Embed Size (px)
Citation preview

Rev Med Liege 2005; 60 : Supp. I : 66-70
Les génophotodermatoses sont des maladiesgénétiques dont un des signes majeurs est unesensibilité exacerbée à l’action des radiationsactiniques.
DÉFICIT DE SYNTHÈSE DE LA MÉLANINE.
A) ALBINISME
L’albinisme oculo-cutané comprend plusd’une dizaine de types cliniques différents quisont rares et de transmission autosomique réces-sive. Ces affections se caractérisent par undéfaut partiel ou total de la synthèse ou du trans-fert de la mélanine avec hypopigmentationsévère de la peau, des phanères et des yeux. Laphotosensibilité et le risque d’apparition detumeurs cutanées malignes sont élevés. La pré-sence d’anomalies immunitaires ou des pla-quettes définissent sur un plan clinique dessous-types encore plus rares tels que les syn-dromes de Hermansky-Pudlak, de Pruniéras-Griscelli et de Chediak-Higashi.
L’albinisme de type 1 est dû à des mutationsdirectes du gène de la tyrosinase dont la fonctiondevient déficitaire. Dans les sous-types 1B et1TS (thermosensible), l’enzyme conserve uneactivité résiduelle avec respectivement synthèsede phaeomélanine caractérisant l’albinismejaune, ou synthèse d’eumélanine au-dessous de35°C avec pigmentation limitée aux zonesfroides de la peau des extrémités.
Dans le type 2 qui est le plus fréquent, l’acti-vité de la tyrosinase est normale. Les mutationstouchent le gène codant pour la protéine P inter-venant dans le transport des mélanosomes etpeut-être aussi celui de la tyrosine, substrat de latyrosinase.
Le type 3, lui aussi tyrosinase positif, est dû àdes anomalies de la protéine TRP1 (tyrosinase-related protein) impliquée dans la synthèse de lamélanine.
Les autres sous-types cliniques sont d’unetrès grande rareté. La prise en charge de cespatients est difficile, associant une photo-protec-tion stricte, une surveillance étroite pour dépis-ter l’apparition de tumeurs malignes, et la priseéventuelle de rétinoïdes oraux.
DÉFICIENCES DE LA RÉPARATION DE
L’ADN
A) XERODERMA PIGMENTOSUM
Définition
Le xeroderma pigmentosum est un ensembled’affections héréditaires, autosomiques réces-sives, caractérisées par une sensibilité patholo-gique aux UV, liée à un déficit des systèmesenzymatiques de réparation de lésions acquisesde l’ADN (1). Le mécanisme biochimique fon-damental est l’absence de réparation des muta-tions de type CC ---> TT d’origine génotoxique,chimique et surtout physique par les UV. Cesmutations qui surviennent au hasard peuventtoucher des gènes-clés du fonctionnement cellu-laire, notamment des proto-oncogènes, maiségalement des gènes suppresseurs de tumeurs,tels que la protéine p53 (2). Toutefois, l’anoma-lie de réparation n’est probablement pas le seulmécanisme en cause dans la carcinogenèsepuisque des anomalies similaires dans le syn-drome de Cockayne et dans la trichothiodystro-phie ne s’accompagnent pas de tumeurscutanées. Il existe donc probablement d’autresfacteurs, en particulier immunitaires portant surles fonctions des lymphocytes natural-killer(NK) ou des altérations de la détoxification desERO génotoxiques par baisse de l’activité de lasuperoxyde-dismutase. Dans les différents sous-groupes de xeroderma pigmentosum, l’anomaliegénétique qui sous-tend le défaut de réparationest présente dans toutes les cellules de l’orga-nisme, mais se traduit surtout par un tableaupathologique cutané. D’autres anomaliesinternes sont toutefois possibles, notammentneurologiques dans le groupe de complémenta-tion A.
(1) Chef de Laboratoire, (2) Assistant de Recherche,(3) Chargé de Cours, Chef de Service, CHU du SartTilman, Service de Dermatopathologie
GÉNOPHOTODERMATOSES
RÉSUMÉ : Les génophotodermatoses peuvent résulter d’undéficit de synthèse de mélanine, d’une déficience de réparationde l’ADN ou d’une photosensibilité congénitale.MOTS-CLÉS : Albinisme - Protoprophyrie/erythropoïétique - Xero-derma pigmentosum
GENOPHOTODERMATOSES
SUMMARY : Genophotodermatoses can result from a defect inmelanin synthesis, a defect in DNA repair or a congenital pho-tosensibility.KEYWORDS : Albinism - Erythropoietic protoporphyria - Xero-derma pigmentosum
J.E. ARRESE (1), F. HENRY (2), G.E. PIÉRARD (3)
66
GÉNOPHOTODERMATOSES
Rev Med Liege 2005; 60 : Supp. I : 66-70 67
Présentation clinique
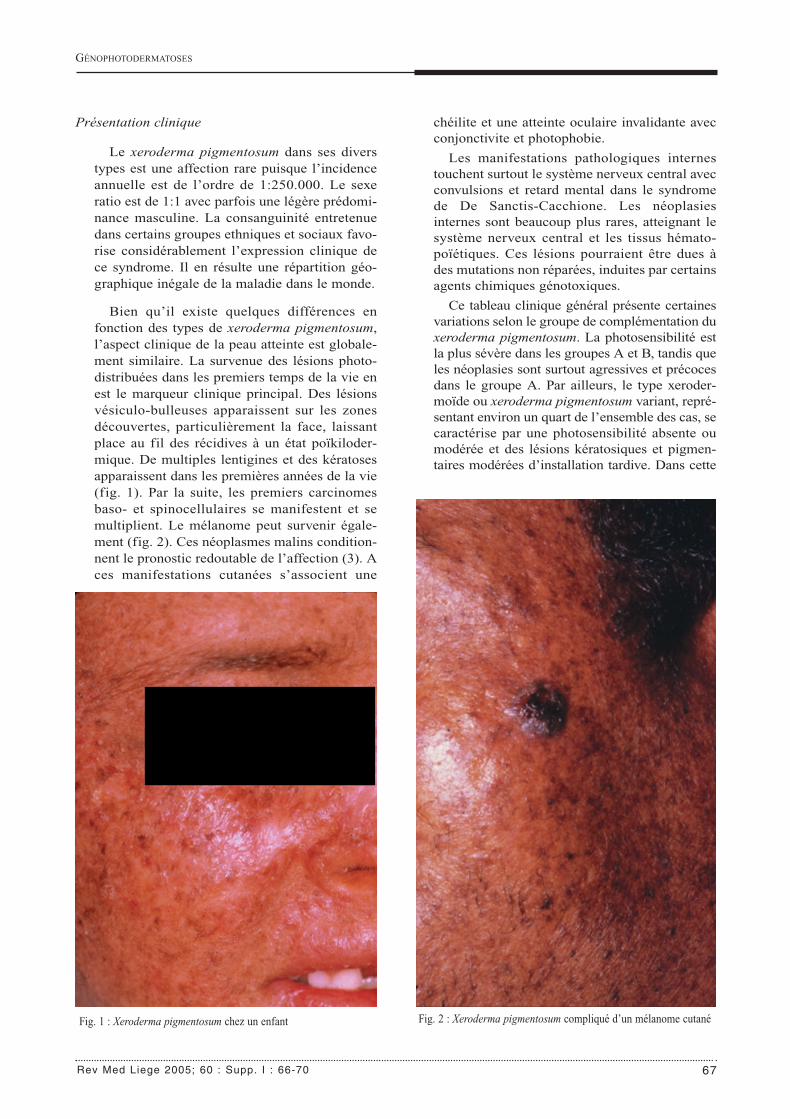
Le xeroderma pigmentosum dans ses diverstypes est une affection rare puisque l’incidenceannuelle est de l’ordre de 1:250.000. Le sexeratio est de 1:1 avec parfois une légère prédomi-nance masculine. La consanguinité entretenuedans certains groupes ethniques et sociaux favo-rise considérablement l’expression clinique dece syndrome. Il en résulte une répartition géo-graphique inégale de la maladie dans le monde.
Bien qu’il existe quelques différences enfonction des types de xeroderma pigmentosum,l’aspect clinique de la peau atteinte est globale-ment similaire. La survenue des lésions photo-distribuées dans les premiers temps de la vie enest le marqueur clinique principal. Des lésionsvésiculo-bulleuses apparaissent sur les zonesdécouvertes, particulièrement la face, laissantplace au fil des récidives à un état poïkiloder-mique. De multiples lentigines et des kératosesapparaissent dans les premières années de la vie(fig. 1). Par la suite, les premiers carcinomesbaso- et spinocellulaires se manifestent et semultiplient. Le mélanome peut survenir égale-ment (fig. 2). Ces néoplasmes malins condition-nent le pronostic redoutable de l’affection (3). Aces manifestations cutanées s’associent une
chéilite et une atteinte oculaire invalidante avecconjonctivite et photophobie.
Les manifestations pathologiques internestouchent surtout le système nerveux central avecconvulsions et retard mental dans le syndromede De Sanctis-Cacchione. Les néoplasiesinternes sont beaucoup plus rares, atteignant lesystème nerveux central et les tissus hémato-poïétiques. Ces lésions pourraient être dues àdes mutations non réparées, induites par certainsagents chimiques génotoxiques.
Ce tableau clinique général présente certainesvariations selon le groupe de complémentation duxeroderma pigmentosum. La photosensibilité estla plus sévère dans les groupes A et B, tandis queles néoplasies sont surtout agressives et précocesdans le groupe A. Par ailleurs, le type xeroder-moïde ou xeroderma pigmentosum variant, repré-sentant environ un quart de l’ensemble des cas, secaractérise par une photosensibilité absente oumodérée et des lésions kératosiques et pigmen-taires modérées d’installation tardive. Dans cette
Fig. 1 : Xeroderma pigmentosum chez un enfant Fig. 2 : Xeroderma pigmentosum compliqué d’un mélanome cutané

affection, les carcinomes basocellulaires n’appa-raissent qu’après 40 ans.
Toutes les formes de xeroderma pigmentosumbénéficient d’une photoprotection stricte. Lescaroténoïdes oraux et des topiques renfermantdes enzymes anti-oxydantes proches de la super-oxyde dismutase ont été proposés. Certainspatients pourraient bénéficier d’un traitementpréventif par rétinoïdes oraux. La dose néces-saire pour une protection efficace vis-à-vis del’apparition de tumeurs est souvent élevée aveccomme corrolaire une tolérance médiocre. Desgreffes de kératinocytes autologues, après cor-rection in vitro du déficit génétique, font partiedu nouvel arsenal de thérapie génique.
Diagnostic biologique
Le diagnostic biologique du xeroderma pig-mentosum se fait en trois étapes. D’abord, untrouble de l’excision-réparation des lésions UVinduites de l’ADN est mis en évidence sur leslymphocytes sanguins. Le taux d’excision-répa-ration est plus ou moins réduit selon les groupesde complémentation et la réduction est surtoutmarquée dans le groupe A. En revanche, le tauxd’excision-réparation est normal dans le xero-derma pigmentosum de type variant.
La deuxième étape est constituée par la déter-mination du groupe de complémentation. Septgroupes de complémentation classés de A à Gdéfinissent les anomalies de réparation pré-réplicative. Le groupe variant identifie les ano-malies post-réplicatives. Ces groupes sontdéfinis en fonction du type de cellules néces-saire dans des modèles de fusion cellulaire pourcorriger le défaut de réparation présent dans lescellules du patient.
Enfin, la troisième étape qui est du domainede la recherche fondamentale détermine lalésion génétique. Les sept types classiques dexeroderma pigmentosum et le xeroderma pig-mentosum variant ont été associés à des altéra-tions génétiques précises. Le type A résulted’une mutation du gène XPA codant pour uneprotéine confirmant la présence de lésions del’ADN après leur détection précoce par la pro-téine issue du gène lésé dans le xeroderma pig-mentosum du groupe C. Le type B, toujoursassocié à un phénotype de type syndrome deCockayne, est lié à des anomalies du gèneERCC-3 (Excision-Reparation Cross Comple-mentation), qui code pour une protéine de typehélicase, sous-unité du complexe TFIIH detranscription de l’ADN. Le type C est lié à desanomalies du gène XPC qui code pour une pro-téine de détection précoce des lésions de l’ADN.Le type D dépend d’une mutation du gène
ERCC-2 qui code pour une hélicase du com-plexe TFIIH. Le type E correspond à des ano-malies du gène XPE qui intervient dans lareconnaissance de l’ADN lésé par les UV. Letype F est dû à des altérations du gène ERCC-4ou ERCC-11 qui code pour une exonucléase 5’par rapport à une lésion de l’ADN. Le type G estla conséquence des altérations du gène ERCC-5qui code pour une exonucléase agissant en 3’. Lexeroderma pigmentosum variant est associé àdes altérations d’un gène codant pour une ADNpolymérase qui permet la poursuite de la dupli-cation de l’ADN malgré la présence de dimèresde thymine.
B) SYNDROME DE COCKAYNE
Le syndrome de Cockayne est une poïkiloder-mie congénitale de transmission autosomiquerécessive. Les signes cliniques apparaissent leplus souvent vers l’âge de 6 mois et se caractéri-sent par des anomalies cutanées, oculaires, neu-rologiques et squelettiques, associées à un retardstaturo-pondéral et psycho-intellectuel. Lessignes cutanés sont dominés par une photosensi-bilité constante n’aboutissant pas à la formationde tumeurs cutanées. Il s’y associe une dégéné-rescence pigmentaire progressive de la rétine,éventuellement associée à une cataracte, unesurdité et une spasticité avec ataxie. Le tableaudysmorphique est caractérisé par un visagepseudo-sénile, une microcéphalie, un progna-tisme, un nez pointu et des oreilles parfois pro-éminentes rappelant le faciès de Mickey Mouse.Les membres sont longs avec des extrémitésélargies.
Le syndrome de Cockayne résulte d’un défautde réparation de l’ADN, qui porte électivementsur les gènes activement transcrits. Aucun défi-cit immunitaire n’est présent contrairement auxeroderma pigmentosum. Ce syndrome est hété-rogène sur le plan génétique puisque 3 groupesde complémentation ont été définis. Le groupeA dépend d’anomalies du gène CSA situé sur lechromosome 5 et codant pour une protéine com-portant un domaine de répétition de motifs WD(tryptophane-arginine) qui interagit avec ERCC-2, 3 et 6 au sein du complexe transcriptionnelTFIIH. Le groupe B dépend de mutations dugène ERCC-6 localisé en 10q11-21. Le groupeC est dû à des mutations du même gène impliquédans le xeroderma pigmentosum du groupe B(ERCC-3), mais avec un profil mutationnel pro-bablement différent touchant d’autres régions dugène. Cette hétérogénéité génétique est encorecompliquée par le fait que certains patients ontun phénotype à la fois xeroderma pigmentosumtype G et syndrome de Cockayne, avec une
J.E. ARRESE ET COLL.
Rev Med Liege 2005; 60 : Supp. I : 66-7068

GÉNOPHOTODERMATOSES
Rev Med Liege 2005; 60 : Supp. I : 66-70 69
amputation importante de la protéine ERCC-5qui emporte des domaines fonctionnels, proba-blement séparés, dont l’altération est respon-sable, d’une part, du défaut de réparation“tumorigène” du xeroderma pigmentosum et,d’autre part, du syndrome de Cockayne.
L’évolution est fatale à brève échéance et iln’existe pas de traitement spécifique de ce syn-drome hormis la photoprotection qui s’imposeprécocement, associée aux différentes prises encharge particulières vis-à-vis d’anomalies ocu-laires, squelettiques et neurologiques.
C) TRICHOTHIODYSTROPHIE
La dénomination trichothiodystrophieregroupe des affections cliniquement et biologi-quement hétérogènes, de transmission autoso-mique récessive qui associent des anomalies desstructures neuro-ectodermiques, en particulierdes cheveux (4). La caractéristique commune àtoutes ces affections est une dystrophie pilaireavec des cheveux cassants (B, brittle hair) qui seraréfient progressivement avec l’âge. Leuraspect “tigré” régulier est particulièrement bienmis en évidence à l’examen en lumière polari-sée. Le contenu en acides aminés soufrés estconsidérablement diminué dans les phanères,notamment dans les cheveux. Diverses autresanomalies sont parfois présentes, notammentune photosensibilité (P), un aspect ichtyosi-forme (I), un retard intellectuel (I) et psychomo-teur, une diminution (D) de la fertilité, et unretard staturo-pondéral (S). L’ensemble de cesanomalies caractérise les entités définies par lesacronymes PIBIDS et IBIDS selon l’existenceou non d’une photosensibilité qui est présentedans près de la moitié des cas. D’autres anoma-lies ectodermiques sont parfois associées. Ellespeuvent consister en une dysplasie unguéale,une cataracte bilatérale, des anomalies dentaires,une dysmorphie faciale, des bronchestasies etdes infections à répétition, notamment des voiesaériennes supérieures.
Une anomalie de la réparation des lésions UVinduites de l’ADN est présente chez les patientssouffrant d’une photosensibilité anormale. Lespatients atteints de PIBIDS se répartissent en 3groupes de complémentation génétique. L’ori-gine génétique du groupe A n’est pas définie.Les groupes B et C présentent des anomaliesgénétiques identiques au xeroderma pigmento-sum respectivement de type B (ERCC-3) et D(ERCC-2), mais avec un profil différent demutation. Des mutations de ces deux gènes sontégalement constatées chez certains patients pré-sentant un phénotype mixte de type xerodermapigmentosum et syndrome de Cockayne. Il est
donc hautement probable que les syndromes detype trichothiodystrophie soient en fait en rap-port avec une anomalie de la transcription assu-rée par le complexe TFIIH.
Le pronostic de ces syndromes est variable,conditionné en priorité par l’importance duretard psychomoteur. En l’absence de traitementétiologique, la sévérité potentielle de l’atteinteneurologique incite à procéder au diagnosticanténatal en cas d’antécédent familial.
D) SYNDROME DE BLOOM
Le syndrome de Bloom est une poïkilodermiecongénitale rare, à transmission autosomiquerécessive, associant un retard statural à débutintra-utérin, une photosensibilité précoce etmajeure qui se complique de lésions facialesérythémato-croûteuses, progressivement télan-giectasiques, et un déficit immunitaire à l’ori-gine d’infections respiratoires à répétition. Unedysmorphie faciale associant un nez long etétroit avec une dolichocéphalie est relativementfréquente. Les lésions cutanées s’atténuent avecl’âge. Cependant, près de 20% des patientsdéveloppent des tumeurs malignes, notammentdes hémopathies et des carcinomes digestifs.
Le syndrome de Bloom exprime des muta-tions du gène BLM codant pour une hélicase dugroupe RecQ. Il se caractérise par une instabilitéet une fragilité chromosomiques vis-à-vis del’action d’un grand nombre d’agents géno-toxiques, probablement par défaut de réparationde l’ADN et par un taux élevé d’échange deschromatides-soeurs lors de la méiose.
E) SYNDROME DE ROTHMUND-THOMSON
Le syndrome de Rothmund-Thomson est unepoïkilodermie congénitale qui se manifeste parl’apparition très précoce d’un érythème télan-giectasique avec atrophie des joues et du front.L’éruption s’étend ensuite à l’ensemble duvisage, au cou, aux membres supérieurs, auxcuisses et aux fesses. La photosensibilité esthabituelle. L’exposition solaire peut provoquerl’apparition de bulles et favorise l’extension dela dermatose. D’autres anomalies incluent unecataracte, une hypotrichose, des onychodystro-phies, un retard statural et parfois des anomaliesosseuses. L’affection s’atténue avec l’âge et restede bon pronostic vital. Chez certains patients, undéfaut de réparation des lésions UV-induites aété attribué à des mutations de l’hélicase RecQ4.

PHOTOSENSIBILITÉ CONGÉNITALE
A) PROTOPORPHYRIE ÉRYTHROPOÏÉTIQUE
La protoporphyrie érythropoïétique est uneaffection génétique transmise sur le mode autoso-mique dominant. Elle se manifeste chez le jeuneenfant par des poussées érythémato-œdémateuseset prurigineuses des zones photo-exposées. Cer-taines lésions deviennent purpuriques ou vési-culo-bulleuses. La répétition des poussées aboutità la formation de cicatrices atrophiques. D’autreszones du tégument apparaissent épaissies par uneinfiltration cireuse (fig. 3). Des lithiases biliairesriches en prophyrines peuvent conduire à une cir-rhose cholestatique.
L’anomalie enzymatique touchant au métabo-lisme des porphyrines consiste en un déficit dela ferrochélatase.
La photoprotection et la prise orale de ß-caro-ténoïde limitent la sévérité des manifestationscutanées.
B) MALADIE DE HARTNUP
La maladie se transmet sur un mode autoso-mique récessif. La carence d’absorption du tryp-tophane conduit à un déficit de synthèse de
nicotinamide et de sérotonine. Dès lors, lestroubles du métabolisme du tryptophane rencon-trés dans la maladie de Hartnup aboutissent àune carence en acide nicotinique (vitamine B3ou PP) ou avec accumulation de chromophoresanormaux (5). Le tableau est habituellementcelui d’un érythème pellagroïde, se traduisantpar des poussées de photosensibilité, aboutissantà un érythème sombre avec atrophie de la peauqui devient hyperpigmentée et couverte desquames grises.
Chez des individus plus âgés, l’association designes cutanés similaires à des troubles neurolo-giques et digestifs constitue la pellagre, excep-tionnelle dans les pays développés, et liée à unecarence polyvitaminique.
RÉFÉRENCES
1. Mc Gregor WG.— DNA repair, DNA replication, andUV mutagenesis. J Invest Dermatol Symp Proc, 1999, 4,1-5.
2. Cleaver JE.— Common pathways for ultraviolet skincarcinogenesis in the repair and replication defectivegroups of xeroderma pigmentosum. J Dermatol Sci,2000, 23, 1-11.
3. Fazaa B, Piérard-Franchimont C, Zghal M, et al.—-Nuclear morphometry in xeroderma pigmentosum-asso-ciated malignant melanomas. Am J Dermatopathol,1994, 16, 611-614.
4. Itin PH, Sarasin A, Pittelkow MR.— Trichothiodystro-phy : update on the sulfur-deficient brittle hair syn-dromes. J Am Acad Dermatol, 2001, 44, 891-920.
5. Galadari E, Hadi S, Sabarinathan K.— Hartnup’sdisease. Int J Dermatol, 1993, 32, 904.
J.E. ARRESE ET COLL.
Rev Med Liege 2005; 60 : Supp. I : 66-7070
Les demandes de t i rés à par t sont à adresser au Dr. J.E. Arrese, Service de Dermatopathologie, CHUdu Sart Tilman, 4000 Liège. E-mail : [email protected]. 3 : Infiltration cutanée de la photoporphyrie érythropoïétique









![Des sites du Limousin aux autres sites Rapport GEP · dossier [Recom1]. Il s’agira en particulier, pour donner l’impulsion nécessaire, de préciser dès que possible le processus](https://img.pdfslide.fr/doc/110x75/60106ffcca1e35456f22c7d2/des-sites-du-limousin-aux-autres-sites-rapport-gep-dossier-recom1-il-saagira.jpg)



















