Embed Size (px)
Citation preview

Erreurs diagnostiques en maladies métaboliques: à propos de 5 cas
Anaïs Brassier Hôpital Necker- Enfants Malades Paris
Centre de référence des Maladies Héréditaires du Métabolisme de l’enfant et de l’adulte
Ma.M.E.A SFEIM 21/06/2013

Raphael (1)
• 3ème enfant de parents non apparentés • PN= 4060g (95ep); TN=50 cm; PC=36 cm • H3: première tétée/ dextro=0.76 g/L • H26: enfant geignard, polypnéique avec signes de lutte, dextro blanc,
débord hépatique à 2TD • Bilan : pH=7.11; bicarbonates : 3.6 mmoles/L, lactates=20 mmoles/L; Na
= 147; K=5.2; chlore=103 (TA= 39), Acétest positif à 2 croix • NFS: N; TP=63% • Bilan infectieux négatif • Traitement par perfusion glucosé et bicarbonates de sodium • Normalisation de l’acidose lactique en 24h

Raphael (2) • Explorations métaboliques:
• CAA pl: augmentation isolée de l’alanine (704 µmol/L) et de la proline (827 µmol/L )
• CAO urinaire: acide lactique à 60 888 µmol/mmol créat • Ionogramme sanguin, bilan hépatique, NH3 normaux • Profil des acylcarnitines normal
• Examen clinique strictement normal au décours de l’épisode • Cycle glycémie/lactates:
Avant bib 14h
Avant bib 17h
Avant bib 20h
Avant bib 23h
Avant bib 2h
Glycémie (mmoles/L)
6.9 4.5 6.5 6.0 5.5
Lactates (moles/L)
6.6 2 5.6 2 1.5

Raphael (3)
• Épreuve de tolérance au jeune H6
• Au total: pas d’hypoglycémie,
persistance d’une hyperlactatémie modérée
• À 6 mois et 1 an : croissance
normale/ DPM normale/ 4 repas/jour et lactates normaux
Jeûne H4
Jeûne H6
Lactates
(mmol/L)
2.98 3.92
Pyruvate (mmol/L)
0.14 0.19
L/P 21.3 20.6
AGL (mmol/L) 0.23 0.26
3 OH butyrate sodium(mmol/L)
<0.02 <0.02
Acétoacétate (mmol/L)
0.06 0.08
Glycémie (mmol/L)
3.5 3.0

Raphael (4)
• 14 mois: contexte de GEA • Déshydratation aigue (-15 %); troubles de la conscience; hépatomégalie à 2 TD • Acidose lactique majeure (pH = 6.97; bicarbonates = 4 mmoles/L; lactates = 18
mmoles/L; TA = 31;ASAT= 104 UI/L; ALAT = 95 UI/L; TP:68 %, NH3 normale • CAO urinaire: hyperlactatémie et cétose • Remplissage et perfusion glucosé + bicarbonates; dichloroacétate, vitaminothérapie
par biotine, thiamine, carnitine • Régression de l’acidose lactique en 48h et récupération clinique totale • Bilan d’extension:
IRM cérébrale/Spectro N echo cœur N Biopsie musculaire : déficit partiel du complexe II de la CR
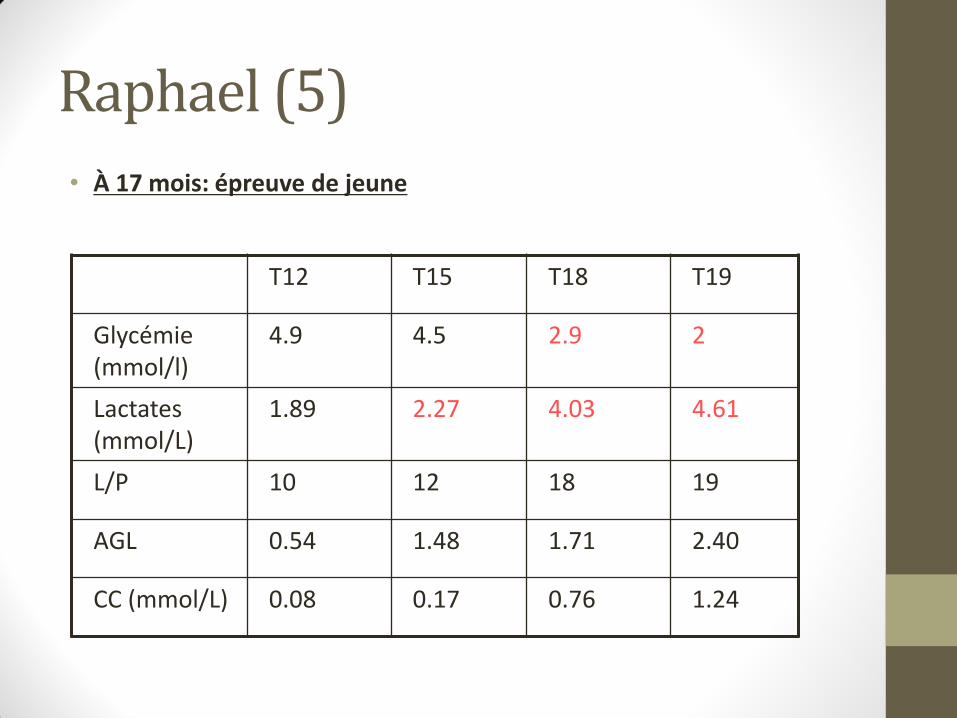
Raphael (5)
• À 17 mois: épreuve de jeune
T12 T15 T18 T19
Glycémie (mmol/l)
4.9 4.5 2.9 2
Lactates (mmol/L)
1.89 2.27 4.03 4.61
L/P 10 12 18 19
AGL 0.54 1.48 1.71 2.40
CC (mmol/L) 0.08 0.17 0.76 1.24

Raphael (6)
• Puis 11 épisodes de décompensations (entre 2 ans et 6 ans) en contexte de jeune: tableau d’acidose lactique avec ou sans hypoglycémie et hépatomégalie
• Entre les épisodes: DPM normal; croissance staturo-pondérale normale,
pas d’hépatomégalie • Persistance d’une hyperlactatémie modérée entre 2 et 3 mmoles/L • À l’âge de 8 ans: « relecture » du dossier: déficit plutôt généralisé de la
CR (déficit secondaire ?); absence de déplétion de l’ADN mt • Suspicion déficit néoglucogénèse
• Diagnostic de déficit en F 1.6 di Phosphatase ( activité enzymatique sur lymphocytes effondrée et confirmation moléculaire )

Melissa (1)
• 2ème enfant de parents apparentés, cousins germains, d’ origine Turque
• Frère aîné en bonne santé
• Grossesse normale
• Accouchement à terme, eutrophe, APGAR 10/10
• Atrésie duodénale opérée à J3 de vie
• DPM normal

Melissa (2)
• 14 mois: GEA à Rotavirus avec déshydratation sévère, somnolence, hépatomégalie modérée et dyspnée d’acidose
• Bilan biologique: acidose lactique sévère (pH= 6.98; bicarbonates= 3 mmoles/L;
lactates= 18 mmoles/L), glycémie à 3 mmoles/L; insuffisance hépatique avec TP = 27 %; Insuffisance rénale fonctionnelle; cytolyse 2N; cétonurie, NH3 ?
• Amélioration avec perfusion glucosé et bicarbonates de sodium • Bilan métabolique: CAA plasmatique (hyperlalaninémie), CAO urinaire (CC ++ et lactaturie), profil
des acylcarnitines normal Points redox: hyperlactatémie permanente modérée avec rapport L/P à 25 Biopsie musculaire: enzymologie de la CR normale Reste du bilan normal (échographie cardiaque, examen ophtalmo, IRM
cérébrale…)

Melissa (3)
• Deuxième épisode identique de coma 1 mois après sans facteur déclenchant identifié
• Poursuite du bilan:
Biopsie hépatique: normale (histo et CR)
Activité pyruvate carboxylase sur fibroblastes normale
• 12 épisodes de décompensation avec acidose lactique profonde au cours d’infection avec vomissements résolutifs en 2 à 3 jours avec perfusion de glucosé et de bicarbonates. Pas de notion de glycémie avant perfusion…

Melissa (4)
• Bilan métabolique: Cycle glycémie/lactates: glycémies normales; hyperlactatémie
modérée permanente entre 2.4 et 3.7 mmoles/L sans hyperpyruvicémie
Séquençage PDHA1: normal Activité E3 normale
• Déficit en fructose 1.6 diphosphatase (dosage enzymatique et
confirmation moléculaire) • Régime d’exclusion fructose et Maizena au coucher • Actuellement 7 ans: va bien /DPM normal/ pas de nouvelle
décompensation

Sacha (1)
• Premier enfant de parents non apparentés en BS
• Développement jugé normal jusqu’à 10 mois
• 10 mois: épisode de régression des acquisitions avec hypotonie globale (perte de la station assise) et troubles de la déglutition en contexte fébrile; Récupération progressive
• Hyperlactacidémie à 4 mmol/l, hyperlactatorrachie à 5.3 mmol/l
• IRM cérébrale normale
• Biopsie musculaire: déficit profond du complexe I de la CR
• Diagnostic:
Cytopathie mitochondriale avec déficit du complexe I

IRM à 2 ans Pic de lactate Atteinte isolée des Pallidum
Evolution inhabituelle: à 2 ans Développement psychomoteur # normal 5 épisodes de régression avec récupération lente, mais complète 2 Episodes de dystonie paroxystique MIG
Sacha (2)
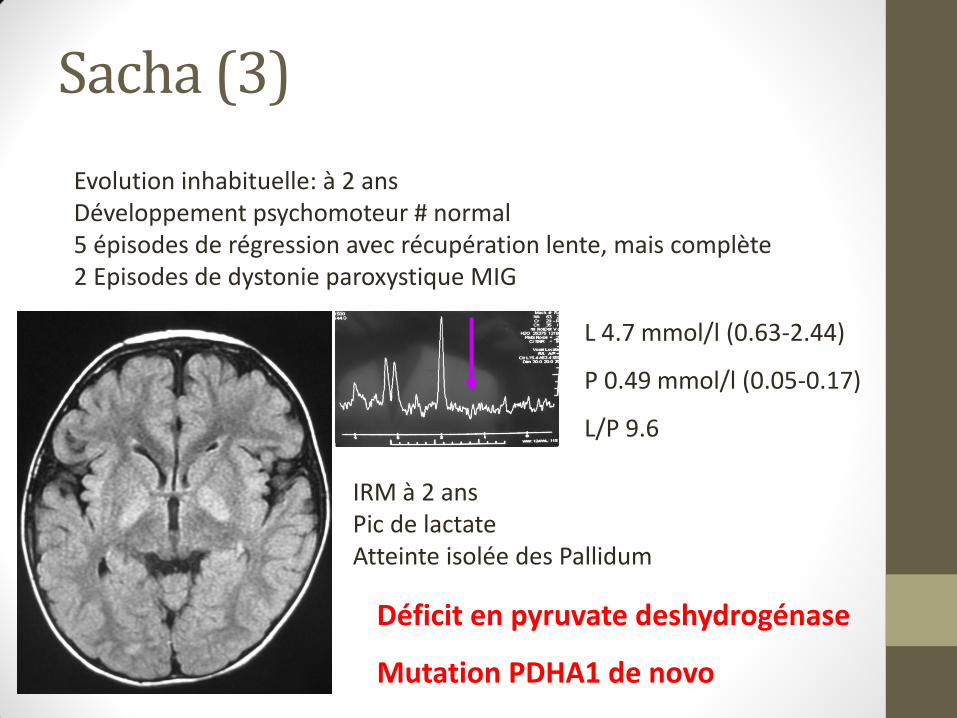
IRM à 2 ans Pic de lactate Atteinte isolée des Pallidum
Déficit en pyruvate deshydrogénase
Mutation PDHA1 de novo
Evolution inhabituelle: à 2 ans Développement psychomoteur # normal 5 épisodes de régression avec récupération lente, mais complète 2 Episodes de dystonie paroxystique MIG
L 4.7 mmol/l (0.63-2.44)
P 0.49 mmol/l (0.05-0.17)
L/P 9.6
Sacha (3)

- Né 40SA PN 3870g TN 51,5 PC 36
- Développement psychomoteur dit normal
- À 23 mois: vomissements répétés depuis 24h/ apathie/ polypnée
Examen clinique:
- déshydratation
- dyspnée de Kussmaul
- examen neurologique normal
- pas d’hépatomégalie
- conscience fluctuante
Biologie:
- Na 148 K 4,2 Cl 118
- Bicar 4,5, TA= 30
- pH= 7,12
- lactates= 11,3 mmol/L
- Ammoniémie: 70 µmol/L
- Glycémie: 1,5g/l
- Corps cétoniques: +++
- Pas d’insuffisance hépatique
-NFS: normale
Loan (1)

A jeun Ap repas
lactate 2,3 3,2
pyruvate 0,12 0,18
L/P 19 18
3OHbutyrate 0,16 0,09
acétoacétate 0,11 0,08
3OHB/AA 1,5 1,1
CAA:
glutamine normale
proline et alanine: normales
CAO:
lactaturie+++
βOH butyrate acétoacétate (cétose+++)
Mais activité pyruvate carboxylase effondrée (non détectable)
déficit en PC (confirmation moléculaire: 2
substitutions à l’état hétérozygote)
CR sur fibroblastes: déficit complexe 4
Loan (2)

Layana (1)
• Née à 41SA eutrophe
• Fièvre maternelle , liquide teinté
• Fièvre à 3 semaines de vie/ AEG/diminution prise alimentaire, Hypotonie globale, HMG sans splénomégalie: Suspicion IMF
• Bilan biologique: cholestase et cytolyse, pas d’IHC, acidose tubulaire, CAA pl et CAO urinaire normales, Profil acylcarnitines normal

Layana (2): 2 mois et demi
• Atteinte multiviscérale • Atteinte rénale: acidose tubulaire
• Atteinte hépatique: HMG avec cholestase et cytolyse
• Atteinte neurologique: retard psychomoteur, atteinte de la SB et atrophie olivopontocérebelleuse
• Atteinte hématologique: CIVD
• Bilan : • Biopsie hépatique: aspect de fibrose portale modérée; cholestase et
stéatose modérée
• WB: electrophorèse de la transferrine: anormale à deux reprises en faveur CDG Syndrome
• classé « CDG I x »

Layana (3): évolution
• 3 ans: crises convulsives (dépakine, dihydan)
• RPM important (à 8 ans: marche instable, dit quelques mots, propreté non acquise, gribouille)
• Troubles du comportement à partir de 7 ans
• Bilan (8 ans): disparition tubulopathie et atteinte hépatique; ERG et FO normaux

Layana (4): évolution
• IRM cérébrale: leucodystrophie importante, pas d’atrophie cérebelleuse
• Dosage polyols urinaires: pic de galactitol à 213 mmol/mol créat (N 2-9) et du galactose urinaire à 59 mmol/mol créat (0-18)
• Confirmation de galatosémie (déficit en GAL 1P et 2 mutations hétérozygotes) avec CDG secondaire…
• Pas de régime alimentaire car Gal 1P normal (3,5 mg/100 ml)
• Normalisation du WB

Au total: une maladie (ou “anomalie”) métabolique peut en cacher une autre…
• Penser au déficit de la néoglucogénèse
déficit en F 1,6 diphosphatase = tableau d’acidose lactique sévère en contexte de jeûne prolongé avec hépatomégalie et +/-cétose même si l’HYPOGLYCEMIE n’est pas au premier plan. DPM le plus souvent normal
• Les déficits de la CR mitochondriale
Diganostic “ d’élimination”
Ne pas s’arrêter à l’enzymologie de la CR
Confirmation moléculaire du diagnostic si possible
Garder à l’esprit les fréquents déficits secondaires de la CR
• Cela est vrai aussi pour le diagnostic de CDG syndrome…penser au trouble de la glycosylation secondaire

Remerciements • L’équipe de métabolisme de Necker: Médecins: P de Lonlay, V Valayannopoulos, G Touati, JB Arnoux Diététiciennes: M Assoun, S Dubois, S Leverge L’équipe paramédicale
• Les biochimistes: D Rabier, C Ottolenghi
• Dr A Boutron (Bicêtre) • Dr M Brivet
• Pr N Seta (Bichat)
• Et toutes les personnes qui se sont occupées des enfants

QUIZZ • Fille 7 ans, pas d’ATCD familiaux
• Diarrhées chroniques (3 selles molles à liquides/jour) depuis la naissance mais croissance staturopondérale régulière sur la moyenne. Bilan allergo, copro, fécalogramme, stéatorrhée, bilan maladie coeliaque: négatif …
• Cataracte sous capsulaire postérieure bilatérale diagnostiquée à l’âge de 6 ans
• Très léger décalage des acquisitions (marche à 16 mois, scolarité normale, mais pataude et lente, petits troubles coordination)
• Examen clinique strictement normal
• Bilan:
• Iono sang, bilan hépatique, EPP, vit A,D,E, TG/cholestérol : N
• CAA pl, CAO urinaire, point redox : N
• CDG: négatif

QUIZZ (suite)
• Diagnostic fait par les parents…
• Dosage de cholestanol plasmatique : 81 μmol/L (N<10)
• Confirmation moléculaire de xanthomatose cérébrotendineuse
• IRM cérébrale normale
• Traitement par acide chénodésoxycholique (disparition de la diarrhée)





























