Embed Size (px)
Citation preview

La régulation du transport du glucose dans le muscle squelettique
L’implication des protéines AMPK et iNOS
Thèse
Emmanuelle St-Amand
Doctorat en physiologie-endocrinologie
Philosophiae doctor (Ph.D.)
Québec, Canada
© Emmanuelle St-Amand, 2015


iii
Résumé
Le tissu musculaire squelettique contribue considérablement au maintien de l’homéostasie du
glucose chez l’humain. Que ce soit en situation postprandiale ou lors d’un travail musculaire, le
muscle squelettique capte de grandes quantités de glucose sanguin à des fins d’entreposage ou de
production d’énergie. Les voies de signalisation cellulaire impliquées dans la régulation du transport
du glucose à l’intérieur de la cellule musculaire sont nombreuses et complexes. En présence de
désordres physiologiques et/ou métaboliques, de nombreux médiateurs chimiques et enzymatiques
peuvent interagir avec les différentes protéines de ces voies de signalisation et entraîner des
perturbations importantes au niveau de l’homéostasie du glucose.
Notre première étude nous a permis de confirmer l’implication de la protéine AMPK dans le transport
du glucose induit par la contraction musculaire. L’AMPK est un senseur énergétique important activé
dans le muscle squelettique au cours d’un effort physique. Toutefois, son rôle dans le transport du
glucose induit par la contraction musculaire demeure controversé. Grâce à un modèle murin
d’invalidation génétique de l’AMPK spécifique au tissu musculaire et à l’élaboration d’un protocole de
contraction ex vivo approprié, nous avons établi l’importance de l’AMPK dans la régulation du
transport du glucose.
Notre seconde étude nous a permis de démontrer que l’incubation ex vivo prolongée du muscle
épitrochléen modifie l’expression du transporteur de glucose GLUT1. Nous avons également observé
l’induction de la protéine iNOS et la production du NO. Parallèlement, nous avons mesuré une
augmentation de l’expression de GLUT1 à la suite d’une exposition au NO dans un modèle de
cellules musculaires ainsi qu’une augmentation du transport basal du glucose. L’ensemble de nos
résultats nous permet de consolider le lien causal entre la production du NO et la modulation de
l’expression de GLUT1 et potentiellement, le développement de perturbations au niveau du
métabolisme du glucose musculaire.


v
Table des matières
RÉSUMÉ ............................................................................................................................................ III
TABLE DES MATIERES ..................................................................................................................... V
LISTE DES TABLEAUX ..................................................................................................................... IX
LISTE DES FIGURES ........................................................................................................................ XI
LISTE DES ABRÉVIATIONS ET DES SIGLES ............................................................................... XIII
REMERCIEMENTS .......................................................................................................................... XIX
AVANT-PROPOS ............................................................................................................................. XXI
INTRODUCTION ................................................................................................................................. 1
1 LE TRANSPORT DU GLUCOSE DANS LE MUSCLE SQUELETTIQUE ................................... 1 1.1 LE MUSCLE SQUELETTIQUE ...................................................................................................... 1
1.1.1 Anatomie et structure ....................................................................................................... 1
1.1.2 Type de fibres musculaires .............................................................................................. 2
1.2 TRANSPORTEURS DE GLUCOSE ................................................................................................ 3
1.2.1 GLUT1 ............................................................................................................................. 4
1.2.2 GLUT4 ............................................................................................................................. 5
1.3 VOIE DE SIGNALISATION DE L’INSULINE ..................................................................................... 7
1.3.1 Récepteur de l’insuline ..................................................................................................... 7
1.3.2 Substrat du récepteur de l’insuline ................................................................................... 8
1.3.3 Phosphatidylinositol 3-kinase ........................................................................................... 9
1.3.4 Protéine kinase B ou Akt ................................................................................................ 10
1.3.5 Substrats d’Akt ............................................................................................................... 10
1.3.5.1 TBC1D4 ................................................................................................................. 11
1.3.5.2 TBC1D1 ................................................................................................................. 12
1.3.6 Rac1 .............................................................................................................................. 14
1.4 VOIES DE SIGNALISATION DE LA CONTRACTION MUSCULAIRE ..................................................... 14
1.4.1 Calcium .......................................................................................................................... 14
1.4.2 Protéine Kinase C .......................................................................................................... 15
1.4.3 Les protéines kinases activées par les facteurs mitogènes ........................................... 16
1.4.4 Monoxyde d’azote .......................................................................................................... 17
1.4.5 Espèces réactives de l’oxygène ..................................................................................... 17
1.4.6 Rac1 .............................................................................................................................. 18
1.4.7 Substrats d’Akt ............................................................................................................... 18
1.4.7.1 TBC1D4 ................................................................................................................. 18
1.4.7.2 TBC1D1 ................................................................................................................. 19

vi
2 AMPK ........................................................................................................................................ 20 2.1 STRUCTURE ET EXPRESSION ................................................................................................. 21
2.2 RÉGULATION DE L’AMPK ...................................................................................................... 23
2.2.1 Nucléotides de l’adénine ................................................................................................ 23
2.2.2 AMPKK .......................................................................................................................... 24
2.2.2.1 LKB1 ...................................................................................................................... 24
2.2.2.2 CAMKK .................................................................................................................. 25
2.2.2.3 TAK1 ...................................................................................................................... 26
2.3 ACTIVATION DE L’AMPK ....................................................................................................... 26
2.3.1 Activateurs pharmacologiques ....................................................................................... 26
2.3.1.1 AICAR .................................................................................................................... 27
2.3.1.2 Metformine ............................................................................................................. 27
2.3.1.3 Thiazolidinediones ................................................................................................. 28
2.3.1.4 A-769662 ............................................................................................................... 28
2.3.1.5 Composés naturels ................................................................................................ 29
2.3.2 Activation par la contraction musculaire ......................................................................... 29
2.3.2.1 Statut énergétique .................................................................................................. 30
2.3.2.2 Calcium .................................................................................................................. 30
2.3.2.3 Espèces réactives de l’oxygène ............................................................................. 31
2.3.2.4 Interleukine-6 ......................................................................................................... 31
2.4 RÉGULATION DU TRANSPORT DU GLUCOSE DANS LE MUSCLE SQUELETTIQUE ............................. 32
2.4.1 Modèles transgéniques des sous-unités catalytiques de l’AMPK ................................... 32
2.4.1.1 Souris AMPK KD1 .................................................................................................. 32
2.4.1.2 Souris AMPK α2i TG .............................................................................................. 34
2.4.1.3 Souris AMPKα1/α2 KO .......................................................................................... 34
2.4.1.4 Souris AMPKα1α2 mdKO ...................................................................................... 35
2.4.2 Modèles transgéniques des sous-unités régulatrices de l’AMPK ................................... 35
2.4.2.1 Souris AMPKγ3 KO ................................................................................................ 35
2.4.2.2 Souris AMPKβ1/β2 KO ........................................................................................... 36
2.4.2.3 Souris AMPKβ1/β2M-KO ....................................................................................... 36
3 RÉSISTANCE À L’INSULINE MUSCULAIRE ........................................................................... 38 3.1 DÉFINITION .......................................................................................................................... 38
3.2 DÉFAUT DE LA VOIE DE SIGNALISATION DE L’INSULINE ............................................................... 38
3.2.1 Récepteur à l’insuline ..................................................................................................... 39
3.2.2 Substrat du récepteur à l’insuline 1 ................................................................................ 39
3.2.3 Phosphatidylinositol 3-kinase ......................................................................................... 39
3.2.4 Protéine kinase B/Akt ..................................................................................................... 40
3.3 LIPOTOXICITÉ ....................................................................................................................... 40
3.3.1 Acyl-coenzyme-A ........................................................................................................... 41
3.3.2 Diacylglycérol ................................................................................................................. 41
3.3.3 Céramides ...................................................................................................................... 42

vii
3.4 INFLAMMATION ..................................................................................................................... 43
3.4.1 LPS ................................................................................................................................ 44
3.4.2 TNFα .............................................................................................................................. 45
3.4.3 Interleukine-6 ................................................................................................................. 46
3.4.4 Macrophages ................................................................................................................. 48
4 INOS .......................................................................................................................................... 48 4.1 STRUCTURE ET EXPRESSION ................................................................................................. 48
4.2 RÉGULATION DE L’EXPRESSION D’INOS ................................................................................. 49
4.3 INOS ET LA RÉSISTANCE À L’INSULINE MUSCULAIRE................................................................. 50
4.3.1 Nitration ......................................................................................................................... 52
4.3.2 S-nitrosylation ................................................................................................................ 53
5 OBJECTIFS DE RECHERCHE ................................................................................................. 54
CHAPITRE 1 ..................................................................................................................................... 57 RÉSUMÉ .......................................................................................................................................... 58
ABSTRACT ....................................................................................................................................... 59
INTRODUCTION ................................................................................................................................. 60
EXPERIMENTAL PROCEDURES ............................................................................................................ 62
RESULTS ......................................................................................................................................... 66
DISCUSSION .................................................................................................................................... 68
ACKNOWLEDGEMENTS ...................................................................................................................... 71
REFERENCES ................................................................................................................................... 72
LEGENDS TO FIGURES ....................................................................................................................... 75
CHAPITRE 2 ..................................................................................................................................... 81 RÉSUMÉ .......................................................................................................................................... 82
ABSTRACT ....................................................................................................................................... 83
INTRODUCTION ................................................................................................................................. 84
EXPERIMENTAL PROCEDURES ............................................................................................................ 86
RESULTS ......................................................................................................................................... 89
DISCUSSION .................................................................................................................................... 91
REFERENCES ................................................................................................................................... 94
LEGENDS TO FIGURES ....................................................................................................................... 97
CONCLUSION ................................................................................................................................. 103 LE RÔLE DE L’AMPK DANS LE TRANSPORT DU GLUCOSE INDUIT PAR LA CONTRACTION MUSCULAIRE ...... 103
L’INCUBATION À LONG TERME DU MUSCLE ÉPITROCHLÉEN CAUSE L’INDUCTION D’INOS ET L’AUGMENTATION
DE L’EXPRESSION DE GLUT1 .......................................................................................................... 109
BIBLIOGRAPHIE ............................................................................................................................ 115


ix
Liste des tableaux
INTRODUCTION
TABLEAU 1 CLASSIFICATION DES GLUTS ...................................................................................... 4
TABLEAU 2 EXPRESSION DES DIFFÉRENTS ISOFORMES DE L'AMPK DANS LES MUSCLES
EDL ET SOLÉAIRE DE SOURIS ............................................................................................... 21


xi
Liste des Figures
INTRODUCTION
FIGURE 1 ORGANISATION STRUCTURELLE D'UNE FIBRE MUSCULAIRE ................................... 2
FIGURE 2 STRUCTURE DE L’IR ........................................................................................................ 8
FIGURE 3 STRUCTURE D'IRS1 ......................................................................................................... 9
FIGURE 4 STRUCTURE DE LA PI3K DE CLASSE IA ........................................................................ 9
FIGURE 5 STRUCTURE DE AS160/TBC1D4 ET TBC1D1 ............................................................... 13
FIGURE 6 VOIE DE SIGNALISATION DE L'INSULINE .................................................................... 13
FIGURE 7 VOIES DE SIGNALISATION DE LA CONTRACTION MUSCULAIRE ............................. 20
FIGURE 8 STRUCTURE DE L'AMPK ............................................................................................... 23
FIGURE 9 RÉGULATION DE L'AMPK .............................................................................................. 26
FIGURE 10 STRUCTURE D'INOS .................................................................................................... 49
FIGURE 11 MODIFICATIONS NITROSATIVES DE LA VOIE DE SIGNALISATION DE L'INSULINE54
CHAPITRE 1
FIGURE 1 LACK OF AMPKα2 ACTIVATION AND GLUCOSE TRANSPORT STIMULATION BY
AICAR IN EDL OF AMPK-KD MICE .......................................................................................... 77
FIGURE 2 FORCE PRODUCTION AND FATIGABILITY OF EDL MUSCLES OF WT AND KD-
AMPKα2 .................................................................................................................................... 78
FIGURE 3 EFFECTS OF DIFFERENT STIMULATION FREQUENCIES ON AMPKα
PHOSPHORYLATION IN EDL FROM WT MICE ....................................................................... 79
FIGURE 4 LACK OF AMPKα2 ACTIVITY REDUCES CONTRACTION-MEDIATED GLUCOSE
TRANSPORT IN EDL FROM KD-AMPKα2 MICE ..................................................................... 80
CHAPITRE 2
FIGURE 1 LONG-TERM INCUBATION OF EPITROCHLEARIS MUSCLE MODULATES NOS
EXPRESSION ........................................................................................................................... 98
FIGURE 2 TIME COURSE OF INCUBATION-MEDIATED INDUCTION OF INOS AND GLUT1 ...... 99
FIGURE 3 INOS INDUCTION CAUSES GLUT1 EXPRESSION IN EPITROCHLEARIS MUSCLE . 100
FIGURE 4 NO INDUCTION INCREASE GLUT1 EXPRESSION AND BASAL GLUCOSE UPTAKE
INL6 MUSCLE CELLS ............................................................................................................. 101


xiii
Liste des abréviations et des sigles
ACC Acétyl-coenzyme A carboxylase
Acyl-Coa Acyl-coenzyme A
Adn Adiponectine
ADP Adénosine diphosphate
AGL Acides gras libres
AICAR 5-aminoimidazole-4-carboxamide ribonucléotide
Akt De l’anglais Ak transforming
AMP Adénosine monophosphate
AMPK Kinase activée par l’AMP
AMPKK AMPK kinase
ARK De l’anglais AMPK-related kinase family
ARNm Acide ribonucléique messager
AS160 De l’anglais Akt susbtrat of 160kDa
ATP Adénosine triphosphate
BH4 Tétrahydrobioptérine
C2C12 Cellules musculaires de souris
Ca2+ Calcium
CAM Calmoduline
CAMK Kinase Ca2+/calmoduline dépendante de l’anglais Ca2+/calmodulin
kinase
CAMKK De l’anglais Ca2+/calmodulin dependent protein kinase kinase
CCL2 Chimiokine C-C de type 2 de l’anglais Chemokine ligand2
CCR Récepteur de chimiokine de la famille C-C de l’anglais C-C
chemokine receptor type 2
CBS Cystathionine β-synthase
C-terminal Carboxyl-terminal
DAG Diacylglycérol
DGAT1 Diglycéride acyltransférases1
EDL Long extenseur des orteils du latin extensor digitorum longus
FAD Flavine adénine mononucléotide
FAS Acide gras synthase de l’anglais Fatty acid synthase
FMN Flavine mononucléotide
G6Pase Glucose-6-phoshatase
GAP De l’anglais GTPase-activating protein
GDP Guanosine diphosphate
GLUT Transporteur de glucose de l’anglais glucose transporter
GP Glycogène phosphorylase
GTP Guanosine triphosphate
H2O2 Peroxyde d’oxygène

xiv
HeLa Cellules cancéreuses humaines
HMG-CoA reductase Hydroxyméthylglutaryl-CoA réductase
HMIT Cotransporteur de proton/myoinositol de l’anglais Proton Myo-
inositol Transporter
HuR De l’anglais Hu antegen R
KD De l’anglais Kinase dead
IκB Inhibiteur de kappa B
IKK Kinase iκB de l’anglais IκB kinase
IL Interleukine
INF Interféron
IR Récepteur de l’insuline de l’anglais Insulin receptor
IRS1 Substrat du récepeteur de l’insuline de l’anglais Insulin receptor
substrate
JNK Kinase c-Jun-N-terminale de l’anglais c-Jun-n-terminal kinase
KI De l’anglais Knockin
KO De l’anglais Knockout
L6 Cellules musculaires de rat
LKB1 De l’anglais Liver kinase B1
L-NAME NG-Nitro-L-arginine-methyl
L-NMMA L-N-monométhyl-arginine
LPS Lipopolysaccharide
MAPK Kinase activée par les facteurs mitogènes de l’anglais Mitogen-
activated protein kinase
MAP3K De l’anglais Mitogen-activated protein kinase kinase kinase
MEF Cellules embryonnaires fibroblastiques de souris
mTORC2 Complexe 2 de la cible de la rapamycine chez les mammifères de
l’anglais mammalian target of rapamycin
NADPH Nicotinamide adénine dinucléotide phosphate
NF-κB Facteur nucléaire kappaB de l’anglais Nuclear factoe kappaB
NO Monoxyde d’azote de l’anglais Nitric oxide
NOS Synthase de monoxyde d’azote de l’anglais Nitric oxide synthase
N-terminal Amino-terminal
O2 Oxygène
O2- Superoxyde
ONOO- Peroxynitrite
PDK1 Kinase phosphoinositide-dépendente de l’anglais phosphoinositide-
dependent kinase1
PEPCK Phosphoénolpyruvate carboxykinase
PH De l’anglais pleckstrin homology
PI Phosphoinositols
PI3K Phosphatidylinositol 3-kinase

xv
PIP2 Phosphoinositide-4, 5-biophosphate
PIP3 Phosphoinositide-3, 4, 5-triphosphate
PKB Protéine kinase B
PKC Protéine kinase C
PTB De l’anglais phosphotyrosine binding
PP2A/2C Protéine phosphatases
PPARγ Récepteur activé par les proliférateurs de peroxysomes gamma
RAC1 De l’anglais Ras-related C3 botulinum toxin substrate1
RNS Espèces réactives de l’azote de l’anglais Reactive nitrogen species
ROS Espèces réactives de l’oxygène de l’anglais reactive oxygen species
Ser Sérine
SH De l’anglais Src homology
SNF1 De l’anglais Sucrose non-fermenting1
SphK1 Kinase sphingosine1 de l’anglais sphingosine kinase1
SPT1 Sérine palmitoyltransférase
STAT3 De l’anglais Signal transducer and activator of transcription 3
TA Tibial antérieur
TAK1 De l’anglais Transforming growthn factor-β- activated kinase1
TBC1D1 De l’anglais TBC1 (tre-2/USP6, BUB2, cdc16) domain family,
member 1
TBC1D4 De l’anglais TBC1 (tre-2/USP6, BUB2, cdc16) domain family,
member 4
Thr Thréonine
TNF Facteur de nécrose tumoral de l’anglais Tumoral necrosis factor
TLR4 Récepteur de type Toll4 de l’anglais Toll like receptor4
Tubule-t Tubule transverse
Tyr Tyrosine
VL Vaste lateral
W-7 N-(6-aminohexyl)-5-chloro-1-naphthalenessulfonamide
ZMP 5-aminoimidazole-4-carboxamide-1-β-ribofuranosyl monophosphate


xvii
À mes parents,
À mes frères et sœurs


xix
Remerciements
Je tiens tout d’abord à remercier mon directeur de recherche, le Dr. André Marette, pour m’avoir
donné la chance de faire partie de son équipe. Je tiens également à lui dire merci pour les
opportunités uniques qu’il m’a permis de vivre tout au long de mes études supérieures.
Je tiens à dire merci à mon codirecteur, le Dr. Claude Côté, pour son soutien, sa disponibilité, ses
encouragements et ses conseils jusqu’à la toute fin de ce long parcours.
Merci à tous mes collègues passés et présents du laboratoire Marette. Votre support et votre aide
tout au long de ces années furent très appréciés. Un merci tout particulier à Bruno Marcotte et Marie-
Julie Dubois sans qui toutes ces années n’auraient pas été les mêmes. Un gros merci à Geneviève
Pilon, la responsable de mon projet, une collègue, mais avant tout et par-dessus tout, une amie.
Un merci tout particulier à ma famille pour leur soutien et leurs encouragements.
Finalement, merci aux organismes subventionnaires qui ont rendu possible mes études: Diabète
Québec, le CRIUCPQ, le FQRNT, le CDA et l’Université Paris Descartes.


xxi
Avant-propos
CHAPITRE 1 Ce manuscrit fut publié dans la revue American Journal of Physiology Endocrinology & Metabolism. N. Lefort*, E. St-Amand*, S. Morasse, CH. Côté, A. Marette. The alpha subunit of AMPK is essential for submaximal contraction-mediated glucose transport in skeletal muscle in vitro. American Journal of Physiology Endocrinology Metabolism 2008; 295:1447-1454. * Les auteurs ont collaboré également à la rédaction du manuscrit. Pour cette étude, j’ai contribué à la collecte de l’ensemble des données publiées en collaboration
avec Nathalie Lefort. J’ai participé à la rédaction finale du manuscrit.
CHAPITRE 2 Ce manuscrit est en préparation. Pour cette étude, j’ai contribué à la collecte des données en
collaboration avec Geneviève Pilon et Kathleen Lemieux. J’ai participé à la rédaction du manuscrit
avec l’aide de Phillip J White.
Outre les deux manuscrits présentés de cette thèse, j’ai participé à la collecte de données des quatre
articles suivants:
P. J. White, P. St-Pierre, A. Charbonneau, P. Mitchell, E. St-Amand, B. Marcotte, A. Marette. Protectin DX alleviates insulin resistance by activating a myokine-liver glucoregulatory axis. Nature Medecine, 2014, 20(6):664-9.
L. Lantier, J. Fentz, R. Mounier, J. Leclerc, J. T. Treebak, C. Pehmøller, N. Sanz, I. Sakakibara, E. Saint-Amand, S. Rimbaud, P. Maire, A. Marette, R. Ventura-Clapier, A. Ferry, J. F.P. Wojtaszewski, M. Foretz, B. Viollet. AMPK controls exercise endurance, mitochondrial oxidative capacity and skeletal muscle integrity. 2014 Jul;28(7):3211-24.
E. Xu,M. P. Forest, M. Schawb, R. K. Avramoglu , E. St-Amand, A. Z. Caron, K. Bellmann, M. Shum, G. Voisin, M. Paquet, A. Montoubis, E. Lévy, K. A. Siminovitch, B. G. Neel, N. Beauchemin, A. Marette. Hepatocyte-specific Ptpn6 deletion promotes insulin-sensitive hepatic steatosis in diet-induced obesity. Hepatology2014, 59(5):1803-15
M. Sanchez, C. Darimont, V. Drapeau, S. Emady-Azar, L. Philippe, C. Ammon-Zuffrey, G. Chevrier, E. St-Amand, A. Marette, J. Doré, A. Tremblay. Effect of Lactobacillus rhamnosus CGMCC1.3724 supplementation on weight loss and maintenance in obese men and women. British Journal of Nutrition 2014, 111(8):1507-19.


1
INTRODUCTION
1 Le transport du glucose dans le muscle
squelettique
1.1 Le muscle squelettique
Chez l’humain, le muscle squelettique représente environ quarante à cinquante pourcent de la
masse corporelle totale. Bien qu’on en dénombre plus de six cents, chaque muscle est un organe
bien distinct. L’une des fonctions du muscle squelettique est la mise en mouvement des os du
squelette, caractéristique qui lui a valu son nom. De plus, il possède la capacité de transformer
l’énergie chimique, l’adénosine triphosphate (ATP), en énergie mécanique, la contraction musculaire.
Ceci lui permet de développer la force nécessaire à la production du mouvement.
1.1.1 Anatomie et structure
Un muscle est un ensemble de fibres musculaires regroupés et maintenus ensemble par un tissu
conjonctif. Le nombre de fibres musculaires qui constitue un muscle varie selon la fonction de celui-
ci. De plus, ces fibres ont différentes grosseurs et différentes longueurs. Chez l’adulte, leur diamètre
peut varier de 10μm à 100μm tandis que leur longueur peut atteindre jusqu’à 30cm, bien que la
majorité soit de l’ordre de 10cm. Chaque fibre musculaire est entourée d’une membrane plasmique,
nommée le sarcolemme, qui la relie à l’os via le tendon. De nombreuses myofibrilles, éléments
contractiles du muscle squelettique, composent chacune des fibres musculaires. Chaque myofibrille
est entourée d’un réseau de tubules transverses (tubules-T) et de tubules longitudinaux, mieux
connus sous le nom de réticulum sarcoplasmique. Les tubules-T sont de minuscules invaginations
qui s’enfoncent transversalement dans chacune des fibres musculaires à partir du sarcolemme. Les
tubules-T permettent la propagation des potentiels d’action qui engendrent la contraction musculaire
et ils servent également de canaux de transport pour différentes molécules telles le glucose et
l’oxygène. Le réticulum sarcoplasmique est parallèle aux myofibrilles. Les extrémités du réticulum
sarcoplasmique se nomment les citernes terminales. Elles emmagasinent les ions calcium (Ca2+)
essentiels à la contraction musculaire. Une triade est l’ensemble formé d’un tubule-T et de deux
citernes terminales du réticulum sarcoplasmique [1]. L’organisation structurelle est représentée à la
figure 1.
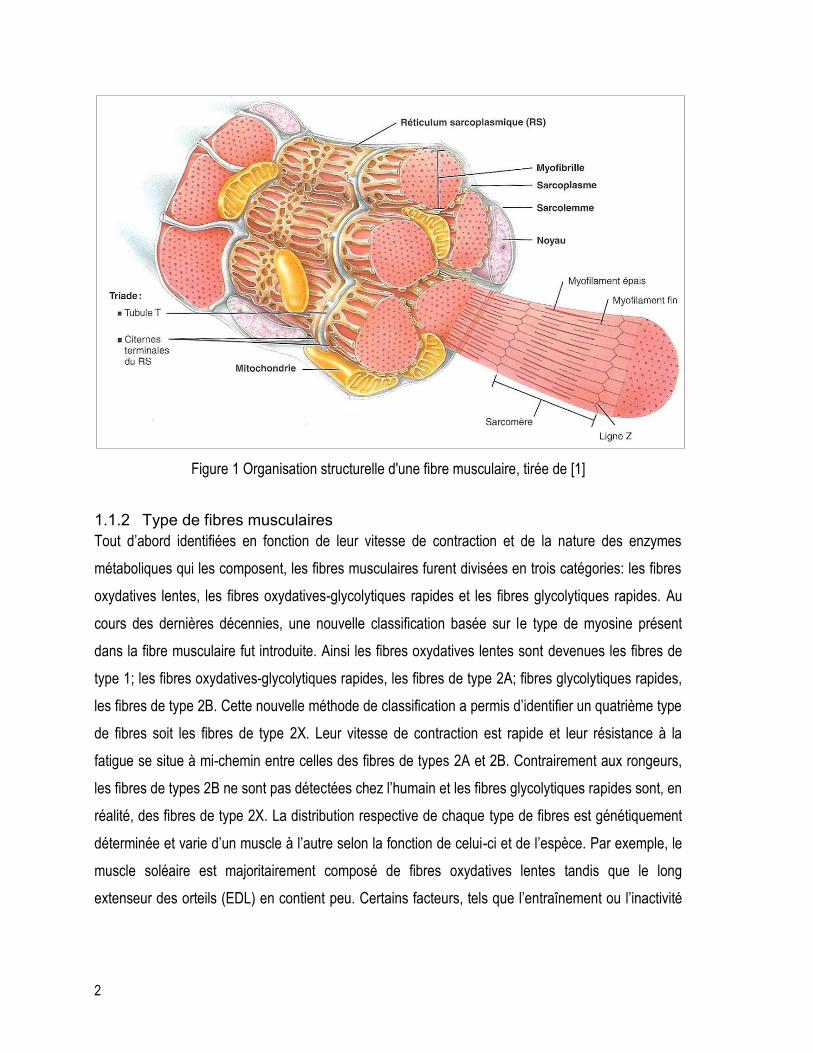
2
Figure 1 Organisation structurelle d'une fibre musculaire, tirée de [1]
1.1.2 Type de fibres musculaires
Tout d’abord identifiées en fonction de leur vitesse de contraction et de la nature des enzymes
métaboliques qui les composent, les fibres musculaires furent divisées en trois catégories: les fibres
oxydatives lentes, les fibres oxydatives-glycolytiques rapides et les fibres glycolytiques rapides. Au
cours des dernières décennies, une nouvelle classification basée sur le type de myosine présent
dans la fibre musculaire fut introduite. Ainsi les fibres oxydatives lentes sont devenues les fibres de
type 1; les fibres oxydatives-glycolytiques rapides, les fibres de type 2A; fibres glycolytiques rapides,
les fibres de type 2B. Cette nouvelle méthode de classification a permis d’identifier un quatrième type
de fibres soit les fibres de type 2X. Leur vitesse de contraction est rapide et leur résistance à la
fatigue se situe à mi-chemin entre celles des fibres de types 2A et 2B. Contrairement aux rongeurs,
les fibres de types 2B ne sont pas détectées chez l’humain et les fibres glycolytiques rapides sont, en
réalité, des fibres de type 2X. La distribution respective de chaque type de fibres est génétiquement
déterminée et varie d’un muscle à l’autre selon la fonction de celui-ci et de l’espèce. Par exemple, le
muscle soléaire est majoritairement composé de fibres oxydatives lentes tandis que le long
extenseur des orteils (EDL) en contient peu. Certains facteurs, tels que l’entraînement ou l’inactivité

3
physique, peuvent également faire varier la proportion des types de fibres retrouvés dans un muscle
[2].
Les fibres oxydatives lentes se caractérisent par leur petit diamètre et leur couleur rouge. Elles
contiennent une grande quantité de myoglobine et de nombreuses mitochondries. Ainsi, elles
produisent l’ATP nécessaire à la contraction musculaire principalement par la respiration cellulaire
d’où leur qualificatif de fibres oxydatives. Ce type de fibres développe peu de puissance musculaire,
mais il est très résistant à la fatigue ce qui lui permet d’effectuer des contractions soutenues et
prolongées nécessaires à la stabilisation de la posture et aux activités d’endurance musculaire tel
que le marathon [1].
Les fibres oxydatives-glycolytiques rapides se caractérisent par un diamètre moyen comparativement
aux deux autres types de fibres et par une couleur rouge-violet. Puisqu’elles possèdent une bonne
quantité de mitochondries et de glycogène intramusculaire, elles utilisent, à la fois, la respiration
cellulaire et la glycolyse anaérobie pour produire de l’ATP. Leur vitesse de contraction est rapide et
leur résistance à la fatigue est modérée. Elles sont donc sollicitées lors d’activités telles que la
marche et le sprint [1].
Finalement, les fibres glycolytiques rapides sont de couleur blanche et elles se caractérisent par la
grosseur de leur diamètre. Elles possèdent peu de mitochondries, mais un contenu élevé en
glycogène intramusculaire. La production d’ATP se fait essentiellement par glycolyse anaérobie. Leur
vitesse de contraction est rapide et leur nombre élevé en myofibrilles leur permet de développer la
puissance musculaire nécessaire aux mouvements explosifs, mais de courte durée. Ainsi, les fibres
glycolytiques rapides se fatiguent rapidement [1].
1.2 Transporteurs de glucose
Outre sa capacité à produire un mouvement, le muscle squelettique joue un rôle primordial dans le
maintien de l’homéostasie énergétique. Le principal substrat énergétique du muscle squelettique est
le glucose. À jeun, le muscle squelettique capte environ soixante-cinq pourcent du glucose sanguin
produit par le foie tandis qu’en situation post prandiale, environ quatre-vingt pourcent du transport du
glucose stimulé par l’insuline se fait au niveau du muscle squelettique chez l’humain [3].

4
La captation du glucose sanguin par le muscle squelettique se fait par diffusion facilitée et elle
nécessite la présence des transporteurs de glucose (GLUT) au niveau du sarcolemme et des
tubules-T. Douze transporteurs de glucose (GLUT1–GLUT12) ainsi que le cotransporteur de
proton/myoinositol (HMIT) furent identifiés. Les GLUTs sont des protéines possédant douze
domaines transmembranaires. L’expression tissulaire est spécifique à chaque GLUT ainsi que son
affinité aux différents saccharides. Les GLUTs sont subdivisés en trois classes. Cette classification
est essentiellement basée sur la séquence d’acides aminés et l’affinité pour le glucose des différents
GLUTs. La classe I comprend les GLUT1-4; la classe II, les GLUT5, GLUT7, GLUT9 et GLUT11. Par
conséquent, la classe III comprend tous les autres GLUTs ainsi que le HMIT [4, 5]. Les différents
GLUTs sont présentés dans le tableau 1. En ce qui concerne le tissu musculaire squelettique,
GLUT1 et GLUT4 sont les transporteurs de glucose les plus étudiés à ce jour.
Tableau 1 Classification des GLUTs, tiré de [4]
1.2.1 GLUT1
La protéine GLUT1 est exprimée de façon ubiquitaire tant chez l’humain que chez l’animal. Certaines
études ont observé un contenu élevé en GLUT1 dans le muscle squelettique à la naissance et au
cours des premières semaines de vie chez le rat. Toutefois, il semble que l’expression de GLUT1
chute de façon drastique au cours du développement musculaire [6, 7]. D’ailleurs, les niveaux d’acide
ribonucléique messager (ARNm) de GLUT1 sont faiblement détectés dans le muscle vaste latéral
(VL) adulte chez l’humain [8]. Des études d’immunofluorescence chez le rat ont permis de d’établir
que la localisation de GLUT1 dans le muscle squelettique est restreinte à la surface de la membrane
plasmique, plus particulièrement au niveau du sarcolemme [9, 10]. De plus, il fut suggéré que les

5
fibres musculaires oxydatives aient un contenu plus élevé en GLUT1 que les fibres musculaires
glycolytiques chez le rat [9, 11]. Cependant, certaines études n’ont observé aucune variation dans
l’expression de GLUT1 entre les différents types de fibres chez ce même modèle animal [12, 13].
L’expression et l’activité de GLUT1 varient en réponse à diverses conditions métaboliques.
L’exposition prolongée des cellules musculaires L6 de rat (cellules L6) à de fortes concentrations de
glucose diminue l’expression des GLUT1 à la surface de la membrane plasmique tandis que
l’absence de glucose provoque le phénomène inverse [14]. De plus, une exposition de vingt-quatre
heures à un cocktail de cytokines combiné à une endotoxine, le lipopolysaccharide (LPS), augmente
l’expression de GLUT1 dans les cellules L6. Le même phénomène fut rapporté lors d’une exposition
à un stress oxydatif induit par du peroxyde d’oxygène (H2O2) [15, 16]. Chez l’humain, une diminution
de la protéine GLUT1 fut observée dans le muscle VL des individus atteints de diabète de type 2
tandis qu’une augmentation de l’ARNm de GLUT1 fut mesurée dans les cellules musculaires
d’individus soumis à huit semaines d’entraînement aérobie [17, 18].
L’implication de GLUT1 dans transport basal de glucose fut rapidement démontrée. En effet, une
forte augmentation du transport basal de glucose fut observée dans des cellules L6 surexprimant
GLUT1 [19]. Des résultats similaires furent rapportés dans les muscles épitrochléen et soléaire de
souris transgéniques surexprimant GLUT1 ainsi que dans le muscle gastrocnémien de rat pour
lequel la protéine fut surexprimée après électroporation [20-22]. Néanmoins, il fut rapporté que
l’utilisation d’un inhibiteur de GLUT4, l’indinavir, diminue de quatre-vingt pourcent le transport basal
de glucose dans les cellules musculaires L6 enrichies de protéines GLUT4 (L6GLUT4myc) [23]. Par
ailleurs, chez les souris génétiquement invalidées pour GLUT4 dans le tissu musculaire, une
diminution importante du transport basal de glucose fut observée dans les muscles EDL et soléaire
[24, 25]. L’ensemble de ces résultats suggère que GLUT1 n’est pas seul responsable du transport
basal dans les cellules musculaires. La protéine GLUT4 semble également impliquée.
1.2.2 GLUT4
Contrairement à GLUT1, l’expression de GLUT4 croît au cours du développement musculaire et
devient le transporteur de glucose dominant dans le muscle squelettique adulte [6-8]. Sa localisation
intracellulaire en condition basale et sa translocation au niveau du sarcolemme et plus

6
particulièrement, au niveau des tubules-T en présence d’insuline le distingue de tous les autres
GLUTs. En effet, des études en immunofluorescence ont permis de localiser GLUT4 à la surface du
sarcolemme ainsi qu’à l’intérieur de la cellule musculaire de rat. Il fut démontré qu’en condition
basale, le rapport GLUT4:GLUT1 à la surface du sarcolemme est de 4:1. Ce rapport augmente à 7:1
à la suite d’une infusion d’insuline in situ au niveau des muscles des membres inférieurs chez le rat
[9, 10]. Selon plusieurs études réalisées chez les rongeurs, les fibres musculaires oxydatives
seraient plus riches en GLUT4 que les fibres musculaires glycolytiques [9, 11-13, 26-28].
L’utilisation de modèles de souris transgéniques a permis d’établir l’importance de cette protéine
dans la régulation du transport du glucose stimulé par l’insuline. En effet, une résistance à l’insuline
périphérique ainsi qu’une propension au diabète furent observées chez la souris où l’expression de
GLUT4 est partiellement réduite dans le muscle squelettique et dans le tissu adipeux (GLUT4+/-) [29,
30]. La réinsertion de la protéine GLUT4 dans le muscle squelettique de la souris GLUT4+/- rétablit la
sensibilité à l’insuline ainsi que la tolérance au glucose à des niveaux normaux [31]. De plus, chez la
souris génétiquement invalidée pour GLUT4 spécifiquement dans le tissu musculaire (GLUT4 KO
muscle), une résistance à l’insuline sévère et une intolérance au glucose furent rapportées [24]. Par
ailleurs, le transport du glucose stimulé par l’insuline ex vivo dans les muscles EDL et soléaire est
aboli chez la souris GLUT4 KO muscle et chez la souris qui présente une invalidation totale de
GLUT4 (GLUT4-/-) [24, 25]. En contrepartie, la souris qui surexprime GLUT4 dans le muscle
squelettique est caractérisée par une augmentation de sa sensibilité à l’insuline ainsi que par une
tolérance au glucose améliorée comparativement à la souris contrôle [32, 33].
Fait intéressant, il fut également observé que l’insuline induit la translocation des GLUT12 des
vésicules cytoplasmiques vers la membrane plasmique dans le muscle squelettique humain [34].
Néanmoins, des études complémentaires seront nécessaires pour définir le rôle exact et le
mécanisme d’action des GLUT12.
Parallèlement à l’insuline, la contraction musculaire engendre aussi la translocation des GLUT4 des
vésicules cytoplasmiques vers la membrane plasmique et les tubules-T. De plus, tant chez l’humain
que chez l’animal, les études ont démontré une augmentation de l’expression de l’ARNm ainsi que
de la protéine GLUT4 dans le muscle squelettique immédiatement après une seule séance

7
d’exercice aérobie chez le sujet entraîné ou non [35-39]. L’importance de GLUT4 dans les effets de
la contraction musculaire fut démontrée par l’abolition du transport du glucose induit par un protocole
de stimulation électrique (contraction ex vivo) dans les muscles EDL et soléaire des souris GLUT4-/-
et des souris GLUT4 KO [24, 25]. De plus, une réduction marquée du métabolisme du glucose fut
observée dans les muscles soléaire, gastrocnémien et VL chez la souris GLUT4-/- en réponse à un
exercice in vivo sur tapis roulant [40]. L’analyse du contenu musculaire en GLUT4 de différents
muscles squelettiques chez le rat a démontré que la stimulation maximale du transport du glucose
induit par la contraction musculaire ex vivo corrèle positivement avec la quantité de GLUT4 présent
dans le muscle. Cette même étude a démontré que la contraction musculaire et une stimulation à
l’insuline ex vivo ont des effets additifs sur le transport du glucose dans ces mêmes muscles
squelettiques [26].
Ainsi, l’insuline et la contraction musculaire stimulent le transport du glucose dans le muscle
squelettique grâce à la participation des GLUT4. Néanmoins, ces deux stimuli physiologiques
agissent par des voies de signalisation bien distinctes.
1.3 Voie de signalisation de l’insuline
L’insuline est une petite hormone synthétisée et sécrétée par les cellules β des îlots de Langerhans
du pancréas en réponse à une augmentation de la glycémie. Le rôle de l’insuline est de normaliser la
glycémie. Ainsi, l’augmentation de la concentration plasmatique d’insuline entraîne la translocation
des GLUT4 et par conséquent, la captation du glucose sanguin par le muscle squelettique et le tissu
adipeux. Dans le muscle squelettique, l’insuline entraîne la translocation des GLUT4 par la voie de
signalisation de la phosphatidylinositol 3-kinase (PI3K) [41].
1.3.1 Récepteur de l’insuline
Le récepteur à l’insuline (IR de l’anglais insulin receptor) appartient à la famille des récepteurs de
facteurs de croissance. Il forme un complexe hétérotétradimère composé de deux sous-unités α liées
aux deux sous-unités β par des ponts disulfures. Les sous-unités alpha (α) sont situées sur la
membrane plasmique extracellulaire et elles possèdent un domaine de liaison à l’insuline. Les sous-
unités bêta (β) sont transmembranaires et elles possèdent une activité tyrosine kinase dans leur
domaine intracellulaire. La liaison de l’hormone aux sous-unités α entraîne un changement de

8
conformation de l’IR et par conséquent, induit l’autophosphorylation des sous-unités β sur les résidus
tyrosine 1158, 1163, 1162 (Tyr1158, Tyr1163, Tyr1162). La phosphorylation de ces résidus est
essentielle à l’activation de l’IR et à la transmission du signal insulinique. En effet,
l’autophosphorylation des sous-unités β permet l’activation complète du domaine tyrosine kinase et la
phosphorylation d’autres tyrosines présentes sur les chaînes β. La phosphorylation du résidu tyrosine
960 ou 972 (Tyr960/972) dans le domaine juxta membranaire de la sous-unité β permet l’ancrage
des substrats du récepteur de l’insuline [42-44]. La structure de l’IR est représentée à la figure 2.
Figure 2 Structure de l’IR, tirée de [45]
TM: Domaine Transmembranaire; JM: Domaine Juxta Membranaire; TK: Domaine tyrosine kinase;
CT: Région C-Terminale
1.3.2 Substrat du récepteur de l’insuline
La famille des substrats du récepteur de l’insuline (IRS de l’anglais insulin receptor substrat)
comprend six membres, IRS1-6 [46-50]. Bien que les protéines IRS1 et IRS2 soient toutes deux
impliquées dans la signalisation de l’insuline, IRS1 serait celle responsable du transport du glucose
stimulé par l’insuline au niveau du muscle squelettique [51-53]. La région amino-terminale (N-
terminale) de la protéine IRS1 contient un domaine pleckstrin homology (PH) adjacent au domaine
de liaison à la phosphotyrosine (PTB). Le domaine PH permet à la protéine IRS1 de se lier aux
phosphoinositols (PI) à la membrane plasmique près de l’IR tandis que le domaine PTB est
responsable de la liaison de la protéine IRS1 au résidu Tyr960/972 de l’IR [54-56]. Cette liaison
entraîne la phosphorylation des résidus tyrosine contenus dans la région carboxy-terminale (C-

9
terminale) de la protéine IRS1. Ces nouveaux sites de liaisons sont reconnus, à leur tour, par des
protéines contenant un domaine src homology 2 (SH2) dont la sous-unité régulatrice p85 de la
phosphatidylinositol 3-kinase (PI3K) reconnue pour son implication dans la transmission du signal
insulinique [57]. La structure d’IRS1 est représentée à la figure 3.
Figure 3 Structure d'IRS1, tirée et modifiée de [58]
1.3.3 Phosphatidylinositol 3-kinase
La protéine IRS1 se lie à la phosphatidylinositol 3-kinase (PI3K) de classe IA, un complexe
hétérodimère composé d’une sous-unité régulatrice, la p85, et d’une sous-unité catalytique, la p110.
Il existe deux isoformes de la sous-unité p85, les isoformes α et β. La sous-unité p85 possède un
domaine SH3 et deux domaines SH2 [59-61]. Ces derniers servent, entre autre chose, à lier la p85 à
IRS1 [62]. Cette liaison permet le recrutement de la sous-unité catalytique p110. Il existe également
trois isoformes de la sous-unité p110, les isoformes α, β et delta (δ). Toutefois, l’isoforme δ ne
semble pas être exprimée dans le muscle squelettique [63-66]. L’activation de la PI3K résulte de la
liaison de ces deux sous-unités. La structure de la PI3K de classe 1A est représentée à la figure 4.
Figure 4 Structure de la PI3K de classe IA, tirée de [67]
La PI3K activée se lie aux phospholipides membranaires. La phosphorylation des phospholipides
membranaires par la PI3K permet la conversion du phosphoinositide-4,5-biphosphate (PIP2) en
phosphoinositide-3, 4, 5-triphosphate (PIP3). Les premières évidences de l’importance de la PI3K
dans la voie de signalisation de l’insuline dans le muscle squelettique furent révélées par l’utilisation
d’un inhibiteur de la PI3K, la wortmannine. En effet, il fut démontré qu’un traitement à la wortmannine

10
inhibe la translocation des GLUT4 et le transport du glucose stimulés par l’insuline dans les cellules
musculaires L6 et les muscles épitrochléen et soléaire de rat [68, 69]. Le PIP3 produit par la PI3K
interagit avec le domaine PH de la protéine kinase B/Akt.
1.3.4 Protéine kinase B ou Akt
La protéine kinase B (PKB), communément nommée Akt, est une sérine/thréonine kinase dont il
existe trois isoformes, Akt1(PKBα), Akt2 (PKBβ) et Akt3 (PKB gamma (γ)) [70-73]. L’utilisation de
modèles murins transgéniques a démontré que chaque isoforme joue un rôle bien distinct dans
divers processus biologiques. Il est suggéré que la protéine Akt2 soit l’isoforme impliquée dans le
maintien de l’homéostasie du glucose par l’insuline dans le muscle squelettique [74, 75].
Le recrutement d’Akt à la membrane plasmique par les PIP3 provoque un changement de
conformation de la protéine démasquant ainsi ses sites de phosphorylation sur la thréonine 308
(Thr308) et la sérine 473 (Ser473). L’activation maximale d’Akt implique la phosphorylation de ces
deux sites par deux intermédiaires distincts [76, 77]. La phosphoinositide-dépendante kinase 1
(PDK1), également activée par PIP3, phosphoryle la Thr308 située dans le domaine catalytique
d’Akt. Ensuite, le complexe 2 de la cible de la rapamycine chez les mammifères (mTORC2)
phosphoryle la Ser473 située dans le domaine hydrophobe d’Akt [78, 79]. L’activation d’Akt mène à
la translocation des GLUT4 dans le muscle squelettique par l’intermédiaire des substrats d’Akt soient
les protéines TBC1D4 et TBC1D1.
1.3.5 Substrats d’Akt
Les protéines TBC1D4 et TBC1D1 sont codées par le même gène et elles possèdent une activité
Rab GTPase. Au cours de la dernière décennie, plusieurs études ont suggéré que ses substrats
d’Akt seraient le filon manquant entre l’activation d’Akt par l’insuline et la translocation des GLUT4.
Récemment, une étude a rapporté l’implication du substrat d’Akt Girdin dans la régulation du signal
de l’insuline dans les cellules musculaires. En effet, l’utilisation de petits ARN interférents pour la
protéine Girdin diminue la phosphorylation de la Thr308 d’Akt en réponse à une stimulation à
l’insuline dans les cellules musculaires de souris C2C12 (cellules C2C12) tandis qu’une
surexpression de la protéine produit l’effet inverse [80]. Toutefois, des études complémentaires

11
seront nécessaires afin de définir le rôle exact et le mécanisme d’action de la protéine Girdin dans le
transport du glucose musculaire.
1.3.5.1 TBC1D4
La protéine TBC1D4, également nommée AS160 (de l’anglais Akt substrat of 160kDa), fut le premier
substrat d’Akt découvert pour son implication dans la translocation des GLUT4 dans les cellules
musculaires. Elle possède deux domaines PTB dans la région N-terminale ainsi qu’un domaine de
liaison à la calmoduline (CBD de l’anglais calmodulin-binding domaine) et un domaine GTPase-
activating protein (GAP) dans la région C-terminale [81, 82]. La protéine TBC1D4 possède plusieurs
sites de phosphorylation dont le motif est reconnu par Akt. La transfection d’un plasmide
surexprimant une forme mutante des résidus sérine 318, 588, 751 et thréonine 642 (Ser318, Ser588,
Ser751 et Thr642) de TBC1D4 dans les cellules L6 a permis de démontrer le rôle de cette protéine
dans la translocation des GLUT4. En effet, une abolition de la translocation des GLUT4 à la
membrane plasmique fut observée en réponse à une stimulation à l’insuline dans ces conditions [83].
Des résultats similaires avaient été obtenus chez la souris. En effet, il fut rapporté que l’introduction
d’un plasmide mutant pour ces mêmes résidus de TBC1D4 dans le muscle tibial antérieur (TA) par
électroporation diminue le transport du glucose stimulé par l’insuline. Par ailleurs, le transport du
glucose était complètement inhibé lorsque le plasmide introduit contenait à la fois des mutants de
quatre sites de phosphorylation et un mutant qui inhibe l’activité Rab GTPase [84]. De plus, la souris
TBC1D4 T649 KI, génétiquement modifiée pour l’invalidation du résidu Thr649, présente une
intolérance au glucose et une résistance à l’insuline. L’abolition partielle du transport du glucose
stimulé par l’insuline occasionnée par une diminution de la translocation des GLUT4 dans le muscle
squelettique des souris TBC1D4 T649 KI serait la cause de ces perturbations au niveau de la
régulation du métabolisme du glucose. Fait intéressant, la diminution du transport du glucose stimulé
par l’insuline est plus marquée dans le muscle soléaire comparativement au muscle EDL [85]. Cette
observation concorde avec le fait que la protéine TBC1D4 fut rapportée pour être plus exprimée dans
les muscles davantage oxydatifs comparativement à ceux glycolytiques [86].
Chez l’humain, il fut rapporté qu’une stimulation à l’insuline entraîne la phosphorylation des résidus
sérine 318, 341, 588 et 751 de TBC1D4 dans le muscle squelettique. Ces résultats suggèrent

12
l’implication de la protéine TBC1D4 dans la voie de signalisation de l’insuline chez l’humain telle
qu’observée chez la souris [87].
1.3.5.2 TBC1D1
La protéine TBC1D1 est un paralogue de TBC1D4. Quarante-sept pourcent de leur séquence est
commune et la grande majorité des similitudes que partagent ces deux protéines se situe au niveau
du domaine GAP [88]. Tout comme TBC1D4, la protéine TBC1D1 contient deux domaines PTB dans
la région N-terminale ainsi qu’un domaine CBD et un domaine GAP dans la région C-terminale.
TBC1D1 possède également plusieurs sites de phosphorylation dont le motif est reconnu par Akt.
Entre autre chose, une augmentation de la phosphorylation des résidus sérine 489, thréonine 499,
sérine 501 et thréonine 590 (Ser489, Thr499, Ser501, Thr590) de TBC1D1 fut rapportée en réponse
à un traitement à l’insuline dans les cellules C2C12 et L6, respectivement [89, 90]. Chez la souris,
une stimulation à l’insuline entraîne la phosphorylation des résidus Thr590 et thréonine 253 (Thr253)
dans le muscle TA. Toutefois, la Thr253 n’est pas présente chez l’humain. Ainsi, il est proposé que la
phosphorylation du résidu Thr590 chez la souris, Thr596 chez l’humain, par une stimulation à
l’insuline serait responsable de la régulation de l’activité Rab GTPase dans le domaine GAP de
TBC1D1. Le résidu Thr590 est l’équivalent du résidu Thr649 retrouvé sur la protéine TBC1D4 [86,
91]. De plus, une diminution du transport du glucose en réponse à un traitement à l’insuline ex vivo
chez différents modèles d’invalidation génétique de l’activité Rab GTPase de TBC1D1 fut observée
dans le muscle EDL de souris [92-94]. L’ensemble de ces résultats suggère un rôle pour TBC1D1
dans la translocation des GLUT4 induite par l’insuline. Toutefois, une étude chez l’humain n’a noté
aucune augmentation de la phosphorylation du résidu Thr596 de TBC1D1 dans le muscle VL à la
suite d’une infusion d’insuline [95]. Les structures des substrats d’Akt TBC1D4 et TBC1D1 sont
représentés à la figure 5.
Chez la souris, il est suggéré que la protéine TBC1D1 soit principalement exprimée dans le tissu
musculaire, et plus particulièrement dans les muscles davantage glycolytiques, contrairement à
TBC1D4 [86].

13
Figure 5 Structure de AS160/TBC1D4 et TBC1D1, tirée de [96]
Ainsi, il est proposé que la phosphorylation des protéines TBC1D4 sur le résidu Thr642 et de
TBC1D1 sur le résidu Thr596 augmente la liaison de celles-ci avec les protéines 14-3-3 et inactive
les substrats d’Akt. En effet, il est suggéré que cette interaction entre TBC1D4 et les protéines 14-3-3
serait responsable de l’inhibition de l’activité Rab GTPase dans le domaine GAP dont le rôle est
l’hydrolyse de la guanosine triphosphate (GTP) en guanosine diphosphate (GDP) par les protéines
Rab. Cette inhibition de l’activité Rab GTPase a pour conséquence l’augmentation du nombre de
protéines Rab actives, c’est-à-dire l’augmentation du nombre de protéines Rab liées à la GTP (Rab-
GTP) comparativement à sa forme inactive liée à la GDP (Rab-GDP). L’activation des protéines Rab
entraîne la translocation des GLUT4 des vésicules cytoplasmiques vers la membrane plasmique et
les tubules-T [97]. Néanmoins, la liaison de TBC1D1 aux protéines 14-3-3 ne semble pas induite par
la phosphorylation de la Thr590 [90]. La figure 6 résume la voie de signalisation de l’insuline dans la
cellule musculaire.
Figure 6 Voie de signalisation de l'insuline, tirée et modifiée de [96]

14
1.3.6 Rac1
Au cours des dernières années, certaines études ont démontré l’implication d’une nouvelle protéine
dans le transport du glucose stimulé par l’insuline, soit Rac1 (De l’anglais Ras-related C3 botulinum
toxin substrate 1). Il fut d’abord suggéré que l’insuline entraîne une réorganisation structurale des
filaments d’actine et que cette modification participe à la translocation des GLUT4 via la PI3K dans
les cellules L6 [98, 99]. Par la suite, l’implication de la protéine Rac1 dans le réarrangement du
cytosquelette des filaments d’actine et dans la translocation des GLUT4 stimulée par l’insuline fut
démontrée dans les cellules L6 et le muscle gastrocnémien de souris [100-102]. Récemment, il fut
proposé que les protéines Rac1 et Akt agissent de manière indépendante sur la translocation des
GLUT4 stimulée par l’insuline bien que toutes deux soient dépendantes de l’activation de la PI3K
[103, 104]. À l’inverse, d’autres études stipulent qu’Akt2 régule l’activité de Rac1 [105, 106].
1.4 Voies de signalisation de la contraction musculaire
Contrairement à la voie de signalisation de l’insuline où plusieurs étapes menant à la translocation
des GLUT4 furent largement étudiées, le mécanisme d’action par lequel la contraction musculaire
induit le transport du glucose demeure encore à être élucidé. Ceci dit, la translocation des GLUT4 au
sarcolemme et aux tubules-T est essentielle au transport du glucose induit par la contraction
musculaire et plusieurs médiateurs furent suggérés pour participer à ce phénomène.
1.4.1 Calcium
Les premières évidences de l’implication du calcium dans le transport du glucose lors de la
contraction musculaire furent démontrées par l’utilisation de la caféine. En effet, Clausen et ses
collaborateurs rapportèrent l’augmentation de la tension musculaire, des concentrations
cytoplasmiques de Ca2+ et du transport du glucose, tel qu’observé lors de la contraction musculaire,
dans le muscle soléaire de rat incubé en présence de caféine [107]. Par la suite, il fut démontré que
l’incubation du muscle épitrochléen de rat en présence de faibles concentrations de caféine provoque
le relâchement de Ca2+ des citernes terminales du réticulum endoplasmique ainsi que l’augmentation
du transport du glucose sans toutefois, induire une augmentation de la tension musculaire. Des
résultats similaires furent obtenus avec l’utilisation d’un composé chimique, le N-(6-aminohexyl)-5-
chloro-1-naphthalenesulfonamide (W-7), qui provoque le relâchement de Ca2+ des citernes externes
du réticulum endoplasmique. À cet instant, il fut suggéré que le calcium stimule le transport du

15
glucose dans le muscle squelettique par une voie, du moins partiellement indépendante de celle de
la contraction musculaire [108]. De plus, des études subséquentes ont rapporté que la caféine
augmentait l’activité de la kinase activée par l’adénosine monophosphate (AMPK), plus
particulièrement de l’isoforme AMPKα1, dans les muscles squelettiques de souris et de rats. Par
ailleurs, l’effet de la caféine sur le transport du glucose est altéré chez la souris surexprimant une
forme inactive de l’AMPK dans les tissus musculaires [109-111]. L’ensemble de ces résultats
suggère que l’augmentation du transport du glucose induit par le relâchement de Ca2+ par la caféine
serait dépendante de l’AMPK.
Toutefois, il fut également démontré que l’ajout de dantrolène, un myorelaxant qui inhibe le
relâchement de Ca2+ par le réticulum endoplasmique, dans le milieu d’incubation diminue le transport
du glucose induit par la contraction musculaire ex vivo dans le muscle soléaire de rat. Des résultats
similaires furent obtenus dans les muscles épitrochléen et soléaire de rat avec l’ajout d’inhibiteurs
des protéines kinases Ca2+/calmoduline-dépendante (CAMK), le KN62 et le KN93, suggérant
l’implication de la voie Ca2+/calmoduline (Ca2+/CaM) dans le transport du glucose induit par la
contraction musculaire [112, 113]. De plus, il fut démontré que la protéine CAMKII, dont l’activation
dépend de la formation du complexe Ca2+/CaM, est fortement exprimée dans le muscle squelettique
humain [114-116]. Son activité ainsi que la phosphorylation du résidu thréonine 287 (Thr287) dans le
muscle VL sont rapidement augmentées et maintenues en réponse à un exercice musculaire modéré
sur ergocycle ainsi que lors d’un sprint [117, 118]. Finalement, l’introduction d’un plasmide invalidant
la CAMKII par électroporation dans le muscle TA de souris diminue le transport du glucose induit par
une contraction musculaire ex vivo. Ni l’expression ni la phosphorylation de l’AMPK n’est affectée par
l’invalidation de la CAMKII [119]. L’ensemble de ces résultats suggère l’implication du Ca2+ dans la
régulation du transport du glucose induit par la contraction musculaire et ce, indépendamment de
l’AMPK contrairement à ce qui fut observé avec la caféine.
1.4.2 Protéine Kinase C
Des études préliminaires ont rapporté une augmentation de la translocation de la protéine kinase C
(PKC) et par conséquent, de son activité dans le muscle gastrocnémien de rat à la suite d’une
contraction in situ et ex vivo [120, 121]. De plus, les résultats obtenus par l’utilisation d’un inhibiteur
de la PKC, la calphostine C, suggérèrent que celle-ci participe au transport du glucose induit par la

16
contraction musculaire in situ chez le rat [122, 123]. La protéine kinase C (PKC) est une
sérine/thréonine kinase qui comporte plusieurs isoformes regroupées en trois catégories: les PCK
conventionnelles, les nouvelles PKC et les PKC atypiques. La classe des PKC conventionnelles
comprend 3 isoformes, α, β et γ. L’isoforme PKCα est la plus abondante dans le muscle squelettique.
Toutefois, le transport du glucose induit par une contraction musculaire ex vivo chez la souris
génétiquement modifiée pour l’invalidation de la PKCα est semblable à celui de la souris contrôle. De
plus, l’effet des inhibiteurs de la classe des PKC conventionnelles, dont la calphostine C, sur le
transport du glucose est le même chez les deux phénotypes de souris [124].
Comparativement aux résultats obtenus chez les modèles animaux, aucune augmentation de
l’activité des PKC conventionnelles ne fut observée à la suite d’une séance d’exercice modéré sur
ergocycle dans le muscle VL chez l’humain. Par contre, une augmentation de l’activité des PKC
atypiques fut mesurée [125-127]. La classe des PKC atypiques comprend deux isoformes, zêta (ζ) et
lambda (λ). L’expression de l’isoforme PKCλ, l’homologue de l’isoforme PKCι chez l’humain,
prédomine dans le muscle squelettique de souris. Une étude a rapporté que l’invalidation génétique
de l’isoforme PKCλ dans le muscle squelettique de souris n’affecte pas le transport du glucose induit
par un exercice sur tapis roulant suggérant que la PKCλ n’est pas un médiateur de la régulation du
transport du glucose induit par la contraction musculaire [128].
1.4.3 Les protéines kinases activées par les facteurs mitogènes
Parmi les protéines kinases activées par les facteurs mitogènes (MAPK), la MAPK3 (ou ERK1), la
MAPK1 (ou ERK2), la p38 MAPK et la protéine kinase c-Jun-N-terminale (JNK) sont activées par la
contraction musculaire. Néanmoins, il fut rapporté que l’invalidation chimique ou génétique des
protéines ERK1, EKR2 et JNK n’altère pas le transport du glucose induit par la contraction
musculaire [129-131].
En contrepartie, l’utilisation d’un l’inhibiteur de la p38 MAPK, le SB203580, suggéra l’implication de
cette protéine dans le transport du glucose induit par la contraction musculaire. En effet, une
diminution du transport du glucose fut observée dans les muscles EDL et soléaire de souris soumis à
une stimulation électrique en présence de SB203580 [132]. Par contre, des études subséquentes ont
démontré que le SB203580 se liait directement au GLUT4 rendant ainsi laborieuse l’interprétation

17
des résultats obtenus par l’utilisation de cet inhibiteur [133, 134]. De plus, il fut rapporté que la
surexpression de l’isoforme p38MAPKγ par électroporation dans le muscle TA de souris augmente le
transport basal du glucose, mais le transport du glucose induit par la contraction musculaire tend à
diminuer. Encore une fois, l’interprétation de ces résultats demeure ardue puisque la surexpression
de la p38MAPKγ diminue l’expression des GLUT4 [135].
1.4.4 Monoxyde d’azote
La synthase de monoxyde d’azote (NOS de l’anglais nitric oxyde synthase) est une protéine qui
produit le monoxyde d’azote (NO) à partir de la L-arginine et de l’oxygène [136]. Il existe trois formes:
la forme neurale (nNOS), la forme endothéliale (eNOS) et la forme inductible (iNOS). Le muscle
squelettique exprime chacune des trois isoformes. Néanmoins, cette dernière est très peu exprimée
dans le muscle squelettique sain [137-142]. La contraction musculaire augmente l’activité de la sous-
unité μ de nNOS (nNOSμ) et de eNOS dans le muscle squelettique de rat tandis que seule l’isoforme
nNOSμ est augmentée dans le muscle squelettique humain [143, 144]. Il fut démontré que
l’administration d’un compétiteur de L-arginine qui inhibe la production de NO, le L-N-monométhyl-
arginine (L-NMMA), diminue la captation du glucose au cours d’un exercice modéré sur ergocycle
chez l’humain. À l’inverse, l’infusion simultanée de L-arginine rétablit la captation du glucose à des
niveaux semblables à ceux des sujets contrôles [145]. Toutefois, il fut rapporté que l’infusion de L-
NMMA au cours d’un travail musculaire de faible intensité n’affecte pas le transport du glucose dans
le muscle VL chez l’humain [146]. Des études chez le rat et la souris ont également démontré une
réduction du transport du glucose induit par la contraction musculaire ex vivo et in situ ainsi que par
un exercice sur tapis roulant en présence d’inhibiteurs de NOS. Fait intéressant, l’étude menée chez
la souris suggère que l’effet du NO sur le transport du glucose serait plus important dans le muscle
EDL comparativement au muscle soléaire [147-150]. En contrepartie, certaines études chez les
animaux n’ont rapporté aucune altération du transport du glucose induit par la contraction musculaire
en présence d’inhibiteurs de NOS [151, 152].
1.4.5 Espèces réactives de l’oxygène
Les espèces réactives de l’oxygène (ROS de l’anglais reactive oxygen species) sont continuellement
produites, entre autre, par le muscle squelettique et leur production s’accroît lors de la contraction
musculaire. La production de ROS par le muscle squelettique est associée à la fatigue musculaire.

18
D’ailleurs, l’utilisation d’un antioxydant, le N-acétylcystéine (NAC), retarde la fatigue musculaire lors
d’exercices physiques [153]. Une étude a démontré que la présence de NAC dans le milieu
d’incubation lors d’une contraction musculaire ex vivo diminue le transport de glucose de moitié dans
le muscle EDL de souris [154]. Par contre, des études menées chez l’humain et le rat ont démontré
que l’infusion de NAC simultanément à un travail musculaire d’intensité modérée n’affecte pas le
transport du glucose induit par ce dernier [155, 156].
1.4.6 Rac1
Parallèlement à l’insuline, il fut récemment rapporté qu’un exercice physique sur tapis roulant active
Rac1 dans les muscles gastrocnémien et soléaire chez la souris et chez l’humain. De plus, le
transport du glucose induit par un protocole de contraction musculaire ex vivo est diminué dans les
muscles EDL et soléaire des souris génétiquement invalidées pour Rac1. Il s’agit d’une nouvelle
piste qui demande à être approfondie afin de préciser le rôle de Rac1 dans le transport du glucose
induit par la contraction musculaire [157].
1.4.7 Substrats d’Akt
Au cours des dernières années, plusieurs études ont suggéré que les TBC1D4 et TBC1D1 sont le
point de convergence entre la voie de signalisation de l’insuline et la voie de signalisation de la
contraction musculaire qui mène à la translocation des GLUT4 et par conséquent, l’entrée massive
de glucose dans la cellule musculaire.
1.4.7.1 TBC1D4
Chez l’humain, plusieurs études ont démontré une augmentation de la phosphorylation de TBC1D4
en réponse à des exercices d’endurance d’intensité modérée comparativement à des exercices de
courte durée à haute intensité ou à des exercices de résistance musculaire. En effet, il fut rapporté
que la protéine TBC1D4 est phosphorylée dans le muscle VL chez l’humain à la suite d’une séance
d’entraînement modéré sur ergocycle, mais cette augmentation de la phosphorylation de TBC1D4
n’est observable qu’à partir de quarante minutes d’exercice. Bien qu’aucune augmentation de
TBC1D4 ne soit mesurée immédiatement à l’arrêt d’exercices de courte durée ou de résistance
musculaire, des études suggèrent une augmentation de la phosphorylation de certaines sérines
(Ser318, Ser341, Ser751) dans les heures subséquentes [87, 158-161]. Des résultats similaires

19
furent observés à la suite de deux heures de nage chez le rat. Plus intéressant encore, la
phosphorylation de TBC1D4 était toujours mesurable dans les vingt-sept heures suivant l’arrêt de
l’exercice [162, 163].
Chez la souris surexprimant une forme mutante des Ser318, Ser588, Ser751 et Thr642 de TBC1D4
(4P), une diminution du transport du glucose induit par la contraction musculaire fut observée
suggérant l’implication de cette protéine [84]. Il fut également rapporté que l’introduction d’un
plasmide mutant pour le domaine liant à la calmoduline (de l’anglais calmodulin binding domain,
CBD) de TBC1D4 dans le muscle TA de souris par électroporation diminue le transport du glucose
induit par la contraction musculaire. De plus, la combinaison des deux plasmides mutants (4P+CBD)
n’a pas d’effet additif sur la diminution du transport du glucose [164]. Néanmoins, le transport du
glucose induit par la contraction musculaire n’est pas affecté chez la souris TBC1D4 T649 KI [165].
Aussi, l’inhibition de la phosphorylation de TBC1D4 par un inhibiteur de la PI3K, la wortmannine,
n’altère pas le transport du glucose induit par une contraction musculaire ex vivo dans le muscle
épitrochléen de rat [166].
Il est important de prendre en compte que dans les études précédentes chez des modèles murins, la
phosphorylation de TBC1D4 est augmentée à la suite de protocoles d’exercices musculaires de
courte durée, ce qui ne semble pas être nécessairement le cas dans les études effectuées chez
l’humain.
1.4.7.2 TBC1D1
La phosphorylation de TBC1D1 est également augmentée par des protocoles de contraction
musculaire in situ ou ex vivo chez le rat et contrairement à TBC1D4, la phosphorylation de TBC1D1
n’est pas inhibée par la wortmannine [86, 166]. Autre différence notoire, en plus de posséder un site
de phosophorylation dont le motif est reconnu par Akt (Thr590/Thr596), TBC1D1 possède des sites
de phosphorylation dont le motif est reconnu par l’AMPK soient la sérine 231 (Ser231), la sérine 660
(Ser660) et la sérine 700 (Ser700) chez la souris. Il fut démontré qu’un protocole de contraction ex
vivo et in situ chez la souris augmente significativement la phosphorylation de la Ser231 et de la
Ser660 dans les muscles EDL et TA. La phosphorylation de la Ser700 tend également à augmenter
tandis la phosphorylation de la Thr590 par la contraction musculaire semble accrue dans une étude

20
et inchangée dans une autre [91, 167]. De plus, une étude chez l’humain a démontré que la
phosphorylation de la Sérine 237, équivalent de la Ser231 chez la souris, est augmentée par des
exercices de courte durée (30 secondes-20 minutes) et d’intensité modérée à très élevée [168].
L’introduction d’un plasmide mutant de TBC1D1 pour les sites de phosphorylation reconnus par
l’AMPK par électroporation dans le muscle TA de souris diminue significativement le transport du
glucose induit par un protocole de contraction musculaire in situ [91, 169]. De plus, l’invalidation
génétique du gène TBC1D1 chez la souris diminue également le transport du glucose induit par un le
5-aminoimidazole-4-carboxamide ribonucléotide (AICAR) dans le muscle EDL de souris, un
activateur pharmacologique de l’AMPK connu pour induire la translocation des GLUT4 [92]. Ces
résultats suggèrent une interaction entre TBC1D1 et l’AMPK dans la régulation du transport de
glucose dans le muscle squelettique. D’ailleurs, certaines évidences tendent à démontrer l’implication
de l’AMPK dans le transport du glucose induit par la contraction musculaire.
La figure 7 représente les différentes protéines proposées pour participer au transport du glucose
induit par la contraction musculaire.
Figure 7 Voies de signalisation de la contraction musculaire, tirée de [170]
2 AMPK L’année 1973 est associée à la découverte de l’AMPK alors que deux études indépendantes
rapportèrent l’existence d’une kinase pouvant phosphoryler et inhiber l’acétyl-coenzyme A
21
carboxylase (ACC) et l'hydroxyméthylglutaryl-CoA réductase (HMG-CoA reductase), deux enzymes
impliquées, respectivement, dans la synthèse des acides gras et du cholestérol [171, 172]. Il faudra
attendre 1989 pour que Carling et ses collaborateurs purifient cette enzyme et démontrent que cette
kinase responsable de la phosphorylation de l’ACC était en fait, la même que celle responsable de la
phosphorylation de l’HMG-CoA reductase [173]. Cette enzyme devint la kinase activée par l’AMP
(AMPK). Rapidement, l’AMPK fut reconnue comme un joueur clé dans la régulation du métabolisme
énergétique.
2.1 Structure et expression
L’AMPK est un sérine/thréonine kinase composée d’une sous-unité catalytique α et de deux sous-
unités régulatrices, β et γ [174, 175]. Le complexe hétérotrimère formé par ces trois sous-unités est
essentiel à la stabilité et l’activation de l’AMPK [176-178]. Chaque sous-unité présente différentes
isoformes (α1, α2, β1, β2, γ1, γ2, γ3) [178-182]. L’AMPK est exprimée de façon ubiquitaire dans tout
l’organisme, mais les complexes formés par les différentes isoformes varient selon le tissu et
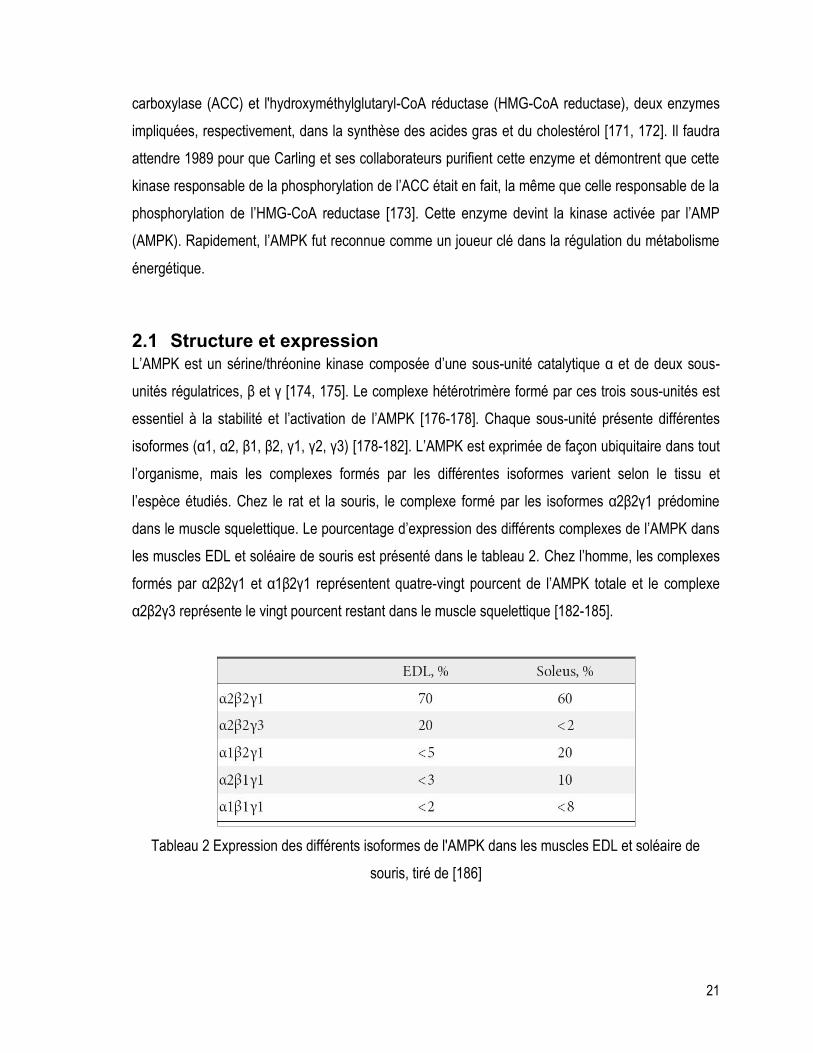
l’espèce étudiés. Chez le rat et la souris, le complexe formé par les isoformes α2β2γ1 prédomine
dans le muscle squelettique. Le pourcentage d’expression des différents complexes de l’AMPK dans
les muscles EDL et soléaire de souris est présenté dans le tableau 2. Chez l’homme, les complexes
formés par α2β2γ1 et α1β2γ1 représentent quatre-vingt pourcent de l’AMPK totale et le complexe
α2β2γ3 représente le vingt pourcent restant dans le muscle squelettique [182-185].
Tableau 2 Expression des différents isoformes de l'AMPK dans les muscles EDL et soléaire de
souris, tiré de [186]

22
La sous-unité α contient un domaine catalytique où se trouve la boucle d’activation du résidu
thréonine 172 (Thr172) situé dans la région N-terminale. De plus, elle possède un domaine
autoinhibiteur et un domaine globulaire situé dans la région C-terminale qui interagit avec le domaine
C-terminal de l’isoforme β afin de former le complexe hétérotrimère [187-195]. La sous-unité α
contient également d’autres sites de phosphorylation dont la sérine 485 (Ser485) sur l’isoforme α1 et
son homologue sur l’isoforme α2, la sérine 491(Ser491) [196]. Les isoformes α1 et α2 sont toutes
deux exprimées dans le muscle squelettique de rat, mais l’isoforme α2 prédomine [197]. Des études
suggèrent que l’isoforme α2 serait localisée à l’intérieur et à l’extérieur du noyau cellulaire tandis que
l’isoforme α1 serait principalement localisée à l’extérieur du noyau cellulaire. Néanmoins, l’absence
totale de l’isoforme α1 à l’intérieur du noyau cellulaire n’est pas encore démontrée [198, 199].
La sous-unité β contient un site de myristoylation situé dans le domaine N-terminal, un module
central de liaison des carbohydrates, plus connu sous le nom de site de liaison du glycogène, et un
domaine C-terminal qui se lie aux sous-unités α et γ afin de former le complexe hétérotrimère [152,
176, 189, 190, 194-196, 200-206]. La sous-unité β possède également des sites de phosphorylation
dont les résidus sérine 24 (Ser24), sérine 25 (Ser25), sérine 108 (Ser108) et sérine 182 (Ser182)
[200]. Selon une étude, les isoformes β1 et β2 seraient localisées à l’intérieur et à l’extérieur du
noyau cellulaire [198]. Bien que chaque isoforme soit fortement exprimée, l’isoforme β2 prédomine
dans le muscle squelettique tant chez le rat que chez l’homme [181-185].
La sous-unité γ contient une région N-terminale avec ou non une extension de longueur variable
selon l’isoforme, un site d’interaction avec la sous-unité β et deux domaines Bateman formés par
deux paires de séquences cystathionine β-synthase (CBS) situés dans sa région C-terminale. On
dénombre donc quatre sites de CBS [190, 207-211]. L’isoforme γ1 s’exprime de façon ubiquitaire
tandis que l’expression de l’isoforme γ2 se limite aux muscles squelettiques et au muscle cardiaque.
L’isoforme γ3 ne s’exprime que dans le muscle squelettique [182, 208, 212]. La localisation de
l’isoforme γ1 serait davantage nucléaire comparativement aux deux autres isoformes [213].
Les structures des sous-unités de l’AMPK sont représentées dans la figure 8.

23
Figure 8 Structure de l'AMPK, tirée de [214]
2.2 Régulation de l’AMPK
2.2.1 Nucléotides de l’adénine
Tel que son nom l’indique, l’AMPK est une kinase activée par l’adénosine monophosphate (AMP),
mais plus particulièrement par l’augmentation du rapport AMP: ATP. L’AMP est un ribonucléotide
contenant des résidus d’adénine telles que l’adénosine diphosphate (ADP) et l’adénosine
triphosphate (ATP). En condition basale, les niveaux d’AMP sont relativement faibles puisque
l’adénylate kinase favorise la synthèse d’ADP.
ATP+AMP ↔ 2ADP
Néanmoins, la déplétion accélérée de l’ATP par un stress métabolique provoque l’augmentation du
rapport ADP: ATP et par conséquent, l’adénylate kinase favorise la réaction inverse, c’est-à-dire la
synthèse d’ATP et d’AMP. Ainsi, il y a augmentation des niveaux d’AMP et par le fait même, du
rapport AMP: ATP [215, 216].
Les quatre sites de CBS que contient l’isoforme γ facilitent la liaison des nucléotides de l’adénine.
Les sites CBS1 et 3 peuvent lier chacun des nucléotides tandis que le site CBS4 ne se lie qu’à
l’AMP. Des études suggèrent qu’en condition basale, les sites CBS1 et 3 seraient liés à l’ATP tandis
qu’en présence d’un stress métabolique, l’ADP et l’AMP compétitionneraient pour les sites de liaison
CBS1 et 3. La liaison de l’ADP ou de l’AMP à la sous-unité γ entraîne un changement de

24
conformation du complexe hétérotrimère et ainsi active l’AMPK [190, 191, 211]. L’activation
allostérique de l’AMPK dévoile le site de phosphorylation du résidu Thr172 et le rend ainsi
inaccessible aux protéines phosphatases 2A et 2C (PP2A et PP2C) responsables de la
déphosphorylation de la sous-unité catalytique α [191, 205, 217-220].
2.2.2 AMPKK
La phosphorylation du résidu Thr172 sur la boucle de la sous-unité α par les AMPK kinases
(AMPKK) est essentielle à l’activation maximale de l’AMPK [221]. L’identification des deux premières
AMPKK, la liver kinase B1 (LKB1) et la Ca2+/calmodulin dependent protein kinase kinase (CAMKK),
fit suite à la démonstration par Sutherland et ses collaborateurs de trois kinases Tos3, Sak1 et Elm1
responsables de l’activation du complexe sucrose non-fermenting 1 (SNF1), ortologue de l’AMPK
chez la levure [222-224]. Outre le fait que la séquence du domaine catalytique de la LKB1
correspondait à celle des trois kinases identifiées chez la levure, LKB1 présentait des similitudes de
séquences sur la boucle d’activation de la Thr172 laissant présager un rôle en amont de l’AMPK
[224, 225].
2.2.2.1 LKB1
LKB1 est une sérine/thréonine kinase qui fut d’abord identifiée comme un gène suppresseur de
tumeurs dont la mutation est associée au syndrome de Peutz-Jeghers [226, 227]. Hawley et ses
collaborateurs furent les premiers à démontrer que la LKB1 active directement l’AMPK grâce à
l’utilisation de cellules cancéreuses humaines (HeLa). L’activation de l’AMPK par la phenformine, un
médicament de la famille des biguanides, et par l’AICAR est abolie dans les cellules HeLa
génétiquement modifiées pour le gène LKB1. La réintroduction du gène actif de la LKB1 dans les
cellules HeLa génétiquement modifiées rétablit l’activation de l’AMPK par ces deux composés [223].
Des résultats similaires furent observés dans des cellules embryonnaires fibroblastiques de souris
(MEF) génétiquement invalidées pour LKB1 [223-227].
LKB1 forme un complexe hétérotrimère avec les protéines STRAD (α/β) et MO25 (α/β) [228, 229]. Il
fut démontré que LKB1 serait responsable de la phosphorylation et de l’activation de treize kinases
dont l’AMPK, maintenant regroupées sous le nom de l’AMPK-related kinase family (ARK) [230].
Récemment, il fut proposé qu’un changement de localisation subcellulaire de LKB1 soit responsable

25
de la phosphorylation du résidu Thr172 d’AMPK dans le muscle squelettique et dans les cellules L6
[231, 232]. De plus, il fut démontré que l’invalidation génétique du site de farnélysation du résidu
cystéine433 de la protéine LKB1 chez la souris (LKB1C433S/C433S KI) diminue l’activité basale de
l’AMPK ainsi que son activation pharmacologique (AICAR) et physiologique (contraction musculaire)
dans le muscle EDL [233]. Ces résultats corroborent ceux obtenus chez la souris invalidée
génétiquement pour LKB1 dans le muscle squelettique où une forte diminution de l’activation de
l’AMPK par AICAR, la phenformine et la contraction musculaire avaient été démontrée. Cette même
étude suggéra que l’activation de l’isoforme AMPKα2 serait davantage dépendante de LKB1
comparativement à l’isoforme AMPKα1 [234].
2.2.2.2 CAMKK
L’activation de l’AMPK par la CAMKK fut étudiée pour la première fois en 1995. Néanmoins, les
résultats obtenus ne parvinrent pas à conclure que la CAMKK activait l’AMPK de façon significative
[219]. Il fallut attendre une décennie pour que des études démontrent le contraire. En effet,
l’activation de l’AMPK par le A23187, un ionophore calcique, fut rapportée dans les cellules HeLa.
Cette activation était bloquée par le STO-609, un inhibiteur spécifique de la CAMKK [235]. Deux
autres études indépendantes rapportèrent également l’activation de l’AMPK par l’ionomycine, un
ionophore calcique sélectif. Cette activation était abolie par la présence de STO-609, mais maintenue
dans les cellules HeLa génétiquement invalidées pour LKB1 [236, 237]. Ces études permirent de
reconnaître la CAMKK comme une seconde AMPKK.
Il existe deux isoformes de la CAMKK, la CAMKKα et la CAMKKβ. Les études démontrent la
présence de l’isoforme CAMKKα dans le muscle squelettique des modèles animaux tandis qu’une
première étude avait échoué à détecter la présence de l’isoforme CAMKKβ. Néanmoins, des études
subséquentes l’ont détecté [238-240]. Entre autre chose, l’augmentation de l’activité de la CAMKKβ
fut observée lors du développement hypertrophique des fibres musculaires des muscles
gastrocnémien et soléaire en réponse à un programme d’entraînement en résistance [241]. De plus,
il fut rapporté que la surexpression d’une forme constitutivement active de la CAMKKα dans le
muscle squelettique de souris augmente la phosphorylation du résidu Thr172 de l’AMPK [242].

26
2.2.2.3 TAK1
Momcilovic et ses collaborateurs furent les premiers à démontrer la régulation de l’AMPK par la
Transforming growth factor-β- activated kinase 1 (TAK1) [243]. TAK1 est une sérine/thréonine kinase
appartenant à la famille des protéines kinases activées par les facteurs mitogènes (MAP3K) [244].
TAK1 est une protéine qui contient un domaine de liaison à une protéine adaptatrice TAB1 dans sa
région N-terminale et un domaine de liaison aux protéines adaptatrices TAB2 et TAB3 dans sa région
C-terminale [245, 246].
TAK1 est sensible aux variations du rapport AMP: ATP induites par un traitement AICAR ou
metformine, deux activateurs de l’AMPK. Entre autre chose, il fut démontré que l’inhibition de TAK1
dans le muscle cardiaque prévient la phosphorylation de l’AMPK sur la Thr172 par un traitement à la
metformine [247]. Néanmoins, l’implication de TAK1 dans la régulation de l’activité de l’AMPK dans le
muscle squelettique reste à démontrer. L’expression des protéines TAK1 et TAB1 fut rapportée dans
le muscle squelettique de souris [241].
Les différentes protéines proposées pour réguler l’AMPK sont représentées à la figure 9.
Figure 9 Régulation de l'AMPK, tirée et modifiée de [248]
2.3 Activation de l’AMPK
2.3.1 Activateurs pharmacologiques
Afin de mieux comprendre les mécanismes qui régissent l’activation de l’AMPK, mais également son
rôle dans l’organisme, l’emploi d’agents pharmacologiques fut indispensable. Parmi ces agents, nous

27
y retrouvons des composés chimiques, mais également des médicaments et des produits naturels
utilisés depuis de nombreuses années bien avant la découverte de l’AMPK.
2.3.1.1 AICAR
Le groupe de Van de Berghe fut le premier à utiliser l’AICAR afin d’activer l’AMPK [249]. L’AICAR est
un nucléoside transporté à l’intérieur de la cellule par les transporteurs d’adénosine où il sera
converti en 5-aminoimidazole-4-carboxamide-1-β-ribofuranosyl monophosphate (ZMP) par
l’adénosine kinase [250]. Le ZMP mime l’effet de l’AMP de par sa capacité à se lier sur le site CBS3
de la sous-unité AMPKγ. Cette liaison active l’AMPK, mais son pouvoir activateur est beaucoup plus
faible que celui de l’AMP [251]. De nombreuses études rapportent l’activation de l’AMPK par AICAR
dans le muscle squelettique in vitro et in vivo. Néanmoins, l’AICAR n’est pas un activateur spécifique
de l’AMPK. Entre autre chose, il fut également démontré que l’AICAR active la glycogène
phosphorylase (GP) et la fructose-1,6-bisphosphatase [252, 253].
2.3.1.2 Metformine
La metformine est un médicament de la famille des biguanides utilisé pour le traitement du diabète
de type 2 depuis 1957. La metformine est reconnue pour diminuer la glycémie, le taux de lipides
sanguins ainsi qu’augmenter la sensibilité à l’insuline chez les patients atteints de diabète de type 2
[254]. Bien que son mécanisme d’action ne soit pas totalement élucidé, sa capacité à activer l’AMPK
fut démontrée par plusieurs groupes. Malgré le fait que la principale cible de la metformine semble le
foie, il fut également rapporté que la metformine active l’AMPK dans le muscle squelettique de rat
ainsi que dans le muscle squelettique humain [255, 256].
Il fut également démontré que la metformine inhibe le complexe 1 de la chaîne respiratoire suggérant
une activation de l’AMPK déclenchée par l’augmentation des rapports ADP: ATP et AMP: ATP [257,
258]. Cette hypothèse fut corroborée récemment par une étude qui observa une augmentation du
rapport ADP: ATP par la metformine. De plus, aucune activation de l’AMPK par la metformine ne fut
mesurée dans les cellules génétiquement modifiées pour le site CBS3 de la sous-unité AMPK γ
[259]. Aussi, l’invalidation de LKB1 dans le muscle squelettique de souris prévient l’activation de
l’isoforme AMPKα2 par la phenformine bien que l’activité de LKB1 ne soit modulée ni par la
metformine ni par la phenformine [234, 260].

28
2.3.1.3 Thiazolidinediones
La rosiglitazone, la pioglitazone ainsi que la troglitazone font partie des thiazolidinediones, une
famille de médicaments dont certains sont prescrits dans le traitement du diabète de type 2 [261-
263]. Les thiazolidinediones sont connues pour leurs effets hypoglycémiants [264]. L’activation du
récepteur des proliférateurs de peroxysomes gamma (PPARγ) suivie du relâchement d’adiponectine,
une hormone sécrétée par le tissu adipeux, est connue pour améliorer la sensibilité à l’insuline chez
des animaux insulino-résistants traités au pioglitazone. La sécrétion d’adiponectine en réponse à
l’activation des PPARγ serait un mécanisme d’action responsable des effets bénéfiques des
thiazolidinediones au niveau du foie, mais non au niveau du muscle squelettique [265].
Une activation rapide de l’AMPK fut observée dans le muscle squelettique de rat traité à la
troglitazone ainsi qu’à la pioglitazone [261]. Selon certaines études, l’inhibition du complexe 1 de la
chaîne respiratoire et par conséquent, l’augmentation du rapport ADP: ATP serait partiellement
responsable de cette activation tel qu’observé dans le cas de la metformine [259, 262, 266]. De plus,
il fut rapporté que l’activation de l’AMPK par les thiazolidinediones dans le muscle squelettique des
souris génétiquement invalidées pour l’adiponectine (AdnKO) est diminuée. Toutefois, les niveaux
initiaux de la phosphorylation ainsi que l’activité de l’AMPK tendent à être augmentés chez les souris
AdnKO [267]. Finalement, il fut récemment observé que l’adiponectine entraîne la translocation de
LKB1 du noyau cellulaire vers le cytoplasme et par conséquent, l’activation de l’AMPK dans les
cellules L6 [232].
2.3.1.4 A-769662
En 2006, les laboratoires Abbott ont caractérisé un nouvel activateur synthétique de l’AMPK nommé
A-769662, une petite molécule de la famille des thiénopyridines. À l’instar de la metformine et des
thiazolidinediones, l’étude préliminaire menée sur cette nouvelle molécule démontra des résultats
prometteurs pour un éventuel traitement du diabète de type 2. En effet, l’injection intrapéritonéale de
cette molécule augmenta l’oxydation des lipides chez le rat sain. Chez un modèle de souris obèses,
l’injection de l’A-769662 diminua le gain de poids corporel, les niveaux sanguins de glucose et de
triglycérides ainsi que les niveaux d’expression hépatique de la phosphoénolpyruvate carboxykinase
(PEPCK), de la glucose-6-phoshatase (G6Pase) et de l’acide gras synthase (FAS de l’anglais fatty
acid synthase) [268]. En 2012, des résultats similaires furent rapportés par l’injection de salicylate, un

29
médicament utilisé pour abaisser la fièvre et diminuer l’inflammation. Le salicylate activerait
directement l’AMPK de la même manière que l’A-769662 [269].
Il fut proposé que l’A-769662 active la Ser108 de la sous-unité AMPKβ1 [270, 271]. Cette liaison
entraînerait un changement de conformation du complexe hétérotrimère, activerait l’AMPK,
indépendamment de l’AMP, et empêcherait sa déphosphorylation par les phosphatases PP2C [272].
Néanmoins, la biodisponibilité du composé A-769662 dans le muscle squelettique est très faible
comparativement aux autres tissus, particulièrement au niveau du foie [268]. L’activation de la sous-
unité AMPKβ1 par le A-769662 fut observée dans les muscles EDL et soléaire chez la souris [273].
2.3.1.5 Composés naturels
Au cours des dernières années, plusieurs composés naturels furent l’objet d’études dus, entre autre
chose, à leur capacité à activer l’AMPK. Parmi ceux-ci, il fut démontré que le resvératrol, un
polyphénol de la classe des stilbènes, augmente la phosphorylation et l’activité de l’AMPK dans le
muscle squelettique de la souris et de l’humain [274, 275]. De plus, la berbérine, une molécule
extraite des végétaux fréquemment utilisée dans la médecine traditionnelle chinoise pour traiter le
diabète de type 2, fut rapportée pour activer l’AMPK dans les cellules musculaires L6 ainsi que dans
les muscles épitrochléen et soléaire de rat [276, 277]. Il fut suggéré que, le resvératrol et la
berbérine, activent l’AMPK par l’augmentation du rapport AMP: ATP causée par l’inhibition de l’ATP
synthétase de type F1 et du complexe 1 de la chaîne respiratoire, respectivement [278, 279].
2.3.2 Activation par la contraction musculaire
L’AMPK est un senseur du métabolisme énergétique qui a pour fonction de maintenir l’homéostasie
cellulaire. Ainsi, toutes variations physiologiques des rapports AMP: ATP et ADP: ATP causées par
un stress énergétique activent l’AMPK. L’activation de l’AMPK stimule les voies cataboliques
impliquées dans la synthèse d’ATP. L’hypoxie, l’ischémie, la restriction calorique, les chocs
osmotiques et thermiques ainsi que l’exercice physique sont tous des facteurs physiologiques de
stress énergétiques.
L’une des découvertes majeures entourant l’AMPK fut son activation par la contraction musculaire
ainsi que par l’exercice physique tant chez le rongeur que chez l’humain [280-285]. Chez l’humain,

30
un exercice physique active en premier lieu, le complexe AMPKα2β2γ3 dans le muscle squelettique
suivi du complexe AMPKα2β2γ1 lorsque l’exercice se prolonge au-delà de soixante minutes [159,
286]. Lors de la contraction musculaire, de nombreux facteurs peuvent expliquer l’activation de
l’AMPK dont un changement du statut énergétique qui se traduit par une augmentation du rapport
AMP: ATP, l’augmentation des niveaux intracellulaires de Ca2+, l’induction des espèces réactives de
l’oxygène (ROS) ainsi que le relâchement d’hormones, de cytokines et de facteurs de croissance.
2.3.2.1 Statut énergétique
La contraction musculaire augmente considérablement la demande énergétique [287]. Cette grande
consommation d’ATP par la contraction musculaire entraîne une augmentation du ratio AMP: ATP et
par conséquent, l’activation allostérique de l’AMPK de par la liaison de l’AMP à la sous-unité γ.
Certaines études suggèrent que LKB1 serait l’AMPKK responsable de la phosphorylation de la
Thr172 et ainsi de l’activation maximale de l’AMPK par la contraction musculaire. En effet, l’activation
de l’AMPKα2 est considérablement inhibée dans les muscles TA et EDL des souris LKB1 KO bien
que les rapports AMP: ATP et ADP: ATP soient augmentés en réponse à des contractions
musculaires de haute intensité [234, 288, 289]. Pour sa part, l’augmentation de l’activité de l’isoforme
AMPKα1 par la contraction musculaire demeure controversée. Il fut rapporté que ni une contraction
musculaire ex vivo ni une contraction musculaire in situ n’augmente significativement l’activité de
l’isoforme AMPKα1 dans les muscles EDL, TA et gastrocnémien [234, 290]. Néanmoins, une autre
étude a démontré l’augmentation de l’activité de l’isoforme AMPKα1 en réponse à une contraction ex
vivo de courte durée et de faible intensité dans le muscle épitrochléen de rat. Aucun changement du
rapport AMP: ATP ne fut observé lors de ce protocole de contraction [291].
2.3.2.2 Calcium
Tel que précédemment discuté dans la section 1.4.1, lors de la contraction musculaire, une grande
quantité de Ca2+ contenu dans les citernes terminales du réticulum sarcoplasmique est relâchée
dans le sarcoplasme. L’interaction entre le Ca2+ et la calmoduline active une autre AMPKK, la
CAMKK. Il fut démontré qu’un protocole de contraction musculaire in situ augmente l’activité de la
CAMKKβ dans le muscle gastrocnémien de rat. De plus, cette même étude a rapporté une diminution
de l’activité de l’isoforme AMPKα2 induite par la contraction musculaire en présence d’un inhibiteur
spécifique de la CAMKK, le STO-609 [239]. Des résultats similaires furent obtenus avec l’utilisation

31
du KN-93, un inhibiteur de la Ca2+/CaM. En effet, il fut également rapporté que la phosphorylation de
la Thr172 et l’activité des deux isoformes AMPKα sont inhibées par la contraction musculaire ex vivo
de faible intensité et de courte durée dans les muscles EDL et soléaire de souris traitées
préalablement avec le KN-93 [238].
2.3.2.3 Espèces réactives de l’oxygène
Certains groupes de recherche ont également étudié l’activation de l’AMPK par les ROS lors de la
contraction musculaire. Une augmentation de l’activité de l’AMPKα1 dans le muscle épitrochléen de
rat fut rapportée lors d’un traitement au H2O2 bien qu’aucune modification du ratio AMP: ATP ne fut
observée. Cet effet fut bloqué par un antioxydant, le N-acétylcystéine [292]. Dans le même ordre
d’idée, une réduction de cinquante pourcent de la phosphorylation et de l’activité de l’AMPK fut
démontrée lors d’un protocole de contraction musculaire ex vivo dans le muscle EDL de souris
incubé en présence de N-acétylcystéine [154].
2.3.2.4 Interleukine-6
Finalement, la contraction musculaire induit également l’augmentation des niveaux sanguins de
certaines hormones, de facteurs de croissance et de cytokines dont l’interleukine-6 (IL-6). IL-6 est
une cytokine impliquée dans le système immunitaire et le processus inflammatoire dont le rôle
physiologique demeure controversé. Certaines études rapportent un rôle pro-inflammatoire tandis
que d’autres suggèrent plutôt l’inverse. En effet, depuis quelques années, IL-6 est également
reconnue comme une myokine, c’est-à-dire une cytokine induite et relâchée par le muscle
squelettique. Par ailleurs, une augmentation des concentrations plasmatiques en IL-6 fut observée à
la suite d’une contraction musculaire tant chez l’humain que chez le rongeur [293-295]. Une
augmentation de la phosphorylation de l’AMPK fut démontrée dans le muscle EDL de rat traité avec
IL6 ex vivo. Une forte diminution de la phosphorylation de l’AMPK fut également observée à la suite
d’un exercice physique chez la souris dont le gène IL6 fut génétiquement invalidé (IL6 KO)
comparativement à la souris contrôle [295]. De plus, une augmentation des niveaux intracellulaires
de Ca2+ fut rapportée à la suite d’un traitement avec IL6 dans des cellules musculaires de souris
suggérant l’implication de la CAMKK dans l’activation de l’AMPK par IL6 [296].

32
2.4 Régulation du transport du glucose dans le muscle
squelettique
Dès 1997, Merrill et ses collaborateurs démontrèrent que l’AMPK est impliquée dans le transport du
glucose stimulé par AICAR dans le muscle squelettique. Ils observèrent une augmentation de
l’AMPK ainsi qu’une augmentation du transport du glucose dans le muscle gastrocnémien de rat à la
suite d’une perfusion des membres inférieurs avec l’AICAR [297]. Ces résultats furent corroborés par
un autre groupe à la suite d’un traitement AICAR ex vivo du muscle épitrochléen de rat. De plus,
cette même étude démontra une augmentation de l’activité de l’AMPK en réponse à une stimulation
électrique ex vivo du muscle épitrochléen ainsi qu’une augmentation du transport du glucose. La
stimulation électrique du muscle épitrochléen suivie d’un traitement à l’insuline présentait des effets
additifs au niveau du transport du glucose tandis qu’une stimulation électrique suivie d’un traitement
AICAR n’en présentait pas [298]. En 2001, Mu et ses collaborateurs créèrent le premier modèle
murin transgénique d’invalidation de l’AMPK spécifique aux muscles squelettiques et cardiaque en
surexprimant une forme inactive de l’isoforme AMPKα2 inhibant ainsi son activité (souris AMPK
KD1). Le transport du glucose induit par l’AICAR fut aboli dans le muscle EDL des souris AMPK KD1.
Ils observèrent également une diminution partielle du transport du glucose en réponse à une
stimulation électrique ex vivo et à une stimulation électrique in situ dans le muscle EDL des souris
AMPK KD1 [299]. L’ensemble de ces résultats suggérèrent un rôle pour l’AMPK dans le transport du
glucose dans le muscle squelettique. Les études qui ont utilisé d’autres activateurs
pharmacologiques de l’AMPK dont la metformine et le A-769662 ont d’autant plus démontré
certaines évidences de l’implication de l’AMPK dans les effets de ces composés sur le transport du
glucose. Néanmoins, l’implication de l’AMPK dans le transport du glucose induit par la contraction
musculaire demeure controversée. Plusieurs modèles transgéniques de l’AMPK furent générés afin
de répondre à cette question.
2.4.1 Modèles transgéniques des sous-unités catalytiques de l’AMPK
2.4.1.1 Souris AMPK KD1
Tel que précédemment mentionné, le premier modèle transgénique de l’AMPK (AMPK KD1) fut
généré par Mu et ses collaborateurs. Il s’agit d’une surexpression d’une forme inactive de l’isoforme
AMPKα2 spécifique aux muscles squelettiques et cardiaque inhibant ainsi l’activité AMPKα2. Chez
ce modèle, le transport du glucose induit par un traitement AICAR ex vivo est inhibé dans le muscle

33
EDL. Cette donnée suggéra que l’isoforme AMPKα2 soit responsable des effets bénéfiques d’un
traitement AICAR au niveau du transport du glucose dans le muscle squelettique. Une diminution de
quarante pourcent du transport du glucose fut également observée dans le muscle EDL de la souris
AMPK KD1 comparativement au muscle contrôle en réponse à un protocole de contraction ex vivo.
Le transgène n’a aucun effet sur le transport du glucose stimulé par l’insuline. De plus, une
diminution de trente pourcent du transport du glucose fut rapportée dans le muscle EDL de la souris
AMPK KD1 en réponse à un protocole de contraction in situ. Le transport du glucose dans le muscle
soléaire est également diminué de façon significative, mais dans une moindre mesure. Il est à noter
que ce modèle transgénique présente également une force musculaire absolue réduite ainsi qu’une
diminution de l’activité physique volontaire quotidienne.[299, 300].
Récemment, une seconde étude a également démontré un défaut au niveau du transport du glucose
dans le muscle gastrocnémien de la souris AMPK KD1 en réponse à un protocole de contraction
musculaire in situ [301]. Toutefois, un autre groupe n’a observé aucun défaut chez cette même lignée
de souris dans les muscles EDL et soléaire à la suite d’un protocole de contraction ex vivo [149]. En
complément, une étude a voulu tester le rôle de l’AMPK dans le transport du glucose induit par un
exercice physique in vivo sur tapis roulant. Trois protocoles d’exercices furent élaborés. Le premier
protocole était identique pour les deux génotypes de souris. Néanmoins, puisque la souris AMPK
KD1 présente une diminution de sa capacité physique à l’exercice, les deux autres protocoles furent
élaborés en fonction du pourcentage de la vitesse maximale prédéterminée pour chaque sujet, soit
trente pourcent et soixante-dix pourcent. Aucune différence ne fut observée au niveau de la clairance
du glucose dans les muscles quadriceps et gastrocnémien en réponse à chacun des trois protocoles.
Les seules différences significatives rapportées entre les deux génotypes furent des niveaux de
glycogène plus faibles avant et après chaque protocole d’exercice chez la souris AMPK KD1 ainsi
qu’une diminution du contenu musculaire en GLUT4. Toutefois, le pourcentage de GLUT4 qui subit
une translocation à la membrane en réponse au protocole d’exercice est similaire chez les deux
génotypes [302]. D’un autre côté, une toute autre conclusion fut rapportée par un groupe ayant
également testé les souris AMPK KD1 sur tapis roulant. En effet, une diminution de la captation du
glucose dans le muscle gastrocnémien de ces souris fut rapportée en réponse à un protocole
d’exercice sur tapis roulant comparativement aux souris contrôles [303].

34
2.4.1.2 Souris AMPK α2i TG
Une autre lignée transgénique d’invalidation de la sous-unité catalytique α2 dans le muscle
squelettique fut générée, soit la souris AMPK α2i TG. L’activité basale de l’isoforme α2 ainsi que son
activation par un traitement AICAR dans le muscle EDL sont fortement réduites par le transgène.
Néanmoins, l’activité basale de l’isoforme α1 n’est pas affectée par le transgène et son activation par
un traitement AICAR est partiellement diminuée. Le transport du glucose induit par un traitement
AICAR est aboli dans le muscle EDL des souris. En complément, le muscle EDL des souris AMPK
α21TG fut soumis à un premier protocole de contraction ex vivo. Une faible diminution, mais
significative, du transport du glucose fut observée. Néanmoins, tel qu’observé chez la souris AMPK
KD1, les souris AMPK α2i TG génèrent une force musculaire absolue plus faible que les souris
contrôles. Afin de contrer ce facteur, certains paramètres du protocole de contraction musculaire
furent modifiés dont le voltage qui fut ajusté à la baisse pour les souris contrôles afin que la force
générée par le muscle EDL soit identique à celle des souris AMPK α2i TG. Sous ces nouveaux
paramètres, le transport du glucose induit par la contraction musculaire dans le muscle EDL est
identique chez les deux génotypes de souris. Des résultats identiques furent obtenus à la suite d’un
protocole de contraction musculaire in situ. L’ensemble des résultats obtenus par ce modèle
transgénique suggèrent que l’AMPK n’est pas impliquée dans le transport du glucose induit par la
contraction musculaire [290].
2.4.1.3 Souris AMPKα1/α2 KO
Des modèles transgéniques de suppression totale des sous-unités catalytiques α1 et α2 dans tout
l’organisme furent également générés (AMPK α2KO et AMPK α1KO). Chez la souris AMPK KOα2,
l’effet hypoglycémiant en réponse à un traitement AICAR est diminué. De plus, cette souris se
caractérise par une intolérance au glucose et une résistance à l’insuline comparativement à la souris
contrôle. Sans surprise, le transport du glucose induit par un traitement AICAR est inhibé dans les
muscles EDL et soléaire des souris AMPK α2KO. Néanmoins, aucun défaut au niveau du transport
du glucose ne fut observé dans le muscle EDL et soléaire en réponse à un protocole de contraction
ex vivo chez les souris AMPK α2KO. Par contre, il faut tenir compte de l’augmentation de
l’expression de l’isoforme α1 dans les muscles EDL et soléaire des souris AMPK α2KO. De plus, il fut
rapporté que l’activité de l’AMPKα1 est doublée à la suite du protocole de contraction

35
comparativement à la souris contrôle. Ces résultats suggèrent une compensation de l’isoforme α1
dans les muscles de la souris AMPK KOα2 [304, 305].
Comparativement aux souris AMPK KOα2, aucune intolérance au glucose et aucune résistance à
l’insuline ne furent observées chez les souris AMPK KOα1. De plus, le transport du glucose stimulé
par un traitement AICAR n’est pas altéré dans les muscles EDL et soléaire par l’absence de
l’isoforme AMPKα1. Néanmoins, une diminution d’environ vingt-cinq pourcent du transport du
glucose fut mesurée dans le muscle soléaire des souris AMPK KOα1 en réponse au protocole de
contraction ex vivo [305].
2.4.1.4 Souris AMPKα1α2 mdKO
Récemment, un nouveau modèle transgénique d’invalidation concomitante des sous-unités
catalytiques α1 et α2 spécifique au tissu musculaire (AMPKα1α2 mdKO) fut étudié. Tel que
préalablement décrit chez d’autres modèles d’invalidation génétique de l’AMPK, les souris
AMPKα1α2 mdKO présentent une diminution de l’activité physique volontaire quotidienne, une
diminution de la durée d’un exercice sur tapis roulant ainsi qu’une diminution de la force maximale
absolue des muscles soléaire et TA. À la suite d’un protocole de contraction ex vivo, aucune
différence au niveau du transport du glucose ne fut observée dans le muscle EDL des souris
AMPKα1α2 mdKO comparativement aux souris contrôles tant chez les femelles que chez les mâles.
Des résultats similaires furent obtenus pour ce qui est du muscle soléaire chez les souris AMPKα1α2
mdKO femelles. Toutefois, le transport du glucose induit par une contraction musculaire ex vivo est
réduit dans le muscle soléaire de ces mêmes souris de sexe masculin [306].
2.4.2 Modèles transgéniques des sous-unités régulatrices de l’AMPK
2.4.2.1 Souris AMPKγ3 KO
Barnes et ses collaborateurs ont généré un modèle transgénique de suppression de la sous-unité
AMPKγ3, soit la souris AMPK γ3KO. Ni l’isoforme γ1 ni l’isoforme γ2 ne semble affectée par le
transgène de l’isoforme γ3 et aucune compensation ne fut observée dans le muscle squelettique à
l’état basal. La souris AMPK γ3KO présente une tolérance au glucose semblable à celle de la souris
contrôle ainsi que des niveaux normaux d’insuline. Bien que la phosphorylation de l’AMPK soit
augmentée par un traitement AICAR, le transport du glucose stimulé par ce traitement est

36
complétement inhibé dans le muscle EDL des souris AMPK γ3KO tel qu’observé pour les modèles
transgéniques de la sous-unité catalytique α2. Toutefois, aucune réduction du transport du glucose
induit par la contraction musculaire ne fut observée dans le muscle EDL des souris AMPK γ3KO à la
suite d’un protocole de contraction ex vivo. Il est à noter que l’augmentation de la phosphorylation de
l’AMPK en réponse au protocole de contraction est similaire à celle obtenue chez les souris contrôles
[307].
2.4.2.2 Souris AMPKβ1/β2 KO
Deux modèles transgéniques de suppression totale des isoformes AMPKβ1 (AMPKβ1 KO) et
AMPKβ2 (AMPKβ2 KO) furent générés. La souris AMPKβ1 KO se caractérise par une adiposité
inférieure à la souris contrôle soumise tant à une diète standard qu’à une diète riche en gras. Ces
résultats s’expliquent par une consommation alimentaire plus faible chez la souris AMPKβ1 KO. Il fut
observé que l’ablation totale de l’isoforme β1 entraîne une réduction de quatre-vingt-dix pourcent de
l’activité basale de l’AMPK dans le foie. Néanmoins, aucune différence ne fut mesurée dans les
muscles squelettiques et cardiaque [308].
En contrepartie, chez la souris AMPKβ2 KO, une diminution de l’expression et de l’activité des deux
sous-unités α fut observée malgré une augmentation de l’expression de l’isoforme β1 dans les
muscles VL et EDL. Le transport du glucose stimulé par un traitement AICAR est aboli dans le
muscle EDL de la souris AMPKβ2 KO tel qu’observé lors de la suppression des isoformes α2 et γ3.
De plus, la souris AMPKβ2 KO se caractérise par une diminution de sa vitesse maximale ainsi
qu’une diminution de son endurance lors d’un exercice sur tapis roulant. Toutefois, aucun défaut ne
fut rapporté en ce qui a trait au transport du glucose induit par un protocole de contraction musculaire
ex vivo dans le muscle EDL de la souris AMPKβ2 KO [309]. L’ensemble de ces résultats suggère
que l’isoforme β2 joue un rôle prédominant dans le muscle squelettique comparativement à
l’isoforme β1.
2.4.2.3 Souris AMPKβ1/β2M-KO
Afin de mieux comprendre l’implication de la sous-unité AMPKβ, trois modèles transgéniques de
suppression des différentes isoformes spécifiques au muscle (AMPKβ1M-KO, AMPKβ2M-KO et
AMPKβ1β2M-KO) furent étudiés.

37
Chez la souris AMPKβ2M-KO, spécifique au tissu musculaire, l’expression des isoformes α1 et β1
demeure inchangée par le transgène, mais l’expression de l’isoforme α2 n’est plus détectable dans le
muscle squelettique. Cette suppression des niveaux d’expression des isoformes α2 et β2 se traduit
par des niveaux initiaux réduits de la phosphorylation de la Thr172 sans pour autant empêcher son
augmentation par un protocole de contraction ex vivo. La souris AMPKβ2M-KO présente une
intolérance à l’exercice comparable à la souris AMPK KD1. Néanmoins, aucune réduction du
transport du glucose induit par un protocole de contraction musculaire ex vivo ne fut observée dans
le muscle EDL. Aucun phénotype particulier ne fut observé chez la souris AMPKβ1M-KO.
La souris AMPKβ1β2M-KO fut générée à partir du croisement des souris AMPK β1M-KO et
AMPKβ2M-KO. Dans le muscle squelettique, l’expression des isoformes α2 et β2 est nulle tandis que
l’expression des isoformes α1 et β1 est réduite de soixante-dix et quarante-cinq pourcent,
respectivement. L’ensemble de ces modifications se traduit par une inhibition de la phosphorylation
de la Thr172 tant en condition basale qu’en réponse à un protocole de contraction ex vivo.
Comparativement aux souris transgéniques AMPK KD1 et AMPK β2M-KO, la souris AMPK β1β2M-
KO est fortement intolérante à l’exercice sur tapis roulant en ce qui a trait à la vitesse maximale et à
la distance parcourue. À la suite d’un protocole sur tapis roulant, la clairance du glucose sanguin
observée dans les muscles gastrocnémien et soléaire est fortement inhibée chez la souris AMPK
β1β2M-KO comparativement à la souris contrôle. De plus, le transport du glucose induit par un
protocole de contraction musculaire ex vivo établi en fonction de chaque muscle est réduit de
soixante-dix pourcent dans le muscle EDL et de quarante-cinq pourcent dans le muscle soléaire. En
parallèle, la phosphorylation de TBC1D1 par la contraction musculaire est fortement diminuée dans
le muscle EDL des souris AMPK β1β2M-KO. Les résultats obtenus par ce modèle transgénique
suggèrent l’implication de l’AMPK dans le transport du glucose induit par la contraction musculaire
ainsi qu’une interaction avec TBC1D1, responsable de la translocation des GLUT4 [310].
Finalement, dépendamment et/ou indépendamment de l’AMPK, le transport du glucose induit par la
contraction musculaire demeure fonctionnel chez les individus résistants à l’insuline
comparativement à celui stimulé par l’insuline. Il s’agit donc d’une avenue intéressante dans la
recherche d’un traitement pour contrer la résistance à l’insuline.

38
3 Résistance à l’insuline musculaire
3.1 Définition
La résistance à l’insuline se définit comme une diminution de la réponse des tissus sensibles à
l’insuline qui se traduit par une réduction du transport du glucose dans le muscle squelettique et le
tissu adipeux, une diminution de la suppression de la production hépatique de glucose et une
augmentation de la lipolyse du tissu adipeux. La résistance à l’insuline se caractérise, entre autre
chose, par une hyperinsulinémie à jeun, une hyperglycémie à jeun et postprandiale, une
hyperlipidémie, une intolérance au glucose, une augmentation de l’hémoglobine glycosylée (HbA1c)
et une augmentation de nombreux marqueurs inflammatoires plasmatiques.
De nombreux mécanismes furent suggérés pour contribuer au développement de la résistance à
l’insuline et la majorité d’entre eux sont associés à l’augmentation du tissu adipeux et plus
particulièrement, du tissu adipeux viscéral [311, 312]. Des études ont observé une résistance à
l’insuline des tissus périphériques associée à l’augmentation de l’adiposité bien avant l’apparition de
l’hyperglycémie à jeun et postprandiale, indice utilisé pour diagnostiquer la résistance à l’insuline
chez l’humain. Par ailleurs, il fut rapporté qu’une augmentation de trois pourcent du tissu adipeux à la
suite d’une augmentation de l’apport calorique quotidienne résulte en une hyperinsulinémie à jeun et
une diminution de la sensibilité à l’insuline postprandiale chez de jeunes adultes en santé malgré
l’absence d’hyperglycémie [313]. De plus, il fut démontré qu’une diminution de la masse adipeuse
augmente la sensibilité à l’insuline chez les individus obèses tolérants au glucose [314]. Ces résultats
suggèrent que l’augmentation de l’adiposité contribue à résistance à l’insuline des tissus
périphériques dont le muscle squelettique.
3.2 Défaut de la voie de signalisation de l’insuline
Puisque le muscle squelettique est responsable de quatre-vingts pourcent de la captation du glucose
en situation post prandiale, une diminution de sa sensibilité à l’insuline entraîne des perturbations
métaboliques majeures et peut mener, entre autre chose, à l’apparition du diabète de type 2. Un
défaut au niveau de la cascade de signalisation de l’insuline est principalement à l’origine de la
diminution du transport du glucose dans le muscle squelettique.

39
3.2.1 Récepteur à l’insuline
Une diminution, ou non, de la liaison de l’insuline à son récepteur fut rapportée dans le muscle
squelettique d’individus obèses et diabétiques [315-317]. Similairement, certaines études ont
démontré une diminution de l’activité tyrosine de l’IR tandis que d’autres n’ont observé aucune
différence entre les sujets [316-322]. Néanmoins, chez les individus diabétiques, une réduction de
l’expression de l’IR ne serait pas la cause de la diminution de son activité tyrosine [318]. Toutefois,
une étude in vitro a démontré que l’incubation de cellules musculaires isolées à partir du muscle
soléaire de rat en présence d’acide palmitique, un acide gras saturé, diminue l’expression et l’activité
de l’IR [323].
3.2.2 Substrat du récepteur à l’insuline 1
Des études ont démontré une diminution de la phosphorylation des résidus tyrosine d’IRS-1 dans le
muscle VL des individus obèses et diabétiques comparativement aux sujets sains ainsi qu’une
diminution de la liaison d’IRS-1 à la sous-unité régulatrice p85 de la PI3K [320, 324, 325]. De plus,
une augmentation de la phosphorylation du résidu sérine 312 (Ser312), l’équivalent de la sérine 307
(Ser307) chez la souris et le rat, fut rapportée chez les individus résistants à l’insuline [326]. La
phosphorylation du résidu sérine 1101 (Ser1101) d’IRS-1 dans le muscle VL humain fut également
associée à la résistance à l’insuline ainsi que la phosphorylation de la sérine 636 (Ser636) observée
dans des cultures primaires de cellules musculaires d’individus atteints de diabète de type 2 [327,
328]. Par ailleurs, il fut rapporté qu’une diète riche en gras augmente la phosphorylation des
Ser636/639 dans le muscle EDL de rat [329]. Ainsi, il fut suggéré que la phosphorylation des résidus
sérine d’IRS-1 empêche la phosphorylation des résidus tyrosine de cette protéine bloquant ainsi sa
liaison à la PI3K. D’ailleurs, l’invalidation des résidus Ser302, Ser307 et Ser612 dans le muscle
squelettique de souris prévient le développement de la résistance à l’insuline induite par une diète
riche en gras [330]. Il est également suggéré que la dégradation d’IRS-1 contribue au défaut de la
voie de signalisation de l’insuline. Néanmoins les mécanismes moléculaires responsables de cette
dégradation restent à être démontrés.
3.2.3 Phosphatidylinositol 3-kinase
Une diminution de l’activité de la PI3K associée à IRS-1 fut observée dans le muscle VL d’individus
obèses et d’individus atteints de diabète de type 2 ainsi qu’à la suite d’une infusion de lipides [325,

40
331, 332]. L’activation de la PI3K dépend de l’interaction entre ses deux sous-unités. De façon
inattendue, il fut démontré que l’invalidation génétique totale de la sous-unité régulatrice p85α et
p85β améliore la sensibilité à l’insuline systémique, entre autre chose, par une augmentation du
transport du glucose musculaire [333, 334]. À la suite de la démonstration de ces résultats, il fut
suggéré que l’activation de la PI3K dépend du rapport entre les deux sous-unités, p85 et p110 [335].
Toutefois, il demeure que l’invalidation génétique des sous-unités p85 et p110 de la PIK3
spécifiquement dans le muscle squelettique de souris résulte en une résistance à l’insuline
musculaire et une intolérance au glucose systémique [336]. En somme, les mécanismes de
régulation de l’activité PI3K associée à IRS-1 demandent à être davantage étudiés.
3.2.4 Protéine kinase B/Akt
Certaines études ont démontré une réduction de l’activité d’Akt dans le muscle VL d’individus obèses
ainsi que dans le muscle gastrocnémien de rats génétiquement obèses tandis que d’autres n’ont
révélé aucun défaut de l’activité d’Akt tant chez l’humain que chez le rongeur [325, 332, 337, 338].
Malgré ces résultats contradictoires, il n’en demeure pas moins que la phosphorylation des résidus
Ser473 et Thr308 est diminuée dans le muscle squelettique en présence de résistance à l’insuline.
Ceci dit, certaines études ont suggéré que la phosphorylation du résidu Ser473 ne serait pas
corrélée nécessairement à l’activité d’Akt [339, 340].
3.3 Lipotoxicité
La lipotoxicité se définit par une accumulation ectopique de lipides, entre autre lieu, dans le muscle
squelettique causée par un dysfonctionnement du métabolisme des lipides menant à une
augmentation de la concentration plasmatique des acides gras libres (AGL). Il est maintenant bien
établi que l’augmentation aigue et chronique des concentrations plasmatiques d’AGL, en lien avec
l’augmentation du tissu adipeux, contribue à l’apparition de la résistance à l’insuline musculaire. En
effet, quatre heures d’infusion de lipides chez des individus sains résultent en une résistance à
l’insuline musculaire. De plus, la sévérité de la résistance à l’insuline musculaire est dose-
dépendante des concentrations plasmatiques d’AGL [341, 342]. Le développement de la résistance à
l’insuline musculaire causée par des concentrations plasmatiques élevées d’AGL serait associé à
l’augmentation des lipides intramyocellulaires, et plus particulièrement à l’accumulation des

41
métabolites issus des acides gras dont l’acyl-coenzyme-A (acyl-CoA), le diacylglycérol (DAG) et les
céramides dans les cellules musculaires [343-345].
3.3.1 Acyl-coenzyme-A
L’acyl-CoA est un métabolite formé d’un acide gras à chaîne longue et d’une coenzyme A impliqué
dans la dégradation des acides gras par la bêta oxydation (β-oxydation). Une corrélation négative fut
démontrée entre la sensibilité à l’insuline et l’accumulation d’acyl-CoA dans le muscle VL chez
l’humain. L’amélioration de la sensibilité à l’insuline observée par l’administration d’acipimox, un
médicament qui a pour mode d’action d’inhiber la lipolyse, est concomitante à une diminution des
niveaux plasmatiques d’AGL ainsi qu’à une diminution du contenu musculaire d’acyl-CoA chez des
individus atteints de diabète de type 2. De plus, l’infusion de lipides et trois semaines de diète riche
en gras induisent également l’accumulation d’acyl-CoA dans le muscle gastrocnémien de rat. Entre
autre, il fut démontré que l’infusion de lipides résulte en une augmentation du contenu musculaire en
acyl-CoA et en DAG ainsi qu’en une activation de la protéine kinase C thêta (PKCθ) et en la
phosphorylation du résidu sérine 307 (Ser307) d’IRS-1. Ces résultats suggèrent que l’acyl-CoA
contribue à la résistance à l’insuline musculaire en activant directement, ou par l’intermédiaire d’un
autre métabolite, la PKCθ inhibant ainsi la capacité d’IRS-1 à recruter et à activer la PI3K [346-349].
3.3.2 Diacylglycérol
L’infusion de lipides ainsi qu’une diète riche en gras résultent en une augmentation du contenu
musculaire en diacylglycérol (DAG). Le DAG, également nommé diglycéride, est un métabolite formé
de résidus d’acide gras liés à un résidu de glycérol. Il s’agit d’un second messager impliqué dans le
métabolisme des triglycérides et des phospholipides. La diglycéride acyltransférase1 (DGAT1) est
l’enzyme responsable de la synthèse des triglycérides à partir du DAG. Il fut démontré que la
surexpression d’une forme active de la DAGT1 dans le muscle squelettique de souris prévient
l’accumulation musculaire en DAG et le développement de la résistance à l’insuline induite par une
diète riche en gras. Ces résultats suggèrent que l’augmentation du contenu musculaire en DAG
contribue à la résistance à l’insuline. De plus, il fut suggéré que le DAG participe à la résistance à
l’insuline via l’activation de la PKCθ [347, 350-352].

42
Il fut démontré que l’infusion de lipides résulte en une augmentation du contenu musculaire en DAG
en concomitance avec une activation de la PKCθ, une augmentation de la phosphorylation du résidu
Ser307 d’IRS-1 et par conséquent, une diminution de la phosphorylation des résidus tyrosine d’IRS-1
et une diminution de l’activité de la PI3K. En dépit de l’accumulation de lipides intramyocellulaires,
l’invalidation de la PKCθ dans le muscle squelettique de souris prévient la déphosphorylation d’IRS-1
sur ses résidus tyrosine et le développement de la résistance à l’insuline induite par une diète riche
en gras. De plus, il fut démontré que l’invalidation des résidus Ser302, Ser307 et Ser612 dans le
muscle squelettique prévient le développement de la résistance à l’insuline induit par une diète riche
en gras malgré l’accumulation de lipides intramyocellulaires. Toutefois, le mécanisme par lequel
l’activation de la PKCθ mène à la phosphorylation de la Ser307 d’IRS-1 reste à démontrer [330, 353].
3.3.3 Céramides
Outre l’accumulation d’acyl-CoA et de DAG, certaines études ont rapporté une augmentation du
contenu musculaire en céramides tant chez le rat que chez l’humain obèse résistant à l’insuline [354-
356]. De plus, il fut rapporté que l’infusion de lipides chez l’humain résulte aussi en une augmentation
du contenu en céramides [357]. À l’inverse, certaines études n’ont mesuré aucun changement du
contenu musculaire en céramides à la suite d’une infusion de lipides chez l’humain et le rat [347,
352]. Le céramide est un métabolite de la famille des sphingolipides formé d’un acide gras à longue
chaîne et d’un alcool aminé, la sphingosine. Il s’agit d’un second messager essentiel à la synthèse
de la plupart des sphingolipides. L’administration de myriocine, un inhibiteur de la sérine
palmitoyltransférase 1 (SPT1), une enzyme responsable de la synthèse des céramides, fut rapportée
pour prévenir l’accumulation du contenu musculaire en céramides ainsi que le développement de la
résistance à l’insuline induite par une diète riche en gras ou une infusion de lipides [358, 359]. De
plus, la surexpression de la kinase sphingosine 1 (SphK1), impliquée dans le catabolisme des
céramides, prévient également l’accumulation musculaire en céramides et améliore la sensibilité à
l’insuline à la suite d’une diète riche en gras [360].
Il fut observé que l’accumulation du contenu musculaire en céramides inhibe la cascade de
signalisation de l’insuline par la déphosphorylation du résidu Thr308 d’Akt et par conséquent, sa
translocation à la membrane plasmique. En effet, il fut démontré que le traitement des cellules L6
avec un analogue des céramides, le céramide C2, inhibe le transport du glucose stimulé par l’insuline

43
dû à l’absence de translocation d’Akt. L’inhibition de la synthèse des céramides prévient cette
absence de translocation d’Akt. Des résultats similaires furent obtenus en réponse à l’administration
de myriocine [358, 361, 362]. De plus, il fut démontré que les céramides activent la PKCζ et les
PP2A qui seraient responsables de la déphosphorylation du résidu Thr308 d’Akt. Enfin, un dérivé des
céramides, le glycosylcéramide pourrait également inhiber l’activité tyrosine de l’IR [363-365].
L’ensemble de ces résultats suggère que les céramides contribuent au développement de la
résistance à l’insuline musculaire causée par l’augmentation des concentrations plasmatiques en
AGL.
Finalement, il fut proposé que l’activation de la voie de signalisation des récepteurs de type Toll4
(TLR4) en réponse à une infusion d’acides gras saturés serait responsable de la biosynthèse des
céramides via l’activation de la protéine kinase IκB beta (IKKβ) dans le muscle squelettique. Des
médiateurs pro-inflammatoires, le lipopolysaccharide (LPS) et le facteur de nécrose tumorale alpha
(TNFα), furent également rapportés pour induire la biosynthèse des céramides [366]. Ces résultats
suggèrent la présence d’interactions entre la lipotoxicité et l’activation des voies inflammatoires dans
le développement de la résistance à l’insuline musculaire.
3.4 Inflammation
Outre une augmentation des lipides intramyocellulaires causée par des concentrations plasmatiques
élevées en AGL, l’obésité se caractérise par un état inflammatoire chronique. Il fut proposé que
l’inflammation débute dans le tissu adipeux où il se produit une infiltration massive de macrophages
[367, 368]. L’infiltration de macrophages pourrait être une conséquence d’une hypoxie causée par
l’expansion du tissu adipeux [369-371]. Il fut proposé que le tissu adipeux, et plus particulièrement
les macrophages présents dans celui-ci, sécrète des cytokines pro-inflammatoires dans la circulation
sanguine participant ainsi à l’inflammation systémique associée à l’obésité. Entre autre chose, des
concentrations plasmatiques élevées des cytokines pro-inflammatoires TNFα, interleukine 1β (IL-1β)
et IL-6 sont associées à la résistance à l’insuline induite par l’obésité [372, 373]. Récemment, il fut
également suggéré que l’endotoxémie métabolique, c’est-à-dire l’augmentation des concentrations
plasmatiques de l’endotoxine LPS, participe au développement de l’inflammation chronique associée
à l’obésité [374].

44
Les myocytes expriment différents récepteurs de cytokines dont les récepteurs 1 et 2 d’IL-1, le
récepteur d’IL-6, les récepteurs 1 et 2 de TNFα, le récepteur d’interféron γ (INFγ) et les récepteurs
des chimiokines de la famille CC dont CCR2, CCR4 et CCR10 [375, 376]. La présence de TNFα
dans le milieu d’incubation augmente l’expression des récepteurs d’IL-6 et de TNFα tandis qu’un
cocktail de cytokines (TNFα et INFγ) combiné à du LPS augmente l’expression des récepteurs d’IL-1,
d’IL6, d’INFγ et de TNFα dans les cellules L6 [376]. Ce cocktail de cytokines/LPS induit une
résistance à l’insuline [15]. De plus, l’administration de LPS augmente également l’expression des
récepteurs d’IL-6 et de TNFα dans le muscle épitrochléen de rat [376]. Ces résultats suggèrent que
le muscle squelettique est sensible à l’inflammation systémique induite par l’obésité et que
l’augmentation plasmatique de cytokines pro-inflammatoires puisse contribuer au développement de
la résistance à l’insuline musculaire.
3.4.1 LPS
Les concentrations plasmatiques de l’endotoxine LPS sont de deux à trois fois plus élevées chez la
souris soumise à une diète riche en gras comparativement à la souris soumise à une diète standard
[374]. Le LPS est la composante principale de la membrane externe de bactéries à gram négatif
présentes dans le microbiote intestinal. Il fut démontré que la nature de la diète alimentaire influence
la composition bactérienne du microbiote de même que l’intégrité des parois intestinales [377].
D’ailleurs, une diète riche en gras augmente la perméabilité de la paroi intestinale et par conséquent,
facilite la migration des produits bactériens, dont le LPS, vers la circulation sanguine [378]. Il fut
rapporté que l’infusion chronique d’une faible dose de LPS, comparable aux concentrations
plasmatiques mesurées à la suite d’une diète riche en gras, pour une durée de quatre semaines chez
la souris induit une augmentation du tissu adipeux viscéral comparable à celle observée pour la
même période de temps à la suite d’une diète riche en gras. De plus, une augmentation de
l’expression des cytokines IL-1 et TNFα comparable à celle obtenue à la suite d’une diète riche en
gras fut démontrée dans le muscle squelettique de souris en réponse à ce traitement [374]. Des
résultats similaires furent observés conséquemment à l’infusion de LPS dans le muscle VL chez
l’humain [379]. Ces données suggèrent une interaction entre le LPS et le développement de
l’inflammation associé à l’obésité.

45
L’inhibition et l’invalidation génétique du récepteur de type Toll4 (TLR4) ont permis d’identifier l’un
des mécanismes d’action du LPS dans le muscle squelettique. En effet, la présence d’un inhibiteur
spécifique de TLR4, le TAK-242, lors d’un traitement au LPS dans les cellules L6 prévient l’activation
de la voie du facteur nucléaire kappa B (NF-κB) et le développement de la résistance à l’insuline
[380]. Des résultats similaires furent observés dans un modèle de cellules musculaires humaines. De
plus, l’augmentation la phosphorylation de la kinase c-Jun N-terminale (JNK), impliquée dans la
réponse inflammatoire, ainsi que l’augmentation de l’expression d’IL-6 et de la chimiokine ligand 2
(CCL2) furent observées en réponse au traitement au LPS [381]. Il fut également rapporté que le
LPS active la voie NF-κB [382]. NF-κB est un facteur de transcription de gènes impliqués dans la
réponse inflammatoire, via l’activation des protéines kinase IκB α et β (IKKα/β) et la dégradation des
protéines inhibitrices de NF-κB α et β (IκBα/α). Par ailleurs, il fut démontré que l’activation de la voie
NF-κB par le LPS induit la transcription et la production de médiateurs inflammatoires tels le TNFα et
l’IL-6 dans les cellules C2C12 et le muscle quadriceps de souris [383]. Ainsi, l’induction de cytokines
et chimiokines par le LPS dans le muscle squelettique pourrait exacerber la réponse inflammatoire
systémique liée à l’obésité et participer au recrutement de macrophages au niveau des muscles
squelettiques.
Fait intéressant, il fut rapporté qu’un protocole d’exercice physique chronique diminue les
concentrations plasmatiques de LPS ainsi que l’ARNm des récepteurs TLR4 dans le muscle
gastrocnémien chez le rat soumis à une diète riche en gras. Ces modifications sont accompagnées
par une diminution de la phosphorylation de JNK, d’IKKβ et du résidu Ser307 d’Akt [384].
3.4.2 TNFα
Le facteur de nécrose tumorale α est une glycoprotéine de la famille des cytokines impliquée dans la
réaction inflammatoire. Il fut rapporté que l’ajout du TNFα dans le milieu de culture de cellules
musculaires primaires de souris prévient la phosphorylation tyrosine de l’IR ainsi que l’association
d’IRS-1 à la sous-unité p85 de la PI3K induite par l’insuline [385]. De plus, l’infusion de TNFα
pendant quatre heures induit une résistance à l’insuline dans le muscle VL chez l’humain. Une
augmentation de la phosphorylation de JNK en concomitance avec une augmentation de la
phosphorylation du résidu Ser312 d’IRS-1 et une diminution de la phosphorylation de AS160 furent
observées à la suite d’une infusion de TNFα [386]. Ces résultats suggèrent que les effets néfastes du

46
TNFα pourraient être expliqués par une activation de JNK1. Il fut établi que JNK1 est une kinase
impliquée dans la voie de signalisation du stress cellulaire et dans la transcription de gènes, eux-
mêmes impliqués dans la réponse inflammatoire. L’invalidation génétique globale de JNK prévient la
résistance à l’insuline induite par une diète riche en gras chez la souris [387]. Au niveau musculaire,
il fut démontré que la surexpression d’une forme constitutivement active de JNK par électroporation
augmente la phosphorylation sérine d’IRS1, diminue la phosphorylation de la Tyr1361 d’IRS1 et des
résidus Ser473 et Thr308 d’Akt dans le muscle TA de souris diminuant ainsi la clairance du glucose
[388].
Outre la voie JNK, le TNFα est connu pour activer la voie NF-κB. Il fut rapporté qu’un traitement au
TNFα diminue le contenu en IKKβ dans des cellules musculaires primaires humaines. L’utilisation de
petits ARN interférents pour IKKβ prévient la diminution de la phosphorylation des résidus Ser473 et
Thr308 d’Akt ainsi que la diminution du transport du glucose stimulé par l’insuline. Ces résultats
suggèrent l’implication de la voie IKK/NF-κB dans la résistance à l’insuline induite par TNFα [389].
Toutefois, il fut rapporté que l’invalidation génétique globale de TNFα ne prévient pas l’accumulation
de lipides et l’apparition de marqueurs inflammatoires dans le muscle squelettique de souris
soumises à une diète riche en gras, contrairement aux résultats obtenus dans le foie de ces mêmes
souris [390]. Ces résultats suggèrent que les concentrations plasmatiques élevées de TNFα
associées à l’obésité n’influencent pas l’activation de la réponse inflammatoire dans le muscle
squelettique. Ainsi, d’autres médiateurs, dont les acides gras et le LPS, seraient responsables de
l’activation des voies JNK et IKK/NFκ-B observée dans le muscle squelettique résistant à l’insuline
[352, 381, 382, 391, 392].
3.4.3 Interleukine-6
Chez l’humain, il fut démontré que l’augmentation des concentrations plasmatiques d’IL-6 est
corrélée à l’augmentation de l’adiposité [393-395]. Récemment, il fut rapporté que les concentrations
artérielles et veineuses d’IL-6 sont négativement corrélées avec le transport du glucose dans les
muscles de l’avant-bras chez les individus obèses comparativement aux individus contrôles [373]. De
plus, une diminution de la sensibilité à l’insuline systémique fut observée à la suite d’une infusion
aigue d’IL-6 chez la souris. Cette perte de sensibilité à l’insuline correspond à une réduction de
quarante pourcent du transport du glucose ainsi qu’à une diminution de l’activité PI3K associée à

47
IRS-1 dans le muscle gastrocnémien. Une augmentation des acyl-Coa et de la phosphorylation
tyrosine du facteur de transcription STAT3 fut également observée dans le muscle quadriceps
conséquemment à l’infusion d’IL-6 [396]. Toutefois, une étude précédente n’avait révélé aucun défaut
au niveau de la voie signalétique de l’insuline dans le muscle squelettique de souris à la suite d’une
infusion chronique d’IL-6 [397]. De plus, des augmentations du transport basal du glucose et de celui
stimulé à l’insuline ainsi que la translocation des GLUT4 furent observées dans les cellules L6
traitées deux heures avec l’IL-6 [398].
Il fut également démontré qu’un traitement de trois heures avec l’IL-6 suivi d’une stimulation à
l’insuline a des effets additifs sur le transport du glucose ainsi que sur la phosphorylation de TBC1D4
dans les cellules C2C12. Néanmoins, le transport du glucose, la phosphorylation du résidu Ser473
d’Akt et la phosphorylation de TBC1D4 stimulés par l’insuline sont inhibés après vingt-quatre de
traitement avec l’IL-6. Ces défauts observés dans la voie de signalisation de l’insuline sont associés
à une augmentation de la phosphorylation de JNK et du résidu Ser307 d’IRS-1. L’inhibition de JNK
par un agent pharmacologique ou par de petits ARN interférents rétablit le transport du glucose
stimulé par l’insuline en présence d’IL-6 dans les cellules C2C12. Ces effets furent aussi obtenus
chez la souris. En effet, quarante-huit heures d’infusion d’IL-6 inhibent la phosphorylation d’Akt dans
le muscle squelettique de souris et induit une hyperglycémie systémique à court terme. Cependant,
une augmentation de la sensibilité à l’insuline fut observée après trois heures d’infusion [399].
En somme, le rôle d’IL-6 dans le développement de la résistance à l’insuline musculaire demeure
controversé. Récemment, nous avons démontré que la production et la sécrétion d’IL-6 par le muscle
gastrocnémien conséquemment à une infusion de la protectine DX améliorent la sensibilité à
l’insuline chez des modèles de souris résistantes à l’insuline [400]. Par ailleurs, tel que mentionné à
la section 2.3.2.4, l’exercice physique provoque l’induction et le relâchement d’IL-6 par les fibres
musculaires. Fait intéressant, tandis que les concentrations plasmatiques basales d’IL-6 sont plus
élevées chez les individus obèses et résistants à l’insuline, les concentrations plasmatiques basales
d’IL-6 sont plus faibles chez les individus entraînés comparativement aux individus sédentaires [401].

48
3.4.4 Macrophages
Les macrophages sont des cellules provenant de la différenciation des monocytes impliqués dans la
défense immunitaire. La présence d’une infiltration de macrophages dans le muscle squelettique
causée par l’obésité est beaucoup moins évidente que celle observée dans le tissu adipeux. Malgré
tout, certaines études tendent à démontrer que le phénomène est aussi présent dans le muscle
squelettique [402]. En effet, une étude a démontré une augmentation du marqueur de macrophage
CD68 dans le muscle VL d’individus obèses. L’augmentation de CD68 est corrélée positivement à
l’indice de masse corporelle [403]. De plus, il fut démontré que la mise en culture de cellules
musculaires humaines isolées à partir du muscle VL avec des macrophages en présence d’acide
palmitique induit une augmentation de l’expression des cytokines TNFα, IL-6 et IL-1β. De plus, une
diminution d’IκBα, une augmentation de la phosphorylation de JNK et une diminution de la
phosphorylation du résidu Ser473 d’Akt furent également observées dans ces conditions [402].
Certaines études ont aussi rapporté l’augmentation des marqueurs de macrophages CD68, F4/F80+
et CD11c+ dans les muscles gastrocnémien et quadriceps de souris soumises à une diète riche en
gras en concomitance avec une augmentation des cytokines TNFα et IL-6 et du récepteur de
chimiokine CCR2 [404, 405]. De plus, une diminution du contenu musculaire en macrophages ainsi
que de l’expression de CD68, de la chimiokine CCL2 et de TNFα fut observé à la suite d’une
déplétion en cellules CD11c+ [406]. Ces résultats suggèrent que l’obésité résulte en une infiltration de
macrophages dans le muscle squelettique. L’infiltration de macrophages pourrait contribuer à
l’activation de la réponse inflammatoire et au développement de la résistance à l’insuline musculaire
par une action paracrine.
Les cytokines TNFα, IL-6 ainsi que l’endotoxine LPS et les macrophages ont tous en commun la
capacité d’induire l’expression d’iNOS, la forme inductible du monoxyde d’azote.
4 iNOS
4.1 Structure et expression
La forme inductible de la synthase du monoxyde d’azote (iNOS) fait partie de la famille des enzymes
responsables de la production endogène NO, c’est-à-dire la conversion de l’oxygène (O2) et de la L-
arginine en NO et en L-citruline [136]. La protéine iNOS est un complexe tétradimère constitué de
deux NOS monomères et deux calmodulines. La protéine iNOS contient deux domaines catalytiques.

49
Le domaine oxygénase dans la région N-terminale contient des sites de liaison à la
tétrahydrobioptérine (BH4), à l’hème et à L-arginine. Le domaine réductase dans la région C-
terminale contient les sites de liaison à la flavine adénine mononucléotide (FAD), à la flavine
mononucléotide (FMN) et au nicotinamide adénine dinucléotide phosphate (NADPH) [407-409].
L’interaction de la calmoduline avec iNOS est essentielle à son activité. Contrairement à eNOS et
nNOS, l’enzyme iNOS se lie de façon constitutive à la calmoduline, ce qui en fait une forme
indépendante de la présence du Ca2+ [410, 411]. La structure d’iNOS est représentée à la figure 10.
Figure 10 Structure d'iNOS, tirée de [412]
D’abord découverte dans les macrophages, iNOS s’exprime dans différents tissus dont le muscle
squelettique [413]. Son expression n’est pas détectée, ou très peu, dans le muscle squelettique sain
contrairement aux formes constitutivement actives nNOS et eNOS. En contrepartie, la présence de
facteurs inflammatoires entraîne l’expression d’iNOS qui à son tour, génère de grandes quantités de
NO de façon continue jusqu’à la dégradation de l’enzyme [414]. Bien qu’iNOS joue un rôle primordial
dans la défense immunitaire, son induction chronique est associée à plusieurs conditions
pathophysiologiques, dont la résistance à l’insuline liée à l’obésité [415-421].
4.2 Régulation de l’expression d’iNOS
De nombreux facteurs inflammatoires mènent à l’induction d’iNOS dont l’endotoxine LPS, les
cytokines IFNγ, IL-1β, IL-6 et TNFα. La combinaison de deux ou plusieurs de ces facteurs semble
nécessaire pour activer les voies signalétiques impliquées dans l’induction de l’ARNm d’iNOS dans
les cellules C2C12 et L6 [15, 141, 422]. Toutefois, le LPS seul induit l’expression d’iNOS dans les
muscles soléaire, EDL et gastrocnémien de rat en réponse à une stimulation LPS in vivo et ex vivo
[423, 424].

50
Peu d’études furent menées sur les mécanismes d’action par lesquels le LPS et/ou les cytokines
induisent l’expression d’iNOS dans le muscle squelettique. Néanmoins, il fut rapporté que l’IFNγ en
combinaison avec l’IL-1β induisent l’expression d’iNOS dans les cellules L6 par l’augmentation des
récepteurs d’IL-1 par l’INFγ. De plus, cette même étude a également démontré que l’inhibition de la
voie NF-κB par un antioxydant, le PDTC, inhibe l’expression d’iNOS tandis que l’inhibition de ERK1/2
par l’agent pharmacologique PD98059 réduit partiellement l’expression d’iNOS [425]. Une seconde
étude a également observé l’inhibition de l’expression d’iNOS par un traitement TNFα/INFγ dans les
cellules C2C12 en présence de la surexpression d’un mutant négatif de NF-κB. En complément, la
synthèse d’iNOS requiert la présence d’une protéine liant son ARN, la protéine Hu antegen R (HuR).
En effet, il fut démontré que l’inhibition de la protéine HuR par l’utilisation de petits ARN interférents
dans les cellules C2C12 prévient l’induction d’iNOS par le traitement TNFα/INFγ [426]. L’ensemble
de ces résultats suggère que la voie NF-κB ainsi que la protéine HuR sont des médiateurs de la
régulation de l’expression d’iNOS dans le muscle squelettique par les cytokines.
Outre les cytokines, il fut rapporté que les acides gras saturés induisent également l’expression
d’iNOS. En effet, un traitement avec de l’acide palmitique, un acide gras saturé, induit l’expression
d’iNOS et active la voie NF-κB dans les cellules musculaires humaines [427].
Fait intéressant, il fut démontré que l’activation de l’AMPK par des agents pharmacologiques (AICAR,
metformine, troglitazone) inhibe la production de NO et l’expression d’iNOS dans les cellules L6
traitées avec du LPS et des cytokines. L’administration intrapéritonéale d’AICAR prévient également
la production de NO induite par l’injection de LPS dans les muscles TA et gastrocnémien de rat [424].
Des résultats similaires furent obtenus avec un traitement au resvératrol, un composé naturel qui
active l’AMPK. En effet, l’administration intrapéritonéale de resvératrol précédant une injection de
LPS prévient l’induction d’iNOS dans le muscle gastrocnémien de souris. Par ailleurs, la suppression
de l’AMPK par de petits ARN interférents dans les cellules L6 prévient les effets inhibiteurs du
resvératrol sur iNOS [428].
4.3 iNOS et la résistance à l’insuline musculaire
L’induction d’iNOS dans le muscle squelettique fut rapportée dans plusieurs modèles d’obésité et de
résistance à l’insuline. En effet, l’expression d’iNOS fut observée dans le muscle gastrocnémien de

51
souris génétiquement obèses (ob/ob) ainsi que de souris soumises à une diète riche en gras [416,
429]. Des résultats similaires furent rapportés dans le muscle soléaire de rats soumis à une diète
riche en gras et dans le muscle gastrocnémien de rats génétiquement obèses [416, 429, 430]. Chez
l’humain, une étude a rapporté une augmentation de l’expression de la protéine iNOS dans le muscle
quadriceps d’individus atteints de diabète de type 2 comparativement aux sujets contrôles [431].
Toutefois, une étude récente a échoué à détecter une augmentation significative de l’expression
d’iNOS dans le muscle VL de sujets résistants à l’insuline comparativement aux sujets contrôles
[432].
Des études de notre laboratoire ont démontré que l’induction d’iNOS par une combinaison de
LPS/cytokines dans les cellules L6 diminuait le transport du glucose stimulé par l’insuline. L’ajout
d’un inhibiteur de NOS, le NG-Nitro-L-arginine-methyl ester (L-NAME), prévient la production de NO
par iNOS et la diminution transport du glucose stimulé par l’insuline [15]. Des résultats similaires
furent obtenus par l’ajout d’un donneur de NO, le GEA 5024, dans les muscles EDL et soléaire de rat
ainsi que dans les cellules L6 suggérant ainsi un rôle pour iNOS dans la régulation du transport du
glucose stimulé par l’insuline dans le muscle squelettique [423]. Par la suite, il fut rapporté que
l’invalidation génétique totale d’iNOS prévient le développement de la résistance à l’insuline
musculaire induite par une diète riche en gras malgré la présence d’obésité. En effet, la délétion
d’iNOS prévient la diminution de l’activité PI3K et de la phosphorylation du résidu Ser473 d’Akt
induite par une diète riche en gras [416]. Ces résultats furent corroborés par des études
subséquentes chez l’animal. La délétion génétique et l’usage d’un inhibiteur pharmacologique
d’iNOS, le L-NIL, améliorent le signal insulinique dans le muscle gastrocnémien de souris ob/ob
[430]. De plus, l’invalidation génétique totale d’iNOS prévient la diminution du transport du glucose,
de l’activité de la PI3K ainsi que de la phosphorylation du résidu Ser473 d’Akt dans le muscle
gastrocnémien de souris soumises à une infusion de lipides en dépit des niveaux élevés en cytokines
[433].
Fait intéressant, il fut rapporté que l’inhibition d’iNOS par des activateurs pharmacologiques de
l’AMPK prévient également la diminution de l’activité PI3K par le cocktail LPS/cytokines dans les
cellules L6 [424]. En contrepartie, une étude récente a rapporté que l’activation de l’AMPK par
l’AICAR lors d’une infusion de lipides ne réduit pas l’expression d’iNOS et n’améliore pas la

52
sensibilité à l’insuline systémique. Néanmoins, une forte expression d’iNOS fut observée dans le
muscle gastrocnémien de la souris AMPK KD1 en condition basale [434].
Il fut suggéré que les modifications nitrosatives de la voie de signalisation de l’insuline par le NO
produit par iNOS expliqueraient la résistance à l’insuline musculaire observée.
4.3.1 Nitration
La nitration résulte en une liaison covalente entre un ou plusieurs groupements nitro (NO2)
triatomiques à l’anneau phénol des résidus de tyrosine [435, 436]. Toutefois, il demeure un
processus sélectif et seul un nombre restreint de résidus de tyrosine sont sujets à la nitration [437]. Il
fut démontré que non seulement la nitration survient à la suite de l’interaction entre des protéines
avec le NO, mais également à la suite d’interactions entre les protéines et les espèces réactives de
l’azote (de l’anglais reactive nitrogen species, RNS), des intermédiaires secondaires du NO.
Néanmoins, le NO ne semble pas un joueur majeur dans le processus de nitration. Le peroxynitrite
(ONOO-) représente un puissant oxydant capable de nitration. Le ONOO- résulte de l’interaction
entre le NO et l’anion superoxide (O2-) [438].
Des concentrations élevées en nitrotyrosines furent rapportées dans le plasma et le muscle
squelettique d’individus atteints de diabètes de type 2 suggérant une production accrue de ONOO-
chez ces derniers [431, 439]. Des résultats similaires furent observés dans le muscle soléaire de
souris soumises à une diète riche en gras. Il fut démontré que l’administration de FeTTPS, un agent
qui provoque la dégradation du ONOO-, prévient l’augmentation du contenu musculaire en
nitrotyrosines ainsi que la diminution de la phosphorylation du résidu Ser347 d’Akt et du transport du
glucose stimulé par l’insuline dans le muscle soléaire de souris soumises à une diète riche en gras
[440]. De plus, une étude subséquente a démontré que l’administration intrapéritonéale d’un donneur
de ONOO-, le 3-morpholinosydnonimine hydrochloride (SIN-1), résulte en la nitration de l’IRβ, d’IRS-
1 et d’Akt dans le muscle squelettique de souris. La nitration de l’IRβ et d’IRS-1 fut également
observée dans le muscle squelettique des souris soumises à une diète riche en gras.
L’administration de FeTTPS renverse la nitration de ces protéines [441]. Finalement, il fut rapporté
que l’endotoxine LPS induit la nitration d’IRS-1 dans les cellules L6. L’utilisation d’un inhibiteur
d’iNOS, le 1400W, prévient cette induction [442]. L’ensemble de ces résultats suggèrent qu’iNOS

53
contribue, entre autre chose, à la résistance à l’insuline musculaire via la nitration des protéines
impliquées dans la voie de signalisation de l’insuline.
4.3.2 S-nitrosylation
La S-nitrosylation résulte en une liaison covalente entre un composé nitroso diatomique et le
groupement thiol-SH (groupement sulfhydryle) d’une cystéine à la suite d’une réaction
d’oxydoréduction [443]. À l’instar de la nitration, la S-nitrosylation demeure restreinte à un nombre
sélectif de cystéines [444]. Les trioxydes d’azote (N2O3) générés à la suite de la réaction entre deux
atomes d’azote avec l’oxygène constituent les RNS capables de S-nitrosylation [445].
Une induction de la S-nitrosylation d’Akt fut observée dans le muscle gastrocnémien de souris
génétiquement diabétiques (db/db) comparativement aux souris contrôles. La S-nitrosylation d’Akt
est corrélée à une diminution de la phosphorylation du résidu Thr308 ainsi qu’à une diminution de
l’activité d’Akt [446]. De plus, la S-nitrosylation de l’IRβ, d’IRS-1 et d’Akt fut également rapportée
dans le muscle soléaire de souris ob/ob et de rats soumis à une diète riche en gras. L’inhibition
d’iNOS par des oligonucléotides renverse la S-nitrosylation de ces protéines dans le muscle soléaire
des souri ob/ob de même qu’un traitement à la rosiglitazone chez le rat soumis à une diète riche en
gras [429]. Des résultats similaires furent obtenus en réponse à un traitement avec l’endotoxine LPS.
En effet, le LPS induit également la S-nitrosylation de l’IRβ, d’IRS-1 et d’Akt dans le muscle soléaire
de souris. L’invalidation génétique d’iNOS prévient la S-nitrosylation de ces protéines [447]. Ces
résultats suggèrent qu’iNOS contribue également à la résistance à l’insuline musculaire via la S-
nitrosylation des protéines impliquées dans la voie de signalisation de l’insuline. La figure 11 résume
les modifications nitrosatives de la voie de signalisation de l'insuline.
Fait intéressant, ces mêmes auteurs ont rapporté une diminution de la S-nitrosylation de l’IRβ, d’IRS-
1 et d’Akt dans le muscle gastrocnémien de rats induite par une diète riche en gras à la suite d’un
protocole d’exercice aigu. Cette diminution de la S-nitrosylation des protéines impliquées dans la voie
de signalisation de l’insuline est corrélée à une augmentation de la sensibilité à l’insuline systémique,
une diminution d’iNOS et une activation de l’AMPK dans le muscle gastrocnémien [448].

54
Figure 11 Modifications nitrosatives de la voie de signalisation de l'insuline, tirée et modifiée de [449]
Y-NO2: Nitration; S-NO: S-Nitrolysation
5 Objectifs de recherche Que ce soit à la suite d’un repas (augmentation des niveaux plasmatique d’insuline) ou lors d’un
exercice physique (contraction musculaire), le tissu musculaire squelettique capte de grandes
quantités de glucose grâce à la translocation des GUT4 à la membrane plasmique et aux tubules-T.
Les mécanismes moléculaires responsables de la translocation des GLUT4 à la membrane
plasmique de la cellule musculaire sont bien définis lors d’une stimulation à l’insuline, mais ceux
impliqués dans la régulation du transport du glucose induit par la contraction musculaire restent à
être élucidés.
Certaines évidences tendent à démontrer l’implication de la protéine AMPK dans le transport du
glucose induit par la contraction musculaire. Toutefois, encore aujourd’hui, son rôle demeure
controversé. La diminution de la force absolue observée au niveau des muscles squelettiques des
différents modèles transgéniques pour la protéine AMPK comparativement aux souris contrôles rend
laborieuse l’interprétation des résultats obtenus. Puisque le transport du glucose est proportionnel au
niveau de force développé par le muscle squelettique, nous nous sommes penchés sur l’élaboration
d’un protocole de stimulation électrique (contraction ex vivo) qui permettrait de produire des forces
semblables entre les souris AMPK KD1 et les souris contrôles pour un même stimulus. L’élaboration

55
de ce protocole nous a permis de clarifier le rôle de l’AMPK dans le transport du glucose induit par la
contraction musculaire.
Notre laboratoire s’intéresse également à la modulation du transport du glucose dans la cellule
musculaire par le NO produit par la protéine iNOS. En effet, nous avons démontré que l’induction
d’iNOS et par conséquent, la production de NO par un cocktail de cytokines combiné à la LPS
modulent l’expression de GLUT1 et de GLUT4 et diminuent le transport du glucose stimulé par
l’insuline dans les cellules L6. Des résultats préliminaires obtenus dans le muscle épitrochléen de rat
suggèrent que l’incubation prolongée d’un muscle squelettique induit l’expression d’iNOS. Nous nous
sommes donc intéressés à l’impact de l’induction d’iNOS sur l’expression des transporteurs de
glucose lors d’une incubation prolongée du muscle épitrochléen. Nous nous sommes également
penchés sur l’impact de l’induction d’iNOS sur le transport du glucose lors d’une incubation
prolongée du muscle épitrochléen.


57
CHAPITRE 1
The α subunit of AMPK is essential for submaximal contraction-mediated glucose transport in skeletal muscle in vitro
Natalie Simard-Lefort1,3+, Emmanuelle St-Amand1,3+, Sébastien Morasse1,3, Claude H. Côté2,3, and André Marette1,3 *
Departments of 1Anatomy and Physiology and 2of Rehabilitation, and 3Lipid Research Unit, Laval University Hospital Research Center, Québec, Canada
Running title: AMPK is essential for contraction-induced glucose transport
+ These authors have contributed equally to this work.
*Address correspondence to: Dr. André Marette Department of Physiology & Lipid Research Unit Laval University Hospital Research Center 2705, Laurier Bld Ste-Foy, (Québec), Canada, G1V 4G2 Tel: (418) 656-4141 ext. 47549 Fax: (418) 654-2176 E-mail:[email protected]

58
Résumé
L’AMPK est une sérine/thréonine kinase clé dans la régulation du transport du glucose dans le
muscle squelettique. Toutefois, son implication dans le transport du glucose induit par la contraction
musculaire demeure controversée. Une réduction du transport du glucose induit par la contraction
musculaire fut observée dans le muscle EDL de souris surexprimant une forme inactive de l’isoforme
AMPKα2 (AMPK KD1). Néanmoins, une diminution de la force maximale du muscle EDL fut
également rapportée chez la souris AMPK KD1 évoquant la possibilité que la réduction du transport
du glucose observée chez la souris AMPK KD1 puisse s’expliquer par cette diminution de la force
musculaire. L’étude des courbes force/fréquence lors d’une stimulation électrique du muscle EDL a
permis d’établir que pour des fréquences ≤ 50Hz, la force générée par les souris AMPK KD1 est
comparable à celle des souris sauvages. Par ailleurs, l’activation de l’AMPK est maximale à une
fréquence de 50Hz. Chez la souris sauvage, une stimulation électrique du muscle EDL à une
fréquence de 50Hz sur une période de 2 minutes (200ms, 1/s, 30V) induit une activation de l’AMPK
et une augmentation du transport du glucose de 2 fois supérieur à celui observé en condition basale
chez la souris sauvage. Contrairement à la souris sauvage, aucune activation de l’AMPK n’est
observée dans le muscle EDL des souris AMPK KD1 à la suite du protocole de stimulation électrique.
De plus, le transport du glucose est réduit de 50% comparativement à celui mesuré chez la souris
sauvage. L’ensemble de ces résultats suggère un rôle essentiel de l’AMPK dans le transport du
glucose induit par la contraction musculaire ainsi que l’implication de mécanismes indépendants de
l’AMPK.

59
Abstract
AMPK is a key signaling pathway in the regulation of skeletal muscle glucose uptake but its role in
mediating contraction-induced glucose transport is still debated. The effect of contraction on glucose
transport is impaired in EDL muscle of transgenic mice expressing a kinase-dead, dominant negative
form of the AMPK α2 subunit (KD-α2AMPK mice). However, maximal force production is reduced in
this muscle, raising the possibility that the defect in glucose transport was due to a secondary
decrease in force production and not impaired AMPK α2 activity. Generating force: frequency curves
revealed that muscle force production is matched between wild-type (WT) and KD-α2AMPK mice at
frequencies ≤ 50 Hz. Moreover, AMPK activation is already maximal at 50 Hz in muscles of WT mice.
When EDL muscles from WT mice were stimulated at a frequency of 50 Hz for 2 min (200ms train,
1/s, 30V), contraction caused a ~3.5-fold activation of AMPKα2 activity and a ~2-fold stimulation of
glucose uptake. Conversely, while force production was similar in EDL of KD-α2AMPK animals, no
effect of contraction was observed on AMPKα2 activity and glucose uptake stimulation was reduced
by 50% (P<0.01) As expected, AICAR caused a 2.3-fold stimulation of AMPKα2 activity and a 1.7-
fold increase in glucose uptake in EDL from wild-type (WT) mice while no effect was detected in
muscle from KD-α2AMPK mice. These data demonstrate that AMPK activation is essential for both
AICAR and submaximal contraction-induced glucose transport in skeletal muscle, but that AMPK-
independent mechanisms are also involved.

60
Introduction
5'-AMP-activated protein kinase (AMPK) is a member of a metabolite-sensing protein kinase family
that responds to modulations in cellular energy levels (6, 14). When AMPK "senses" decreased
energy stores, it acts to switch off ATP-consuming pathways and switch on alternative pathways for
ATP regeneration. AMPK is a ubiquitous heterotrimer comprised of a catalytic α, and regulatory β
and γ subunits. It is thought to be a key actor in the alternative, noninsulin-mediated, pathways
leading to glucose uptake in skeletal muscle (13). As AMPK is potently activated by muscular
contraction, it is also believed to represent a key signaling molecule for exercise-induced glucose
transport (15, 28, 29, 35, 38). In rats, exercise predominantly increases the activity of the α2-subunit
of AMPK while tetanic contractions ex vivo increase both the α1 and α2 AMPK subunits (26).
Exercise performed at 65-70% VO2max preferentially stimulates AMPKα2 in human muscle whereas
activation of AMPKα1 at this intensity is still controversial (4, 7, 39). In type 2 diabetic patients,
exercise at 70% VO2max stimulates AMPKα2 activity to an extent similar to that observed in healthy
subjects (25), making AMPK an attractive target for glucose-lowering pharmacological therapies.
Transgenic models have proven essential in elucidating the role of AMPKα2 in the alternative
glucose transport pathway. Mu et al. (24) first showed that muscle-specific expression of a dominant
inhibitory mutant of AMPK in transgenic mouse (KD-α2AMPK) reduced contraction-induced glucose
uptake by 30-40% whereas it fully abrogated the stimulatory effect of AICAR (a pharmacological
activator of AMPK) or hypoxia on glucose transport. Knockout (KO) mouse models for individual
AMPKα subunits were also generated. Whereas α1-AMPK KO mice exhibited normal glucose
homeostasis, mice lacking the α2-AMPK showed impaired insulin secretion and in vivo insulin
resistance (36, 37). However, α2-AMPK KO mice also exhibited increased adrenergic tone which
could explain both decreased insulin secretion and insulin resistance. Surprisingly, isolated muscles
from α2-AMPK KO mice failed to respond to AICAR but exhibited normal response to contraction-
induced glucose uptake (20). However, this is likely explained by a compensatory increase in
expression and activity of the α1-AMPK subunit. This compensatory response of the α1-AMPK
subunit was not observed in muscle-specific expression of a dominant inhibitory mutant of α2-AMPK
(24). Interestingly, γ3-AMPK KO mice also display impaired AICAR- but not contraction-induced
glucose uptake (2). In this case, no compensatory increases in other γ or α-subunits were observed.

61
Thus, there is still some debate as to the exact role of AMPK in contraction or exercise-induced
glucose uptake in skeletal muscle.
A recent report suggests that contraction-mediated glucose transport is fully independent of AMPKα2
since it is not ablated in a second murine model expressing an inactive form of AMPKα2 in skeletal
muscle (α2i TG) (12). This conclusion relies on the premise that when force generation was matched
between muscles of α2i TG mice and their wild-type (WT) littermates, no deficit in contraction-
induced glucose transport was observed. To match force production between WT and α2i TG mice,
Fujii and colleagues opted to use a non supramaximal stimulation voltage (8.4-21V) in the WT group
as compared to a supramaximal voltage (i.e. >30V) to stimulate α2i TG muscles. However, this
strategy has the inherent limitation of leading to differential recruitment in muscle fibers even though
force production is matched between the genotypes. In the present study, a contraction stimulation
protocol was designed to match force production between WT and KD-α2AMPK mice without
compromising the voltage stimulation parameters in the WT mice and resulting in comparable fiber
recruitment in both groups. This allowed us to investigate the role of AMPK in contraction-mediated
glucose transport under conditions of similar fiber recruitment and muscle force responses, and thus
test the hypothesis that AMPK is essential for contraction-induced glucose transport. When force
development between KD-α2AMPK and WT EDL muscles is matched, while using identical
stimulation voltage and contraction protocols, glucose transport is diminished by half in EDL of KD-
α2AMPK mice. Our data are thus consistent with a major role for AMPK in contraction-induced
glucose transport.

62
Experimental procedures
Materials
AICAR was purchased from Toronto Research Chemicals (Toronto, Canada). [-32P] ATP, protein A-
and G-Sepharose, and antimouse or antirabbit immunoglobulin G conjugated to horseradish
peroxidase were purchased from Amersham Pharmacia Biotech (Baie d’Urfé, Canada). SAMS
peptide was synthesized at the Eastern Quebec Proteomics Centre at the CHUL Research Centre.
Protein A/G PLUS-Agarose and anti-AMPKα2 (C-20) were from Santa Cruz Biotechnology (Santa
Cruz, CA). Polyclonal antibodies used for immunoblotting: anti-phospho-AMPKα1/α2 (pThr172), anti-
phospho-ACC recognizing both α and β isoforms (pSer79), an anti-AMPKα1 and anti-AMPKα2
antibody from Cell Signaling (Beverly, MA, USA) and anti-cmyc (9E10) was purchased from Santa
Cruz Biotechnology. All common chemicals were from Sigma and of the highest analytical grade.
Animals
All animal handling and treatments were approved and followed the guidelines set by Laval University
Hospital Research Centre Animal Care and Handling Committee. Mice were housed 2-5 per cage
under a 12-hr light/dark cycle in animal quarters at 22°C and allowed unlimited access to standard
rodent food and water WT and KD-α2AMPK mice (20-25 g) were generated by an established colony
at the CHUL Research Center, from original breeders generously provided by Dr. Morris Birnbaum
(University of Pennsylvania). Food was withdrawn 15-18 hours prior to experiments. Mice were
anesthetized (50mg/kg sodium pentobarbital, i.p.) and were given the analgesic buprenorphine
(0.1mg/kg, i.p.). Extensor digitorum longus (EDL) muscles were carefully dissected and either
mounted vertically between two platinum electrodes for contraction studies or on a plastic support for
AICAR treatments.
In vitro contraction studies
EDLs were incubated in vitro and allowed to equilibrate for 15 min in 29°C oxygenated (95% O2/5%
CO2) Krebs buffer (137mM NaCl, 5mM KCl, 1mM MgSO4, 1mM NaH2PO4, 2mM CaCl2, 24mM
NaHCO3, 8mM glucose and 32mM mannitol) at which time the optimal length (Lo) (the muscle length
which allows maximal twitch tension) was determined using 5 twitches. Muscles were incubated at
this length for an additional 30 min. The force produced was visualized and analyzed on a digital
oscilloscope (Hewlett-Packard) throughout the contraction protocol. A force: frequency curve was

63
obtained on a set of muscles not used for glucose transport studies by using supramaximal voltage
(30-35 volts) and 200ms train stimulations at frequencies ranging between 10 and 300Hz with 2 min
rest in between each contraction. The contraction protocol used for the contraction-stimulated
glucose uptake (30V, 50Hz, 200ms train, 1/s), which is near-physiological conditions (42), was
performed during the last 2 min of the 30 min incubation. For immunoblotting studies and kinase
activity assays, EDLs were frozen in liquid nitrogen immediately at the end of the protocol and stored
at -80°C until processed.
AICAR treatment
EDLs were allowed to recover from the surgery for 30 min in oxygenated Krebs buffer supplemented
with 8mM glucose and 32mM mannitol. EDLs were then incubated 30 min in 2mM AICAR in Krebs
buffer in which the osmolarity was reduced to account for the contribution of AICAR (8mM glucose
and 30mM mannitol). AICAR stock solution was prepared in Krebs buffer lacking glucose and
mannitol and the same volume of this buffer was added to basal muscles. Immediately after the
protocol, EDLs underwent glucose transport measurement or were frozen in liquid nitrogen and
stored at -80°C until processing.
In vitro glucose transport measures
Following the appropriate treatments (i.e. contraction or AICAR), EDLs were mounted on a plastic
support if not previously done. Extracellular glucose was removed during a 10 min wash in Krebs
buffer with 40mM mannitol. Glucose transport was then measured via the incorporation of
radiolabeled [2-[1,2 3H(N)]-deoxyglucose (DG)] during 20 min, as previously described (21). In brief,
the muscles were incubated in 4 ml of Krebs buffer containing 8 mM 2-[3H]-DG (1.125μCi/ml), 32 mM
[14C]mannitol (0.15μCi/ml), 2 mM sodium pyruvate, and 0.1% BSA at 29°C. Muscles were quickly
dried, frozen in liquid nitrogen and kept at -80°C. 2-[3H]-DG incorporation was determined by adding
1 mL of H2O to the muscles, incubating in boiling water for 10 min and centrifuging at 10 000g for 10
min. Aliquots of the supernatant and the radioactive incubation media were read using a Liquid
Scintillation Analyzer (Tri-Carb 2800TR, Perkin-Elmer) for quantification of 3H and 14C. Results were
expressed as µmol glucose/g•h.

64
Kinase assays
AMPK activity. EDLs were ground to a powder and resuspended in lysis buffer (30mM HEPES, pH
7.4, 2.5mM EGTA, 3mM EDTA, 70mM KCl, 0.1% NP-40, 20mM β-glycerophosphate, 20mM NaF,
2mM sodium tetrapyrophosphate, 1mM Na3VO4, 200uM phenylmethylsulfonylfluoride, 1mM
benzamidine, 1:1000 protease inhibitor cocktail (Sigma)). Insoluble material was removed by
centrifugation at 10 000g for 10 min. Protein concentration was determined using the DC Assay (Bio-
Rad Laboratories, Inc., Richmond, CA). 200µg of total protein lysate was added to protein A/G-
Sepharose coupled to anti-AMPKα2 (2µg) or anti- AMPKα1 (1µg) antibodies. Following an overnight
incubation, beads with immunocomplexes were washed extensively: 4X with 500uL lysis buffer with
500mM NaCl and 2X with 500uL reaction buffer (40mM HEPES, pH 7.4, 80mM NaCl, 5mM MgCl2,
1mM DTT). Immunocomplexes were incubated, in the presence of 50uL reaction mix (200uM SAMS
peptide, 100uM ATP, 240uM AMP, 1μCi [-32P] ATP, completed with reaction buffer) 10 min at 37°C
at 850rpm. 30µL of the reaction mix was blotted on p81 Filter paper (Whatman International Ltd,
England). Filter papers were washed 5 x 20 min with 1% phosphoric acid to remove excess
radioactivity not bound to the peptide. Remaining radioactivity on Filter papers was quantified using a
Liquid Scintillation Analyzer (Tri-Carb 2800 TR, Perkin Elmer).
Western blotting
50µg of muscle protein lysate was resolved by 7.5% SDS-PAGE, transferred to a PVDF membrane
and analyzed by western blot. Blots were blocked with 5% non-fat dry milk in 10 mM Tris pH 7.5, 150
mM NaCl, and 0.04% Igepal and 0.02% NP-40, incubated with primary antibody overnight at 4°C (in
5% non-fat milk or 1-3% BSA); and then incubated with a secondary horseradish-conjugated
antibody for 1 hr at room temperature. Immunoreactive bands were visualized by enhanced
chemiluminescence (Immubilon Western, Millipore Corporation, MA, USA) and quantified by
ImageQuant TL software. Relevant primary antibodies used: anti-phospho-AMPKα, (1:1000 dilution),
anti-phospho-ACC (1:1000 dilution), anti-c-Myc (1:500 dilution), anti-AMPKα1 (1:1000), and anti-
AMPKα2 (1:1000).

65
Data analysis
All results are presented as mean ± SEM. Statistical analysis of results was performed either by one
or two-way analysis of variance and the assumptions of these models were systematically tested
each time. Differences were considered to be statistically significant at P<0.05.

66
Results
Lack of AMPKα activation and glucose transport stimulation by AICAR in EDL from KD-
α2AMPK mice.
As shown previously (24), the mutated form of AMPKα2 is tagged with a c-Myc epitope which
contributes to the slower electrophoretic mobility of this isoform on SDS-PAGE (Fig. 1A). As expected
the endogenous α2 subunit was fully replaced by the KD-dominant inhibitory mutant in the KD-
α2AMPK mice. The c-Myc epitope was solely expressed in muscles, as expected from its targeted
expression in this tissue. Analysis of EDL muscles from several animals also revealed that the forced
expression of the KD-dominant inhibitory mutant of α2AMPK also reduced the expression of the
AMPKα1 subunit by 66% (rel. Densit. Units of WT: 0.67±0.16 versus KD: 0.23±0.17, mean±SE, P <
0.05). This is consistent with the observations of Mu et al. (24) who reported that the forced
overexpression of the KD AMPKα2 leads to displacement of the endogenous α1/α2 subunits from the
heterotrimer followed by degradation of the free alpha subunits.
We first assessed the effect of the AMPK pharmacological activator AICAR on EDL muscle of WT
and KD-α2AMPK mice. AICAR induced the phosphorylation of AMPKαThr172 (Fig. 1B and 1C) and
its downstream target ACCSer79 (Fig. 1B and 1D) but these effects were markedly reduced in
muscles from KD-α2AMPK mice. The elevated basal phosphorylation of AMPKα in muscle of KD-
α2AMPK mice has been reported previously (24) and may be considered a compensatory
mechanism attempting to increase the activity of AMPK under fasting conditions. Direct
measurements of AMPK kinase activity confirmed that expression of the kinase-dead form of AMPKα
functions as a dominant inhibitory protein in muscle, blunting the AICAR-induced activation of both
the α1 and α2 AMPK isoforms (Fig. 1E). Incubation of WT EDL muscles with 2 mM AICAR led to a
~1.7-fold increase in glucose transport (P<0.01) but AICAR failed to increase glucose transport in
EDL of KD-α2AMPK mice (Fig. 1F). These experiments confirm that the AMPK pathway is blunted in
muscle of the KD-α2AMPK mouse, and thus makes it an appropriate model for the study of AMPK-
dependent processes in skeletal muscle.
Effects of different stimulation frequencies on muscle force and AMPK activation
In order to test the role of AMPK in contraction-induced glucose transport it is imperative that the
force produced by the WT muscle during electrical stimulation is matched to that of the KD-α2AMPK

67
muscle. As shown in figure 2A, EDL muscles from WT mice have a significantly higher maximal
absolute tetanic force compared to muscles of KD-α2AMPK mice. The deficit in force generation of
the EDL from KD-α2AMPK mice only became significant in the upper end of the force: frequency
curve. At frequencies ≤ 50Hz, absolute force generation of KD-α2AMPK EDL was similar to that of
WT EDL (Fig. 2A). When muscles were electrically stimulated in vitro at 50Hz, no significant
difference was observed in the force generation or muscle fatigability over a 2-min period (Area under
curve : KD-α2AMPK: 94.4±7.2 vs WT: 107.0±7.4, NS) (Fig. 2B). Furthermore, we found that
AMPKαThr172 phosphorylation is already maximally induced at 50Hz (Fig. 3), indicating that this
stimulation frequency is relevant to investigate the role of AMPK signaling in glucose uptake
stimulation by contraction. Therefore, a 2-min 50Hz electrical stimulation protocol was employed in
order to study the contribution of AMPK in contraction-mediated glucose transport under matched
force production.
Lack of AMPKα activity reduces contraction-induced glucose transport in EDL muscles from
KD-α2AMPK mice.
Phosphorylation of both AMPKThr172 and ACCSer79 were increased following submaximal
contraction of WT mouse muscles at 50Hz whereas these responses were markedly blunted in EDL
of KD-α2AMPK mice (Figs. 4A-C). A ~3.5-fold stimulation of AMPKα2 activity was observed in
electrically-stimulated EDL muscles from WT mice, whereas contraction failed to increase AMPKα2
activity in EDL of KD-α2AMPK animals (Fig. 4D). Contraction also enhanced AMPKα1 activity in WT
muscle but this effect was also abrogated in the KD- α2AMPK mice. We next triggered contraction of
EDL muscles from both genotypes (50Hz, 200ms stimulation, 1/s, supramaximal voltage) and
measured glucose uptake stimulation. After 2 min, glucose transport was stimulated 2.1-fold over
basal in WT EDL muscles, whereas it was only stimulated 1.6-fold in muscles of KD-α2AMPK mice
(Fig. 4E). Thus, glucose transport stimulation by contraction was reduced by 50% (P<0.01 vs WT
muscles) in KD-α2AMPK muscles when compared to WT mice.

68
Discussion
Early studies showed that exercise (29) and electrically-stimulated contractions (15, 28, 35, 38)
increase the activity of AMPK in muscle. Moreover, several studies have shown that AICAR
stimulates glucose transport in fast-twitch muscles and that its effect is not additive to that of
contraction (3, 15, 23, 26). Further genetic evidence for a role of AMPK in contraction-induced
glucose uptake came from the work of Mu et al.(24) who showed that expression of a dominant
inhibitory mutant of AMPK in KD-α2AMPK muscle reduced contraction-induced glucose uptake by
30-40% whereas it fully abrogated the stimulatory effect of AICAR or hypoxia on glucose transport.
Using another mouse model expressing an inactive form of AMPKα2 in muscle, Fujii et al. (12)
recently suggested that the stimulatory effect of contraction on glucose transport is fully independent
of AMPKα2. This is based on the finding that when force generation was matched between muscles
of α2i TG mice and their WT littermates, no deficit in contraction-induced glucose transport was
observed. The need to match force production is related to the fact that mouse models lacking an
active α2AMPK have a reduced capacity for maximal force production, a finding we have confirmed
in KD-α2AMPK mice (see Fig. 2). However, the conclusion of Fujii et al. (12) relies on the premise
that contraction-mediated glucose transport is proportional to force development (1, 17) but this is in
fact a matter of debate as early and more recent studies indicate that work load plays little, if any, role
in contraction-mediated glucose transport (16, 33). Of more concern is that Fujii and colleagues
opted to use a non supramaximal voltage (8.4-21V) in the WT group in order to match force
production to the level of the α2i TG animals that were stimulated with a supramaximal voltage (i.e.
>30V). However, during electrical stimulation of isolated muscles ex vivo and particularly in the
context of measuring glucose uptake, it is imperative that the voltage remains supramaximal in order
to ensure activation of all (especially the deepest) muscle fibers (9). Thus the option of reducing the
stimulation voltage in the WT mice would inevitably lead to a lesser recruitment of the deepest
muscle fibers, even though the work load was equal to that of α2i TG muscle.
To circumvent these limitations, we designed a contraction stimulation protocol to match force
production between WT and KD-α2AMPK Tg mice without compromising the voltage stimulation
parameters in the WT mice and thus fiber recruitment remained similar between groups. This was
made possible by stimulating both WT and KD-α2AMPK EDL muscles at a lower frequency (50Hz),
where genotypic differences in absolute force production were no longer observed (Fig. 2). This

69
allowed us to investigate the role of AMPK in contraction-mediated glucose transport under
conditions of equivalent electrical stimulation and muscle force responses. When muscle force
development between genotypes is matched, using identical stimulation voltage and contraction
protocols, glucose transport stimulation is diminished by half in EDL of KD-α2AMPK mice as
compared to WT littermates. Our data are therefore entirely consistent with a significant role for
AMPK in submaximal contraction-induced glucose transport.
AMPK is currently believed to be primarily involved in glucose transport stimulation following high
intensity contraction or strenuous exercise. This is partly based on the observation that AMPK is
absolutely required for hypoxia-induced glucose uptake (24); (32) since oxygen availability may be
reduced during strenuous exercise and intense muscle contraction. However, this dogma is now
challenged by recent data showing that AMPK is activated by low-intensity contraction of muscles in
vitro. Indeed, Toyoda et al. recently reported that the α1-subunit, but not the α2 subunit, of AMPK is
activated by a very low-intensity 2 min electrical stimulus (1-2 Hz) of epitrochlearis muscle (34). Both
isoforms were activated when the frequency was increased to > 5 Hz or when the duration of the
stimulus was > 15 min. Furthermore, Jensen et al. (18) found that both the α1 and α2-subunits of
AMPK can be activated in soleus and EDL muscles using a mild, low-work load tetanic contraction
protocol for 2-10 min where a high frequency (100 Hz) but a low train stimulation (1 s trains every 15
s) is applied. When this paper was under review, Jensen et al. (19) also reported that activation of the
α1 AMPK, but not the α2 AMPK subunit, is necessary for twitch-stimulated (2 Hz, 2 min) glucose
transport in soleus muscle. Our own protocol consisted of a middle-range frequency (50 Hz)
combined with a more intense tetanic stimulation rate (0.2 s trains every s) for 2 min. Whatever the
protocol used in these different studies, the general concept that can be derived from these data is
that both the α1 and α2-subunits of AMPK can be activated by low intensity contraction protocols
(lower tetanic or reduced frequency) and our data in KD-α2AMPK mice further indicate that this
activation is required for at least partial stimulation of glucose transport. These data also suggest that
compensatory activation of the α1 AMPK subunit may have contributed to maintain contraction-
induced glucose transport in muscle of α2 AMPK knockout mice (20). However, in muscle of KD-
α2AMPK mice, the forced expression of the α2AMPK dominant negative inhibitory mutant also
blunted the activation of the α1AMPK subunit by contraction (Fig. 4D), suggesting that the non-

70
abrogated portion of the contraction-induced glucose transport in EDL of KD-α2AMPK mice is not
explained by a compensatory increased activation of the α1AMPK subunit.
Our finding that submaximal contraction-induced glucose uptake is only partially inhibited in muscle
expressing a dominant-negative AMPK is in agreement with earlier work from Mu et al. (24) using a
more intense contraction stimulus in the same model and with the growing concept that AMPK-
independent mechanisms are also required for full stimulation of hexose uptake during contraction.
AMPK-independent mechanisms are also essential for GLUT4 translocation to the muscle cell
surface. Indeed, we previously found that AMPK activation with AICAR increases GLUT4
translocation to the plasma membrane but fails to induce recruitment of the transporter to T-tubules
(22), which represent 60% of the total cell surface area in skeletal muscle fibers (8, 10). Since we
have reported that exercise and contraction stimulate GLUT4 translocation to both the plasma
membrane and the T-tubules (30, 31), it is conceivable that contraction induces GLUT4 mobilization
to the tubular elements through an AMPK-independent mechanism.
The nature of AMPK-independent pathways remains elusive. It was suggested that Ca2+/calmodulin-
dependent protein kinase (CaMK) may represent one such pathway as inhibitors (e.g. KN-93) of this
kinase were reported to reduce contraction-induced glucose uptake during intense electrical
stimulation of fast- and slow-twitch muscles without affecting AMPK activation (40, 41). However, KN-
93 was recently shown to inhibit AMPK activity and glucose uptake stimulation at the onset (i.e. 2
min) of mild tetanic contraction of EDL muscle and further studies strontly suggested that low-
intensity contraction rapidly activates CaMK kinase upstream of AMPK (18). More recently, Raney
and Turcotte (27) also showed that KN-93 inhibits contraction-induced AMPK activity, glucose uptake
as well as lipid uptake and oxidation in perfused rat hindlimb muscles. Thus it is unlikely that CaMK
represents the AMPK-independent pathway that is stimulated by our contraction protocol which is of
short duration (2 min) and of submaximal intensity. Other molecules that have recently emerged as
potential contraction-induced metabolic signals in skeletal muscle are neuregulins, a family of growth
factors belonging to the epidermal growth factor family (5, 11). Guma and colleagues recently
reported that neuregulins are released by a Ca2+-dependent metalloproteinase activity, which
causes phosphorylation of the type I tyrosine kinase ErbB receptors. They further showed that
recombinant active neuregulin-1 (HRG) increases glucose transport in myocytes and rat skeletal

71
muscle. Both HRG and contraction-induced glucose uptake in isolated muscles are blunted by ErbB4
blockade. Importantly, HRG does not increase AMPK activity and AICAR-induced glucose transport
is not affected by ErbB4 blockade, strongly suggesting that neuregulins mediate at least part of
contraction-mediated glucose uptake through an AMPK-independent mechanism. It will be interesting
to investigate in future studies whether the neuregulin/ErbB4 pathway is activated by submaximal
contractions and contributes to glucose uptake stimulation in muscle of KD-α2AMPK mice.
In summary, the present study shows that when force development is matched between electrically-
stimulated EDL muscles from WT and KD-α2AMPK mice, contraction-induced glucose transport is
diminished by half in muscle of KD-α2AMPK mice. These results are thus consistent with a major role
for AMPK in contraction-induced glucose transport in vitro. Our data also support the contribution of
AMPK-independent mechanisms to the stimulation of glucose transport by contraction.
Acknowledgements
This work was supported by a grant from the Canadian Diabetes Association to A.M. A.M. is the
recipient of a National Researcher Award of the “Fonds de la Recherche en Santé du Québec”. N.L.
and E. St-A were supported by studentships from the National Sciences and Engineering Research
Council of Canada and Association Diabète Québec. We thank Dr. Morrie Birnbaum for generously
providing KD-AMPKα2 mouse breeders.

72
References
1. Aslesen R, Engebretsen EM, Franch J, and Jensen J. Glucose uptake and metabolic stress in rat muscles stimulated electrically with different protocols. J Appl Physiol 91: 1237-1244, 2001. 2. Barnes BR, Marklund S, Steiler TL, Walter M, Hjalm G, Amarger V, Mahlapuu M, Leng Y, Johansson C, Galuska D, Lindgren K, Abrink M, Stapleton D, Zierath JR, and Andersson L. The 5'-AMP-activated protein kinase gamma3 isoform has a key role in carbohydrate and lipid metabolism in glycolytic skeletal muscle. The Journal of biological chemistry 279: 38441-38447, 2004. 3. Bergeron R, Russell RR, 3rd, Young LH, Ren JM, Marcucci M, Lee A, and Shulman GI. Effect of AMPK activation on muscle glucose metabolism in conscious rats. The American journal of physiology 276: E938-944, 1999. 4. Birk JB, and Wojtaszewski JF. Predominant alpha2/beta2/gamma3 AMPK activation during exercise in human skeletal muscle. The Journal of physiology 577: 1021-1032, 2006. 5. Canto C, Chibalin AV, Barnes BR, Glund S, Suarez E, Ryder JW, Palacin M, Zierath JR, Zorzano A, and Guma A. Neuregulins mediate calcium-induced glucose transport during muscle contraction. The Journal of biological chemistry 281: 21690-21697, 2006. 6. Carling D. The AMP-activated protein kinase cascade--a unifying system for energy control. Trends in biochemical sciences 29: 18-24, 2004. 7. Chen ZP, McConell GK, Michell BJ, Snow RJ, Canny BJ, and Kemp BE. AMPK signaling in contracting human skeletal muscle: acetyl-CoA carboxylase and NO synthase phosphorylation. American journal of physiology 279: E1202-1206, 2000. 8. Cullen MJ, Hollingworth S, and Marshall MW. A comparative study of the transverse tubular system of the rat extensor digitorum longus and soleus muscles. Journal of anatomy 138 ( Pt 2): 297-308, 1984. 9. Egginton S, and Hudlicka O. Selective long-term electrical stimulation of fast glycolytic fibres increases capillary supply but not oxidative enzyme activity in rat skeletal muscles. Experimental physiology 85: 567-573, 2000. 10. Eisenberg BR. Quantitative ultrastructure of mammalian skeletal muscle. In: Handbook of Physiology: Skeletal Muscle. Bethesda, MD: Am Physiol Soc, 1983, p. 73-112. 11. Falls DL. Neuregulins: functions, forms, and signaling strategies. Experimental cell research 284: 14-30, 2003. 12. Fujii N, Hirshman MF, Kane EM, Ho RC, Peter LE, Seifert MM, and Goodyear LJ. AMP-activated protein kinase alpha2 activity is not essential for contraction- and hyperosmolarity-induced glucose transport in skeletal muscle. The Journal of biological chemistry 280: 39033-39041, 2005. 13. Fujii N, Jessen N, and Goodyear LJ. AMP-activated protein kinase and the regulation of glucose transport. American journal of physiology 291: E867-877, 2006. 14. Hardie DG. AMP-activated protein kinase: a key system mediating metabolic responses to exercise. Medicine and science in sports and exercise 36: 28-34, 2004. 15. Hayashi T, Hirshman MF, Kurth EJ, Winder WW, and Goodyear LJ. Evidence for 5' AMP-activated protein kinase mediation of the effect of muscle contraction on glucose transport. Diabetes 47: 1369-1373, 1998. 16. Holloszy JO, and Narahara HT. Studies of tissue permeability. X. Changes in permeability to 3-methylglucose associated with contraction of isolated frog muscle. The Journal of biological chemistry 240: 3493-3500, 1965. 17. Ihlemann J, Ploug T, and Galbo H. Effect of force development on contraction induced glucose transport in fast twitch rat muscle. Acta physiologica Scandinavica 171: 439-444, 2001.

73
18. Jensen TE, Rose AJ, Jorgensen SB, Brandt N, Schjerling P, Wojtaszewski JF, and Richter EA. Possible CaMKK-dependent regulation of AMPK phosphorylation and glucose uptake at the onset of mild tetanic skeletal muscle contraction. American journal of physiology 292: E1308-1317, 2007. 19. Jensen TE, Schjerling P, Viollet B, Wojtaszewski JF, and Richter EA. AMPK alpha1 activation is required for stimulation of glucose uptake by twitch contraction, but not by H2O2, in mouse skeletal muscle. PLoS ONE 3: e2102, 2008. 20. Jorgensen SB, Viollet B, Andreelli F, Frosig C, Birk JB, Schjerling P, Vaulont S, Richter EA, and Wojtaszewski JF. Knockout of the alpha2 but not alpha1 5'-AMP-activated protein kinase isoform abolishes 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranosidebut not contraction-induced glucose uptake in skeletal muscle. The Journal of biological chemistry 279: 1070-1079, 2004. 21. Kapur S, Bedard S, Marcotte B, Cote CH, and Marette A. Expression of nitric oxide synthase in skeletal muscle: a novel role for nitric oxide as a modulator of insulin action. Diabetes 46: 1691-1700, 1997. 22. Lemieux K, Konrad D, Klip A, and Marette A. The AMP-activated protein kinase activator AICAR does not induce GLUT4 translocation to transverse tubules but stimulates glucose uptake and p38 mitogen-activated protein kinases alpha and beta in skeletal muscle. Faseb J 17: 1658-1665, 2003. 23. Merrill GF, Kurth EJ, Hardie DG, and Winder WW. AICA riboside increases AMP-activated protein kinase, fatty acid oxidation, and glucose uptake in rat muscle. The American journal of physiology 273: E1107-1112, 1997. 24. Mu J, Brozinick JT, Jr., Valladares O, Bucan M, and Birnbaum MJ. A role for AMP-activated protein kinase in contraction- and hypoxia-regulated glucose transport in skeletal muscle. Molecular cell 7: 1085-1094, 2001. 25. Musi N, Fujii N, Hirshman MF, Ekberg I, Froberg S, Ljungqvist O, Thorell A, and Goodyear LJ. AMP-activated protein kinase (AMPK) is activated in muscle of subjects with type 2 diabetes during exercise. Diabetes 50: 921-927, 2001. 26. Musi N, Hayashi T, Fujii N, Hirshman MF, Witters LA, and Goodyear LJ. AMP-activated protein kinase activity and glucose uptake in rat skeletal muscle. American journal of physiology 280: E677-684, 2001. 27. Raney MA, and Turcotte LP. Evidence for the involvement of CaMKII and AMPK in Ca2+-dependent signaling pathways regulating FA uptake and oxidation in contracting rodent muscle. J Appl Physiol 104: 1366-1373, 2008. 28. Rasmussen BB, Hancock CR, and Winder WW. Postexercise recovery of skeletal muscle malonyl-CoA, acetyl-CoA carboxylase, and AMP-activated protein kinase. J Appl Physiol 85: 1629-1634, 1998. 29. Rasmussen BB, and Winder WW. Effect of exercise intensity on skeletal muscle malonyl-CoA and acetyl-CoA carboxylase. J Appl Physiol 83: 1104-1109, 1997. 30. Roy D, Johannsson E, Bonen A, and Marette A. Electrical stimulation induces fiber type-specific translocation of GLUT-4 to T tubules in skeletal muscle. The American journal of physiology 273: E688-694, 1997. 31. Roy D, and Marette A. Exercise induces the translocation of GLUT4 to transverse tubules from an intracellular pool in rat skeletal muscle. Biochemical and biophysical research communications 223: 147-152, 1996. 32. Russell RR, 3rd, Li J, Coven DL, Pypaert M, Zechner C, Palmeri M, Giordano FJ, Mu J, Birnbaum MJ, and Young LH. AMP-activated protein kinase mediates ischemic glucose uptake and

74
prevents postischemic cardiac dysfunction, apoptosis, and injury. The Journal of clinical investigation 114: 495-503, 2004. 33. Sandstrom ME, Zhang SJ, Westerblad H, and Katz A. Mechanical load plays little role in contraction-mediated glucose transport in mouse skeletal muscle. The Journal of physiology 579: 527-534, 2007. 34. Toyoda T, Tanaka S, Ebihara K, Masuzaki H, Hosoda K, Sato K, Fushiki T, Nakao K, and Hayashi T. Low-intensity contraction activates the alpha1-isoform of 5'-AMP-activated protein kinase in rat skeletal muscle. American journal of physiology 290: E583-590, 2006. 35. Vavvas D, Apazidis A, Saha AK, Gamble J, Patel A, Kemp BE, Witters LA, and Ruderman NB. Contraction-induced changes in acetyl-CoA carboxylase and 5'-AMP-activated kinase in skeletal muscle. The Journal of biological chemistry 272: 13255-13261, 1997. 36. Viollet B, Andreelli F, Jorgensen SB, Perrin C, Flamez D, Mu J, Wojtaszewski JF, Schuit FC, Birnbaum M, Richter E, Burcelin R, and Vaulont S. Physiological role of AMP-activated protein kinase (AMPK): insights from knockout mouse models. Biochemical Society transactions 31: 216-219, 2003. 37. Viollet B, Andreelli F, Jorgensen SB, Perrin C, Geloen A, Flamez D, Mu J, Lenzner C, Baud O, Bennoun M, Gomas E, Nicolas G, Wojtaszewski JF, Kahn A, Carling D, Schuit FC, Birnbaum MJ, Richter EA, Burcelin R, and Vaulont S. The AMP-activated protein kinase alpha2 catalytic subunit controls whole-body insulin sensitivity. The Journal of clinical investigation 111: 91-98, 2003. 38. Winder WW, and Hardie DG. Inactivation of acetyl-CoA carboxylase and activation of AMP-activated protein kinase in muscle during exercise. The American journal of physiology 270: E299-304, 1996. 39. Wojtaszewski JF, Nielsen P, Hansen BF, Richter EA, and Kiens B. Isoform-specific and exercise intensity-dependent activation of 5'-AMP-activated protein kinase in human skeletal muscle. The Journal of physiology 528 Pt 1: 221-226, 2000. 40. Wright DC, Geiger PC, Holloszy JO, and Han DH. Contraction- and hypoxia-stimulated glucose transport is mediated by a Ca2+-dependent mechanism in slow-twitch rat soleus muscle. American journal of physiology 288: E1062-1066, 2005. 41. Wright DC, Hucker KA, Holloszy JO, and Han DH. Ca2+ and AMPK both mediate stimulation of glucose transport by muscle contractions. Diabetes 53: 330-335, 2004. 42. Zhang SJ, Andersson DC, Sandstrom ME, Westerblad H, and Katz A. Cross bridges account for only 20% of total ATP consumption during submaximal isometric contraction in mouse fast-twitch skeletal muscle. Am J Physiol Cell Physiol 291: C147-154, 2006.

75
Legends to figures
Figure 1. Lack of AMPKα2 activation and glucose transport stimulation by AICAR in EDL of
AMPK-KD mice. (A) Western blots for AMPKα1, α2 and c-Myc expression in different tissues (heart
[H], adipose tissue [A], liver [L], gastrocnemius [G], tibialis [T], soleus [S] and EDL [E]) of wild-type
(WT) and muscle-specific AMPKα2 kinase-dead (KD) Tg mice. Results are representative of 5
mice/group. The AMPKα2 transgene contains a cmyc epitope tag, contributing to the slower
electrophoretic migration of AMPKα2 in KD mice. (B) Representative western blots for
pAMPKαThr172 and pACCSer79 levels. Equal loading for western blots was verified by α-tubulin
content in each lane. (C-F) Isolated EDL muscles from WT(■) and AMPKα2-KD Tg(□) were
stimulated for 30 min with 2mM AICAR. (C-D) Quantification of pAMPKαThr172 and pACCSer79
western blots, n= 6-8 mice/group, (E) AICAR-stimulated AMPKα1 or α2 kinase activity in muscle
lysates, n=3-7 mice/group. (F) AICAR-stimulated glucose transport, n=5 mice/group. Results are
presented as mean ± SEM. *P<0.05, **P<0.01 versus basal in same genotype; §P<0.5, §§P<0.01
versus EDL from WT mice in same condition. nd : non-detectable
Figure 2. Force production and fatigability of EDL muscles of WT and KD-AMPKα2 mice. (A)
Force generation-frequency curves. Muscles were allowed to recover 15 min at which time the Lo
was determined by delivering 5 twitches. Muscles were allowed to rest for 30 min. The force
generated at increasing frequency stimulation was measured every 2 min. §P<0.05, §§P<0.01 versus
EDL from WT mice in same condition. (B) Electrical stimulation was performed during the last 2 min
of a 30 min incubation. Force generation was measured every fourth contraction during a 50Hz
(200ms train, 30V and 1/s rate), 2 min electrical stimulation in WT (●) and KD-AMPKα2 (○) EDL
muscles. The areas under the curve for WT (107.0±7.4) and KD-AMPKα2 (94.4±7.2) were not
significantly different (NS). No significant difference was observed in the Lo between WT and KD
EDL muscles (data not shown). Results presented as the mean ± SEM from n=8-9 mice/group.
Figure 3. Effects of different stimulation frequencies on AMPKα phosphorylation in EDL from
WT mice. Representative western blots and quantification for pAMPKαThr172 are shown. Muscles
were allowed to recover 15 min at which time the Lo was determined by delivering 5 twitches.

76
Electrical stimulation was performed during the last 2 min of a 30 min incubation at frequencies of 20,
40, 50 and 100 Hz. Results are presented as mean ± SEM of n=6 mice/group. *P<0.05 versus basal.
Figure 4. Lack of AMPKα2 activity reduces contraction-mediated glucose transport in EDL
from KD-AMPKα2 mice. EDLs were stimulated to contract for 2-min at 50Hz. The electrical
stimulation was performed during the last 2 min of a 30 min incubation as described in methods and
muscles were used for the following analyses. (A) Representative blots for pAMPKαThr172 and
pACCSer79 levels as detected by immunoblotting. Equal loading was verified by α-tubulin content in
each lane. (B-C) Quantification of pAMPKαThr172 and pACCSer79 immunoblots, n=4 mice/group.
*P<0.05 versus basal in same genotype. (D) Contraction-mediated AMPKα1 and α2 kinase activity,
n=5-11 mice/group. (E) Contraction-stimulated glucose transport in WT and KD-AMPKα2 EDL
muscles. Glucose transport was measured as described in Methods. n=8-9 mice/group. Results are
presented as mean ± SEM. *P<0.05, **P<0.01, ***P<0.001 versus basal condition from same
genotype, §P<0.05, §§P<0.01 versus WT mice in same condition. nd: non-detectable
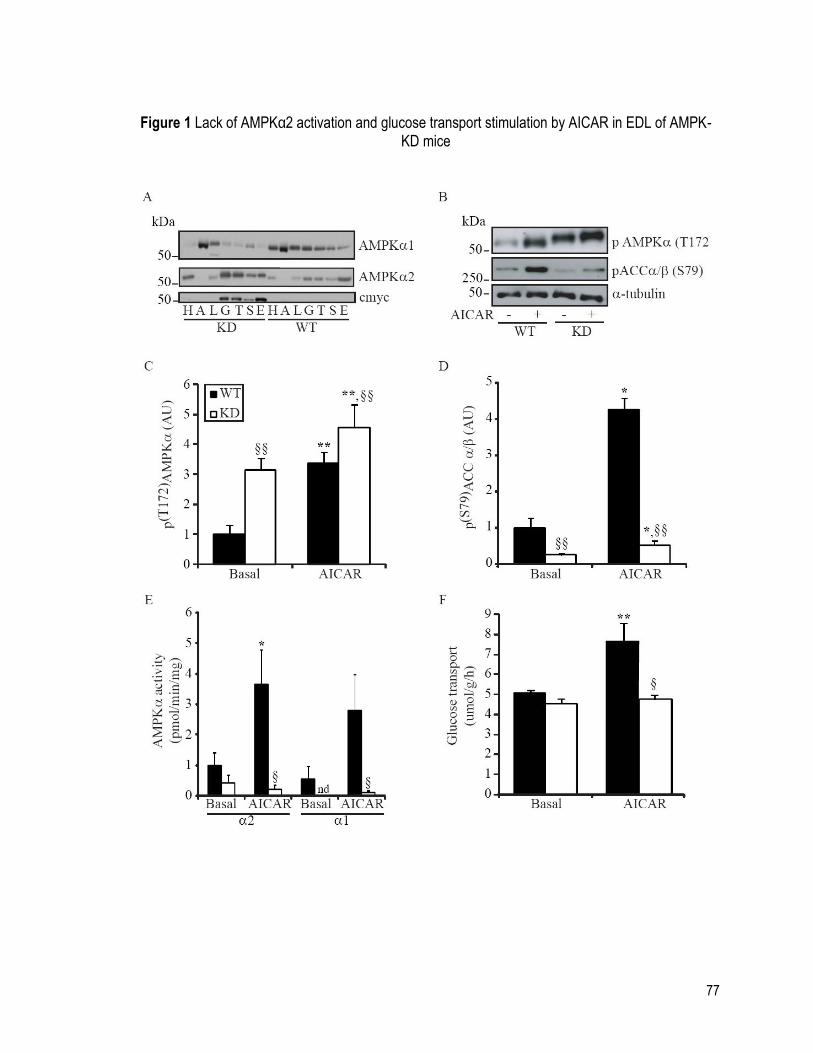
77
Figure 1 Lack of AMPKα2 activation and glucose transport stimulation by AICAR in EDL of AMPK-KD mice
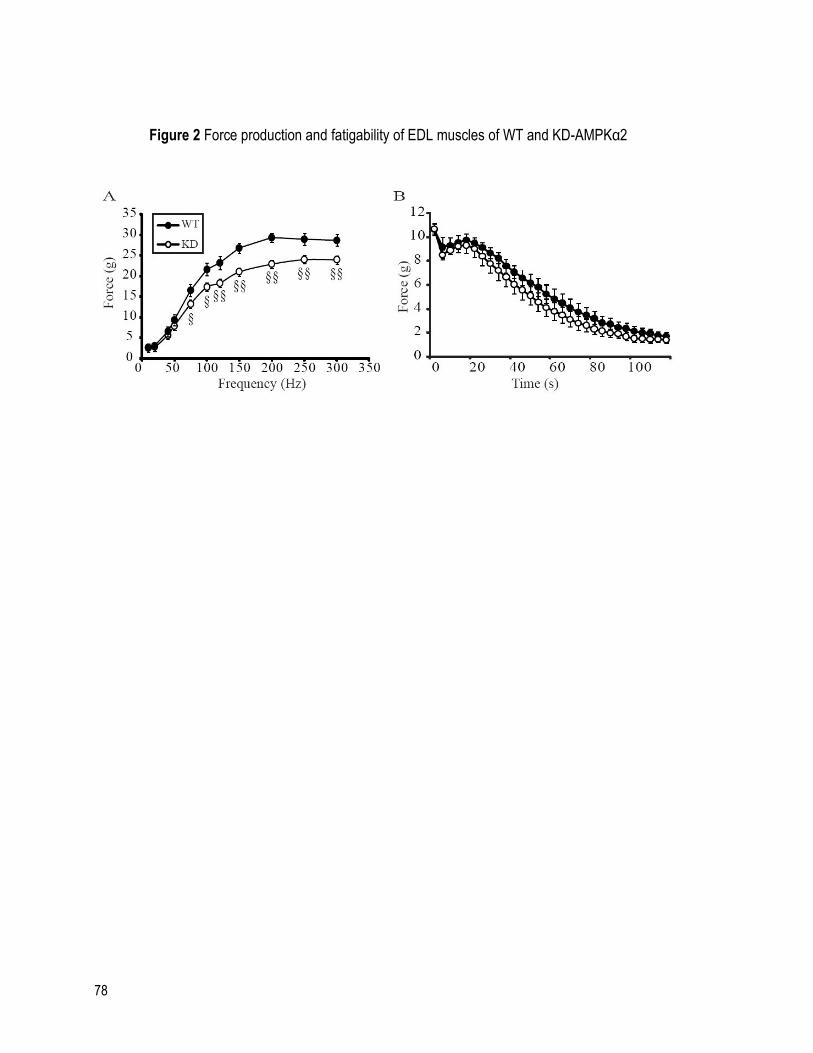
78
Figure 2 Force production and fatigability of EDL muscles of WT and KD-AMPKα2
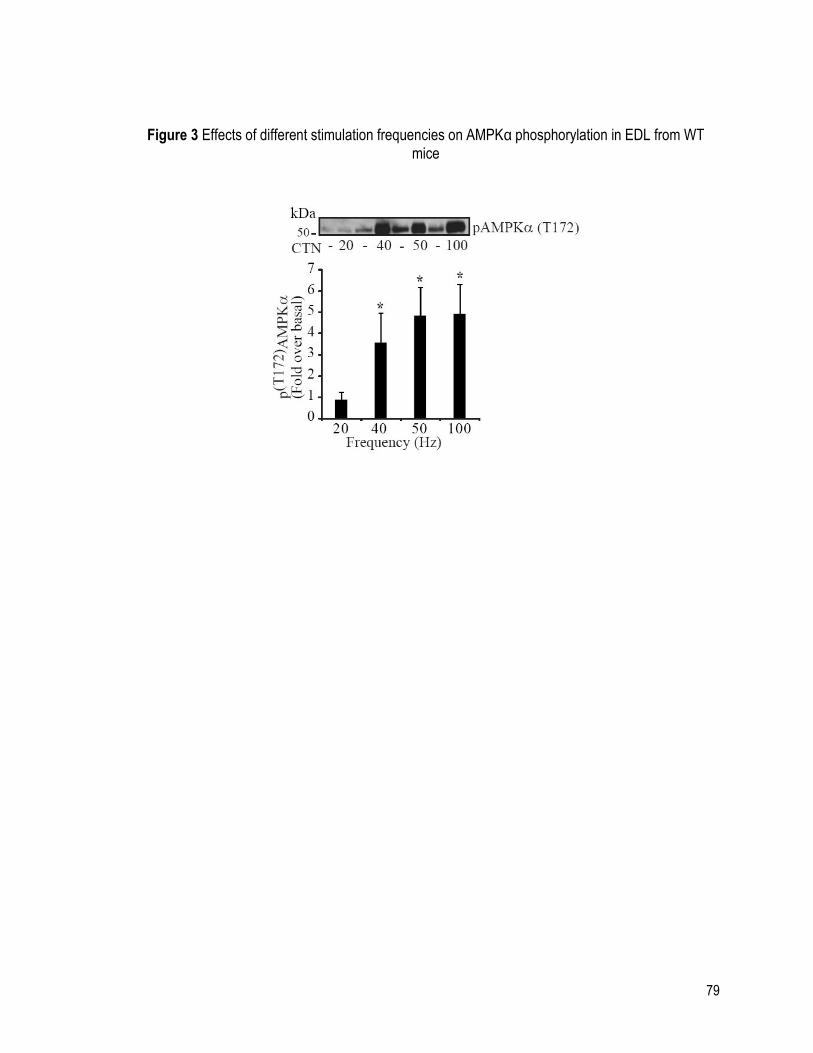
79
Figure 3 Effects of different stimulation frequencies on AMPKα phosphorylation in EDL from WT mice
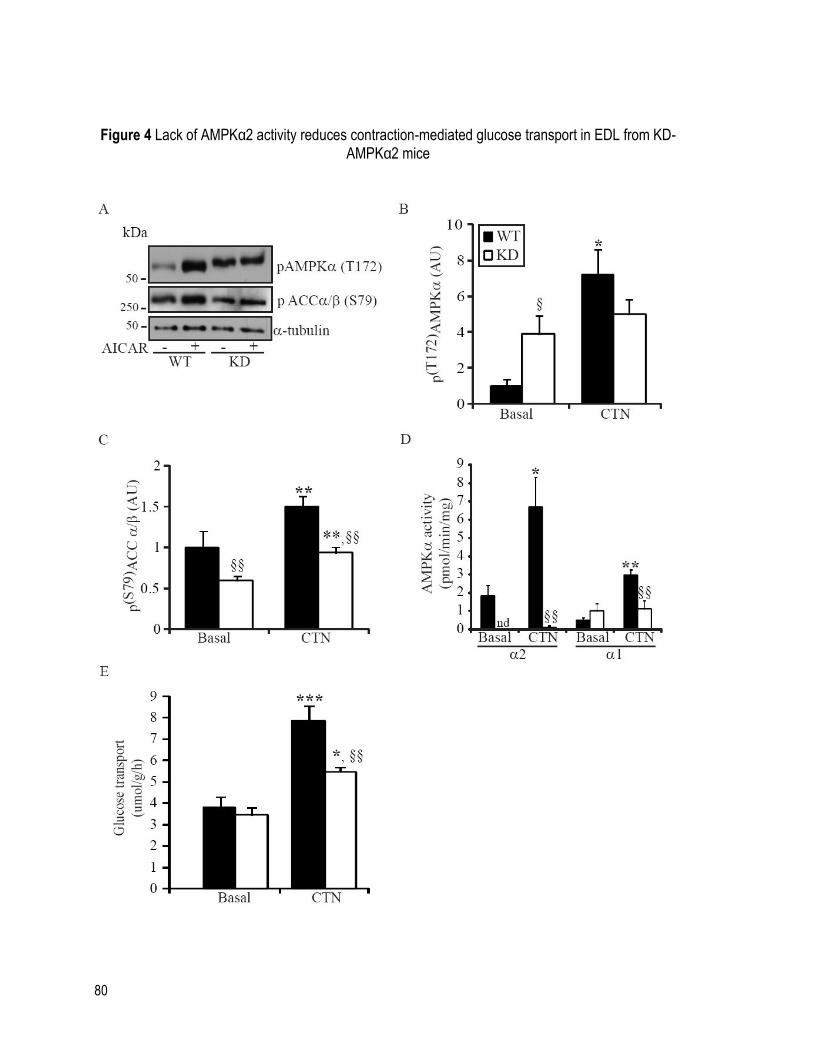
80
Figure 4 Lack of AMPKα2 activity reduces contraction-mediated glucose transport in EDL from KD-AMPKα2 mice

81
CHAPITRE 2
Long-term incubation of epitrochlearis muscle causes iNOS induction and nitric oxide production:
potential impact on muscle glucose transport
Emmanuelle St-Amand, Geneviève Pilon, Phillip J. White, Kathleen Lemieux, and André Marette*
Department of Medicine, Quebec Heart and Lung Institute (Laval Hospital), Pavillon Marguerite
d'Youville, Ste-Foy, Québec, Canada, G1V 4G5, and Laval University Hospital Research Center,
Metabolism, Vascular and Renal Health Axis, Quebec,
Canada G1V 4G2
* To whom correspondence should be addressed at: The Quebec Heart and Lung Institute Hôpital Laval, Pavillon Marguerite d'Youville, Bureau Y4340 Ste-Foy, Québec, Canada, G1V 4G5 Tel 418-656-8711 (ext. 3781) Fax 418-656-4749 Email: [email protected]

82
Résumé
L’incubation ex vivo de différents muscles squelettiques est une technique régulièrement utilisée
dans de nombreuses études. Toutefois, il fut rapporté que l’incubation prolongée du muscle
épitrochléen de rat altère le transport du glucose. Les mécanismes responsables de ces
perturbations métaboliques ne sont pas bien définis. Il est possible que l’incubation prolongée ex vivo
induise un stress et par conséquent, la production de facteurs inflammatoires dont la forme inductible
de monoxyde d’azote synthase (iNOS). En effet, l’induction de la protéine iNOS est connue pour
altérer le métabolisme du glucose dans le muscle squelettique. Les résultats de cette étude nous ont
permis d’observer qu’effectivement, dix heures d’incubation ex vivo du muscle épitrochléen de rat
entraînent une augmentation de l’expression de l’ARNm et de la protéine iNOS ainsi qu’une
importante production de NO. L’induction d’iNOS est également associée à une augmentation de
l’expression des transporteurs de glucose GLUT1. La présence d’un inhibiteur pharmacologique
d’iNOS, le 1400W, dans le milieu d’incubation prévient la surexpression des GLUT1. De plus,
l’utilisation d’un donneur de NO, le NOC-15, dans des cellules musculaires L6 a permis de confirmer
le lien causal entre la production de NO et l’augmentation de l’expression des GLUT1. Afin d’évaluer
les altérations métaboliques causées par l’induction d’iNOS dans le muscle épitrochléen lors d’une
incubation prolongée, nous avons mesuré le transport basal de glucose et celui stimulé à l’insuline
après une période de 10 heures. Aucune altération du transport basal du glucose ni de celui stimulé
par l’insuline ne fut observée à la suite de 10 heures d’incubation ex vivo du muscle épitrochléen.
Toutefois, le traitement de cellules musculaires L6 pendant 24 heures avec le NOC-15 résulte en une
augmentation de la captation du glucose en condition basale et par conséquent, une diminution du
transport du glucose stimulé par l’insuline. L’ensemble de ces résultats suggèrent que l’incubation
prolongée ex vivo d’un muscle squelettique entraîne la production de facteurs inflammatoires, qui
pourraient potentiellement participer au développement de perturbations métaboliques.

83
Abstract
Long-term incubation of isolated skeletal muscle has been reported to alter the regulation of glucose
uptake but the underlying mechanisms remain unclear. We hypothesized that stress associated with
long-term skeletal muscle incubation might trigger inducible nitric oxide (NO) synthase (iNOS) that
could impact upon both basal and insulin-stimulated glucose metabolism. Here we observed that
prolonged (10 hours) epitrochlearis muscle incubation leads to iNOS mRNA and protein induction, as
well as NO production in the incubation medium. These changes were associated with increased
iNOS activity and reduced constitutive NOS (cNOS) activity. iNOS induction in the incubation model
was associated with an increase in glucose transporter 1 (GLUT1) expression that was prevented by
the presence of a specific iNOS inhibitor (1400W) in the incubation medium. Importantly, these data
were upheld in L6 skeletal muscle cells where the NO donor NOC-15 also increased GLUT1
expression. We investigated whether iNOS induction alters glucose transport by measuring 2-
deoxyglucose (2-DG) uptake in epitrochlearis muscle following the 30 minute or 10 hour incubation
periods. Unexpectedly, long-term muscle incubation did not impair either basal or insulin-stimulated
glucose transport despite iNOS induction. However, 24 hours NOC-15 treatment in L6 skeletal
muscle cells did result in a significant dose dependent increase in basal glucose transport and
decreased of insulin stimulated glucose transport. Taken together, these results suggest that long-
term skeletal muscle incubation causes iNOS induction and leads to increased GLUT1 expression.
Induction of this pro inflammatory marker could potentially impact metabolic pathways during long
term muscle incubation. These findings provide futher support for reports of abnormal skeletal muscle
responses during long-term incubation and call for caution when using this technique for metabolic
studies.

84
Introduction
The isolated skeletal muscle preparation is a useful technique that permits the study of skeletal
muscle metabolism in a highly controlled environment ex vivo (6). This technique is widely employed
for the study of skeletal muscle responses to hormones and metabolic substrates and has played an
instrumental role in the development of our understanding of the etiology of skeletal muscle insulin
resistance. The utility of this technique for such studies relies on the premise that the viability of
skeletal muscle preparations can be maintained throughout the duration of the experimental
procedure with minimal impact on metabolic pathways. Indeed, Alkhateeb et al. have shown that the
viability of isolated soleus muscle can be maintained for 18h ex vivo without altering the expression of
the glucose transporter, GLUT4, and the fatty acid transporters FAT/CD36 and FABPpm (2). Despite
these findings, it has also been reported that long term skeletal muscle incubation may perturb
glucose metabolism by increasing basal glucose transport. However, the mechanism underlying this
effect of muscle incubation is poorly understood (14). Since such a metabolic perturbation may limit
the interpretation of findings derived from studies using this widely employed technique it is important
to fully understand the contributing mechanisms.
The radical gas nitric oxide (NO) is an essential player in an ever–growing number of cellular
processes. This diffusible and reactive molecular messenger is fundamental for the regulation of
vasodilatation, neurotransmission and immune responses. The synthesis of NO is generated by the
oxidation of nitrogen atoms from L-arginine by at least three isoforms of nitric oxide synthase (NOS):
the endothelial form (eNOS or NOS3), the neuronal form (nNOS or NOS1) and the inducible form
(iNOS or NOS2) (37). While eNOS and nNOS are expressed constitutively and transiently activated
by an increase in intracellular Ca2+, iNOS activity is relatively calcium independent and its prolonged
activation plays an important role in host defense. Indeed, iNOS is expressed at very low levels under
normal conditions; however, inflammatory cytokines and bacterial endotoxins markedly stimulate
iNOS expression (33). Although this mechanism likely evolved to protect the host from bacterial or
viral invaders, it is well accepted that uncontrolled iNOS induction is directly involved in the etiology of
many disease states. Indeed, iNOS has been associated with the necrosis of pancreatic islets,
hypotension, Huntington’s disease, Alzheimer’s disease, rheumatoid arthritis, and inflammatory
bowel disease (1, 9, 20, 22, 23).

85
Adding to these unfavorable effects of iNOS induction, we have demonstrated a role for iNOS in the
etiology of insulin resistance (3, 18, 28, 29). We have shown that iNOS knockout mice are protected
against high fat diet-induced insulin resistance (28) and these results have since been confirmed by
Noronha et al. (24). It has also been reported that treatment with the iNOS inhibitor L-NIL reverses
fasting hyperglycemia and restores hyperinsulinemia in ob/ob mice (11, 32). Furthermore we have
shown that iNOS induction by cytokines and LPS in L6 muscle cells causes insulin resistance by
blunting insulin-induced IRS-1 associated PI3-kinase activity, a key signaling event in insulin’s
metabolic action (29). iNOS has been detected in aorta and skeletal muscle of high-fat fed mice, in
skeletal muscle, liver and adipose tissue of ob/ob mice, in heart of diabetic Zucker rats and in skeletal
muscle and platelets from type 2 diabetic subjects (7, 24, 34). It is conceivable that induction of iNOS
as a result of stress associated with muscle denervation could also contribute to the altered glucose
metabolism in isolated skeletal muscles incubated for prolonged periods of time. However to the best
of our knowledge the effect of long term muscle incubation on iNOS expression and NO production
has not been investigated.
In the present study, we show that prolonged muscle incubation gives rise to iNOS induction causing
nitric oxide accumulation in the medium. Our findings point out a limitation for long-term muscle
incubation and further support a role for iNOS as a mediator of GLUT1 expression.

86
Experimental procedures
Materials
1400W was from Biomol Research Laboratories (Plymouth, PA). ADP-Sepharose 4B beads were
from Amersham Biosciences, and the Renaissance enhanced chemiluminescence kit was from
PerkinElmer Life Sciences (Boston, MA). Monoclonal antibodies against iNOS and eNOS and the
polyclonal antibody against nNOS were obtained from Transduction Laboratories (Mississauga,
Canada). IRS-1 antibody (C-20) was obtained from Santa Cruz Biotechnology (Santa Cruz, CA).
NOC-15 was from Alexis (San Diego, Calif., USA). [3H]-L-arginine, [3H]2-deoxy-D-glucose and
[14C]mannitol were was from Amersham (Oakville, Ont., Canada) and Dowex AG50W-X8 from Bio-
Rad (Mississauga, Ont., Canada). α-Minimum essential medium (α-MEM), fetal bovine serum and
other tissue-culture products were obtained from Gibco BRL. Human insulin (Humulin R) was
purchased from Eli Lilly. 2,3-diaminonaphthotriazole (DAN) was purchased from Aldrich chemicals
(Milwaukee, WI) and all others products were from Sigma Chemicals (St. Louis, MO).
Animals
This study was approved by the Animal Care and Handling Committee of Laval University. Male
Sprague-Dawley rats (175–200 g) purchased from Charles River (Montreal, Qc, Canada) were
maintained on a 12 h dark and light schedule, and fed ad libitum with Purina rat chow.
Muscle isolation and incubation
Rats were anesthetised by a ketamine/xylazine injection after over-night fasting. The epitrochlearis
muscles were rapidly isolated and immediately flash frozen in liquid nitrogen or preincubated for 30
min in 4 ml of Krebs-Ringer bicarbonate (KRB) buffer pH 7.4 at 30˚C, containing 2 mM sodium
pyruvate (KRB-P). Muscles were then incubated for the appropriate times (up to 12 hours) in the KRB
buffer supplement containing 0.1% BSA and amino acids at concentrations similar to those found in
plasma (Asn 70µM, Thr 300µM, Ser 280µM, Asp 15µM, Glu 200 µM, Pro 180 µM, Gln 600 µM, Gly
400 µM, Ala 350 µM, Cys 40 µM, Met 70 µM, phe 1000 µM, Ile 100 µM, Leu 170 µM, Lys 600 µM,
His 80 µM, Trp 120 µM, Arg 80 µM, Val 200 µM, Tau 200 µM, Orn 90 µM, Cit 80 µM.). The
incubation medium was changed every two hours and was under constant oxygenation via
continuous bubbling of 95% O2-5% CO2. After the appropriate incubation period, the muscles were

87
extensively washed in KRB and frozen in liquid nitrogen, or kept for in vitro glucose uptake
determinations.
In vitro glucose uptake in isolated muscle
Glucose transport in isolated epitrochlearis muscles was measured using the glucose analogue 2-
[3H]2-deoxy-D-glucose as described previously (18).
Cell Culture
A line of L6 skeletal muscle cells (kind gift of Dr. Amira Klip, Hospital for Sick Children, Toronto, ON,
Canada) clonally selected for high fusion potential was used in the present study. The L6 cell line
was derived from neonatal rat thigh skeletal muscle cells and retains many morphological,
biochemical, and metabolic characteristics of skeletal muscle. Cells were grown and maintained in
monolayer culture in -MEM containing 2% (v/v) fetal bovine serum and 1% (v/v) antibiotic/antimycotic
solution (10000 units/ml penicillin, 10000 µg/ml streptomycin and 25 µg/ml amphotericin B) in an
atmosphere of 5% CO2 at 37°C. Next, different doses of NOC-15 (1μM, 5μM, 10μM, 25μM, 50μM,
100μM) were added to the appropriate wells for 24 hours. After the treatment, cells were washed and
glucose uptake assay and protein extraction were performed.
Glucose uptake assay
2-Deoxyglucose uptake was determined as previously described (3). Briefly, L6 myotubes were
incubated with or without NOC-15 for 24h, followed by the addition of insulin 100nM (or medium
alone) for an additional 30 min. Cells were rinsed once with glucose-free Hepes-buffered saline
solution, pH 7.4 (140 mM NaCl, 20 mM Hepes/Na, 5 mM KCl, 2.5 mM MgSO4 and 1 mM CaCl2), and
were subsequently incubated for 8 min with 10 µM 2-deoxy-D-glucose containing 0.3 µCi/ml 2-
deoxy-D-[3H]glucose in the same buffer. After the incubation in transport medium, cells were rinsed
three times with ice-cold saline solution, and then disrupted by adding 50 mM NaOH. Cell-associated
radioactivity was determined by scintillation counting. Protein concentrations were determined by the
bicinchoninic acid method (Pierce), and the results were expressed in pmol/min per mg. Glucose
uptake values were corrected for non-carrier-mediated transport by measuring hexose uptake in the
presence of 10 µM cytochalasin B.

88
Western blotting
Protein extraction was performed using (20 mM Tris (pH 7.5), 150 mM NaCl, 10% glycerol, 1% Triton
X-100, 1 mM EDTA, 2 mM Na3VO4, 1 mM phenylmethylsulfonylfluoride, 10 µg/ml aprotinin, and 10
µg/ml leupeptin), and insoluble material was removed by centrifugation at 12,000 x g. For western
blotting, protein extracts were resolved by SDS-PAGE and transferred to a polyvinylidene difluoride
(PVDF) membrane. Blots were blocked with 5% non-fat dry milk in 10 mM Tris (pH 7.5), 150 mM
NaCl, and 0.05% Tween 20; incubated with primary antibody; and then incubated with a secondary
horseradish-conjugated antibody. Immunoreactive bands were visualized by enhanced
chemiluminescence.
RNA analysis
Total cellular RNA was isolated using guanidinium thiocyanate-phenol-chloroform extraction and
iNOS, eNOS, nNOS, GLUT4 and GLUT1 mRNAs were measured by semi-quantitative RT-PCR as
described previously (3, 27).
Nitric oxide synthase activity
Nitric oxide synthase activity was quantified by the conversion of [3H]-L-arginine to [3H]-L-citrulline as
described previously (3, 28).
NO production
NO production was measured by fluorometric detection of both nitrite and nitrate products (NOx)
levels in plasma and incubation medium. Nitrate was reduced to nitrite using nitrate reductase and
the NADPH regenerating system as described previously (30) . NOx levels were determined by
measuring the fluorescence of 2,3-diaminonaphthotriazole DAN at ex of 360 nm and em of 450 nm.
Data Analysis
The effects of the treatments were compared by an analysis of variance followed by Fisher's post hoc
test. Differences were considered to be statistically significant at P < 0.05

89
Results
Long-term incubation of epitrochlearis muscle modulates NOS expression
We speculated that stress associated with muscle incubation may lead to induction of the iNOS
enzyme. Rat epitrochlearis muscles were isolated and snap frozen immediately in liquid nitrogen or
incubated for 10 hours and then snap frozen. In figure 1 we show that long-term muscle incubation
leads to robust iNOS mRNA induction (Fig. 1A) and protein expression (Fig. 1B). In contrast, the two
calcium dependent constitutive NOS, eNOS and nNOS, were down-regulated following muscle
incubation. This alteration in NOS expression was associated with shifts in NOS activity. Indeed,
constitutive NOS activity (e.g. eNOS/nNOS-dependent) was reduced by long-term skeletal muscle
incubation while iNOS activity was up-regulated under the same conditions (Fig. 1C). We also
measured NO2 accumulation, an index of NO production, in the incubation medium. In accordance
with the iNOS expression and activity data we observed that NO production was ~ 4 times higher in
long-term incubated muscle medium compared to the control muscles that were only pre-incubated
for 30 min (Fig.1D). These results suggest that long-term incubation of skeletal muscle enhances
iNOS expression and activity.
Time course of iNOS and GLUT1 induction
To further characterize iNOS modulation by muscle incubation, we examined the time course of iNOS
induction. Muscles were incubated and frozen every hour for up to 12 hours. In muscles incubated for
3 hours, a slight iNOS induction was observed both at the protein (Fig. 2A) and mRNA (Fig. 2B)
levels, followed by a stronger induction at ≥ 4 hours. We have previously reported that iNOS
induction by cytokines and LPS increased the expression of the glucose transporter-1 (GLUT1),
leading to increased basal glucose transport (3). We thus next investigated whether GLUT1 was
associated with iNOS induction in the incubated epitrochlearis. GLUT1 expression was indeed
induced during the muscle incubation and the time course of its expression closely followed the time
course of iNOS expression (Fig. 2B).
iNOS induction causes GLUT1 expression in epitrochlearis muscle
To clarify the link between iNOS induction and GLUT1 expression, we measured the expression of
GLUT1 in long-term incubated muscles in the presence or absence of 1400W, a specific iNOS

90
inhibitor. GLUT1 mRNA expression was induced by long-term skeletal muscle incubation but this
response was repressed by the presence of the iNOS inhibitor (Fig. 3A). Importantly, no changes in
GLUT4 expression were observed in incubated muscle treated with 1400W (Fig. 3B). These results
confirm GLUT1 expression is dependent on iNOS induction. We next sought to determine whether
iNOS induction leads to alterations of glucose uptake in these long-term incubated muscles. We
measured basal and insulin-stimulated glucose transport in muscle incubated following the 30 minute
or 10 hour incubation periods. Despite elevated GLUT1 expression, basal glucose transport was not
significantly different in long-term incubated muscle compared to short-term incubated muscle. (Fig.
3C) Moreover, insulin was found to increase glucose transport (~1.5 fold) in both conditions and there
was no difference observed between the 30 minute and 10 hour incubations. (Fig. 3C-D)
NO induction increases GLUT1 expression and basal glucose uptake in L6 muscle cells
To further test the role of high NO output in mediating GLUT1 expression and glucose transport, we
looked at the effects of NOC-15, a chemical NO donor, in L6 muscle cells after 24 hour. Increasing
doses of NOC-15 caused a dose dependent increase in GLUT1 mRNA expression (Fig. 4A). The
induction of GLUT1 mRNA corresponded with GLUT1 protein expression at 50 and 100 µM of NOC-
15 (Fig. 4B). As in incubated muscle, no effect of the NO donor was observed on GLUT4 mRNA
expression in this cellular model (Fig. 4A). We next sought to determine whether NO leads to
alterations of glucose uptake in these cells, we looked at the effects of NOC-15 on basal and insulin-
stimulated glucose uptake. L6 myocytes were incubated with increasing doses of NOC-15 (1μM,
5μM, 25μM) for 24h, followed by measurement of glucose uptake. Basal glucose uptake was
augmented by the NO donor while insulin-stimulated glucose uptake was reduced (Fig. 4C and 4D).
In contrast to our observations in incubated muscles, the presence of NO in the incubation media for
24 hour causes perturbations of glucose metabolism in L6 myocytes.

91
Discussion
We have previously established a causal link between iNOS induction and skeletal muscle insulin
resistance. Using known inducers of iNOS, such as inflammatory cytokines and LPS, we showed that
iNOS induction impairs insulin stimulated glucose transport in myocytes and rat muscle (3, 18).
Furthermore, at the genetic level, we demonstrated that targeted disruption of iNOS prevents obesity-
linked skeletal muscle insulin resistance caused by high fat feeding (28). In the present study, despite
a strong iNOS induction in rat epitrochlearis muscle and NO release, neither basal nor insulin-
stimulated glucose uptake were altered after a 10 hour ex vivo incubation Here we observed that,
long-term skeletal muscle incubation induces iNOS, giving rise to GLUT1 expression. However,
under these conditions, we observed that basal glucose uptake was not altered and insulin mediated
glucose transport was comparable following a 10 hour incubation period. However, 24 hour of
exposure to NOC-15, a NO donor, lead to GLUT1 overexpression that was associated with increased
basal glucose and reduced insulin-stimulated glucose uptake in L6 myocytes. According to the
results observed in L6 cells, we could hypothesize that a longer incubation time, and thus a longer
NO exposure would result in a significant increase in glucose uptake in epitrochlearis muscle.
Therefore the time limitations of long term muscle incubation may explain the difference observed on
glucose metabolism between isolated skeletal muscle and cultured muscle cells. Taken together,
these results provide further understanding of the impact of iNOS on insulin sensitivity and glucose
metabolism.
The elevated GLUT1 protein observed in long-term incubated muscles is in line with the findings of a
previous study (19). Here, it had been proposed that the increase in basal glucose uptake in
incubated muscle could be due to a greater amount of glucose reaching the cells by diffusion
compared to the amount of glucose available from capillaries within the muscle in vivo. Furthermore,
it was also proposed that a less-than optimal supply of oxygen to the incubated muscle could give
rise to regional hypoxia and an increase in basal glucose uptake (6). However, it is well appreciated
that GLUT1 is induced in stressful conditions such as oxidative stress, denervation, hyperosmolarity,
glucose deprivation and hypoxia (5, 8, 16). Indeed, in stressful situations, where energy is critical for
survival, GLUT1 is mobilized to fulfill essential functions and ensure host defense. It is noteworthy
that such stress situations have also been linked to insulin resistance (25, 36).

92
One might consider that long-term incubation of isolated muscles as a stressful process since it
involves muscle denervation and prolonged isolation from the physiological milieu. Accordingly, it has
been demonstrated that situations such as crush injuries, shear stress or oxidative stress, also lead
to iNOS induction (12, 21, 31). Under these circumstances, cellular damage or stress may induce
cytokine expression leading to iNOS induction. Interestingly, it has been shown that heat shock
protein 70 (HSP70), which is produced during cellular stress, enhances cytokine-induced iNOS
expression by activating p38 MAP kinase (4). In our model, muscle denervation may have caused
enough stress to increase cytokine expression and/or activate stress-signaling pathways leading to
iNOS induction. Importantly, high NO concentrations are known to activate GLUT1 in different
models. In fact, we have previously reported that cytokine and LPS treatment increases glucose
transport in L6 muscle cells via GLUT1 induction (3). Furthermore, the NO donors sodium
nitroprusside (SNP) and diethylenetriamine (DETA) induce GLUT1 in HUVEC cells (26). However, to
the best of our knowledge, the potential role of NO as an inducer of GLUT1 in isolated intact muscle
has never been investigated. Here, we provide novel complementary evidence that iNOS induction
leads to GLUT1 overexpression since the iNOS inhibitor 1400W blunted GLUT1 protein expression.
We also observed a decreased expression and activity of constitutive NOS isoforms following long-
term skeletal muscle incubation. This downregulation could be explained by the effect of high NO
output from iNOS. Indeed, some studies have reported that NO inhibits NOS activity through a
negative feedback loop and that nNOS and eNOS are more sensitive than iNOS to the inhibitory
action of NO (13). Our finding of reduced expression of eNOS and nNOS is consistent with previous
observations that iNOS induction in endothelial cells reduces the expression of eNOS (10). It is thus
possible that this mechanism has evolved to temporarily inhibit cNOS in an effort to keep cofactors
available for iNOS during infection.
While previous studies on denervated and incubated muscles reported a decrease in GLUT4
expression (17), we did not observe any change in the expression of this glucose transporter. This
apparent discrepancy may be explained by the composition of the muscle fiber type. Following
denervation, Handberg et al. reported a much greater GLUT4 decrement in oxidative red muscle
(type I) compared to (type IIB) glycolytic white muscle (15). Thus, the fact that epitrochlearis is a fast

93
twitch glycolytic muscle could explain why we did not observe a reduction of GLUT4 expression in
our study. However, Alkhateeb et al. (2) failed to detect a reduction in GLUT4 expression after long
term incubation of the soleus muscle. Taken together, our data are more consistent with the
hypothesis that long-term skeletal muscle incubation modifies glucose metabolism by upregulation of
GLUT1 expression. Importantly, since the induction of iNOS and GLUT1 are clearly evident after just
6 hours of muscle incubation. These data illustrate a potential limitation of using prolonged skeletal
muscle incubation to study glucose metabolism ex vivo.
In summary, our data clearly demonstrate that long-term skeletal muscle incubation causes robust
iNOS induction leading to elevated GLUT1 expression that could impact glucose metabolism. Taken
together these results provide a potential molecular basis for the alterations in glucose utilization that
manifest in patients suffering from denervated muscle following injuries. Moreover our findings
provide important considerations for researchers planning long-term incubation studies with isolated
muscle preparations.

94
References
1. Aguilera P, Chanez-Cardenas ME, Floriano-Sanchez E, Barrera D, Santamaria A, Sanchez-Gonzalez DJ, Perez-Severiano F, Pedraza-Chaverri J, and Jimenez PD. Time-related changes in constitutive and inducible nitric oxide synthases in the rat striatum in a model of Huntington's disease. Neurotoxicology 28: 1200-1207, 2007. 2. Alkhateeb H, Chabowski A, and Bonen A. Viability of the isolated soleus muscle during long-term incubation. Appl Physiol Nutr Metab 31: 467-476, 2006. 3. Bedard S, Marcotte B, and Marette A. Cytokines modulate glucose transport in skeletal muscle by inducing the expression of inducible nitric oxide synthase. Biochem J 325 ( Pt 2): 487-493, 1997. 4. Bellmann K, Burkart V, Bruckhoff J, Kolb H, and Landry J. p38-dependent enhancement of cytokine-induced nitric-oxide synthase gene expression by heat shock protein 70. J Biol Chem 275: 18172-18179, 2000. 5. Boado RJ, and Pardridge WM. Glucose deprivation and hypoxia increase the expression of the GLUT1 glucose transporter via a specific mRNA cis-acting regulatory element. J Neurochem 80: 552-554, 2002. 6. Bonen A, Clark MG, and Henriksen EJ. Experimental approaches in muscle metabolism: hindlimb perfusion and isolated muscle incubations. Am J Physiol 266: E1-16, 1994. 7. Carvalho-Filho MA, Ueno M, Hirabara SM, Seabra AB, Carvalheira JB, de Oliveira MG, Velloso LA, Curi R, and Saad MJ. S-nitrosation of the insulin receptor, insulin receptor substrate 1, and protein kinase B/Akt: a novel mechanism of insulin resistance. Diabetes 54: 959-967, 2005. 8. Coderre L, Monfar MM, Chen KS, Heydrick SJ, Kurowski TG, Ruderman NB, and Pilch PF. Alteration in the expression of GLUT-1 and GLUT-4 protein and messenger RNA levels in denervated rat muscles. Endocrinology 131: 1821-1825, 1992. 9. Cuzzocrea S. Role of nitric oxide and reactive oxygen species in arthritis. Curr Pharm Des 12: 3551-3570, 2006. 10. de Frutos T, de Miguel LS, Garcia-Duran M, Gonzalez-Fernandez F, Rodriguez-Feo JA, Monton M, Guerra J, Farre J, Casado S, and Lopez-Farre A. NO from smooth muscle cells decreases NOS expression in endothelial cells: role of TNF-alpha. Am J Physiol 277: H1317-1325, 1999. 11. Fujimoto M, Shimizu N, Kunii K, Martyn JA, Ueki K, and Kaneki M. A role for iNOS in fasting hyperglycemia and impaired insulin signaling in the liver of obese diabetic mice. Diabetes 54: 1340-1348, 2005. 12. Gosgnach W, Messika-Zeitoun D, Gonzalez W, Philipe M, and Michel JB. Shear stress induces iNOS expression in cultured smooth muscle cells: role of oxidative stress. Am J Physiol Cell Physiol 279: C1880-1888, 2000. 13. Griscavage JM, Hobbs AJ, and Ignarro LJ. Negative modulation of nitric oxide synthase by nitric oxide and nitroso compounds. Adv Pharmacol 34: 215-234, 1995. 14. Gulve EA, Cartee GD, and Holloszy JO. Prolonged incubation of skeletal muscle in vitro: prevention of increases in glucose transport. Am J Physiol 261: C154-160, 1991. 15. Handberg A, Megeney LA, McCullagh KJ, Kayser L, Han XX, and Bonen A. Reciprocal GLUT-1 and GLUT-4 expression and glucose transport in denervated muscles. Am J Physiol 271: E50-57, 1996. 16. Hwang DY, and Ismail-Beigi F. Control of Glut1 promoter activity under basal conditions and in response to hyperosmolarity: role of Sp1. Am J Physiol Cell Physiol 290: C337-344, 2006.

95
17. Jensen EB, Zheng D, Russell RA, Bassel-Duby R, Williams RS, Olson AL, and Dohm GL. Regulation of GLUT4 expression in denervated skeletal muscle. Am J Physiol Regul Integr Comp Physiol 296: R1820-1828, 2009. 18. Kapur S, Bedard S, Marcotte B, Cote CH, and Marette A. Expression of nitric oxide synthase in skeletal muscle: a novel role for nitric oxide as a modulator of insulin action. Diabetes 46: 1691-1700, 1997. 19. Kern M, Tapscott EB, Snider RD, and Dohm GL. Differences in glucose transport rates between perfused and in vitro incubated muscles. Horm Metab Res 22: 366-368, 1990. 20. Kolios G, Valatas V, and Ward SG. Nitric oxide in inflammatory bowel disease: a universal messenger in an unsolved puzzle. Immunology 113: 427-437, 2004. 21. Kuo PC, Abe KY, and Schroeder RA. Oxidative stress increases hepatocyte iNOS gene transcription and promoter activity. Biochem Biophys Res Commun 234: 289-292, 1997. 22. Li F, and Mahato RI. iNOS gene silencing prevents inflammatory cytokine-induced beta-cell apoptosis. Mol Pharm 5: 407-417, 2008. 23. Malinski T. Nitric oxide and nitroxidative stress in Alzheimer's disease. J Alzheimers Dis 11: 207-218, 2007. 24. Noronha BT, Li JM, Wheatcroft SB, Shah AM, and Kearney MT. Inducible nitric oxide synthase has divergent effects on vascular and metabolic function in obesity. Diabetes 54: 1082-1089, 2005. 25. O'Donnell CP. Metabolic consequences of intermittent hypoxia. Adv Exp Med Biol 618: 41-49, 2007. 26. Paik JY, Lee KH, Ko BH, Choe YS, Choi Y, and Kim BT. Nitric oxide stimulates 18F-FDG uptake in human endothelial cells through increased hexokinase activity and GLUT1 expression. J Nucl Med 46: 365-370, 2005. 27. Perreault M, Dombrowski L, and Marette A. Mechanism of impaired nitric oxide synthase activity in skeletal muscle of streptozotocin-induced diabetic rats. Diabetologia 43: 427-437, 2000. 28. Perreault M, and Marette A. Targeted disruption of inducible nitric oxide synthase protects against obesity-linked insulin resistance in muscle. Nat Med 7: 1138-1143, 2001. 29. Pilon G, Dallaire P, and Marette A. Inhibition of inducible nitric-oxide synthase by activators of AMP-activated protein kinase: a new mechanism of action of insulin-sensitizing drugs. J Biol Chem 279: 20767-20774, 2004. 30. Rao AM, Dogan A, Hatcher JF, and Dempsey RJ. Fluorometric assay of nitrite and nitrate in brain tissue after traumatic brain injury and cerebral ischemia. Brain Res 793: 265-270, 1998. 31. Rubinstein I, Abassi Z, Coleman R, Milman F, Winaver J, and Better OS. Involvement of nitric oxide system in experimental muscle crush injury. J Clin Invest 101: 1325-1333, 1998. 32. Sugita H, Fujimoto M, Yasukawa T, Shimizu N, Sugita M, Yasuhara S, Martyn JA, and Kaneki M. Inducible nitric-oxide synthase and NO donor induce insulin receptor substrate-1 degradation in skeletal muscle cells. J Biol Chem 280: 14203-14211, 2005. 33. Suschek CV, Schnorr O, and Kolb-Bachofen V. The role of iNOS in chronic inflammatory processes in vivo: is it damage-promoting, protective, or active at all? Curr Mol Med 4: 763-775, 2004. 34. Torres SH, De Sanctis JB, de LBM, Hernandez N, and Finol HJ. Inflammation and nitric oxide production in skeletal muscle of type 2 diabetic patients. J Endocrinol 181: 419-427, 2004. 35. Tremblay F, Lavigne C, Jacques H, and Marette A. Dietary cod protein restores insulin-induced activation of phosphatidylinositol 3-kinase/Akt and GLUT4 translocation to the T-tubules in skeletal muscle of high-fat-fed obese rats. Diabetes 52: 29-37, 2003.

96
36. Turinsky J, and Damrau-Abney A. Akt1 kinase and dynamics of insulin resistance in denervated muscles in vivo. Am J Physiol 275: R1425-1430, 1998. 37. Wahl SM, McCartney-Francis N, Chan J, Dionne R, Ta L, and Orenstein JM. Nitric oxide in experimental joint inflammation. Benefit or detriment? Cells Tissues Organs 174: 26-33, 2003.

97
Legends to figures
Figure 1. Long-term incubation of epitrochlearis muscle modulates NOS expression. Rat
epitrochlearis muscles were isolated and frozen immediately or incubated for 10h before being
frozen. A) RT-PCR of iNOS, eNOS, nNOS and GAPDH mRNA. B) immunoblot of iNOS, eNOS and
nNOS protein C) NOS activity in non incubated (CTL) or incubated muscle (INC). D) NO production
in the incubation medium was measured by nitrite/nitrate (NOx) accumulation. Results in C and D
represent the mean± SD of 2-3 independent experiments. * P<0.05 vs corresponding CTL values.
Figure 2. Time course of incubation-mediated induction of iNOS and GLUT1. Rat epitrochlearis
muscles were removed and frozen immediately or incubated for the indicated hours before being
frozen. Muscle were processed for protein or RNA isolation and used for western blot analyses of
iNOS protein (A) or RT-PCR determinations of iNOS and GLUT1 mRNAs (B). Immunoblots and gels
are representative at 2 independent time-course experiments.
Figure 3. iNOS induction causes GLUT1 expression in epitrochlearis muscle. (A) and (B) show
GLUT1 and GLUT4 mRNA expression, respectively, following muscle incubation in the presence or
absence of specific iNOS inhibitor, 1400W (0.1M) . (C) Basal (BA) and insulin-stimulated (INS)
glucose transport were measured in muscle that was incubated following a 30 min or 10 hours
period. (D) Insulin-stimulated glucose transport is expressed as fold stimulation over basal. Results
represent the mean ± SE of 5-7 independent experiments. * P<0.05 vs corresponding CTL values. #
P<0.05 vs corresponding INC (C) or Insulin (E) values.
Figure 4. NO induction increase GLUT1 expression and basal glucose uptake in L6 muscle
cells. L6 muscle cells were incubated with increasing doses of the chemical NO donor NOC-15. (A)
GLUT1 and GLUT4 were measured by RT-PCR (B) GLUT1 protein expression was assessed by
western blotting. (C) Basal and (D) insulin-stimulated 2-DG uptake were measured in L6 cells
pretreated for 24 hours with increasing doses of NOC-15. Results in C and D represent the mean±
SE of 6-7 independent experiments. * P<0.05 vs cells not exposed to NOC-15.
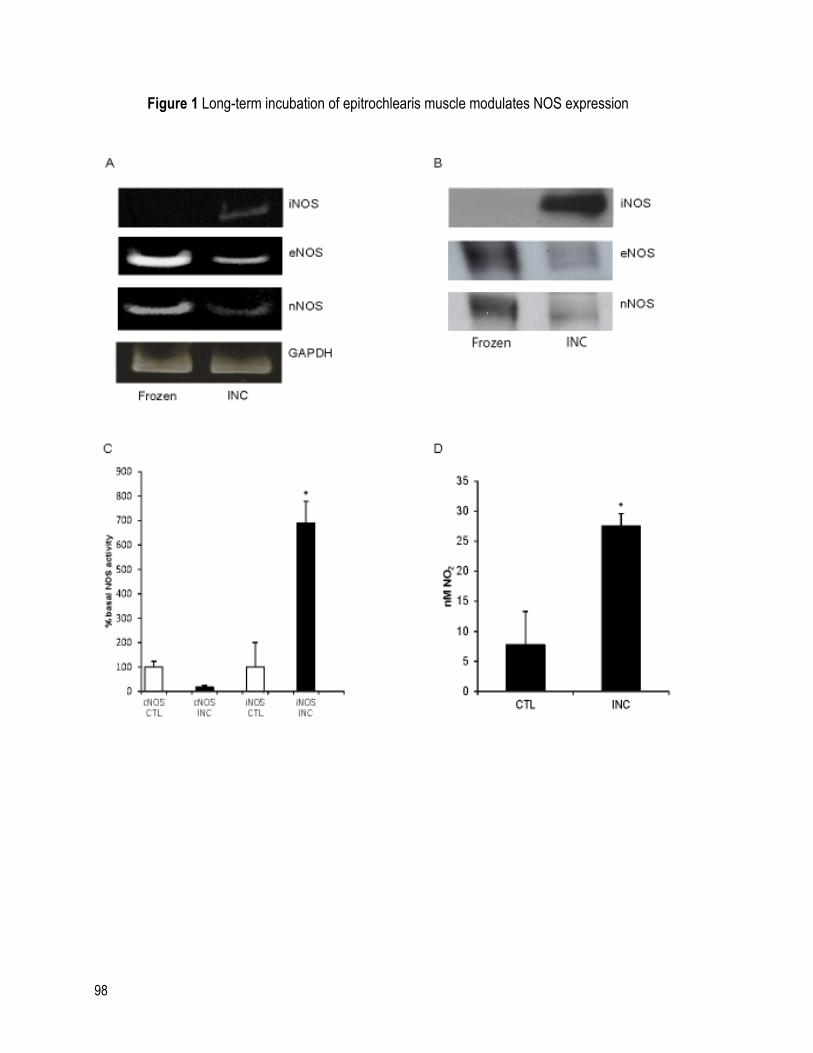
98
Figure 1 Long-term incubation of epitrochlearis muscle modulates NOS expression

99
Figure 2 Time course of incubation-mediated induction of iNOS and GLUT1
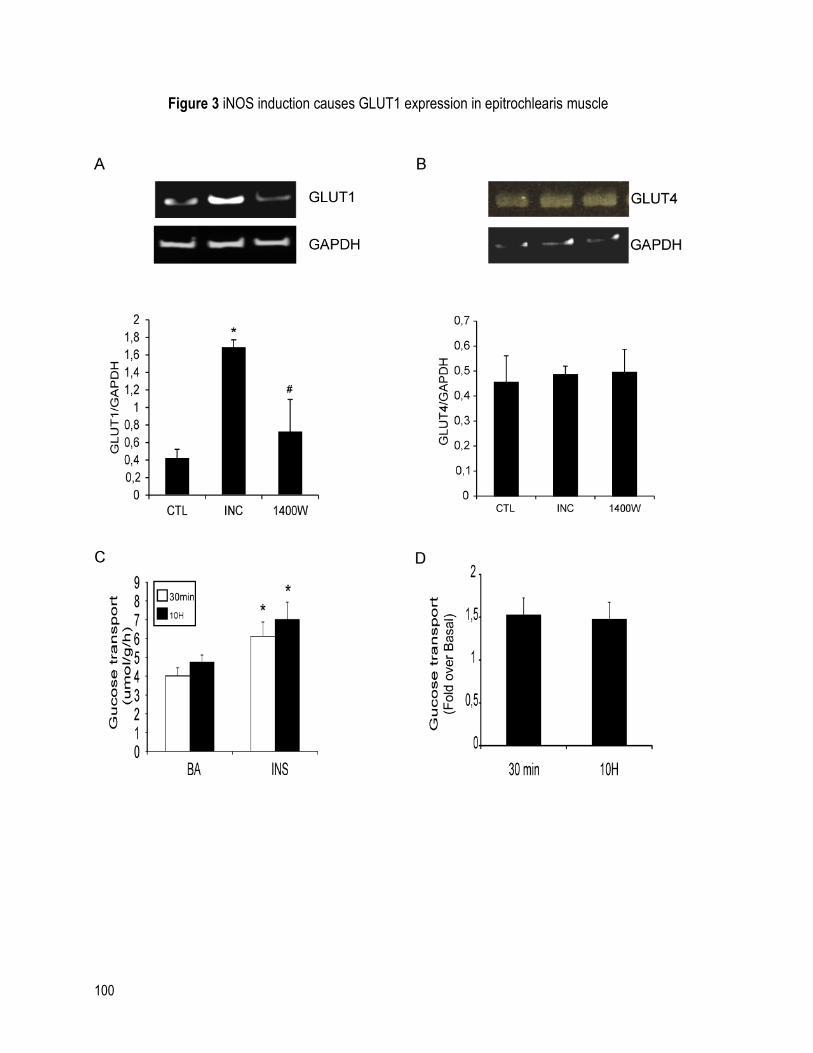
100
Figure 3 iNOS induction causes GLUT1 expression in epitrochlearis muscle
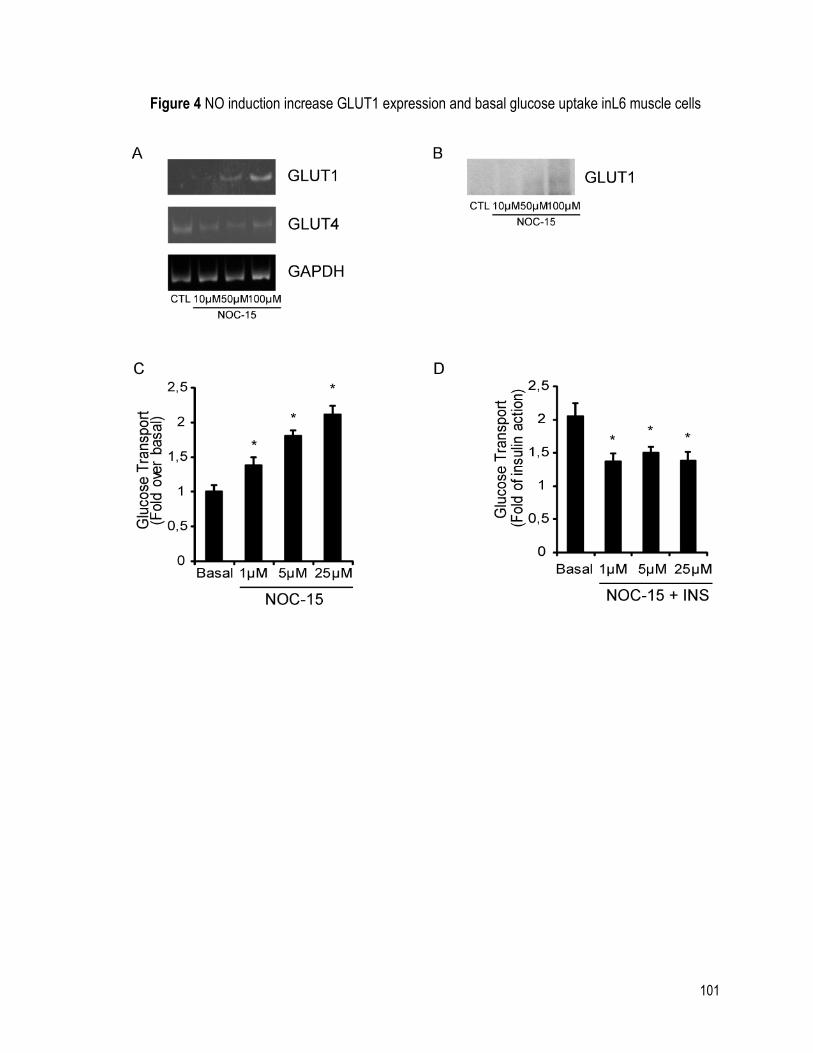
101
Figure 4 NO induction increase GLUT1 expression and basal glucose uptake inL6 muscle cells


103
CONCLUSION
Le tissu musculaire squelettique représente l’organe le plus volumineux du corps humain. Outre sa
capacité à mettre en mouvement les os du squelette, il joue un rôle primordial dans le maintien de
l’homéostasie énergétique. Chez l’humain, en condition à jeun, le muscle squelettique capte environ
soixante-cinq pourcent du glucose sanguin produit par le foie tandis qu’en situation post prandiale,
environ quatre-vingt pourcent du transport du glucose stimulé par l’insuline se font au niveau du
muscle squelettique. Le glucose est le substrat énergétique principal du muscle squelettique. Ainsi, il
n’est plus à démontrer qu’une diminution de la sensibilité aux effets de l’insuline au niveau du muscle
squelettique entraîne des perturbations métaboliques majeures.
Les travaux de notre laboratoire ont permis de révéler l’implication de différentes protéines, dont
l’AMPK et iNOS, dans la régulation du transport du glucose dans le muscle squelettique.
Le rôle de l’AMPK dans le transport du glucose induit par la
contraction musculaire
Tel que décrit précédemment, de nombreuses études ont rapporté l’implication de la kinase activée
par l’adénosine monophosphate (AMPK) dans la régulation du métabolisme énergétique au cours
des dernières années. En effet, l’AMPK est sensible aux variations du rapport AMP/ADP : ATP, un
indicateur du statut énergétique cellulaire. L’augmentation du rapport ADP : ATP, et plus
particulièrement l’augmentation du rapport AMP : ATP, par un stress physiologique active l’AMPK.
Rapidement, il fut démontré que l’exercice physique induit l’activation de l’AMPK dans le muscle
squelettique. De plus, l’utilisation d’un activateur pharmacologique de l’AMPK, l’AICAR, suggéra
l’implication de l’AMPK dans la régulation du transport du glucose dans la cellule musculaire.
Toutefois, encore à ce jour et malgré de nombreux modèles décrits à la section 2.4, le rôle de
l’AMPK dans la régulation du transport du glucose lors de la contraction musculaire demeure
controversé.
L’arrivée du premier modèle transgénique d’invalidation de la sous-unité α2 de l’AMPK (AMPK KD1),
spécifique au tissu musculaire, permit d’observer une réduction du transport du glucose à la suite de

104
protocoles de contraction ex vivo et in situ. Cependant, ce modèle animal se révéla intolérant à
l’exercice physique et une étude subséquente démontra une diminution de la force absolue des
muscles EDL et soléaire [299, 300]. Considérant que le transport du glucose induit par la contraction
musculaire est proportionnel à la force développée par le muscle, l’interprétation des résultats
préalablement obtenus devint discutable.
Tels nos prédécesseurs et la majorité des études subséquentes, nous avons observé une diminution
de la force maximale absolue du muscle EDL des souris AMPK KD1. Afin de confirmer ou non
l’implication de l’AMPK dans la régulation du transport du glucose induit par la contraction
musculaire, nous avons élaboré un protocole de stimulation électrique ex vivo au cours duquel les
muscles EDL des souris AMPK KD1 et des souris contrôles développaient une force équivalente
pour un même stimulus donné. Sous ces conditions expérimentales, nous avons obtenu une
diminution de cinquante pourcent de la captation du glucose par le muscle EDL à la suite du
protocole de contraction musculaire ex vivo. Ainsi, nos résultats suggèrent un rôle essentiel de
l’AMPK dans la captation du glucose lors de la contraction musculaire bien que des voies alternatives
soient également impliquées. De plus, notre étude a démontré l’importance du choix du protocole de
contraction musculaire lors de la comparaison de deux phénotypes musculaires différents.
Des études subséquentes au cours desquelles d’autres protocoles de contraction musculaire et/ou
d’autres modèles d’invalidation génétique de l’AMPK furent utilisés rapportèrent des conclusions à la
fois similaires et contradictoires. L’ensemble de ces études ne permet toujours pas de confirmer ou
non l’implication de l’AMPK dans le transport du glucose induit par la contraction musculaire.
Chacune d’entre elles apporta son lot de réponses et d’interrogations. Le choix du type (ex vivo, in
situ, in vivo), de la durée et de l’intensité de la contraction musculaire ainsi que et le phénotype
musculaire généré par l’invalidation génétique de la protéine doivent être considérés lors de
l’interprétation des résultats. En effet, sur un total de douze études portant sur l’implication de l’AMPK
dans le transport du glucose au cours d’un protocole de contraction musculaire, neuf modèles
différents d’invalidation génétique de l’AMPK furent utilisés. Ceci dit, toutes ces études s’entendent
sur la présence de voies AMPK-indépendantes dans le transport du glucose induit par la contraction
musculaire. Par ailleurs, la section 1.4 présente diverses autres protéines proposées pour réguler le
transport du glucose induit par la contraction musculaire.

105
L’entrée du glucose dans les myocytes au cours d’un exercice physique dépend à la fois de la
disponibilité du glucose sanguin, du transport du glucose par l’intermédiaire des GLUT4 et de la
phosphorylation du glucose à l’intérieur de la cellule. Chez l’humain, au cours d’un exercice
physique, le flux sanguin en direction des muscles squelettiques peut augmenter jusqu’à vingt fois ce
qui accroit considérablement la quantité de glucose disponible [450]. De plus, l’exercice physique
augmente le recrutement de capillaires permettant d’augmenter les échanges entre la circulation
sanguine et la cellule musculaire tant chez l’humain que chez l’animal [451-454]. Une diminution du
nombre de capillaires au niveau du muscle chez la souris AMPK KD1 fut rapportée. De plus, une
réduction de l’expression de nNOSμ ainsi que de l’activité totale de NOS fut observée dans les
muscles gastrocnémien et VL de la souris AMPK KD1 [303]. Par ailleurs, une diminution de la
phosphorylation du résidu sérine 1446 (Ser1446) de nNOSμ fut rapportée dans le muscle TA de la
souris AMPK β1β2M KO parallèlement à une réduction de la densité de capillaires et à une
augmentation de l’agrégation plaquettaire [455]. Le nNOSμ est connu pour être, du moins
partiellement, responsable de la vasodilation des artérioles lors de la contraction musculaire [456].
Ces altérations vasculaires pourraient contribuer à la diminution du transport du glucose observée
chez la souris AMPK KD1 dans certaines études.
Outre la disponibilité du glucose sanguin, la translocation des GLUT4 à la membrane plasmique et
aux tubules-T régule la captation du glucose au cours d’un exercice physique. Chez les rongeurs,
une relation directe entre le contenu en GLUT4 et le transport du glucose lors d’un protocole de
contraction musculaire ex vivo fut rapportée ainsi que lors d’un exercice musculaire d’extension de la
jambe chez l’humain [26, 457]. Ces résultats suggèrent que le contenu en GLUT4 régule directement
le transport du glucose lors d’une contraction musculaire. Toutefois, aucune différence du contenu
musculaire en GLUT4 ne fut observée dans les différents modèles d’invalidation génétique de
l’AMPK, de sorte que la perte des différentes sous-unités de l’AMPK n’influence pas l’expression de
GLUT4. De plus, aucun défaut de la translocation de GLUT4 induite par la contraction musculaire ne
fut rapporté chez la souris AMPK α2iTg. Toutefois, la translocation des GLUT4 à la suite d’une
stimulation AICAR est abolie chez ce même modèle suggérant l’implication de l’AMPK dans la
mobilisation des GLUT4 dans la cellule musculaire [458].

106
Par ailleurs, le contenu musculaire en glycogène semble également influer sur la translocation des
GLUT4 au cours de la contraction. En effet, il fut observé que le contenu en glycogène musculaire et
la translocation des GLUT4 lors d’un exercice physique sont corrélés négativement chez le rat. Ainsi,
plus les réserves en glycogène musculaire sont diminuées, plus il y a translocation de GLUT4 et vice
versa [459]. Il fut rapporté que le contenu musculaire en glycogène tant au repos qu’à la suite d’un
protocole de contraction musculaire, est réduit dans la majorité des modèles d’invalidation génétique
de l’AMPK. En présence d’un déficit en glycogène musculaire, une augmentation plus importante de
la translocation de GLUT4 par des voies indépendantes de l’AMPK pourrait contribuer à masquer le
défaut observé au niveau du transport du glucose induit par la contraction musculaire en l’absence
de l’AMPK dans certaines études.
Une fois à l’intérieur de la cellule musculaire, le glucose est phosphorylé en glucose-6-phosphate
(G6P) par l’intervention de l’hexokinase II (HKII). Certaines études suggèrent que chez la souris, la
phosphorylation du glucose serait une étape limitante dans la captation du glucose par le muscle
squelettique lors d’un exercice physique. La surexpression de HKII augmente la captation du glucose
induite par la contraction musculaire tandis que l’effet inverse fut observé chez la souris partiellement
invalidée pour la HKII (souris HKII+/-) [460, 461]. Des résultats non publiés de notre laboratoire ont
démontré une diminution de l’ARNm de la HKII dans le muscle EDL des souris AMPK KD1. Une
diminution de l’expression de la HKII pourrait contribuer au défaut observé au niveau du transport du
glucose chez la souris AMPK KD1 dans certaines études. Toutefois, cette observation ne fut pas
corroborée par d’autres études. Par ailleurs, chez l’humain, cette étape, la phosphorylation du
glucose, semble s’avérer beaucoup moins limitante dans la régulation du transport du glucose dans
le muscle squelettique au cours d’un exercice physique. Le glucose sanguin semble constituer
l’apport énergétique primordial chez la souris lors de la contraction musculaire en comparaison au
glycogène musculaire. En effet, les réserves de glycogène musculaire sont beaucoup plus faibles
chez la souris comparativement à l’humain. De plus, l’invalidation génétique de la synthase du
glycogène qui résulte en l’absence de glycogène musculaire n’altère pas les capacités physiques de
la souris lors d’un exercice sur tapis roulant tandis qu’il est bien connu que des niveaux bas en
glycogène musculaire limitent les performances physiques chez l’humain [462, 463].

107
Finalement, tous les modèles murins d’invalidation de l’AMPK utilisés se caractérisent par une
intolérance à l’exercice qui se traduit par une diminution de la vitesse maximale et de la distance
parcourue sur tapis roulant ainsi qu’une diminution de l’activité physique volontaire. Les études
s’entendent de plus en plus pour dire que l’invalidation génétique de l’AMPK altère la fonction
mitochondriale de la cellule musculaire, et plus particulièrement au niveau de la chaîne de transport
des électrons. Un défaut de la fonction mitochondriale ainsi qu’une diminution des échanges entre le
myocyte et la circulation sanguine sont des facteurs qui peuvent contribuer à réduire la production
d’énergie nécessaire au maintien d’une activité contractile normale chez la souris déficiente en
AMPK. De plus, des dommages ultrastructuraux au niveau de la cellule musculaire ainsi que la
présence de fibres musculaires nécrosées et pourvues de noyaux centraux furent récemment
rapportés dans les muscles TA et gastrocnémien des souris AMPKα1α2 mdKO et AMPKβ1β2M KO
[306, 455]. L’ensemble de ces résultats suggère l’implication de l’AMPK dans le maintien de
l’intégrité du tissu musculaire squelettique. De plus, ces nouvelles données peuvent contribuer à
expliquer la diminution de la force maximale absolue observée chez la souris génétiquement
invalidée pour l’AMPK.
Somme toute, bien que l’implication de l’AMPK dans la régulation du transport du glucose induit par
la contraction musculaire demeure controversée, ces nombreuses études ont permis de révéler
l’importance de l’AMPK, à bien des égards, dans le tissu musculaire squelettique. De plus, il ne faut
pas négliger que tous les modèles utilisés sont invalidés pour l’AMPK depuis leur développement
embryonnaire. La présence de mécanismes compensatoires afin de contrecarrer les impacts
physiologiques et métaboliques occasionnés par la perte de l’AMPK ne peut être écartée dans
l’interprétation des résultats de chacune de ces études.
Du point de vue des perspectives, les résultats obtenus par Abbott et ses collaborateurs laissent
suggérer que l’implication de l’AMPK dans le transport du glucose induit par la contraction musculaire
serait dépendante de la durée de la contraction [464]. Il serait donc pertinent de comparer les
résultats que nous avons obtenus, soit une diminution de la captation du glucose à la suite d’une
contraction d’intensité élevée d’une durée de deux minutes, à différents protocoles de contraction
d’intensité et de durée variable. L’apport des différents substrats énergétiques utilisés au cours d’un
exercice physique varie en fonction de l’intensité et de la durée de celui-ci, il serait donc intéressant

108
de clarifier l’implication de l’AMPK dans la régulation du transport du glucose en fonction de ces
mêmes paramètres.
De plus, nous ne pouvons exclure la présence de mécanismes compensatoires, connus ou non, lors
de l’utilisation d’un modèle murin transgénique, telle la souris AMPK KD1 que nous avons utilisée.
Par ailleurs, Dzamko et ses collaborateurs ont démontré que la phosphorylation de plusieurs kinases
est plus importante chez la souris AMPK KD1 en réponse à une contraction musculaire
comparativement aux souris contrôles [465]. Ainsi, l’invalidation génétique de l’AMPK depuis le
développement embryonnaire n’est sans doute pas le modèle idéal pour étudier son rôle dans le
métabolisme du glucose au cours d’une contraction musculaire. Au cours des dernières années, un
nouveau modèle d’invalidation a vu le jour. Il s’agit de l’invalidation génétique conditionnelle qui
consiste à contrôler le moment de l’induction de la mutation grâce, entre autre, à l’utilisation de
protéines de fusion sur un gène spécifique. Cette nouvelle approche d’invalidation pourrait nous
permettre d’invalider le gène de l’AMPK au moment voulu, c’est-à-dire juste avant d’entamer des
protocoles de contractions musculaires/d’exercice physique.
Il est probable que des mécanismes compensatoires pourraient également survenir chez ces
modèles d’invalidation conditionnelle. Ainsi, des études phosphoprotéomiques sur des muscles
préalablement contractés de souris contrôles et/ou de souris transgéniques, suivant différents
protocoles de contraction, pourraient nous donner des pistes sur de nouvelles kinases phosphorylées
au cours d’un exercice physique. Des études complémentaires seraient nécessaires afin de clarifier
le rôle de ces protéines dans la régulation du transport du glucose induit par la contraction
musculaire. Ceci dit, il est primordial de garder à l’esprit qu’il s’agit de modèles murins et qu’il existe
des différences importantes entre la souris et l’homme, entre autre chose, au niveau du métabolisme
du glucose.

109
L’incubation à long terme du muscle épitrochléen cause
l’induction d’iNOS et l’augmentation de l’expression de GLUT1
Tel que pour la contraction musculaire par stimulation électrique, l’utilisation de la technique
d’incubation ex vivo d’un muscle squelettique procure certains avantages. En effet, elle permet
d’étudier les impacts de différents traitements sur diverses fonctions musculaires tout en éliminant les
facteurs exogènes qui pourraient influencer l’interprétation des résultats. Toutefois, des changements
au niveau du métabolisme énergétique musculaire furent observés lors d’une incubation ex vivo
prolongée, et plus particulièrement au niveau du transport du glucose [466]. Bien que les modalités
d’incubation soient maximisées pour simuler les conditions physiologiques environnantes du muscle
squelettique à l’intérieur de l’organisme, il n’en demeure pas moins que l’extraction du muscle, en
soi, représente un stress physiologique.
Il est bien admis que la réponse physiologique à un stress modifie le métabolisme glucidique. De
nombreuses études ont démontré la présence d’une résistance à l’insuline des tissus périphériques
simultanément à une augmentation du transport basal de glucose à la suite de divers stress dont les
accidents traumatiques, les états septiques et post opératoires ainsi qu’à la suite d’un exercice
physique [467-470]. La libération de cytokines en présence d’un stress est l’un des mécanismes
proposés pour expliquer ces changements observés au niveau du métabolisme du glucose.
L’implication de la voie de synthèse du NO dans l’action des cytokines est fortement suggérée.
Le NO joue un rôle essentiel dans divers processus cellulaires dont la vasodilatation des vaisseaux
sanguins, la transmission du signal nerveux et la réponse immunitaire. Toutefois, la surproduction de
NO par l’induction prolongée d’iNOS est associée à diverses conditions pathologiques dont la
résistance à l’insuline musculaire. Par ailleurs, nous avons précédemment démontré que l’induction
d’iNOS par un cocktail de cytokines/LPS modifie l’expression des transporteurs de glucose GLUT1 et
GLUT4 dans les cellules L6. L’ensemble de ces modifications altère le transport basal de glucose et
cause une résistance à l’insuline. L’utilisation d’un inhibiteur de NOS, le L-NAME, rétablit l’expression
de GLUT1 à des niveaux basaux dans ce modèle cellulaire suggérant une interaction entre iNOS et
la régulation de l’expression de GLUT1 [15].

110
À la suite d’une période de dix heures d’incubation ex vivo, nous avons observé une augmentation
de l’expression de la protéine iNOS dans le muscle épitrochléen de rat. Cette induction est
concomitante à une diminution de l’expression des isoformes eNOS et nNOS. Il est possible que la
diminution de l’expression des isoformes eNOS et nNOS soit attribuable à l’augmentation de
l’expression de iNOS. La protéine iNOS est connue pour induire une grande quantité de NO
comparativement aux deux autres isoformes. Conséquement, nous avons mesuré une augmentation
des concentrations de NO dans le milieu d’incubation. De plus, nous avons démontré que l’induction
d’iNOS cause une surexpression de GLUT1 lors de l’incubation ex vivo prolongée du muscle
épitrochléen de rat. L’ajout d’un inhibiteur spécifique d’iNOS, le 1400W, rétablit l’expression de
GLUT1 à des niveaux basaux. Toutefois, l’expression de GLUT4 n’est pas altérée en réponse à une
incubation prolongée du muscle épitrochléen. Ceci dit, nous n’avons détecté aucun défaut au niveau
du transport basal de glucose et de celui stimulé par un traitement à l’insuline à la suite d’une période
de dix heures d’incubation, malgré l’induction d’iNOS.
Afin de tester l’impact d’une exposition à de fortes concentrations de NO sur une plus longue période
de temps, nous avons traité des cellules musculaires L6 avec un donneur de NO, le NOC-15, pour
une période de vingt-quatre heures. Tel qu’observé lors d’une incubation ex vivo de dix heures du
muscle épitrochléen, nous avons observé une augmentation de l’expression de GLUT1, mais aucune
variation de l’expression de GLUT4. Cette surexpression de GLUT1 dans les cellules L6 se traduit
par une augmentation dose-dépendante significative du transport basal de glucose.
L’ensemble de ces résultats suggère que l’incubation ex vivo prolongée du muscle épitrochléen induit
l’expression d’iNOS et par conséquence, accroît la synthèse de NO. La production de NO entraîne
une augmentation de l’expression de GLUT1 sans toutefois, modifier de façon significative le
transport du glucose au cours d’une incubation sur une période de dix heures. Néanmoins, un
traitement de vingt-quatre heures avec un donneur de NO dans les cellules musculaires L6 résulte
en une surexpression de GLUT1 et une augmentation significative du transport basal de glucose. Les
résultats obtenus grâce au modèle cellulaire nous permettent de renforcer le lien causal entre la
synthèse de NO et la régulation de l’expression de GLUT1.

111
Dans notre modèle expérimental, nous observons une induction nette de l’expression d’iNOS dans le
muscle épitrochléen de rat à partir de la quatrième heure d’incubation ex vivo. Des résultats
similaires furent obtenus chez la souris au cours d’une étude subséquente. En effet, nous avons
mesuré une augmentation de l’ARNm d’iNOS dans les muscles EDL et soléaire de souris à la suite
d’une incubation ex vivo d’une durée de quatre heures. À ce jour, les cytokines et les acides gras
sont les deux stimuli connus pour induire iNOS dans la cellule musculaire. Le muscle squelettique
contient plusieurs types de cellules autres que les myocytes dont des cellules immunes résidentes
parmi lesquelles on retrouve des macrophages. Il est possible que ceux-ci puissent libérer des
cytokines en réponse au stress occasionné par l’isolation du muscle (la dénervation, la rupture des
tendons, l’interruption vasculaire) et par conséquent, stimuler l’induction d’iNOS. Des expériences
complémentaires seront nécessaires pour confirmer cette hypothèse. Néanmoins, il demeure que
l’induction d’iNOS stimule la synthèse de NO ce qui résulte en une augmentation de l’expression de
GLUT1 dans le muscle squelettique au cours d’une incubation prolongée ex vivo. L’interaction entre
le NO et l’expression de GLUT1 fut rapportée dans d’autres modèles. Les mécanismes responsables
de cette interaction demandent à être davantage étudiés. Toutefois, il fut récemment démontré que la
protéine ATM (de l’anglais ataxia telangiectasia mutated) régule l’expression de GLUT1 à la
membrane plasmique dans les cellules musculaires L6 [471]. La protéine ATM est une
sérine/thréonine appartenant à la famille des PI3K connue pour son rôle dans la réparation des
dommages au niveau de l’acide désoxyribonucléique (ADN). Son implication dans la régulation du
transport du glucose dans les cellules musculaires fut nouvellement proposée [472, 473]. De plus,
différents modèles cellulaires suggèrent que le NO active la protéine ATM. Il est possible que cette
dernière participe à l’augmentation de l’expression de GLUT1 en réponse à l’accumulation de NO
dans notre étude. Des expériences supplémentaires seront nécessaires afin de valider cette
hypothèse.
Contrairement aux résultats préalablement publiés par notre laboratoire, l’induction d’iNOS dans
notre présent modèle expérimental n’entraîne pas de variations de l’expression de GLUT4. Une
étude a récemment rapporté l’implication du facteur induit par l’hypoxie 1α (HIF-1α, de l’anglais
hypoxia inductible factor 1α) dans la translocation des GLUT4 à la membrane plasmique et dans le
transport du glucose lors d’une stimulation à l’insuline dans les cellules musculaires C2C12. En effet,
l’inhibition de HIF-1α abolit le transport du glucose stimulé par l’insuline ainsi que la translocation des

112
GLUT4. À l’inverse, une forme constitutivement active de HIF-1α entraîne, au contraire, une
translocation des GLUT4 à la membrane plasmique comparable à celle induite par une stimulation à
l’insuline [474]. La protéine HIF-1α est un facteur de transcription activé en réponse à une diminution
d’oxygène à l’intérieur des cellules [475]. Toutefois, il est également suggéré que la protéine HIF-1α
participe à la régulation de diverses fonctions cellulaires en condition de normoxie. Entre autre
chose, il fut démontré que la cytokine TNFα module l’activité de HIF1α dans les cellules épithéliales
alvéolaires en absence d’hypoxie [476]. Des résultats préliminaires de notre laboratoire proposent
une augmentation de l’ARNm de HIF-1α dans le muscle épitrochléen de rat à la suite de dix heures
d’incubation ex vivo. Ainsi, il est concevable que l’activation de la protéine HIF-1α dans notre modèle
expérimental soit responsable du maintien de l’expression de GLUT4 et du transport du glucose
stimulé par l’insuline en dépit de l’induction d’iNOS et de la production de NO. Des expériences
supplémentaires seront nécessaires afin de valider cette hypothèse. De plus, afin de déterminer la
présence ou non d’hypoxie dans notre modèle expérimental, il serait pertinent d’effectuer d’autres
mesures que celle de HIF-1α.
Ainsi, en dépit de l’induction d’iNOS lors d’une incubation ex vivo prolongée du muscle épitrochléen,
la surexpression des GLUT1 ne résulte pas en une augmentation du transport basal de glucose et la
production de NO n’affecte pas l’expression des GLUT4. Toutefois, une exposition de vingt-quatre
heures à un donneur de NO induit une augmentation du transport basal de glucose dans les cellules
musculaires L6. Il est envisageable que l’incubation ex vivo du muscle épitrochléen sur une plus
longue période résulte en une augmentation significative du transport basal de glucose occasionnée
par une accumulation plus importante de NO tel qu’observée dans les cellules musculaires L6
traitées avec le NOC-15.
Finalement, il fut démontré que la résistance à l’insuline observée en situation de stress
postopératoire est proportionnelle à l’intensité du stress initial [477]. Il est également possible que le
stress induit par la technique d’incubation ex vivo d’un muscle squelettique ne soit pas assez
important pour induire des modifications majeures au niveau du métabolisme du glucose malgré la
présence de marqueurs inflammatoires. Il n’en demeure pas moins que la présence possible d’une
réaction inflammatoire doit être prise en considération lors du choix de cette technique.

113
Du point de vue des perspectives, il est concevable que l’augmentation de GLUT1 en présence de
fortes concentrations de NO soit un mécanisme de défense cellulaire au cours d’une réaction
inflammatoire. Entre autre chose, il fut démontré que l’incubation d’un muscle ex vivo induit la
production d’espèces réactives d’oxygène (ROS) dont l’anion superoxyde (O2-) et le peroxyde
d’hydrogène (H2O2) [478]. L’interaction entre l’O2- et le NO forme le peroxynitrite, un puissant
oxydant, capable de nitration sur différentes protéines de la cascade de signalisation de l’insuline
dans le muscle squelettique tel que décrit à la section 4.3.1. Plus encore, il fut récemment démontré
que GLUT1 régule la production de ROS et prévient ainsi la diminution du transport du glucose
stimulé par l’insuline. [479]. À la lumière de ces données nouvelles, il serait pertinent de mesurer les
niveaux de ROS dans notre modèle expérimental, de trouver les conditions optimales d’incubation
pour minimiser la production de ROS et ainsi clarifier l’impact de tels changements sur l’expression
de GLUT1. Ceci dit, il serait également opportun d’identifier la source de production d’iNOS et ainsi
développer de meilleures conditions d’incubation pour de futurs protocoles expérimentaux. Ces
données nous permettraient d’en connaître davantage sur la régulation d’iNOS dans le muscle
squelettique.
En conclusion, le tissu musculaire squelettique est un organe important dans le maintien de
l’homéostasie du glucose. La régulation du métabolisme du glucose dans le muscle squelettique fait
intervenir des nombreux mécanismes. Plusieurs conditions physiologiques et métaboliques
interagissent avec ceux-ci et peuvent engendrer des perturbations métaboliques majeures.
Néanmoins, et heureusement, la pratique régulière d’activité physique permet de prévenir plusieurs
effets métaboliques indésirables et procure, entre autres, de nombreux effets bénéfiques sur la santé
du métabolisme musculaire.


115
BIBLIOGRAPHIE
1. Tortora, G.J., Derrickson, B., Principes d'Anatomie et de Physiologie. 2e Édition ed. 2007, Canada: Éditions du Renouveau Pédagogique INC.
2. Schiaffino, S. and C. Reggiani, Fiber types in mammalian skeletal muscles. Physiological reviews, 2011. 91(4): p. 1447-531.
3. DeFronzo, R.A., et al., The effect of insulin on the disposal of intravenous glucose. Results from indirect calorimetry and hepatic and femoral venous catheterization. Diabetes, 1981. 30(12): p. 1000-7.
4. Wood, I.S. and P. Trayhurn, Glucose transporters (GLUT and SGLT): expanded families of sugar transport proteins. Br J Nutr, 2003. 89(1): p. 3-9.
5. Joost, H.G. and B. Thorens, The extended GLUT-family of sugar/polyol transport facilitators: nomenclature, sequence characteristics, and potential function of its novel members (review). Mol Membr Biol, 2001. 18(4): p. 247-56.
6. Sadiq, H.F., et al., Intra-uterine growth restriction differentially regulates perinatal brain and skeletal muscle glucose transporters. Brain Res, 1999. 823(1-2): p. 96-103.
7. Schroeder, R.E., et al., Effect of maternal diabetes upon fetal rat myocardial and skeletal muscle glucose transporters. Pediatr Res, 1997. 41(1): p. 11-9.
8. Stuart, C.A., et al., Hexose transporter mRNAs for GLUT4, GLUT5, and GLUT12 predominate in human muscle. American journal of physiology. Endocrinology and metabolism, 2006. 291(5): p. E1067-73.
9. Marette, A., et al., Abundance, localization, and insulin-induced translocation of glucose transporters in red and white muscle. The American journal of physiology, 1992. 263(2 Pt 1): p. C443-52.
10. Wang, W., et al., Insulin unmasks a COOH-terminal Glut4 epitope and increases glucose transport across T-tubules in skeletal muscle. J Cell Biol, 1996. 135(2): p. 415-30.
11. Goodyear, L.J., et al., Glucose transporter number, activity, and isoform content in plasma membranes of red and white skeletal muscle. Am J Physiol, 1991. 261(5 Pt 1): p. E556-61.
12. Kern, M., et al., Insulin responsiveness in skeletal muscle is determined by glucose transporter (Glut4) protein level. The Biochemical journal, 1990. 270(2): p. 397-400.
13. Duehlmeier, R., et al., Distribution patterns of the glucose transporters GLUT4 and GLUT1 in skeletal muscles of rats (Rattus norvegicus), pigs (Sus scrofa), cows (Bos taurus), adult goats, goat kids (Capra hircus), and camels (Camelus dromedarius). Comp Biochem Physiol A Mol Integr Physiol, 2007. 146(2): p. 274-82.
14. Mitsumoto, Y., et al., Differential expression of the GLUT1 and GLUT4 glucose transporters during differentiation of L6 muscle cells. Biochemical and biophysical research communications, 1991. 175(2): p. 652-9.
15. Bedard, S., B. Marcotte, and A. Marette, Cytokines modulate glucose transport in skeletal muscle by inducing the expression of inducible nitric oxide synthase. The Biochemical journal, 1997. 325 ( Pt 2): p. 487-93.
16. Kozlovsky, N., et al., Transcriptional activation of the Glut1 gene in response to oxidative stress in L6 myotubes. The Journal of biological chemistry, 1997. 272(52): p. 33367-72.
17. Ciaraldi, T.P., et al., Skeletal muscle GLUT1 transporter protein expression and basal leg glucose uptake are reduced in type 2 diabetes. The Journal of clinical endocrinology and metabolism, 2005. 90(1): p. 352-8.

116
18. Bourlier, V., et al., Enhanced glucose metabolism is preserved in cultured primary myotubes from obese donors in response to exercise training. The Journal of clinical endocrinology and metabolism, 2013. 98(9): p. 3739-47.
19. Robinson, R., et al., Glucose transport in L6 myoblasts overexpressing GLUT1 and GLUT4. The Journal of biological chemistry, 1993. 268(29): p. 22119-26.
20. Cleasby, M.E., et al., Acute bidirectional manipulation of muscle glucose uptake by in vivo electrotransfer of constructs targeting glucose transporter genes. Diabetes, 2005. 54(9): p. 2702-11.
21. Marshall, B.A., et al., Germline manipulation of glucose homeostasis via alteration of glucose transporter levels in skeletal muscle. The Journal of biological chemistry, 1993. 268(25): p. 18442-5.
22. Hansen, P.A., et al., Dissociation of GLUT4 translocation and insulin-stimulated glucose transport in transgenic mice overexpressing GLUT1 in skeletal muscle. The Journal of biological chemistry, 1998. 273(29): p. 18173-9.
23. Rudich, A., et al., Indinavir uncovers different contributions of GLUT4 and GLUT1 towards glucose uptake in muscle and fat cells and tissues. Diabetologia, 2003. 46(5): p. 649-58.
24. Zisman, A., et al., Targeted disruption of the glucose transporter 4 selectively in muscle causes insulin resistance and glucose intolerance. Nat Med, 2000. 6(8): p. 924-8.
25. Ryder, J.W., et al., Postexercise glucose uptake and glycogen synthesis in skeletal muscle from GLUT4-deficient mice. FASEB journal : official publication of the Federation of American Societies for Experimental Biology, 1999. 13(15): p. 2246-56.
26. Henriksen, E.J., et al., Glucose transporter protein content and glucose transport capacity in rat skeletal muscles. Am J Physiol, 1990. 259(4 Pt 1): p. E593-8.
27. Megeney, L.A., et al., Effects of muscle activity and fiber composition on glucose transport and GLUT-4. Am J Physiol, 1993. 264(4 Pt 1): p. E583-93.
28. Richardson, J.M., et al., Differential regulation of glucose transporter activity and expression in red and white skeletal muscle. J Biol Chem, 1991. 266(19): p. 12690-4.
29. Rossetti, L., et al., Peripheral but not hepatic insulin resistance in mice with one disrupted allele of the glucose transporter type 4 (GLUT4) gene. J Clin Invest, 1997. 100(7): p. 1831-9.
30. Stenbit, A.E., et al., GLUT4 heterozygous knockout mice develop muscle insulin resistance and diabetes. Nat Med, 1997. 3(10): p. 1096-101.
31. Tsao, T.S., et al., Prevention of insulin resistance and diabetes in mice heterozygous for GLUT4 ablation by transgenic complementation of GLUT4 in skeletal muscle. Diabetes, 1999. 48(4): p. 775-82.
32. Tsao, T.S., et al., Enhanced insulin action due to targeted GLUT4 overexpression exclusively in muscle. Diabetes, 1996. 45(1): p. 28-36.
33. Tsao, T.S., et al., Metabolic adaptations in skeletal muscle overexpressing GLUT4: effects on muscle and physical activity. FASEB journal : official publication of the Federation of American Societies for Experimental Biology, 2001. 15(6): p. 958-69.
34. Stuart, C.A., et al., Insulin-stimulated translocation of glucose transporter (GLUT) 12 parallels that of GLUT4 in normal muscle. The Journal of clinical endocrinology and metabolism, 2009. 94(9): p. 3535-42.
35. Douen, A.G., et al., Exercise induces recruitment of the "insulin-responsive glucose transporter". Evidence for distinct intracellular insulin- and exercise-recruitable transporter pools in skeletal muscle. The Journal of biological chemistry, 1990. 265(23): p. 13427-30.

117
36. Neufer, P.D. and G.L. Dohm, Exercise induces a transient increase in transcription of the GLUT-4 gene in skeletal muscle. The American journal of physiology, 1993. 265(6 Pt 1): p. C1597-603.
37. Ren, J.M., et al., Exercise induces rapid increases in GLUT4 expression, glucose transport capacity, and insulin-stimulated glycogen storage in muscle. The Journal of biological chemistry, 1994. 269(20): p. 14396-401.
38. Kraniou, Y., et al., Effects of exercise on GLUT-4 and glycogenin gene expression in human skeletal muscle. Journal of applied physiology, 2000. 88(2): p. 794-6.
39. Greiwe, J.S., J.O. Holloszy, and C.F. Semenkovich, Exercise induces lipoprotein lipase and GLUT-4 protein in muscle independent of adrenergic-receptor signaling. Journal of applied physiology, 2000. 89(1): p. 176-81.
40. Fueger, P.T., et al., Glucose kinetics and exercise tolerance in mice lacking the GLUT4 glucose transporter. The Journal of physiology, 2007. 582(Pt 2): p. 801-12.
41. Tremblay, F., M.J. Dubois, and A. Marette, Regulation of GLUT4 traffic and function by insulin and contraction in skeletal muscle. Front Biosci, 2003. 8: p. d1072-84.
42. Rosen, O.M., Insulin receptor as a tyrosine protein kinase. Ann N Y Acad Sci, 1986. 463: p. 13-9.
43. Ellis, L., et al., Replacement of insulin receptor tyrosine residues 1162 and 1163 compromises insulin-stimulated kinase activity and uptake of 2-deoxyglucose. Cell, 1986. 45(5): p. 721-32.
44. White, M.F., et al., Mutation of the insulin receptor at tyrosine 960 inhibits signal transmission but does not affect its tyrosine kinase activity. Cell, 1988. 54(5): p. 641-9.
45. Youngren, J.F., Regulation of insulin receptor function. Cell Mol Life Sci, 2007. 64(7-8): p. 873-91.
46. Sun, X.J., et al., Structure of the insulin receptor substrate IRS-1 defines a unique signal transduction protein. Nature, 1991. 352(6330): p. 73-7.
47. Sun, X.J., et al., Role of IRS-2 in insulin and cytokine signalling. Nature, 1995. 377(6545): p. 173-7.
48. Lavan, B.E., W.S. Lane, and G.E. Lienhard, The 60-kDa phosphotyrosine protein in insulin-treated adipocytes is a new member of the insulin receptor substrate family. J Biol Chem, 1997. 272(17): p. 11439-43.
49. Lavan, B.E., et al., A novel 160-kDa phosphotyrosine protein in insulin-treated embryonic kidney cells is a new member of the insulin receptor substrate family. J Biol Chem, 1997. 272(34): p. 21403-7.
50. Cai, D., et al., Two new substrates in insulin signaling, IRS5/DOK4 and IRS6/DOK5. J Biol Chem, 2003. 278(28): p. 25323-30.
51. Kido, Y., et al., Tissue-specific insulin resistance in mice with mutations in the insulin receptor, IRS-1, and IRS-2. J Clin Invest, 2000. 105(2): p. 199-205.
52. Huang, C., et al., Differential contribution of insulin receptor substrates 1 versus 2 to insulin signaling and glucose uptake in l6 myotubes. J Biol Chem, 2005. 280(19): p. 19426-35.
53. Long, Y.C., et al., Insulin receptor substrates Irs1 and Irs2 coordinate skeletal muscle growth and metabolism via the Akt and AMPK pathways. Mol Cell Biol, 2011. 31(3): p. 430-41.
54. Burks, D.J., et al., Heterologous pleckstrin homology domains do not couple IRS-1 to the insulin receptor. The Journal of biological chemistry, 1997. 272(44): p. 27716-21.
55. Yenush, L., et al., The pleckstrin homology and phosphotyrosine binding domains of insulin receptor substrate 1 mediate inhibition of apoptosis by insulin. Molecular and cellular biology, 1998. 18(11): p. 6784-94.

118
56. Yenush, L., et al., The pleckstrin homology domain is the principal link between the insulin receptor and IRS-1. J Biol Chem, 1996. 271(39): p. 24300-6.
57. Backer, J.M., et al., Phosphatidylinositol 3'-kinase is activated by association with IRS-1 during insulin stimulation. EMBO J, 1992. 11(9): p. 3469-79.
58. Taniguchi, C.M., B. Emanuelli, and C.R. Kahn, Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol, 2006. 7(2): p. 85-96.
59. Escobedo, J.A., et al., cDNA cloning of a novel 85 kd protein that has SH2 domains and regulates binding of PI3-kinase to the PDGF beta-receptor. Cell, 1991. 65(1): p. 75-82.
60. Skolnik, E.Y., et al., Cloning of PI3 kinase-associated p85 utilizing a novel method for expression/cloning of target proteins for receptor tyrosine kinases. Cell, 1991. 65(1): p. 83-90.
61. Otsu, M., et al., Characterization of two 85 kd proteins that associate with receptor tyrosine kinases, middle-T/pp60c-src complexes, and PI3-kinase. Cell, 1991. 65(1): p. 91-104.
62. Antonetti, D.A., P. Algenstaedt, and C.R. Kahn, Insulin receptor substrate 1 binds two novel splice variants of the regulatory subunit of phosphatidylinositol 3-kinase in muscle and brain. Mol Cell Biol, 1996. 16(5): p. 2195-203.
63. Hiles, I.D., et al., Phosphatidylinositol 3-kinase: structure and expression of the 110 kd catalytic subunit. Cell, 1992. 70(3): p. 419-29.
64. Hu, P., et al., Cloning of a novel, ubiquitously expressed human phosphatidylinositol 3-kinase and identification of its binding site on p85. Mol Cell Biol, 1993. 13(12): p. 7677-88.
65. Vanhaesebroeck, B., et al., P110delta, a novel phosphoinositide 3-kinase in leukocytes. Proc Natl Acad Sci U S A, 1997. 94(9): p. 4330-5.
66. Chantry, D., et al., p110delta, a novel phosphatidylinositol 3-kinase catalytic subunit that associates with p85 and is expressed predominantly in leukocytes. J Biol Chem, 1997. 272(31): p. 19236-41.
67. Vanhaesebroeck, B., et al., The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol, 2010. 11(5): p. 329-41.
68. Tsakiridis, T., et al., Multiple roles of phosphatidylinositol 3-kinase in regulation of glucose transport, amino acid transport, and glucose transporters in L6 skeletal muscle cells. Endocrinology, 1995. 136(10): p. 4315-22.
69. Brozinick, J.T., Jr. and M.J. Birnbaum, Insulin, but not contraction, activates Akt/PKB in isolated rat skeletal muscle. J Biol Chem, 1998. 273(24): p. 14679-82.
70. Jones, P.F., T. Jakubowicz, and B.A. Hemmings, Molecular cloning of a second form of rac protein kinase. Cell Regul, 1991. 2(12): p. 1001-9.
71. Altomare, D.A., et al., Cloning, chromosomal localization and expression analysis of the mouse Akt2 oncogene. Oncogene, 1995. 11(6): p. 1055-60.
72. Brodbeck, D., P. Cron, and B.A. Hemmings, A human protein kinase Bgamma with regulatory phosphorylation sites in the activation loop and in the C-terminal hydrophobic domain. J Biol Chem, 1999. 274(14): p. 9133-6.
73. Nakatani, K., et al., Identification of a human Akt3 (protein kinase B gamma) which contains the regulatory serine phosphorylation site. Biochem Biophys Res Commun, 1999. 257(3): p. 906-10.
74. Cho, H., et al., Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta). Science, 2001. 292(5522): p. 1728-31.
75. Garofalo, R.S., et al., Severe diabetes, age-dependent loss of adipose tissue, and mild growth deficiency in mice lacking Akt2/PKB beta. J Clin Invest, 2003. 112(2): p. 197-208.

119
76. Alessi, D.R., et al., Mechanism of activation of protein kinase B by insulin and IGF-1. The EMBO journal, 1996. 15(23): p. 6541-51.
77. Milburn, C.C., et al., Binding of phosphatidylinositol 3,4,5-trisphosphate to the pleckstrin homology domain of protein kinase B induces a conformational change. Biochem J, 2003. 375(Pt 3): p. 531-8.
78. Alessi, D.R., et al., Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Current biology : CB, 1997. 7(4): p. 261-9.
79. Sarbassov, D.D., et al., Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science, 2005. 307(5712): p. 1098-101.
80. Hartung, A., et al., The Akt substrate Girdin is a regulator of insulin signaling in myoblast cells. Biochimica et biophysica acta, 2013. 1833(12): p. 2803-11.
81. Kane, S., et al., A method to identify serine kinase substrates. Akt phosphorylates a novel adipocyte protein with a Rab GTPase-activating protein (GAP) domain. The Journal of biological chemistry, 2002. 277(25): p. 22115-8.
82. Kane, S. and G.E. Lienhard, Calmodulin binds to the Rab GTPase activating protein required for insulin-stimulated GLUT4 translocation. Biochem Biophys Res Commun, 2005. 335(1): p. 175-80.
83. Thong, F.S., P.J. Bilan, and A. Klip, The Rab GTPase-activating protein AS160 integrates Akt, protein kinase C, and AMP-activated protein kinase signals regulating GLUT4 traffic. Diabetes, 2007. 56(2): p. 414-23.
84. Kramer, H.F., et al., AS160 regulates insulin- and contraction-stimulated glucose uptake in mouse skeletal muscle. The Journal of biological chemistry, 2006. 281(42): p. 31478-85.
85. Chen, S., et al., Mice with AS160/TBC1D4-Thr649Ala knockin mutation are glucose intolerant with reduced insulin sensitivity and altered GLUT4 trafficking. Cell metabolism, 2011. 13(1): p. 68-79.
86. Taylor, E.B., et al., Discovery of TBC1D1 as an insulin-, AICAR-, and contraction-stimulated signaling nexus in mouse skeletal muscle. The Journal of biological chemistry, 2008. 283(15): p. 9787-96.
87. Treebak, J.T., et al., Potential role of TBC1D4 in enhanced post-exercise insulin action in human skeletal muscle. Diabetologia, 2009. 52(5): p. 891-900.
88. Roach, W.G., et al., Substrate specificity and effect on GLUT4 translocation of the Rab GTPase-activating protein Tbc1d1. The Biochemical journal, 2007. 403(2): p. 353-8.
89. Peck, G.R., et al., Insulin-stimulated phosphorylation of the Rab GTPase-activating protein TBC1D1 regulates GLUT4 translocation. The Journal of biological chemistry, 2009. 284(44): p. 30016-23.
90. Chen, S., et al., Complementary regulation of TBC1D1 and AS160 by growth factors, insulin and AMPK activators. The Biochemical journal, 2008. 409(2): p. 449-59.
91. Vichaiwong, K., et al., Contraction regulates site-specific phosphorylation of TBC1D1 in skeletal muscle. The Biochemical journal, 2010. 431(2): p. 311-20.
92. Dokas, J., et al., Conventional knockout of Tbc1d1 in mice impairs insulin- and AICAR-stimulated glucose uptake in skeletal muscle. Endocrinology, 2013. 154(10): p. 3502-14.
93. Chadt, A., et al., Tbc1d1 mutation in lean mouse strain confers leanness and protects from diet-induced obesity. Nat Genet, 2008. 40(11): p. 1354-9.
94. Szekeres, F., et al., The Rab-GTPase-activating protein TBC1D1 regulates skeletal muscle glucose metabolism. Am J Physiol Endocrinol Metab, 2012. 303(4): p. E524-33.

120
95. Middelbeek, R.J., et al., Insulin stimulation regulates AS160 and TBC1D1 phosphorylation sites in human skeletal muscle. Nutr Diabetes, 2013. 3: p. e74.
96. Sakamoto, K. and G.D. Holman, Emerging role for AS160/TBC1D4 and TBC1D1 in the regulation of GLUT4 traffic. Am J Physiol Endocrinol Metab, 2008. 295(1): p. E29-37.
97. Ramm, G., et al., A role for 14-3-3 in insulin-stimulated GLUT4 translocation through its interaction with the RabGAP AS160. The Journal of biological chemistry, 2006. 281(39): p. 29174-80.
98. Khayat, Z.A., et al., Insulin-induced actin filament remodeling colocalizes actin with phosphatidylinositol 3-kinase and GLUT4 in L6 myotubes. Journal of cell science, 2000. 113 Pt 2: p. 279-90.
99. Brozinick, J.T., Jr., et al., Disruption of cortical actin in skeletal muscle demonstrates an essential role of the cytoskeleton in glucose transporter 4 translocation in insulin-sensitive tissues. The Journal of biological chemistry, 2004. 279(39): p. 40699-706.
100. Ueda, S., T. Kataoka, and T. Satoh, Activation of the small GTPase Rac1 by a specific guanine-nucleotide-exchange factor suffices to induce glucose uptake into skeletal-muscle cells. Biology of the cell / under the auspices of the European Cell Biology Organization, 2008. 100(11): p. 645-57.
101. JeBailey, L., et al., Skeletal muscle cells and adipocytes differ in their reliance on TC10 and Rac for insulin-induced actin remodeling. Mol Endocrinol, 2004. 18(2): p. 359-72.
102. Ueda, S., et al., Crucial role of the small GTPase Rac1 in insulin-stimulated translocation of glucose transporter 4 to the mouse skeletal muscle sarcolemma. FASEB J, 2010. 24(7): p. 2254-61.
103. Sylow, L., et al., Rac1 signaling is required for insulin-stimulated glucose uptake and is dysregulated in insulin-resistant murine and human skeletal muscle. Diabetes, 2013. 62(6): p. 1865-75.
104. Sylow, L., et al., Akt and Rac1 signaling are jointly required for insulin-stimulated glucose uptake in skeletal muscle and downregulated in insulin resistance. Cell Signal, 2014. 26(2): p. 323-31.
105. Nozaki, S., et al., Akt2 regulates Rac1 activity in the insulin-dependent signaling pathway leading to GLUT4 translocation to the plasma membrane in skeletal muscle cells. Cell Signal, 2013. 25(6): p. 1361-71.
106. Takenaka, N., et al., A critical role of the small GTPase Rac1 in Akt2-mediated GLUT4 translocation in mouse skeletal muscle. FEBS J, 2014. 281(5): p. 1493-504.
107. Clausen, T., J. Elbrink, and A.B. Dahl-Hansen, The relationship between the transport of glucose and cations across cell membranes in isolated tissues. IX. The role of cellular calcium in the activation of the glucose transport system in rat soleus muscle. Biochimica et biophysica acta, 1975. 375(2): p. 292-308.
108. Youn, J.H., E.A. Gulve, and J.O. Holloszy, Calcium stimulates glucose transport in skeletal muscle by a pathway independent of contraction. The American journal of physiology, 1991. 260(3 Pt 1): p. C555-61.
109. Jensen, T.E., et al., Caffeine-induced Ca(2+) release increases AMPK-dependent glucose uptake in rodent soleus muscle. American journal of physiology. Endocrinology and metabolism, 2007. 293(1): p. E286-92.
110. Raney, M.A. and L.P. Turcotte, Evidence for the involvement of CaMKII and AMPK in Ca2+-dependent signaling pathways regulating FA uptake and oxidation in contracting rodent muscle. Journal of applied physiology, 2008. 104(5): p. 1366-73.

121
111. Egawa, T., et al., Caffeine activates preferentially alpha1-isoform of 5'AMP-activated protein kinase in rat skeletal muscle. Acta physiologica, 2011. 201(2): p. 227-38.
112. Wright, D.C., et al., Ca2+ and AMPK both mediate stimulation of glucose transport by muscle contractions. Diabetes, 2004. 53(2): p. 330-5.
113. Wright, D.C., et al., Contraction- and hypoxia-stimulated glucose transport is mediated by a Ca2+-dependent mechanism in slow-twitch rat soleus muscle. American journal of physiology. Endocrinology and metabolism, 2005. 288(6): p. E1062-6.
114. Chin, D. and A.R. Means, Calmodulin: a prototypical calcium sensor. Trends in cell biology, 2000. 10(8): p. 322-8.
115. Colbran, R.J., et al., Regulatory interactions of the calmodulin-binding, inhibitory, and autophosphorylation domains of Ca2+/calmodulin-dependent protein kinase II. The Journal of biological chemistry, 1988. 263(34): p. 18145-51.
116. Payne, M.E., et al., Calcium/calmodulin-dependent protein kinase II. Characterization of distinct calmodulin binding and inhibitory domains. The Journal of biological chemistry, 1988. 263(15): p. 7190-5.
117. Rose, A.J., B. Kiens, and E.A. Richter, Ca2+-calmodulin-dependent protein kinase expression and signalling in skeletal muscle during exercise. The Journal of physiology, 2006. 574(Pt 3): p. 889-903.
118. Serpiello, F.R., et al., Performance and physiological responses to repeated-sprint exercise: a novel multiple-set approach. European journal of applied physiology, 2011. 111(4): p. 669-78.
119. Witczak, C.A., et al., CaMKII regulates contraction- but not insulin-induced glucose uptake in mouse skeletal muscle. American journal of physiology. Endocrinology and metabolism, 2010. 298(6): p. E1150-60.
120. Richter, E.A., et al., Contraction-associated translocation of protein kinase C in rat skeletal muscle. FEBS letters, 1987. 217(2): p. 232-6.
121. Cleland, P.J., et al., Exercise-induced translocation of protein kinase C and production of diacylglycerol and phosphatidic acid in rat skeletal muscle in vivo. Relationship to changes in glucose transport. The Journal of biological chemistry, 1989. 264(30): p. 17704-11.
122. Wojtaszewski, J.F., et al., Hypoxia and contractions do not utilize the same signaling mechanism in stimulating skeletal muscle glucose transport. Biochimica et biophysica acta, 1998. 1380(3): p. 396-404.
123. Ihlemann, J., H. Galbo, and T. Ploug, Calphostin C is an inhibitor of contraction, but not insulin-stimulated glucose transport, in skeletal muscle. Acta physiologica Scandinavica, 1999. 167(1): p. 69-75.
124. Jensen, T.E., et al., Knockout of the predominant conventional PKC isoform, PKCalpha, in mouse skeletal muscle does not affect contraction-stimulated glucose uptake. American journal of physiology. Endocrinology and metabolism, 2009. 297(2): p. E340-8.
125. Rose, A.J., et al., Effect of exercise on protein kinase C activity and localization in human skeletal muscle. The Journal of physiology, 2004. 561(Pt 3): p. 861-70.
126. Richter, E.A., et al., Differential effect of bicycling exercise intensity on activity and phosphorylation of atypical protein kinase C and extracellular signal-regulated protein kinase in skeletal muscle. The Journal of physiology, 2004. 560(Pt 3): p. 909-18.
127. Nielsen, J.N., et al., Increased atypical PKC activity in endurance-trained human skeletal muscle. Biochem Biophys Res Commun, 2003. 312(4): p. 1147-53.

122
128. Sajan, M.P., et al., AICAR and metformin, but not exercise, increase muscle glucose transport through AMPK-, ERK-, and PDK1-dependent activation of atypical PKC. American journal of physiology. Endocrinology and metabolism, 2010. 298(2): p. E179-92.
129. Wojtaszewski, J.F., et al., Differential regulation of MAP kinase by contraction and insulin in skeletal muscle: metabolic implications. The American journal of physiology, 1999. 277(4 Pt 1): p. E724-32.
130. Hayashi, T., et al., Skeletal muscle contractile activity in vitro stimulates mitogen-activated protein kinase signaling. The American journal of physiology, 1999. 277(4 Pt 1): p. C701-7.
131. Witczak, C.A., et al., JNK1 deficiency does not enhance muscle glucose metabolism in lean mice. Biochemical and biophysical research communications, 2006. 350(4): p. 1063-8.
132. Somwar, R., et al., Activation of p38 mitogen-activated protein kinase alpha and beta by insulin and contraction in rat skeletal muscle: potential role in the stimulation of glucose transport. Diabetes, 2000. 49(11): p. 1794-800.
133. Antonescu, C.N., et al., Reduction of insulin-stimulated glucose uptake in L6 myotubes by the protein kinase inhibitor SB203580 is independent of p38MAPK activity. Endocrinology, 2005. 146(9): p. 3773-81.
134. Ribe, D., et al., Endofacial competitive inhibition of glucose transporter-4 intrinsic activity by the mitogen-activated protein kinase inhibitor SB203580. Endocrinology, 2005. 146(4): p. 1713-7.
135. Ho, R.C., et al., p38gamma MAPK regulation of glucose transporter expression and glucose uptake in L6 myotubes and mouse skeletal muscle. American journal of physiology. Regulatory, integrative and comparative physiology, 2004. 286(2): p. R342-9.
136. Nathan, C., Nitric oxide as a secretory product of mammalian cells. FASEB J, 1992. 6(12): p. 3051-64.
137. Kobzik, L., et al., Nitric oxide in skeletal muscle. Nature, 1994. 372(6506): p. 546-8. 138. Kobzik, L., et al., Endothelial type nitric oxide synthase in skeletal muscle fibers:
mitochondrial relationships. Biochem Biophys Res Commun, 1995. 211(2): p. 375-81. 139. Grozdanovic, Z., et al., Species-independent expression of nitric oxide synthase in the
sarcolemma region of visceral and somatic striated muscle fibers. Cell Tissue Res, 1995. 281(3): p. 493-9.
140. Nakane, M., et al., Cloned human brain nitric oxide synthase is highly expressed in skeletal muscle. FEBS Lett, 1993. 316(2): p. 175-80.
141. Williams, G., et al., Cytokine-induced expression of nitric oxide synthase in C2C12 skeletal muscle myocytes. Am J Physiol, 1994. 267(4 Pt 2): p. R1020-5.
142. Gath, I., et al., Inducible NO synthase II and neuronal NO synthase I are constitutively expressed in different structures of guinea pig skeletal muscle: implications for contractile function. FASEB J, 1996. 10(14): p. 1614-20.
143. Balon, T.W. and J.L. Nadler, Evidence that nitric oxide increases glucose transport in skeletal muscle. Journal of applied physiology, 1997. 82(1): p. 359-63.
144. McConell, G.K., et al., Skeletal muscle nNOS mu protein content is increased by exercise training in humans. American journal of physiology. Regulatory, integrative and comparative physiology, 2007. 293(2): p. R821-8.
145. Bradley, S.J., B.A. Kingwell, and G.K. McConell, Nitric oxide synthase inhibition reduces leg glucose uptake but not blood flow during dynamic exercise in humans. Diabetes, 1999. 48(9): p. 1815-21.
146. Heinonen, I., et al., Effect of nitric oxide synthase inhibition on the exchange of glucose and fatty acids in human skeletal muscle. Nutrition & metabolism, 2013. 10(1): p. 43.

123
147. Ross, R.M., et al., Local nitric oxide synthase inhibition reduces skeletal muscle glucose uptake but not capillary blood flow during in situ muscle contraction in rats. Diabetes, 2007. 56(12): p. 2885-92.
148. Roberts, C.K., et al., Acute exercise increases nitric oxide synthase activity in skeletal muscle. The American journal of physiology, 1999. 277(2 Pt 1): p. E390-4.
149. Merry, T.L., et al., Skeletal muscle glucose uptake during contraction is regulated by nitric oxide and ROS independently of AMPK. American journal of physiology. Endocrinology and metabolism, 2010. 298(3): p. E577-85.
150. Stephens, T.J., et al., 5'-aminoimidazole-4-carboxyamide-ribonucleoside-activated glucose transport is not prevented by nitric oxide synthase inhibition in rat isolated skeletal muscle. Clinical and experimental pharmacology & physiology, 2004. 31(7): p. 419-23.
151. Higaki, Y., et al., Nitric oxide increases glucose uptake through a mechanism that is distinct from the insulin and contraction pathways in rat skeletal muscle. Diabetes, 2001. 50(2): p. 241-7.
152. Etgen, G.J., Jr., D.A. Fryburg, and E.M. Gibbs, Nitric oxide stimulates skeletal muscle glucose transport through a calcium/contraction- and phosphatidylinositol-3-kinase-independent pathway. Diabetes, 1997. 46(11): p. 1915-9.
153. Reid, M.B., Free radicals and muscle fatigue: Of ROS, canaries, and the IOC. Free radical biology & medicine, 2008. 44(2): p. 169-79.
154. Sandstrom, M.E., et al., Role of reactive oxygen species in contraction-mediated glucose transport in mouse skeletal muscle. The Journal of physiology, 2006. 575(Pt 1): p. 251-62.
155. Merry, T.L., et al., N-Acetylcysteine infusion does not affect glucose disposal during prolonged moderate-intensity exercise in humans. The Journal of physiology, 2010. 588(Pt 9): p. 1623-34.
156. Merry, T.L., et al., Local hindlimb antioxidant infusion does not affect muscle glucose uptake during in situ contractions in rat. Journal of applied physiology, 2010. 108(5): p. 1275-83.
157. Sylow, L., et al., Rac1 is a novel regulator of contraction-stimulated glucose uptake in skeletal muscle. Diabetes, 2013. 62(4): p. 1139-51.
158. Deshmukh, A., et al., Exercise-induced phosphorylation of the novel Akt substrates AS160 and filamin A in human skeletal muscle. Diabetes, 2006. 55(6): p. 1776-82.
159. Treebak, J.T., et al., AS160 phosphorylation is associated with activation of alpha2beta2gamma1- but not alpha2beta2gamma3-AMPK trimeric complex in skeletal muscle during exercise in humans. American journal of physiology. Endocrinology and metabolism, 2007. 292(3): p. E715-22.
160. Sriwijitkamol, A., et al., Effect of acute exercise on AMPK signaling in skeletal muscle of subjects with type 2 diabetes: a time-course and dose-response study. Diabetes, 2007. 56(3): p. 836-48.
161. Dreyer, H.C., et al., Resistance exercise increases human skeletal muscle AS160/TBC1D4 phosphorylation in association with enhanced leg glucose uptake during postexercise recovery. Journal of applied physiology, 2008. 105(6): p. 1967-74.
162. Arias, E.B., et al., Prior exercise increases phosphorylation of Akt substrate of 160 kDa (AS160) in rat skeletal muscle. American journal of physiology. Endocrinology and metabolism, 2007. 292(4): p. E1191-200.
163. Funai, K., et al., Increased AS160 phosphorylation, but not TBC1D1 phosphorylation, with increased postexercise insulin sensitivity in rat skeletal muscle. American journal of physiology. Endocrinology and metabolism, 2009. 297(1): p. E242-51.

124
164. Kramer, H.F., et al., Calmodulin-binding domain of AS160 regulates contraction- but not insulin-stimulated glucose uptake in skeletal muscle. Diabetes, 2007. 56(12): p. 2854-62.
165. Ducommun, S., et al., Thr649Ala-AS160 knock-in mutation does not impair contraction/AICAR-induced glucose transport in mouse muscle. American journal of physiology. Endocrinology and metabolism, 2012. 302(9): p. E1036-43.
166. Funai, K. and G.D. Cartee, Inhibition of contraction-stimulated AMP-activated protein kinase inhibits contraction-stimulated increases in PAS-TBC1D1 and glucose transport without altering PAS-AS160 in rat skeletal muscle. Diabetes, 2009. 58(5): p. 1096-104.
167. Pehmoller, C., et al., Genetic disruption of AMPK signaling abolishes both contraction- and insulin-stimulated TBC1D1 phosphorylation and 14-3-3 binding in mouse skeletal muscle. American journal of physiology. Endocrinology and metabolism, 2009. 297(3): p. E665-75.
168. Frosig, C., et al., Exercise-induced TBC1D1 Ser237 phosphorylation and 14-3-3 protein binding capacity in human skeletal muscle. The Journal of physiology, 2010. 588(Pt 22): p. 4539-48.
169. An, D., et al., TBC1D1 regulates insulin- and contraction-induced glucose transport in mouse skeletal muscle. Diabetes, 2010. 59(6): p. 1358-65.
170. Richter, E.A. and M. Hargreaves, Exercise, GLUT4, and skeletal muscle glucose uptake. Physiol Rev, 2013. 93(3): p. 993-1017.
171. Carlson, C.A. and K.H. Kim, Regulation of hepatic acetyl coenzyme A carboxylase by phosphorylation and dephosphorylation. The Journal of biological chemistry, 1973. 248(1): p. 378-80.
172. Beg, Z.H., D.W. Allmann, and D.M. Gibson, Modulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity with cAMP and wth protein fractions of rat liver cytosol. Biochemical and biophysical research communications, 1973. 54(4): p. 1362-9.
173. Carling, D., et al., Purification and characterization of the AMP-activated protein kinase. Copurification of acetyl-CoA carboxylase kinase and 3-hydroxy-3-methylglutaryl-CoA reductase kinase activities. European journal of biochemistry / FEBS, 1989. 186(1-2): p. 129-36.
174. Mitchelhill, K.I., et al., Mammalian AMP-activated protein kinase shares structural and functional homology with the catalytic domain of yeast Snf1 protein kinase. The Journal of biological chemistry, 1994. 269(4): p. 2361-4.
175. Davies, S.P., et al., Purification of the AMP-activated protein kinase on ATP-gamma-sepharose and analysis of its subunit structure. European journal of biochemistry / FEBS, 1994. 223(2): p. 351-7.
176. Woods, A., et al., Characterization of AMP-activated protein kinase beta and gamma subunits. Assembly of the heterotrimeric complex in vitro. The Journal of biological chemistry, 1996. 271(17): p. 10282-90.
177. Dyck, J.R., et al., Regulation of 5'-AMP-activated protein kinase activity by the noncatalytic beta and gamma subunits. The Journal of biological chemistry, 1996. 271(30): p. 17798-803.
178. Woods, A., et al., The alpha1 and alpha2 isoforms of the AMP-activated protein kinase have similar activities in rat liver but exhibit differences in substrate specificity in vitro. FEBS letters, 1996. 397(2-3): p. 347-51.
179. Stapleton, D., et al., Mammalian AMP-activated protein kinase subfamily. The Journal of biological chemistry, 1996. 271(2): p. 611-4.
180. Stapleton, D., et al., AMP-activated protein kinase isoenzyme family: subunit structure and chromosomal location. FEBS letters, 1997. 409(3): p. 452-6.

125
181. Thornton, C., M.A. Snowden, and D. Carling, Identification of a novel AMP-activated protein kinase beta subunit isoform that is highly expressed in skeletal muscle. The Journal of biological chemistry, 1998. 273(20): p. 12443-50.
182. Cheung, P.C., et al., Characterization of AMP-activated protein kinase gamma-subunit isoforms and their role in AMP binding. The Biochemical journal, 2000. 346 Pt 3: p. 659-69.
183. Wojtaszewski, J.F., et al., 5'AMP activated protein kinase expression in human skeletal muscle: effects of strength training and type 2 diabetes. The Journal of physiology, 2005. 564(Pt 2): p. 563-73.
184. Chen, Z., et al., Expression of the AMP-activated protein kinase beta1 and beta2 subunits in skeletal muscle. FEBS letters, 1999. 460(2): p. 343-8.
185. Frosig, C., et al., 5'-AMP-activated protein kinase activity and protein expression are regulated by endurance training in human skeletal muscle. American journal of physiology. Endocrinology and metabolism, 2004. 286(3): p. E411-7.
186. O'Neill, H.M., AMPK and Exercise: Glucose Uptake and Insulin Sensitivity. Diabetes Metab J, 2013. 37(1): p. 1-21.
187. Johnson, L.N., M.E. Noble, and D.J. Owen, Active and inactive protein kinases: structural basis for regulation. Cell, 1996. 85(2): p. 149-58.
188. Crute, B.E., et al., Functional domains of the alpha1 catalytic subunit of the AMP-activated protein kinase. The Journal of biological chemistry, 1998. 273(52): p. 35347-54.
189. Hudson, E.R., et al., A novel domain in AMP-activated protein kinase causes glycogen storage bodies similar to those seen in hereditary cardiac arrhythmias. Current biology : CB, 2003. 13(10): p. 861-6.
190. Xiao, B., et al., Structural basis for AMP binding to mammalian AMP-activated protein kinase. Nature, 2007. 449(7161): p. 496-500.
191. Xiao, B., et al., Structure of mammalian AMPK and its regulation by ADP. Nature, 2011. 472(7342): p. 230-3.
192. Pang, T., et al., Conserved alpha-helix acts as autoinhibitory sequence in AMP-activated protein kinase alpha subunits. The Journal of biological chemistry, 2007. 282(1): p. 495-506.
193. Iseli, T.J., et al., AMP-activated protein kinase beta subunit tethers alpha and gamma subunits via its C-terminal sequence (186-270). The Journal of biological chemistry, 2005. 280(14): p. 13395-400.
194. Iseli, T.J., et al., AMP-activated protein kinase subunit interactions: beta1:gamma1 association requires beta1 Thr-263 and Tyr-267. The Journal of biological chemistry, 2008. 283(8): p. 4799-807.
195. Towler, M.C. and D.G. Hardie, AMP-activated protein kinase in metabolic control and insulin signaling. Circulation research, 2007. 100(3): p. 328-41.
196. Woods, A., et al., Identification of phosphorylation sites in AMP-activated protein kinase (AMPK) for upstream AMPK kinases and study of their roles by site-directed mutagenesis. The Journal of biological chemistry, 2003. 278(31): p. 28434-42.
197. Verhoeven, A.J., et al., The AMP-activated protein kinase gene is highly expressed in rat skeletal muscle. Alternative splicing and tissue distribution of the mRNA. Eur J Biochem, 1995. 228(2): p. 236-43.
198. Salt, I., et al., AMP-activated protein kinase: greater AMP dependence, and preferential nuclear localization, of complexes containing the alpha2 isoform. The Biochemical journal, 1998. 334 ( Pt 1): p. 177-87.

126
199. Ai, H., et al., Effect of fiber type and nutritional state on AICAR- and contraction-stimulated glucose transport in rat muscle. American journal of physiology. Endocrinology and metabolism, 2002. 282(6): p. E1291-300.
200. Warden, S.M., et al., Post-translational modifications of the beta-1 subunit of AMP-activated protein kinase affect enzyme activity and cellular localization. The Biochemical journal, 2001. 354(Pt 2): p. 275-83.
201. Mitchelhill, K.I., et al., Posttranslational modifications of the 5'-AMP-activated protein kinase beta1 subunit. The Journal of biological chemistry, 1997. 272(39): p. 24475-9.
202. Polekhina, G., et al., AMPK beta subunit targets metabolic stress sensing to glycogen. Current biology : CB, 2003. 13(10): p. 867-71.
203. Amodeo, G.A., M.J. Rudolph, and L. Tong, Crystal structure of the heterotrimer core of Saccharomyces cerevisiae AMPK homologue SNF1. Nature, 2007. 449(7161): p. 492-5.
204. Townley, R. and L. Shapiro, Crystal structures of the adenylate sensor from fission yeast AMP-activated protein kinase. Science, 2007. 315(5819): p. 1726-9.
205. Oakhill, J.S., et al., beta-Subunit myristoylation is the gatekeeper for initiating metabolic stress sensing by AMP-activated protein kinase (AMPK). Proceedings of the National Academy of Sciences of the United States of America, 2010. 107(45): p. 19237-41.
206. Bendayan, M., et al., Association of AMP-activated protein kinase subunits with glycogen particles as revealed in situ by immunoelectron microscopy. The journal of histochemistry and cytochemistry : official journal of the Histochemistry Society, 2009. 57(10): p. 963-71.
207. Lang, T., et al., Molecular cloning, genomic organization, and mapping of PRKAG2, a heart abundant gamma2 subunit of 5'-AMP-activated protein kinase, to human chromosome 7q36. Genomics, 2000. 70(2): p. 258-63.
208. Yu, H., et al., Cloning and characterization of mouse 5'-AMP-activated protein kinase gamma3 subunit. American journal of physiology. Cell physiology, 2004. 286(2): p. C283-92.
209. Viana, R., et al., A conserved sequence immediately N-terminal to the Bateman domains in AMP-activated protein kinase gamma subunits is required for the interaction with the beta subunits. The Journal of biological chemistry, 2007. 282(22): p. 16117-25.
210. Kemp, B.E., Bateman domains and adenosine derivatives form a binding contract. The Journal of clinical investigation, 2004. 113(2): p. 182-4.
211. Scott, J.W., et al., CBS domains form energy-sensing modules whose binding of adenosine ligands is disrupted by disease mutations. The Journal of clinical investigation, 2004. 113(2): p. 274-84.
212. Milan, D., et al., A mutation in PRKAG3 associated with excess glycogen content in pig skeletal muscle. Science, 2000. 288(5469): p. 1248-51.
213. Turnley, A.M., et al., Cellular distribution and developmental expression of AMP-activated protein kinase isoforms in mouse central nervous system. Journal of neurochemistry, 1999. 72(4): p. 1707-16.
214. Hardie, D.G., AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol, 2007. 8(10): p. 774-85.
215. Hardie, D.G., D. Carling, and S.J. Gamblin, AMP-activated protein kinase: also regulated by ADP? Trends in biochemical sciences, 2011. 36(9): p. 470-7.
216. Hardie, D.G., F.A. Ross, and S.A. Hawley, AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nature reviews. Molecular cell biology, 2012. 13(4): p. 251-62.

127
217. Gowans, G.J., et al., AMP is a true physiological regulator of AMP-activated protein kinase by both allosteric activation and enhancing net phosphorylation. Cell metabolism, 2013. 18(4): p. 556-66.
218. Davies, S.P., et al., 5'-AMP inhibits dephosphorylation, as well as promoting phosphorylation, of the AMP-activated protein kinase. Studies using bacterially expressed human protein phosphatase-2C alpha and native bovine protein phosphatase-2AC. FEBS letters, 1995. 377(3): p. 421-5.
219. Hawley, S.A., et al., 5'-AMP activates the AMP-activated protein kinase cascade, and Ca2+/calmodulin activates the calmodulin-dependent protein kinase I cascade, via three independent mechanisms. The Journal of biological chemistry, 1995. 270(45): p. 27186-91.
220. Oakhill, J.S., et al., AMPK is a direct adenylate charge-regulated protein kinase. Science, 2011. 332(6036): p. 1433-5.
221. Hawley, S.A., et al., Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. The Journal of biological chemistry, 1996. 271(44): p. 27879-87.
222. Sutherland, C.M., et al., Elm1p is one of three upstream kinases for the Saccharomyces cerevisiae SNF1 complex. Current biology : CB, 2003. 13(15): p. 1299-305.
223. Hawley, S.A., et al., Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. Journal of biology, 2003. 2(4): p. 28.
224. Hong, S.P., et al., Activation of yeast Snf1 and mammalian AMP-activated protein kinase by upstream kinases. Proceedings of the National Academy of Sciences of the United States of America, 2003. 100(15): p. 8839-43.
225. Shaw, R.J., et al., The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proceedings of the National Academy of Sciences of the United States of America, 2004. 101(10): p. 3329-35.
226. Hemminki, A., et al., A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature, 1998. 391(6663): p. 184-7.
227. Jenne, D.E., et al., Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nature genetics, 1998. 18(1): p. 38-43.
228. Baas, A.F., et al., Activation of the tumour suppressor kinase LKB1 by the STE20-like pseudokinase STRAD. The EMBO journal, 2003. 22(12): p. 3062-72.
229. Boudeau, J., et al., MO25alpha/beta interact with STRADalpha/beta enhancing their ability to bind, activate and localize LKB1 in the cytoplasm. The EMBO journal, 2003. 22(19): p. 5102-14.
230. Lizcano, J.M., et al., LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. The EMBO journal, 2004. 23(4): p. 833-43.
231. Yamada, E., et al., Fyn-dependent regulation of energy expenditure and body weight is mediated by tyrosine phosphorylation of LKB1. Cell Metab, 2010. 11(2): p. 113-24.
232. Vu, V., et al., Globular adiponectin induces LKB1/AMPK-dependent glucose uptake via actin cytoskeleton remodeling. J Mol Endocrinol, 2013. 51(1): p. 155-65.
233. Houde, V.P., et al., Investigation of role that LKB1 Ser431 phosphorylation and Cys433 farnesylation play by mouse knock-in analysis reveals an unexpected role of prenylation in regulating AMPK activity. The Biochemical journal, 2013.
234. Sakamoto, K., et al., Deficiency of LKB1 in skeletal muscle prevents AMPK activation and glucose uptake during contraction. The EMBO journal, 2005. 24(10): p. 1810-20.

128
235. Hawley, S.A., et al., Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell metabolism, 2005. 2(1): p. 9-19.
236. Woods, A., et al., Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell metabolism, 2005. 2(1): p. 21-33.
237. Hurley, R.L., et al., The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. The Journal of biological chemistry, 2005. 280(32): p. 29060-6.
238. Jensen, T.E., et al., Possible CaMKK-dependent regulation of AMPK phosphorylation and glucose uptake at the onset of mild tetanic skeletal muscle contraction. American journal of physiology. Endocrinology and metabolism, 2007. 292(5): p. E1308-17.
239. Abbott, M.J., A.M. Edelman, and L.P. Turcotte, CaMKK is an upstream signal of AMP-activated protein kinase in regulation of substrate metabolism in contracting skeletal muscle. American journal of physiology. Regulatory, integrative and comparative physiology, 2009. 297(6): p. R1724-32.
240. Kitani, T., S. Okuno, and H. Fujisawa, Molecular cloning of Ca2+/calmodulin-dependent protein kinase kinase beta. Journal of biochemistry, 1997. 122(1): p. 243-50.
241. McGee, S.L., et al., Normal hypertrophy accompanied by phosphoryation and activation of AMP-activated protein kinase alpha1 following overload in LKB1 knockout mice. The Journal of physiology, 2008. 586(6): p. 1731-41.
242. Witczak, C.A., et al., Ca2+/calmodulin-dependent protein kinase kinase-alpha regulates skeletal muscle glucose uptake independent of AMP-activated protein kinase and Akt activation. Diabetes, 2007. 56(5): p. 1403-9.
243. Momcilovic, M., S.P. Hong, and M. Carlson, Mammalian TAK1 activates Snf1 protein kinase in yeast and phosphorylates AMP-activated protein kinase in vitro. The Journal of biological chemistry, 2006. 281(35): p. 25336-43.
244. Yamaguchi, K., et al., Identification of a member of the MAPKKK family as a potential mediator of TGF-beta signal transduction. Science, 1995. 270(5244): p. 2008-11.
245. Shibuya, H., et al., TAB1: an activator of the TAK1 MAPKKK in TGF-beta signal transduction. Science, 1996. 272(5265): p. 1179-82.
246. Besse, A., et al., TAK1-dependent signaling requires functional interaction with TAB2/TAB3. The Journal of biological chemistry, 2007. 282(6): p. 3918-28.
247. Xie, M., et al., A pivotal role for endogenous TGF-beta-activated kinase-1 in the LKB1/AMP-activated protein kinase energy-sensor pathway. Proceedings of the National Academy of Sciences of the United States of America, 2006. 103(46): p. 17378-83.
248. Jorgensen, S.B. and A.J. Rose, How is AMPK activity regulated in skeletal muscles during exercise? Front Biosci, 2008. 13: p. 5589-604.
249. Henin, N., et al., Inhibition of fatty acid and cholesterol synthesis by stimulation of AMP-activated protein kinase. FASEB journal : official publication of the Federation of American Societies for Experimental Biology, 1995. 9(7): p. 541-6.
250. Sabina, R.L., D. Patterson, and E.W. Holmes, 5-Amino-4-imidazolecarboxamide riboside (Z-riboside) metabolism in eukaryotic cells. The Journal of biological chemistry, 1985. 260(10): p. 6107-14.
251. Day, P., et al., Structure of a CBS-domain pair from the regulatory gamma1 subunit of human AMPK in complex with AMP and ZMP. Acta crystallographica. Section D, Biological crystallography, 2007. 63(Pt 5): p. 587-96.

129
252. Longnus, S.L., et al., 5-Aminoimidazole-4-carboxamide 1-beta -D-ribofuranoside (AICAR) stimulates myocardial glycogenolysis by allosteric mechanisms. American journal of physiology. Regulatory, integrative and comparative physiology, 2003. 284(4): p. R936-44.
253. Vincent, M.F., et al., Hypoglycaemic effect of AICAriboside in mice. Diabetologia, 1996. 39(10): p. 1148-55.
254. Knowler, W.C., et al., Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. The New England journal of medicine, 2002. 346(6): p. 393-403.
255. Zhou, G., et al., Role of AMP-activated protein kinase in mechanism of metformin action. The Journal of clinical investigation, 2001. 108(8): p. 1167-74.
256. Musi, N., et al., Metformin increases AMP-activated protein kinase activity in skeletal muscle of subjects with type 2 diabetes. Diabetes, 2002. 51(7): p. 2074-81.
257. Owen, M.R., E. Doran, and A.P. Halestrap, Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. The Biochemical journal, 2000. 348 Pt 3: p. 607-14.
258. El-Mir, M.Y., et al., Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. The Journal of biological chemistry, 2000. 275(1): p. 223-8.
259. Hawley, S.A., et al., Use of cells expressing gamma subunit variants to identify diverse mechanisms of AMPK activation. Cell metabolism, 2010. 11(6): p. 554-65.
260. Sakamoto, K., et al., Activity of LKB1 and AMPK-related kinases in skeletal muscle: effects of contraction, phenformin, and AICAR. American journal of physiology. Endocrinology and metabolism, 2004. 287(2): p. E310-7.
261. LeBrasseur, N.K., et al., Thiazolidinediones can rapidly activate AMP-activated protein kinase in mammalian tissues. American journal of physiology. Endocrinology and metabolism, 2006. 291(1): p. E175-81.
262. Fryer, L.G., A. Parbu-Patel, and D. Carling, The Anti-diabetic drugs rosiglitazone and metformin stimulate AMP-activated protein kinase through distinct signaling pathways. The Journal of biological chemistry, 2002. 277(28): p. 25226-32.
263. Saha, A.K., et al., Pioglitazone treatment activates AMP-activated protein kinase in rat liver and adipose tissue in vivo. Biochemical and biophysical research communications, 2004. 314(2): p. 580-5.
264. Patel, J., R.J. Anderson, and E.B. Rappaport, Rosiglitazone monotherapy improves glycaemic control in patients with type 2 diabetes: a twelve-week, randomized, placebo-controlled study. Diabetes, obesity & metabolism, 1999. 1(3): p. 165-72.
265. Kubota, N., et al., Pioglitazone ameliorates insulin resistance and diabetes by both adiponectin-dependent and -independent pathways. The Journal of biological chemistry, 2006. 281(13): p. 8748-55.
266. Brunmair, B., et al., Thiazolidinediones, like metformin, inhibit respiratory complex I: a common mechanism contributing to their antidiabetic actions? Diabetes, 2004. 53(4): p. 1052-9.
267. Nawrocki, A.R., et al., Mice lacking adiponectin show decreased hepatic insulin sensitivity and reduced responsiveness to peroxisome proliferator-activated receptor gamma agonists. The Journal of biological chemistry, 2006. 281(5): p. 2654-60.
268. Cool, B., et al., Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell metabolism, 2006. 3(6): p. 403-16.

130
269. Hawley, S.A., et al., The ancient drug salicylate directly activates AMP-activated protein kinase. Science, 2012. 336(6083): p. 918-22.
270. Scott, J.W., et al., Thienopyridone drugs are selective activators of AMP-activated protein kinase beta1-containing complexes. Chemistry & biology, 2008. 15(11): p. 1220-30.
271. Sanders, M.J., et al., Defining the mechanism of activation of AMP-activated protein kinase by the small molecule A-769662, a member of the thienopyridone family. The Journal of biological chemistry, 2007. 282(45): p. 32539-48.
272. Goransson, O., et al., Mechanism of action of A-769662, a valuable tool for activation of AMP-activated protein kinase. The Journal of biological chemistry, 2007. 282(45): p. 32549-60.
273. Treebak, J.T., et al., A-769662 activates AMPK beta1-containing complexes but induces glucose uptake through a PI3-kinase-dependent pathway in mouse skeletal muscle. American journal of physiology. Cell physiology, 2009. 297(4): p. C1041-52.
274. Baur, J.A., et al., Resveratrol improves health and survival of mice on a high-calorie diet. Nature, 2006. 444(7117): p. 337-42.
275. Timmers, S., et al., Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans. Cell metabolism, 2011. 14(5): p. 612-22.
276. Lee, Y.S., et al., Berberine, a natural plant product, activates AMP-activated protein kinase with beneficial metabolic effects in diabetic and insulin-resistant states. Diabetes, 2006. 55(8): p. 2256-64.
277. Ma, X., et al., Berberine-induced activation of 5'-adenosine monophosphate-activated protein kinase and glucose transport in rat skeletal muscles. Metabolism: clinical and experimental, 2010. 59(11): p. 1619-27.
278. Gledhill, J.R., et al., Mechanism of inhibition of bovine F1-ATPase by resveratrol and related polyphenols. Proceedings of the National Academy of Sciences of the United States of America, 2007. 104(34): p. 13632-7.
279. Turner, N., et al., Berberine and its more biologically available derivative, dihydroberberine, inhibit mitochondrial respiratory complex I: a mechanism for the action of berberine to activate AMP-activated protein kinase and improve insulin action. Diabetes, 2008. 57(5): p. 1414-8.
280. Winder, W.W. and D.G. Hardie, Inactivation of acetyl-CoA carboxylase and activation of AMP-activated protein kinase in muscle during exercise. The American journal of physiology, 1996. 270(2 Pt 1): p. E299-304.
281. Chen, Z.P., et al., AMPK signaling in contracting human skeletal muscle: acetyl-CoA carboxylase and NO synthase phosphorylation. American journal of physiology. Endocrinology and metabolism, 2000. 279(5): p. E1202-6.
282. Hutber, C.A., D.G. Hardie, and W.W. Winder, Electrical stimulation inactivates muscle acetyl-CoA carboxylase and increases AMP-activated protein kinase. The American journal of physiology, 1997. 272(2 Pt 1): p. E262-6.
283. Vavvas, D., et al., Contraction-induced changes in acetyl-CoA carboxylase and 5'-AMP-activated kinase in skeletal muscle. The Journal of biological chemistry, 1997. 272(20): p. 13255-61.
284. Fujii, N., et al., Exercise induces isoform-specific increase in 5'AMP-activated protein kinase activity in human skeletal muscle. Biochemical and biophysical research communications, 2000. 273(3): p. 1150-5.

131
285. Wojtaszewski, J.F., et al., Isoform-specific and exercise intensity-dependent activation of 5'-AMP-activated protein kinase in human skeletal muscle. The Journal of physiology, 2000. 528 Pt 1: p. 221-6.
286. Birk, J.B. and J.F. Wojtaszewski, Predominant alpha2/beta2/gamma3 AMPK activation during exercise in human skeletal muscle. The Journal of physiology, 2006. 577(Pt 3): p. 1021-32.
287. Sahlin, K., M. Tonkonogi, and K. Soderlund, Energy supply and muscle fatigue in humans. Acta physiologica Scandinavica, 1998. 162(3): p. 261-6.
288. Koh, H.J., et al., Skeletal muscle-selective knockout of LKB1 increases insulin sensitivity, improves glucose homeostasis, and decreases TRB3. Molecular and cellular biology, 2006. 26(22): p. 8217-27.
289. Thomson, D.M., et al., Skeletal muscle and heart LKB1 deficiency causes decreased voluntary running and reduced muscle mitochondrial marker enzyme expression in mice. American journal of physiology. Endocrinology and metabolism, 2007. 292(1): p. E196-202.
290. Fujii, N., et al., AMP-activated protein kinase alpha2 activity is not essential for contraction- and hyperosmolarity-induced glucose transport in skeletal muscle. The Journal of biological chemistry, 2005. 280(47): p. 39033-41.
291. Toyoda, T., et al., Low-intensity contraction activates the alpha1-isoform of 5'-AMP-activated protein kinase in rat skeletal muscle. American journal of physiology. Endocrinology and metabolism, 2006. 290(3): p. E583-90.
292. Toyoda, T., et al., Possible involvement of the alpha1 isoform of 5'AMP-activated protein kinase in oxidative stress-stimulated glucose transport in skeletal muscle. American journal of physiology. Endocrinology and metabolism, 2004. 287(1): p. E166-73.
293. Keller, C., et al., Transcriptional activation of the IL-6 gene in human contracting skeletal muscle: influence of muscle glycogen content. FASEB journal : official publication of the Federation of American Societies for Experimental Biology, 2001. 15(14): p. 2748-50.
294. Penkowa, M., et al., Immunohistochemical detection of interleukin-6 in human skeletal muscle fibers following exercise. FASEB journal : official publication of the Federation of American Societies for Experimental Biology, 2003. 17(14): p. 2166-8.
295. Kelly, M., et al., AMPK activity is diminished in tissues of IL-6 knockout mice: the effect of exercise. Biochemical and biophysical research communications, 2004. 320(2): p. 449-54.
296. Weigert, C., et al., Upregulation of IL-6 mRNA by IL-6 in skeletal muscle cells: role of IL-6 mRNA stabilization and Ca2+-dependent mechanisms. American journal of physiology. Cell physiology, 2007. 293(3): p. C1139-47.
297. Merrill, G.F., et al., AICA riboside increases AMP-activated protein kinase, fatty acid oxidation, and glucose uptake in rat muscle. The American journal of physiology, 1997. 273(6 Pt 1): p. E1107-12.
298. Hayashi, T., et al., Evidence for 5' AMP-activated protein kinase mediation of the effect of muscle contraction on glucose transport. Diabetes, 1998. 47(8): p. 1369-73.
299. Mu, J., et al., A role for AMP-activated protein kinase in contraction- and hypoxia-regulated glucose transport in skeletal muscle. Molecular cell, 2001. 7(5): p. 1085-94.
300. Mu, J., E.R. Barton, and M.J. Birnbaum, Selective suppression of AMP-activated protein kinase in skeletal muscle: update on 'lazy mice'. Biochem Soc Trans, 2003. 31(Pt 1): p. 236-41.
301. Abbott, M.J., L.D. Bogachus, and L.P. Turcotte, AMPKalpha2 deficiency uncovers time dependency in the regulation of contraction-induced palmitate and glucose uptake in mouse muscle. Journal of applied physiology, 2011. 111(1): p. 125-34.

132
302. Maarbjerg, S.J., et al., Genetic impairment of AMPKalpha2 signaling does not reduce muscle glucose uptake during treadmill exercise in mice. American journal of physiology. Endocrinology and metabolism, 2009. 297(4): p. E924-34.
303. Lee-Young, R.S., et al., Skeletal muscle AMP-activated protein kinase is essential for the metabolic response to exercise in vivo. The Journal of biological chemistry, 2009. 284(36): p. 23925-34.
304. Viollet, B., et al., The AMP-activated protein kinase alpha2 catalytic subunit controls whole-body insulin sensitivity. The Journal of clinical investigation, 2003. 111(1): p. 91-8.
305. Jorgensen, S.B., et al., Knockout of the alpha2 but not alpha1 5'-AMP-activated protein kinase isoform abolishes 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranosidebut not contraction-induced glucose uptake in skeletal muscle. The Journal of biological chemistry, 2004. 279(2): p. 1070-9.
306. Lantier, L., et al., AMPK controls exercise endurance, mitochondrial oxidative capacity, and skeletal muscle integrity. FASEB J, 2014.
307. Barnes, B.R., et al., The 5'-AMP-activated protein kinase gamma3 isoform has a key role in carbohydrate and lipid metabolism in glycolytic skeletal muscle. The Journal of biological chemistry, 2004. 279(37): p. 38441-7.
308. Dzamko, N., et al., AMPK beta1 deletion reduces appetite, preventing obesity and hepatic insulin resistance. The Journal of biological chemistry, 2010. 285(1): p. 115-22.
309. Steinberg, G.R., et al., Whole body deletion of AMP-activated protein kinase {beta}2 reduces muscle AMPK activity and exercise capacity. The Journal of biological chemistry, 2010. 285(48): p. 37198-209.
310. O'Neill, H.M., et al., AMP-activated protein kinase (AMPK) beta1beta2 muscle null mice reveal an essential role for AMPK in maintaining mitochondrial content and glucose uptake during exercise. Proceedings of the National Academy of Sciences of the United States of America, 2011. 108(38): p. 16092-7.
311. Carey, D.G., et al., Abdominal fat and insulin resistance in normal and overweight women: Direct measurements reveal a strong relationship in subjects at both low and high risk of NIDDM. Diabetes, 1996. 45(5): p. 633-8.
312. Cnop, M., et al., The concurrent accumulation of intra-abdominal and subcutaneous fat explains the association between insulin resistance and plasma leptin concentrations : distinct metabolic effects of two fat compartments. Diabetes, 2002. 51(4): p. 1005-15.
313. Erdmann, J., et al., Development of hyperinsulinemia and insulin resistance during the early stage of weight gain. American journal of physiology. Endocrinology and metabolism, 2008. 294(3): p. E568-75.
314. Van Cromphaut, S.J., I. Vanhorebeek, and G. Van den Berghe, Glucose metabolism and insulin resistance in sepsis. Current pharmaceutical design, 2008. 14(19): p. 1887-99.
315. Ciaraldi, T.P., et al., Insulin and insulin-like growth factor-1 action on human skeletal muscle: preferential effects of insulin-like growth factor-1 in type 2 diabetic subjects. Metabolism, 2002. 51(9): p. 1171-9.
316. Caro, J.F., et al., Insulin receptor kinase in human skeletal muscle from obese subjects with and without noninsulin dependent diabetes. The Journal of clinical investigation, 1987. 79(5): p. 1330-7.
317. Maegawa, H., et al., Impaired autophosphorylation of insulin receptors from abdominal skeletal muscles in nonobese subjects with NIDDM. Diabetes, 1991. 40(7): p. 815-9.

133
318. Arner, P., et al., Defective insulin receptor tyrosine kinase in human skeletal muscle in obesity and type 2 (non-insulin-dependent) diabetes mellitus. Diabetologia, 1987. 30(6): p. 437-40.
319. Nolan, J.J., et al., Role of human skeletal muscle insulin receptor kinase in the in vivo insulin resistance of noninsulin-dependent diabetes mellitus and obesity. The Journal of clinical endocrinology and metabolism, 1994. 78(2): p. 471-7.
320. Krook, A., et al., Characterization of signal transduction and glucose transport in skeletal muscle from type 2 diabetic patients. Diabetes, 2000. 49(2): p. 284-92.
321. Klein, H.H., et al., Elevation of serum insulin concentration during euglycemic hyperinsulinemic clamp studies leads to similar activation of insulin receptor kinase in skeletal muscle of subjects with and without NIDDM. Diabetes, 1995. 44(11): p. 1310-7.
322. Meyer, M.M., et al., Insulin signalling in skeletal muscle of subjects with or without Type II-diabetes and first degree relatives of patients with the disease. Diabetologia, 2002. 45(6): p. 813-22.
323. Dey, D., et al., Inhibition of insulin receptor gene expression and insulin signaling by fatty acid: interplay of PKC isoforms therein. Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology, 2005. 16(4-6): p. 217-28.
324. Cusi, K., et al., Insulin resistance differentially affects the PI 3-kinase- and MAP kinase-mediated signaling in human muscle. The Journal of clinical investigation, 2000. 105(3): p. 311-20.
325. Kim, Y.B., et al., Normal insulin-dependent activation of Akt/protein kinase B, with diminished activation of phosphoinositide 3-kinase, in muscle in type 2 diabetes. The Journal of clinical investigation, 1999. 104(6): p. 733-41.
326. Morino, K., et al., Reduced mitochondrial density and increased IRS-1 serine phosphorylation in muscle of insulin-resistant offspring of type 2 diabetic parents. The Journal of clinical investigation, 2005. 115(12): p. 3587-93.
327. Tremblay, F., et al., Identification of IRS-1 Ser-1101 as a target of S6K1 in nutrient- and obesity-induced insulin resistance. Proceedings of the National Academy of Sciences of the United States of America, 2007. 104(35): p. 14056-61.
328. Bouzakri, K., et al., Reduced activation of phosphatidylinositol-3 kinase and increased serine 636 phosphorylation of insulin receptor substrate-1 in primary culture of skeletal muscle cells from patients with type 2 diabetes. Diabetes, 2003. 52(6): p. 1319-25.
329. Khamzina, L., et al., Increased activation of the mammalian target of rapamycin pathway in liver and skeletal muscle of obese rats: possible involvement in obesity-linked insulin resistance. Endocrinology, 2005. 146(3): p. 1473-81.
330. Morino, K., et al., Muscle-specific IRS-1 Ser->Ala transgenic mice are protected from fat-induced insulin resistance in skeletal muscle. Diabetes, 2008. 57(10): p. 2644-51.
331. Bjornholm, M., et al., Insulin receptor substrate-1 phosphorylation and phosphatidylinositol 3-kinase activity in skeletal muscle from NIDDM subjects after in vivo insulin stimulation. Diabetes, 1997. 46(3): p. 524-7.
332. Kruszynska, Y.T., et al., Fatty acid-induced insulin resistance: decreased muscle PI3K activation but unchanged Akt phosphorylation. The Journal of clinical endocrinology and metabolism, 2002. 87(1): p. 226-34.
333. Terauchi, Y., et al., Increased insulin sensitivity and hypoglycaemia in mice lacking the p85 alpha subunit of phosphoinositide 3-kinase. Nature genetics, 1999. 21(2): p. 230-5.

134
334. Ueki, K., et al., Increased insulin sensitivity in mice lacking p85beta subunit of phosphoinositide 3-kinase. Proceedings of the National Academy of Sciences of the United States of America, 2002. 99(1): p. 419-24.
335. Ueki, K., et al., Molecular balance between the regulatory and catalytic subunits of phosphoinositide 3-kinase regulates cell signaling and survival. Molecular and cellular biology, 2002. 22(3): p. 965-77.
336. Luo, J., et al., Loss of class IA PI3K signaling in muscle leads to impaired muscle growth, insulin response, and hyperlipidemia. Cell metabolism, 2006. 3(5): p. 355-66.
337. Kim, Y.B., et al., Divergent regulation of Akt1 and Akt2 isoforms in insulin target tissues of obese Zucker rats. Diabetes, 2000. 49(5): p. 847-56.
338. Brozinick, J.T., Jr., B.R. Roberts, and G.L. Dohm, Defective signaling through Akt-2 and -3 but not Akt-1 in insulin-resistant human skeletal muscle: potential role in insulin resistance. Diabetes, 2003. 52(4): p. 935-41.
339. Jacinto, E., et al., SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell, 2006. 127(1): p. 125-37.
340. Moore, S.F., R.W. Hunter, and I. Hers, mTORC2 protein complex-mediated Akt (Protein Kinase B) Serine 473 Phosphorylation is not required for Akt1 activity in human platelets [corrected]. J Biol Chem, 2011. 286(28): p. 24553-60.
341. Roden, M., et al., Mechanism of free fatty acid-induced insulin resistance in humans. The Journal of clinical investigation, 1996. 97(12): p. 2859-65.
342. Belfort, R., et al., Dose-response effect of elevated plasma free fatty acid on insulin signaling. Diabetes, 2005. 54(6): p. 1640-8.
343. Boden, G., et al., Effects of acute changes of plasma free fatty acids on intramyocellular fat content and insulin resistance in healthy subjects. Diabetes, 2001. 50(7): p. 1612-7.
344. Storlien, L.H., et al., Influence of dietary fat composition on development of insulin resistance in rats. Relationship to muscle triglyceride and omega-3 fatty acids in muscle phospholipid. Diabetes, 1991. 40(2): p. 280-9.
345. Pagliassotti, M.J., et al., Tissue oxidative capacity, fuel stores and skeletal muscle fatty acid composition in obesity-prone and obesity-resistant rats. Obesity research, 1995. 3(5): p. 459-64.
346. Bajaj, M., et al., Effect of a sustained reduction in plasma free fatty acid concentration on intramuscular long-chain fatty Acyl-CoAs and insulin action in type 2 diabetic patients. Diabetes, 2005. 54(11): p. 3148-53.
347. Yu, C., et al., Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. The Journal of biological chemistry, 2002. 277(52): p. 50230-6.
348. Chalkley, S.M., et al., Five-hour fatty acid elevation increases muscle lipids and impairs glycogen synthesis in the rat. Metabolism: clinical and experimental, 1998. 47(9): p. 1121-6.
349. Ellis, B.A., et al., Long-chain acyl-CoA esters as indicators of lipid metabolism and insulin sensitivity in rat and human muscle. American journal of physiology. Endocrinology and metabolism, 2000. 279(3): p. E554-60.
350. Griffin, M.E., et al., Free fatty acid-induced insulin resistance is associated with activation of protein kinase C theta and alterations in the insulin signaling cascade. Diabetes, 1999. 48(6): p. 1270-4.
351. Liu, L., et al., Upregulation of myocellular DGAT1 augments triglyceride synthesis in skeletal muscle and protects against fat-induced insulin resistance. The Journal of clinical investigation, 2007. 117(6): p. 1679-89.

135
352. Itani, S.I., et al., Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IkappaB-alpha. Diabetes, 2002. 51(7): p. 2005-11.
353. Kim, J.K., et al., PKC-theta knockout mice are protected from fat-induced insulin resistance. The Journal of clinical investigation, 2004. 114(6): p. 823-7.
354. Turinsky, J., D.M. O'Sullivan, and B.P. Bayly, 1,2-Diacylglycerol and ceramide levels in insulin-resistant tissues of the rat in vivo. The Journal of biological chemistry, 1990. 265(28): p. 16880-5.
355. Adams, J.M., 2nd, et al., Ceramide content is increased in skeletal muscle from obese insulin-resistant humans. Diabetes, 2004. 53(1): p. 25-31.
356. Straczkowski, M., et al., Increased skeletal muscle ceramide level in men at risk of developing type 2 diabetes. Diabetologia, 2007. 50(11): p. 2366-73.
357. Straczkowski, M., et al., Relationship between insulin sensitivity and sphingomyelin signaling pathway in human skeletal muscle. Diabetes, 2004. 53(5): p. 1215-21.
358. Ussher, J.R., et al., Inhibition of de novo ceramide synthesis reverses diet-induced insulin resistance and enhances whole-body oxygen consumption. Diabetes, 2010. 59(10): p. 2453-64.
359. Holland, W.L., et al., Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell metabolism, 2007. 5(3): p. 167-79.
360. Bruce, C.R., et al., Overexpression of sphingosine kinase 1 prevents ceramide accumulation and ameliorates muscle insulin resistance in high-fat diet-fed mice. Diabetes, 2012. 61(12): p. 3148-55.
361. Hajduch, E., et al., Ceramide impairs the insulin-dependent membrane recruitment of protein kinase B leading to a loss in downstream signalling in L6 skeletal muscle cells. Diabetologia, 2001. 44(2): p. 173-83.
362. Chavez, J.A. and S.A. Summers, Characterizing the effects of saturated fatty acids on insulin signaling and ceramide and diacylglycerol accumulation in 3T3-L1 adipocytes and C2C12 myotubes. Archives of biochemistry and biophysics, 2003. 419(2): p. 101-9.
363. Summers, S.A., Sphingolipids and insulin resistance: the five Ws. Current opinion in lipidology, 2010. 21(2): p. 128-35.
364. Holland, W.L. and S.A. Summers, Sphingolipids, insulin resistance, and metabolic disease: new insights from in vivo manipulation of sphingolipid metabolism. Endocrine reviews, 2008. 29(4): p. 381-402.
365. Lipina, C. and H.S. Hundal, Sphingolipids: agents provocateurs in the pathogenesis of insulin resistance. Diabetologia, 2011. 54(7): p. 1596-607.
366. Holland, W.L., et al., Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J Clin Invest, 2011. 121(5): p. 1858-70.
367. Weisberg, S.P., et al., Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest, 2003. 112(12): p. 1796-808.
368. Xu, H., et al., Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest, 2003. 112(12): p. 1821-30.
369. Ye, J., et al., Hypoxia is a potential risk factor for chronic inflammation and adiponectin reduction in adipose tissue of ob/ob and dietary obese mice. American journal of physiology. Endocrinology and metabolism, 2007. 293(4): p. E1118-28.
370. Rausch, M.E., et al., Obesity in C57BL/6J mice is characterized by adipose tissue hypoxia and cytotoxic T-cell infiltration. International journal of obesity, 2008. 32(3): p. 451-63.

136
371. Regazzetti, C., et al., Hypoxia decreases insulin signaling pathways in adipocytes. Diabetes, 2009. 58(1): p. 95-103.
372. Hotamisligil, G.S., N.S. Shargill, and B.M. Spiegelman, Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science, 1993. 259(5091): p. 87-91.
373. Mitrou, P., et al., Skeletal muscle insulin resistance in morbid obesity: the role of interleukin-6 and leptin. Experimental and clinical endocrinology & diabetes : official journal, German Society of Endocrinology [and] German Diabetes Association, 2011. 119(8): p. 484-9.
374. Cani, P.D., et al., Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes, 2007. 56(7): p. 1761-72.
375. Sell, H., U. Kaiser, and J. Eckel, Expression of chemokine receptors in insulin-resistant human skeletal muscle cells. Hormone and metabolic research = Hormon- und Stoffwechselforschung = Hormones et metabolisme, 2007. 39(4): p. 244-9.
376. Zhang, Y., et al., Cytokines and endotoxin induce cytokine receptors in skeletal muscle. American journal of physiology. Endocrinology and metabolism, 2000. 279(1): p. E196-205.
377. Turnbaugh, P.J., et al., The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Science translational medicine, 2009. 1(6): p. 6ra14.
378. Cani, P.D., et al., Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes, 2008. 57(6): p. 1470-81.
379. Andreasen, A.S., et al., Type 2 diabetes is associated with altered NF-kappaB DNA binding activity, JNK phosphorylation, and AMPK phosphorylation in skeletal muscle after LPS. PloS one, 2011. 6(9): p. e23999.
380. Hussey, S.E., et al., TAK-242, a small-molecule inhibitor of Toll-like receptor 4 signalling, unveils similarities and differences in lipopolysaccharide- and lipid-induced inflammation and insulin resistance in muscle cells. Bioscience reports, 2013. 33(1): p. 37-47.
381. Liang, H., et al., Effect of lipopolysaccharide on inflammation and insulin action in human muscle. PloS one, 2013. 8(5): p. e63983.
382. Holland, W.L., et al., Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. The Journal of clinical investigation, 2011. 121(5): p. 1858-70.
383. Frost, R.A., G.J. Nystrom, and C.H. Lang, Lipopolysaccharide regulates proinflammatory cytokine expression in mouse myoblasts and skeletal muscle. American journal of physiology. Regulatory, integrative and comparative physiology, 2002. 283(3): p. R698-709.
384. Oliveira, A.G., et al., Physical exercise reduces circulating lipopolysaccharide and TLR4 activation and improves insulin signaling in tissues of DIO rats. Diabetes, 2011. 60(3): p. 784-96.
385. Rosenzweig, T., et al., Differential effects of tumor necrosis factor-alpha on protein kinase C isoforms alpha and delta mediate inhibition of insulin receptor signaling. Diabetes, 2002. 51(6): p. 1921-30.
386. Plomgaard, P., et al., Tumor necrosis factor-alpha induces skeletal muscle insulin resistance in healthy human subjects via inhibition of Akt substrate 160 phosphorylation. Diabetes, 2005. 54(10): p. 2939-45.
387. Hirosumi, J., et al., A central role for JNK in obesity and insulin resistance. Nature, 2002. 420(6913): p. 333-6.

137
388. Henstridge, D.C., et al., Skeletal muscle-specific overproduction of constitutively activated c-Jun N-terminal kinase (JNK) induces insulin resistance in mice. Diabetologia, 2012. 55(10): p. 2769-78.
389. Austin, R.L., et al., siRNA-mediated reduction of inhibitor of nuclear factor-kappaB kinase prevents tumor necrosis factor-alpha-induced insulin resistance in human skeletal muscle. Diabetes, 2008. 57(8): p. 2066-73.
390. Salles, J., et al., TNFalpha gene knockout differentially affects lipid deposition in liver and skeletal muscle of high-fat-diet mice. The Journal of nutritional biochemistry, 2012. 23(12): p. 1685-93.
391. Zhang, J., et al., Overactivation of NF-kappaB impairs insulin sensitivity and mediates palmitate-induced insulin resistance in C2C12 skeletal muscle cells. Endocrine, 2010. 37(1): p. 157-66.
392. Sinha, S., et al., Fatty acid-induced insulin resistance in L6 myotubes is prevented by inhibition of activation and nuclear localization of nuclear factor kappa B. The Journal of biological chemistry, 2004. 279(40): p. 41294-301.
393. Bastard, J.P., et al., Elevated levels of interleukin 6 are reduced in serum and subcutaneous adipose tissue of obese women after weight loss. The Journal of clinical endocrinology and metabolism, 2000. 85(9): p. 3338-42.
394. Carey, A.L., et al., Interleukin-6 and tumor necrosis factor-alpha are not increased in patients with Type 2 diabetes: evidence that plasma interleukin-6 is related to fat mass and not insulin responsiveness. Diabetologia, 2004. 47(6): p. 1029-37.
395. Vozarova, B., et al., Circulating interleukin-6 in relation to adiposity, insulin action, and insulin secretion. Obesity research, 2001. 9(7): p. 414-7.
396. Kim, H.J., et al., Differential effects of interleukin-6 and -10 on skeletal muscle and liver insulin action in vivo. Diabetes, 2004. 53(4): p. 1060-7.
397. Klover, P.J., et al., Chronic exposure to interleukin-6 causes hepatic insulin resistance in mice. Diabetes, 2003. 52(11): p. 2784-9.
398. Carey, A.L., et al., Interleukin-6 increases insulin-stimulated glucose disposal in humans and glucose uptake and fatty acid oxidation in vitro via AMP-activated protein kinase. Diabetes, 2006. 55(10): p. 2688-97.
399. Nieto-Vazquez, I., et al., Dual role of interleukin-6 in regulating insulin sensitivity in murine skeletal muscle. Diabetes, 2008. 57(12): p. 3211-21.
400. White, P.J., et al., Protectin DX alleviates insulin resistance by activating a myokine-liver glucoregulatory axis. Nat Med, 2014.
401. Fischer, C.P., Interleukin-6 in acute exercise and training: what is the biological relevance? Exercise immunology review, 2006. 12: p. 6-33.
402. Varma, V., et al., Muscle inflammatory response and insulin resistance: synergistic interaction between macrophages and fatty acids leads to impaired insulin action. American journal of physiology. Endocrinology and metabolism, 2009. 296(6): p. E1300-10.
403. Fink, L.N., et al., Pro-Inflammatory macrophages increase in skeletal muscle of high fat-Fed mice and correlate with metabolic risk markers in humans. Obesity, 2013.
404. Nguyen, M.T., et al., A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via Toll-like receptors 2 and 4 and JNK-dependent pathways. The Journal of biological chemistry, 2007. 282(48): p. 35279-92.
405. Hong, E.G., et al., Interleukin-10 prevents diet-induced insulin resistance by attenuating macrophage and cytokine response in skeletal muscle. Diabetes, 2009. 58(11): p. 2525-35.

138
406. Patsouris, D., et al., Ablation of CD11c-positive cells normalizes insulin sensitivity in obese insulin resistant animals. Cell metabolism, 2008. 8(4): p. 301-9.
407. McMillan, K. and B.S. Masters, Prokaryotic expression of the heme- and flavin-binding domains of rat neuronal nitric oxide synthase as distinct polypeptides: identification of the heme-binding proximal thiolate ligand as cysteine-415. Biochemistry, 1995. 34(11): p. 3686-93.
408. Richards, M.K. and M.A. Marletta, Characterization of neuronal nitric oxide synthase and a C415H mutant, purified from a baculovirus overexpression system. Biochemistry, 1994. 33(49): p. 14723-32.
409. Ghosh, D.K. and D.J. Stuehr, Macrophage NO synthase: characterization of isolated oxygenase and reductase domains reveals a head-to-head subunit interaction. Biochemistry, 1995. 34(3): p. 801-7.
410. Bredt, D.S. and S.H. Snyder, Isolation of nitric oxide synthetase, a calmodulin-requiring enzyme. Proceedings of the National Academy of Sciences of the United States of America, 1990. 87(2): p. 682-5.
411. Hauschildt, S., et al., Induction and activity of NO synthase in bone-marrow-derived macrophages are independent of Ca2+. The Biochemical journal, 1990. 270(2): p. 351-6.
412. Tengan, C.H., G.S. Rodrigues, and R.O. Godinho, Nitric oxide in skeletal muscle: role on mitochondrial biogenesis and function. Int J Mol Sci, 2012. 13(12): p. 17160-84.
413. Rao, K.M., Molecular mechanisms regulating iNOS expression in various cell types. Journal of toxicology and environmental health. Part B, Critical reviews, 2000. 3(1): p. 27-58.
414. Nathan, C. and Q.W. Xie, Regulation of biosynthesis of nitric oxide. The Journal of biological chemistry, 1994. 269(19): p. 13725-8.
415. Geller, D.A. and T.R. Billiar, Molecular biology of nitric oxide synthases. Cancer metastasis reviews, 1998. 17(1): p. 7-23.
416. Perreault, M. and A. Marette, Targeted disruption of inducible nitric oxide synthase protects against obesity-linked insulin resistance in muscle. Nature medicine, 2001. 7(10): p. 1138-43.
417. Behr-Roussel, D., et al., Effect of chronic treatment with the inducible nitric oxide synthase inhibitor N-iminoethyl-L-lysine or with L-arginine on progression of coronary and aortic atherosclerosis in hypercholesterolemic rabbits. Circulation, 2000. 102(9): p. 1033-8.
418. Cromheeke, K.M., et al., Inducible nitric oxide synthase colocalizes with signs of lipid oxidation/peroxidation in human atherosclerotic plaques. Cardiovascular research, 1999. 43(3): p. 744-54.
419. Shimabukuro, M., et al., Role of nitric oxide in obesity-induced beta cell disease. The Journal of clinical investigation, 1997. 100(2): p. 290-5.
420. Shimabukuro, M., et al., Fatty acid-induced beta cell apoptosis: a link between obesity and diabetes. Proceedings of the National Academy of Sciences of the United States of America, 1998. 95(5): p. 2498-502.
421. Zhou, Y.T., et al., Lipotoxic heart disease in obese rats: implications for human obesity. Proceedings of the National Academy of Sciences of the United States of America, 2000. 97(4): p. 1784-9.
422. Okuda, S., et al., Regulation of inducible nitric oxide synthase expression in L6 rat skeletal muscle cells. Am J Physiol, 1997. 272(1 Pt 1): p. C35-40.
423. Kapur, S., et al., Expression of nitric oxide synthase in skeletal muscle: a novel role for nitric oxide as a modulator of insulin action. Diabetes, 1997. 46(11): p. 1691-700.

139
424. Pilon, G., P. Dallaire, and A. Marette, Inhibition of inducible nitric-oxide synthase by activators of AMP-activated protein kinase: a new mechanism of action of insulin-sensitizing drugs. The Journal of biological chemistry, 2004. 279(20): p. 20767-74.
425. Adams, V., et al., Induction of iNOS expression in skeletal muscle by IL-1beta and NFkappaB activation: an in vitro and in vivo study. Cardiovascular research, 2002. 54(1): p. 95-104.
426. Di Marco, S., et al., NF-kappa B-mediated MyoD decay during muscle wasting requires nitric oxide synthase mRNA stabilization, HuR protein, and nitric oxide release. Molecular and cellular biology, 2005. 25(15): p. 6533-45.
427. Lambertucci, R.H., et al., The effects of palmitic acid on nitric oxide production by rat skeletal muscle: mechanism via superoxide and iNOS activation. Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology, 2012. 30(5): p. 1169-80.
428. Centeno-Baez, C., P. Dallaire, and A. Marette, Resveratrol inhibition of inducible nitric oxide synthase in skeletal muscle involves AMPK but not SIRT1. American journal of physiology. Endocrinology and metabolism, 2011. 301(5): p. E922-30.
429. Carvalho-Filho, M.A., et al., S-nitrosation of the insulin receptor, insulin receptor substrate 1, and protein kinase B/Akt: a novel mechanism of insulin resistance. Diabetes, 2005. 54(4): p. 959-67.
430. Sugita, H., et al., Inducible nitric-oxide synthase and NO donor induce insulin receptor substrate-1 degradation in skeletal muscle cells. The Journal of biological chemistry, 2005. 280(14): p. 14203-11.
431. Torres, S.H., et al., Inflammation and nitric oxide production in skeletal muscle of type 2 diabetic patients. The Journal of endocrinology, 2004. 181(3): p. 419-27.
432. Kraus, R.M., et al., Obesity, insulin resistance, and skeletal muscle nitric oxide synthase. Journal of applied physiology, 2012. 113(5): p. 758-65.
433. Cha, H.N., et al., Lack of inducible nitric oxide synthase prevents lipid-induced skeletal muscle insulin resistance without attenuating cytokine level. Journal of pharmacological sciences, 2011. 117(2): p. 77-86.
434. Lee-Young, R.S., et al., AMP-activated protein kinase (AMPK)alpha2 plays a role in determining the cellular fate of glucose in insulin-resistant mouse skeletal muscle. Diabetologia, 2013. 56(3): p. 608-17.
435. Ischiropoulos, H., Protein tyrosine nitration--an update. Archives of biochemistry and biophysics, 2009. 484(2): p. 117-21.
436. Ischiropoulos, H., Biological selectivity and functional aspects of protein tyrosine nitration. Biochemical and biophysical research communications, 2003. 305(3): p. 776-83.
437. Ara, J., et al., Inactivation of tyrosine hydroxylase by nitration following exposure to peroxynitrite and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Proceedings of the National Academy of Sciences of the United States of America, 1998. 95(13): p. 7659-63.
438. Ischiropoulos, H., Biological tyrosine nitration: a pathophysiological function of nitric oxide and reactive oxygen species. Archives of biochemistry and biophysics, 1998. 356(1): p. 1-11.
439. Ceriello, A., et al., Detection of nitrotyrosine in the diabetic plasma: evidence of oxidative stress. Diabetologia, 2001. 44(7): p. 834-8.
440. Duplain, H., et al., Stimulation of peroxynitrite catalysis improves insulin sensitivity in high fat diet-fed mice. The Journal of physiology, 2008. 586(16): p. 4011-6.
441. Zhou, J. and K. Huang, Peroxynitrite mediates muscle insulin resistance in mice via nitration of IRbeta/IRS-1 and Akt. Toxicology and applied pharmacology, 2009. 241(1): p. 101-10.

140
442. Pilon, G., et al., Endotoxin mediated-iNOS induction causes insulin resistance via ONOO(-) induced tyrosine nitration of IRS-1 in skeletal muscle. PloS one, 2010. 5(12): p. e15912.
443. Hess, D.T., et al., Protein S-nitrosylation: purview and parameters. Nature reviews. Molecular cell biology, 2005. 6(2): p. 150-66.
444. Stamler, J.S., S. Lamas, and F.C. Fang, Nitrosylation. the prototypic redox-based signaling mechanism. Cell, 2001. 106(6): p. 675-83.
445. Wink, D.A., et al., Reactions of the bioregulatory agent nitric oxide in oxygenated aqueous media: determination of the kinetics for oxidation and nitrosation by intermediates generated in the NO/O2 reaction. Chemical research in toxicology, 1993. 6(1): p. 23-7.
446. Yasukawa, T., et al., S-nitrosylation-dependent inactivation of Akt/protein kinase B in insulin resistance. The Journal of biological chemistry, 2005. 280(9): p. 7511-8.
447. Carvalho-Filho, M.A., et al., Targeted disruption of iNOS prevents LPS-induced S-nitrosation of IRbeta/IRS-1 and Akt and insulin resistance in muscle of mice. American journal of physiology. Endocrinology and metabolism, 2006. 291(3): p. E476-82.
448. Pauli, J.R., et al., Acute physical exercise reverses S-nitrosation of the insulin receptor, insulin receptor substrate 1 and protein kinase B/Akt in diet-induced obese Wistar rats. The Journal of physiology, 2008. 586(2): p. 659-71.
449. White, P.J., et al., Nitrosative modifications of protein and lipid signaling molecules by reactive nitrogen species. Am J Physiol Endocrinol Metab, 2010. 299(6): p. E868-78.
450. Andersen, P. and B. Saltin, Maximal perfusion of skeletal muscle in man. J Physiol, 1985. 366: p. 233-49.
451. Dawson, D., et al., Vascular recruitment in skeletal muscle during exercise and hyperinsulinemia assessed by contrast ultrasound. Am J Physiol Endocrinol Metab, 2002. 282(3): p. E714-20.
452. Inyard, A.C., et al., Contraction stimulates nitric oxide independent microvascular recruitment and increases muscle insulin uptake. Diabetes, 2007. 56(9): p. 2194-200.
453. Sjoberg, K.A., et al., A new method to study changes in microvascular blood volume in muscle and adipose tissue: real-time imaging in humans and rat. Am J Physiol Heart Circ Physiol, 2011. 301(2): p. H450-8.
454. Vincent, M.A., et al., Mixed meal and light exercise each recruit muscle capillaries in healthy humans. Am J Physiol Endocrinol Metab, 2006. 290(6): p. E1191-7.
455. Thomas, M.M., et al., Muscle-specific AMPK beta1beta2-null mice display a myopathy due to loss of capillary density in nonpostural muscles. FASEB J, 2014.
456. Lau, K.S., et al., nNOS and eNOS modulate cGMP formation and vascular response in contracting fast-twitch skeletal muscle. Physiol Genomics, 2000. 2(1): p. 21-7.
457. Kristiansen, S., et al., Glucose uptake is increased in trained vs. untrained muscle during heavy exercise. J Appl Physiol (1985), 2000. 89(3): p. 1151-8.
458. Lauritzen, H.P., et al., Kinetics of contraction-induced GLUT4 translocation in skeletal muscle fibers from living mice. Diabetes, 2010. 59(9): p. 2134-44.
459. Derave, W., et al., Contraction-stimulated muscle glucose transport and GLUT-4 surface content are dependent on glycogen content. Am J Physiol, 1999. 277(6 Pt 1): p. E1103-10.
460. Fueger, P.T., et al., Hexokinase II partial knockout impairs exercise-stimulated glucose uptake in oxidative muscles of mice. Am J Physiol Endocrinol Metab, 2003. 285(5): p. E958-63.
461. Fueger, P.T., et al., Control of exercise-stimulated muscle glucose uptake by GLUT4 is dependent on glucose phosphorylation capacity in the conscious mouse. J Biol Chem, 2004. 279(49): p. 50956-61.

141
462. Pederson, B.A., et al., Exercise capacity of mice genetically lacking muscle glycogen synthase: in mice, muscle glycogen is not essential for exercise. J Biol Chem, 2005. 280(17): p. 17260-5.
463. Bergstrom, J., et al., Diet, muscle glycogen and physical performance. Acta Physiol Scand, 1967. 71(2): p. 140-50.
464. Abbott, M.J., L.D. Bogachus, and L.P. Turcotte, AMPKalpha2 deficiency uncovers time dependency in the regulation of contraction-induced palmitate and glucose uptake in mouse muscle. J Appl Physiol (1985), 2011. 111(1): p. 125-34.
465. Dzamko, N., et al., AMPK-independent pathways regulate skeletal muscle fatty acid oxidation. J Physiol, 2008. 586(Pt 23): p. 5819-31.
466. Gulve, E.A., G.D. Cartee, and J.O. Holloszy, Prolonged incubation of skeletal muscle in vitro: prevention of increases in glucose transport. Am J Physiol, 1991. 261(1 Pt 1): p. C154-60.
467. Henderson, A.A., et al., Dose-response relationships for the effects of insulin on glucose and fat metabolism in injured patients and control subjects. Clin Sci (Lond), 1991. 80(1): p. 25-32.
468. Little, R.A., et al., The disposal of intravenous glucose studied using glucose and insulin clamp techniques in sepsis and trauma in man. Acta Anaesthesiol Belg, 1987. 38(4): p. 275-9.
469. Thorell, A., J. Nygren, and O. Ljungqvist, Insulin resistance: a marker of surgical stress. Curr Opin Clin Nutr Metab Care, 1999. 2(1): p. 69-78.
470. Del Aguila, L.F., et al., Muscle damage impairs insulin stimulation of IRS-1, PI 3-kinase, and Akt-kinase in human skeletal muscle. Am J Physiol Endocrinol Metab, 2000. 279(1): p. E206-12.
471. Andrisse, S., et al., ATM and GLUT1-S490 phosphorylation regulate GLUT1 mediated transport in skeletal muscle. PLoS One, 2013. 8(6): p. e66027.
472. Halaby, M.J., et al., ATM protein kinase mediates full activation of Akt and regulates glucose transporter 4 translocation by insulin in muscle cells. Cell Signal, 2008. 20(8): p. 1555-63.
473. Jeong, I., et al., Role of ataxia telangiectasia mutated in insulin signalling of muscle-derived cell lines and mouse soleus. Acta Physiol (Oxf), 2010. 198(4): p. 465-75.
474. Sakagami, H., et al., Loss of HIF-1alpha impairs GLUT4 translocation and glucose uptake by the skeletal muscle cells. Am J Physiol Endocrinol Metab, 2014. 306(9): p. E1065-76.
475. Schofield, C.J. and P.J. Ratcliffe, Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol, 2004. 5(5): p. 343-54.
476. Haddad, J.J. and S.C. Land, A non-hypoxic, ROS-sensitive pathway mediates TNF-alpha-dependent regulation of HIF-1alpha. FEBS Lett, 2001. 505(2): p. 269-74.
477. Thorell, A., et al., Insulin resistance after abdominal surgery. Br J Surg, 1994. 81(1): p. 59-63.
478. Reid, M.B., et al., Reactive oxygen in skeletal muscle. I. Intracellular oxidant kinetics and fatigue in vitro. J Appl Physiol (1985), 1992. 73(5): p. 1797-804.
479. Andrisse, S., et al., Role of GLUT1 in regulation of reactive oxygen species. Redox Biol, 2014. 2: p. 764-71.





























