Embed Size (px)
Citation preview

Ce livre rassemble les textes et les conférences des 20es Journées francophones d’électroneuromyographie (ENMG) qui se sont dé-roulées à Strasbourg du 1er au 3 juin 2016.
L’ENMG, prolongement direct de la clinique, reste encore au-jourd’hui en 2016 un outil diagnostique précieux et irremplaçable dans le domaine des maladies neuromusculaires, malgré la concur-rence croissante des nouvelles techniques d’imagerie, d’analyse génétique et d’immunologie. Les auteurs passent ici en revue les données fondamentales, les techniques nouvelles d’investigation, les étiologies émergentes de maladies neuromusculaires et les stra-tégies d’exploration utiles en pratique quotidienne.
Ce livre est destiné à tous ceux qui s’intéressent à l’ENMG et aux maladies neuromusculaires, qu’ils soient neurologues, rhumatolo-gues, rééducateurs ou pédiatres.
Le coordinateur
ANDONI ECHANIZ-LAGUNA est neurologue, neurophysiologiste et docteur en neurosciences. Praticien hospitalier aux hôpitaux uni-versitaires de Strasbourg, il travaille au sein du Centre de Référence des Maladies Neuromusculaires Grand-Est (CERNEST). Il a fait de ces maladies, en particulier les myopathies et les pathologies du nerf périphérique, son domaine de surspécialité.
ENMG2016ISBN : 978-2-35327-343-0
www.deboecksuperieur.com
Publics Neurologues Neurophysiologistes Rhumatologues Médecins de MPR
s u p é r i e u r
ENM
G 20
16
Sou
s la
dir
ecti
on d
’And
oni E
chan
iz-L
agun
a
ENMG 2016 20es Journées francophones d’électroneuromyographie
Sous la direction d’Andoni Echaniz-Laguna


ENMG 2016
Dirigé par Andoni ECHANIZ-LAGUNA
s u p é r i e u r

De Boeck Supérieur04, rue de la Michodière75002 ParisTél. : 01.72.36.41.60
© De Boeck Supérieur SA, 2016Rue du Bosquet, 7B-1348 Louvain-la-Neuve
Tous droits réservés pour tous pays.Il est interdit, sauf accord préalable et écrit de l’éditeur, de reproduire (notamment par photocopie) partiellement ou totalement le présent ouvrage, de le stocker dans une banque de données ou de le communiquer au public, sous quelque forme ou de quelque manière que ce soit.
Imprimé en Belgique
Dépôt légal :Bibliothèque royale de Belgique, Bruxelles : 2016/13647/098Bibliothèque nationale, Paris : mai 2016ISBN : 978-2-35327-343-0
Pour toute information sur notre fonds et les nouveautés
dans votre domaine de spécialisation, consultez notre site web :
www.deboecksuperieur.com

Sommaire
Introduction ........................................................................................................... V
1. Guillain-Barré syndrome: from 1916 to 2016 (Hugh J. Willison)............. 1
2. Bilan étiologique d’une polyneuropathie axonale : jusqu’où aller ? (Laurent Magy) ............................................................................................... 11
3. Pièges et difficultés de l’ENMG du sujet âgé (Timothée Lenglet) ..........25
4. Les neuropathies d’origine professionnelle (Arnaud Lacour) .................45
5. Les complications neurologiques de la chirurgie pelvienne (Jean-Jacques Labat, Amélie Levesque, Marie-Aimée Perrouin-Verbe, Stéphane Ploteau) ..................................... 61
6. Le périnée après une chirurgie digestive (Anne-Geneviève Herbaut) ... 79
7. Les lésions traumatiques des nerfs périnéaux (Philippe Clavert, Philippe Adam)............................................................... 87
8. Le périnée après une chirurgie rachidienne (Maria Carmelita Scheiber-Nogueira) ...................................................... 101
9. Intérêt de l’électro-neuromyogramme dans les atteintes radiculaires du membre supérieur (Philippe Petiot) .....................................................107
10. ENMG et lombo-sciatalgie : quelle utilité ? (Jean-Philippe Camdessanché)................................................................... 119
11. ENMG et myalgies : quelle utilité ? (Émilien Delmont) .........................129
12. Neuropathies sensitives de l’enfant : démarche diagnostique (Cyril Gitiaux) ...............................................................................................139
13. L’ENMG dans les atteintes motoneuronales de l’enfant : quelle place face à la génétique ? (Matthew Pitt) ...................................... 149

IV
ENMG 2016
14. Orientation diagnostique devant une neuropathie démyélinisante de l’enfant (Armelle Magot) ........................................................................ 167
15. EMG neurogène ou myogène : comment distinguer ? (Yann Péréon) ................................................................................................ 187
16. Quelle stratégie ENMG devant un patient suspect de polyneuropathie ? (Thierry Kuntzer) ....................................................199
17. Y a-t-il des contre-indications à l’examen ENMG ? (François Charles Wang) ............................................................................. 211
18. Comment rédiger un compte rendu d’examen électromyographique (Emmanuel Fournier) ..................................................................................225
19. Pour la biopsie musculaire dans les myopathies inflammatoires (Shahram Attarian) ......................................................................................239
20. Contre la biopsie musculaire dans les myopathies inflammatoires (Olivier Benveniste) ......................................................................................255
21. Polyneuropathie et maladie de Parkinson : quel lien ? (Christine Tranchant) ..................................................................................257
22. L’ENMG dans les syndromes cérébelleux d’origine génétique : quelle utilité ? (Mathieu Anheim) ..............................................................267
23. Ce qu’il ne fallait pas manquer depuis Saint-Étienne 2014 (Jean Puget) ....................................................................................................279
Table des matières ...............................................................................................289

V
Introduction
« The times they are a-changin » chantait Bob Dylan en 1964. Aujourd’hui, en 2016, les temps changent toujours aussi vite et la médecine, la neurologie et l’électroneuromyographie (ENMG) suivent le mouvement.
En préparant le programme du congrès de la Société francophone d’électroneuromyographie (SFENMG) à Strasbourg, j’ai voulu rester au plus près de l’actualité et de ces changements qui nous obligent, quelquefois bruta-lement, à modifier notre pratique quotidienne. Ces évolutions se déclinent sur un versant sociétal et technique, mais elles concernent également les pratiques médicales et les patients eux-mêmes :
− La société se transforme. Prenant exemple sur le modèle américain, la judiciarisation de la société s’accélère et tout résultat d’examen com-plémentaire est aujourd’hui susceptible d’être porté devant un tribu-nal. Dans mon activité d’expertise, combien de fois me suis-je fait la réflexion que bon nombre de procédures judiciaires auraient pu être évitées si le compte-rendu d’ENMG incriminé avait été un peu plus détaillé dans son analyse, un peu plus clair dans ses conclusions ou, au contraire, un peu plus mesuré dans son propos… Dans ce contexte, j’ai demandé à E. Fournier, F. Wang et A. Lacour de traiter de la théma-tique du compte-rendu d’ENMG, des contre-indications de l’ENMG et de la problématique des neuropathies d’origine professionnelle.
− Les techniques évoluent. L’analyse d’ADN à haut-débit, les auto-anti-corps et les nouveaux traitements issus de l’ingénierie moléculaire bousculent nos habitudes et redéfinissent en permanence les indica-tions de l’examen ENMG et de la biopsie neuromusculaire. Dans ce contexte, il m’a paru crucial de consacrer plusieurs chapitres de ce livre aux neuropathies des maladies génétiques ainsi qu’aux indications de la biopsie musculaire dans les myopathies inflammatoires (M. Anheim, C. Tranchant, S. Attarian, O. Benveniste)

VI
ENMG 2016
− Les pratiques médicales se modifient. Nos autorités de tutelle nous demandent de plus en plus d’efficacité, de calculabilité, de prédictibi-lité, de traçabilité et de contrôle. Les « procédures », les « checklists » et les « bilans standards » se généralisent et cette « McDonaldisation » de nos pratiques, bien que souvent discutable et contre-productive, n’en reste pas moins une réalité avec laquelle il faut composer au quo-tidien (Dorsey, 2016). Dans ce contexte, il m’a paru utile de demander à T. Kuntzer, L. Magy, J.-P. Camdessanché, C. Petiot et E. Delmont de nous donner un cadre de référence « minimal » pour réaliser un bilan ENMG le plus pertinent possible devant un patient avec une polyneu-ropathie, des myalgies diffuses ou une atteinte radiculaire.
− Les patients changent. Le vieillissement de la population est une réa-lité que nous vivons maintenant au quotidien. Au CHU de Strasbourg, 30 % des examens ENMG sont réalisés chez des patients de plus de 70 ans et 18 % des myopathies sont diagnostiquées après l’âge de 70 ans (Echaniz-Laguna, 2010). Dans ce contexte, il m’est apparu fondamental de consacrer un chapitre de cet ouvrage aux pièges et aux difficultés de l’ENMG chez le sujet âgé (T. Lenglet).
Outre les thématiques abordées ci-dessus, plusieurs chapitres de cet ouvrage sont consacrés à l’ENMG pédiatrique (A. Magot, C. Gitiaux et M. Pitt) et à l’ENMG périnéal (J.-J. Labat, A.-G. Herbaut, M.-C. Scheiber-Nogueira et P. Adam).
Enfin, 2016 est l’année du centenaire de la description du syndrome de Guillain-Barré : cet ouvrage est donc l’occasion de faire le point, à l’aube du xxie siècle, sur la physiopathologie de cette redoutable affection du système nerveux périphérique (H. WILLISON).
Bonne lecture à tous !
Andoni Echaniz-LagunaPrésident du Comité local d’organisation
17 mars 2016
Références
Dorsey ER, Ritzer G. The McDonaldization of Medicine. JAMA Neurol 2016 ; 73 : 15-6.
Echaniz-Laguna A, Mohr M, Lannes B, Tranchant C. Myopathies in the elderly : a hospital-based study. Neuromuscul Disord 2010; 20:443-7.

1
1. Guillain-Barré syndrome: from 1916 to 2016
Hugh J. Willison, PhD, FRCP
College of Biomedical, Veterinary and Life Sciences, University of Glasgow, Scotland

2
ENMG 2016
Introduction
Our mechanistic understanding of the Guillain-Barré syndromes (GBS) has advanced in leaps and bounds since the now classic publication from 1916 by Guillain, Barré and Strohl. At this 20es Journées Francophones d’ENMG we celebrate this centenary point, and reflect on the past, enjoy the present and anticipate the future. In particular we recognize the greatness of Jean-Alexandre Barré and his contribution to Neurology in Strasbourg where this meeting is held. In virtually every sphere of medicine applicable to GBS, knowledge has been transformed by astonishing discoveries and pivotal points of progress. From the fields of epidemiology, causation, infectious precedents, biomarker science, immunopathogenesis, treatment and outcome, we steadily and surely move forward with new information on a yearly basis. Yet, some of the points made in the original 1916 paper have resolutely stood the test of time, such is their resistance to advancement! Notably, the albumino- cytologic dissociation in the cerebrospinal fluid still remains a cornerstone of GBS diag-nostics. Beyond this simple measurement of CSF total protein levels, we have no widely used additional applications of CSF analysis in clinical practice to inform either diagnosis or clinical outcome. Whether this reflects the brilliant completeness of the originally proposed test, or our failings to yet refine it, may require a further 100 years of research. Such is the pace of progress, we would expect new CSF tests that monitor disease severity and activity, such as levels of neurofilament protein released from degenerating axons, to be in clinical use well before the bicentenary.
1. A brief summary of progress in GBS over the last 100 years
During the course of preparations within the Inflammatory Neuropathy Consortium (INC) for the GBS centenary year, John Goodfellow and I have edited a monograph on GBS that comprises around 65 chapters across a broad range of subjects outlining the major advances that have been achieved over the last century. This is freely available as an e-book down-load from the Peripheral Nerve Society website (www.pnsociety.com). In the introduction, co-written with David Cornblath, we highlight some of the landmarks which are discussed in detail in the subsequent chapters. Not for-getting Octave Landry’s prior clinical description, the Guillain-Barré-Strohl syndrome was notably distinguishable from acute poliomyelitis which also caused acute onset weakness but exhibited asymmetry, variable recovery, and

3
Guillain-Barré syndrome: from 1916 to 2016
an inflammatory CSF. As time passed, the disease became best known by the names of its first two authors. Strohl’s name receded from the roll-call, and Landry’s prior description also became increasingly overlooked as the current usage took its place in the medical eponyms hall of fame. Thus, the Landry-Guillain-Barré-Strohl syndrome became the Guillain-Barré syndrome, more commonly just referred to as GBS. GBS is now the most common cause of acute onset flaccid paralysis in the Western world as polio has receded due to vaccination. A notable point is that in our post-polio world, we shouldn’t ignore other presumed infectious causes of acute flaccid paralysis that are widely seen clinically in some geographical regions, yet receive little investi-gative or public health attention.
Epidemiological studies from many quarters – England, Italy, India, China, Asia, Japan and others have concluded that GBS has a steady world-wide incidence of 2 cases per 100,000 of population per year. On this basis, one can estimate there are over 100,000 cases per annum worldwide. Yet in some geographical regions, notably Africa, we have very little information at all on incidence, except to know that it certainly occurs. The International GBS Outcome Study (IGOS) aims to address some of these global dispar-ities. IGOS is a multinational collaborative effort led by Bart Jacobs in the Rotterdam GBS group currently involving over 1200 GBS patients, all con-tributing to a substantial collection of clinical, electrophysiological, biobanked sera and DNA samples, and outcome data. This will be a rich source of study for years to come and represents many of the best principles of the new age of international cooperation, data pooling, bioinformatics and high-through-put screening. Indeed from 2 soldiers to 1200 globally gathered GBS cases is a remarkable step forward we can be hugely proud of.
Clinical advances particularly worthy of highlighting include formes frustes of GBS that originally were not thought to be classic GBS, (i.e. an acute inflammatory demyelinating sensory-motor polyneuropathy, AIDP) but were eventually realized to be part of the spectrum of GBS. These disorders, includ-ing Miller Fisher syndrome (MFS) and Acute Motor Axonal Neuropathy (AMAN) were gateway advances and have indeed taught us much about the classic syndrome. The discovery of anti-GQ1b antibodies in MFS by Atsuro Chiba, working with Susumu Kusunoki was an extraordinary step forward into deepening our understanding of GBS pathogenesis, such is the power of the MFS/GQ1b association. The recognition of the AMAN variant, initially by Tom Feasby and colleagues in Canada, represented a paradigm shift with fundamental repercussions, further reinforced by discovering the relationship with Campylobacter jejuni infection and anti-GM1/GD1a antibodies, pio-neered by Nobuhiro Yuki’s group in Japan.

4
ENMG 2016
Progress in investigating and diagnosing GBS has focused on defining the key clinical and electrophysiological characteristics: in what still remains a very clinically-orientated diagnosis. The highly cited diagnostic criteria laid out in 1990 by Art Asbury and David Cornblath remained a gold standard for several decades, with refinements and modifications now coming forward as we understand more about the pathogenesis of GBS. Whilst invasive patho-logical studies are rarely conducted, they led to the first ideas that GBS was an auto-immune disorder mediated by both the humoral and cellular com-partments of the immune system. One of the major limitations in advancing understanding is the inability to serially examine peripheral nerve pathology during the course of the disease, or indeed prior to clinical onset. Clinical neurophysiology has helped hugely to refine the pathophysiological under-standing of GBS, notably the distinction between AIDP and AMAN, more recently leading to the concept of the node of Ranvier as a major site of action in the evolution of the disease. Refined imaging modalities that are becom-ing increasingly available in routine clinical settings promise to provide new insights.
Basic research, including animal models, immunology and neurosci-ence, autoantibodies, infectious antecedent events and genetic susceptibility has advanced considerably beyond the early observations in the 1950’s on experimental allergic neuritis (EAN). Through this we are building a clearer picture of the totality of GBS and its variants. As such, our current under-standing of GBS involves the so-called molecular mimicry hypothesis: an auto-immune disorder in which a genetically susceptible individual comes into contact with a specific inciting agent and then develops antibodies against that agent. These antibodies mistakenly identify the nerve as similar and attack it. Whilst this molecular mimicry hypothesis clearly accounts for the AMAN and MFS variants, less is known about the precise mechanisms operating in AIDP.
Advances in treatment are the cornerstone of patient care and the fun-damental goal of all research efforts. The current standards of care, meticu-lously discovered through randomized controlled clinical trials led from the USA, France, UK and the Netherlands during the 1980-90s comprise plasma exchange and intravenous immunoglobulin therapy. The roles of evidence–based therapeutic pioneers in the GBS landscape, such as Pieter van Doorn and Richard Hughes in this effort cannot be under-estimated. Equally impor-tant is the place of supportive care as critically necessary in the overall man-agement, steadily rising in quality with the advent of intensive therapy units with mechanical ventilation, and rehabilitation practices. We are all highly aware that no new treatment has been approved for GBS since the 1990’s;

5
Guillain-Barré syndrome: from 1916 to 2016
current ongoing clinical trials of complement inhibition in the UK and Japan offer the expectation that this situation may change, at least for the AMAN form of GBS . In the absence of directly efficacious new therapies, we have still come far in the provision of rehabilitation services and our ability to predict outcomes. However we also need better outcome measures for our clinical trials that mimic what patients think about themselves and their recovery. An area that requires much more research is in symptom relief, notably pain which beleaguers many GBS patients both in the acute phase and long term, where reliance on opiates is still prominent. Considering that opium was first used over 5000 years ago for pain relief and has little changed since then, GBS seems very young at 100 years old.
2. An historical commentary on acute motor axonal neuropathy
Today’s presentation will outline in more detail some of the specific advances in our understanding of AMAN; this work was also presented at the retirement symposium held in Baltimore in January 2011 for Dr. Jack Griffin of Johns Hopkins University who pioneered the human studies that revealed the pathological intricacies of this disorder. Segmental demyelina-tion with loss of myelin internodes and inflammatory cell infiltrates leading to conduction block was the accepted pathophysiological conceptualization of GBS up until the late 20th century, as clinically manifested under the umbrella term AIDP. The myelin protein directed T and B cell-mediated experimental allergic neuritis (EAN) models provided much of the experimental support for such a view. However, translating the immune response directed against myelin proteins into human studies had failed to consistently yield equivalent results to those observed in rodent EAN models. At this point, the putative peripheral nerve antigens driving human GBS thus remained undefined.
The concept that primary axonal forms of GBS might exist pre-dates the ‘China experience’, conducted by physicians in the Second Teaching Hospital, Shijiazhuang, Hebei Province and neurologists, pathologists, microbiologists, electrophysiologists and immunologists in the USA and European centers. The AMAN idea first gained credence in the mid-1980s when Tom Feasby and colleagues described the clinical and pathological features of ‘axonal GBS’. However the relative rarity of these axonal cases in Western research centers, combined with the paucity of pathological material from early GBS and variant cases, made for slow progress in advancing any pathophysiological concepts. Around the time Feasby described AMAN, anti-glycolipid antibodies were

6
ENMG 2016
discovered in paraproteinaemic neuropathy and multifocal motor neuropa-thy, and the stage was thus set for also seeking these in GBS and its variants, first being observed through studies in Dick Quarles’ lab at NIH, Bethesda in the late 1980s. When GQ1b ganglioside was found to be the biomarker for MFS, it followed that seeking similar glycolipid antibody markers in other GBS sub-forms, including AMAN would be rewarded. Around this time, Yuki and colleagues described anti-GM1 ganglioside antibodies in Japanese cases of AMAN associated with preceding Campylobacter jejuni infection and it was soon clear that Campylobacter was also the preceding infection in the Chinese cases, thereby firmly establishing the molecular mimicry hypothesis.
At this point, in the early to mid-1990s, the identification of a major epidemic of AMAN that could be comprehensively studied provided a wealth of human biological material to explore these evolving concepts about GBS pathogenesis. This opportunity was rapidly and effectively exploited over a 5 year period. After the first descriptions of the clinical, electrophysiological and preliminary pathological findings underpinning AMAN, the definitive paper was published by Griffin and colleagues in 1996 that describes what remains the contemporary view of the immunopathological procession of AMAN. In this pathological study on spinal roots and nerves from autopsy cases of the acute phase of AMAN, degenerating motor fibres were observed, accompanied by extensive Wallerian degeneration. Some motor fibres had lengthening of the node of Ranvier accompanied by condensation of the axonal cytoplasm. Demyelination was infrequent or absent. A striking finding was the presence of infiltrating macrophages with extensive processes in the periaxonal space abutting the nodal and internodal axolemma and displac-ing the adaxonal Scwhann cell membrane and myelin sheath. The findings demonstrated by immunocytochemistry were of very intense IgG and com-plement C3d and C5b-9 (membrane attack complex, MAC) deposits bound to the nodal and internodal axolemma in the periaxonal space.
In conjunction with ideas supported by other data accumulating in the field, these pathological findings indicated the likely sequence of events that is still held today. At the time of this first pathological paper in 1996, the pre-vailing view as stated above was that GM1 and its corresponding anti-GM1 antibody were likely to be the major axonal targets. However, shortly after, it emerged that many of the Chinese AMAN cases harbored anti-GD1a anti-bodies, in addition to anti-GM1. Such antibodies were similarly being dis-covered in Japanese AMAN cohorts. Contemporaneous with this description of the pathology of AMAN in the ventral roots, Griffin and colleagues also demonstrated that very distal motor axonal degeneration might take place in AMAN, and that this could also be an explanation for the rapid recovery that

7
Guillain-Barré syndrome: from 1916 to 2016
sometimes occurs. Using motor point biopsy from an AMAN case they were able to demonstrate denervated neuromuscular junctions and reduced intra-muscular nerve fiber densities, speculating that if confined to this site, these changes could be rapidly reversible through axonal regeneration over rela-tively short distances. The rapid progress in defining the anti-glycolipid anti-body signature in AMAN, notably GM1 and GD1a antibodies, allowed ideas on target localization to be incorporated into disease modeling. With these mechanistic ideas in place, animal experiments were then conducted by a vari-ety of groups to provide support for the pathological findings in man, and to explore therapeutic possibilities that might block these effector pathways.
In studies dating back to the 1970s, Yoshi Nagai, the Saida’s and others provided good evidence that anti-glycolipid antibodies (to GM1 and galac-tocerebroside respectively) could mediate peripheral nerve injury in animal models, notably the rabbit. Over the subsequent 20 years, many studies inves-tigated such pathogenic pathways in different model systems. With the advent of AMAN as a new human disorder and its pathological description, atten-tion particularly focused on the node of Ranvier and motor nerve terminal as potential target sites. In 2001, Yuki and colleagues recapitulated some of the key pathological findings of Griffin in rabbits sensitised with ganglio-sides including GM1. In these animals, high anti-GM1 IgG antibody titres were observed, accompanied by a flaccid limb weakness. Peripheral nerves showed predominant Wallerian-like degeneration with neither lymphocytic infiltration nor demyelination, whereas IgG was deposited on ventral root axons. Thus a rabbit model of AMAN was established. This group, notably Kei Susuki latterly working in Matt Rasband’s laboratory at Baylor College, Houston, went on to demonstrate macrophage infiltration into the periaxonal space, nodal complement deposits and the molecular events that might lead to axonal conduction block, including disappearance of sodium channel immu-noreactivity at the node of Ranvier.
Initial studies by my group followed up the Griffin pathology and related data by focusing on the motor nerve terminal as a target site, using AMAN-associated anti-GM1 and -GD1a antibodies derived from both humans and mice immunized with Campylobacter jejuni saccharides in which tolerance was overcome by using ganglioside-deficient mouse strains. We showed that the motor nerve terminal was indeed a vulnerable site for anti-ganglioside antibody attack that resulted in complement fixation. Deposition of MAC pores allowed uncontrolled calcium ingress triggering a sequence of destruc-tive events, including calpain activation, with subsequent paralysis. In our studies on the motor nerve nodes of Ranvier, using mice passively immu-nized with monoclonal anti-GD1a antibodies and supplemented with human

8
ENMG 2016
complement, severe functional and pathological changes were observed over very short time frames of a few hours. One crucial experimental modification was to study mice expressing abnormally large amounts of GD1a; made pos-sible through the targeted deletion of a biosynthetic enzyme in the ganglio-side pathway, GD3 synthase. These mice also developed a highly destructive motor nerve terminal lesion in response to anti-GD1a and human comple-ment delivery, which from a functional perspective, disguised more proximal injury to the node of Ranvier.
By using perineural current recordings, rather than relying on end plate potential recordings (that were abolished due to end plate injury), we were able to demonstrate that the node of Ranvier in this model was functionally paralyzed, and this was accompanied by a complete loss of Na channel immu-nostaining. The lesion site was richly decorated in both the passively trans-ferred anti-GD1a antibodies and complement activation products, including membrane attack complex (MAC). We speculated that ingress of calcium through MAC pores resulting in calpain activation was a major pathological event, similar to that we had previously observed at the motor nerve terminal in our model of MFS. Inhibition of calpain, using an inhibitor provided by Jonathon Glass and colleagues, confirmed this, and afforded complete preser-vation of Na channel integrity, consistent with these channels being a known calpain substrate. Calpain inhibition was insufficient to protect the node from electrophysiological failure, a finding we interpreted as being due to uncon-trolled water and salt flux through the nodal MAC pores resulting in a failure to maintain electrical potentials across the axolemmal membrane.
The crucial finding that resonates with the human pathological studies was that inhibition of MAC assembly using an inhibitor of C5 had a major neuroprotective effect, completely preventing any structural and functional changes at the node of Ranvier, as we also found previously at the motor nerve terminal. Thus a major pathway of acute pathological injury mediated by anti-glycolipid antibodies in GBS and variants converges at MAC. The development of novel therapeutics that prevent MAC formation thus repre-sent a clear direction for the clinical trials that are now underway in the UK and Japan.

9
Guillain-Barré syndrome: from 1916 to 2016
Concluding remarks
One often reflects in these historical accounts on what our forbearers, Landry, Guillain, Barré and Strohl, might have thought about this new knowl-edge had they been alive to witness its genesis. Their memorable achieve-ments, based largely on clinical acumen and rudimentary investigations, are justly praised through the persistence of the eponymous term, GBS. Still, it is quite clear the last century of GBS research promises to be equaled by things to come, and we all eagerly expect great advances amongst the young transla-tional researchers of today who will improve the lot of our patients worldwide who are afflicted by GBS.
Further readingGBS100: Celebrating a century of progress in Guillain-Barre syndrome. 2016.
Edited by Hugh J Willison and John A Goodfellow, University of Glasgow, UK. Publisher: Peripheral Nerve Society, USA (available as an e-book from the PNS website).
Willison HJ, Jacobs BC, van Doorn PA. Guillain-Barré syndrome. Lancet 2016; in press.


11
2. Bilan étiologique d’une polyneuropathie axonale : jusqu’où aller ?
laurent Magy
Service et Laboratoire de Neurologie, Centre de Référence « Neuropathies périphériques rares », CHU Limoges, 2 avenue Martin Luther King, 87 042 Limoges Cedex

12
ENMG 2016
Introduction
La découverte d’une neuropathie périphérique chronique est une cir-constance clinique très fréquente en pratique neurologique. Les études épi-démiologiques diffèrent par leur méthodologie, mais une revue systématique récente estime la prévalence des neuropathies chroniques à 1 % dans la popu-lation générale et jusqu’à 7 % chez les sujets âgés (Hanewinckel, van Oijen, Ikram, et van Doorn, 2015). Selon les études, 12 à 49 % des cas correspon-draient à des polyneuropathies axonales idiopathiques chroniques (PAIC). Ainsi, une analyse en population récente montre que 26% des cas incidents de polyneuropathies chroniques sont idiopathiques (Visser, Notermans, Linssen, van den Berg, et Vrancken, 2015). De plus, les études longitudinales de patients présentant une PAIC montrent qu’ils ont une neuropathie d’évo-lutivité lente, et qu’ils développent peu de handicap même si leur qualité de vie peut être significativement altérée (Notermans et al., 1994).
Pour ces raisons, la mise en évidence d’une polyneuropathie axonale chronique aboutit le plus souvent après un bilan minimal à une impasse dia-gnostique rapide et à une prise en charge frustrante, principalement basée sur le traitement du symptôme douloureux et la réassurance du patient. Cet article a pour objet de délimiter les contours des polyneuropathies axonales chroniques, d’en déterminer quelques diagnostics différentiels et pièges dia-gnostics et d’insister sur les drapeaux rouges qui doivent alerter le clinicien sur une cause potentiellement curable ou sur une neuropathie à risque évolutif.
1. Qu’est-ce qu’une polyneuropathie axonale ?
1.1. Éléments cliniques et pièges diagnostiques
Cliniquement une polyneuropathie (axonale) chronique se caractérise le plus souvent par une atteinte à prédominance distale et aux membres infé-rieurs, d’installation et de progression symétrique (par définition), plus sensi-tive que motrice. L’anamnèse s’attachera à préciser au mieux l’ancienneté des troubles et surtout le mode d’installation. L’expérience montre que les patients sous-estiment fréquemment l’ancienneté des premiers troubles sensitifs et la découverte d’un rapport d’électroneuromyogramme ancien dans un dossier apporté par le patient qui avait « oublié » l’année de début des symptômes est souvent un élément précieux pour attester de la chronicité de l’atteinte. De la même façon, le support d’un accompagnant (conjoint ou autre) permet

13
Bilan étiologique d’une polyneuropathie axonale : jusqu’où aller ?
fréquemment « d’allonger » de plusieurs mois ou années la durée des symp-tômes et signes cliniques.
L’évolutivité de la neuropathie est également un élément crucial et une atteinte sensitive ascendante jusqu’aux genoux ou une atteinte motrice, dans la première année d’évolution, doit absolument faire rechercher une cause (toxique ou inflammatoire notamment). De la même façon, l’anamnèse doit s’attacher à rechercher un début asymétrique ou multifocal, car une mono-neuropathie multiple peut confluer à un stade avancé (Magy et Vallat, 2008). Un début des symptômes aux membres supérieurs ou un début concomi-tant aux 4 membres doit faire rejeter de principe la possibilité d’une PAIC, et s’interroger sur une polyradiculonévrite inflammatoire démyélinisante chronique ou une neuronopathie sensitive (Camdessanché et Antoine, 2014; Viala et al., 2010). Il en est de même de la présence d’une ataxie au premier plan du tableau clinique, qui devra faire évoquer systématiquement une neu-ronopathie sensitive, une neuropathie carentielle (B12) ou par toxicité vitami-nique (B6), ou une neuropathie démyélinisante acquise (cf ci-après). Enfin, une polyneuropathie à nette prédominance motrice, surtout en l’absence de troubles sensitifs subjectifs, doit faire suspecter de principe une neuropathie héréditaire sensitivomotrice et mener une enquête familiale systématique (Teunissen et al., 1997). Il faut souligner d’ailleurs que, quel que soit le profil de la neuropathie, la recherche d’antécédents familiaux, et au besoin, l’examen clinique (et électrophysiologique) des apparentés, doit être la règle, tant les neuropathies héréditaires peuvent avoir une présentation polymorphe, allant bien au-delà du classique phénotype de la maladie de Charcot-Marie-Tooth (Rossor, Evans, et Reilly, 2015).
Enfin, le neurologue est un clinicien avant tout, et la présence d’une polyneuropathie justifie, quelle qu’en soit la présentation, un examen géné-ral (peau, phanères, adénopathies, organomégalie, etc.) et un interrogatoire précis (antécédents, exposition à des toxiques, mode de vie, etc.).
1.2. Comment définir une polyneuropathie axonale au plan électrophysiologique ?
Le plus souvent, la distinction entre polyneuropathie axonale chro-nique et polyneuropathie démyélinisante ne pose pas de problème au plan électrophysiologique et une polyneuropathie axonale se définira en fait négati-vement par la présence d’anomalies des amplitudes sensitives, ou sensitives et motrices sans altération significative des vitesses de conduction ni des latences distales et proximales. En pratique, la préoccupation du praticien sera essen-tiellement d’éliminer la possibilité d’une neuropathie démyélinisante acquise

14
ENMG 2016
telle qu’une polyradiculonévrite inflammatoire démyélinisante chronique (PIDC) ou une polyneuropathie liée à une IgM monoclonale à activité anti-MAG (PN-MAG), ou d’une neuropathie démyélinisante héréditaire telle qu’une maladie de Charcot-Marie-Tooth de type 1 (CMT1).
Sur le plan théorique, définir une neuropathie axonale reviendra donc à éliminer une neuropathie démyélinisante, ce qui implique bien sûr de connaître les paramètres électrophysiologiques définissant la démyélinisa-tion. De ce point de vue, la plupart des critères actuellement utilisés ont été dérivés de l’article fondateur publié en 1989 par Albers et Kelly, évaluant sur la conduction distale (latences motrices distales) intermédiaire (vitesses de conduction motrice, blocs et dispersions) et proximale (latences des ondes F) les modifications minimales nécessaires à définir une neuropathie acquise comme démyélinisante (Albers et Kelly, 1989). Par la suite, de nombreuses publications affinèrent ces critères, en ajoutant aux anomalies classiques l’ana-lyse de la durée des potentiels moteurs comme critère supplémentaire (Isose et al., 2009; Joint Task Force of the EFNS and the PNS, 2010).
On voit bien que la difficulté résidera dans de nombreux cas, dans l’in-terprétation d’anomalies électrophysiologiques ne permettant pas de façon certaine de définir une neuropathie comme démyélinisante, mais trop impor-tantes en termes de ralentissement pour conclure à une neuropathie axonale. Dans ce cas de figure assez fréquent, le clinicien/neurophysiologiste doit éviter absolument d’utiliser le terme de neuropathie axono-myélinique ou mixte, qui ne recouvre aucune entité particulière, et ne débouche sur aucune enquête étiologique ciblée. En fait, des vitesses « intermédiaires » peuvent cor-respondre théoriquement au CMT lié à l’X (Dubourg et al., 2001), mais égale-ment à des neuropathies axonales avec perte importante en fibres rapides, ou à des neuropathies démyélinisantes anciennes avec prépondérance de fibres remyélinisées, à conduction ralentie.
Une difficulté supplémentaire consiste à ne pas accorder un poids trop important à des anomalies électriques suggérant une démyélinisation, mais siégeant dans les zones d’étroitesse anatomique résultant en des ralentisse-ments uniquement focaux (nerf fibulaire au col du péroné, nerf médian au canal carpien, nerf ulnaire au coude). Pour les latences motrices distales, il n’est pas inutile de rappeler que des extrémités froides peuvent diminuer la vitesse de conduction, et qu’il faudra bien sûr s’assurer que la tempéra-ture cutanée est suffisante (en pratique supérieure à 32°C) pour assurer une conduction « normale ». De la même façon, de nombreux patients présentent des anomalies des ondes F, qui peuvent être l’unique critère « de démyélini-sation » dans l’exploration d’une neuropathie axonale. Il faudra tenir compte d’anomalies rachidiennes éventuelles d’une part, et envisager d’autre part

15
Bilan étiologique d’une polyneuropathie axonale : jusqu’où aller ?
que ces anomalies proximales, même en l’absence de facteur compressif, ne témoignent pas forcément d’une démyélinisation proximale, mais plutôt de modifications de l’excitabilité axonale comme cela peut être montré dans les neuropathies diabétiques (Fisher, 2014).
Sur le plan technique, on rappellera qu’il est bien sûr essentiel dans les neuropathies en apparence distales, d’examiner les membres supérieurs. Ainsi, la recherche de signes de démyélinisation pour le diagnostic de PIDC est grandement facilitée par une étude exhaustive de la conduction aux membres supérieurs, ce d’autant que la perte axonale est déjà conséquente aux membres inférieurs (Rajabally, Jacob, et Hbahbih, 2005). Dans quelques cas, des tech-niques alternatives telles que la triple stimulation pour rechercher des blocs de conduction très proximaux se révèleront utiles (Attarian et al., 2015).
2. Qu’est-ce qu’une polyneuropathie axonale idiopathique chronique (PAIC) ?
Le concept de PAIC a notamment été développé par l’équipe de Nicolette Notermans dans les années 90 (Notermans et al., 1993, 1994). En effet, cette décennie était particulièrement propice à l’émergence d’un tel concept en raison de l’avancée des méthodes diagnostiques, notamment électrophysio-logiques, et de l’émergence une ou deux décennies plus tôt des concepts de PIDC et de neuropathies associées aux anticorps anti-MAG (Dyck et al., 1975; Latov, Hays, et Sherman, 1988). Plus loin dans le passé, les neuropathies de causes indéterminées incluaient en fait toutes les neuropathies considérées comme idiopathiques, y compris démyélinisantes (Prineas, 1970), et plusieurs publications montraient qu’un bilan poussé permettait dans une grande majo-rité des cas de préciser une étiologie (Dyck, Oviatt, et Lambert, 1981; McLeod, Tuck, Pollard, Cameron, et Walsh, 1984).
Dans le travail initial de l’équipe hollandaise portant sur 75 patients avec diagnostic de PAIC, il est intéressant de noter qu’avaient été exclus les patients pour lesquelles une cause inflammatoire avait été évoquée (sans pré-cision sur les critères utilisés), et qu’en cas de signes de démyélinisation (sans que soit précisée leur définition) ou d’hyperprotéinorachie même modérée, une biopsie nerveuse était pratiquée, excluant les patients de l’étude en cas de signes inflammatoires (Notermans et al., 1993). Au plan clinique, certains patients de cette étude avaient des symptômes et des signes cliniques qu’on considérerait actuellement comme atypiques pour une PAIC, tels qu’une pré-dominance motrice (pouvant suggérer une neuropathie héréditaire) ou un début sensitif aux membres supérieurs (suggérant une possible neuronopathie

16
ENMG 2016
sensitive ou une PIDC). De façon générale l’évolution était considérée comme bénigne, mais certains patients avaient quand même des troubles de l’équi-libre et nécessitaient une aide à la marche. L’intérêt du travail mené par cette équipe résida surtout dans la durée de suivi, puisque la totalité des patients put bénéficier d’un suivi clinique pendant 5 ans. Quatre patients (5%) reçurent finalement un diagnostic étiologique autre : PIDC dans un cas, neuropathie héréditaire dans 2 cas, et neuropathie alcoolique dans un cas, suggérant qu’un simple suivi clinique, sans répéter les examens complémentaires, était suffi-sant pour porter le diagnostic de PAIC (Notermans et al., 1994).
La question actuellement en suspens reste bien sûr celle de l’étiologie ou plutôt des étiologies des PAIC, qui ne constituent probablement pas une entité parfaitement homogène. Outre les expositions à des toxiques environnemen-taux qui peuvent dans certains cas constituer une piste possible, voire sérieuse, le mode de vie « occidental » et ses conséquences comme le syndrome méta-bolique peuvent constituer une association, sinon une cause possible (Visser, Vrancken, van der Schouw, van den Berg, et Notermans, 2013). De la même façon, certains travaux montrent l’hypertriglycéridémie (éventuellement asso-cié à un certain hyperinsulinisme) comme facteur plus fréquemment retrouvé chez des sujets avec PAIC que chez des témoins (Hughes et al., 2004). De plus, les triglycérides sembleraient agir comme cofacteur dans les neuropathies diabétiques, ce qui renforce l’idée de leur implication dans les neuropathies (Wiggin et al., 2009). Quant aux facteurs vasculaires qu’il semble logique d’incriminer, leur implication réelle dans le développement de neuropathies chroniques reste à démontrer. Finalement, l’ensemble des données actuelle-ment disponibles suggère que des facteurs environnementaux multiples, dont certains seraient relativement corrigibles et d’autres pas, pourraient interagir avec une prédisposition génétique pour favoriser le développement des PAIC. De nombreux travaux restent donc à mener pour déterminer de façon plus précise ces facteurs, et surtout tenter de les corriger pour apprécier leur impli-cation dans cette pathologie fréquente.
3. Y a-t-il des diagnostics différentiels d’une PAIC ?
La question qui se posera devant un patient ayant un tableau électro-clinique de PAIC, sera donc de savoir quelles étiologies peuvent être évoquées, quels éléments cliniques et électriques devront amener à rechercher une étio-logie de façon « active », et jusqu’où aller dans les examens complémentaires. Schématiquement, on s’attachera à éliminer des causes curables plus ou moins fréquentes telles que PIDC, vascularites, amylose, sarcoïdose, maladie

17
Bilan étiologique d’une polyneuropathie axonale : jusqu’où aller ?
de Lyme, syndrome de Gougerot Sjögren, et des associations pour lesquelles le lien de causalité est parfois difficile à établir telles que les gammapathies monoclonales.
La recherche d’une PIDC devant un tableau de PAIC se discutera notamment devant certains éléments atypiques tels qu’un début aux membres supérieurs ou une ataxie marquée, et la biopsie nerveuse, dans certains cas ciblés, pourra être utile (Vallat et al., 2003). Parfois, des anomalies électro-physiologiques n’atteignant pas les critères de démyélinisation, mais ne permettant pas de conclure formellement à une axonopathie, amèneront fina-lement à un diagnostic de PIDC, et surtout à une réponse thérapeutique (Chin et al., 2004). De multiples situations amènent en fait à évoquer la possibilité d’une PIDC devant un tableau initialement qualifié de PAIC (Ayrignac et al., 2013). Les situations cliniques au cours desquelles une PIDC est dite atypique ont été listées dans un travail collaboratif français et on s’y référera pour plus de précisions (French CIDP Study Group, 2008).
Classiquement, les vascularites systémiques primitives ou secondaires se traduiront lorsqu’elles comportent une atteinte neurologique périphérique, par un tableau de mononeuropathie multiple. Néanmoins, dans certains cas, une neuropathie sensitive symétrique révèle une vascularite. La douleur constitue alors un signe cardinal important, même si elle n’est pas retrouvée de façon constante (Seo, Ryan, Claussen, Thomas, et Oh, 2004). En fait, c’est souvent l’évolution clinique rapide sur quelques mois qui permettra d’évo-quer une vascularite, qui sera généralement confirmée ou fortement suspec-tée, et amènera à une corticothérapie (Vrancken, Notermans, Jansen, Wokke, et Said, 2004). L’absence de contexte systémique n’éliminera pas d’emblée le diagnostic de vascularite, notamment dans les cas de vascularites limitées aux nerfs périphériques (Üçeyler, Geng, Reiners, Toyka, et Sommer, 2015).
Une neuropathie amyloïde héréditaire doit être évoquée dans de très nombreuses situations cliniques, tant le spectre phénotypique de cette affec-tion est large, et là encore, c’est l’évolutivité clinique qui sera souvent détermi-nante pour en évoquer le diagnostic (Adams et al., 2015). Toutefois, certains travaux montrent que des patients arrivent au diagnostic parfois plusieurs années après les premiers symptômes de la maladie dans les formes tardives de neuropathies amyloïdes familiales par mutation de la transthyrétine, ce qui doit inciter à ne pas rejeter systématiquement ce diagnostic, y compris en cas d’évolution en apparence torpide (Koike et al., 2011). La recherche de dysau-tonomie au moins clinique devra de toute façon être systématique devant toute neuropathie axonale chronique.
La question de l’association d’une neuropathie au syndrome de Gougerot-Sjögren (SGS) est récurrente et des phénotypes électro-cliniques

18
ENMG 2016
variés semblent pouvoir être liés à cette maladie auto-immune (Grant, Hunder, Homburger, et Dyck, 1997; Mellgren, Conn, Stevens, et Dyck, 1989). Plus spé-cifiquement, dans un contexte de neuropathie axonale chronique considérée comme idiopathique, la découverte d’un SGS est possible, et sa recherche sys-tématique doit donc être recommandée (van Dijk et al., 1997), même si les implications thérapeutiques en sont pour l’instant incertaines.
Enfin, la mise en évidence d’une gammapathie monoclonale (GM) au cours de l’exploration d’une neuropathie de cause indéterminée n’est pas rare (Kelly, Kyle, O’Brien, et Dyck, 1981). La causalité de la GM est toutefois difficile à affirmer dans de nombreux cas et les liens entre les 2 pathologies peuvent se révéler directs ou indirects (Vallat et al., 1996). Le caractère fortuit de l’association avec un tableau clinique de PAIC sera en fait souvent retenu comme l’a montré un travail prospectif hollandais, ce d’autant que la préva-lence des GM dites de signification indéterminée (MGUS) augmente avec l’âge (Notermans et al., 1996). On soulignera là encore qu’il est indispensable d’examiner les membres supérieurs d’un point de vue électrophysiologique, car de nombreux cas de neuropathies associées à une IgM à activité anti-MAG ont une telle perte axonale distale aux membres inférieurs qu’il n’est pas rare de voir retenir un diagnostic initial de neuropathie axonale après un examen incomplet chez ces patients.
4. Quel bilan de première ligne ?
La Haute autorité de santé (HAS) définit un bilan dit de première ligne, qui s’adresse de toute évidence au médecin généraliste, et qui est destinée à dépis-ter les causes considérées comme fréquentes de polyneuropathies chroniques telles que l’éthylisme chronique, le diabète, l’insuffisance rénale, l’hypothyroïdie. Pour information, les recommandations de la HAS figurent à l’adresse internet suivante : http://www.has-sante.fr/portail/upload/docs/application/pdf/diagnos-tic_neuropathies_peripheriques_recommandations.pdf.
En fait, le neurologue/neurophysiologiste, lorsqu’il sera amené à éva-luer un patient présentant une neuropathie axonale chronique, disposera généralement des éléments de ce bilan de première ligne minimal. En dehors d’éléments cliniques et/ou neurophysiologiques atypiques, ce bilan initial sera utilement complété par une radiographie de thorax de face, des sérologies des hépatites et de la maladie de Lyme (VIH selon le contexte), un bilan d’auto-immunité minimal, une immunofixation du sérum, une recherche de cryo-globulinémie et une biopsie des glandes salivaires accessoires, examen très rentable dans de nombreuses situations (amylose, sarcoïdose, SGS).

19
Bilan étiologique d’une polyneuropathie axonale : jusqu’où aller ?
5. Pour qui, quand, et comment aller plus loin ?
Les arguments cliniques devant faire systématiquement remettre en cause le diagnostic de PAIC ont été développés plus haut. Ils comprennent principalement des éléments anamnestiques d’évolutivité, la présence de cer-taines maladies associées, et des particularités de l’examen clinique tels qu’un début aux membres supérieurs. De la même façon, certains critères neurophy-siologiques comme des aspects de dispersion temporelle, des anomalies plus marquées aux membres supérieurs qu’aux membres inférieurs, doivent faire remettre en cause le diagnostic de PAIC. Tous ces éléments sont détaillés dans le tableau 1.
La question temporelle dans la remise en cause du diagnostic de PAIC est très importante et conditionnée par l’évolutivité de la neuropathie. Sans donner de fenêtre temporelle précise, on peut considérer qu’une évolutivité notable sur 6 mois à un an doit faire pratiquer dans un délai de 18 mois maxi-mum des examens complémentaires à la recherche d’un diagnostic alternatif, avant de voir apparaître une perte axonale trop importante et définitive. Dans les cas les plus chroniques, une réévaluation clinique périodique une fois par an pendant les 5 premières années, permettra généralement de voir émerger une étiologie, mais après ce délai, il est peu probable comme on l’a vu plus haut, de faire un diagnostic utile.
La panoplie des examens complémentaires utiles en seconde intention est très vaste et passe par des examens biologiques, neurophysiologiques, neu-ropathologiques et d’imagerie. Il est très difficile de modéliser de façon simple un algorithme décisionnel selon les différentes situations cliniques, mais on se référera à quelques exemples donnés dans le tableau 1 à titre indicatif. Dans tous les cas, la décision de recourir à des examens complémentaires éven-tuellement invasifs devra faire au mieux l’objet d’une discussion collégiale. L’implication d’un centre de référence peut alors se révéler précieuse pour aider le cas échéant à guider la démarche thérapeutique.
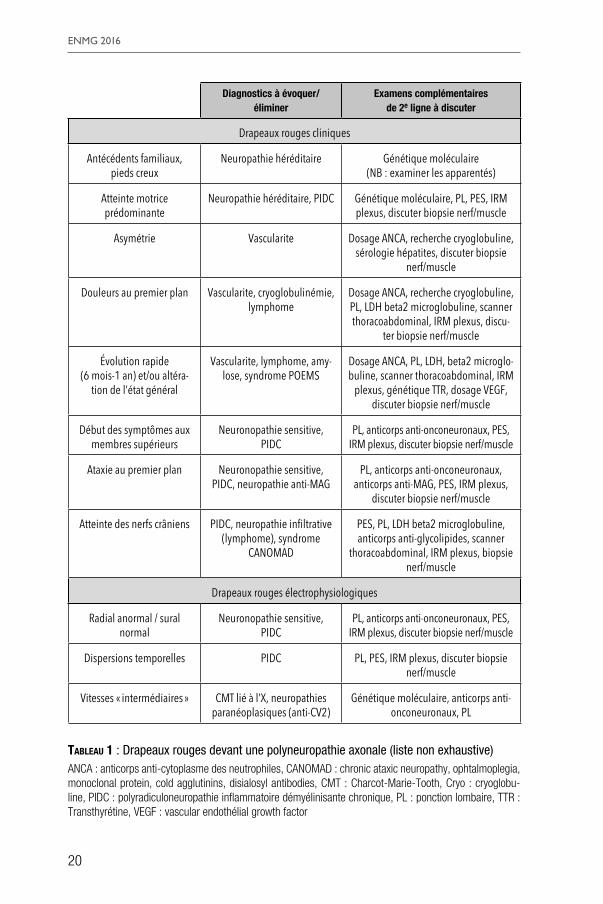
20
ENMG 2016
Diagnostics à évoquer/éliminer
Examens complémentaires de 2e ligne à discuter
Drapeaux rouges cliniques
Antécédents familiaux, pieds creux
Neuropathie héréditaire Génétique moléculaire (NB : examiner les apparentés)
Atteinte motrice prédominante
Neuropathie héréditaire, PIDC Génétique moléculaire, PL, PES, IRM plexus, discuter biopsie nerf/muscle
Asymétrie Vascularite Dosage ANCA, recherche cryoglobuline, sérologie hépatites, discuter biopsie
nerf/muscle
Douleurs au premier plan Vascularite, cryoglobulinémie, lymphome
Dosage ANCA, recherche cryoglobuline, PL, LDH beta2 microglobuline, scanner thoracoabdominal, IRM plexus, discu-
ter biopsie nerf/muscle
Évolution rapide (6 mois-1 an) et/ou altéra-
tion de l’état général
Vascularite, lymphome, amy-lose, syndrome POEMS
Dosage ANCA, PL, LDH, beta2 microglo-buline, scanner thoracoabdominal, IRM
plexus, génétique TTR, dosage VEGF, discuter biopsie nerf/muscle
Début des symptômes aux membres supérieurs
Neuronopathie sensitive, PIDC
PL, anticorps anti-onconeuronaux, PES, IRM plexus, discuter biopsie nerf/muscle
Ataxie au premier plan Neuronopathie sensitive, PIDC, neuropathie anti-MAG
PL, anticorps anti-onconeuronaux, anticorps anti-MAG, PES, IRM plexus,
discuter biopsie nerf/muscle
Atteinte des nerfs crâniens PIDC, neuropathie infiltrative (lymphome), syndrome
CANOMAD
PES, PL, LDH beta2 microglobuline, anticorps anti-glycolipides, scanner
thoracoabdominal, IRM plexus, biopsie nerf/muscle
Drapeaux rouges électrophysiologiques
Radial anormal / sural normal
Neuronopathie sensitive, PIDC
PL, anticorps anti-onconeuronaux, PES, IRM plexus, discuter biopsie nerf/muscle
Dispersions temporelles PIDC PL, PES, IRM plexus, discuter biopsie nerf/muscle
Vitesses « intermédiaires » CMT lié à l’X, neuropathies paranéoplasiques (anti-CV2)
Génétique moléculaire, anticorps anti-onconeuronaux, PL
Tableau 1 : Drapeaux rouges devant une polyneuropathie axonale (liste non exhaustive)ANCA : anticorps anti-cytoplasme des neutrophiles, CANOMAD : chronic ataxic neuropathy, ophtalmoplegia, monoclonal protein, cold agglutinins, disialosyl antibodies, CMT : Charcot-Marie-Tooth, Cryo : cryoglobu-line, PIDC : polyradiculoneuropathie inflammatoire démyélinisante chronique, PL : ponction lombaire, TTR : Transthyrétine, VEGF : vascular endothélial growth factor

21
Bilan étiologique d’une polyneuropathie axonale : jusqu’où aller ?
Conclusion (provisoire)
Les polyneuropathies axonales idiopathiques chroniques sont des neu-ropathies d’apparition relativement tardives, mais ne peuvent en aucun cas être considérées comme liées à un vieillissement normal. Identifier leurs fac-teurs de risque constitue un défi, tant l’approche des maladies multifactorielles reste complexe, et l’interaction gènes/environnement difficile à étudier.
Le diagnostic de PAIC reste donc éminemment frustrant pour le méde-cin, mais finalement d’une certaine façon rassurant pour les patients. En effet, si sa physiopathologie reste obscure, de nombreuses données cliniques de suivi à long terme montrent que son évolution est le plus souvent bénigne. Il faudra toutefois toujours, devant un patient ayant reçu ce diagnostic, garder un esprit ouvert et un œil attentif, en ayant en tête un certain nombre de diagnostics dif-férentiels utiles. Dans cette perspective, certains « drapeaux rouges » doivent systématiquement faire remettre en cause le diagnostic de PAIC, et discuter au cas par cas certains examens complémentaires, parfois invasifs, mais utilisés de façon rationnelle.
RéférencesAdams, D., Coelho, T., Obici, L., Merlini, G., Mincheva, Z., Suanprasert,
N., … Dyck, P. J. (2015). Rapid progression of familial amyloidotic polyneuropathy: a multinational natural history study. Neurology, 85(8), 675–682.
Albers, J. W., et Kelly, J. J. (1989). Acquired inflammatory demyelinating polyneuropathies: clinical and electrodiagnostic features. Muscle et Nerve, 12(6), 435–451.
Attarian, S., Franques, J., Elisabeth, J., Trébuchon, A., Duclos, Y., Wybrecht, D., … Pouget, J. (2015). Triple-stimulation technique improves the diagnosis of chronic inflammatory demyelinating polyradiculoneuropathy. Muscle et Nerve.
Ayrignac, X., Viala, K., Koutlidis, R. M., Taïeb, G., Stojkovic, T., Musset, L., … Bouche, P. (2013). Sensory chronic inflammatory demyelinating polyneuropathy: an under-recognized entity? Muscle et Nerve, 48(5), 727–732.
Camdessanché, J.-P., et Antoine, J.-C. (2014). [Sensory neuronopathy: diagnostic strategy]. Presse Médicale (Paris, France: 1983), 43(11), 1185–1188.
Chin, R. L., Latov, N., Sander, H. W., Hays, A. P., Croul, S. E., Magda, P., et Brannagan, T. H. (2004). Sensory CIDP presenting as cryptogenic sensory polyneuropathy. Journal of the Peripheral Nervous System: JPNS, 9(3), 132–137.
Dubourg, O., Tardieu, S., Birouk, N., Gouider, R., Léger, J. M., Maisonobe, T., … LeGuern, E. (2001). Clinical, electrophysiological and molecular genetic characteristics

22
ENMG 2016
of 93 patients with X-linked Charcot-Marie-Tooth disease. Brain: A Journal of Neurology, 124(Pt 10), 1958–1967.
Dyck, P. J., Lais, A. C., Ohta, M., Bastron, J. A., Okazaki, H., et Groover, R. V. (1975). Chronic inflammatory polyradiculoneuropathy. Mayo Clinic Proceedings, 50(11), 621–637.
Dyck, P. J., Oviatt, K. F., et Lambert, E. H. (1981). Intensive evaluation of referred unclassified neuropathies yields improved diagnosis. Annals of Neurology, 10(3), 222–226.
Fisher, M. A. (2014). F-waves in diabetes mellitus: Answers and questions. Muscle et Nerve, 49(6), 783–785.
French CIDP Study Group. (2008). Recommendations on diagnostic strategies for chronic inflammatory demyelinating polyradiculoneuropathy. Journal of Neurology, Neurosurgery, and Psychiatry, 79(2), 115–118.
Grant, I. A., Hunder, G. G., Homburger, H. A., et Dyck, P. J. (1997). Peripheral neuropathy associated with sicca complex. Neurology, 48(4), 855–862.
Hanewinckel, R., van Oijen, M., Ikram, M. A., et van Doorn, P. A. (2015). The epidemiology and risk factors of chronic polyneuropathy. European Journal of Epidemiology.
Hughes, R. a. C., Umapathi, T., Gray, I. A., Gregson, N. A., Noori, M., Pannala, A. S., … Swan, A. V. (2004). A controlled investigation of the cause of chronic idiopathic axonal polyneuropathy. Brain: A Journal of Neurology, 127(Pt 8), 1723–1730.
Isose, S., Kuwabara, S., Kokubun, N., Sato, Y., Mori, M., Shibuya, K., … Tokyo Metropolitan Neuromuscular Electrodiagnosis Study Group. (2009). Utility of the distal compound muscle action potential duration for diagnosis of demyelinating neuropathies. Journal of the Peripheral Nervous System: JPNS, 14(3), 151–158.
Joint Task Force of the EFNS and the PNS. (2010). European Federation of Neurological Societies/Peripheral Nerve Society Guideline on management of chronic inflammatory demyelinating polyradiculoneuropathy: report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society--First Revision. Journal of the Peripheral Nervous System: JPNS, 15(1), 1–9.
Kelly, J. J., Kyle, R. A., O’Brien, P. C., et Dyck, P. J. (1981). Prevalence of monoclonal protein in peripheral neuropathy. Neurology, 31(11), 1480–1483.
Koike, H., Hashimoto, R., Tomita, M., Kawagashira, Y., Iijima, M., Tanaka, F., et Sobue, G. (2011). Diagnosis of sporadic transthyretin Val30Met familial amyloid polyneuropathy: a practical analysis. Amyloid: The International Journal of Experimental and Clinical Investigation: The Official Journal of the International Society of Amyloidosis, 18(2), 53–62.
Latov, N., Hays, A. P., et Sherman, W. H. (1988). Peripheral neuropathy and anti-MAG antibodies. Critical Reviews in Neurobiology, 3(4), 301–332.

23
Bilan étiologique d’une polyneuropathie axonale : jusqu’où aller ?
Magy, L., et Vallat, J.-M. (2008). [Clinical and electrophysiologic aspects of peripheral nervous system vasculitis]. Revue Neurologique, 164 Spec No 2, F118–125.
McLeod, J. G., Tuck, R. R., Pollard, J. D., Cameron, J., et Walsh, J. C. (1984). Chronic polyneuropathy of undetermined cause. Journal of Neurology, Neurosurgery, and Psychiatry, 47(5), 530–535.
Mellgren, S. I., Conn, D. L., Stevens, J. C., et Dyck, P. J. (1989). Peripheral neuropathy in primary Sjögren’s syndrome. Neurology, 39(3), 390–394.
Notermans, N. C., Wokke, J. H., Franssen, H., van der Graaf, Y., Vermeulen, M., van den Berg, L. H., … Jennekens, F. G. (1993). Chronic idiopathic polyneuropathy presenting in middle or old age: a clinical and electrophysiological study of 75 patients. Journal of Neurology, Neurosurgery, and Psychiatry, 56(10), 1066–1071.
Notermans, N. C., Wokke, J. H., van den Berg, L. H., van der Graaf, Y., Franssen, H., Teunissen, L. L., et Lokhorst, H. M. (1996). Chronic idiopathic axonal polyneuropathy. Comparison of patients with and without monoclonal gammopathy. Brain: A Journal of Neurology, 119 ( Pt 2), 421–427.
Notermans, N. C., Wokke, J. H., van der Graaf, Y., Franssen, H., van Dijk, G. W., et Jennekens, F. G. (1994). Chronic idiopathic axonal polyneuropathy: a five year follow up. Journal of Neurology, Neurosurgery, and Psychiatry, 57(12), 1525–1527.
Prineas, J. (1970). Polyneuropathies of undetermined cause. Acta Neurologica Scandinavica. Supplementum, 44, 1–72.
Rajabally, Y. A., Jacob, S., et Hbahbih, M. (2005). Optimizing the use of electrophysiology in the diagnosis of chronic inflammatory demyelinating polyneuropathy: a study of 20 cases. Journal of the Peripheral Nervous System: JPNS, 10(3), 282–292.
Rossor, A. M., Evans, M. R. B., et Reilly, M. M. (2015). A practical approach to the genetic neuropathies. Practical Neurology, 15(3), 187–198.
Seo, J.-H., Ryan, H. F., Claussen, G. C., Thomas, T. D., et Oh, S. J. (2004). Sensory neuropathy in vasculitis: a clinical, pathologic, and electrophysiologic study. Neurology, 63(5), 874–878.
Teunissen, L. L., Notermans, N. C., Franssen, H., van der Graaf, Y., Oey, P. L., Linssen, W. H., … Wokke, J. H. (1997). Differences between hereditary motor and sensory neuropathy type 2 and chronic idiopathic axonal neuropathy. A clinical and electrophysiological study. Brain: A Journal of Neurology, 120 ( Pt 6), 955–962.
Üçeyler, N., Geng, A., Reiners, K., Toyka, K. V., et Sommer, C. (2015). Non-systemic vasculitic neuropathy: single-center follow-up of 60 patients. Journal of Neurology, 262(9), 2092–2100.
Vallat, J. M., Jauberteau, M. O., Bordessoule, D., Yardin, C., Preux, P. M., et Couratier, P. (1996). Link between peripheral neuropathy and monoclonal dysglobulinemia: a study of 66 cases. Journal of the Neurological Sciences, 137(2), 124–130.

24
ENMG 2016
Vallat, J.-M., Tabaraud, F., Magy, L., Torny, F., Bernet-Bernady, P., Macian, F., et Couratier, P. (2003). Diagnostic value of nerve biopsy for atypical chronic inflammatory demyelinating polyneuropathy: evaluation of eight cases. Muscle et Nerve, 27(4), 478–485.
van Dijk, G. W., Notermans, N. C., Kater, L., Kruize, A. A., Linssen, W. H., et Wokke, J. H. (1997). Sjögren’s syndrome in patients with chronic idiopathic axonal polyneuropathy. Journal of Neurology, Neurosurgery, and Psychiatry, 63(3), 376–378.
Viala, K., Maisonobe, T., Stojkovic, T., Koutlidis, R., Ayrignac, X., Musset, L., … Bouche, P. (2010). A current view of the diagnosis, clinical variants, response to treatment and prognosis of chronic inflammatory demyelinating polyradiculoneuropathy. Journal of the Peripheral Nervous System: JPNS, 15(1), 50–56.
Visser, N. A., Notermans, N. C., Linssen, R. S. N., van den Berg, L. H., et Vrancken, A. F. J. E. (2015). Incidence of polyneuropathy in Utrecht, the Netherlands. Neurology, 84(3), 259–264.
Visser, N. A., Vrancken, A. F. J. E., van der Schouw, Y. T., van den Berg, L. H., et Notermans, N. C. (2013). Chronic idiopathic axonal polyneuropathy is associated with the metabolic syndrome. Diabetes Care, 36(4), 817–822.
Vrancken, A. F. J. E., Notermans, N. C., Jansen, G. H., Wokke, J. H. J., et Said, G. (2004). Progressive idiopathic axonal neuropathy--a comparative clinical and histopathological study with vasculitic neuropathy. Journal of Neurology, 251(3), 269–278.
Wiggin, T. D., Sullivan, K. A., Pop-Busui, R., Amato, A., Sima, A. A. F., et Feldman, E. L. (2009). Elevated triglycerides correlate with progression of diabetic neuropathy. Diabetes, 58(7), 1634–1640.

25
3. Pièges et difficultés de l’ENMG du sujet âgé
Timothée lengleT
Centre de Référence Neuromusculaire, Hôpital de la Pitié-Salpétrière, APHP, Paris, France

26
ENMG 2016
Introduction
L’ENMG aux extrêmes de la vie est toujours source d’une certaine appré-hension pour le praticien. Avec actuellement 9 % de la population âgée de plus de 75 ans et une prévision de 12 % pour 2030, il s’agit pourtant d’une situation déjà récurrente de la pratique de l’ENMG et qui n’aura de cesse de croitre dans les décennies à venir. Sans compter certaines difficultés techniques inhé-rentes au sujet vieillissant, l’ENMG du sujet âgé est en cela déconcertant que les données qui en sont issues sortent volontiers des normes, classiquement établies chez des sujets de 20 à 60 ans. Il se place également au-delà de la vision schématique d’un simple « prolongement de l’examen clinique » avec la mise en évidence quasi systématique au cours de l’examen d’anomalies fortuites. Dans cette revue nous nous intéresserons plus particulièrement au contexte de sujets âgés pour qui se pose la question d’une pathologie neuromusculaire évolutive avec toute la difficulté d’en établir les signes neurophysiologiques évocateurs au-delà du « bruit de fond » induit par le contexte polypatholo-gique et des comorbidités classiquement relevées dans cette population.
Après avoir décrit les conséquences fonctionnelles et neurophysio-logiques du vieillissement neuromusculaire, nous établirons les enjeux de l’explo ration ENMG chez le sujet âgé et donnerons des recommandations sur sa pratique en routine clinique.
1. Contexte du sujet âgé
Définir le sujet âgé est déjà en soi une difficulté. En effet, le vieillissement ne surgit pas soudainement à partir d’un certain âge, il s’agit d’un processus lié au temps entraînant une perte progressive d’adaptabilité des organismes sous l’influence de multiples facteurs tels que l’hérédité, les habitudes d’alimenta-tion, l’environnement social et familial… Selon l’OMS, le sujet âgé présente un âge civil supérieur à 65 ans, mais rien n’est plus éloigné d’un sujet vieillissant, mais indemne de pathologie au long cours qu’un vieillard fragile présentant de multiples handicaps. La définition d’une personne âgée dépend ainsi du contexte et en médecine gériatrique on oppose schématiquement : le vieillis-sement réussi sans pathologie, sans retentissement des modifications phy-siologiques, avec des capacités fonctionnelles entretenues donc préservées ; le vieillissement usuel, sans pathologie, mais avec une atteinte physiologique d’organes dont résulte un sujet fragile pouvant devenir dépendant à l’occa-sion d’une affection intercurrente ; et le vieillissement pathologique, avec pathologies et handicaps sources d’une dépendance le plus souvent définitive.

27
Pièges et difficultés de l’ENMG du sujet âgé
Les problématiques de la gériatrie (polypathologie, perte d’autonomie, fragi-lité) concernent de fait relativement peu d’individus âgés de 65-70 ans (les « young-old » selon la terminologie anglaise), mais bien souvent ceux de 75 ans et plus (« middle-old », voire « oldest-old » pour les plus de 85 ans).
2. Vieillissement physiologique du nerf et des muscles, conséquences fonctionnelles et traduction sur l’ENMG standard
Le système neuromusculaire dans son ensemble est soumis aux contraintes du vieillissement normal.
2.1. Vieillissement du nerf et traduction ENMG
L’âge influence l’état morphologique et fonctionnel du système nerveux périphérique avec une perte progressive en fibres myéliniques et amyéliniques. S’y associe la survenue d’anomalies structurelles de la gaine de myéline, mais également fonctionnelles impliquant les processus de démyélinisation et remyélinisation. Ces dernières résulteraient d’une baisse de l’expression avec l’âge des protéines majeures de la myéline (P0, PM22, MBP) alors que la perte axonale s’expliquerait par une réduction au niveau du nerf périphérique de l’expression et du transport axonal des protéines du cytosquelette (Verdú, Ceballos, Vilches, Navarro, 2000). En outre, les capacités de repousse axonale et de réinnervation collatérale perdurent au cours de la vie, mais tendent à devenir retardées et à perdre progressivement en efficacité limitant en cas de lésion nerveuse les capacités de récupération fonctionnelle (Kovacic, Sketelj, Bajrović, 2009).
Sur le plan clinique, ce vieillissement se traduit de façon inconstante sur l’examen des grosses fibres par des anomalies de la sensibilité vibratoire et du sens de position du gros orteil et des réflexes achilléens respectivement per-turbés et abolis chez plus de la moitié des sujets de 80 ans et plus, et volontiers associés des symptômes subjectifs à type de paresthésies et crampes (Bouche, Cattelin, Saint-Jean, Léger, Queslati, Guez et al., 1993). Quant aux altérations des petites fibres, elles impliquent essentiellement les fibres Aδ et s’accom-pagnent entre autres d’une altération des seuils de détection thermique et de la douleur (Kemp, Després, Pebayle, Dufour, 2014). Plusieurs études de cohorte majoritairement transversales et combinant recensement épidémio-logique, données cliniques et neurophysiologiques ont permis d’établir la

28
ENMG 2016
relation entre la dégradation de la fonction nerveuse périphérique et le reten-tissement fonctionnel sur la qualité de la marche, l’équilibre, la baisse de la mobilité et l’impact négatif sur les activités de la vie courante (Resnick, Vinik, Schwartz, Leveille, Brancati, Balfour et al., 2000 ; Guralnik, Ferrucci, Pieper, Leveille, Markides, Ostir et al., 2000 ; Ward, Boudreau, Caserotti, Harris, Zivkovic, Goodpaster, et al., 2015). Cette relation est d’autant plus forte quand elle implique un nerf fragilisé par un contexte diabétique (Strotmeyer, de Rekeneire, Schwartz, Faulkner, Resnick, 2008), mais s’observe également chez la personne âgée indemne de toute autre cause de neuropathie (Baldereschi, Inzitari, Di Carlo, Farchi, Scafato, Inzitari, 2007 ; Gregg, Sorlie, Paulose-Ram, Gu, Eberhardt, Wolz, et al., 1999, 2000 ; Hoffman, Staff, Robb, St Sauver, Dyck, Klein, 2015). Récemment, une analyse longitudinale effectuée sur une cohorte américaine de 1680 sujets indique que l’altération nerveuse périphé-rique, définie sur le plan moteur à partir d’une conduction motrice du nerf péronier et sur le plan sensitif par des symptômes subjectifs et une évaluation de la sensibilité vibratoire et au monofilament, concernait 55 % des sujets et était un facteur indépendant de survenue ultérieure d’un handicap moteur (Ward, Caserotti, Faulkner, Boudreau, Zivkovic, Lee et al., 2013).
Sur le plan neurophysiologique de nombreuses études ont pu établir une altération des vitesses de conduction motrices et plus encore sensitives, ralentissements dont on discute le caractère linéaire ou non linéaire avec l’âge (Bouche, Cattelin, Saint-Jean, Léger, Queslati, Guez et al., 1993 ; Dorfman, Bosley, 1979 ; Tong, Werner, Franzblau, 2004) et sans que l’on sache avec cer-titude s’ils reflètent une atteinte myélinique primitive ou plus certainement une réduction du diamètre axonal associée à des altérations secondaires de la gaine de myéline (Chase, Engelhardt, Adinolfi, Chirwa, 1992). Globalement, on admet comme physiologique une perte de 1 à 2 m/s par décennie au-delà de 60 ans qui se reportera sur l’ensemble des données de l’étude de conduc-tion. Également la perte axonale se traduira par une baisse progressive et glo-bale à prédominance distale des amplitudes sensitives sur l’ENMG (Fujimaki, Kuwabara, Sato, Isose, Shibuya, Sekiguchi et al., 2009). Cette perte axonale sen-sitive apparait relativement plus précoce et marquée que la baisse des ampli-tudes motrices sans doute du fait de capacités de réinnervation collatérale par sprouting qui vient durablement compenser la perte pourtant très significative avec l’âge en unités motrices fonctionnelles (McComas, 2013). Ces dernières années, les variations de l’examen de conduction liées au vieillissement ont été partiellement appréhendées par les études de cohortes. On peut à titre d’exemple se référer aux données communiquées tout récemment par l’équipe de Rotterdam de PA Van Doorn avec l’étude de plus de 800 sujets de 45 ans et plus dépourvus de neuropathie chez qui on retrouve une baisse de 1,48 µV

29
Pièges et difficultés de l’ENMG du sujet âgé
tous les 5 ans du potentiel d’action sensitif du sural (et de 0,93 m/s de la vitesse de conduction sensitive), et de 0,35 mV sur l’amplitude motrice du nerf péro-nier sur la même période. Et ce déclin était significativement plus prononcé pour les sujets de plus grande taille (Hanewinckel, Drenthen, Hofman, Ikram, van Doorn, 2015). Pour autant, ces variations établies à l’échelle de population sont difficiles à appréhender au niveau individuel et il n’est pas rare en routine clinique que les données de l’étude de conduction demeurent tout à fait dans les normes de laboratoire y compris chez des sujets de 80 ans et plus.
2.2. Vieillissement musculaire et traduction ENMG
Le terme de sarcopénie du grec « sarx » (chair) et « penia » (pauvreté) a été introduit dès 1988 par Irvin Rosenberg, médecin nutritionniste améri-cain lequel voyait dans la perte musculaire squelettique l’une des modifica-tions les plus spectaculaires de l’organisme du sujet âgé (Rosenberg, 1989). Mais ce n’est qu’à l’aube des années 2010 qu’une définition plus consensuelle de la sarcopénie a émergée en associant à la perte insidieuse et progressive de la masse musculaire squelettique, une conséquence clinique sous la forme d’une baisse de la force musculaire et des performances physiques (Cruz-Jentoft, Baeyens, Bauer, Boirie, Cederholm, Landi et al., 2010). Elle résulte d’une mécanique complexe dépendante à la fois du nerf et du muscle et conju-guant à degré variable d’un individu à l’autre : une décroissance du nombre de neurones moteurs source de dénervation musculaire chronique ; des modi-fications du statut hormonal en particulier des hormones anabolisantes ; une baisse de l’acti vité physique ; des modifications des habitudes alimentaires avec réduction des apports protéiques ; une dysfonction mitochondriale favorisant l’apoptose (Morley, Baumgartner, Roubenoff, Mayer, Nair, 2001). En résultent des perturbations de la transmission neuromusculaire, de l’orga-nisation musculaire et de sa composition en fibres (perte en fibres de type 2 au 1er plan), du couplage excitation-contraction, et du métabolisme musculaire (Doherty, 2003 ; Edström, Altun, Bergman, Johnson, Kullberg, Ramírez-León et al., 2007 ; Ryall, Schertzer, Lynch, 2008).
On estime ainsi à plus de 50 % la masse musculaire perdue au-delà de 80 ans et cette perte s’accompagne d’un déclin progressif de l’intensité de la contraction musculaire de l’ordre de 15 % par décade dès 50 ans et atteignant environ 30 % après 70 ans (Metter, Conwit, Tobin, Fozard, 1997 ; American College of Sports Medicine Position Stand, 1998). Les conséquences fonction-nelles de cette perte de force sont désormais également bien établies : alté-ration de la mobilité, perte d’autonomie, et inévitablement augmentation de la morbidité (Dufour, Hannan, Murabito, Kiel, McLean, 2013 ; Xue, Walston,

30
ENMG 2016
Fried, Beamer, 2011 ; Marsh, Rejeski, Espeland, Miller, Church, Fielding et al., 2011 ; Visser, Goodpaster, Kritchevsky, Newman, Nevitt, Rubin et al., 2005). À ce titre, la sarcopénie est maintenant reconnue comme un facteur clé de la physiopathologie du syndrome de fragilité de la personne âgée et perçue comme un enjeu majeur de santé publique (Morley, Haren, Rolland, Kim, 2006). En effet, son coût engendré aux États-Unis est de 18 milliards de dollars sur la seule année 2000 et un soin qui permettrait une baisse de la prévalence de la sarcopénie de seulement 10 % serait source d’une économie annuelle de plus de 1 milliard de dollars (Janssen, Shepard, Katzmarzyk, Roubenoff, 2004). Cependant, la difficulté initiale à établir une définition consensuelle et la complexité des modulateurs potentiels de la sarcopénie ont nui au déve-loppement de molécules à même de prévenir la détérioration progressive de la masse musculaire (McLean, Kiel, 2015). Cette prévention repose actuelle-ment uniquement sur des exercices de résistance musculaire avec une effica-cité variable et difficilement applicables au-delà de 80 ans (Liu, Latham, 2009). Mais un certain nombre d’approches innovantes sont en cours d’investiga-tions qu’elles ciblent la myostatine, facteur de croissance limitant la croissance des tissus musculaires (Becker, Lord, Studenski, Warden, Fielding, Recknor et al., 2015), agissent sur les facteurs hormonaux (Velders, Diel, 2013), ou sur la composante neurogène via par exemple un inhibiteur des histones déacé-tylases (Walsh, Bhattacharya, Sataranatarajan, Qaisar, Sloane, Rahman et al., 2015).
Si les conséquences du vieillissement musculaire sont désormais bien appréhendées en imagerie conventionnelle (IRM, tomodensitométrie voire ultrasonographie) sous la forme d’une diminution du volume et de l’épais-seur musculaire associée à une augmentation de la quantité de graisse intra- et intermusculaire (Wattjes, Fischer, 2013), il n’y a pas en revanche en l’état de signature neurophysiologique de la sarcopénie du moins au niveau de l’ENMG standard. On retient tout au plus la démonstration de sa composante neurogène avec une réduction significative chez le sujet âgé sarcopénique du nombre d’unités motrices évalué par une technique désormais répandue de comptage d’unités motrices (MUNIX) par ailleurs bien corrélée à la perte de force progressive (Kaya, Nakazawa, Hoffman, Clark, 2013 ; Drey, Krieger, Sieber, Bauer, Hettwer, Bertsch, 2014).

31
Pièges et difficultés de l’ENMG du sujet âgé
3. Prise en compte du contexte polypathologique et des comorbidités du sujet âgé
Le terme polypathologie désigne la présence simultanée d’au moins 2 maladies chroniques (c’est-à-dire nécessitant des soins sur le long terme chez le même individu). Une enquête de l’INSEE effectuée en 1991-92 estimait qu’il concernait plus de 90 % des sujets âgés de 70 ans et plus et actuellement un sujet de 80 ans déclare en moyenne 7 pathologies chroniques auxquelles s’associe naturellement le contexte d’une polymédication. Certaines de ces maladies chroniques (ex : diabète qui concerne 10-20 % des sujets de 75 ans et plus, dysthyroïdie, insuffisance rénale…) pourront, seules ou en association, avoir un retentissement neuromusculaire direct ou par l’intermédiaire de trai-tements à potentialité neurotoxique (cancer, VIH, pathologies cardiovascu-laires…) et se traduire par des anomalies de l’ENMG.
La comorbidité désigne la présence d’un ou plusieurs troubles asso-ciés à une pathologie primaire qui sans constituer une maladie en soi va être de nature à influencer la présentation et éventuellement la sévérité de cette pathologie primaire. C’est là encore un contexte classique du sujet âgé volon-tiers mis en évidence sur l’ENMG dans le bilan d’une pathologie neuromuscu-laire. On citera les syndromes canalaires tels que le syndrome du canal carpien dont l’incidence et la sévérité n’ont de cesse d’augmenter avec l’âge (English, Gwynne-Jones, 2015), et il en va naturellement de même pour ce qui concerne la pathologie radiculaire cervicale ou lombaire (Schoenfeld, George, Bader, Caram, 2012 ; Schoenfeld, Laughlin, Bader, Bono, 2012), et du risque de cooc-currence de ces troubles (Lo, Chou, Meng, Chen, Juan, Ho et al., 2012).
4. ENMG du sujet âgé fragile : quels enjeux ?
Pour répondre à cette question, il est utile de se placer dans une problé-matique de la médecine gériatrique et d’appréhender le vieillissement comme une source de diminution des réserves fonctionnelles éventuellement aggravée du fait de pathologies chroniques surajoutées et des comorbidités, et plaçant le sujet dans un état instable susceptible de « décompenser » en cas de pathologie intercurrente. Ce raisonnement gériatrique se traduit schématiquement dans le « 1 + 2 + 3 » proposé en 1984 par le médecin gériatre JP Bouchon et qui peut s’appliquer à la situation de la pathologie neuromusculaire en gériatrie (voir figure 1).
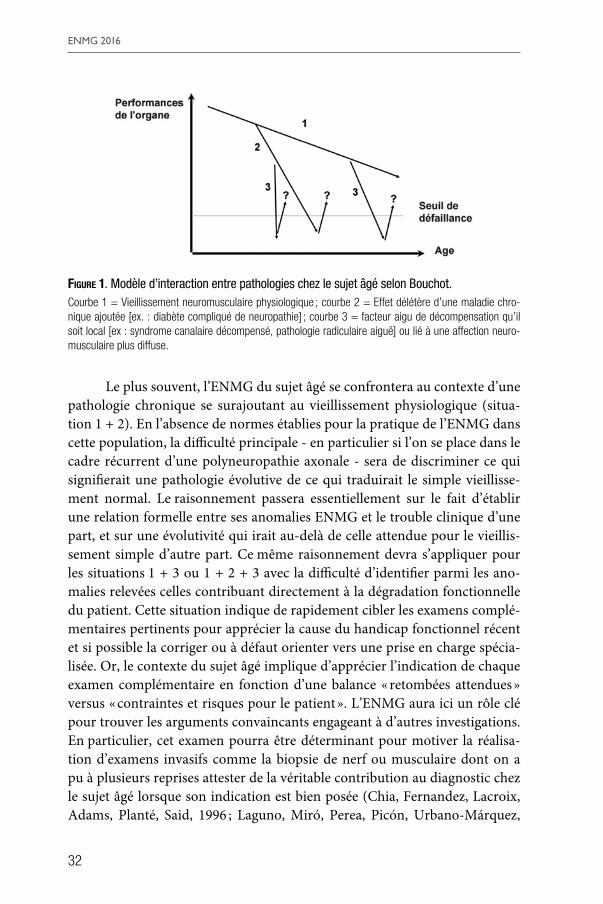
32
ENMG 2016
Figure 1. Modèle d’interaction entre pathologies chez le sujet âgé selon Bouchot.Courbe 1 = Vieillissement neuromusculaire physiologique ; courbe 2 = Effet délétère d’une maladie chro-nique ajoutée [ex. : diabète compliqué de neuropathie] ; courbe 3 = facteur aigu de décompensation qu’il soit local [ex : syndrome canalaire décompensé, pathologie radiculaire aiguë] ou lié à une affection neuro-musculaire plus diffuse.
Le plus souvent, l’ENMG du sujet âgé se confrontera au contexte d’une pathologie chronique se surajoutant au vieillissement physiologique (situa-tion 1 + 2). En l’absence de normes établies pour la pratique de l’ENMG dans cette population, la difficulté principale - en particulier si l’on se place dans le cadre récurrent d’une polyneuropathie axonale - sera de discriminer ce qui signifierait une pathologie évolutive de ce qui traduirait le simple vieillisse-ment normal. Le raisonnement passera essentiellement sur le fait d’établir une relation formelle entre ses anomalies ENMG et le trouble clinique d’une part, et sur une évolutivité qui irait au-delà de celle attendue pour le vieillis-sement simple d’autre part. Ce même raisonnement devra s’appliquer pour les situations 1 + 3 ou 1 + 2 + 3 avec la difficulté d’identifier parmi les ano-malies relevées celles contribuant directement à la dégradation fonctionnelle du patient. Cette situation indique de rapidement cibler les examens complé-mentaires pertinents pour apprécier la cause du handicap fonctionnel récent et si possible la corriger ou à défaut orienter vers une prise en charge spécia-lisée. Or, le contexte du sujet âgé implique d’apprécier l’indication de chaque examen complémentaire en fonction d’une balance « retombées attendues » versus « contraintes et risques pour le patient ». L’ENMG aura ici un rôle clé pour trouver les arguments convaincants engageant à d’autres investigations. En particulier, cet examen pourra être déterminant pour motiver la réalisa-tion d’examens invasifs comme la biopsie de nerf ou musculaire dont on a pu à plusieurs reprises attester de la véritable contribution au diagnostic chez le sujet âgé lorsque son indication est bien posée (Chia, Fernandez, Lacroix, Adams, Planté, Said, 1996 ; Laguno, Miró, Perea, Picón, Urbano-Márquez,

33
Pièges et difficultés de l’ENMG du sujet âgé
Grau, 2002 ; Anish, Nagappa, Mahadevan, Taly, 2015). Ainsi l’âge ne doit pas isolément être un prétexte à limiter les investigations et l’ENMG est essentiel pour en motiver la poursuite.
5. ENMG du sujet âgé : quelles pathologies neuromusculaires ?
L’ensemble des pathologies neuromusculaires de l’adulte peut se ren-contrer chez le sujet âgé et bon nombre d’entre elles voient même leur inci-dence augmenter avec l’âge. Leurs caractéristiques cliniques diffèrent souvent peu du sujet plus jeune et c’est en général le contexte polypathologique et de comorbidité qui contribue à leur difficulté diagnostique et éventuellement thé-rapeutique. Les traitements des pathologies chroniques et comorbidités sont eux-mêmes volontiers pourvoyeurs de pathologie du nerf (ex. : traitements anticancéreux) ou du muscle (ex. : médicaments anti-cholestérol).
Au-delà d’un retard diagnostic le risque est parfois celui d’un sous-dia-gnostic comme cela a pu être établi pour la myasthénie du sujet âgé (Vincent, Clover, Buckley, Grimley Evans, Rothwell, 2003), dont la présentation est pourtant tout à fait superposable à celles du sujet jeune, quoique plus volon-tiers limitée à une expression oculaire, et en dépit d’une plus grande fré-quence de la séropositivité pour les anticorps anti-récepteurs à l’acétylcholine (Zivković, Clemens, Lacomis, 2012). Un âge de début avancé d’une patholo-gie contribue également à en altérer le pronostic fonctionnel et/ou la survie comme pour la sclérose latérale amyotrophique (Tanaka, Yoshikura, Harada, Yamada, Koumura, Sakurai et al., 2012), pathologie pour laquelle la difficulté principale sur le plan ENMG consistera à faire émerger le cadre d’une atteinte neurogène motrice évolutive dans un contexte où la conduction motrice et surtout sensitive se révèleront le plus souvent anormales.
La prévalence des polyneuropathies se majore indiscutablement avec l’âge et atteint 6,6 % des personnes âgées de 60 ans (Hoffman, Staff, Robb, St Sauver, Dyck, Klein, 2015), contre 1 % dans la population générale (Hanewinckel, van Oijen, Ikram, van Doorn, 2015). Parallèlement, la part de polyneuropathie demeurant idiopathique augmente également atteignant près de 40 % pour les sujets de 80 ans et plus (Verghese, Bieri, Gellido, Schaumburg, Herskovitz, 2001). Ces polyneuropathies qualifiées selon les auteurs de « sen-sitive cryptogénique » (CSPN, Wolfe, Barohn, 1998) ou de polyneuropathie chronique axonale idiopathique (CIAP, Notermans, Wokke, van der Graaf, Franssen, van Dijk, Jennekens, 1994) se traduisent typiquement par l’appa-rition vers 60 ans ou au-delà, et plus volontiers chez l’homme, d’un trouble

34
ENMG 2016
sensitif symétrique des extrémités inférieures marqué par un engourdisse-ment et des paresthésies, volontiers plus désagréable que franchement dou-loureux et associé ou non à une faiblesse motrice distale demeurant limitée. Le trouble peut éventuellement toucher les bras, mais garde toujours un pat-tern longueur-dépendant. Sa progression est lente et source d’un faible han-dicap (Wolfe, Baker, Amato, Jackson, Nations, Saperstein et al., 1999). Cette entité constitue sans doute un groupe assez hétérogène, et récemment une étude suggère une certaine association avec le syndrome métabolique (into-lérance au glucose, dyslipidémie obésité, HTA) (Visser, Vrancken, van der Schouw, van den Berg, Notermans, 2013). Dans le contexte du sujet âgé sa pré-sentation clinique (aréflexie achilléenne, altération de la sensibilité vibratoire et thermoalgique) et ENMG (baisse voire abolition du potentiel sensitif sural et du potentiel moteur péronier en recueil pédieux), parait particulièrement difficile à discriminer de celle attendue dans le cadre du simple vieillissement. Mais plus que la distinction neuropathie du vieillissement versus idiopathique ce qui importe est surtout de ne pas méconnaitre les causes classiques de polyneuropathie axonale. Le diabète constitue de loin l’étiologie la plus fré-quente, mais la recherche d’un contexte éthylique, carentiel, toxique, dysglo-bulinémique, infectieux ou inflammatoire sera systématiquement étayée sur un bilan biologique de 1re intention. Et en l’absence de diagnostic, certains points d’appel tels qu’un trouble douloureux ou dystaunomique au 1er plan ou au contraire franchement ataxique, des signes moteurs dépassant les extrémi-tés inférieures, une distribution asymétrique, ou des signes d’évolutivité mani-feste sur 2 examens menés à quelques mois d’intervalle, nécessiteront d’élargir le bilan. Lorsque l’exploration de la neuropathie indique la biopsie de nerf, et en particulier en cas de présentation asymétrique, elle aboutit majoritai-rement au diagnostic de vascularite, avec une proportion non négligeable de patients (de 25 à 40 %) pour qui cette vascularite demeure restreinte au sys-tème nerveux périphérique (Chia, Fernandez, Lacroix, Adams, Planté, Said, 1996 ; Kararizou, Davaki, Karandreas, Davou, Vassilopoulos, 2006).
Des patients âgés peuvent exprimer tardivement une neuropathie héré-ditaire. Les progrès de la biologie moléculaire permettent d’élucider un cer-tain nombre de cas de patients avec des neuropathies d’emblée évocatrices de Charcot-Marie-Tooth tardif avec ou sans histoire familiale, le plus sou-vent dans des formes axonales (Bennett, Lawson, Brickell, Isaacs, Seltzer, Lipe et al., 2008). Et dans le champ des neuropathies héréditaires on insistera plus particulièrement sur la neuropathie amyloïde liée à une mutation du gène de la transthyrétine (TTR) qui dans la population française survient à un âge volontiers tardif et avec une présentation atypique qui rend son diagnostic difficile (Mariani, Lozeron, Théaudin, Mincheva, Signate, Ducot et al., 2015).

35
Pièges et difficultés de l’ENMG du sujet âgé
En pratique la recherche de mutation TTR devrait être très systématique en cas de neuropathie manifestement évolutive et plus particulièrement en cas de signes de dysautonomie.
Concernant les polyneuropathies dysimmunitaires inflammatoires qui ont le mérite de pouvoir faire l’objet de traitements spécifiques, on retient que le syndrome de Guillain Barré et ses variantes s’observent à tout âge avec un pronostic plus péjoratif chez le sujet âgé (Chiò, Cocito, Leone, Giordana, Mora, Mutani, 2003). De même, établir un diagnostic de polyneuropathie inflamma-toire démyélinisante chronique (PIDC) après 65 ans n’a rien d’exceptionnel (Chiò, Cocito, Bottacchi, Buffa, Leone, Plano et al., 2007), mais pourra consti-tuer un défi diagnostique, l’examen clinique et ENMG se révélant volontiers insuffisants pour l’affirmer. Et il convient d’évoquer plus spécifiquement les neuropathies liées à une IgM avec activité anti-MAG, associées ou non à une hémopathie, dont l’âge de début moyen se situe après 60 ans et avec un âge au diagnostic qui va souvent bien au-delà (Luigetti, Padua, Mazza, Rossini, Sabatelli, Lo Monaco, 2013). Elle est reconnaissable sur le plan ENMG par son pattern de démyélinisation distale particulier, mais son diagnostic peut-être difficile en cas de perte axonale importante et de coexistence de syndromes du canal carpien.
Dans le champ de la pathologie musculaire peu d’étude se sont spécifi-quement intéressées au contexte particulier du sujet âgé avec toute la difficulté de considérer le cadre d’une myopathie de révélation tardive comme source des symptômes musculaires lorsque le sujet vieillissant se plaint de douleurs, de faiblesse, d’une intolérance à l’effort ou de douleurs musculo-squelettiques (Laguno, Miró, Perea, Picón, Urbano-Márquez, Grau, 2002). Une étude menée en centre spécialisé indique pourtant que près de 20 % des patients avec un diagnostic de myopathie prouvée sur biopsie musculaire sont âgés de 70 ans et plus (Echaniz-Laguna, Mohr, Lannes, Tranchant, 2010). Dans la série d’Echa-niz-Laguna et al., les points d’appel classiques de type élévation des CPK et/ou anomalies myogènes sur l’EMG sont retrouvés dans les mêmes proportions que chez les 18-69 ans, mais on note chez les patients âgés une prépondérance des douleurs musculaires. Un patient sur 2 présentait une myopathie inflam-matoire majoritairement de type dermato- ou poly-myosite associée chez plus d’un tiers à une tumeur maligne, qu’il convient de rechercher attentivement dans cette population (Echaniz-Laguna, Mohr, Lannes, Tranchant, 2010). Parmi les myopathies d’évolution plus chronique, on retient principalement la myosite à inclusions dont l’étude de l’histoire naturelle chez 136 patients indique qu’un âge de début élevé est associé à un risque plus élevé de décès et à une progression plus rapide vers le handicap (Benveniste, Guiguet, Freebody, Dubourg, Squier, Maisonobe et al., 2011). Mais il n’est pas exceptionnel dans

36
ENMG 2016
ce cadre de pathologie chronique d’identifier une myopathie génétique par-fois difficile à distinguer du vieillissement musculaire normal qu’il s’agisse de certaines dystrophies des ceintures ou de la dystrophie myotonique de type 2 (Palmio, Udd, 2014). D’aucuns estiment même que la dystrophie musculaire oculopharyngée et les dystrophies myotoniques de type 1 et 2 traduiraient en fait une exacerbation des mécanismes du vieillissement musculaire normal (Raz et Raz, 2014 ; Mateos-Aierdi, Goicoechea, Aiastui, Fernández-Torrón, Garcia-Puga, Matheu et al., 2015). L’imagerie musculaire pourra aider dans cette distinction vieillissement normal/pathologie, mais également l’ENMG constituera un examen de choix pour documenter une atteinte myopathique caractérisée et guider la réalisation d’une biopsie musculaire et/ou un diagnos-tic génétique.
6. Conseils pour la pratique de l’ENMG chez le sujet âgé fragilisé ou dépendant en situation polypathologique
Les conseils prodigués ici sont le fruit d’une pratique régulière de l’ENMG du sujet âgé dans le service avec plus 1000 examens réalisés chez des patients de 70 ans et plus sur la seule période de janvier 2014 à décembre 2015.
6.1. Interrogatoire et examen clinique
Encore plus que pour tout autre patient il s’agit ici d’un temps indis-pensable pour la bonne conduite de l’examen d’autant que l’ENMG consti-tue parfois également le temps d’un 1er contact avec un neurologue clinicien. La question posée pour l’examen ENMG est volontiers moins précise que dans la pratique conventionnelle avec des portes d’entrée génériques de type bilan d’une altération de l’état général, d’un amaigrissement, de dou-leurs diffuses, d’un trouble de la marche ou de chutes à répétition. Pourtant le praticien se heurte volontiers à un interrogatoire peu informatif, flou chez des patients à l’état cognitif parfois altéré. Si le sujet est accompagné, il sera utile d’élargir d’emblée cet interrogatoire à l’entourage familial ou aux inter-venants sociosanitaires. Il s’agira pour le praticien de faire le tri dans l’his-toire du patient et dans un 1er temps de renseigner au mieux les antécédents médicaux et comorbi dités connues (diabète, traitement neurotoxique passé, contexte rachidien, syndromes canalaires opérés…) qui influenceront les données de l’examen ENMG à venir. Dans un deuxième temps, tenant compte

37
Pièges et difficultés de l’ENMG du sujet âgé
d’un examen clinique volontiers très « riche », il faudra orienter questions et examen sur les signes pertinents pour orienter vers l’hypothèse d’une affec-tion neuromusculaire récemment acquise. Si le mode d’installation ne peut être précisé, on essaiera d’établir quelques repères temporels (« Depuis quand la canne ? » « Depuis quand le fauteuil roulant ? »). Devant un trouble moteur on n’oubliera pas de poser quelques questions clés telles que la coexistence de crampes, de myalgies le caractère fluctuant du trouble et on recherchera acti-vement la présence de fasciculations. Des douleurs étant volontiers présentes pour de multiples raisons, on essaiera d’en préciser des caractéristiques mus-culaires, ou leur participation neuropathique au besoin en s’aidant des items de l’échelle DN4 (Bouhassira, Attal, Alchaar, Boureau, Brochet, Bruxelle et al., 2005).
Également, on relèvera d’emblée les contraintes techniques de nature à limiter faisabilité et interprétation de l’étude de conduction : lymphœdème, ulcères variqueux, botte sclérodermiforme et autres pathologies cutanées fréquentes du sujet âgé, membre amputé ou inexplorable du fait de soins locaux, perfusions…). Et de même pour l’examen de détection : trouble de la conscience, agitation, état cognitif ne permettant pas l’obtention d’un repos ou d’une participation volontaire, présence d’un générateur d’impulsion qui sera source d’artéfacts permanents.
6.2. Examen de stimulo-détection
L’enjeu va être ici de renseigner au mieux l’existence de signes de perte axonale ou d’anomalie démyélinisantes non attribuables au contexte vieillisse-ment-polypathologie-comorbidités. Pour la problématique d’une neuropathie axonale, mais également pour établir la significativité d’éventuels ralentis-sements de la conduction distale ou tronculaire il pourra être nécessaire de compléter l’examen standard en dehors des zones soumises au vieillissement physiologique (extrémités inférieures) ou à la coexistence de syndromes cana-laires (membres supérieurs). On proposera par exemple l’étude de conduction étagée du nerf fibulaire commun en recueil tibial antérieur ou du nerf médian en recueil fléchisseur radial du carpe. Pour ce qui concerne la conduction sen-sitive aux membres supérieurs elle ne se limitera pas à l’étude du nerf médian et ulnaire, mais elle impliquera également au minimum le nerf radial qui sera utile à la fois pour évaluer la sévérité de la perte axonale dans le contexte d’une polyneuropathie axonale par l’intermédiaire du ratio d’amplitude sural/radial (Rutkove, Kothari, Raynor, Levy, Fadic, Nardin, 1997 ; Overbeek, van Alfen, Bor, Zwarts, 2005), mais également pour renseigner une éventuelle inversion du ratio sensitif dans le cadre d’une PIDC ou d’une neuronopathie sensitive.

38
ENMG 2016
L’étude bilatérale comparative systématique des nerfs moteur et sensitif est hautement conseillée pour déceler une éventuelle asymétrie de la présen-tation électrique qui si elle va dans le même sens qu’une asymétrie clinique et qu’elle s’associe à des signes d’évolutivité dans le même territoire en détection, constituera un critère de choix pour aller plus loin dans l’exploration d’une possible mononeuropathie multiple avec l’indication éventuelle à une biopsie neuromusculaire.
Sur le plan technique, au-delà du soin habituel apporté pour la correc-tion d’éventuelle baisse de la température cutanée des extrémités (fréquente chez le sujet âgé) et la préparation de la peau (abrasée, dégraissée, nettoyée), on n’hésitera pas à recourir, en l’absence de réponse en surface qu’on estime-rait liée à la présence d’œdèmes, à l’usage d’électrodes-aiguilles concentriques qui permettent de se rapprocher du nerf et de bénéficier d’une baisse de l’im-pédance cutanée.
6.3. Étude de jonction neuromusculaire
Elle sera naturellement systématique en cas de contexte clinique évoca-teur (trouble oculomoteur, ptosis, fluctuations…), mais au-delà la recherche d’un bloc de jonction neuromusculaire devrait être de principe proposée en cas de trouble moteur mal défini ou demeurant non expliqué sur la base de l’examen clinique et de la stimulo-détection. Le sujet âgé étant décrit comme volontiers plus intolérant à la répétition des stimulations, on pourra se limiter à 5 stimulations délivrées à 3 Hz sur chaque couple testé des 2 côtés et en cas de doute quant à l’existence d’un décrément on pourra réitérer le test avec 10 stimulations.
6.4. Examen de détection
Le but est de bien faire reposer le raisonnement à partir de l’étude rigoureuse des tracés d’effort dans les zones cliniquement déficitaires pour en établir l’origine neuropathique ou myopathique et non à partir des muscles piqués à distance de ce déficit qui risqueraient de se révéler pathologiques pour d’autres raisons. Au repos au-delà de l’existence de potentiels de fibrilla-tions ou de pointes positives on recherchera activement la présence d’activités spontanées qui traduiraient d’emblée l’existence d’une affection neuromus-culaire par exemple la présence de salves myotoniques pour le diagnostic de dystrophie myotonique, ou évocatrice comme les potentiels de fasciculations.

289
Table des matières
Introduction ...................................................................................V
1. Guillain-Barré syndrome: from 1916 to 2016 .......................11. A brief summary of progress in GBS over the last 100 years .................... 22. An historical commentary on acute motor axonal neuropathy ............... 5
2. Bilan étiologique d’une polyneuropathie axonale : jusqu’où aller ? .......................................................................11
1. Qu’est-ce qu’une polyneuropathie axonale ? .............................................. 121.1. Éléments cliniques et pièges diagnostiques .......................................................... 121.2. Comment définir une polyneuropathie axonale
au plan électrophysiologique ? ................................................................................ 132. Qu’est-ce qu’une polyneuropathie axonale idiopathique chronique ? .... 153. Y a-t-il des diagnostics différentiels d’une PAIC ? ..................................... 164. Quel bilan de première ligne ? ...................................................................... 185. Pour qui, quand, et comment aller plus loin ? ............................................ 19
3. Pièges et difficultés de l’ENMG du sujet âgé ........................251. Contexte du sujet âgé ..................................................................................... 262. Vieillissement physiologique du nerf et des muscles, conséquences
fonctionnelles et traduction sur l’ENMG standard .................................. 272.1. Vieillissement du nerf et traduction ENMG ........................................................ 272.2. Vieillissement musculaire et traduction ENMG ................................................. 29
3. Prise en compte du contexte polypathologique et des comorbidités du sujet âgé ....................................................................................................... 31
4. ENMG du sujet âgé fragile : quels enjeux ? ................................................. 315. ENMG du sujet âgé : quelles pathologies neuromusculaires ? ................ 336. Conseils pour la pratique de l’ENMG chez le sujet âgé fragilisé
ou dépendant en situation polypathologique ............................................. 36

290
ENMG 2016
6.1. Interrogatoire et examen clinique .......................................................................... 366.2. Examen de stimulo-détection ................................................................................. 376.3. Étude de jonction neuromusculaire ....................................................................... 386.4. Examen de détection ................................................................................................ 386.5. Conclure l’examen ..................................................................................................... 396.6. Reconduire l’examen ................................................................................................ 39
4. Les neuropathies d’origine professionnelle .........................451. Neuropathies liées à un facteur mécanique ............................................... 47
1.1. Troubles musculo-squelettiques : généralités ....................................................... 481.2. Syndromes canalaires ............................................................................................... 481.3. Neuropathies liées aux vibrations .......................................................................... 511.4. Rôle de l’ENMG ........................................................................................................ 52
2. Neuropathies toxiques ................................................................................... 522.1. Généralités ................................................................................................................. 522.2. Métaux lourds ............................................................................................................ 532.3. Les solvants ................................................................................................................ 552.4. Les pesticides ............................................................................................................. 562.5. Gaz............................................................................................................................... 56
3. Neuropathies liées à un agent biologique ................................................... 56
5. Les complications neurologiques de la chirurgie pelvienne ....611. Les signes évocateurs d’une atteinte neurologique.................................... 62
1.1. Nerfs somatiques ....................................................................................................... 631.2. Nerfs végétatifs .......................................................................................................... 65
2. Les atteintes des nerfs somatiques ............................................................... 662.1. Nerfs ilio-inguinal, ilio-hypogastrique, génito-fémoral, cutané latéral
de la cuisse ................................................................................................................. 662.2. Les atteintes du nerf pudendal ................................................................................ 672.3. Les atteintes du nerf fémoral ................................................................................... 692.4. Les atteintes du nerf obturateur .............................................................................. 69
3. Les atteintes neuro-végétatives .................................................................... 713.1. Les troubles de la contraction vésicale .................................................................. 713.2. Les troubles de l’éjaculation .................................................................................... 713.3. Les troubles de l’érection .......................................................................................... 713.4. Les constipations ...................................................................................................... 72
4. Approche thérapeutique ................................................................................ 724.1. Nerfs somatiques ....................................................................................................... 72

291
Table des matières
6. Le périnée après une chirurgie digestive ..............................791. Quels sont les types d’intervention postérieure pouvant donner
des séquelles neurologiques ? ........................................................................ 802. Quelles sont les questions posées par le chirurgien digestif
au neurophysiologiste ? .................................................................................. 823. Mise en évidence de l’atteinte du système nerveux autonome ................ 824. Mise en évidence de l’atteinte du nerf pudendal
et de ses différentes branches ........................................................................ 824.1. Innervation du SAE .................................................................................................. 824.2. Innervation du BC .................................................................................................... 834.3. Innervation du SUE .................................................................................................. 834.4. Innervation du PR .................................................................................................... 834.5. L’électromyographie .................................................................................................. 83
5. La mesure de la latence des réflexes sacrés ................................................. 85
7. Les lésions traumatiques des nerfs périnéaux .....................871. Mécanismes lésionnels ................................................................................... 88
1.1. Traumatismes ouverts .............................................................................................. 881.2. Traumatismes fermés ............................................................................................... 89
2. Les syndromes canalaires .............................................................................. 892.1. Syndromes canalaires des nerfs ilio-hypogastriques et ilio-inguinal ............... 892.2. Le syndrome canalaire du nerf cutané latéral de la cuisse ................................. 902.3. Le syndrome de compression du nerf obturateur ............................................... 922.4. La compression du nerf pudendal ......................................................................... 952.5. Les névralgies de la région glutéale ........................................................................ 97
8. Le périnée après une chirurgie rachidienne ....................... 1011. Généralités .....................................................................................................1022. Le syndrome de la queue de cheval post-chirurgical ..............................103
9. Intérêt de l’électro-neuromyogramme dans les atteintes radiculaires du membre supérieur ..................................... 107
1. Données générales ........................................................................................1082. Rappel sur la sémiologie électrophysiologique des souffrances
radiculaires ....................................................................................................1093. Diagnostic topographique ...........................................................................1124. Intérêt pronostique .......................................................................................1145. Diagnostic différentiel ..................................................................................115

292
ENMG 2016
10. ENMG et lombo-sciatalgie : quelle utilité ? ........................ 1191. La stimulodétection ......................................................................................120
1.1. Les vitesses de conduction motrice ......................................................................1201.2. Les vitesses de conduction sensitives ...................................................................1211.3. L’étude des ondes F ..................................................................................................1211.4. L’étude du réflexe H.................................................................................................1211.5. L’étude du réflexe T .................................................................................................122
2. L’examen de détection à l’aiguille ..............................................................1222.1. Cartographie EMG .................................................................................................1232.2. Les muscles paraspinaux ........................................................................................1232.3. Étude des muscles proximaux ...............................................................................1242.4. Les activités de repos ..............................................................................................1242.5. Les anomalies du recrutement ..............................................................................1242.6. L’analyse des potentiels d’unité motrice ..............................................................125
3. Stratégie diagnostique ..................................................................................1253.1. Racine L2 ..................................................................................................................1253.2. Racine L3 ..................................................................................................................1253.3. Racine L4 ..................................................................................................................1263.4. Racine L5 ..................................................................................................................1263.5. Racine S1 ..................................................................................................................126
11. ENMG et myalgies : quelle utilité ? .................................... 1291. Myalgies et myopathies ................................................................................1302. Hyperexcitabilité neuromusculaire et trouble de la repolarisation
des fibres musculaires ..................................................................................1322.1. Myotonie ..................................................................................................................1322.2. Contractures ...........................................................................................................1342.3. Hyperexcitabilité des fibres nerveuses, syndrome d’Isaacs ..............................1342.4. Hyperexcitabilité des neurones moteurs périphériques ...................................135
3. Myalgies : neuropathies périphériques, plexopathies et maladies du neurone moteur .......................................................................................136
4. Myalgies non neurologiques .......................................................................1375. Un protocole ENMG guidé par la clinique ..............................................137
12. Neuropathies sensitives de l’enfant : démarche diagnostique ....................................................... 139
1. L’étude de la conduction nerveuse sensitive chez l’enfant......................1401.1. Particularités pédiatriques de l’étude de la conduction nerveuse sensitive ...140

293
Table des matières
1.2. Les limites de l’exploration électrophysiologique des neuropathies sensitives ....................................................................................141
2. Quelles sont les pathologies associées à une neuropathie sensitive « pure » chez l’enfant ? ...................................................................................1422.1. Neuropathies sensitives dégénératives et héréditaires ......................................1422.2. Neuropathies sensitives acquises ..........................................................................145
3. En pratique, quelle est la démarche diagnostique devant une neuropathie sensitive chez l’enfant ? .....................................145
4. Conclusion ....................................................................................................146
13. L’ENMG dans les atteintes motoneuronales de l’enfant : quelle place face à la génétique ? ......................................... 149
1. Les maladies du motoneurone chez l’enfant ............................................1501.1. Génétique .................................................................................................................1501.2. Les causes acquises de troubles du motoneurone .............................................153
2. Présentations cliniques ................................................................................1562.1. Distribution de l’atteinte motrice .........................................................................1562.2. Symptomatologie ....................................................................................................1562.3. Le diagnostic différentiel des maladies du motoneurone ................................156
3. Diagnostic ......................................................................................................1573.1. ENMG ......................................................................................................................1573.2. Méthodologie de l’EMG ........................................................................................1583.3. Avantages et inconvénients de l’EMG .................................................................159
4. Diagnostic par test génétique ......................................................................1604.1. Avantages .................................................................................................................1604.2. Inconvénients ..........................................................................................................160
5. Comment l’EMG et les tests génétiques devraient-ils être associés ? ...1615.1. Si un phénotype de maladie génétique est cliniquement reconnaissable ......1615.2. S’il n’y a pas de phénotype évident de maladie génétique ................................161
14. Orientation diagnostique devant une neuropathie démyélinisante de l’enfant .................................................. 167
1. Considérations préalables ............................................................................1681.1. Valeurs normales chez l’enfant ..............................................................................1681.2. Orientation étiologique ..........................................................................................169
2. Les neuropathies acquises ...........................................................................1702.1. Le syndrome de Guillain-Barré ............................................................................1702.2. Les polyradiculonévrites inflammatoires chroniques (PIDC) ........................171
3. Les neuropathies d’origine génétique non métaboliques .......................173

294
ENMG 2016
3.1. Les maladies de Charcot-Marie-Tooth (CMT) ..................................................1733.2. Neuropathie par hypersensibilité à la pression (HNPP) ..................................178
4. Les neuropathies métaboliques ..................................................................1794.1. Leucodystrophie métachromatique .....................................................................1794.2. Maladie de Krabbe ..................................................................................................1794.3. Maladie de Nieman-Pick .......................................................................................1824.4. Maladie de Refsum .................................................................................................1824.5. Hyperoxalurie ..........................................................................................................1824.6. Maladie de Tangier .................................................................................................1834.7. Adrénoleucodystrophie ou adrénomyéloneuropathie ......................................1834.8. Maladies mitochondriales avec neuropathie démyélinisante ..........................183
15. EMG neurogène ou myogène : comment distinguer ? ...... 1871. Qu’est-ce qu’un tracé typiquement neurogène ou typiquement
myogène ? .......................................................................................................1881.1. Tracé neurogène ......................................................................................................1881.2. Tracé myogène.........................................................................................................190
2. Pourquoi y a-t-il des situations compliquées ? ..........................................1912.1. Activités spontanées ...............................................................................................1912.2. Caractéristiques des PUM .....................................................................................1922.3. Anomalies du recrutement ....................................................................................193
3. Quels outils supplémentaires ? ....................................................................1944. Sur qui compter ? ..........................................................................................196
16. Quelle stratégie ENMG devant un patient suspect de polyneuropathie ? ........................................................... 199
1. Quel protocole électrophysiologique la HAS a-t-elle proposé ? .............2002. Après les recommandations de la HAS, quels critères
de démyélinisation ? .....................................................................................2023. Des protocoles spécifiques lors de suspicion de PIDC ? .........................2034. Quelles autres stratégies pour quelles autres neuropathies ? .................2045. Protocoles stratégiques proposés ................................................................205
17. Y a-t-il des contre-indications à l’examen ENMG ? .......... 2111. Risque infectieux ..........................................................................................212
1.1. Données de la littérature ........................................................................................2131.2. Contre-indications à la pratique de l’ENMG .....................................................214
2. Risque d’hématome ......................................................................................2142.1. Données de la littérature ........................................................................................215

295
Table des matières
2.2. Contre-indications à la pratique de l’ENMG .....................................................2163. Risque d’électrocution .................................................................................217
3.1. Cas particulier des patients en réanimation .......................................................2173.2. Recommandations pour limiter le risque ...........................................................2173.3. Contre-indications à la pratique de l’ENMG .....................................................218
4. Risque de dysfonctionnement d’un appareillage implanté ....................2184.1. Données de la littérature ........................................................................................2184.2. Recommandations pour limiter le risque ...........................................................2194.3. Contre-indications à la pratique de l’ENMG .....................................................219
18. Comment rédiger un compte rendu d’examen électromyographique .......................................................... 225
1. Le résumé clinique ........................................................................................2271.1. Tableau sémiologique clinique .............................................................................2271.2. Histoire et contexte de l’affection, antécédents ..................................................2281.3. Examens complémentaires et traitements ..........................................................228
2. Tableaux de résultats des différentes techniques .....................................2282.1. Étude de la conduction nerveuse motrice et proximale ...................................2292.2. Étude de la conduction nerveuse sensitive .........................................................2292.3. Étude de la transmission neuromusculaire et de l’excitabilité musculaire .....2302.4. Électromyographie (de détection) .......................................................................230
3. Interprétation des résultats et rédaction de la conclusion ......................2303.1. Description des résultats ........................................................................................2313.2. Interprétation et conclusion finale .......................................................................2333.3. Nécessité parfois d’une lettre d’accompagnement .............................................234
4. Comment énoncer les difficultés de conclusion ? ....................................2354.1. Hésitations diagnostiques ......................................................................................2354.2. Problèmes de pathologies associées .....................................................................2354.3. Anomalies asymptomatiques ................................................................................2364.4. Tableaux incomplets ...............................................................................................2364.5. Absence d’anomalie ................................................................................................236
5. Au total… .......................................................................................................237
19. Pour la biopsie musculaire dans les myopathies inflammatoires..................................................................... 239
1. Les caractéristiques générales des MII ......................................................2422. Les auto-anticorps associés aux MII..........................................................2433. La biopsie musculaire dans les MII ...........................................................244

296
ENMG 2016
4. Critères diagnostiques des MII ...................................................................2445. Glossaire .........................................................................................................252
20. Contre la biopsie musculaire dans les myopathies inflammatoires..................................................................... 255
21. Polyneuropathie et maladie de Parkinson : quel lien ? ...... 2571. Épidémiologie ................................................................................................2582. Sémiologie ......................................................................................................2583. Facteurs de risque .........................................................................................2594. Physiopathologie ...........................................................................................2605. Traitement ......................................................................................................2616. Prévention ......................................................................................................262
22. L’ENMG dans les syndromes cérébelleux d’origine génétique : quelle utilité ? .................................................... 267
1. Les ataxies cérébelleuses autosomiques récessives (ACAR) .................2701.1. Les ACAR caractérisées par une ataxie spino-cérébelleuse, cordonale
postérieure et par une neuronopathie sensitive pure (ganglionopathie) .......2711.2. Les ACAR caractérisées par une ataxie cérébelleuse avec neuropathie
axonale sensitivo-motrice .....................................................................................2721.3. Les ACAR caractérisées par une ataxie cérébelleuse avec une neuropathie
axonale sensitivo-motrice et éventuelle composante démyélinisante associée ........................................................................................273
2. Les ataxies cérébelleuses autosomiques dominantes (ADCA) .............2743. Les ataxies cérébelleuses liées à l’X ............................................................2754. Les cytopathies mitochondriales liées à des mutations de l’ADN
mitochondrial ...............................................................................................275
23. Ce qu’il ne fallait pas manquer depuis Saint-Étienne 2014 .................................................. 279
1. Actualités des syndromes d’hyperexcitabilité nerveuse .........................2802. Les neuropathies démyélinisantes inflammatoires : les critères,
toujours les critères… ...................................................................................2823. L’estimation du nombre d’unités motrices renait de ses cendres ..........284


Ce livre rassemble les textes et les conférences des 20es Journées francophones d’électroneuromyographie (ENMG) qui se sont dé-roulées à Strasbourg du 1er au 3 juin 2016.
L’ENMG, prolongement direct de la clinique, reste encore au-jourd’hui en 2016 un outil diagnostique précieux et irremplaçable dans le domaine des maladies neuromusculaires, malgré la concur-rence croissante des nouvelles techniques d’imagerie, d’analyse génétique et d’immunologie. Les auteurs passent ici en revue les données fondamentales, les techniques nouvelles d’investigation, les étiologies émergentes de maladies neuromusculaires et les stra-tégies d’exploration utiles en pratique quotidienne.
Ce livre est destiné à tous ceux qui s’intéressent à l’ENMG et aux maladies neuromusculaires, qu’ils soient neurologues, rhumatolo-gues, rééducateurs ou pédiatres.
Le coordinateur
ANDONI ECHANIZ-LAGUNA est neurologue, neurophysiologiste et docteur en neurosciences. Praticien hospitalier aux hôpitaux uni-versitaires de Strasbourg, il travaille au sein du Centre de Référence des Maladies Neuromusculaires Grand-Est (CERNEST). Il a fait de ces maladies, en particulier les myopathies et les pathologies du nerf périphérique, son domaine de surspécialité.
ENMG2016ISBN : 978-2-35327-343-0
www.deboecksuperieur.com
Publics Neurologues Neurophysiologistes Rhumatologues Médecins de MPR
s u p é r i e u r
ENM
G 20
16
Sou
s la
dir
ecti
on d
’And
oni E
chan
iz-L
agun
aENMG 2016
20es Journées francophones d’électroneuromyographie
Sous la direction d’Andoni Echaniz-Laguna