Embed Size (px)
Citation preview

VETAGRO SUP CAMPUS VETERINAIRE DE LYON
Année 2013-2014 Thèse n°
Les utilisations de la thérapie génique
THESE
Présentée à l’UNIVERSITE CLAUDE-BERNARD - LYON I (Médecine - Pharmacie)
et soutenue publiquement le Lundi 27 Octobre 2014 pour obtenir le grade de Docteur Vétérinaire
par
HELLEZ Justine
Née le 20 mai 1989 à Seclin (59)

2
3
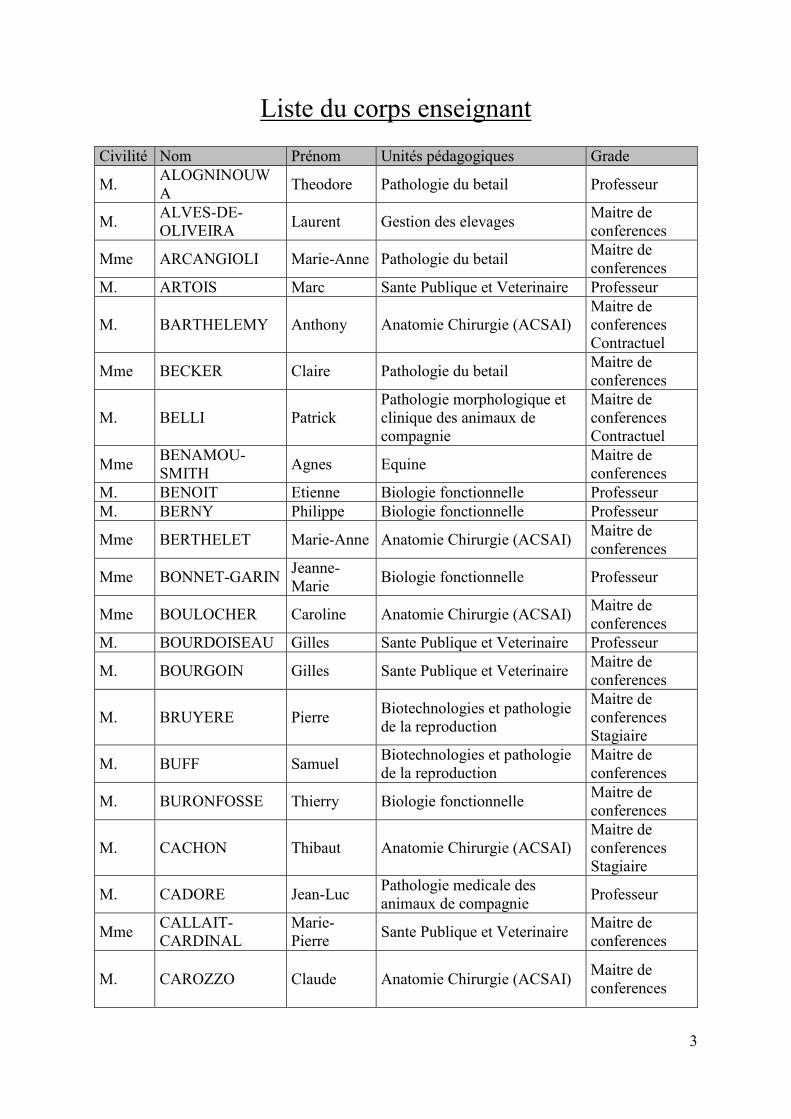
Liste du corps enseignant Civilité Nom Prénom Unités pédagogiques Grade
M. ALOGNINOUWA Theodore Pathologie du betail Professeur
M. ALVES-DE-OLIVEIRA Laurent Gestion des elevages Maitre de
conferences
Mme ARCANGIOLI Marie-Anne Pathologie du betail Maitre de conferences
M. ARTOIS Marc Sante Publique et Veterinaire Professeur
M. BARTHELEMY Anthony Anatomie Chirurgie (ACSAI) Maitre de conferences Contractuel
Mme BECKER Claire Pathologie du betail Maitre de conferences
M. BELLI Patrick Pathologie morphologique et clinique des animaux de compagnie
Maitre de conferences Contractuel
Mme BENAMOU-SMITH Agnes Equine Maitre de
conferences M. BENOIT Etienne Biologie fonctionnelle Professeur M. BERNY Philippe Biologie fonctionnelle Professeur
Mme BERTHELET Marie-Anne Anatomie Chirurgie (ACSAI) Maitre de conferences
Mme BONNET-GARIN Jeanne-Marie Biologie fonctionnelle Professeur
Mme BOULOCHER Caroline Anatomie Chirurgie (ACSAI) Maitre de conferences
M. BOURDOISEAU Gilles Sante Publique et Veterinaire Professeur
M. BOURGOIN Gilles Sante Publique et Veterinaire Maitre de conferences
M. BRUYERE Pierre Biotechnologies et pathologie de la reproduction
Maitre de conferences Stagiaire
M. BUFF Samuel Biotechnologies et pathologie de la reproduction
Maitre de conferences
M. BURONFOSSE Thierry Biologie fonctionnelle Maitre de conferences
M. CACHON Thibaut Anatomie Chirurgie (ACSAI) Maitre de conferences Stagiaire
M. CADORE Jean-Luc Pathologie medicale des animaux de compagnie Professeur
Mme CALLAIT-CARDINAL
Marie-Pierre Sante Publique et Veterinaire Maitre de
conferences
M. CAROZZO Claude Anatomie Chirurgie (ACSAI) Maitre de conferences
4
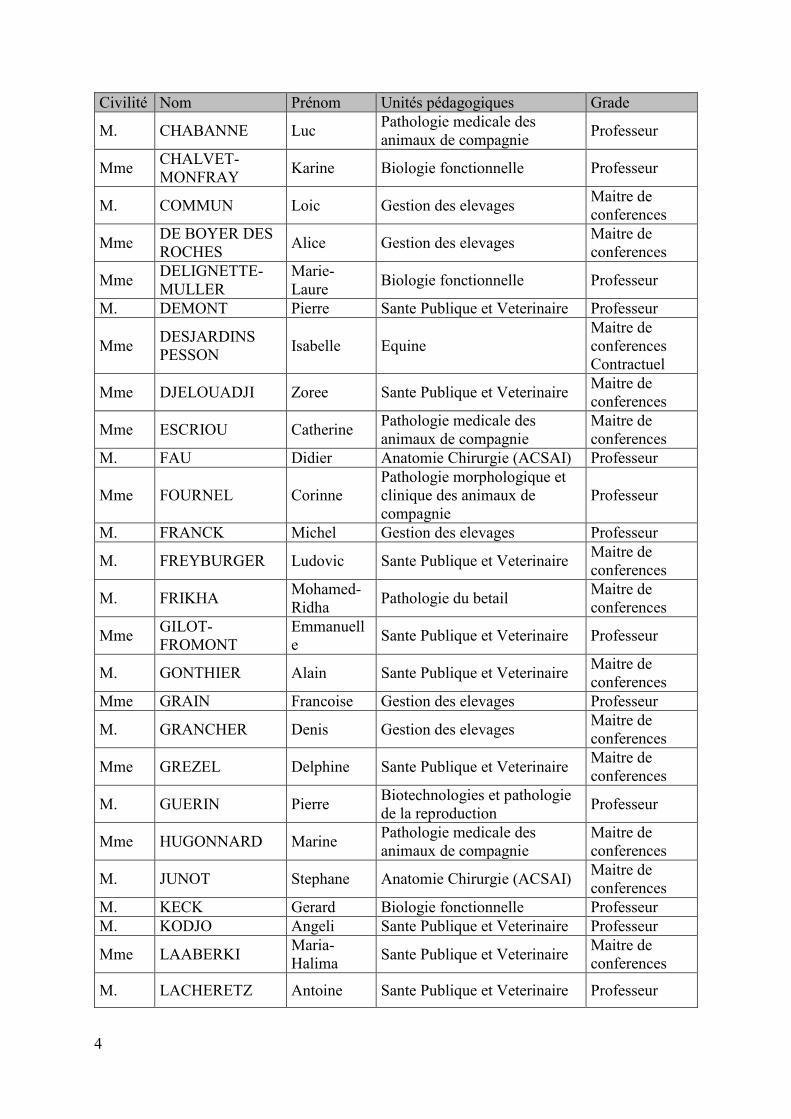
Civilité Nom Prénom Unités pédagogiques Grade
M. CHABANNE Luc Pathologie medicale des animaux de compagnie Professeur
Mme CHALVET-MONFRAY Karine Biologie fonctionnelle Professeur
M. COMMUN Loic Gestion des elevages Maitre de conferences
Mme DE BOYER DES ROCHES Alice Gestion des elevages Maitre de
conferences
Mme DELIGNETTE-MULLER
Marie-Laure Biologie fonctionnelle Professeur
M. DEMONT Pierre Sante Publique et Veterinaire Professeur
Mme DESJARDINS PESSON Isabelle Equine
Maitre de conferences Contractuel
Mme DJELOUADJI Zoree Sante Publique et Veterinaire Maitre de conferences
Mme ESCRIOU Catherine Pathologie medicale des animaux de compagnie
Maitre de conferences
M. FAU Didier Anatomie Chirurgie (ACSAI) Professeur
Mme FOURNEL Corinne Pathologie morphologique et clinique des animaux de compagnie
Professeur
M. FRANCK Michel Gestion des elevages Professeur
M. FREYBURGER Ludovic Sante Publique et Veterinaire Maitre de conferences
M. FRIKHA Mohamed-Ridha Pathologie du betail Maitre de
conferences
Mme GILOT-FROMONT
Emmanuelle Sante Publique et Veterinaire Professeur
M. GONTHIER Alain Sante Publique et Veterinaire Maitre de conferences
Mme GRAIN Francoise Gestion des elevages Professeur
M. GRANCHER Denis Gestion des elevages Maitre de conferences
Mme GREZEL Delphine Sante Publique et Veterinaire Maitre de conferences
M. GUERIN Pierre Biotechnologies et pathologie de la reproduction Professeur
Mme HUGONNARD Marine Pathologie medicale des animaux de compagnie
Maitre de conferences
M. JUNOT Stephane Anatomie Chirurgie (ACSAI) Maitre de conferences
M. KECK Gerard Biologie fonctionnelle Professeur M. KODJO Angeli Sante Publique et Veterinaire Professeur
Mme LAABERKI Maria-Halima Sante Publique et Veterinaire Maitre de
conferences
M. LACHERETZ Antoine Sante Publique et Veterinaire Professeur
5
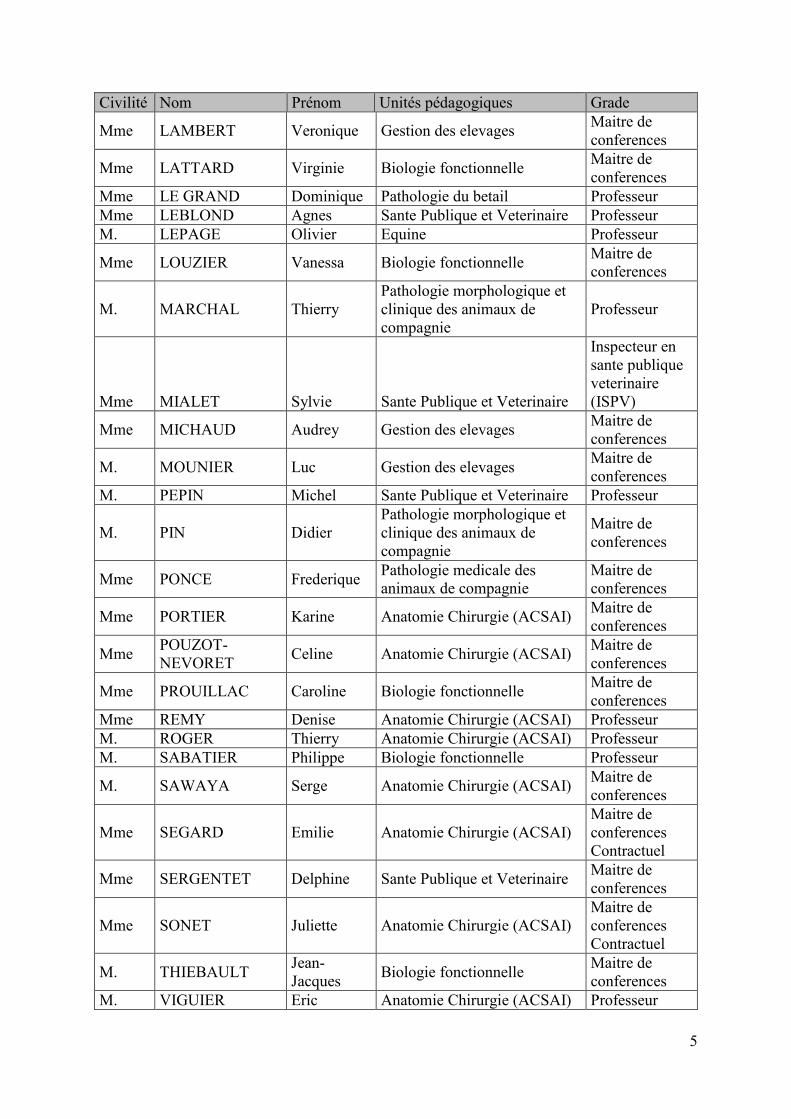
Civilité Nom Prénom Unités pédagogiques Grade
Mme LAMBERT Veronique Gestion des elevages Maitre de conferences
Mme LATTARD Virginie Biologie fonctionnelle Maitre de conferences
Mme LE GRAND Dominique Pathologie du betail Professeur Mme LEBLOND Agnes Sante Publique et Veterinaire Professeur M. LEPAGE Olivier Equine Professeur
Mme LOUZIER Vanessa Biologie fonctionnelle Maitre de conferences
M. MARCHAL Thierry Pathologie morphologique et clinique des animaux de compagnie
Professeur
Mme MIALET Sylvie Sante Publique et Veterinaire
Inspecteur en sante publique veterinaire (ISPV)
Mme MICHAUD Audrey Gestion des elevages Maitre de conferences
M. MOUNIER Luc Gestion des elevages Maitre de conferences
M. PEPIN Michel Sante Publique et Veterinaire Professeur
M. PIN Didier Pathologie morphologique et clinique des animaux de compagnie
Maitre de conferences
Mme PONCE Frederique Pathologie medicale des animaux de compagnie
Maitre de conferences
Mme PORTIER Karine Anatomie Chirurgie (ACSAI) Maitre de conferences
Mme POUZOT-NEVORET Celine Anatomie Chirurgie (ACSAI) Maitre de
conferences
Mme PROUILLAC Caroline Biologie fonctionnelle Maitre de conferences
Mme REMY Denise Anatomie Chirurgie (ACSAI) Professeur M. ROGER Thierry Anatomie Chirurgie (ACSAI) Professeur M. SABATIER Philippe Biologie fonctionnelle Professeur
M. SAWAYA Serge Anatomie Chirurgie (ACSAI) Maitre de conferences
Mme SEGARD Emilie Anatomie Chirurgie (ACSAI) Maitre de conferences Contractuel
Mme SERGENTET Delphine Sante Publique et Veterinaire Maitre de conferences
Mme SONET Juliette Anatomie Chirurgie (ACSAI) Maitre de conferences Contractuel
M. THIEBAULT Jean-Jacques Biologie fonctionnelle Maitre de
conferences M. VIGUIER Eric Anatomie Chirurgie (ACSAI) Professeur

6
Civilité Nom Prénom Unités pédagogiques Grade
Mme VIRIEUX-WATRELOT Dorothee
Pathologie morphologique et clinique des animaux de compagnie
Maitre de conferences Contractuel
M. ZENNER Lionel Sante Publique et Veterinaire Professeur

7
Remerciements
A M. le professeur Damien SANLAVILLE, de la faculté de Médecine de Lyon, Pour avoir accepté de présider ce jury de thèse,
Hommages respectueux.
A Mme Véronique LAMBERT, de VetAgro Sup, Campus Lyon, Pour avoir accepté d’encadrer et de corriger ce travail de thèse,
Sincères remerciements.
A Mme Virginie LATTARD, de VetAgro Sup, Campus Lyon, Pour avoir accepté de participer à ce jury de thèse et de juger mon travail,
Sincères remerciements.

8

9
A mes parents,
A ma sœur,
A mes grands-parents et au reste de la famille,
A ma future belle-famille,
A mes amis,
A toi, mon amour.
Merci de votre présence et de votre soutien, Au cours de ces années,
Passées et futures.

10

11
Table des matières
LISTE DU CORPS ENSEIGNANT 3
REMERCIEMENTS 7
TABLE DES ILLUSTRATIONS 15
TABLE DES TABLEAUX 16
LISTE DES ABREVIATIONS 17
INTRODUCTION 19
PARTIE 1 : LES PRINCIPES DE LA THERAPIE GENIQUE 21
I. Lignée germinale versus lignée somatique 21
II. Modalités d’utilisation de la thérapie génique 21 1. Addition d’un gène 21 2. Réparation des gènes 22 3. Modification de l’expression des gènes 25
a. Extinction génique 26 i. Interférence de l’ARN 26 ii. ARN anti-sens 29 iii. Ribozymes et ADNzymes (ou désoxyribozymes) 30 iv. Autres molécules régulatrices 30
b. Expression forcée 31 c. Autres modifications 32
III. Modalités d’administration de la thérapie génique 32 1. In vivo 32 2. Ciblage de l’expression du transgène 32 3. Ex vivo 33
IV. Les vecteurs 34 1. Les vecteurs viraux 35
a. Les Adénovirus 35 b. Les Adeno-Associated virus 37 c. Les Retroviridae 38
i. Les rétrovirus simples 39 ii. Les lentivirus 39

12
2. Les vecteurs non-viraux 40 a. Les vecteurs chimiques 41
i. Les lipides cationiques 41 ii. Les polymères cationiques 42 iii. Les polymères biodégradables 43 iv. Les nanotubes de carbone 43 v. Autres nanoparticules non virales 44
b. Les acides nucléiques nus 45 i. Micro injection 46 ii. Gene gun 46 iii. Technique hydrodynamique 46 iv. Electroporation 47 v. Nucléofection 47 vi. Sonoporation 48
3. Perspectives d’améliorations 50 a. Les nanoparticules magnétiques 50 b. Les systèmes transposon/transposase : Sleeping Beauty 51 c. Les aptamères 52
4. Conclusions 55
V. Les maladies 55
PARTIE 2 : EXEMPLES D’APPLICATIONS DE LA THERAPIE GENIQUE 57
I. Les maladies génétiques 57 1. L’hémophilie A 57
a. La maladie 57 b. Les modèles animaux 58
i. La souris 58 ii. Le chien 58 iii. Les nouveaux modèles 59
c. Intérêt de la thérapie génique 60 d. Utilisation de la thérapie génique 60
i. Les premiers essais cliniques 61 ii. Nouvelles orientations 63
e. Conclusion 68 2. La myodystrophie de Duchenne 69
a. La maladie 69 b. Les modèles animaux 71
i. La souris 71 ii. Le chien 71 iii. Les autres modèles 72

13
c. Utilisation de la thérapie génique 72 i. Remplacement du gène 72 ii. Le saut d’exon 74 iii. Modification de l’expression des gènes 75
d. Conclusion 76
II. Les maladies acquises : l’insuffisance cardiaque 77 1. Epidémiologie et physiopathologie 77 2. Intérêt de la thérapie génique 77
a. Les modèles animaux 78 i. Le rat 78 ii. La souris 78 iii. Le cochon 78 iv. Le lapin 78
b. Les vecteurs 79 i. Les vecteurs viraux 79 ii. Les vecteurs non-viraux 80 iii. Ciblage et régulation de l’expression 80
c. Les modalités d’administration 80 i. Perfusion via l’artère ou la veine coronaire 80 ii. Injection locale myocardique et péricardique 81 iii. Microbulles et ultrasons 82
d. Les gènes cibles 82 i. Le système β-adrénergique 83 ii. Circulation du calcium 83 iii. Autres cibles 84
3. Conclusion 85
III. Les maladies infectieuses : le SIDA 87 1. L’infection par le VIH et ses conséquences 87
a. Cycle de réplication 87 b. Génome du VIH 88
2. Intérêt de la thérapie génique 89 a. Traitement actuel 89 b. Modèles animaux 90 c. Le « patient berlinois » 91
3. Utilisation de la thérapie génique 91 a. Cibles de la thérapie génique 91 b. Modalités d’administration 92 c. Techniques de la thérapie génique anti-VIH 92
i. Enzymes spécifiques 92 ii. Molécules d’ARN 94 iii. Inhibiteurs protéiques 95
4. Conclusions 95

14
IV. La thérapie génique anticancéreuse 97 1. Thérapie cytotoxique 97
a. Gènes induisant le suicide cellulaire 97 b. Reprogrammation des cellules tumorales 98 c. Extinction génique 99
i. Les siARN 99 ii. Les microARN 99
d. La virothérapie 100 2. Thérapie anti-angiogénique 101 3. Immunothérapie 102
a. Modification des cellules de l’immunité 103 i. Les récepteurs des lymphocytes T 103 ii. Les récepteurs chimériques 104
b. Modifications des cellules tumorales 105 4. Ciblage des cellules tumorales 106 5. Conclusions 108
V. Et en médecine vétérinaire ? 109
DISCUSSION 111
CONCLUSION .
BIBLIOGRAPHIE 117

15
Table des illustrations Figure 1 : Conséquences de l’intégration d’un transgène au sein d’un génome hôte d’après HACKETT et al. (2007) ........................................................................................................... 22
Figure 2 : Recombinaison homologue. A : spontanée, B : Intervention d’une nucléase en doigts de Zinc, D’après PESSACH et NOTARANGELO (2011) ...................................................... 23
Figure 3 : Correspondance RVD / nucléotide d’après VALTON et al. (2014) ....................... 24
Figure 4 : Structure et mise en place des TALEN sur l’ADN d’après VALTON (2014) ........ 24
Figure 5 : Fonctionnement schématique du système CRISPR, d’après BONDY-DENOMY et DAVIDSON (2014) ................................................................................................................. 25
Figure 6 : Schéma de la synthèse des microARN d’après ARBUTHNOT et THOMPSON (2008) ....................................................................................................................................... 27
Figure 7 : Mécanisme d’action des siARN, d’après LI Z. et RANA (2011) ........................... 28
Figure 8 : Schématisation de l’expérience menée par Brown et son équipe ............................ 29
Figure 9 : A. Liaisons Watson-Crick, B. Liaisons Hoogsteen, d’après GHOSAL et al. (2006) .................................................................................................................................................. 31
Figure 10 : Mécanismes des systèmes « Tet Off » / « Tet On » d’après NAIDOO et YOUNG (2012) ....................................................................................................................................... 33
Figure 11 : Concept de thérapie génique in vivo et ex vivo d’après KAUFMANN et al. (2013) .................................................................................................................................................. 34
Figure 12 : Mode de pénétration de l’adénovirus HAd5 d’après COUGHLAN et al. (2010) . 36
Figure 13 : cycle de vie des rétrovirus d’après SPENCER et PALMARINI (2012) ............... 38
Figure 14 : Représentation des barrières qui limitent l’utilisation de vecteurs non viraux in vivo
d’après AL-DOSARI et GAO (2009) ...................................................................................... 41
Figure 15 : Combinaisons à base de nanotubes de carbone étudiées d’après BATES et KOSTARELOS (2013) ............................................................................................................ 44
Figure 16 : Schématisation de la réaction des microbulles aux ultrasons d’après LENTACKER et al. (2013) .............................................................................................................................. 48
Figure 17 : Effets physiques de la cavitation stable d’après LENTACKER et al. (2013) ....... 49
Figure 18 : Effets physiques de la cavitation inertielle d’après LENTACKER et al. (2013) .. 49
Figure 19 : Schématisation du fonctionnement d’un vecteur magnétique codant pour l’expression d’une protéine fluorescente d’après ZHENG et al. (2012) .................................. 50
Figure 20 : Schématisation du fonctionnement d’un système transposon/transposase d’après (Skipper, 2013) ......................................................................................................................... 52
Figure 21 : Schématisation de la dystrophine et du complexe dystrophine/glycoprotéines membranaires d’après DOUGLAS et WOOD (2013) ............................................................. 70
Figure 22 : Schématisation de trois différents gènes de mini-dystrophine d’après WANG B. et
al. (2000) .................................................................................................................................. 73
Figure 23 : Principe de l’utilisation des ONA lors d’une mutation provoquant la délétion de l’exon 50 d’après – DOUGLAS et WOOD (2013) .................................................................. 74
Figure 24 : Techniques de perfusion via les artères ou veines coronaires d’après HAJJAR (2013) .................................................................................................................................................. 81

16
Figure 25 : Techniques d’injection locale d’après HAJJAR (2013) ........................................ 81
Figure 26 : Schématisation du cardiomyocyte d’après HAJJAR (2013) ................................. 84
Figure 27 : Cycle de réplication du VIH d’après PANDHARE et DASH (2011) ................... 88
Figure 28 : Organisation du génome du VIH d’après COSTIN (2007) ................................... 89
Figure 29 : Schématisation de la création de lymphocytes génétiquement modifiés d’après SCHMITT et al. (2009) .......................................................................................................... 104
Figure 30 : Effets anti-tumoraux de l’interleukine 12 d’après LASEK et al. (2014) ............ 105
Figure 31 : Schématisation du mécanisme de l’empreinte génomique .................................. 107
Table des tableaux Tableau 1 : Comparaison des vecteurs en thérapie génique ..................................................... 54
Tableau 2 : Procédures pour induire une insuffisance cardiaque d’après ZARAGOZA et al. (2011) ....................................................................................................................................... 79

17
Liste des abréviations AAV Adeno-Associated Virus
AC Adényl Cyclase
ADN Acide désoxyribonucléique
ADNc ADN complémentaire
AMP Adénosine monophosphate
AMPc Adénosine monophosphate cyclique
ANSES Agence Nationale de Sécurité Sanitaire
ARN Acide ribonucléique
ARNm ARN messager
ATP Adénosine triphosphate
BDD B-Domain-Deleted
CAR Récepteur aux Coxsakievirus et Adénovirus
CMH Complexe Majeur d'Histocompatibilité
CMV Cytomegalovirus
CPA Cellule Présentatrice d’Antigènes
CRISPR Clustered Regulary Interspaced Short Palindromic Repeats
CXCR Récepteur aux chémokines
DMD Myodystrophie de Duchenne
F8 Gène codant pour le facteur VIII
FDA Food and Drugs Administration
fVIII Facteur VIII
fVIIIa Facteur VIII Activé
FvW Facteur de Von Willebrand
HA Hémophilie A
IRM Imagerie par Résonnance Magnétique
kb Kilobases
KO Knock-Out
LTR Long Terminal Repeat
NILV Non Integrating Lentivirus
ONA Oligonucléotide Antisens
PANAM Polyamidoamine
PEI Polyéthylènimine
RISC Complexe d'extinction induit par l'ARN

18
Rnase Ribonucléase
SELEX Systematic Evolution of Ligands by Exponential Enrichment
siARN Short Interfering ARN
SIDA Syndrome d'ImmunoDéficience Acquise
SIV Virus de l'Immunodéficience Simienne
TALEN Transcription Activator-Like Effector Nuclease
VHS Virus Herpès Simplex
VIH Virus de l'Immunodéficience Humaine
VSV Virus de la Stomatite Vésiculeuse

19
Introduction La thérapie génique est une biotechnologie basée sur le transfert de matériel génétique. Elle consiste à utiliser une séquence d’acides nucléiques (le plus souvent un gène, appelé transgène) afin de traiter une maladie ou ses symptômes. Cette séquence est insérée dans les cellules du sujet malade (qu’on appelle alors cellules transfectées) afin qu’elles produisent elles-mêmes le médicament à effet thérapeutique. Pour réaliser cela, on construit une séquence d’acides nucléiques, appelée cassette génique, via des techniques de recombinaison de l’ADN. On y insère le transgène et tout ce qui sera nécessaire à la cellule cible afin qu’elle puisse l’exprimer (promoteur, autres séquences régulatrices, etc.). Cette cassette génique est ensuite prise en charge par un vecteur qui va la délivrer au sein des noyaux des cellules d’intérêt. On peut aussi utliser, à la place d’une cassette génique, d’autres types d’acides nucléiques comme des ARN créés par synthèse chimique. Le concept de thérapie génique n’est pas récent. Dès les années 70, avec l’exploration et la cartographie du génome humain, on pense qu’il est possible de guérir d’une maladie génétique en modifiant les allèles mutés (réparation de la mutation ou adjonction de l’allèle sauvage) - ESCORS et BRECKPOT (2010). La technique de transfert génétique in vitro est maitrisée dans les années 80. La recherche en thérapie génique se tourne donc en premier lieu vers les syndromes d’immnuodéficience congénitaux ou héréditaires. En effet, dans ces maladies, les cellules atteintes sont des cellules circulantes, qu’on peut prélever, mettre en culture et réinjecter après transfection. Les premiers vecteurs à avoir été utilsés sont les rétrovirus en raison de leur capacité à insérer leur génome de façon stable dans les cellules qu’ils infectent. Le premier essai clinique approuvé a lieu en 1991. Il concerne un syndrome d’immunodéficience, la déficience en adénoside déaminase qui est une enzyme nécessaire au métabolisme des purines. Cet essai clinique implique deux patients. Le vecteur utilisé est un rétrovirus transportant le gène pour l’adénoside déaminase. Les résultats sont mitigés mais l’essai clinique ne met en évidence aucun effet secondaire. La recherche en thérapie génique est fortement remise en question lors d’un autre essai clinique conduit en 1999 : des enfants immunodéficients (atteints d’immunodéficience combinée sévère liée au chromosome X) sont traités par thérapie génique via un vecteur rétroviral. Les premiers résultats sont très encourageants (reconstitution d’une population immunitaire normale), mais trois ans après l’essai clinique, deux des enfants traités développent une leucémie dont on détermine qu’elle est due à l’insertion du transgène dans le génome des cellules souches sanguines. L’un des deux décéde des suites de sa leucémie – PESSACH et NOTARANGELO (2011). A partir de ce moment-là, les recherches en thérapie génique se portent vers d’autres vecteurs afin de mieux contrôler le lieu d’insertion du transgène. Malgré cet échec, la thérapie génique s’est bien développée dans différents domaines. Aujourd’hui, la thérapie génique est appliquée

20
à de nombreuses maladies, génétiques comme acquises, telles que l’insuffisance cardiaque, le SIDA et les cancers. L’application de la thérapie génique aux maladies acquises repose sur la compréhension des mécanismes moléculaires impliqués dans le développement de la maladie et l’identification de cibles pouvant être modifiées par modulation de l’expression génique. Les principes de la thérapie génique seront présentés dans une première partie afin de présenter les stratégies et les techniques nécessaires à la mise en place d’un traitement de thérapie génique. Nous verrons que plusieurs paramètres sont à prendre en compte avant de créer un traitement de thérapie génique. Il faut choisir le type de modification à apporter au génome, les modalités d’administration ainsi que le vecteur qui transportera la cassette génétique. Nous illustrerons ensuite ces principes à travers plusieurs maladies à l’étude à l’heure actuelle.

21
Partie 1 : Les principes de la thérapie génique Nous décrirons dans cette partie les notions de base nécessaires à la compréhension du fonctionnement de la thérapie génique.
I. Lignée germinale versus lignée somatique La thérapie génique a été créée pour pouvoir s’appliquer à toutes les cellules eucaryotes, aux cellules germinales comme aux cellules somatiques. Par transfection des cellules de la lignée germinale, il serait possible d’éliminer certaines maladies de la descendance des sujets traités. S’il est théoriquement possible de le réaliser, cela soulève trop de problèmes éthiques pour que la recherche s’y intéresse vraiment et lève les barrières technologiques actuelles. Pour le moment, la thérapie génique humaine intéresse exclusivement les cellules somatiques. Les techniques de thérapie génique germinale sont uniquement utilisée en transgénèse animale, chez les modèles animaux, afin de créer de contrôler leur génotype pour les besoins de la recherche – SMITH (2004). Un décret interdisant la thérapie génique germinale humaine a été édité par le Conseil de l’Europe en 1991 mais chaque pays possède sa propre législation à ce sujet. Par exemple, en Allemagne, l’utilisation de la thérapie génique sur les cellules germinales est considérée comme un crime – JIN (2008). A l’opposé, la FDA (Food and Drug Administration, équivalent nord-américain de l’ANSES) a autorisé l’utilisation de la thérapie génique sur la lignée germinale aux Etats-Unis. II. Modalités d’utilisation de la thérapie génique
1. Addition d’un gène
L’addition d’un gène consiste à insérer dans le noyau cellulaire ou directement dans le génome de la cellule hôte un nouveau gène, qu’on appelle transgène. Dans la plupart des cas, l’expression du transgène permet à la cellule de produire son propre « médicament ». Cette méthode ne fonctionne que si la maladie est due à une protéine non ou peu fonctionnelle. Le transgène peut rester en dehors du génome : il se comporte alors comme un épisome, se réplique de façon indépendante, mais peut être perdu lors des divisions cellulaires. Ou bien, il peut être inséré dans le génome par le vecteur. Il est alors important de pouvoir contrôler le site d’insertion (par exemple dans un « safe-harbor » c’est-à-dire, une région de l’ADN qui peut être modifiée sans crainte de provoquer des mutations) afin de ne pas modifier des gènes fonctionnels – PESSACH et NOTARANGELO (2011) – PEREZ-LUZ et DIAZ-NIDO (2010). La figure 1 présente les différentes possibilités d’intégration du transgène et leurs conséquences.

22
Figure 1 : Conséquences de l’intégration d’un transgène au sein d’un génome hôte d’après
HACKETT et al. (2007)
1. L’intégration au sein de l’hétérochromatine ne permet généralement pas l’expression du transgène et n’a aucune conséquence sur le génome de l’hôte.
2. L’intégration au sein de l’euchromatine, entre les gènes est la situation idéale. Le transgène peut s’exprimer sans modification de l’expression du génome de l’hôte.
3. L’intégration au sein d’une région régulatrice de la transcription peut avoir plusieurs conséquences. Le transgène s’exprime dans la majorité des cas mais son expression peut être modifiée par les éléments régulateurs environnants. En parallèle, les éléments régulateurs présents dans la cassette génique peuvent modifier l’expression des gènes environnants voire empêcher l’expression d’un gène actif.
4. L’intégration au sein d’une région transcrite d’un gène (ici, gène X) permet l’expression du transgène mais peut bloquer l’expression du gène X ou provoquer la production d’une protéine mutée à partir de ce gène.
Les situations 3 et 4 sont les plus dangereuses car elles peuvent conduire à de graves mutations et à l’apparition de cellules tumorales. Il est donc crucial de pouvoir contrôler les sites d’insertion des transgènes par l’étude et la sélection des vecteurs.
2. Réparation des gènes La réparation des gènes consiste à utiliser une séquence d’ADN ou d’ARN pour modifier le génome de la cellule hôte et corriger l’anomalie d’expression du gène à l’origine de la maladie. Plusieurs techniques utilisant l’allèle entier sont à l’étude – PESSACH et NOTARANGELO (2011) :
Remplacement de l’allèle muté par recombinaison homologue spontanée avec l’allèle normal. Dans ce cas, on injecte dans les cellules cibles des allèles sauvages sans autre molécule pour aider à la recombinaison homologue. Cependant, la fréquence de recombinaison homologue spontanée est très faible (inférieure à 1/106 cellules modifiées). (Figure 2, A)
Remplacement des deux allèles par insertion d’une séquence d’ADN double brin sur le même locus pour permettre une régulation normale du transgène. On utilise alors des
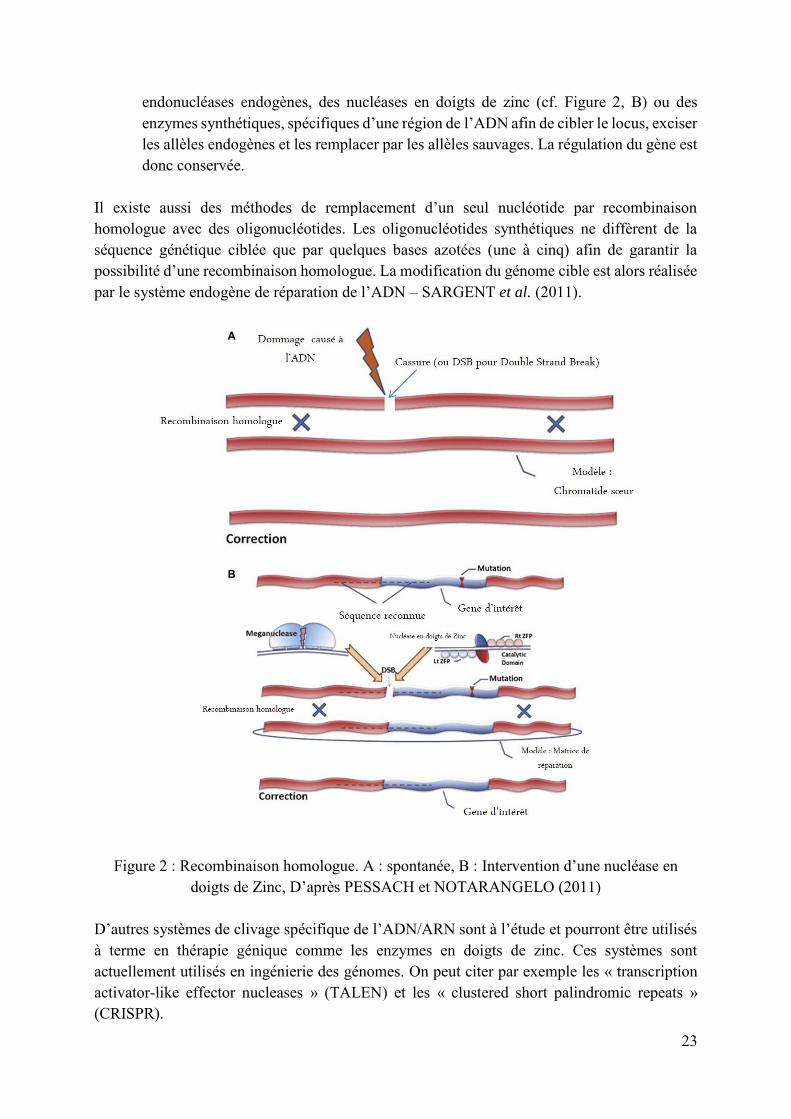
23
endonucléases endogènes, des nucléases en doigts de zinc (cf. Figure 2, B) ou des enzymes synthétiques, spécifiques d’une région de l’ADN afin de cibler le locus, exciser les allèles endogènes et les remplacer par les allèles sauvages. La régulation du gène est donc conservée.
Il existe aussi des méthodes de remplacement d’un seul nucléotide par recombinaison homologue avec des oligonucléotides. Les oligonucléotides synthétiques ne diffèrent de la séquence génétique ciblée que par quelques bases azotées (une à cinq) afin de garantir la possibilité d’une recombinaison homologue. La modification du génome cible est alors réalisée par le système endogène de réparation de l’ADN – SARGENT et al. (2011).
Figure 2 : Recombinaison homologue. A : spontanée, B : Intervention d’une nucléase en doigts de Zinc, D’après PESSACH et NOTARANGELO (2011)
D’autres systèmes de clivage spécifique de l’ADN/ARN sont à l’étude et pourront être utilisés à terme en thérapie génique comme les enzymes en doigts de zinc. Ces systèmes sont actuellement utilisés en ingénierie des génomes. On peut citer par exemple les « transcription activator-like effector nucleases » (TALEN) et les « clustered short palindromic repeats » (CRISPR).

24
Les TALEN sont des protéines chimériques contenant un domaine capable de se lier à l’ADN de manière spécifique (appelé TALE) et un domaine catalytique non spécifique (une nucléase). Le domaine se liant à l’ADN est dérivé de protéines appelées TALE pour Transcription Activator-Like Effectors présentes chez les bactéries du genre Xanthomonas. Ces bactéries sont des parasites végétaux ; les TALE leur permettent d’induire l’expression de certains gènes chez leur hôte. Le domaine se liant à l’ADN des TALE est composé de 13 à 28 unités de répétition de 34 acides aminés chacune. Ces unités de répétition ne se différencient que par deux résidus cruciaux en position 12 et 13 (appelés repeat variable di-residue ou RVD) qui déterminent le nucléotide auquel l’unité se lie. Il est cependant nécessaire que le premier nucléotide reconnu soit une thymine (appelée T0) pour permettre l’activité des TALE. La figure 3 montre le code de correspondance entre les résidus et les nucléotides.
Figure 3 : Correspondance RVD / nucléotide d’après VALTON et al. (2014)
Légende H : Histidine D : Acide aspartique N : Asparagine
G : Glycine I : Isoleucine
Le site catalytique est issu de la nucléase Fok1 isolée chez la bactérie Flavobacterium
okeanokoites. Cette nucléase fonctionnant en dimère afin de pouvoir créer une cassure sur les deux brins de l’ADN, les TALEN vont toujours par paire et reconnaissent deux séquences d’ADN distantes de 14 à 30 nucléotides (séquence « espace »). La figure 4 schématise la structure des TALEN.
Figure 4 : Structure et mise en place des TALEN sur l’ADN d’après VALTON (2014)

25
Il semblerait que les TALEN soient plus spécifiques que les enzymes en doigts de zinc : leur code de liaison à l’ADN est simple et le nombre des unités de répétition peut être augmenté afin de garantir la spécificité du site de clivage – TAKASU et al. (2013) – HOLKERS (2013). Les CRISPR sont des séquences d’ADN non codantes. Elles appartiennent au système de défense immunitaire des bactéries et des archées. Lors d’une infection par un virus ou un bactériophage, il y a insertion d’une séquence d’ADN étrangère au génome de la bactérie ou de l’archée ce qui active les CRISPR. Le système CRISPR fonctionne en trois phases – RICHTER et al. (2012) :
Premièrement, il faut qu’il y ait insertion de l’ADN étranger au sein de la séquence des CRISPR.
Cette insertion déclenche la transcription des CRISPR en ARN (appelés crARN) par des protéines associées aux CRISPR ou Cas.
D’autres protéines Cas prennent en charge les crARN et forment un complexe capable de dégrader l’ADN étranger.
La figure 5 montre le fonctionnement schématique du système CRISPR.
Figure 5 : Fonctionnement schématique du système CRISPR, d’après BONDY-DENOMY et
DAVIDSON (2014) Il est donc possible de modifier ces systèmes pour diriger l’action de dégradation vers le site de la mutation responsable de la maladie – MALI et al. (2013).
3. Modification de l’expression des gènes Il est aussi possible de modifier l’expression des gènes en les empêchant de s’exprimer ou en forçant leur expression.

26
a. Extinction génique
i. Interférence de l’ARN
L’interférence de l’ARN est un mécanisme de régulation de l’expression génique présent chez de nombreux eucaryotes. L’interférence de l’ARN provoque la dégradation d’un ARNm cible par une enzyme dirigée par un ARN non codant, complémentaire de l’ARNm cible. Ce mécanisme a été découvert en 1998 par Fire et son équipe – FIRE et al. (1998). Cette équipe a mis en évidence le potentiel des ARN dans l’extinction génique en injectant des ARN double brin chez des nématodes (Caenorhabditis elegans). On a découvert en 1993 chez le nématode Caenorhabditis elegans le premier microARN appelé l’ARN lin-4 – LIM et al. (2003). Ces microARN interviennent dans différents processus cellulaires comme la mitose, l’apoptose et le développement des différents tissus (cerveau, cœur, ...) ainsi que dans le mécanisme d’interférence de l’ARN. On en dénombre plus de 500 chez l’Homme et les chercheurs suspectent qu’il en existe plus d’un millier – MARQUEZ et McCAFFREY (2008). Les gènes des microARN appartiennent à la partie non codante du génome. Un premier pri-microARN est transcrit à partir du gène microARN par l’ARN polymérase II (Pol II). Il est ensuite clivé dans le noyau en pré-microARN par un complexe enzymatique (Drosha et DGCR8) puis exporté dans le cytoplasme via le transporteur Exp5. Il est alors à nouveau clivé par une enzyme spécifique appelée « Dicer » (complexée avec la protéine TRBP qui se lie au pré-microARN) pour devenir le microARN définitif (20 à 25 nucléotides). La figure 6 illustre la synthèse des microARN.
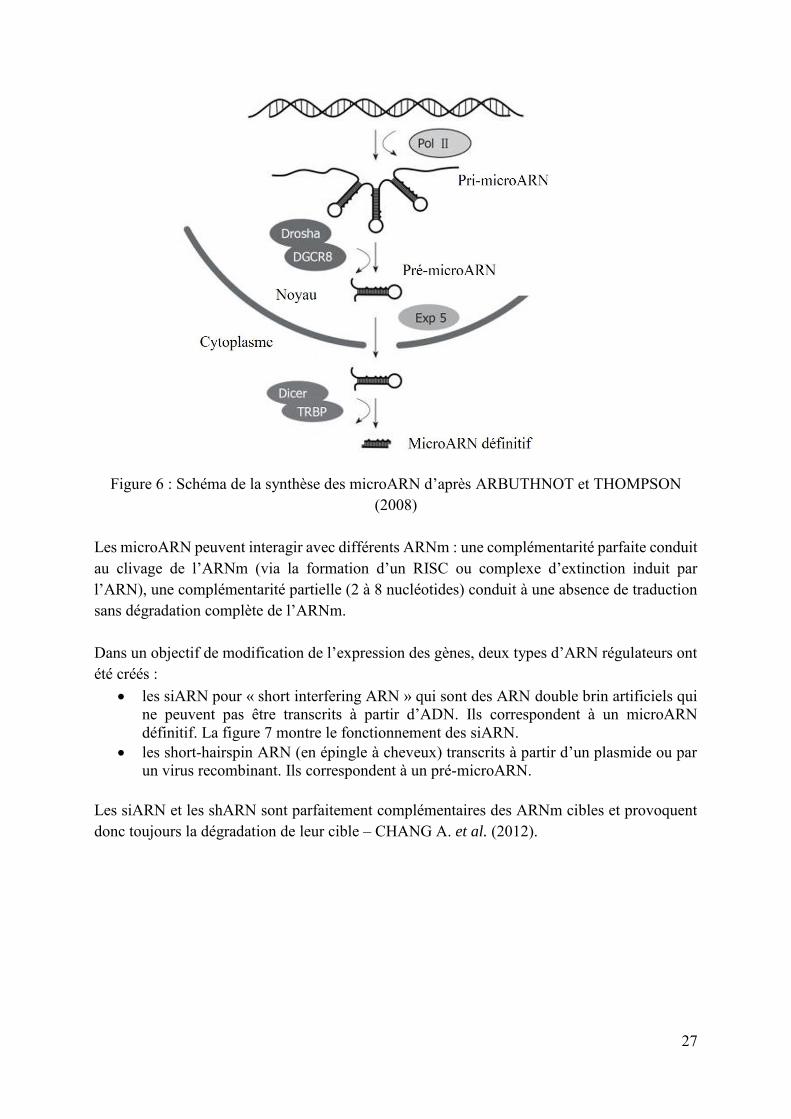
27
Figure 6 : Schéma de la synthèse des microARN d’après ARBUTHNOT et THOMPSON
(2008) Les microARN peuvent interagir avec différents ARNm : une complémentarité parfaite conduit au clivage de l’ARNm (via la formation d’un RISC ou complexe d’extinction induit par l’ARN), une complémentarité partielle (2 à 8 nucléotides) conduit à une absence de traduction sans dégradation complète de l’ARNm. Dans un objectif de modification de l’expression des gènes, deux types d’ARN régulateurs ont été créés :
les siARN pour « short interfering ARN » qui sont des ARN double brin artificiels qui ne peuvent pas être transcrits à partir d’ADN. Ils correspondent à un microARN définitif. La figure 7 montre le fonctionnement des siARN.
les short-hairspin ARN (en épingle à cheveux) transcrits à partir d’un plasmide ou par un virus recombinant. Ils correspondent à un pré-microARN.
Les siARN et les shARN sont parfaitement complémentaires des ARNm cibles et provoquent donc toujours la dégradation de leur cible – CHANG A. et al. (2012).
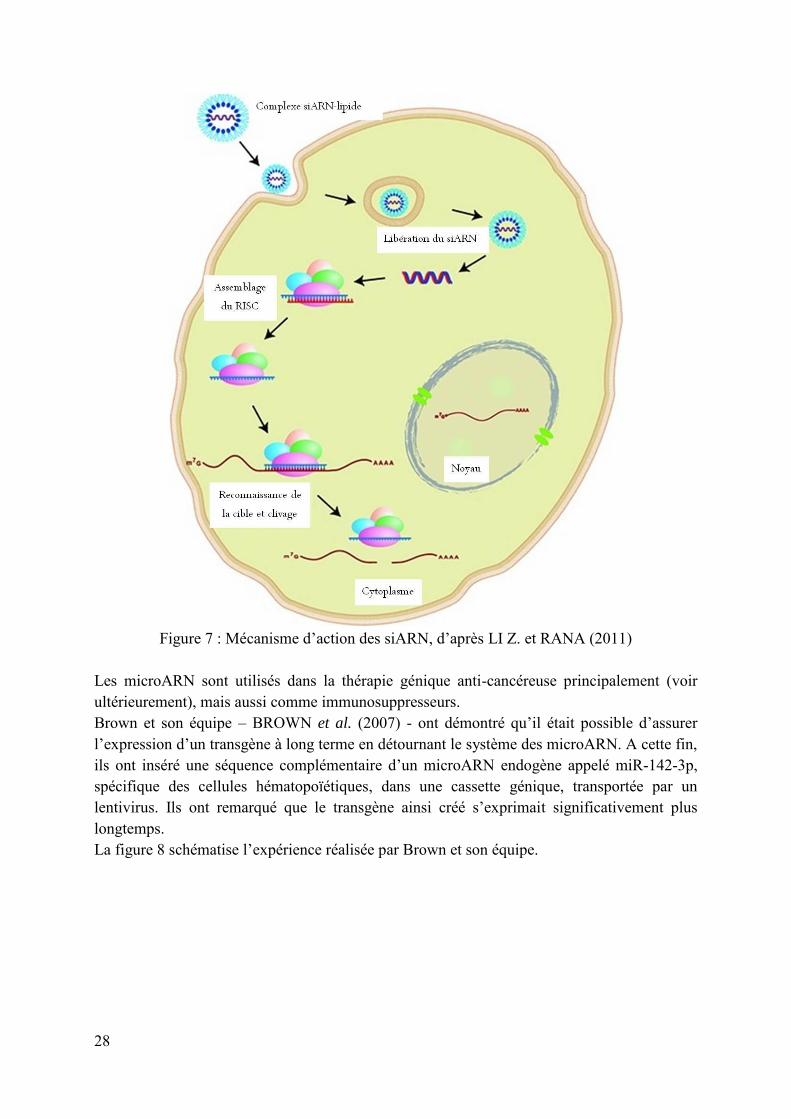
28
Figure 7 : Mécanisme d’action des siARN, d’après LI Z. et RANA (2011)
Les microARN sont utilisés dans la thérapie génique anti-cancéreuse principalement (voir ultérieurement), mais aussi comme immunosuppresseurs. Brown et son équipe – BROWN et al. (2007) - ont démontré qu’il était possible d’assurer l’expression d’un transgène à long terme en détournant le système des microARN. A cette fin, ils ont inséré une séquence complémentaire d’un microARN endogène appelé miR-142-3p, spécifique des cellules hématopoïétiques, dans une cassette génique, transportée par un lentivirus. Ils ont remarqué que le transgène ainsi créé s’exprimait significativement plus longtemps. La figure 8 schématise l’expérience réalisée par Brown et son équipe.
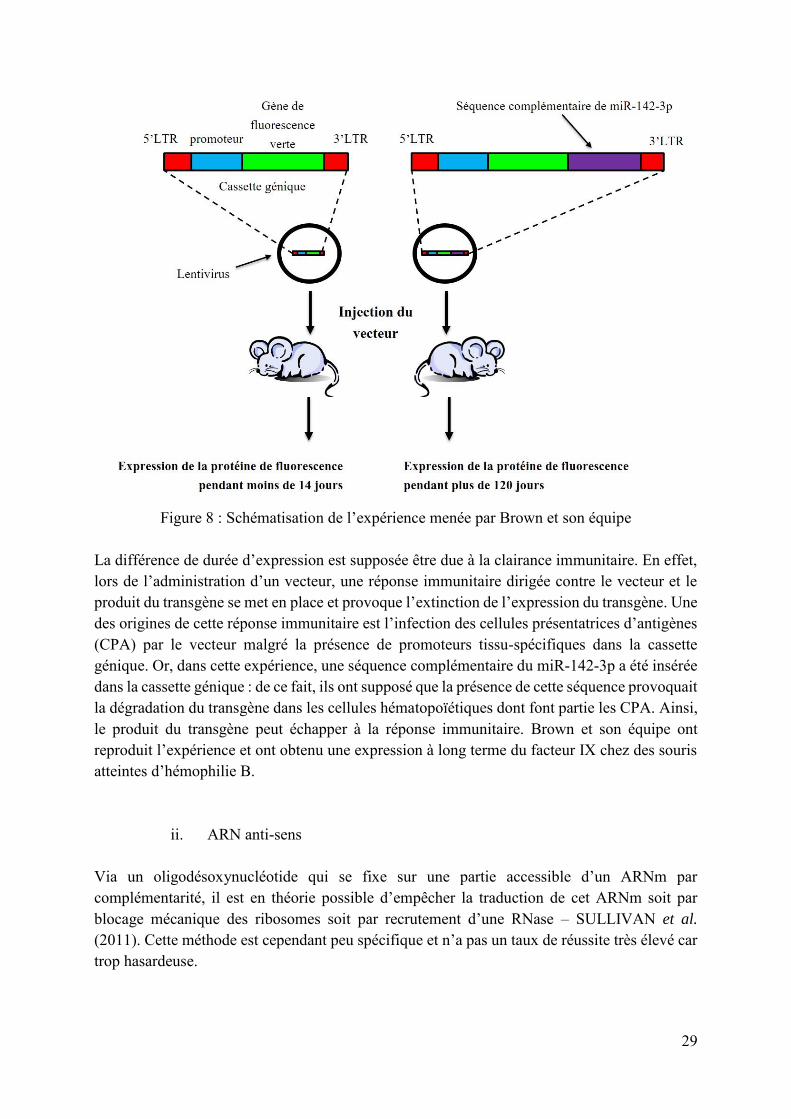
29
Figure 8 : Schématisation de l’expérience menée par Brown et son équipe
La différence de durée d’expression est supposée être due à la clairance immunitaire. En effet, lors de l’administration d’un vecteur, une réponse immunitaire dirigée contre le vecteur et le produit du transgène se met en place et provoque l’extinction de l’expression du transgène. Une des origines de cette réponse immunitaire est l’infection des cellules présentatrices d’antigènes (CPA) par le vecteur malgré la présence de promoteurs tissu-spécifiques dans la cassette génique. Or, dans cette expérience, une séquence complémentaire du miR-142-3p a été insérée dans la cassette génique : de ce fait, ils ont supposé que la présence de cette séquence provoquait la dégradation du transgène dans les cellules hématopoïétiques dont font partie les CPA. Ainsi, le produit du transgène peut échapper à la réponse immunitaire. Brown et son équipe ont reproduit l’expérience et ont obtenu une expression à long terme du facteur IX chez des souris atteintes d’hémophilie B.
ii. ARN anti-sens Via un oligodésoxynucléotide qui se fixe sur une partie accessible d’un ARNm par complémentarité, il est en théorie possible d’empêcher la traduction de cet ARNm soit par blocage mécanique des ribosomes soit par recrutement d’une RNase – SULLIVAN et al. (2011). Cette méthode est cependant peu spécifique et n’a pas un taux de réussite très élevé car trop hasardeuse.

30
iii. Ribozymes et ADNzymes (ou désoxyribozymes) Ce sont des acides nucléiques capables de cliver de façon spécifique de l’ARN. Ils se lient par complémentarité à un ARNm et le clivent en un site spécifique, empêchant ainsi la traduction. Il est possible de les encoder et de les transcrire à partir d’ADN – BARAR et OMIDI (2012).
iv. Autres molécules régulatrices De nouveaux outils pour moduler l’expression génique ont été créés, la plupart à base d’ARN comme les « ARN switches ». Ce sont des molécules d’ARN formées d’un domaine sensitif, capable de reconnaître certains signaux cellulaires (présence de certaines molécules, de certaines séquences génétiques voire d’une modification d’un paramètre physique comme la température ou le pH intracellulaire) et d’un domaine acteur, capable de moduler l’expression génique – CHANG A.L. et al. (2012). Il existe aussi un autre domaine de recherche pour les outils de modulation de l’expression génique : les molécules d’ADN formées par des liaisons de type Hoogsteen. La plupart des liaisons entre les bases azotées sont des liaisons hydrogènes de type Watson-Crick (cf. Figure 9, A). Or, il existe un autre type de liaisons hydrogènes appelées liaisons de type Hoogsteen (cf. Figure 9, B). Ces liaisons permettent de former différentes sortes de structures tridimensionnelles de l’ADN, autres que la double hélice. Il a été prouvé que les G quadruplexes (structures formées par quatre brins d’ADN au sein des régions riches en guanine, capables de se lier par des liaisons de type Hoogsteen à d’autres molécules d’ADN) interviennent dans la régulation de certains oncogènes – LIN et al. (2013). Les triplexes (une double hélice d’ADN liée à un troisième brin d’ADN ou d’ARN par des liaisons de type Hoogsteen) sont aussi impliqués dans certains mécanismes endogènes d’extinction génique – GHOSAL et al. (2006).

31
Figure 9 : A. Liaisons Watson-Crick, B. Liaisons Hoogsteen, d’après GHOSAL et al. (2006)
Suite à ces études, des molécules utilisant les liaisons de type Hoogsteen ont été développées comme les « polypurine reverse Hoogsteen hairpins ». Ce sont des molécules d’ADN formées par deux chaines de purines antiparallèles reliées par une boucle de cinq thymidines. Les liaisons à l’intérieur des chaines sont des liaisons de type Hoogsteen inversées. Ces molécules sont capables d’interférer avec les gènes et d’en modifier l’expression par liaison avec le gène cible ou l’ARNm, ce qui diminue l’expression du gène cible – RODRIGUEZ et al. (2013).
b. Expression forcée L’expression forcée est le fait de provoquer l’expression de certains gènes d’un individu en insérant des promoteurs. On prendra ici l’exemple des thalassémies β. Les thalassémies β sont des anémies héréditaires dues à une production réduite ou absente de chaînes β-globines qui composent l’hémoglobine (formule : α2β2). De ce fait, les chaînes α-globines sont en surnombre et ne peuvent pas toutes se lier à des chaînes β ; elles précipitent sous forme d’homotétramères α4 et empêchent la maturation des précurseurs des hématies, ce qui conduit à leur apoptose et donc à une anémie. Le phénotype des malades dépend de leur génotype (homozygotes ou hétérozygotes pour une mutation du gène codant pour la chaîne β) mais aussi de leur capacité à produire des chaînes γ-globines, présentes lors de la vie fœtale. En effet, les chaînes γ sont capables de se lier aux chaînes α libres et de former l’hémoglobine fœtale (formule : α2γ2), et ainsi de diminuer la gravité des symptômes de la maladie. Dans le traitement des thalassémies β par thérapie génique, une des pistes poursuivies est l’insertion d’une cassette génique comportant le gène de la γ-globine ainsi que son promoteur

32
spécifique dans le génome des personnes malades. Cela provoquerait la production de cette globine par les cellules transduites et permettrait de réduire les symptômes de la maladie. Les essais cliniques in vitro sont prometteurs – PAPANIKOLAOU et al. (2012).
c. Autres modifications Nous verrons dans la deuxième partie de cette thèse, dans le chapitre concernant la myodystrophie de Duchenne, qu’il est possible de provoquer un « saut d’exon » lors de l’épissage des ARN pré-messagers afin de modifier la protéine traduite à partir de cet ARNm et de réduire les symptômes de la maladie – DIETZ (2010). On remarque que, pour chaque maladie, les techniques développées précédemment sont adaptées afin de traiter la maladie avec la meilleure efficacité. Ainsi, la recherche sur ces maladies conduit régulièrement à la création de nouvelles techniques de thérapie génique. III. Modalités d’administration de la thérapie génique
1. In vivo L’administration in vivo consiste à injecter le vecteur par voies intraveineuse, sous-cutanée ou intramusculaire directement chez l’hôte. Le principal problème est alors le contrôle de l’adressage de la séquence d’ADN d’intérêt uniquement dans les cellules cibles. Cette voie permettrait néanmoins l’utilisation de la thérapie génique à grande échelle et à moindre coût. Les études sur les vecteurs cherchent donc à les rendre utilisables et efficaces en thérapie in
vivo. On peut également injecter le vecteur directement dans le tissu cible. On appelle cette administration in situ. Cependant, le taux de transfection reste faible car le vecteur diffuse peu, ce qui est le plus souvent incompatible avec une guérison complète, sauf dans le cas de tumeur localisée (cf. chapitre sur la thérapie génique anticancéreuse) – NAYEROSSADAT et al. (2012).
2. Ciblage de l’expression du transgène Lors d’administrations in vivo, il est nécessaire de faire pénétrer le gène d’intérêt uniquement dans les cellules cibles et donc de pouvoir cibler l’utilisation de la thérapie génique. Le ciblage peut être réalisé à plusieurs niveaux :
- Au niveau de la séquence d’ADN : Il est possible d’associer un promoteur tissu-spécifique au transgène. Malheureusement, peu de promoteurs spécifiques d’un seul tissu sont connus et l’expression qui suit l’utilisation d’un tel vecteur est souvent limitée en quantité, parfois trop faible pour avoir un effet thérapeutique.

33
- Au niveau des vecteurs : Certains vecteurs, comme les virus, ont un tropisme naturel mais il est souvent très large. La plupart des vecteurs sont modifiables afin de leur faire porter des protéines de surface ciblant spécifiquement un tissu (ou pseudotypage, cf. ultérieurement).
- Il est aussi possible de moduler à distance l’expression d’un transgène en utilisant par exemple des promoteurs réagissant à la présence ou à l’absence d’une molécule. On prendra comme exemple les systèmes « Tet Off » / « Tet On ».
Les systèmes « Tet Off » / « Tet On » sont dérivés de l’opéron bactérien de résistance à la tétracycline. Des trans-activateurs (rTA) sont produits de façon constitutive à partir de la fusion de la protéine tet-répresseur (tetR) et d’une protéine virale (VP16). Dans le système « Tet Off » (Figure 10, a), en absence de doxycycline (Dox), les trans-activateurs se lient à la séquence tet-opératrice (TetO) et provoquent l’activation du promoteur du cytomegalovirus (CMV) ainsi que l’expression du transgène. La doxycycline se lie aux trans-activateurs, les empêchant ainsi d’activer TetO. Le système « Tet On » fonctionne à l’inverse (Figure 10, b) – NAIDOO et YOUNG (2012). Le système « Tet Off » (expression en absence d’antibiotiques) est particulièrement intéressant pour la thérapie génique alors qu’en transgénèse animale, c’est surtout le système « Tet On » (expression en présence d’antibiotiques) qui est utilisé.
Figure 10 : Mécanismes des systèmes « Tet Off » / « Tet On » d’après NAIDOO et YOUNG
(2012) On ne développera pas plus ici les techniques de ciblage car elles sont spécifiques à chaque couple maladie/vecteur.
3. Ex vivo L’administration ex vivo se déroule en trois étapes :
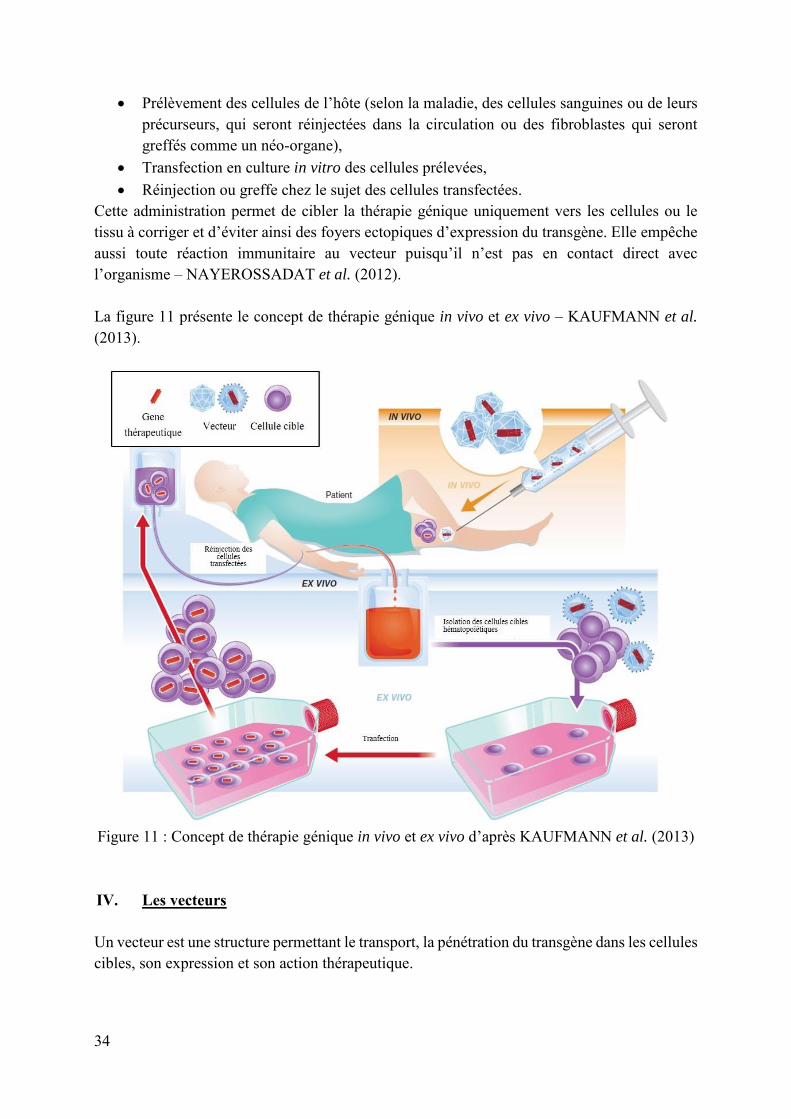
34
Prélèvement des cellules de l’hôte (selon la maladie, des cellules sanguines ou de leurs précurseurs, qui seront réinjectées dans la circulation ou des fibroblastes qui seront greffés comme un néo-organe),
Transfection en culture in vitro des cellules prélevées, Réinjection ou greffe chez le sujet des cellules transfectées.
Cette administration permet de cibler la thérapie génique uniquement vers les cellules ou le tissu à corriger et d’éviter ainsi des foyers ectopiques d’expression du transgène. Elle empêche aussi toute réaction immunitaire au vecteur puisqu’il n’est pas en contact direct avec l’organisme – NAYEROSSADAT et al. (2012). La figure 11 présente le concept de thérapie génique in vivo et ex vivo – KAUFMANN et al. (2013).
Figure 11 : Concept de thérapie génique in vivo et ex vivo d’après KAUFMANN et al. (2013) IV. Les vecteurs Un vecteur est une structure permettant le transport, la pénétration du transgène dans les cellules cibles, son expression et son action thérapeutique.

35
Le vecteur idéal a plusieurs propriétés – PEREZ-LUZ et DIAZ-NIDO (2010) – MANJILA et
al. (2013) : Culture/production facile et de faible coût, Faible immunogénicité, Non cytotoxique ou pathogène, Possibilité d’insérer une séquence d’ADN relativement longue, Protection de la séquence insérée contre les nucléases, Capacité d’infection des cellules quiescentes comme en division, Ciblage des cellules à transfecter possible, Ciblage du site d’insertion ou insertion en un site non mutagène, Expression à long terme du transgène chez l’hôte.
A l’heure actuelle, on peut classer les vecteurs en deux types : les vecteurs viraux, les vecteurs non viraux.
1. Les vecteurs viraux Les virus sont naturellement capables de pénétrer dans les cellules et d’y injecter leur propre génome afin de se répliquer. En réussissant à supprimer les gènes responsables de la pathogénicité des virus sans modifier leurs capacités de pénétration, ces derniers sont devenus des vecteurs de choix pour le transport et la livraison des gènes en thérapie génique. Le principal risque de l’utilisation de virus en tant que vecteurs est leur pathogénicité résiduelle pour l’hôte, malgré les délétions des gènes à effet pathogène, et le risque qu’ils le redeviennent par mutation. Enfin, le principal défaut des virus est leur immunogénicité – PEREZ-LUZ et DIEZ-NIDO (2010). De nombreux virus sont utilisés en thérapie génique (Adénovirus, Lentivirus, Adeno-Associated Virus ou AAV, Herpesvirus, …) et chaque famille de virus a ses avantages et ses inconvénients. Nous allons prendre ici quelques exemples de virus utilisés en thérapie génique.
a. Les Adénovirus Les adénovirus sont des virus nus à ADN double brin. Leurs caractéristiques sont :
- Taille des virions entre 80 et 120nm de diamètre, - Capside icosaédrique, - Génome linéaire et long de 26 à 44 kilobases (kb).
Il existe plus de cent sérotypes découverts à ce jour dont 51 infectant l’Homme. Les adénovirus figurent parmi les vecteurs les plus utilisés en thérapie génique et le sérotype 5 (HAd5) est le plus étudié – SHARMA et al. (2010). Les adénovirus s’attachent aux cellules via un récepteur appelé CAR (pour Coxsackievirus and Adenovirus Receptor). La protéine virale VI permet l’échappement de l’endosome. Les dynéines cytoplasmiques permettent le transport de la capside le long des microtubules
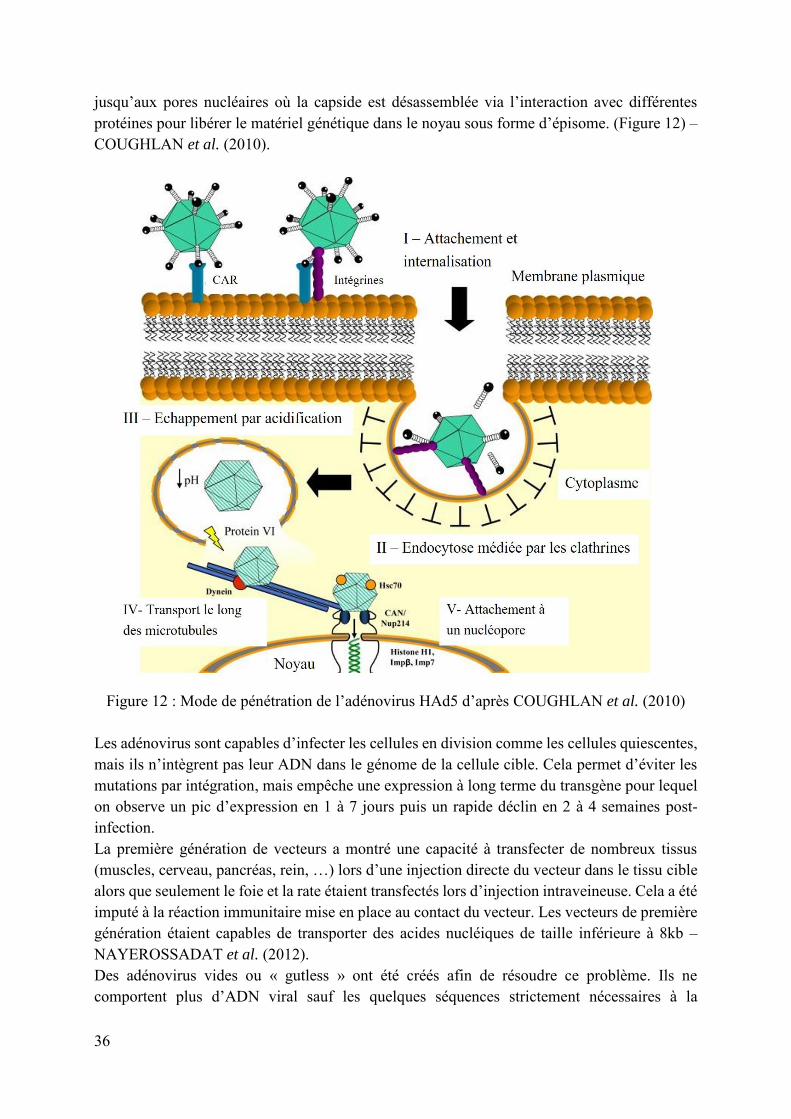
36
jusqu’aux pores nucléaires où la capside est désassemblée via l’interaction avec différentes protéines pour libérer le matériel génétique dans le noyau sous forme d’épisome. (Figure 12) – COUGHLAN et al. (2010).
Figure 12 : Mode de pénétration de l’adénovirus HAd5 d’après COUGHLAN et al. (2010)
Les adénovirus sont capables d’infecter les cellules en division comme les cellules quiescentes, mais ils n’intègrent pas leur ADN dans le génome de la cellule cible. Cela permet d’éviter les mutations par intégration, mais empêche une expression à long terme du transgène pour lequel on observe un pic d’expression en 1 à 7 jours puis un rapide déclin en 2 à 4 semaines post-infection. La première génération de vecteurs a montré une capacité à transfecter de nombreux tissus (muscles, cerveau, pancréas, rein, …) lors d’une injection directe du vecteur dans le tissu cible alors que seulement le foie et la rate étaient transfectés lors d’injection intraveineuse. Cela a été imputé à la réaction immunitaire mise en place au contact du vecteur. Les vecteurs de première génération étaient capables de transporter des acides nucléiques de taille inférieure à 8kb – NAYEROSSADAT et al. (2012). Des adénovirus vides ou « gutless » ont été créés afin de résoudre ce problème. Ils ne comportent plus d’ADN viral sauf les quelques séquences strictement nécessaires à la

37
réplication du génome. Ces adénovirus sont produits en présence de cellules transcomplémentantes ou cellules « helper » qui leur procurent les protéines virales nécessaires à la construction des virions – KAMIMURA et al. (2011). Ces virus sont capables de transporter des transgènes de plus grande taille (jusqu’à 38kb) et l’expression du transgène se fait à plus longue échéance (une étude a montré une expression pendant plus de 2 ans dans le foie de babouins) – LENTZ et al. (2012). De nombreuses études sont réalisées afin de moduler le tropisme des adénovirus via l’identification des récepteurs cellulaires et viraux impliqués dans la pénétration cellulaire et dans la réponse immunitaire. En particulier, l’utilisation des adénovirus en thérapie génique anticancéreuse est compliquée par le fait que la plupart des cellules cancéreuses n’expriment pas le récepteur CAR – SHARMA et al. (2010). Ces recherches sont donc essentielles pour pouvoir utiliser les adénovirus comme vecteurs dans le traitement des maladies acquises.
b. Les Adeno-Associated virus Les Adeno-Associated Virus (ou AAV) sont des virus incapables de se répliquer sans un virus « helper » tel que les adénovirus, les herpesvirus, … Ce sont de petits virus (18-26 nm) nus à ADN simple brin. A l’état naturel, ils sont capables de s’intégrer dans le génome humain, préférentiellement dans un site spécifique sur le chromosome 19 grâce à des protéines virales codées par leur gène REP.
Le taux d’intégration spontanée est faible et quasiment nul quand le gène REP est supprimé. Dans ce cas, l’intégration perd aussi sa spécificité de site – LENTZ et al. (2012) – LI H. et al. (2011). Leur matériel se comporte la plupart du temps comme un épisome et reste en dehors du génome de la cellule infectée. Les vecteurs créés à partir d’AAV sont capables de transporter des acides nucléiques de taille inférieure à 4,8kb – NAYEROSSADAT et al. (2012). Il a été démontré que l’expression du transgène était longue (jusqu’à 9 ans chez des chiens). Il existe de nombreux sérotypes peu toxiques pour l’organisme et chaque sérotype cible préférentiellement différents organes (par exemple, le sérotype 2 infecte préférentiellement les cellules musculaires et les neurones) – KAMIMURA et al. (2011). Ils sont donc tout indiqués pour servir de vecteurs en thérapie génique des maladies affectant l’appareil musculaire ou le système nerveux. Le principal inconvénient est qu’il faut injecter une grande quantité de virus afin d’obtenir une réponse thérapeutique significative et que cela provoque chez l’hôte une forte réaction immunitaire – ZHONG et al. (2012), d’autant plus précoce que ces virus sont très répandus dans la population humaine ou animale. Une étude réalisée en 2010 par Sylvie BOUTIN et son équipe révèle que 60% de la population humaine possède des anticorps anti-AAV1 et anti-AAV2 – BOUTIN et al. (2012). De nombreuses études sont en cours afin de trouver les sérotypes qui permettront une meilleure efficacité de transfection (en quantité) tout en limitant leur immunogénicité – SEN et al. (2013).
38
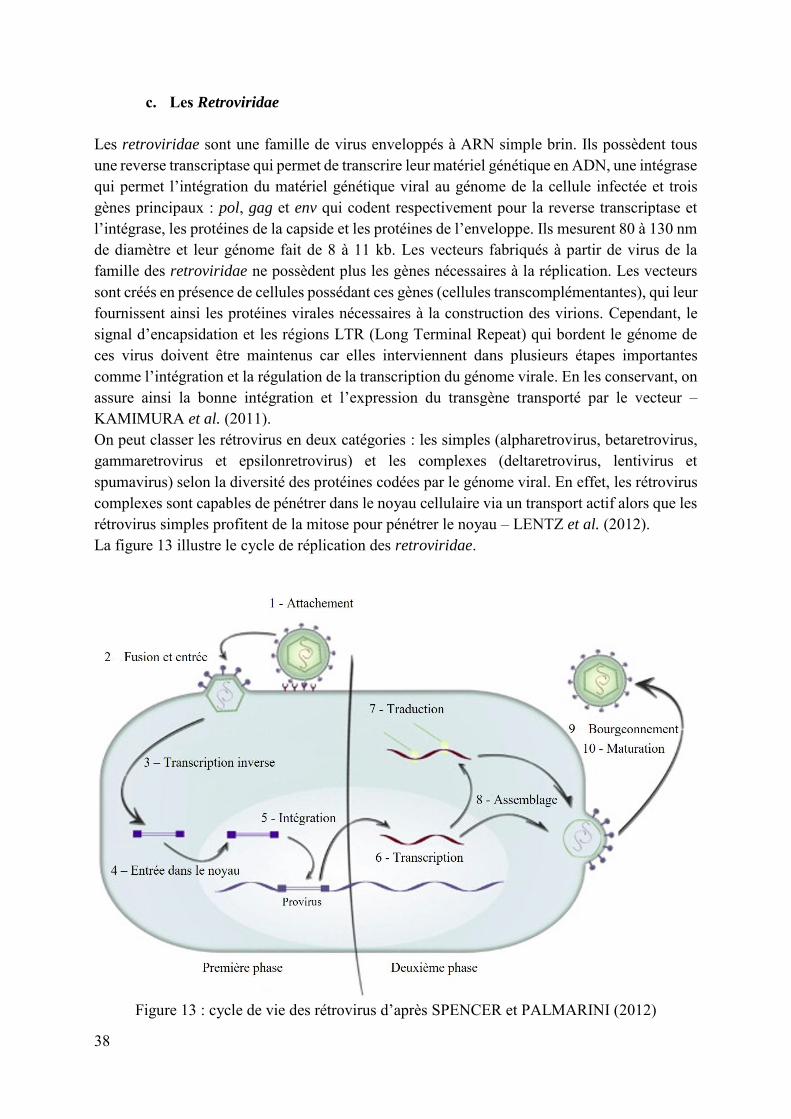
c. Les Retroviridae Les retroviridae sont une famille de virus enveloppés à ARN simple brin. Ils possèdent tous une reverse transcriptase qui permet de transcrire leur matériel génétique en ADN, une intégrase qui permet l’intégration du matériel génétique viral au génome de la cellule infectée et trois gènes principaux : pol, gag et env qui codent respectivement pour la reverse transcriptase et l’intégrase, les protéines de la capside et les protéines de l’enveloppe. Ils mesurent 80 à 130 nm de diamètre et leur génome fait de 8 à 11 kb. Les vecteurs fabriqués à partir de virus de la famille des retroviridae ne possèdent plus les gènes nécessaires à la réplication. Les vecteurs sont créés en présence de cellules possédant ces gènes (cellules transcomplémentantes), qui leur fournissent ainsi les protéines virales nécessaires à la construction des virions. Cependant, le signal d’encapsidation et les régions LTR (Long Terminal Repeat) qui bordent le génome de ces virus doivent être maintenus car elles interviennent dans plusieurs étapes importantes comme l’intégration et la régulation de la transcription du génome virale. En les conservant, on assure ainsi la bonne intégration et l’expression du transgène transporté par le vecteur – KAMIMURA et al. (2011). On peut classer les rétrovirus en deux catégories : les simples (alpharetrovirus, betaretrovirus, gammaretrovirus et epsilonretrovirus) et les complexes (deltaretrovirus, lentivirus et spumavirus) selon la diversité des protéines codées par le génome viral. En effet, les rétrovirus complexes sont capables de pénétrer dans le noyau cellulaire via un transport actif alors que les rétrovirus simples profitent de la mitose pour pénétrer le noyau – LENTZ et al. (2012). La figure 13 illustre le cycle de réplication des retroviridae.
Figure 13 : cycle de vie des rétrovirus d’après SPENCER et PALMARINI (2012)

39
Dans le cas des virus enveloppés, et particulièrement dans le cas des retroviridae, on peut modifier le tropisme du vecteur viral en le pseudotypant. Le pseudotypage est une technique qui consiste à modifier les glycoprotéines de surface des vecteurs viraux afin de modifier leur tropisme tissulaire. Cette technique est dérivée de la modification des enveloppes des virus lorsque deux types de virus (ou plus) infectent la même cellule. Lors du bourgeonnement, les virions des deux types se retrouvent porteurs de ses propres glycoprotéines ainsi que de celles de l’autre type de virus. On peut donc attribuer à un vecteur viral enveloppé la glycoprotéine que l’on souhaite pour pouvoir maîtriser son tropisme. A ce jour, les glycoprotéines les plus utilisées en pseudotypage sont les glycoprotéines du virus de la stomatite vésiculeuse (VSV-G). Ce virus possède un tropisme très large (pratiquement tous les types cellulaires peuvent être infectés par un virus de la stomatite vésiculeuse) et une enveloppe très stable qui permet l’utilisation de l’ultracentrifugation pour concentrer le vecteur viral. Il existe d’autres types de pseudotypage qui ne seront pas développés ici – CRONIN et
al. (2005).
i. Les rétrovirus simples Les rétrovirus simples ont été très utilisés au début des essais cliniques en thérapie génique comme vecteur car ils permettent une intégration stable du transgène dans le génome de l’hôte. Ils ne peuvent cependant infecter que des cellules en division. Cela en fait des vecteurs tout indiqués dans les thérapies géniques ex vivo. Plusieurs virus ont été utilisés avec succès comme vecteur en thérapie génique comme le virus de la leucémie murine de Moloney. Les vecteurs créés à partir de rétrovirus ont une capacité de 8kb - NAYEROSSADAT et al. (2012). Leurs inconvénients sont cependant majeurs : ils sont immunogènes (comme la majorité des vecteurs viraux) et peuvent provoquer le déclenchement de cancers par activation d’un oncogène ou l’inactivation d’un gène répresseur de tumeurs lors de l’insertion du transgène dans le génome cellulaire. Cela a été le cas lors de l’essai clinique conduit sur des enfants atteints du syndrome d’immunodéficience combiné sévère lié à l’X (SCID X) en 2000. Onze enfants ont reçu par thérapie génique ex vivo le gène codant pour la chaîne γ du récepteur aux interleukines et ont été guéris du SCID X. Malheureusement, plusieurs cas de leucémies ont été rapportés dans les trois ans qui ont suivi l’essai clinique. Le déclenchement de la leucémie a été attribué à l’insertion du transgène au sein d’un proto-oncogène - ESCORS et BRECKPOT (2010). Les rétrovirus simples sont donc relégués au second plan jusqu’à ce qu’on sache maîtriser leur lieu d’insertion dans le génome hôte.
ii. Les lentivirus Au sein des retroviridae, les lentivirus (rétrovirus complexes) sont particulièrement étudiés en ce moment comme vecteurs en thérapie génique.

40
On utilise en thérapie génique des « self-inactivating » lentivirus (ou SIN lentivirus) dont le génome a été modifié afin de les rendre incapables de se répliquer. Les lentivirus ont la capacité d’infecter des cellules quiescentes grâce à des protéines virales capables de faire pénétrer leur matériel génétique dans le noyau des cellules cibles via les pores nucléaires - KAMIMURA et al. (2011) - ce qui en fait des vecteurs de choix pour toutes les maladies impliquant des cellules hautement différenciées et qui se divisent peu, comme les neurones. Et comme tous les rétrovirus, ils sont capables d’insérer leur génome dans celui de la cellule hôte ce qui permet une expression à long terme du transgène. Cependant, cette insertion dans le génome des cellules hôtes a déjà eu pour résultat de provoquer des mutations chez les cellules cibles. Néanmoins, les lentivirus seraient moins susceptibles de provoquer ces mutations par insertion - ESCORS et BRECKPOT (2010). En effet, ils insèrent naturellement leur matériel génétique près des sites de transcription actifs alors que les autres rétrovirus ont plutôt tendance à s’intégrer près des promoteurs, ce qui augmente le risque d’oncogénèse – KAMIMURA et al. (2011). Il est aussi possible d’utiliser des lentivirus qui ne peuvent pas intégrer leur matériel génétique : ce sont les « Non Integrating LentiViruses » (NILVs). Dans ce cas-là, le transgène reste sous forme d’épisome dans le noyau cellulaire et l’expression à long terme du transgène est liée à l’état de quiescence des cellules infectées - LENTZ et al. (2012). Tout comme les rétrovirus simples, les vecteurs construits à partir de lentivirus ont une capacité de 8kb – NAYEROSSADAT et al. (2012). Malheureusement, les lentivirus induisent une forte réponse immunitaire de l’hôte à l’encontre du transgène ce qui diminue la durée de vie des cellules corrigées et donc l’effet thérapeutique. Une des pistes d’étude serait l’utilisation de microRNA pour induire une immunotolérance en empêchant l’expression du transgène dans les cellules présentatrices d’antigènes – DUFAIT et
al. (2012).
2. Les vecteurs non-viraux Les vecteurs non-viraux correspondent à une autre catégorie de vecteurs qui ont été développés pour répondre au besoin de disposer de vecteurs non pathogènes à la différence des virus. Leur production est souvent plus facile et moins coûteuse, mais les taux de transfection restent inférieurs à ceux atteints lors de l’utilisation d’un vecteur viral. Les vecteurs non viraux doivent lever de nombreuses barrières afin de pouvoir être utilisés in
vivo (Figure 14). Ils doivent avoir une demi-vie assez longue dans le système circulatoire et aussi pouvoir traverser la membrane plasmique. D’autres vecteurs non viraux provoquent une endocytose. Il est alors nécessaire que le vecteur puisse se libérer de l’endosome. Une fois dans le cytoplasme, le vecteur non-viral doit libérer le transgène et/ou le guider jusqu’au noyau cellulaire. Les mécanismes du fonctionnement de ces vecteurs ne sont pas encore tous élucidés (en particulier, le passage des acides nucléiques dans le noyau cellulaire après décomplexation) – GREEN (2012). Cependant, seule une très petite quantité des molécules d’acides nucléiques du vecteur arrive à destination dans le noyau d’une cellule cible viable. Le taux de transfection réussie avec expression du transgène est très faible par rapport à celui obtenu avec des vecteurs viraux, en particulier lorsque les cellules cibles ne sont pas en division.

41
Figure 14 : Représentation des barrières qui limitent l’utilisation de vecteurs non viraux in
vivo d’après AL-DOSARI et GAO (2009) A la différence des virus, la plupart des vecteurs non-viraux ne sont pas naturellement spécifiques d’un type cellulaire et cela reste le plus grand défi à relever pour cette famille de vecteurs si on veut pouvoir l’utiliser in vivo.
a. Les vecteurs chimiques
i. Les lipides cationiques Les lipides cationiques sont des lipides formés par une chaine hydrophobe et une tête hydrophile chargée positivement. Ce type de lipides s’est révélé être le plus efficace dans la fabrication de vecteurs en thérapie génique. Il est possible de former avec ces lipides des liposomes qui sont des bicouches lipidiques sous forme de micelles. L’ADN chargé négativement interagit avec les charges positives des lipides et crée spontanément des lipocomplexes ou lipoplexes. A la différence de la production de vecteurs viraux, les lipides cationiques sont très peu coûteux à produire. Ils ne sont pas pathogènes et provoquent une faible réponse immunitaire. Ils sont capables de transporter de grande quantité d’ADN (voire des chromosomes entiers) et peuvent

42
être modifiés afin de cibler des cellules spécifiques via des lipoprotéines ou des anticorps insérés dans la bicouche lipidique – KAMIMURA et al. (2011) – KONG et al. (2012). Cependant, on observe une certaine cytotoxicité lors de fusion avec les membranes cellulaires (plasmique, endosomale ou lysosomale) due à la présence du groupement hydrophile chargé positivement, le plus souvent une amine tertiaire ou quaternaire. A cause du mécanisme de clairance hépatique (demi-vie très courte des liposomes), on n’observe qu’un faible taux de transfection – SARKER et al. (2013). Lors d’injection systémique, les liposomes peuvent provoquer des réactions d’hypersensibilité. L’addition de certains composés chimiques (comme le polyéthylène glycol) permet de réduire leur toxicité tout en les stabilisant – MIELE et al. (2012). Des liposomes sont actuellement fabriqués et vendus par différents laboratoires : la Lipofectamine 2000 ® est un liposome commercialisé par le laboratoire Life Technologies, fréquemment utilisé comme gold standard lors d’études sur un nouveau vecteur non viral.
ii. Les polymères cationiques Les polymères cationiques sont des assemblages de macromolécules (comme des hydrocarbures, des monosaccharides, …) modifiés par différentes réactions chimiques afin de posséder les groupements chimiques nécessaires à leur fonction (ici, des groupements cationiques). Ils forment des polyplexes quand ils sont associés à de l’ADN. Ils sont, à la différence des lipides cationiques, totalement solubles dans l’eau et ont l’avantage de pouvoir condenser l’ADN en des particules de petite taille, ce qui semble permettre une transfection plus efficace – LV et al. (2006). Les polymères à base de chaines hydrocarbonées hydrophobiques se sont montrés les plus aptes au transport d’acides nucléiques. Les polymères sont des molécules faciles à modifier par l’ajout ou le retrait de différents groupements ce qui en fait un sujet d’étude très vaste. Il est possible, comme avec les liposomes, de leur adjoindre des groupements permettant le ciblage tissulaire. Les groupements amine et arginine semblent être déterminants dans le mécanisme d’échappement aux endosomes même si le mécanisme reste encore obscur – GREEN (2012) – LEE et al. (2012). Cependant, plus leur capacité à tranfecter les cellules augmente (présence de groupes hydroxyl, le ratio charges positives/ADN, longueur des chaînes de carbone …), plus leur toxicité est élevée. Des moyens pour diminuer cette cytotoxicité sont à l’étude comme l’utilisation de polyéthylène glycol pour entourer les nanoparticules – ZHANG et al. (2012). De nombreux polymères sont à l’étude, le polyéthylènimine (PEI) et le polypropylènimine étant les plus étudiés. Des travaux ayant pour objectif de les modifier pour diminuer leur toxicité et augmenter leur capacité de transfection sont en cours – MANJILA et al. (2013). D’autres études se concentrent sur les polymères à base de chaines aliphatiques de carbonates (succession de groupes - O – C (O) – O) – XU et al. (2013).

43
iii. Les polymères biodégradables Il est démontré que les polymères biodégradables (comme les poly-amido-amines) ont une plus faible cytotoxicité que les polymères et lipides cationiques. Afin de créer des polymères biodégradables, il faut créer des liaisons hydrolytiques ou bioréductibles au sein du polymère notamment par l’addition de groupement disulfide – LEE et al. (2012). Cela réduit jusqu’à cent fois la toxicité du polymère par rapport à un polymère classique comme le polyéthylènimine. Il est également possible d’utiliser des macromolécules biocompatibles. Nous prendrons ici deux exemples bien étudiés. Le chitosane (dérivé de la chitine) est un polysaccharide très étudié car biodégradable, biocompatible et qui possède des charges positives. Cependant, sans modification il reste peu performant comme vecteur. Mais associé à différents groupements, le chitosane devient un support très prometteur – MAO et al. (2010). La gélatine (dérivée du collagène animal) est déjà très utilisée dans l’industrie pharmaceutique en raison de ses caractères biocompatible et biodégradable. Elle est approuvée par la FDA et est très peu chère à produire. Des études ont montré qu’il était possible de créer des nanoparticules de gélatine capables de transporter des acides nucléiques (par encapsulation, complexation avec des groupements de surface ou par attraction électrostatique). Des études in
vitro et in vivo ont montré le potentiel de la gélatine comme vecteur non viral par rapport à l’ADN nu et la Lipofectamine®. Cependant, la majorité de la gélatine produite vient de l’industrie animale ce qui rend ce produit susceptible de transmettre des encéphalopathies spongiformes subaigües transmissibles. De la gélatine humaine recombinante est disponible sur le marché et considérée comme moins à risque. Cette gélatine est plus homogène et semble mieux convenir à la préparation de nanoparticules. A l’heure actuelle, la gélatine est surtout étudiée pour le transport de médicaments de nature autre que les acides nucléiques – ELZOGHBY (2013). Une différence majeure apparaît dans le transport des acides nucléiques : les siARN et autres oligonucléotides bénéficient d’un relargage rapide dans le cytoplasme alors que l’ADN doit être protégé des nucléases cytoplasmiques le plus longtemps possible afin d’être efficacement transporté jusqu’au noyau. Ainsi, deux types de biodégradabilité sont à l’étude : une rapide (grâce par exemple aux groupements disulfides qui permettent une libération de l’acide nucléique lors du passage du vecteur en milieu intracellulaire) et une plus progressive – GREEN (2012).
iv. Les nanotubes de carbone Les nanotubes de carbone sont des structures cylindriques formées par des feuilles de graphite concentriques. Leurs dimensions sont de 1 nanomètre de diamètre et de quelques dizaines à quelques centaines de micromètres de longueur. On les classe en deux catégories selon le nombre de couches de graphite (une ou plusieurs).

44
Afin de pouvoir remplir leur rôle de vecteur et principalement afin de pouvoir se lier aux acides nucléiques, les nanotubes de carbone doivent être modifiés en surface par différentes réactions chimiques – HUANG et al. (2013). De nombreuses études ont déjà été menées afin de déterminer la modification la plus efficace et la moins toxique (Figure 15). Différents groupements cationiques ont été utilisés : des polymères comme le PEI, des polymères dendritiques (cf. paragraphe suivant), des lipides cationiques et autres nanoparticules. Leurs mécanismes d’action précis sont toujours inconnus.
Figure 15 : Combinaisons à base de nanotubes de carbone étudiées d’après BATES et
KOSTARELOS (2013) L’efficacité des nanotubes de carbone a surtout été démontrée in vitro et d’autres études sur la toxicité doivent être réalisées avant que le nanotube modifié devienne le vecteur non viral de choix en thérapie génique – BATES et KOSTARELOS (2013).
v. Autres nanoparticules non virales De très nombreuses nanoparticules synthétiques ou naturelles sont à l’étude comme potentiel vecteur en thérapie génique. Seuls quelques exemples intéressants seront développés ici.

45
Les polymères dendritiques (ou dendrimères) : Ce sont des macromolécules synthétiques, de l’ordre de grandeur du nanomètre, construites en trois dimensions. Il est possible d’y attacher un gène par liaison covalente ou par liaison électrostatique car ils possèdent de nombreux groupes hydrophobes, hydrophiles, cationiques et anioniques qu’on peut modifier – MANDEVILLE et al. (2012). Les dendrimères de poly-amido-amines ou PANAM sont actuellement les dendrimères les plus prometteurs de par leur biocompatibilité et leur capacité à transfecter les cellules – NAM et al. (2008). Les nanoparticules de phosphate de calcium : Le plus grand avantage de ces nanoparticules est qu’elles sont totalement biocompatibles et biodégradables car le phosphate de calcium est un élément présent chez les animaux et l’Homme sous forme d’os. Des études in vitro ont montré la capacité des nanoparticules de phosphate de calcium à transporter des acides nucléiques et à transfecter les cellules. Ces nanoparticules nécessitent une préparation spéciale afin d’éviter la précipitation ou l’agglomération et des modifications de surface pour les stabiliser in vivo – CAO X. et al. (2011). Les nanoparticules d’or : Ces nanoparticules ont la caractéristique de pouvoir relâcher les acides nucléiques qu’elles transportent quand elles sont soumises à une onde lumineuse (800 à 1200nm de longueur d’onde) ou une onde électrique. Elles sont aussi non toxiques et capables de protéger les acides nucléiques transportés contre les nucléases endogènes – PISSUWAN et al. (2011). Les nanoparticules à double couche hydroxyde : C’est une famille de cristaux dont la formule générale est [MII
nMIII(OH)2+2n]+(Am−)1/m × H2O (n = 2–4) où MII est un cation métallique divalent, MIII un cation métallique trivalent et Am- un anion. Ces nanoparticules présentent une grande biocompatibilité, une faible cytotoxicité et une capacité à relâcher leur chargement en fonction du pH. Elles sont capables de lier des acides nucléiques par liaison électrostatique. Ces cristaux peuvent prendre différentes formes. Les formes hexagonales semblent rester dans le cytoplasme alors que les formes circulaires passent la membrane nucléaire. Leur stabilité reste à étudier afin de pouvoir les utiliser in vivo – LADEWIG et al. (2010).
b. Les acides nucléiques nus L’utilisation d’acides nucléiques dits nus (car non liés à une autre molécule) comme vecteur non viral en thérapie génique est pratiquée depuis plus de vingt ans. L’ADN nu ne provoque pas de réponse immunitaire et est facile à produire via des systèmes bactériens et les techniques d’ADN recombinant. On crée alors des plasmides recombinants contenant une origine de réplication, un promoteur et la séquence d’intérêt. Une des limites principales de l’utilisation

46
des acides nucléiques nus est qu’ils ont une demi-vie très courte en raison de l’action des nucléases endogènes. L’autre limite est que l’expression du transgène est transitoire la plupart du temps. Plusieurs hypothèses ont été formulées à ce sujet – YAN et al. (2012) :
Activation du système immunitaire contre le plasmide par des séquences d’ADN bactérien résiduelles (qui ne présentent pas de méthylation)
Inactivation du promoteur s’il est viral. Les promoteurs viraux sont très utilisés (par exemple, la séquence LTR des rétrovirus) car il est démontré que ces derniers provoquent une expression massive du transgène à la différence des promoteurs tissu-spécifique – HERWEIJER et WOLFF (2003).
Nombreuses mitoses induisant la perte du transgène non intégré Grâce à des méthodes physiques pour améliorer la transfection, le taux d’expression du transgène peut atteindre celui d’une transfection par vecteur viral – WOLFF et BUDKER (2005).
i. Micro injection Cette technique consiste à injecter directement les transgènes nus par le biais d’une solution d’acides nucléiques dans le cytoplasme ou le noyau des cellules grâce à des aiguilles de 0.5 à 5 µm de diamètre et un microscope optique spécialisé appelé micromanipulateur. Cette technique est lourde à réaliser et se retrouve donc reléguée derrière les autres méthodes physiques. Elle est aujourd’hui surtout utilisée en transgénèse animale – MANJILA et al. (2013).
ii. Gene gun La technique du « gene-gun » (ou pistolet à gènes) consiste à injecter dans les tissus cibles, à très grande vitesse, grâce à un gaz pressurisé, des microparticules d’or ou de tungstène (1 à 3 µm de diamètre) recouvertes d’acides nucléiques – NAYEROSSADAT et al. (2012). Cette méthode a été développée pour la création de plantes transgéniques dans les années 80. Cependant, il a été démontré que l’utilisation de cette méthode provoquait une réponse immunitaire plus importante que celle induite par la micro-injection d’ADN et ce, avec un apport d’une quantité d’acide nucléique moindre. Actuellement, la technique du « gene-gun » est essentiellement utilisée dans la vaccination à partir de gènes ou vaccination ADN – MANJILA et al. (2013).
iii. Technique hydrodynamique Cette technique consiste à injecter très rapidement un grand volume de sérum physiologique contenant une grande quantité d’acides nucléiques par voie intraveineuse (les quantités dépendent du modèle animal choisi). Cette technique a été initialement décrite en utilisant la

47
veine de la queue comme voie intraveineuse. Le mécanisme est le suivant : l’injection rapide d’un bolus provoque une congestion du cœur ce qui fait refluer le sang et le bolus dans le système veineux (veine cave inférieure et veine porte principalement). L’augmentation de la pression veineuse provoque une dilatation des fenestrations des capillaires sanguins irrigant le foie et perméabilise les membranes plasmiques permettant ainsi l’entrée des acides nucléiques. Tous les paramètres systémiques reviennent à la normale en quelques minutes et les membranes plasmiques sont restaurées en 48 à 72h. Par cette technique, les hépatocytes sont les cellules qui présentent l’expression génétique du transgène la plus importante. Des cellules du cœur, des poumons, de la rate et des reins montrent aussi une expression, mais 10 000 fois inférieure aux hépatocytes. Cette technique est aussi utilisable pour transfecter d’autres tissus comme des muscles en bloquant la circulation sanguine le temps de l’injection. On peut utiliser des plasmides d’ADN ou d’autres molécules (siARN, oligonucléotides, …) Le taux de transfection est de 30% à 40%, ce qui est le taux le plus important décrit en utilisant un vecteur non viral – BONAMASSA et al. (2011).
iv. Electroporation L’électroporation consiste à appliquer un courant électrique à un tissu pour faciliter l’entrée des acides nucléiques dans les cellules en créant des pores membranaires. Il a été démontré que les courtes impulsions (100µs) de haut voltage permettaient de perméabiliser les membranes plasmiques et que les longues impulsions (100ms) de bas voltage permettaient de guider l’ADN dans les cellules. Le voltage dépend du type d’application (directe ou en transcutané), du modèle d’étude ainsi que du tissu (les hépatocytes semblent résister à un courant plus fort que les cellules musculaires), sachant qu’il doit rester assez faible afin de ne pas détruire les cellules transfectées. Cette technique est principalement utilisée in
vitro. L’utilisation in vivo se limite aux cellules cutanées – CUKJATI et al. (2007).
v. Nucléofection Dérivée de la recherche sur l’électroporation, cette technique est actuellement la méthode électrique la plus sûre. Par l’association d’impulsions électriques avec une solution spécifique pour le transport des acides nucléiques, cette technique transporte les acides nucléiques directement dans le noyau des cellules. Elle a le grand avantage de permettre la transfection de cellules se divisant peu ou quiescentes et elle est très reproductible. Des kits de nucléofection sont disponibles sur le marché pour une utilisation in vitro uniquement (Exemple : Nucleofector®) – GRESCH et al. (2004).

48
vi. Sonoporation La sonoporation est le nom donné au mécanisme provoquant la formation de pores sur la membrane plasmique sous l’effet des ultrasons. L’utilisation de microbulles est désormais presque incontournable car elles permettent d’augmenter le taux de réussite de cette méthode. Les microbulles sont des particules remplies de gaz délimitées par une couche protéique ou lipidique. Ces microbulles peuvent être utilisées comme vecteur et être modifiées afin de pouvoir transporter des acides nucléiques (par exemple, en incorporant des lipides cationiques) et de pouvoir cibler les cellules d’intérêt (par l’insertion d’anticorps). Les microbulles réagissent de deux façons aux ultrasons. A basse fréquence, les microbulles oscillent (expansion/rétrécissement) de façon linéaire jusqu’à entrer en résonance et osciller de façon stable. On appelle cela la cavitation stable. A haute fréquence, les microbulles oscillent de plus en plus vite jusqu’à l‘effondrement des microbulles à cause de l’inertie du liquide environnant. On appelle cela la cavitation inertielle. (Figure 16)
Figure 16 : Schématisation de la réaction des microbulles aux ultrasons d’après
LENTACKER et al. (2013) Les effets physiques sur la membrane plasmique des deux types de cavitation sont représentés sur les figures 17 et 18.

49
Figure 17 : Effets physiques de la cavitation stable d’après LENTACKER et al. (2013)
Figure 18 : Effets physiques de la cavitation inertielle d’après LENTACKER et al. (2013)
Les deux types de cavitation provoquent aussi la formation de résidus super-oxydes qui modifient les canaux ioniques et entrainent la formation de pores par peroxydation des phospholipides membranaires. De plus, il semble que les ultrasons augmentent le nombre d’endocytose/exocytose par modification de la concentration intracellulaire en calcium et par modification du cytosquelette – LENTACKER et al. (2013). La modification des microbulles est essentielle afin de perfectionner la sonoporation et d’en faire une méthode efficace de transfert d’acides nucléiques.

50
3. Perspectives d’améliorations En parallèle de l’amélioration de la structure des vecteurs (viraux et non viraux), la recherche s’intéresse aussi à des méthodes permettant d’augmenter le taux de transfection et la stabilité de l’expression du transgène tout en diminuant la toxicité en faisant appel à différents systèmes. Voici quelques exemples de méthodes prometteuses. Il est à noter que ces molécules peuvent aussi être utilisées seules comme vecteur.
a. Les nanoparticules magnétiques Il est possible d’attacher un vecteur (viral ou non) à une nanoparticule magnétique telle que la magnétite(Fe304) ou maghémite (Fe2O3) (sous forme de cristal de 5 à 20 nm). Il est alors possible de guider le vecteur (et l’ADN qu’il transporte) injecté par voie intraveineuse ou intrapéritonéale par un aimant positionné au niveau du site ciblé et qui crée un champ magnétique localisé – LI C. et al. (2012). Zheng et al ont montré qu’il était possible de créer un vecteur stable à partir de PEI et d’oxyde de Fer magnétique et que ce vecteur pénétrait les cellules par voie d’endocytose (Figure 19).
Figure 19 : Schématisation du fonctionnement d’un vecteur magnétique codant pour
l’expression d’une protéine fluorescente d’après ZHENG et al. (2012)

51
Wang et son équipe ont prouvé que leur vecteur magnétique, fabriqué à partir de PEI, chitosane et de nanoparticules d’oxyde de Fer incluses dans des micelles, était non toxique pour les cellules transfectées, biocompatible et sûr en utilisation in vivo – WANG C. et al. (2012). Mannell et al ont démontré que l’utilisation de nanoparticules magnétiques en association avec la technique de sonoporation permettait de guider les microbulles vers le tissu cible et d’augmenter ainsi le taux de réussite de cette technique – MANNELL et al. (2012). L’utilisation d’un champ magnétique a aussi pour effet d’augmenter le taux de transfection par agrégation en un cours laps de temps de très nombreux vecteurs autour des cellules cibles ce qui augmente la probabilité qu’un vecteur pénètre la cellule. Les mécanismes de pénétration ne semblent pas modifiés par le champ magnétique. L’utilisation d’un champ pulsé par rapport à un champ statique augmenterait encore plus le taux de transfection. L’étude de l’utilisation d’un champ magnétique alternatif doit encore être approfondie – PLANK et al. (2011). Cette méthode, qui en est à ses débuts, serait une solution parfaite pour le ciblage des tissus à traiter lors d’utilisation de vecteurs non viraux ainsi que pour réduire la quantité de vecteurs nécessaires à une transfection efficace (de fortes doses de vecteurs provoquant le plus souvent une toxicité ou une réponse immunitaire) – LI C. et al. (2012).
b. Les systèmes transposon/transposase : Sleeping Beauty Le système Sleeping Beauty est l’association d’un transposon (séquence d’ADN mobile) avec une transposase, enzyme qui catalyse l’excision et la réintégration du transposon. Ce système a été découvert chez la souris. La Figure 20 montre le fonctionnement du système transposon/transposase.

52
Figure 20 : Schématisation du fonctionnement d’un système transposon/transposase d’après
(Skipper, 2013) Associé à une séquence d’ADN sous la forme d’un plasmide, le système Sleeping Beauty est capable d’intégrer la séquence génétique d’intérêt dans le génome de la cellule transfectée – JUNG et al. (2013). Afin de parfaire ce système, de nouvelles transposases plus efficaces ont été développées. Le système amélioré s’appelle Sleeping Beauty hyperactif – FIELD et al.
(2013). D’autres systèmes transposons/transposase (Tol2, PiggyBac) sont connus, mais semblent moins efficaces que le système Sleeping Beauty – PESSACH et NOTARANGELO (2011). L’avantage de ce système, par rapport aux vecteurs viraux et aux autres systèmes transposon/transposase est que son intégration semble être dirigée vers des lieux sûrs (« safeharbor »), loin des gènes transcrits et que son promoteur/enhancer est peu actif ce qui empêche la surexpression de gènes environnants.
c. Les aptamères Les aptamères sont des séquences d’acides nucléiques, simple brin, de petite taille capables de se lier spécifiquement à des protéines ou à d’autres cibles cellulaires. Leurs séquences sont conçues à partir de banques de séquences et sélectionnées par différentes méthodes de sélection comme SELEX (Systematic Evolution of Ligands by Exponential enrichment) ou plus récemment AptaBid. Ils ont l’avantage de ne pas déclencher de réponse immunitaire, mais ont

53
une demi-vie courte due à l’action des nucléases. Il est possible de les modifier (modification des nucléotides en fin de séquence afin qu’ils ne soient pas reconnus par les endonucléases, utilisation de nucléotides modifiés dans la séquence comme les 2’-O pyrimidines modifiées) afin d’allonger leur demi-vie. Les aptamères peuvent être utilisés comme vecteurs en thérapie génique grâce à leur grande spécificité et leur capacité à se lier à des protéines membranaires permettant leur internalisation. Cependant, ils ne peuvent transporter que des séquences nucléiques de petite taille (comme les siARN). Les aptamères peuvent aussi être utilisés pour renforcer ou créer une spécificité de tissu : positionnés en surface d’un vecteur, ils peuvent agir comme le feraient des anticorps en ciblant une protéine de surface vis-à-vis de laquelle ils présentent une affinité. Leur dernière utilisation consiste à les employer en tant qu’outil de thérapie génique car ils peuvent, en se liant, inhiber les fonctions de certaines protéines, telles que les facteurs angiogéniques – NI et al. (2011). Kotula et son équipe ont démontré in vitro que l’utilisation de chimères comprenant un aptamère et une protéine nucléolaire appelée la nucléoline, permettait une livraison efficace de la séquence nucléique d’intérêt directement dans le noyau cellulaire – KOTULA et al. (2012). Le tableau 1 compare les vecteurs utilisés en thérapie génique et présentés dans cette thèse.
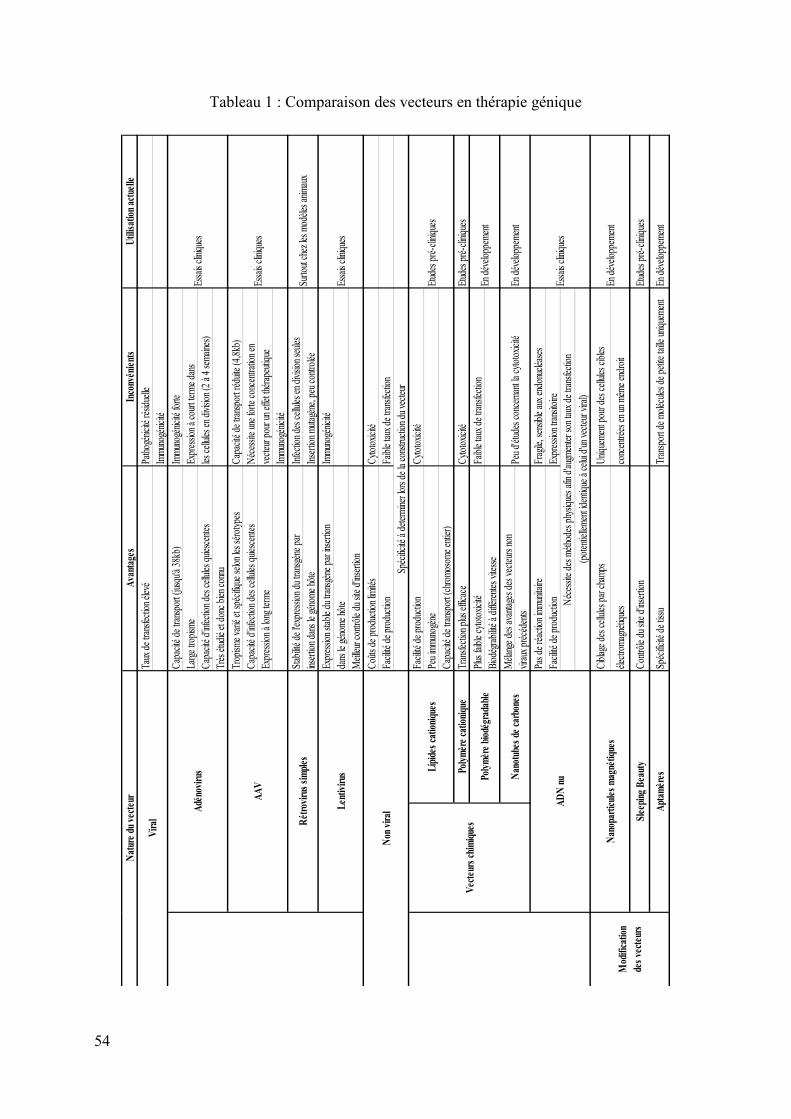
54
Tableau 1 : Comparaison des vecteurs en thérapie génique
Avan
tages
Inco
nvén
ients
Utilis
ation
actue
lleTa
ux de
transf
ectio
n élev
éPa
thogé
nicité
résid
uelle
Immu
nogé
nicité
Capa
cité d
e tran
sport
(jusq
u'à 38
kb)
Immu
nogé
nicité
forte
Large
tropis
meEx
pressi
on à
court
term
e dan
sCa
pacit
é d'inf
ectio
n des
cellul
es qu
iesce
ntes
les ce
llules
en di
vision
(2 à
4 sem
aines)
Très
étudié
et do
nc bi
en co
nnu
Trop
isme v
arié e
t spé
cifiqu
e selo
n les
séroty
pes
Capa
cité d
e tran
sport
rédu
ite (4
,8kb)
Capa
cité d
'infec
tion d
es ce
llules
quies
cente
sNé
cessi
te un
e fort
e con
centr
ation
en
Expre
ssion
à lon
g term
eve
cteur
pour
un ef
fet th
érape
utiqu
eIm
muno
génic
ité
Stabil
ité de
l'exp
ressio
n du t
ransgè
ne pa
rInf
ectio
n des
cellul
es en
divis
ion se
ules
insert
ion da
ns le
géno
me hô
teIns
ertion
muta
gène
, peu
contr
olée
Expre
ssion
stab
le du
transg
ène p
ar ins
ertion
Im
muno
génic
itéda
ns le
géno
me hô
teM
eilleu
r con
trôle
du sit
e d'ins
ertion
Coûts
de pr
oduc
tion l
imité
sCy
totox
icité
Facili
té de
prod
uctio
nFa
ible t
aux d
e tran
sfecti
on
Facili
té de
prod
uctio
nCy
totox
icité
Peu i
mmun
ogèn
eCa
pacit
é de t
ransp
ort (c
hromo
some
entie
r)Po
lymèr
e cati
oniqu
eTr
ansfe
ction
plus
effica
ceCy
totox
icité
Etude
s pré-
cliniqu
esPlu
s faib
le cy
totox
icité
Faibl
e tau
x de t
ransfe
ction
Biodé
grabil
ité à
différ
entes
vites
seM
élang
e des
avan
tages
des v
ecteu
rs no
nvir
aux p
récéd
ents
Pas d
e réa
ction
immu
nitair
eFr
agile,
sensi
ble au
x end
onuc
léases
Facili
té de
prod
uctio
nEx
pressi
on tra
nsitoi
re
Cibla
ge de
s cellu
les pa
r cha
mps
Uniqu
emen
t pou
r des
cellul
es cib
les
électr
omag
nétiq
ues
conc
entré
es en
un m
ême e
ndroi
t
Contr
ôle du
site d
'insert
ionEtu
des p
ré-clin
iques
Spéc
ificité
de tis
suTr
ansp
ort de
molé
cules
de pe
tite ta
ille un
iquem
ent
En dé
velop
peme
nt
En dé
velop
peme
nt
Essai
s clini
ques
Nano
parti
cules
mag
nétiq
ues
En dé
velop
peme
nt
Spéc
ificité
à de
termi
ner lo
rs de
la co
nstruc
tion d
u vec
teur
Nano
tubes
de ca
rbon
esPe
u d'ét
udes
conc
ernan
t la cy
totox
icité
En dé
velop
peme
nt
Néce
ssite
des m
éthod
es ph
ysiqu
es afi
n d'au
gmen
ter so
n tau
x de t
ransfe
ction
Essai
s clini
ques
Essai
s clini
ques
Surto
ut ch
ez les
mod
èles a
nimau
x
Essai
s clini
ques
Etude
s pré-
cliniqu
es
ADN
nu
Sleep
ing B
eauty
Aptam
ères
Mod
ificati
onde
s vec
teurs
AAV
Rétro
virus
simp
les
Lenti
virus
Vecte
urs c
himiqu
es
Lipid
es ca
tioniq
ues
Polym
ère b
iodég
rada
ble
(poten
tiellem
ent id
entiq
ue à
celui
d'un v
ecteu
r vira
l)
Vira
l
Natur
e du v
ecteu
r
Non v
iralAd
énov
irus

55
4. Conclusions La recherche se poursuit afin de trouver de nouvelles molécules capables d’assurer la fonction de vecteurs en thérapie génique ainsi que l’optimisation de nouvelles méthodes améliorant l’efficacité de ces vecteurs. Il est évident que le vecteur idéal n’existe pas (encore), qu’il sera sûrement spécifique d’un type de maladie et que sa création nécessitera la combinaison de plusieurs vecteurs existants avec des méthodes d’amélioration. Actuellement, les vecteurs les plus utilisés en essais cliniques sont les adénovirus (23,5%), les rétrovirus (19,1%) et l’ADN nu ou les plasmides (17,7%) – Charts & Tables (2014). On note ainsi que les vecteurs les plus utilisés ne sont pas forcément les plus efficaces, mais les plus étudiés, essentiellement du fait de leur innocuité et de leurs effets secondaires bien connus ou maîtrisés. Ainsi, des médicaments de thérapie génique sont désormais disponibles sur le marché du médicament :
Gendicine® est un adénovirus associé au gène p53, gène impliqué dans la réparation de l’ADN, disponible uniquement sur le marché du médicament chinois depuis 2004 pour les patients atteint de carcinomes des cellules squameuses de la tête et du cou. Si aucun effet secondaire n’a été rapporté, l’efficacité de ce traitement est remise en question – KAUFMANN et al. (2013).
Glybera® est un AAV associé à un des allèles codant pour la lipoprotéine lipase disponible sur le marché du médicament en Europe depuis 2012 pour les patients atteints d’hyperlipidémie de type I (déficience en lipoprotéine lipase). On peut cependant se demander si ce médicament est vraiment efficace car la population atteinte est tellement réduite que les essais cliniques ne permettent pas toujours d’avoir des résultats numériquement probants même s’ils sont statistiquement valables – BÜNING (2013).
V. Les maladies A l’origine, la thérapie génique a été conçue dans le but de réparer les gènes dans certaines maladies congénitales ou héréditaires telles que l’hémophilie et les désordres immunitaires congénitaux. L’idée de base était de guérir les malades en réparant le gène impliqué dans leur maladie – ESCORS et BRECKPOT (2010). Cependant, s’il est assez simple d’apporter un allèle sauvage pour remplacer un allèle mutant de ce même gène qui n’exprime pas de protéine fonctionnelle, il est plus compliqué d’inhiber un allèle mutant qui code pour une protéine anormale ou pathogène. Ainsi, de nombreuses maladies génétiques provoquées par la présence d’une telle protéine sont souvent réfractaires à une addition de gène. De nouvelles techniques ont été développées afin de répondre à ce nouvel élément comme les techniques de réparation des gènes. Avec le développement des outils de thérapie génique, il est devenu évident qu’on pouvait l’appliquer à d’autres maladies que les affections congénitales telles que les cancers et les maladies dégénératives de toutes sortes, de l’insuffisance cardiaque à la maladie de Parkinson – KAUFMANN et al. (2013).

56
Les cibles principales de la thérapie génique sont aujourd’hui les maladies acquises dont les cancers. Actuellement, les essais cliniques concernant les cancers représentent 63,8% des essais cliniques en thérapie génique – Charts & Tables (2014).

57
Partie 2 : Exemples d’applications de la thérapie génique
Les techniques de thérapie génique se développent grâce aux confrontations avec les maladies auxquelles on veut l’appliquer. On prendra ici quelques exemples choisis parmi les maladies les plus étudiées afin de mettre en application les techniques évoquées dans la première partie. Actuellement, les essais cliniques en thérapie génique concernent les maladies cancéreuses à 63,8%, les maladies monogéniques à 8,9%, les maladies infectieuses à 8,2% et les maladies cardiaques à 8,1% - Charts & Tables (2014).
I. Les maladies génétiques Les maladies génétiques, congénitales et/ou héréditaires, sont les maladies pour lesquelles la thérapie génique a été initialement créée. Si elles sont reléguées loin derrière les cancers en tant que sujet d’étude, la thérapie génique reste la plupart du temps la meilleure alternative thérapeutique (voire la seule) pour ces maladies.
1. L’hémophilie A
a. La maladie L’hémophilie est un trouble de la coagulation héréditaire, récessif et lié au chromosome X. Cette maladie monogénique est due à l’absence de certains facteurs de coagulation fonctionnels. Il existe deux types principaux d’hémophilie : l’hémophilie A, due à un manque en facteur VIII (fVIII) et l’hémophilie B, due à un manque en facteur IX. L’hémophilie A (HA) est la plus courante et touche un garçon sur 7500 naissances à travers le monde – DOERING et al. (2009). On peut classer l’hémophilie A en trois classes selon la quantité de facteurs VIII circulante :
<1% de la quantité normale (100 à 200ng/mL de plasma) : hémophilie A sévère >1% et <5% : hémophilie A modérée >5% et <30% : hémophilie A bénigne
Les symptômes de cette maladie sont des risques d’hémorragie accrus lors de traumatismes et dans la forme sévère, des saignements spontanés dans les articulations et les tissus mous qui provoquent des problèmes articulaires graves et qui peuvent être mortels – DOERING et SPENCER (2009). Le traitement actuel consiste en des perfusions de dérivés de plasma humain ou de facteurs VIII recombinants. Pour un patient atteint de HA sévère, ces perfusions doivent être répétées plusieurs fois par semaine. Ce traitement, bien que lourd, est efficace, mais présente un problème majeur : la formation d’inhibiteurs. Ce sont des anticorps anti-facteur VIII, qui se

58
lient aux facteurs circulants et les inactivent. 20% à 30% des patients traités pour la première fois développent des inhibiteurs – WALSH et BATT (2013). Le gène F8 qui code pour le facteur VIII se situe sur le bras long du chromosome X en région Xq28. Il mesure 186 kb et possède 26 exons – CHUAH et al. (1998). Le facteur VIII est composé de deux chaînes : une chaîne lourde formée par les domaines A1-A2-B et une chaîne légère formée par les domaines A3-C1-C2. Le rôle du domaine B est peu connu, mais il a été prouvé que sa délétion ne modifiait pas l’activité du facteur VIII. Dans la cascade de la coagulation, le facteur VIII est clivé par la thrombine en une forme active composée de trois sous-unités : A1, A2 et A3-C1-C2, appelée fVIIIa (facteur VIII activé). Dans la circulation sanguine, le facteur VIII est stabilisé par le facteur de Von Willebrand (FvW) et a une demi-vie courte (12h) – WALSH et BATT (2013).
b. Les modèles animaux Il existe de nombreux modèles animaux disponibles pour étudier l’hémophilie A ainsi que les traitements proposés. Chacun a ses avantages et ses inconvénients.
i. La souris La souris est un modèle d’étude très pratique : entretien facile, nombreux sujets disponibles, reproduction rapide, etc. Le premier modèle de souris pour l’hémophilie A a été créé dans les années 90. Deux types de souris transgéniques knock-out (KO) dirigés respectivement vers les exons 16 et 17 (tous deux dans le domaine A3 de la chaîne légère) ont été utilisés pour créer des modèles murins atteints d’hémophilie A sévère. Les souris KO pour l’exon 16 possèdent une protéine tronquée et celles KO pour l’exon 17 possèdent une protéine tronquée ou partiellement détruite. Les deux types ne sont pas différenciables au niveau du phénotype. Au contraire des humains atteints de HA sévère, les souris ne présentent pas de saignements spontanés fréquents. Cependant, le modèle murin présente l’avantage d’être modifiable : il est possible de créer une grande variété de lignées de souris atteintes de HA provenant de divers fonds génétiques, c’est-à-dire présentant une grande variété de capacités de leur réponse immunitaire (voire aucune pour les lignées Nude) afin de permettre l’étude des traitements dans différents contextes de réponse immunitaire – SABATINO et al. (2012).
ii. Le chien Le modèle canin est utilisé depuis le milieu du XXème siècle dans l’étude de l’hémophilie. De nombreuses études ont permis de valider l’extrapolation à l’Homme des résultats des études sur le chien et le modèle canin apparaît pour beaucoup comme le passage obligatoire en essais pré-cliniques pour le développement de nouveaux traitements pour l’hémophilie.

59
La colonie de Chapel Hill (Caroline du Nord, Etats-Unis) est la plus ancienne (1947). Elle a pour origine des setters irlandais qui présentaient naturellement un déficit en facteur VIII. Une autre lignée est maintenue à l’Université Queens au Canada. Ces deux lignées présentent une inversion de séquence au sein du gène F8 ce qui provoque une transcription aberrante, tout comme ce qui est rencontré chez plus de la moitié des humains atteints d’hémophilie A. Les chiens atteints de HA sévère présentent des saignements spontanés fréquents dans les articulations et les tissus mous. De plus, une des lignées produit des inhibiteurs en cas de traitement classique, ce qui en fait un très bon modèle d’étude pour tester de nouveaux traitements qui ne provoqueraient pas de réaction immunitaire de type inhibiteur – SABATINO et al. (2012). Le chien a une durée de vie beaucoup plus longue que celle de la souris ce qui autorise des études à long terme (sur dix ans environ) en particulier pour les études sur la durée de l’expression du transgène et les effets indésirables.
iii. Les nouveaux modèles Deux nouveaux modèles d’études, le mouton et le cochon, sont proposés afin de répondre à différents impératifs. En effet, une nouvelle piste d’étude en thérapie génique pour l’hémophilie A est la production de facteurs VIII par les plaquettes. Or, il se trouve que, tout comme l’Homme, le mouton possède dans ses plaquettes, des facteurs de Von Willebrand, qui aident à stabiliser les facteurs VIII. Une ancienne lignée de moutons a été recréée par Porada et son équipe en 2009 à partir de semence congelée. Cette lignée avait été maintenue entre 1979 et 1982 par l’Institut Fédéral de Technologie suisse qui l’avait laissée s’éteindre faute de moyens, ne conservant que quelques paillettes de semence. Les moutons produits à partir de cette semence présentent une déficience en facteur VIII suite à une modification de la séquence de l’exon 14, ce qui produit un changement de cadre de lecture et l’apparition d’un codon stop prématuré. Il s’agit de la première espèce, à part l’Homme, où ce type de mutation est découvert – PORADA et al. (2010). Le mouton est aussi un modèle d’étude adapté à l’expérimentation in utero d’une part grâce aux ressemblances entre les gestations humaines et ovines et d’autre part par la maîtrise des techniques d’expérimentation sur les fœtus ovins – PORADA et ALMEIDA-PORADA (2012). Le cochon est un modèle animal attractif car il possède un système de coagulation très proche de celui de l’Homme et le facteur VIII porcin a déjà été utilisé efficacement pour traiter des patients hémophiles possédant des inhibiteurs. Kashiwakura et son équipe ont créé, par une technique de transgénèse animale via le clonage, des cochons KO pour le gène F8. Ils ont tout d’abord créé une lignée de fibroblastes porcins KO pour le gène F8, puis ont transféré le noyau de ces fibroblastes dans des ovocytes receveurs énucléés. Ils ont ainsi obtenu 4 porcelets qui sont malheureusement tous morts en moins de deux mois (les deux premiers de cause incertaine et les deux suivants par hémorragie interne). De nouvelles études sont nécessaires afin de produire en quantité suffisante des cochons hémophiles capables de survivre au-delà de deux

60
mois et de vérifier la validité du modèle porcin avant de pouvoir l’utiliser en routine – KASHIWAKURA et al. (2012). Il faut cependant noter que, quel que soit le modèle animal, même le primate non-humain, il ne peut reproduire toutes les réactions qui seront observées chez l’Homme. Les nombreux essais pré-cliniques ont permis de démontrer que l’Homme est beaucoup moins immunotolérant que ne le sont les modèles animaux (et en particulier, le chien) et que ses réactions immunitaires sont plus graves et invalidantes. Cela explique le décalage important entre la recherche et les retombées thérapeutiques et la raison pour laquelle de nombreux essais pré-cliniques prometteurs n’aboutissent jamais à des produits viables chez l’Homme.
c. Intérêt de la thérapie génique L’hémophilie A présente plusieurs caractéristiques qui en font une cible parfaite pour la thérapie génique – DOERING et SPENCER (2009) – KAY et HIGH (1999).
La thérapie génique est une technique très coûteuse. Cependant, le coût actuel du traitement d’un patient atteint de la forme sévère est de 100 000 $ à 300 000 $ par an aux Etats-Unis, ce qui motive la recherche pour trouver un traitement à moindre coût.
Le traitement actuel est très contraignant, lourd, accessible à une petite proportion de la population malade et présente un effet secondaire qui le rend inefficace (formation d’inhibiteurs). Une thérapeutique alternative est donc plus que bienvenue.
La fenêtre thérapeutique est large : une élévation même légère de la quantité de protéines circulantes permet une diminution importante des symptômes et cette quantité peut atteindre 150% de la quantité normale sans provoquer d’effets secondaires délétères.
Le facteur VIII est classiquement produit par le foie, mais d’autres types cellulaires sont capables de le produire, tels que les cellules endothéliales, les fibroblastes et les cellules musculaires. Il suffit théoriquement que le tissu transfecté puisse délivrer la protéine dans la circulation sanguine.
Il existe des techniques éprouvées afin de contrôler la présence du facteur VIII (test ELISA) et la réussite du traitement (tests de coagulation, thromboélastographie, etc.)
Il existe des modèles animaux permettant les études pré-cliniques nécessaires au développement d’un traitement.
d. Utilisation de la thérapie génique Le premier écueil sur lequel s’est heurté la thérapie génique pour l’hémophilie A est la taille du gène F8. L’ADN complémentaire (ADNc) créé en 1984 mesure plus de 9kb – WALSH et BATT (2013) - (l’ARNm mesure 9048 nucléotides – DOERING et SPENCER (2009)). Or, seuls les vecteurs créés à partir d’adénovirus « gutless » ou les vecteurs non viraux sont capables de prendre en charge une séquence aussi longue. Mais ces derniers n’ont été développés qu’à

61
la fin des années 90. Aussi, d’autres techniques ont été mises en œuvre pour permettre de réduire la taille de la séquence à prendre en charge. La principale méthode utilisée est la délétion de la partie codant pour le domaine B. L’ADNc ainsi produit (appelée ADNc BDD-fVIII pour B-Domain-Deleted) mesure 4.6kb et peut être pris en charge par des vecteurs viraux conventionnels – CHUAH et al. (1998). Un autre souci est que des parties de la séquence de l’ADNc fVIII humain provoquent une répression de la production de fVIII. De plus, le facteur VIII a une demi-vie courte et sa stabilité dépend de sa conjugaison avec des facteurs de Von Willebrand. Pour toutes ces raisons, et encore plus que dans les autres maladies, il est primordial que les techniques de thérapie génique permettent la production de facteurs VIII en grande quantité afin d’atteindre une concentration de protéines circulantes à dose thérapeutique – CHUAH et
al. (1998). Différentes méthodes de thérapie génique ont ainsi été explorées afin de déterminer laquelle permettrait une correction à long-terme des troubles de la coagulation chez les patients atteints d’hémophilie A.
i. Les premiers essais cliniques Les rétrovirus simples Le premier vecteur utilisé en thérapie génique pour l’hémophilie A est un rétrovirus simple : le virus de la leucémie murine de Moloney dont l’efficacité in vitro à transporter et à insérer le transgène du facteur VIII à des cellules a été démontrée en 1990 – ISRAEL et KAUFMAN (1990). Ce virus avait déjà fait ses preuves dans le traitement d’enfants atteints du syndrome d’immunodéficience sévère lié à l’X. Il est capable de transfecter de nombreux types cellulaires, mais a besoin d’une division cellulaire afin de pénétrer le noyau – DOERING et SPENCER (2009). Des essais ex vivo ont permis de démontrer qu’il était nécessaire que les cellules transduites, greffées en néo-organe soient en contact avec la circulation sanguine : une greffe sous-cutanée ne permet pas de modifier la quantité de fVIII circulant malgré la production effective par le néo-organe. Un premier succès a été constaté par la transfection ex vivo de fibroblastes humains greffés en néo-organe dans la cavité péritonéale de souris immunodéficientes. Une élévation majeure de la concentration sanguine en fVIII a été constatée (jusqu’à 100ng/mL), mais s’est révélée être de courte durée (diminution jusqu’à la concentration basale en deux semaines) sûrement due à la mort des cellules du néo-organe – CHUAH et al. (1998). D’autres types cellulaires ont été utilisés, en particulier les cellules de la moelle osseuse. La greffe de moelle osseuse est une technique éprouvée et ces cellules sont en contact direct avec la circulation sanguine. Alors que les cellules de la lignée hématopoïétique n’expriment qu’une faible quantité de facteur VIII, les cellules mésenchymateuses sont capables d’en produire une quantité thérapeutique. Des études in vitro ont montré une concentration égale à 30ng/mL ce qui est suffisant pour passer d’un phénotype de HA sévère à un phénotype de HA bénigne –

62
CHUAH et al. (1998). Cependant, aucun essai in vivo n’a eu de résultat durable ce qui a été imputé à une probable inhibition de l’expression du transgène – DOERING et SPENCER (2009). L’utilisation in vivo des rétrovirus simples a posé de nombreux problèmes principalement liés au fait que les rétrovirus simples ne peuvent transfecter que des cellules en division active. Or les hépatocytes n’ont pas un fort taux de mitoses. Une des premières méthodes a donc été l’hépatectomie partielle avant l’injection du vecteur, ce qui provoque une régénération du foie et donc la division des hépatocytes restants. D’autres méthodes moins invasives ont été ensuite développées, comme l’utilisation de facteurs de croissance ou de modèles animaux en croissance. Il a aussi été remarqué qu’il était nécessaire de concentrer le vecteur. Lors de la préparation, seuls 2 vecteurs sur 10 possèdent le transgène. Il existe donc un fort taux de particules virales « vides » dépourvues de l’acide nucléique thérapeutique. Des techniques ont été développées afin d’augmenter ce ratio. Par exemple, on a remarqué que les virus pseudotypés avec les glycoprotéines du virus de la stomatite vésiculeuse (VSV-G) étaient très stables et supportaient sans altération une ultracentrifugation à la différence d’autres virus enveloppés comme le virus de Moloney. En créant un virus de Moloney ainsi pseudotypé, on a pu utiliser l’ultracentrifugation pour concentrer le vecteur – KAY et HIGH (1999). On peut ainsi injecter une même quantité de vecteurs tout en améliorant l’efficacité de la transfection via l’infection virale – CHUAH et al. (1998). Un essai clinique de phase I a été autorisé au début des années 2000. Powell et son équipe - POWELL et al. (2003) - ont utilisé un vecteur produit à partir du virus de la leucémie murine défectif pour la réplication (pas de gènes codant pour les protéines virales, ce qui rend le vecteur incapable de se répliquer). Ce vecteur transportait un ADNc humain BDD-fVIII et le promoteur viral du virus de la leucémie murine. Ils l’ont injecté par voie intraveineuse. Cette technique avait été éprouvée dans des essais précliniques chez le chien et le lapin et avait démontré une certaine efficacité : élévation de la concentration en fVIII pendant 6 mois. Cependant, des chiens avaient développé des anticorps neutralisants. Les résultats de cet essai clinique ont été mitigés :
Aucun effet secondaire n’a été observé sur un an de suivi. Neuf semaines post-traitement, un des patients a été positif pour la présence d’ADN viral dans son sperme, mais ce cas est resté isolé : aucun autre patient n’a présenté ce symptôme et le patient en cause a été testé négatif toutes les fois suivantes.
Malgré la présence d’ADN viral dans des cellules sanguines plus d’un an après le traitement, aucun des patients n’a présenté d’élévation durable de la concentration en fVIII. Cependant, certains ont présenté des pics de production (sur quelques jours seulement et en faible quantité) et aussi moins d’épisodes de saignements spontanés.
Aucune conclusion n’a pu être retirée de cet essai clinique car il n’y avait pas de groupe placebo.

63
Les adénovirus Les adénovirus, à la différence des rétrovirus simples, sont capables d’infecter des cellules quiescentes. Cependant, leur matériel génétique reste épisomal et les vecteurs créés à partir d’adénovirus provoquent une forte réponse immunitaire dirigée contre les protéines virales. Plusieurs études ont cependant choisi l’utilisation d’un tel vecteur. A chaque fois, on a constaté une élévation majeure de la concentration en fVIII qui ne s’est maintenue que pendant quelques jours à quelques semaines suivant les études et le modèle animal. Chez le chien, on a constaté la synthèse d’inhibiteurs à la suite de l’infection. Chez tous les autres modèles animaux, on a observé une hépatite avec élévation des paramètres hépatiques et une thrombocytopénie. Ces effets secondaires sont à mettre en lien avec la diminution de la concentration en fVIII. L’utilisation de vecteurs « gutless » a permis de réduire ces effets secondaires chez la souris, mais pas chez les autres modèles animaux : une concentration de 3x1012 particules virales (vp) par kilogramme, qui est la concentration minimale permettant la production de fVIII en quantité mesurable, provoque une toxicité hépatique et hématologique transitoire. Un essai clinique a été réalisé chez un patient unique. On lui a injecté un vecteur adénoviral « gutless » transportant l’ADNc total du gène F8 ainsi que le promoteur de l’albumine humaine qui est spécifique du foie à la concentration de 4,3 vp/kg. Une élévation de la concentration en fVIII a été notée (>1%) pendant plusieurs mois, mais l’essai a dû être stoppé à cause des effets secondaires : fièvre, courbatures, maux de tête, élévation des paramètres hépatiques et thrombocytopénie, effets secondaires indésirables liés au vecteur lui-même et à la réponse de l’hôte consécutive à son utilisation – DOERING et SPENCER (2009). L’ADN nu L’électroporation d’ADN nu a également été utilisée dans un essai clinique incluant douze patients. La technique utilisée était la transfection ex vivo de fibroblastes par l’ADNc BDD-fVIII et le promoteur de la fibronectine humaine. Les fibroblastes efficacement transfectés ont alors été implantés en néo-organe dans l’omentum des patients sous anesthésie générale. Aucun effet secondaire ni la synthèse d’inhibiteurs n’ont été rapportés. Mais, la concentration en fVIII est restée égale ou légèrement supérieure à celle avant la chirurgie. Cependant, un moindre besoin en traitement classique a été noté ainsi qu’une diminution de la fréquence des saignements spontanés jusqu’à 10 mois après l’implantation du néo-organe. Comme précédemment, peu de conclusions peuvent être tirées de cet essai clinique réalisé sans groupe placebo et non poursuivi au-delà de la phase I – DOERING et SPENCER (2009).
ii. Nouvelles orientations Depuis ces trois essais cliniques datant du début du XXIème siècle, aucune stratégie thérapeutique n’a été développée au point d’être testée chez l’Homme. Cependant, de nombreuses pistes sont à l’étude. Elles en sont, pour la plupart, au stade des essais précliniques.

64
Un nouveau transgène Des recherches se sont orientées vers la synthèse d’un transgène plus efficace. On a vu précédemment que l’ADNc du gène F8 comporte des séquences régulatrices qui diminuent la transcription du transgène. Il est aussi rapporté que, comme pour la plupart des protéines sécrétées, la sécrétion du facteur VIII est régulée par l’appareil de Golgi. Une première méthode permettant la synthèse d’un transgène plus efficace est la délétion de la séquence codant pour le domaine B. Mais, si cela augmente la quantité d’ARNm transcrits, cela diminue la quantité de fVIII sécrétés, probablement due à une moindre quantité de groupements N-glycane et donc à une moindre prise en charge par l’appareil de Golgi. Une étude a montré in vitro que l’addition d’une partie de la séquence codant pour le domaine B (et plus spécifiquement, les séquences codant pour des sites de glycosylation) permettait d’augmenter la sécrétion, mais cela n’a pas été démontré avec l’utilisation d’un vecteur. Il a aussi été démontré que l’insertion de l’intron 1 du gène codant pour le facteur de coagulation IX en lieu et place des introns 1 et 13 du gène F8 permet de multiplier la production de fVIII par treize par rapport à l’utilisation d’un ADNc BDD-fVIII classique. Cependant, par cette stratégie, la sécrétion reste limitée. Une seconde approche consiste à modifier l’interaction de l’ADNm avec les protéines chaperones du réticulum endoplasmique. Une mutation dans la séquence codant pour le domaine A1 a permis de multiplier l’expression du facteur VIII par trois. La méthode la plus récente consiste à remplacer les séquences codant pour les domaines A1 et A3 par les séquences du gène F8 porcin. En effet, il a été démontré in vitro que l’ADNc BDD-fVIII porcin permet la synthèse de dix à cent fois plus de facteurs VIII que l’ADNc humain et que cette différence vient essentiellement des séquences codant pour les domaines A1 et A3. Le mécanisme n’est pas encore élucidé, mais cette méthode est, à ce jour, la plus efficace et la plus prometteuse. Le transgène est appelé HP-fVIII – DOERING et al. (2009). Il reste maintenant à utiliser ces nouveaux transgènes en thérapie génique. De nouveaux vecteurs
Utilisation des AAV Les AAV ont fait leurs preuves dans le traitement de l’hémophilie B. La recherche s’est donc orientée naturellement vers l’utilisation de ces vecteurs pour l’hémophilie A. Les deux problèmes principaux sont la taille du transgène par rapport à la capacité du vecteur et le risque très élevé que le sujet développe des inhibiteurs suite à la thérapie (comme avec le traitement de base actuel). Des études ont montré que les sérotypes 8 et 9 des AAV étaient capables d’infecter les hépatocytes et étaient plus efficaces que les autres sérotypes. Deux types d’approches sont utilisées : la première consiste à séparer l’expression des deux chaînes en administrant deux vecteurs l’un portant la séquence codant pour la chaîne légère (A3-C1-C2) et l’autre celle pour la chaîne lourde (domaine A1-A2-B). La seconde approche est l’utilisation de l’ADNc BDD-fVIII (mesurant 4,6kb) avec un nombre limité de séquences régulatrices. Il a cependant été décrit que les AAV sont capables de

65
transporter des séquences plus importantes et qu’il est possible de fabriquer des vecteurs AAV hébergeant une séquence supérieure à leur capacité de transport théorique, et ce en quantité suffisante pour réaliser une thérapie génique – LU H. (2008). Sabatino et son équipe - SABATINO et al. (2011) - ont montré que ces deux méthodes permettent d’obtenir une quantité suffisante de fVIII circulants pour traiter des chiens atteints de HA. Selon la dose administrée, ces chiens sont passés d’un phénotype sévère à un phénotype modéré ou bénin durant toute la période d’observation (33 mois en moyenne, au maximum 68 mois pour un des chiens). Ils ont observé une réduction des saignements spontanés chez les chiens traités par rapport aux chiens témoins. Aucun des chiens traités n’a développé d’inhibiteur, qu’importe la méthode utilisée. L’administration du vecteur s’est faite par voie intraveineuse périphérique ou par injection dans la veine porte. Aucune différence n’a été montrée entre ces deux voies d’administration. Dans le traitement de l’hémophilie B, on a remarqué que le traitement avec une thérapie génique à base d’AAV provoquait une hépatite réactionnelle. Cela n’a pas été observé dans l’étude de Sabatino et de son équipe - SABATINO et al. (2011). D’après cette étude, on peut conclure que les AAV sont des vecteurs prometteurs pour le traitement de l’hémophilie A. Cependant, le facteur VIII canin est plus stable et a une activité supérieure au facteur VIII humain. Et les essais cliniques précédents ont montré que les humains ont une réaction immunitaire plus forte que les autres espèces. Il reste donc encore de nombreuses recherches à effectuer, principalement sur la sécurité et l’efficacité du vecteur et du transgène humain, avant qu’un essai clinique ne soit accepté – MARKUSIC et HERZOG (2013).
Utilisation des lentivirus Les lentivirus sont capables de transfecter les cellules quiescentes (comme les hépatocytes) et d’intégrer leur génome dans l’ADN de la cellule cible (ce qui permet une expression à long terme), loin des promoteurs, réduisant ainsi le risque de mutation par insertion. Ils sont aussi capables de transporter des séquences de 8 kb. Ces caractéristiques en font des candidats pour la thérapie génique de l’hémophilie A. Des études ont cherché à augmenter la quantité de fVIII circulants en améliorant les lentivirus. Une séquence (« woodchuck post-transcriptional regulatory element » ou WPRE) est régulièrement insérée après le transgène afin d’augmenter son expression lors de l’utilisation de lentivirus (de 2 à 5 fois plus que sans le WPRE). Cette augmentation est due à une augmentation de l’exportation des ARNm et potentiellement à une facilitation de la transcription. Cependant, l’action du WPRE dépend à la fois du type cellulaire transfecté et du promoteur utilisé. De fait, dans certaines conditions, l’adjonction du WPRE peut diminuer l’expression du transgène. De plus, le WPRE code pour les soixante premiers acides aminés d’un oncogène. Son utilisation est donc remise en question. Johnston et son équipe ont montré que dans le cas particuliers de l’hémophilie A (utilisation de deux systèmes à base de lentivirus) le WPRE n’a aucune influence sur l’expression du transgène HP-fVIII – JOHNSTON et al. (2013). Dans cette étude, le système le plus efficace, qui a permis la sécrétion par les souris traitées d’une quantité thérapeutique de fVIII, est un système utilisant un vecteur produit à partir

66
d’un virus de l’immunodéficience simienne avec le promoteur du cytomegalovirus (CMV) et le transgène HP-fVIII.
Utilisation de vecteurs non viraux Bowman et son équipe - BOWMAN et al. (2008) - ont étudié la possibilité de créer un traitement administrable par voie orale. Ils ont créé des nanoparticules de chitosane encapsulant un plasmide contenant l’ADNc BDD-fVIII. Ils ont montré que des souris KO pour le gène F8 nourries avec ces nanoparticules présentaient des niveaux d’activité du facteur VIII thérapeutique (1 à 4%) pendant un mois. Le transgène a été retrouvé dans le tissu gastro-intestinal, le foie, la rate et d’autres tissus. Cette étude met en évidence que la voie d’administration per os est utilisable dans le traitement de l’hémophilie A par thérapie génique. D’autres études sont nécessaires au développement de cette technique qui présente de nombreux écueils (protection du plasmide à travers le tube digestif, transport du tube digestif aux tissus, relargage du plasmide dans le cytoplasme, protection contre les nucléases cytoplasmiques, etc). Aucun article développant cette technique n’est paru depuis ce qui laisse à penser que l’administration per os présente trop de problèmes pour déboucher sur un réel traitement. L’utilisation du système Sleeping Beauty a aussi été rapportée avec une certaine efficacité (niveau d’expression du facteur VIII thérapeutique pendant 50 semaines chez des souris KO) – KREN et al. (2009). Cependant, de même que pour l’utilisation du chitosane, la communauté scientifique semble penser que cette méthode a moins de potentiel que les vecteurs viraux car aucun article proposant cette technique n’est paru depuis. De nouvelles méthodes
Approche plaquettaire Une nouvelle approche du traitement de l’hémophilie A par la thérapie génique est le ciblage des plaquettes. Les plaquettes ne sont pas connues pour produire ni pour stocker le facteur VIII, mais elles produisent et stockent des facteurs de von Willebrand qui permettent de stabiliser les facteurs VIII. Plusieurs études ont établi que l’expression du facteur VIII par les plaquettes était possible et qu’elle permet de diminuer le nombre d’épisodes de saignements spontanés. Dans toutes ces études, des cellules de la moelle osseuse ont été prélevées et transfectées ex
vivo par des lentivirus transportant l’ADN cBDD-fVIII. Deux promoteurs spécifiques des mégacaryocytes ont été utilisés (le promoteur de l’intégrine αIIb et celui de la glycoprotéine Ibα). Les cellules transfectées ont été ensuite greffées chez le donneur. Pour ces interventions, les sujets étaient sous traitement conventionnel (transfusion de fVIII recombinants pour éviter les hémorragies lors des chirurgies). Montgomery a montré que plus de 60% des souris traitées survivaient au test de la queue coupée avec seulement 5% de leur moelle osseuse transfectée. La présence d’inhibiteurs ne modifie pas l’action des facteurs VIII sécrétés par les plaquettes et ces derniers ne provoquent pas de

67
développement d’inhibiteurs chez les souris et les chiens traités – MONTGOMERY et SHI (2010) – DU et al. (2013).
Traitement in utero L’hémophilie A peut être détectée chez le fœtus tôt dans la gestation. Elle est donc candidate au traitement in utero. Le traitement in utero présente de nombreux avantages : il permettrait de diminuer voire supprimer les besoins en traitement classique par correction phénotypique dès la naissance. Outre le gain financier, les enfants atteints d’hémophilie A auraient un confort de vie bien meilleur, ainsi qu’à l’âge adulte. Le traitement in utero augmente l’efficacité de la thérapie génique : les tissus cibles sont tous en croissance ce qui permet l’utilisation de tous les vecteurs viraux (et pas seulement ceux capables de transfecter les cellules quiescentes) et la transduction d’un petit nombre de cellules peut conduire, par division des cellules transduites, à la constitution d’un tissu assez riche en cellules exprimant la protéine fVIII pour avoir un effet thérapeutique – PORADA et ALMEIDA-PORADA (2012). De plus, en intervenant avant la mise en place du système immunitaire du fœtus, on peut le rendre immunotolérant au produit de la thérapie génique, ce qui, dans le cas de l’hémophilie A, empêcherait la formation d’inhibiteurs. Cependant, il a été prouvé que les cellules germinales étaient, elles aussi, transduites ce qui pose de nombreux problèmes éthiques. Porada et son équipe ont montré dans leur étude que le risque de transmettre le transgène à la descendance était plus faible que le taux de mutation normal chez l’Homme. On peut aussi se demander l’impact qu’aurait une immunotolérance au vecteur viral utilisé. Il faut aussi préciser que la transduction de cellules en division n’a pas que des avantages : cela provoque aussi la perte des transgènes non intégrés dans l’ADN cellulaire. L’utilisation des AAV dans cet optique est donc compliquée, mais reste possible comme l’ont démontré Hu et son équipe – HU et LIPSHUTZ (2012). Les souris transfectées ont présenté un déclin de l’activité du facteur VIII jusqu’à une stabilisation à un niveau thérapeutique qui a perduré pendant toute l’étude (22 mois). Le traitement in utero est déjà utilisé dans d’autres maladies, mais la thérapie génique n’a pour l’instant jamais été utilisée in utero chez l’Homme. Les essais sur les modèles animaux sont très encourageants, mais encore trop peu nombreux pour obtenir l’autorisation de lancer les essais cliniques sur des fœtus humains. Traitement des patients possédant des inhibiteurs et prévention 20 à 30% des patients atteints d’hémophilie A développent des inhibiteurs. Le traitement actuel (appelé induction d’une immunotolérance) consiste à perfuser tous les jours sur plusieurs mois les patients avec des facteurs VIII. Cela conduit à une disparition des inhibiteurs et à l’installation d’une immunotolérance au facteur VIII dans deux tiers des cas. Outre le faible succès de cette méthode, elle est très invasive, lourde à subir et très chère. La thérapie génique offre des solutions. Pour prévenir la formation d’inhibiteurs, on peut utiliser le traitement in utero (cf. précédemment) et le ciblage de l’expression du transgène. En effet, la synthèse d’inhibiteurs est provoquée par la présentation du facteur VIII par les cellules présentatrices d’antigènes

68
(CPA) aux lymphocytes. En restreignant l’expression du transgène aux hépatocytes uniquement, on empêche cette présentation. Ce ciblage peut être réalisé par pseudotypage des vecteurs viraux, par insertion d’un promoteur spécifique ou d’un microARN spécifique des CPA – MIAO (2011). Pour traiter les patients possédant déjà des inhibiteurs, l’approche plaquettaire (cf. précédemment) ou la modification des cellules de l’immunité seraient intéressantes. En effet, une équipe de recherche - SU et al. (2011) - a transfecté ex vivo des cellules dendritiques murines afin qu’elles expriment sur leur surface membranaire les antigènes du facteur VIII et qu’elles induisent l’immunotolérance à cet antigène lors sa présentation aux lymphocytes. Une fois transplantées chez des souris possédant des inhibiteurs, on remarque une absence de réponse immunitaire à l’injection de facteur VIII recombinant. Pour l’instant, ces techniques n’en sont encore qu’aux études précliniques et ne peuvent actuellement pas bénéficier aux patients possédant des inhibiteurs.
e. Conclusion La thérapie génique pour l’hémophilie A a subi de nombreux échecs, en partie dus à une inadéquation entre les réactions des modèles animaux et celles de l’espèce humaine. La création d’un nouveau transgène ainsi que l’amélioration des vecteurs, et en particulier les AAV qui ont fait leurs preuves dans le traitement de l’hémophilie B, laisse à penser que de nouveaux essais cliniques seront acceptés et donneront des résultats si ce n’est probants, du moins exploitables. La thérapie génique offre une alternative thérapeutique pertinente : le protocole thérapeutique est beaucoup plus léger que celui actuellement utilisé et le traitement des patients possédant des inhibiteurs est possible et apparemment plus efficace que le protocole actuel. De plus, la thérapie génique semble pouvoir éviter à long terme la synthèse de ces inhibiteurs, réglant ainsi définitivement le problème. L’utilisation de vecteurs non viraux reste anecdotique malgré les possibilités offertes par ces vecteurs (faible immunogénicité, possibilité d’administration per
os, etc).

69
2. La myodystrophie de Duchenne
a. La maladie La myodystrophie de Duchenne (DMD) est une maladie récessive liée à l’X due à l’absence de dystrophine dans les cellules musculaires (striées, lisses et cardiaques). Cette maladie touche environ un garçon sur 3500. Dans un tiers des cas, la mutation est congénitale. Le reste du temps, elle est héréditaire. Les symptômes apparaissent dans la petite enfance avec des retards moteurs importants et, dans un cas sur trois, on observe aussi des troubles cognitifs. Les enfants atteints de DMD perdent la capacité de se déplacer à l’adolescence. L’espérance de vie sans traitement est de moins de vingt ans. L’analyse histologique révèle une infiltration fibro-graisseuse des muscles – HOFFMAN et al. (1986) – DOUGLAS et WOOD (2013). Les patients atteints de DMD développent aussi des cardiomyopathies dilatées ou présentent des arythmies qui peuvent conduire à une insuffisance cardiaque ou à des arrêts cardiaques spontanés – LAI et DUAN (2012). La mort est la plupart du temps due à une défaillance cardio-respiratoire par infiltration du diaphragme et des autres muscles respiratoires ce qui les rend inefficaces. Le traitement actuel consiste en une corticothérapie et des mesures de soutien lors de la détérioration de l’état du malade – MENDELL et al. (2012). Il a permis d’améliorer la qualité de vie des malades et d’allonger leur survie de quelques années parfois. Le gène de la dystrophine se situe sur le chromosome X en position Xp21. Il s’agit du plus long gène humain connu à ce jour. Il mesure 2500 kb et contient 79 exons. L’ADNc du gène DMD mesure 14kb. Au moins sept isomères (dystrophine cérébrale, rétinienne, etc.) peuvent être produits à partir de ce gène grâce à des promoteurs intra-gènes. Malgré la grande diversité des mutations observées, au plan phénotypique, seules les dystrophines musculaires sont affectées et parfois certaines dystrophines cérébrales (ce qui conduit à des troubles cognitifs). L’isomère présent dans les cellules musculaires striées squelettiques est long de 3685 acides aminés et pèse 14 kDa. La dystrophine est un élément essentiel du cytosquelette des cellules musculaires. Elle met en lien différentes protéines cytoplasmiques et transmembranaires comme le montre la figure 21.
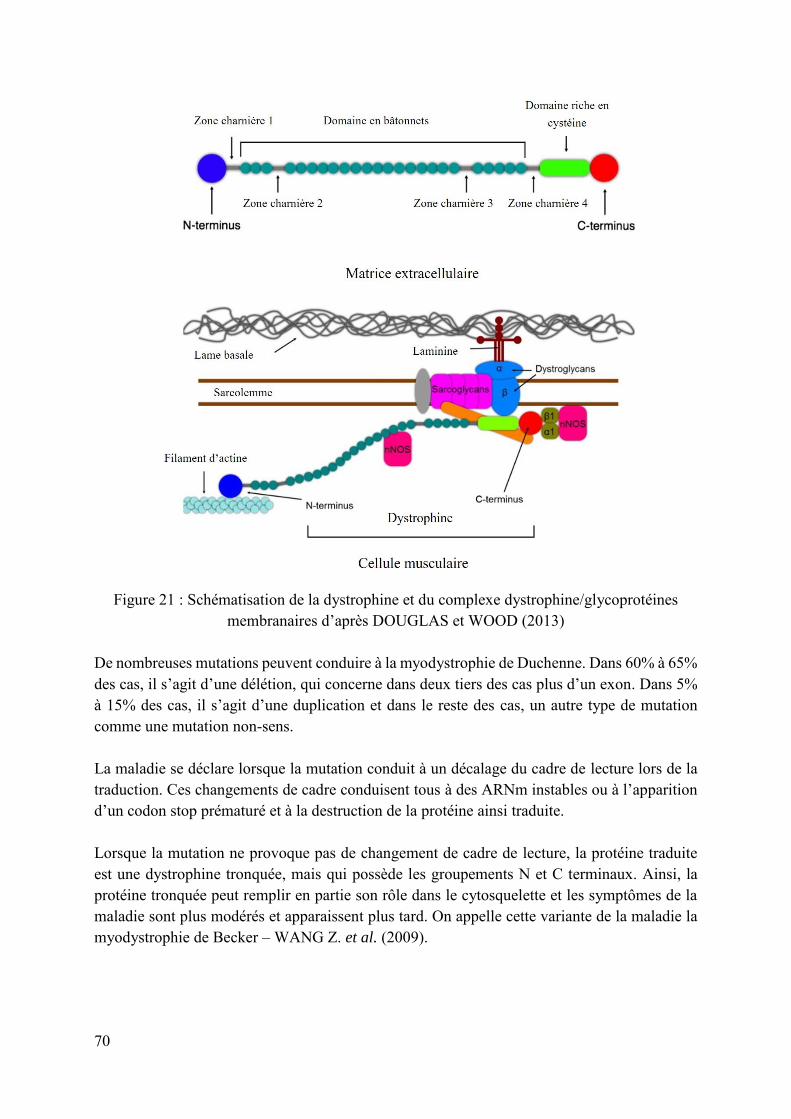
70
Figure 21 : Schématisation de la dystrophine et du complexe dystrophine/glycoprotéines
membranaires d’après DOUGLAS et WOOD (2013) De nombreuses mutations peuvent conduire à la myodystrophie de Duchenne. Dans 60% à 65% des cas, il s’agit d’une délétion, qui concerne dans deux tiers des cas plus d’un exon. Dans 5% à 15% des cas, il s’agit d’une duplication et dans le reste des cas, un autre type de mutation comme une mutation non-sens. La maladie se déclare lorsque la mutation conduit à un décalage du cadre de lecture lors de la traduction. Ces changements de cadre conduisent tous à des ARNm instables ou à l’apparition d’un codon stop prématuré et à la destruction de la protéine ainsi traduite. Lorsque la mutation ne provoque pas de changement de cadre de lecture, la protéine traduite est une dystrophine tronquée, mais qui possède les groupements N et C terminaux. Ainsi, la protéine tronquée peut remplir en partie son rôle dans le cytosquelette et les symptômes de la maladie sont plus modérés et apparaissent plus tard. On appelle cette variante de la maladie la myodystrophie de Becker – WANG Z. et al. (2009).

71
b. Les modèles animaux Il existe deux principaux modèles animaux de myodystrophie présents à l’état naturel : la souris et le chien.
i. La souris La souris mdx présente une mutation ponctuelle sur l’exon 23 du gène de la dystrophine murine ce qui conduit à l’apparition d’un codon stop. Cependant, le modèle murin ne présente pas les mêmes symptômes que les humains : les souris malades présentent des symptômes modérés, en particulier en regard du développement des cardiomyopathies. Les souris malades ont une durée de vie inférieure de 20% à une souris normale. Il existe d’autres modèles murins créés artificiellement : la souris mdx5cv qui présente aussi une mutation sur l’exon 10 et la souris déficiente en dystrophine et utrophine (protéine similaire à la dystrophine, exprimée essentiellement durant la vie fœtale). Malgré ces différences importantes, la souris reste un modèle d’étude pratique de par l’entretien facile d’une colonie de grande taille – LIEW et KANG (2013).
ii. Le chien Au contraire de la souris mdx, le chien cxmd présente des symptômes très similaires à l’Homme. Les lésions musculaires débutent lors du développement in utero. Les premiers symptômes apparaissent vers l’âge de 6 à 8 semaines. La mort survient par défaillance cardio-respiratoire à différents âges (survie de quelques jours lors de forme natale fulminante, à quelques mois et parfois de 2 à 4 ans). Cette progression de la maladie en fait des modèles d’études très intéressants pour tester l’efficacité des traitements. Cependant, le maintien d’une colonie est très coûteux et requiert beaucoup de soins. Les chiens sont donc plus difficilement utilisés que les souris – WANG Z. et al. (2009). Plusieurs races sont utilisées comme modèles (Golden Retriever, Rottweiler, Braque Allemand, Welsh Corgi et Cavalier King Charles principalement) présentant toutes des mutations différentes. Le Golden Retriever est la race la plus étudiée. Les Golden Retriever malades présentent une mutation ponctuelle sur le site d’épissage de l’intron 6 ce qui provoque un saut d’exon (exon 7) et modifie le cadre de lecture. L’ARNm présente alors un codon stop prématuré et la protéine traduite est non fonctionnelle. De nombreuses études ont permis de mettre en place des tests fonctionnels, biochimiques et d’imagerie permettant d’évaluer la progression de la maladie chez le Golden Retriever. De ce fait, cette race est particulièrement bien connue et on peut ainsi vérifier avec précision l’efficacité d’un traitement. A ce jour, le Golden Retriever a été utilisé dans la majorité des essais pré-cliniques, dans des études pharmacologiques, cellulaires comme génétiques – KORNEGAY et al. (2012).

72
iii. Les autres modèles D’autres modèles animaux sont utilisés dans l’étude de la myodystrophie de Duchenne. Il s’agit du poisson zèbre (Danio rerio), du hamster et du chat. Ces derniers ne sont pas utilisés comme modèles en thérapie génique, c’est pourquoi ils ne seront que cités ici. Le poisson zèbre est utilisé pour l’étude des gènes et des molécules pouvant avoir un avantage thérapeutique et le hamster comme modèle d’étude des cardiodystrophies – BERGER et CURRIE (2012) – LAI et DUAN (2012). Le cas du chat est particulier. Une dystrophie musculaire a été identifiée chez le chat en 1986, caractérisée par un manque de dystrophie dans les cellules musculaires striées squelettiques et cardiaques principalement. Les chats dystrophiques présentent un phénotype unique : hypertrophie musculaire, calcification de la langue, mégaœsophage, cardiomyopathie dilatée, insuffisance rénale, … A cause de ce phénotype très éloigné du phénotype humain, le chat n’est pas réellement utilisé comme modèle d’étude dans le développement d’un traitement – NAKAMURA et TAKEDA (2011).
c. Utilisation de la thérapie génique La thérapie génique est une alternative thérapeutique attrayante. Cependant, la myodystrophie de Duchenne présente des particularités qui la rendent difficile à traiter par cette méthode.
i. Remplacement du gène Le gène de la dystrophine est le plus grand gène humain connu et l’ADNc créé à partir de ce gène (14 kb) ne peut être transporté que par des adénovirus « gutless ». Cependant, les AAV sont privilégiés comme vecteurs dans cette maladie de par leur tropisme naturel pour les muscles, leur persistance dans le muscle sain et leur innocuité – MENDELL et al. (2012). Les lentivirus sont aussi considérés comme vecteurs d’intérêt, mais le risque de mutagénèse rend leur utilisation sujette à caution – PICHAVANT et al. (2011). On a donc créé des gènes modifiés de plus petite taille, pouvant être pris en charge par des vecteurs conventionnels. Miniaturisation du gène de la dystrophine Des chercheurs ont remarqué une mutation intéressante chez des patients atteints de la myodystrophie de Becker : une délétion des exons 17 à 48 (ce qui représente une délétion de 48% de la séquence codante du gène) n’entraîne une perte de la capacité à se déplacer qu’à un âge tardif (un malade est resté ambulatoire jusqu’à 61 ans) – MENDELL et al. (2012). En effet, cette délétion concerne principalement le domaine en bâtonnets, partie impliquée dans des rôles mineurs de la dystrophine. L’ARNm est donc transcrit normalement, sans modification du cadre de lecture. Les gènes de mini-dystrophine (mDYS) ou micro-dystrophine (µDYS) selon les auteurs (et la longueur du transgène) sont construits en supprimant des exons codant pour le domaine en bâtonnets et la partie C-terminale (cf Figure 22).

73
Figure 22 : Schématisation de trois différents gènes de mini-dystrophine d’après WANG B. et
al. (2000) Des études ont montrés l’efficacité du mDYS chez la souris : l’injection dans le muscle gastrocnémien d’un AAV portant un mDYS avec le promoteur du CMV ou de la créatinine kinase (spécifique du muscle) permet une expression du transgène à moyen terme (de 2 à 6 mois), augmente la force de contraction isométrique du muscle et protège contre les déchirures musculaires. Ces essais pré-cliniques ont permis la mise en place d’un essai clinique de phase I. Bowles et son équipe ont utilisé un AAV recombinant créé à partir d’un AAV-2 modifié par 5 mutations présentes chez les AAV-1 ce qui octroie à leur vecteur (appelée AAV-2.5) le tropisme accru de l’AAV-1 pour les cellules musculaires, une immunogénicité diminuée (pas de réaction croisée avec les antigènes des virus parents) et les caractéristiques de l’AAV-2 (innocuité démontrée, purification facile et maîtrisée) – BOWLES et al. (2011). Ils ont utilisé le promoteur du CMV et le gène mDYS Δ3990 (cf. Figure 22). Ils ont injecté dans un muscle biceps de 6 patients atteint de DMD le vecteur avec le transgène et dans le muscle contro-latéral du sérum physiologique ou des capsides de AAV-2.5 vides. Les résultats ont montrés que, bien que le gène ait été inséré avec succès dans les fibres musculaires, pratiquement aucune cellule n’exprimait la minidystrophine. Des analyses histochimiques ont montré la présence d’une réponse immunitaire dirigée contre la mini-dystrophine via des épitopes codés par des exons absents de la dystrophine endogène des patients. De plus, chez un des patients, on a montré que des fibres révertantes (qui présentent une nouvelle mutation permettant l’expression de la dystrophine) provoquaient la mise en place d’une réponse immunitaire contre la dystrophine exprimée par ces cellules et non une immunotolérance. Cet essai clinique montre l’importance de la sélection des sujets par rapport au transgène utilisé – MENDELL et al. (2012). Plasmide du gène entier Un essai clinique utilisant un plasmide contenant l’ADNc de la dystrophine a été réalisé en 2004. L’expression du transgène à la suite de l’injection du plasmide a été très faible voire nulle,

74
et en tous cas, inférieure au seuil nécessaire à une amélioration du phénotype. Ceci est probablement dû à une très faible efficacité de la transfection par injection seule. D’autres techniques ont été utilisées afin d’améliorer la transfection : la technique hydrodynamique et l’électroporation. Des résultats intéressants ont été démontrés chez le chien. Mais ces méthodes semblent trop coûteuses à développer pour prendre le pas sur l’utilisation de vecteurs viraux. De plus, tous les muscles ne peuvent pas être traités par ces techniques : l’électroporation nécessite une intervention locale pour chaque muscle et la technique hydrodynamique ne peut être réalisée que sur un bras ou une jambe mais pas pour la tête ou le tronc – PICHAVANT et al. (2011).
ii. Le saut d’exon Le saut d’exon est une technique de modification de l’expression génique qui consiste à masquer les sites d’épissage ou des sites régulateurs de l’épissage de certains exons cibles, afin qu’ils ne soient pas excisés. On peut ainsi rétablir le cadre de lecture de l’ARNm et permettre l’expression d’une dystrophine tronquée mais en partie fonctionnelle, comme avec la technique de miniaturisation de la dystrophine. Afin d’atteindre ce résultat, des oligonucléotides antisens (ONA) mesurant entre 20 et 30 nucléotides ont été créés. Virtuellement, toutes les mutations peuvent bénéficier de cette technique, mais chaque mutation demande son ONA spécifique. Ainsi, la recherche s’est concentrée sur les ONA qui bénéficieraient au plus grand nombre. La figure 23 présente un exemple de l’utilisation du saut d’exon.
Figure 23 : Principe de l’utilisation des ONA lors d’une mutation provoquant la délétion de
l’exon 50 d’après – DOUGLAS et WOOD (2013)

75
Cependant cette technique ne permet de sauter qu’un seul exon. Seuls 64% des patients atteints de DMD pourraient être traités ainsi. En effet certaines mutations requièrent le saut de deux exons minimum afin de rétablir le cadre de lecture. Afin de réaliser ce saut, plusieurs ONA doivent être utilisés. Des essais cliniques chez des souris mdx ont montré la possibilité de rétablir l’expression de la dystrophine en provoquant le saut des exons 45 à 55 par un mélange de plusieurs ONA. Les mutations par duplication peuvent aussi bénéficier de cette technique, mais il a été montré que certaines y sont réfractaires – DOUGLAS et WOOD (2013). Toujours chez la souris, deux types d’ONA ont été utilisées dans le saut d’exon : le 2’ O-methyl-ribo-oligonucléotide-phosphorothioate (2’OMe) et le phosphorodiamidate morpholino oligomère (PMO). Deux essais cliniques ont été réalisés utilisant chacun une des deux molécules citées plus haut et visant au saut de l’exon 51. Dans une première étude, ils ont directement injecté les ONA dans un muscle puis, dans une seconde étude, par voie systémique. Les essais cliniques sont toujours en cours et en sont à la phase IIb pour celui utilisant un PMO – DUAN (2011). Dans les deux cas, l’expression d’une dystrophine a été montrée dans 60 à 100% des fibres musculaires selon la dose administrée. Aucun effet secondaire n’a été observé – MENDELL et
al. (2012). Des recherches ont été faites concernant l’utilisation des AAV pour délivrer les ONA aux fibres musculaires. Des résultats intéressants ont été démontrés chez la souris – OKADA et TAKEDA (2013). Cependant, les essais cliniques en cours semblent prouver qu’il n’est pas nécessaire d’ajouter un vecteur, sauf peut-être pour pouvoir traiter les cellules cardiaques.
iii. Modification de l’expression des gènes Surexpression de l’utrophine L’utrophine est une molécule semblable à la dystrophine et qui peut suppléer certaines fonctions de la dystrophine. Elle est principalement exprimée lors de la vie fœtale puis seulement dans les jonctions neuromusculaires et musculo-tendineuses. Il a été démontré chez des souris mdx que la surexpression de l’utrophine (trois à quatre fois plus que la normale) permettait la normalisation du phénotype – PICHAVANT et al. (2011). L’utrophine peut être surexprimée par l’administration de certaines molécules, mais la recherche s’oriente vers la miniaturisation du gène de l’utrophine afin de pouvoir utiliser la thérapie génique pour surexprimer ce gène – DUAN (2011). Autres pistes Il existe d’autres protéines qui pourraient améliorer le phénotype des patients atteints de DMD comme la laminine, les intégrines, etc. Par exemple, la laminine-111, normalement uniquement exprimée lors de la vie fœtale, a été étudiée dans le traitement de la DMD. Son efficacité a été prouvée chez des souris mdx : l’injection de laminine-111 permet d’accroître la résistance et la force des muscles malgré l’absence de dystrophine – GOUDENEGE et al. (2010).

76
Les gènes codant ces protéines sont aussi à l’étude et permettraient de diminuer les symptômes de la maladie. Ces pistes pourraient compléter la thérapie génique actuelle – DUAN (2011).
d. Conclusion La myodystrophie de Duchenne est une maladie incurable. Le traitement actuel est peu efficace et provoque des effets secondaires à surveiller. La thérapie génique a été développée sous différentes formes afin d’obtenir un traitement valable. On observe cependant qu’aucune piste actuellement à l’étude ne permet de soigner la maladie, mais en réduit seulement les symptômes. Le traitement qui paraît le plus prometteur est le saut d’exon. Les essais cliniques sont en cours afin d’aboutir à un traitement fiable. Pourtant, cette technique doit être modulée en fonction des mutations présentes chez le patient. De nombreuses études seront encore nécessaires afin que tous puissent profiter d’un traitement adapté. Il est à noter que la thérapie génique utilisant les vecteurs viraux provoque des réactions immunitaires difficiles à contrer, rendant ainsi cette technique infructueuse pour l’instant. L’utilisation de microARN permettant la régulation de l’immunité pourrait pallier ce problème. Cependant, pour l’instant, l’utilisation de vecteurs viraux semble être la seule option permettant de transfecter tous les muscles en même temps.

77
II. Les maladies acquises : l’insuffisance cardiaque Les maladies acquises ou dégénératives peuvent également être traitées par thérapie génique. Encore faut-il connaître et comprendre le mécanisme de ces maladies qui provoque l’apparition des symptômes. On prendra ici l’exemple de l’insuffisance cardiaque pour illustrer l’usage de la thérapie génique dans le traitement des maladies acquises.
1. Epidémiologie et physiopathologie L’insuffisance cardiaque gauche se caractérise par une incapacité du cœur à faire face aux besoins en oxygène et nutriments des organes en ne réussissant pas à maintenir un débit sanguin suffisant pour irriguer l’organisme. On ne parlera ici que de l’insuffisance cardiaque gauche qui est la plus fréquente et qui est sous-entendue lorsqu’on ne précise pas le type. L’insuffisance cardiaque a plusieurs causes, congénitales et dégénératives bien que les premières d’entre elles soient les maladies coronariennes, le diabète et l’hypertension artérielle. L’insuffisance cardiaque est systolique (défaut de contraction du ventricule gauche) dans 60% des cas et diastolique (défaut de remplissage du ventricule gauche) dans les autres cas. Cette maladie est une des causes majeures de décès dans le monde. Le nombre de personnes touchées augmente chaque année, notamment en raison du vieillissement de la population. Lors d’insuffisance cardiaque, le système rénine-angiotensine-aldostérone est activé comme mécanisme compensateur pour maintenir la perfusion des organes nobles (cerveau, cœur, poumons) en provoquant une vasoconstriction périphérique. Cela provoque, à terme, un remodelage et une modification de la géométrie du cœur. Au niveau de la cellule cardiaque, on note une altération de l’excitation cellulaire, de l’expression de la chaîne lourde de la myosine, une moindre sensibilité aux β-adrénergiques et une hypertrophie. On remarque aussi une dégradation et une fibrose de la matrice extracellulaire. Les symptômes sont variés (fatigue, essoufflement, etc). Une personne sur cinq meurt dans l’année du diagnostic par décompensation cardiaque – CHAANINE et al. (2010). Actuellement, le traitement repose sur une trithérapie à base d’inhibiteur de l’enzyme de conversion de l’angiotensine (IECA), de β-bloquant et de diurétique. Ce traitement réduit les symptômes et allonge l’espérance de vie, mais reste imparfait et incapable de restaurer efficacement le fonctionnement cardiaque.
2. Intérêt de la thérapie génique Grâce à la compréhension des mécanismes moléculaires impliqués dans le dysfonctionnement de la cellule cardiaque lors d’insuffisance cardiaque, la thérapie génique est aujourd’hui une alternative thérapeutique en développement. Le principe de la thérapie génique dans le traitement de l’insuffisance cardiaque est le ciblage des mécanismes moléculaires permettant de réduire ou d’inverser les modifications subies par le cœur. Comme dit précédemment, l’insuffisance cardiaque a plusieurs causes. De fait, les

78
solutions de thérapie génique devront s’adapter à l’étiologie de la maladie. Cependant, une fois la maladie installée, certaines modifications moléculaires s’opèrent et sont identiques quelle que soit l’étiologie. C’est donc sur ces mécanismes que se concentrent actuellement les stratégies de recherche – VINGE et al. (2008).
a. Les modèles animaux De nombreux modèles animaux sont utilisés pour comprendre et analyser les mécanismes mis en place lors d’insuffisance cardiaque ainsi que pour étudier les solutions thérapeutiques – ZARAGOZA et al. (2011).
i. Le rat Le rat possède de nombreuses qualités en tant que modèle d’expérimentation, pour la plupart identiques à celles du modèle murin (rapidité de reproduction, facilité d’entretien, sujets en grand nombre disponibles, etc), mais sa plus grande taille facilite les chirurgies et autres contrôles macroscopiques du cœur.
ii. La souris La taille de la souris constitue un inconvénient majeur dans l’étude de l’insuffisance cardiaque. Cependant, la possibilité de créer des sujets génétiquement modifiés met la souris au premier plan des études en thérapie génique. Ces modèles, bien que très couramment utilisés en recherche, présentent de grandes différences avec l’Homme : morphologie du cœur, vascularisation, fréquence, besoin en oxygène ce qui les rend inaptes à reproduire certains aspects de la maladie et de son développement.
iii. Le cochon Le premier modèle animal de grande taille à avoir été utilisé fut le chien, mais le cochon présente des caractéristiques qui en font un meilleur modèle, en particulier son schéma de vascularisation très proche de celui de l’Homme. Cependant, comme tous les modèles de grande taille, son entretien provoque un surcoût par rapport aux autres modèles.
iv. Le lapin Le lapin représente un compromis entre les rongeurs et le cochon. Il présente aussi des caractéristiques comparables à l’Homme comme la composition en protéines de la membrane du réticulum sarcoplasmique. De plus, il existe un modèle naturel de lapin présentant une insuffisance cardiaque à la suite d’un infarctus du myocarde : le lapin WHHLMI.
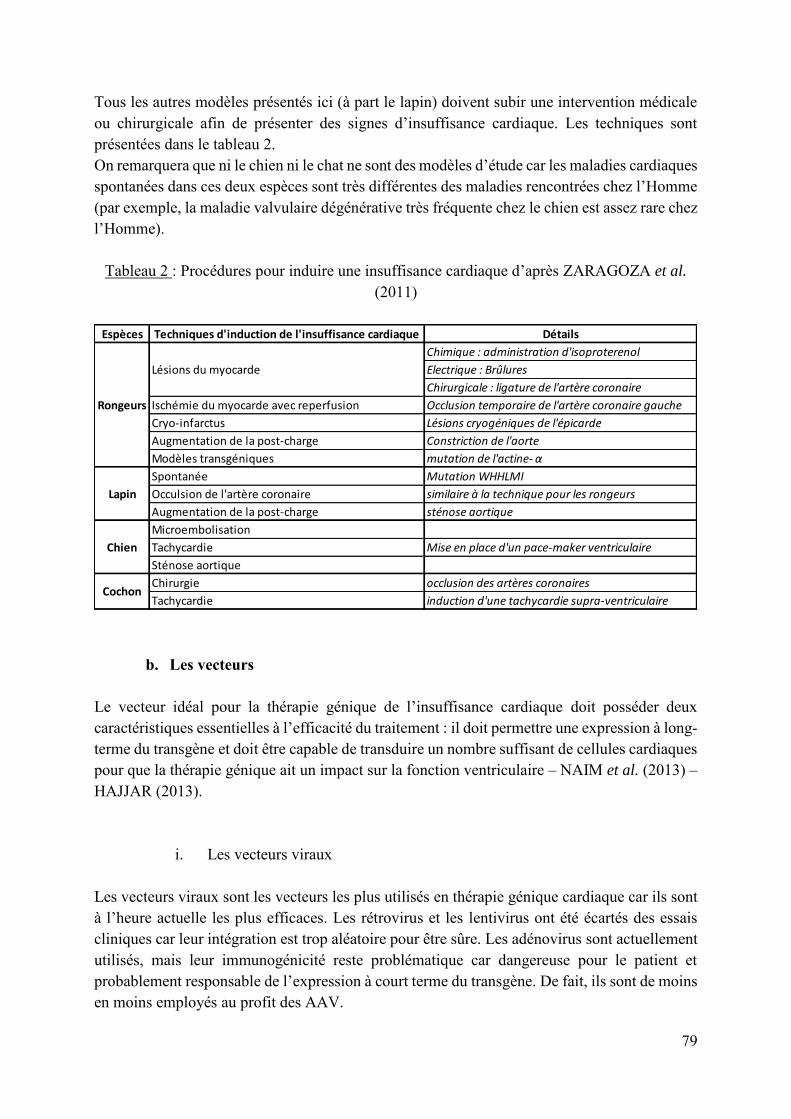
79
Tous les autres modèles présentés ici (à part le lapin) doivent subir une intervention médicale ou chirurgicale afin de présenter des signes d’insuffisance cardiaque. Les techniques sont présentées dans le tableau 2. On remarquera que ni le chien ni le chat ne sont des modèles d’étude car les maladies cardiaques spontanées dans ces deux espèces sont très différentes des maladies rencontrées chez l’Homme (par exemple, la maladie valvulaire dégénérative très fréquente chez le chien est assez rare chez l’Homme).
Tableau 2 : Procédures pour induire une insuffisance cardiaque d’après ZARAGOZA et al. (2011)
b. Les vecteurs Le vecteur idéal pour la thérapie génique de l’insuffisance cardiaque doit posséder deux caractéristiques essentielles à l’efficacité du traitement : il doit permettre une expression à long-terme du transgène et doit être capable de transduire un nombre suffisant de cellules cardiaques pour que la thérapie génique ait un impact sur la fonction ventriculaire – NAIM et al. (2013) – HAJJAR (2013).
i. Les vecteurs viraux Les vecteurs viraux sont les vecteurs les plus utilisés en thérapie génique cardiaque car ils sont à l’heure actuelle les plus efficaces. Les rétrovirus et les lentivirus ont été écartés des essais cliniques car leur intégration est trop aléatoire pour être sûre. Les adénovirus sont actuellement utilisés, mais leur immunogénicité reste problématique car dangereuse pour le patient et probablement responsable de l’expression à court terme du transgène. De fait, ils sont de moins en moins employés au profit des AAV.
Espèces Techniques d'induction de l'insuffisance cardiaque Détails
Chimique : administration d'isoproterenol
Electrique : Brûlures
Chirurgicale : ligature de l'artère coronaire
Ischémie du myocarde avec reperfusion Occlusion temporaire de l'artère coronaire gauche
Cryo-infarctus Lésions cryogéniques de l'épicarde
Augmentation de la post-charge Constriction de l'aorte
Modèles transgéniques mutation de l'actine- α
Spontanée Mutation WHHLMI
Occulsion de l'artère coronaire similaire à la technique pour les rongeurs
Augmentation de la post-charge sténose aortique
Microembolisation
Tachycardie Mise en place d'un pace-maker ventriculaire
Sténose aortique
Chirurgie occlusion des artères coronaires
Tachycardie induction d'une tachycardie supra-ventriculaire
Rongeurs
Lésions du myocarde
Lapin
Chien
Cochon

80
Certains sérotypes des AAV ont un tropisme pour les cellules cardiaques, en particulier les sérotypes 1, 6, 8 et 9. Cependant, ce tropisme n’est que partiel et les AAV infectent en même temps d’autres tissus comme le foie. Des modifications de la capside visent à réduire ces infections erratiques. L’expression du transgène est stable au cours du temps, mais une partie de la population présente des anticorps neutralisants ce qui les exclut des essais cliniques – NAIM et al. (2013).
ii. Les vecteurs non-viraux L’utilisation de vecteurs non-viraux dans la thérapie génique de l’insuffisance cardiaque n’en est qu’à ses débuts. De nombreuses pistes sont étudiées, mais aucune ne présente assez d’avantages pour être développée au-delà du stade de l’expérimentation animale. Le problème majeur est le faible taux de transfection et une expression transitoire du transgène – NAIM et
al. (2013).
iii. Ciblage et régulation de l’expression Afin de renforcer ou de créer un tropisme pour les cellules cardiaques, différents promoteurs peuvent être insérés dans la cassette génique tels que les promoteurs de la chaîne lourde de la myosine α, de la chaîne légère de la myosine kinase 2 ou de la troponine T. Certains promoteurs (comme les promoteurs des facteurs induits par l’hypoxémie) permettent de réguler l’expression du transgène uniquement lors de stress ou d’hypoxémie du myocarde.
c. Les modalités d’administration
i. Perfusion via l’artère ou la veine coronaire La cathétérisation de l’artère coronaire (figure 24, A) est une technique bien maîtrisée, peu invasive et plutôt sûre. Elle permet une répartition homogène du vecteur dans le myocarde, mais la vitesse de circulation du sang n’autorise qu’un court temps de contact. L’occlusion de la veine coronaire est possible par ballonisation et permet alors d’augmenter le temps de séjour du vecteur dans le cœur (figure 24, B). Il est aussi possible d’inverser la technique en injectant le vecteur dans la veine coronaire et en obstruant l’artère. Cette modalité est plus sûre pour les patients présentant une maladie coronarienne. Un by-pass peut aussi être mis en place durant l’intervention : le cœur est alors isolé du reste de la circulation et le vecteur peut rester en contact plus longtemps. De plus, l’ischémie cardiaque provoquée augmente la transfection. Des études sur le mouton ont prouvé l’efficacité de cette méthode par rapport aux autres car elle réduit fortement l’expression extracardiaque du transgène, mais le risque d’œdème du myocarde et d’hémorragie est non négligeable – NAIM et al. (2013).
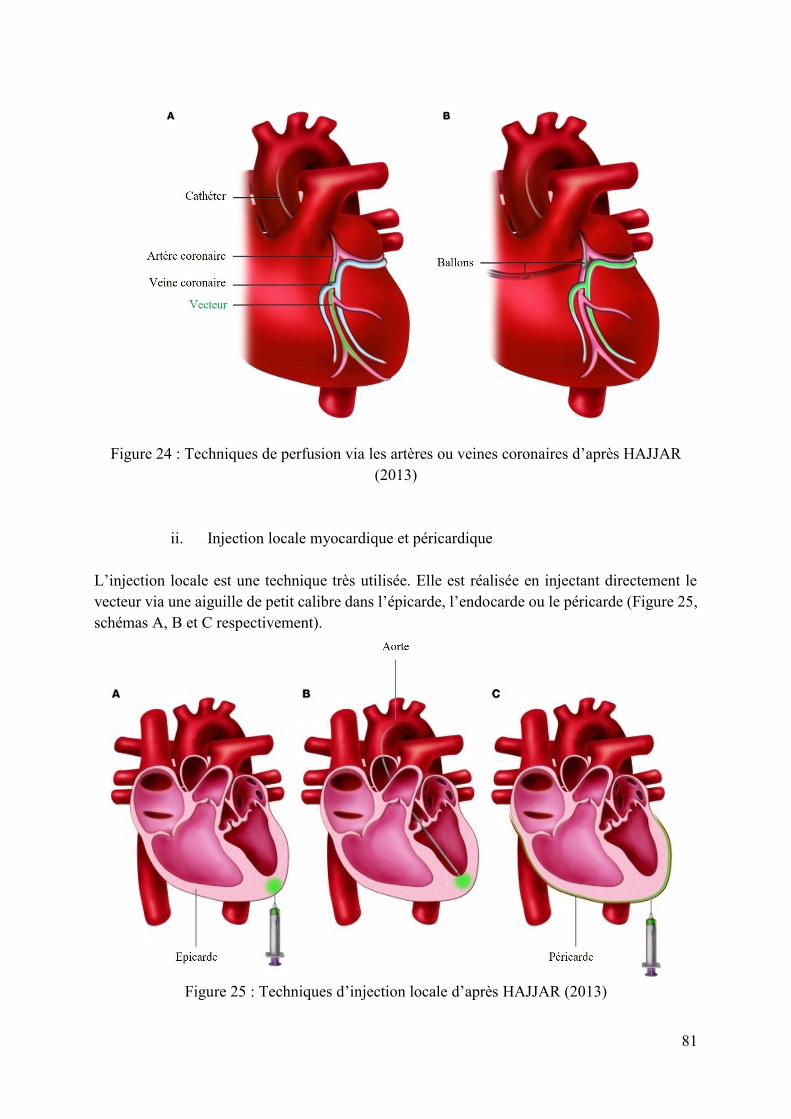
81
Figure 24 : Techniques de perfusion via les artères ou veines coronaires d’après HAJJAR
(2013)
ii. Injection locale myocardique et péricardique L’injection locale est une technique très utilisée. Elle est réalisée en injectant directement le vecteur via une aiguille de petit calibre dans l’épicarde, l’endocarde ou le péricarde (Figure 25, schémas A, B et C respectivement).
Figure 25 : Techniques d’injection locale d’après HAJJAR (2013)

82
Cette technique présente trois avantages majeurs : elle permet de traverser la barrière endothéliale, de minimiser le contact avec les anticorps présents dans le sang et de réduire l’expression extracardiaque du transgène. La méthode la plus simple et la plus invasive consiste à réaliser l’injection lors d’une thoracotomie : elle permet cependant d’observer directement le lieu de l’injection (en particulier, en périphérie d’un infarctus) (Figure 25, A). Il est aussi possible de réaliser une injection via un cathéter possédant une aiguille rétractable. Une surveillance par imagerie est alors nécessaire (Figure 25, B). L’injection péricardique permet de mettre en contact le vecteur avec toute la surface du cœur. (Figure 25, C) Elle est réalisable par voie sous-xyphoïdienne sous contrôle échographique (par exemple, par une sonde intravasculaire). Cependant, la transfection ne se fait que dans les cellules superficielles du myocarde et il existe une expression extracardiaque, probablement due à la résorption rapide du fluide intra-péricardique par le système lymphatique. Il est possible d’administrer en parallèle de l’intervention des agents pharmacologiques (comme des enzymes protéolytiques ou du polyéthylènimine) dont on a prouvé qu’ils augmentaient la profondeur de pénétration du vecteur dans le myocarde. De fait, cette méthode est encore peu utilisée – NAIM et al. (2013).
iii. Microbulles et ultrasons Une méthode émergente est l’utilisation de microbulles (cf. Partie 1, IV, 2, b, vi). Il est possible d’insérer un vecteur viral dans une microbulle, de l’injecter par voie intraveineuse et de la faire exploser au niveau du cœur par application d’ultrasons. Ainsi, on peut protéger le vecteur des anticorps circulants, le diriger vers le tissu cible et augmenter la transfection par l’application des ultrasons (créations de pores). L’efficacité de l’utilisation des microbulles a été prouvée dans de nombreuses études sur des animaux d’expérimentation – CHEN et al. (2013). Cependant, cette technique crée des pores dans l’endothélium et le risque associé à ses lésions permanentes ou transitoires n’est pas connu – HAJJAR (2013). Pour l’instant, l’utilisation de microbulles est cantonnée aux modèles animaux jusqu’à ce que la sécurité de cette technique ait été évaluée.
d. Les gènes cibles Grâce à la connaissance des mécanismes moléculaires impliqués dans le fonctionnement de la cellule cardiaque, il a été possible d’identifier des protéines capables d’améliorer, voire de rétablir le fonctionnement normal de la cellule cardiaque lors d’insuffisance cardiaque.

83
i. Le système β-adrénergique On a vu plus haut que, lors d’insuffisance cardiaque, les cellules sont moins sensibles aux signaux du système sympathique. Cette désensibilisation se fait via une diminution du nombre de récepteurs β2 adrénergiques (β-AR) à la surface de la cellule. Cela provoque une perte d’inotropisme. Il y a cinquante ans, on a décidé de lutter contre cette perte d’inotropisme avec des molécules β agonistes. Cependant, ces molécules avaient plus d’effets délétères que bénéfiques à long terme, d’où l’arrêt de ce traitement et l’utilisation de β bloquants – HO et al. (2010). Ces dernières années, on a étudié la surexpression du gène codant pour les β-AR chez les modèles animaux et on a montré qu’elle permettait un travail du cœur plus efficace – KAIROUZ et al. (2012). Une autre cible est une des molécules intervenant dans le rétrocontrôle négatif, le GRK2 (pour récepteur kinase couplé à la protéine G). Cette molécule phosphoryle les β-AR ce qui empêche la liaison avec l’adrénaline ou la noradrénaline – RENGO et al. (2011). Un peptide, le βARKct, est capable d’inhiber le GKR2. Des études chez les rongeurs ont montré que l’insertion du transgène codant pour ce peptide améliorait la contraction du ventricule gauche – RENGO et
al. (2009). L’adényl-cyclase de type VI est aussi une molécule capable d’inhiber le GRK2. Une étude chez le cochon a montré une amélioration de la fonction cardiaque avec une augmentation de la capacité à régénérer l’AMPc (adénosine monophosphate cyclique) qui est un messager intracellulaire impliqué dans la contraction musculaire – NAIM et al. (2013).
ii. Circulation du calcium Lors d’insuffisance cardiaque, la circulation du calcium dans les cellules cardiaques est altérée : on remarque une moindre activité de la SERCA2a (Sarco-Endoplasmic Reticulum Ca2+-ATPase) qui est une pompe calcique située dans la membrane du réticulum sarcoplasmique permettant la rentrée du calcium intracellulaire dans le réticulum sarcoplasmique lors de la diastole. Il a été démontré chez tous les modèles animaux que la transduction des cellules cardiaques avec le gène codant pour la SERCA2a permettait d’améliorer la contractilité cardiaque, de rétablir les déformations subies par le cœur et sa consommation énergétique, de diminuer le risque d’arythmies et d’augmenter le flux sanguin dans le système coronaire grâce à l’activation de la synthase endothéliale d’oxyde nitrique par les cellules endothéliales des artères coronaires – KAIROUZ et al. (2012). Trois autres mécanismes permettent d’augmenter l’activité de la SERCA2a – NAIM et al. (2013):
Le phospholamban (PLB), lorsqu’il est phosphorylé, augmente l’activité de la SERCA 2a. Il a été montré lors d’insuffisance cardiaque que le PP1 (protéine phosphatase 1), inhibiteur du phospholamban par déphosphorylation, était surexprimé et que le PP1-I,
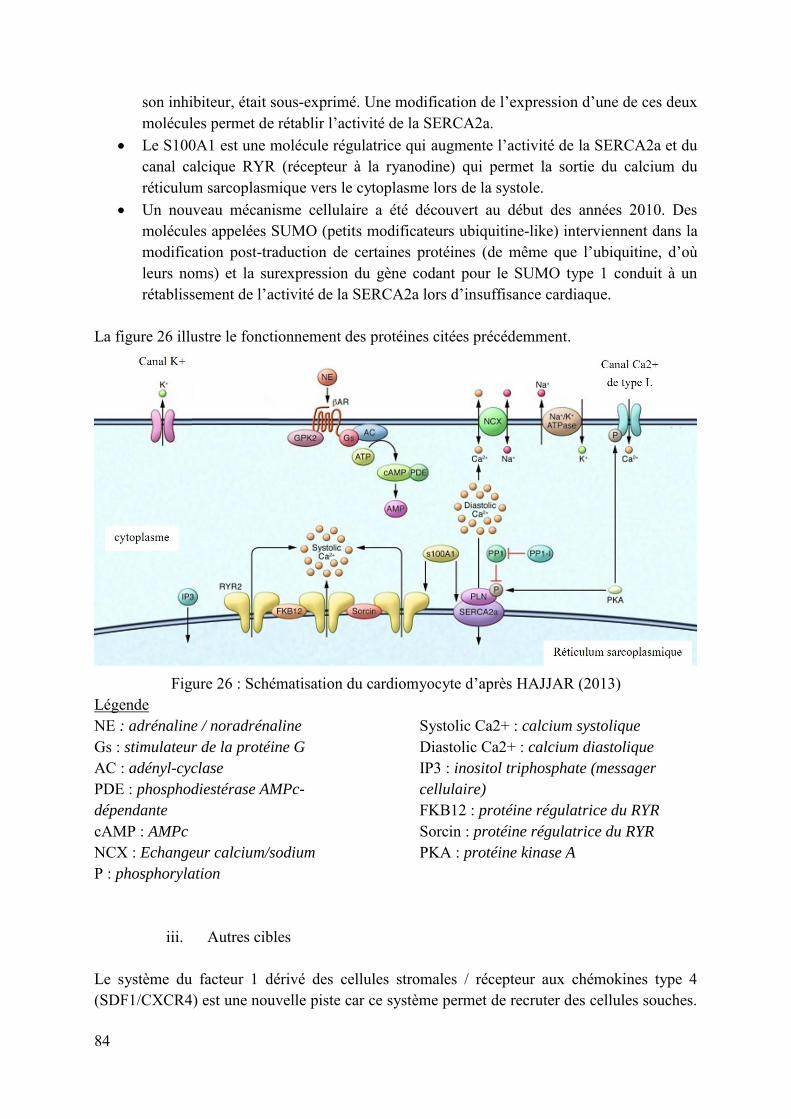
84
son inhibiteur, était sous-exprimé. Une modification de l’expression d’une de ces deux molécules permet de rétablir l’activité de la SERCA2a.
Le S100A1 est une molécule régulatrice qui augmente l’activité de la SERCA2a et du canal calcique RYR (récepteur à la ryanodine) qui permet la sortie du calcium du réticulum sarcoplasmique vers le cytoplasme lors de la systole.
Un nouveau mécanisme cellulaire a été découvert au début des années 2010. Des molécules appelées SUMO (petits modificateurs ubiquitine-like) interviennent dans la modification post-traduction de certaines protéines (de même que l’ubiquitine, d’où leurs noms) et la surexpression du gène codant pour le SUMO type 1 conduit à un rétablissement de l’activité de la SERCA2a lors d’insuffisance cardiaque.
La figure 26 illustre le fonctionnement des protéines citées précédemment.
Figure 26 : Schématisation du cardiomyocyte d’après HAJJAR (2013)
Légende NE : adrénaline / noradrénaline Gs : stimulateur de la protéine G AC : adényl-cyclase PDE : phosphodiestérase AMPc-
dépendante
cAMP : AMPc NCX : Echangeur calcium/sodium P : phosphorylation
Systolic Ca2+ : calcium systolique Diastolic Ca2+ : calcium diastolique IP3 : inositol triphosphate (messager
cellulaire) FKB12 : protéine régulatrice du RYR Sorcin : protéine régulatrice du RYR
PKA : protéine kinase A
iii. Autres cibles Le système du facteur 1 dérivé des cellules stromales / récepteur aux chémokines type 4 (SDF1/CXCR4) est une nouvelle piste car ce système permet de recruter des cellules souches.

85
De plus, il a été démontré chez des souris que ces deux molécules avaient des effets individuels sur le cœur. Des études sont en cours pour étudier l’intérêt de ce système (ou de ces molécules individuellement) dans le traitement de l’insuffisance cardiaque par thérapie génique – NAIM et al. (2013).
3. Conclusion Le traitement actuel de l’insuffisance cardiaque est efficace, mais ne permet pas d’inverser les dommages subis par le cœur. La thérapie génique permettrait cela. En ce moment, trois essais cliniques principaux sont en cours : un essai de phase I étudiant le système AAV6 – SERCA2a par rapport à un placebo, et deux essais de phase II étudiant le système AAV1 – SERCA2a. Les essais sont prometteurs. D’autres essais cliniques impliquant d’autres cibles sont en cours (par exemple le système Adénovirus 5 – Adenyl cyclase type VI) – NAIM et al. (2013). La thérapie génique pour l’insuffisance cardiaque commence à être assez développée pour qu’on puisse espérer un traitement efficace (ou en tous cas, plus efficace que le traitement actuel) dans les décennies qui viennent.

86

87
III. Les maladies infectieuses : le SIDA On trouve aussi parmi les nouvelles cibles de la thérapie génique, les maladies infectieuses au premier rang desquelles le Syndrome d’ImmunoDéficience Acquise ou SIDA provoqué par l’infection par le virus de l’immunodéficience humaine (VIH).
1. L’infection par le VIH et ses conséquences Le VIH a été découvert au début des années 80. Depuis sa découverte, on estime à plus de 65 millions le nombre de personnes infectées par le virus à travers le monde et à plus de 32 millions le nombre de décès liés au SIDA. Selon l’OMS, 34 millions de personnes sont aujourd’hui infectées et chaque année, 2,5 millions de personnes contractent la maladie. La transmission du virus se fait par relation sexuelle, échange de sang (transfusion ou échange de seringues chez les toxicomanes) et de la mère à l’enfant (pendant la gestation ou par l’allaitement). La maladie se déroule en trois phases : virémie aigüe, phase de latence (qui peut durer plusieurs années) et SIDA qu’on décline en plusieurs niveaux selon le comptage des lymphocytes T CD4+. La personne décède le plus souvent d’une affection secondaire et plus rarement d’un cancer ou d’une autre forme de l’infection par le VIH (neuropathie) – COSTIN (2007).
a. Cycle de réplication Le VIH est un lentivirus, de la famille des retroviridae. Il infecte les cellules porteuses du récepteur CD4, principalement les lymphocytes T CD4+, les macrophages (et certaines cellules du système nerveux central) et provoque ainsi une immunodéficience. On distingue aujourd’hui deux sous-types : le VIH-1 qui est le plus répandu et le VIH-2 qui diffèrent par une protéine régulatrice. Cependant, il existe de nombreux variants du VIH dus à la capacité du virus à muter lors de l’étape de transcription inverse (la transcriptase inverse réalise environ une erreur toutes les 104 bases azotées). Le virion est enveloppé et contient deux copies de son génome sous forme d’ARN simple brin d’une dizaine de kilobases ainsi que plusieurs protéines virales. La figure 27 montre le cycle de réplication du VIH. Entre les deux phases, on décrit une phase de latence pendant laquelle le provirus ne s’exprime pas. Cette latence a une durée variable qui peut aller jusqu’à plusieurs années.
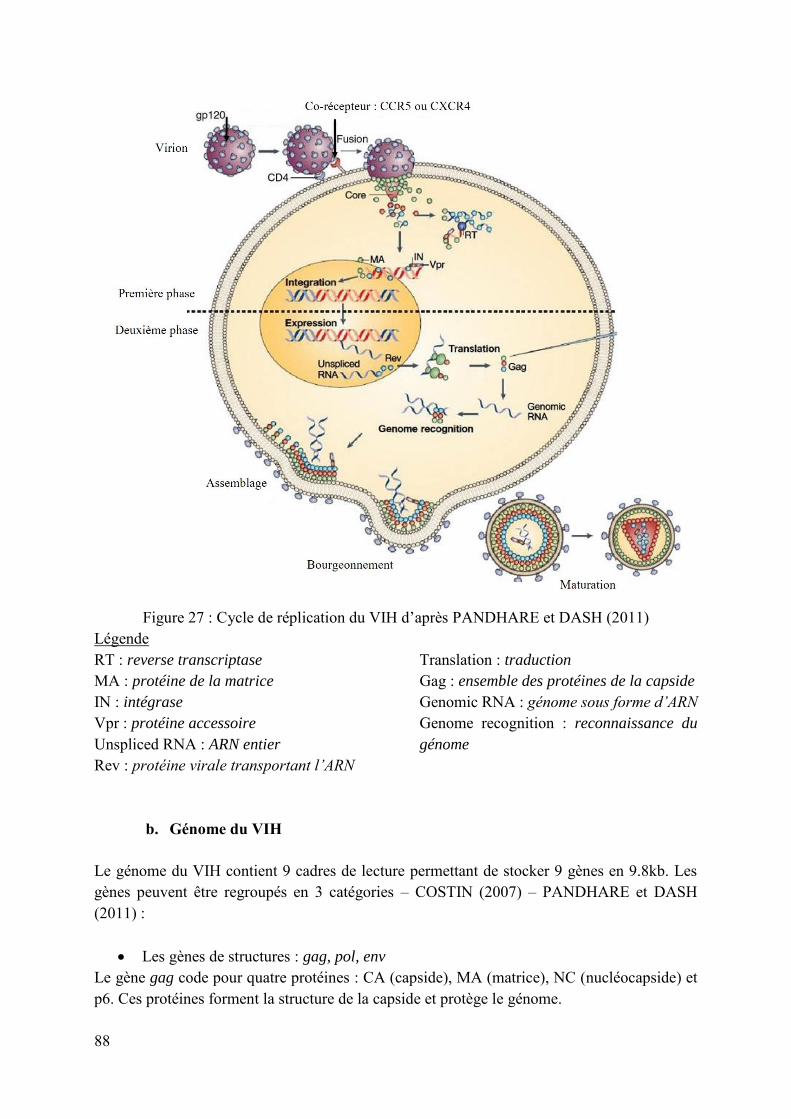
88
Figure 27 : Cycle de réplication du VIH d’après PANDHARE et DASH (2011)
Légende RT : reverse transcriptase
MA : protéine de la matrice IN : intégrase Vpr : protéine accessoire Unspliced RNA : ARN entier Rev : protéine virale transportant l’ARN
Translation : traduction Gag : ensemble des protéines de la capside Genomic RNA : génome sous forme d’ARN
Genome recognition : reconnaissance du
génome
b. Génome du VIH Le génome du VIH contient 9 cadres de lecture permettant de stocker 9 gènes en 9.8kb. Les gènes peuvent être regroupés en 3 catégories – COSTIN (2007) – PANDHARE et DASH (2011) :
Les gènes de structures : gag, pol, env Le gène gag code pour quatre protéines : CA (capside), MA (matrice), NC (nucléocapside) et p6. Ces protéines forment la structure de la capside et protège le génome.

89
Le gène pol code pour trois enzymes : RT (reverse transcriptase), IN (intégrase) et PR (protéase). La protéase permet la maturation des protéines virales afin de les rendre fonctionnelles. Le gène env code pour deux glycoprotéines de surface qui permettent l’attachement puis la fusion de la membrane virale avec la membrane cellulaire : SU (surface) ou gp120 et TM (transmembranaire) ou gp41
Les gènes régulateurs :rev et tat La protéine Rev permet la sortie du noyau cellulaire de l’ARN entier, qui va devenir le génome des nouveaux virions, via une liaison avec une partie de l’ARN viral appelée élément de réponse au Rev (ou RRE). La protéine Tat est un activateur de la transcription qui se lie sur le domaine TAR (transactivating region) et assure que les ARN produits sont entiers.
Les gènes accessoires : nef, vpr, vpu et vif La protéine Nef augmente l’infectiosité du VIH. C’est un perturbateur des flux de potassium. La protéine Vpr permet au VIH d’infecter les cellules quiescentes en agissant comme un facteur de transport nucléocytoplasmique. La protéine Vpu réduit la production de récepteurs CD4 via une dégradation des ARNm par des molécules ubiquitine-like. La protéine Vif semble essentielle à la réplication du virus dans les cellules de la lignée blanche, peut-être à cause de la reconnaissance d’un facteur intracellulaire. La figure 28 montre l’agencement de ces différents gènes dans le génome viral.
Figure 28 : Organisation du génome du VIH d’après COSTIN (2007)
2. Intérêt de la thérapie génique
a. Traitement actuel Le traitement actuel repose sur l’administration combinée de plusieurs molécules visant différents mécanismes du cycle viral. Il existe une trentaine de molécules disponibles inhibant quatre phases du cycle : des inhibiteurs de la reverse transcriptase, de la protéase et de l’intégrase ainsi que deux molécules capables d’empêcher les nouveaux virions d’infecter des cellules (modifications des protéines de surface gp120 et gp41).

90
Le plus souvent, on associe en trithérapie :
- Un inhibiteur nucléosidique de la reverse transcriptase - Un inhibiteur non nucléosidique de la reverse transcriptase - Un inhibiteur de la protéase
On a remarqué que des résistances apparaissaient très rapidement lors de monothérapie – ENGELMAN et CHEREPANOV (2012). Ce traitement antirétroviral appelé par les anglo-saxons « Highly active anti-retroviral therapy » ou HAART a révolutionné la vie des patients infectés, mais ne va pas sans un certain nombre d’inconvénients. Le traitement antirétroviral peut être toxique et nécessite une adaptation des dosages de chaque molécule à chaque patient selon sa sensibilité. Cela conduit parfois à une mauvaise observance du traitement d’autant plus qu’il s’agit d’un traitement à vie. Malheureusement, malgré la trithérapie, des résistances sont toujours observées et conduisent à un échec du traitement. Il convient de préciser que le traitement n’est pas curatif, mais vise à limiter la charge virale – ROSSI et al. (2007). Une nouvelle approche est donc nécessaire et la thérapie génique a été développée dans cette optique.
b. Modèles animaux Le VIH pose problème quant à l’identification d’un modèle animal convenable. Parmi tous les animaux d’expérimentation, seul le chimpanzé permet au VIH de se répliquer, mais le chimpanzé ne développe pas de SIDA à la suite de l’infection – HAIGWOOD (2009). Les études sur le VIH ont donc d’abord été réalisées en étudiant les virus de la même famille et en particulier, le virus de l’immunodéficience simienne (SIV) chez différents primates non humains – LINT et al. (2013). Cependant, les primates non humains sont rarement disponibles pour la recherche et donc d’autres espèces ont été utilisées comme le chat et son FIV (Feline immuodeficiency virus ou virus de l’immunodéficience féline) ou les bovins et leur BIV (Bovine immuodeficiency virus ou virus de l’immunodéficience bovine). Mais, il existe des différences importantes entre les autres lentivirus et le VIH ainsi que certaines caractéristiques de la physiopathologie du virus qui empêchaient l’étude de nouveaux traitements. Il est aussi à noter qu’il n’existe pas de virus de l’immunodéficience murine. On a donc cherché à créer un modèle animal capable d’être infecté par le VIH. La souris étant un modèle d’étude privilégié, les chercheurs ont créé une souris « humanisée ». Il existe plusieurs types de souris « humanisées » :
- des souris qui possèdent certains trangènes humains, mais un génome principalement murin,
- des souris immunodéficientes transplantées avec des cellules ou des tissus humains (comme le thymus ou le foie),
- des souris immunodéficientes greffées avec des précurseurs hématopoïétiques des cellules CD34+,

91
- une combinaison des trois types précédents. Le modèle le plus intéressant est la souris greffée avec des précurseurs hématopoïétiques des cellules CD34+ car ces souris produisent des cellules lymphoïdes humaines capables d’être infectées par le VIH – DENTON et GARCIA (2011). Une autre solution a été de créer une chimère entre le SIV et le VIH afin que ce nouveau virus soit capable d’infecter un primate non humain, mais qu’il provoque les mêmes symptômes que le VIH et possède les mêmes mécanismes physiopathologiques. On a donc créé le SHIV qui possède l’enveloppe du VIH ainsi que les gènes TAT et REV et qui est capable d’infecter le macaque, un primate non humain très utilisé en expérimentation. Ce virus provoque une perte majeure des lymphocytes CD4+ ainsi qu’une immunodéficience. Cette chimère permet l’étude de molécules inhibant la fusion membranaire et les gènes TAT et REV. Cependant, les mécanismes physiopathologiques et le cycle de réplication viral ne sont pas identiques à ceux du VIH – TROBRIDGE et KIEM (2010). De nouvelles chimères ont été créées afin de pouvoir évaluer d’autres molécules inhibant d’autres mécanismes – WADFORD et al. (2014).
c. Le « patient berlinois » En 2007, un patient atteint d’une leucémie due à l’infection par le VIH a été transplanté avec la moelle osseuse d’un donneur mutant pour le gène du co-récepteur CCR5 (mutation CCR5Δ32 : délétion de 32 paires de bases conduisant à un changement de cadre de lecture et à la production d’une protéine non fonctionnelle). Cette greffe a conduit à l’élimination du virus chez ce patient malgré l’arrêt du traitement antirétroviral. Or, on sait que la plupart des souches de VIH ont besoin du récepteur CCR5 afin de pouvoir fusionner avec les cellules cibles. On a donc supposé que la mutation CCR5Δ32 ne permettait pas au virus de détourner le récepteur. Transférer cette mutation a donc protégé les nouvelles cellules greffées de l’infection par le VIH. Ce traitement de thérapie cellulaire équivaut à une thérapie génique ex vivo où on aurait transféré le gène CCR5Δ32 aux cellules de la moelle osseuse prélevées sur le patient et qu’on aurait réinjectées. Si ce patient a été effectivement guéri du SIDA, il a dû subir une ablation totale de la moelle osseuse par destruction de ses cellules souches par chimiothérapie, puis une greffe, ce qui rend le protocole trop lourd et dangereux pour pouvoir être appliqué à tous les patients. De plus, la mutation est rare et il serait impossible de trouver des donneurs portant cette mutation, dont les cellules seraient compatibles avec tous les patients atteints du SIDA – ALLERS et al. (2011). Cet exemple illustre cependant que la thérapie génique est une option valable dans le traitement de l’infection par le VIH.
3. Utilisation de la thérapie génique
a. Cibles de la thérapie génique Il existe deux types de cibles : les cibles virales et les cibles endogènes – BURNETT et al. (2012).

92
Les cibles virales sont tous les éléments viraux nécessaires à la réplication du virus. Cependant, ces cibles peuvent muter et devenir résistantes au traitement. On peut classer les inhibiteurs de ces cibles en trois catégories : inhibition des étapes précédant l’intégration du génome viral (Classe I), inhibition de l’expression virale (Classe II) et inhibition de l’assemblage ou du bourgeonnement (Classe III). Les cibles endogènes sont toutes les molécules cellulaires dont le virus a besoin pour sa réplication. Au contraire des cibles virales, elles sont moins à même de muter et leurs modifications peuvent protéger la cellule contre l’infection par le VIH. Mais, les cibles endogènes doivent être non essentielles à la survie de la cellule. C’est pourquoi, par exemple, bien que le récepteur CD4 soit une cible intéressante car sa mutation empêcherait le VIH de pénétrer dans les cellules, ce récepteur a une fonction importante dans le système immunitaire. Il est donc impossible de le prendre pour cible.
b. Modalités d’administration La thérapie génique anti-VIH se fait principalement ex vivo. Les modalités d’administration visent évidemment les lymphocytes T CD4+, mais aussi leurs précurseurs, les cellules hématopoïétiques CD34+. En effet, la thérapie génique anti-VIH ne cible pas les cellules productrices de virions qui vont mourir, mais les cellules indemnes ou en phase de latence afin de les protéger. Le problème qui est rencontré avec l’utilisation des lymphocytes CD4+ est qu’ils sont déjà spécifiques de certains antigènes. Ne traiter que cette population pourrait conduire à un appauvrissement de la mémoire antigénique essentielle au fonctionnement du système immunitaire. Les cellules hématopoïétiques ont l’avantage d’être encore naïves au niveau immunitaire (elles ne possèdent que les récepteurs de la reconnaissance du soi), elles sont donc plus attractives. De plus, en traitant ces cellules, on rend tous les nouveaux lymphocytes à venir résistants au VIH – ROSSI et al. (2007) – KITCHEN et al. (2011) – ALLERS et al. (2011).
c. Techniques de la thérapie génique anti-VIH
i. Enzymes spécifiques Une des stratégies de thérapie génique consiste à modifier les gènes grâce à des enzymes capables de découper des gènes spécifiques. Lors de la réparation de l’ADN à la suite de cette rupture, des mutations apparaissent. On peut diriger ces enzymes contre le génome viral ou contre des cibles endogènes non essentielles à la survie de la cellule afin de rendre impossible l’expression de ces gènes. Trois enzymes sont actuellement à l’étude : les enzymes en doigts de zinc, les TALEN et les CRISPR – MANJUNATH et al. (2013).

93
Les enzymes en doigts de zinc sont celles pour lesquelles les connaissances sont les plus avancées. Elles ont été étudiées dans la rupture des gènes CCR5, CXCR4 et de l’ADN proviral intégré dans le génome cellulaire. Le récepteur CCR5 est le co-récepteur le plus souvent utilisé par les souches de VIH-1. Il n’est pas essentiel au fonctionnement cellulaire ce qui en fait une cible idéale pour conférer une résistance aux cellules vis-à-vis de l’infection par le VIH. Des études in vitro ont montré que des enzymes en doigts de zinc spécifiques du gène CCR5 étaient capables de couper ce gène dans 50% des cellules exposées et avec moins de 5% d’erreur. En effet, cette enzyme peut aussi, par erreur, couper le gène codant pour le récepteur CCR2. Il existe aussi des souches de VIH-1 qui utilisent comme co-récepteur le récepteur CXCR4. Des enzymes en doigts de zinc spécifiques du gène CXCR4 ont été créées. Cependant, pour que la résistance à l’infection soit complète, des chercheurs ont créé des enzymes en doigts de zinc spécifiques du gène CXCR4 qui insèrent aussi un gène codant pour un inhibiteur à la place du gène CCR5. Les deux co-récepteurs sont ainsi inutilisables ce qui bloque l’entrée de toutes les souches de VIH-1 connues. Afin de guérir les malades, empêcher l’entrée du virus ne suffit pas, il faut aussi supprimer le génome viral des cellules infectées, mais en phase de latence. Des enzymes en doigts de zinc ont été créées dans ce but avec différentes cibles : les gènes structuraux (tous ou un seul) et les séquences LTR 5’ et 3’ qui servent d’éléments régulateurs. Les résultats in vitro sont encourageants, mais ces cellules en latence sont rares et donc difficiles à cibler in vivo. Les enzymes en doigts de zinc peuvent être délivrées aux cellules par des adénovirus, des lentivirus ou par nucléofection. Cependant, l’expression à long terme provoquée par l’intégration du transgène lors de l’utilisation d’un lentivirus semble être contre-productive car elle augmenterait le nombre d’erreurs commises par l’enzyme. Les TALEN sont également à l’étude. Lors d’une étude in vitro comparative, les TALEN ont montré la même capacité à couper le gène CCR5 (environ 45% des cellules), mais ont prouvé une cytotoxicité moindre associée à un taux d’erreurs inférieur. Il n’a pas été possible de délivrer les TALEN par un vecteur lentivirus, mais cela reste possible avec un adénovirus malgré la taille importante du transgène. Des études sont nécessaires afin de déterminer la fiabilité des TALEN et si leur utilisation est sûre. Les CRISPR n’en sont qu’à leur tout début. Selon les premières études, leur capacité à couper spécifiquement le gène CCR5 est semblable à celles des TALEN et des enzymes en doigts de zinc, mais leur cytotoxicité est encore inférieure à celle des TALEN. Les CRISPR sont aussi plus faciles à produire que les deux autres enzymes, mais de nombreuses études seront nécessaires avant que les CRISPR ne supplantent les enzymes en doigts de zinc. Pour l’instant, deux essais cliniques de phases I et deux essais cliniques de phase I/II sont en cours. Ils utilisent tous un adénovirus transportant l’enzyme en doigts de zinc spécifique du gène CCR5 et l’étudient sur différentes populations – BURNETT et al. (2012). Les premiers résultats montrent que les cellules transfectées ex vivo sont inoffensives et persistent dans le temps. Cependant aucun résultat sur l’éradication du VIH ne pourra être montré tant que les

94
patients seront sous traitement antirétroviral dont l’arrêt pose des problèmes éthiques – MANJUNATH et al. (2013).
ii. Molécules d’ARN Ribozymes Les ribozymes ont été utilisés en thérapie génique anti-VIH pour éteindre l’expression des gènes viraux. Plusieurs gènes ont été pris pour cibles : TAT, REV et les séquences LTR – ROSSI et al. (2007). Trois essais cliniques utilisant un rétrovirus comme vecteur (deux sur des lymphocytes CD4+ et un sur des cellules CD34+) ont été réalisés. Ils ont prouvé la sécurité de l’utilisation du vecteur et des ribozymes ainsi que la faisabilité de l’expression d’un ribozyme par un transgène, mais aucun n’a montré une réelle efficacité thérapeutique du traitement (pas de différence significative avec les cellules infectées par le vecteur vide). ARN antisens Un essai clinique utilisant un trangène codant pour un ARN antisens dirigé contre l’ARN viral Env et un vecteur appelé VRX496 a été réalisé. Ce vecteur est un lentivirus modifié qui ne possède aucun gène viral, mais qui est capable de se répliquer uniquement dans les cellules déjà infectées par une souche sauvage du VIH-1. La thérapie génique a été réalisée ex vivo sur des lymphocytes CD4+. Mais, de nouveau, aucune amélioration du système immunitaire n’a été mise en évidence – BURNETT et al. (2012). Aptamères Deux aptamères spécifiques des régions RRE et TAR respectivement sont arrivés aux essais cliniques. Deux rétrovirus ont été utilisés comme vecteurs et la thérapie génique a été réalisée ex vivo sur des cellules CD34+. Aucun effet secondaire n’a été observé, mais le nombre de leucocytes exprimant les aptamères un an après la thérapie était très faible – KOHN et al. (1999). siARN et shARN L’interférence de l’ARN peut aussi être utilisée pour empêcher la réplication des virus en ciblant des ARNm viraux ou endogènes (particulièrement CCR5 et CXCR4). Les siARN et les shARN ont fait leurs preuves in vitro dans la thérapie génique anti-VIH. Ils diffèrent principalement par le fait que les siARN doivent être apportés par des vecteurs non viraux et ont une expression à court terme car ils ne peuvent pas être transcrits à partir d’ADN. Les shARN, au contraire, peuvent être insérés dans le génome cellulaire sous forme de transgène par des vecteurs viraux, ce qui rend leur expression plus ou moins stable dans le temps. Les essais cliniques ont pour l’instant démontré l’innocuité des deux techniques. Reste à voir leur efficacité in vivo : des essais pré-cliniques sont en cours sur des macaques et un essai clinique utilisant un shARN a été autorisé – SUBRAMANYA et al. (2010) – BURNETT et al. (2012).

95
iii. Inhibiteurs protéiques
La protéine C46 est un inhibiteur de la fusion membranaire. Elle est dérivée de la protéine virale gp41. Des essais cliniques utilisant un rétrovirus comme vecteur ont montré l’innocuité du traitement et l’expression de la protéine pendant plus d’un an. Les essais précliniques prouvent l’efficacité de cette protéine. De plus, elle peut être exprimée de façon à pouvoir être sécrétée et à pouvoir protéger les cellules environnantes. Cependant, l’action de la protéine C46 peut être inhibée par évolution et sélection des membranes virales – BURNETT et al. (2012) – CHUNG et al. (2013). On a découvert chez l’Homme et les autres espèces animales l’existence de facteurs de restriction capables d’inhiber le VIH. Deux facteurs sont particulièrement étudiés : APOBEC3G et TRIM. Les mécanismes conduisant à l’inhibition du VIH ne sont pas encore élucidés. Les essais précliniques utilisant un lentivirus comme vecteur et le facteur TRIM étant concluants, des essais cliniques sont en préparation. Au contraire, les études sont contradictoires au sujet du facteur APOBEC3G : certaines études tendent à prouver que ce facteur provoque la mutation du VIH et le rend plus rapidement résistant au traitement actuel – CHAN et al. (2014).
4. Conclusions Malgré les différentes stratégies développées, aucune n’a encore donné de résultats probants quant à son efficacité chez l’Homme. De plus en plus, les chercheurs se tournent vers une combinaison de ces différentes stratégies ce qui permettrait en plus de pallier une éventuelle mutation du virus qui le rendrait résistant à une des stratégies employées. Pour l’instant un seul essai clinique de ce genre a été réalisé. Les chercheurs ont utilisé un lentivirus pour délivrer un aptamère, un shARN et un ribozyme. La thérapie a été bien supportée par les quatre patients et les transgènes ont été exprimés pendant au moins 36 mois chez un des patients. Mais les preuves de l’efficacité de cette méthode ne sont pas encore faites – BURNETT et al. (2012) – CHUNG et al. (2013).

96

97
IV. La thérapie génique anticancéreuse Les cancers ont tous une base génétique, bien que moins de 5% d’entre eux soient véritablement héréditaires. La tumorisation des cellules provient d’un ensemble de mutations sur des gènes régulateurs clés ; la thérapie génique a donc sa place dans le traitement des cancers – RANKI et HEMMINKI (2010). La thérapie génique anticancéreuse a un objectif différent des autres applications de la thérapie génique ; elle vise à détruire les cellules cibles. Les techniques décrites précédemment sont détournées afin d’obtenir ce résultat – BARAR et OMIDI (2012). Nous décrirons ici les différentes stratégies développées en thérapie génique anticancéreuse.
1. Thérapie cytotoxique
a. Gènes induisant le suicide cellulaire L’une des premières stratégies de thérapie génique anti-cancéreuse à avoir été développée est la thérapie génique induisant le suicide cellulaire. Cette approche consiste à insérer dans les cellules cancéreuses un gène codant pour un toxique ou pour une enzyme capable de transformer une molécule inoffensive (pro-drogue) en une molécule toxique (drogue). Les deux systèmes de base sont la cytosine désaminase / 5-fluorocytosine et la thymidine kinase / ganciclovir – ZAROGOULIDIS et al. (2013).
Le gène de la cytosine désaminase est un gène bactérien présent chez Escherichia coli (E. coli). La cytosine désaminase transforme la 5-fluorocytosine en 5-fluorouracil. A partir de là, les enzymes endogènes vont créer trois métabolites toxiques. L’effet cytotoxique provient de trois mécanismes : liaisons des dérivés fluorés avec l’ARN, formation de complexes avec l’ADN et inhibition de la thymidylate synthase, impliquée dans la synthèse de la thymidine.
Le gène de la thimidine kinase provient du virus Herpès Simplex. La thymidine kinase convertit le ganciclovir en ganciclovir monophosphate qui est transformé par les enzymes endogènes en ganciclovir triphosphate. L’effet cytotoxique provient du fait que le ganciclovir triphosphate allonge la durée des phases S et G2 du cycle cellulaire, ce qui provoque la mort cellulaire par augmentation de la durée de division cellulaire.
Il existe d’autres systèmes enzymatiques comme la désoxycytidine kinase humaine / cytarabine ou la phosphorylase des bases puriques de E. coli / analogues d’une base purique qui ne seront pas développés ici – ARDIANI et al. (2012). Les vecteurs utilisés pour transfecter les cellules avec ces enzymes diffèrent selon la tumeur étudiée. Des études ont été réalisées avec des vecteurs viraux (comme les adénovirus) et des vecteurs non viraux (comme les nanoparticules de phosphate de calcium). Pour l’instant, aucun vecteur n’a prouvé sa supériorité par rapport aux autres.

98
Avec cette technique, on a observé un effet de proche en proche : des cellules à proximité des cellules traitées entrent en apoptose alors qu’elles n’ont pas été infectées. Cinq explications à ce phénomène ont été proposées :
- libération de molécules cytotoxiques par les cellules en apoptose, - passage par les jonctions gap, - transport passif, - stimulation du microenvironnement, - endocytose de vésicules apoptotiques.
Cet effet, encore mal compris, est très recherché car la thérapie génique actuelle ne permet pas de transfecter toutes cellules cibles, mais seulement une partie plus ou moins importante selon le type de vecteur utilisé et l’intensité de la réponse immunitaire du sujet – ZAROGOULIDIS et al. (2013).
b. Reprogrammation des cellules tumorales On a observé que dans de nombreux processus tumoraux, des gènes suppresseurs de tumeurs sont mutés et ne fonctionnent pas normalement. Ces mutations permettent aux cellules tumorales d’échapper à l’apoptose et de proliférer plus rapidement. Ces cellules mutantes se montrent plus agressives que les autres cellules tumorales ce qui péjore le pronostic. Une stratégie de thérapie génique consiste à transfecter les cellules tumorales avec une copie normale de ces gènes afin de rétablir le fonctionnement cellulaire normale et provoquer l’apoptose de ces cellules mutantes. Trois gènes principaux sont étudiés : le gène P53, le gène P16 et le gène PTEN (pour phosphatase and tensin homolog, qui code pour une phosphatase antagoniste des phosphatidyl-inositol-3 kinases) – KWIATKOWSKA et al. (2013). Le gène p53 est le principal gène suppresseur de tumeur. Il code pour la protéine P53 qui intervient dans la supervision de la progression du cycle cellulaire, dans les réparations de l’ADN et dans la mise en place de l’apoptose lors de stress ou de dommages cellulaires importants. Le gène P53 est muté dans la majorité des cellules tumorales dans l’espèce humaine. Des études ont montré que la transfection des cellules tumorales mutantes avec l’allèle du gène P53 sauvage permettait de stopper la croissance de la tumeur, accroître la durée de survie du patient et augmenter le taux d’apoptose parmi les cellules tumorales. Prabha et son équipe ont démontré cela sur des souris présentant une allogreffe de cellules tumorales de cancer du sein en utilisant comme vecteur un polymère biocompatible, le poly D,L-lactide-co-glycolide par voie intraveineuse – PRABHA et al. (2012). Le gène p16 est aussi impliqué dans la régulation du cycle cellulaire : il code pour la protéine P16 qui arrête le cycle entre la phase G1 et la phase S. La surexpression de ce gène dans les cellules tumorales provoque une augmentation du taux d’apoptose et la sénescence des cellules, contribuant ainsi à stopper la croissance de la tumeur – LU Y. et al. (2012).

99
Le gène pten est impliqué dans plusieurs mécanismes de la vie cellulaire (apoptose, arrêt du cycle de réplication, migration cellulaire, prolifération, etc.). La surexpression du gène pten stoppe la prolifération des cellules tumorales par arrêt du cycle cellulaire en phase G1 et en provoquant l’apoptose – LIU et al. (2012). De nombreux essais précliniques ont prouvé l’efficacité de ces antioncogènes dans le traitement de différentes tumeurs (cancer du sein, tumeurs cérébrales, tumeurs hépatiques). Il reste à convertir ces études en essais cliniques chez l’Homme.
c. Extinction génique L’extinction génique anticancéreuse consiste aussi à rétablir le fonctionnement cellulaire normal afin que les cellules tumorales entrent en apoptose.
i. Les siARN Les siARN sont utilisés dans la thérapie génique anticancéreuse afin de diminuer l’expression des gènes impliqués dans l’oncogenèse comme les gènes régulant le cycle cellulaire, la sénescence, la résistance à la chimiothérapie par exemple. Les siARN ont un potentiel thérapeutique important, mais leur utilisation in vivo pose plusieurs problèmes. La molécule nue est fragile car susceptible à l’action des enzymes endogènes. De plus, les siARN, bien que conçus pour être spécifiques, agissent aussi sur d’autres cibles non désirées de façon non prédictible. Ils peuvent présenter une cytotoxicité par trois mécanismes principaux – MIELE et
al. (2012) : - saturation du système de l’interférence de l’ARN (complexe RISC, principalement), - compétition avec les microARN endogènes, - stimulation du système immunitaire.
Plusieurs essais cliniques utilisant des siARN sont en cours : un essai clinique de phase I étudie un siARN dirigé contre l’oncogène KRAS dans l’adénocarcinome des canaux pancréatiques. Le vecteur utilisé est un polymère biodégradable. L’efficacité du système a été prouvée dans les essais précliniques, mais pas encore chez l’Homme – GUO et al. (2013).
ii. Les microARN Un certain nombre d’études ont montré que les microARN sont impliqués dans la tumorisation et son inhibition. Par exemple, la mutation au niveau du locus 13q14 sur le chromosome 13 provoque la perte de deux microARN (miR-15a et miR-16-1) et est associée au développement d’un type de leucémie. Il a été prouvé que ces deux microARN régulent l’expression de la protéine Bcl-2, une protéine anti-apoptotique surexprimée dans ce type de leucémie. Les microARN peuvent aussi provoquer l’apparition de tumeurs lorsque certains d’entre eux sont surexprimés.

100
Ces découvertes font des microARN une cible de thérapie génique anticancéreuse. Deux approches sont considérées : extinction des microARN tumorigènes et surexpression des microARN anti-tumoraux. L’extinction des microARN se fait par complémentarité. Différentes molécules peuvent inhiber les microARN en se liant à eux par complémentarité, ainsi les microARN ne peuvent se lier à leur cible. Sont utilisés :
- des oligonucléotides complémentaires (ou oligonucléotides antisens) des pri-microARN, des pré-microARN ou des microARN matures,
- des polymères synthétiques appelés des « acides peptidiques nucléiques » ou APN similaires à l’ARN ou à l’ADN, mais avec une structure basée sur des groupements amines plutôt que sur des sucres.
- Des éponges à microARN : on utilise alors une molécule similaire à un ARNm qui possède plusieurs sites compatibles avec différents microARN ce qui permet d’inhiber plusieurs microARN en les accrochant avec une seule molécule.
Deux stratégies permettent de surexprimer des microARN : utilisation d’imitations de microARN ou d’ADN (plasmide ou génome viral). Les imitations de microARN sont des ARN double brin avec un brin principal identique au microARN mature (une fois clivé par l’enzyme Dicer) et un brin accessoire plus ou moins complémentaire avec le brin principal. Il existe aussi des imitations composées d’un seul brin d’ARN, mais ces molécules sont moins efficaces. Les imitations de microARN doivent être modifiées afin d’être résistantes aux nucléases : l’adjonction de groupements méthyl ou aromatiques allonge leur demi-vie intracellulaire. L’ADN utilisé code pour des shARN (similaire à un pré-microARN) ou pour des pri-microARN. Ainsi, l’enzyme Dicer peut les prendre en charge et ces molécules remplissent le rôle de microARN. Cependant, les shARN ou les pri-microARN ne sont pas entièrement spécifiques et peuvent réguler d’autres protéines que les protéines cibles. Leur synthèse doit donc être très rigoureuse afin de limiter l’action potentiellement néfaste sur d’autres protéines. Actuellement, l’utilisation d’oligonucléotides anti-sens et d’imitations de microARN sont les approches les plus développées : elles ont déjà démontré leur efficacité en essais in vitro et in
vivo sur des modèles animaux. Le choix du vecteur est très important car ces molécules sont fragiles – COSTA et PEDROSO DE LIMA (2013).
d. La virothérapie La virothérapie consiste à infecter les cellules tumorales avec des virus oncolytiques afin qu’ils lysent les cellules tumorales. En virothérapie, les virus sont à la fois les vecteurs et l’outil thérapeutique grâce à leur génome. Les virus peuvent transporter en même temps des molécules thérapeutiques pour compléter leur effet – RANKI et al. (2010).

101
Les virus utilisés dépendent de la tumeur cible. Par exemple, on utilisera des virus neurotropes pour cibler des tumeurs cérébrales. Il faut aussi considérer le mode de pénétration du virus dans les cellules. En effet, les adénovirus utilisent le récepteur CAR. Mais ce récepteur est peu exprimé en surface des cellules tumorales : il faut donc modifier la surface du virus (pseudotypage) afin qu’il reconnaisse d’autres récepteurs, et en particulier, des récepteurs spécifiques de la tumeur cible. Les virus oncolytiques doivent aussi être modifiés afin de rendre leur utilisation sûre pour le patient. Des gènes sont supprimés pour rendre le virus spécifique des cellules tumorales, non cytotoxique pour les autres cellules et ainsi libérer de la place pour le transport d’un transgène. On prendra ici l’exemple du virus Herpès simplex (VHS) dans le traitement du glioblastome (tumeur primitive cérébrale la plus fréquente qui touche des cellules gliales). Le VHS est neurotrope et son génome peut être modifié afin de le rendre spécifique des cellules tumorales. De plus, il existe des molécules antivirales très efficaces contre ce virus ce qui rend son usage assez sûr. Trois VHS ont été développés dans le traitement du glioblastome. Tous sont délétés pour le gène responsable de la neurovirulence et deux d’entre eux ne possède pas le gène leur permettant d’infecter les cellules quiescentes. Des essais cliniques ont démontré que leur utilisation était sûre, mais l’efficacité observée lors des essais in vitro n’a pas été retrouvée. De nombreux autres virus sont étudiés dans le cadre de la virothérapie contre le glioblastome, mais aucun n’a fait la preuve de son efficacité en essai clinique – MURPHY et RABKIN (2013).
2. Thérapie anti-angiogénique La néo-vascularisation est régulée par l’équilibre entre les facteurs pro et anti-angiogenèse. Les cellules tumorales se divisent trop rapidement par rapport à la vascularisation ce qui conduit à l’hypoxémie des nouvelles cellules tumorales qui produisent alors de nombreux facteurs angiogéniques. La tumeur peut alors continuer à se développer. La thérapie anti-angiogénique consiste à empêcher cette néo-vascularisation, mais elle ne permet que très rarement de réduire la taille de la tumeur. La plupart du temps, cette approche ne fait que ralentir la croissance de la tumeur. La thérapie anti-angiogénique doit donc être utilisée en parallèle d'une autre stratégie. Afin d’atteindre cet objectif, la thérapie génique offre plusieurs options : surexpression des facteurs anti-angiogéniques, inhibition des facteurs angiogéniques, ou modifications des cellules endothéliales irrigant la tumeur. De nombreux facteurs anti-angiogéniques ont été identifiés. Deux d’entre eux semblent être plus efficaces que les autres : l’angiostatine et l’endostatine qui sont tous les deux des résidus de la protéolyse de la matrice extra-cellulaire (collagène et plasminogène respectivement). Des AAV transportant l’ADNc codant ces molécules ont été testés sur des xénogreffes (glioblastome) chez des souris par injection directe dans la tumeur et ont prouvé leur efficacité en diminuant la vitesse de croissance de la tumeur jusqu’à 90% - CASTRO et al. (2011).

102
On a aussi identifié les facteurs angiogéniques produits par les tumeurs. Le facteur principal semble être le facteur de croissance de l’endothélium vasculaire (VEGF en anglais). Plusieurs techniques ont été utilisées pour diminuer l’expression de ce gène par les cellules tumorales : insertion d’un ADNc anti-sens du VEGF, d’un shARN spécifique du VEGF, etc. Ces techniques ont été efficaces chez les modèles animaux étudiés – CASTRO et al. (2011). Une autre approche anti-angiogénique consiste à modifier les cellules endothéliales irrigant la tumeur. Ces cellules ont des spécificités qui les rendent différentes des cellules endothéliales normales. Des études ont montré qu’en plus, les cellules endothéliales irrigant les tumeurs n’ont pas toutes la même origine : certaines sont des cellules tumorales transformées en cellules endothéliales, d’autres proviennent de la moelle osseuse, d’autres encore sont des cellules mésenchymateuses différenciées en cellules endothéliales. Cela rend le ciblage de ces vaisseaux sanguins plus difficile qu’il n’était prévu à l’origine. D’autres études sont nécessaires avant de pouvoir cibler spécifiquement les cellules endothéliales irrigant les tumeurs – DUDLEY (2012). Certains travaux ont cependant montré qu’il était possible de modifier ces cellules. Ils ont utilisé des promoteurs spécifiques des cellules endothéliales normales avec injection locale du vecteur. Un des grands axes de recherche est le récepteur au VEGF. Il est possible de faire exprimer aux cellules une version soluble de ce récepteur, ce dernier est alors sécrété et est beaucoup plus affin pour le VEGF, mais ne permet pas la transduction du signal – DONG et NÖR (2009). Les résultats sur des souris d’expérimentation ont été encourageants, mais d’autres études ont montré que l’inhibition du VEGF amenait les cellules endothéliales à choisir d’autres voies et à exprimer d’autres facteurs de croissance ce qui les rend finalement insensibles aux thérapies anti-VEGF – DUDLEY (2012). D’autres facteurs de croissance ont été découverts par hasard. C’est le cas de la protéine S100A4 exprimée dans les cellules endothéliales irrigant les tumeurs, étudiée pour son rôle dans la dissémination métastatique. Une étude ayant utilisé un siARN dirigé contre cette protéine a montré que cela ralentissait la croissance de la tumeur et la formation de nouveaux capillaires – OCHIYA et al. (2014). Malgré les obstacles, cette voie n’est pas abandonnée mais nécessite des recherches plus approfondies.
3. Immunothérapie Les cellules tumorales disposent de nombreux outils qui leur permettent d’échapper au système immunitaire. Par exemple, les cellules tumorales expriment peu de complexes majeurs de l’histocompatibilité (CMH) dont ont besoin les lymphocytes pour reconnaître les cellules. Les cellules tumorales sont aussi capables de sécréter des facteurs immunosuppressifs limitant le nombre de cellules de l’immunité présentes dans le microenvironnement de la tumeur. Le manque de co-stimulation lors de la reconnaissance d’un antigène tumoral par les lymphocytes T peut aussi conduire à une immunotolérance voire une anergie, ce qui protège d’autant plus la tumeur contre le système immunitaire – COLLINS et al. (2010).

103
L’immunothérapie consiste à modifier les cellules de l’immunité ou les cellules tumorales afin de rendre le système immunitaire du patient à même de détruire seul les cellules tumorales.
a. Modification des cellules de l’immunité Les cellules impliquées dans l’immunité anti-tumorale sont entre autres les lymphocytes T cytotoxiques. Il existe aujourd’hui deux approches similaires permettant à ces lymphocytes de mieux reconnaître les cellules tumorales afin de les détruire.
i. Les récepteurs des lymphocytes T On sélectionne dans la population lymphocytaire du patient les lymphocytes présentant un récepteur spécifique de la tumeur étudiée, le plus affin possible (cf. Figure 29). On analyse ensuite le récepteur afin de créer un ADNc. Les récepteurs des lymphocytes sont composés de deux chaînes α et β qui doivent être exprimées en même temps et s’assembler par paire : il s’agit du plus grand défi de cette approche. Afin d’atteindre ce résultat, il est possible d’encoder les deux chaînes avec une seule séquence en supprimant les introns. Pour augmenter le taux d’appariement, différentes techniques ont été utilisées comme le remplacement d’une séquence des deux chaînes par son homologue murin ou en créant d’autres ponts disulfures ce qui rend les chaînes plus affines les unes pour les autres. Le mauvais appariement pose problème car on crée alors un récepteur nouveau dont on ne connait pas la spécificité ni les conséquences sur le fonctionnement de la cellule. Une cassette génique est créée et est insérée via un vecteur (le plus souvent un rétrovirus, capable d’insérer la séquence directement dans le génome de la cellule cible) dans les lymphocytes (ou dans les cellules souches hématopoïétiques) du receveur. Les lymphocytes transduits sont ensuite réinjectés chez le receveur.

104
Figure 29 : Schématisation de la création de lymphocytes génétiquement modifiés d’après
SCHMITT et al. (2009) Des essais cliniques utilisant cette technique sont en cours afin d’assurer l’innocuité de ce traitement – KURNET et al. (2013).
ii. Les récepteurs chimériques Semblable à la stratégie précédente, la création de récepteurs chimériques répond à une lacune des récepteurs endogènes : ils sont rares, difficiles à trouver et plus sujets à une immunotolérance car peut-être déjà présents chez le patient. Des chercheurs ont alors créé des récepteurs chimériques possédant un domaine de liaison synthétique à un antigène tumoral et un domaine endogène permettant la transduction du signal lors de la liaison avec l’antigène. De fait, ces récepteurs peuvent reconnaître beaucoup plus de cibles que les récepteurs endogènes car ils ne sont pas limités aux seuls antigènes protéiques. De la même façon que précédemment, on peut ensuite transfecter des lymphocytes avec la cassette génique codant pour ce récepteur et les réinjecter chez le patient. Des essais cliniques ont montré qu’il y avait peu de réponse immunitaire aux lymphocytes transfectés malgré la présence d’un récepteur étranger – RAMOS et DOTTI (2011). Grada et son équipe ont créé un récepteur capable de reconnaître deux antigènes ce qui augmente la spécificité et diminue le risque d’échappement de la tumeur au traitement – GRADA et al. (2013).
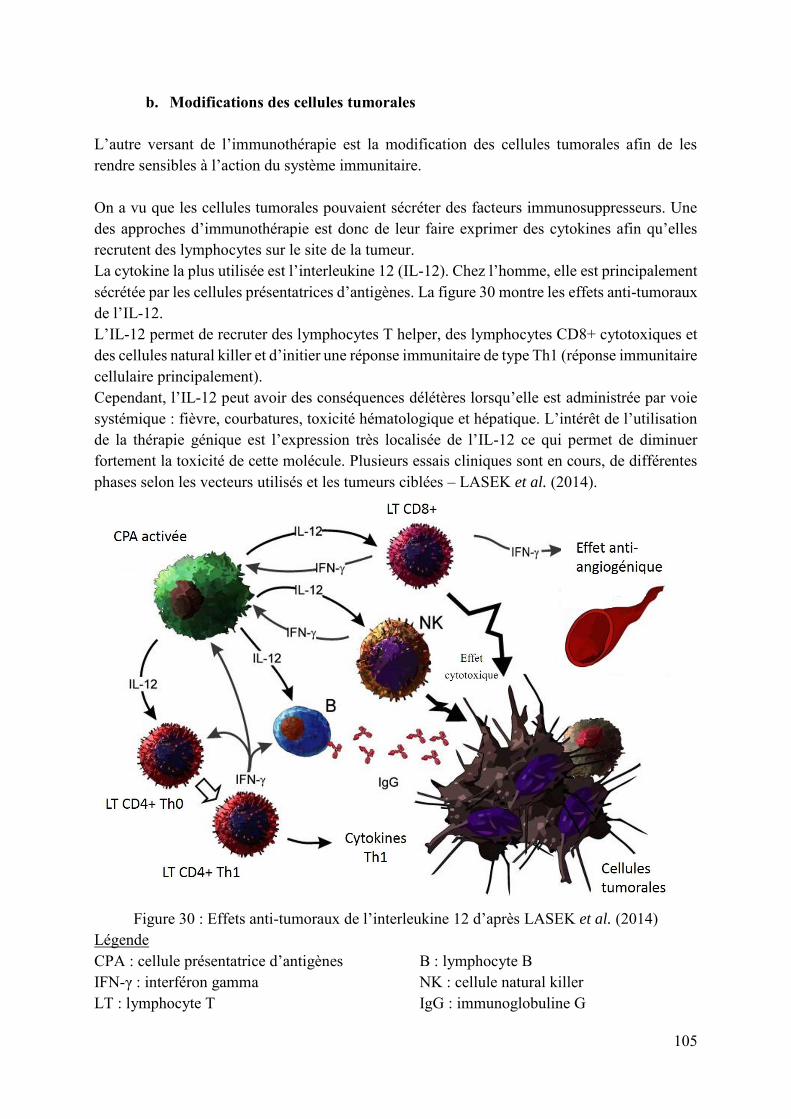
105
b. Modifications des cellules tumorales L’autre versant de l’immunothérapie est la modification des cellules tumorales afin de les rendre sensibles à l’action du système immunitaire. On a vu que les cellules tumorales pouvaient sécréter des facteurs immunosuppresseurs. Une des approches d’immunothérapie est donc de leur faire exprimer des cytokines afin qu’elles recrutent des lymphocytes sur le site de la tumeur. La cytokine la plus utilisée est l’interleukine 12 (IL-12). Chez l’homme, elle est principalement sécrétée par les cellules présentatrices d’antigènes. La figure 30 montre les effets anti-tumoraux de l’IL-12. L’IL-12 permet de recruter des lymphocytes T helper, des lymphocytes CD8+ cytotoxiques et des cellules natural killer et d’initier une réponse immunitaire de type Th1 (réponse immunitaire cellulaire principalement). Cependant, l’IL-12 peut avoir des conséquences délétères lorsqu’elle est administrée par voie systémique : fièvre, courbatures, toxicité hématologique et hépatique. L’intérêt de l’utilisation de la thérapie génique est l’expression très localisée de l’IL-12 ce qui permet de diminuer fortement la toxicité de cette molécule. Plusieurs essais cliniques sont en cours, de différentes phases selon les vecteurs utilisés et les tumeurs ciblées – LASEK et al. (2014).
Figure 30 : Effets anti-tumoraux de l’interleukine 12 d’après LASEK et al. (2014)
Légende CPA : cellule présentatrice d’antigènes IFN-γ : interféron gamma LT : lymphocyte T
B : lymphocyte B NK : cellule natural killer IgG : immunoglobuline G

106
D’autres cytokines peuvent être exprimées par les cellules tumorales, comme le facteur de stimulation des colonies de macrophages/granulocytes qui joue un rôle important dans la différenciation des phagocytes. Les cellules tumorales échappent aussi au système immunitaire par manque de co-stimultation lors de la reconnaissance d’un antigène par des lymphocytes qui ne peuvent plus alors déclencher de réponse cytotoxique. Il est possible de transfecter les cellules tumorales avec des ligands qui permettent la co-stimulation nécessaire comme le facteur B7-1 qui se lie au récepteur CD28 des lymphocytes T. Le récepteur CD28 est considéré comme le principal co-stimulateur des lymphocytes T – COLLINS et al. (2010). D’autres molécules ont été étudiées pour leur effet immunostimulant, comme le ligand FasL. Le système Fas est une des voies d’apoptose. Lorsque le récepteur Fas est lié à son ligand FasL (normalement exprimé dans les cellules de l’immunité), la cellule possédant le récepteur entre en apoptose. Cependant, de nombreuses cellules tumorales ne possèdent plus le récepteur Fas ou la voie d’apoptose associée en état de fonction. Mais, l’expression par les cellules tumorales du ligand FasL provoque, en particulier, l’apoptose des polynucléaires neutrophiles présents dans le voisinage de la tumeur. Par leurs apoptoses, ils déclenchent une réponse inflammatoire puissante qui empêche la croissance de la tumeur et peut arriver au rejet de la tumeur par recrutement des lymphocytes (cela a été démontré chez des souris). Chez le chien atteint d’ostéosarcome, on n’observe pas de rejet de la tumeur, mais une réponse inflammatoire importante, une nécrose et une fibrose de la tumeur majorées par rapport au groupe témoin. On observe aussi un allongement de la durée de survie – MODIANO et al. (2012).
4. Ciblage des cellules tumorales Le ciblage des cellules cibles est une part importante de la réflexion en thérapie génique, mais revêt un aspect particulier en thérapie génique anticancéreuse car, comme on l’a vu, le but de la thérapie est de tuer les cellules tumorales. Il faut donc restreindre au maximum l’expression du transgène aux seules cellules tumorales afin de limiter la cytotoxicité du traitement et donc ses effets secondaires. Nous prendrons ici deux exemples afin d’illustrer le ciblage des cellules tumorales en thérapie génique. Il est possible d’utiliser des aptamères afin de rendre le vecteur spécifique des cellules tumorales étudiées. Les aptamères ont l’avantage d’être faciles à produire, stables et peu cytotoxiques ou immunogéniques. Dans le cancer de la prostate, on a identifié un récepteur cellulaire appelé PSMA, comme une cible attractive : en effet, ce récepteur est très exprimé en surface des cellules cancéreuses et est capable d’internaliser certains ligands. On a donc créé des aptamères spécifiques de ce récepteur. Ils ont été utilisés pour guider et augmenter le taux
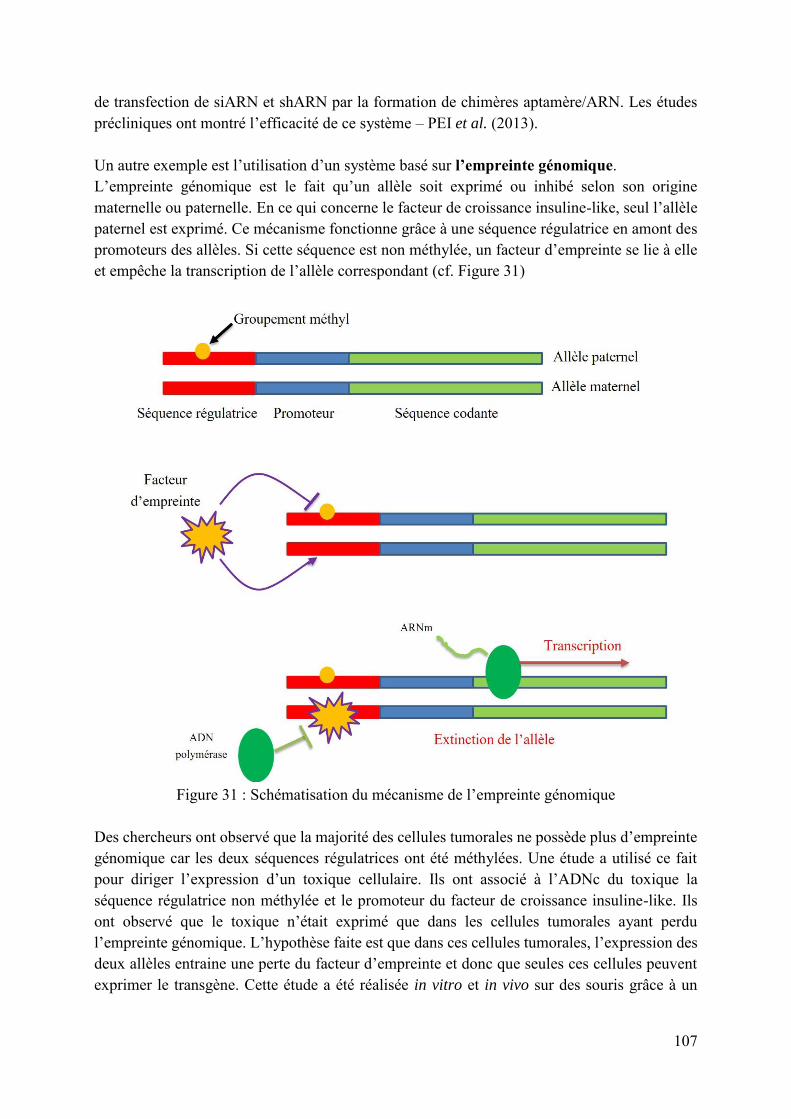
107
de transfection de siARN et shARN par la formation de chimères aptamère/ARN. Les études précliniques ont montré l’efficacité de ce système – PEI et al. (2013). Un autre exemple est l’utilisation d’un système basé sur l’empreinte génomique. L’empreinte génomique est le fait qu’un allèle soit exprimé ou inhibé selon son origine maternelle ou paternelle. En ce qui concerne le facteur de croissance insuline-like, seul l’allèle paternel est exprimé. Ce mécanisme fonctionne grâce à une séquence régulatrice en amont des promoteurs des allèles. Si cette séquence est non méthylée, un facteur d’empreinte se lie à elle et empêche la transcription de l’allèle correspondant (cf. Figure 31)
Figure 31 : Schématisation du mécanisme de l’empreinte génomique
Des chercheurs ont observé que la majorité des cellules tumorales ne possède plus d’empreinte génomique car les deux séquences régulatrices ont été méthylées. Une étude a utilisé ce fait pour diriger l’expression d’un toxique cellulaire. Ils ont associé à l’ADNc du toxique la séquence régulatrice non méthylée et le promoteur du facteur de croissance insuline-like. Ils ont observé que le toxique n’était exprimé que dans les cellules tumorales ayant perdu l’empreinte génomique. L’hypothèse faite est que dans ces cellules tumorales, l’expression des deux allèles entraine une perte du facteur d’empreinte et donc que seules ces cellules peuvent exprimer le transgène. Cette étude a été réalisée in vitro et in vivo sur des souris grâce à un

108
adénovirus. Cette méthode semble prometteuse si elle fonctionne chez l’Homme – PAN et al. (2013).
5. Conclusions La thérapie génique anticancéreuse utilise toutes les stratégies développées en thérapie génique. Malheureusement, la plupart n’ont pas montré d’efficacité visualisée in vitro ou en essais précliniques. Des progrès sont nécessaires avant l’avènement d’une approche réellement efficace. Plusieurs transgènes peuvent être combinés afin d’attaquer les cellules tumorales sur différents fronts en même temps (facteur anti-angiogénique dans un virus oncolytique ou avec un gène induisant le suicide, etc.). La thérapie génique peut aussi servir de complément thérapeutique. En effet, on peut, par thérapie génique, modifier les cellules tumorales afin de les rendre plus sensibles à la radiothérapie – KALIBEROV et BUSCHBAUM (2012). L’avancement des recherches est différent selon le type de cancer considéré, mais un médicament est actuellement disponible sur le marché pharmaceutique chinois : le Gendicine® (adénovirus/gène p53) pour le carcinome des cellules squameuses. De nombreux essais cliniques de différentes phases sont en cours.

109
V. Et en médecine vétérinaire ? Il est évident que la thérapie génique actuelle ne concerne les animaux qu’en tant que modèles d’études. Cependant, certains pensent que la thérapie génique viendra un jour compléter l’arsenal thérapeutique des vétérinaires. L’oncologie semble être la seule discipline qui pourrait se voir dotée de ce nouvel outil qu’est la thérapie génique. Selon Breen, les animaux domestiques présentent naturellement des tumeurs comparables à celles de l’Homme. Ils peuvent donc être utilisés comme animaux d’expérimentation et à cette occasion, dévoiler une thérapie utilisable en médecine vétérinaire – BREEN (2009). Les essais précliniques ont permis de mettre en évidence que de nombreuses techniques de thérapie génique fonctionnent chez nos animaux domestiques. On peut prendre par exemple le cas du fibrosarcome du chat. Des essais cliniques ont montrés l’efficacité de traitement de thérapie génique à base d’IL-12, d’interféron γ, de facteur de stimulation des colonies de macrophages/granulocytes et de gènes induisant le suicide cellulaire – PAVLIN et al. (2012). Nous prendrons ici l’exemple de l’utilisation du gène de l’interleukine 2 (IL-2), une cytokine immunostimulatrice. Jourdier et son équipe ont utilisé des poxvirus comme vecteurs : le vecteur ALVAC, basé sur un canarypox et le vecteur NYVAC, basé sur un virus de la vaccine atténué, qui sont des vecteurs dont l’innocuité a été prouvée, même chez l’Homme. Cette équipe a injecté ces vecteurs in situ (c’est-à-dire au sein de la tumeur) à des chats atteints d’un fibrosarcome. Une première étude a permis de démontrer que le vecteur ne se diffusait pas et restait localisé à la tumeur. Cela implique que seules les cellules tumorales peuvent être infectées par cette technique, ce qui permet de réduire la toxicité de l’IL-2. En effet, comme toutes les cytokines, une expression systémique importante déclenche une réponse immunitaire délétère. On observe que l’expression de l’IL-2 se maintient pendant au moins 4 jours au sein de la tumeur. On constate que la tumeur réduit de taille dans 50% des cas, ce qui est associé avec une infiltration de la tumeur par cellules immunitaires, une production d’IFN-γ voire dans certains cas, la mise en place d’une réponse immunitaire anti-tumorale spécifique. Le suivi des chats traités par thérapie génique montre que le traitement permet de réduire le nombre de récidives par deux dans l’année suivant l’exérèse de la tumeur et la radiothérapie – JOURDIER et al. (2003). Cette technique pourrait à terme être utilisée en routine pour réduire la taille des tumeurs avant chirurgie, limiter le risque de récidive et ralentir la croissance de tumeur non opérable. L’efficacité de cette technique n’est pas démontrée sur les métastases. De même, comme décrit précédemment (§3-modification des cellules tumorales), l’utilisation du ligand FasL dans le traitement de l’ostéosarcome du chien induit une importante réponse inflammatoire associée à une nécrose et une fibrose de la tumeur –MODIANO et al. (2012). Depuis plusieurs années, on constate que les propriétaires d’animaux de compagnie sont non seulement prêts (financièrement et psychologiquement) mais également demandeurs à ce que l’on utilise des outils thérapeutiques jusqu’alors réservés aux humains. La chimiothérapie et la radiothérapie, mais aussi des moyens d’imagerie comme le scanner et l’IRM sont devenus

110
pratique courrante chez nos animaux de compagnie. Il n‘est donc pas impensable que certaines techniques de thérapie génique soient mises à la disposition des vétérinaires. L’oncologie en médecine vétérinaire est amenée à se développer toujours plus parce que la population des animaux de compagnie vieillit, que les examens complémentaires à notre disposition permettent de diagnostiquer les tumeurs et que les propriétaires sont aujourd’hui demandeurs d’un traitement permettant de prolonger la vie de leurs animaux de compagnie. Il est cependant nécessaire de préciser que, compte tenu des frais liés à la création d’un traitement de thérapie génique, les seules maladies animales qui pourront être traitées ainsi seront les maladies utilisées comme modèle pour l’Homme, comme celles présentées dans cette thèse. Il convient aussi de préciser que les maladies génétiques animales ne bénéficieront sans doute jamais de ce type de traitement car on préfère éliminer de la reproduction les animaux porteurs des allèles mutés afin d’assainir la population.

111
Discussion La thérapie génique est une technologie qui permet de traiter les maladies en modifiant le génome des individus par adjonction de gènes, réparation des gènes et modification de l’expression génique (extinction ou expression forcée). Si le principe est simple, son application est beaucoup plus complexe. Aujourd’hui, la thérapie génique a démontré une efficacité dans les essais in vitro de façon certaine, chez les modèles animaux dans la plupart des cas mais pas chez l’Homme. Comparée aux modèles animaux, l’Homme est une espèce beaucoup plus difficile à traiter par thérapie génique, notamment en raisonde son système peu immunotolérant par rapport à celui des modèles animaux. La réponse immunitaire à la thérapie, forte dans l’espèce humaine, ne permet pas au traitement d’être efficace, et ce pour plusieurs raisons. Tout d’abord, un faible nombre de vecteurs atteignent efficacement les cellules cibles car ils sont neutralisés. Les protéines nouvellement traduites peuvent ensuite être détruites avant même de pouvoir exercer leur rôle curatif, comme par exemple dans le cas des dystrophines tronquées lors du traitement de la myodystrophie de Duchenne. Enfin, les cellules transfectées qui expriment une protéine jusqu’ici étrangère à l’organisme peuvent être détruites lorsqu’elles sont reconnues comme anormales par le système immunitaire. Par exemple, dans le traitement de l’hémophilie A, peu de cellules transfectées ex
vivo survivent en raison de la répose immunitaire cellulaire vis-à-vis de ces cellules. Ceci explique en partie pourquoi des méthodes éprouvées en essais pré-clinques se soldent par un échec lors des premiers essais cliniques chez l’Homme. D’autre part, la thérapie génique présente trois limitations majeures : le ciblage des cellules d’intérêt, le taux de transfection et l’expression génique à moyen/long terme. Sans ciblage des cellules, la thérapie génique peut être sans effet dans le meilleur des cas, ou toxique dans le pire, en particulier en thérapie génique anticancéreuse cytotoxique. Dans ce contexte, il est primordial que seules les cellules tumorales soient transfectées afin qu’elles soient les seules à être détruites. En revanche, si les cellules cibles ne sont pas transfectées, aucun résultat thérapeutique n’est observé. Le ciblage cellulaire permet également de limiter la réponse immunitaire induite par l’expression d’une nouvelle protéine et / ou par le contact avec le vecteur, en particulier, les vecteurs viraux. En effet, il a été démontré que si les cellules présentatrices d’antigènes n’étaient pas transfectées, la réponse immunitaire à la thérapie génique était très faible voire absente – MARQUEZ et McCAFFREY (2008). Un autre point critique en thérapie génique est le taux de transfection. Il ne suffit pas que les cellules cibles soient transfectées, il faut qu’elles le soient en nombre assez important pour pouvoir espérer une modification du phénotype.

112
On a vu, dDans le contexte de la thérapie génique appliquée à l’insuffisance cardiaque, certaines méthodes d’administration ne permettent pas de transfecter un nombre significatif de cellules pour que le fonctionnement normal du cœur soit rétabli, en raison d’un temps de contact entre le vecteur et les cellules trop court, ou d’une injection trop localisée (épicarde). Même dans le cas de l’hémophilie A où on a montré qu’une petite augmentation de la concentration en fVIII circulant permettait de modifier le phénotype, un nombre assez important de cellules doivent être transfectées pour obtenir ce résultat. Dans la myodystrophie de Duchenne, toutes les cellules musculaires sont affectées par l’absence de dystrophine. Il faut donc que la majorité d’entre elles soient transfectées pour espérer obtenir un effet thérapeutique. Il en est de même pour la thérapie génique appliquée au SIDA et la thérapie génique anticancéreuse. Dans la lutte contre le SIDA, un grand nombre de cellules CD34+ doivent être transfectées afin de pouvoir assurer au malade un système immunitaire fonctionnel (mémoire antigénique complète) lui permettant de s’immuniser contre le VIH. En thérapie anticancéreuse cytotoxique, l’objectif est de détruire toutes les cellules tumorales, d’où la nécessité de transfecter la majorité d’entre elles, même s’il existe un effet de proche en proche. Enfin, dans la plupart des cas, l’expression du transgène doit se faire à long terme. En effet, mis à part lors de l’utilisation d’outils en vue de réparer le génome (type enzymes en doigts de zinc), l’expression à long terme est le garant de la guérison de la maladie. Cela est vrai pour toutes les maladies où on rétablit le fonctionnement normal de la cellule. Lors de l’extinction du transgène, la cellule revient à son fonctionnement antérieur. L’expression à long terme est recherchée dans pratiquement toutes les stratégies présentées dans ce manuscrit, à part pour la thérapie génique anticancéreuse. La thérapie génique anticancéreuse diffère par le fait que l’expression du transgène doit être juste suffisament longue pour permettre la destruction de toutes les cellules cancéreuses. Afin de pallier ces limitations, de nombreuses techniques ont été développées comme la création de nouveaux vecteurs plus spécifiques (utilisation d’aptamères, pseudotypage et modifications des protéines d’entrée des vecteurs viraux, direction des vecteurs par champs magnétiques, etc.) et permettant une expression à long terme (insertion dans le génome cellulaire principalement, par des lentivirus et le système Sleeping Beauty, transfection des cellules quiescentes, etc.). Cependant, aucune méthode n’est à ce jour plus performante que les autres et chaque maladie apporte son lot de restrictions. On remarquera que les vecteurs viraux sont, pour le moment, préférés aux vecteurs non viraux en raison de leur taux de transfection supérieur en efficacité. Si un certain nombre d‘études sont en cours pour améliorer le rendement des vecteurs non viraux, pratiquement aucun n’est utilisé en essai clinique : seuls 23% des essais en cours utilisent un vecteur non viral (ADN nu ou liposomes) ; les autres emploient tous un vecteur viral – Charts & Tables (2014). Dans le cas spécifique des maladies affectant les cellules circulantes sanguines, on a vu qu’il était plus facile de réaliser une thérapie génique efficace sur des cellules souches hématopoïétiques. Ces dernières sont transfectées ex vivo (ciblage parfait), sélectionnées afin

113
de ne garder que les cellules exprimant le transgène puis cultiveées pour pouvoir en réinjecter le maximum (taux de transfection de 100%). Les cellules souches réinjectées vont alors pouvoir coloniser leur tissu et compartiment de destination (sang et moelle osseuse). L’utilisation de cellules CD34 dans la thérapie génique antirétrovirale a été decrite. On rappellera que cette méthode n’est pas des plus efficaces car les cellules souches non transfectées n’étant pas détruites, les cellules transfectées ne représentent qu’une petite fraction de la population immunitaire. Il faut aussi considérer le risque potentiel de rejet de la greffe. Les cellules souches mésenchymateuses sont aussi utilisées dans la thérapie génique appliquée à l’hémophilie A. C’est à ce niveau que se fait le lien avec un nouvel axe de recherche très prometteur : la thérapie cellulaire. La thérapie cellulaire consiste à soigner par l’apport de cellules souches pluripotentes, capables de réparer les dommages causés par la maladie. C’est une des pistes poursuivies dans le traitement de l’insuffisance cardiaque avec l’emploi du système SDF1/CXCR4 qui permet de recruter des cellules souches. La modification des cellules souches par la thérapie génique est attractive, mais présente le risque majeur de déclencher des tumeurs par modification du génome de cellules non différenciées et en phase active du cycle cellulaire. L’association des deux approches thérapie cellulaire et thérapie génique reste un sujet sensible qui nécessitera de nombreuses études afin d’en prouver l’innocuité à long terme – SNG et LUFKIN (2012).

114

115
Conclusion
La thérapie génique est un outil thérapeutique très puissant car il peut être appliqué à un large éventail de maladies (génétiques, acquises, infectieuses et tumorales). La thérapie génique est passée en cinquante ans du statut de projet théorique à une réalité pratique. Plus de 1900 essais cliniques ont été réalisés à ce jour, mais seuls deux traitements de thérapie génique sont autorisés dans le monde (Gendicine® et Glybera®) – KAUFMANN et al. (2013). La thérapie génique est développée pour des maladies dont le traitement actuel est trop contraignant ou trop peu efficace. Avec la thérapie génique, on peut aujourd’hui espérer soigner des maladies incurables comme par exemple, le SIDA. Pour d’autres maladies telles les maladies génétiques qui touchent principalement des enfants et dont la durée de vie est très écourtée par la maladie, la thérapie génique représente la seule chance de survie. Afin d’améliorer les techniques de thérapie génique, une compréhension accrue des mécanismes cellulaires, notamment de l’expression génétique et des voies de signalisation intracellulaire, est nécessaire. Chaque maladie apporte son lot de spécificités auxquelles la thérapie génique doit s’adapter ; de fait, aucune recette universelle n’existe et les recherches doivent souvent recommencer du début pour chaque approche de traitement par thérapie génique. De nombreux efforts sont accomplis afin de produire des systèmes de thérapie génique dont l’administration ne nécessite pas d’hospitalisation (injection parentérale unique voire administration per os). Le coût du traitement est aussi un paramètre important à considérer pour que la thérapie génique soit accessible à tous. Les vecteurs viraux coûtent en général plus chers à produire que les vecteurs non viraux. Mais tant que ceux-ci n’auront pas fait la preuve de leur supériorité, les vecteurs viraux demeureront les vecteurs les plus utilisés. La médecine vétérinaire n’est pas proche de bénéficier des apports de la thérapie génique, même si celle-ci aboutit à des traitements efficaces. On peut néanmoins espérer que, grâce aux essais précliniques réalisés chez nos animaux de compagnie, la thérapie génique soit un jour une solution thérapeutique disponible pour les vétérinaires. Cela concernera uniquement les affections tumorales, voire certaines maladies acquises comme l’insuffisance cardiaque ou l’arthrose puisque les maladies génétiques sont éradiquées de la population par sélection à la reproduction. Au vu de la médication actuelle de la population féline et du coût que représenterait un tel traitement, le SIDA du chat ne sera sûrement jamais traité par thérapie génique.

116
La thérapie génique constitue un nouveau marché du médicament où on n’utilise plus de molécules pharmaceutiques mais des acides nucléiques. Les conditions à l’ouverture de ce marché sont bien évidemment la réussite des essais cliniques et l’assurance de l’innocuité de ce traitement qui soulèvent, à juste titre, de nombreuses questions éthiques. Thèse de Mme HELLEZ Justine Le Professeur responsable Le Directeur général VetAgro Sup campus vétérinaire VetAgro Sup Le Président de la thèse Vu et permis d’imprimer Lyon, le Le Président de l’Université, Professeur F.N GILLY

117
Bibliographie AL-DOSARI M. S., GAO X. (2009) Non viral gene delivery : principles, limitations and recent progress, American Association of Pharmaceutical Scientists Journal, 11, (4), 671-681 ALLERS K. et al. (2011) Evidence for the cure of HIV infection by CCR5 delta32/delta32 stem cell transplantation, Blood, 117, 2791-2799 ARBUTHNOT P., THOMPSON L.J. (2008) Harnessing the RNA interference pathway to advance treatment and prevention of hepatocellular carcinoma, World Jounral of Gastroenterology, 14, (11), 1670-1681 ARDIANI A. et al. (2012) Enzymes to die for : exploiting nucleotide metabolizing enzymes for cancer gene therapy, Current Gene Therapy, 12, (2), 77-91 BARAR J., OMIDI Y. (2012) Translational approaches toward cancer gene therapy : hurdles and hopes, BioImpacts, 2, (3), 127-143 BATES K., KOSTARELOS K. (2013) Carbon nanotubes as vectors for gene therapy : past achievements, present challenges and future goals, Advanced Drug Delivery Reviews, 65, 2023-2033 BERGER J., CURRIE P.D. (2012) Zebrafish models flex their muscles to shed light on muscular dystrophies, Disease models & mechanisms, 5, (6), 726-732 BONAMASSA B., HAI L., LIU D. (2011) Hydrodynamic gene delivery and its applications in pharmaceutical research, Pharmaceutical Research, 24, (4), 694-701 BONDY-DENOMY J., DAVIDSON A.R. (2014) To acquire or resist : the complex biological effects of CRISPR-Cas systems, Cell, 22, (4), 218-225 BOUTIN S. et al. (2010) Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population implications for gene therapy using AAV vectors, Human Gene Therapy, 21, (6), 704-712 BOWLES D.E. et al. (2012) Phase 1 gene therapy for Duchenne muscular dystrophy using a translational optimized AAV vector, Molecular therapy, 20, (2), 443-455

118
BOWMAN K., SARKAR R., RAUT S., LEONG K.W. (2008) Gene transfer to hemophilia A mice via oral delivery of FVIII-chitosan nanoparticles, Journal of Controlled Release, 132, 252-259 BREEN M. (2009) Update on genomics in veterinary oncology, Topics in companion animal medecine, 24, (3), 113-121 BROWN B.D. et al. (2007) A microRNA-regulated lentiviral vector mediates stable correction of hemophilia B mice, Blood, 110, 13, 4144-4153 BÜNING H. (2013) Gene therapy enters the pharma market : the short story of a long journey, EMBO Molecular Medicine, 5, 1-3 BURNETT J.C., ZAIA J.A., ROSSI J.J. (2012) Creating genetic resistance to HIV, Current Opinion in Immunology, 24, (5), 625-632 CAO B., QIU P., MAO C. (2013) Mesoporous Iron Oxide Nanoparticles Prepared by Polyacrylic Acid Etching and Their Application in Gene Delivery to Mesenchymal Stem Cells, Microscopy research and technique, 76, (9), 936-941 CAO X. et al. (2011) Encapsulation of plasmid DNA in calcium phosphate nanoparticles: stem cell uptake and gene transfer efficiency, International Jounal of Nanomedecine, 6, 3335-3349 CASTRO M.G. et al. (2011) Gene therapy and targeted toxins for glioma, Current Gene Therapy, 11, (3), 155-180 CHAANINE A., KALMAN J., HAJJAR R. (2010) Cardiac Gene Therapy, Seminars in Thoracic and Cardiovascular Surgery, 22, (2), 127-139 CHAN E., TOWERS G.J., QASIM W. (2014) Gene therapy strategies to exploit TRIM derived restriction factors against HIV-1, Viruses, 6, 243-263 CHANG A.L., WOLF J.J., SMOLKE C.D. (2012) Synthetic RNA switches as a tool for temporal and spatial control over gene expression, Current Opinion in Biotechnology, 23, 679-688 CHANG C. et al. (2011) Structural Diversity Repertoire of Gene Silencing Small Interfering RNAs, Nucleic Acid Therapeutics, 21, (3), 125-131

119
CHEN Z-Y., LIN Y., YANG F., JIANG L., GE S.P. (2013) Gene therapy for cardiovascular disease mediated by ultrasound and microbubbles, Cardiovascular Ultrasound, 11, 1-14 CHUAH M.K.L., COLLEN D., VANDENDRIESSCHE T. (1998) Gene therapy for hemophilia : hopes and hurdles, Critical Reviews in Oncology/Hematology, 28, 153-171 CHUNG J., DiGIUSTO D.L., ROSSI J.J. (2013) Combinatorial RNA-based gene therapy for the treatment of HIV/AIDS, Expert Opinion on Biological Therapy, 13, (3), 437-445 COHEN-HAGUENAUER O. (2001) La thérapie génique, Editions TEC & DOC, Paris, 709p COLLINS S.A. et al. (2010) AAV2-mediated in vivo immune gene therapy of solid tumours, Genetic vaccines and therapy, 8, 1-13 COSTA P.M., PEDROSO DE LIMA M.C. (2013) MicroRNAs as molecular targets for cancer therapy : On the modulation of microRNA expression, Pharmaceuticals, 6, 1195-1220 COSTIN J.M. (2007) Cytopathic mechanisms of HIV-1, Virology Journal, 4, 100-122 COUGHLAN L. et al. (2010) Tropisme-modification strategies for targeted gene delivery using adenoviral vectors, Viruses, 2, 2290-2355 CRONIN J., ZHANG X.-Y., REISER J. (2005) Altering the tropism of lentiviral vectors through pseudotyping, Current Gene Therapy, 5, (4), 387-398 CUKJATI D., BATIUSKAITE D., ANDRE F., MIKLAVČIČ D., MIR L.M. (2007) Real time electroporation control for accurate and safe in vivo non-viral gene therapy, Bioelectochemistry, 70, 501-507 DENTON P.W., GARCIA J.V. (2011) Humanized mouse models of HIV infection, Aids Review, 13, (3), 135-148 DIETZ H. C. (2010) New therapeutic approaches to Mendelian disorders, Genomic Medecine, 363, 852-863

120
DOERING C. et al. (2009) Directed engineering of a high-expression chimeric transgene as a strategy for gene therapy of hemophilia A, Molecular therapy, 17, (7), 1145-1154 DOERING C., SPENCER H.T. (2009) Advancements in gene transfer-based therapy for hemophilia A, Expert Review of Hematology, 2, (6), 673-683 DONG Z., NÖR J.E. (2009) Transcriptional targeting of tumor endothelial cells for gene therapy, Advanced Drug Delivery Reviews, 61, (7-8), 542-553 DOUGLAS A.G.L., WOOD M.J.A. (2013) Splicing therapy for neuromuscular diseases, Molecular and cellular neurosciences, 56, (100), 169-185 DU L.M. et al. (2013) Platelet-targeted gene therapy with human factor VIII establishes haemostasis in dogs with haemophilia A, Nature Communications, 4, 1-11 DUAN D. (2011) Duchenne muscular dystrophy gene therapy : Lost in translation ?, Research and reports in biology, 2011, (2), 31-42 DUDLEY A.C. (2012) Tumor endothelial cells, Cold Spring Harbor Perspectives in Medicine, 2, 1-18 DUFAIT I. et al (2012) Retroviral and lentiviral vectors for the induction of immunological tolerance, Scientifica, 2012, 1-14 ELZOGHBY A.O. (2013) Gelatin-based nanoparticles as drug and gene delivery systems : reviewing three decades of research, Journal of Controlled Release, 172, 1075-1091 ENGELMAN A., CHEREPANOV P. (2012) The structural biology of HIV-1 : mechanistic and therapeutic insights, Nature Review Microbiology, 10, (4), 279-290 ESCORS D., BRECKPOT K. (2010) Lentiviral vectors in gene therapy : their current status ans future potential, Archivum Immunologiae et Therapiae Experimentalis, 58, (2), 107-119 FIELD A-C. et al. (2013) Comparison of lentiviral dans Sleeping Beauty mediated ab T cell receptor gene transfer, Plos One, 8, (6), 1-9

121
FIRE A. et al. (1998) Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans, Nature, 391, 806-811 Gene Therapy Clinical Trials Worldwide (page consultée en août 2014), Charts & Tables Adresse URL : http://www.wiley.com/legacy/wileychi/genmed/clinical/ GHOSAL G., MUNIYAPPA K. (2006) Hoogsteen base-pairing revisited : resolving a role in normal biological processes and human diseases, Biochemical and Biophysical Research Communications, 343, 1-7 GOUDENEGE S. et al. (2010) Laminin-111 : A potential therapeutic agent for Duchenne muscular dystrophy, Molecular Therapy, 18, (12), 2155-2163 GRADA Z. et al. (2013) TanCAR : A novel bispecific chimeric antigen receptor for cancer immuntherapy, Molecular therapy - Nucleic Acids, 2, 1-11 GREEN J.J. (2012) Nanoparticules for intracellular nucleic acid delivery, Annals of Biomedical Engineering, 40, (7), 1408-1418 GRESCH O. et al. (2004) New non-viral method for gene transfer into primary cells, Methods, 33, 151-163 GUO W., CHEN W., YU W., HUANG W., DENG W. (2013) Small interfering RNA-based molecular therapy of cancers, Chinese Journal of Cancer, 32, (9), 488-493 HACKETTC.S., GEURTS A.M., HACKETT P.B. (2007) Predicting preferential DNA vector insertion sites: implications for functional genomics and gene therapy, Genome Biology, 8, (supplément I), 1-17 HAIGWOOD N.L. (2009) Update on animal models for HIV research, European Journal of Immunology, 39, (8), 1994-1999 HAJJAR R.J. (2013) Potential of gene therapy as a treatment for heart failure, The journal of Clinical Investigation, 123, (1), 53-61 HERWEIJER H., WOLFF J.A. (2003) Progress and prospects: naked DNA gene transfer and therapy, Gene Therapy, 10, 453-458

122
HO D., YAN L., IWATSUBO K., VATNER D.E., VATNER S.F. (2010) Modulation of β-adrenergic receptor signaling in heart failure and longevity : targeting adenylyl cyclase type 5, Heart Failure Review, 15, (5), 495-512 HOFFMAN E.P., BROWN R.H., KUNKEL L.M. (1987) Dystrophin : The protein product of Duchenne muscular dystrophy locus, Cell, 51, 919-928 HOLKERS M. (2013) Differential integrity of TALE nuclease genes following adenoviral and lentiviral vector gene transfer into human cells, Nucleic Acids Research, 41, (5), 1-14 HU C.,LIPSHUTZ G.S. (2012) AAV-based neonatal gene therapy for hemophilia A : Long-term correction and avoidance of immune responses in mice, Gene Therapy, 19, (12), 1166-1176 HUANG Y. et al. (2013) Delivery of small interfering RNAs in human cervical cancer cells by polyethylenimine-functionalized carbon nanotubes, Nanoscale Research Letters, 8, 267-278 ISRAEL D.I., KAUFMAN R.J. (1990) Retroviral-mediated transfer and amplification of a functional human factor VIII gene, Blood, 75, (5), 1074-1080 JIN X., YANG Y-D., LI Y-M. (2008) Gene therapy : regulations, ethics and its practicalities in liver disease, World Jounral of Gastroenterology, 14, (15), 2303-2307 JOHNSTON J.M., DENNING G., DOERING C.B., TRENT SPENCER H. (2013) Generation of an optimized lentiviral vector encoding a hign-expression factor VIII transgene for gene therapy of hemophilia A, Gene Therapy, 20, (6), 607-615 JOURDIER T.M. et al. (2003) Local immunotherapy of spontaneous feline fibrosarcomas using recombinant poxviruses expressing interleukin 2 (IL2), Gene Therapy, 10, 2126-2132 JUNG S. et al. (2013) Sleeping Beauty transposon system harboring HRAS, c-Myc and shp53 induces sarcomatoid carcinomas in mouse skin, Oncology Reports, 29, 1293-1298

123
KAIROUZ V., LIPSKAIA L., HAJJAR R.J., CHEMALY E.R. (2012) Molecular targets in heart failure gene therapy : current controversies and translational perspectives, Annals of the New York Academy of Sciences , 1254, 42-50 KALIBEROV S.A., BUSCHBAUM D.J. (2012) Cancer treatment with gene therapy and radiation therapy, Advanced Cancer Research, 115, 221-263 KAMIMURA K., SUDA T., ZHANG G., LIU D. (2011) Advances in gene delivery systems, Pharmaceutical Medecine, 25, (5), 293-306 KASHIWAKURA Y. et al. (2012) Porcine model of hemophilia A, Plos One, 11, (7), 1-6 KAUFMANN K. B., BÜNING H., GALY A., SCHAMBACH A., GREZ M. (2013) Gene therapy on the move, EMBO Molecular Medicine, 5, 1642-1661 KAY M., HIGH K. (1999) Gene therapy for the hemophilias, Proceedings of the National Academy of Sciences, 96, 9973-9975 KITCHEN S.G., SHIMIZU S., AN D.S. (2011) Stem cell based anti-HIV gene therapy, Virology, 411, (2), 260-272 KOHN D.B. et al. (1999) A clinical trial of retroviral-mediated transfer of a rev-responsive element decoy gene into CD34+ cells from the bone marrow of human deficiency virus-1 infected children, Blood, 94, 368-371 KONG F., ZHOU F., GE L., LIU X., WANG Y. (2012) Mannosylated liposomes for targeted gene delivery, International Journal of Nanomedecine, 7, 1079-1089 KORNEGAY J.N. et al. (2012) Canine models of Duchenne muscular dystrophy and their use in therapeutic strategies, Mammalian Genome, 23, 85-108 KOTULA J. W. et al. (2012) Aptamer-mediated delivery of splice-switching oligonucleotides to nuclei of cancer cells, Nucleic Acid Therapeutics, 22, (3), 187-195 KREN B.T. et al. (2009) Nanocapsule-delivered Sleeping Beauty mediates therapeutic factor VIII expression in liver sinusoidal endothelial cells of hemophilia A mice, The journal of Clinical Investigation, 119, (7), 2086-2099

124
KUNERT A. et al. (2013) TCR-engineered T cells meet new challenges to treat solid tumors : choice of antigen, T cell fitness, and sensitization of tumor milieu, Frontiers in Immunology, 4, 1-16 KWIATKOWSKA A., NANDHU M.S., BEHERA P., CHIOCCA E.A., VIAPIANO M.S. (2013) Strategies in gene therapy for glioblastoma, Cancers, 5, 1271-1305 LADEWIG K., NIEBERT M., XU Z.P., GRAY P.P., LU G.Q.M. (2010) Efficient si RNA delivery to mammalian cells using layered double hydroxide nanoparticles, Biomaterials, 31, 1821-1829 LAI Y., DUAN D. (2012) Progress in gene therapy of dystrophic heart disease, Gene Therapy, 19, (6), 678-685 LASEK W., ZAGOŻDŻON R., JAKOBISIAK M. (2014) Interleukin 12 : still a promising candidate for tumor immunotherapy ?, Cancer Immunology, Immunotherapy, 63, 419-435 LEE Y. et al. (2012) Human Erythropoietin Gene Delivery Using an Arginine-grafted Bioreducible Polymer System, Molecular therapy, 20, (7), 1360-1366 LENTACKER I., DE COCK I., DECKERS R., DE SMEDT S.C., MOONEN C.T.W. (2013) Understanding ultrasound induced sonoporation: Definitions and underlying mechanisms, Advanced Drug Delivery Reviews, (à paraître) LENTZ T.B., GRAY S.J., SAMULSKI J. (2012) Viral vectors for gene delivery to the central nervous system, Neurobiology of Disease, 48, (2), 179-188 LI C., LI L., KEATES A.C. (2012) Targetting cancer gene therapy with magnetic nanoparticles, Oncotarget, 3, (4), 365-370 LI H. et al. (2011) Assessing the potential for aav vector genotoxicity in a murine model, Blood, 117, (12), 3311-3319 LI Z., RANA T.M. (2012) Molecular mechanisms of RNA-triggered gene silencing machineries, Accounts of chemical research, 45, (7), 1122-1131

125
LIEW W.K.M., KANG P.B. (2013) Decent developments in the treatment of Duchenne muscular dystrophy and spinal muscular atrophy, Therapeutic Advances in Neurological Disorders, 6, (3), 147-160 LIM L.P. et al. (2003) The microRNAs of Caenorhabditis elegans, Genes & Development, 17, 991-1008 LIN J. et al. (2012) Stabilization of G-quadruplex DNA by C-5-methyl-cytosine in bcl-2 promoter: Implications for epigenetic regulation, Biochemical and Biophysical Research Communications, 433, 368-373 LINT C.V., BOUCHAT S., MARCELLO A. (2013) HIV-1 transcription and latency : an update, Retrovirology, 10, 67-105 LIU Z. et al. (2012) Mannan-modified Ad5-PTEN treatment combined with docetaxel improves the therapeutic effect in H22 tumor-bearing mice, International Journal of Nanomedecine, 7, 5039-5049 LU H. et al. (2008) Complete correction of hemophilia A with adeno-associated viral vectos containing a full-size expression cassette, Human Gene Therapy, 19, (6), 648-654 LU Y., ZHANG X., ZHANG J. (2012) Inhibition of breast tumor cell growth by ectopic expression of p16/INK4A via combined effects of cell cycle arrest, senescence and apoptotic induction, and angiogenesis inhibition, Journal of Cancer, 3, 333-344 LV H., ZHANG S., WANG B., CUI S., YAN J. (2006) Toxicity of cationic lipids and cationic polymers in gene delivery, Journal of Controlled Release, 114, 100-109 MALI P. et al. (2013) RNA-guided human genome engineering via Cas9, Science, 339, (6121), 823-826 MANDEVILLE J-S., BOURASSA P., THOMAS T.J., TAJMIR-RIAHI H-A. (2012) Biogenic and synthetic polyamines bind cationic dendrimers, Plos One, 7, (4) MANJILA S. B. et al. (2013) Novel gene delivery systems, International Journal of pharmaceutical investigation, 3, (1), 1-7

126
MANJUNATH N., YI G., DANG Y., SHANKAR P. (2013) Newer gene editing technologies toward HIV gene therapy, Viruses, 5, 2748-2766 MANNELL H. et al. (2012) Site directed vascular gene delivery in vivo by ultrasonic destruction of magnetic nanoparticle coated microbubbles, Nanomedecine : Nanotechnology, Biology and Medecine, 8, 1309-1318 MAO S., SUN W., KISSEL T. (2009) Chitosan-based formulations for delivery of DNA and siRNA, Advanced Drug Delivery Reviews, 62, 12-27 MARKUSIC D., HERZOG R.W. (2013) Liver-directed adeno-associated viral gene therapy for hemophilia, Jounal of genetic syndromes & gene therapy, 1, 1-9 MARQUEZ R.T., McCAFFREY A.P. (2008) Advances in microRNAs : Implications for gene therapists, Human Gene Therapy, 19, (1), 27-38 MENDELL J.R. et al. (2012) Gene therapy for muscular dystrophy : Lessons learned and path forward, Neuroscience Letters, 527, (2), 90-99 MIAO C.H. (2010) Immunomodulation for inhibitors in hemophilia A : the important role of Treg cells, Expert Review of Hematology, 3, (4), 469-483 MIELE E. et al. (2012) Nanoparticle-based delivery of small interfering RNA: challenges for cancer therapy, International Jounal of Nanomedecine, 7, 3637-3657 MIRAMBEAU G., LYONNAIS S., GORELICK R.J. (2010) Features, processing states and heterologous protein interactions in modulation of retroviral nucleocapsid protein function, RNA Biology, 7, (6), 724-734 MODIANO J.F. et al. (2012) Inflammation, Apoptosis, and necrosis induced by neoadjuvant fas ligand gene therapy improves survival of dogs with spontaneous bone cancer, Molecular therapy, 20, (12), 2234-2243 MONTGOMERY R.R., SHI Q. (2010) Alternative stratégies for gene therapy of hemophilia, Hematology / the Education Program of the American Society of Hematology, 2010, 197-202 MURPHY A.M., RABKIN S.D. (2013) Current status of gene therapy for brain tumors, Translational Research, 161, (4), 339-354

127
NAIDOO J., YOUNG D. (2012) Gene regulation systems for gene therapy applications in the central nervous system, Neurology Research International, 2012, 1-10 NAIM C., YEREVANIAN A., HAJJAR R.J. (2013) Gene therapy for heart failure : where do we stand ?, Current Cardiology Report, 15, (2), 1-16 NAKAMURA A., TAKEDA S. (2011) Mammalian models of Duchenne muscular dystrophy : Pathological characteristics and therapeutic applications, Journal of Biomedecine and Biotechnology, 2011, 1-8 NAM H.Y. et al. (2009) Biodegradable PANAM ester for enhanced transfection efficiency with low cytotoxicity, Biomaterials, 30, 665-673 NAYEROSSADAT N., MAEDEH T., ABAS ALI P. (2012) Viral and nonviral delivery systems for gene therapy, Advanced biomedical research, 1, 3-27 NI X., CASTANARES M., MUKHERJEE A., LUPOLD S.E. (2011) Nucleic acid aptamers : clinical applications and promising new horizons, Current Medicinal Chemistry, 18, (27), 4206-4214 OCHIYA T., TAKENAGA K., ENDO H. (2014) Silencing of S100A4, a metastasis-associated protein, in endothelial cells inhibits tumor angiogenesis and growth, Angiogenesis, 17, 17-26 OKADA T., TAKEDA S. (2013) Current challenges and future directions in recombinant AAV-mediated gene therapy of Duchenne muscular dystrophy, Pharmaceuticals, 2013, (6), 813-836 PAN Y. et al. (2013) Gene therapy for cancer through adenovirus vector-mediated expression of the Ad5 early region gene 1A based on loss of IGF2 imprinting, Oncology Reports, 30, 1814-1822 PANDHARE J., DASH C. (2011) A prospective on drug abused epigenetics and HIV-1 replication, Life Science, 88, (21-22), 995-999 PAPANIKOLAOU E. et al. (2012) The New Self-Inactivating Lentiviral Vector for Thalassemia Gene Therapy Combining Two HPFH Activating Elements Corrects Human Thalassemic Hematopoietic Stem Cells, Human Gene Therapy, 23, 15-31

128
PAVLIN D., CEMAZAR M., GREGOR S., TOZON N. (2012) IL-12 based gene therapy in veterinary medicine, Journal of translational medecine, 10, 234-245 PEI X., ZHANG J., LIU J. (2014) Clinical applications of nucleic acid aptamers in cancer (Review), Molecular and clinical oncology, 2, 341-348 PEREZ-LUZ S., DIAZ-NIDO J. (2010) Prospects for the Use of Artificial Chromosomes and Minichromosomes-Like Episomes in Gene Therapy, Journal of Biomedecine and Biotechnology, 2010, (4), 1-16 PESSACH I. M., NOTARANGELO L. D. (2011) Gene therapy for primary immunodeficiencies : looking ahead, towards gene correction, Journal of allergy and clinical immunology, 127, (6), 1344-1350 PICHAVANT C. et al. (2011) Current status of pharmaceutical and genetic therapeutic approaches to treat DMD, Molecular therapy, 19, (5), 830-840 PISSUWAN D., NIIDOME T., CORTIE M.B. (2011) The forthcoming applications of gold nanoparticles in drug and gene delivery systems, Journal of Controlled Release, 149, 65-71 PLANK C., ZELPHATI O., MYKHAYLYK O. (2011) Magnetically enhanced nucleic acid delivery. Ten years of magnetofection—Progress and prospects, Advanced Drug Delivery Reviews, 63, 1300-1331 PORADA C.D. et al. (2010) Clinical and molecular characterization of a re-established line of sheep exhibiting hemophilia A, Journal of Thrombosis and Haemostasis, 8, (2), 276-285 PORADA C.D., ALMEIDA-PORADA G. (2012) Treatment of hemophilia A in utero and postnatally using sheep as model for cell and gene delivery, Journal of genetic syndromes and gene therapy, supplément 1, 1-26 POWELL J.S. et al. (2003) Phase 1 trial of FVIII gene transfer for severe hemophilia A using a retroviral construct administered by peripheral intravenous infusion, Blood, 102, 2038-2045 PRABHA S., SHARMA B., LABHASETWAR V. (2012) Inhibition of tumor angiogenesis and growth by nanoparticle-mediated p53 gene therapy in mice, Cancer Gene Therapy, 19, (8), 530-537

129
RAMOS C., DOTTI G. (2011) Chimeric Antigen Receptor (CAR)-engineered lymphocytes for cancer therapy, Expert opinion on biological therapy, 11, (7), 855-873 RANKI T., HEMMINKI A. (2010) Serotype chimeric human adenoviruses for cancer gene therapy, Viruses, 2, 2196-2212 RENGO G. et al. (2009) Myocardial Adeno-Associated Virus Serotype 6-βARKct Gene Therapy Improves Cardiac Function and Normalizes the Neurohormonal Axis in Chronic Heart Failure, Circulation, 119, (1), 89-98 RENGO G., LYMPEROPOULOS A., LEOSCO D., KOCH W. (2011) GRK2 as a Novel Gene Therapy Target in Heart Failure, Journal of molecular and cellular cardiology, 50, (5), 785-792 RICHTER C., CHANG J.T., FINERAN P.C. (2012) Function and regulation of Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) / CRISPR Associated (Cas) systems, Viruses, 4, 2291-2311 RODRIGUEZ L. et al. (2013) Polyurine reverse Hoogsteen hairpins as a gene therapy tool against survivin in human prostate cancer PC3 cells in vitro and in vivo, Biochemical Pharmacology, 86, 1541-1554 ROSSI J.J., JUNE C.H., KOHN D.B. (2007) Genetic therapies against HIV, Nature Biotechnology, 25, (12), 1444-1454 SABATINO D.E. et al. (2011) Efficacy and safety of long-term prophylaxis in severe hemophilia A dogs following liver gene therapy using AAV vectors, Molecular therapy, 19, (3), 442-449 SABATINO D.E. et al. (2012) Animal models of hemophilia, Progress in Molecular Biology and Translational Science, 105, 151-209 SARGENT R. G., KIM S., GRUENERT D. C. (2011) Oligo/polynucleotide-based gene modification : strategies and therapeutic potential, Oligonucleotides, 21, (2), 55-75 SARKER S. et al. (2013) Arginine-based cationic liposomes for efficient in vitro plasmid DNA delivery with low cytotoxicity, International Jounal of Nanomedecine, 8,1361-1375

130
SCHMITT T.M., RAGNARSSON G.B., GREENBERG P.D. (2009) T cell receptor gene therapy for cancer, Human Gene Therapy, 20, 1240-1248 SEN D. et al. (2013) Improved adeno-associated virus serotype 1 and 5 vectors for gene therapy, Scientific Reports, 3, 1832-1838 SHARMA A., LI X., BANGARI D.S., MITTAL S.K. (2009) Adenovirus receptors and their implications in gene delivery, Virus Research, 143, (2), 184-194 SHEN H., MITTAL V., FERRARI M., CHANG J. (2013) Delivery of gene silencing agents for breast cancer therapy, Breast Cancer Research, 15, 1-8 SKIPPER A.A., ANDERSEN P.R., SHARMA N., MIKKELSEN J.G. (2013) DNA transposon-based gene vehicles - scenes from a evolutionary drive, Journal of Biomedical Science, 20, 92-113 SMITH K.R. (2004) Gene Therapy: The Potential Applicability of Gene Transfer Technology to the Human Germline, International journal of medical sciences, 1, (2), 76-91 SNG J., LUFKIN T. (2012) Emerging Stem Cell Therapies : Treatment, Safety and Biology, Stem Cells International, 2012, 1-9 SPENCER T.E., PALMARINI M. (2012) Application of next generation sequencing in mammalian embryogenomics: Lessons learned from endogenous betaretroviruses of sheep, Animal Reproduction Science, 134, 95-103 SU R-J. et al. (2011) Suppression of the immune response to fVIII in hemophilia A mice by transgène modified tolerogenic dendritic cells, Molecular therapy, 19, (10), 1896-1904 SUBRAMANYA S., KIM S-S., MANJUNATH N., SHANKAR P. (2010) RNA interference-based therapeutics for human immunodeficiency virus HIV-1 treatment : synthetic siRNA or vector-based shRNA ?, Expert Opinion on Biological Therapy, 10, (2), 201-213 SULLIVAN J.M. et al. (2011) Variables and stratégies in development of therapeutic post-transcriptional gene silencing agents, Jounrnal of Ophtalmology, 2011, 1-31

131
TAKASU Y. et al. (2013) Efficient TALEN construction for Bombyx mori Gene targeting, Plos One, 8, (9), 1-11 TROBRIDGE G.D., KIEM H-P. (2010) Large animal models of hematopoietic stem cell gene therapy, Gene Therapy, 17, (8), 939-948 VALTON J. et al. (2014) Efficient strategies for TALEN-mediated genome editing in mammalian cell lines, Methods, article en presse VINGE L.E., RAAKE P.W., KOCH W. (2008) Gene therapy in Heart Failure, Circulation Research, 102, (12), 1458-1470 WADFORD D.A. et al. (2014) Variationof human immunodeficiency virus type 1 reverse transcriptase within the simian immunodeficiency virus genome of RT-SHIV, Plos One, 9, (1), 1-14 WALSH C.E., BATT K.M. (2013) Hemophilia clinical gene therapy : brief review, Translational Research, 161, 307-312 WANG B., LI J., XIAO X (2000) Adeno-associated virus vector carrying human minidystrophin genes effectively ameliorates muscular dystrophy in mdx mouse model, Proceedings of the National Academy of Sciences, 97, (25), 13714-13719 WANG C. et al. (2012) Dual-purpose magnetic micelles for MRI and gene delivery, Journal of Controlled Release, 163, 82-92 WANG Z., CHAMBERLAIN J.S., TAPSCOTT S.J., STORB R. (2009) Gene therapy in large animal models of muscular dystrophy, Institute of Laboratory Animal Resources Journal, 50, (2), 187-198 WOLFF J.A., BUDKER V. (2005) The mechanism of naked DNA uptake and expression, Advances in Genetics, 54, 1-20 XU J., FENG E., SONG J. (2014) Renaissance of aliphatic polycarbonates : New techniques and biomedical applications, Journal of Applied Polymer Science, 131, (5), 1-16 YAN S. et al. (2012) High levels of gene expression in the hepatocytes of adult mice, neonatal mice and tree shrews via retro-orbital sinus hydrodynamic injections of naked plasmid DNA, Journal of Controlled Release, 161, 763-771

132
ZARAGOZA C. et al. (2011) Animal models of cardiovascular diseases, Journal of Biomedecine and Biotechnology, 2011, 1-13 ZAROGOULIDIS P. et al. (2013) Suicide gene therapy for cancer - Current strategies, Journal of genetic syndromes and gene therapy, 4, 1-29 ZHANG Y., SATTERLEE A., HUANG L. (2012) In vivo gene delivery by nonviral vectors : overcoming hurdles ?, Molecular therapy, 20, (7), 1298-1304 ZHENG et al. (2012) Preparation and characterization of magnetic gene vectors for targeting gene delivery, Applied Surface Science, 259, 201-207 ZHONG L. et al. (2012) Development of Novel Recombinant AAV Vectors and Strategies for the Potential Gene Therapy of Hemophilia, Journal of genetic syndromes and gene therapy, 10, (supplément), 1-16

133
NOM PRENOM : HELLEZ Justine TITRE : Les utilisations de la thérapie génique Thèse d’Etat de Doctorat Vétérinaire : Lyon, le 27 Octobre 2014 RESUME : La thérapie génique est une biotechnologie qui utilise des acides nucléiques afin de traiter les maladies génétiques comme acquises. La stratégie de thérapie génique consiste à insérer un ou des acides nucléiques dans les cellules d’un individu malade, via un vecteur. Ces acides nucléiques doivent permettre à la cellule de produire le médicament nécessaire à sa guérison. De nombreuses approches ont été développées à cette fin. Différents types d’acides nucléiques peuvent être utilisés (allèles, oligonucléotides, ARN, aptamères, …). Chaque type d’acides nucléiques permet une approche différente de la modification du génome nécessaire à la guérison (ajout d’un allèle fonctionnel, réparation du gène, extinction génique ou expression forcée). La voie d’administration de la thérapie génique est aussi un axe de recherche important. Elle doit permettre au traitement de thérapie génique d’accéder à un nombre suffisant de cellules malades afin d’obtenir un effet thérapeutique. Enfin, de nombreux vecteurs ont été créés. On peut les classer en deux types : les vecteurs viraux, créés à partir d’un ou plusieurs virus et les vecteurs non viraux, fabriqués à partir de différentes molécules (lipides, protéines, minéraux, …). On peut aussi utiliser les acides nucléiques sans autre molécule : on parle alors d’acides nucléiques nus. Chaque maladie présente ses particularités propres ce qui oblige à développer pour chacune d’entre elles un traitement de thérapie génique unique. A ce jour, deux traitements de thérapie génique sont disponibles sur le marché du médicament. Cependant, la thérapie génique en est encore aux essais cliniques pour quasi-totalité des maladies auxquelles on essaie de l’appliquer. MOTS CLES : - Thérapie génique - Insuffisance cardiaque - Maladies héréditaires - Modèles animaux - Molécules recombinées JURY : Président : Monsieur le Professeur SANLAVILLE Damien 1er Assesseur : Madame LAMBERT Véronique, Maître de Conférences 2ème Assesseur : Madame LATTARD Virginie, Maître de Conférences DATE DE SOUTENANCE : Le 27 Octobre 2014 ADRESSE DE L’AUTEUR : 3 rue de la Roche Durand, Appartement 113 22360 LANGUEUX





























