Embed Size (px)
Citation preview

69
Chapitre 5
Progrès dansles pathologies
mitochondriales
Annabelle Chaussenot,Agnès Rötig,
Véronique Paquis-Flucklinger
Points essentiels
☞ Les maladies mitochondriales touchent envi-ron 2,5 personnes sur 10 000 et sont aujourd’huiconsidérées comme les plus fréquentes des mala-dies métaboliques. Elles représentent un grouped’atteintes dont le dénominateur commun est undysfonctionnement de la chaîne respiratoire et setraduisent donc par un déficit énergétique.
☞ Les patients sont des enfants ou des adulteset une maladie mitochondriale peut se manifes-ter à tout âge, de la période néonatale jusqu’àune période avancée de la vie.
☞ Les mitochondries étant présentes dans tou-tes les cellules, une pathologie mitochondrialepeut toucher n’importe quel tissu ou organe.L’expression clinique de ces affections est donctrès hétérogène (encéphalomyopathie, retardmental, épilepsie, diabète, cardiomyopathie,surdité, cécité, insuffisance hépatique…). Une« association illégitime » de symptômes qui vonts’additionner sur un mode évolutif doit fairesuspecter une pathologie mitochondriale.
☞ Les maladies mitochondriales sont hétérogè-nes sur le plan génétique, liées soit à des muta-tions de l’ADN mitochondrial, soit à des muta-tions dans des gènes nucléaires qui restent àidentifier chez la majorité des patients. L’identi-fication des mutations responsables est impor-tante pour le diagnostic mais également pour leconseil génétique et le diagnostic prénatal.
☞ Le pronostic est généralement péjoratif, par-ticulièrement dans les formes de début précoce,et le traitement reste essentiellement symptomati-que à ce jour.
Les maladies mitochondriales sont considé-rées comme les plus fréquentes des maladiesmétaboliques, avec une fréquence d’environ2,5 personnes sur 10 000. Elles sont carac-térisées par un dysfonctionnement de lachaîne respiratoire et se traduisent par undéficit énergétique.Les mitochondries étant présentes dans tou-tes les cellules, une pathologie mitochon-driale peut toucher n’importe quel tissu ouorgane. Elle peut également se manifester àtout âge, de la période néonatale jusqu’à unâge avancé de la vie.
5669_ Page 69 Vendredi, 4. mars 2011 12:09 12> STDI FrameMaker noir

70
Le diagnostic est difficile et complexe, du faitde la grande hétérogénéité des présentationscliniques (encéphalopathie, épilepsie, dia-bète, surdité, cécité, cardiomyopathie, insuf-fisance hépatique…) et le grand nombre degènes impliqués. Une « association illégitime »de symptômes qui vont s’additionner sur unmode évolutif doit faire suspecter une patho-logie mitochondriale. La démarche diagnos-tique repose sur des examens d’orientation eta été améliorée par les progrès réalisés dans ledomaine de la neuro-imagerie. L’hypothèsediagnostique est confirmée par des analysesspécifiques (enzymologiques, moléculaires ethistologiques) devant être réalisées sur le tissuqui exprime le déficit.Les maladies mitochondriales sont hétérogè-nes sur le plan génétique, liées soit à desmutations de l’ADN mitochondrial, soit à desmutations dans des gènes nucléaires, quirestent à identifier chez la majorité despatients. L’identification des mutations res-ponsables est importante pour le diagnosticmais également pour le conseil génétique etle diagnostic prénatal. Là encore, les progrèsrécents dans les techniques d’analyses del’ADN mitochondrial d’une part, et dansl’identification de nombreux gènes nucléai-res d’autre part, ont permis d’améliorer laprise en charge des patients et des familles.
Enfin, le pronostic est généralement péjora-tif, particulièrement dans les formes de débutprécoce, et le traitement reste essentiellementsymptomatique à ce jour.
I Rappel sur la phosphorylation oxydative
A Chaîne respiratoire mitochondriale
La mitochondrie occupe une place cen-trale dans le métabolisme intermédiaire. Elleest le siège de nombreuses réactions du cata-bolisme cellulaire, telles celles conduisant àl’oxydation des acides gras (β-oxydation),des acides carboxyliques dérivant des sucres(cycle de Krebs) ou des acides aminés. Elleassure également la respiration cellulaire etfournit une grande partie de l’énergie néces-saire au fonctionnement de la cellule grâce àla phosphorylation oxydative, qui fait inter-venir, d’une part, des réactions d’oxydationaboutissant à une consommation d’oxygèneet, d’autre part, une réaction de phosphoryla-tion de l’ADP intramitochondrial en ATP. Laphosphorylation oxydative est réalisée dansla chaîne respiratoire (CR) au niveau de lamembrane mitochondriale interne (fig. 5.1).
Figure 5.1 La chaîne respiratoire mitochondriale.
Membraneinterne
Matrice
CoQ
O2FumarateSuccinate
ATP ADP
PiFlux de protonsFlux d’électrons
NAD+
+ H+NADH
Cytc
CI
H+ H+ H+ H+
CII CIII CIV CV
Membrane externe
5669_ Page 70 Vendredi, 4. mars 2011 12:09 12> STDI FrameMaker noir

71
La CR est constituée de 5 complexes fonc-tionnant principalement comme des trans-porteurs d’électrons :— le complexe I (NADH-coenzyme Q réduc-
tase) est constitué par plus de 40 sous-uni-tés différentes et transfère les équivalentsréduits du NADH au coenzyme Q (CoQ) ;
— le complexe II (succinate-CoQ réductase)comporte 4 sous-unités et transfère leséquivalents réduits du FADH2 vers le CoQ ;
— le complexe III (ubiquinol cytochrome créductase) est constitué par 11 sous-uni-tés et transporte les électrons de l’ubiqui-none vers le cytochrome c ;
— le complexe IV (cytochrome c oxydase,ou COX) est composé par 2 cytochromes(a et a3), 2 atomes de cuivre et 13 sous-unités protéiques. Il catalyse le transfertdes équivalents réduits du cytochrome cjusqu’à l’accepteur final qu’est l’oxygène.L’énergie générée par l’oxydation desdifférents constituants de la CR entraînel’expulsion d’ions H+ dans l’espace inter-membranaire au niveau des comple-xes I, III et IV, qui génère une différencede potentiel électrochimique ;
— le complexe V, ou ATPase (14 sous-uni-tés), permet l’entrée des protons dans lamatrice mitochondriale et utilise l’éner-gie libérée par ce flux d’ions H+ poursynthétiser de l’ATP à partir de l’ADP etdu phosphate inorganique.
B Génétique mitochondrialeTous les complexes de la CR ont un dou-
ble contrôle génétique, principalement par legénome nucléaire mais également par l’ADNmitochondrial (ADNmt), à l’exception ducomplexe II qui est exclusivement nucléaire.
1 Génome mitochondrial
Le génome mitochondrial est une molé-cule d’ADN circulaire double brin de16 569 paires de bases, localisée dans lamatrice mitochondriale. Chaque moléculecomporte un brin dit lourd (ou H pour Heavy),car riche en résidus guanine, et un brin léger(ou L pour Light). Chaque mitochondrie com-
porte plusieurs molécules d’ADNmt. Chaquemolécule possède 37 gènes codant pour2 ARN ribosomiques (ARNr 12S et 16S),22 ARN de transfert (ARNt) et 13 sous-unitésprotéiques : 7 appartiennent au complexe I(ND1-ND6, dont ND4L), 1 au complexe III(cytb), 3 au complexe IV (COXI-COXIII) et 2au complexe V (ATPase 6 et 8). La D-Loop,seule région non codante, comporte notam-ment l’origine de réplication du brin lourd etles promoteurs des 2 brins.
La réplication, la transcription et la traduc-tion de l’ADNmt se déroulent dans la matricemitochondriale. Selon le modèle de Clayton,la réplication débute avec la synthèse du brinlourd au niveau de son origine de réplicationet progresse dans le sens des aiguilles d’unemontre. Lorsque l’origine de réplication dubrin léger est atteinte et se retrouve exposéesous forme simple brin, le second brin estalors répliqué en sens inverse à partir du brinléger. Ce modèle de réplication est dit « bidi-rectionnel et asynchrone ». Un autre modèleest également proposé chez les mammifèresà partir de multiples origines de réplication.Les 2 brins d’ADNmt sont transcrits à partir depromoteurs spécifiques en ARNs polycistro-niques dont la maturation génère des ARNr,des ARNt et des ARN messagers (ARNm). LesARNm sont traduits dans la matrice mitochon-driale selon un code génétique différent ducode universel, avec une machinerie spécifi-que à la mitochondrie.
Deux spécificités de l’ADNmt sont parti-culièrement importantes pour la compréhen-sion des pathologies mitochondriales. D’unepart, la transmission de l’ADNmt est d’originematernelle. Les femmes transmettent leurADNmt à leurs enfants, alors que les hommesne le transmettent théoriquement pas. Parailleurs, au cours des mitoses, les mitochon-dries sont réparties au hasard dans les cellulesfilles (ségrégation mitotique). Si une cellulemère comporte 2 types d’ADNmt, il est pos-sible qu’au bout d’un certain nombre de divi-sions cellulaires, elle n’ait retenu qu’un seultype d’ADNmt (population homoplasmique).Mais elle peut aussi avoir retenu les 2 typesd’ADNmt, se traduisant, par la présence
5669_ Page 71 Vendredi, 4. mars 2011 12:09 12> STDI FrameMaker noir

72
dans un même tissu, de mitochondries por-tant des molécules d’ADNmt sauvages etmutées (population hétéroplasmique). Celasignifie que chez un même malade, le pour-centage d’ADNmt muté varie d’un type cel-lulaire à l’autre, un pourcentage d’ADNmtmuté élevé étant généralement retrouvé dansle tissu qui exprime le déficit.
2 Gènes nucléaires
Les études réalisées chez la levure mon-trent que le fonctionnement de la CR est appa-remment contrôlé par plus de 1 000 gènes dif-férents, qui sont tous des candidats potentielspour les pathologies mitochondriales. Lesgènes nucléaires codent pour :— la majorité des sous-unités protéiques de
la CR (traduites dans le cytoplasme puisimportées dans la mitochondrie) ;
— de nombreuses protéines impliquéesdans l’assemblage et le maintien des dif-férents complexes de la CR ;
— les protéines impliquées dans la traduc-tion mitochondriale, telles que les protéi-nes ribosomales, les aminoacyl-ARNt syn-thétases, les enzymes de modification desARNt et les facteurs d’élongation et determinaison ;
— toutes les protéines impliquées dans lemaintien et la stabilité de l’ADNmt : laréplication et la réparation de l’ADNmtsont sous le contrôle d’un grand nombrede protéines, telles que l’ADN polymé-rase gamma, seule ADN polymérase pré-sente dans la mitochondrie, ou l’hélicasemitochondriale Twinkle ;
— des protéines impliquées dans la bioge-nèse mitochondriale : dans la morphologiemitochondriale, des enzymes antioxydan-tes, des chaperonnes, des transporteurs…
II Présentations cliniques des maladies mitochondriales
Une maladie mitochondriale peut semanifester à tout âge, de la période néona-tale jusqu’à une période avancée de la vie.Les mitochondries étant présentes dans
toutes les cellules, une pathologie mitochon-driale peut toucher n’importe quel tissu ouorgane. L’expression clinique de ces affec-tions est donc très hétérogène (encéphalo-myopathie, retard mental, épilepsie, dia-bète, cardiomyopathie, surdité, cécité,insuffisance hépatique…) (tab. 5.1). Une« association illégitime » de symptômes quivont s’additionner sur un mode évolutif doitfaire suspecter une pathologie mitochon-driale. Mais il existe aussi des atteintesisolées subaiguës, comme dans l’atrophieoptique de Leber, par exemple. Les présen-tations cliniques étant très différentes, le dia-gnostic est souvent difficile. Enfin, le pro-nostic de ces affections est généralementpéjoratif, particulièrement dans les formesà début précoce. Néanmoins des amélio-rations, voire des guérisons spontanées,comme par exemple dans l’atrophie optiquede Leber ou dans des atteintes hépatiques,ont été rapportées et le pronostic est souventimprévisible et propre à chaque patient.
III Stratégie diagnostique des maladies mitochondriales
La démarche diagnostique repose sur desexplorations indirectes et des explorationstissulaires (enzymologiques, moléculaires ethistologiques) (fig. 5.2). Les progrès récentsles plus significatifs reposent, d’une part, surla mise en place de nouvelles méthodesd’étude de l’ADNmt et, d’autre part, sur l’iden-tification de nouveaux gènes nucléaires.
A Explorations indirectes
1 Bilan métabolique
Une hyperlactatémie persistante(> 2,5 mM), une élévation du rapport lac-tate/pyruvate (L/P > 20) et du rapport descorps cétoniques 3-hydroxybutyrate/acéto-acétate (3OHB/AcAc > 2) font suspecter unemaladie mitochondriale, en particulier enpériode néonatale. Ces rapports reflètentle statut d’oxydoréduction, respectivement
5669_ Page 72 Vendredi, 4. mars 2011 12:09 12> STDI FrameMaker noir
73
dans le cytoplasme et les mitochondries, etsont importants pour les diagnostics différen-tiels d’acidose lactique congénitale. En effet,dans un déficit en pyruvate déshydrogénase(PDH), le rapport L/P sera normal voireabaissé (L/P < 10). Ces dosages sanguins doi-vent être réalisés au repos, répétés au cours
de la journée, à jeun et en postprandial, afinde démasquer une hyperlactacidémie latenteet/ou une hypercétonémie paradoxale. Unehyperlactatorachie est également évocatricedans les formes neurologiques. L’excrétionurinaire des dérivés intermédiaires du cyclede Krebs et/ou de l’acide 3-méthylglutaconi-
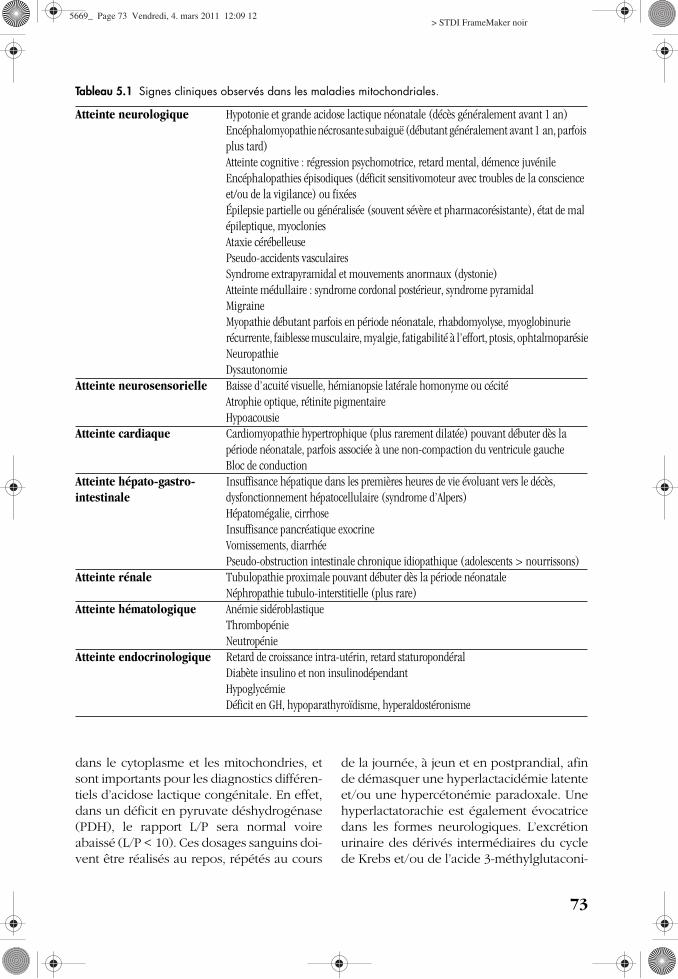
Tableau 5.1 Signes cliniques observés dans les maladies mitochondriales.
Atteinte neurologique Hypotonie et grande acidose lactique néonatale (décès généralement avant 1 an)Encéphalomyopathie nécrosante subaiguë (débutant généralement avant 1 an, parfois plus tard)Atteinte cognitive : régression psychomotrice, retard mental, démence juvénileEncéphalopathies épisodiques (déficit sensitivomoteur avec troubles de la conscience et/ou de la vigilance) ou fixéesÉpilepsie partielle ou généralisée (souvent sévère et pharmacorésistante), état de mal épileptique, myocloniesAtaxie cérébelleusePseudo-accidents vasculairesSyndrome extrapyramidal et mouvements anormaux (dystonie)Atteinte médullaire : syndrome cordonal postérieur, syndrome pyramidalMigraineMyopathie débutant parfois en période néonatale, rhabdomyolyse, myoglobinurie récurrente, faiblesse musculaire, myalgie, fatigabilité à l’effort, ptosis, ophtalmoparésieNeuropathieDysautonomie
Atteinte neurosensorielle Baisse d’acuité visuelle, hémianopsie latérale homonyme ou cécitéAtrophie optique, rétinite pigmentaireHypoacousie
Atteinte cardiaque Cardiomyopathie hypertrophique (plus rarement dilatée) pouvant débuter dès la période néonatale, parfois associée à une non-compaction du ventricule gaucheBloc de conduction
Atteinte hépato-gastro-intestinale
Insuffisance hépatique dans les premières heures de vie évoluant vers le décès, dysfonctionnement hépatocellulaire (syndrome d’Alpers)Hépatomégalie, cirrhoseInsuffisance pancréatique exocrineVomissements, diarrhéePseudo-obstruction intestinale chronique idiopathique (adolescents > nourrissons)
Atteinte rénale Tubulopathie proximale pouvant débuter dès la période néonataleNéphropathie tubulo-interstitielle (plus rare)
Atteinte hématologique Anémie sidéroblastiqueThrombopénieNeutropénie
Atteinte endocrinologique Retard de croissance intra-utérin, retard staturopondéralDiabète insulino et non insulinodépendantHypoglycémieDéficit en GH, hypoparathyroïdisme, hyperaldostéronisme
5669_ Page 73 Vendredi, 4. mars 2011 12:09 12> STDI FrameMaker noir

74
que, bien que non spécifique, est souventobservée dans les déficits de la CR. La chro-matographie des acides aminés sanguinspeut révéler indirectement une hyperlactaci-témie par élévation de l’alanine et de la pro-line et, occasionnellement, une hyperméthio-ninémie. Une hypocitrullinémie, égalementnon spécifique, a été notamment rapportéedans les déficits en complexe V causés par lamutation NARP. Par ailleurs, de nombreusessituations peuvent empêcher la détectiond’un déficit d’oxydoréduction dans leplasma : une tubulopathie proximale (baissede la lactatémie et augmentation de la lacta-
turie), un diabète sucré (diminution del’entrée du pyruvate dans le cycle de Krebs)ou une atteinte de la CR tissu-spécifique sansrépercussion significative sur l’état d’oxydo-réduction plasmatique. L’absence de ces ano-malies n’élimine pas le diagnostic et ne doitpas conduire à l’arrêt des explorations. Eneffet, en cas de suspicion de maladie mito-chondriale, un bilan « d’extension » systéma-tique de tous les organes et tissus potentiel-lement impliqués doit être réalisé à larecherche d’autres atteintes associées : exa-men ophtalmologique, auditif, échographiecardiaque… (tab. 5.2).
Figure 5.2 Stratégie diagnostique.
Clinique et examens paracliniques évocateurs de pathologies mitochondriales :Biologie :– élévation du lactate et du rapport L/P, hyperlactatorachie– élévation des dérivés du cycle de Krebs– bilan métabolique sans argument pour une autre pathologie métaboliqueIRM cérébrale : hypersignaux des NGC, pseudo-strokes, pic de lactates à la spectro IRM
Biopsie tissulaireMuscle, foie, rein, fibroblastes…
Analyse moléculaire Enzymologie Histologie
Recherche desmutations les plus
fréquentes
3243 : MELAS8344 : MERRF8993 : NARP
Délétionunique
(syndrome deKeams-Sayre,
PEO)
Délétionsmultiples
Délétionde l’ADNmt
Déplétionde l’ADNmt
Déficits combinésou multiples
de la CR
Déficit isoléde la CR :
complèxe I, II, III,IV ou V
Surcharge lipidique,RRF, fibres COX
négatives
Analyse des gènes nucléaires d’instabilité de l’ADNmt seIon le tableau clinique : POLG1, POLG2, ANTI, PEO1, DGUOK, MPV17, TK2, RRM2B, SUCLG1,
SUCLA2, TP… sur prélèvement sanguin
Analyse des gènes mitochondriaux (tissu) ou nucléaires (sang) correspondant aux sous-unités
du complexe déficitaire et/ou à la clinique,ou étude exhaustive de l’ADNmt par Surveyor
ou séquençage
–IRlaPrélèvement sanguin
• Recherche d’une délétion de l’ADNmt(syndrome de Pearson) ou des mutationsresponsables de l’atrophie optique de Leber• Recherche des mutations fréquentes maisla rentabilité est faible
5669_ Page 74 Vendredi, 4. mars 2011 12:09 12> STDI FrameMaker noir

75
2 Imagerie par résonance magnétique (IRM)
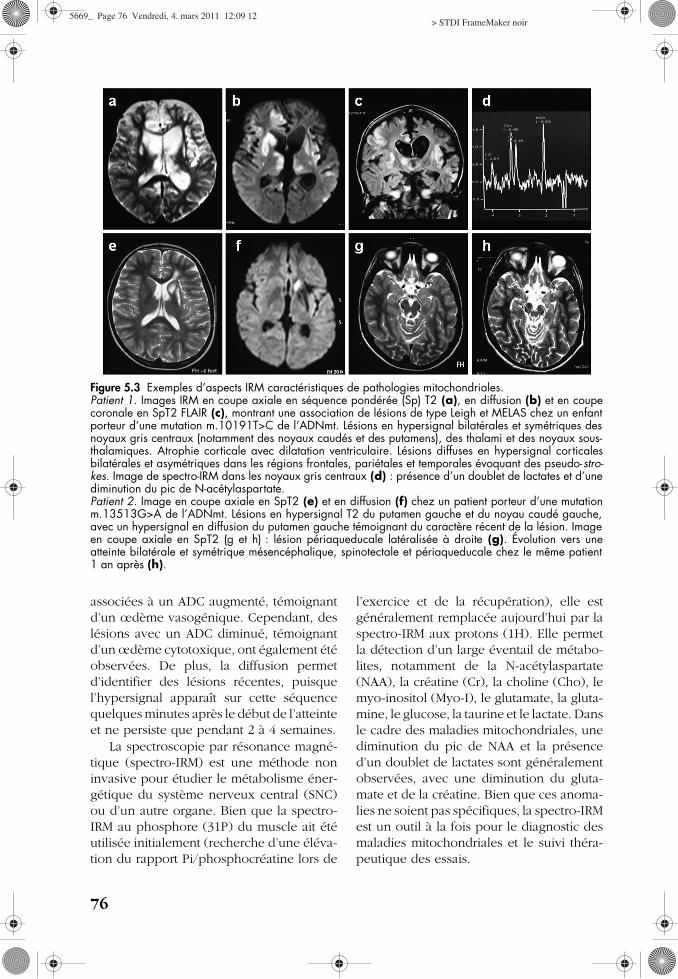
Les caractéristiques neuroradiologiquesdes maladies mitochondriales sont très varia-bles. L’IRM est l’examen le plus performanten sensibilité et en résolution pour identifierles atteintes encéphaliques. La tomodensito-métrie reste utile pour visualiser les calcifica-tions, qui sont uniquement visibles enséquence pondérée (Sp) T2* à l’IRM. Lesséquences IRM classiques apportent desinformations structurelles, et les techniquesd’imagerie plus récentes, comme les séquen-ces en diffusion et la spectroscopie, amènentdes données fonctionnelles et métaboliquessupplémentaires (fig. 5.3).
a Imagerie conventionnelle
Dans les maladies mitochondriales àexpression neurologique, des caractéristi-ques IRM communes ont été observées defaçon isolée ou associée [5.17] :— des anomalies bilatérales et symétriques de
la substance grise (corticale ou des noyauxgris centraux) et du tronc cérébral ;
— des anomalies de la substance blanchediffuses, à type de leucodystrophie oud’hypersignaux non spécifiques ;
— une atrophie cérébrale et cérébelleuse ;— des calcifications cérébrales.
À la phase aiguë et subaiguë, les lésionsapparaissent hyperintenses en SpT2 (témoinde l’œdème) et parfois hyperintenses en SpT1.Les lésions vont ensuite diminuer de taille,voire disparaître, avec souvent une atrophievisible de la structure concernée. La survenuede nouvelles lésions ou la réapparitiond’anciennes lésions sur une imagerie ulté-rieure peuvent donner un aspect de migrationdes lésions. Par ailleurs, la séquence T2 FLAIR(fluide atténué inversion récupération) est plussensible pour identifier certaines lésions,notamment périventriculaires, chez les enfantsde plus de 1 an.
b Apport des nouvelles techniques
L’imagerie de diffusion et le coefficientapparent de diffusion (ADC) permettent depréciser la nature des hypersignaux en SpT2,orientant ainsi la démarche diagnostique. Eneffet, ces séquences permettent de différen-cier l’œdème vasogénique, fréquemmentassocié aux lésions mitochondriales, del’œdème cytotoxique, observé dans lesinfarctus cérébraux. Les lésions mitochon-driales en hypersignal en diffusion sont
Tableau 5.2 Investigations à réaliser devant une suspicion de maladie mitochondriale.
Bilan métaboliqueRapport d’oxydoréduction (L/P et 3OHB/AcAc) à jeun et en post-prandialLactaturie et lactatorachieChromatographie des acides aminés plasmatiques et urinairesChromatographie des acides organiques urinairesProfil des acylcarnitines, carnitine libre et totale plasmatique et urinaireÉpreuve fonctionnelle (test de charge en glucose) rarement réaliséeBilan d’extensionOphtalmologique : FO, ERG, PEVCardiaque : échographie, ECGHépatique : transaminases, protéines de la coagulationPancréatique : recherche de stéatorrhée, dosage de l’élastase fécaleRénale : recherche de polyurie/polydipsie, glycosurie, pH urinaire, ionogramme sanguin et urinaireMusculaire : dosage des CPK, EMG, testing musculaire, étude en spectro-IRM (31P)Cérébrale : IRM cérébrale avec spectro-IRM, EEG
L/P : lactate/pyruvate ; 3OHB/AcAc : 3-hydroxybutyrate sur acétoacétate ; FO : fond d’œil ; ERG : électrorétinogramme ;PEV : potentiels évoqués visuels ; ECG : électrocardiogramme ; CPK : créatine phosphokinase ; EMG : électromyogramme ;EEG : électroencéphalogramme.
5669_ Page 75 Vendredi, 4. mars 2011 12:09 12> STDI FrameMaker noir
76
associées à un ADC augmenté, témoignantd’un œdème vasogénique. Cependant, deslésions avec un ADC diminué, témoignantd’un œdème cytotoxique, ont également étéobservées. De plus, la diffusion permetd’identifier des lésions récentes, puisquel’hypersignal apparaît sur cette séquencequelques minutes après le début de l’atteinteet ne persiste que pendant 2 à 4 semaines.
La spectroscopie par résonance magné-tique (spectro-IRM) est une méthode noninvasive pour étudier le métabolisme éner-gétique du système nerveux central (SNC)ou d’un autre organe. Bien que la spectro-IRM au phosphore (31P) du muscle ait étéutilisée initialement (recherche d’une éléva-tion du rapport Pi/phosphocréatine lors de
l’exercice et de la récupération), elle estgénéralement remplacée aujourd’hui par laspectro-IRM aux protons (1H). Elle permetla détection d’un large éventail de métabo-lites, notamment de la N-acétylaspartate(NAA), la créatine (Cr), la choline (Cho), lemyo-inositol (Myo-I), le glutamate, la gluta-mine, le glucose, la taurine et le lactate. Dansle cadre des maladies mitochondriales, unediminution du pic de NAA et la présenced’un doublet de lactates sont généralementobservées, avec une diminution du gluta-mate et de la créatine. Bien que ces anoma-lies ne soient pas spécifiques, la spectro-IRMest un outil à la fois pour le diagnostic desmaladies mitochondriales et le suivi théra-peutique des essais.
Figure 5.3 Exemples d’aspects IRM caractéristiques de pathologies mitochondriales.Patient 1. Images IRM en coupe axiale en séquence pondérée (Sp) T2 (a), en diffusion (b) et en coupecoronale en SpT2 FLAIR (c), montrant une association de lésions de type Leigh et MELAS chez un enfantporteur d’une mutation m.10191T>C de l’ADNmt. Lésions en hypersignal bilatérales et symétriques desnoyaux gris centraux (notamment des noyaux caudés et des putamens), des thalami et des noyaux sous-thalamiques. Atrophie corticale avec dilatation ventriculaire. Lésions diffuses en hypersignal corticalesbilatérales et asymétriques dans les régions frontales, pariétales et temporales évoquant des pseudo-stro-kes. Image de spectro-IRM dans les noyaux gris centraux (d) : présence d’un doublet de lactates et d’unediminution du pic de N-acétylaspartate. Patient 2. Image en coupe axiale en SpT2 (e) et en diffusion (f) chez un patient porteur d’une mutationm.13513G>A de l’ADNmt. Lésions en hypersignal T2 du putamen gauche et du noyau caudé gauche,avec un hypersignal en diffusion du putamen gauche témoignant du caractère récent de la lésion. Imageen coupe axiale en SpT2 (g et h) : lésion périaqueducale latéralisée à droite (g). Évolution vers uneatteinte bilatérale et symétrique mésencéphalique, spinotectale et périaqueducale chez le même patient1 an après (h).
5669_ Page 76 Vendredi, 4. mars 2011 12:09 12> STDI FrameMaker noir
77
c Anomalies IRM rencontrées dans certains syndromesLes caractéristiques et la localisation des
lésions IRM sont un élément déterminantpour le diagnostic des syndromes de Leigh etMELAS (tab. 5.3) [5.17]. Dans le syndrome deLeigh, les noyaux gris centraux sont sélecti-vement vulnérables à une défaillance dumétabolisme énergétique, et des hypersi-
gnaux en SpT2 sont communément observésdans le putamen, le pallidum et/ou le noyaucaudé. Cependant, ces lésions ne sont passpécifiques des déficits primaires de la CR, etpeuvent également être vues lors d’uneintoxication au monoxyde de carbone, d’unetoxicité de la bilirubine, d’un déficit enbiotinidase, des troubles du métabolisme desacides gras ou d’aciduries organiques [5.17].
Tableau 5.3 Caractéristiques IRM selon le phénotype.
Acidose lactique sévère infantile
Atrophie corticale avec un corps calleux mince visible à l’échographie et des lésions kystiques des ganglions de la baseAvec mutation m.3243A>G de l’ADNmt : lésions bilatérales du striatum, occipitales ou atrophie cérébrale
Syndrome de Leigh Lésions en hyposignal T1 et hypersignal T2 spin-écho et FLAIRAtteinte bilatérale et symétrique évocatrice, mais non systématiqueAtteintes des NGC (putamen +++, noyau caudé, pallidum), du thalamus, du mésencéphale (noyau rouge, substantia nigra et région périaqueducale), du tronc cérébral et du cervelet (noyaux dentelés)Atrophie des structures impliquées, associée à une atrophie cérébrale diffuse et/ou cérébelleusePlus rare : atteinte de la SB périventriculaire ou leucodystrophie, atteinte du corps calleux et des capsules internes, retard de myélinisationSpectro-IRM : diminution du NAA et présence d’un doublet de lactatesGène SURF1 : atteinte des noyaux sous-thalamiques, du tronc cérébral (bulbe), du cervelet (pédoncules cérébelleux inférieurs, noyaux dentelés), de la substantia nigra et du tractus tegmental central, mais l’atteinte des NGC est plus rareGène SDHA : leucodystrophie avec un pic de succinate caractéristique dans la SB cérébrale et cérébelleuse en spectro-IRM
Syndrome de Kearns-Sayre/PEO
Leucodystrophie (atteinte de la SB sous-corticale qui est épargnée dans la plupart des autres leucodystrophies)Atteinte des NGC (± calcifications), du thalamus et du mésencéphaleAtrophie corticale, du cervelet et du tronc cérébral
MELAS Association de lésions corticales de topographie non vasculaire, souvent occipitales (± prise de contraste), et de lésions des NGC (± calcifications)En phase aiguë : hypersignal en séquence de diffusion puis disparition des lésions ou apparition d’une atrophiePic de lactates dans les régions affectées en spectro-IRMAtrophie du cervelet, leucoencéphalopathie (rare)
NARP Atrophie pontocérébelleuse, pic de lactates en spectro-IRMCaractéristiques mimant celles du syndrome de Leigh, du MELAS, de l’encéphalomyélite aiguë disséminée ou d’une leucomalacie périventriculaire
LHON Hypersignaux de la SBMNGIE Leucoencéphalopathie épargnant le corps calleuxDéficit en coenzyme Q10 Atrophie marquée du cervelet, ± agénésie du corps calleux
PEO : ophtalmoplégie externe progressive ; MELAS : encéphalomyopathie mitochondriale avec acidose lactique et épisodes de stroke-like ; NARP : neuro-pathie ataxiante et rétinite pigmentaire ; LHON : neuropathie optique héréditaire de Leber ; MNGIE : encéphalopathie mitochondriale gastro-intestinale ;NGC : noyaux gris centraux ; SB : substance blanche ; NAA : N-acétylaspartate.
5669_ Page 77 Vendredi, 4. mars 2011 12:09 12> STDI FrameMaker noir

78
Dans le MELAS, les lésions focales de« pseudo-strokes » ont une distribution nonvasculaire et sont préférentiellement occipi-tales. Des aspects de leucodystrophie ont éténotamment associés au phénotype NARP, liéà la mutation m.8993T>G. Une étude récentesur les caractéristiques IRM chez les patientsporteurs d’un déficit en complexe I a montrél’existence, d’une part, d’aspects communs,comme les atteintes du tronc cérébral et laprésence de pic de lactates et, d’autre part,des caractéristiques IRM évocatrices dugénome impliqué [5.28]. En effet, les lésionsdes noyaux sous-thalamiques, périaqueduca-les et des colliculi étaient particulièrementprésentes avec les mutations de l’ADNmt. Laprésence d’une leucodystrophie (plutôt cavi-taire) et l’atteinte des noyaux dorsaux dutronc cérébral ont été observées uniquementchez les patients avec des mutations de gènesnucléaires [5.28].
B Explorations tissulairesLe diagnostic de maladie mitochondriale
repose sur un ensemble d’arguments parmilesquels la mise en évidence d’un déficit bio-chimique de la CR est un élément prépon-dérant. L’exploration enzymologique doitêtre réalisée si possible sur le tissu atteint(muscle, foie, rein…).
1 Explorations enzymologiques de la CR
Les tests diagnostiques comprennent desétudes en polarographie et spectrophotomé-trie, qui fournissent des informations diffé-rentes mais complémentaires. Les étudespolarographiques consistent à mesurer laconsommation d’oxygène dans des fractionsenrichies en mitochondries en utilisant uneélectrode Clarke, en présence de différentssubstrats oxydatifs. La limite de cette techni-que est l’utilisation obligatoire de prélève-ments frais. Les études spectrophotométri-ques consistent à doser l’activité enzymatiquedes différents complexes de la chaîne respi-ratoire, isolés ou combinés. Ces analyses,effectuées sur des homogénats de tissus, peu-vent être réalisées à partir d’une petite quan-
tité de matériel (1-20 mg), obtenue à partir debiopsies hépatique, rénale, endomyocardi-que ou de fibroblastes en culture. Les échan-tillons doivent être immédiatement congelésen azote liquide et conservés à – 80 °C. Letissu à étudier est celui qui exprime clinique-ment la maladie mais, en général, le muscleet la peau sont les tissus les plus accessibles.Le foie doit être prélevé en cas d’atteintehépatique ou d’atteinte neurologique avecépilepsie associée.
Les déficits peuvent être isolés ou com-binés. Le déficit en complexe I est le plus fré-quemment observé (30 % des patients). Undéficit combiné en complexes I, III et IVpeut orienter vers une déplétion de l’ADNmtou une mutation sur un gène ARNt del’ADNmt. Une même présentation cliniquepeut résulter d’un déficit en différents com-plexes et, inversement, un même déficit peutêtre à l’origine de présentations cliniquestrès différentes. Par exemple, le syndromede Leigh peut être associé à un déficit isoléou combiné de différents complexes de laCR et lié à des mutations dans des gènesmitochondriaux ou nucléaires.
2 Examen anatomopathologique
Il permet d’orienter le diagnostic en objec-tivant sur la biopsie musculaire une surchargelipidique, des agrégats mitochondriaux sous-sarcolemniques et des fibres COX-négativesà la coloration COX-SDH. La présence defibres ragged-red (RRF) après coloration autrichrome de Gomori (accumulation de mito-chondries anormales péri et intermyofibrillai-res) est extrêmement évocatrice mais n’estque très rarement retrouvée chez les jeunesenfants. Des anomalies ultrastructurales sontégalement observées en microscopie électro-nique (mitochondries globulaires, inclusionsparacristallines et crêtes mitochondrialesanormales). Une stéatose, une cirrhose, unefibrose, une hémosidérose, une proliférationdes canaux biliaires, voire un effondrementde l’architecture lobulaire peuvent être obser-vés sur la biopsie hépatique. Ces anomaliessont évocatrices mais non spécifiques. De
5669_ Page 78 Vendredi, 4. mars 2011 12:09 12> STDI FrameMaker noir

79
plus, l’absence de toute anomalie histologi-que ne doit pas faire réfuter le diagnostic.
C Explorations génétiquesLes maladies mitochondriales sont hétéro-
gènes sur le plan génétique et tous les modesde transmission sont possibles : cas sporadi-ques, transmission maternelle par mutationsde l’ADNmt, transmission autosomale réces-sive ou dominante et transmission liée à l’X parmutations dans des gènes nucléaires.
1 Anomalies de l’ADNmt
Elles sont retrouvées chez 10 à 15 % despatients atteints de maladies mitochondrialeset sont responsables de nombreux syndromes(tab. 5.4). Dans la majorité des cas, les ano-malies de l’ADNmt sont hétéroplasmiques etle nombre de molécules mutées est supérieurà celui des molécules sauvages dans le tissuatteint. La recherche de mutations de l’ADNmtdoit donc être réalisée, lorsque cela est possi-ble, dans le tissu qui exprime le déficit (biopsiemusculaire, hépatique voire rénale ou endo-myocardique). L’absence d’une mutation dansles leucocytes ne permet pas d’éliminer sa pré-sence dans d’autres tissus.
a Grands réarrangements de l’ADNmt
Les délétions de l’ADNmt sont retrouvéesprincipalement dans les syndromes deKearns-Sayre, de Pearson et dans les oph-talmoplégies externes progressives (PEO).Leur taille et leur position peuvent être varia-bles mais elles portent généralement sur plu-sieurs kilobases en emportant des gènescodant pour des sous-unités protéiques etdes ARNt. Elles sont présentes à l’état hété-roplasmique dans le muscle, à l’exceptiondu syndrome de Pearson où la délétion estretrouvée dans tous les tissus. Les bornes deces délétions sont constituées par desséquences répétées. Une délétion de4 977 paires de bases (pb) dite « commune »est retrouvée dans 30 % des cas, encadréepar une séquence répétée directe de 13 pb.Ces réarrangements surviennent générale-ment de novo au cours de l’oogenèse ou du
développement embryonnaire précoce, cequi explique que ces syndromes soientmajoritairement sporadiques.
b Mutations ponctuelles
• Dans des gènes codant pour des sous-unités protéiques
L’atrophie optique héréditaire de Leber(LHON) est caractérisée par une baisse rapideet bilatérale de la vision centrale par atteintedu nerf optique, touchant préférentiellementles hommes. La mutation responsable estgénéralement présente à l’état homoplasmi-que dans les leucocytes. Il existe 3 mutationsrécurrentes qui sont retrouvées chez90 % des patients, toutes dans des gènescodant pour des sous-unités du complexe I(m.11778G>A, m.3460G>A, m.14484T>C),mais d’autres mutations peuvent être respon-sables [5.37].
Une mutation dans le gène codant pourl’ATPase 6 (m.8993T>G) est égalementretrouvée de manière récurrente chez les patientsatteints d’un syndrome NARP/Leigh.La mutation est présente à l’état hétéroplas-mique et la gravité du tableau clinique estliée au pourcentage d’ADNmt muté présentdans les tissus. Le syndrome NARP (Neuro-pathic muscle weakness, Ataxia, RetinitisPigmentosa) débute après 5 ans et est carac-térisé par une neuropathie sensitivomotrice,une ataxie et une rétinite pigmentaire. L’évo-lution est marquée par l’apparition de signespyramidaux, extrapyramidaux, de convul-sions et d’une démence progressive. Le syn-drome de Leigh est une encéphalomyopa-thie subaiguë nécrosante caractérisée pardes épisodes récurrents de régression psy-chomotrice avec une atteinte des noyauxgris centraux, du tronc cérébral et de la subs-tance blanche, débutant généralement aucours de la première année de vie et rapi-dement fatale. Dans une même famille, unpatient atteint d’un syndrome de Leigh auraplus de 90 % d’ADNmt muté dans les tissus,alors qu’un patient présentant un syndromeNARP présentera un pourcentage de molé-cules mutées inférieur.
5669_ Page 79 Vendredi, 4. mars 2011 12:09 12> STDI FrameMaker noir
80
De nombreuses mutations dans desgènes codant pour des sous-unités descomplexes I, III et IV ont été rapportées.Elles sont responsables d’une grande variétéde tableaux cliniques et sont majoritaire-ment hétéroplasmiques. Elles peuvent sur-
venir de novo mais sont principalement héri-tées selon un mode maternel. Les mères sontalors porteuses d’une quantité faible demolécules mutées (10-20 %), et les enfantsatteints peuvent présenter plus de 90 % demolécules mutées dans le tissu atteint.
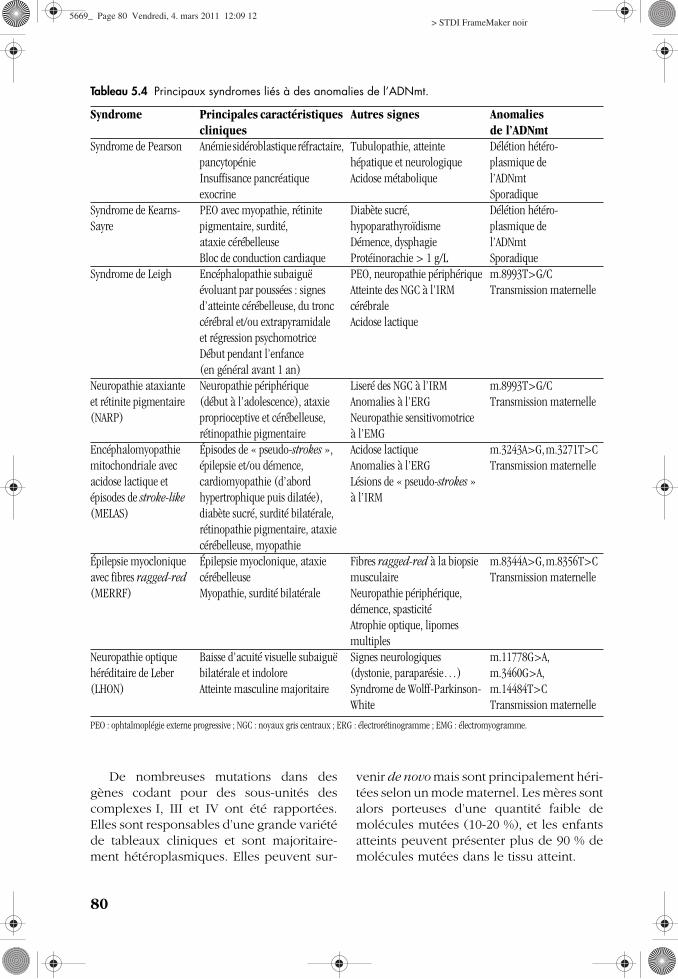
Tableau 5.4 Principaux syndromes liés à des anomalies de l’ADNmt.
Syndrome Principales caractéristiques cliniques
Autres signes Anomalies de l’ADNmt
Syndrome de Pearson Anémie sidéroblastique réfractaire, pancytopénieInsuffisance pancréatique exocrine
Tubulopathie, atteinte hépatique et neurologiqueAcidose métabolique
Délétion hétéro-plasmique del’ADNmtSporadique
Syndrome de Kearns-Sayre
PEO avec myopathie, rétinite pigmentaire, surdité, ataxie cérébelleuseBloc de conduction cardiaque
Diabète sucré, hypoparathyroïdismeDémence, dysphagieProtéinorachie > 1 g/L
Délétion hétéro-plasmique del’ADNmtSporadique
Syndrome de Leigh Encéphalopathie subaiguë évoluant par poussées : signes d’atteinte cérébelleuse, du tronc cérébral et/ou extrapyramidale et régression psychomotriceDébut pendant l’enfance (en général avant 1 an)
PEO, neuropathie périphériqueAtteinte des NGC à l’IRM cérébraleAcidose lactique
m.8993T>G/CTransmission maternelle
Neuropathie ataxiante et rétinite pigmentaire (NARP)
Neuropathie périphérique (début à l’adolescence), ataxie proprioceptive et cérébelleuse, rétinopathie pigmentaire
Liseré des NGC à l’IRMAnomalies à l’ERGNeuropathie sensitivomotrice à l’EMG
m.8993T>G/CTransmission maternelle
Encéphalomyopathie mitochondriale avec acidose lactique et épisodes de stroke-like (MELAS)
Épisodes de « pseudo-strokes », épilepsie et/ou démence, cardiomyopathie (d’abord hypertrophique puis dilatée), diabète sucré, surdité bilatérale, rétinopathie pigmentaire, ataxie cérébelleuse, myopathie
Acidose lactiqueAnomalies à l’ERGLésions de « pseudo-strokes » à l’IRM
m.3243A>G, m.3271T>CTransmission maternelle
Épilepsie myoclonique avec fibres ragged-red (MERRF)
Épilepsie myoclonique, ataxie cérébelleuseMyopathie, surdité bilatérale
Fibres ragged-red à la biopsie musculaireNeuropathie périphérique, démence, spasticitéAtrophie optique, lipomes multiples
m.8344A>G, m.8356T>CTransmission maternelle
Neuropathie optique héréditaire de Leber (LHON)
Baisse d’acuité visuelle subaiguë bilatérale et indoloreAtteinte masculine majoritaire
Signes neurologiques (dystonie, paraparésie…)Syndrome de Wolff-Parkinson-White
m.11778G>A, m.3460G>A, m.14484T>CTransmission maternelle
PEO : ophtalmoplégie externe progressive ; NGC : noyaux gris centraux ; ERG : électrorétinogramme ; EMG : électromyogramme.
5669_ Page 80 Vendredi, 4. mars 2011 12:09 12> STDI FrameMaker noir

81
• Dans des gènes codant pour des ARN de transfert ou ribosomaux
La mutation m.3243A>G dans le gènecodant pour l’ARNtleu est responsable du syn-drome MELAS (Mitochondrial Encephalomyo-pathy with Lactic Acidosis and Stroke-like epi-sodes syndrome). La symptomatologie débutedans l’enfance et est caractérisée par des épi-sodes récurrents de migraines et vomisse-ments, avec déficit neurologique évoquant unaccident vasculaire cérébral, et sur l’imageriecérébrale des lésions non vasculaires. Le syn-drome MELAS peut être secondaire à d’autresmutations telles que la m.3271T>C. À l’inverse,la mutation m.3243A>G est également respon-sable du syndrome MIDD (Maternally Inheri-ted Diabetes and Deafness), associant un dia-bète sans surpoids et une surdité prédominantsur les fréquences aiguës [5.26]. La mutationm.8344A>G dans le gène codant pour unARNtlys est retrouvée chez 80 % des patientsprésentant un syndrome MERRF (MyoclonusEpilepsy with Ragged-Red Fibers). Biend’autres mutations dans des ARNt ou des ARNront été décrites dans différentes familles(http://www.mitomap.org/MITOMAP) [5.37].
c Screening de l’ADNmt
Lorsque les principales mutations ont étééliminées, une analyse plus complète del’ADNmt peut être réalisée. Le séquençagesystématique de l’ADNmt est compliqué parla taille importante de l’ADNmt (16,5 kb) etl’hétéroplasmie. De nouvelles techniques ontdonc été développées. La méthode Surveyor(Transgenomic) utilise une endonucléase quireconnaît et clive les mésappariements del’ADN double brin. Elle permet ainsi d’identi-fier les mutations hétéroplasmiques del’ADNmt [5.2, 5.3]. L’analyse complémentaireréalisée grâce à une puce de reséquençageAffymetrix (GeneChip Mitochondrial Rese-quencing 2.0 Array) permet d’identifier lesmutations homoplasmiques de l’ADNmt [5.29].
2 Mutations dans des gènes nucléaires
Le protéome mitochondrial comprendplus de 1 000 protéines codées par desgènes nucléaires qui sont tous des candidats
potentiels pour les maladies mitochondria-les. Seule une cinquantaine de gènes res-ponsables est connue à ce jour mais ce nom-bre est en augmentation constante [5.41].
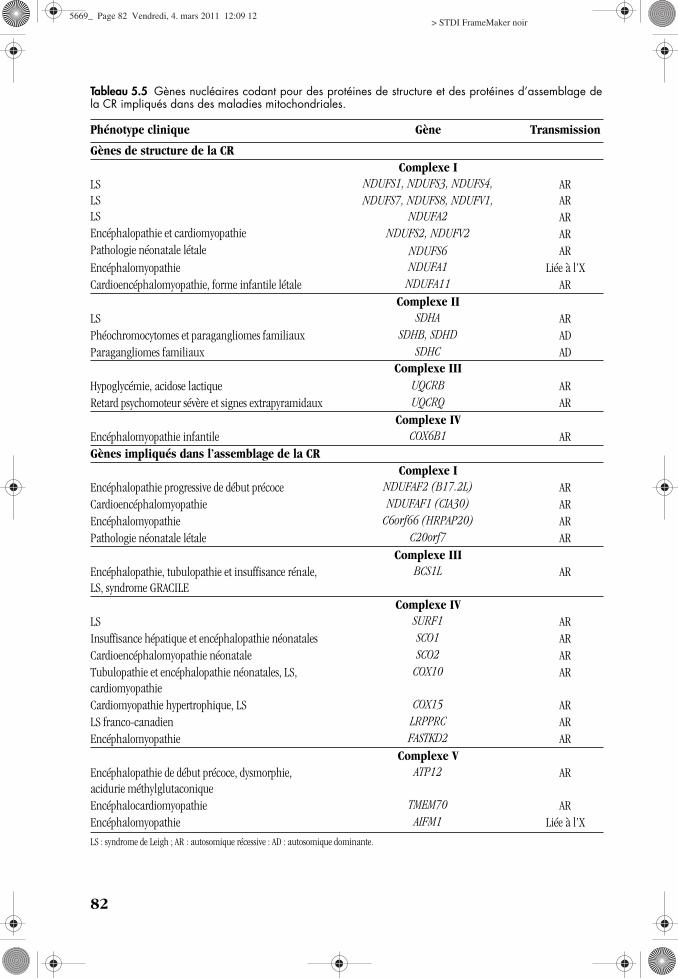
a Gènes de structure de la CRDes mutations pathogènes ont été décri-
tes dans 12 des gènes de structure ducomplexe I (tab. 5.5). Elles entraînent dans lamajorité des cas un syndrome de Leigh ouun tableau neurologique, associé ou nonà une cardiomyopathie [5.13]. L’implicationdu gène SDHA (sous-unité flavoprotéique) aété décrite dans le syndrome de Leigh associéà un déficit en complexe II [5.6]. Parmi lesgènes des 10 sous-unités nucléaires du com-plexe III, 2 (UQCRB et UQCRQ) ont été impli-qués respectivement dans un tableau d’hypo-glycémie avec acidose lactique et dans unphénotype neurologique avec retard psycho-moteur sévère et signes extrapyramidaux[5.4]. Seul un gène de structure du com-plexe IV (COX6B1), responsable d’uneencéphalomyopathie sévère, a été identifié[5.31] et, à ce jour, aucune mutation dans ungène de structure du complexe V n’a pu êtremise en évidence.
b Gènes d’assemblage de la CRPlusieurs gènes d’assemblage des com-
plexes I et IV ont été impliqués respective-ment dans des tableaux neurologiques etdans des atteintes multisystémiques (neuro-logiques, cardiaques, hépatiques et rénales)(tab. 5.5). Des mutations dans le gèneBCS1L, qui code pour un facteur d’assem-blage du complexe III, ont été décrites dansun tableau associant une encéphalopathie,une insuffisance hépatique et une tubulopa-thie ainsi que dans le syndrome GRACILE(retard de croissance, aminoacidurie, cho-lestase, surcharge en fer et décès précoce).De la même façon, deux gènes d’assemblagedu complexe V (ATP12, TMEM70) ont étéidentifiés [5.10, 5.11].
c Gènes impliqués dans la traduction mitochondrialeLes gènes identifiés appartiennent à 3 clas-
ses fonctionnelles différentes (tab. 5.6) : ceux
5669_ Page 81 Vendredi, 4. mars 2011 12:09 12> STDI FrameMaker noir
82
Tableau 5.5 Gènes nucléaires codant pour des protéines de structure et des protéines d’assemblage dela CR impliqués dans des maladies mitochondriales.
Phénotype clinique Gène Transmission
Gènes de structure de la CRComplexe I
LS NDUFS1, NDUFS3, NDUFS4, ARLS NDUFS7, NDUFS8, NDUFV1, ARLS NDUFA2 AREncéphalopathie et cardiomyopathie NDUFS2, NDUFV2 ARPathologie néonatale létale NDUFS6 AREncéphalomyopathie NDUFA1 Liée à l’XCardioencéphalomyopathie, forme infantile létale NDUFA11 AR
Complexe IILS SDHA ARPhéochromocytomes et paragangliomes familiaux SDHB, SDHD ADParagangliomes familiaux SDHC AD
Complexe IIIHypoglycémie, acidose lactique UQCRB ARRetard psychomoteur sévère et signes extrapyramidaux UQCRQ AR
Complexe IVEncéphalomyopathie infantile COX6B1 ARGènes impliqués dans l’assemblage de la CR
Complexe IEncéphalopathie progressive de début précoce NDUFAF2 (B17.2L) ARCardioencéphalomyopathie NDUFAF1 (CIA30) AREncéphalomyopathie C6orf66 (HRPAP20) ARPathologie néonatale létale C20orf7 AR
Complexe IIIEncéphalopathie, tubulopathie et insuffisance rénale, LS, syndrome GRACILE
BCS1L AR
Complexe IVLS SURF1 ARInsuffisance hépatique et encéphalopathie néonatales SCO1 ARCardioencéphalomyopathie néonatale SCO2 ARTubulopathie et encéphalopathie néonatales, LS, cardiomyopathie
COX10 AR
Cardiomyopathie hypertrophique, LS COX15 ARLS franco-canadien LRPPRC AREncéphalomyopathie FASTKD2 AR
Complexe VEncéphalopathie de début précoce, dysmorphie, acidurie méthylglutaconique
ATP12 AR
Encéphalocardiomyopathie TMEM70 AREncéphalomyopathie AIFM1 Liée à l’X
LS : syndrome de Leigh ; AR : autosomique récessive : AD : autosomique dominante.
5669_ Page 82 Vendredi, 4. mars 2011 12:09 12> STDI FrameMaker noir

83
codant pour des protéines des mitoribosomes,ceux impliqués dans la maturation des ARNtmitochondriaux et ceux codant pour des fac-teurs d’élongation [5.19, 5.41, 5.42].
d Gènes impliqués dans le métabolisme de l’ADNLes gènes identifiés à ce jour peuvent être
classés en 2 catégories : ceux impliquésdans la réplication de l’ADNmt et ceux régu-lant le pool de nucléotides (tab. 5.6).
Les ophtalmoplégies externes de transmis-sion autosomale dominante (AdPEO) débu-tent en général à l’âge adulte, et sont associéesà des délétions multiples de l’ADNmt, retrou-vées uniquement dans le muscle. Plus rare-
ment, la transmission est autosomale récessive(ArPEO). Plusieurs gènes ont été impliquésdans les AdPEO : POLG1 et POLG2 (sous-unités catalytique et accessoire de la polymé-rase γ) [5.30], PEO1 (hélicase Twinkle), ANT1(translocase ADP/ATP mitochondriale) etOPA1 (GTPase impliquée dans la fusion mito-chondriale) [5.1].
Le syndrome MNGIE (MitochondrialNeuro-Gastro-Intestinal Encephalopathy) estcaractérisé par une PEO, un ptosis, des trou-bles de la motilité intestinale avec cachexie,une neuropathie, une myopathie et une leu-codystrophie diffuse. Les patients ont desdélétions multiples et/ou une déplétion dansle muscle secondaires à des mutations dans
Tableau 5.6 Gènes nucléaires codant pour des protéines impliquées dans la traduction mitochondriale etdans la stabilité de l’ADNmt.
Phénotype clinique Gène Transmission
Gènes impliqués dans la traduction mitochondrialeMitoribosome
Agénésie du corps calleux, dysmorphie et acidose lactique néonatale fataleŒdème cutané, cardiomyopathie et tubulopathie
MRPS16
MRPS22
AR
ARMaturation des ARNt mitochondriaux
Myopathie, acidose lactique et anémie sidéroblastiqueAtteinte hépatique
PUS1
TRMU
AR
ARFacteurs d’élongation
Leucodystrophie infantile macrokystique avec micropolygyrieEncéphalomyopathie, cardiomyopathie hypertrophiqueEncéphalopathie sévère avec acidose lactique/atteinte hépatique
EFTu
EFTs
EFG1
AR
AR
AR
Gènes impliqués dans la stabilité de l’ADNmtAdPEOArPEOMNGIEAtteinte hépatocérébrale, syndrome d’AlpersMyopathieMyopathie avec déplétion très sévèreEncéphalopathie néonatale avec acidémie méthylmalonique
POLG1, POLG2, PEO1, ANT1, OPA1, RRM2BPOLG1
TPPOLG1, PEO1, DGUOK, MPV17
TK2RRM2B
SUCLG1, SUCLA2
ADARARARARARAR
LS : syndrome de Leigh ; AdPEO : ophtalmoplégie externe de transmission autosomale dominante ; ArPEO : ophtalmoplégie externe de transmission auto-somale récessive ; AR : autosomique récessive ; AD : autosomique dominante.
5669_ Page 83 Vendredi, 4. mars 2011 12:09 12> STDI FrameMaker noir

84
le gène TP, qui code pour la thymidine phos-phorylase, enzyme cytosolique impliquéedans l’équilibre intramitochondrial du poolde déoxynucléotides (dNTP).
Le syndrome de déplétion de l’ADNmt a étédécrit dans différentes formes cliniques : myo-pathique, encéphalomyopathique ou hépato-cérébrale, qui affectent généralement de jeunesenfants. Un déficit enzymologique (tissu-spéci-fique) touchant les complexes dépendant del’ADNmt et respectant le complexe II peut évo-quer une déplétion, qui sera confirmée par ladiminution du niveau d’ADNmt, généralementinférieur à 10 % de la valeur normale. À ce jour,8 gènes, dont certains sont également respon-sables de délétions multiples de l’ADNmt, ontété identifiés. Une forme hépatocérébrale [5.15]ou un syndrome d’Alpers (épilepsie pharma-corésistante, insuffisance hépatique et retardpsychomoteur) [5.33] doivent faire rechercherdes mutations dans les gènes POLG1, PEO1(codant pour Twinkle), DGUOK (codant pourla déoxyguanosine kinase mitochondriale,impliquée dans l’équilibre du pool mitochon-drial de dNTP) ou MPV17, dont la fonctionest inconnue [5.38]. Une forme myopathiquedoit orienter vers le gène TK2, codant pourla thymidine kinase, également impliquée dansl’équilibre du pool de dNTP mais égalementvers le gène RRM2B, particulièrement lorsquela déplétion est très sévère (ADNmt correspon-dant à 1 ou 2 % de la valeur normale) [5.8].Le gène RRM2B code pour la petite sous-unitéde la ribonucléotide réductase cytosolique quicatalyse la synthèse des dNTP à partir des NTP.Enfin, des mutations dans les gènes codant pourdes sous-unités de la succinyl-CoA synthase(SUCLA2, SUCLG1) sont responsables de syn-drome de déplétion de l’ADNmt avec acidémieméthylmalonique modérée [5.14, 5.34].
IV Prise en charge et traitement des maladies mitochondriales
Le traitement des maladies mitochon-driales est essentiellement symptomatique,à l’exception des maladies mitochondriales
par déficit primaire en coenzyme Q10 (CoQ)[5.36].
A Traitement spécifique : coenzyme Q10
Le CoQ est efficace dans les déficits pri-maires de la biosynthèse du CoQ quand ilest introduit précocement et à forte dose. Laprésentation clinique de ces pathologies estvariable : syndrome de Leigh, maladie mul-tisystémique avec néphropathie prédomi-nante, épisodes de rhabdomyolyse avecconvulsions, ataxie avec ou sans épilepsie,et myopathie pure [5.25]. En revanche, l’effi-cacité du coenzyme Q10 dans les autresmaladies de la CR est variable, et les résultatsdes essais contrôlés randomisés ont donnédes résultats contradictoires [5.9].
B Prise en charge
1 Régime cétogène
Un régime pauvre en carbohydrates etriche en lipides (60 à 70 % de la ration calo-rique) est recommandé dans les déficits encomplexe I associés à une hyperlactacidé-mie. En effet, l’apport important de glucoseest déconseillé car il ne peut être utilisé dufait du bloc enzymatique. Il faut égalementéviter tout jeûne prolongé.
2 Traitements symptomatiques
De nombreux agents pharmacologiquesont été essayés dans les maladies mitochon-driales, mais les bénéfices ont été limités etaucun n’a apporté la preuve de son efficacité.Les quelques essais cliniques randomisés endouble aveugle réalisés ont donné des résul-tats peu concluants ou contradictoires, ne per-mettant pas d’établir des recommandationsthérapeutiques [5.9]. Bien qu’il existe des casrapportés anecdotiques de l’efficacité dedivers agents (riboflavine, succinate, L-carni-tine, acide alpha-lipoïque et vitamines C, Eet K), l’hétérogénéité clinique et l’évolutionimprévisible des maladies mitochondriales,faite souvent de rechutes et de rémissions,
5669_ Page 84 Vendredi, 4. mars 2011 12:09 12> STDI FrameMaker noir

85
rend très difficile l’interprétation de l’efficacitéd’un agent chez un seul individu. Récemment,de nouvelles approches pharmacologiquesont émergé visant à stimuler la biogenèse mito-chondriale via le coactivateur transcriptionnelPGC1a, avec des agents comme le bézafibrateet le resvératrol [5.5, 5.24, 5.39].
a Correction des acidoses lactiques aiguës ou chroniques
Les bicarbonates peuvent être utiliséspour corriger des acidoses lactiques aiguësou chroniques. Le dichloroacétate (DCA), enmaintenant la pyruvate déshydrogénasekinase dans son état activé, réduit la pro-duction de lactates. Le DCA peut être effi-cace dans les états d’acidose aiguë, mais sonutilisation est limitée par sa toxicité, respon-sable de neuropathie périphérique, régres-sive à l’arrêt du DCA [5.20].
b Élimination ou neutralisation de métabolites toxiques
L’utilisation de la dialyse rénale, pouréliminer l’accumulation de thymidine etdésoxyuridine dans le plasma des patientsMNGIE, ou de diurétiques pour augmenter leurexcrétion rénale, n’a pas été concluante [5.40].
c Substitution enzymatique ou de métabolites
Les stratégies thérapeutiques visant àremplacer l’activité de la thymidine phos-phorylase (TP) par des transfusions répétéesde plaquettes [5.27] ou par l’administrationde TP encapsulée dans des globules rouges[5.32] n’ont entraîné que des réductions tran-sitoires du niveau de thymidine plasmati-que. La transplantation allogénique de cel-lules souches a été la méthode la plusefficace pour restaurer l’activité de la TPchez les patients MNGIE, mais elle s’estaccompagnée d’une mortalité élevée, vrai-semblablement liée au stade de cachexieavancé des malades [5.18]. La transplantationhépatique, utilisée pour traiter des déficitsen déoxyguanosine kinase, n’a permisd’obtenir que des bénéfices très limités[5.12].
d Traitement par L-arginine dans le MELAS
Dans les « pseudo-strokes » du MELAS, ilexiste un dysfonctionnement endothélial,une altération segmentaire de la vasodilata-tion des artères intracérébrales et une pro-duction d’espèces réactives oxygénées(ROS). De récentes études suggèrent que lathérapie par L-arginine serait efficace dans laprévention et le traitement de ces épisodes.L’arginine, précurseur de l’acide nitrique, agi-rait principalement en favorisant la microvas-cularisation cérébrale, en augmentant le débitsanguin par vasodilatation et en préservantles cellules endothéliales par diminution deslésions médiées par les radicaux libres [5.23].Des perfusions de L-arginine, instaurées dansles 30 minutes après le début des signes, amé-liorent significativement les symptômes enphase aiguë [5.22]. Par ailleurs, une supplé-mentation orale par L-arginine au long coursen périodes intercritiques semble améliorerles fonctions endothéliales, normaliser lestaux plasmatiques de L-arginine et diminuerde manière significative la fréquence et lagravité des « pseudo-strokes » [5.21].
e Supplémentation en acide folique
Dans le syndrome de Kearns-Sayre, desdéficits secondaires en folates dans le LCR ontdéjà été rapportés, mais on ne connaît pas leurprévalence dans l’ensemble des maladiesmitochondriales. En cas de suspicion de défi-cit central en folates, le dosage doit être réalisédans le LCR car le taux de folates plasmatiquene reflète pas exactement le statut en folatesdu SNC. Récemment, une réponse cliniquerapide à l’acide folinique a été rapportée chezun garçon de 8 ans porteur d’une délétion del’ADNmt avec une leucoencéphalopathie etune carence en acide folique [5.35]. Il fautnoter que, contrairement aux folates, l’acidefolinique traverse la barrière hématoencé-phalique.
f Contre-indications médicamenteuses
Certains médicaments ont des consé-quences négatives sur le fonctionnement dela CR, contre-indiquant ou nécessitant des
5669_ Page 85 Vendredi, 4. mars 2011 12:09 12> STDI FrameMaker noir

86
précautions dans leur utilisation chez despatients atteints de maladies mitochondriales.Certaines substances ont une action inhibi-trice sur la synthèse des protéines mitochon-driales, comme les inhibiteurs nucléosidiquesde la transcriptase inverse, l’interféron α oule chloramphénicol. D’autres agissent direc-tement sur la β-oxydation ou la séquestrationdu coenzyme A, comme le valproate desodium, l’aspirine, les anti-inflammatoiresnon stéroïdiens et les tétracyclines, et peuvententraîner des insuffisances hépatiques sévè-res. Les anesthésiants ont également des inte-ractions complexes avec la mitochondrie. Lerisque de PRIS (Propofol Infusion Syndrome)contre-indique l’utilisation du propofol encontinu. Il faut également être prudent dansl’utilisation de l’halothane, de l’isoflurane etdes curares, notamment la succinylcholine.Les risques liés aux vaccinations sont égale-ment discutés, notamment dans les formesneurologiques. Les vaccins pourraient eneffet favoriser la survenue de poussées dansle syndrome de Leigh.
V Conseil génétique et diagnostic prénatal
Du fait de l’hétérogénéité génétique desmaladies mitochondriales, si l’identificationde la mutation responsable est indispensa-ble pour guider le conseil génétique, elle nepermet pas toujours de proposer un dia-gnostic prénatal fiable. S’il s’agit de muta-tions dans un gène nucléaire, la transmissionde la pathologie se fera selon un mode men-délien. Le risque pourra être fixé précisé-ment et la recherche des mutations se ferasur prélèvement de villosités choriales à11 semaines d’aménorrhée (SA) dans de très
bonnes conditions de fiabilité. Les délétionsuniques de l’ADNmt sont généralement spo-radiques. Si la mère du cas index n’est pasporteuse de la délétion, le risque de trans-mission est très faible. Un DPN sur liquideamniotique sera proposé à 16 SA. En cas demutation ponctuelle hétéroplasmique del’ADNmt, si la mutation est présente dans lesleucocytes de la mère du cas index, le risquede récurrence est élevé mais très difficile àdéterminer avec précision. La fiabilité duDPN est peu satisfaisante du fait de la notiond’hétéroplasmie. En fonction de la volontédu couple, il est possible de proposer plu-sieurs prélèvements (à 11, 16 et 20 SA) afind’analyser l’évolution du taux d’hétéro-plasmie au cours de la grossesse dans destypes cellulaires différents. Un pourcentaged’ADNmt mutant inférieur à 20 % ou supé-rieur à 80 % devrait prédire un risqued’expression de la maladie faible ou élevé,respectivement [5.7]. Les résultats intermé-diaires ont une valeur prédictive encoremoins certaine. Si la mutation est absentedans différents tissus de la mère du cas index(cellules épithéliales urinaires, leucocytes,frottis buccal) ainsi que chez les apparentésmaternels, le risque de récurrence est faibleet il s’agit vraisemblablement d’une mutationde novo. Un DPN sur liquide amniotique seraproposé à 16 SA. Pour les mutations ponc-tuelles homoplasmiques (LHON), le conseilgénétique est également difficile car denombreux facteurs encore inconnus sontimpliqués dans l’expressivité de la maladie.Actuellement, seul le don d’ovocytes permetthéoriquement d’empêcher la transmissionmaternelle d’une maladie mitochondriale.Toutefois, certaines études préliminairesconcernant les mutations NARP et MELASsuggèrent que le diagnostic préimplanta-toire serait une alternative intéressante[5.16]. Enfin, un homme atteint n’a aucunrisque de transmettre une anomalie del’ADNmt à sa descendance.
5669_ Page 86 Vendredi, 4. mars 2011 12:09 12> STDI FrameMaker noir

87
Conclusion
Malgré les progrès réalisés, le diagnosticdes maladies mitochondriales reste difficile,notamment du fait de leur hétérogénéité cli-nique et génétique. L’identification des nom-breux gènes nucléaires responsables est unobjectif majeur car, outre l’intérêt diagnosti-que et du conseil génétique, elle permettra demieux comprendre les mécanismes physio-pathologiques responsables de ces maladies.Cette compréhension est un préalable indis-pensable au développement de traitementsspécifiques, actuellement inexistants, dansdes pathologies dont le pronostic est sombre.
Références[5.1] Amati-Bonneau P, Valentino ML, Reynier
P, Gallardo ME, Bornstein B, Boissiere Aet al. OPA1 mutations induce mitochon-drial DNA instability and optic atrophy“plus” phenotypes. Brain 2008 ; 131 :338-51.
[5.2] Bannwarth S, Procaccio V, Paquis-Fluc-klinger V. Surveyor Nuclease : a newstrategy for a rapid identification of hete-roplasmic mitochondrial DNA mutationsin patients with respiratory chain defects.Hum Mutat 2005 ; 25 : 575-82.
[5.3] Bannwarth S, Procaccio V, Paquis-Fluc-klinger V. Rapid identification of unknownheteroplasmic mutations across the entirehuman mitochondrial genome with mis-match-specific Surveyor Nuclease. NatProtoc 2006 ; 1 : 2037-47.
[5.4] Barel O, Shorer Z, Flusser H, Ofir R,Narkis G, Finer G et al. Mitochondrialcomplex III deficiency associated with ahomozygous mutation in UQCRQ. Am JHum Genet 2008 ; 82 : 1211-6.
[5.5] Bastin J, Aubey F, Rötig A, Munnich A,Djouadi F. Activation of peroxisome pro-liferator-activated receptor pathway sti-mulates the mitochondrial respiratorychain and can correct deficiencies inpatients’ cells lacking its components. JClin Endocrinol Metab 2008 ; 93 : 1433-41.
[5.6] Baysal BE. Clinical and molecular pro-gress in hereditary paraganglioma. J MedGenet 2008 ; 45 : 689-94.
[5.7] Bouchet C, Steffann J, Corcos J, MonnotS, Paquis V, Rotig A et al. Prenatal diagno-sis of MELAS syndrome : contribution tounderstanding mitochondrial DNA segre-gation during human embryo fetal deve-lopment. J Med Genet 2006 ; 43 : 788-92.
[5.8] Bourdon A, Minai L, Serre V, Jais JP, SarziE, Aubert S et al. Mutation of RRM2B,encoding p53-controlled ribonucleotidereductase (p53R2), causes severe mito-chondrial DNA depletion. Nat Genet2007 ; 39 : 776-80.
[5.9] Chinnery P, Majamaa K, Turnbull D,Thorburn D. Treatment for mitochon-drial disorders. Cochrane Database SystRev 2006 : CD004426.
[5.10] Cizkova A, Stranecky V, Mayr JA, Tesa-rova M, Havlickova V, Paul J et al.TMEM70 mutations cause isolated ATPsynthase deficiency and neonatal mito-chondrial encephalocardiomyopathy.Nat Genet 2008 ; 40 : 1288-90.
[5.11] De Meirleir L, Seneca S, Lissens W, DeClercq I, Eyskens F, Gerlo E et al. Respi-ratory chain complex V deficiency due toa mutation in the assembly gene ATP12.J Med Genet 2004 ; 41 : 120-4.
[5.12] Dimmock DP, Dunn JK, Feigenbaum A,Rupar A, Horvath R, Freisinger P et al.Abnormal neurological features predictpoor survival and should preclude livertransplantation in patients with deoxy-guanosine kinase deficiency. LiverTranspl 2008 ; 14 : 1480-5.
[5.13] Distelmaier F, Koopman WJ, Van denHeuvel LP, Rodenburg RJ, Mayatepek E,Willems PH et al. Mitochondrial com-plex I deficiency : from organelle dys-function to clinical disease. Brain 2009 ;132 : 833-42.
[5.14] Elpeleg O, Miller C, Hershkovitz E,Bitner-Glindzicz M, Bondi-Rubinstein G,Rahman S et al. Deficiency of the ADP-forming succinyl-CoA synthase activity isassociated with encephalomyopathy andmitochondrial DNA depletion. Am J HumGenet 2005 ; 76 : 1081-6.
[5.15] Ferrari G, Lamantea E, Donati A, FilostoM, Briem E, Carrara F et al. Infantilehepatocerebral syndromes associatedwith mutations in the mitochondrial DNApolymerase-gamma A. Brain 2005 ; 128 :723-31.
5669_ Page 87 Vendredi, 4. mars 2011 12:09 12> STDI FrameMaker noir

88
[5.16] Feyereisen E, Steffann J, Romana S,Lelorc’h M, Ray P, Kerbrat V et al. Fiveyears’ experience of preimplantationgenetic diagnosis in the Parisian Center :outcome of the first 441 started cycles.Fertil Steril 2007 ; 87 : 60-73.
[5.17] Haas R, Dietrich R. Neuroimaging ofmitochondrial disorders. Mitochondrion2004 ; 4 : 471-90.
[5.18] Hirano M, Marti R, Casali C, Tadesse S,Uldrick T, Fine B et al. Allogeneic stemcell transplantation corrects biochemicalderangements in MNGIE. Neurology2006 ; 67 : 1458-60.
[5.19] Jacobs HT, Turnbull DM. Nuclear genesand mitochondrial translation : a newclass of genetic disease. Trends Genet2005 ; 21 : 312-4.
[5.20] Kaufmann P, Engelstad K, Wei Y, JhungS, Sano MC, Shungu DC et al. Dichloroa-cetate causes toxic neuropathy inMELAS : a randomized, controlled clini-cal trial. Neurology 2006 ; 66 : 324-30.
[5.21] Koga Y, Akita Y, Junko N, Yatsuga S,Povalko N, Fukiyama R et al. Endothelialdysfunction in MELAS improved byL-arginine supplementation. Neurology2006 ; 13 : 1766-9.
[5.22] Koga Y, Akita Y, Nishioka J, Yatsuga S,Povalko N, Tanabe Y et al. L-arginineimproves the symptoms of stroke-likeepisodes in MELAS. Neurology 2005 ;64 : 710-2.
[5.23] Koga Y, Akita Y, Nishioka J, YatsugaS,Povalko N, Katayama K et al. MELAS andL-arginine therapy. Mitochondrion 2007 ;7 : 133-9.
[5.24] Lagouge M, Argmann C, Gerhart-Hines Z,Meziane H, Lerin C, Daussin F et al. Res-veratrol improves mitochondrial func-tion and protects against metabolicdisease by activating SIRT1 and PGC-1 alpha. Cell 2006 ; 127 : 1109-22.
[5.25] Lalani SR, Vladutiu GD, Plunkett K, LotzeTE, Adesina AM, Scaglia F. Isolated mito-chondrial myopathy associated withmuscle coenzyme Q10 deficiency. ArchNeurol 2005 ; 62 : 317-20.
[5.26] Laloi-Michelin M, Meas T, Ambonville C,Bellanné-Chantelot C, Beaufils S, MassinP et al. The clinical variability of mater-nally inherited diabetes and deafness isassociated with the degree of hetero-plasmy in blood leukocytes. J Clin Endo-crinol Metab 2009 ; 94 : 3025-30.
[5.27] Lara MC, Weiss B, Illa I, Madoz P, MassuetL, Andreu AL et al. Infusion of plateletstransiently reduces nucleoside overloadin MNGIE. Neurology 2006 ; 67 : 1461-3.
[5.28] Lèbre AS, Rio M, Faivre-d’Arcier L,Vernerey D, Landrieu P, Slama A et al. Acommon pattern of brain MRI imaging inmitochondrial diseases with complex Ideficiency. J Med Genet 2010 [Epubahead of print].
[5.29] Lévêque M, Marlin S, Jonard L, ProcaccioV, Reynier P, Amati-Bonneau P et al.Whole mitochondrial genome screeningin maternally inherited non-syndromichearing impairment using a microarrayresequencing mitochondrial DNA chip.Eur J Hum Genet 2007 ; 15 : 1145-55.
[5.30] Longley MJ, Clark S, Yu Wai Man C, Hud-son G, Durham SE, Taylor RW et al.Mutant POLG2 disrupts DNA polymerasegamma subunits and causes progressiveexternal ophthalmoplegia. Am J HumGenet 2006 ; 78 : 1026-34.
[5.31] Massa V, Fernandez-Vizarra E, Alsha-hwan S, Bakhsh E, Goffrini P, Ferrero Iet al. Severe infantile encephalomyopa-thy caused by a mutation in COX6B1, anucleus-encoded subunit of cytochromec oxidase. Am J Hum Genet 2008 ; 82 :1281-9.
[5.32] Moran NF, Bain MD, Muqit MM, Bax BE.Carrier erythrocyte entrapped thymidinephosphorylase therapy for MNGIE. Neu-rology 2008 ; 71 : 686-8.
[5.33] Naviaux RK, Nguyen KV. POLG muta-tions associated with Alpers' syndromeand mitochondrial DNA depletion. AnnNeurol 2004 ; 55 : 706-12.
[5.34] Ostergaard E, Christensen E, KristensenE, Mogensen B, Duno M, Shoubridge EAet al. Deficiency of the alpha subunit ofsuccinate-coenzyme A ligase causes fatalinfantile lactic acidosis with mitochon-drial DNA depletion. Am J Hum Genet2007 ; 81 : 383-7.
[5.35] Pineda M, Ormazabal A, Lopez-GallardoE, Nascimento A, Solano A, Herrero MDet al. Cerebral folate deficiency andleukoencephalopathy caused by a mito-chondrial DNA deletion. Ann Neurol2006 ; 59 : 394-8.
[5.36] Rahman S, Hanna MG. Diagnosis andtherapy in neuromuscular disorders :diagnosis and new treatments in mito-chondrial diseases. J Neurol NeurosurgPsychiatry 2009 ; 80 : 943-53.
5669_ Page 88 Vendredi, 4. mars 2011 12:09 12> STDI FrameMaker noir

89
[5.37] Ruiz-Pesini E, Lott MT, Procaccio V,Poole JC, Brandon MC, Mishmar D et al.An enhanced MITOMAP with a globalmtDNA mutational phylogeny. NucleicAcids Res 2007 ; 35 : D823-8.
[5.38] Spinazzola A, Viscomi C, Fernandez-Vizarra E, Carrara F, D’Adamo P, Calvo Set al. MPV17 encodes an inner mitochon-drial membrane protein and is mutatedin infantile hepatic mitochondrial DNAdepletion. Nat Genet 2006 ; 38 : 570-5.
[5.39] Wenz T, Diaz F, Spiegelman BM, MoraesCT. Activation of the PPAR/PGC-1alphapathway prevents a bioenergetic deficitand effectively improves a mitochondrialmyopathy phenotype. Cell Metab 2008 ;8 : 249-56.
[5.40] Yavuz H, Ozel A, Christensen M, Chris-tensen E, Schwatrz M, Elmaci M et al.Treatment of mitochondrial neuro-gastrointestinal encephalomyopathywith dialysis. Arch Neurol 2007 ; 64 :435-8.
[5.41] Zhu X, Peng X, Guan MX, Yan Q.Pathogenic mutations of nuclear genesassociated with mitochondrial disorders.Acta Biochim Biophys Sin 2009 ; 41 :179-87.
[5.42] Zeharia A, Shaag A, Pappo O, Mager-Heckel AM, Saada A, Beinat M et al.Acute infantile liver failure due to muta-tions in the TRMU gene. Am J Hum Genet2009 ; 85 : 401-7.
5669_ Page 89 Vendredi, 4. mars 2011 12:09 12> STDI FrameMaker noir





























