clés : Myélofi brose – Mutations – Séquençage – Épigénétique – Pronostic – JAK2.
Since the discovery in 2005 of the JAK2 V617F mutation in non-CML MPN, many somatic mutations have been identifi ed in myeloid malignancies over the last years, involving MPL, LNK, CBL, NF1, RAS, ASXL1, EZH2, SUZ12, TET2, DNMT3A, IDH1/2, SF3B1 and SRSF2 genes. Recently, CALR gene mutations solved the MPN molecular puzzle. The study of these mutations in myelofi brosis aims to identify new biomarkers for diagnosis, prognosis and treatment, in order to develop a targeted therapeutic management of patients with non-CML MPN.
Keywords: Myelofibrosis – Mutations – Sequencing –Epigenetic – Prognosis – JAK2.
L es syndromes myéloprolifératifs (SMP) BCR-ABL négatifs regroupent la polyglobulie pri-mitive encore appelée maladie de Vaquez ou
Polycythemia Vera (PV), la thrombocytémie essentielle (TE) et la myélo fi brose (MF). La myélofi brose est divisée en 2 catégories : la MF primitive (MFP) et la MF secon-daire à une PV ou à une TE (MF post-PV/TE). La myélofi -brose est plus rare et de pronostic plus grave que les PV et les TE, avec une médiane de survie inférieure à 6 ans. Les causes principales de décès sont la transformation en leucémie aiguë, les complications vasculaires et les infections. Les SMP sont des hémopathies clonales qui proviennent de la transformation d’une cellule souche hématopoïétique. La détection d’anomalies molécu-laires permet d’affi rmer la clonalité de la maladie.En 2005, la découverte de la mutation somatique JAK2 V617F située dans l’exon 14 du gène JAK2 a permis une avancée majeure en termes de compréhension de la physiopathologie des SMP (1). Cette mutation est res-ponsable de l’activation constitutive de l’activité kinase de JAK2 et favorise la voie JAK/STAT mais également les
voies de signalisation PI3K et MAPK. La mutation JAK2 V617F est retrouvée dans près de 95 % des PV (des muta-tions sur l’exon 12 de JAK2 sont retrouvées dans 3 % des cas), et dans environ la moitié des cas de TE et de MFP.Les SMP BCR-ABL négatifs qui ne présentent pas la mutation JAK2 V617F (et dont le caryotype est normal le plus souvent) représentent près de 50 % des SMP et constituent un groupe pour lequel il est nécessaire de mieux comprendre les mécanismes moléculaires. La compréhension des altérations génétiques par la caracté-risation des gènes d’intérêt constitue un enjeu prioritaire pour la défi nition de nouvelles cibles thérapeutiques.
Huit ans après la découverte de la mutation JAK2 V617F…
Le recours à l’analyse pangénomique par les techniques de CGH array (Comparative Genomic Hybridization array) et de SNP array (Single Nucleotide Polymorphism array) a permis d’identifi er de nouvelles altérations génomiques.
* Laboratoire de biopatho-logie, unité d’hématologie cellulaire et d’oncologie moléculaire ; centre de recherche en cancérologie de Marseille, Institut Paoli-Calmettes ; UMR1068 Inserm, Marseille.

Myélofibrose
d o s s i e r
Correspondances en Onco-Hématologie - Vol. IX - n° 1 - janvier-février 20142424
Les techniques classiques de séquençage (Sanger) et, plus récemment, le séquençage de nouvelle génération (New Generation Sequencing [NGS]) ont mis en évidence de nouvelles mutations dans les SMP. Ces mutations aff ectent des gènes codant pour des protéines qui appartiennent à 5 catégories diff érentes selon leur fonction cellulaire.
✓ Des protéines des voies de signalisation des facteurs de croissance hématopoïétiques : MPL (Myeloproliferative Leukemia virus), CBL (Casitas B-lineage Lymphoma), LNK (Lymphocyte-specifi c adaptator pro-tein) [2], NF1 (Neurofi bromin 1) [3].
✓ Des régulateurs épigénétiques impliqués dans la méthylation/déméthylation de l’ADN : DNMT3A (DNA cytosine Methyltransferase 3) [4], IDH1, IDH2 (Isocitrate Dehydrogenase 1 et 2) [2] et TET2 (Ten-Eleven Translocation oncogene family member 2) [5].
✓ Des régulateurs épigénétiques impliqués dans la méthylation des histones : ASXL1 (Additional Sex Combs-like 1) [6], EZH2 (Enhancer of Zeste Homolog 2), SUZ12 (Suppressor of Zeste 2 homolog) [2].
✓ Des composants du spliceosome impliqués dans la maturation de l’ARN : SF3B1 (RNA Splicing Factor 3B, subunit 1) [3, 7], SRSF2 (Serine/arginine-Rich Splicing Factor 2) [8].
✓ Des gènes suppresseurs de tumeur (TP53) [2].Les voies de signalisation des facteurs de croissance hématopoïétiques sont majoritairement impliquées dans la physiopathologie des SMP avec les mutations des gènes JAK2, MPL et LNK spécifi ques des SMP, alors que les mutations de CBL et RAS sont retrouvées aussi dans les autres hémopathies myéloïdes (leucémies aiguës myéloïdes [LAM] et syndromes myélodyspla-siques [SMD]). Les gènes impliquant les autres fonctions cellulaires (ASXL1, EZH2, SUZ12, TET2, DNMT3A, IDH1/2, SF3B1 et SRSF2) sont mutés dans les SMP, les LAM et les SMD. Une question persistait il y a encore quelques semaines : existe-t-il des mutations spécifi ques des SMP sans mutation de JAK2 ou de MPL ? Deux études parues en décembre 2013 (9, 10) ont montré, grâce au séquençage à grande échelle, la présence de mutations récurrentes du gène de la calréticuline (CALR) dans les SMP. Pour la première fois, CALR est impliqué dans les cancers, et cette découverte majeure vient compléter la caractérisation moléculaire des SMP.
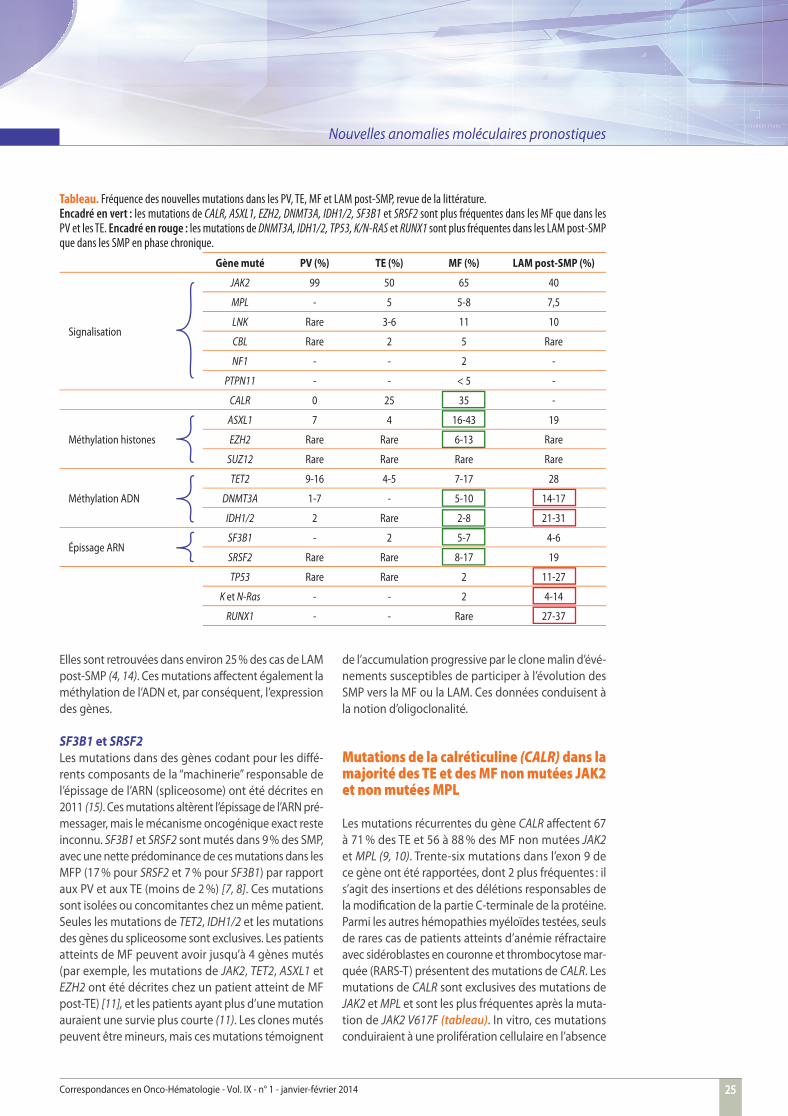
La fréquence de ces nouvelles mutations est notable dans les MF
Ces 2 dernières années, les études réalisées sur la fré-quence de ces nouvelles mutations au sein des SMP
ont montré une nette diff érence entre les PV, les TE et les MF. En eff et, en dehors de la mutation de TET2 − qui est présente de façon homogène au sein des 3 types de SMP −, les mutations aff ectant les gènes impliqués dans les modifi cations épigénétiques (ASXL1, EZH2, IDH1/2 et DNMT3A) et les gènes impliqués dans la maturation de l’ARN (SF3B1 et SRSF2) sont plus fréquentes dans les MF (3, 8, 11) que dans les TE et les PV (tableau).
ASXL1 et EZH2En 2009, des mutations dans l’exon 12 d’ASXL1 ont été décrites dans les SMP (6). Par la suite, d’autres études ont confi rmé ces résultats, montrant une nette prédomi-nance de ces mutations dans les MF comparativement aux PV et aux TE. En eff et, M. Brecqueville et al. ont montré, sur une série de 148 patients atteints de SMP (30 PV, 53 TE et 65 MF), que 13,5 % des cas présentaient une mutation dans l’exon 12 d’ASXL1 : 25 % des cas (16 sur 65) de MF, 4 % des cas (2 sur 53) de TE et 7 % des cas (2 sur 30) de PV (3, 11). La fréquence de ces mutations dans les MF est variable selon les séries (de 16 à 43 % des cas) [2].ASXL1 régule les modifi cations épigénétiques telles que la méthylation des histones et interagit avec divers répresseurs et activateurs de la transcription. Son rôle dans la leucémogenèse est mal connu. Toutefois, de récentes données ont montré qu’ASXL1 interagit avec 2 composants du complexe Polycomb 2 (PRC2), EZH2 et SUZ12, impliqués dans la répression de la transcrip-tion via la méthylation de l’histone 3 sur la lysine 27 (H3K27). Ces 2 protéines sont également mutées dans les SMP, avec une fréquence de 3 à 13 % pour EZH2 dans les MF (2), et plus rarement pour SUZ12 (12). Le rôle d’ASXL1 pourrait être de favoriser le recrutement du complexe PRC2 au niveau des gènes HOXA impliqués dans la prolifération cellulaire. ASXL1 pourrait aussi s’associer à BAP1 (une déubiquitinase nucléaire) dans un autre complexe de régulation épigénétique. Chez la souris, la perte de fonction du gène ASXL1 aboutit à un phénotype de SMD (13).
TET2, IDH1/2 et DNMT3ALes mutations de TET2 sont retrouvées dans 12 % des cas de SMP (16 % des MF, 8 % des TE et 10 % des PV) [3, 5]. TET2 est une dioxygénase qui catalyse l’hydroxylation de la 5-méthylcytosine (5-mC) en 5-hydroxyméthylcyto-sine (5-hmC), conduisant à une déméthylation de l’ADN après conversion de la 5-hmC en cytosine.Les mutations de IDH1/2 et DNMT3A sont plus fré-quentes dans les MFP (4 à 8 % et 10 à 15 %, respec-tivement) et très rares dans les TE et les PV (1 à 3 %). Elles sont clairement associées à l’évolution en LAM.
Correspondances en Onco-Hématologie - Vol. IX - n° 1 - janvier-février 2014 2525
Nouvelles anomalies moléculaires pronostiques
Elles sont retrouvées dans environ 25 % des cas de LAM post-SMP (4, 14). Ces mutations aff ectent également la méthylation de l’ADN et, par conséquent, l’expression des gènes.
SF3B1 et SRSF2Les mutations dans des gènes codant pour les diff é-rents composants de la “machinerie” responsable de l’épissage de l’ARN (spliceosome) ont été décrites en 2011 (15). Ces mutations altèrent l’épissage de l’ARN pré-messager, mais le mécanisme oncogénique exact reste inconnu. SF3B1 et SRSF2 sont mutés dans 9 % des SMP, avec une nette prédominance de ces mutations dans les MFP (17 % pour SRSF2 et 7 % pour SF3B1) par rapport aux PV et aux TE (moins de 2 %) [7, 8]. Ces mutations sont isolées ou concomitantes chez un même patient. Seules les mutations de TET2, IDH1/2 et les mutations des gènes du spliceosome sont exclusives. Les patients atteints de MF peuvent avoir jusqu’à 4 gènes mutés (par exemple, les mutations de JAK2, TET2, ASXL1 et EZH2 ont été décrites chez un patient atteint de MF post-TE) [11], et les patients ayant plus d’une mutation auraient une survie plus courte (11). Les clones mutés peuvent être mineurs, mais ces mutations témoignent
de l’accumulation progressive par le clone malin d’évé-nements susceptibles de participer à l’évolution des SMP vers la MF ou la LAM. Ces données conduisent à la notion d’oligoclonalité.
Mutations de la calréticuline (CALR) dans la majorité des TE et des MF non mutées JAK2 et non mutées MPL
Les mutations récurrentes du gène CALR aff ectent 67 à 71 % des TE et 56 à 88 % des MF non mutées JAK2 et MPL (9, 10). Trente-six mutations dans l’exon 9 de ce gène ont été rapportées, dont 2 plus fréquentes : il s’agit des insertions et des délétions responsables de la modifi cation de la partie C-terminale de la protéine. Parmi les autres hémopathies myéloïdes testées, seuls de rares cas de patients atteints d’anémie réfractaire avec sidéroblastes en couronne et thrombocytose mar-quée (RARS-T) présentent des mutations de CALR. Les mutations de CALR sont exclusives des mutations de JAK2 et MPL et sont les plus fréquentes après la muta-tion de JAK2 V617F (tableau). In vitro, ces mutations conduiraient à une prolifération cellulaire en l’absence
Tableau. Fréquence des nouvelles mutations dans les PV, TE, MF et LAM post-SMP, revue de la littérature. Encadré en vert : les mutations de CALR, ASXL1, EZH2, DNMT3A, IDH1/2, SF3B1 et SRSF2 sont plus fréquentes dans les MF que dans les PV et les TE. Encadré en rouge : les mutations de DNMT3A, IDH1/2, TP53, K/N-RAS et RUNX1 sont plus fréquentes dans les LAM post-SMP que dans les SMP en phase chronique.
Gène muté PV (%) TE (%) MF (%) LAM post-SMP (%)
Signalisation
JAK2 99 50 65 40
MPL - 5 5-8 7,5
LNK Rare 3-6 11 10
CBL Rare 2 5 Rare
NF1 - - 2 -
PTPN11 - - < 5 -
CALR 0 25 35 -
Méthylation histones
ASXL1 7 4 16-43 19
EZH2 Rare Rare 6-13 Rare
SUZ12 Rare Rare Rare Rare
Méthylation ADN
TET2 9-16 4-5 7-17 28
DNMT3A 1-7 - 5-10 14-17
IDH1/2 2 Rare 2-8 21-31
Épissage ARNSF3B1 - 2 5-7 4-6
SRSF2 Rare Rare 8-17 19
TP53 Rare Rare 2 11-27
K et N-Ras - - 2 4-14
RUNX1 - - Rare 27-37

Myélofibrose
d o s s i e r
Correspondances en Onco-Hématologie - Vol. IX - n° 1 - janvier-février 20142626
de cytokines. Cette prolifération serait sensible aux anti-JAK2, ce qui suggère l’implication de la voie JAK-STAT (9).
Impact pronostique de ces nouvelles mutations dans les MF
Actuellement, l’allogreff e est le seul traitement curatif dans les MF. Toutefois, cette option thérapeutique ne peut être envisagée que chez un faible nombre de patients, et elle est associée à une morbidité et à une mortalité non négligeables. Les autres alternatives thérapeutiques sont les agents stimulant l’érythro-poïèse, les androgènes, le thalidomide, l’hydroxy-urée, la splénectomie, la radiothérapie splénique et, plus récemment, les inhibiteurs de JAK2, qui traitent la splénomégalie et les signes généraux. Aucun de ces traitements ne réduit cependant la fi brose médullaire et aucun ne conduit à une rémission moléculaire. La décision thérapeutique repose sur les scores pronos-tiques des MF tels que l’IPSS (International Prognostic Scoring System) [16], qui s’applique au diagnostic, et le DIPPS (Dynamic IPSS) [17], qui s’applique à tout moment durant l’évolution de la maladie. Récemment, le DIPPS-plus a été proposé, avec 3 facteurs de risque supplé-mentaires :
✓ le taux de plaquettes inférieur à 100 G/L ; ✓ les besoins transfusionnels de globules rouges ; ✓ un caryotype défavorable (18).
Au fi nal, ces 3 scores pronostiques permettent de clas-ser les patients en 4 catégories de risque en fonction des courbes de survie : les patients de faible risque, de risque intermédiaire 1 et 2, et de haut risque.Les résultats récents concernant l’impact pronostique des nouvelles mutations sont très intéressants. Alors que les mutations de TET2 n’ont pas d’impact sur la survie des patients, plusieurs études ont montré que les mutations des gènes ASXL1, IDH1/2, EZH2 et SRSF2 sont associées à un mauvais pronostic (3, 8, 11, 19, 20). En eff et, une récente étude portant sur une cohorte de 879 patients atteints de MFP (483 patients européens et 396 patients issus de la Mayo Clinic) montre que les mutations de ASXL1, SRSF2 et EZH2 sont associées à une survie plus courte (20), confi rmant des résultats prélimi-naires publiés (3, 8, 11, 19). Cette même étude montre également un eff et sur le risque d’évolution en leucé-mie aiguë, qui est plus élevé chez les patients mutés pour ASXL1, SRSF2 et IDH1/2 (20). De plus, A. Teff eri et al. ont montré, sur une cohorte de 301 patients atteints de MFP, que les mutations de IDH1/2 sont de mauvais pronostic, avec un impact sur la survie et le risque de transformation en leucémie (14). Ces résultats sont en accord avec ceux des études dans les SMD et les LAM (21, 22). Les mutations de ASXL1, IDH1/2, EZH2 et SRSF2 seraient des facteurs de risque indépendants du score pronostique des MFP (8, 14, 19, 20) et permettraient d’identifi er des patients atteints de MFP à risque pré-coce de décès ou de transformation en leucémie aiguë.
Figure. Nouvelles mutations : quand les rechercher ? Proposition de classifi cation moléculaire pour les SMP non LMC au diagnostic. Mutations des gènes ASXL1, SRSF2, EZH2 et IDH1/2 : facteurs pronostiques indépendants ?
Suspicion PV, TE, MFP
Suspicion PV
Confirmation SMP
Confirmation PV
ConfirmationTE/MFP
Suspicion TE Suspicion MFP
Mutations CALR/MPLW515
Discussion nouveauxmarqueurs moléculaires Discussion nouveaux
marqueurs moléculaires
TET2, DNMT3A, CBL, LNK, SF3B1, PTPN11
JAK2 V617F
+
TET2, ASXL1
JAK2 exon 12
ASXL1, SRSF2, EZH2, IDH1/2
Autres gènes ? (NGS)
ASXL1, SRSF2, EZH2, IDH1/2
Nouveau score pronostique ?Orientation thérapeutique ?
+
+
–
–
–

Correspondances en Onco-Hématologie - Vol. IX - n° 1 - janvier-février 2014 2727
Nouvelles anomalies moléculaires pronostiques
D’après l’étude de T. Klampfl et al. (9), les patients avec des mutations de CALR auraient un moindre risque de thrombose et une meilleure survie que les patients mutés JAK2.
Conclusion
Des études complémentaires seront nécessaires afi n d’élucider les mécanismes d’activation de la voie JAK-STAT par les mutants de la calréticuline dans les cellules myéloïdes. En attendant, la recherche des mutations de CALR/JAK2/MPL lors du diagnostic des TE et des MF devient un outil important. La preuve de la clonalité sera à présent possible pour la presque totalité des MF. Même si la recherche des autres nouvelles mutations
n’est pour l’instant pas réalisée en routine, leur étude au moment du diagnostic dans les MF mais également dans les PV et les TE dans le cadre de la recherche grâce au NGS prend tout son sens (fi gure). En eff et, les études prospectives concernant ces mutations permettront de confi rmer leur impact sur l’évolution de la mala-die, avec notamment le risque d’évolution d’une TE ou d’une PV en MF et le risque d’évolution en LAM ; elles permettront également d’évaluer un éventuel impact sur le risque vasculaire. Ces mutations aff ectant la régu-lation épigénétique, il serait intéressant de confronter le statut mutationnel des patients avec la réponse aux traitements tels que les anti-JAK2 en association avec les agents déméthylants ou les inhibiteurs des histones déacéthylases. L’objectif, à terme, étant de prévoir la sensibilité des patients aux nouveaux traitements. ■
1. James C, Ugo V, Le Couédic JP et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycy-thaemia vera. Nature 2005;434(7037):1144-8.
2. Vainchenker W, Delhommeau F, Constantinescu SN et al. New mutations and pathogenesis of myeloproliferative neo-plasms. Blood 2011;118(7):1723-35.
3. Brecqueville M, Rey J, Bertucci F et al. Mutation analysis of ASXL1, CBL, DNMT3A, IDH1, IDH2, JAK2, MPL, NF1, SF3B1, SUZ12, and TET2 in myeloproliferative neoplasms. Genes Chromosomes Cancer 2012;51(8):743-55.
4. Stegelmann F, Bullinger L, Schlenk RF et al. DNMT3A mutations in myeloproliferative neoplasms. Leukemia 2011;25(7):1217-9.
5. Delhommeau F, Dupont S, Della Valle V et al. Mutation in TET2 in myeloid cancers. N Engl J Med 2009;360(22):2289-301.
6. Carbuccia N, Murati A, Trouplin V et al. Mutations of ASXL1 gene in myeloproliferative neoplasms. Leukemia 2009;23(11):2183-6.
7. Lasho TL, Finke CM, Hanson CA et al. SF3B1 mutations in primary myelofi brosis: clinical, histopathology and genetic correlates among 155 patients. Leukemia 2012;26(5):1135-7.
8. Lasho TL, Jimma T, Finke CM et al. SRSF2 mutations in pri-mary myelofi brosis: signifi cant clustering with IDH mutations and independent association with inferior overall and leuke-mia-free survival. Blood 2012;120(20):4168-71.
9. Klampfl T, Gisslinger H, Harutyunyan AS et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med 2013;369(25):2379-90.
10. Nangalia J, Massie CE, Baxter EJ et al. Somatic CALR muta-tions in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med 2013;369(25):2391-405.
11. Brecqueville M, Rey J, Devillier R et al. Array compara-tive genomic hybridization and sequencing of 23 genes in 80 patients with myelofibrosis at chronic or acute phase. Haematologica 2014;99(1):37-45.
12. Brecqueville M, Cervera N, Adélaïde J et al. Mutations and deletions of the SUZ12 polycomb gene in myeloproliferative neoplasms. Blood Cancer J 2011;1(8):e33.
13. Abdel-Wahab O, Dey A. The ASXL-BAP1 axis: new fac-tors in myelopoiesis, cancer and epigenetics. Leukemia 2013;27(1):10-5.
14. Teff eri A, Jimma T, Sulai NH et al. IDH mutations in primary myelofi brosis predict leukemic transformation and shortened survival: clinical evidence for leukemogenic collaboration with JAK2 V617F. Leukemia 2012;26(3):475-80.
15. Yoshida K, Sanada M, Shiraishi Y et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011;478(7367):64-9.
16. Cervantes F, Dupriez B, Pereira A et al. New prognostic scoring system for primary myelofi brosis based on a study of
the International Working Group for Myelofi brosis Research and Treatment. Blood 2009;113(13):2895-901.
17. Passamonti F, Cervantes F, Vannucchi AM et al. Dynamic International Prognostic Scoring System (DIPSS) predicts pro-gression to acute myeloid leukemia in primary myelofi brosis. Blood 2010;116(15):2857-8.
18. Gangat N, Caramazza D, Vaidya R et al. DIPSS plus: a refi ned Dynamic International Prognostic Scoring System for primary myelofi brosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J Clin Oncol 2011;29(4):392-7.
19. Guglielmelli P, Biamonte F, Score J et al. EZH2 muta-tional status predicts poor survival in myelofi brosis. Blood 2011;118(19):5227-34.
20. Vannucchi AM, Lasho TL, Guglielmelli P et al. Mutations and prognosis in primar y myelofibrosis. Leukemia 2013;27(9):1861-9.
21. Gelsi-Boyer V, Brecqueville M, Devillier R et al. Mutations in ASXL1 are associated with poor prognosis across the spectrum of malignant myeloid diseases. J Hematol Oncol 2012;5:12.
22. Schnittger S, Eder C, Jeromin S et al. ASXL1 exon 12 muta-tions are frequent in AML with intermediate risk karyotype and are independently associated with an adverse outcome. Leukemia 2013;27(1):82-91.
R é f é r e n c e s
A. Murati déclare ne pas avoir de liens d’intérêts.





























