Embed Size (px)
Citation preview
Biologie Moleculaire-2016 1 http://mysite.science.uottawa.ca/jbasso/moleculaire/accueil.htm

Biologie Moleculaire-2016 2
DIRECTIVES GÉNÉRALES
1. La présence dans le laboratoire est obligatoire. S'il vous plaît, être à l'heure.
2. Des chaussures et vêtements appropriés doivent être portés en tout temps.
3. Laisser vos vêtements d'extérieur, sacs à dos, et toutes autres matières étrangères dans les
casiers à l'extérieur du laboratoire. Il est fortement recommandé que vous ayez un cadenas.
Nous ne somme pas responsable pour les objets perdus ou volés.
4. Porter une blouse de laboratoire et des gants en tout temps quand vous travaillez dans le
labo.
5. Enlever vos gants chaque fois que vous sortez du labo.
6. Enlever vos gants quand vous utilisez notre ou votre ordinateur.
7. Toujours placer les pipettes utilisées, les embouts, tubes de microcentrifuge et autres
matériaux dans les sacs biohazard fournis afin qu'ils puissent être éliminés de façon
appropriée. Ne pas jeter de déchets dans les sacs d'autoclaves.
8. Ne jamais se lécher les doigts, ou mettre vos doigts dans votre bouche.
9. Ne pas manger ou boire dans le laboratoire.
10. Pas de radios, lecteurs MP3, de lecteurs de CD dans le laboratoire.
11. Aucune utilisation des téléphones cellulaires ou des textos en laboratoire.
12. Aviser l'A.E. ou un instructeur de tout accident, même mineur.
13. Aviser l'A.E. ou un instructeur de tout bris ou malfonctionnement de l’équipement fourni.
Matériel que vous DEVEZ avoir pour travailler dans le laboratoire de microbiologie :
Une blouse de laboratoire
Un crayon-feutre à pointe fine permanent, de préférence noir, pour l'étiquetage.
Un cahier de notes pour enregistrer vos résultats. N'importe quel type est acceptable. Ne gaspillez
pas votre argent
Une clef A USB pour sauvegarder vos images
Une calculatrice. L’utilisation des calculatrices sur les cellulaires n’est pas permise.
Facultatif, mais fortement recommandé :
Aviser l'instructeur de tout problème médical de sorte que des accommodations appropriées
peuvent être prises. Par exemple, les allergies, le diabète, l'hypoglycémie, l'épilepsie, les blessures
apparentes, daltonisme, etc.
Aviser l'instructeur de tout besoin particulier dont vous pourriez avoir besoin de sorte que les
accommodations appropriées peuvent être prises. Par exemple, si vous écrivez vos examens avec
SASS.

Biologie Moleculaire-2016 3
Horaire Exercice 1 13 janv. Concentrations et Dilutions
Digestions par enzymes de restriction et électrophorèse sur gel d’agarose
Exercice 2 20 janv. Projet I: Vérifier la carte de restriction d’une insertion d’ADN
Projet II: Mutagénèse dirigée de GFP
Isolation d’ADN plasmidique
PCR
Exercice 3 27 janv. Projet I: Vérifier la carte de restriction d’une insertion d’ADN
Projet II : Mutagénèse dirigée de GFP
Électrophorèse des amplicons de PCR de GFP
Digestion des amplicons de PCR de GFP et de pGFPuv
Ligature des amplicons de GFP
Exercice 4 3 févr. Projet I: Vérifier la carte de restriction d’une insertion d’ADN (XhoI) Projet II : Mutagénèse dirigée de GFP
Transformation des mélanges de ligatures
Projet III: Empreintes génomiques
Isolation d’ADN génomique humain des cellules de joues
Amplification par PCR du VNTR ApoC2
Amplification par PCR du RFLP ApoB
Exercice 5 10 févr. Projet II : Mutagénèse dirigée de GFP
Criblage par PCR des transformants
Analyse des produits du PCR de colonies
Repiquage
Projet III: Empreintes génomiques
Projet IV: Contrôle transcriptionnel de GFP par le promoteur LacZ
Isolation d’ARN d’E.coli
SEMAINE D’ÉTUDE 14-20 févr.
MI-SESSION 24 févr.

Biologie Moleculaire-2016 4
Exercice 6 2 mars Projet IV: Contrôle transcriptionnel de GFP par le promoteur LacZ
Électrophorèse sur gel d’ARN
Transfert northern
Exercice 7 9 mars Projet IV: Contrôle transcriptionnel de GFP par le promoteur LacZ
Hybridation northern
Projet V: RT-PCR du la protéine ribosomale URP1 de la levure
Isolation d’ARN de la levure
RT-PCR
Exercice 8 16 mars Projet II : Mutagénèse dirigée de GFP
Évaluation de l’activité de GFP
Projet IV: Contrôle transcriptionnel de GFP par le promoteur LacZ
Immunodétection des northerns
Projet V: RT-PCR du la protéine ribosomale URP1 de la levure
Analyse des réactions de RT-PCR
EXAMEN FINAL PRATIQUE 30 mars
EXAMEN FINAL THÉORIQUE 6 avr.
Biologie Moleculaire-2016 5
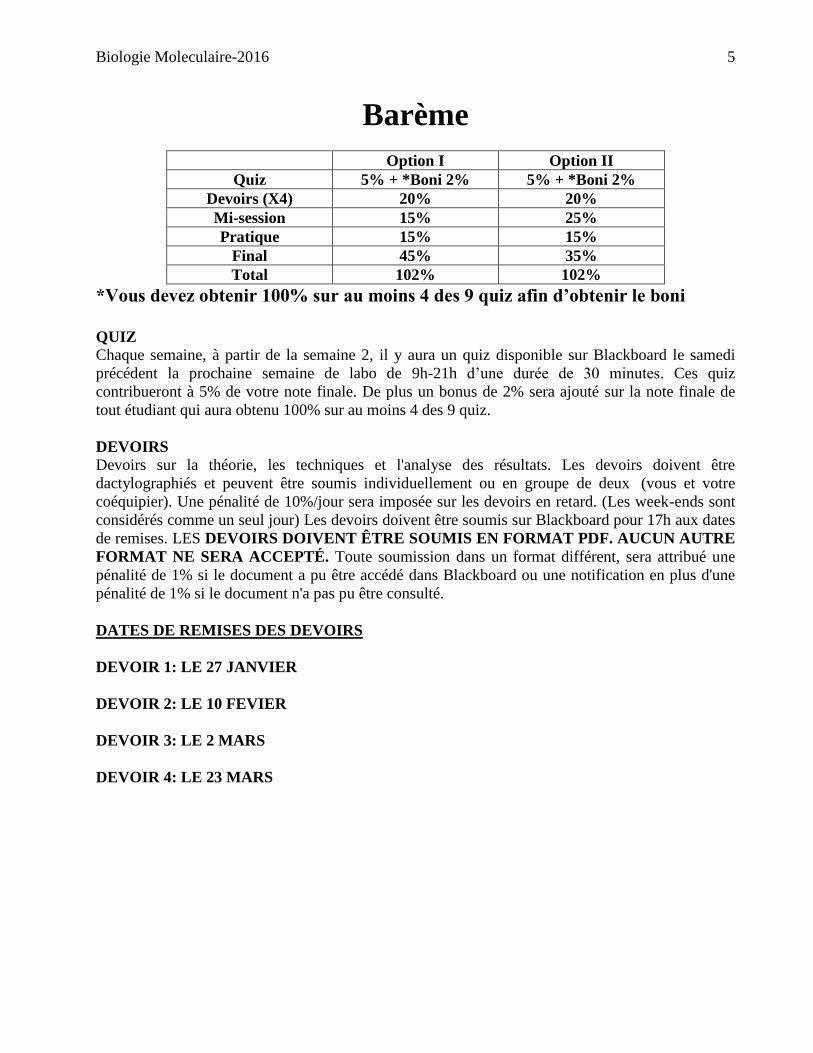
Barème
Option I Option II
Quiz 5% + *Boni 2% 5% + *Boni 2%
Devoirs (X4) 20% 20%
Mi-session 15% 25%
Pratique 15% 15%
Final 45% 35%
Total 102% 102%
*Vous devez obtenir 100% sur au moins 4 des 9 quiz afin d’obtenir le boni
QUIZ
Chaque semaine, à partir de la semaine 2, il y aura un quiz disponible sur Blackboard le samedi
précédent la prochaine semaine de labo de 9h-21h d’une durée de 30 minutes. Ces quiz
contribueront à 5% de votre note finale. De plus un bonus de 2% sera ajouté sur la note finale de
tout étudiant qui aura obtenu 100% sur au moins 4 des 9 quiz.
DEVOIRS
Devoirs sur la théorie, les techniques et l'analyse des résultats. Les devoirs doivent être
dactylographiés et peuvent être soumis individuellement ou en groupe de deux (vous et votre
coéquipier). Une pénalité de 10%/jour sera imposée sur les devoirs en retard. (Les week-ends sont
considérés comme un seul jour) Les devoirs doivent être soumis sur Blackboard pour 17h aux dates
de remises. LES DEVOIRS DOIVENT ÊTRE SOUMIS EN FORMAT PDF. AUCUN AUTRE
FORMAT NE SERA ACCEPTÉ. Toute soumission dans un format différent, sera attribué une
pénalité de 1% si le document a pu être accédé dans Blackboard ou une notification en plus d'une
pénalité de 1% si le document n'a pas pu être consulté.
DATES DE REMISES DES DEVOIRS
DEVOIR 1: LE 27 JANVIER
DEVOIR 2: LE 10 FEVIER
DEVOIR 3: LE 2 MARS
DEVOIR 4: LE 23 MARS

Biologie Moleculaire-2016 6
Examens écrits
Tous les examens sont à livre ouvert. L'accès à l'internet est permis pour l'examen de mi-
session et l'examen final.
Le format de l'examen de mi-session (2.5 heures) sera comme suit:
8 problèmes de calculs (16 points)
1 problème de calcul incluant une composante pratique. (4 points)
5 exercices de bioinfo (5 points)
5 questions théoriques sur la bioinfo et les procédures moléculaires (5 points)
2 sur 3 problèmes avec une emphase sur l'analyse de données et la conception expérimentale
(10 points)
L'examen final sera récapitulatif et le format (3 heures) sera comme suit:
5 problèmes de calculs (10 points)
10 exercices de bioinfo (10 points)
5 questions théoriques sur la bioinfo et les procédures moléculaires (5 points)
3 sur 4 problèmes sur l'analyse de données et la conception expérimentale (15 points)
Examen pratique
Comme pour tous les examens, cet examen est à livre ouvert et sera donné sur une période de 2
heures. Les étudiants devront se présenter au labo, SUR UNE BASE INDIVIDUELLE, pour
exécuter des tâches qu'ils ont faites de façon routinière durant la session. Celles-ci pourraient
inclure la préparation de réactions de PCR, la préparation de solutions, faire des digestions par
enzymes de restrictions, l'isolation d'ADN, migration de gels d'agarose, etc.. L'évaluation sera
fondée sur les résultats obtenus et non pas sur l'approche ou la performance durant la période
d'examen.

Biologie Moleculaire-2016 7
Exercice 1
Qu'est-ce qu’on fait aujourd’hui !
Concentrations et dilutions
Digestions par enzymes de restriction et électrophorèse sur gel
d’agarose

Biologie Moleculaire-2016 8
Introduction aux concentrations Une propriété très importante au sujet des solutions qui doit être adressée c’est la concentration.
En général, la concentration indique la quantité d’un soluté dans une quantité donnée d’une
solution. Pour travailler avec des concentrations vous devez tenir à l’esprit les distinctions entre le
soluté, le solvant et la solution.
Puisque différentes quantités de solutés peuvent être dissoutes dans une solution, la concentration
est une propriété variable. Nous avons donc besoin d’une façon numérique pour indiquer comment
concentrée est une solution. Une variété de différentes façons ont été développées pour calculer et
exprimer les concentrations des solutions.
Ceci peut être fait par pourcentage en utilisant des mesures du poids (masse) ou le volume ou les
deux. Alternativement, on peut aussi utiliser des mesures qui sont propres à la façon que les
composés chimiques réagissent entre eux (moles).
Dans les pages qui suivent, plusieurs types de concentrations seront présentés. Elles incluent le
pourcentage par volume, le pourcentage par poids, le pourcentage du poids/volume, la
molarité (l’utilisation principale des concentrations chimiques) et poids/volume.
Vous obtiendrez de l’expérience avec plusieurs des façons d’établir la concentration d’une solution.
Vous pouvez préparer une solution à partir des matières brutes et mesurer chacune des composantes
qui doivent être présentes dans la solution. Vous pouvez préparer une solution en diluant une
solution préexistante. Si une solution est colorée, vous pouvez déterminer sa concentration en
mesurant l’intensité de la couleur par colorimétrie.
POURCENTAGE
L’utilisation des pourcentages est une façon commune pour exprimer la concentration d’une
solution. C’est une approche simple qui indique la quantité d’un composé par 100. Les
pourcentages peuvent être calculés en utilisant les volumes ainsi que les poids, ou les deux. Une
façon, que vous connaissez, sans doute, d’exprimer les concentrations est le pourcentage par
volume. Une autre façon est le pourcentage par poids. Enfin, une autre façon est un hybride
appelé le pourcentage par poids/volume.
Le pourcentage par volume est habituellement utilisé quand la solution est préparée en
mélangeant deux liquides.
Par exemple, l’alcool à friction est habituellement
70% par volume d’alcool isopropyl. Ceci veut dire
que 100 mL de solution contient 70 mL d’alcool
isopropyl. Ceci veut aussi dire qu’un litre (ou 1000
mL) de cette solution contient 700 mL d’alcool
isopropyl et assez d’eau pour compléter le volume
à un total de 1 litre ou 1000 mL.
Pourcentage par volume = volume du soluté
volume de solution x 100

Biologie Moleculaire-2016 9
Le pourcentage par poids est une façon d’exprimer la concentration d’une solution comme le
poids du soluté/poids de la solution.
Pourcentage par poids = Poids du soluté
Poids de la solution x 100
À titre d’exemple, considérons une solution
de 12% NaCl par poids. Une telle solution
aurait 12 grammes de NaCl pour chaque 100
grammes de solution. Pour préparer une telle
solution, vous pourriez peser 12 grammes de
NaCl, et ensuite ajouter 88 grammes d’eau,
afin que la masse totale de la solution soit de
100 grammes. Puisque les masses sont
conservées, les masses des composantes de
la solution s’additionneront à la masse totale
de la solution.
12 % NaCl solution = 12 g NaCl
100 g solution
12 g NaCl
(12 g NaCl + 88 g eau) = 12% NaCl solution
Afin de calculer le pourcentage par poids ou le pourcentage par masse d’une solution, vous devez
diviser la masse du soluté par la masse de la solution (la somme combinée de la masse du soluté et
du solvant) et ensuite multipliée par 100 pour la changer en pourcentage.
Pourcentage par poids/volume
Une autre variante des concentrations par pourcentage est le pourcentage poids/volume ou le
pourcentage masse/volume. Cette variante mesure la quantité de soluté en grammes, mais mesure la
quantité de la solution en millilitres. Un exemple serait une solution de 5% NaCl (m/v). Elle
contient 5 g de NaCl pour chaque 100 mL de solution.
Pourcentage par volume = Poids du soluté (en g)
Volume de solution (en mL) x 100
Cette façon est la manière la plus couramment utilisée dans ce cours pour exprimer les solutions en
pourcentages.

Biologie Moleculaire-2016 10
MOLARITÉ
Une autre façon d’exprimer une concentration
s’appelle la molarité. La molarité c’est le nombre
de moles de soluté dissout dans un litre de
solution. Les unités sont donc moles par litre,
plus précisément, c’est les moles de soluté par
litre de solution.
Molarité = moles de soluté
litre de solution
Plutôt d’écrire moles par litre, l’abréviation “M” est utilisée pour ces unités. Alors, quand vous
voyez la lettre M cela signifie molarité et représente moles par litre (pas seulement moles). Vous
devez faire très attention de distinguer entre moles et molarité. "Moles" est une mesure de la
quantité que vous avez d’une composante; "molarité" mesure la concentration de la composante.
Alors quand un problème vous est donné, qui stipule que la concentration d’une solution est 0.1 M,
ceci veut dire qu’elle possède 0.1 mole pour chaque litre de solution; cela ne veut pas dire que c’est
0.1 mole.
POIDS/VOLUME
Cette façon d’exprimer une concentration est très semblable à celle des pourcentages et représente
une des façons les plus populaires utilisées par les biologistes moléculaires. Contrairement au
pourcentage, la concentration est exprimée par quelconque volume l’utilisateur désire utiliser. Plus
communément, ces concentrations sont exprimées par une unité de mesure. Par exemple, par 1 mL
ou 1 µL ou 1L, etc. Essentiellement, ces expressions représentent la masse d’un soluté dans une
quantité donnée de la solution. Par exemple, une solution qui est à une concentration de 1mg/mL
contient 1mg de soluté dans 1 mL de solution.
LES RAPPORTS
Toutes les façons décrites ci-dessus pour exprimer les concentrations se font en fonction du volume
de la solution qui est la somme du volume du soluté et du solvant. Une manière courante que
plusieurs microbiologistes et chimistes utilisent pour d’exprimer les concentrations est les rapports.
Dans ce cas-ci, la relation entre le soluté et le solvant est exprimée indépendamment de la solution.
Par exemple, nous pourrions dire que le rapport entre un soluté est un solvant est de 2 pour 1 avec la
notation 2 :1. Ceci indique que pour deux parties du soluté il y a une partie du solvant. Donc, trois
parties total de solution!

Biologie Moleculaire-2016 11
Les dilutions et l’utilisation des micropipetteurs
Les dilutions : Savoir préparer des dilutions est essentiel pour la préparation de plusieurs réactifs, mélanges
réactionnels, et solutions utilisées dans les laboratoires de biologie moléculaire. Essentiellement, les
dilutions servent à réduire la concentration initiale du composé afin d’atteindre une nouvelle
concentration voulue. Les solutions préparées par l’entremise de dilutions peuvent être composées
d’un ou de plusieurs composés. Le nombre d’ingrédients dans la solution à être préparée
n’influence aucunement la formulation des dilutions. Un bref survol de la préparation des dilutions
est présenté ci-dessous. Assurez-vous de bien comprendre leurs préparations, car vous serez appelé
à les préparer tout au cours de la session ainsi que pour l’examen final.
La compréhension de la formulation des dilutions comporte trois concepts. La concentration, le
facteur de dilution, et la dilution.
Une concentration se définit comme la quantité d'un composé pour un volume total de solution.
Puisqu'une quantité peut être exprimée de plusieurs façons, les concentrations sont exprimées
comme l'unité de mesure de la quantité du composé/volume total et final.
E.x. Gram/Litre
Molécules/Litre
Moles/Litre
Etc.
Le facteur de dilution représente la multiple arithmétique par laquelle une concentration initiale
doit être divisée afin d'obtenir la concentration finale désirée. Par exemple, si une solution contient
30 grammes de caféine par 1L de solution et que vous désirez réduire la concentration de caféine à
0.3 Grammes/Litre vous devrez diviser la concentration initiale par 100, ce qui représente le facteur
de dilution. Vous pouvez utiliser la formule suivante afin de déterminer un facteur de dilution.
Le facteur de dilution = Concentration initiale ou Ce que j’ai
Concentration finale Ce que je désire
La dilution représente la fraction du composé étant examiné. Par exemple, dans le problème
précédent, une dilution de 1/100 a été effectuée. La dilution est exprimée comme une fraction de 1
sur le facteur de dilution. C.a.d. que la solution initiale est représentée comme une fraction de
l’original sur le total. Par exemple si vous avez déterminé qu’une dilution de 1/100 doit être
préparée, cela veut dire qu’un centième de la nouvelle solution doit être représenté par la solution
originale. Donc pour un volume total de disons 2mL, 0.02 mL doit être représenté par la solution
originale.

Biologie Moleculaire-2016 12
Préparer une solution qui requière la dilution de plus d’un ingrédient :
Le principe pour la formulation d’une solution qui nécessite la dilution de plus d’un composé est la
même que pour une solution avec seulement qu’une composante. La seule différence est le volume
de solvant qui doit être ajouté.
Disons par exemple que l’on désire préparer 10mL d’une solution de 0.1M à partir d’une solution
mère de 2M.
Pour calculer le facteur de dilution : Ce que j’ai = 2M = 20
Ce que je désire 0.1M
Donc la dilution requise est de 1/20 ce qui veut dire qu’un vingtième du total doit être représenté
par la solution originale.
Donc pour préparer 10mL on doit ajouter 0.5mL de la solution mère et compléter avec 9.5mL de
solvant.
Si la solution à préparer inclut une deuxième composante; disons que celle-ci doit être à une
concentration finale de 0.5 M et que la solution mère est de 3M.
Encore une fois pour calculer le facteur de dilution : Ce que j’ai = 3M = 6
Ce que je désire 0.5M
Donc la dilution requise est de 1/6. Ce qui veut dire qu’un sixième du total doit être représenté par
la solution originale.
Donc pour préparer 10mL on doit ajouter 0.5mL de la première solution, 1.7mL de la deuxième
solution et puis complété avec 7.8mL de solvant.
Si les explications ci-haut ne sont pas suffisantes, vous pouvez consulter le site web suivant :
http://www.wellesley.edu/Biology/Concepts/HtmL/dilutions.html

Biologie Moleculaire-2016
13
Utilisation des micropipetteurs
Les micropipettes sont des outils indispensables dans les laboratoires de biologie moléculaire
modernes. Quelle est l’exactitude des vôtres? Quelles sont leurs précisions? Quelle est la
différence entre l’exactitude et la précision?
Dans le cas de l’exactitude, il s’agit de la performance comparativement à une valeur de référence.
Tandis que la précision est une indication, de la fiabilité ou la répétitivité est ne dépend pas
nécessairement d’une référence. Ces deux attributs sont indépendants l’un de l’autre. Il est possible
d’avoir un instrument qui est précisément inexact (ou exactement imprécis)!
Analogie d’une cible pour illustrer l’exactitude et la précision
Dans cet exemple, le centre de la cible représente la référence à laquelle l’exactitude est évaluée.
Afin d’évaluer de façon simultanée l’exactitude et la précision de vos pipettes vous devez faire des
mesures multiples afin de calculer le % d’erreur de la moyenne et l’écart type des mesures pour
chacune de pipettes.
Directives pour l’utilisation des micropipetteurs Gilson
Vous avez des micropipettes de trios grandeurs. La P-1000, la P-200 et la P-20. Le numéro du
modèle indique le volume maximal en microlitres. Initialement, vous devrez déterminer quelle
pipette utiliser sous des circonstances données. Par exemple, si vous devez transférer 0.18 mL vous
aurez probablement besoin de la P-200 puisque 0.18 mL = 180 µL car la P-200 fera la mesure de ce
volume avec une exactitude et une précision plus grande que ne le ferait la P-1000.
Règles:
Toujours utiliser un embout jetable. La P-20 et la P-200 utilisent les plus petits embouts et
la P-1000 utilise les plus gros.
Ne jamais aspirer du liquide dans le baril blanc de la pipette.
Ne jamais déposer la pipette quand il y a du fluide dans l’embout. Le fluide pourrait
accidentellement se retrouver dans le baril.
Ne jamais tourner le réglage de volume au-dessus ou en dessous de l’étendue prévue.
Afin de maximiser la précision, toujours utiliser la pipette de plus petit volume qui
correspond au volume total donné.
1. Régler le volume désiré:
Ajustez le volume à un réglage légèrement plus élevé que celui désiré, et ensuite
retournez-le à celui désiré.
2. Placer un embout:
Pousser l’embout fermement avec une motion de torsion. L’embout doit créer un
joint étanche avec le baril de la pipette.
Exact et précis Précis & inexact Imprécis & inexact

Biologie Moleculaire-2016
14
3. Peser le piston jusqu’au premier arrêt.
4. Introduire l’embout dans le liquide que vous désirez transférer. Pas trop loin, juste un peu
sous la surface.
5. Lentement, relâchez le piston.
6. Tout en retirant l’embout, toucher le côté de la paroi du tube afin de retirer l’excédent de
fluide de l’extérieur.
7. Afin de livrer, peser sur le piston jusqu’au premier arrêt, puis jusqu’au fond.
Ne jamais livrer de très petits volumes dans le vide. Toujours livrer dans un liquide
ou sur la paroi du tube, de telle façon à ce que la force d’adhésion va retirer le
liquide livré de l’embout.
8. Avec le piston, toujours complètement au fond, retirer l’embout du liquide.
RAPPELEZ-VOUS QUE VOS MICROPIPETTEURS SONT DES INSTRUMENTS
DISPENDIEUX!
Exercice de dilutions avec les micropipetteurs (CET EXERCICE DOIT ÊTRE FAIT PAR CHAQUE PERSONNE)
Nous avons inclus cet exercice afin que chacun se familiarise avec la préparation des dilutions,
l’utilisation du micropipetteur et des embouts. (TOUJOURS ÉTIQUETER VOS TUBES!!)
Matériaux: Solution I (1% Composé “A” (m/v); P.M. 800g/mole)
Solution II (1.2M Composé “B” P.M. 60g/mole; Densité: 1.6g/mL)
Méthode:
1. Préparer 1mL de chacune des solutions suivantes des solutions mères ci-dessus.
a. Une solution de 1.5mM du composé “A”.
b. Une solution de 0.36% (m/v) du composé “B”.
c. Une solution de 6% (v/v) de la solution I.
d. Une solution qui contient 0.5mg du composé “A” et 0.1% (v/v) du composé “B”.
e. Une solution qui représente le rapport suivant : solution I : solution II : eau : 2 :1 :2
2. Transférer 100 µL de chacune des solutions aux puits appropriés d’une plaque de 96 puits tel
qu’indiqué ci-dessous:
Plan de la plaque à 96 puits (une plaque/table)
Soln. a Soln. b Soln. c Soln. d Soln. e Personne 1 Groupe 1
Soln. a Soln. b Soln. c Soln. d Soln. e Personne 2
Soln. a Soln. b Soln. c Soln. d Soln. e Personne 1 Groupe 2
Soln. a Soln. b Soln. c Soln. d Soln. e Personne 2
Soln. a Soln. b Soln. c Soln. d Soln. e Personne 1 Groupe 3
Soln. a Soln. b Soln. c Soln. d Soln. e Personne 2
Soln. a Soln. b Soln. c Soln. d Soln. e Personne 1 Groupe 4
Soln. a Soln. b Soln. c Soln. d Soln. e Personne 2

Biologie Moleculaire-2016
15
Encore des dilutions: déterminer la concentration d'ADN (Groupes de 2) Comme avec la plupart des composés, la concentration d'acides nucléiques peut être déterminée par
spectrophotométrie. Pour ce faire, l'absorbance du composé est déterminée à la longueur d'onde
d'absorption maximale. Dans le cas des acides nucléiques, c'est dans la gamme des UV à une
longueur d'onde de 260 nm. Dans l'exercice suivant, vous allez préparer des échantillons à
différentes concentrations connues d'ADN et ensuite utiliser les valeurs d'absorbance obtenues pour
générer une courbe étalon représentant l'absorbance Vs concentration d'ADN (PAS QUANTITÉ).
La courbe étalon vous permettra alors de déterminer la concentration d'un échantillon d'ADN
inconnu ainsi que la détermination de la relation entre la concentration de l'ADN et l'absorbance à
une longueur d'onde de 260 nm. Plus précisément, vous souhaitez déterminer quelle concentration
de l'ADN (en µg/mL ou ng/µl) est égale à une absorbance de 1.0.
Matériaux
ADN de sperme de saumon (200µL à 0.5mg/mL)
ADN de sperme de saumon (200µL de concentration inconnue)
Méthode:
1. Préparer 500µL dans de l'eau de chacune des solutions standards d'ADN suivantes à partir de
l'échantillon d'ADN de concentration connue: 0.0, 0.05, 0.025, 0.01, 0.005 and 0.0025mg/mL.
2. Préparer des échantillons de 500µL dans l'eau représentant des dilutions de 1/4 et 1/10 de
l'échantillon d'ADN inconnu.
3. Transférer 200µL de chacun des solutions d'ADN standards dans la plaque tel qu'indiqué dans
le plan ci-dessous.
4. Transférer 200µL de chacune des solutions d'ADN inconnue dans la plaque tel qu'indiqué dans
le plan ci-dessous.
5. Mesurer l'absorbance à 260nm.
Plan de la plaque d'essai d'ADN
0.0 0.05 0.025 0.01 0.005 0.0025 Vide INC
1/4
INC
1/10 Groupe 1
0.0 0.05 0.025 0.01 0.005 0.0025 Vide INC
1/4
INC
1/10 Groupe 2
0.0 0.05 0.025 0.01 0.005 0.0025 Vide INC
1/4
INC
1/10 Groupe 3
0.0 0.05 0.025 0.01 0.005 0.0025 Vide INC
1/4
INC
1/10 Groupe 4
0.0 0.05 0.025 0.01 0.005 0.0025 Vide INC
1/4
INC
1/10 Groupe 5
0.0 0.05 0.025 0.01 0.005 0.0025 Vide INC
1/4
INC
1/10 Groupe 6
0.0 0.05 0.025 0.01 0.005 0.0025 Vide INC
1/4
INC
1/10 Groupe 7
0.0 0.05 0.025 0.01 0.005 0.0025 Vide INC
1/4
INC
1/10 Groupe 8

Biologie Moleculaire-2016
16
Digestions par enzymes de restrictions & électrophorèse sur gel d’agarose
(Groupes de 2) Au milieu des années 1970, le domaine de la biologie moléculaire a connu une croissance
importante grâce à l’utilisation des endonucléases de restriction pour le clivage d’ADN à des sites
spécifiques. Il existe maintenant plusieurs centaines d’enzymes de restriction différentes disponibles
commercialement. Vous pouvez trouver de l’information détaillée sur plusieurs d’entre elles
(incluant celles que vous utiliserez dans ce cours) dans des catalogues de produits commerciaux.
Leurs spécificités les rendent particulièrement utile pour plusieurs tâches incluant entre autres la
cartographie de l’ADN, le clonage et le sous clonage de l’ADN, etc. Le but de ce premier
exercice est de faire des digestions simples de votre ADN afin de répondre aux questions
suivantes :
Quelles enzymes coupent dans l’insertion?
Quelles enzymes ne coupent pas dans l’insertion?
Quelle est la taille de l’insertion?
Quelles sont les positions possibles des différents sites de restrictions?
Le fragment d’ADN a été inséré dans quel site de restriction du SCM?
Pour de plus amples informations au sujet des enzymes de restrictions consultez le site Web
suivant : NOTEZ : VOUS ÊTES RESPONSABLE POUR CETTE INFORMATION!!
http://askabiologist.asu.edu/expstuff/mamajis/restriction/restriction.html
Électrophorèse sur gel d’agarose L’électrophorèse sur gel d’agarose implique la séparation de molécules d’ADN d’après leurs tailles
et leurs conformations. La migration électrophorétique d’un fragment d’ADN dans l’agarose est
inversement proportionnelle au logarithme de son poids moléculaire (ceci est vrai seulement pour
certaines gammes de taille sous des conditions définies). [Conseil! Quand vous estimez la taille de
fragments de restrictions, gardez à l’esprit la précision requise, spécifiquement le nombre de
chiffres significatifs! La concentration de l’agarose utilisée dépend de l’éventail des tailles étant
étudiées; pour la séparation d’ADN linéaires entre 0.5 kb et 10 kb, un gel d’agarose de 1% est
habituellement utilisé. Suite à l’électrophorèse, les gels sont visualisés sous des ultras violets et
photographiés avec une caméra numérique. L’intensité de la fluorescence est proportionnelle à la
quantité (et la longueur) de l’ADN linéaire. Cette méthode peut être utilisée pour obtenir une
estimation de la quantité d’ADN dans les échantillons.
L’électrophorèse sur gel d’agarose utilise un courant à haute tension ce qui peut présenter un
danger. Le boîtier qui contient le gel est muni d’un mécanisme de connexion spécial pour des
raisons de sécurité. Les connexions doivent donc être retirées avant de pouvoir ouvrir le couvercle.
COUPEZ TOUJOURS L’ALIMENTATION AVANT DE DÉBRANCHER L’APPAREIL.
Le bromure d’éthidium, qui est utilisé comme colorant des gels d’agarose afin de pouvoir observer
l’ADN sous lumière UV, est un agent cancérigène potentiel. Toujours porter des gants pour
manipuler tout ce qui en contient. La source de lumière UV est aussi très dangereuse pour la peau et
surtout pour vos yeux, assurez-vous donc de bien vous protéger (gants, sarrau, masques) lorsque
vous observez vos gels d’agarose.

Biologie Moleculaire-2016
17
Chaque appareil a deux supports de gels et trois peignes (8-puits, 10 puits et 14-puits contenants
environ 14μL, 12 μL et 6μL respectivement). Rappelez-vous que les gels à 8 et 10 puits contiennent
à peu près deux fois plus d’ADN que les gels à 14 puits à cause de la taille de leurs puits. De plus, si
vous désirez visualiser des petits fragments (<500bp), qui sont moins intense, vous devriez
considérer utiliser plus d'ADN et peut-être une plus forte concentration d’agarose- ex. 1.5%, au lieu
du 1% standard) afin d’obtenir une meilleure résolution.
Méthode:
A. Préparation du gel d’agarose (peigne à 8 puits)
Matériaux :
Agarose
10X TBE
1. Préparer dans votre cylindre gradué 200mL de tampon TBE à une concentration finale de 1X.
Assurez-vous de bien mélanger.
2. Mélanger la quantité appropriée d’agarose pour obtenir une concentration finale de 1.0% m/v
dans 25mL de tampon TBE de 1X.
3. Pour dissoudre l’agarose, chauffé aux micro-ondes (25-45 secs.). Recouvrir le flacon d’une
pellicule de plastique afin de minimiser l’évaporation). Lorsque l’agarose sera complètement
dissout, laissez refroidir jusqu’à 50-60oC (à peu près 5 minutes).
4. Ajoutez 50 L d’une solution mère de bromure d’éthidium à 1mg/mL (ATTENTION!
CANCÉRIGÈNE!) et bien mélanger.
5. Versez l’agarose dans le support à gel. Après avoir versé le gel, placer le peigne de 8 puits
(volume qui peut être accommodé est approx. 14µL) et enlever toutes les bulles d’airs (avec un
embout jaune de micropipetteur). Refermez le couvercle de l’appareil et laissez solidifier le gel
pour au moins 15-20 minutes.
6. Une fois le gel solidifié, retirez les joints noirs puis versez une quantité suffisante du tampon
TBE 1X dans l’appareil pour recouvrir le gel d’environ 0.5cm.
7. Enlevez lentement le peigne.

Biologie Moleculaire-2016
18
B. Conseils pour vérifier l’opération adéquate de votre gel
8. Branchez les électrodes de façon à ce que la cathode (électrode négative) soit à l’extrémité qui
représente l’origine du gel. Les échantillons vont migrer au travers du gel vers l’anode
(électrode positive). (ATTENTION! HAUTE TENSION!).
9. Régler l’appareil pour ~ 100V. Allumez l’appareil. Vérifier que votre intensité est dans les
environs de 40-55 mA. Si cela n’est pas le cas, il y a un problème.
Problèmes possibles :
Le tampon de migration est à la mauvaise concentration
Le tampon dans le gel est à la mauvaise concentration
Vous avez oublié de mettre le tampon dans le gel
C. Analyse des digestions par enzymes de restrictions
10. Charger les échantillons d’ADN suivants. L’inconnu est un recombinant avec le vecteur pUC9:
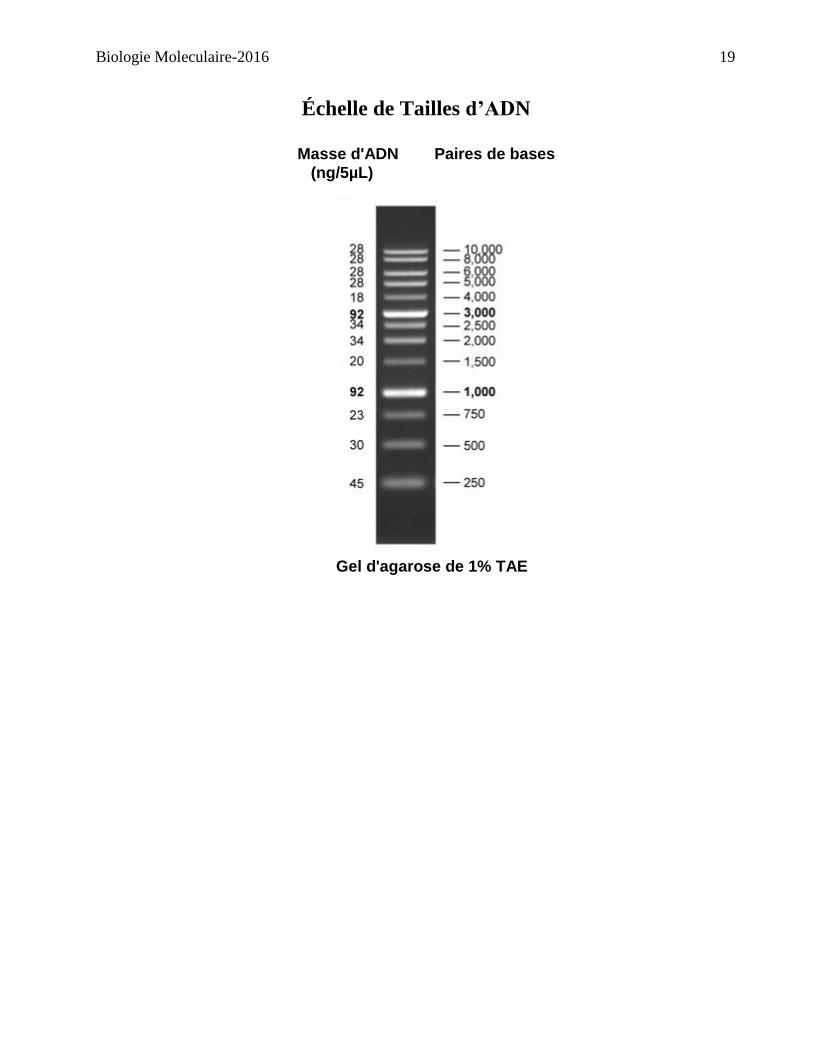
a. Échelle de poids moléculaire de 1Kpb (5µL)
b. Plasmide recombinant pUC9 non digéré 5µL
c. Plasmide recombinant pUC9 digéré avec BamHI, 5µL
d. Plasmide recombinant pUC9 digéré avec EcoRI, 5µL
e. Plasmide recombinant pUC9 digéré avec HindIII, 5µL
f. Plasmide recombinant pUC9 digéré avec EcoRI + HindIII, 5µL
g. Plasmide recombinant pUC9 digéré avec PstI, 5µL
h. Plasmide pUC9 digéré avec BamHI, 5µL
11. Faire la migration à 100V pour approx. 45 minutes, puis demander à un aide enseignant de faire
la prise d’une photo.
Biologie Moleculaire-2016
19
Échelle de Tailles d’ADN
Masse d'ADN Paires de bases
(ng/5µL)
Gel d'agarose de 1% TAE
Biologie Moleculaire-2016
20

Biologie Moleculaire-2016
21
Exercice 2
Qu'est-ce qu’on fait aujourd’hui !
Projet I: Vérifier la carte de restriction d’une insertion d’ADN
Projet II: Mutagénèse dirigée de GFP
Isolation d’ADN plasmidique
PCR

Biologie Moleculaire-2016
22
Les enzymes de restriction et l’électrophorèse sur gel d'agarose Suite à la plupart des digestions, il est nécessaire de répondre aux questions suivantes :
L’ADN a-t-il été digéré?
Combien de fois est-ce que l’enzyme a coupé?
Quelles sont les tailles des fragments générés?
Est-ce que la digestion est complète ou partielle?
Les échantillons d’ADN sont-ils complètement digérés? S’il y a seulement eu clivage partiel de
l’ADN (exemple de cause possible : réduction de l’activité enzymatique à cause de conditions de
réactions non appropriées; impuretés dans l’ADN qui aurait inhibé l’enzyme), les fragments à
migration plus lente (qui représentent les fragments non clivés contenant un site pour l’enzyme
utilisée) sont habituellement présents à des proportions inférieures aux prédictions
stœchiométriques. Autrement dit, puisque l’intensité de la fluorescence UV est proportionnelle à la
quantité de bromure d’éthidium liée (et donc la quantité/longueur de l’ADN), ces fragments sont
moins intenses qu’ils le seraient s’ils étaient présents en quantité équimolaire aux fragments
complètement digérés.
Est-ce que les tailles calculées des fragments de restriction sont conformes? Si vous effectuez la
migration de différentes digestions d’ADN cloné, il est important de vérifier que la somme des
tailles des fragments calculées est à peu près égale pour chacune des voies de migration (c. a. d. la
taille de la molécule d’ADN intacte). Puisque l’estimation de la taille à l’aide des marqueurs de
taille n’est pas exacte (et que l’erreur associée aux fragments se trouvant dans la partie non linéaire
de la courbe étalon est plus grande), il n’y a habituellement qu’un accord approximatif (une
différence d’environ 200-300 pb pour un plasmide recombinant de 5 Kpb). Si vous obtenez de plus
grandes différences, considérez les points suivants: Y aurait-il co-migration de plusieurs fragments?
(Observez l’intensité relative de la fluorescence.) Y aurait-il des produits de digestion partielle
inclus dans vos calculs? Y aurait-il plusieurs fragments de faible masse moléculaire (donc de faible
intensité que vous avez de la difficulté à voir)? Les marqueurs utilisés pour évaluer la taille de
molécules d’ADN linéaire sont-ils aussi linéaires? Rappelez-vous que les molécules d’ADN
linéaires, circulaires déroulées et bicaténaires superenroulées ont toutes des propriétés de migrations
différentes sous nos conditions d’électrophorèse sur gel d’agarose. Puisque les marqueurs de tailles
que nous allons utiliser sont linéaires, peuvent-ils être utilisés pour estimer la taille de l’ADN d’un
plasmide recombinant non coupée?

Biologie Moleculaire-2016
23
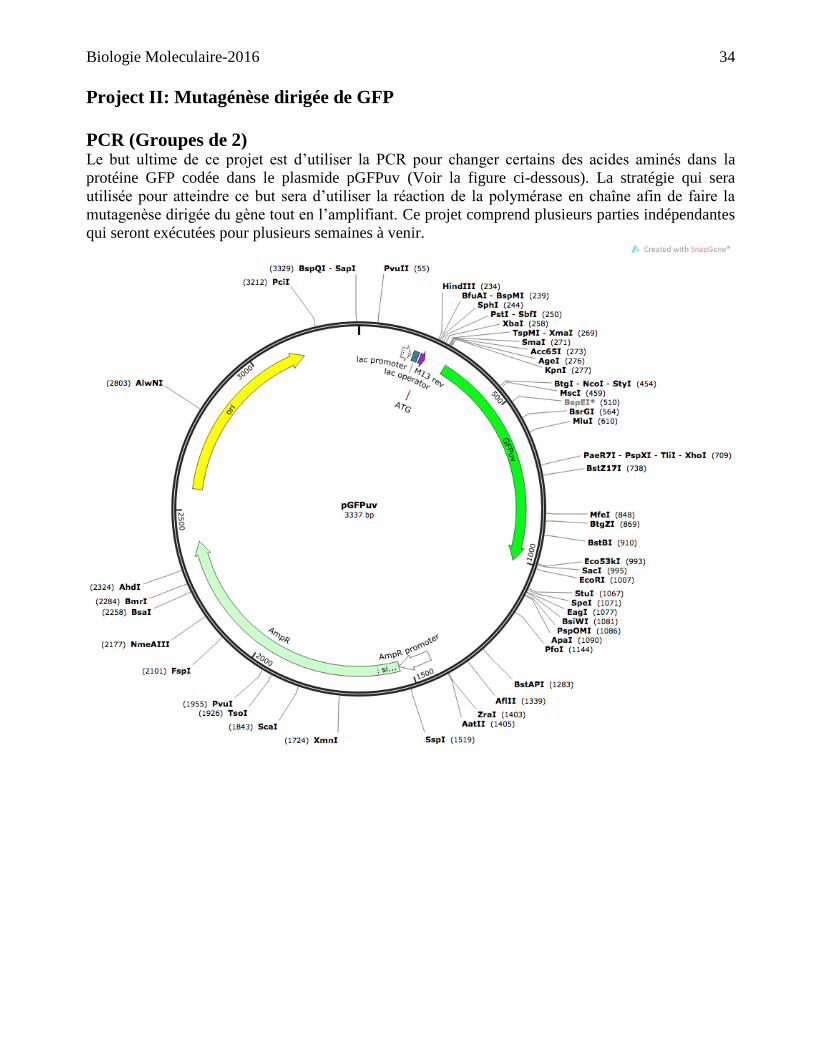
Projet I : vérifier la carte de restriction d’une insertion d’ADN (Groupes de 2) Le but de ce projet est d’obtenir une compréhension de la technique de cartographie par des
enzymes de restrictions. Dans les semaines à venir, vous aurez à utiliser des approches
expérimentales et de bio-informatiques pour caractériser un fragment d’ADN cloné. Chaque groupe
de deux aura à travailler avec un plasmide qui contient une insertion qui représente un des gènes
listé sous la rubrique "séquences" > gène inconnu" sur la page web de ce cours. Parmi un groupe de
4 (par bout de table) les deux groupes de deux possèderont le même inconnu, mais dans des
orientations opposées. Ces insertions furent toutes obtenues d’une librairie génomique créée dans le
vecteur de clonage pUC19. Essentiellement l’ADN génomique d’un organisme a été isolé, digéré,
après quoi une ligature des fragments générés a été faite dans le vecteur pUC19 linéarisé avec une
enzyme appropriée dont le site de restriction se retrouve dans le site de clonage multiple. (Voir la
figure sur la page suivante).
Les buts de ce projet sont :
Utiliser une approche expérimentale pour: o Déterminer le site d’insertion
o Vérifier la taille de l’insertion
o Vérifier l’orientation de l’insertion
o Déterminer la carte de restriction
Conseils pour l'utilisation des enzymes de restrictions Gardez toujours les solutions mères d’enzymes sur glace lorsqu’elles ne sont pas au congélateur à
-20oC (ce qui devrait être le temps le plus court possible).
L’enzyme est toujours la dernière composante ajoutée au mélange réactionnel et elle est ajoutée
directement à la solution dans le fond du tube plutôt que par goutte sur le bord du tube.
Assurez-vous que la solution est bien mélangée (ex. en donnant des petits coups sur le tube avec
votre doigt). S’il reste des gouttes sur le bord du tube, centrifugez brièvement les tubes. Vous
pouvez aussi les mélanger en utilisant le vortex (à moins que vous utilisiez de l’ADN génomique de
taille moléculaire élevée qui risque de se fragmenter).
Ne touchez jamais la pointe des embouts de pipette avec vos doigts ou avec autre chose que la
solution que vous voulez transférer.
Toujours utiliser un nouvel embout pour chaque opération et jeter les embouts utilisés aussitôt.
En accord avec les LIGNES DIRECTRICES SUR LE MATÉRIEL BIOLOGIQUE
DANGEREUX, tout le matériel jetable (embouts de micropipette, tubes de microcentrifugeuse,
etc.) utilisé lors de travaux avec de l’ADN recombinant et des cellules hôtes bactériennes doit être
placé dans des récipients à déchets spéciaux (c’est-à-dire, les boites de déchets à vos postes de
travail). Vous allez transférer ces déchets dans des sacs orangés qui seront autoclavés avant d’être
jetés.

Biologie Moleculaire-2016
24
Les enzymes suivantes ne coupent pas pUC19 : AdeI, AloI, ApaI, AscI, BaeI, BbvCI, BclI, BcuI, BglII, BoxI, BpiI, BplI,
Bpu10I, Bpu1102I, BsaAI, BsaBI, BseRI, BsgI, BshTI, BsmFI, Bsp68I, Bsp119I,
Bsp120I, Bsp1407I, BspTI, Bst1107I, BstXI, Bsu15I, BtrI, Cfr42I, CpoI, DsaI,
Eco32I, Eco47III, Eco52I, Eco72I, Eco81I, Eco91I, Eco105I, Eco130I, Eco147I,
FseI, Kpn2I, KspAI, MlsI, MluI, Mph1103I, MssI, MunI, Mva1269I, NcoI, NheI,
NotI, PacI, PauI, PdiI, Pfl23II, PsiI, Psp5II, PsyI, SacII, SanDI, SexAI, SfiI,
SgfI, SgrAI, SmiI, SrfI, SstII, Van91I, XagI, XcmI, XhoI, XmaJI.
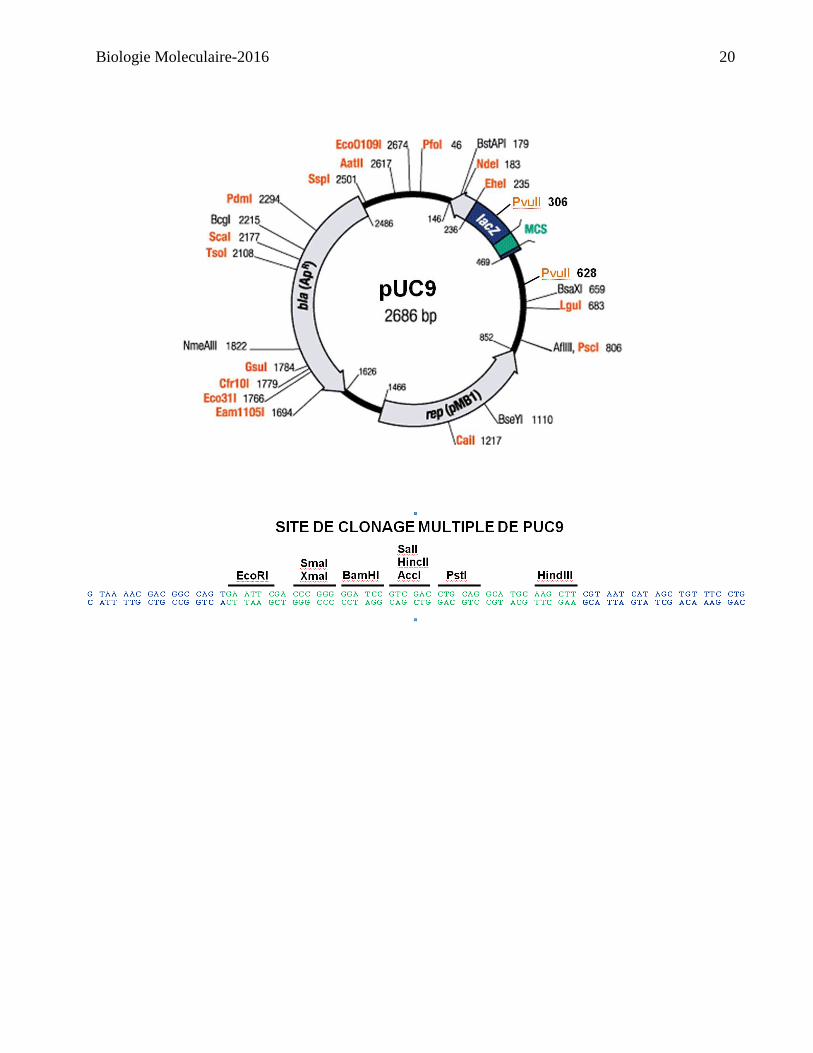
Les vecteurs pUC sont de petits plasmides, dont le nombre de copies maintenues est élevé qui
possèdent un site de clonage multiple (SCM), l’origine de réplication pMB1 nécessaire à la
réplication du plasmide (source – plasmide pBR322), et le gène bla, qui code pour la bêta-
lactamase, qui confère une résistance à l’ampicilline (source – plasmide pBR322). Noter que tous
les sites de restriction dans le site de clonage multiple se retrouvent seulement qu’une fois dans le
plasmide.
PvuII
PvuII
Univ. forward primer
Univ. reverse primer

Biologie Moleculaire-2016
25
Conseils pour le clivage d’ADN plasmidiques
Quantité d’ADN à utiliser:
La facilité que vous aurez à détecter vos fragments de restrictions (par fluorescence UV après une
coloration au bromure d’éthidium où l’intensité de la fluorescence est proportionnelle à la masse de
l’ADN dans un fragment) dépend du montant d’ADN utilisé (et des paramètres tels que l’épaisseur
du gel, la précision des bandes, etc.). On utilise habituellement entre 200-600ng d’ADN
plasmidique/puits pour une digestion avec une seule enzyme. Pour des digestions qui génèreront
plusieurs fragments, vous pourriez avoir besoin de faire la restriction de 400-1000ng afin de
pouvoir déceler les plus petits fragments (POURQUOI??)
Après avoir sorti les échantillons d’ADN du congélateur, assurez-vous de les dégeler complètement
avant de les prélever avec une pipette (pour obtenir la quantité adéquate d’ADN dans un volume
donné).
Vos digestions seront faites en présence d’un tampon commercial fourni par la compagnie
Fermentas. La concentration de sel idéale varie d’une enzyme de restriction à l’autre et vous
pouvez l’obtenir en utilisant différentes formulations du tampon.
Certaines enzymes requièrent des températures d’incubation autre que 37oC. Notez également que
les enzymes de restrictions qui reconnaissent les mêmes séquences s’appellent des isoschizomères
(ex. SacI et SstII). Il y a aussi des enzymes qui portent des noms très semblables (EcoRI, EcoRV)
mais qui reconnaissent des séquences distinctes (donc, soyez certain de bien lire les étiquettes sur
les tubes d’enzymes).
En principe, 1 unité d’enzyme de restriction digère complètement 1 g d’ADN purifié en 1
heure. Par contre, puisque nous utilisons souvent des préparations d’ADN brutes, nous
augmentons souvent la quantité d’enzyme utilisé et dans le cas d’ADN génomique complexe, les
temps d’incubation sont habituellement prolongés aussi. Typiquement, les enzymes de restrictions
sont fournies à des concentrations de 5-10 unités/l.
Le volume final d’enzyme ne devrait pas excéder 1/10 du volume réactionnel total, puisque le
glycérol dans la solution stock d’enzyme, que l’on ajoute pour empêcher la formation de cristaux
lors de la congélation, peut inhiber la réaction. Notez aussi que les solutions qui contiennent du
glycérol sont plus difficiles à manipuler avec des pipettes que des solutions aqueuses, soyez
attentifs aux volumes dans vos embouts de pipette.
Pour un survol de la cartographie par d’enzymes de restriction, consultez les sites Web
suivants:
http://faculty.plattsburgh.edu/donald.slish/RestMap/RestMapTutorial.html
http://www.vivo.colostate.edu/hbooks/genetics/biotech/enzymes/maps.html
http://wps.prenhall.com/esm_klug_essentials_5/17/4576/1171606.cw/content/index.html

Biologie Moleculaire-2016
26
Liste d’enzymes de restriction disponibles
ENZYME SITE
BamHI G▼
GATCC
HindIII A▼
AGCTT
PstI CTGCA▼
G
PvuII CAG▼
CTG
ScaI AGT▼
ACT
XhoI C▼
TCGAG
▼
- indique le lien phosphodiester clivé

Biologie Moleculaire-2016
27
Matériaux et Méthodes :
Certaines des enzymes de restrictions que vous utiliserez se retrouvent dans votre boîte au
congélateur. Si elles ne sont pas dans votre boîte, elles sont dans la boîte au congélateur du
groupe en face de vous sur le même bout de banc que vous.
Votre plasmide recombinant inconnu, à une concentration de 100g/mL est dans votre boîte
au congélateur.
Les tampons 10X d’enzymes de restrictions sont dans votre boîte au congélateur.
Préparation de vos digestions : Puisque les différentes enzymes requièrent différents tampons, vous aurez à être flexible lorsque
vous ferez une digestion de restriction. Préparez un tableau en indiquant toutes les composantes que
vous avez l’intention d’ajouter et leurs volumes avant de débuter. Lorsque vous ajouterez une des
composantes à votre tube étiqueté, rayez-la de votre tableau. Choisir le tampon qui permet d’avoir
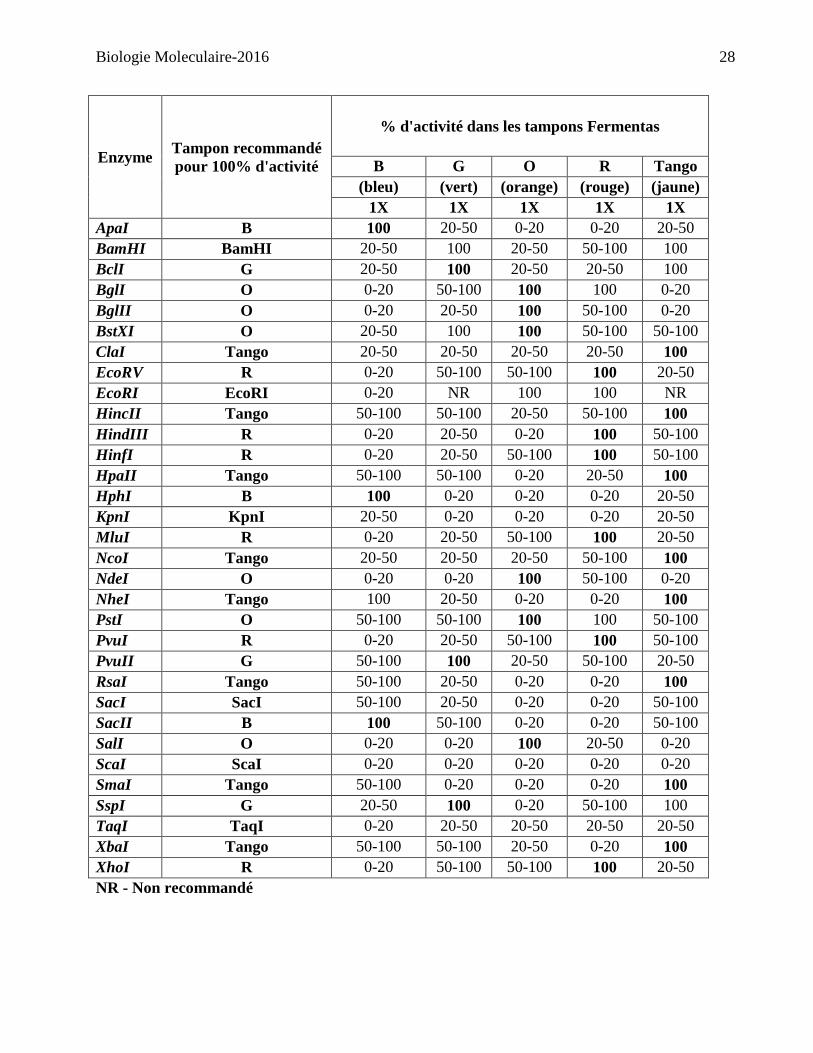
100% d’activité avec l’enzyme appropriée. (Consulter le tableau sur la page suivante)
Mélange réactionnel d’une digestion simple typique de 30µL:
Ingrédients Concentration finale/µL
ADN (100ng/µL) 250ng total
Tampon de restriction 10X 1X
Enzyme (10 unités/µL) 10 unités
Eau Compléter à 30µL
Biologie Moleculaire-2016
28
Enzyme Tampon recommandé
pour 100% d'activité
% d'activité dans les tampons Fermentas
B G O R Tango
(bleu) (vert) (orange) (rouge) (jaune)
1X 1X 1X 1X 1X
ApaI B 100 20-50 0-20 0-20 20-50
BamHI BamHI 20-50 100 20-50 50-100 100
BclI G 20-50 100 20-50 20-50 100
BglI O 0-20 50-100 100 100 0-20
BglII O 0-20 20-50 100 50-100 0-20
BstXI O 20-50 100 100 50-100 50-100
ClaI Tango 20-50 20-50 20-50 20-50 100
EcoRV R 0-20 50-100 50-100 100 20-50
EcoRI EcoRI 0-20 NR 100 100 NR
HincII Tango 50-100 50-100 20-50 50-100 100
HindIII R 0-20 20-50 0-20 100 50-100
HinfI R 0-20 20-50 50-100 100 50-100
HpaII Tango 50-100 50-100 0-20 20-50 100
HphI B 100 0-20 0-20 0-20 20-50
KpnI KpnI 20-50 0-20 0-20 0-20 20-50
MluI R 0-20 20-50 50-100 100 20-50
NcoI Tango 20-50 20-50 20-50 50-100 100
NdeI O 0-20 0-20 100 50-100 0-20
NheI Tango 100 20-50 0-20 0-20 100
PstI O 50-100 50-100 100 100 50-100
PvuI R 0-20 20-50 50-100 100 50-100
PvuII G 50-100 100 20-50 50-100 20-50
RsaI Tango 50-100 20-50 0-20 0-20 100
SacI SacI 50-100 20-50 0-20 0-20 50-100
SacII B 100 50-100 0-20 0-20 50-100
SalI O 0-20 0-20 100 20-50 0-20
ScaI ScaI 0-20 0-20 0-20 0-20 0-20
SmaI Tango 50-100 0-20 0-20 0-20 100
SspI G 20-50 100 0-20 50-100 100
TaqI TaqI 0-20 20-50 20-50 20-50 20-50
XbaI Tango 50-100 50-100 20-50 0-20 100
XhoI R 0-20 50-100 50-100 100 20-50
NR - Non recommandé

Biologie Moleculaire-2016
29
Vos Digestions :
1. Préparer les digestions par enzymes de restrictions suivantes de 0.25g d’ADN.
BamHI
HindIII
PstI
PvuII
ScaI
2. Préparer un témoin de digestion, qui contient toutes les composantes, sauf pour l’enzyme.
3. Incuber à 37oC pour 60 minutes.
4. Durant l’incubation des digestions faire un gel d’agarose de 1.0% avec du bromure d’éthidium.
5. Après la période d’incubation, transférer 5L de vos digestions a des nouveaux tubes étiquetés
de façon appropriés et ensuite ajouter du tampon de chargement 5X à chacune d’entre elles afin
d’obtenir une concentration d’au moins 1X.
6. Entreposer les restants de chacune de vos digestions (étiquetées de façon appropriées) à -20oC.
7. Charger les échantillons qui contiennent le tampon de chargement dans le gel.
8. Charger 5μL du vecteur pUC9 linéarisé qui a été préparé pour vous.
9. Charger 5μL de l’échelle de poids moléculaire.
10. Aussi charger sur ce gel 5 μL, n’incluant pas le tampon de chargement, du plasmid pGFPuv
isolé par la méthode de lyse alcaline et la méthode de Qiagen. (Sera fait plus tard)
11. Faire l’électrophorèse à 100V.
12. Après l’électrophorèse, examiner votre gel aux ultras violets et prendre une photo pour votre
analyse.
Analyse de restriction avec des digestions doubles Après l’analyse de vos digestions, vous devriez avoir une liste d’enzymes de restrictions qui
coupent à l’intérieur du fragment cloné ainsi que les tailles des fragments produits par ces
digestions. D’après cette information, vous avez généré une carte de restriction préliminaire (ou
plusieurs cartes, s’il n’y avait pas une carte unique non ambigüe).
La semaine prochaine vous devrez utiliser ses données et vérifier la validité de la carte propose.
Votre but est d’utiliser des digestions doubles afin de confirmer aussi précisément que possible la
position des sites de restrictions à l’intérieur de l’insertion et de résoudre toutes ambiguïtés.
Un exemple simple vous démontre l’approche. Supposons qu’après la première digestion avec une
seule enzyme vous avez trouvé un site de restriction dans l’insertion pour l’enzyme X et aucun site
de restriction pour l’enzyme EcoR1. Sachant qu’EcoR1 se retrouve dans le site de clonage
multiple, une digestion double avec EcoR1 et X vous donnera la distance entre X et le site connu
d’EcoR1.
Pour le laboratoire la semaine prochaine vous devez déterminer quelles digestions doubles vous
permettront de cartographier le plus précisément possible tous les sites de restrictions qui se
retrouvent dans votre insertion. Notez que vous pouvez utiliser une combinaison de n'importe
lesquelles des enzymes utilisées dans le labo de cette semaine.

Biologie Moleculaire-2016
30
Projet II: Mutagénèse dirigée de GFP
Isolation d’ADN plasmidique (Groupes de 2)
La plupart des méthodes utilisées pour la purification de plasmide se basent sur la disruption des
cellules en présence de dénaturants puissants. La disruption peut se faire par des cycles de gel et de
dégel, le broyage ou par une lyse chimique en utilisant des solutions fortement alcalines. Les
dénaturants sont essentiels afin d’inactiver les nucléases endogènes et exogènes qui dégraderaient
l’ADN. Examinez les différentes composantes du tampon d’extraction et déterminez la fonction de
chaque composé chimique.
Les plasmides sont des réplicons bactériens extrachromosomiques circulaires non obligatoires
L’isolation d’ADN plasmidique nécessite la séparation de cet ADN de l’ADN chromosomique,
ainsi que des polysaccharides, les lipides et les protéines de la cellule. Les manipulations
subséquentes, tout particulièrement les modifications enzymatiques, nécessitent que l’ADN soit
dépourvu de ces impuretés.
Purification d’ADN plasmidique par lyse alcaline
Dans cette procédure, la lyse des cellules est accomplie avec une solution fortement alcaline
(NaOH) et les protéines sont dénaturées à l’aide d’une solution fortement alcaline et d’un détergent
(SDS). Les complexes de détergent sont ensuite précipités avec un sel neutralisant (KOAC). Le
plasmide est séparé de l’ADN bactérien en vertu de sa stabilité relative dans des conditions
alcalines. De laisser le plasmide dans la solution alcaline trop longtemps détruira le plasmide aussi.
De plus, le chromosome est lié aux membranes et sera précipité par le sel et le détergent. Il est donc
important de ne pas mélanger la solution de façon trop vigoureuse, car ceci aurait pour effet de
relâcher le chromosome qui est piégé. Le plasmide est plus petit et demeure donc libre en solution.
La solution de plasmide est séparée des débris cellulaires par une centrifugation, puis concentrée
par une précipitation à l’alcool.

Biologie Moleculaire-2016
31
A. Préparation de vos solutions:
Préparer 1 mL des solutions I & II ainsi que le TE à partir des solutions mères suivantes :
10N NaOH
10% (m/v) SDS
0.5M glucose
1M Tris-Cl (pH 8.0)
0.5M EDTA
3 M KOAc pH 5
Isopropanol
ARNase 10mg/mL
Solution I: 50mM Glucose (tampon)
25mM Tris-Cl (pH 8.0) (tampon)
10mM EDTA (pH 8.0) (Chélateur)
Solution II: 0.2N NaOH (Alcalin)
1% SDS (Détergent)
T.E.: 10mM Tris-Cl pH 8.0
1mM EDTA pH 8.0
B. Protocole pour l’isolation du plasmide pGFPuv: 1. Centrifuger à vitesse maximale 1.5mL de la suspension d’E.coli/plasmide qui vous a été donnée
dans la microcentrifugeuse pour une durée de 1 minute.
2. Déverser le surnageant sans déranger le culot. Utiliser une micropipette pour enlever le
surnageant résiduel.
3. Ajouter 200μL de la Solution I au culot et le suspendre sur le vortex.
4. Ajouter 400μL de la solution II. Fermer le tube et mélanger par inversion.
5. Ajouter 300μL d’une solution glacée de KOAc. Bien mélangé et gardé sur glace pour 5 minutes.
6. Centrifuger dans la microcentrifuge à vitesse maximale pour 5-10 minutes. Ceci précipite les
protéines et l’ADN chromosomique le long de la paroi du tube.
7. Transférer 700µL du surnageant à un nouveau tube de microcentrifuge. Ajouter un volume égal
d’isopropanol. Fermer le tube et mélanger par des inversions rapides des tubes.
8. Centrifuger les tubes à vitesse maximale pour 5 minutes. Déverser le surnageant.
9. Le culot blanc au fond du tube contient l’ADN plasmidique et de l’ARN.
10. Suspendre le culot dans 50μL de TE pH8.0.
11. Ajouter 1μL d’une solution de 10mg/mL d’ARNase et incuber à 37oC 5-10 min.
12. Vous aurez besoin d’un échantillon de cette préparation pour une autre section de cet exercice
de labo.

Biologie Moleculaire-2016
32
Purification d'ADN plasmidique avec le kit de QIAGEN Une autre façon de purifier les plasmides consiste à utiliser des kits commerciaux tels que celui offert
par Qiagen. À la base, cette technique de purification est fondée sur la méthode de lyse alcaline que
vous avez exécutée précédemment. Afin de pouvoir comparer les deux méthodes, vous allez répéter
la purification à partir de la même culture bactérienne avec le kit de Qiagen. À la fin de la procédure,
vous devriez être capable de comparer les deux méthodes en ce qui a trait aux similarités et les
différences.
Étape 1
Étapes 2-5
Étapes 6-7
Étapes 8-9
Étape 10
Culot bactérien
Suspension
Lyse
Neutralisation
Liaison
Lavage
Élution
ADN plasmidique pure

Biologie Moleculaire-2016
33
Protocole pour l’isolation du plasmide pGFPuv 1. Obtenir la culture de 1.5mL d'E.coli avec un plasmide qui a été préparé pour vous et récolter les
cellules par une centrifugation à vitesse maximale pour 1 minute. Déversez-le surnageant.
2. Suspendre au vortex le culot de cellules dans 250µL de tampon P1 (+ ARNase) et transférer dans
un tube de microcentrifugeuse. Vous ne devriez pas avoir d’agrégats de cellules après cette
étape.
3. Ajoutez 250µL de Tampon P2 (qui contient du NaOH/SDS) et inversez doucement le tube 4-6
fois afin de mélanger. Ne passez pas le mélange au vortex puisque ceci causerait un déchirement
de l’ADN génomique. Si nécessaire, continuez à inverser le tube jusqu’à ce que la solution
devienne visqueuse et un peu claire. Ne pas dépasser 5 min, car l’ADN plasmidique risquerait
de se dénaturer irréversiblement.
4. Ajoutez 350µL du tampon N3 (neutralisation, tampon à forte concentration de sel) et inversez
immédiatement le tube doucement 4-6 fois. La solution devrait devenir trouble.
5. Centrifugez à vitesse maximale 10 min. Un culot compact blanc va se former au fond du tube
avec le “lysat éclairci” au-dessus.
6. Utilisez une pipette pour transférer le surnageant de l’étape 5 à une colonne QIAprep placée dans
un tube de récupération de 2mL.
7. Centrifugez 60 sec. à vitesse maximale. Jetez le volume écoulé dans les déchets organiques.
8. Lavez la colonne QIAprep en ajoutant 0.375mL de tampon PE (qui contient de l’éthanol) et
centrifugez 60 secs. Jetez l’écoulement (déchets organiques). Répétez une deuxième fois.
9. Jetez l’écoulement (déchets organiques), transférez dans un tube à centrifugeuse avec le
capuchon coupé et centrifugez pour 1 min additionnelle afin d’éliminer le tampon de lavage
résiduel.
IMPORTANT: L’éthanol résiduel du tampon PE peut inhiber des réactions enzymatiques
subséquentes. Les étapes 8 et 9 sont donc très importantes.
10. Placez la colonne QIAprep dans un tube à microcentrifugeuse (1.5mL) propre avec capuchon
coupé. Afin d’éluer l’ADN, ajoutez 50 µL de tampon EB (tampon d’élution = 10 mM Tris-HCl,
pH 8.5) et centrifugez 1 min.
11. Transférez dans un tube de microcentrifugeuse étiqueté. Assurez-vous de bien étiqueter et
d’entreposer cette préparation puisque vous en aurez besoin dans des expériences ultérieur
et pour l’examen pratique de labo! Entreposez dans votre boîte à -20oC
Biologie Moleculaire-2016
34
Project II: Mutagénèse dirigée de GFP
PCR (Groupes de 2) Le but ultime de ce projet est d’utiliser la PCR pour changer certains des acides aminés dans la
protéine GFP codée dans le plasmide pGFPuv (Voir la figure ci-dessous). La stratégie qui sera
utilisée pour atteindre ce but sera d’utiliser la réaction de la polymérase en chaîne afin de faire la
mutagenèse dirigée du gène tout en l’amplifiant. Ce projet comprend plusieurs parties indépendantes
qui seront exécutées pour plusieurs semaines à venir.

Biologie Moleculaire-2016
35
Des amorces ont été conçues afin de faire la mutagenèse et amplifier une partie de la séquence
codante du gène GFP qui se retrouve dans le plasmide pGFPuv. Les amorces qui seront utilisées sont
indiquées ci-dessous. Notez que plusieurs ont été incluses afin de faciliter le clonage et le criblage
subséquent. Consulter les pages web suivantes pour rafraîchir vos connaissances sur la PCR:
http://www.maxanim.com/genetics/PCR/PCR.htm
http://www.dnalc.org/resources/animations/pcr.html
Amorce “Forward”: GFPfor
GCCAAGCTTGCATGCCTGCAGATCGAC
Caractéristiques:
Un site HindIII indiqué en italique et souligné sera utilisé pour le clonage.
Un changement d’une base, indiqué en rouge, souligné et en caractère gras, représente une mutation
silencieuse qui abolit un site de restriction SalI.
Amorces “Reverse”:
GFPrev-1
TGTACTCGAGTTTGTGTCGGAGAATGTTTCCATC
GFPrev-2
TGTACTCGAGTTTGTGTCAGAGAATGTTTCCATC
GFPrev-3
TGTACTCGAGTTTGTGTGCGAGAATGTTTCCATC
GFPrev-4
TGTACTCGAGTTTGTG*CCGAGAATGTTTCCATC Caractéristiques:
Un site XhoI est indiqué en italique et souligné qui sera utilisé pour le clonage.
GFPrev-1 possède une substitution d’une base, indiquée en rouge, soulignée et en caractère gras,
change un codon de la glycine à l’arginine.
GFPrev-2 possède une substitution d’une base, indiquée en rouge, soulignée et en caractère gras, qui
crée un codon d’arrête.
GFPrev-3 possède une substitution d’une base, indiquée en rouge, soulignée et en caractère gras,
change un codon de la glycine à l’alanine.
GFPrev-4 possède une délétion d’une base indiquée en rouge, soulignée et en caractère gras, qui crée
un décalage du cadre de lecture de -1.
Biologie Moleculaire-2016
36
Method:
Préparation de la matrice d’ADN pGFPuv: 1. Obtenez la préparation de pGFPuv isolée par la méthode de Qiagen.
2. Diluer un échantillon de votre préparation par un facteur de 100.
3. Utiliser 5µL de la matrice diluée pour votre réaction de PCR.
Préparation de la réaction de PCR :
Préparez vos réactions de PCR dans un volume réactionnel total de 50µL. Dans des tubes de PCR
étiquetés (utilisez des tubes de PCR à paroi mince de 200 µL), ajoutez les ingrédients dans l’ordre
indiqué dans le tableau ci-dessous. Pour chaque composante ajoutée, utilisez un nouvel embout
autoclavé. Notez : la polymérase Taq sera ajoutée par l’aide enseignant.
Ingrédients Conc. mère Conc. finale Volume
Eau Compléter à 50µL
Tampon PCR Taq 10X 1X
Amorce GFPfor 2µM 0.2µM
*Amorce GFPrev assignée 2µM 0.2µM
MgCl2 50mM 1.5mM
dNTP 2mM 200µM
pGFPuv - 5µL
Polymérase Taq 5 unités/µL 0.05 unités/µL
*Assurez-vous de noter quelle amorce GFP reverse vous a été assignée!
Une fois que votre mélange réactionnel est complété, remettez-le à l’aide enseignant. Votre
échantillon vous sera retourné la semaine prochaine.
Conditions d’amplifications par PCR:
1. 1 cycle de 5min, 94oC pour dénaturer.
2. 30 cycles de 30sec, 94oC dénaturation; 1 minute à 68
oC appariement et extension.
3. 1 cycle de 5min, 72oC.
4. Refroidir à 4oC, indéfinie.

Biologie Moleculaire-2016
37
Exercice 3
Qu’est qu’on fait aujourd’hui !
Projet I: Vérifier la carte de restriction d’une insertion d’ADN
Projet II : Mutagénèse dirigée de GFP
Électrophorèse des amplicons de PCR de GFP
Digestion des amplicons de PCR de GFP et de pGFPuv
Ligature des amplicons de GFP

Biologie Moleculaire-2016
38
Projet I : Vérifier la carte de restriction d’une insertion d’ADN Vous devriez déjà avoir déterminé quelles digestions doubles vous désirez faire pour l’expérience de
cette semaine. Notez que pour chaque digestion double vous devrez aussi faire la migration des
digestions simples correspondantes sur votre gel. Par exemple si vous désirez faire une digestion
double BamHI-EcoRI, votre gel devrait aussi inclure des digestions simples BamHI et EcoRI pour
faciliter l’analyse.
Préparation de vos digestions : 1. Préparer vos digestions telles que précédemment. Puisque différentes enzymes requièrent
différents tampons, vous devrez être flexible afin de faire vos digestions. Si les deux enzymes
fonctionnent dans le même tampon, ajoutez simplement 1.0L de chacune des enzymes. Par
contre, si les deux enzymes fonctionnent dans des tampons différents, consulter le tableau de
compatibilité afin de déterminer le tampon approprié pour une activité maximale des deux
enzymes.
2. Incuber vos digestions pour 60 minutes à 37oC.
3. Migrer vos digestions tel que précédemment sur un gel d’agarose de 1%. Assurez-vous d’utiliser
le peigne approprié; celui qui fournira le nombre minimal de puits requis.

Biologie Moleculaire-2016
39
Projet II : Mutagenèse dirigée de GFP (Groupes de 2)
Électrophorèse des amplicons de PCR de GFP La semaine passée vous avez fait un PCR pour amplifier et faire la mutagenèse dirigée du gène GFP.
Cette semaine vous utiliserez l’électrophorèse sur gel d’agarose afin de vérifier la réussite de
l’amplification avant d’initier le clonage.
1. Récupérez vos réactions de PCR
2. Transférez 8µL de votre produit de PCR et placez-le dans un nouveau tube, puis ajoutez 2.5µL
de tampon de chargement 5X. Entreposer le restant sur glace pour des expériences subséquentes.
3. Charger vos échantillons sur un gel d’agarose de 1% précoulé qui contient du bromure
d’éthidium et une échelle de poids moléculaire appropriée.
4. Observer sous les UV. Prendre une photo pour l’analyse.
Digestion des amplicons de PCR de GFP et de pGFPuv Les produits de PCR amplifiés et analysés par électrophorèse sur gel peuvent maintenant être clonés
dans un vecteur approprié. Rappelez-vous que durant votre réaction de PCR des sites de restriction
étaient présents dans chacune des amorces afin de permettre le clonage directionnel du produit de
PCR dans un vecteur. Maintenant, nous devrons digérer le vecteur ainsi que le produit de PCR afin
de générer des extrémités compatibles pour la ligature subséquente. Puisque le PCR, les digestions et
la ligature sont faites dans différents tampons, nous devrons purifier l’ADN entre ces étapes afin
d’optimiser la prochaine réaction. La suite des étapes est:
Réaction de PCR (entreposer dans le congélateur)
Purification des amplicons et de pGFPuv
Purification des produits de PCR digérés avec HindIII et XhoI
Ligature des amplicons dans pGFPuv pour les transformations subséquentes

Biologie Moleculaire-2016
40
Purification avec le kit de purification QIAQUICK des amplicons digérés
Méthode: (Groupes de 2 assignés)
1. Ajoutez 5 volumes de tampon PB à 1 volume de la réaction de PCR et mélangez par inversion.
2. Placez une colonne “spin” QIAQUICK dans un tube de récupération de 2mL.
3. Pour lier l’ADN, appliquez l’échantillon sur la colonne QIAQUICK et centrifugez 1 min.
4. Jetez l’éluât dans les DÉCHETS ORGANIQUES et replacez la colonne “spin” QIAQUICK
dans le même tube.
5. Ajoutez 0.375mL de tampon PE à la colonne et centrifugez 1 minute afin de laver.
6. Jeter l’éluât dans les DÉCHETS ORGANIQUES.
7. Lavez encore une fois en ajoutant 0.375mL de tampon PE à la colonne et centrifugez 1 minute.
8. Jetez l’éluât dans les DÉCHETS ORGANIQUES. Replacez la colonne QIAQUICK dans le
même tube. Centrifugez 1 minute. Cette centrifugation élimine les résidus d’éthanol du tampon
PE qui pourrait éluer avec l’ADN et interférer avec les étapes subséquentes.
9. Placez la colonne QIAQUICK dans un nouveau tube à microcentrifugeuse de 1.5mL étiqueté.
10. Afin d’éluer l’ADN, ajoutez 30µL du tampon EB (10 mM Tris-HCl, pH 8.5) au centre de la
colonne QIAQUICK et centrifugez 1 minute.
11. En utilisant un nouvel embout de pipette, transférez le liquide récupéré au centre de la colonne
QIAQUICK et centrifugez une deuxième fois pour 1 minute.
12. Transférez le liquide récupéré dans un tube à microcentrifugeuse de 1.5mL étiqueté et entreposez
jusqu’au besoin. (Si vous n’étiez pas capable de récupérer 30.0µL, ajoutez assez du tampon
d’élution afin de compléter le volume à 30.0µL).
Digestions de l’amplico de GFP ou du vecteur pGFPuv
Méthode: (Groupes de 2 assignés)
Un des groupes de deux fera la digestion de l’amplicon de GFP tandis que l’autre groupe de
deux fera la digestion du plasmide pGFPuv.
1. Préparer comme suit un mélange réactionnel de 50uL pour faire une digestion double HindIII-
XhoI du produit de PCR de GFP ainsi que du plasmide pGFPuv purifié par la méthode de Qiagen
à la semaine 2.
Volume PCR Volume Vecteur
Produit de PCR de GFP 10uL ---------
OU plasmide pGFPuv --------- 5uL
Tampon de restriction rouge 10X 1X 1X
HindIII 1uL 1uL
XhoI 1uL 1uL
Eau Compléter à 50uL Compléter à 50uL
2. Faire la digestion à 37oC pour 1 heure.

Biologie Moleculaire-2016
41
Purification avec le kit de purification QIAQUICK des amplicons ou de pGFPuv digérés
Méthode: (Groupes de 2 assignés)
1. Ajoutez 5 volumes de tampon PB à 1 volume de la réaction de restriction et mélangez par
inversion.
2. Placez une colonne “spin” QIAQUICK dans un tube de récupération de 2mL.
3. Pour lier l’ADN, appliquez l’échantillon sur la colonne QIAQUICK et centrifugez 1 min.
4. Jetez l’éluât dans les DÉCHETS ORGANIQUES et replacez la colonne “spin” QIAQUICK
dans le même tube.
5. Ajoutez 0.375mL de tampon PE à la colonne et centrifugez 1 minute afin de laver.
6. Jeter l’éluât dans les DÉCHETS ORGANIQUES.
7. Lavez encore une fois en ajoutant 0.375mL de tampon PE à la colonne et centrifugez 1 minute.
8. Jetez l’éluât dans les DÉCHETS ORGANIQUES. Replacez la colonne QIAQUICK dans le
même tube. Centrifugez 1 minute. Cette centrifugation élimine les résidus d’éthanol du tampon
PE qui pourrait éluer avec l’ADN et interférer avec les étapes subséquentes.
9. Placez la colonne QIAQUICK dans un nouveau tube à microcentrifugeuse de 1.5mL étiqueté.
10. Afin d’éluer l’ADN, ajoutez 30µL du tampon EB (10 mM Tris-HCl, pH 8.5) au centre de la
colonne QIAQUICK et centrifugez 1 minute.
11. En utilisant un nouvel embout de pipette, transférez le liquide récupéré au centre de la colonne
QIAQUICK et centrifugez une deuxième fois pour 1 minute.
12. Transférez le liquide récupéré dans un tube à microcentrifugeuse de 1.5mL étiqueté et entreposez
jusqu’au besoin. (Si vous n’étiez pas capable de récupérer 30.0µL, ajoutez assez du tampon
d’élution afin de compléter le volume à 30.0µL).
Biologie Moleculaire-2016
42
Protocole pour la Purification QIAquick
Produit de PCR
ou
Morceau de gel solubilisé
ou
Réaction enzymatique
Liaison
Lavage
Élution

Biologie Moleculaire-2016
43
Projet II: Mutagénèse dirigée de GFP (Groupes de 4)
Ligature des amplicons de GFP
Ayant digéré l’amplicon de PCR de GFP et le vecteur pGFPuv vous procéderez maintenant avec la
ligature. Votre but ultime est de substituer la séquence sauvage de GFP dans le plasmide avec celle
que sur laquelle vous avez fait la mutagénèse par PCR.
Ingrédient Tube 1 Tube 2
Plasmide pGFPuv digéré 5.0L 5.0L
Tampon de ligature 10 X 2.0L 2.0L
Amplicon de PCR de GFP 5.0L 0.0L
Eau ajouter de l’eau pour compléter le volume à 19.0L
Ligase 1.0L 1.0L
DONNER VOS ÉCHANTILLONS ÉTIQUETÉS À VOTRE AIDE ENSEIGNANT
Les ligatures seront incubées à la température de la pièce jusqu'à demain puis entreposées à -20oC

Biologie Moleculaire-2016
44
Exercice 4
Qu'est-ce qu’on fait aujourd'hui!
Projet I: Vérifier la carte de restriction d’une insertion d’ADN (XhoI)
Projet II : Mutagénèse dirigée de GFP
Transformation des ligatures
Projet III: Empreintes génomiques
Isolation d’ADN génomique humain des cellules de joues
Amplification par PCR du VNTR ApoC2
Amplification par PCR du RFLP ApoB

Biologie Moleculaire-2016
45
Projet I : Vérifier la carte de restriction d’une insertion d’ADN (XhoI) Après l’analyse de vos résultats, vous avez pu déterminer précisément la position de tous les sites de
restrictions dans l'insertion. Maintenant, vous utiliserez cette information pour cartographier un
nouveau site de restriction; XhoI. Il a été découvert que cette enzyme coupe une fois dans l'insertion.
Concevoir une expérience appropriée pour déterminer la position précise du site XhoI.
Projet II : Mutagenèse dirigée de GFP
Transformation des mélanges de ligatures (Groupes de 4) La semaine dernière, vous avez fait des ligatures afin d’introduire vos amplicons dans pUC19. Afin
d’isoler, d’amplifier et de maintenir les recombinants voulus vous allez maintenant introduire ces
plasmides recombinant dans E.coli. Vous allez transformer des cellules d’Escherichia coli XL-1 avec
vos ligatures de la semaine passée. E. coli n’incorpore habituellement pas facilement l’ADN mais ces
cellules ont subi un traitement au CaCl2 afin de modifier leur paroi cellulaire. Les cellules que vous
allez recevoir sont donc COMPÉTENTES pour la transformation d’ADN. Mais, même avec ce
traitement, seulement qu’un faible pourcentage des cellules prendront de l’ADN. Afin d’identifier
celles-ci, un marqueur de sélection, la résistance à l’ampicilline, est inclus dans le vecteur. Donc,
seulement les cellules qui auront incorporé un plasmide circulaire, qui peut être répliqué et maintenu,
exprimeront cette résistance et seront capables de croître sur des géloses qui contiennent de
l’ampicilline. Toutes les cellules qui n’auront pas incorporé d’ADN ou qui auront incorporé de
l’ADN linéaire n’exprimeront pas cette résistance est ne pourront pas croître sur des géloses
sélectives.
Matériaux :
Cellules d’E.coli XL-1 compétentes
Géloses YT-AMP: (supplémentées avec 50μg/mL d’ampicilline pour la sélection).
Méthode :
1. Étiqueter 2 tubes jetables (tubes “snap-cap” de polypropylène de 15mL) et entreposés sur glace.
2. Transférer 90µL de cellules dans chaque tube, en vous assurant de ne pas contaminer les parois du
tube avec votre pipette.
3. Ajoutez 5µL de la ligature appropriée au tube étiqueté. Mélangez en remuant, et garder 30
minutes sur glace.
4. Incubez pour exactement 45 secondes dans un bain d’eau à 42ºC. Ce choc thermique est
nécessaire afin d’obtenir une bonne efficacité de transformation, mais si vous gardez les cellules à
42ºC pour trop longtemps, elles vont mourir. Ensuite, transférez immédiatement sur glace.
5. Ajoutez 0.9mL de bouillon SOC préchauffé (37ºC). Incubez 1 heure à 37
ºC en mélangeant afin de
permettre l’expression du gène de résistance à l’antibiotique.
6. Étalez 100µL et 200L de chacun des mélanges de transformation sur des géloses YT-AMP et
incubez toute la nuit à 37°C. Assurez-vous que vos plaques sont clairement étiquetées et placées à
l’envers dans l’incubateur à 37°C pour la soirée.
Les témoins de transformation suivants seront faits par vos aides enseignants:
Cellules traitées avec de l’ADN plasmidique (~5 pg de pUC circulaire)
Toujours utiliser les contenants à déchets de biohazard pour jeter les déchets microbiens et
d’ADN recombinant.

Biologie Moleculaire-2016
46
Projet III : Empreintes génomiques (Groupes de 2) L’analyse de polymorphismes de nucléotides simples (SNP) est devenue chose courante pour
identifier des génotypes de maladies et autres.
VNTR
Le principe de la variation évolutionnaire parmi une population est une des fondations de la biologie.
Ces variations sont le résultat de différences subtiles dans la séquence d'ADN des individus d'une
espèce donnée. Les variations surviennent souvent à cause d'erreurs de duplication de courtes
séquences de nucléotides où seulement qu'une copie était présente avant la réplication. Ceci résulte
en une répétition en tandem de la séquence originale. Si cette erreur survient encore une fois dans
une autre ronde de réplication, alors trois copies de la séquence seront en tandem (figure). Ces
répétitions en tandem font partie de nos chromosomes et sont donc héritées d'après le principe de la
génétique de Mendel. Au cours des siècles, le nombre de répétitions en tandem a augmenté ce qui
fait que chacun d'entre nous a hérité un nombre variable de répétitions en tandem (VNTRs) à
plusieurs loci dans nos génomes. On peut penser à un VNTR comme un loci avec chaque nombre de
répétitions d'une unité particulière étant analogue à différents allèles. Donc, chaque être humain (sauf
pour les vrais jumeaux) possède une combinaison unique de VNTRs et ces allèles peuvent être
utilisés dans les études de populations ou pour identifier un individu particulier.
Figure. Illustration du nombre de répétitions en tandem variable (VNTRs). Un simple brin d'ADN
du même locus de trois différents individus est montré. Dans cette région, le trinucléotide répété
CAT est présent une, deux ou trois fois ce qui donne des allèles de trois différentes longueurs.
RFLP
Un autre type de polymorphisme qui survient est appelé un polymorphisme de restriction de longueur
(RFLP). Ces polymorphismes surviennent quand un nucléotide unique est changé dans un site de
restriction, ce qui l'abolit ou le change. En conséquence, une région donnée du génome peut avoir un
site de restriction qui est absent dans la même région d'une autre copie du génome ou dans le génome
d'un autre individu.
Dans ce projet, vous déterminerez votre génotype de deux polymorphismes, un VNTR et un RFLP,
associé avec les gènes ApoC2 et ApoB respectivement.

Biologie Moleculaire-2016
47
Isolation d’ADN génomique des cellules de joues:
1. Une personne de chaque groupe de deux devrait utiliser un écouvillon stérile afin de se frotter
l’intérieur d’une des joues six fois. Sans tourner l’écouvillon, le déplacer de l’autre côté et frotter
l’intérieur de l’autre joue six fois.
2. Placer la portion de coton de l’écouvillon dans un tube de microcentrifuge de 1.5mL. Ensuite,
briser la tige juste au-dessus du coton pour qu’il tombe dans le tube.
3. Ajouter 400µL de PBS 1X pH 7.4 dans le tube.
4. Ajouter 20µL de protéinase K puis 400µL du tampon AL. Mélangé immédiatement au vortex
pour 15 secs. Ne pas ajouter la protéinase K directement au tampon AL.
5. Incuber à 56oC pour 10 min. Centrifuger brièvement pour récolter le liquide sur les parois.
6. Ajouter 400µL d’éthanol à 99%. Mélanger sur le vortex pour 8 à 10 sec. Centrifuger brièvement
pour récolter le liquide sur les parois.
7. Appliquer 600µL de votre mélange à la colonne. Fermer le capuchon afin d’éviter des aérosols et
centrifuger à 8k rpm pour 1min. Changer le tube de collecte.
8. Appliquer le restant de votre mélange à la colonne. Fermer le capuchon afin d’éviter des aérosols
et centrifuger à 8k rpm pour 1min. Changer le tube de collecte.
9. Ajouter 500µL du tampon AW1. Fermer et centrifuger à 8k rpm pour 1min. Changer le tube de
collecte.
10. Ajouter 500µL du tampon AW2. Fermer et centrifuger à vitesse maximale pour 3min. Changer le
tube de collecte.
11. Centrifuger à vitesse maximale pour 1 min afin de retirer le tampon résiduel et prévenir le
transfère.
12. Placer une colonne QIAprep dans un nouveau tube de 1.5mL de microcentrifuge duquel vous
avez coupé le capuchon. Ajouter 150µL du tampon AE à la colonne. Attendre 1 min puis
centrifuger à 8k rpm pour 1 min afin d’éluer.
13. Transférer l'ADN élué à un nouveau tube de microcentrifuge étiqueté et entreposé pour une
utilisation ultérieure.
Biologie Moleculaire-2016
48
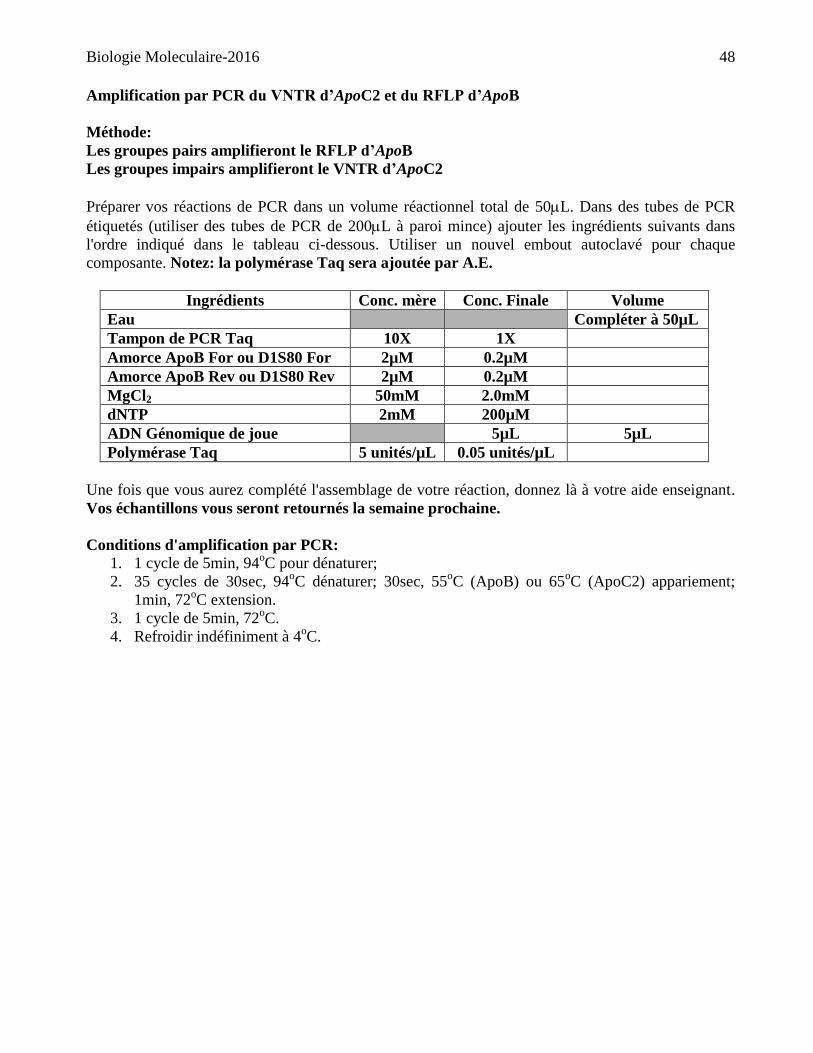
Amplification par PCR du VNTR d’ApoC2 et du RFLP d’ApoB
Méthode:
Les groupes pairs amplifieront le RFLP d’ApoB
Les groupes impairs amplifieront le VNTR d’ApoC2
Préparer vos réactions de PCR dans un volume réactionnel total de 50L. Dans des tubes de PCR
étiquetés (utiliser des tubes de PCR de 200L à paroi mince) ajouter les ingrédients suivants dans
l'ordre indiqué dans le tableau ci-dessous. Utiliser un nouvel embout autoclavé pour chaque
composante. Notez: la polymérase Taq sera ajoutée par A.E.
Ingrédients Conc. mère Conc. Finale Volume
Eau Compléter à 50µL
Tampon de PCR Taq 10X 1X
Amorce ApoB For ou D1S80 For 2µM 0.2µM
Amorce ApoB Rev ou D1S80 Rev 2µM 0.2µM
MgCl2 50mM 2.0mM
dNTP 2mM 200µM
ADN Génomique de joue 5µL 5µL
Polymérase Taq 5 unités/µL 0.05 unités/µL
Une fois que vous aurez complété l'assemblage de votre réaction, donnez là à votre aide enseignant.
Vos échantillons vous seront retournés la semaine prochaine.
Conditions d'amplification par PCR:
1. 1 cycle de 5min, 94oC pour dénaturer;
2. 35 cycles de 30sec, 94oC dénaturer; 30sec, 55
oC (ApoB) ou 65
oC (ApoC2) appariement;
1min, 72oC extension.
3. 1 cycle de 5min, 72oC.
4. Refroidir indéfiniment à 4oC.

Biologie Moleculaire-2016
49
Exercice 5
Qu'est-ce qu’on fait aujourd’hui !
Projet I: Mutagénèse dirigée de GFP
Criblage par PCR des transformants
Analyse des produits de la PCR de colonies
Repiquage
Projet III: Empreintes génomiques
Projet IV: Contrôle transcriptionnel de GFP par le promoteur de LacZ
Isolation d’ARN d’E.coli

Biologie Moleculaire-2016
50
Projet II : Mutagenèse dirigée de GFP
Criblage par PCR des transformants (Groupes de 2) Si vos transformations ont été une réussite, vous allez voir plusieurs colonies représentant des clones
indépendants d’E.coli qui possèdent des plasmides avec ou sans insertions. Votre première tâche
consiste à compter toutes les colonies sur toutes les géloses des ligatures, incluant les témoins de
ligatures et de transformation. Enregistrer le nombre de colonies sur chacune des géloses.
ASSUREZ-VOUS DE GARDER UN RAPPORT PRÉCIS DES DONNÉES; CELLES-CI NE
VOUS SERONT PAS DONNÉES À UNE DATE ULTÉRIEURE!!
Une fois les comptes enregistrés vous utiliserez la PCR et une digestion par enzyme de restriction
pour cribler les recombinants pour des insertions (et visualiser les produits de PCR suite à une
électrophorèse).
Méthode:
1. Étiqueter 8 tubes de microcentifuge de 1-8 et faire une grille sur une gélose YT+ amp étiquetée
de 1-8.
2. Ajouter 50L d’eau a chacun des tubes. En utilisant un embout de pipette stérile, ramasser une
des colonies sur la gélose qui représente la transformation du plasmide + insertion. Strier sur le
quadrant étiquetée1 de la plaque, puis placer l’embout dans le tube correspondant étiqueté 1.
3. Répéter la procédure ci-dessus avec 7 colonies supplémentaires. Placer la gélose à l’endroit
désigné pour qu’elle soit incubée.
4. Mélanger brièvement au vortex chacun des 8 tubes contenants les embouts pour suspendre la
colonie (Tenir l’embout pendant que vous mélangez).
5. Retirez les embouts de chaque tube, et bouillir pour 5 minutes. Placez sur glace pour 1 minute.
6. Centrifuger à vitesse maximale pour 5 minutes. Vous utiliserez le surnageant pour votre analyse
par PCR.
7. Préparer un cocktail pour 9 réactions de PCR de 20L. GARDER SUR GLACE. Ci-dessous est
la recette pour une réaction de 20l.
Réactions de PCR
Solution Conc. mère Conc. finale
Eau L pour un volume final de 20 L
Tampon Taq 10X 1 X
Amorce MutScreenFor 2 M 0.2 M
Amorce MutScreenRev 2 M 0.2 M
MgC12 50 mM 2.5 mM
dNTPs 2mM 200 M
ADN Pol. Taq 5U/µL 2.5U
8. Distribuer 19L du cocktail à chacun des 8 tubes de PCR étiquetés (SUR GLACE)
9. Ajouter 1L de chacun des surnageant des colonies obtenus à l’étape 6 aux tubes de PCR
appropriés.
10. Préparer un gel d’agarose de 1.25% contenant du bromure d’éthidium.

Biologie Moleculaire-2016
51
Conditions de PCR:
i. 1 cycle de 5 min, 94oC pour dénaturer;
ii. 30 cycles de 30s, 94oC pour dénaturer; 1 min. à 68
oC pour l’appariement et l’extension.
iii. 1 cycle de 5 min, 72oC
iv. Refroidir à 4oC.
Digestion par enzyme de restriction des produits de PCR :
1. Obtenir vos réactions de PCR.
2. Préparer 8 digestions par l’enzyme SalI, une pour chacune de vos réactions de PCR, dans un
volume final de 20 µL qui inclut 5 µL de chacune de vos réactions de PCR.
3. Préparer un témoin non-digéré en utilisant n’importe lesquels de vos 8 réactions de PCR.
4. Digérer pour une heure.
5. Après la digestion, ajouter 5 µL de tampon de chargement à chaque échantillon.
6. Charger 10 µL de chaque mélange, ainsi que l’échelle de poids moléculaire, sur votre gel
d’agarose de 1.25%. Migrer pour 1.5 heures à 100 V.
7. Prendre une photo de votre gel.
Projet III : Empreintes génomiques (Groupes de 2)
Gène ApoB:
1. Obtenir vos réactions de PCR du gène ApoB.
2. Dans le cas du gène ApoB, utiliser 10µl de la réaction de PCR pour préparer une digestion par
enzyme de restriction avec l'enzyme EcoRI dans un volume final de 20µl.
3. Un groupe de la classe préparera aussi un témoin non digéré.
4. Après avoir fait la digestion pour une heure, ajoutée du tampon de chargement et chargée sur le
gel précoulé.
5. Une fois que tout le monde aura chargé le gel, ceux-ci seront migrés et une photo sera prise pour
votre analyse.
Gène ApoC2 1. Obtenir vos réactions de PCR du gène ApoC2.
2. Ajouter du tampon de chargement à un échantillon de 10µl de votre réaction de PCR.
3. Charger sur le gel précoulé.
4. Une fois que tout le monde aura chargé le gel, ceux-ci seront migrés et une photo sera prise pour
votre analyse.

Biologie Moleculaire-2016
52
Projet IV: contrôle transcriptionnel de GFP par le promoteur LacZ
Isolation d’ARN d’E.coli Cette semaine vous initierez des expériences qui vous permettront d’examiner la régulation
transcriptionnelle du gène Mel1 qui code pour l’enzyme alpha galactosidase. Vous examinerez
l’abondance relative du transcrit de Mel1 sous différentes conditions de croissances. Deux méthodes
seront utilisées. La première, est l’analyse northern. Cette technique implique l’isolation, la
séparation et le transfert de l’ARN plutôt que de l’ADN. La deuxième technique qui pourrait être
utilisée est le RT-PCR qui sera discuté plus loin.
Isolation d’ARN totaux:
Comme dans toutes expériences, la purification du matériel de départ est cruciale. Vous allez isoler
de l’ARN de cellules d’E.coli et utiliser ensuite la spectrophotométrie afin d’estimer la concentration
et la pureté de l’ARN.
Les manipulations de l’ARN sont plus exigeantes que les manipulations de l’ADN puisque l’ARN est
moins stable et plus susceptible à la dégradation. Vous devez donc faire très attention de ne pas
contaminer aucune des solutions.
Toujours porter des gants!! Méthode:
Chaque groupe de 2 sera assigné une des conditions de croissance suivante à partir de laquelle vous
ferrez l’isolation d’ARN:
Culture crue dans du LB.
Culture crue dans du LB + glucose.
Culture crue dans du LB + lactose.
Culture crue dans du LB + IPTG.
1. Centrifuger a vitesse maximale pour 1 minute 1.5mL de la culture bactérienne assignée.
2. Retirer le surnageant, puis ajouter 0.5mL du réactif RNAzol. Suspendre le culot au vortex.
3. Incuber pour 5 minutes à 65oC.
4. Ajouter 0.2 mL de chloroforme, mélanger vigoureusement au vortex pour 15 sec et garder à la
température de la pièce pour 2-3 min.
5. Centrifuger à vitesse maximale pour 5 min.
6. Transférer approximativement 75% de la phase aqueuse supérieure (sans déranger les phases) à
un nouveau tube de microcentrifuge et ajouter 1 volume d’isopropanol.
7. Entreposer à la température de la pièce pour 5 minutes.
8. Centrifuger à vitesse maximale pour 5 minutes. Retirer le surnageant.
9. Brièvement laisser le culot sécher à l’air ambiant.
10. Suspendre le culot dans de la formamide.
RNAzol:
1.86 M guanidine isothiocyanate
12 mM sodium citrate
87 mM sodium acetate pH 4.0
0.37% sarcosyl
42 mM 2-mercaptoethanol

Biologie Moleculaire-2016
53
Déterminer la concentration d’ARN et la quantité récupérée par spectrophotométrie.
1. Préparer une dilution de votre échantillon d’ARN de 1/20 avec de l’eau dans un volume final de
100 µL et mesurer l’absorbance aux longueurs d’onde de 260 nm et 280 nm en utilisant de l’eau
comme témoin.
2. Vous pouvez estimer la concentration d’ARN en utilisant le rapport suivant (pour de l’ARN
simple brin): absorbance de 1.0 à 260 nm est équivalent à une concentration d’ARN de 40
g/mL Si le rapport A260/A280 >1.90, l’ARN est relativement pure et sans protéine.
3. Calculer la quantité d’ARN récupérée.

Biologie Moleculaire-2016
54
Exercice 6
Qu'est-ce qu’on fait aujourd’hui !
Projet IV: Contrôle transcriptionnel de GFP par le promoteur LacZ
Électrophorèse sur gel d’ARN
Transfert northern

Biologie Moleculaire-2016
55
Projet IV : Contrôle transcriptionnel de GFP par le promoteur LacZ
Électrophorèse sur gel d’ARN On peut utiliser l’analyse Northern afin de détecter une seule espèce d’ARN à l’intérieur d’une
population d’ARN total provenant d’un organisme (ou organite). Avant qu’un transfert Northern
puisse être fait, les molécules d’ARN sont séparées en fonction de leurs tailles sur un gel d’agarose
dénaturant. Le type de gel d’ARN les plus couramment utilisés est celui fait de formaldéhyde. Ce
type de gel procure des conditions dénaturantes qui empêchent l’appariement des bases, réduisant
ainsi la formation de structures secondaires chez les molécules d’ARN. Ceci est important puisque,
contrairement aux fragments d’ADN bicaténaires qui ont la même conformation malgré les
différentes longueurs, les ARN monocaténaires peuvent avoir différentes conformations en raison de
l’appariement de bases à l’intérieur du brin. De telles structures secondaires affecteraient la
migration électrophorétique et rendraient l’estimation de la taille imprécise.
Matériaux: Les ribonucléases sont partout! Afin de réduire le risque de dégradation de l’ARN, portez des gants
et conservez vos échantillons sur glace.
Le formaldéhyde et la formamide sont des composés chimiques toxiques. Portez des gants et
travaillez sous la hotte autant que possible.
Méthodes :
A. Préparation d’un gel d’agarose au formaldéhyde (Un gel par groupes de 4)
1. Combinez les composantes suivantes pour la préparation d’un gel d’agarose-formaldéhyde de
1.5% (25mL) (en utilisant le peigne à 8 puits):
Tampon MOPS 5X, pH 7.0 5 mL
H20 15.5 mL
Agarose 0.375 g
2. Dissoudre l’agarose dans le four à micro-ondes. Laissez refroidir un peu. Ensuite, sous la hotte,
ajoutez 4.5mL de formaldéhyde (solution 37%) (ATTENTION! VAPEURS VOLATILES).
Mélangez en remuant et coulez le gel immédiatement.
3. Fermer le couvercle et laisser le gel solidifier au moins 30 minutes.

Biologie Moleculaire-2016
56
B. Préparation des échantillons d’ARN (par groupes de 4) Vous allez préparer quatre échantillons d’ARN différents. Il est important que chaque voie de
migration du gel contienne la même quantité d’ARN.
Vous aurez besoin des échantillons d’ARN suivants :
Un échantillon d’ARN totaux de cultures crues dans le Lb
Un échantillon d’ARN totaux de cultures crues dans le LB + glucose
Un échantillon d’ARN totaux de cultures crues dans le LB + lactose
Un échantillon d’ARN totaux de cultures crues dans le LB + IPTG
1. Afin de préparer les échantillons d’ARN pour la migration sur le gel, mélangez les composantes
suivantes en utilisant des tubes étiquetés SUR GLACE:
Volume final = 30 µL
Échantillon d’ARN: µL (Ce volume d’échantillon devrait contenir 5 g d’ARN dans un
volume maximum de 9L)
Tampon de chargement: 21µL (Tampon de chargement pour l’ARN et non pas pour l’ADN)
Eau (jusqu’à 30 µl): µL
2. Incubez vos échantillons 10 minutes à 70ºC et ensuite, placez rapidement les échantillons sur
glace. S’il y a du liquide sur le bord du tube, centrifugez quelques secondes.
3. Chaque groupe de 4 doit préparer un gel comme suit : Charger 14 µL des échantillons d’ARN des
différentes conditions de croissances sur un gel comme suit :
VOIES ÉCHANTILLONS
1 Vide
2 Vide
3 ARN de cultures crues dans le LB
4 ARN de cultures crues dans le LB + glucose
5 ARN de cultures crues dans le LB + lactose
6 ARN de cultures crues dans le LB + IPTG
7 Vide
8 Vide
Ceci génèrera un gel qui contient les échantillons de deux groupes de 4 et des gels duplicata par table.

Biologie Moleculaire-2016
57
C. Électrophorèse sur gel 1. Le tampon de migration sera du MOPS 1X. Faire migrer le gel à 80 V (courant de ~ 80 mA)
jusqu’à ce que le colorant bleu de bromophénol dépasse la moitié du gel.
2. Enlevez le gel du boîtier d’électrophorèse, examinez sous les UV et prenez une photo.
Notez : Les gels de formaldéhydes sont plus FRAGILES que les gels non dénaturants, faites
très attention!
3. Lavez le gel 2 fois avec 5 volumes d’eau, et ensuite 1 fois en utilisant 2 volumes de 10X SSC
pour environ 5 minutes chaque fois. Ceci est nécessaire afin d’enlever le formaldéhyde qui
préviendrait l’hybridation.
D. Transfert capillaire (par groupes de 4)
On vous fournira une membrane de nylon ainsi que des papiers-filtres coupés de la même taille que le
gel. Éviter autant que possible de manipuler la membrane avec vos doigts.
1. Étiqueter une extrémité de la membrane avec le jour du labo, votre no de groupe, et « ARN ».
2. Tremper la membrane dans de l’eau distillée et ensuite dans du 2X SSC.
3. Assembler votre transfert northern tel qu’illustré sur la page suivante. Assurez-vous que la
périphérie de votre gel est entourée avec de la pellicule de plastique afin d’éviter un court-circuit.
4. Après le transfert, la membrane est séchée, étiquetée, fixée aux UV et entreposée dans un sac de
plastique.
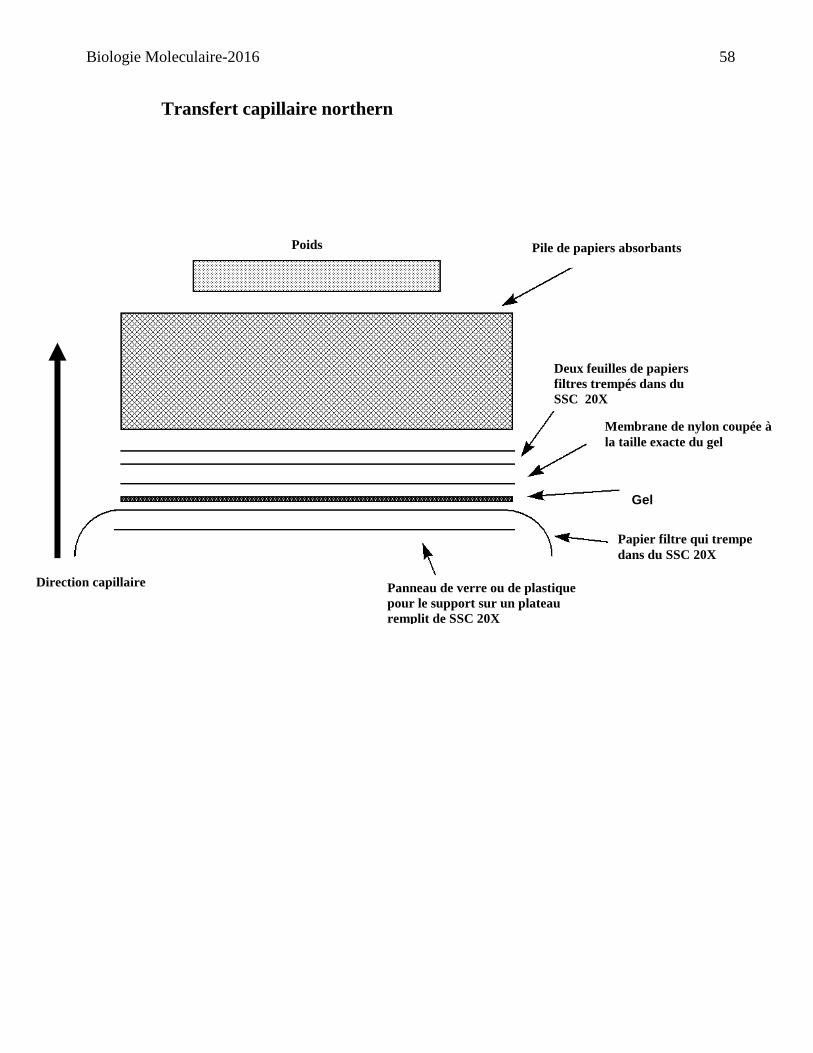
Biologie Moleculaire-2016
58
Transfert capillaire northern
Membrane de nylon coupée à
la taille exacte du gel
Gel
Deux feuilles de papiers
filtres trempés dans du
SSC 20X
Papier filtre qui trempe
dans du SSC 20X
Panneau de verre ou de plastique
pour le support sur un plateau
remplit de SSC 20X
Pile de papiers absorbants Poids
Direction capillaire

Biologie Moleculaire-2016
59
Exercice 7
Qu'est-ce qu’on fait aujourd’hui !
Projet IV: Contrôle transcriptionnel de GFP par le promoteur lacZ
Hybridations northern
Projet V: RT-PCR de la protéine ribosomale URP1 de la levure
Isolation d’ARN de levure
RT-PCR

Biologie Moleculaire-2016
60
Projet IV : Contrôle transcriptionnel de GFP par le promoteur LacZ
Hybridation northern (Groupes de 4) Dans les dernières semaines, vous avez généré un Northern. Cette semaine vous allez préparer les
membranes pour l’hybridation avec une sonde étiquetée avec Dig.
Informations générales sur l’hybridation d’acides nucléiques:
Avant de faire l’hybridation de la sonde à la membrane, une étape de pré hybridation est faite. Cette
étape est nécessaire afin de réduire le bruit de fond et l’hybridation non spécifique. Le bruit de
fond se définit comme la liaison non spécifique de la sonde à la membrane et elle n’implique pas
d’hybridation. L’hybridation non spécifique représente l’hybridation de la sonde à des séquences
qui possèdent un faible pourcentage d’identité. Afin de réduire ces deux évènements, les membranes
sont initialement assujetties à un réactif de blocage qui inclut habituellement une concentration
élevée d’ADN non spécifique, tel que de l’ADN de sperme de saumon. Le but de ce réactif est de
saturer tous les sites de liaisons sur la membrane ainsi que les sites d’hybridations non spécifiques sur
les acides nucléiques.
Suite à l’étape de pré hybridation, les membranes sont assujetties à la sonde étiquetée en présence du
réactif de blocage. Dépendant des conditions de stringence, la sonde va facilement faire compétition
avec des liaisons non spécifiques et donc s’hybrider de façon spécifique aux séquences
complémentaires.
L’excédant de sondes, qui sont non hybridées ou qui sont faiblement hybridée à des séquences avec
une complémentarité faible, sera enlevée par une série de lavages de stringences variées. Notez que la
stringence est déterminée par la température et la concentration de sels. Ces deux paramètres
déterminent le niveau d’homologie qui est requis pour permettre l’hybridation. Une stringence plus
faible représente des conditions d’hybridation qui sont plus permissives et permet donc l’hybridation
avec des séquences dont le niveau de complémentarité est plus faible. De façon inverse, une
stringence élevée est moins permissive et exige donc un niveau de complémentarité élevé afin de
permettre la formation d’hybrides stables.

Biologie Moleculaire-2016
61
Préhybridation
Sur chaque table, vous devriez avoir des membranes duplicatas de northern.
Solution de Préhybridation/Hybridation :
5X SSC (20X SSC: 3M NaCl et 0.3M NaCitrate)
0.02% sodium dodecyl sulfate (SDS)
20mM sodium maléate, pH 7.5
4M Urée
100μg/mL ADN de sperme de saumon
1. Placer votre membrane northern dans un tube Falcon de 50mL étiquetés clairement avec votre
numéro de groupe. Ajoutez 10mL du mélange de préhybridation et fermez en serrant fortement.
2. Introduire vos tubes dans les bouteilles de verres du four d’hybridation. Assurez-vous de ne pas
égratigner les parois intérieures des bouteilles de verre.
3. Positionnez le tube en verre dans le four à hybridation. Assurez-vous que les tubes sont
balancés en plaçant un deuxième tube en verre dans la position opposée de la roue.
4. Faites tourner lentement (position no3) à 42
oC pour un minimum de 1 heure.
Hybridations
À présent, vous êtes prêts pour l’hybridation avec la sonde Mel1. Mais avant de pouvoir faire
l’hybridation, l’ADN marqué doit être dénaturé. Assurez-vous d’enregistrer quelle sonde vous a
été assignée!
1. Faites bouillir la sonde pour 3 min et refroidissez là rapidement sur glace. Centrifugez brièvement
afin que l’échantillon s’accumule au fond du tube.
Préparation de vos hybridations
2. Déverser la solution de préhybridation des tubes contenants votre membrane northern.
3. Ajouter immédiatement 10mL de solution fraîche d’hybridation à chacun des tubes.
4. Ensuite, ajouter soigneusement 50µL de la sonde dénaturée de GFP ou de bla (tel qu’assigné)
directement au liquide d’hybridation de la membrane du northern. Évitez tout contact direct entre
la sonde concentrée et la membrane. Fermez les tubes fermement et installez-les dans le four à
hybridation tel qu’auparavant.
5. Faire l’hybridation de vos membranes northerns à 42oC toute la nuit.
APRÈS L’HYBRIDATION LES MEMBRANES SERONT LAVÉES PAR LES A.E. PUIS
ENTREPOSÉES POUR UNE UTILISATION ULTERIEUR

Biologie Moleculaire-2016
62
Projet V: RT-PCR de la protéine ribosomale URP1 de la levure (Groupes de 2)
Isolation d’ARN de levure (Les étapes 1-3 ont été faites pour vous)
1. Obtenir 10 mL d’une culture de levure.
2. Centrifuger pour 10 minutes à 7 000 rpm.
3. Jeter le surnageant.
4. Suspendre le culot dans 200μL de tampon de lyse.
5. Transférer à un nouveau tube avec un bouchon à vis qui contient des billes de verres et 200μL de
phénol:chloroform:isoamyl alcool (25:24:1).
6. Mélanger au vortex pendant 2 minutes et ensuite placer sur glace pour 2 minutes.
7. Répéter l’étape 6 deux fois de plus.
8. Centrifuger à vitesse maximale pour 5 minutes. Transférer la phase aqueuse supérieure à un
nouveau tube.
9. Ajouter 0.1 volume de 3M NaOAc et 1mL d’ETOH de 99%, bien mélangé par inversion et
centrifuger à vitesse maximale pour 2 minutes.
10. Jeter le surnageant et suspendre le culot dans 100μL d’eau traitée au DEPC.
11. Déterminer le rendement et la pureté.
RT-PCR Une autre méthode qui peut être utilisée pour comparer l’abondance relative d’un transcrit sous
différentes conditions est le RT-PCR. Contrairement à une analyse northern, cette méthode est plus
sensible et idéale pour l’analyse de transcrits faiblement exprimés.
Contrairement aux réactions de PCR traditionnelles, tel que celle que vous avez faite avec l’ADN
génomique, le RT-PCR utilise l'ARN plutôt que l'ADN comme matrice. Par contre, puisque l’ADN
polymérase thermostable utilisé dans la réaction de PCR fonctionne seulement avec une matrice
d'ADN, l'ARN doit préalablement être transcrit en ADN. Ceci est accompli par un ADN polymérase
ARN-dépendante, la transcriptase inverse (RT). La transcriptase inverse est utilisée pour synthétiser
un ADN complémentaire (ADNc) simple-brin à partir de l'ARN d'intérêt. Une amorce oligo dT sera
utilisée pour cette réaction. Une fois que l'ADNc a été généré, une réaction de PCR standard peut être
utilisée pour amplifier la séquence d’ADN complémentaire à l’ARNm.
Pour un survol sur la technique de RT-PCR consultez le site web suivant :
http://www.bio.davidson.edu/courses/Immunology/Flash/RT_PCR.html

Biologie Moleculaire-2016
63
Méthode
Chaque groupe exécutera les 5 réactions suivantes :
Réaction de PCR avec l’ARN traité avec l’ADNase non traité avec la RT
Réaction de PCR de la réaction de RT sur l’ADN traité avec l’ADNase
Réaction de PCR de la réaction de RT sur l’ADN NON traité avec l’ADNase
Réaction de PCR de l’ARN Non traité avec soit l’ADNase ou la RT
Réaction de PCR sans ARN (pas de matrice)
Préparation des échantillons d’ARN pour les réactions de RT
Préparer une dilution de votre ARN comme suit : Cette dilution sera ensuite utilisée comme votre
source d’ARN pour toutes les réactions requises décrites dans cette section.
4.0g de votre échantillon d’ARN
5.0L du tampon 10X de l’ADNase I
Eau jusqu’à un volume final de 48L
Traitement de l’ARN total avec ADNase :
1. Avant de faire votre réaction de RT, vous devez vous assurer d'éliminer tout ADN contaminant
de vos échantillons d'ARN. Dans un volume final de 25L, ajouter les composantes suivantes:
24L de votre échantillon d’ARN dilué
1L de l’ADNase I
2. Incubez 15 minutes à la température de la pièce. Après le traitement à l'ADNase, inactivez
l'enzyme en ajoutant 2.5µL d’EDTA 25mM et en incubant 5 minutes à 65ºC. Refroidir sur glace.
Réactions de transcriptase inverse :
3. Préparer votre réaction de RT dans des tubes de PCR tel qu’indiqué ci-dessous. Utiliser un nouvel
embout autoclavé pour chacune des composantes ajoutées.
4. Mettre 19.0L d’eau DEPC dans chacun de deux tubes étiquetés : +ADNase et -ADNase.
5. Ajouter 4.0L d’amorce oligo-dT à chaque tube.
6. Ajouter 4.0L d’un mélange de dNTP à 2mM à chaque tube.
7. Ajouter 2L de votre ARN traité à l’ADNase au tube étiqueté + ADNase.
8. Ajouter 2L de votre dilution d’ARN non traité au tube étiqueté -ADNase.
9. Permettre l’appariement de l’amorce oligo-dT en incubant vos mélanges pré-réactionnels de RT à
70oC pour 5 minutes.
10. Laisser vos tubes refroidir sur votre table de travail pour 5 minutes.
11. Ajouter 8.0L du tampon RT 5X à chacun des tubes.
12. Ajouter 2.0L de DTT à 100mM pour obtenir une concentration finale de 5mM à chacun des
tubes.
13. Ajouter 1.0 L de la transcriptase inverse de MMLV à 200U/L pour obtenir une concentration
finale de 5U/L à chacun des tubes.
14. Incuber à 42oC pour 60 minutes.
15. Inactiver la transcriptase inverse en incubant le mélange réactionnel à 70oC pour 15 minutes.

Biologie Moleculaire-2016
64
Réactions de PCR :
1. Après les réactions de transcriptase inverse, préparer les réactions de PCR suivantes dans 6 tubes
étiquetés de façon appropriés :
PCR d’ADN génomique de la levure (Sera fourni)
PCR de l’ARN traité avec l’ADNase, mais non traité avec la RT
PCR de la réaction de RT sur l’ARN traité avec l’ADNase
PCR de la réaction de RT sur l’ARN NON traité avec l’ADNase
PCR de l’ARN non traité avec soit l’ADNase et la RT
Réaction de PCR sans ARN (Pas de matrice)
Amorces :
URP (FOR) GTGCAATTTCAGGCATTACCG
URP (REV) CTGGGGCCAAAGTTTGAGGAAC
Échantillon Conc. mère. Conc. finale
Eau l pour un volume final de 49L
Tampon de la polymérase Taq 10 X 1X
Amorce URP (For) 2M 0.2M
Amorce URP (Rev) 2M 0.2M
MgCl2 50mM 2.0mM
dNTP 2mM 200M
Polymérase Taq 5unités/µL 2.5U/réaction
2. Ajouter 1L de chacune des matrices appropriés ou de l’eau à chacun des mélanges de PCR
appropriés. Une fois vos mélanges réactionnels complétés, entreposés les dans le bac à glace à
l’avant pour faire le PCR.
3. Vos réactions de PCR vous seront retournées la semaine prochaine.
Vos PCR seront effectuées sous les conditions suivantes:
i. 1 cycle: 5 min, 94oC pour dénaturer;
ii. 30 cycles: 30 sec, 94oC pour dénaturer; 30 sec, 58
oC l’appariement; 30 sec, 72
oC pour élongation.
iii. 1 cycle: 5 min, 72oC.
iv. Refroidir à 4oC.

Biologie Moleculaire-2016
65
Exercice 8
Qu'est-ce qu’on fait aujourd’hui !
Projet II : Mutagénèse dirigée de GFP
Évaluation de l’activité de GFP
Projet IV: Contrôle transcriptionnel de GFP par le promoteur LacZ
Immunodétection des northerns
Projet V: RT-PCR du la protéine ribosomale URP1 de la levure
Analyse des réactions de RT-PCR

Biologie Moleculaire-2016
66
Projet II: Mutagénèse dirigée de GFP
Évaluation de l’activité de GFP
Projet IV : Contrôle transcriptionnel de GFP par le promoteur LacZ
Immunodétection des membranes northerns: Les hybridations des northerns étant faites, vous allez maintenant suivre une procédure pour la
détection des hybrides. Notez que les membranes ont été lavées pour vous afin de retirer la sonde
libre ainsi que celle qui s’est faiblement hybridée.
Tous les traitements se font à la température de la pièce.
1. Placer toutes les membranes dans un petit sac de plastique.
2. Ajouter 15mL de tampon d’acide maléique 1X. Incuber 15 min avec agitation. Jeter la solution.
3. Ajouter 20mL de solution de blocage. Incuber 30 min avec agitation. Jeter la solution.
4. Ajouter 10mL de solution d’anticorps. Incuber 30 min avec agitation. Jetez la solution.
5. Transférer les membranes dans le contenant de Tupperware. NE LAISSEZ PAS LES
MEMBRANES SÉCHER! Ajouter 50mL de tampon de lavage. Incuber 15 min avec agitation.
Jeter la solution et répéter l’étape 5 une fois de plus.
6. Ajouter 50mL de tampon d’acide maléique. Incuber 15 min avec agitation. Jeter la solution.
7. Ajouter 10mL de tampon de détection. Incuber 5 min avec agitation. Jeter la solution.
8. Les prochaines étapes doivent être faites rapidement et de façons coordonnés. Aviser votre aide
enseignant ou le technicien avant de procéder. Mettre 2.5mL de la solution CDP-Star dans un
coin du contenant et faire glisser la solution sur la membrane pour la recouvrir. NE METTEZ
PAS la solution directement sur la membrane. Incuber 5 minutes à l’obscurité dans votre tiroir.
9. Placer la membrane avec le côté contenant l’ARN vers le haut sur un papier Whatman.
10. Placer 2 membranes avec le côté contenant l’ARN vers le haut dans l’appareil photo. Prendre une
photo de la réaction de chimioluminescence (exposition de 2 à 10 minutes).

Biologie Moleculaire-2016
67
Project V: RT-PCR de la protéine ribosomale URP1 de la levure
Analyse des réactions de RT-PCR 1. Couler un gel d’agarose de 1.5%.
2. Obtenir vos réactions de RT-PCR du congélateur.
3. Ajouter la quantité appropriée du tampon de chargement d’ADN à 10 µL d’échantillon de
chacune de vos réactions.
4. Charger et migrer à 80V.
5. Après la migration, visualiser sous les UV et prendre une photo.
Biologie Moleculaire-2016
68
Annexes
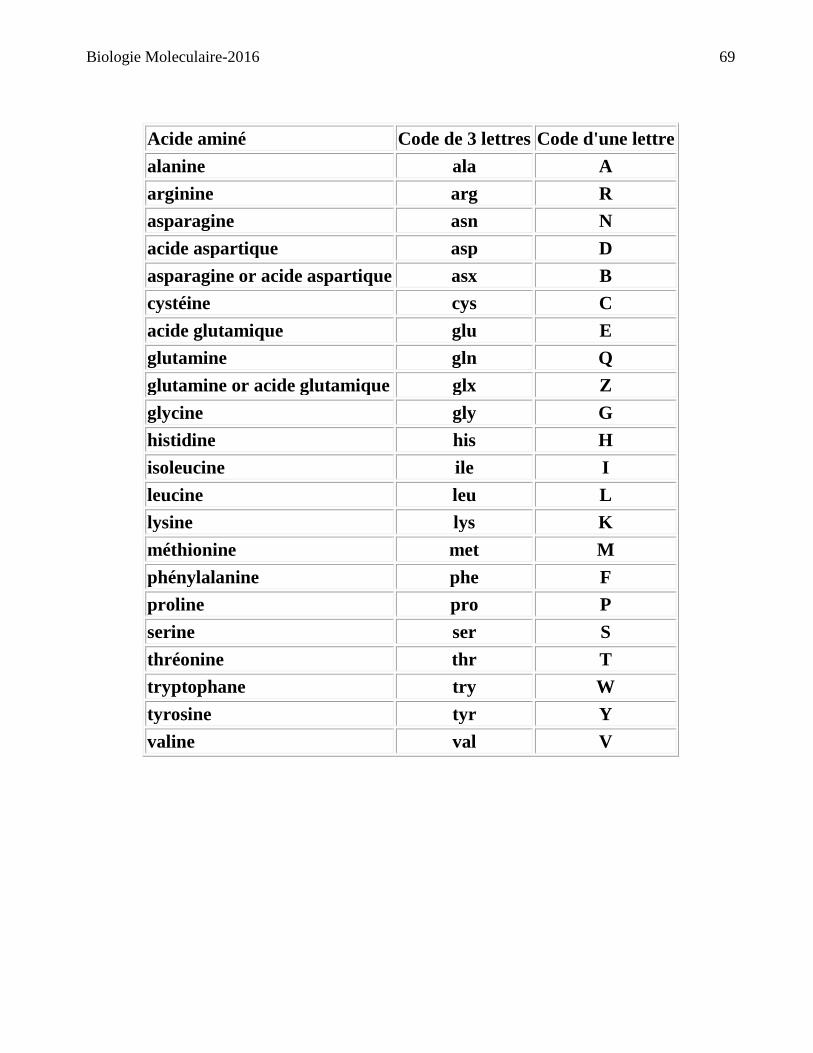
Biologie Moleculaire-2016
69
Acide aminé Code de 3 lettres Code d'une lettre
alanine ala A
arginine arg R
asparagine asn N
acide aspartique asp D
asparagine or acide aspartique asx B
cystéine cys C
acide glutamique glu E
glutamine gln Q
glutamine or acide glutamique glx Z
glycine gly G
histidine his H
isoleucine ile I
leucine leu L
lysine lys K
méthionine met M
phénylalanine phe F
proline pro P
serine ser S
thréonine thr T
tryptophane try W
tyrosine tyr Y
valine val V
Biologie Moleculaire-2016
70
Conversions Spectrophotométrique
1 unité A 260 d'ADN double brin = 50 µg/ml
1 unité A 260 d'ADN simple brin = 33 µg/ml
1 unité A 260 d'ARN simple brin= 40 µg/ml





























