Embed Size (px)
Citation preview

TTHHEESSEEEn vue de l'obtention du
DOCTORAT DE L’UNIVERSITÉ DE TOULOUSEDOCTORAT DE L’UNIVERSITÉ DE TOULOUSE
Délivré par l'Université Paul Sabatier - Toulouse IIIDiscipline ou spécialité : Biologie Moléculaire et Cellulaire
JURYM. Jean-Jacques DIAZ Directeur de Recherche Rapporteur
Mme Sylvie HERMANN-LE DENMAT Maître de Conférences Rapportrice M. Sébastien FRIBOURG Chargé de Recherche ExaminateurM. Michel WEBER Directeur de Recherche Président du jury
M. Pierre-Emmanuel GLEIZES Professeur Directeur de Thèse
Ecole doctorale : Biologie-Santé-BiotechnologiesUnité de recherche : Laboratoire de Biologie Moléculaire Eucaryote
Directeur(s) de Thèse : Prof. Pierre-Emmanuel GLEIZESRapporteurs :
Présentée et soutenue par Almass-Houd AGUISSA TOURELe 18 Juin 2008
Titre :
Bases moléculaires de l'anémie de Diamond-Blackfan :étude structure-fonction de la protéine ribosomique RPS19 chez Saccharomyces cerevisiae
1

2

Remerciements
Ce travail a été effectué au Laboratoire de Biologie Moléculaire Eucaryotes (LBME) au sein
de l’équipe Dynamique fonctionnelle du noyau et transport des ARN dirigée par Monsieur
Pierre-Emmanuel Gleizes, Professeur à l’Université Paul Sabatier et à l’Institut
Universitaire de France. Il est le fruit de la contribution de plusieurs personnes, sans qui sa
réalisation n’aurait été possible. Je tiens à remercier Pierre-Emmanuel Gleizes pour m’avoir
donné la chance de faire mes preuves dans le monde de la recherche scientifique. Ses précieux
et judicieux conseils m’ont toujours encouragé à aller de l’avant dans ma future carrière. Son
dynamisme et ses compétences scientifiques m'ont permis de mener à bien cette étude et au
cours de ces sept années passées au LBME, pas un instant où je me suis senti abandonné à la
paillasse ou à mon sort. J’ai été vraiment gaté par son soutien permanent et total. Merci
infiniment d’avoir cru en moi!
Je tiens à remercier sincèrement les membres du jury, qui ont accepté d’évaluer mon travail de
thèse.
Merci à M. Michel Weber, Directeur de Recherche au LBME, d’avoir accepté de présider le
jury de cette thèse. Je remercie Mme Sylvie Hermann-le Denmat, Maître de Conférences à
l’ENS et Université Paris XI, et Jean-Jacques Diaz, directeur de recherche au CGMC,
Villeurbanne, d’avoir accepté d’être les rapporteurs de ce manuscrit. Leurs remarques et
suggestions lors de la lecture de mon rapport m’ont permis d’apporter des améliorations à la
qualité de ce dernier. Merci également à M. Sébastien Fribourg, Chargé de Recherche à
l’IECB-Pessac, pour avoir accepté d’examiner mon mémoire et de faire partie de mon jury de
thèse après cette fructueuse collaboration.
Je tiens à remercier aussi Jacqueline Depeyre, dont les grandes qualités humaines m’ont
permis de ne pas trop sentir le poids du travail et de mener à bout cette thèse.
3

Merci à Philipp Milkereit et Isabelle Léger, qui ont été des bons amis depuis mon année de
Master Recherche. Je tiens également à les remercier pour la confiance et la sympathie qu’ils
m’ont témoignées au cours de ces années de thèse.
A Guillaume Stahl, avec qui j’ai partagé le Bureau et la paillasse et dont l’aide précieuse m’a
été indispensable sur le plan scientifique, sans oublier ses conseils avisés en informatique.
Merci également à Valérie Choesmel, Marlène Faubladier, Marie-françoise O’Donohue,
Coralie Carron, Célia Plisson, Jacques Rouquette, Sébastien Ferreira-cerca et tous les autres
qui étaient de passage dans l’équipe et qui m’ont tous permis d’effectuer cette thèse dans de
très bonnes et très agréables conditions de travail .
Je n’oublie pas non plus les autres parmi lesquels je remercierai plus particulièrement Nicole
Gas, Marie-Ange Dupont et Nacer Benmeradi.
Je tiens à remercier l’ensemble du personnel du LBME et plus particulièrement Cathy
Rousseau et Safi Sylla pour leur gentillesse et leur efficacité.
Je dois aussi remercier les organismes CNRS et FRM pour leur soutien financier au cours de
mes années de thèse.
Je tiens à remercier les amis, thésards ou non qui m’ont aidé au cours des ces années de thèse,
je pense en particulier à mon frère et ami de tous les temps, Mahamadou Adama Maiga .
Merci aussi à tous mes collègues et amis de longue date. Je leur exprime ma profonde
sympathie et leur souhaite beaucoup de bien.
Un remerciement particulier pour mes familles Toulousaines du 57, rue Boyssonne ; de la rue
Ferdinand Bebel et de la rue des Myosotis qui ont fait que je me suis toujours senti chez moi
dans l’agréable ville rose de la France.
Finalement, j’adresse un grand merci à toute ma famille qui a toujours été présente lorsque
j’en ai eu besoin.
4

SOMMAIRE
5

6

SOMMAIRE
RESUME 11
SUMMARY 13
ABREVIATIONS 15
INTRODUCTION 17
A. L'ANEMIE DE DIAMOND-BLACKFAN : UN DESORDRE DE LABIOGENESE DES RIBOSOMES 21
A.1. L’Anémie de Diamond-Blackfan ou ADB 22
A.2. Génétique de l'ADB 25A.2.1. Mutation du gène RPS19 25A.2.2. De nouvelles protéines ribosomiques impliquées dans l'ADB 29
A.3. La biogenèse du ribosome 31A.3.1. Voie de maturation de l’ARNr 31A.3.2. Maturation des particules pré-40S 35A.3.3. Rôle des protéines ribosomiques dans la maturation des particules pré-40S 39
A.4. La protéine RPS19 41A.4.1. Fonction de RPS19 dans la biogenèse des ribosomes 41A.4.2. Partenaires protéiques de la protéine RPS19 42
A.5. Modèles de l’ADB fondés sur la perte de fonction de RPS19 44
A.6. Altération de la biogenèse des ribosomes chez les patients 45
A.7. Les érythrocytes victimes d'un stress ribosomique? 46
A.8. Le nucléole multi-fonctionnel 48
B. MECANISMES MOLECULAIRES DE L'IMPORT NUCLEAIRE DESPROTEINES 51
B.1 Les complexes de pore nucléaire 51
B.2. Les voies d’import nucléaire 52
B.3. La voie d'import classique 53B.3.1. Assemblage du complexe d’import 56B.3.2. Translocation à travers les CPN 58B.3.3. Désassemblage du complexe d’import 59
7

B.3.3.1. Dissociation du complexe importine α/β 59 B.3.3.2. Relargage du cargo. 62B.3.4. Recyclage des importines 63
B.4. Import des protéines ribosomiques 65
RESULTATS 69
ETUDE STRUCTURE-FONCTION DE LA PROTEINE RPS19 71
INTERACTION IN VITRO DE RPS19 AVEC L’ARN RIBOSOMIQUE 83
1. Etude in vitro de l’interaction RPS19 avec le motif 1537-1598 de l’ARNr 18S 832. Etude de l’effet des mutations ADB sur l’interaction de RPS19 avec l’ARN poly-U 873. Conclusion 90
L’IMPORT NUCLEAIRE DE RPS19 EST-IL AFFECTE DANS L’ANEMIE DEDIAMOND-BLACKFAN ? 93
DISCUSSION 121
MATERIEL ET METHODES 135
1. Souches et conditions de culture 1371.1. Souches de bactéries 1371.2. Conditions de croissance 137
2. Transformation bactérienne et isolement de l’ADN plasmidique 1372.1. Bactéries compétentes et transformation 1372.2. Extraction de l’ADN plasmidique de bactéries 139
3. Souches de levure et conditions de culture 1403.1. Souches de levure 1403.2. Conditions de croissance 141
4. Transformation de levure et isolement de l’ADN génomique 1424.1. Transformation de la levure S. cerevisiae 1424.2. Co-transformation et clonage par recombinaison 1424.3. Préparation de l’ADN de S. cerevisiae et isolement du plasmide obtenu par recombinaison 143
5. Manipulations de l’ADN 1445.1. Réaction de PCR 1445.2. Réaction RT-PCR 1445.3. Réaction PCR sur colonie 1455.4. Électrophorèse de l’ADN sur gel d’agarose 1455.5. Isolement d’ADN sur gel d’agarose 1455.6. Digestion de l’ADN par une enzyme de restriction 1465.7. Ligature 146
6. Construction plasmidique 1466.1. Mutagenèse dirigée 1476.2. Clonage du gène RPS19 humain (hsaRPS19) 1486.3. Clonage du gène RPS19 de P. abyssi (PyARPS19) 149
8

6.4. Localisation cellulaire par observation de la GFP 1496.5. Observation de la GFP après fixation des cellules 150
7. Le système du double-hybride chez la levure 1507.1. Principe 1507.2. Construction des vecteurs 151
8. Etude de l'interaction in vitro par retard sur gel (EMSA) 1528.1. Principe 1528.2. Source de l’ARN 1528.3. Transcription in vitro et marquage des ARN 1528.4. Interaction ARN-protéine 153
9. Purification de ribosome sur gradient de sucrose 1539.1. Culture des cellules 1539.2. fractionnement sur gradient 1549.3. Extraction protéique des fractions 1549.4. Extration des ARN 155
10. Immunoprécipitation des protéines étiquetées TAP-TAG sur billes IgG-Sepharose 15510.1. Culture des cellules 15510.2. Immunoprécipitation 15610.3. Extraction des ARN 15710.4. Northern blot 158
BIBLIOGRAPHIE 163
ANNEXE : ETUDE DE LA PROTEINE RPS24 177
I. Test de complementation fonctionnelle chez Saccharomyces cerevisiae 193
II. Recherche de mutants thermosensibles (ts) de Rps19p 195
III. Cas des mutants W53R et S60E 197
TABLEAUX
Tableau 3. Souches de bactéries E. Coli 137
Tableau 4. Souches de levure S. cerevisiae 141
Tableau 5. Plasmides utilisés pour l’étude de la localisation cellulaire chez les mutants d’importine 151
Tableau 6. Plasmides utilisés pour les expériences de double-hybride 158
Tableau 7. Plasmides étiquetés TAP utilisés pour purifier les ribosomes 159
Tableau 8. Plasmides utilisés pour la complémentation fonctionnelle 160
Tableau 9. Plasmides utilisés pour l’étude de la localisation cellulaire 161-162
Tableau 10. Plasmides utilisés pour la production de protéines recombinantes et latranscription in vitro de différents fragments de l'ARN 18S 162
9

10

RESUME
L'anémie de Diamond-Blackfan (ADB) est une érythroblastopénie congénitale rare associée à
des mutations mono-alléliques dans plusieurs gènes de protéine ribosomique. Cette liaison
suggère une relation causale entre l’altération de la biogenèse ou de la fonction des ribosomes et
cette pathologie.
Le gène RPS19 est le plus fréquement muté (25% des patients). Pour comprendre l'impact des
mutations, en particulier les mutations ponctuelles faux-sens, nous avons engagé une étude
structure-fonction de l’homologue de la protéine RPS19 chez la levure Saccharomyces
cerevisiae. Parallèlement, nous avons mené un travail collaboratif pour déterminer la structure
cristalline de l'homologue de RPS19 de l'archeae Pyrococcus abyssi. Nos résultats distinguent
deux types de mutations : certaines, enfouies dans la structure, affectent le repliement et la
stabilité de la protéine, tandis que d'autres, en surface de la protéine, inhibent l’incorporation de
RPS19 dans les particules pré-ribosomiques 40S.
Par ailleurs, nous avons déterminé la séquence de localisation nucléaire et les déterminants
moléculaires de l'adressage nucléaire de RPS19. Les différentes mutations de RPS19
n’interfèrent pas avec ce mécanisme de transport.
Ainsi, les mutations faux-sens de la protéine RPS19 affectent en premier lieu la capacité de la
protéine à être incorporée dans les pré-ribosomes, ce qui altère la biogenèse des ribosomes. Ceci
pourrait d'une part activer une réponse de stress et d'autre part conduire à un déficit en
ribosomes, deux facteurs qui pourraient être fatals dans certains processus physiologiques
comme l'érythropoïèse. Ce mécanisme peut être étendu à toute protéine ribosomique mutée dans
l'ADB.
11

12

SUMMARY
Diamond-Blackfan Anaemia (DBA) is a rare congenital erythroblastopenia associated with
mono-allelic mutations in several ribosomal protein genes. This linkage suggests a causal
relationship between alteration of ribosome biogenesis or functions and this pathology.
The RPS19 gene is the most frequently mutated (25% of patients). To understand the impact of
the mutations, especially missense mutations, we undertook a structure-function study of RPS19
homolog in yeast Saccharomyces cerevisiae and we conducted a collaborative work to
determine the crystal structure of archeae Pyrococcus abyssi RPS19. Our results distinguish two
types of mutations: some affect residues buried in the structure and alter the protein folding and
stability, while others change amino acids at the protein surface and prevent incorporation of
RPS19 into 40S pre-ribosomal particles.
In addition, we determined the nuclear localization sequence and the molecular determinants of
RPS19 transport to the nucleus. Mutations linked to DBA do not interfere with the nuclear
localization of the protein.
Thus, missense mutations in RPS19 primarily affect the ability of the protein to be incorporated
into pre-ribosomes, which alters ribosome biogenesis. This could on the one hand activate a
stress response and on the other hand lead to ribosome shortage, two events that could be fatal
to some physiological processes like erythropoiesis. A similar mechanism can be proposed for
any ribosomal protein mutated to the DBA.
13

14

ABREVIATIONS
ADB Anémie de Diamond-Blackfan
ADN Acide Désoxyribonucléique
ARN Acide ribonucléique
ARNm ARN messager
ARNr ARN ribosomique
RNP Ribonucléoparticule
SnoRNPs Small nucleolar ribonucleoproteins
IGS Intergenic sequences
ITS Internal transcribed spacer
ETS External transcribed spacer
NMD Nonsense mediated decay
NLS Nuclear localization sequence
HNSCC Human head and Neck Squamous Cell Carcinoma cell lines
eIF2 Eukaryotic Initiation Factor 2
FGF-2 Basic fibroblast growth factor
ADP Adénosine di-phosphate
ATP Adénosine tri-phosphate
GDP Guanosine di-phosphate
GTP Guanosine tri-phosphate
GFP Green fluorescent protein
Ran Ras-related Nuclear protein
RanBP2 Ran binding protein 2
RanGAP Ran GTPase Activating protein
RanGEF Ran guanosine exchange factor
CPN Complexe de pore nucléaire
PEG Poly(ethylene glycol)
BFU-e Burst forming units- erythroid
CFU-e Colony forming units- erythroid
EPO Erythropoietin
GM-CSF Granulocyte/ macrophage- colony stimulating factor
KDa kilodaltons
MDa Megadaltons
15

16

INTRODUCTION
17

Figure 1. Etapes de l'érythropoïèse
Les BFU-E sont des progéniteurs précoces alors que les CFU-E sont plus tardifs. Leur différenciation s'accom-pagne d'une réduction de la taille et une dimunition progressive du rapport noyau/ cytoplasme. Vers la fin de la différenciation, on assiste à une synthèse progressive d'hémoglobine avant l'expulsion du noyau et matura-tion finale.
18

L'anémie de Diamond-Blackfan ou ADB est une érythroblastopénie congénitale rare
caractérisée par une grande hétérogénéité phénotypique et génétique et une faible
quantité de précurseurs érythroïdes dans la moelle osseuse. En plus de l'anémie, plus de
50% de patients ADB ont une petite stature et/ou diverses anomalies physiques. C’est
seulement en 1999 qu'un gène a été lié à cette maladie, connue depuis les années 1930.
De manière surprenante, il s'agissait d'un gène de protéine ribosomique, RPS19, dont on
montra la mutation chez 25% des patients. L'identification du gène affecté dans une
maladie génétique peut être la clef permettant une meilleure compréhension de la
pathologie et le développement de nouvelles thérapies. Dans un désordre congénital
comme l'ADB, caractérisé par une affection spécifique des cellules érythroïdes, on
attendait l’implication d’un régulateur de l'érythropoïèse : interleukine, récepteur, ou
élément d'une voie de signalisation. Au démarrage de cette thèse en 2004, le gène
RPS19 restait le seul gène associé à l'ADB. Mais récemment, des mutations dans
plusieurs autres gènes ont été découvertes, toutes dans des gènes de protéines
ribosomiques. L'ADB est donc le signe d'un lien physiologique inattendu entre
biogenèse des ribosomes et érythropoïèse (Figure 1).
Les gènes identifiés établissent l’ADB comme un désordre ribosomique. Ce travail de
thèse est principalement une étude structure-fonction de la protéine ribosomique RPS19
pour tenter d’élucider l'impact des mutations de RPS19 dans l'ADB et de comprendre le
rôle de la protéine dans la maladie. Ce travail présente l’effet des différentes mutations
associées à l'ADB au cours des événements successifs de la biogenèse des ribosomes
(import nucléaire, incorporation dans la petite sous-unité 40S, interaction avec l’ARNr).
A cette fin, nous avons étudié la protéine RPS19 au niveau moléculaire grâce à sa
structure cristalline.
19

Pour aborder ces questions, nous avons choisi d'étudier l'homologue de RPS19 chez la
levure S. cerevisiae. Cette levure est non seulement un modèle de choix pour
développer une approche de génétique moléculaire, mais c'est également l'organisme
eucaryote chez lequel on connaît le mieux la biogenèse des ribosomes. En plus du degré
significatif de conservation de la protéine ribosomique RPS19 entre la levure S.
cerevisiae et H. sapiens, les étapes de la biogenèse des ribosomes présentent de fortes
similitudes entre ces organismes.
Dans cette introduction, nous aborderons dans un premier temps une présentation de
l'anémie de Diamond-Blackfan en donnant des éléments de référence de la biogenèse
sur la maturation et l’assemblage de la particule pré-40S, processus dans lequel RPS19
est impliquée. Dans un deuxième temps, nous procéderons à un exposé des mécanismes
de l'import nucléaire des protéines, une partie de cette thèse ayant trait au transport
nucléocytoplasmique de RPS19.
20

A. L'ANEMIE DE DIAMOND-BLACKFAN : UN DESORDRE DE LA
BIOGENESE DES RIBOSOMES
La biogenèse des ribosomes est l'activité métabolique la plus consommatrice d'énergie
dans une cellule qui prolifère. La synthèse, la maturation et l'assemblage des 4 ARN et
des 79 protéines ribosomiques en deux sous-unités mobilisent environ la moitié de
l'activité transcriptionnelle d'une cellule, et les ARNm codant les protéines
ribosomiques comptent pour la moitié des ARNm cellulaires. La synthèse des
ribosomes est étroitement connectée à la régulation du cycle cellulaire. Malgré son
caractère central, peu de pathologies ont été liées à la mutation de gènes impliqués dans
ce processus. Récemment, des maladies génétiques rares ont été associées soit à la
mutation de gènes de protéines ribosomiques, soit à celle de gènes codant un des 300
facteurs en trans requis pour la maturation des pré-ribosomes. De manière surprenante,
elles correspondent toutes à des dysplasies myéloïdes : anémie de Diamond-Blackfan
(gènes de protéines ribosomiques), syndrome 5q- (gène de la protéine ribosomique
RPS14), syndrome de Shwachman-Diamond (gène SDBS), syndrome de Treacher-
Collins (gène TCOF1), dyskératose congenitale (gène Dyskérine), cartilage-hair
hypoplasia (gène RMRP). Ces résultats font apparaître un lien particulier et inattendu
entre la biogenèse des ribosomes, un mécanisme ubiquitaire, et le tissu
hématopoïétique. Cette relation est la plus évidente dans le cas de l'anémie de Diamond-
Blackfan, puisque plusieurs gènes de protéines ribosomiques sont mutés dans cette
pathologie.
21

A.1. L’Anémie de Diamond-Blackfan ou ADB
L'Anémie de Diamond-Blackfan ou ADB (Diamond-Blackfan Anemia), a été rapportée
pour la première fois en 1936 par H.W. Josephs (Josephs HW, 1936) et 2 ans plus tard
par L.K. Diamond et K.D. Blackfan (Diamond and Blackfan, 1938), d’où le nom de la
maladie. C'est une maladie hétérogène qui a pour principal signe clinique un arrêt de
l'érythropoïèse, même si les autres lignées hématopoïétiques sont habituellement
épargnées. Elle touche environ 7 enfants par million de naissances vivantes en France.
L’anémie qui s’installe progressivement en quelques semaines est rarement découverte
à la naissance ou dans les premiers jours de la vie. En effet, moins de 10% des cas sont
révélés à la naissance alors que 90% des cas le sont avant l’âge de 3 mois ;
l’érythropoïèse fœtale est le plus souvent préservée. Dans 10-20% des cas, l’un des
deux parents est atteint (Willig et al., 2000).
Un ensemble de signes cliniques et biologiques caractérisent la maladie. L’absence ou
la rareté des précurseurs érythroïdes (moins de 5 % des éléments nucléés) dans une
moelle normocellulaire est un argument majeur pour le diagnostic de l’ADB (Willig et
al., 1999b). Les malformations qui concernent 40% des cas sont très diverses dans leur
localisation et leur sévérité (Chen et al., 2005; Leblanc et al., 2003; Willig et al.,
1999b). Elles concernent surtout la face, les membres, la sphère urogénitale, le cœur et
la moelle épinière. La majorité des patients présente une petite stature. Au niveau
cellulaire et moléculaire, on note un taux bas ou une absence totale de réticulocytes, une
macrocytose des érythrocytes, une augmentation du taux d’hémoglobine F et de
l’adénosine déaminase érythrocytaire, eADA (Lipton et al., 2006). Par ailleurs, le taux
d’incidence des leucémies est élevé, notamment la leucémie myéloïde aiguë, et on
22

constate également une légère augmentation de l'incidence des tumeurs solides (Lipton
et al., 2001).
L’ADB résulterait d’un défaut intrinsèque des progéniteurs érythroïdes médullaires
(Lipton et al., 1986; Tsai et al., 1989). Les cultures cellulaires ont été d’un apport
majeur dans l’élucidation des mécanismes de la maladie. En effet, les colonies
érythroïdes obtenues in vitro en méthylcellulose à partir de cellules médullaires ou des
progéniteurs circulants de patients atteints d’ADB sont de petite taille comparés aux
témoins (Ohene-Abuakwa et al., 2005). Ces travaux ont permis la mise en évidence du
blocage partiel de la différenciation érythroïde en le situant in vitro après le stade de
progéniteurs érythroïdes immatures ou burst forming units (BFU-e) (Bagnara et al.,
1991). La capacité de prolifération des colony forming units (CFU-e), plus en aval dans
la différenciation, est diminuée (Halperin and Freedman, 1989; Lipton et al., 1986).
Récemment, une étude dans un système de culture a montré que le blocage de la
différenciation érythroïde au cours de l’ADB se situe entre la phase d’indépendance à
l’érythropoïétine (EPO) et la phase EPO-dépendante (Ohene-Abuakwa et al., 2005). Le
taux sérique de l’EPO est élevé dans l’ADB (Kalmanti et al., 1993).
Plusieurs données expérimentales et quelques données cliniques élargissent le défaut de
prolifération cellulaire à une lignée (granulo-macrophagique) (McGuckin et al., 1995;
Santucci et al . , 1999) voire deux autres l ignées (granulo-
macrophagique/mégacaryocytaire) (Giri et al., 2000), ce qui expliquerait certaines
évolutions de la maladie vers l’hypoplasie médullaire globale. En présence de G-CSF
(granulocyte-cell stimulating factor), les cellules CD34(+)CD38(-) de patients ADB ont
un défaut de clonogénicité (Hamaguchi et al., 2003) qui s’accentue avec l’âge des
patients et l’évolution de l’ADB (Casadevall et al., 1994; McGuckin et al., 1995).
23

Figure 2. Identification du premier gène candidat RPS19 sur le chromosome 19Des délétions chez trois patients ont permis de réduire la zone potentiellement impliquée dans l'ADB à 1 Mb sur le chromosome 19. La région de translocation a été ensuite clonée et tous les gènes s'y trouvant ont été par la suite séquencés, ce qui a permis d'identifier le gène qui code pour la protéine ribosomique Rps19 comme le premier gène associé à l'Anémie de Diamond-blackfan, ADB.
D'après Gustavsson et al. (1998), Am J Hum Genet. 63: 1388-1395
Figure 3. Mutations hétérogènes et mono-allélique du gène RPS19 chez les patients ADBLes 6 exons du gène RPS19 sont représentés avec pour chacun le numéro du nucléotide et l'acide aminé du début et de la fin. L'ATG est au début du deuxième exon et les différents types de mutations sont indiqués sur chacun des exons.
D'après, Willig et al., 1999a
24

A.2. Génétique de l'ADB
A.2.1. Mutation du gène RPS19
L’ADB est sporadique dans 75 % des cas (Willig et al., 1999b). Dans 10-20% des cas,
l’un des deux parents est atteint (Willig et al., 2000). Dans les formes familiales, la
transmission est le plus souvent dominante, même si des transmissions récessives ont pu
être évoquées. La découverte, en 1997, chez une patiente suédoise, d’un caryotype
anormal avec translocation équilibrée, 46, XX, t(X,19)(p21,q13) a permis d’incriminer
le chromosome 19 (Gustavsson et al., 1997a). Les analyses de liaison génétique dans les
familles multiplex identifièrent grâce à la transmission des marqueurs microsatellites
une zone de 1,8 Mb où se situait le gène d’intérêt (Gustavsson et al., 1997b).
Le premier gène impliqué dans l’ADB a été identifié après réduction de la zone
chromosomique à 1 Mb, grâce en particulier à la découverte de délétions dans des
formes sporadiques et le clonage de la translocation 19q13 (Figure 2). Il s’agit du gène
RPS19, codant pour une protéine ribosomique (Draptchinskaia et al., 1999). Ce gène de
6 exons dont le premier est non codant mesure 11 Kb chez H. sapiens (Draptchinskaia
et al., 1999). Le promoteur comprend deux régions intervenant dans l’activité
promotrice globale avec effet synergique (Da Costa et al., 2003). L’ARNm comporte
une ORF de 435 nucléotides et code pour une protéine de 145 acides aminés. La
présence d’une mutation du gène RPS19 a été détectée chez seulement 25 % des
patients (Willig et al., 1999a). Ce pourcentage reste stable quel que soit le pays
(Draptchinskaia et al., 1999; Ramenghi et al., 2000; Willig et al., 1999a). De même, il
existe une grande diversité de mutations dans RPS19 associées à l’ADB (Figure 3,
Tableau 1). Ainsi, on a répertorié des mutations dans la région promotrice, dans les
25
26
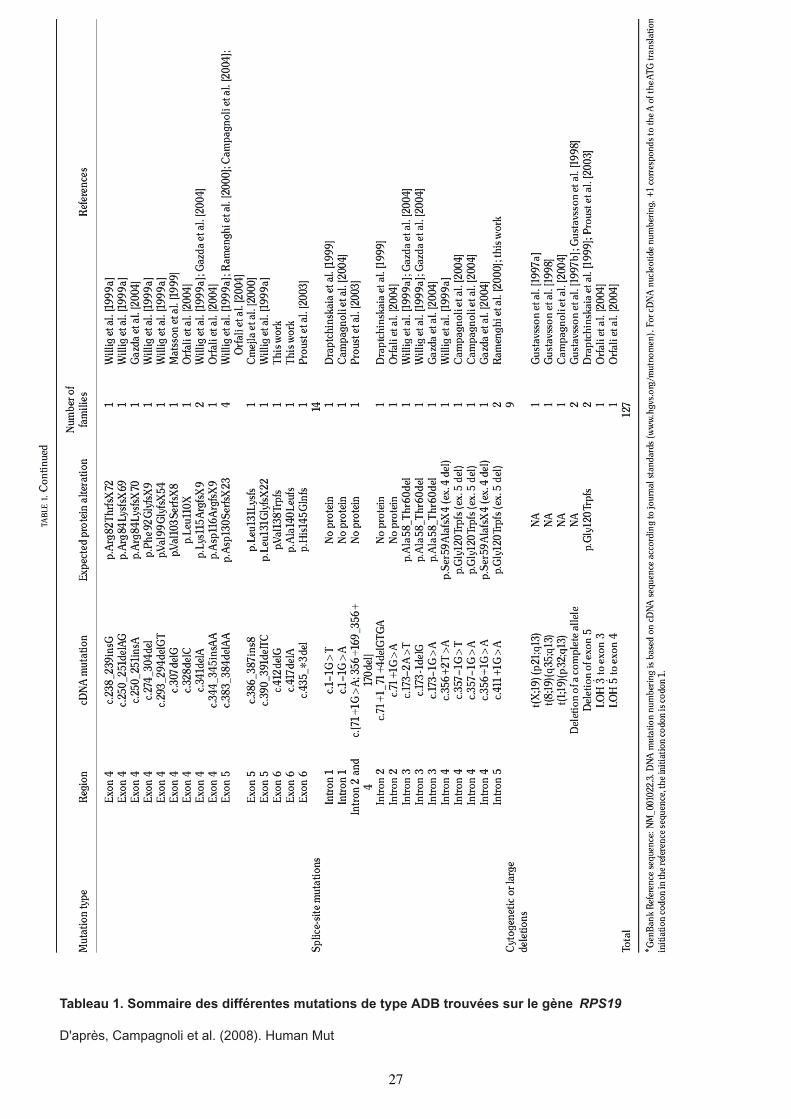
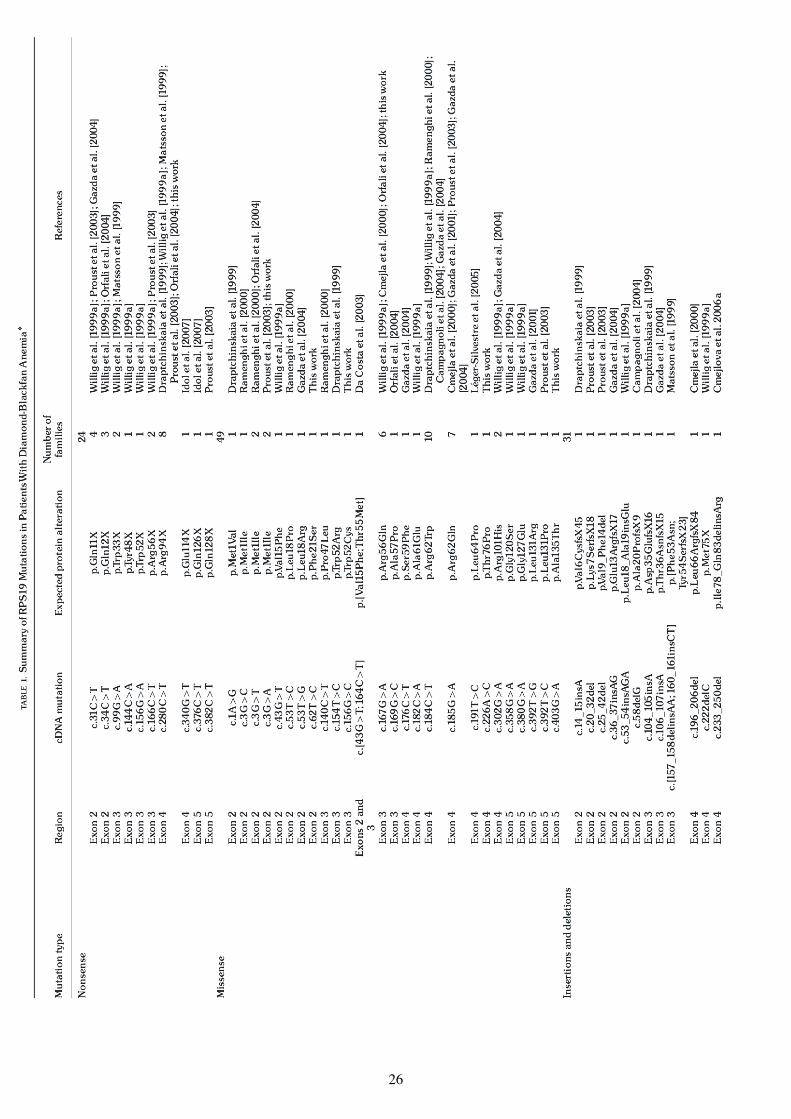
Tableau 1. Sommaire des différentes mutations de type ADB trouvées sur le gène RPS19
D'après, Campagnoli et al. (2008). Human Mut
27

séquences introniques, et sur l’ensemble de la séquence codante : mutations faux-sens,
mutations non sens, ainsi que des insertions et des délétions (Draptchinskaia et al.,
1999; Gazda et al., 2004; Matsson et al., 1999; Willig et al., 1999a). Une zone
préférentielle (hot spot) de mutations faux-sens a été distinguée dans la région codant
pour les acides aminés 52 à 62, sans que pour l’instant n’émerge une hypothèse
physiopathologique concernant ce site (Willig et al., 1999a).
Aucune corrélation entre le phénotype et le génotype n’a pu être clairement démontrée à
ce jour. Cependant, les délétions semblent être associées à des formes plus graves de
l’ADB chez certains patients (Ramenghi et al., 2000). Il a pu être démontré dans des
fibroblastes primaires de patients et dans les lignées lymphoblastoïdes immortalisées
que les mutations non sens de RPS19 responsables d’un stop prématuré ou d’une
abolition du codon stop entraînent une dégradation rapide de ces transcrits anormaux
par le système NMD (nonsense mediated decay and non stop decay) (Chatr-Aryamontri
et al., 2004). Ce système contrôle-qualité présent chez les eucaryotes, assure
l’élimination rapide des ARNm anormaux, qui seraient dommageables pour la
traduction protéique. L’implication de ce système renforce l’hypothèse d’un mécanisme
d’haploinsuffisance de la protéine RPS19 dans l’ADB : soit l’allèle normal serait seul
exprimé, soit le niveau d’expression des deux allèles serait variable selon la mise en jeu
du système NMD. Ce mécanisme pourrait rendre compte des variations évolutives si
fréquemment observées chez un individu au cours de la vie et de l’existence de
phénotypes silencieux définis par une élévation de l’eADA et/ou une mutation du gène
RPS19 sans qu’il y ait d’anémie. L’élévation de l’eADA est liée à la maladie (Willig et
al., 1998). Les familles dans lesquelles un des parents a une élévation d’eADA sont
désormais classées comme des familles à transmission dominante (Willig et al., 1998).
Il existerait un ou des effets modulateurs génétiques ou d’autre nature qui rendraient
28

l’ADB plus ou moins pénétrante. Cette notion de phénotype silencieux et le caractère
imprévisible de l’expression clinique, de la réponse thérapeutique et de l’évolution de la
maladie rendent le conseil génétique et le diagnostic anténatal plus compliqué. Il a
cependant été réalisé aux Etats-Unis des diagnostics pré-implantatoires, dans certains
cas d’ADB avec établissement de la compatibilité HLA (Verlinsky et al., 2004).
A.2.2. De nouvelles protéines ribosomiques impliquées dans l'ADB
Très récemment (à la fin de cette thèse), d'autres gènes de protéines ribosomiques ont
été liés à l'ADB. L'analyse cartographique pangénomique des SNP (Single Nucleotide
Polymorphism) sur 215 familles a révélé d'autres loci en lien avec l'ADB : une région
de 18 Mb sur le chromosome 8 (8p23.3-22) et des régions plus petites sur les
chromosomes 6 et 10. Dans le chromosome 10, le séquençage du gène de la protéine
ribosomique RPS24 a mis en évidence chez trois patients, deux mutations non-sens et
une délétion de la région codante qui ne sont pas trouvées dans deux cents génomes
contrôles (Gazda et al., 2006). Indépendamment, le séquençage systématique de
plusieurs gènes de protéines chez des patients a montré la présence de mutations
hétérozygotes dans les gènes RPS17 (Cmejla et al., 2007), RPL5, RPL11 (Farrar et al.,
2007; Gazda et al., 2007) et RPL35a (Farrar et al., 2007). Les mutations des gènes
RPS24, RPS17, et RPL35a touchent moins de 2% des patients, alors que RPL5 et
RPL11 sont affectés chez 10% et 6% des patients respectivement. Il est notable que ces
gènes codent aussi bien des protéines de la sous-unité 40S (gènes RPS) que des
protéines de la sous unité 60S (gènes RPL). Sans être aussi complètes que celles de
RPS19, ces études récentes élargissent le nombre de protéines ribosomiques liées à
l'ADB et confortent l'hypothèse d'un lien entre la biogenèse ou la fonction du ribosome
29

Figure 4. Vue protéomique de la voie d'assemblage du ribosomeLa particule pré-ribosomique 90S contient le pré-ARNr 35S et le SnoRNP U3. Les clivages succes-sifs aux sites A0, A1 et A2 aboutissent à la séparation des particules pré-40S et pré-60S. On peut distinguer des complexes de maturation précoce (E), intermédiaire (M) et tardif (L). Sur ce shéma, on distingue également des GTPases potentielles (vert), des ARN-hélicases potentielles (rouge), ainsi que des composants requis pour le transport (souligné).
D'après, Fatica et al. (2002). Curr Opin Cell Biol. 14: 313-318
30

et la pathogenèse de cette maladie. De plus, l'association très récente d'une
érythroblastopénie acquise, le syndrome 5q- , à la mutation du gène de la protéine
ribosomique RPS14 (Ebert et al., 2008) vient renforcer s'il en était besoin cette relation
physiologique particulière entre biogenèse des ribosomes et érythropoïèse .
A.3. La biogenèse du ribosome
Les deux sous-unités formant le ribosome sont constituées de 4 ARNr et de 79 protéines
ribosomiques. Leur biogenèse se fait au cours d’un processus très complexe qui fait
intervenir environ 200 facteurs en trans ( protéines non ribosomiques et snoRNPs).
L'étude la plus complète de la maturation du pré-ARN ribosomique (pré-ARNr) a été
réalisée chez la levure S. cerevisiae. De plus, des études récentes chez la levure
employant des techniques de purification par affinité combinées à la spectrométrie de
masse ont permis la caractérisation des particules pré-ribosomiques, conduisant à une
meilleure compréhension de la biogenèse des ribosomes (Fromont-Racine et al., 2003;
Granneman and Baserga, 2004; Johnson et al., 2002; Kressler et al., 1999; Nazar, 2004;
Tschochner and Hurt, 2003; Venema and Tollervey, 1999) (Figure 4). Bien que la
connaissance de la biogenèse des ribosomes progresse chez les mammifères, je
m'attacherai ici au modèle de la levure pour discuter la formation de la particule pré-40S
dans laquelle intervient RPS19.
A.3.1. Voie de maturation de l’ARNr
La formation de ribosomes fonctionnels commence par la synthèse nucléolaire des pré-
ARNr par les ARN polymérases I et III. Chez S. cerevisiae, le premier ARN
31

Figure 5. La voie de maturation de l’ARNr chez S. cerevisiaeA. Unité de transcription des gènes ribosomiques : le pré-ARNr 35S comprend les séquences des trois ARNr matures 18S, 5.8S et 25S et est synthétisé par l’ARN pol I alors que le gène de l’ARNr 5S est indépendamment synthétisé par l’ARN pol III dans le sens inverse du gène du pré-ARNr 35S.B. Etapes de maturation des pré-ARNr : Chez S. cerevisiae, le pré-ARNr 35S est clivé au site A0 pour aboutir au pré-ARNr 33S qui donnera le pré-ARNr 32S après clivage au site A1. Ce dernier est à son tour clivé au site A2 pour donner les pré-ARNr 20S et 27SA2. Le pré-ARNr 20S va être clivé pour donner l’ARNr 18S mature après son export vers le cytoplasme où il subit une diméthylation. Le prè-ARNr 27SA2 va être maturé via deux voies différentes pour donner les ARNr matures de 5.8S et 25S.
D'après, Fromont-Racine et al., 2003
32

intermédiaire détectable correspond à l’ARNr 35S transcrit à partir de séquences
d’ADNr. Ces gènes, présents à raison de 150 ou 200 copies sur le chromosome XII,
sont séparés par des séquences intergéniques non transcrites ou IGS (InterGenic
Sequences) de 2 à 30 kb. L'ARNr 35S contient les régions transcrites externes 5’ et 3’
ETS (External Transcribed Spacer) ainsi que les ARNr matures de 18S, 5,8S et 25S
séparés par les séquences transcrites internes ITS1 et ITS2 (Internal Transcribed
Spacer). Au microscope électronique, la transcription très active de ces gènes
ribosomiques structurés en tandem est visualisée sous la forme de structures appelées
" arbres de Miller" ou "de Noêl" visibles après étalement de chromatine (French et al.,
2003; Miller and Beatty, 1969). L’ARNr 18S est le composant ARN de la petite sous-
unité 40S tandis que les ARNr 5,8S, 25S et l’ARNr 5S constitueront le composant
ARNr de la grande sous-unité 60S (Figure 5, A). L’ARNr 5S étant indépendamment
synthétisé par l’ARN polymérase III.
Le pré-ARNr 35S est successivement clivé dans la région 5’ ETS au site A0 (générant le
pré-ARNr 33S), au site A1 à l'extrémité 5’ de l’ARNr mature 18S (générant le pré-ARNr
32S) et au site A2 dans l’ITS1 (générant les pré-ARNr 20S et 27SA2). Les étapes
ultérieures de maturation de l’ARNr 20S se produisent dans le cytoplasme par le clivage
au site D pour produire de l’ARNr mature 18S tandis que la maturation des pré-ARNr
27SA2 en ARNr 5,8S et 25S s’effectue dans le noyau par deux voies alternatives :
environ 85% de la population 27SA2 est clivé au site A3 dans l’ITS1 par la RNase MRP,
rapidement suivie d’une maturation exonucléolytique 5’-3’ au site B1S par Rat1p; 15%
de la population 27SA2 est directement clivé au site B1L. Le clivage au site B2, à
l’extrémité 3’ du 25S, se produit simultanément avec le clivage au site B1. Les deux
formes de 27SB (27SBS et 27SBL) sont maturées suivant des voies identiques
impliquant le clivage endonucléolytique de l'ITS2 au point C2 et la maturation
33

Figure 6. Maturation des sous-unités ribosomiques chez S. cerevisiae.La particule précoce 90S à laquelle appartient le snoRNP U3 contenant le processome est clivée au site A2 pour donner les sous-unités 40S et 60S. Le processome se dissocie des particules prè-40S en maturation. La plupart des facteurs s’associent au prè-60S pendant ou après le clivage au site A2. Pendant le passage vers le pore nucléaire, plusieurs facteurs se dissocient du pré-60S à des étapes précises, alors qu’un certain nombre de protéines s’associent de manière transitoire pendant les étapes tardives. Pour acquérir la compétence d’export, un facteur (en rouge), se lie aux sous-unités. La maturation finale a lieu après le passage à travers le pore nucléaire.
D'après, Tschochner and Hurt, 2003
34

exonucléolytique aux sites C2 et E. La digestion exonucléolytique 3’-5’ au site E, dans
la partie 3' de l'ARNr 5.8S est assurée par l'exosome (Figure 5, B).
Chez des mutants, le clivage prématuré du pré-ARNr 35S au site A3 produit un
intermédiaire aberrant, l'ARNr 23S. L'ARNr 22S est accumulé quand les clivages se
produisent aux sites A0 et A3 en l'absence de clivage aux sites A1 et A2 (Venema and
Tollervey, 1999). En absence du clivage A2, les particules 40S ne sont plus produites
tandis que les particules 60S peuvent être constituées par des clivages alternatifs, par
exemple au site A3 (Venema and Tollervey, 1999). En revanche, les mutations inhibant
la maturation aux sites A0 et A2 ont peu d'impact sur la synthèse des ARNr 5.8S et 25S
tandis que l'inverse n'est pas vrai. La mutation de la plupart des facteurs requis pour la
synthèse du 60S retarde les clivages précoces du site A0 au site A2 avec une production
concomitante d’ARNr 23S aberrant.
De manière concomitante avec la transcription des ARNr et les étapes précoces de
clivage, des particules ribonucléoprotéiques nucléolaires (snoRNP) guident et
catalysent la méthylation et la pseudouridylation des pré-ARNr (Granneman and
Baserga, 2004).
A.3.2. Maturation des particules pré-40S
Après le clivage du pré-ribosome 90S, la petite sous-unité de 40S a une composition
moins complexe que celle de la grande sous-unité de 60S. Les particules pré-40S sont
majoritairement cytoplasmiques : après clivage au point A2, elles sont rapidement
exportées du noyau vers le cytoplasme, où a lieu la maturation finale. Les facteurs
impliqués dans les clivages A0, A1 et A2 s’associent au pré-ARNr 35S et précèdent
l'assemblage des facteurs du pré-60S. La structure formée par ces facteurs précoces sur
35

les ARN naissant a été appelée "processome" (Dragon et al., 2002). Néanmoins, la
composition protéique de cette structure correspond à celle de la particule 90S
(Bernstein et al., 2004; Dragon et al., 2002; Grandi et al., 2002). La plupart des facteurs
associés au pré-ARNr 35S, à quelques exceptions près, n'accompagnent pas les
particules 40S naissantes dans le cytoplasme (Figure 6). Mis à part le pré-ARNr 20S et
les protéines ribosomiques, on compte peu de facteurs non ribosomiques dans cette
particule: Rio2p, Enp1p, Tsr1p, Dim1p, Dim2p, Nob1p, Rrp12p, Hrr25p, Nop14p,
Asc1p, Bud23p et Ltv1p (Schafer et al., 2003). Parmi ces facteurs certains sont déjà
présents dans la particule 90S : Enp1p, Rrp12p, Nop14p. A l'heure actuelle, la fonction
exacte de ces facteurs reste à découvrir.
On connaît mal le clivage au site D qui convertit le pré-ARNr 20S en ARNr 18S
mature. Ce clivage serait probablement effectué par une endonucléase qui pourrait être
la protéine Nob1p (Fatica et al., 2003; Fatica et al., 2004) ou la protéine Fab7p
(Granneman et al., 2005). Récemment, quelques facteurs affectant spécifiquement cette
réaction ont été trouvés. Ainsi, RIO1/RRP10 a été isolé dans un crible de létalité
synthétique avec un allèle mutant de GAR1, un gène requis pour la production de
l’ARNr 18S et la pseudouridylation d’ARNr (Vanrobays et al., 2001). Rio2p, identifié
sur la base de son homologie avec Rio1p forme avec ce dernier une nouvelle famille de
protéines sérine kinases. L'activité kinase a été démontrée in vitro pour Rio1p qui
phosphoryle plusieurs substrats dont Rio1p lui-même (Angermayr and Bandlow, 2002).
D'ailleurs, des mutations au sein des résidus d'acides aminés conservés, prédits pour
former le site actif, suppriment aussi bien l'activité kinase que la fonction de la protéine
in vivo (Angermayr and Bandlow, 2002). On ne connaît pas encore les cibles de la
phosphorylation de Rio1p, même si Rps31p ou Rps6p, deux protéines ribosomiques de
36

la particule 40S du ribosome, connues pour être phosphorylées chez la levure sont
citées pour être les candidats potentiels (Ficarro et al., 2002).
Rio1p et Rio2p sont trouvées dans le cytoplasme en association avec des complexes
ayant un coefficient de sédimentation d’environ 40S contenant le pré-ARNr 20S
(Vanrobays et al., 2003; Vanrobays et al., 2001). En absence d’une de ces protéines, les
cellules accumulent le pré-ARNr 20S dans le cytoplasme montrant que la maturation du
20S n'est pas requise pour l'export des particules pré-40S. Des expériences de
localisation utilisant le mutant thermosensible crm1-1 dans des conditions non
permissives suggèrent que Rio2p et Rio1p font la navette entre le noyau et le
cytoplasme bien que leur localisation à l’état d'équilibre soit principalement
cytoplasmique (Vanrobays et al., 2003). Ces résultats suggèrent que les deux protéines
se lient à la particule pré-40S nucléaire et accompagnent la particule dans le cytoplasme.
Si Rio2p a été repérée dans des particules pré-40S purifiées, Rio1p en revanche n'a pas
été identifiée dans ces purifications bien que l'analyse des ribosomes sur gradient de
sucrose indique son association avec les particules 40S (Vanrobays et al., 2001).
Les protéines Rio ne sont pas les seuls facteurs associés à la particule pré-40S qui soient
nécessaires à la maturation de l’ARNr 20S. La perte de fonction de la protéine Tsr1p
aboutit également à l'accumulation du pré-ARNr 20S (Gelperin et al., 2001; Leger-
Silvestre et al., 2004). Bien que Tsr1p ait été identifiée dans quelques particules 90S, les
complexes purifiés en étiquetant Tsr1p contiennent la plupart des composants des
particules pré-40S (Gavin et al., 2002). C'est également le cas pour Ltv1p (Schafer et
al., 2006; Schafer et al., 2003). Une souche portant une mutation cryo-sensible de Ltv1p
présente une accumulation spécifique du pré-ARNr 20S (Seiser et al., 2006). Il a été
proposé que Ltv1p et Tsr1p participent au transport de la particule pré-40S (Schafer et
al., 2003; Seiser et al., 2006). Cependant, Ltv1p n'est pas une protéine essentielle et ne
37

peut pas jouer un rôle majeur dans ce processus. Par ailleurs, les résultats de notre
laboratoire ont montré que les particules pré-40S s'accumulent dans le cytoplasme en
absence de Tsr1p (Leger-Silvestre et al., 2004), en contradiction avec les résultats
précédemment présentés.
On dispose de peu d'informations sur la dynamique des facteurs pré-ribosomiques. A
l'équilibre, Tsr1p est majoritairement cytoplasmique, en accord avec son association
avec des particules pré-40S tardives (Leger-Silvestre et al., 2004). Néanmoins, en cas
d'interruption de la maturation ou du transport des particules pré-40S, Tsr1p s'accumule
dans le noyau ; ce qui suggère qu'elle fait la navette entre le cytoplasme et le noyau. En
revanche, Rio2p reste localisée dans le cytoplasme (Leger-Silvestre et al., 2004). Rio2p
possède des séquences d'export nucléaire susceptibles de médier son exclusion du noyau
par Crm1p (discuté par Schäffer et al., 2003). Certains facteurs de la particule pré-40S
viennent précocement s’associer avec les particules 90S, c’est le cas de Enp1p. De fait,
la perte de fonction de Enp1p entraine un défaut de clivage au site A2, ce qui provoque
l'accumulation de pré-ARNr 21S (Chen et al., 2003) dans le nucléole (Léger-Silvestre,
2004).
Il est possible que la maturation du pré-ARNr 20S en ARNr 18S soit liée à l'initiation de
la traduction. Ainsi, la délétion du gène codant la protéine Hcr1p, une protéine
cytoplasmique ayant des liens génétique et physique avec eIF3 et le complexe
d'initiation de la traduction, entraine un retard de maturation du pré-ARNr 20S (Valasek
et al., 2001). Par ailleurs, l'hélicase Ded1p, requise pour l'initiation de la traduction de
tous les ARNm chez la levure, est associée aux particules pré-40S (Schafer et al., 2003).
Cependant, il n'existe pas pour l'instant d'argument direct supportant un couplage entre
maturation du pré-ARNr 20S et l'initiation de la traduction.
38

A.3.3. Rôle des protéines ribosomiques dans la maturation des particules
pré-40S
Les protéines ribosomiques sont recrutées à différents moments de la maturation.
Certaines s'associent avec le pré-ARNr naissant au cours de la transcription alors que
d'autres sont incorporées très tardivement dans le processus. Au cours de la maturation,
les facteurs non ribosomiques doivent être recrutés à des sites spécifiques de la surface
de la particule ribosomique. Les protéines ribosomiques et l’ARNr, doivent jouer un
rôle important dans ce processus de recrutement soit en servant de surface d'atterrissage
à ces facteurs, soit en formant des sous-complexes avec ces facteurs qui sont alors
recrutés sur la particule en cours de maturation. En outre, les protéines ribosomiques
peuvent jouer un rôle actif dans des étapes distinctes de la biogenèse de la particule 40S
du ribosome. L'ordre du recrutement des protéines ribosomiques et leur(s) fonction(s)
dans les pré-ribosomes chez les eucaryotes sont encore mal connus, mais des données
récentes ont apporté des éclaircissements sur ce sujet.
En effet, la perte de fonction de certaines protéines ribosomiques affecte le clivage du
pré-ARNr 20S : c’est le cas de Rps0p, Rps14p et Rps21p (Ford et al., 1999; Jakovljevic
et al., 2004; Tabb-Massey et al., 2003). Une étude en cryomicroscopie électronique a
permis de localiser la partie C-terminale de Rps14p à proximité de l’extrémité 3’ de
l’ARNr 18S (Spahn et al., 2001). Rps0p se trouve également au voisinage de ce
domaine. La délétion de la queue C-terminale de Rps14p conduit à l'accumulation
cytoplasmique du pré-ARNr 20S, soutenant un rôle de ce domaine dans la maturation
(Jakovljevic et al., 2004). Bien que la séquence primaire de Rps14p ne montre pas
d'homologie avec des domaines nucléases connus, il se peut qu’elle soit requise pour
induire une conformation du pré-ARNr 20S, ce qui permet le clivage, ou le recrutement
de l’enzyme de maturation du pré-ARNr 20S. En cohérence avec cette dernière
39

hypothèse, on a observé une interaction directe entre Rps14p et Fab7p, une des
endonucléases potentielles pour le clivage du pré-ARNr 20S (Granneman et al., 2005).
On peut noter par ailleurs que l'interruption de l'expression de Rps14p aboutit à un
défaut de maturation de l'ETS1 (Ferreira-Cerca et al., 2005). Cette protéine est donc
nécessaire à différents niveaux dans le processus de maturation.
Certaines protéines ribosomiques sont nécessaires pour des étapes plus en amont de la
biogenèse de la particule 40S; c’est le cas de Rps19p (Leger-Silvestre et al., 2005),
Rps18p et Rps15p (Leger-Silvestre et al., 2004). La perte d'expression de Rps15p
aboutit à l'accumulation de particules pré-40S correctement assemblées dans le
nucléoplasme, ce qui suggère fortement un rôle de cette protéine dans l'export nucléaire
de ces RNP. En absence de Rps18p et Rps19p, les levures présentent un défaut complet
de clivage au point A2, ce qui indique un rôle de ces protéines en amont de l'export
(Leger-Silvestre et al., 2005). Les particules pré-40S aberrantes contenant du pré-ARNr
21S, formées en l'absence de Rps19p, ne recrutent pas correctement les facteurs pré-
ribosomiques Enp1p, Tsr1p ou Rio2p (voir plus loin).
Récemment, une étude systématique du rôle des protéines ribosomiques RPS dans la
biogenèse de la sous-unité 40S a permis leur classification en plusieurs groupes avec
des rôles distincts dans la maturation des particules 40S (Ferreira-Cerca et al., 2005).
Des souches dans lesquelles l’une ou l’autre des protéines ribosomiques peuvent être
exprimées sous contrôle d'un promoteur GAL ont été construites et utilisées pour étudier
la perte de fonction de chacune d'entre elles. Certaines protéines, Rps1p, Rps6p, Rps8p,
Rps11p, Rps13p, Rps14p, Rps16p, Rps23p, Rps24p, Rps27p sont nécessaires pour la
maturation de l'ETS1. D'autres au contraire sont impliquées dans des étapes plus
tardives. Ainsi, Rps0p, Rps2p, Rps3p, Rps10p, Rps15p et Rps26p semblent nécessaires
pour l'export nucléaire et/ou la maturation cytoplasmique des particules pré-40S.
40

L'implication de plusieurs protéines ribosomiques dans la maturation du pré-ARNr 20S
pourrait entrer en jeu dans le contrôle qualité de la biogenèse de la sous-unité 40S.
Suivant cette hypothèse, le clivage du pré-ARNr 20S se produirait seulement si toutes
les étapes de maturation antérieures se sont réalisées. Les protéines ribosomiques
pourraient servir de marqueurs dans ce processus. Enfin, d'après cette étude, deux
protéines ribosomiques, Rps12p et Rps25p, ne sont pas essentielles pour la viabilité
cellulaire.
A.4. La protéine RPS19
A.4.1. Fonction de RPS19 dans la biogenèse des ribosomes
Dans notre équipe, Rps19p (homologue de RPS19 chez S. cerevisiae) a été identifiée au
départ dans un crible génétique comme une protéine candidate pour jouer un rôle dans
l’export des particules pré-ribosomiques. Du fait de son implication dans une maladie
génétique rare et de son rôle potentiel dans la maturation de la particule pré-40S,
l'analyse fonctionnelle de la protéine a été entreprise en construisant une souche dans
laquelle la protéine est exprimée de manière conditionnelle chez S. cerevisiae sous
contrôle d'un promoteur GAL (Leger-Silvestre et al., 2005). La protéine Rps19p est
essentielle à la viabilité cellulaire. L'arrêt de sa synthèse entraine un défaut massif de
production de la petite sous-unité ribosomique. De fait, la perte de Rps19p bloque le
clivage du précurseur de l’ARNr 18S au site A2 : les pré-ARNr 20S et 27S-A2 ne sont
plus produits. Le clivage A3 permet néanmoins de cliver l'ITS1 et la production de sous-
unité 60S n'est pas significativement altérée. De plus, le pré-ARNr 21S n'est pas maturé
en ARNr 18S : les particules pré-ribosomiques aberrantes contenant le pré-ARNr 21S
41

s’accumulent dans le noyau cellulaire et sont dégradées. La perte de fonction de Rps19
affecte donc spécifiquement la maturation de la petite sous-unité ribosomique.
Le défaut de clivage en A2 s'accompagne d'un défaut de recrutement de facteurs
spécifiques des particules pré-40S. Ceci a été montré non seulement pour des facteurs
tardifs (Rio2p, Tsr1p), mais également pour Enp1p qui s'assemble à la particule 90S. De
fait, la perte de fonction de Enp1p dans le mutant thermosensible enp1-1 entraine
également un défaut de clivage en A2 (Chen et al., 2003), ce qui suggère que le défaut
de recrutement de Enp1p pourrait participer au phénotype observé en absence de
Rps19p. Par ailleurs, on peut noter que la protéine Rps18p est la seule protéine de la
sous-unité 40S dont la perte de fonction conduit à un phénotype similaire à celui
observé en absence de Rps19p. Ces deux protéines sont localisées sur la tête de la sous-
unité 40S.
La fonction de Rps19p a également été abordée dans des cellules humaines HeLa en
utilisant des siRNA (Choesmel et al., 2007) ou dans la lignée érythroleucémique TF-1
(Flygare et al., 2007). L'inhibition de la synthèse de Rps19p conduit à un retard du
clivage de l'ITS1 et à l'accumulation du pré-ARNr 21S, l'équivalent du pré-ARNr 20S
chez la levure. Les particules pré-40S contenant le pré-ARNr 21S sont dégradées dans
le noyau. Ce phénotype (clivage anormal du 21S, défaut de maturation en ARNr 18S)
rappelle fortement celui observé chez la levure. Il est possible que le clivage dans l'ITS1
ait lieu à un site alternatif, mais le manque de connaissance des sites de clivage chez les
vertébrés n'a pas permis de répondre à cette question pour l'instant.
A.4.2. Partenaires protéiques de la protéine RPS19
Le rôle de RPS19 en dehors de son implication dans la biogenèse des ribosomes reste à
définir notamment en ce qui concerne l’érythropoïèse. Cette protéine ribosomique
42

appartient à la petite sous-unité 40S du ribosome. Outre son rôle dans l’ADB, il a été
montré que l’ARNm de RPS19 est surexprimé dans les carcinomes de cerveau
(Scheurle et al., 2000), de colon (Kondoh et al., 1992) et de léiomyome utérin (myome)
(Li et al., 2002). Inversement, il est sous-exprimé dans les carcinomes pancréatiques
(Andersson et al., 1999) et les lignées de cellules humaines HNSCC (Sengpiel et al.,
2004). Des expériences d’interaction protéique (cross-linking) ou d’inhibition de liaison
montrent que RPS19, au niveau du ribosome, lie le facteur d’initiation de la traduction
eIF-2 (Bommer et al., 1988). RPS19 semble être impliquée directement dans les étapes
initiales de la synthèse protéique. De plus, eIF-2 subit une régulation qui est spécifique
de la lignée érythroïde via l’heme regulated inhibitor (HRI) qui phosphoryle la sérine
51 de la sous-unité α de l’eIF-2 (Zhan et al., 2002). La fonction non-ribosomique la plus
documentée est l’implication des dimères de RPS19 dans le chimiotactisme des
monocytes dans des liquides articulaires de patients atteints de polyarthrite rhumatoïde
(Shrestha et al., 1999).
Les partenaires protéiques de RPS19 connus à ce jour se résument au facteur de
croissance fibroblastique-2 (FGF-2) qui a un effet sur la croissance, la différenciation et
la migration cellulaire (Soulet et al., 2001) et plus récemment au proto-oncogène Pim-1,
une sérine thréonine kinase qui phosphoryle et régule SOCS-1, un inhibiteur de
l’activation de STAT5 (Chiocchetti et al., 2005), qui est aussi impliqué dans la
transformation cellulaire et surexprimé dans de nombreux cancers (Bachmann and
Moroy, 2005). Pim-1 phosphoryle RPS19 in vitro et in vivo et s’associe aux ribosomes,
montrant que RPS19 est capable de relier les voies de transduction du signal avec la
machinerie traductionnelle (Chiocchetti et al., 2005). Cette dernière interaction
démontrerait pour la première fois un lien entre RPS19 et des voies de signalisation
intervenant directement dans l’érythropoïèse.
43

A.5. Modèles de l’ADB fondés sur la perte de fonction de RPS19
Les modèles animaux n’ont pas été, pour le moment, d’une grande aide dans la
compréhension de la maladie et du rôle de RPS19. Certaines mutations des protéines
ribosomiques chez Drosophila Melanogaster sont responsables d’un syndrome «
Minute » (Lambertsson, 1998) défini par diverses malformations : petite taille, retard du
développement larvaire, infertilité, et diverses anomalies morphologiques concernant
l’abdomen, la cuticule et les antennes. Le syndrome « Minute » est de transmission
dominante et l’homozygotie est létale. La sévérité est inversement proportionnelle à la
quantité en ARNm d’où la notion d’haploinsuffisance. Il n’existe pas de syndrome «
Minute » naturel de RPS19, mais on a pu produire un modèle de drosophile déficiente
en RPS19 en introduisant un élément P dans la région promotrice du gène qui est situé
sur le chromosome X. Il ne s’agit pas d’un véritable syndrome « Minute » mais d’un
syndrome apparenté car la perte des deux allèles chez la femelle ou d’un seul chez le
mâle est nécessaire à l’expression d’un phénotype (Dr Jason Fixler, Maria Daniella
Arturi Foundation, 4th international consensus conference, New York, 2003).
En dehors du modèle de la drosophile, l’inactivation génique de rps19 par
recombinaison homologue chez la souris n’a abouti à aucun phénotype à l’état
hétérozygote et l’homozygotie est létale à un stade très précoce préimplantatoire
(Matsson et al., 2004). L’absence de modèles animaux pertinents a conduit à
l’utilisation d’autres outils moléculaires pour reproduire en partie le phénotype de
l’ADB et notamment les petits ARN à interférence (siRNA) (Ebert et al., 2005; Flygare
et al., 2005). Après infection de cellules CD34+ médullaires isolées, le siRNA
spécifique de l’ARNm du gène RPS19 diminue in vitro la prolifération et la
44

différenciation érythroïde (Ebert et al., 2005; Flygare et al., 2005). Cette anomalie est
bien spécifique de l’inhibition de l’ARNm de rps19 par le siRNA puisqu’un lentivirus
porteur du transcrit du gène RPS19 sauvage conduit à la restauration d’un niveau
d’expression normal de RPS19 et d’une prolifération et d’une différenciation normales
des précurseurs érythroïdes (Flygare et al., 2005). Cette étude a permis de reproduire in
vitro le phénotype cellulaire de l’ADB et de relier ainsi directement l’atteinte de
l’érythropoïèse à la diminution du niveau d’expression de RPS19.
A.6. Altération de la biogenèse des ribosomes chez les patients
En accord avec les données obtenues in vitro avec des siRNA, des cellules de patients
souffrant d'ADB présentent un défaut de maturation du pré-ARNr très comparable à
l'effet des siRNA, même si l'intensité en est diminuée. Ainsi, des précurseurs
hématopoiétiques, des fibroblastes de peau, ou encore des lignées lymphoblastoïdes
établies à partir de patients portant des mutations dans RPS19 présentent un défaut de
conversion du pré-ARN ribosomique 21S en ARN 18S-E (Choesmel et al., 2007;
Flygare et al., 2007; Idol et al., 2007). Dans les fibroblastes de peau, ce retard de
maturation observé par northern-blot et pulse-chase s'accompagne d'une désorganisation
de l'ultrastructure des nucléoles (Choesmel et al., 2007). De la même manière, des
lignées lymphoblastoïdes issues de patients affectés dans RPS24 montrent un taux élevé
de l’ARNr 30S, en accord avec le phénotype observé après inhibition de l'expression de
RPS24 dans des cellules en culture (Choesmel et al., 2008). La présence de ce retard de
maturation dans des fibroblastes de peau comme dans des cellules de la moëlle osseuse
est en accord avec la nature ubiquitaire de la biogenèse des ribosomes. De fait, les
45

malformations développementales dont souffrent certains patients atteints d'ADB
indiquent que l'effet pathogène des mutations ne se limite pas à l'hématopoïèse.
A.7. Les érythrocytes victimes d'un stress ribosomique?
La notion de « stress ribosomique », un stress lié à un défaut de la biogenèse des
ribosomes, a été largement documentée. La perturbation de la biogenèse des ribosomes
provoque un arrêt du cycle cellulaire à la transition G1/S. Ainsi, dans des cellules en
culture, ce phénomène a été observé en inhibant la transcription des gènes ribosomiques
par l'ARN polymerase I avec de l'actinomycine D (Dai and Lu, 2004; Dai et al., 2004;
Lohrum et al., 2003) ou en surexprimant des mutants dominants-négatifs de protéines
impliquées dans la maturation de la sous-unité 60S (Pestov et al., 2001). Cette voie de
stress a été également observée en traitant les cellules au 5-fluorouracile (Sun et al.,
2007) ou en surexprimant la protéine nucléolaire nucléostémine (Dai et al., 2008). In
vivo, la délétion conditionnelle du gène RPS6 dans des lymphocytes entraine une forte
augmentation de l'apoptose des cellules au cours de leur activation dans les organes
lymphoïdes suivant un mécanisme dépendant de p53 (Sulic et al., 2005). D'autres
exemples in vivo montrent que l'altération de la biogenèse des ribosomes se traduit par
des défauts de contrôle du cycle cellulaire (Anderson et al., 2007; Azuma et al., 2006).
Les mécanismes de réponse à un stress nucléolaire sont encore peu connus, mais deux
pistes ont été plus particulièrement explorées. La première nous amène du côté de la
protéine suppresseur de tumeur p53 dont l'activité bloque la transition G1/S. Dans les
conditions normales, p53 est présente dans les cellules à un niveau à peine détectable.
Après exposition des cellules à diverses formes de stress exogènes (tels que des
dommages d'ADN suite à un choc thermique) p53 est stabilisée et active la transcription
46

de la protéine p21, un inhibiteur des cyclines D1-CDK4 et E1-CDK2, entraînant l'arrêt
du cycle cellulaire en phase G1. La stabilité de la protéine p53 chez les mammifères est
principalement régulée dans les cellules non-transformées par l'effet de deux protéines,
HDM2 et p14ARF chez l'homme, MDM2 et p19ARF chez la souris. HDM2 fonctionne
comme une ligase d'ubiquitine E3 pour p53, ce qui entraine la dégradation de p53 par le
protéasome. Divers stimuli, incluant les voies de stress et les signaux oncogènes,
augmentent l'expression de p14ARF, qui s'associe alors à HDM2 pour inhiber
l'ubiquitination, l'export nucléaire et la dégradation de p53. Il a été proposé que p14ARF
séquestre physiquement HDM2 dans les nucléoles, stabilisant de ce fait p53 dans le
noyau (Visintin and Amon, 2000; Wsierska-Gadek and Horky, 2003). Plusieurs études
ont montré que des protéines ribosomiques de la grande et de la petite sous-unité (L5,
L11, L23, S7) peuvent également activer la protéine p53 en se liant à la protéine HDM2
(Bhat et al., 2004; Dai and Lu, 2004; Dai et al., 2006; Dai et al., 2004; He and Sun,
2007; Marechal et al., 1994; Sun et al., 2007). Comme pour ARF, la liaison de protéines
ribosomiques à HDM2 inhibe son interaction avec sa cible, ce qui aboutit à la
stabilisation de p53 et à l'arrêt du cycle cellulaire à la transition G1/S. Ainsi, un défaut
de synthèse ou de maturation des ribosomes pourrait conduire à l'augmentation de la
quantité de ces protéines ribosomiques libres dans le noyau et à la séquestration de
HDM2.
La deuxième piste concerne la protéine ARF (p19ARF chez la souris, p14ARF chez
l'homme), un autre inhibiteur de la progression en G1. Bien que ARF puisse activer p53
en séquestrant HDM2, cette protéine est également capable de bloquer la progression en
G1 dans des cellules n'exprimant pas p53 (Carnero et al., 2000). Par ailleurs, ARF
intervient dans la biogenèse des ribosomes à deux étapes distinctes : la transcription par
l'ARN polymérase I et la maturation du pré-ARNr (Ayrault et al., 2006). Une
47

interaction entre ARF et la topoisomérase I a été observée au niveau du promoteur des
gènes ribosomiques (Ayrault et al., 2004). ARF peut également interagir avec le facteur
de transcription UBF. Par ailleurs, ARF est associée avec la protéine B23, une
endonucléase requise pour la maturation de la grande sous-unité (Bertwistle et al., 2004;
Sugimoto et al., 2003). De fait, ARF est retrouvée dans les particules pré-60S. Il a été
proposé que la surexpression de p14ARF dans des cellules d'ostéosarcome U2OS
entraîne la dégradation de B23 et un délai dans la maturation du pré-ARN ribosomique
(Itahana et al., 2003). Néanmoins, ces résultats sont débattus, d'autres équipes n'ayant
pas observé de dégradation de B23 après expression d’ARF (Rizos et al., 2006). Enfin,
une troisième piste a été récemment ouverte en montrant que la protéine ribosomique
RPL11 inhibe directement l'activité du facteur de transcription c-Myc (Dai et al., 2007).
La transcription des protéines ribosomiques étant activée par c-Myc, ces auteurs
envisagent que RPL11 agit sur c-Myc dans le cadre d'une boucle de régulation négative
impliquée dans la régulation de la prolifération cellulaire.
A.8. Le nucléole multi-fonctionnel
Plus généralement, le nucléole apparaît aujourd'hui comme un domaine central dans la
prolifération cellulaire, au-delà de sa fonction dans la biogenèse des ribosomes. Cet
élargissement fonctionnel du nucléole (certains auteurs parlent de "multifunctional
nucleolus") est fondé entre autres sur la détection dans le domaine de protéines n'ayant
pas de lien direct avec la biogenèse des ribosomes (Andersen et al., 2002; Boisvert et
al., 2007; Coute et al., 2006; Pederson, 1998; Pendle et al., 2005; Scherl et al., 2002).
Ainsi, l'analyse de la composition de nucléoles isolés par analyse protéomique a montré
qu'il contient des protéines régulatrices du cycle et de la différenciation cellulaire. Par
48

ailleurs, le nucléole est également impliqué dans des événements de contrôle et de
régulation du cycle cellulaire (Dez and Tollervey, 2004; Visintin and Amon, 2000). De
plus, des facteurs pré-ribosomiques jouent un rôle direct dans la réplication (Du and
Stillman, 2002; Zhang et al., 2002), de même que plusieurs protéines ribosomiques
entrent en jeu dans des voies de signalisation (Wan et al., 2007). Ces données
connectent la biogenèse des ribosomes aux autres mécanismes de l'expression génique
et à la régulation du destin cellulaire. Bien que les mécanismes sous-jacents à ce
concept soient encore flous, le nucléole apparait comme un domaine senseur de stress
dans lequel sont concentrées des protéines permettant de répondre à des agressions ou
des dérèglements (Olson, 2004). Ainsi l'activation de p53 en réponse à l'exposition aux
UV requiert que le nucléole ait été irradié (Rubbi and Milner, 2003).
Ces données indiquent qu’il existe une coordination étroite entre la régulation de la
croissance cellulaire (éventuellement par p53) et les réponses cellulaires aux
dysfonctionnements de la biogénèse des ribosomes. On peut dès lors envisager qu'un
défaut partiel chronique de la biogenèse des ribosomes, consécutif à la mutation d'un
alléle d'un gène de protéine ribosomique, aboutisse à une pathologie. La question de la
spécificité du tissu lésé reste entière : pourquoi la lignée érythrocytaire dans le cas de
l'ADB? Par ailleurs, explorer l'hypothèse d'une réponse à un stress ribosomique dans le
cas de l'ADB demande entre autre que l'on connaisse l'impact des mutations sur la
fonction de la protéine mutée et sur la biogenèse des ribosomes. C'est en partie l'objet de
cette thèse dans le cas des mutations de RPS19.
49

Figure 7. Le complexe de pore nucléaire et des domaines FGChez les eucaryotes, le CPN correspond à une perforation de l’enveloppe nucléaire et sert de conduit acqueux connectant le nucléoplasme au cytoplasme des cellules. Les motifs FG séparés par des domaines non structurés correspondent aux répétitions Phe-Gly des nucléoporines et forment des filaments flexibles à travers la structure du CPN.
D'après, Patel et al. (2007). Cell. 129: 83-96
50

B. MECANISMES MOLECULAIRES DE L’IMPORT NUCLEAIRE DES
PROTEINES
La séparation spatiale de la transcription et de la traduction a fourni aux eucaryotes des
mécanismes très efficaces de contrôle de l'expression génique, ce qui rend nécessaire un
transport sélectif entre les compartiments nucléaire et cytoplasmique pour maintenir la
composition distincte de chaque compartiment. En conséquence, l'enveloppe nucléaire
constitue une barrière cruciale à travers laquelle des protéines et les ARN doivent être
transportés de façon régulée. En plus du transfert massif des protéines nucléaires
constitutives après leur synthèse dans le cytoplasme, les changements de l'expression
génique requièrent généralement une entrée contrôlée de molécules régulatrices comme
les facteurs de transcription. Le transport des macromolécules à travers l'enveloppe
nucléaire par les complexes de pore nucléaire (CPN) est assuré par des facteurs de
transport solubles qui font la navette entre le cytoplasme et le noyau (Chook and Blobel,
2001; Conti and Izaurralde, 2001; Conti et al., 2006; Fahrenkrog and Aebi, 2003;
Gorlich and Kutay, 1999; Macara, 2001; Madrid and Weis, 2006; Mosammaparast and
Pemberton, 2004; Pemberton and Paschal, 2005; Stewart, 2007; Stewart et al., 2001;
Weis, 2003).
B.1 Les complexes de pore nucléaire
Les CPN (Figure 7) sont des structures multiprotéiques qui perforent l'enveloppe
nucléaire et forment un canal pour l'échange bidirectionnelle des molécules entre le
cytoplasme et le noyau par un canal central qui a un diamètre de 25-30 nm (Chook and
Blobel, 1999). Les CPN sont constitués de plusieurs copies d’environ 30 protéines
51

différentes collectivement appelées nucléoporines (Patel et al., 2007; Cronshaw et al.,
2002; Rout et al., 2000; Rout and Wente, 1994). Le poids moléculaire du CPN est
estimée à 50 MDa chez S. cerevisiae, et 120 MDa chez les vertébrés. On distingue trois
niveaux dans sa structure : une couronne centrale, ancrée dans l'enveloppe nucléaire; un
anneau cytoplasmique, duquel s'étendent des structures filamenteuses; et un anneau
nucléaire formant un panier. L'ancrage à l’enveloppe est assurée par quatre
nucléoporines : toutes les autres nucléoporines sont des protéines solubles. L'intérieur
du complexe de pore est tapissée de nucléoporines présentant une structure flexible et
riche en répétition phénylalanine–glycine, ou nucléoporines FG. Certaines
nucléoporines FG se situent également sur l'anneau cytoplasmique. Ces nucléoporines
forment une barrière sélective pour le passage du pore. Plusieurs modèles ont été
proposés: certains auteurs évoquent une brosse moléculaire qui "repoussent" les
molécules sous l'effet de l'agitation brownienne; d'autres ont proposés que les
répétitions FG hydrophobes soient engagées dans des interactions intermoléculaires et
conduisent à la formation d'un gel de nucléoporines au cœur du pore. Dans les deux cas,
le franchissement du pore requiert une interaction avec les nucléoporines, soit pour
surmonter la barrière entropique que constitue la brosse, soit pour désorganiser
localement le gel de nucléoporines FG. Cette interaction est assurée par les facteurs de
transport, encore appelés caryophérines ou importines/exportines.
B.2. Les voies d’import nucléaire
Il y a différentes voies d'import nucléaire de protéines, mais elles partagent plusieurs
dispositifs communs. Elles sont basées sur une série concertée d’interactions protéine-
protéine par lesquelles des "cargo" (terme anglais désignant les molécules transportées,
52

« le chargement ») sont identifiées dans le cytoplasme, transférées à travers les CPN et
libérées dans le noyau. Dans chaque voie, les protéines sont ciblées pour l'import
nucléaire grâce à des motifs courts appelés séquence (ou signal) de localisation
nucléaire (NLS). Il y a différentes classes de NLS et chacune est reconnue par les
composants spécifiques d'une voie. Chez la levure, on compte 13 caryophérines β et 1
caryophérine α. La voie classique d’import nucléaire des protéines utilise l’importine β
(Kap95p chez S. cerevisiae) comme transporteur. Elle concerne une gamme variée de
cargo et a été étudiée de manière substantielle. Dans cette partie, nous abordons dans un
premier temps la voie d’import classique pour mieux comprendre les différents
évènements affectant cette voie de manière générale, afin d’étudier dans un second
temps la voie d’import utilisée par les protéines ribosomiques. Cette dernière utilise des
transporteurs différents qui ont des NLS distinctes de celles observées dans la voie
classique.
B.3. La voie d'import classique
Le cycle classique d'import nucléaire des protéines (Gorlich and Kutay, 1999), qui
assure le transport de 100-1.000 cargo par minute et par CPN (Ribbeck and Gorlich,
2001) est le résultat d'une série d’interactions protéine-protéine. Des protéines cargo
avec une NLS classique (voir plus loin la définition) sont transportées par l’importine β,
qui les lie par l’intermédiaire d’une protéine adaptatrice, l’importine α. En interagissant
avec le complexe de pore nucléaire, l'importine β facilite le mouvement à travers les
CPN. Dans le noyau, RanGTP se lie à l'importine β, dissociant le complexe d'import et
libérant le cargo. L’importine β complexée à RanGTP est recyclée dans le cytoplasme.
L'importine α est également exportée vers le cytoplasme après liaison avec l'exportine
53
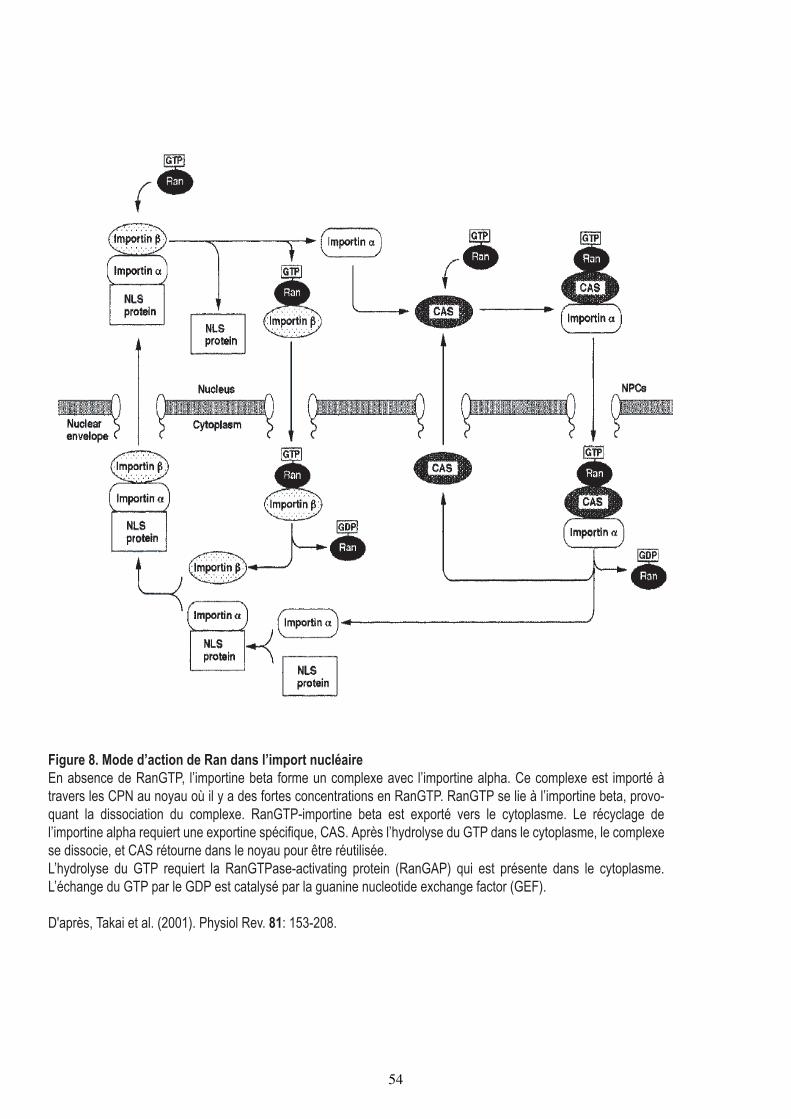
Figure 8. Mode d’action de Ran dans l’import nucléaireEn absence de RanGTP, l’importine beta forme un complexe avec l’importine alpha. Ce complexe est importé à travers les CPN au noyau où il y a des fortes concentrations en RanGTP. RanGTP se lie à l’importine beta, provo-quant la dissociation du complexe. RanGTP-importine beta est exporté vers le cytoplasme. Le récyclage de l’importine alpha requiert une exportine spécifique, CAS. Après l’hydrolyse du GTP dans le cytoplasme, le complexe se dissocie, et CAS rétourne dans le noyau pour être réutilisée.L’hydrolyse du GTP requiert la RanGTPase-activating protein (RanGAP) qui est présente dans le cytoplasme. L’échange du GTP par le GDP est catalysé par la guanine nucleotide exchange factor (GEF).
D'après, Takai et al. (2001). Physiol Rev. 81: 153-208.
54

CAS et RanGTP. Dans le cytoplasme, RanGAP cytoplasmique (Ran GTPase activating
protein) stimule l'activité GTPasique de Ran, ce qui entraine l'hydrolyse du GTP et
dissocie les importines. Elles sont ainsi disponibles pour un autre cycle d'import (Figure
8).
Le cycle d'import nucléaire des protéines est basé sur une série d'interactions
soigneusement orchestrée et qui doit être contrôlée avec précision dans l'espace et le
temps. La reconnaissance moléculaire est cruciale pour le cycle, aussi bien pour les
transporteurs reconnaissant leurs cargo que pour les partenaires de Ran (Ran-GAP et
Ran-GEF) qui permettent de distinguer les compartiments nucléaire et cytoplasmique.
En effet, les interactions entre les cargo et les transporteurs sont orchestrées par l'état
nucléotidique de Ran, qui oscille entre liaison au GDP et au GTP. L'état nucléotidique
de Ran est contrôlé par son facteur d’échange nucléotidique guanine (RanGEF), qui
catalyse la recharge de RanGDP en GTP, et le RanGAP, qui stimule l'hydrolyse du
GTP. RanGEF est nucléaire et RanGAP est cytoplasmique : par conséquent la fraction
nucléaire Ran est liée au GTP dans le noyau, tandis que Ran dans le cytoplasme est
complexée au GDP. RanGDP est transporté dans le noyau par un transporteur
spécifique, NTF2 (Ribbeck et al., 1998; Smith et al., 1998), et est rechargé en GTP par
la RanGEF dans le noyau. La conformation de Ran change nettement avec l'état
nucléotidique. Ce changement structural détermine l'association de Ran avec les
caryophérines et dirige de ce fait l'interaction des facteurs de transport de la famille des
importines β avec leur cargo. Ce mécanisme permet d'assurer la directionalité du
transport. L'énergie du GTP hydrolysé par Ran est donc employée pour réguler la
formation de ces complexes protéiques.
Le cycle d'import nucléaire des protéines peut donc être subdivisé en quatre étapes :
l’assemblage du cargo au complexe d'import dans le cytoplasme; la translocation à
55

travers les CPN; le désassemblage du complexe d’import dans le noyau et le recyclage
de l'importine.
B.3.1. Assemblage du complexe d’import
Les NLS classiques contiennent un ou deux clusters de résidus basiques. Les NLS
monopartites, comme c’est le cas chez SV40, ont un simple cluster de 4-5 résidus
basiques, tandis que les NLS bipartites, comme pour la nucléoplasmine, ont un
deuxième cluster basique localisé aux environs de 10-12 résidus en aval du premier
cluster. La reconnaissance moléculaire de ces NLS est cruciale pour la formation du
complexe d'import et est médiée par des sites spécifiques de l’importine α (Conti and
Kuriyan, 2000; Conti et al., 1998; Fontes et al., 2000). L’importine α est constituée
d'une répétition de motifs Armadillo (ARM) générant une molécule en forme de
banane, et les sites de liaison aux NLS sont formés d'une rangée de Trp, Asn et des
résidus acides situés dans un sillon sur la surface concave intérieure. Le site de liaison
primaire pour les NLS monopartites couvre un site majeur formé de 1 à 4 répétitions de
motifs ARM et un site secondaire (le site mineur) couvrant 6-8 répétitions de motifs
ARM (Conti and Kuriyan, 2000; Conti et al., 1998; Fontes et al., 2000). Avec les NLS
bipartites, le plus grand cluster basique occupe le site majeur, et le plus petit cluster
basique occupe le site mineur. Les chaînes basiques latérales des NLS font une série
d’interactions spécifiques avec les principaux résidus le long de la surface intérieure de
l'importine α : des interactions électrostatiques, comme attendu de la présence de
résidus basiques dans ces NLS, mais également des interactions hydrophobes au niveau
des chaines latérales des lysines. Généralement, la NLS s'adapte au site de liaison et
ainsi est habituellement présente comme boucle de surface non structurée dans la
56

protéine cargo, bien que la NLS soit parfois formée à partir de régions disparates d'une
protéine (Rodriguez et al., 2006).
La partie N- terminale de l'importine α se lie à l'importine β par un domaine connu pour
se lier à l’importine β (IBB, pour importine β -Binding domain) (Gorlich et al., 1996;
Weis et al., 1996), qui facilite également le relargage du cargo dans le noyau (Kobe,
1999). Le domaine IBB contient un cluster de résidus basiques qui est semblable à une
NLS et qui peut se lier aux sites de liaison aux NLS de l’importine β (Kobe, 1999;
Matsuura and Stewart, 2004). Par conséquent, en plus de la connexion de l'importine α
à l’importine β, le domaine IBB a un rôle d’autoinhibition (Kobe, 1999) de sorte qu’en
absence de liaison à l’importine β, il rentre en compétition avec les NLS pour la liaison
à l'importine α et contribue ainsi au relargage du cargo. En accord avec cette hypothèse,
les constructions d'importine α manquant le domaine IBB (ΔIBB-importine α )
présentent une affinité sensiblement plus haute pour les NLS que celle de la protéine
entière (Goldfarb et al., 2004; Matsuura et al., 2003; Matsuura and Stewart, 2005).
Cependant, l'importine α présente habituellement une affinité plus élevée pour les NLS
que pour le domaine IBB (Goldfarb et al., 2004). Quand le domaine IBB se lie à
l'importine β , il ne peut plus entrer en compétition avec les NLS pour se lier à
l'importine α. Par conséquent, dans le cytoplasme, les cargo contenant les NLS se lient
probablement principalement au complexe importine α /β plutôt qu’à la seule importine
α. Habituellement, les NLS ont une affinité d’environ 10 nM pour le ΔIBB-importine α
ou pour le complexe importine α /β (Goldfarb et al., 2004; Matsuura et al., 2003;
Matsuura and Stewart, 2005) et le taux d’import nucléaire est en corrélation avec la
force de liaison à l’importine α (Hodel et al., 2006).
Les protéines ne possédant pas de NLS classique se lient directement aux caryophérines
β (Cingolani et al., 2002; Lee et al., 2003), plutôt qu’à l'importine α. Le mécanisme par
57

lequel l’importine α reconnaît les NLS classiques est différent de celui par lequel les
NLS non classiques sont directement reconnues par d’autres caryophérines β.
B.3.2. Translocation à travers les CPN
Les macromolécules qui ont un poids moléculaire supérieur à 40 kD sont exclues des
CPN, et seulement celles qui se lient aux transporteurs peuvent traverser le CPN. Par
ailleurs, même si les plus petites molécules peuvent simplement diffuser à travers les
CPN, leur transport efficace requiert également l'association avec une caryophérine.
Cependant, le mécanisme par lequel les complexes cargo/transporteur sont transportés à
travers les CPN, de même que celui par lequel d'autres molécules en sont exclues
demeurent controversés. Des études faites avec des molécules fluorescentes simples
(Yang et al., 2004) indiquent que le mouvement des complexes cargo/transporteur à
travers les CPN est rapide et bidirectionnel, ressemblant à un échange aléatoire, et en
général, semble être un processus de premier ordre dans lequel le taux est limité par la
sortie du cargo. Par conséquent, la directionnalité du transport n'est pas imposée par le
pore lui-même, mais par la dissociation du complexe d’import cargo/importine induite
par RanGTP dans le noyau. La translocation des complexes cargo/importine est plus
rapide que la translocation des importines seules, et il semble y avoir de multiple voies
parallèles de transport (Kubitscheck et al., 2005). Le temps d’import et l'efficacité de
transport semblent dépendre directement de la concentration en importine β, et de la
vitesse de formation des complexes cargo/importines (Timney et al., 2006; Yang and
Musser, 2006). La nature des molécules transportées par les pores nucléaires est
principalement déterminée par les transporteurs, dont chacun lie une gamme spécifique
58

des cargo, bien qu'il y ait parfois un certain recouvrement; quelques cargo pouvant être
transportés par plus d'un transporteur.
B.3.3. Désassemblage du complexe d’import
RanGTP nucléaire dissocie le complexe d’import cargo/importine et impose de ce fait la
directionnalité dans le processus de transport. La diffusion rapide du complexe
cargo /importine au sein des CPN (Yang and Musser, 2006) maintient un équilibre
rapide à travers le CPN; le déplacement du complexe du côté nucléaire du CPN par sa
dissociation par RanGTP aura comme conséquence un écoulement net du complexe
cargo /importine du cytoplasme au noyau. En effet, bien que l'énergie thermique
(mouvement brownien) permette au cargo de se lier à l'importine et de diffuser à travers
le CPN, le cargo ne peut pas retourner dans le cytoplasme après qu'il se soit dissocié de
son importine dans le noyau. Ainsi, RanGTP dirige le mouvement brownien du
complexe cargo/importine.
B.3.3.1. Dissociation du complexe importine α/β
La liaison de RanGTP à l'importine β dissocie le complexe importine α /β, permettant le
relargage du cargo. Il y a seulement un chevauchement limité entre les sites de liaison à
RanGTP et au domaine IBB sur l'importine β. RanGTP libère principalement le
domaine IBB de l'importine α en induisant un changement conformationnel dans
l'importine β, plutôt que par compétition. Ce changement conformationnel augmente la
hauteur hélicoïdale de l'importine β , libérant de ce fait le domaine IBB par un
mecanisme allostérique (Vetter et al., 1999 ; Lee et al., 2005). Dans le complexe
importine β/domaine IBB (Lee et al., 2005), il y a une interaction électrostatique
59

Figure 9. Désassemblage du complexe d’import(a) Structure du complexe importine beta: RanGTP chez S. cerevisiae montrant comment importine beta (en jaune) s'enroule autour Ran (bleu) lié au GTP (cyan). La boucle I de Ran est en rouge et la boucle II est en vert.(b) Représentation schématique de (a). Les répétitions HEAT de l’importine beta sous forme de blocs jaunes (H1-19). Les résidus qui constituent l'interface entre l’importine beta (jaune) et RanGTP (bleu clair) sont indiqués (les résidus de la boucle I de Ran sont en rouge et ceux de la boucle II sont en vert). RanGTP se lie à trois sites différents.
D'après, Lee et al., 2005
60

étendue entre le domaine basique IBB et les résidus acides qui forment un patch continu
négativement chargé sur la surface intérieure de l'importine β. Cette interaction étendue
requiert un jeu précis entre la hauteur hélicoïdale de l'importine β et l’hélice α apporté
par le domaine IBB, de sorte que l'importine β entoure le domaine IBB. Le changement
conformationnel de l'importine β provoqué par la liaison de RanGTP perturbe la
géométrie de ce complexe et empêche par conséquent leur interaction efficace (Lee et
al., 2005).
L’importine β est constituée de 19 répétitions HEAT (H1-19), dont chacune contenant
deux hélices α qui vont s’empiler ensemble pour produire une molécule hélicoïdale
ovale (Cingolani et al., 1999; Lee et al., 2005; Liu and Stewart, 2005). RanGTP se lie à
trois sites sur l’importine β (Lee et al., 2005; Vetter et al., 1999) (Figure 9). La boucle
II lie la partie N-terminale de l'importine β à une région qui entoure des répétitions
HEAT (1-4) et qui est commune à toutes les caryophérines β (Gorlich et al., 1997). Une
interaction similaire de la boucle II est observée dans les complexes de RanGTP avec la
transportine (Chook and Blobel, 1999) ou l'exportine CAS (Matsuura and Stewart,
2004). Le second site de liaison à l'importine β implique une interaction principalement
électrostatique avec une boucle dans la répétition HEAT 8 (Lee et al., 2005; Vetter et
al., 1999). Le troisième site de liaison de RanGTP implique la boucle I qui interagit
avec les répétitions HEAT (12-15) et est crucial pour produire un changement
conformationnel qui augmente la hauteur hélicoidale (Lee et al., 2005). Dans le
complexe importine β/IBB, le chemin suivi par l'importine β hélicoidale s'opposerait à
celui de Ran. La liaison de RanGTP peut donc seulement être adaptée si la chaîne de
l'importine β se déplace, lequel produit alternativement l'augmentation de la hauteur
hélicoïdale qui libère le domaine IBB. Une interaction électrostatique entre les résidus
Lys37 et Lys152 de Ran avec un cluster de résidus négativement chargés sur l'importine
61

β est centrale pour cette interaction et, bien que la rupture de cette interaction avec
K37D/K152A-Ran n'empêche pas la liaison de RanGTP à l'importine β, elle inhibe le
relargage du domaine IBB (Lee et al., 2005).
B.3.3.2. Relargage du cargo.
Bien que la dissociation de l'importine α de l'importine β rende l’import irréversible, il
est encore nécessaire de dissocier le cargo de l'importine α . Une fois libéré de
l'importine β par RanGTP, le domaine IBB rentre en compétition avec la NLS du cargo
pour se lier à l'importine α, diminuant de ce fait l'affinité du cargo pour l'importine α et
faciliter ainsi le relargage (Kobe, 1999) par un mécanisme d’autoinhibition. Un
mécanisme analogue a été proposé pour le relargage des NLS M9 de la transportine
même si une boucle dans la répétition HEAT 8 de la transportine pourrait entrer en
compétition avec le site de liaison de la NLS après son relargage par RanGTP (Gorlich
et al., 1997; Lee et al., 2006).
Néanmoins, la forte affinité de l'importine α pour les NLS classiques ( ≈ 10 nM) est
incompatible avec ses taux de transport mesurés d’environ 100-1.000 par seconde par
CPN (Timney et al., 2006). Les nucléoporines FG situées à la face nucléaire des CPN,
tel que Nup1p et Nup2p de S. cerevisiae, accélèrent le désassemblage du complexe
d’import importine α /β /cargo (Gilchrist et al., 2002; Goldfarb et al., 2004; Liu and
Stewart, 2005; Matsuura and Stewart, 2005). Nup1p a une affinité plus élevée pour le
complexe d’import importine α/β que d'autres nucléoporines (Liu and Stewart, 2005)
(Gilchrist et al., 2002) et ainsi concentre le complexe d'import à la face nucléaire du
pore ce qui favorise la liaison à RanGTP. De même, parce qu'il peut se lier à la fois à
RanGTP et CAS, l'exportine de l'importine α, Nup2p peut également augmenter le taux
62

de désassemblage du complexe d’import en favorisant l'assemblage du complexe de
recyclage importine α/CAS/RanGTP (Matsuura et al., 2003).
Par ailleurs, Nup2p déplace également activement les NLS monopartite et bipartite de
l'importine α (Gilchrist et al., 2002; Matsuura et al., 2003; Matsuura and Stewart, 2005).
Des études structurales montrent que Nup2p/NUP50 se lie à deux sites de l'importine α
: un site de haute affinité à la partie C-terminale de l'importine α et un site de faible
affinité qui recouvre les sites de liaison aux NLS. La liaison de Nup2/NUP50 au site de
haute affinité de la partie C-terminale de l'importine α augmente considérablement la
concentration locale du domaine Nup2/NUP50 qui se lie au site de faible affinité et
accélère ainsi la dissociation NLS par un mécanisme analogue à celui proposé pour le
domaine IBB (Matsuura and Stewart, 2005).
B.3.4. Recyclage des importines
Après le désassemblage du complexe d’import par RanGTP, les importines doivent être
recyclées dans le cytoplasme. L’importine β est recyclée, complexée avec RanGTP,
tandis que l'importine α est activement exportée par l'exportine CAS. La structure du
complexe d’export CAS/importine α /RanGTP de S. cerevisiae (Matsuura et al., 2003)
montre que CAS recouvre à la fois RanGTP et l'importine α avec une interface
d'interaction exceptionnellement grande. L’exportine CAS est structuralement
semblable à l'importine β et est basée sur une répétition de 19 motifs HEAT. Dans le
complexe, le domaine IBB se lie aux sites de liaison NLS de l'importine α, et des études
de mutagénèse (Matsuura et al., 2003) ont montré que cette liaison est cruciale pour la
formation du complexe d’export. Des mutations qui interfèrent avec la liaison du
domaine IBB à d’autres sites NLS ou à CAS empêchent la formation du complexe
63

d'export. Une conséquence importante du rôle central du domaine IBB dans la
formation du complexe d'export est que le complexe peut seulement se former quand le
cargo est libéré de l’importine α. Ce dispositif empêche que l'importine α retourne dans
le cytoplasme portant toujours le cargo (Matsuura et al., 2003). Cependant, le domaine
IBB doit également déplacer la liaison de Nup2/NUP50 à l'importine α, et ceci est
rendu plus difficile par la liaison de ces nucléoporines à l'importine α avec une affinité
plus élevée que les NLS (Matsuura and Stewart, 2005). Il a été proposé (Matsuura and
Stewart, 2005) que la clef à ce dilemme est la manière dont CAS se lie à l'importine α.
Il semble que Nup2/NUP50 soit enlevé dans un mécanisme en deux étapes qui est
l'inverse de la manière par laquelle Nup2/NUP50 déplace les NLS. CAS déplacerait
d'abord Nup2/NUP50 du site de haute affinité de la partie C-terminale de l'importine α,
facilitant alors le déplacement Nup2/NUP50 des sites de liaison aux NLS par le
domaine IBB. Bien que pour la simplicité toutes ces interactions aient été considérées
en tant qu'étapes séparées, il est probable que les séries d'interactions à la face
nucléoplasmique du CPN impliquées dans le désassemblage du complexe d’import et le
recyclage de l’importine se produisent d'une façon concertée avec des nucléoporines tels
que Nup1 et Nup2 fonctionnant comme un échafaudage pour coordonner le processus.
Une fois que les complexes importine β / RanGTP et CAS / RanGTP /importine α
retournent dans le cytoplasme par les CPN, RanGAP cytoplasmique (ainsi que la
protéine accessoire RanBP1, qui enlève RanGTP de l'importine β) (Bischoff and
Gorlich, 1997) stimule l'hydrolyse du GTP qui relargue Ran de l'importine β et de CAS.
Les importines sont alors disponibles pour un autre cycle d’import. Bien qu'il ne soit pas
documenté en détail, il est probable que le relargage de l'importine et le désassemblage
du complexe d’export puissent également être coordonnés par des nucléoporines
localisées sur la face cytoplasmique du CPN (tel que RanBP2, qui a le potentiel de
64

liaison à RanGAP, l'importine β et Ran), liant les différents composants et les réunissant
d'une façon coordonnée.
La conformation de CAS libre produit après hydrolyse de RanGTP (Cook et al., 2005)
montre un changement marqué dans la hauteur hélicoïdale comparé avec le complexe
d’export CAS / importine α / RanGTP (Matsuura and Stewart, 2004). Une fois libéré de
Ran et de l'importine α dans le cytoplasme, CAS prend une conformation fermée dans
laquelle les résidus de la partie N-terminale se lient à une région près du centre de la
molécule (Cook et al., 2005). Cette conformation fermée contraste avec la
conformation plus ouverte adoptée dans le complexe d'export et est probablement
rigide, analogue à la forme plus rigide que l'importine β assume une fois complexée au
RanGTP (Conti et al., 2006). La différence importante de la hauteur hélicoïdale
observée entre les deux états fonctionnels de CAS indique une flexibilité
conformationnelle semblable à celle de l'importine β (Conti et al., 2006).
B.4. Import des protéines ribosomiques
Les protéines ribosomiques représentent une fraction très importante des protéines
importées dans le noyau. Ainsi, dans une culture de levure en phase exponentielle de
croissance qui double leur contenu ribosomique toutes les 1.5 heures, chaque cellule
doit importer environ 150.000 protéines ribosomiques par minute à travers l'enveloppe
nucléaire (tout en exportant simultanément environ 4.000 sous-unités ribosomiques par
minute (Warner, 1999).
65

Tableau 2. Liste exhaustive des caryophérines beta
D'après, Pemberton et al., 2005
66

Une voie majeure et distincte d'import nucléaire est assurée par un membre de la famille
des β- caryophérines de levure, Kap123p/caryopherin β4 dont l'homologue de vertébrés
le plus proche est l'importine 5 (Rout et al., 1997; Schlenstedt et al., 1997; Sydorskyy et
al., 2003; Timney et al., 2006) (Tableau 2). Kap123p se lie directement à ses substrats
d'import, un ensemble de protéines ribosomiques. C'est la caryophérine la plus
abondante chez la levure (Timney et al., 2006). Ce n'est pas une protéine essentielle,
mais son absence entraine une baisse importante de la cinétique d'import nucléaire d'une
protéine rapportrice fusionnée à la NLS de la protéine ribosomique Rpl25p. En accord
avec le caractère non-essentiel de Kap123p, un autre membre de la famille β,
Pse1p/Kap121p/caryopherin β3, peut fonctionnellement remplacer Kap123p (Moroianu,
1998; Rout et al., 1997). Kap123p comme Kap121p peut diriger in vivo la β -
galactosidase dans le noyau une fois fusionée à la NLS de la protéine ribosomique
Rpl25p (Rout et al., 1997). Bien que Kap123p lie beaucoup de protéines ribosomiques,
elle ne s'associe pas aux ribosomes matures, très probablement parce que les NLS se
retrouvent masqués dans ces derniers. Il faut noter que la liaison à Kap123p a été
démontrée formellement pour l'instant pour un nombre très restreint de protéines
ribosomiques. Par ailleurs, l'importine Kap108p peut également se lier à des protéines
ribosomiques (Rosenblum et al., 1997).
Chez les mammifères, il a été proposé que chaque protéine ribosomique peut être
reconnue par plusieurs importines différentes (Jakel and Gorlich, 1998). Ainsi, la
protéine RPL23A, homologue de la protéine Rpl25p, s'associe à quatre importines
différentes. Le domaine de liaison de RPL23A à ces quatre transporteurs semble le
même. Ces importines sont capables de médier l'import nucléaire de RPL23A dans le
noyau de cellules semi-perméabilisées. De la même manière, la protéine RPS2 se lie à
plusieurs importines (Antoine et al., 2005). La protéine RPL12 en revanche est
67

transportée par l'importine 11 (Plafker and Macara, 2002), une importine pour laquelle
on connaît très peu de substrats. L'importine 11 ne se lie pas à RPL23A, et a contrario
RPL12 ne semble pas être importée efficacement par d'autres importines que l'importine
β. La diversité de ces situations laisse penser que l'import nucléaire des protéines
ribosomiques implique de nombreux mécanismes.
A ce jour, les voies de transport nucléocytoplasmiques empruntées par des protéines
ribosomiques n'ont été étudiées que pour un très petit nombre d'entre elles. Par ailleurs,
on ne connaît les NLS que d'une poignée de protéines ribosomiques, chez la levure
comme chez les mammifères. Bien qu'il ait été proposé une séquence consensus pour
ces NLS non conventionnels (Timmers et al., 1999), pour l'instant sans grand support
expérimental, on ne connaît pas bien les déterminants de ces séquences qui permettent
leur reconnaissance par les importines.
68

RESULTATS
69

70

I. ETUDE STRUCTURE-FONCTION DE LA PROTEINE RPS19
La protéine ribosomique RPS19 est un acteur de la biogenèse des ribosomes impliquée
dans l’anémie de Diamond-Blackfan. Chez 25% de patients ADB, une mutation dans le
gène codant la protéine est associée à la pathologie, ce qui a fait d’elle la première
protéine ribosomique impliquée dans une maladie génétique.
Dans notre équipe, il a été montré chez la levure S. cerevisiae que Rps19p est impliquée
dans le clivage du précurseur de l’ARNr 18S au site A2. La perte de fonction de Rps19
affecte spécifiquement la production de la petite sous-unité ribosomique et la rend
incompétente pour son export : en absence de Rps19p, le pré-ARNr 21S, qui est un
précurseur aberrant de l’ARNr 18S, s’accumule dans le noyau cellulaire et plus
particulièrement dans le nucléole.
Dans cette partie du travail, nous avons effectué une étude structure-fonction de la
protéine Rps19p. Ce travail a été effectué en collaboration avec le groupe de Sébastien
Fribourg (IECB-Pessac, France) pour étudier la structure de RPS19. Nous avons cloné
la séquence codante de l’homologue de RPS19 chez l’archaéon hyperthermophile
Pyrococcus abyssi afin de l’exprimer dans E. coli. Sa structure a été résolue par Lynn
Gregory, ce qui nous a permis de localiser les différentes mutations affectant RPS19
chez les patients ADB, et de distinguer deux classes :
- des mutations qui affectent les acides aminés à l’intérieur de la protéine et qui vont
avoir un impact sur le repliement et la stabilité de la protéine,
- des mutations qui se trouvent en surface de la protéine et qui définissent deux
domaines distincts : un sillon qui correspond au hot spot de mutation; et un patch
d'acides aminés basiques qui pourrait interagir avec des acides nucléiques. Ces
71

mutations de classe II sont susceptibles d’intervenir dans les interactions de la protéine
avec ses partenaires.
Pour vérifier l’impact des mutations de classe II, nous avons étudié l’incorporation de la
protéine Rps19p dans les ribosomes matures chez la levure S. cerevisiae. Pour cela, les
sous-unités ribosomiques ont été isolées sur gradient de sucrose pour évaluer
l'association de la protéine aux ribosomes grâce à l’analyse biochimique des fractions
contenant la sous-unité 40S, les ribosomes 80S et les polysomes. De plus, nous avons
directement regardé l’association de différents mutants avec les pré-ARNr. L’effet des
mutations sur la croissance cellulaire a également été étudié.
Nos résultats indiquent que les domaines affectés par les mutations en surface de la
protéine sont essentiels pour l’incorporation de Rps19p dans les pré-ribosomes. Nous
concluons que les mutations faux-sens de la protéine Rps19 dans l’ADB empêchent
l’incorporation de la protéine dans les ribosomes en affectant des surfaces d’interaction
intermoléculaire ou en affectant la stabilité de la protéine.
72

Published online 28 July 2007 Nucleic Acids Research, 2007, Vol. 35, No. 17 5913–5921doi:10.1093/nar/gkm626
Molecular basis of Diamond–Blackfan anemia:structure and function analysis of RPS19Lynn A. Gregory1,2, Almass-Houd Aguissa-Toure3, Noel Pinaud1,2,
Pierre Legrand4, Pierre-Emmanuel Gleizes3,* and Sebastien Fribourg1,2,*
1INSERM U869, Institut Europeen de Chimie et Biologie, 2 rue Robert Escarpit Pessac, F-33607, 2UniversiteVictor Segalen, Bordeaux 2, F-33076, 3Laboratoire de Biologie Moleculaire des eucaryotes (UMR5099) andInstitut d’Exploration Fonctionnelle des Genomes (IFR109), CNRS and Universite Paul Sabatier, 118 route deNarbonne F-31062 Toulouse and 4Synchrotron SOLEIL L’Orme des Merisiers, Saint Aubin- BP48, 91192 Gif surYvette Cedex, France
Received July 6, 2007; Revised and Accepted July 30, 2007 EBI Accession No. 2v7f
ABSTRACT
Diamond–Blackfan anemia (DBA) is a rare congeni-tal disease linked to mutations in the ribosomalprotein genes rps19, rps24 and rps17. It belongs tothe emerging class of ribosomal disorders. Tounderstand the impact of DBA mutations onRPS19 function, we have solved the crystal struc-ture of RPS19 from Pyrococcus abyssi. The proteinforms a five a-helix bundle organized around acentral amphipathic a-helix, which corresponds tothe DBA mutation hot spot. From the structure, weclassify DBA mutations relative to their respectiveimpact on protein folding (class I) or on surfaceproperties (class II). Class II mutations cluster intotwo conserved basic patches. In vivo analysis inyeast demonstrates an essential role for class IIresidues in the incorporation into pre-40S ribosomalparticles. This data indicate that missense muta-tions in DBA primarily affect the capacity of theprotein to be incorporated into pre-ribosomes, thusblocking maturation of the pre-40S particles.
INTRODUCTION
Diamond–Blackfan anemia (DBA) is a rare congenitaldisorder characterized by the defective differentiation ofpro-erythroblasts, the precursors of red blood cells.Patients suffer severe anemia and display heterogeneousclinical features including malformations, growth failureand predisposition to cancer (1,2). Linkage analysis hasrevealed that a quarter of all DBA reported cases are
connected to the heterozygous mutation of the geneencoding the ribosomal protein RPS19 (3,4). The RPS19protein is a component of the 40S ribosomal subunit andbelongs to a family of ribosomal proteins restricted toeukaryotes and archea. It is essential for yeast viabilityand for early stages of development in mice (5,6).Disruption as well as point mutations of the rps19 genein yeast and human cells affect maturation of the pre-ribosomal RNA (pre-rRNA) and block production of the40S ribosomal subunits (5,7–9). Why the mutation ofa ribosomal protein primarily affects pro-erythroblastdifferentiation remains a central question. However,recent linkage of a two other ribosomal protein genes,rps24 and rps17, to DBA (10,11) strongly supports thehypothesis that DBA is the consequence of a ribosomaldisorder (8,12).Over 60 different mutations affecting the rps19 gene
have been reported, including deletions, insertions, frame-shifts, premature stop codons and missense mutations(13–15). Some mutations, like very early stop codons ormodification of the promoter clearly result in RPS19haplodeficiency by hampering synthesis of RPS19 fromthe mutated allele. However, for more subtle mutationslike missense mutations, the question arises as towhether they affect the folding of the protein or whetherthey are milder mutations affecting the function whilepreserving the overall fold. Since there is no homolog ofRPS19 in bacteria, for which high-resolution structuresof the small ribosomal subunit are available, the structureof RPS19 and its precise location within the 40S subunitremain unknown. The crystal structure of RPS19from Pyroccocus abyssi presented herein fills this gapand provides a rationale for the impact of RPS19mutations in DBA.
Correspondence may also be addressed to Pierre-Emmanuel Gleizes. Tel/Fax: 00 33 5 61 33 59 26/58 86, Email: [email protected]
The authors wish it to be known that, in their opinion, the first two authors should be regarded as joint First Authors.
*To whom correspondence should be addressed. Tel: 00 33 5 40 00 30 63; Fax: 00 33 5 40 00 30 68; Email: [email protected]
� 2007 The Author(s)
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/
by-nc/2.0/uk/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
73

MATERIAL AND METHODS
Protein expression, purification and crystallization
Pyrococcus abyssi RPS19 cDNA was cloned into apET-15b (Novagen) modified plasmid. The expressionwas carried out in BL21 (DE3) Rosetta cells (Novagen) at158C. Bacterial cells were sonicated and centrifuged for30min at 50 000 g. The supernatant was heated up for20min at 508C and centrifuged for another 30min at50 000 g. After incubation and elution from the cobalt-affinity resin, the tag was cleaved from the protein by anovernight digest with 1/200 (w/w) ratio with TEV. A stepof purification on Hi-S (Pharmacia) was carried out andthe protein was eluted at about 600mM NaCl. Theprotein was concentrated to up to 10mg/ml in 50mMTris–HCl pH 7.5, 600mM NaCl. Crystals were obtainedat 208C by the hanging drop vapor diffusion method bymixing equal amounts of the protein solution and of areservoir composed of 30–36% PEG 2000 MME, 100mMTris–HCl pH 6.8–7.5 over a couple days to a size of50� 200� 200 microns. They diffracted to 1.15 A onsynchrotron beamline and belonged to the space groupP212121 with cell dimensions a¼ 32 A, b¼ 57 A, c¼ 82 Aand contained one molecule per asymmetric unit and 48%solvent.
Structure solution and refinement
Native and derivative data were collected at the ESRFsynchrotron and processed with XDS (16). Data collectionstatistics are shown in Table 1. A HgBr2 derivativeresulting from a 12-h soak in 2.5% (v/v) HgBr2 (preparedfrom a saturated solution) was collected at the LIII-edgeof Hg. Derivative data set was combined with the mostisomorphous native data set, SHELXD and SHARP wereused to locate and refine the heavy atom site position(17,18). The resulting phases had an overall figure of merit(FOM) of 0.33 at a resolution of 2.0 A. After solventflattening with Solomon (SHARP), automatic buildingwas carried out with ARP/wARP (19). Out of a total of150 residues, 125 were initially placed. Further buildingand refinement cycles were carried out with Coot andREFMAC (20,21). Last cycles of refinement were carriedout including hydrogens and using individual anisotropicB factors.The final model has a good stereochemistry with an
R-free value of 15.5% and a R-factor value of 13.7%(Table 1).
Analysis of RPS19 mutations in yeast
Site-directed mutagenesis on yeast RPS19 was performedby PCR. For complementation experiments, the mutatedalleles were subcloned into vector pFL38-Ps15 (URA3),downstream of the constitutive RPS15 promoter. Theresulting plasmids were introduced in the ura3�GAL-RPS19 strain (5). Cells were cultured in liquidsynthetic medium containing galactose, but no uracil, andspotted at different densities on agar plates using the samemedium, with either galactose or glucose as the carbonsource to modulate RPS19 expression.
To evaluate incorporation of wild-type or mutatedRPS19A into ribosomes, the corresponding open readingframes were also cloned in frame downstream the TAP tagcoding sequence in the pFL38-Ps15 vector and expressedin Euroscarf strain Y06271 (rps19A::KanMX, RPS19B).Whole cell extracts were fractionated by ultracentrifuga-tion on sucrose gradient for ribosome analysis. Twohundred milliliters of yeast culture were grown in YPDmedium to an OD600 of 0.5 and cycloheximide was addedat a final concentration of 100 mg/ml. After 10minincubation, yeast cells were harvested by centrifugationat 5000 r.p.m and washed in 20ml ice-cold bufferA [20mM HEPES (pH 7.5), 10mM KCl, 5mM MgCl2,1mM EGTA, 1mM DTT and 100 mg/ml cycloheximide].Cells were broken with glass beads and re-suspendedin 150 ml buffer A. The suspension was clarified bycentrifugation for 5min at 10 000 r.p.m. An amount ofthe extract corresponding to 1mg of protein was loadedon a 10.5ml 10–50% sucrose gradient in buffer A withoutcycloheximide and centrifuged for 12 h at 26 000 r.p.m in aSW41 rotor. A gradient collector (AKTAprime—Amersham Biosciences), in combination with aPharmacia UV-detector LKB.UV-M II, was used torecord the UV profile. Twenty 0.5ml fractions werecollected and 150 ml of each fraction were slot-blottedon nitrocellulose membrane to detect TAP-taggedRPS19A. The membrane was then incubated with
Table 1. Crystallographic data phasing and refinement statistics
Native 1 Native 2 Hg derivative
Space group P212121 P212121 P212121
Cell dimensionsa, b, c (A) 32.70, 57.43,
80.2031.78, 57.78,
81.0331.58, 57.39,
81.58a, b, g (8) 90.0, 90.0,
90.090.0, 90.0,
90.090.0, 90.0,
90.0X-ray source ID14-4 ID29 ID29Wavelength (A) 0.9840 1.0332 1.00932Resolution (A) 28–1.15 25–1.9 20–1.91Total measurements 375 444
(18 387)75 962
(7898)78 175 (9427)
Unique reflections 49 876(5 511)
19 967 (2147) 21 251 (2664)
Redundancy 7.5 (3.3) 3.8 (3.7) 3.7 (3.5)Completeness (%) 91.5 (72.3) 87.9 (66.0) 94.1 (82.6)Rsym (%) 5.5 (21.2) 6.8 (29.1) 5.6 (15.7)Riso 23.4 (26.0)a
(Native 2 versus Hg derivative) (%)I/� 22.25 (6.12) 14.01 (4.43) 15.48 (7.64)FOM (acentric) 0.330Refinement statisticsNumber of residues 140Number of water
molecules186
Number of ions 2RMSD �2 bonds (A) 0.019RMSD �2 angles (8) 1.818Rwork (%) 13.73Rfree (%) 15.49
Statistics for high resolution bin (1.15–1.25 A, 1.9–2.0 A) are indicatedin parentheses.aHigh resolution bin (2.5–2.6 A) are indicated in parentheses.
5914 Nucleic Acids Research, 2007, Vol. 35, No. 17
74

peroxidase anti-peroxidase complexes (Sigma), which wererevealed by chemoluminescence (ECL, GE Healthcare).After scanning of the film to obtain a digital image,the labeling intensity for each fraction was quantifiedby densitometry using MetaMorph (Universal Imaging).The UV profiles were superimposed with the blotquantifications to obtain Figure 5.
Ribosomal RNAs were co-immunoprecipitated withRPS19-TAP and analysed as published previously (22,23).Pre-rRNAs were detected on northern blots with probeD-A2 (50-GAAATCTCTCACCGTTTGGAATAGC-30)and A2-A3 (50-ATGAAAACTCCACAGTG-30).
RESULTS AND DISCUSSION
Overall structure of RPS19
The structure of RPS19 from P. abyssi (RPS19Pa) wasdetermined by SIRAS on a mercury derivative and refinedagainst the best native data set. The final model has anR-free value of 15.5% (Table 1) and comprises residues2 to 150 with the exception of two disordered loops
between residues 37–44 and 79–84 (Figure 1a). Thestructure of RPS19 is almost entirely a-helical and foldsaround a five a-helix bundle. Knowing that human RPS19(RPS19Hs) shares 36% identity and 57% similarity withP. abyssi RPS19 (RPS19Pa) sequence (Figure 1c), we mayassume that they display a similar fold. In the rest ofthe text, RPS19 residues in P. abyssi, Saccharomycescerevisiae and Homo sapiens are labeled ‘Pa’, ‘Sc’ and ‘Hs’respectively (Table 2).
Two distinct classes of mutations in DBA patients clusteraround an amphipathic helix
Fourteen amino acids of RPS19 are the targets of missensemutations in DBA patients. Although these mutationsare spread along the entire primary sequence (Figure 1c),the structure of RPS19Pa shows that most missensemutations cluster within or around the a-helix 3(Figure 1a). This a-helix, made of residues 50–65 ofRPS19Pa (52–67 in human numbering), corresponds to themutation hot spot. It is located at a central position in the
K RF I KAL E A EK P I E
TV V V K K EW K L Q D W R L V I
R NR HA ER YKAGGS I A Q AAGF V .GK VITPKGRSFLDK AT LKKELEEIIPELKKY.... YGG G P R Q L V GR G LD A V HF V L E EK G QRD RI Q V HF V K L E DK P G QRD RI Q K V HF K L E EK P G QRD RI Q K V HF K E EK P G D RI
RQSN M S SRGSKS A V A SLKM D S . KLTPQG LD AG VAAASKKQH.......... R RN H S CRAADGAA A A HARL H D . KLSSIG LD AN IVFKQRDAAKQTGPIVISK N RR A N QTSAGNCL AV Q KIKW H D K ILSKQGRK LD ATSLRSSGQQA........... K HF V L Q D G QRD V
RQRN M S SRGSKS A RV A GLKM DQD . KLTPQ G VAAANKKH...........
S RN SR P CKSSGG A HI Q TMNI LDTK . KITSSG LDQ AGRIAAAV.............. K V I K L E E P G QRD RI Q A SR R YKHIDASGS N V A KIGI IS K . RISENG LD AA TLEEDE.............
YGG G P R V Q L L V GR G LD A YGG G P A R A Q L A V GR G LD A YGG G P R AVQ L I V GR G LD A
YGG G P RRV Q L L V GR G LD A
...MA Y PGDLL ERV QR EIPEI P APF TGRH RL E E . W Y V I R DGPV ERLRTY G TVK V E V A F K GK KV EW D K LA D N F A H LR VG KI
D A L P VK E P W Y R AS R Y G IK V E F K GK KV EW D K LA D D F AA H R IG KV D A L P VK E P W Y R AS R Y G G TVK I A F K GK KV D D K LA D D F H R VG KI
D A L P VK E P W Y R AS R Y G
D A L P VK E P W Y R AS R Y G .MP V DQHAVTK V V KT L QM I TAKF Y P . V C IL L H SPA SIT MTRATS DQH ATKSI H KS V S L LGVN V P . T LA L F .PA AFK G TVK V E V A K GK W D K LA D D Y A LR VG
G TVK V E V A F K GK KV EW D K LA D N F A H LR VG KI
MAT K SPH F K Y AH RS IEL L T I TGKL Y P . I MA KV GGL AFRR
.MP V NQQ F R L A KS L V T LAKH Y EN. T TV L GGA SMT
D A L P VK E P W Y R AS R Y G G V V D I A F GK V D M D E F A H MR VG K .MP VS R AAQ F N Y S QRQ LE GYV I TSSGN P Q A G K VA I KQV KLN
TT 1 10 20 30 40 50 60 70
IL
80 90 100 110 120 130 140 150 80 90 100 110 120 130 140 150
YGG G P RHI Q LQ M V GR G LD A
η1 α1 η2 α2 α3
α5 β1 β2 α6
.MP V NQQ F R L A KS L V T LAKH Y EN. T TA L GGA SMT
TT
YGG G P R Q L V GR G LD A YGG G P R Q L V GR G LD A
R56QS59F
C
N
P47L
I60
L131R
G127EL64P
Rps19 P. abyssi Rps19 H. sapiensRps19 X. laevisRps19 D. melanogasterRps19 C. elegansRps19 A. thalianaRps19 S. cerevisiae
Rps19 P. abyssiRps19 H. sapiensRps19 X. laevisRps19 D. melanogasterRps19 C. elegansRps19 A. thaliana Rps19 S. cerevisiae
76787878787979
150143145154143140144
R62W/Q
(c)
(a)
G118G125
L129
V13
V16
P45
R54S57
R60V62
P. abyssi numbering
H. sapiens numbering
H. sapiens numbering
1 10 20 30 40 50 60 70
80 90 100 110 120 130 140 150
L18P
G120S
A61S/EL59
V15F
37
44
79
84
∆78-8376-81
P. abyssi numbering
∆9-147-12
GG
HOT SPOTGY
Y
Y S RRWEVKPLAD
L P
LLLL
DDDD
AAAA
AL
PPPP
PVK
VKVKVK
EEEEE
YYYYY
YYYYY
GGGG
GGY
W RRRRRR
SSSSSS
R
RR
RRR
WWWWWEVKD A
VK
I
V
I
VVTTV
YY
VLLV
AAA
A
L
LLLLL
I
V
V
V
YGGYGGYGGYGGYGGYGG G
GGGGG
PPPPPP
R
RRRRR
Q LQ LQ LQ LQ LQ L
V RV RV RV RV RV R
DD AD AD AD AD AG
A
AD RQ L VRYGG G P
(b)
W50W52R
R56Q
S59F
P47L
L131R
L64P
R62W/Q
L129
P45
R54
S57
R60
V62A61S/EL59
V15F
W50W52R
W51I58
R61
PPPPPPP
R101HR99
L18P
V13
V16
∆9-147-12
A57PV55
A57PV55
V
AAA
AAAAAA
AA
α4
C lass I
C lass II
GGGGGGG
G127QG125
α1
α2
α3
α4
α5
α6
β2
β1
A
Figure 1. RPS19 structure and sequence alignment. (a) Overall structure of RPS19 and point mutations found in DBA patients. Residues labeled ingreen and red correspond to structural residues (class I) and to solvent accessible residues (class II), respectively. Green and red numbers correspondto human numbering. Gray numbers correspond to P. abyssi numbering. Deletions encountered in DBA patients are shown in orange. Dashed linescorrespond to disordered loops in the structure. (b) The hot spot of mutations. Mutated residues found in and around the hot spot of mutation areshown as in (a). (c) Sequence alignment of RPS19 orthologs. RPS19 ortholog sequences have been retrieved and aligned using PipeAlign programsuite (31). Class I (boxed in green) and class II (boxed in red) residues are displayed. Deletions encountered in DBA patients are show as orangeboxes. Secondary sequence elements are shown on top of the sequence.
Nucleic Acids Research, 2007, Vol. 35, No. 17 5915
75

structure where it bridges a-helices 1 and 6 on one sidewith a-helices 4 and 5 on the other side (Figure 1a).The apolar side of a-helix 3 is engaged into hydrophobicinteractions with residues of the neighboring a-helices,thus forming the hydrophobic core of the protein(Figure 1b). Strikingly, three mutations on a-helix 3(A57PHs, A61S/EHs, L64PHs), two on a-helix 1 (V15FHs
and L18PHs) and two on a-helix 6 (G127EHs and
L131RHs) affect residues involved in this hydrophobiccore (green residues on Figure 1). Thus, although distanton the primary sequence, these seven amino acids arefunctionally related and are involved in the folding andthe stability of the protein.
In contrast, mutations P47LHs, W52RHs, R56QHs,S59FHs, R62W/QHs, R101HHs and G120SHs affect resi-dues located on the surface of RPS19 (red residues onFigure 1). Remarkably, these residues show a much higherdegree of inter-species conservation than the amino acidswithin the hydrophobic core (Figure 1c). These residuesare located within two highly conserved basic patchesat the surface of the protein (Figure 2). On the polar faceof a-helix 3, four exposed mutated residues (W52Hs,R56Hs, S59Hs and R62Hs) form the floor of a central basicgroove (patch A on Figure 2a–c). In addition, residuesfrom a-helices 4 and 5 and from the b-sheet definethe conserved patch B (Figure 2d–f). It comprisesresidues R99Pa (R101Hs), K100Pa (R102Hs), Q103Pa
(Q105Hs), K113Pa (K115Hs), G118Pa (G120Hs) andR119Pa (R121Hs). DBA mutations R101HHs andG120SHs affect this conserved surface. Interestingly, bothconserved surfaces have a positive electrostatic charge:patch A harbors a localized positive charge (Figure 2c),whereas patch B is embedded in a larger positive area(Figure 2f).
Based on these observations, we propose to subdivideDBA mutations into two classes: class I encompasses
R56Q
S59F
R62W/Q
R54
S57
R60
W50W52R
R56Q
S59F
C
N
R62W/Q
(a)
G118
R54
S57
R60
G120S
R101HR99
(b)
W50W52R
G118G120S
R101HR99
G120S
(c)
(d)
R121R119
Q105Q103
90˚
C
0 100conservation
90˚ 90˚
(e) (f)
Patch A (hot spot)
R56Q
S59FR54
S57
W50W52R
R101HR99
R121R119
Q105Q103
R62W/QR60
R102K100
K115K113
K115K113
H63R61
H91R89
(−) (+)charge
R101H
Patch A (hot spot)
Patch BPatch B
R99
G118
Figure 2. Inter-species conservation and electrostatic surface properties of RPS19. Panels a–c and panels d–f are presented under the sameorientation, respectively. (a) and (d) Ribbon diagrams of RPS19. (b) and (e) Surface residue conservation of RPS19. Surface conservation wascalculated based on the sequence alignment shown in Figure 1 and using ConSurf server with default parameters (32). Conservation is displayedaccording to white (non-conserved) to purple (100% conservation). Conserved regions are circled in black. Numbering follows the same rule as inFigure 1. (c) and (f) Electrostatic charge distribution on the surface of RPS19 was calculated using APBS program default parameters (33). Highlyconserved and charged areas are boxed.
Table 2. Amino acid correspondence in Human, yeast and P. abyssi
P, abyssi Yeast Human DBA mutation Class
V13 I15 V15 V15F IV16 Y18 L18 L18P IP45 P47 P47 P47L IIW50 W53 W52 W52R IIR54 R57 R56 R56Q IIV55 A58 A57 A57P IS57 S60 S59 S59F IIL59 A62 A61 A61S/E IR60 R63 R62 R62W/Q IIV62 I65 L64 L64P IR99 R102 R101 R101H IIG118 G121 G120 G120S IIR119 R122 R120 -G125 G128 G127 G127E IS127 R130 R129 -L129 L132 L131 L131R IK131 R134 R133 -
5916 Nucleic Acids Research, 2007, Vol. 35, No. 17
76

structural residues affecting the folding of the protein;class II mutations affect surface residues and would impairthe function of RPS19 without altering its overall fold.It should be noted that the Val13Pa (V15FHs) and W50Pa
(W52RHs) are partially involved in Van der Waalscontacts with neighboring atoms and are also partiallyexposed to the solvent. Class I mutations affecting proteinfolding are predicted to render the protein unstable.Indeed, when expressed in COS cells, mutant RPS19V15FHs and G127EHs were shown to be present at lowlevel, in contrast to class II mutants R56QHs, R62WHs
whose level was similar to that of wild-type RPS19 (24,25).Along this line, a recent report confirms and extendsthese results by showing that class I mutants V15FHs,L18PHs, A57PHs, A61S/EHs, G127EHs and L131PHs arerapidly degraded by the proteasome when expressed inmammalian cells, whereas class II mutants P47LHs,W52RHs, R56QHs, R62WHs, R62QHs, R101HHs, areG120SHs are more stable (26). Thus, the classificationproposed here perfectly correlates with the instabilityobserved for class I mutants expressed in cultured cells.
The conserved basic patches are critical for RPS19 function
In order to test whether the conserved surface areas areessential for the function RPS19, we performed functionalcomplementation in yeast. RPS19 is encoded by twogenes, RPS19A and RPS19B, in S. cerevisiae. DBA-likemutations, as well as systematic R to E mutations thatalter the charge of these basic conserved areas, wereintroduced by site-directed mutagenesis into the RPS19Agene. The ability of these mutant alleles to support growthwas evaluated in a rps19A� rps19B� mutant strainconditionally expressing the wild-type RPS19A undercontrol of a galactose promoter. As shown in Figure 3,
DBA mutations R57QSc, R63QSc and R102HSc hada drastic effect on RPS19 capacity to support cellgrowth in glucose, whereas mutation R63WSc partiallysupported growth. Similarly, mutation to glutamic acid ofR57Sc, S60Sc and R63Sc in patch A and R102Sc, R122Sc inpatch B, completely abolished cell growth. In contrast, thedouble mutation R130E-R134E, outside of the conservedpatches, had little impact on viability. These results showthe crucial role of basic patches A and B in RPS19function.As already observed with this assay (23), among class I
mutations, I65PSc (L64PHs) was not viable, I15FSc
(V15FHs) strongly affected RPS19 function, whereassubstitution A62SSc (A61SHs) had little impact on cellgrowth, which was also the case for class II mutationG121SSc (G120SHs). Although affecting highly conservedresidues, these two latter mutations may have more criticaleffects in human than in yeast.
Requirement of patches A and B for RPS19 incorporationinto pre-ribosomes
In order to further characterize the loss-of-functionassociated with mutation of surface residues, we testedwhether class II mutations affected RPS19 incorporationinto pre-ribosomes. We thus analysed association ofRPS19 mutants with pre-ribosomal RNAs in yeast. TAPtagged wild-type and mutant alleles of RPS19A wereexpressed in place of the endogenous RPS19A gene in arps19A�/RPS19Bþ strain. Production of RPS19 in thisstrain is insufficient and pre-rRNA processing is partiallydefective (5), which mimics RPS19 haploinsufficiency inDBA patients. Using the complementation assaydescribed in Figure 3, we checked that the RPS19-TAPprotein remained functional (data not shown).
WT R57Q R57E R63EA62S R63W R63Q R102E R122E R102H G121SI15F I65P S60E_
GAL
GLC
Conserved Patch A (mutation hot spot) Conserved Patch B
Class II mutationsClass I mutations
R130E-R134E
Figure 3. Effect of mutations on RPS19 function in yeast. Capacity of various RPS19 mutants to complement RPS19 expression knockdown wastested in a yeast strain expressing wild-type RPS19 under control of a GAL promoter. Expression of wild-type RPS19 gene is shutdown upontransfer to glucose containing medium whereas transcription of the mutant forms is driven by a constitutive promoter. Cells were spotted at threedifferent densities.
Nucleic Acids Research, 2007, Vol. 35, No. 17 5917
77

When captured on IgG Sepharose, the wild-typeRPS19-TAP and the G121SSc mutant co-precipitatedwith the 18S and 25S rRNAs (Figure 4b, lower panel),indicating incorporation into mature ribosomes(not dissociated under our experimental conditions).Detection of the precursors of the 18S rRNA, by
northern blot analysis with the D-A2 probe, showedco-precipitation of the 20S pre-rRNA (Figure 4a, upperpanel), the direct precursor to the 18S rRNA (Figure 4b).This indicates the presence of these proteins in pre-40Sparticles. The 32S and 35S pre-rRNAs, which are part ofthe early 90S pre-ribosomes, were undetectable or close to
WT WTR57Q
R57ER63E
TAPI15F
R102E G121SR122E
20S
32S35S
20S
32S35S
20S
32S35S
20S
32S35S
INPUT
I. P.
PROBE: D-A2
WT R57QR57E
R63EI15F R102E G121S
R122E
RPS19-T AP(INPUT)
TAP
WTR57Q
R57ER63E
TAP WTI15F
R102E G121SR122E
25S18S
25S18S
25S18S
25S18S
WTR57E
R63ER102E
R122ETAP
WTR57E
R63ER102E
R122ETAP
21S
32S35S
25S18S
27S-A2
PROBE: A2-A3
21S
32S35S
27S-A2
25S18S
18S
D
A0A1 A2A3 EC2 C1D
18S 25S5.8S5′ 3′
ITS1 ITS2ETS1 ETS2
A3 EC2
25S5.8S
B C1
35S
A2A3 EC2D
18S 25S5.8S
C132S
27S-A 220S
(c)
(a)
(b)
(d)
18S
21S EC2
25S5.8S
B C1 27S-A 3A2D
Pre-40S Pre-60S
90S pre-ribosomes
40S rib. subunit 60S rib. subunit
Pre-40S Pre-60S
rps19 −WT
60S rib. subunitNo maturation
A0A1 A2A3 EC2 C1D
18S 25S5.8S5′ 3′
ITS1 ITS2ETS1 ETS235S
A2A3 EC2D
18S 25S5.8S
C132S
90S pre-ribosomes
Figure 4. Co-immunoprecipitation of precursor and mature ribosomal RNAs with RPS19-TAP. (a) Schematic of pre-rRNA processing in wild-type(top panel) or rps19 deficient cells (lower panel). Letters above pre-rRNAs indicate cleavage sites. (b, c) TAP-tagged wild-type or mutated RPS19Aexpressed in rps19A�/RPS19B yeast cells were isolated with IgG Sepharose beads. Pre-rRNAs in the whole cell extract (‘Input’) and the isolatedmaterial (‘I.P.’) were analyzed by northern blot with probes complementary to segment D-A2 (panel B) or to segment A2-A3 (panel C). The mature18S and 25S rRNAs were detected by ethidium bromide staining. The three panels correspond to independent experiments. In panels A and C,expression of the sole TAP tag was used as a negative control (TAP). The precipitation background level was also evaluated by detecting the 27S-Bpre-rRNA with a probe complementary to the ITS2 (data not shown):50.1% was co-precipitated in panel A and C,50.3% in panel B, as measuredby phosphorimaging. The portion of co-precipitated 20S, 18S and 25S RNAs did not significantly exceed the background value in all panels, exceptfor the wild-type and G121S forms (1–3% of co-precipitation). (d) Levels of TAP-tagged proteins in inputs (corresponding to panel A) were analyzedby western blot with peroxidase anti-peroxidase antibody complexes, which bind to protein A.
5918 Nucleic Acids Research, 2007, Vol. 35, No. 17
78

background in the precipitated fractions, depending onthe experiments.
Mutations in basic patch A (R57ESc, R57QSc, R63ESc)or basic patch B (R102ESc, R122ESc) reduced theamount of co-precipitated mature 18S and 25S rRNAsto background level (Figure 4b and c, lower panel).A similar result was obtained with class I mutation I15FSc.In addition, these proteins did not co-purify with 20Spre-rRNA (Figure 4b). This result was not correlated withdifferences in protein expression levels of the variousmutants (Figure 4d). Cleavage of the 32S pre-rRNA in theearly 90S pre-ribosomal particles normally yields the 20S
and 27S-A2 pre-rRNAs, included in the pre-40S and pre-60S particles, respectively (Figure 4a, left panel).Deficiency in RPS19 was shown to block cleavage at siteA2 (5); alternative cleavage at site A3 produces the 21Sand 27S-A3 pre-rRNAs, as illustrated in figure 4a(right panel). We thus considered the hypothesisthat RPS19 mutants were incorporated into ill-maturedpre-40S particles containing 21S pre-rRNA instead of 20S.As expected, a significant increase in the amount of 21Spre-rRNA, paralleled by a drop in the 27S-A2 pre-rRNAlevel, was detected in cells expressing RPS19-TAP withmutations R57ESc, R63ESc, R102ESc or R122ESc
R122ER102E R130E-R134EWT
R57Q R57E S60E R63E
A62S G121SWT I15F
WT
5 10 15 20
5 10 15 20 5 10 15 20 5 10 15 20 5 10 15 20
5 10 15 20 5 10 15 20 5 10 15 20 5 10 15 20
5 10 15 20 5 10 15 20 5 10 15 20 5 10 15 20
5 10 15 20
TAP
TOP BOTTOMFRACTIONS
OD 262 nm
RPS19
80S
POLYSOMESFree 40S subunits
Free 60S subunits
(a)
(b)
(c)
Figure 5. Analysis of wild-type and mutant RPS19 association to ribosomes on sucrose gradient.Ribosomes from rps19A�/RPS19B yeast strainsexpressing wild-type or mutated forms of RPS19A fused to the TAP tag were separated on sucrose gradient as described in material and methods.RPS19-TAP was detected in gradient fractions on slot blots with peroxidase. Panels a, b and c correspond to three series of experiments. Experimentswere performed two or three times with similar results. Expression of the sole TAP tag was used as a negative control (TAP).
Nucleic Acids Research, 2007, Vol. 35, No. 17 5919
79

(Figure 4c, ‘input’). However, the 21S pre-rRNA did notco-precipitate with mutated RPS19-TAP (Figure 4c,‘I. P.’). Thus, mutation of the basic patches A and Bprevents incorporation of RPS19 into pre-ribosomalparticles.These data were fully consistent with the analysis of
RPS19 association with ribosomes on sucrose gradients.As shown in Figure 5a, the wild-type TAP-RPS19 proteinwas mostly present in the fractions containing the40S subunits, the 80S ribosomes and the polysomes,indicating very efficient incorporation into maturesubunits. In contrast, mutations in conserved patches Aand B resulted in a large amount of free protein at the topof the gradient, with no preferential associationwith mature ribosomes (Figure 5a,b). Noticeably, part ofthese mutated forms of RPS19 was present in high-densityfractions. The A62SSc and G121SSc mutants, as well asdouble mutant R130ESc/R134ESc (outside of the con-served basic patches), which all support growth in RPS19depleted yeast, showed an association profile very close tothat of wild-type RPS19 (Figure 5c). The I15FSc mutation,which does not totally abrogate RPS19 function in yeast,showed partial association with mature ribosomes(Figure 5c).These results indicate that the solvent accessible
residues in the mutation hot spot (patch A) or in basicpatch B play a critical role in the incorporation of RPS19into pre-ribosomes. This may involve directinteraction with pre-ribosomal RNA, as supported bythe positive charge of these domains, and/or withribosomal proteins.In conclusion, we propose that DBA missense muta-
tions primarily result in RPS19 haplodeficiency, byimpacting either its folding and its stability, or its capacityto engage intermolecular interactions. The DBA mutationhot spot appears as a key structural element of RPS19involved both in the constitution of the hydrophobic coreand in a highly conserved surface. Impairment ofribosome biogenesis resulting from RPS19 haplodeficiencymay have two direct consequences. First, a low rate ofsmall ribosomal subunit production may limit the celltranslation capacity (25). Second, alteration of ribosomebiogenesis, and subsequently of the nucleolus organization(7), may be perceived by the cell as a ‘nucleolar stress’and favor cell cycle arrest (27–29) These phenomena,alone or in combination, could prevent differentiation ofpro-erythroblasts (7,12,30). In addition, our data do notexclude a dominant negative effect of the missensemutants, upstream or aside of association with pre-ribosomes, especially in the case of the more stable classII mutants. Presence of some mutants in high-densityfractions of the sucrose gradient suggests that they may beengaged in multi-molecular complexes, different fromribosomes, and whose composition remains to be deter-mined. Establishing genotype/phenotype relationships hasproved difficult in DBA and this remains true whenconsidering the relative clinical impacts of class I and classII mutations. Availability of RPS19 crystal structureshould be decisive to design new strategies to understandRPS19 function and its role in erythropoiesis.
PDB code: PDB coordinates and structure factors havebeen deposited at the PDB under accession number 2v7f.
ACKNOWLEDGEMENTS
We are grateful to Marlene Faubladier, Guillaume Stahl,Valerie Choesmel and Isabelle Leger-Silvestre for scientificdiscussion and technical support, to Steven Ellis (Univ.Louisville, USA) for sharing yeast strains, and to the staffmembers of ID29 and ID14-4 beamlines at ESRF, France.This work was funded by the Agence Nationale pour laRecherche (RIBODBA project) and supported by post-doctoral fellowships from the Conseil Regionald’Aquitaine (L.A.G.) and INSERM (S.F.), and by aCNRS doctoral fellowship (A-H.A-T.). Funding to paythe Open Access publication charges for this article wasprovided by the Agence Nationale pour la Recherche(RIBODBA project).
Conflict of interest statement. None declared.
REFERENCES
1. Da Costa,L., Willig,T.N., Fixler,J., Mohandas,N. and Tchernia,G.(2001) Diamond-Blackfan anemia. Curr. Opin. Pediatr., 13, 10–15.
2. Willig,T.N., Gazda,H. and Sieff,C.A. (2000) Diamond-Blackfananemia. Curr. Opin. Hematol., 7, 85–94.
3. Gustavsson,P., Garelli,E., Draptchinskaia,N., Ball,S., Willig,T.N.,Tentler,D., Dianzani,I., Punnett,H.H., Shafer,F.E. et al. (1998)Identification of microdeletions spanning the Diamond-Blackfananemia locus on 19q13 and evidence for genetic heterogeneity.Am. J. Hum. Genet., 63, 1388–1395.
4. Draptchinskaia,N., Gustavsson,P., Andersson,B., Pettersson,M.,Willig,T.N., Dianzani,I., Ball,S., Tchernia,G., Klar,J. et al. (1999)The gene encoding ribosomal protein S19 is mutated inDiamond-Blackfan anaemia. Nat. Genet., 21, 169–175.
5. Leger-Silvestre,I., Caffrey,J.M., Dawaliby,R., Alvarez-Arias,D.A.,Gas,N., Bertolone,S.J., Gleizes,P.E. and Ellis,S.R. (2005) Specificrole for yeast homologs of the Diamond Blackfananemia-associated Rps19 protein in ribosome synthesis.J. Biol. Chem., 280, 38177–38185.
6. Matsson,H., Davey,E.J., Draptchinskaia,N., Hamaguchi,I.,Ooka,A., Leveen,P., Forsberg,E., Karlsson,S. and Dahl,N. (2004)Targeted disruption of the ribosomal protein S19 gene is lethal priorto implantation. Mol. Cell. Biol., 24, 4032–4037.
7. Choesmel,V., Bacqueville,D., Rouquette,J., Noaillac-Depeyre,J.,Fribourg,S., Cretien,A., Leblanc,T., Tchernia,G., Da Costa,L. et al.(2007) Impaired ribosome biogenesis in Diamond-Blackfan anemia.Blood, 109, 1275–1283.
8. Flygare,J., Aspesi,A., Bailey,J.C., Miyake,K., Caffrey,J.M.,Karlsson,S. and Ellis,S.R. (2007) Human RPS19, the gene mutatedin Diamond-Blackfan anemia, encodes a ribosomal proteinrequired for the maturation of 40S ribosomal subunits. Blood, 109,980–986.
9. Idol,R.A., Robledo,S., Du,H.Y., Crimmins,D.L., Wilson,D.B.,Ladenson,J.H., Bessler,M. and Mason,P.J. (2007) Cells depletedfor RPS19, a protein associated with Diamond Blackfan anemia,show defects in 18S ribosomal RNA synthesis and small ribosomalsubunit production. Blood Cells Mol. Dis., 39, 35–43.
10. Gazda,H.T., Grabowska,A., Merida-Long,L.B., Latawiec,E.,Schneider,H.E., Lipton,J.M., Vlachos,A., Atsidaftos,E., Ball,S.E.et al. (2006) Ribosomal protein S24 gene is mutated in Diamond-Blackfan anemia. Am. J. Hum. Genet., 79, 1110–1118.
11. Cmejla,R., Cmejlova,J., Handrkova,H., Petrak,J. andPospisilova,D. (2007) Ribosomal protein S17 gene (RPS17) ismutated in Diamond-Blackfan anemia. Hum. Mutat. (in press).
12. Ellis,S.R. and Massey,A.T. (2006) Diamond Blackfan anemia: aparadigm for a ribosome-based disease. Med. Hypotheses, 66,643–648.
5920 Nucleic Acids Research, 2007, Vol. 35, No. 17
80

13. Campagnoli,M.F., Garelli,E., Quarello,P., Carando,A., Varotto,S.,Nobili,B., Longoni,D., Pecile,V., Zecca,M. et al. (2004) Molecularbasis of Diamond-Blackfan anemia: new findings from the Italianregistry and a review of the literature. Haematologica, 89, 480–489.
14. Willig,T.N., Draptchinskaia,N., Dianzani,I., Ball,S., Niemeyer,C.,Ramenghi,U., Orfali,K., Gustavsson,P., Garelli,E. et al. (1999)Mutations in ribosomal protein S19 gene and Diamond Blackfananemia: wide variations in phenotypic expression. Blood, 94,4294–4306.
15. Gazda,H.T. and Sieff,C.A. (2006) Recent insights into thepathogenesis of Diamond-Blackfan anaemia. Br. J. Haematol., 135,149–157.
16. Kabsch,W. (1993) Automatic processing of rotation diffractiondata from crystals of initially unknown symmetry and cellconstants. J. Appl. Cryst., 26, 795–800.
17. Schneider,T.R. and Sheldrick,G.M. (2002) Substructure solutionwith SHELXD. Acta Crystallogr. D Biol. Crystallogr., 58,1772–1779.
18. De La Fortelle,E. and Bricogne,G. (1997) Maximum-likelihoodheavy-atom parameter refinement for multiple isomorphous repla-cement and multiwavelength anomalous diffraction methods.Methods Enzymol., 472, 472–494.
19. Cohen,S.X., Morris,R.J., Fernandez,F.J., Ben Jelloul,M.,Kakaris,M., Parthasarathy,V., Lamzin,V.S., Kleywegt,G.J. andPerrakis,A. (2004) Towards complete validated models in the nextgeneration of ARP/wARP. Acta Crystallogr. D Biol. Crystallogr.,60, 2222–2229.
20. Emsley,P. and Cowtan,K. (2004) Coot: model-building tools formolecular graphics. Acta Crystallogr, D Biol, Crystallogr., 60,2126–2132.
21. Winn,M.D., Isupov,M.N. and Murshudov,G.N. (2001) Use ofTLS parameters to model anisotropic displacements inmacromolecular refinement. Acta Crystallogr. D Biol. Crystallogr.,57, 122–133.
22. Dez,C., Noaillac-Depeyre,J., Caizergues-Ferrer,M. and Henry,Y.(2002) Naf1p, an essential nucleoplasmic factor specificallyrequired for accumulation of box H/ACA small nucleolar RNPs.Mol. Cell. Biol., 22, 7053–7065.
23. Leger-Silvestre,I., Milkereit,P., Ferreira-Cerca,S., Saveanu,C.,Rousselle,J.C., Choesmel,V., Guinefoleau,C., Gas,N. and
Gleizes,P.E. (2004) The ribosomal protein Rps15p is required fornuclear exit of the 40S subunit precursors in yeast. EMBO J., 23,2336–2347.
24. Da Costa,L., Tchernia,G., Gascard,P., Lo,A., Meerpohl,J.,Niemeyer,C., Chasis,J.A., Fixler,J. and Mohandas,N. (2003)Nucleolar localization of RPS19 protein in normal cells andmislocalization due to mutations in the nucleolar localization signalsin 2 Diamond-Blackfan anemia patients: potential insights intopathophysiology. Blood, 101, 5039–5045.
25. Cmejlova,J., Dolezalova,L., Pospisilova,D., Petrtylova,K., Petrak,J.and Cmejla,R. (2006) Translational efficiency in patients withDiamond-Blackfan anemia. Haematologica, 91, 1456–1464.
26. Angelini,M., Cannata,S., Mercaldo,V., Gibello,L., Santoro,C.,Dianzani,I. and Loreni,F. (2007) Missense mutationsassociated to Diamond-Blackfan Anemia affect the assembly ofribosomal protein S19 into the ribosome. Hum. Mol. Genet., 16,1720–1727.
27. Olson,M.O. (2004) Sensing cellular stress: another new function forthe nucleolus? Sci STKE, 2004, pe10.
28. Pestov,D.G., Strezoska,Z. and Lau,L.F. (2001) Evidence ofp53-dependent cross-talk between ribosome biogenesis and the cellcycle: effects of nucleolar protein Bop1 on G(1)/S transition. Mol.Cell. Biol., 21, 4246–4255.
29. Rubbi,C.P. and Milner,J. (2003) p53 is a chromatin accessibilityfactor for nucleotide excision repair of DNA damage. EMBO J., 22,975–986.
30. Flygare,J. and Karlsson,S. (2007) Diamond-Blackfan anemia:erythropoiesis lost in translation. Blood, 109, 3152–3154.
31. Plewniak,F., Bianchetti,L., Brelivet,Y., Carles,A., Chalmel,F.,Lecompte,O., Mochel,T., Moulinier,L., Muller,A. et al. (2003)PipeAlign: a new toolkit for protein family analysis. Nucleic AcidsRes., 31, 3829–3832.
32. Landau,M., Mayrose,I., Rosenberg,Y., Glaser,F., Martz,E.,Pupko,T. and Ben-Tal,N. (2005) ConSurf 2005: the projection ofevolutionary conservation scores of residues on protein structures.Nucleic Acids Res., 33, W299–W302.
33. Baker,N.A., Sept,D., Joseph,S., Holst,M.J. and McCammon,J.A.(2001) Electrostatics of nanosystems: application tomicrotubules and the ribosome. Proc Natl Acad. Sci. USA, 98,10037–10041.
Nucleic Acids Research, 2007, Vol. 35, No. 17 5921
81
Figure II-1: Structure secondaire de l'ARNr 18S chez Homo sapiens.L'ARNr 18S est constitué de 4 domaines distincts: le domaine 5' (1-662 nt), le domaine central (663-1195 nt), le domaine majeur 3' (1196-1698 nt) et le domaine mineur 3' (1699-1870 nt). La structure en tige boucle du domaine majeur 3' correspondant à la séquence nucléotidique primaire 1537-1598 de l'ARNr 18S est proposée pour interagir avec RPS19. Elle correspond à la portion encadrée du domaine majeur 3' et elle est utilisée ici pour tester l'interaction de l'ARNr 18S avec les formes sauvage et mutées de RPS19.
82

II. INTERACTION IN VITRO DE RPS19 AVEC L’ARN RIBOSOMIQUE
L’étude structure-fonction de RPS19 que nous avons réalisé a montré que les mutations
associées à l’ADB affectent son incorporation dans les pré-ribosomes, en particulier en
altérant des surfaces basiques. Une hypothèse envisageable est la perte d’interaction
avec l’ARN ribosomique. Pour examiner cette hypothèse, nous avons cherché à
caractériser l’interaction de RPS19 avec l’ARN in vitro. Suite à l’étude chez S.
cerevisiae de la protéine nucléolaire Nep1p, impliquée dans la maturation de l’ARNr
18S (Buchhaupt et al., 2006), il a été proposé que RPS19 interagisse avec la séquence
1537 à 1598 du 3’ domaine majeur de l’ARNr 18S qui se structure en tige boucle
(Figure II-1). Nous avons donc choisi de tester l’interaction de la protéine RPS19
humaine avec ce motif tige-boucle formée par les nucléotides 1537 à 1598 dans l’ARN
18S humain par des expériences de retard sur gel en vue d’analyser l’effet des
différentes mutations ADB dans l’interaction RPS19-ARNr 18S.
1. Etude in vitro de l’interaction RPS19 avec le motif 1537-1598 de l’ARNr 18S
Pour étudier in vitro l’interaction de RPS19 avec la tige boucle 1537-1598 de l’ARNr
18S, la protéine RPS19 de H. sapiens a été produite et purifiée chez E. coli par le
laboratoire de Sébastien Fribourg à Bordeaux (Figure II-2,A). Nous avons cloné la
séquence codant la tige-boucle dans un vecteur portant un promoteur T7. L’ARN a été
synthétisé par transcription in vitro et purifié sur colonne avant d’être marqué à son
extrémité 3’ au 32P.
83

WTS59
FR133E
R62QR56Q
K115EQ105E
R101H
70
55
15
27
35
100
60 20 6.7 2.2 60 20 6.7 2.2 060 20 6.7 2.2
160 16 1.6
Rps19p (pmol)
ARNt (pmol)
1 2 3 4 5 6 7 8 9 10 11 12 13
ARN
Rps19p-ARN
Figure II-2. Interaction Rps19p-ARN(A) Les formes recombinantes sauvage et mutées de RPS19 produites chez E. Coli. (B) Interaction de Rps19 sauvage avec le motif 1537-1598 de l'ARNr 18S. Nous avons utilisé des concentrations décroissantes en protéine RPS19 ( 60, 20, 6.7, 2.2 et 0 pmol) en présence de 2.5 fmol d'ARN marqué à des concentrations différentes en ARNt (160, 16 et 1.6 pmol) qui est le compétiteur des interactions non spécifiques. L'interaction ne donne des complexes qu'avec des très faibles quantités d'ARNt compétiteur.
B
A
84

Nous avons testé la capacité de RPS19 à se lier à l’ARN par EMSA (Electrophoretic
Mobility Shift Assay). Pour cela, 2.5 fmol de transcrits ARN marqué au 32P ont été
incubés à 25°C avec des quantités décroissantes de RPS19 (60, 20, 6.7 et 2.2 pmol).
L’ARNt est utilisé ici comme agent compétiteur des réactions de faible affinité et trois
quantités d’ARNt ont été testées. Les complexes RPS19-ARN ont été séparés sur gel de
polyacrilamide non dénaturant à 4°C.
L’expérience montre la formation d’un complexe RPS19-ARN, accompagné d’une
baisse de l’ARN libre, pour la quantité la plus élevée en protéine RPS19, soit 60 pmol
(Figure II-2,B, pistes 1, 5 et 9). Un complexe semble également se former pour 20 pmol
de protéines, mais seulement quand on utilise une faible quantité d’ARNt qui est l’agent
compétiteur (Figure II-2,B, piste 10). L’utilisation de 6.7 ou 2.2 pmol de protéines n’est
pas suffisante pour la formation de complexe RPS19-ARN (Figure II-2,B, pistes 3-4, 7-
8 et 11-12) et donne un résultat équivalent à l’échantillon ne contenant pas de protéine
(Figure II-2,B, piste 13). Cette expérience ne permet pas de conclure à une interaction
forte et spécifique de RPS19 avec la tige-boucle 1537-1598 de l’ARNr 18S.
L’absence de complexe observée dès la concentration de 20 pmol en protéine RPS19
pourrait être dûe en partie à la force du compétiteur. En effet, une forte affinité de
RPS19 pour l’ARNt déplacerait le complexe dans l’autre sens. Pour vérifier cette
hypothèse, nous avons répété l’expérience précédente avec différents compétiteurs :
ARN poly-A, ARN poly-C et ARN poly-U. Le résultat de cette expérience présenté
dans la Figure II-3 montre que la présence d’ARN poly-U (Figure II-3, pistes 10-12)
empêche la formation du complexe précédemment observé. Inversement, l’ARN poly-A
(Figure II-3, pistes 4-6) conduit à un résultat sensiblement équivalent à celui obtenu
avec l’ARNt. Quand à l’ARN poly-C (Figure II-3, pistes 7-9), il semble montrer un
effet intermédiaire.
85

1 2 3 4 5 6 7 8 9 10 11 12 13
ARNt ARN poly-A ARN poly-C ARN poly-U
60 20 6.7 60 20 6.760 20 6.760 20 6.7 0Rps19p (pmol)
ARN (160 pmol)
Rps19p-ARN
ARN
Figure II-3: Choix de compéti-teurs pour l'interaction Rps19p-ARN.Test de l'interaction Rps19p-ARN avec trois concentrations différentes en protéine Rps19 (60, 20 et 6.7 pmol) et 2.5 fmol d'ARN marqué en présence de 4 compétiteurs différents.L'ARN poly-U (pistes 10-12) montre une très grande affinité pour l'ARN. En effet, aucun complexe Rps19p-ARN ne se forme lorsque de l'ARN poly-U est utilisé comme compétiteur. Par contre, l'ARN poly-A (pistes 4-6) apparaît comme un bon compétiteur de l'interaction Rps19p-ARN.
160 16 1.6
60 20 6.7 60 20 6.7 60 20 6.7 2.2 0
1 2 3 4 5 6 7 8 9 10 11
RPS19-ARN poly-U
ARN poly-U
ARN poly-A (pmol)
RPS19 (pmol)
Figure II-4: Interaction RPS19-ARN poly-UTest de l'interaction Rps19p-ARN poly-U avec trois concentrations différentes en protéine Rps19 ( 60, 20 et 6.7 pmol). L'interaction est faite en présence de 2.5 fmol d'ARN poly-U marqué avec trois dilutions différentes d'ARN poly-A (160, 16 et 1.6 pmol) qui est utilisé comme compétiteur des interactions non spécifiques . L'ARN poly-U montre une affinité pour Rps19p. Cette affinité s'améliore lorsqu'on abaisse la quantité du compétiteur (pistes 4-10). En effet, un complexe Rps19p-ARN poly-U se forme avec une dimunition d'ARN poly-U libre (pistes 7-10) lorsqu'on utilise la faible quantité d'ARN poly-A compétiteur (1.6 pmol).
86

Ces données ne confirment pas l’hypothèse d’une forte affinité de RPS19 pour l’ARNt.
En revanche, elles suggèrent une plus forte affinité de RPS19 pour l’ARN poly-U. Nous
avons tiré partie de cette observation en prenant de l’ARN poly-U comme matrice pour
la liaison de RPS19. Nous avons donc examiné directement l’interaction de RPS19 avec
un oligo-nucléotide poly-U marqué au 32P en utilisant l’ARN poly-A comme agent
compétiteur (Figure II-4). Nous avons utilisé 2.5 fmol d’ARN poly-U marqué au 32P
afin de tester son interaction avec la protéine RPS19 utilisée à des dilutions
décroissantes (60, 20, 6.7 et 2.2 pmol) en présence de 1.6 pmol d’ARN poly-A. Comme
le montrent les expériences présentées dans la Figure II-4, un complexe RPS19-ARN
poly-U est formé pour des quantités de protéines plus faibles comparativement à celles
qui sont effectuées avec la tige-boucle 1537-1598, indiquant une meilleure affinité de
RPS19 pour l’ARN poly-U (Figure II-4, pistes 7-10). Dans la suite de ce travail, nous
avons donc choisi d’utiliser l’ARN poly-U comme substrat d’interaction.
2. Etude de l’effet des mutations ADB sur l’interaction de RPS19 avec l’ARN
poly-U
Nous avons vu qu’in vitro, RPS19 sauvage et la tige boucle 1537-1598 du domaine
majeur 3’ de l’ARNr 18S ne reconstituent pas efficacement le complexe RPS19-ARN,
ce qui ne permet pas de poursuivre le test d’interaction avec les formes mutées. Par
contre, il y a bien eu formation de complexe RPS19-ARN poly-U. Afin de mieux
analyser l’interaction de l’ARN avec les formes sauvage et mutées de RPS19, nous
avons utilisé de l’ARN poly-U marqué au 32P à la place de la tige boucle 1537-1598 du
3’ domaine majeur de l’ARNr 18S. On peut noter que l’ARN poly-U est généralement
87

A B
C D E
Rps19p-ARN Poly-U
Rps19p-ARN Poly-U
ARN Poly-U
ARN Poly-U
ARN Poly-U
Rps19p-ARN Poly-U
1 2 3 4 5 6 7 8 9 10 11 1 2 3 4 5 6 7 8 9 10 11
1 2 3 4 5 6 7 8 9 10 11 1 2 3 4 5 6 7 8 9 10 1 2 3 4 5 6 7 8
Rps19p ( ug)1 0.33 0.11 1 0.33 0.11 1 0.33 0.11 1 0 1 0.33 0.11 1 0.33 0.11 1 0.33 0.11 1 0Rps19p ( ug)
1 0.33 0.11 1 0.33 0.11 1 0Rps19p ( ug) 1 0.33 0.11 1 0.33 0.11 1 0.33 0.11 1 0 1 0.33 0.11 0.03 1 0.33 0.11 0.03 1 0
Figure II-5. Les mutations DBA n'altèrent pas la liaison de RPS19 à l'ARN poly-U.Des concentrations décroissantes ( 1, 0.33 et 0.11 ug ) de protéine Rps19 ont été utilisées pour tester l'interaction avec 2.5 fmol d'ARN poly-U en présence de 1.6 pmol d'ARN poly-A utilisé comme compétiteur. Les mutants R56Q (A), R62Q (B) et Q106E (D) montrent une affinité pour l'ARN poly-U qui peut être estimée équivalente à celle montrée par RPS19 sauvage. Par contre, les mutations R101H (B), K115E (C), R133E (D) et R121E (E) montrent une affinité qui apparaît plus forte pour l'ARN poly-U comparée à celle du sauvage. Le mutant S59F (A) intera-git également avec l'ARN poly-U même si il montre deux complexes superposés de taille différente.
WT R56Q S59F BSA WT R62Q R101H BSA
BSABSA BSAWT WTWTK115E Q106E R133E R121E
88

utilisé pour analyser l’interaction d’une protéine avec l’ARN lorsque aucune séquence
spécifique n’est connue.
Plusieurs formes mutées de RPS19 humaine ont été produites chez E. coli par le
laboratoire de Sébastien Fribourg (Figure II-2,A). Les mutations portaient sur des
résidus à la surface de l’hélice 3 formant le sillon central (patch basique 1 : S59F,
R56Q, R62Q) et des acides aminés du deuxième domaine basique (R101H, Q105E,
K115E, K121E). Une autre mutation concerne la lysine 133 (K133E) dans l’hélice C-
terminale. Nous avons analysé l’interaction de ces protéines avec 2.5 fmol d’ARN poly-
U en présence de 1.6 pmol d’ARN poly-A utilisé comme agent compétiteur des
interactions de faible affinité (Figure II-5).
L’expérience nous a montré qu’in vitro, des complexes RPS19-ARN se constituent
entre les formes sauvage et mutées de RPS19 et l’ARN poly-U. Contrairement à l’effet
attendu, aucune des mutations étudiées ici n’altère l’interaction de RPS19 avec l’ARN
(Figure II-5). Les mutants présentent une affinité pour l’ARN équivalente, voire
supérieure à celle de la forme sauvage dans la mesure où des complexes sont observés
pour de plus faibles quantités de protéine. En effet, les formes portant les mutations
R56Q (Figure II-5,A, pistes 4-6), R62Q (Figure II-5,B, pistes 4-6) et Q106E (Figure II-
5,D, pistes 4-6) montrent la même affinité pour l’ARN poly-U que la forme sauvage.
Par ailleurs, d’autres mutations semblent entrainer une plus grande affinité pour l’ARN
poly-U: c’est le cas des mutations S59F (Figure II-5,A, pistes 7-9), R121E (Figure II-
5,E, pistes 5-8), R101H (Figure II-5,B, pistes 7-9), K115E (Figure II-5,C, pistes 4-6) et
R133E (Figure II-5,D, pistes 7-9). La mutation S59F (Figure II-5,A, pistes 7-9)
constitue un cas particulier avec l’observation de deux complexes de taille relativement
différente. Ces données indiquent que les mutations ADB n’altèrent pas la capacité de
RPS19 à interagir avec l’ARN poly-U in vitro.
89

3. Conclusion
Dans ce travail, nous avons effectué une étude in vitro des interactions RPS19-ARN
avec des protéines recombinantes purifiées chez E. coli afin d’analyser l’effet des
mutations ADB sur cette activité. Dans ces conditions, nous avons montré l’existence
d’une interaction RPS19-ARN même si une conclusion sur l’effet qu’auraient les
différentes mutations n’a pu être rapportée.
En effet, la formation d’un complexe entre RPS19 et l’ARN poly-U confirme que la
protéine est capable de se lier à l’ARN. En revanche, le site potentiel de liaison proposé
par Buchhaupt et al. (2006) ne s’est pas révélé être un bon substrat in vitro. Outre la
nature hypothétique de ce domaine d’interaction, on peut noter qu’il ne présente pas de
segment d’uracyles répétées. Nos résultats n’excluent pas formellement que cette tige
boucle soit le site de liaison in vivo. L’intervention de chaperones, ou la nécessité d’une
structure particulière de l’ARN non reproduite in vitro, pourrait permettre une
interaction de plus forte affinité.
Les expériences chez la levure S. cerevisiae montrent que les mutations ADB
empêchent l’incorporation de Rps19p dans les particules pré-40S. Les résultats
d’interaction in vitro avec l’ARN ne mettent pas en évidence un impact direct des
mutations sur la liaison de RPS19 avec un ARN poly U.
En effet, les mutations R56Q et R62Q localisées dans le hot spot des mutations DBA
ainsi que la mutation R101H du patch basique B altèrent la croissance cellulaire chez S.
cerevisiae. D’après la structure de RPS19, ces résidus sont exposés au solvant et
seraient potentiellement impliqués dans les interactions de la protéine avec l’ARN. Il a
été également montré que ces mutations affectent l’incorporation de la protéine dans les
ribosomes matures. Avec ces trois mutations, nous nous attendions à une altération de
90

l’interaction avec l’ARN. De manière surprenante, les complexes formés après
l’interaction indiquent une affinité supérieure ou égale de ces dernières pour l’ARN
poly U comparativement à la forme non mutée.
In vivo, le complexe Rps19p-ARNr 18S fait partie d’un ensemble macromoléculaire
plus complexe. Dans ces conditions, la protéine Rps19p pourrait adopter une
conformation particulière pour interagir avec l’ARNr grâce à des partenaires protéiques
qui la structurent dans le ribosome. Par ailleurs, nous savons que la fonction de l’ARNr
18S est étroitement liée à son organisation générale. Dans ce cas, l’absence de tout
facteur peut avoir des conséquences sur l’interaction de la protéine avec l’ARNr.
L’interaction Rps19p-ARN peut être inhibée soit en destabilisant la structure de
l’ARNr, soit en empêchant son interaction avec des facteurs. Or, ces conditions ne sont
pas assurées in vitro, ce qui complique l’analyse.
La structure complète de l’ARNr 18S doit lui conférer une flexibilité suffisante pour
s’adapter à la structure des protéines ribosomiques et ainsi effectuer une interaction
correcte. In vitro, la séquence 1537-1598 qui code pour une boucle de l’ARNr 18S
pourrait ne pas suffire pour effectuer cette interaction. Ceci peut expliquer pourquoi
nous avons obtenu un complexe quelque soit la mutation analysée.
Par ces expériences, nous avons montré qu’il existe une affinité de la protéine RPS19
pour l’ARN, in vitro. En revanche, il n’a pas été possible pour nous de conclure quant à
l’effet des différentes mutations ADB sur l’interaction de la protéine avec l’ARN.
91

92

III. L’IMPORT NUCLEAIRE DE RPS19 EST-IL AFFECTE DANS
L’ANEMIE DE DIAMOND-BLACKFAN ?
La fonction de Rps19p dans la biogenèse des ribosomes est étroitement liée à sa
capacité d’incorporation dans les ribosomes. Comme la plupart des protéines
ribosomiques, Rps19p, une fois synthétisée dans le cytoplasme, doit être importée dans
le noyau pour s’associer aux pré-ribosomes en cours de maturation. Les protéines sont
importées dans le noyau de manière constitutive ou en réponse à un signal grâce à un
signal de localisation nucléaire (Nuclear Localization Sequence : NLS). Il existe
différents types de NLS avec des séquences consensus généralement faibles et
caractérisées par des acides aminés basiques. Les séquences NLS sont reconnues par
des importines qui servent de transporteur à travers le complexe de pore nucléaire. Une
mutation affectant cette séquence de localisation nucléaire, pourrait interférer avec la
localisation nucléaire de Rps19p. Cette protéine mutée, incapable d’import nucléaire
sera bloqué dans le cytoplasme, d’où un défaut d’incorporation dans la sous-unité
ribosomique, affectant toute la biogenèse du ribosome. Par ailleurs, des données
obtenues dans des cellules humaines suggéraient que certaines mutations pouvaient
altérer l'import nucléaire de la protéine Rps19 (Da Costa et al., 2003).
Il était donc nécessaire de trouver la séquence de localisation nucléaire de Rps19p pour
répondre à cette question. Pour cela, nous avons fusionné différents fragments de
Rps19p à la GFP (green fluorescent protein) pour observer la localisation nucléaire ou
cytoplasmique de ces derniers au microscope à fluorescence. C’est ainsi que nous avons
déterminé la région contenant la NLS de Rps19p. Parallèlement, nous avons analysé la
93

localisation cellulaire des protéines exprimant les différentes mutations ADB. Enfin,
grâce à différents mutants de caryophérines, nous avons cherché les importines qui
assurent l’import nucléaire de la protéine.
Dans cette étude, nous avons montré que la séquence de localisation nucléaire de
Rps19p est incluse dans la région codant les acides aminés 67 à 97. Ce domaine n'est
pas la cible de mutations ADB et, réciproquement, les différentes mutations liées à
l’ADB n’interfèrent pas avec la localisation nucléaire de la protéine. Le croisement de
ces données avec la structure cristalline obtenue dans l’étude précédente indique que la
NLS se trouve au niveau d’une boucle qui n'est pas bien structurée dans le cristal. Il
s’agit d’un des seuls exemples montrant la position d’une NLS dans une protéine
ribosomique
Nos travaux précisent également les déterminants moléculaires de l'adressage nucléaire
de Rps19p, en particulier les caryophérines mises en jeu.
94

Nuclear localization of Rps19p, a ribosomal protein linked to Diamond-
Blackfan Anemia
Almass-Houd Aguissa-Touré and Pierre-Emmanuel Gleizes
Laboratoire de Biologie Moléculaire Eucaryote, CNRS and Université Paul Sabatier, Toulouse, France
Correspondence to: Pierre-Emmanuel Gleizes
LBME – CNRS, 118 route de Narbonne, F-31062 Toulouse cedex
Phone : +33 561 335 926 / Fax : +33 561 335 886
95

96

ABSTRACT
The ribosomal protein RPS19 is mutated in Diamond Blackfan anemia, a rare congenital disease
affecting differentiation of erythroid progenitors. To get insight into the intracellular dynamics of this
protein, we studied Rps19p nuclear transport in Saccharomyces cerevisiae. Mutational analysis
indicated that amino acids 67 to 97 constitute the minimum domain sufficient to drive nuclear import
of GFP and contains a NLS in the context of the protein. However, deletions within this domain do not
abrogate Rps19p nuclear translocation, indicating redundant transport mechanisms. Consistent with
the structure of known NLSs, the 67-97 stretch mostly corresponds to a loosely folded loop, as
deduced from the crystal structure of Rps19p homolog, in P. abyssi, and contains a potential PY NLS.
Efficient nuclear translocation of Rps19p depends on karyopherin Kap121p/Pse1p and Kap104p, and
to some extent on Kap123p (karyopherin-ß3), but not on Kap95p (karyopherin-ß1). Consistently, the
67-97 domain of Rps19p interacts with Kap121p in a two-hybrid assay. Introduction in S. cerevisiae
Rps19p of DBA-like mutations does not alter nuclear translocation. These data suggest that nuclear
import of RPS19 is a robust process ensured by different mechanisms, which is unlikely to be
perturbed by missense point mutations. In addition, this work provides one of the few views of the
position of the NLS in the three dimensional structure of a ribosomal protein.
97

98

INTRODUCTION
Nuclear transport of macromolecules across the nuclear envelop is ensured by beta-karyopherins, a
family of soluble proteins comprising 13 members in yeast Saccharomyces cerevisiae (Pemberton and
Pascal, 2005). Beta-karyopherin involved in nuclear import, also called beta-importins, bind their
cargos either directly, or through the alpha-karyophenrin Kap60p. The versatility of karyopherin-betas
and possible combination with Kap60p leads to multiple nuclear import pathways. Karyopherins
Kap123p and Kap121p/Pse1p have been involved in nuclear import of ribosomal proteins in yeast. A
number of ribosomal proteins were shown to bind to these karyopherins in vitro (Rout et al., 1997;
Schlenstedt et al., 1997; Sydorskyy et al., 2003; Timney et al., 2006). Efficient nuclear localization of
reporter proteins fused to ribosomal protein NLS was shown to require Kap123p, which is the most
abundant karyopherin (Rout et al., 1997; Schlenstedt et al., 1997; Sydorskyy et al., 2003; Timney et
al., 2006). However, overexpression of Kap121p can complement deletion of the KAP123 gene. In
addition, mutation of beta-karyopherins Sxm1p/Kap108p, Nmd5p/Kap119p and Kap121p in a
kap123∆ background results in a synthetic fitness defect or synthetic lethality, suggesting that these
karyopherins partially overlap with Kap123p function (Seedorf and Silver, 1997; Sydorskyy et al.,
2003). Consistently, some ribosomal proteins were found to bind to Kap108p (Rosenblum et al.,
1997). Similarly, in mammalian cells, numerous importins can bind ribosomal proteins and mediate
their nuclear import in vitro (Antoine et al., 2005; Jakel and Gorlich, 1998).
The ribosomal protein RPS19 is mutated in 25% of the patients suffering Diamond-Blackfan anemia
(DBA) (Draptchinskaia et al., 1999). This rare congenital disease is characterized by the absence of
pro-erythroblasts in bone marrow due to the arrest of differentiation of erythroid progenitors (Flygare
and Karlsson, 2007; Gazda and Sieff, 2006). The recent identification of mutations in two other
ribosomal protein genes, RPS24 (Gazda et al., 2006) and RPS17 (Cmejla et al., 2007), in a fraction of
the patients has strengthened the hypothesis that this pathology is primarily due to a defect in
ribosome assembly and/or function. Indeed, cells derived from DBA patients have defects in
maturation of ribosomal RNA (Choesmel et al., 2007; Choesmel et al., 2008; Flygare et al., 2007; Idol
et al., 2007). The tissue-specificity of the pathology remains unexplained given the ubiquitous
99

function of ribosomes. Indeed, DBA patients frequently suffer additional developmental abnormalities
and often present a short stature.
Mutations in RPS19 in DBA are heterozygous and it is currently admitted that they result in
haploinsufficiency (Campagnoli et al., 2008; Willig et al., 1999). They include partial deletions of the
ORF, splicing site mutations, premature stop codons, but the most frequent mutations are missense
point mutations. Characterization of the missense mutants by overexpression in simian COS cells or in
human HeLa cells has shown that they split into two groups relative to their localization: some are
essentially detected in the cytoplasm, whereas other are detected in the nucleolus (Angelini et al.,
2007; Da Costa et al., 2003). Although ribosomal proteins are cytoplasmic proteins once associated to
mature ribosomal subunits, the vast majority of them are first imported into the nucleus after synthesis
for assembly into nascent pre-ribosomal particles in the nucleolus, where most of ribosome biogenesis
takes place. It has been proposed that the absence of some RPS19 mutants of the nucleus is due to
deficient nuclear import (Da Costa et al., 2003).
The number of nuclear localization sequences experimentally determined in ribosomal proteins
remains limited. The identified sequences do not correspond to "classical" monopartite or bipartite
NLSs made of basic amino-acid clusters: although they often include basic residues, they are
heterogenous in size and composition. A consensus sequence for yeast ribosomal NLSs was proposed
(Stuger et al., 2000; Timmers et al., 1999), but still lacks a strong experimental support. Here, we
have characterized an NLS in RPS19 from yeast Saccharomyces cerevisiae as well as transport
receptors involved in nuclear import of this protein. In addition, we have examined the impact of
mutations found in DBA patients on this transport process.
100

MATERIAL & METHODS
Yeast strains
The strains used for this work are described in table 1.
Construction of GFP reporter fusions
The entire 435-bp open reading frame of S. cerevisiae RPS19A without the intron found in genomic
DNA was produced from S. cerevisiae total DNA by PCR using the Phusion High-Fidelity DNA
polymerase (Finnzymes, New England Biolabs) and appropriate primers. The cDNA encoding
ribosomal protein RPS19 was produced from P. abyssi total DNA (kindly provided by Dr. Annie
Mougin, LBME, Toulouse, France). The PCR products were subcloned in frame downstream of the
GFP coding sequence into vector pFL38-GAL which contains a GAL1/10-CYC1 promoter (Leger-
Silvestre et al., 2004). Alternatively, the same cDNAs without stop codon were subcloned in frame at
the 5' extremity of the GFP cDNA into the same vector. In some case, the ribosomal protein gene
RPS15 promoter was used in place of the GAL1/10 promoter. Deletion constructs were generated by
PCR using appropriate primers and subcloned in frame upstream of the GFP coding sequence.
Mutation of various amino acids into alanine in the sequence encoding domain 67-97, deletions and
introduction of DBA mutations in RPS19A was performed using site-directed mutagenesis based on
two-step PCR (Barik, 1995). All constructs were verified by DNA sequencing (Sequencing facility of
the IFR30, INSERM U563–CPTP, CHU Purpan- Toulouse, France).
Fluorescence Microscopy
The wild-type BY4742 yeast strain, strains disrupted for RPS19A and strains encoding temperature-
sensitive importin mutants were transformed with plasmids encoding GFP fusion proteins. Cells
expressing GFP-tagged proteins under control of the GAL promoter were grown in selective media at
30 °C for 2- 3 days in the presence of glucose and subsequently shifted to 2% galactose containing
medium for 8 h at 30°C to induce expression of the fusion protein. Strains carrying a temperature-
sensitive mutation were grown at 37 °C for 3 hours before observation. The localization of the fusion
101
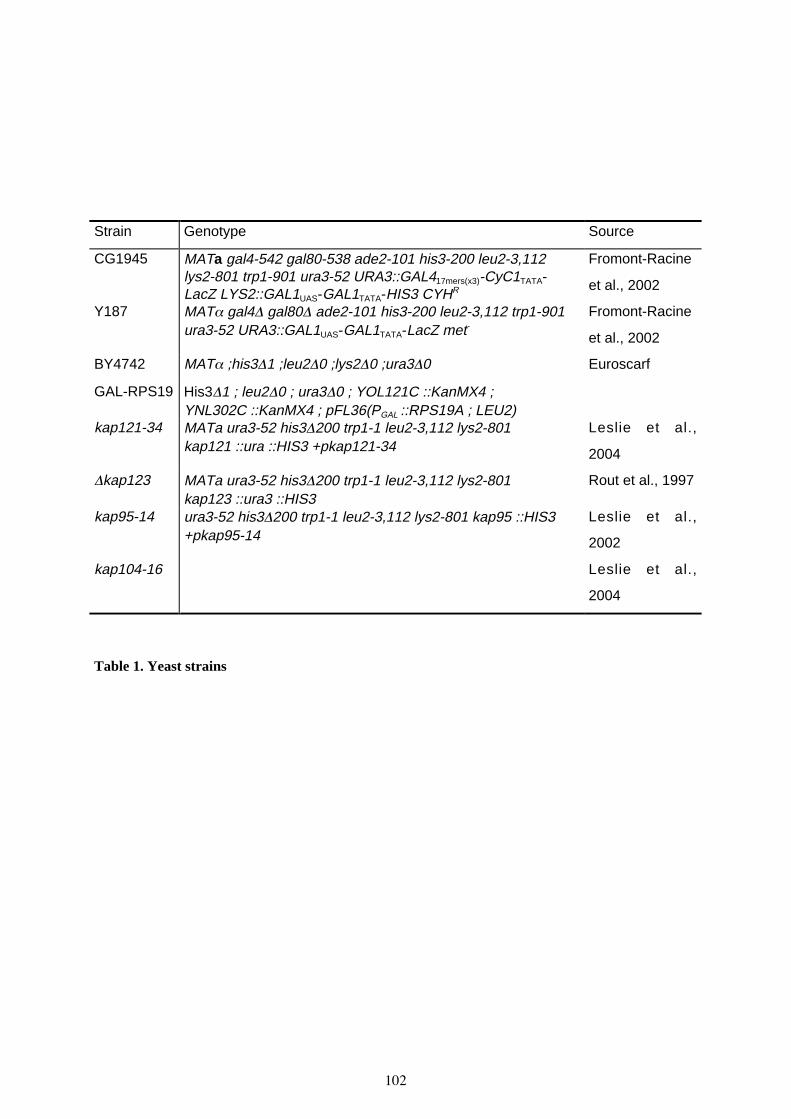
Strain Genotype Source
CG1945 MATa gal4-542 gal80-538 ade2-101 his3-200 leu2-3,112lys2-801 trp1-901 ura3-52 URA3::GAL417mers(x3)-CyC1TATA-LacZ LYS2::GAL1UAS-GAL1TATA-HIS3 CYHR
Fromont-Racine
et al., 2002Y187 MATα gal4Δ gal80Δ ade2-101 his3-200 leu2-3,112 trp1-901
ura3-52 URA3::GAL1UAS-GAL1TATA-LacZ met-Fromont-Racine
et al., 2002BY4742 MATα ;his3Δ1 ;leu2Δ0 ;lys2Δ0 ;ura3Δ0 Euroscarf
GAL-RPS19 His3Δ1 ; leu2Δ0 ; ura3Δ0 ; YOL121C ::KanMX4 ;YNL302C ::KanMX4 ; pFL36(PGAL ::RPS19A ; LEU2)
kap121-34 MATa ura3-52 his3Δ200 trp1-1 leu2-3,112 lys2-801kap121 ::ura ::HIS3 +pkap121-34
Leslie et al.,
2004Δkap123 MATa ura3-52 his3Δ200 trp1-1 leu2-3,112 lys2-801
kap123 ::ura3 ::HIS3Rout et al., 1997
kap95-14 ura3-52 his3Δ200 trp1-1 leu2-3,112 lys2-801 kap95 ::HIS3+pkap95-14
Leslie et al.,2002
kap104-16 Leslie et al.,2004
Table 1. Yeast strains
102

proteins was determined by fluorescence microscopy. Cellular DNA was stained for 30 min by
addition of 1 µg/ml 4',6'-diamidino-2-phenylindole (DAPI) to the cell suspension. The GFP chimeras
were visualized directly by fluorescence microscopy using the Metamorph 6.2 software (Universal
Imaging) and processed with Adobe Photoshop CS.
Two-hybrid interaction analysis
For the two-hybrid analysis, full length or truncated yeast RPS19, as well as its counterparts in H.
sapiens and P. abyssi, were cloned into pAS2ΔΔ (TRP1) in frame with the GAL4 DNA binding
domain and transformed into yeast strain CG1945. Fragments 1550-3270 of KAP121 ORF and 1670-
3147 of NMD5 ORF in frame with the GAL4 transactivation domain in the vector pACTIIst (LEU2)
were kindly provided by Dr. Fromont-Racine Micheline (Institut Pasteur, Paris, France) and
transformed in strain Y187. The two-hybrid procedure was performed as described (Fromont-Racine
et al., 1997).
Sequence analysis
Sequences of proteins of the RPS19 family were obtained from the EMBL/ESI InterPro database and
handled in JalView (Clamp et al., 2004).
103

WT GFP
N84∆ N97∆ N109∆
∆16C ∆53C ∆77C ∆90C
67-97 77-97
WT
∆16C
∆53C
∆77C
∆90C
N52∆
N84∆
N97∆
N109∆
67-97
77-97
+
+
+
+
+
+
-
-
-
-
Preferentialnuclear
localization
- (mitochondria)
A
B
H.s. 66-96
Figure 1. Determination of Rps19p NLS by mutational analysis. (A) Schematic overview of the Rps19p deletion mutants generated for this study. This proteins were fused to the C-terminus of GFP and expressed under control of the GAL promoter. (B) Intracellular localization of Rps19p deletion mutants fused to GFP. Cells were grown overnight with 2% galactose in YNB medium before observation. Domains 67-97 in yeast Rps19p and 66-96 in human RPS19 (H.s. 66-96) were sufficient to drive GFP to the nucleus.
104

RESULTS
The 67-97 domain from yeast Rps19Ap is sufficient to drive nuclear import of GFP
The NLS of Rps19 was determined by sequential deletions in the Rps19 fusion protein, starting from
the N- or the C- terminus of Rps19 (Figure 1A). As shown in figure 1, deletion of the N terminal
domain up to amino-acid 77 did not abrogate nuclear localization, with exception of the ∆53C
construct. The ∆53C protein was detected in elongated cytoplasmic structures stained with DAPI (not
shown) that we identified as mitochondria. Indeed, the amphipatic helix starting at position 53 is
predicted to be a strong mitochondrial import sequence if located at the very N-terminus of the protein
(http://ihg.gsf.de/ihg/mitoprot.html) (Claros and Vincens, 1996). Further deletion of the N-terminal
domain up to residue 77 restored nuclear localization. However, deletion of 84 amino-acids resulted in
homogenous distribution of Rps19 in the cell, indicating the loss of the NLS. Conversely, deletion
from the C-terminus retained nuclear localization up to amino-acid 97. From these data, we inferred
that the NLS was located between amino acids 77 and 97. However, the 77-97 domain was not
sufficient to promote nuclear localization of GFP. Extending the N-teminus to amino acid 67 (67-97)
restored NLS activity, whereas domain 77-109 was unable to drive GFP into the nucleus. Attempts to
reduce the size of the minimum sufficient domain 67-97 proved unsuccessful. Finally, we expressed
the homologous domain from human RPS19 (a.a. 66 to 96) fused to GFP in S. cerevisiae. As shown in
figure 2C, this domain was also sufficient to drive GFP accumulation in the nucleus. Thus, domain 67-
97 is the minimal domain from yeast Rps19p having an autonomous NLS activity and this property is
conserved in its human homolog.
Structure of the NLS in Rps19
The crystal structure of RPS19 homolog from the hyperthermophilic archeon Pyrococcus abyssi was
recently solved [Gregory, 2007]. Given the high similarity with yeast RPS19, it is reasonable to
assume that the two proteins adopt the same fold. We used this structure to generate a model of S.
cerevisiae RPS19 (Figure 2). This model is very close to the archeal structure with a shorter C-
105

S97
M67
N77
S97
M67
N77
N
C
120°
NLS
NLS
Figure 2. Position of the NLS containing domain in Rps19p. A model of S. cerevisiae Rps19p was generated according to the structure of its homolog in Pyrococcus abyssi using Swiss-Model (http://swissmodel.expasy .org/). The 67-97 domain corresponds to a loop-helix-loop motif colored in pink. The 77-97 segment is depicted in dark pink. Amino-acids targeted by misense mutations in DBA are colored in blue.
106

terminal alpha helix. As shown on figure 2, the domain 77-97 mostly corresponds to a loop (loop 3 in
P. abyssi RPS19) whose structure is partially unresolved is the crystal structure, which indicates a high
flexibility. This loop is flanked by two alpha-helices. In addition to this loop (a.a. 81 to 95), the
minimal domain driving GFP into the nucleus (a.a. 67 to 97) includes a short N-terminal loop and an
alpha-helix (a.a. 73 to 80). It contains several lysines and arginines, but there is no cluster of basic
amino acids resembling a classical NLS. Most importantly, this domain is exposed at the surface of
the protein and is accessible to karyopherins.
Functional dissection of Rps19p NLS
To identify residues involved in nuclear tranport, we postulated that these amino acids would be more
conserved in eukaryotes than in archeons. As shown in Figure 3A, sequence alignment of domains
homologous to Rps19p 67-97 shows similar identity scores in eukaryotes and archaeons and the
consensus sequences in both kingdoms are very close. However, similarity is much higher in
eukaryotes when compared to archeaons. More specifically, four segments within domain 67-97
display higher conservation in eukaryotes (figure 3A and 3B). Interestingly, segments III and IV
contain a putative PY NLS (Lee et al., 2006), a sequence proposed to be a consensus NLS for
transportin in vertebrates and recently defined as a Kap104p interacting sequence in yeast (K/R/HX2-
5PY) (Lange et al., 2008; Lee et al., 2006). It is of note however that tyrosine 91 in this motif is not
conserved at this position in most other eukaryotes (Figure 3A and 3C).
We examined the involvement of these conserved amino acids in nuclear transport by point-
mutagenesis of the 67-97 domain fused to GFP (figure 3B). Mutation of arginine 68 (segment I) or
triple substitution of leucine 76, lysine 78 and leucine 79 (segment II) to alanine affected nuclear
accumulation of the reporter protein, but the nuclear signal remained higher than in the cytoplasm. In
contrast, substitution of arginine 84 and lysine 86 (segment III) abrogated nuclear accumulation,
indicating an essential role of this segment in nuclear import. Interestingly, these two basic residues
are present in P. abyssi RPS19, whereas segments I, II and IV are not conserved relative to S.
cerevisiae (Figure 3C). When wild-type full-length P. abyssi RPS19 was expressed in yeast, it showed
no preferential localization in the nucleus, indicating that it contains no functional NLS (Figure 3C).
107

.
MRKQVGVGKLNKLYGGAKSRGVRPYKHIDAS +MAKQVGVGKLNKLYGGAKSRGVRPYKHIDAS +/-MRKQVGVGKANAAYGGAKSRGVRPYKHIDAS +/-MRKQVGVGKLNKLYGGAASAGVRPYKHIDAS -
Similarity
Consensus
4+ 0 5 5+ 9 5 4 7 4 9 9 7 8 7 4 7 6 7 * 4 2+ 2 9 7 3 3 8 5
L R G+V GV G S L R K I Y GGR K R NGV R P S H F C R A S
2 0 4 2 7 * 9 4 2 4 6 4 1 * 8 * 3 3 4 5 * 2 3 3 2 3 2 1 4 8 6
I DGP V G I E R L R S V Y GGR K NR G S+ P E H F+ K G S
Archaea Eukaryotes
I II III IV I II III IV
nuclearlocalizationI II III IV
67 . . 97
70%
77
67-97 R68A L76A, K78A, L79A R84A, K86A
R68A
L76A, K78A, L79A
R84A, K86A
67-97
A
B
C
RPS19Sc RPS19ePa RPS19ePa (88-95)Sc
MRKQVGVGKLNKLYGGAKSRGVRPYKHIDAS LRGGAGVGSMTKIYGGRQRNGVMPSHFSRGSLDGPVGIERLRTYYGGRKNRGHAPERFYKAG
I II III IV
67 97S. cerevisiae
H. sapiens
P. abyssi
66
64
96
94
Figure 3. Functional dissection of domain 67-97. (A) Domains corresponding to amino-acids 67-97 in yeast Rps19p in eukaryotic and archeal homologs were aligned and consensus sequences were determined using WebLogo (Crooks et al., 2004) or JalView (Clamp et al., 2004). Functional conservation (similarity) was determined with JalView. Boxes indicate segments I to IV with higher conservation in eukaryotes than in prokaryotes (B) Point-mutation analysis of residues in segments I, II and III. Amino acids in these segments were substituted to alanines in the 67-97 domain fused to GFP and expressed in yeast under control of the RPS15 promoter. Residues corresponding to a putative PY NS are underlined. (C) Exchange of amino-acids 85-92 in full-length P. abyssi RPS19 with corresponding amino-acids 88-95 from S. cerevisiae Rps19p results in nuclear localization of the prokaryotic protein. All proteins in (C) are expressed under control of the GAL promoter.
108

WFYKRAASVARHIYMRKQVGVGKLNKLYGGAKSRGVRPYKHIDASGSINRK
∆(75-80) ∆(79-84) ∆(86-91) ∆(91-96)
S. cerevisiae Rps19p67 97
A
B
..
79-84 86-9191-96
75-80 84-90
RPS19Sc
∆(84-90)
Figure 4. Deletions in domain 67-97 do not affect nuclear localization of Rps19p. (A) Several deletions of 5-6 amino acids were introduced in yeast full-length Rps19p fused to GFP. (B) None of the deletions affected nuclear accumulation of the protein. All proteins
were expressed under control of the GAL promoter.
109

kap123∆ kap95-14 ts
37°C 25°C 37°CBY474225°C
pse1-1 ts
25°C 37°Ckap104-16 ts
25°C 37°C
Rps19-GFP
Nop1-GFP
25°C
Rps19-GFP
Figure 5. Karyopherins required for Rps19p nuclear import. Rps19-GFP was expressed in strains with constitutive (kap123∆) or thermosensitive loss-of-function of various karyopherins under control of the RPS15 promoter. Cells were observed at 25°C or after a 3 hour shift to 37°C. Nop1-GFP, a Kap121p/Pse1p substrate, is shown as a control.
110

We took advantage of this by replacing amino acids 86-93 in P. abyssi RPS19, which include segment
IV, by the corresponding 88-95 domain from S. cerevisiae, thus creating a loop very homologous to
loop 81-95 in S. cerevisiae. The hybrid protein concentrated in the nucleus, and more specifically in
the nucleolus (Figure 3C). These results demonstrate that loop 81-95 contains an NLS activity in the
context of the protein and indicate that amino acids 84 to 95, which include a potential PY NLS, are
crucial for this activity.
We next examined whether amino acids in loop 81-95 were strictly necessary for Rps19p nuclear
translocation. Several deletions of 5-6 contiguous amino-acids were introduced in Rps19p, spanning
domain 75-96 (Figure 4A). However, none of these deletions abrogated nuclear localization, including
those affecting arginine 84 and lysine 86 (Figure 4B). Thus, although the results above indicate that
this loop is crucial for the autonomous NLS activity of the 67-97 domain and is sufficient to drive
nuclear transport of a prokaryotic homolog in yeast, it is not strictly necessary for Rps19p nuclear
import. This result strongly suggests that translocation of Rps19p to the nucleus is ensured by several
mechanisms.
Nuclear import of Rps19 requires karyopherins Kap104p and Kap121p
Nuclear import of several ribosomal proteins or reporter proteins fused to ribosomal NLSs was shown
to primarily depend on Kap123p and Kap121p/Pse1p. To get some insight into the kayopherins
involved in Rps19p tranport, we expressed the Rps19-GFP fusion protein in cells bearing mutated
alleles of KAP95, KAP104, KAP121, and KAP123. As shown in figure 5, deletion of KAP123 did not
suppress nuclear localization of Rps19-GFP, although more signal was present in the cytoplasm. This
result was expected as Kap123p is not an essential protein and can be functionally substituted by other
karyopherin like Kap121p. However, in strains bearing KAP121 thermosensitive alleles pse1-1
(Seedorf and Silver, 1997) or kap121-34 (Leslie et al., 2004), Rps19-GFP did not accumulate in the
nucleus under nonpermissive conditions (37°C). In contrast, nuclear signal persisted in kap95-14 cells
(Leslie et al., 2002). The presence of a putative PY NLS also prompted us to test the involvement of
Kap104p. Interestingly, nuclear transport of Rps19p was also strongly affected at 37°C in
thermosensitive kap104-16 mutant cells. As a control, we checked that nuclear import of Nop1-GFP,
111

Sc RPS19
- Leu - Leu/Trp/His
∆77C
N97∆
- Trp
121PAK5DMN
rotcev_ 121PAK
5DMNrotcev
121PAK5DMN
rotcev_ _
_
Pa S19e / (88-95)Sc
Pa S19e
Hs RPS19
N84∆
∆90C
67-97
77-97
_ _ _121PAK
121PAK
121PAKrotcev
rotcevrotcev
Sc RPS19
A
B
Figure 6. Two-hybrid interaction of Rps19p with Kap121p/Pse1p. Full-length or truncated versions of yeast RPS19A, human RPS19, or P. abyssi RPS19 were cloned in frame with GAL4 DNA binding domain in vector pAS2∆∆ (TRP1). NMD5 and KAP121 ORFs truncated of a large 5' part were fused with GAL4 transactivation domain in pACTII (LEU2). Interaction of a karyopherin with a version of RPS19 activates of the GAL4 promoter, which drives transcription of the reporter HIS3 gene.
112

which is mediated by Kap121p/Pse1p, was not affected in the kap104-16 strain; as expected, it was
impaired in pse1-1 cells. These data indicate that unlike previously described for other ribosomal
proteins, Kap121p is necessary for nuclear import of Rps19p, even in the presence of Kap123p. In
addition, efficient nuclear import of Rps19p also involves Kap104p.
Interaction of Rps19 with Kap121p involves domain 67-97
We next examined the interaction between Rps19 and karyopherin Kap121p with the two-hybrid
system. The RPS19 full-length or partial ORF and part of the KAP121 ORF were fused downstream
the GAL4 DNA binding domain and the GAL4 DNA activating domain, respectively. The two fusion
genes were introduced in separate his3∆ strains of different mating type. Diploids resulting from
crossing the two strains were tested on histidine-depleted medium, the HIS3 gene being under control
of the GAL4 promoter. This truncated allele of KAP121 misses the 5' part encoding the first 515
amino acids of the protein. It was initially isolated in a two-hybrid screen performed with ribosomal
protein Rps15p as a bait (M. Fromont-Racine, P-E Gleizes, unpublished results). By analogy with
importin-beta1 and transportin, the N-terminal part of Kap121p is predicted to be essential for binding
of Ran-GTP, whereas the 574 C-terminal residues left in the construct should rather be involved in
NLS binding. Consistent with the requirement of Kap121p for nuclear localization of Rps19p-GFP,
Rps19p and Kap121p were found to interact in this system (Figure 6). In contrast, Pyroccocus abyssi
RPS19 did not interact with Kap121p, consistent with the absence of NLS in this protein. Binding to
Kap121p was specific since Rps19p did not interact with the C-terminal 492 amino acids of
karyopherin Nmd5p. Interaction of Rps19p N-terminal and C-terminal deletion mutants with Kap121p
in this assay was correlated with their NLS activity. Hence, deletion mutants ∆77C and N97∆
promoted transcription of the HIS3 gene, whereas mutant ∆90C could not. As an exception, the N84∆
deletion mutant, which does not efficiently drive GFP into the nucleus, appeared to interact with
Kap121p. Finally, the 67-97 domain, but not the 77-97 segment, yielded a positive signal. In addition,
interaction with Kap121p was also observed for P. abyssi RPS19 after substitution with amino-acids
88-95 from S. cerevisiae Rps19p (Figure 6B), consistent with its nuclear localization (see Figure 3C).
113

WT I15F A62S R63W
R63Q G121S G128Q ∆(79-84)
Figure 7. DBA mutations have no major impact on Rps19p nuclear transport. Yeast Rps19p fused to GFP and containing DBA-like muta-tions was expressed under control of the GAL promoter in wild-type yeast.
114

These data are fully consistent with the presence of a binding site for Kap121p within domain 67-97 of
S. cerevisiae Rps19p and strongly support the hypothesis that this sequence has an NLS activity.
Rps19 missense or deletion mutations found in DBA patients do not alter nuclear localization
No point missense mutations have been observed in this domain in DBA. However, Cmejlova et al.
recently reported the deletion of amino acids 78-83 in a DBA patient and found no alteration of the
mutant protein nuclear localization. This domain is made of the most conserved amino-acid stretch in
RPS19.
The impact of DBA related missense mutations on nuclear localization was examined by reproducing
these mutations in the S. cerevisiae RPS19A gene fused to the GFP cDNA. These mutations affected
the N-terminus (I15F), the mutation hot-spot (A62S, R63W, R63Q), or the C-terminal helix (G121S,
G128Q). The I15F, A62S, G121S and G128Q mutations affected amino acids buried in the protein
and likely to be involved in Rps19p folding and stability, whereas Arginine 63 is located in a
conserved basic groove at the surface of the protein (Gregory et al., 2007). The mutant constructs were
introduced in wild-type yeast cells and observed by fluorescence microscopy. As shown in figure 7, all
these mutants showed a clear nuclear localization similar to the wild-type protein. Thus, none of these
DBA missense mutations strongly affects nuclear import. This is consistent with the observation that
expression of mutants I15F, A62S, R63W or G121S partially complement deletion of the two genes
encoding Rps19p in yeast (Gregory et al., 2007; Leger-Silvestre et al., 2005).
Finally, an in-frame deletion leading to the absence of amino-acids 78 to 83 in human RPS19 was
recently described (Cmejlova et al., 2006). This mutant protein was found to localize in the nucleus
when expressed in mammalian cells. In accordance with this result, introduction of this deletion in
yeast Rps19p (∆79-84) had no effect on nuclear accumulation, as already shown in Figure 4, even
though this deletion directly affects the NLS characterized herein.
Although we cannot exclude that these mutations affect the kinetics of nuclear import, these results do
not support the idea that transport of Rps19p to the nucleus is altered by DBA mutations.
115

DISCUSSION
From the data presented here, we conclude that eukaryotic Rps19 contains a NLS between
amino acids 67 and 97. This domain in P. abyssi Rps19 crystal structure corresponds to a loop-helix-
loop structure (loop 2/helix 4/loop 3). The second loop in this motif is likely to be highly flexible, as
indicated by the indetermination of its structure in the crystal. It is exposed to solvent at the surface of
the protein, which allows interaction with karyopherins. NLSs have been determined only for a
handful of ribosomal proteins. A tentative consensus has been proposed (Stuger et al., 2000), but is not
supported by our results in Rps19. In contrast to other NLS identified in ribosomal proteins, the
content of this NLS in basic residues is limited, although arginines and lysines are clear important for
nuclear localization. However, this domain contains the consensus sequence defined for a PY NLS.
PY NLSs were defined as the consensus sequence recognized by transportin/Kap-beta2, on the basis
of the crystal structure of this nucleoporin complexed to the NLS of hnRNP A1 (Lee et al., 2006). This
sequence was also shown to mediate binding of the RNA-binding protein Hpr1p to Kap104p, the yeast
homolog transportin/ importin β2 (Lange et al., 2008). Consistent with the presence of this sequence in
domain 67-97, transport of RPS19-GFP was highly perturbed in kap104-16 cells, even at permissive
temperature. In addition, modification of the core of this PY NLS is sufficient to abolish the
autonomous nuclear localization activity of this domain. These data strongly support a direct role of
Kap104p in nuclear import of Rps19p, although direct binding of the 67-97 domain to Kap104p
remains to be shown. Interestingly, the tyrosine of this motif in yeast Rps19p (Y91) is not conserved
in other eukaryotic homologs where one finds a serine, an asparagine or an alanine. In contrast,
conservation of the two following amino acids (K92, F93) is much higher. Thus, it might be
interesting to re-evaluate the consensus for transportin/Kap104p binding sequences in light of
interspecies sequence comparison.
Our data suggest that there is more than one nuclear transport pathway for Rps19, since none
of the partial deletions made in loop 3 abrogated nuclear localization. Despite an extensive mutational
study, we cannot exclude that the protein contains a second NLS made of amino-acids that are
clustered on the surface of the folded protein, although distant in the primary structure. Such an NLS
116

has already been described for exemple in xenopus RPL5 (Claussen et al., 1999) and is difficult to
detect with a deletion strategy. The presence of multiple NLSs was reported in several ribosomal
proteins . Alternatively, Rps19 could also piggy back after association with partner proteins, like
another ribosomal protein or pre-ribosomal proteins that may chaperone Rps19 incorporation into pre-
ribosomes. However, we rather favor the possibility that domain 67-97 is recognized by several
importins with different structural requirements. Indeed, nuclear localization activity of this domain
requires both Kap121p and Kap104p and two-hybrid interactions indicates that it interacts with
Kap121p. Interestingly, overlap of Kap121p and Kap104p function was already observed in the case
of Nop1p GAR domain (Leslie et al., 2004).
It has been proposed that, in contrast to most nuclear proteins, ribosomal proteins may be able
to take several paths to the nucleus, in order to ensure to high transport rate needed to assemble
ribosomal subunits in proliferating cells. Indeed, in mammalian cells, nuclear translocation of
ribosomal proteins can be mediated by several karyopherin-betas in semi-permeabilized cell assays,
and the NLS of RPL23a (homolog of yeast Rpl25p) was shown to bind to importin-beta, transportin,
importin 5 and importin 7. (Jakel and Gorlich, 1998). In yeast, it was reported that Rpl25p NLS can
function with Kap123p and Kap121p, but not with Kap104p or Kap95p (Rout et al., 1997; Timney et
al., 2006). In addition, Rpl25p binds to Kap121p and Kap123p in vitro, but not to Kap104p
(Schlenstedt et al., 1997). Recently, Timney et al. (2006) have used in situ transport kinetics analysis
to demonstrate that nuclear import of GFP fused to the NLS of Rpl25p primarily depends on
karyopherin Kap123p, the most abundant karyopherin in yeast. This observation correlates with the
preferential recognition of ribosomal proteins by Kap123p in overlay assays (Rout et al., 1997). Thus,
from the example of Rpl25p, it seems that the nuclear import pathways in yeast are not as redundant
for ribosomal proteins as suggested in mammalian cells. However, in the case of Rps19p, loss-of-
function of Kap121p has a much more drastic effect on nuclear accumulation than deletion of
Kap123p, which challenges the conclusions established with Rpl25p. Our data rather suggest that
distinct ribosomal proteins may be imported according to different mechanisms. Functional study of a
larger number of ribosomal NLSs is needed to explore this hypothesis.
117

None of the DBA mutations introduced in the yeast Rps19 perturbed nuclear localization. This
result extends previous data obtained with the human protein in culture cells. Although some missense
mutations were found to prevent localization of Rps19 in the nucleolus, none were shown to inhibit
transport to the nucleus. It must be noted that neither these experiments nor ours in yeast were suitable
to evaluate the import rate of the protein, which may be affected by some of the mutations. Since our
experiments indicate that even deletions within the NLS loop do not abrogate nuclear transport, one
may also hypothesize that missense mutations in this region do not have enough impact on the protein
function to be pathogenic. This sequence is different from the nucleolar localization sequences defined
by Da Costa et al. (2003) in human Rps19, which where not sufficient for nuclear import of a reporter
protein. However, absence of nuclear localization in this early work may be re-interpreted as protein
instability rather than defective transport, a conclusion supported by recent data (Angelini et al., 2007;
Crétien et al., submitted for publication).
AKNOWLEDGEMENTS
The authors are indebted to Rick Wozniak, Pamela Silver, Anita Corbett, Annie Mougin and
Micheline Fromont-Racine for providing strains and reagents. They also wish to thank Guillaume
Stahl for scientific and technical advising and Yves Quentin for his help with sequence analysis.
AMAT was supported by the CNRS and the Fondation pour la Recherche Médicale. This study was
funded by the French National Research Agency (ANR – RIBODBA project).
118

REFERENCES
Angelini M, Cannata S, Mercaldo V, Gibello L, Santoro C, Dianzani I, Loreni F (2007) Missensemutations associated with Diamond-Blackfan anemia affect the assembly of ribosomal proteinS19 into the ribosome. Hum Mol Genet 16: 1720-1727
Antoine M, Reimers K, Wirz W, Gressner AM, Muller R, Kiefer P (2005) Identification of anunconventional nuclear localization signal in human ribosomal protein S2. Biochem BiophysRes Commun 335: 146-153
Barik S (1995) Site-directed mutagenesis by double polymerase chain reaction. Mol Biotechnol 3: 1-7Campagnoli MF, Ramenghi U, Armiraglio M, Quarello P, Garelli E, Carando A, Avondo F, Pavesi E,
Fribourg S, Gleizes PE, Loreni F, Dianzani I (2008) RPS19 mutations in patients withDiamond-Blackfan anemia. Hum Mutat
Choesmel V, Bacqueville D, Rouquette J, Noaillac-Depeyre J, Fribourg S, Cretien A, Leblanc T,Tchernia G, Da Costa L, Gleizes PE (2007) Impaired ribosome biogenesis in Diamond-Blackfan anemia. Blood 109: 1275-1283
Choesmel V, Fribourg S, Aguissa-Toure AH, Pinaud N, Legrand P, Gazda HT, Gleizes PE (2008)Mutation of ribosomal protein RPS24 in Diamond-Blackfan anemia results in a ribosomebiogenesis disorder. Hum Mol Genet 17: 1253-1263
Clamp M, Cuff J, Searle SM, Barton GJ (2004) The Jalview Java Alignment Editor. Bioinformatics20: 426-427
Claros MG, Vincens P (1996) Computational method to predict mitochondrially imported proteins andtheir targeting sequences. Eur. J. Biochem. 241: 779-786
Claussen M, Rudt F, Pieler T (1999) Functional modules in ribosomal protein L5 forribonucleoprotein complex formation and nucleocytoplasmic transport. J Biol Chem 274:33951-33958
Cmejla R, Cmejlova J, Handrkova H, Petrak J, Pospisilova D (2007) Ribosomal protein S17 gene(RPS17) is mutated in Diamond-Blackfan anemia. Hum Mutat 28: 1178-1182
Cmejlova J, Cerna Z, Votava T, Pospisilova D, Cmejla R (2006) Identification of a new in-framedeletion of six amino acids in ribosomal protein S19 in a patient with Diamond-Blackfananemia. Blood Cells Mol Dis 36: 337-341
Crooks GE, Hon G, Chandonia JM, Brenner SE (2004) WebLogo: A sequence logo generator.Genome Research 14: 1188-1190
Da Costa L, Tchernia G, Gascard P, Lo A, Meerpohl J, Niemeyer C, Chasis JA, Fixler J, Mohandas N(2003) Nucleolar localization of RPS19 protein in normal cells and mislocalization due tomutations in the nucleolar localization signals in 2 Diamond-Blackfan anemia patients:potential insights into pathophysiology. Blood 101: 5039-5045
Draptchinskaia N, Gustavsson P, Andersson B, Pettersson M, Willig TN, Dianzani I, Ball S, TcherniaG, Klar J, Matsson H, Tentler D, Mohandas N, Carlsson B, Dahl N (1999) The gene encodingribosomal protein S19 is mutated in Diamond-Blackfan anaemia. Nat Genet 21: 169-175
Flygare J, Aspesi A, Bailey JC, Miyake K, Caffrey JM, Karlsson S, Ellis SR (2007) Human RPS19,the gene mutated in Diamond-Blackfan anemia, encodes a ribosomal protein required for thematuration of 40S ribosomal subunits. Blood 109: 980-986
Flygare J, Karlsson S (2007) Diamond-Blackfan anemia: erythropoiesis lost in translation. Blood 109:3152-3154
Fromont-Racine M, Rain JC, Legrain P (1997) Toward a functional analysis of the yeast genomethrough exhaustive two-hybrid screens. Nat Genet 16: 277-282
Gazda HT, Grabowska A, Merida-Long LB, Latawiec E, Schneider HE, Lipton JM, Vlachos A,Atsidaftos E, Ball SE, Orfali KA, Niewiadomska E, Da Costa L, Tchernia G, Niemeyer C,Meerpohl JJ, Stahl J, Schratt G, Glader B, Backer K, Wong C, Nathan DG, Beggs AH, SieffCA (2006) Ribosomal protein S24 gene is mutated in Diamond-Blackfan anemia. Am J HumGenet 79: 1110-1118
Gazda HT, Sieff CA (2006) Recent insights into the pathogenesis of Diamond-Blackfan anaemia. Br JHaematol 135: 149-157
119

Gregory LA, Aguissa-Toure AH, Pinaud N, Legrand P, Gleizes PE, Fribourg S (2007) Molecular basisof Diamond-Blackfan anemia: structure and function analysis of RPS19. Nucleic Acids Res35: 5913-5921
Idol RA, Robledo S, Du HY, Crimmins DL, Wilson DB, Ladenson JH, Bessler M, Mason PJ (2007)Cells depleted for RPS19, a protein associated with Diamond Blackfan Anemia, show defectsin 18S ribosomal RNA synthesis and small ribosomal subunit production. Blood Cells MolDis 39: 35-43
Jakel S, Gorlich D (1998) Importin beta, transportin, RanBP5 and RanBP7 mediate nuclear import ofribosomal proteins in mammalian cells. Embo J 17: 4491-4502
Lange A, Mills RE, Devine SE, Corbett AH (2008) A PY-NLS nuclear targeting signal is required fornuclear localization and function of the Saccharomyces cerevisiae mRNA-binding protein,Hrp1. J Biol Chem
Lee BJ, Cansizoglu AE, Suel KE, Louis TH, Zhang Z, Chook YM (2006) Rules for nuclearlocalization sequence recognition by karyopherin beta 2. Cell 126: 543-558
Leger-Silvestre I, Caffrey JM, Dawaliby R, Alvarez-Arias DA, Gas N, Bertolone SJ, Gleizes PE, EllisSR (2005) Specific Role for Yeast Homologs of the Diamond Blackfan Anemia-associatedRps19 Protein in Ribosome Synthesis. J Biol Chem 280: 38177-38185
Leger-Silvestre I, Milkereit P, Ferreira-Cerca S, Saveanu C, Rousselle JC, Choesmel V, GuinefoleauC, Gas N, Gleizes PE (2004) The ribosomal protein Rps15p is required for nuclear exit of the40S subunit precursors in yeast. Embo J 23: 2336-2347
Leslie DM, Grill B, Rout MP, Wozniak RW, Aitchison JD (2002) Kap121p-mediated nuclear importis required for mating and cellular differentiation in yeast. Mol Cell Biol 22: 2544-2555
Leslie DM, Zhang W, Timney BL, Chait BT, Rout MP, Wozniak RW, Aitchison JD (2004)Characterization of karyopherin cargoes reveals unique mechanisms of Kap121p-mediatednuclear import. Mol Cell Biol 24: 8487-8503
Rosenblum JS, Pemberton LF, Blobel G (1997) A nuclear import pathway for a protein involved intRNA maturation. J Cell Biol 139: 1655-1661
Rout MP, Blobel G, Aitchison JD (1997) A distinct nuclear import pathway used by ribosomalproteins. Cell 89: 715-725
Schlenstedt G, Smirnova E, Deane R, Solsbacher J, Kutay U, Görlich D, Ponstingl H, Bischoff FR(1997) Yrb4p, a yeast ran-GTP-binding protein involved in import of ribosomal protein L25into the nucleus. EMBO J 16: 6237-6249
Seedorf M, Silver PA (1997) Importin/karyopherin protein family members required for mRNAexport from the nucleus. Proc Natl Acad Sci U S A 94: 8590-8595
Stuger R, Timmers AC, Raué HA, van 't Riet J. (2000) Nuclear import of ribosomal proteins: evidencefor a novel type of nuclear localization signal. In Garett, R.A., Douthwaite, S.R., Lilijas, A.,Matheson, A.T., Moore, P.B. and Noller, H.F. (eds.), The Ribosome: Structure, Function,Antibiotics, and Cellular Interactions. ASM Press, Washington, D.C.
Sydorskyy Y, Dilworth DJ, Yi EC, Goodlett DR, Wozniak RW, Aitchison JD (2003) Intersection ofthe Kap123p-mediated nuclear import and ribosome export pathways. Mol Cell Biol 23:2042-2054
Timmers AC, Stuger R, Schaap PJ, van 't Riet J, Raué HA (1999) Nuclear and nucleolar localizationof Saccharomyces cerevisiae ribosomal proteins S22 and S25. FEBS Lett 452: 335-340
Timney BL, Tetenbaum-Novatt J, Agate DS, Williams R, Zhang W, Chait BT, Rout MP (2006)Simple kinetic relationships and nonspecific competition govern nuclear import rates in vivo.J Cell Biol 175: 579-593
Willig TN, Draptchinskaia N, Dianzani I, Ball S, Niemeyer C, Ramenghi U, Orfali K, Gustavsson P,Garelli E, Brusco A, Tiemann C, Perignon JL, Bouchier C, Cicchiello L, Dahl N, MohandasN, Tchernia G (1999) Mutations in ribosomal protein S19 gene and diamond blackfan anemia:wide variations in phenotypic expression. Blood 94: 4294-4306
120

DISCUSSION
121

122

Sur le plan génétique, l'ADB est caractérisée par des mutations monoalléliques d'un
gène de protéine ribosomique, le plus fréquemment le gène RPS19. Il est couramment
admis que ces mutations conduisent à une situation d'haplo-insuffisance : la protéine
fonctionnelle, produite à partir d'un seul allèle, est en quantité insuffisante. Ceci est
assez facile à envisager dans le cas de mutations drastiques comme des codons stop
précoces, des mutations des sites d'épissage, des mutations du promoteur ou des
délétions chromosomiques. Néanmoins, la majorité des mutations de RPS19 dans
l'ADB sont de type faux-sens. Etant donné la grande variété de ce type de mutations, on
peut se demander si elles ont un impact commun sur la fonction de la protéine. Par
ailleurs, on ne peut pas écarter l'hypothèse que ces protéines mutées altèrent directement
la biogenèse des ribosomes ou la traduction en ayant par exemple un effet dominant-
négatif.
La résolution de la structure de RPS19 facilitée par un homologue d'archaéon
Notre étude des relations structure-fonction au sein de la protéine ribosomique RPS19
visait à comprendre l'impact des mutations sur la fonction de la protéine. Pour répondre
à cette question, nous avons mené un travail collaboratif pour d'une part déterminer la
structure de la protéine RPS19 et d'autre part réaliser des études fonctionnelles. Le
choix de l'homologue de RPS19 chez l'archaéon hyperthermophile Pyrococcus abyssi
s'est imposé à la suite de différents essais de cristallisation menés dans l'équipe de
Sébastien Fribourg avec la protéine humaine. Malheureusement, cette protéine est
extrêmement soluble et ne cristallise pas, une expérience partagée par une autre équipe
(Alan Warren, communication personnelle). Par ailleurs, la protéine de S. cerevisiae
n'est pas facilement produite dans E. coli. La protéine RPS19 de P. abyssi s'est avérée
123

au contraire très adaptée à cette étude. Le taux de similarité élevé de cette protéine avec
ses homologues eucaryotes laisse présager une structure commune à ces protéines.
Cependant, il est possible que la protéine d'archaeon présente des variations de structure
assurant une conformation plus stable que la protéine humaine. De fait, les résultats
préliminaires de la détermination de la structure de la protéine humaine par RMN
indiquent qu'il s'agit d'une protéine très flexible ne comportant que quatre hélices
(Buenders et al., 2008)
La structure de la protéine RPS19 révèle l'impact des mutations dans l'ADB
L'obtention de la structure de RPS19 a permis d'examiner directement la position des
résidus mutés dans l'ADB. Deux classes ont pu être distinguées : d'une part, des résidus
hydrophobes orientés vers l'intérieur de la structure et susceptibles d'être importants
pour le repliement et la stabilité conformationnelle de la protéine (classe I); d'autre part,
des résidus en surface de la protéine potentiellement engagés dans des interactions
intermoléculaires (classe II).
Cette distinction amène à prédire que les mutations de classe I induisent une instabilité
de la protéine. De fait, ceci a été vérifié dans des cellules en culture. En 2003, Da Costa
et al. ont montré que le niveau de protéine détectée par western-blot était nettement plus
faible pour les mutants de classe I V15F et G127Q que pour les mutants de classe II
R56Q et R62W, après transfection de cellules COS avec des plasmides permettant
l'expression de ces protéines étiquetées. Ces données ont ensuite été étendues par
Angelini et al. (2007) qui ont utilisé la même approche pour étudier la stabilité d'un
nombre de mutations plus élevé. Leurs résultats montrent une corrélation parfaite entre
l'instabilité des protéines mutées et l'appartenance des mutations à la classe I. Les
124

protéines portant les mutations V15F, L18P, A57P, A61E et G127E (classe I) sont plus
instables que celles portant les mutations P47L, W52R, R56Q, R62Q, R62W et R101H
(classe II). Cette instabilité est due à l'action du protéasome comme l'indique
l'augmentation du taux de protéine en réponse au MG-132, un inhibiteur du protéasome
(Angelini et al., 2007). Donc l'effet des mutations de classe I sur la stabilité de RPS19
est bien susceptible d'aboutir à une sous-production de protéine RPS19.
Nous montrons ici que les mutations de classe II touchent des domaines basiques en
surface de RPS19 qui jouent un rôle important dans l'assemblage de RPS19 avec les
pré-ribosomes chez la levure. En accord avec ces données, des mutants de RPS19 de
classe II exprimés dans des cellules embryonnaires humaines HEK293 en culture ne
sont pas retrouvés dans les ribosomes matures (Angelini et al., 2007). Nos résultats
indiquent que l'absence de RPS19 dans les ribosomes cytoplasmiques est due à un
défaut d'incorporation dans les particules pré-40S dans le noyau. Il était également
envisageable que les protéines mutées soient incorporées dans des particules pré-40S et
bloquent leur maturation, entrainant leur rétention dans le noyau et leur dégradation.
Cependant, les formes mutées de Rps19p que nous avons examinées ne co-précipitent
pas avec l'ARN 21S qui composent les particules pré-40S aberrantes chez la levure.
Nous favorisons donc l'hypothèse selon laquelle les mutations de RPS19 de classe II
interfèrent avec des interactions intermoléculaires importantes pour son incorporation
dans les particules pré-40S.
Toutes les mutations faux-sens entrainent une haplo-insuffisance
Comme pour les mutants de classe I, les protéines portant des mutations de classe II ne
sont donc pas fonctionelles pour l'assemblage des particules pré-40S, ce qui produit un
125

phénotype équivalent à un mutant nul. Cette similitude phénotypique au niveau
moléculaire avec des types de mutations entrainant un défaut de synthèse (faux-sens,
sites d'épissage, délétion mono-allélique) est probablement une des raisons de l'absence
de corrélation génotype-phénotype parmi les patients portant une mutation dans RPS19.
Cette conclusion est aussi supportée par la nature du défaut de maturation du pré-ARNr
de cellules issus de patients portant diverses mutations non-sens dans RPS19 : le patron
des pré-ARNr est le même quelque soit la mutation et correspond à celui observé en
inhibant la synthèse de RPS19 avec des siRNA. On peut bien parler dans tous les cas
d'haplo-insuffisance.
Certaines mutations sont hypomorphes chez la levure
Il convient toutefois de nuancer cette conclusion, car certaines mutations de Rps19p
n'entrainent pas une perte de fonction totale chez la levure. En effet, des mutants de
classe I (I15F, A62S, G121S, et G128Q) et de classe II (R63Q, R63W, R101H)
peuvent complémenter à des degrés divers la perte d'expression de RPS19 et supporter
la croissance cellulaire (cette thèse; Leger-Silvestre et al., 2005). Dans le cas des
mutations A62S, G121S et G128Q, la complémentation est même très efficace. De fait,
nous avons vérifié ici que le mutant G121S s'associe avec le pré-ANRr 20S et que ces
trois mutants sont présents dans des ribosomes matures.
Il est possible que ces trois mutations de classe I n'aient pas un effet destabilisant
important dans la protéine Rps19p de S. cerevisiae. En effet, la conformation de la
protéine de levure pourrait être plus robuste grâce à des interactions intramoléculaires
supplémentaires. Par ailleurs, la cinétique de transport et d'incorporation des protéines
ribosomiques pourrait être plus rapide chez la levure. La vitesse de biogenèse des
ribosomes y est beaucoup plus élevée que dans des cellules de mammifères, comme le
126

montrent classiquement l'analyse de la maturation des pré-ARN ribosomiques en pulse-
chase. Une instabilité conformationnelle mineure aurait plus de chance de ne pas être
détectée avant incorporation de la protéine dans les particules pré-40S chez la levure. A
contrario, il est également possible que la dégradation des protéines ribosomiques libres
soit plus rapide dans les cellules de mammifère. A ce propos, on peut noter que la
régulation de l'expression des protéines ribosomiques est principalement liée au niveau
de transcription chez les levures et à la traduction chez les vertébrés. Il est donc possible
qu'il y ait aussi des différences importantes dans la régulation post-traductionnelle de la
demi-vie des protéines.
Concernant les mutations de classe II R63Q/W et R101H, il est possible qu'elles
perturbent moins la fonction de ces résidus dans la protéine de levure. La protéine
RPS19 humaine ne complémente que partiellement la perte de fonction de Rps19p chez
la levure. Ceci sous-entend que les deux protéines ne mettent pas en jeu exactement le
même type d'interactions avec leurs partenaires.
En dépit des hypothèses énoncées ci-dessus pour expliquer l'impact limité de certaines
mutations sur la protéine Rps19p de levure, ces données de complémentation suggèrent
que certains allèles mutés de RPS19 dans l'ADB pourraient rester en partie fonctionnels
et donner des protéines présentes dans des ribosomes matures, même à un taux faible. Il
est alors possible d'envisager une approche thérapeutique fondée sur l'inhibition de la
dégradation des protéines Rps19p mutées par des approches pharmacologiques. Cette
idée a été avancée par l'équipe de Lydie Da Costa qui a proposé l'emploi du
Borthezomib, un inhibiteur du protéasome déjà utilisé en thérapie anti-cancéreuse.
Cependant, il reste à montrer que l'inhibition de la dégradation des formes mutées de
RPS19 rétablit leur fonctionnalité. En utilisant un autre inhibiteur du protéasome, le
MG132, Angelini et ses collègues n'ont pas observé de restauration de l'incorporation
127

des protéines mutées dans les ribosomes. Mais il est difficile de tirer une conclusion
définitive de ces expériences car l'inhibition du protéasome a aussi pour effet d'altérer
directement la biogenèse des ribosomes (Stavreva et al., 2006). Des expériences
complémentaires sont donc nécessaires pour valider ou écarter cette idée.
La localisation nucléaire de RPS19 est un mécanisme robuste
L'étude du transport nucléaire de Rps19p chez la levure nous a permis à la fois de
déterminer les éléments structuraux de Rps19p impliqués dans cette fonction et plus
largement de questionner les travaux antérieurs sur l'import nucléaire des protéines
ribosomiques. Le transport de Rps19p dans le noyau apparaît comme un mécanisme
complexe assuré par différentes voies. Plusieurs arguments convergents soutiennent la
présence d'une NLS dans la région 67-97 que nous avons isolée : (1) elle est suffisante
pour le transport d'une protéine rapportrice dans le noyau; (2) elle interagit avec une
caryophérine en double-hybride; (3) elle est présente dans une boucle flexible qui
s'étend à la surface de Rps19p; (4) elle contient un motif consensus de PY NLS (5) la
substitution de la région homologue dans la protéine RPS19 de P. abyssi par quelques
acides aminés de cette séquence est suffisante pour concentrer la protéine procaryote
dans le noyau. Pourtant, la perte de fonction de cette séquence n'est pas suffisante pour
empêcher l'import nucléaire de Rps19p. On ne peut donc pas exclure qu'il existe un ou
plusieurs autres motifs de localisation nucléaire formés par des acides aminés éloignés
dans la séquence primaire et difficiles à détecter par une approche mutationnelle
classique. Rps19p pourrait également entrer dans le noyau en s'associant dans le
cytoplasme à des partenaires protéiques possédant une NLS. Cette hypothèse est
particulièrement séduisante, car elle sous-entend que Rps19p serait capable de
128

s'assembler avec d'autres protéines dans un pré-complexe. Il sera donc intéressant de
vérifier l'existence d'un tel pré-complexe par purification d'affinité en utilisant les
versions étiquetées de RPS19 construites au cours de cette thèse, ce qui pourrait aboutir
à l'identification de partenaires fonctionnels. Ces données montrent en tout cas que le
transport de Rps19p est un mécanisme robuste. De fait, il n'est pas altéré par les
mutations de type ADB, y compris par la délétion d'une partie du domaine NLS
(Cmejlova et al., 2006).
Dans des cellules humaines, les mutants de RPS19 fusionnés à la GFP sont également
adressés dans le noyau, mais seuls les mutants de classe II sont détectés dans le nucléole
(Angelini et al., 2007; Da Costa et al., 2003). Cette forte corrélation entre la localisation
nucléolaire et stabilité permet de proposer une ré-interprétation des données de Da
Costa et al., (2003), qui concluait à la présence de séquences de localisation
nucléaire/nucléolaire aux extrémités N- et C- terminales de RPS19. Il est plus
vraisemblable que l'absence de ces mutants par délétion dans le nucléole tenait à leur
instabilité, et non pas à la perte de signaux de localisation. En accord avec cette
hypothèse, des données récentes de cette équipe indiquent que des formes de RPS19
présentant un codon stop prématuré, donc tronquées dans leur partie C-terminale, sont
adressées dans le noyau de cellules COS lorsque le protéasome est inactivé (Aurore
Crétien et Lydie Da Costa, communication personnelle). De plus, l'adressage nucléaire
ne fonctionne plus lorsque leur taille est inférieure à une centaine d’acides aminés; il
s'agit de fait de la taille minimum pour inclure l'équivalent du domaine 67-97 dans la
protéine. Ces données suggèrent donc fortement que cette séquence joue un rôle
identique dans des cellules de mammifère.
129

La voie d'import nucléaire de Rps19p diffère de celle d'autres protéines
ribosomiques
Les travaux sur les voies d'import nucléaire des protéines ribosomiques ont conduit à
deux hypothèses, parfois présentées comme des différences entre levure et mammifère.
D'une part, il a été proposé que les protéines ribosomiques de levure étaient
majoritairement importées dans le noyau par une caryophérine particulière, Kap123p
(Rout et al., 1997; Timney et al., 2006). D'autre part, il est couramment admis que les
protéines ribosomiques de mammifère peuvent être transportées par plusieurs
caryophérines différentes (Jakel and Gorlich, 1998; Jakel et al., 2002). Ici, nous
montrons que le transport de Rps19p dépend de Kap121p et de Kap104p. Ceci n'exclut
pas la participation de Kap123p, mais elle n'apparaît pas comme une importine majeure
de ce processus. A notre connaissance, c'est la première protéine ribosomique dont le
transport nucléaire dépend de Kap121p. Dans le cas de la NLS de la protéine Rpl25p,
Kap121p peut suppléer la perte de Kap123p, mais avec une efficacité moins grande
(Rout et al., 1997; Schlenstedt et al., 1997; Timney et al., 2006). Nos résultats indiquent
que le transport de Rps19p suit une voie différente et spécifique, à l'image de la protéine
RPL12 humaine qui est transportée exclusivement par l'importine 11 (homologue de
Kap120p) (Plafker and Macara, 2002; Patel et al., 2002). Il est donc probable que la
réalité est plus complexe que ce qui a été envisagé jusqu'ici : alors que certaines
protéines ribosomiques seraient importées par une large gamme de caryophérines,
d'autres en revanche suivraient une voie de transport spécifique. Cette diversité pourrait
fournir un niveau de régulation particulier dans la biogenèse des ribosomes. Par
exemple, des caryophérines particulières pourraient présider au transport de protéines
ribosomiques impliquées dans des étapes de contrôle-qualité, une hypothèse proposée
pour RPL12 (Plafker and Macara, 2002). Une régulation spécifique de ces voies de
130

transport permettrait de moduler la biogenèse des ribosomes. Dans le cas de Rps19p, il
reste à comprendre l'implication conjointe de deux caryophérines Kap104p et Kap121p :
s'agit-il d'un dimère de caryophérines, comme cela a été démontré précédemment pour
l'histone H1 qui est importé par l'hétérodimère importine-β/importine 7 (Jakel and
Gorlich, 1998)? Pour répondre à cette question, il sera nécessaire d'examiner plus
directement in vitro l'interaction de Rps19p avec ces caryophérines.
Conclusion
Les données présentées dans cette étude supportent l'hypothèse selon laquelle les
mutations de RPS19 dans l'ADB aboutissent à une production insuffisante de protéine
fonctionnelle capable d'être incorporée dans les particules pré-ribosomiques. Cette
conclusion est en accord avec les données récentes montrant que des cellules de
mammifères issues de patients et portant différents types de mutations dans RPS19
présentent un défaut de maturation des ribosomes similaire à celui observé en inhibant
la synthèse de RPS19 avec des siRNA (Choesmel et al., 2007; Flygare et al., 2007; Idol
et al., 2007). La même situation est observée dans le cas de RPS24 dont les mutations
dans l'ADB, récemment décrites, aboutissent à la perte de fonction de la protéine
(Choesmel et al., 2008; Gazda et al., 2006).
L'association de l'haplo-insuffisance de plusieurs gènes de protéines ribosomiques à
l'ADB suggère que la mutation de ces différents gènes altère un mécanisme commun.
En étendant les résultats obtenus avec RPS19 dans ce travail aux autres protéines
ribosomiques, il apparaît que la limitation du taux de ces protéines ribosomiques va
avoir deux conséquences génériques : la perturbation de la maturation du pré-ARNr et
un déficit de production de ribosomes, deux phénomènes qui peuvent interférer avec la
131

progession du cycle cellulaire ou la différenciation (Figure 10). En effet, comme cela a
été développé dans l'introduction, un défaut de maturation des ribosomes est susceptible
d'entraîner une réponse au stress ribosomique. Des protéines ribosomiques libres
peuvent séquestrer HDM2 et stabiliser p53, entraînant la synthèse de protéines
inhibitrices des cyclines. Ce mécanisme est probablement relayé par d'autres voies de
régulation qui connecte la biogenèse des ribosomes et la progression dans le cycle
cellulaire, comme par exemple le contrôle de la transcritption de facteurs prolifératifs
comme c-myc (Dai et al., 2007). La présence constante d'un stress ribosomique pourrait
élever le seuil de sensibilité des cellules à d'autres stress et devenir fatale dans des
circonstances physiologiques particulières, comme la différenciation érythropoïétique.
A ce premier phénomène peuvent venir s'ajouter des défauts de traduction liés à la
limitation du nombre de ribosomes. Ce déséquilibre pourrait être critique pour la
synthèse de protéines régulatrices en altérant par exemple l'initiation dans des sites
d'entrée interne des ribosomes (Yoon et al., 2006), en modifiant la capacité d'initier la
traduction sur des codons alternatifs ou en dégradant la fidélité de la traduction. Il a été
proposé par Yoon et al. (2006) que les maladies génétiques liées à un défaut de
biogenèse des ribosomes pourrait être la conséquence d’un défaut de traduction d’un
sous-ensemble spécifique d’ARN messagers. La nature de ces ARNm pourrait être à
l’origine de la spécificité tissulaire, voire cellulaire, du mécanisme physiopathologique.
De manière moins subtile, un déficit en ribosomes pourrait s'avérer critique dans des
situations de fort besoin en synthèse protéique. Ainsi, les érythroblastes doivent
produire de très grandes quantités d'hémoglobine. Le nombre important de division
qu'ils subissent pendant l'érythropoïèse pourrait rendre plus critique le déficit des
capacités traductionnelles. Or, un défaut partiel de synthèse d'hémoglobine aboutit à
l'apoptose des cellules, en particulier du fait du déséquilibre avec la production de
132

noyau hème qui devient toxique pour la cellule lorsqu'il s'accumule sous forme libre.
Cette situation pourrait donc conduire à l'arrêt de la prolifération et de la différentiation
des érythroblastes.
Cette dernière hypothèse est séduisante, car elle rend compte de la spécificité tissulaire
de la maladie. Néanmoins, elle n'a pas pour l'instant de soutien expérimental fort. La
diversité des protéines ribosomiques mutées chez les patients est un argument solide en
faveur d'une réponse générique à un défaut de biogenèse des ribosomes. Il n'est
cependant pas exclu que ces protéines aient des relations particulières entre elles. Ainsi
RPL5 et RPL11 sont deux partenaires de l'ARNr 5S avec lequel elles forment un pré-
complexe (Zhang et al., 2007) et sont l'une et l'autre capables d'activer p53. Les
protéines ribosomiques mutées dans l'ADB pourraient être impliquées dans des étapes
particulières de la maturation des pré-ribosomes liées à des voies de stress. Des
arguments indirects en faveur d'un lien fonctionnel entre RPS19 et RPS24 ont été
proposés par notre équipe (Choesmel et al., 2008). Il est certain que l'étude des
mécanismes physiopathologiques de l'ADB nous amènera à faire progresser notre
connaissance des mécanismes de la biogenèse des ribosomes et de son intégration dans
la physiologie de la cellule.
133

134

MATERIEL ET METHODES
135

136

1. Souches et conditions de culture
1.1. Souches de bactériesLes clonages ont été effectués dans différentes souches d’Escherichia coli présentées autableau 3.
Tableau 3. Souches de bactéries E. Coli
1.2. Conditions de croissance
Le milieu de culture utilisé pour toutes les souches de bactéries de cette étude se nomme
LB (Lysogeny Broth). Il est composé de 1% de tryptone, de 1% de NaCl et de 0,5%
d’extrait de levure. Pour un milieu solide, on ajoute de l’agar à une concentration de
2%. Les différentes cultures sont incubées à 37oC et sous agitation pour les milieux
liquides (bouillons). Le temps d’incubation varie habituellement de 12 à 16 heures. Les
bactéries qui contiennent des plasmides sont sélectionnées grâce à la présence du gène
conférant la résistance à l’ampicilline (100 µg/ml), la kanamycine (250 µg/ml) ou le
chloramphénicol (200 µg/ml).
2. Transformation bactérienne et isolement de l’ADN plasmidique
2.1. Bactéries compétentes et transformation
Pour préparer les bactérie compétentes, 2 ml de milieu LB sont inoculés avec une
colonie de DH5α provenant d’une boîte LB. Ensuite, 500 ml de milieu LB sont
inoculés avec 0.5 ml de culture parvenue en phase stationnaire. La culture est incubée à
137

37oC sous agitation rapide jusqu’à l’obtention d’une D.O. 600 nm située entre 0.6 et 0.8.
Les manipulations sont toujours effectuées avec des solutions stériles et préalablement
refroidies sur la glace. La culture est alors répartie dans des tubes de 250 ml et refroidie
dans la glace pendant 10 min. Les cellules sont culottées par centrifugation à 4oC,
pendant 10 min et à 5000 rpm. Les cellules sont remises en suspension dans 250 ml de
10% glycérol (v/v,H2O) et centrifugées 5 min à 5000 rpm. Après un second lavage par
suspension dans 25 ml de 10% glycérol, les cellules électrocompétentes sont finalement
resuspendues dans 2.5 ml de 10% glycérol, puis des aliquots de 200 µ l de la
resuspension sont congelés à -80oC. Elles peuvent être conservées au moins un an dans
ces conditions. Lors de la transformation, les cellules électrocompétentes sont
décongelées sur la glace. On les met en présence de l’ADN (environ 100 ng) puis
directement dans la cuvette d’électroporation. Un choc électrique est appliqué avec le
programme Ec2 (BioRad MicroPulser). Les cellules sont récupérées dans un tube
Eppendorf par ajout de 1 ml de milieu LB + 0.4% glucose. On laisse incuber les cellules
à 37oC pendant 1 heure sans agitation avant de les étaler sur un milieu LB solide
contenant de l’antibiotique requis.
Pour ce qui est des cellules chimiocompétentes, on procède différemment, par un
traitement au chlorure de calcium. Pour ce faire, 500 ml de milieu LB sont inoculés
avec 0.5 ml d’une culture en phase stationnaire. La culture est incubée à 37oC sous
agitation rapide jusqu’à l’obtention d’une D.O. 600 nm comprise entre 0.3 et 0.8. Pour une
D.O. 600 nm = 0.66, on a 2.7 108 cellules/ml (Avec D.O. 600 nm = 1, on a donc 4.108
cellules/ml). Les cellules sont déposées sur la glace pendant 10 min, puis centrifugées
pendant 10 min à 3000 rpm à 4oC. Le culot est remis en suspension pendant 30 min à
4°C dans 125 ml d’une solution de CaCl2 100 mM stérile, préalablement refroidie, il y a
par la suite une centrifugation de 10 min à 3000 rpm à 4oC suivie d’une remise en
138

suspension du culot dans 25 ml de la même solution de CaCl2 à 4°C pendant 20 min.
Les cellules sont toujours gardées sur la glace. Les cellules ainsi perméabilisées au
CaCl2 sont finalement récupérées par centrifugation avant d’êtres reprises dans du CaCl2
100 mM/ 15% glycérol. Les cellules chimiocompétentes sont alors réparties en aliquots
de 200 µl puis congelées dans de l’azote liquide et gardés à – 80°C.
Lors de la transformation, environ 2.108 cellules chimiocompétentes sont décongelées
sur la glace. On les met en présence de 100 ng d’ADN pendant une heure. Un choc
thermique de 5 min à 37oC ou d’une minute à 42oC est ensuite effectué. Les cellules
sont déposées sur la glace pendant environ 3 min avant d’ajouter 0.5 ml de milieu LB.
On laisse incuber les cellules à 37oC pendant 1 heure sans agitation avant de les étaler
sur un milieu LB solide contenant de l’antibiotique requis. Si le plasmide utilisé est
pBluescript, on peut sélectionner les colonies blanches en étalant sur le milieu 50 µl de
X-gal 2% et 50 µl d’IPTG 100 mM. Les boîtes sont finalement incubées pendant 24
heures à 37oC.
2.2. Extraction de l’ADN plasmidique de bactéries
2.2.1. METHODE TENS
Tous les plasmides obtenus à la suite d’une transformation ont été isolés pour
vérification par la méthode rapide d’isolement de l’ADN plasmidique. Les
transformants sont tout d’abord repiqués dans 2 ml de milieu LB avec de l’antibiotique
pour une incubation sous agitation à 37oC pour toute la nuit. Par la suite, 1.5 ml de
chaque culture sont transférés dans un tube Eppendorf de 1.5 ml et centrifugés 10
secondes à 13000 rpm dans une microcentrifugeuse. On élimine le surnageant en
gardant de quoi ressuspendre le culot en vortexant. On ajoute 300 µl de tampon TENS
(SDS 0.5%, NaOH 0.1M et EDTA 1 mM) et on agite en retournant le tube jusqu’à ce
139

que le mélange devienne clair et visqueux. On neutralise ensuite avec 150 µl d’acétate
de sodium (NaAc 3M pH 5.2) par agitation pour obtenir un mélange fluide et
floconneux. On centrifuge à vitesse maximale pendant 2 min et on récupère le
surnageant. On précipite l’ADN en ajoutant 900 µl d’alcool éthylique 100% et en
déposant les tubes sur la glace 10 min. Finalement on centrifuge les tubes pendant 5 min
à 4°C. On peut effectuer un autre lavage avec 500 µl d’alcool éthylique 70%. Le culot
d’ADN est enfin dissout dans 50 µl de tampon TE (Tris-HCl 10 mM pH 8, EDTA 1mM
pH 8.0). Le plasmide est vérifié par digestion de 1 à 3 µl de la préparation avec 1U
d’enzyme dans un volume final de 15 µl pendant 30 à 45 min. On inclue 10 ng de
RNase A dans 30 µl de tampon de charge juste au moment de charger le gel.
2.2.2. METHODE PAR COLONNE QIAGEN
Tous les plasmides utilisés pour les clonages et qui ont été envoyés pour séquençage ont
été isolés à l’aide de colonne QIAGEN provenant du QIAprepR Spin Miniprep Kit. Le
protocole suivi est celui décrit dans le manuel d’utilisation du constructeur.
3. Souches de levure et conditions de culture
3.1. Souches de levure
Le génotype des souches de levure S. cerevisiae utilisées dans cette étude est décrit dans
le tableau 4.
140

Souche Génotype Source
CG1945 MATa gal4-542 gal80-538 ade2-101 his3-200 leu2-3,112 lys2-801 trp1-901 ura3-52 URA3::GAL417mers(x3)-CyC1TATA-LacZ LYS2::GAL1UAS-GAL1TATA-HIS3 CYHR
Fromont-Racineet al, 2002
Y187 MATα gal4Δ gal80Δ ade2-101 his3-200 leu2-3,112 trp1-901 ura3-52URA3::GAL1UAS-GAL1TATA-LacZ met-
Fromont-Racineet al, 2002
BY4742 MATα ;his3Δ1 ;leu2Δ0 ;lys2Δ0 ;ura3Δ0 EuroscarfY06271(rps19AΔ)
BY4741 ; Mat a ; his3Δ1 ; leu2Δ0 ; met15Δ0 ; ura3Δ0 ;YOL121C ::KanMX4
Euroscarf
Y11142(rps19BΔ)
BY4742 ; Matα ; his3Δ1 ; leu2Δ0 ; lys2Δ0 ; ura3Δ0 ; YNL302C ::KanMX4
Euroscarf
GAL-RPS19 His3Δ1 ; leu2Δ0 ; ura3Δ0 ; YOL121C ::KanMX4 ;YNL302C ::KanMX4 ; pFL38(PGAL ::RPS19A ; URA3)
Leger-silvestreet al, 2004
GAL-RPS19 His3Δ1 ; leu2Δ0 ; ura3Δ0 ; YOL121C ::KanMX4 ;YNL302C ::KanMX4 ; pFL36(PGAL ::RPS19A ; LEU2)
Leger-silvestreet al, 2004
kap121-34 MATa ura3-52 his3Δ200 trp1-1 leu2-3,112 lys2-801kap121 ::ura ::HIS3 +pkap121-34
Leslie et al,2004
Δkap123 MATa ura3-52 his3Δ200 trp1-1 leu2-3,112 lys2-801kap123 ::ura3 ::HIS3
Rout et al, 1997
kap95-14 ura3-52 his3Δ200 trp1-1 leu2-3,112 lys2-801 kap95 ::HIS3 +pkap95-14
Leslie et al,2004
kap104-16 Leslie et al,2004
Tableau 4. Souches de levure S. Cerevisiae
3.2. Conditions de croissance
Le milieu de croissance riche et non sélectif pour les souches de levure S. cerevisiae
sans plasmide est le YPD. Il est composé de 1% d’extrait de levure, 2% de tryptone et
2% de glucose. La sélection pour les différentes auxotrophies a été effectuée à l’aide
d’un milieu minimal composé de 0.67% de milieu azoté sans acide aminé (Yeast
nitrogen base without amino acids), 2% de glucose et 0.2% d’un mélange d’acides
aminés essentiels (adénine, arginine, histidine, leucine, lysine, tryptophane, et uracile)
sans la présence de l’acide aminé utilisé pour la sélection. Lorsque les milieux sont
solides, 2% d’agar est ajouté. Très souvent, on utilise le milieu casaminoacide qui
correspond à un hydrolysat de caséine. La croissance optimale de S. cerevisiae est à
30oC, à moins d’indication contraire, et l’incubation se fait sous agitation.
141

4. Transformation de levure et isolement de l’ADN génomique
4.1. Transformation de la levure S. cerevisiae
Les différentes souches ont été transformées avec les plasmides à l’aide de la méthode
utilisant l’acétate de lithium, LiAc (Kaiser, 1994). Pour ce faire, un volume de 2 ml
d’une culture en phase exponentielle de croissance (D.O. 600 nm = 0.6) est transféré dans
un tube Eppendorf de 2 ml et centrifugé dans une microcentrifugeuse. On enlève le
surnageant et on ressuspend les cellules dans 0.5 ml de 0.1M d’acétate de lithium. Les
cellules sont finalement remises en suspension dans 40 µl de 0.1M LiAc. Un volume de
3 µ l d’ADN de sperme de saumon à une concentration de 10 mg/ml préalablement
bouilli est ajouté. On ajoute 168 µl de la solution (PEG 4000 50%, acétate de lithium
0,1 M, Tris-HCl 10 mM, EDTA 1 mM, pH 7.0). La quantité d’ADN plasmidique
utilisée pour la transformation est habituellement de 100 ng. Il est important de
vortexer vigoureusement avant d’incuber pendant 45 min à 30°C. Par la suite, on incube
les cellules à 42°C pendant 12 min. On lave les cellules par suspension dans 450 µl de
NaCl 9‰, puis on centrifuge à 13000 rpm pendant 15 secondes. Les cellules sont
finalement reprises dans 100 µl NaCl 9‰. Les levures ainsi transformées sont étalées
sur un milieu « Synthetic Dropout (SD) » (milieu contenant tous les acides aminés
essentiels pour la croissance des levures en absence de l’acide aminé utilisé pour la
sélection) et incubées à 30°C jusqu’à l’obtention de colonies en se référant toujours aux
témoins positifs et négatifs.
4.2. Co-transformation et clonage par recombinaison
Une grande partie des plasmides utilisés au cours de cette thèse a été construite par
recombinaison chez la levure S. cerevisiae. Pour cela, nous utilisons le même protocole
de transformation cité en paragraphe 4.1. On utilise un vecteur rendu linéaire (par
142

double digestion enzymatique ou par simple digestion suivie d’une déphosphorylation
de l’extrémité 3’) et l’ADN à cloner contenant des séquences de recombinaison
homologue aux deux extrémités. La co-transformation de la levure par ces 2 séquences
permet le clonage par recombinaison homologue.
4.3. Préparation de l’ADN de S. cerevisiae et isolement du plasmide obtenu
par recombinaison
Pour isoler l’ADN génomique de différentes souches de levure, 1 ml d’une culture de
levure en phase stationnaire est centrifugé dans un tube Eppendorf de 1.5 ml à 12000
rpm pendant 1 minute à température ambiante. Après avoir éliminé le surnageant, on
ressuspend les cellules dans 100 µl de tampon d’extraction (Triton X-100 4% (V/V),
LiCl 2.5 M, Tris-HCl 50 mM pH 8.0, EDTA 62.5 mM). On ajoute 100 µl d’un mélange
de phénol:chloroforme:alcool isoamylique (25:24:1) et environ 200 mg de billes de
verre (0.45-0.5 mm de diamètre). On agite vigoureusement le mélange au vortex
pendant 5 min à 4°C pour casser la paroi des cellules. Le mélange est centrifugé 2 min à
vitesse maximale et on récupère 50 µl de la phase aqueuse dans un tube propre. On
précipite l’ADN avec 125 µl d’éthanol pendant 5 min dans la glace, puis le tube est
centrifugé à 13000 rpm pendant 2 min à 4°C. Finalement, on re-solubilise l’ADN dans
20 µl de tampon TE. Si on veut obtenir un ADN génomique exempté d’ARN, on peut le
traiter avec de 10 ng de RNase A pendant 15 min à 68oC, suivi d’une étape de
précipitation au phénol:chloroforme avant l’étape de resolubilisation finale. On utilise
25% de la ressuspension pour transformer les bactéries. La dernière étape consiste à
faire une minipréparation plasmidique en vue de vérifier le plasmide isolé.
143

5. Manipulations de l’ADN
5.1. Réaction de PCR
Le mélange des réactions de PCR (polymerase chain reaction) se compose comme suit :
0.5 µl de chacun des oligonucléotides à une concentration de 10 µM; 10 ng d’ADN
plasmidique ou 50 ng d’ADN génomique de S. cerevisiae , 10 µ l du tampon 5X
contenant du MgCl2 7.5 mM, 1 µl de dNTP 20 mM, compléter le volume à 50 µl, et
finalement 0.5 µl d’ADN polymérase Phusion (Finnzymes, New England Biolabs). Les
cycles d’élongation débutent par une période de dénaturation de 2 min à 98oC puis un
premier programme de 5 cycles; 10 secondes à 98oC, 15 secondes à la température
d’appariement des amorces (44°C pour la plupart) et 10 secondes à 72oC. Un second
programme de 25 cycles utilisant une température d’appariement des amorces plus
élévée que celle utilisée dans les 5 premiers cycles (50°C pour la plupart) s’ensuit. Avec
la Phusion, on utilise 5°C au dessus de la Tm. La réaction de PCR se termine par une
période d’élongation de 2 min à 72oC.
5.2. Réaction RT-PCR
La RT-PCR qui a été mis au point pour utiliser l’ARN comme matrice d’amplification
de la PCR s’effectue en deux étapes. Dans un premier temps, on prend 1 à 2 µg d’ARN
totaux et 1 µg d’oligo dT, le tout dans un volume final de 18.5 µl qu’on incube à 70°C
pendant 10 min. Ce mélange est ensuite incubé dans la glace pendant 5 min. Dans un
second temps, on ajoute à ce mélange, 1.5 µl de dNTP 20 mM, 6 µl de tampon AMV-
RT 5X, 1 µl de Rnasin, et 3 µl d’enzyme AMV-RT (Finnzymes). Ce volume final de 30
µl est alors incubé pendant 2 heures dans un bain marie à 42°C. Finalement, on effectue
144

une PCR classique en utilisant comme matrice un dixième du produit RT-PCR total (3
µl).
5.3. Réaction PCR sur colonie
Une petite portion de la colonie est ressuspendue dans 50 µl de solution de zymolyase
60 U/ml (6 mg de zymolyase 100 T dans 10 ml H2O) et incubée à 37°C pendant 30 min
puis à 95°C pendant 10 min. Finalement, on utilise un dixième du volume total (5 µl),
comme matrice pour une PCR classique.
5.4. Électrophorèse de l’ADN sur gel d’agarose
Les électrophorèses ont été effectuées sur des gels d’agarose à 1%. L’agarose est dissout
dans du TAE 0.5X. Du bromure d’éthidium 0.5 µg/ml est ajouté à l’agarose juste avant
sa solidification afin de visualiser l’ADN sur une plaque UV. On ajoute à l’échantillon
du tampon de charge 6X (bleu de bromophénol 0.25%, glycérol 30%) à une
concentration finale de 1X. L’ADN peut migrer dans le gel à l’aide d’une cellule
électrophorétique remplie avec du tampon TAE 0.5X. La tension appliquée est de 100
volts et le temps de migration peut être variable. Le marqueur de poids moléculaire
utilisé est le 1 Kb DNA ladder de Promega.
5.5. Isolement d’ADN sur gel d’agarose
La concentration du gel permettant l’isolement d’ADN est de 1% d’agarose. On fait
migrer l’ADN jusqu’à identification de la bande d’intérêt qui est coupée à l’aide d’une
lame tranchante sur un illuminateur UV. Le fragment d’ADN est finalement purifié sur
colonne GFX suivant le protocole GFX PCR DNA and Gel Band Purification Kit de
Amersham Biosciences.
145

5.6. Digestion de l’ADN par une enzyme de restriction
La digestion de l’ADN est généralement effectuée pour réaliser des clonages
plasmidiques. On ajoute à l’ADN à digérer le tampon approprié et 1 unité d’enzyme
par µg d’ADN à digérer dans un volume final de 25 µl. La digestion s’effectue
généralement à 37oC pendant une à deux heures. On procède à des digestions
séquentielles lorsqu’on fait intervenir plus d’une enzyme, et que ces dernières n’ont pas
la même activité enzymatique ou qu’il y est une incompatibilité de tampon de digestion.
5.7. Ligature
La réaction de ligature se fait entre le vecteur digéré (environ 50 à 100 ng d’ADN
plasmidique) et une quantité trois fois supérieur d’ADN à insérer. On quantifie l’ADN
sur gel d’agarose par référence à l’intensité des marqueurs de poids moléculaire. Par la
suite, le fragment d’ADN et le vecteur sont mis en présence de 1.5 µl de tampon de
ligature 10X, 1 µl de la ligase provenant du bactériophage T4 et d’eau distillée stérile
pour un volume total de 15 µl. La réaction s’effectue 4 heures à la température ambiante
ou bien toute la nuit à 16oC. Finalement, 5 µl de cette réaction sont utilisés pour
transformer la souche de bactérie.
6. Construction plasmidique
Les différents plasmides utilisés dans cette étude sont décrits aux tableaux 5-10. Tout
d’abord, la séquence correspondant au gène (ou le fragment de gène) à cloner est
amplifiée par PCR grâce à des oligonucléotides sens et antisens contenant soit des sites
de restriction enzymatique, soit des séquences de recombinaison homologue aux deux
146

extrémités. Ce produit de PCR est ensuite purifié et quantifié en vue de son clonage
dans un vecteur soit par restriction enzymatique, soit par recombinaison homologue.
6.1. Mutagenèse dirigée
Les différentes mutations ont été introduites dans les oligonucléotides ayant servi à
amplifier par PCR la séquence contenant une mutation quelconque dans le gène RPS19.
Cette séquence est ensuite clonée dans le vecteur plasmidique par recombinaison
homologue chez la levure S. cerevisiae.
6.1.1. OLIGONUCLEOTIDES UTILISES
La mutagenèse dirigée fait intervenir deux couples d’oligonucléotides. On utilise le
couple 1 d’oligonucléotides complémentaires contenant la mutation désirée et possédant
une séquence de 15 nucléotides des deux côtés de la mutation. Le couple 2
d’oligonucléotides qui flanque la séquence à cloner est conçu de manière à permettre
son clonage par recombinaison homologue dans le vecteur choisi. Des séquences d’une
longueur de 27 nucléotides sont utilisées pour la recombinaison homologue chez S.
cerevisiae. On effectue une première série de 2 PCR où on utilise les couples
oligonucléotide sens 1/ oligonucléotide non sens 2 et oligonucléotide sens 2/
oligonucléotide non sens 1. Les deux produits issus de cette première série de PCR sont
purifiés sur gel et mélangés en quantités équimolaires pour être utilisé comme matrice
dans la deuxième série de PCR utilisant les oligonucléotides du couple 2. C’est ce
produit de PCR qui est utilisé pour le clonage par recombinaison homologue chez S.
cerevisiae.
147

6.1.2. REACTION
Les mutations sur RPS19 ont été effectuées à partir d’un programme classique de PCR
utilisant le gène RPS19 comme matrice. Pour cela, on effectue une première série de
PCR où on utilise deux couples d’oligonucléotides différents. Le premier couple
comprend l’oligonucléotide sens contenant la mutation et l’oligonucléotide de
l’extremité 3’ du gène RPS19 et contenant la séquence de recombinaison avec le
vecteur, tandis que le second couple comprend l’oligonucléotide antisens contenant la
mutation et l’oligonucléotide de l’extremité 5’ du gène RPS19 et contenant la séquence
de recombinaison avec le vecteur. Les deux produits de PCR sont purifiés sur colonne
GFX après distinction des fragments sur gel d’agarose. Une quantité équimolaire des
deux produits de PCR est ensuite utilisée comme matrice pour amplifier le gène RPS19
contenant la mutation en présence des oligonucléotides en amont et en aval du gène
RPS19 et contenant les séquences de recombinaison dans le vecteur de clonage.
Finalement, le produit issu de cette dernière PCR est utilisé pour la transformation de la
levure S. cerevisiae suivant le protocole de co-transformation (voir paragraphe 4.2).
L’ADN plasmidique des colonies obtenues est isolé. Deux plasmides sont par la suite
purifiés sur colonne et envoyés au séquençage pour vérification de la mutation.
6.2. Clonage du gène RPS19 humain (hsaRPS19)
L’ADN complémentaire du gène RPS19 humain a été cloné dans le vecteur pS15-Term
par RT-PCR à partir des ARN totaux humains et grâce aux oligonucléotides Hs19-F
(sens) et Hs19-R (non sens). Le plasmide pS15-hsaRPS19 est ensuite transformé dans la
souche E. coli DH5α. La séquence du gène codant hsaRPS19 est alors soit amplifiée par
PCR soit isolé sur gel à partir de ce plasmide par digestion enzymatique pour être
148

ensuite inséré dans différents vecteurs selon les expériences. Il y a eu ensuite
vérification par séquençage.
6.3. Clonage du gène RPS19 de P. abyssi (PyARPS19)
Le gène codant pour la protéine RPS19 de P. abyssi a été cloné dans le vecteur
d’expression pET15b (N-His) après amplification par PCR. Cette PCR est réalisée sur
l’ADN génomique de P. abyssi gracieusement offert par Annie Mougin (CNRS-LBME,
Toulouse) et en utilisant les oligonucléotides PyA-F (sens) et PyA-R (non sens). La
séquence de PyARPS19 a été vérifiée par séquençage après clonage.
6.4. Localisation cellulaire par observation de la GFP
Une partie importante de cette étude a été l’observation de la localisation cellulaire de
différentes protéines dont Rps19p grâce au microscope à fluorescence. Pour cela, nous
avons analysé la localisation cellulaire des protéines exprimées en fusion traductionnelle
avec la GFP (Green Fluorescent Protein) grâce au plasmide pGal-GFP (Cormack et al.,
1997). Le gène RPS19 est amplifié sans le codon stop pour permettre l’expression de la
protéine de fusion RPS19p-GFP. La construction a été séquencée pour s’assurer que la
séquence des deux protéines est en phase traductionnelle.
Ce type d’observation permet essentiellement de vérifier la localisation cellulaire des
protéines. Habituellement, les cellules en phase exponentielle de croissance à la
température désirée sont observées directement dans leur milieu de culture après
montage de 5 µl de la culture entre lame et lamelle.
149

6.5. Observation de la GFP après fixation des cellules
L’observation de la fluorescence se fait souvent après fixation des cellules. Pour ce
faire, les cellules sont cultivées dans un volume de 10 ml jusqu’à la phase exponentielle
de croissance. Puis, elles sont centrifugées pour être reprises dans un volume finale de 3
ml. Les cellules sont alors fixées par ajout direct de formaldéhyde à 4% final dans le
milieu pendant 30 min à la température ambiante. On les lave à deux reprises avec du
PBS avant de les resuspendre dans 1 ml de PBS. Finalement, 5 µl de la suspension sont
placées entre lame et lamelle pour être observées au microscope à fluorescence.
7. Le système du double-hybride chez la levure
7.1. Principe
Le système du double-hybride chez la levure permet d’identifier in vivo, une interaction
protéine-protéine par reconstitution d’un activateur de transcription chimérique (GAL4)
chez la levure S. cerevisiae. GAL4 est formé d’un domaine de liaison à l’ADN et d’un
domaine d’activation qui, lorsqu’ils interagissent permettent l’activation de la
transcription.
Expérimentalement, deux souches de signes sexuels différents sont transformées par
deux plasmides différents. Dans notre cas, la souche CG1945 est transformée par le
plasmide pAS2ΔΔ-X dans lequel sont clonées les séquences qui codant pour des
protéines telles que la forme recombinante RPS19 fusionnée au domaine de liaison à
GAL4 et portant le gène de sélection au tryptophane (TRP1), tandis que la souche
Y187 est transformée par le plasmide pACT2-Y portant le gène de sélection leucine 2
(LEU2) et dans lequel ont été cloné les séquences codant pour les importines telle que
Kap121. La première étape consiste à strier sur des milieux sélectifs, des transformants
150

issus des deux souches. Les deux souches sont ensuite pré-croisées sur milieu riche
YPD. Les cellules ayant poussé sur ce milieu riche sont finalement étalées sur un milieu
sans tryptophane, leucine et histidine pour analyser l’interaction protéine-protéine.
L’interaction des deux protéines va permettre la reconstitution du transactivateur GAL4
et la transcription subséquente du gène rapporteur HIS3 permettant la synthèse
d’histidine. Il y a interaction protéine-protéine lorsque ces colonies poussent sur milieu
déficient en histidine.
7.2. Construction des vecteurs
Le vecteur GAL4-BD pAS2ΔΔ portant le gène de sélection TRP1 et le vecteur GAL4-
AD pACT2 portant le gène de sélection LEU2 ont été utilisés pour les études de double-
hybride. Les séquences codant les importines Pse1 et Nmd5 sont clonées dans le vecteur
GAL4-AD pACT2 par le laboratoire de Micheline Fromont-Racine (Institut Pasteur,
Paris). Les séquences codant les différentes protéines dont l’interaction avec les
importines était étudiée dans ce travail ont été clonées dans le vecteur GAL4-BD
pAS2ΔΔ après amplification par PCR.
Nom Réference Marqueur Source
pS15-(67_97)Sc-GFP pAAT-66 URA3 Cette étude
pS15-(67_97)R68A-GFP pAAT-67 URA3 Cette étude
pS15-(67_97)L76AK78AL79A-GFP pAAT-68 URA3 Cette étude
pS15-(67_97)K84AR86A-GFP pAAT-69 URA3 Cette étude
pS15-(67_97)P90AK92AH93A-GFP pAAT-70 URA3 Cette étude
pS15-(RPS19)Hsa-GFP pAAT-71 URA3 Cette étude
pS15-(67_97)Hsa-GFP pAAT-72 URA3 Cette étude
pS15- RPS19Sc(66-92)Pa-GFP pAAT-73 URA3 Cette étude
pS15- RPS19Sc(80-92)Pa-GFP pAAT-74 URA3 Cette étude
Tableau 5. Plasmides utilisés pour l’étude de la localisation cellulaire chez lesmutants d’importine
151

8. Etude des interactions in vitro par retard sur gel (EMSA)
8.1. Principe
La technique de retard sur gel ou EMSA (Electrophoretic Mobility Shift Assay) basée
sur le retard de migration dans un gel natif de polyacrylamide de courtes séquences
d'oligonucléotides en présence de protéines (ou de complexes protéiques) ayant la
propriété de reconnaître spécifiquement la séquence d'intérêt. C’est une méthode utilisée
pour étudier in vitro les interactions ARN-protéine. La variation de migration des
sondes complexées aux protéines par rapport aux sondes libres est suivie grâce au
marquage radioactif au 32P des sondes nucléotidiques. Les complexes ARN-protéine
migrent plus lentement que la sonde radioactive. La spécificité de reconnaissance des
sondes marquées peut être vérifiée par l'ajout en excès d’une sonde identique mais non
radioactif ou de tout autre oligonucléotide capable d’entrer en compétition avec la forme
radioactive.
8.2. Source de l’ARN
L’ADN codant pour la séquence de l’ARN est cloné dans le vecteur pGEMT (promega)
contenant le promoteur de l’ARN polymérase T7. Dans certains cas, on a utilisé un
oligonucléotide sens contenant la séquence du promoteur T7 pour directement amplifier
le fragment d’ADN codant pour la séquence d’ARN par PCR. Ce fragment d’ADN a été
directement utilisé pour la transcription de l’ARN.
8.3. Transcription in vitro et marquage des ARN
La transcription se fait par l’ARN polymérase T7. Pour cela, on utilise 1 microgramme
d’ADN plasmidique linéarisé par simple digestion enzymatique ou 100 nanogrammes
de produit PCR. L’ARN issu de la transcription est finalement purifié sur gel 8%
152

polyacrylamide (19 :1) 7 M urée. On utilise ensuite 20 pmol de l’ARN qui va être
marqué à son extrémité 3’ en présence de 20 µCi de 32P-pCp grâce à la T4 RNA ligase.
8.4. Interaction ARN-protéine
Les ARN marqués pCp sont dénaturés dans de l’eau à 70°C pendant 10 min et placés
sur de la glace pendant 10 min. Les ARN sont ensuite dilués dans du tampon TMK (30
mM Tris-Hcl, pH 7.4, 2 mM MgCl2 et 150 mM KCl) froid de façon à obtenir une
concentration en ARN d’environ 0.5 fmol/ µl et incubés à la température de la pièce
pendant 1 heure. Environ 2.5 fmol d’ARN marqués au 32P sont mélangés avec la
quantité requise de protéines recombinantes (RPS19 sauvage ou muté) dans 20 µl de
TMK avec 0.1% Triton X-100, 1 mM DTT, 20% glycérol, 100 ng/ µl E.Coli tRNA
(Roche), 100 ng/ µl BSA. Le mélange est incubé à la température de la pièce pendant 15
min avant d’être soumis à une électrophorèse dans un gel natif de polyacrylamide
contenant 6% de polyacrylamide, 50 mM Trizma-base, 50 mM glycine, 2 mM MgCl2..
Du glycérol à 5% final peut être ajouté au gel utilisé pour une bonne migration des
complexes. L’électrophorèse se fait dans un tampon de 50 mM Trizma-base, 50 mM
glycine, 2 mM MgCl2.à 4°C et à 20 mA. La visualisation de l’ARN libre ou complexé
aux protéines se fait par visualisation des bandes correspondantes à la radioactivité
grâce au logiciel Image Gauge (Fuji Film) du PhosphorImager (FLA3000IR
PhosphorImager, Fuji).
9. Purification de ribosome sur gradient de sucrose
9.1. Culture des cellules
Les ribosomes sont isolés par centrifugation sur gradient de sucrose. Pour cela, la
souche RPS19B (transformée par le plasmide pS15-TAP-RPS19A exprimant la forme
153

sauvage ou mutée de RPS19A) est cultivée dans un milieu complet jusqu’à une D.O600 nm
= 0.5. De la cycloheximide (antibiotique qui bloque la synthèse protéique) est ajoutée à
la culture à une concentration finale de 100 µg/ ml. Après 10 min d’incubation, les
cellules sont récoltées par centrifugation à 5000 rpm pendant 10 min et à 4°C. Les
cellules sont ensuite lavées avec 20 ml de tampon A froid (20 mM HEPES (pH 7.5), 10
mM KCl, 5 mM MgCl2, 1 mM EGTA, 1 mM DTT et 100 µg/ ml final de
cycloheximide). Finalement, 1 g de cellules sont remises en suspension dans 1.5 ml de
tampon A.
9.2. Fractionnement sur gradient
La lyse des cellules se fait avec 1.4 g de billes de verre (0.5 mm) pour 0.8 ml de la
suspension dans des tubes Eppendorf de 2 ml. La paroi des cellules est cassée en
vortexant pendant 10 min à 8°C. 150 µl de tampon A sont ajoutés à la suspension qui
est ensuite centrifugée à 10 000 rpm pendant 5 min. Après quantification des protéines
par la méthode de Bradford, 1 mg d’extrait total est chargé sur un gradient de sucrose
(10 à 50%) dans du tampon A sans cycloheximide (10.5 ml) et centrifugé à 26 000 rpm
pendant 12 heures dans un rotor SW41. Un collecteur de gradient (ÄKTAprime –
Amersham Biosciences) est utilisé en combinaison avec un détecteur d’UV (Pharmacia
UV-detector LKB.UV-M II). 20 fractions de 0.5 ml sont collectées dans des tubes
Eppendorf.
9.3. Extraction protéique des fractions
Cette extraction se fait sur 250 µl de chaque fraction auxquels on ajoute 30 µl d’acide
trichloroacétique (TCA) 100% et 25 µl de déoxycholate (DOC) 2%. Ce mélange est
bien vortexé et placé sur la glace pendant 10 min. On centrifuge à 14000 rpm pendant
154

10 min à 4°C pour éliminer le surnageant et prendre le culot dans 50 µl de tampon 2X
SDS PP (130 mM Tris-HCl (pH 8), 4.6% SDS, 20% glycérol, 0.02% bleu de
bromophénol et 0.2% DTT ). L’extrait est vortexé avant d’être chauffé à 95°C pendant
5 min. Finalement, l’extrait est revortexé avant d’être congelé à -20°C.
9.4. Extration des ARN
Cette extraction se fait également sur 250 µl de chaque fraction auxquels on ajoute 30
µl de SDS 10% dans 1M Tris-HCl (pH 7.5) et 750 µl de phénol-Chloroforme. Par deux
fois, ce premier mélange est vortexé puis incubé à 65°C pendant 1 minute. Il est ensuite
centrifugé à 14000 rpm pendant 1 minute à la température de la pièce pour récupérer
200 µl de surnageant. Après ajout de 500 µl de phénol-chloroforme, le mélange est
vortexé et incubé à 65°C pendant 1 minute. Finalement, après 1 minute de
centrifugation, 150 µl de la phase aqueuse est récupérée. A cette phase aqueuse, on
ajoute 20 µg de glycogène, 15 µl de 3M NaAc pH 5.2 et 475 µl d’éthanol froid 100%
pour précipiter les ARN à –20°C pendant 30 min. Finalement, on centrifuge à 14000
rpm pendant 10 min à 4°C et le culot est repris dans 15 µl de formamide. On chauffe
pendant 15 min à 65°C et on vortexe pour récupérer les ARN qui peuvent être conservés
à – 80°C.
10. Immunoprécipitation des protéines étiquetées TAP-TAG sur billes IgG-
Sepharose
10.1. Culture des cellules
La souche RPS19B transformée par le plasmide pS15-TAP-RPS19A (exprimant la
forme sauvage ou mutée de RPS19A en fusion avec l’étiquette TAP) est mise en culture
155

dans un volume de 500 ml jusqu’à une DO600 = 0.7-1. Les cellules sont gardées pendant
10 min dans la glace avant d’être centrifugées à 5000 rpm pendant 10 min à 4°C. Le
culot est resuspendu dans 250 ml H2O froide puis centrifugé à 5000 rpm pendant 10
min. On lave une dernière fois dans 15 ml H2O froide pour finalement resuspendre 1 g
de cellules dans 1.5 ml de tampon A200 (200 mM KCl, 20 mM Tris (pH 8), 5 mM
MgAc, 1 mM DTT, 1X PMSF/Benzamidin et 0.5U/µl Rnasin). Les cellules sont
finalement cassées avec 1.4 g de billes de verre pour 0.8 ml de suspension en vortexant
à 4°C pendant 10 min. 150 µl de tampon A200 est ajouté à la suspension qui est
récupérée dans un nouveau tube Eppendorf après centrifugation à 14000 rpm pendant 5
min. L’extrait provenant des différents tubes est récupéré dans un seul tube pour être
quantifié par dosage des protéines par la méthode de « Bradford ».
10.2. Immunoprécipitation
Elle se fait dans des colonnes chromatographiques Poly-PrepR (Bio-Rad) abondemment
lavées avec de l’eau bi-distillée. On coule 150 µl de billes IgG Sepharose dans la
colonne. On lave une première fois avec 10 ml d’eau bi-distillée, puis deux fois
successivement avec 10 ml de tampon A200 (contenant 0.2% de triton). On bouche la
colonne et on récupère les billes IgG sepharose dans un tube Eppendorf de 1.5 ml avec
400 µl de tampon A200.
Pour chaque extrait protéique, on dépose une quantité de l’ordre de 40 mg. On dépose le
même volume d’extrait (1000 µl) sur chaque colonne, en ajustant avec du tampon A200,
sans oublier d’ajuster la concentration finale en triton de l’extrait à 0.2%. On conserve
de l’extrait correspondant à l’INPUT pour les analyses ultérieures en Northern blot et/ou
Western blot. Puis, on coule l’extrait dans le tube eppendorf contenant les billes IgG
Sepharose et on incube l’ensemble sous agitation douce pendant 1H à 4° C.
156

Après l’incubation, on ajoute 1 ml de tampon A200 dans la colonne bouchée, ensuite on
l’ouvre et on ajoute immédiatement les billes d’IgG Sépharose bien ressuspendues, puis
on laisse s’écouler la totalité du volume. Tous les rinçages se font en laissant le liquide
s’écouler sur les parois pour récupérer le maximum d’extrait. On lave une fois avec 1 ml
de tampon A200, puis 5 fois successivement avec 2 ml de tampon A200 et enfin avec
10 ml de tampon A200. Au dernier lavage, on laisse tout s’écouler, on ferme la colonne
et on ajoute 1 ml de tampon A200. Finalement, on ressuspend avec précaution avant de
transvaser les billes IgG Sepharose dans des tubes Eppendorf. Généralement, on utilise
10% pour l’analyse des protéines et 90% pour l’analyse des ARN. Pour récupérer les
billes IgG Sepharose, on centrifuge à 4000 rpm pendant 1 min à 4° C pour éliminer le
surnageant et garder les billes qui peuvent être stockées à – 20° C avant leur utilisation.
10.3. Extraction des ARN
On effectue une extraction parallèle pour l’INPUT (200 µg d’extrait) et les billes IgG
sepharose. On ressuspend les billes IgG dans 500 µl de tampon AE (10 mM EDTA, 50
mM NaAc pH 5.3) en ajustant le volume de l’input à 500 µl avec le même tampon AE.
On ajoute 50 µl de 10% SDS et 500 µl de tampon phénol/ AE et on vortexe pendant 4
min à 65° C. On incube brièvement pendant 2 min dans la glace et on centrifuge à
14000 rpm pendant 2 min à 4° C. Dans un premier temps, on transfert, 3 fois 150 µl de
surnageant dans de nouveaux tubes Eppendorf et on ajoute 500 µl de tampon phénol/
AE, puis on vortexe et on centrifuge à 14000 rpm pendant 30 sec à 4° C. Dans un
second temps, on récupère, 3 fois 130 µl de surnageant dans des nouveaux tubes et on
ajoute 500 µl de chloroforme, puis on vortexe et on centrifuge à 14000 rpm pendant 30
sec à 4° C. Finalement, on récupère 3 fois 100 µl du surnageant dans des nouveaux
tubes Eppendorf. On précipite pendant 5 min à – 20° C après avoir ajouté 750 µl d’
157

éthanol froid, 30 µl 3M d’acetate de sodium à pH 5.2 et 4 µl de glycogène à 10 mg/ml
pour la fraction contenant les billes. On centrifuge à 14000 rpm pendant 5 min à 4° C
pour resuspendre l’ARN dans 50 µl de formamide. On peut le stocker à – 80° C.
10.4. Northern blot
L’ARN issu des immunoprécipitations est séparé par migration sur gel d’électrophorèse
agarose/ formaldéhyde 1% pendant 5 min à 140V puis pendant 4 heures à 100V avant
d’être transféré sur membrane Hybond-N (Amersham). Après le cross-link des ARN sur
la membrane par une lumière UV et leur pré-hybridation dans du tampon
d’hybridation ( 25ml : SDS 10% 0,625ml, SSC 20X 7,5ml, Denharts 20X 2,5ml,
tRNA 10mg/ml 2,25ul, H2O 14,3ml), la membrane est séchée quelques min à la
température ambiante avant son hybridation avec une sonde ARN marquée par du 32P à
son extremité 3’.
Nom Réference Marqueur Source
pACTIIst LEU2 Fromont-Racine et al., 1997
pACT-Kap121 LEU2 Fromont-Racine et al., 1997
pACT-NMD5 LEU2 Fromont-Racine et al., 1997
pAS2ΔΔ TRP1 Fromont-Racine et al., 1997
pAS2ΔΔ-RPS19Hs pAAT-1 TRP1 Cette étude
pAS2ΔΔ-RPS19Sc pAAT-2 TRP1 Cette étude
pAS2ΔΔ-RPS19Pa pAAT-3 TRP1 Cette étude
pAS2ΔΔ-RPS19Pa(86-93)Sc pAAT-4 TRP1 Cette étude
pAS2ΔΔ-N84Δ(RPS19)Sc pAAT-5 TRP1 Cette étude
pAS2ΔΔ-N97Δ(RPS19)Sc pAAT-6 TRP1 Cette étude
pAS2ΔΔ-Δ53C(RPS19)Sc pAAT-7 TRP1 Cette étude
pAS2ΔΔ-Δ77C(RPS19)Sc pAAT-8 TRP1 Cette étude
pAS2ΔΔ-Δ90C(RPS19)Sc pAAT-9 TRP1 Cette étude
pAS2ΔΔ-77_97(RPS19)Sc pAAT-10 TRP1 Cette étude
pAS2ΔΔ-67_97(RPS19)Sc pAAT-11 TRP1 Cette étude
Tableau 6. Plasmides utilisés pour les expériences de double-hybride
158

Nom Réference Marqueur Source
pS15-TAP-Term pAAT-109 URA3 Cette étude
pS15-TAP-RPS19Sc-Term pAAT-110 URA3 Cette étude
pS15-TAP-A62S-Term pAAT-111 URA3 Cette étude
pS15-TAP-I15F-Term pAAT-112 URA3 Cette étude
pS15-TAP-R63W-Term pAAT-113 URA3 Cette étude
pS15-TAP-R63Q-Term pAAT-114 URA3 Cette étude
pS15-TAP-W53R-Term pAAT-115 URA3 Cette étude
pS15-TAP-I65P-Term pAAT-116 URA3 Cette étude
pS15-TAP-R57Q-Term pAAT-117 URA3 Cette étude
pS15-TAP-R57E-Term pAAT-118 URA3 Cette étude
pS15-TAP-R63E-Term pAAT-119 URA3 Cette étude
pS15-TAP-G121S-Term pAAT-120 URA3 Cette étude
pS15-TAP-G128Q-Term pAAT-121 URA3 Cette étude
pS15-TAP-R102H-Term pAAT-122 URA3 Cette étude
pS15-TAP-S60E-Term pAAT-123 URA3 Cette étude
pS15-TAP-R102E-Term pAAT-124 URA3 Cette étude
pS15-TAP-R122E-Term pAAT-125 URA3 Cette étude
pS15-TAP-R130-134E-Term pAAT-126 URA3 Cette étude
pS15-TAP-Q106E-Term pAAT-127 URA3 Cette étude
pS15-TAP-RPS19Pa-Term pAAT-128 URA3 Cette étude
Tableau 7. Plasmides étiquetés TAP utilisés pour purifier les ribosomes
159
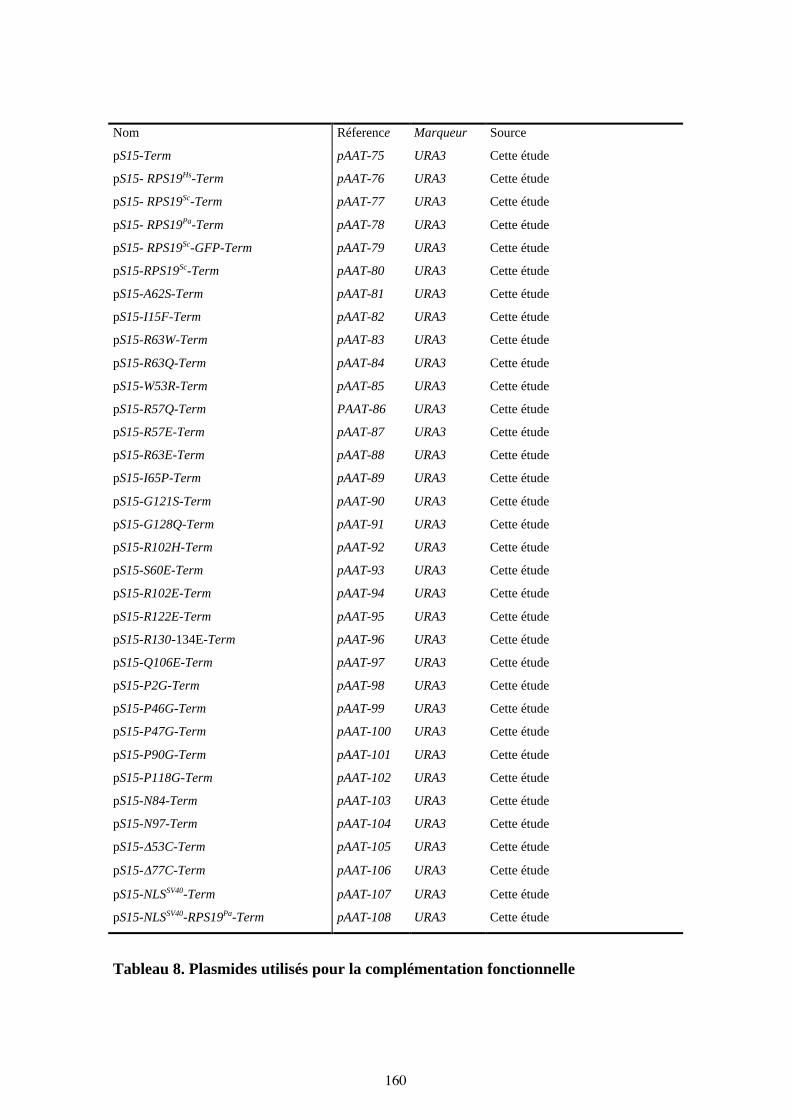
Nom Réference Marqueur Source
pS15-Term pAAT-75 URA3 Cette étude
pS15- RPS19Hs-Term pAAT-76 URA3 Cette étude
pS15- RPS19Sc-Term pAAT-77 URA3 Cette étude
pS15- RPS19Pa-Term pAAT-78 URA3 Cette étude
pS15- RPS19Sc-GFP-Term pAAT-79 URA3 Cette étude
pS15-RPS19Sc-Term pAAT-80 URA3 Cette étude
pS15-A62S-Term pAAT-81 URA3 Cette étude
pS15-I15F-Term pAAT-82 URA3 Cette étude
pS15-R63W-Term pAAT-83 URA3 Cette étude
pS15-R63Q-Term pAAT-84 URA3 Cette étude
pS15-W53R-Term pAAT-85 URA3 Cette étude
pS15-R57Q-Term PAAT-86 URA3 Cette étude
pS15-R57E-Term pAAT-87 URA3 Cette étude
pS15-R63E-Term pAAT-88 URA3 Cette étude
pS15-I65P-Term pAAT-89 URA3 Cette étude
pS15-G121S-Term pAAT-90 URA3 Cette étude
pS15-G128Q-Term pAAT-91 URA3 Cette étude
pS15-R102H-Term pAAT-92 URA3 Cette étude
pS15-S60E-Term pAAT-93 URA3 Cette étude
pS15-R102E-Term pAAT-94 URA3 Cette étude
pS15-R122E-Term pAAT-95 URA3 Cette étude
pS15-R130-134E-Term pAAT-96 URA3 Cette étude
pS15-Q106E-Term pAAT-97 URA3 Cette étude
pS15-P2G-Term pAAT-98 URA3 Cette étude
pS15-P46G-Term pAAT-99 URA3 Cette étude
pS15-P47G-Term pAAT-100 URA3 Cette étude
pS15-P90G-Term pAAT-101 URA3 Cette étude
pS15-P118G-Term pAAT-102 URA3 Cette étude
pS15-N84-Term pAAT-103 URA3 Cette étude
pS15-N97-Term pAAT-104 URA3 Cette étude
pS15-Δ53C-Term pAAT-105 URA3 Cette étude
pS15-Δ77C-Term pAAT-106 URA3 Cette étude
pS15-NLSSV40-Term pAAT-107 URA3 Cette étude
pS15-NLSSV40-RPS19Pa-Term pAAT-108 URA3 Cette étude
Tableau 8. Plasmides utilisés pour la complémentation fonctionnelle
160

Tableau 9. Plasmides utilisés pour l’étude de la localisation cellulaire
Nom Réference Marqueur Source
pGAL-GFP URA3 Leger-Sylvestre et al., 2004
pGAL- RPS19Sc-GFP pAAT-12 URA3 Cette étude
pGAL- RPS19Pa-GFP pAAT-13 URA3 Cette étude
pGAL- RPS19Pa(86-93)Sc-GFP pAAT-14 URA3 Cette étude
pGAL-N15Δ-GFP pAAT-15 URA3 Cette étude
pGAL-N52Δ-GFP pAAT-16 URA3 Cette étude
pGAL-N84Δ-GFP pAAT-17 URA3 Cette étude
pGAL-N89Δ-GFP pAAT-18 URA3 Cette étude
pGAL-N92Δ-GFP pAAT-19 URA3 Cette étude
pGAL-N97Δ-GFP pAAT-20 URA3 Cette étude
pGAL-N109Δ-GFP pAAT-21 URA3 Cette étude
pGAL-Δ16C-GFP pAAT-22 URA3 Cette étude
pGAL-Δ53C-GFP pAAT-23 URA3 Cette étude
pGAL-Δ67C-GFP pAAT-24 URA3 Cette étude
pGAL-Δ77C-GFP pAAT-25 URA3 Cette étude
pGAL-Δ80C-GFP pAAT-26 URA3 Cette étude
pGAL-Δ84C-GFP pAAT-27 URA3 Cette étude
pGAL-Δ90C-GFP pAAT-28 URA3 Cette étude
pGAL-Δ103C-GFP pAAT-29 URA3 Cette étude
pGAL-Δ128C-GFP pAAT-30 URA3 Cette étude
pGAL-N74Δ81C-GFP pAAT-31 URA3 Cette étude
pGAL-N78Δ85C-GFP pAAT-32 URA3 Cette étude
pGAL-N83Δ91C-GFP pAAT-33 URA3 Cette étude
pGAL-N85Δ92C-GFP pAAT-34 URA3 Cette étude
pGAL-N86Δ93C-GFP pAAT-35 URA3 Cette étude
pGAL-N90Δ97C-GFP pAAT-36 URA3 Cette étude
pGAL-(16_109)-GFP pAAT-37 URA3 Cette étude
pGAL-(53_109)-GFP pAAT-38 URA3 Cette étude
pGAL-(56_109)-GFP pAAT-39 URA3 Cette étude
pGAL-(67_109)-GFP pAAT-40 URA3 Cette étude
pGAL-(77_109)-GFP pAAT-41 URA3 Cette étude
pGAL-(80_109)-GFP pAAT-42 URA3 Cette étude
pGAL-(90_109)-GFP pAAT-43 URA3 Cette étude
pGAL-(98_109)-GFP pAAT-44 URA3 Cette étude
pGAL-(56_97)-GFP pAAT-45 URA3 Cette étude
pGAL-(77_92)-GFP pAAT-46 URA3 Cette étude
161

pGAL-(67_92)-GFP pAAT-47 URA3 Cette étude
pGAL-(77_89)-GFP pAAT-48 URA3 Cette étude
pGAL-(67_89)-GFP pAAT-49 URA3 Cette étude
pGAL-(67_84)-GFP pAAT-50 URA3 Cette étude
pGAL-(77_89)-GFP pAAT-51 URA3 Cette étude
pGAL-(77_89)-GFP pAAT-52 URA3 Cette étude
pGAL-(77_89)-GFP pAAT-53 URA3 Cette étude
pGAL-(77_97)-GFP pAAT-54 URA3 Cette étude
pGAL-(67_97)-GFP pAAT-55 URA3 Cette étude
pGAL-A62S-GFP pAAT-56 URA3 Cette étude
pGAL-I15F-GFP pAAT-57 URA3 Cette étude
pGAL-R63W-GFP pAAT-58 URA3 Cette étude
pGAL-R63Q-GFP pAAT-59 URA3 Cette étude
pGAL-W53R-GFP pAAT-60 URA3 Cette étude
pGAL-R57Q-GFP pAAT-61 URA3 Cette étude
pGAL-I65P-GFP pAAT-62 URA3 Cette étude
pGAL-G121S-GFP pAAT-63 URA3 Cette étude
pGAL-G128Q-GFP pAAT-64 URA3 Cette étude
pGAL-R102H-GFP pAAT-65 URA3 Cette étude
Tableau 9 (suite). Plasmides utilisés pour l’étude de la localisation cellulaire
Nom Réference Marqueur Source
pET15-RPS19Hs pAAT-129 Cette étude
pET15-RPS19Sc pAAT-130 Cette étude
pET15-RPS19Pa pAAT-131 Cette étude
pET15-RPS24Pa pAAT-132 Cette étude
pET15-RPS24Hs pAAT-133 Cette étude
pET15-RPS5Hs pAAT-134 Cette étude
pET15-RPS16Hs pAAT-135 Cette étude
pET15-RPS18Hs pAAT-136 Cette étude
pGEM-F1-HsARN 18S pAAT-137 Cette étude
pGEM-F2-HsARN 18S pAAT-138 Cette étude
pGEM-F3-HsARN 18S pAAT-139 Cette étude
pGEM-F4-HsARN 18S pAAT-140 Cette étude
Tableau 10. Plasmides utilisés pour la production de protéines recombinantes et latranscription in vitro de différents fragments de l'ARN 18S
162

BIBLIOGRAPHIE
163

164

BIBLIOGRAPHIE
1. Andersen JS, Lyon CE, Fox AH, Leung AK, Lam YW, Steen H, Mann M, LamondAI (2002) Directed proteomic analysis of the human nucleolus. Curr Biol 12: 1-11
2. Anderson SJ, Lauritsen JP, Hartman MG, Foushee AM, Lefebvre JM, Shinton SA,Gerhardt B, Hardy RR, Oravecz T, Wiest DL (2007) Ablation of ribosomalprotein L22 selectively impairs alphabeta T cell development by activation of ap53-dependent checkpoint. Immunity 26: 759-772
3. Angelini M, Cannata S, Mercaldo V, Gibello L, Santoro C, Dianzani I, Loreni F(2007) Missense mutations associated with Diamond-Blackfan anemia affect theassembly of ribosomal protein S19 into the ribosome. Hum Mol Genet 16:1720-1727
4. Angermayr M, Bandlow W (2002) RIO1, an extraordinary novel protein kinase.FEBS Lett 524: 31-36
5. Antoine M, Reimers K, Wirz W, Gressner AM, Muller R, Kiefer P (2005)Identification of an unconventional nuclear localization signal in humanribosomal protein S2. Biochem Biophys Res Commun 335: 146-153
6. Ayrault O, Andrique L, Larsen CJ, Seite P (2004) Human Arf tumor suppressorspecifically interacts with chromatin containing the promoter of rRNA genes.Oncogene 23: 8097-8104
7. Ayrault O, Andrique L, Larsen CJ, Seite P (2006) [The negative regulation ofribosome biogenesis: a new Arf-dependent pathway controlling cellproliferation?]. Med Sci (Paris) 22: 519-524
8. Azuma M, Toyama R, Laver E, Dawid IB (2006) Perturbation of rRNA synthesis inthe bap28 mutation leads to apoptosis mediated by p53 in the zebrafish centralnervous system. J Biol Chem 281: 13309-13316
9. Bachmann M, Moroy T (2005) The serine/threonine kinase Pim-1. Int J BiochemCell Biol 37: 726-730
10. Bagnara G, Zauli G, Vitale L, Rosito P, Vecchi V, Paolucci G, Avanzi G,Ramenghi U, Timeus F, Gabutti V (1991) In vitro growth and regulation of bonemarrow enriched CD34+ hematopoietic progenitors in Diamond-blackfanAnemia. Blood 78: 2203-2210
11. Bernstein KA, Gallagher JE, Mitchell BM, Granneman S, Baserga SJ (2004) Thesmall-subunit processome is a ribosome assembly intermediate. Eukaryot Cell3: 1619-1626
12. Bertwistle D, Sugimoto M, Sherr CJ (2004) Physical and functional interactions ofthe Arf tumor suppressor protein with nucleophosmin/B23. Mol Cell Biol 24:985-996
13. Bhat KP, Itahana K, Jin A, Zhang Y (2004) Essential role of ribosomal proteinL11 in mediating growth inhibition-induced p53 activation. Embo J 23: 2402-2412
14. Bischoff FR, Gorlich D (1997) RanBP1 is crucial for the release of RanGTP fromimportin beta-related nuclear transport factors. FEBS Lett 419: 249-254
15. Boisvert FM, van Koningsbruggen S, Navascues J, Lamond AI (2007) Themultifunctional nucleolus. Nat Rev Mol Cell Biol 8: 574-585
165

16. Buchhaupt M, Meyer B, Kotter P, Entian KD (2006) Genetic evidence for 18SrRNA binding and an Rps19p assembly function of yeast nucleolar proteinNep1p. Mol Genet Genomics 276: 273-284
17. Buenders T, Tonkin L, Freund S, Bycroft M, Warren A. (2008) Structural andgenetic studies in Diamond Blackfan Anemia. 9th Annual Diamond BlackfanAnemia International Consensus Conference., New York.
18. Carnero A, Hudson JD, Price CM, Beach DH (2000) p16INK4A and p19ARF actin overlapping pathways in cellular immortalization. Nat Cell Biol 2: 148-155
19. Casadevall N, Croisille L, Auffray I, Tchernia G, Coulombel L (1994) Age-relatedalterations in erythroid and granulopoietic progenitors in Diamond-Blackfananaemia. Br J Haematol 87: 369-375
20. Chatr-Aryamontri A, Angelini M, Garelli E, Tchernia G, Ramenghi U, Dianzani I,Loreni F (2004) Nonsense-mediated and nonstop decay of ribosomal proteinS19 mRNA in Diamond-Blackfan anemia. Hum Mutat 24: 526-533
21. Chen S, Warszawski J, Bader-Meunier B, Tchernia G, Da Costa L, Marie I,Dommergues JP (2005) Diamond-blackfan anemia and growth status: theFrench registry. J Pediatr 147: 669-673
22. Chen W, Bucaria J, Band DA, Sutton A, Sternglanz R (2003) Enp1, a yeastprotein associated with U3 and U14 snoRNAs, is required for pre-rRNAprocessing and 40S subunit synthesis. Nucleic Acids Res 31: 690-699
23. Chiocchetti A, Gibello L, Carando A, Aspesi A, Secco P, Garelli E, Loreni F,Angelini M, Biava A, Dahl N, Dianzani U, Ramenghi U, Santoro C, Dianzani I(2005) Interactions between RPS19, mutated in Diamond-Blackfan anemia, andthe PIM-1 oncoprotein. Haematologica 90: 1453-1462
24. Choesmel V, Bacqueville D, Rouquette J, Noaillac-Depeyre J, Fribourg S, CretienA, Leblanc T, Tchernia G, Da Costa L, Gleizes PE (2007) Impaired ribosomebiogenesis in Diamond-Blackfan anemia. Blood 109: 1275-1283
25. Choesmel V, Fribourg S, Aguissa-Toure AH, Pinaud N, Legrand P, Gazda HT,Gleizes PE (2008) Mutation of ribosomal protein RPS24 in Diamond-Blackfananemia results in a ribosome biogenesis disorder. Hum Mol Genet 17: 1253-1263
26. Chook YM, Blobel G (1999) Structure of the nuclear transport complexkaryopherin-beta2-Ran x GppNHp. Nature 399: 230-237
27. Chook YM, Blobel G (2001) Karyopherins and nuclear import. Curr Opin StructBiol 11: 703-715
28. Cingolani G, Bednenko J, Gillespie MT, Gerace L (2002) Molecular basis for therecognition of a nonclassical nuclear localization signal by importin beta. MolCell 10: 1345-1353
29. Cingolani G, Petosa C, Weis K, Muller CW (1999) Structure of importin-betabound to the IBB domain of importin-alpha. Nature 399: 221-229
30. Cmejla R, Cmejlova J, Handrkova H, PetraK J, Pospisilova D (2007) Ribosomalprotein S17 gene (RPS17) is mutated in Diamond-Blackfan Anemia. Humanmutation 28: 1178-1182
31. Cmejlova J, Cerna Z, Votava T, Pospisilova D, Cmejla R (2006) Identification ofa new in-frame deletion of six amino acids in ribosomal protein S19 in a patientwith Diamond-Blackfan anemia. Blood Cells Mol Dis 36: 337-341
32. Conti E, Izaurralde E (2001) Nucleocytoplasmic transport enters the atomic age.Curr Opin Cell Biol 13: 310-319
166

33. Conti E, Kuriyan J (2000) Crystallographic analysis of the specific yet versatilerecognition of distinct nuclear localization signals by karyopherin alpha.Structure 8: 329-338
34. Conti E, Muller CW, Stewart M (2006) Karyopherin flexibility innucleocytoplasmic transport. Curr Opin Struct Biol 16: 237-244
35. Conti E, Uy M, Leighton L, Blobel G, Kuriyan J (1998) Crystallographic analysisof the recognition of a nuclear localization signal by the nuclear import factorkaryopherin alpha. Cell 94: 193-204
36. Cook A, Fernandez E, Lindner D, Ebert J, Schlenstedt G, Conti E (2005) Thestructure of the nuclear export receptor Cse1 in its cytosolic state reveals aclosed conformation incompatible with cargo binding. Mol Cell 18: 355-367
37. Cormack BP, Bertram G, Egerton M, Gow NA, Falkow S, Brown AJ (1997)Yeast-enhanced green fluorescent protein (yEGFP)a reporter of gene expressionin Candida albicans. Microbiology 143 (Pt 2): 303-311
38. Coute Y, Burgess JA, Diaz JJ, Chichester C, Lisacek F, Greco A, Sanchez JC(2006) Deciphering the human nucleolar proteome. Mass Spectrom Rev 25:215-234
39. Cronshaw JM, Krutchinsky AN, Zhang W, Chait BT, Matunis MJ (2002)Proteomic analysis of the mammalian nuclear pore complex. J Cell Biol 158:915-927
40. Da Costa L, Narla G, Willig TN, Peters LL, Parra M, Fixler J, Tchernia G,Mohandas N (2003) Ribosomal protein S19 expression during erythroiddifferentiation. Blood 101: 318-324
41. Dai MS, Arnold H, Sun XX, Sears R, Lu H (2007) Inhibition of c-Myc activity byribosomal protein L11. Embo J 26: 3332-3345
42. Dai MS, Lu H (2004) Inhibition of MDM2-mediated p53 ubiquitination anddegradation by ribosomal protein L5. J Biol Chem 279: 44475-44482
43. Dai MS, Shi D, Jin Y, Sun XX, Zhang Y, Grossman SR, Lu H (2006) Regulationof the MDM2-p53 pathway by ribosomal protein L11 involves a post-ubiquitination mechanism. J Biol Chem 281: 24304-24313
44. Dai MS, Sun XX, Lu H (2008) Aberrant expression of nucleostemin activates p53and induces cell cycle arrest via inhibition of MDM2. Mol Cell Biol
45. Dai MS, Zeng SX, Jin Y, Sun XX, David L, Lu H (2004) Ribosomal protein L23activates p53 by inhibiting MDM2 function in response to ribosomalperturbation but not to translation inhibition. Mol Cell Biol 24: 7654-7668
46. Dez C, Tollervey D (2004) Ribosome synthesis meets the cell cycle. Curr OpinMicrobiol 7: 631-637
47. Diamond L, Blackfan K (1938) Hypoplastic anemia. Am J Dis Child. 56: 464-46748. Dragon F, Gallagher JE, Compagnone-Post PA, Mitchell BM, Porwancher KA,
Wehner KA, Wormsley S, Settlage RE, Shabanowitz J, Osheim Y, Beyer AL,Hunt DF, Baserga SJ (2002) A large nucleolar U3 ribonucleoprotein required for18S ribosomal RNA biogenesis. Nature 417: 967-970
49. Draptchinskaia N, Gustavsson P, Andersson B, Pettersson M, Willig TN, DianzaniI, Ball S, Tchernia G, Klar J, Matsson H, Tentler D, Mohandas N, Carlsson B,Dahl N (1999) The gene encoding ribosomal protein S19 is mutated inDiamond-Blackfan anaemia. Nat Genet 21: 169-175
50. Du YC, Stillman B (2002) Yph1p, an ORC-interacting protein: potential linksbetween cell proliferation control, DNA replication, and ribosome biogenesis.Cell 109: 835-848
167

51. Ebert BL, Lee MM, Pretz JL, Subramanian A, Mak R, Golub TR, Sieff CA (2005)An RNA interference model of RPS19 deficiency in Diamond-Blackfan anemiarecapitulates defective hematopoiesis and rescue by dexamethasone:identification of dexamethasone-responsive genes by microarray. Blood 105:4620-4626
52. Ebert BL, Pretz J, Bosco J, Chang CY, Tamayo P, Galili N, Raza A, Root DE,Attar E, Ellis SR, Golub TR (2008) Identification of RPS14 as a 5q- syndromegene by RNA interference screen. Nature 451: 335-339
53. Fahrenkrog B, Aebi U (2003) The nuclear pore complex: nucleocytoplasmictransport and beyond. Nat Rev Mol Cell Biol 4: 757-766
54. Farrar JE, Nater M, Caywood E, McDevitt M, Kowalski J, Takemoto C, Talbot C,Meltzer P, Esposito D, Beggs AH, Schnierder HE, Grabowska A, Ball SE,Niewiadomska E, Sieff CA, Vlachos A, Atsidaftos E, Ellis SR, Lipton JM,Gazda HT, R.J. A (2007) A large ribosomal subunit protein abnormality inDiamond-Blackfan Anemia (DBA). Blood (ASH Meeting Abstracts) 110: 422
55. Fatica A, Oeffinger M, Dlakic M, Tollervey D (2003) Nob1p is required forcleavage of the 3' end of 18S rRNA. Mol Cell Biol 23: 1798-1807
56. Fatica A, Tollervey D, Dlakic M (2004) PIN domain of Nob1p is required for D-site cleavage in 20S pre-rRNA. Rna 10: 1698-1701
57. Ferreira-Cerca S, Poll G, Gleizes PE, Tschochner H, Milkereit P (2005) Roles ofeukaryotic ribosomal proteins in maturation and transport of pre-18S rRNA andribosome function. Mol Cell 20: 263-275
58. Ficarro SB, McCleland ML, Stukenberg PT, Burke DJ, Ross MM, Shabanowitz J,Hunt DF, White FM (2002) Phosphoproteome analysis by mass spectrometryand its application to Saccharomyces cerevisiae. Nat Biotechnol 20: 301-305
59. Flygare J, Aspesi A, Bailey JC, Miyake K, Caffrey JM, Karlsson S, Ellis SR(2007) Human RPS19, the gene mutated in Diamond-Blackfan anemia, encodesa ribosomal protein required for the maturation of 40S ribosomal subunits.Blood 109: 980-986
60. Flygare J, Kiefer T, Miyake K, Utsugisawa T, Hamaguchi I, Da Costa L, RichterJ, Davey EJ, Matsson H, Dahl N, Wiznerowicz M, Trono D, Karlsson S (2005)Deficiency of ribosomal protein S19 in CD34+ cells generated by siRNA blockserythroid development and mimics defects seen in Diamond-Blackfan anemia.Blood 105: 4627-4634
61. Fontes MR, Teh T, Kobe B (2000) Structural basis of recognition of monopartiteand bipartite nuclear localization sequences by mammalian importin-alpha. JMol Biol 297: 1183-1194
62. Ford CL, Randal-Whitis L, Ellis SR (1999) Yeast proteins related to thep40/laminin receptor precursor are required for 20S ribosomal RNA processingand the maturation of 40S ribosomal subunits. Cancer Res 59: 704-710
63. French SL, Osheim YN, Cioci F, Nomura M, Beyer AL (2003) In exponentiallygrowing Saccharomyces cerevisiae cells, rRNA synthesis is determined by thesummed RNA polymerase I loading rate rather than by the number of activegenes. Mol Cell Biol 23: 1558-1568
64. Fromont-Racine M, Senger B, Saveanu C, Fasiolo F (2003) Ribosome assembly ineukaryotes. Gene 313: 17-42
65. Gavin AC, Bosche M, Krause R, Grandi P, Marzioch M, Bauer A, Schultz J, RickJM, Michon AM, Cruciat CM, Remor M, Hofert C, Schelder M, Brajenovic M,Ruffner H, Merino A, Klein K, Hudak M, Dickson D, Rudi T, Gnau V, BauchA, Bastuck S, Huhse B, Leutwein C, Heurtier MA, Copley RR, Edelmann A,
168

Querfurth E, Rybin V, Drewes G, Raida M, Bouwmeester T, Bork P, SeraphinB, Kuster B, Neubauer G, Superti-Furga G (2002) Functional organization of theyeast proteome by systematic analysis of protein complexes. Nature 415: 141-147
66. Gazda HT, Grabowska A, Merida-Long LB, Latawiec E, Schneider HE, LiptonJM, Vlachos A, Atsidaftos E, Ball SE, Orfali KA, Niewiadomska E, Da Costa L,Tchernia G, Niemeyer C, Meerpohl JJ, Stahl J, Schratt G, Glader B, Backer K,Wong C, Nathan DG, Beggs AH, Sieff CA (2006) Ribosomal protein S24 geneis mutated in Diamond-Blackfan anemia. Am J Hum Genet 79: 1110-1118
67. Gazda HT, Sheen MR, Darras N, Shneider H, Sieff CA, Ball SE, NiewiadomskaE, Newburger PE, Atsidaftos E, Vlachos A, Lipton JM, Beggs AH (2007)Mutations of the genes for Ribosomal Rroteins L5 and L11are common cause ofDiamond-Blackfan Anemia. Blood (ASH Meeting Abstracts) 110: 241
68. Gazda HT, Zhong R, Long L, Niewiadomska E, Lipton JM, Ploszynska A, ZauchaJM, Vlachos A, Atsidaftos E, Viskochil DH, Niemeyer CM, Meerpohl JJ,Rokicka-Milewska R, Pospisilova D, Wiktor-Jedrzejczak W, Nathan DG, BeggsAH, Sieff CA (2004) RNA and protein evidence for haplo-insufficiency inDiamond-Blackfan anaemia patients with RPS19 mutations. Br J Haematol127: 105-113
69. Gelperin D, Horton L, Beckman J, Hensold J, Lemmon SK (2001) Bms1p, a novelGTP-binding protein, and the related Tsr1p are required for distinct steps of 40Sribosome biogenesis in yeast. Rna 7: 1268-1283
70. Gilchrist D, Mykytka B, Rexach M (2002) Accelerating the rate of disassembly ofkaryopherin.cargo complexes. J Biol Chem 277: 18161-18172
71. Giri N, Kang E, Tisdale JF, Follman D, Rivera M, Schwartz GN, Kim S, YoungNS, Rick ME, Dunbar CE (2000) Clinical and laboratory evidence for atrilineage haematopoietic defect in patients with refractory Diamond-Blackfananaemia. Br J Haematol 108: 167-175
72. Goldfarb DS, Corbett AH, Mason DA, Harreman MT, Adam SA (2004) Importinalpha: a multipurpose nuclear-transport receptor. Trends Cell Biol 14: 505-514
73. Gorlich D, Dabrowski M, Bischoff FR, Kutay U, Bork P, Hartmann E, Prehn S,Izaurralde E (1997) A novel class of RanGTP binding proteins. J Cell Biol 138:65-80
74. Gorlich D, Henklein P, Laskey RA, Hartmann E (1996) A 41 amino acid motif inimportin-alpha confers binding to importin-beta and hence transit into thenucleus. Embo J 15: 1810-1817
75. Gorlich D, Kutay U (1999) Transport between the cell nucleus and the cytoplasm.Annu Rev Cell Dev Biol 15: 607-660
76. Grandi P, Rybin V, Bassler J, Petfalski E, Strauss D, Marzioch M, Schafer T,Kuster B, Tschochner H, Tollervey D, Gavin AC, Hurt E (2002) 90S pre-ribosomes include the 35S pre-rRNA, the U3 snoRNP, and 40S subunitprocessing factors but predominantly lack 60S synthesis factors. Mol Cell 10:105-115
77. Granneman S, Baserga SJ (2004) Ribosome biogenesis: of knobs and RNAprocessing. Exp Cell Res 296: 43-50
78. Granneman S, Nandineni MR, Baserga SJ (2005) The putative NTPase Fap7mediates cytoplasmic 20S pre-rRNA processing through a direct interaction withRps14. Mol Cell Biol 25: 10352-10364
169

79. Gustavsson P, Skeppner G, Johansson B, Berg T, Gordon L, Kreuger A, Dahl N(1997a) Diamond-Blackfan anaemia in a girl with a de novo balanced reciprocalX;19 translocation. J Med Genet 34: 779-782
80. Gustavsson P, Willing TN, van Haeringen A, Tchernia G, Dianzani I, Donner M,Elinder G, Henter JI, Nilsson PG, Gordon L, Skeppner G, van't Veer-Korthof L,Kreuger A, Dahl N (1997b) Diamond-Blackfan anaemia: genetic homogeneityfor a gene on chromosome 19q13 restricted to 1.8 Mb. Nat Genet 16: 368-371
81. Halperin D, Freedman M (1989) Diamond-Blackfan Anemia: etiology,pathophysiology, and treatment. Am J Pediatr Hematol Oncol 11: 4
82. Hamaguchi I, Flygare J, Nishiura H, Brun AC, Ooka A, Kiefer T, Ma Z, Dahl N,Richter J, Karlsson S (2003) Proliferation deficiency of multipotenthematopoietic progenitors in ribosomal protein S19 (RPS19)-deficient diamond-Blackfan anemia improves following RPS19 gene transfer. Mol Ther 7: 613-622
83. He H, Sun Y (2007) Ribosomal protein S27L is a direct p53 target that regulatesapoptosis. Oncogene 26: 2707-2716
84. Hodel AE, Harreman MT, Pulliam KF, Harben ME, Holmes JS, Hodel MR,Berland KM, Corbett AH (2006) Nuclear localization signal receptor affinitycorrelates with in vivo localization in Saccharomyces cerevisiae. J Biol Chem281: 23545-23556
85. Idol RA, Robledo S, Du HY, Crimmins DL, Wilson DB, Ladenson JH, Bessler M,Mason PJ (2007) Cells depleted for RPS19, a protein associated with DiamondBlackfan Anemia, show defects in 18S ribosomal RNA synthesis and smallribosomal subunit production. Blood Cells Mol Dis 39: 35-43
86. Itahana K, Bhat KP, Jin A, Itahana Y, Hawke D, Kobayashi R, Zhang Y (2003)Tumor suppressor ARF degrades B23, a nucleolar protein involved in ribosomebiogenesis and cell proliferation. Mol Cell 12: 1151-1164
87. Jakel S, Gorlich D (1998) Importin beta, transportin, RanBP5 and RanBP7mediate nuclear import of ribosomal proteins in mammalian cells. Embo J 17:4491-4502
88. Jakel S, Mingot JM, Schwarzmaier P, Hartmann E, Gorlich D (2002) Importinsfulfil a dual function as nuclear import receptors and cytoplasmic chaperones forexposed basic domains. Embo J 21: 377-386
89. Jakovljevic J, de Mayolo PA, Miles TD, Nguyen TM, Leger-Silvestre I, Gas N,Woolford JL, Jr. (2004) The carboxy-terminal extension of yeast ribosomalprotein S14 is necessary for maturation of 43S preribosomes. Mol Cell 14: 331-342
90. Johnson AW, Lund E, Dahlberg J (2002) Nuclear export of ribosomal subunits.Trends Biochem Sci 27: 580-585
91. Kaiser C, Michaelis S, Mitchell, A. (1994) Methods in Yeast Genetics. ColdSpring Harbor Laboratory Press: 201-202
92. Kalmanti M, Kalmantis T, Dimitriou H, Kattamis C (1993) Correlation of in vitroenhancement of erythropoiesis and clinical response to steroids in Diamond-Blackfan anaemia. Haematologia (Budap) 25: 263-269
93. Kobe B (1999) Autoinhibition by an internal nuclear localization signal revealedby the crystal structure of mammalian importin alpha. Nat Struct Biol 6: 388-397
94. Kondoh N, Schweinfest CW, Henderson KW, Papas TS (1992) Differentialexpression of S19 ribosomal protein, laminin-binding protein, and human
170

lymphocyte antigen class I messenger RNAs associated with colon carcinomaprogression and differentiation. Cancer Res 52: 791-796
95. Kressler D, Doere M, Rojo M, Linder P (1999) Synthetic lethality withconditional dbp6 alleles identifies rsa1p, a nucleoplasmic protein involved in theassembly of 60S ribosomal subunits. Mol Cell Biol 19: 8633-8645
96. Kubitscheck U, Grunwald D, Hoekstra A, Rohleder D, Kues T, Siebrasse JP,Peters R (2005) Nuclear transport of single molecules: dwell times at the nuclearpore complex. J Cell Biol 168: 233-243
97. Lambertsson A (1998) The minute genes in Drosophila and their molecularfunctions. Adv Genet 38: 69-134
98. Leblanc T, Gluckman E, Brauner R (2003) Growth hormone deficiency caused bypituitary stalk interruption in Diamond-Blackfan anemia. J Pediatr 142: 358
99. Lee BJ, Cansizoglu AE, Suel KE, Louis TH, Zhang Z, Chook YM (2006) Rulesfor nuclear localization sequence recognition by karyopherin beta 2. Cell 126:543-558
100. Lee SJ, Matsuura Y, Liu SM, Stewart M (2005) Structural basis for nuclearimport complex dissociation by RanGTP. Nature 435: 693-696
101. Lee SJ, Sekimoto T, Yamashita E, Nagoshi E, Nakagawa A, Imamoto N,Yoshimura M, Sakai H, Chong KT, Tsukihara T, Yoneda Y (2003) Thestructure of importin-beta bound to SREBP-2: nuclear import of a transcriptionfactor. Science 302: 1571-1575
102. Leger-Silvestre I, Caffrey JM, Dawaliby R, Alvarez-Arias DA, Gas N, BertoloneSJ, Gleizes PE, Ellis SR (2005) Specific Role for Yeast Homologs of theDiamond Blackfan Anemia-associated Rps19 Protein in Ribosome Synthesis. JBiol Chem 280: 38177-38185
103. Leger-Silvestre I, Milkereit P, Ferreira-Cerca S, Saveanu C, Rousselle JC,Choesmel V, Guinefoleau C, Gas N, Gleizes PE (2004) The ribosomal proteinRps15p is required for nuclear exit of the 40S subunit precursors in yeast. EmboJ 23: 2336-2347
104. Li B, Sun M, He B, Yu J, Zhang YD, Zhang YL (2002) Identification ofdifferentially expressed genes in human uterine leiomyomas using differentialdisplay. Cell Res 12: 39-45
105. Lipton J, Kudisch M, Gross R, Nathan D (1986) Defective erythroid progenitordiferentiation system in congenital hypoplastic (Diamond-Blackfan) anemia.Blood 67: 962-968
106. Lipton JM, Atsidaftos E, Zyskind I, Vlachos A (2006) Improving clinical careand elucidating the pathophysiology of Diamond Blackfan anemia: an updatefrom the Diamond Blackfan Anemia Registry. Pediatr Blood Cancer 46: 558-564
107. Lipton JM, Federman N, Khabbaze Y, Schwartz CL, Hilliard LM, Clark JI,Vlachos A (2001) Osteogenic sarcoma associated with Diamond-Blackfananemia: a report from the Diamond-Blackfan Anemia Registry. J PediatrHematol Oncol 23: 39-44
108. Liu SM, Stewart M (2005) Structural basis for the high-affinity binding ofnucleoporin Nup1p to the Saccharomyces cerevisiae importin-beta homologue,Kap95p. J Mol Biol 349: 515-525
109. Lohrum MA, Ludwig RL, Kubbutat MH, Hanlon M, Vousden KH (2003)Regulation of HDM2 activity by the ribosomal protein L11. Cancer Cell 3:577-587
171

110. Macara IG (2001) Transport into and out of the nucleus. Microbiol Mol Biol Rev65: 570-594
111. Madrid AS, Weis K (2006) Nuclear transport is becoming crystal clear.Chromosoma 115: 98-109
112. Marechal V, Elenbaas B, Piette J, Nicolas JC, Levine AJ (1994) The ribosomalL5 protein is associated with mdm-2 and mdm-2-p53 complexes. Mol Cell Biol14: 7414-7420
113. Matsson H, Davey EJ, Draptchinskaia N, Hamaguchi I, Ooka A, Leveen P,Forsberg E, Karlsson S, Dahl N (2004) Targeted disruption of the ribosomalprotein S19 gene is lethal prior to implantation. Mol Cell Biol 24: 4032-4037
114. Matsson H, Klar J, Draptchinskaia N, Gustavsson P, Carlsson B, Bowers D, deBont E, Dahl N (1999) Truncating ribosomal protein S19 mutations and variableclinical expression in Diamond-Blackfan anemia. Hum Genet 105: 496-500
115. Matsuura Y, Lange A, Harreman MT, Corbett AH, Stewart M (2003) Structuralbasis for Nup2p function in cargo release and karyopherin recycling in nuclearimport. Embo J 22: 5358-5369
116. Matsuura Y, Stewart M (2004) Structural basis for the assembly of a nuclearexport complex. Nature 432: 872-877
117. Matsuura Y, Stewart M (2005) Nup50/Npap60 function in nuclear protein importcomplex disassembly and importin recycling. Embo J 24: 3681-3689
118. McGuckin CP, Ball SE, Gordon-Smith EC (1995) Diamond-Blackfan anaemia:three patterns of in vitro response to haemopoietic growth factors. Br JHaematol 89: 457-464
119. Miller OL, Jr Beatty BR. (1969) Visualization of nucleolar genes. Sciences 164:955-957
120. Moroianu J (1998) Distinct nuclear import and export pathways mediated bymembers of the karyopherin beta family. J Cell Biochem 70: 231-239
121. Mosammaparast N, Pemberton LF (2004) Karyopherins: from nuclear-transportmediators to nuclear-function regulators. Trends Cell Biol 14: 547-556
122. Nazar RN (2004) Ribosomal RNA processing and ribosome biogenesis ineukaryotes. IUBMB Life 56: 457-465
123. Ohene-Abuakwa Y, Orfali KA, Marius C, Ball SE (2005) Two-phase culture inDiamond Blackfan anemia: localization of erythroid defect. Blood 105: 838-846
124. Olson MO (2004) Sensing cellular stress: another new function for thenucleolus? Sci STKE 2004: pe10
125. Pederson T (1998) The plurifunctional nucleolus. Nucleic Acids Res 26: 3871-3876
126. Pemberton LF, Paschal BM (2005) Mechanisms of receptor-mediated nuclearimport and nuclear export. Traffic 6: 187-198
127. Pendle AF, Clark GP, Boon R, Lewandowska D, Lam YW, Andersen J, MannM, Lamond AI, Brown JW, Shaw PJ (2005) Proteomic analysis of theArabidopsis nucleolus suggests novel nucleolar functions. Mol Biol Cell 16:260-269
128. Pestov DG, Strezoska Z, Lau LF (2001) Evidence of p53-dependent cross-talkbetween ribosome biogenesis and the cell cycle: effects of nucleolar proteinBop1 on G(1)/S transition. Mol Cell Biol 21: 4246-4255
129. Plafker SM, Macara IG (2002) Ribosomal protein L12 uses a distinct nuclearimport pathway mediated by importin 11. Mol Cell Biol 22: 1266-1275
130. Ramenghi U, Campagnoli MF, Garelli E, Carando A, Brusco A, Bagnara GP,Strippoli P, Izzi GC, Brandalise S, Riccardi R, Dianzani I (2000) Diamond-
172

Blackfan anemia: report of seven further mutations in the RPS19 gene andevidence of mutation heterogeneity in the Italian population. Blood Cells MolDis 26: 417-422
131. Ribbeck K, Gorlich D (2001) Kinetic analysis of translocation through nuclearpore complexes. Embo J 20: 1320-1330
132. Ribbeck K, Lipowsky G, Kent HM, Stewart M, Gorlich D (1998) NTF2 mediatesnuclear import of Ran. Embo J 17: 6587-6598
133. Rizos H, McKenzie HA, Ayub AL, Woodruff S, Becker TM, Scurr LL, Stahl J,Kefford RF (2006) Physical and functional interaction of the p14ARF tumorsuppressor with ribosomes. J Biol Chem 281: 38080-38088
134. Rodriguez M, Benito A, Tubert P, Castro J, Ribo M, Beaumelle B, Vilanova M(2006) A cytotoxic ribonuclease variant with a discontinuous nuclearlocalization signal constituted by basic residues scattered over three areas of themolecule. J Mol Biol 360: 548-557
135. Rosenblum JS, Pemberton LF, Blobel G (1997) A nuclear import pathway for aprotein involved in tRNA maturation. J Cell Biol 139: 1655-1661
136. Rout MP, Aitchison JD, Suprapto A, Hjertaas K, Zhao Y, Chait BT (2000) Theyeast nuclear pore complex: composition, architecture, and transportmechanism. J Cell Biol 148: 635-651
137. Rout MP, Blobel G, Aitchison JD (1997) A distinct nuclear import pathway usedby ribosomal proteins. Cell 89: 715-725
138. Rout MP, Wente SR (1994) Pores for thought: nuclear pore complex proteins.Trends Cell Biol 4: 357-365
139. Rubbi CP, Milner J (2003) Disruption of the nucleolus mediates stabilization ofp53 in response to DNA damage and other stresses. Embo J 22: 6068-6077
140. Santucci MA, Bagnara GP, Strippoli P, Bonsi L, Vitale L, Tonelli R, Locatelli F,Gabutti V, Ramenghi U, D'Avanzo M, Paolucci G, Rosito P, Pession A,Freedman MH (1999) Long-term bone marrow cultures in Diamond-Blackfananemia reveal a defect of both granulomacrophage and erythroid progenitors.Exp Hematol 27: 9-18
141. Schafer T, Maco B, Petfalski E, Tollervey D, Bottcher B, Aebi U, Hurt E (2006)Hrr25-dependent phosphorylation state regulates organization of the pre-40Ssubunit. Nature 441: 651-655
142. Schafer T, Strauss D, Petfalski E, Tollervey D, Hurt E (2003) The path fromnucleolar 90S to cytoplasmic 40S pre-ribosomes. Embo J 22: 1370-1380
143. Scherl A, Coute Y, Deon C, Calle A, Kindbeiter K, Sanchez JC, Greco A,Hochstrasser D, Diaz JJ (2002) Functional proteomic analysis of humannucleolus. Mol Biol Cell 13: 4100-4109
144. Scheurle D, DeYoung MP, Binninger DM, Page H, Jahanzeb M, Narayanan R(2000) Cancer gene discovery using digital differential display. Cancer Res 60:4037-4043
145. Schlenstedt G, Smirnova E, Deane R, Solsbacher J, Kutay U, Görlich D,Ponstingl H, Bischoff FR (1997) Yrb4p, a yeast ran-GTP-binding proteininvolved in import of ribosomal protein L25 into the nucleus. EMBO J 16:6237-6249
146. Seiser RM, Sundberg AE, Wollam BJ, Zobel-Thropp P, Baldwin K, Spector MD,Lycan DE (2006) Ltv1 is required for efficient nuclear export of the ribosomalsmall subunit in Saccharomyces cerevisiae. Genetics 174: 679-691
173

147. Sengpiel V, Rost T, Gorogh T, Rathcke IO, Werner JA (2004) S19-mRNAexpression in squamous cell carcinomas of the upper aerodigestive tract.Anticancer Res 24: 2161-2167
148. Shrestha A, Horino K, Nishiura H, Yamamoto T (1999) Acquired immuneresponse as a consequence of the macrophage-dependent apoptotic cellclearance and role of the monocyte chemotactic S19 ribosomal protein dimer inthis connection. Lab Invest 79: 1629-1642
149. Smith A, Brownawell A, Macara IG (1998) Nuclear import of Ran is mediatedby the transport factor NTF2. Curr Biol 8: 1403-1406
150. Soulet F, Al Saati T, Roga S, Amalric F, Bouche G (2001) Fibroblast growthfactor-2 interacts with free ribosomal protein S19. Biochem Biophys ResCommun 289: 591-596
151. Spahn CM, Beckmann R, Eswar N, Penczek PA, Sali A, Blobel G, Frank J(2001) Structure of the 80S ribosome from Saccharomyces cerevisiae--tRNA-ribosome and subunit-subunit interactions. Cell 107: 373-386
152. Stavreva DA, Kawasaki M, Dundr M, Koberna K, Muller WG, Tsujimura-Takahashi T, Komatsu W, Hayano T, Isobe T, Raska I, Misteli T, Takahashi N,McNally JG (2006) Potential roles for ubiquitin and the proteasome duringribosome biogenesis. Mol Cell Biol 26: 5131-5145
153. Stewart M (2007) Molecular mechanism of the nuclear protein import cycle. NatRev Mol Cell Biol 8: 195-208
154. Stewart M, Baker RP, Bayliss R, Clayton L, Grant RP, Littlewood T, MatsuuraY (2001) Molecular mechanism of translocation through nuclear pore complexesduring nuclear protein import. FEBS Lett 498: 145-149
155. Sugimoto M, Kuo ML, Roussel MF, Sherr CJ (2003) Nucleolar Arf tumorsuppressor inhibits ribosomal RNA processing. Mol Cell 11: 415-424
156. Sulic S, Panic L, Barkic M, Mercep M, Uzelac M, Volarevic S (2005)Inactivation of S6 ribosomal protein gene in T lymphocytes activates a p53-dependent checkpoint response. Genes Dev 19: 3070-3082
157. Sun XX, Dai MS, Lu H (2007) 5-fluorouracil activation of p53 involves anMDM2-ribosomal protein interaction. J Biol Chem 282: 8052-8059
158. Sydorskyy Y, Dilworth DJ, Yi EC, Goodlett DR, Wozniak RW, Aitchison JD(2003) Intersection of the Kap123p-mediated nuclear import and ribosomeexport pathways. Mol Cell Biol 23: 2042-2054
159. Tabb-Massey A, Caffrey JM, Logsden P, Taylor S, Trent JO, Ellis SR (2003)Ribosomal proteins Rps0 and Rps21 of Saccharomyces cerevisiae haveoverlapping functions in the maturation of the 3' end of 18S rRNA. NucleicAcids Res 31: 6798-6805
160. Timmers AC, Stuger R, Schaap PJ, van 't Riet J, Raué HA (1999) Nuclear andnucleolar localization of Saccharomyces cerevisiae ribosomal proteins S22 andS25. FEBS Lett 452: 335-340
161. Timney BL, Tetenbaum-Novatt J, Agate DS, Williams R, Zhang W, Chait BT,Rout MP (2006) Simple kinetic relationships and nonspecific competitiongovern nuclear import rates in vivo. J Cell Biol 175: 579-593
162. Tsai P, Arkin S, Lipton J (1989) An intrinsic progenitor defect in Diamond-Blackfan anemia. Br J Haematol 73: 112-120
163. Tschochner H, Hurt E (2003) Pre-ribosomes on the road from the nucleolus tothe cytoplasm. Trends Cell Biol 13: 255-263
174

164. Valasek L, Hasek J, Nielsen KH, Hinnebusch AG (2001) Dual function ofeIF3j/Hcr1p in processing 20 S pre-rRNA and translation initiation. J Biol Chem276: 43351-43360
165. Vanrobays E, Gelugne JP, Gleizes PE, Caizergues-Ferrer M (2003) Latecytoplasmic maturation of the small ribosomal subunit requires RIO proteins inSaccharomyces cerevisiae. Mol Cell Biol 23: 2083-2095
166. Vanrobays E, Gleizes PE, Bousquet-Antonelli C, Noaillac-Depeyre J,Caizergues-Ferrer M, Gelugne JP (2001) Processing of 20S pre-rRNA to 18Sribosomal RNA in yeast requires Rrp10p, an essential non-ribosomalcytoplasmic protein. Embo J 20: 4204-4213
167. Venema J, Tollervey D (1999) Ribosome synthesis in Saccharomyces cerevisiae.Annu Rev Genet 33: 261-311
168. Verlinsky Y, Rechitsky S, Sharapova T, Morris R, Taranissi M, Kuliev A (2004)Preimplantation HLA testing. Jama 291: 2079-2085
169. Vetter IR, Arndt A, Kutay U, Gorlich D, Wittinghofer A (1999) Structural viewof the Ran-Importin beta interaction at 2.3 A resolution. Cell 97: 635-646
170. Visintin R, Amon A (2000) The nucleolus: the magician's hat for cell cycletricks. Curr Opin Cell Biol 12: 372-377
171. Wan F, Anderson DE, Barnitz RA, Snow A, Bidere N, Zheng L, Hegde V, LamLT, Staudt LM, Levens D, Deutsch WA, Lenardo MJ (2007) Ribosomal proteinS3: a KH domain subunit in NF-kappaB complexes that mediates selective generegulation. Cell 131: 927-939
172. Warner JR (1999) The economics of ribosome biosynthesis in yeast. TrendsBiochem Sci 24: 437-440
173. Weis K (2003) Regulating access to the genome: nucleocytoplasmic transportthroughout the cell cycle. Cell 112: 441-451
174. Weis K, Ryder U, Lamond AI (1996) The conserved amino-terminal domain ofhSRP1 alpha is essential for nuclear protein import. Embo J 15: 1818-1825
175. Willig TN, Draptchinskaia N, Dianzani I, Ball S, Niemeyer C, Ramenghi U,Orfali K, Gustavsson P, Garelli E, Brusco A, Tiemann C, Perignon JL, BouchierC, Cicchiello L, Dahl N, Mohandas N, Tchernia G (1999a) Mutations inribosomal protein S19 gene and diamond blackfan anemia: wide variations inphenotypic expression. Blood 94: 4294-4306
176. Willig TN, Gazda H, Sieff CA (2000) Diamond-Blackfan anemia. Curr OpinHematol 7: 85-94
177. Willig TN, Niemeyer CM, Leblanc T, Tiemann C, Robert A, Budde J,Lambiliotte A, Kohne E, Souillet G, Eber S, Stephan JL, Girot R, Bordigoni P,Cornu G, Blanche S, Guillard JM, Mohandas N, Tchernia G (1999b)Identification of new prognosis factors from the clinical and epidemiologicanalysis of a registry of 229 Diamond-Blackfan anemia patients. DBA group ofSociete d'Hematologie et d'Immunologie Pediatrique (SHIP), Gesellshaft furPadiatrische Onkologie und Hamatologie (GPOH), and the European Society forPediatric Hematology and Immunology (ESPHI). Pediatr Res 46: 553-561
178. Willig TN, Perignon JL, Gustavsson P, Gane P, Draptchinskaya N, Testard H,Girot R, Debre M, Stephan JL, Chenel C, Cartron JP, Dahl N, Tchernia G(1998) High adenosine deaminase level among healthy probands of DiamondBlackfan anemia (DBA) cosegregates with the DBA gene region onchromosome 19q13. The DBA Working Group of Societe d'ImmunologiePediatrique (SHIP). Blood 92: 4422-4427
175

179. Wsierska-Gadek J, Horky M (2003) How the nucleolar sequestration of p53protein or its interplayers contributes to its (re)-activation. Ann N Y Acad Sci1010: 266-272
180. Yang W, Gelles J, Musser SM (2004) Imaging of single-molecule translocationthrough nuclear pore complexes. Proc Natl Acad Sci U S A 101: 12887-12892
181. Yang W, Musser SM (2006) Nuclear import time and transport efficiency dependon importin beta concentration. J Cell Biol 174: 951-961
182. Yoon A, Peng G, Brandenburger Y, Zollo O, Xu W, Rego E, Ruggero D (2006)Impaired control of IRES-mediated translation in X-linked dyskeratosiscongenita. Science 12: 902-906
183. Zhan K, Vattem KM, Bauer BN, Dever TE, Chen JJ, Wek RC (2002)Phosphorylation of eukaryotic initiation factor 2 by heme-regulated inhibitorkinase-related protein kinases in Schizosaccharomyces pombe is important forresistance to environmental stresses. Mol Cell Biol 22: 7134-7146
184. Zhang J, Harnpicharnchai P, Jakovljevic J, Tang L, Guo Y, Oeffinger M, RoutMP, Hiley SL, Hughes T, Woolford JL, Jr. (2007) Assembly factors Rpf2 andRrs1 recruit 5S rRNA and ribosomal proteins rpL5 and rpL11 into nascentribosomes. Genes Dev 21: 2580-2592
185. Zhang Y, Yu Z, Fu X, Liang C (2002) Noc3p, a bHLH protein, plays an integralrole in the initiation of DNA replication in budding yeast. Cell 109: 849-860
176

ANNEXE
177

178

ETUDE DE LA PROTEINE RPS24
En 1999, les mutations trouvées dans le gène codant la protéine ribosomique Rps19p ne
concernaient que 25% des patients ADB. En 2006, des expériences d'analyse génétique
ont montré le lien entre l’anémie de Diamond-Blackfan et le gène RPS24 chez environ
2% de patients (Gazda et al., 2006). La mutation de ce gène qui code pour une protéine
de la petite sous-unité du ribosome a ainsi renforcé l’idée que l’ADB soit liée à un
désordre ribosomique.
A l’époque, nous étions sur le point de resoudre la structure cristalline de l’homologue
Rps19p chez P. abyssi. Et parallèlement au travail effectué dans l’équipe pour
comprendre l’implication de Rps24p dans la biogenèse des ribosomes (chez la levure S.
cerevisiae) et l’ADB (sur des cellules humaines), un travail collaboratif a été relancé
avec Sébastien Fribourg (IECB, Bordeaux) en vue de cristalliser la structure de
l’homologue RPS24 de P. abyssi.
Ma participation à ce travail a été de cloner l’homologue de RPS24 de P. abyssi pour la
produire chez E. coli.
La structure, rapidement résolue par Sébastien Fribourg montre une forte homologie
structurale avec la protéine ribosomique RPL23 de Thermus thermophilus. Elle contient
un domaine caractéristique de liaison à l’ARN.
L’analyse fonctionnelle de Rps24p humaine montre qu’elle est requise pour les clivages
dans le 5’ ETS, alors que Rps19p est impliquée dans la maturation de l’ITS1. Malgré
cette différence, les deux protéines pourraient agir de manière coordonnée dans la
maturation du pré-ARNr.
179

180

Mutation of ribosomal protein RPS24 inDiamond-Blackfan anemia results in a ribosomebiogenesis disorder
Valerie Choesmel1,2, Sebastien Fribourg3,4, Almass-Houd Aguissa-Toure1,2, Noel Pinaud3,4,
Pierre Legrand5, Hanna T. Gazda6,7 and Pierre-Emmanuel Gleizes1,2,�
1Laboratoire de Biologie Moleculaire Eucaryote, Universite de Toulouse, 31062 Toulouse, France, 2CNRS, UMR
5099, 31062 Toulouse, France, 3INSERM U869, Institut Europeen de Chimie et Biologie, F-33607 Pessac, France,4Universite Victor Segalen, Bordeaux 2, F-33076 Bordeaux, France, 5Synchrotron SOLEIL, 91192 Gif sur Yvette
Cedex, France, 6Division of Genetics and Program in Genomics, Children’s Hospital Boston, Boston, MA, USA and7Harvard Medical School, Boston, MA, USA
Received November 12, 2007; Revised and Accepted January 16, 2008
Diamond-Blackfan anemia (DBA) is a rare congenital disease affecting erythroid precursor differentiation.DBA is emerging as a paradigm for a new class of pathologies potentially linked to disorders in ribosomebiogenesis. Three genes encoding ribosomal proteins have been associated to DBA: after RPS19, mutationsin genes RPS24 and RPS17 were recently identified in a fraction of the patients. Here, we show that cells frompatients carrying mutations in RPS24 have defective pre-rRNA maturation, as in the case of RPS19 mutations.However, in contrast to RPS19 involvement in the maturation of the internal transcribed spacer 1, RPS24 isrequired for processing of the 50 external transcribed spacer. Remarkably, epistasis experiments with smallinterfering RNAs indicate that the functions of RPS19 and RPS24 in pre-rRNA processing are connected.Resolution of the crystal structure of RPS24e from the archeon Pyroccocus abyssi reveals domains ofRPS24 potentially involved in interactions with pre-ribosomes. Based on these data, we discuss theimpact of RPS24 mutations and speculate that RPS19 and RPS24 cooperate at a particular stage of ribosomebiogenesis connected to a cell cycle checkpoint, thus affecting differentiation of erythroid precursors as wellas developmental processes.
INTRODUCTION
Although they play a central role in cell metabolism, ribosomalproteins have been linked to genetic diseases only recently. TheRPS19 (ribosomal protein S19) gene was first to be associatedwith a rare congenital red cell disease, Diamond-Blackfananemia (DBA) (1). This pathology is characterized by a severeerythroblastopenia that results from early arrest ofpro-erythroblast differentiation (2–4). DBA patients alsoexhibit a number of heterogeneous clinical features (shortstature, webbed neck, cranio-facial, skeletal, kidney, and heartabnormalities), consistent with the ubiquitous nature of RPS19expression and of ribosome biogenesis. Heterozygous mutationsof RPS19 affect �25% of the DBA patients. Why RPS19
deficiency has a prime effect on erythropoiesis remainsunclear (5,6). It has been suspected that RPS19 might have anextra-ribosomal function important for pro-erythroblasts, butno solid evidence has substantiated this hypothesis so far,despite the characterization of several proteins interacting withRPS19 (7–9). Alternatively (although not exclusively),pro-erythroblast differentiation may be critically sensitive topartial deficiency of the translation machinery biogenesis orfunction. Indeed, this latter possibility has recently gainedstrong support from the finding that two other ribosomalproteins, RPS24 and RPS17, are mutated in �2% of DBApatients (10,11). Thus, so far, three autosomal genes encodingribosomal proteins have been linked to this acute defect inerythropoiesis.
�To whom correspondence should be addressed at: LBME-CNRS, 118 route de Narbonne, F-31062 Toulouse Cedex, France. Tel: þ33 561335926;Fax: þ33 561335886; Email: [email protected]
# The Author 2008. Published by Oxford University Press. All rights reserved.For Permissions, please email: [email protected]
Human Molecular Genetics, 2008, Vol. 17, No. 9 1253–1263doi:10.1093/hmg/ddn015Advance Access published on January 29, 2008
181

We and others have shown that RPS19 is critical for matu-ration of the 40S ribosomal subunit in yeast (12) and inmammals (13–15). Knockdown of RPS19 expression withsmall interfering RNAs (siRNAs) delays pre-rRNA cleavagein the internal-transcribed spacer 1 (ITS1; Fig. 1) and preventssubsequent maturation of the 18S rRNA precursor 30 end intopre-40S particles. A similar defect, although milder, isdetected in cells from patients bearing mutations in RPS19.Consistently, DBA-related mutations in RPS19 hamper itsincorporation into ribosomal subunits (16,17). Perturbationof ribosome biogenesis is known to trigger cell-cycle arrestand apoptosis signaling pathways, as observed upon RNA-polymerase I inhibition (18,19) or expression of mutatedribosome biogenesis factors (20,21). A constitutive defect inribosome production in DBA patients may thus put the cellsunder constant stress, which would render some cell types,especially pro-erythroblasts, very sensitive to cell-cyclearrest signals.
Sub-optimal production of ribosomes, or alteration of aribosomal protein function in the 40S subunit, might alsohave direct consequences on translation efficiency and regu-lation, which pro-erythroblasts may be particularly sensitiveto. Recent data indicate a lower protein synthesis rate in acti-vated lymphocytes from DBA patients (22). In addition,RPS19 was found to interact with the translation initiationfactor eIF2-a (23), and with the serine-threonine kinasePIM-1 in translating ribosomes (7), which may be related toa function in translation regulation.
The recent discovery that RPS24 is mutated in a fraction ofpatients brings an opportunity to further challenge the linkbetween ribosome biogenesis and DBA (10). Although,RPS24 homolog in yeast Saccharomyces cerevisiae wasfound essential for pre-rRNA processing (24), its function in
metazoan cells has not been described yet. In addition,whether there is a functional link between RPS19 andRPS24 is unknown. One may wonder to which degree thetwo proteins cooperate in ribosome biogenesis, and whetherthe lack of function of any of them may alter a particularstep connected to a cell-cycle checkpoint. The data presentedhere show that RPS24 bears structural similarity with RNA-binding proteins and is as essential as RPS19 for productionof the small ribosomal subunit. However, unlike RPS19, it isinvolved in processing of the pre-rRNA 50-externaltranscribed spacer (50-ETS). Accordingly, lymphoblastoidcells derived from DBA patients bearing mutations in RPS24and RPS19 display different pre-rRNA processing defects.Double knockdown of these proteins suppresses the 50-ETSprocessing defect observed upon the lack of RPS24, whichsuggests a functional relationship between the two ribosomalproteins.
RESULTS
Alteration of pre-rRNA processing in DBA patients withmutations in RPS24
To analyze the impact of RPS24 mutation on pre-rRNA pro-cessing in DBA, we examined the pre-rRNA maturationpattern in lymphoblastoid cell lines (LCLs) derived fromDBA patients with mutations in RPS24 (Q106stop, R16stop,del E2-Q22) (10) and in RPS19 (R62Q, del exon2, delA58-T60) (25). Total RNAs were submitted to northern blot-ting and analyzed with a probe to the 50-ITS1 probe specificof the 18S rRNA precursors (Fig. 2). Cells with mutations inRPS19 showed increased levels of 21S and 41S pre-rRNAswhen compared with control cells (Fig. 2A lanes 4–6). This
Figure 1. Pre-rRNA processing pathways in human cells. Within the primary transcript, the mature 18S, 5.8S and 28S rRNAs are flanked by external transcribedspacers (50- and 30-ETS) and separated by internal transcribed spacers (ITS1 and ITS2). Numbers and arrowheads above the 45S pre-rRNA indicate cleavagesites. Two alternative processing pathways are defined by the order of cleavage at sites 1 and 2. Early removal of the 50-ETS in pathway A yields the 41S pre-cursor. This RNA is then cleaved within the ITS1 to generate the 21S and 32S species that are precursors to the RNA components of the large and small ribo-somal subunits, respectively. In pathway B, cleavage within the ITS1 comes first, leading to the formation of 30S and 32S pre-rRNAs. Adapted from Hadjiolovaet al. (55) and Rouquette et al. (30).
1254 Human Molecular Genetics, 2008, Vol. 17, No. 9
182

phenotype, characterized by high 21S/18S-E and 41S/30Sratios (Fig. 2B), corresponds to delayed maturation of thepre-40S subunits and reproduces previous observations inprimary hematopoietic cells or skin fibroblasts from DBApatients affected in this gene, in LCLs bearing mutations inRPS19, or in rps19 siRNA-treated HeLa cells (13–15). Thethree LCLs with mutations in RPS24 also showed a clearalteration of pre-rRNA processing, but with distinct defects:the level of 41S pre-rRNA was lower than in control cells,whereas the 30S species accumulated (Fig. 2A, lanes 7–9),resulting in a lower 41S/30S ratio (Fig. 2B). In parallel,there was less 21S and 18S-E pre-rRNAs than in controlcells. Similar analysis of the 5.8S/28S RNA maturationpathway with a probe complementary to the ITS2 showedno significant difference with control cells (not shown).
These data indicate that the mutations in RPS24 found inDBA patients also affect maturation of the 18S rRNA, butnot at the same stage as mutations in RPS19.
RPS24 is essential for production of the 40S subunit
To directly study the consequences of RPS24 loss-of-functionon ribosome biogenesis, we designed two siRNAs (rps24-1and rps24-2) that were used to transfect HeLa cells. TheRPS24 mRNA level was reduced over 10 times by bothsiRNAs, as measured by quantitative reverse transcriptase-polymerase chain reaction (qRT-PCR) (Fig. 3A). Analysis ofthe translational machinery of cells depleted of RPS24 onsucrose gradient showed a drastic loss of free 40S subunitsparalleled by a strong accumulation of free 60S subunits andreduced levels of 80S monosomes and polysomes (Fig. 3B).This profile is indicative of a strong defect of 40S subunit syn-thesis. Thus, like RPS19, RPS24 is essential for the productionof the small ribosomal subunit.
RPS24 is required for processing of the 50-ETS
We next examined pre-rRNA processing in cells depleted ofRPS24 by pulse-chase labeling with L-[methyl-3H]methionine,which serves as a methyl group donor for methylation of pre-rRNAs by C/D box snoRNPs. Cells were labeled for 30 min,and chase with cold methionine was performed for up to90 min. As seen in Figure 4A (see also Fig. 6C), cellstreated with rps24-1 or rps24-2 siRNAs displayed a differentprocessing pattern when compared with control cells. Moststrikingly, there was no formation of the 41S pre-rRNA,which normally results from the cleavage of the 50-ETS(Fig. 1, pathway A). In addition, the 21S pre-rRNA, whosegeneration requires cleavage of the 50-ETS (Fig. 1, pathwayB), was also absent, and no 18S rRNA formed in the time-course of the experiment. In contrast, the kinetics of 28SrRNA formation was equivalent to that in control cells.These observations are consistent with a function of RPS24in the maturation of the 50-ETS.
This hypothesis was further supported by detection of pre-rRNAs by northern blot with the 50-ITS1 probe (Fig. 4B). InRPS24-depleted cells, we observed a strong reduction in thelevels of several 18S rRNA precursors, namely the 41S, 21Sand 18S-E pre-rRNAs. In parallel, there was a build-up ofthe amount of 30S pre-rRNA, as well as a mild increase in45S pre-rRNA. Accumulation of 30S pre-rRNA, correlatedwith the absence of the 41S, 21S and 18S-E species indicatesstrong inhibition of cleavage of the 50-ETS, as well as block-age of the subsequent processing of the ITS1 at the 30 end ofthe 18S rRNA. Consistent with the absence of 18S-Epre-rRNA, which is normally exported from the nucleus, fluor-escence in situ hybridization (FISH) with the same probeshowed a strong reduction of the cytoplasmic labeling relativeto control cells (Fig. 4C). Pre-18S rRNAs retained in thenucleus were detected in the nucleolus as well as in smallnucleoplasmic dots.
The pattern of the precursors to the 60S subunit rRNAs, asdetected with a probe to the ITS2, was unchanged (not shown).The amounts of 28S and 5.8S rRNAs were the same in rps24siRNA-treated cells as in control cells, whereas the 18S/28S
Figure 2. Alteration of pre-rRNA processing in lymphoblastoid cells derivedfrom Diamond-Blackfan anemia (DBA) patients with mutations in RPS24 andRPS19. (A) RNAs from lymphoblastoid cells derived from control individualsand DBA patients were analyzed by northern blot with a probe hybridizing tothe 50 extremity of the internal transcribed spacer 1 (50-ITS1) to detect the pre-cursors to the 18S rRNA. (B) Ratios between different pre-rRNAs were eval-uated for each lane by phosphorimaging. Each bar represents the average(+SD) of the values determined for the three corresponding lymphoblastoidcell lines. Variations of these ratios in cells from DBA patients relative tocontrol cells are indicated on the bars. The results were analyzed with Stu-dent’s t-test (�P , 0.05; ��P , 0.01; n.s., not significant). This experimentwas performed twice with similar results.
Human Molecular Genetics, 2008, Vol. 17, No. 9 1255
183
ratio was lower, which points towards a ribosome biogenesisdefect primarily affecting formation of the 40S subunit. Thechanges in pre-rRNA pattern observed with rps24 siRNAsare similar, albeit more pronounced, to those observed incells derived from RPS24-deficient DBA patient, fromwhich we infer that the pre-rRNA processing defect observedin patient-derived cells is the direct consequence of RPS24loss-of-function.
Down-regulation of RPS19 level in RPS24-depleted cells
Because RPS19 and RPS24 are both involved in DBA, onemay suspect a specific functional link between the twoproteins. Interestingly, RPS19 protein levels in cells fromDBA patients carrying a mutation in RPS24 were reportedto be lower than in control cells (10). In accordance to thisobservation, we found a strong reduction of RPS19 proteinlevel in cells treated with rps24-1 siRNAs on western blots(Fig. 5A). Down-regulation of the protein level was notbecause of a decrease in RNA synthesis or stability, sincethe amount of RPS19 mRNA was unchanged in these cells(Fig. 5B). However, this effect was not specific to rps24siRNAs, since knockdown of RPS15 led to a similar drop inthe level of RPS19. Thus, we propose that loss of RPS19 inRPS24-depleted cells is primarily because of impaired syn-thesis of the 40S ribosomal subunit, which hampers stabiliz-ation of ribosomal proteins in mature ribosomal subunits.
RPS24 and RPS19 exert coordinated functions in ribosomebiogenesis
The pre-rRNA processing defects observed upon knockdownof RPS19 or RPS24 expression indicate that these two proteinsare involved in different cleavage steps: RPS19 is mostlyrequired for processing of the ITS1, whereas RPS24 is necess-ary to cleave the 50-ETS. Previous studies in yeast and in ver-tebrates have established that processing of the 50-ETS and theITS1 are spatially and temporally coupled (26–29). To get
insights into the functional relationship between RPS24 andRPS19 in ribosome biogenesis, we combined rps19 andrps24 siRNAs to knockdown the expression of both proteins(Fig. 6). As a control, we also transfected each of thesesiRNAs with siRNAs directed against the ribosomal proteinRPS15, which is required for nuclear export of the pre-40Sparticles and for the last processing step of the 18S rRNA atits 30 end (30). Indeed, knockdown of RPS24 or RPS19 hada dominant effect on pre-rRNA processing when RPS15 wasco-depleted (Fig. 6A, lanes 6 and 7), consistent with RPS19and RPS24 acting upstream of RPS15 in the 40S subunit bio-genesis process. But surprisingly, depletion of RPS19 togetherwith RPS24 suppressed most of the 50-ETS processing defectobserved with rps24 siRNAs, as indicated by the presence of41S and 21S pre-rRNA (Fig. 6A, compare lanes 3 and 5;quantifications on Fig. 6B). Accumulation of 21S pre-rRNAand low levels of 18S-E pre-rRNA recapitulated the ITS1 pro-cessing defect observed upon knockdown of RPS19expression alone (Fig. 6A, lane 2). This result was reproducedwith various combinations of two rps24 and two rps19siRNAs (not shown). As shown in Figure 5B, the RPS24mRNA was still reduced by over 10-fold when rps24 andrps19 siRNAs were co-transfected. Restoration of the50-ETS processing in cells depleted of both RPS24 andRPS19 was confirmed by pulse-chase experiments withL-[methyl-3H]methionine (Fig. 6C): the cells produced 43S,41S and 21S pre-rRNAs, but did not process the 21Spre-rRNA into 18S-E RNA, as in rps19 siRNA-treated cells.These data show that depletion of RPS19 suppresses the phe-notype induced by RPS24 knockdown, which stronglysuggests that RPS19 and RPS24 exert tight inter-dependentfunctions in pre-rRNA processing.
Structure of RPS24e
The three DBA-related mutations in RPS24 reported so faraffect the protein in different ways. The most drastic,R16stop, results in early translation arrest and may be
Figure 3. RPS24 expression knockdown by siRNAs strongly affects the translational apparatus. (A) Knockdown of RPS24 mRNA level in HeLa cells upontransfection with siRNAs rps24-1 and rps24-2 was measured by quantitative reverse transcriptase-polymerase chain reaction. (B) Analysis on sucrose gradientof the ribosomal subunits from HeLa cells 48 h after transfection with the rps24-1 siRNA shows a strong imbalance between free 60S and 40S subunits, reflectinga specific defect of the 40S subunit production. The 18S, 28S and 18S-E rRNAs were detected in the gradient fractions by northern blot with probes 18S, 28S and50-ITS1, respectively (30). Control cells were treated according to the transfection protocol, but without siRNAs.
1256 Human Molecular Genetics, 2008, Vol. 17, No. 9
184

equivalent to a null allele. Deletion N2-Q22 and mutationQ106stop are expected to yield proteins truncated of their N-terminal and C-terminal domains, respectively. To get insightsinto the structural domains affected by mutations in RPS24,we solved the crystal structure of protein RPS24e from thehyperthermophilic archeon Pyroccocus abyssi. RPS24e ishomologous to the human RPS24 (27% identity and 57.5%similarity) and the two proteins are thus expected to harbora similar fold. The crystal structure was determined at 1.9 Aresolution and refined to an Rfree of 26.6% and an R-factorof 22.1% (Table 1).
The protein RPS24e is built around a four-stranded anti-parallel b-sheet surrounded by three short a-helices(Fig. 7A). Structural similarity search performed with the
DALI program (31) revealed homology with the prokaryoticribosomal protein L23 (Fig. 7B), a component of the largeribosomal subunit, as observed in the crystal structure oflarge ribosomal subunits from archaea Haloarcula marismor-tui (PDB code 1FFK, 1S72) (32,33) and bacteria Deinococcusradiodurans (PDB code 2D3O) (34). The L23 family of pro-teins includes RPL25 in Saccharomyces cerevisiae andRPL23a in mammals. Prokaryotic L23 binds to the 23S ribo-somal RNA through its babb structure, which recalls theRNA recognition motif (RRM) of snRNP protein U1A (33),and also interacts with ribosomal protein L29 (Fig. 7C).Presence of a very similar babb substructure in RPS24strongly points towards a putative RRM made of b-strands2-3-4 and a-helix 1 (amino acids 18 to 79 in human
Figure 4. Depletion of RPS24 blocks processing of the 50-external transcribed spacer (50-ETS). (A) Pulse-chase analysis of rRNA synthesis withL-[methyl-3H]methionine in HeLa cells 48 h after transfection with rps24 siRNAs. Formation of the 43S, 41S, 21S pre-rRNAs is blocked and no 18Smature rRNA is produced. (B) Detection of the precursors to the 18S rRNA by northern blot with probe 50-ITS1. The graph shows the intensity profiles foreach lane as measured by phosphorimaging. The table contains the variations of the levels of different RNAs in the RPS24-depleted cells relative to thecells transfected with the scramble siRNA, in two independent experiments. The intensity profiles and the amount of the pre-rRNAs reported in the tablewere normalized with respect to the amount of 28S rRNA. (C) Detection by fluorescence in situ hybridization (FISH) of pre-18S RNAs in RPS24-depletedcells shows reduced cytoplasmic labeling and small nucleoplasmic dots. Cells treated with siRNAs targeting RPS15 synthesis display a different phenotype,characterized by nucleoplasmic retention of 18S-E pre-rRNA (30). The FISH pictures are displayed with inverted contrast and high saturation of the darkgray levels to enhance the cytoplasmic signal (gamma ¼ 0.7). These experiments were performed at least three times with similar results.
Human Molecular Genetics, 2008, Vol. 17, No. 9 1257
185

RPS24). The structure of this region is likely to be affected bythe deletion of residues N2 to Q22, which compose b-strands 1and 2 (Fig. 7A and D). In addition, the high sequence conser-vation of the N-terminal N2-Q22 domain between P. abyssi, S.cerevisiae and Homo sapiens (Fig. 7D) strongly argues for anessential contribution to the protein structure and function.Interestingly, the C-terminal domain missing in theQ106stop mutant is only found in eukaryotes. This domain,which is not present in the structure presented here, may notbe essential for protein folding, but rather involved in intermo-lecular interactions with eukaryote-specific molecules.
DISCUSSION
The data presented here reinforce the hypothesis that DBA islinked to defective ribosome biogenesis and bring furtherinsights into the mechanisms underlying the physiopathologyof this disease. First, we find that mutations in RPS24 inDBA patients are associated with altered pre-rRNA proces-sing. Lymphoblastoid cells derived from DBA patients withmutated RPS24 display a clear defect in pre-rRNA maturation,although distinct from the phenotype observed in cells derivedfrom patients with mutations in RPS19. Consistently, knock-down of RPS24 expression with siRNAs perturbs processingof the 50-ETS and maturation of the 30 end of the 18SrRNA. Three cleavage sites were characterized in the 50-ETSof mammalian pre-rRNA: the early cleavage site to becleaved first (around nucleotides 415/422 in human 47Spre-rRNA) (35); site 1 at the 50 end of the 18S; and the A0site (around nucleotide 1643), which seems to be processed
Figure 5. Low level of RPS19 in RPS24-depleted cells. (A) Western blotanalysis of RPS19 level in HeLa cells transfected with siRNAs targetingexpression of RPS19, RPS24 or RPS15. A marked diminution of RPS19 isobserved in all three cases. (B) Measurement by qRT-PCR of the decreasein the ribosomal protein mRNA levels in the same cells as in (A). This exper-iment was repeated twice with similar results.
Figure 6. Co-depletion of RPS19 suppresses the 50-external transcribedspacer (50-ETS) processing defect observed in RPS24-depleted cells.(A) Northern blot analysis of pre-rRNAs with the 50-internal transcribedspacer 1 (ITS1) probe in HeLa cells transfected with siRNAs targetingexpression of RPS19, RPS24 or RPS15. (B) Variations of the pre-rRNAlevels in these cells relative to cells transfected with the scramble siRNAwere measured by phosphorimaging on the Northern blot. Measurementsin each lane were normalized with respect to the amount of 28S rRNA.Results of two independent experiments are reported. (C) Pulse-chase exper-iment with L-[methyl-3H]methionine shows 41S pre-rRNA synthesis in cellsdepleted of both RPS19 and RPS24, when compared with RPS24 knock-down only, indicating restoration of 50-ETS processing.
1258 Human Molecular Genetics, 2008, Vol. 17, No. 9
186

either more slowly or quasi-simultaneously with site 1 (30).Similar to the yeast S. cerevisiae, depletion of Rps24p(RPS24 homolog) blocks maturation of the 50-ETS, as wellas cleavage of the ITS1 at site A2 (24) and results in strongaccumulation of ‘23S’ pre-rRNA, which may be seen as theequivalent of the mammalian 30S pre-rRNA. Thus, as forRPS19, the function of RPS24 in ribosome biogenesis is con-served from yeast to mammals.
The fact that the pre-rRNA processing pattern in alllymphoblastoid cells matches the one observed in rps24siRNA-treated cells strongly suggests that the three mutationsaffecting RPS24 in DBA patients yield haploinsufficiency.Besides mutation R16stop, which may be equivalent to anull allele, mutation Q106stop and deletion of E2-Q22 alsointerrupt RPS24 function in ribosome biogenesis. The crystalstructure of RPS24e reveals a strong structural homologywith the L23 family of ribosomal proteins (L23 in prokaryotes,RPL25 in yeast, RPL23a in mammals): these proteins notonly interact with rRNA and ribosomal protein L29, but alsoprovide docking sites for the trigger factor and the signalrecognition particle (36,37). Structural superimposition withL23 suggests a putative RNA-binding surface in RPS24.Deletion of 20 amino acids between E2 and Q22 is expected,at least, to alter this surface, hampering association with thepre-rRNA, and most probably to destabilize the structure ofthe whole protein, leading to its degradation. In contrast, the C-terminal domain absent in mutant Q106stop does not seem tobe required for the folding of the protein, since it is absent inthe archaeal RPS24e. This module may instead be involvedin additional interactions in eukaryotic pre-ribosomes, eitherwith the pre-ribosomal RNA or with some of the numerouspre-ribosomal factors required for ribosome biogenesis.Alternatively, this lysine-rich domain may contain the
nuclear localization signal of the protein. In parallel, the prema-ture stop codon in the mRNA encoding this mutant may alsotrigger nonsense-mediated decay. Further work is needed tosort out these hypotheses.
Loss-of-function of pre-ribosomal factors and ribosomalproteins of the large and the small subunits were shown toactivate p53 and trigger cell cycle arrest and apoptosis(18–21). This stress response, together with deficient trans-lation because of a low ribosome number, is likely to play adirect role in a pathological process involving interruption ofdifferentiation. Remarkably, knockdown of different proteinsinvolved in ribosome biogenesis affect distinct cell types invivo. Hence, homozygous knockout of ribosomal proteinRPL22 prevents ab T-cell development in mouse (38),whereas deficiency of protein SDBS, which is required formaturation of the 60S ribosomal subunit, primarily affectsneutrophil differentiation in Shwachman-Diamond syndrome(39,40). Thus, the involvement of RPS19 and RPS24 inDBA, characterized by a differentiation defect in yet anothercell lineage (pro-erythroblasts), calls for a closer functionallink between the two proteins. By immunoelectron microscopyon isolated rat 40S subunits, RPS19 was localized on the‘head’ of the particle and RPS24 at some distance on the‘body’ (41), and the two proteins were found to assembleinto pre-40S particles independently from one another inyeast (42). These observations do not support a direct inter-action of the two proteins within the ribosome. However,one cannot formally rule out that they interact physically orsimply functionally within the pre-ribosomal particles duringthe maturation process.
In this respect, the connection reported here between theactivities of the two proteins in pre-rRNA processing is aninteresting clue. Depletion of RPS19 suppresses, in largepart, the 50-ETS processing defect induced by RPS24siRNAs, which reveals a functional interaction. RPS19depletion strongly affects processing of the ITS1: slower clea-vage at site 2, alternative cleavage of the ITS1, inhibition ofthe 18S rRNA 30 end processing (13–15,43). Epistasis ofRPS19 on RPS24 is thus counter-intuitive, since RPS19 isnot necessary for processing of the 50-ETS. However, it iswell established, in yeast as in vertebrates, that the 50-ETSand the ITS1 are processed in a coordinated manner (26,44–46). RPS19 and RPS24 may function in concert, togetherwith factors known to be involved in coupling the processingof the 50-ETS and ITS1, like the snoRNP U3. Different modelsof the functional interaction between RPS19 and RPS24 maybe proposed. For example, the two proteins could chaperonea structural transition of the pre-rRNA. This conformationalchange would not be directly required for processing of the50-ETS, which would thus occur in the absence of the two pro-teins. However, absence of RPS24 alone would lead to anaberrant structure incompatible with cleavage of the 50-ETS.Alternatively, RPS19 and RPS24 could regulate the bindingto the pre-ribosomal particle of an early pre-ribosomalfactor, whose dissociation would be a pre-requisite to proces-sing of the 50-ETS. Recruitment of this factor would involveRPS19, whereas RPS24 would be needed for its dissociation.Absence of RPS24 would lead to accumulation of aberrantpre-ribosomal particles containing the non-dissociated factorand unable to cleave the 50-ETS.
Table 1. Crystallographic data phasing and refinement statistics
Se-Met derivative
Space group P212121
Cell dimensionsa, b, c (A) 66.81, 85.63, 42.11a, b, g (8) 90.0, 90.0, 90.0X-ray source ID14-4Wavelength (A) 0.9793Resolution (A) 50–1.90Total measurements 101,890 (13,468)Unique reflections 19,214 (2,861)Redundancy 5.3 (4.7)Completeness (%) 97.5 (91.3)Rsym (%) 6.2 (31.2)I/s 15.3 (4.8)FOM (acentric) 0.288
Refinement statisticsPhi, Psi angles
Most favored (%) 99.6Additionally allowed (%) 0.4Protein residues 189Water molecules 97Rwork (%) 22.13Rfree (%) 26.61
Numbers in brackets refer to the highest resolution shell (1.90–2.02 A).
Human Molecular Genetics, 2008, Vol. 17, No. 9 1259
187

These data lead us to propose that RPS19 and RPS24cooperate at a particular stage in ribosome biogenesis vali-dated by a checkpoint, whose non-compliance would signalcell cycle arrest. This signal, in combination with additionalstresses, would be deleterious to differentiation of erythroid
progenitors, which require rapid proliferation, or to develop-mental processes. Analysis of new genes involved in DBA,like RPS17, may confirm or invalidate this hypothesis,depending on their relationship with RPS19 and RPS24. Inthis respect, it is of note that RPS17 is located on the head
Figure 7. Structure of RPS24e from Pyrococcus abyssi. (A) The structure of P. abyssi RPS24e is displayed with rainbow colors from blue for N-terminus to red forC-terminus. Secondary structure elements are labeled. The position of mutation R16stop and deletion of N2-Q22 found in human RPS24 are shown in red. (B) Super-position of ribbon models of P. abyssi RPS24e (rainbow) and Haloarcula marismortui L23 (red). (C) Structural environment of L23 in the large subunit of the ribo-some from H. marismortui as described in Klein et al. (33). (D) Sequence alignment of RPS24e and its orthologues was performed using the PipeAlign suite (56). Thesequences of P. abyssi RPS24e and H. marismortui RPL23 were aligned according to the structural alignment. Secondary structure elements for RPS24e and RPL23are above and below the alignment, respectively. The mutations found in human RPS24 in Diamond-Blackfan anemia patients are shown in red.
1260 Human Molecular Genetics, 2008, Vol. 17, No. 9
188

of the 40S subunit, in close proximity to RPS19 with which itmay directly interact (41).
MATERIALS AND METHODS
Tissue culture
Epstein-Barr Virus (EBV)-transformed LCLs were cultured inRPMI 1640 medium supplemented with 15% fetal bovineserum (FBS), 100 U/ml penicillin, 100 mg/ml streptomycinand 0.292 mg/ml L-glutamine (all from Invitrogen, Carlsbad,California) at 378C in 5% CO2. Total RNAs were isolatedfrom LCLs using RNA isolation kit (Qiagen, Valencia, CA,USA).
Human cervical carcinoma HeLa cells were maintained inculture in Dulbecco’s Modified Eagle Medium (DMEM) sup-plemented with 10% fetal calf serum (FCS), 1 mM sodium pyr-uvate, 100 U/ml penicillin and 100 mg/ml streptomycin (allfrom Invitrogen) in 5% CO2 at 378C.
Small interfering RNAs
Two different 21-mer siRNAs were designed to knockdownexpression of the human RPS24 [GenBank accession no.NM_033022 (full-length transcript)]: 50-CACCGGAUGUCAUCUUUGUdTdT-30 (rps24-1 siRNA), 50-GGAAACAAAUGGUCAUUGAdTdT-30 (rps24-2 siRNA). The siRNAs tar-geting expression of RPS15 and RPS19 were described else-where (13,30). All siRNAs were purchased from Eurogentec(Seraing, Belgium) and transfected using electrotransforma-tion as described previously (30).
Fractionation and analysis of ribosomes by sucrose densitygradient centrifugation
Forty-eight hours after transfection with siRNAs, HeLa cellswere treated with 100 mg/ml cycloheximide (Sigma, Sigma-Aldrich Co., Saint-Louis, Missouri) for 10 min, fractionatedand the cytoplasmic fraction was analyzed on a sucrose gradi-ent as described previously (30).
Quantitative reverse transcriptase-polymerase chainreaction
The RPS24 gene down-regulation induced by RNA interfer-ence was analyzed by quantitative PCR taking GAPDH asan internal control to normalize the RPS24 expression. TotalRNAs were isolated using TRIzol reagent (Invitrogen), andfirst-strand synthesis of cDNAs was performed on 2 mg ofRNA at 428C for 2 h using AMV reverse transcriptase as rec-ommended by the supplier (Promega, Madison, WI, USA).The sequences of primers used for PCR were the follow-ing—for RPS24: forward, 50-CCGACTACTTCAGAGGAAACAAA-30; reverse, 50-GCCACCACCAAAATGAGTTCT-30;for GAPDH: forward, 50-TGCACCACCAACTGCTTAG-30;reverse, 50-GTTCAGCTCAGGGATGACC-30. Real-timePCR was carried out using Platinum SYBR Green qPCRSuperMix-UDG (Invitrogen) according to the manufacturers0
instructions, on a ICyclerQIM (Bio-Rad, Hercules, CA,USA). After initial denaturation at 958C for 3 min, 40 cycles
were performed with the following conditions: 958C for20 s; 568C for 30 s and 728C for 20 s. The fluorescentsignals were measured after each extension step at 788C. Tri-plicate PCR reactions were prepared for each sample andmeasurements were independently repeated twice. Data wereanalyzed using the IQCycler version software. The concen-tration of the gene-specific mRNA in treated cells relative tountreated cells was calculated by subtracting the normalizedCt values obtained for untreated cells from those obtainedfrom treated samples (DDCt ¼ DCt, treated—DCt, untreated)and the relative concentration was determined (2–DDCt).
Analysis of pre-rRNAs
Pre-rRNA analysis by northern blot was performed asdescribed previously (47) with probes 18S, 28S, 50-ITS1,ITS2b and ITS2-d/e and actin (30). For detection of theITS2, the ITS2-b and ITS2-d/e probes were mixed in equalamounts. The protocol of FISH with the 50-ITS1 probe wasreported in Rouquette et al. (30).
For pulse-chase analysis, cells were transfected with rps24-1and rps24-2 siRNAs using electroporation and plated at adensity of 1.5� 105 cells in 12-well dishes. Two days later,they were pre-incubated for 30 min in serum-free methionine-free medium and then incubated for 30 min in 0.5 ml of thesame medium containing 25 mCi L-[methyl-3H]methionine(50 mCi/ml). The cells were then chased in non-radioactivemedium containing cold methionine (30 mg/ml) for varioustimes, after which total RNAs were isolated using TRIzol (Invi-trogen). RNAs were separated on a 1% agarose gel, transferredto nylon membrane and exposed to a film using an intensifyingscreen (Transcreen LE, Amersham).
Protein expression, purification and crystallization
The full-length cDNA of P. abyssi RPS24e was cloned into amodified pET-15b allowing production of an N-terminallyHis-tagged fused protein in Escherichia coli (48). Afterinitial growth at 378C, cell cultures were cooled down to158C and protein production was induced with 0.5 mM isopro-pyl beta D galactopyranoside (IPTG) overnight. Purificationwas carried out after cell lysis by centrifugation at 48C for1 h at 50,000 g. The supernatant was incubated at 608C for30 min and further centrifuged. The supernatant was thenincubated in batch with HIS-Select affinity resin (Sigma).The eluate was loaded on a HiQ-Sepharose. The protein wasconcentrated up to 13 mg/ml in 25 mM Tris–HCl of pH 7.5and 100 mM NaCl. Crystallization of RPS24e was carriedout at room temperature using sitting-drop vapor diffusionby mixing one volume of protein solution with one volumeof 40% PEG 400, 100 mM sodium acetate pH 4.2 and185 mM MgCl2 of reservoir solution (Nextal). Crystals weredirectly flash-frozen in liquid nitrogen for data collection.Data were processed with XDS (49). Data collection andphasing statistics are shown in Table 1.
Structure solution
Phase information was obtained by a single-wavelength anom-alous dispersion (SAD) experiment at a resolution of 1.9 A on
Human Molecular Genetics, 2008, Vol. 17, No. 9 1261
189

a Se-Met-substituted protein crystal. Seven Se sites werelocated using SHELXD (50) and initial phases were improvedwith SHARP/AutoSHARP (51). The model was automaticallybuilt with Arp/Warp (52) and further improved manually withCoot (53). Model refinement was achieved with REFMAC5(54). The final model has a Rfree value of 26.6% and Rwork
value of 22.1%. The crystallographic asymmetric unit containstwo molecules of RPS24e. Both molecules have a very similarstructure with an RMS deviation of 0.9 A. The final modelincludes most of the RPS24 amino acids with the exceptionof the C-terminal residues for both molecules (respectively,seven residues for molecule A and five residues for moleculeB). Four amino acids coming from the His-tag are also visibleon molecule A. The crystal structure of RPS24e from P. abyssiis similar to the structure of RPS24e from archeon Methano-sarcina mazei determined by nuclear magnetic resonancespectroscopy (unpublished, PDB code 1XN9) with the excep-tion of the C-terminal a-helix. This difference could beaccounted for protein rearrangement upon crystallizationsince this area of P. abyssi RPS24e is involved in crystal con-tacts with neighboring molecules. The atomic coordinates andstructure factor have been deposited in the Protein Data Bank(accession code 2v94).
ACKNOWLEDGEMENTS
The authors wish to thank Coralie Carron and Marlene Faubla-dier for scientific discussion and technical assistance. Theauthors are grateful to the Daniella Maria Arturi Foundationfor stimulating research collaborations in the field of DBA.
Conflict of interest statement. None declared.
FUNDING
This project was supported by the Agence Nationale de laRecherche (ANR—RIBODBA project), the CNRS and theUniversity of Toulouse. This project was also supported bya research grant from The Diamond-Blackfan Anemia Foun-dation (to HTG).
REFERENCES
1. Draptchinskaia, N., Gustavsson, P., Andersson, B., Pettersson, M., Willig,T.N., Dianzani, I., Ball, S., Tchernia, G., Klar, J., Matsson, H. et al. (1999)The gene encoding ribosomal protein S19 is mutated inDiamond-Blackfan anaemia. Nat. Genet., 21, 169–175.
2. Da Costa, L., Willig, T.N., Fixler, J., Mohandas, N. and Tchernia, G.(2001) Diamond-Blackfan anemia. Curr. Opin. Pediatr., 13, 10–15.
3. Willig, T.N., Gazda, H. and Sieff, C.A. (2000) Diamond-Blackfan anemia.Curr. Opin. Hematol., 7, 85–94.
4. Gazda, H.T. and Sieff, C.A. (2006) Recent insights into the pathogenesisof Diamond-Blackfan anaemia. Br. J. Haematol., 135, 149–157.
5. Flygare, J. and Karlsson, S. (2007) Diamond-Blackfan anemia:erythropoiesis lost in translation. Blood, 109, 3152–3154.
6. Liu, J.M. and Ellis, S.R. (2006) Ribosomes and marrow failure:coincidental association or molecular paradigm? Blood, 107, 4583–4588.
7. Chiocchetti, A., Gibello, L., Carando, A., Aspesi, A., Secco, P., Garelli,E., Loreni, F., Angelini, M., Biava, A., Dahl, N. et al. (2005) Interactionsbetween RPS19, mutated in Diamond-Blackfan anemia, and the PIM-1oncoprotein. Haematologica, 90, 1453–1462.
8. Soulet, F., Al Saati, T., Roga, S., Amalric, F. and Bouche, G. (2001)Fibroblast growth factor-2 interacts with free ribosomal protein S19.Biochem. Biophys. Res. Commun., 289, 591–596.
9. Orru, S., Aspesi, A., Armiraglio, M., Caterino, M., Loreni, F., Ruoppolo,M., Santoro, C. and Dianzani, I. (2007) Analysis of the ribosomal proteinS19 interactome. Mol. Cell. Proteomics, 6, 382–393.
10. Gazda, H.T., Grabowska, A., Merida-Long, L.B., Latawiec, E., Schneider,H.E., Lipton, J.M., Vlachos, A., Atsidaftos, E., Ball, S.E., Orfali, K.A. et al.
(2006) Ribosomal protein S24 gene is mutated in Diamond-Blackfananemia. Am. J. Hum. Genet., 79, 1110–1118.
11. Cmejla, R., Cmejlova, J., Handrkova, H., Petrak, J. and Pospisilova, D.(2007) Ribosomal protein S17 gene (RPS17) is mutated inDiamond-Blackfan anemia. Hum. Mutat., 28, 1178–1182.
12. Leger-Silvestre, I., Caffrey, J.M., Dawaliby, R., Alvarez-Arias, D.A., Gas,N., Bertolone, S.J., Gleizes, P.E. and Ellis, S.R. (2005) Specific role foryeast homologs of the Diamond Blackfan anemia-associated Rps19protein in ribosome synthesis. J. Biol. Chem., 280, 38177–38185.
13. Choesmel, V., Bacqueville, D., Rouquette, J., Noaillac-Depeyre, J.,Fribourg, S., Cretien, A., Leblanc, T., Tchernia, G., Da Costa, L. andGleizes, P.E. (2007) Impaired ribosome biogenesis in Diamond-Blackfananemia. Blood, 109, 1275–1283.
14. Flygare, J., Aspesi, A., Bailey, J.C., Miyake, K., Caffrey, J.M., Karlsson,S. and Ellis, S.R. (2007) Human RPS19, the gene mutated inDiamond-Blackfan anemia, encodes a ribosomal protein required for thematuration of 40S ribosomal subunits. Blood, 109, 980–986.
15. Idol, R.A., Robledo, S., Du, H.Y., Crimmins, D.L., Wilson, D.B.,Ladenson, J.H., Bessler, M. and Mason, P.J. (2007) Cells depleted forRPS19, a protein associated with Diamond-Blackfan anemia, showdefects in 18S ribosomal RNA synthesis and small ribosomal subunitproduction. Blood Cells Mol. Dis., 39, 35–43.
16. Angelini, M., Cannata, S., Mercaldo, V., Gibello, L., Santoro, C.,Dianzani, I. and Loreni, F. (2007) Missense mutations associated withDiamond-Blackfan anemia affect the assembly of ribosomal protein S19into the ribosome. Hum. Mol. Genet., 16, 1720–1727.
17. Gregory, L.A., Aguissa-Toure, A.H., Pinaud, N., Legrand, P., Gleizes,P.E. and Fribourg, S. (2007) Molecular basis of Diamond-Blackfananemia: structure and function analysis of RPS19. Nucleic Acids Res., 35,5913–5921.
18. Dai, M.S. and Lu, H. (2004) Inhibition of MDM2-mediated p53ubiquitination and degradation by ribosomal protein L5. J. Biol. Chem.,279, 44475–44482.
19. Lohrum, M.A., Ludwig, R.L., Kubbutat, M.H., Hanlon, M. and Vousden,K.H. (2003) Regulation of HDM2 activity by the ribosomal protein L11.Cancer Cell, 3, 577–587.
20. Pestov, D.G., Strezoska, Z. and Lau, L.F. (2001) Evidence ofp53-dependent cross-talk between ribosome biogenesis and the cell cycle:effects of nucleolar protein Bop1 on G(1)/S transition. Mol. Cell. Biol., 21,4246–4255.
21. Azuma, M., Toyama, R., Laver, E. and Dawid, I.B. (2006) Perturbationof rRNA synthesis in the bap28 mutation leads to apoptosis mediatedby p53 in the zebrafish central nervous system. J. Biol. Chem., 281,13309–13316.
22. Cmejlova, J., Dolezalova, L., Pospisilova, D., Petrtylova, K., Petrak, J.and Cmejla, R. (2006) Translational efficiency in patients withDiamond-Blackfan anemia. Haematologica, 91, 1456–1464.
23. Bommer, U.A., Stahl, J., Henske, A., Lutsch, G. and Bielka, H. (1988)Identification of proteins of the 40S ribosomal subunit involved ininteraction with initiation factor eIF-2 in the quaternary initiation complexby means of monospecific antibodies. FEBS Lett., 233, 114–118.
24. Ferreira-Cerca, S., Poll, G., Gleizes, P.E., Tschochner, H. and Milkereit,P. (2005) Roles of eukaryotic ribosomal proteins in maturation andtransport of pre-18S rRNA and ribosome function. Mol. Cell, 20,263–275.
25. Gazda, H.T., Zhong, R., Long, L., Niewiadomska, E., Lipton, J.M.,Ploszynska, A., Zaucha, J.M., Vlachos, A., Atsidaftos, E., Viskochil, D.H.et al. (2004) RNA and protein evidence for haplo-insufficiency inDiamond-Blackfan anaemia patients with RPS19 mutations.Br. J. Haematol., 127, 105–113.
26. Borovjagin, A.V. and Gerbi, S.A. (2001) Xenopus U3 snoRNA GAC-BoxA0 and Box A sequences play distinct functional roles in rRNAprocessing. Mol. Cell. Biol., 21, 6210–6221.
27. Gerbi, S.A. and Borovjagin, A.V. (2004) Pre-ribosomal RNA processingin multicellular organisms. Olson, M.O.J. (ed), The Nucleolus,Eurekah.com and Kluwer Academic/Plenum Publishers, pp. 170–198.
28. Venema, J. and Tollervey, D. (1999) Ribosome synthesis inSaccharomyces cerevisiae. Annu. Rev. Genet., 33, 261–311.
1262 Human Molecular Genetics, 2008, Vol. 17, No. 9
190

29. Venema, J., Henry, Y. and Tollervey, D. (1995) Two distinct recognitionsignals define the site of endonucleolytic cleavage at the 50-end of yeast18S rRNA. EMBO J., 14, 4883–4892.
30. Rouquette, J., Choesmel, V. and Gleizes, P.E. (2005) Nuclear export andcytoplasmic processing of precursors to the 40S ribosomal subunits inmammalian cells. EMBO J., 24, 2862–2872.
31. Holm, L. and Park, J. (2000) DaliLite workbench for protein structurecomparison. Bioinformatics, 16, 566–567.
32. Ban, N., Nissen, P., Hansen, J., Moore, P.B. and Steitz, T.A. (2000) Thecomplete atomic structure of the large ribosomal subunit at 2.4 Aresolution. Science, 289, 905–920.
33. Klein, D.J., Moore, P.B. and Steitz, T.A. (2004) The roles of ribosomalproteins in the structure assembly, and evolution of the large ribosomalsubunit. J. Mol. Biol., 340, 141–177.
34. Schlunzen, F., Wilson, D.N., Tian, P., Harms, J.M., McInnes, S.J.,Hansen, H.A., Albrecht, R., Buerger, J., Wilbanks, S.M. and Fucini, P.(2005) The binding mode of the trigger factor on the ribosome:implications for protein folding and SRP interaction. Structure, 13,1685–1694.
35. Kass, S., Craig, N. and Sollner-Webb, B. (1987) Primary processing ofmammalian rRNA involves two adjacent cleavages and is not speciesspecific. Mol. Cell. Biol., 7, 2891–2898.
36. Gu, S.Q., Peske, F., Wieden, H.J., Rodnina, M.V. and Wintermeyer, W.(2003) The signal recognition particle binds to protein L23 at the peptideexit of the Escherichia coli ribosome. RNA, 9, 566–573.
37. Nissen, P., Hansen, J., Ban, N., Moore, P.B. and Steitz, T.A. (2000) Thestructural basis of ribosome activity in peptide bond synthesis. Science,289, 920–930.
38. Anderson, S.J., Lauritsen, J.P., Hartman, M.G., Foushee, A.M., Lefebvre,J.M., Shinton, S.A., Gerhardt, B., Hardy, R.R., Oravecz, T. and Wiest,D.L. (2007) Ablation of ribosomal protein L22 selectively impairsalphabeta T cell development by activation of a p53-dependentcheckpoint. Immunity, 26, 759–772.
39. Menne, T.F., Goyenechea, B., Sanchez-Puig, N., Wong, C.C., Tonkin,L.M., Ancliff, P.J., Brost, R.L., Costanzo, M., Boone, C. and Warren, A.J.(2007) The Shwachman-Bodian-Diamond syndrome protein mediatestranslational activation of ribosomes in yeast. Nat. Genet., 39, 486–495.
40. Elghetany, M.T. and Alter, B.P. (2002) p53 protein overexpression inbone marrow biopsies of patients with Shwachman-Diamond syndromehas a prevalence similar to that of patients with refractory anemia. Arch.Pathol. Lab. Med., 126, 452–455.
41. Lutsch, G., Stahl, J., Kargel, H.J., Noll, F. and Bielka, H. (1990)Immunoelectron microscopic studies on the location of ribosomal proteinson the surface of the 40S ribosomal subunit from rat liver. Eur. J. CellBiol., 51, 140–150.
42. Ferreira-Cerca, S., Poll, G., Kuhn, H., Neuder, A., Jakob, S., Tschochner,H. and Milkereit, P. (2007) Analysis of the in vivo assembly pathway ofeukaryotic 40S ribosomal proteins. Mol. Cell, 28, 446–457.
43. Leger-Silvestre, I., Milkereit, P., Ferreira-Cerca, S., Saveanu, C.,Rousselle, J.C., Choesmel, V., Guinefoleau, C., Gas, N. and Gleizes, P.E.
(2004) The ribosomal protein Rps15p is required for nuclear exit of the40S subunit precursors in yeast. EMBO J., 23, 2336–2347.
44. Beltrame, M. and Tollervey, D. (1995) Base pairing between U3 and thepre-ribosomal RNA is required for 18S rRNA synthesis. EMBO J., 14,4350–4356.
45. Borovjagin, A.V. and Gerbi, S.A. (1999) U3 small nucleolar RNA isessential for cleavage at sites 1, 2 and 3 in pre-rRNA and determineswhich rRNA processing pathway is taken in Xenopus oocytes. J. Mol.
Biol., 286, 1347–1363.
46. Hughes, J.M. and Ares, M., Jr (1991) Depletion of U3 small nucleolarRNA inhibits cleavage in the 50 external transcribed spacer of yeastpre-ribosomal RNA and impairs formation of 18S ribosomal RNA. EMBO
J., 10, 4231–4239.
47. Vanrobays, E., Gleizes, P.E., Bousquet-Antonelli, C., Noaillac-Depeyre,J., Caizergues-Ferrer, M. and Gelugne, J.P. (2001) Processing of 20Spre-rRNA to 18S ribosomal RNA in yeast requires Rrp10p, an essentialnon-ribosomal cytoplasmic protein. EMBO J., 20, 4204–4213.
48. Romier, C., Ben Jelloul, M., Albeck, S., Buchwald, G., Busso, D., Celie,P.H., Christodoulou, E., De Marco, V., van Gerwen, S., Knipscheer, P. et
al. (2006) Co-expression of protein complexes in prokaryotic andeukaryotic hosts: experimental procedures, database tracking and casestudies. Acta Crystallogr. D Biol. Crystallogr., 62, 1232–1242.
49. Kabsch, W. (1993) Automatic processing of rotation diffraction data fromcrystals of initially unknown symmetry and cell constants. J. Appl. Cryst.,26, 795–800.
50. Schneider, T.R. and Sheldrick, G.M. (2002) Substructure solution withSHELXD. Acta Crystallogr. D Biol. Crystallogr., 58, 1772–1779.
51. De La Fortelle, E. and Bricogne, G. (1997) Maximum-likelihood
heavy-atom parameter refinement for multiple isomorphous replacementand multiwavelength anomalous diffraction methods. Methods Enzymol.,472, 472–494.
52. Perrakis, A., Harkiolaki, M., Wilson, K.S. and Lamzin, V.S. (2001) ARP/wARP and molecular replacement. Acta Crystallogr. D Biol. Crystallogr.,57, 1445–1450.
53. Emsley, P. and Cowtan, K. (2004) Coot: model-building tools for
molecular graphics. Acta Crystallogr. D Biol. Crystallogr., 60,2126–2132.
54. Winn, M.D., Isupov, M.N. and Murshudov, G.N. (2001) Use of TLSparameters to model anisotropic displacements in macromolecular
refinement. Acta Crystallogr. D Biol. Crystallogr., 57, 122–133.
55. Hadjiolova, K.V., Nicoloso, M., Mazan, S., Hadjiolov, A.A. andBachellerie, J.P. (1993) Alternative pre-rRNA processing pathways in
human cells and their alteration by cycloheximide inhibition of proteinsynthesis. Eur. J. Biochem., 212, 211–215.
56. Plewniak, F., Bianchetti, L., Brelivet, Y., Carles, A., Chalmel, F.,Lecompte, O., Mochel, T., Moulinier, L., Muller, A., Muller, J. et al.
(2003) PipeAlign: a new toolkit for protein family analysis. Nucleic Acids
Res., 31, 3829–3832.
Human Molecular Genetics, 2008, Vol. 17, No. 9 1263
191

WT
TAP-WT
TAP Vecteur
Vecteur
GAL GLC
Hsa
PyA
WT
GAL GLC
WT-GFP
WT
GAL GLC A B
C
Figure S1. Test de complémentation fonctionnelle chez S. cerevisiae.A. TAP-WT (l'étiquette TAP) n'interfère pas avec l'expression de la protéine. Il en est de même pour l'étiquette GFP (WT-GFP en B). Ceci est démontré par la complémentation fonctionnelle de la perte d'expression de la protéine Rps19p sur milieu GLC (glucose). En C, nous montrons que la protéine humaine (Hsa) est capable de complémenter fonctionnellement la perte de son homologue chez S. cerevisiae, ce qui n'est pas le cas de la protéine de P. abyssi (PyA).
Vecteur
192

I. Test de complémentation fonctionnelle chez S. cerevisiae
Dans ce travail de thèse, nous avons étudié l’homologue de la protéine RPS19 humaine chez
la levure S. cerevisiae. Des tests de complémentation fonctionnelle ont été effectués pour
analyser la conservation fonctionnelle de la protéine RPS19 entre différentes espèces afin de
valider un modèle d’étude. Ce test de complémentation fonctionnelle a été réalisé chez la
levure S. cerevisiae.
Pour cela, nous avons utilisé une souche qui a subi une délétion bi-allélique des gènes codant
RPS19A et RPS19B et dont la viabilité est assurée par l’expression de la copie RPS19A sous
contrôle du promoteur GAL1/10-CYC1. Cette souche est ensuite transformée par un plasmide
contenant la séquence qui code pour la protéine dont on veut étudier la complémentation
fonctionnelle après passage sur milieu glucose. Sur le milieu glucose on exprime uniquement
la copie dont on veut étudier l’effet sur la viabilité cellulaire car le promoteur GAL1/10-CYC1
est inactivé.
Ainsi, nous avons montré que la protéine RPS19 humaine complémente la perte de fonction
de son homologue chez la levure S. cerevisiae, même si cette complémentation fonctionnelle
n’est pas totale (Figure S1,C). A contrario, la protéine de Pyrococcus abyssi n’a pas été
capable d’assurer la perte de fonction de son homologue chez la levure (Figure S1,C). Par les
mêmes expériences nous avons montré que l’étiquette GFP fusionée en C-ter n’interfère pas
avec la fonction de la protéine (Figure S1,B). Il en est de même pour l’étiquette TAP qui a été
fusionée en N-ter de la protéine RPS19 (Figure S1,A).
193

Vecteur
P46G
P90G
P118G
WT
GAL GLU
Figure S2. Test de complémentation fonctionnelle chez la levure S. cerevisiae et recher-che de mutants ts. La mutation des 5 résidues proline se trouvant dans la protéine RPS19A de levure par des résidues glycine ne confère pas à la protéine un phénotype thermosensible (ts). Ici, nous montrons seulement 3 mutations. Toutes les 5 mutations n'altèrent pas la crois-sance de la levure comme le montre le test de complémentation fonctionnelle, ce qui nous a permis de tester leur thermosensibilité à 37°C.
194

II. Recherche de mutants thermosensibles (ts) de Rps19p
Au cours de la thèse, nous avons cherché à générer un mutant ts pour une analyse
fonctionnelle de la protéine. Il s’agit d’une stratégie qui est basée sur l’altération fonctionnelle
de la protéine par introduction d’une mutation affectant la fonction de la protéine à certaines
températures. Pour cela, nous avons cherché à introduire ces mutations par PCR en utilisant
des conditions optimisées pour augmenter le taux d’erreur de la Taq polymérase : ajout de
DMSO à 10% final dans la réaction de PCR. Ce produit de PCR est ensuite cloné par
recombinaison dans un vecteur pour être exprimé sous contrôle d’un promoteur endogène
dans la souche où la viabilité cellulaire est assurée par l’expression de la copie RPS19A grâce
au promoteur GAL1/10-CYC1. Le produit de cette transformation est étalé sur milieu glucose
de manière à faire pousser la souche à 25°C par la seule copie de RPS19A issue de la
recombinaison. Parmi ces transformants, nous recherchons ceux qui ne sont pas capables de
pousser à température restrictive (37°C) tout en ayant la capacité de pousser à température
permissive (25°C) : il s’agit du phénotype ts.
Cette stratégie ne nous a pas permis d’obtenir la mutation qui conduit au phénotype ts et nous
avons alors décidé de muter systématiquement tous les résidus proline (P2, P46, P47, P90 et
P118) par des résidus glycine de manière à affecter la structure de la protéine Rps19p. Cette
séconde stratégie n’a pas permis de générer un mutant ts : aucune des 5 mutations ne conduit
à un phénotype ts comme le montre la figure S2.
195

Input I.P
35S32S
20S
25S
18S
WT W53R S60E TAP WT W53R S60E TAP
Figure S3. Co-immunoprécipitation des ARN ribosomiques avec RPS19-TAP. RPS19A sauvage ou muté , fusionné avec l'étiquette TAP est exprimé dans la souche de levure RPS19Adelta/RPS19B puis isolé sur colonne IgG sepharose. Les pré-ARNr isolés dans l'extrait cellulaire total (Input) et le matériel isolé (IP) sont analysés par northern blot avec la sonde complé-mentaire D-A2 alors que les ARNr matures 18S et 25S sont détectés par coloration au bromure d'éthidium. L'expression de l'étiquetteTAP seule est utilisée comme contrôle négatif.
196

III. Cas des mutations W53R et S60E
Nous avons montré que les mutations W53R et S60E ont un effet direct sur la fonction de la
protéine Rps19p. En effet, nous avons observé une perte totale de la viabilité cellulaire
consécutive à la mutation W53R. Il a été montré que la mutation S60E également affecte la
croissance cellulaire chez S. cerevisiae ( Gregory et al., 2007). Ici, nous montrons par analyse
northern blot avec la sonde D-A2 que la forme RPS19A-TAP co-précipite avec le pré-ARNr
20S qui est le précurseur de l’ARNr 18S (Figure S3), confirmant son incorporation dans les
ribosomes matures. Dans la fraction IP, nous avons une absence de co-précipitation du pré-
ARNr 20S avec les formes W53R-TAP et S60E-TAP contrairement à la forme non mutée
(WT). Les mutations W53R et S60E affectent la capacité de la protéine à s’incorporer dans
les ribosomes matures.
197

Vecteur
86-91-GFP
75-80-GFP
79-84-GFP
WT
GAL GLU
Figure S4. Test de complémentation fonctionnelle chez la levure S. cerevisiae. Complémentation fonctionnelle avec trois délétions effectuées dans la région 67-97 correspondant à la NLS. Toutes ces délétions affectent la croissance de la levure même s'ils n'ont pas eu d'impact sur la localisation de la protéine.
198

Molecular basis of Diamond-Blackfan anemia: stucture-function study of ribosomal protein
RPS19 in Saccharomyces cerevisiae
Diamond-Blackfan Anaemia (DBA) is a rare congenital erythroblastopenia associated with mono-
allelic mutations in several ribosomal protein genes. This linkage suggests a causal relationship
between alteration of ribosome biogenesis or functions and this pathology.
The RPS19 gene is the most frequently mutated (25% of patients). To understand the impact of the
mutations, especially missense mutations, we undertook a structure-function study of RPS19 homolog
in yeast Saccharomyces cerevisiae and we conducted a collaborative work to determine the crystal
structure of archeae Pyrococcus abyssi RPS19. Our results distinguish two types of mutations: some
affect residues buried in the structure and alter the protein folding and stability, while others change
amino acids at the protein surface and prevent incorporation of RPS19 into 40S pre-ribosomal
particles.
In addition, we determined the nuclear localization sequence and the molecular determinants of RPS19
transport to the nucleus. Mutations linked to DBA do not interfere with the nuclear localization of the
protein.
Thus, missense mutations in RPS19 primarily affect the ability of the protein to be incorporated into
pre-ribosomes, which alters ribosome biogenesis. This could on the one hand activate a stress response
and on the other hand lead to ribosome shortage, two events that could be fatal to some physiological
processes like erythropoiesis. A similar mechanism can be proposed for any ribosomal protein mutated
to the DBA.
199

Auteur : Almass-Houd AGUISSA TOURE
Titre : Bases moléculaires de l’anémie de Diamond-Blackfan : étude structure-fonction de la protéine ribosomique RPS19 chez Saccharomyces cerevisiae
Directeur de thèse : Prof. Pierre-Emmanuel GLEIZES
Soutenance à l’Université Paul Sabatier- Toulouse III : Salle de conférence de l’IEFG, Bât IBCG, 118
route de Narbonne, 31062 Toulouse cedex ; Mercredi 18 juin 2008 à 14h30.
L'anémie de Diamond-Blackfan (ADB) est une érythroblastopénie congénitale rare associée à des
mutations mono-alléliques dans plusieurs gènes de protéine ribosomique. Cette liaison suggère une
relation causale entre l’altération de la biogenèse ou de la fonction des ribosomes et cette pathologie.
Le gène RPS19 est le plus fréquemment muté (25% des patients). Pour comprendre l'impact des
mutations, en particulier les mutations ponctuelles faux-sens, nous avons engagé une étude structure-
fonction de l’homologue de la protéine RPS19 chez la levure Saccharomyces cerevisiae.
Parallèlement, nous avons mené un travail collaboratif pour déterminer la structure cristalline de
l'homologue de RPS19 de l'archeae Pyrococcus abyssi. Nos résultats distinguent deux types de
mutations : certaines, enfouies dans la structure, affectent le repliement et la stabilité de la protéine,
tandis que d'autres, en surface de la protéine, inhibent l’incorporation de RPS19 dans les particules
pré-ribosomiques 40S.
Par ailleurs, nous avons déterminé la séquence de localisation nucléaire et les déterminants
moléculaires de l'adressage nucléaire de RPS19. Les différentes mutations de RPS19 n’interfèrent pas
avec ce mécanisme de transport.
Ainsi, les mutations faux-sens de la protéine RPS19 affectent en premier lieu la capacité de la protéine
à être incorporée dans les pré-ribosomes, ce qui altère la biogenèse des ribosomes. Ceci pourrait d'une
part activer une réponse de stress et d'autre part conduire à un déficit en ribosomes, deux facteurs qui
pourraient être fatals dans certains processus physiologiques comme l'érythropoïèse. Ce mécanisme
peut être étendu à toute protéine ribosomique mutée dans l'ADB.
Mots clefs : Anémie de Diamond-Blackfan, Maladie génétique, Erythropoièse, Ribosome, Protéine
ribosomique, Structure, Trafic nucléocytoplasmique, NLS.
Discipline : Biologie Moléculaire et Cellulaire
Laboratoire : Laboratoire de Biologie Moléculaire Eucaryote
118, route de Narbonne, 31062 Toulouse cedex
200





























