Embed Size (px)
Citation preview

1
Cours : Génomique fonctionnelle et structurale
Niveau : Master 1 de Génétique
Nombre de crédits : 4
Session : 1er Semestre
Horaire : Mardi et Jeudi 8h – 12h et 14 h – 18h
Locaux : Plateau de Médecine
Enseignants : Dr GONEDELE BI Sery Ernest
Dr TIAN BI T. Yves Nathan
BUTS DU COURS
Le cours de génomique fonctionnelle et structurale est conçu de façon à atteindre les principaux buts suivants :
1. Permettre aux étudiants d’acquérir de bases théoriques et pratiques en génomique;
2. Sensibiliser les étudiants aux recherches de pointe dans le domaine de la génomique;
3. Permettre aux étudiants d'obtenir les connaissances nécessaires pour être autonomes dans le traitement de données en génomique.
UFR BIOSCIENCES 22 BP 582 Abidjan 22 Tél. /Fax : 22 44 44 73

2
1. La génomique : présentation générale
Depuis la découverte de son rôle comme support de l''hérédité, l'ADN est sans doute devenue la molécule la plus étudiée au monde. L'information qu'elle renferme est scrutée lettre à lettre pour emplir de gigantesques banques de données. Mais la recherche fondamentale n'est pas la seule à s'y intéresser. De nombreuses applications technologiques se servent de cette "molécule de la vie", complétant l'attirail des biotechnologies. Après la génétique est donc venu le temps de la génomique. L’avènement de la génomique en tant que nouvelle discipline biologique repose essentiellement sur des progrès techniques importants réalisés au cours des années 90. Ces techniques comblent, en effet, en grande partie un fossé qui séparait ce qu’on pouvait identifier au niveau génétique, de ce qu’on pouvait analyser au niveau moléculaire. Classiquement, on savait d’un côté identifier et localiser des caractères génétiques sur les cartes chromosomiques, et de l’autre étudier et modifier des gènes in vitro. Au cours des dix ou quinze dernières années, sont apparues des méthodologies qui permettent de relier ces deux niveaux d’analyse : on peut par exemple plus facilement isoler les gènes à la base des caractères localisés sur les cartes génétiques. Un aspect important des approches de génomique est l’objectif d’exhaustivité qu’elles affichent. Si l’ambition de la génomique structurale est de décrire l’organisation des chromosomes et dresser l’inventaire des gènes qu’ils contiennent, celle de la génomique fonctionnelle est d’attribuer un rôle biologique à ces gènes, de déterminer la façon dont ils sont régulés et leurs interactions. La génomique n’est donc qu’une façon différente d’aborder la génétique, avec des ambitions nouvelles quant aux questions que l’on pose et aux outils qu’on met en place pour y répondre.

3
ECUE 1 : Génomique fonctionnelle
La génétique fonctionnelle s’intéresse à l’expression des gènes, et surtout aux ARN messagers (ARNm), première étape de la cascade conduisant des gènes aux protéines puis aux caractères phénotypiques, par l’intermédiaire des différents métabolismes. Ces dernières années, il est apparu nécessaire d'accéder à des analyses systématiques de “ tous ” les gènes exprimés dans un type cellulaire ou un tissu donné, à un état biologique précis ; ces analyses constituent la génomique fonctionnelle. Elles ont conduit à la notion de “ transcriptome ” ou ensemble des ARNm exprimés par un type cellulaire ou un tissu, et par extension, dans tout l'organisme d'une espèce donnée. Cette approche est d’autant plus intéressante que les études ciblées sur des gènes identifiés participant à des métabolismes spécifiés ont montré leurs limites. L’objectif de telles recherches est d’identifier les gènes qui interviennent dans l’expression d’une fonction, d’un caractère et de comprendre les mécanismes de régulation mis en oeuvre par l’animal dans les diverses situations physiopathologiques auxquelles il est soumis.

4
CHAPITRE 1 : Outils de la génomique fonctionnelle
Introduction
Depuis les années 1980, les biologistes étudient finement l'expression de certains gènes qu'ils ont pu identifier. Cette approche artisanale va s'amplifier et venir compléter la nécessaire description systématique des gènes des organismes vivants.
Les séquences de gènes identifiés peuvent être utilisées comme des sondes moléculaires pour déterminer à quelles périodes de la vie de l'organisme et dans quels organes ces gènes sont exprimés. Cette approche systématique est devenue nécessaire. Un nombre croissant de gènes dont les fonctions étaient totalement inconnues deviendra disponible. Il reste à établir les patrons d'expression de tous ces gènes pour tenter de définir leur rôle. En pratique, l'opération consiste à évaluer systématiquement dans quels types cellulaires sont exprimés les 25 000 gènes humains par exemple. Le patron d'expression d'un génome est appelé le transcriptome. Avec la fin du xxe siècle, le génie génétique est entré dans sa maturité. Les techniques de base, définies dans le courant des années 1960, se sont perfectionnées, simplifiées et en partie automatisées. Les plus importantes sont : le clonage de gènes, l'amplification de gènes (PCR, pour Polymerase Chain Reaction), la détermination de la séquence des bases (nucléotides) des gènes, la synthèse chimique des fragments de gène ou de gènes entiers, la mutation des gènes isolés, la construction de gènes fonctionnels et le transfert de gènes dans des cellules, ou des organismes entiers, pour observer les effets produits ou pour exploiter les organismes modifiés de la sorte. Ainsi, la génomique, c'est-à-dire l'étude des ensembles de gènes qui caractérisent les différentes espèces et définissent leur génome, est passée d'une phase descriptive à une phase d'expérimentation fonctionnelle. Les études de génomique fonctionnelle reposent sur les méthodologies de mesure des niveaux d’expression de “ l’ensemble ” des gènes d’un tissu ou d’un type cellulaire, en réponse à différentes situations biologiques, pathologiques ou non , ou pour différents génotypes.
1. Les puces à ADN (microarray)
Cette technologie a été développée dans le laboratoire de Pat Brown à l’Université de Stanford (Schena, Shalon et al. 1995; Eisen and Brown 1999) et consiste à déposer les sondes sur une lame de verre recouverte d’une substance interagissant de façon stable avec l’ADN. Le support en verre permet de réaliser, grâce à un robot, des dépôts très rapprochés à l’origine d’une densité élevée (6.000 sondes différentes sur moins de 2 cm2 pour une puce du génome de la levure). Les cibles sont marquées le plus souvent par des molécules fluorescentes qui émettent dans des longueurs d’ondes différentes, permettant de comparer directement les expressions de deux populations de cellules, en une seule hybridation. Au final, une mesure d’expression différentielle pour chacun des gènes est obtenue (Figure 1).
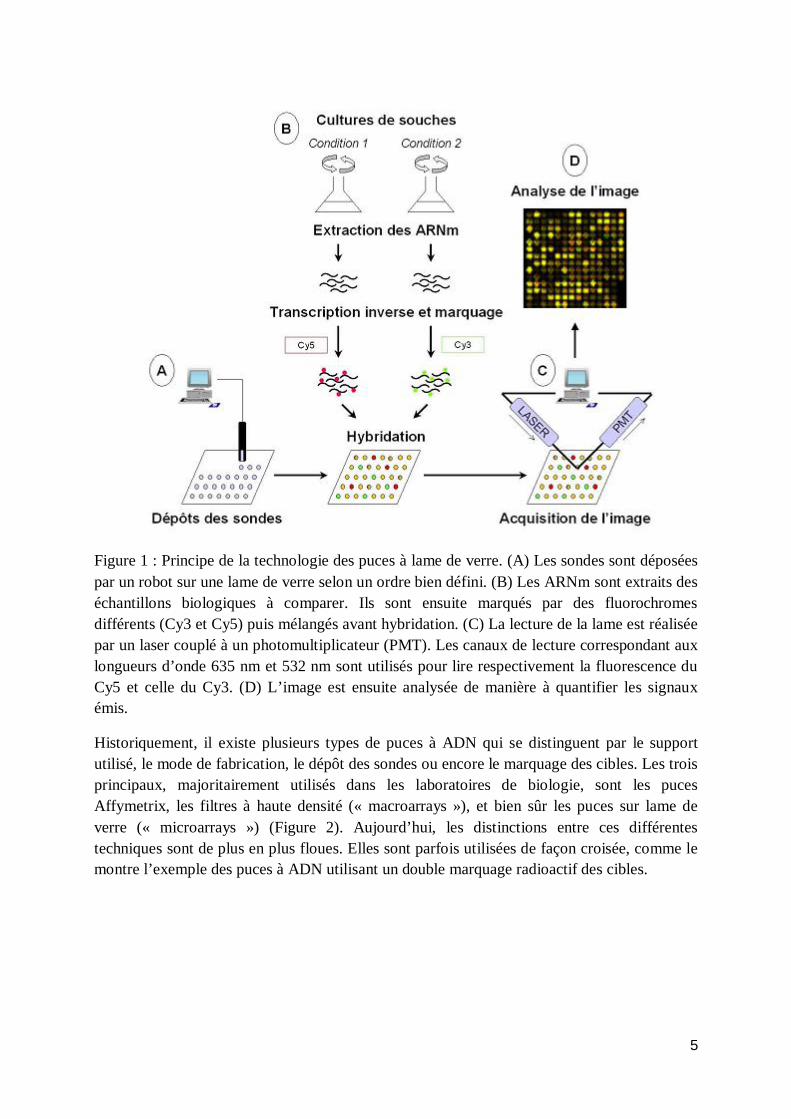
5
Figure 1 : Principe de la technologie des puces à lame de verre. (A) Les sondes sont déposées par un robot sur une lame de verre selon un ordre bien défini. (B) Les ARNm sont extraits des échantillons biologiques à comparer. Ils sont ensuite marqués par des fluorochromes différents (Cy3 et Cy5) puis mélangés avant hybridation. (C) La lecture de la lame est réalisée par un laser couplé à un photomultiplicateur (PMT). Les canaux de lecture correspondant aux longueurs d’onde 635 nm et 532 nm sont utilisés pour lire respectivement la fluorescence du Cy5 et celle du Cy3. (D) L’image est ensuite analysée de manière à quantifier les signaux émis.
Historiquement, il existe plusieurs types de puces à ADN qui se distinguent par le support utilisé, le mode de fabrication, le dépôt des sondes ou encore le marquage des cibles. Les trois principaux, majoritairement utilisés dans les laboratoires de biologie, sont les puces Affymetrix, les filtres à haute densité (« macroarrays »), et bien sûr les puces sur lame de verre (« microarrays ») (Figure 2). Aujourd’hui, les distinctions entre ces différentes techniques sont de plus en plus floues. Elles sont parfois utilisées de façon croisée, comme le montre l’exemple des puces à ADN utilisant un double marquage radioactif des cibles.

6
Figure 2 : Les trois principaux types de puces à ADN, majoritairement utilisés dans les laboratoires. Ces différentes puces se distinguent par la nature du support utilisé, le mode de fabrication et de dépôt des sondes ou encore le marquage des cibles.
I.2. Les EST (Expressed Sequence Tag)
La technique de séquençage d'EST est apparue au début des années 90 chez l'humain et a ensuite été appliquée à un grand nombre d'espèces. On dénombre 44 actuellement, plus de 22 millions d'EST sur 740 espèces au NCBI (dbEST). Les EST sont des séquences générées automatiquement et en simple lecture, à partir de banques d'ADNc (molécule d'ADN rétrotranscrite à partir d'une population d'ARNm). Les banques d'ADNc contiennent des dizaines de milliers de clones représentant une photographie instantanée de l'expression des gènes d'un tissu dans une condition physiologique précise. Le faible relatif coût du séquençage d'EST en fait le moyen le plus attractif pour avoir une description du ranscriptome. Le séquençage d'EST à partir de l'extrémité 5' des ADNc clonés directionnellement a initialement été favorisé. En effet, les séquences 5' sont à même de contenir plus de région codante que celles obtenues à partir de l'extrémité 3' qui contient une partie significative de région non traduite (UTR). A l'heure actuelle, le séquençage à partir de l'extrémité 3' est également pris en compte car il permet d'obtenir plus de séquences uniques et peut être utilisé pour distinguer les gènes paralogues.
3. Le séquençage à haut débit
La méthode de Sanger automatisée est considérée comme étant la stratégie de séquençage haut-débit de première génération. Depuis cinq ans, de nouvelles méthodes ont été mises au point parmi lesquelles le pyroséquençage haut-débit, les techniques de terminaison cyclique réversible (par Solexa/Illumina et Helicos) ou encore le séquençage par ligation (SOliD).

7
Elles ont pour point commun de fournir un grand nombre de données à une vitesse telle que des projets de séquençage demandant plusieurs années avec la technique de Sanger peuvent théoriquement être achevés en quelques semaines. De plus le coût requis pour séquencer un génome est nettement plus réduit. Malgré tous ces avantages, les nouvelles technologies de séquençage présentent certains biais qui rendent leur utilisation complexe. a) Le pyroséquençage haut-débit Le pyroséquençage est une technique de séquençage basée sur la détection du pyrophosphate relâché lors de la réaction de polymérisation de l’ADN. Les nucléotides sont ajoutés les uns après les autres dans un ordre défini. Lorsqu’un nouveau nucléotide est ajouté, l’ATP sulfurylase va utiliser le pyrophosphate relâché lors de la polymérisation pour générer de l’ATP. Cet ATP sera utilisé par la luciférase pour oxyder la luciférine en oxyluciférine et émettre de la lumière (Figure 2). C’est ce signal lumineux qui est détecté par une caméra puis traduit en chromatogramme. A l’aide de cette technique, il est possible de séquencer 500 millions de bases en 10 heures environ, ce qui en fait la technique de séquençage haut-débit commercialisée la plus rapide. Cette vitesse de séquençage peut être atteinte grâce à des améliorations constantes de la processivité des enzymes utilisées lors de la réaction. La grande quantité de séquences générées par cette technique vient en partie de la préparation des échantillons. Chaque fragment est isolé puis amplifié par PCR sur une microbille qui sera ensuite déposée dans le puits d’une plaque de microtitration (il est possible de charger 1 à 2 millions de billes sur une même plaque) (Figure 3). La rapidité de chaque cycle de séquençage permet d’obtenir des lectures d’une taille moyenne de 400 pb. Si un même nucléotide est incorporé dans le même cycle, un signal lumineux proportionnel au nombre de nucléotides est décelé. Au-delà de 4 à 5 nucléotides, le caractère proportionnel du signal est perdu. Ces séquences homopolymériques représentent la source majeure d’erreurs de séquençage par cette méthode, comme l’ont estimé Huse et al. (2007). Ils attribuent 39% des erreurs de type insertions/délétions à ce contexte nucléotidique spécifique.
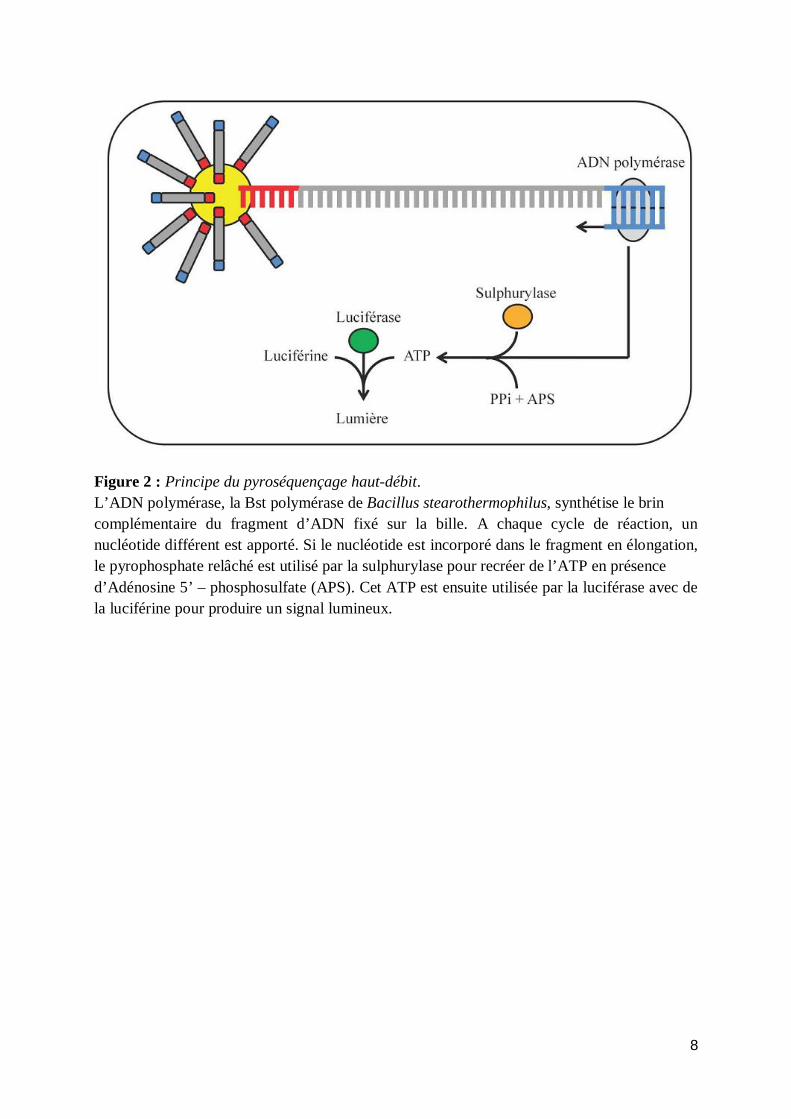
8
Figure 2 : Principe du pyroséquençage haut-débit. L’ADN polymérase, la Bst polymérase de Bacillus stearothermophilus, synthétise le brin complémentaire du fragment d’ADN fixé sur la bille. A chaque cycle de réaction, un nucléotide différent est apporté. Si le nucléotide est incorporé dans le fragment en élongation, le pyrophosphate relâché est utilisé par la sulphurylase pour recréer de l’ATP en présence d’Adénosine 5’ – phosphosulfate (APS). Cet ATP est ensuite utilisée par la luciférase avec de la luciférine pour produire un signal lumineux.

9
Figure 3 : Préparation des fragments d’ADN pour le pyroséquençage haut-débit. A : L’ADN à séquencer est physiquement cassé par nébulisation en fragments de 400 à 1000 pb. B : Des séquences adaptatrices sont ajoutées aux extrémités de chaque fragment obtenu. C : L’amplification des fragments se fait par une PCR en émulsion (le milieu est composé d’eau et d’huile). Chaque bille est isolée dans une micelle (qui va agir comme un microréacteur) avec un unique fragment d’ADN. D : Les fragments possédant un adaptateur sont immobilisés à l’état simple brin sur une bille pour être amplifiés. Chaque bille porte le même fragment d’ADN en plusieurs millions de copies. b) Le séquençage par terminaison cyclique réversible d’Illumina/Solexa Dans le principe de séquençage d’ADN commercialisé par Illumina/Solexa, une ADN polymérase ajoute un nucléotide modifié porteur d’une molécule fluorescente et d’un terminateur labile (figure 4). Chaque nucléotide est porteur d’un fluorochrome spécifique. Suite à l’incorporation, le milieu réactionnel est nettoyé pour supprimer les nucléotides non-incorporés qui sont en excès. Les fluorochromes sont ensuite excités, les émissions lumineuses sont détectées par le séquenceur puis traduites en une séquence. A la fin de

10
chacun des cycles, le fluorochrome et le terminateur en 3’ sont clivés pour que la réaction de séquençage puisse continuer. Par cette méthode, il est possible de séquencer 18 à 35 milliards de bases en 4 à 9 jours. La grande quantité de données générées par cette méthode est due à la préparation des fragments. Chaque fragment est isolé sur une plaque de verre puis amplifié par une réaction de PCR (figure 5). Par cette méthode, il est possible de produire 100 à 200 millions de matrices pouvant être séquencées sur une même lame. Bien que cette technique de séquençage soit à l’heure actuelle l’une des plus utilisées, elle présente plusieurs biais notables. Le principal problème de cette technique est la taille relativement faible des lectures générées, entre 35 et 100 pb, comparées à celles générées par le pyroséquençage, jusqu’à 400 pb, et de la méthode Sanger, jusqu’à 1 kb. L’autre biais de cette technique est la diminution de la fiabilité du séquençage aux extrémités des lectures. Dohm et al. (2008) ont émis l’hypothèse que l’accumulation d’erreurs à l’extrémité 3’ des lectures était due au phénomène de déphasage, qui correspond à l’extension incomplète ou l’addition de multiples nucléotides au fragment séquencé. Ainsi, plus le nombre de cycles augmente, plus les décalages dans la séquence s’accumulent conduisant ainsi à une augmentation du bruit de fluorescence et une interprétation erronée des signaux lumineux.
Figure 4 : Principe du séquençage Illumina/Solexa.

11
A : Des nucléotides marqués à l’aide de fluorochromes différents sont apportés au mélange réactionnel. Une ADN polymérase va les insérer dans le brin complémentaire du fragment séquencé. La présence en 3’ des nucléotides ne permet l’incorporation que d’un seul nucléotide. B : Les fluorochromes sont excités grâce à des lasers permettant l’émission d’un signal lumineux, spécifique à chaque nucléotide, qui sera détecté par une caméra. C : Exemple de signaux détectés par la caméra du séquenceur. Des programmes informatiques vont traduire les signaux en séquence nucléotidique. D : Après détection, le fluorochrome et le terminateur de chaque nucléotides sont retirés pour permettre l’initiation d’un nouveau cycle.
Figure 5 : Préparation des fragments d’ADN pour le séquençage Solexa/Illumina. A : L’ADN à séquencer est fragmenté par nébulisation en fragments d’environ 400 à 500 pb. B : Des séquences adaptatrices sont ajoutées aux extrémités des fragments. C : Les fragments à séquencer sous forme simple brin sont immobilisés sur une plaque de verre à l’aide d’amorces s’hybridant avec un des adaptateurs. D : L’amplification des fragments se fait par pontage à l’aide de l’ADN polymérase. Cela permet l’amplification localisée d’un fragment unique en plusieurs centaines de millions de fragments identiques, appelés « polonie ».

12
Liens Internet et références bibliographiques
Adams et al. 1991. Complementary DNA sequencing: expressed sequence tags and human genome project. Science 252, 1651 - 1656
Boguski et al. 1993. dbEST-database for expressed sequence tags. Nat. Genet. 4, 332 – 333.
Diehl et al. 2001. Manufacturing DNA microarrays of high spot homogeneity and reduced background signal. Nuc. Acids Res. 29, e38
Gibson & Muse. 2004. Précis de génomique",
Glaser P. 2005. Les puces à ADN vont-elles révolutionner l’identification des bactéries? Médecine/Sciences 21 :539-544
Metzker ML. 2010. Sequencing technologies - the next generation. Nat Rev Genet. Jan;11(1):31-46.
Nagaraj et al. 2007. A hitchhiker's guide to expressed sequence tag (EST) analysis. Brief Bioinform. 8, 6 - 21
Pontius et al. 2003. UniGene : a unified view of the transcriptome. The NCBI Handbook - Bethesda (MD) - National Center for Biotechnology Information
dbEST: http://www.ncbi.nlm.nih.gov/dbEST/index.html

13
Chapitre 2 : Annotations fonctionnelles et familles de gène
Introduction
Dans ce chapitre, nous abordons le problème de l’identification de régions codantes. Historiquement, c’est l’un des premiers problèmes sur lequel s’est penchée la communauté bioinformatique. Ce problème constitue une partie importante de l’annotation structurale des génomes : où se trouvent les gènes codant pour des protéines ? Quelle est leur structure ?
Pour répondre à ces questions, les informations utilisées peuvent être de nature différente : la présence de signaux qui balisent et concourent à la structure du gène et à son expression, le contenu de la séquence codante qui peut comporter des biais de composition, ou la similarité avec d’autres molécules connues. Nous regroupons ces informations en deux groupes : les informations intrinsèques, c’est-à-dire les informations contenues dans la séquence nucléique considérée, et les informations extrinsèques, c’est-à-dire les informations obtenues par comparaison de la séquence nucléique d’intérêt avec des séquences déjà connues. Cela donne lieu à deux types d’approche de prédiction : les approches ab initio et les approches par homologie de séquences.
I. Prédiction de gènes : méthodes ab initio
Les approches ab initio ont pour objectif de prédire l’ensemble des gènes présents dans une séquence nucléique sans autre connaissance extérieure. Pour cela, elles tirent parti des signaux présents dans la séquence et des biais de composition des séquences codantes.
1) Le cadre ouvert de lecture
Le premier signal qui peut être exploité provient simplement des bornes des gènes, en particulier pour les organismes procaryotes. Un élément essentiel d’un gène codant est son cadre ouvert de lecture, débutant par un codon START suivi d’un enchaînement ininterrompu de codons et terminé par un codon STOP. Cette information est souvent suffisante pour identifier une bonne partie des gènes au niveau génomique quand le cadre ouvert de lecture est significativement long. L’absence de codon STOP peut cependant être statistiquement peu significative pour des séquences courtes ou lorsque la fréquence de ces codons est localement fortement réduite. A cause de l’épissage chez les eucaryotes et certaines bactéries, rechercher au niveau génomique un codon STOP après un codon START dans le même cadre de lecture n’a pas de sens à cause de la présence des introns.
2) Les autres signaux liés à la structure du gène
Des signaux plus fins que la détection d’un cadre ouvert de lecture peuvent être utilisés pour identifier les séquences codantes. plusieurs signaux balisent et concourent à la structuration d’un gène, que celui-ci soit d’origine eucaryote ou procaryote. Ces signaux interviennent dans les processus mis en œuvre lors de l’expression du gène. Les régions en amont et en aval de la région codante des gènes contiennent ainsi des sites de fixation de facteurs de transcription, le

14
site d’initiation de la transcription, le signal de poly-adénylation, etc. Chez les eucaryotes, d’autres signaux sont présents dans la région codante des gènes et servent à guider l’épissage des introns. La grande majorité de ces signaux sont des motifs relativement courts, dont la longueur est inférieure à une vingtaine de nucléotides. Ce sont des motifs approchés, établis à partir d’un certain nombre d’observations. Ils doivent donc être décrits au moyen d’une représentation plus flexible qu’une simple séquence. Cela pose la question du modèle choisi pour les représenter.
3) Les biais de composition de la séquence codante
A côté des signaux liés à la structure d’un gène, on peut utiliser la composition de la séquence codante elle-même. De façon générale, les régions codantes ont des asymétries et périodicités qui facilitent leur distinction des autres régions. Ces caractéristiques sont propres à chaque espèce. Les premières analyses de régions codantes ont révélé chez certaines bactéries un biais dans l’usage des codons. Du fait de la redondance du code génétique, des codons différents codent pour le même acide aminé. Pour coder un acide aminé, plusieurs organismes affichent une préférence marquée pour un ou plusieurs codons. Pour un organisme, cette préférence est en partie corrélée `a l’abondance de copies des gènes d’ARN de transfert correspondants dans son génome. Plusieurs autres mesures ont par la suite été testées pour caractériser les régions codantes : la fréquence d’apparition des nucléotides, des hexamers, la périodicité d’occurrence des nucléotides, etc. L’étude menée par Fickett [FT92] a permis de mettre en évidence que la fréquence d’apparition des hexamers est la mesure qui discrimine le mieux les régions codantes.
II. Les approches par homologie de séquence
Les méthodes par homologie de séquences utilisent comme première et principale source d’information la similarité avec des séquences connues et déjà annotées. L’hypothèse de travail est la suivante : deux séquences significativement similaires ont généralement une fonction identique ou proche. En effet, durant l’´evolution, les séquences fonctionnelles, a fortiori les séquences codantes, sont soumises `a une contrainte fonctionnelle. Sous cette contrainte, les séquences codantes tendent à être plus conservées entre les espèces que les séquences non fonctionnelles. D’après cette assertion, les informations disponibles à propos d’une séquence connue peuvent être transférées à toute séquence significativement similaire.
Au moins trois types de séquences sont susceptibles d’apporter de l’information pour détecter des régions codantes dans une séquence nucléique : des séquences de protéines, des séquences d’ARN ou d’EST et des s´equences génomiques annotées.

15
Analyse comparative des gènes de β-actine de l'homme et de la carpe
1) Similarité avec des séquences peptidiques
La manière la plus simple et la plus utilisée pour déterminer si une séquence nucléique est codante est de chercher à identifier la protéine qu’elle code, si celle-ci est connue, ou une protéine homologue dans le cas contraire.
Les banques de données les plus utilisées pour effectuer ce type de recherche sont SwissProt et PIR, car elles contiennent exclusivement des séquences de protéines vérifiées expérimentalement. En complément, d’autres banques proposent des TrEMBL contient ainsi uniquement les traductions automatiques des séquences annotées codantes du projet Ensembl où toutes les séquences déjà présentes dans SwissProt sont exclues. Afin de simplifier les recherches, UniProt réunit les banques SwissProt et TrEMBL. Au même titre qu’UniProt, RefSeq propose des séquences vérifiées expérimentalement et des séquences issues de l’annotation automatique basée sur les données et l’expertise des membres du NCBI.
Pour effectuer la recherche dans toutes ces banques, on a recours en première approche `a des algorithmes d’alignement deux à deux, tels que Fasta et Blast. Il existe plusieurs déclinaisons de ces méthodes qui permettent de fouiller une banque de séquences protéiques `a partir d’une séquence nucléique, telles que FastX, FastY et BlastX. L’idée est de comparer indépendamment les six traductions potentielles d’une séquence nucléique contre toutes les séquences d’acides aminés contenues d’une banque.
2) Similarité avec des séquences transcrites
Le second type de séquences auquel on peut faire appel pour identifier des régions codantes sont les séquences d’ARN matures, ou des fragments d’ARN matures. Pour des raisons expérimentales de séquençage, la majorité des séquences d’ARN présentes dans les banques de données comme RefSeq ou dbEST se trouvent sous forme d’ADN complémentaires. Ces

16
ADN complémentaires, notés ADNc, sont obtenus par transcription inverse d’ARN matures. Il existe plusieurs protocoles pour obtenir les séquences d’ADNc rétrotranscrits. Le séquençage “classique” d’un ADNc permet d’en obtenir la séquence complète et ce de manière fiable. Avant que soit mis au point ce protocole, le séquençage des ARN messagers se faisait par un protocole à haut débit moins fiable. Ce protocole consiste à séquencer quelques centaines de nucléotides en une seule fois à chaque extrémité d’un ADNc. Ces fragments nommés des EST, acronyme pour Expressed Sequence Tags, ne représentent donc qu’une information partielle par rapport à la taille de certains ADNc qui peuvent atteindre plusieurs milliers de nucléotides.
3) Similarité avec des séquences génomiques
La similarité avec des séquences génomiques peut également permettre l’identification de régions codantes, même si ces génomes ne sont pas annotés. L’idée est que sous la pression de sélection (section 1.4) les séquences codantes présentent un niveau de conservation plus élevé que les régions non fonctionnelles. Plusieurs protocoles de recherche peuvent être envisagés : une comparaison intra-génomique à la recherche de séquences paralogues, c’est-à-dire de séquences homologues au sein du même génome, ou une comparaison inter-génomique pour trouver des séquences orthologues, c’est-à-dire des séquences homologues chez d’autres organismes. Les comparaisons de séquences peuvent être réalisées au niveau nucléotidique ou au niveau peptidique, en traduisant “à la volée” selon les six cadres de lecture possibles. Quelque soit le niveau de comparaison utilisé, l’exploitation des résultats est relativement plus laborieuse que la comparaison avec des banques de protéines ou d’ARN messagers. En effet, les résultats sont ici bruit´es par la présence d’autres types de séquences conservées dans les génomes que des séquences codantes : des séquences non codantes fonctionnelles telles que des gènes à ARN non-codants ou des éléments répétés, des séquences régulatrices, etc. De plus, la détection d’une ou plusieurs séquences significativement similaires à une séquence nucléique d’intérêt dépend essentiellement des génomes utilisés pour les comparaisons. Dans les faits, les résultats que l’on peut espérer obtenir de comparaisons inter-génomiques varient selon les distances évolutives qui séparent les organismes dont les séquences sont comparées. Entre deux espèces distantes, il est plus facile de discriminer les régions codantes car celles-ci seront significativement plus conservées que le reste des séquences génomiques. Inversement sur deux espèces proches dont les séquences génomiques complètes sont globalement ressemblantes, il est plus difficile de distinguer les régions plus conservées. Enfin, plus la distance évolutive entre les espèces comparées est élevée, plus la recherche de séquences similaires dépend de la sensibilité de la méthode d’alignement utilisée. Le choix de la méthode d’alignement et son paramétrage sont donc des critères importants qui influencent la qualité des résultats obtenus lorsqu’on compare des espèces séparées par une distance évolutive élevée.
III. Les approches par analyse comparative
Les approches de prédiction de gènes codants par analyse comparative travaillent sur des alignements de deux ou plusieurs séquences. L’originalité de ces méthodes est de caractériser des biais liés à l’évolution des séquences codantes observables entre un couple ou une famille

17
de séquences. Ces biais peuvent être de plusieurs nature : la synténie, c’est-à-dire la conservation de l’ordre des gènes entre génomes, un biais de conservation de certaines régions ou encore la caractérisation d’un biais dans les mutations entre des séquences codantes homologues. Syncod est la première méthode qui exploite réellement les biais de mutations entre des séquences codantes homologues. La méthode calcule un ratio entre les mutations silencieuses et les mutations faux-sens observables entre deux cadres ouverts de lecture alignés avec Blast. Les séquences correspondantes sont identifiées comme des séquences codantes homologues si ce ratio est significativement plus élevé que ce qu’on pourrait observer par hasard sur des séquences ayant le même pourcentage d’identité.
Liens Internet et références bibliographiques
Velculescu VE, Zhang L, Vogelstein B, Kinzler KW. 1995. Serial analysis of gene
expression. Science, 270(5235):484-487.
NCBI : http://www.ncbi.nlm.nih.gov/projects/mapview/.
Ensembl : http://www.ensembl.org.
SwissProt http://www.ebi.ac.uk/uniprot/
TrEMBL
UniProt : http://www.uniprot.org/
RefSeq : https://www.ncbi.nlm.nih.gov/refseq/rsg/

18
ECUE 2 : Génomique structurale
Chapitre 1 : Structure des protéines et bases de données structurales
I. Structure des protéines Les protéines peuvent être décrites selon quatre niveaux d’organisation structurale. Une séquence linéaire d’acides aminés, formant une chaîne polypeptidique, constitue la structure primaire de la protéine. Cette structure qui ressemble à un chapelet de « perles » d’acides aminés, est le squelette de la molécule de protéine. Ce squelette se tord et se repli sur lui-même pour donner des niveaux d’organisation moléculaires plus complexes (structures secondaire, tertiaire et quaternaire).
1. Structure primaire et variabilité des protéines
La structure primaire des protéines est représentée par la séquence d’acides aminés qui se lient de manière à former une chaîne polypeptidique. Les propriétés uniques de chaque protéine dépendent des types d’acides aminés qui la composent et de leur séquence. On peut considérer les 20 acides aminés comme un « alphabet » de 20 lettres, utilisé pour construire des « mots » (les protéines). De même, qu’on peut changer le sens d’un mot en remplaçant une lettre par une autre (faire -- foire), on peut créer une nouvelle protéine de fonction différente en remplaçant un acide aminé ou en changeant sa position. Parfois, le nouveau mot n’a aucun sens (faire -- faore), tout comme il arrive que les changements de la combinaison des acides aminés donnent des protéines non fonctionnelles. Exemple : les hémoglobines pathologiques humaines (hémoglobines falciforme S et anémiante C) ne diffèrent de l’hémoglobine normale qu’au niveau du sixième résidu de la chaîne β (remplacement de l’acide glutamique respectivement par la valine et la lysine). Les êtres vivants renferment des milliers de protéines différentes, aux propriétés fonctionnelles distinctes, toutes construites à partir d’une vingtaine d’acides aminés. 2. Structure tridimensionnelle des protéines a) Structure secondaire Les protéines n'existent pas sous forme de chaînes linéaires d'acides aminés : elles se tordent et se replient sur elles-mêmes. C'est leur structure secondaire. La structure secondaire la plus courante est celle de l’hélice alpha (α). Dans l'hélice alpha, la chaîne primaire s'enroule sur ellemême puis est stabilisée par des liaisons hydrogène entre les groupes NH et CO, à tous les quatre acides aminés environ. Le feuillet plissé bêta (β) est une autre structure secondaire, où les chaînes polypeptidiques primaires ne s'enroulent pas mais se lient côte à côte au moyen de liaisons hydrogène et forment une sorte d'échelle pliante. Dans ce type de structure secondaire, les liaisons hydrogène peuvent unir différentes parties d'une même chaîne qui s'est repliée sur elle-même en accordéon ou encore différentes chaînes polypeptidiques. Dans les hélices alpha, les
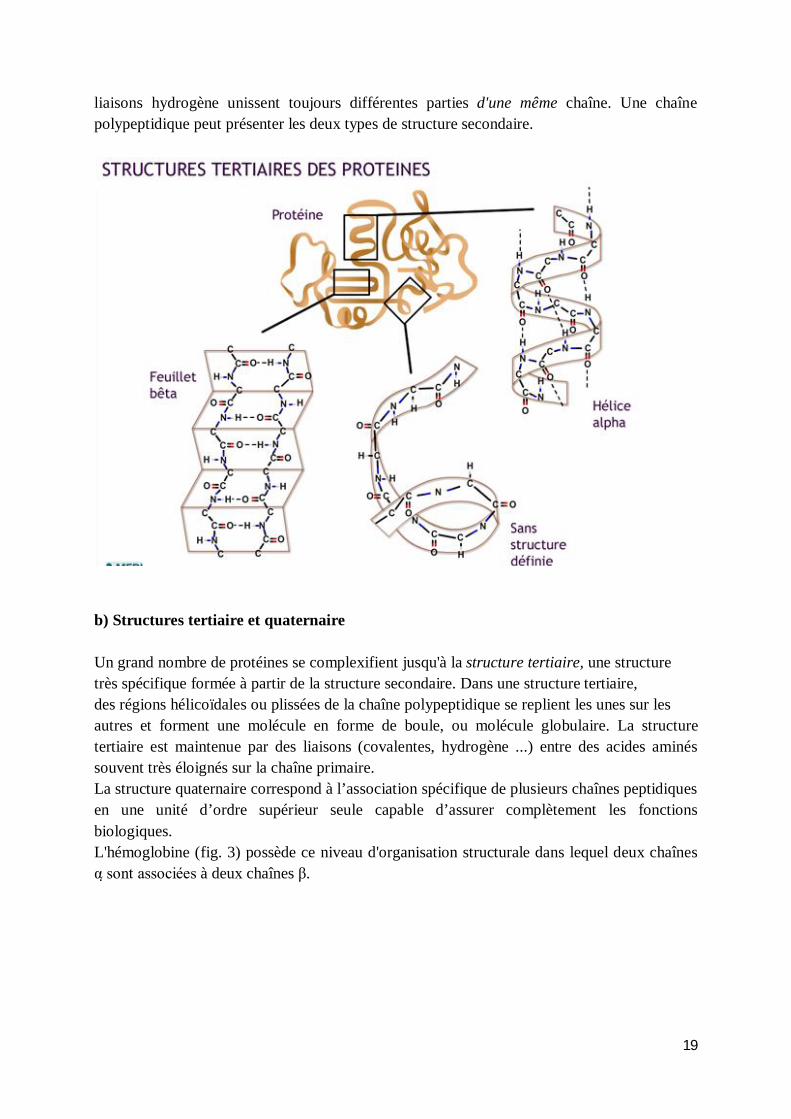
19
liaisons hydrogène unissent toujours différentes parties d'une même chaîne. Une chaîne polypeptidique peut présenter les deux types de structure secondaire.
b) Structures tertiaire et quaternaire Un grand nombre de protéines se complexifient jusqu'à la structure tertiaire, une structure très spécifique formée à partir de la structure secondaire. Dans une structure tertiaire, des régions hélicoïdales ou plissées de la chaîne polypeptidique se replient les unes sur les autres et forment une molécule en forme de boule, ou molécule globulaire. La structure tertiaire est maintenue par des liaisons (covalentes, hydrogène ...) entre des acides aminés souvent très éloignés sur la chaîne primaire. La structure quaternaire correspond à l’association spécifique de plusieurs chaînes peptidiques en une unité d’ordre supérieur seule capable d’assurer complètement les fonctions biologiques. L'hémoglobine (fig. 3) possède ce niveau d'organisation structurale dans lequel deux chaînes α sont associées à deux chaînes β.

20
Figure 3: Structure quaternaire de l’hémoglobine
II. Les bases de données structurales
1) PDB : Protein Data Bank
Les structures des protéines déterminées expérimentalement sont regroupées dans la Protein Data Bank (PDB ; http://www.rcsb.org/PDB/), banque de données crée en 1971. Depuis, sa croissance a été exponentielle, et elle compte actuellement plus de 30 000 structures.
De nombreuses protéines de la PDB présentent des taux d’identité de séquence élevés lorsqu’elles sont alignées deux à deux. Lorsque ce taux d’identité est supérieur à un seuil de 25% à 30%, les protéines ont une forte probabilité d’adopter une même structure 3D. Cette redondance est très gênante pour certaines analyses car elle engendre un biais. En ne considérant que les protéines présentant moins de 25% d’identité de séquence, la banque résultante, alors dite non redondante, ne contient plus qu’environ 2500 structures protéiques.
Enfin, bien que l’évolution du nombre d’entrées dans la PDB soit considérable, elle reste cependant nettement inférieure à celle des banques de données de séquences. Les difficultés de mise en œuvre des méthodes expérimentales de détermination des structures protéiques sont responsables de ce constat, malgré les différents progrès techniques et les tentatives d’automatisation.
2) Classifications des structures protéiques connues
Différentes bases de données ont été développées autour de la PDB établissant une classification des structures protéiques. Les deux principales, SCOP et CATH, sont décrites ci-dessous. Ces classifications visent à rendre compte des relations évolutives entre les protéines. Deux protéines homologues peuvent être identifiées par une forte similitude de séquence, à laquelle est généralement associée une forte similitude de structure. Toutefois, lorsque la similitude de séquence n’est plus détectable entre deux protéines homologues, elles peuvent néanmoins conserver un repliement similaire. En effet, les protéines présentant une relation évolutive distante ont généralement une structure mieux conservée que la séquence.

21
a) SCOP
La base de données SCOP (Structural Classification of Proteins) est une classification hiérarchique des protéines basée sur les ressemblances de leurs structures 3D et sur leurs relations évolutives. SCOP repose essentiellement sur une inspection visuelle des structures protéiques, la classification est réalisée manuellement. Les structures peuvent être constituées d’un ou de plusieurs domaines structurellement indépendants : l’unité de classification utilisée dans SCOP est le domaine.
SCOP présente quatre niveaux hiérarchiques majeurs : (i) famille, (ii) super-famille, (iii) repliement, et (iv) classe. Une famille correspond à un ensemble de domaines protéiques présentant un fort taux d’identité de séquence (supérieur à 30%) ou des structures et fonctions extrêmement proches, et donc pour lesquels une origine évolutive commune est fortement supposée. Une super-famille regroupe des familles dont les protéines présentent des structures et des fonctions similaires, suggérant une origine évolutive commune plus distante entre les différentes familles. Un repliement correspond à des super-familles ayant une même structure de cœur : les mêmes éléments de structure secondaire, arrangés et connectés de manière similaire. Enfin, en fonction de la composition en structures secondaires, quatre principales classes de domaines protéiques sont distinguées : tout alpha (domaines constitués majoritairement d’hélices α), tout bêta (domaines constitués majoritairement de brins β), alpha et bêta (noté α / β ; domaines constitués d’une alternance d’hélices α et de brins β), et enfin alpha plus bêta (noté α + β ; domaines constitués de régions séparées en hélices α et en brins β). SCOP est accessible à l’adresse suivante : http://scop.mrc-lmb.cam.ac.uk/scop.
b) CATH
CATH (Class, Architecture, Topology, Homologous superfamily) correspond également à une classification hiérarchique des domaines protéiques sur la base de leurs similitudes structurales et de leurs relations évolutives. Contrairement à SCOP, elle est réalisée de manière semi-automatique et regroupe les domaines protéiques en quatre niveaux majeurs : (i) Classe protéique, (ii) Architecture, (iii) Topologie, (iv) Homologie (Figure 1-17). La classe protéique dépend de la composition en éléments de structure secondaire et de leur arrangement. L’architecture décrit l’orientation des structures secondaires dans l’espace. La topologie prend en compte la manière dont sont connectés les éléments de structure secondaire. Le dernier niveau, correspondant à celui des super-familles homologues, regroupe des domaines protéiques pour lesquels un ancêtre commun est fortement supposé. La base de données CATH est accessible à l’adresse suivante : http://www.biochem.ucl.ac.uk/bsm/cath_new/
Liens Internet et références bibliographiques
Anfinsen et al. 1961. Proc. Natl. Acad. Sci. USA 47, 1309 - 1314

22
Branden C. & Tooze J. 1996. Introduction à la structure des protéines - - ed. De Boeck Université
Carl-Ivar Brändén & John Tooze. 1996. Introduction à la structure des protéines, De Boeck Université, Bruxelles.
Koga et al. 2012. Principles for designing ideal protein structures. Nature 491, 222 - 227
Lubert Stryer, Jeremy Mark Berg, John L. Tymoczko. 2003. Biochimie, Flammarion, « Médecine-Sciences », Paris, 5e éd.
Ramachandran & Sassiekharan. 1968. Conformation of polypeptides and proteins. Adv. Prot.Chem. 28, p 283 - 437
PDB : Protein Data Bank : http://www.rcsb.org/pdb/home/home.do
SCOP : Structural Classification of Proteins : http://scop.mrc-lmb.cam.ac.uk/scop/

23
Chapitre 2 : Prédiction de la structure des protéines
Introduction
La fonction biologique d'une protéine est intimement liée aux conformations que peut adopter la macromolécule. Par contraste avec la plupart des polymères synthétiques comme le plastique, une protéine n'existe généralement qu'en un seul état natif (autrement dit, de plus basse énergie). Ces états natifs sont atteints dans des conditions typiquement trouvées dans les cellules des êtres vivants (pH quasi-neutre, des températures de 20 à 40 °C., en solution aqueuse). Les protéines peuvent être dénaturées (autrement dit, perdre leur structure tridimensionnelle native) en les plaçant dans des conditions sortant de ce cadre. Toutefois, ce processus est souvent réversible, et de telles molécules peuvent retrouver leur état natif sous des conditions contrôlées. Le rôle pratique de la prédiction de la structure tertiaire de protéines devient de plus en plus important, car nous sommes arrivés à un point où la Science découvre bien plus rapidement de nouvelles structures primaires de protéines (notamment dans le cadre du Projet Génome Humain) qu'elle n'est capable d'en déterminer les structures tertiaires respectives de manière expérimentale (principalement par cristallographie à rayons X et spectroscopie à résonance magnétique nucléaire).
1) Prédiction de la structure d’une protéine à partir de sa séquence
Face aux limites des méthodes expérimentales de détermination des structures protéiques, les méthodes bioinformatiques de prédiction constituent une alternative particulièrement intéressante. Elles reposent sur l’hypothèse d’Anfinsen qui stipule que la structure 3D d’une protéine devrait être déterminée par sa séquence en acides aminés.
Avant d’aborder les différentes méthodes de prédiction des structures tertiaires, il convient de s’intéresser aux très nombreuses méthodes développées pour prédire les structures secondaires des protéines à partir de la séquence. Ce type de prédiction apporte des informations structurales importantes et est souvent considéré comme une première étape vers la prédiction de la structure tertiaire.
2) Prédiction de la structure secondaire
Les méthodes de prédiction des structures secondaires reposent principalement sur l’observation que les différents acides aminés ont des préférences pour certaines structures secondaires. Pour une séquence cible, l’objectif va donc être de prédire pour chaque résidu son état conformationnel. Le taux de prédiction calculé est généralement noté Q3 (3 états possibles : hélice α, brin β ou boucle).
Les premières méthodes de prédiction des structures secondaires développées sont des méthodes statistiques. Il convient ici de rappeler la méthode de Chou et Fasman reposant sur le calcul pour chaque acide aminé d’une probabilité pour chacun des états de structure secondaire. Un taux de prédiction de l’ordre de 52% était obtenu.

24
Différentes améliorations ont ensuite été proposées par des méthodes prenant en compte diverses informations, par exemple sur les résidus environnants), la classe structurale prédite, ou sur des peptides similaires. Les valeurs de Q3 augmentèrent alors jusqu’à environ 65%. Par la suite, des progrès considérables ont été réalisés par des méthodes ne reposant plus sur la seule séquence cible mais utilisant un alignement multiple de séquences similaires. De même, l’utilisation de méthodes d’apprentissage comme les réseaux de neurones a fortement contribué à l’augmentation des taux de prédiction. Ainsi, il convient de citer des méthodes comme PHD, PSIPRED, ou encore SSpro. Les taux de prédiction actuels atteignent des valeurs de l’ordre de 75-80%. Des consensus de plusieurs méthodes peuvent être utilisés et contribuent généralement à l’augmentation des taux de prédiction.
3) Prédiction de la structure tertiaire
Lorsqu’il s’agit de prédire la structure tertiaire d’une protéine à partir de sa séquence en acides aminés, la tâche est beaucoup plus difficile que pour les structures secondaires, et les performances réalisées loin d’être aussi satisfaisantes. Les méthodes de prédiction in silico de la structure 3D des protéines à partir de la séquence en acides aminés sont généralement regroupées en trois grandes catégories : la modélisation par homologie (ou comparative), les méthodes de reconnaissance de repliement (ou d’enfilage) et les méthodes ab initio / de novo. Le choix de la méthode dépend de l’existence ou non dans la PDB d’une protéine de séquence similaire à celle de la protéine à modéliser (ou protéine homologue), et du taux d’identité de séquences entre ces protéines (homologie plus ou moins distante). Des problèmes majeurs sont liés à l’évaluation des performances des méthodes de prédiction développées et à leur comparaison. Une solution à ces problèmes a été la mise en place d’une réunion nommée CASP (Critical Assessment of Structure Prediction) qui a lieu tous les deux ans. Cette dernière est née d’une volonté de tester la fiabilité des méthodes développées en évaluant leurs performances sur un même jeu de protéines, et avec les mêmes critères. Elle consiste à proposer aux différents groupes de recherche d’appliquer leurs méthodes sur des protéines dont la structure vient d’être déterminée expérimentalement mais n’est pas encore déposée dans la PDB, ni publiée. Les méthodes sont donc appliquées en aveugle. A la fin de la compétition, les différents modèles proposés sont évalués par rapport à la vraie structure 3D. La première compétition CASP (ou CASP1) date de 1994. Les résultats publiés les plus récents sont ceux de CASP10 qui a eu lieu en 2012. Cette compétition permet d’évaluer les progrès réalisés au cours des années dans les différentes catégories de méthodes de prédiction de structure. Liens Internet et références bibliographiques
Bairoch A. and Apweiler,R. (2000) The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res., 28, 45–48.
Jones D.T. (1999) Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol., 292, 195–202.
Rost B. (2001) Protein secondary structure prediction continues to rise. J. Struct. Biol., 134, 204–218.

25
CASP : http://predictioncenter.org/
ExPASy (Portail de ressources bioinformatiques sur les protéines): http://www.expasy.org/tools/

26
TD-TP de Génomique fonctionnelle et structurale
Bases de données génomiques Exercice 1
1. Rechercher une séquence de mRNA par mot-clé dans Genbank nucléique, via Entrez (visualisez la fiche genbank et les annotations: numéro GI? numéro d'accession?).
2. Recherchez le produit de ce gène dans Swissprot via SRS. 3. Comparez les fiches dans ces 2 banques de données. Vérifiez consistence (traduction
Genbank = séquence Swissprot)
Exemples: (facteur de transcription TFIIB de C. elegans).
Exercice 2
1. Rendez-vous sur le site du NCBI. 2. Retrouvez les génomes bactériens séquencés, avec leur classification phylogénétique 3. Choisissez E.coli K-12. Cliquez sur le lien "refseq" (numéro commençant par NC) et
cherchez dans la carte génomique la région de l'opéron lactose (lacA, lacY, lacZ,..) 4. A quelle position dans le génome se trouve l'opéron? Quels sont les gènes en amont et
en aval de l'opéron?
Exercice 3
1. Recherchez dans la base Ensembl Human toutes les entrées ayant un rapport avec la maladie d'Alzheimer.
2. Notez les gènes impliqués (combien?) ainsi que les liens OMIM (Base de données de gènes humains associés à des maladies)
3. Retrouvez sur le génome le gène d'apolipoprotéine. Affichez le fragment génomique. Quelle est la longueur du gène? Combien a-t-il d'exons?
4. Récupérez la séquence génomique de 10kb en 5' du gène ("flat file" avec annotations "gene information"). Regardez le fichier produit. Un gène doit se trouver annoté dans cette région.
Recherche dans les banques par similitude de séquence
Exercice 4 Sur le serveur du NCBI, trouvez l'outil BLAST (option basic blast)
o Choisissez le programme blastp contre la banque SWISSPROT

27
o Copiez collez la séquence du prion humain anormal (CJD) puis exécutez la recherche
o Quelles famille (s) de protéines trouvez-vous? o Quelle est l'étendue des pourcentages d'identités rapportés par BLAST?
2. Répétez la recherche contre la banque nr (non-redondante) o Différence entre nr et SWISSPROT?
Alignement multiple de séquences
Exercice 5
o Dans les résultats du BLAST du prion contre SWISSPROT, sélectionnez 10 protéines prions, allant des plus proches aux plus éloignées Obtenez chaque séquence au format FASTA (dans la fiche ENTREZ d'une séquence, changez le format en haut à gauche de Default à FASTA puis cliquez sur Display)
o Ajoutez au fur et à mesure chaque séquence dans un fichier texte (par exemple dans Notepad)
o Sauvez votre liste de séquence au format FASTA 2. Sur le serveur du PBIL, trouvez l'outil CLUSTALW 3. Copiez-collez les 10 séquences au format FASTA du fichier texte
o Alignez les 10 séquences o Repérez les régions conservées/divergentes o Comment trouver une explication fonctionnelle/structurale aux régions
conservées? 4. Sauvegardez cet alignement sur votre disque. Imprimez le si possible
Annotations
Exercice 6
a. Extraire l’information concernant la protéine P36914. Entrez cet identifiant dans la boîte de requête et cliquez « Search ».
b. Ouvrez la fiche correspondante (http://www.uniprot.org/uniprot/P36914)
c. Consultez les annotations pour comprendre la fonction de la protéine.
d. Prêtez une attention particulière à la rubrique « Sequence annotation (features) », qui vous indique les positions des domaines fonctionnels.
e. Dans la section références croisées (« cross-references »), vous trouverez des liens vers la description du domaine CBM20 dans deux bases de données de domaines protéiques (Prosite et Cazy), qui vous indiqueront la fonction de ce domaine.

28
Projet Etudiant
1. Analysez la séquence inconnue suivante:
>cDNA inconnu AATGCAAGTGCATGCATGCATGCATCGGATCGTACGGATTGCAGTTCGGATTCATAATAA ATGCGTAAAAACAGTAGTTTCACTAGTTTCAAAAGTTGCATAATACTTGCTGTTCTTCTT GTTTACCCTAACAGTATGGCTGTTTTCGCTGTTGCTGCTGACGGTATACCTTTCCCTTAC CACGCTAAATACAGTAACGGTGCTATAAGTCCTCTTCACGTTACTCAAAGTAGTGGTAAC AGTAGTGTTAAAGCTGAATGGGAACAATGGAAAAGTGCTCACATAACTAGTGACCTTAAC GGTGCTGGTGGTTACAAATACGTTCAACGTGACATAAACGGTAACACTGACGGTGTTAGT GAAGGTCTTGGTTACGGTCTTATAGCTACTGTTTGCTTCAACGGTGCTGACAGTAACGCT CAAACTCTTTACGACGGTCTTTACAAATACGTTAAAAGTTTCCCTAGTGCTAACAACCCT AACCTTATGGGTTGGCACATAAACAGTAGTAACAACATAACTGAAAAAGACGACGGTATA GGTGCTGCTACTGACGCTGACGAAGACATAGCTGTTAGTCTTATACTTGCTCACAAAAAA TGGGGTACTAGTGGTAAAATAAACTACCTTAAAGCTGCTCGTGACTACATAAACAAAAAC ATATACGCTAAAATGGTTGAACCTAACAACTACACTCTTAAACTTGGTGACATGTGGGGT GGTAACGACTTCAAAAACGCTACTCGTCCTAGTTACTTCGCTCCTGCTCACCTTCGTATA TTCTACGCTTACACTGGTGACAAAGGTTGGATAAACGTTGCTAACAAACTTTACACTACT GTTAACGAAGTTCGTAACAAATACGCTCCTAAAACTGGTCTTCTTCCTGACTGGTGCGCT GCTAACGGTACTCCTGAAAGTGGTCAAAGTTTCGACTACGACTACGACGCTTGCCGTGTT CAACTTCGTACTGCTATAGACTACAGTTGGTACGGTGACGCTCGTGCTGCTGCTCAAAGT GACAAAATGAACAGTTTCATAGCTGCTGACACTGCTAAAAACCCTAGTAACATAAAAGAC GGTTACACTCTTAACGGTAGTAAAATAAGTAGTAACCACAGTGCTAGTTTCTACAGTCCT

29
GCTGCTGCTGCTGCTATGACTGGTACTAACACTGCTTTCGCTAAATGGATAAACAGTGGT TGGGACAAAGTTAAAGACAGTAAAAAATACGGTTACTACGGTGACAGTCTTAAAATGCTT ATAATGCTTTACATAACTGGTAACTTCCCTAACCCTCTTAGTGACCTTAGTAGTCAACCT AGTCCTGGTGACCTTAACGGTGACGGTGAAATAGACGAACTTGACATAGCTGCTCTTAAA AAAGCTATACTTAAACAAAGTACTAGTAACATAAACCTTACTAACGCTGACATGAACCGT
GACGGTGCTATAGACGCTAGTGACTTCGCTATACTTAAAGTTTACCTTTAAT
Rédigez un rapport de synthèse (1 page max) rassemblant les résultats principaux des analyses ainsi que vos hypothèses et conclusions sur la fonction possible de la séquence inconnue