Embed Size (px)
Citation preview

Pr. Jean-Louis Stéphan, Service de Pédiatrie - CHU Saint-Etienne
Cahier n°1


- 1 -
Som
ma
ire
Cahier n°1
01- Introduction à l’étude des DIP 3
Quand suspecter une anomalie
constitutionnelle de l’immunité ? 4
Comment affirmer simplement
l’existence d’un DIP ? 7
02- Les déficits syndromiques 14
L’anomalie de Di George 14
Le cartilage-hair hypoplasia 22
Le syndrome de Netherton 26
Les candidoses muco-cutanées
chroniques (CMC) 30
Les syndromes Hyper IgE (HIE) 31
Les syndromes d’instabilité
chromosomique ou réparatoses 44
03- Annexes 60
Glossaire 60
Valeurs de référence 76
04- Références 77

ADG : Anomalie de Di George (ou DG)
AFP : Alpha-fœtoprotéine
ALPS : Auto-immune LymphoproliferativeSyndrome (syndrome lymphoprolifératif avec auto-immunité)
APECED : Autoimmune Polyendocrinopathy -Candidiasis - Ectodermal - Dystrophy
ARNr : ARN ribosomaux
ATM : Serine/threonine protein kinaseAtaxiatelangiectasia Mutated
BCG : Bacille de Calmette et Guérin
BrdU : Bromodéoxyuridine
CEREDIH : Centre de Référence DéficitsImmunitaires Héréditaires
CHH : Cartilage-hair Hypoplasia
CMC : Candidoses Muco-cutanées Chroniques
CMF : Cytométrie de Flux
CV : Charge Virale
DICV : Déficit Immunitaire Commun Variable
DIP : Déficits Immunitaires Primitifs
EBV : Epstein-Barr Virus
FISH : Hybridation in situ en Fluorescence
FRH : Fièvres Récurrentes Héréditaires
Greffe de MO : Greffe de Moelle Osseuse
HIE : Syndromes Hyper IgE
HSV : Herpes simplex virus
ICF : Déficit Immunitaire, Instabilité Centromérique,anomalies Faciales
IPEX : Immune dysregulation, Polyendocrinopathy,Enteropathy, X-linked
IRM : Imagerie par Résonance Magnétique
LEKTI : Lymphoepithelial Kazal Type relatedInhibitor
LT : Lymphocyte T
LTa : Lymphotoxine a
NK : Natural Killer
MBL : Mannose-Binding Lectin
MIM : Online Mendelian Inheritance in Man
MLPA : Multiplex Ligation-dependent ProbeAmplification
MNI : Mononucléose Infectieuse
MO : Microscopie Optique
Nbs : Syndrome de Nijmegen (Nijmegen BreakageSyndrome)
nbt : Test au Nitrobleu de Tétrazolium
PCR : Polymerase Chain Amplification
PN : Polynucléaires
RA : Acide Rétinoïque
RMRP : Ribonuclease Mitochondrial RNA Processing
ROR : Retinoïd Related Orphan Receptor
SLP : Syndrome Lymphoprolifératif
SNC : Système Nerveux Central
snoRNAs : Small Nucleolar RNA
TDM : Tomodensitométrie
TRECS : T cell Receptor Excision Circles
UV : Ultra-violets
Ca : Calcémie
CRP : C-Réactive Protéine
Fe : Fer sérique
GB : Globules Blancs
G/l : Giga/litre
Hb : Hémoglobine
LCR : Liquide Céphalorachidien
Ly : Lymphocytes
Na : Natrémie
PLT : Plaquettes
PN : Polynucléaires Neutrophiles
VGM :Volume Globulaire Moyen
Termes de biologieTermes de biologie
Liste des abréviations Liste des abréviations
- 2 -

DEF
AU
TS G
ENET
IQU
ES D
E LA
REP
ON
SE IM
MU
NIT
AIR
EIn
trod
uctio
n à
l’étu
de d
es D
IP
01
année avec un DIP etqu’environ 5000 patients,enfants et adultesvivraient avec un teldéficit.3,4
En outre, la prise encharge a d’autant plusde chance d’être efficacequ’elle sera mise en placeprécocement. C’est le casde la substitution par
immunoglobulines dans les anomalies de la fonctionhumorale, de la greffe de moelle dans les déficits Tou de l’antibiothérapie prophylactique. La correctiondu défaut génétique par transfert de gène est mêmedésormais possible et en cours d’évaluation pourun petit nombre d’entre eux.5
Avant d’illustrer les formes syndromiques de DIP*
qui constitueront l’objet de ce 1er cahier, quelques«pistes » pour le diagnostic des DIP et des autreserreurs innées de l’immunité seront brièvementexposées.
Les anomalies constitutionnelles del’immunité sont multiples et varient dansleur traduction clinique. Les déficitsimmunitaires primitifs (DIP) sontcaractérisés par une atteinte qualitativeet/ou quantitative du système immunitaire,entraînant une susceptibilité accrue auxinfections. Plus de 300 maladies sontdécrites et des mutations répertoriéesdans quelques 180 gènes, et de trèsnombreux mécanismes impliqués(cf répartition des différents DIP).1,2 Avecles techniques de séquençage génomeentier et exome entier le rythme despublications s'est accéléré et l'on peutprédire l'élucidation de centaines de DIPdans les 10 ans à venir (JL Casanova).1,2
Leur meilleure définition à l'échelonmoléculaire permet aujourd’hui d’envisager,pour un grand nombre, un conseil génétiqueet un diagnostic prénatal. La prévalencedans la population générale est assezmal connue et en France on estimequ’environ 150 enfants naissent chaque
Le diagnostic des DIP est une véritable urgence car ces affections mettent en jeu le pronostic vital en raison de la susceptibilité aux infections ou des manifestations auto-immunes graves,auxquelles elles exposent.
- 3 -
Introduction à l’étudedes DIP
55%22%
5%
10%
8%
Déficits humoraux
Déficits en lymphocytes T
Déficits en cellules phagocytaires
Anomalies du complément
Autres
Répartition des différents DIP (2011) :Données de la SociétéEuropéenne des DéficitsImmunitaires -ESID-http://www.esid.org/statistics.php?sub=
* Cf. glossaire page 60

- 4 -
DEF
AU
TS G
ENET
IQU
ES D
E LA
REP
ON
SE IM
MU
NIT
AIR
EIn
trod
uctio
n à
l’étu
de d
es D
IP
01
7 8 9Mycose cutanée ou muqueuse
récidivante ou chronique
Nécessité d’un traitement
antibiotique par voie IV
2 infections sévères
dans l’année
4 5 6Plus de 3 pneumonies Ralentissement de la croissance
1 2 33Plus de 8 otites par an Plus de 2 sinusites par an Plus de 2 mois de traitement
antibiotique par an
Les signes cliniques d’alerte
10
Attention : L’un ou l’autre de ces signes cliniques peuvent laisser penser à un déficit
immunitaire primitif, sans toutefois qu’il en soit systématiquement ainsi.
En cas de suspicion de déficit immunitaire primitif, nous vous conseillons
de consulter le centre de compétences.
connu dans la famille
Premiers examens diagnostics devant une suspicion clinique
• Dosage pondéral des immunoglobulines (IgG, IgA, IgM).
• Numération Formule Sanguine (valeurs du taux de lymphocytes à interpréter selon l’âge) + frottis.
• Sérologie post vaccinale et/ou post infectieuse.
Il n’y a aucune indication de réaliser un dosage des sous-classes d’immunoglobulines G
avant l’âge de 18 mois.
C'est la récurrence et la précocité desinfections qui doivent interpeler le praticien,surtout si ces infections revêtent un caractèreinhabituel, telles celles à pathogèneopportuniste [Aspergillus, Cytomegalovirus,Pneumocystis jiroveci (ex carinii), Toxoplasmagondii), un muguet récalcitrant malgré untraitement bien conduit, une pneumopathieinterstitielle ou une infection grave par EBV(Epstein-Barr Virus).
De façon peut-être plus inattendue, les anomaliesde différenciation des lymphocytes B et T ainsique celles du complément rendent les patientsparticulièrement susceptibles aux manifestationsauto-immunes (cytopénie autoimmune, vascularite),même en l’absence d’infection. Elles prédominentdans les syndromes de dysrégulation immunitaire(IPEX, APECED et ALPS)*. Les défauts génétiquesde la cytotoxicité se révèlent par un syndromed’activation lymphohistiocytaire (lymphohistiocytoses,DI associés à un albinisme).
Le DIP peut passer au second plan derrière unesémiologie qui «appartient» à une autre spécialitépédiatrique : syndromes malformatifs, troublesneurologiques, affections dermatologiques ouosseuses. Ce sont les formes syndromiquesdes déficits immunitaires.
Certains de ces enfants peuvent développer unenéoplasie (gènes impliqués dans la régulation du cyclecellulaire et dans la réparation des dommagesgénotoxiques). Les hémopathies lymphoïdes etles tumeurs solides concernent aussi l’adulte atteintde Déficit Immunitaire Commun Variable (DICV).
Les fièvres récurrentes héréditaires (FRH) sont desmaladies génétiques caractérisées par un syndromeinflammatoire en l'absence de stigmate d'auto-immunité (absence d’auto-anticorps et de cellules Tantigène-spécifiques et sans déclenchement parun antigène). Elles sont caractérisées par des accèsrécurrents de durée variable, de douleurs abdominaleset/ou articulaires et des signes cutanés, et une sensibilitétrès variable aux différentes thérapeutiques proposées.Avec d'autres maladies chroniques inflammatoirestelles que la maladie de Crohn, les FRH constituentle groupe des maladies baptisées «auto-inflammatoires».
* IPEX : Immune dysregulation, Polyendocrinopathy, Enteropathy, X-linked ; APECED : Autoimmune Polyendocrinopathy - Candidiasis - Ectodermal - Dystrophy ; ALPS : Auto-immune Lymphoproliferative Syndrome (syndrome lymphoprolifératif avec auto-immunité)
Remerciements à l’association IRIS
constitutionnelle de l'immunité ? (6,7)
1.1 Quand suspecter une anomalie 1.1 Quand suspecter une anomalieconstitutionnelle de l'immunité ? (6,7)

Les prises en charge diagnostique et thérapeutique des DIP sont coordonnées par unréseau spécialisé de médecins cliniciens (pédiatrie et adultes) et de biologistes. Ce réseauest coordonné par le Centre de Référence Déficits Immunitaires Héréditaires (CEREDIH)et un comité scientifique national. Le CEREDIH propose des consultations d’expertscliniques, des explorations immunologiques et des examens génétiques (à viséediagnostique ou à la suite d’un conseil génétique) et collige l’ensemble des observationssur le territoire national pour l’étude de l’épidémiologie des DIP. http://www.ceredih.fr/
Introduction à l’étudedes DIP
Qua
nd sus
pecter une
ano
malie cons
titu
tionn
elle de l'immun
ité ?
- 5 -
L'interrogatoire de la famille et du carnetde santé donnent des informations trèsprécieuses sur la généalogie familiale (cancers,histoire familiale de décès dans la petite enfance,parents apparentés, sexe des patients qui sontéventuellement atteints). Quelles sont lesvaccinations réalisées (pour l’étude ultérieuredes sérologies post-vaccinales), le BCG a-t-ilété fait (vaccin vivant…) ? L’enfant a-t-ilprésenté les maladies éruptives classiquesde la petite enfance (varicelle, mononucléoseinfectieuse…) ?
Il faut distinguer la situation de « l'enfanttoujours malade », qui fréquente la crèche,enchifrené, présentant certes des infectionsrépétées de la sphère ORL ou broncho-pulmonaire mais toujours bénignes et plutôtsaisonnières. L'évaluation clinique est essentielle;l'enfant est en forme, garde une courbe staturo-pondérale parfaitement normale : c'est en fait« l'immaturité immunitaire», la disparitionnormale des immunoglobulines maternellestransmises de façon passive, le terrain allergique,l’hypertrophie des végétations adénoïdes,le reflux gastro-œsophagien qui sous-tendentcette situation (observation page suivante).
Certains DIP peuvent se révéler très tardivement,à l’adolescence ou chez l’adulte, sous des masquesdivers (granulomes pulmonaires ou hépatospléniques,cytopénies, lymphomes, infections sévères à HPV(Human Polyomavirus), maladie «coeliaque-like»,hyperplasie nodulaire lymphoïde digestive, dilatationdes bronches).8

Exemple d’observation
Très beau petit garçon âgé de 8 mois, bien dodu, adressé enconsultation pour de nombreuses infections annotées régulièrementsur le carnet de santé. L’examen clinique est rigoureusementnormal. La courbe staturo-pondérale est irréprochable. Ces infectionsrécurrentes sont relativement bénignes, n’ayant jamais entraînéune hospitalisation et n’ayant aucune conséquence sur sa trophicité.
Le dosage pondéral des immunoglobulines montre unehypogammaglobulinémie IgG = 2g/l ; IgA = 0,10g/l ; IgM = 0,35g/lportant sur essentiellement les IgG. Les IgA sont présentes, les IgMsont normales pour son âge. Les anticorps anti-tétaniques sontprésents à un titre protecteur. Le chiffre de lymphocytes est normal.
Le terrain allergique avec une suspicion d’allergie aux protéines du lait de vache et une authentiqueallergie dans la famille, au moins chez une grand-mère, jouent sûrement un rôle délétère. Il n’ya aucune prise en charge spécifique et ces constatations permettent de rassurer les parents sansrégler le problème pour autant. On peut aussi discuter l’ablation des végétations avec l’ORL.Cette « hypogammaglobulinémie physiologique » devrait en fait se corriger dans les mois(ou années…) qui suivent.
- 6 -
DEF
AU
TS G
ENET
IQU
ES D
E LA
REP
ON
SE IM
MU
NIT
AIR
EIn
trod
uctio
n à
l’étu
de d
es D
IP
01
L’association IRIS représente en France les patients atteints d’un déficit immunitaire primitif.
Les objectifs d’IRIS :
• Développer l’information sur les déficits immunitaires primitifs et leurs traitements,les circuits de prise de prise en charge et l’état de la recherche améliorer le diagnostic.
• Soutenir les patients et leurs familles, vaincre l’isolement, permettre des échanges,organiser des spectacles et animations pour soutenir les enfants durant l’hospitalisation(Soutien de LFB BIOMEDICAMENTS), représenter les patients auprès des pouvoirs publics,laboratoires pharmaceutiques sur les questions sociales et de santé.
• Encourager la recherche et l’information des médecins.
Contact : e-mail : [email protected] ; tél : 03 29 83 48 34 ; site : http://www.associationiris.org
l’existence d'un DIP ?(9,10)
Introduction à l’étudedes DIP
Commen
t affirm
er sim
plem
ent l’e
xisten
ce d'un DIP ?
- 7 -
Les données de l'examen clinique comme la nature des problèmes infectieux (diarrhée chronique,pneumopathie, abcès contenant du pus, ou lésions purement nécrotiques), la présence ou nonde ganglions, l’hypertrophie des amygdales, l’existence d’une fièvre ou non, un syndrome dysmorphique,une génodermatose, etc. sont complétées par une série d’analyses.
1.2.1 Les examens à pratiquer en première intention
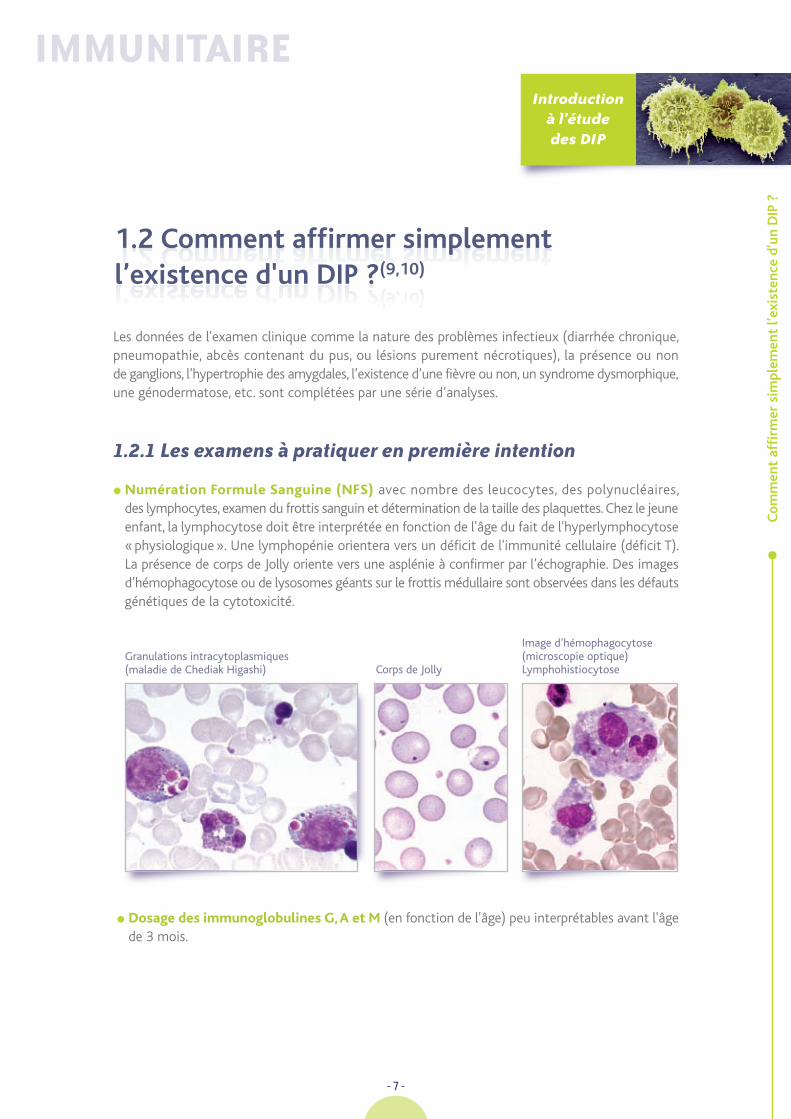
� Numération Formule Sanguine (NFS) avec nombre des leucocytes, des polynucléaires,des lymphocytes, examen du frottis sanguin et détermination de la taille des plaquettes. Chez le jeuneenfant, la lymphocytose doit être interprétée en fonction de l'âge du fait de l'hyperlymphocytose«physiologique». Une lymphopénie orientera vers un déficit de l'immunité cellulaire (déficit T).La présence de corps de Jolly oriente vers une asplénie à confirmer par l’échographie. Des imagesd’hémophagocytose ou de lysosomes géants sur le frottis médullaire sont observées dans les défautsgénétiques de la cytotoxicité.
� Dosage des immunoglobulines G, A et M (en fonction de l'âge) peu interprétables avant l'âgede 3 mois.
1.2 Comment affirmer simplement1.2 Comment affirmer simplementl’existence d'un DIP ?(9,10)
Image d’hémophagocytose (microscopie optique) Lymphohistiocytose
Granulations intracytoplasmiques (maladie de Chediak Higashi) Corps de Jolly

- 8 -
DEF
AU
TS G
ENET
IQU
ES D
E LA
REP
ON
SE IM
MU
NIT
AIR
EIn
trod
uctio
n à
l’étu
de d
es D
IP
01
� Titre des anticorps vaccinaux éventuellement après un rappel vaccinal [les anticorps anti-tétaniqueset anti-diphtériques sont de classe IgG1 dirigés contre des protéines (réponse T-dépendante),les anticorps anti-pneumococciques (après vaccin 23-valent non conjugué) et anti-méningococciques Aet C (vaccin méningocoque A et C polyosidique) sont des IgG2 anti-polysaccharides (PS)(réponse T-indépendante). Leur absence identifie les enfants qui ont un défaut spécifique de productiond’anticorps anti-PS, les taux sériques d’IgGAM sont normaux ainsi que les sous-classes. Ils présententalors des infections invasives à Streptococcus pneumoniae récurrentes.
� Les allo-hémagglutinines naturelles de groupe sanguin sont d’isotype IgM pour les enfants de groupeA, B et O. Elles ne sont pas évaluables avant l'âge de deux ans.
� L'étude des sérologies après une infection prouvée (varicelle, pneumocoque, Herpès) permet aussid'apprécier les capacités de production des anticorps spécifiques.
� L'ensemble des sérologies doit être interprété avec prudence dans les six premiers mois de la vieen raison de la persistance des immunoglobulines maternelles.
� Le dosage des sous-classes d’IgG (non interprétable avant 18 mois) si le taux de base est normalet si l’enfant présente un tableau évocateur de déficit immunitaire humoral (bronchectasie, bactériesencapsulées) ou un déficit en IgA. Un déficit en sous-classe peut s’associer à un DIP complexe (telqu’une ataxie-télangiectasie).
� La recherche d’une cytopénie auto-immune (test de Coombs érythrocytaire notamment).
� Radiographie du thorax (voit-on l’ombre du thymus ? existe-t-il une pneumopathie interstitielle ?)
� In vivo, tests d'hypersensibilité retardée : intradermoréaction à la tuberculine (si l’enfant a été vaccinépar le Bacille de Calmette et Guérin (BCG)), Intradermoréactions (IDR) à la candidine, à la phytohémagglutinine(PHA) sont devenues désuètes.
A gauche, radiographie de thorax normal : le thymus est bien visible (flèches). A droite : déficit immunitaire combinésévère (défaut de Jak3) : le thymus n’est pas visible ce qui est confirmé par l’échographie.
La très large majorité des DI « classiques » est vite éliminée sur la normalitéde ce bilan très simple, surtout si l’enfant est « irréprochable » sur le plande sa croissance pondérale et son examen clinique et en regard de la bénignitédes infections, très souvent ORL (comme dans l’exemple 1).

Introduction à l’étudedes DIP
- 9 -
Orienté par la clinique, après un éventuel avis auprès d’un médecin immunologiste, le praticienpeut aussi demander :
� Un phénotypage lymphocytaire (étude des sous-populations lymphocytaires T, B, NK encytométrie de flux (CMF), à interpréter en valeur absolue et selon l'âge de l’enfant) en fonctionde la gravité de la situation clinique présentée. La molécule CD3 est exprimée par les lymphocytesT qui se répartissent en deux sous-populations : CD4 + et CD8 +. Les molécules CD19 et CD20sont spécifiques des lymphocytes B. Les cellules Natural Killer (NK) expriment les molécules CD56et CD16. La numération des lymphocytes T, des sous-populations lymphocytaires T, deslymphocytes B et NK doit être interprétée en valeur absolue et selon l’âge du patient 11
(voir tableaux en annexes). Elle est complétée par l’étude de populations T particulières, naïves etmémoires, en fonction du tableau présenté, notamment lorsque l’on suspecte un déficit immunitairecellulaire avec lymphocytes T présents : syndrome d’Omenn et syndrome de Di George. De même,le phénotype B sera complété par l’étude des populations B naïves : CD27-IgM+D+ et des Bmémoires : CD27+IgM- dans certains DIP humoraux. L’étude de la fonction des LT est évaluéepar les réponses prolifératives in vitro (« tests de transformation lymphoblastique »), réalisabledans quelques laboratoires.
� Un dosage du complément hémolytique, le CH50 (confirmé par le dosage spécifique desdifférents composants) devant des infections répétées à germes encapsulés, surtout du genreNeisseria. La voie alterne du complément étudiée par le dosage de « l'Alternative Pathway»(AP50) est, en cas d'anomalie, complétée par le dosage des différents composés (notammentpour le déficit en properdine lié à l’X, non exploré par le «CH50»).
Commen
t affirm
er sim
plem
ent l’e
xisten
ce d'un DIP ?
Voie classiqueCH 100/50
Voie lectineCH 100/50, MBL*
Voie alterneAH 100/50
C2C1 C3bC4
C3
C5b-9Complexe d’attaque membranaire
C5C6C7C8C9
C4 BC2 D
P
Bd
C4a
C1 INHIBITEUR
H, IC3dC3a
C5asC5b-9
Détermination de l’activité anti-complémentaire
* MBL : mannose binding lectine
Les infections invasives récidivantesà méningocoque doivent faire évoquerun déficit en composé terminal ducomplément (C5-C9). Un déficit en C3se révèle par des infections sévèresà bactéries encapsulées. Les défautsde protéines régulatrices (H, I, MCP)sont associés aux syndromes urémiqueset hémolytiques atypiques. Les défautsdes composés « précoces » C1, C2, C4constituent des facteurs de risquegénétique du lupus systémique.

- 10 -
DEF
AU
TS G
ENET
IQU
ES D
E LA
REP
ON
SE IM
MU
NIT
AIR
EIn
trod
uctio
n à
l’étu
de d
es D
IP
01
D’autres tests explorent la capacité de bactéricidie, telle que la chimioluminescence qui mesure l’émissiond’énergie lumineuse émise par l’explosion oxydative des phagocytes, grâce à un compteur à scintillationou plus récemment la cytométrie de flux DHR qui étudie l’activité de la NADPH oxidase (après activation,la dihydrorhodamine 123 se transforme en un composé fluorescent).12
L’analyse d’un cheveu en microscopie optique entre lame et lamelle peut s’avérer très utile quandon suspecte un défaut génétique de la cytotoxicité ou un syndrome de Netherton (voir plus loin). C’estun examen très simple.
� Un test au nitrobleu de tétrazolium, ou « test au nbt », pour explorer l’explosion oxydative dupolynucléaire.
Maladie de Griscelli : répartition anormale de la mélanine dans la tige pilaire
Dépigmentation des phanères et des sourcils (maladie de Griscelli type 2)
Chez un enfant qui a présenté une infection sévère, « insolite », la normalitéde l’ensemble des explorations immunologiques ci-dessus doit inciterà rechercher un défaut génétique de l’immunité innée.
A B
La réduction du NBT visible en microscopie optique (MO) colore les polynucléaires (PN) en bleu noir (A), en (B) patientatteint de granulomatose septique chronique, une maladie enzymatique des PN incapables de fabriquer les radicauxoxydants bactéricides et donc de réduire le nitrobleu.
Remerciements : Marie-José Stasia - Grenoble
Introduction à l’étudedes DIP
- 11 -
Axes diagnostiques Examens utiles Hypothèses diagnostiques
Commen
t affirm
er sim
plem
ent l’e
xisten
ce d'un DIP ?
1.2.2 Grands tableaux cliniques et examens de première intention
Anomalies génétiques de la cytotoxicité
• Syndrome d’activation lymphohistiocytaire
• ± Trouble de la pigmentation
• Expression de la perforine et recherche des mutations dans les autres gènes de la cytotoxicité
• Etude des cheveux en microscopieoptique
• Frottis sanguin (recherche de lysosomes géants)
• Charge virale EBV • Sous-populations NKT/ recherche des mutations dans les gènes XLP1et 2 et ITK
• Lymphohistiocytoses familiales
• Albinisme partiel et déficitimmunitaire (maladie de Griscelli,maladie de Chédiak Higashi)
• Maladie de Purtilo
exemple : lymphohistiocytose familiale (défaut de perforine)
Déficits des cellules phagocytaires
• Infections à Aspergillus spp.• Infections bactériennes invasives
• Numération des polynucléaires• Etudes fonctionnelles (NBT,chimioluminescence, DHR)
• Expression des molécules d’adhésion en CMF
• Neutropénies congénitales• Granulomatoses septiques chroniques
• Déficit d’expression des moléculesd’adhésion
exemple : omphalite révélant un déficit en protéine d’adhésion leucocytaire
- 12 -
DEF
AU
TS G
ENET
IQU
ES D
E LA
REP
ON
SE IM
MU
NIT
AIR
EIn
trod
uctio
n à
l’étu
de d
es D
IP
01
Axes diagnostiques Examens utiles Hypothèses diagnostiques
Déficits du complément et asplénie
• Infections récurrentes à germesencapsulés (Haemophilus, S. pneumoniae, Neisseria)
• Dosage du CH50• Dosage de l’«alternative pathway»(AP50)
Echographie de la rate Corps de Jolly sur le frottis
• Déficits des différents composés du complément précisés par des dosages spécifiques
• Asplénie
exemple : infection invasive à pneumocoque et asplénie
Déficits humoraux
• Sinusites• Bronchectasies• Otites à répétition• Infections invasives à bactéries à multiplication extracellulaire
• Infections digestives à enterobactéries et G. intestinalis
• Encéphalites à Enterovirus
• Dosage des immunoglobulinessériques
• Numération des lymphocytes B dans le sang périphérique
• Dosage des sous-classesd'immunoglobulines après l'âge de 18 mois
• IgM sériques normales ou élevéescontrastant avec IgG et IgAeffondrées
Dans tous les cas : • Étude des sérologies vaccinales aprèsvaccination et des sérologies aprèsinfections certaines, à interpréteravec prudence avant l'âge de 6 mois(persistance des immunoglobulinesmaternelles)
• Agammaglobulinémie de Bruton liéeà l’X et formes autosomalesrécessives
• Déficits dissociés enimmunoglobulines : le plus fréquent= déficit en IgG2 et IgG4
• Syndromes «hyper IgM» liés à une anomalie de la commutationisotypique
exemple : sinusite à répétition, bronchectasies révélant une agammaglobulinémie

A côté des déficits immunitaires «classiques», un certain nombre de formes très sévèresde maladies infectieuses de l'enfant (et de l’adulte), telles que les infections à germeencapsulé (défaut en IRAK4/MyD88**), les encéphalites herpétiques, les infectionsà mycobactéries environnementales (cf page 42) révèlent certains défauts génétiques(et notamment de l’immunité innée) : ces enfants ont la particularité d’être vulnérablesà un spectre étroit de microorganismes. L’exploration de ces patients est complexeet ne peut être confiée qu’à un nombre restreint de laboratoires de recherche.Les méthodes de séquençage haut débit permettent de séquencer en quelques joursplusieurs gigabases d'ADN et d'identifier ainsi très rapidement de 'nouveaux' DIP dansdes tableaux d' infections insolites.
Introduction à l’étudedes DIP
- 13 -
Axes diagnostiques Examens utiles Hypothèses diagnostiques
Commen
t affirm
er sim
plem
ent l’e
xisten
ce d'un DIP ?
Déficits de l’immunité cellulaire T
• Infections virales • Diarrhée sévère• Précocité des infections• Mauvaise courbe pondérale• Pneumocystose
• Numération lymphocytaire et phénotypage des lymphocytes du sang périphérique
• Étude fonctionnelle deslymphocytes T : réponsesprolifératives in vitro avec mitogèneset antigènes spécifiques notammentvaccinaux
Anomalie de la différenciationlymphocytaire T :• Déficits immunitaires combinéssévères (DICS)
Selon les symptômes associés :
• Recherche d'une auto-immunité• Expression des moléculesd'histocompatibilité
• FISH*
• Caryotype
Déficits immunitaires T sansanomalies de la différenciation : • Syndrome de Wiskott Aldrich• Déficit de l’expression des moléculesd'histocompatibilité de classe II
• Syndrome de Di George• Ataxie-télangiectasie
exemple : déficit immunitaire combiné sévère révélé par une varicelle sévère (défaut en chaîne gamma commune)
* FISH : Hybridation in situ en fluorescence** Cf. glossaire page 60
!

- 14 -
DEF
AU
TS G
ENET
IQU
ES D
E LA
REP
ON
SE IM
MU
NIT
AIR
ELe
s dé
ficits
syn
drom
ique
s 02
Certains Déficits Primitifs ont une présentation originale qui « appartient» à une autre spécialité pédiatrique : le DI est alors relégué au second plan ou d'installation tardive, masqué par un tableau clinique caractéristique :troubles de la marche (ataxie-télangiectasie), thrombopénie/auto-immunité (syndrome de Wiskott-Aldrich), cardiopathie/ hypocalcémienéonatale (anomalie de Di George), eczéma, abcès froids (syndromesHyper IgE), nanisme avec chondrodystrophie (cartilage hair hypoplasia)pour ne citer que les principaux.
les déficitssyndromiques
2.1 L'anomalie de Di George (OMIM # 192430)2.1 L'anomalie de Di George (OMIM # 192430)
L’anomalie de Di George (ADG) : une aberration de programmedu développement des 3e et 4e arcs branchiaux par aneuploïdiesegmentaire.
L’anomalie de Di George est caractérisée par une anomalie de migration des cellules des crêtes neuralesvers les 3ème et 4ème poches branchiales avec une absence de développement du thymus conduisanttrès exceptionnellement à une alymphocytose T, associée ou non à des malformations cardiaquescono-troncales (hypoplasie de l'arc aortique, défauts septaux), à une absence des parathyroïdes(hypocalcémie néonatale) et à des anomalies faciales (fente palatine, troubles fonctionnels du carrefouroropharyngé). (Revue dans 13)
Les déficitssyndromiques
- 15 -
L'an
omalie de Di G
eorge
Les enfants ont habituellement une morphologiefaciale assez caractéristique avec un hypertélorisme,des oreilles assez larges, implantées bas, unediminution de la formation de l'hélix, un philtrumcourt. (Voir figures). En fait, la plupart des patientsont un déficit immunitaire relativement modéréet ne développent jamais d'infections opportunistesmettant en jeu le pronostic vital. Les infectionsvirales sont souvent fréquentes et prolongéesnotamment sinusites et otites : ce sont plutôtles anomalies anatomiques du palais quiconduisent à une fréquence élevée des infections.Certains enfants ont une diminution de capacité
à répondre aux antigènes polysaccharidiques.Comme dans d'autres déficits immunitairescellulaires, il existe une augmentation du risquede maladies auto-immunes (thrombopénie auto-immune, arthrites inflammatoires, maladiecœliaque) et une susceptibilité à développer unsyndrome lymphoprolifératif lié à l’EBV. Le défautlymphocytaire T résulte d'une masse thymiqueinsuffisante mais la persistance de résidus thymiquesdans des endroits tels que la langue, la thyroïde,et même l'oreille moyenne peut sans doutepermettre une fonction lymphocytaire Tcompétente.
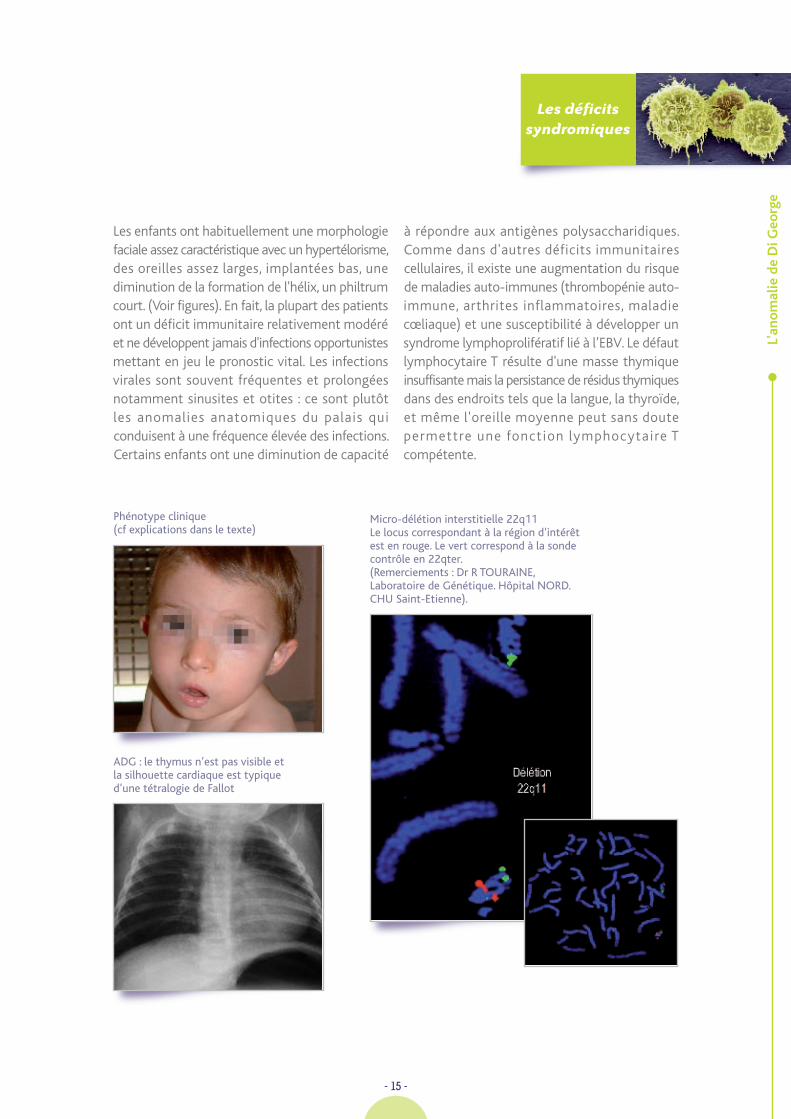
Phénotype clinique (cf explications dans le texte)
ADG : le thymus n’est pas visible etla silhouette cardiaque est typiqued’une tétralogie de Fallot
Micro-délétion interstitielle 22q11 Le locus correspondant à la région d’intérêtest en rouge. Le vert correspond à la sondecontrôle en 22qter.(Remerciements : Dr R TOURAINE,Laboratoire de Génétique. Hôpital NORD.CHU Saint-Etienne).

Il existe beaucoup de similitudes cliniques entre l’anomalie de Di George,le syndrome vélocardiofacial et les anomalies cono-troncales, d'où parfoisl’acronyme CATCH 22 pour désigner cette pathologie de gènes de contiguïté :anomalies cardiaques, faciès anormal, hypoplasie thymique, fente palatine(cleft) et hypocalcémie ; ces syndromes cliniques proches sont associéshabituellement à une délétion hémizygote de la bande 22q11.2 (voir plusloin). Certains enfants ont la délétion et ne rentrent pas dans un cadresyndromique bien précis tandis que d'autres ont le syndrome décrit parDi George mais n'ont pas la délétion.14,18
- 16 -
DEF
AU
TS G
ENET
IQU
ES D
E LA
REP
ON
SE IM
MU
NIT
AIR
ELe
s dé
ficits
syn
drom
ique
s02
Bases génétiques du phénotype15
La délétion 22q11.2 à l’état haploïde est associéeà la majorité des anomalies de Di George.Elle résulte d’un évènement méïotique derecombinaison liée à la présence d’élémentsde répétition recombinogènes (duplicons*) dansune région génétique, de fait instable. Elle faitde l’ADG une pathologie de gènes de contiguïtéet dans la bande 22 q11, il y a plus de 35 gènes.Il existe une très grande hétérogénéité de laprésentation clinique, avec l'intervention denombreux gènes modificateurs encore inconnus.C’est une situation d’haplo-insuffisance du gèneTBX1 qui constitue le déterminant majeurdes anomalies cardiaques, thymiques et
des parathyroïdes comme l’a montré une sériede mutations conditionnelles chez la souris.Le retard mental, les problèmes psychiatriques,squelettiques et rénaux sont fréquents. TBX1 estexprimé dans le cerveau et le sclérotome impliquédans la différenciation de certaines structures dela colonne vertébrale. Le phénotype Di Georgepeut être aussi observé chez les enfants présentantune fœtopathie induite par des tératogèneset notamment les rétinoïdes [l'acide rétinoïqueest un répresseur de l'expression de TBX1]ou un diabète maternel. La fréquence de la micro-délétion 22 q11.2 est de 1/3000 naissances.13,14
* Cf. glossaire page 60

Les déficitssyndromiques
L'an
omalie de Di G
eorge
- 17 -
Prise en charge
** FISH : Hybridation in situ en fluorescence*** Cf. glossaire page 60
Le diagnostic
Le souvent, les enfants atteints d’une ADG ontun taux de lymphocytes T modérément abaissé,avec un pronostic immunitaire excellent à longterme. Un petit nombre ont des anomalieshumorales : réduction quantitative des IgA,hypogammaglobulinémie, altération de laréponse vaccinale. L’antibioprophylaxie contrele Pneumocystis est indiquée en cas delymphopénie marquée pendant la premièreannée. Un phénotypage lymphocytaire T (CD3,4,8)sera complété par l’étude des populations TCD4 naïves et mémoires respectivement,CD31+CD45RA+RO-CD4+ et CD45RA-RO+CD4+,
des réponses prolifératives aux antigèneset aux mitogènes, et un dosage pondéraldes immunoglobulines. Les Di George completsavec alymphocytose T persistante et absencede réponse proliférative doivent être référés dansun Centre spécialisé pour discuter leur priseen charge et notamment une greffe de tissuthymique, ce sont de véritables déficits immunitairescombinés sévères.16 En dehors des problèmesinfectieux, les enfants justifient d’une priseen charge multidisciplinaire (cardiopathie, troublesdu langage, retard cognitif).
L’anomalie de Di George repose sur l’associationde 2 (ou 3) signes cliniques parmi les suivants : déficitcellulaire ± hypoplasie thymique, hypocalcémie± hypoparathyroïdie, malformation cardiaquecono-troncale.
Le diagnostic des formes délétionnelles reposesur la mise en évidence de la délétion encytogénétique de haute résolution, remplacéepar l'étude en fluorescence des chromosomesmétaphasiques avec la sonde commercialiséeTuple1 («FISH** ») et la technique de MLPA***
Multiplex Ligation-dependent Probe Amplificationpour les plus petites délétions. (Voir page 20)
La majorité des délétions 22q11 sont desévénements sporadiques qui ne surviennent quechez les individus atteints. L'incidence des casfamiliaux est environ de 10 %.17 Il est importantd'examiner les parents d'un enfant atteint à la
recherche d’une anomalie dysmorphique subtile,une voix nasonnée, un déficit cognitif. Les parentsqui présentent la micro-délétion interstitiellepeuvent bénéficier alors d'un conseil génétiqueet d’un diagnostic prénatal ; il est impossiblede prédire le phénotype de l’enfant à naître s’il(ou elle) est porteur. Il n'y a pas de corrélationentre la taille de la délétion interstitielle surle chromosome 22 et la pénétrance et la sévéritédu phénotype clinique. La variabilité intra-familialeest significative et le phénotype parental n'estpas prédictif du tableau observé chez l'enfant.La surveillance échographique pendant la grossesseest de mise, à la recherche d'une fente palatine,d'anomalie rénale ou d'une malformation cardiaque.Le conseil génétique est particulièrement difficilechez les enfants qui présentent une forme nondélétionnelle. Une minorité d'enfants présentantle syndrome peut avoir d'autres anomalies quela microdélétion 22q11.18

- 18 -
DEF
AU
TS G
ENET
IQU
ES D
E LA
REP
ON
SE IM
MU
NIT
AIR
ELe
s dé
ficits
syn
drom
ique
s02
Observation clinique
Cette fillette d’origine marocaine est la 4ème d'une fratrie de six et les parents ne sont pas apparentés.C’est à l’âge de 12 mois que l’on porte le diagnostic de maladie cœliaque après une diarrhéede «retour» avec les examens suivants : Protéines : 45g/l ; Ca : 2,09 mmol/l ; Fe : 4 µmol/l ; GB :23,5 G/l dont 45% lymphocytes ; IgA indosable ; IgG et IgM dans des valeurs normales pourl’âge. IgG anti-gliadine +ve ; anti-endomysium +ve. La biopsie digestive montre une atrophievillositaire complète et les mesures diététiques ad hoc restaurent une vitesse de croissancesatisfaisante avec prise de 2,5 kg en six mois. La compliance au régime reste très imparfaite dansles 3 premières années avec la persistance des auto-Ac, une hyposidérémie et une hypocalcémie.Elle présente à + 3 A une convulsion fébrile ; une bactériémie à méningocoque W. 135 sansméningite est documentée. La suite de son histoire sera émaillée de nombreuses pousséesd’otorrhée chronique avec vaste perforation tympanique bilatérale entraînant une surditéde perception, un retard de langage qui bénéficiera un temps d’un appareil de conduction osseuseet d’un soutien orthophonique. A 5 ans, le régime est parfaitement suivi, le statut du fer estnormal ainsi que l’ionogramme sanguin. Elle présentera une fièvre typhoïde non compliquéedurant des vacances au Maroc.
Elle est hospitalisée à l’âge de 7 ans pour une pâleur, une asthénie considérable, des sueurs et unefièvre à 40°C. La rate est très « tumorale », il existe de très nombreuses adénopathies pathologiquesdans les creux axillaires et dans les plis inguinaux. On retrouve les éléments suivants GB : 3,8 G/l ;PN : 1,198 G/l ; Ly 2,3 G/l ; plaquettes : 119 G/l ; Hb : 9,6 g/dl ; VGM : 67µ3 ; CRP : 31 mg/l ; LDH :2X ; Fe 2 µmol/l ; IgA indosable ; IgG : 8,3 g/l ; IgM : 15 g/l ; I : 2,3 g/l ; Na : 135 mmol/l ; Protides :69 g/l ; Ca : 2,09 mmol/l ; transaminases : 36/30 U/l ; triglycérides : 2,3 g/l. Sérologie EBV :VCA 320, EBNA-f. Charge virale EB : 2000 cp/ml ; CD3 : 790 /µl ; CD4 : 348/µl ; CD8 : 397/µl.
Quel diagnostic portez-vous initialement ?
Il s’agit très probablement d’une mononucléose infectieuse (MNI) comme en témoignela positivité des Ac anti-VCA et la charge virale EBV. Cette MNI est inhabituelle par l’existenced’un syndrome lymphoprolifératif (SLP) et une hypergammaglobulinémie d’isotype IgMparticulièrement marquée. Les cytopénies, le fibrinogène peu élevé dans le contexte plaidaientpour un syndrome d’activation lymphohistiocytaire associé, mais le frottis médullaire ne montraitpas, en l’occurrence, d’images d’hémophagocytose. La biopsie ganglionnaire objectivaitune «simple» hyperplasie lymphoïde sans anomalie de l’architecture. Le caryotype médullaireet ganglionnaire ne montrait pas de maladie clonale.

Les déficitssyndromiques
- 19 -
L’importance des symptômes a conduit à la prescription d’une corticothérapie et l’évolutiona été tout à fait simple. La baisse progressive des stéroïdes a précipité deux épisodes de « rechute »à 3 mois et 6 mois, la reprise d’une corticothérapie minimale induisant très facilement une rémission.L’enfant restait en excellent état général.
L’enfant a été initialement suivie en CAMSP (Centre d’Action Médico-Sociale Précoce) puis accueillieen IME (Institut Médico-Educatif). La sérologie EBV persistait inchangée, déséquilibrée avecl’absence d’Ac anti-EBNA. La CV EBV demeurera ensuite nulle. Le phénotypage lymphocytaireà + 9A montre CD3 : 2362/µl ; CD4 : 999/µl ; CD8 : 1083/µl ; les Ac vaccinaux sont à un titre protecteur.A 11 ans, elle présente à nouveau un épisode de SLP très fébrile qui conduit à une hospitalisation.
Les examens paracliniques montrent GB : 2,52 G/l ; PN : 1,21 G/l ; L : 0,93 G/l ; Hb : 12 g/dl ;Plaquettes : 49 G/l ; I : 2,4g/l ; Na : 138 mmol/l ; Créatinine : 56 µmol/l ; Ca : 1,56 mmol/l ;Triglycérides : 0,95 g/l ; IgA : 0,05 g/l ; IgG : 16 g/l ; IgM : 0,64 g/l ; LCR : normal ; myélogramme :quelques images d’hémophagocytose. CV EBV nulle ; Ac anti-EBNA négatifs. L’échographie montreune rate micronodulaire.
La sérologie EBV reste déséquilibrée et l’évolution est scandée par despoussées de syndrome lymphoprolifératif B, très corticosensibles. Cettesituation suggère un déficit de l’immunité cellulaire. Quelle anomaliemarquée dans la biologie de routine est la clé de cette énigme diagnostique ?
Aspect micronodulaire du parenchyme splénique.Aorte déroulée à droite (confirmée par l’échographie cardiaque).
L'an
omalie de Di G
eorge
- 20 -
DEF
AU
TS G
ENET
IQU
ES D
E LA
REP
ON
SE IM
MU
NIT
AIR
ELe
s dé
ficits
syn
drom
ique
s02
Réponse
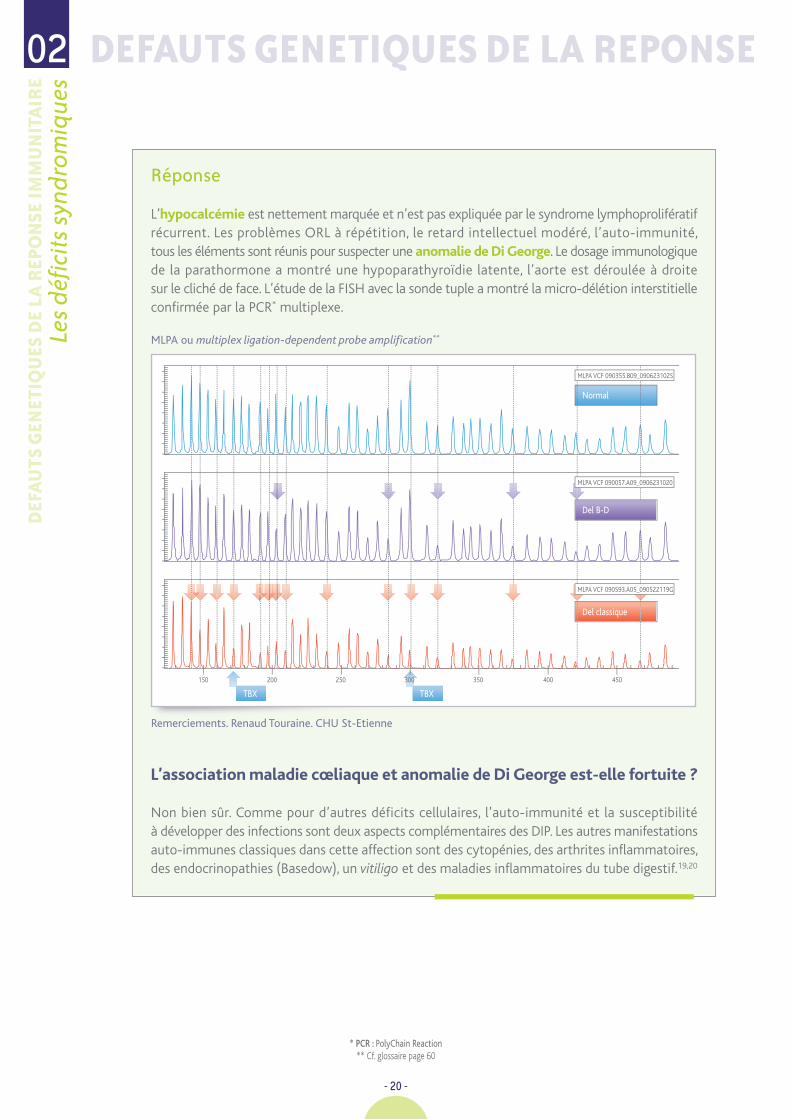
L’hypocalcémie est nettement marquée et n’est pas expliquée par le syndrome lymphoprolifératifrécurrent. Les problèmes ORL à répétition, le retard intellectuel modéré, l’auto-immunité,tous les éléments sont réunis pour suspecter une anomalie de Di George. Le dosage immunologiquede la parathormone a montré une hypoparathyroïdie latente, l’aorte est déroulée à droitesur le cliché de face. L’étude de la FISH avec la sonde tuple a montré la micro-délétion interstitielleconfirmée par la PCR* multiplexe.
L’association maladie cœliaque et anomalie de Di George est-elle fortuite ?
Non bien sûr. Comme pour d’autres déficits cellulaires, l’auto-immunité et la susceptibilitéà développer des infections sont deux aspects complémentaires des DIP. Les autres manifestationsauto-immunes classiques dans cette affection sont des cytopénies, des arthrites inflammatoires,des endocrinopathies (Basedow), un vitiligo et des maladies inflammatoires du tube digestif.19,20
Normal
TBXTBX
Del B-D
150 200 250 300 350 400 450
Del classique
MLPA VCF 090593.A05_090522119G
MLPA VCF 090057.A09_0906231020
MLPA VCF 090355.809_090623102S
* PCR : PolyChain Reaction** Cf. glossaire page 60
MLPA ou multiplex ligation-dependent probe amplification**
Remerciements. Renaud Touraine. CHU St-Etienne
Liens utiles :Recommandations pour la surveillance immunitairedes patients atteints d'un syndrome de Di George.www.ceredih.fr/documents/Recos_DiGeorge_v3.pdf
Les déficitssyndromiques
- 21 -
* Cf. glossaire page 60
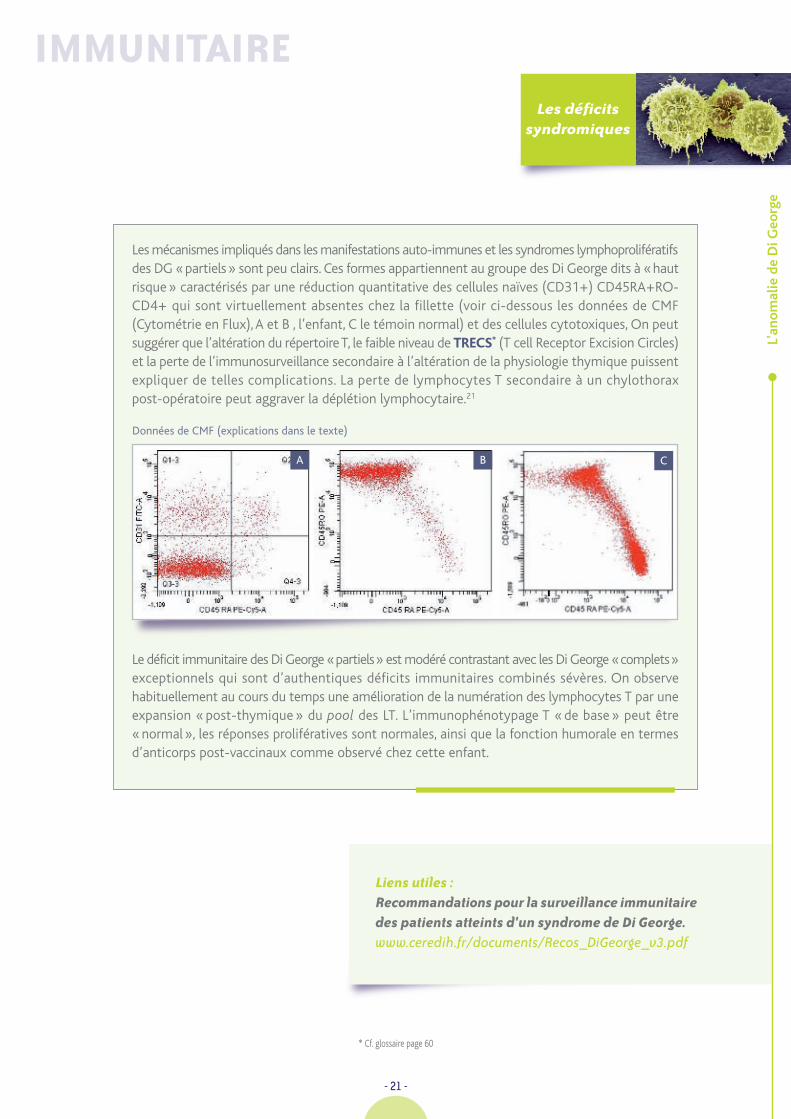
Les mécanismes impliqués dans les manifestations auto-immunes et les syndromes lymphoprolifératifsdes DG «partiels» sont peu clairs. Ces formes appartiennent au groupe des Di George dits à «hautrisque» caractérisés par une réduction quantitative des cellules naïves (CD31+) CD45RA+RO-CD4+ qui sont virtuellement absentes chez la fillette (voir ci-dessous les données de CMF(Cytométrie en Flux), A et B , l’enfant, C le témoin normal) et des cellules cytotoxiques, On peutsuggérer que l’altération du répertoire T, le faible niveau de TRECS* (T cell Receptor Excision Circles)et la perte de l’immunosurveillance secondaire à l’altération de la physiologie thymique puissentexpliquer de telles complications. La perte de lymphocytes T secondaire à un chylothoraxpost-opératoire peut aggraver la déplétion lymphocytaire.21
Le déficit immunitaire des Di George «partiels» est modéré contrastant avec les Di George «complets»exceptionnels qui sont d’authentiques déficits immunitaires combinés sévères. On observehabituellement au cours du temps une amélioration de la numération des lymphocytes T par uneexpansion «post-thymique» du pool des LT. L’immunophénotypage T «de base» peut être«normal», les réponses prolifératives sont normales, ainsi que la fonction humorale en termesd’anticorps post-vaccinaux comme observé chez cette enfant.
A B C
Données de CMF (explications dans le texte)
L'an
omalie de Di G
eorge
- 22 -
DEF
AU
TS G
ENET
IQU
ES D
E LA
REP
ON
SE IM
MU
NIT
AIR
ELe
s dé
ficits
syn
drom
ique
s02
2.2 Cartilage-hair hypoplasia (CHH) (OMIM # 250250)2.2 Cartilage-hair hypoplasia (CHH) (OMIM # 250250)
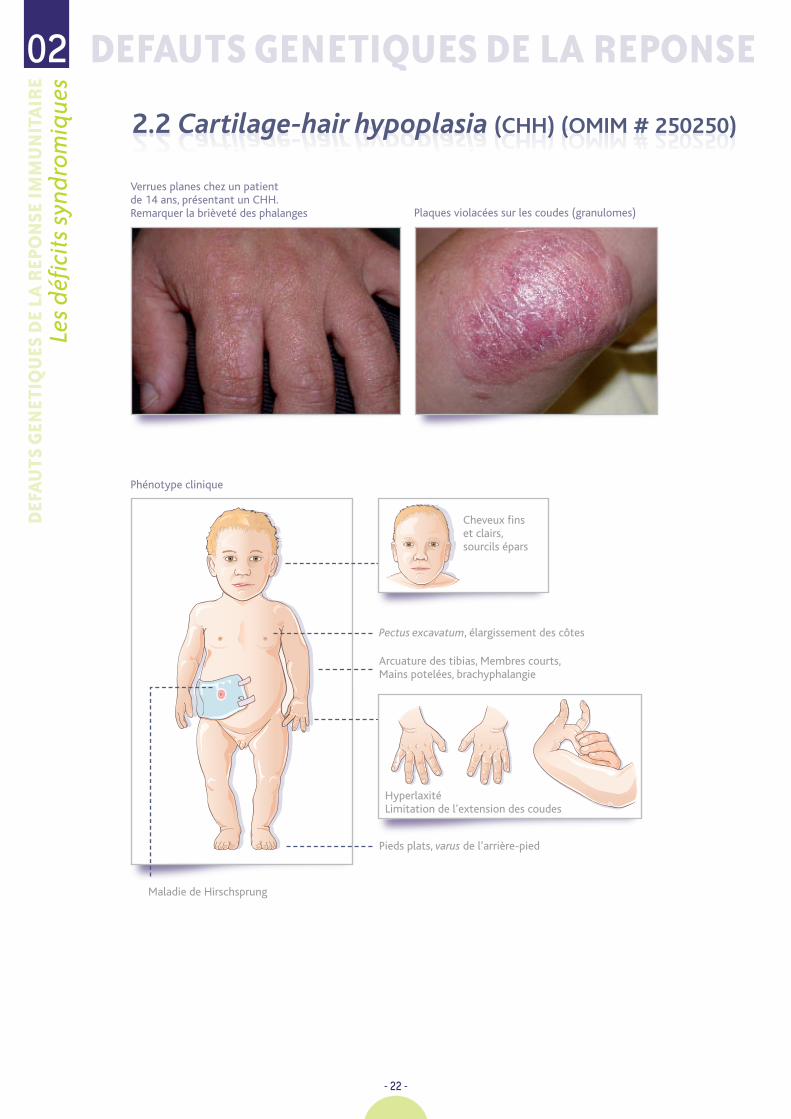
Verrues planes chez un patient de 14 ans, présentant un CHH. Remarquer la brièveté des phalanges
Phénotype clinique
Pectus excavatum, élargissement des côtes
Pieds plats, varus de l’arrière-pied
Maladie de Hirschsprung
Arcuature des tibias, Membres courts, Mains potelées, brachyphalangie
Plaques violacées sur les coudes (granulomes)
Cheveux fins et clairs,sourcils épars
HyperlaxitéLimitation de l’extension des coudes

Les déficitssyndromiques
- 23 -
Car
tilage
-hair hy
poplas
ia
Le CHH : un défaut génétique de la biogenèse des ribosomes22
Le nucléole est le site de transcription des ARN ribosomaux (ARNr), leur processing et leur assemblage.Les cellules de mammifères contiennent 5 à 10 millions de ribosomes qui doivent être synthétiséschaque fois que la cellule se divise. Le nucléole est ainsi une usine de production ribosomale,pour remplir des besoins très importants pour la production efficace des ARNr et l'assemblagedes différentes sous-unités ribosomales.
Les protéines ribosomales sont exportées ducytoplasme vers le noyau puis commencent àse rassembler sur l'ARNr précurseur avant samaturation par clivage. Au fur et à mesure duprocessing de ce pré ARN-r, d'autres protéinesribosomales, et l'ARN r 5S synthétisés ailleursdans le noyau, s'assemblent pour constituer lesparticules pré-ribosomales. Les dernières étapes dela maturation vont suivre l'exportation des particulesdans le cytoplasme pour produire les 2 sous-unités40S et 60S des ribosomes eucaryotes (schéma).
L'assemblage des différentes sous-unités ribosomales
Nucléole (n) en ME (microscopie électronique)
Noyau ADNr
Pré-ARNr
5S ARNr
28S
Nucléole
Protéinesribosomales
Cytoplasme
Sous-unité 60SSous-unité 40S
n
5,8S
Particulespréribosomales
Pré-18S
18S
5S
28S
5,8S5S

- 24 -
DEF
AU
TS G
ENET
IQU
ES D
E LA
REP
ON
SE IM
MU
NIT
AIR
ELe
s dé
ficits
syn
drom
ique
s02
Le clivage du pré-ARNr nécessite l'action de protéineset d'un grand nombre d'ARN nucléolaires(« snoRNAs» : small nucleolar RNA) qui sontcomplexés pour former des snoRNP, «à la manière»des snRNP du splicéosome impliqués dansla maturation des ARN messagers. Certaines de cesparticules sont responsables du clivage du précurseurcommun. D'autres guident la modificationdes bases, la méthylation de certains résidus riboses,la formation des pseudouridines par exemple.
Trois syndromes sont associés à des défautsgénétiques de la biogenèse des ribosomes :le cartilage-hair hypoplasia (CHH), l’érythroblastopéniede Blackfan-Diamond et le syndrome de Schwachmanqui sera abordé plus tard avec les neutropéniesconstitutionnelles.
Le CHH : un DIP très variable associé à une chondrodysplasiemétaphysaire
Le CHH associe une dysplasie métaphysaireresponsable d'une insuffisance staturale avecmembres courts, un déficit immunitaire T + B trèsvariable et des anomalies très particulières descheveux. Cette maladie génétique se transmetsur un mode autosomique récessif et a été d’abordrapportée dans la population Amish de Pennsylvaniepar Victor Mc Kusick en 1965.23
Le diagnostic anténatal peut être suspecté surdes données échographiques (raccourcissementdes fémurs à partir de 17 semaines). Le poidsde naissance est normal mais les membres sontcourts. Dans la petite enfance, en observe des pliscutanés excessifs dans le cou ou dans les extrémités.L’insuffisance staturale est due à un raccourcissementdysharmonieux des extrémités. Les mains sontpetites, épaisses avec des ongles courts. Lesarticulations sont laxes. Les cheveux et les sourcilssont blancs ou jaunes, épars et fins. L’alopécie estparfois marquée. Sur les radiographies, les centresd'ossification sont normaux, mais il existe des zonesde sclérose irrégulière avec des zones kystiques,des métaphyses élargies, rappelant le rachitismeou un déficit en adénosine déaminase.
Les patients présentent d’autres problèmes :anémie et neutropénie, problèmes digestifs avecmalabsorption, maladie de Hirschsprung. Il existe
une augmentation de l'incidence des cancerslymphoïdes.24
Le déficit immunitaire qui concerne essentiellementle compartiment T est très hétérogène. La plupartdes patients ont une susceptibilité assez limitéeaux infections et mènent une vie normale. D'autrespeuvent présenter un véritable tableau de déficitimmunitaire combiné sévère (parfois même sousla forme d’un syndrome d’Omenn) avec unesensibilité très importante aux infections qui fontindiquer une greffe de MO (moelle osseuse). Celle-ci ne corrige pas les symptômes osseux.25
On observe habituellement une lymphopénie T(parfois une lymphopénie CD8 isolée) et unediminution des réponses prolifératives. On décritaussi un déficit quantitatif en IgA et en sous-classesd’IgG chez certains patients. L'altération de l’immunitécellulaire entraîne des risques de varicelle sévère,classique dans ce DIP. L'altération de la fonctionhumorale chez certains patients contribueà la fréquence des infections ORL et respiratoires.Certains peuvent présenter des manifestationsauto-immunes telles qu'une anémie hémolytiqueauto-immune, une neutropénie ou une hyperthyroïdie.Le déficit immunitaire concerne plutôt les mutationsqui conduisent à une activité altérée du clivage dela Cycline B* et des anomalies du cycle cellulaire
* Cf. glossaire page 60

Les déficitssyndromiques
- 25 -
et un phénotype squelettique modéré. Les anomaliessquelettiques peuvent manquer chez des enfantsqui présentent alors un tableau de déficit combinéT/B « isolé». La dysplasie osseuse est beaucoup
plus sévère chez les patients qui présententdes mutations affectant de façon prédominantela biogenèse des ribosomes.25
Génétique
En 2001, il a été établi que le CHH est lié àdes mutations récessives dans le gène RMRP(ribonuclease mitochondrial RNA processing) quicode pour un ARN entrant dans la compositiond’un complexe multi-protéique impliqué dansle clivage des ARN ribosomaux et la maturationdes amorces ARN de la mitochondrie (une étapecritique de la réplication de l’ADN mitochondrial).Ce complexe ribonucléoprotéique RNase -MRPest localisé principalement dans le nucléole etparticipe à la biogenèse des ribosomes (cf. figurep. 22). Il effectue le clivage endonucléiquede l'ARN ribosomal qui conduit à la générationde l'ARN ribosomal 5,8S. De plus, le complexeparticipe à la régulation du cycle cellulaire*
en clivant l'ARN de la cycline*. C'est un gèneextrêmement polymorphe malgré le degréimportant de conservation dans les espèces eton a fait l'hypothèse que ces polymorphismespuissent jouer un rôle de modif icateurdu phénotype. Plus de 60 mutations différentesont été décrites. Les mutations entraînent l'instabilitéet la destruction rapide de cet ARN.22,25
La mutation la plus habituelle chez les Finlandais(fréquence : 1/23000 naissances vivantes ; tauxd’hétérozygotie : 1/76)25 et dans la communautéAmish est une substitution homozygote 70 AGdans le domaine catalytique présumé. Les autrespatients habituellement ont une mutation nulledans un des allèles et une mutation ponctuellepar exemple dans l'autre. Il y a toujours une expression
résiduelle de la protéine, et il est probable quel'absence d'expression de la ribonucléoprotéinesoit létale. Il n'y a pas de corrélation entre letaux d'expression et le phénotype clinique.La physiopathologie moléculaire des maladieshumaines associées aux mutations ducomplexe RMRP n'est pas définie enl’absence de modèle animal. Les mutationsdu gène RMRP sont associées à 3 autreschondrodystrophies génotypiques : la dysplasiemétaphysaire sans hypotrichose (OMIM # 250460),la dysplasie cyphomélique (OMIM % 211350)et la dysplasie anauxétique (OMIM # 607095).25
Ces différentes maladies génétiques diffèrent selonl'atteinte du squelette et la fréquence desanomalies extra-squelettiques. Il existe unecorrélation phénotype/génotype et les différentesmutations distinctes du gène RMRP affectent defaçon différente la fonction de la RNase MRPmulti-protéique. Le cycle cellulaire est anormalcomme en témoigne un déficit de la phase G1.Ces anomalies concernent les fibroblastes et laformation in vitro des colonies érythrocytaires,myéloïdes, mégacaryocytaires suggérant uneanomalie intrinsèque commune de proliférationdes cellules souches hématopoïétiques. Lesmécanismes moléculaires impliqués dans le déficitimmunitaire sont peu clairs et impliquent uneréduction des LT naïfs, un excès d’apoptose deseffecteurs T, une instabilité chromosomique.25
* Cf. glossaire page 60
Car
tilage
-hair hy
poplas
ia
- 26 -
DEF
AU
TS G
ENET
IQU
ES D
E LA
REP
ON
SE IM
MU
NIT
AIR
ELe
s dé
ficits
syn
drom
ique
s02
2.3 Le syndrome de Netherton 2.3 Le syndrome de Netherton
Observation clinique
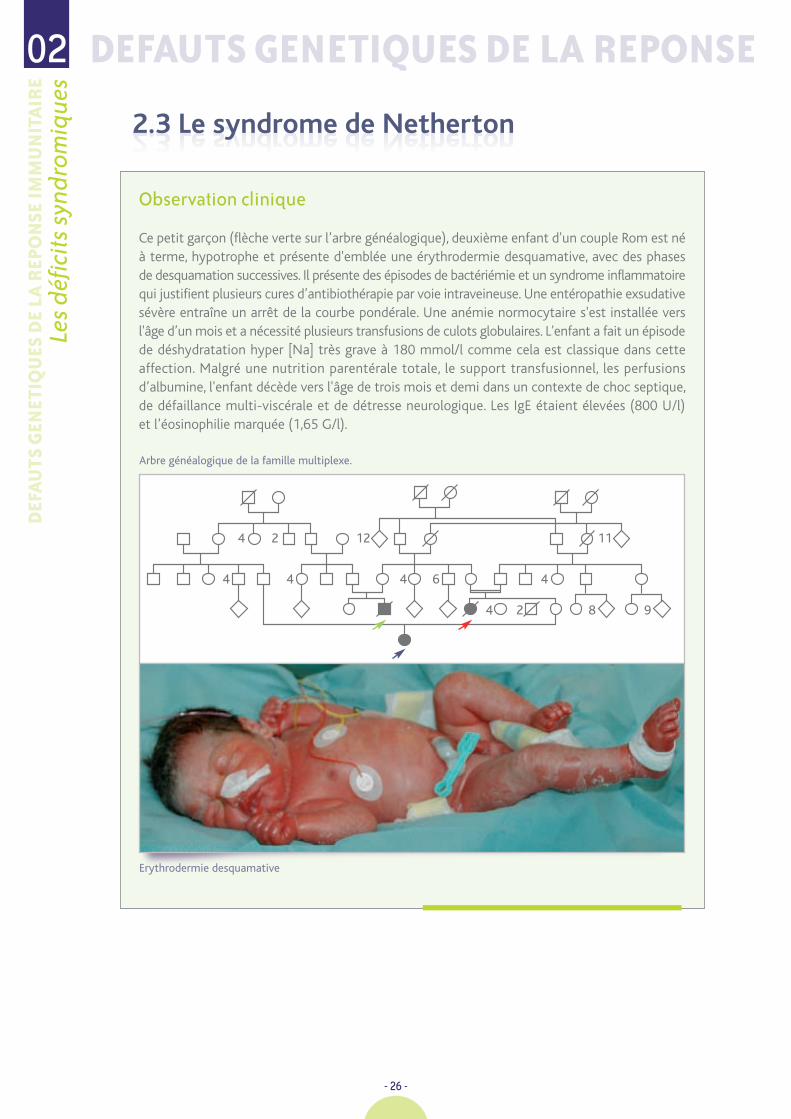
Ce petit garçon (flèche verte sur l’arbre généalogique), deuxième enfant d'un couple Rom est néà terme, hypotrophe et présente d'emblée une érythrodermie desquamative, avec des phasesde desquamation successives. Il présente des épisodes de bactériémie et un syndrome inflammatoirequi justifient plusieurs cures d’antibiothérapie par voie intraveineuse. Une entéropathie exsudativesévère entraîne un arrêt de la courbe pondérale. Une anémie normocytaire s'est installée versl'âge d’un mois et a nécessité plusieurs transfusions de culots globulaires. L'enfant a fait un épisodede déshydratation hyper [Na] très grave à 180 mmol/l comme cela est classique dans cetteaffection. Malgré une nutrition parentérale totale, le support transfusionnel, les perfusionsd’albumine, l'enfant décède vers l'âge de trois mois et demi dans un contexte de choc septique,de défaillance multi-viscérale et de détresse neurologique. Les IgE étaient élevées (800 U/l)et l’éosinophilie marquée (1,65 G/l).
4
4 4 4 6 4
4 2 8 9
12 112
Arbre généalogique de la famille multiplexe.
Erythrodermie desquamative

Les déficitssyndromiques
- 27 -
Le syn
drome de
Net
herton
Une cousine germaine de cet enfant (flèche bleue) était décédée vers l'âge de cinq mois, aprèsune histoire clinique assez similaire, dans un tableau de détresse respiratoire infectieuse.Le diagnostic d'érythrodermie ichtyosiforme congénitale non bulleuse avait été porté alors chezcette enfant.
La biopsie de peau (à gauche) ne montre pas de fixation de l’anticorps anti-LEKT1 en immunohistochimie(à droite, contrôle positif).
Quel est votre diagnostic ? Le diagnostic de syndrome de Netherton est suspecté sur les données d'immunohistochimie cutanée.Une analyse moléculaire du gène SPINK5 a montré une mutation dans l’intron 15, 1431+12G>Aen double dose chez le nourrisson tandis que les parents étaient porteurs à l'état hétérozygote.
Le troisième cas est celui d'une première enfant d'uncouple de jeunes parents apparentés de cette famille(cf arbre généalogique flèche rouge). Dès la naissancele diagnostic est évoqué devant une érythrodermiedesquamative dans le contexte familial. L'évolutionde la maladie, aggravée par la prématurité (naissanceà 33 semaines) a conduit à un décès précoce versl'âge de deux mois. L'évolution cutanée s'est faite parphases desquamatives entrecoupées de phasesd'amélioration grâce à l'application de vaseline stérilepluriquotidienne et de bains antiseptiques. Associée
à la desquamation, s'est installée une chute précoce et diffuse des cheveux aboutissant à une alopéciecomplète à un mois. Cette petite fille a présenté plusieurs épisodes de bactériémie : Staphylococcusaureus, Pseudomonas aeruginosa, Candida albicansmalgré une antibiothérapie large associée à desantifongiques et une immunosubstitution. Comme les deux autres enfants, la fillette a développédes oedèmes, une hypoprotidémie, un syndrome d'entéropathie exsudative sévère, une pancytopénieprogressive associée à un taux d'IgE élevé à 190 U/l.
Biopsie cutanée : A : absence d’immunomarquage anti-LEKT1 ; B : (à droite) contrôle positif
Erythrodermie diffuse néonatale (seconde observation)
A B

- 28 -
02
Quel examen très simple permettrait de faire le diagnostic en quelquesminutes ?
L’examen du cheveu montre un aspect trèsparticulier baptisé trichorrhexis invaginataou une appellation très suggestive en microscopieoptique (MO) de « cheveu en bambou »(ci-contre).
La mutation 1431+12G>A d'une seule base dans l'intron 15, 12 bases en amont du site accepteurd’épissage entraîne l'apparition d'un nouveau site accepteur dont l’utilisation conduit à une extensionen 3 prime de l’exon 16 avec décalage du cadre de lecture et l'apparition d'un codon stopprématuré dans l’exon 16. Il s'agit d'une mutation nulle qui, à l'état homozygote, entraîneune absence de protéine.
«Cheveu en bambou»
Desquamation en lambeaux Larges squames du visage et du scalp
Exon 15 Exon 15
Site accepteurAlternatif
GT.
GT ....
GG AG
AG .TAA
STOP
DEF
AU
TS G
ENET
IQU
ES D
E LA
REP
ON
SE IM
MU
NIT
AIR
ELe
s dé
ficits
syn
drom
ique
s
Remerciements à Dr Yline Montiel-Capri, Pr Pierre Dechelotte et Dr Etienne Merlin (CHU de Clermont-Ferrand) pourcette observation.

Introduction à l’étudedes DIP
- 29 -
Le syndrome de Comèl-Netherton est unegénodermatose très rare, autosomale et récessive,caractérisée par une ichtyose congénitale, un aspecttrès particulier de la tige pilaire « en bambou»(trichorrhexis invaginata ou «bamboo-hair» desAnglo-Saxons) et une atopie marquée. Cettemaladie est létale dans la première année de viepour 20 % des patients. Le diagnostic est souventposé à la naissance devant une érythrodermie etl'évolution vers une ichtyose. Le diagnostic estaisément posé en microscopie optique sur l'aspectcaractéristique du cheveu. L'atopie est fortementmarquée avec une hyper-réactivité bronchique, uneczéma, des allergies alimentaires, et des pousséesd'angioœdème avec Hyper IgE et éosinophilie.L'enfant présente habituellement une entéropathieexsudative, avec une mauvaise courbe pondérale,des troubles hydro-électrolytiques, un retard mentalet une susceptibilité aux infections.26
Le gène code pour une protéine exprimée parles cellules épithéliales de la peau, les muqueuseset les corpuscules de Hassal du thymus : LEKTI pourLymphoepithelial Kazal Type related Inhibitor.La protéine joue un rôle central dans le processusde la desquamation normale.27 Cette maladievéritablement multi-systémique est associéeà une anomalie de fonctionnement du systèmeimmunitaire inné et adaptatif. L’étude des sous-populations lymphocytaires montre habituellementune diminution du taux des cellules mémoire Bcommutées CD19+CD27+IgM-IgD- et des cellules B
de la zone marginale 19+ IgD+ 27+. Cette dernièrepopulation joue un rôle dans la défense contreles bactéries encapsulées et les réponses anti-polysaccharidiques sont altérées. Les réponsesprolifératives aux antigènes sont normales.La cytotoxicité NK est anormale contrastant avecdes sous-populations lymphocytaires NK normalesen nombre. Des cytokines pro-inflammatoires sontaugmentées de façon significative : interleukineIL-1b, IL-12, TNF-a, GM, IL-1 RA ainsi que lescytokines de type TH2 : IL-4 et 5. L’immunosubstitutionaméliore le statut des patients notammentl'inflammation cutanée, la fragilité des phanères,les infections récurrentes et la qualité de vie.Certains adultes vont présenter des cancers cutanés.Bien que la protéine soit exprimée dans lescorpuscules de Hassal, les patients testés ont unrépertoire TCR Vbêta normal, et des émigrantsthymiques CD4+45RA+ en nombre normal.Le déficit de la cytotoxicité NK correspond peutêtre à une anomalie de la maturation des cellulesNK qui nécessite un contact avec les cellulesépithéliales. La cytotoxicité NK est amélioréepar la prise en charge thérapeutique. Les cellulesNK des patients semblent capables de monterune réponse cytotoxique à corréler au fait queles patients ne présentent pas les infectionscaractéristiques des déficits NK, notammentles infections à virus du groupe HSV (Herpes simplexvirus).28
Le syn
drome de
Net
herton

- 30 -
DEF
AU
TS G
ENET
IQU
ES D
E LA
REP
ON
SE IM
MU
NIT
AIR
ELe
s dé
ficits
syn
drom
ique
s02
2.4 Les candidoses muco-cutanées chroniques (CMC)292.4 Les candidoses muco-cutanées chroniques (CMC)29
* Cf. glossaire page 60
Les candidoses muqueuses chroniques sont observéesdans l'infection par le VIH, les déficitsimmunitaires T tels que les déficits immunitairescombinés sévères et le défaut de STAT 3 (syndromehyper IgE, page suivante). Mais une susceptibilitéparticulière à Candida sp est observée de façonisolée dans les Candidoses Muco-cutanéesChroniques (CMC). Les cas sont habituellementsporadiques mais des agrégations de cas familiauxavec une transmission dominante ou récessive ontété décrites. Les CMC constituent un aspect importantdu phénotype APECED (Auto-immune Poly-Endocrinopathie-Candidose-EctodermiqueDystrophie). L'APECED est une maladie monogéniquepar mutations bialléliques du gène AIRE en 21q22.3impliqué dans la délétion clonale thymique.L’exploration d’une très large famille iranienne de58 membres sur 3 générations atteinte de «CMC»isolée a conduit à l’identification d’une régioncandidate en 9q puis d’une mutation en 295,entraînant un stop prématuré dans CARD9(Cf. schéma ci-dessous).30 CARD9 est impliquéedans la signalisation intracellulaire du récepteurtransmembranaire Dectin 1 de l’immunité innéequi lie les ß-glucans de la paroi de certains fungi,la production de cytokines pro-inflammatoireset la différenciation des LT TH17* impliqués dansla défense antifungique chez l'homme. Le déficit en dectine est un déficit immunitairemodéré décrit dans une famille hollandaise cheztrois filles qui présentaient une candidose vaginalerécurrente, une onychomycose. Une mutationhomozygote non-sens dans dectine était retrouvéechez ces enfants.31
À la différence des pertes de fonction dans le domaineSRC ou le domaine de binding au DNA de STAT 1qui conduit à un tableau de susceptibilité génétiqueaux infections à mycobactéries et aux virus,les mutations du domaine coiled-coil de STAT1sont associées à une susceptibilité aux infectionsfongiques cutanéo-muqueuses. Ces mutationss'exercent par un gain de fonction qui conduità une déphosphorylation de STAT 1 activé et sonaccumulation dans le noyau. Les patients présententune candidose cutanéo-muqueuse dominante avecune candidose chronique oropharyngée sévère
et une dermatophytose associée, des phénomènesauto-immuns hypothyroïdies et hépatites auto-immunes. Ces mutations avec gain de fonctionentraînent un biais de la réponse immunitaire versla synthèse de cytokines qui inhibent la générationdes cellules Th17.32
Les neutropénies congénitales sévères,le neutrophile jouant le rôle ultime dans l'éliminationdes fungi se compliquent aussi volontiers par descandidoses sévères chroniques. Deux étiologiesgénétiques du syndrome CMC isolé viennent d'êtreélucidées : une mutation dominante autosomaledans le gène codant pour IL 17-F conduisant àun déficit de cette cytokine et une mutationrécessive dans le gène codant pour le récepteur Ade l'interleukine 17. Ces observations confirment
le rôle central de la voieIL-17 pour la réponseimmune muqueuseanti-Candida.33
Les récepteurs de type C lectine (à gauche) et les TLR (à droite) sontles principaux récepteurs aux fungi. Dectin-1 se lie aux b-glucans etla signalisation se fait via Syk et CARD9 pour induire une réponsecellulaire telle que l’explosion oxydative et la production de cytokine.Dectin-2 qui interagit avec la formation mycélienne (hyphe) s’associeavec FcgR pour déclencher la production de cytokines.MannoseR : mannose récepteur ; ITAM : immunoreceptor tyrosine-based activation motif ; Lyn and Fyn kinases ; TIR : Toll-IL-1 récepteur.
Candidose linguale
Les déficitssyndromiques
Les ca
ndidose
s muc
ocu
tané
es chr
oniqu
es
- 31 -
Observation clinique
Cette enfant âgée de sept ans est admise aux urgences pour une gêne respiratoire et une touximportante : la radiographie pulmonaire met en évidence une pneumopathie. L'enfant est traitéepar antibiothérapie par voie intraveineuse. L'interrogatoire a posteriori révèle l'existence d'une touxgrasse persistante depuis quelques mois. On apprend aussi qu'elle avait été drainée d'un abcèsde la fesse droite quelques mois auparavant, alors qu'elle n'avait pas de fièvre à ce moment-làet la culture a montré un staphylocoque doré (photographies). L'examen de la courbe staturo-pondérale montre une cassure progressive observée entre 4 ans et 7 ans avec une taille actuellequi se situe entre -2DS (déviation standard) et -3 DS et un poids entre -2DS et -3DS. Le carnetde santé est annoté de nombreux problèmes ORL et pulmonaires survenus dès l'âge de trois moistandis que le développement psychomoteur est normal. Elle est porteuse de nombreux molluscumcontagiosum. Il existe une hyperlaxité ligamentaire.
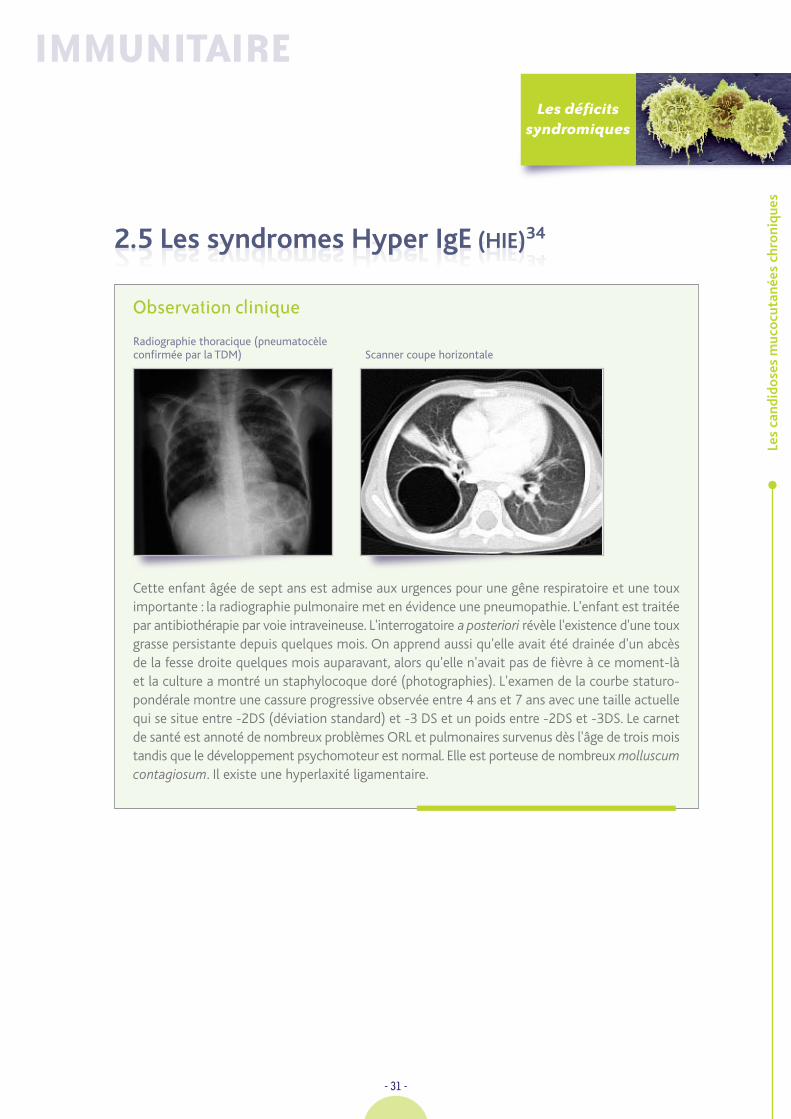
2.5 Les syndromes Hyper IgE (HIE)342.5 Les syndromes Hyper IgE (HIE)34
Scanner coupe horizontaleRadiographie thoracique (pneumatocèleconfirmée par la TDM)

- 32 -
DEF
AU
TS G
ENET
IQU
ES D
E LA
REP
ON
SE IM
MU
NIT
AIR
ELe
s dé
ficits
syn
drom
ique
s02
1 : persistance des dents lactéales ; 2 : abcès froid de la fesse ; 3 : pneumatocèle (TDM) ; 4 : hyperlaxité
1 2
3 4
L'enfant est très menue avec des lésions de dermatite atopique au niveau des plis de flexion,en particulier au niveau du coude, qui sont le siège de nombreux molluscum. Il n'y a pas de syndromedysmorphique évident du visage. Les lésions au niveau de la convexité des fesses sont séquellaires.L'auscultation révèle de nombreux râles ronflants avec une toux grasse. Les cheveux sont épars.Très bon développement psychomoteur. Les parents ne sont pas apparentés.
Quelle anomalie sur la radiographie de thorax n'a pas manqué de vouséchapper ?
Il existe une image de « bulle » claire dans le lobe supérieur droit, qui est tout à fait caractéristiqued'une pneumatocèle. La TDM (tomodensitométrie) précise qu’il s’agit d’une lésion cavitaireà paroi fine présentant un aspect discrètement polylobé à la partie haute et externe, située dansle segment apico-dorsal du lobe inférieur droit. L'IRM (Imagerie par Résonance Magnétique)montre l’existence de plusieurs images nodulaires infra-centimétriques en hyper-signal T2bilatérales, disséminées dans la substance blanche péri-ventriculaire. Le Z Score mesurépar ostéodensitométrie est très abaissé au niveau du rachis lombaire.
Les déficitssyndromiques
Les sy
ndro
mes
Hyp
er Ig
E (H
IE)
- 33 -
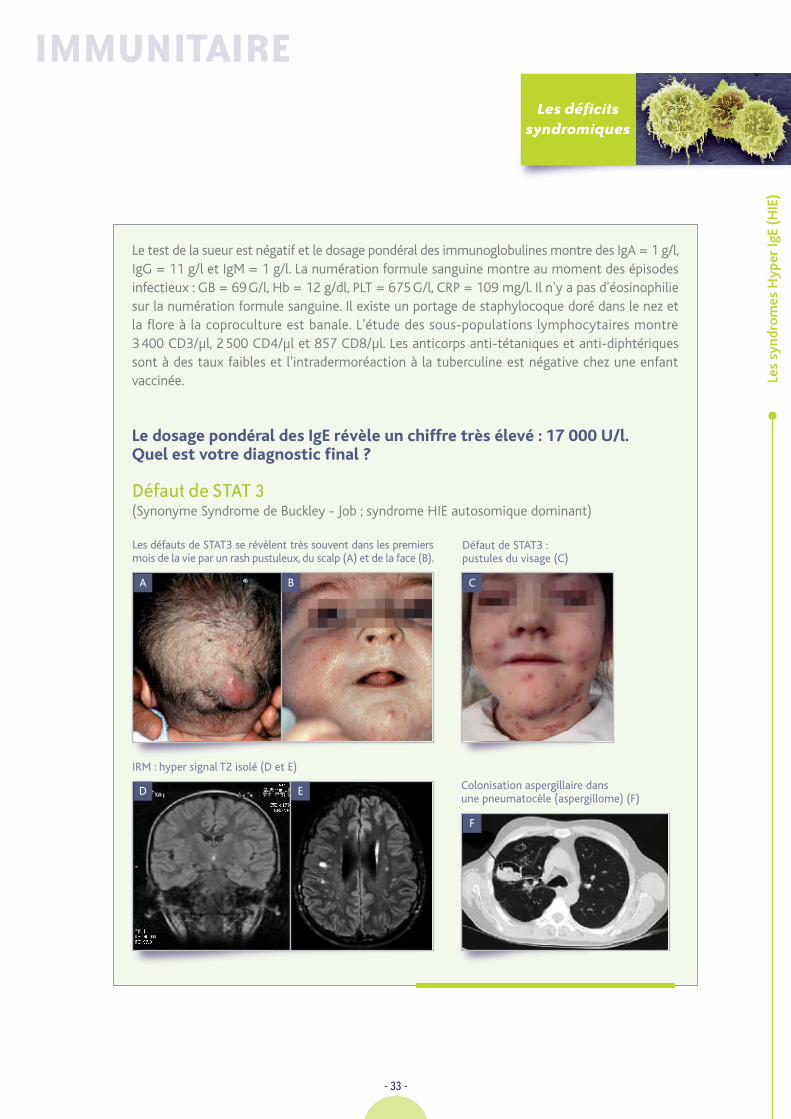
Le test de la sueur est négatif et le dosage pondéral des immunoglobulines montre des IgA = 1 g/l,IgG = 11 g/l et IgM = 1 g/l. La numération formule sanguine montre au moment des épisodesinfectieux : GB = 69G/l, Hb = 12 g/dl, PLT = 675G/l, CRP = 109 mg/l. Il n'y a pas d'éosinophiliesur la numération formule sanguine. Il existe un portage de staphylocoque doré dans le nez etla flore à la coproculture est banale. L'étude des sous-populations lymphocytaires montre3400 CD3/µl, 2500 CD4/µl et 857 CD8/µl. Les anticorps anti-tétaniques et anti-diphtériquessont à des taux faibles et l'intradermoréaction à la tuberculine est négative chez une enfantvaccinée.
Le dosage pondéral des IgE révèle un chiffre très élevé : 17 000 U/l. Quel est votre diagnostic final ?
Défaut de STAT 3(Synonyme Syndrome de Buckley - Job ; syndrome HIE autosomique dominant)
Les défauts de STAT3 se révèlent très souvent dans les premiersmois de la vie par un rash pustuleux, du scalp (A) et de la face (B).
IRM : hyper signal T2 isolé (D et E)
Défaut de STAT3 : pustules du visage (C)
A
D
CB
E Colonisation aspergillaire dans une pneumatocèle (aspergillome) (F)
F
- 34 -
DEF
AU
TS G
ENET
IQU
ES D
E LA
REP
ON
SE IM
MU
NIT
AIR
ELe
s dé
ficits
syn
drom
ique
s02
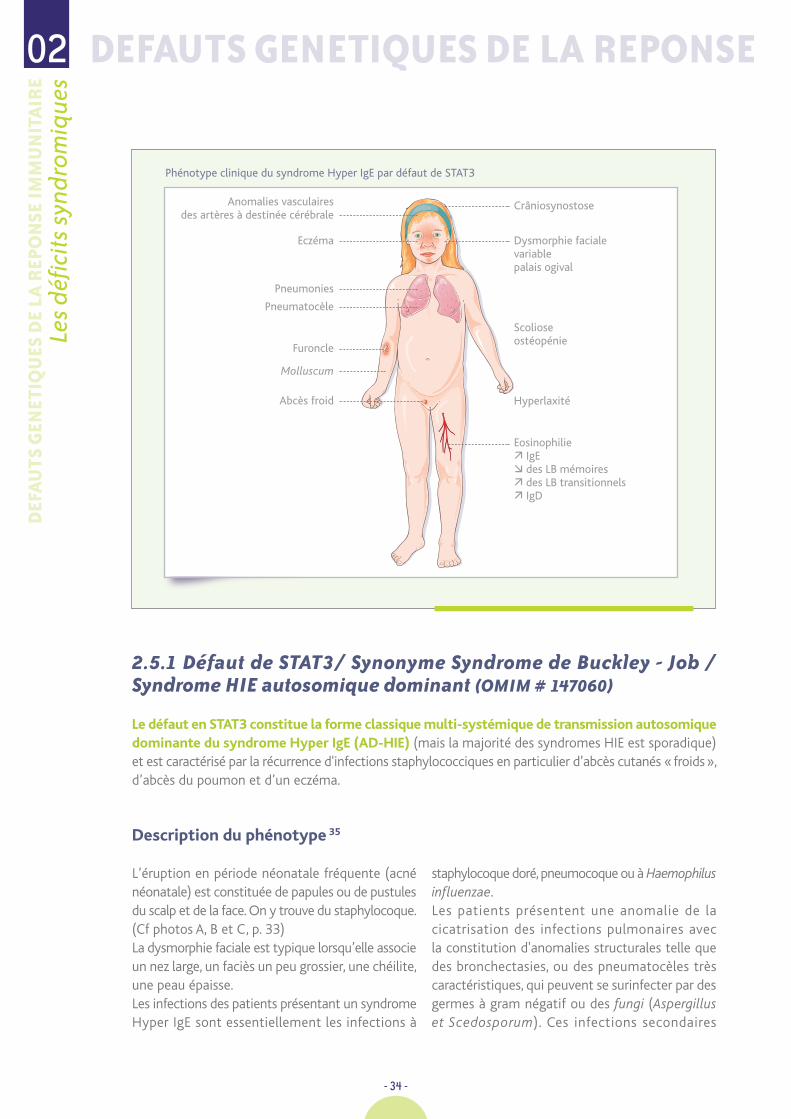
Phénotype clinique du syndrome Hyper IgE par défaut de STAT3
Eczéma Dysmorphie facialevariablepalais ogival
Scolioseostéopénie
Hyperlaxité
Eosinophilie IgE des LB mémoires des LB transitionnels IgD
Pneumonies
Pneumatocèle
Furoncle
Molluscum
Abcès froid
CrâniosynostoseAnomalies vasculaires des artères à destinée cérébrale
2.5.1 Défaut de STAT3/ Synonyme Syndrome de Buckley - Job /Syndrome HIE autosomique dominant (OMIM # 147060)
Le défaut en STAT3 constitue la forme classique multi-systémique de transmission autosomiquedominante du syndrome Hyper IgE (AD-HIE) (mais la majorité des syndromes HIE est sporadique)et est caractérisé par la récurrence d'infections staphylococciques en particulier d’abcès cutanés «froids»,d’abcès du poumon et d’un eczéma.
Description du phénotype 35
L’éruption en période néonatale fréquente (acnénéonatale) est constituée de papules ou de pustulesdu scalp et de la face. On y trouve du staphylocoque.(Cf photos A, B et C, p. 33)La dysmorphie faciale est typique lorsqu’elle associeun nez large, un faciès un peu grossier, une chéilite,une peau épaisse.Les infections des patients présentant un syndromeHyper IgE sont essentiellement les infections à
staphylocoque doré, pneumocoque ou àHaemophilusinfluenzae. Les patients présentent une anomalie de lacicatrisation des infections pulmonaires avecla constitution d'anomalies structurales telle quedes bronchectasies, ou des pneumatocèles trèscaractéristiques, qui peuvent se surinfecter par desgermes à gram négatif ou des fungi (Aspergilluset Scedosporum). Ces infections secondaires
Les déficitssyndromiques
- 35 -
habituellement sont indolentes et difficilesà traiter. Elles présentent d'autre part un risqueévolutif vers la rupture dans les vaisseauxpulmonaires, entraînant une hémoptysiefoudroyante, ou une dissémination fongiquedans le système nerveux central. Les anévrismes artériels des carotides ou descoronaires constituent une complication récemmentreconnue à l'âge adulte du déficit en STAT3.Des infarctus lacunaires sont souvent relevéssur l'IRM et sans conséquence clinique. De même,
on a décrit des anomalies des signaux (hyperintensité T2, cf. IRM p. 34) de découverte fortuite.Des fractures pathologiques récurrentes liéesà une réduction de la masse osseuse surviennentchez la moitié des patients ainsi qu'une scoliose. La réduction de la résorption des racines primitivesdes dents lactéales entraîne une rétention des dentslactées.Les patients sont à risque de développer un néoplasiesecondaire (lymphomes et adénocarcinomespulmonaires).
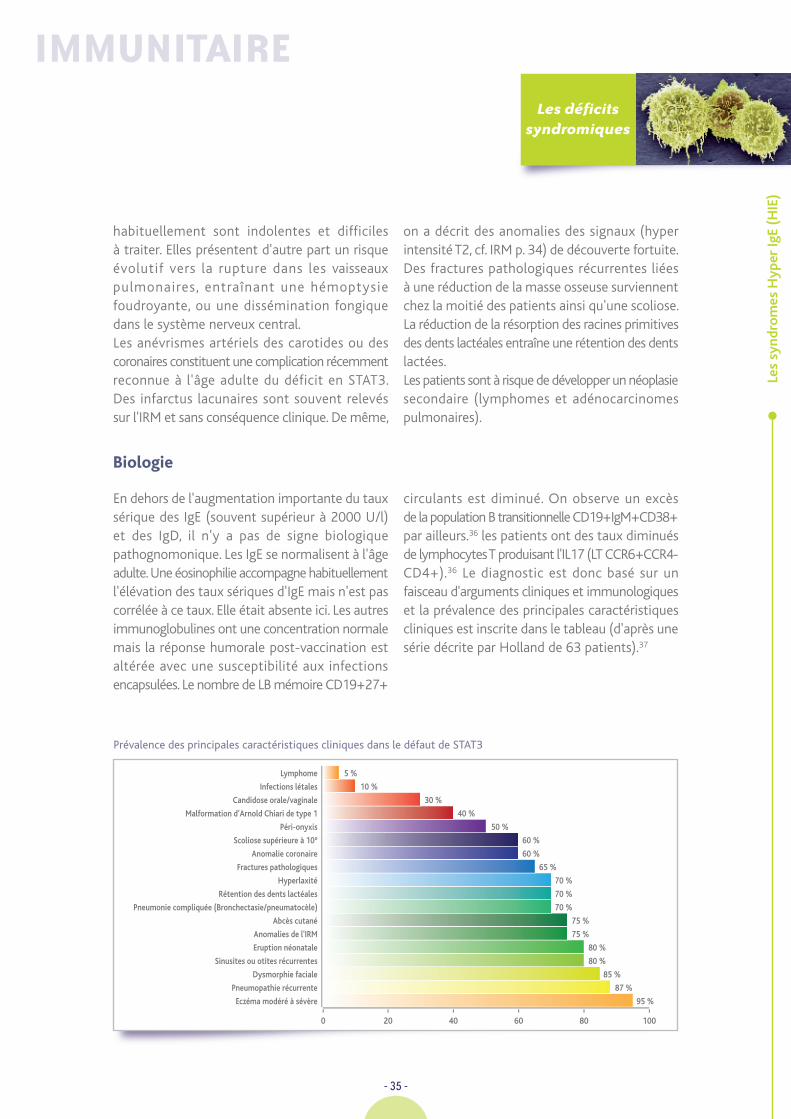
Biologie
0 20 40 60 80 100
Lymphome 5 %
Infections létales 10 %
Candidose orale/vaginale 30 %
Malformation d’Arnold Chiari de type 1 40 %
Péri-onyxis 50 %
Scoliose supérieure à 10° 60 %
Anomalie coronaire 60 %
Fractures pathologiques 65 %
Hyperlaxité 70 %
Rétention des dents lactéales 70 %
Pneumonie compliquée (Bronchectasie/pneumatocèle) 70 %
Abcès cutané 75 %
Anomalies de l'IRM 75 %
Eruption néonatale 80 %
Sinusites ou otites récurrentes 80 %
Dysmorphie faciale 85 %
Pneumopathie récurrente 87 %
Eczéma modéré à sévère 95 %
En dehors de l'augmentation importante du tauxsérique des IgE (souvent supérieur à 2000 U/l)et des IgD, il n'y a pas de signe biologiquepathognomonique. Les IgE se normalisent à l'âgeadulte. Une éosinophilie accompagne habituellementl'élévation des taux sériques d'IgE mais n'est pascorrélée à ce taux. Elle était absente ici. Les autresimmunoglobulines ont une concentration normalemais la réponse humorale post-vaccination estaltérée avec une susceptibilité aux infectionsencapsulées. Le nombre de LB mémoire CD19+27+
circulants est diminué. On observe un excèsde la population B transitionnelle CD19+IgM+CD38+par ailleurs.36 les patients ont des taux diminuésde lymphocytes T produisant l'IL17 (LT CCR6+CCR4-CD4+).36 Le diagnostic est donc basé sur unfaisceau d'arguments cliniques et immunologiqueset la prévalence des principales caractéristiquescliniques est inscrite dans le tableau (d'après unesérie décrite par Holland de 63 patients).37
Prévalence des principales caractéristiques cliniques dans le défaut de STAT3
Les sy
ndro
mes
Hyp
er Ig
E (H
IE)

- 36 -
DEF
AU
TS G
ENET
IQU
ES D
E LA
REP
ON
SE IM
MU
NIT
AIR
ELe
s dé
ficits
syn
drom
ique
s02
Des mutations de STAT3 ont été identifiées dans la plupart des cas de syndromeHyper IgE de type autosomal dominant.
Immunologie
IFNα/β IL-12 IL-23 IL-10 IL-6
STAT1STAT2
STAT4 STAT3STAT4
STAT3 STAT3
IFNAR1 IFNAR2 p35 p40 p40 p19
IL-12Rβ2 IL-12Rβ1 IL-23RIL-10R2 IL-10R1 IL-6R
gp130
Jak1Tyk2 Tyk2Tyk2
Tyk2Tyk2Jak1 Jak1Jak1Jak2 Jak2 Jak2
Les mutations ont été trouvées dans deux hotspots* du gène STAT3, le domaine SH2 (interactionprotéine-protéine) et le domaine liant l'ADN.Les mutations observées dans différents groupesethniques sont identiques (mutations récurrentesde novo). Il s'agit de mutations faux sens, oudélétionnelles sans décalage de cadre de lectureconduisant à la production d'une protéine mutantetronquée. Les mutations humaines de STAT3entraînent un effet dominant négatif* surla version normale du gène, à corréler avec le modede transmission dominante de l’affection.37
Chez la souris, le défaut hétérozygote de STAT3n’entraîne pas de conséquence, tandis que les animauxinvalidés pour STAT3 meurent durant l’embryogenèse.L'invalidation tissu-spécifique du gène STAT3,notamment dans l'épithélium pulmonaire, montre
une inflammation excessive, une augmentationdes espaces aériens après exposition à l'oxygène,réminiscentes des pneumatocèles du patient HIE.Les animaux invalidés pour STAT3 dans le tissucérébral ont des anomalies du remodelage vasculaire,sans doute un modèle pertinent pour l'étude desanévrismes et des artères coronaires du syndromeHIE.37
STAT3 joue un rôle important dans la différenciationdes ostéoblastes et des ostéoclastes in vitroet l'invalidation de STAT3 chez la souris dans lesostéoblastes ou les ostéoclastes entraîne uneostéoporose marquée. Les ostéoclastes différenciésà partir des monocytes circulants de patientsprésentant un défaut de STAT3 ont une activité derésorption osseuse accrue par rapport aux contrôles.
* Cf. glossaire page 60
C'est l'anomalie de signalisation de l'IL-11 via STAT3 qui estresponsable de la crâniosynostose et des dents surnuméraires.Une mutation faux sens dans le récepteur de l'IL-11 vient en effetd'être rapportée et qui rend compte de ces manifestations isolées.38
Les récepteurs aux cytokines et lesmolécules Jak associées (Jak1, Jak2,and TYK-2) et les facteurs STATS. STAT3 est associé aux récepteursde l’IL-23, IL-10, IL-6 et IL-11 non figuréici. Les mutations de TYK-2, JAK3,STAT1, and STAT5b sont associéesà des DIP ce qui souligne le caractèrecentral des voies JAK/STAT dansde nombreux systèmes biologiques.D’après Minegishi.39

Les déficitssyndromiques
- 37 -
Le gène STAT3 est localisé sur la bande 17q21. STAT3 est l'un des 7 facteurs de transcription de la famille STAT. La voieSTAT / JAK permet la transduction du signal pour un nombre important de récepteurs aux cytokines conduisantà la transcription/traduction des gènes cible. Les protéines STAT se lient aux tyrosine-kinases JAK associées aux portionsintracellulaires des récepteurs aux cytokines. Quand la cytokine appropriée se lie à son récepteur, la kinase Jak phosphorylele récepteur et subséquemment la protéine STAT. Celle-ci se dissocie alors du complexe JAK/récepteur et constituedes homo-dimères au sein du cytosol. Ce dimère de STAT3 transloque dans le noyau, se lie aux séquences promotricesdes gènes cibles et active la transcription. Cette activité est bloquée par la déphosphorylation de STAT. 40
Cytokines
Récepteurs de cytokine
JakPhosphorylation
Phosphorylation
PhosphataseTranscription
STAT3
Les sy
ndro
mes
Hyp
er Ig
E (H
IE)

- 38 -
DEF
AU
TS G
ENET
IQU
ES D
E LA
REP
ON
SE IM
MU
NIT
AIR
ELe
s dé
ficits
syn
drom
ique
s02
STAT3 est indispensable pour la différenciationdes TH17* : les cellules CD4 des patients atteintsne génèrent pas de TH17 par défaut d'expressiondu régulateur de transcription spécifique appeléRORgt (ROR pour rétinoïd related orphan receptor)via STAT3.39 Ces cellules TH17 sont importantespour l'immunité muqueuse et épithéliale,notamment contre les bactéries extracellulaireset les champignons, de plus ces sous-populationslymphocytaires CD4+ jouent un rôle central dans
la mobilisation des neutrophiles en induisant,par l’épithélium, la production de chimiokines.
Les cytokines IL-22 et IL-17 produites par les TH17conduisent à la production par les kératinocytesdes défensines (peptides anti-microbiens) importantesdans l'immunité cutanée. La salive des patients estpauvre en peptides fungicides anti-Candida induitspar l'IL-17 et les candidoses muqueuses sontfréquentes.37
A : Défense normale B : Défaut de STAT3
* Cf. glossaire page 60
Les cellules dendritiques produisent l’IL-1, l’IL-6 et l’IL-23 qui promeuvent la différentiation des TH17. La signalisation IL-6est dépendante de STAT3. La signalisation IL-21/ IL-23 est médiée par STAT3 en partie. Les TH17 sécrètent IL-17 A et F quidéclenchent la synthèse de chimiokines CXC par les cellules épithéliales. Ces chimiokines recrutent les PN pour la destructiondes pathogènes extracellulaires. L’IL-22 sécrétée par les TH17 conduit aussi à la sécrétion par les cellules épithéliales des peptides antimicrobiens. Dans le syndrome HIE, la diminution de transcription de RORgt et de la différenciation des TH17 altère la défense des cellules épithéliales contre les bactéries et les champignons extracellulaires.C'est l'altération de l'immunité via IL-6 qui rend compte de la susceptibilité aux infections staphylococciques cutanées.85 % de la cohorte française a développé une infection chronique par Candida.36 Le rôle spécifique des lymphocytes Tproducteurs de IL-17 dans l'immunité anti-Candida albicans est à nouveau illustré dans cette maladie. (Voir précédemmentdiscussion sur les étiologies génétiques des candidoses cutanéo-muqueuses.
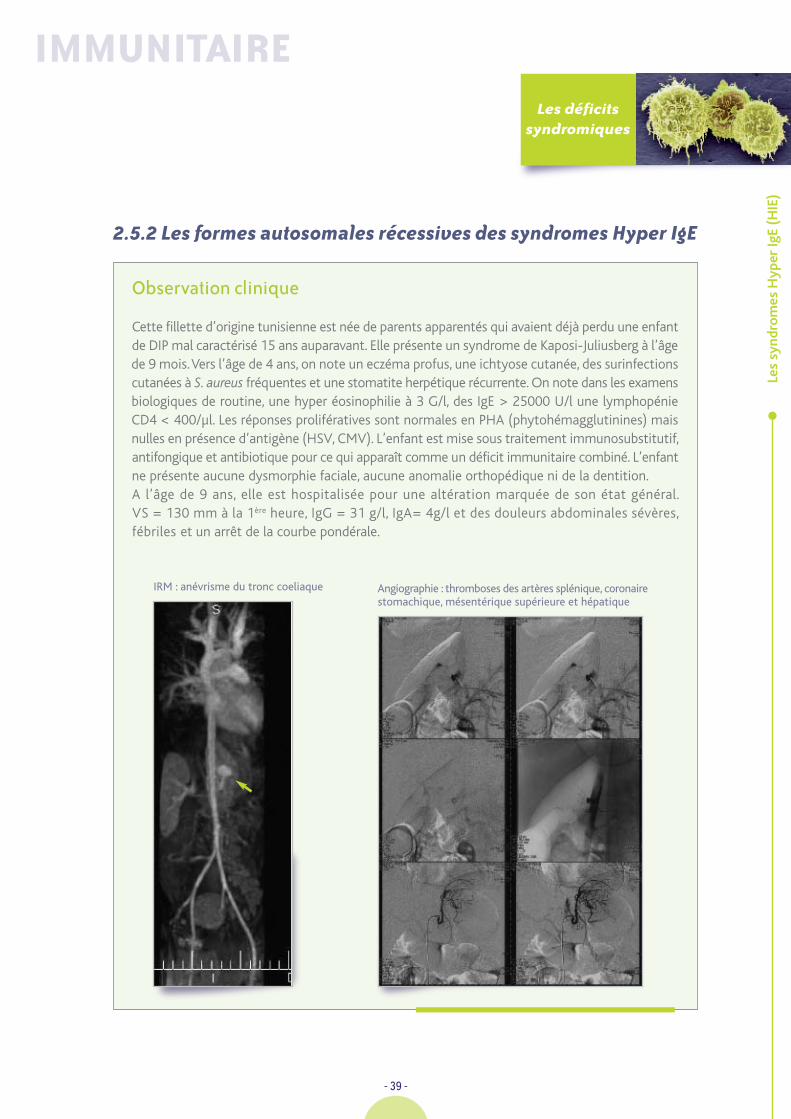
Les déficitssyndromiques
- 39 -
2.5.2 Les formes autosomales récessives des syndromes Hyper IgE
Observation clinique
Cette fillette d’origine tunisienne est née de parents apparentés qui avaient déjà perdu une enfantde DIP mal caractérisé 15 ans auparavant. Elle présente un syndrome de Kaposi-Juliusberg à l’âgede 9 mois. Vers l’âge de 4 ans, on note un eczéma profus, une ichtyose cutanée, des surinfectionscutanées à S. aureus fréquentes et une stomatite herpétique récurrente. On note dans les examensbiologiques de routine, une hyper éosinophilie à 3 G/l, des IgE > 25000 U/l une lymphopénieCD4 < 400/µl. Les réponses prolifératives sont normales en PHA (phytohémagglutinines) maisnulles en présence d’antigène (HSV, CMV). L’enfant est mise sous traitement immunosubstitutif,antifongique et antibiotique pour ce qui apparaît comme un déficit immunitaire combiné. L’enfantne présente aucune dysmorphie faciale, aucune anomalie orthopédique ni de la dentition. A l’âge de 9 ans, elle est hospitalisée pour une altération marquée de son état général. VS = 130 mm à la 1ère heure, IgG = 31 g/l, IgA= 4g/l et des douleurs abdominales sévères,fébriles et un arrêt de la courbe pondérale.
IRM : anévrisme du tronc coeliaque Angiographie : thromboses des artères splénique, coronairestomachique, mésentérique supérieure et hépatique
Les sy
ndro
mes
Hyp
er Ig
E (H
IE)

- 40 -
DEF
AU
TS G
ENET
IQU
ES D
E LA
REP
ON
SE IM
MU
NIT
AIR
ELe
s dé
ficits
syn
drom
ique
s02
Il existe une vascularite des gros vaisseauxartériels digestifs, avec un anévrisme du tronccœliaque et de multiples thromboses des artèressplénique, coronaire stomachique, mésentériquesupérieure et hépatique. Le PET scan objectiveune activité métabolique anormale au niveaude l’artère cœliaque. Cette vascularite des gros vaisseaux sera traitéepar immunosuppresseur et corticothérapie.
L’évolution à 7 semaines est marquée parun syndrome lymphoprolifératif EBV+ avec denombreux nodules intratissulaires disséminés.La charge virale EBV est à 80 000 copies/mlet à la biopsie d’un nodule rénal on objectiveune prolifération B immunoblastique exprimantLMP2A et CD20. Le traitement immunosuppresseurest diminué, puis l’enfant est traitée par anticorpsmonoclonal.
Après mise en rémission, elle est greffée avecsa sœur HLA compatible vers l’âge de 9,5 ans.La prise est rapide et le chimérisme complet eststable avec 2 ans de recul avec une reconstitutionhématologique et immunitaire satisfaisante.
Quel diagnostic évoquez-vous ici ?L’Hyper IgE, l’eczéma profus, les infectionsrécurrentes à HSV sont très suggestives d’unsyndrome HIE autosomal récessif compliquéici par une vascularite abdominale très sévère.
Comment le confirmer ? L’étude en array CGH a montré une large délétionhomozygote de 400 kb emportant le gène DOCK8confirmant le diagnostic de défaut génétiqueen DOCK8 dont la fonction hypothétique estillustrée dans le schéma page suivante.
Doppler : thrombose dans le tronc cœliaque
TDM : nodule pulmonaire sous-pleural
TDM : nodules intratissulaires disséminés bien visiblesici sur les reins

Introduction à l’étudedes DIP
- 41 -
Les formes autosomales récessives constituentune petite fraction des syndromes HIE.
Si les taux sériques des IgE sont habituellementcomparables, avec une éosinophilie habituellementtrès marquée, la susceptibilité aux infections viralescutanées est remarquable : molluscum contagiosum,Herpès virus et VZV. Il n'y a pas de constitutionde pneumatocèle dans ces formes ni de manifestationssquelettiques, aucun problème dentaire et aucunedysmorphie faciale. Les formes « autosomalesrécessives de syndrome Hyper IgE» constituent
un cadre hétérogène sur le plan génétique. Deuxdéfauts génétiques ont été élucidés : le défautde DOCK8 et les mutations récessives de TYK2.Sur un plan nosologique, leur description dansle cadre des syndromes HIE est remise enquestion. Reflet d'une dysrégulation T dans cesmaladies, une élévation des IgE sérique peut êtreobservée dans de nombreux déficits immunitaireset notamment le syndrome de Wiskott Aldrich,le syndrome d'Omenn déjà évoqué, l'anomalie deDiGeorge, l'IPEX, et le syndrome de Comel-Netherton.
Fonction hypothétique de DOCK8.DOCK8 fonctionne possiblement comme un «guanine exchanging factor» qui convertit les GTPases Cdc42 et Rac1en configuration active. Celle-ci autorise le réarrangement des filaments d’actine, la formation des lamellipodes parl’intermédiaire de WASP (la protéine codée par le gène du sd de Wiskott Aldrich) puis la migration cellulaire, lacroissance, l’adhésion et la formation de la synapse immunologique. Dans un modèle de souris invalidées pour DOCK8,il a été montré récemment l'importance de cette protéine pour la migration des cellules dendritiques interstitielles.41
Redessiné d’après : 42Les lamellipodes sont des extensions membranaires qui permettent le mouvement de la cellule par protrusion,adhésion et rétraction.
Remerciements : V. Barlogis et C. Galambrun. Hôpital La Timone Enfants, Service d’Hématologie Pédiatrique, Marseille Le
s sy
ndro
mes
Hyp
er Ig
E (H
IE)

- 42 -
DEF
AU
TS G
ENET
IQU
ES D
E LA
REP
ON
SE IM
MU
NIT
AIR
ELe
s dé
ficits
syn
drom
ique
s02
Mutation homozygote dans le gène de la tyrosine kinase : TYK-2 (OMIM # 611521)
La description par Minegishi d’une forme récessivede « syndrome Hyper IgE » liée à une délétionhomozygote dans le gène de la tyrosine kinase :TYK-243 a précédé l’élucidation du défaut de STAT3par le même chercheur. Ce patient unique présentaitpar ailleurs des infections à BCG et à salmonelles,comme on les rencontre dans les syndromesde susceptibilité mendélienne aux infections àmycobactérie par anomalies de la boucle IFN-g/IL-12.La tyrosine kinase TYK-2 est une kinase appartenantà la famille des kinases Jak intervenant dans
la signalisation des cytokines : interféron de type 1,interleukines IL-6, IL-10, IL-12 et IL-23. Ce patientprésentait une susceptibilité aux infections viralesprobablement en raison d'un défaut de signalisationdes interférons de type 1. L'absence de TYK-2entraîne un défaut de signalisation de l’IL-12conduisant à un défaut de la production d'IFN-gavec comme conséquence une susceptibilité auxinfections bactériennes intracellulaires.
Les erreurs innées de la boucle IL-12/23-IFN-g entraînent une susceptibilité mendélienne aux infections en mycobactéries.Le schéma indique les voies de production de cytokines et la coopération entre les cellules monocytaires et les cellulesdendritiques et les populations lymphocytaires NK et T. La boucle IL-12/23-IFN-g et la voie CD40-CD40 Ligand qui permettentla coopération NK/T et les monocytes et les dendritiques sont indispensables pour la protection contre les infectionsmycobactéries chez l'homme. Les mutations de NEMO modifient la voie dépendante de CD40-NEMO. Les protéines pourlesquelles les mutations dans les gènes correspondants ont été associées à une susceptibilité particulière aux infectionsà mycobactéries sont signalées en bleu. Non figuré dans le schéma, le défaut d'ISG15 qui agit comme une cytokine qui induit la sécrétion d'IFN-g vient d'êtrerécemment rapporté comme une nouvelle forme récessive de susceptibilité mendélienne aux infections à mycobactéries.Les mutations ont été identifiées par séquençage exome entier. Encore un exemple de la puissance de cette technologiedans le domaine des DIP. Les phénotypes cliniques et immunologiques sont proches des patients présentant un défautd'IL-12 p40, et d'IL-12 Rb1. La boucle ISG15-IFN-g opére principalement entre les PN et les cellules NK et peut réaliserainsi la voie complémentaire « innée » de la circuiterie IL-12-IFN-g de l'immunité adaptative qui concerne les phagocytesmononucléés et les LT. 44
Monocytes / cellules dendritiques Lymphocytes T / cellules NK

Introduction à l’étudedes DIP
- 43 -
Un deuxième malade TYK-2 -/-45 vient d'être identifié mais qui, à la différence du premier rapportépar Minegishi, ne présente pas de « syndrome Hyper IgE» mais souffre de BCGite disséminéeet de neurobrucellose. La réponse aux interférons de type1 est altérée mais non abolie, et l’enfanta présenté une infection herpétique limitée. A la différence du premier patient japonais, celui-ci n’apas présenté d'infection à pyogène typique des syndromes Hyper IgE ni d'eczéma.
Ainsi, le défaut génétique de TYK-2 ne conduit pas nécessairement à un « syndrome Hyper IgE » maisdoit être recherché chez des patients présentant un déficit immunitaire caractérisé par des infectionsà germes intracellulaires (mycobactéries, salmonelle ou Brucella) et des infections virales cutanées(HSV).
Déficit en DOCK8 (OMIM # 243700)
Des mutations bialléliques (délétions HZ ou mutationshétérozygotes composites) dans le gène DOCK8 ont étérapportées par 2 groupes42,46 dans un DIP associant des infectionsORL et pulmonaires, des infections virales cutanées récurrenteset sévères notamment à HSV et VZV, Papillomavirus et molluscum(figure) et une atopie marquée souvent surinfectée. Un spectretrès étendu d’infections suggère un déficit combiné de l’immunitéadaptative. Des encéphalites à virus JC et des vascularitesdu SNC sont aussi rapportées. Il n’y a jamais de pneumatocèle.Des taux élevés d’IgE, une éosinophilie, une lymphopénie T/Bet des altérations de la fonction humorale sont retrouvés chezles patients avec une hypo IgM chez la plupart, et une baissede l’immunité vaccinale. Les réponses prolifératives CD4+et CD8+ sont très altérées suggérant un rôle majeur de cette
protéine dans l’activation lymphocytaire. Certains présentent une diminution des TH17 impliquésdans la défense antifungique et contre les bactéries extracellulaires, expliquant les candidosescutanéo-muqueuses de ces patients. Le défaut en DOCK8 prédispose au cancer (carcinomes,hémopathies lymphoïdes). La fonction de DOCK8 est inconnue, mais cette protéine qui appartientà la famille des guanine exchange factors régule le réarrangement du cytosquelette, la migration,l’adhésion cellulaire. D’autres DIP tels que le syndrome de Wiskott-Aldrich sont liés à un défautgénétique en protéine de l’organisation du cytosquelette. Les caractéristiques immunologiquesdu défaut en DOCK8 doivent le faire classer dans les déficits immunitaires combinés. Certainspatients ne présentent pas d''hyper IgE.47
Molluscum diffus (défaut de DOCK8),un trait clinique qui permet de distinguerles défauts de STAT3 (syndrome Hyper IgEdominant) et les défauts de DOCK8(formes récessives de sd HIE à considérerplutôt comme un déficit immunitairecombiné).
Les sy
ndro
mes
Hyp
er Ig
E (H
IE)

- 44 -
DEF
AU
TS G
ENET
IQU
ES D
E LA
REP
ON
SE IM
MU
NIT
AIR
ELe
s dé
ficits
syn
drom
ique
s02
2.5.3 Quelques données thérapeutiques dans les défauts de STAT3
Un certain nombre de syndromes sont caractérisés par l'existence d’aberrations chromosomiques,spontanées ou induites par les radiations ionisantes. Les gènes impliqués codent des protéines constituantun réseau multimoléculaire d'une complexité inouïe, responsables du maintien de l'intégrité du génome :régulation du cycle cellulaire, résolution des cassures double brin. Ce sont les réparatoses ou maladiescassantes. Il faut citer l'ataxie-télangiectasie, l'anémie de Fanconi (qui sera traitée avec les neutropénies),le syndrome de Bloom et d’autres affections très exceptionnelles : le syndrome ataxie-télangiectasielike, les défauts de ligase 4, Cernunos et de RAD50. L'instabilité génomique explique la prédispositionde ces différentes affections aux néoplasies.
chromosomique ou réparatoses2.6 Les syndromes d’instabilité2.6 Les syndromes d’instabilitéchromosomique ou réparatoses
* Pour une information complète se reporter au Dictionnaire des Spécialités Pharmaceutiques
Pour éviter les infections superficielles et invasivesà staphylocoque doré, l'hygiène cutanée estfondamentale. Une antibiothérapie dissuasivesous forme de TMP-SMZ* est proposée pourla prévention des infections cutanées et pulmonaires.L'antibiothérapie antifongique pour prévenirl'aspergillose est logique, ainsi que pour la préventionde la candidose cutanée ou muqueuse. Le traitementdes infections pulmonaires est adapté à l'écologiemicrobienne. L'attention des praticiens doit êtreattirée sur le fait que ces enfants ne présententpas de fièvre en cas de surinfection pulmonaire.L’immunosubstitution peut diminuer la récurrence
des infections chez certains enfants qui présententde plus une susceptibilité aux infections àpneumocoques. La prise en charge de l'ostéoporoseet des fractures doit être précisée à l'avenir toutcomme la prise en charge des anévrismes.48,66
Le caractère multi-systémique complexe du déficiten STAT3 justifie une prise en charge multidisciplinaire.Les défauts de DOCK8 sont d'évolution bien plusgrave et justifient d’une greffe de moelle osseuseaprès contrôle des problèmes infectieux etdysimmunitaires dans ce DIP qui apparaît plutôtcomme un déficit cellulaire combiné.47

Introduction à l’étudedes DIP
- 45 -
2.6.1 Le syndrome de BLOOM (OMIM # 210900)
Observation clinique
Une enfant de 4 ans est adressée pour des infections ORL à répétition et une croissance pondéraleinsuffisante. Cette petite fille présente un érythème du visage, qui s'accentue l'été au soleil. Elle mesure 91 cm(- 2,5 DS) et pèse 11 kg. Il existe de multiples taches cutanées café au lait, diffuses, de formeirrégulière, trois ayant plus de 2 cm, les autres étant autour de 0,5 cm avec, de façon associée,des macules achromiques des jambes et du dos de la main. Le visage est particulier avec unehypoplasie du massif facial. Le reste de l'examen clinique est normal. Il existe des adénopathiesbanales des plis inguinaux et les amygdales sont bien vues. Le développement psychomoteurest satisfaisant. La voix est « haut perchée ».
Ses parents, non apparentés sont d'origine française. Elle avait été hospitalisée à l'âge de 17 moispour une gastro-entérite et on avait noté alors un retard staturo-pondéral à - 3 DS. C'est à 20 moisque sont apparues les taches café au lait. Le dosage pondéral des immunoglobulines montre desIgG à 6,9 g/l, des IgA à 0,19 g/l, des IgM à 0,23 g/l. Les anticorps vaccinaux sont à un titre nonprotecteur malgré un rappel de vaccination. L'intradermoréaction à la tuberculine est négativemalgré un vaccin anti-tuberculeux récent. La numération formule sanguine montre :leucocytes = 7 ,97 G/l, hémoglobine = 12,5 g/dl, plaquettes = 371 G/l. L'âge osseux est en rapport avec l'âge chronologique. La petite sœur de deux ans a une morphologiecomplètement différente.
Quel examen demandez-vous chez cette enfant qui présente unephotosensibilité cutanée, un déficit immunitaire et une dyspigmentation ?
Réponse
Un caryotype constitutionnelL'étude des échanges des chromatides sœurs montre un taux anormalement élevé d'échangespar rapport au témoin (malade : plus de 60 échanges par cellule, témoin : environ 15 échanges).Le caryotype montre par ailleurs un taux élevé spontané de cassures chromosomiques, environ19 %, avec notamment des cassures transversales des chromosomes au niveau centromérique.
Les sy
ndro
mes
d’in
stab
ilité
chr
omoso
mique
s ou répa
rato
ses

- 46 -
DEF
AU
TS G
ENET
IQU
ES D
E LA
REP
ON
SE IM
MU
NIT
AIR
EIn
trod
uctio
n à
l’étu
de d
es D
IP
01
Il s’agit d’un syndrome de BLOOM.Le déficit immunitaire est variable et porte éventuellement sur une diminution des trois isotypesdes immunoglobulines et sur la réponse proliférative des LT et la réponse anticorps. Comme pourl'ataxie-télangiectasie, il n'y a pas de corrélation stricte entre les anomalies constatées in vitroet la susceptibilité réelle aux infections. Le phénotype du syndrome de Bloom est caractéristique. La confirmation est apportéepar la démonstration cytogénétique : l'augmentation du taux d'échange des chromatides sœurs.
Petite taille, faciès anguleux, hypoplasie malaire,micrognathie, érythème solaire et anomalie dela pigmentation, avec des plages d’hypo et d’hyper- pigmentation au niveau du tronc sont les traitsphénotypiques caractéristiques du syndromede Bloom.
La culture des lymphoblastes et des fibroblastes objective l'instabilitégénomique avec de nombreux gaps chromatiniens, cassures,et réarrangements. Les cellules montrent une tendance trèsimportante à l'échange de matériel entre les chromatides de chacundes chromosomes d'une même paire pour former des figures quadri-radiales (cf. cercle) dans des régions homologues ou entre leschromatides sœurs d'un chromosome donné (milieu contenant duBrdU pour leur visualisation donnant un aspect caractéristiquecomme ci-contre) (Cliché Fabienne Prieur, laboratoire de cytogénétique,CHU de Saint-Etienne).
A B
Description du phénotype clinique49
Ce syndrome est parfois confondu avec le nanisme de Silver-Russel mais la petite taille est ici harmonieuseavec dolichocéphalie. C'est probablement l'instabilité génomique qui est responsable de la réductiondu capital cellulaire et de la petite taille. Le visage est caractéristique, avec un massif crânial étroit,anguleux, une hypoplasie malaire et mandibulaire. Il existe une réduction de la masse graisseuse.Une sensibilité particulière aux UV (ultra-violets) se traduit par un érythème solaire parfois télangiectasiquedu visage ou des parties découvertes s'accompagnant souvent de lésions fissuraires ou de perlèche(comme dans le cas de cette patiente).
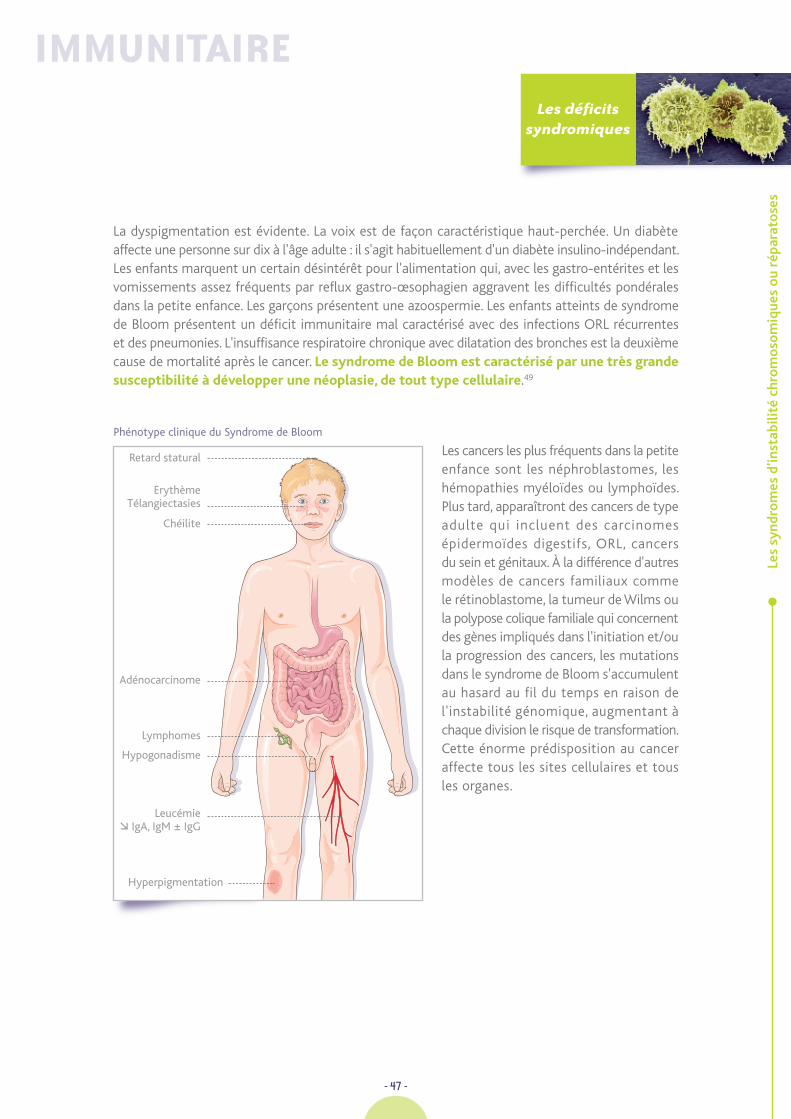
Les déficitssyndromiques
- 47 -
La dyspigmentation est évidente. La voix est de façon caractéristique haut-perchée. Un diabèteaffecte une personne sur dix à l'âge adulte : il s'agit habituellement d'un diabète insulino-indépendant.Les enfants marquent un certain désintérêt pour l'alimentation qui, avec les gastro-entérites et lesvomissements assez fréquents par reflux gastro-œsophagien aggravent les difficultés pondéralesdans la petite enfance. Les garçons présentent une azoospermie. Les enfants atteints de syndromede Bloom présentent un déficit immunitaire mal caractérisé avec des infections ORL récurrenteset des pneumonies. L'insuffisance respiratoire chronique avec dilatation des bronches est la deuxièmecause de mortalité après le cancer. Le syndrome de Bloom est caractérisé par une très grandesusceptibilité à développer une néoplasie, de tout type cellulaire.49
Les cancers les plus fréquents dans la petiteenfance sont les néphroblastomes, leshémopathies myéloïdes ou lymphoïdes.Plus tard, apparaîtront des cancers de typeadulte qui incluent des carcinomesépidermoïdes digestifs, ORL, cancersdu sein et génitaux. À la différence d'autresmodèles de cancers familiaux commele rétinoblastome, la tumeur de Wilms oula polypose colique familiale qui concernentdes gènes impliqués dans l'initiation et/oula progression des cancers, les mutationsdans le syndrome de Bloom s'accumulentau hasard au fil du temps en raison del'instabilité génomique, augmentant àchaque division le risque de transformation.Cette énorme prédisposition au canceraffecte tous les sites cellulaires et tousles organes.
Phénotype clinique du Syndrome de Bloom
Chéilite
Adénocarcinome
ErythèmeTélangiectasies
Lymphomes
Hypogonadisme
Hyperpigmentation
Retard statural
Leucémie IgA, IgM ± IgG
Les sy
ndro
mes
d’in
stab
ilité
chr
omoso
mique
s ou répa
rato
ses

- 48 -
DEF
AU
TS G
ENET
IQU
ES D
E LA
REP
ON
SE IM
MU
NIT
AIR
EIn
trod
uctio
n à
l’étu
de d
es D
IP
01
Génétique 49
Le déficit immunitaire
La maladie est monogénique et se transmet selonun mode autosomique récessif. Le gène BLM(en 15q26.1) code pour une protéine nucléaire quicontient des motifs caractéristiques des hélicasesRecQ. L’étude des fonctions de cette protéineapparaît comme une donnée essentielle pourcomprendre la façon dont le génome est maintenude façon stable pendant toute la vie, de l’œuf aux1014 cellules qui constituent l’organisme entier.La protéine du syndrome de Werner a des homologiesde séquences avec BLM, ces protéines ontprobablement des rôles très similaires dansle métabolisme de l'ADN.
Le phénotype Bloom est produit, soit par unehomozygotie, soit par une hétérozygotie composite.Les mutations sont nulles. Un allèle particuliercomportant une délétion de 6bp et une insertionde 7bp en 2281 résultant en une protéine tronquéea une fréquence très élevée (1%) par effetfondateur, dans la population juive ashkénaze. 49,50
Dans toutes les cellules somatiques, le génomeest particulièrement instable avec une tendanceimportante à muter. Ainsi les mutations s'accumulentavec le temps et ces lésions moléculaires sont
visibles au caryotype sous la forme de gapschromatiniens, de cassures, de réarrangementset de mutations dans certains loci spécifiques.Les cellules montrent une tendance très importanteà l'échange de matériel entre les chromatidesde chacun des chromosomes d'une même pairepour former des figures quadri-radiales dansdes régions homologues ou entre les chromatidessœurs d'un chromosome donné (dans du milieucontenant du BrdU (Bromodéoxyuridine) pour leurvisualisation).
Certains patients hétérozygotes composites sontdes mosaïques. Une recombinaison intra-géniquepeut générer un allèle normal fonctionnel sur l'undes chromosomes 15 qui, s'il ségrège à la mitoseavec la chromatide non recombinante de l'autre15, corrige le phénotype cellulaire.
Historiquement, les figures quadri-radiales ontconstitué la première preuve cytogénétiquede l'existence d'une recombinaison homologuedans les cellules somatiques de mammifères sousla forme de crossing-over somatique.
L'hypogammaglobulinémie est présente chezla plupart des patients, prédominant sur l'un ou l'autredes isotypes d'immunoglobulines, et la productiond'anticorps vaccinaux ou post-infectieux estcependant le plus souvent normale. L'étudedes sous-populations lymphocytaires T, B, NKest normale ainsi que les réponses prolifératives.Le processus de recombinaison somatiqueV(D)J et de mutation somatique des gènes des
immunoglobulines est normal. Ainsi le déficitimmunitaire, d'ailleurs assez modéré, n'est pas biencompris. 49,50
BLM constitue une part du complexe protéiquequi coordonne les activités nécessaires pourla maintenance de l'intégrité du génomedurant le processus de réplication.
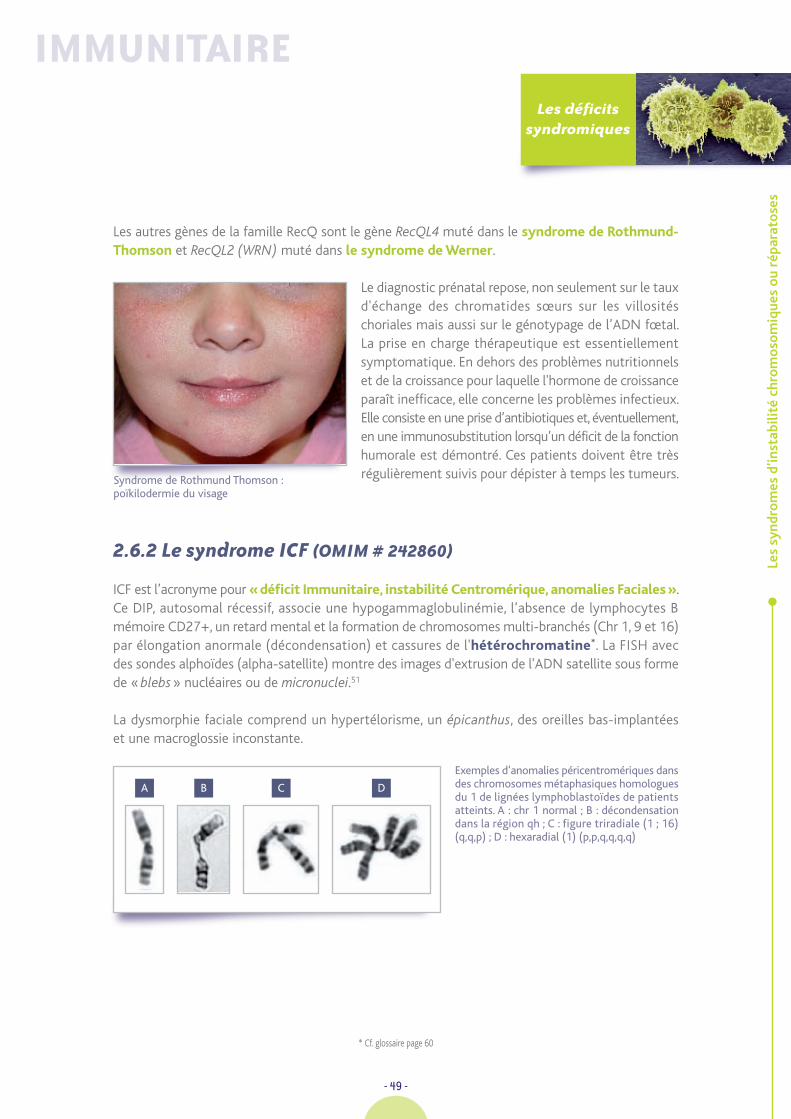
Les déficitssyndromiques
- 49 -
Les autres gènes de la famille RecQ sont le gène RecQL4 muté dans le syndrome de Rothmund-Thomson et RecQL2 (WRN) muté dans le syndrome de Werner.
Le diagnostic prénatal repose, non seulement sur le tauxd'échange des chromatides sœurs sur les villositéschoriales mais aussi sur le génotypage de l’ADN fœtal.La prise en charge thérapeutique est essentiellementsymptomatique. En dehors des problèmes nutritionnelset de la croissance pour laquelle l'hormone de croissanceparaît inefficace, elle concerne les problèmes infectieux.Elle consiste en une prise d’antibiotiques et, éventuellement,en une immunosubstitution lorsqu’un déficit de la fonctionhumorale est démontré. Ces patients doivent être trèsrégulièrement suivis pour dépister à temps les tumeurs.
2.6.2 Le syndrome ICF (OMIM # 242860)
ICF est l’acronyme pour «déficit Immunitaire, instabilité Centromérique, anomalies Faciales».Ce DIP, autosomal récessif, associe une hypogammaglobulinémie, l’absence de lymphocytes Bmémoire CD27+, un retard mental et la formation de chromosomes multi-branchés (Chr 1, 9 et 16)par élongation anormale (décondensation) et cassures de l'hétérochromatine*. La FISH avecdes sondes alphoïdes (alpha-satellite) montre des images d'extrusion de l'ADN satellite sous formede « blebs» nucléaires ou de micronuclei.51
La dysmorphie faciale comprend un hypertélorisme, un épicanthus, des oreilles bas-implantéeset une macroglossie inconstante.
Exemples d’anomalies péricentromériques dansdes chromosomes métaphasiques homologuesdu 1 de lignées lymphoblastoïdes de patientsatteints. A : chr 1 normal ; B : décondensationdans la région qh ; C : figure triradiale (1 ; 16)(q,q,p) ; D : hexaradial (1) (p,p,q,q,q,q)
Syndrome de Rothmund Thomson : poïkilodermie du visage
A C DBLe
s sy
ndro
mes
d’in
stab
ilité
chr
omoso
mique
s ou répa
rato
ses
* Cf. glossaire page 60

- 50 -
DEF
AU
TS G
ENET
IQU
ES D
E LA
REP
ON
SE IM
MU
NIT
AIR
ELe
s dé
ficits
syn
drom
ique
s02
ICF constitue une pathologie moléculaire «à plusieurs étages»
ICF -syndrome ICF- est une maladie mendélienne caractériséepar une méthylation aberrante de l'ADN liée à des mutationsdu gène DNMT3B. Son impact est multiple et étagé, passantpar une hypométhylation du DNA satellite de l’hétérochromatinepéricentromérique de certains chromosomes (Chr 1, 9, 16).
Epicanthus
Oreilles basimplantées
Anomalies chromosomiques structurales du chromosome 1 et micronuclei (métaphaseet interphase). Fish multi-couleur avec en vertle chr. 1, et en rouge le chr. 16.
La méthylation de l 'ADN est une marqueépigénétique de la répression transcriptionnelle,essentielle pour le maintien de la structure de lachromatine et la stabilité du génome ; elle est sousle contrôle de trois enzymes méthyltransférases :DNMT1, DNMT3A and DNMT3B. Les histonespeuvent être aussi méthylées et cette modificationest impliquée dans le contrôle de la transcription,constituant ainsi une sorte de « code », appelécode des histones, dont la lecture permetde prédire si le gène où est situé le nucléosomes'exprime ou non. Les mutations germinalesdu gène de la DNMT3B - DNA méthyltransférase3B- (localisation = chromosome 20q11.2), une DNAcytosine-méthyl transférase, entraînent le syndromeICF, caractérisé par des anomalies de la mitose et
une instabilité génomique. L'interaction entre lesprotéines de la région centromérique et péri-centromérique et cette enzyme est critique pourla méthylation de l'ADN satellite et la régulationdu code des histones. 52
Des mutations de la DNMT3B résultent dansun défaut de méthylation de l'hétérochromatineconstitutive juxta-centromérique des chromosomes1, 9 et 16 et des effets pléïotropes sur des ciblesnon encore identifiées. Les mutations sont desfaux-sens ou entraînent des anomalies d’épissage,les patients étant homozygotes ou hétérozygotescomposites. Les allèles sont «nuls» car conduisantà la perte complète de la fonction enzymatique.

Le diagnostic en CMF : l’absence de LB 19+27+ 51-53
Le déficit immunitaire porte sur la fonction humorale :les LB sont présents, contrastant avec un tauxd'immunoglobulines sériques très bas. Les LBde phénotype «mémoire» CD19+27+ sont absents.Les patients présentent des infections du tractusrespiratoire (germes rencontrés = staphylocoque,pyocyanique, klebsielle, pneumocoque). Les infectionsavec le virus JC, les candidoses et les pneumocystosessuggèrent une anomalie de l’immunité cellulaireassociée : ICF est un déficit lymphocytaire combinéT+B. Les problèmes gastro-intestinaux sont fréquentsà type de diarrhée chronique nécessitant parfoisune nutrition parentérale. Les patients ont un risquede développer une hémopathie maligne, une maladiede Hodgkin ou un syndrome myélodysplasique.Le retard des acquisitions cognitives n'est pas constantet un tiers des enfants présentent des aptitudesintellectuelles normales. Le syndrome dysmorphiquepeut être totalement absent. La prévention des infectionsrespiratoires repose sur l’immunosubstitution et uneantibiothérapie préventive. 51,53
Les déficitssyndromiques
- 51 -
CD 27
CD 19
Patient : absence de lymphocyte B CD19+,27+
Patient témoin
Les sy
ndro
mes
d’in
stab
ilité
chr
omoso
mique
s ou répa
rato
ses

- 52 -
DEF
AU
TS G
ENET
IQU
ES D
E LA
REP
ON
SE IM
MU
NIT
AIR
ELe
s dé
ficits
syn
drom
ique
s02
2.6.3 L’ataxie-télangiectasie (OMIM # 208900)
Phénotype clinique de l’AT
Caryotype : détail.L’AT est une maladie cassante, les anomalies chromosomiquesspontanées affectent les chromosomes 7 et 14, au niveaudes régions génétiques des immunoglobulines et du récepteur Tsuggérant ainsi un rôle central de la protéine ATM dansla reconnaissance et la réparation des cassures double brinau cours de l'ontogénie des lymphocytes T et B. Remerciement R Touraine. CHU St Etienne
Télangiectasies cutanées et oculaires
AtaxieNystagmus
Lymphomes
Infectionspulmonaires et sinusiennes
Taches café au lait
Granulomes
* Cf. glossaire page 60
Les télangiectasies oculaires, tortueuses, larges,bien visibles sur la conjonctive bulbairesont caractéristiques. Ces lésions traduisentune sénescence prématurée des cellulesendothéliales, par sensibilité exagérée austress oxydatif. Leur apparition est tardive(4 à 6 mois).
Les cassures double brin sont introduites dans l’ADNau cours d'un certain nombre de processusphysiologiques tels que la lymphopoïèse(recombinaison V(D)J*), la méïose et la réplicationde l'ADN, mais aussi au cours de l'infection viraleet de la transformation néoplasique. Ces dommagesgéniques constituent une menace permanente pourl'intégrité du génome et donc pour la survie del'organisme.Les cellules eucaryotes ont développé plusieurssystèmes de surveillance très sensibles pour réparerde telles lésions. Dès que ce type de lésionsmoléculaires est détecté, une machinerie très
complexe de protéines impliquées dans le contrôledu cycle cellulaire* est activée qui conduit à l'arrêtdu cycle à chacun des points de contrôle ou pointsde restriction = check-points* ce qui « laissele temps» de réparer ces dommages génotoxiques.ATM* pour serine/threonine protein kinase ataxia-telangiectasia mutated joue un rôle fondamentaldans la régulation du cycle et le maintien de l’intégritéde l’ADN en participant à la réparation. Les mutationsbilalléliques du gène ATM entraînent un syndromed’instabilité génomique associé à un déficitimmunitaire, l’ataxie-télangiectasie. 54
IRM du cervelet. L'ataxie est le signe le plus précoce de l'ataxie télangiectasie.L’IRM montre une atrophie cérébelleuse. Celle-ci reflèteune perte progressive des cellules de Purkinje et des cellulesgranuleuses.

Phénotype clinique55
L’AT est caractérisée par un déficit immunitaire combiné, une atteinte neurologique (ataxie secondaireà la dégénérescence des cellules de Purkinje probablement par un processus d'apoptose) responsablede troubles précoces de la marche, une dysgénésie gonadique et une hypersensibilité des cellulesaux radiations ionisantes avec une susceptibilité accrue à développer un cancer. La prévalenceen France est de 1/150000 enfants.56 Cette affection est incurable.
L'ataxie est le plus souvent le symptôme inaugural et devientévident lorsque que l'enfant commence à marcher à la finde la première année de la vie. Les mouvements involontaires,la démarche ataxique, les difficultés d'élocution deviennentévidentes au fur et à mesure du temps. La motilité oculaire estanormale avec une asynergie oculocéphalogyre (ou «contraversionoculaire»). Ces anomalies correspondent à une dégénérescencecérébelleuse corticale qui prédomine sur les cellules de Purkinjeet les cellules granuleuses. Les anomalies dégénératives concernentaussi des noyaux gris centraux. Des télangiectasies sont bienvisibles sur les sclérotiques, au niveau des oreilles, ou sur le visageavec une disposition parfois en « aile de papillon ». Les enfantsprésentent des infections récurrentes de la sphère ORLavec otites, sinusites fréquentes et pneumonies à répétitionse compliquant de bronchectasies et fibrose pulmonaire,conduisant à l’insuffisance respiratoire. Il existe un retard
de taille habituellement évident chez les adolescents avec l'intrication de plusieurs facteurs :l’inappétence, les complications infectieuses chroniques, l'hypogonadisme. Il existe un retard pubertaire,avec un défaut de spermatogenèse, l'absence ou l'hypoplasie des ovaires chez la fille. Les patientsont un risque de développer un diabète par insulinorésistance.55
L'instabilité chromosomique constitue dans ce syndrome une susceptibilité importante à développerune leucémie ou un lymphome (maladie de Hodgkin ou lymphome non hodgkinien). Elle a concerné22 % des patients de la série française du CEREDIH avec un âge moyen au diagnostic de 14 ans ;47 ont développé une néoplasie lymphoïde ; six enfants ont développé un carcinome à un âge plustardif. Les patients avec des mutations nulles de ATM ont un risque plus élevé d’hémopathie maligneet les mutations hypomorphes* ont un risque de décès par infections respiratoires. 56
Les parents hétérozygotes ont une susceptibilité à développer un cancer plus importantque la population générale de l’ordre de 3,5 à 3,8 fois pour tous types de cancer, et pour la mamanun risque de cancer du sein 5 fois plus élevé. 55
Les déficitssyndromiques
- 53 -
AT : bronchectasies
* Cf. glossaire page 60
Les sy
ndro
mes
d’in
stab
ilité
chr
omoso
mique
s ou répa
rato
ses

- 54 -
DEF
AU
TS G
ENET
IQU
ES D
E LA
REP
ON
SE IM
MU
NIT
AIR
ELe
s dé
ficits
syn
drom
ique
s02
Biologie
Le déficit immunitaire est variable. Il existehabituellement une hypogammaglobulinémie,dont l'importance est corrélée à la fréquencedes infections respiratoires : absence d’IgA, déficiten IgE, réduction des sous-classes en IgG2 et IgG4.Les anomalies humorales ne sont pas liées à uneréduction de la numération des lymphocytes Bmais à une anomalie de la maturation avec undéfaut de réponse aux antigènes vaccinaux, variabled'un patient à un autre. La réponse aux antigènespolysaccharidiques peut également être médiocre.55
Le déficit immunitaire pourrait résulter d'une diminutionde la fidélité du processus de recombinaison V(D)J.Il existe parfois une hyper IgM avec une hypo IgAet hypo IgG, probablement liées à une anomaliede la commutation de classe. La récurrence des infectionsassociée à ces anomalies biologiques oriente versun syndrome hyper IgM.
Il existe un défaut de développement thymiquereconnu anciennement à l'autopsie des patients(hypoplasie thymique) avec un défaut fonctionneldes LT et une anomalie de réponse in vitro à différentsmitogènes. Il existe une réduction quantitativedes sous-populations lymphocytaires T etnotamment des CD4 naïfs. Les patients présententune augmentation relative des lymphocytes T,gamma / delta. Les émigrants thymiques mesuréspar les TRECS* sont diminués ainsi que la numérationdes cellules mémoires 45RO+.67
Les lymphocytes périphériques des patients montrentdes réarrangements spontanés des chromosomes7 et 14.
Des anomalies des tests hépatiques sont fréquentes,avec augmentation des phosphatases alcalines, destransaminases et des alpha-fœtoprotéines (AFP).
Le diagnostic du syndrome d’ataxie-télangiectasieest essentiellement clinique. Pour le pédiatreneurologue, avant l'apparition des télangiectasies,le diagnostic différentiel est celui d'une ataxie,un syndrome de Friedreich, les tumeurs du systèmenerveux, les maladies métaboliques, les neuropathieshéréditaires comme la maladie de Charcot-Marie-Tooth. L'absence des IgA sériques et l'élévationde l’alpha-fœtoprotéine sont caractéristiques maisnon spécifiques. La présence sur le caryotypede remaniement des chromosomes 7 et 14 estégalement très évocatrice. L'étude de la radiosensibilitédes lignées lymphoblastoïdes des patients a longtempsconstitué l'examen de référence. Le diagnosticgénotypique direct est rendu difficile par la trèsgrande variation des lésions moléculaires dansun très grand gène. La plupart des mutationsentraînent un défaut d'expression de la protéineATM dans les cellules en immunoBlot. 55 Le canceret les infections sévères invasives restent les deuxcauses indépendantes de décès chez les enfantsatteints d’AT. Aucun traitement n'est capablede freiner la dégénérescence neurologique.En cas de déficit immunitaire clinique, etd'hypogammaglobulinémie avec anomalie de lafonction humorale, le patient peut bénéficierd’une immunosubstitution.55,56
* Cf. glossaire page 60
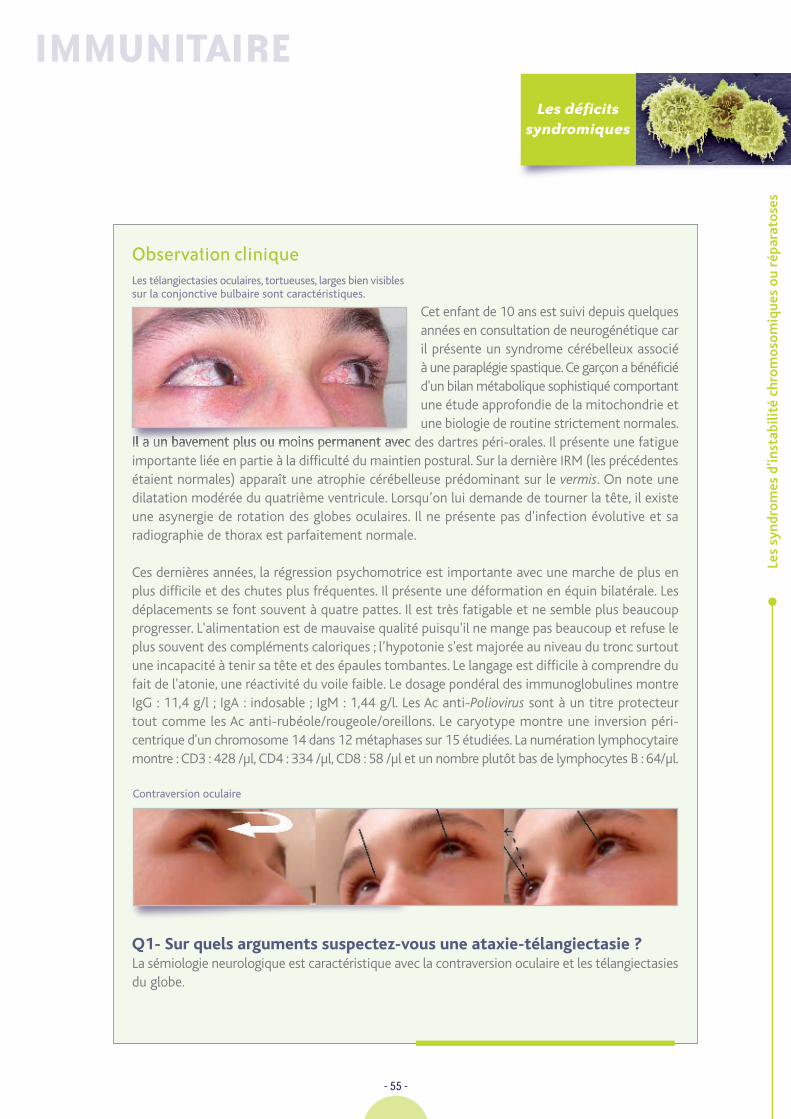
Observation clinique
Cet enfant de 10 ans est suivi depuis quelquesannées en consultation de neurogénétique caril présente un syndrome cérébelleux associéà une paraplégie spastique. Ce garçon a bénéficiéd'un bilan métabolique sophistiqué comportantune étude approfondie de la mitochondrie etune biologie de routine strictement normales.
Il a un bavement plus ou moins permanent avec des dartres péri-orales. Il présente une fatigueimportante liée en partie à la difficulté du maintien postural. Sur la dernière IRM (les précédentesétaient normales) apparaît une atrophie cérébelleuse prédominant sur le vermis. On note unedilatation modérée du quatrième ventricule. Lorsqu’on lui demande de tourner la tête, il existeune asynergie de rotation des globes oculaires. Il ne présente pas d'infection évolutive et saradiographie de thorax est parfaitement normale.
Ces dernières années, la régression psychomotrice est importante avec une marche de plus enplus difficile et des chutes plus fréquentes. Il présente une déformation en équin bilatérale. Lesdéplacements se font souvent à quatre pattes. Il est très fatigable et ne semble plus beaucoupprogresser. L'alimentation est de mauvaise qualité puisqu'il ne mange pas beaucoup et refuse leplus souvent des compléments caloriques ; l’hypotonie s'est majorée au niveau du tronc surtoutune incapacité à tenir sa tête et des épaules tombantes. Le langage est difficile à comprendre dufait de l'atonie, une réactivité du voile faible. Le dosage pondéral des immunoglobulines montreIgG : 11,4 g/l ; IgA : indosable ; IgM : 1,44 g/l. Les Ac anti-Poliovirus sont à un titre protecteurtout comme les Ac anti-rubéole/rougeole/oreillons. Le caryotype montre une inversion péri-centrique d'un chromosome 14 dans 12 métaphases sur 15 étudiées. La numération lymphocytairemontre : CD3 : 428 /µl, CD4 : 334 /µl, CD8 : 58 /µl et un nombre plutôt bas de lymphocytes B : 64/µl.
Q1- Sur quels arguments suspectez-vous une ataxie-télangiectasie ? La sémiologie neurologique est caractéristique avec la contraversion oculaire et les télangiectasiesdu globe.
Les déficitssyndromiques
- 55 -
Contraversion oculaire
Les télangiectasies oculaires, tortueuses, larges bien visiblessur la conjonctive bulbaire sont caractéristiques.
Les sy
ndro
mes
d’in
stab
ilité
chr
omoso
mique
s ou répa
rato
ses

- 56 -
DEF
AU
TS G
ENET
IQU
ES D
E LA
REP
ON
SE IM
MU
NIT
AIR
ELe
s dé
ficits
syn
drom
ique
s02
Q2- Quel examen biologique permettra rapidement de confirmer vosimpressions ? Le dosage de l’alpha-fœtoprotéine (AFP) sérique. Les AFP étaient ici à 112 kUI/l. Le taux des AFPreste relativement stable avec le temps et il n'y a pas de corrélation avec la sévérité de la maladie.Les autres étiologies de l'élévation des AFP chez l'enfant : la tyrosinémie héréditaire, l’hépatoblastomeet la persistance héréditaire de l'alpha-foetoprotéine, asymptomatique, autosomal récessive.
Q3- Commentez le bilan immunitaireL'absence des immunoglobulines d'isotype IgA est l'anomalie la plus fréquente de l'immunitéhumorale rapportée dans l'ataxie-télangiectasie et concerne 2/3 des patients.56
Le patient est lymphopénique avec une diminution des lymphocytes T. La réponse proliférativeaux antigènes et aux mitogènes était normale. La réponse humorale vis-à-vis des antigènesprotéiques et des vaccins polysaccharidiques était conservée. Il existe des anomalies cytogénétiquesspontanées typiques sur les lymphocytes circulants.
Le diagnostic d’AT a été prouvé par le génotypage avec l’identification de 2 mutations(c.2727insT/919stop ; c.3802delG/1268stop) très probablement associées à une perte de fonctionbiologique de la protéine correspondante et pouvant être considérées comme responsablesde l'augmentation du risque tumoral.
2.6.4 Le syndrome de Nijmegen 57
(Nijmegen Breakage syndrome) (OMIM # 251260)
Cette maladie autosomale récessive associe un retard mental modéré, un déficit immunitaire combiné,une sensibilité particulière aux radiations ionisantes. La dysmorphie faciale est mal caractérisée, le frontest habituellement fuyant, le nez long, la micrognathie s’associe à des fentes palpébrales obliques enhaut.
Le cancer est la principale cause de mortalité des patients atteints de ce syndrome. Il s’agit essentiellementde lymphomes de tous types, mais il y a des observations anecdotiques de gliome du SNC (systèmenerveux central), de rhabdomyosarcome ou de médulloblastome.57 Les hétérozygotes ont aussi unesusceptibilité accrue à développer un cancer comme les parents hétérozygotes pour une mutationd’ATM. La plupart des patients présentent des infections respiratoires récurrentes compliquées debronchectasie et parfois des manifestations auto-immunes telles qu’une thrombopénie, une anémiehémolytique auto-immune ou un vitiligo. Le taux d’AFP est normal.

Le gène NBS1 cloné en 1998 situé dans la bande 8q21est constitué de 16 exons s’étendant sur 50 kilobaseset code pour la nibrine* qui recrute le complexeMRN* au site de cassure. La délétion de 5 bp dansNBS1 en 657 résultant en une protéine tronquée estl’anomalie moléculaire la plus fréquente. Les cellulesde l’ataxie-télangiectasie et du NBS comportent desphénotypes cellulaires très comparables avec uneradiosensibilité in vitro, une instabilité chromosomiqueet des anomalies du cycle cellulaire. L’irradiationentraîne notamment des cassures simple-brin et descassures double-brin. Le caryotype sanguin montrehabituellement des réarrangements illégitimesimpliquant les chromosomes 7 et 14. L’instabilitégénomique est démontrée au laboratoire par uneaugmentation de la mortalité des différents typescellulaires (lignées lymphoblastoïdes ou fibroblastes
cutanés) après irradiation. L’étude du cycle cellulaire en cytométrie de flux est comparable auxrésultats des cellules d’ataxie-télangiectasie avec un défaut du contrôle au point de restriction G1/S.L’ImmunoBlot* démontre l’absence de la protéine nibrine dans les lignées lymphoblastoïdes.
Étant donné les similitudes cliniques, le syndrome NBS était considéré comme un « variant »de l’ataxie-télangiectasie. Si ces deux maladies génétiques sont bien distinctes, les protéines respectivescodées par les deux gènes sont cependant impliquées dans un même pathway activé en réponseà un dommage génomique.
Le déficit immunitaire est combiné avec une a/hypo- gammaglobulinémie chez un 1/3 des patients.Ceux qui ont un dosage des IgG normal, ont parfois un déficit en sous classe IgG2/ IgG4.L'immunophénotypage est caractérisé par une lymphopénie T avec un excès de cellules mémoireet une diminution des cellules naïves CD4+ CD45+RA. 58
Chromosomes métaphasiques d’un patient atteintde syndrome de Nijmegen : cassures spontanées etfusion (flèche)
Les déficitssyndromiques
- 57 -
* Cf. glossaire page 60
Les sy
ndro
mes
d’in
stab
ilité
chr
omoso
mique
s ou répa
rato
ses
7 ans - Granulomes et plaques atrophiques.Histoire d’infections pulmonaires à répétitiondepuis la petite enfance. Microcéphalie, retardde croissance et de langage. Lymphopénie Tmodérée. Altération de la réponse proliférative Taux anti-CD3 et aux ag. c.657_661del5 dans legène NBS1. Diagnostic final de Nijmegenbreakage syndrome

- 58 -
DEF
AU
TS G
ENET
IQU
ES D
E LA
REP
ON
SE IM
MU
NIT
AIR
ELe
s dé
ficits
syn
drom
ique
s02
Les autres syndromes d’instabilité chromosomique proches de l’AT
� Le phénotype du défaut de Cernunos est un déficit immunitaire combiné avec uneradiosensibilité excessive, des anomalies importantes de la recombinaison V(D)J etune microcéphalie. Les patients présentent un retard de croissance intra-utérin, parfois desmalformations urogénitales et des cytopénies auto-immunes. L'étude des sous-populationslymphocytaires objective une lymphopénie T et B, épargnant la population NK. Le comptedes lymphocytes B décline avec le temps et la plupart présente une hypogammaglobulinémieprédominant sur les IgG et IgA, tandis que les taux sériques d'IgM sont fluctuants. Les sous-populations lymphocytaires T sont composées uniquement de cellules mémoires dont lesréponses prolifératives sont diminuées. L'association d'une microcéphalie, d'une absence delymphocytes T naïfs et la disparition progressive des lymphocytes B circulants indique undéfaut de la maturation du système immunitaire comme dans le déficit en Lig4 (voir plusloin) et le syndrome de Nijmegen.59
� Des mutations dans le gène LIG4 présentant un syndrome proche du Nijmegen ont étérapportées. Le déficit en ligase 4 ressemble sur le plan du phénotype au syndrome de Nijmegen avecune radiosensibilité cellulaire marquée. Le diagnostic différentiel est le nanisme de Seckelet l’anémie de Fanconi. Le phénotype du déficit en ligase 4 est caractérisé par une microcéphalie et des anomaliesdu développement intellectuel, une pancytopénie et des problèmes cutanés divers : psoriasis,verrues disséminées.60 Mais le défaut de lig4 constitue également une étiologie génétiquede SCID T-B-NK+ radiosensible.61
� Le déficit en RAD50 est caractérisé par une instabilité chromosomique spontanée,une radiosensibilité, des anomalies de la phosphorylation de la protéine NBS, une anomaliede la constitution du complexe de surveillance. Le phénotype clinique ressemble au syndromede Nijmegen avec une microcéphalie, un retard de taille, des anomalies des chromosomes 7et 14, une hypersensibilité aux radiations ionisantes, un retard intellectuel, une spasticité,une discrète ataxie et des anomalies de la pigmentation. Le dosage pondéral desimmunoglobulines, les sous-populations lymphocytaires et les réponses prolifératives sontnormales.62
� Le syndrome AT like correspond à des mutations de la protéine MRE 11 et le tableau cliniqueressemble au syndrome ataxie-télangiectasie qui partage l’apraxie oculomotrice, l’hypoplasiedu cervelet, les anomalies cytogénétiques et l’hypersensibilité aux radiations ionisantes.Les patients ne présentent pas de déficit immunitaire ni de télangiectasie oculaire. Ils peuventdévelopper un adénocarcinome bronchique. 63
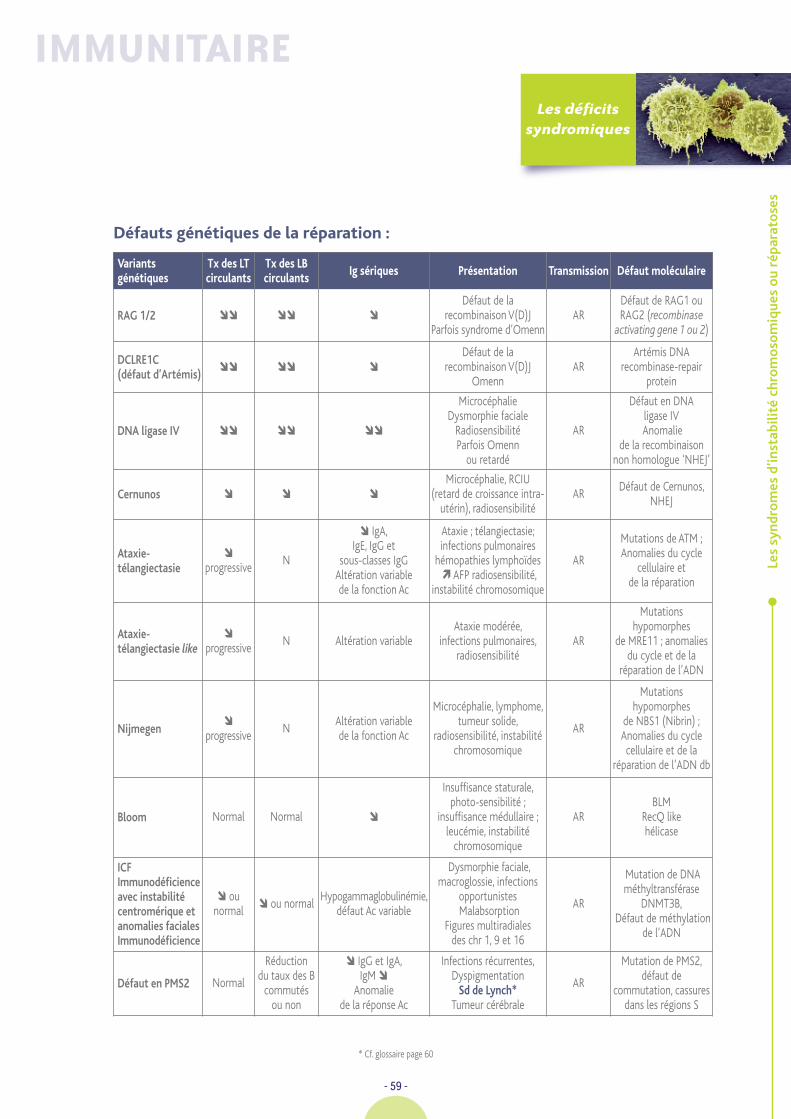
Les déficitssyndromiques
- 59 -
Variantsgénétiques
Tx des LTcirculants
Tx des LBcirculants
Ig sériques Présentation Transmission Défaut moléculaire
RAG 1/2
Défaut de la recombinaison V(D)J
Parfois syndrome d’OmennAR
Défaut de RAG1 ouRAG2 (recombinase
activating gene 1 ou 2)
DCLRE1C(défaut d’Artémis)
Défaut de la recombinaison V(D)J
OmennAR
Artémis DNArecombinase-repair
protein
DNA ligase IV
Microcéphalie Dysmorphie facialeRadiosensibilitéParfois Omennou retardé
AR
Défaut en DNA ligase IV Anomalie
de la recombinaisonnon homologue ‘NHEJ’
Cernunos
Microcéphalie, RCIU (retard de croissance intra-utérin), radiosensibilité
ARDéfaut de Cernunos,
NHEJ
Ataxie-télangiectasie
progressiveN
IgA,IgE, IgG et
sous-classes IgGAltération variable de la fonction Ac
Ataxie ; télangiectasie;infections pulmonaireshémopathies lymphoïdes AFP radiosensibilité,
instabilité chromosomique
AR
Mutations de ATM ;Anomalies du cycle
cellulaire et de la réparation
Ataxie-télangiectasie like
progressiveN Altération variable
Ataxie modérée, infections pulmonaires,
radiosensibilitéAR
Mutationshypomorphes
de MRE11 ; anomaliesdu cycle et de la
réparation de l’ADN
Nijmegen
progressiveN
Altération variable de la fonction Ac
Microcéphalie, lymphome,tumeur solide,
radiosensibilité, instabilitéchromosomique
AR
Mutationshypomorphes
de NBS1 (Nibrin) ;Anomalies du cyclecellulaire et de la
réparation de l’ADN db
Bloom Normal Normal
Insuffisance staturale,photo-sensibilité ;
insuffisance médullaire ;leucémie, instabilitéchromosomique
ARBLM
RecQ likehélicase
ICF Immunodéficienceavec instabilitécentromérique etanomalies facialesImmunodéficience
ounormal
ou normalHypogammaglobulinémie,
défaut Ac variable
Dysmorphie faciale,macroglossie, infections
opportunistesMalabsorption
Figures multiradiales des chr 1, 9 et 16
AR
Mutation de DNAméthyltransférase
DNMT3B,Défaut de méthylation
de l’ADN
Défaut en PMS2 Normal
Réduction du taux des Bcommutés ou non
IgG et IgA, IgM
Anomalie de la réponse Ac
Infections récurrentes,DyspigmentationSd de Lynch*
Tumeur cérébrale
AR
Mutation de PMS2,défaut de
commutation, cassuresdans les régions S
Défauts génétiques de la réparation :
Les sy
ndro
mes
d’in
stab
ilité
chr
omoso
mique
s ou répa
rato
ses
* Cf. glossaire page 60

- 60 -
Les déficitssyndromiques
DEF
AU
TS G
ENET
IQU
ES D
E LA
REP
ON
SE IM
MU
NIT
AIR
EAn
nexe
s 03
Annexes
ADN satelliteRépétition de très nombreuses copies d’une séquence courte. Il est principalement situé au niveau descentromères.
ATM (serine/threonine protein kinase ataxia-telangiectasia mutated)Joue un rôle fondamental dans la régulation du cycle et le maintien de l’intégrité de l’ADN en participantà la réparation.
ATM active la réponse cellulaire aux dommagesgénotoxiques mais aussi participe à la résolutiondes cassures double brin générées au cours de larecombinaison V(D)J* (voir ce terme).
ATM est présent sous forme d'un dimère inactif.L'introduction de cassures double brin active ATMen stimulant son autophosphorylation ce qui conduità la dissociation des dimères inactifs. ATM (et ATR pourRAD3-related, une autre kinase, pour les cassures simplebrin) active alors les protéines de régulation du cyclecellulaire : ATM et ATR phosphorylent et activentles kinases CHK2 et CHK1. Ces 2 kinases phosphorylentà leur tour et inactivent les Cdc phosphatases, celles-là
même qui sont nécessaires pour la déphosphorylation des phosphores inhibiteurs des Cdk. La progressionvers la mitose est bloquée et la cellule est en arrêt en G1. (Voir schéma sur le cycle cellulaire*).
M
G1
G2
S
STOP STOP
STOPSTOP
Contrôle du cycle cellulaire
Non alignement duchromosome
ADN endommagéou non répliqué
ADN endommagéou non répliqué
ADNendommagé
3.1 Glossaire3.1 Glossaire
* Cf. glossaire page 60

- 61 -
Glossaire
Annexes
Kua
b
c
d
ADN-PKcs
ADN polymérase µ
ADN ligase IVXRCC4
Artémis
ATM et réparation - MRNATM participe aussi aux processus de réparation. Le complexe protéique très conservé MRN quicomprend les trois partenaires NBS1/RAD50/MRE11 joue un rôle central dans les phases les plusprécoces de reconnaissance de la cassure.
Centromères Les centromères des chromosomes jouent un rôle central dans la mitose et la méïose et permettentla séparation des chromosomes.
p p p
H 2AX
pp
pp
monomèreATM actif
MRE11
RAD50NBS1 MRN complexe
dimère ATM inactif
monomère ATM actif
H 2AX
BRCA
1
CHK2
SMC1
p
p
p
pp
p
53BP
1
Les monomères de la protéine ATM phosphorylentNBS1 (protéine du sd de Nijmegen ) and H2AX(histone 2A family, member X) . Cette dernièreprotéine modifiée permet aux complexes MRNde se stabiliser au niveau des cassures puis derecruter différentes protéines ici dessinées en bleu (BRCA1 (breast cancer 1, early onset) and 53BP1(p53-binding protein), CHK2 (checkpoint kinase2 homologue) et SMC1 (structural maintenance-of-chromosomes-1-like 1).
La recombinaison non homologue « NHEJ »L'étape la plus précoce est la fixation del'holoenzyme : la DNA PK avec la fixation des2 sous-unités Ku 70 / Ku80 et les DNA PKcs(= Sous-unités catalytiques de la DNA PK). PuisRecrutement d’ Artémis, XRCC4 (X-ray-repaircross-complementing protein , mutée dansle Xeroderma pigmentosum), DNA ligase IV andDNA polymérase au site de la cassure) ; puisles sous-unités catalytiques actives phosphorylentet activent les molécules cibles Ku, XRCC4.L’activité endonucléase de Artémis est activée etcette enzyme peut résoudre certaines contraintes,telles que les bouts francs d’ADN et les structuresen épingle à cheveux. La polymérase µ élonguela chaîne et « comble » les « trous » , la ligaseIV achève la ligation en formant un complexe avecXRCC4 et Cernunos non figuré ici.

- 62 -
DEF
AU
TS G
ENET
IQU
ES D
E LA
REP
ON
SE IM
MU
NIT
AIR
EAn
nexe
s 03
Check-points (synonyme : points de restriction)Les facteurs de croissance et les signaux de danger ou de dommages génotoxiques inhibent la progressiondu cycle, ce qui joue un rôle très important pour maintenir l'intégrité du génome. Ces différents check-points ou points de restriction donnent du temps à la cellule pour réparer les erreurs, avant la réplicationde l'ADN et la division cellulaire.
Les kinases CHK2 et CHK1 bloquent l'activité des phosphatases impliquées dans l'activation des complexesCdk/CYCLINE ; ces phosphatases enlèvent les radicaux phosphoryles inhibiteurs. En phase G1 et S CHK2et CHK1 inactivent la phosphatase Cdc25A entraînant l'inhibition de Cdk2. Au check-point G2, CHK2et CHK1 inhibent la phosphatase cdc25C responsable de l'activation de Cdk1/Cycline B : la progressionvers la mitose est alors bloquée et les cellules sont arrêtées en phase G2.
Cycle cellulaire et cyclines
Le contrôle du cycle cellulaire des cellules eucaryotes fait alors intervenir une série de protéines kinasesconservées, à chacune des transitions G1/S et G2/M. La régulation se fait par un jeu de phosphorylation/déphosphorylation. Les études chez la levure et chez les batraciens ont montré le rôle fondamentalde ces cascades de phosphorylation dans la régulation du cycle. Les régulateurs sont constituésde 2 sous-unités, la protéine kinase Cdk (pour Cyclin dependant kinase) et une sous-unité régulatricenécessaire pour son activité catalytique : la cycline.
Cdk1 forme un complexe avec la cycline B durant la phase G2. Cdk1 est phosphorylée sur la thréonine 161 et sur la Tyrosine15, résidus phosphoryles inhibiteurs. La déphosphorylation active Cdk1 et permet la transition G2 M (mitose). Il s’ensuivraune dégradation protéolytique de la cycline B à la fin de la mitose.
Mitose
Cycline B
Thr 14
MPFactive
Tyr 15Thr 161
Déphosphorylation
Déphosphorylation
PhosphorylationCdk1
Cdk1
Cdk1Cdk1
Cdk1
Cycline B Cycline B
Cycline B
Synthèse de la cycline B
Dégradation de la cycline B

Annexes
- 63 -
Glossaire
En page 62 est illustrée la cycline B synthétisée puis formant un complexe avec CdK1 pendantla phase G2 du cycle. Au fur et à mesure que ces complexes se forment, Cdk1 est phosphoryléeen deux positions régulatrices critiques : la thréonine 161 et la tyrosine 15 et la thréonine 14 adjacente.La phosphorylation de la tyrosine 15 catalysée par une kinase appelée Wee1 inhibe l'activité kinasede CdK1 et conduit à l'accumulation des complexes pendant la phase G2. La transition G2/M estdéclenchée par l'activation du complexe par une déphosphorylation de la thréonine 14 et de la tyrosine15 par une phosphatase. Ainsi activée, la protéine kinase Cdk1 phosphoryle à son tour toute unesérie de protéines cibles qui initient les premiers événements de la phase M. L'activité de CdK1déclenche la dégradation de la cycline B par ubiquitination et protéolyse. Ceci conduit à l’activationde la kinase, l'arrêt de la mitose, la diakinése et le retour à l'interphase.
Le cycle cellulaire des eucaryotes est ainsi contrôlé par de multiples cyclines, ainsi que des kinasesproches de Cdk1. Ces différents membres de cette famille s'associent avec des cyclines spécifiquesà chacune des transitions : la progression G1 S est régulée principalement par Cdk2, 4 et 6 enassociation avec les cyclines D et E. La figure résume les rôles des complexes cyclines/cdks. Il existeune certaine redondance entre les différentes cyclines et les cdks, un membre pouvant compenserl'absence de l'autre.
L'activité de ces protéines kinase durant la progression du cycle cellulaire est aussi régulée par la fixationde protéines inhibitrices = les CKI pour Cdk inhibitors.
Déficits immunitaires syndromiques(ou formes syndromiques de DIP) Dans ce cadre hétérogène, baptisé « DI autres, bien définis » ou « Other well defined ID syndromes »de la classification internationale 2, on place des DIP au phénotype clinique étendu (atteinteneurologique, retard mental, anomalies osseuses et des phanères, dysmorphie). Il s’agit pour la plupartde maladies monogéniques, de métaboloses ou d’anomalies chromosomiques mais certaines de cesaffections complexes ne sont pas encore élucidées sur le plan moléculaire.
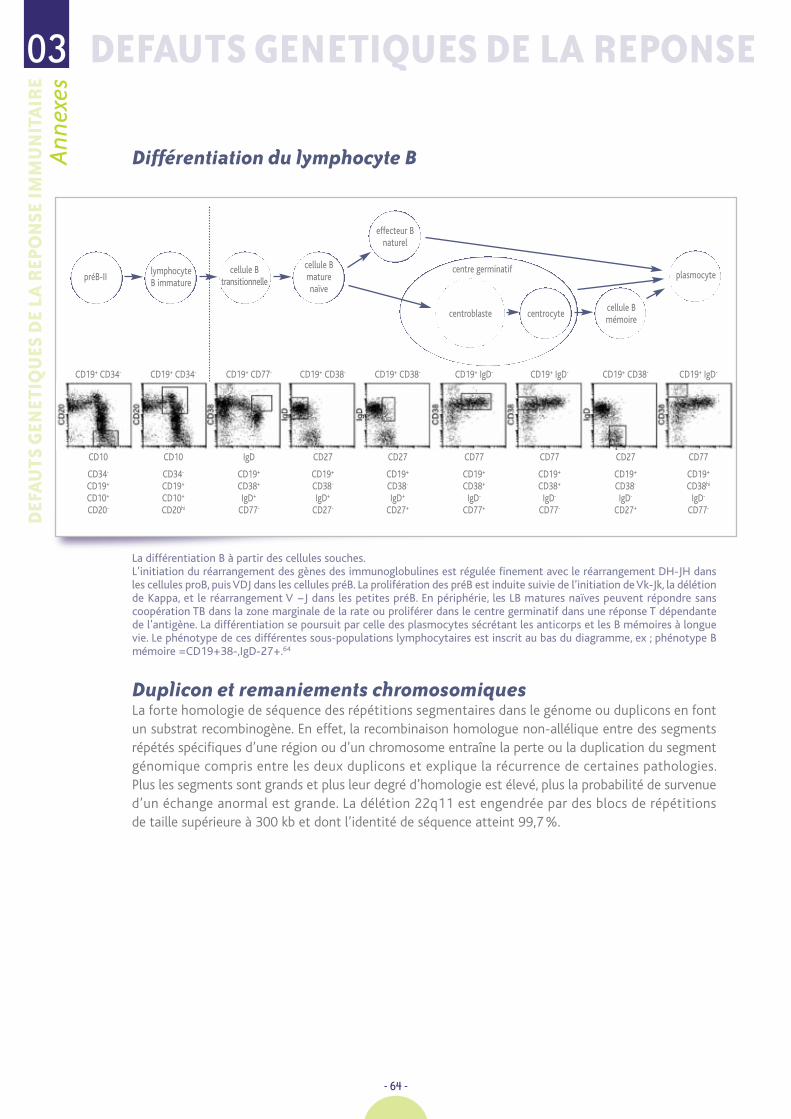
- 64 -
DEF
AU
TS G
ENET
IQU
ES D
E LA
REP
ON
SE IM
MU
NIT
AIR
EAn
nexe
s 03
Différentiation du lymphocyte B
La différentiation B à partir des cellules souches.L’initiation du réarrangement des gènes des immunoglobulines est régulée finement avec le réarrangement DH-JH dansles cellules proB, puis VDJ dans les cellules préB. La prolifération des préB est induite suivie de l’initiation de Vk-Jk, la délétionde Kappa, et le réarrangement V −J dans les petites préB. En périphérie, les LB matures naïves peuvent répondre sanscoopération TB dans la zone marginale de la rate ou proliférer dans le centre germinatif dans une réponse T dépendantede l’antigène. La différentiation se poursuit par celle des plasmocytes sécrétant les anticorps et les B mémoires à longuevie. Le phénotype de ces différentes sous-populations lymphocytaires est inscrit au bas du diagramme, ex ; phénotype Bmémoire =CD19+38-,IgD-27+.64
Duplicon et remaniements chromosomiquesLa forte homologie de séquence des répétitions segmentaires dans le génome ou duplicons en fontun substrat recombinogène. En effet, la recombinaison homologue non-allélique entre des segmentsrépétés spécifiques d’une région ou d’un chromosome entraîne la perte ou la duplication du segmentgénomique compris entre les deux duplicons et explique la récurrence de certaines pathologies.Plus les segments sont grands et plus leur degré d’homologie est élevé, plus la probabilité de survenued’un échange anormal est grande. La délétion 22q11 est engendrée par des blocs de répétitionsde taille supérieure à 300 kb et dont l’identité de séquence atteint 99,7%.
préB-II
CD19+ CD34- CD19+ CD34- CD19+ CD77- CD19+ CD38- CD19+ CD38- CD19+ CD38-CD19+ IgD- CD19+ IgD- CD19+ IgD-
CD10 CD10 IgD CD27 CD27 CD27CD77 CD77 CD77
CD34-
CD19+
CD10+
CD20-
CD34-
CD19+
CD10+
CD20hi
CD19+
CD38+
IgD+
CD77-
CD19+
CD38-
IgD+
CD27-
CD19+
CD38-
IgD+
CD27+
CD19+
CD38-
IgD-
CD27+
CD19+
CD38+
IgD-
CD77+
CD19+
CD38+
IgD-
CD77-
CD19+
CD38hi
IgD-
CD77-
lymphocyteB immature
cellule Btransitionnelle
cellule Bmature naïve
effecteur Bnaturel
centre germinatif
cellule Bmémoire
plasmocyte
centroblaste centrocyte

Annexes
- 65 -
Glossaire
Encéphalite herpétiqueL'encéphalite herpétique est la forme la plus commune d'encéphalite virale sporadique avecune incidence approximative de 1 à 2 pour 250 000 par an. Le pic de fréquence se situe entre l'âgede trois mois et six ans. Au cours de la primo-infection par HSV1, le virus atteint le système nerveuxcentral via les nerfs olfactifs et le trijumeau. Il n’y a pas de virémie, ni d’atteinte d’organe en dehorsdu système nerveux. Le traitement par antiviral diminue la mortalité mais le pronostic est sombreen raison de séquelles importantes chez la plupart des survivants et en particulier chez le jeuneenfant.
Ces éléments contrastent avec le caractère ubiquitaire du virus « inoffensif » pour la populationhumaine. L'encéphalite herpétique de l'enfant n'est pas associée à un déficit immunitaire classique.La première étiologie génétique de cette condition a été élucidée par JL Casanova avec la descriptiond'une forme autosomale récessive de défaut génétique en UNC 93B qui abolit la signalisation deTLR3, et la forme dominante de défaut de TLR3. Ces 2 défauts affectent spécifiquement la signalisationde la voie Toll- R3. Il n'y a pas de production d'interféron de type 1 en réponse à la stimulation de lavoie TLR3 par les fragments RNA double brin du virus HSV dans le système nerveux central. D'autresgènes ont été impliqués plus récemment par la même équipe : TRIF chez un patient de transmissiondominante, TRAF3 : 1 patient (dominant) et TBK1 pour deux patients (transmission dominante).
Ainsi, il apparaît que l'encéphalite herpétique du très jeune nourrisson résulte d'une collectionde défauts génétiques d'un seul gène, mais liés entre eux sur un plan fonctionnel.
Effet dominant négatifLes mutations avec un effet dominant négatif concernent des situations particulières, où ce n'estpas le taux absolu de protéines qui est important mais son taux relatif par rapport à un partenaire :dans cette relation tout déséquilibre quantitatif est pathologique et peut être assimilé à un effetde titration. C’est le cas pour le défaut de STAT3 dominant.
La mutation en simple dose suffit à entraîner la maladie chez les hétérozygotes parce que la mutationconduit à une protéine dysfonctionnelle dont l'effet n'est pas contrebalancé par la version normalede la protéine émanant de l'allèle non muté. Lorsque les homodimères STAT3 se formentchez les patients, 75% de ces complexes sont non fonctionnels.
Par le truchement de mutations aux effets différents sur la protéine, un même gène peut donnerlieu à une pathologie récessive avec perte de fonction, ou dominante si gain de fonction. Nousretrouverons cette notion avec les défauts génétiques de l’IFNg récepteur R1/R2 qui conduitau syndrome de susceptibilité génétique aux mycobactéries environnementales décrit par JL Casanova.

- 66 -
DEF
AU
TS G
ENET
IQU
ES D
E LA
REP
ON
SE IM
MU
NIT
AIR
EAn
nexe
s 03
Etude du cycle cellulaire en cytométrie de flux (CMF)
En CMF, on étudie le cycle cellulaire avec l’iodure de propidiumqui marque l’ADN. La plupart des cellules se trouvent en G1parce que c’est l’étape la plus longue. En G1, les cellules sontdiploïdes (2n). En phase S, l’ADN commence à se dupliqueralors l’intensité de la fluorescence augmente puis atteindrale double de la valeur de G1 dans les cellules en G2 et M qui ontcomplètement dupliqué leur ADN (cellules tétraploïdes : 4n).
Haplo-insuffisanceDans certains cas, la protéine est produite en quantité limitante et il suffit d'un seul allèle avec pertede fonction pour entraîner une pathologie : on dit qu'il y a haplo-insuffisance et la maladie se manifesteà l'état hétérozygote par un effet de dose. La transmission est de type dominant.
HémizygotieSi le gène est porté par le chromosome X, seuls les sujets de sexe masculin étant haploïdes pour ce chromosomesont atteints (hémizygotie) et les femmes hétérozygotes sont, par suite de l'inactivation aléatoirede l'un des X (lyonisation), constituées par une mosaïque de cellules normales et de cellules déficientes.
HétérochromatineRégion du génome où le DNA existe sous forme hypercondensée et non exprimée, se répliquanttardivement au cours du cycle cellulaire. Il en existe deux catégories : l’hétérochromatine constitutive(centromères et bras courts de certains chromosomes acrocentriques) et l’hétérochromatine facultative.L'hétérochromatine constitutive reste constamment sous une forme condensée et ne contient pasde gène fonctionnel. L’ADN de l'hétérochromatine constitutive est constitué de séquences courteshautement répétitives ou ADN satellite. Les séquences qui correspondent à cette chromatine sontregroupées principalement près des centromères de la plupart des chromosomes (elles correspondentaux bandes « C » du caryotype).
Hétérochromatine constitutiveL’ADN des centromères reste très compact et ne contient pas de gène. Il constitue une grande partde l'hétérochromatine constitutive. Chez l'homme, l'organisation est très complexe, elle varied'un chromosome à l'autre. Certaines séquences comme les alpha-satellites ou alphoïdes sontretrouvés dans tous les chromosomes. L'ADN alpha-satellite correspond à la répétition en tandemd'une séquence de 171 paires de bases. L'ADN centromérique est associé à des protéines dont une sérieappelée CLIP qui sont associées au kinétochore qui permet au chromosome de se fixer au fuseauet de migrer le long de celui-ci lors de l'anaphase. Certaines de ces protéines sont reconnues parles auto-anticorps retrouvés chez des malades atteints de maladies systémiques comme le lupus.
Intensité de fluorescence
G0-G1 G2-M
S

Annexes
- 67 -
Glossaire
Hot spotSite où la fréquence des mutations est anormalement élevée.
Hypomorphes (mutations)Mutation qui n’abolit pas complètement la fonction du produit du gène et qui s’oppose aux mutationsnulles ou amorphes.
ImmunoBlot (ting) ou Western Blot :
Il permet d'identifier une protéine donnée dans unlysat cellulaire. Les cellules subissent une dégradationpar un agent détergent pour solubiliser toutesles protéines cellulaires et le lysat est placé sur un geld’électrophorèse pour séparer les différentes protéinesselon leur taille. Les protéines sont transférées du gelà une membrane de nitrocellulose. Elles sont alorsdétectées par les anticorps spécifiques : ceux-ci sontrévélés par des anticorps anti-immunoglobulinesmarqués par des radio-isotopes ou un enzyme.
L’irradiation des lignées contrôles (CTRL) permetl’expression de la nibrine (NBN). L’expressionde RAD50 est normale chez les patients Nijmegen.L’expression de la ß-actine sert de contrôle interne.
IRAK4 Les déficits autosomiques récessifs en IRAK4 et en MyD88 sontdes déficits immunitaires de description récente. MyD88 est unemolécule adaptatrice cytosolique qui permet la signalisation desToll like récepteurs et des récepteurs de l'interleukine-1 via lecouplage avec le complexe kinase IRAK formé de 4 sous-unités.(schéma). Cette voie conduit à la synthèse des cytokinesinflammatoires telles que IL-1 b, IL-6, IL-8, TNF-a, IFN-a, b et λ.
Les défauts génétiques en IRAK4 et MyD88 sont des phénocopiessur le plan immunologique. Les leucocytes des patients ont desanomalies de réponse à la plupart des agonistes des voies TLRet IL-1R. Les patients atteints ont une susceptibilité aux bactériesinvasives surtout à gram positif : Streptococcus pneumoniae,Staphylococcus aureus et quelques bactéries à gram négatif telsque Pseudomonas aeruginosa et Shigella. Ces patients présententune résistance normale aux infections fongiques, parasitaires et
Sur le même ImmunoBlot : CTRL = contrôle ; RAD 50-/-= défaut de RAD50 ; Nijmegen ; AT (ataxie télangiectasie).Les lignées des patients sont irradiées à 6 Gray(symbole +) ou non (symbole -).Aucune bande n’est visible pour les lignées RAD50-/-.
Interaction MyD88/IRAK4

- 68 -
DEF
AU
TS G
ENET
IQU
ES D
E LA
REP
ON
SE IM
MU
NIT
AIR
EAn
nexe
s 03
virales, ainsi qu'à la plupart des autres bactéries. Un fait particulièrement marquant est l'améliorationdu déficit immunitaire avec l'âge. Les autres DIP qui sont associés aux infections bactériennes invasivesà Streptococcus pneumoniae concernent l’opsonisation des bactéries et la phagocytose splénique.C'est la plupart des déficits T et B, les asplénies, les défauts de C3, les composants de la voie classiqueet de la voie alterne du complément.
MéthylationElle protège le génome contre les lésions moléculaires qui peuvent résulter de recombinaison entreséquences répétitives (translocations, réarrangements illégitimes, délétions).
MLPA (Multiplex Ligation-dependent Probe Amplification) Cette procédure multiplexe est dérivée de l’OLApour Oligo nucléotide Ligation Assay et permetde détecter des duplications et des délétionsd’exons ce que ne repèrerait pas une PCR encondition standard. Le principe : un coupled’oligonucléotides adjacents, spécifiques dechaque exon et qui contiennent aux extrémités5’ et 3’ les séquences (noires) homologuesà celles des amorces de PCR, s’hybride sur l’ADNà tester et le « gap » entre les 2 est comblépar une réaction de ligation. (enzyme : DNAligase) pour former une seule molécule. Cetteligation ne se produit que si l’hybridation estparfaite. Une séquence variable est ajoutée dansla partie 3’ de la sonde (« stuffer ») (en vert)
qui sert à différencier les produits de ligation en fonction de la taille après amplification. L’une desamorces est marquée par un fluorophore et la détection des fragments amplifiés se fait par migrationsur un automate de séquençage. Les résultats sont analysés par un logiciel dédié. Si une délétion estretrouvée au niveau de la partie 5’, 3’ ou les 2, la ligation ne peut avoir lieu. La MLPA est utilisée dansla détection des aneuploïdies, des délétions/duplications d’exon, la détection des anomalies des exonsdes gènes BRCA 1 et BRCA 2 du cancer du sein héréditaire par exemple.
NibrineLa Nibrine joue un rôle important dans l'activation du complexe MRN* (voir ce terme) impliqué dansle traitement des cassures double-brin de l'ADN au cours de la recombinaison V(D)J ou de la commutationde classe mais surtout dans la réparation des dommages génotoxiques sous l'effet des agents mutagènestels que les radiations ionisantes et les produits radio-mimétiques.
OMIM (Online Mendelian Inheritance in Man)Le catalogue des maladies mendéliennes établi par Victor McKusick et qui constitue une véritableencyclopédie pour les généticiens est accessible en ligne (Online Mendelian Inheritance in Man/MIM).Il contient des données phénotypiques cartographiques moléculaires et bibliographiques. Elle estconstamment mise à jour. http://www.ncbi.nlm.nih.gov/omim
La ligation crée la matricepour la réaction de PCR
Figure 1
5’ 3’
5’3’
Figure 2Mismatch, pas de ligationpas de matrice
* Cf. glossaire page 60

Annexes
- 69 -
Dans les cellules de mammifères, le stop de la transition G1/Sest aussi provoqué par l'action d'une protéine, la p53 qui estphosphorylée par ATM et CHK2. La phosphorylation stabilisela protéine p53 qui l'empêche de se dégrader rapidementet augmente rapidement son taux, en réponse aux dommagesdu DNA. La protéine p53 est un facteur de transcriptionet conduit à l'induction de l'inhibiteur p21. Celui-ci inhibe
les complexes Cdk2/cycline Eet conduit à l 'arrêt ducycle cellulaire en G1. Lesmutations du gène p53 sontles altérations génétiquesles plus fréquentes descancers humains ce quiillustre le rôle central de larégulation du cycle cellulairepour de nombreux organismesmulticellulaires.
P53Dans les cellules de mammifères, le stop de la transition G1/S (check-pointG1/S ou point de restriction)du cycle cellulaire est sous le contrôle de la p53 qui est phosphorylée par ATM et CHK2. Laphosphorylation stabilise la protéine p53, l'empêche de se dégrader rapidement et augmenterapidement son taux en réponse aux dommages de l’ADN.
La protéine p53 est un facteur de transcription et conduit à l'induction de l'inhibiteur p21. Celui-ciinhibe les complexes Cdk2/cycline E et conduit à l'arrêt du cycle cellulaire en G1. L'oncogène p53est fréquemment muté dans un certain nombre de cancers. La perte de la fonction de p53 entraînel'accumulation de dommages génotoxiques et de l'instabilité générale du génome cellulaire, étapeimportante dans le développement du cancer. Les mutations du gène p53 sont les altérationsgénétiques les plus fréquentes des cancers humains ce qui illustre le rôle central de la régulationdu cycle cellulaire pour de nombreux organismes multicellulaires. Les cellules irradiées des patientsAT ont une activation de p53 anormale et un défaut du contrôle G1-S.
L'arrêt du cycle est déclenché par les protéines kinases ATMet ATR protéines kinases, composantes d'un édifice moléculairequi reconnaît l'ADN endommagé ou non répliqué. ATM estactivée principalement par les cassures double-brin, ATR parles cassures simples brins et l'ADN non répliqué. Une foisactivées, ATM et ATR phosphorylent et activent les kinasesCHK2 et CHK1 qui arrêtent le cycle.
Glossaire

- 70 -
DEF
AU
TS G
ENET
IQU
ES D
E LA
REP
ON
SE IM
MU
NIT
AIR
EAn
nexe
s 03
Perte de fonction (voir aussi Gain de fonction)Perte ou gain de fonction : une explication moléculaire de la dominance et de la récessivité.Les conséquences primaires des mutations sur les protéines sont qualitatives ou quantitatives ou les deux ;il en résulte au final une perte de fonction totale ou partielle. Les mutations avec perte de fonctiontouchant un gène autosomique sont récessives si à l'état hétérozygote la production de protéines parl'allèle normal suffit pour maintenir une fonction normale (on parle aussi d’haplo-suffisance : erreursinnées du métabolisme récessives) ; pour que la pathologie apparaisse, il faut que les deux allèles soientmutés, soit identiquement (homozygotie), soit différemment (hétérozygotie composite).
Radiosensibilité
L’étude de la radiosensibilité des fibroblastes (figure ci-contre) des patientsatteints de syndrome d’instabilité chromosomique est réalisée encomptant les colonies survivant à des doses progressives d’irradiation1-5 gray. La survie est exprimée en terme de pourcentage de cellulesirradiées/ non irradiées. On obtient ce genre de courbe où sont figuréesà titre d’exemple des lignées d’un contrôle, des cellules du patientexploré, et des cellules d’un patient présentant une AT : ataxie-télangiectasie.CTRL : patient contrôle.
Recombinaison V(D)J
100
CTRL
surv
ie (%
)
AT
Pt
10
1
0,1
0 1 2 3 4 5 6
VH
recombinaison V(D)J
a
b
recombinaison V(D)J
RSS RSS
«Joint codant»
Joint signal
Epingle à cheveuxRag1/Rag2
DNA-PKcsKu70/Ku80Artemis
XRCC4, DNA-LigIVCernunnos
NHEJ
VH DH JH JH Cµ
VH VH DH JH Cµ
VH DH
DHDH
JH
JH
DH JH
DH JH
DH JH
DH JHJH
Cµ
7 799
7 799
7 799
99
77
Organisation du locus IgH et processus de réarrangement
RSS : Recombination Signal Sequence = signal de recombinaison

Annexes
- 71 -
Glossaire
Signal de recombinaison :
heptamère
12segmD
VH
D
segment J
J
C
G
C
G
C
G
CA
T
AA
T
G
C
G
C
séquence CACAGTG
12
12
12
12
12
23
23
23
2323
heptamère
heptamère heptamère
séquence CACAGTG
séquence ACAAAAACC
séquence ACAAAAACC
séquence ACAAAAACC
nonamère
nonamère
nonamère nonamère
C
G
C
G
C
G
C
G
C
G
C
A
T
A
T
AA
T
G
C
G
C
G
C
G
C
A
T
A
T
C
G
C
G
C
C
G
C C
G
A
T
A
T
A
T
A
T
A
T
A G
C
G
C
A
T
A
T
A
T
A
T
A
C
G
C
G
C
G
A
T
A
T
A
T
G
C
G
C
T
A
T
A
T
A
T
A
T
C
G
A
T
Recombinaison V(D)J
En raison d'un vaste répertoire d'antigènes, la nature a sélectionné un mécanisme de diversificationpour le système immunitaire qui est basé sur l'association de segments de gènes, variable (V)diversité (D) et joint (J) qui codent pour le domaine variable des immunoglobulines et du récepteurT pour l'antigène.
La recombinaison V(D)J est un processus de réarrangement somatique restreint à la lignée lymphoïde.Non seulement, ce mécanisme est responsable de la diversité, mais elle constitue une étape centralepour le développement du système immunitaire. Dans le cas des lymphocytes B, il existe 2 mécanismes supplémentaires, déclenchés après reconnaissancede l'antigène, qui optimisent la réponse anticorps. Il s'agit de la commutation de classe qui permetà un domaine variable réarrangé d’une IgH de s’associer avec une région constante différentece qui conduit à la production d'isotypes différents, IgG, IgA, ou IgE. De plus, les domaines variablesdes immunoglobulines augmentent leur affinité pour l'antigène, grâce à l'accumulationde mutations somatiques dans les gènes V. Les anomalies de la commutation et de la générationdes mutations somatiques conduisent aux syndromes hyper IgM. À la fois chez l'homme et chez l'animal, les anomalies de la recombinaison entraînent un arrêtde la maturation lymphocytaire T et B et constituent un sous-groupe de déficits immunitairescombinés sévères. Les anomalies de régulation de la recombinaison V(D)J sont parfois associéesau développement des hémopathies lymphoïdes.
La recombinaison est un processus enzymatique multi-étape.

- 72 -
DEF
AU
TS G
ENET
IQU
ES D
E LA
REP
ON
SE IM
MU
NIT
AIR
EAn
nexe
s 03
� une cassure double-brin aux limites des séquences de recombinaison (RSS) canoniques conduisantà la constitution d’épingle à cheveux au niveau des deux brins codants, sur le chromosome et l'excisionde matériel chromosomique, à bouts francs. Ces séquences canoniques sont symbolisés par 7 et 9(il s’agit de l’heptamère CACAGTG et du nonamère ACAAAAACC).
� La deuxième étape est prise en charge par la machinerie de réparation de l'ADN avec au moinsl'intervention de six facteurs, le complexe DNA-PK (Dna Dependent Protein Kinase) qui comporteles sous-unités Ku70/Ku80 et la sous-unité catalytique qui va identifier les lésions du DNA par la fixationdes sous-unités Ku aux extrémités de l'ADN. Enfin, le « processing » des épingles à cheveux parune enzyme baptisée ‘Artémis’, et la réparation par la XRCC4/DNA ligase IV. Un défaut de l'un ou l'autrede ces composants interrompt la réaction de recombinaison V(D)J et conduit pour certains à un phénotypede SCID. Pendant la phase finale de la recombinaison, l'enzyme TDT augmente la diversité en ajoutantdes nucléotides entre les deux brins codants.
Le complexe d'enzymes qui agissent de concert pour réaliser la recombinaison somatique V(D)J s'appellela recombinase. Les produits de deux gènes RAG1 et 2 sont les composants spécifiques de cetterecombinase ; cette paire de gènes est exprimée uniquement dans les lymphocytes en développement,au moment où ils sont engagés pour assembler les récepteurs aux antigènes. Ils sont essentiels pourla recombinaison somatique. Lorsque l'on transfecte des cellules de lignées non lymphoïdes tellesdes fibroblastes avec ces 2 gènes, on confère aux fibroblastes la propriété de réarranger des segmentsexogènes d'ADN à condition qu’ils contiennent des séquences de recombinaison appropriées; c'estd'ailleurs comme cela que les 2 gènes RAG1 et RAG2 ont été clonés.
Les mécanismes moléculaires du réarrangement de l'ADN et de la recombinaison V(D)J sont illustrésdans les figures. Les signaux de recombinaison CACAGTG (heptamère) et ACAACCCCC (nonamère)espacés de 12 et 23 (« spacers») paires de base sont rapprochées par des interactions qui se jouententre protéines qui vont reconnaître spécifiquement la longueur du spacer. C'est la règle 12/23de la recombinaison V(D)J.
Les molécules ADN sont cassées en 2 puis se reforment avec une configuration différente. Les bordsdes séquences «heptamère» se rejoignent précisément tête-bêche pour former un « joint-signal »,pièce circulaire de matériel ADN extra-chromosomique ; cette pièce circulaire sera perdue au coursde la division cellulaire. Ils constituent les TRECS* pour le réarrangement du récepteur T. Les TRECSsont dilués avec le temps lorsque les LT proliférent après contact antigénique. Les cellules T naïves quisont récemment émigrées du thymus ont des taux bien plus élevés de TRECS que les LT « âgés »,ayant rencontré l’Ag. L’évaluation des TRECS peut se faire en cytométrie de flux (populationsCD4+CD45RA+CD31) en particulier pour confirmer un syndrome de Di George ou pour monitorerla reconstitution immunologique après greffe de MO. La quantification des TRECS à partir des échantillonsde sang prélevés en période néonatale sur papier de Guthrie proposée récemment dans 2 états auxEtats-Unis se révèlent un test de dépistage pour les déficits T et notamment des SCID.Les segments V et J qui restent sur le chromosome vont se rejoindre pour former le « joint-codant ».La jonction est imprécise et par conséquent génère de la variabilité additionnelle pour la séquencede la région V des Ig.
* Cf. glossaire page 60

Annexes
- 73 -
Glossaire
JD
Palindrome :« P nucléotides »
Palindrome :« P nucléotides »
TDT : « N nucléotides »
TDT
A
T T
T T
T
AA
T TT
Les nucléotides P et N introduisent de la diversité entre les segments de gènes au cours du réarrangement.
La première réaction est une coupure qui nécessite l'activité coordonnée de RAG 1 et 2 quireconnaissent les séquences de recombinaison spécifiques. La règle 12/23 est respectée selondes mécanismes encore inconnus. L’activité endo-nucléase de la protéine RAG 1 entraîne une cassuresimple brin de l'ADN en 5’ de chaque séquence de recombinaison en laissant un groupe OH en 3'de l'extrémité de chaque segment codant. Le groupe OH 3' hydrolyse la liaison phospho-di-ester surl'autre brin. Ce processus crée une cassure double-brin à bords francs aux extrémités des deux signauxheptamères. Des extrémités sont maintenues fermement par le complexe RAG et d'autres enzymesde réparation jusqu'à ce que le joint soit complété. Les deux signaux de recombinaison spécifiquesont joints pour former le « joint-signal».
La formation du « joint-codant» est plus complexe :
� Ouverture de l'épingle à cheveux par une cassure simple-brin. Cette cassure peut survenirà des endroits différents de l'épingle à cheveux ce qui conduit à une variabilité de séquencessupplémentaires.
� Les enzymes de réparation du complexe modifient l'épingle à cheveux ouverte en enlevantdes nucléotides par son activité exonucléotidase, ajoutant des nucléotides de façon aléatoirepar l'activité transférase terminale TDT.
� Enfin les enzymes ligases comme la DNA ligase IV joignent les brins pour générer un ADN double-brin continu reconstituant le chromosome, en intégrant la séquence combinée.
D
idem
C
GA
T OH
A
T
A
T
T T
D
J
C
GA
T
T T
A
T
A
T
JDC
GA
T
Formation du « joint-signal»

- 74 -
DEF
AU
TS G
ENET
IQU
ES D
E LA
REP
ON
SE IM
MU
NIT
AIR
EAn
nexe
s 03
Sous-populations lymphocytaires T CD4, TH1, TH2, TH 17 et TRegLes sous-populations lymphocytaires T auxiliaires comportent les TH1 qui produisent l'interféron-gamma(IFNg). Ce sont les effecteurs de l'hypersensibilité retardée, ils activent les macrophages et sont impliquésdans la défense contre les bactéries intracellulaires.
Les TH2 produisent les cytokines suivantes : IL-4, IL-5, IL-13, et IL-25, ils sont impliqués dans la productiond'IgE, l'inflammation médiée par les éosinophiles, la clairance des helminthes.
A ces 2 sous-populations classiques, s'ajoutent les TH17, baptisés ainsi parce qu'ils produisent IL-17,protègent l'hôte contre les bactéries extracellulaires et les champignons. Les TH17 sont les effecteursde certaines situations d’auto-immunité.
IL-17 initie le recrutement des polynucléaires neutrophiles, constituant ainsi un lien entre l'immunitéadaptative et l'immunité innée. TGF-b, IL-23, et les cytokines pro-inflammatoires IL-1b and IL-6 sontles médiateurs essentiels de la différentiation TH17 chez l'homme et sont nécessaires pour l’expressiond’IL-17, IL-23 R, et RORgt.
L'exposition de CD4 naïfs à TGF-b et IL-2 entraîne la différentiation des lymphocytes en cellules auxfonctions suppressives, les T régulateurs, TReg, avec l'expression du facteur de transcription : Forkheadbox Protein 3 (Foxp3). Les TReg jouent un rôle important dans le maintien de la tolérance en supprimantl'activation des effecteurs T auto-réactifs. Les mutations de Fox p3 sont associées au syndrome IPEXqui sera abordé dans un autre cahier sur les syndromes de dysrégulation immunitaire.
Th1 Th2
Treg Th17
Th2Th1
Treg Th17
GATA3STAT5
RORγtSTAT3
T-betSTAT4
IFN-γIL-2LTα
IFN-γIL-4
IFN-γIL-4IL-12
IFN-γIL-12
TGF-βIL-2RA
IL-4IL-2
TGF-βIL-6IL-21IL-23IL-6
IL-21
IL-4IL-5IL-13IL-25
TGF-βIL-10IL-35
IL-17AIL-17FIL-21IL-22
FOXP3STAT5
Différentiation des CD4 «drivée» par les différentes cytokines et parl’expression des facteurs de transcription correspondants.
Chacune de ces quatre sous-populationslymphocytaires CD4 génère des cytokinesqui contrôlent leur fonction biologique.Au-dessous de chaque cellule sont indiquésen encadré, les différents facteurs de transcriptionimpliqués dans le développement et le maintiende chaque sous-population. En vert, les cytokinesqui favorisent le développement de chaquesous-population de CD4, en rouge les cytokinesqui en bloquent le développement. LTa :lymphotoxine a ; RA acide rétinoïque.

Annexes
- 75 -
Glossaire
Syndrome de LynchLe cancer colorectal est héréditaire dans 5% des cas environ. Les 2 formes principales de cancerhéréditaire sont la polypose adénomateuse familiale et le cancer héréditaire du colon sans polyposeou syndrome de Lynch. Ce dernier apparaît chez le sujet jeune ; sa transmission est autosomiquedominante. Il est dû à des mutations dans les gènes codant pour des protéines de la réparation desmésappariements. Les gènes touchés le plus souvent sont MSH2, MLH1 et plus rarement PMS1,PMS2 et MSH6. Les effets des mutations ne se limitent pas au cancer colorectal, car on retrouveaussi des cancers de l’utérus, des ovaires, de l’estomac, du tractus urinaire, de l’intestin grêle et dutractus biliaire.
Syndrome d’Omenn Ces nourrissons présentent une érythrodermie généralisée avec pachydermie et alopécie, un retardpondéral, une diarrhée chronique, une hépato splénomégalie et des adénopathies. Une éosinophiliesouvent marquée, une hypoprotidémie, une absence de lymphocytes B contrastant avec un nombrevariable de lymphocytes T mémoire, activés, CD45RO, oligoclonaux mais non fonctionnels caractérisentcette forme très particulière de déficit immunitaire combiné sévère. Le diagnostic est souvent retardéet confondu avec un « eczéma » sévère ou une dermatite de Leiner-Moussous. Cette affection estrapidement fatale en l'absence de greffe de moelle osseuse. Les lymphocytes T correspondent àl'amplification de quelques clones dans la périphérie en présence d'antigènes du soi. Ce syndrome
ne doit pas être confondu avec uneGVH materno- foetale d’expressioncutanée sévère (recherche d’unmicrochimérisme maternel par FISHou VNTR, étude des populationsCD45RO/CD4 et CD45RO/CD8). Ils’agit habituellement de mutationshypomorphes des enzymes de larecombinaison (RAG1 et 2, Artémis)ou d’un défaut partiel de IL-7 RA.
TRECS (voir aussi recombinaison V(D)J)T cell Receptor Excision Circles : constitue un marqueur moléculaire des lymphocytes T récemmentémigrés du thymus.
Syndrome d'Omenn.Erythrodermie, alopécie
Syndrome d'Omenn.Pachydermie
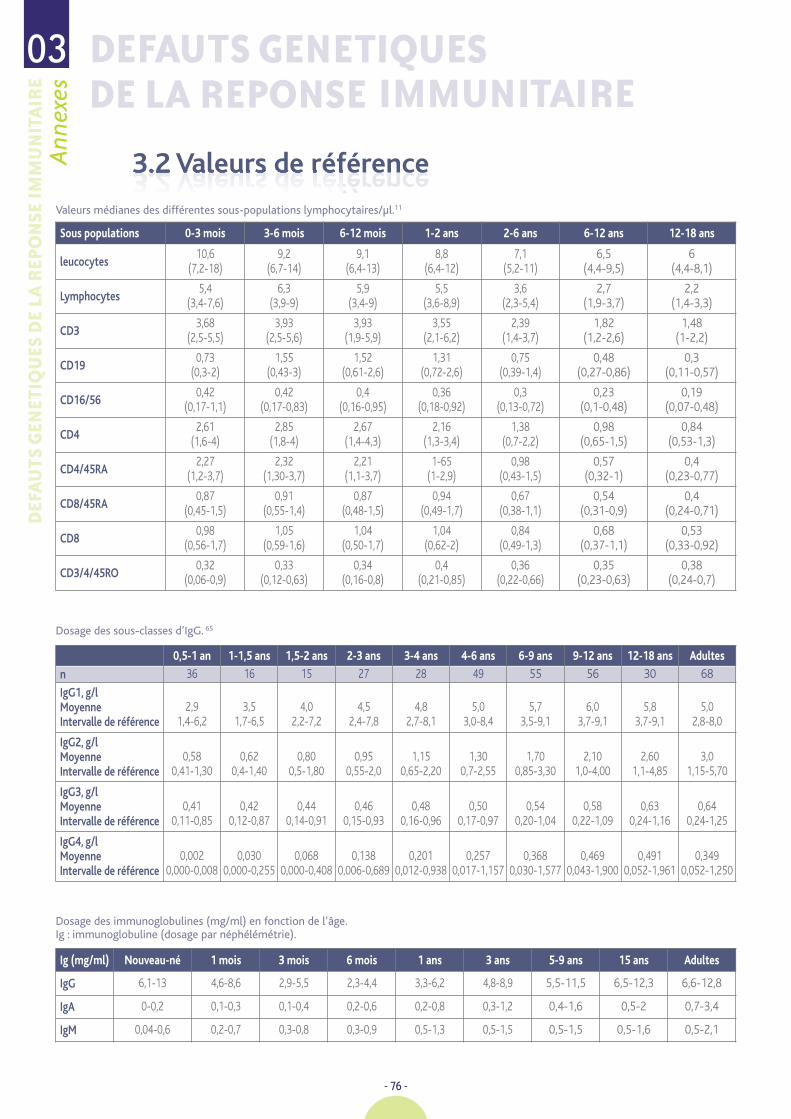
- 76 -
DEF
AU
TS G
ENET
IQU
ES D
E LA
REP
ON
SE IM
MU
NIT
AIR
EAn
nexe
s 03
Dosage des immunoglobulines (mg/ml) en fonction de l’âge.Ig : immunoglobuline (dosage par néphélémétrie).
Ig (mg/ml) Nouveau-né 1 mois 3 mois 6 mois 1 ans 3 ans 5-9 ans 15 ans Adultes
IgG 6,1-13 4,6-8,6 2,9-5,5 2,3-4,4 3,3-6,2 4,8-8,9 5,5-11,5 6,5-12,3 6,6-12,8
IgA 0-0,2 0,1-0,3 0,1-0,4 0,2-0,6 0,2-0,8 0,3-1,2 0,4-1,6 0,5-2 0,7-3,4
IgM 0,04-0,6 0,2-0,7 0,3-0,8 0,3-0,9 0,5-1,3 0,5-1,5 0,5-1,5 0,5-1,6 0,5-2,1
3.2 Valeurs de référence3.2 Valeurs de référence
Valeurs médianes des différentes sous-populations lymphocytaires/µl.11
Sous populations 0-3 mois 3-6 mois 6-12 mois 1-2 ans 2-6 ans 6-12 ans 12-18 ans
leucocytes10,6
(7,2-18)9,2
(6,7-14)9,1
(6,4-13)8,8
(6,4-12)7,1
(5,2-11)6,5
(4,4-9,5)6
(4,4-8,1)
Lymphocytes5,4
(3,4-7,6)6,3
(3,9-9)5,9
(3,4-9)5,5
(3,6-8,9)3,6
(2,3-5,4)2,7
(1,9-3,7)2,2
(1,4-3,3)
CD33,68
(2,5-5,5)3,93
(2,5-5,6)3,93
(1,9-5,9)3,55
(2,1-6,2)2,39
(1,4-3,7)1,82
(1,2-2,6)1,48(1-2,2)
CD190,73(0,3-2)
1,55(0,43-3)
1,52(0,61-2,6)
1,31(0,72-2,6)
0,75(0,39-1,4)
0,48(0,27-0,86)
0,3(0,11-0,57)
CD16/560,42
(0,17-1,1)0,42
(0,17-0,83)0,4
(0,16-0,95)0,36
(0,18-0,92)0,3
(0,13-0,72)0,23
(0,1-0,48)0,19
(0,07-0,48)
CD42,61(1,6-4)
2,85(1,8-4)
2,67(1,4-4,3)
2,16(1,3-3,4)
1,38(0,7-2,2)
0,98(0,65-1,5)
0,84(0,53-1,3)
CD4/45RA2,27
(1,2-3,7)2,32
(1,30-3,7)2,21
(1,1-3,7)1-65(1-2,9)
0,98(0,43-1,5)
0,57(0,32-1)
0,4(0,23-0,77)
CD8/45RA0,87
(0,45-1,5)0,91
(0,55-1,4)0,87
(0,48-1,5)0,94
(0,49-1,7)0,67
(0,38-1,1)0,54
(0,31-0,9)0,4
(0,24-0,71)
CD80,98
(0,56-1,7)1,05
(0,59-1,6)1,04
(0,50-1,7)1,04
(0,62-2)0,84
(0,49-1,3)0,68
(0,37-1,1)0,53
(0,33-0,92)
CD3/4/45RO0,32
(0,06-0,9)0,33
(0,12-0,63)0,34
(0,16-0,8)0,4
(0,21-0,85)0,36
(0,22-0,66)0,35
(0,23-0,63)0,38
(0,24-0,7)
Dosage des sous-classes d’IgG. 65
0,5-1 an 1-1,5 ans 1,5-2 ans 2-3 ans 3-4 ans 4-6 ans 6-9 ans 9-12 ans 12-18 ans Adultes
n 36 16 15 27 28 49 55 56 30 68
IgG1, g/lMoyenneIntervalle de référence
2,91,4-6,2
3,51,7-6,5
4,02,2-7,2
4,52,4-7,8
4,82,7-8,1
5,03,0-8,4
5,73,5-9,1
6,03,7-9,1
5,83,7-9,1
5,02,8-8,0
IgG2, g/lMoyenneIntervalle de référence
0,580,41-1,30
0,620,4-1,40
0,800,5-1,80
0,950,55-2,0
1,150,65-2,20
1,300,7-2,55
1,700,85-3,30
2,101,0-4,00
2,601,1-4,85
3,01,15-5,70
IgG3, g/lMoyenneIntervalle de référence
0,410,11-0,85
0,420,12-0,87
0,440,14-0,91
0,460,15-0,93
0,480,16-0,96
0,500,17-0,97
0,540,20-1,04
0,580,22-1,09
0,630,24-1,16
0,640,24-1,25
IgG4, g/lMoyenneIntervalle de référence
0,0020,000-0,008
0,0300,000-0,255
0,0680,000-0,408
0,1380,006-0,689
0,2010,012-0,938
0,2570,017-1,157
0,3680,030-1,577
0,4690,043-1,900
0,4910,052-1,961
0,3490,052-1,250

- 77 -
DEF
AU
TS G
ENET
IQU
ES D
E LA
REP
ON
SE IM
MU
NIT
AIR
ERé
fére
nces
04
Références
1. Conley ME, et al. Definition of primary immunodeficiency in 2011: a "trialogue" among friends. Ann N Y Acad Sci. 2011Nov;1238:1-6.
2. Al-Herz W, Bousfiha A, Casanova JL, et al. Primary immunodeficiency diseases: an update on the classification from theInternational Union of Immunological Societies Expert committee for primary immunodeficiency. Front Immunol 2011;54:1–26.
3. source : http://www.associationiris.org/infos-medicales-et-traitements/les-deficits-immunitaires. http://www.ceredih.fr.
4. CEREDIH: The French PID study group. The French national registry of primary immunodeficiency diseases. Clin Immunol.2010;135:264-72.
5. Fischer A, et al. Gene therapy for primary adaptive immune deficiencies. J Allergy Clin Immunol. 2011;127:1356-9.
6. De Vries E. Clinical Working Party of the European Society for Immunodeficiencies (ESID). Patient-centred screening for primaryimmunodeficiency: a multi-stage diagnostic protocol designed for non-immunologists. Clin Exp Immunol. 2006;145:204-14.
7. Notarangelo LD. Primary immunodeficiencies. J Allergy Clin Immunol. 2010 ;125(2 Suppl 2).
8. Bussone G, et al. Late onset of primary immune deficiencies Presse Med. 2010 ;39:196-207.
9. Oliveira JB, et al. Laboratory evaluation of primary immunodeficiencies. J Allergy Clin Immunol. 2010;125 (2 Suppl 2):S297-305.
10. Picard C. Comment explorer un déficit immunitaire héréditaire ? Rev Prat 2007 ; 57 : 1671-6.
11. Shearer WT, et al. Lymphocyte subsets in healthy children from birth through 18 years of age: the Pediatric AIDS ClinicalTrials Group P1009 study. J Allergy Clin Immunol. 2003 ;112:973-80.
12. Joao B. Oliveira et al. Applications of flow cytometry for the study of primary immune deficiencies. Current Opinion inAllergy and Clinical Immunology 2008, 8:499–509.
13. Sullivan KE. Chromosome 22q11.2 deletion syndrome: DiGeorge syndrome/velocardiofacial Syndrome. Immunol AllergyClin North Am. 2008;28:353-66.
14. Kobrynski LJ, et al. Velocardiofacial syndrome, DiGeorge syndrome: the chromosome 22q11.2 deletion syndromes. Lancet.2007;370:1443-52.
15. Emanuel BS. Molecular mechanisms and diagnosis of chromosome 22q11.2 rearrangements. Dev Disabil Res Rev. 2008;14:11-8.
16. Markert ML, et al. Review of 54 patients with complete DiGeorge anomaly enrolled in protocols for thymus transplantation:outcome of 44 consecutive transplants. Blood. 2007 ;109:4539-47.
17. Thompson PW, et al. Frequency of inherited deletions of 22q11. J Med Genet 1998;35:789.
18. Rope AF, et al. DiGeorge anomaly in the absence of chromosome 22q11.2 deletion. J Pediatr. 2009 ;155:560-5.
19. McLean-Tooke A, et al. Immunodeficiency and autoimmunity in 22q11.2 deletion syndrome. Scand J Immunol. 2007 ;66:1-7.
20. Digilio MC et al. Screening for celiac disease in patients with deletion 22q11.2 (DiGeorge/velo-cardio-facial syndrome).Am J Med Genet A. 2003, 121:286-8.
21. Eberle P, et al. Persistent low thymic activity and non-cardiac mortality in children with chromosome 22q11.2 microdeletionand partial DiGeorge syndrome. Clin Exp Immunol 2009 ;155:189-98.
22. Ganapathi KA, et al. Ribosomal dysfunction and inherited marrow failure. Br J Haematol. 2008 ;141376-87.

- 78 -
DEF
AU
TS G
ENET
IQU
ES D
E LA
REP
ON
SE IM
MU
NIT
AIR
ERé
fére
nces
04
23. Thiel CT. Cartilage-Hair Hypoplasia - Anauxetic Dysplasia Spectrum Disorders. In: Pagon RA, Bird TD, Dolan CR, StephensK, Adam MP, editors. GeneReviews™ [Internet]. Seattle (WA): University of Washington, Seattle; 1993-. 2012 Mar 15 source :http://www.ncbi.nlm.nih.gov/books/NBK84550/.
24. Taskinen M, et al. Extended follow-up of the Finnish cartilage-hair hypoplasia cohort confirms high incidence of non-Hodgkin lymphoma and basal cell carcinoma. Am J Med Genet A. 2008;146A:2370–5.
25. Notarangelo LD, et al. Cartilage-hair hypoplasia: molecular basis and heterogeneity of the immunological phenotype.Current Opinion in Allergy and Clinical Immunology 2008, 8:534–539.
26. De Felipe I, et al. Comel-Netherton syndrome. A diagnostic challenge. Br J Dermatol. 1997;137:468-9.
27. Sprecher E, et al. The spectrum of pathogenic mutations in SPINK5 in 19 families with Netherton syndrome: implicationsfor mutation detection and first case of prenatal diagnosis. J Invest Dermatol. 2001 ;117:179-87.
28. Renner ED et al. Comèl-Netherton syndrome defined as primary immunodeficiency. J Allergy Clin Immunol. 2009;124:536-43.
29. Engelhardt KR, Grimbacher B. Mendelian traits causing susceptibility to mucocutaneous fungal infections in human subjects.J Allergy Clin Immunol. 2012;129:294-305.
30. Glocker EO et al. A Homozygous CARD9 Mutation in a Family with Susceptibility to Fungal Infections. N Engl J Med2009;361:1727-35.
31. Ferwerda B, Human dectin-1 deficiency and mucocutaneous fungal infections. N Engl J Med. 2009 Oct 29;361(18):1760-7.
32. Liu L, et al. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneouscandidiasis. J Exp Med. 2011;208:1635-48.
33. Puel A, et al. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science. 2011Apr 1;332(6025):65-8.
34. Grimbacher B, Puck JM, Holland SM (2007) Hyper-IgE syndrome. In: Ochs H, Smith HIE, Puck JM (eds) PrimaryImmunodeficiency Diseases: a Molecular and Genetic Approach, 2 ed. Oxford University Press, NY, pp 496-504.
35. Kathryn J. Sowerwine, Hyper-IgE syndrome update Ann. N.Y. Acad. Sci. 1250 (2012) 25–32.
36. Chandesris MO, et al. Autosomal Dominant STAT3 Deficiency and Hyper-IgE Syndrome: Molecular, Cellular, and ClinicalFeatures From a French National Survey. Medicine (Baltimore). 2012;91:e1-e19.
37. Freeman AF, Holland SM. Clinical manifestations, etiology, and pathogenesis of the hyper-IgE syndromes. Pediatr Res. 2009May;65(5 Pt 2):32R-37R.
38. Nieminen P, et al. Inactivation of IL11 signaling causes craniosynostosis, delayed tooth eruption, and supernumerary teeth.Am J Hum Genet. 2011 15;89(1):67-81.
39. Minegishi Y. Hyper-IgE syndrome. Current Opinion in Immunology 2009, 21:487-492.
40. Casanova JL, et al D.Inborn errors of human JAKs and STATs. Immunity. 2012 20;36:515-28.
41. Harada Y, et al. DOCK8 is a Cdc42 activator critical for interstitial dendritic cell migration during immune responses. Blood.2012;119:4451-61.
42. Engelhardt K et al. Large deletions and point mutations involving the dedicator of cytokinesis 8 (DOCK8) in the autosomal-recessive form of hyper-IgE syndrome. J Allergy Clin Immunol. 2009; 124:1289-302.
43. Minegishi Y, et al. Human tyrosine kinase 2 deficiency reveals its requisite roles in multiple cytokine signals involved ininnate and acquired immunity. Immunity 2006;25:745-55.
44. Bogunovic D, et al. Mycobacterial Disease and Impaired IFN-γ Immunity in Humans with Inherited ISG15 Deficiency.Science. 2012 337:1684-88.
45. Kilic S et al. A patient with tyrosine kinase 2 deficiency without hyper-IgE syndrome. J Pediatr. 2012;160:1055-7.
46. Zhang Q, et al. Combined immunodeficiency associated with DOCK8 mutations. N Engl J Med. 2009; 361:2046–55.
47. Su HC, et al. DOCK8 deficiency. Ann N Y Acad Sci. 2011;1246:26-33.
48. Chandesris MO, et al. Frequent and widespread vascular abnormalities in human signal transducer and activator of transcription3 deficiency. Circ Cardiovasc Genet. 2012 1;5:25-34.

Références
Référen
ces
- 79 -
49. Sanz MM, German J. Bloom's Syndrome. 2006 Mar 22 [Updated 2010 Aug 24]. In: Pagon RA, Bird TD, Dolan CR, et al.,editors. GeneReviews™ [Internet]. Seattle (WA): University of Washington, Seattle; 1993-. Source : http://www.ncbi.nlm.nih.gov/books/NBK1398/
50. Tikoo S, Sengupta S. Time to bloom. Genome Integr 2010 4;1:14.
51. Ehrlich M, et al.Immunodeficiency, centromeric region instability, facial anomalies syndrome (ICF).Orphanet J Rare Dis.2006 1;1:2.
52. Matarazzo MR et al. Lessons from two human chromatin diseases, ICF syndrome and Rett syndrome. Int. J BiochemCell Biol. 2009 ; 41:117-26.
53. Hagleitner MM,et al. Clinical spectrum of immunodeficiency, centromeric instability and facial dysmorphism (ICFsyndrome). J Med Genet. 2008 ;45: :93-9.
54. Lavin MF. Ataxia-telangiectasia: from a rare disorder to a paradigm for cell signalling and cancer. Nat Rev Mol Cell Biol.2008 ;9:759-69.
55. Gatti R. Ataxia-Telangiectasia. 1999 Mar 19 [Updated 2010 Mar 11]. In: Pagon RA, Bird TD, Dolan CR, et al., editors.GeneReviews™ [Internet]. Seattle (WA): University of Washington, Seattle; 1993-. source: http://www.ncbi.nlm.nih.gov/books/NBK26468/
56. Micol R, et al. CEREDIH Network Investigators. Morbidity and mortality from ataxia-telangiectasia are associated withATM genotype. J Allergy Clin Immunol. 2011 ;128:382-9.e1.
57. Concannon P, Gatti R. Nijmegen Breakage Syndrome. 1999 May 17 [Updated 2011 Mar 1]. In: Pagon RA, Bird TD,Dolan CR, et al., editors. GeneReviews™ [Internet]. Seattle (WA): University of Washington, Seattle; 1993-. Source :http://www.ncbi.nlm.nih.gov/books/NBK1176/
58. Michałkiewicz J,et al. Abnormalities in the T and NK lymphocyte phenotype in patients with Nijmegen breakagesyndrome. Clin Exp Immunol 2003 ;134:482-90.
59. Buck D, et al. Cernunnos, a novel nonhomologous end-joining factor, is mutated in human immunodeficiency withmicrocephaly. Cell. 2006;124:287-99.
60. O'Driscoll M, et al. DNA ligase IV mutations identified in patients exhibiting developmental delay and immunodeficiency.Mol Cell. 2001; 8: 1175–85.
61. Van der Burg, M., et al. A new type of radiosensitive T-B-NK+ severe combined immunodeficiency caused by a LIG4mutation. J. Clin. Invest. 116: 137-145, 2006.
62. Waltes R, et al. Human RAD50 deficiency in a Nijmegen breakage syndrome-like disorder. Am J Hum Genet. 2009;84:605-16.
63. Uchisaka N, et al. Two brothers with ataxia-telangiectasia-like disorder with lung adenocarcinoma. J Pediatr. 2009 ;155:435-8.
64. Van Zelm MC, et al Replication history of B lymphocytes reveals homeostatic proliferation and extensive antigen-induced B cell expansion. J Exp Med. 2007 19;204:645-55.
65. Schauer U et al. IgG Subclass Concentrations in Certified Reference Material 470 and Reference Values for Childrenand Adults Determined with The Binding Site Reagents. Clinical Chemistry 2003 49:1924-1929
66. Freeman A F and Holland S M. The Hyper IgE Syndromes. Immunol Allergy Clin North Am. 2008 May ; 28(2): 277(14 pages).
67. Nowak-Wegrzyn A., Crawford T O., Winkelstein JA., Carson K A., and Lederman H M. Immunodeficiency and InfectionsIn Ataxia-Telangiectasia. J Pediatr 2004;144:505-11.

- 80 -
DEF
AU
TS G
ENET
IQU
ES D
E LA
REP
ON
SE IM
MU
NIT
AIR
ERé
fére
nces
04
• Janeway’s Immunobiology (2011) - 8e Edition - Kenneth M. Murphy, Paul Travers, Mark Walport - Ed GarlandScience.
• Human Molecular Genetics (2010) - Tom Strachan et Andrew Read - 4e Edition - Ed Garland Science.
• Primary Immunodeficiency Diseases : A molecular and Genetic Approach - Second Edition - Hans D. Ochs, I.Edvard Smith, Jennifer M. Puck - Ed Oxford university Press (2006).
• Biologie moléculaire et médecine - Jean-Claude Kaplan, Marc Delpech - Ed. Flammarion fms (2007).
• The Cell: A molecular Approach - 5e Ed. - Geoffrey M. Cooper et Robert E. Hausman 2009 - Ed. ASM Press andSinauer Associates.
Livres de référenceLivres de référence

LFB BIOMEDICAMENTSS.A. au capital de 150 000 000 Euros - 491 371 167 RCS EVRY
3, avenue des Tropiques - BP 305 - Les Ulis - 91958 Courtaboeuf Cedex - FranceTéléphone : +33 (0)1 69 82 70 10 - Fax : +33 (0)1 69 07 19 03
www.lfb.fr
65575 - AVRIL 2013 - SeeYouSoonOnTheMoon
Pour toute demande d'Information Médicale, contactez le 01 69 82 70 04





























