Embed Size (px)
Citation preview

N° d’ordre 03ISAL 0067 Année 2003
Thèse
Etude des propriétés rhéologiques et de l’état de dispersion de suspensions PDMS/Silice
Présentée devant L’Institut National des Sciences Appliquées de Lyon
Pour obtenir
Le grade de Docteur
École doctorale : École Doctorale Matériaux de Lyon Spécialité : Matériaux Polymères et Composites
Par
Jean-Noël PAQUIEN Ingénieur EEIGM
Soutenue le 3 Décembre 2003 devant la Commission d’examen
Jury de soutenance :
J. GALY Chargé de recherche CNRS LMM/IMP-INSA de Lyon J-F. GERARD Professeur LMM/IMP-INSA de Lyon C. MARTELET Professeur ECL A. POUCHELON Ingénieur de Recherche Rhodia Silicones J.F. TASSIN Professeur Université du Maine H. VAN DAMME Professeur ESPCI Membres invités : G. COSTA Directeur de recherche ISMAC – Genova
Département des Etudes doctorales 3
INSA DE LYON DEPARTEMENT DES ETUDES DOCTORALES MARS 03
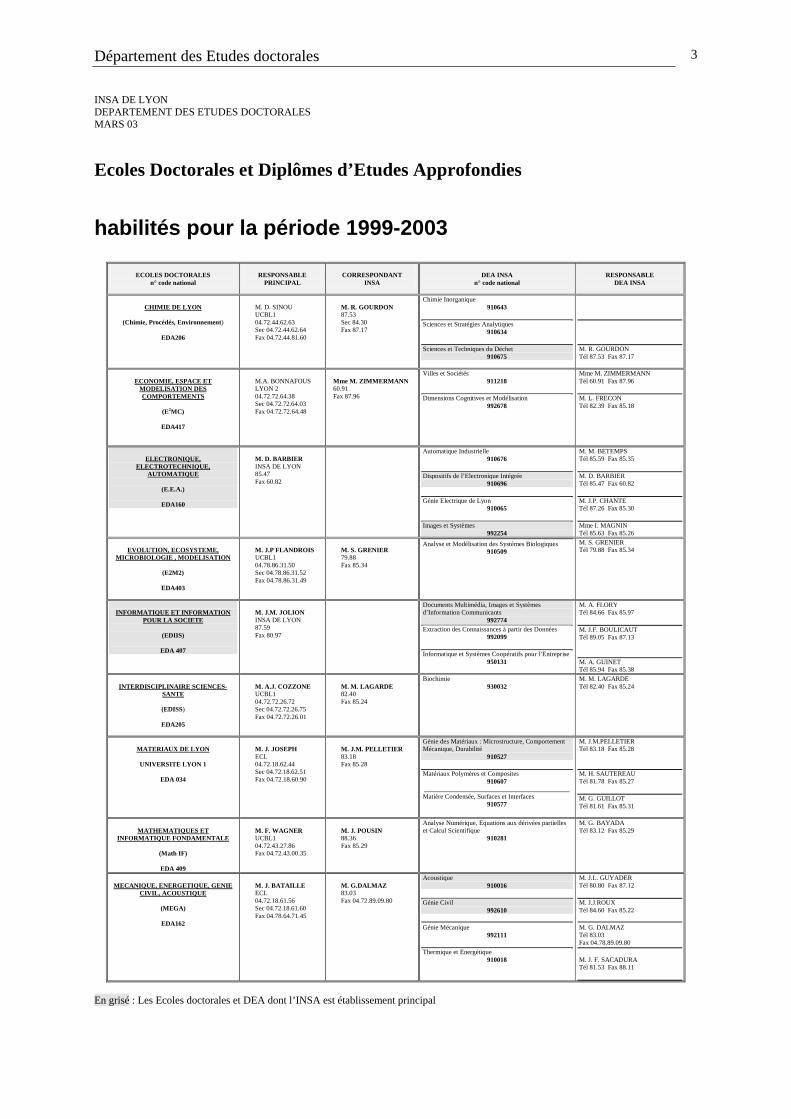
Ecoles Doctorales et Diplômes d’Etudes Approfondies
habilités pour la période 1999-2003
ECOLES DOCTORALES
n° code national
RESPONSABLE
PRINCIPAL
CORRESPONDANT
INSA
DEA INSA
n° code national
RESPONSABLE
DEA INSA
CHIMIE DE LYON
(Chimie, Procédés, Environnement)
EDA206
M. D. SINOU UCBL1 04.72.44.62.63 Sec 04.72.44.62.64 Fax 04.72.44.81.60
M. R. GOURDON 87.53 Sec 84.30 Fax 87.17
Chimie Inorganique 910643
Sciences et Stratégies Analytiques
910634
Sciences et Techniques du Déchet 910675
M. R. GOURDON Tél 87.53 Fax 87.17
ECONOMIE, ESPACE ET
MODELISATION DES COMPORTEMENTS
(E2MC)
EDA417
M.A. BONNAFOUS LYON 2 04.72.72.64.38 Sec 04.72.72.64.03 Fax 04.72.72.64.48
Mme M. ZIMMERMANN 60.91 Fax 87.96
Villes et Sociétés 911218
Dimensions Cognitives et Modélisation
992678
Mme M. ZIMMERMANN Tél 60.91 Fax 87.96 M. L. FRECON Tél 82.39 Fax 85.18
ELECTRONIQUE,
ELECTROTECHNIQUE, AUTOMATIQUE
(E.E.A.)
EDA160
M. D. BARBIER INSA DE LYON 85.47 Fax 60.82
Automatique Industrielle 910676
Dispositifs de l’Electronique Intégrée
910696
Génie Electrique de Lyon 910065
Images et Systèmes
992254
M. M. BETEMPS Tél 85.59 Fax 85.35 M. D. BARBIER Tél 85.47 Fax 60.82 M. J.P. CHANTE Tél 87.26 Fax 85.30 Mme I. MAGNIN Tél 85.63 Fax 85.26
EVOLUTION, ECOSYSTEME,
MICROBIOLOGIE , MODELISATION
(E2M2)
EDA403
M. J.P FLANDROIS UCBL1 04.78.86.31.50 Sec 04.78.86.31.52 Fax 04.78.86.31.49
M. S. GRENIER 79.88 Fax 85.34
Analyse et Modélisation des Systèmes Biologiques 910509
M. S. GRENIER Tél 79.88 Fax 85.34
INFORMATIQUE ET INFORMATION
POUR LA SOCIETE
(EDIIS)
EDA 407
M. J.M. JOLION INSA DE LYON 87.59 Fax 80.97
Documents Multimédia, Images et Systèmes d’Information Communicants
992774 Extraction des Connaissances à partir des Données
992099
Informatique et Systèmes Coopératifs pour l’Entreprise 950131
M. A. FLORY Tél 84.66 Fax 85.97 M. J.F. BOULICAUT Tél 89.05 Fax 87.13 M. A. GUINET Tél 85.94 Fax 85.38
INTERDISCIPLINAIRE SCIENCES-
SANTE
(EDISS)
EDA205
M. A.J. COZZONE UCBL1 04.72.72.26.72 Sec 04.72.72.26.75 Fax 04.72.72.26.01
M. M. LAGARDE 82.40 Fax 85.24
Biochimie 930032
M. M. LAGARDE Tél 82.40 Fax 85.24
MATERIAUX DE LYON
UNIVERSITE LYON 1
EDA 034
M. J. JOSEPH ECL 04.72.18.62.44 Sec 04.72.18.62.51 Fax 04.72.18.60.90
M. J.M. PELLETIER 83.18 Fax 85.28
Génie des Matériaux : Microstructure, Comportement Mécanique, Durabilité
910527
Matériaux Polymères et Composites 910607
____________________________________________ Matière Condensée, Surfaces et Interfaces
910577
M. J.M.PELLETIER Tél 83.18 Fax 85.28 M. H. SAUTEREAU Tél 81.78 Fax 85.27 M. G. GUILLOT Tél 81.61 Fax 85.31
MATHEMATIQUES ET
INFORMATIQUE FONDAMENTALE
(Math IF)
EDA 409
M. F. WAGNER UCBL1 04.72.43.27.86 Fax 04.72.43.00.35
M. J. POUSIN 88.36 Fax 85.29
Analyse Numérique, Equations aux dérivées partielles et Calcul Scientifique
910281
M. G. BAYADA Tél 83.12 Fax 85.29
MECANIQUE, ENERGETIQUE, GENIE
CIVIL, ACOUSTIQUE
(MEGA)
EDA162
M. J. BATAILLE ECL 04.72.18.61.56 Sec 04.72.18.61.60 Fax 04.78.64.71.45
M. G.DALMAZ 83.03 Fax 04.72.89.09.80
Acoustique 910016
Génie Civil
992610 Génie Mécanique
992111
Thermique et Energétique 910018
M. J.L. GUYADER Tél 80.80 Fax 87.12 M. J.J.ROUX Tél 84.60 Fax 85.22 M. G. DALMAZ Tél 83.03 Fax 04.78.89.09.80 M. J. F. SACADURA Tél 81.53 Fax 88.11
En grisé : Les Ecoles doctorales et DEA dont l’INSA est établissement principal

Liste des Professeurs 4
MAI 2003
INSTITUT NATIONAL DES SCIENCES APPLIQUEES DE LYON Directeur : STORCK A. Professeurs : AUDISIO S. PHYSICOCHIMIE INDUSTRIELLE BABOT D. CONT. NON DESTR. PAR RAYONNEMENTS IONISANTS BABOUX J.C. GEMPPM*** BALLAND B. PHYSIQUE DE LA MATIERE BAPTISTE P. PRODUCTIQUE ET INFORMATIQUE DES SYSTEMES MANUFACTURIERS BARBIER D. PHYSIQUE DE LA MATIERE BASTIDE J.P. LAEPSI**** BAYADA G. MECANIQUE DES CONTACTS BENADDA B. LAEPSI**** BETEMPS M. AUTOMATIQUE INDUSTRIELLE BIENNIER F. PRODUCTIQUE ET INFORMATIQUE DES SYSTEMES MANUFACTURIERS BLANCHARD J.M. LAEPSI**** BOISSON C. VIBRATIONS-ACOUSTIQUE BOIVIN M. (Prof. émérite) MECANIQUE DES SOLIDES BOTTA H. UNITE DE RECHERCHE EN GENIE CIVIL - Développement Urbain BOTTA-ZIMMERMANN M. (Mme) UNITE DE RECHERCHE EN GENIE CIVIL - Développement Urbain BOULAYE G. (Prof. émérite) INFORMATIQUE BOYER J.C. MECANIQUE DES SOLIDES BRAU J. CENTRE DE THERMIQUE DE LYON - Thermique du bâtiment BREMOND G. PHYSIQUE DE LA MATIERE BRISSAUD M. GENIE ELECTRIQUE ET FERROELECTRICITE BRUNET M. MECANIQUE DES SOLIDES BRUNIE L. INGENIERIE DES SYSTEMES D’INFORMATION BUREAU J.C. CEGELY* CAVAILLE J.Y. GEMPPM*** CHANTE J.P. CEGELY*- Composants de puissance et applications CHOCAT B. UNITE DE RECHERCHE EN GENIE CIVIL - Hydrologie urbaine COMBESCURE A. MECANIQUE DES CONTACTS COUSIN M. UNITE DE RECHERCHE EN GENIE CIVIL - Structures DAUMAS F. (Mme) CENTRE DE THERMIQUE DE LYON - Energétique et Thermique DOUTHEAU A. CHIMIE ORGANIQUE DUFOUR R. MECANIQUE DES STRUCTURES DUPUY J.C. PHYSIQUE DE LA MATIERE EMPTOZ H. RECONNAISSANCE DE FORMES ET VISION ESNOUF C. GEMPPM*** EYRAUD L. (Prof. émérite) GENIE ELECTRIQUE ET FERROELECTRICITE FANTOZZI G. GEMPPM*** FAVREL J. PRODUCTIQUE ET INFORMATIQUE DES SYSTEMES MANUFACTURIERS FAYARD J.M. BIOLOGIE FONCTIONNELLE, INSECTES ET INTERACTIONS FAYET M. MECANIQUE DES SOLIDES FERRARIS-BESSO G. MECANIQUE DES STRUCTURES FLAMAND L. MECANIQUE DES CONTACTS FLORY A. INGENIERIE DES SYSTEMES D’INFORMATIONS FOUGERES R. GEMPPM*** FOUQUET F. GEMPPM*** FRECON L. REGROUPEMENT DES ENSEIGNANTS CHERCHEURS ISOLES GERARD J.F. INGENIERIE DES MATERIAUX POLYMERES GERMAIN P. LAEPSI**** GIMENEZ G. CREATIS** GOBIN P.F. (Prof. émérite) GEMPPM*** GONNARD P. GENIE ELECTRIQUE ET FERROELECTRICITE GONTRAND M. PHYSIQUE DE LA MATIERE GOUTTE R. (Prof. émérite) CREATIS** GOUJON L. GEMPPM*** GOURDON R. LAEPSI****. GRANGE G. GENIE ELECTRIQUE ET FERROELECTRICITE GUENIN G. GEMPPM*** GUICHARDANT M. BIOCHIMIE ET PHARMACOLOGIE GUILLOT G. PHYSIQUE DE LA MATIERE GUINET A. PRODUCTIQUE ET INFORMATIQUE DES SYSTEMES MANUFACTURIERS GUYADER J.L. VIBRATIONS-ACOUSTIQUE GUYOMAR D. GENIE ELECTRIQUE ET FERROELECTRICITE HEIBIG A. MATHEMATIQUE APPLIQUEES DE LYON JACQUET-RICHARDET G. MECANIQUE DES STRUCTURES JAYET Y. GEMPPM*** JOLION J.M. RECONNAISSANCE DE FORMES ET VISION JULLIEN J.F. UNITE DE RECHERCHE EN GENIE CIVIL - Structures JUTARD A. (Prof. émérite) AUTOMATIQUE INDUSTRIELLE KASTNER R. UNITE DE RECHERCHE EN GENIE CIVIL - Géotechnique KOULOUMDJIAN J. INGENIERIE DES SYSTEMES D’INFORMATION LAGARDE M. BIOCHIMIE ET PHARMACOLOGIE LALANNE M. (Prof. émérite) MECANIQUE DES STRUCTURES LALLEMAND A. CENTRE DE THERMIQUE DE LYON - Energétique et thermique LALLEMAND M. (Mme) CENTRE DE THERMIQUE DE LYON - Energétique et thermique LAUGIER A. PHYSIQUE DE LA MATIERE

Liste des Professeurs 5
Mai 2003 LAUGIER C. BIOCHIMIE ET PHARMACOLOGIE LAURINI R. INFORMATIQUE EN IMAGE ET SYSTEMES D’INFORMATION LEJEUNE P. UNITE MICROBIOLOGIE ET GENETIQUE LUBRECHT A. MECANIQUE DES CONTACTS MASSARD N. INTERACTION COLLABORATIVE TELEFORMATION TELEACTIVITE MAZILLE H. PHYSICOCHIMIE INDUSTRIELLE MERLE P. GEMPPM*** MERLIN J. GEMPPM*** MIGNOTTE A. (Mle) INGENIERIE, INFORMATIQUE INDUSTRIELLE MILLET J.P. PHYSICOCHIMIE INDUSTRIELLE MIRAMOND M. UNITE DE RECHERCHE EN GENIE CIVIL - Hydrologie urbaine MOREL R. MECANIQUE DES FLUIDES ET D’ACOUSTIQUES MOSZKOWICZ P. LAEPSI**** NARDON P. (Prof. émérite) BIOLOGIE FONCTIONNELLE, INSECTES ET INTERACTIONS NIEL E. AUTOMATIQUE INDUSTRIELLE NORTIER P. DREP ODET C. CREATIS** OTTERBEIN M. (Prof. émérite) LAEPSI****
PARIZET E. VIBRATIONS-ACOUSTIQUE PASCAULT J.P. INGENIERIE DES MATERIAUX POLYMERES PAVIC G. VIBRATIONS-ACOUSTIQUE PELLETIER J.M. GEMPPM*** PERA J. UNITE DE RECHERCHE EN GENIE CIVIL - Matériaux PERRIAT P. GEMPPM*** PERRIN J. INTERACTION COLLABORATIVE TELEFORMATION TELEACTIVITE PINARD P. (Prof. émérite) PHYSIQUE DE LA MATIERE PINON J.M. INGENIERIE DES SYSTEMES D’INFORMATION PONCET A. PHYSIQUE DE LA MATIERE POUSIN J. MODELISATION MATHEMATIQUE ET CALCUL SCIENTIFIQUE PREVOT P. INTERACTION COLLABORATIVE TELEFORMATION TELEACTIVITE PROST R. CREATIS** RAYNAUD M. CENTRE DE THERMIQUE DE LYON - Transferts Interfaces et Matériaux REDARCE H. AUTOMATIQUE INDUSTRIELLE RETIF J-M. CEGELY* REYNOUARD J.M. UNITE DE RECHERCHE EN GENIE CIVIL - Structures RIGAL J.F. MECANIQUE DES SOLIDES RIEUTORD E. (Prof. émérite) MECANIQUE DES FLUIDES ROBERT-BAUDOUY J. (Mme) (Prof. émérite) GENETIQUE MOLECULAIRE DES MICROORGANISMES ROUBY D. GEMPPM*** ROUX J.J. CENTRE DE THERMIQUE DE LYON – Thermique de l’Habitat RUBEL P. INGENIERIE DES SYSTEMES D’INFORMATION SACADURA J.F. CENTRE DE THERMIQUE DE LYON - Transferts Interfaces et Matériaux SAUTEREAU H. INGENIERIE DES MATERIAUX POLYMERES SCAVARDA S. AUTOMATIQUE INDUSTRIELLE
SOUIFI A. PHYSIQUE DE LA MATIERE SOUROUILLE J.L. INGENIERIE INFORMATIQUE INDUSTRIELLE THOMASSET D. AUTOMATIQUE INDUSTRIELLE THUDEROZ C. ESCHIL – Equipe Sciences Humaines de l’Insa de Lyon UBEDA S. CENTRE D’INNOV. EN TELECOM ET INTEGRATION DE SERVICES VELEX P. MECANIQUE DES CONTACTS VIGIER G. GEMPPM*** VINCENT A. GEMPPM*** VRAY D. CREATIS** VUILLERMOZ P.L. (Prof. émérite) PHYSIQUE DE LA MATIERE Directeurs de recherche C.N.R.S. : BAIETTO-CARNEIRO M-C. (Mme) MECANIQUE DES CONTACTS ET DES SOLIDES BERTHIER Y. MECANIQUE DES CONTACTS CONDEMINE G. UNITE MICROBIOLOGIE ET GENETIQUE COTTE-PATAT N. (Mme) UNITE MICROBIOLOGIE ET GENETIQUE ESCUDIE D. (Mme) CENTRE DE THERMIQUE DE LYON FRANCIOSI P. GEMPPM*** MANDRAND M.A. (Mme) UNITE MICROBIOLOGIE ET GENETIQUE POUSIN G. BIOLOGIE ET PHARMACOLOGIE ROCHE A. INGENIERIE DES MATERIAUX POLYMERES SEGUELA A. GEMPPM*** Directeurs de recherche I.N.R.A. : FEBVAY G. BIOLOGIE FONCTIONNELLE, INSECTES ET INTERACTIONS GRENIER S. BIOLOGIE FONCTIONNELLE, INSECTES ET INTERACTIONS RAHBE Y. BIOLOGIE FONCTIONNELLE, INSECTES ET INTERACTIONS Directeurs de recherche I.N.S.E.R.M. : PRIGENT A.F. (Mme) BIOLOGIE ET PHARMACOLOGIE MAGNIN I. (Mme) CREATIS** * CEGELY CENTRE DE GENIE ELECTRIQUE DE LYON ** CREATIS CENTRE DE RECHERCHE ET D’APPLICATIONS EN TRAITEMENT DE L’IMAGE ET DU SIGNAL ***GEMPPM GROUPE D'ETUDE METALLURGIE PHYSIQUE ET PHYSIQUE DES MATERIAUX ****LAEPSI LABORATOIRE D’ANALYSE ENVIRONNEMENTALE DES PROCEDES ET SYSTEMES INDUSTRIELS


ETUDE DES PROPRIETES RHEOLOGIQUES ET DE L’ETAT DE DISPERSION DE SUSPENSIONS PDMS/SILICE
RESUME : Le but de cette étude est de comprendre comment les interactions PDMS-silice contrôlent la rhéologie de ces systèmes et d’étudier des additifs polymères permettant de modifier la rhéologie de ces suspensions.
Ces interactions ont été modulées par réaction contrôlée de l’hexaméthyldisilazane avec la surface de la silice. Après une étude approfondie du greffage, la rhéologie des suspensions a été étudiée afin de connaître l’influence des caractéristiques de la silice et du procédé de mise en œuvre.
L’effet d’additifs polymère sur la rhéologie des suspensions a été évalué par mesures de contrainte seuil en mode statique. Un mécanisme de structuration des additifs par séparation de phase avec le milieu PDMS et adsorption à la surface de la silice a été proposé, permettant d’interpréter les résultats obtenus. Cette étude a permis de mieux comprendre les paramètres clés contrôlant les propriétés rhéologiques des suspensions PDMS/Silice et d’identifier les microstructures des additifs conduisant aux propriétés rhéologiques souhaitées. MOTS CLES : PDMS – silice – greffage – HMDS – rhéologie – interactions – suspensions – contrainte seuil
RHEOLOGICAL PROPERTIES AND DISPERSION OF PDMS/SILICA SUSPENSIONS
ABSTRACT The aim of the study is to understand how PDMS – silica interactions control the rheology of these systems and to study polymeric additives which modify the rheology of these suspensions. These interactions were modulated by controlled reaction of hexamethyldisilazane with silica surface. After a study of silica grafting, the rheology of suspensions was investigated in order to determine the influence of silica characteristics and processing. The impact of polymeric additives on suspensions rheology was evaluated by yield stress measurements in static mode. A mechanism of additives structuration by phase separation with PDMS medium and adsorption on silica surface was proposed to interpret the results. This study allows a better knowledge of the key parameters controlling the PDMS/silica suspensions rheology and an identification of additives microstructures leading to desired rheological properties. KEYWORDS : PDMS – silica – grafting – HMDS – rheology – interactions – suspensions – yield stress

Remerciements 7
A ma famille
A Catherine

Remerciements 8

Remerciements 9
REMERCIEMENTS
Ce travail a été effectué au Laboratoire des Matériaux Macromoléculaires au sein de l’UMR 5627, en collaboration avec le laboratoire Synthèse et Formulation de la société Rhodia silicones. J’adresse mes remerciements à Jean-François Gérard et à Jocelyne Galy, pour la confiance qu’ils m’ont accordé tout au long de ce projet, ainsi que pour leurs précieux conseils et discussions. Je suis sensible à l’intérêt que Messieurs J.F. Tassin et H. Van Damme, professeurs à l’Université du Maine et à l’Ecole Supérieure de Physique et de Chimie Industrielles, ont porté à ce travail en acceptant d’en être les rapporteurs. Merci également à N. Jaffrezic-Renault, directeur de recherche CNRS à l’Ecole Centrale de Lyon, d’avoir accepté de participer au jury. Je tiens à remercier la société Rhodia qui est à l’initiative de ce sujet de thèse, et plus particulièrement A. Pouchelon pour son encadrement au cours de ces trois années, J.-M. Pujol pour son suivi, Pascale Meynard pour son aide dans le laboratoire Synthèse et formulation et son immense gentillesse, Ludovic Odoni pour l’intérêt et la confiance portés à mon travail. Cette étude faisant l’objet d’une bourse BDI CNRS je souhaite remercier le CNRS en la personne de Jean-Claude Bernier, directeur du département Sciences Chimiques. Je remercie vivement le Pr. Slusarski et l’ensemble des membres de son laboratoire pour leur acceuil à l’Institut des Polymères de l’Université polytechnique de lodz, ainsi que Piotr Glab et Lucas Kaczmarek avec lesquels j’ai partagé de très bons moments à Lodz durant ce séjour. Cette collaboration s’est faite dans le cadre du programme PICS CNRS n°617 dont je tiens à remercier les responsables : Andrzej Galeski (CMMS, Académie des Sciences Polonaises, Lodz) et Gisèle Boiteux (UCB Lyon I, UMR 5627 CNRS).

Remerciements 10
Je voudrais exprimer ma gratitude au Dr. Costa de m’avoir acceuilli à Gênes au sein de son laboratoire ISMAC du CNR, et à Lucia Conzatti, Maila Castellano, Luciano Falqui pour leur aide scientifique et technique et le séjour formidable qu’ils m’ont permit de passer. Mes remerciements vont également au Pr. Turturro du département Chimie et Chimie Industrielle de l’Université de Gênes pour le suivi de mes expériences d’IGC. Je tiens à dire combien l’aide de Najiba Douja pendant son ATER m’a été précieuse, en particulier concernant les analyses RMN du solide, pour lesquelles je remercie également M.F. Laurro (Service RMN/FRPL), C. Lorentz (IRC) et F. Lefebvre (LCOMS). Un grand merci aussi à toutes les personnes qui m’ont apporté leur aide : Annie Rivoire (Centre Technologique des Microstructures) pour la microscopie electronique à transmission, Jannick Duchet (LMM) pour le greffage de silice et l’AFM, le Pr. Bernango et Batoule Smatti (Centre de Quantimétrie de la Faculté de Médecine de Lyon) pour l’analyse d’image, Vincent Martin (LMOPS) pour les experiences de diffusion de lumière, Philippe Vergne (LMC-INSA Lyon) pour les essais de rhéologie, Yann Mollard pour son travail d’ATER en modélisation moléculaire, complémentaire de mon sujet. Pour finir mes pensées vont à toutes les personnes du laboratoire qui m’ont apporté leur aide et leur amitié durant ces trois années passées au laboratoire.


Sommaire 11
SOMMAIRE INTRODUCTION GENERALE...............................................................15 CHAPITRE I : GENERALITES…………………………………………..19 I.1- LES SILICONES…………………………………………………………………….20 I.1.1- PRÉSENTATION GÉNÉRALE…………………………………………………………………………………20
I.1.2- LE POLYDIMÉTHYLSILOXANE (PDMS)…………………………………………………………………..23
I.1.3- LES ÉLASTOMÈRES SILICONES…………………………………………………………………………….25 I.2- LA SILICE COLLOIDALE………………………………………………………..28 I.2.1- INTRODUCTION………………………………………………………………………………..………………28
I.2.2- LES SILICES DE SYNTHÈSE………………………………………………………………………………….28
I.2.3- SURFACE ET CHIMIE DE SURFACE DES SILICES...…………………………………..………..…..34 I.3- CONCLUSIONS……………………………………………………………………..49 CHAPITRE II : MODIFICATION CHIMIQUE DE SURFACES DE SILICE PAR L’HEXAMETHYLDISILAZANE ET CARACTERISATION DU GREFFAGE……..………………………55 II.1- INTRODUCTION…………………………………………………………………..56 II.2- ETUDE BIBLIOGRAPHIQUE……………………………………………………57 II.2.1- MODIFICATION CHIMIQUE DE LA SURFACE DE LA SILICE PAR GREFFAGE D’UN ORGANOSILANE…………...…………………..……………………………………………..57
II.2.2- TAUX DE GREFFAGE DES SURFACES GREFFÉES PAR RÉACTION AVEC L’HMDS…………60
II.2.3- CONCLUSIONS…………………………………………………………………………………………………63 II.3- PARTIE EXPÉRIMENTALE……………………………………………………..65 II.3.1- MATÉRIAUX UTILISÉS…………………………………………………..…………………………………..65
II.3.2- MODIFICATION CHIMIQUE PAR VOIE SOLVANT (EX-SITU)……………………..………………65 II.3.3- MODIFICATION CHIMIQUE PAR VOIE GAZEUSE (EX-SITU)………..…………………………...67

Sommaire 12
II.3.4- MODIFICATION CHIMIQUE AU SEIN DES SUSPENSIONS PDMS/SILICE ( VOIE IN-SITU)..69
II.3.5- CARACTÉRISATION DES SURFACES MODIFIÉES…………………………………………………...70 II.4- RÉSULTATS ET DISCUSSION………………………………………………….72 II.4.1- EVOLUTION DU GREFFAGE EN FONCTION DE LA QUANTITÉ DE SILANE INTRODUIT ET COMPARAISON DES TAUX DE GREFFAGE VOIE SOLVANT/VOIE GAZEUSE….........................72
II.4.2- COMPARAISON DES TAUX DE GREFFAGE DES SILICES MODIFIÉE IN-SITU ET EX-SITU…..….…………………………………………………………………………………………...84 II.5- CONCLUSIONS……………………………………………………………………89 CHAPITRE III : ETUDE DES PROPRIETES RHEOLOGIQUES ET DE L’ETAT DE DISPERSION DE SUSPENSIONS PDMS/SILICE…..93 III.1- INTRODUCTION…………………………………………………………………94 III.2- ETUDE BIBLIOGRAPHIQUE…………………………………………………..95 III.2.1- DÉFINITIONS………………………………………………………………………………………………….95
III.2.2- ETAT DE DISPERSION ET PROPRIÉTÉS RHÉOLOGIQUES DES SUSPENSIONS PDMS/SILICE……………………………………..………………………………….…………….97
III.2.3- CONCLUSIONS……………………………………..………………………………………………………106 III.3- PARTIE EXPÉRIMENTALE…………………………………………………..107 III.3.1- MATÉRIAUX…………………………………………………..……………………………………………..107
III.3.2- MISE EN ŒUVRE DES SUSPENSIONS PDMS/SILICE…………………………………………..…107
III.3.3- CARACTÉRISATION DES SUSPENSIONS…………………………………………………………..…111 III.4- RÉSULTATS ET DISCUSSION……………………..…………………………115 III.4.1- DESCRIPTION DU COMPORTEMENT RHÉOLOGIQUE GÉNÉRAL DES SUSPENSIONS PDMS/SILICE……………………………………………………..………………………………115
III.4.2- COMPORTEMENT RHÉOLOGIQUE ET ÉTAT DE DISPERSION DES SUSPENSIONS RÉALISÉES IN-SITU EN 1 ÉTAPE (SÉRIE Z)…………………………..…………………..121
III.4.3- INFLUENCE DU PROCÉDÉ DE MISE EN ŒUVRE DES SUSPENSIONS…………………..….134
III.4.4- INFLUENCE DE LA MASSE MOLAIRE DU PDMS SUR LES PROPRIÉTÉS RHÉOLOGIQUES DES SUSPENSIONS……………………………………………………..…139
III.4.5- MODÉLISATION RHÉOLOGIQUE…………………………………………………………………..….140 III.5- CONCLUSIONS………………………………………………………………….148

Sommaire 13
CHAPITRE IV : POLYMERES MODIFICATEURS DE LA RHEOLOGIE DES SUSPENSIONS PDMS/SILICE……………....156 IV.1- INTRODUCTION………………………………………………………………..157 IV.2- ETUDE BIBLIOGRAPHIQUE…………………………………………………159 IV.3- PARTIE EXPERIMENTALE…………………………………………………..164 IV.3.1- ADDITIFS UTILISES ET NOMENCLATURE…………………………………………..………………164
IV.3.2- MODE D’INTRODUCTION DES ADDITIFS ET CARACTERISATION DES GELS FORMES…………………………………………………………………..……………………………..168 IV.4- RESULTATS ET DISCUSSION………………………………………………..170 IV.4.1- DESCRIPTION GENERALE DES GELS…………………………………………………..…………….170
IV.4.2- INFLUENCE DES PARAMETRES DE LA FORMULATION…………………………..…………….177
IV.4.3- INFLUENCE DE LA MICROSTRUCTURE DES ADDITIFS…………………………..…………….184
IV.4.4- ETUDE PAR MICROSCOPIE DES SUSPENSIONS ET MECANISMES DE STRUCTURATION PROPOSES…………………………….……………………..……….195
IV.4.5- MESURE DES ENERGIES D’INTERACTIONS…………………………………………………...……209 IV.5- CONCLUSIONS ET PERSPECTIVES………………………………………...238 CONCLUSION GENERALE……………………………….…………….243 ANNEXES……………………………………………….………………….247 ANNEXE A : DILUTION DES SUSPENSIONS………………………………………………………..…………246
ANNEXE B : MESURES RHEOLOGIQUES……………………………………………………..……………….247
ANNEXE C : MICROSCOPIES TEM ET AFM………………………………………………...…………………249
ANNEXE D : DIFFUSION DYNAMIQUE DE LA LUMIERE……………………...............................…..253
ANNEXE E : CONTRAINTES SEUIL DES SUSPENSIONS (SERIE Z)…………..………………………….256
ANNEXE F : DETERMINATION DES PENTES ET DES FRACTIONS VOLUMIQUES CRITIQUES....257
ANNEXE G : ANALYSE DE LA COMPOSITION CHIMIQUE DES COPOLYMERES PDMS / POLYETHER…………………………………..………………………………………260

Sommaire 14

INTRODUCTION GENERALE

Introduction générale
15
La rhéologie des suspensions constitue un sujet de recherche important dans le domaine de la rhéologie, puisqu’elle conditionne une large gamme d’applications industrielles, telles que les ciments, les peintures, les encres… En ce qui concerne les matériaux silicones, les suspensions de silice dans les polydiméthylsiloxanes (PDMS) constituent la base des formulations de pâtes et élastomères silicones, représentant un grand nombre d’applications. Les pâtes sont principalement utilisées comme produits d’étanchéité et d’isolation électrique, comme revêtements protecteurs et comme agents de démoulage. Les élastomères silicones servent au collage, au moulage, à assurer l’étanchéité et la protection… Le rôle de la silice est d’améliorer les propriétés mécaniques du polymère : la résistance en traction par exemple passe de 0,3 à 14 MPa lorsque le réseau élastomère initialement non chargé est renforcé par de la silice [1]. La rhéologie des suspensions PDMS/silice est déterminante vis-à-vis des propriétés finales des mélanges. De nombreux paramètres régissent ces propriétés rhéologiques, comme la nature des constituants (masse molaire et fonctionnalité du PDMS, type de silice…) ou le procédé de mise en œuvre de celles-ci. Cependant l’un des facteurs essentiels est le degré d’interaction entre les chaînes PDMS et la charge de silice. Ces interactions initialement fortes sont dues à la formation de liaisons hydrogène entre les atomes d’oxygène des chaînes PDMS et les groupements hydroxyle de surface de la silice (Figure 1) [2]. Elles conduisent au renforcement mécanique des élastomères mais aussi à un certain nombre de difficultés, en particulier des problèmes de mise en œuvre liés à une viscosité trop importante et des problèmes de vieillissement des élastomères. L’une des solutions consiste alors à modifier la surface de la silice pour diminuer le nombre de groupements hydroxyle de surface et donc l’intensité des interactions. Cette modification est généralement réalisée par réaction (greffage) d’un organosilane avec les groupements silanol de surface de la silice pour conduire à ‘écranter’ ces interactions par liaisons H. En effet, la silice déployant une grande surface spécifique (environ 200 m2.g-1), les interactions aux interfaces deviennent essentielles.
Figure 1: Interactions PDMS - silice par liaison hydrogène
Afin de mieux comprendre l’influence de la modification de surface de la silice sur les propriétés des mélanges, nous avons réalisé des systèmes modèles PDMS - silice, c’est-à-dire
Si
O
Si Si
H
Silice
chaîne PDMS OSi
OSi
OSi
O
O OO O
O
H
O
H

Introduction générale
16
simplifiés par rapport aux formulations industrielles, avec des silices de degrés de modification de surface (hydrophobisation) contrôlés. Ceci nous permettra de mettre en évidence la corrélation entre le degré de modification des silices et les propriétés rhéologiques des mélanges, une étape importante pour pouvoir ensuite prévoir et contrôler la rhéologie complexe de ces suspensions PDMS-silice. Nous verrons aussi comment l’état de dispersion de la silice au sein du PDMS est affecté lorsque les interactions sont modulées et comment relier cet état de dispersion à la rhéologie des mélanges. Obtenir des silices de taux de greffage contrôlé étant crucial dans le cadre de ces objectifs, cette étape a fait l’objet d’une étude approfondie du greffage, sa caractérisation et la quantification des taux de greffage. Cette partie nous permet d’approfondir nos connaissances sur les propriétés physico-chimiques de la silice et sur le greffage de silice, en comparant le greffage in-situ de la silice, c'est-à-dire réalisé directement au sein des mélanges PDMS - silice, avec le greffage ex-situ de la silice, réalisé avant incorporation au PDMS. D’autre part, ces systèmes modèles constituent la base de la suite de notre travail sur l’étude des polymères modificateurs de la rhéologie des mélanges. Ces additifs ont pour but de conférer à des suspensions PDMS/silice initialement fluides, une contrainte à seuil permettant au produit de ne pas s’écouler. L’aspect pratique est d’éviter un écoulement lors de l’application de ces formulations sur des surfaces non horizontales. L’objectif est d’identifier les mécanismes d’action de ces molécules ou macromolécules, ainsi que les paramètres moléculaires pertinents (structure chimique, fonctionnalité…) qui permettront ensuite de classer les additifs et prévoir leur efficacité, ceci en corrélation avec les paramètres initiaux des mélanges (taux de greffage, fraction volumique de silice). Ce travail doit donc permettre dans un premier temps de mieux comprendre et prévoir les propriétés rhéologiques des suspensions PDMS/silice, en fonction du taux de greffage et de la fraction volumique de silice, puis d’évaluer et classer des additifs pour une modification ultérieure de la rhéologie des suspensions, ce qui passe par la compréhension de leur mode d’action. En conséquence, ce mémoire est organisé de la manière suivante : Le chapitre I est destiné à rappeler les notions de base sur les élastomères silicones, le PDMS et la silice, puis sur les suspensions PDMS/silice en mettant l’accent sur les interactions polymère - charge. Le chapitre II concerne l’étude de la modification chimique de surfaces de silice par réaction des groupements hydroxyle de surface avec un organosilane, plus précisément l’hexaméthyldisilazane (HMDS), et la caractérisation des surfaces modifiées par plusieurs

Introduction générale
17
techniques d’analyse : infrarouge à transformée de Fourier (IRTF), analyse élémentaire du carbone, résonance magnétique nucléaire du silicium solide (RMN 29Si CP/MAS), mesures de mouillage. Plusieurs procédures de greffage seront envisagées : i/des silices greffées in situ au sein du PDMS, aussi celles-ci seront analysées après extraction des suspensions, ii/ des silices greffées ex-situ, c’est à dire en solution ou par voie gazeuse. Le chapitre III aborde l’élaboration et la caractérisation des suspensions PDMS/silice. Les propriétés rhéologiques des mélanges, caractérisées en régime statique et dynamique, seront corrélées à la fraction volumique et au taux de greffage de silice, afin de modéliser le comportement des suspensions PDMS/silice. La microstructure des mélanges observée par microscopie électronique à transmission (TEM) et microscopie à force atomique (AFM), sera reliée aux paramètres de formulation et aux propriétés rhéologiques. Enfin, le chapitre IV sera consacré aux polymères modificateurs de la rhéologie initiale des suspensions. Ces polymères, souvent des copolymères de structure complexe, à base de polyoxyéthylène (POE), polyoxypropylène (POP) et polydiméthylsiloxane (PDMS), seront modélisés par des homopolymères POE, POP et PDMS, ayant des groupements de fin de chaine de natures différentes, afin de comprendre dans un premier temps quels sont les paramètres microstructuraux déterminant leur aptitude à influencer le comportement rhéologique des suspensions. Cette étude s’accompagnera d’une compréhension des mécanismes d’action de ces additifs au sein des suspensions PDMS/silice. Pour cela, plusieurs approches seront abordées. Nous verrons comment l’étude de la morphologie par microscopie électronique à transmission et optique (confocale et fluorescence), peut nous aider à comprendre comment ces polymères interagissent avec les suspensions initiales et quelle organisation des structures ils sont susceptibles de former. Une approche par analyse des énergies d’adsorption, via la chromatographie gazeuse en phase inverse, et par mesure des énergies de surface des additifs viendra compléter la compréhension de ces phénomènes.
Références bibliographiques 1. Schorsch G., Les applications des élastomères de silicones, Caoutchoucs et Plastiques,
1993, Vol. 720, pp.58. 2. Cohen-Addad J.P., Adsorption of PDMS on bare fumed silica surfaces, in: Papirer E.,
Adsorption on Silica Surfaces, New-York: Marcel Dekker, 2000, pp.621-643.

Introduction générale
18

CHAPITRE I : GENERALITES

Chapitre I : Généralités
19
CHAPITRE I : GENERALITES I.1- LES SILICONES ......................................................................................................................................... 20
I.1.1- PRÉSENTATION GÉNÉRALE ...................................................................................................................... 20 I.1.2- LE POLYDIMÉTHYLSILOXANE (PDMS).................................................................................................... 23 I.1.3- LES ÉLASTOMÈRES SILICONES ................................................................................................................. 25
I.1.3.1 – Les élastomères vulcanisables à froid (EVF) ................................................................................ 25 I.1.3.2 – Les élastomères vulcanisables à chaud (EVC).............................................................................. 27
I.2- LA SILICE COLLOIDALE........................................................................................................................ 28 I.2.1- INTRODUCTION........................................................................................................................................ 28 I.2.2- LES SILICES DE SYNTHÈSE ....................................................................................................................... 28
I.2.2.1- Modes de préparation des silices.................................................................................................... 29 a) Procédé sol-gel ..................................................................................................................................................... 29 b) Silices de précipitation ......................................................................................................................................... 30 c) Silices de pyrohydrolyse (fumed silica)................................................................................................................ 30 d) Autres voies de synthèse ...................................................................................................................................... 32
I.2.2.2- Propriétés physiques des silices...................................................................................................... 32 I.2.3- SURFACE ET CHIMIE DE SURFACE DES SILICES ......................................................................................... 34
I.2.3.1– Groupements chimiques de surface................................................................................................ 34 I.2.3.2- Déshydratation et déshydroxylation ............................................................................................... 36 I.2.3.3- Techniques de caractérisation des surfaces de silice...................................................................... 37
a) Caractérisation par spectroscopie infrarouge........................................................................................................ 37 b) Caractérisation par RMN 29Si CP/MAS ............................................................................................................... 39 c) Autres techniques d’analyse ................................................................................................................................. 41
I.2.3.4- Quantification du nombre de silanols ............................................................................................. 43 I.2.3.5- La distribution des différents types de silanols ............................................................................... 45
I.3- CONCLUSIONS .......................................................................................................................................... 49

Chapitre I : Généralités
20
I.1- LES SILICONES I.1.1- Présentation générale Les silicones (ou polyorganosiloxanes) ont une structure chimique basée sur une alternance d’atomes de silicium et d’oxygène (Figure I - 1). C’est par la présence de silicium et par la liaison Si-O que les silicones se distinguent des autres polymères organiques. Cette liaison est à l’origine de leur nom : silicones, contraction de silicon ketones, par analogie avec les cétones. Les liaisons covalentes que le silicium créé avec l’oxygène pour former le squelette des macromolécules sont exceptionnellement stables. Ceci conduit pour les polydiméthylsiloxanes (PDMS) à des propriétés de résistance à haute température, aux rayons UV et IR, et à de nombreuses agressions extérieures, combinées à un remarquable pouvoir d’étalement. Leur point faible réside dans leurs propriétés mécaniques moins bonnes que celles des polymères organiques [1]. Les progrès scientifiques essentiels qui ont permis la préparation des silicones ont été presque exclusivement réalisés en Europe, entre 1823, date ou le silicium a été pour la première fois isolé par Berzelius, et 1935. Mais c’est aux Etats-Unis que les applications industrielles ont été entrevues, dès 1937, et qu’un procédé industriel d’accès aux chlorosilanes, les matières premières des silicones, a été breveté en 1941.
Figure I - 1: Formule générale des polymères silicones [2]
Les chlorosilanes sont obtenus par réaction entre le silicium en poudre (extrait de la silice naturelle par un processus électrochimique) et le chlorure de méthyle, issu lui-même de la réaction entre l’acide chlorhydrique et le méthanol (Figure I - 2).
Figure I - 2: Synthèse directe des chlorosilanes [3]
Les catalyseurs utilisés sont des métaux, en particulier le cuivre. Les méthylchlorosilanes, utilisés pour la synthèse des méthylsilicones comme le
Si O
R
R'
( )n
Si + 2CH3ClCu, 300°C
(CH3)2SiCl2

Chapitre I : Généralités
21
polydiméthylsiloxane, sont préparés par réaction à 300°C du silicium avec le chlorure de méthyle, en présence de cuivre (environ 10% en poids par rapport au silicium) [3]. D’autres métaux sont également parfois ajoutés (aluminium, zinc) pour augmenter encore le rendement de la réaction. Les différents chlorosilanes obtenus (diméthyldichlorosilanes, méthyltrichlorosilanes, méthyldichlorosilanes, triméthylchlorosilanes…) sont ensuite séparés par distillation [4]. L’hydrolyse des chlorosilanes conduit à la formation de silanols, qui vont réagir entre eux pour former des organosiloxanes, constitués d’enchaînements Si-O-Si (Figure I - 3).
Figure I - 3: Réaction d’hydrolyse des chlorosilanes [3]
La polycondensation des prépolymères ainsi obtenus permet d’augmenter les masses molaires, pour obtenir des polymères silicones. On peut distinguer i/ les huiles et gommes qui sont des polymères linéaires, et qui se différencient par leur masse molaire, ou viscosité (huiles : 10-3 à 2,5.103 Pa.s-1, gommes : 2,5.103 à 2.104 Pa.s-1), ii/ les élastomères qui sont des réseaux tridimensionnels obtenus par réticulation d’huiles ou de gommes réactives, c’est-à-dire possédant des groupements fonctionnels (Si-H par exemple). Des composés cycliques sont également synthétisés lors de la réaction de polycondensation, et sont alors séparés des polymères linéaires par distillation [1]. Les gommes sont généralement obtenues par polymérisation des siloxanes cycliques. On peut aussi classer les polymères silicones suivant la nomenclature M, D, T, Q (Figure I - 4) basée sur la nature des motifs constituant leur structure. Les motifs monofonctionnels, représentés par le symbole M, sont présents en fin de chaîne des polymères. Les motifs difonctionnels, de symbole D, constituent le squelette des chaînes linéaires ou des composés cycliques. Les motifs trifonctionnels, de symbole T, permettent d’obtenir des élastomères (réseaux tridimensionnels). Enfin, les motifs tétrafonctionnels, représentés par le symbole Q, conduisent à des réseaux tridimensionnels encore plus rigides [1]. Dans cette nomenclature les polymères linéaires peuvent s’écrire MDxM où x est le degré de polymérisation et les composés cycliques sous la forme Dx où x est le nombre d’atomes de silicium formant le cycle. La Figure I - 5 présente quelques exemples de nomenclature des structures silicones.
R2SiCl22 + 4H2O 2 [R2Si(OH)2] + 4HCl
+ 2 H2OSi O
R
R
Si
R
R
O )(

Chapitre I : Généralités
22
Figure I - 4 : Nomenclature M, D, T, Q des différents motifs siloxane [1]
(a) (b) (c) Figure I - 5 : Exemples de nomenclature M, D, T, Q de structures silicones : (a) huile MD2M ; (b) composé
cyclique D4, (c) structure MDT présente dans les élastomères
La nature des groupements pendants R et R’ peut être très variée. On distingue les groupements non fonctionnels tels que les groupements alkyles et aryles (méthyle, éthyle, phényle…) des groupements fonctionnels, de type Si-X où X est le groupement fonctionnel (par exemple Si-H, Si-OH, Si-CH=CH2, Si-Cl, Si-OR, Si-NR2…) ou de type Si-R-X, où R est un groupement alkyle et X un groupement fonctionnel (par exemple Si-R-OH, Si-R-SH, Si-RCO2H…) [5]. La substitution des groupements R et R’ n’est que partielle. La nature des groupements R et R’ des polyorganosiloxanes a une importance sur les propriétés de ces polymères, comme par exemple la température de transition vitreuse et l’énergie de surface. Les températures de transition vitreuse de plusieurs polyorganosiloxanes de natures de groupements R et R’ différents sont données au Tableau I - 1 [6]. Deux effets sont responsables des variations de transition vitreuse observés, d’une part le volume occupé par le groupement pendant et d’autre part le volume libre généré par le mouvement de ce groupement. Lorsque le volume du groupement augmente la flexibilité de la chaîne diminue ce qui conduit à une température de transition vitreuse plus élevée. C’est cet effet qui permet d’expliquer la température de transition vitreuse de plus en plus élevée lorsque l’on passe d’un groupement R’ = H à un groupement R’ = CH3 plus volumineux (en gardant R = CH3)
Si
M D
OR
R
R
Si OO
R
R
Si OO
O
R
T
Si OO
O
O
Q
O Si
R
R
O Si O Si
R
R
R
R
RSi
R
R
R
O Si
R
O
RSi
R
O
R
Si SiO
R
R
R
R
O Si
R
R
O Si O
R
O
Si
R
R
R
Chapitre I : Généralités
23
[2]. Lorsque le volume libre augmente la mobilité de la chaîne augmente ce qui diminue la température de transition vitreuse. C’est ce qui permet d’expliquer qu’un groupement R’ = C2H5 conduit à un polymère température de transition vitreuse plus basse qu’un polymère possédant un groupement R’ = CH3 (en gardant R = CH3), en raison de son plus grand volume libre. Lorsque les deux effets s’ajoutent le classement devient plus difficile à établir, et on s’aperçoit par exemple qu’en augmentant la longueur de la chaîne alkyle l’effet de volume occupé par le groupement pendant devient prépondérant puisque la température de transition vitreuse augmente [6].
R R’ Tg (°C) CH3 CH3 -123 C2H5 C2H5 -139 C3H7 C3H7 -109 C4H9 C4H9 -116 C6H5 C6H5 49 CH3 H -138 CH3 C2H5 -135 CH3 C3H7 -120 CH3 C8H17 -92 CH3 CH2CH2CF3 -70 CH3 C6H5 -33
Tableau I - 1 : Températures de transition vitreuse de polyorganosiloxanes en fonction de la nature des groupements pendants R et R’ [6]
L’énergie de surface dépend elle aussi des groupements pendants. Celle-ci augmente avec le volume des groupements, ainsi par exemple le poly(3,3,3-trifluoropropyl-méthylsiloxane) présente à l’état liquide une tension de surface de 24 mN.m-1 supérieure à celle du polydiméthylsiloxane (21 mN.m-1) [5]. I.1.2- Le polydiméthylsiloxane (PDMS) Le PDMS est le plus courant des polyorganosiloxanes. Son unité de répétition possède une masse molaire de 74 g (Figure I - 6a). Sa conformation la plus stable est la conformation dans laquelle tous les groupements méthyle sont en position trans (Figure I - 6b). Les angles de valence des liaisons C-Si-C, Si-O-Si, O-Si-O et H-C-H sont de 112°, 143°, 110° et 109° respectivement. Les longueurs de liaisons sont 0,188, 0,163 et 0,110 nm pour les liaisons Si-C, Si-O et C-H. Le grand angle de valence de la liaison Si-O-Si, la distance interatomique Si-

Chapitre I : Généralités
24
O élevée et l’absence de substituant sur l’atome d’oxygène conduit à une chaîne flexible et mobile. En outre, celle-ci se trouve protégée de toute association avec des molécules voisines par les groupes méthyle apolaires.
Figure I - 6: Le polydiméthylsiloxane (PDMS) : (a) structure chimique [1] ; (b) représentation spatiale, pour n = 4, fins de chaînes méthyles
Cette mobilité théorique est confirmée expérimentalement par la température de transition vitreuse (Tg) particulièrement faible du PDMS (-123°C). La flexibilité et la mobilité de la chaîne PDMS entraînent [1, 2, 5] : - une faible évolution des propriétés physiques (viscosité, propriétés diélectriques…) avec la température - une solubilité et une perméabilité aux gaz relativement élevées (en particulier à la vapeur d’eau) - un certain caractère amphiphile ou amphipathique également dû à la légère polarité de la liaison Si-O combiné à la présence de groupements apolaires méthyle sur la chaîne PDMS. D’autre part, la présence de groupements apolaires et hydrophobes confèrent à la chaîne PDMS des caractéristiques spécifiques : - de faibles interactions intermoléculaires et donc une faible énergie cohésive, expliquent à la fois leur fluidité malgré leur masse molaire élevée et leur aptitude à l’étalement, mais aussi un faible coefficient de friction monomérique, à la base du caractère lubrifiant de ces produits. - une bonne tenue thermique
Si
CH3
CH3
O )(n
(a)
(b)
Chapitre I : Généralités
25
- la faible tension de surface des PDMS explique l’utilisation des silicones comme agents de démoulage ou dans le traitement des papiers utilisés pour la protection des surfaces autocollantes. Le Tableau I - 2 résume les principales caractéristiques des PDMS.
Caractéristiques PDMS Densité 0,97
Température de transition vitreuse (°C) -123 Tension superficielle (mN.m-1) 21
Indice de réfraction 1,4 Coefficient de dilatation volumique 9,45.10-4
Tableau I - 2 : Caractéristiques principales du PDMS [1, 4]
Ces huiles silicones possèdent avant tout une stabilité thermique importante entre -70°C et +250°C et un très bon pouvoir d’étalement dû à une faible tension superficielle. Elles possèdent également de bonnes propriétés diélectriques, une résistance importante au vieillissement, à l’oxydation et à l’hydrolyse. Elles sont donc employées comme fluides hydrauliques, diélectriques, de chauffage ou de refroidissement, comme lubrifiants, additifs pour peintures… Elles sont également employées en cosmétique, pharmacie et médecine [1]. I.1.3- Les élastomères silicones Lorsque les applications nécessitent des propriétés mécaniques supérieures à celles des huiles seules ou une bonne cohésion du matériau, on fait appel aux élastomères silicones. Ceux-ci sont formulés à partir de chaînes linéaires réactives auxquelles est ajouté un agent réticulant et des charges minérales telles que la silice, conférant au matériau de bonnes propriétés mécaniques telles que l’élasticité, l’amortissement, la résistance au déchirement. Les élastomères silicones sont généralement à base de PDMS et peuvent se différencier par leur système de réticulation. I.1.3.1 – Les élastomères vulcanisables à froid (EVF) Les EVF sont des élastomères formulés à partir d’huiles réactives de degré de polymérisation variable. La réticulation s’effectue à température ambiante grâce à un agent réticulant réagissant avec les groupements réactifs des chaînes PDMS et à un catalyseur qui

Chapitre I : Généralités
26
permet de contrôler la réticulation. L’élastomère est soit monocomposant soit bicomposant, avec une des deux parties contenant le catalyseur [1]. Dans le cas d’un monocomposant la réaction est activée par l’humidité de l’air, et conduit à la réticulation de l’élastomère (Figure I - 7).
Figure I - 7: Réticulation des EVF monocomposants [1]
Dans le cas d’un système bicomposant la réticulation n’est pas activée par l’humidité atmosphérique. La réaction débute lorsque les deux composants sont mis en présence, et plusieurs modes de réticulation sont possibles : Réticulation par hydrosilylation : cette réaction d’addition repose sur la capacité du groupe hydrogénosilane à réagir avec une double liaison carbone - carbone en présence d’un catalyseur. La réaction a lieu à température ambiante, généralement en présence d’un dérivé de platine (Figure I - 8). Elle ne conduit pas à la formation de sous-produits.
Figure I - 8: Réticulation des EVF bicomposants par réaction d’hydrosilylation [1]
Réticulation par réaction d’un hydrogénosilane avec un silanol, en présence de sel d’étain utilisé comme catalyseur (Figure I - 9). L’hydrogène libéré peut servir à fabriquer des mousses [4].
Figure I - 9: Réticulation des EVF bicomposants par réaction avec un hydrogénosilane [1]
Réticulation par condensation des silanols sur des fonctions alcoxy, en présence de sel d’étain (Figure I - 10).
Si OR SiRO+ H2O + Sn Si O Si + H2O
Si H + SiCHH2C Pt Si CH2 CH2 Si
Si OH SiH+ Si O Si + H2
sel d'étain

Chapitre I : Généralités
27
Figure I - 10: Réticulation des EVF bicomposants par condensation [1]
I.1.3.2 – Les élastomères vulcanisables à chaud (EVC) Les EVC s’obtiennent par réticulation à chaud de gommes silicones en présence de peroxydes organiques, suivant le mécanisme radicalaire présenté à la Figure I - 11. Décomposition du peroxyde : Formation de radicaux libres : Réticulation :
Figure I - 11: Mécanisme de réticulation des EVC [1]
Si OH + Si
OC2H5
CH3
OC2H5
C2H5O Si
OC2H5
CH3
OC2H5
OSi + C2H5OHsel d'étain
Si
CH
O
CH2
( ) Si
CH
O
CH2
( )
Si
CH
O
CH2
Si
CH2
O
CH
( )
Si
CH
O
H2C
Si
CH
O
H2C
( )
RRR
R
Si
CH
O
CH2
( ) +
.RO Si
CH
O
CH
( ) + ROH
R O O R 2 RO

Chapitre I : Généralités
28
I.2- LA SILICE COLLOIDALE I.2.1- Introduction
La silice, ou dioxyde de silicium, de formule générale SiO2, est composée de l’enchaînement de tétraèdres SiO4 liés entre eux. Elle peut être d’origine naturelle ou synthétique, amorphe ou cristalline.
La silice se trouve sous forme naturelle dans des minéraux tels que le quartz, ou dans les végétaux. Dans sa forme naturelle, la silice se présente principalement sous forme cristalline. Plusieurs phases peuvent exister, en fonction de la température, de la pression et du degré d’hydratation. Ainsi, à pression atmosphérique la silice cristalline anhydre se présente sous quatre phases différentes en fonction de la température. Le quartz est la forme la plus stable en dessous de 1143K, la tridymite entre 1 143K et 1 743K, la cristobalite entre 1 743K et 1 973K. Au dessus de 1 973K la cristobalite se transforme en verre de silice amorphe [7, 8]. La structure cristalline implique une structure ordonnée et dense, ainsi la surface active qui participe aux interactions chimiques ou physiques avec d’autres composés est limitée à la surface externe des particules cristallines. La surface spécifique est dans ce cas équivalente à la surface géométrique. Les silices synthétiques amorphes et divisées conduisent à des surfaces spécifiques plus importantes en raison de leur géométrie (arrangement aléatoire des tétraèdres SiO4) et de leur porosité. Ceci leur confère des propriétés de surface intéressantes, notamment en terme de réactivité ou d’interactions physico-chimiques. Nous nous limiterons ici à la description des silices de synthèse, principalement amorphes. I.2.2- Les silices de synthèse La Figure I - 12 présente les différentes catégories de silice de synthèse classées suivant leur mode de préparation. Après avoir rappelé ces différents modes de préparation, les propriétés des différentes silices de synthèse seront comparées.

Chapitre I : Généralités
29
Figure I - 12: Principales catégories de silices de synthèse [9]
I.2.2.1- Modes de préparation des silices a) Procédé sol-gel Le procédé sol-gel est celui qui est le mieux décrit dans la littérature. Un de ses avantages est qu’il conduit à un produit pur et homogène à faibles températures. Le procédé est basé sur la condensation de molécules de Si(OH)4 pour former un réseau siloxane. Un silicate tel que le silicate de sodium est couramment utilisé comme matière première, même si plus récemment l’utilisation d’alcoxysilanes de formule générale Si(OR)4 (R pouvant être CH3, C2H5, ou C3H7) a pris de l’importance [7]. C’est par l’hydrolyse de ces molécules que l’on obtient des silanols qui peuvent ensuite donner naissance par condensation à des liaisons siloxane (Figure I - 13). L’hydrolyse et la condensation ont lieu simultanément dans la solution aqueuse, formant des particules stables de taille colloïdale.
Figure I - 13: Formation de liaisons siloxanes par condensation de silanols [7]
La réaction de condensation peut être influencée par l’addition d’un électrolyte ou en
changeant le pH de la solution. On peut ainsi favoriser au choix la croissance des particules ou le pontage des particules pour donner naissance à des chaînes. Dans ce dernier cas on peut, à partir d’un certain point, passer d’un sol à un gel, ou l’on a un comportement élastique. Ce gel est appelé hydrogel, ou alcogel dans le cas ou le solvant utilisé est un alcool. La structure de l’hydrogel est contrôlée par la température, le pH du milieu, la nature du solvant, le type d’électrolyte employé et le type de silicate ou alcoxyde. Les hydrogels sont ensuite séchés pour obtenir des xerogels. Lors du séchage, le liquide remplissant les pores de la structure est peu à peu évaporé. Les pores se resserrent en
Si OH + SiHO Si O Si + H2O
Silicesde p yrolyse
Solides élaborés par fusion
Silices obtenues par voie hum ide
Arc électr ique Si lices de py rohy drolys e
Plas maSilic es de
précipitationGels de si lice V erres de silice
Silicesde p yrolyse
Solides élaborés par fusion
Silices obtenues par voie hum ide
Arc électr ique Si lices de py rohy drolys e
Arc électr ique Si lices de py rohy drolys e
Plas maSilic es de
précipitationGels de si liceSilic es de
précipitationGels de si lice V erres de silice

Chapitre I : Généralités
30
raison des forces capillaires exercées par ce liquide. On obtient alors un xerogel, de porosité réduite. Les alcogels peuvent donner naissance, dans des conditions de séchage supercritiques, à des aérogels. Il s’agit de gels présentant des volumes de pores très importants (jusqu’à 98% du volume total). Cette caractéristique est due au fait que le resserrement des pores par attraction capillaire est dans ce cas impossible. Enfin pour passer des xerogels (ou aerogels) à des matériaux solides et stables, une étape de traitement thermique est nécessaire [7]. Les silices obtenues par procédé sol-gel sont utilisées pour la fabrication de films, de fibres, de poudres, de composites ou encore de matériaux poreux. Les applications les plus intéressantes sont celles qui nécessitent la formation d’objets purs et homogènes, de porosité contrôlée, à faible température [10]. b) Silices de précipitation
Les silices de précipitation ont été développées au début des années 1940 comme charge de renforcement blanche pour les caoutchoucs. L’usage de ces silices a constamment augmenté depuis, et 75% de la production est encore destinée au renforcement des caoutchoucs [10]. Les silices de précipitation sont obtenues par acidification d’une solution de silicate de sodium en présence d’acide sulfurique ou d’un mélange de dioxyde de carbone et d’acide chlorhydrique (Figure I - 14).
Figure I - 14: Préparation des silices de précipitation [10]
Durant la préparation ce ces silices, l’état de gel est évité, et des particules individuelles de silices sont obtenues. c) Silices de pyrohydrolyse (fumed silica) Les silices de pyrohydrolyse ont été préparées pour la première fois par le chimiste allemand Klopfer en 1941, en essayant de créer une charge de renforcement de couleur blanche pour les
Na2OXSiO2 + H2SO4 XSiO2 + Na2SO4 + H2O

Chapitre I : Généralités
31
caoutchoucs. Le procédé est basé sur l’hydrolyse à haute température, dans une flamme, du tétrachlorure de silicium SiCl4, suivant la réaction décrite à la Figure I - 15.
Figure I - 15: Réaction d’hydrolyse du tétrachlorure de silicium [9]
Cette réaction chimique libère une quantité très importante de chaleur qui est évacuée
dans une zone de refroidissement. Le seul sous-produit formé est l’acide chlorhydrique qui est séparé de la silice en sortie du procédé (Figure I - 16). Celui-ci peut ensuite être recyclé pour former, par réaction avec le silicium, du tétrachlorure de silicium.
Ce procédé conduit à des silices très pures. Le mélange gazeux étant homogène, les conditions de formation sont les mêmes pour chaque particule. Il en résulte notamment une distribution relativement étroite en diamètre de particules primaires (agrégation et agglomération dans la flamme elle même). De plus, en faisant varier la concentration des produits réactionnels, la température de la flamme et le temps de séjour de la silice dans la chambre de combustion, il est possible d’influencer dans de larges limites la taille des particules, leur répartition, les surfaces spécifiques et l’état de surface des silices [9].
Figure I - 16: Schéma fonctionnel de fabrication de la silice de pyrohydrolyse [9]
2H2 + O2
SiCl4 + 2H2O
2H2 + O2 + SiCl4
2H2O
SiO2 + 4HCl
SiO2 + 4HCl

Chapitre I : Généralités
32
d) Autres voies de synthèse Des silices de pyrolyse peuvent être également synthétisées par un procédé à l’arc électrique, par réduction du quartz à 2000°C en présence de coke : le monoxyde de silicium ainsi créé est ensuite oxydé par l’air, pour former de la silice. Ce procédé est très peu employé en raison de son coût élevé et des propriétés moins intéressantes de cette silice en comparaison avec la silice de pyrohydrolyse. Enfin, la projection plasma est une technique également très peu employée [10]. I.2.2.2- Propriétés physiques des silices Les propriétés physiques des silices dépendent essentiellement de leur procédé d’élaboration, que nous venons de décrire. Les caractéristiques physiques déterminantes sont en particulier la surface spécifique, la taille des particules primaires et des agrégats, ainsi que la porosité. Le Tableau I - 3 rassemble les principales caractéristiques physiques des silices en fonction de leur mode de préparation.
Tableau I - 3: Comparaison des propriétés physiques des silices [11]
Les différences de propriétés physiques des silices dépendent fortement de la manière dont les particules sont agrégées ou agglomérées. En effet, on peut distinguer trois échelles de structure dans les silices. Les particules primaires, dont la taille varie de 3 à 500 nm de diamètre, n’existent pas en pratique à l’échelle individuelle car aussitôt formées pendant le procédé de fabrication, elles s’agrègent sous l’effet de forces colloïdales, pour donner

Chapitre I : Généralités
33
naissance à une seconde structure de particules secondaires, les agrégats. Ce phénomène est irréversible, et même sous l’action de forces de cisaillement, il est impossible de casser cette structure secondaire. Dans certaines conditions ces agrégats peuvent à leur tour se lier pour former une structure tertiaire constituée d’agglomérats, dont la taille peut atteindre plusieurs dizaines de microns. Ces agglomérats peuvent être séparés dans certaines conditions sous l’effet de forces de cisaillement et retourner à l’état d’agrégats. Ces trois niveaux d’échelle sont schématisés à la Figure I - 17, dans le cas de silices de pyrohydrolyse qui feront l’objet de cette étude.
Figure I - 17: Structure multi-échelle des silices de pyrohydrolyse [12]
La taille des particules primaires, ainsi que la densité et le degré d’agrégation et
d’agglomération détermine la porosité et la surface spécifique des silices. L’agglomération des silices de pyrohydrolyse sous forme de chaînes conduit à une structure non poreuse. Ces silices sont finement divisées et ne présentent pas de surface interne. Un exemple de structure observée au microscope électronique à transmission (TEM) est présenté à la Figure I - 18. On peut observer que les particules primaires sont pratiquement sphériques et qu’elles forment un réseau lâche.
Figure I - 18: Observation de la structure d’une silice de pyrohydrolyse (S = 380 m2.g-1) par TEM [7]
shear stress Irreversible !

Chapitre I : Généralités
34
Plus les particules primaires sont petites, plus la formation d’agrégats est marquée. La taille des particules primaires influence également la surface spécifique : plus la taille des particules primaires est importante, plus la surface spécifique est faible [9]. Les gels de silice possèdent des surfaces spécifiques plus élevées que les silices de pyrohydrolyse, en raison de leur porosité. Une majeure partie de cette surface est donc interne, ce qui est à prendre en compte lors du choix de la silice. Par exemple pour le renforcement des élastomères les propriétés proviennent du rôle joué par la surface externe ce qui explique que l’on n’utilise pas les gels de silice. I.2.3- Surface et chimie de surface des silices
Les particules de silice peuvent être considérées comme des polymères d’acide silicique, constitués de tétraèdres de SiO4 interconnectés. A la surface plusieurs espèces chimiques sont présentes et jouent un rôle important dans les interactions de la silice avec d’autres composés, comme par exemple les chaînes de PDMS. L’étude de la surface de la silice consiste à déterminer le type d’espèces présentes en surface et à les quantifier, ainsi qu’à déterminer les quantités d’eau physisorbés. Ici nous présentons les caractéristiques physico-chimiques essentielles de la surface de la silice permettant d’étudier par la suite la chimie de modification de la silice. I.2.3.1– Groupements chimiques de surface A la surface, la structure de la silice se termine soit par un groupement siloxane Si-O-Si où l’oxygène est à la surface, soit par un groupement silanol Si-OH. Il existe trois types de silanols, représentés à la Figure I - 19.
Figure I - 19: Les trois différents types de silanols présents à la surface de la silice [7]
Si
OH
Si
OH
Si
OH
Si
OOHH
silanol isolé silanols vicinaux silanols géminés

Chapitre I : Généralités
35
Les silanols libres, ou isolés, possèdent un atome de silicium dont trois des liaisons sont rattachées à la silice et la quatrième au groupement hydroxyle. Les silanols vicinaux, ou silanols liés, sont deux groupements hydroxyle, chacun rattachés à un atome de silicium différent, qui sont assez proches pour se lier entre eux par liaison hydrogène. Enfin, les silanols géminaux sont deux groupements hydroxyle portés par le même atome de silicium. Les silanols géminaux sont trop proches pour créer une liaison hydrogène entre eux, tandis qu’à l’inverse les silanols libres sont trop loin pour interagir. On peut également classer les espèces présentes à la surface de la silice par une terminologie Qn définie en chimie du silicium, qui classe les groupements par le nombre n d’atomes d’oxygène de la silice, liés à l’atome central de silicium. La Figure I - 20 présente les différents groupements de surface classés de cette manière. Les silanols libres et vicinaux sont rattachés à des atomes de silicium Q3, tandis que les silanols géminaux sont portés par des siliciums Q2.
Figure I - 20: Différents groupements silanol de surface répertoriés suivant la nomenclature Qn [13]
Les groupements siloxane ne jouent aucun rôle dans les phénomènes d’adsorption ni dans la réactivité de la surface, par contre en présence d’eau les liaisons siloxane sont susceptibles de s’hydrolyser pour former des groupements silanol. L’eau, qui est toujours présente à la surface de la silice, même après séchage, peut donc non seulement être physisorbée à la surface de la silice, mais également réagir avec les groupements siloxane de surface, lors du processus de réhydroxylation de la silice, décrit dans le paragraphe suivant.
Si
OH
OH
O
Si
O
OHO
SiO
O
OH
O
Si
O
OO
SiO
O
O
H
H
Si
O
OO
SiO
O
O
H
H
O
H
H
O
SiO
O
O Si
Q2
Q4
Q3
O
O
O

Chapitre I : Généralités
36
I.2.3.2- Déshydratation et déshydroxylation
Les termes de déshydratation et de déshydroxylation sont souvent confondus dans la littérature car ces deux processus sont difficiles à dissocier. Lors d’une élévation de température, la déshydratation correspond à l’élimination de l’eau physisorbée tandis que la déshydroxylation représente la condensation de groupements hydroxyle pour former une liaison siloxane (Figure I - 21).
Figure I - 21: Réactions de déshydratation et déshydroxylation [7]
La température à laquelle la déshydratation est complète est difficile à déterminer sans ambiguïté. Celle-ci dépend de la porosité, de la taille et de la morphologie des pores et diffère suivant les auteurs. Iler [14] parvient à la conclusion que les molécules d’eau sont complètement éliminées sous vide à température ambiante ou à 150°C à pression atmosphérique. Pour lui, comme pour Gregg et Sing [15], la meilleure façon est le séchage sous vide à basse température car cela n’affecte pas les groupements hydroxyle de surface. De Boer montre qu’à 120°C sous pression atmosphérique toute l’eau physisorbée est déjà éliminée, mais qu’à 110°C la silice retient encore de l’eau si l’air est humide. Pour Okkerse l’élimination de l’eau ne peut se faire à 120°C que si la silice ne contient pas de micropores, autrement elle peut retenir l’eau jusqu’à 180°C [16]. Pour des silices non microporeuses, on peut donc estimer que la déshydratation est complète entre 120°C et 150°C. Au delà de ces températures commence la déshydroxylation. Le processus de réhydroxylation a lieu en deux étapes. Les molécules d’eau s’adsorbent dans un premier temps sur les silanols, puis cette eau adsorbée va réagir avec un groupement siloxane pour former deux nouveaux groupements silanols. Les silices qui ont été exposées à des températures supérieures à 400°C ne peuvent se réhydroxyler spontanément, il est alors nécessaire de les placer dans l’eau à température ambiante pour qu’elles soient complètement réhydroxylées. Plus la silice a été déhydroxylée, plus le temps nécessaire à la réhydroxylation sera long. Ce temps peut cependant être diminué en utilisant de l’eau à ébullition, mais il faut alors prendre garde aux changements de structure ou de surface
Si O
H
O H
H
OSi
H
- H2OSi O
H
OSi
H
- H2O Si
SiO
Chapitre I : Généralités
37
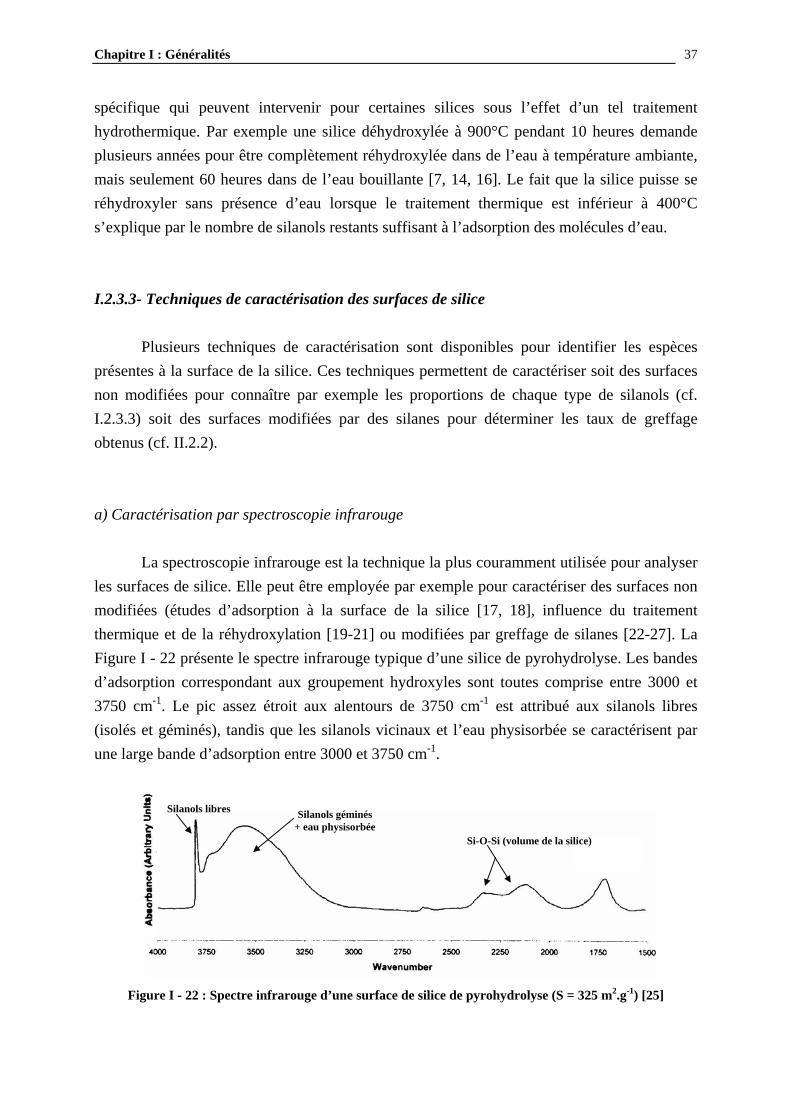
spécifique qui peuvent intervenir pour certaines silices sous l’effet d’un tel traitement hydrothermique. Par exemple une silice déhydroxylée à 900°C pendant 10 heures demande plusieurs années pour être complètement réhydroxylée dans de l’eau à température ambiante, mais seulement 60 heures dans de l’eau bouillante [7, 14, 16]. Le fait que la silice puisse se réhydroxyler sans présence d’eau lorsque le traitement thermique est inférieur à 400°C s’explique par le nombre de silanols restants suffisant à l’adsorption des molécules d’eau. I.2.3.3- Techniques de caractérisation des surfaces de silice Plusieurs techniques de caractérisation sont disponibles pour identifier les espèces présentes à la surface de la silice. Ces techniques permettent de caractériser soit des surfaces non modifiées pour connaître par exemple les proportions de chaque type de silanols (cf. I.2.3.3) soit des surfaces modifiées par des silanes pour déterminer les taux de greffage obtenus (cf. II.2.2). a) Caractérisation par spectroscopie infrarouge La spectroscopie infrarouge est la technique la plus couramment utilisée pour analyser les surfaces de silice. Elle peut être employée par exemple pour caractériser des surfaces non modifiées (études d’adsorption à la surface de la silice [17, 18], influence du traitement thermique et de la réhydroxylation [19-21] ou modifiées par greffage de silanes [22-27]. La Figure I - 22 présente le spectre infrarouge typique d’une silice de pyrohydrolyse. Les bandes d’adsorption correspondant aux groupement hydroxyles sont toutes comprise entre 3000 et 3750 cm-1. Le pic assez étroit aux alentours de 3750 cm-1 est attribué aux silanols libres (isolés et géminés), tandis que les silanols vicinaux et l’eau physisorbée se caractérisent par une large bande d’adsorption entre 3000 et 3750 cm-1.
Figure I - 22 : Spectre infrarouge d’une surface de silice de pyrohydrolyse (S = 325 m2.g-1) [25]
Silanols géminés + eau physisorbée
Silanols libres
Si-O-Si (volume de la silice)
Chapitre I : Généralités
38
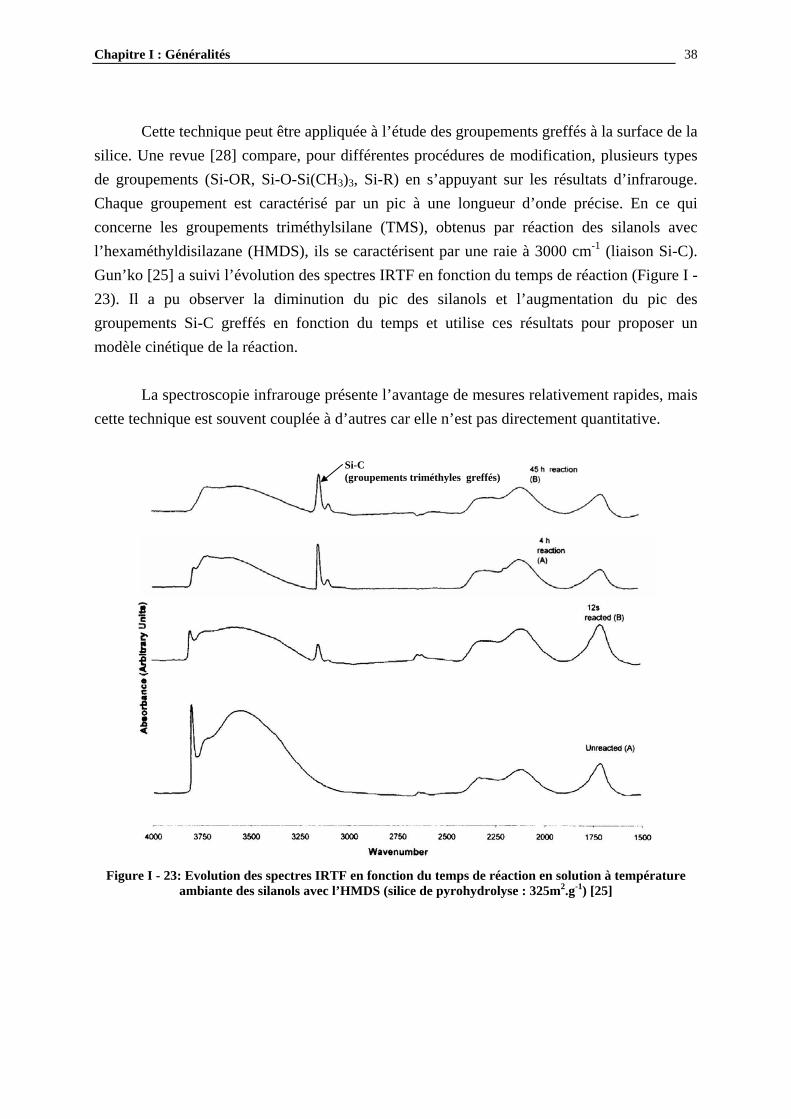
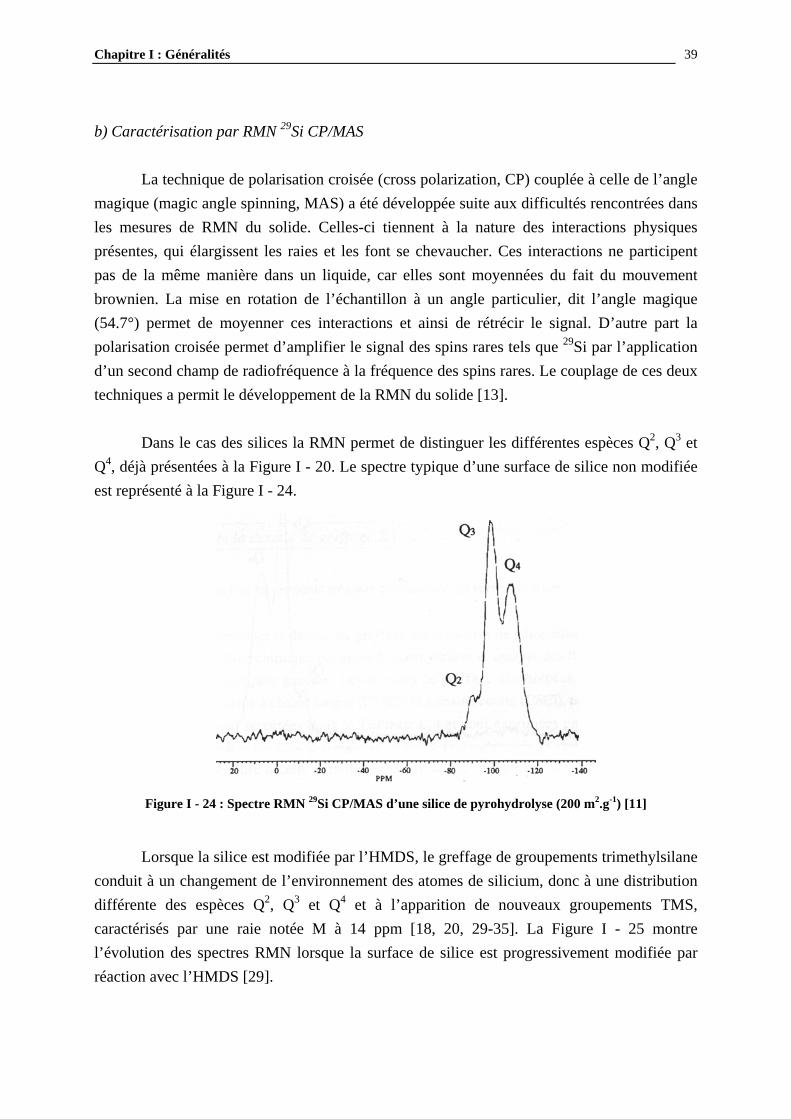
Cette technique peut être appliquée à l’étude des groupements greffés à la surface de la silice. Une revue [28] compare, pour différentes procédures de modification, plusieurs types de groupements (Si-OR, Si-O-Si(CH3)3, Si-R) en s’appuyant sur les résultats d’infrarouge. Chaque groupement est caractérisé par un pic à une longueur d’onde précise. En ce qui concerne les groupements triméthylsilane (TMS), obtenus par réaction des silanols avec l’hexaméthyldisilazane (HMDS), ils se caractérisent par une raie à 3000 cm-1 (liaison Si-C). Gun’ko [25] a suivi l’évolution des spectres IRTF en fonction du temps de réaction (Figure I - 23). Il a pu observer la diminution du pic des silanols et l’augmentation du pic des groupements Si-C greffés en fonction du temps et utilise ces résultats pour proposer un modèle cinétique de la réaction. La spectroscopie infrarouge présente l’avantage de mesures relativement rapides, mais cette technique est souvent couplée à d’autres car elle n’est pas directement quantitative.
Figure I - 23: Evolution des spectres IRTF en fonction du temps de réaction en solution à température ambiante des silanols avec l’HMDS (silice de pyrohydrolyse : 325m2.g-1) [25]
Si-C (groupements triméthyles greffés)
Chapitre I : Généralités
39
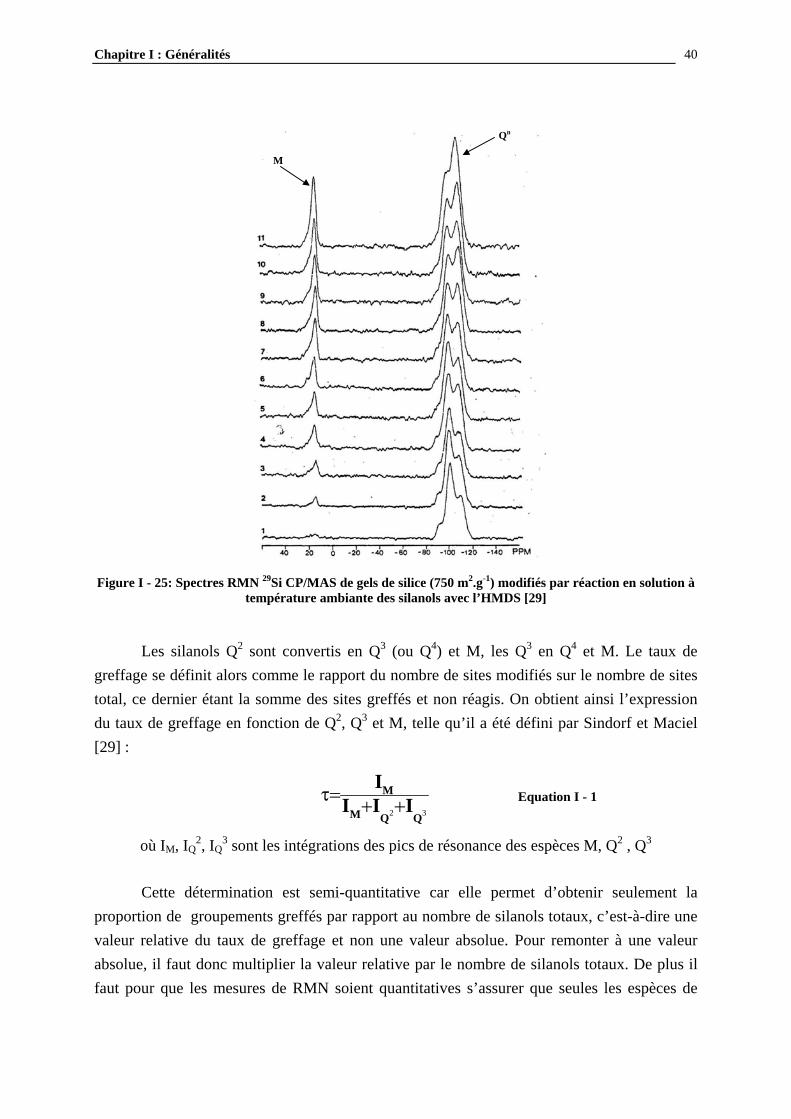
b) Caractérisation par RMN 29Si CP/MAS La technique de polarisation croisée (cross polarization, CP) couplée à celle de l’angle magique (magic angle spinning, MAS) a été développée suite aux difficultés rencontrées dans les mesures de RMN du solide. Celles-ci tiennent à la nature des interactions physiques présentes, qui élargissent les raies et les font se chevaucher. Ces interactions ne participent pas de la même manière dans un liquide, car elles sont moyennées du fait du mouvement brownien. La mise en rotation de l’échantillon à un angle particulier, dit l’angle magique (54.7°) permet de moyenner ces interactions et ainsi de rétrécir le signal. D’autre part la polarisation croisée permet d’amplifier le signal des spins rares tels que 29Si par l’application d’un second champ de radiofréquence à la fréquence des spins rares. Le couplage de ces deux techniques a permit le développement de la RMN du solide [13]. Dans le cas des silices la RMN permet de distinguer les différentes espèces Q2, Q3 et Q4, déjà présentées à la Figure I - 20. Le spectre typique d’une surface de silice non modifiée est représenté à la Figure I - 24.
Figure I - 24 : Spectre RMN 29Si CP/MAS d’une silice de pyrohydrolyse (200 m2.g-1) [11]
Lorsque la silice est modifiée par l’HMDS, le greffage de groupements trimethylsilane conduit à un changement de l’environnement des atomes de silicium, donc à une distribution différente des espèces Q2, Q3 et Q4 et à l’apparition de nouveaux groupements TMS, caractérisés par une raie notée M à 14 ppm [18, 20, 29-35]. La Figure I - 25 montre l’évolution des spectres RMN lorsque la surface de silice est progressivement modifiée par réaction avec l’HMDS [29].
Chapitre I : Généralités
40
Figure I - 25: Spectres RMN 29Si CP/MAS de gels de silice (750 m2.g-1) modifiés par réaction en solution à
température ambiante des silanols avec l’HMDS [29]
Les silanols Q2 sont convertis en Q3 (ou Q4) et M, les Q3 en Q4 et M. Le taux de greffage se définit alors comme le rapport du nombre de sites modifiés sur le nombre de sites total, ce dernier étant la somme des sites greffés et non réagis. On obtient ainsi l’expression du taux de greffage en fonction de Q2, Q3 et M, telle qu’il a été défini par Sindorf et Maciel [29] : Equation I - 1
où IM, IQ
2, IQ3 sont les intégrations des pics de résonance des espèces M, Q2 , Q3
Cette détermination est semi-quantitative car elle permet d’obtenir seulement la proportion de groupements greffés par rapport au nombre de silanols totaux, c’est-à-dire une valeur relative du taux de greffage et non une valeur absolue. Pour remonter à une valeur absolue, il faut donc multiplier la valeur relative par le nombre de silanols totaux. De plus il faut pour que les mesures de RMN soient quantitatives s’assurer que seules les espèces de
M
Qn
32 QQM
MIII
I++=τ

Chapitre I : Généralités
41
surface soient prises en compte (et non le volume de la silice), et pour cela utiliser des temps de contact appropriés [29, 35, 36] c) Autres techniques d’analyse L’analyse thermogravimétrique (ATG) est une technique également adaptée à la détermination des groupements de surface de la silice. Il a été montré [37, 38] que le nombre de silanols de surface pouvait être estimé à partir de la perte de poids des échantillons lors d’une montée lente en température (environ 5°C/min) jusque 1250°C. La principale difficulté réside dans la séparation des phénomènes de déshydratation (départ de l’eau physisorbée) et de déshydroxylation (départ des silanols) qui n’est pas toujours évidente. Ek [37] considère que la perte de masse est due jusque 200°C à l’eau physisorbée et entre 200 et 1250°C aux silanols. Ces résultats sont également dépendants de la rampe de température appliquée. Lorsque la surface de silice est modifiée par réaction des silanols avec un silane, la perte de poids mesurée par ATG permet alors de calculer le pourcentage de silane greffé, définit par la relation [39] : Equation I - 2
où τ est le pourcentage de silane greffé, wi et wf les masses initiales et finales de l’échantillon de silice, respectivement. L’analyse élémentaire du carbone permet également de caractériser de manière quantitative le greffage, étant donné que seules les espèces greffées contiennent du carbone. Dans le cas du greffage de silice par réaction avec l’HMDS, le taux de greffage (TMS/nm2), noté τ, peut être calculé à partir du taux de carbone présent dans l’échantillon : Equation I - 3
avec C : fraction massique de carbone et Nc : nombre d’atomes de carbone par site TMS (c’est à dire 3). Cette technique suppose qu’aucune autre espèce contenant du carbone ne soit adsorbée à la surface. De plus la présence de silane résiduel doit être évitée pour ne pas surestimer le
C
a
SNCN
2010.12=τ
( )f
fi
www −
=100
τ

Chapitre I : Généralités
42
taux de greffage. Cela montre l’intérêt de coupler cette technique avec d’autres moyens d’analyse qui permettront de vérifier si ces hypothèses sont vérifiées. Une mesure indirecte du greffage peut être obtenue par la chromatographie gazeuse en phase inverse. Les interactions entre la silice et des composés sondes sont mesurés. Lorsque la silice est modifiée chimiquement (par des groupements hydrophobes), les interactions changent et l’on peut ainsi caractériser le greffage. La Figure I - 28 montre que l’indice d’interaction spécifique de la silice de pyrohydrolyse A200 (non modifiée) est largement supérieur que celui d’une silice modifiée par le diméthyldichlorosilane (R972), en raison de son plus grand nombre de silanols de surface. Cette technique sera décrite plus en détails au paragraphe IV.2.2. Figure I - 26: Comparaison des interactions spécifiques entre une silice non modifiée (A200) et greffée par
réaction avec le diméthyldichlorosilane (R972) [40]
Enfin une approche complémentaire pour évaluer le greffage des silices est la mesure de mouillage. Lorsque la silice est modifiée, la surface, initialement hydrophile, devient hydrophobe. Cette propriété peut être évaluée simplement par mesure de l’angle de contact de la surface de la silice avec l’eau. Pour cela une surface plane de silice a été réalisée par compaction. Une goutte d’eau est déposée à la surface, et l’angle de contact défini à partir des dimensions de la goutte (Figure I - 27). L’angle de contact peut donc être défini par la relation [11] : Equation I - 4
Lorsque les surfaces sont modifiées l’affinité de la surface avec l’eau diminue, et l’angle de contact augmente, jusqu’à des valeurs comprises entre 110 et 130°C [41].
( )Dharctg 22=θ

Chapitre I : Généralités
43
Figure I - 27 : Mesure des dimensions de la goutte posée
I.2.3.4- Quantification du nombre de silanols
Les silanols présents en surface sont responsables de la réactivité de la silice et de ses interactions avec d’autres molécules. Ainsi il est important de connaître quel est leur nombre et comment la quantité de silanols varie avec la température. Beaucoup d’études ont été menées sur ce sujet par plusieurs approches. Il est possible de calculer une valeur théorique à partir de modèles cristallographiques de la silice. L’approche expérimentale fait intervenir soit des techniques chimiques, en faisant réagir des espèces chimiques avec les silanols de surface de la silice et en quantifiant ensuite le nombre de molécules greffées, soit des techniques physiques, telles que la spectroscopie infrarouge à transformée de Fourier (IRTF), l’analyse thermogravimétrique (ATG), etc. Les différentes valeurs obtenues par ces trois approches pour les silices de pyrohydrolyse sont répertoriées au Tableau I - 4.
Référence Surface spécifique
(m2.g-1) Technique d’analyse
Nombre de silanols (OH.nm-2)
[16] moyenne Echange avec le deutérium 4,6 [19] 325 ATG 3,1 [42] 230 ATG + dosage TMCS 4,1 [43] 380 ATG 2,6 [37] 200 ATG et 1H NMR 4,4 [44] 200 Dosage avec le TEA 4,1 [45] 200 Titrage acide base 1,9 [46] 200 Dosage méthanol 2,9
Tableau I - 4: Concentration surfacique en silanols de surface de silices de pyrohydrolyse rapportées dans la littérature
θ
θ
D
hθ
θ
D
h

Chapitre I : Généralités
44
Le calcul théorique réalisé à partir du plan (111) d’une face octaédrique de la β-cristobalite a conduit à la valeur de 4,55 OH/nm2 [7]. La même valeur a été trouvée en considérant le plan (100) de la cristobalite [47]. Cette silice cristalline a été prise comme référence car sa densité et son indice de réfraction sont très proches de ceux de la silice amorphe. L’inconvénient de ce calcul est qu’il ne permet pas d’estimer l’évolution du nombre de silanols en fonction de la température, il permet donc seulement d’estimer le nombre de silanols d’une silice complètement hydroxylée. Les techniques chimiques font intervenir des espèces réactives telles que trichlorure de bore, des organosilanes ou des composés organométalliques [44]. Le nombre de silanols est déterminé par dosage des espèces fixées à la surface de la silice ou par le volume gazeux dégagé par la réaction. Cette approche est cependant dépendante de la réactivité de l’espèce chimique choisie. En particulier certains composés peuvent réagir sélectivement avec certains types de silanols. Par ailleurs, l’encombrement stérique de la molécule fixée et l’accessibilité des silanols jouent un rôle capital dans ces réactions de dosage. On peut alors s’interroger sur la signification du nombre de silanols obtenu. Il peut s’agir du nombre total de silanols, mais on considèrera plutôt un nombre de silanols accessibles (libres) selon la méthode de dosage considérée. L’existence de ces différents paramètres explique pour une grande part les différences observées au Tableau 3 et rend difficile une quantification en absolu de la densité surfacique de silanols. Comme nous l’avons vu l’IRTF est l’une des méthodes les plus rencontrées pour analyser les groupements de surface de la silice, cependant cette technique s’avère limitée pour la détermination quantitative du nombre de silanols, car elle ne permet pas de séparer les silanols de surface de l’eau physisorbée. La RMN 29Si CP/MAS, présentée au paragraphe précédent permet en revanche une estimation du nombre de silanols [30], tout comme l’ATG [37, 38].
L’une des études les plus conséquentes sur la quantification du nombre de silanols reste cependant celle réalisée par Zhuravlev [16], par réaction d’échange avec le deutérium et mesures par spectrométrie de masse. Cette méthode a l’avantage que seuls les silanols de surface sont pris en compte et non pas les silanols internes ni l’eau adsorbée. Cette étude a été menée sur plus de 100 échantillons, avec des silices de nature différente : gel de silice, silice de pyrohydrolyse, verre poreux, et de caractéristiques différentes. En particulier on a fait varier les surfaces spécifiques entre 5 et 1000 m2.g-1. De plus, des silices commerciales tout comme des silices préparées en laboratoire ont été étudiées. Il ressort de cette étude que la concentration en silanols de surface peut être considérée comme étant une constante physico-
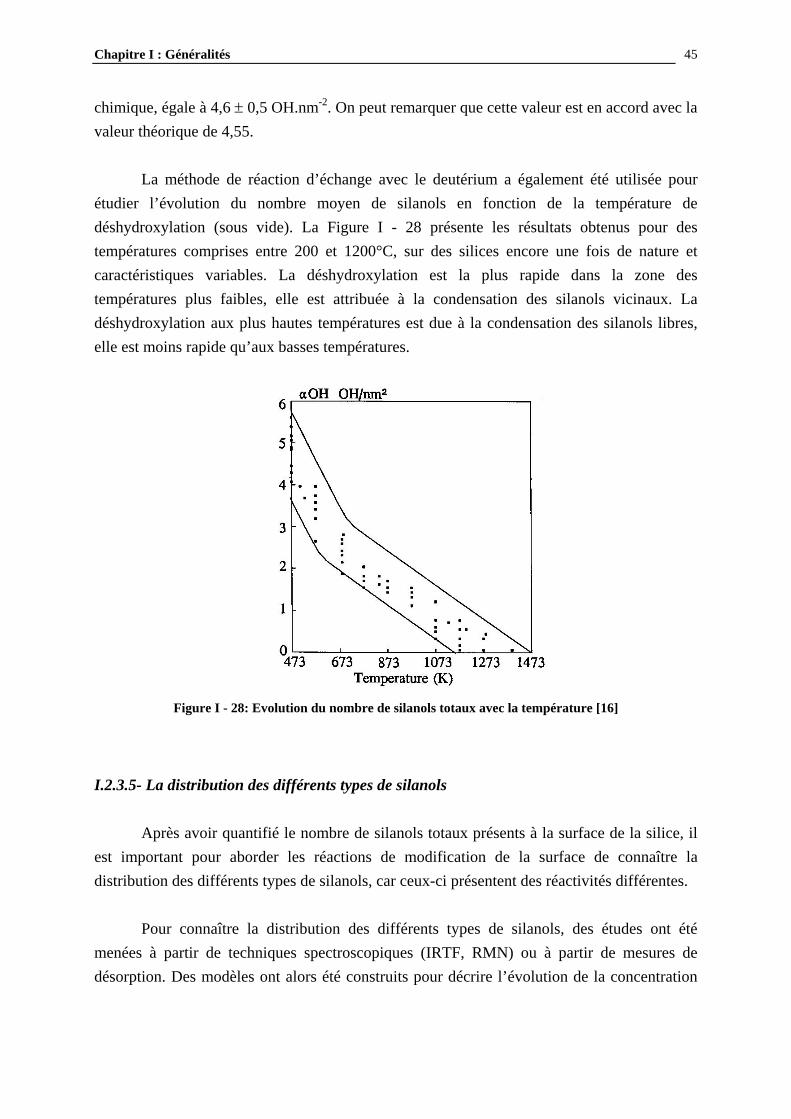
Chapitre I : Généralités
45
chimique, égale à 4,6 ± 0,5 OH.nm-2. On peut remarquer que cette valeur est en accord avec la valeur théorique de 4,55. La méthode de réaction d’échange avec le deutérium a également été utilisée pour étudier l’évolution du nombre moyen de silanols en fonction de la température de déshydroxylation (sous vide). La Figure I - 28 présente les résultats obtenus pour des températures comprises entre 200 et 1200°C, sur des silices encore une fois de nature et caractéristiques variables. La déshydroxylation est la plus rapide dans la zone des températures plus faibles, elle est attribuée à la condensation des silanols vicinaux. La déshydroxylation aux plus hautes températures est due à la condensation des silanols libres, elle est moins rapide qu’aux basses températures.
Figure I - 28: Evolution du nombre de silanols totaux avec la température [16]
I.2.3.5- La distribution des différents types de silanols Après avoir quantifié le nombre de silanols totaux présents à la surface de la silice, il est important pour aborder les réactions de modification de la surface de connaître la distribution des différents types de silanols, car ceux-ci présentent des réactivités différentes. Pour connaître la distribution des différents types de silanols, des études ont été menées à partir de techniques spectroscopiques (IRTF, RMN) ou à partir de mesures de désorption. Des modèles ont alors été construits pour décrire l’évolution de la concentration

Chapitre I : Généralités
46
des différents types de silanols en fonction de la température. Nous présentons brièvement certains de ces modèles et terminerons par une comparaison de leurs estimations. Le modèle le plus simple consiste à déterminer la concentration des différents silanols à partir de l’intégration de l’aire de chaque bande de vibration correspondant en infrarouge [48, 49]. Les valeurs sont normées par rapport à une bande de référence. On peut ainsi comparer les proportions de silanols libres et de silanols vicinaux, sachant que les silanols libres incluent à la fois les silanols isolés et les silanols géminés. Il a ainsi pu être montré qu’à basse température la surface est presque entièrement constituée de silanols vicinaux, puis lorsque la température augmente une partie de ces silanols vicinaux est convertie en silanols libres. A partir de 600°C, il ne reste plus que des silanols libres, dont la concentration est comprise entre 1 et 2/nm2. Ce modèle reste approximatif en raison du recouvrement des différentes bandes d’absorption et sous-estime le nombre de silanols libres à basse température. Une autre méthode est basée sur l’analyse de la bande maximum des silanols libres [50]. En effet, il a été observé que l’intensité de cette bande évolue en fonction de la température et que sa longueur d’onde peut être corrélée à la population de silanols libres et vicinaux. A partir de la variation de ce pic, ce modèle permet donc de remonter à la proportion de silanols libres et vicinaux. L’approche par la RMN du silicium en solide (RMN CP/MAS 29Si) est complémentaire de l’IRTF. Alors que la spectroscopie infrarouge permet de distinguer les silanols libres (isolés ou géminaux) des silanols vicinaux, la RMN du silicium fait la distinction entre silanols simples (isolés ou vicinaux), portés par un silicium Q3 et silanols doubles (géminaux) portés par un silicium Q2. Il a ainsi été montré que la proportion de silanols géminaux à température ambiante est de 15% [30]. Zhuravlev [16], après avoir déterminé le nombre de silanols totaux à la surface de la silice, a également étudié les proportions de chaque type de silanols par réaction d’échange avec le deutérium et par spectroscopie infrarouge. Au delà de 400°C, seuls des silanols libres sont présents et à partir des coefficients d’adsorption des silanols à la surface, pour des températures supérieures à 400°C, il est donc possible de calculer la concentration en silanols libres. A partir ce ces valeurs et du nombre de silanols totaux, le nombre de silanols vicinaux et de ponts siloxanes peuvent ensuite être déterminés. Pour conclure, la Figure I - 29 montre l’évolution des silanols libres (isolés et géminaux) en fonction de la température pour les différents modèles. Le modèle de référence est celui de Zhuravlev, avec une précision de 0,5 OH/nm2 qui est la précision sur la mesure du nombre de silanols totaux. Presque tous les modèles s’inscrivent dans cette plage de
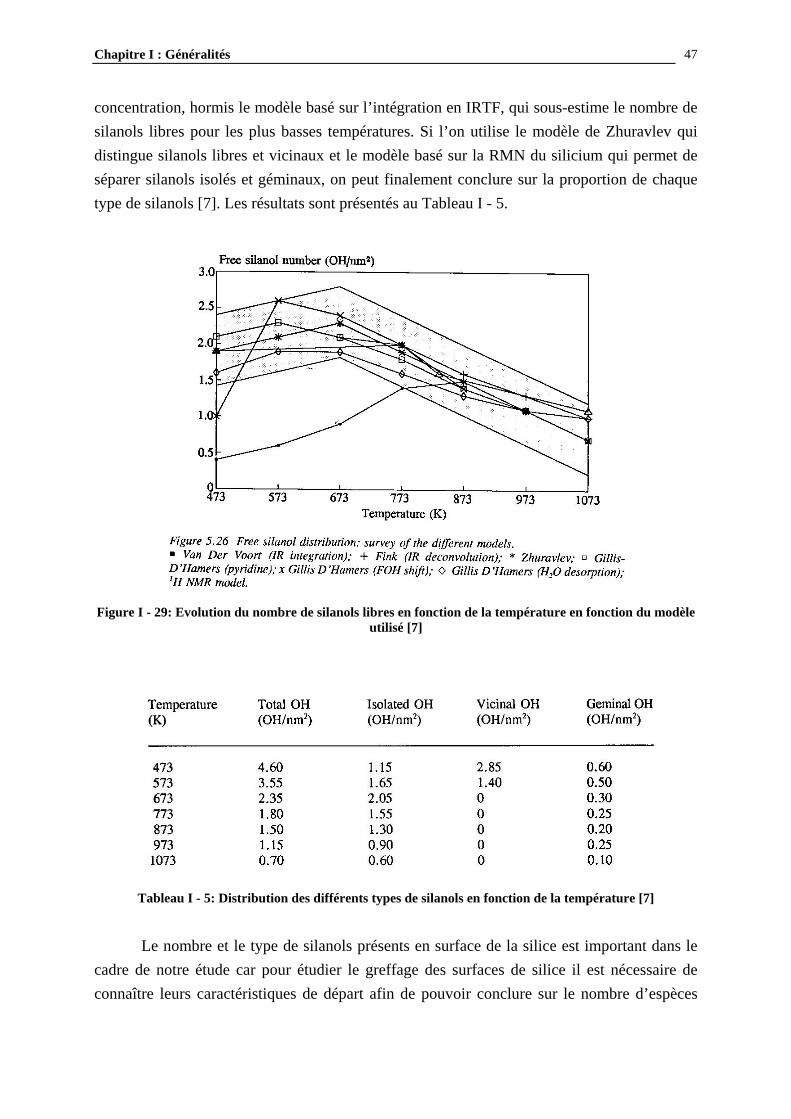
Chapitre I : Généralités
47
concentration, hormis le modèle basé sur l’intégration en IRTF, qui sous-estime le nombre de silanols libres pour les plus basses températures. Si l’on utilise le modèle de Zhuravlev qui distingue silanols libres et vicinaux et le modèle basé sur la RMN du silicium qui permet de séparer silanols isolés et géminaux, on peut finalement conclure sur la proportion de chaque type de silanols [7]. Les résultats sont présentés au Tableau I - 5.
Figure I - 29: Evolution du nombre de silanols libres en fonction de la température en fonction du modèle utilisé [7]
Tableau I - 5: Distribution des différents types de silanols en fonction de la température [7]
Le nombre et le type de silanols présents en surface de la silice est important dans le cadre de notre étude car pour étudier le greffage des surfaces de silice il est nécessaire de connaître leurs caractéristiques de départ afin de pouvoir conclure sur le nombre d’espèces

Chapitre I : Généralités
48
greffées et de silanols résiduels. Cette analyse doit être la plus fine possible car les propriétés des mélanges PDMS/silice dépendent fortement de cette modification de surface, comme nous le verrons au chapitre III.

Chapitre I : Généralités
49
I.3- CONCLUSIONS Lorsque des charges de silice sont incorporées au PDMS, on obtient des mélanges qui sont des suspensions de silice au sein d’une matrice PDMS. Les propriétés rhéologiques du PDMS changent complètement lors de l’ajout des nanocharges de silice, de part les fortes interactions entre les chaînes PDMS et la silice qui modifient fortement la dynamique de celles-ci. Il est admis que ces deux constituants interagissent par liaison hydrogène entre les silanols de surface de la silice et les atomes d’oxygène de la chaîne PDMS. Ce phénomène d’adsorption des chaînes a été étudié par Cohen-Addad [51]. Il a montré en particulier que la vitesse d’adsorption dépend de la racine carrée de la masse moléculaire du PDMS et de la concentration en silice. Ces études font appel à la notion de polymère lié, défini comme la fraction de polymère adsorbée sur les particules de silice, qui ne peut être extraite contrairement à la fraction libre de polymère non adsorbée. Lorsque la fraction de silice augmente, l’agglomération des nanoparticules au sein des suspensions conduit à de très fortes viscosités. Les premiers modèles qui faisaient l’hypothèse d’un contact direct entre les agrégats [52] ont été complétés par la suite en envisageant également des enchevêtrements entre des chaînes adsorbées et/ou des chaînes non adsorbées (Figure I - 30).
Figure I - 30: Interactions PDMS – silice [53]
Ces interactions polymère – charge, dont l’intensité peut être modulée par greffage de la surface de la silice, contrôlent donc les morphologies des suspensions et les propriétés rhéologiques qui en découlent comme nous le verrons en détails au chapitre III. Ainsi le greffage de la surface de la silice, qui joue un rôle tout particulier sur les propriétés des mélanges PDMS-silice, fait l’objet du chapitre suivant.
Si
O
Si Si
H
Silice
chaîne PDMS OSi
OSi
OSi
O
O OO O
O
H
O
H

Chapitre I : Généralités
50
Références bibliographiques
1. Carette L., Pouchol J.M., Silicones, in: Traité plastiques et composites: Techniques de
l'ingénieur, 2001, A3475, pp.1-18.
2. Dvornic P.R., Lenz R.W., Polysiloxanes, in: High temperature siloxane elastomers, Basel: Huthig & Wepf, 1990, pp.25-83.
3. Rochow E., Introduction à la chimie des silicones, Paris: Dunod, 1957, 181p.
4. Hardman B., Torkelson A., Silicones, in: Encyclopedia of polymer science and engineering, New-York: John Wiley & Sons, 1989, pp.204-308.
5. Kricheldorf H.R., Burger C., Silicon in polymer synthesis, Heidelberg: Springer, 1996, 494p.
6. Clarson S.J., Depolymerization, degradation and thermal properties of siloxane polymers, in: Clarson S.J., Sernlyen J.A., Siloxane polymers, Upper Saddle River: Prentice hall, 1993,
7. Vansant E.F., Van Der Voort P., Vrancken K.C., Characterization and Chemical Modification of the Silica Surface, Amsterdam: Elsevier, 1996, 556p.
8. Legrand A.P., On the silica edge, Chapitre 1, in: The surface properties of silicas, New-York: John Wiley, 1998,
9. Günther M., Horst F., Principes fondamentaux de l'Aerosil, Frankfurt: Tech. Bull. Pigments N°11, Degussa Corporation Data Sheet, 4th Edition, 1992, 79p.
10. Wason S.K., Synthetic silicas, Chapitre 9, in: Katz H.S., Milewski J.V., Handbook of fillers for plastics, New-York: Van Nostrand Reinhold, 1987, pp.165-201.
11. Duchet J., Système modèle polyéthylène/verre: rôle de chaînes connectrices greffees sur l'adhésion, Thèse de Doctorat, Université Claude Bernard, Lyon I, 1996, 310p.
12. Wacker, Production of Wacker HDK, [en ligne]. Disponible sur : http://www.wacker.com/internet/noc/Products/P_Pyrogene_Kiesels/ (consulté le 18.08.2003)
13. Legrand A.P., Introduction à la résonnance magnétique nucléaire haute résolution dans les solides : principe, possibilités, limitations, J. Chim. Phys., 1987, Vol. 10, pp.1204-1215.
14. Iler R.K., The chemistry of silica : solubility, polymerization, colloid and surface properties, and biochemistry, New-York: John Wiley & Sons, 1979, 866p.
15. Gregg S.J., Sing K.S.W., Adsorption, surface area, and porosity, London: Academic Press, 1967, 371p.

Chapitre I : Généralités
51
16. Zhuravlev L.T., The surface chemistry of amorphous silica. Zhuravlev model, Colloids Surf., A, 2000, Vol. 173, pp.1-38.
17. Bermudez V.M., Infrared study of boron trichloride chemisorbed on silica gel, J. Phys. Chem., 1971, Vol. 75 (21), pp.3249-3257.
18. Haukka S., Lakomaa E.-L., Root A., IR and NMR study of the chemisorption of TiCl4 on silica, J. Phys. Chem., 1993, Vol. 97, pp.5085-5094.
19. Morrow B.A., McFarlan A.J., Infrared and Gravimetric Study of an Aerosil and a precipitated silica using chemical and H/D exchange probes, Langmuir, 1991, Vol. 7, pp.1695-1701.
20. Zhao X.S., Lu G.Q., Whittaker A.K., Millar G.J., Zhu H.Y., Comprehensive study of surface chemistry of MCM-41 using 29Si CP/MAS NMR, FTIR, Pyridine-TPD, and TGA, J. Phys. Chem. B, 1997, Vol. 101, pp.6525-6531.
21. Dugas V., Chevalier Y., Surface hydroxylation and silane grafting on fumed and thermal silica, J. Colloid Interface Sci., 2003, Vol. 264, pp.354-361.
22. Tripp C.P., Hair M.L., An infrared study of the reaction of octadecyltrichlorosilane with silica, Langmuir, 1992, Vol. 8, pp.1120-1126.
23. Tripp C.P., Hair M.L., Direct observation of the surface bonds between self-assembled monolayers of octadecyltrichlorosilane and silica surfaces : a low-frequency IR study at the solid/liquid interface, Langmuir, 1995, Vol. 11, pp.1215-1219.
24. Bjelopavlic M., Singh P.K., El-Shall H., Moudgil B.M., Role of surface molecular architecture and energetics of hydrogen bonding sites in adsorption of polymers and surfactants, J. Colloid Interface Sci., 2000, Vol. 226, pp.159-165.
25. Gun'ko V.M., Vedamuthu M.S., Henderson G.L., Blitz J.P., Mechanism and kinetics of Hexamethyldisilazane reaction with a fumed silica surface, J. Colloid Interface Sci., 2000, Vol. 228, pp.157-170.
26. Snyder L.R., Ward J.W., The surface structure of porous silica, J. Phys. Chem., 1966, Vol. 70 (12), pp.3941-3952.
27. Haukka S., Root A., The reaction of hexamethyldisilazane and subsequent oxidation of trimethylsilyl groups on silica studied by solid-state NMR and FT-IR, J. Phys. Chem., 1994, Vol. 98, pp.1695-1703.
28. Chmielowiec J., Morrow B.A., Alkylation of silica surfaces, J. Colloid Interface Sci., 1983, Vol. 94 (2), pp.319-327.
29. Sindorf, D.W., Maciel E., CP/MAS 29Si NMR Study of silica gel using trimethylsilane bonding as a probe of surface geometry and reactivity, J. Phys. Chem., 1982, Vol. 86, pp.5208-5219.
30. Sindorf, D.W., Maciel E., Si29 nuclear magnetic resonance study of hydroxyl sites on dehydrated silica gel surfaces, using silylation as a probe, J. Phys. Chem., 1983, Vol. 87 (26), pp.5516-5521.

Chapitre I : Généralités
52
31. Köhler J., Chase D.B., Farlee R.D., Vega A.J., Kirkland J.J., Comprehensive characterization of some silica-based stationary phases for high-performance liquid chromatography, J. Chromatogr., 1986, Vol. 352, pp.275-305.
32. Huang W., Huang Y., Yu Y., Trimethylsilylated silica as rheology modifier for silicone resins, Chin. J. Polym. Sci., 2000, Vol. 18 (1), pp.51-55.
33. Suratwala T.I., Hanna M.L., Miller E.L., Whitman P.K., Thomas I.M., Ehrmann P.R., Maxwell R.S., Burnham A.K., Surface chemistry and trimethylsilyl functionalization of stöber silica sols, J. Non-Crystalline Solids, 2003, Vol. 316, pp.349-363.
34. Sutra P., Fajula F., Brunel D., Lentz P., 29Si and 13C MAS NMR characterization of surface modification of micelle-templated silicas during the grafting of organic moieties and end-capping, Colloids Surf., A, 1999, Vol. 158, pp.21-27.
35. Petiaud R., Caractérisation de silices greffées. Détermination des taux de greffage et étude des silanols après greffage par RMN sur solides du silicium, J. Chim. Phys., 1994, Vol. 91, pp.895-900.
36. Bauer F., Freyer A., Ernst H., Gläsel H.-J., Mehnert R., Application of temperature-programmed oxidation, multinuclear MAS NMR and DRIFT spectroscopy to the surface characterization of modified silica nanoparticles, Appl. Surf. Sci., 2001, Vol. 179, pp.118-121.
37. Ek S., Root A., Peussa M., Niinistö L.,, Determination of the hydroxyl group content in silica by thermogravimetry and a comparison with 1H MAS NMR results, Thermochim. Acta, 2001, Vol. 379, pp.201-212.
38. Mueller R., Kammler H.K., Wegner K., Pratsinis S.E., Determination of functional groups (OH-) on the surface of nanoparticles by thermogravimetric analysis, Silica 2001, Mulhouse, France.
39. Ramos M.A., Gil M.H., Schacht E., Matthys G., Mondelaers W., Figueiredo M.M., Physical and chemical characterisation of some silicas and silica derivatives, Powder Technology, 1998, Vol. 99, pp.79-85.
40. Zumbrum M.A., Acid/Base properties of fumed silica fillers used in silicone elastomers, XVIth Annual Meeting of the Adhesion Society, Inc, 1993, Williamsburgh, Virginia, U.S.A.
41. Belyakova L.A., Varvarin A.M., Surface properties of silica gels modified with hydrophobic groups, Colloids Surf., A, 1999, Vol. 154, pp.285-294.
42. Buzek F., Rathousky J., Stoechiometry and kinetics of the reaction of silica with organosilicon compounds, J. Colloid Interface Sci., 1981, Vol. 79 (1), pp.47-55.
43. Mueller R., Kammler H.K., Wegner K., Pratsinis S.E., Determination of functional groups (OH-) on the surface of nanoparticles by thermogravimetric analysis, Silica 2001, 2001, Mulhouse, France.

Chapitre I : Généralités
53
44. Tsubokawa N., Saitoh K., Shirai Y., Surface grafting of polymers onto ultrafine silica : cationic polymerization initiated by benzylium perchlorate groups introduced onto ultrafine silica surface, Polym. Bull., 1995, Vol. 35 (4), pp.399-406.
45. Barthel H., Surface interactions of dimethylsiloxy group-modified fumed silica, Colloids Surf., A, 1995, Vol. 101, pp.217-226.
46. Vidal A., et al., Hydroxyles des silices divisées, Adv. Colloid Interface Sci., 1990, Vol. 33, pp.91-330.
47. Peri J.B., Hensley, A.L., The surface structure of silica gel, J. Phys. Chem., 1968, Vol. 72 (8), pp.2926-2933.
48. Van Der Voort P., Gillis-D'Hamers I., Vansant E.F., Estimation of the distribution of surface hydroxyl groups on silica gel, using chemical modification with trichlorosilane, J. Chem. Soc. Faraday Trans., 1990, Vol. 86 (22), pp.3751-3755.
49. Gillis-D'Hamers I., Cornelissens I., Vrancken K.C., Van Der Voort P., Vansant E.F., Modelling of the hydroxyl group population using an energetic analysis of the temperature-programmed desorption of pyridine from silica gel, J. Chem. Soc. Faraday Trans., 1992, Vol. 88 (5), pp.723-727.
50. Gillis-D'Hamers I., Cornelissens I., Vrancken K.C., Van Der Voort P., Vansant E.F., Fourier-transform infrared photo-acoustic spectroscopy study of the free hydroxyl group vibration, J. Chem. Soc. Faraday Trans., 1992, Vol. 88 (14), pp.2047-2050.
51. Cohen-Addad J.P., Adsorption of PDMS on bare fumed silica surfaces, in: Papirer E., Adsorption on Silica Surfaces, New-York: Marcel Dekker, 2000, pp.621-643.
52. Chahal R.S., St Pierre L.E., Interfacial phenomena in macromolecular systems. II. Relaxation modulus of uncross-linked silica-polydimethylsiloxane composites, Macromolecules, 1969, Vol. 2 (2),
53. Aranguren M.I., Mora E., DeGroot Jr. J.V., Macosko C.W., Effect of reinforcing fillers on the rheology of polymer melts, J. Rheol., 1992, Vol. 36 (6), pp.1165-1182.

Chapitre I : Généralités
54

CHAPITRE II : MODIFICATION CHIMIQUE DE
SURFACES DE SILICE PAR L’HEXAMETHYLDISILAZANE ET
CARACTERISATION DU GREFFAGE

Chapitre II : Modification chimique de surfaces de silice par l’hexaméthyldisilazane
55
CHAPITRE II : MODIFICATION CHIMIQUE DE SURFACES DE SILICE PAR
L’HEXAMETHYLDISILAZANE ET CARACTERISATION DU GREFFAGE
II.1- INTRODUCTION ...................................................................................................................................... 56 II.2- ETUDE BIBLIOGRAPHIQUE................................................................................................................. 57
II.2.1- MODIFICATION CHIMIQUE DE LA SURFACE DE LA SILICE PAR GREFFAGE D’UN ORGANOSILANE ............. 57 II.2.1.1- Modification chimique par l’hexaméthyldisilazane....................................................................... 57 II.2.1.2- Procédures de modification........................................................................................................... 58
a) Modification chimique en solution....................................................................................................................... 59 b) Modification chimique en phase gazeuse ............................................................................................................. 60
II.2.2- TAUX DE GREFFAGE DES SURFACES GREFFÉES PAR RÉACTION AVEC L’HMDS ...................................... 60 II.2.3- CONCLUSIONS ........................................................................................................................................ 63
II.3- PARTIE EXPÉRIMENTALE................................................................................................................... 65 II.3.1- MATÉRIAUX UTILISÉS ............................................................................................................................ 65 II.3.2- MODIFICATION CHIMIQUE PAR VOIE SOLVANT (EX-SITU) ....................................................................... 65 II.3.3- MODIFICATION CHIMIQUE PAR VOIE GAZEUSE (EX-SITU) ....................................................................... 67 II.3.4- MODIFICATION CHIMIQUE AU SEIN DES SUSPENSIONS PDMS/SILICE ( VOIE IN-SITU).............................. 69 II.3.5- CARACTÉRISATION DES SURFACES MODIFIÉES ....................................................................................... 70
II.3.5.1- Analyse élémentaire du carbone.................................................................................................... 70 II.3.5.2- Spectroscopie Infrarouge à Transformée de Fourier (IRTF) ........................................................ 70 II.3.5.3- Résonance magnétique nucléaire CP/MAS 29Si............................................................................. 70 II.3.5.4- Mesures de mouillage .................................................................................................................... 71
II.4- RÉSULTATS ET DISCUSSION............................................................................................................... 72 II.4.1- EVOLUTION DU GREFFAGE EN FONCTION DE LA QUANTITÉ DE SILANE INTRODUIT ET COMPARAISON DES TAUX DE GREFFAGE VOIE SOLVANT/VOIE GAZEUSE........................................................................................... 72
II.4.1.1- Suivi par spectroscopie IRTF ........................................................................................................ 72 II.4.1.2- Suivi par RMN 29Si CP/MAS.......................................................................................................... 76 II.4.1.3- Suivi par mesure des angles de contact eau/silice......................................................................... 80 II.4.1.4- Suivi par analyse élémentaire........................................................................................................ 82 II.4.1.5- Comparaison entre les techniques de caractérisation et analyse des taux de greffage................. 82
II.4.2- COMPARAISON DES TAUX DE GREFFAGE DES SILICES MODIFIÉES IN-SITU ET EX-SITU .............................. 84 II.5- CONCLUSIONS......................................................................................................................................... 89

Chapitre II : Modification chimique de surfaces de silice par l’hexaméthyldisilazane
56
II.1- INTRODUCTION
Ce chapitre est consacré à l’étude de la modification de la surface de la silice par l’HMDS, et à la caractérisation des surfaces greffées. C’est une étape essentielle pour pouvoir faire varier de manière précise le taux de silanols à la surface de la silice et donc le degré d’interaction entre la silice et le PDMS dans les suspensions, ce qui va conditionner les propriétés finales des matériaux. Après avoir rappelé les principes et procédures expérimentales de modification chimique de la surface de la silice, nous présenterons les matériaux utilisés et les techniques de caractérisation choisies pour étudier le greffage. L’évolution du greffage sera suivie en fonction de la quantité d’HMDS introduite et l’influence de la procédure de greffage sur le nombre de sites modifiés sera établie. Des informations complémentaires seront recueillies par l’utilisation de plusieurs techniques d’analyse : spectroscopie IRTF, analyse élémentaire, RMN du solide (RMN 29Si CP/MAS) et mesures d’énergies de surface. En particulier, le couplage IRTF/analyse élémentaire permet de mettre au point une procédure rapide et précise pour évaluer le taux de greffage de silices modifiées. Enfin, les silices modifiées ex-situ seront comparées aux silices modifiées in-situ au sein des suspensions PDMS/silice par le même organosilane.

Chapitre II : Modification chimique de surfaces de silice par l’hexaméthyldisilazane
57
II.2- ETUDE BIBLIOGRAPHIQUE II.2.1- Modification chimique de la surface de la silice par greffage d’un organosilane Les applications de la silice sont basées sur ses propriétés telles que son module, sa surface spécifique et la réactivité de sa surface, etc. Ces caractéristiques peuvent être combinées à son aptitude à moduler les interactions physico-chimiques spécifiques par modification chimique de sa surface. Par exemple, dans le cas présent, il s’agit de combiner les propriétés de renforcement mécanique des élastomères silicones avec des interactions PDMS/silices modulées garantissant une viscosité modérée des suspensions. Parmi les molécules qui peuvent être utilisées pour modifier la surface de la silice, les organosilanes sont les plus utilisés. Leur formule générale est RnSiX4-n , où n est compris entre 1 et 3. R est un ligand organique, comme une chaîne alkyle ou une chaîne courte portant un groupement organofonctionnel, et X représente une fonction hydrolysable (chlore, amine, ethoxy…). Ces composés ont l’avantage par rapport aux composés organiques de pouvoir se lier par plusieurs mécanismes. Les forces électrocinétiques peuvent permettre une attraction ou une répulsion à grande distance, tandis qu’à une échelle de distance inférieure, les silanols permettent d’établir des forces de Van Der Waals, des liaisons hydrogène et covalentes. En combinant toutes ces possibilités en une seule molécule les organosilanes sont particulièrement adaptés à la modification de surface de la silice [1]. II.2.1.1- Modification chimique par l’hexaméthyldisilazane Parmi les organosilanes, nous allons nous intéresser aux aminosilanes, et en particulier à l’hexaméthyldisilazane, noté HMDS. Les aminosilanes sont particulièrement intéressants car comme nous allons le voir la fonction amine catalyse le greffage covalent du silane avec la surface. L’HMDS ne suit pas la structure générale des organosilanes puisque le groupe X (la fonction NH), porte deux groupements Si-R3, en l’occurrence deux Si-(CH3)3 (Figure II - 1). En raison de sa grande réactivité, l’HMDS a été utilisé pour sonder la surface de la silice, notamment pour déterminer les caractéristiques des silanols de surface [2], les propriétés d’adsorption de la silice [3] ou les énergies de surface [4]. L’HMDS, qui réagit avec les silanols de surface de la silice, conduit au remplacement d’une partie de ces silanols par des groupements triméthylsilane, notés TMS avec formation d’ammoniac. Le bilan de la réaction est présenté à la Figure II - 2 [1, 5, 6].

Chapitre II : Modification chimique de surfaces de silice par l’hexaméthyldisilazane
58
Figure II - 1: Formule chimique de l’hexaméthyldisilazane (HMDS)
Figure II - 2: Réaction de l’HMDS avec les silanols de surface de la silice [1, 5, 6]
Le taux de greffage et le type de silanols ayant réagis seront abordés dans la partie concernant la caractérisation des surfaces. En ce qui concerne le mécanisme de réaction, il a été montré qu’il pouvait se diviser en deux étapes [5]. La première est la réaction de l’HMDS avec un premier silanol de surface qui conduit à la formation d’un triméthylsilane greffé à la surface et d’un composé intermédiaire triméthylaminosilane. Ce composé va ensuite réagir au cours d’une deuxième étape avec un second silanol de surface pour former un autre triméthylsilane et de l’ammoniac (Figure II - 3). La première étape est celle qui détermine la vitesse de réaction. L’énergie d’activation a été calculée de manière théorique [5] et expérimentale [7], conduisant à une valeur autour de 70 kJ/mol.
Figure II - 3: Mécanisme de réaction en deux étapes de l’HMDS avec la surface de la silice [5]
II.2.1.2- Procédures de modification
Nous décrivons dans ce paragraphe les deux types de procédures utilisées généralement pour le greffage de silanes, la modification chimique en solution et en phase vapeur, en reportant les procédures utilisées dans le cas précis de l’HMDS.
SiH3CHN Si CH3
CH3
CH3
CH3
CH3
+Si O Si O Si
OO OH H
Si O Si O Si OOOO O
HH H H H+ NH3
Si O Si O SiOO O
Si O Si O Si OOOO O
SiH H
CH3
H3C CH3H H
SiH3C CH3
CH3
HSiH3CHN Si CH3
CH3
CH3
CH3
CH3
+Si O Si O Si
OO OH H
Si O Si O Si OOOO O
HH H H H+ NH3
Si O Si O SiOO O
Si O Si O Si OOOO O
SiH H
CH3
H3C CH3H H
SiH3C CH3
CH3
HSiH3CHN Si CH3
CH3
CH3
CH3
CH3
+Si O Si O Si
OO OH H
Si O Si O Si OOOO O
HH H H H+ NH3
Si O Si O SiOO O
Si O Si O Si OOOO O
SiH H
CH3
H3C CH3H H
SiH3C CH3
CH3
H
1ère étape : 2ème étape :
SiH3CHN Si CH3
CH3
CH3
CH3
CH3

Chapitre II : Modification chimique de surfaces de silice par l’hexaméthyldisilazane
59
a) Modification chimique en solution On distingue la modification chimique en milieux aqueux (eau ou hydroalcoolique) de la modification chimique en milieu organique (solvant organique). La modification chimique en milieu aqueux se déroule en trois étapes : i/ les silanes sont d’abord hydrolysés, et les silanols ainsi créés peuvent interagir entre espèces hydrolysées. ii/ on assiste alors à la condensation de ces groupements pour former une liaison siloxane. iii/ ensuite les molécules condensées vont se lier avec la surface de la silice, par réaction des fonctions silanol des silanes avec les silanols de surface de la silice. La vitesse d’hydrolyse des silanes dépend de leur groupement organique. Par exemple des acycloxysilanes ou aminosilanes sont hydrolysées en quelques minutes dans l’eau, en raison du caractère acide ou basique de leurs groupements fonctionnels, tandis que plusieurs heures seront nécessaires dans le cas d’autres alkoxysilanes [1]. Pour ces derniers, un catalyseur acide ou basique pourra être utilisé pour accélérer leur hydrolyse. En solution, à pH acide les réactions d’hydrolyse sont catalysées et les réactions de condensation ralentie [1, 8]. Une manière de réaliser la condensation sans hydrolyse est de catalyser la réaction avec de l’ammoniac. Des taux de greffage jusqu’à douze fois supérieurs ont pu être observés lorsque la réaction est catalysée de cette manière [1]. Les aminosilanes, qui contiennent déjà une fonction amine dans leur structure, permettent donc des réactions autocatalysées. C’est ce qui peut expliquer en partie la réactivité importante de l’HMDS vis-à-vis de la surface de la silice. En raison de sa facilité de mise en œuvre, la modification chimique en solution a fait l’objet de nombreuses études. En ce qui concerne l’HMDS, la plupart des modifications chimiques ont été réalisés en solution dans le toluène, à température ambiante [5, 9] ou à des températures plus élevées, à reflux à 110°C [10]. Certains auteurs [9] ajoutent également une faible quantité d’eau (5% en poids par rapport à la silice), mais cela n’est à priori pas nécessaire puisque la réaction de condensation est déjà catalysée par la fonction amine de l’HMDS. Enfin, des études ont été menées avec d’autres solvants, tels que le n-hexane [3] ou l’éthanol [11]. Les temps de réaction sont très différents d’une étude à l’autre (20 minutes à plusieurs jours), ainsi le temps de réaction devra être optimisé en fonction des résultats du greffage.

Chapitre II : Modification chimique de surfaces de silice par l’hexaméthyldisilazane
60
b) Modification chimique en phase gazeuse Dans le cas d’une modification chimique en phase gazeuse, la réaction s’effectue dans un réacteur contenant la silice à modifier. Le silane est introduit soit directement sous forme gazeuse, soit sous forme de solution ou le solvant sera rapidement évaporé. Si la silice est bien agitée dans le réacteur, on obtient un greffage homogène et plus reproductif que par modification chimique en solution. Concernant le procédé, on peut utiliser un système dynamique où le courant gazeux réactif traverse le réacteur ou un système statique où un volume de gaz prédéterminé réagit avec la silice. La principale différence entre ces deux types de procédures réside dans le contrôle du mécanisme de réaction. Dans des conditions statiques, la vitesse de réaction est contrôlée de manière thermodynamique. En régime dynamique, le procédé est sous contrôle cinétique [1]. L’HMDS étant un composé volatil, il se prête facilement à ce type de procédure en phase gazeuse. Plusieurs études ont été menées, pour des températures de réaction de la température ambiante à plus de 200°C [6, 12-17]. II.2.2- Taux de greffage des surfaces greffées par réaction avec l’HMDS Les surfaces modifiées par des silanes peuvent être caractérisées à l’aide des techniques expérimentales présentées au paragraphe I.2.3.3. Nous présentons ici les principaux résultats obtenus dans le cas de surfaces modifiées par l’HMDS et une comparaison des taux de greffages ainsi déterminés. La détermination de la valeur du taux de greffage absolu, c’est à dire le nombre de silanols remplacés par des groupements TMS, a fait l’objet de nombreuses études, pour connaître sa valeur maximum lorsque l’HMDS réagit avec la silice. Sindorf [2] a remarqué que malgré la réactivité importante de l’HMDS, les taux de greffage restaient limités par rapport au nombre de silanols initiaux. Sur des gels de silice, il trouve des taux de greffage, déterminés par perte de masse, de 1,69 à 2,55 TMS/nm2 suivant les procédures de greffage employées. Il explique cette limitation par l’encombrement stérique des groupements TMS, et calcule alors de façon théorique l’encombrement stérique suivant deux modèles géométriques. La valeur maximale calculée est de 2,1 et 2,8 TMS/nm2 suivant le modèle adopté. Sindorf a aussi montré que la réaction est d’abord très rapide, conduisant au greffage de 1,5 TMS/nm2, valeur confirmée par Snyder [18], puis la réaction ralentit en raison de l’encombrement stérique. Le taux de greffage dépend également du temps de réaction. Gun’ko [5] montre que la réaction n’est complète qu’après 45 heures, à température ambiante. Cependant, le temps

Chapitre II : Modification chimique de surfaces de silice par l’hexaméthyldisilazane
61
de réaction nécessaire pour atteindre un greffage maximum, reporté par différent auteurs, est très variable, de 30 minutes [10] à 72 heures [3]. Dans une autre étude [19], Sindorf étudie l’effet de la déshydratation de la silice sur la fraction de silanols convertis en groupements TMS. Cette fraction augmente avec la température de déshydratation, puisque le nombre de silanols à convertir est plus faible, mais à partir de 560°C, la fraction diminue, ce qui s’explique par la diminution de la réactivité de la silice à ces hautes températures. La diminution de la réactivité des silanols à haute température est confirmée par Buzek [13] qui observe que le taux de greffage, voisin de 1,6 TMS/nm2 jusqu’à 600°C, chute à 1,27 TMS/nm2 à 800°C. Hertl et Hair [7] expliquent que l’HMDS ne réagit qu’avec les silanols libres, laissant les silanols vicinaux intacts. Ainsi, le taux de greffage maximum ne serait pas uniquement déterminé par l’encombrement stérique, mais également par la plus faible réactivité des silanols vicinaux. Morrow et Mc Farlan [16], qui trouvent un taux de greffage de 1,42 TMS/nm2 sur les 3,1 silanols totaux de l’Aerosil 150, affirment en revanche qu’une partie des silanols vicinaux réagissent également. On peut également calculer les taux de greffage relatifs aux silanols géminés et aux silanols libres [2]. Pour des gels de silice, le taux de greffage moyen déduit des mesures RMN 29Si CP/MAS est de 42%, avec des proportions différentes suivant le type de silanols. Le taux de greffage moyen des silanols libres est de 39%, tandis que celui des silanols géminés est de 65%. Il en est déduit que la réactivité vis-à-vis de l’HMDS des silanols géminés est plus importante que celle des silanols libres. A partir des taux de greffage en valeur relative, et de la mesure des taux de greffage en valeur absolue par perte de masse, le nombre total de silanols de surface a été calculé. Les valeurs trouvées, comprises entre 4,5 et 5,5 OH/nm2, sont en accord avec la constante de Zhuravlev, 4,6 OH/nm2. Reprenant une méthodologie similaire, Köhler [20] a également déterminé des taux de greffage sur des silices greffées TMS, par différentes procédures, et trouve des résultats cohérents avec ceux obtenus par d’autres techniques d’analyse (ATG, analyse élémentaire), ce qui permet de valider l’approche semi-quantitative utilisée en RMN 29Si CP/MAS. Enfin, cette technique a également été utilisée pour mesurer les taux de greffage de silices en solution (sols) modifiés par l’HMDS. Des taux de greffage de 5 à 33% ont été trouvés, suivant la manière dont les sols avaient été synthétisés et suivant le temps de réaction avec l’HMDS. Un taux de greffage relatif similaire, de 33%, est reporté par Huang [11] pour des sols également. Takei [3], après avoir déterminé le taux de greffage de silices poreuses modifiées avec différentes concentrations en groupements TMS, a étudié le phénomène d’adsorption de l’eau. Il a montré que même une fois ces surfaces modifiées, les molécules d’eau peuvent venir s’adsorber sur les silanols restants, entre les groupements TMS. Le volume occupé par les
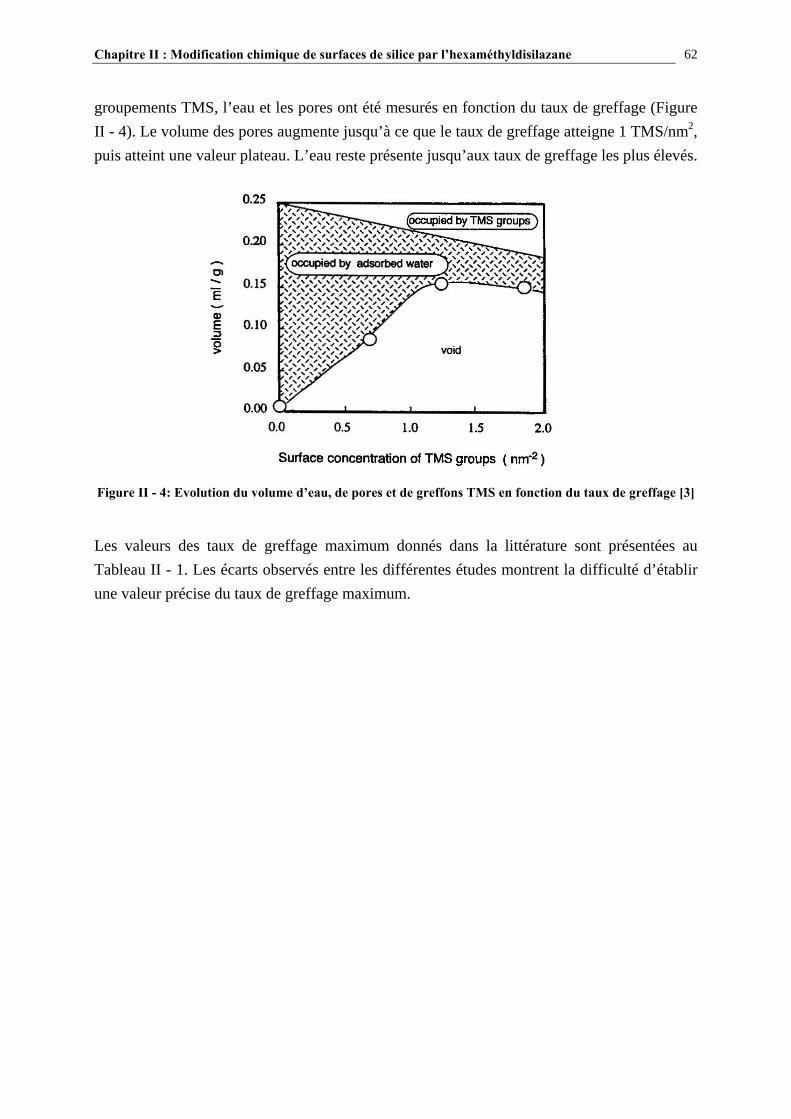
Chapitre II : Modification chimique de surfaces de silice par l’hexaméthyldisilazane
62
groupements TMS, l’eau et les pores ont été mesurés en fonction du taux de greffage (Figure II - 4). Le volume des pores augmente jusqu’à ce que le taux de greffage atteigne 1 TMS/nm2, puis atteint une valeur plateau. L’eau reste présente jusqu’aux taux de greffage les plus élevés. Figure II - 4: Evolution du volume d’eau, de pores et de greffons TMS en fonction du taux de greffage [3]
Les valeurs des taux de greffage maximum donnés dans la littérature sont présentées au Tableau II - 1. Les écarts observés entre les différentes études montrent la difficulté d’établir une valeur précise du taux de greffage maximum.
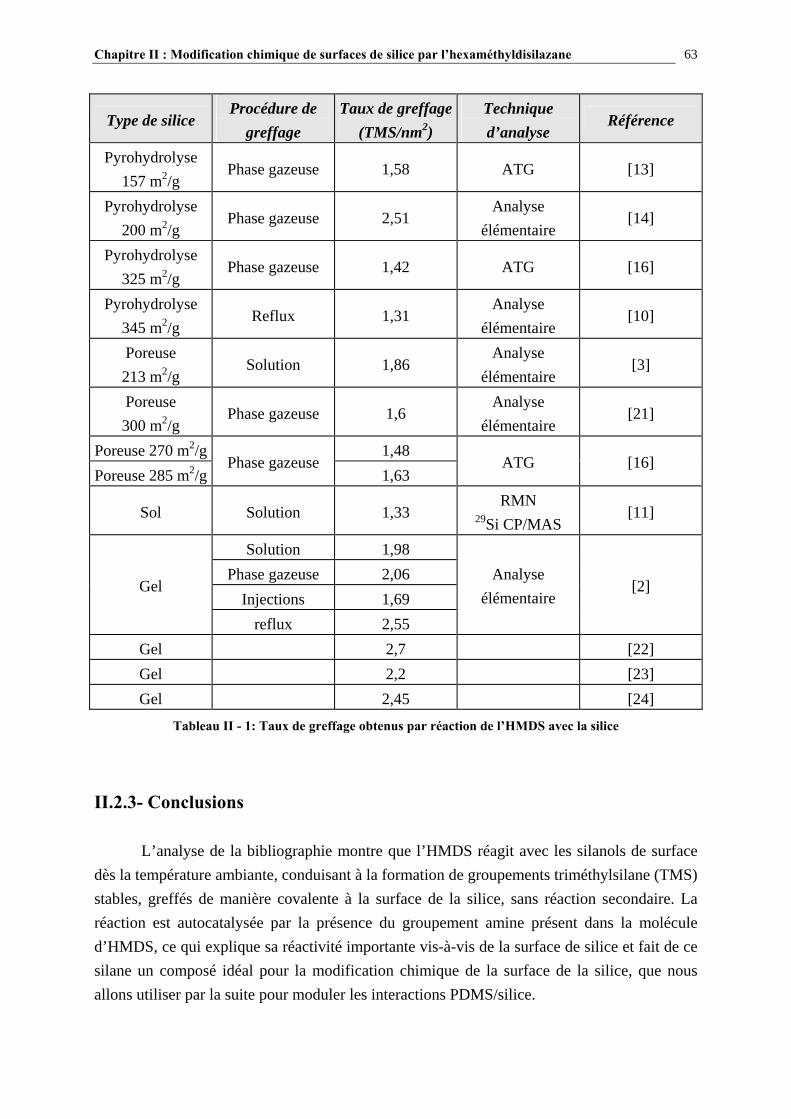
Chapitre II : Modification chimique de surfaces de silice par l’hexaméthyldisilazane
63
Type de silice Procédure de
greffage Taux de greffage
(TMS/nm2) Technique d’analyse
Référence
Pyrohydrolyse 157 m2/g
Phase gazeuse 1,58 ATG [13]
Pyrohydrolyse 200 m2/g
Phase gazeuse 2,51 Analyse
élémentaire [14]
Pyrohydrolyse 325 m2/g
Phase gazeuse 1,42 ATG [16]
Pyrohydrolyse 345 m2/g
Reflux 1,31 Analyse
élémentaire [10]
Poreuse 213 m2/g
Solution 1,86 Analyse
élémentaire [3]
Poreuse 300 m2/g
Phase gazeuse 1,6 Analyse
élémentaire [21]
Poreuse 270 m2/g 1,48 Poreuse 285 m2/g
Phase gazeuse 1,63
ATG [16]
Sol Solution 1,33 RMN
29Si CP/MAS [11]
Solution 1,98 Phase gazeuse 2,06
Injections 1,69 Gel
reflux 2,55
Analyse élémentaire
[2]
Gel 2,7 [22] Gel 2,2 [23] Gel 2,45 [24]
Tableau II - 1: Taux de greffage obtenus par réaction de l’HMDS avec la silice
II.2.3- Conclusions L’analyse de la bibliographie montre que l’HMDS réagit avec les silanols de surface dès la température ambiante, conduisant à la formation de groupements triméthylsilane (TMS) stables, greffés de manière covalente à la surface de la silice, sans réaction secondaire. La réaction est autocatalysée par la présence du groupement amine présent dans la molécule d’HMDS, ce qui explique sa réactivité importante vis-à-vis de la surface de silice et fait de ce silane un composé idéal pour la modification chimique de la surface de la silice, que nous allons utiliser par la suite pour moduler les interactions PDMS/silice.

Chapitre II : Modification chimique de surfaces de silice par l’hexaméthyldisilazane
64
La caractérisation des espèces TMS greffées peut se faire à l’aide des différentes
techniques d’analyse présentées précédemment (IRTF, RMN 29Si CP/MAS, analyse élémentaire du carbone…) et de nombreux résultats ont déjà été reportés par différents auteurs. Cependant, les taux de greffage donnés dans la littérature varient énormément suivant le type de silice, les conditions de greffage et les techniques utilisées. L’étude qui suit se focalise donc plutôt sur un seul type de silice (silice de pyrohydrolyse A200), modifiée par différentes concentrations d’HMDS. En couplant plusieurs techniques de caractérisation, nous pourrons ainsi conclure sur l’évolution du greffage et la valeur maximale du taux de greffage atteint. Les trois procédures de modification chimique de la surface de la silice que nous avons développé dans le cadre de ce travail, en solution et par voie gazeuse (ex-situ) et en milieu PDMS (in-situ) seront successivement présentées, après une brève description du type de silice utilisée et de l’HMDS.
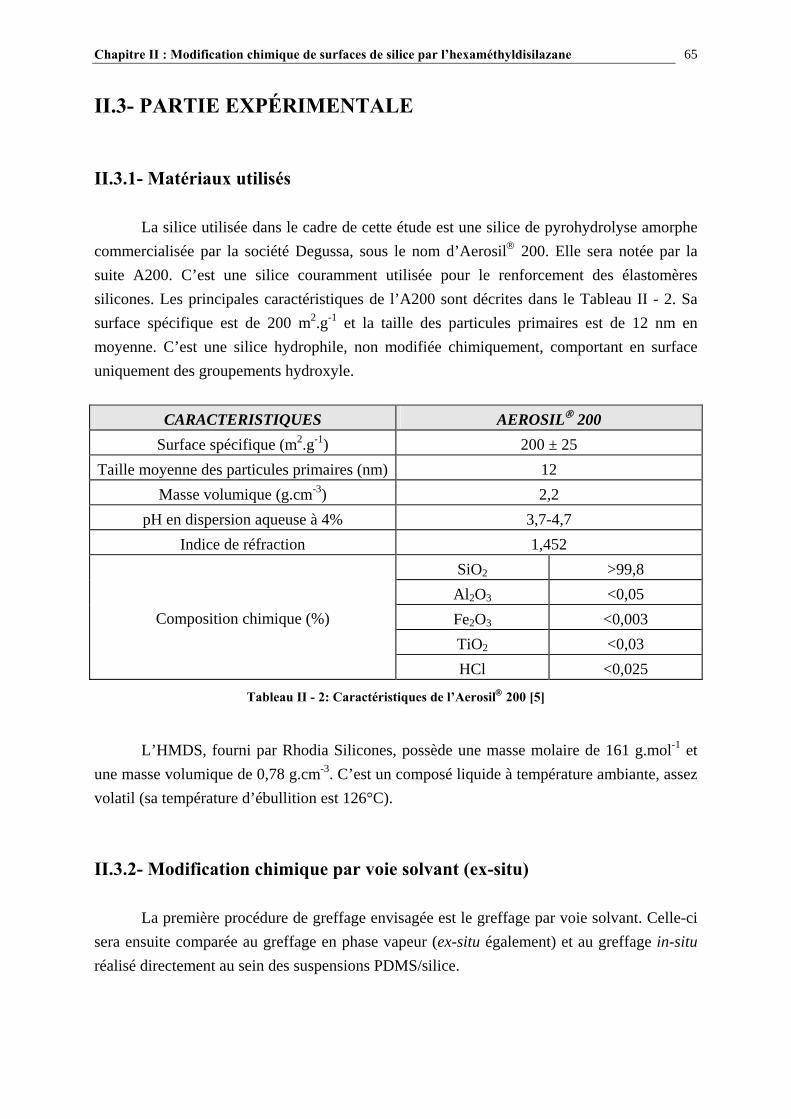
Chapitre II : Modification chimique de surfaces de silice par l’hexaméthyldisilazane
65
II.3- PARTIE EXPÉRIMENTALE II.3.1- Matériaux utilisés
La silice utilisée dans le cadre de cette étude est une silice de pyrohydrolyse amorphe commercialisée par la société Degussa, sous le nom d’Aerosil® 200. Elle sera notée par la suite A200. C’est une silice couramment utilisée pour le renforcement des élastomères silicones. Les principales caractéristiques de l’A200 sont décrites dans le Tableau II - 2. Sa surface spécifique est de 200 m2.g-1 et la taille des particules primaires est de 12 nm en moyenne. C’est une silice hydrophile, non modifiée chimiquement, comportant en surface uniquement des groupements hydroxyle.
CARACTERISTIQUES AEROSIL® 200 Surface spécifique (m2.g-1) 200 ± 25
Taille moyenne des particules primaires (nm) 12 Masse volumique (g.cm-3) 2,2
pH en dispersion aqueuse à 4% 3,7-4,7 Indice de réfraction 1,452
SiO2 >99,8 Al2O3 <0,05 Fe2O3 <0,003 TiO2 <0,03
Composition chimique (%)
HCl <0,025
Tableau II - 2: Caractéristiques de l’Aerosil® 200 [5]
L’HMDS, fourni par Rhodia Silicones, possède une masse molaire de 161 g.mol-1 et
une masse volumique de 0,78 g.cm-3. C’est un composé liquide à température ambiante, assez volatil (sa température d’ébullition est 126°C). II.3.2- Modification chimique par voie solvant (ex-situ) La première procédure de greffage envisagée est le greffage par voie solvant. Celle-ci sera ensuite comparée au greffage en phase vapeur (ex-situ également) et au greffage in-situ réalisé directement au sein des suspensions PDMS/silice.

Chapitre II : Modification chimique de surfaces de silice par l’hexaméthyldisilazane
66
La méthode de greffage par voie solvant consiste en trois étapes que nous allons détailler pour expliquer le mode opératoire optimisé qui a été adopté : i/ la préparation de la silice ii/ La réaction de greffage iii/ L’extraction des espèces physisorbées et le post-traitement de la silice i/ Comme nous l’avons vu au chapitre I, l’état initial de la silice (conditions de stockage, pré-traitement) est très important car le degré d’hydroxylation, c’est-à-dire les quantités de silanols et d’eau physisorbée à la surface, en est dépendant et va donc influencer les réactions de greffage. Pour partir d’un même état initial, la silice a été placée avant chaque réaction de greffage en étuve à 120°C pendant 1 heure. La température de 120°C a été également choisie pour éviter le phénomène de déshydroxylation qui commence à partir de 150°C comme cela a été montré précédemment (cf. paragraphe I.3.3.2). Ce séchage permet d’éliminer en grande partie l’eau physisorbée. La silice est alors placée sous atmosphère inerte d’azote. ii/ Le schéma du montage expérimental pour le greffage par voie solvant, inspiré des travaux décrits dans la partie bibliographique, est représenté sur la Figure II - 5. Le toluène a été initialement employé comme solvant, mais il a été montré que le toluène pouvait être absorbé en surface de la silice par de fortes interactions π-OH qui rendent difficile son élimination par séchage [25]. Le dichlorométhane a alors été utilisé comme solvant vecteur pour réaliser cette étude. La silice est placée en solution à 1% en poids dans le dichlorométhane. Dans le cas du greffage d’aminosilanes comme l’HMDS, l’eau n’est pas nécessaire à l’activation de la réaction, qui est catalysée par la fonction amine. Au contraire, la présence d’eau peut gêner la réaction des silanols avec l’HMDS, c’est pourquoi le montage a été placé sous un courant d’azote pour conserver une atmosphère inerte. La quantité de silane introduite est calculée en fonction du rapport stœchiométrique HMDS/silanol, noté r, calculé en supposant 4,6 OH/nm2 (constante de Kiselev-Zhuravlev) et en considérant que chaque molécule d’HMDS conduit à deux groupements triméthylsilane pouvant être greffés. L’Equation II - 1 permet de calculer r en fonction de la quantité de silice à greffer et de la quantité de silane introduit. La réaction se déroule sous agitation ultrasons, à température ambiante car l’HMDS est déjà très réactif vis-à-vis de la silice à cette température. Des temps de greffage de 15 minutes à 48 heures ont été étudiés, pour définir une durée de greffage optimale de 2 heures, sur la base des analyses réalisées par spectroscopie infrarouge. iii/ Enfin la dernière étape consiste à évaporer le solvant ainsi que l’excès éventuel d’HMDS puis à sécher les silices à 120°C en étuve. Lors de ce séchage, les espèces HMDS

Chapitre II : Modification chimique de surfaces de silice par l’hexaméthyldisilazane
67
non réagies (c’est-à-dire adsorbées) et les sous produits sont éliminés compte tenue de la faible tension de vapeur de l’HMDS.
Figure II - 5: Montage expérimental pour le greffage en solution de la silice A200 par l’hexaméthyldisilazane HMDS (volume du réacteur : 250 ml, volume de la solution : 150 ml)
Equation II - 1
Avec Na : Nombre d’Avogadro mHMDS : Masse d’HMDS MHMDS : Masse molaire de l’HMDS msilice : Masse de silice S : Surface spécifique de la silice Le Tableau II - 3 récapitule les échantillons réalisés par modification chimique par voie solvant, pour différentes quantités d’HMDS.
Quantité d’HMDS ( % en poids par rapport à la silice)
3 6 12 24 36 48
Rapport stœchiométrique HMDS/silanol correspondant, r
0,25 0,5 1 2 3 4
Tableau II - 3: Echantillons réalisés par modification chimique par voie solvant
II.3.3- Modification chimique par voie gazeuse (ex-situ) La modification chimique par voie gazeuse a été utilisée pour pouvoir comparer les taux de greffage obtenus avec ceux obtenus avec les autres protocoles de greffage et surtout pouvoir produire des quantités suffisantes de silice greffées nécessaires à la réalisation de suspensions PDMS/silice.
Silice en solution+ HMDS
Entrée d’azote
Sortie d’azote
Bain à ultrasons
HMDSsilice
HMDSa
MSmmNr 1810.6,4
2=

Chapitre II : Modification chimique de surfaces de silice par l’hexaméthyldisilazane
68
Ne disposant pas au départ de l’équipement nécessaire à la modification chimique en phase vapeur, un montage a été élaboré en adaptant un dessiccateur en verre, comme réacteur dans lequel la silice va être greffée. Au départ un flux d’azote entraîne l’HMDS sous forme gazeuse dans le bas du dessiccateur, où le flux gazeux est répartie dans l’enceinte réactionnelle à l’aide d’un fritté (porosité = 100 µm), ce qui assure un courant homogène du réactif sur toute la section. Au sein du réacteur, l’HMDS réagit avec la silice, qui est agitée par des pâles afin de garantir une exposition homogène de toute la silice au flux gazeux. Enfin, l’azote, l’excès d’HMDS et l’ammoniac formé sont évacués par le haut du montage et piégés dans un bulleur, ce dernier permettant également de contrôler le débit d’azote. L’ensemble du dispositif est décrit à la Figure II - 6.
Figure II - 6: Schéma du protocole de modification chimique par greffage d’HMDS en phase vapeur (volume du réacteur : 5400 cm3 ; quantité maximum de silice traitée : 90g)
Le temps de réaction dans un tel procédé est déterminé par le temps nécessaire pour volatiliser l’HMDS et passer au sein du réacteur, ainsi ce temps de réaction est dépendant du débit d’azote imposé. Des essais ont été menés pour définir un débit d’azote assurant un greffage maximum par rapport à la quantité d’HMDS introduit. L’homogénéité du traitement par ce procédé a été vérifié par deux méthodes. Une première estimation de la dispersion de la silice au sein du réacteur consiste à introduire quelques grammes de noir de carbone avec la silice. Après agitation, le noir de carbone est parfaitement dispersé au sein de la silice car on peut observer une poudre grise homogène à l’échelle macroscopique. Une évaluation plus fine consiste à prélever après réaction de la silice modifiée en différents points du réacteur. Après analyse IRTF, il a été montré qu’aucune différence ne pouvait être observée entre les différents prélèvements. Ainsi, on peut considérer que le dispositif mis au point permet d’assurer une modification chimique par voie gazeuse homogène au moins à l’échelle du prélèvement sur tout le volume réactionnel.
N2Silice
N2 + HMDS résiduel
HMDSFritté
Tige d’agitation
N2Silice
N2 + HMDS résiduel
HMDSFritté
Tige d’agitation

Chapitre II : Modification chimique de surfaces de silice par l’hexaméthyldisilazane
69
L’ensemble des échantillons réalisés par cette procédure est présenté au Tableau II - 4.
Quantité d’HMDS (% en poids par rapport à la silice)
1,5 3 4,5 6 9 12 18 24 36 48
Rapport stœchiométrique HMDS/silanol correspondant, r
0,12 0,25 0,37 0,5 0,75 1 1,5 2 3 4
Tableau II - 4: Echantillons réalisés par modification chimique en phase vapeur
II.3.4- Modification chimique au sein des suspensions PDMS/silice ( voie in-situ) La modification chimique de la silice peut également s’effectuer simultanément à la réalisation des suspensions PDMS/silice. On peut considérer qu’il s’agit d’une modification par voie solvant de haute viscosité, le milieu réactionnel étant le PDMS. Cependant de nombreuses différences interviennent : la viscosité du milieu plus importante, les interactions PDMS-silice, etc. Cette procédure a donc été utilisée afin de comparer les taux de greffage obtenus avec les autres protocoles. Par ailleurs c’est le procédé qui est utilisé au niveau industriel pour la production des élastomères silicones renforcés par la silice. La préparation des suspensions en présence d’HMDS est décrite au paragraphe III.3.2.1. Les silices ont été extraites des suspensions par centrifugation pour les concentrer avant analyse du taux de greffage. La séparation du PDMS de la silice est difficile en raison des fortes interactions entre ces deux composés. Les suspensions concentrées ont été alors mises en solution dans le cyclohexane, bon solvant des PDMS, puis centrifugées à nouveau. On récupère ainsi la silice avec du PDMS résiduel présent. Les conditions de centrifugation, en particulier la vitesse et le temps de centrifugation, et la concentration des solutions sont capitales pour obtenir des silices avec une quantité de PDMS résiduel la plus faible possible. Ces conditions ont été optimisées même si elles peuvent varier d’une formulation à une autre. Le choix des paramètres est le suivant : 2 heures de centrifugation à vitesse de 5 000 tours/min de solutions diluées à 15% en poids dans le cyclohexane. Les échantillons utilisés pour l’analyse du greffage sont donnés dans le Tableau II - 5, avec le rapport stœchiométrique HMDS/silanol correspondant et la nomenclature utilisée pour définir les suspensions.
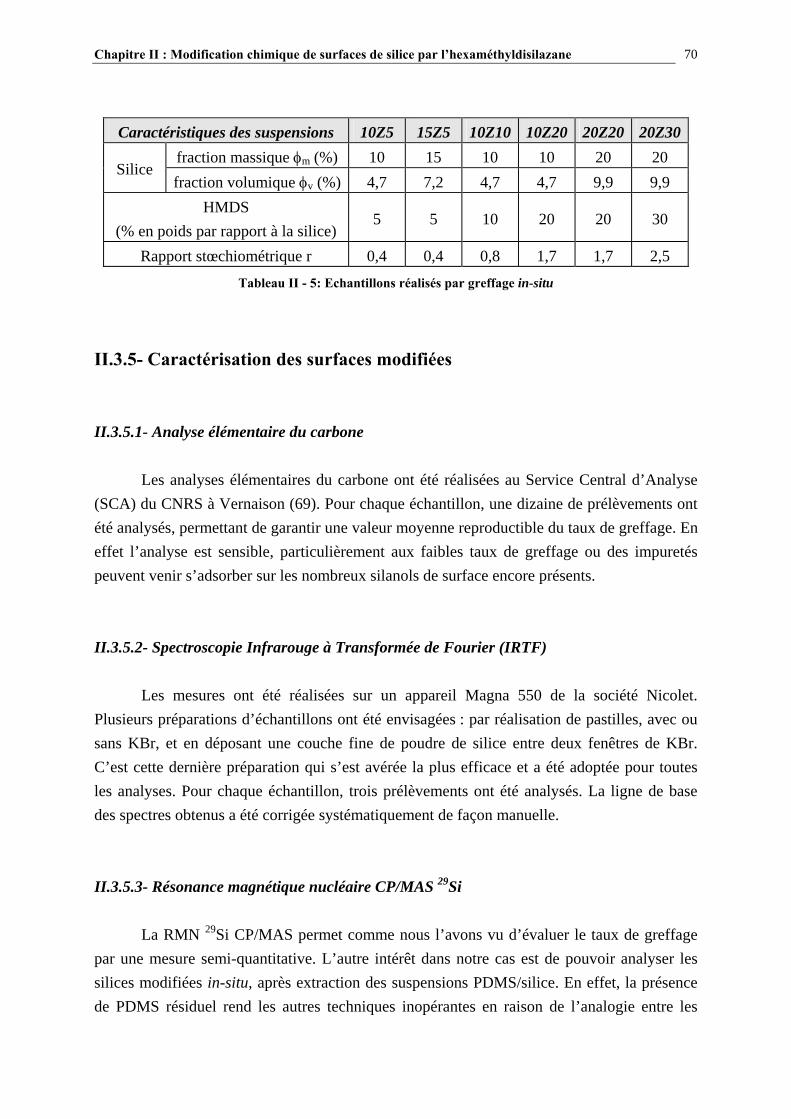
Chapitre II : Modification chimique de surfaces de silice par l’hexaméthyldisilazane
70
Caractéristiques des suspensions 10Z5 15Z5 10Z10 10Z20 20Z20 20Z30
fraction massique φm (%) 10 15 10 10 20 20 Silice
fraction volumique φv (%) 4,7 7,2 4,7 4,7 9,9 9,9 HMDS
(% en poids par rapport à la silice) 5 5 10 20 20 30
Rapport stœchiométrique r 0,4 0,4 0,8 1,7 1,7 2,5 Tableau II - 5: Echantillons réalisés par greffage in-situ
II.3.5- Caractérisation des surfaces modifiées II.3.5.1- Analyse élémentaire du carbone Les analyses élémentaires du carbone ont été réalisées au Service Central d’Analyse (SCA) du CNRS à Vernaison (69). Pour chaque échantillon, une dizaine de prélèvements ont été analysés, permettant de garantir une valeur moyenne reproductible du taux de greffage. En effet l’analyse est sensible, particulièrement aux faibles taux de greffage ou des impuretés peuvent venir s’adsorber sur les nombreux silanols de surface encore présents. II.3.5.2- Spectroscopie Infrarouge à Transformée de Fourier (IRTF) Les mesures ont été réalisées sur un appareil Magna 550 de la société Nicolet. Plusieurs préparations d’échantillons ont été envisagées : par réalisation de pastilles, avec ou sans KBr, et en déposant une couche fine de poudre de silice entre deux fenêtres de KBr. C’est cette dernière préparation qui s’est avérée la plus efficace et a été adoptée pour toutes les analyses. Pour chaque échantillon, trois prélèvements ont été analysés. La ligne de base des spectres obtenus a été corrigée systématiquement de façon manuelle. II.3.5.3- Résonance magnétique nucléaire CP/MAS 29Si La RMN 29Si CP/MAS permet comme nous l’avons vu d’évaluer le taux de greffage par une mesure semi-quantitative. L’autre intérêt dans notre cas est de pouvoir analyser les silices modifiées in-situ, après extraction des suspensions PDMS/silice. En effet, la présence de PDMS résiduel rend les autres techniques inopérantes en raison de l’analogie entre les

Chapitre II : Modification chimique de surfaces de silice par l’hexaméthyldisilazane
71
groupements TMS greffés et les groupements diméthylsilane du PDMS. Seule la RMN 29Si CP/MAS permet de séparer ces deux espèces de même nature par leurs déplacements chimiques différents. Les spectres de RMN 29Si CP/MAS ont été obtenus à partir d’un spectromètre DSX400 (400 MHz) de la société Bruker, à 5 kHz avec un temps de contact de 5 ms. L’aire des pics Q2 et Q3 ont été calculées à partir de la déconvolution du massif des Q2, Q3, Q4, par un logiciel de déconvolution. Les silices ont été compactées par immersion dans un solvant suivi d’un séchage à 120°C. Cette étape est nécessaire afin de pouvoir placer suffisamment de silice dans le rotor de l’appareil. II.3.5.4- Mesures de mouillage Un appareil Digidrop de la société GBX a été utilisé pour mesurer automatiquement l’angle de contact. L’image du profil de la goutte est enregistrée informatiquement par une caméra, suivi du calcul de l’angle de contact. Les mesures ont été réalisées sur plusieurs gouttes, avec plusieurs surfaces par échantillon (préparées par compactage de la poudre), pour s’affranchir des problèmes de reproductibilité. Ces mesures sont parfois sensibles et peuvent varier en fonction de la rugosité de la surface. D’autre part, pour les surfaces peu modifiées, le fort caractère hydrophile de la surface conduit à des mesures délicates car l’eau s’adsorbe rapidement à la surface (l’angle de mouillage diminue continuellement avec le temps lors de la pénétration de l’eau par capillarité dans les pores). Aussi les mesures d’angles de contact pour les faibles taux de greffage sont entachées d’une erreur importante.

Chapitre II : Modification chimique de surfaces de silice par l’hexaméthyldisilazane
72
II.4- RÉSULTATS ET DISCUSSION II.4.1- Evolution du greffage en fonction de la quantité de silane introduit et comparaison des taux de greffage voie solvant/voie gazeuse Dans un premier temps nous nous intéressons aux silices modifiées ex-situ, par voie solvant et par voie gazeuse. L’objectif de cette partie est de mettre en place une méthodologie permettant de suivre l’évolution du greffage en fonction de la quantité d’HMDS introduite et de caractériser de manière précise le taux de greffage des silices pour ces deux procédures de greffage. II.4.1.1- Suivi par spectroscopie IRTF
La silice A200 non modifiée a d’abord été caractérisée. La Figure II - 7 a montre le spectre infrarouge obtenu sur la plage 1500-4000 cm-1 où les pics correspondant aux groupements de surface sont mis en évidence. La bande d’adsorption comprise entre 3 000 et 3 750 cm-1 est due aux groupements hydroxyle de surface. Parmi eux, les silanols libres (isolés et géminés) se distinguent nettement par un pic assez étroit à 3 750 cm-1. Le reste de la bande d’adsorption est du aux autres types de groupements hydroxyle, à savoir les silanols vicinaux et l’eau physisorbée. Cette dernière est facilement mise en évidence : en comparant le spectre de la silice avant et après passage à l’étuve à 120°C, on observe une large diminution de la bande comprise entre 3 000 et 3 750 cm-1, ce qui confirme la forte adsorption de l’eau par la silice non modifiée (caractère hydrophile). La double bande comprise entre 1 750 et 2 100 cm-1 est attribuée aux liaisons Si-O-Si du volume de la silice qui n’est donc peu modifiée lors du greffage de surface de la silice. Cette bande sera alors utilisée comme référence par la suite.
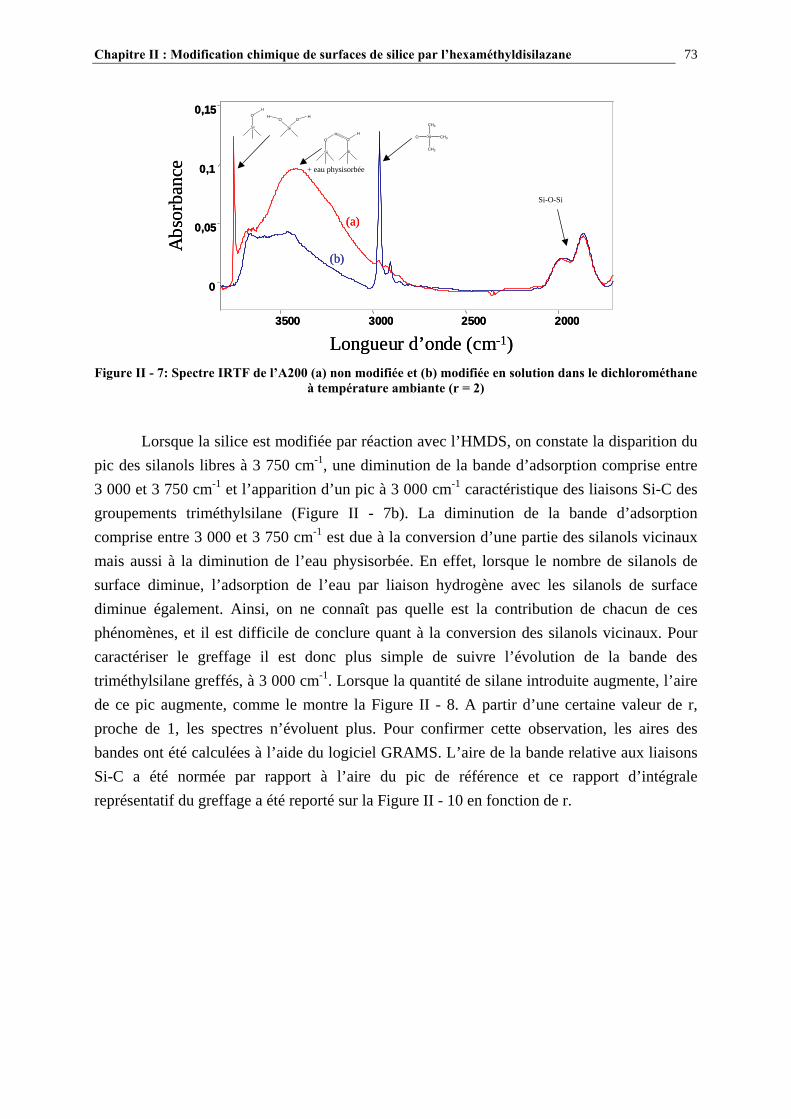
Chapitre II : Modification chimique de surfaces de silice par l’hexaméthyldisilazane
73
Figure II - 7: Spectre IRTF de l’A200 (a) non modifiée et (b) modifiée en solution dans le dichlorométhane
à température ambiante (r = 2)
Lorsque la silice est modifiée par réaction avec l’HMDS, on constate la disparition du pic des silanols libres à 3 750 cm-1, une diminution de la bande d’adsorption comprise entre 3 000 et 3 750 cm-1 et l’apparition d’un pic à 3 000 cm-1 caractéristique des liaisons Si-C des groupements triméthylsilane (Figure II - 7b). La diminution de la bande d’adsorption comprise entre 3 000 et 3 750 cm-1 est due à la conversion d’une partie des silanols vicinaux mais aussi à la diminution de l’eau physisorbée. En effet, lorsque le nombre de silanols de surface diminue, l’adsorption de l’eau par liaison hydrogène avec les silanols de surface diminue également. Ainsi, on ne connaît pas quelle est la contribution de chacun de ces phénomènes, et il est difficile de conclure quant à la conversion des silanols vicinaux. Pour caractériser le greffage il est donc plus simple de suivre l’évolution de la bande des triméthylsilane greffés, à 3 000 cm-1. Lorsque la quantité de silane introduite augmente, l’aire de ce pic augmente, comme le montre la Figure II - 8. A partir d’une certaine valeur de r, proche de 1, les spectres n’évoluent plus. Pour confirmer cette observation, les aires des bandes ont été calculées à l’aide du logiciel GRAMS. L’aire de la bande relative aux liaisons Si-C a été normée par rapport à l’aire du pic de référence et ce rapport d’intégrale représentatif du greffage a été reporté sur la Figure II - 10 en fonction de r.
0
0,05
0,1
0,15
3500 3000 2500 2000
Abs
orba
nce
Longueur d’onde (cm-1)
(b)
(a)
0
0,05
0,1
0,15
3500 3000 2500 2000
Abs
orba
nce
Longueur d’onde (cm-1)
(b)
(a)
Si
OOHH
Si
OH
Si
OH
Si
OH
+ eau physisorbée
Si CH3O
CH3
CH3
Si-O-Si
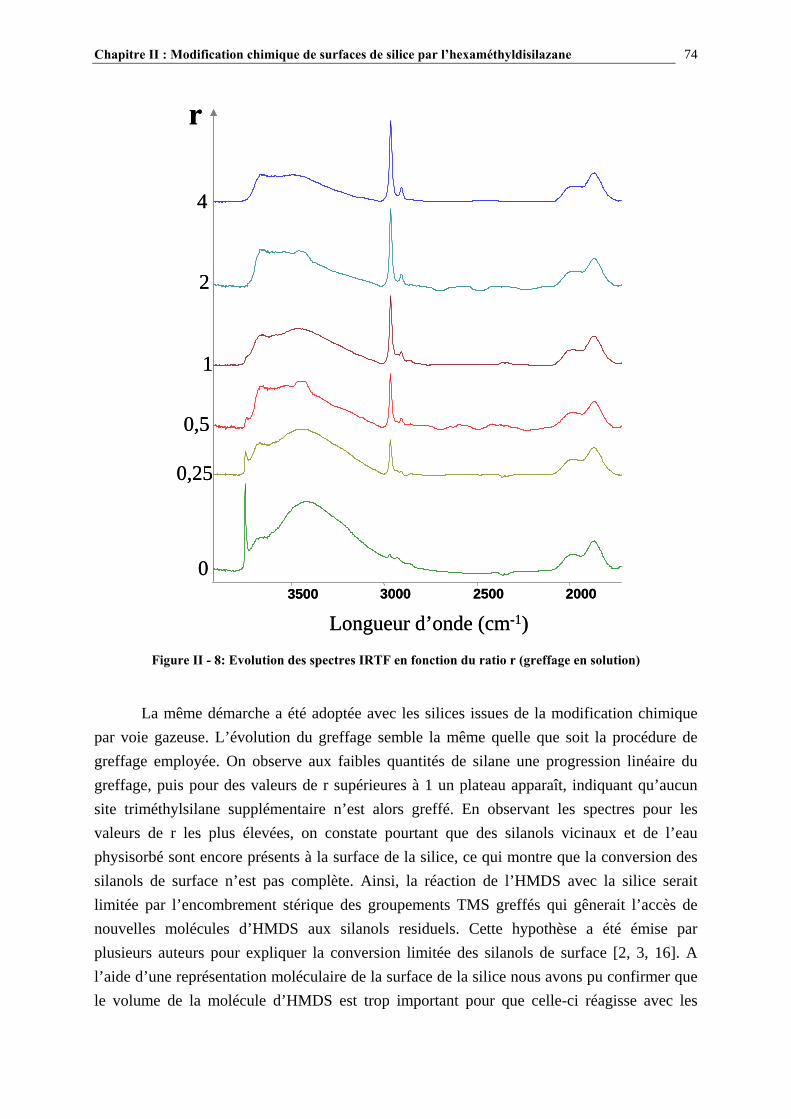
Chapitre II : Modification chimique de surfaces de silice par l’hexaméthyldisilazane
74
Figure II - 8: Evolution des spectres IRTF en fonction du ratio r (greffage en solution)
La même démarche a été adoptée avec les silices issues de la modification chimique par voie gazeuse. L’évolution du greffage semble la même quelle que soit la procédure de greffage employée. On observe aux faibles quantités de silane une progression linéaire du greffage, puis pour des valeurs de r supérieures à 1 un plateau apparaît, indiquant qu’aucun site triméthylsilane supplémentaire n’est alors greffé. En observant les spectres pour les valeurs de r les plus élevées, on constate pourtant que des silanols vicinaux et de l’eau physisorbé sont encore présents à la surface de la silice, ce qui montre que la conversion des silanols de surface n’est pas complète. Ainsi, la réaction de l’HMDS avec la silice serait limitée par l’encombrement stérique des groupements TMS greffés qui gênerait l’accès de nouvelles molécules d’HMDS aux silanols residuels. Cette hypothèse a été émise par plusieurs auteurs pour expliquer la conversion limitée des silanols de surface [2, 3, 16]. A l’aide d’une représentation moléculaire de la surface de la silice nous avons pu confirmer que le volume de la molécule d’HMDS est trop important pour que celle-ci réagisse avec les
3500 3000 2500 2000
r
4
2
1
0,5
0,25
0
Longueur d’onde (cm-1)3500 3000 2500 2000
r
4
2
1
0,5
0,25
0
Longueur d’onde (cm-1)
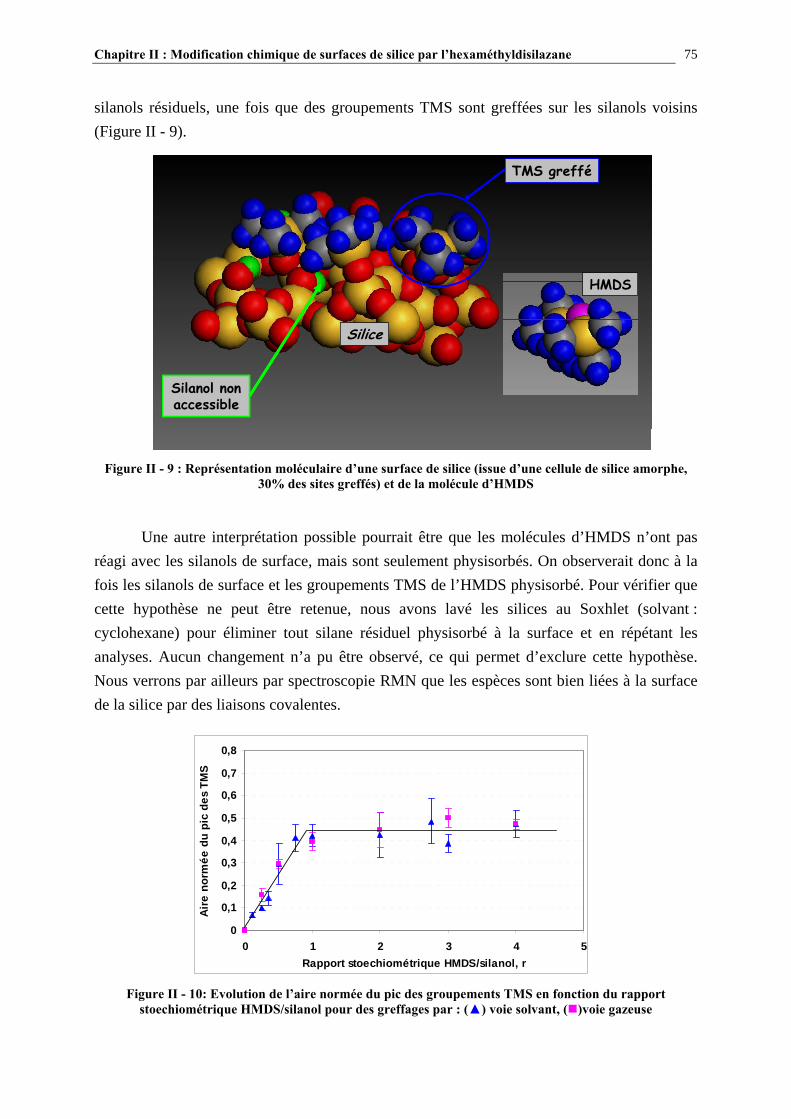
Chapitre II : Modification chimique de surfaces de silice par l’hexaméthyldisilazane
75
silanols résiduels, une fois que des groupements TMS sont greffées sur les silanols voisins (Figure II - 9).
Figure II - 9 : Représentation moléculaire d’une surface de silice (issue d’une cellule de silice amorphe, 30% des sites greffés) et de la molécule d’HMDS
Une autre interprétation possible pourrait être que les molécules d’HMDS n’ont pas réagi avec les silanols de surface, mais sont seulement physisorbés. On observerait donc à la fois les silanols de surface et les groupements TMS de l’HMDS physisorbé. Pour vérifier que cette hypothèse ne peut être retenue, nous avons lavé les silices au Soxhlet (solvant : cyclohexane) pour éliminer tout silane résiduel physisorbé à la surface et en répétant les analyses. Aucun changement n’a pu être observé, ce qui permet d’exclure cette hypothèse. Nous verrons par ailleurs par spectroscopie RMN que les espèces sont bien liées à la surface de la silice par des liaisons covalentes.
Figure II - 10: Evolution de l’aire normée du pic des groupements TMS en fonction du rapport stoechiométrique HMDS/silanol pour des greffages par : (▲) voie solvant, ( )voie gazeuse
0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0 1 2 3 4 5Rapport stoechiométrique HMDS/silanol, r
Aire
nor
mée
du
pic
des
TMS
TMS grefféTMS greffé
HMDS
Silanol non accessibleSilanol non accessible
Silice

Chapitre II : Modification chimique de surfaces de silice par l’hexaméthyldisilazane
76
Les évolutions comparables des rapports d’intégrale pour les deux types de procédure montrent que ce n’est pas le procédé qui est le facteur limitant de la réaction, puisque dans les deux cas on atteint un plateau pour les mêmes quantités de silane. La réactivité et l’accessibilité des sites sont donc les mêmes quel que soit le type de procédure employé. En ce qui concerne la reproductibilité des mesures, l’incertitude sur chaque échantillon est liée non seulement à l’incertitude due à la mesure, mais également au calcul du rapport d’intégrale et à la correction au préalable de la ligne de base de chaque spectre. On note cependant une dispersion des résultats relativement faible. En conclusion, l’IRTF apporte déjà de nombreuses informations sur le mode de greffage, même si cette technique reste dans un premier temps uniquement qualitative. En la couplant à une technique quantitative telle que l’analyse élémentaire, nous verrons qu’il sera possible de calibrer l’IRTF afin d’obtenir des résultats quantitatifs du taux surfacique d’espèces TMS greffées. II.4.1.2- Suivi par RMN 29Si CP/MAS La RMN 29Si CP/MAS permet de vérifier si des liaisons O-Si-(CH3)3 ont bien été formées. Le spectre de la silice A200 avant modification est représenté sur la Figure II - 11. Le massif des espèces Q2, Q3 et Q4 est compris entre –120 ppm et –80 ppm. Lorsque la silice est modifiée on observe l’apparition à 14 ppm d’une raie de résonnance supplémentaire caractéristique des silicium liés à des groupements TMS (noté M), comme le montre la Figure II - 12 présentant les spectres des silices modifiées par voie solvant et par voie gazeuse pour un large excès d’HMDS (r = 2).
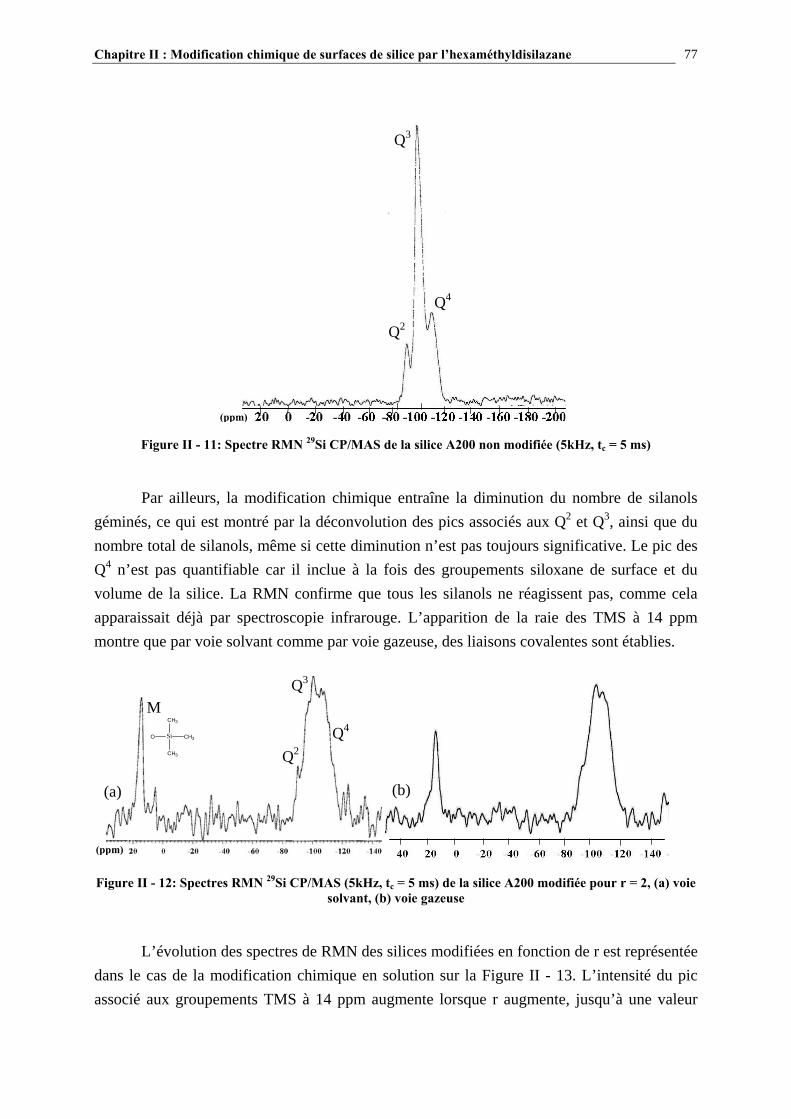
Chapitre II : Modification chimique de surfaces de silice par l’hexaméthyldisilazane
77
Figure II - 11: Spectre RMN 29Si CP/MAS de la silice A200 non modifiée (5kHz, tc = 5 ms)
Par ailleurs, la modification chimique entraîne la diminution du nombre de silanols géminés, ce qui est montré par la déconvolution des pics associés aux Q2 et Q3, ainsi que du nombre total de silanols, même si cette diminution n’est pas toujours significative. Le pic des Q4 n’est pas quantifiable car il inclue à la fois des groupements siloxane de surface et du volume de la silice. La RMN confirme que tous les silanols ne réagissent pas, comme cela apparaissait déjà par spectroscopie infrarouge. L’apparition de la raie des TMS à 14 ppm montre que par voie solvant comme par voie gazeuse, des liaisons covalentes sont établies. Figure II - 12: Spectres RMN 29Si CP/MAS (5kHz, tc = 5 ms) de la silice A200 modifiée pour r = 2, (a) voie
solvant, (b) voie gazeuse
L’évolution des spectres de RMN des silices modifiées en fonction de r est représentée dans le cas de la modification chimique en solution sur la Figure II - 13. L’intensité du pic associé aux groupements TMS à 14 ppm augmente lorsque r augmente, jusqu’à une valeur
(b) (a)
(ppm)
(ppm)
Si CH3O
CH3
CH3
Q2
Q3
Q4
Q2
Q3
Q4
M

Chapitre II : Modification chimique de surfaces de silice par l’hexaméthyldisilazane
78
limite. A partir de la déconvolution des pics Q2 et Q3, les taux de greffage ont été calculés. Ces valeurs ont ensuite été reportées en fonction de r sur la Figure II - 14. L’évolution du taux de greffage est identique pour les deux types de modification chimique et correspond à l’évolution observée par IRTF. L’incertitude sur les valeurs aux faibles taux de greffage est due à la difficulté d’évaluer avec précision l’aire de la raie de résonance associée aux groupements TMS car celle-ci étant peu intense, elle se confond avec le bruit. La valeur maximale du taux de greffage est de 0,8 TMS/nm2, valeur obtenue à partir de r proche de 1. Ce taux de greffage maximum semble donc sous-estimé vis à vis des valeurs reportées dans la littérature et des résultats d’analyse élémentaire que nous verrons plus loin. Ceci peut s’expliquer par la surestimation des silanols de surface due à la prise en compte d’une partie des silanols internes de la silice.
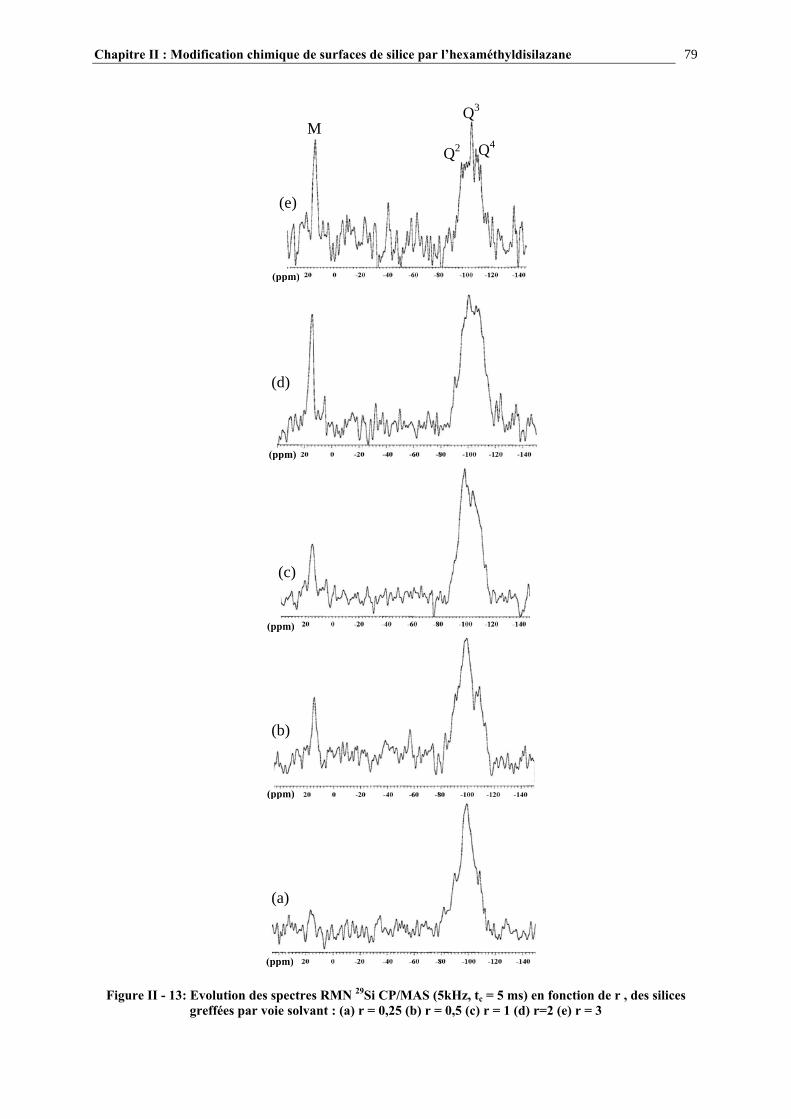
Chapitre II : Modification chimique de surfaces de silice par l’hexaméthyldisilazane
79
Figure II - 13: Evolution des spectres RMN 29Si CP/MAS (5kHz, tc = 5 ms) en fonction de r , des silices greffées par voie solvant : (a) r = 0,25 (b) r = 0,5 (c) r = 1 (d) r=2 (e) r = 3
(a)
(b)
(c)
(d)
(e)
(ppm)
(ppm)
(ppm)
(ppm)
(ppm)
Q2
Q3
Q4M
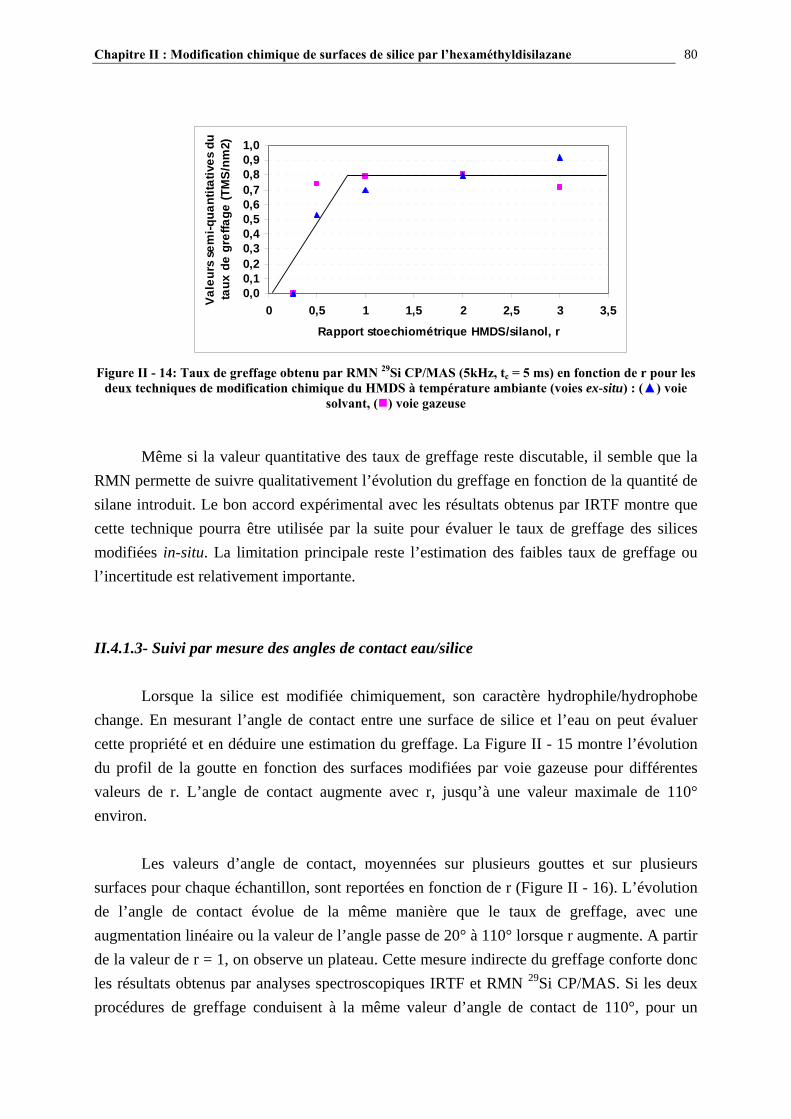
Chapitre II : Modification chimique de surfaces de silice par l’hexaméthyldisilazane
80
Figure II - 14: Taux de greffage obtenu par RMN 29Si CP/MAS (5kHz, tc = 5 ms) en fonction de r pour les
deux techniques de modification chimique du HMDS à température ambiante (voies ex-situ) : (▲) voie solvant, ( ) voie gazeuse
Même si la valeur quantitative des taux de greffage reste discutable, il semble que la RMN permette de suivre qualitativement l’évolution du greffage en fonction de la quantité de silane introduit. Le bon accord expérimental avec les résultats obtenus par IRTF montre que cette technique pourra être utilisée par la suite pour évaluer le taux de greffage des silices modifiées in-situ. La limitation principale reste l’estimation des faibles taux de greffage ou l’incertitude est relativement importante. II.4.1.3- Suivi par mesure des angles de contact eau/silice Lorsque la silice est modifiée chimiquement, son caractère hydrophile/hydrophobe change. En mesurant l’angle de contact entre une surface de silice et l’eau on peut évaluer cette propriété et en déduire une estimation du greffage. La Figure II - 15 montre l’évolution du profil de la goutte en fonction des surfaces modifiées par voie gazeuse pour différentes valeurs de r. L’angle de contact augmente avec r, jusqu’à une valeur maximale de 110° environ. Les valeurs d’angle de contact, moyennées sur plusieurs gouttes et sur plusieurs surfaces pour chaque échantillon, sont reportées en fonction de r (Figure II - 16). L’évolution de l’angle de contact évolue de la même manière que le taux de greffage, avec une augmentation linéaire ou la valeur de l’angle passe de 20° à 110° lorsque r augmente. A partir de la valeur de r = 1, on observe un plateau. Cette mesure indirecte du greffage conforte donc les résultats obtenus par analyses spectroscopiques IRTF et RMN 29Si CP/MAS. Si les deux procédures de greffage conduisent à la même valeur d’angle de contact de 110°, pour un
0,00,10,20,30,40,50,60,70,80,91,0
0 0,5 1 1,5 2 2,5 3 3,5
Rapport stoechiométrique HMDS/silanol, r
Vale
urs
sem
i-qua
ntita
tives
du
taux
de
gref
fage
(TM
S/nm
2)
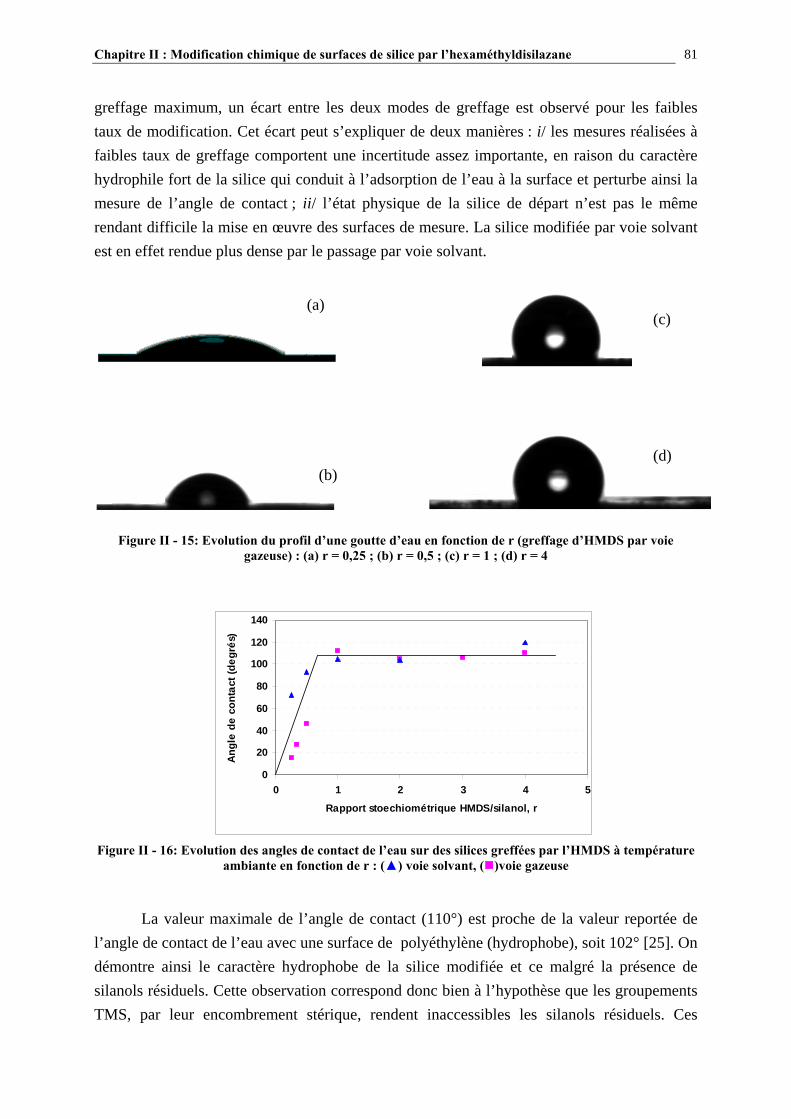
Chapitre II : Modification chimique de surfaces de silice par l’hexaméthyldisilazane
81
greffage maximum, un écart entre les deux modes de greffage est observé pour les faibles taux de modification. Cet écart peut s’expliquer de deux manières : i/ les mesures réalisées à faibles taux de greffage comportent une incertitude assez importante, en raison du caractère hydrophile fort de la silice qui conduit à l’adsorption de l’eau à la surface et perturbe ainsi la mesure de l’angle de contact ; ii/ l’état physique de la silice de départ n’est pas le même rendant difficile la mise en œuvre des surfaces de mesure. La silice modifiée par voie solvant est en effet rendue plus dense par le passage par voie solvant.
Figure II - 15: Evolution du profil d’une goutte d’eau en fonction de r (greffage d’HMDS par voie gazeuse) : (a) r = 0,25 ; (b) r = 0,5 ; (c) r = 1 ; (d) r = 4
Figure II - 16: Evolution des angles de contact de l’eau sur des silices greffées par l’HMDS à température
ambiante en fonction de r : (▲) voie solvant, ( )voie gazeuse
La valeur maximale de l’angle de contact (110°) est proche de la valeur reportée de l’angle de contact de l’eau avec une surface de polyéthylène (hydrophobe), soit 102° [25]. On démontre ainsi le caractère hydrophobe de la silice modifiée et ce malgré la présence de silanols résiduels. Cette observation correspond donc bien à l’hypothèse que les groupements TMS, par leur encombrement stérique, rendent inaccessibles les silanols résiduels. Ces
(a)
(b)
(c)
(d)
0
20
40
60
80
100
120
140
0 1 2 3 4 5
Rapport stoechiométrique HMDS/silanol, r
Angl
e de
con
tact
(deg
rés)
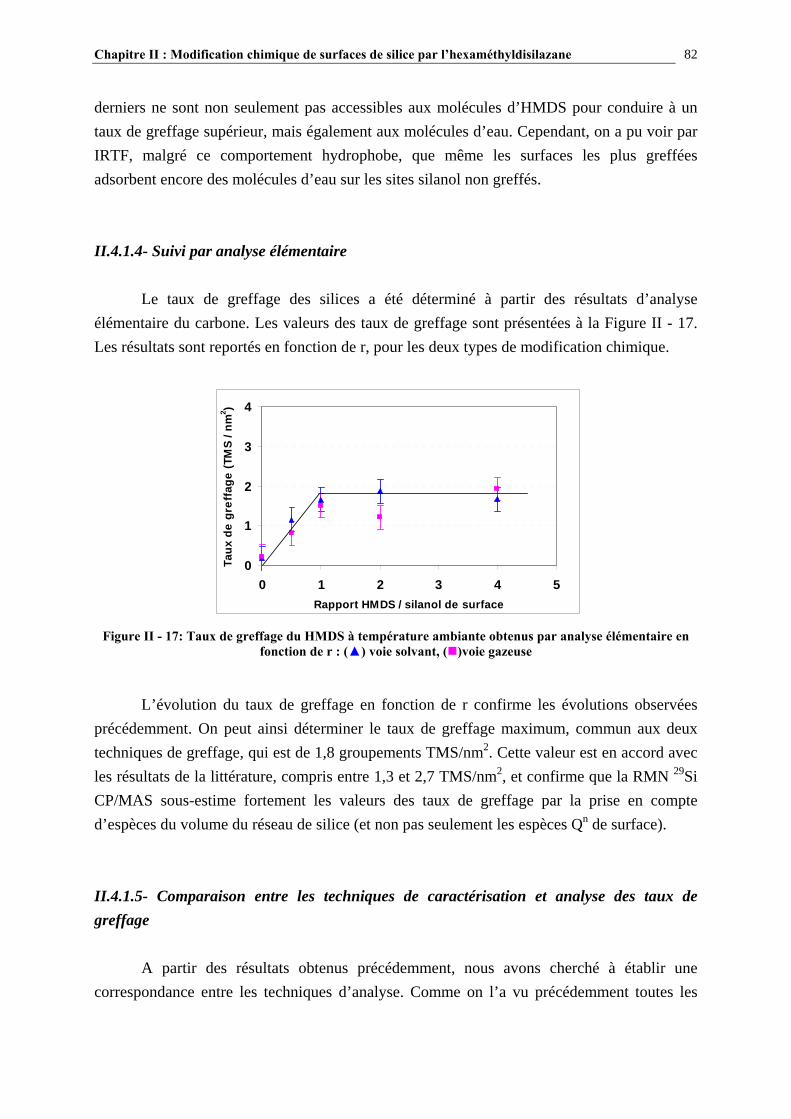
Chapitre II : Modification chimique de surfaces de silice par l’hexaméthyldisilazane
82
derniers ne sont non seulement pas accessibles aux molécules d’HMDS pour conduire à un taux de greffage supérieur, mais également aux molécules d’eau. Cependant, on a pu voir par IRTF, malgré ce comportement hydrophobe, que même les surfaces les plus greffées adsorbent encore des molécules d’eau sur les sites silanol non greffés. II.4.1.4- Suivi par analyse élémentaire Le taux de greffage des silices a été déterminé à partir des résultats d’analyse élémentaire du carbone. Les valeurs des taux de greffage sont présentées à la Figure II - 17. Les résultats sont reportés en fonction de r, pour les deux types de modification chimique.
Figure II - 17: Taux de greffage du HMDS à température ambiante obtenus par analyse élémentaire en fonction de r : (▲) voie solvant, ( )voie gazeuse
L’évolution du taux de greffage en fonction de r confirme les évolutions observées précédemment. On peut ainsi déterminer le taux de greffage maximum, commun aux deux techniques de greffage, qui est de 1,8 groupements TMS/nm2. Cette valeur est en accord avec les résultats de la littérature, compris entre 1,3 et 2,7 TMS/nm2, et confirme que la RMN 29Si CP/MAS sous-estime fortement les valeurs des taux de greffage par la prise en compte d’espèces du volume du réseau de silice (et non pas seulement les espèces Qn de surface). II.4.1.5- Comparaison entre les techniques de caractérisation et analyse des taux de greffage A partir des résultats obtenus précédemment, nous avons cherché à établir une correspondance entre les techniques d’analyse. Comme on l’a vu précédemment toutes les
0
1
2
3
4
0 1 2 3 4 5Rapport HMDS / silanol de surface
Taux
de
gref
fage
(TM
S / n
m2 )
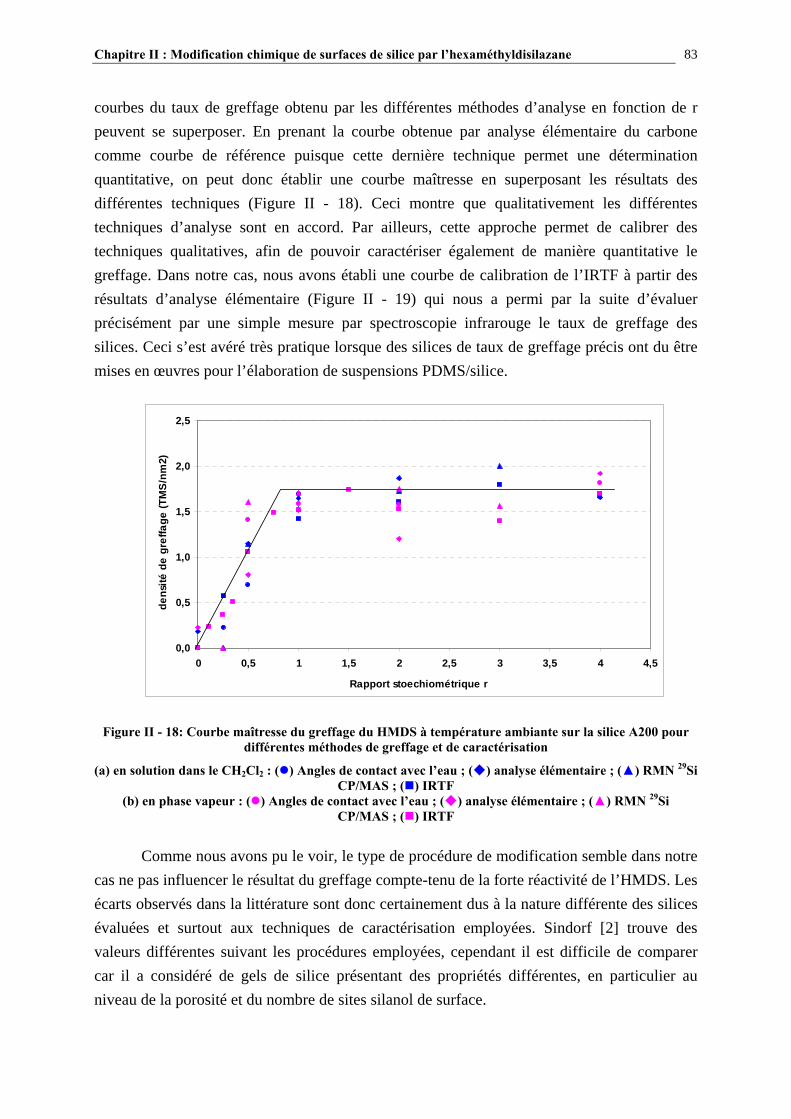
Chapitre II : Modification chimique de surfaces de silice par l’hexaméthyldisilazane
83
courbes du taux de greffage obtenu par les différentes méthodes d’analyse en fonction de r peuvent se superposer. En prenant la courbe obtenue par analyse élémentaire du carbone comme courbe de référence puisque cette dernière technique permet une détermination quantitative, on peut donc établir une courbe maîtresse en superposant les résultats des différentes techniques (Figure II - 18). Ceci montre que qualitativement les différentes techniques d’analyse sont en accord. Par ailleurs, cette approche permet de calibrer des techniques qualitatives, afin de pouvoir caractériser également de manière quantitative le greffage. Dans notre cas, nous avons établi une courbe de calibration de l’IRTF à partir des résultats d’analyse élémentaire (Figure II - 19) qui nous a permi par la suite d’évaluer précisément par une simple mesure par spectroscopie infrarouge le taux de greffage des silices. Ceci s’est avéré très pratique lorsque des silices de taux de greffage précis ont du être mises en œuvres pour l’élaboration de suspensions PDMS/silice.
Figure II - 18: Courbe maîtresse du greffage du HMDS à température ambiante sur la silice A200 pour différentes méthodes de greffage et de caractérisation
(a) en solution dans le CH2Cl2 : ( ) Angles de contact avec l’eau ; ( ) analyse élémentaire ; (▲) RMN 29Si CP/MAS ; ( ) IRTF
(b) en phase vapeur : ( ) Angles de contact avec l’eau ; ( ) analyse élémentaire ; (▲) RMN 29Si CP/MAS ; ( ) IRTF
Comme nous avons pu le voir, le type de procédure de modification semble dans notre cas ne pas influencer le résultat du greffage compte-tenu de la forte réactivité de l’HMDS. Les écarts observés dans la littérature sont donc certainement dus à la nature différente des silices évaluées et surtout aux techniques de caractérisation employées. Sindorf [2] trouve des valeurs différentes suivant les procédures employées, cependant il est difficile de comparer car il a considéré de gels de silice présentant des propriétés différentes, en particulier au niveau de la porosité et du nombre de sites silanol de surface.
0,0
0,5
1,0
1,5
2,0
2,5
0 0,5 1 1,5 2 2,5 3 3,5 4 4,5
Rapport stoechiométrique r
dens
ité d
e gr
effa
ge (T
MS/
nm2)
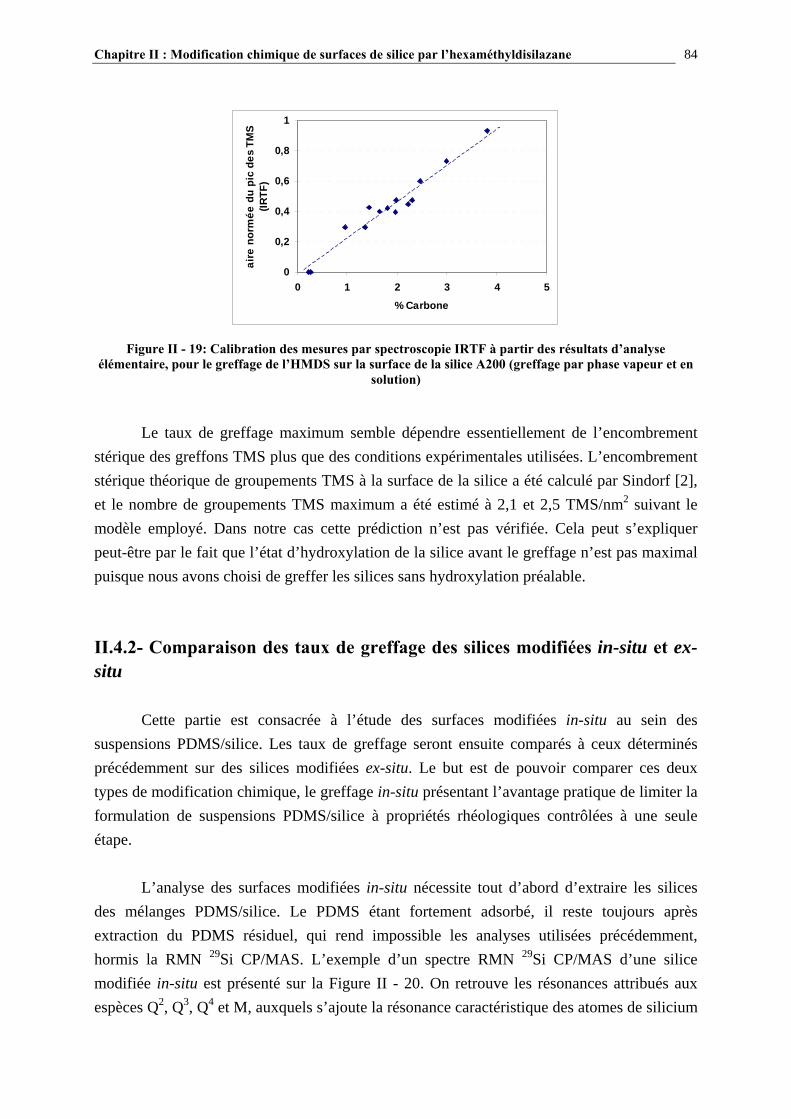
Chapitre II : Modification chimique de surfaces de silice par l’hexaméthyldisilazane
84
Figure II - 19: Calibration des mesures par spectroscopie IRTF à partir des résultats d’analyse élémentaire, pour le greffage de l’HMDS sur la surface de la silice A200 (greffage par phase vapeur et en
solution)
Le taux de greffage maximum semble dépendre essentiellement de l’encombrement stérique des greffons TMS plus que des conditions expérimentales utilisées. L’encombrement stérique théorique de groupements TMS à la surface de la silice a été calculé par Sindorf [2], et le nombre de groupements TMS maximum a été estimé à 2,1 et 2,5 TMS/nm2 suivant le modèle employé. Dans notre cas cette prédiction n’est pas vérifiée. Cela peut s’expliquer peut-être par le fait que l’état d’hydroxylation de la silice avant le greffage n’est pas maximal puisque nous avons choisi de greffer les silices sans hydroxylation préalable. II.4.2- Comparaison des taux de greffage des silices modifiées in-situ et ex-situ
Cette partie est consacrée à l’étude des surfaces modifiées in-situ au sein des suspensions PDMS/silice. Les taux de greffage seront ensuite comparés à ceux déterminés précédemment sur des silices modifiées ex-situ. Le but est de pouvoir comparer ces deux types de modification chimique, le greffage in-situ présentant l’avantage pratique de limiter la formulation de suspensions PDMS/silice à propriétés rhéologiques contrôlées à une seule étape. L’analyse des surfaces modifiées in-situ nécessite tout d’abord d’extraire les silices des mélanges PDMS/silice. Le PDMS étant fortement adsorbé, il reste toujours après extraction du PDMS résiduel, qui rend impossible les analyses utilisées précédemment, hormis la RMN 29Si CP/MAS. L’exemple d’un spectre RMN 29Si CP/MAS d’une silice modifiée in-situ est présenté sur la Figure II - 20. On retrouve les résonances attribués aux espèces Q2, Q3, Q4 et M, auxquels s’ajoute la résonance caractéristique des atomes de silicium
0
0,2
0,4
0,6
0,8
1
0 1 2 3 4 5
% Carbone
aire
nor
mée
du
pic
des
TMS
(IRTF
)
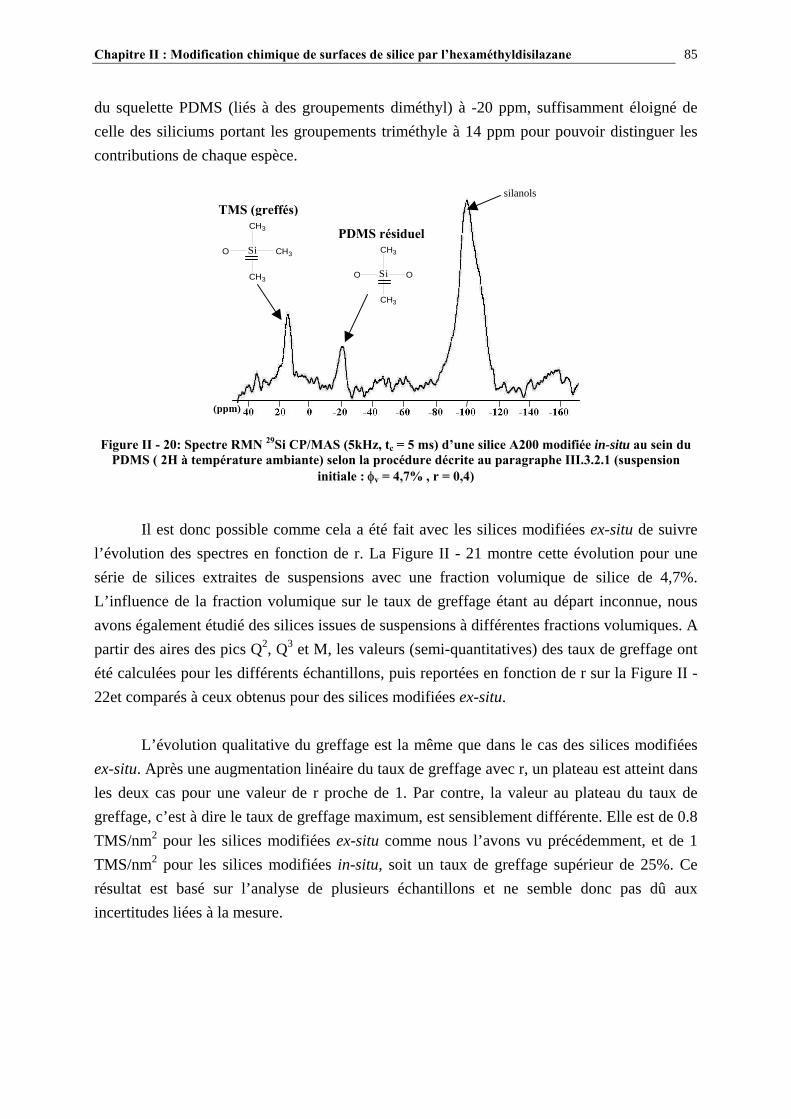
Chapitre II : Modification chimique de surfaces de silice par l’hexaméthyldisilazane
85
du squelette PDMS (liés à des groupements diméthyl) à -20 ppm, suffisamment éloigné de celle des siliciums portant les groupements triméthyle à 14 ppm pour pouvoir distinguer les contributions de chaque espèce. Figure II - 20: Spectre RMN 29Si CP/MAS (5kHz, tc = 5 ms) d’une silice A200 modifiée in-situ au sein du
PDMS ( 2H à température ambiante) selon la procédure décrite au paragraphe III.3.2.1 (suspension initiale : φv = 4,7% , r = 0,4)
Il est donc possible comme cela a été fait avec les silices modifiées ex-situ de suivre l’évolution des spectres en fonction de r. La Figure II - 21 montre cette évolution pour une série de silices extraites de suspensions avec une fraction volumique de silice de 4,7%. L’influence de la fraction volumique sur le taux de greffage étant au départ inconnue, nous avons également étudié des silices issues de suspensions à différentes fractions volumiques. A partir des aires des pics Q2, Q3 et M, les valeurs (semi-quantitatives) des taux de greffage ont été calculées pour les différents échantillons, puis reportées en fonction de r sur la Figure II - 22et comparés à ceux obtenus pour des silices modifiées ex-situ. L’évolution qualitative du greffage est la même que dans le cas des silices modifiées ex-situ. Après une augmentation linéaire du taux de greffage avec r, un plateau est atteint dans les deux cas pour une valeur de r proche de 1. Par contre, la valeur au plateau du taux de greffage, c’est à dire le taux de greffage maximum, est sensiblement différente. Elle est de 0.8 TMS/nm2 pour les silices modifiées ex-situ comme nous l’avons vu précédemment, et de 1 TMS/nm2 pour les silices modifiées in-situ, soit un taux de greffage supérieur de 25%. Ce résultat est basé sur l’analyse de plusieurs échantillons et ne semble donc pas dû aux incertitudes liées à la mesure.
Si OO
CH3
CH3
Si CH3O
CH3
CH3
TMS (greffés)
PDMS résiduel
(ppm)
silanols
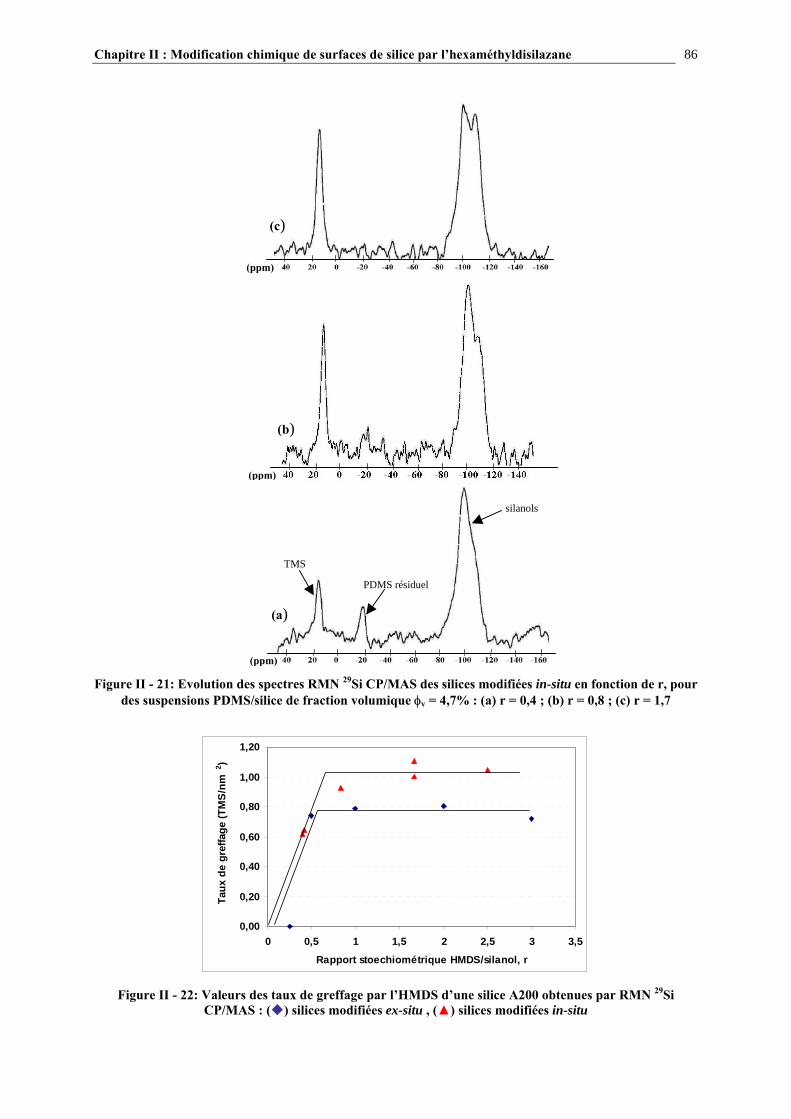
Chapitre II : Modification chimique de surfaces de silice par l’hexaméthyldisilazane
86
Figure II - 21: Evolution des spectres RMN 29Si CP/MAS des silices modifiées in-situ en fonction de r, pour
des suspensions PDMS/silice de fraction volumique φv = 4,7% : (a) r = 0,4 ; (b) r = 0,8 ; (c) r = 1,7
Figure II - 22: Valeurs des taux de greffage par l’HMDS d’une silice A200 obtenues par RMN 29Si CP/MAS : ( ) silices modifiées ex-situ , (▲) silices modifiées in-situ
(a)
(b)
(c)
(ppm)
(ppm)
(ppm)
silanols
PDMS résiduel
TMS
0,00
0,20
0,40
0,60
0,80
1,00
1,20
0 0,5 1 1,5 2 2,5 3 3,5
Rapport stoechiométrique HMDS/silanol, r
Taux
de
gref
fage
(TM
S/nm
2 )

Chapitre II : Modification chimique de surfaces de silice par l’hexaméthyldisilazane
87
Ce résultat est surprenant car on pourrait penser que la forte adsorption du PDMS à la surface limite l’accès des molécules d’HMDS lors de la modification in-situ. De plus en milieu PDMS la mobilité des espèces est réduite, ce qui ne favorise pas non plus la réaction. Il semblerait donc que ces aspects ne jouent pas un rôle majeur, peut être en raison de l’importante réactivité de l’HMDS vis-à-vis de la surface. Les taux de greffage plus élevés dans le cas de silices modifiées in-situ ont finalement été expliquées par la présence d’ammoniac, formé pendant la réaction de l’HMDS avec les silanols de surface. Le rôle de l’ammoniac formé au cours de la réaction de l’HMDS avec les silanols de surface est controversé. Nawrocki [26] montre par des mesures de teneur en azote que l’ammoniac formé ne s’adsorbe pas à la surface de la silice. Il confirme ce résultat en saturant la surface de silice d’ammoniac et en chauffant à reflux dans l’hexadecane, sans trouver de trace d’azote après le traitement thermique. En revanche des analyses par infrarouge [24, 27] ont montré que de nouveaux pics apparaissaient dans le spectre des silices lorsque des ajouts d’ammoniac sont effectués. Ces pics sont attribués i/ à des groupements NH3 adsorbés à la surface de la silice (1400 cm-1), ii/ à des groupements Si-NH2 lorsque l’ammoniac réagit avec la silice (1550, 3447 et 3525 cm-1). La réaction de l’ammoniac avec la surface de la silice peut s’écrire de deux manières (Figure II - 23) suivant le type de groupements qui réagissent (groupements silanol et siloxane) [1].
Figure II - 23 : Réaction de l’ammoniac avec la surface de la silice [1]
La réaction de l’ammoniac avec les groupements siloxane implique la formation de nouveaux groupements silanol disponibles pour le greffage d’HMDS. Dans le cas des silices modifiées ex-situ, la modification chimique s’effectue en système ouvert, c’est à dire que l’ammoniac formé ne reste pas dans le milieu réactionnel. Ce n’est pas le cas pour les silices modifiées in-situ. L’ammoniac peut donc réagir avec la silice, et ce d’autant plus qu’à la fin de l’introduction de la silice la température est augmentée (150°C). On peut remarquer également que le taux de greffage ne dépend pas de façon évidente de la fraction volumique de silice du mélange initial. Les deux échantillons réalisés à r = 0,4,
Si OH + NH3 Si NH2 + H2O
Si O NH3 Si NH2 +Si + Si OH

Chapitre II : Modification chimique de surfaces de silice par l’hexaméthyldisilazane
88
pour φv = 4,7% et φv = 7,2% présentent un taux de greffage très voisin, tandis qu’une différence un peu plus importante existe pour r = 1,7 entre les taux de greffage des silices issues des suspensions à φv = 4,7% et φv = 9,9%. Cette différence reste cependant peu significative compte-tenu des incertitudes liées à ce type de mesure.

Chapitre II : Modification chimique de surfaces de silice par l’hexaméthyldisilazane
89
II.5- CONCLUSIONS Ce chapitre était consacré à la modification chimique de la surface de la silice de pyrohydrolyse A200 par l’HMDS. Les surfaces ont été modifiées soit ex-situ en solution ou en phase vapeur, soit in-situ au sein des suspensions PDMS/silice. L’évolution du greffage en fonction du rapport HMDS/silanols de surface r, a été suivie par plusieurs techniques de caractérisation : l’analyse élémentaire du carbone, la spectroscopie infrarouge, la RMN 29Si CP/MAS et des mesures de mouillage par l’eau. Les deux types de procédure de modification ex-situ ont tout d’abord été comparées. La conversion des silanols de surfaces en groupements TMS évolue dans un premier temps linéairement en fonction de r, puis dans un second temps atteint un plateau pour r environ égal à 1. La valeur du taux de greffage maximum obtenue au plateau est de 1,8 TMS/nm2. Cette valeur résulte de l’analyse élémentaire du carbone. Aucune différence n’est observée entre les deux procédures. La conversion des silanols, suivie par infrarouge, montre la présence de silanols résiduels même pour les taux de greffage les plus élevés. Cette limitation peut s’expliquer par l’encombrement stérique des groupements TMS, déjà reporté dans des études antérieures. La RMN 29Si CP/MAS a permis d’une part de montrer que les espèces greffées sont liées de manière covalente à la surface, et d’autre part de donner une mesure semi-quantitative du greffage. Il semble cependant que la RMN sous-estime les taux de greffage en comparaison à l’analyse élémentaire du carbone. Ceci peut s’expliquer par la prise en compte d’une partie des silanols internes qui conduit à une surestimation du nombre initial de silanols totaux. En revanche un bon accord qualitatif entre les différentes techniques d’analyse a été montré, en ce qui concerne l’évolution du greffage en fonction de r. Par ailleurs la mise au point d’un dispositif de modification chimique en phase gazeuse va nous servir à la réalisation de suspensions PDMS/silice, qui nécessitent des quantités de silice greffée assez importantes. Ces silices devant être de taux de greffage contrôlé, la spectroscopie infrarouge a été calibrée à partir des résultats précédents de l’analyse élémentaire du carbone, pour pouvoir obtenir de manière rapide et fiable une mesure précise du taux de greffage des silices. Enfin, les silices modifiées in-situ ont été analysées par RMN 29Si CP/MAS, qui est la seule technique restant disponible dans ce cas. En effet la présence de PDMS résiduel à la surface, après extraction des suspensions, rend les autres techniques de caractérisation inutilisables. Les mesures semi-quantitatives du greffage par RMN ont montré que leur taux de greffage était supérieur de 25% à celui des silices modifiées ex-situ. Cette différence a été attribuée à l’ammoniac formé au cours de la réaction qui reste au sein du milieu réactionnel contrairement au cas des procédés ex-situ. L’ammoniac catalyse la réaction et est susceptible

Chapitre II : Modification chimique de surfaces de silice par l’hexaméthyldisilazane
90
de réagir avec des groupements siloxane, pour former des groupements hydroxyles supplémentaires, disponibles pour réagir avec l’HMDS.

Chapitre II : Modification chimique de surfaces de silice par l’hexaméthyldisilazane
91
Références bibliographiques
1. Vansant E.F., Van Der Voort P., Vrancken K.C., Characterization and Chemical
Modification of the Silica Surface, Amsterdam: Elsevier, 1996, 556p.
2. Sindorf, D.W., Maciel E., CP/MAS 29Si NMR Study of silica gel using trimethylsilane bonding as a probe of surface geometry and reactivity, J. Phys. Chem., 1982, Vol. 86, pp.5208-5219.
3. Takei T., Yamazaki A., Watanabe T., Chikazawa M., Water adsorption properties on porous silica glass surface modified by trimethylsilyl groups, J. Colloid Interface Sci., 1997, Vol. 188, pp.409-414.
4. Zumbrum M.A., Acid/Base properties of fumed silica fillers used in silicone elastomers, XVIth Annual Meeting of the Adhesion Society, Inc, 1993, Williamsburgh, Virginia, U.S.A.
5. Gun'ko V.M., Vedamuthu M.S., Henderson G.L., Blitz J.P., Mechanism and kinetics of Hexamethyldisilazane reaction with a fumed silica surface, J. Colloid Interface Sci., 2000, Vol. 228, pp.157-170.
6. Haukka S., Root A., The reaction of hexamethyldisilazane and subsequent oxidation of trimethylsilyl groups on silica studied by solid-state NMR and FT-IR, J. Phys. Chem., 1994, Vol. 98, pp.1695-1703.
7. Hertl W., Hair M.L., Reaction of hexamethyldisilazane with silica, J. Phys. Chem., 1971, Vol. 75 (14), pp.2181-2185.
8. Plueddeman E.P., Chemistry of silane coupling agents, in: Silane coupling agents, New-York: Plenum Press, 1982, pp.29-48.
9. Maxson M.T., Lee C.L., Effects of fumed silica treated with functional disilazanes on silicone elastomers properties, Rubber Chem. Technol., 1982, Vol. 55, pp.233-244.
10. Chmielowiec J., Morrow B.A., Alkylation of silica surfaces, J. Colloid Interface Sci., 1983, Vol. 94 (2), pp.319-327.
11. Huang W., Huang Y., Yu Y., Trimethylsilylated silica as rheology modifier for silicone resins, Chin. J. Polym. Sci., 2000, Vol. 18 (1), pp.51-55.
12. Slavov S.V., Sanger A.R., Chuang K.T.,, Mechanism of silation of silica with hexamethyldisilazane, J. Phys. Chem. B, 2000, Vol. 104, pp.983-989.
13. Buzek F., Rathousky J., Stoechiometry and kinetics of the reaction of silica with organosilicon compounds, J. Colloid Interface Sci., 1981, Vol. 79 (1), pp.47-55.
14. Barthel H., Surface interactions of dimethylsiloxy group-modified fumed silica, Colloids Surf., A, 1995, Vol. 101, pp.217-226.

Chapitre II : Modification chimique de surfaces de silice par l’hexaméthyldisilazane
92
15. Belyakova L.A., Varvarin A.M., Surface properties of silica gels modified with hydrophobic groups, Colloids Surf., A, 1999, Vol. 154, pp.285-294.
16. Morrow B.A., McFarlan A.J., Infrared and Gravimetric Study of an Aerosil and a precipitated silica using chemical and H/D exchange probes, Langmuir, 1991, Vol. 7, pp.1695-1701.
17. Sutra P., Fajula F., Brunel D., Lentz P., 29Si and 13C MAS NMR characterization of surface modification of micelle-templated silicas during the grafting of organic moieties and end-capping, Colloids Surf., A, 1999, Vol. 158, pp.21-27.
18. Snyder L.R., Ward J.W., The surface structure of porous silica, J. Phys. Chem., 1966, Vol. 70 (12), pp.3941-3952.
19. Sindorf, D.W., Maciel E., Si29 nuclear magnetic resonance study of hydroxyl sites on dehydrated silica gel surfaces, using silylation as a probe, J. Phys. Chem., 1983, Vol. 87 (26), pp.5516-5521.
20. Köhler J., Chase D.B., Farlee R.D., Vega A.J., Kirkland J.J., Comprehensive characterization of some silica-based stationary phases for high-performance liquid chromatography, J. Chromatogr., 1986, Vol. 352, pp.275-305.
21. Haukka S., Lakomaa E.-L., Root A., IR and NMR study of the chemisorption of TiCl4 on silica, J. Phys. Chem., 1993, Vol. 97, pp.5085-5094.
22. Unger K.K., Becker N., Roumeliotis P., Recent developments in the evaluation of chemically bonded silica packings for liquid chromatography, J. Chromatogr., 1976, Vol. 125, pp.115-127.
23. Lowen W.K., Broge E.C., Effects of dehydration and chemisorbed materials on the surface properties of amorphous silica, J. Phys. Chem., 1961, Vol. 65 (16), pp.16-19.
24. Zettlemoyer A.C., Hsing H.H., Water on organosilane-treated silica surfaces, J. Colloid Interface Sci., 1977, Vol. 58 (2), pp.263-274.
25. Duchet J., Système modèle polyéthylène/verre: rôle de chaînes connectrices greffees sur l'adhésion, Thèse de Doctorat, Université Claude Bernard, Lyon I, 1996, 310p.
26. Nawrocki J., Modification of silica with mixtures of hexamethylcyclotrisilazane and hexamethyldisilazane, Chromatographia, 1985, Vol. 20 (5), pp.308-312.
27. Morrow B.A., Coody I.A., Lee L.S.M., Infrared studies of reactions on oxide surfaces.7. Mechanism of the adsorption of water and ammonia on dehydroxylated silica, J. Phys. Chem., 1976, Vol. 80 (25), pp.2761-2767.

CHAPITRE III : ETUDE DES PROPRIETES RHEOLOGIQUES ET DE L’ETAT DE
DISPERSION DE SUSPENSIONS PDMS / SILICE

Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
93
CHAPITRE III : ETUDE DES PROPRIETES RHEOLOGIQUES ET DE L’ETAT DE
DISPERSION DE SUSPENSIONS PDMS/SILICE III.1- INTRODUCTION..................................................................................................................................... 94 III.2- ETUDE BIBLIOGRAPHIQUE ............................................................................................................... 95
III.2.1- DÉFINITIONS ......................................................................................................................................... 95 a) Comportement rhéologique en régime dynamique (faibles déformations) ........................................................... 95 b) Comportement rhéologique en régime statique.................................................................................................... 96
III.2.2- ETAT DE DISPERSION ET PROPRIÉTÉS RHÉOLOGIQUES DES SUSPENSIONS PDMS/SILICE ........................ 97 III.2.3- CONCLUSIONS..................................................................................................................................... 106
III.3- PARTIE EXPÉRIMENTALE ............................................................................................................... 107 III.3.1- MATÉRIAUX........................................................................................................................................ 107 III.3.2- MISE EN ŒUVRE DES SUSPENSIONS PDMS/SILICE............................................................................... 107
II.3.2.1- Procédés in-situ ........................................................................................................................... 108 a) Procédé in-situ en 1 étape (Z)............................................................................................................................. 108 b) Procédé in-situ en 2 étapes (F) ........................................................................................................................... 109 c) Mélanges obtenus par dilution (ZD)................................................................................................................... 110
III.3.2.2- Procédé ex-situ........................................................................................................................... 110 III.3.3- CARACTÉRISATION DES SUSPENSIONS................................................................................................. 111
III.3.3.1- Caractérisation des propriétés rhéologiques ............................................................................. 111 a) Appareillage et géométries ................................................................................................................................. 111 b) Mise au point des protocoles d’essais................................................................................................................. 111
III.3.3.2- Caractérisation de la microstructure des suspensions............................................................... 113 a) Microscopie électronique à transmission (TEM)................................................................................................ 113 b) Microscopie à force atomique (AFM) ................................................................................................................ 113 c) Diffusion dynamique de la lumière (DDL)......................................................................................................... 114
III.4- RÉSULTATS ET DISCUSSION ........................................................................................................... 115 III.4.1- DESCRIPTION DU COMPORTEMENT RHÉOLOGIQUE GÉNÉRAL DES SUSPENSIONS PDMS/SILICE ........... 115
III.4.1.1- Mise en évidence du comportement viscoélastique non-linéaire des suspensions ..................... 115 III.4.1.2- Etude rhéologique en régime dynamique ................................................................................... 116 III.4.1.3- Etude rhéologique en régime statique........................................................................................ 119
a) Mise en évidence d’un caractère thixotrope ....................................................................................................... 119 III.4.2- COMPORTEMENT RHÉOLOGIQUE ET ÉTAT DE DISPERSION DES SUSPENSIONS RÉALISÉES IN-SITU EN 1 ÉTAPE (SÉRIE Z).............................................................................................................................................. 121
III.4.2.1- Evolution du comportement rhéologique et de l’état de dispersion des suspensions PDMS/silice en fonction du taux de greffage de la silice ............................................................................................... 121 III.4.2.2- Evolution des suspensions en fonction de la fraction volumique de silice ................................. 127 III.4.2.3- Suspensions obtenues par dilution (série D) .............................................................................. 131 III.4.2.4- Diagramme de phases des suspensions – Transition liquide - gel ............................................. 133
III.4.3- INFLUENCE DU PROCÉDÉ DE MISE EN ŒUVRE DES SUSPENSIONS.......................................................... 134 III.4.3.1- Comparaison in-situ (Z) / ex-situ (T).......................................................................................... 134 III.4.3.2- Comparaison in-situ 1 étape (Z) / in-situ 2 étapes (F) ............................................................... 138
III.4.4- INFLUENCE DE LA MASSE MOLAIRE DU PDMS SUR LES PROPRIÉTÉS RHÉOLOGIQUES DES SUSPENSIONS........................................................................................................................................................................ 139 III.4.5- MODÉLISATION RHÉOLOGIQUE ........................................................................................................... 140
III.4.5.1- Calcul des dimensions fractales D et des fractions volumiques critiques φv* à partir des mesures
rhéologiques .............................................................................................................................................. 141 III.4.5.2- Calcul de la taille des objets fractals R...................................................................................... 143 III.4.5.3- Comparaison avec les résultats obtenus par diffusion dynamique de la lumière....................... 144
III.5- CONCLUSIONS ..................................................................................................................................... 148

Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
94
III.1- INTRODUCTION Ce chapitre a pour objet l’étude du comportement rhéologique des suspensions PDMS/silice. L’objectif général est de comprendre comment ces propriétés sont contrôlées par les morphologies des suspensions, qui dépendent des interactions PDMS/silice, gouvernées par le taux de greffage des silices. Nous nous appuierons pour cela sur les résultats du chapitre II concernant l’analyse du greffage des silices. Après une présentation de la bibliographie sur ce sujet puis des matériaux et techniques expérimentales utilisés, le comportement rhéologique général des suspensions sera étudié. Les suspensions réalisées in-situ seront tout d’abord considérées pour déterminer l’influence des paramètres de formulation (fraction volumique et taux de greffage) sur la morphologie des dispersions et les propriétés rhéologiques qui en découlent (modules rhéologiques, contraintes à seuil). L’analyse des contraintes à seuil des différentes formulations conduit à un diagramme de transition liquide-gel permettant de représenter la transition sol-gel en fonction des paramètres de la formulation. Des suspensions obtenues par différentes voies de mise en œuvre seront ensuite examinées. Pour les suspensions réalisées in-situ, deux voies de mise en œuvre seront considérées : i/ en 1 étape, avec introduction de l’HMDS dès le début du malaxage PDMS/silice, ii/ en 2 étapes, avec introduction de l’HMDS en fin de malaxage. Les suspensions réalisées à partir de silices greffées in-situ en milieu PDMS seront comparées aux suspensions où le greffage des silices a lieu ex-situ par voie gazeuse. Enfin, les résultats de rhéologie obtenus sur les suspensions réalisées in-situ seront exploités à l’aide d’un modèle physique d’objets fractals semi-dilués. Ce modèle permet de prévoir l’évolution du module G’ en fonction de la fraction volumique, pour différents taux de greffage, et d’en déduire les dimensions fractales des différents réseaux de silice. Ce chapitre est une base de compréhension pour l’étude ultérieure des modifications rhéologiques des suspensions PDMS/silice introduites par ajout d’additifs, qui fait l’objet du chapitre IV.

Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
95
III.2- ETUDE BIBLIOGRAPHIQUE III.2.1- Définitions a) Comportement rhéologique en régime dynamique (faibles déformations) Lorsqu’une déformation de cisaillement sinusoïdale est appliquée à un matériau présentant un comportement viscoélastique linéaire, la contrainte correspondante est également sinusoïdale, mais présente un déphasage δ. On peut définir la déformation γ(t) et la contrainte σ (t) par les relations [1] : Equation III - 1
où γ0 et τ0 sont les amplitudes maximales de la déformation et de la contrainte, respectivement et t est le temps.
Le module complexe G∗ est défini à tout instant comme le rapport de la contrainte sur la déformation, soit : Equation III - 2
Ce module de cisaillement peut se décomposer en une partie réelle G’, appelé module
de conservation, et une partie imaginaire G’’ appelé module de perte : avec , Equation III - 3
Le facteur de perte se définit comme le rapport du module de perte sur le module de conservation : Equation III - 4
La viscosité complexe η*, qui peut se décomposer en une partie réelle η’ et une partie imaginaire η’’, peut également être définie à partir des modules complexes [2] : avec et Equation III - 5
tiet ϖγγ 0)( =( )δϖττ += tiet 0)(
( )( )
δ
γτ
γτ ie
ttG
0
0==∗
GiGG ′′+′=∗ δγτ cos
0
0=′G δγτ sin
0
0=′′G
GG
′′′
=δtan
''' Giηηη +=∗
ωη G ′′
=′ω
η G′=′′
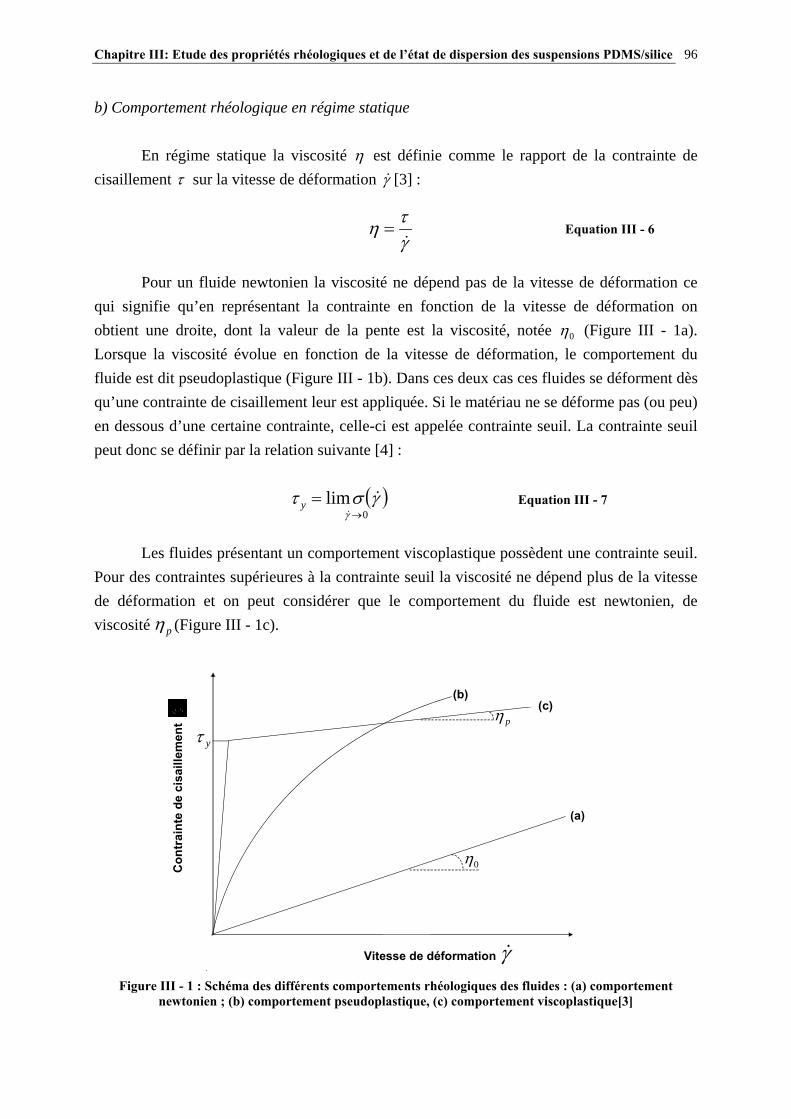
Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
96
b) Comportement rhéologique en régime statique En régime statique la viscosité η est définie comme le rapport de la contrainte de cisaillement τ sur la vitesse de déformation γ& [3] : Equation III - 6
Pour un fluide newtonien la viscosité ne dépend pas de la vitesse de déformation ce qui signifie qu’en représentant la contrainte en fonction de la vitesse de déformation on obtient une droite, dont la valeur de la pente est la viscosité, notée 0η (Figure III - 1a). Lorsque la viscosité évolue en fonction de la vitesse de déformation, le comportement du fluide est dit pseudoplastique (Figure III - 1b). Dans ces deux cas ces fluides se déforment dès qu’une contrainte de cisaillement leur est appliquée. Si le matériau ne se déforme pas (ou peu) en dessous d’une certaine contrainte, celle-ci est appelée contrainte seuil. La contrainte seuil peut donc se définir par la relation suivante [4] : Equation III - 7
Les fluides présentant un comportement viscoplastique possèdent une contrainte seuil. Pour des contraintes supérieures à la contrainte seuil la viscosité ne dépend plus de la vitesse de déformation et on peut considérer que le comportement du fluide est newtonien, de viscosité (Figure III - 1c).
Figure III - 1 : Schéma des différents comportements rhéologiques des fluides : (a) comportement newtonien ; (b) comportement pseudoplastique, (c) comportement viscoplastique[3]
γτη&
=
( )0
lim→
=γ
γστ&
&y
pη
γ&Vitesse de déformation
Con
trai
nte
de c
isai
llem
ent
yτ
(a)
(b)
0η
pη(c)

Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
97
Le comportement thixotrope est un phénomène dépendant du temps. La thixotropie peut se définir comme étant la diminution de la viscosité en fonction du temps, sous cisaillement et le retour à la viscosité initiale lorsque le cisaillement est interrompu [5]. Il est mis en évidence en effectuant une expérience de charge (vitesse de déformation croissante) et décharge (vitesse de déformation décroissante), en représentant la contrainte de cisaillement en fonction de la vitesse de déformation. Trois cas de figure existent : fluide thixotrope ne possédant pas de contrainte seuil (Figure III - 2a), fluide thixotrope possédant une contrainte à seuil initiale non modifiée (Figure III - 2b), fluide thixotrope possédant une contrainte à seuil initiale modifiée (Figure III - 2c). Figure III - 2 : Schémas illustrant le comportement thixotrope dans différentes situations : (a) fluide sans contrainte à seuil ; (b) fluide possédant une contrainte à seuil initiale non modifiée ; (c) fluide présentant
une contrainte à seuil initiale modifiée [6]
Dans le cadre des suspensions PDMS/silice étudiées seuls les deux premiers cas ont été rencontrés. D’autre part, nous avons pu observé que les suspensions présentant une contrainte seuil ont systématiquement un comportement thixotrope, mais la réciproque n’est pas vraie. III.2.2- Etat de dispersion et propriétés rhéologiques des suspensions PDMS/silice Les propriétés de renforcement des charges dépendent de leur état de dispersion au sein de l’élastomère. Les interactions polymère-charge qui conduisent à l’agglomération des suspensions définissent la morphologie finale des matériaux et ainsi leur propriétés rhéologiques. L’analyse de la dispersion des suspensions colloïdales doit être faite à différentes échelles compte-tenu de la morphologie des nanoparticules dans le milieu de suspension : particules élémentaires, agrégats, agglomérats. Il faut donc utiliser des outils différents suivant les caractéristiques recherchées.
γ&
τ
γ&
τ
γ&
τ
(a) (b) (c)

Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
98
La dispersion des charges peut être étudiée à l’échelle des agglomérats par microscopie optique ou à balayage, mais pour une observation à l’échelle des agrégats les techniques les mieux adaptées sont la microscopie électronique à transmission (TEM) et plus récemment la microscopie à force atomique (AFM). Le réseau de silice observé en TEM peut être formé d’aggrégats connectés les uns aux autres pour former au-delà du seuil de percolation un réseau de nanoparticules (Figure III - 3). Ce phénomène d’agglomération est comme les travaux de Chahal [7] le montraient dépendant de l’intensité des interactions polymère/charge. Cela a été confirmé plus récemment par comparaison de la taille des agglomérats de silice de précipitation, non greffée et greffée par réaction avec l’HMDS. Ces micrographies (Figure III - 4) ont été obtenues par microscopie électronique à balayage (MEB) une technique originale de fracture à froid des échantillons [8]. L’observation de la microstructure des suspensions a été également utilisée pour juger du vieillissement de celles-ci [9].
Figure III - 3: Dispersion des suspensions PDMS/silice observée par TEM : (a) morphologie observéejuste au dessous du seuil de percolation, φv = 7% ; (b) morphologie observée au-
delà du seuil de percolation, φv = 14% [10]
Figure III - 4: Agglomération des suspensions PDMS/silice de précipitation observée par MEB (a) silice non greffée (b) silice greffée par réaction avec l’HMDS [8]
Avec le développement récent de l’AFM, cette technique permet maintenant de visualiser les dispersions de silice, avec une préparation des échantillons plus aisée qu’en TEM [11], [12]. Le potentiel de l’AFM en contraste de phase en mode ‘tapping’ a été
(a) (b)

Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
99
démontré par une étude comparative sur plusieurs élastomères silicone chargés par de la silice [13], des images obtenues en AFM et TEM montrant un bon accord entre ces deux techniques. Une méthode indirecte pour étudier la dispersion des suspensions PDMS/silice et les interactions polymère-charge est la rhéologie, complémentaire de l’observation de la dispersion par microscopie. L’une des premières études réalisée sur les suspensions PDMS/silice concerne l’influence de la nature du greffage de la silice sur l’évolution des propriétés mécaniques en fonction de la fraction volumique [7]. Il est montré que le module de relaxation E0 est proportionnel au logarithme de la fraction volumique (Figure III - 5), et ce quel que soit le greffage de la silice: Equation III - 8
Plusieurs études [14-18] se sont ensuite intéressées aux changements de comportement rhéologique lors de l’introduction de silice dans des PDMS, en particulier le phénomène de ‘dépassement’ de contrainte que l’on peut observer lors d’un test de cisaillement, en représentant la viscosité en fonction du temps. Il a été montré que ce ‘dépassement de contrainte’ est très sensible à l’histoire du matériau, par exemple le temps de repos entre le précisaillement et le début du test (Figure III - 6), mais également de la masse molaire du PDMS étudié comme milieu de suspension. Ceci montre la sensibilité des mesures rhéologiques dans le cas de suspensions complexes telles que les suspensions PDMS/silice, présentant des comportements non linéaires et dépendant du temps.
Figure III - 5: Evolution du module de conservation en fonction de la fraction volumique [7]
05,50 vE φ∝
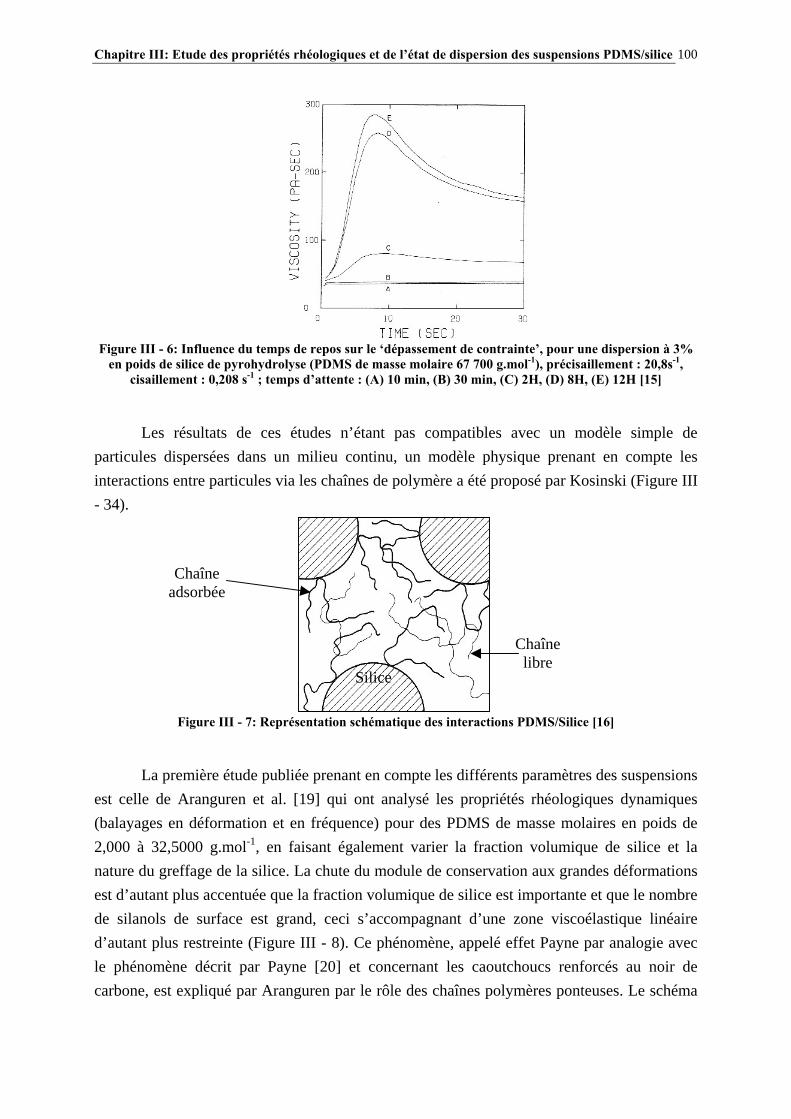
Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
100
Figure III - 6: Influence du temps de repos sur le ‘dépassement de contrainte’, pour une dispersion à 3%
en poids de silice de pyrohydrolyse (PDMS de masse molaire 67 700 g.mol-1), précisaillement : 20,8s-1, cisaillement : 0,208 s-1 ; temps d’attente : (A) 10 min, (B) 30 min, (C) 2H, (D) 8H, (E) 12H [15]
Les résultats de ces études n’étant pas compatibles avec un modèle simple de particules dispersées dans un milieu continu, un modèle physique prenant en compte les interactions entre particules via les chaînes de polymère a été proposé par Kosinski (Figure III - 34).
Figure III - 7: Représentation schématique des interactions PDMS/Silice [16]
La première étude publiée prenant en compte les différents paramètres des suspensions est celle de Aranguren et al. [19] qui ont analysé les propriétés rhéologiques dynamiques (balayages en déformation et en fréquence) pour des PDMS de masse molaires en poids de 2,000 à 32,5000 g.mol-1, en faisant également varier la fraction volumique de silice et la nature du greffage de la silice. La chute du module de conservation aux grandes déformations est d’autant plus accentuée que la fraction volumique de silice est importante et que le nombre de silanols de surface est grand, ceci s’accompagnant d’une zone viscoélastique linéaire d’autant plus restreinte (Figure III - 8). Ce phénomène, appelé effet Payne par analogie avec le phénomène décrit par Payne [20] et concernant les caoutchoucs renforcés au noir de carbone, est expliqué par Aranguren par le rôle des chaînes polymères ponteuses. Le schéma
Chaîne adsorbée
Chaîne libre
Silice
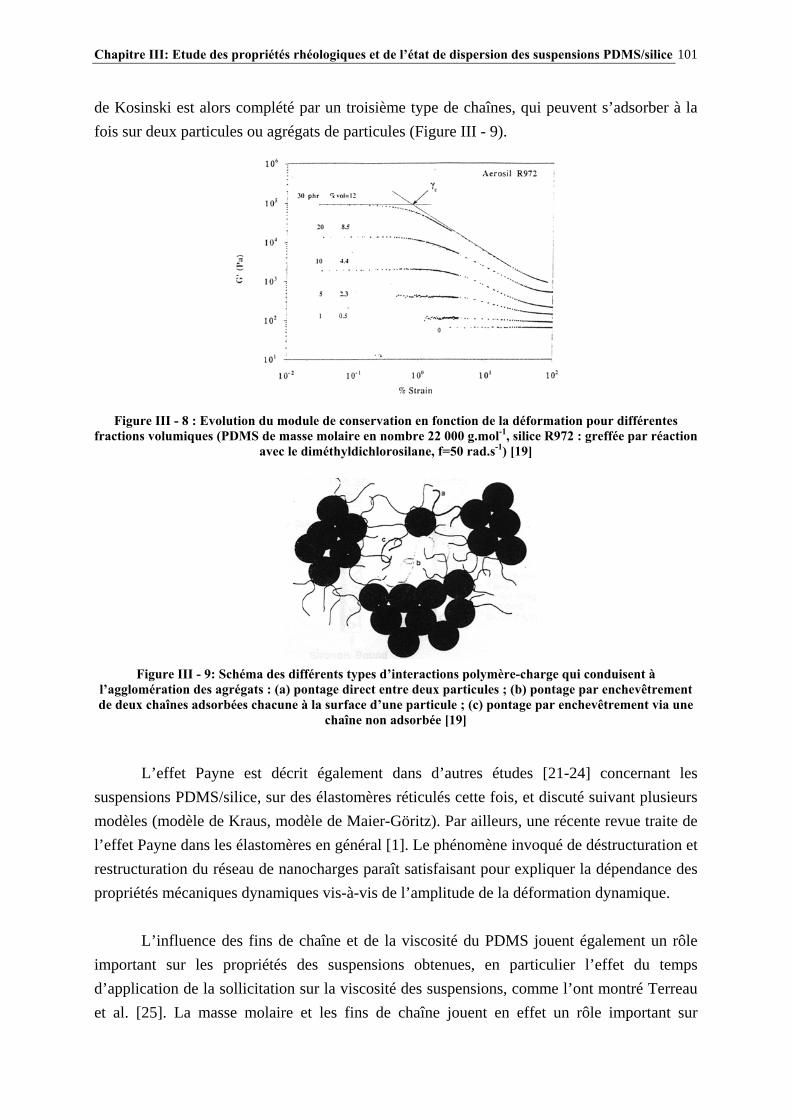
Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
101
de Kosinski est alors complété par un troisième type de chaînes, qui peuvent s’adsorber à la fois sur deux particules ou agrégats de particules (Figure III - 9).
Figure III - 8 : Evolution du module de conservation en fonction de la déformation pour différentes fractions volumiques (PDMS de masse molaire en nombre 22 000 g.mol-1, silice R972 : greffée par réaction
avec le diméthyldichlorosilane, f=50 rad.s-1) [19]
Figure III - 9: Schéma des différents types d’interactions polymère-charge qui conduisent à l’agglomération des agrégats : (a) pontage direct entre deux particules ; (b) pontage par enchevêtrement de deux chaînes adsorbées chacune à la surface d’une particule ; (c) pontage par enchevêtrement via une
chaîne non adsorbée [19]
L’effet Payne est décrit également dans d’autres études [21-24] concernant les suspensions PDMS/silice, sur des élastomères réticulés cette fois, et discuté suivant plusieurs modèles (modèle de Kraus, modèle de Maier-Göritz). Par ailleurs, une récente revue traite de l’effet Payne dans les élastomères en général [1]. Le phénomène invoqué de déstructuration et restructuration du réseau de nanocharges paraît satisfaisant pour expliquer la dépendance des propriétés mécaniques dynamiques vis-à-vis de l’amplitude de la déformation dynamique. L’influence des fins de chaîne et de la viscosité du PDMS jouent également un rôle important sur les propriétés des suspensions obtenues, en particulier l’effet du temps d’application de la sollicitation sur la viscosité des suspensions, comme l’ont montré Terreau et al. [25]. La masse molaire et les fins de chaîne jouent en effet un rôle important sur

Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
102
l’adsorption de la chaîne et influencent donc le comportement rhéologique de ces suspensions. Cette étude confirme l’importance du phénomène de vieillissement des suspensions, déjà montrée par plusieurs auteurs [9, 26-30]. Les modèles classiques de prévision de la viscosité (ou des modules) reposent sur la loi d’Einstein [31, 32] :
Equation III - 9
où η0 est la viscosité de la matrice seule et φv la fraction volumique de la charge
Cette relation n’est valable que pour les très faibles fractions volumiques, et ne prend
pas en compte les interactions polymère/charge, ainsi que les effets de volume occlus. Le volume occlus correspond au polymère piégé à l’interieur des agglomérats de silice. Celui-ci conduit à une augmentation artificielle de la fraction volumique de la charge, puisque le volume apparent de l’agglomérat, comprenant le volume de la charge et le volume occlus de polymère, est supérieur au volume effectif, c’est-à-dire le volume de la charge seule. Des modèles dérivés de la loi d’Einstein tentent de prendre en compte ces effets en introduisant des facteurs correctifs [33-38]. Citons en exemple le modèle de Sato et Furukawa [37] qui prend en compte l’adhésion entre le polymère et la charge par l’intermédiaire d’un facteur d’adhésion k : Equation III - 10
k = 0 dans le cas d’une adhésion parfaite k = 1 dans le cas d’une adhésion nulle
Cependant dans le cadre des suspensions PDMS/silice ces modèles sous-estiment la viscosité (ou le module) car ils ne prennent pas en compte l’aggrégation et la percolation de la silice. D’autres modèles plus pertinents ont été développés, par analogie avec les noirs de carbone [39], sur la base de la théorie de la percolation. Un réseau de charges de silice percolées au sein de l’élastomère est considéré et des lois d’échelle basées sur les travaux de De Gennes [40] ont été proposées pour décrire le comportement de suspensions de silices [41]. Ces lois peuvent s’écrire sous la forme générale :
⎥⎥⎥⎥
⎦
⎤
⎢⎢⎢⎢
⎣
⎡
⎟⎟⎟
⎠
⎞
⎜⎜⎜
⎝
⎛
+−
−+
⎟⎠⎞
⎜⎝⎛ −
−⎟⎟⎟
⎠
⎞
⎜⎜⎜
⎝
⎛
⎟⎟⎟
⎠
⎞
⎜⎜⎜
⎝
⎛
+−
−+−
⎟⎟⎟
⎠
⎞
⎜⎜⎜
⎝
⎛
−+=
32
31
32
31
31
32
32
31
32
31
32
31
32
01
1
131
131
1211
vv
vv
v
v
vv
vvvv kkEE
φφ
φφ
φ
φ
φφ
φφφ
φ
φ
( )vφηη 5,210 +=
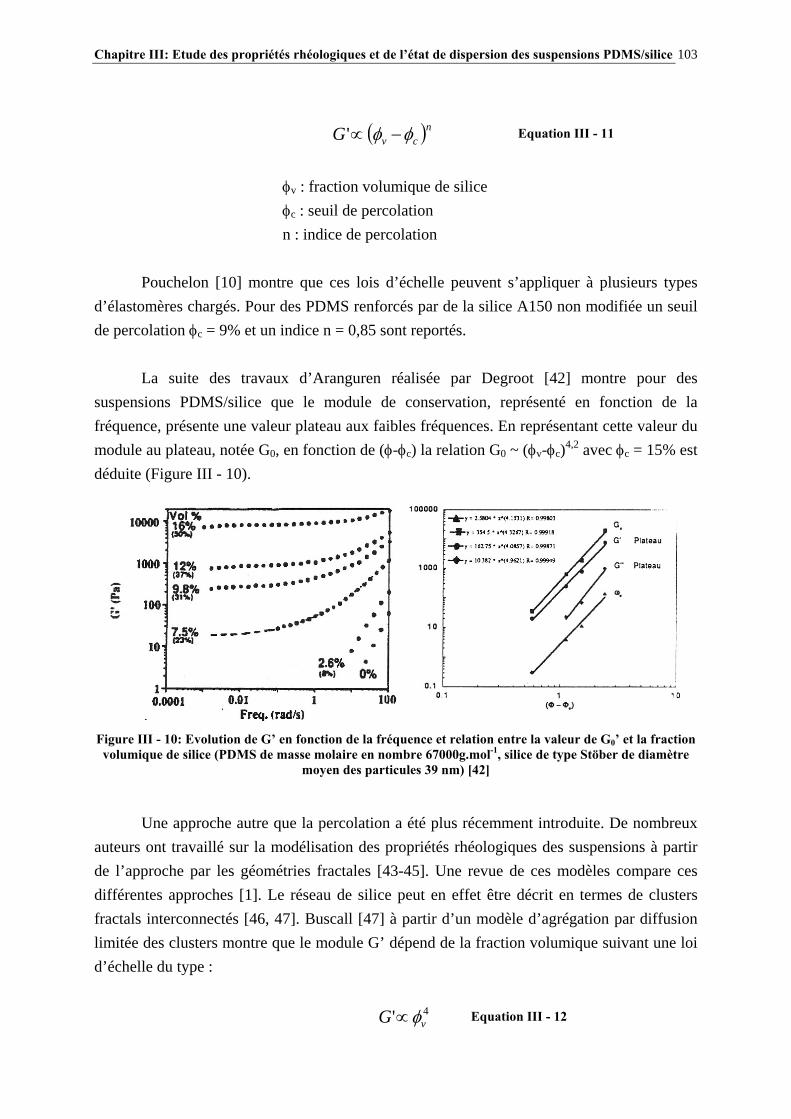
Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
103
Equation III - 11
φv : fraction volumique de silice φc : seuil de percolation n : indice de percolation Pouchelon [10] montre que ces lois d’échelle peuvent s’appliquer à plusieurs types d’élastomères chargés. Pour des PDMS renforcés par de la silice A150 non modifiée un seuil de percolation φc = 9% et un indice n = 0,85 sont reportés. La suite des travaux d’Aranguren réalisée par Degroot [42] montre pour des suspensions PDMS/silice que le module de conservation, représenté en fonction de la fréquence, présente une valeur plateau aux faibles fréquences. En représentant cette valeur du module au plateau, notée G0, en fonction de (φ-φc) la relation G0 ~ (φv-φc)4,2 avec φc = 15% est déduite (Figure III - 10). Figure III - 10: Evolution de G’ en fonction de la fréquence et relation entre la valeur de G0’ et la fraction volumique de silice (PDMS de masse molaire en nombre 67000g.mol-1, silice de type Stöber de diamètre
moyen des particules 39 nm) [42]
Une approche autre que la percolation a été plus récemment introduite. De nombreux auteurs ont travaillé sur la modélisation des propriétés rhéologiques des suspensions à partir de l’approche par les géométries fractales [43-45]. Une revue de ces modèles compare ces différentes approches [1]. Le réseau de silice peut en effet être décrit en termes de clusters fractals interconnectés [46, 47]. Buscall [47] à partir d’un modèle d’agrégation par diffusion limitée des clusters montre que le module G’ dépend de la fraction volumique suivant une loi d’échelle du type : Equation III - 12
( )ncvG φφ −∝'
4' vG φ∝

Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
104
Khan [48] obtient une relation identique mais avec un exposant dépendant du milieu de suspension considéré. Pour une huile minérale (400 g.mol-1) l’indice de percolation a pour valeur 4 tandis que pour un polypropylène glycol (700 g.mol-1) de même viscosité, l’exposant est 6. L’exposant ne semble pas dépendre de la chimie de surface de la silice, par contre les valeurs de G’ augmentent lorsque le nombre de groupements hydroxyle de surface est plus élevé. Le module G’ peut ainsi être relié à la chimie de surface et au milieu de suspension, par l’intermédiaire des paramètres de solubilité. A partir des résultats obtenus sur neuf types de silices et deux polymères (PPG et PEG) une corrélation entre le module G’ et les paramètres de solubilité des groupements de surface et du milieu de suspension est obtenue sous la forme [49, 50] : Equation III - 13
δs : paramètre de solubilité des groupements de surface δs : paramètre de solubilité du milieu de suspension La nature fractale du réseau de silice au sein des suspensions a pu être également observée par Dorget en microscopie et confirmée par des mesures de diffusion [51]. A partir de ces observations, un modèle physique a été proposé, décrivant la formation du réseau de silice par interpénétration d’objets fractals. Les différentes fractions volumiques se traduiraient alors par une interpénétration plus ou moins étroite de ces objets. Lors d’un écoulement il n’y aurait pas rupture de ces objets mais ‘désinterpénétration’. L’écoulement serait donc localisé au niveau de ces structures fractales sans rupture de liens particule-particule et non à l’échelle de l’interface PDMS-silice. Les objets fractals sont constitués d’un certains nombre de particules primaires de silice, de rayon a, associées au sein d’une structure de dimension fractale donnée, notée D (Figure III - 34). Ces objets ont un rayon R donné par la relation [52] : Equation III - 14
Lorsque la fraction volumique de silice augmente, on passe progressivement d’une suspension ‘diluée’ où les objets sont individualisés à une suspension de type semi-diluée, où les objets deviennent interpénétrés par analogie avec des solutions de polymère (Figure III - 12).
DaNR 1=
( )2' msG δδ −∝

Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
105
Figure III - 11 : Schéma d’un objet fractal, de rayon R, de dimension fractale D, contenant N particules de
rayon a
Figure III - 12 : Passage d’une suspension diluée à une suspension semi-diluée pour la fraction volumique
critique φv*
La fraction volumique critique à partir de laquelle la suspension peut être considérée comme de type semi-diluée est définie par la relation : Equation III - 15
A partir de ce modèle physique, l’évolution du module G0’ en fonction de la fraction volumique de silice peut être estimée en fonction des caractéristiques des objets fractals et de la fraction volumique critique φv
* : Equation III - 16
où k est la constante de Boltzmann, T la température et α, β des constantes
La constante α n’a pas de signification physique et indique seulement qu’il existe une relation de proportionnalité entre G0’ et D
v−35φ . En revanche la constante β est un coefficient
permettant de prendre en compte la rigidité de la structure [4].
3−∗ ⎟
⎠⎞
⎜⎝⎛=
D
v aRφ
⇔⎟⎟⎠
⎞⎜⎜⎝
⎛≈
−
∗
D
v
v
RkTG
35
30' φφβ D
vG −≈ 350' αφ
φv
φv < φv* φv > φv
*
Suspension diluée Suspension semi-diluée
φv
φv < φv* φv > φv
*
Suspension diluée Suspension semi-diluée
N particules de rayon a
Objet fractal de rayon R
N particules de rayon a
Objet fractal de rayon R

Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
106
Ce modèle conduit dans le cas des suspensions étudiées par Dorget à la relation suivante [4], très similaire à celle trouvée par Buscall et Khan [47] : Equation III - 17
G0’ est la valeur au temps longs (c’est-à-dire aux basses fréquences) lors d’un balayage en fréquence, dans le domaine viscoélastique linéaire. A partir de mesures de diffusion de lumière, la fraction volumique critique φv
∗ a alors été calculée. Celle-ci est très faible, de l’ordre de 1%.. Ce modèle est intéressant car il permet une approche physique des résultats rhéologiques et permet d’accéder à partir des mesures rhéologiques à la dimension fractale des objets. C’est pourquoi ce modèle a été utilisé pour la suite de notre étude. III.2.3- Conclusions L’analyse de la bibliographie montre que les interactions polymère-charge contrôlent à la fois l’état de dispersion des suspensions et les propriétés rhéologiques qui en résultent. Des approches phénoménologiques font intervenir le rôle des chaînes de polymères qui pontent les particules, directement ou indirectement, modifiant la rhéologie finale des suspensions. Lorsque la silice est greffée la rhéologie est modifiée comme l’ont bien montré Aranguren et al. Cependant, le rôle du greffage sur la modification des suspensions n’a été que partiellement étudié. En particulier le rôle du taux de greffage, pour une même nature de greffon, sur les propriétés des suspensions reste à déterminer. Existe-t-il par exemple un taux de greffage critique vis-à-vis de la dispersion et des propriétés rhéologiques ? Par ailleurs le greffage n’est pas pris en compte dans la modélisation du module G0
’ en fonction de la fraction volumique. Or la géométrie des agrégats de silice est certainement changée lorsque les interactions sont modifiées. Ce chapitre est donc destiné à clarifier l’influence du greffage des silices sur les propriétés des suspensions, pour pouvoir prédire en particulier le comportement rhéologique des suspensions en fonction de la fraction volumique, tout en tenant compte du taux de greffage des silices. Nous nous appuierons pour cela sur le modèle d’objets fractals semi-dilués développé par Dorget [51], qui permet une approche physique du problème. Par ailleurs l’influence du procédé de greffage (greffage in-situ et ex-situ) sur les propriétés finales des suspensions, qui n’a jamais été étudiée, viendra compléter cette étude.
2,40 ' vG φ∝
Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
107
III.3- PARTIE EXPÉRIMENTALE III.3.1- Matériaux Les suspensions PDMS/silice ont été réalisées avec des huiles PDMS à fins de chaîne vinyle, confiées par Rhodia Silicones. Ces PDMS ont des masses molaires différentes, déterminées par GPC (Tableau III - 1), mais l’étude porte principalement sur le PDMS noté H1500V qui possède une masse molaire en poids de 41 000 g.mol-1 (Tableau III - 2). Le taux de vinyle correspond aux fins de chaînes vinyles des PDMS. Les nanoparticules considérées sont des silices A200 déjà présentées au chapitre II, modifiées partiellement par réaction avec l’HMDS pour des concentrations variables en HMDS.
Nomenclature Viscosité (mPa.s) à 25°C Masse molaire Mw (g.mol-1) H600V 600 25 000
H1500V 1 500 41 000 H10000V 10 000 70 000
Tableau III - 1: Masses molaires et viscosités (à 25°C) des différentes huiles PDMS étudiées
Caractéristiques H1500V
Viscosité (mPa.s) à 25°C 1 500 Masse molaire Mw (g.mol-1) 41 000 Indice de polymolécularité Ip 1,88 Taux de vinyle (% massique) 0,24
Tableau III - 2: Caractéristiques de l’huile PDMS H1500V
III.3.2- Mise en œuvre des suspensions PDMS/silice Les procédés de mise en œuvre, qui incluent également le greffage de silice dans le cas des procédés in-situ, sont présentés dans ce paragraphe. On peut distinguer d’une part les procédés in-situ où la silice est greffée au sein des mélanges, avec introduction de l’HMDS avant ou après incorporation de la silice, et d’autre part le procédé ex-situ où la silice est préalablement modifiée par réaction avec l’HMDS en phase gazeuse puis incorporée au PDMS. Toutes les suspensions ont été mises en œuvre dans les laboratoires de Rhodia Silicones, dans un malaxeur de 1 litre (MEILI) avec une vitesse de rotation des pâles constante de 12,5 tours par minute. La quantité totale de matière introduite dans le malaxeur est de 750g afin d’obtenir un remplissage optimal. Enfin des suspensions ont été réalisées par

Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
108
dilution dans le PDMS d’une première suspension PDMS/silice plus concentrée en silice. La Figure III - 13 résume de manière schématique toutes ces procédures de mise en œuvre, et la nomenclature qui a été adoptée pour chaque procédé.
Figure III - 13 : Schéma et nomenclature des différentes procédures de mise en œuvre des suspensions PDMS/silice
II.3.2.1- Procédés in-situ a) Procédé in-situ en 1 étape (Z) Ce procédé a été le plus fréquemment utilisé. L’huile est tout d’abord introduite dans le malaxeur, avec l’HMDS et une faible quantité d’eau (30% en poids par rapport à la quantité d’HMDS), qui catalyse la réaction de l’HMDS avec la surface de la silice. Ces trois composants sont mélangés à température ambiante pendant 5 minutes puis la silice est incorporée au PDMS par ajouts successifs d’une dizaine de grammes. L’incorporation de la silice varie ainsi de 20 minutes à 1 heure suivant la fraction volumique à atteindre. Le mélange passe d’un état de fluide visqueux à une pâte dont la viscosité peut devenir très importante suivant la fraction volumique de silice et la concentration en silane. Une fois toute la silice incorporée, une phase de mélange supplémentaire de 2 heures intervient afin de laisser la réaction entre l’HMDS et la surface de silice se terminer, puis le mélange est porté pendant deux heures supplémentaires à 100°C sous vide pour évaporer l’eau et l’ammoniac
1
2
PROCEDE EX-SITU
A200
+HMDS
A200 greffée
Nomenclature : TPDMS
A200 greffée
PROCEDES IN-SITU
Nomenclature : Z
1
Nomenclature : F
2
HMDS
PDMS + A200
1PDMS
A200
PDMS+HMDS
A200
Nomenclature : ZD
Dilution
1
2
PROCEDE EX-SITU
A200
+HMDS
A200 greffée
Nomenclature : TPDMS
A200 greffée
PROCEDE EX-SITU
A200
+HMDS
A200 greffée
Nomenclature : TPDMS
A200 greffée
PROCEDES IN-SITU
Nomenclature : Z
1
Nomenclature : F
2
HMDS
PDMS + A200
1PDMS
A200
PDMS+HMDS
A200
Nomenclature : ZD
Dilution
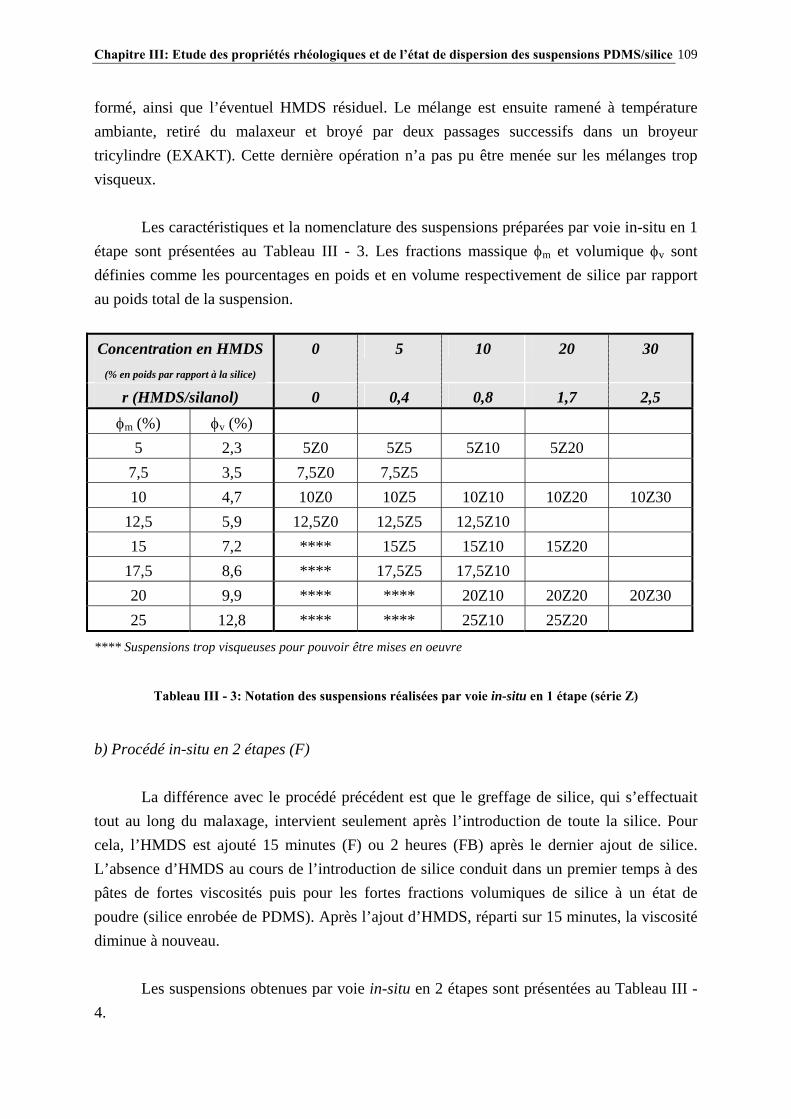
Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
109
formé, ainsi que l’éventuel HMDS résiduel. Le mélange est ensuite ramené à température ambiante, retiré du malaxeur et broyé par deux passages successifs dans un broyeur tricylindre (EXAKT). Cette dernière opération n’a pas pu être menée sur les mélanges trop visqueux.
Les caractéristiques et la nomenclature des suspensions préparées par voie in-situ en 1 étape sont présentées au Tableau III - 3. Les fractions massique φm et volumique φv sont définies comme les pourcentages en poids et en volume respectivement de silice par rapport au poids total de la suspension.
Concentration en HMDS
(% en poids par rapport à la silice)
0 5 10 20 30
r (HMDS/silanol) 0 0,4 0,8 1,7 2,5 φm (%) φv (%)
5 2,3 5Z0 5Z5 5Z10 5Z20 7,5 3,5 7,5Z0 7,5Z5 10 4,7 10Z0 10Z5 10Z10 10Z20 10Z30
12,5 5,9 12,5Z0 12,5Z5 12,5Z10 15 7,2 **** 15Z5 15Z10 15Z20
17,5 8,6 **** 17,5Z5 17,5Z10 20 9,9 **** **** 20Z10 20Z20 20Z30 25 12,8 **** **** 25Z10 25Z20
**** Suspensions trop visqueuses pour pouvoir être mises en oeuvre
Tableau III - 3: Notation des suspensions réalisées par voie in-situ en 1 étape (série Z)
b) Procédé in-situ en 2 étapes (F) La différence avec le procédé précédent est que le greffage de silice, qui s’effectuait tout au long du malaxage, intervient seulement après l’introduction de toute la silice. Pour cela, l’HMDS est ajouté 15 minutes (F) ou 2 heures (FB) après le dernier ajout de silice. L’absence d’HMDS au cours de l’introduction de silice conduit dans un premier temps à des pâtes de fortes viscosités puis pour les fortes fractions volumiques de silice à un état de poudre (silice enrobée de PDMS). Après l’ajout d’HMDS, réparti sur 15 minutes, la viscosité diminue à nouveau. Les suspensions obtenues par voie in-situ en 2 étapes sont présentées au Tableau III - 4.
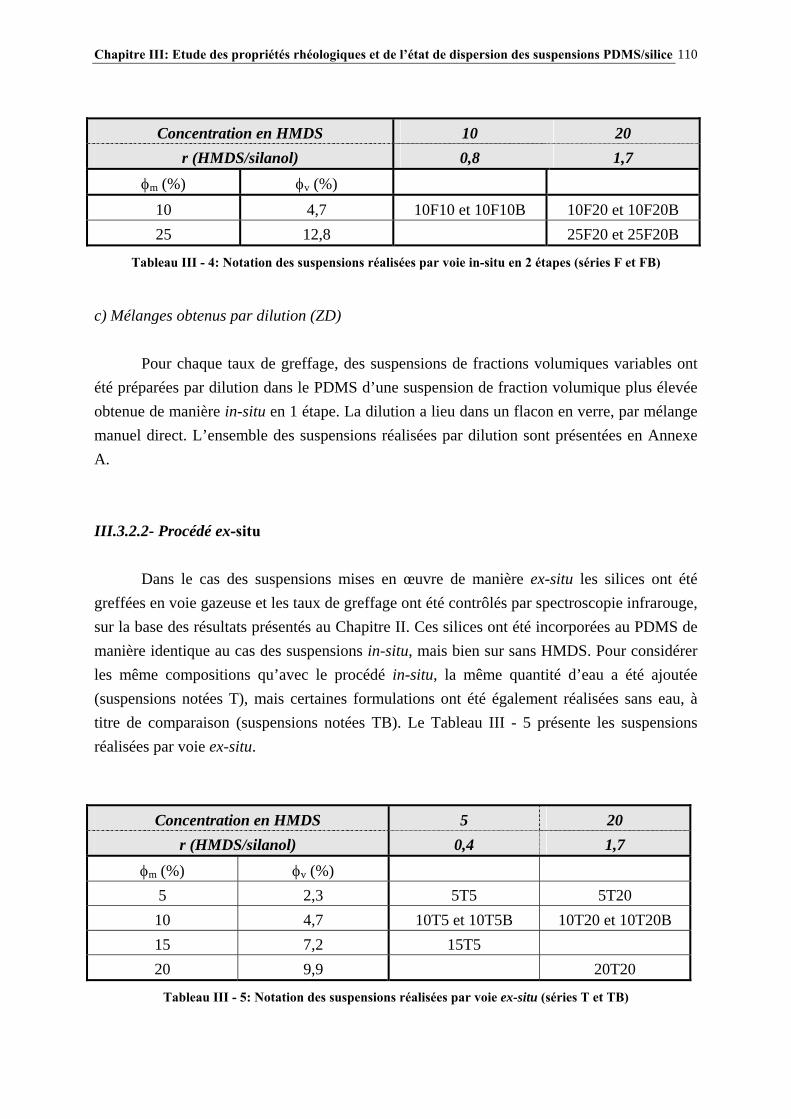
Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
110
Concentration en HMDS 10 20
r (HMDS/silanol) 0,8 1,7 φm (%) φv (%)
10 4,7 10F10 et 10F10B 10F20 et 10F20B 25 12,8 25F20 et 25F20B
Tableau III - 4: Notation des suspensions réalisées par voie in-situ en 2 étapes (séries F et FB)
c) Mélanges obtenus par dilution (ZD) Pour chaque taux de greffage, des suspensions de fractions volumiques variables ont été préparées par dilution dans le PDMS d’une suspension de fraction volumique plus élevée obtenue de manière in-situ en 1 étape. La dilution a lieu dans un flacon en verre, par mélange manuel direct. L’ensemble des suspensions réalisées par dilution sont présentées en Annexe A. III.3.2.2- Procédé ex-situ Dans le cas des suspensions mises en œuvre de manière ex-situ les silices ont été greffées en voie gazeuse et les taux de greffage ont été contrôlés par spectroscopie infrarouge, sur la base des résultats présentés au Chapitre II. Ces silices ont été incorporées au PDMS de manière identique au cas des suspensions in-situ, mais bien sur sans HMDS. Pour considérer les même compositions qu’avec le procédé in-situ, la même quantité d’eau a été ajoutée (suspensions notées T), mais certaines formulations ont été également réalisées sans eau, à titre de comparaison (suspensions notées TB). Le Tableau III - 5 présente les suspensions réalisées par voie ex-situ.
Concentration en HMDS 5 20 r (HMDS/silanol) 0,4 1,7
φm (%) φv (%) 5 2,3 5T5 5T20
10 4,7 10T5 et 10T5B 10T20 et 10T20B 15 7,2 15T5 20 9,9 20T20
Tableau III - 5: Notation des suspensions réalisées par voie ex-situ (séries T et TB)

Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
111
III.3.3- Caractérisation des suspensions III.3.3.1- Caractérisation des propriétés rhéologiques a) Appareillage et géométries La rhéologie des suspensions a été étudiée principalement en régime dynamique, en utilisant des appareils à déformation imposée et à contrainte imposée. Les géométries utilisées dépendent des suspensions étudiées. La géométrie cône-plan est théoriquement la mieux adaptée car elle permet d’appliquer une déformation en cisaillement homogène au sein de l’échantillon, mais pour des forts taux de silice le contact des nanoparticules dans l’entrefer perturbe les mesures. Pour les forts taux de charge et les faibles concentrations en HMDS, une géométrie plan-plan a donc été plutôt utilisée (cf. Annexe B). Toutes les mesures rhéologiques réalisées au cours de cette étude sont réalisés à la température constante de 25°C, les suspensions PDMS/silice étant peu sensibles à l’effet de la température dans ce domaine. b) Mise au point des protocoles d’essais Mesures en régime dynamique En régime dynamique, des balayages en déformation à fréquence imposée et des balayages en fréquence à contrainte imposée ont été principalement réalisés. Un premier point délicat concerne la mise en place de l’échantillon. Une attention particulière doit être portée à cette étape pour de ne pas déstructurer l’échantillon ce qui affecterait les résultats du test (cf. Annexe B). Pour évaluer l’impact de la mise en place de l’échantillon sur la déstructuration des suspensions, les modules dynamiques complexes G’ et G’’ ont été enregistrés en fonction du temps à déformation et fréquence fixes. La Figure III - 14 montre que suivant les formulations le temps nécessaire pour la stabilisation du module G’, caractéristique du temps nécessaire pour l’atteinte d’une structuration à l’équilibre est différent, mais se situe en moyenne autour de 20 minutes. Ce temps d’attente entre la mise en place de l’échantillon et le début des mesures a donc été adopté pour la suite des essais. Par ailleurs, un autre point critique est l’évolution des mélanges au cours du temps. En effet le vieillissement des suspensions a été décrit comme étant principalement la
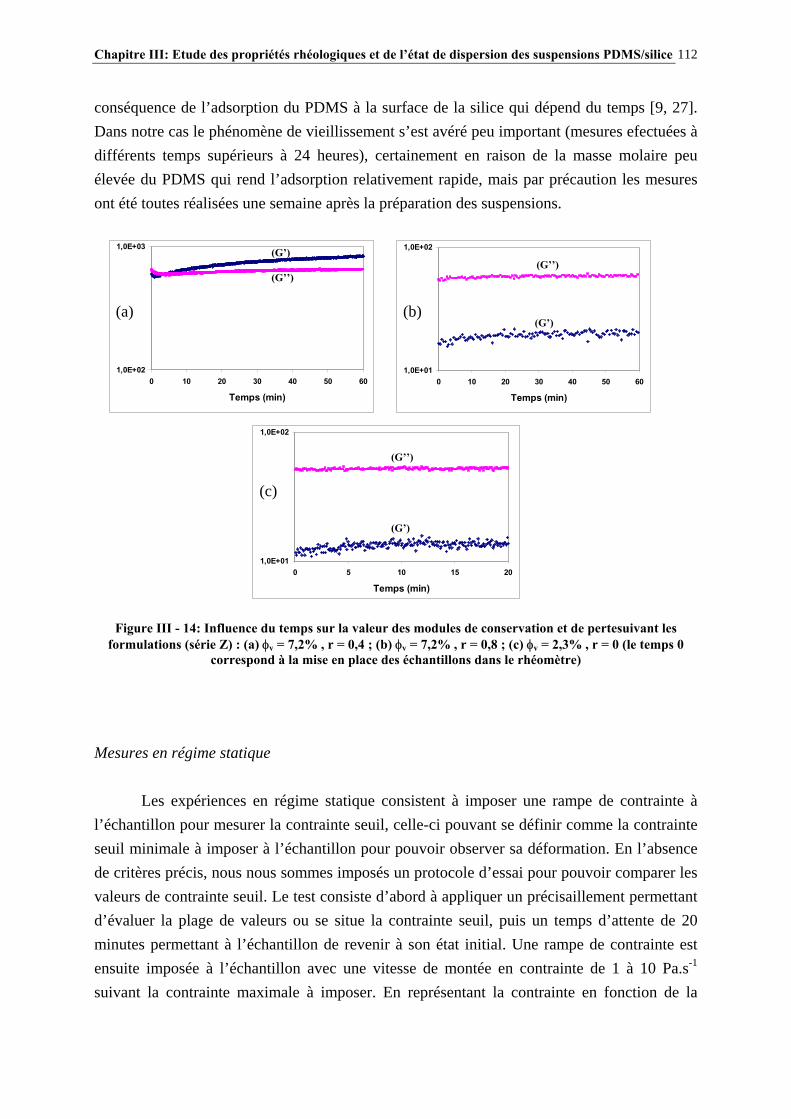
Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
112
conséquence de l’adsorption du PDMS à la surface de la silice qui dépend du temps [9, 27]. Dans notre cas le phénomène de vieillissement s’est avéré peu important (mesures efectuées à différents temps supérieurs à 24 heures), certainement en raison de la masse molaire peu élevée du PDMS qui rend l’adsorption relativement rapide, mais par précaution les mesures ont été toutes réalisées une semaine après la préparation des suspensions.
Figure III - 14: Influence du temps sur la valeur des modules de conservation et de pertesuivant les formulations (série Z) : (a) φv = 7,2% , r = 0,4 ; (b) φv = 7,2% , r = 0,8 ; (c) φv = 2,3% , r = 0 (le temps 0
correspond à la mise en place des échantillons dans le rhéomètre)
Mesures en régime statique Les expériences en régime statique consistent à imposer une rampe de contrainte à l’échantillon pour mesurer la contrainte seuil, celle-ci pouvant se définir comme la contrainte seuil minimale à imposer à l’échantillon pour pouvoir observer sa déformation. En l’absence de critères précis, nous nous sommes imposés un protocole d’essai pour pouvoir comparer les valeurs de contrainte seuil. Le test consiste d’abord à appliquer un précisaillement permettant d’évaluer la plage de valeurs ou se situe la contrainte seuil, puis un temps d’attente de 20 minutes permettant à l’échantillon de revenir à son état initial. Une rampe de contrainte est ensuite imposée à l’échantillon avec une vitesse de montée en contrainte de 1 à 10 Pa.s-1 suivant la contrainte maximale à imposer. En représentant la contrainte en fonction de la
1,0E+02
1,0E+03
0 10 20 30 40 50 60
Temps (min)
1,0E+01
1,0E+02
0 10 20 30 40 50 60
Temps (min)
1,0E+01
1,0E+02
0 5 10 15 20
Temps (min)
(a) (b)
(c)
(G’)
(G’’)
(G’)
(G’’)
(G’)
(G’’)
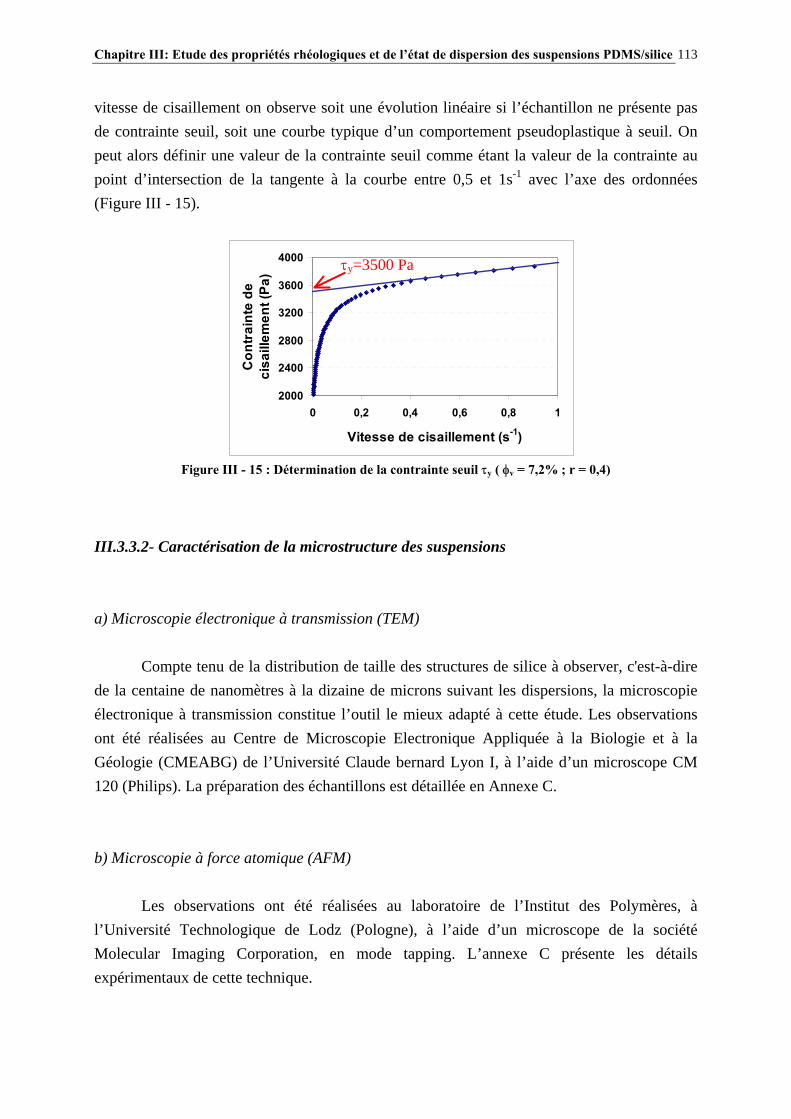
Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
113
vitesse de cisaillement on observe soit une évolution linéaire si l’échantillon ne présente pas de contrainte seuil, soit une courbe typique d’un comportement pseudoplastique à seuil. On peut alors définir une valeur de la contrainte seuil comme étant la valeur de la contrainte au point d’intersection de la tangente à la courbe entre 0,5 et 1s-1 avec l’axe des ordonnées (Figure III - 15).
Figure III - 15 : Détermination de la contrainte seuil τy ( φv = 7,2% ; r = 0,4)
III.3.3.2- Caractérisation de la microstructure des suspensions a) Microscopie électronique à transmission (TEM) Compte tenu de la distribution de taille des structures de silice à observer, c'est-à-dire de la centaine de nanomètres à la dizaine de microns suivant les dispersions, la microscopie électronique à transmission constitue l’outil le mieux adapté à cette étude. Les observations ont été réalisées au Centre de Microscopie Electronique Appliquée à la Biologie et à la Géologie (CMEABG) de l’Université Claude bernard Lyon I, à l’aide d’un microscope CM 120 (Philips). La préparation des échantillons est détaillée en Annexe C. b) Microscopie à force atomique (AFM) Les observations ont été réalisées au laboratoire de l’Institut des Polymères, à l’Université Technologique de Lodz (Pologne), à l’aide d’un microscope de la société Molecular Imaging Corporation, en mode tapping. L’annexe C présente les détails expérimentaux de cette technique.
2000
2400
2800
3200
3600
4000
0 0,2 0,4 0,6 0,8 1
Vitesse de cisaillement (s-1)
Con
trai
nte
de
cisa
illem
ent (
Pa) τy=3500 Pa

Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
114
c) Diffusion dynamique de la lumière (DDL) Cette technique a été utilisée pour caractériser la taille des agrégats de silice. Comme ces objets sont enchevêtrés et forment un réseau dans la suspension nous avons dilué les suspensions dans le cyclohexane, bon solvant des silicones, afin de séparer les objets élémentaires et pouvoir en mesurer la taille. Les mesures ont été réalisées au Laboratoire des Matériaux Organiques à Propriétés Spécifiques (LMOPS) du CNRS de Solaize, avec un laser Spectra Physics Series 2000, de longueur d’onde 514 nm et un angle de mesure de 90°. Les détails de cette technique sont précisés en Annexe D.
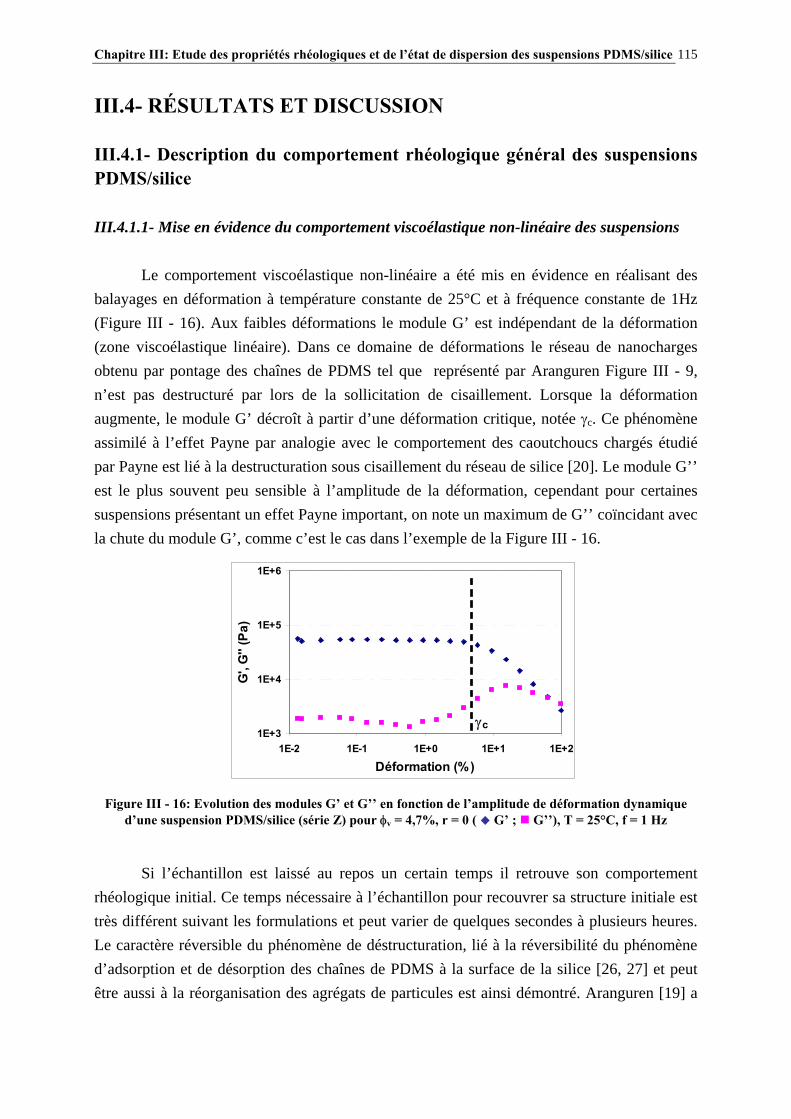
Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
115
III.4- RÉSULTATS ET DISCUSSION III.4.1- Description du comportement rhéologique général des suspensions PDMS/silice III.4.1.1- Mise en évidence du comportement viscoélastique non-linéaire des suspensions Le comportement viscoélastique non-linéaire a été mis en évidence en réalisant des balayages en déformation à température constante de 25°C et à fréquence constante de 1Hz (Figure III - 16). Aux faibles déformations le module G’ est indépendant de la déformation (zone viscoélastique linéaire). Dans ce domaine de déformations le réseau de nanocharges obtenu par pontage des chaînes de PDMS tel que représenté par Aranguren Figure III - 9, n’est pas destructuré par lors de la sollicitation de cisaillement. Lorsque la déformation augmente, le module G’ décroît à partir d’une déformation critique, notée γc. Ce phénomène assimilé à l’effet Payne par analogie avec le comportement des caoutchoucs chargés étudié par Payne est lié à la destructuration sous cisaillement du réseau de silice [20]. Le module G’’ est le plus souvent peu sensible à l’amplitude de la déformation, cependant pour certaines suspensions présentant un effet Payne important, on note un maximum de G’’ coïncidant avec la chute du module G’, comme c’est le cas dans l’exemple de la Figure III - 16.
Figure III - 16: Evolution des modules G’ et G’’ en fonction de l’amplitude de déformation dynamique d’une suspension PDMS/silice (série Z) pour φv = 4,7%, r = 0 ( G’ ; G’’), T = 25°C, f = 1 Hz
Si l’échantillon est laissé au repos un certain temps il retrouve son comportement rhéologique initial. Ce temps nécessaire à l’échantillon pour recouvrer sa structure initiale est très différent suivant les formulations et peut varier de quelques secondes à plusieurs heures. Le caractère réversible du phénomène de déstructuration, lié à la réversibilité du phénomène d’adsorption et de désorption des chaînes de PDMS à la surface de la silice [26, 27] et peut être aussi à la réorganisation des agrégats de particules est ainsi démontré. Aranguren [19] a
1E+3
1E+4
1E+5
1E+6
1E-2 1E-1 1E+0 1E+1 1E+2
Déformation (%)
G',
G''
(Pa)
γc
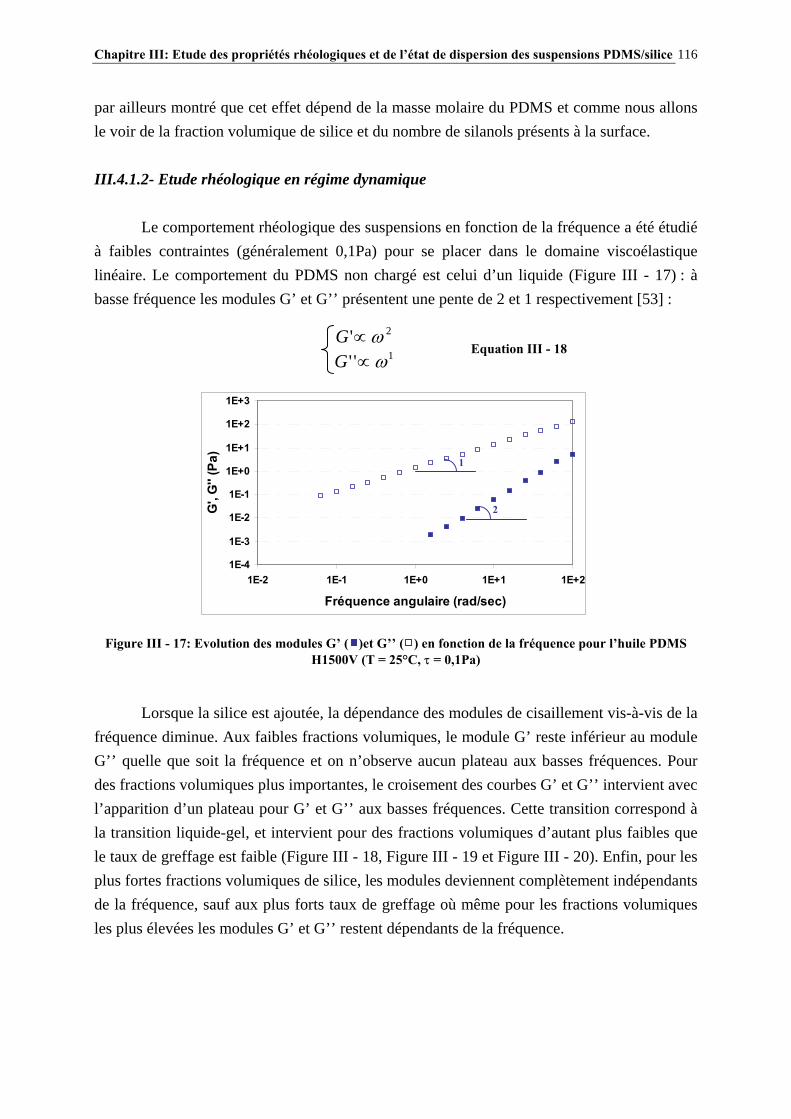
Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
116
par ailleurs montré que cet effet dépend de la masse molaire du PDMS et comme nous allons le voir de la fraction volumique de silice et du nombre de silanols présents à la surface. III.4.1.2- Etude rhéologique en régime dynamique Le comportement rhéologique des suspensions en fonction de la fréquence a été étudié à faibles contraintes (généralement 0,1Pa) pour se placer dans le domaine viscoélastique linéaire. Le comportement du PDMS non chargé est celui d’un liquide (Figure III - 17) : à basse fréquence les modules G’ et G’’ présentent une pente de 2 et 1 respectivement [53] : Equation III - 18
Figure III - 17: Evolution des modules G’ ( )et G’’ ( ) en fonction de la fréquence pour l’huile PDMS H1500V (T = 25°C, τ = 0,1Pa)
Lorsque la silice est ajoutée, la dépendance des modules de cisaillement vis-à-vis de la fréquence diminue. Aux faibles fractions volumiques, le module G’ reste inférieur au module G’’ quelle que soit la fréquence et on n’observe aucun plateau aux basses fréquences. Pour des fractions volumiques plus importantes, le croisement des courbes G’ et G’’ intervient avec l’apparition d’un plateau pour G’ et G’’ aux basses fréquences. Cette transition correspond à la transition liquide-gel, et intervient pour des fractions volumiques d’autant plus faibles que le taux de greffage est faible (Figure III - 18, Figure III - 19 et Figure III - 20). Enfin, pour les plus fortes fractions volumiques de silice, les modules deviennent complètement indépendants de la fréquence, sauf aux plus forts taux de greffage où même pour les fractions volumiques les plus élevées les modules G’ et G’’ restent dépendants de la fréquence.
1E-4
1E-3
1E-2
1E-1
1E+0
1E+1
1E+2
1E+3
1E-2 1E-1 1E+0 1E+1 1E+2
Fréquence angulaire (rad/sec)
G',
G''
(Pa)
2' ω∝G1'' ω∝G
1
2
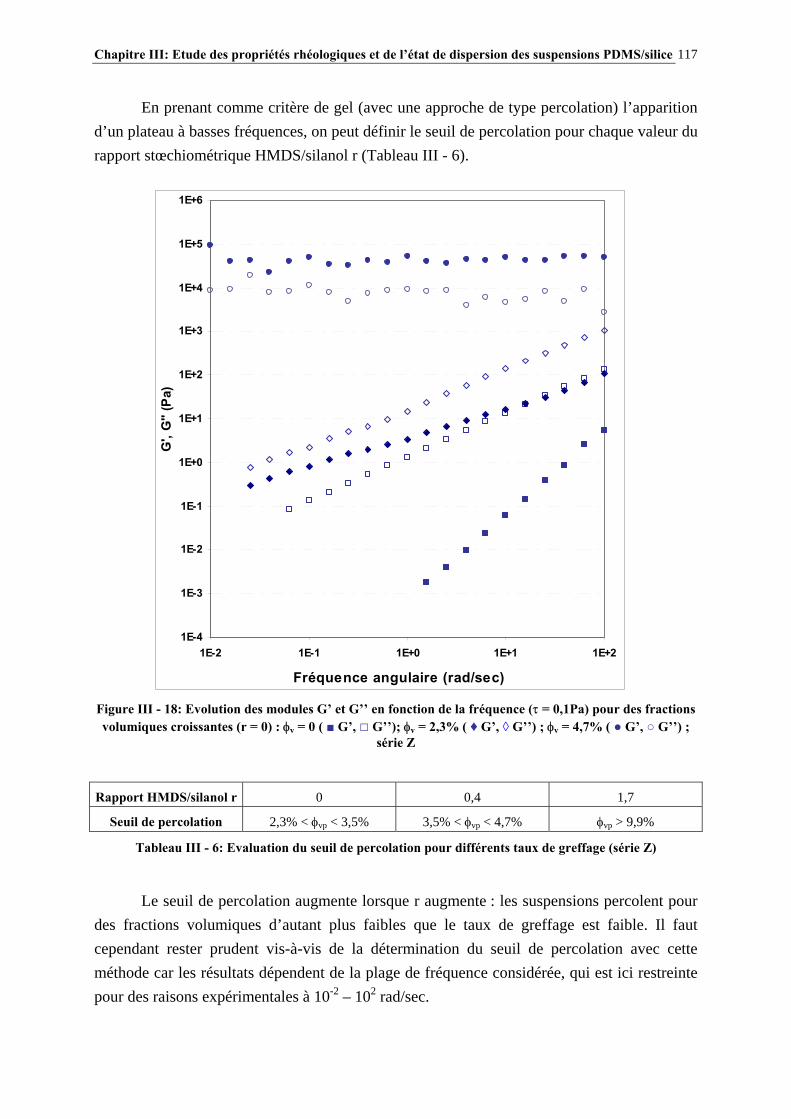
Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
117
En prenant comme critère de gel (avec une approche de type percolation) l’apparition d’un plateau à basses fréquences, on peut définir le seuil de percolation pour chaque valeur du rapport stœchiométrique HMDS/silanol r (Tableau III - 6). Figure III - 18: Evolution des modules G’ et G’’ en fonction de la fréquence (τ = 0,1Pa) pour des fractions volumiques croissantes (r = 0) : φv = 0 ( ■ G’, □ G’’); φv = 2,3% ( ♦ G’, ◊ G’’) ; φv = 4,7% ( ● G’, ○ G’’) ;
série Z
Rapport HMDS/silanol r 0 0,4 1,7
Seuil de percolation 2,3% < φvp < 3,5% 3,5% < φvp < 4,7% φvp > 9,9%
Tableau III - 6: Evaluation du seuil de percolation pour différents taux de greffage (série Z)
Le seuil de percolation augmente lorsque r augmente : les suspensions percolent pour des fractions volumiques d’autant plus faibles que le taux de greffage est faible. Il faut cependant rester prudent vis-à-vis de la détermination du seuil de percolation avec cette méthode car les résultats dépendent de la plage de fréquence considérée, qui est ici restreinte pour des raisons expérimentales à 10-2 – 102 rad/sec.
1E-4
1E-3
1E-2
1E-1
1E+0
1E+1
1E+2
1E+3
1E+4
1E+5
1E+6
1E-2 1E-1 1E+0 1E+1 1E+2
Fréquence angulaire (rad/sec)
G',
G''
(Pa)
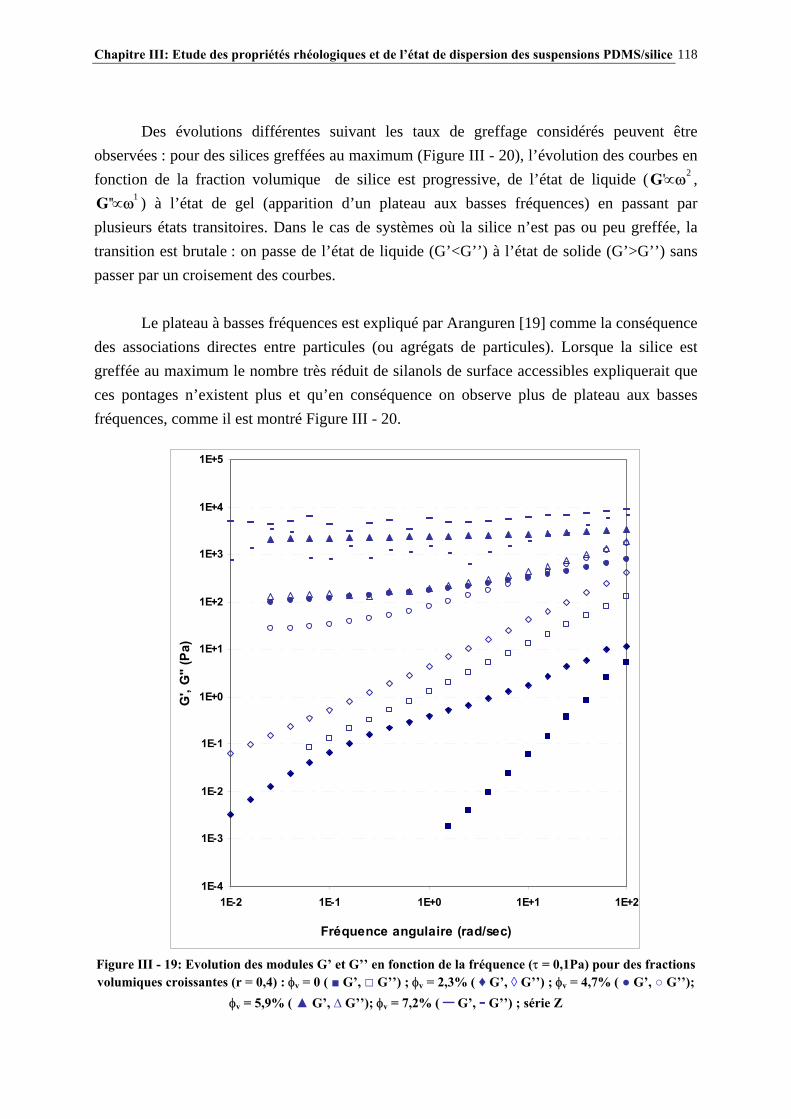
Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
118
Des évolutions différentes suivant les taux de greffage considérés peuvent être observées : pour des silices greffées au maximum (Figure III - 20), l’évolution des courbes en fonction de la fraction volumique de silice est progressive, de l’état de liquide ( 2' ω∝G ,
1'' ω∝G ) à l’état de gel (apparition d’un plateau aux basses fréquences) en passant par plusieurs états transitoires. Dans le cas de systèmes où la silice n’est pas ou peu greffée, la transition est brutale : on passe de l’état de liquide (G’<G’’) à l’état de solide (G’>G’’) sans passer par un croisement des courbes. Le plateau à basses fréquences est expliqué par Aranguren [19] comme la conséquence des associations directes entre particules (ou agrégats de particules). Lorsque la silice est greffée au maximum le nombre très réduit de silanols de surface accessibles expliquerait que ces pontages n’existent plus et qu’en conséquence on observe plus de plateau aux basses fréquences, comme il est montré Figure III - 20. Figure III - 19: Evolution des modules G’ et G’’ en fonction de la fréquence (τ = 0,1Pa) pour des fractions volumiques croissantes (r = 0,4) : φv = 0 ( ■ G’, □ G’’) ; φv = 2,3% ( ♦ G’, ◊ G’’) ; φv = 4,7% ( ● G’, ○ G’’);
φv = 5,9% ( ▲ G’, ∆ G’’); φv = 7,2% ( ─ G’, - G’’) ; série Z
1E-4
1E-3
1E-2
1E-1
1E+0
1E+1
1E+2
1E+3
1E+4
1E+5
1E-2 1E-1 1E+0 1E+1 1E+2
Fréquence angulaire (rad/sec)
G',
G''
(Pa)
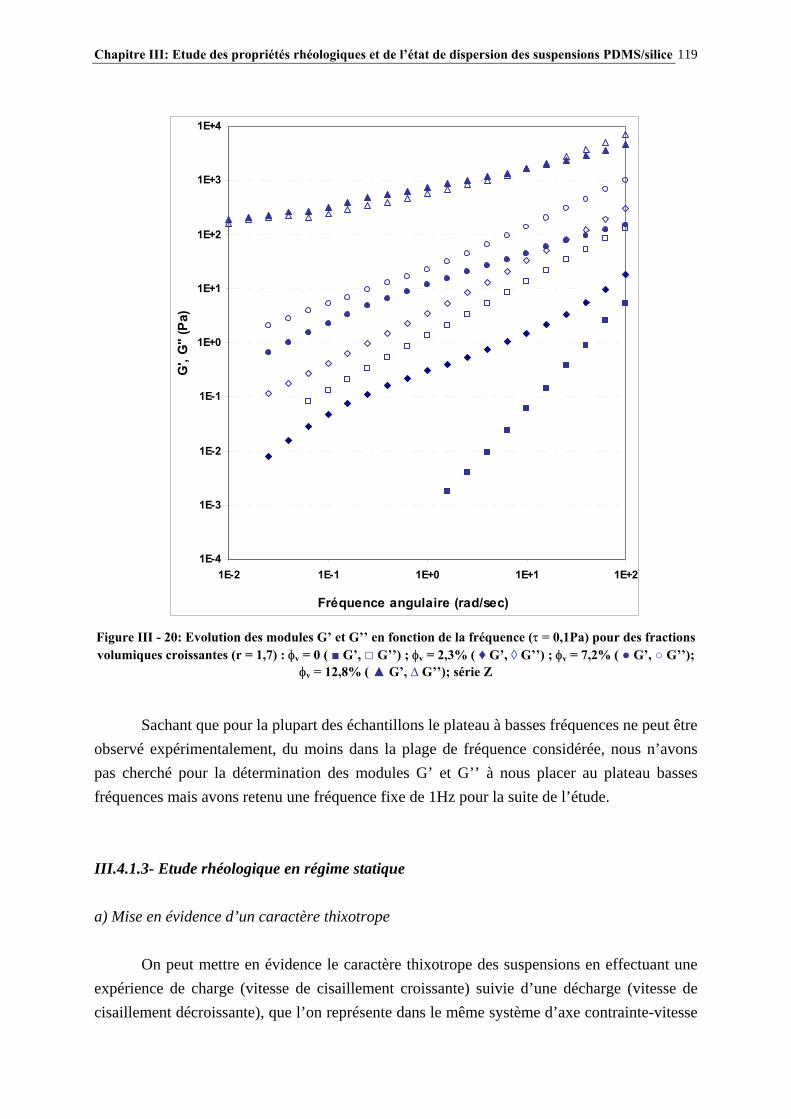
Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
119
Figure III - 20: Evolution des modules G’ et G’’ en fonction de la fréquence (τ = 0,1Pa) pour des fractions volumiques croissantes (r = 1,7) : φv = 0 ( ■ G’, □ G’’) ; φv = 2,3% ( ♦ G’, ◊ G’’) ; φv = 7,2% ( ● G’, ○ G’’);
φv = 12,8% ( ▲ G’, ∆ G’’); série Z
Sachant que pour la plupart des échantillons le plateau à basses fréquences ne peut être observé expérimentalement, du moins dans la plage de fréquence considérée, nous n’avons pas cherché pour la détermination des modules G’ et G’’ à nous placer au plateau basses fréquences mais avons retenu une fréquence fixe de 1Hz pour la suite de l’étude. III.4.1.3- Etude rhéologique en régime statique a) Mise en évidence d’un caractère thixotrope On peut mettre en évidence le caractère thixotrope des suspensions en effectuant une expérience de charge (vitesse de cisaillement croissante) suivie d’une décharge (vitesse de cisaillement décroissante), que l’on représente dans le même système d’axe contrainte-vitesse
1E-4
1E-3
1E-2
1E-1
1E+0
1E+1
1E+2
1E+3
1E+4
1E-2 1E-1 1E+0 1E+1 1E+2
Fréquence angulaire (rad/sec)
G',
G''
(Pa)
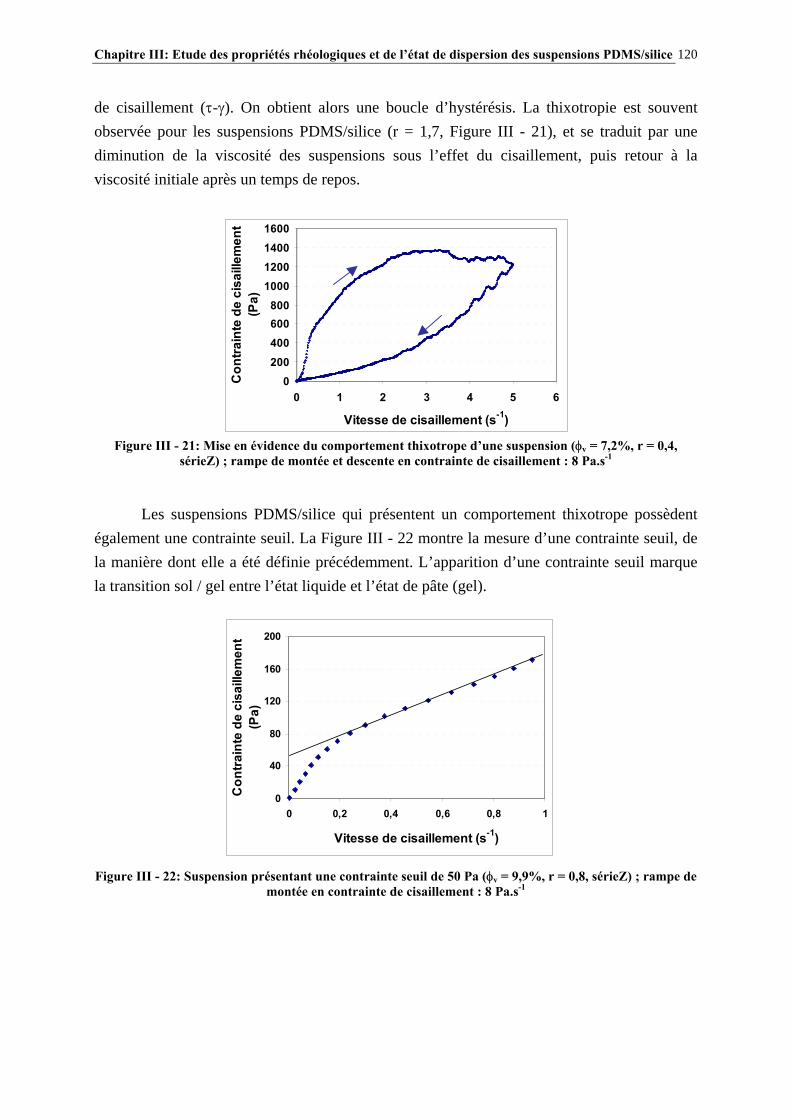
Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
120
de cisaillement (τ-γ). On obtient alors une boucle d’hystérésis. La thixotropie est souvent observée pour les suspensions PDMS/silice (r = 1,7, Figure III - 21), et se traduit par une diminution de la viscosité des suspensions sous l’effet du cisaillement, puis retour à la viscosité initiale après un temps de repos.
Figure III - 21: Mise en évidence du comportement thixotrope d’une suspension (φv = 7,2%, r = 0,4, sérieZ) ; rampe de montée et descente en contrainte de cisaillement : 8 Pa.s-1
Les suspensions PDMS/silice qui présentent un comportement thixotrope possèdent également une contrainte seuil. La Figure III - 22 montre la mesure d’une contrainte seuil, de la manière dont elle a été définie précédemment. L’apparition d’une contrainte seuil marque la transition sol / gel entre l’état liquide et l’état de pâte (gel). Figure III - 22: Suspension présentant une contrainte seuil de 50 Pa (φv = 9,9%, r = 0,8, sérieZ) ; rampe de
montée en contrainte de cisaillement : 8 Pa.s-1
0200400600800
1000120014001600
0 1 2 3 4 5 6
Vitesse de cisaillement (s-1)
Con
trai
nte
de c
isai
llem
ent
(Pa)
0
40
80
120
160
200
0 0,2 0,4 0,6 0,8 1
Vitesse de cisaillement (s-1)
Con
train
te d
e ci
saill
emen
t (P
a)
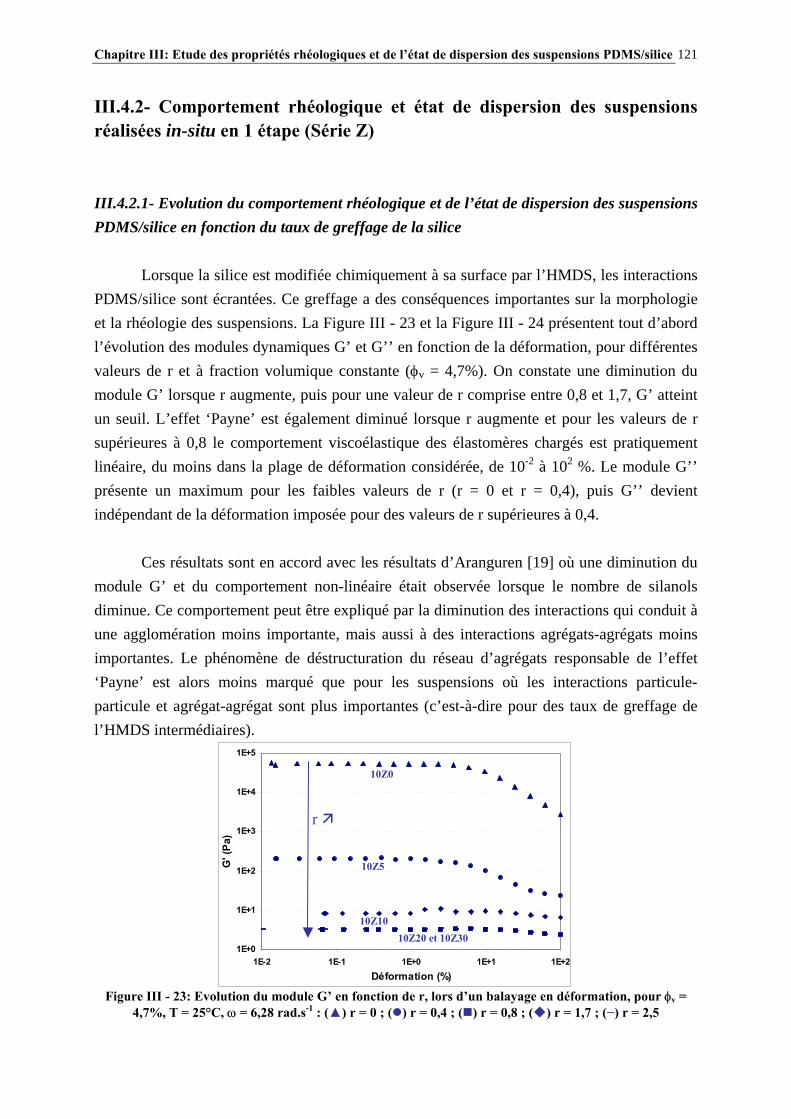
Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
121
III.4.2- Comportement rhéologique et état de dispersion des suspensions réalisées in-situ en 1 étape (Série Z) III.4.2.1- Evolution du comportement rhéologique et de l’état de dispersion des suspensions PDMS/silice en fonction du taux de greffage de la silice Lorsque la silice est modifiée chimiquement à sa surface par l’HMDS, les interactions PDMS/silice sont écrantées. Ce greffage a des conséquences importantes sur la morphologie et la rhéologie des suspensions. La Figure III - 23 et la Figure III - 24 présentent tout d’abord l’évolution des modules dynamiques G’ et G’’ en fonction de la déformation, pour différentes valeurs de r et à fraction volumique constante (φv = 4,7%). On constate une diminution du module G’ lorsque r augmente, puis pour une valeur de r comprise entre 0,8 et 1,7, G’ atteint un seuil. L’effet ‘Payne’ est également diminué lorsque r augmente et pour les valeurs de r supérieures à 0,8 le comportement viscoélastique des élastomères chargés est pratiquement linéaire, du moins dans la plage de déformation considérée, de 10-2 à 102 %. Le module G’’ présente un maximum pour les faibles valeurs de r (r = 0 et r = 0,4), puis G’’ devient indépendant de la déformation imposée pour des valeurs de r supérieures à 0,4. Ces résultats sont en accord avec les résultats d’Aranguren [19] où une diminution du module G’ et du comportement non-linéaire était observée lorsque le nombre de silanols diminue. Ce comportement peut être expliqué par la diminution des interactions qui conduit à une agglomération moins importante, mais aussi à des interactions agrégats-agrégats moins importantes. Le phénomène de déstructuration du réseau d’agrégats responsable de l’effet ‘Payne’ est alors moins marqué que pour les suspensions où les interactions particule-particule et agrégat-agrégat sont plus importantes (c’est-à-dire pour des taux de greffage de l’HMDS intermédiaires).
Figure III - 23: Evolution du module G’ en fonction de r, lors d’un balayage en déformation, pour φv = 4,7%, T = 25°C, ω = 6,28 rad.s-1 : (▲) r = 0 ; ( ) r = 0,4 ; ( ) r = 0,8 ; ( ) r = 1,7 ; (−) r = 2,5
1E+0
1E+1
1E+2
1E+3
1E+4
1E+5
1E-2 1E-1 1E+0 1E+1 1E+2Déformation (%)
G' (
Pa)
r
10Z0
10Z5
10Z10 10Z20 et 10Z30
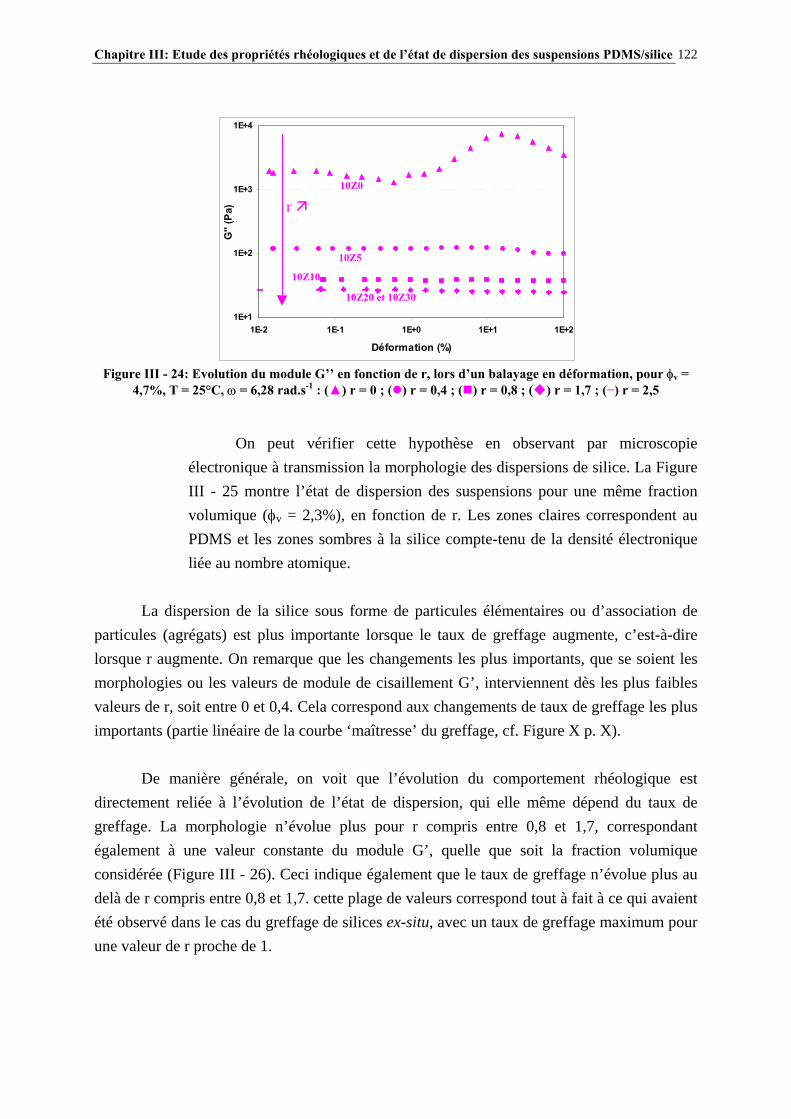
Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
122
Figure III - 24: Evolution du module G’’ en fonction de r, lors d’un balayage en déformation, pour φv = 4,7%, T = 25°C, ω = 6,28 rad.s-1 : (▲) r = 0 ; ( ) r = 0,4 ; ( ) r = 0,8 ; ( ) r = 1,7 ; (−) r = 2,5
On peut vérifier cette hypothèse en observant par microscopie électronique à transmission la morphologie des dispersions de silice. La Figure III - 25 montre l’état de dispersion des suspensions pour une même fraction volumique (φv = 2,3%), en fonction de r. Les zones claires correspondent au PDMS et les zones sombres à la silice compte-tenu de la densité électronique liée au nombre atomique.
La dispersion de la silice sous forme de particules élémentaires ou d’association de particules (agrégats) est plus importante lorsque le taux de greffage augmente, c’est-à-dire lorsque r augmente. On remarque que les changements les plus importants, que se soient les morphologies ou les valeurs de module de cisaillement G’, interviennent dès les plus faibles valeurs de r, soit entre 0 et 0,4. Cela correspond aux changements de taux de greffage les plus importants (partie linéaire de la courbe ‘maîtresse’ du greffage, cf. Figure X p. X). De manière générale, on voit que l’évolution du comportement rhéologique est directement reliée à l’évolution de l’état de dispersion, qui elle même dépend du taux de greffage. La morphologie n’évolue plus pour r compris entre 0,8 et 1,7, correspondant également à une valeur constante du module G’, quelle que soit la fraction volumique considérée (Figure III - 26). Ceci indique également que le taux de greffage n’évolue plus au delà de r compris entre 0,8 et 1,7. cette plage de valeurs correspond tout à fait à ce qui avaient été observé dans le cas du greffage de silices ex-situ, avec un taux de greffage maximum pour une valeur de r proche de 1.
1E+1
1E+2
1E+3
1E+4
1E-2 1E-1 1E+0 1E+1 1E+2
Déformation (%)
G''
(Pa) r
10Z0
10Z5
10Z10
10Z20 et 10Z30

Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
123
Figure III - 25 : Evolution de la morphologie des suspensions (série Z, φv = 4,7%) en fonction de r : (a) 5Z0 : r = 0 ; (b) 5Z5 : r = 0,4 ; (c) 5Z10 : r = 0,8 ; (d) 5Z20 : r = 1,7
200 nm
1 µm 200 nm
2 µm
200 nm
200 nm
1 µm
1 µm
5Z0 (a)
5Z5 (b)
5Z10 (c)
5Z20 (d)
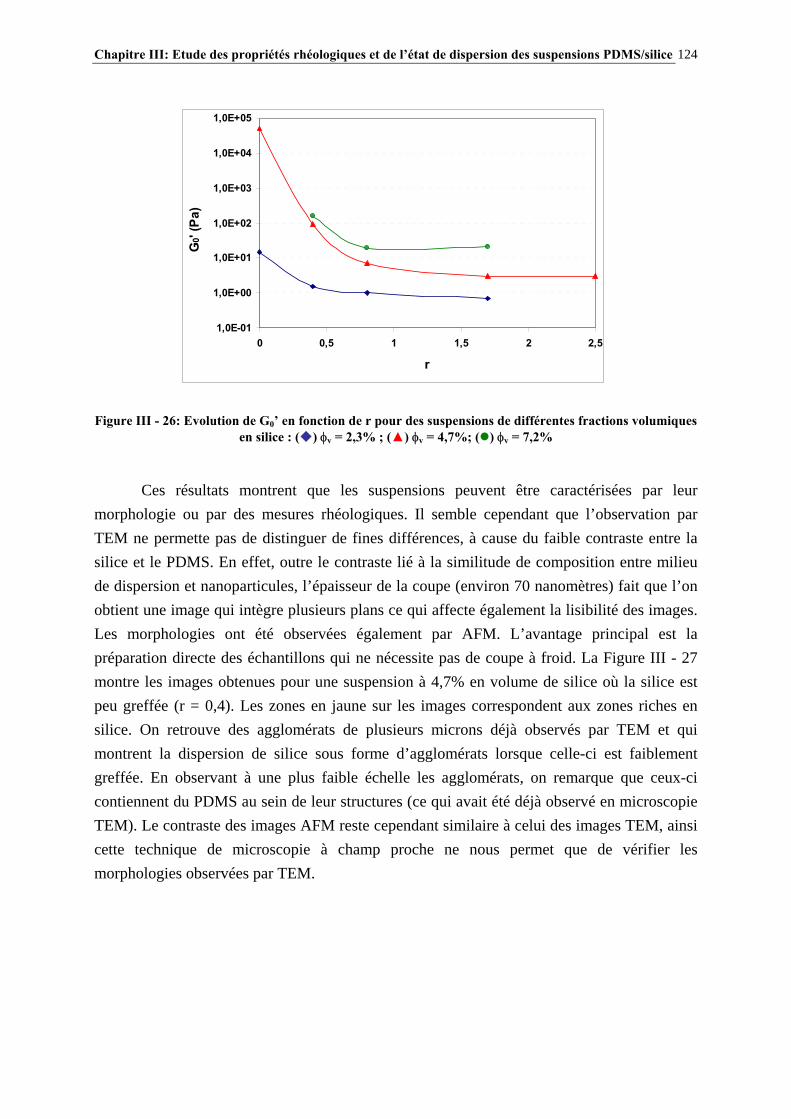
Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
124
Figure III - 26: Evolution de G0’ en fonction de r pour des suspensions de différentes fractions volumiques
en silice : ( ) φv = 2,3% ; (▲) φv = 4,7%; ( ) φv = 7,2%
Ces résultats montrent que les suspensions peuvent être caractérisées par leur morphologie ou par des mesures rhéologiques. Il semble cependant que l’observation par TEM ne permette pas de distinguer de fines différences, à cause du faible contraste entre la silice et le PDMS. En effet, outre le contraste lié à la similitude de composition entre milieu de dispersion et nanoparticules, l’épaisseur de la coupe (environ 70 nanomètres) fait que l’on obtient une image qui intègre plusieurs plans ce qui affecte également la lisibilité des images. Les morphologies ont été observées également par AFM. L’avantage principal est la préparation directe des échantillons qui ne nécessite pas de coupe à froid. La Figure III - 27 montre les images obtenues pour une suspension à 4,7% en volume de silice où la silice est peu greffée (r = 0,4). Les zones en jaune sur les images correspondent aux zones riches en silice. On retrouve des agglomérats de plusieurs microns déjà observés par TEM et qui montrent la dispersion de silice sous forme d’agglomérats lorsque celle-ci est faiblement greffée. En observant à une plus faible échelle les agglomérats, on remarque que ceux-ci contiennent du PDMS au sein de leur structures (ce qui avait été déjà observé en microscopie TEM). Le contraste des images AFM reste cependant similaire à celui des images TEM, ainsi cette technique de microscopie à champ proche ne nous permet que de vérifier les morphologies observées par TEM.
1,0E-01
1,0E+00
1,0E+01
1,0E+02
1,0E+03
1,0E+04
1,0E+05
0 0,5 1 1,5 2 2,5
r
G0'
(Pa)
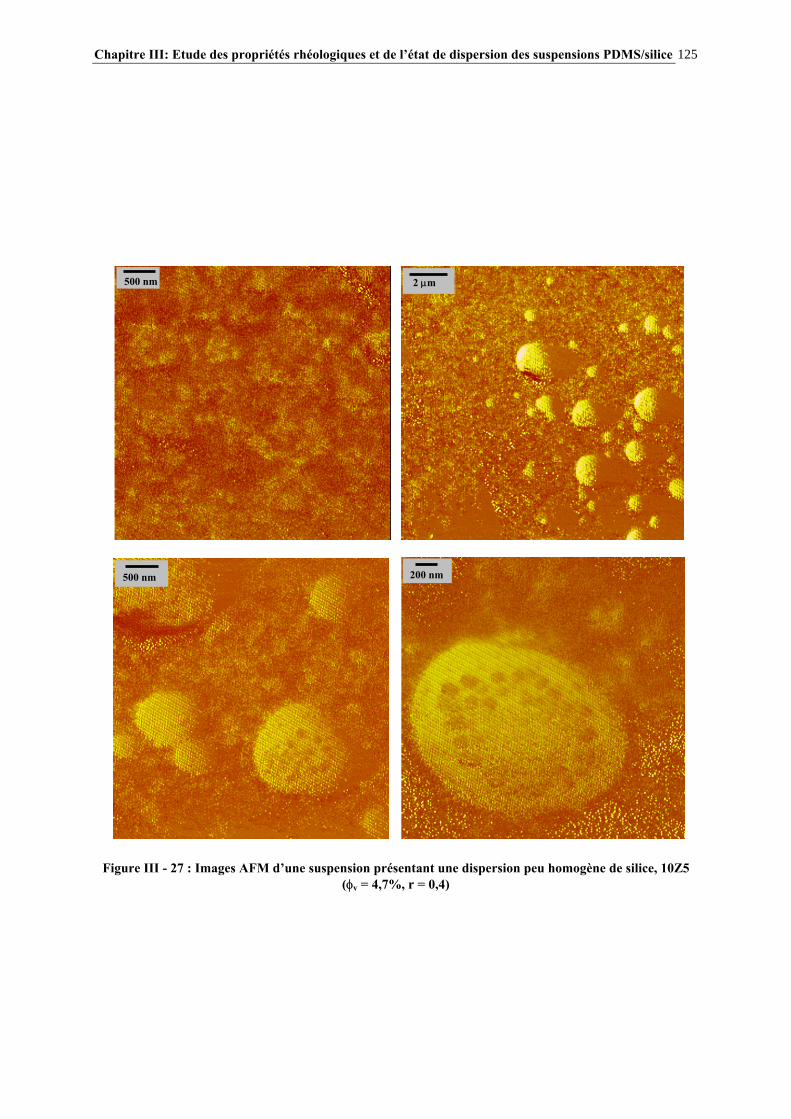
Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
125
Figure III - 27 : Images AFM d’une suspension présentant une dispersion peu homogène de silice, 10Z5 (φv = 4,7%, r = 0,4)
500 nm 2 µm
500 nm 200 nm
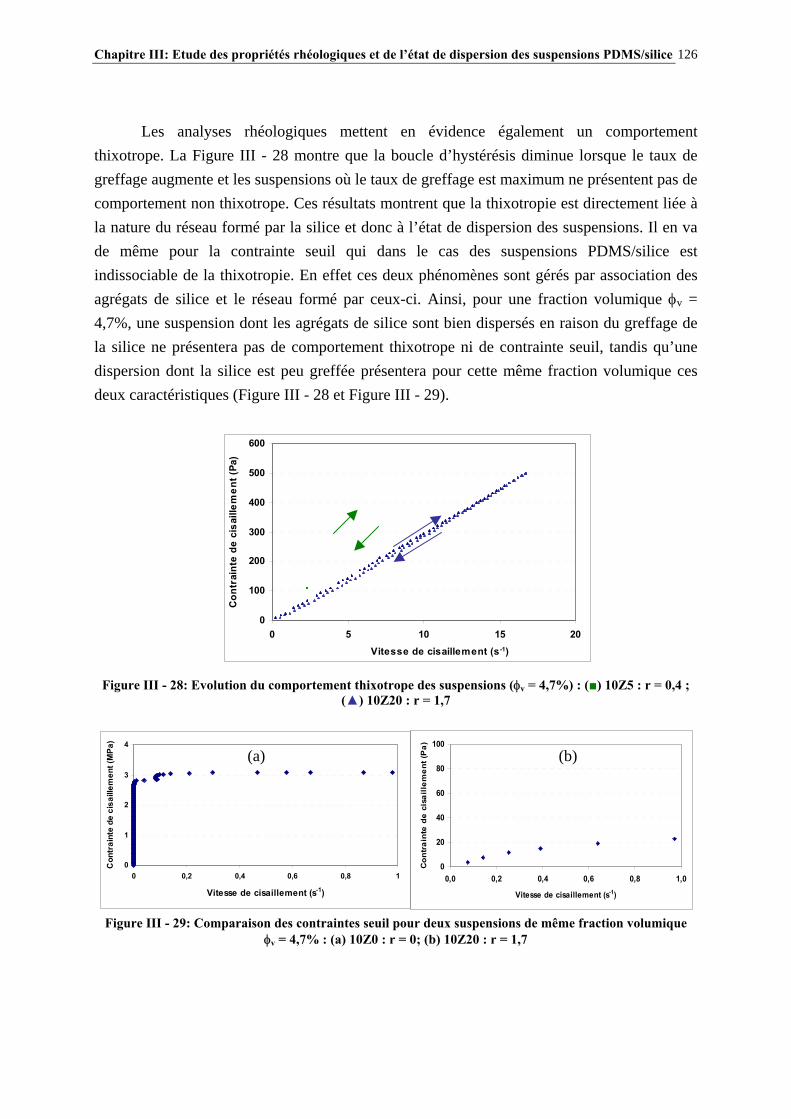
Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
126
Les analyses rhéologiques mettent en évidence également un comportement thixotrope. La Figure III - 28 montre que la boucle d’hystérésis diminue lorsque le taux de greffage augmente et les suspensions où le taux de greffage est maximum ne présentent pas de comportement non thixotrope. Ces résultats montrent que la thixotropie est directement liée à la nature du réseau formé par la silice et donc à l’état de dispersion des suspensions. Il en va de même pour la contrainte seuil qui dans le cas des suspensions PDMS/silice est indissociable de la thixotropie. En effet ces deux phénomènes sont gérés par association des agrégats de silice et le réseau formé par ceux-ci. Ainsi, pour une fraction volumique φv = 4,7%, une suspension dont les agrégats de silice sont bien dispersés en raison du greffage de la silice ne présentera pas de comportement thixotrope ni de contrainte seuil, tandis qu’une dispersion dont la silice est peu greffée présentera pour cette même fraction volumique ces deux caractéristiques (Figure III - 28 et Figure III - 29).
Figure III - 28: Evolution du comportement thixotrope des suspensions (φv = 4,7%) : (■) 10Z5 : r = 0,4 ; (▲) 10Z20 : r = 1,7
Figure III - 29: Comparaison des contraintes seuil pour deux suspensions de même fraction volumique φv = 4,7% : (a) 10Z0 : r = 0; (b) 10Z20 : r = 1,7
0
1
2
3
4
0 0,2 0,4 0,6 0,8 1
Vitesse de cisaillement (s-1)
Con
trai
nte
de c
isai
llem
ent (
MPa
)
0
100
200
300
400
500
600
0 5 10 15 20Vitesse de cisaillement (s-1)
Con
trai
nte
de c
isai
llem
ent (
Pa)
0
20
40
60
80
100
0,0 0,2 0,4 0,6 0,8 1,0
Vitesse de cisaillement (s-1)
Con
trai
nte
de c
isai
llem
ent (
Pa)
(a) (b)
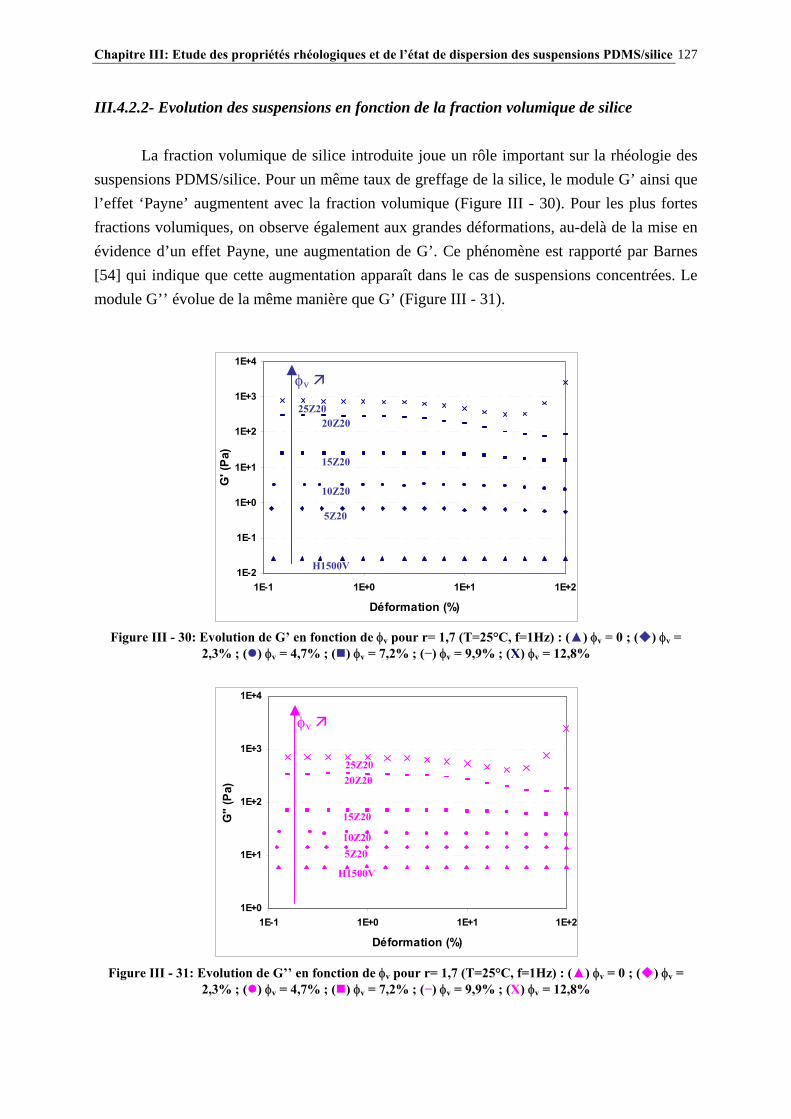
Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
127
III.4.2.2- Evolution des suspensions en fonction de la fraction volumique de silice La fraction volumique de silice introduite joue un rôle important sur la rhéologie des suspensions PDMS/silice. Pour un même taux de greffage de la silice, le module G’ ainsi que l’effet ‘Payne’ augmentent avec la fraction volumique (Figure III - 30). Pour les plus fortes fractions volumiques, on observe également aux grandes déformations, au-delà de la mise en évidence d’un effet Payne, une augmentation de G’. Ce phénomène est rapporté par Barnes [54] qui indique que cette augmentation apparaît dans le cas de suspensions concentrées. Le module G’’ évolue de la même manière que G’ (Figure III - 31).
Figure III - 30: Evolution de G’ en fonction de φv pour r= 1,7 (T=25°C, f=1Hz) : (▲) φv = 0 ; ( ) φv = 2,3% ; ( ) φv = 4,7% ; ( ) φv = 7,2% ; (−) φv = 9,9% ; (X) φv = 12,8%
Figure III - 31: Evolution de G’’ en fonction de φv pour r= 1,7 (T=25°C, f=1Hz) : (▲) φv = 0 ; ( ) φv = 2,3% ; ( ) φv = 4,7% ; ( ) φv = 7,2% ; (−) φv = 9,9% ; (X) φv = 12,8%
1E-2
1E-1
1E+0
1E+1
1E+2
1E+3
1E+4
1E-1 1E+0 1E+1 1E+2
Déformation (%)
G' (
Pa)
1E+0
1E+1
1E+2
1E+3
1E+4
1E-1 1E+0 1E+1 1E+2
Déformation (%)
G''
(Pa)
φv
φv
H1500V
5Z20
10Z20
15Z20
20Z20 25Z20
5Z20
H1500V
10Z20
15Z20
20Z20 25Z20

Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
128
Les morphologies des suspensions obtenues pour différentes fractions volumiques ont été observées par microscopie TEM. La Figure III - 32 et la Figure III - 33 représentent l’évolution des morphologies en fonction de la fraction volumique pour des silices non greffées et des silices greffées avec un taux de greffage maximum (r>1, cf. Chapitre II). Lorsque la silice n’est pas greffée, on observe dès les plus faibles fractions volumiques (φv = 0,4%) la formation d’agglomérats de la taille du micron (Figure III - 32a). Lorsque la fraction volumique augmente, la taille des agglomérats augmente pour finalement former un réseau constitué de gros agglomérats. On obtient des images possédant un bon contraste entre les zones riches en silice (agglomérats) et les zones sans silice (Figure III - 32c). Dans le cas d’une silice greffée, la situation est différente. A faible fraction volumique, les agrégats de silice sont relativement bien dispersés et les structures formées ont une taille allant de 200 à 800 nm (Figure III - 33a). Lorsque la fraction volumique en silice augmente la silice reste bien dispersée même aux plus fortes fractions volumiques (Figure III - 33d). Le contraste des micrographies TEM est par conséquence assez faible car la silice est répartie de manière homogène. Ces deux figures illustrent bien l’évolution des morphologies en fonction de la fraction volumique de silice introduite, et le rôle essentiel du greffage de la silice.
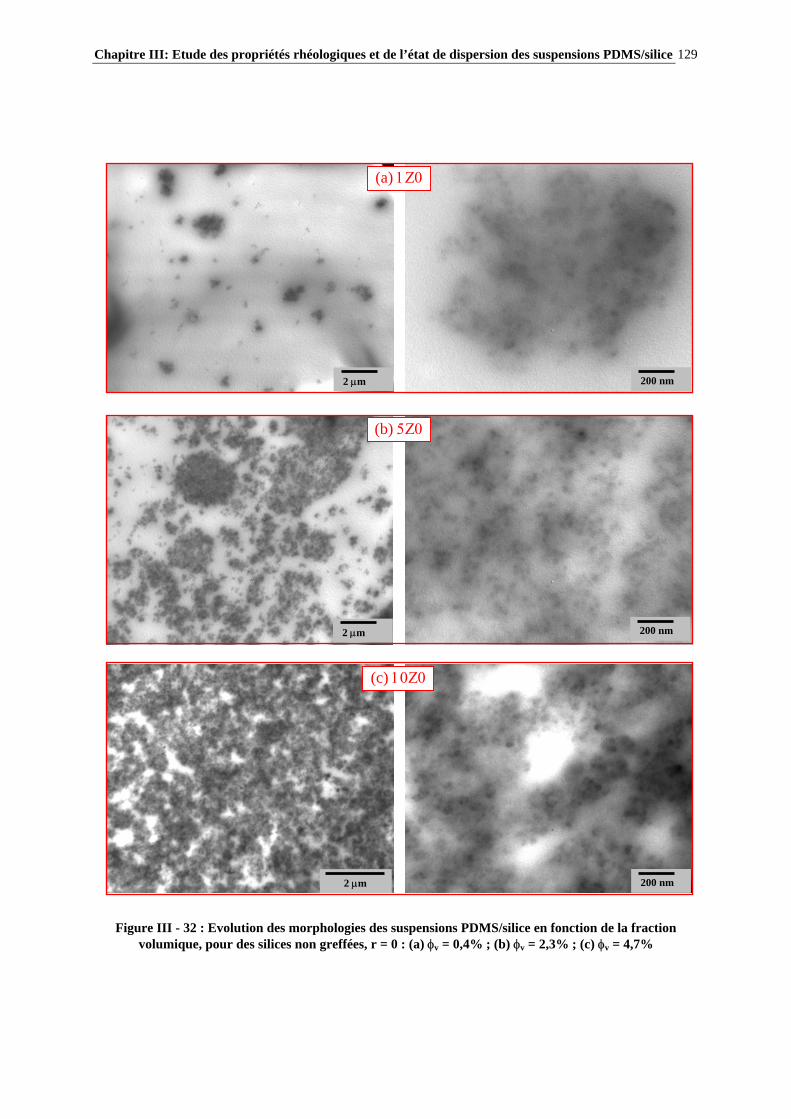
Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice 129
Figure III - 32 : Evolution des morphologies des suspensions PDMS/silice en fonction de la fraction
volumique, pour des silices non greffées, r = 0 : (a) φv = 0,4% ; (b) φv = 2,3% ; (c) φv = 4,7%
2 µm 200 nm
2 µm 200 nm
2 µm 200 nm
(c) 10Ζ0
(b) 5Ζ0
(a) 1Ζ0

Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice 130
Figure III - 33: Evolution des morphologies des suspensions PDMS/silice en fonction de la fraction
volumique, pour des silices greffées au maximum (r=1,7) : (a) φv = 0,4% ; (b) φv = 2,3% ; (c) φv = 4,7%
2 µm
2 µm
2 µm
200 nm
200 nm
200 nm
(a) 1Ζ20
(b) 5Ζ20
(c) 10Ζ20
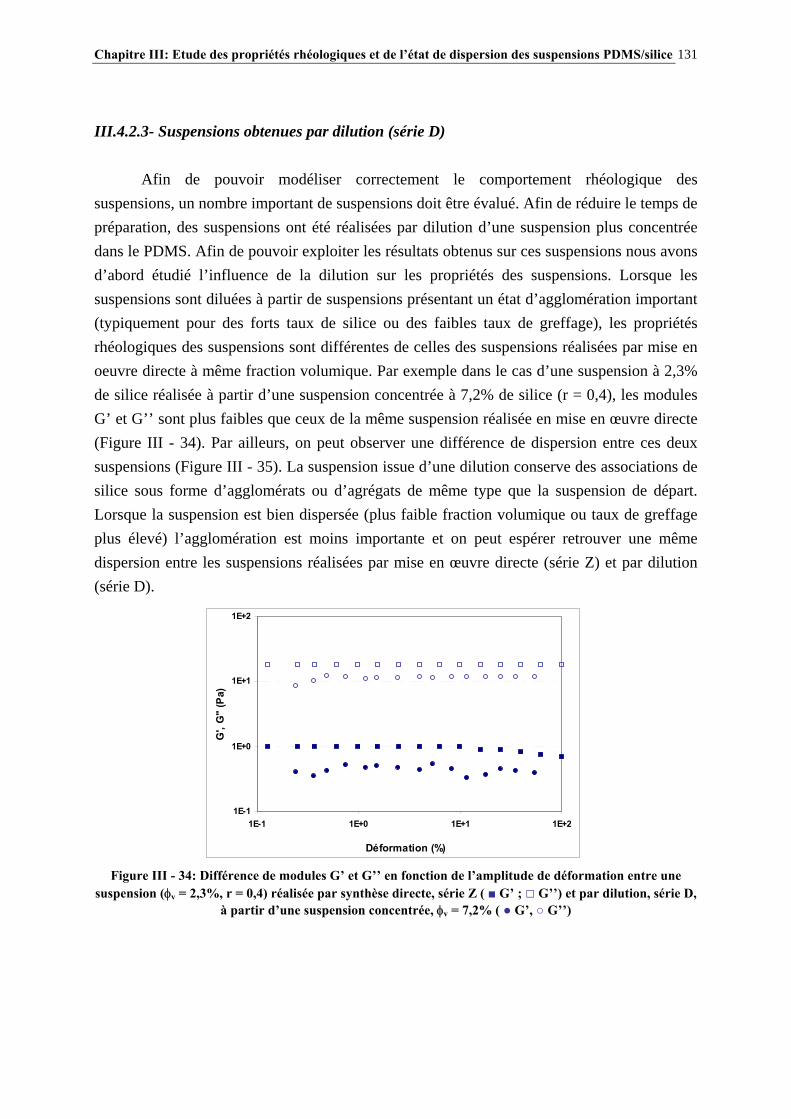
Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
131
III.4.2.3- Suspensions obtenues par dilution (série D) Afin de pouvoir modéliser correctement le comportement rhéologique des suspensions, un nombre important de suspensions doit être évalué. Afin de réduire le temps de préparation, des suspensions ont été réalisées par dilution d’une suspension plus concentrée dans le PDMS. Afin de pouvoir exploiter les résultats obtenus sur ces suspensions nous avons d’abord étudié l’influence de la dilution sur les propriétés des suspensions. Lorsque les suspensions sont diluées à partir de suspensions présentant un état d’agglomération important (typiquement pour des forts taux de silice ou des faibles taux de greffage), les propriétés rhéologiques des suspensions sont différentes de celles des suspensions réalisées par mise en oeuvre directe à même fraction volumique. Par exemple dans le cas d’une suspension à 2,3% de silice réalisée à partir d’une suspension concentrée à 7,2% de silice (r = 0,4), les modules G’ et G’’ sont plus faibles que ceux de la même suspension réalisée en mise en œuvre directe (Figure III - 34). Par ailleurs, on peut observer une différence de dispersion entre ces deux suspensions (Figure III - 35). La suspension issue d’une dilution conserve des associations de silice sous forme d’agglomérats ou d’agrégats de même type que la suspension de départ. Lorsque la suspension est bien dispersée (plus faible fraction volumique ou taux de greffage plus élevé) l’agglomération est moins importante et on peut espérer retrouver une même dispersion entre les suspensions réalisées par mise en œuvre directe (série Z) et par dilution (série D).
Figure III - 34: Différence de modules G’ et G’’ en fonction de l’amplitude de déformation entre une suspension (φv = 2,3%, r = 0,4) réalisée par synthèse directe, série Z ( ■ G’ ; □ G’’) et par dilution, série D,
à partir d’une suspension concentrée, φv = 7,2% ( ● G’, ○ G’’)
1E-1
1E+0
1E+1
1E+2
1E-1 1E+0 1E+1 1E+2
Déformation (%)
G',
G''
(Pa)
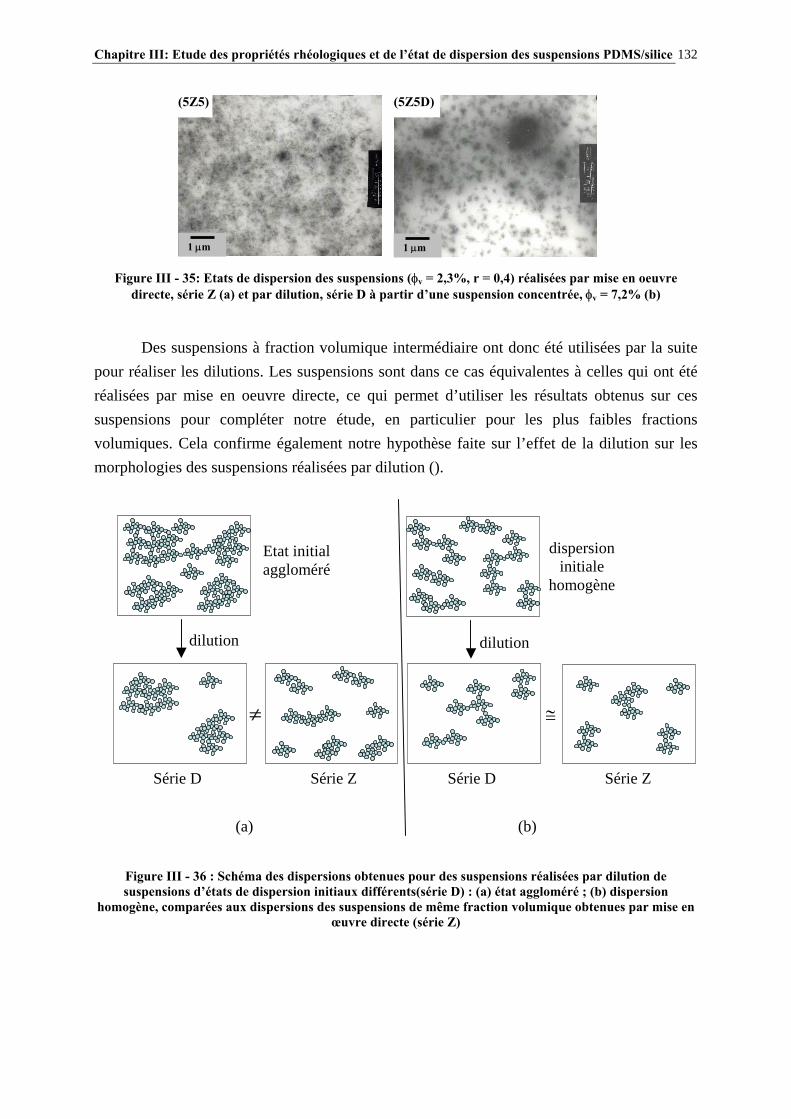
Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
132
Figure III - 35: Etats de dispersion des suspensions (φv = 2,3%, r = 0,4) réalisées par mise en oeuvre directe, série Z (a) et par dilution, série D à partir d’une suspension concentrée, φv = 7,2% (b)
Des suspensions à fraction volumique intermédiaire ont donc été utilisées par la suite pour réaliser les dilutions. Les suspensions sont dans ce cas équivalentes à celles qui ont été réalisées par mise en oeuvre directe, ce qui permet d’utiliser les résultats obtenus sur ces suspensions pour compléter notre étude, en particulier pour les plus faibles fractions volumiques. Cela confirme également notre hypothèse faite sur l’effet de la dilution sur les morphologies des suspensions réalisées par dilution (). Série D Série Z Série D Série Z (a) (b)
Figure III - 36 : Schéma des dispersions obtenues pour des suspensions réalisées par dilution de suspensions d’états de dispersion initiaux différents(série D) : (a) état aggloméré ; (b) dispersion
homogène, comparées aux dispersions des suspensions de même fraction volumique obtenues par mise en œuvre directe (série Z)
(5Z5) (5Z5D)
1 µm 1 µm
dilution
≠ ≅
dilution
Etat initial aggloméré
dispersion initiale
homogène
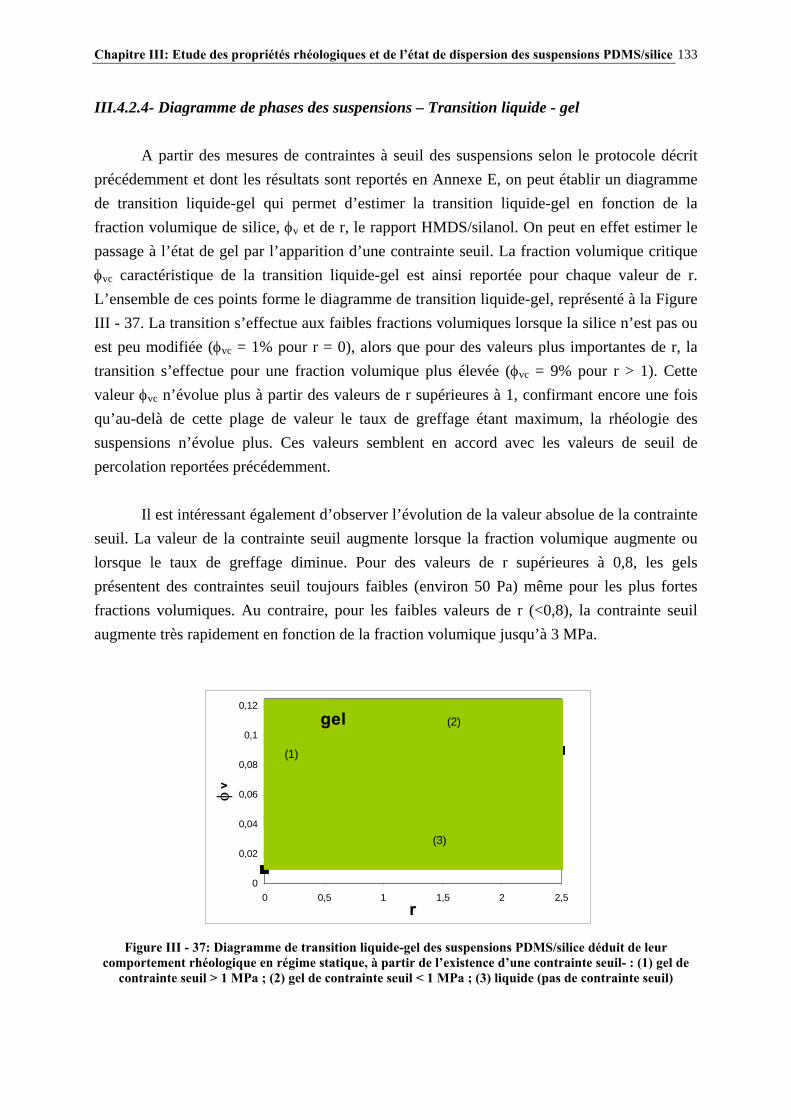
Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
133
III.4.2.4- Diagramme de phases des suspensions – Transition liquide - gel A partir des mesures de contraintes à seuil des suspensions selon le protocole décrit précédemment et dont les résultats sont reportés en Annexe E, on peut établir un diagramme de transition liquide-gel qui permet d’estimer la transition liquide-gel en fonction de la fraction volumique de silice, φv et de r, le rapport HMDS/silanol. On peut en effet estimer le passage à l’état de gel par l’apparition d’une contrainte seuil. La fraction volumique critique φvc caractéristique de la transition liquide-gel est ainsi reportée pour chaque valeur de r. L’ensemble de ces points forme le diagramme de transition liquide-gel, représenté à la Figure III - 37. La transition s’effectue aux faibles fractions volumiques lorsque la silice n’est pas ou est peu modifiée (φvc = 1% pour r = 0), alors que pour des valeurs plus importantes de r, la transition s’effectue pour une fraction volumique plus élevée (φvc = 9% pour r > 1). Cette valeur φvc n’évolue plus à partir des valeurs de r supérieures à 1, confirmant encore une fois qu’au-delà de cette plage de valeur le taux de greffage étant maximum, la rhéologie des suspensions n’évolue plus. Ces valeurs semblent en accord avec les valeurs de seuil de percolation reportées précédemment. Il est intéressant également d’observer l’évolution de la valeur absolue de la contrainte seuil. La valeur de la contrainte seuil augmente lorsque la fraction volumique augmente ou lorsque le taux de greffage diminue. Pour des valeurs de r supérieures à 0,8, les gels présentent des contraintes seuil toujours faibles (environ 50 Pa) même pour les plus fortes fractions volumiques. Au contraire, pour les faibles valeurs de r (<0,8), la contrainte seuil augmente très rapidement en fonction de la fraction volumique jusqu’à 3 MPa.
Figure III - 37: Diagramme de transition liquide-gel des suspensions PDMS/silice déduit de leur comportement rhéologique en régime statique, à partir de l’existence d’une contrainte seuil- : (1) gel de
contrainte seuil > 1 MPa ; (2) gel de contrainte seuil < 1 MPa ; (3) liquide (pas de contrainte seuil)
0
0,02
0,04
0,06
0,08
0,1
0,12
0 0,5 1 1,5 2 2,5r
φv
liquide
gel
(1)
(2)
(3)

Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
134
Ce diagramme permet donc de prévoir l’état de la suspension (liquide ou gel) suivant les deux paramètres principaux des formulations (φv et r) et d’estimer la valeur de sa contrainte seuil. III.4.3- Influence du procédé de mise en œuvre des suspensions III.4.3.1- Comparaison in-situ (Z) / ex-situ (T) Nous avons conclu au Chapitre II que les silices modifiées in-situ présentaient un taux de greffage maximum supérieur à celui des silices greffées ex-situ. Le but de cette partie est maintenant de comparer les propriétés (rhéologie et état de dispersion) des suspensions préparées à partir de silices issues de ces deux types de méthode de greffage. Nous avons considéré deux rapports stœchiométriques r : i/ r = 0,4 où nous nous situons dans la partie linéaire de la courbe de greffage; ii/ r = 1,7 où le taux de greffage est maximum : le taux de greffage ne dépend alors plus de r (cf. chapitre II). La Figure III - 38 montre l’évolution du module G0’ en fonction de la fraction volumique pour les différentes voies de mise en œuvre, pour un faible taux de greffage (r =0,4). Aucune différence majeure n’apparaît entre les différentes voies de greffage, ce qui peut s’expliquer par l’évolution similaire du taux de greffage en fonction de r pour des taux de greffage faibles, entre le procédé ex-situ et in-situ. L’état de dispersion est également similaire par comparaison des micrographies TEM, ce qui indique que la silice est dispersée de la même manière lorsqu’elle est greffée avant d’être introduite dans le PDMS que lorsqu’elle est greffée après avoir été additionnée au PDMS (voie in-situ). Cependant, le mélange paraît plus difficile à réaliser lorsque la silice est moins hydrophile. Ce phénomène est accentué pour les suspensions réalisées à r = 1,7. Dans ce cas, un temps de mélange supérieur est nécessaire, en particulier pour les premiers ajouts de silice. La Figure III - 39 montre l’évolution du module G0’en fonction de la fraction volumique de silice pour les différentes voies de greffage. Les suspensions réalisées ex-situ présentent cette fois une valeur de module différente des suspensions réalisées in-situ. Aux fractions volumiques faibles ou modérées (φv <7,2%), G0’ est plus important dans le cas des suspensions réalisées ex-situ, ce qui peut s’expliquer par leur taux de greffage plus faible, comme montré au Chapitre II.
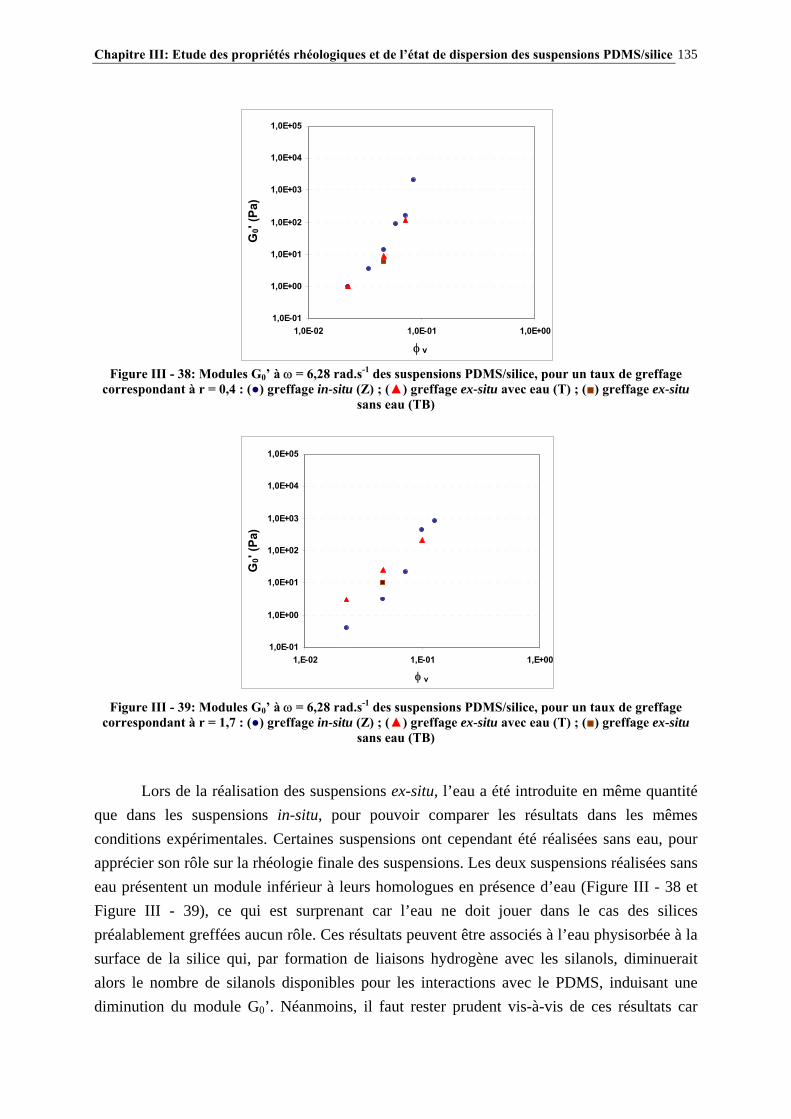
Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
135
Figure III - 38: Modules G0’ à ω = 6,28 rad.s-1 des suspensions PDMS/silice, pour un taux de greffage correspondant à r = 0,4 : (●) greffage in-situ (Z) ; (▲) greffage ex-situ avec eau (T) ; (■) greffage ex-situ
sans eau (TB)
Figure III - 39: Modules G0’ à ω = 6,28 rad.s-1 des suspensions PDMS/silice, pour un taux de greffage correspondant à r = 1,7 : (●) greffage in-situ (Z) ; (▲) greffage ex-situ avec eau (T) ; (■) greffage ex-situ
sans eau (TB)
Lors de la réalisation des suspensions ex-situ, l’eau a été introduite en même quantité que dans les suspensions in-situ, pour pouvoir comparer les résultats dans les mêmes conditions expérimentales. Certaines suspensions ont cependant été réalisées sans eau, pour apprécier son rôle sur la rhéologie finale des suspensions. Les deux suspensions réalisées sans eau présentent un module inférieur à leurs homologues en présence d’eau (Figure III - 38 et Figure III - 39), ce qui est surprenant car l’eau ne doit jouer dans le cas des silices préalablement greffées aucun rôle. Ces résultats peuvent être associés à l’eau physisorbée à la surface de la silice qui, par formation de liaisons hydrogène avec les silanols, diminuerait alors le nombre de silanols disponibles pour les interactions avec le PDMS, induisant une diminution du module G0’. Néanmoins, il faut rester prudent vis-à-vis de ces résultats car
1,0E-01
1,0E+00
1,0E+01
1,0E+02
1,0E+03
1,0E+04
1,0E+05
1,0E-02 1,0E-01 1,0E+00
φ v
G0'
(Pa)
1,0E-01
1,0E+00
1,0E+01
1,0E+02
1,0E+03
1,0E+04
1,0E+05
1,E-02 1,E-01 1,E+00
φ v
G0'
(Pa)

Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
136
seulement deux suspensions ont été réalisées sans eau. Une étude plus approfondie sur le rôle de l’eau de manière générale serait utile pour bien comprendre les phénomènes mis en jeu. Nous avons vu précédemment lors de l’étude des silices réalisées in-situ que lorsque le taux de greffage est plus faible la dispersion est plus hétérogène et le module G0’ augmente. En considérant les suspensions ex-situ dont le taux de greffage maximum des silices est plus faible que celui des silices des suspensions in-situ, on peut raisonner de la même manière et conclure que le module G0’ sera plus important (c’est ce que l’on constate) et que la dispersion sera plus hétérogène. En comparant les dispersions (φv = 2,3%, r = 1,7) il est difficile de mettre en évidence des différences nettes entre les suspensions réalisées in-situ et ex-situ (Figure III - 40). Il faudrait pour cela une analyse quantitative des dispersions, que nous avons essayé de mettre en place par analyse d’image. Cependant, deux difficultés majeures sont apparues : d’une part le contraste électronique entre les phases PDMS et silice est faible ce qui conduit à des images où la résolution n’est pas suffisante pour une analyse d’image, d’autre part dès que la fraction volumique est supérieure à quelques pourcents le contraste diminue, du au fait que l’image intègre la morphologie sur une épaisseur importante (épaisseur de la coupe 70 nm, diamètre d’une particule élémentaire 12 nm). La microscopie TEM ne permet donc pas d’évaluer de manière aussi fine que la rhéologie les différences entre les suspensions, mais permet d’observer des tendances lors de changements importants.

Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
137
Figure III - 40: Dispersions de silice au sein d’une huile PDMS observées par microscopie électronique à transmission pour une fraction volumique φv de 2,3% et un rapport HMDS/silanol r de 1,7 ; (a) Greffage
in-situ (Z), (b) Greffage ex-situ (T)
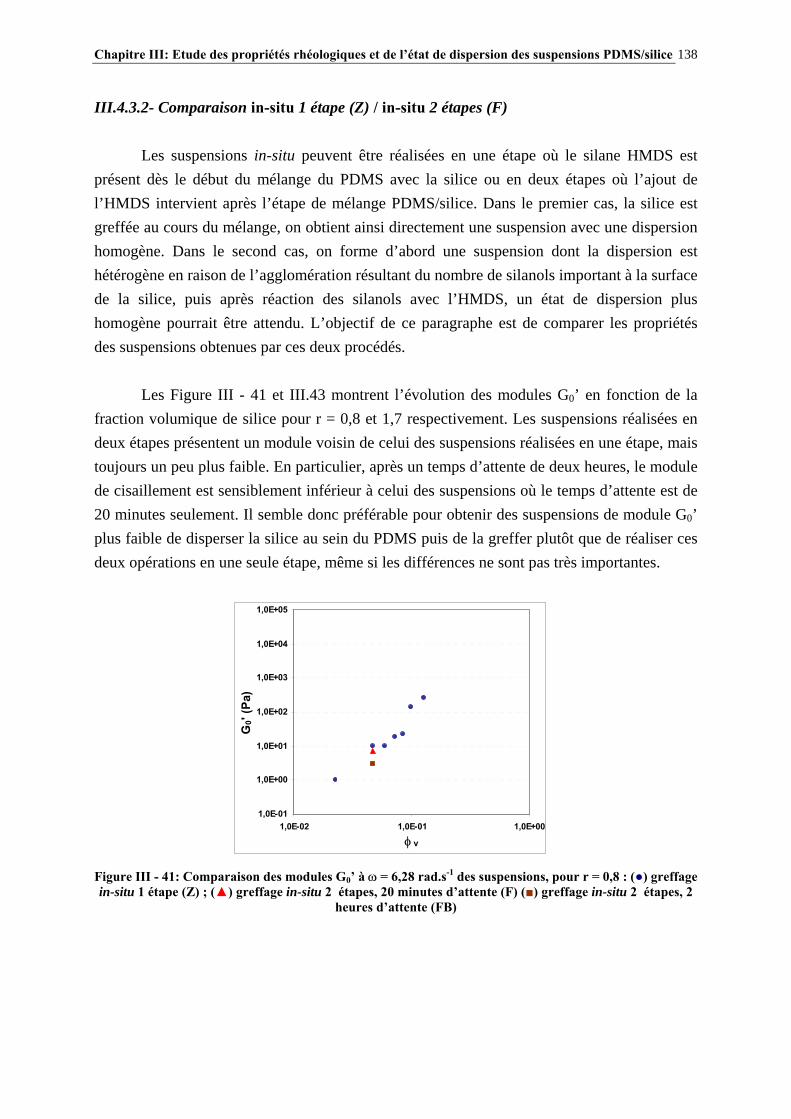
Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
138
III.4.3.2- Comparaison in-situ 1 étape (Z) / in-situ 2 étapes (F) Les suspensions in-situ peuvent être réalisées en une étape où le silane HMDS est présent dès le début du mélange du PDMS avec la silice ou en deux étapes où l’ajout de l’HMDS intervient après l’étape de mélange PDMS/silice. Dans le premier cas, la silice est greffée au cours du mélange, on obtient ainsi directement une suspension avec une dispersion homogène. Dans le second cas, on forme d’abord une suspension dont la dispersion est hétérogène en raison de l’agglomération résultant du nombre de silanols important à la surface de la silice, puis après réaction des silanols avec l’HMDS, un état de dispersion plus homogène pourrait être attendu. L’objectif de ce paragraphe est de comparer les propriétés des suspensions obtenues par ces deux procédés. Les Figure III - 41 et III.43 montrent l’évolution des modules G0’ en fonction de la fraction volumique de silice pour r = 0,8 et 1,7 respectivement. Les suspensions réalisées en deux étapes présentent un module voisin de celui des suspensions réalisées en une étape, mais toujours un peu plus faible. En particulier, après un temps d’attente de deux heures, le module de cisaillement est sensiblement inférieur à celui des suspensions où le temps d’attente est de 20 minutes seulement. Il semble donc préférable pour obtenir des suspensions de module G0’ plus faible de disperser la silice au sein du PDMS puis de la greffer plutôt que de réaliser ces deux opérations en une seule étape, même si les différences ne sont pas très importantes. Figure III - 41: Comparaison des modules G0’ à ω = 6,28 rad.s-1 des suspensions, pour r = 0,8 : (●) greffage in-situ 1 étape (Z) ; (▲) greffage in-situ 2 étapes, 20 minutes d’attente (F) (■) greffage in-situ 2 étapes, 2
heures d’attente (FB)
1,0E-01
1,0E+00
1,0E+01
1,0E+02
1,0E+03
1,0E+04
1,0E+05
1,0E-02 1,0E-01 1,0E+00
φ v
G0'
(Pa)
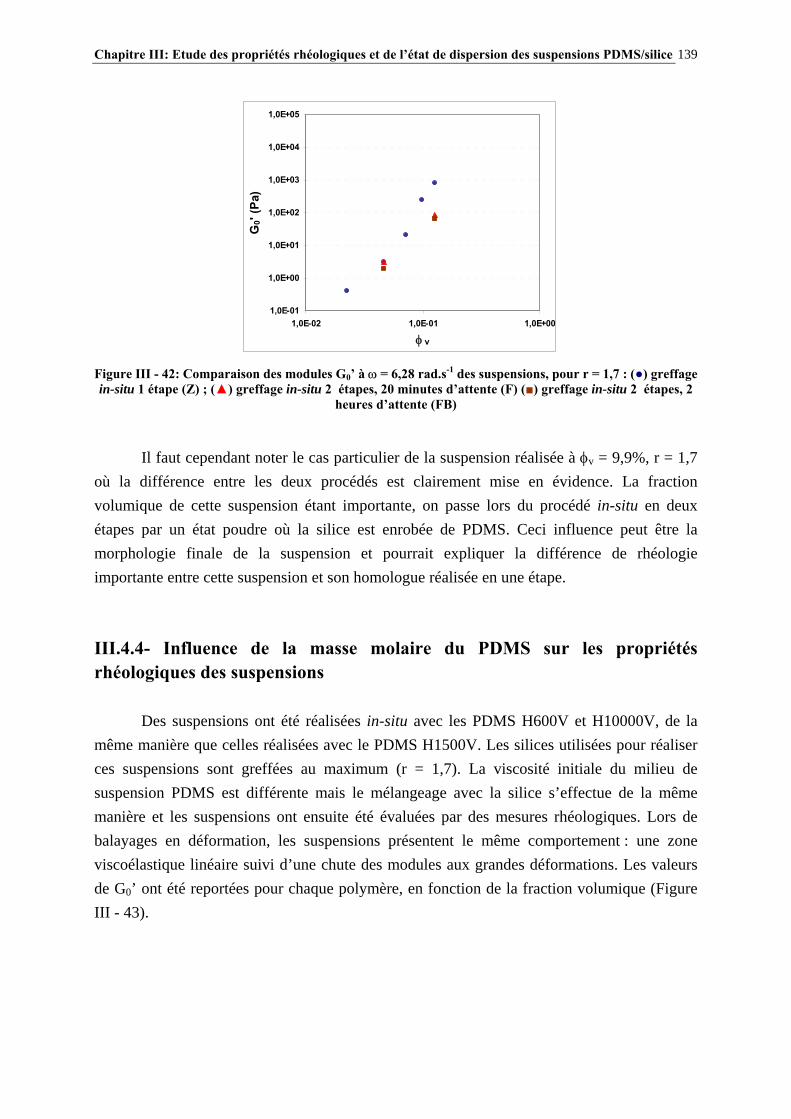
Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
139
Figure III - 42: Comparaison des modules G0’ à ω = 6,28 rad.s-1 des suspensions, pour r = 1,7 : (●) greffage in-situ 1 étape (Z) ; (▲) greffage in-situ 2 étapes, 20 minutes d’attente (F) (■) greffage in-situ 2 étapes, 2
heures d’attente (FB)
Il faut cependant noter le cas particulier de la suspension réalisée à φv = 9,9%, r = 1,7 où la différence entre les deux procédés est clairement mise en évidence. La fraction volumique de cette suspension étant importante, on passe lors du procédé in-situ en deux étapes par un état poudre où la silice est enrobée de PDMS. Ceci influence peut être la morphologie finale de la suspension et pourrait expliquer la différence de rhéologie importante entre cette suspension et son homologue réalisée en une étape. III.4.4- Influence de la masse molaire du PDMS sur les propriétés rhéologiques des suspensions Des suspensions ont été réalisées in-situ avec les PDMS H600V et H10000V, de la même manière que celles réalisées avec le PDMS H1500V. Les silices utilisées pour réaliser ces suspensions sont greffées au maximum (r = 1,7). La viscosité initiale du milieu de suspension PDMS est différente mais le mélangeage avec la silice s’effectue de la même manière et les suspensions ont ensuite été évaluées par des mesures rhéologiques. Lors de balayages en déformation, les suspensions présentent le même comportement : une zone viscoélastique linéaire suivi d’une chute des modules aux grandes déformations. Les valeurs de G0’ ont été reportées pour chaque polymère, en fonction de la fraction volumique (Figure III - 43).
1,0E-01
1,0E+00
1,0E+01
1,0E+02
1,0E+03
1,0E+04
1,0E+05
1,0E-02 1,0E-01 1,0E+00
φ v
G0'
(Pa)
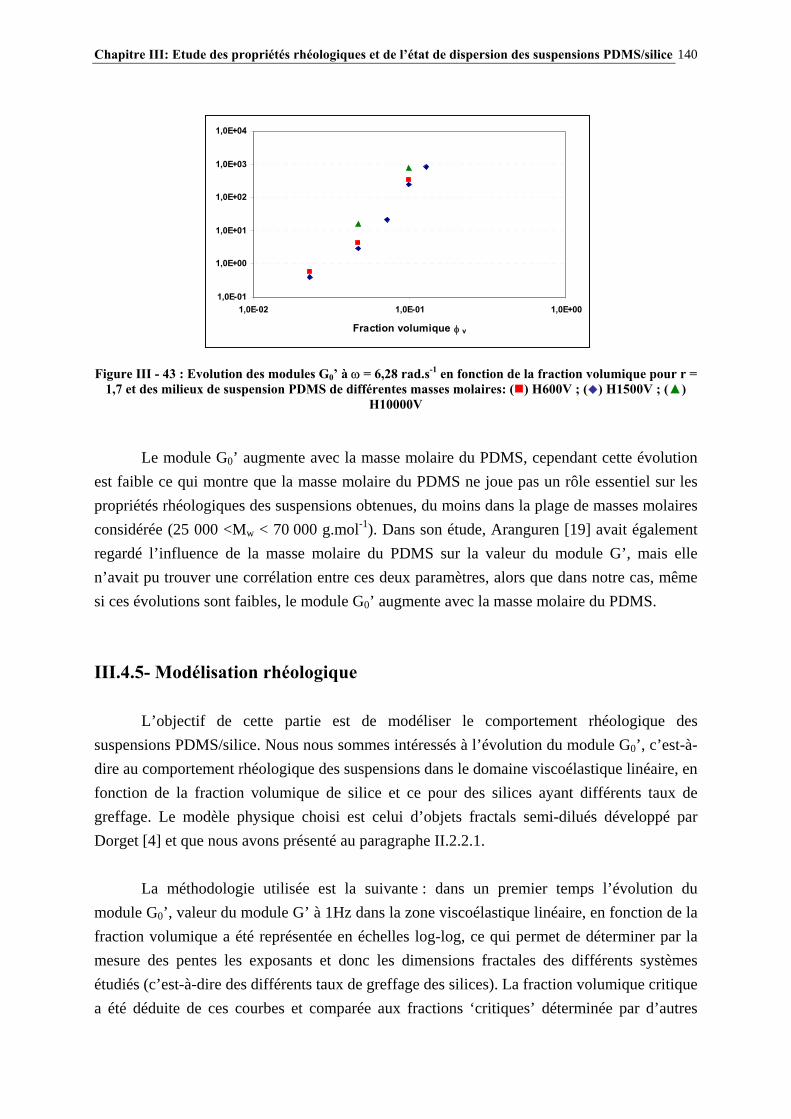
Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
140
Figure III - 43 : Evolution des modules G0’ à ω = 6,28 rad.s-1 en fonction de la fraction volumique pour r =
1,7 et des milieux de suspension PDMS de différentes masses molaires: ( ) H600V ; ( ) H1500V ; (▲) H10000V
Le module G0’ augmente avec la masse molaire du PDMS, cependant cette évolution est faible ce qui montre que la masse molaire du PDMS ne joue pas un rôle essentiel sur les propriétés rhéologiques des suspensions obtenues, du moins dans la plage de masses molaires considérée (25 000 <Mw < 70 000 g.mol-1). Dans son étude, Aranguren [19] avait également regardé l’influence de la masse molaire du PDMS sur la valeur du module G’, mais elle n’avait pu trouver une corrélation entre ces deux paramètres, alors que dans notre cas, même si ces évolutions sont faibles, le module G0’ augmente avec la masse molaire du PDMS. III.4.5- Modélisation rhéologique L’objectif de cette partie est de modéliser le comportement rhéologique des suspensions PDMS/silice. Nous nous sommes intéressés à l’évolution du module G0’, c’est-à-dire au comportement rhéologique des suspensions dans le domaine viscoélastique linéaire, en fonction de la fraction volumique de silice et ce pour des silices ayant différents taux de greffage. Le modèle physique choisi est celui d’objets fractals semi-dilués développé par Dorget [4] et que nous avons présenté au paragraphe II.2.2.1. La méthodologie utilisée est la suivante : dans un premier temps l’évolution du module G0’, valeur du module G’ à 1Hz dans la zone viscoélastique linéaire, en fonction de la fraction volumique a été représentée en échelles log-log, ce qui permet de déterminer par la mesure des pentes les exposants et donc les dimensions fractales des différents systèmes étudiés (c’est-à-dire des différents taux de greffage des silices). La fraction volumique critique a été déduite de ces courbes et comparée aux fractions ‘critiques’ déterminée par d’autres
1,0E-01
1,0E+00
1,0E+01
1,0E+02
1,0E+03
1,0E+04
1,0E-02 1,0E-01 1,0E+00
Fraction volumique φ v

Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
141
méthodes. A partir des valeurs de dimension fractale et de fraction volumique critique, la taille des objets fractals a pu être calculée. Ces résultats issus du modèle physique seront enfin comparés aux résultats obtenus expérimentalement par diffusion de lumière. L’ensemble de la méthodologie adoptée dans cette partie est présenté Figure III - 44. Figure III - 44: Méthodologie adoptée pour la modélisation des comportements rhéologiques à partir du
modèle de Dorget
III.4.5.1- Calcul des dimensions fractales D et des fractions volumiques critiques φv
* à partir des mesures rhéologiques La Figure III - 45 montre l’évolution du module G0’ en fonction de la fraction volumique pour les différentes valeurs de r. Il s’agit ici des suspensions réalisées in-situ en une étape (Z et D). Les courbes sont divisées en deux parties : une partie où la dépendance du module G0’ vis à vis de la fraction volumique reste faible (suspension diluée) puis une partie où cette dépendance est plus importante (suspension semi-diluée). En régime semi-dilué, on obtient dans cette représentation log-log des droites, ce qui prouve que l’on a bien une dépendance suivant une loi puissance. La dimension fractale D a été calculée à partir de la valeur de la pente n de ces droites. Celle-ci a été déterminée pour chaque valeur de r (Annexe F). La pente n diminue lorsque r augmente, ainsi la dimension fractale diminue également en fonction de r (Figure III - 46). Les valeurs de dimension fractale sont comprises entre D = 1,4 pour r > 1 et D = 2,3 pour r = 0. L’évolution de la pente n et de la dimension fractale en fonction de r se corrèlent de manière évidente avec l’évolution du taux de greffage en fonction de r (cf. Chapitre II), c’est-à-dire une évolution jusque r = 1 suivi d’une valeur plateau obtenue simultanément à partir de cette valeur de r. Lorsque la silice n’est pas greffée la dimension fractale des objets correspond à la dimension fractale reportée pour un réseau de silice, c’est-à-dire entre 2,1 et 2,5 [55, 56]. Lorsque la silice est greffée la dimension fractale des objets diminue ce qui correspond à des objets plus ‘ouverts’. Dans ses travaux, Dorget
1- Détermination à partir des mesures rhéologiques de φv* et D
R
DG0’
Log-log
Y=Axn
2- Calcul de R
n
3- Confrontation avec l’expérience
Mesure de R par diffusion de la lumière
ConfrontationD
Confrontationφv* φv
φv*
1- Détermination à partir des mesures rhéologiques de φv* et D
R
DG0’
Log-log
Y=Axn
2- Calcul de R
n
3- Confrontation avec l’expérience
Mesure de R par diffusion de la lumière
ConfrontationD
Confrontationφv* φv
φv*
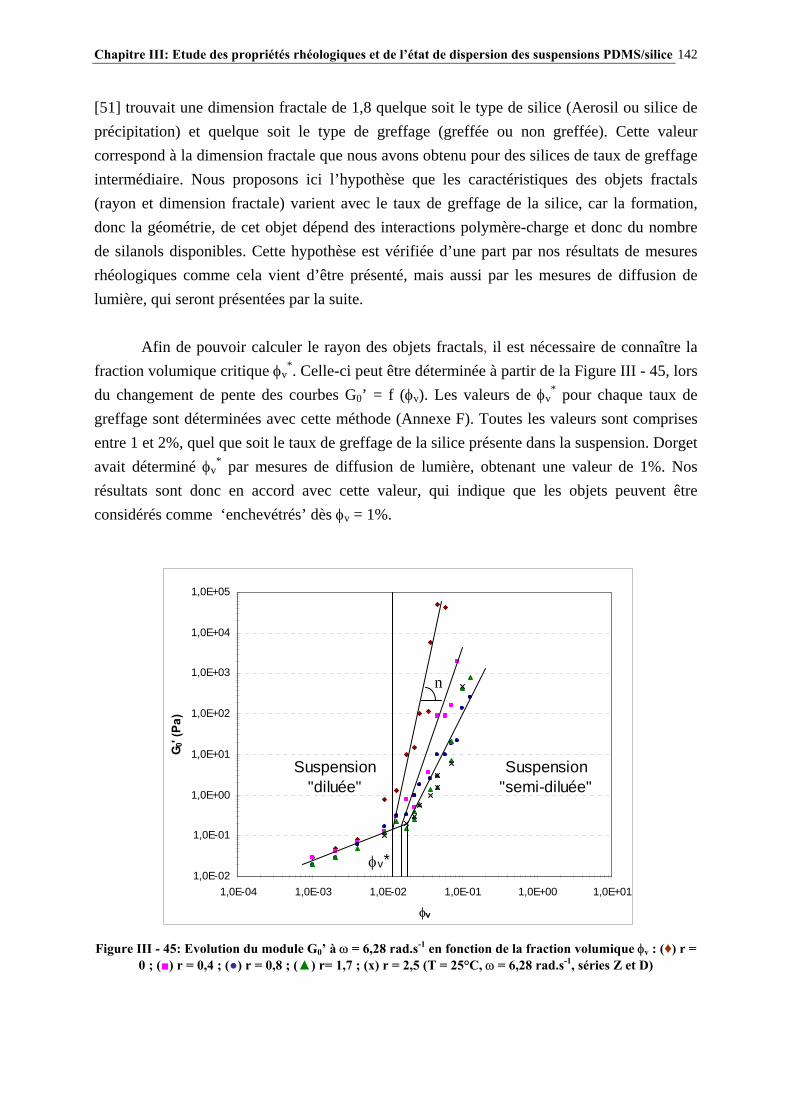
Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
142
[51] trouvait une dimension fractale de 1,8 quelque soit le type de silice (Aerosil ou silice de précipitation) et quelque soit le type de greffage (greffée ou non greffée). Cette valeur correspond à la dimension fractale que nous avons obtenu pour des silices de taux de greffage intermédiaire. Nous proposons ici l’hypothèse que les caractéristiques des objets fractals (rayon et dimension fractale) varient avec le taux de greffage de la silice, car la formation, donc la géométrie, de cet objet dépend des interactions polymère-charge et donc du nombre de silanols disponibles. Cette hypothèse est vérifiée d’une part par nos résultats de mesures rhéologiques comme cela vient d’être présenté, mais aussi par les mesures de diffusion de lumière, qui seront présentées par la suite. Afin de pouvoir calculer le rayon des objets fractals, il est nécessaire de connaître la fraction volumique critique φv
*. Celle-ci peut être déterminée à partir de la Figure III - 45, lors du changement de pente des courbes G0’ = f (φv). Les valeurs de φv
* pour chaque taux de greffage sont déterminées avec cette méthode (Annexe F). Toutes les valeurs sont comprises entre 1 et 2%, quel que soit le taux de greffage de la silice présente dans la suspension. Dorget avait déterminé φv
* par mesures de diffusion de lumière, obtenant une valeur de 1%. Nos résultats sont donc en accord avec cette valeur, qui indique que les objets peuvent être considérés comme ‘enchevétrés’ dès φv = 1%.
Figure III - 45: Evolution du module G0’ à ω = 6,28 rad.s-1 en fonction de la fraction volumique φv : (♦) r = 0 ; (■) r = 0,4 ; (●) r = 0,8 ; (▲) r= 1,7 ; (x) r = 2,5 (T = 25°C, ω = 6,28 rad.s-1, séries Z et D)
1,0E-02
1,0E-01
1,0E+00
1,0E+01
1,0E+02
1,0E+03
1,0E+04
1,0E+05
1,0E-04 1,0E-03 1,0E-02 1,0E-01 1,0E+00 1,0E+01
φv
G 0' (
Pa)
φv*
n
Suspension"diluée"
Suspension"semi-diluée"
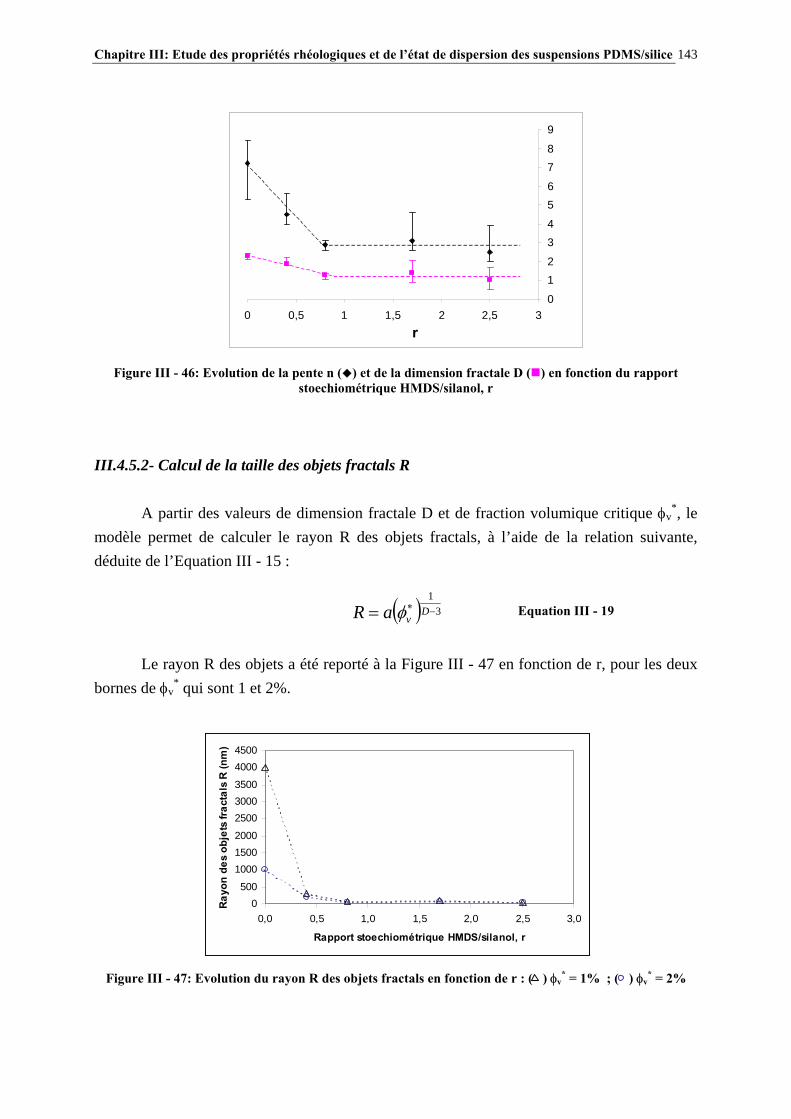
Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
143
Figure III - 46: Evolution de la pente n ( ) et de la dimension fractale D ( ) en fonction du rapport stoechiométrique HMDS/silanol, r
III.4.5.2- Calcul de la taille des objets fractals R A partir des valeurs de dimension fractale D et de fraction volumique critique φv
*, le modèle permet de calculer le rayon R des objets fractals, à l’aide de la relation suivante, déduite de l’Equation III - 15 : Equation III - 19
Le rayon R des objets a été reporté à la Figure III - 47 en fonction de r, pour les deux bornes de φv
* qui sont 1 et 2%.
Figure III - 47: Evolution du rayon R des objets fractals en fonction de r : ( ) φv* = 1% ; ( ) φv
* = 2%
0
12
34
56
78
9
0 0,5 1 1,5 2 2,5 3r
( ) 31−∗= D
vaR φ
0500
100015002000
250030003500
40004500
0,0 0,5 1,0 1,5 2,0 2,5 3,0
Rapport stoechiométrique HMDS/silanol, r
Ray
on d
es o
bjet
s fra
ctal
s R
(nm
)

Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
144
Le rayon R des objets diminue fortement dès que le taux de greffage augmente. Ce phénomène peut s’expliquer par l’agrégation qui devient moins importante lorsque le nombre de silanols de surface diminue, ce qui conduirait à des objets plus petits. Le rayon des objets fractals passe du micromètre à la cinquantaine de nanomètres lorsque r passe de 0 à 2,5. Cela montre l’importance de la chimie de surface sur l’agrégation de la silice, qui avait déjà été observée précédemment. Les résultats sont peu différents suivant la valeur de φv
* considérée, entre 1 et 2%. Le plus grand écart se situe pour les silices non greffées (r = 0) où le rayon varie de 1 à 4 micromètres. Ces ordres de grandeur des objets sont en accord avec les observations TEM réalisées précédemment. La même conclusion est donnée que taille des objets diminue lorsque le taux de greffage augmente. D’autre part l’évolution du rayon R suit encore une fois l’évolution du taux de greffage, atteignant une valeur plateau pour le même rapport stœchiométrique r = 1. Le rayon R varie également avec r de la même manière que la dimension fractale : une forte diminution dès que la silice est greffée puis on atteint une valeur limite correspondant au taux de greffage maximum. C’est entre 0 < r < 0,4 que les changements les plus importants se produisent, ce qui correspond bien aux résultats concernant l’étude de la rhéologie et de la morphologie des suspensions, où les changements les plus clairs étaient également observés entre 0 < r < 0,4. C’est dans cette zone que les caractéristiques physico-chimiques de la silice et donc les interactions polymère charge sont le plus modifiées. III.4.5.3- Comparaison avec les résultats obtenus par diffusion dynamique de la lumière Les rayons des objets élémentaires ont été mesurés par diffusion dynamique de la lumière. Pour avoir accès aux dimensions de ces objets par la technique de diffusion de la lumière (permettant d’accéder à des tailles d’objets du type de celles mises en évidence précédemment – c’est-à-dire quelques micromètres à quelques dizaines de micromètres-), il faut tout d’abord les individualiser par dilution dans un bon solvant du milieu de dispersion PDMS, le cyclohexane. En effet, les mesures de diffusion de lumière ne peuvent s’effectuer en milieu PDMS car la viscosité de la suspension et la concentration sont trop importantes. Cette méthodologie suppose que la dilution dans le cyclohexane permettent d’individualiser seulement les objets constituant les ‘briques élémentaires’ de la dispersion et ne détruise pas de structures. La distribution de taille des objets a été calculée à partir des mesures de diffusion de lumière dynamique à différents taux de dilution. Les histogrammes présentés Figure III - 48 montrent l’évolution de la distribution des tailles d’objets élémentaires en fonction de la
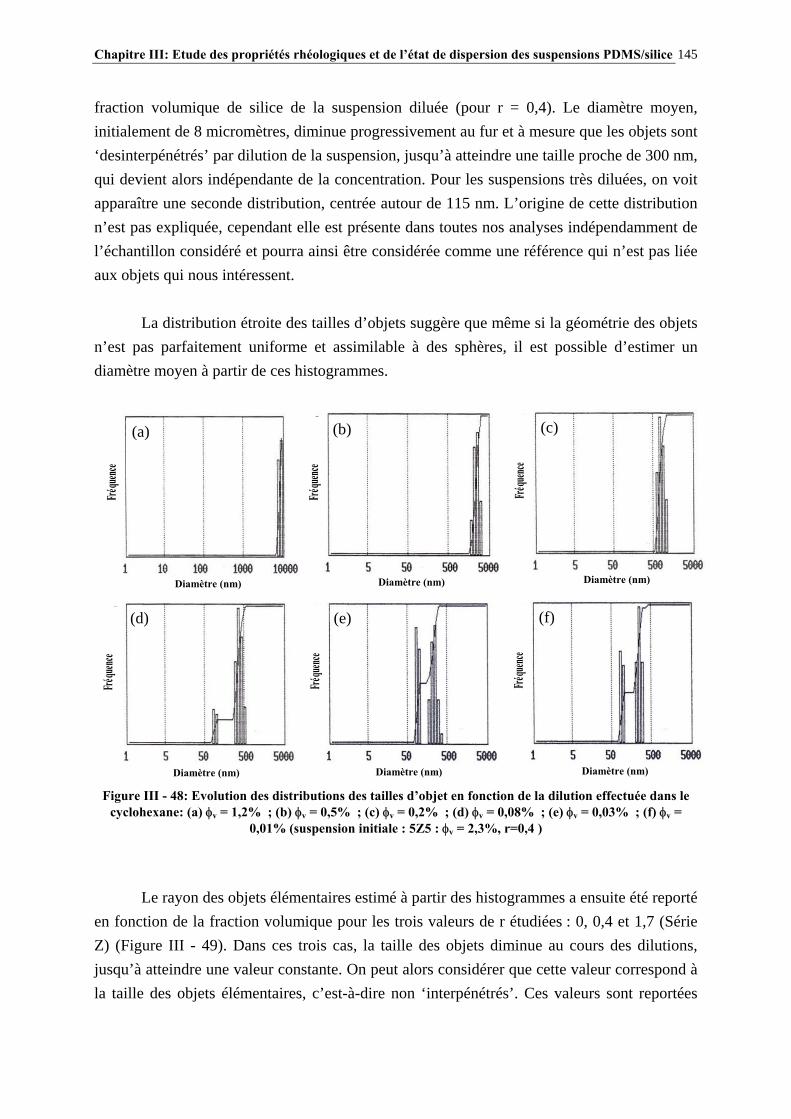
Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
145
fraction volumique de silice de la suspension diluée (pour r = 0,4). Le diamètre moyen, initialement de 8 micromètres, diminue progressivement au fur et à mesure que les objets sont ‘desinterpénétrés’ par dilution de la suspension, jusqu’à atteindre une taille proche de 300 nm, qui devient alors indépendante de la concentration. Pour les suspensions très diluées, on voit apparaître une seconde distribution, centrée autour de 115 nm. L’origine de cette distribution n’est pas expliquée, cependant elle est présente dans toutes nos analyses indépendamment de l’échantillon considéré et pourra ainsi être considérée comme une référence qui n’est pas liée aux objets qui nous intéressent. La distribution étroite des tailles d’objets suggère que même si la géométrie des objets n’est pas parfaitement uniforme et assimilable à des sphères, il est possible d’estimer un diamètre moyen à partir de ces histogrammes.
Figure III - 48: Evolution des distributions des tailles d’objet en fonction de la dilution effectuée dans le cyclohexane: (a) φv = 1,2% ; (b) φv = 0,5% ; (c) φv = 0,2% ; (d) φv = 0,08% ; (e) φv = 0,03% ; (f) φv =
0,01% (suspension initiale : 5Z5 : φv = 2,3%, r=0,4 )
Le rayon des objets élémentaires estimé à partir des histogrammes a ensuite été reporté en fonction de la fraction volumique pour les trois valeurs de r étudiées : 0, 0,4 et 1,7 (Série Z) (Figure III - 49). Dans ces trois cas, la taille des objets diminue au cours des dilutions, jusqu’à atteindre une valeur constante. On peut alors considérer que cette valeur correspond à la taille des objets élémentaires, c’est-à-dire non ‘interpénétrés’. Ces valeurs sont reportées
Diamètre (nm)
Fréqu
ence
Fr équ
ence
Diamètre (nm) Diamètre (nm)
Diamètre (nm) Diamètre (nm) Diamètre (nm)
Fréqu
ence
Fr équ
ence
Fr équ
ence
Fr équ
ence
(a) (b) (c)
(d) (e) (f)
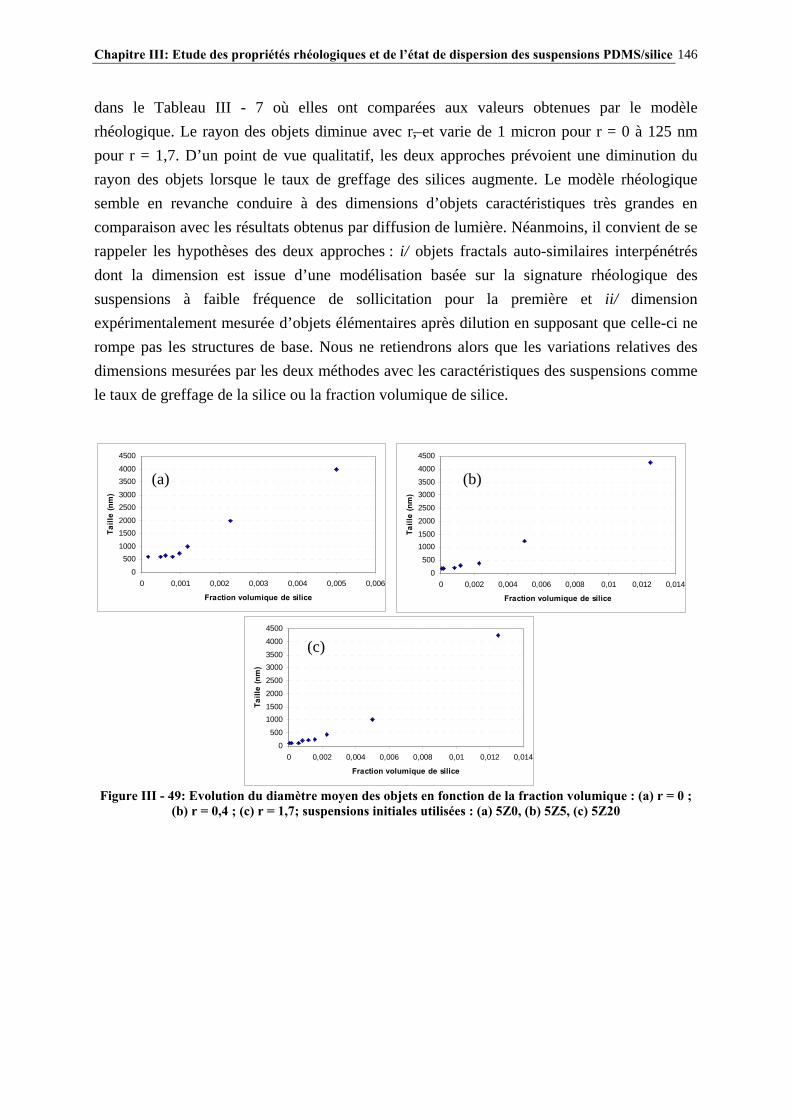
Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
146
dans le Tableau III - 7 où elles ont comparées aux valeurs obtenues par le modèle rhéologique. Le rayon des objets diminue avec r, et varie de 1 micron pour r = 0 à 125 nm pour r = 1,7. D’un point de vue qualitatif, les deux approches prévoient une diminution du rayon des objets lorsque le taux de greffage des silices augmente. Le modèle rhéologique semble en revanche conduire à des dimensions d’objets caractéristiques très grandes en comparaison avec les résultats obtenus par diffusion de lumière. Néanmoins, il convient de se rappeler les hypothèses des deux approches : i/ objets fractals auto-similaires interpénétrés dont la dimension est issue d’une modélisation basée sur la signature rhéologique des suspensions à faible fréquence de sollicitation pour la première et ii/ dimension expérimentalement mesurée d’objets élémentaires après dilution en supposant que celle-ci ne rompe pas les structures de base. Nous ne retiendrons alors que les variations relatives des dimensions mesurées par les deux méthodes avec les caractéristiques des suspensions comme le taux de greffage de la silice ou la fraction volumique de silice. Figure III - 49: Evolution du diamètre moyen des objets en fonction de la fraction volumique : (a) r = 0 ;
(b) r = 0,4 ; (c) r = 1,7; suspensions initiales utilisées : (a) 5Z0, (b) 5Z5, (c) 5Z20
0
5001000
15002000
25003000
35004000
4500
0 0,001 0,002 0,003 0,004 0,005 0,006
Fraction volumique de silice
Taill
e (n
m)
0
500
10001500
2000
2500
30003500
4000
4500
0 0,002 0,004 0,006 0,008 0,01 0,012 0,014
Fraction volumique de silice
Taill
e (n
m)
0
500
10001500
2000
2500
30003500
4000
4500
0 0,002 0,004 0,006 0,008 0,01 0,012 0,014
Fraction volumique de silice
Taill
e (n
m)
(a) (b)
(c)
Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
147
Modélisation rhéologique Méthode Diffusion de lumière
φv* = 1% φv
* = 2% R (nm) , r = 0 600 4000 1000
R (nm) , r = 0,4 175 300 200 R (nm) , r = 1,7 125 90 60 Tableau III - 7: Rayons des objets élémentaires obtenus par mesures de diffusion de lumière et par
modélisation rhéologique (Série Z)
Enfin à partir des résultats de diffusion de lumière (valeur de R), il est possible de recalculer la dimension fractale D des objets en s’appuyant sur le modèle rhéologique : Equation III - 20
Les dimensions fractales obtenues par les deux approches (rhéologie et diffusion de lumière) sont comparées au Tableau III - 8. Les évolutions sont similaires, la dimension fractale D diminue lorsque r augmente, cependant les résultats de diffusion de lumière conduisent à des variations plus faibles de la dimension fractale en fonction de r, que ce soit pour des fractions volumiques critiques de 1 ou 2%. On s’aperçoit également que les valeurs de dimension fractale dépendent de façon non négligeable de la fraction volumique critique, celle-ci étant difficile à déterminer précisément.
Diffusion de lumière Méthode
φv* = 1% φv
* = 2% Rhéologie
D , r = 0 2,04 2,18 2,3 D , r = 0,4 1,70 1,90 1,9 D, r = 1,7 1,57 1,78 1,4
Tableau III - 8: Dimensions fractales obtenues à partir du modèle développé par Dorget (en considérant les rayons des objets élémentaires déterminés par diffusion de lumière et par rhéologie)
( )⎟⎠⎞
⎜⎝⎛
+=
aR
D v
log
log3*φ

Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
148
III.5- CONCLUSIONS L’analyse des suspensions réalisées avec une méthode in-situ nous a permis de corréler le comportement rhéologique et la morphologie des suspensions au taux de greffage et à la fraction volumique de silice. Lorsque la fraction volumique augmente ou que le taux de greffage diminue, l’agrégation des particules de silice dans les suspensions est plus importante, ce qui conduit à un module G0’, module de cisaillement aux faibles déformations à ω = 6,28 rad.s-1, plus élevé et à un domaine viscoélastique linéaire plus étroit. La transition liquide-gel, qui peut être estimée par des mesures de contrainte seuil, apparaît pour des fractions volumiques plus faibles lorsque le taux de greffage est faible. A l’aide de ces résultats, un diagramme de transition liquide-gel des suspensions en fonction des deux paramètres principaux: fraction volumique φv et rapport stœchiométrique HMDS/silanol r a pu être établi. Aux forts taux de greffage, les suspensions réalisées à partir de silices greffées ex-situ présentent un module G0’ plus élevé que les suspensions dont les silices ont été greffées in-situ en milieu PDMS, en raison du plus faible taux de greffage atteint pour celles-ci. Cependant, les différences de morphologies entre ces deux types de suspensions sont peu évidentes et difficilement quantifiables par analyse d’image en microscopie électronique à transmission. La rhéologie semble donc un outil plus sensible que la microscopie pour évaluer la dispersion des silices au sein de suspensions (même si ces deux techniques restent complémentaires), permettant de mettre en évidence des différences d’états de dispersion peu importantes. L’influence du mode d’introduction du silane HMDS a pu être évaluée pour les suspensions réalisées par la méthode dite in-situ, en comparant les modules G0’ des suspensions lorsque l’HMDS est introduit avant le mélangeage du PDMS avec la silice et après celui-ci. Les différences restent faibles mais il semble que l’on obtient un module G0’ légèrement inférieur lorsque l’HMDS est introduit dans un deuxième temps, en particulier lorsque le temps d’attente entre la fin du mélangeage et l’ajout d’HMDS est important (2 heures). A partir des mesures rhéologiques réalisées sur les suspensions où la silice est greffée in-situ, l’évolution du module G0’ (valeur du module G’ dans la zone viscoélastique linéaire) en fonction de la fraction volumique a été représentée pour différents taux de greffage. La dépendance du module G0’ vis-à vis de la fraction volumique diminue lorsque les taux de greffage augmentent. Ces résultats ont été interprétés à l’aide d’un modèle de suspensions d’objets fractals semi-dilués qui rend compte de l’évolution du module G0’ en fonction de la

Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
149
fraction volumique et de la dimension fractale des objets. Nous avons ainsi pu déduire la fraction volumique critique à partir de laquelle les objets sont ‘désinterpénétrés’ (c’est-à-dire n’interagissant plus) et la dimension fractale de ces objets. La fraction volumique critique a été estimée entre 1 et 2% suivant les systèmes en accord avec des travaux publiés précédemment et la dimension fractale, qui diminue lorsque le taux de greffage augmente, est comprise entre D = 2,3 pour r = 0 et D = 1,4 pour r > 1. Une tentative de quantification du diamètre des objets élémentaires constitutifs de la morphologie a été faite par diffusion de la lumière en considérant des dilutions des suspensions afin d’individualiser ces structures primaires. En conclusion, ce chapitre permet de bien comprendre les propriétés (rhéologie et état de dispersion) des suspensions PDMS/silice suivant les paramètres des formulations (fraction volumique φv et rapport stœchiométrique HMDS/silanol r) et suivant les procédés de mise en œuvre (greffage in-situ et ex-situ). Ce sont ces bases qui permettront ensuite de comprendre (Chapitre IV), l’effet des additifs polymères qui pourront être ajoutés à ces suspensions pour en modifier les propriétés rhéologiques.

Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
150
Références bibliographiques
1. Heinrich G., Klüppel M., Recent Advances in the theory of filler networking in
elastomers, Adv. Polym. Sci., 2002, Vol. 160, pp.1-44.
2. Macosko C.W., Ch.3 : Linear viscoelasticity, in: Rheology - Principles, measurements, and applications, New-York: VCH, 1994, pp.109-126.
3. Macosko C.W., Ch.2 : Viscous liquid, in: Rheology - Principles, measurements, and applications, New-York: VCH, 1994, pp.65-108.
4. Dorget M., Propriétés rhéologiques des composés silice/silicone, Thèse de Doctorat, Université Joseph Fourier, Grenoble, 1995, 256p.
5. Mewis J., Thixotropy-a general review, J. Non-Newtonian Fluid Mechanics, 1979, Vol. 6, pp.1-20.
6. Couarraze G., Grossiord J.L., Variation du comportement rheologique d'écoulement avec le temps, in: Initiation à la rhéologie, Paris: Tec & Doc Lavoisier, 1983, pp.66-72.
7. Chahal R.S., St Pierre L.E., Interfacial phenomena in macromolecular systems. II. Relaxation modulus of uncross-linked silica-polydimethylsiloxane composites, Macromolecules, 1969, Vol. 2 (2),
8. Kobayashi Y., Hori Y., Aoki T., Observation of the dispersion of silica in silicone rubber, Int. Polym. Sci. Tech., 2000, Vol. 27 (4),
9. DeGroot Jr J.V., Macosko C.W., Aging phenomena in silica-filled PDMS, J. Colloid Interface Sci., 1999, Vol. 217, pp.86-93.
10. Pouchelon A., Vondracek P., Semiempirical relationships between properties and loading in filled elastomers, Rubber Chem. Technol., 1986, Vol. 62 (5), pp.788-799.
11. Jokinen M., Györvary E., Rosenholm J.B., Viscoelastic characterization of three different sol-gel derived silica gels, Colloids Surf., A, 1998, Vol. 141, pp.205-216.
12. Feng J., Weng L.-T., Chan C.-M., Xhie J., Li L., Imaging of sub-surface nano particles by tapping-mode atomic force microscopy, Polymer, 2001, Vol. 42, pp.2259-2262.
13. Clement F., Lapra A., Bokobza L., Monnerie L., Menez P., Atomic force microscopy investigation of filled elastomers and comparison with transmission electron microscopy - application to silica-filled silicone elastomers, Polymer, 2001, Vol. 42, pp.6259-6270.
14. Cantu T.S., Caruthers J.M., Viscometric properties of PDMS filled with spherical silica particles, J. Appl. Polym. Sci., 1982, Vol. 27, pp.3079-3088.

Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
151
15. Kosinski L.E., Caruthers J.M., Rheological properties of PDMS filled with fumed silica : II. Stress relaxation and stress growth, J. Non-Newtonian Fluid Mechanics, 1985, Vol. 17, pp.69-89.
16. Kosinski L.E., Caruthers J.M., The effect of molecular weight on the rheological properties of PDMS filled with fumed silica, Rheol. Acta, 1986, Vol. 25, pp.153-160.
17. Ziegelbaur R.S., Caruthers J.M., Rheological properties of PDMS filled with fumed silica : I. Hysteresis behaviour, J. Non-Newtonian Fluid Mechanics, 1985, Vol. 17, pp.45-68.
18. Hadjistamov D., The viscoelastic properties of filled silicone fluids, Rheology, 1993, Vol. 93, pp.113-119.
19. Aranguren M.I., Mora E., DeGroot Jr. J.V., Macosko C.W., Effect of reinforcing fillers on the rheology of polymer melts, J. Rheol., 1992, Vol. 36 (6), pp.1165-1182.
20. Payne A.R., The dynamic properties of carbon black loaded natural rubber vulcanizates. Part II, J. Appl. Polym. Sci., 1962, Vol. 6 (21), pp.368-372.
21. Clement F., Etude des mécanismes de renforcement dans les réseaux de PDMS charges silice, Thèse de Doctorat, Université Paris VI, 1999,
22. Clement F., Bokobza L., Monnerie L., Varlet J.,, Non-linear behavior of silica-filled silicone elastomers, Eurofillers'99, 1999, Villeurbanne, France.
23. Bokobza L., Reinforcement of elastomeric networks by fillers, Macromol. Symp., 2001, Vol. 171, pp.163-170.
24. Bokobza L., Reinforcement of elastomeric networks by fillers, Macromol. Symp., 2001, Vol. 169, pp.243-260.
25. Terreau C., Vandereecken P., Carlier F., Rheological analysis of silica-silicone interactions, XIIIth International Congress on Rheology, 2000, Cambridge, UK.
26. Levresse P., Feke D.L., Manas-Zloczower I., Analysis of the formation of bound PDMS on silica, Polymer, 1998, Vol. 39 (17), pp.3919-3924.
27. Cohen-Addad J.P., Adsorption of PDMS on bare fumed silica surfaces, in: Papirer E., Adsorption on Silica Surfaces, New-York: Marcel Dekker, 2000, pp.621-643.
28. Cosgrove T., Turner M.J., Thomas D.R., The adsorption of PDMS onto silica from the melt, Polymer, 1997, Vol. 38 (15), pp.3885-3892.
29. Osaheni J.A., Truby K.E., Silvi N., The influence of filler morphology and surface hydroxyls on crepe-hardening of uncured silicone elastomers, Eurofillers'99, 1999, Villeurbanne, France.
30. Silvi N., Osaheni J.A., Characterization of polymer-filler interactions in highly filled silicone elastomers, Eurofillers'99, 1999, Villeurbanne, France.

Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
152
31. Einstein A., Eine neue bestimmung der moleküldimensionen, Annalen der Physik, 1906, Vol. 19, pp.289-306.
32. Einstein A., Berichtigung zu meiner arbeit : eine neue bestimmung des moleküldimensionen, Annalen der Physik, 1911, Vol. 34, pp.591-592.
33. Smallwood H.M., Limiting law of the reinforcement of rubber, J. Appl. Physics, 1944, Vol. 15, pp.758-766.
34. Guth E., Theory of filler reinforcement, J. Appl. Physics, 1945, Vol. 16, pp.20-25.
35. Mooney M., The viscosity of a concentrated suspension of spherical particles, J. Colloid Sci., 1951, Vol. 6, pp.162-170.
36. Kerner E.H., The elastic and thermo-elastic properties of composite media, Proc. Phys. Soc., 1956, Vol. 69B, pp.808-813.
37. Sato T., Furukawa J., A molecular theory of filler reinforcement based upon the conception of internal deformation, Rubber Chem. Techn., 1963, Vol. 36, pp.1081-1097.
38. Nielsen L.E., Simple theory of stress-strain properties of filled polymers, J. Appl. Polym. Sci., 1966, Vol. 10, pp.97-103.
39. Amari T., Uesugi K., Suzuki H., Viscoelastic properties of carbon black suspension as a flocculated percolation system, Prog. Org. Coat., 1997, Vol. 31, pp.171-178.
40. De Gennes P.G., Scaling concepts in polymer physics, New-York: Cornell University Press, 1979, 324p.
41. Mall S., Russel W.B., Effective medium approximation for an elastic network model of flocculated suspensions, J. Rheol., 1987, Vol. 31, pp.651-689.
42. DeGroot Jr J.V., Macosko C.W., Dynamic properties of model filled polymer melts in the linear viscoelastic region, XIth Int. Congr. On Rheology, 1992, Brussels, Belgium.
43. Shiyan A.A., Viscosity for fractal suspensions:dependence on fractal dimensionality, Physics letter A, 1996, Vol. 220, pp.117-119.
44. Uriev N.B., Ladyzhinsky I. Ya., Fractal models in rheology of colloidal gels, Colloids Surf., A, 1996, Vol. 108, pp.1-11.
45. Lapasin R., Grassi M., Pricl S., Rheological modeling of fractal and dense suspensions, Chem. Eng. J., 1996, Vol. 64, pp.99-106.
46. Chen M., Russel W.B., Characteristics of flocculated silica dispersions, J. Colloid Interface Sci., 1991, Vol. 141 (2), pp.564-577.
47. Buscall R., Mills P.D.A., Scaling behaviour of the rheology of aggregate networks formed from colloidal particles, J. Chem. Soc. Faraday Trans., 1988, Vol. 84 (12), pp.4249-4260.

Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
153
48. Khan S.A., Zoeller N.J., Dynamic rheological behavior of flocculated fumed silica suspensions, J. Rheol., 1993, Vol. 37 (6), pp.1225-1235.
49. Walls H.J., Zhou J., Yerian J.A., Fedkiw P.S., Khan S.A., Stowe M.K., Baker J.L., Fumed silica-based composite polymer electrolytes: synthesis, rheology, and electrochemistry, J. Power Sources, 2000, Vol. 89, pp.156-162.
50. Raghavan S.R., Hou J., Baker G.L., Khan S.A., Colloidal interactions between particles with tethered nonpolar chains dispersed in polar media:direct correlation between dynamic rheology and interaction parameters, Langmuir, 2000, Vol. 16, pp.1066-1077.
51. Piau J.M., Dorget M., Palierne J.-F., Shear elasticity and yield stress of silica-silicone physical gels : fractal approach, J. Rheol., 1999, Vol. 43 (2), pp.305-314.
52. Daoud M., Van Damme H., Fractales, in: Daoud M., Williams C., La juste argile, Les Ulis: Les Editions de Physique, 1995, pp.43-84.
53. Muller R., Gerard E., Dugand P., Rempp P., Gnanou Y., Rheological characterization of the gel point : a new interpretation, Macromolecules, 1991, Vol. 24, pp.1321-1326.
54. Barnes H.A., Hutton J.F., Walters K., Rheology of suspensions, in: An introduction to rheology, Amsterdam: Elsevier, 1989, pp.115-140.
55. Aubert C., Cannell D.S., Restructuring of colloidal silica aggregates, Phys. Rev. Lett., 1986, Vol. 56, pp.738-741.
56. Papirer E., Balard H., Vergelati C., Adsorption on Silica Surfaces, Surface energetics of Silica Investigated by Inverse Gas Chromatography, in: Surfactant Science Series, Vol. 90, New-York: Marcel Dekker, 2000, pp.205-241.

Chapitre III: Etude des propriétés rhéologiques et de l’état de dispersion des suspensions PDMS/silice
154

CHAPITRE IV : POLYMERES MODIFICATEURS DE LA RHEOLOGIE DES
SUSPENSIONS PDMS /SILICE

Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
155
CHAPITRE IV : POLYMERES MODIFICATEURS DE LA RHEOLOGIE DES SUSPENSIONS
PDMS/SILICE IV.1- INTRODUCTION................................................................................................................................. 157
IV.2- ETUDE BIBLIOGRAPHIQUE ........................................................................................................... 159
IV.3- PARTIE EXPÉRIMENTALE ............................................................................................................. 164 IV.3.1- ADDITIFS UTILISÉS ET NOMENCLATURE ............................................................................................ 164
IV.3.1.1- Copolymères PDMS/Polyéther................................................................................................. 164 IV.3.1.2- Homopolymères........................................................................................................................ 164
a) Polydiméthylsiloxanes (PDMS) ....................................................................................................................... 164 Fournisseur ............................................................................................................................................. 166
NOMENCLATURE ............................................................................................................................................ 166 b) Polyoxydes d’éthylène (POE) téléchéliques .................................................................................................... 167 c) Polyoxydes de propylène (POP) téléchéliques................................................................................................. 167
IV.3.2- MODE D’INTRODUCTION DES ADDITIFS ET CARACTÉRISATION DES GELS FORMÉS............................. 168 IV.3.2.1- Mode d’introduction des additifs ............................................................................................. 168 IV.3.2.2- Caractérisation des propriétés rhéologiques ........................................................................... 169 IV.3.2.3- Caractérisation des morphologies ........................................................................................... 169
IV.4- RÉSULTATS ET DISCUSSION ......................................................................................................... 170 IV.4.1- DESCRIPTION GÉNÉRALE DES GELS ................................................................................................... 170
IV.4.1.1- Comportement rhéologique des gels ........................................................................................ 170 IV.4.1.2- Essais préliminaires : influence des paramètres principaux.................................................... 172
a) Influence des paramètres d’élaboration des suspensions additivées................................................................. 173 b) Influence de la concentration en additif ........................................................................................................... 175
IV.4.2- INFLUENCE DES PARAMÈTRES DE LA FORMULATION ......................................................................... 177 IV.4.2.1 – Influence de la fraction volumique de silice / diagramme de transition liquide-gel .............. 177 IV.4.2.2 – Influence du taux de greffage de la silice ............................................................................... 179 IV.4.2.3 – Influence du procédé de mise en œuvre des suspensions PDMS/silice................................... 182
IV.4.3- INFLUENCE DE LA MICROSTRUCTURE DES ADDITIFS.......................................................................... 184 IV.4.3.1 – Homopolymères (PDMS, POE, POP) .................................................................................... 184
a) Influence de la nature chimique de l’additif ..................................................................................................... 184 b) Influence des fins de chaîne de l’additif........................................................................................................... 186 c) Influence de la masse molaire .......................................................................................................................... 189
IV.4.3.2- Copolymères PDMS/Polyéther................................................................................................. 191 a) Influence de la masse molaire .......................................................................................................................... 191 b) Influence de la composition chimique.............................................................................................................. 192 c) Comparaison des copolymères avec les homopolymères sur la contrainte seuil des suspensions.................... 194
IV.4.4- ETUDE PAR MICROSCOPIE DES SUSPENSIONS ET MÉCANISMES DE STRUCTURATION PROPOSÉS .......... 195 IV.4.4.1- Introduction.............................................................................................................................. 195 IV.4.4.2- Observation des structures par microscopies optique et électronique..................................... 196 IV.4.4.3- Observation de la structuration des suspensions par fluorescence ......................................... 198 IV.4.4.4- Mécanismes proposés............................................................................................................... 204
IV.4.5- MESURE DES ÉNERGIES D’INTERACTIONS ......................................................................................... 209 IV.4.5.1- Chromatographie gazeuse en phase inverse (IGC).................................................................. 209

Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
156
a) Principe ............................................................................................................................................................ 209 b) Méthodologie d’analyse des résultats............................................................................................................... 209 c) Etudes antérieures ............................................................................................................................................ 212 d) Expérimental .................................................................................................................................................... 214 e) Résultats et discussion...................................................................................................................................... 216
IV.4.5.2- Mesures des énergies de surface et des énergies interfaciales................................................. 227 a) expérimental ..................................................................................................................................................... 227 c) Résultats et discussion...................................................................................................................................... 229
IV.5- CONCLUSIONS ET PERSPECTIVES .............................................................................................. 238

Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
157
IV.1- INTRODUCTION Les suspensions PDMS/silice, dont les propriétés ont été décrites au chapitre précédent, ont volontairement un comportement liquide pour une mise en œuvre aisée. Or pour certaines applications sur le marché du moulage on souhaite un produit qui ne s’écoule pas, c’est-à-dire une suspension présentant une contrainte seuil. Une solution consiste à ajouter à la suspension fluide une faible quantité d’additif (quelques pourcents) qui va modifier la suspension, lui conférant les propriétés d’un gel. Ces additifs, généralement des copolymères ‘peigne’ PDMS/polyéther sont cependant parfois inefficaces dans le cas de certaines formulations industrielles. Ce chapitre est consacré à l’étude du phénomène de gélification de suspensions PDMS/silice par ajout de polymères modificateurs. Il a pour double objectif la compréhension du mécanisme de formation de ces gels et la mise en évidence des relations structure-propriétés des gels en fonction de la formulation initiale des suspensions et de la microstructure des additifs utilisés. Le but final est la détermination des paramètres clefs influençant la formation des gels et l’identification des additifs les plus efficaces suivant les formulations, pour revenir éventuellement à la synthèse de nouveaux additifs aux microstructures contrôlées. Les suspensions PDMS/silice étudiées au cours de ce chapitre sont celles dont les silices ont été modifiées in-situ par réaction avec l’HMDS en milieu PDMS (séries Z). La partie bibliographique sera consacrée d’une part aux gels présentant des analogies avec les gels PDMS/silice/polyéther que nous étudions et d’autre part au potentiel de la chromatographie gazeuse en phase inverse (IGC) pour évaluer les interactions polymère/silice. Après avoir présenté les différents additifs évalués et les techniques de caractérisation des gels (rhéologie, microscopie…), les méthodes de détermination des énergies de surface seront détaillées. L’influence des différents paramètres expérimentaux et de la concentration en additif sur la rhéologie des gels sera tout d’abord examinée, puis la relation entre les paramètres de la formulation des suspensions initiales et les contraintes seuil des gels formés sera explicitée, ainsi que la relation entre la microstructure des additifs et les contraintes seuil des gels. Ces résultats seront complétés par une étude de la microstructure des gels observée par microscopies optique, électronique et par fluorescence, conduisant à la proposition d’un nouveau mécanisme de formation des gels, sur la base de ces résultats. Enfin, nous verrons comment les interactions dans ce système ternaire PDMS/silice/polyéther peuvent être

Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
158
évaluées à partir du concept d’énergies de surface, apportant des informations complémentaires sur les mécanismes de formation des gels.

Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
159
IV.2- ETUDE BIBLIOGRAPHIQUE Sur le marché du moulage, pouvoir obtenir par l’ajout d’additifs une suspension présentant une contrainte seuil (gel) est important pour certaines applications, comme par exemple les produits devant être appliqués sur une surface non horizontale [1]. Les additifs utilisés sont généralement des copolymères ‘peigne’ PDMS/polyéther dont la structure chimique générale est présentée à la Figure IV - 1. Ces additifs sont macroscopiquement ‘miscibles’ à température ambiante avec la suspension, mais moins avec le PDMS seul, en raison de l’incompatibilité PDMS/polyéther.
Figure IV - 1 : Microstructure des copolymères PDMS/polyéther
L’ajout de ces additifs, dont la concentration peut varier de 1 à 5% en poids par rapport à la suspension initiale, permet de passer en quelques secondes d’agitation d’une suspension initiale fluide, c'est-à-dire d’un comportement liquide newtonien, à un comportement de gel avec une contrainte seuil pouvant atteindre jusque plusieurs milliers de Pascal pour certaines formulations. D’un point de vue théorique les mécanismes de formation des gels dans ces formulations sont mal connus. Le mécanisme envisagé est le pontage des agrégats de silice par l’intermédiaire des molécules d’additifs pontant les nanoparticules [1]. Les liens s’établiraient par liaison hydrogène entre d’une part les silanols de surface de la silice et d’autre part soit les fins de chaînes (OH, NH2) soit les groupements éther des polymères ajoutés, ce qui est représenté schématiquement à la Figure IV - 2. Dans certaines formulations industrielles, l’état de gel ne peut être atteint par ajout de ces additifs et une connaissance plus approfondie du mécanisme d’action de ces molécules est nécessaire. Pour cela nous allons raisonner sur des molécules plus simples, en particulier des homopolymères poly(oxyde d’éthylène) (POE).
R
Si O Si
CH3
O
CH3
CH3
( ) ( )m n
CH2 CH2O )xO( CH2 CH
CH3)y
(R=

Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
160
Figure IV - 2 : Mécanisme d’action envisagé des additifs polymères au sein des suspensions PDMS/silice ( pontage par les fins de chaîne, pontage par les groupements éther présents dans la chaîne)
A notre connaissance ces systèmes PDMS/POE/silice n’ont pas encore été étudiés, par contre un certain nombre de travaux ont été menés sur le système POE/silice et le phénomène d’agglomération qui mène à la formation de gels [2-5]. Le POE est un polymère couramment utilisé pour étudier les phénomènes d’adsorption des macromolécules à la surface de la silice, car il s’adsorbe facilement par liaison hydrogène entre les atomes d’oxygène des groupements éther (plus éventuellement par ses fins de chaînes) et les silanols de surface de la silice. L’énergie libre de chaque liaison hydrogène est faible (environ 0,5kT) mais appliqué à chaque unité monomère en contact avec la surface l’énergie d’adsorption totale de la macromolécule est assez importante [2]. Cependant, il a été montré que toutes les liaisons hydrogène ne possèdent pas la même énergie, en fonction du type de silanol (isolé, géminé, vicinal). Ceci s’explique par leur différence d’acidité de Bronsted (tendance à donner des protons) : les silanols les plus acides forment des liaisons hydrogène plus fortes [6, 7]. L’adsorption du POE à la surface de la silice en milieu aqueux a été étudiée par des mesures d’adsorption. La quantité de polymère adsorbé et l’épaisseur de la couche à la surface des particules de silices ont été déterminées et reliées à la masse molaire du POE, par les relations suivantes [3] : Equation IV - 1
où : Q : quantité de POE adsorbée (g.m-2)
M : masse molaire en poids du POE (g.mol-1) d : épaisseur de la couche adsorbée (nm)
A peu près comme pour les autres polymères, l’épaisseur de la couche augmente donc faiblement avec la masse molaire du POE.
Chaîne polymère (additif)
Silice
PDMSChaîne polymère (additif)
Silice
PDMS
54,0Md ∝
11,0410.5,2 MQ ×= −
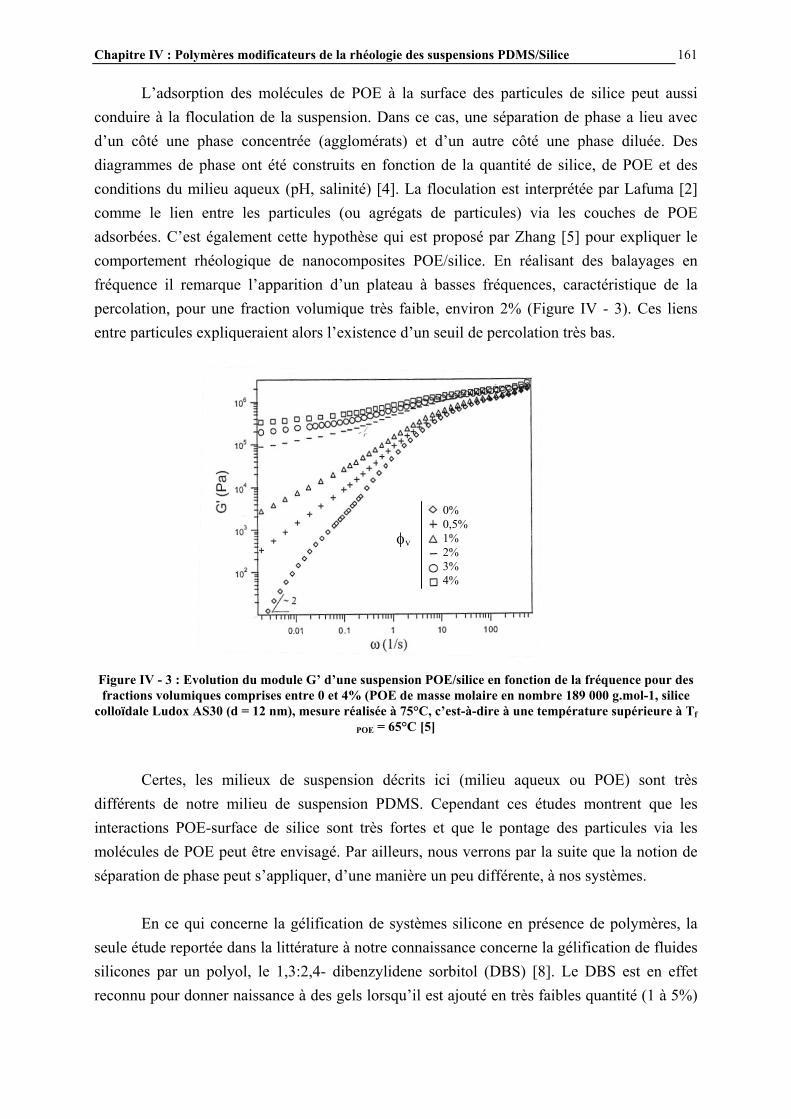
Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
161
L’adsorption des molécules de POE à la surface des particules de silice peut aussi conduire à la floculation de la suspension. Dans ce cas, une séparation de phase a lieu avec d’un côté une phase concentrée (agglomérats) et d’un autre côté une phase diluée. Des diagrammes de phase ont été construits en fonction de la quantité de silice, de POE et des conditions du milieu aqueux (pH, salinité) [4]. La floculation est interprétée par Lafuma [2] comme le lien entre les particules (ou agrégats de particules) via les couches de POE adsorbées. C’est également cette hypothèse qui est proposé par Zhang [5] pour expliquer le comportement rhéologique de nanocomposites POE/silice. En réalisant des balayages en fréquence il remarque l’apparition d’un plateau à basses fréquences, caractéristique de la percolation, pour une fraction volumique très faible, environ 2% (Figure IV - 3). Ces liens entre particules expliqueraient alors l’existence d’un seuil de percolation très bas.
Figure IV - 3 : Evolution du module G’ d’une suspension POE/silice en fonction de la fréquence pour des fractions volumiques comprises entre 0 et 4% (POE de masse molaire en nombre 189 000 g.mol-1, silice
colloïdale Ludox AS30 (d = 12 nm), mesure réalisée à 75°C, c’est-à-dire à une température supérieure à Tf POE = 65°C [5]
Certes, les milieux de suspension décrits ici (milieu aqueux ou POE) sont très différents de notre milieu de suspension PDMS. Cependant ces études montrent que les interactions POE-surface de silice sont très fortes et que le pontage des particules via les molécules de POE peut être envisagé. Par ailleurs, nous verrons par la suite que la notion de séparation de phase peut s’appliquer, d’une manière un peu différente, à nos systèmes. En ce qui concerne la gélification de systèmes silicone en présence de polymères, la seule étude reportée dans la littérature à notre connaissance concerne la gélification de fluides silicones par un polyol, le 1,3:2,4- dibenzylidene sorbitol (DBS) [8]. Le DBS est en effet reconnu pour donner naissance à des gels lorsqu’il est ajouté en très faibles quantité (1 à 5%)
0% 0,5% 1% 2% 3% 4%
φv

Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
162
dans des liquides organiques. Ce type de composés, appelés ‘organo-gelators’ ou gélificateurs, peut s’autoassocier par différents types de liaisons (liaison hydrogène, liaison de coordination organométallique, transfert d’électron…) pour former par séparation de phase une structure continue par assemblage supramoléculaire, responsable de la gélification du liquide. L’ajout se fait à chaud pour rendre miscible le gélificateur avec le milieu organique et c’est au refroidissement que le gélificateur se structure et conduit à un gel du milieu [9]. Les structures formées sont souvent des fibrilles avec des diamètres variant de quelques nanomètres à la centaine de nanomètres suivant les systèmes [9, 10]. Ces gels sont thermoréversibles, c'est-à-dire que l’état liquide peut être retrouvé lorsque la température est augmentée. Le DBS peut donc donner naissance à ce même type de gel dans un liquide silicone. L’incorporation peut être obtenue soit par élévation de température, comme décrit précédemment dans le cas des milieux organiques, soit à l’aide d’un cosolvant (N-méthylpyrrolidone). La Figure IV - 4 présente la structure de gels DBS-PDMS et DBS-D4 obtenus par solubilisation en température et refroidissement puis observation en microscopie électronique à balayage. Ces gels présentent des régions opaques constitués de macrofibres (µm) et des zones claires constitués de fibrilles de la taille de plusieurs dizaines de nm. Lorsque les gels sont obtenus à l’aide d’un cosolvant seules les zones claires sont présentes. Figure IV - 4 : Images de microscopie électronique à balayage montrant la structure des gels formés par
ajout de 1% de DBS dans un milieu silicone (a) D4 ; (b) PDMS de viscosité 1,5 cSt [8]
Moins de 4% de DBS sont nécessaires pour obtenir ces structures. Dans notre cas la teneur en additif sera de cet ordre de grandeur, mais la question du mécanisme de formation du gel est plus délicate en raison de la présence des charges de silice, comme cela sera discuté au cours de ce chapitre, puisqu’alors les interactions PDMS ou DBS/silice entrent en compétition avec l’assemblage supramoléculaire du DBS. Ainsi, ce sont les interactions entre
(a) (b)

Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
163
les trois constituants (PDMS, silice, additif) qui vont gérer l’organisation des mélanges et la formation ou non de gels. En conclusion, la formation de gels dans les systèmes PDMS/silice par ajout de polymère comme le POE n’est pas encore décrite dans la littérature, cependant nous pouvons nous appuyer sur deux types de systèmes proche du notre, dont la transition liquide-gel a été rapportée : les suspensions POE/silice et les gels PDMS/DBS. L’étude des systèmes POE/silice montre que le POE s’adsorbe très facilement à la surface de la silice ce qui conduit sous certaines conditions à la formation d’un gel avec moins de 4% de silice, par pontage des particules de silice via les chaînes de POE adsorbées. Ces interactions très fortes seront donc à prendre en compte pour la compréhension de nos propres systèmes. La floculation de ces suspensions fait également intervenir la notion de séparation de phase, àlaquelle on peut également faire appel pour l’interprétation de la formation des gels PDMS/DBS (avec l’autoassemblage des molécules de DBS). Nous verrons que cette notion importante de séparation de phase doit être également prise en compte pour expliquer le comportement de systèmes ternaires PDMS/silice/POE. Dans les systèmes PDMS/DBS une quantité très faible en DBS, seulement 1%, est nécessaire pour former une structure percolée par assemblage supramoléculaire, et donc un gel. Dans ces deux types de gels l’ajout de quelques pourcents seulement de polymère ou de silice est nécessaire. Il y a donc des similitudes entre ces gels et les notres, même si nos systèmes sont plus complexes par la présence de trois constituants (PDMS, POE, silice) au lieu de deux.

Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
164
IV.3- PARTIE EXPÉRIMENTALE IV.3.1- Additifs utilisés et nomenclature IV.3.1.1- Copolymères PDMS/Polyéther Les copolymères utilisés sont des copolymères ‘peigne’ PDMS/POE ou PDMS/POE/POP. Ce sont des polymères commerciaux dont la structure chimique générale a déjà été présentée à la Figure IV - 1. Afin de caractériser de manière précise la microstructure de chacun des copolymères (proportion de chaque bloc) des analyses RMN 1H liquide ont été réalisées au Laboratoire des Matériaux Organiques à Propriétés Spécifiques (service RMN/FRPL) à Solaize, sur certains copolymères dont la composition chimique n’était pas connue précisément. Un spectre RMN typique obtenu pour un copolymère PDMS/POE/POP est présenté à la Figure IV - 5. Les attributions des différents pics de résonance sont données au Tableau IV - 1. A partir de l’aire des pics de résonance la composition chimique des copolymères a été déterminée. L’ensemble des spectres et la méthode de calcul sont présentés en Annexe G. Le Tableau IV - 2 et le Tableau IV - 3 récapitulent la composition chimique et la nomenclature des copolymères PDMS/POE et PDMS/POE/POP qui seront utilisés au cours de ce travail. Les paramètres structuraux que nous pouvons faire varier sont donc la masse molaire, de 600 à 30 000 g.mol-1 et la proportion de chaque bloc (PDMS, POE, POP). IV.3.1.2- Homopolymères a) Polydiméthylsiloxanes (PDMS) Des PDMS de différentes masses molaires et de différentes fins de chaîne ont été étudiés. Le Tableau IV - 4 récapitule les caractéristiques de ces différents additifs.
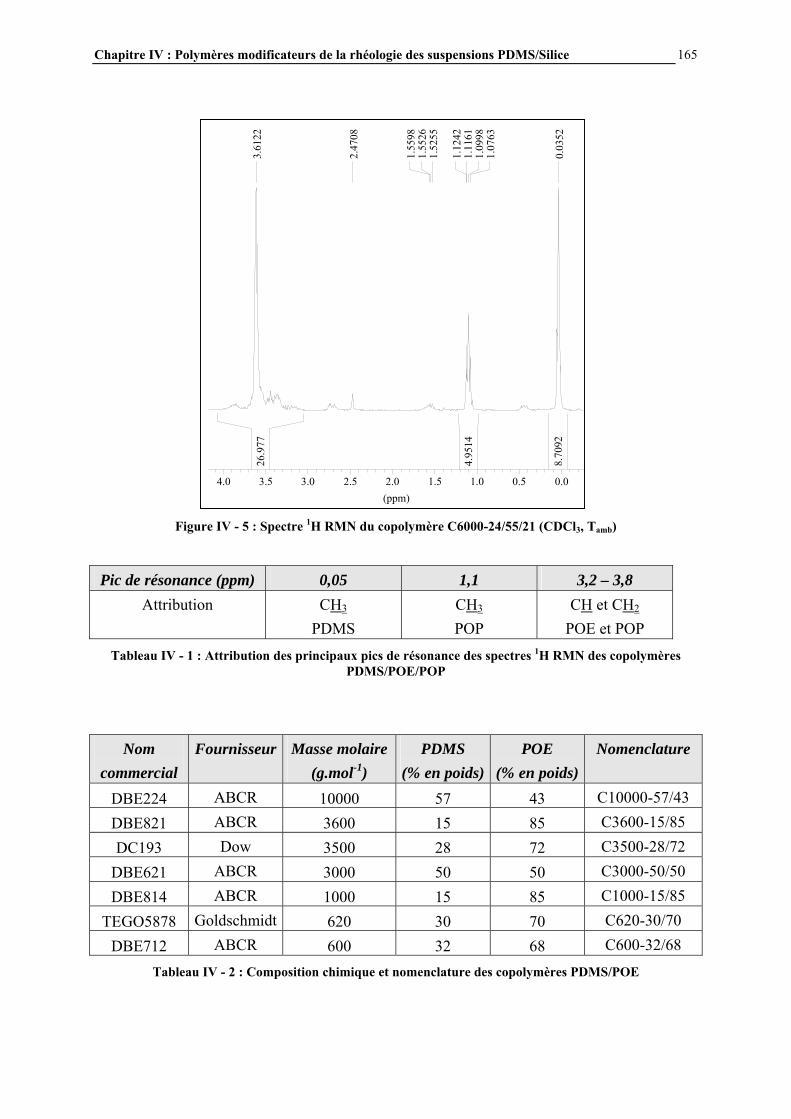
Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
165
Figure IV - 5 : Spectre 1H RMN du copolymère C6000-24/55/21 (CDCl3, Tamb)
Pic de résonance (ppm) 0,05 1,1 3,2 – 3,8
Attribution CH3 PDMS
CH3 POP
CH et CH2 POE et POP
Tableau IV - 1 : Attribution des principaux pics de résonance des spectres 1H RMN des copolymères PDMS/POE/POP
Nom commercial
Fournisseur Masse molaire (g.mol-1)
PDMS (% en poids)
POE (% en poids)
Nomenclature
DBE224 ABCR 10000 57 43 C10000-57/43 DBE821 ABCR 3600 15 85 C3600-15/85 DC193 Dow 3500 28 72 C3500-28/72
DBE621 ABCR 3000 50 50 C3000-50/50 DBE814 ABCR 1000 15 85 C1000-15/85
TEGO5878 Goldschmidt 620 30 70 C620-30/70 DBE712 ABCR 600 32 68 C600-32/68
Tableau IV - 2 : Composition chimique et nomenclature des copolymères PDMS/POE
26.9
77
4.95
14
8.70
92
3.61
22
2.47
08
1.55
981.
5526
1.52
55
1.12
421.
1161
1.09
981.
0763
0.03
52
(ppm)0.00.51.01.52.02.53.03.54.0
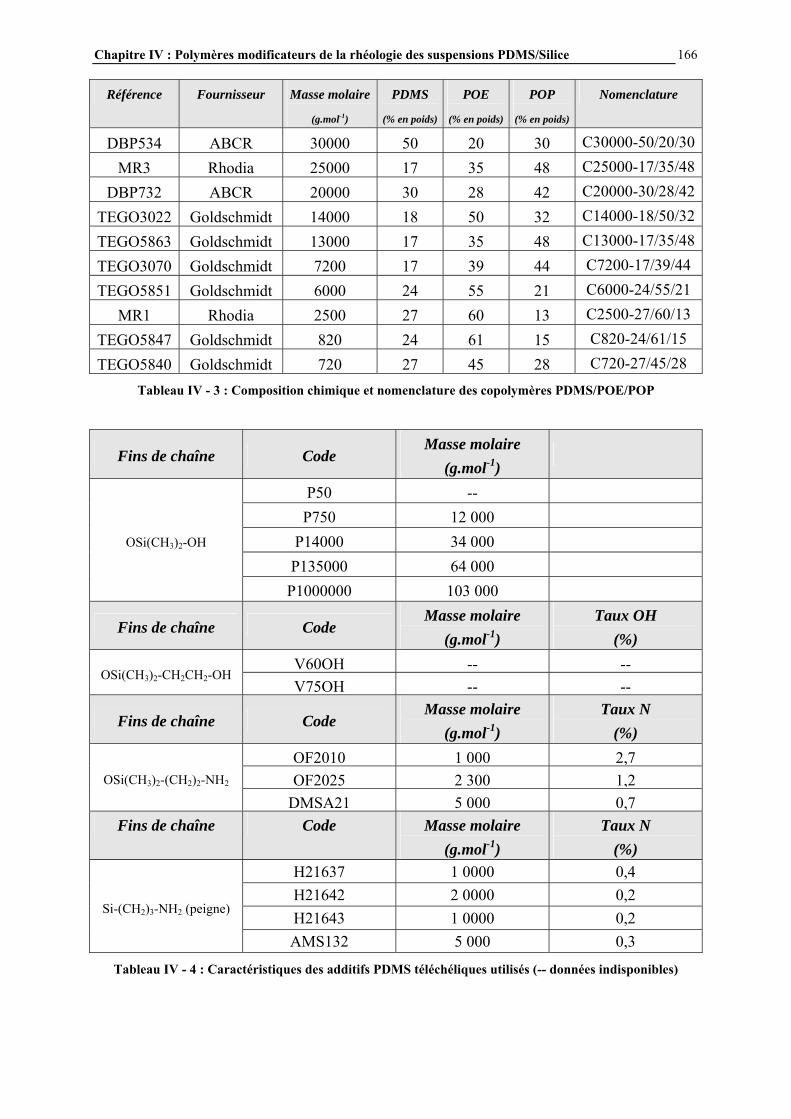
Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
166
Référence Fournisseur Masse molaire
(g.mol-1)
PDMS
(% en poids)
POE
(% en poids)
POP
(% en poids)
Nomenclature
DBP534 ABCR 30000 50 20 30 C30000-50/20/30MR3 Rhodia 25000 17 35 48 C25000-17/35/48
DBP732 ABCR 20000 30 28 42 C20000-30/28/42TEGO3022 Goldschmidt 14000 18 50 32 C14000-18/50/32TEGO5863 Goldschmidt 13000 17 35 48 C13000-17/35/48TEGO3070 Goldschmidt 7200 17 39 44 C7200-17/39/44 TEGO5851 Goldschmidt 6000 24 55 21 C6000-24/55/21
MR1 Rhodia 2500 27 60 13 C2500-27/60/13 TEGO5847 Goldschmidt 820 24 61 15 C820-24/61/15 TEGO5840 Goldschmidt 720 27 45 28 C720-27/45/28
Tableau IV - 3 : Composition chimique et nomenclature des copolymères PDMS/POE/POP
Fins de chaîne Code Masse molaire
(g.mol-1)
P50 -- P750 12 000
P14000 34 000 P135000 64 000
OSi(CH3)2-OH
P1000000 103 000
Fins de chaîne Code Masse molaire
(g.mol-1) Taux OH
(%) V60OH -- --
OSi(CH3)2-CH2CH2-OH V75OH -- --
Fins de chaîne Code Masse molaire
(g.mol-1) Taux N
(%) OF2010 1 000 2,7 OF2025 2 300 1,2 OSi(CH3)2-(CH2)2-NH2
DMSA21 5 000 0,7 Fins de chaîne Code Masse molaire
(g.mol-1) Taux N
(%) H21637 1 0000 0,4 H21642 2 0000 0,2 H21643 1 0000 0,2 Si-(CH2)3-NH2 (peigne)
AMS132 5 000 0,3
Tableau IV - 4 : Caractéristiques des additifs PDMS téléchéliques utilisés (-- données indisponibles)
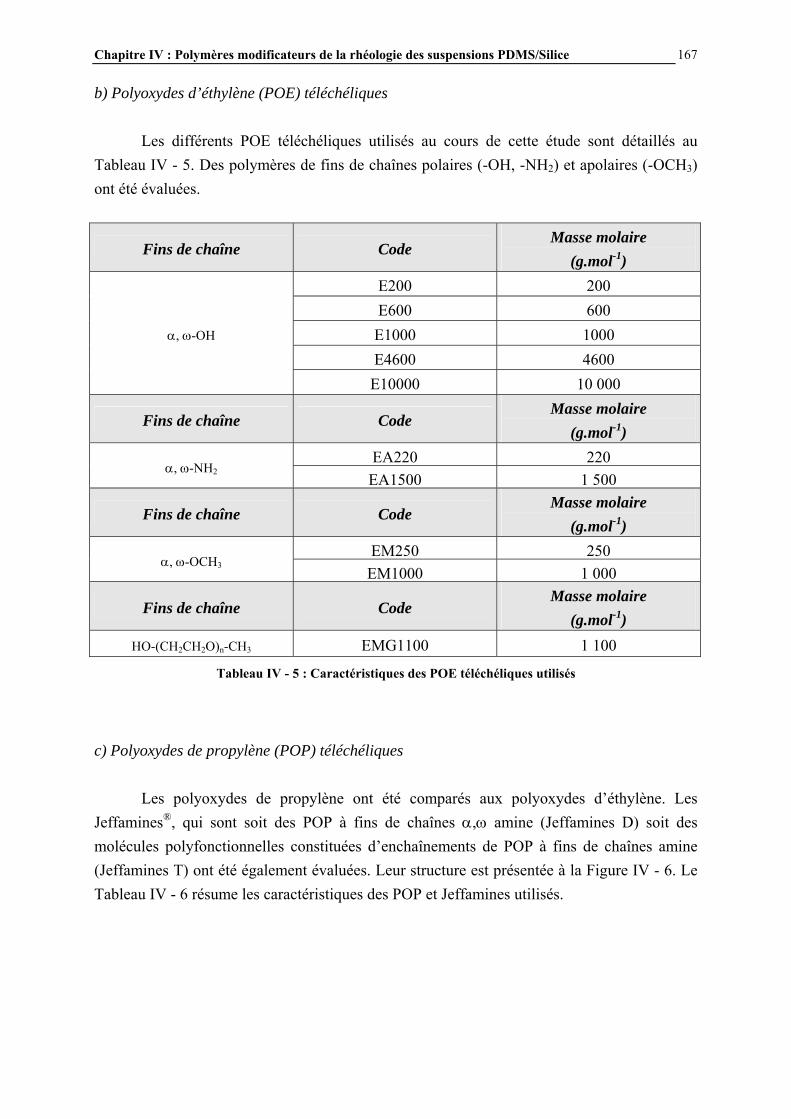
Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
167
b) Polyoxydes d’éthylène (POE) téléchéliques Les différents POE téléchéliques utilisés au cours de cette étude sont détaillés au Tableau IV - 5. Des polymères de fins de chaînes polaires (-OH, -NH2) et apolaires (-OCH3) ont été évaluées.
Fins de chaîne Code Masse molaire
(g.mol-1) E200 200 E600 600
E1000 1000 E4600 4600
α, ω-OH
E10000 10 000
Fins de chaîne Code Masse molaire
(g.mol-1) EA220 220
α, ω-NH2 EA1500 1 500
Fins de chaîne Code Masse molaire
(g.mol-1) EM250 250
α, ω-OCH3 EM1000 1 000
Fins de chaîne Code Masse molaire
(g.mol-1) HO-(CH2CH2O)n-CH3 EMG1100 1 100
Tableau IV - 5 : Caractéristiques des POE téléchéliques utilisés
c) Polyoxydes de propylène (POP) téléchéliques Les polyoxydes de propylène ont été comparés aux polyoxydes d’éthylène. Les Jeffamines®, qui sont soit des POP à fins de chaînes α,ω amine (Jeffamines D) soit des molécules polyfonctionnelles constituées d’enchaînements de POP à fins de chaînes amine (Jeffamines T) ont été également évaluées. Leur structure est présentée à la Figure IV - 6. Le Tableau IV - 6 résume les caractéristiques des POP et Jeffamines utilisés.
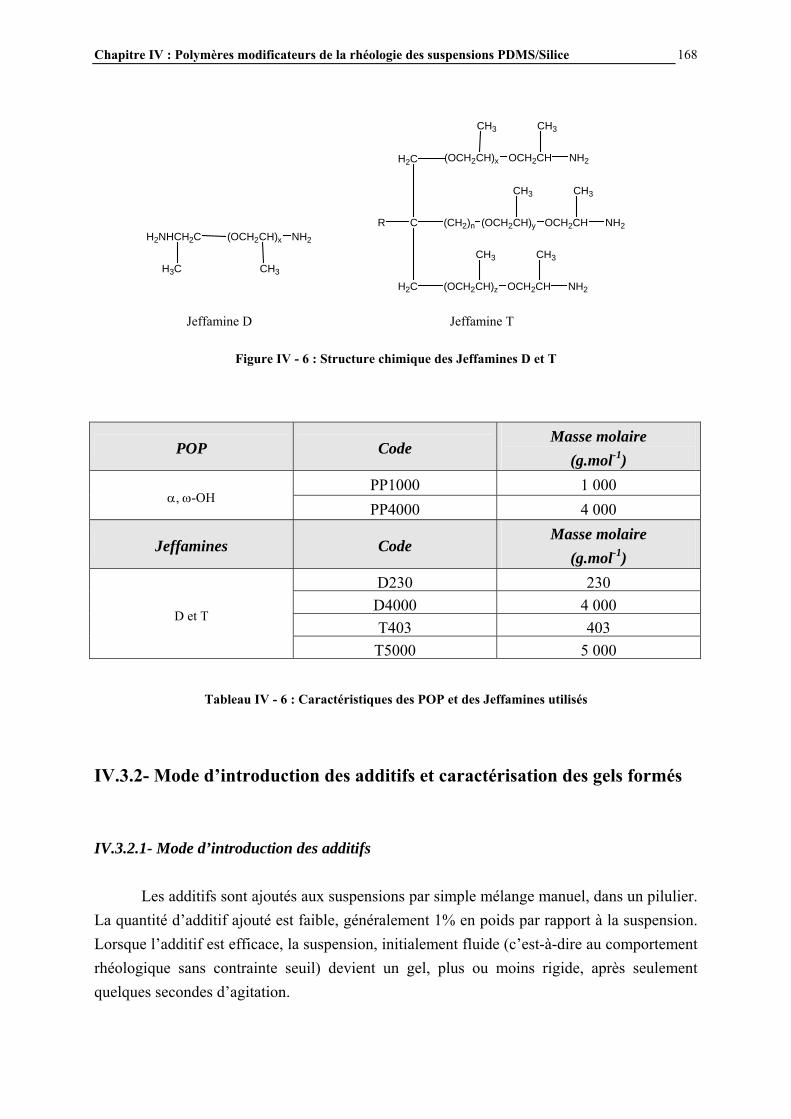
Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
168
Figure IV - 6 : Structure chimique des Jeffamines D et T
POP Code Masse molaire
(g.mol-1) PP1000 1 000
α, ω-OH PP4000 4 000
Jeffamines Code Masse molaire
(g.mol-1) D230 230
D4000 4 000 T403 403
D et T
T5000 5 000
Tableau IV - 6 : Caractéristiques des POP et des Jeffamines utilisés
IV.3.2- Mode d’introduction des additifs et caractérisation des gels formés IV.3.2.1- Mode d’introduction des additifs Les additifs sont ajoutés aux suspensions par simple mélange manuel, dans un pilulier. La quantité d’additif ajouté est faible, généralement 1% en poids par rapport à la suspension. Lorsque l’additif est efficace, la suspension, initialement fluide (c’est-à-dire au comportement rhéologique sans contrainte seuil) devient un gel, plus ou moins rigide, après seulement quelques secondes d’agitation.
H2NHCH2C (OCH2CH)x NH2
CH3H3C
(OCH2CH)x OCH2CH
C
H2C
(CH2)nR (OCH2CH)y OCH2CH
CH3 CH3
NH2
(OCH2CH)z
CH3
OCH2CH NH2
CH3
H2C
CH3
NH2
CH3
Jeffamine D Jeffamine T

Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
169
IV.3.2.2- Caractérisation des propriétés rhéologiques Les gels obtenus ont été caractérisés par des mesures rhéologiques en mode statique (mesures de contraintes seuil) et en mode dynamique (balayages en déformation et en fréquence), avec les mêmes méthodologies que celles décrites au chapitre III. Etant donné la plus forte viscosité des gels comparée à celle des suspensions initiales, une géométrie plan-plan a été principalement utilisée. IV.3.2.3- Caractérisation des morphologies La microstructure des gels a été observée à plusieurs niveaux d’échelle : aux faibles grossissements par microscopie optique (LEICA 12POLS) et microscopie optique confocale (LEICA PCS SP2), qui permet de travailler également en fluorescence. A des plus forts grossissements, la microscopie électronique à transmission a été employée, avec la même technique de préparation des échantillons que celle présentée au chapitre III. La préparation est facilitée dans le cas des gels en raison de leur viscosité initiale plus élevée.
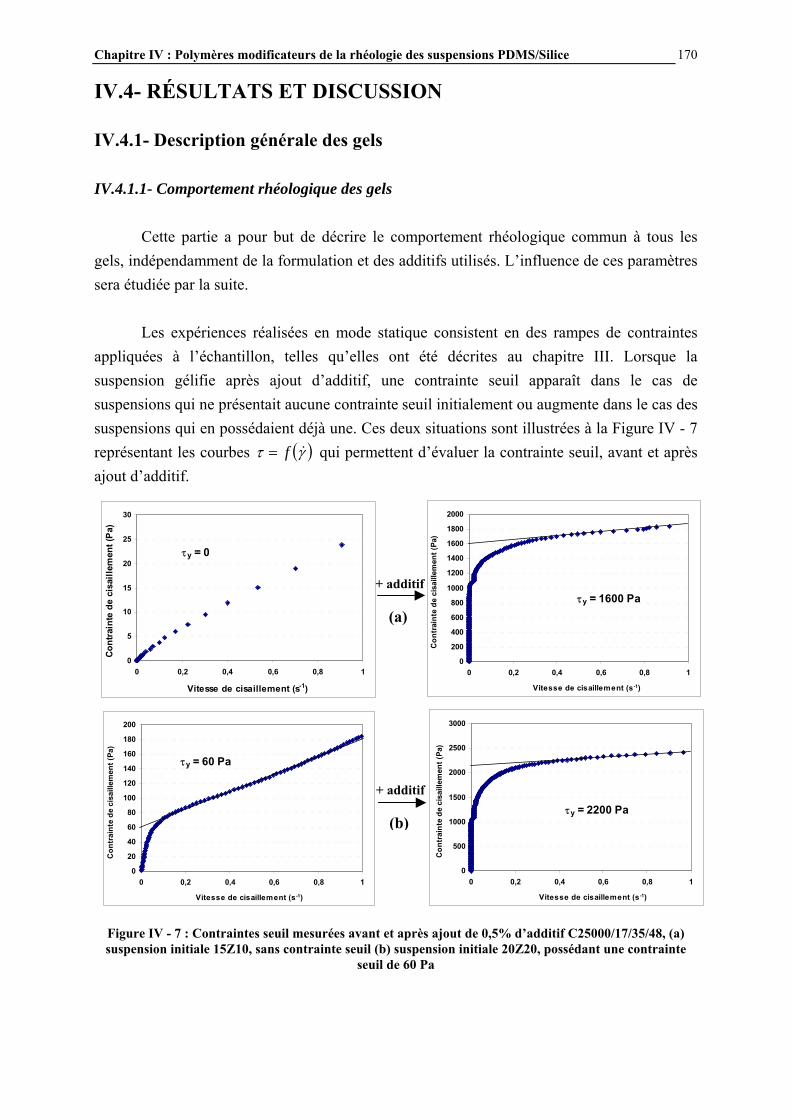
Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
170
IV.4- RÉSULTATS ET DISCUSSION IV.4.1- Description générale des gels IV.4.1.1- Comportement rhéologique des gels Cette partie a pour but de décrire le comportement rhéologique commun à tous les gels, indépendamment de la formulation et des additifs utilisés. L’influence de ces paramètres sera étudiée par la suite. Les expériences réalisées en mode statique consistent en des rampes de contraintes appliquées à l’échantillon, telles qu’elles ont été décrites au chapitre III. Lorsque la suspension gélifie après ajout d’additif, une contrainte seuil apparaît dans le cas de suspensions qui ne présentait aucune contrainte seuil initialement ou augmente dans le cas des suspensions qui en possédaient déjà une. Ces deux situations sont illustrées à la Figure IV - 7 représentant les courbes ( )γτ &f= qui permettent d’évaluer la contrainte seuil, avant et après ajout d’additif.
Figure IV - 7 : Contraintes seuil mesurées avant et après ajout de 0,5% d’additif C25000/17/35/48, (a) suspension initiale 15Z10, sans contrainte seuil (b) suspension initiale 20Z20, possédant une contrainte
seuil de 60 Pa
+ additif (a)
+ additif (b)
0
5
10
15
20
25
30
0 0,2 0,4 0,6 0,8 1
Vitesse de cisaillement (s-1)
Cont
rain
te d
e ci
saill
emen
t (Pa
)
τy = 0
0
200
400
600
800
1000
1200
1400
1600
1800
2000
0 0,2 0,4 0,6 0,8 1
Vitesse de cisaillement (s-1)
Con
trai
nte
de c
isai
llem
ent (
Pa)
τy = 1600 Pa
0
20
40
60
80
100
120
140
160
180
200
0 0,2 0,4 0,6 0,8 1
Vitesse de cisaillement (s-1)
Con
trai
nte
de c
isai
llem
ent (
Pa)
τy = 60 Pa
0
500
1000
1500
2000
2500
3000
0 0,2 0,4 0,6 0,8 1
Vitesse de cisaillement (s-1)
Con
trai
nte
de c
isai
llem
ent (
Pa)
τy = 2200 Pa
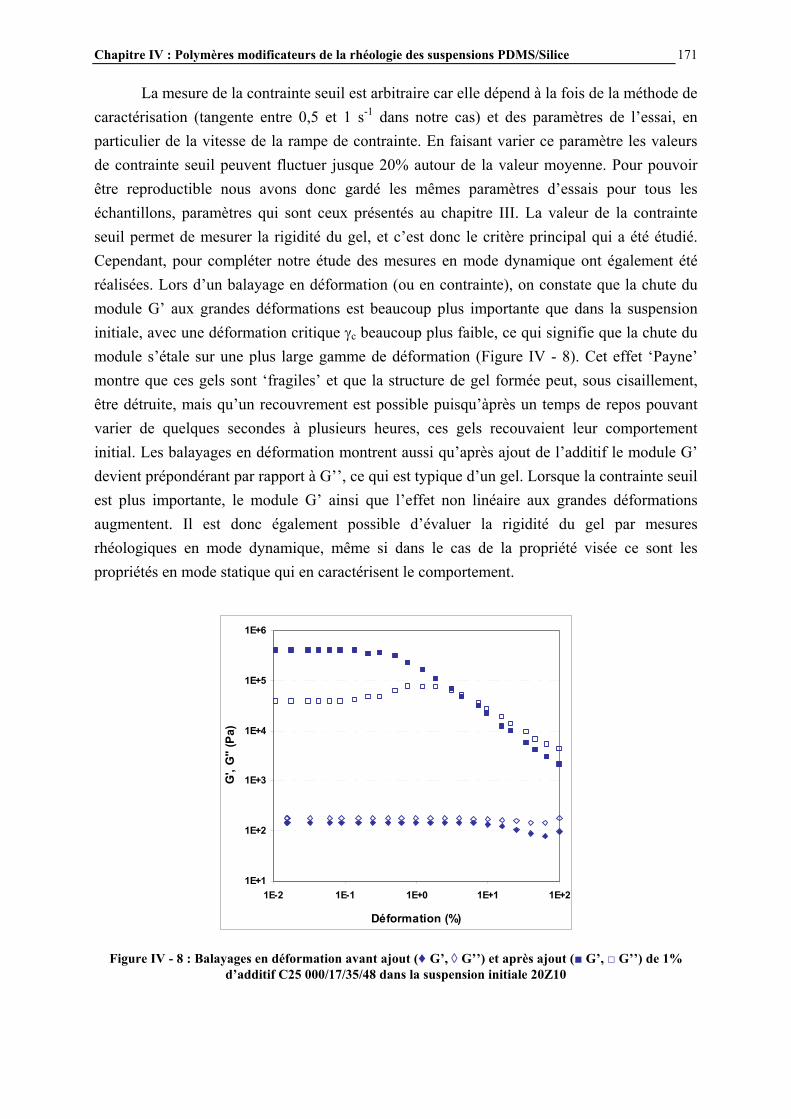
Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
171
La mesure de la contrainte seuil est arbitraire car elle dépend à la fois de la méthode de caractérisation (tangente entre 0,5 et 1 s-1 dans notre cas) et des paramètres de l’essai, en particulier de la vitesse de la rampe de contrainte. En faisant varier ce paramètre les valeurs de contrainte seuil peuvent fluctuer jusque 20% autour de la valeur moyenne. Pour pouvoir être reproductible nous avons donc gardé les mêmes paramètres d’essais pour tous les échantillons, paramètres qui sont ceux présentés au chapitre III. La valeur de la contrainte seuil permet de mesurer la rigidité du gel, et c’est donc le critère principal qui a été étudié. Cependant, pour compléter notre étude des mesures en mode dynamique ont également été réalisées. Lors d’un balayage en déformation (ou en contrainte), on constate que la chute du module G’ aux grandes déformations est beaucoup plus importante que dans la suspension initiale, avec une déformation critique γc beaucoup plus faible, ce qui signifie que la chute du module s’étale sur une plus large gamme de déformation (Figure IV - 8). Cet effet ‘Payne’ montre que ces gels sont ‘fragiles’ et que la structure de gel formée peut, sous cisaillement, être détruite, mais qu’un recouvrement est possible puisqu’àprès un temps de repos pouvant varier de quelques secondes à plusieurs heures, ces gels recouvaient leur comportement initial. Les balayages en déformation montrent aussi qu’après ajout de l’additif le module G’ devient prépondérant par rapport à G’’, ce qui est typique d’un gel. Lorsque la contrainte seuil est plus importante, le module G’ ainsi que l’effet non linéaire aux grandes déformations augmentent. Il est donc également possible d’évaluer la rigidité du gel par mesures rhéologiques en mode dynamique, même si dans le cas de la propriété visée ce sont les propriétés en mode statique qui en caractérisent le comportement.
Figure IV - 8 : Balayages en déformation avant ajout (♦ G’, ◊ G’’) et après ajout (■ G’, □ G’’) de 1% d’additif C25 000/17/35/48 dans la suspension initiale 20Z10
1E+1
1E+2
1E+3
1E+4
1E+5
1E+6
1E-2 1E-1 1E+0 1E+1 1E+2
Déformation (%)
G',
G''
(Pa)
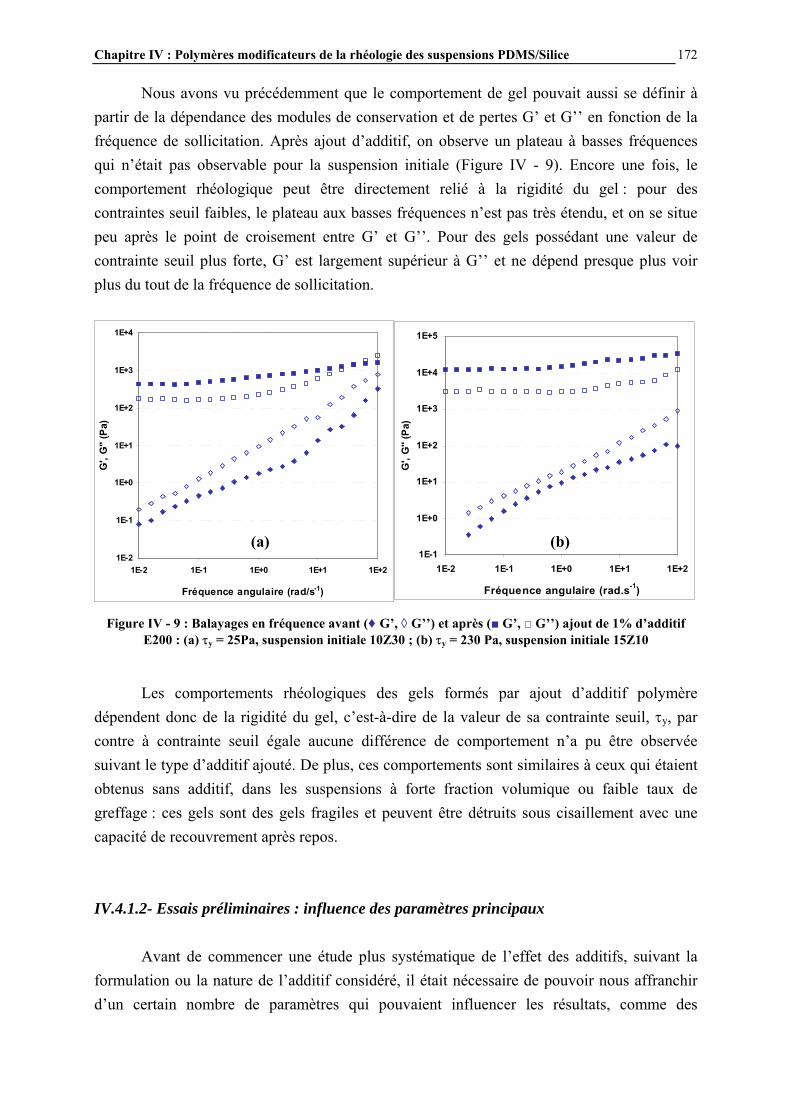
Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
172
Nous avons vu précédemment que le comportement de gel pouvait aussi se définir à partir de la dépendance des modules de conservation et de pertes G’ et G’’ en fonction de la fréquence de sollicitation. Après ajout d’additif, on observe un plateau à basses fréquences qui n’était pas observable pour la suspension initiale (Figure IV - 9). Encore une fois, le comportement rhéologique peut être directement relié à la rigidité du gel : pour des contraintes seuil faibles, le plateau aux basses fréquences n’est pas très étendu, et on se situe peu après le point de croisement entre G’ et G’’. Pour des gels possédant une valeur de contrainte seuil plus forte, G’ est largement supérieur à G’’ et ne dépend presque plus voir plus du tout de la fréquence de sollicitation.
Figure IV - 9 : Balayages en fréquence avant (♦ G’, ◊ G’’) et après (■ G’, □ G’’) ajout de 1% d’additif E200 : (a) τy = 25Pa, suspension initiale 10Z30 ; (b) τy = 230 Pa, suspension initiale 15Z10
Les comportements rhéologiques des gels formés par ajout d’additif polymère dépendent donc de la rigidité du gel, c’est-à-dire de la valeur de sa contrainte seuil, τy, par contre à contrainte seuil égale aucune différence de comportement n’a pu être observée suivant le type d’additif ajouté. De plus, ces comportements sont similaires à ceux qui étaient obtenus sans additif, dans les suspensions à forte fraction volumique ou faible taux de greffage : ces gels sont des gels fragiles et peuvent être détruits sous cisaillement avec une capacité de recouvrement après repos. IV.4.1.2- Essais préliminaires : influence des paramètres principaux Avant de commencer une étude plus systématique de l’effet des additifs, suivant la formulation ou la nature de l’additif considéré, il était nécessaire de pouvoir nous affranchir d’un certain nombre de paramètres qui pouvaient influencer les résultats, comme des
1E-1
1E+0
1E+1
1E+2
1E+3
1E+4
1E+5
1E-2 1E-1 1E+0 1E+1 1E+2
Fréquence angulaire (rad.s-1)
G',
G''
(Pa)
1E-2
1E-1
1E+0
1E+1
1E+2
1E+3
1E+4
1E-2 1E-1 1E+0 1E+1 1E+2
Fréquence angulaire (rad/s-1)
G',
G''
(Pa)
(a) (b)
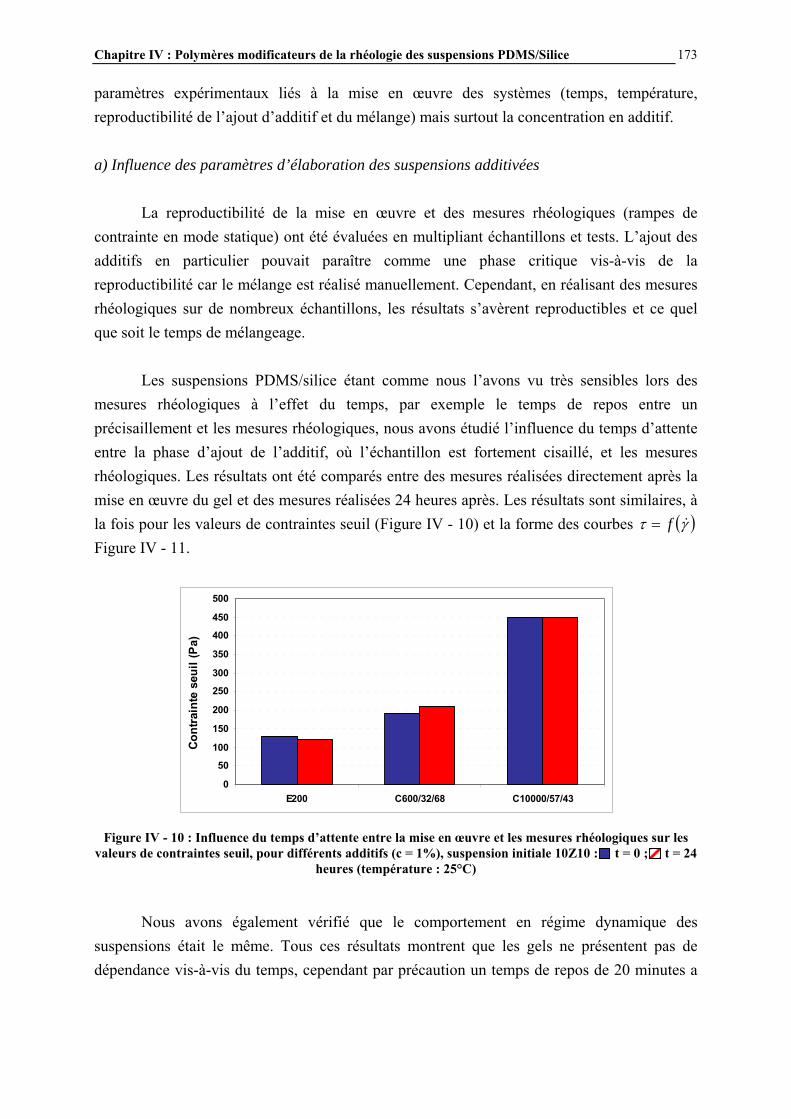
Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
173
paramètres expérimentaux liés à la mise en œuvre des systèmes (temps, température, reproductibilité de l’ajout d’additif et du mélange) mais surtout la concentration en additif. a) Influence des paramètres d’élaboration des suspensions additivées La reproductibilité de la mise en œuvre et des mesures rhéologiques (rampes de contrainte en mode statique) ont été évaluées en multipliant échantillons et tests. L’ajout des additifs en particulier pouvait paraître comme une phase critique vis-à-vis de la reproductibilité car le mélange est réalisé manuellement. Cependant, en réalisant des mesures rhéologiques sur de nombreux échantillons, les résultats s’avèrent reproductibles et ce quel que soit le temps de mélangeage. Les suspensions PDMS/silice étant comme nous l’avons vu très sensibles lors des mesures rhéologiques à l’effet du temps, par exemple le temps de repos entre un précisaillement et les mesures rhéologiques, nous avons étudié l’influence du temps d’attente entre la phase d’ajout de l’additif, où l’échantillon est fortement cisaillé, et les mesures rhéologiques. Les résultats ont été comparés entre des mesures réalisées directement après la mise en œuvre du gel et des mesures réalisées 24 heures après. Les résultats sont similaires, à la fois pour les valeurs de contraintes seuil (Figure IV - 10) et la forme des courbes ( )γτ &f= Figure IV - 11.
Figure IV - 10 : Influence du temps d’attente entre la mise en œuvre et les mesures rhéologiques sur les valeurs de contraintes seuil, pour différents additifs (c = 1%), suspension initiale 10Z10 : t = 0 ; t = 24
heures (température : 25°C)
Nous avons également vérifié que le comportement en régime dynamique des suspensions était le même. Tous ces résultats montrent que les gels ne présentent pas de dépendance vis-à-vis du temps, cependant par précaution un temps de repos de 20 minutes a
0
50
100
150
200
250
300
350
400
450
500
E200 C600/32/68 C10000/57/43
Cont
rain
te s
euil
(Pa)
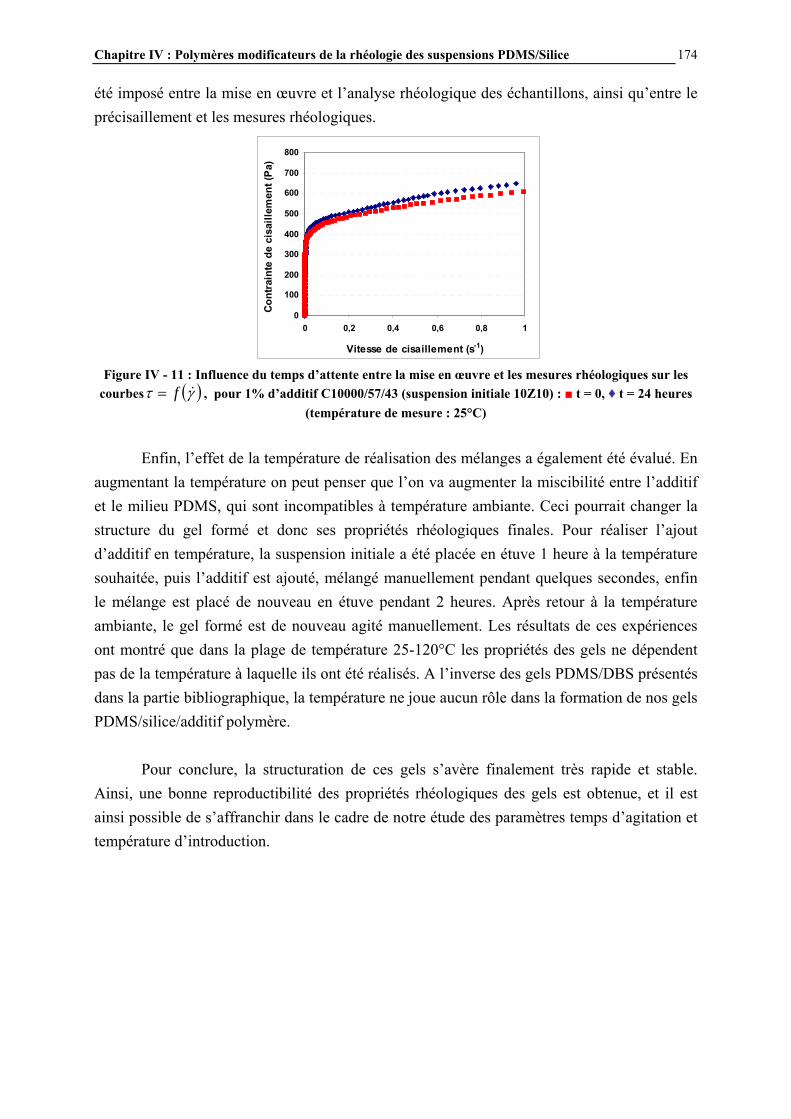
Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
174
été imposé entre la mise en œuvre et l’analyse rhéologique des échantillons, ainsi qu’entre le précisaillement et les mesures rhéologiques.
Figure IV - 11 : Influence du temps d’attente entre la mise en œuvre et les mesures rhéologiques sur les courbes ( )γτ &f= , pour 1% d’additif C10000/57/43 (suspension initiale 10Z10) : ■ t = 0, ♦ t = 24 heures
(température de mesure : 25°C)
Enfin, l’effet de la température de réalisation des mélanges a également été évalué. En augmentant la température on peut penser que l’on va augmenter la miscibilité entre l’additif et le milieu PDMS, qui sont incompatibles à température ambiante. Ceci pourrait changer la structure du gel formé et donc ses propriétés rhéologiques finales. Pour réaliser l’ajout d’additif en température, la suspension initiale a été placée en étuve 1 heure à la température souhaitée, puis l’additif est ajouté, mélangé manuellement pendant quelques secondes, enfin le mélange est placé de nouveau en étuve pendant 2 heures. Après retour à la température ambiante, le gel formé est de nouveau agité manuellement. Les résultats de ces expériences ont montré que dans la plage de température 25-120°C les propriétés des gels ne dépendent pas de la température à laquelle ils ont été réalisés. A l’inverse des gels PDMS/DBS présentés dans la partie bibliographique, la température ne joue aucun rôle dans la formation de nos gels PDMS/silice/additif polymère. Pour conclure, la structuration de ces gels s’avère finalement très rapide et stable. Ainsi, une bonne reproductibilité des propriétés rhéologiques des gels est obtenue, et il est ainsi possible de s’affranchir dans le cadre de notre étude des paramètres temps d’agitation et température d’introduction.
0
100
200
300
400
500
600
700
800
0 0,2 0,4 0,6 0,8 1
Vitesse de cisaillement (s-1)
Con
trai
nte
de c
isai
llem
ent (
Pa)

Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
175
b) Influence de la concentration en additif Les valeurs de contrainte seuil reportées plus loin sont les valeurs obtenues pour des concentrations en additif de 1% en poids par rapport au poids de la suspension. Cependant, la variation de la contrainte seuil en fonction de la concentration en additif doit être prise en compte car la valeur maximum de la contrainte seuil n’est pas forcément obtenue pour la même concentration en additif, suivant les formulations et le type d’additif. Nous avons étudié l’évolution de la contrainte seuil en fonction de la concentration en additif pour deux copolymères PDMS/POE C620-30/70 et C10000-57/43/0 et un homopolymère POE, E200, pour différentes formulations (Figure IV - 12, Figure IV - 13 et Figure IV - 14). La concentration en additif est exprimée en pourcentage en poids par rapport à la suspension initiale. Un échantillon différent a été réalisé pour chaque concentration en additif. La dépendance de la contrainte seuil avec la concentration en additif polymère présente un maximum, qui correspond à la contrainte seuil maximale, caractéristique d’une formulation et d’un additif. Ce maximum intervient pour une concentration critique très faible, de 0,2 à 5%. Deux comportements se distinguent suivant la nature de l’additif: dans le cas des homopolymères la concentration critique est très faible (0,2%) et la valeur de la contrainte seuil est constante après avoir atteint son maximum. Dans le cas des copolymères, la concentration critique est généralement plus élevée et la contrainte seuil diminue pour les concentrations élevées. Ce phénomène peut s’expliquer par une séparation de phase PDMS/additif. En effet, une fois que le maximum d’additif s’est adsorbé à la surface de la silice par l’intermédiaire des groupements POE, l’excès d’additif restant se trouve dans le milieu PDMS avec lequel il n’est pas miscible, et se sépare sous forme de particules d’additif. Ce phénomène de séparation de phase a été confirmé par la turbidité du gel, habituellement transparent, qui devient trouble (diffusion de la lumière par les particules micrométriques d’additif). En faisant l’hypothèse que la contrainte seuil maximale est atteinte lorsque tous les sites hydroxyle de surface de la silice sont saturés par adsorption du POE, la concentration critique devrait diminuer lorsque le nombre de sites silanol disponibles est plus faible, c'est-à-dire pour des fractions volumiques plus faibles ou des taux de greffage plus élevés. Ce type de corrélation n’a cependant pas pu être déduite de nos résultats, ainsi la valeur de la concentration critique ne semble pas pouvoir être déduite de manière simple suivant la formulation ou l’additif utilisé. Pour la suite de l’étude, nous avons considéré une concentration de 1% en additif, valeur la plus proche de l’ensemble des concentrations critiques (c’est-à-dire pour lesquelles une valeur maximale de contrainte seuil est atteinte).
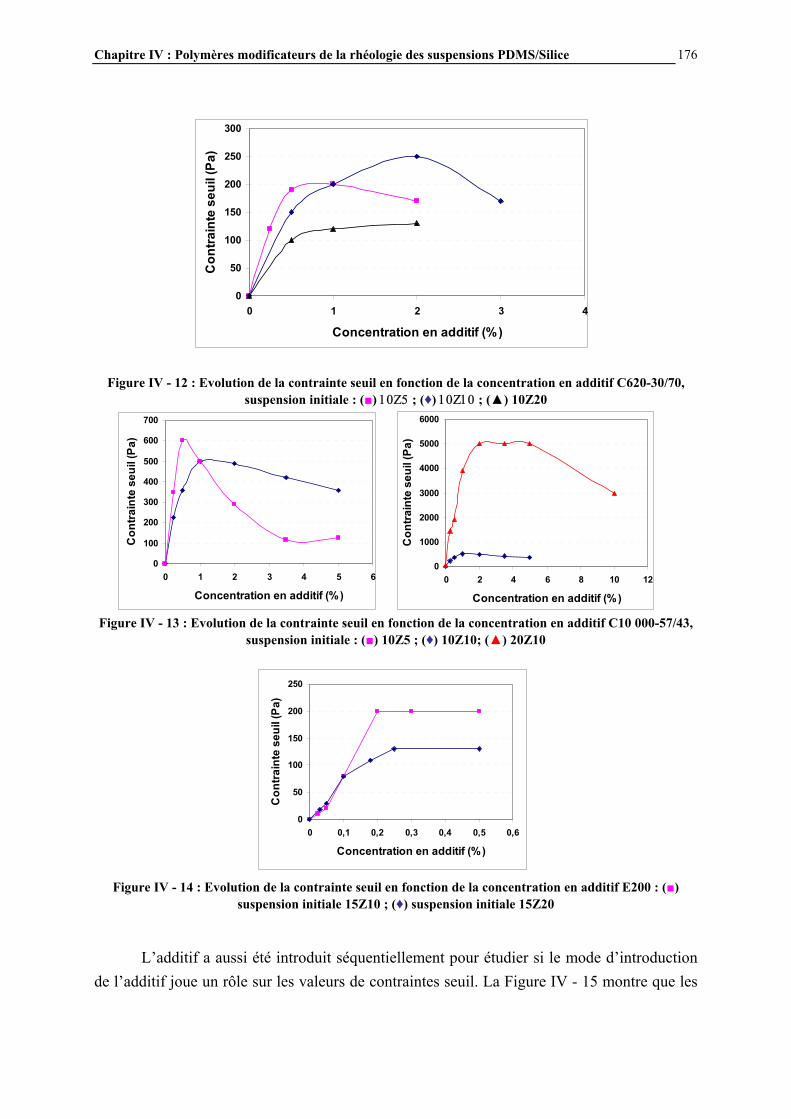
Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
176
Figure IV - 12 : Evolution de la contrainte seuil en fonction de la concentration en additif C620-30/70, suspension initiale : (■) 10Ζ5 ; (♦) 10Ζ10 ; (▲) 10Z20
Figure IV - 13 : Evolution de la contrainte seuil en fonction de la concentration en additif C10 000-57/43,
suspension initiale : (■) 10Z5 ; (♦) 10Z10; (▲) 20Z10
Figure IV - 14 : Evolution de la contrainte seuil en fonction de la concentration en additif E200 : (■) suspension initiale 15Z10 ; (♦) suspension initiale 15Z20
L’additif a aussi été introduit séquentiellement pour étudier si le mode d’introduction de l’additif joue un rôle sur les valeurs de contraintes seuil. La Figure IV - 15 montre que les
0
50
100
150
200
250
300
0 1 2 3 4
Concentration en additif (%)
Con
train
te s
euil
(Pa)
0
100
200
300
400
500
600
700
0 1 2 3 4 5 6
Concentration en additif (%)
Con
train
te s
euil
(Pa)
0
1000
2000
3000
4000
5000
6000
0 2 4 6 8 10 12
Concentration en additif (%)
Con
train
te s
euil
(Pa)
0
50
100
150
200
250
0 0,1 0,2 0,3 0,4 0,5 0,6
Concentration en additif (%)
Con
train
te s
euil
(Pa)
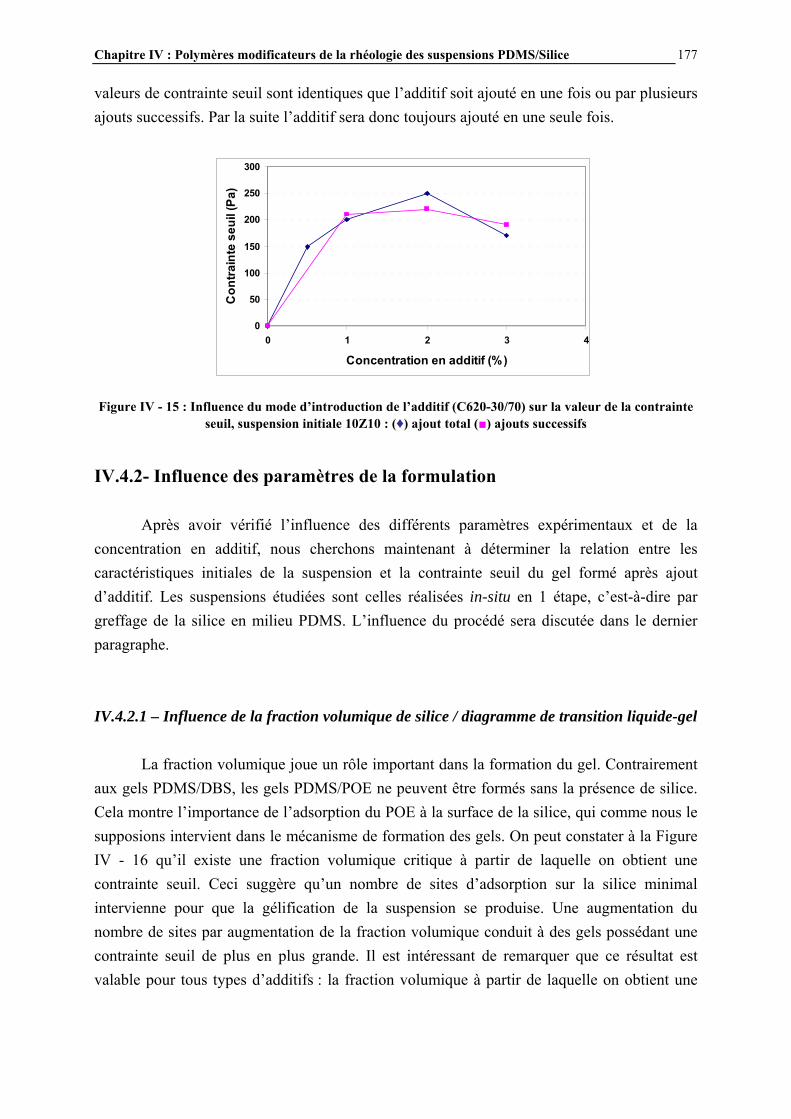
Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
177
valeurs de contrainte seuil sont identiques que l’additif soit ajouté en une fois ou par plusieurs ajouts successifs. Par la suite l’additif sera donc toujours ajouté en une seule fois. Figure IV - 15 : Influence du mode d’introduction de l’additif (C620-30/70) sur la valeur de la contrainte
seuil, suspension initiale 10Z10 : (♦) ajout total (■) ajouts successifs
IV.4.2- Influence des paramètres de la formulation Après avoir vérifié l’influence des différents paramètres expérimentaux et de la concentration en additif, nous cherchons maintenant à déterminer la relation entre les caractéristiques initiales de la suspension et la contrainte seuil du gel formé après ajout d’additif. Les suspensions étudiées sont celles réalisées in-situ en 1 étape, c’est-à-dire par greffage de la silice en milieu PDMS. L’influence du procédé sera discutée dans le dernier paragraphe. IV.4.2.1 – Influence de la fraction volumique de silice / diagramme de transition liquide-gel La fraction volumique joue un rôle important dans la formation du gel. Contrairement aux gels PDMS/DBS, les gels PDMS/POE ne peuvent être formés sans la présence de silice. Cela montre l’importance de l’adsorption du POE à la surface de la silice, qui comme nous le supposions intervient dans le mécanisme de formation des gels. On peut constater à la Figure IV - 16 qu’il existe une fraction volumique critique à partir de laquelle on obtient une contrainte seuil. Ceci suggère qu’un nombre de sites d’adsorption sur la silice minimal intervienne pour que la gélification de la suspension se produise. Une augmentation du nombre de sites par augmentation de la fraction volumique conduit à des gels possédant une contrainte seuil de plus en plus grande. Il est intéressant de remarquer que ce résultat est valable pour tous types d’additifs : la fraction volumique à partir de laquelle on obtient une
0
50
100
150
200
250
300
0 1 2 3 4
Concentration en additif (%)
Con
train
te s
euil
(Pa)
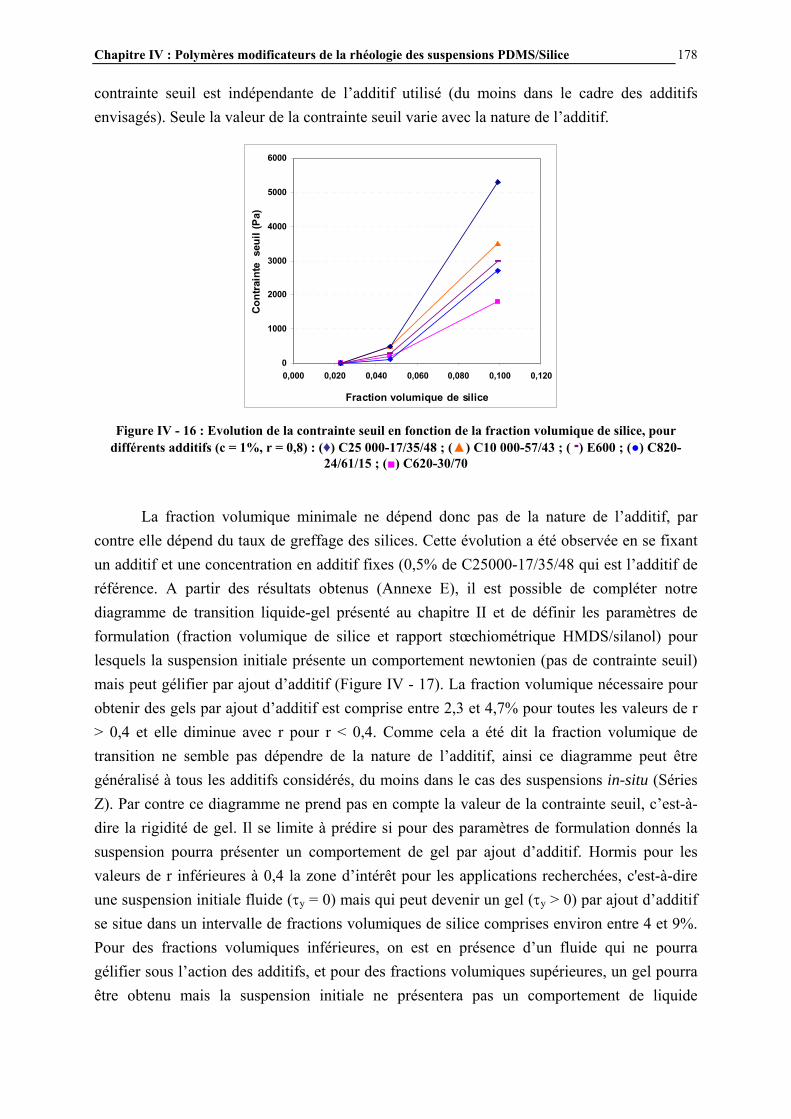
Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
178
contrainte seuil est indépendante de l’additif utilisé (du moins dans le cadre des additifs envisagés). Seule la valeur de la contrainte seuil varie avec la nature de l’additif.
Figure IV - 16 : Evolution de la contrainte seuil en fonction de la fraction volumique de silice, pour différents additifs (c = 1%, r = 0,8) : (♦) C25 000-17/35/48 ; (▲) C10 000-57/43 ; ( ־) E600 ; (●) C820-
24/61/15 ; (■) C620-30/70
La fraction volumique minimale ne dépend donc pas de la nature de l’additif, par contre elle dépend du taux de greffage des silices. Cette évolution a été observée en se fixant un additif et une concentration en additif fixes (0,5% de C25000-17/35/48 qui est l’additif de référence. A partir des résultats obtenus (Annexe E), il est possible de compléter notre diagramme de transition liquide-gel présenté au chapitre II et de définir les paramètres de formulation (fraction volumique de silice et rapport stœchiométrique HMDS/silanol) pour lesquels la suspension initiale présente un comportement newtonien (pas de contrainte seuil) mais peut gélifier par ajout d’additif (Figure IV - 17). La fraction volumique nécessaire pour obtenir des gels par ajout d’additif est comprise entre 2,3 et 4,7% pour toutes les valeurs de r > 0,4 et elle diminue avec r pour r < 0,4. Comme cela a été dit la fraction volumique de transition ne semble pas dépendre de la nature de l’additif, ainsi ce diagramme peut être généralisé à tous les additifs considérés, du moins dans le cas des suspensions in-situ (Séries Z). Par contre ce diagramme ne prend pas en compte la valeur de la contrainte seuil, c’est-à-dire la rigidité de gel. Il se limite à prédire si pour des paramètres de formulation donnés la suspension pourra présenter un comportement de gel par ajout d’additif. Hormis pour les valeurs de r inférieures à 0,4 la zone d’intérêt pour les applications recherchées, c'est-à-dire une suspension initiale fluide (τy = 0) mais qui peut devenir un gel (τy > 0) par ajout d’additif se situe dans un intervalle de fractions volumiques de silice comprises environ entre 4 et 9%. Pour des fractions volumiques inférieures, on est en présence d’un fluide qui ne pourra gélifier sous l’action des additifs, et pour des fractions volumiques supérieures, un gel pourra être obtenu mais la suspension initiale ne présentera pas un comportement de liquide
0
1000
2000
3000
4000
5000
6000
0,000 0,020 0,040 0,060 0,080 0,100 0,120
Fraction volumique de silice
Cont
rain
te s
euil
(Pa)
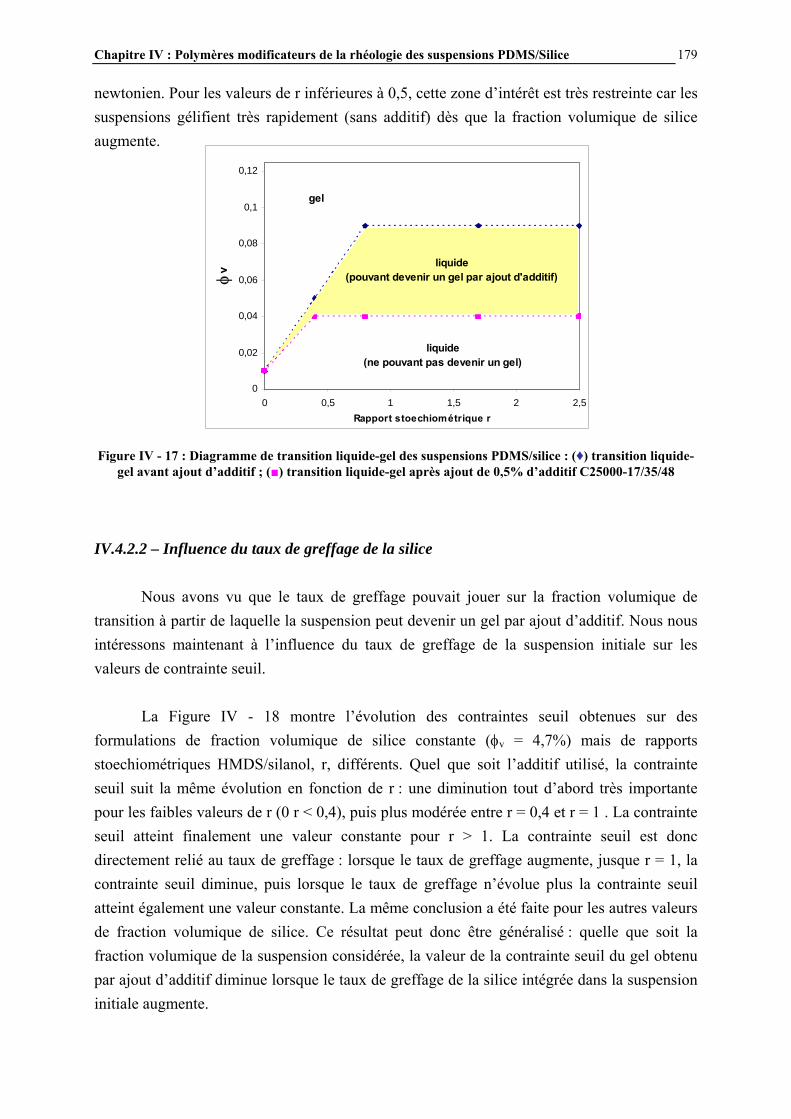
Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
179
newtonien. Pour les valeurs de r inférieures à 0,5, cette zone d’intérêt est très restreinte car les suspensions gélifient très rapidement (sans additif) dès que la fraction volumique de silice augmente.
Figure IV - 17 : Diagramme de transition liquide-gel des suspensions PDMS/silice : (♦) transition liquide-gel avant ajout d’additif ; (■) transition liquide-gel après ajout de 0,5% d’additif C25000-17/35/48
IV.4.2.2 – Influence du taux de greffage de la silice Nous avons vu que le taux de greffage pouvait jouer sur la fraction volumique de transition à partir de laquelle la suspension peut devenir un gel par ajout d’additif. Nous nous intéressons maintenant à l’influence du taux de greffage de la suspension initiale sur les valeurs de contrainte seuil. La Figure IV - 18 montre l’évolution des contraintes seuil obtenues sur des formulations de fraction volumique de silice constante (φv = 4,7%) mais de rapports stoechiométriques HMDS/silanol, r, différents. Quel que soit l’additif utilisé, la contrainte seuil suit la même évolution en fonction de r : une diminution tout d’abord très importante pour les faibles valeurs de r (0 r < 0,4), puis plus modérée entre r = 0,4 et r = 1 . La contrainte seuil atteint finalement une valeur constante pour r > 1. La contrainte seuil est donc directement relié au taux de greffage : lorsque le taux de greffage augmente, jusque r = 1, la contrainte seuil diminue, puis lorsque le taux de greffage n’évolue plus la contrainte seuil atteint également une valeur constante. La même conclusion a été faite pour les autres valeurs de fraction volumique de silice. Ce résultat peut donc être généralisé : quelle que soit la fraction volumique de la suspension considérée, la valeur de la contrainte seuil du gel obtenu par ajout d’additif diminue lorsque le taux de greffage de la silice intégrée dans la suspension initiale augmente.
0
0,02
0,04
0,06
0,08
0,1
0,12
0 0,5 1 1,5 2 2,5Rapport stoechiométrique r
φv
gel
liquide(ne pouvant pas devenir un gel)
liquide(pouvant devenir un gel par ajout d'additif)
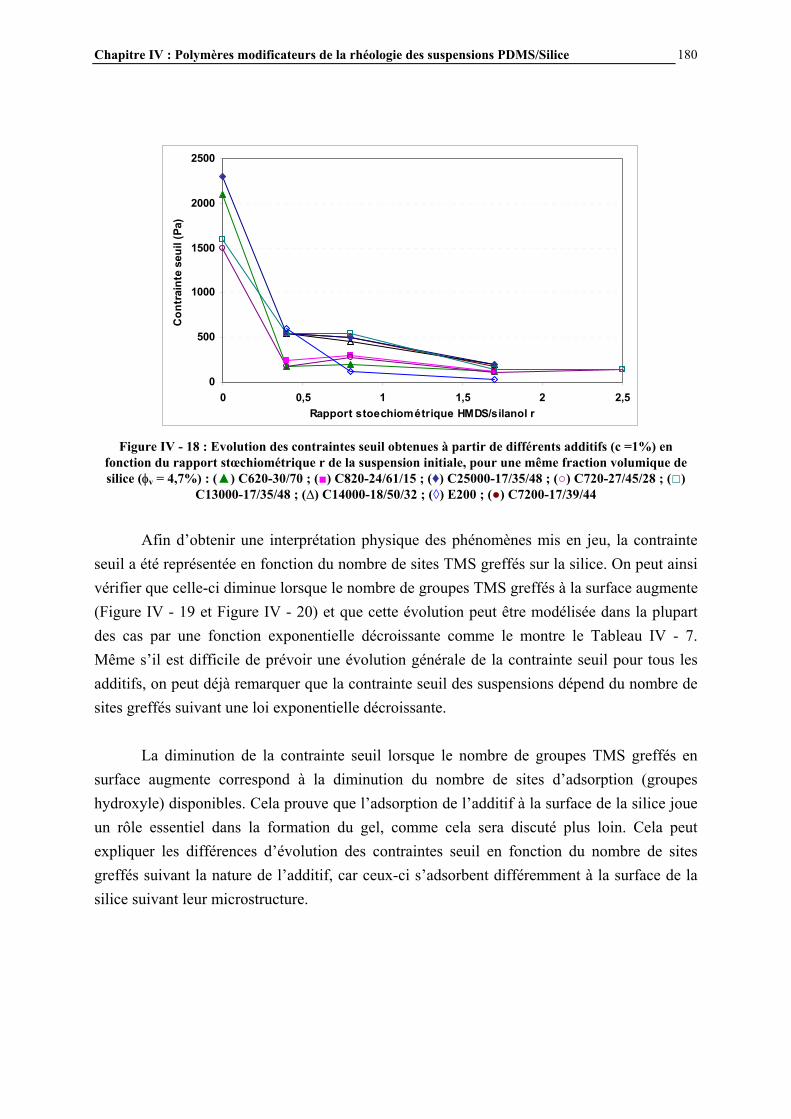
Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
180
Figure IV - 18 : Evolution des contraintes seuil obtenues à partir de différents additifs (c =1%) en fonction du rapport stœchiométrique r de la suspension initiale, pour une même fraction volumique de silice (φv = 4,7%) : (▲) C620-30/70 ; (■) C820-24/61/15 ; (♦) C25000-17/35/48 ; (○) C720-27/45/28 ; (□)
C13000-17/35/48 ; (∆) C14000-18/50/32 ; (◊) E200 ; (●) C7200-17/39/44
Afin d’obtenir une interprétation physique des phénomènes mis en jeu, la contrainte seuil a été représentée en fonction du nombre de sites TMS greffés sur la silice. On peut ainsi vérifier que celle-ci diminue lorsque le nombre de groupes TMS greffés à la surface augmente (Figure IV - 19 et Figure IV - 20) et que cette évolution peut être modélisée dans la plupart des cas par une fonction exponentielle décroissante comme le montre le Tableau IV - 7. Même s’il est difficile de prévoir une évolution générale de la contrainte seuil pour tous les additifs, on peut déjà remarquer que la contrainte seuil des suspensions dépend du nombre de sites greffés suivant une loi exponentielle décroissante. La diminution de la contrainte seuil lorsque le nombre de groupes TMS greffés en surface augmente correspond à la diminution du nombre de sites d’adsorption (groupes hydroxyle) disponibles. Cela prouve que l’adsorption de l’additif à la surface de la silice joue un rôle essentiel dans la formation du gel, comme cela sera discuté plus loin. Cela peut expliquer les différences d’évolution des contraintes seuil en fonction du nombre de sites greffés suivant la nature de l’additif, car ceux-ci s’adsorbent différemment à la surface de la silice suivant leur microstructure.
0
500
1000
1500
2000
2500
0 0,5 1 1,5 2 2,5Rapport stoechiométrique HMDS/silanol r
Con
trai
nte
seui
l (Pa
)
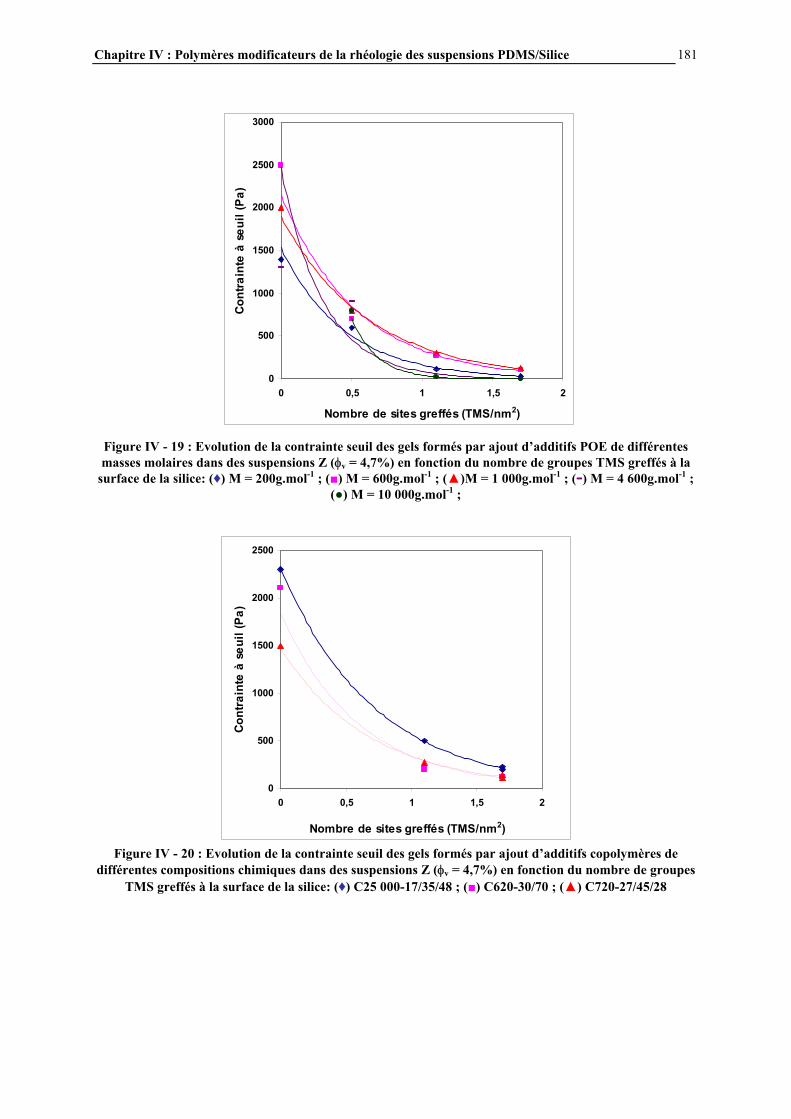
Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
181
Figure IV - 19 : Evolution de la contrainte seuil des gels formés par ajout d’additifs POE de différentes masses molaires dans des suspensions Z (φv = 4,7%) en fonction du nombre de groupes TMS greffés à la
surface de la silice: (♦) M = 200g.mol-1 ; (■) M = 600g.mol-1 ; (▲)M = 1 000g.mol-1 ; (-) M = 4 600g.mol-1 ; (●) M = 10 000g.mol-1 ;
Figure IV - 20 : Evolution de la contrainte seuil des gels formés par ajout d’additifs copolymères de différentes compositions chimiques dans des suspensions Z (φv = 4,7%) en fonction du nombre de groupes
TMS greffés à la surface de la silice: (♦) C25 000-17/35/48 ; (■) C620-30/70 ; (▲) C720-27/45/28
0
500
1000
1500
2000
2500
3000
0 0,5 1 1,5 2
Nombre de sites greffés (TMS/nm2)
Con
train
te à
seu
il (P
a)
0
500
1000
1500
2000
2500
0 0,5 1 1,5 2
Nombre de sites greffés (TMS/nm2)
Cont
rain
te à
seu
il (P
a)
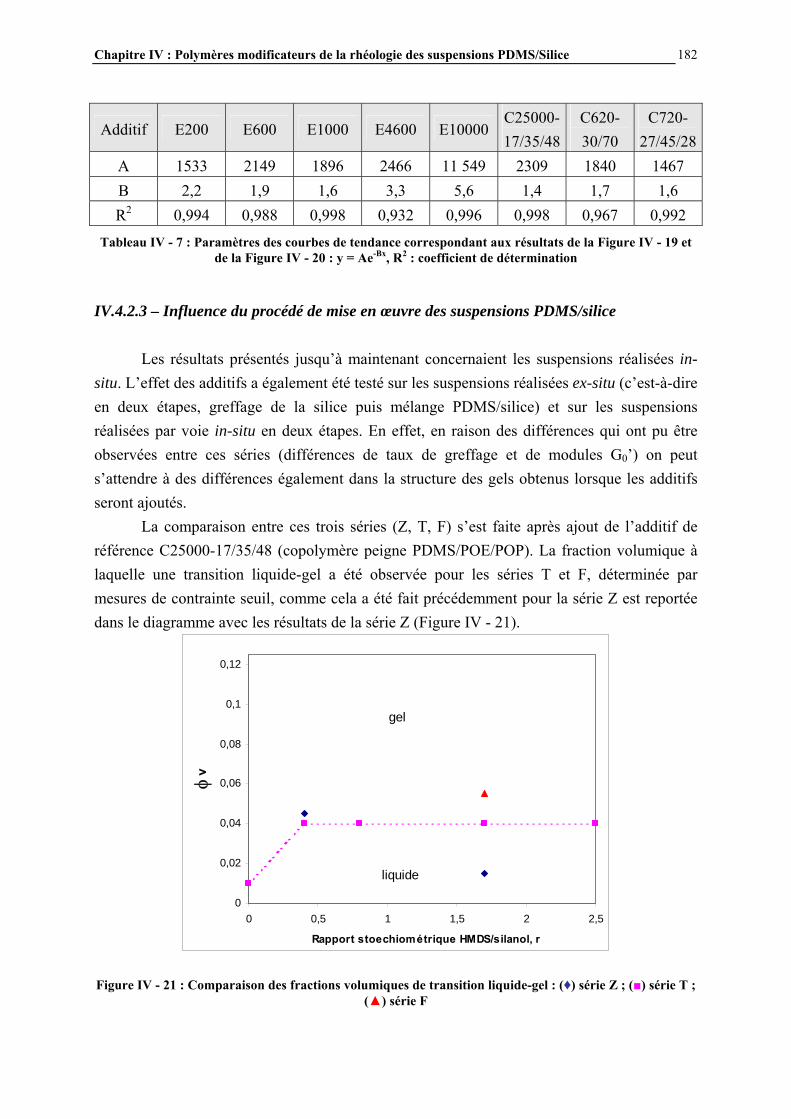
Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
182
Additif E200 E600 E1000 E4600 E10000 C25000-17/35/48
C620-30/70
C720-27/45/28
A 1533 2149 1896 2466 11 549 2309 1840 1467 B 2,2 1,9 1,6 3,3 5,6 1,4 1,7 1,6 R2 0,994 0,988 0,998 0,932 0,996 0,998 0,967 0,992
Tableau IV - 7 : Paramètres des courbes de tendance correspondant aux résultats de la Figure IV - 19 et de la Figure IV - 20 : y = Ae-Bx, R2 : coefficient de détermination
IV.4.2.3 – Influence du procédé de mise en œuvre des suspensions PDMS/silice Les résultats présentés jusqu’à maintenant concernaient les suspensions réalisées in-situ. L’effet des additifs a également été testé sur les suspensions réalisées ex-situ (c’est-à-dire en deux étapes, greffage de la silice puis mélange PDMS/silice) et sur les suspensions réalisées par voie in-situ en deux étapes. En effet, en raison des différences qui ont pu être observées entre ces séries (différences de taux de greffage et de modules G0’) on peut s’attendre à des différences également dans la structure des gels obtenus lorsque les additifs seront ajoutés. La comparaison entre ces trois séries (Z, T, F) s’est faite après ajout de l’additif de référence C25000-17/35/48 (copolymère peigne PDMS/POE/POP). La fraction volumique à laquelle une transition liquide-gel a été observée pour les séries T et F, déterminée par mesures de contrainte seuil, comme cela a été fait précédemment pour la série Z est reportée dans le diagramme avec les résultats de la série Z (Figure IV - 21).
Figure IV - 21 : Comparaison des fractions volumiques de transition liquide-gel : (♦) série Z ; (■) série T ; (▲) série F
0
0,02
0,04
0,06
0,08
0,1
0,12
0 0,5 1 1,5 2 2,5
Rapport stoechiométrique HMDS/silanol, r
φv
gel
liquide

Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
183
Pour r = 0,4, c’est-à-dire une hydrophobisation partielle de la silice, la série T présente une fraction volumique de transition voisine de celle de la série Z, ce qui paraît cohérent avec les observations faites au chapitre III indiquant qu’aux faibles taux de greffage, il n’y a pas de différences de taux de greffage ni de module G0’ entre ces deux séries. La fraction volumique de transition de la série F n’a pas pu être calculée car nous ne disposons que d’un seul échantillon à ce taux de greffage. Pour r = 1,7 des différences entre les séries Z et T apparaissent. La fraction volumique de transition s’effectue à des fractions volumiques de silice significativement plus basses dans le cas des suspensions T où les silices sont greffées ex-situ : même à la plus basse fraction volumique de 2,3%, la suspension peut devenir un gel par ajout d’additif. Ce comportement surprenant peut s’expliquer par le plus faible taux de greffage des silices modifiées ex-situ, comme nous l’avons montré, cependant la différence de comportement face aux additifs de cette suspension reste très importante par rapport à la différence de taux de greffage somme toute modérée entre les séries Z et T. De plus, il est surprenant de voir que la fraction volumique de transition pour la série T est plus faible lorsque le taux de greffage est plus élevé, ce qui est contraire aux observations faites sur la série Z. Cependant, le module G0’ des suspensions T à r = 1,7 était plus élevé que ces mêmes suspensions à r = 0,4. Ceci expliquerait que le gel soit atteint pour des fractions volumiques plus faibles à r = 1,7. En ce qui concerne la série F, c’est-à-dire les suspensions où la silice est greffée in-situ en deux étapes, avec ajout de l’HMDS après le mélange PDMS/silice, la transition liquide-gel intervient pour une fraction volumique similaire mais tout de même légèrement supérieure. Ce résultat se corrèle très bien avec nos observations précédentes où le module G0’ était légèrement plus faible pour ce type de suspensions, ce qui laissait supposer un taux de greffage légèrement supérieur. La transition interviendrait donc pour une fraction volumique supérieure en raison de ce taux de greffage plus élevé (en comparaison avec la série Z). Ces résultats permettent de confirmer qu’il existe des différences de comportement entre les trois séries de suspensions Z, T et F, et montrent encore une fois l’importance du taux de greffage sur la gélification des suspensions par ajout d’additif polymère. Par ailleurs, il est maintenant possible d’entrevoir une interprétation sur le comportement particulier des suspensions industrielles qui ne gélifient pas sous l’effet des additifs polymère. En effet, dans ces suspensions le taux de greffage de la silice est supposé plus élevé que dans les autres formulations industrielles qui peuvent gélifier [1]. Cette différence de taux de greffage pourrait expliquer ce comportement particulier ; nous en reparlerons après avoir étudié plus en détail les mécanismes de formation des gels.
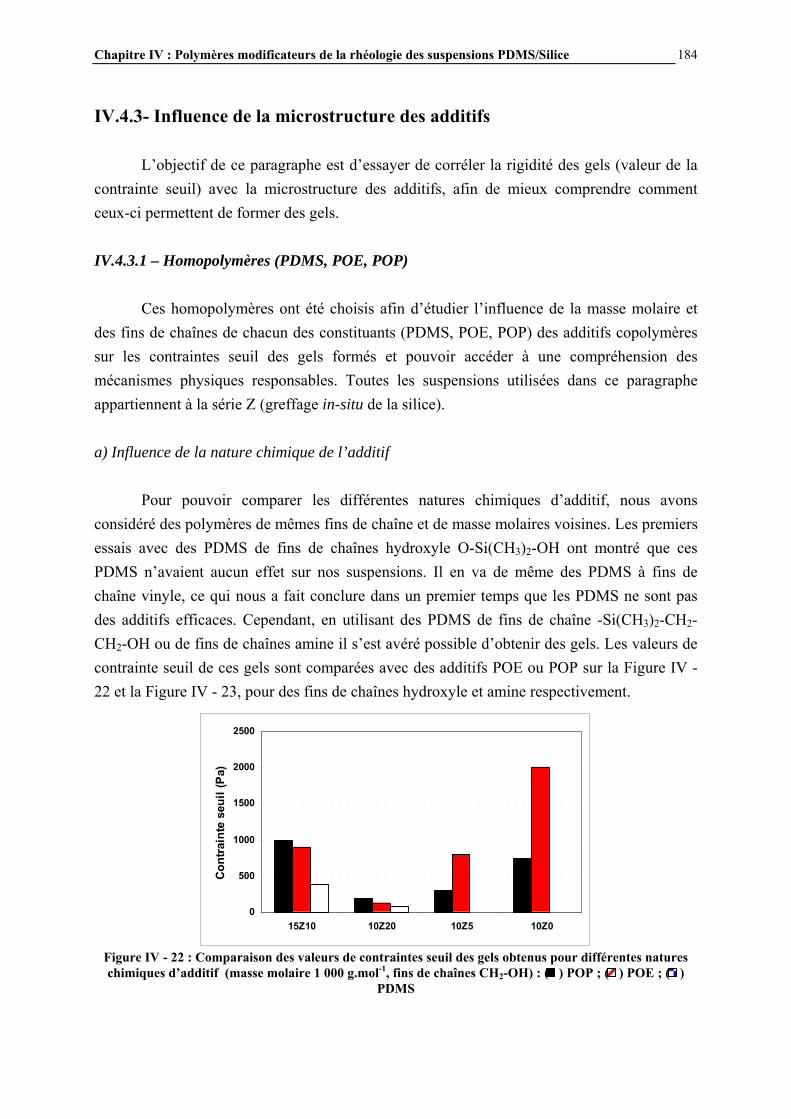
Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
184
IV.4.3- Influence de la microstructure des additifs L’objectif de ce paragraphe est d’essayer de corréler la rigidité des gels (valeur de la contrainte seuil) avec la microstructure des additifs, afin de mieux comprendre comment ceux-ci permettent de former des gels. IV.4.3.1 – Homopolymères (PDMS, POE, POP) Ces homopolymères ont été choisis afin d’étudier l’influence de la masse molaire et des fins de chaînes de chacun des constituants (PDMS, POE, POP) des additifs copolymères sur les contraintes seuil des gels formés et pouvoir accéder à une compréhension des mécanismes physiques responsables. Toutes les suspensions utilisées dans ce paragraphe appartiennent à la série Z (greffage in-situ de la silice). a) Influence de la nature chimique de l’additif Pour pouvoir comparer les différentes natures chimiques d’additif, nous avons considéré des polymères de mêmes fins de chaîne et de masse molaires voisines. Les premiers essais avec des PDMS de fins de chaînes hydroxyle O-Si(CH3)2-OH ont montré que ces PDMS n’avaient aucun effet sur nos suspensions. Il en va de même des PDMS à fins de chaîne vinyle, ce qui nous a fait conclure dans un premier temps que les PDMS ne sont pas des additifs efficaces. Cependant, en utilisant des PDMS de fins de chaîne -Si(CH3)2-CH2-CH2-OH ou de fins de chaînes amine il s’est avéré possible d’obtenir des gels. Les valeurs de contrainte seuil de ces gels sont comparées avec des additifs POE ou POP sur la Figure IV - 22 et la Figure IV - 23, pour des fins de chaînes hydroxyle et amine respectivement.
Figure IV - 22 : Comparaison des valeurs de contraintes seuil des gels obtenus pour différentes natures chimiques d’additif (masse molaire 1 000 g.mol-1, fins de chaînes CH2-OH) : ( ) POP ; ( ) POE ; ( )
PDMS
0
500
1000
1500
2000
2500
15Z10 10Z20 10Z5 10Z0
Con
train
te s
euil
(Pa)
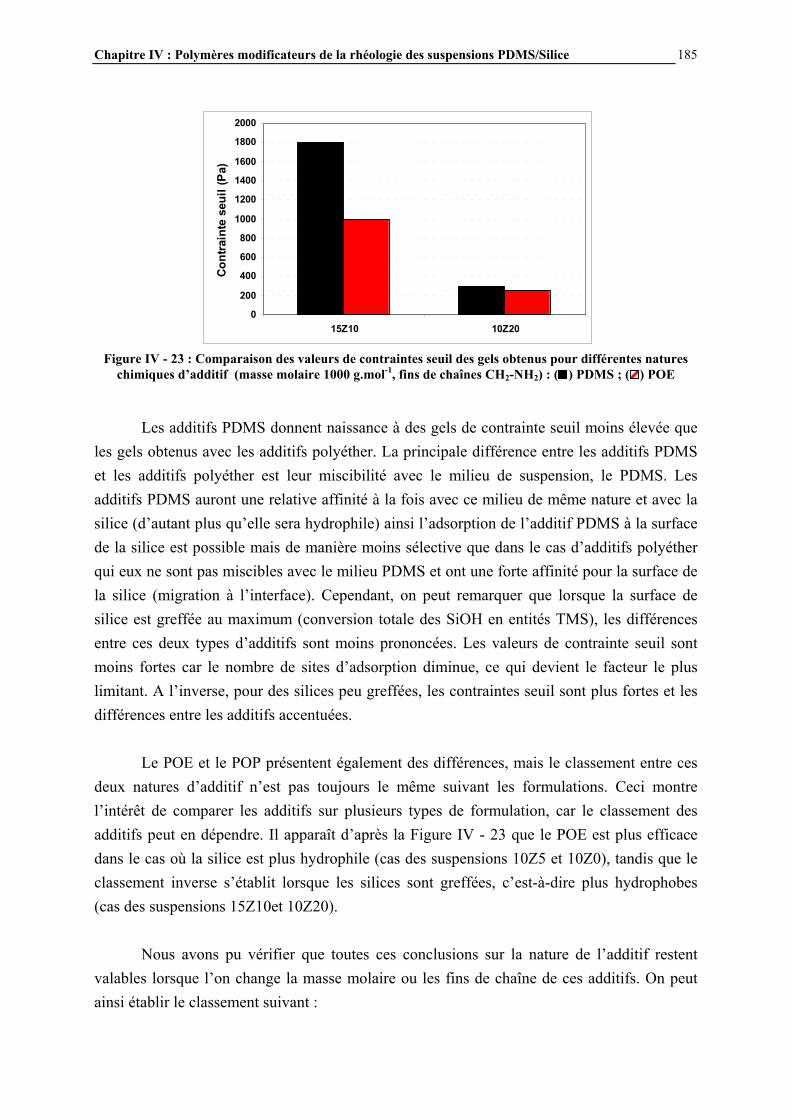
Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
185
Figure IV - 23 : Comparaison des valeurs de contraintes seuil des gels obtenus pour différentes natures chimiques d’additif (masse molaire 1000 g.mol-1, fins de chaînes CH2-NH2) : ( ) PDMS ; ( ) POE
Les additifs PDMS donnent naissance à des gels de contrainte seuil moins élevée que les gels obtenus avec les additifs polyéther. La principale différence entre les additifs PDMS et les additifs polyéther est leur miscibilité avec le milieu de suspension, le PDMS. Les additifs PDMS auront une relative affinité à la fois avec ce milieu de même nature et avec la silice (d’autant plus qu’elle sera hydrophile) ainsi l’adsorption de l’additif PDMS à la surface de la silice est possible mais de manière moins sélective que dans le cas d’additifs polyéther qui eux ne sont pas miscibles avec le milieu PDMS et ont une forte affinité pour la surface de la silice (migration à l’interface). Cependant, on peut remarquer que lorsque la surface de silice est greffée au maximum (conversion totale des SiOH en entités TMS), les différences entre ces deux types d’additifs sont moins prononcées. Les valeurs de contrainte seuil sont moins fortes car le nombre de sites d’adsorption diminue, ce qui devient le facteur le plus limitant. A l’inverse, pour des silices peu greffées, les contraintes seuil sont plus fortes et les différences entre les additifs accentuées. Le POE et le POP présentent également des différences, mais le classement entre ces deux natures d’additif n’est pas toujours le même suivant les formulations. Ceci montre l’intérêt de comparer les additifs sur plusieurs types de formulation, car le classement des additifs peut en dépendre. Il apparaît d’après la Figure IV - 23 que le POE est plus efficace dans le cas où la silice est plus hydrophile (cas des suspensions 10Z5 et 10Z0), tandis que le classement inverse s’établit lorsque les silices sont greffées, c’est-à-dire plus hydrophobes (cas des suspensions 15Z10et 10Z20). Nous avons pu vérifier que toutes ces conclusions sur la nature de l’additif restent valables lorsque l’on change la masse molaire ou les fins de chaîne de ces additifs. On peut ainsi établir le classement suivant :
0
200
400
600
800
1000
1200
1400
1600
1800
2000
15Z10 10Z20
Con
trai
nte
seui
l (P
a)

Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
186
PDMS<POP<POE lorsque la silice n’est pas ou peu greffée (r ≤ 0,4)
PDMS<POE<POP lorsque la silice est fortement greffée (r > 0,8) b) Influence des fins de chaîne de l’additif Dans le cas des additifs PDMS, l’influence des fins de chaîne est nette puisque comme nous l’avons vu des PDMS à fins de chaîne amine ou hydroxyle (CH2CH2OH) permettent d’obtenir des gels, alors que des PDMS à fins de chaîne vinyle ou hydroxyle (OSi(CH3)2-OH) s’avèrent inefficaces. Ceci montre que dans le cas des PDMS la fonctionnalité du polymère est prépondérante sur l’effet de la nature chimique de la chaîne macromoléculaire. Lorsque les fins de chaînes sont polaires, l’affinité de l’additif PDMS pour la surface de la silice est certainement accrue, ce qui pourrait expliquer ces différences de comportement. En ce qui concerne les additifs PDMS qui conduisent à des comportements de gels, les valeurs de contrainte seuil obtenues ont été comparées en fonction de la nature de la fin de chaîne. Les PDMS à fins de chaîne amine, en fins de chaînes α,ω ou ramifications (peignes) présentent des valeurs de contrainte seuil largement plus élevées que les PDMS à fins de chaîne hydroxyle (Figure IV - 24). Cette différence suggère une affinité plus forte de ces polymères vers la surface de la silice, et ce quel que soit le taux de greffage de la silice. On en déduit une plus grande affinité des groupements amine que des groupements hydroxyle vis-à-vis des surfaces de silice. Ce point sera développé plus tard dans la discussion. L’influence de la fonctionnalité du polymère est moins importante dans le cas des polyéthers (Figure IV - 25). Seuls les POE à fins de chaîne amine se détachent largement des autres types de fins de chaîne, et ce même pour les plus forts taux de greffage où la contrainte seuil chute considérablement. Ce résultat confirme que les groupes amine ont des interactions très fortes avec la silice, plus fortes même que des groupements hydroxyle. Hormis les POE à fins de chaîne amine, les autres types de fonctionnalité ne modifient pas notablement la valeur de la contrainte seuil, ce qui peut paraître surprenant. En particulier des fins de chaînes polaires (OH) et apolaires (OCH3) ont le même effet. Ces résultats peuvent s’expliquer par le rôle plus important du squelette de la chaîne qui participe aussi à l’adsorption des molécules de POE à la surface de la silice, via les oxygènes du motif OCH2CH2. Cependant, en faisant l’hypothèse que le rôle des fins de chaînes est négligeable devant le rôle du squelette de la chaîne, il est difficile à ce moment d’expliquer pourquoi les POE à fins de chaînes amine ont un effet accru sur la contrainte seuil. En fait la microstructure de la chaîne et les fins de chaînes jouent certainement tous les deux un rôle important dans le mécanisme de formation des gels.
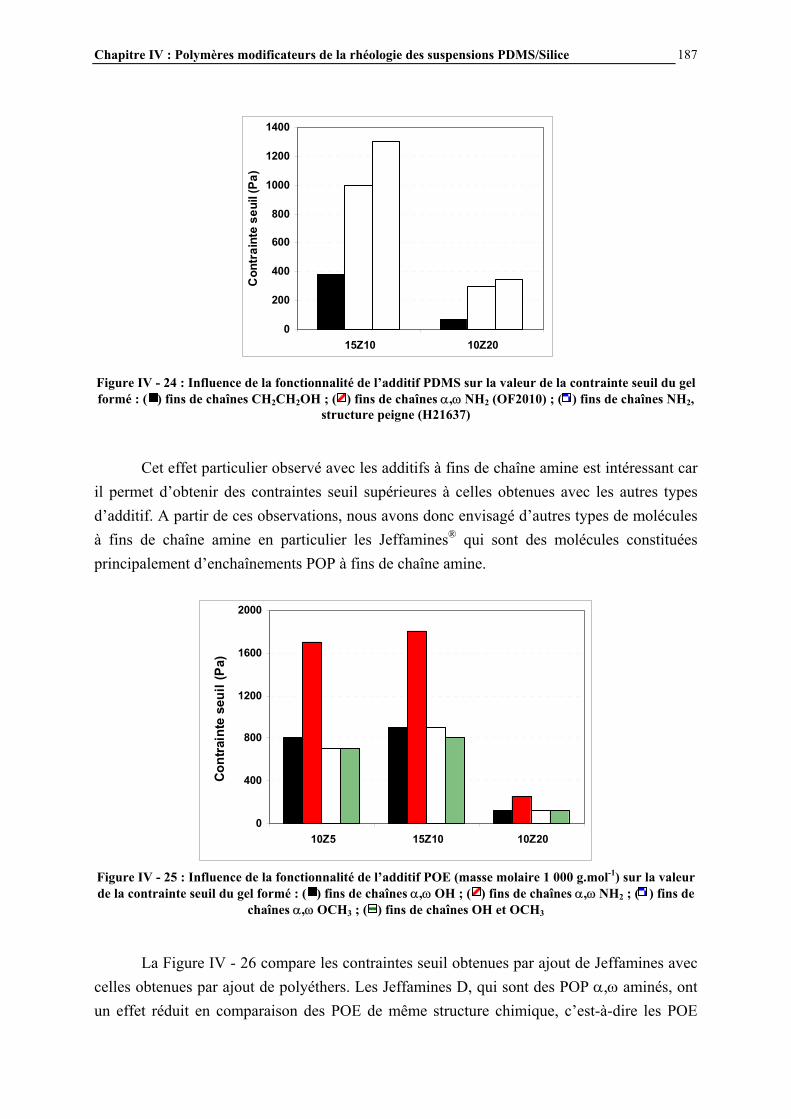
Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
187
Figure IV - 24 : Influence de la fonctionnalité de l’additif PDMS sur la valeur de la contrainte seuil du gel formé : ( ) fins de chaînes CH2CH2OH ; ( ) fins de chaînes α,ω NH2 (OF2010) ; ( ) fins de chaînes NH2,
structure peigne (H21637)
Cet effet particulier observé avec les additifs à fins de chaîne amine est intéressant car il permet d’obtenir des contraintes seuil supérieures à celles obtenues avec les autres types d’additif. A partir de ces observations, nous avons donc envisagé d’autres types de molécules à fins de chaîne amine en particulier les Jeffamines® qui sont des molécules constituées principalement d’enchaînements POP à fins de chaîne amine. Figure IV - 25 : Influence de la fonctionnalité de l’additif POE (masse molaire 1 000 g.mol-1) sur la valeur de la contrainte seuil du gel formé : ( ) fins de chaînes α,ω OH ; ( ) fins de chaînes α,ω NH2 ; ( ) fins de
chaînes α,ω OCH3 ; ( ) fins de chaînes OH et OCH3
La Figure IV - 26 compare les contraintes seuil obtenues par ajout de Jeffamines avec celles obtenues par ajout de polyéthers. Les Jeffamines D, qui sont des POP α,ω aminés, ont un effet réduit en comparaison des POE de même structure chimique, c’est-à-dire les POE
0
200
400
600
800
1000
1200
1400
15Z10 10Z20
Con
trai
nte
seui
l (Pa
)
0
400
800
1200
1600
2000
10Z5 15Z10 10Z20
Cont
rain
te s
euil
(Pa)
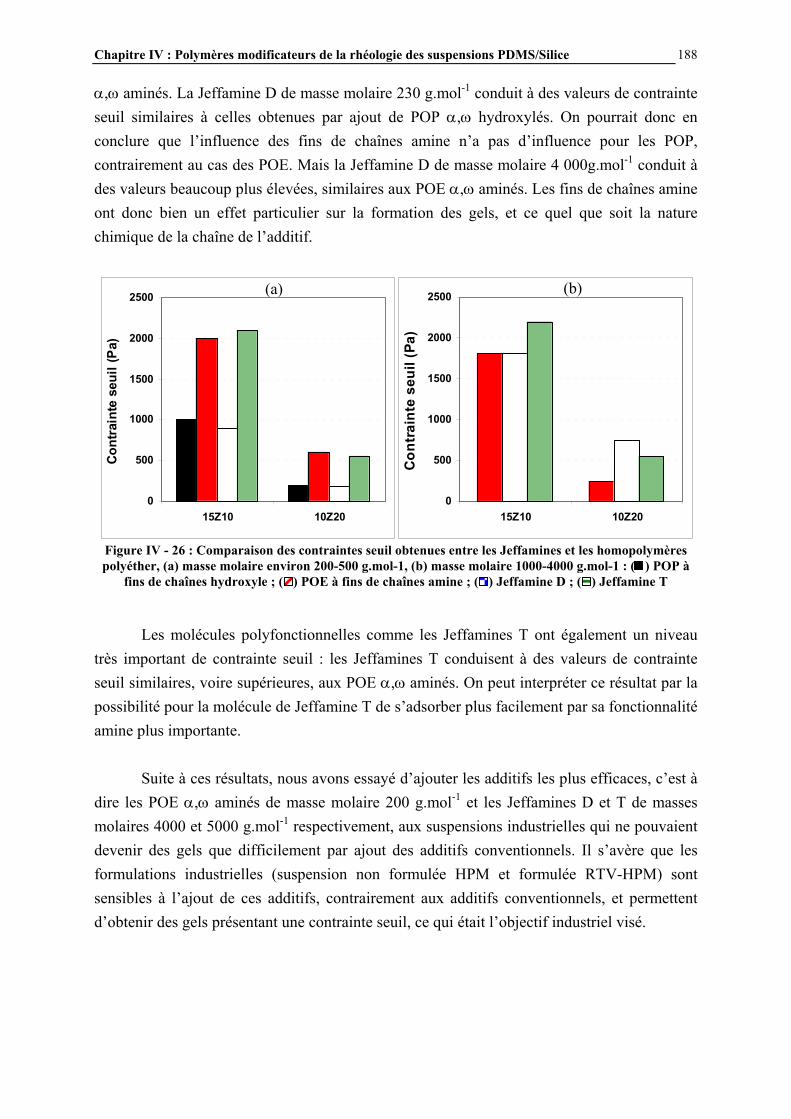
Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
188
α,ω aminés. La Jeffamine D de masse molaire 230 g.mol-1 conduit à des valeurs de contrainte seuil similaires à celles obtenues par ajout de POP α,ω hydroxylés. On pourrait donc en conclure que l’influence des fins de chaînes amine n’a pas d’influence pour les POP, contrairement au cas des POE. Mais la Jeffamine D de masse molaire 4 000g.mol-1 conduit à des valeurs beaucoup plus élevées, similaires aux POE α,ω aminés. Les fins de chaînes amine ont donc bien un effet particulier sur la formation des gels, et ce quel que soit la nature chimique de la chaîne de l’additif.
Figure IV - 26 : Comparaison des contraintes seuil obtenues entre les Jeffamines et les homopolymères polyéther, (a) masse molaire environ 200-500 g.mol-1, (b) masse molaire 1000-4000 g.mol-1 : ( ) POP à
fins de chaînes hydroxyle ; ( ) POE à fins de chaînes amine ; ( ) Jeffamine D ; ( ) Jeffamine T
Les molécules polyfonctionnelles comme les Jeffamines T ont également un niveau très important de contrainte seuil : les Jeffamines T conduisent à des valeurs de contrainte seuil similaires, voire supérieures, aux POE α,ω aminés. On peut interpréter ce résultat par la possibilité pour la molécule de Jeffamine T de s’adsorber plus facilement par sa fonctionnalité amine plus importante. Suite à ces résultats, nous avons essayé d’ajouter les additifs les plus efficaces, c’est à dire les POE α,ω aminés de masse molaire 200 g.mol-1 et les Jeffamines D et T de masses molaires 4000 et 5000 g.mol-1 respectivement, aux suspensions industrielles qui ne pouvaient devenir des gels que difficilement par ajout des additifs conventionnels. Il s’avère que les formulations industrielles (suspension non formulée HPM et formulée RTV-HPM) sont sensibles à l’ajout de ces additifs, contrairement aux additifs conventionnels, et permettent d’obtenir des gels présentant une contrainte seuil, ce qui était l’objectif industriel visé.
0
500
1000
1500
2000
2500
15Z10 10Z20
Con
trai
nte
seui
l (P
a)
0
500
1000
1500
2000
2500
15Z10 10Z20
Con
trai
nte
seui
l (Pa
)
(a) (b)

Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
189
c) Influence de la masse molaire L’effet de la masse molaire de l’additif est assez complexe car ce paramètre influence lui-même d’autres caractéristiques intervenant dans le mécanisme de gélification étudié. Lorsque l’on augmente la masse molaire de l’additif, on change, en effet, sa mobilité et en conséquence sa capacité de diffuser aux interfaces PDMS-silice. Mais on change également, dans le cas simple de molécules à fins de chaînes (α,ω), le taux de fins de chaînes, qui diminue lorsque la masse molaire augmente. Et ceci a aussi pour conséquence de changer son énergie de surface donc sa miscibilité dans le milieu PDMS et le nombre d’interactions possibles avec la surface de la silice. Au contraire, l’augmentation de la masse molaire permet d’avoir des chaînes plus longues pontant les particules puisque de longueur suffisante. Il est ainsi difficile d’estimer quel va être l’influence de la masse molaire sachant que l’on a vu que l’efficacité des additifs dépend à la fois de la microstructure de la chaîne et des fins de chaînes. Nous avons donc commencé par étudier l’influence de la masse molaire sur les additifs les plus simples que sont les POE α,ω fonctionnalisés. La Figure IV - 27 montre l’évolution de la contrainte seuil des suspensions en fonction de la masse molaire de l’additif POE α,ω hydroxylé, pour différentes formulations. Une première remarque générale est qu’il existe quelle que soit la formulation considérée une masse molaire optimale où la contrainte seuil est maximale. Après avoir atteint ce maximum, la contrainte seuil diminue de nouveau. Pour des masses molaires très importantes on peut même avoir dans le cas de certaines suspensions une contrainte seuil très faible voir présenter un comportement newtonien. La contrainte seuil maximale est atteinte pour une masse molaire faible, voisine de 1000 g.mol-1, sauf pour la formulation 10Z5 où l’optimum se situe vers 5 000 g.mol-1. Il est en fait surprenant de voir que l’optimum soit le même pour toutes les formulations, quel que soit le taux de greffage ou la fraction volumique de silice. Cela montre qu’il existe, indépendamment de la formulation considérée, un compromis entre l’aptitude à la diffusion dans le fondu liée à la masse molaire et le nombre de fins de chaînes, ce qui définit une microstructure optimale pour laquelle la contrainte seuil sera maximale. Le fait que la masse molaire optimale soit si faible suggère que la mobilité et les fins de chaînes sont des paramètres plus importants. Cet argument sera développé dans la discussion ultérieure du mécanisme d’action des additifs. Dans le but de pouvoir généraliser ces résultats, nous avons étudié d’autres additifs, mais ne disposant pas toujours de composés de masses molaires très différentes, seulement quelques tendances peuvent être dégagées. La Figure IV - 28 montre l’évolution de la
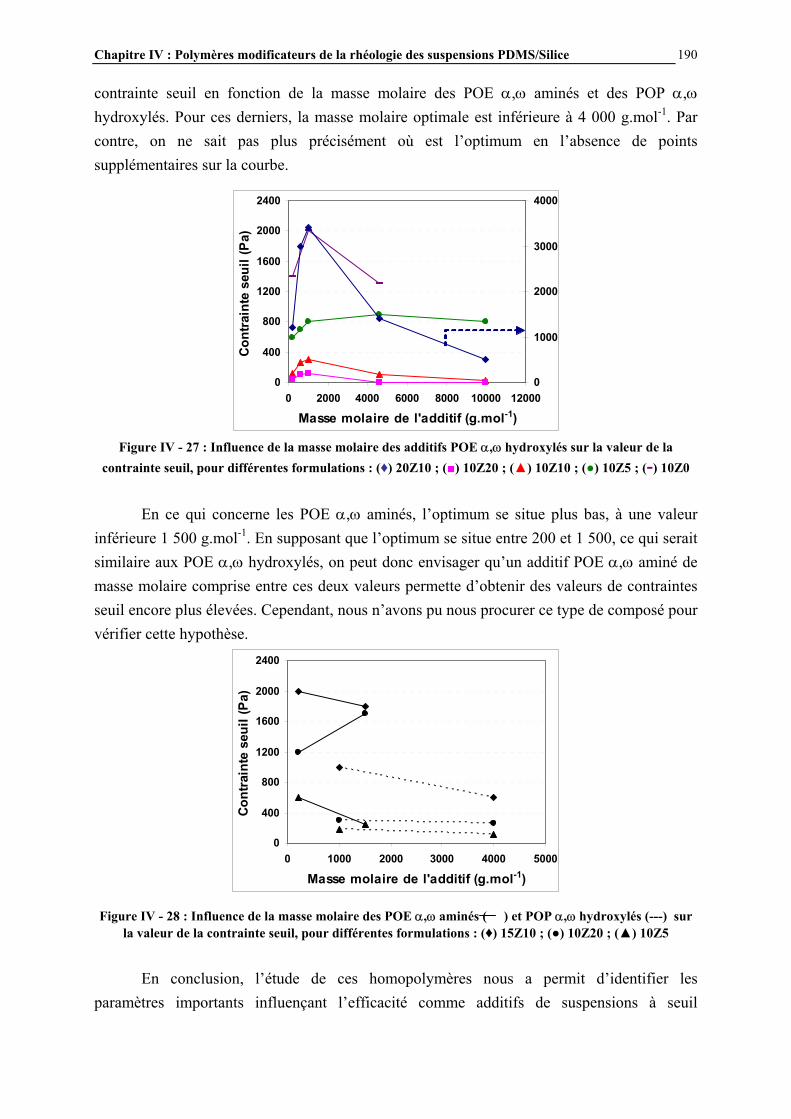
Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
190
contrainte seuil en fonction de la masse molaire des POE α,ω aminés et des POP α,ω hydroxylés. Pour ces derniers, la masse molaire optimale est inférieure à 4 000 g.mol-1. Par contre, on ne sait pas plus précisément où est l’optimum en l’absence de points supplémentaires sur la courbe.
Figure IV - 27 : Influence de la masse molaire des additifs POE α,ω hydroxylés sur la valeur de la contrainte seuil, pour différentes formulations : (♦) 20Z10 ; (■) 10Z20 ; (▲) 10Z10 ; (●) 10Z5 ; (-) 10Z0
En ce qui concerne les POE α,ω aminés, l’optimum se situe plus bas, à une valeur inférieure 1 500 g.mol-1. En supposant que l’optimum se situe entre 200 et 1 500, ce qui serait similaire aux POE α,ω hydroxylés, on peut donc envisager qu’un additif POE α,ω aminé de masse molaire comprise entre ces deux valeurs permette d’obtenir des valeurs de contraintes seuil encore plus élevées. Cependant, nous n’avons pu nous procurer ce type de composé pour vérifier cette hypothèse. Figure IV - 28 : Influence de la masse molaire des POE α,ω aminés ( ) et POP α,ω hydroxylés (---) sur
la valeur de la contrainte seuil, pour différentes formulations : (♦) 15Z10 ; (●) 10Z20 ; (▲) 10Z5
En conclusion, l’étude de ces homopolymères nous a permit d’identifier les paramètres importants influençant l’efficacité comme additifs de suspensions à seuil
0
400
800
1200
1600
2000
2400
0 2000 4000 6000 8000 10000 12000
Masse molaire de l'additif (g.mol-1)
Con
train
te s
euil
(Pa)
0
1000
2000
3000
4000
0
400
800
1200
1600
2000
2400
0 1000 2000 3000 4000 5000
Masse molaire de l'additif (g.mol-1)
Cont
rain
te s
euil
(Pa)

Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
191
PDMS/silice, afin d’en optimiser la structure. Les POE de masses molaires faibles (environ 200-1 000 g.mol-1), à fins de chaînes α,ω aminés semblent être les plus efficaces, ainsi que les Jeffamines D et T de grandes masses molaires (4 000 à 5 000 g.mol-1). IV.4.3.2- Copolymères PDMS/Polyéther Après les résultats obtenus sur les homopolymères fonctionnels PDMS, POE et POP, le même type d’étude a été réalisée sur les copolymères peigne PDMS-b-polyéther. Ces molécules ont une microstructure complexe et le but ici est seulement de dégager des grandes tendances concernant l’influence des principaux paramètres moléculaires (masse molaire, proportions de chaque bloc), ainsi que de comparer leur efficacité avec les homopolymères déjà étudiés. a) Influence de la masse molaire L’influence de la masse molaire du copolymère sur la contrainte seuil du gel formé a été déterminée sur deux formulations de même fraction volumique (φv = 4,7%) mais de taux de greffage différents (r = 0,4 et r = 1,7). Afin de pouvoir obtenir suffisamment de données, plusieurs additifs de compositions chimiques différentes ont été étudiés (Figure IV - 29). La courbe représentant chaque série d’additifs de même composition chimique ne comporte que peu de valeurs, cependant en observant l’ensemble des résultats il est possible de dégager quelques tendances. Lorsque la silice est peu greffée (formulation 10Z5), on retrouve un comportement similaire à celui décrit dans le cas des homopolymères POE, c’est-à-dire une forte augmentation de la contrainte seuil avec la masse molaire de l’additif, jusqu’à une masse molaire critique proche de 1 500 g.mol-1. Il s’agit d’une valeur très approximative car nous ne disposons que de quelques points, cependant au delà de cette valeur il semblerait que la contrainte seuil n’augmente plus. Les additifs copolymères se distinguent ici des homopolymères car la contrainte seuil ne diminue pas de nouveau lorsque la masse molaire augmente : un plateau est atteint. Dans le cas d’une silice greffée au maximum (formulation 10Z20), la contrainte seuil, comprise entre 100 et 200 Pa, ne semble pas être beaucoup affectée par la masse molaire de l’additif.
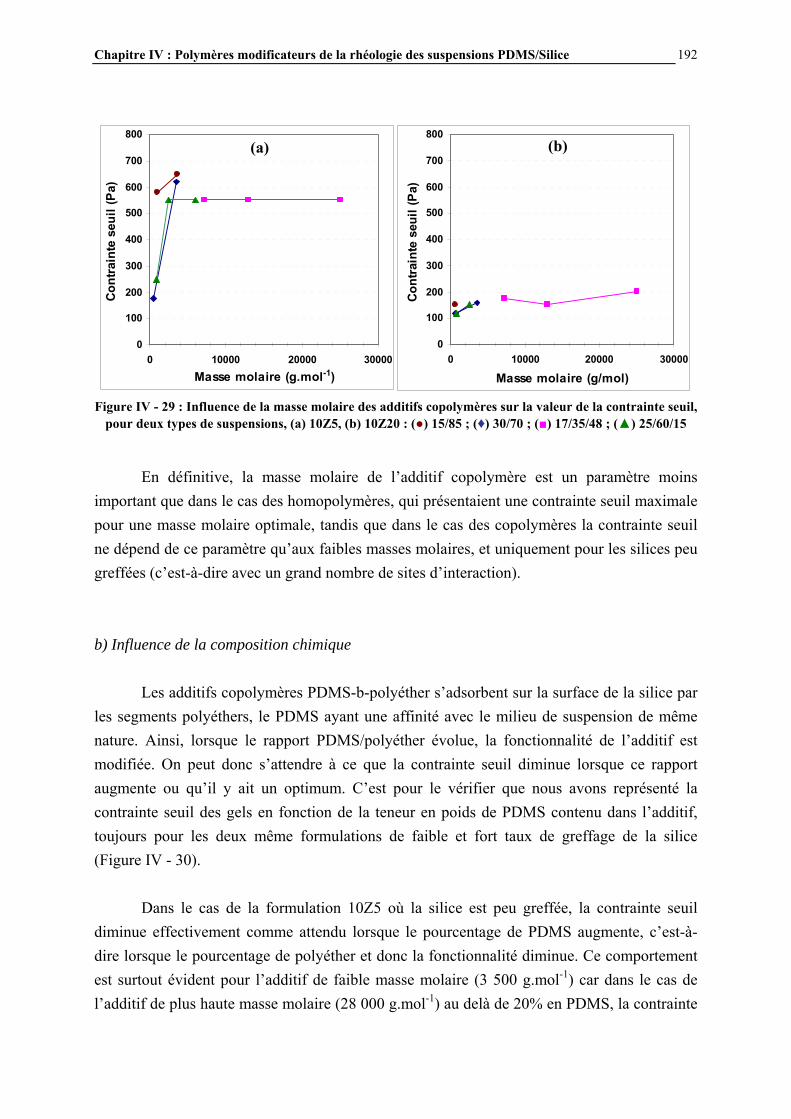
Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
192
Figure IV - 29 : Influence de la masse molaire des additifs copolymères sur la valeur de la contrainte seuil,
pour deux types de suspensions, (a) 10Z5, (b) 10Z20 : (●) 15/85 ; (♦) 30/70 ; (■) 17/35/48 ; (▲) 25/60/15
En définitive, la masse molaire de l’additif copolymère est un paramètre moins important que dans le cas des homopolymères, qui présentaient une contrainte seuil maximale pour une masse molaire optimale, tandis que dans le cas des copolymères la contrainte seuil ne dépend de ce paramètre qu’aux faibles masses molaires, et uniquement pour les silices peu greffées (c’est-à-dire avec un grand nombre de sites d’interaction). b) Influence de la composition chimique Les additifs copolymères PDMS-b-polyéther s’adsorbent sur la surface de la silice par les segments polyéthers, le PDMS ayant une affinité avec le milieu de suspension de même nature. Ainsi, lorsque le rapport PDMS/polyéther évolue, la fonctionnalité de l’additif est modifiée. On peut donc s’attendre à ce que la contrainte seuil diminue lorsque ce rapport augmente ou qu’il y ait un optimum. C’est pour le vérifier que nous avons représenté la contrainte seuil des gels en fonction de la teneur en poids de PDMS contenu dans l’additif, toujours pour les deux même formulations de faible et fort taux de greffage de la silice (Figure IV - 30). Dans le cas de la formulation 10Z5 où la silice est peu greffée, la contrainte seuil diminue effectivement comme attendu lorsque le pourcentage de PDMS augmente, c’est-à-dire lorsque le pourcentage de polyéther et donc la fonctionnalité diminue. Ce comportement est surtout évident pour l’additif de faible masse molaire (3 500 g.mol-1) car dans le cas de l’additif de plus haute masse molaire (28 000 g.mol-1) au delà de 20% en PDMS, la contrainte
0
100
200
300
400
500
600
700
800
0 10000 20000 30000
Masse molaire (g/mol)
Con
train
te s
euil
(Pa)
0
100
200
300
400
500
600
700
800
0 10000 20000 30000Masse molaire (g.mol-1)
Cont
rain
te s
euil
(Pa)
(a) (b)
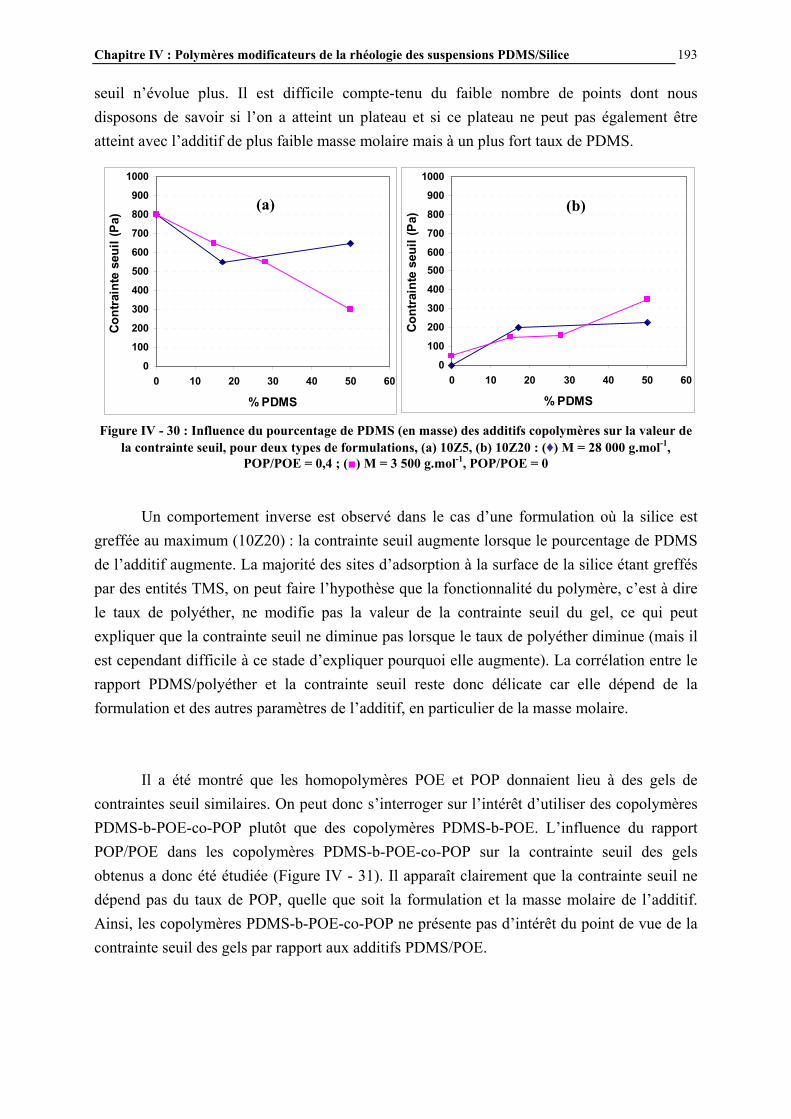
Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
193
seuil n’évolue plus. Il est difficile compte-tenu du faible nombre de points dont nous disposons de savoir si l’on a atteint un plateau et si ce plateau ne peut pas également être atteint avec l’additif de plus faible masse molaire mais à un plus fort taux de PDMS. Figure IV - 30 : Influence du pourcentage de PDMS (en masse) des additifs copolymères sur la valeur de
la contrainte seuil, pour deux types de formulations, (a) 10Z5, (b) 10Z20 : (♦) M = 28 000 g.mol-1, POP/POE = 0,4 ; (■) M = 3 500 g.mol-1, POP/POE = 0
Un comportement inverse est observé dans le cas d’une formulation où la silice est greffée au maximum (10Z20) : la contrainte seuil augmente lorsque le pourcentage de PDMS de l’additif augmente. La majorité des sites d’adsorption à la surface de la silice étant greffés par des entités TMS, on peut faire l’hypothèse que la fonctionnalité du polymère, c’est à dire le taux de polyéther, ne modifie pas la valeur de la contrainte seuil du gel, ce qui peut expliquer que la contrainte seuil ne diminue pas lorsque le taux de polyéther diminue (mais il est cependant difficile à ce stade d’expliquer pourquoi elle augmente). La corrélation entre le rapport PDMS/polyéther et la contrainte seuil reste donc délicate car elle dépend de la formulation et des autres paramètres de l’additif, en particulier de la masse molaire. Il a été montré que les homopolymères POE et POP donnaient lieu à des gels de contraintes seuil similaires. On peut donc s’interroger sur l’intérêt d’utiliser des copolymères PDMS-b-POE-co-POP plutôt que des copolymères PDMS-b-POE. L’influence du rapport POP/POE dans les copolymères PDMS-b-POE-co-POP sur la contrainte seuil des gels obtenus a donc été étudiée (Figure IV - 31). Il apparaît clairement que la contrainte seuil ne dépend pas du taux de POP, quelle que soit la formulation et la masse molaire de l’additif. Ainsi, les copolymères PDMS-b-POE-co-POP ne présente pas d’intérêt du point de vue de la contrainte seuil des gels par rapport aux additifs PDMS/POE.
0
100
200
300
400
500600
700
800
900
1000
0 10 20 30 40 50 60
% PDMSCo
ntra
inte
seu
il (P
a)
0
100
200
300
400
500
600
700
800
900
1000
0 10 20 30 40 50 60
% PDMS
Cont
rain
te s
euil
(Pa)
(a) (b)
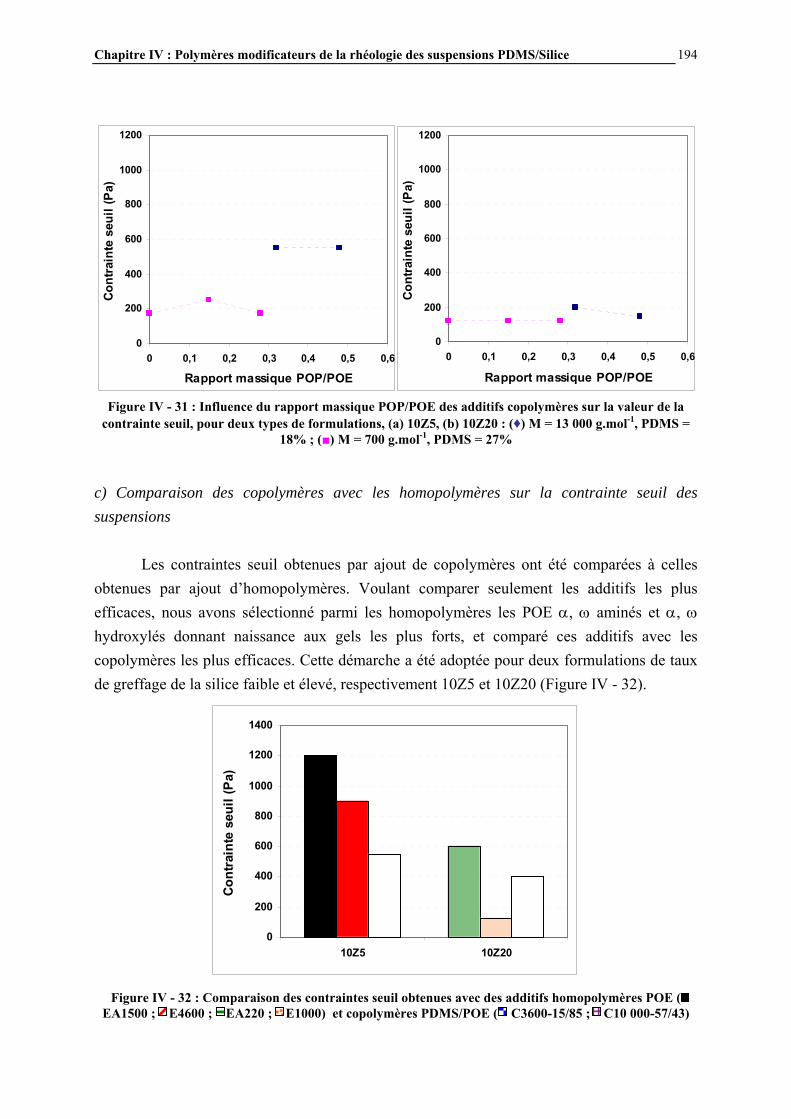
Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
194
Figure IV - 31 : Influence du rapport massique POP/POE des additifs copolymères sur la valeur de la contrainte seuil, pour deux types de formulations, (a) 10Z5, (b) 10Z20 : (♦) M = 13 000 g.mol-1, PDMS =
18% ; (■) M = 700 g.mol-1, PDMS = 27%
c) Comparaison des copolymères avec les homopolymères sur la contrainte seuil des suspensions Les contraintes seuil obtenues par ajout de copolymères ont été comparées à celles obtenues par ajout d’homopolymères. Voulant comparer seulement les additifs les plus efficaces, nous avons sélectionné parmi les homopolymères les POE α, ω aminés et α, ω hydroxylés donnant naissance aux gels les plus forts, et comparé ces additifs avec les copolymères les plus efficaces. Cette démarche a été adoptée pour deux formulations de taux de greffage de la silice faible et élevé, respectivement 10Z5 et 10Z20 (Figure IV - 32).
Figure IV - 32 : Comparaison des contraintes seuil obtenues avec des additifs homopolymères POE ( EA1500 ; E4600 ; EA220 ; E1000) et copolymères PDMS/POE ( C3600-15/85 ; C10 000-57/43)
0
200
400
600
800
1000
1200
0 0,1 0,2 0,3 0,4 0,5 0,6
Rapport massique POP/POE
Cont
rain
te s
euil
(Pa)
0
200
400
600
800
1000
1200
0 0,1 0,2 0,3 0,4 0,5 0,6
Rapport massique POP/POE
Con
train
te s
euil
(Pa)
0
200
400
600
800
1000
1200
1400
10Z5 10Z20
Con
trai
nte
seui
l (Pa
)

Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
195
Pour la formulation où la silice est peu greffée (10Z5) les copolymères ont une efficacité inférieure aux homopolymères POE, quelle que soit la fonctionnalité du POE. Dans le cas de la formulation 10Z20 où la silice est greffée au maximum, les copolymères sont toujours moins efficaces que les POE à fins de chaînes amine mais plus efficaces que les POE à fins de chaînes hydroxyle. Cette formulation représente un cas important car elle est représentative du cas industriel où les silices sont greffées avec un taux de greffage maximum. Cette comparaison montre que les copolymères ne sont pas systématiquement les additifs les plus efficaces et qu’en choisissant un homopolymère POE de masse molaire et de fonctionnalité adaptées des gels plus rigides peuvent être obtenus, même pour des taux de greffage élevés de la silice incorporée. IV.4.4- Etude par microscopie des suspensions et mécanismes de structuration proposés IV.4.4.1- Introduction Après avoir étudié la relation entre la structure des additifs et la contrainte seuil des gels formés, nous nous intéressons dans ce paragraphe au mécanisme de formation des gels. Le mécanisme supposé jusqu’alors dans ce travail opère par pontage des agrégats de particules de silice par les molécules de POE (ou d’autres additifs) adsorbés à la surface des particules (Figure IV - 2). Une première observation issue des travaux précédents vient tout d’abord remettre en cause ce mécanisme. Pour pouvoir ponter les agrégats de silice, il faut logiquement que la longueur des chaînes d’additifs soit égale ou supérieure à la distance inter-agrégats. En prenant le cas simple de molécules de POE, nous avons vu que des POE de faible masse molaire (200g.mol-1) permettent d’obtenir des gels dans des suspensions possédant une fraction volumique de silice de 2,7%. En supposant les particules de silice individuelles réparties dans un réseau parfait cubique centré, la distance entre particules à cette fraction volumique est de 14 nm. En réalité, la distance inter-agrégats est supérieure puisque les particules sont agrégées. La longueur d’une chaîne de POE de masse molaire 200 g.mol-1 étant environ de 1,3 nm, la longueur de cette molécule est donc au minimum 10 fois trop faible pour pouvoir ponter deux agrégats de silice. Si le phénomène d’adsorption du POE à la surface de la silice n’est pas à remettre en cause, le mécanisme de formation du gel semble en revanche être différent d’un pontage direct inter-agrégats. Pour pouvoir déterminer le mécanisme réel, il faut comprendre comment ces additifs polymère se structurent dans les

Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
196
suspensions PDMS/silice. La microstructure des gels a ainsi été étudiée par différentes techniques de microscopie. IV.4.4.2- Observation des structures par microscopies optique et électronique Une substance peut se définir comme un gel si elle possède une structure continue à l’échelle macroscopique et un comportement rhéologique présentant une réponse élastique [9]. Partant de cette définition, une structure continue a été créée au sein des suspensions PDMS/silice lors de l’ajout du polymère modificateur puisqu’un gel (du point de vue rhéologique) est formé. Pour observer cette structure, les gels ont été observés d’abord à faible grossissement par microscopie optique. Une suspension présentant une forte fraction volumique de silice et un fort taux de greffage (φv = 9,9%, r = 1,7) a ainsi été étudiée. Avant ajout d’additif polymère, aucune structure à cette échelle d’observation n’est présente, les clichés de microscopie optique ne mettent rien en évidence (Figure IV - 33a). Après ajout d’1% d’additif copolymère C25000-17/35/48 on constate une structuration assez nette de l’échantillon, avec des structures de la taille de quelques microns environ (Figure IV - 33b). Cette structuration est également observée lors de l’ajout d’autres types de polymères, et pour différentes formulations. La structuration de l’échantillon est d’autant plus nette que le gel est rigide, c’est-à-dire que la contrainte seuil est élevée. Ainsi, pour les additifs peu efficaces, ou pour les suspensions qui conduisent à des gels faibles, c’est-à-dire à faible fraction volumique de silice et fort taux de greffage, ces structures ne peuvent même pas être détectées. A l’inverse, la Figure IV - 34 montre le cliché de microscopie d’un gel rigide, obtenu avec l’additif POE à fins de chaînes α, ω amine particulièrement efficace comme nous l’avons vu précédemment. Le contraste est renforcé et la structuration semble plus importante encore que dans l’exemple précédent. La question qui se pose à partir de ces clichés de microscopie est : quelle doit être l’interprétation de ces images ? Ces structures sont-elles des agglomérats de silice constitués par pontage via les molécules de polymère modificateur, des zones correspondant à de l’additif seul (séparation de phase) ou une structure mixte silice/POE ? En effet on observe un contraste de phase difficile à interpréter sans l’aide d’indices supplémentaires. Si les structures observées sont de la silice, une différence importante devrait exister entre la morphologie de la suspension initiale et celle de la suspension finale après ajout d’additif. L’observation des gels par microscopie électronique à transmission montre cependant que la dispersion de silice n’est pas modifiée après ajout d’additif, comme le témoigne la Figure IV - 35.
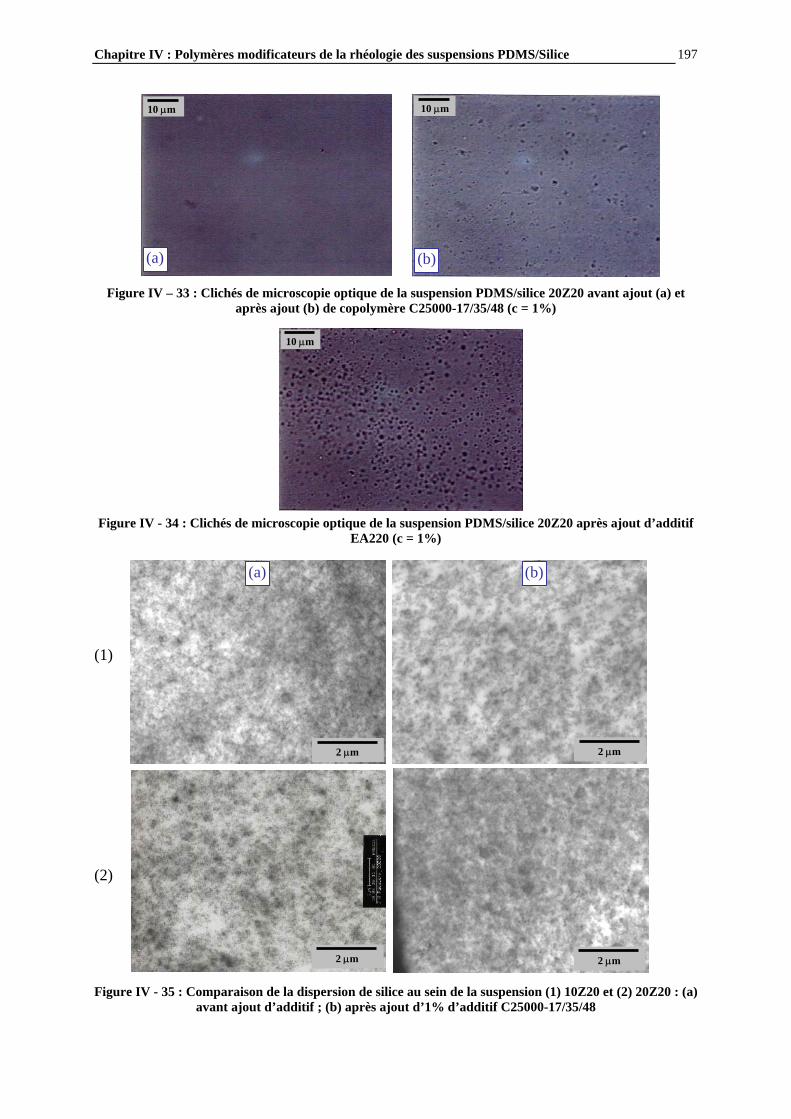
Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
197
Figure IV – 33 : Clichés de microscopie optique de la suspension PDMS/silice 20Z20 avant ajout (a) et après ajout (b) de copolymère C25000-17/35/48 (c = 1%)
Figure IV - 34 : Clichés de microscopie optique de la suspension PDMS/silice 20Z20 après ajout d’additif
EA220 (c = 1%)
(1) (2) Figure IV - 35 : Comparaison de la dispersion de silice au sein de la suspension (1) 10Z20 et (2) 20Z20 : (a)
avant ajout d’additif ; (b) après ajout d’1% d’additif C25000-17/35/48
(a) (b)
2 µm
2 µm
10 µm 10 µm
10 µm
2 µm
2 µm
(a) (b)

Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
198
Il semble donc que cette structuration observée par microscopie optique ne soit pas due à un changement de dispersion de la silice. Pour pouvoir comprendre quelle est l’origine de ce phénomène, il nous faut visualiser le polymère modificateur pour comprendre comment celui-ci s’organise au sein des suspensions et comment il génère ces structures à grande échelle observées par microscopie optique. IV.4.4.3- Observation de la structuration des suspensions par fluorescence L’additif peut être visualisé en lui ajoutant un marqueur fluorescent. Pour commencer avec un cas simple le POE à fins de chaînes α, ω hydroxylé, de masse molaire 200 g.mol-1 (E200) a été choisi. En plus de sa structure chimique simple, cet additif s’est avéré particulièrement adapté à l’étude par fluorescence en raison de la bonne miscibilité du marqueur fluorescent, la Rhodamine B (Figure IV - 36) avec cet additif, ce qui n’est pas le cas par exemple du POE à fins de chaînes α, ω amine. La Rhodamine b étant un composé polaire, elle est miscible en général avec les composés comme les POE et POP.
Figure IV - 36 : Structure chimique du marqueur fluorescent, la Rhodamine B
De plus le POE E200 n’est pas miscible avec le PDMS, ce qui permet d’augmenter les chances que la Rhodamine B ne migre pas dans le milieu de suspension PDMS et donc de ne visualiser que l’additif. Il faut également être sur que la Rhodamine B ne migre pas aux interfaces avec la silice. Pour cela nous avons réalisé des mélanges témoins : des suspensions PDMS/silice sans POE mais contenant de la Rhodamine B. Il a ainsi été vérifié que la Rhodamine B était répartie de manière homogène au sein de la suspension et n’avait pas d’affinité particulière avec la surface de la silice. Ces vérifications étant faites, les suspensions ont été observées par fluorescence après ajout du POE marqué azvec la Rhodamine B. L’ajout du POE marqué s’effectue de la même manière que l’ajout du POE non marqué et nous avons vérifié que le marqueur n’intervient pas sur l’effet de l’additif polymère sur la suspension : les valeurs de contrainte seuil sont ainsi les mêmes que le POE soit marqué ou non. Les clichés de microscopie en fluorescence révèlent alors les structures que forment le POE (sur les clichés de microscopie en fluorescence, le POE marqué apparaît en blanc).

Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
199
Après avoir étudié plusieurs suspensions de taux de greffage et de fraction volumique de silice différents, les morphologies peuvent se résumer à quatre cas de figure (Figure IV - 37) : i/ Le premier cas correspond à des suspensions qui ne gélifient pas, en raison de leur paramètres de formulation (φv et r, cf. le diagramme de transition liquide-gel en présence d’additif). Le POE se structure alors sous forme de gouttelettes, de la taille de plusieurs dizaines de microns (Figure IV - 37a). Il y a simplement séparation de phase entre le milieu PDMS et l’additif POE. Le même type de microstructure est observé lorsque l’additif POE est ajouté au PDMS, sans silice. Le POE se structure alors sous forme de gouttelettes de même taille que celles observées dans les suspensions qui ne gélifient pas, confirmant la séparation de phase PDMS/POE. ii/ Le second cas est celui de structures un peu plus fines, que l’on rencontre dans les gels qui sont généralement de rigidité faible ou intermédiaire (Figure IV - 37b). Le POE est certainement adsorbé à la surface de la silice, ce qui explique la différence avec la structure de gouttelettes précédemment observée. On perçoit que ces structures fines sont sans doute percolées à une plus faible échelle d’observation. En effet, même si à partir de ce cliché de microscopie, on ne voit pas de structure continue, en zoomant on fait apparaître au fur et à mesure des structures de plus en plus fines, que l’on observait pas dans un premier temps à un grossissement plus faible. Le fait que cet échantillon soit un gel nous porte à croire également que les structures forment un continuum de POE à plus faible échelle. iii/ Dans le troisième cas (Figure IV - 37c), la structure de POE formée est complètement différente, formant cette fois de manière évidente une phase continue de POE au sein de l’échantillon. Le contraste est moins net et le POE apparaît de couleur grise plutôt que blanche, contrairement aux autres clichés de microscopie. Ces structures sont obtenues lorsque les gels ont une forte rigidité. iv/ Enfin dans le dernier cas on peut avoir des structures mixtes, constituées à la fois d’une phase continue (en gris) de POE et de structures plus fines et plus blanches (d). Ce sont également dans les gels rigides que l’on observe ce type de structuration. Il peut paraître surprenant de voir l’importance de ces structures POE en comparaison des 1% qui ont seulement été ajoutés aux suspensions. Cependant, en se rappelant les études qui ont été menées sur les gels PDMS/DBS, un réseau de fibrilles très important constitué de DBS avait pu être observé, pour des concentrations en DBS de 1% [8, 9]. Il est donc tout à fait concevable d’imaginer de la même façon une structure de POE aussi importante avec 1% de POE dans les suspensions PDMS/silice. Dans le cas présent, un simple mécanisme de

Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
200
séparation de phase est avancé alors que pour le DBS la structuration s’effectue par assemblage supramoléculaire. Les différents types de structures POE peuvent être directement reliés à la morphologie initiale des suspensions PDMS/silice et donc aux paramètres de la formulation. Le cas (a) apparaît lorsque la suspension n’a pas franchi la transition liquide-gel, c’est-à-dire lorsque la fraction volumique est faible. On peut l’obtenir quel que soit le taux de greffage de la silice mais pour des silices greffées on peut le rencontrer jusque des fractions volumiques plus élevées, 5% environ (cf. diagramme de transition liquide-gel). Le cas (b) correspond à des gels de rigidité intermédiaire ou élevée, où la dispersion initiale de silice est homogène. Ce cas concerne donc les suspensions dont le taux de greffage et la fraction volumique de silice sont élevés. Pour les suspensions dont le taux de greffage des silices est nul, le gel obtenu est très rigide et la transition liquide-gel brutale ; la morphologie observée au delà de la transition sol gel correspond au cas (c). Enfin le cas (d) correspond à des gels de rigidité intermédiaire, correspondant à des fractions volumiques et des taux de greffage intermédiaires. Les morphologies obtenues peuvent être placées suivant les paramètres de la formulation, dans le diagramme de transition liquide-gel. Ceci montre que la formation du gel est directement liée à la structuration de la phase POE dans l’échantillon, celle-ci étant dépendante du nombre de sites d’adsorption disponibles, à savoir la fraction volumique et le taux de greffage de la silice.

Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
201
Figure IV - 37 : Différents types de structures POE formées au sein des suspensions PDMS/silice, (a) 10Z20, (b) 10Z10, (c) 10Z0, (d) 15Z5
Figure IV - 38 : Morphologies observées par microscopie optique en mode fluorescent, en fonction du diagramme de transition liquide-gel des suspensions PDMS/silice en présence d’additif polymère
0
0,02
0,04
0,06
0,08
0,1
0,12
0 0,5 1 1,5 2 2,5
Rapport stoechiométrique HMDS/silanol, r
φv
gel
liquide
(c)
(b)(d)
(a)
(a) (b)
(c) (d)
50 µm 50 µm
50 µm 50 µm

Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
202
Pour vérifier que ces structures POE observées par fluorescence correspondent bien aux structures initiales observées par microscopie optique, les gels ont été observés par microscopie optique simultanément en mode fluorescent et en mode non fluorescent. Les deux images sont ensuite comparées. La Figure IV - 39 montre que les microstructures observées dans ces deux modes sont identiques et ce pour deux cas de figures : une morphologie constituée de fines structures POE qui percolent et une morphologie où le POE forme une phase continue plus distincte. Dans le premier cas, le contraste est important entre les zones riches en POE et les zones ne contenant pas de POE. La structure est très nette et la corrélation entre les images en mode fluorescent et en mode non fluorescent est parfaite. Dans le second cas la structure est moins marquée et il est plus difficile de corréler les deux images. Cependant certaines zones très riches en POE (zones apparaissant très claires sur la micrographie en mode fluorescent) se retrouvent parfaitement à la même localisation en mode non fluorescent, ce qui ne laisse aucun doute sur la correspondance entre les deux images. La corrélation entre ces deux types d’images montre que la structure responsable de la formation du gel est bien une structure constituée de POE. Cependant, l’inconvénient de ce type d’images est que l’on ne visualise pas la structure de silice. Est-elle contenue dans les structures POE ? A priori on peut penser que oui puisque le POE a tendance à venir s’adsorber fortement à sa surface. On peut alors penser que l’on a une structure continue de POE, celle-ci étant créée lorsque le POE s’adsorbe à la surface de la silice. Cette hypothèse sera développée dans le paragraphe suivant. Compte-tenu des différences de propriétés des gels suivant les additifs utilisés, il n’est pas évident que les gels soient formés de la même manière pour tous les additifs. La microscopie en mode fluorescent a donc été utilisée pour d’autres types d’additifs. Cependant, un certain nombre de difficultés sont apparues en changeant la nature de l’additif. En ce qui concerne les additifs PDMS, il n’est pas possible de les marquer spécifiquement puisqu’ils sont de même nature que le milieu de suspension. Nous avons donc étudié les additifs POE de différentes fins de chaînes et les copolymères PDMS/POE. Pour ces derniers, la présence de PDMS a également posé des problèmes et les résultats obtenus n’ont pu être exploités en raison du très faible contraste des images. Les additifs POE à fins de chaînes α, ω OCH3 en revanche ont pu être étudiés, même si le contraste s’est révélé être moins bon qu’avec le POE à fins de chaînes α, ω hydroxyle (Figure IV - 40). Il semble que la structuration du POE ait lieu de la même manière que précédemment, avec une structure fine lorsque la suspension initiale est homogène (forts taux de greffage) et une structure où la phase continue se développe à une plus grande échelle lorsque la suspension initiale n’est pas homogène (faibles taux de greffage). Le mécanisme semble donc inchangé lorsque les fins de chaînes sont différentes. Le POE à fins de chaînes α, ω amine n’a par contre pas pu être étudié car il semble interagir avec la Rhodamine B. En effet, lorsque la Rhodamine B est mélangée à ce

Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
203
type de POE elle perd son aspect fluorescent. Cependant nous faisons l’hypothèse que le mécanisme est le même pour les différents POE.
Figure IV - 39 : Comparaison des microstructures entre les images de microscopie optique (a) en mode fluorescent et (b) en mode non fluorescent
Figure IV - 40 : Images de microscopie optique en mode fluorescent après marquage du POE à fins de chaînes α, ω OCH3 : (a) formulation initiale 10Z5 ; (b) formulation initiale 20Z20
15Z20
10Z0
50 µm 50 µm
50 µm 50 µm
50 µm 50 µm

Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
204
IV.4.4.4- Mécanismes proposés Le mécanisme de formation des gels envisagé initialement était le pontage des particules ou agrégats de particules de silice par les chaînes de polymères (additifs), qui conduirait à la formation d’un réseau continu responsable de la transition liquide-gel. Après avoir étudié plus en détails l’influence des paramètres de la formulation et de la structure des additifs sur les caractéristiques des gels, ce modèle a été remis en cause. D’une part des polymères de longueur de chaîne beaucoup plus faible que la distance inter-agrégats se sont avérés efficaces, ce qui n’est pas compatible avec le mécanisme envisagé, d’autre part la nature chimique du polymère joue un rôle considérable sur la rigidité du gel (en particulier les différences sont importantes entre les PDMS et les POE), ce qui n’est pas du tout pris en compte par cette approche. Jusqu’alors, seule la percolation d’agrégats de silice a été envisagée. Cependant, la structuration de la suspension n’est pas obligatoirement due à la percolation des agrégats de silice, il est aussi possible que l’additif forme une structure continue contenant elle-même les agrégats de silice. C’est par exemple le cas de gels PDMS/DBS où le DBS forme par assemblage supramoléculaire une structure au sein du milieu PDMS, même lorsqu’il est introduit à hauteur de 1%. Ceci nous a amené à revoir notre interprétation de la formation des gels, et grâce au suivi par marquage fluorescent des additifs POE, nous proposons un nouveau mécanisme de formation des gels PDMS/silice/POE. Ce modèle s’appuie sur les observations faites par microscopie optique et électronique dont nous venons de discuter les résultats, sur les résultats de l’étude structure-propriétés des additifs présentée dans ce chapitre, ainsi que sur des analogies avec des travaux antérieurs. Il est développé dans le cas d’homopolymères POE qui sont les additifs qui ont été étudiés par marquage fluorescent, pour des suspensions PDMS/silice colloïdale greffée in-situ. Lorsque l’on étudie le système binaire PDMS / 1% de POE, le POE qui n’est pas miscible avec le PDMS forme de larges gouttelettes (séparation de phase liquide-liquide PDMS/POE). Lorsque le PDMS est en présence d’une faible quantité de silice, et que l’on ajoute 1% de POE, on est cette fois dans un système ternaire polymère de suspension/polymère additif/charge. Le POE a cette fois ci des affinités avec la surface de la silice mais reste non miscible avec le milieu de suspension PDMS. On entre alors dans une compétition entre ces deux types d’interactions. A faible fraction volumique de silice (c’est-à-dire une faible quantité de surface d’interaction déployée), le POE reste sous forme de gouttelettes et ne s’adsorbe pas ou peu à la surface de la silice, c’est ce qui a été montré par fluorescence (Figure IV - 37 a) :

Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
205
l’organisation du système ternaire n’est pas modifiée, les trois phases silice, PDMS et POE sont séparées. Lorsque la fraction volumique augmente, on rompt alors cet équilibre puisque les distances d’interaction deviennent faibles et permettent la diffusion aux interfaces de l’additif polymère et le POE va s’adsorber préférentiellement à la surface de la silice, plutôt que de former des gouttelettes. La question peut se poser de savoir si le POE peut vraiment s’adsorber à la surface de la silice, sachant que le PDMS est lui-même déjà adsorbé à la surface de la silice. Cependant, l’adsorption du PDMS est réversible et le POE a de fortes affinités avec les surfaces de silice. Ces chaînes de POE sont courtes dans le cas de nos additifs et possèdent donc une mobilité importante dans le milieu. Les chaînes de POE peuvent donc facilement remplacer les chaînes de PDMS à la surface de la silice. Lorsque la fraction volumique de silice augmente le POE s’adsorbe donc à la surface de la silice et on peut faire l’analogie avec une étude sur la modification de l’organisation de systèmes polymère/polymère par l’ajout de charges de silice décrite récemment par Lipatov [11]. Dans des mélanges polyéthylène chloré (CPE) / copolymère éthylène-vinyl acétate (EVA), la température de séparation de phase est modifiée lors de l’ajout de silice, en raison du changement de paramètres d’interaction dans le système ternaire ainsi formé. De même, dans notre cas, les images de fluorescence montrent bien le changement d’organisation du POE dans la suspension lorsqu’une quantité de silice suffisante est présente. L’hypothèse que nous faisons ici pour expliquer la formation du gel est que le POE, adsorbé à la surface des agrégats de particules de silice, n’ayant pas d’affinité avec le milieu PDMS, forme une structure continue dans l’échantillon. Ce serait donc la silice, de par sa dispersion dans la suspension initiale, son nombre de sites disponibles pour l’adsorption du POE (dépendant de la fraction volumique et du taux de greffage) qui conditionnerait l’organisation de cette structure POE et donc la caractéristique des gels. La structure percolante donnant lieu à une réponse élastique (G’>G’’) est une phase POE-silice donc de fraction volumique φsilice + φadditif . On peut voir aussi une analogie entre notre système PDMS/POE/silice et les gels PDMS/DBS. Dans les deux cas on a une structure continue formée d’un polymère n’ayant pas d’affinité avec le milieu PDMS, qui conduit à la formation d’un gel. Seule la raison pour laquelle cette structure est formée diffère. Dans le cas du DBS, c’est par autoassociation des molécules de DBS et par séparation de phase avec le PDMS. Dans notre cas c’est par adsorption du POE à la surface de la silice et par séparation de phase avec le PDMS. La silice comme nous l’avons dit joue ici uniquement le rôle d’aide à la structuration du POE. Cette hypothèse qui est faite sur le mécanisme de formation des gels, dans le cas de POE, est confortée par les images de fluorescence qui ont démontré la structuration de l’additif POE. Ainsi, on peut représenter ce mécanisme par les schémas

Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
206
présentés à la Figure IV - 41, illustrés par des clichés de microscopie optique en mode fluorescent faisant apparaître la structure du POE, en accord avec le mécanisme proposé. Le second schéma, Figure IV - 41 b, explique les microstructures observées pour des gels rigides, dans des suspensions où le taux de greffage de la silice est faible, c’est-à-dire pour lesquelles la dispersion de silice est hétérogène et le nombre de sites d’adsorption disponibles pour le POE élevé (cas le plus favorable pour la formation de gels). Les zones plus foncées sur le schéma correspondent à des zones plus riches en POE, comme cela apparaît en microscopie en mode fluorescent. On peut alors concevoir qu’il s’agit de zones plus denses en silices (agglomérats) ou bien des zones où la silice est moins bien greffée par rapport à l’ensemble de l’échantillon, ce qui favoriserait l’adsorption du POE. Ce schéma peut être adapté au cas où la suspension initiale est homogène (taux de greffage élevés, Figure IV - 41 c). Dans ce cas, il est plus difficile de voir si la structure est réellement continue, car à l’échelle d’observation de la microscopie optique, les objets apparaissent plutôt séparés. A plus fort grossissement il est possible de faire apparaître au fur et à mesure de nouveaux objets encore plus petits, ce qui peut laisser penser que la structure est continue mais à une plus petite échelle. Le schéma envisagé dans ce cas consiste en des zones riches en POE autour d’agrégats de silice, qui apparaissent clairement en mode fluorescent et une structure continue de POE pontant les agrégats puisque l’échantillon a un comportement de gel. Il est difficile par contre de savoir si ce POE est pur (comme nous l’avons représenté sur le schéma) ou également adsorbé sur de plus petits agrégats de silice non visualisables compte-tenu de leur taille. L’étude de fluorescence étant réalisée par microscopie optique, nous sommes limités à un grossissement trop faible pour pouvoir observer ces plus petits objets. La microstructure du gel dans le premier cas (taux de greffage faible) conduit à des gels plus rigides que dans le second cas, ce qui montre que la structure formée est plus rigide. On conçoit également que plus la fraction volumique de silice sera importante plus le réseau formé sera important et le gel rigide. Ceci est conforme aux valeurs de contraintes seuil qui montrent que celle-ci augmente lorsque la fraction volumique augmente.
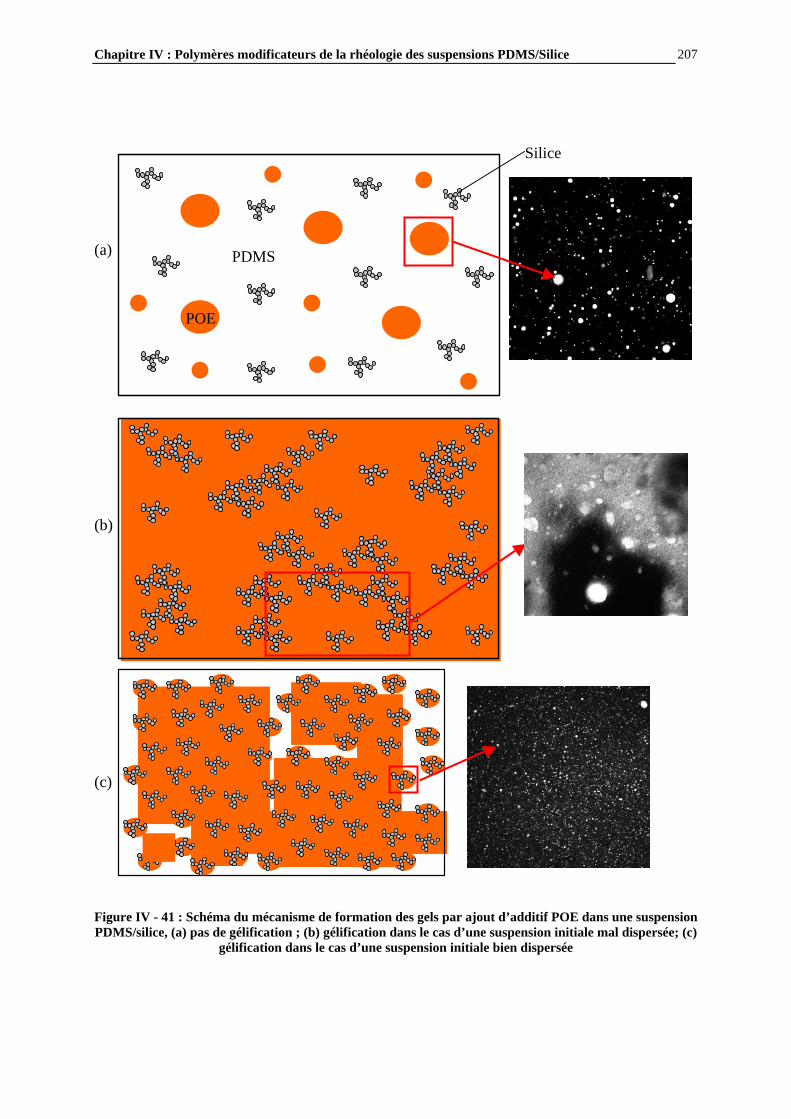
Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
207
(a) (b) (c) Figure IV - 41 : Schéma du mécanisme de formation des gels par ajout d’additif POE dans une suspension PDMS/silice, (a) pas de gélification ; (b) gélification dans le cas d’une suspension initiale mal dispersée; (c)
gélification dans le cas d’une suspension initiale bien dispersée
PDMS
POE
Silice

Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
208
Ce mécanisme proposé permet d’interpréter un certain nombre de résultats. Il faut cependant rappeler que celui-ci n’a été validé que pour les additifs POE. Il n’est pas sur que les additifs PDMS et copolymères PDMS/polyéther mettent également en jeu ce mécanisme. Concernant les paramètres de la formulation initiale, on comprend mieux pourquoi ceux-ci influencent l’efficacité des additifs, puisque la structuration du POE dépend directement de la fraction volumique de silice et du taux de greffage. Ce mécanisme explique aussi pourquoi les additifs de masse molaire peu élevée (200-4 000 g.mol-1) sont les plus efficaces. En effet la longueur de la chaîne polymère en elle-même n’a pas d’importance dans ce type de mécanisme, par contre la mobilité du polymère, accrue lorsque sa masse molaire est faible, est capitale puisque la diffusion des molécules POE dans le milieu PDMS aux interfaces avec les particules de silice est le phénomène principal conduisant à la structuration du POE et donc à la formation des gels. Le rôle des fins de chaînes peut aussi être envisagé d’une nouvelle manière. A supposer que le mécanisme soit le même pour les POE de différentes fins de chaînes, le rôle majeur de celles-ci n’est sûrement pas le fait de pouvoir créer des liaisons hydrogène ou non avec les silanols de surface de la silice, mais de modifier les paramètres d’interaction entre le POE et la silice. En effet les fins de chaînes d’un polymère peuvent modifier son énergie de surface (en particulier dans les chaînes courtes où les fins de chaînes sont loin d’être négligeables) et donc les interactions polymère-charge. La preuve qu’il ne faut pas raisonner en termes de liaisons hydrogène établissant les pontages est que les POE à fins de chaînes hydroxyle et méthyle ont la même efficacité et pourtant le POE à fins de chaînes méthyle ne peut former de liaisons hydrogène par ses fins de chaîne. A l’inverse le POE à fins de chaîne amine qui peut former des liaisons hydrogène de la même manière que le POE à fins de chaînes hydroxyles possède une efficacité accrue. Ainsi, si un mécanisme de formation des gels satisfaisant a pu être proposé, il nous reste à comprendre l’influence des fins de chaînes des additifs sur leur efficacité. Pour cela une étude des énergies de surface et des énergies interfaciales est proposée dans le paragraphe suivant.

Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
209
IV.4.5- Mesure des énergies d’interactions IV.4.5.1- Chromatographie gazeuse en phase inverse (IGC) a) Principe La chromatographie gazeuse en phase inverse (IGC) est une technique qui permet de quantifier les interactions entre la surface d’un composé solide (phase stationnaire) et des composés sondes. On peut alors remonter aux caractéristiques du solide si on utilise des composés sondes connus, ou comparer plusieurs composés sondes caractéristiques (segments) des constituants réels du milieu à étudier, pour une surface de caractéristiques connues. Le principe consiste à remplir une colonne du solide à étudier et de sélectionner des composés sonde. Ceux-si sont injectés dans la colonne où ils sont transportés par un gaz vecteur (He par exemple), en faibles quantités pour pouvoir travailler en conditions de dilution infinie (interactions entre molécules sonde alors négligées). A partir de la mesure de leur temps de rétention dans la colonne, on peut remonter à différentes caractéristiques du solide ou des interactions solide – composés sonde. Nous présentons ci-dessous la méthodologie décrite dans la littérature pour calculer la composante dispersive de l’énergie de surface d’une silice et le paramètre d’interaction spécifique silice – composés sonde [12-20]. b) Méthodologie d’analyse des résultats b1) Calcul de la composante dispersive de l’énergie de surface d’une silice L’énergie de surface peut se décomposer en une composante dispersive (interactions de London) et une composante spécifique (polaire), notées d
sγ et psγ respectivement :
ps
dss γγγ += Equation IV - 2
La composante dispersive de l’énergie de surface de la silice peut être calculée en injectant une série de n-alcanes par la méthode de Dorris et Gray [12]. En effet, les alcanes interagissent uniquement par interactions dispersives, en raison de leur caractère apolaire. A partir de leur temps de rétention dans la colonne, le volume de rétention de chaque alcane est calculé à l’aide de l’Equation IV - 3 :

Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
210
Equation IV - 3
avec tr le temps de rétention du composé sonde, tm le temps de rétention de référence (méthane par exemple), D, le flux du gaz vecteur, Pw, la pression de vapeur de l’eau à la température du fluxmètre, P0, la pression au fluxmètre, Tc, la température dans la colonne, Tf, la température du fluxmètre et j, le facteur de James-Martin, défini par la relation : Equation IV - 4
où Pin et Pout sont les pressions à l’entrée et à la sortie de la colonne respectivement. L’énergie libre d’adsorption, notée 0
aG∆ est ensuite calculée à partir du volume de rétention VN : Equation IV - 5
avec S, la surface spécifique de la silice, g la masse de silice contenue dans la colonne et C une constante égale à 2,99.108 m-1. L’énergie libre d’adsorption 0
aG∆ des alcanes évolue linéairement en fonction du nombre de carbones n. La valeur de l’énergie libre d’adsorption d’un groupe CH2 , notée
2CHaG∆ , peut donc être déterminé à partir de la courbe 0
aG∆ = f (n) tel que le montre la Figure IV - 42.
Figure IV - 42 : Principe de détermination de 2CHaG∆ à partir de la courbe 0
aG∆ = f (n)
Enfin, on peut en déduire la valeur de la composante dispersive de l’énergie de surface :
( )f
cwmrN T
TPPttDjV ⎟⎟
⎠
⎞⎜⎜⎝
⎛−−=
0
1
⎥⎥⎦
⎤
⎢⎢⎣
⎡−⎟⎟
⎠
⎞⎜⎜⎝
⎛
⎥⎥⎦
⎤
⎢⎢⎣
⎡−⎟⎟
⎠
⎞⎜⎜⎝
⎛
=
1
1
23
3
2
out
in
out
in
PP
PP
j
⎟⎟⎠
⎞⎜⎜⎝
⎛−=∆
SgCVRTG N
a ln0
2CHaG∆
0aG∆
n

Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
211
γCH2 : énergie de surface d’un groupe CH2
aCH2 : aire superficielle d’un groupe CH2 Equation IV - 6
N : nombre d’Avogadro La valeur de γCH2 peut être calculée par la relation empirique [13] : Equation IV - 7
b2) Calcul du paramètre d’interaction spécifique surface de silice / composés sonde Les composés apolaires, tels que le polyéthylène, n’échangent que des interactions dispersives, tandis qu’un composé polaire, comme l’acétone échange des interactions dispersives et spécifiques. Pour évaluer la composante spécifique des interactions d’un composé par IGC, on soustrait alors à l’énergie libre d’adsorption de ce composé la composante dispersive des interactions. Cette dernière correspond à l’énergie libre d’adsorption d’un alcane de même paramètre moléculaire que le composé sonde. Ceci définit le paramètre d’interaction spécifique, p
sI ,du composé sonde avec la surface de la silice. Il peut être représenté graphiquement en représentant 0
aG∆ en fonction du paramètre moléculaire (Figure IV - 43). Les alcanes s’alignent sur une droite et le paramètre d’interaction spécifique correspond à l’écart entre 0
aG∆ du composé sonde et la droite des alcanes.
Figure IV - 43 : Détermination du paramètre d’interaction spécifique d’un composé sonde
Le paramètre moléculaire choisi dépend des auteurs : pression partielle [14-16], indice topologique [12, 17] ou aire superficielle [13, 18] de la molécule. Dans le cadre de cette étude nous avons utilisé comme paramètre moléculaire l’aire superficielle, qui peut être déterminée à partir de la densité et la masse molaire des molécules par la relation empirique [21] :
2
2
2
22
1
⎥⎥⎦
⎤
⎢⎢⎣
⎡ ∆=
CH
CHa
CH
ds Na
Gγ
γ
( )cCH T−+= 20058,06,352
γ
0aG∆
Droite des alcanes
Composé sonde
psI
Paramètre moléculaire

Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
212
a : aire superficielle (nm2)
M : masse molaire de la molécule (g.mol-1) Equation IV - 8
d : densité de la molécule (g.cm-3) N : nombre d’Avogadro c) Etudes antérieures La composante dispersive de l’énergie de surface des silices de pyrohydrolyse a été déterminée par IGC pour différentes silices de pyrohydrolyse. A 110°C, les valeurs sont toutes comprises entre 47 et 74 mJ/m2, une plage de valeurs assez restreinte en raison de la faible variation de structure de ce type de silice. Pour l’Aerosil 200 la valeur de d
sγ à 110°C est de 50 mJ/m2 [12, 15]. d
sγ diminue linéairement lorsque la température augmente tant que la température reste dans un domaine où la surface ne subit pas de modifications chimiques importantes. L’influence du traitement thermique sur les caractéristiques de surface de la silice a pu être mise en évidence par IGC (température de mesure = 110°C) [12, 15]. d
sγ augmente avec la température de traitement de 67,6 mJ/m2 à 150°C jusque 87,8 mJ/m2 à 650°C, en raison du phénomène de déshydroxylation, qui conduit à la formation de liaisons Si-O-Si d’énergie plus forte. En ce qui concerne les interactions spécifiques elles diminuent lorsque la température de traitement augmente, en accord avec la diminution des espèces polaires que sont les silanols de surface et l’eau physisorbée. Plusieurs études ont été menées pour évaluer l’influence du greffage de la silice par des groupements diméthylsilane (DMS) ou triméthylsilane (TMS) sur la composante dispersive de l’énergie de surface d
sγ et sur le paramètre d’interaction spécifique psI avec des
composés sonde [12, 15, 16, 19]. Il est montré que dsγ diminue progressivement lorsque le
taux de greffage augmente, ce qui s’explique par le remplacement des groupements hydroxyles, possédant des électrons libres, par des groupements méthyles dont les interactions dispersives sont moins fortes. Le Tableau IV - 8 montre l’évolution de d
sγ en fonction de l’espèce greffée, du taux de greffage et de la température d’analyse. Les évolutions sont à peu près similaires quelle que soit la nature du greffon (DMS ou TMS) ou la température d’analyse. Les valeurs sont plus fortes lorsque la température est plus faible, mais la diminution relative reste du même ordre de grandeur, soit environ -30% entre une surface non greffée et greffée au maximum. Cette diminution reste modérée en comparaison de la chute du paramètre d’interaction spécifique. En effet celui-ci dépend de la polarité des espèces et il
32
1610.091,1 ⎟⎠⎞
⎜⎝⎛=
NdMa
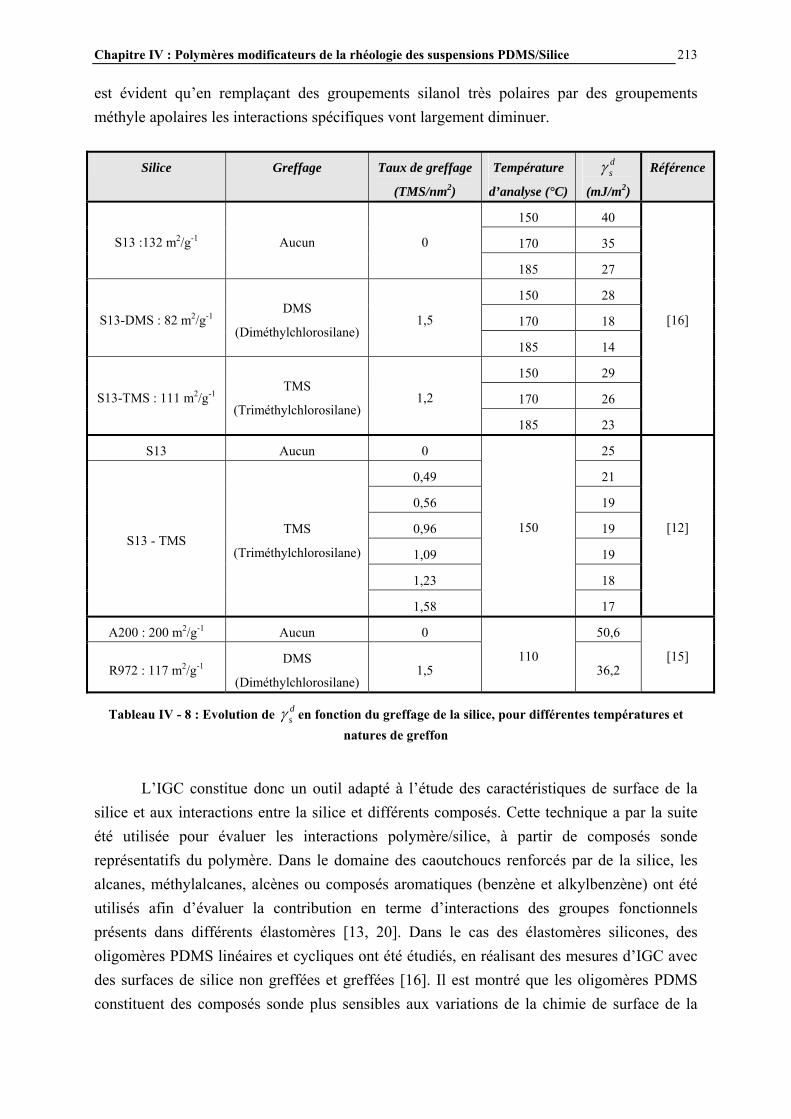
Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
213
est évident qu’en remplaçant des groupements silanol très polaires par des groupements méthyle apolaires les interactions spécifiques vont largement diminuer.
Silice Greffage Taux de greffage
(TMS/nm2)
Température
d’analyse (°C)
dsγ
(mJ/m2)
Référence
150 40
170 35 S13 :132 m2/g-1 Aucun 0
185 27
150 28
170 18 S13-DMS : 82 m2/g-1 DMS
(Diméthylchlorosilane) 1,5
185 14
150 29
170 26 S13-TMS : 111 m2/g-1 TMS
(Triméthylchlorosilane) 1,2
185 23
[16]
S13 Aucun 0 25
0,49 21
0,56 19
0,96 19
1,09 19
1,23 18
S13 - TMS TMS
(Triméthylchlorosilane)
1,58
150
17
[12]
A200 : 200 m2/g-1 Aucun 0 50,6
R972 : 117 m2/g-1 DMS
(Diméthylchlorosilane) 1,5
110 36,2
[15]
Tableau IV - 8 : Evolution de dsγ en fonction du greffage de la silice, pour différentes températures et
natures de greffon
L’IGC constitue donc un outil adapté à l’étude des caractéristiques de surface de la silice et aux interactions entre la silice et différents composés. Cette technique a par la suite été utilisée pour évaluer les interactions polymère/silice, à partir de composés sonde représentatifs du polymère. Dans le domaine des caoutchoucs renforcés par de la silice, les alcanes, méthylalcanes, alcènes ou composés aromatiques (benzène et alkylbenzène) ont été utilisés afin d’évaluer la contribution en terme d’interactions des groupes fonctionnels présents dans différents élastomères [13, 20]. Dans le cas des élastomères silicones, des oligomères PDMS linéaires et cycliques ont été étudiés, en réalisant des mesures d’IGC avec des surfaces de silice non greffées et greffées [16]. Il est montré que les oligomères PDMS constituent des composés sonde plus sensibles aux variations de la chimie de surface de la
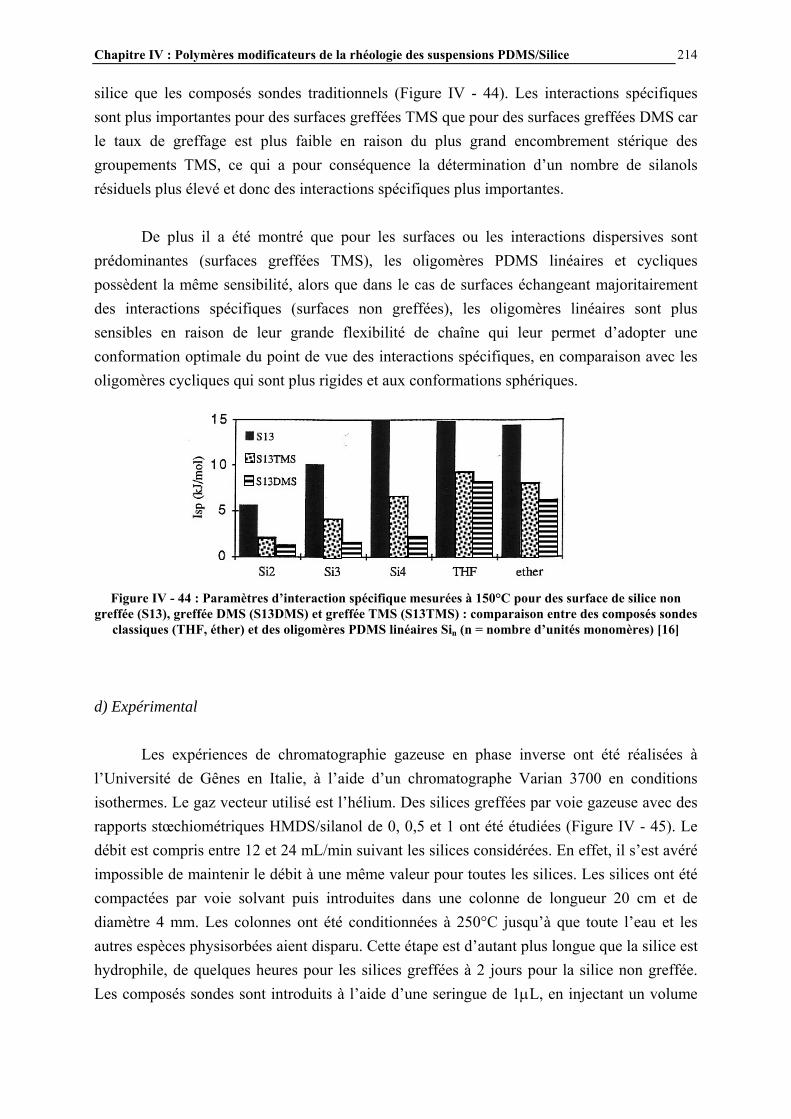
Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
214
silice que les composés sondes traditionnels (Figure IV - 44). Les interactions spécifiques sont plus importantes pour des surfaces greffées TMS que pour des surfaces greffées DMS car le taux de greffage est plus faible en raison du plus grand encombrement stérique des groupements TMS, ce qui a pour conséquence la détermination d’un nombre de silanols résiduels plus élevé et donc des interactions spécifiques plus importantes. De plus il a été montré que pour les surfaces ou les interactions dispersives sont prédominantes (surfaces greffées TMS), les oligomères PDMS linéaires et cycliques possèdent la même sensibilité, alors que dans le cas de surfaces échangeant majoritairement des interactions spécifiques (surfaces non greffées), les oligomères linéaires sont plus sensibles en raison de leur grande flexibilité de chaîne qui leur permet d’adopter une conformation optimale du point de vue des interactions spécifiques, en comparaison avec les oligomères cycliques qui sont plus rigides et aux conformations sphériques.
Figure IV - 44 : Paramètres d’interaction spécifique mesurées à 150°C pour des surface de silice non greffée (S13), greffée DMS (S13DMS) et greffée TMS (S13TMS) : comparaison entre des composés sondes
classiques (THF, éther) et des oligomères PDMS linéaires Sin (n = nombre d’unités monomères) [16]
d) Expérimental Les expériences de chromatographie gazeuse en phase inverse ont été réalisées à l’Université de Gênes en Italie, à l’aide d’un chromatographe Varian 3700 en conditions isothermes. Le gaz vecteur utilisé est l’hélium. Des silices greffées par voie gazeuse avec des rapports stœchiométriques HMDS/silanol de 0, 0,5 et 1 ont été étudiées (Figure IV - 45). Le débit est compris entre 12 et 24 mL/min suivant les silices considérées. En effet, il s’est avéré impossible de maintenir le débit à une même valeur pour toutes les silices. Les silices ont été compactées par voie solvant puis introduites dans une colonne de longueur 20 cm et de diamètre 4 mm. Les colonnes ont été conditionnées à 250°C jusqu’à que toute l’eau et les autres espèces physisorbées aient disparu. Cette étape est d’autant plus longue que la silice est hydrophile, de quelques heures pour les silices greffées à 2 jours pour la silice non greffée. Les composés sondes sont introduits à l’aide d’une seringue de 1µL, en injectant un volume
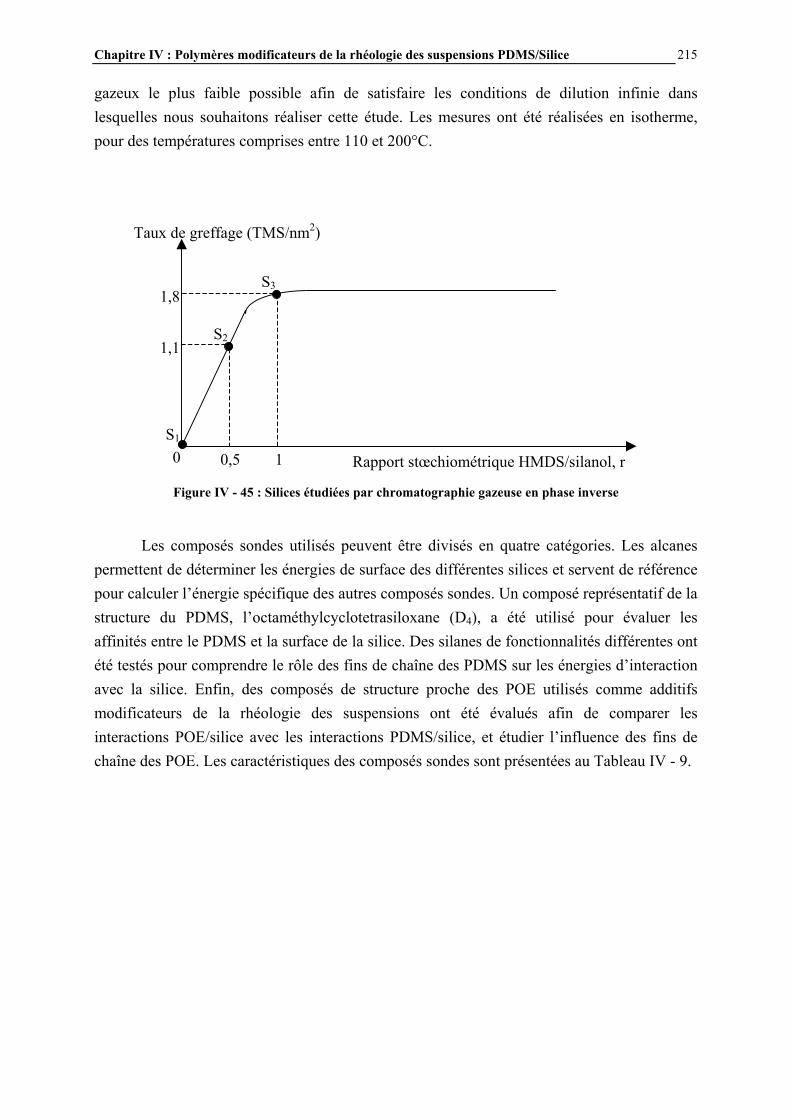
Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
215
gazeux le plus faible possible afin de satisfaire les conditions de dilution infinie dans lesquelles nous souhaitons réaliser cette étude. Les mesures ont été réalisées en isotherme, pour des températures comprises entre 110 et 200°C.
Figure IV - 45 : Silices étudiées par chromatographie gazeuse en phase inverse
Les composés sondes utilisés peuvent être divisés en quatre catégories. Les alcanes permettent de déterminer les énergies de surface des différentes silices et servent de référence pour calculer l’énergie spécifique des autres composés sondes. Un composé représentatif de la structure du PDMS, l’octaméthylcyclotetrasiloxane (D4), a été utilisé pour évaluer les affinités entre le PDMS et la surface de la silice. Des silanes de fonctionnalités différentes ont été testés pour comprendre le rôle des fins de chaîne des PDMS sur les énergies d’interaction avec la silice. Enfin, des composés de structure proche des POE utilisés comme additifs modificateurs de la rhéologie des suspensions ont été évalués afin de comparer les interactions POE/silice avec les interactions PDMS/silice, et étudier l’influence des fins de chaîne des POE. Les caractéristiques des composés sondes sont présentées au Tableau IV - 9.
0 0,5 1 Rapport stœchiométrique HMDS/silanol, r
Taux de greffage (TMS/nm2)
S1
S2
S3
1,1
1,8
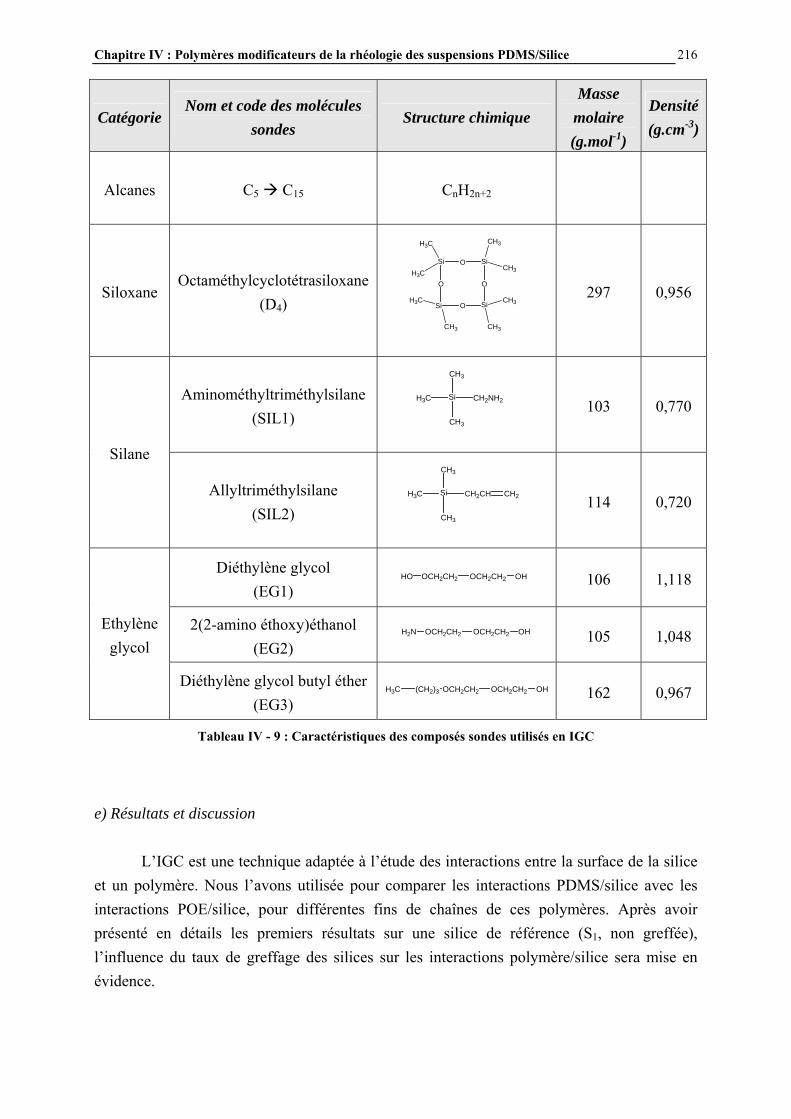
Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
216
Catégorie Nom et code des molécules
sondes Structure chimique
Masse molaire (g.mol-1)
Densité (g.cm-3)
Alcanes
C5 C15
CnH2n+2
Siloxane Octaméthylcyclotétrasiloxane
(D4)
297 0,956
Aminométhyltriméthylsilane (SIL1)
103 0,770
Silane
Allyltriméthylsilane (SIL2)
114 0,720
Diéthylène glycol (EG1)
106 1,118
2(2-amino éthoxy)éthanol (EG2)
105 1,048 Ethylène
glycol
Diéthylène glycol butyl éther (EG3)
162 0,967
Tableau IV - 9 : Caractéristiques des composés sondes utilisés en IGC
e) Résultats et discussion L’IGC est une technique adaptée à l’étude des interactions entre la surface de la silice et un polymère. Nous l’avons utilisée pour comparer les interactions PDMS/silice avec les interactions POE/silice, pour différentes fins de chaînes de ces polymères. Après avoir présenté en détails les premiers résultats sur une silice de référence (S1, non greffée), l’influence du taux de greffage des silices sur les interactions polymère/silice sera mise en évidence.
Si O Si
OO
Si O Si
CH3
CH3
H3C
H3C
CH3
CH3CH3
H3C
H3C Si
CH3
CH3
CH2NH2
H3C Si
CH3
CH3
CH2CH CH2
(CH2)3 OCH2CH2H3C OCH2CH2 OH
OCH2CH2 OCH2CH2 OHHO
OCH2CH2 OCH2CH2 OHH2N
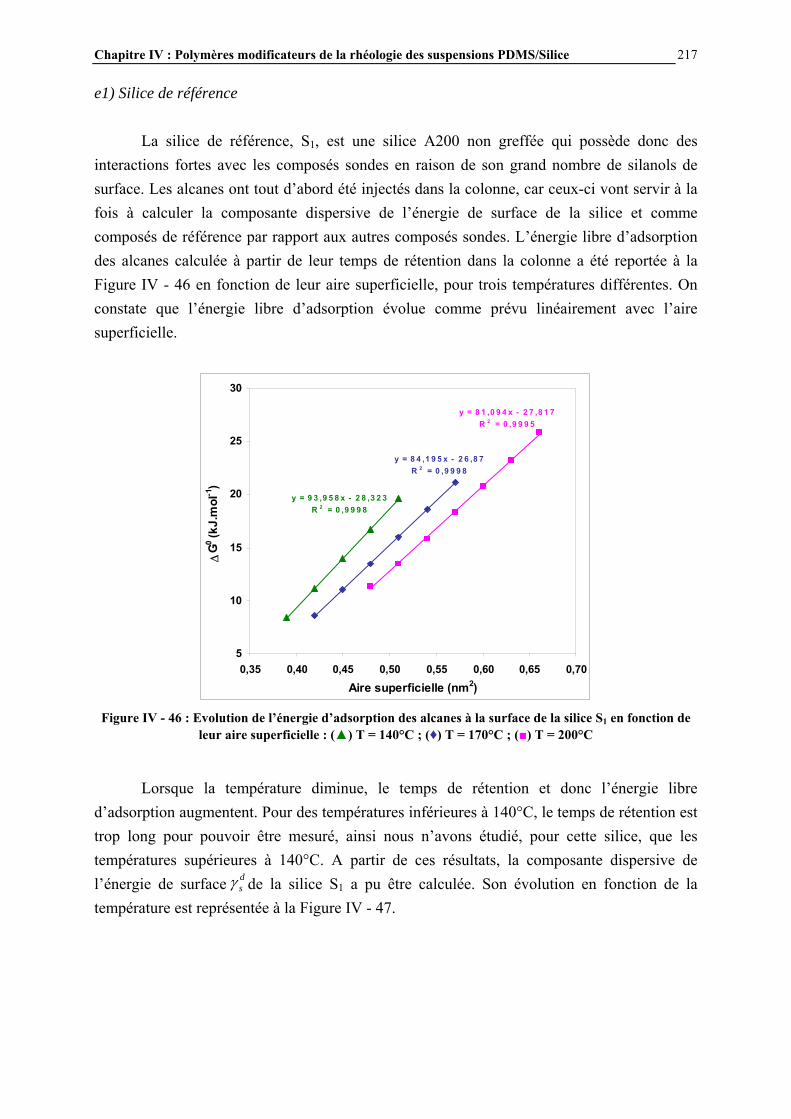
Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
217
e1) Silice de référence La silice de référence, S1, est une silice A200 non greffée qui possède donc des interactions fortes avec les composés sondes en raison de son grand nombre de silanols de surface. Les alcanes ont tout d’abord été injectés dans la colonne, car ceux-ci vont servir à la fois à calculer la composante dispersive de l’énergie de surface de la silice et comme composés de référence par rapport aux autres composés sondes. L’énergie libre d’adsorption des alcanes calculée à partir de leur temps de rétention dans la colonne a été reportée à la Figure IV - 46 en fonction de leur aire superficielle, pour trois températures différentes. On constate que l’énergie libre d’adsorption évolue comme prévu linéairement avec l’aire superficielle.
Figure IV - 46 : Evolution de l’énergie d’adsorption des alcanes à la surface de la silice S1 en fonction de leur aire superficielle : (▲) T = 140°C ; (♦) T = 170°C ; (■) T = 200°C
Lorsque la température diminue, le temps de rétention et donc l’énergie libre d’adsorption augmentent. Pour des températures inférieures à 140°C, le temps de rétention est trop long pour pouvoir être mesuré, ainsi nous n’avons étudié, pour cette silice, que les températures supérieures à 140°C. A partir de ces résultats, la composante dispersive de l’énergie de surface de la silice S1 a pu être calculée. Son évolution en fonction de la température est représentée à la Figure IV - 47.
y = 8 1 ,0 9 4 x - 2 7 ,8 1 7R 2 = 0 ,9 9 9 5
y = 8 4 ,1 9 5 x - 2 6 ,8 7R 2 = 0 ,9 9 9 8
y = 9 3 ,9 5 8 x - 2 8 ,3 2 3R 2 = 0 ,9 9 9 8
5
10
15
20
25
30
0,35 0,40 0,45 0,50 0,55 0,60 0,65 0,70Aire superficielle (nm2)
∆G0 (k
J.m
ol-1
)
dsγ
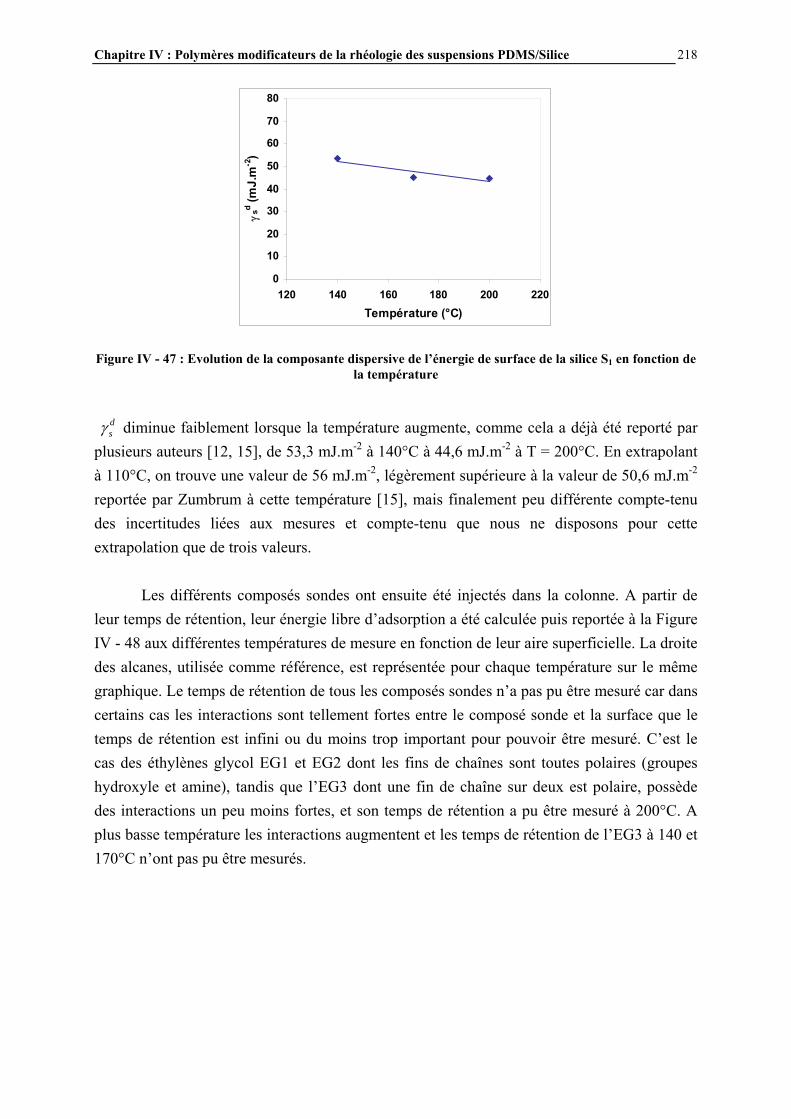
Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
218
Figure IV - 47 : Evolution de la composante dispersive de l’énergie de surface de la silice S1 en fonction de
la température
d
sγ diminue faiblement lorsque la température augmente, comme cela a déjà été reporté par plusieurs auteurs [12, 15], de 53,3 mJ.m-2 à 140°C à 44,6 mJ.m-2 à T = 200°C. En extrapolant à 110°C, on trouve une valeur de 56 mJ.m-2, légèrement supérieure à la valeur de 50,6 mJ.m-2 reportée par Zumbrum à cette température [15], mais finalement peu différente compte-tenu des incertitudes liées aux mesures et compte-tenu que nous ne disposons pour cette extrapolation que de trois valeurs. Les différents composés sondes ont ensuite été injectés dans la colonne. A partir de leur temps de rétention, leur énergie libre d’adsorption a été calculée puis reportée à la Figure IV - 48 aux différentes températures de mesure en fonction de leur aire superficielle. La droite des alcanes, utilisée comme référence, est représentée pour chaque température sur le même graphique. Le temps de rétention de tous les composés sondes n’a pas pu être mesuré car dans certains cas les interactions sont tellement fortes entre le composé sonde et la surface que le temps de rétention est infini ou du moins trop important pour pouvoir être mesuré. C’est le cas des éthylènes glycol EG1 et EG2 dont les fins de chaînes sont toutes polaires (groupes hydroxyle et amine), tandis que l’EG3 dont une fin de chaîne sur deux est polaire, possède des interactions un peu moins fortes, et son temps de rétention a pu être mesuré à 200°C. A plus basse température les interactions augmentent et les temps de rétention de l’EG3 à 140 et 170°C n’ont pas pu être mesurés.
0
10
20
30
40
50
60
70
80
120 140 160 180 200 220Température (°C)
γ sd (m
J.m
-2)
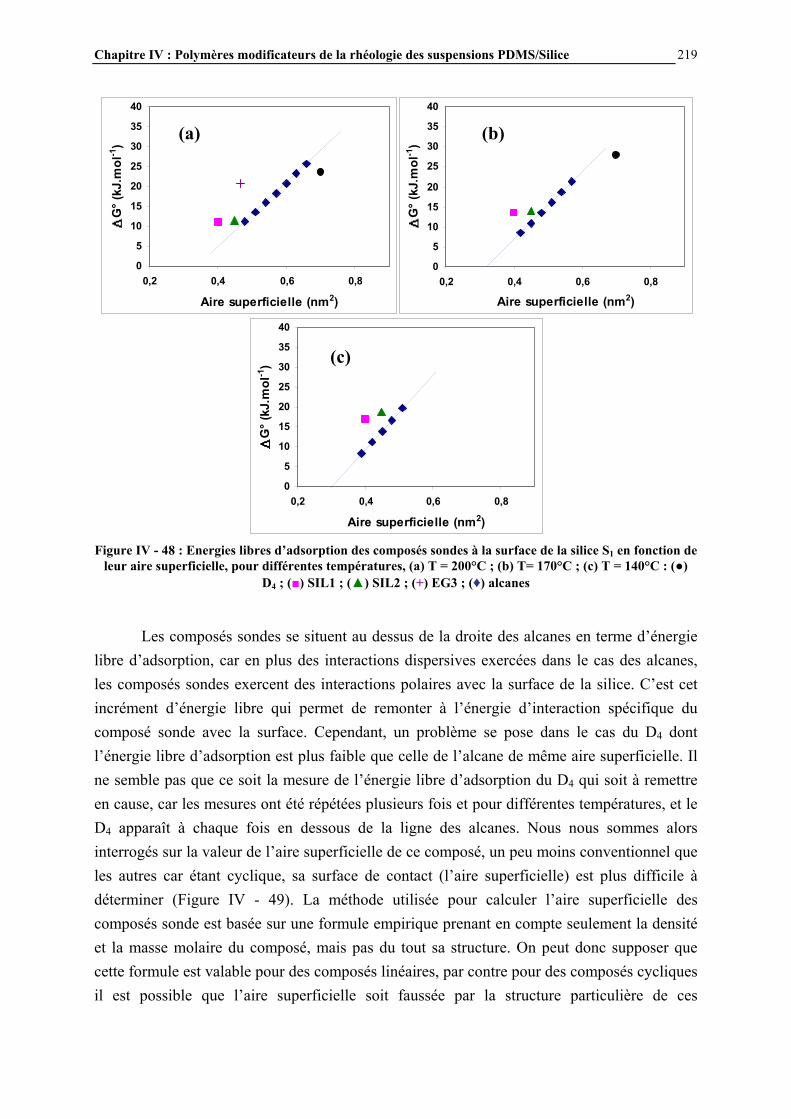
Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
219
Figure IV - 48 : Energies libres d’adsorption des composés sondes à la surface de la silice S1 en fonction de
leur aire superficielle, pour différentes températures, (a) T = 200°C ; (b) T= 170°C ; (c) T = 140°C : (●) D4 ; (■) SIL1 ; (▲) SIL2 ; (+) EG3 ; (♦) alcanes
Les composés sondes se situent au dessus de la droite des alcanes en terme d’énergie libre d’adsorption, car en plus des interactions dispersives exercées dans le cas des alcanes, les composés sondes exercent des interactions polaires avec la surface de la silice. C’est cet incrément d’énergie libre qui permet de remonter à l’énergie d’interaction spécifique du composé sonde avec la surface. Cependant, un problème se pose dans le cas du D4 dont l’énergie libre d’adsorption est plus faible que celle de l’alcane de même aire superficielle. Il ne semble pas que ce soit la mesure de l’énergie libre d’adsorption du D4 qui soit à remettre en cause, car les mesures ont été répétées plusieurs fois et pour différentes températures, et le D4 apparaît à chaque fois en dessous de la ligne des alcanes. Nous nous sommes alors interrogés sur la valeur de l’aire superficielle de ce composé, un peu moins conventionnel que les autres car étant cyclique, sa surface de contact (l’aire superficielle) est plus difficile à déterminer (Figure IV - 49). La méthode utilisée pour calculer l’aire superficielle des composés sonde est basée sur une formule empirique prenant en compte seulement la densité et la masse molaire du composé, mais pas du tout sa structure. On peut donc supposer que cette formule est valable pour des composés linéaires, par contre pour des composés cycliques il est possible que l’aire superficielle soit faussée par la structure particulière de ces
0
5
10
15
20
25
30
35
40
0,2 0,4 0,6 0,8
Aire superficielle (nm2)
G° (
kJ.m
ol-1
)
0
5
10
15
20
25
30
35
40
0,2 0,4 0,6 0,8
Aire superficielle (nm2)
G°
(kJ.
mol
-1)
0
5
10
15
20
25
30
35
40
0,2 0,4 0,6 0,8
Aire superficielle (nm2)
G°
(kJ.
mol
-1)
(a) (b)
(c)

Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
220
composés, qui peuvent s’adsorber suivant différentes configurations. Nous avons alors essayé d’estimer l’aire superficielle de la molécule de D4 par des méthodes géométriques, à partir du volume de la molécule ou de la surface formée par le plan principal (Si-0)4. Ces deux méthodes sont illustrées à la Figure IV - 50. La méthode de calcul à partir du volume de la molécule est basée sur le fait que la molécule de D4 est presque sphérique. La mesure du volume de la molécule permet de remonter au rayon équivalent de la sphère à laquelle peut être assimilée la molécule, et l’aire superficielle peut être estimée par le disque dont le rayon est celui de la sphère (a). On peut également considérer que le plan d’adsorption principal de la molécule de D4 est le plan constitué des motifs Si-0, et l’aire superficielle de la molécule peut alors être estimée par la surface formée par ce plan (b). Enfin, des mesures d’IGC utilisant la molécule de D4 comme composé sonde ont été décrites dans la littérature [16]. Le paramètre moléculaire utilisé n’est pas l’aire superficielle mais la pression partielle des composés. Cependant, en sachant que le paramètre moléculaire du D4 donné dans cette étude est le même que celui de l’alcane C10, on en déduit que les aires superficielles de ces deux composés sont identiques. Connaissant l’aire superficielle du C10, on en déduit celle du D4, c’est-à-dire 0,51. Le Tableau IV - 10 compare les valeurs d’aire superficielle du D4 obtenues suivant ces différentes méthodes de détermination.
Figure IV - 49 : Représentations spatiales de la molécule de D4
Figure IV - 50 : Méthodes de calcul de l’aire superficielle du D4 à partir de considérations géométriques :
(a) à partir du volume de la molécule ; (b) à partir de la surface du plan principal
a
r
(a) (b)
a
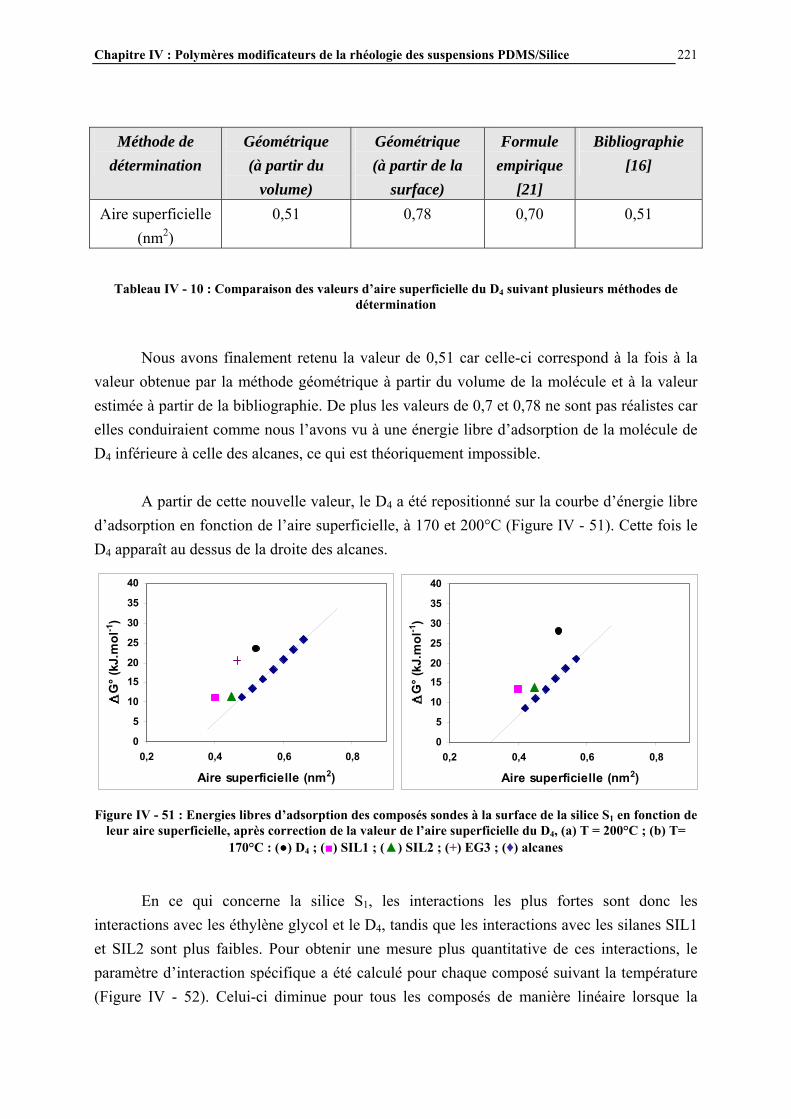
Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
221
Méthode de détermination
Géométrique (à partir du
volume)
Géométrique (à partir de la
surface)
Formule empirique
[21]
Bibliographie [16]
Aire superficielle (nm2)
0,51 0,78 0,70 0,51
Tableau IV - 10 : Comparaison des valeurs d’aire superficielle du D4 suivant plusieurs méthodes de
détermination
Nous avons finalement retenu la valeur de 0,51 car celle-ci correspond à la fois à la valeur obtenue par la méthode géométrique à partir du volume de la molécule et à la valeur estimée à partir de la bibliographie. De plus les valeurs de 0,7 et 0,78 ne sont pas réalistes car elles conduiraient comme nous l’avons vu à une énergie libre d’adsorption de la molécule de D4 inférieure à celle des alcanes, ce qui est théoriquement impossible. A partir de cette nouvelle valeur, le D4 a été repositionné sur la courbe d’énergie libre d’adsorption en fonction de l’aire superficielle, à 170 et 200°C (Figure IV - 51). Cette fois le D4 apparaît au dessus de la droite des alcanes. Figure IV - 51 : Energies libres d’adsorption des composés sondes à la surface de la silice S1 en fonction de
leur aire superficielle, après correction de la valeur de l’aire superficielle du D4, (a) T = 200°C ; (b) T= 170°C : (●) D4 ; (■) SIL1 ; (▲) SIL2 ; (+) EG3 ; (♦) alcanes
En ce qui concerne la silice S1, les interactions les plus fortes sont donc les interactions avec les éthylène glycol et le D4, tandis que les interactions avec les silanes SIL1 et SIL2 sont plus faibles. Pour obtenir une mesure plus quantitative de ces interactions, le paramètre d’interaction spécifique a été calculé pour chaque composé suivant la température (Figure IV - 52). Celui-ci diminue pour tous les composés de manière linéaire lorsque la
0
5
10
15
20
25
30
35
40
0,2 0,4 0,6 0,8
Aire superficielle (nm2)
G° (
kJ.m
ol-1
)
0
5
10
15
20
25
30
35
40
0,2 0,4 0,6 0,8
Aire superficielle (nm2)
G°
(kJ.
mol
-1)
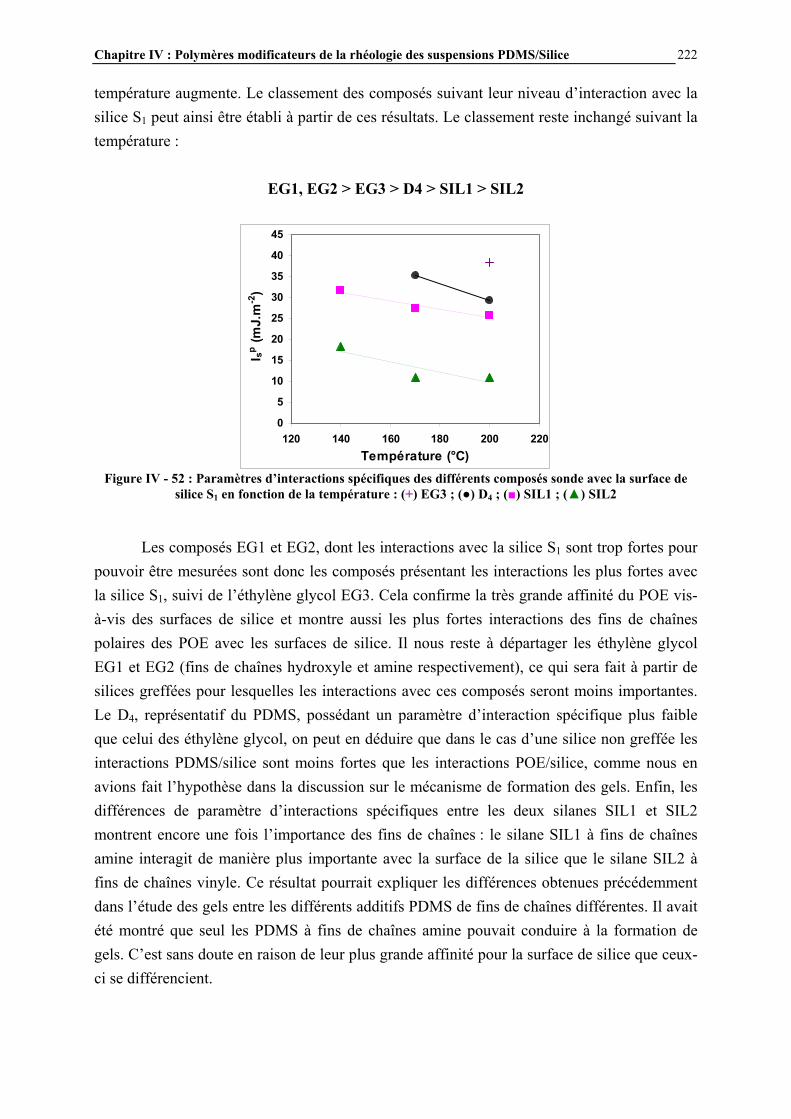
Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
222
température augmente. Le classement des composés suivant leur niveau d’interaction avec la silice S1 peut ainsi être établi à partir de ces résultats. Le classement reste inchangé suivant la température :
EG1, EG2 > EG3 > D4 > SIL1 > SIL2
Figure IV - 52 : Paramètres d’interactions spécifiques des différents composés sonde avec la surface de silice S1 en fonction de la température : (+) EG3 ; (●) D4 ; (■) SIL1 ; (▲) SIL2
Les composés EG1 et EG2, dont les interactions avec la silice S1 sont trop fortes pour pouvoir être mesurées sont donc les composés présentant les interactions les plus fortes avec la silice S1, suivi de l’éthylène glycol EG3. Cela confirme la très grande affinité du POE vis-à-vis des surfaces de silice et montre aussi les plus fortes interactions des fins de chaînes polaires des POE avec les surfaces de silice. Il nous reste à départager les éthylène glycol EG1 et EG2 (fins de chaînes hydroxyle et amine respectivement), ce qui sera fait à partir de silices greffées pour lesquelles les interactions avec ces composés seront moins importantes. Le D4, représentatif du PDMS, possédant un paramètre d’interaction spécifique plus faible que celui des éthylène glycol, on peut en déduire que dans le cas d’une silice non greffée les interactions PDMS/silice sont moins fortes que les interactions POE/silice, comme nous en avions fait l’hypothèse dans la discussion sur le mécanisme de formation des gels. Enfin, les différences de paramètre d’interactions spécifiques entre les deux silanes SIL1 et SIL2 montrent encore une fois l’importance des fins de chaînes : le silane SIL1 à fins de chaînes amine interagit de manière plus importante avec la surface de la silice que le silane SIL2 à fins de chaînes vinyle. Ce résultat pourrait expliquer les différences obtenues précédemment dans l’étude des gels entre les différents additifs PDMS de fins de chaînes différentes. Il avait été montré que seul les PDMS à fins de chaînes amine pouvait conduire à la formation de gels. C’est sans doute en raison de leur plus grande affinité pour la surface de silice que ceux-ci se différencient.
0
5
10
15
20
25
30
35
40
45
120 140 160 180 200 220Température (°C)
I sp (m
J.m
-2)
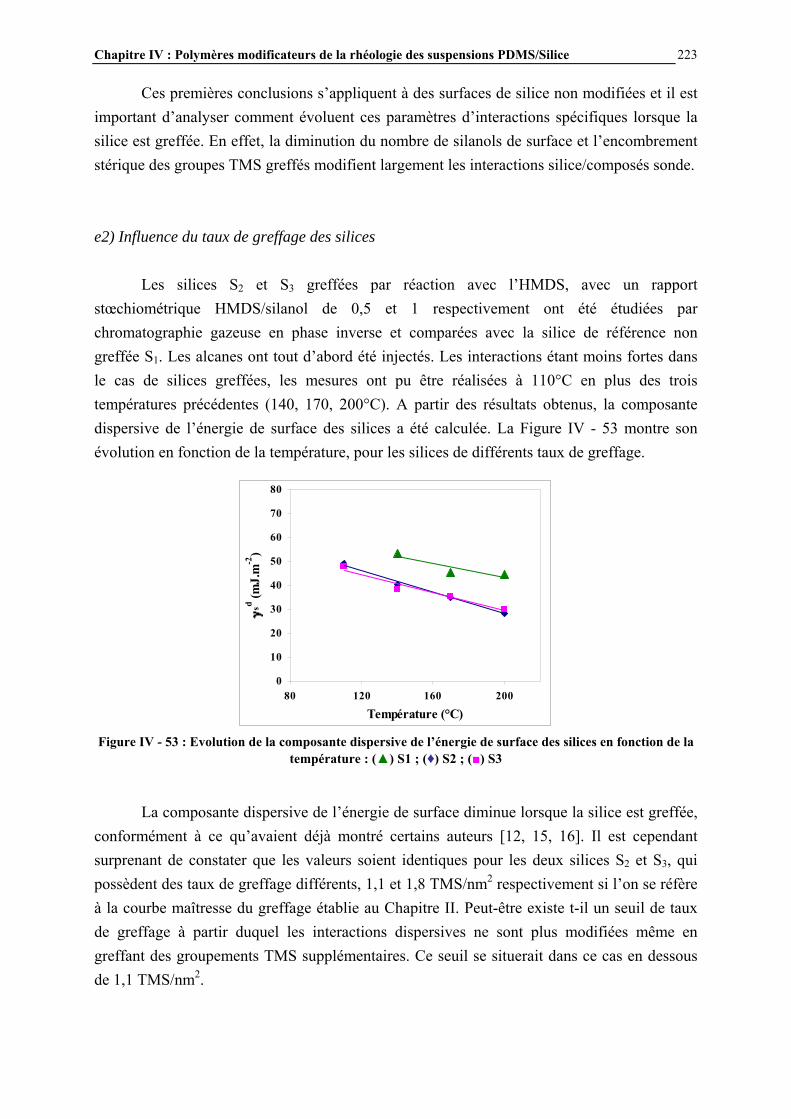
Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
223
Ces premières conclusions s’appliquent à des surfaces de silice non modifiées et il est important d’analyser comment évoluent ces paramètres d’interactions spécifiques lorsque la silice est greffée. En effet, la diminution du nombre de silanols de surface et l’encombrement stérique des groupes TMS greffés modifient largement les interactions silice/composés sonde. e2) Influence du taux de greffage des silices Les silices S2 et S3 greffées par réaction avec l’HMDS, avec un rapport stœchiométrique HMDS/silanol de 0,5 et 1 respectivement ont été étudiées par chromatographie gazeuse en phase inverse et comparées avec la silice de référence non greffée S1. Les alcanes ont tout d’abord été injectés. Les interactions étant moins fortes dans le cas de silices greffées, les mesures ont pu être réalisées à 110°C en plus des trois températures précédentes (140, 170, 200°C). A partir des résultats obtenus, la composante dispersive de l’énergie de surface des silices a été calculée. La Figure IV - 53 montre son évolution en fonction de la température, pour les silices de différents taux de greffage. Figure IV - 53 : Evolution de la composante dispersive de l’énergie de surface des silices en fonction de la
température : (▲) S1 ; (♦) S2 ; (■) S3
La composante dispersive de l’énergie de surface diminue lorsque la silice est greffée, conformément à ce qu’avaient déjà montré certains auteurs [12, 15, 16]. Il est cependant surprenant de constater que les valeurs soient identiques pour les deux silices S2 et S3, qui possèdent des taux de greffage différents, 1,1 et 1,8 TMS/nm2 respectivement si l’on se réfère à la courbe maîtresse du greffage établie au Chapitre II. Peut-être existe t-il un seuil de taux de greffage à partir duquel les interactions dispersives ne sont plus modifiées même en greffant des groupements TMS supplémentaires. Ce seuil se situerait dans ce cas en dessous de 1,1 TMS/nm2.
0
10
20
30
40
50
60
70
80
80 120 160 200
Température (°C)
sd (mJ.
m-2
)
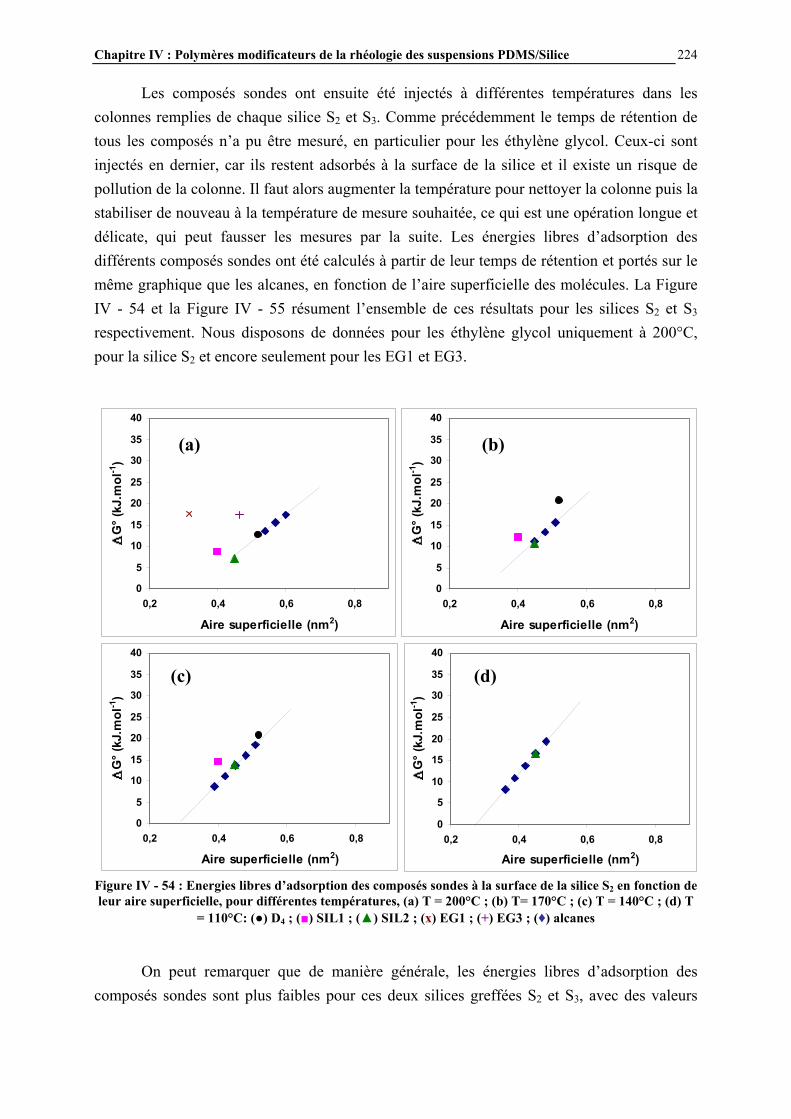
Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
224
Les composés sondes ont ensuite été injectés à différentes températures dans les colonnes remplies de chaque silice S2 et S3. Comme précédemment le temps de rétention de tous les composés n’a pu être mesuré, en particulier pour les éthylène glycol. Ceux-ci sont injectés en dernier, car ils restent adsorbés à la surface de la silice et il existe un risque de pollution de la colonne. Il faut alors augmenter la température pour nettoyer la colonne puis la stabiliser de nouveau à la température de mesure souhaitée, ce qui est une opération longue et délicate, qui peut fausser les mesures par la suite. Les énergies libres d’adsorption des différents composés sondes ont été calculés à partir de leur temps de rétention et portés sur le même graphique que les alcanes, en fonction de l’aire superficielle des molécules. La Figure IV - 54 et la Figure IV - 55 résument l’ensemble de ces résultats pour les silices S2 et S3 respectivement. Nous disposons de données pour les éthylène glycol uniquement à 200°C, pour la silice S2 et encore seulement pour les EG1 et EG3. Figure IV - 54 : Energies libres d’adsorption des composés sondes à la surface de la silice S2 en fonction de leur aire superficielle, pour différentes températures, (a) T = 200°C ; (b) T= 170°C ; (c) T = 140°C ; (d) T
= 110°C: (●) D4 ; (■) SIL1 ; (▲) SIL2 ; (x) EG1 ; (+) EG3 ; (♦) alcanes
On peut remarquer que de manière générale, les énergies libres d’adsorption des composés sondes sont plus faibles pour ces deux silices greffées S2 et S3, avec des valeurs
0
5
10
15
20
25
30
35
40
0,2 0,4 0,6 0,8
Aire superficielle (nm2)
G° (
kJ.m
ol-1
)
0
5
10
15
20
25
30
35
40
0,2 0,4 0,6 0,8
Aire superficielle (nm2)
G° (
kJ.m
ol-1
)
0
5
10
15
20
25
30
35
40
0,2 0,4 0,6 0,8
Aire superficielle (nm2)
G°
(kJ.
mol
-1)
0
5
10
15
20
25
30
35
40
0,2 0,4 0,6 0,8
Aire superficielle (nm2)
G° (
kJ.m
ol-1
)
(a) (b)
(c) (d)
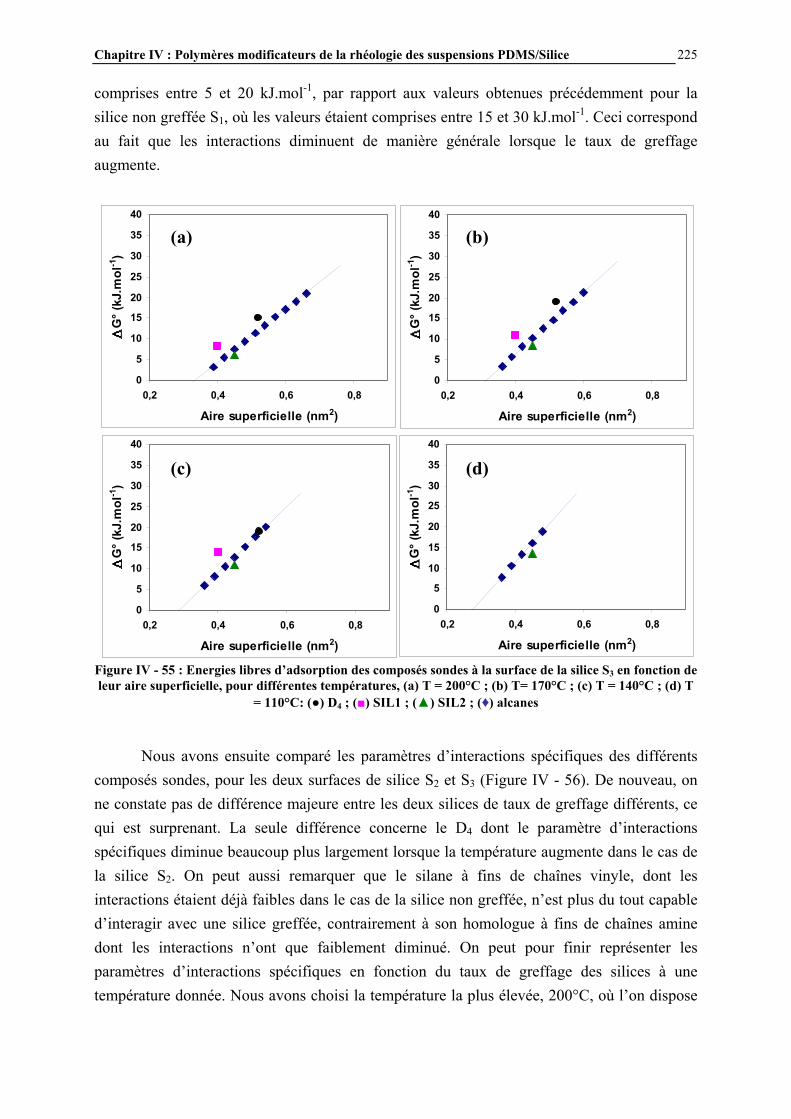
Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
225
comprises entre 5 et 20 kJ.mol-1, par rapport aux valeurs obtenues précédemment pour la silice non greffée S1, où les valeurs étaient comprises entre 15 et 30 kJ.mol-1. Ceci correspond au fait que les interactions diminuent de manière générale lorsque le taux de greffage augmente. Figure IV - 55 : Energies libres d’adsorption des composés sondes à la surface de la silice S3 en fonction de leur aire superficielle, pour différentes températures, (a) T = 200°C ; (b) T= 170°C ; (c) T = 140°C ; (d) T
= 110°C: (●) D4 ; (■) SIL1 ; (▲) SIL2 ; (♦) alcanes
Nous avons ensuite comparé les paramètres d’interactions spécifiques des différents composés sondes, pour les deux surfaces de silice S2 et S3 (Figure IV - 56). De nouveau, on ne constate pas de différence majeure entre les deux silices de taux de greffage différents, ce qui est surprenant. La seule différence concerne le D4 dont le paramètre d’interactions spécifiques diminue beaucoup plus largement lorsque la température augmente dans le cas de la silice S2. On peut aussi remarquer que le silane à fins de chaînes vinyle, dont les interactions étaient déjà faibles dans le cas de la silice non greffée, n’est plus du tout capable d’interagir avec une silice greffée, contrairement à son homologue à fins de chaînes amine dont les interactions n’ont que faiblement diminué. On peut pour finir représenter les paramètres d’interactions spécifiques en fonction du taux de greffage des silices à une température donnée. Nous avons choisi la température la plus élevée, 200°C, où l’on dispose
0
5
10
15
20
25
30
35
40
0,2 0,4 0,6 0,8
Aire superficielle (nm2)
G°
(kJ.
mol
-1)
0
5
10
15
20
25
30
35
40
0,2 0,4 0,6 0,8
Aire superficielle (nm2)G
° (k
J.m
ol-1
)
0
5
10
15
20
25
30
35
40
0,2 0,4 0,6 0,8
Aire superficielle (nm2)
G°
(kJ.
mol
-1)
0
5
10
15
20
25
30
35
40
0,2 0,4 0,6 0,8
Aire superficielle (nm2)
G° (
kJ.m
ol-1
)
(a) (b)
(c) (d)
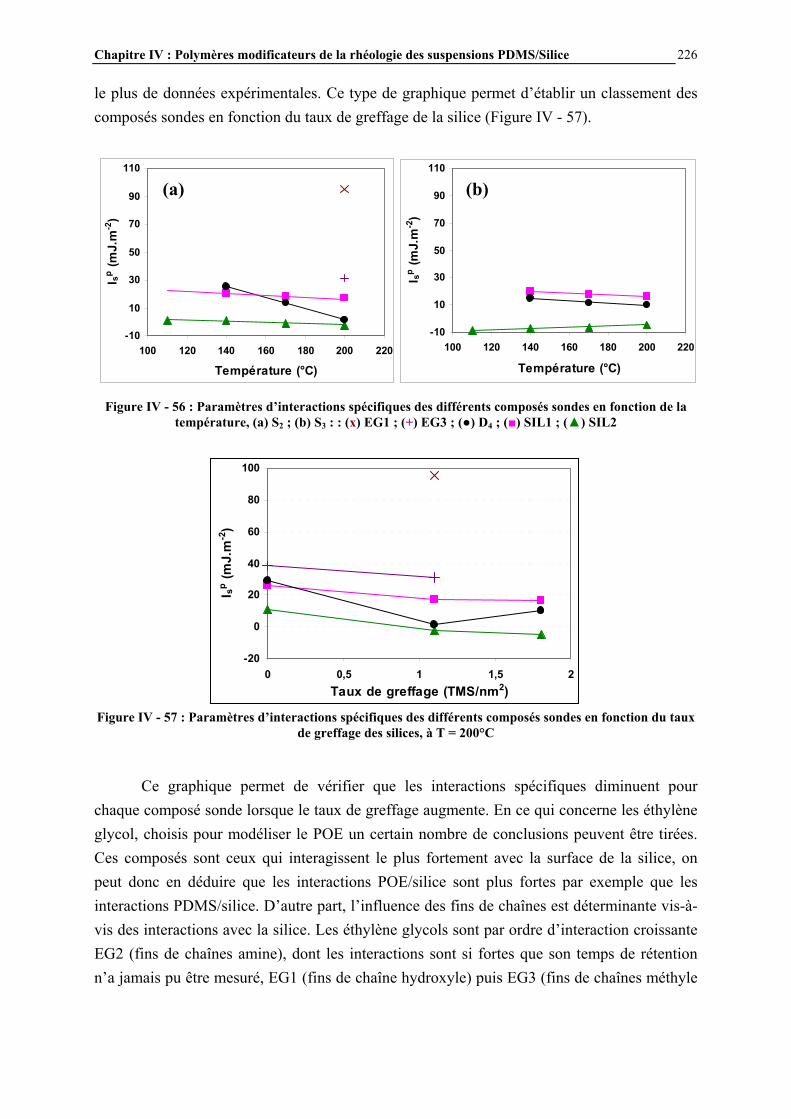
Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
226
le plus de données expérimentales. Ce type de graphique permet d’établir un classement des composés sondes en fonction du taux de greffage de la silice (Figure IV - 57).
Figure IV - 56 : Paramètres d’interactions spécifiques des différents composés sondes en fonction de la température, (a) S2 ; (b) S3 : : (x) EG1 ; (+) EG3 ; (●) D4 ; (■) SIL1 ; (▲) SIL2
Figure IV - 57 : Paramètres d’interactions spécifiques des différents composés sondes en fonction du taux
de greffage des silices, à T = 200°C
Ce graphique permet de vérifier que les interactions spécifiques diminuent pour chaque composé sonde lorsque le taux de greffage augmente. En ce qui concerne les éthylène glycol, choisis pour modéliser le POE un certain nombre de conclusions peuvent être tirées. Ces composés sont ceux qui interagissent le plus fortement avec la surface de la silice, on peut donc en déduire que les interactions POE/silice sont plus fortes par exemple que les interactions PDMS/silice. D’autre part, l’influence des fins de chaînes est déterminante vis-à-vis des interactions avec la silice. Les éthylène glycols sont par ordre d’interaction croissante EG2 (fins de chaînes amine), dont les interactions sont si fortes que son temps de rétention n’a jamais pu être mesuré, EG1 (fins de chaîne hydroxyle) puis EG3 (fins de chaînes méthyle
-10
10
30
50
70
90
110
100 120 140 160 180 200 220
Température (°C)
I sp (m
J.m
-2)
-10
10
30
50
70
90
110
100 120 140 160 180 200 220
Température (°C)
I sp (m
J.m
-2)
(a) (b)
-20
0
20
40
60
80
100
0 0,5 1 1,5 2Taux de greffage (TMS/nm2)
I sp (m
J.m
-2)

Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
227
et hydroxyle).Ceci nous permet de classer les fins de chaînes POE suivant leur affinité pour la surface de silice : POE : NH2>OH>Me La plus grande affinité des groupements amine vis-à-vis de la surface de la silice a également été confirmée par la comparaison de silanes SIL1 (fins de chaînes amine) et SIL2 (fins de chaînes vinyle). Le paramètre d’interaction spécifique de SIL1 est plus élevé, en particulier lorsque la silice est greffée car le paramètre d’interaction spécifique de SIL1 diminue seulement faiblement avec le taux de greffage de la silice, tandis que celui de SIL2 devient nul. Cela prouve que les groupements amine sont capables malgré l’encombrement stérique des groupements TMS, de venir s’adsorber sur les silanols de surface restants. Ces fortes interactions expliquent sans doute que les additifs polymères (PDMS ou POE) à fins de chaînes amine sont plus efficaces pour former des gels dans les suspensions PDMS/silice, car le mécanisme de formation des gels proposé fait intervenir l’affinité des polymères modificateurs pour les surfaces de silice. IV.4.5.2- Mesures des énergies de surface et des énergies interfaciales La chromatographie gazeuse en phase inverse permet d’évaluer les interactions entre les polymères modificateurs et la surface de la silice. Cependant le mécanisme de formation des gels dans les suspensions PDMS/silice fait également intervenir les interfaces PDMS/polymère modificateur. La raison de l’efficacité des additifs POE, hormis leur affinité pour la surface de la silice, est leur non miscibilité avec le milieu de suspension PDMS. Ce phénomène peut être appréhendé par les énergies de surface des PDMS et des POE, ainsi que par les énergies interfaciales PDMS/POE, permettant d’évaluer les affinités entre ces deux polymères. La mesure des énergies de surface et des énergies interfaciales fait l’objet de ce dernier paragraphe. a) expérimental a1) Technique de la goutte pendante Cette technique est basée sur la mesure des dimensions d’une goutte du liquide à caractériser à l’extrémité d’un capillaire. Lorsque la goutte, en équilibre au bout d’un capillaire, atteint un état stable, ses dimensions sont mesurées à partir de l’image du profil de
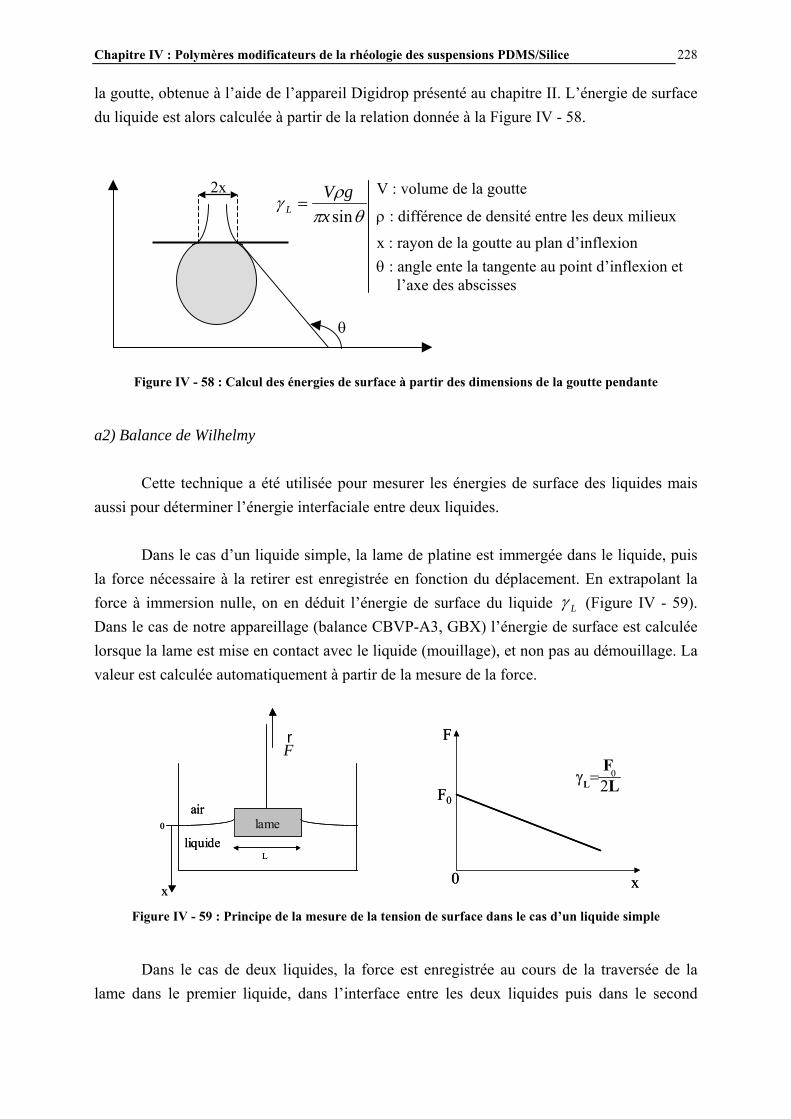
Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
228
la goutte, obtenue à l’aide de l’appareil Digidrop présenté au chapitre II. L’énergie de surface du liquide est alors calculée à partir de la relation donnée à la Figure IV - 58.
V : volume de la goutte
ρ : différence de densité entre les deux milieux
x : rayon de la goutte au plan d’inflexion θ : angle ente la tangente au point d’inflexion et l’axe des abscisses
Figure IV - 58 : Calcul des énergies de surface à partir des dimensions de la goutte pendante
a2) Balance de Wilhelmy Cette technique a été utilisée pour mesurer les énergies de surface des liquides mais aussi pour déterminer l’énergie interfaciale entre deux liquides. Dans le cas d’un liquide simple, la lame de platine est immergée dans le liquide, puis la force nécessaire à la retirer est enregistrée en fonction du déplacement. En extrapolant la force à immersion nulle, on en déduit l’énergie de surface du liquide Lγ (Figure IV - 59). Dans le cas de notre appareillage (balance CBVP-A3, GBX) l’énergie de surface est calculée lorsque la lame est mise en contact avec le liquide (mouillage), et non pas au démouillage. La valeur est calculée automatiquement à partir de la mesure de la force.
Figure IV - 59 : Principe de la mesure de la tension de surface dans le cas d’un liquide simple
Dans le cas de deux liquides, la force est enregistrée au cours de la traversée de la lame dans le premier liquide, dans l’interface entre les deux liquides puis dans le second
θ
2x
θπργsinx
gVL =
liquidelame
air
L
0
x
liquidelame
air
L
0
x
F0
x0
F
F0
x0
FFr
LF
L 20=γ
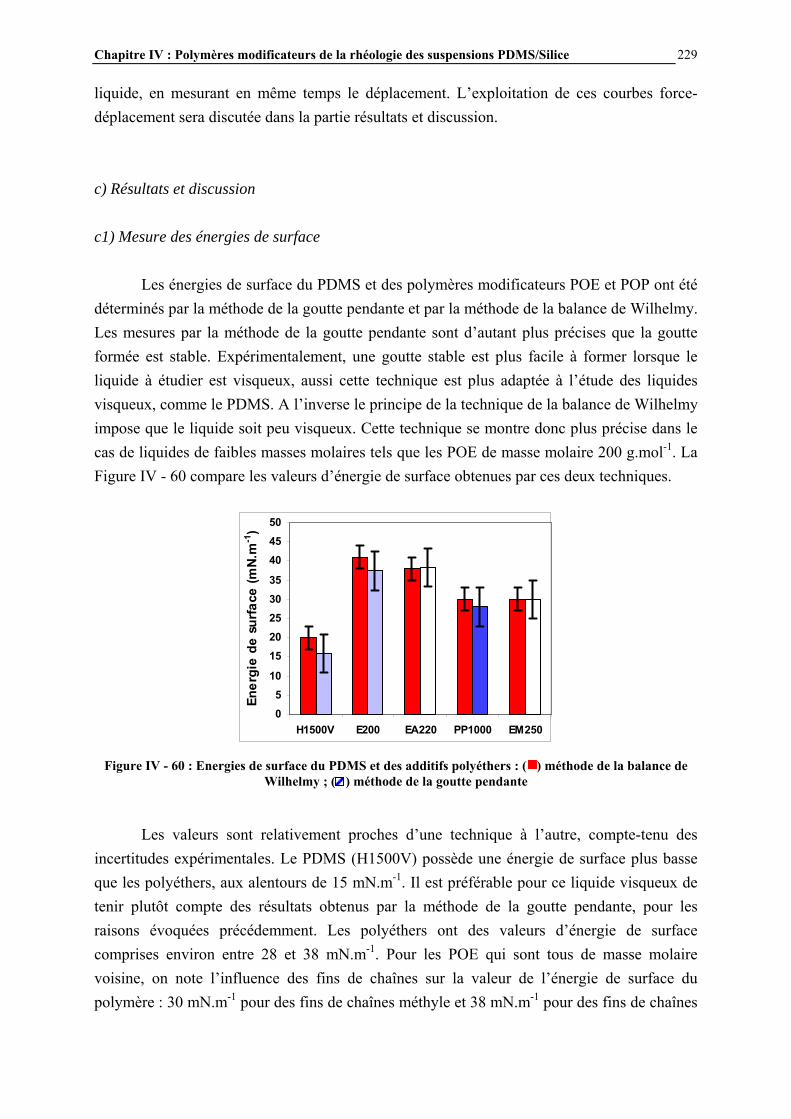
Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
229
liquide, en mesurant en même temps le déplacement. L’exploitation de ces courbes force-déplacement sera discutée dans la partie résultats et discussion. c) Résultats et discussion c1) Mesure des énergies de surface Les énergies de surface du PDMS et des polymères modificateurs POE et POP ont été déterminés par la méthode de la goutte pendante et par la méthode de la balance de Wilhelmy. Les mesures par la méthode de la goutte pendante sont d’autant plus précises que la goutte formée est stable. Expérimentalement, une goutte stable est plus facile à former lorsque le liquide à étudier est visqueux, aussi cette technique est plus adaptée à l’étude des liquides visqueux, comme le PDMS. A l’inverse le principe de la technique de la balance de Wilhelmy impose que le liquide soit peu visqueux. Cette technique se montre donc plus précise dans le cas de liquides de faibles masses molaires tels que les POE de masse molaire 200 g.mol-1. La Figure IV - 60 compare les valeurs d’énergie de surface obtenues par ces deux techniques.
Figure IV - 60 : Energies de surface du PDMS et des additifs polyéthers : ( ) méthode de la balance de Wilhelmy ; ( ) méthode de la goutte pendante
Les valeurs sont relativement proches d’une technique à l’autre, compte-tenu des incertitudes expérimentales. Le PDMS (H1500V) possède une énergie de surface plus basse que les polyéthers, aux alentours de 15 mN.m-1. Il est préférable pour ce liquide visqueux de tenir plutôt compte des résultats obtenus par la méthode de la goutte pendante, pour les raisons évoquées précédemment. Les polyéthers ont des valeurs d’énergie de surface comprises environ entre 28 et 38 mN.m-1. Pour les POE qui sont tous de masse molaire voisine, on note l’influence des fins de chaînes sur la valeur de l’énergie de surface du polymère : 30 mN.m-1 pour des fins de chaînes méthyle et 38 mN.m-1 pour des fins de chaînes
05
10
1520253035
404550
H1500V E200 EA220 PP1000 EM250
Ener
gie
de s
urfa
ce (m
N.m
-1)

Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
230
polaires (hydroxyle et amine). Le POP a une tension de surface plus faible de 28 mN.m-1 , mais cette valeur ne peut pas être comparée directement au POE car la masse molaire du POP est plus importante que la masse molaire du POE. Ces valeurs peuvent ensuite être comparées aux valeurs d’énergies de surface de silices hydrophiles et hydrophobes. L’énergie de surface de la silice hydrophile est équivalente à l’énergie de surface de l’eau, soit 72,8 mN.m-1, tandis que l’énergie de surface d’une silice hydrophobe peut être estimée à 30 mN.m-1 [22]. On retrouve d’ailleurs des valeurs similaires en mesurant les énergies de surface de wafers de silicium vierges (71 mN.m-1) et greffés par réaction avec l’HMDS (38 mN.m-1). Pour qu’une surface solide soit mouillée par un liquide il faut que l’énergie de surface du liquide soit inférieure à celle du solide. Les surfaces de silices hydrophiles sont donc mouillés à la fois par le PDMS et les additifs polyéthers. Dans le cas d’une silice hydrophobe, seul le PDMS mouille la surface. c2) Mesure des énergies interfaciales Les énergies interfaciales entre le PDMS et les polymères modificateurs peuvent être calculées à partir de leurs énergies de surface, ou mesurées expérimentalement par la méthode de la balance de Wilhelmy à deux liquides. Pour calculer l’énergie interfaciale entre deux liquides la relation suivante a été utilisée [23] : Equation IV - 9
où est l’énergie interfaciale entre les liquides 1 et 2 , l’énergie de surface, et les composantes dispersive et polaire de l’énergie de surface, respectivement. Il faut donc connaître les composantes polaire et dispersive de l’énergie de surface des liquides pour pouvoir calculer la tension interfaciale entre ces deux liquides. Pour cela une méthode consiste à mesurer l’angle de contact entre le liquide dont les composantes dispersives et polaires de l’énergie de surface sont à déterminer et une surface dont l’énergie et ses composantes dispersives et polaires sont connues. La surface de référence choisie est une surface de polystyrène réalisée par dépôt d’une couche fine sur un wafer de silicium (spray coating). Les composantes dispersives et polaires de son énergie de surface devant être connues, celles-ci ont été déterminés par mesures d’angle de contact avec des liquides de référence (diiodométhane, alphabromonaphtalène, eau). On peut à partir de l’angle de contact remonter aux composantes dispersives et polaires de la surface de polystyrène, par l’approche
NDNDDD21212112 22 γγγγγγγ −−+=
12γ γ Dγ NDγ
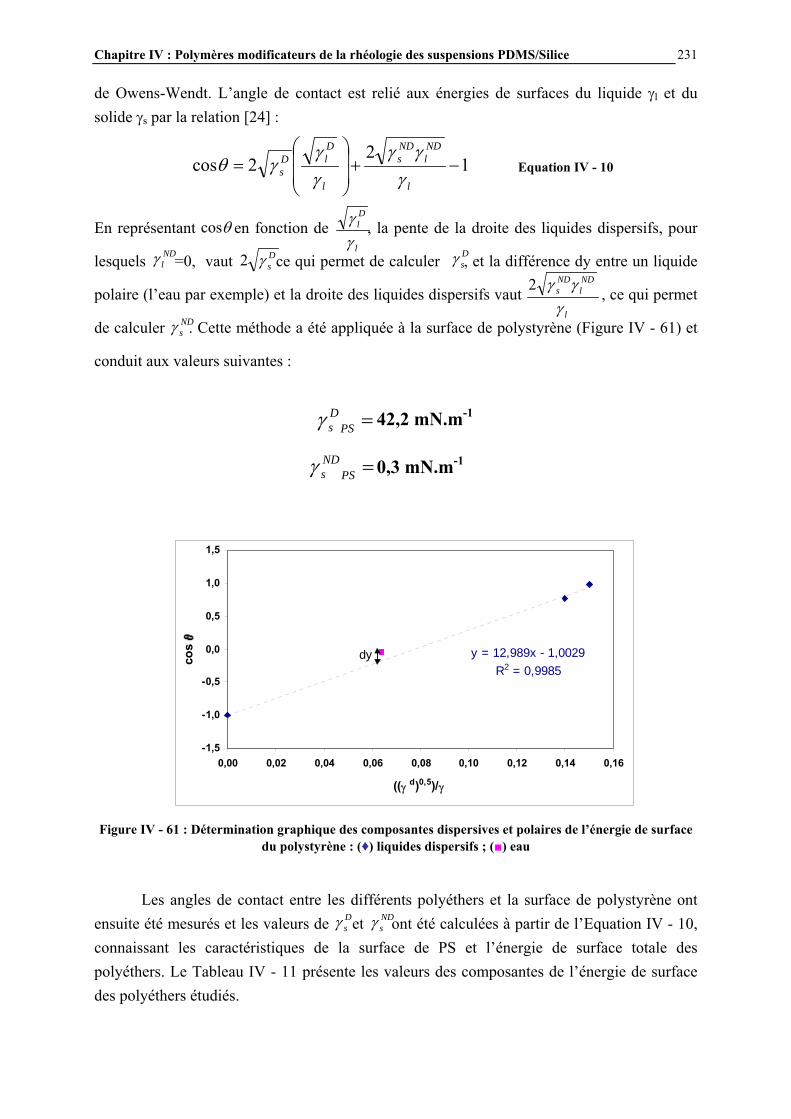
Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
231
de Owens-Wendt. L’angle de contact est relié aux énergies de surfaces du liquide γl et du solide γs par la relation [24] : Equation IV - 10
En représentant en fonction de , la pente de la droite des liquides dispersifs, pour
lesquels =0, vaut ce qui permet de calculer , et la différence dy entre un liquide
polaire (l’eau par exemple) et la droite des liquides dispersifs vaut , ce qui permet
de calculer . Cette méthode a été appliquée à la surface de polystyrène (Figure IV - 61) et
conduit aux valeurs suivantes :
42,2 mN.m-1 0,3 mN.m-1 Figure IV - 61 : Détermination graphique des composantes dispersives et polaires de l’énergie de surface
du polystyrène : (♦) liquides dispersifs ; (■) eau
Les angles de contact entre les différents polyéthers et la surface de polystyrène ont ensuite été mesurés et les valeurs de et ont été calculées à partir de l’Equation IV - 10, connaissant les caractéristiques de la surface de PS et l’énergie de surface totale des polyéthers. Le Tableau IV - 11 présente les valeurs des composantes de l’énergie de surface des polyéthers étudiés.
y = 12,989x - 1,0029R2 = 0,9985
-1,5
-1,0
-0,5
0,0
0,5
1,0
1,5
0,00 0,02 0,04 0,06 0,08 0,10 0,12 0,14 0,16
((γ d)0,5)/γ
cos dy
θcos
12
2cos −+⎟⎟
⎠
⎞
⎜⎜
⎝
⎛=
l
NDl
NDs
l
DlD
s γγγ
γγ
γθ
l
Dl
γγ
Dsγ2 D
sγ
l
NDl
NDs
γγγ2
NDsγ
NDlγ
=PSDsγ
=PSNDsγ
Dsγ ND
sγ

Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
232
Additif polyéther ( mN.m-1) ( mN.m-1) ( mN.m-1)
E200 38 20,9 17,1 EA220 38 26 12 EM250 30 18,6 11,4 PP1000 30 18,1 11,9
Tableau IV - 11 : Valeur des composantes dispersives et polaires des énergies de surface des polyéthers étudiés
A énergie de surface égale, on note l’influence des fins de chaînes sur les composantes polaires des énergies de surface : 17,1 mN.m-1 pour des POE à fins de chaînes hydroxyle contre 12 mN.m-1 pour des POE à fins de chaînes amine : les fins de chaînes hydroxyle sont plus polaires que les fins de chaîne amine.
En ce qui concerne le PDMS cette méthode de détermination donne des résultats peu réalistes, et nous avons préféré utiliser les valeurs données dans la littérature [25] : 17,5 mN.m-1 2,7 mN.m-1 A partir de l’énergie de surface et de ses composantes dispersives et polaires des différents polymères, on peut estimer l’énergie interfaciale entre le PDMS et les différents polyéthers à partir de l’Equation IV - 9. On peut aussi évaluer les tensions interfaciales POE/silice (silice non greffée) en utilisant comme composantes d’énergie de surface de la silice celles de l’eau, à savoir [24]: 72,8 mN.m-1 21,8 mN.m-1 51 mN.m-1 L’ensemble des tensions interfaciales ainsi calculées sont reportées au Tableau IV - 12.
=PDMSDlγ
=PDMSNDlγ
NDlγD
lγlγ
=eaulγ=eau
Dlγ
=eauNDlγ
Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
233
Polyéther γ polyéther/PDMS ( mN.m-1) γ polyéther/silice ( mN.m-1) E200 6,9 9
EA220 4,6 13,7 EM250 25,4 14,3 PP1000 3,7 13,8
Tableau IV - 12 : Energies interfaciales des systèmes ternaires PDMS / Polyéther /silice
Plus l’énergie interfaciale est faible, plus l’affinité entre les deux composés est grande. D’après ces résultats, l’affinité des polyéthers est plus prononcée pour la surface de silice que pour le PDMS dans le cas de l’additif EM250 (POE fins de chaînes méthyle) alors que l’inverse est observé pour les autres additifs. Ce résultat est surprenant car on s’attendait à ce que tous ces additifs, non miscibles avec le PDMS possèdent une énergie interfaciale plus faible avec la silice qu’avec le PDMS. En comparant les différents polyéthers, leur affinité pour la surface de silice augmente lorsque la polarité des fins de chaînes augmente, mais le POE à fins de chaîne amine (EA220) se différencie peu du POE à fins de chaîne méthyle (EM250) alors que pourtant nous avons observé en IGC qu’il avait une affinité particulière pour la surface de silice. Les énergies interfaciales entre les polyéthers et le PDMS ont été en parallèle mesurées par la technique de la balance de Wilhelmy à deux liquides. La force est mesurée en fonction du déplacement d lorsque la lame traverse successivement les deux liquides. A partir de la valeur de la force on peut calculer la tension interfaciale entre les deux liquides. Avant cela, nous avons testé la méthode sur un seul liquide, ce qui permet de remonter à son énergie de surface. Les courbes force-déplacement pour l’eau, le PDMS H1500V et le POE E200 sont présentées à la Figure IV - 62. En extrapolant ces courbes à déplacement nul, on obtient la valeur de la force pour laquelle γl = F/2L où L est la largeur de la lame. On en déduit les valeurs de γl pour l’eau (70 mN.m-1), le PDMS H1500V (18,8 mN.m-1) et le POE E200 (42,5mN.m-1). Ces valeurs sont relativement proches des valeurs obtenues par les autres méthodes (goutte pendante et balance de Wilhelmy en mode automatique), ce qui montre que cette approche, qui va maintenant être étendue aux tensions interfaciales entre deux liquides, permet d’obtenir une estimation cohérente des énergies de surface.
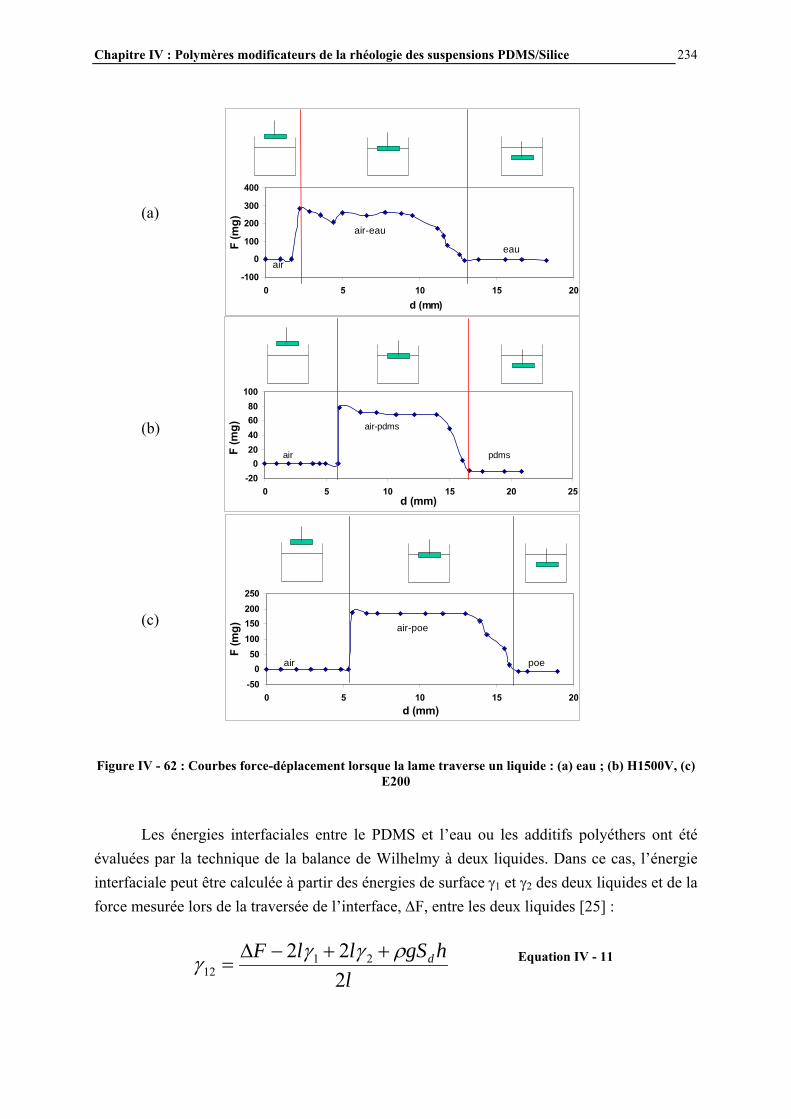
Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
234
(a) (b) (c) Figure IV - 62 : Courbes force-déplacement lorsque la lame traverse un liquide : (a) eau ; (b) H1500V, (c)
E200
Les énergies interfaciales entre le PDMS et l’eau ou les additifs polyéthers ont été évaluées par la technique de la balance de Wilhelmy à deux liquides. Dans ce cas, l’énergie interfaciale peut être calculée à partir des énergies de surface γ1 et γ2 des deux liquides et de la force mesurée lors de la traversée de l’interface, ∆F, entre les deux liquides [25] : Equation IV - 11
lhgSllF d
222 21
12ργγγ ++−∆
=
-200
20406080
100
0 5 10 15 20 25d (mm)
F (m
g)
air
air-pdms
pdms
-500
50100150200250
0 5 10 15 20d (mm)
F (m
g)
air
air-poe
poe
-100
0
100
200
300
400
0 5 10 15 20d (mm)
F (m
g)
air
air-eau
eau

Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
235
où l et Sd sont la longueur et l’aire de la section droite de la lame, respectivement ; ρ la masse volumique du liquide 1 (le moins dense) et h la hauteur du liquide 1. Dans le cas de notre lame de platine Sd est très faible donc le terme hgSdρ peut être négligé ce qui conduit à l’expression simplifiée de l’énergie interfaciale : Equation IV - 12
A partir des courbes force-déplacement présentées Figure IV - 63, les énergies interfaciales entre le PDMS et l’eau, puis entre le PDMS et les différents polyéthers de l’étude ont été calculées. Ces valeurs sont comparées aux valeurs théoriques obtenues par l’Equation IV - 9 au Tableau IV - 13. Les valeurs d’énergies interfaciales entre les polyéthers et l’eau n’ont pu être mesurées expérimentalement car ces deux liquides étant miscibles on ne peut créer une interface.
lllF
222 21
12γγγ +−∆
=
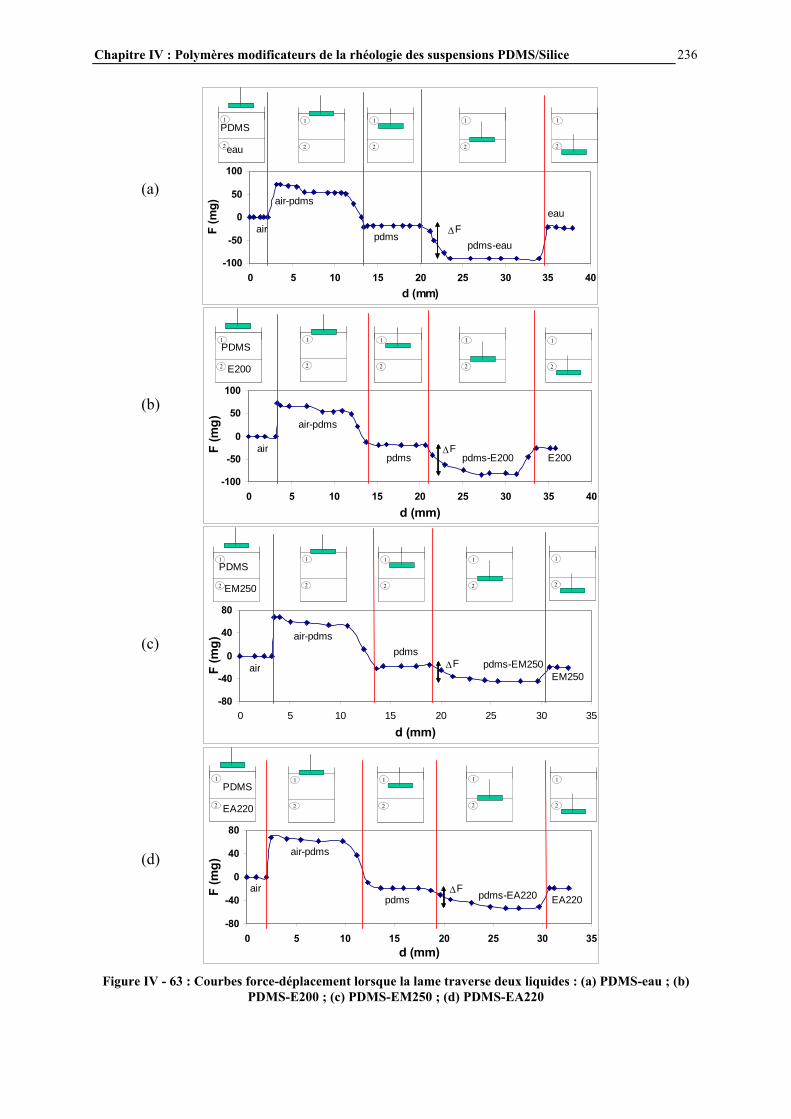
Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
236
(a) (b) (c) (d)
Figure IV - 63 : Courbes force-déplacement lorsque la lame traverse deux liquides : (a) PDMS-eau ; (b) PDMS-E200 ; (c) PDMS-EM250 ; (d) PDMS-EA220
-100
-50
0
50
100
0 5 10 15 20 25 30 35 40d (mm)
F (m
g)air
pdms-eaupdms
air-pdmseau
2
1
2
1
2
1
2
1
2
1
2
1
2
1
2
1
2
1
2
1PDMS
eau
∆F
-100
-50
0
50
100
0 5 10 15 20 25 30 35 40d (mm)
F (m
g)
airpdms-E200pdms
air-pdms
E200
2
1
2
1
2
1
2
1
2
1
2
1
2
1
2
1
2
1
2
1PDMS
E200
∆F
-80
-40
0
40
80
0 5 10 15 20 25 30 35
d (mm)
F (m
g)
air pdms-EM250pdms
air-pdms
EM250
2
1
2
1
2
1
2
1
2
1
2
1
2
1
2
1
2
1
2
1PDMS
EM250
∆F
-80
-40
0
40
80
0 5 10 15 20 25 30 35d (mm)
F (m
g)
airpdms-EA220pdms
air-pdms
EA220
2
1
2
1
2
1
2
1
2
1
2
1
2
1
2
1
2
1
2
1PDMS
EA220
∆F
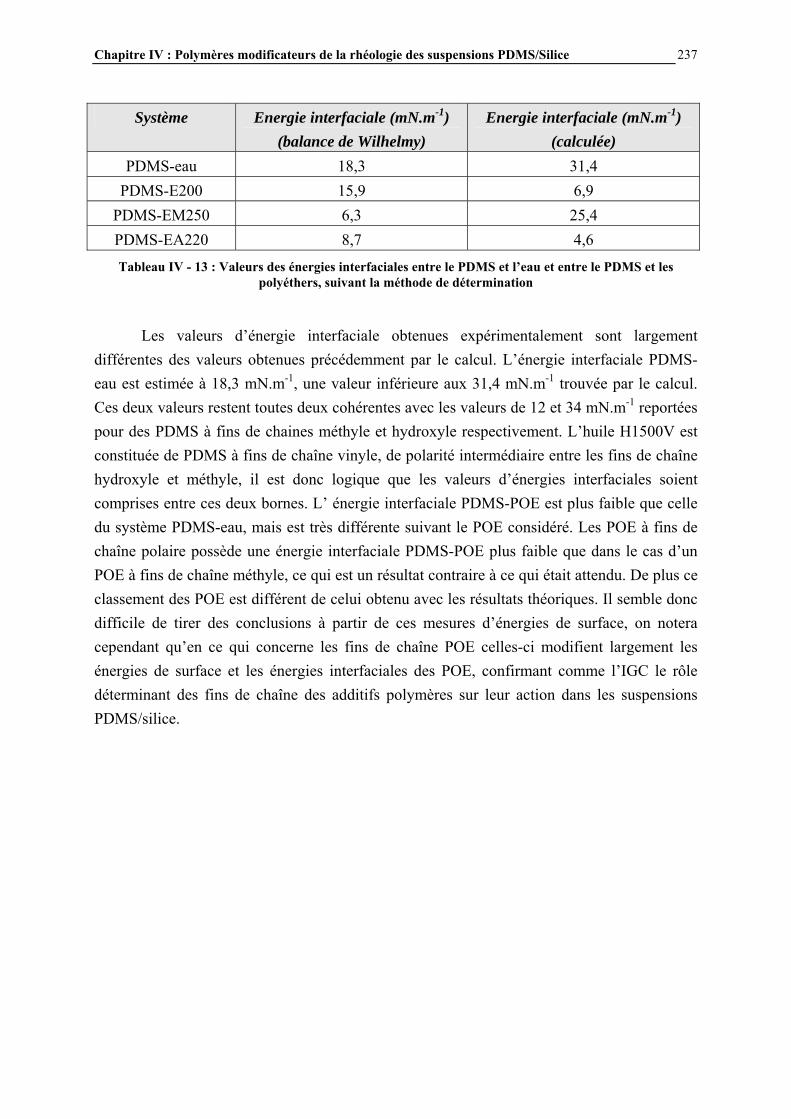
Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
237
Système Energie interfaciale (mN.m-1)
(balance de Wilhelmy) Energie interfaciale (mN.m-1)
(calculée) PDMS-eau 18,3 31,4
PDMS-E200 15,9 6,9 PDMS-EM250 6,3 25,4 PDMS-EA220 8,7 4,6
Tableau IV - 13 : Valeurs des énergies interfaciales entre le PDMS et l’eau et entre le PDMS et les polyéthers, suivant la méthode de détermination
Les valeurs d’énergie interfaciale obtenues expérimentalement sont largement différentes des valeurs obtenues précédemment par le calcul. L’énergie interfaciale PDMS-eau est estimée à 18,3 mN.m-1, une valeur inférieure aux 31,4 mN.m-1 trouvée par le calcul. Ces deux valeurs restent toutes deux cohérentes avec les valeurs de 12 et 34 mN.m-1 reportées pour des PDMS à fins de chaines méthyle et hydroxyle respectivement. L’huile H1500V est constituée de PDMS à fins de chaîne vinyle, de polarité intermédiaire entre les fins de chaîne hydroxyle et méthyle, il est donc logique que les valeurs d’énergies interfaciales soient comprises entre ces deux bornes. L’ énergie interfaciale PDMS-POE est plus faible que celle du système PDMS-eau, mais est très différente suivant le POE considéré. Les POE à fins de chaîne polaire possède une énergie interfaciale PDMS-POE plus faible que dans le cas d’un POE à fins de chaîne méthyle, ce qui est un résultat contraire à ce qui était attendu. De plus ce classement des POE est différent de celui obtenu avec les résultats théoriques. Il semble donc difficile de tirer des conclusions à partir de ces mesures d’énergies de surface, on notera cependant qu’en ce qui concerne les fins de chaîne POE celles-ci modifient largement les énergies de surface et les énergies interfaciales des POE, confirmant comme l’IGC le rôle déterminant des fins de chaîne des additifs polymères sur leur action dans les suspensions PDMS/silice.

Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
238
IV.5- CONCLUSIONS ET PERSPECTIVES Ce chapitre avait pour objet l’étude des gels formés lors de l’ajout d’additifs polymères dans les suspensions PDMS/silice. Ces gels se caractérisent par l’apparition d’une contrainte seuil, mesurée en mode statique en imposant une rampe de contrainte à l’échantillon. Cette contrainte seuil, caractéristique de la rigidité du gel, est le principal critère qui a été utilisé pour évaluer les gels, même si nous avons également vu que la formation du gel peut être suivie par mesures rhéologiques en mode dynamique, les suspensions présentant un module de conservation G’ supérieur au module de perte G’’ et un effet Payne plus important que pour la suspension initiale, avec un plateau basses fréquences étendu sur une large plage de fréquence. La présence de l’effet Payne montre que ces gels sont des gels fragiles qui peuvent être détruits sous cisaillement, reprenant leur structure initiale au repos Avant d’étudier la relation entre les paramètres de formulations ou la structure des additifs et les propriétés des gels, nous avons évalué l’influence d’un certain nombre de paramètres expérimentaux sur les propriétés des gels obtenus. Il apparaît que les gels PDMS/silice/additif sont très stables et ne sont sensibles ni au temps ni à la température de mise en œuvre. L’évolution de la contrainte seuil en fonction de la concentration en additif a également été évaluée. Une concentration optimale existe pour chaque formulation et chaque type d’additif, pour laquelle la contrainte seuil est maximale. Celle-ci varie de 0,2 à 5% mais se situe généralement autour de 1%. La concentration en additif pour la suite de l’étude a donc été fixée à 1%. Les valeurs de contraintes seuil peuvent être directement reliées aux paramètres de la formulation des suspensions initiales. La contrainte seuil augmente avec la fraction volumique de silice et augmente également lorsque le taux de greffage des silices diminue, ceci quel que soit l’additif. Il existe une fraction volumique de transition à partir de laquelle on peut former un gel par ajout d’additif. Celle-ci dépend encore une fois du taux de greffage des silices : plus les silices sont greffées, plus la fraction volumique de transition est élevée. Par contre elle ne dépend pas du type d’additif, du moins dans la gamme d’additifs évalués au cours de cette étude. Nous avons ainsi pu compléter le diagramme de transition liquide-gel réalisé à partir des résultats du chapitre II sur les suspensions PDMS-silice sans additif avec cette fois les résultats obtenus après ajout d’additif. Ce diagramme permet de définir une fenêtre de valeurs pour les deux paramètres de formulations principaux que sont la fraction volumique et le taux de greffage des silices, pour laquelle les suspensions initiales sont fluides (pas de contrainte seuil) mais peuvent gélifier sous l’action des additifs polymères. Cette propriété permet de remplir le cahier des charges imposé pour certaines applications sur le marché du moulage (application du produit sur une surface non horizontale). Par ailleurs le diagramme de phase est modifié lorsque les propriétés initiales des suspensions ne sont pas

Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
239
identiques (en particulier le taux de greffage) comme c’est le cas pour nos formulations Z, T et F. Ceci pourrait expliquer les différences de comportement face aux additifs entre les différentes formulations industrielles. Nous avons ensuite étudié l’influence de la microstructure de l’additif sur les valeurs de contraintes seuil des gels obtenus. Le squelette de la chaîne joue un rôle important, tout comme les fins de chaînes. Les polyéthers (POE et POP) sont de manière générale plus efficaces que les PDMS (à fins de chaînes et masses molaires comparables). Les fins de chaînes polaires (hydroxyle, amine) confèrent également une plus grande efficacité aux additifs polymères, cependant les fins de chaînes amine se distinguent particulièrement par les valeurs élevées de contrainte seuil qu’elles permettent d’obtenir, que ce soit les POE, POP ou PDMS. En ce qui concerne les additifs copolymères, ils ne sont pas systématiquement plus efficaces que les homopolymères, en particulier en comparaison des homopolymères à fins de chaînes amine. Les gels obtenus par ajout de copolymères PDMS/POE/POP présentent des valeurs de contrainte seuil similaires à ceux obtenus par ajout de copolymères PDMS/POE. Afin de pouvoir interpréter tous ces résultats, le mécanisme de formation des gels a été étudié. Pour cela la microstructure des gels a été observée par microscopies optique et électronique. Lorsque l’additif polymère est ajouté aux suspensions PDMS/silice, une structuration de l’échantillon apparaît. Elle ne correspond pas à un changement de la dispersion de silice au sein de la suspension, qui comme nous avons pu le vérifier par microscopie électronique à transmission n’est pas modifiée par l’ajout de l’additif polymère. En suivant par fluorescence les additifs POE dans les suspensions PDMS/silice par microscopie optique, il peut être montré à partir d’un phénomène de séparation de phases que la structuration observée correspond à la structuration de l’additif POE. Nous avons alors proposé à partir de ces résultats le mécanisme suivant pour expliquer la formation des gels : le POE, non miscible avec le PDMS, forme une structure continue dans la suspension, en s’adsorbant à la surface de la silice. Si le nombre de sites d’adsorption est trop faible (fraction volumique faible ou taux de greffage élevé), le POE reste sous forme de gouttelettes dans la suspension : aucun gel n’est formé. Ce mécanisme explique alors le diagramme de transition liquide-gel décrit précédemment, c’est-à-dire le rôle de la fraction volumique et du taux de greffage des silices. Enfin, en considérant ce mécanisme le rôle des fins de chaînes est envisagé différemment, non pas en terme de possibilité de liaison-H assurant ou non un pontage des particules de silice envisagé initialement, mais en termes d’affinités ou d’étalement avec la surface de la silice. En effet, dans le mécanisme proposé la formation du gel dépend de la structuration du POE, qui elle-même dépend de l’adsorption du POE à la surface de la silice. Ainsi les POE qui auront les interactions les plus fortes avec la surface de la silice seront plus efficaces. Nous avons pu montrer par chromatographie gazeuse en phase inverse (IGC) que les composés à fins de chaînes amine présentaient effectivement des

Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
240
interactions très fortes avec la silice, ce qui permet d’expliquer leur efficacité particulière à comparer des additifs possédant d’autres natures de fins de chaînes. Ainsi, ce chapitre nous a permi d’identifier les mécanismes de base responsables de la formation des gel et de corréler la rigidité des gels avec les paramètres de formulation des suspensions initiales et la structure des additifs (nature du squelette et des fins de chaînes). Le mécanisme proposé est cependant limité pour l’instant aux additifs POE et devra être élargi aux autres types d’additifs (PDMS, copolymères PDMS/polyéther…). De plus, il serait utile de vérifier que lors de la formation d’une structure continue de POE celui-ci est véritablement adsorbé à la surface de la silice. En effet, il est difficile de visualiser à la fois le POE et la silice : en microscopie électronique à transmission seul un contraste entre la silice et le POE est possible et par microscopie optique en mode fluorescent, on observe uniquement le POE marqué. Une solution consisterait donc à marquer à la fois la silice et le POE avec deux marqueurs fluorescents différents.

Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
241
Références bibliographiques
1. Jackson S., Thixotropie des RTV HPM, St-Fons: Rapport interne Rhodia Silicones,
1999, 13p.
2. Lafuma F., Wong K., Cabane B., Bridging of colloidal particles through adsorbed polymers, J. Colloid Interface Sci., 1991, Vol. 143 (1), pp.9-21.
3. Wind B., Killmann E., Adsorption of polyethylene oxide on surface modified silica-stability of bare and covered particles in suspension, Colloid Polym. Sci., 1998, Vol. 276, pp.903-912.
4. Liu S.F., Legrand V., Gourmand M., Lafuma F., Audebert R., General phase and rheological behavior of silica/peo/water systems, Colloids Surf., A, 1996, Vol. 111, pp.139-145.
5. Zhang Q., Archer L.A., Poly(ethylene oxide)/silica nanocomposites : structure and rheology, Langmuir, 2002, Vol. 18, pp.10435-10442.
6. Mathur S., Moudgil M., Adsorption mechanism(s) of poly(ethylene oxide) on oxide surfaces, J. Colloid Interface Sci., 1997, Vol. 196, pp.92-98.
7. Bjelopavlic M., Singh P.K., El-Shall H., Moudgil B.M., Role of surface molecular architecture and energetics of hydrogen bonding sites in adsorption of polymers and surfactants, J. Colloid Interface Sci., 2000, Vol. 226, pp.159-165.
8. Smith J.M., Katsoulis D., Gelation of silicone fluids using 1,3:2,4-dibenzylidene sorbitol, J. Mat. Chem., 1995, Vol. 5 (11), pp.1899-1903.
9. Terech P., Weiss R.G., Low Molecular Mass Gelators of Organic Liquids and the Properties of their gels, Chem. Rev., 1997, Vol. 97, pp.3133-3159.
10. Van Esch J., Schoonbeck F., De Loos Maaike, Kooijman H., Spek A.L., Kellogg R.M., Feringa B.L., Cyclic bis-urea compounds as gelators for organic solvents, Chem. Eur. J., 1999, Vol. 5 (3), pp.937-950.
11. Lipatov Y.S., Nesterov A.E., Ignatova T.D., Nesterov D.A., Effect of polymer-filler surface interactions on the phase separation in polymer blends, Polymer, 2002, Vol. 43, pp.875-880.
12. Papirer E., Balard H., Vergelati C., Adsorption on Silica Surfaces, Surface energetics of Silica Investigated by Inverse Gas Chromatography, in: Surfactant Science Series, Vol. 90, New-York: Marcel Dekker, 2000, pp.205-241.
13. Castellano M., Falqui L., Costa G., Turturro A., Valenti B., Castello G., Investigation on elastomer-silica interactions by inverse gas chromatography and image analysis aided transmission electron microscopy, J. Macromol. Sci., 2002, Vol. B41 (3), pp.451-471.

Chapitre IV : Polymères modificateurs de la rhéologie des suspensions PDMS/Silice
242
14. Donnet J.B., Li Y.J., Wang T.K., Balard H., Burns G.T., Adsorption energy of silica xerogel surfaces by inverse gas and liquid chromatography, Silica 2001, Mulhouse, France.
15. Zumbrum M.A., Acid/Base properties of fumed silica fillers used in silicone elastomers, XVIth Annual Meeting of the Adhesion Society, Inc, 1993, Williamsburgh, Virginia, U.S.A.
16. Khalfi A., Papirer E., Balard H., Barthel H., Heinemann M.G., Characterization of silylated silicas by inverse gas chromatography : modelization of the PDMS monomer unit / surface interactions using PDMS oligomers as probes, J. Colloid Interface Sci., 1996, Vol. 184, pp.586-593.
17. Brendlé E., Papirer E., A new topological index for molecular probes used in inverse gas chromatography for the surface nanorugosity evaluation - 1. Method of evaluation, J. Colloid Interface Sci., 1997, Vol. 194, pp.207-216.
18. Hamdi B., Kessaissia Z., Donnet J.B., Wang T.K., IGC characterization of surface energy and morphology of two natural fillers:Kieselguhr and bentonite, Ann. Chim. Sci. Mat., 2000, Vol. 25, pp.481-494.
19. Balard H., Papirer E., Coupling of IGC and controlled surface modification : a powerful method to characterise the filler surface topology, Eurofillers'99, 1999, Villeurbanne, France.
20. Zhang W., Leonov A.I., IGC study of filler-filler and filler-rubber interactions in silica-filled compounds, J. Appl. Polym. Sci., 2001, Vol. 81, pp.2517-2530.
21. Emmet P.H., Brunnaur S., The use of low temperature van der waals adsorption isotherms in determining the surface area of iron synthetic ammonia catalysts, J. Am. Chem. Soc., 1937, Vol. 59, pp.1553-1564.
22. Barthel H., Surface interactions of dimethylsiloxy group-modified fumed silica, Colloids Surf., A, 1995, Vol. 101, pp.217-226.
23. Wu S., Surface and interfacial tensions of polymers, oligomers, plasticizers, and organic pigments, in: Brandrup J., Immergut E.H., Polymer Handbook, New-York: John Wiley & Sons, 1989, pp.VI 411-434.
24. Germain C., Caractérisation superficielle d'encres et de polymères dans le but de prévoir leur adhérence, Thèse de Doctorat, Université Claude Bernard, Lyon I, 1994, 280p.
25. Bessaha N., Adhésion de matériaux polyuréthanes : application aux propulseurs à base de propergols solides, Thèse de Doctorat, Université de Haute-Alsace, Mulhouse, 1995, 195p.

CONCLUSION GENERALE

Conclusion générale
243
L’objectif de notre étude a été de comprendre comment la fraction volumique et le taux de greffage de la silice influencent les propriétés rhéologiques de suspensions PDMS/silice et comment contrôler ces propriétés par ajout d’un additif polymère. Notre travail s’est déroulé en trois grandes étapes : i/ étude du greffage de la silice par réaction avec l’hexaméthyldisilazane (HMDS), ii/ étude des propriétés rhéologiques des suspensions en fonction de la fraction volumique et du greffage de la silice (nombre de sites d’interaction), ainsi que du procédé de mise en œuvre des suspensions, iii/ évaluation des additifs polymères pour la modification des propriétés rhéologiques des suspensions initiales et compréhension de leur mécanisme d’action. La première partie concernant le greffage de la silice, c’est-à-dire la modulation du nombre de sites d’interaction de surface par hydrophobisation, a permis de quantifier le greffage et de comparer les différentes procédures expérimentales : i/ greffage ex-situ, en solution ou par voie gaz, ii/ greffage in-situ, c’est-à-dire en milieude haute viscosité PDMS. Les deux procédures ex-situ conduisent à des résultats similaires : la conversion des silanols de surface en groupements TMS, évaluée par différentes techniques d’analyse (infrarouge à transformée de Fourier, analyse élémentaire du carbone, résonance magnétique nucléaire RMN 29Si CP/MAS, mesures de mouillage) évolue d’abord linéairement avec le rapport stoechiométrique HMDS/silanol, puis atteint un plateau pour un rapport de 1 environ. La valeur du taux de greffage maximum obtenue par analyse élémentaire du carbone est de 1,8 TMS/nm2. La limitation de la conversion des silanols est due à l’encombrement stérique des groupements TMS greffés. Les silices greffées in-situ ont été extraites des mélanges PDMS/silice et également analysées par RMN 29Si CP/MAS. Leur taux de greffage supérieur de 25% par rapport aux silices greffées ex-situ peut être attribué à la présence dans ce cas dans le milieu réactionnel de l’ammoniac formé au cours de la réaction, qui peut réagir avec les siloxanes de surface pour former des groupements hydroxyle supplémentaires disponibles pour réagir avec l’HMDS. Le comportement rhéologique et la morphologie des suspensions a pu être relié au taux de greffage et à la fraction volumique de silice. Lorsque la fraction volumique augmente ou que le taux de greffage diminue, l’agrégation des particules ou agrégats de particules est plus importante, ce qui conduit à un module de cisaillement aux faibles déformations, G0’, plus élevé, ainsi qu’à un domaine viscoélastique linéaire plus étroit. Aux forts taux de greffage , les suspensions réalisées à partir de silices greffées ex-situ présentent un module G0’ plus élevé que les suspensions dont les silices ont été greffées in-situ en milieu PDMS, en raison du plus faible taux de greffage atteint pour celles-ci. Cependant, peu de différences dans la morphologie des suspensions ont pu être observées, ce qui nous permet de conclure

Conclusion générale
244
que la rhéologie est un outil complémentaire de la microscopie électronique à transmission, adapté à l’évaluation de la dispersion des suspensions, c’est-à-dire de l’architecture des agrégats de silice à différentes échelles. En ce qui concerne le procédé de mise en œuvre des suspensions, l’introduction d’HMDS après avoir dans un premier temps réalisé le mélange PDMS-silice permet d’obtenir des suspensions de module G0’ légèrement inférieur au module des suspensions où l’HMDS est introduit dès le début du mélange PDMS-silice. L’évolution du module G0’ en fonction de la fraction volumique de silice pour différents taux de greffage a été représentée et un modèle de suspensions d’objets fractals semi-dilués a été choisi pour interpréter les résultats. La dimension fractale des objets diminue lorsque le taux de greffage augmente, ce qui a été confirmé par des mesures de la taille des objets élémentaires par diffusion de lumière. La fraction volumique critique de silice à partir de laquelle les objets sont ‘interpénétrés’ (suspension semi-diluée) a pu ainsi être estimée entre 1 et 2%. Lors de l’ajout (environ 1%) d’additifs polymère à séquences hydrophiles dans les suspensions PDMS/silice initialement de faible viscosité, celles-ci présentent un comportement de gel, avec apparition d’une contrainte seuil. Ces gels sont fragiles car ils peuvent être facilement détruits sous cisaillement, reprenant leur structure initiale au repos. Il existe une fraction volumique minimale de silice à partir de laquelle les additifs peuvent conférer aux suspensions ce comportement de gel (suspension à seuil), et qui dépend également du taux de greffage. La contrainte seuil augmente quel que soit l’additif utilisé lorsque la fraction volumique de silice de la suspension augmente ou que le taux de greffage de la silice diminue. Cette corrélation entre la contrainte seuil et les paramètres de la formulation conduit à la représentation d’un diagramme de transition liquide-gel en fonction de la fraction volumique et du taux de greffage de la silice, permettant de déterminer une fenêtre de valeurs pour laquelle la suspension initiale est fluide mais peut devenir une suspension à seuil lors de l’ajout d’additif. Cette propriété permet de remplir le cahier des charges de certaines applications (application du produit sur une surface non horizontale dans le domaine du moulage). Nous avons étudié également l’influence de la microstructure des additifs sur les valeurs de contrainte seuil des gels (suspensions à seuil) obtenus. Les polyéthers sont de manière générale plus efficaces que les PDMS, et il apparaît que la nature des fins de chaînes des additifs joue un rôle prépondérant sur l’efficacité des additifs. Les fins de chaînes polaires, et en particulier les amines, confèrent aux additifs des propriétés remarquables vis-à-vis des contraintes seuil des suspensions additivées. Les POE et POP à fins de chaîne amine apportent une solution technique aux problème de certaines formulations industrielles qui, après ajout d’additifs conventionnels, ne présentaient pas le comportement de suspension à seuil souhaité.

Conclusion générale
245
Nous nous sommes intéressés en parallèle au mécanisme d’action de ces additifs, constitués de PDMS et/ou de polyéthers (polyoxyde d’éthylène POE et polyoxyde de propylène POP). La structuration observée par microscopie optique dans les gels n’étant pas attribuée à la silice (pas de changement de la dispersion de silice avant et après ajout d’additif polymère), il a été montré par fluorescence que ceux-ci sont responsables, par séparation de phase avec le milieu, de la structuration observée et donc du comportement de gel conféré à la suspension. Nous avons émis l’hypothèse que le POE , non miscible avec le milieu PDMS, forme une structure continue dans la suspension, en s’adsorbant à la surface de la silice avec laquelle de fortes interactions sont créées. Ce mécanisme permet d’interpréter les résultats précédents (dépendance vis-à-vis des caractéristiques de la silice : taux de greffage et fraction volumique) et permet aussi d’expliquer l’efficacité de différents additifs, en terme d’interactions silice-additif. Ces interactions ont été évaluées par chromatographie gazeuse en phase inverse (IGC) et par mesures d’énergies interfaciales, confirmant le rôle tout particulier des fins de chaîne amine qui interagissent fortement avec la surface de la silice. Le mécanisme proposé ne s’applique pour l’instant que dans le cadre des additifs POE qui ont pu être suivis par fluorescence au cours de cette étude. Il serait intéressant par la suite de voir si ce mécanisme peut être généralisé aux autres types d’additifs ou si ceux-ci agissent par le biais d’un autre mécanisme, en particulier dans le cas des additifs PDMS. Ce travail permet une nouvelle approche dans la manière d’obtenir une suspension présentant une contrainte seuil, par ajout d’additifs polymères. Une architecture adaptée de ces polymères en termes de sites d’interaction et de miscibilité avec le milieu doit pour cela être recherchée, ce qui peut nécessiter la synthèse de nouveaux polymères ou copolymères avec une microstructure chimique adaptée. On peut enfin envisager de généraliser cette démarche à des suspensions dont le milieu serait différent du PDMS.

Conclusion générale
246

Annexes 247
ANNEXES

Annexes
247
ANNEXES ANNEXE A : DILUTION DES SUSPENSIONS ......................................................................................... 248
ANNEXE B : MESURES RHÉOLOGIQUES ............................................................................................. 249
ANNEXE C : MICROSCOPIES TEM ET AFM......................................................................................... 251
ANNEXE D : DIFFUSION DYNAMIQUE DE LA LUMIÈRE................................................................. 255
ANNEXE E : CONTRAINTES SEUIL DES SUSPENSIONS (SÉRIE Z) ................................................ 258
ANNEXE F : DÉTERMINATION DES PENTES N ET DES FRACTIONS VOLUMIQUES CRITIQUES φV
* ..................................................................................................................................................................... 259
ANNEXE G : ANALYSE DE LA COMPOSITION CHIMIQUE DES COPOLYMÈRES PDMS / POLYÉTHER ................................................................................................................................................. 262
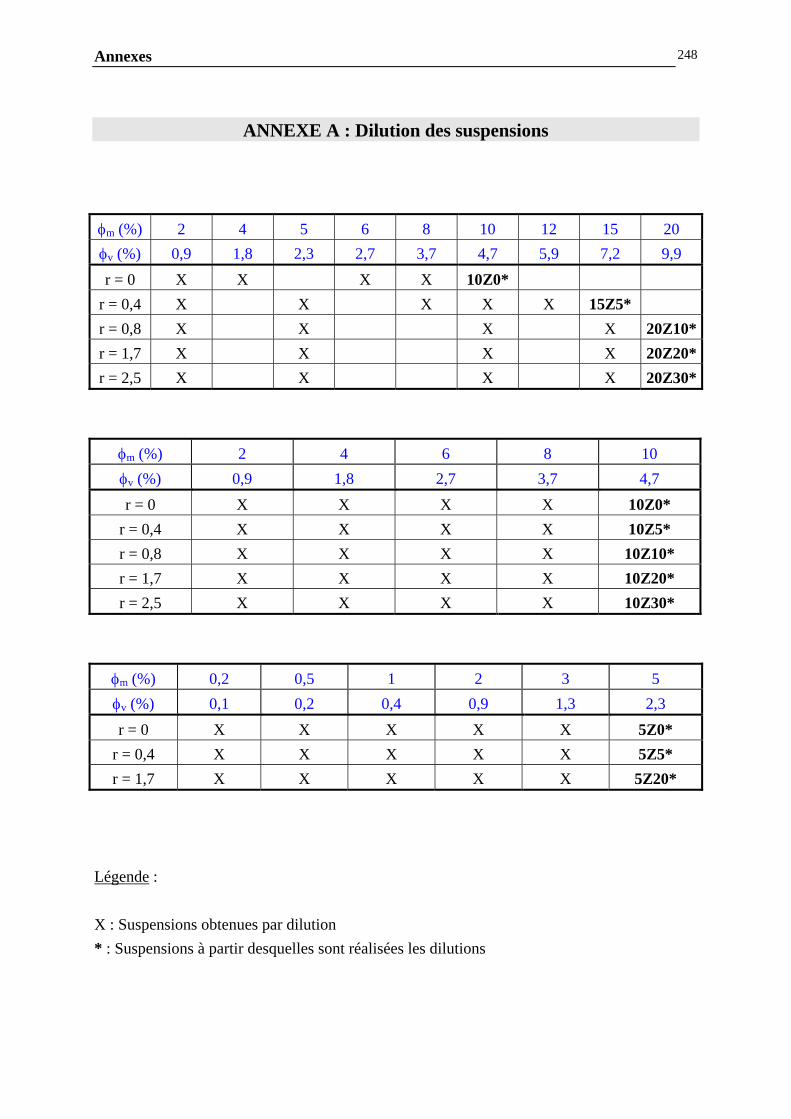
Annexes
248
ANNEXE A : Dilution des suspensions
φm (%) 2 4 5 6 8 10 12 15 20 φv (%) 0,9 1,8 2,3 2,7 3,7 4,7 5,9 7,2 9,9 r = 0 X X X X 10Z0*
r = 0,4 X X X X X 15Z5* r = 0,8 X X X X 20Z10*r = 1,7 X X X X 20Z20*r = 2,5 X X X X 20Z30*
φm (%) 2 4 6 8 10 φv (%) 0,9 1,8 2,7 3,7 4,7 r = 0 X X X X 10Z0*
r = 0,4 X X X X 10Z5* r = 0,8 X X X X 10Z10* r = 1,7 X X X X 10Z20* r = 2,5 X X X X 10Z30*
φm (%) 0,2 0,5 1 2 3 5 φv (%) 0,1 0,2 0,4 0,9 1,3 2,3 r = 0 X X X X X 5Z0*
r = 0,4 X X X X X 5Z5* r = 1,7 X X X X X 5Z20*
Légende : X : Suspensions obtenues par dilution * : Suspensions à partir desquelles sont réalisées les dilutions

Annexes
249
ANNEXE B : Mesures rhéologiques
A) Mesures rhéologiques en mode dynamique Les mesures ont été réalisées avec des rhéomètres à déformation imposée pour les balayages en déformations (RDA II, Rheometrics) et à contrainte imposée pour les balayages en fréquence (AR 1000, TA Instruments). Deux types de géométries ont été utilisés, une géométrie cône-plan et une géométrie plan-plan (Figure A - 1). La géométrie cône-plan n’est pas adaptée aux échantillons de forte viscosité car l’entrefer est fixe, ainsi elle a été utilisée uniquement pour les suspensions de faibles viscosités. Pour les échantillons de viscosité plus élevée la géométrie plan-plan a été plutôt utilisée. Le réglage de l’entrefer permet de s’adapter à la viscosité de la suspension. Généralement un entrefer de 1 mm a été utilisé, pour un diamètre de plateaux de 25 mm sur le RDA II et 20 mm sur l’AR1000. Figure A - 1 : Schéma des deux types de géométrie utilisées sur le RDA II pour les mesures rhéologiques
en mode dynamique
Afin de vérifier que ces deux géométries donnent les mêmes résultats, plusieurs suspensions de viscosités intermédiaires ont été testées par balayage en déformation. Les résultats montrent que les écarts entre les deux géométries sont faibles, et équivalents aux incertitudes de mesure. Les résultats ne dépendent donc pas de la géométrie considérée. Afin d’éviter de perturber la structure de la suspension, le dépôt de l’échantillon sur le plateau doit être réalisé avec précaution, c’est pourquoi une compression lente et régulière est appliquée lors du réglage de l’entrefer, ce dernier étant ajusté en fonction de la viscosité de l’échantillon (dans le cas d’une géométrie plan-plan) afin de ne pas imposer une contrainte normale trop importante. Les balayages en fréquence ont été réalisés à faible contrainte pour se placer dans le domaine viscoélastique linéaire. Cette zone est d’autant plus restreinte que la fréquence est
D
h D = 25 mm
h=1mm
D
hα D = 40 mm
α = 0,02 radian
h = 50 µm
Géométrie cône-plan Géométrie plan-plan
D
h D = 25 mm
h=1mm
D
hα D = 40 mm
α = 0,02 radian
h = 50 µm
Géométrie cône-plan Géométrie plan-plan

Annexes
250
faible, ainsi pour les mesures à faible fréquence, il est peut-être délicat de rester en régime linéaire. B) Mesures rhéologiques en mode statique Les mesures en mode statique ont été réalisées avec des rhéomètres à contrainte imposée (RS 2000, Rheometrics et AR 1000, TA Instruments). Deux géométries plan-plan similaires ont été employées (diamètre de 25 mm avec le SR2000 et 20 mm avec l’AR1000) et un entrefer généralement de 1 mm.

Annexes
251
ANNEXE C : Microscopies TEM et AFM
A) Microscopie électronique à transmission (TEM) La difficulté principale est d’obtenir une coupe fine et présentant une cohésion suffisante pour pouvoir être observée. En effet, les suspensions à étudier n’ont pas de tenue mécanique et la température de transition vitreuse du PDMS étant très faible (-123°C), il est impossible de les couper à froid. Afin de pouvoir obtenir ces coupes, il a donc été nécessaire dans un premier temps de réticuler les échantillons. Pour ne pas risquer de modifier la dispersion de silice au sein du PDMS, une réticulation par irradiation a été choisie plutôt qu’une réticulation chimique. Les essais de réticulation ont été réalisés au sein de la société Ionisos, par rayonnements β et γ pour plusieurs doses d’irradiation. Le temps d’exposition et la dose nécessaire pour obtenir une cohésion suffisante du matériau étant plus faible pour les rayons β, ceux-ci ont été finalement choisis pour notre étude. Une dose de 100 kGy a été appliquée à tous les échantillons. Après cette réticulation les matériaux restent néanmoins très souples et une coupe à froid – cryomicrotomie- à environ -150°C a été nécessaire pour obtenir une coupe fine d’environ 70 nm B) Microscopie à force atomique (AFM) Principe de fonctionnement Cette technique repose sur la mesure des forces d’interactions entre la pointe du microscope et l’échantillon. Lorsque la pointe se rapproche de la surface une force répulsive se crée entre les atomes de la pointe et ceux de l’échantillon. Ces forces répulsives s’expriment pour de très faibles distances interatomiques et sont donc très sensibles aux variations de distance entre la pointe et l’échantillon. Les variations de force imposées par le relief sont alors exploitées pour enregistrer des images. L’AFM nécessite l’utilisation d’une pointe très fine (tip) de forme pyramidale, placée à l’extrémité d’un levier (cantilever), qui vient balayer la surface. Le levier se courbe en réponse à la force s’exerçant entre les atomes de l’extrémité de la pointe et ceux de la surface étudiée. La lumière du laser focalisé à son extrémité est réfléchie sur les photodiodes et les modifications d’inclinaison du levier sont alors mesurées par la différence de signal entre les

Annexes
252
deux photodiodes, permettant de remonter à la force entre la pointe et l’échantillon (Figure III - 1). L’échantillon est placé sur un tube piézo-électrique assurant les déplacements tridimensionnels (x,y,z). Pendant l’acquisition la pointe balaie une portion carrée de la surface de taille définie par l’utilisateur et correspondant à un déplacement de l’échantillon dans le plan (x,y). L’ensemble du montage est placé sur une table à coussins d’air, de façon à l’isoler des vibrations.
Figure III - 1: Schéma de principe du système de détection
Les différents modes de fonctionnement Le mode contact est le premier à avoir été développé. La pointe est maintenue en contact avec la surface de manière permanente pendant le balayage. L’interaction pointe/surface est alors répulsive. On peut travailler soit à déplacement constant (mode force) soit à force constante (mode hauteur). Bien que les forces utilisées par l’AFM en mode contact soient faibles, elles suffisent à détruire les échantillons mous ou fragiles. De plus cette force est appliquée en continu. C’est pour diminuer ces forces que le mode contact intermittent (tapping mode) a été développé. Dans ce mode la pointe est maintenue à faible distance (5 à 10 nm) au dessus de la surface. On détecte alors des forces faiblement attractives de Van der Waals entre l’échantillon et la pointe. Le levier est mis en vibration à sa fréquence de résonance afin de ramener la pointe vers la surface de façon intermittente. Le gradient de force dû à l’interaction pointe / surface modifie la constante de force du levier et donc sa fréquence de résonance. Ce changement de fréquence de résonance au cours du balayage induit une variation de l’amplitude d’oscillation (Figure III - 2). Pour maintenir cette
Support du cantilever
Echantillon
Scanner piézo-électrique XYZ
Faisceau laser
Photodiodes
Diode laser
Levier
Pointe
Miroir
Support du cantilever
Echantillon
Scanner piézo-électrique XYZ
Faisceau laser
Photodiodes
Diode laser
Levier
Pointe
Miroir

Annexes
253
amplitude constante , il faut modifier la position verticale z du piézo-électrique. On obtient alors une image topographique en balayant la pointe au dessus de la surface. Ce mode est utilisé surtout dans le domaine des polymères, et s’avère particulièrement adapté à nos élastomères chargés qui sont des matériaux mous.
Figure III - 2: Variation de l’amplitude d’oscillation de la pointe en mode tapping
Préparation des échantillons Dans le cas de l’AFM les échantillons ne nécessitent aucune préparation particulière, si ce n’est une surface plane. Dans notre cas les échantillons utilisés sont ceux qui ont été réticulés pour les coupes TEM mais il s’est avéré qu’ils étaient pour la plupart à la fois trop mous pour pouvoir préparer une surface plane, et trop collants ce qui provoque l’attachement de la pointe à la surface. Une réticulation supplémentaire a donc été réalisée par irradiations, mais n’étant pas suffisante nous avons eu recours à la réticulation chimique par réaction avec le peroxyde de benzoyle (en solution dans le cyclohexane à 1,4%), suivi d’un séchage à 100°C pendant 4H en étuve. Appareillage Les observations ont été réalisées sur un microscope de la société Molecular Imaging Corporation, à l’Institut des polymères de l’Université Technologique de Lodz (Pologne). Le levier utilisé est un NSC 11/50 (Ultrasharp), dont les dimensions sont données à la Figure A - 2. La pointe notée B de ce levier a été utilisée pour cette étude.
Amplitude libre A0
Amplitude réduite Ar
Amplitude libre A0
Amplitude réduite Ar

Annexes
254
Figure A - 2: Caractéristiques techniques du levier et de la pointe utilisés
A
B
3,4 mm
1,6 mm
Caractéristiques de la pointe B :
- Longueur : 90 µm- Largeur : 60 µm- Epaisseur : 2 µm- Fréquence de résonance : 315 kHz
A
B
3,4 mm
1,6 mm
Caractéristiques de la pointe B :
- Longueur : 90 µm- Largeur : 60 µm- Epaisseur : 2 µm- Fréquence de résonance : 315 kHz
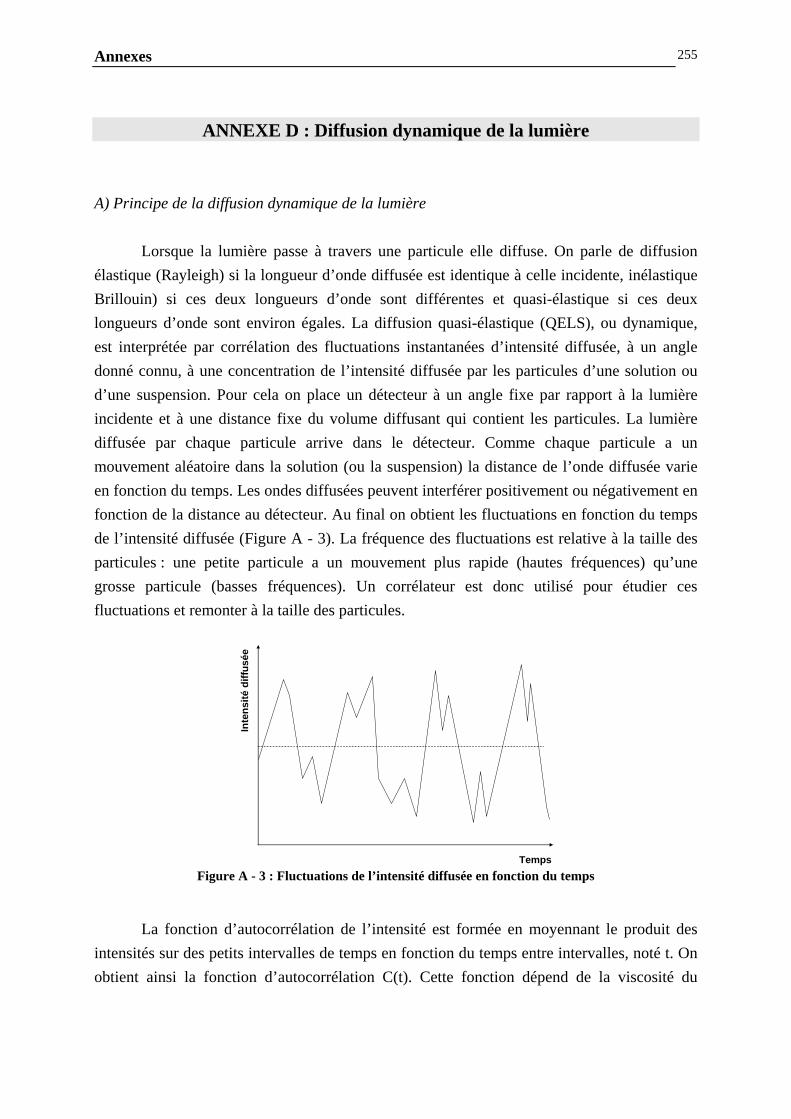
Annexes
255
ANNEXE D : Diffusion dynamique de la lumière
A) Principe de la diffusion dynamique de la lumière Lorsque la lumière passe à travers une particule elle diffuse. On parle de diffusion élastique (Rayleigh) si la longueur d’onde diffusée est identique à celle incidente, inélastique Brillouin) si ces deux longueurs d’onde sont différentes et quasi-élastique si ces deux longueurs d’onde sont environ égales. La diffusion quasi-élastique (QELS), ou dynamique, est interprétée par corrélation des fluctuations instantanées d’intensité diffusée, à un angle donné connu, à une concentration de l’intensité diffusée par les particules d’une solution ou d’une suspension. Pour cela on place un détecteur à un angle fixe par rapport à la lumière incidente et à une distance fixe du volume diffusant qui contient les particules. La lumière diffusée par chaque particule arrive dans le détecteur. Comme chaque particule a un mouvement aléatoire dans la solution (ou la suspension) la distance de l’onde diffusée varie en fonction du temps. Les ondes diffusées peuvent interférer positivement ou négativement en fonction de la distance au détecteur. Au final on obtient les fluctuations en fonction du temps de l’intensité diffusée (Figure A - 3). La fréquence des fluctuations est relative à la taille des particules : une petite particule a un mouvement plus rapide (hautes fréquences) qu’une grosse particule (basses fréquences). Un corrélateur est donc utilisé pour étudier ces fluctuations et remonter à la taille des particules.
Figure A - 3 : Fluctuations de l’intensité diffusée en fonction du temps
La fonction d’autocorrélation de l’intensité est formée en moyennant le produit des intensités sur des petits intervalles de temps en fonction du temps entre intervalles, noté t. On obtient ainsi la fonction d’autocorrélation C(t). Cette fonction dépend de la viscosité du
Temps
Inte
nsité
diff
usée

Annexes
256
milieu, de l’indice de réfraction, de la température et de la taille des particules. Elle est plus ou moins complexe, et plusieurs formes sont donc possibles. B) Appareillage Le schéma du montage est représenté à la Figure A - 4. Un laser Spectra Physics série 2000 a été utilisé avec une longueur d’onde de 514 nm et une puissance ajustée selon la concentration en particules diffusantes. Le photomultiplicateur et analyseur Brookvaven BI-8000 a été placé à 90°.
Figure A - 4 : Schéma du montage de diffusion dynamique de la lumière
C) Méthode de calcul du diamètre moyen des particules La fonction de corrélation peut se mettre sous la forme suivante (non negative linear square) : Avec : temps de corrélation : q : vecteur d’onde :
Laser
Corrélateur Photomultiplicateur
Lentille
Echantillon
Lentille
BtAtC +Γ−= )2exp()(
2sin4 1 θλπ −= nq
2qD×=ΓΓ

Annexes
257
D : constante de diffusion : n : indice de réfraction du diluant λ : longueur d’onde du faisceau laser θ : angle de mesure kB : constante de Boltzmann T : température (K) η : viscosité du diluant d : diamètre des particules On en déduit la relation entre le diamètre des particules et le temps de corrélation, qui permet de calculer le diamètre moyen des particules :
dTkD B
πη3=
Γ=
πη3
2Tqkd B
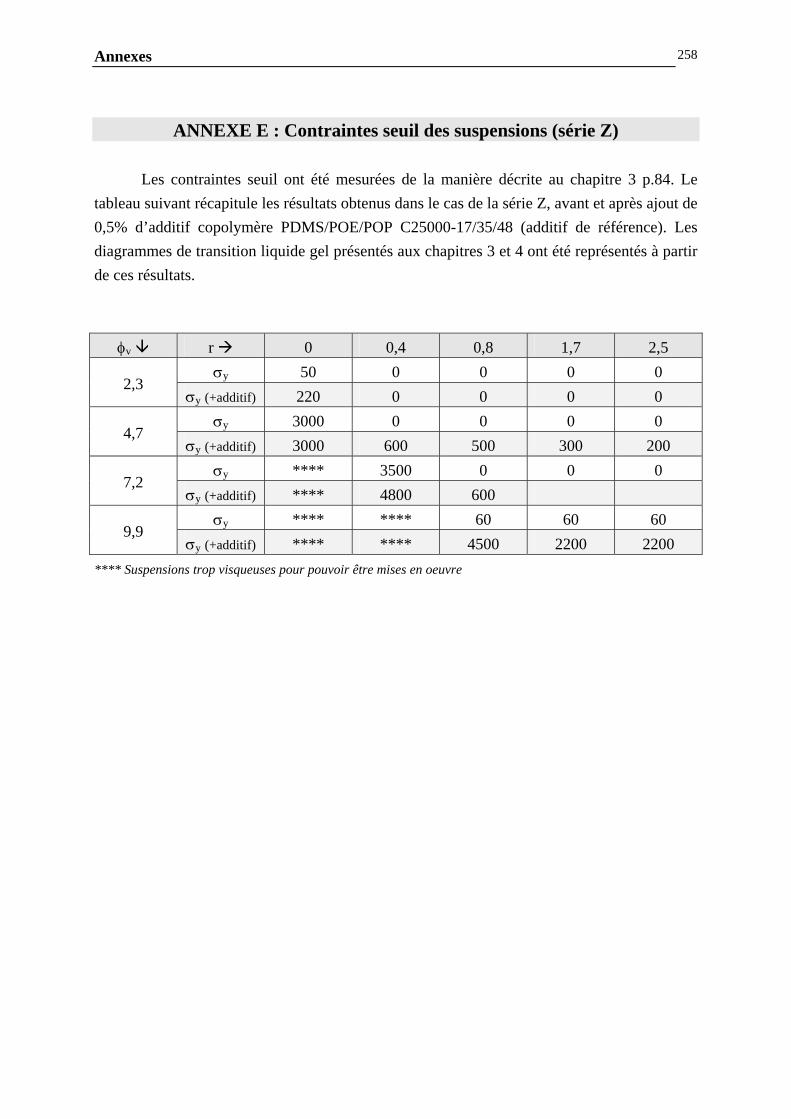
Annexes
258
ANNEXE E : Contraintes seuil des suspensions (série Z)
Les contraintes seuil ont été mesurées de la manière décrite au chapitre 3 p.84. Le tableau suivant récapitule les résultats obtenus dans le cas de la série Z, avant et après ajout de 0,5% d’additif copolymère PDMS/POE/POP C25000-17/35/48 (additif de référence). Les diagrammes de transition liquide gel présentés aux chapitres 3 et 4 ont été représentés à partir de ces résultats.
φv r 0 0,4 0,8 1,7 2,5 σy 50 0 0 0 0
2,3 σy (+additif) 220 0 0 0 0
σy 3000 0 0 0 0 4,7
σy (+additif) 3000 600 500 300 200 σy **** 3500 0 0 0
7,2 σy (+additif) **** 4800 600
σy **** **** 60 60 60 9,9
σy (+additif) **** **** 4500 2200 2200 **** Suspensions trop visqueuses pour pouvoir être mises en oeuvre
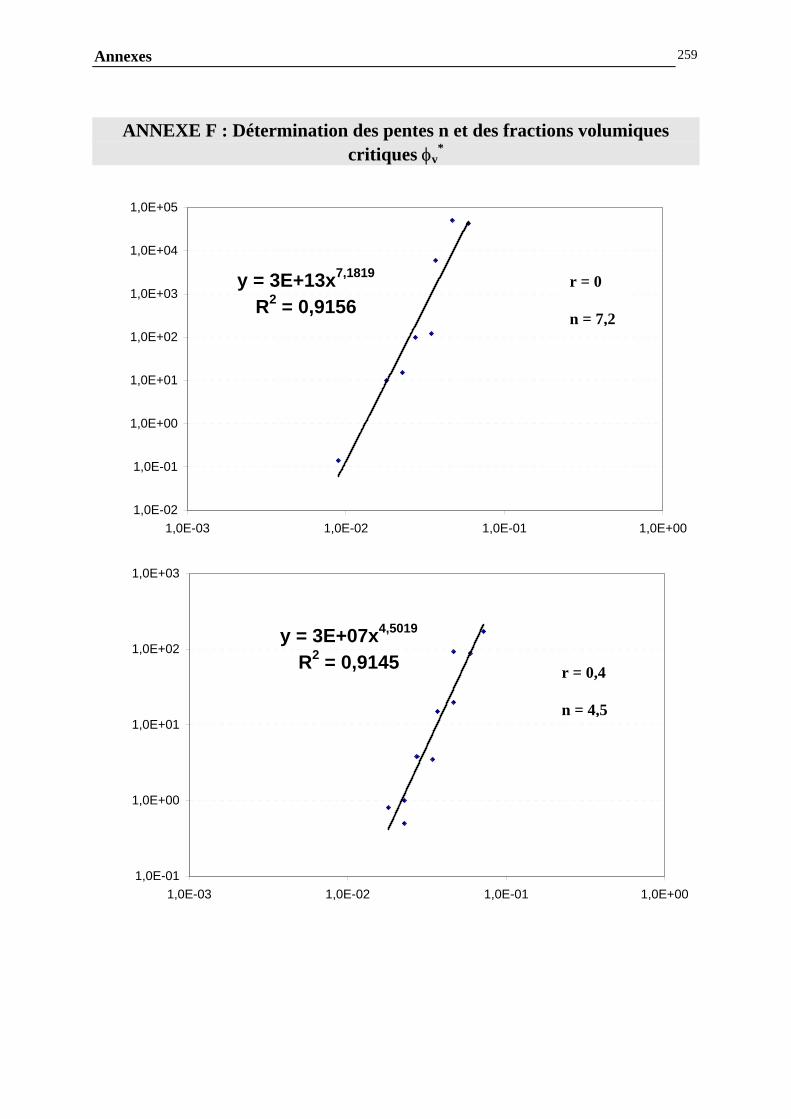
Annexes
259
ANNEXE F : Détermination des pentes n et des fractions volumiques
critiques φv*
y = 3E+13x7,1819
R2 = 0,9156
1,0E-02
1,0E-01
1,0E+00
1,0E+01
1,0E+02
1,0E+03
1,0E+04
1,0E+05
1,0E-03 1,0E-02 1,0E-01 1,0E+00
r = 0 n = 7,2
y = 3E+07x4,5019
R2 = 0,9145
1,0E-01
1,0E+00
1,0E+01
1,0E+02
1,0E+03
1,0E-03 1,0E-02 1,0E-01 1,0E+00
r = 0,4 n = 4,5
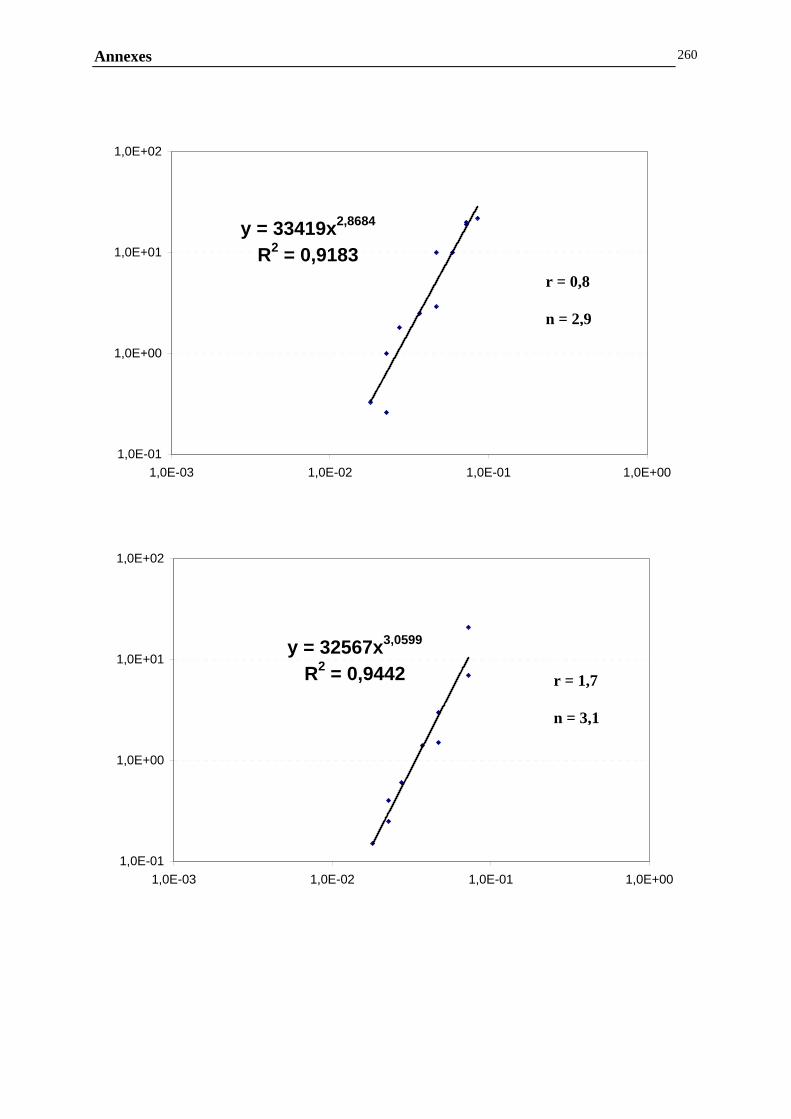
Annexes
260
y = 32567x3,0599
R2 = 0,9442
1,0E-01
1,0E+00
1,0E+01
1,0E+02
1,0E-03 1,0E-02 1,0E-01 1,0E+00
y = 33419x2,8684
R2 = 0,9183
1,0E-01
1,0E+00
1,0E+01
1,0E+02
1,0E-03 1,0E-02 1,0E-01 1,0E+00
r = 0,8 n = 2,9
r = 1,7 n = 3,1
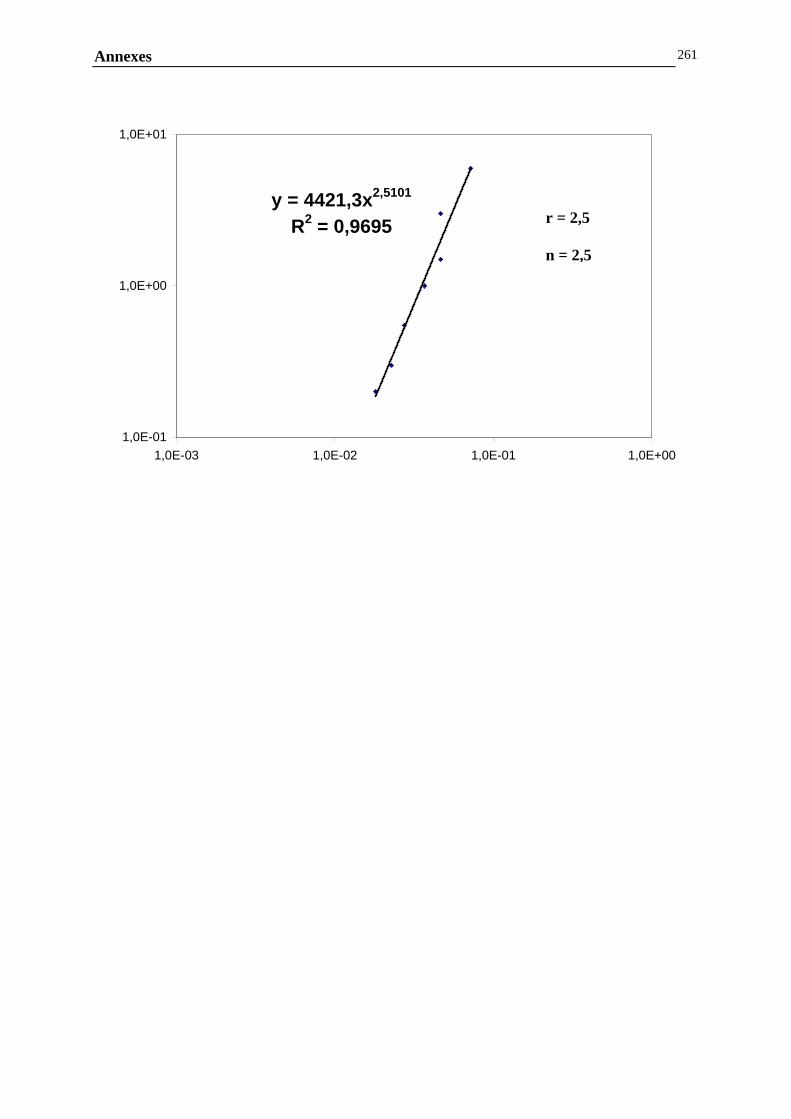
Annexes
261
y = 4421,3x2,5101
R2 = 0,9695
1,0E-01
1,0E+00
1,0E+01
1,0E-03 1,0E-02 1,0E-01 1,0E+00
r = 2,5 n = 2,5

Annexes
262
ANNEXE G : Analyse de la composition chimique des copolymères PDMS /
Polyéther A/ Méthode de calcul Soit A1, A2 et A3 les intégrales des pics de résonance à 0,05, 1,1 et 3.2-3,8 ppm respectivement. Soit X, Y et Z le nombre de motifs PDMS, POP et POE respectivement. Soit M1, M2 et M3 les masses correspondantes des motifs PDMS, POP et POE A1 correspond à l’aire des protons des méthyles du PDMS soit 6X protons A2 correspond à l’aire des protons des méthyles du POP soit 3Y protons A3 correspond à l’aire des protons des CH2 du POE, ceux des CH2 et CH du POP soit 3Y+4Z protons On en déduie : A1=6X ⇒ X=A1/6 A2=3Y ⇒ Y=A2/3 A3=3Y+4Z ⇒ Z=(A3-A2)/4 Les proportions de chaque bloc (en masse) sont définies de la manière suivante : % PDMS = M1/(M1+M2+M3) avec M1 = 74X % POP = M2/(M1+ M2+ M3) avec M2 = 44Y %POE = M3/(M1+ M2+ M3) avec M3 = 58Z On en déduit les relations :
% PDMS = ( )458344
674
674
2321
1
AAAA
A−++
% POP = ( )458344
674
374
2321
2
AAAA
A−++
% POE = ( )
( )458344
674
4582321
23
AAAA
AA
−++
−
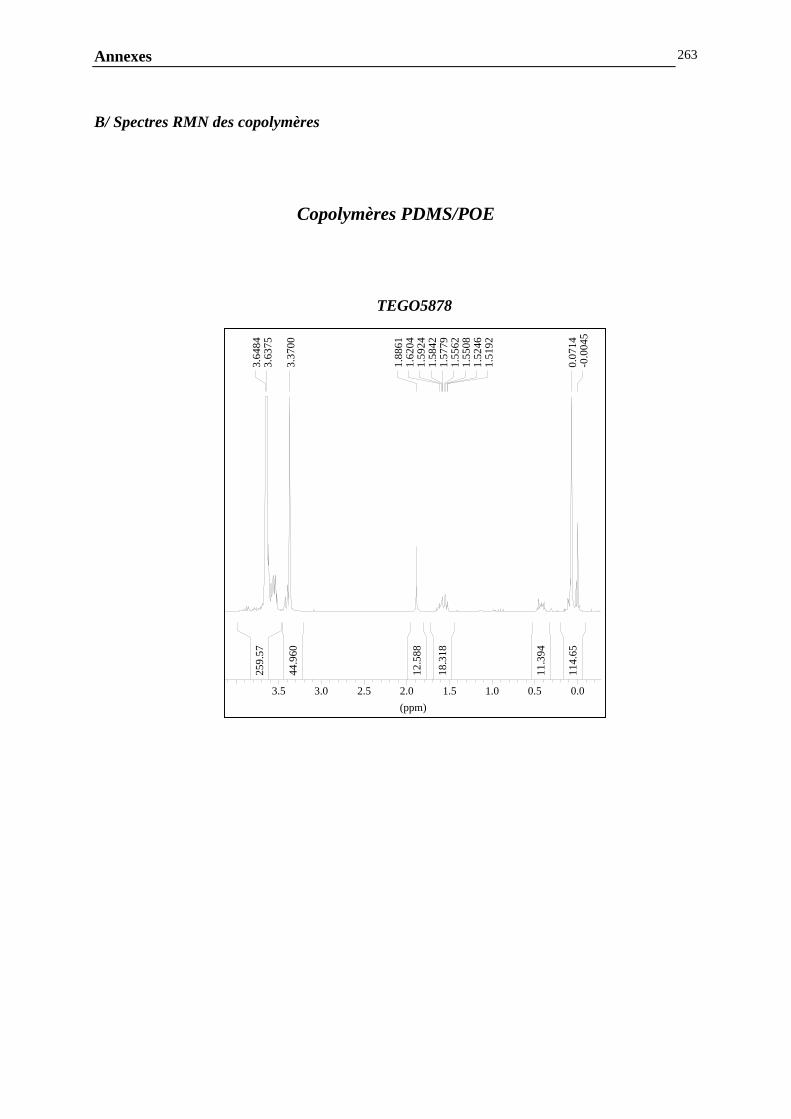
Annexes
263
B/ Spectres RMN des copolymères
Copolymères PDMS/POE TEGO5878
259.
57
44.9
60
12.5
88
18.3
18
11.3
94
114.
65
3.64
843.
6375
3.37
00
1.88
611.
6204
1.59
241.
5842
1.57
791.
5562
1.55
081.
5246
1.51
92
0.07
14-0
.004
5
(ppm)0.00.51.01.52.02.53.03.5
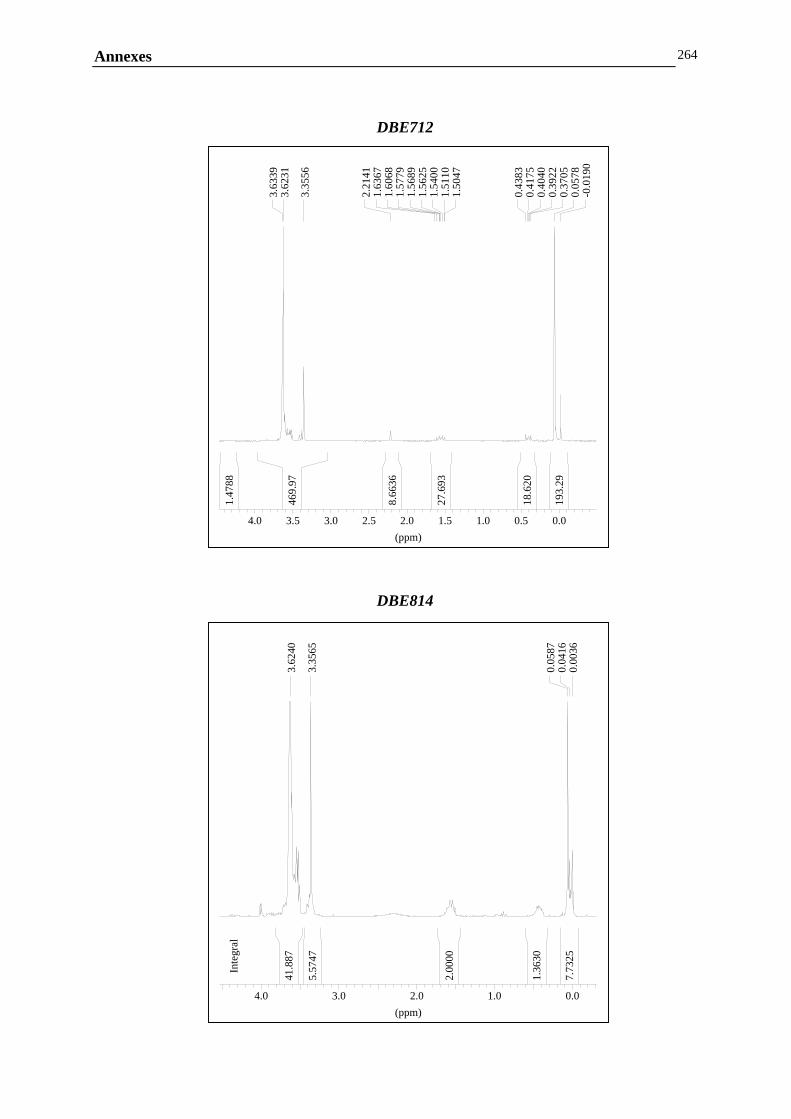
Annexes
264
DBE712 DBE814
1.47
88
469.
97
8.66
36
27.6
93
18.6
20
193.
29
3.63
393.
6231
3.35
56
2.21
411.
6367
1.60
681.
5779
1.56
891.
5625
1.54
001.
5110
1.50
47
0.43
830.
4175
0.40
400.
3922
0.37
050.
0578
-0.0
190
(ppm)0.00.51.01.52.02.53.03.54.0
41.8
87
5.57
47
2.00
00
1.36
30
7.73
25
Inte
gral
3.62
40
3.35
65
0.05
870.
0416
0.00
36
(ppm)0.01.02.03.04.0
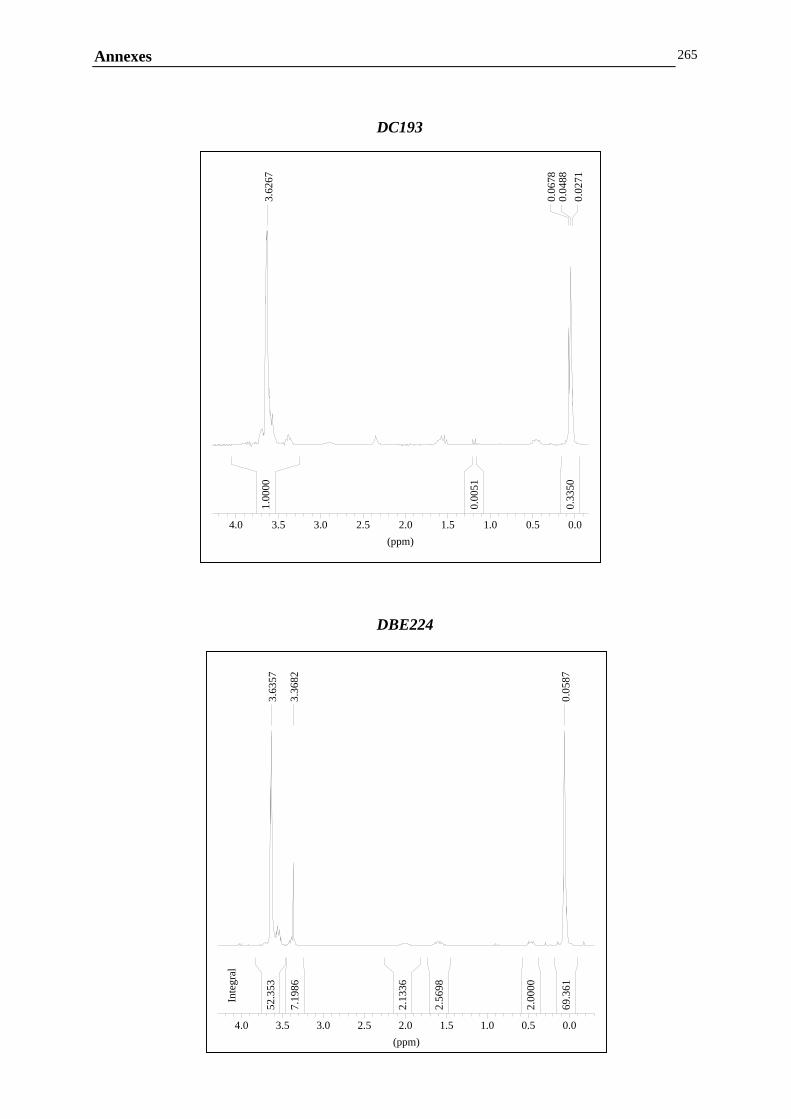
Annexes
265
DC193 DBE224
1.00
00
0.00
51
0.33
50
3.62
67
0.06
780.
0488
0.02
71
(ppm)0.00.51.01.52.02.53.03.54.0
52.3
53
7.19
86
2.13
36
2.56
98
2.00
00
69.3
61
Inte
gral
3.63
57
3.36
82
0.05
87
(ppm)0.00.51.01.52.02.53.03.54.0
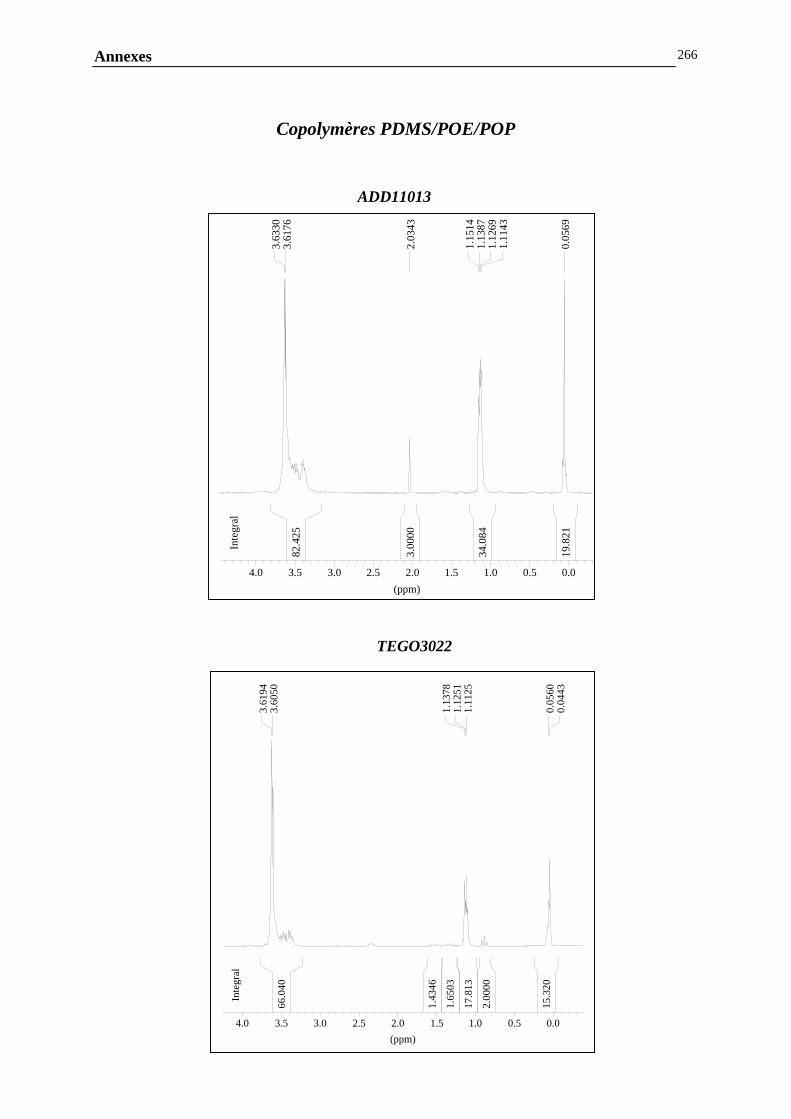
Annexes
266
Copolymères PDMS/POE/POP
ADD11013 TEGO3022
82.4
25
3.00
00
34.0
84
19.8
21
Inte
gral
3.63
303.
6176
2.03
43
1.15
141.
1387
1.12
691.
1143
0.05
69
(ppm)0.00.51.01.52.02.53.03.54.0
66.0
40
1.43
46
1.65
03
17.8
13
2.00
00
15.3
20
Inte
gral
3.61
943.
6050
1.13
781.
1251
1.11
25
0.05
600.
0443
(ppm)0.00.51.01.52.02.53.03.54.0
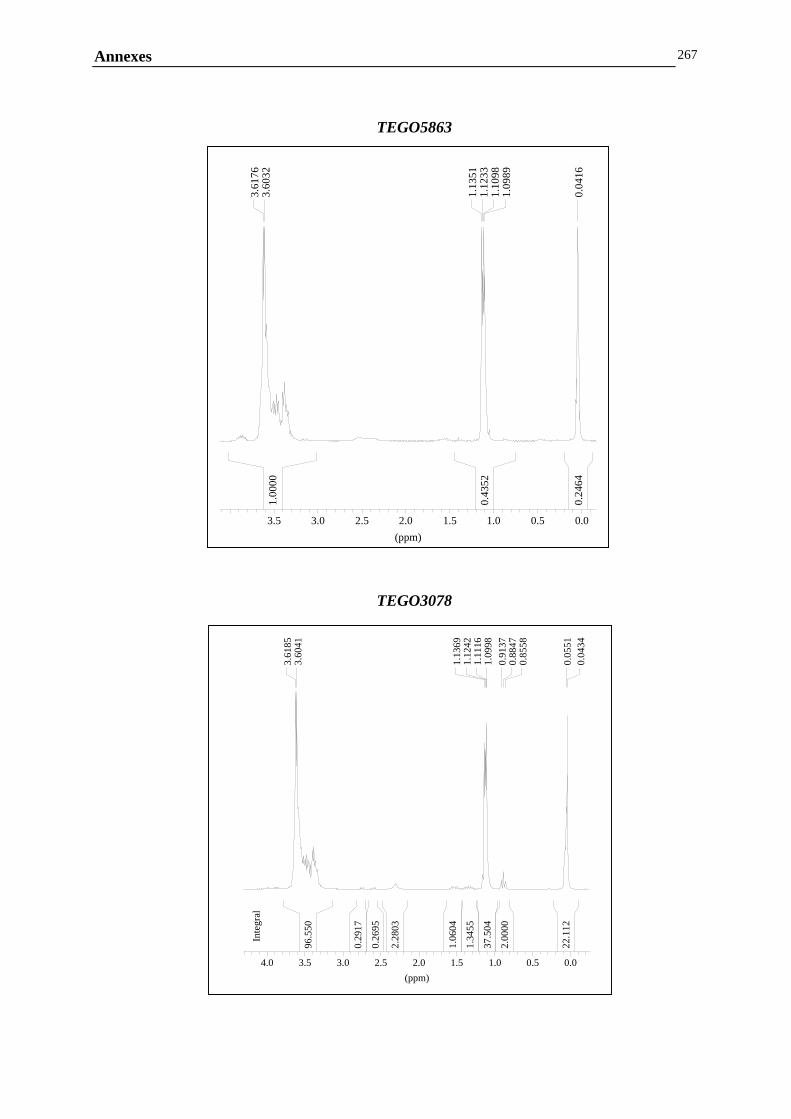
Annexes
267
TEGO5863 TEGO3078
1.00
00
0.43
52
0.24
64
3.61
763.
6032
1.13
511.
1233
1.10
981.
0989
0.04
16
(ppm)0.00.51.01.52.02.53.03.5
96.5
50
0.29
17
0.26
95
2.28
03
1.06
04
1.34
55
37.5
04
2.00
00
22.1
12
Inte
gral
3.61
853.
6041
1.13
691.
1242
1.11
161.
0998
0.91
370.
8847
0.85
58
0.05
510.
0434
(ppm)0.00.51.01.52.02.53.03.54.0
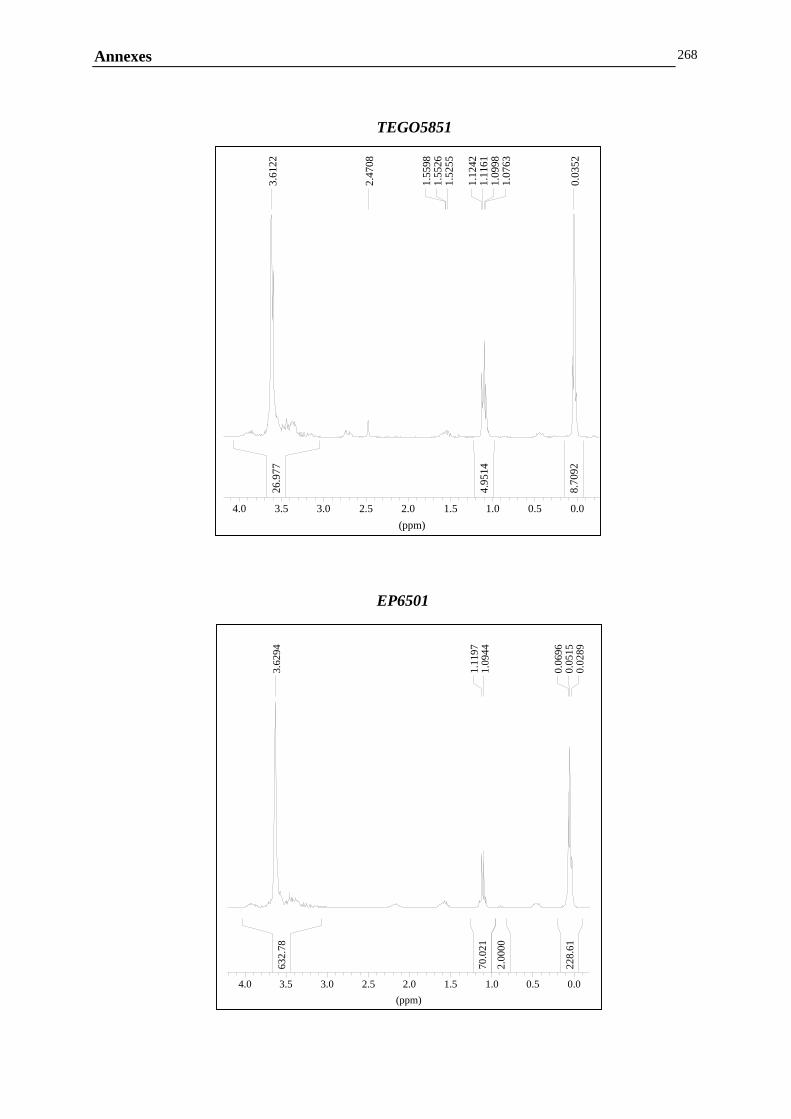
Annexes
268
TEGO5851 EP6501
26.9
77
4.95
14
8.70
92
3.61
22
2.47
08
1.55
981.
5526
1.52
55
1.12
421.
1161
1.09
981.
0763
0.03
52
(ppm)0.00.51.01.52.02.53.03.54.0
632.
78
70.0
21
2.00
00
228.
61
3.62
94
1.11
971.
0944
0.06
960.
0515
0.02
89
(ppm)0.00.51.01.52.02.53.03.54.0
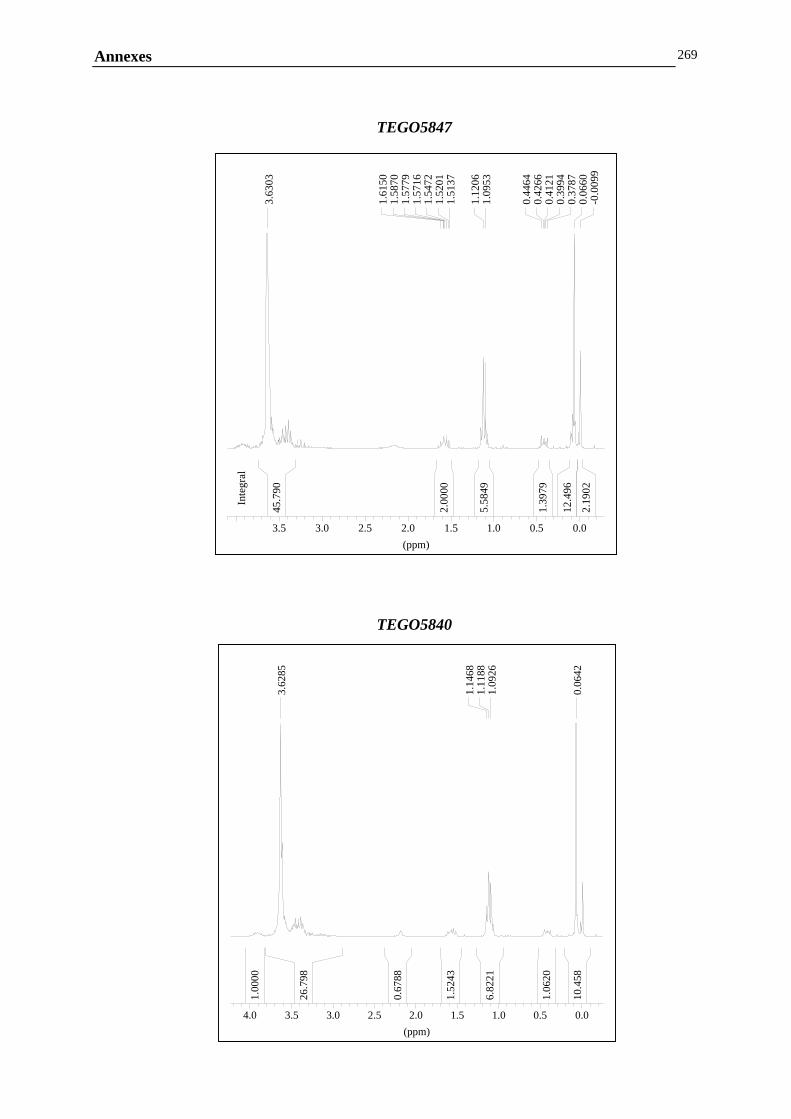
Annexes
269
TEGO5847 TEGO5840
45.7
90
2.00
00
5.58
49
1.39
79
12.4
96
2.19
02
Inte
gral
3.63
03
1.61
501.
5870
1.57
791.
5716
1.54
721.
5201
1.51
37
1.12
061.
0953
0.44
640.
4266
0.41
210.
3994
0.37
870.
0660
-0.0
099
(ppm)0.00.51.01.52.02.53.03.5
1.00
00
26.7
98
0.67
88
1.52
43
6.82
21
1.06
20
10.4
58
3.62
85
1.14
681.
1188
1.09
26
0.06
42
(ppm)0.00.51.01.52.02.53.03.54.0

Annexes
270

FOLIO ADMINISTRATIF
THESE SOUTENUE DEVANT L'INSTITUT NATIONAL DES SCIENCES APPLIQUEES DE LYON
NOM : PAQUIEN DATE de SOUTENANCE : 3 Décembre 2003 (avec précision du nom de jeune fille, le cas échéant) Prénoms : Jean-Noël TITRE : ETUDE DES PROPRIETES RHEOLOGIQUES ET DE L’ETAT DE DISPERSION DE SUSPENSIONS PDMS/SILICE NATURE : Doctorat Numéro d'ordre : 03 ISAL Ecole doctorale : Matériaux Spécialité : Matériaux Polymères et Composites Cote B.I.U. - Lyon : T 50/210/19 / et bis CLASSE : RESUME : Le but de cette étude est de comprendre comment les interactions PDMS-silice contrôlent la rhéologie de ces systèmes et d’étudier des additifs polymères permettant de modifier la rhéologie de ces suspensions. Ces interactions ont été modulées par réaction contrôlée de l’hexaméthyldisilazane avec la surface de la silice. Après une étude approfondie du greffage, la rhéologie des suspensions a été étudiée afin de connaître l’influence des caractéristiques de la silice et du procédé de mise en œuvre. L’effet d’additifs polymère sur la rhéologie des suspensions a été évalué par mesures de contrainte seuil en mode statique. Un mécanisme de structuration des additifs par séparation de phase avec le milieu PDMS et adsorption à la surface de la silice a été proposé, permettant d’interpréter les résultats obtenus. Cette étude a permis de mieux comprendre les paramètres clés contrôlant les propriétés rhéologiques des suspensions PDMS/Silice et d’identifier les microstructures des additifs conduisant aux propriétés rhéologiques souhaitées. MOTS-CLES : PDMS – silice – greffage – HMDS – rhéologie – interactions – suspensions – contrainte seuil Laboratoire (s) de recherches : Laboratoire des Matériaux Macromoléculaires (INSA, UMR5627) Directeur de thèse: J.-F. GERARD, Professeur INSA J. GALY, Chargé de Recherche CNRS Président de jury : C. MARTELET, Composition du jury : J.-F. TASSIN, H. VAN DAMME, J.-F. GERARD, J. GALY, A. POUCHELON





























