Embed Size (px)
Citation preview

Correspondances en Onco-Théranostic - Vol. V - n° 1 - janvier-février-mars 201626
d o s s i e r t h é m a t i q u e
Tumeurs cérébrales
Tumeurs épendymaires Ependymomas Audrey Rousseau*, Catherine Godfraind**
* Département de pathologie cellulaire et tissulaire et UMR U1066-
MINT, CHU d'Angers. ** Service d’anatomo-
pathologie, CHU Gabriel-Montpied
et EA4677 ERTICa, Clermont-Ferrand.
RÉ
SU
MÉ
Su
mm
ar
y
» Les épendymomes sont des tumeurs dérivées de cellules souches périventriculaires. Bien que histologiquement semblables, ils constituent des entités (épi)génétiques distinctes selon leur localisation et l’âge du patient. Ainsi, la fusion C11orf95-RELA est retrouvée dans 70 % des épendymomes supratentoriels pédiatriques, et un profi l hyperméthylé dans certains épendymomes de la fosse postérieure. Les enfants sont le plus souvent atteints de lésions intracrâniennes aux profi ls génomiques équilibrés, alors que les adultes sont atteints d’épendymomes médullaires présentant de nombreux déséquilibres chromosomiques. Le gain du chromosome 1q est le seul facteur pronostique péjoratif reconnu. La qualité de l’exérèse chirurgicale reste l’élément pronostique décisif. Il n’y a pas actuellement de thérapie ciblée effi cace reconnue.
Mots-clés : Épendymome – Fusion RELA – Gain 1q – Déséquilibres génomiques – Hyperméthylation.
Ependymomas are rare glial tumors originating from cancer stem cells located in the vicinity of the ependymal lining. While morphologically alike, ependymomas demonstrate (epi)genetic anomalies varying according to the patient’s age and the tumor’s location. Supratentorial ependymomas harbor C11orf95-RELA fusion. A number of posterior fossa ependymomas display a hypermethylated phenotype. Most paediatric ependymomas show balanced genomes whereas adult ependymomas exhibit numerous chromosomal imbalances. Gain of 1q is the only recognized poor prognostic factor. At the opposite, complete surgical resection is associated with better prognosis. Targeted therapies have yet to be developed.
Keywords: Ependymoma – RELA fusion – 1q gain – Chromosomal imbalances – CIMP.
L es épendymomes se développent à tous les âges de la vie et à tous les niveaux de l’axe cranio spinal, avec cependant une prédilection intra crânienne
chez les enfants, et médullaire chez les adultes. En dehors du système nerveux, on en décrit dans la région sacrococcygienne, le médiastin et l’ovaire. Si histologi-quement ces tumeurs présentent des caractéristiques similaires quelle que soit leur localisation anatomique, (épi)génétiquement elles sont distinctes. Ces tumeurs sont incurables chez 45 % des patients. La chirurgie reste l’arme thérapeutique de choix. La radiothérapie vient en complément. Aujourd’hui, il n’y a pas de thérapie ciblée effi cace.
Caractéristiques histologiques
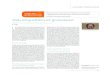
Les tumeurs épendymaires sont dérivées de cellules souches localisées en bordure du système ventri-culaire (1) . Ces dernières peuvent se diff érencier en plu-sieurs types cellulaires qui constituent le système nerveux, notamment en cellules épendymaires. À l’état physio-logique, ces cellules possèdent des cils (fi gures 1A et 1B) dont le témoin est la positivité intracytoplasmique en
do t de l’EMA (Epithelial Membrane Antigen) [fi gure 1C] . Ce marquage constitue une des caractéristiques micros-copiques des tumeurs épendymaires. Une autre est la présence de pseudorosettes périvasculaires (fi gure 1D) qui sont des régions uniquement constituées de pro-longements cellulaires disposés de façon radiaire autour des sections vasculaires. Ces zones anucléées alternent de façon caractéristique avec des régions nucléées (fi gure 1E) . Plus rarement, les épendymomes présentent d’authentiques rosettes épendymaires (creusées d’une lumière centrale) [fi gure 1F] et des tubes épendymaires. Ces critères morphologiques ne sont cependant pas limités aux tumeurs épendymaires. En eff et, on observe des pseudorosettes périvasculaires dans les tumeurs papillaires de la région pinéale (TPRP) et les gliomes angiocentriques, et des dots intracytoplasmiques d’EMA dans ces dernières ainsi que dans des astrocytomes diff us. L’immunoréactivité contre la vimentine et la GFAP (Glial Fibrillary Acidic Protein) , autre caractéristique des tumeurs épendymaires, s’observe aussi dans les astrocytomes et les TPRP. Contrairement aux astrocytomes, les tumeurs épendymaires n’expriment pas, en général, le facteur de transcription Olig2 et, à la diff érence des TPRP et des tumeurs plexuelles, elles n’expriment pas les cytokératines.
0026_COO 26 21/03/2016 14:36:12

Correspondances en Onco-Théranostic - Vol. V - n° 1 - janvier-février-mars 2016 27
Tumeurs épendymaires
Figure. Les épendymomes sont des tumeurs dérivées des cellules qui bordent le système ventriculaire (B) et, plus particulièrement, des cellules souches ayant un phénotype de cellules gliales radiaires (J) . Histologiquement, ces tumeurs présentent un aspect lobulé, très marqué dans les subépendymomes (G) . Les pseudo-rosettes périvasculaires (D et E) , les rosettes épendymaires (F) et un marquage cytoplasmique pour l’EMA (C) , subrogé de l’existence de cils (A) , les caractérisent. Leur profi l génétique varie en fonction de l’âge du patient et de la localisation tumorale. Les épendymomes supratentoriels sont porteurs de translocation entre C11orf95 et, le plus souvent, RELA entraînant une activation de la voie NF-κB détectable en immunohistochimie (I) . L’amplifi cation de 1q25 est pronostique (K) .
Notch
Hox
A
CIMP ±
B C
D E F
I
G
H J K
0027_COO 27 21/03/2016 14:36:13

Correspondances en Onco-Théranostic - Vol. V - n° 1 - janvier-février-mars 201628
d o s s i e r t h é m a t i q u e
Tumeurs cérébrales
Entités tumorales
Subépendymomes Les subépendymomes sont des tumeurs de grade I selon la classifi cation de l’Organisation mondiale de la santé (OMS) [fi gure 1G, p. 27] localisées dans les régions périventriculaires. Peu symptomatiques, ces tumeurs sont en général de découverte fortuite. Elles sont macro- et microscopiquement lobulées. Elles sont pauci cellulaires avec des régions nucléées alternant avec de larges zones anucléées fi brillaires, parfois microkystiques. Leurs vaisseaux ont très souvent des parois hyalinisées, sujettes à dissection et, ainsi, à l’origine d'hémorragies et d'ischémies intratumorales.
Épendymomes myxopapillaires Ces tumeurs, de grade I selon la classification de l’OMS, sont localisées au niveau de la queue de che-val (figure 1H, p. 27) et encapsulées. Leur exérèse chirurgicale sera donc complète, sauf si la croissance tumorale s’accompagne d’une rupture capsulaire entraînant la perte du caractère indolent de la lésion . Histologiquement, elles ont un aspect en cocarde ; un vaisseau central est entouré par une couche de maté-riel mucoïde, elle-même recouverte d’une couronne de cellules gliales généralement effi lées. Les parois vasculaires peuvent aussi être hyalinisées et ainsi provoquer d’importantes hémorragies venant même, histologiquement, à dissimuler la tumeur initiale.
Épendymomes Les épendymomes sont de grade II selon la clas-sification de l'OMS. Cliniquement, ils induisent des symptômes d’hypertension intracrânienne. Histologiquement, ils sont caractérisés par des pseudo-rosettes périvasculaires. Leur densité cellulaire est modérée, les mitoses peu nombreuses, les vaisseaux non proliférants et la nécrose absente. Si la classi-fi cation 2007 de l’OMS reconnaissait 4 variants aux épendymomes, celle de 2016 fait disparaître l’épen-dymome cellulaire. L’OMS a maintenu un variant très rare : l’épendymome papillaire qui pose un problème de diagnostic diff érentiel avec les tumeurs papillaires que l’on rencontre dans le système nerveux : méta-stase, tumeur plexuelle, TPRP ou encore tumeur du sac endolymphatique (2) . Ces tumeurs sont positives pour diverses cytokératines contrairement aux épen-dymomes. Est également resté le variant à cellules claires qui, lui, pose un problème de diagnostic dif-férentiel avec un oligodendrogliome porteur d’une codélétion des bras chromosomiques 1p et 19q, et, éventuellement, avec une métastase de carcinome
à cellules claires. Enfi n, le dernier variant décrit est l’épendymome tanicytique, très rare, qui doit se dis-tinguer du schwannome.
Épendymomes anaplasiques Les épendymomes anaplasiques sont de grade III selon la classifi cation de l’OMS. Ils se diff érencient de ceux de grade II par une densité cellulaire majorée, une activité mitotique importante, la présence d’une prolifération endothéliocapillaire et, enfi n, celle de nécrose. Les cri-tères ainsi défi nis par l’OMS manquent de reproductibi-lité interobservateur et de valeur histopronostique (3) . Cela se comprend du fait de leur manque de précision, mais aussi parce que le grading de l'OMS est fondé sur une logique de croissance tumorale : l’augmen-tation de la cellularité sera suivie par l’apparition de mitoses, puis d’une prolifération endothéliocapillaire, enfi n de nécrose. Or, cette évolution n’est pas une règle constante pour les épendymomes (4) . Enfi n, les épendy momes présentent des anomalies génétiques qui varient en fonction de l’âge du patient et de la localisation tumorale, alors que le grading OMS est identique pour tous les épendymomes.
Épendymome RELA La classifi cation OMS de 2016 introduit une nouvelle entité , l’épendymome RELA-fusion (fi gure 1I, p. 27) [5, 6] décrit dans la partie “Génétique” ci-dessous .
Caractéristiques génétiques
Génétique familiale Les altérations moléculaires qui prédisposent au dévelop pement des tumeurs ont souvent été éclai-rées par la génétique familiale des cancers. Ce n’est pas le cas pour les épendymomes. En eff et, il n’a pas été démontré de rôle de NF2 , TP53 ou d’ APC (Anaphase Promoting Complex) dans les épendymomes spora-diques, alors que de rares cas familiaux ont été associés à des neurofi bromatoses de type 2, des syndromes de Li-Fraumeni ou encore de Turcot.
ADN Les épendymomes ont des anomalies chromo-somiques qui diffèrent en fonction de leur locali-sation le long du névraxe. Ainsi, la perte du 22q et les mutations de NF2 sont plus fréquentes dans les lésions spinales (7) . Les aberrations chromosomiques sont plus nombreuses au niveau du cône terminal, région quasi synonyme d’épendymome myxo-papillaire (8) .
0028_COO 28 21/03/2016 14:36:14

Correspondances en Onco-Théranostic - Vol. V - n° 1 - janvier-février-mars 2016 29
Tumeurs épendymaires
L’âge est également important. Seuls 10 % des épendy-momes de l’adulte ont un caryotype équilibré, contre 40 % chez l’enfant. De plus, les caryotypes pédiatriques aneuploïdes sont souvent associés à des gains du chromo some 1q (9) . La première anomalie récurrente identifi ée dans les épendymomes est une fusion entre C11orf95 (11q13.1), un gène mal caractérisé, et RELA comme partenaire majoritaire, et YAP1 ou MAML1 comme partenaires minoritaires (5, 6). Ces fusions s’observent uniquement dans les épendymomes supratentoriels de l’enfant. RELA (11q13.1) est le principal eff ecteur de la voie de signalisation NF-κB, une voie de l’infl ammation. En cas de fusion, cette dernière est activée (fi gure 1I, p. 27) . Chez des souris allogreff ées, la fusion C11orf95 -RELA est à l’origine du développement d’épendymomes. C’est la seconde altération génétique identifi ée dans la tumorigenèse des épendymomes. La première était l’amplifi cation du gène EphB2 qui, chez des souris Ink4a/Arf−/−, induit des tumeurs dont le profi l d’ex-pression génique correspond chez l’homme à un sous-groupe d’épendymomes supratentoriels (10) .
ARN Les anomalies de l’ARN varient également en fonction de la localisation tumorale. Ainsi, les épendymomes intra-crâniens surexpriment les voies Notch, BMP et SHH , tandis que les épendymomes spinaux surexpriment des gènes de la famille Hox (1, 11, 12) . Ces voies de signalisation jouent un rôle primordial dans le maintien, la prolifération et la dif-férenciation des cellules souches. L’expression diff érentielle de ces voies a aussi été observée au niveau des cellules gliales radiaires, cellules qui jouent un rôle essentiel dans la formation du cortex et peuvent se diff érencier en neurones et en cellules gliales (figure 1J, p. 27) . Cela conduit à l’hypothèse selon laquelle les épendymomes déri-veraient de cellules souches cancéreuses ayant un phénotype de glie radiaire.
Épigénétique Le peu d’anomalies caryotypiques et l’absence de muta-tion des gènes situés dans des régions hémidélétées ont font rechercher des altérations épigénétiques dans les épendymomes. Ainsi, des délétions hétérozygotes sont fréquemment observées en 9p21, sans que CDKN2A/p16INK4A , CDKN2A/p14ARF et CDKN2B soient mutés. En revanche, on observe que les régions promotrices de ces mêmes gènes sont hyperméthylées, et ce, une fois de plus en fonction de l’âge du patient et de la localisation de la tumeur (13) . De même, au niveau de la fosse cérébrale postérieure, 2 sous-groupes d’épendymomes ont été identifiés
en fonction de leur profi l de méthylation : un sous-groupe hyperméthylé, appelé A ou CIMP (CpG Island Methylator Phenotype) , et un sous-groupe non hyper-méthylé, appelé B (14) . Dans le sous-groupe A, les îlots CpG hyperméthylés intéressent principalement les cibles du complexe PRC2 (Polycomb Repressive Complex 2) . Ce complexe inhibe la diff érenciation des cellules souches et maintient le caractère pluripotent des cellules via la triméthylation de l’histone H3 sur la lysine en position 27 (H3K27). D’un point de vue clinicopathologique, les tumeurs du sous-groupe A aff ectent des enfants jeunes, ont un caryotype équili-bré, une localisation latérale, des signes histologiques d’invasion, et surtout un mauvais pronostic. Le gain du 1q est l’altération moléculaire la plus fréquente de ce groupe (15 % des cas). Par opposition, le sous-groupe B est caractérisé par une survenue chez des enfants plus âgés et des adultes, un génome instable, une localisa-tion médiane (IV e ventricule), une absence de signes d’invasion ainsi qu’un pronostic favorable, rejoignant celui des épendymomes de la moelle.
Biomarqueurs
Cibles thérapeutiques Aujourd’hui, il n’y a aucune cible thérapeutique reconnue dans les épendymomes. Les données molé-culaires accumulées apportent néanmoins des pistes. Par exemple, il est histologiquement très rare de voir des fi gures apoptotiques dans les épendymomes. Or, les voies de l’apoptose sont activées dans les épendy-momes de grade III selon la classifi cation de l’OMS. Mais la survivine, une protéine antiapoptotique, y est aussi surexprimée (12) . Un inhibiteur de cette molé-cule pourrait être envisagé. Parallèlement, des agents déméthylant l’ADN, des inhibiteurs du complexe PRC2 ou de la triméthylation de H3K27 pourraient trouver une place dans le traitement des épendymomes de la fosse postérieure du sous-groupe A (14) .
Marqueurs pronostiques Le seul marqueur pronostique qui a été confi rmé par plusieurs équipes indépendantes est l’amplifi cation de la région 1q25 (figure 1K, p. 27) [9, 15] . C’est un marqueur de mauvais pronostic qui est indépendant du degré de résection chirurgicale. Il faut rappeler que, à l’heure actuelle, la qualité de l’exérèse chirurgicale reste le facteur pronostique des épendymomes. Très récemment, la fusion C11orf95-RELA a été associée à un pronostic sombre (6) . Il est cependant nécessaire que cette association soit confi rmée par d’autres groupes.
0029_COO 29 21/03/2016 14:36:15

Correspondances en Onco-Théranostic - Vol. V - n° 1 - janvier-février-mars 201630
d o s s i e r t h é m a t i q u e
Tumeurs cérébrales
En défi nitive, cela illustre le futur de l’épendymome : une histologie pour confi rmer la nature de la lésion et des critères clinicogénétiques pour en établir le pronostic et le traitement.
Conclusion
Parmi les tumeurs épendymaires, les subépen-dymomes et les épendymomes myxopapillaires constituent des entités aux caractéristiques histo-logiques, cliniques et génétiques bien identifi ables.
Les épendy momes, qu’ils soient de grade II ou III, ne se diff érencient histologiquement que par des critères d’agressivité. Ce grading n’a pas de valeur pronostique. Les épendymomes présentent un éventail d’anomalies (épi)génétiques dont les caractéristiques dépendent de la localisation tumorale et de l’âge du patient. Ces aberrations sont aujourd’hui associées à des comportements cliniques et le seront demain à des traitements personnalisés. ■
Remerciements à Mlle K. Silva et au Dr A. Jouvet pour l’illustration d’un épendymome C11orf95-RELA.
Les auteurs dclarent ne pas avoir de liens d’intérêts .
1. Taylor MD, Poppleton H, Fuller C et al. Radial glia cells are candidate stem cells of ependymoma. Cancer Cell 2005;8(4):323-35.
2. Du J, Wang J, Cui Y et al. Clinicopathologic study of endolym-phatic sac tumor (ELST) and diff erential diagnosis of papillary tumors located at the cerebellopontine angle. Neuropathology 2015;35(5):410-20.
3. Ell ison DW, Kocak M, Figarella-Branger D et al. Histopathological grading of pediatric ependymoma: repro-ducibility and clinical relevance in European trial cohorts. J Negat Results Biomed 2011;10:7.
4. Godfraind C. Classifi cation and controversies in pathology of ependymomas. Childs Nerv Syst 2009;25(10):1185-93.
5. Parker M, Mohankumar KM, Punchihewa C et al. C11orf95-RELA fusions drive oncogenic NF-κB signalling in ependymoma. Nature 2014;506(7489):451-5.
6. Pajtler KW, Witt H, Sill M et al. Molecular Classification of Ependymal Tumors across All CNS Compartments, Histopathological Grades, and Age Groups. Cancer Cell 2015;27(5):728-43. 7. Ebert C, von Haken M, Meyer-Puttlitz B et al. Molecular genetic analysis of ependymal tumors. NF2 mutations and chromosome 22q loss occur preferentially in intramedullary spinal ependymomas. Am J Pathol 1999;155(2):627-32. 8. Rousseau A, Idbaih A, Ducray F et al. Specifi c chromosomal imbalances as detected by array CGH in ependymomas in association with tumor location, histological subtype and grade. J Neurooncol 2010;97(3):353-64. 9. Dyer S, Prebble E, Davison V et al. Genomic imbalances in pediatric intracranial ependymomas defi ne clinically relevant groups. Am J Pathol 2002;161(6):2133-41. 10. Johnson RA, Wright KD, Poppleton H et al. Cross-species genomics matches driver mutations and cell compartments to model ependymoma. Nature 2010;466(7306):632-6.
11. Modena P, Lualdi E, Facchinetti F et al. Identifi cation of tumor-specifi c molecular signatures in intracranial ependy-moma and association with clinical characteristics. J Clin Oncol 2006;24(33):5223-33. 12. Palm T, Figarella-Branger D, Chapon F et al. Expression profi ling of ependymomas unravels localization and tumor grade-specifi c tumorigenesis. Cancer 2009;115(17):3955-68. 13. Rousseau E, Ruchoux MM, Scaravilli F et al. CDKN2A, CDKN2B and p14ARF are frequently and diff erentially methy-lated in ependymal tumours. Neuropathol Appl Neurobiol 2003;29(6):574-83. 14. Mack SC, Witt H, Piro RM et al. Epigenomic alterations defi ne lethal CIMP-positive ependymomas of infancy. Nature 2014;506(7489):445-50. 15. Godfraind C, Kaczmarska JM, Kocak M et al. Distinct disease-risk groups in pediatric supratentorial and posterior fossa ependymomas. Acta Neuropathol 2012;124(2):247-57.
R é f é r e n c e s
0030_COO 30 21/03/2016 14:36:16