Embed Size (px)
Citation preview

OLYMPIADES DE LA CHIMIE 2010 -2011
« Chimie et eau »
RECUEIL DES ENONCES
DE TRAVAUX PRATIQUES
ET CORRIGES
ACADEMIE DE NANCY-METZ
Propriété exclusive de l’Association Olympiades de la Chimie en Lorraine
ENSIC -1 rue Grandville - BP 20451 - 54001 Nancy cedex
Contact : Alexandra Gigante email : [email protected] tel 03 83 17 51 11

SOMMAIRE
TP 1 : Etude d’une réaction d’estérification
TP 2 : Fonctionnement d’un adoucisseur d’eau domestique :
“Déminéralisation” d’une eau minérale
TP 3 : Mesures de la pollution organique d’une eau :
Oxydabilité au permanganate de potassium, DCO
TP 4 : Oxydation d’un alcool secondaire
TP 5 : Synthèse de l’hélianthine ou méthylorange
TP 6 : Dosage d’un effluent industriel

TP 1 : Etude d’une réaction d’estérification
TP 1 : Etude d’une réaction d’estérification Page 1
Académie de Nancy - Metz Olympiades de la chimie 2010-2011 « La chimie et l’eau »
TP 1 : Etude d’une réaction d’estérification
1 Introduction L’utilisation d’arômes artificiels est de nos jours l’une des pratiques les plus courantes dans les industries alimentaires et de la parfumerie. Tenter de recréer l’odeur et la saveur si subtiles d’un fruit ou d’une fleur demeure un défi de taille pour le chimiste. Un des procédés utilisés dans ce domaine est l’estérification de Fischer, qui consiste à synthétiser un composé organique nommé « ester » dégageant une odeur fruitée reconnaissable à partir d’un acide carboxylique et d’un alcool. Quelques exemples :
Ester Odeur résultante
éthanoate d’isoamyle banane
éthanoate d’hexyle poire
éthanoate de linalyle bergamote
éthanoate de phényléthyle rose
éthanoate de benzyle jasmin
butanoate d’éthyle ananas
butanoate d’isoamyle pomme
2 Les esters
2.1 Le groupe caractéristique des esters
Un ester est un composé organique qui possède le groupement caractéristique d’atomes suivant :
La formule générale des esters est :
où R désigne une chaîne carbonée ou un atome d’hydrogène et R’ désigne une chaîne carbonée.
C
O
O C
C
O
O R’
R

TP 1 : Etude d’une réaction d’estérification Page 2
Remarques : � Les esters sont présents dans les corps gras qui composent les huiles, les graisses, ainsi que dans les
cires. � Les esters permettent d’élaborer des savons, du glycérol et de nombreux polymères comme le plexiglas.
2.2 Nomenclature des esters
Le nom d’un ester comporte deux parties : � la première partie désigne la chaîne carbonée contenant l’atome de carbone du groupe caractéristique.
Elle dérive du nom de l’acide carboxylique correspondant dans lequel la terminaison « oïque » est remplacée par la terminaison « oate » ;
� la seconde partie désigne la chaîne carbonée liée à l’atome d’oxygène. C’est le groupe alkyle, avec la terminaison « yle ».
Dans le cas de chaîne carbonée ramifiée, les chaînes sont numérotées à partir du groupe caractéristique. Exemple :
i. Donner le nom des esters ci-dessous.
a) b) c)
3 Généralités sur l’estérification 3.1 Réaction de synthèse d’un ester : estérificatio n
Un ester est le produit de la réaction entre un alcool et un acide carboxylique. Cette réaction d’estérification a pour équation :
Remarque : L’eau n’est plus un solvant, mais un produit de la réaction.
3.2 Réaction étudiée
L’acide carboxylique est l’acide éthanoïque (appelé acide acétique).
ii. Donner sa formule semi-développée.
L’alcool est le 3-méthylbutan-1-ol (appelé alcool isoamylique).
iii. Donner sa formule semi-développée.
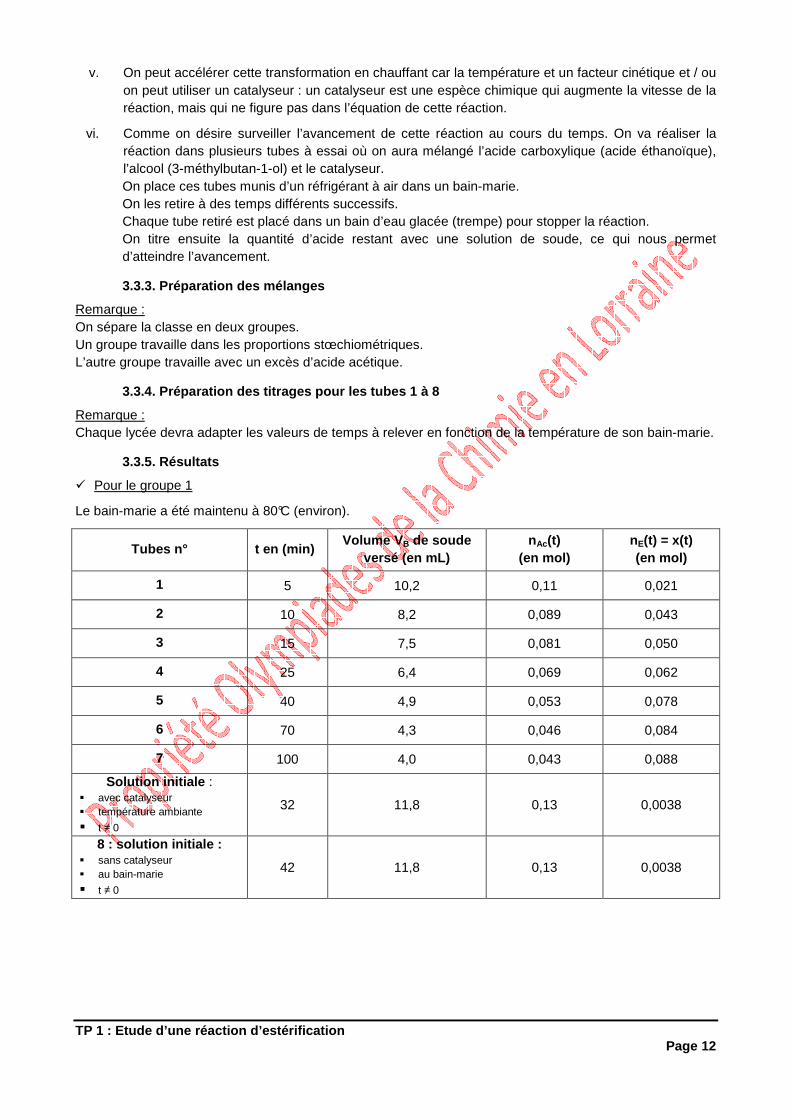
iv. Ecrire la réaction d’estérification correspondante.
C
O
O CH2
H
CH3
1 2
C
O
O CH3
CH2
H3C CH2
12
34
C
O
O CH
CH
H3C
12
3H3C
CH3
CH3
1
2
C
O
O H
R OHR’ C
O
O R’
R+ H2O= +

TP 1 : Etude d’une réaction d’estérification Page 3
3.3 Protocole expérimental
3.3.1 Quelques données
Formule
brute
Masse molaire
(en g.mol -1) Densité
Température d’ébullition
(en °C)
Solubilité dans l’eau
Acide éthanoïque C2H4O2 60 1,05 118 Grande
3-méthylbutan-1-ol C5H12O 88 0,81 132 Faible
Ethanoate de 3-méthylbutyle
C7H14O2 130 0,87 142 Très faible(1)
Cyclohexane C6H12 84 0,78 80,7 Très faible
Eau H2O 18 1,00 100
(1) La solubilité de l’ester dans l’eau salée est encore plus faible.
3.3.2 Réflexion préalable
La transformation étudiée est très lente. Dans les conditions ordinaires, elle peut prendre plusieurs mois pour parvenir à son avancement final.
v. Comment l’accélérer ?
vi. Comment réaliser le suivi de cette réaction au cours du temps ?
3.3.3 Préparation des mélanges
GROUPE 1
� Dans un bécher de 100 mL, verser V = 7,5 mL d’acide éthanoïque glacial à l’aide d’une burette graduée, puis ajouter V’ = 14,1 mL de 3-méthylbutan-1-ol avec une burette graduée.
GROUPE 2
� Dans un bécher de 100 mL, verser V = 15,0 mL d’acide éthanoïque glacial à l’aide d’une burette graduée, puis ajouter V’ = 14,1 mL de 3-méthylbutan-1-ol avec une burette graduée.
POUR LES DEUX GROUPES
� Agiter à l’aide d’une baguette en verre.
� Préparer le bain-marie bouillant.
� Numéroter 8 tubes à essais de 1 à 8 inclus.
� Verser dans le tube n°8 (étude de la réaction en l’ absence de catalyseur), 2,0 mL de mélange prélevé à la pipette munie d’une propipette.
� Ajouter au mélange restant dans le bécher 2 gouttes d’acide sulfurique à 2,5 mol.L-1, qui servira de catalyseur. Agiter à l’aide de la baguette en verre.
� Sans tarder, verser 2,0 mL de mélange ainsi obtenu dans chacun des tubes n°1 à 7.
� Boucher les 8 tubes numérotés de 1 à 8 avec des bouchons percés traversés par de fins tubes de verre bien secs (réfrigérant à air). Conserver le mélange loin de toute source de chaleur.
� Plonger les tubes munis des réfrigérants (tubes 1 à 8) dans le bain-marie bouillant en déclenchant le chronomètre.
� Relever la température du bain-marie et la maintenir constante tout au long de la manipulation.
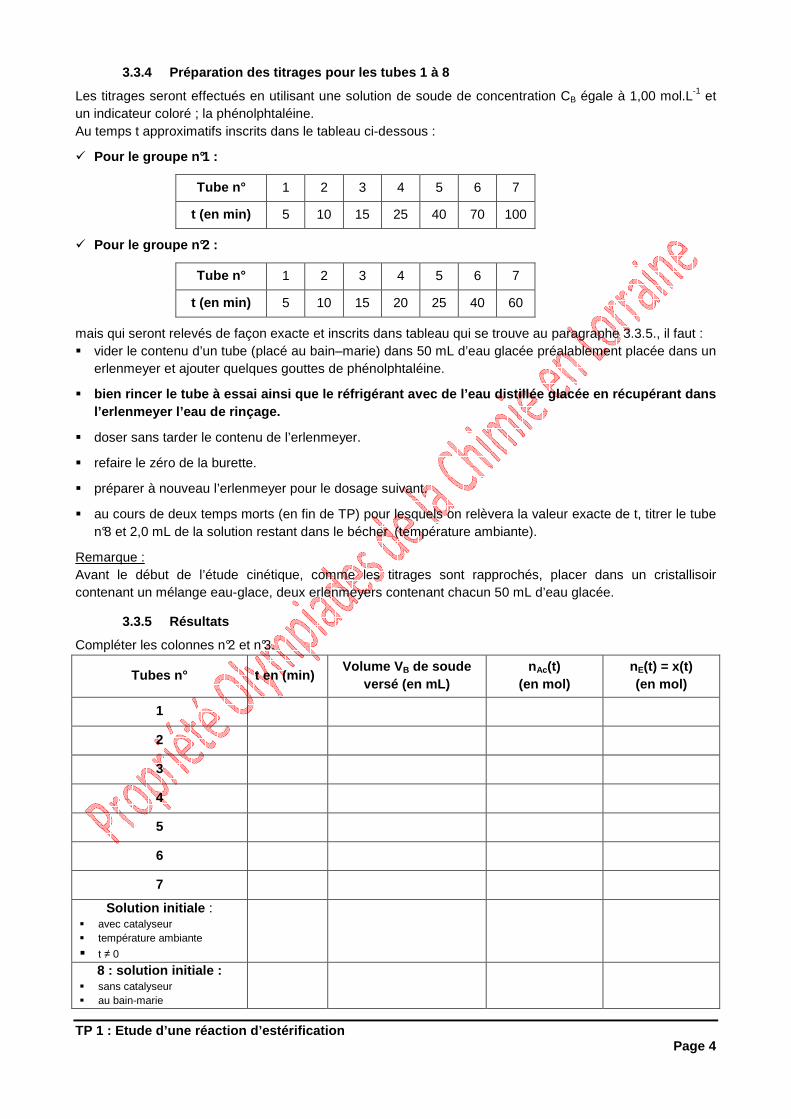
TP 1 : Etude d’une réaction d’estérification Page 4
3.3.4 Préparation des titrages pour les tubes 1 à 8
Les titrages seront effectués en utilisant une solution de soude de concentration CB égale à 1,00 mol.L-1 et un indicateur coloré ; la phénolphtaléine. Au temps t approximatifs inscrits dans le tableau ci-dessous :
� Pour le groupe n°1 :
Tube n° 1 2 3 4 5 6 7
t (en min) 5 10 15 25 40 70 100
� Pour le groupe n°2 :
Tube n° 1 2 3 4 5 6 7
t (en min) 5 10 15 20 25 40 60
mais qui seront relevés de façon exacte et inscrits dans tableau qui se trouve au paragraphe 3.3.5., il faut : � vider le contenu d’un tube (placé au bain–marie) dans 50 mL d’eau glacée préalablement placée dans un
erlenmeyer et ajouter quelques gouttes de phénolphtaléine.
� bien rincer le tube à essai ainsi que le réfrigéran t avec de l’eau distillée glacée en récupérant dans l’erlenmeyer l’eau de rinçage.
� doser sans tarder le contenu de l’erlenmeyer.
� refaire le zéro de la burette.
� préparer à nouveau l’erlenmeyer pour le dosage suivant.
� au cours de deux temps morts (en fin de TP) pour lesquels on relèvera la valeur exacte de t, titrer le tube n°8 et 2,0 mL de la solution restant dans le bécher (température ambiante).
Remarque : Avant le début de l’étude cinétique, comme les titrages sont rapprochés, placer dans un cristallisoir contenant un mélange eau-glace, deux erlenmeyers contenant chacun 50 mL d’eau glacée.
3.3.5 Résultats
Compléter les colonnes n°2 et n°3.
Tubes n° t en (min) Volume V B de soude
versé (en mL) nAc(t)
(en mol) nE(t) = x(t) (en mol)
1
2
3
4
5
6
7
Solution initiale : � avec catalyseur � température ambiante
� t ≠ 0
8 : solution initiale : � sans catalyseur � au bain-marie

TP 1 : Etude d’une réaction d’estérification Page 5
� t ≠ 0
3.3.6 Exploitation des résultats
vii. Calculer, pour le mélange initial, les quantités de matière initiales d’acide éthanoïque, notée nAc0, de 3-méthylbutan-1-ol, notée nAl0.
viii. Le mélange est-il dans les proportions stœchiométriques ?
ix. Faire le schéma légendé du dispositif de titrage.
x. Quelles sont les caractéristiques d’une transformation de titrage ?
xi. Quelles espèces titre-t-on lors du titrage ?
xii. Evaluer le volume de soude nécessaire pour titrer l’acide sulfurique présent dans une prise d’essai. On considérera que le volume d’une goutte représente 0,05 mL. Que peut-on en conclure ?
Rappel : l’acide sulfurique est un diacide, il est capable de libérer deux ions H+.
xiii. En déduire l’équation chimique de la réaction support du titrage.
xiv. Exprimer en fonction de VB, la quantité de matière d’acide éthanoïque à tout instant dans le mélange initial, notée nAc(t). Compléter la colonne n°4 du tableau qui se tro uve au paragraphe 3.3.5.
xv. En déduire l’expression de l’avancement x puis celle de la quantité de matière d’ester dans le mélange initial, notée nE(t) (on s’aidera d’un tableau d’avancement). Compléter la colonne n°5 du tableau qui se trouve au paragraphe 3.3.5
xvi. Tracer les courbes représentant nAc et nE en fonction du temps avec les points 1 à 7, puis prolonger en extrapolant. Conclure.
xvii. Placer sur le graphe les points correspondants à la réaction sans catalyseur (tube n°8) et à la réaction à température ambiante. Conclure.
xviii. Evaluer graphiquement l’avancement de la réaction à l’état final et en déduire le taux d’avancement final de la réaction.
xix. En déduire le rendement de cette transformation ?
xx. Comment évolue la vitesse au cours du temps ?
xxi. Définir la constante d’équilibre de cette réaction et la calculer.

TP 1 : Etude d’une réaction d’estérification Page 6
4 Autre façon d’améliorer le rendement de la réacti on d’estérification
Pour éviter que la réaction d’hydrolyse ne se produise, il faut retirer du milieu réactionnel au fur et à mesure de sa production soit l’ester, soit l’eau. On se propose de synthétiser l’éthanoate de 3-méthylbutyle couramment appelé acétate d’isoamyle (arôme de banane) à l’aide d’un appareil de Dean-Stark (schéma du montage ci-contre) permettant d’éliminer l’eau du mélange réactionnel.
4.1 Protocole expérimental
4.1.1 Etude du montage
Dans le mélange réactionnel initial, on introduit en plus des réactifs usuels (acide carboxylique, alcool et catalyseur), un solvant organique, ici le cyclohexane. Le chauffage du milieu réactionnel entraîne la formation d’un composé mixte (eau-cyclohexane), de température d’ébullition inférieure à celles de tous les autres constituants du mélange. Ce composé mixte monte dans l’appareil de Dean-Stark puis dans le réfrigérant à boules, où il se condense et se retrouve dans le tube vertical gradué préalablement rempli avec du cyclohexane. La séparation eau-cyclohexane se produit : comme l’eau est plus dense que le cyclohexane, elle tombe au fond du tube vertical. Le cyclohexane retourne dans le ballon.
4.1.2 Mode opératoire
4.1.2.1 Synthèse de l’ester avec un montage de Dean -Stark
Dans le ballon, verser 20,0 mL d’acide éthanoïque glacial à l’aide d’une pipette jaugée munie d’une propipette, 38,0 mL de 3-méthylbutan-1-ol avec une burette graduée et 30 mL de cyclohexane avec une éprouvette.
xxii. Calculer les quantités de matière initiales d’acide et d’alcool, notées respectivement nAC0 et nAl0. Est-on dans les proportions stœchiométriques ?
• Ajouter quelques gouttes d’acide sulfurique concentré, qui joue le rôle de catalyseur.
• Ajouter deux grains de pierre ponce.
• Remplir l’appareil de Dean-Stark de cyclohexane de sorte que le niveau affleure le tube coudé descendant.
• Mettre en place le réfrigérant et ouvrir le robinet d’eau.
• Continuer la réaction jusqu’à ce que le volume du liquide dans le tube gradué n’évolue plus.
• Le chauffage étant arrêté, descendre de quelques centimètres le chauffe ballon. Laisser refroidir le ballon à l’air quelques minutes en laissant la circulation d’eau dans le réfrigérant.

TP 1 : Etude d’une réaction d’estérification Page 7
• Evacuer la phase aqueuse du décanteur, après avoir noté son volume.
On trouve Veau = …………… mL xxiii. Calculer la quantité de matière d’eau obtenue et en déduire le rendement de cette transformation
(on pourra s’aider d’un tableau d’avancement).
xxiv. Comparer ce rendement avec l’expérience précédente ? Conclure.
4.1.2.2 Extraction de l’ester formé
Relargage :
• Retirer le ballon du montage.
• Mettre le mélange (sans les grains de pierre ponce) dans une ampoule à décanter.
• Ajouter environ 60 mL d’une solution saturée de chlorure de sodium préalablement refroidie dans un bain eau + glace pilée.
xxv. Pourquoi utilise-t-on une solution saturée de chlorure de sodium ?
• Boucher l’ampoule à décanter et agiter en tenant le bouchon. Le robinet est fréquemment ouvert pour éviter les surpressions et dirigé vers le haut. Laisser décanter.
• Soutirer la phase aqueuse puis laisser la phase organique dans l’ampoule à décanter.
Lavage de la phase organique :
• Laver la phase organique en ajoutant lentement et en agitant constamment l’ampoule environ 60 mL d’une solution d’hydrogénocarbonate de sodium saturée, préalablement refroidie dans un bain eau + glace-pilée.
Attention au dégagement gazeux.
xxvi. Pourquoi les lavages de la phase organique doivent-ils se faire avec des solutions préalablement refroidies dans le bain eau + glace pilée ?
xxvii. Quel est le rôle des lavages avec la solution d’hydrogénocarbonate de sodium ? Quelle est la nature du gaz qui se dégage ?
xxviii. Ecrire l’équation de la réaction associée à la transformation réalisée. Préciser la nature des couples acido-basiques qui participent à la réaction.
• Décanter et recueillir la phase organique.
• Recommencer une fois cette dernière opération.
• Pour terminer, laver la phase organique avec environ 60 mL d’eau refroidie. Décanter et recueillir la phase organique.
• Récupérer la phase organique dans un erlenmeyer bouché.
Séchage :
• Sécher la phase organique avec du sulfate de magnésium anhydre.
• Filtrer sur coton la solution précédente dans un erlenmeyer sec.
5 Conclusion : contrôle d’une réaction chimique Le contrôle vise à améliorer le rendement et la vitesse de la réaction . Pour augmenter le rendement d’une réaction d’estérification, on peut :

TP 1 : Etude d’une réaction d’estérification Page 8
� introduire en excès un des réactifs � éliminer un des produits au fur et à mesure de sa formation.
Pour augmenter la vitesse d’une réaction d’estérification, on peut :
� élever la température du mélange réactionnel � ajouter un catalyseur au milieu réactionnel.

TP 1 : Etude d’une réaction d’estérification Page 9
Académie de Nancy - Metz Olympiades de la chimie 2010-2011 « La chimie et l’eau »
TP 1 : Etude d’une réaction d’estérification LISTE MATERIELS ET PRODUITS
LISTE PRODUITS (en rouge non disponible dans les lycées a priori) Quantités par poste
� acide éthanoïque glacial : 40mL
� 3-méthylbutan-1-ol : 55mL
� acide sulfurique à 2,5 mol.L-1: quelques gouttes.
� phénolphtaléine
� soude à 1,00 mol.L-1 : 200mL
� cyclohexane : 50mL
� solution saturée de chlorure de sodium : 100mL
� solution d’hydrogénocarbonate de sodium saturée : 150mL
� sulfate de magnésium anhydre
LISTE MATERIEL ESTERIFICATION ET TITRAGES
Sous la hotte : • flacon avec de l’acide éthanoïque glacial avec une burette graduée de 25 mL, un bécher pour le
prélèvement et un bécher pour mettre sous la burette : 20 mL par poste. • flacon avec du 3-méthylbutan-1-ol avec une burette graduée de 25 mL, un bécher pour le prélèvement
et un bécher pour mettre sous la burette : 15 mL par poste. • flacon avec de l’acide sulfurique à 2,5 mol.L-1 avec pipette simple et un bécher pour le prélèvement :
quelques gouttes par poste. Au bureau : • 1 tige aimantée pour récupérer les barreaux aimantés. Sur chaque paillasse : • gants et lunettes. • 1 bécher de 100 mL en verre pour faire le mélange initial. • 1 agitateur en verre. • dispositif pour bain-marie : nous avons fait le bain-marie en utilisant une plaque chauffante avec un
bécher de 500 mL. Nous avons mis du coton pour maintenir les tubes à essais dans le bain-marie. • 1 thermomètre pour relever la température du bain-marie. • 1 chronomètre. • 8 tubes à essais avec réfrigérant à air dans porte tube, numérotés de 1 à 8. • 1 crayon pour écrire sur le verre. • 1 pipette jaugée de 2,0 mL ou pipette graduée de 10 mL (test fait avec la pipette graduée) + propipette. • 2 erlenmeyers de 100 mL.

TP 1 : Etude d’une réaction d’estérification Page 10
• 1 burette de 25 mL. • 1 agitateur magnétique avec 2 barreaux aimantés. • 1 flacon compte-gouttes avec de la phénolphtaléine. • 1 fiole contenant 200 mL (par poste)d e soude à 1,00 mol.L-1. • 1 bécher poubelle. • 2 pissettes d’eau distillée glacée (50 mL × 9 dosages) dans un cristallisoir avec eau-glace. • 1 éprouvette graduée de 50 mL. ESTERIFICATION EN UTILISANT UN MONTAGE DE DEAN-STAR K
Sous la hotte : • flacon avec de l’acide éthanoïque glacial avec pipette jaugée de 20 mL + propipette, un bécher de
prélèvement : 20 mL par poste. • flacon avec du 3-méthylbutan-1-ol avec une burette graduée de 50 mL, un bécher pour le prélèvement
et un bécher pour mettre sous la burette : 40 mL par poste. • flacon avec de l’acide sulfurique concentré avec pipette simple et un bécher pour le prélèvement :
quelques gouttes par poste. Au bureau : • grains de pierre ponce. • 1 spatule. Sur chaque paillasse : • gants et lunettes. • 1 chauffe-ballon. • 1 ballon rodé. • 1 valet en liège. • 1 réfrigérant à boules. • 1 montage de Dean Stark • 1 support élévateur. • des clips. • graisse pour rodage. • 1 éprouvette de 50 mL pour mettre sous le tube gradué. • 1 flacon avec 50 mL de cyclohexane par poste. • 1 éprouvette de 50 mL pour prélever le cyclohexane. • 1 bécher de 50 mL pour contenir le cyclohexane. • 1entonnoir à liquide. • 1 ampoule à décanter sur support. • 1 cristallisoir contenant un mélange eau-glace. • 1 flacon contenant 100 mL (par poste) d’une solution saturée de chlorure de sodium refroidie. • 1 flacon contenant 150 mL (par poste) d’une solution d’hydrogénocarbonate de sodium saturée refroidie. • 1 pissette d’eau distillée glacée. • 1 éprouvette de 100 mL. • 1 erlenmeyer bouché de 100 mL pour récupérer la phase organique. • 1 erlenmeyer pour récupérer la phase aqueuse de 250 mL. • 1 flacon contenant du sulfate de magnésium anhydre. • 1 spatule. • 1 entonnoir à liquide sur support. • 1 erlenmeyer sec bouché de 100 mL pour récupérer la phase organique. • coton pour la filtration sur coton.

TP 1 : Etude d’une réaction d’estérification Page 11
Académie de Nancy - Metz Olympiades de la chimie 2010-2011 « La chimie et l’eau »
TP 1 : Etude d’une réaction d’estérification REPONSES AUX QUESTIONS
3.1 Le groupe caractéristique des esters i. Nom des esters
a) : méthanoate d’éthyle
b) : butanoate de méthyle
c) : 2-méthylpropanoate de 1-méthyléthyle.
3.2 Réaction étudiée
L’acide carboxylique est l’acide éthanoïque (appelé acide acétique).
ii. Donner sa formule semi-développée.
L’alcool est le 3-méthylbutan-1-ol (appelé alcool isoamylique).
iii. Donner sa formule semi-développée.
iv. Ecrire la réaction d’estérification correspondante.
Remarque : L’acétate d’isoamyle a une odeur d’arôme de banane.
3.3.2. Réflexion préalable
La transformation étudiée est très lente. Dans les conditions ordinaires, elle peut prendre plusieurs mois pour parvenir à son avancement final.
C
O
OH
CH3
CH3 CH CH2 CH2 OH
CH3
H3C
CH3
OH+
HO
O
CH3 H3C
CH3
O=
O
CH3
+ H2O
3-méthylbutan-1-olou alcool isoamylique
acide éthanoïque ou acide acétique
éthanoate de 3-méthylbutyleou acétate d’isoamyle
eau
TP 1 : Etude d’une réaction d’estérification Page 12
v. On peut accélérer cette transformation en chauffant car la température et un facteur cinétique et / ou on peut utiliser un catalyseur : un catalyseur est une espèce chimique qui augmente la vitesse de la réaction, mais qui ne figure pas dans l’équation de cette réaction.
vi. Comme on désire surveiller l’avancement de cette réaction au cours du temps. On va réaliser la réaction dans plusieurs tubes à essai où on aura mélangé l’acide carboxylique (acide éthanoïque), l’alcool (3-méthylbutan-1-ol) et le catalyseur. On place ces tubes munis d’un réfrigérant à air dans un bain-marie. On les retire à des temps différents successifs. Chaque tube retiré est placé dans un bain d’eau glacée (trempe) pour stopper la réaction. On titre ensuite la quantité d’acide restant avec une solution de soude, ce qui nous permet d’atteindre l’avancement.
3.3.3. Préparation des mélanges
Remarque : On sépare la classe en deux groupes. Un groupe travaille dans les proportions stœchiométriques. L’autre groupe travaille avec un excès d’acide acétique.
3.3.4. Préparation des titrages pour les tubes 1 à 8
Remarque : Chaque lycée devra adapter les valeurs de temps à relever en fonction de la température de son bain-marie.
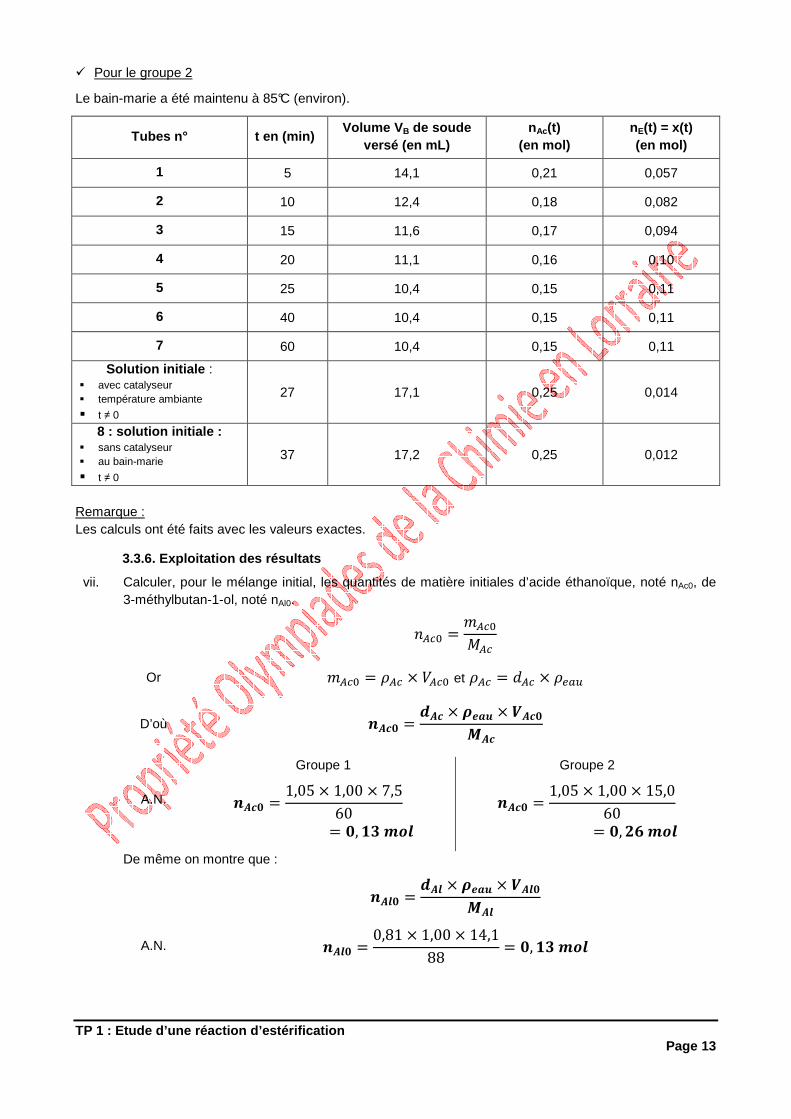
3.3.5. Résultats
� Pour le groupe 1
Le bain-marie a été maintenu à 80°C (environ).
Tubes n° t en (min) Volume V B de soude
versé (en mL) nAc(t)
(en mol) nE(t) = x(t) (en mol)
1 5 10,2 0,11 0,021
2 10 8,2 0,089 0,043
3 15 7,5 0,081 0,050
4 25 6,4 0,069 0,062
5 40 4,9 0,053 0,078
6 70 4,3 0,046 0,084
7 100 4,0 0,043 0,088
Solution initiale : � avec catalyseur � température ambiante
� t ≠ 0
32 11,8 0,13 0,0038
8 : solution initiale : � sans catalyseur � au bain-marie
� t ≠ 0
42 11,8 0,13 0,0038
TP 1 : Etude d’une réaction d’estérification Page 13
� Pour le groupe 2
Le bain-marie a été maintenu à 85°C (environ).
Tubes n° t en (min) Volume V B de soude
versé (en mL) nAc(t)
(en mol) nE(t) = x(t) (en mol)
1 5 14,1 0,21 0,057
2 10 12,4 0,18 0,082
3 15 11,6 0,17 0,094
4 20 11,1 0,16 0,10
5 25 10,4 0,15 0,11
6 40 10,4 0,15 0,11
7 60 10,4 0,15 0,11
Solution initiale : � avec catalyseur � température ambiante
� t ≠ 0
27 17,1 0,25 0,014
8 : solution initiale : � sans catalyseur � au bain-marie
� t ≠ 0
37 17,2 0,25 0,012
Remarque : Les calculs ont été faits avec les valeurs exactes.
3.3.6. Exploitation des résultats
vii. Calculer, pour le mélange initial, les quantités de matière initiales d’acide éthanoïque, noté nAc0, de 3-méthylbutan-1-ol, noté nAl0.
���� �����
���
Or ���� � ��� ��� et ��� � ��� �� �
D’où ���� ���� ���� ����
���
A.N.
Groupe 1
���� �1,05 1,00 7,5
60� �, ! #$%
Groupe 2
���� �1,05 1,00 15,0
60� �, &' #$%
De même on montre que :
��%� ���% ���� ��%�
��%
A.N. ��%� �0,81 1,00 14,1
88� �, ! #$%

TP 1 : Etude d’une réaction d’estérification Page 14
viii. Le mélange est-il dans les proportions stœchiométriques ?
D’après l’équation écrite au paragraphe III.3., on sait qu’une mole d’acide éthanoïque réagit avec une mole de 3-méthylbutan-1-ol.
� Pour le groupe n°1 : On vient de montrer que nAc0 = nAl0. Le mélange initial est donc dans les proportions stœchiométriques .
� Pour le groupe n°2 : On vient de montrer que nAc0 > nAl0. Le mélange initial n’est donc pas dans les proportions stœchiométriques . L’alcool est le réactif limitant .
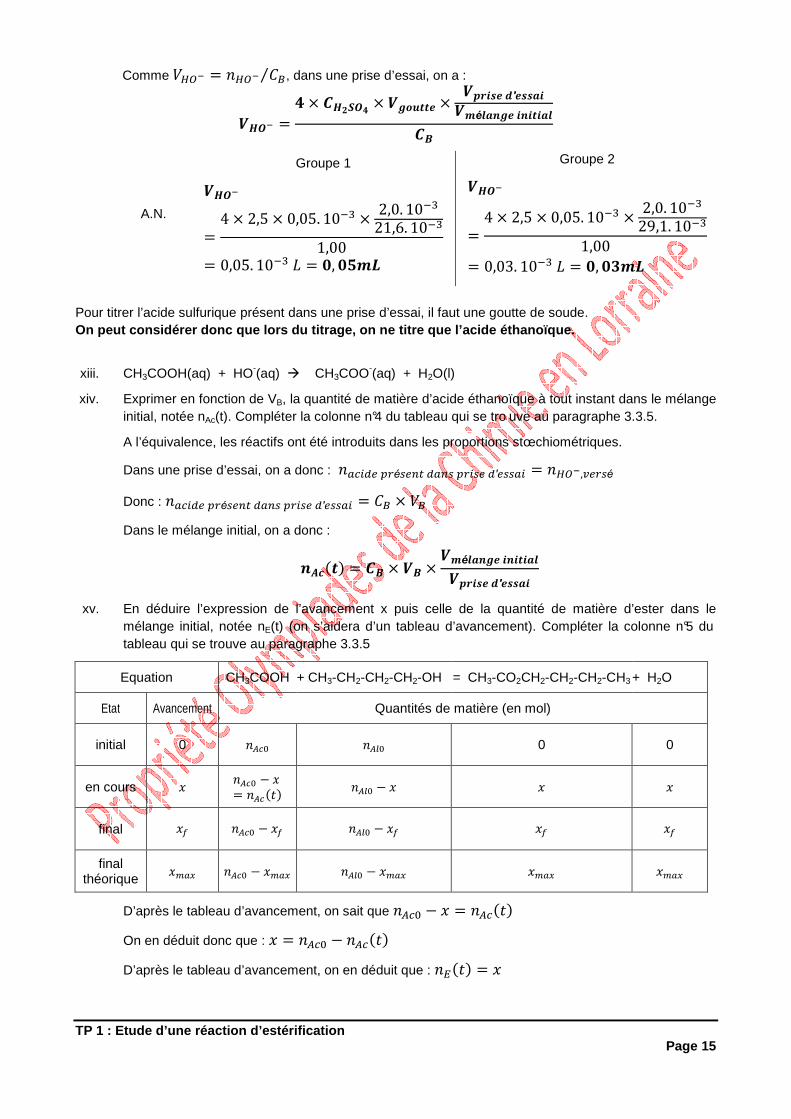
ix. Faire le schéma légendé du dispositif de titrage.
x. Quelles sont les caractéristiques d’une transformation de titrage ?
Une transformation de titrage est rapide, totale et spécifique de l’espèce à titrer.
xi. Quelles espèces titre-t-on lors du titrage ?
On titre l’acide éthanoïque restant et éventuellement l’acide sulfurique (catalyseur).
xii. Evaluer le volume de soude nécessaire pour titrer l’acide sulfurique présent dans une prise d’essai. On considérera que le volume d’une goutte est 0,05 mL. Que peut-on en conclure ?
Les ions H+ réagissent avec les ions HO- selon la réaction suivante :
H+(aq) + HO-(aq) → H2O(l)
A l’équivalence, les réactifs ont été introduits dans les proportions stœchiométriques.
Dans une prise d’essai, on a donc �*+,,-./01�,. � �*23,4�/5é.
Or �*+,,-./01�,. � 2 �*7829
Mais on sait que dans la prise d’essai :
�*7829� :*7829
2 ;0�..� </,5� 1′�55 ,
=é> -;� ,-,., >
On a donc
�*+ � 4 :*7829 ;0�..�
</,5� 1′�55 ,
=é> -;� ,-,., >
Burette graduée contenant une solution de soude de concentration
CB = 1,00 mol.L –1
Agitateur magnétique
Erlenmeyer contenant 2,0 mL du mélange réactionnel + 50 mL d’eau glacée + quelques gouttes de phénolphtaléine
TP 1 : Etude d’une réaction d’estérification Page 15
Comme *23 � �*23 :?⁄ , dans une prise d’essai, on a :
�AB3 �
C DA&EBC �F$�GG�
�HIJK� �′�KK�J
�#é%��F� J�JGJ�%
DL
A.N.
Groupe 1
�AB3
�4 2,5 0,05. 10NO
2,0. 10NO
21,6. 10NO
1,00� 0,05. 10NO P � �, �Q#R
Groupe 2
�AB3
�4 2,5 0,05. 10NO
2,0. 10NO
29,1. 10NO
1,00
� 0,03. 10NO P � �, �!#R
Pour titrer l’acide sulfurique présent dans une prise d’essai, il faut une goutte de soude. On peut considérer donc que lors du titrage, on ne titre que l’acide éthanoïque.
xiii. CH3COOH(aq) + HO-(aq) � CH3COO-(aq) + H2O(l)
xiv. Exprimer en fonction de VB, la quantité de matière d’acide éthanoïque à tout instant dans le mélange initial, notée nAc(t). Compléter la colonne n°4 du tableau qui se tro uve au paragraphe 3.3.5.
A l’équivalence, les réactifs ont été introduits dans les proportions stœchiométriques.
Dans une prise d’essai, on a donc : � �,1� </é5�-. 1 -5 </,5� 1′�55 , � �*23,4�/5é
Donc : � �,1� </é5�-. 1 -5 </,5� 1′�55 , � :? ?
Dans le mélange initial, on a donc :
���UGV � DL �L �#é%��F� J�JGJ�%
�HIJK� �′�KK�J
xv. En déduire l’expression de l’avancement x puis celle de la quantité de matière d’ester dans le mélange initial, notée nE(t) (on s’aidera d’un tableau d’avancement). Compléter la colonne n°5 du tableau qui se trouve au paragraphe 3.3.5
Equation CH3COOH + CH3-CH2-CH2-CH2-OH = CH3-CO2CH2-CH2-CH2-CH3 + H2O
Etat Avancement Quantités de matière (en mol)
initial 0 ���� ��>� 0 0
en cours W ���� X W
� ���UYV ��>� X W W W
final WZ ���� X WZ ��>� X WZ WZ WZ
final théorique
W= [ ���� X W= [ ��>� X W= [ W= [ W= [
D’après le tableau d’avancement, on sait que ���� X W � ���UYV
On en déduit donc que : W � ���� X ���UYV
D’après le tableau d’avancement, on en déduit que : �\UYV � W
TP 1 : Etude d’une réaction d’estérification Page 16
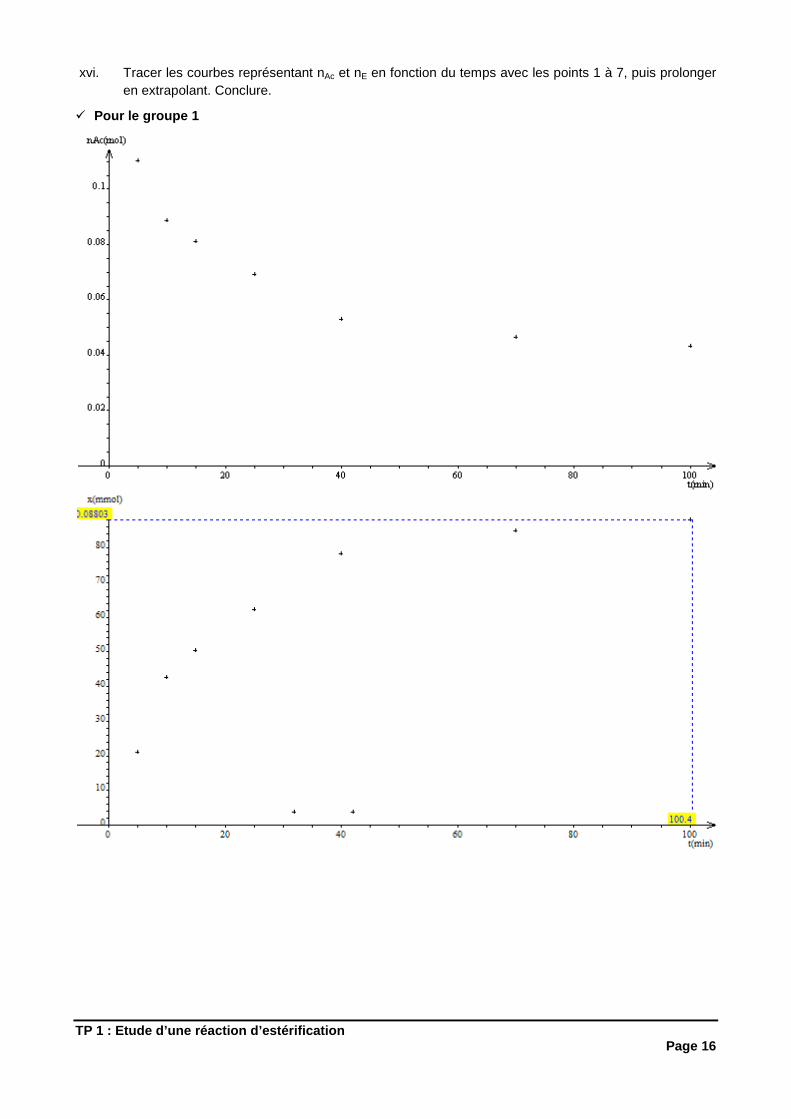
xvi. Tracer les courbes représentant nAc et nE en fonction du temps avec les points 1 à 7, puis prolonger en extrapolant. Conclure.
� Pour le groupe 1
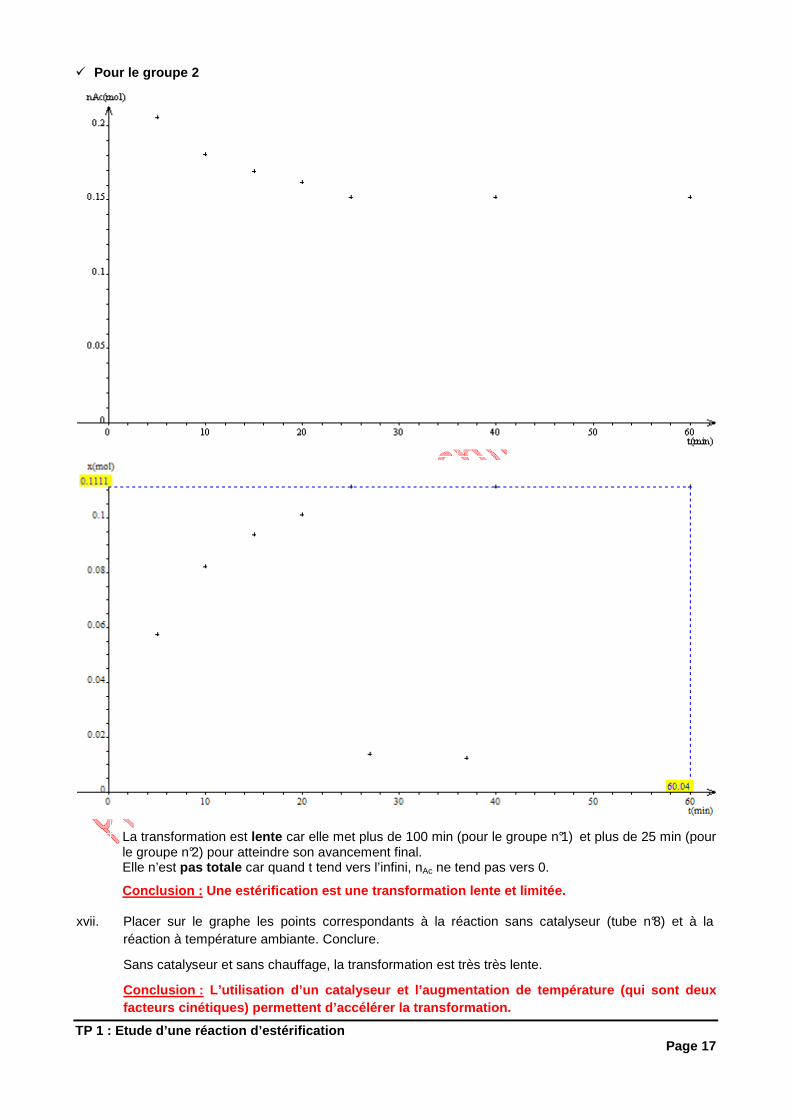
TP 1 : Etude d’une réaction d’estérification Page 17
� Pour le groupe 2
La transformation est lente car elle met plus de 100 min (pour le groupe n°1) et plus de 25 min (pour le groupe n°2) pour atteindre son avancement final. Elle n’est pas totale car quand t tend vers l’infini, nAc ne tend pas vers 0.
Conclusion : Une estérification est une transformation lente et limitée.
xvii. Placer sur le graphe les points correspondants à la réaction sans catalyseur (tube n°8) et à la réaction à température ambiante. Conclure.
Sans catalyseur et sans chauffage, la transformation est très très lente.
Conclusion : L’utilisation d’un catalyseur et l’augmentation de température (qui sont deux facteurs cinétiques) permettent d’accélérer la tran sformation.

TP 1 : Etude d’une réaction d’estérification Page 18
xviii. Evaluer graphiquement l’avancement de la réaction à l’état final et en déduire le taux d’avancement final de la réaction.
D’après le graphe, on en déduit que :
� pour le groupe n°1 :
WZ � 0,088�]^ (valeur de l’asymptote horizontale).
� pour le groupe n°2 :
WZ � 0,11�]^ (valeur de l’asymptote horizontale).
Par définition, on sait que _ � `a `#�`⁄ .
Groupe 1
On est dans les proportions stoechiométriques.
On a donc W= [ � ���� � ��>� et
bc �WZ
����
Groupe 2
L’alcool est le réactif limitant.
On a donc W= [ � ��>� et
bd �WZ
��>�
A.N.
_ �0,088
0,13� 'e%
Remarque : Le calcul a été fait avec la valeur exacte de ����.
_& �0,11
0,13� gQ%
Remarque : Le calcul a été fait avec la valeur exacte de ��>�.
xix. En déduire le rendement de cette transformation ?
Par définition, on sait que
h ��UijYihV�[<é/,=�-. >�
�UijYihV.lé0/,m��
Or �UijYihV�[<é/,=�-. >� � WZ et �UijYihV.lé0/,m�� � W= [
On a donc h � Wn W�oW � b⁄ .
AN :
� Pour le groupe 1 : r 1 = 67 %
Conclusion : Le rendement dépend de la classe de l’alcool. A pa rtir d’un mélange équimolaire d’acide carboxylique et d’alcool, est de :
• environ 67 % pour un alcool primaire • 60 % pour un alcool secondaire • environ 6 % pour un alcool tertiaire
� Pour le groupe 2 : r 2 = 85 %
On remarque r2 > r1.
Conclusion : L’utilisation d’un réactif en excès (généralement le moins couteux) permet d’améliorer le rendement de la réaction d’estérific ation.

TP 1 : Etude d’une réaction d’estérification Page 19
xx. Comment évolue la vitesse au cours du temps ?
Par définition, on sait que pUYV �1
q
�W
�YUYV
Comme q est une constante, pUYV est proportionnel à : �W
�YUYV
�W
�YUYV
représente le coefficient directeur de la tangente à la courbe W � nUYV à l’instant t.
Ce coefficient directeur diminue au cours du temps car la tangente « s’aplatit ».
rUGV diminue au cours du temps.
xxi. Définir la constante d’équilibre de cette réaction et la calculer.
Par définition, on a
s �U�tV�u U����V�u
U���V�u U��%V�u
A.N.
Groupe 1
s �0,088d
U0,13 X 0,088Vd� C,
Groupe 2
s �0,11d
U0,26 X 0,11V U0,13 X 0,11V
� C, � Remarque :
Les calculs ont été faits avec la valeur exacte de ��>� et la valeur exacte de ����. Conclusion : Quelque soit la température et la concentration ini tiale des réactifs, la constante d’équilibre d’une réaction d’estérification vaut toujours 4. Remarques : De manière générale, à chaque équation, on associe une constante d’équilibre K qui ne dépend que de la température. Elle ne dépend pas de la concentration initiale des réactifs. Comme la réaction d’estérification est athermique, K ne dépend pas non plus de la température.
4.1.2.1.Synthèse de l’ester avec un montage de Dean -Stark
Remarques :
Il faut contrôler la température et ne pas chauffer trop fort car il se forme deux azéotropes :
- un azéotrope eau-cylohexane qui bout à 70 °C
- un azéotrope acide acétique - cyclohexane qui bout à 79 °C.
A la place du ballon monocol, vous pouvez mettre un ballon bicol avec un thermomètre pour surveiller la température.
xxii. Calculer les quantités de matière initiales d’acide et d’alcool, notées respectivement nAC0 et nAl0. Est-on dans les proportions stœchiométriques ?
���� ���� ���� ����
���
A.N. ���� �1,05 1,00 20
60� �, !Q#$%

TP 1 : Etude d’une réaction d’estérification Page 20
De même on montre que :
��%� ���% ���� ��%�
��%
A.N. ��%� �0,81 1,00 38
88� �, !Q #$%
D’après l’équation écrite au paragraphe 3.2., on sait qu’une mole d’acide éthanoïque réagit avec une mole de 3-méthylbutan-1-ol.
On vient de montrer que ���� � ��>� On est donc dans les proportions stœchiométriques .
On trouve Veau = 6,3 mL
xxiii. Calculer la quantité de matière d’eau obtenue et en déduire le rendement de cette transformation (on pourra s’aider d’un tableau d’avancement).
On sait que ���� ����� ����
����
A.N. ���� �1,00 6,3
18� �, !Q#$%
On a déjà montré que hO � bO � WZ W= [⁄ avec WZ � ��5.�/ � �� � et W= [ � ��>�
On a donc I! �����
��%�
A.N. I! �0,35
0,35� ��%
xxiv. Comparer ce rendement avec l’expérience précédente ? Conclure.
On remarque que r3 > r1
Conclusion : Lorsqu’on élimine l’eau au fur à mesure, on empêche le quotient de réaction (Q r) d’atteindre la valeur du quotient de réaction à l’équilibre (Q r,éq = K). Le système n’arrive jamais à l’équilibre et continu e d’évoluer dans le sens direct. Le système continuera d’évoluer dans le sens direct jusqu’à épuisement du réactif limitant.
4.1.2.2. Extraction de l’ester formé
Remarques :
Comme nous n’avons pas réussi à caractériser l’ester formé, vous n’êtes pas obligé de faire cette partie. Nous avons essayé de faire une chromatographie (éluant : acétate d’éthyle + cyclohexane), mais nous n’arrivons pas à révéler l’ester (diiode, UV, KMnO4 à 1% dans l’eau, vanilline, H2SO4 conc / méthanol). Nous avons essayé de faire une distillation, mais impossible d’isoler l’ester formé. Grâce à l’odeur qui se dégage, on sait juste que l’on a bien fabriqué de l’acétate d’isoamyle (odeur de banane). Cette partie peut présenter l’avantage d’apprendre à l’élève à traiter une phase organique après une synthèse.

TP 1 : Etude d’une réaction d’estérification Page 21
xxv. Pourquoi utilise-t-on une solution saturée de chlorure de sodium ?
Cette opération, appelée relargage, permet de limiter la solubilité de l’ester dans l’eau. La solubilité de l’ester est plus faible dans de l’eau salée que dans de l’eau pure. D’autre part, la séparation de la phase organique et de la phase aqueuse est meilleure puisque les densités des deux phases sont bien différentes.
xxvi. Pourquoi les lavages de la phase organique doivent-ils se faire avec des solutions préalablement refroidies dans le bain eau + glace pilée ?
En diminuant la température des solutions de lavage, on diminue la solubilité de l’ester dans l’eau. De ce fait, les pertes en ester sont diminuées.
xxvii. Quel est le rôle des lavages avec la solution d’hydrogénocarbonate de sodium ? Quelle est la nature du gaz qui se dégage ?
Les lavages avec la solution d’hydrogénocarbonate de sodium permettent d’éliminer l’excès d’acide éthanoïque. Le gaz obtenu est du dioxyde de carbone.
xxviii. Ecrire l’équation de la réaction associée à la transformation réalisée. Préciser la nature des couples acido-basiques qui participent à la réaction.
CH3COOH(aq) + HCO3-(aq) = CH3COO-(aq) + CO2, H2O
Les deux couples acido-basiques mis en jeu sont : CH3COOH(aq) / CH3COO-(aq) et CO2 ,H2O / HCO3
-(aq).

TP 2 : Fonctionnement d’un adoucisseur d’eau domestique
“Déminéralisation” d’une eau minérale »

TP 2 : Fonctionnement d’un adoucisseur d’eau domest ique “Déminéralisation” d’une eau minérale Page 1
Académie de Nancy - Metz Olympiades de la chimie 2010-2011 « La chimie et l’eau »
TP 2 : Fonctionnement d’un adoucisseur d’eau domestique “Déminéralisation” d’une eau minérale
1 Fonctionnement d’un adoucisseur d’eau domestique 1.1 Pourquoi adoucir l’eau ?
Une eau dure est une eau riche en ions calcium (Ca 2+) et / ou magnésium (Mg 2+). Elle ne présente aucun danger pour la santé et peut donc être consommée en tant qu’eau de boisson Mais elle peut être à l’origine de certains inconvénients tels que l’entartrage (dépôt de carbonate de calcium CaCO3 ou de carbonate de magnésium MgCO3) des appareils dans lesquels l’eau est chauffée (lave-linge, lave-vaisselle…) ou de traces sur des surfaces lavées (baignoires, lavabos, robinetterie). Utilisée lors de la toilette, elle peut être responsable d’une certaine sécheresse de la peau et des cheveux. D’autre part, le savon donne, en présence d’une eau dure, un précipité de carboxylate de calcium (R-COO)2Ca et/ou de magnésium (R-COO)2Mg, donc son pouvoir détergent sera réduit et le dépôt de ces précipités sur les textiles lors de la lessive rend le linge rêche. La dureté d’une eau se mesure en degré hydrotimétrique (°TH).
1.2 Les résines échangeuses d’ions
1.2.1 Constitution
De nombreux adoucisseurs contiennent des résines échangeuses d’ions , et plus précisément des résines échangeuses de cations (ou cationiques ). Une résine échangeuse d’ions est un solide insoluble, qui, au contact d’une solution, peut échanger les ions qu’il contient avec d’autres ions de même signe provenant de la solution. Cette propriété d’échanger les ions était reconnue depuis longtemps à des silico-aluminates naturels appelés « zéolithes » ; dans ces composés, des ions alcalins ou alcalino-terreux sont logés dans les interstices ou les cavités d’un réseau polymère anionique, et peuvent être échangés avec d’autres cations lors du contact avec une solution. Le développement des polymères synthétiques, stables aussi bien vis-à-vis des acides et des bases que des oxydants et des réducteurs, a suscité l’apparition d’échangeurs d’ions artificiels de nature organique. Ils sont constitués d’un réseau macromoléculaire sur lequel sont fixés un très grand nombre de groupements actifs ionisables ; ces groupements portant des charges électriques, retiennent à leur voisinage, par attraction électrostatique, les ions de charges antagonistes qui sont susceptibles d’être échangés. Les résines les plus courantes sont des résines de polymérisation : par exemple, une résine de type polystyrène est un copolymère tridimensionnel formé de styrène et de divinylbenzène (cas des résines Amberlite IR-120 et IRA-400).
La vitesse des échanges doit être aussi rapide que possible, la résine doit donc être finement divisée afin de présenter une grande surface de contact avec la solution ; c’est sous forme de perles très fines que la plupart des échangeurs d’ions sont généralement utilisés.

TP 2 : Fonctionnement d’un adoucisseur d’eau domest ique “Déminéralisation” d’une eau minérale Page 2
Le groupe ionique est introduit dans les résines en général après polymérisation, en substituant un atome de carbone du noyau benzénique.
On distingue les résines cationiques pouvant échanger des cations et les résines anioniques échangeant des anions. Dans les résines cationiques, le groupement actif est un anion de type sulfonate, phosphate ou carboxylate :
R-SO3- R-PO3
2- R-CO2- (R symbolise la résine)
Les résines anioniques sont constituées de groupements ammonium quaternaire : R-N(CH3)3+
Toutes ces résines sont commercialisées sous forme d’acide, de base ou de sel suivant le type.
Une des caractéristiques des résines échangeuses d’ions est la capacité d’échange : c’est le nombre d’équivalents-g d’ions fixés sur 1 kg de résine sèche (ce qui correspond au nombre de moles de groupes actifs).
1.2.2 Mécanisme de l’échange
Lorsqu’on plonge une résine gorgée d’eau dans une solution contenant des ions, ceux-ci traversent les mailles du réseau et diffusent jusqu’aux centres actifs ; par suite une quantité équivalente d’ions fixés initialement sur la résine passent dans la solution ; nous pouvons représenter ce phénomène réversible, dans le cas d’une résine cationique sous forme acide et d’une solution de chlorure de sodium (par exemple) par l’équation :
R-SO3-, H+ + Na+ = R-SO3
-, Na+ + H+
Résine Solution Résine Solution Soit: H+
R + Na+S = Na+
R + H+S
(R = résine; S = solution) La constante d’équilibre associée à l’équation de cette réaction, à une température donnée, est:
K ��Na��� · �H���
�Na��� · �H���

TP 2 : Fonctionnement d’un adoucisseur d’eau domest ique “Déminéralisation” d’une eau minérale Page 3
K est une grandeur caractéristique de la réaction, dépendant de la température, sans dimension. Si K est élevée (supérieure à 104), on peut considérer que la réaction est totale, si bien que les ions Na+ sont remplacés quantitativement par des ions H+ dans la solution. L’équilibre dépend de l’affinité des ions pour la résine ; cette affinité augmente avec la charge (pour des solutions diluées) :
Na+ ≤ Ca2+ ≤ Al3+ ≤ Th4+
Par exemple les ions calcium déplaceront les ions sodium de la résine suivant :
2(R-SO3-, Na+) + Ca2+
S = (R-SO3-)2, Ca2+ + 2Na+
S
L’affinité augmente également avec le diamètre de l’ion solvaté :
Li+ ≤ H+ ≤ Na+ ≤ K+ , NH4+ ≤ Rb+ ≤ Cs+ ≤ Tl+ ≤Ag+
Donc Li+ mis à part, H+ est donc déplacé de la résine par tous les cations.
1.2.3 Régénération d’une résine
L’ion initialement présent dans la résine et qui a été échangé, peut être à nouveau fixé dans la résine en utilisant une solution concentrée de cet ion, ce qui a pour conséquence d’inverser le sens de l’équilibre d’échange. Par exemple, si l’on veut régénérer une résine cationique initialement sous forme sodique ayant fixé des ions calcium, on plongera la résine dans une solution aqueuse saturée en chlorure de sodium:
(R-SO3-)2, Ca2+ + 2Na+
S = 2(R-SO3-, Na+) + Ca2+
S
1.3 Les adoucisseurs d’eau domestiques
1.3.1 Principe de fonctionnement
L’adoucissement d’une eau consiste à remplacer ses cations Ca2+ et Mg2+ par des ions sodium Na+ :
2(R-SO3-, Na+) + Ca2+
S = (R-SO3-)2, Ca2+ + 2Na+
S
2(R-SO3-, Na+) + Mg2+
S = (R-SO3-)2, Mg2+ + 2Na+
S
Les ions sodium, contrairement aux ions calcium et magnésium, ne provoquent pas l’entartrage des canalisations.
1.3.2 Caractéristiques
Un adoucisseur domestique contient un compartiment contenant la résine, un bac à sel (chlorure de sodium sous forme de sel de cuisine), ainsi qu’un système de vannes régulant la circulation de l’eau.

TP 2 : Fonctionnement d’un adoucisseur d’eau domest ique “Déminéralisation” d’une eau minérale Page 4
Encombrement L’appareil et le bac à sel sont en général des cylindres de hauteur de l’ordre de 1 m, et de diamètre variant entre 20 et 50 cm selon la capacité Coût Le prix d’achat varie de 900 à 4000 euros, auquel il faut ajouter environ 200 euros pour l’installation par un installateur agréé, sans compter les aménagements éventuels du réseau de distribution d’eau de la maison que cette installation pourrait nécessiter. Il faut tenir compte d’une augmentation de la consommation moyenne d’eau après l’installation (environ 10%), due aux rinçages de la résine, ainsi que du coût du contrat annuel d’entretien.
Régénération de la résine Lorsque tous les ions sodium de la résine ont été échangés, la résine ne peut reprendre son rôle qu’après avoir été régénérée. La régénération de la résine par une solution aqueuse saturée en sel peut être programmée. Deux types de programmation existent : la régénération volumétrique se fait automatiquement dès qu’un volume d’eau maximal (préétabli lors de la programmation) est passé sur la résine ; la régénération chronométrique se fait automatiquement à intervalles de temps réguliers (entre 1 et 12 jours) sans tenir compte da la consommation en eau. Inconvénients liés à l’utilisation d’un adoucisseur domestique Le principal inconvénient de l’adoucisseur est l’enrichissement de l’eau en sodium surtout pour celle qui en contient déjà naturellement en quantité notable. De telles eaux adoucies peuvent nuire aux personnes souffrant d’hypertension, de problèmes cardiaques, aux femmes enceintes, aux nourrissons et à toutes les personnes soumises à un régime sans sel. D’autre part, une eau contenant trop d’ions sodium peut accroître la corrosion de la tuyauterie domestique. Il est donc fortement conseillé de n’adoucir que les circuits alimentant les équipements où l’eau est chauffée (salle de bains, chauffe-eau, lave-linge, lave-vaisselle), et de consommer en tant qu’eau de boisson de l’eau non adoucie. Il peut également apparaître des problèmes de prolifération bactérienne sur les résines. Un bon entretien de l’adoucisseur domestique (nettoyage et désinfection deux fois par an) permet de limiter la prolifération des bactéries. Bibliographie et sites Internet T.P de préparation aux Olympiades de la Chimie – académie de Nancy-Metz ( 2005-2006) Chimie inorganique et générale F.Brénon-Audat, F.Rafflegeau, D.Prévoteau Chimie Analytique Skoog, West, Holler Chimie Term S Hachette Encyclopédie Universalis BUP : TP résines IZBICKI Micheline http://www.sdea.fr http://www.cile.be http://www.ac-nancy-metz.fr/enseign/physique/CHIM/Chromato01/chromato1.htm
TP 2 : Fonctionnement d’un adoucisseur d’eau domest ique “Déminéralisation” d’une eau minérale Page 5
2 Déminéralisation” d’une eau minérale (Contrexévil le ou Hépar) sur résine échangeuse d’ions
Les eaux minérales sont riches en cations tels que Ca2+, Mg2+, Na+… Nous souhaitons adoucir une eau minérale, c’est-à-dire supprimer ces ions en les fixant sur une résine échangeuse de cations puis réaliser un dosage-acido-basique des ions H+
(aq) cédés par la résine.
2.1 Analyse de l’eau minérale
Vous disposez d’une eau minérale soit « Eau de Contrexéville », soit « Hépar » pour lesquelles les fabricants indiquent les compositions suivantes :
Contrexéville
(mg/L) Hépar (mg/L)
Calcium 486 549
Magnésium 84 119
Sodium 9,1 14,2
Potassium 3,2 non renseigné
Sulfate 1187 1530
Hydrogénocarbonate 403 383,7
Chlorure 10 non renseigné
Nitrate 2,7 4,3
Données : masses molaires en g/mol : H : 1,008 C : 12,011 O : 15,999 Ca : 40,08 Mg : 24,312 Na : 22,989 K : 39,102
2.1.1 Détermination de la teneur en ions calcium et magnésium
La dureté d'une eau correspond à la présence de cations métalliques autres que ceux des métaux alcalins. Dans les eaux naturelles, la dureté est due essentiellement aux ions calcium et magnésium . La dureté totale ou T.H (titre hydrotimétrique) correspond à la teneur globale en ions calcium et magnésium. Différentes unités sont utilisées pour exprimer le T.H: le degré français (d° français), la teneur en mg/L ou en mmol/L de carbonate de calcium. On dispose des correspondances suivantes: 1d° français = 0,1 mmol.L -1 =10 mg de CaCO3 par litre 1 mmol de Ca2+= 40,08 mg de Ca2+ 1 mmol de Mg2+= 24,305 mg de Mg2+
M(CaCO3)= 100,09 g.mol-1
On classe les eaux (autres que les eaux minérales) en différentes catégories: eau très douce T.H de 0 à 5 ° français eau douce T.H de 5 à 15 ° français eau demi-dure T.H de 15 à 25 ° français eau potable T.H< 30 ° eau industrielle 30 °< T.H < 60 °

TP 2 : Fonctionnement d’un adoucisseur d’eau domest ique “Déminéralisation” d’une eau minérale Page 6
2.1.1.1 Principe
L’EDTA est l’ ion Ethylène Diamine TétraAcétate . Il est noté Y4- . Les ions calcium et magnésium sont dosés par une solution d’EDTA disodique selon les réactions d’équation :
Ca2++ H2Y2- = CaY2- + 2 H+
Mg2++ H2Y2- = MgY2- + 2 H+
Pour que la réaction se produise, il faut travailler à un pH voisin de 12 .En effet, les complexes CaY2- et MgY2- sont d’autant plus stables que le pH est élevé. Les complexes formés CaY2- et MgY2- sont incolores. Or, lors d’un dosage, il faut que l’on puisse apprécier l’équivalence. Il est donc nécessaire d’ajouter un indicateur coloré : c’est le noir ériochrome T appelé NET. La forme libre de cet indicateur est de couleur bleue tandis que sa forme complexée avec les ions calcium ou magnésium est de couleur rose- rouge.
2.1.1.2 Mode opératoire
• Placer dans la burette la solution d'EDTA disodique étalonnée (CEDTA # 2,50 . 10-2 mol.L-1).
• Placer dans un erlenmeyer : - une prise d'essai de l'eau minérale E1 = 20 mL - environ 20 mL de tampon ammoniacal - une pointe de spatule de NET
• Chauffer jusqu'à 40 à 60°C
• Titrer jusqu'au virage du rouge sombre au bleu noir. Deux essais concordants sont demandés. Soit V1 le volume équivalent.
2.1.1.3 Exploitation
i. Donner la formule semi-développée de l’EDTA .
ii. Expliquer pourquoi l’équivalence est obtenue lorsque la couleur de la solution n’évolue plus et non pas au début du changement de couleur.
iii. Exprimer la somme des concentrations en ions Mg2+ et Ca2+ de l’eau minérale en fonction des grandeurs connues CEDTA , E1 et V1 . Faire l’application numérique et comparer aux données de l’étiquette.
2.1.2 Détermination de la teneur en ions hydrogénoc arbonate
2.1.2.1 Principe
L'alcalinité d'une eau correspond à la présence d'hydrogénocarbonate, de carbonate et d'hydroxyde. L'alcalinité forte ou titre alcalimétrique T.A correspond à la teneur de l'eau en hydroxyde et carbonate et se mesure assez exactement au virage de la phénolphtaléine. La décoloration se produit lorsqu'il n'y a plus d'ions carbonate en solution, car ils sont transformés en hydrogénocarbonate. L'alcalinité totale ou titre alcalimétrique complet (T.A.C) correspond à la teneur de l'eau en ions hydroxyde, carbonate et hydrogénocarbonate et se mesure au virage de l'hélianthine c'est à dire dans la zone de pH comprise entre 3 et 4,5. Dans ce TP, le dosage sera suivi par pH-métrie.
2.1.2.2 Mode opératoire
• Placer E2 =50 mL de l'eau minérale à étudier dans un becher et ajouter le minimum d'eau déminéralisée bouillie et refroidie à l'abri de l'air pour immerger l'extrémité des électrodes du pH-mètre.
• Placer la solution d'acide chlorhydrique étalonnée de concentration voisine de 0,05 mol.L-1 dans la burette.
• Faire le dosage pH-métrique et tracer pH= f(volume d'acide versé). Soit V2 le volume équivalent.

TP 2 : Fonctionnement d’un adoucisseur d’eau domest ique “Déminéralisation” d’une eau minérale Page 7
2.1.2.3 Exploitation
iv. Exprimer la concentration en ion HCO3- dans l'eau minérale en fonction de CHCl, E2 et V2 .
v. En déduire T.A et T.A.C pour l'eau minérale en mmol.L-1
2.1.3 Détermination de la teneur en autres ions
vi. A l’aide de l’étiquette, dresser la liste de tous les ions présents dans cette eau minérale.
vii. Pour ceux dont la concentration n’a pas été déterminée expérimentalement, convertir la concentration massique en concentration molaire.
2.2 “Déminéralisation” de l’eau minérale
Les expériences suivantes ont pour objectifs : • d’étudier le fonctionnement d’une résine échangeuse d’ions initialement présente sous sa forme acide,
• d’établir la relation bilan entre la concentration des ions H3O+ échangés et les concentrations en Ca2+,
Mg2+ , HCO3- préalablement dosés et Na+ et K+ relevées sur l’étiquette de l’eau minérale .
2.2.1 Principe
Les ions Ca2+ , Mg2+ , K+ et Na+ de l’eau minérale sont remplacés par des ions H+ fournis par une résine échangeuse d’ions fortement acide selon la réaction :
Mn+(solution) + n H+
(résine) = Mn+(résine) + n H+
(solution)
L'eau à analyser est passée sur une résine cationique et l'éluat est titré par une solution d'hydroxyde de sodium de concentration connue voisine de 0,0500 mol. L-1 par pH-métrie.
2.2.2 Préparation de la résine
La résine utilisée est une résine cationique (voir feuille annexe), le groupement actif étant un anion de type sulfonate . Cette résine est commercialisée sous forme de sel : R – SO3Na . • Dans un bécher de 100 mL, immerger 10 g de résine dans de l’acide chlorhydrique 6 mol.L-1 pendant
quelques heures.
viii. Ecrire l’équation de la réaction d’échange ayant lieu entre les cations de la résine et ceux de la solution d’acide chlorhydrique concentré.
Pourquoi utilise-t-on de l’acide chlorhydrique concentré ( 6 mol.L-1 ) ?
• Introduire la résine dans une burette de 25 mL (cette opération aura été réalisée quelques heures auparavant).
• A l’aide de papier pH, évaluer le pH de l’eau distillée.
• Rincer la résine plusieurs fois à l’eau distillée (environ 60 mL d’eau distillée sont nécessaires), jusqu’à ce que le pH de l’éluat soit égal au pH de l’eau distillée.
• Laisser alors éluer jusqu’à ce que le liquide affleure la résine.

TP 2 : Fonctionnement d’un adoucisseur d’eau domest ique “Déminéralisation” d’une eau minérale Page 8
ix. Quel est le rôle des rinçages successifs de la résine à l’eau distillée lors de sa préparation ?
2.2.3 Echange des ions
Dans toutes les opérations qui suivent, la résine d oit toujours rester immergée .
• Préparer un bécher de 100 mL sous la burette afin de recueillir les éluats.
• Introduire à l’aide d’une pipette jaugée E3 = 10 mL d’eau de Contrexéville ou d’Hépar en faisant couler le liquide le long de la paroi de la burette.
• Régler le débit en un goutte-à-goutte lent. Un peu avant que le niveau du liquide n’affleure la résine, relever (papier pH) le pH de l’éluat
2.2.4 Récupération totale de l’éluat
• Lorsque le niveau du liquide affleure la résine, ajouter de l’eau distillée à la pipette (entre 30 et 40 mL ajoutés par fractions de 10 mL), toujours en laissant couler le liquide le long de la paroi de la burette.
• Laisser éluer jusqu’à ce que le pH de l’éluat ait repris sa valeur initiale, celle de l’eau distillée (Faire un contrôle au papier pH).
Cette opération permet de s’assurer que tous les ions H+ libérés par la résine sont “descendus” dans le bécher.
2.2.5 Dosage de l’éluat
• Porter l’éluat à ébullition pendant 5 min. Laisser ensuite refroidir.
• Remplir une burette de 25 mL avec une solution de soude de concentration 2,0.10-2 mol.L-1.
• Placer le bécher contenant l’éluat sous la burette.
• Ajouter dans ce bécher quelques gouttes de bleu de bromothymol.
• Réaliser le dosage.
• Relever la valeur du volume V3 versé à l’équivalence.
2.2.6 Analyse du protocole et exploitation des résu ltats
x. Ecrire les équations des réactions d’échange ayant lieu entre les cations de l’eau et les protons
initialement fixés sur la résine. En déduire le nombre de protons cédés par la résine pour chaque type de cation fixé.
xi. Quel est le pH mesuré au cours de l’élution ? Cela confirme-t-il les prévisions théoriques ?
xii. Lors de l’élution, une réaction acido-basique a lieu entre les protons cédés par la résine et un anion de l’eau minérale. Quel est cet anion ? Ecrire l’équation de la réaction. Comment cette réaction modifie-t-elle la concentration des ions H3O
+ dosés ?
xiii. Pourquoi porter l’éluat à ébullition avant d’effectuer le dosage des ions H+ ?
xiv. Déterminer la concentration [H+]éluat de l’éluat.
xv. Etablir la relation entre [H+ ]éluat et les concentration des ions présents dans l’eau minérales : [Ca2+], [Mg2+] ,[Na+] ,[K+] et [HCO3
-] . xvi. Vérifier cette relation à partir des valeurs déterminées dans le paragraphe 1 .

TP 2 : Fonctionnement d’un adoucisseur d’eau domest ique “Déminéralisation” d’une eau minérale Page 9
Académie de Nancy - Metz Olympiades de la chimie 2010-2011 « La chimie et l’eau »
TP 2 : Fonctionnement d’un adoucisseur d’eau domest ique “Déminéralisation” d’une eau minérale
LISTE MATERIELS ET PRODUITS
Matériel
pH-mètres- millivoltmètres avec électrodes verre + calomel
Burettes (2)
Coton et sable de Fontainebleau
Béchers 100 mL (4)
Pipette jaugée 10 mL
Eprouvette graduée 50 mL, éprouvettes graduées de 100 mL , de 250 mL
thermomètres
1 Agitateur magnétique + 1 plaque chauffante
Barreau aimanté
Papier pH
Produits et solutions (en rouge non disponible dans les lycées a priori)
- Eau de Contrexéville ou Eau Hépar (250 mL par poste)
- NET en trituration dans NaCl ou KNO3
- solution d’EDTA disodique à exactement 0,0250 mol/L : 125 mL par poste
- tampon noté « tampon pH= 10» préparé ainsi : 1 mole de NH3 (ammoniac) et 1 mole de NH4Cl (chlorure
d’ammonium) pour 2 litres de solution ; à placer dans un flacon muni d'un distributeur réglé sur 20 mL (
50 mL par poste)
- solutions tampons pour étalonner les pH-mètres
- acide chlorhydrique de concentration connue voisine de 0,0500 mol/L ( 50 mL par poste)
- Résine Amberlyte IR 120 ( résine cationique sous forme RSO3H , capacité d’échange 5 éq/kg) (10 g par
poste) - Acide chlorhydrique concentré (6 mol.L-1) ( 50 mL par résine)
- Solution de soude à 2,0.10-2 mol.L-1 ( 50 mL par poste)
- Bleu de bromothymol
- Eau distillée

TP 2 : Fonctionnement d’un adoucisseur d’eau domest ique “Déminéralisation” d’une eau minérale Page 10
Académie de Nancy - Metz Olympiades de la chimie 2010-2011 « La chimie et l’eau »
TP 2 : Fonctionnement d’un adoucisseur d’eau domest ique “Déminéralisation” d’une eau minérale
REPONSES AUX QUESTIONS
Réponses aux questions :
i.
ii. Au départ, dans le bécher, l’indicateur forme un complexe rose avec les ions calcium et magnésium
Ca (Ind) 2+ et Mg(Ind)2+ .
On verse l’EDTA dans le bécher, il vient à son tour complexer les ions Ca2+ et Mg2+ . Or les complexes Ca
(Ind) 2+ et Mg(Ind)2+ sont moins stables que les complexes CaY2- et MgY2- donc lorsqu’il n’y a plus d’ions
calcium ou magnésium libres, l’ajout d’EDTA provoque la destruction progressive des ions complexés
roses et formation de complexes incolores . Le NET sous forme libre, de couleur bleue, apparait alors
progressivement dans le bécher.
On atteint l’équivalence lorsque tous les ions Ca2+ et Mg2+ sont complexés par l’EDTA.
Tout le NET se trouve alors sous forme libre et donne la couleur définitive dans le bécher.
iii. [Mg2+ ] + [ Ca2+ ] = CEDTA * V1 / E1
iv. [HCO3- ] = CHCl . V2 / E2
v. T.A.C = [ HCO3- ]
vi. et
vii. (Pour la Contrex) ( [Ca2+] = 486 mg.L-1 = 12,1.10-3 mol.L-1 ; [Mg2+] = 84 mg.L-1 = 3,5 mol.L-1 ; )
[Na+] = 9,1 mg.L-1 = 3,9.10-4 mol.L-1 ; [K+] = 3,2 mg.L-1 = 8,2.10-5 mol.L-1 ;
[HCO3-] = 403 mg.L-1 = 6,61.10-3 mol.L-1.
viii. Les rinçages successifs à l’eau distillée permettent d’éliminer l’excès d’ions H+ sur la résine.

TP 2 : Fonctionnement d’un adoucisseur d’eau domest ique “Déminéralisation” d’une eau minérale Page 11
ix. (R-SO3-, H+) + Na+
S = (R-SO3-, Na+) + H+
S ;
2(R-SO3-, H+) + Mg2+
S = (R-SO3-, Mg2+) + 2H+
S;
2(R-SO3-, H+) + Ca2+
S = (R-SO3-)2, Ca2+ + 2H+
S;
(R-SO3-, H+) + K+
S = (R-SO3-, K+) + H+
S.
x. Au cours de l’élution, le pH de l’éluat est plus faible que celui de l’eau distillée, preuve que la résine
cède des ions H+.
xi. H+(aq) + HCO3
- = CO2(g) + H2O
xii. Porter l’éluat à ébullition permet de chasser le dioxyde de carbone dissous.
xiii. [H+] = Csoude * V3 / E3
xiv. [H+] = 2[Ca2+] + 2[Mg2+] + [Na+] + [K+] - [HCO3-]
xv. Théoriquement, la « somme des cations » donne [H+]théo = 2,50.10-2 mol.L-1.

TP 3 : Mesures de la pollution organique d’une eau :
Oxydabilité au permanganate de potassium, DCO

TP 3 : Mesures de la pollution organique d’une eau Page 1
Académie de Nancy - Metz Olympiades de la chimie 2010-2011 « La chimie et l’eau »
TP 3 : Mesures de la pollution organique d’une eau : Oxydabilité au permanganate de potassium, DCO
1 INTRODUCTION L'évaluation de la pollution organique d'une eau est effectuée dans une station d’épuration avant et après traitement des eaux usées. Les méthodes utilisées dans les stations d’épuration pour purifier l’eau sont le plus souvent calquées sur la nature c'est-à-dire sur l’auto-épuration. L’oxygène dissous dans l’eau joue un rôle primordial dans cette épuration spontanée. La charge de pollution organique d’une eau est fonction de la demande d’oxygène que le milieu exerce afin d’oxyder et donc de minéraliser ces matières organiques. La diversité des matières organiques contenues dans les eaux naturelles ou de rejet, rend la détermination spécifique de chacune d'elles longue, délicate et coûteuse pour un intérêt souvent inutile. Des déterminations plus globales donnent des indications amplement suffisantes grâce à certaines mesures, beaucoup plus faciles à réaliser et à interpréter. Ce sont les tests mesurant le caractère réducteur de l'eau en faisant agir les oxydants. Dans cette idée, on peut déterminer
� l'oxydabilité au permanganate de potassium � la Demande Biochimique en Oxygène (DBO) qui renseigne sur le contenu biodégradable des eaux. � la Demande Chimique en Oxygène (DCO) et le Carbone Organique Total (COT) qui caractérisent
l’ensemble de la charge organique qu’elle soit ou non biodégradable Le principe de ces tests sera présenté ci-dessous dans la partie documentation. La partie expérimentale de cette séance comportera :
� la mesure de l’indice permanganate d’une solution de glucose � la mesure de la DCO de la même solution de glucose par la méthode miniaturisée (méthode
spectrophotométrique)
2 DOCUMENTATION 2.1 L'OXYDABILITE AU PERMANGANATE DE POTASSIUM
2.1.1 Généralités
Ce test appelé aussi "Indice de permanganate"; c'est une mesure conventionnelle utilisée pour juger aussi bien la qualité d'eaux potables que d'eaux brutes telles que les eaux superficielles. Les eaux plus fortement contaminées peuvent être analysées moyennant une étape de pré-dilution Le test préconisé par les normes AFNOR est applicable pour des eaux contenant moins de 300 mg.L-1 d'ions chlorures. Les ions nitrites, fer (II) ou sulfures, oxydables dans les conditions expérimentales sont pris en compte par l'oxydation (matières inorganiques) La méthode n'est pas recommandée pour déterminer la charge des eaux résiduaires pour lesquelles existe un autre test, la DCO (cf. plus loin)

TP 3 : Mesures de la pollution organique d’une eau Page 2
2.1.2 Rappels d'oxydoréduction:
L'ion permanganate de potassium, de formule MnO4- est un oxydant fort.
Selon le milieu, il appartient au couple MnO4- /Mn2+ en milieu acide ou au couple MnO4
- /MnO2 en milieu basique. La force de l'oxydant est un peu différente selon le milieu mais est élevée dans les deux cas.
2.1.3 Conditions expérimentales
Les normes actuelles européennes prévues pour des eaux peu chargées (eaux potables, de piscine, eaux brutes superficielles (pisciculture, baignades, ...) sont établies en milieu acide sulfurique et à chaud. Le temps de contact est défini ; il est de 10 min + 2 min. D'autres essais sont réalisables:
� milieu sulfurique à froid: contact pendant 4 heures � milieu alcalin chaud pendant 10 minutes (selon l'ancienne réglementation)
2.1.4 Principe du dosage
L'échantillon : A un volume défini VPE d'échantillon, on ajoute:
� de l'acide sulfurique à 2 mol.L-1 � un volume V(KMnO4) de solution de permanganate de potassium
On porte la solution au voisinage de l'ébullition (env. 96°C) pendant 10 minutes On ajoute alors un volume Vox (ex 10,0 mL) de solution d'oxalate de sodium ce qui produit la décoloration de la solution (oxalate en excès). L’ion oxalate C2O4
2- est en effet un réducteur dans le couple CO2/ C2O42-.
On dose la solution obtenue par la solution de permanganate jusqu'à coloration rose pale persistante pendant au moins 30 s. Soit Ve le volume versé pour ce dosage Le blanc : Parallèlement à l’échantillon, on effectue un blanc en remplaçant l'eau étudiée par le même volume d'eau distillée. Soit Vo le volume versé pour le blanc. Cet essai est traité de la même façon que l’échantillon.
2.1.5 Expression du résultat
Par convention, le résultat de l'oxydabilité au permanganate de potassium n'est pas donné par rapport à KMnO4 mais par rapport au dioxygène dans le couple rédox O2 / H2O et est exprimé en mg/L de O2 (voir dans la partie expérimentale) ATTENTION: Ceci est une convention ; la significati on du résultat correspond en réalité à une consommation de permanganate et non à une consommat ion en dioxygène.
2.2 DEMANDE CHIMIQUE EN OXYGENE ( DCO )
2.2.1 Généralités
Cette détermination consiste à mesurer la quantité de matières oxydables par le dichromate de potassium en milieu sulfurique chaud. C'est une méthode d'oxydation très puissante qui prend en compte pratiquement la totalité des matières organiques présentes. Comme avec l'oxydabilité au permanganate de potassium, le résultat est exprimé en référence à l'oxygène (mg/L de O2) et ne correspond pas à la quantité de dioxygène consommé réellement mais plutôt une équivalence. Ces mesures sont plus spécialement destinées aux eaux résiduaires, permettant ainsi de déterminer la charge en matières organiques. C'est un des paramètres fondamentaux utilisés par les agences de bassin pour établir les taxes de redevance de la pollution.

TP 3 : Mesures de la pollution organique d’une eau Page 3
2.2.2 Rappels d'oxydoréduction:
Dans cette réaction, le dichromate appartient au couple Cr2O72- / Cr3+
La méthode utilise également une solution de sel de Mohr : c'est un sulfate double de fer (II) et d'ammonium. Dans les réactions d'oxydoréduction, il se comporte comme une solution de sulfate de fer (II),
( Fe2+ + SO42 ) ; le couple rédox est Fe3+ / Fe2+
La ferroïne : C'est un indicateur rédox (indicateur d'oxydoréduction) qui permet de repérer l'équivalence.
2.2.3 Conditions expérimentales / limites de la mét hode
Comme avec l'oxydabilité au permanganate, les ions chlorures interfèrent et faussent le dosage. C'est pourquoi on ajoute le sulfate mercurique (les ions mercure(II) Hg2+ forment un complexe avec les ions chlorure Cl-). Le sulfate d'argent catalyse la réaction. Certaines substances organiques résistent à l'oxydation; c'est le cas par exemple des hydrocarbures linéaires (alcanes) ou le benzène; leur volatilité limite le contact avec l'oxydant
2.2.4 Principe de la technique
L'échantillon : A un volume VPE d'échantillon d'eau dans un erlenmeyer à col rodé, on ajoute:
� 1 g de sulfate mercurique � 5 mL d'acide sulfurique concentré contenant du sulfate d'argent � 25 mL de solution de dichromate de potassium
Après homogénéisation, adapter un réfrigérant pour liquéfier les vapeurs et éviter l’évaporation Maintenir à l'ébullition pendant 2 heures. Laisser refroidir, ajouter quelques gouttes de ferroïne Doser le dichromate restant par la solution de sel de Mohr (volume Ve ) Le blanc : Réalisé et traité en même temps que l'échantillon Le volume VPE d'eau à analyser est remplacé par le même volume d'eau déminéralisée. Le dosage donne un volume Vo.
2.2.5 Variante : méthode miniaturisée ou micrométho de
Compte-tenu de la toxicité des sels de chrome (VI) (caractère cancérigène), les laboratoires utilisent de plus en plus une microméthode, permettant de réduire les quantités de réactifs, d’utiliser des kits en tube à essais contenant déjà l’ensemble des réactifs. La mesure de la DCO ne nécessitera pas le dosage par une solution de sel de Mohr, mais la détection de fera par spectrophotométrie après « digestion » à 150°C (voir dan s la partie expérimentale).
2.3 DEMANDE BIOCHIMIQUE EN OXYGENE ou DBO
L'oxydation des composés organiques biodégradables par les microorganismes entraîne une consommation de dioxygène ; cette consommation est appelée demande biochimique en oxygène, en abrégé: DBO. La mesure de cette DBO permet d'évaluer le contenu d'une eau en matières biodégradables et donc sa qualité ou son degré de pollution. La dégradation de ces matières peut être longue, (plusieurs semaines). D'autre part, l'oxydation des dérivés ammoniaqués et des nitrites en nitrates ("nitrification") absorbe également du dioxygène et ne débute qu'au bout d'une dizaine de jours. L'oxydation biochimique complète, au bout de 21 ou de 28 jours, est appelée DBO 21 / DBO 28 C'est la DBO "ultime" (déterminée beaucoup plus rarement)

TP 3 : Mesures de la pollution organique d’une eau Page 4
La DBO ultime étant trop longue à obtenir, on mesure la quantité de dioxygène consommée pendant 5 jours; c'est la DBO 5. La DBO 5 représente une consommation en oxygène; elle est donc exprimée en mg de O2 consommé pour 1 litre d’eau. On la détermine en faisant incuber pendant 5 jours à 20 + 1 °C, un échantillon d'eau conservé à l'abri de l'air et de la lumière. La DBO5 s'obtient par la différence entre la quantité d'oxygène dissous initialement présent et la quantité d'oxygène dissous restant après l'incubation. L'oxygène dissous est mesuré par dosage iodométrique (dosage d'oxydoréduction) ou par mesure électrochimique (potentiométrique) en utilisant un oxygénomètre. Certaines substances minérales réductrices peuvent être prises en compte immédiatement (ions ferreux, sulfures, sulfites...) ; cette oxydation correspond alors à une demande immédiate en dioxygène que l'on peut inclure ou non dans la DBO. A l'origine, les méthodes utilisées en laboratoire tendaient à reproduire le mieux possible le phénomène d'autoépuration et les conditions moyennes du milieu afin que la DBO trouvée en laboratoire soit la même que celle dans les lacs ou les rivières. En fait, les facteurs influents sont nombreux (ensoleillement, température et ses variations, réoxygénation naturelle...), et les mesures effectuées en laboratoire ne reflètent pas rigoureusement la réalité. Néanmoins, parmi les essais effectués, la DBO est le paramètre qui donne l'idée la plus précise des charges biodégradables. Comme la DCO, la DBO 5 est un des paramètres essentiels qui permet d'apprécier le bon fonctionnement d'une station d'épuration et qui est pris en compte par les Agences de l’Eau pour établir les redevances sur la pollution Si dans une eau, toutes les matières organiques étaient biodégradables, on devrait avoir : DBO21 = DCO. En pratique, le rapport DBO / DCO varie fortement et n'est pas prévisible d'une eau à l'autre.
2.4 CARBONE ORGANIQUE TOTAL ou COT
Certains composés organiques (hydrocarbures aromatiques, acide acétique, etc…) résistent à l'oxydation chimique (par le dichromate par exemple) et ne sont pas pris en compte par la DCO. Il faut donc une méthode plus énergique pour les oxyder complètement. On utilise actuellement des appareils dans lesquels les échantillons subissent une combustion totale à 950°C environ sous l'action de l'oxygène gazeux ou une dégradation complète à température ambiante en présence d’un oxydant les ions peroxodisulfate S2O8
2- et d’un rayonnement UV.
On mesure la quantité de dioxyde de carbone formée et on en déduit la quantité de carbone que contenant l'échantillon. Cette mesure est effectuée par spectrométrie infrarouge. Le dioxyde absorbe fortement ce type de rayonnement comme le montre le spectre ci-contre (l’échelle des abscisses
représente le nombre d’onde σ=1/λexprimé en cm-1).

TP 3 : Mesures de la pollution organique d’une eau Page 5
www.elmhurst.edu/~chm/vchembook/images/irCO2.JPEG
Dans ces conditions, on mesure le carbone total c'est-à-dire organique et minéral (carbonates, hydrogénocarbonates, ...). Pour déterminer le carbone purement organique on utilise une des méthodes suivantes: 1°) Acidifier fortement l'échantillon ce qui transf orme carbonate et hydrogénocarbonates en dioxyde de carbone. On doit ensuite dégazer l'échantillon; pour cela, on fait passer un courant gazeux (azote ou air purifié) ce qui élimine tout le dioxyde de carbone de la phase aqueuse. Le carbone total mesuré correspond alors au carbone organique total. 2°) Certains appareils comportent un deuxième tube à combustion, porté à une température plus basse (150°C) où seul le carbone inorganique est oxydé. O n le mesure donc séparément et on déduit cette valeur du carbone total. A noter cependant que certaines substances organiques fragiles sont oxydées en même temps que le carbone minéral. Le COT s'exprime en mg de carbone par litre d'échantillon ; c'est donc un moyen beaucoup plus direct d'exprimer la teneur en matières organiques. Il existe des relations empiriques permettant de relier la DCO voire la DBO5 au carbone organique total La mesure du COT apporte de précieuses indications quant à la qualité des eaux brutes d'approvisionnement ou des eaux traitées. C'est donc un moyen pratique pour apprécier l'efficacité d'un traitement (ozonation, adsorption sur charbon actif, filtration biologique...) La mesure directe permet d'analyser des échantillons dont la teneur en carbone organique est comprise entre 1 et 500 mg/L. Au-delà de cette valeur, une dilution est nécessaire.
3 PARTIE EXPERIMENTALE 3.1 DETERMINATION DE L’INDICE PERMANGANATE (IP) D’U NE SOLUTION DE
GLUCOSE
Vous disposez de 3 solutions: � Solution G de glucose à 0,800 g.L-1
� Solution P de permanganate de potassium de concentration connue voisine de 0,0200 mol.L-1
� Solution O d’oxalate de sodium (2 Na+ + C2O42-) à environ 0,05 mol.L-1
3.1.1 Expériences
Erlen Essai :
� Introduire dans un erlenmeyer de 250 mL : • Une prise d’essai de 10 mL de solution G de glucose • 90 mL d’eau déminéralisée • 5 mL d’acide sulfurique à 2 mol.L-1
� Porter le mélange à l’ébullition
� Ajouter dans le mélange 20 ml de solution P de permanganate de potassium.
� Maintenir l’ébullition durant 10 min + 15 s
� Retirer le mélange de la plaque et y ajouter 25 mL de solution O d’oxalate de sodium : la solution se décolore
i. Préciser le matériel utilisé pour réaliser ces prélèvements en justifiant votre choix

TP 3 : Mesures de la pollution organique d’une eau Page 6
� Procéder au dosage des solutions par la solution de permanganate de potassium jusqu’à coloration rose persistante. Soit Véq le volume versé à l’équivalence.
Erlen Blanc
� Remplacer les 10 mL de solution de glucose par 10 mL d’eau et procéder comme pour l’erlen essai. Le volume versé à l’équivalence est noté Vo
3.1.2 Exploitation
ii. Ecrire les demi-équations redox correspondant aux couples MnO4-/Mn2+, O2/H2O et montrer que l’ion
oxalate est un réducteur dans le couple CO2/C2O42-.
iii. Du point de vue pouvoir oxydant, quelle est la quantité de dioxygène équivalente à une mole de permanganate ?
iv. En comparant le mode opératoire pour l’erlen Essai et pour l’erlen Blanc, montrer que pour la prise
d’essai ��� de solution G de glucose, les matières oxydables consomment une quantité de
permanganate égale à ���� � �� · �� ��� ��� �� �����.
v. En déduire l’expression littérale de la quantité de permanganate qui serait consommée par litre de solution de glucose
vi. En déduire l’expression littérale la quantité équivalente de dioxygène qui serait consommée par litre de la solution de glucose (en mol de dioxygène par litre de solution)
vii. Montrer que l’indice permanganate �� de la solution de glucose en mg de dioxygène par litre de solution peut s’exprimer par la relation suivante, sachant que la masse molaire du dioxygène est de 32 g.mol-1:
�� � 40 000 �� ��� ��� � ��
���
viii. Faire l’application numérique pour la solution G de glucose. On peut envisager une oxydation totale du glucose de formule C6H12O6 par le dioxygène en dioxyde de carbone et eau.
ix. Ecrire l’équation chimique correspondant à cette transformation.
x. En supposant cette transformation totale, quelle serait la masse de dioxygène nécessaire pour oxyder selon cette réaction 1 litre de la solution G de glucose (M(glucose) = 180 g.mol-1)?
xi. Comparer cette masse avec la valeur obtenue pour �� et conclure.
3.2 MESURE DE LA DCO PAR METHODE MINIATURISEE ( ou MICROMETHODE)
3.2.1 Présentation
Depuis plusieurs années est disponible sur le marché (sociétés Hanna, Hach-Lange) un système miniaturisé pour l’analyse de la DCO comportant :
� Des tubes avec bouchons vissés contenant tous les réactifs nécessaires (HgSO4, H2SO4+ Ag2SO4, K2Cr2O7) en quantités environ 10 fois plus faibles que dans la méthode classique. Il suffit d’introduire l’échantillon (2 mL), de mélanger, de fermer hermétiquement et de placer le tube dans le bloc chauffant. On procède la même façon pour les échantillons, les étalons (solution de DCO connue) et les témoins (eau déminéralisée).
� Un bloc chauffant permettant de chauffer les tubes à 148°C pendant 2 heures (un dispositif plus récent permet même de chauffer à 170°C, ce qui rédu it la durée de chauffage à 15 minutes)

TP 3 : Mesures de la pollution organique d’une eau
� Un spectrophotomètre permettant par lecture directe de connaître la DCO de l’échantillon par mesure de l’absorbance à 610 nm (Hanna) ou 620 nmDCO faible)
Cette méthode est facile d’emploi, réduit de beaucoup les quantités de réactifs nécessaires et dont il faut disposer, limite les problèmes liés à la toxicité de ces réactifs (les sociétés se chargeant de l’étubes usagés). Ses principales limitations par rapport à la méthode classique de mesure de la DCO sont d’une part le faible volume d’échantillon introduit surtout dans le cas d’eaux chargées de matières en suspension où la reproductibilité risque d’être moins bonne et d’autre part, le coût d’achat des réactifs tout préparés. Au cours de cette séance, nous étudierons le principe de cette méthode et l’adapterons au matériel disponible au laboratoire.
3.2.2 Principe de la méthode
Vous disposez des spectres d’absorption, tracés entre 400 et 650 nm, d’une solution de dichromate de potassium ( à une concentration voisine de celle présente dans les tubes avant réaction) d’une solution de chlorure de chrome (III) ( la concentration en chrome (III) est voitous les ions dichromate étaient réduits en ions chrome (III)). (Le blanc est constitué d’eau déminéralisée)
xii. Ecrire les demi-équations redox correspondant aux couples Cr
xiii. Rappeler la loi de Beer-Lambert.
xiv. Montrer que les spectres tracés cides solutions de dichromate de potassium et de chlorure de chrome (III)
xv. Quelle est l’espèce dont on peut mesurer la concentration par me
xvi. Pourquoi, pour des DCO faibles, le fabricant recommande420 nm ?
xvii. Quelles relations peut-on établir e
– la quantité de Cr3+ formée et la quantité d’électrons captée par Crréducteur présent dans l’échantillon.
: Mesures de la pollution organique d’une eau
Un spectrophotomètre permettant par lecture directe de connaître la DCO de l’échantillon par mesure de l’absorbance à 610 nm (Hanna) ou 620 nm (Hach Lange) (ou à 420 nm en cas de
Cette méthode est facile d’emploi, réduit de beaucoup les quantités de réactifs nécessaires et dont il faut disposer, limite les problèmes liés à la toxicité de ces réactifs (les sociétés se chargeant de l’é
Ses principales limitations par rapport à la méthode classique de mesure de la DCO sont d’une part le faible volume d’échantillon introduit surtout dans le cas d’eaux chargées de matières en suspension où la
isque d’être moins bonne et d’autre part, le coût d’achat des réactifs tout préparés.
Au cours de cette séance, nous étudierons le principe de cette méthode et l’adapterons au matériel
Principe de la méthode
pectres d’absorption, tracés entre 400 et 650 nm, d’une solution de dichromate de potassium ( à une concentration voisine de celle présente dans les tubes avant réaction) d’une solution de chlorure de chrome (III) ( la concentration en chrome (III) est voisine de celle que l’on aurait dans les tubes si tous les ions dichromate étaient réduits en ions chrome (III)). (Le blanc est constitué d’eau déminéralisée)
équations redox correspondant aux couples Cr2O72-/Cr3+ et O
Lambert.
Montrer que les spectres tracés ci-dessus permettent de retrouver de façon qualitative les couleurs des solutions de dichromate de potassium et de chlorure de chrome (III)
Quelle est l’espèce dont on peut mesurer la concentration par mesure de l’absorbance à 620 nm
Pourquoi, pour des DCO faibles, le fabricant recommande-t-il de faire une mesure d’absorbance à
on établir entre :
formée et la quantité d’électrons captée par Cr2Oréducteur présent dans l’échantillon.
Page 7
Un spectrophotomètre permettant par lecture directe de connaître la DCO de l’échantillon par (Hach Lange) (ou à 420 nm en cas de
Cette méthode est facile d’emploi, réduit de beaucoup les quantités de réactifs nécessaires et dont il faut disposer, limite les problèmes liés à la toxicité de ces réactifs (les sociétés se chargeant de l’élimination des
Ses principales limitations par rapport à la méthode classique de mesure de la DCO sont d’une part le faible volume d’échantillon introduit surtout dans le cas d’eaux chargées de matières en suspension où la
isque d’être moins bonne et d’autre part, le coût d’achat des réactifs tout préparés.
Au cours de cette séance, nous étudierons le principe de cette méthode et l’adapterons au matériel
pectres d’absorption, tracés entre 400 et 650 nm, d’une solution de dichromate de potassium ( à une concentration voisine de celle présente dans les tubes avant réaction) d’une solution de
sine de celle que l’on aurait dans les tubes si tous les ions dichromate étaient réduits en ions chrome (III)). (Le blanc est constitué d’eau déminéralisée)
et O2/H2O.
dessus permettent de retrouver de façon qualitative les couleurs
sure de l’absorbance à 620 nm ?
il de faire une mesure d’absorbance à
O72- donc cédée par le

TP 3 : Mesures de la pollution organique d’une eau Page 8
– la quantité de Cr3+ formée et la quantité de dioxygène qui capterait la même quantité d’électrons
Si on dispose d’un étalon de DCO connue, on peut donc tracer une courbe d’étalonnage qui représente, en abscisses l’absorbance de la solution contenue dans le tube après chauffage et en ordonnées la DCO (en mg/L de O2) de l’échantillon analysé. Les étalons utilisés sont des solutions d’hydrogénophtalate de potassium (HOOC-C6H4-COO- + K+) de concentration connue donc de DCO connue. Sachant que la réaction d’oxydation a lieu en milieu acide, l’hydrogénophtalate de potassium ( M = 204,1 g.mol-1 ) est transformé en acide phtalique HOOC-C6H4-COOH.
xviii. Ecrire l’équation de la réaction d’oxydation de l’acide phtalique en dioxyde de carbone et eau sous l’action du dioxygène.
xix. Quelle doit être la concentration molaire d’une solution d’hydrogénophtalate de potassium de DCO théorique de 1000 mg.L-1 ?
3.2.3 Expérience
� Préparer 250 mL d’une solution mère d’hydrogénophtalate de potassium de DCO 1000 mg.L-1.
� A partir de cette solution mère, préparer par dilution 50 mL d’une solution fille de DCO 500 mg.L-1
ATTENTION Les solutions contenues dans les kits de DCO contie nnent des solutions de dichromate de potassium en milieu acide sulfurique concentré.
Sur le site de l’INRS, on peut trouver les informations sur le dichromate de potassium anhydre et sur l’acide sulfurique concentré. Ces informations sont fournies en pages 10 et 11 . xx. La fiche du dichromate de potassium présente 2 types de pictogrammes. Pourquoi ?
xxi. Quelle est la signification des pictogrammes (fond blanc encadré rouge) placés dans la colonne centrale de la fiche en page 11.
L’utilisation des kits DCO de fera sous hotte ( ou dans un endroit bien ventilé) avec port de lunettes de sécurité et de gants. � Utiliser 4 tubes de kit DCO en suivant le mode opératoire du fabricant (ajout prudent de 2 mL de solution
à analyser (ATTENTION A L’ECHAUFFEMENT !!,) boucher les tubes hermétiquement puis les homogénéiser) avec respectivement:
� Tube I : l’eau déminéralisée (blanc)
� Tube II : la solution étalon d’hydrogénophtalate de potassium de DCO théorique 1000 mg.L-1
� Tube III : la solution étalon d’hydrogénophtalate de potassium de DCO théorique 500 mg.L-1
� Tube IV : la solution G de glucose
� Boucher les tubes hermétiquement, les homogénéiser, les identifier.
xxii. Comment peut-on expliquer l’échauffement des solutions contenues dans les tubes ?
xxiii. Qu’observe-ton en ce qui concerne la couleur des solutions contenues dans ces tubes ? Pourquoi ?
Comme on ne dispose pas d’un thermostat permettant de chauffer ces tubes à 148 °C pendant 2 heures sans risque de projections, on laissera ces tubes à température ambiante pendant 2 semaines.

TP 3 : Mesures de la pollution organique d’une eau Page 9
� 2 semaines plus tard , comparez l’aspect du tube (I) et des autres tubes.
� Mesurer 2 semaines plus tard les absorbances à 620 nm des tubes I, II, III et IV en prenant les précautions suivantes :
� Dans les tubes de kit DCO, on observe un dépôt solide au fond des tubes et le volume contenu dans ces tubes est restreint, ce qui ne permet pas de rincer les cuves de spectro avec la solution contenue dans les tubes. On prélèvera donc avec une pipette pasteur un peu du liquide contenu dans le tube et on le placera dans la cuve de spectro préalablement rincée et sèche.
� Faire le zéro sur le tube I � Mesurer les absorbances des solutions contenues dans les tubes II, III et IV en suivant les
précautions indiquées ci-dessus Remarque : Les laboratoires d’analyse mettant en œuvre la méthode miniaturisée de mesure de la DCO disposent de spectromètres spécialement équipé pour accueillir des tubes cylindriques (et non pas des cuves parallélépipédiques), ce qui fait qu’en laboratoires d’analyse, il ne sera pas nécessaire de transvaser le contenu des tubes de kit pour faire la mesure de la DCO.
3.2.4 Exploitation des mesures
xxiv. Comparer les absorbances des solutions contenues dans les tubes II et III. Le résultat était-il prévisible ?
xxv. Tracer l’absorbance en fonction de la DCO et en déduire la DCO de la solution G de glucose. L’oxydation du glucose a-t-elle été totale ?
BIBLIOGRAPHIE • « Chimie des eaux » de Monique Tardat-Henry aux éditions Le Griffon d’argile • Documentation sur la méthode miniaturisée de mesure de la DCO fournie par les sociétés Hach
Lange et Hanna ( merci plus particulièrement à M Bick de la société Hanna qui a fourni à titre gracieux un kit de test DCO permettant la mise au point de la manipulation sur la DCO)
• Séance de l’atelier scientifique, lycée L Vincent, année 2009-2010
Edith Antonot

TP 3 : Mesures de la pollution organique d’une eau Page 10
Extrait de la notice d’utilisation des tubes Hanna pour la mesure de la DCO Remarques :
• La longueur d’onde choisie est de 610 nm pour la gamme moyenne (620 nm pour tubes Hach-Lange) mais compte-tenu de l’allure de la courbe d’absorbance des ions chrome (III), les 2 longueurs d’onde conviennent.
• Le blanc est réalisé sur le tube contenant le mélange réactionnel et de l’eau (la courbe d’étalonnage dans ces conditions devrait passer par l’origine)

TP 3 : Mesures de la pollution organique d’une eau Page 11
• Le principe de la méthode et en particulier le choix de la longueur d’onde pour les DCO moyennes et pour les DCO faibles est expliqué.

TP 3 : Mesures de la pollution organique d’une eau Page 12

TP 3 : Mesures de la pollution organique d’une eau Page 13

TP 3 : Mesures de la pollution organique d’une eau Page 14

TP 3 : Mesures de la pollution organique d’une eau Page 15
Académie de Nancy - Metz Olympiades de la chimie 2010-2011 « La chimie et l’eau »
TP 3 : Mesures de la pollution organique d’une eau : Oxydabilité au permanganate de potassium, DCO
LISTE MATERIELS ET PRODUITS
(en rouge non disponible dans les lycées a priori)
Par poste ( manip indice de permanganate) : � solution G de glucose à 0,80 g.L-1 : 100mL
� solution de permanganate de potassium de concentration connue voisine de 0,0200 mol.L-1 : 200mL
� solution d’oxalate de sodium de concentration voisine de 0,05 mol.L-1 : 100mL
� acide sulfurique à environ 2 mol.L-1 : 25mL
� Plaque chauffante
� Thermomètre
� 2 erlenmeyers de 250 mL
� 1 burette
� Pipette jaugée de 10, 20 et 25 mL ( les volumes des solutions de glucose, permanganate de potassium et oxalate de sodium pour la manipulation sur l’indice de permanganate doivent être connues avec précision)
� Eprouvette graduée de 100 mL
Par centre ( manip DCO miniaturisée) � Hydrogénophtalate de potassium
� Un peu d’acide sulfurique au ½
� Spectrophotomètre et cuves de spectro

TP 3 : Mesures de la pollution organique d’une eau Page 16
Académie de Nancy - Metz Olympiades de la chimie 2010-2011 « La chimie et l’eau »
TP 3 : Mesures de la pollution organique d’une eau : Oxydabilité au permanganate de potassium, DCO
COMMENTAIRES PROTOCOLES ET REPONSES AUX QUESTIONS
3.2. DETERMINATION DE L’INDICE PERMANGANATE (IP) D’ UNE SOLUTION D’ HYDROGENOPHTALATE DE POTASSIUM
i. Préciser le matériel utilisé pour réaliser ces prélèvements en justifiant votre choix.
ii. MnO4- + 8 H+ + 5 e-= Mn2+ + 4 H2O
O2+ 4 H++ 4 e-= 2 H2O
2 CO2 + 2 e-= C2O42- :
l’ion oxalate est bien un réducteur dans le couple CO2/C2O42-
iii. 1 mole de permanganate peut capter 5 moles d’électrons. 1 mole de dioxygène peut capter 4 moles d’électrons 1 mole de permanganate de potassium équivaut donc à 1,25 moles de dioxygène (5/4)
iv. Dans l’erlen Essai, la quantité totale de permanganate utilisée (!"#$ % &'() a réagi avec 2
réducteurs : le glucose (volume ��� de la solution G) et les ions oxalate (volume 25 mL de la solution O).
Dans l’erlen Témoin, la quantité totale de permanganate utilisée )!"#$ % &"* a réagi avec un seul réducteur : les ions oxalate ( volume 25 mL de la solution O).
On peut donc en déduire que les matières oxydables contenues dans la prise d’essai ��� de la
solution G de glucose ont consommé un volume ��� � �� de la solution de permanganate de
potassium c'est-à-dire une quantité (en mol) de permanganate égale à ���� � �� · �� ���.
v. La quantité de permanganate qui serait consommée par litre de solution de glucose est donc :
���� � �� · �� ���
���
vi. La quantité équivalente de dioxygène qui serait consommée par litre de solution de glucose est donc :
1,25 ���� � �� · �� ���
���
vii. La masse molaire du dioxygène est de 32000 mg.L-1. Par conséquent l’indice permanganate de la solution G de glucose sera :
�� � 32000 1,25 �� ��� ��� � ��
���� 40 000 �� ���
��� � ��
���

TP 3 : Mesures de la pollution organique d’une eau Page 17
viii. Des essais réalisés avec une solution de glucose à 1 g.L-1, une solution d’oxalate de sodium à 0,050 mol.L-1 et une solution de permanganate de potassium à 0,0220 mol.-1 ont donné :
���= 12,2 mL et ��= 3,0 mL soit �� � 810�1 de O2/litre de solution.
ix. Oxydation totale du glucose
C6H12O6 + 6 O2= 6 CO2+ 6 H2O
L’oxydation d’une mole de glucose nécessite donc 6 mol de dioxygène.
x. Un litre de la solution G de glucose contient 0,80 �1 de glucose soit 0,80 180 ���⁄ de glucose ;
son oxydation consommerait donc 6 0,80 180 ��� ⁄ mol de dioxygène soit une masse de
dioxygène de 6 0,80 180 ⁄ 40000 �1 � 1067�1 (pour une solution de glucose à 1g.L-1,
on obtiendrait 1333�1)
xi. La valeur obtenue pour �� est inférieur à cette valeur théorique ; l’oxydation par le permanganate n’est pas totale.
3.2 MESURE DE LA DCO PAR METHODE MINIATURISEE ( ou MICROMETHODE)
3.2.2. Principe de la méthode
xii. C r2O72- + 14 H++ 6 e-= 2 Cr3++ 7 H2O
O2+ 4 H++ 4 e-= 2 H2O
xiii. A = ε×l×C avec ε : absorbance linéique molaire de l’espèce absorbante, l largeur de la cuve (longueur du trajet optique) et C concentration molaire de la solution
xiv. La solution (I) de dichromate de potassium a son maximum d’absorbance dans le domaine du bleu-violet donc apparaîtra plutôt jaune (jaune orangé ici). La solution (II) de chlorure de chrome (III) absorbe à la fois vers 400-450 nm (domaine du bleu) et vers 600 nm (domaine du rouge), elle ne laissera passer que du vert.
xv. A 620 nm, on mesurera une absorbance due uniquement aux ions chrome (III).
xvi. Remarque : Pour tracer ces spectres, je disposais de chlorure de chrome (III) mais pas de sulfate de chrome (III) d’où le choix effectué pour le tracé de spectres.
Si la DCO est faible, après oxydation par le dichromate, la concentration en chrome (III) des solutions obtenues seront faibles donc l’absorbance à 620 nm faible. Il sera donc préférable de se placer à 420 nm où l’on mesurera essentiellement une absorbance due aux ions dichromate restants (surtout s’il y a très peu de chrome (III) formé).
xvii. Pour 6 moles d’électrons captés par Cr2O72-, il y a 2 moles d’ions Cr3+ libérés.
Pour capter 6 moles d’électrons, il faudrait 1,5 (6/4) moles de O2.
Si 1 mole de Cr 3+ se forme, cela correspond à la consommation de 0,7 5 mole de O 2. Il y a donc proportionnalité entre l’absorbance mesurée à 620 n m et la quantité de dioxygène qui serait consommée par l’échantillon donc proportionnalité é galement avec la DCO.
xviii. HOOC-C6H4-COOH + 15/2 O2 = 8 CO2+ 3 H2O
xix. 1 mole d’hydrogénophtalate de potassium soit 204,11 conduit en milieu acide à 1 ���� d’acide
phtalique qui réagira avec 7,5 ����5 de O2 soit 240,01.
Pour 204,1�1 d’hydrogénophtalate de potassium., il y a consommation de 240,0�1 de O2.
Si on veut préparer une solution de DCO 1000�1. 789, il faut donc que la concentration massique
de la solution d’hydrogénophtalate de potassium soit de 204,1 1000/240 � 850�1. 789

TP 3 : Mesures de la pollution organique d’une eau Page 18
xx. La fiche du dichromate de potassium présente 2 types de pictogrammes car elle a été révisée depuis la mise en place des nouveaux pictogrammes de sécurité (la feuille avec les nouveaux pictogrammes est téléchargeable gratuitement sur le site web de l’INRS et donnée en page 12 de l’énoncé)
xxi. Signification des pictogrammes (
De gauche à droite et de haut en bas : Je fais flamber, je tue, je nuis gravement à la san té Je ronge, je pollue Il pourrait être intéressant de détailler un peu l’affiche.
xxii. Comment peut-on expliquer l’échauffement des solutions contenues dans les tubes ?
xxiii. Résultats obtenus : xxiv. xxv. Lorsque j’ai testé la manipulation, j’ai utilisé comme blanc de l’eau ( je ne disposais pas de la notice
détaillée Hanan qui est figure à la fin du T.P élèves) et j’ai fait des mesures à 620 nm ( longueur d’onde préconisée par Hach Lange) Les mesures d’absorbance à 620 nm (en prenant comme blanc de l’eau) ont donné : � Tube (I) (Blanc) : 0,013 � Tube (II) (solution de DCO théorique 1000 mg.L-1) : 0,316 � Tube (III) : (solution de DCO théorique 500 mg.L-1) : 0,166 � Tube (III) : solution de glucose à 0,8 g.L-1 : 0,286
J’ai fait ces essais en faisant le zéro d’absorbance sur un tube constitué d’eau. On constate que l’absorbance de la solution contenue dans le tube (II) est sensiblement le double de celle contenue dans le tube (III) Les résultats seraient peut-être meilleurs (absorbance du tube Blanc plus proche de zéro) en prenant comme blanc une solution d’acide sulfurique au ½ (ce qui correspond sensiblement à la matrice dans laquelle sont réalisés ces tests). On pourrait également envisager de faire le zéro sur le tube (I). En faisant le zéro sur de l’eau déminéralisée et en utilisant les mesures d’absorbances des tubes (I), (II) et (III), on peut tracer la droite modélisée ci-dessous (elle ne passe pas par l’origine) On peut en déduire la DCO de la solution de glucose: 900 mg.L-1et constater que l’oxydation n’a pas été totale (pour avoir une oxydation totale, il faudrait mesurer le COT).
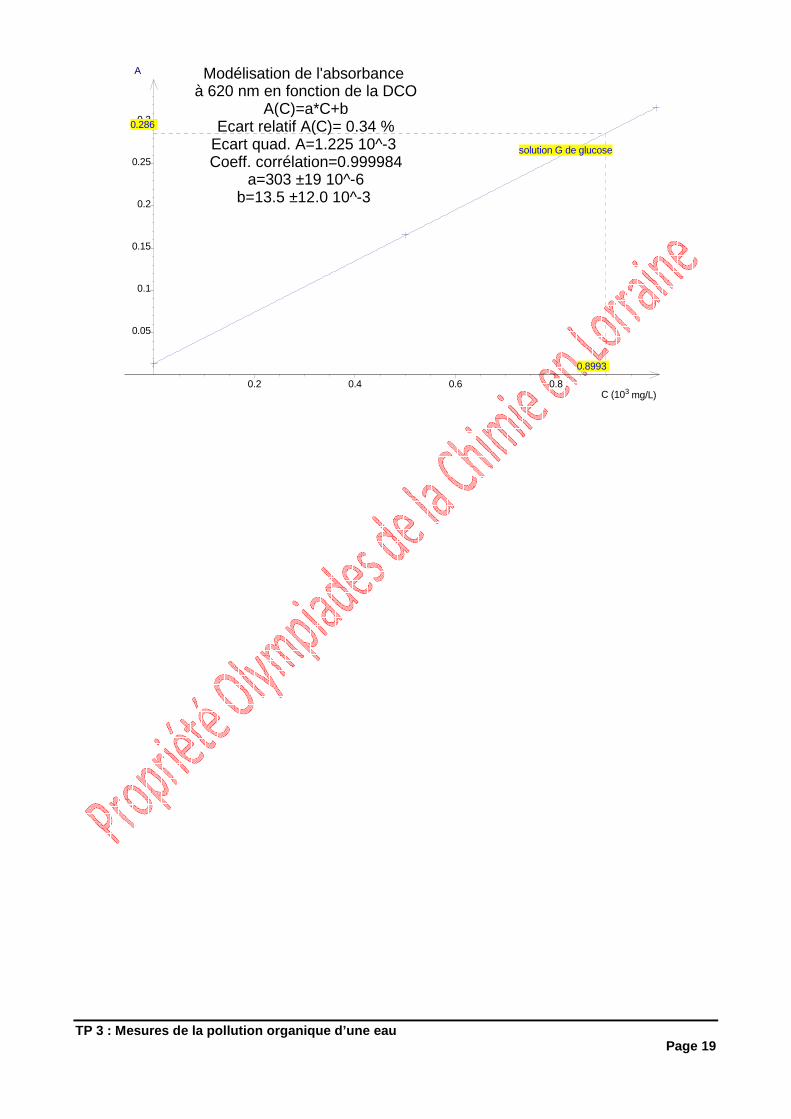
TP 3 : Mesures de la pollution organique d’une eau Page 19
mg/L)3C (100.2 0.4 0.6 0.8
A
0.05
0.1
0.15
0.2
0.25
0.3
Modélisation de l'absorbance à 620 nm en fonction de la DCO
A(C)=a*C+bEcart relatif A(C)= 0.34 %
Ecart quad. A=1.225 10^-3 Coeff. corrélation=0.999984
a=303 ±19 10^-6b=13.5 ±12.0 10^-3
0.286
0.8993
solution G de glucose

TP 4 : Oxydation d’un alcool secondaire

TP 4 : Oxydation d’un alcool secondaire Page 1
Académie de Nancy - Metz Olympiades de la chimie 2010-2011 « La chimie et l’eau »
TP 4 : Oxydation d’un alcool secondaire
1 Principe de la manipulation On envisage l’oxydation d’un alcool secondaire , le diphénylméthanol , par une solution d’eau de Javel. L’équation associée à la réaction qui a lieu est :
OH O
+ ClO- = + Cl- + H2O
(1) diphénylméthanol diphénylméthanone
i. Ecrire les demi-équations d’oxydoréduction des couples : - HClO/Cl- - diphénylméthanone/diphénylméthanol.
En déduire l’équation de la réaction qui est associée à la transformation réalisée dans ce TP Le diphénylméthanol est mis en solution dans l’acétate (éthanoate) d’éthyle. L’eau et l’éthanoate d’éthyle ne sont pas solubles. Les deux réactifs se trouvent donc dans deux phases différentes : la réaction entre les ions hypochlorite et le diphénylméthanol ne peut avoir lieu qu’à l’interface des deux solvants. Pour améliorer le rendement de la réaction, on met en œuvre une catalyse par transfert de phase en ajoutant au milieu réactionnel de l’hydrogénosulfate de tétrabutylammonium.
N HSO4
Les quatre groupements butyle de l’ion tétrabutylammonium lui permettent d’être soluble dans la phase organique. En migrant vers la phase organique l’ion tétrabutylammonium y transférera l’ion hypochlorite ClO- qui se trouvera alors dans la même phase que le diphénylméthanol. La catalyse par transfert de phase a été découverte par le chimiste polonais Makosza en 1965. Elle consiste à réaliser une réaction dans un milieu diphasé organique/aqueux en présence d'un catalyseur pouvant se partager entre les deux milieux, comme un ion ammonium quaternaire R4N+.

TP 4 : Oxydation d’un alcool secondaire Page 2
Le schéma ci-dessous illustre le processus de catalyse par transfert de phase.
Phase organique
Phase aqueuse
N(Bu)4 + ClO + alcool cétone + Cl + N(Bu)4
N(Bu)4 + ClO + HSO4 + Cl
ClO + HSO4 + Cl + N(Bu)4
2 L’eau de Javel L’eau de Javel est un mélange de chlorure et d’hypochlorite de sodium en milieu basique.
2.1 Degré chlorométrique de l'eau de Javel.
Le degré chlorométrique D d’une eau de Javel est un nombre sans dimension défini par la relation :
� � ������� � ����� ��
�
V(Cl2) désignant le volume de dichlore libéré (dans les conditions normales de température et pression) par un volume V d’eau de Javel lors de la réaction d’équation associée : ClO-
aq + Cl- aq + 2 H+ aq = Cl2 g + 2 H2O (2)
Dans les conditions normales de température et de pression (T = 273,15 K et P = 1,013 bar), le volume molaire du dichlore est égal à Vm = 22,4 L.mol-1 Les deux volumes V(Cl2) et V sont exprimés avec la même unité (par exemple le litre). Remarque : V(Cl2) = D × V Un litre d’une eau de Javel de degré chlorométrique D libère un volume de dichlore égal à D litres.
ii. Montrer que la concentration molaire volumique C des ions hypochlorite ClO- dans une eau de Javel
de degré chlorométrique D est donnée par la relation suivante :
� � ���
2.2 Pourcentage de chlore actif de l'eau de Javel
C’est un nombre sans dimension défini par la relation suivante :
%�. �. � ����������������
m(Cl2) désignant la masse de dichlore libéré par une masse m(solution) d’eau de Javel lors de la réaction (2), les deux masses m(Cl2) et m(solution) étant exprimées avec la même unité. iii. Montrer que :
%�. �. � � ������ 100�� � 1000 � � 0,32
�
avec D : degré chlorométrique de l’eau de Javel M(Cl2) = 70,9 g.mol-1 : masse molaire du dichlore Vm = 22,4 L.mol-1 : volume molaire du dichlore dans les CNTP ρ : masse volumique de l’eau de Javel exprimée en g.mL -1.

TP 4 : Oxydation d’un alcool secondaire Page 3
Quelques données relatives aux solutions commerciales :
% de chlore actif
D (°Chl ) chlore actif
g/L Densité
moyenne pH
Concentrés de Javel 9,6 34,88 à 33,68 110,56 à 106,78 1,152 à 1,112 12,5
Eau de Javel 2,6 8,51 à 8,43 26,96 à 26,73 1,037 à 1,028 11,5
3 Expériences préliminaires 3.1 Mise en évidence de la catalyse par transfert d e phase
� Dans un tube à essais, introduire environ 2 mL de dichlorométhane et quelques cristaux de permanganate de potassium.
� Ajouter 2 mL d’eau distillée. Boucher le tube et agiter. Noter les observations.
� Ajouter une pointe de spatule de catalyseur par transfert de phase.
� Boucher le tube et agiter. Noter les observations.
3.2 Dosage de l’eau de Javel
3.2.1 Principe du dosage
La prise d’essai d’eau de la solution d’eau de Javel est mise en présence d’une solution d’iodure de potassium (KI). Le pH de la solution est alors ajusté par ajout d’une solution d’acide éthanoïque de concentration voisine de 2 mol.L-1.
L’équation associée à la réaction d’oxydoréduction qui a lieu dans le milieu réactionel est :
ClO-(aq) + 2 H+(aq) + 2 I-(aq) = I2(aq) + H2O
Le diiode est ensuite dosé par une solution de thiosulfate de sodium jusqu’à disparition de la couleur brune.
I2 aq + 2 S2O32-
aq = 2I- aq + S4O62-
aq
3.2.2 Dosage de la solution S d’eau de Javel utilis ée lors ce cette séance de TP.
La solution S est une solution commerciale à 9,6% de chlore actif diluée 2 fois .
� Diluer 10 fois la solution S. Soit S’ la solution diluée.
� Dans un erlenmeyer contenant 20 mL d’une solution d’iodure de potassium de concentration massique voisine de 100 g.L-1, introduire E=10,0 mL de solution diluée S’ d’eau de Javel puis introduire sous agitation 10 mL d’une solution d’acide acétique de concentration voisine de 2 mol.L-1.
� Doser à l’aide d’une solution de thiosulfate de sodium de concentration molaire CT = 0,100 mol.L-1 jusqu’à décoloration de la solution (ajouter quelques gouttes d’empois d’amidon en fin de dosage pour déterminer le volume V1 à l’équivalence avec précision).
iv. Etablir la relation
���� � 12
�� �� 10
utilisée pour le calcul de la concentration de la solution S d’eau de Javel.
Calculer la concentration ����.

TP 4 : Oxydation d’un alcool secondaire Page 4
4 Synthèse de la diphénylméthanone 4.1 Synthèse en présence et en absence de catalyseu r
On réalise deux milieux réactionnels identiques, l’un contenant en plus le catalyseur.
� Dans deux erlenmeyers A et B introduire : • 100 mL de la solution S d’eau de Javel • 3,68 g de diphénylméthanol • 50 mL d’acétate d’éthyle
� Placer les deux erlenmeyers sous agitation magnétique.
� Introduire 0,68 g d’hydrogénosulfate de tétrabutylammonium dans l’erlenmeyer B.
� Laisser réagir pendant 45 min
4.2 Détermination des concentrations résiduelles en eau de Javel dans les solutions S A et SB
� Au bout de 45 min, prélever 10,0 mL de phase aqueuse SA et SB contenue respectivement dans les erlenmeyers A et B.
� Diluer 10 fois ces prélèvements. Soient S’A et S’B les solutions obtenues.
� Doser l’eau de Javel restant en suivant le même mode opératoire qu’au § 3.2.2. :
• Dans un erlenmeyer contenant 20 mL d’une solution d’iodure de potassium de concentration massique voisine de 100 g.L-1, introduire 10,0 mL de solution diluée S’A (S’B) puis introduire sous agitation 10 mL d’une solution d’acide acétique de concentration voisine de 2 mol.L-1.
• Doser à l’aide d’une solution de thiosulfate de sodium de concentration molaire 0,100 mol.L-1 jusqu’à décoloration de la solution (ajouter quelques gouttes d’empois d’amidon en fin de dosage pour déterminer le volume à l’équivalence avec précision). On notera V 2 le volume à l’équivalence pour la solution S’ A et V3 le volume à l’équivalence pour la solution S’ B.
v. Calculer les concentrations ����� et ���!�
4.3 Comparaison de l’avancement de la réaction en p résence et en absence de catalyseur
vi. Etablir la relation
" � 12
��� # �$� ��
permettant le calcul de l’avancement final x de la réaction d’oxydation du diphénylméthanol (avec Vi = V2 ou V3).
En déduire les valeurs des avancements de la réaction d’oxydation avec et sans catalyseur.
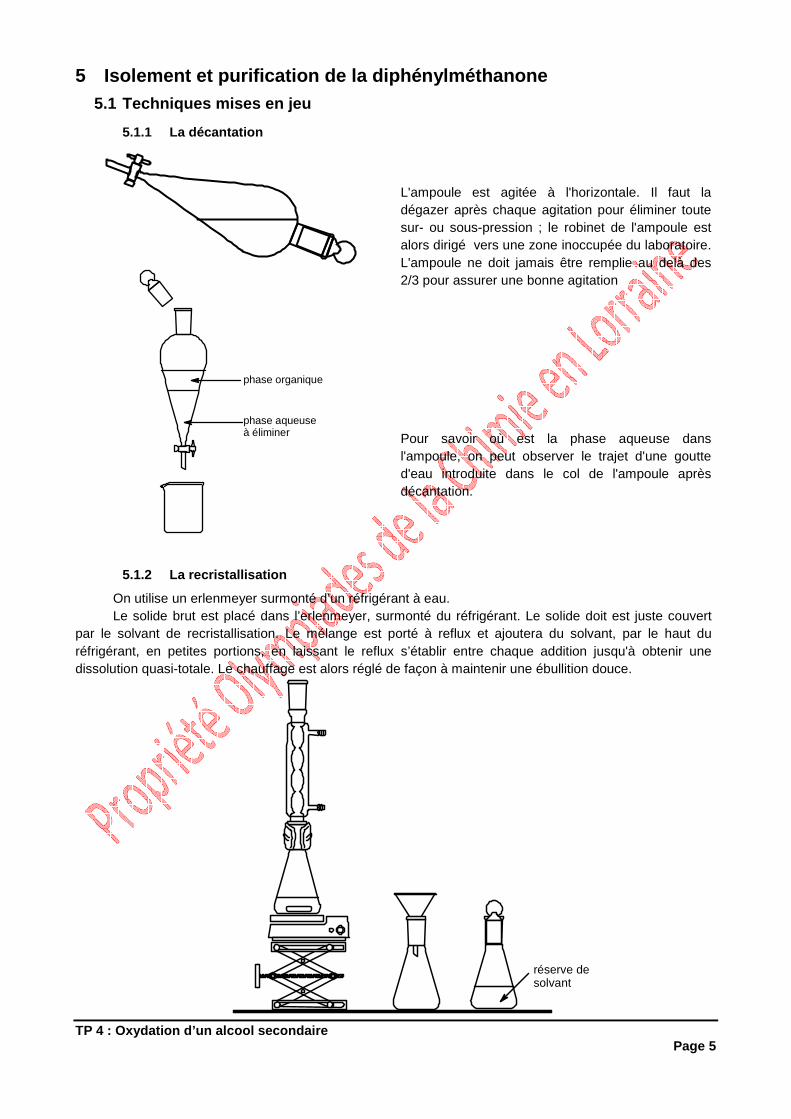
TP 4 : Oxydation d’un alcool secondaire Page 5
5 Isolement et purification de la diphénylméthanone 5.1 Techniques mises en jeu
5.1.1 La décantation
phase organique
phase aqueuseà éliminer
L'ampoule est agitée à l'horizontale. Il faut la dégazer après chaque agitation pour éliminer toute sur- ou sous-pression ; le robinet de l'ampoule est alors dirigé vers une zone inoccupée du laboratoire. L'ampoule ne doit jamais être remplie au delà des 2/3 pour assurer une bonne agitation Pour savoir où est la phase aqueuse dans l'ampoule, on peut observer le trajet d'une goutte d'eau introduite dans le col de l'ampoule après décantation.
5.1.2 La recristallisation
On utilise un erlenmeyer surmonté d’un réfrigérant à eau. Le solide brut est placé dans l’erlenmeyer, surmonté du réfrigérant. Le solide doit est juste couvert
par le solvant de recristallisation. Le mélange est porté à reflux et ajoutera du solvant, par le haut du réfrigérant, en petites portions, en laissant le reflux s’établir entre chaque addition jusqu'à obtenir une dissolution quasi-totale. Le chauffage est alors réglé de façon à maintenir une ébullition douce.
réserve de solvant

TP 4 : Oxydation d’un alcool secondaire Page 6
La solution est, si nécessaire, filtrée rapidement à chaud sur Büchner chaud pour éliminer les impuretés insolubles.
La solution chaude doit alors être refroidie, le produit à purifier cristallise. (Il est préférable de transvaser la solution dans un bécher - préalablement chauffé - d’où il sera plus facile de récupérer les cristaux). La qualité des cristaux obtenus dépend pour une grande part de la vitesse de refroidissement de la solution. Si le refroidissement est trop rapide, les cristaux sont petits et emprisonnent des impuretés Si le refroidissement est trop lent, les cristaux sont gros et emprisonnent du solvant dans le réseau cristallin. Pour obtenir un bon résultat, il faut procéder à un refroidissement lent au contact de l’air ambiant, puis immerger le récipient dans un bain réfrigérant. Evidemment, la température de ce bain ne doit pas être inférieure au point de congélation du solvant. Si la cristallisation n’intervient pas à froid après quelques minutes, on peut l’amorcer par création de points de germination des cristaux organiques autour des micro-éclats de verre obtenus en frottant les parois du bécher ou en introduisant un échantillon de produit cristallisé pur.
Filtration sur Büchner
Le contenu du bécher est alors filtré sur entonnoir Büchner. On lave alors le gâteau avec un peu de
solvant de recristallisation froid (le vide étant coupé ) puis on essore par aspiration. Le gâteau est ensuite séché pour le débarrasser des traces de solvant La séquence des opérations et l’ensemble des précautions à prendre sont les suivantes : - fixer solidement (noix + pinces) la fiole à vide
- employer un filtre dont le diamètre recouvre bien toutes les perforations de l’entonnoir ;
- mouiller le filtre avec un peu de solvant pour obtenir une bonne adhérence sur l’entonnoir et empêcher que des particules de solide ne s’infiltrent sous les bords du papier.
- appliquer le vide en ouvrant le robinet ; il n’est pas nécessaire d’exercer une forte aspiration initiale, car souvent de fines particules obstruent les pores du filtre, ce qui diminue la vitesse de filtration.
- couler d’abord le liquide qui surnage, puis la masse plus dense semi-solide. Tout le solide est transvasé dans l’entonnoir à l’aide d’une spatule ou d’un peu de filtrat
- laver le solide de la manière suivante : interrompre le vide, recouvrir le solide de solvant froid, appliquer de nouveau le vide ; répéter une deuxième fois l’opération.
- en maintenant le vide, essorer en pressant le solide avec un tapon ou un verre de montre.
baguette de verre
vide
pince serréeen fonctionnement

TP 4 : Oxydation d’un alcool secondaire Page 7
5.2 Protocole opératoire
� La phase organique contenue dans l’erlenmeyer B est séparée par décantation (ampoule à décanter).
� Laver deux fois la phase organique avec 40 mL d’une solution saturée de chlorure de sodium
� Laver deux fois la phase organique avec 40 mL d’eau
� Sécher la phase organique avec du sulfate de magnésium
� Eliminer le solvant par distillation simple. En fin de distillation, on travaillera sous pression réduite pour éliminer « complètement » le solvant. Si un évaporateur rotatif est disponible, on pourra l’utiliser pour effectuer cette opération. Le contenu du ballon à distiller se présente sous forme d’une « huile ».
� Transférer le contenu du ballon à distiller dans un bécher. On observe alors l’apparition d’un solide après refroidissement.
� Sécher puis peser le solide brut obtenu. La masse de produit brut est notée m1.
� Recristalliser le solide obtenu dans le minimum d’heptane. On note m2 la masse de produit brut engagé dans la recristallisation et m3 la masse de produit pur obtenu.
vii. Pourquoi les deux premiers lavages se font-ils avec une solution saturée de chlorure de sodium ?
viii. Expliquer, en quelques lignes, le principe de la recristallisation.
ix. Citer un test caractéristique des aldéhydes et des cétones
x. Citer un test caractéristique des aldéhydes.
xi. Etablir les relations littérales permettant le calcul
• du rendement R1 de la synthèse en produit brut
• du rendement R2 de la recristallisation
• du rendement R3 de la synthèse en produit pur
Calculer ces rendements
6 Analyses � Mesurer la température de fusion du produit recristallisé.
� Réaliser une C.C.M sur gel de silice :
• Effectuer trois dépôts : � diphénylméthanol en solution dans le dichlorométhane (fourni par le centre) ; � diphénylméthanone de référence en solution dans le dichlorométhane (fourni par le centre) ; � produit recristallisé dans le dichlorométhane (si la recristallisation n’a pas été faite et si tout le
solvant n’a pas été éliminé, on pourra utiliser une solution de l’huile obtenue dans le dichlorométhane).
• Eluer avec du dichlorométhane
• Révéler sous UV (254 nm).
• Déterminer les valeurs des rapports frontaux Rf
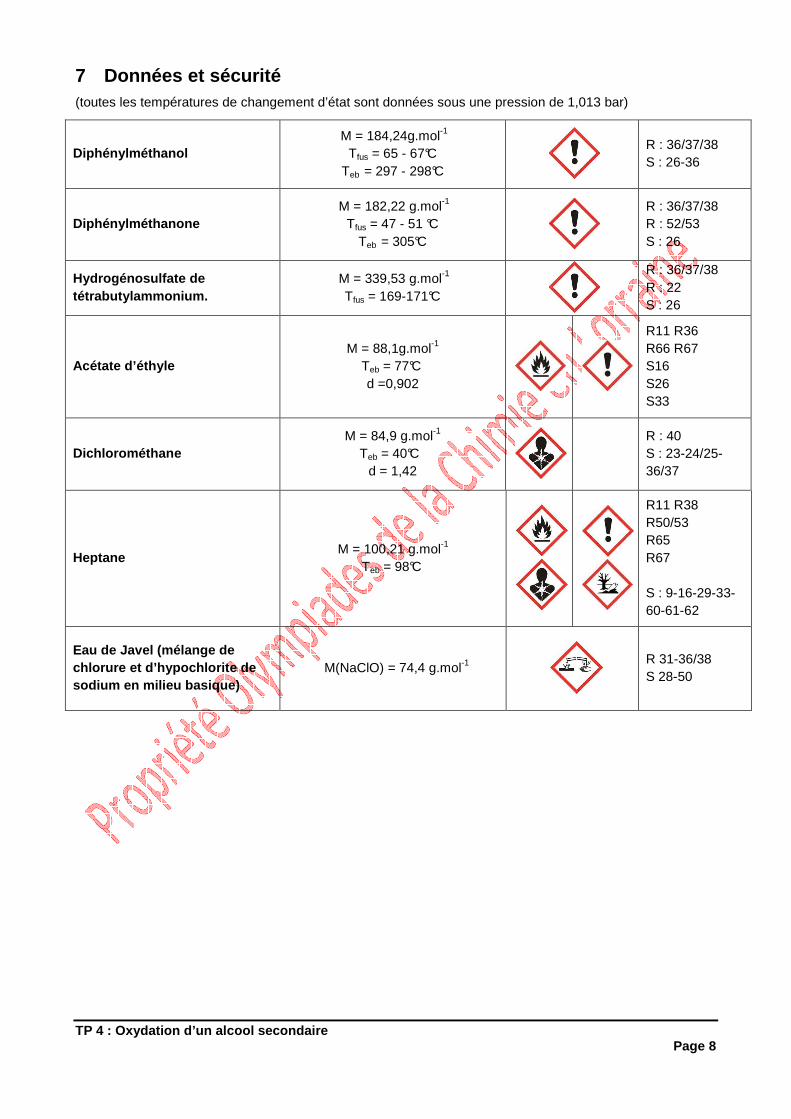
TP 4 : Oxydation d’un alcool secondaire Page 8
7 Données et sécurité (toutes les températures de changement d’état sont données sous une pression de 1,013 bar)
Diphénylméthanol M = 184,24g.mol-1
Tfus = 65 - 67°C Teb = 297 - 298°C
R : 36/37/38 S : 26-36
Diphénylméthanone M = 182,22 g.mol-1
Tfus = 47 - 51 °C Teb = 305°C
R : 36/37/38 R : 52/53 S : 26
Hydrogénosulfate de tétrabutylammonium.
M = 339,53 g.mol-1
Tfus = 169-171°C
R : 36/37/38 R : 22 S : 26
Acétate d’éthyle M = 88,1g.mol-1
Teb = 77°C d =0,902
R11 R36 R66 R67 S16 S26 S33
Dichlorométhane M = 84,9 g.mol-1
Teb = 40°C d = 1,42
R : 40 S : 23-24/25-36/37
Heptane M = 100,21 g.mol-1
Teb = 98°C
R11 R38 R50/53 R65 R67 S : 9-16-29-33-60-61-62
Eau de Javel (mélange de chlorure et d’hypochlorite de sodium en milieu basique)
M(NaClO) = 74,4 g.mol-1
R 31-36/38 S 28-50

TP 4 : Oxydation d’un alcool secondaire Page 9
Académie de Nancy - Metz Olympiades de la chimie 2010-2011 « La chimie et l’eau »
TP 4 : Oxydation d’un alcool secondaire LISTE MATERIELS ET PRODUITS
(en rouge non disponible dans les lycées a priori)
Produits à prévoir par poste de travail :
• Solution S d’eau de Javel préparée en diluant 2 fois une solution d’eau de Javel à 9,6% de chlore actif. :
200 mL
L’eau de Javel à 9,6 % de chlore actif est vendue en berlingots .
• Diphénylméthanol : 4g
• Hydrogénosulfate de tétrabutylammonium : 1g
• Heptane : 20mL
• Ethanoate (acetate) d’éthyle : 100mL
• Dichloromethane : 100ml
• Solution de thiosulfate de sodium de concentration 0,100 mol/L : 100mL
• Thiodène : quelques grammes
• Permanganate de potassium : quelques grammes
• Solution de KI à 100 g/L : 100mL
• Solution d’acide acétique de concentration voisine de 0,2 mol/L : 100mL
• Sulfate de magnésium anhydre : 20g
• 1 plaque CCM à révéler sous UV à 254 nm
Produits à prévoir par centre :
• Diphénylméthanone (benzophénone) : 5g
• Solution étalon de diphényméthanol dans CH2Cl2 pour CCM
• Solution étalon de diphényméthanone dans CH2Cl2 pour CCM

TP 4 : Oxydation d’un alcool secondaire Page 10
Académie de Nancy - Metz Olympiades de la chimie 2010-2011 « La chimie et l’eau »
TP 4 : Oxydation d’un alcool secondaire COMMENTAIRES PROTOCOLES ET REPONSES AUX QUESTIONS
1. Principe de la manipulation i. Ecrire les demi-équations d’oxydoréduction des couples :
- HClO/Cl- : HClO + H+(aq) + 2 e- = H2O + Cl-
- diphénylméthanone/diphénylméthanol : C13H12O = C13H10O + 2H+(aq) + 2 e-
Soit l’équation de la réaction qui est associée à la transformation réalisée dans ce TP C13H12O + HClO = C13H10O + H+(aq) + H2O + Cl-
2. L’eau de Javel
� � ������� � ����� ��
�
V(Cl2) désignant le volume de dichlore libéré (dans les conditions normales de température et pression) par un volume V d’eau de Javel lors de la réaction d’équation associée : ClO-
aq + Cl- aq + 2 H+ aq = Cl2 g + 2 H2O (2)
Dans les conditions normales de température et de pression (T = 273,15 K et P = 1,013 bar), le volume molaire du dichlore est égal à Vm = 22,4 L.mol-1 Les deux volumes V(Cl2) et V sont exprimés avec la même unité (par exemple le litre). Remarque : V(Cl2) = D × V Un litre d’une eau de Javel de degré chlorométrique D libère un volume de dichlore égal à D litres.
ii. Expression de la concentration molaire volumique C des ions hypochlorite ClO-
� � ���%&�� � �����
� � ���
iii. Expression du % de chlore actif
%�. �. � ����� ������ 100� � 1000 � � ������ 100
�� � 1000 � � 0,32�

TP 4 : Oxydation d’un alcool secondaire Page 11
3. Expériences préliminaires
3.2.2. Dosage de la solution S d’eau de Javel utili sée lors ce cette séance de TP.
Pour préparer l’eau de Javel nécessaire à la synthè se, on dilue 2 fois une solution d’eau de Javel à 9,6% de chlore actif. L’eau de Javel à 9,6 % de chlore actif en vendue en berlingots.
iv. Expression de ����
���'� � ���%&� � �(�� � )��%*�&+
2 � �� ��2
���� � ���'� 10 � 12
�� �� 10
A.N. ,- � -., -/0
1�2� � 12
14,1 0,10010 10 � 4, 546/78. 0&-
4. Synthèse de la diphénylméthanone 4.1 Synthèse en présence et en absence de catalyseu r
Il faut compter 3h pour mener à bien cette synthèse . Organisation du travail :
- Mettre en route les deux synthèses (avec et sans catalyseur). Laisser réagir pendant 45 min. L’agita tion doit être efficace.
- Pendant les 45 min :
- Faire le dosage de l’eau de Javel - Faire le montage de distillation simple - Faire l’expérience de mise en évidence des propriét és
du catalyseur par transfert de phase - Préparer l’ampoule à décanter et rassembler les
solutions de lavage
- Au bout des 45 min, prélever les 10,0 mL de chaque phase aqueuse. Préparer les solutions S’ A et S’B qui pourront être dosées pendant la distillation.
Aspect du milieu réactionnel

TP 4 : Oxydation d’un alcool secondaire Page 12
(biphasique) après 45 min.
4.2. Détermination des concentrations résiduelles e n eau de Javel dans les solutions S A et SB
v. Calculer les concentrations ����� et ���!�
A.N. ,9 � -:, 5/0
1�2;� � 12
13,7 0,10010 10 � 4, =>6/78. 0&-
A.N. ,: � ?, :/0
1�2@� � 12
9,3 0,10010 10 � 4, .=6/78. 0&-
4.3. Comparaison de l’avancement de la réaction en présence et en absence de catalyseur
vi. L’avancement de la réaction d’oxydation s’exprime par :
" � ���%&�$B$C$DEF # ���%&�G$BDE
soit " � )���� # ���� �� �! �+ �IJK�L KM�L��L
soit " � 12
��� # �$� �� 10 �IJK�L KM�L��L
Sachant que �NODPF DQRFRPF= 0,1L, on a finalement
soit S � -9
�,- # ,T� 1UV
En déduire les valeurs des avancements de la réaction d’oxydation avec et sans catalyseur.
diphénylméthanol ClO- diphénylméthanone
Quantités initiales en mmol 94 54, 6 4
Quantités finales avec catalyseur 94 # S@ 54, 6 # S@ � .=, = S@
Quantités finales sans catalyseur 94 # S; 54, 6 # S; � =>, 6 S;
On constate que S@ W S;
Rq : on trouve une valeur par excès de l’avancement �S@ W 20//78XX� : une partie des ions
hypochlorite se trouve dans la phase organique !

TP 4 : Oxydation d’un alcool secondaire Page 13
5. Isolement et purification de la diphénylméthanon e - Réaliser les opérations de décantation, lavage et s échage de la phase organique.
- Si on dispose d’un évaporateur rotatif l’éliminatio n du solvant (acétate d’éthyle) sera facile. Sinon
on distille : on pourra utiliser un chauffe ballon ou un bain marie bouillant. Pour éliminer les
dernières traces de solvant j’ai travaillé sous pre ssion réduite. Le solide n’apparaît pas
immédiatement. On transfère l’huile obtenue dans un bécher et on laisse refroidir.
Le montage utilisé ici permet de travailler sous
pression réduite.
Distillation simple. Le cristallisoir contient un
bain d’écailles de graphite à la place d’eau
bouillante
- Pour la recristallisation, on pourra opérer sur la moitié du produit brut.
- Attention à la quantité d’heptane utilisée qui doit être minimale : pour 1,4 g de solide il faut moins
de 5 mL d’heptane ! ! ! On travaillera dans un erle n (ou un ballon) de 50 mL.
- On obtient de belles aiguilles blanches
- Remarque : si – pour des raisons de temps -
on ne parvient pas à isoler la
diphénylméthanone on peut faire la CCM avec
le résidu présent dans le ballon à distiller à la
fin de la distillation. Ce résidu contient la
diphénylméthanone dissoute dans un peu
d’acétate d’éthyle.

TP 4 : Oxydation d’un alcool secondaire Page 14
5.2. Protocole opératoire
vii. Le relargage permet de bien séparer les deux phases du mélange réactionnel : la phase aqueuse et
la phase organique. L’acétate d’éthyle est un peu soluble dans l'eau, il l'est moins dans une solution ionique (solution saturée de chlorure de sodium).
viii. La recristallisation à pour but de purifier un prod uit en le débarrassant des impuretés qu'il pourrait contenir. Cette technique repose sur la différence de solubilité entre les impuretés et le produit pur dans un solvant donné à chaud et à froid.
Sachant qu’en général, la solubilité d’un solide diminue avec la température, la recristallisation consiste donc à : � Dissoudre le produit brut dans le minimum de solvant à ébullition. � Éliminer les impuretés insolubles à chaud (filtration à chaud) � Refroidir la solution. Le produit cristallise seul, en laissant les impuretés en solution dans le
solvant. � Récupérer le solide désiré par filtration sur büchner
ix. Test à la 2,4-D.N.P.H.
x. Test au réactif de TOLLENS ou à la LIQUEUR DE FEHLING
xi. Etablir les relations littérales permettant le calcul
• du rendement R1 de la synthèse en produit brut
Y- � /- Z1-:[-4\⁄)^1-:[-9\+T^T_T`8a
A.N. Masse de produit brut sec : b- � 9, -?c (un deuxième essai a donné b- � 9, ?.c )
Y- � 9, -? ->9, 99⁄4, 494 � =4%
• du rendement R2 de la recristallisation
Y9 � /:/9
A.N. Masse de produit engagée dans la recristallisation: b9 � -, :>c Masse de produit recristallisé: b: � 4, ?5c
Y9 � 4, ?5-, :> � 54%
• du rendement R3 de la synthèse en produit pur
Y: �d/:/-
/9e Z1-:[-4\f
)^1-:[-9\+T^T_T`8a� Y- Y9 � .9%

TP 4 : Oxydation d’un alcool secondaire Page 15
6. Analyses Température de fusion du produit recristallisé (ban c Koffler) : 51°C CCM :
Rf (diphénylméthanone) = 4,25 /7,5 = 0,57
Rf (diphénylméthanol) = 2,5/7,5 = 0,33
De gauche à droite : � diphénylméthanone � diphénylméthanol � produit synthétisé

TP 5 : Synthèse de l’hélianthine ou méthylorange

TP 5 : Synthèse de l’hélianthine ou méthylorange Page 1
Académie de Nancy - Metz Olympiades de la chimie 2010-2011 « La chimie et l’eau »
TP 5 : Synthèse de l’hélianthine ou méthylorange
1 Introduction: De nombreuses substances chimiques prennent en solution aqueuse une couleur qui dépend des conditions de pH. Cette propriété est mise à profit pour déterminer les caractéristiques des solutions en y introduisant l’indicateur sous forme de solution ou bien en utilisant une bandelette de papier imbibée d’indicateur : le papier-pH. Le choix du bon indicateur permet également de repérer l’équivalence au cours des titrages. Dès le dix-septième siècle, Robert Boyle[1] utilise des décoctions de plantes comme indicateurs colorés. Dans son ouvrage « expériences sur les couleurs » il explique en 1663 comment mettre en évidence le caractère acide des solutions à l’aide de sirop de violette. Il suggère de déposer quelques gouttes de sirop sur un papier blanc puis de déposer l’acide sur le papier : la tache de sirop passe du violet au rouge. Il donne ainsi le premier protocole expérimental de test d’acidité à l’aide d’un indicateur coloré et invente du même coup le papier-pH ! À la fin du dix-huitième siècle on commence à utiliser les indicateurs colorés naturels dans les procédés industriels à des fins de contrôles. On constate alors que les solutions acides n’ont pas toutes le même effet sur les couleurs des indicateurs ; cela conduit à la mise au point des premières échelles de comparaison des forces des acides. Il faudra attendre encore un siècle pour développer les premières synthèses d’indicateurs : la phénolphtaléine en 1877 et le méthylorange ou hélianthine en 1878. Le nombre des indicateurs ne cesse de croître et la variété de leur comportement vis-à-vis des différentes solutions acides et basiques est mal comprise jusqu’en 1894, quand Wilhelm Ostwald[2] propose la première théorie des indicateurs. Ostwald émet l’hypothèse suivante : l’indicateur est un acide HIn ou une base faible In- dont les formes dissociées et protonées sont de couleurs différentes. Cette hypothèse s’avère exacte et la définition moderne la reprend en donnant plus de précisions : un indicateur coloré est un acide ou une base faible organique dont la structure interne change lors de l’échange de proton ce qui entraîne une variation de couleur. L'hélianthine est un solide cristallisé de masse molaire. En solution aqueuse, il est utilisé comme indicateur coloré acide-base .La zone de virage de l’hélianthine est comprise entre 3,1 et 4,4 avec la forme acide (HInd) de couleur rouge et la forme basique (Ind-) de couleur jaune-orangée.
La synthèse de l'hélianthine se décompose en deux grandes étapes :
� une réaction de diazotation d'une amine aromatique. � une réaction de copulation sur un dérivé aromatique.
NaO3S NN N

TP 5 : Synthèse de l’hélianthine ou méthylorange Page 2
2 Synthèse de l’hélianthine 2.1 Les réactions de diazotation
Les amines aromatiques Ar-NH2 réagissent avec l'acide nitreux HNO2 pour donner l'ion diazonium Ar-N2
+. Cet ion n'est stable que si la température est inférieure à 5°C sinon il se décompose pour donner le benzophénol Ar-OH et un dégagement de diazote N2. Remarque :
� L'acide nitreux HNO2 se décompose lentement en subissant une dismutation en donnant HNO3 et NO. Il faudra le fabriquer "in situ" en faisant réagir le nitrite de sodium sur l'acide chlorhydrique.
i. Écrire la réaction de dismutation avec les couples HNO3/ HNO2 et HNO2/NO
� Les amines aromatiques sont peu solubles dans l'eau, il faudra les dissoudre au préalable dans une solution d'acide chlorhydrique.
La réaction de diazotation s'effectue en trois étapes :
� Dissolution de l'amine
� Formation de l'acide nitreux par action de l'acide chlorhydrique sur le nitrite de sodium.
� Diazotation. La réaction de diazotation étant légèrement exothermique, il sera nécessaire de refroidir le milieu réactionnel pendant l'addition des réactifs.
2.2 Les réactions de copulation azoïque
Les ions diazonium réagissent dans des réactions de substitution électrophile sur des dérivés aromatiques possédant des groupes donneurs d'électrons (-OH, -NH2, -NRR'......) La copulation se fait en para du groupe donneur. Si la position est déjà occupée, elle se fait en ortho ou pas du tout.
Exemple : avec le phénol
2.3 Protocole opératoire
2.3.1 Diazotation de l'acide sulfanilique
2.3.1.1 Préparation de la solution d'acide 4- amino benzène sulfonique ou acide sulfanilique
� Dans un erlenmeyer de 150 mL, introduire: • 10 mmol d'acide sulfanilique • 10 mL d'eau et 30 g de glace pilée.
� Puis rajouter 1,7 mL d'acide chlorhydrique concentré.
� Mettre sous agitation durant 5 minutes.
OHposition ortho
position para

TP 5 : Synthèse de l’hélianthine ou méthylorange Page 3
2.3.1.2 Préparation de la solution de nitrite de so dium
� Dans un erlenmeyer de 50 mL dissoudre 0,75 g de nitrite de sodium (NaNO2) dans 10 mL d'eau.
� Refroidir cette solution dans la glace.
� Ajouter cette solution goutte à goutte à la solution refroidie d'acide sulfanilique.
Le sel de diazonium précipite. La présence d'acide nitreux est contrôlé avec du papier à l'iodure de potassium (l'apparition d'une coloration violette au niveau du papier indique la présence d'acide nitreux). (Solution A )
� A la fin de l'addition, laisser sous agitation durant 10 min.
2.3.2 Copulation azoïque avec la N,N-diméthylamine
� Dissoudre 1,2 mL de diméthylaniline que l'on place dans un erlenmeyer de 250 mL
� Ajouter 50 mL d'eau, puis 1 mL d'acide acétique glacial. (Solution B)
Sécurité : Attention les amines sont toxiques, opérer sous la hotte, prélever la quantité juste nécessaire, la pipette ayant contenu l'amine sera d isposée dans une éprouvette contenant de l'acide chlorhydrique 1M. � Ajouter lentement la solution A à la solution B, en agitant vigoureusement. Un précipité se forme. � Basifier la solution à l'aide d'une solution de soude à 10 % (vérifier le pH). L'hélianthine orange apparaît. � Dissoudre le précipité formé en chauffant au bain-marie (ne pas dépasser 90 °C) � Après dissolution, ajouter environ 10 g de chlorure de sodium. Continuer à chauffer jusqu'à dissolution. � Laisser ensuite refroidir, puis entourer le bécher de glace pilée. Attendre la cristallisation. � Filtrer le solide sur Büchner, bien essorer.
2.3.3 Recristallisation
� Recristalliser l'hélianthine dans l'eau (solubilité à 20°C = 0,20 g /100 mL). Laisser refroidir.
� Filtrer, sécher le solide. Peser la masse m1 de produit sec.
(On pourra éventuellement ajouter de l'acétone pour faciliter la mise en solution de l'hélianthine)
2.3.4 Question sur le mode opératoire
ii. Ecrire l'équation de la diazotation
iii. Pourquoi travailler à une température inférieure à 5 °C lors de la diazotation ?
iv. Après la diazotation, on vérifie la présence d'acide nitreux en utilisant un papier iodo-amidoné (papier sur lequel sont déposés des ions iodures I- et de l'amidon). Connaissant les potentiels des couples rédox HNO2/NO E°
1 = 1,00 V et I2/ I- E°2 = 0,54 V,
écrire les équations chimiques qui ont lieu s'il reste de l'acide nitreux.
v. Écrire l'équation de copulation azoïque.
vi. Quel est le réactif limitant au cours de cette synthèse ?
vii. Quelle est la masse théorique d'hélianthine ?
viii. Calculer le rendement global de la préparation.

TP 5 : Synthèse de l’hélianthine ou méthylorange Page 4
3 Détermination du pk A de l’hélianthine par spectrophotométrie 3.1 Introduction
3.1.1 La spectrophotométrie
Une source S éclaire un réseau qui disperse la lumière. Une fente F isole un faisceau monochromatique. La position du réseau est réglable pour sélectionner la longueur d'onde λ voulue entre 380 nm et 660 nm pour l'appareil utilisé (le S250PC) ; on ne balaie donc pas toute la gamme du visible. La radiation ainsi obtenue (I0) traverse une cuve contenant la solution colorée à étudier : cette solution absorbe une certaine quantité de lumière. Le reste (I) atteint la cellule de mesure : une photodiode. On mesure ainsi une grandeur appelée absorbance , notée A qui dépend du flux lumineux reçu.
L’absorbance � � ������ �⁄ est la somme de toutes les absorbances de toutes les espèces chimiques en solution.
Dans le cas présent, � � ��� � ����� � ����� � �� ���� ���è��� ����������� ��. 3.1.2 Rappel sur le couple HInd/Ind –
L’hélianthine est un composé organique qui existe sous deux formes : Sa forme acide HIn est rouge en milieu aqueux. Sa base conjuguée In– est jaune-orangée en milieu aqueux. L’équilibre acido-basique est donc :
Hin(aq) + H2O(l) = H3O+ (aq) + In–(aq)
[HIn] et [In–] sont liées par la constante d’acidité du couple HIn / In– :
! � "#$%&'"�(�'"#�('
Cette égalité peut s'écrire sous forme logarithmique avec la notation ) ! � *��� !
) ! � )# * ��� "�(�'"#�('
soit )# � ) ! � ��� "�(�'"#�('
On déterminera dans une solution aqueuse de pH conn u, la valeur du rapport "+,�' "-+,'⁄ ; ce
rapport sera mesuré par spectrophotométrie d’absorp tion, on en déduira le ./0.
3.2 Principe de la détermination du rapport "+,�' "-+,'⁄
La détermination du rapport se fait à partir des valeurs des absorbances des trois solutions suivantes, pour une longueur d’onde donnée. C étant la concentration molaire en soluté de la solution d’hélianthine, on a alors pour toute solution : C = [HIn] + [In-]
Solution 1 Solution 2 Solution 3
monochromateur cellule amplificateur ordinateur S λ λ
I0 I
cuve

TP 5 : Synthèse de l’hélianthine ou méthylorange Page 5
Hélianthine à pH connu ( ≈≈≈≈ 3,5) solution orange
Les deux formes colorées présentes le sont à des concentrations molaires
du même ordre de grandeur. [HIn] 1 + [In –]1 = C Absorbance A 1
Hélianthine à pH ≈≈≈≈ 1 solution rouge
[In–]<<[HIn] Forme majoritaire : HIn
[HIn] 2 ≈≈≈≈ C Absorbance A 2
Hélianthine à pH ≈≈≈≈ 13 solution jaune
[HIn] << [In–] Forme majoritaire In–
[In –]3 ≈≈≈≈ C Absorbance A 3
3.3 Préparation des trois solutions d’hélianthine
Préparer 200 mL d’une solution aqueuse d’hélianthine H à 0,06% en masse. Il faut chauffer légèrement la solution sur une plaque chauffante pour dissoudre l’hélianthine. On peut utiliser l’hélianthine synthétisée pendant la séance ou de l’hélianthine « commerciale »
ix. Calculer la masse d’hélianthine nécessaire (une pesée à 0,01 g près est suffisante)
� Préparer 200 ou 500 mL d’une solution (T) aqueuse d’hydrogénophtalate de potassium à 0,1 mol/L.
x. Calculer les masses d’hydrogénophtalate de potassium nécessaire.
Cette préparation ne sera exécutée qu’après avoir r épondu à la question xi ci-dessous. � A l’aide d’une électrode pH-métrique et d’un pH-mètre préalablement étalonné et d’une solution d’acide
chlorhydrique à 0,1 mol/L, amener cette solution (T) à un pH entre 3,5 et 4. Noter cette valeur pH1 = ….
� Réaliser une dilution au 50ème de la solution H dans trois fioles.
• Solution 1 : dilution sera réalisée avec la solution (T1) d’hydrogénophtalate de potassium de pH1 (Conserver précieusement le reste de la solution d’hydrogénophtalate de potassium de pH1 pour une utilisation ultérieure ; une dizaine de mL est suffisante)
• Solution 2 : dilution réalisée avec une solution d’acide chlorhydrique à 0,1 mol/L.
• Solution 3 : dilution réalisée avec une solution de soude à 0,1 mol/L
xi. Quel matériel (pipette graduée, fiole jaugée) pouvez-vous utiliser pour réaliser chacune des trois
solutions 1, 2,3 ? Une solution plus « coûteuse » en solution, mais plus rapide pourra être privilégiée pour un gain de temps.
3.4 Mesures au spectrophotomètre
Pour chacune des trois solutions, on trace le spectre d'absorption � � 1�2 à l'aide du spectrophotomètre. On utilisera préalablement pour chacune des trois solutions1,2 et 3, un blanc d’hélianthine de pH correspondant. On envoie les spectres obtenus sur 3 pages différentes du même fichier de type "REGRESSI" puis à l'aide du logiciel Regressi on superpose les trois spectres (à l'aide du bouton Coordonnées, (YX), charger la fenêtre Coordonnées du graphe puis cliquer sur la case : Superposition des pages (en cas d'absence de cette option, cliquer sur la case : Autre boite de dialogue coordonnées puis recharger la fenêtre Coordonnées du graphe). Les 3 spectres se trouvent alors superposés sur le même graphe.
A l'aide du curseur Réticule, déterminer les absorbances �3, �4 et �$ pour une longueur d'onde 2 puis les
absorbances �53, �54 et �5$ pour une longueur d'onde 25. (Cf. le modèle de graphe suivant).
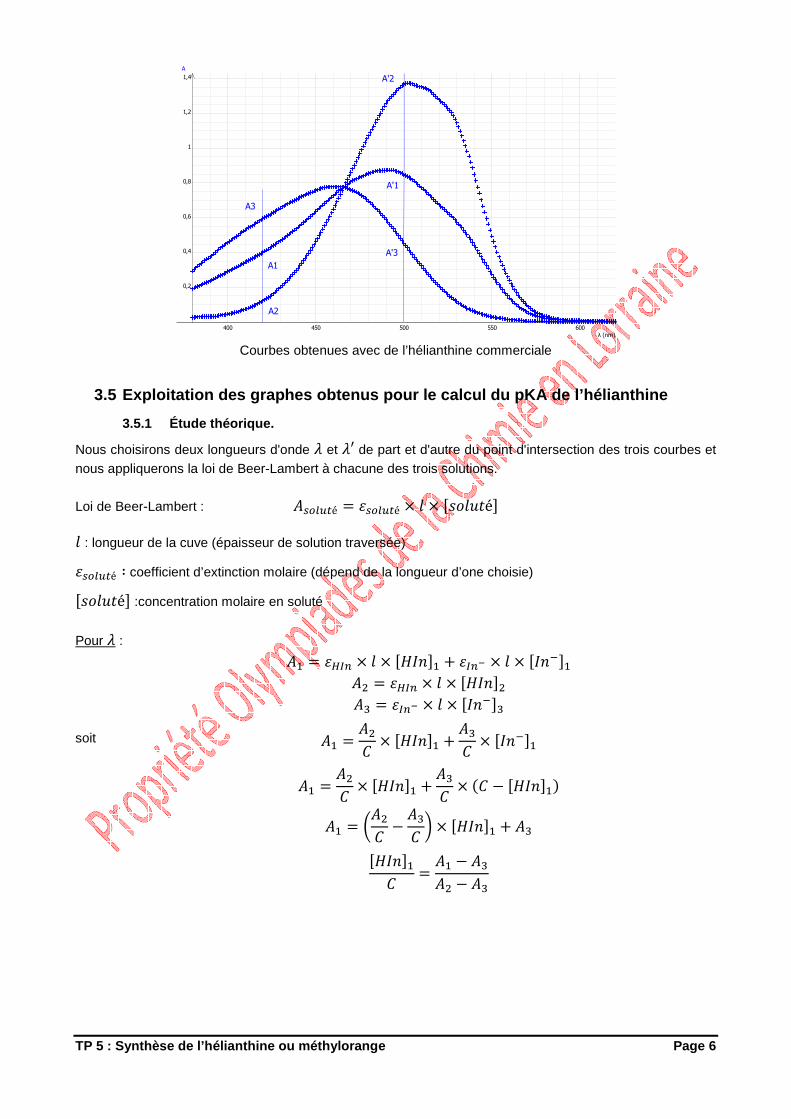
TP 5 : Synthèse de l’hélianthine ou méthylorange Page 6
Courbes obtenues avec de l’hélianthine commerciale
3.5 Exploitation des graphes obtenus pour le calcul du pKA de l’hélianthine
3.5.1 Étude théorique.
Nous choisirons deux longueurs d'onde 2 et 25 de part et d'autre du point d'intersection des trois courbes et nous appliquerons la loi de Beer-Lambert à chacune des trois solutions.
Loi de Beer-Lambert : ���6 �é � 8��6 �é 9 � 9 ":��;<é' � : longueur de la cuve (épaisseur de solution traversée)
8��6 �é = coefficient d’extinction molaire (dépend de la longueur d’one choisie)
":��;<é' :concentration molaire en soluté
Pour 2 :
�3 � 8��� 9 � 9 "#�('3 � 8��� 9 � 9 "�(�'3
�4 � 8��� 9 � 9 "#�('4
�$ � 8��� 9 � 9 "�(�'$
soit �3 � �4> 9 "#�('3 � �$> 9 "�(�'3
�3 � �4> 9 "#�('3 � �$> 9 �> * "#�('3
�3 � ?�4> * �$> @ 9 "#�('3 � �$
"#�('3> � �3 * �$�4 * �$
λ (nm)
400 450 500 550 600
A
0,2
0,4
0,6
0,8
1
1,2
1,4
A1
A2
A3
A'3
A'1
A'2

TP 5 : Synthèse de l’hélianthine ou méthylorange Page 7
Pour 25 :
�53 � 8��� 9 � 9 "#�('3 � 8��� 9 � 9 "�(�'3
�54 � 8��� 9 � 9 "#�('4
�5$ � 8��� 9 � 9 "�(�'$
soit �53 � �54> 9 "#�('3 � �5$> 9 "�(�'3
�53 � �54> 9 �> * "�(�'3 � �5$> 9 "�(�'3
�53 � A�5$> * �54> B 9 "�(�'3 � �54
"�(�'3> � �53 * �54�5$ * �54
donc "�(�'3"#�('3 � �53 * �54�5$ * �54 9 �4 * �$�3 * �$
donc ) ! � )#3 * ��� "�(�'3"#�('3 � )#3 * ��� A�53 * �54�5$ * �54 9 �4 * �$�3 * �$B
3.5.2 Exploitation des résultats.
A partir de la lecture du graphe, compléter le tableau ci-dessous puis calculer le pKA de l’hélianthine La mesure du pH de la solution 1 donne : pH1 =
2 �
�3 �
25 �
�53 �
) ! � �4 � �54 �
�$ � �5$ �
Références bibliographiques : 400 manipulations commentées de chimie organiques vol1 Jean-Pierre BAYLE Tables de chimie Un memento pour laboratoire Jacques Tonneau TP dosage spectro du BBT (Vincent Trossat , lycée Varoquaux) http://culturesciences.chimie.ens.fr/dossiers-experimentale-analyse-article-Indicateurs_pH_Demirdjian.html#ftn.d0e42

TP 5 : Synthèse de l’hélianthine ou méthylorange Page 8
Académie de Nancy - Metz Olympiades de la chimie 2010-2011 « La chimie et l’eau »
TP 5 : Synthèse de l’hélianthine ou méthylorange LISTE MATERIELS ET PRODUITS
Produits et solutions (en rouge non disponible dans les lycées a priori) Synthèse de l’hélianthine * PRODUITS
- acide sulfanilique (MM = 173,19 g.mol-1) : 1,73 g
- nitrite de sodium (MM = 36,45 g.mol-1 , 37 % , d = 1,18, produit toxique et corrosif) : 0,75g
- N, N-diméthylaniline (MM = 121,18 g.mol-1 , d = 0,950) : 1,2mL
(Attention produit toxique, à manipuler sous la hotte)
- acide acétique glacial : 1 mL
- acide chlorhydrique concentré : 1,7mL
- NaOH à 10 % en masse
- acétone
* MATERIEL
- erlenmeyers de 50, 150 mL
- éprouvette graduée de 100 mL
- fiole à vide + büchner
- erlenmeyer de recristallisation
- réfrigérant à boules
- agitateur magnétque chauffant et non chauffant.
- turbulent
- papier pH
- papier iodo-amidonné

TP 5 : Synthèse de l’hélianthine ou méthylorange Page 9
Etude spectrophotométrique de l’hélianthine * PRODUITS
- Hélianthine de synthèse ou commerciale (MM = 327,3 g.mol-1) : 0,12g
- Hydrogénophtalate de potassium KHA (MM = 204,23 g.mol-1) : 4,08g pour 200mL ou 10,21g pour 500 mL ?
- solution d’acide chlorhydrique à 0,1 mol/L : environ 150 ou 300 mL par poste
- solution d’hydroxyde de sodium à 0,1 mol/L : environ 150 ou 300 mL par poste
* MATERIEL
- sabot ou coupelle de pesée (2 par poste)
- spatule
- plaque chauffante (1 ou 2)
- agitateur en verre ou barreau aimanté et agitateur magnétique
- balance de précision
- béchers de prélèvement
- 1 pipette jaugée de 5 mL et 10 mL
- fiole jaugée de 200 mL (1 ou 2 par poste)
- fiole de 500 mL (1 par poste)
- 3 fioles de 250 mL ou 3 fioles de 50 mL (par poste)
Pour la dilution au 1/50 :
� Une dilution pipette de 5 mL + fiole de 250 mL…..plus rapide, plus couteuse en KHA, acide, base
� Deux dilutions (5 mL dans 50 mL puis 10 mL dans 50 mL) moins rapide, moins couteuse
Rq : la solution de KHA amenée à pH 3,5 nécessite d’après « table de chimie p37 » environ 8 mL d’acide chlorhydrique à 0,1 mol/L pour 50 mL de KHA à 0,1 mol/)…Ajout qui permettrait de ne faire que 100mL ou 250 de KHA pour l’utilisation en TP - Spectrophotomètre + cuves + ordinateur
fonction de la dilution au 1/50

TP 5 : Synthèse de l’hélianthine ou méthylorange Page 10
Académie de Nancy - Metz Olympiades de la chimie 2010-2011 « La chimie et l’eau »
TP 5 : Synthèse de l’hélianthine ou méthylorange REPONSES AUX QUESTIONS
2.1 Les réactions de diazotation
i. Écrire la réaction de dismutation avec les couples HNO3/ HNO2 et HNO2/NO
2.3.4. Question sur le mode opératoire
ii. Ecrire l'équation de la diazotation
La réaction de diazotation s'effectue en trois étapes : 1) Dissolution de l'amine :
Ar * NH4 � �H$O& � Cl� � Ar * NH$& � Cl� � H4O
2) Formation de l'acide nitreux par action de l'acide chlorhydrique sur le nitrite de sodium :
�Na& � NO4� � �H$O& � Cl� � HNO4 � H4O � �Na& � Cl�
(L'acide nitreux instable thermodynamiquement, se forme "in situ" par l'ajout d'acide à une solution de nitrite de sodium. Cet acide nitreux évolue par élimination d'une molécule d'eau pour
former le cation nitrosium KL& . Le cation formé réagit ensuite avec le meilleur site nucléophile c'est-à-dire l'atome d'azote de l'acide sulfanilique). 3) Réaction de diazotation :
Ar * NH$& � HNO4 � Ar * N4& � 2H4O
Bilan :
Ar * NH4 � �Na& � NO4� � 2�H$O& � Cl� � NAr * NH$& � Cl�O � �Na& � Cl� � 4H4O
iii. La réaction de diazotation étant légèrement exothermique, il faut refroidir afin d'éviter la destruction de l'ion diazonium formé.
iv. Le papier iodo-amidonné met en évidence l'excès d'acide nitreux. Ce papier renferme des ions iodure I- et de l'amidon. L'acide nitreux oxyde les ions iodure I- en diiode I2 ce qui colore en brun violet le papier.
HNO2 � e* � H��aq S NO � H2O �9 22I* S I2 � 2e*________________________________________________________2HNO2 � 2I* � 2H��aq � I2 � 2NO � 2H2O
TP 5 : Synthèse de l’hélianthine ou méthylorange Page 11
v. Equation de copulation azoïque
vi. Recherche du réactif limitant
Pour la réaction de diazotation :
Quantité introduite Nombre de mole (mmol)
Acide sulfanilique 1,73g 1,73 173 � 10⁄
Nitrite de sodium 0,75g 0,75 69 � 10,9⁄
Acide chlorhydrique 1,7mL 1,18 9 0,37 9 1,17 36,45 � 20,4⁄
D'après l'équation de la réaction de diazotation : (����� � 6b���6�� � � (c�de � (�fdg 2⁄
Donc le nitrite de sodium est en excès. L'acide sulfanilique est le réactif limitant. Il va se former h � ij, j kklm n5olh nopqlhork
Pour la réaction de copulation diazoïque :
Quantité introduite Nombre de mole (mmol)
Aniline 1,2mL 0,95 9 1,2 121,18 � 9,4⁄
Ion diazonium 10
D'après l'équation de la réaction: (��� ���s��� t � (���6���
donc c'est la N, N-diméthylaniline qui sera le réactif limitant .
Il va donc se former h � u, v kklm n5wémophxwohy �z°
vii. Masse théorique d'hélianthine formée
|� � (� 9 }� � 9,4. 10�$ 9 327,33 9 3,077�
viii. Rendement global de la préparation.
Masse expérimentale : m1 = (à préciser)
Volume de soude rajouté pour rendre la solution basique : V = 10 mL
Volume d'eau nécessaire pour la recristallisation : V = 18 mL
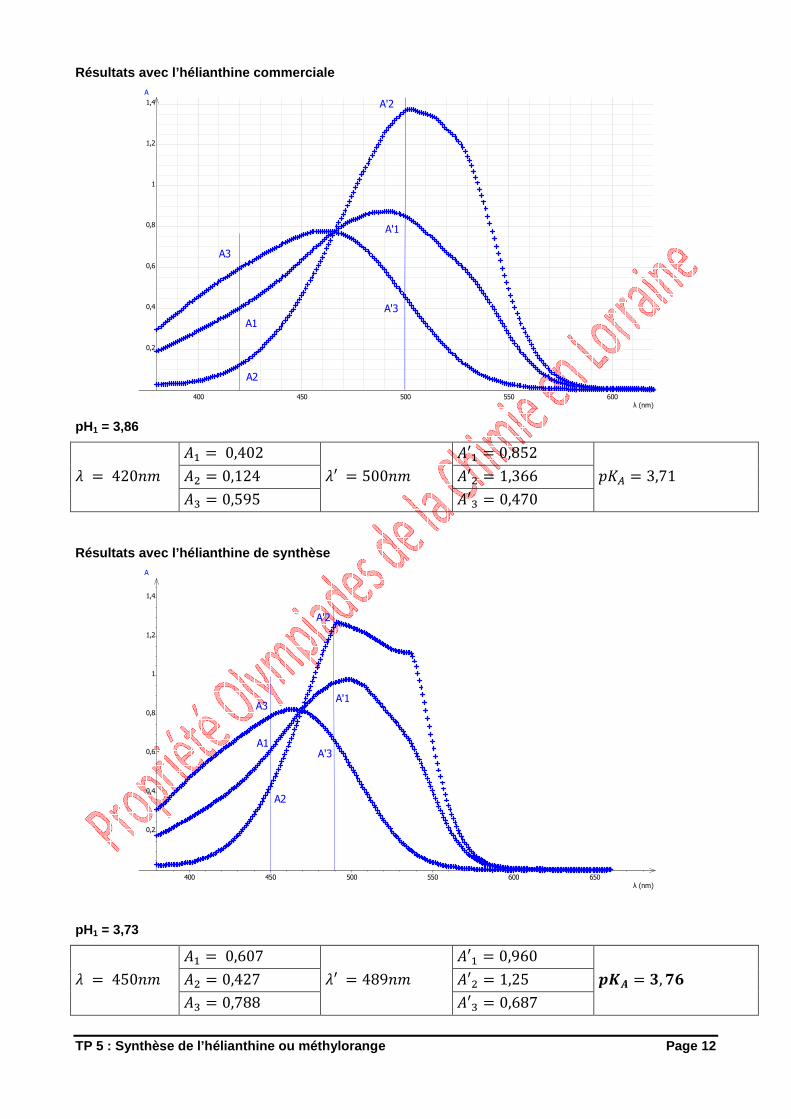
TP 5 : Synthèse de l’hélianthine ou méthylorange Page 12
Résultats avec l’hélianthine commerciale
pH1 = 3,86
2 � 420(|
�3 � 0,402
25 � 500(|
�53 � 0,852
) ! � 3,71 �4 � 0,124 �54 � 1,366
�$ � 0,595 �5$ � 0,470
Résultats avec l’hélianthine de synthèse
pH1 = 3,73
2 � 450(|
�3 � 0,607
25 � 489(|
�53 � 0,960
./0 � �, �� �4 � 0,427 �54 � 1,25
�$ � 0,788 �5$ � 0,687
λ (nm)400 450 500 550 600
A
0,2
0,4
0,6
0,8
1
1,2
1,4
A1
A2
A3
A'3
A'1
A'2
λ (nm)400 450 500 550 600 650
A
0,2
0,4
0,6
0,8
1
1,2
1,4
A2
A1
A3
A'3
A'1
A'2

TP 6 : Dosage d’un effluent industriel

TP 6 : Dosage d’un effluent industriel Page 1
Académie de Nancy - Metz Olympiades de la chimie 2010-2011 « La chimie et l’eau »
TP 6 : Dosage d’un effluent industriel
On s’intéresse ici à la détermination de la composition quantitative d’un effluent aqueux industriel contenant les espèces carbonates �����, phosphates ����� et sulfates �����, ces espèces pouvant être présentes sous différentes formes en fonction du pH de l’effluent. Nous allons voir dans cette manipulation comment il est possible de doser chaque espèce séparément.
1 Dosage par de l’acide chlorhydrique suivi par pH- métrie. On va effectuer un suivi pH métrique de la réaction entre « l’eau industrielle » et une solution d’acide chlorhydrique.
1.1 Courbe attendue et identification des espèces d osées.
On donne pour les acides suivants les valeurs des pKa associés
H2CO3 pKa11 6,4
pKa12 10,3
H2SO4 pKa21 1ère acidité forte
pKa22 2
H3PO4
pKa31 2,1
pKa32 7,2
pKa33 12,4
i. Représenter pour chaque espèce les formes prédominantes sur une échelle de pH
Est représentée ci-dessous la courbe de titrage pH-métrique obtenue.

TP 6 : Dosage d’un effluent industriel Page 2
Utilisant cette courbe et les diagrammes de prédominance de chaque espèce,
ii. Déterminer les formes sous lesquelles coexistent les carbonates, phosphates et sulfates dans chaque grand domaine de pH (avant la 1ère équivalence, entre la 1ère et la 2ème équivalence, après la 2ème équivalence).
iii. En déduire à quoi correspond chacune des deux équivalences.
iv. En déduire quelle espèce ne peut être dosée par cette méthode.
1.2 Procole du dosage pH-métrique
• Dans un bécher de 100 mL, verser 20,0 mL prélevés à la pipette jaugée de l’eau industrielle.
• Plonger la sonde pH-métrique et suivre l’évolution du pH en ajoutant un volume Va d’une solution d’acide chlorhydrique de concentration Ca = 2,00. 10-2 mol.L-1. Va sera compris entre 0 et 25 mL.
• Déterminer les deux volumes VE1 et VE2 correspondant respectivement à la 1ère et la 2ème équivalence.
2 Dosage par une solution de chlorure de baryum sui vi par conductimétrie
2.1 Principe du dosage
Le nitrate de baryum Ba(NO3)2(S) est totalement soluble dans l’eau pour former Ba2+(aq) + 2 NO3-(aq).
Par ailleurs les ions baryum (Ba2+) sont susceptibles de former des précipités avec les carbonates, phosphates et sulfates selon les réactions suivantes :
��� � � ������ � � ������ 3��� � � 2��43�� � � ����������
��� � � ������ � � ������
Le dosage considéré se fait par ajout d’une solution chlorure de baryum dans l’eau industrielle en suivant la conductivité de la solution dont le pH a été au préalable amené à 3 par ajout d’acide.
v. Quel est l’intérêt de fixer le pH à 3 et quelle espèce peut-on ainsi doser ?
0,0
2,0
4,0
6,0
8,0
10,0
12,0
0 5 10 15 20 25 30V HCl
Courbe de titrage pH-métrique

TP 6 : Dosage d’un effluent industriel Page 3
2.2 Protocole du dosage conductimétrique
• Dans bécher de 200 mL, verser 100,0 mL de l’eau industrielle et ajouter 5 mL de solution d’acide chlorhydrique à 0,50 mol/L. Les 100mL peuvent mesurer avec la fiole jaugée mais il faudra récupérer les eaux de rinçage. Vérifier que le pH est proche de 3 sinon l’ajuster.
• Plonger la sonde conductimétrique et suivre l’évolution de la conductivité en ajoutant un volume V d’une solution de nitrate de baryum de concentration CBa = 5,00. 10-2 mol. L-1. V sera compris entre 0 et 20 mL. (bien attendre la stabilisation de la conductivité avant la mesure).
On rappelle que la conductivité d’une solution ionique est donnée par la relation : � � � �����
Avec �� , la conductivité molaire ionique de l’ion i en mS.m2.mol-1 ��, la concentration du l’ion i

TP 6 : Dosage d’un effluent industriel Page 4
On donne les conductivités molaires ioniques à 25°C des ions suivants
ions λ en mS.m2.mol-1 ����� 16,0 �� 12,8 ���� 7,1
vi. Expliquer l’allure de la courbe de dosage obtenue
• Déterminer le volume VE3 correspondant à l’équivalence.
3 Dosage du phosphore par spectrophotométrie 3.1 Principe :
En présence de vanadium (V) et de molybdène (VI), l’acide phosphorique donne en milieu acide un complexe jaune. En milieu acide chlorhydrique à une concentration comprise entre 0,5 à 1,9 mol/L, la coloration se développe assez rapidement et est stable plusieurs semaines. Les concentrations des réactifs doivent être fixées exactement et on les placera dans le blanc car ils absorbent aussi à la longueur d’onde de travail qui a été choisie à 425 nm .
3.2 Protocole
On dispose d’une solution étalon E de dihydrogénophosphate de potassium KH2PO4 de concentration connue égale 2,00. 10-3 mol/L. Préparation du réactif vanadomolybdique juste avant l’emploi ainsi :
• Introduire dans un bécher, dans l’ordre, les volumes étant mesurés à l’éprouvette graduée : � 50 mL d’acide nitrique commercial dilué au ½ ((�) � 50 mL de solution de vanadate d’ammonium à 2,5 g/L � 50 mL de molybdate d’ammonium à 50 g/l
• Homogénéiser le mélange et l’utiliser rapidement. Préparation des solutions à analysées
• Préparer dans des fioles jaugées de 50 mL :
Solution F0 F1 F2 F3 F4 FX
Volume de solution E (fioles F0 à F4) ou de solution de l’eau industrielle (fiole FX) en mL
0 5 10 15 20 5
Acide chlorhydrique au 1/25ème en mL 5 5 5 5 5 5
Réactif phosphovanadomolybdique en mL 15 15 15 15 15 15
Eau q .s.p 50 mL
• Laisser la coloration se développer pendant 15 minutes puis mesurer l’absorbance des solution F1, F2,
F3, F4 et FX en utilisant F0 comme blanc et à 425 nm .
TP 6 : Dosage d’un effluent industriel Page 5
3.3 Exploitation :
vii. Compléter le tableau ci- dessous et tracer la courbe A = f(CP (mol/L) : concentration molaire en phosphore dans les fioles).
Fiole F0 F1 F2 F3 F4 FX
Absorbance à 425 nm
CP (mol/L)
viii. A partir de la concentration en élément phosphore dans FX, calculer la concentration molaire en
élément phosphore dans l’eau industrielle.
4 Détermination de la composition de l’eau industri elle ix. En utilisant l’ensemble des résultats expérimentaux obtenus lors des précédents dosages,
déterminer la composition en carbonates, phosphates et sulfates dans l’eau industrielle en précisant sous quelle(s) forme(s) ils sont présents et en quelles concentrations exprimées en mol/L et en mg/L (les concentrations massiques étant exprimées en équivalents carbonates, phosphore et sulfates respectivement).
On donne les masses molaires suivantes en g/mol : C : 12 ; O : 16 ; P : 31 ; S : 32

TP 6 : Dosage d’un effluent industriel Page 6
Académie de Nancy - Metz Olympiades de la chimie 2010-2011 « La chimie et l’eau »
TP 6 : Dosage d’une eau industrielle LISTE MATERIELS ET PRODUITS
Matériel
Pour les dosages pH-métrique et conductimétrique
- 1 pH mètre + 1 conductimètre
- 1 burette + agitateur
- 1 bécher de 100 mL + 1 bécher de 200mL
- 1 fiole de 100 mL
- 1 pipette calibrée de 10 ou 20 mL
- Pipette plastique de 1 mL
Pour le dosage du phosphore
- 1 éprouvette graduée de 50mL
- 1 bécher de 200mL minimum
- 1 pipette calibrée de 5mL
- 6 fioles de 50mL
- 6 cuves plastiques de spectro
Produits et solutions (en rouge non disponible dans les lycées a priori)
Eau industrielle : prévoir 250mL par poste pour être tranquille, soit par poste
Na2CO3, 10H2O : 214,6mg
NaHCO3 : 147mg
Na2SO4 : 177,5mg
KH2PO4 : 170mg
NaOH : 80mg
- Solution HCl à 0.020 mol/L : 50 mL - Solution HCl à 0.5 mol/L : 10 mL - Solution HCl au 1/25ème : 50 mL - Solution étalon de KH2PO4 à la concentration 2.10-3 mol/L : 100mL (pour être tranquille) soit 27,2mg de
KH2PO4
- Solution de Ba(NO3)2 à la concentration 5.10-2 mol/L : 50mL soit 0,66g de Ba(NO 3)2 solide non hydraté
Réactif vanadomolybdique :
acide nitrique commercial dilué au ½ : 50mL soit 25mL de HNO3 commercial
solution de vanadate d’ammonium à 2,5 g/L : 50mL soit 0,125g de vanadate d'ammonium
molybdate d’ammonium à 50 g/l : 50mL soit 2,5g de molybdate d'ammonium
TP 6 : Dosage d’un effluent industriel Page 7
Académie de Nancy - Metz Olympiades de la chimie 2010-2011 « La chimie et l’eau »
TP 6 : Dosage d’une eau industrielle REPONSES AUX QUESTIONS
Fabrication de l’eau industrielle
Attention : respecter scrupuleusement la quantité d e soude à introduire pour que le pH de la solution
soit conforme aux attentes.
C (mol/L) C (mg/L) M (g/mol)
Na2CO3, 10H2O 0,003 858,42 286,14
NaHCO3 0,007 588 84
Na2SO4 0,005 710 142
KH2PO4 0,005 680 136
NaOH 0,008 320 40
Attention : bien boucher la fiole après préparation de la solution pour éviter tout dégazage de CO 2.
Réponses aux questions :
1 Dosage par de l’acide chlorhydrique suivi par pH- métrie.
i.
ii. Comme le montre la courbe de dosage, le pH initial de la solution est environ égal à 10,4. Ainsi les carbonates sont simultanément sous forme de ����� et de ����� en proportions quasiment égales. Les phosphates sont intégralement sous forme de ������ et les sulfates sous
forme de �����. La valeur du pH à la première équivalence comprise entre 8 et 9 indique clairement que tous les ����� ont disparu. Les carbonates sont donc intégralement sous forme de �����, les phosphates
sous forme de ������ et les sulfates sous forme de �����.
6,4
2
2,1
10,3
7,2 12,4
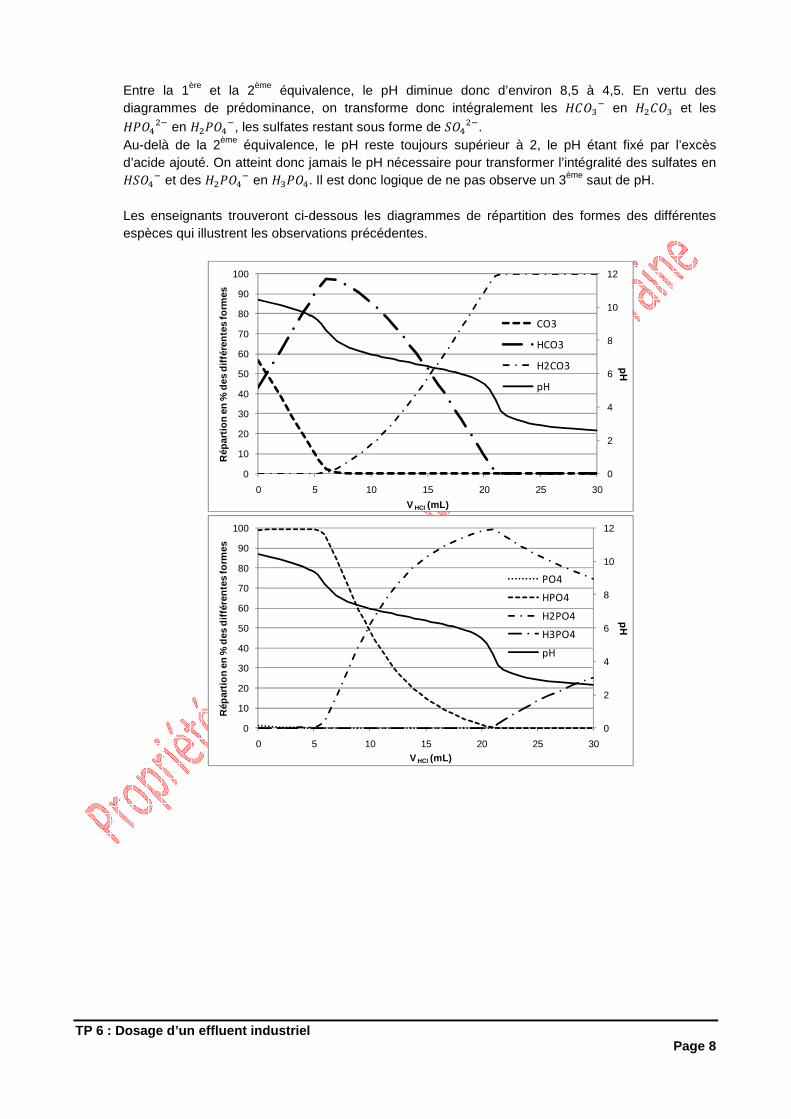
TP 6 : Dosage d’un effluent industriel Page 8
Entre la 1ère et la 2ème équivalence, le pH diminue donc d’environ 8,5 à 4,5. En vertu des diagrammes de prédominance, on transforme donc intégralement les ����� en ����� et les ������ en ������, les sulfates restant sous forme de �����. Au-delà de la 2ème équivalence, le pH reste toujours supérieur à 2, le pH étant fixé par l’excès d’acide ajouté. On atteint donc jamais le pH nécessaire pour transformer l’intégralité des sulfates en ����� et des ������ en �����. Il est donc logique de ne pas observe un 3ème saut de pH. Les enseignants trouveront ci-dessous les diagrammes de répartition des formes des différentes espèces qui illustrent les observations précédentes.
0
2
4
6
8
10
12
0
10
20
30
40
50
60
70
80
90
100
0 5 10 15 20 25 30
pH
Rép
artio
n en
% d
es d
iffér
ente
s fo
rmes
V HCl (mL)
CO3
HCO3
H2CO3
pH
0
2
4
6
8
10
12
0
10
20
30
40
50
60
70
80
90
100
0 5 10 15 20 25 30
pH
Rép
artio
n en
% d
es d
iffér
ente
s fo
rmes
V HCl (mL)
PO4
HPO4
H2PO4
H3PO4
pH

TP 6 : Dosage d’un effluent industriel Page 9
iii. La 1ère équivalence correspond donc au dosage des ions ����� présents initialement dans la solution. La 2ème équivalence correspond au dosage simultané des ions ����� (carbonates total) et
des ions ������ (phosphates total)
iv. Par dosage acido-basique, il n’est pas possible de déterminer la concentration en sulfates.
Est présentée ci-dessous la courbe de dosage pH-métrique obtenue.
ATTENTION : Suite à une erreur dans la masse d’un d es composés introduits lors de la fabrication
de l’eau de synthèse, la composition théorique init iale de cette eau est :
� ���32�� � 5,78. 10�3'(). *�1+���3�, � 6,06. 10�3'(). *�1. / ���32��0(0) � 1,18. 10�2'(). *�1
����42�� � 5. 10�3'(). *�1���42�� � 5. 10�3'(). *�1
L’eau que nous avons utilisée pour les mesures a do nc une concentration totale en carbonates
supérieure à celle préconisée dans le protocole opé ratoire.
En supposant que le 1er saut de pH correspond effectivement au dosage des ����� initialement dans la
solution et que le 2nd saut correspond au dosage des carbonates total et des phosphates, alors les volumes
équivalents attendus seraient : 123 � 5,78'* 40 12� � 22,61'*
Ci-dessous la courbe expérimentale obtenue.
0
2
4
6
8
10
12
0
10
20
30
40
50
60
70
80
90
100
0 5 10 15 20 25 30
pH
Rép
artio
n en
% d
es d
iffér
ente
s fo
rmes
V HCl (mL)
SO4
HSO4
pH

TP 6 : Dosage d’un effluent industriel Page 10
Solution HCl à 0.020 mol/L prise d’essai de 20 mL
VE1= 5.059 mL VE2 = 22,26 mL
Est représentée ci-dessous la courbe théorique simulée du dosage de la même solution établie sans faire aucune approximation.
Qui fournit donc les volumes équivalents suivants : 123 � 6,00'* 40 12� � 22,80'* Ces résultats amènent plusieurs remarques. Tout d’abord, on constate que les volumes équivalents déterminés rigoureusement en cherchant les optimums de la dérivée sur la courbe de dosage exact sont légèrement supérieurs à ceux calculés en supposant que le 1er saut correspond exclusivement au dosage des ����� présents initialement dans l’eau.
Va (mL)5 10 15 20 25 30
pH
2
4
6
8
10
4.432
22.26
8.379
5.059
6,000 22,800
8,62
4,47
0,00
0,20
0,40
0,60
0,80
1,00
1,20
1,40
1,60
1,80
0
2
4
6
8
10
12
0 5 10 15 20 25 30
dp
H/
dVp
H
V (mL)
pH
dpH/dV

TP 6 : Dosage d’un effluent industriel Page 11
On observe un même décalage de l’ordre de 0,2mL. Ce décalage est tout à fait normal si l’on considère les courbes de répartition proposées précédemment. En effet on constate qu’à la 1ère équivalence, la proportion en ����� est légèrement inférieur à 100% et que celle en ����� n’est pas nulle. Cela indique clairement que les dosages des deux basicité des carbonates ne sont pas rigoureusement séparés. En d’autres termes, on commence à doser les ����� avant de terminer le dosage des �����. Quoiqu’il en soit, il n’est pas possible d’éviter le phénomène qui introduit cependant une erreur très faible sur le volume équivalent. Si l’on compare à présent les volumes équivalents déterminés à partir de la courbe expérimentale à ceux déterminés par la courbe théorique, on observe cette fois des écarts plus sensibles. Ces écarts peuvent avoir plusieurs origines :
� un léger écart entre les concentrations introduites et celles théoriquement prévues (nous avons préparé 2 litres de solution et rien n’exclue un léger problème d’homogénéité),
� le pH initial de la solution mesuré est légèrement inférieur à celui attendu (autour de 10 au lieu de 10,3). Ceci peut provenir d’un léger écart de la concentration en soude initialement introduite. Dans ce cas, si le pH initial est légèrement inférieur, à même concentration totale en l’espèce carbonate, la proportion en ions ����� sera légèrement inférieure et de fait le 1er volume équivalent également. Par ailleurs, une imprécision sur la valeur du pH mesuré n’est pas à exclure.
� finalement la plus grande cause d’erreur est liée au repérage du 1er point équivalent. Comme le montre la courbe expérimentale, à la 1ère équivalence, le saut de pH est relativement faible est la détermination du volume équivalent par la méthode des tangentes n’est a priori pas très précise. Il est bien évident que cette détermination avec un pH-mètre couplé à un logiciel qui repère le point équivalent par calcul de la dérivée maximale (comme démontré en théorie) serait meilleure. On est là confronté à un problème de précision de la méthode.
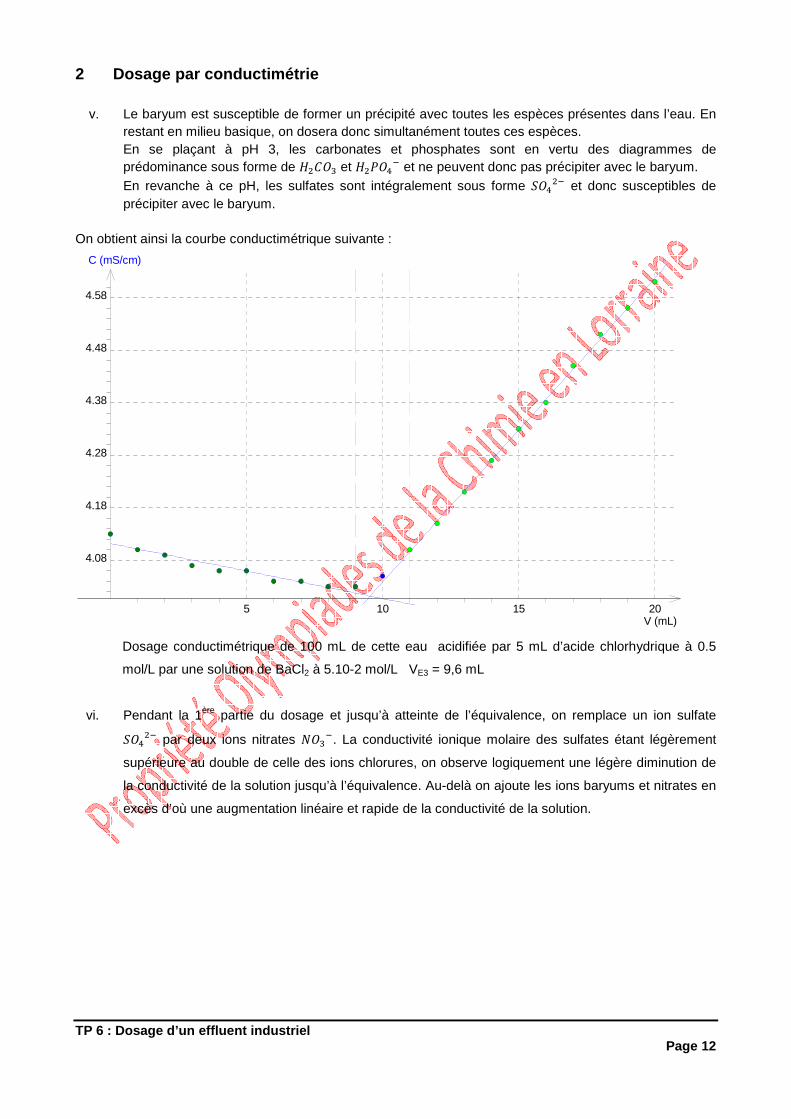
TP 6 : Dosage d’un effluent industriel Page 12
2 Dosage par conductimétrie
v. Le baryum est susceptible de former un précipité avec toutes les espèces présentes dans l’eau. En restant en milieu basique, on dosera donc simultanément toutes ces espèces. En se plaçant à pH 3, les carbonates et phosphates sont en vertu des diagrammes de prédominance sous forme de ����� et ������ et ne peuvent donc pas précipiter avec le baryum. En revanche à ce pH, les sulfates sont intégralement sous forme ����� et donc susceptibles de précipiter avec le baryum.
On obtient ainsi la courbe conductimétrique suivante :
Dosage conductimétrique de 100 mL de cette eau acidifiée par 5 mL d’acide chlorhydrique à 0.5
mol/L par une solution de BaCl2 à 5.10-2 mol/L VE3 = 9,6 mL
vi. Pendant la 1ère partie du dosage et jusqu’à atteinte de l’équivalence, on remplace un ion sulfate ����� par deux ions nitrates ����. La conductivité ionique molaire des sulfates étant légèrement
supérieure au double de celle des ions chlorures, on observe logiquement une légère diminution de
la conductivité de la solution jusqu’à l’équivalence. Au-delà on ajoute les ions baryums et nitrates en
excès d’où une augmentation linéaire et rapide de la conductivité de la solution.
V (mL)5 10 15 20
C (mS/cm)
4.08
4.18
4.28
4.38
4.48
4.58
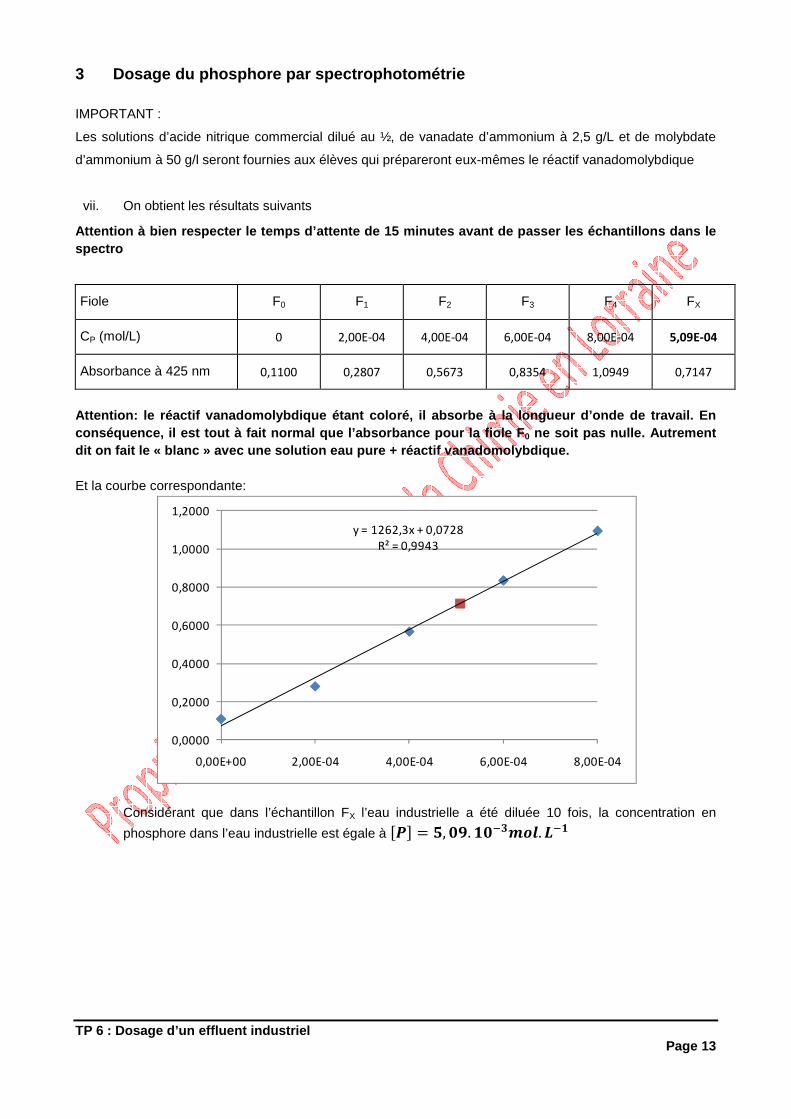
TP 6 : Dosage d’un effluent industriel Page 13
3 Dosage du phosphore par spectrophotométrie
IMPORTANT :
Les solutions d’acide nitrique commercial dilué au ½, de vanadate d’ammonium à 2,5 g/L et de molybdate
d’ammonium à 50 g/l seront fournies aux élèves qui prépareront eux-mêmes le réactif vanadomolybdique
vii. On obtient les résultats suivants
Attention à bien respecter le temps d’attente de 15 minutes avant de passer les échantillons dans le spectro
Fiole F0 F1 F2 F3 F4 FX
CP (mol/L) 0 2,00E-04 4,00E-04 6,00E-04 8,00E-04 5,09E-04
Absorbance à 425 nm 0,1100 0,2807 0,5673 0,8354 1,0949 0,7147
Attention: le réactif vanadomolybdique étant coloré , il absorbe à la longueur d’onde de travail. En conséquence, il est tout à fait normal que l’absorb ance pour la fiole F 0 ne soit pas nulle. Autrement dit on fait le « blanc » avec une solution eau pure + réactif vanadomolybdique. Et la courbe correspondante:
Considérant que dans l’échantillon FX l’eau industrielle a été diluée 10 fois, la concentration en
phosphore dans l’eau industrielle est égale à 567 � 8, 9:. ;9�<=>?. @�;
y = 1262,3x + 0,0728
R² = 0,9943
0,0000
0,2000
0,4000
0,6000
0,8000
1,0000
1,2000
0,00E+00 2,00E-04 4,00E-04 6,00E-04 8,00E-04

TP 6 : Dosage d’un effluent industriel Page 14
4 Composition détaillée de l’eau industrielle
Comme expliquée dans la 1ère partie, les carbonates sont simultanément sous forme de ����� et de ����� en proportions quasiment égales. Les phosphates sont intégralement sous forme de ������
et les sulfates sous forme de �����. En vertu du résultat obtenu par dosage spectrophotométrique, on a :
+������, � 5,09. 10��'(). *�3
soit +������, � 5,09. 10�� B 31C. *�3 � 157,8'C. *�3 4 . �
Le dosage par précipitation au baryum fournit la concentration en sulfates, soit :
+�����, � �DE · 12�1GEH,IJKé
soit +�����, � 5. 10�� · 9,6100 � 4,8. 10��'(). *�3
soit +�����, � 4,8. 10�� B �32 � 4 B 16� � 460,8'C. *�3 4 . ���
Finalement concernant le dosage pH-métrique. 12� � 123 correspond au dosage des carbonates total et des phosphates sous forme de ������
+�����,MJMEN � +������, � �E · �12� � 123�1GEH,IJKé '(). *�3
soit +�����,MJMEN � +������, � 0,02 · �22,26 � 5,06�20� 1,72. 10��'(). *�3
d’où +�����,MJMEN � 1,21. 10��'(). *�3
soit +�����,MJMEN � 1,21. 10�� B �12 � 3 B 16� � 726'C. *�3 4 . ���
Finalement le volume 123 correspond au dosage des carbonates sous forme de �����
+�����, � �E · 1231GEH,IJKé '(). *�3
soit +�����, � 0,02 · 5,0620 � 5,06. 10��'(). *�3
soit +�����, � 5,06. 10�� B �12 � 3 B 16� � 303'C. *�3 4 . ���
On peut donc en déduire aisément la concentration en hydrogénocarbonate, soit
5�����7 � +�����,MJMEN � +�����, � 7,05. 10��'(). *�3
soit 5�����7 � 7,05. 10�� B �12 � 3 B 16� � 423'C. *�3 4 . ���

TP 6 : Dosage d’un effluent industriel Page 15
Si l’on compare les concentrations déterminées expérimentalement avec celles attendues, soient :
� ���32�� � 5,78. 10�3'(). *�1+���3�, � 6,06. 10�3'(). *�1. / ���32��0(0) � 1,18. 10�2'(). *�1
����42�� � 5. 10�3'(). *�1���42�� � 5. 10�3'(). *�1
on peut conclure que :
- la détermination de la concentration en phosphore par spectrophotométrie donne d’excellents résultats
(écart inférieur à 2%),
- la détermination de la concentration en sulfates donne un résultat conforme (écart de l’ordre de 4%).
Notons ici que la détermination du point équivalent est extrêmement sensible à la cinétique de précipitation
du sulfate de baryum (on observe en effet une légère fluctuation de la conductivité de la solution dans le
temps). Il est donc conseillé de ne pas diluer la prise d’essai et donc de respecter rigoureusement le
protocole,
- la détermination de la concentration en carbonates et carbonates total donne des résultats plus médiocres.
Etant donnée la difficulté à repérer la 1ère équivalence, ce résultat n’est somme toute pas vraiment étonnant.
Par ailleurs cette concentration est affectée par l’erreur sur la concentration en phosphates
Précisons néanmoins que les manipulateurs se réservent le droit d’avoir commis quelques imprécisions
dans la fabrication de l’eau industrielle…l’un d’entre eux en particulier n’étant pas un habitué des paillasses.
Il est ainsi légitime de penser qu’avec un maximum de précautions, les résultats seraient bien meilleurs !!!
Finalement le lecteur trouvera ci-dessous la courbe théorique de dosage issue de la simulation pour une eau
industrielle dont la composition respecte scrupuleusement le protocole proposé à savoir :
� ���32�� � 5,78. 10�3'(). *�1+���3�, � 6,06. 10�3'(). *�1. / ���32��0(0) � 1,18. 10�2'(). *�1
����42�� � 5. 10�3'(). *�1���42�� � 5. 10�3'(). *�1
En supposant que le 1er saut de pH correspond effectivement au dosage des ����� initialement dans la
solution et que le 2nd saut correspond au dosage des carbonates total et des phosphates, alors les volumes
équivalents attendus seraient : 123 � 5,69'* 40 12� � 20,69'*
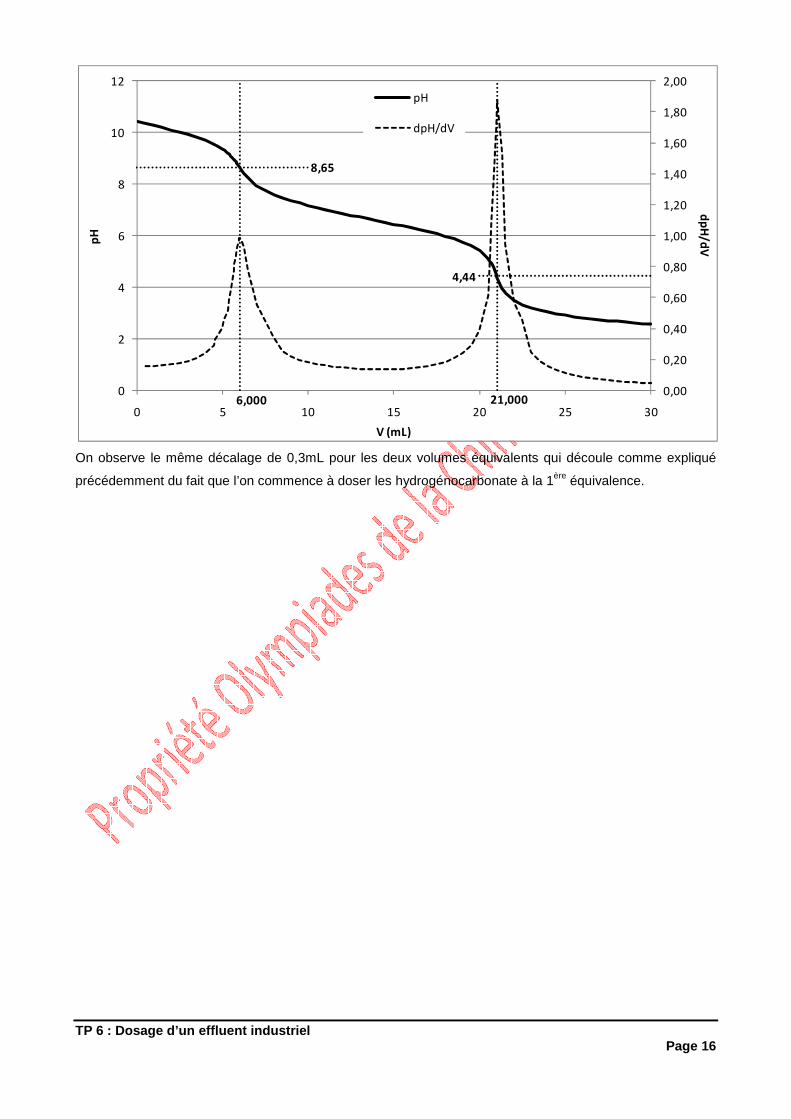
TP 6 : Dosage d’un effluent industriel Page 16
On observe le même décalage de 0,3mL pour les deux volumes équivalents qui découle comme expliqué
précédemment du fait que l’on commence à doser les hydrogénocarbonate à la 1ère équivalence.
6,000 21,000
8,65
4,44
0,00
0,20
0,40
0,60
0,80
1,00
1,20
1,40
1,60
1,80
2,00
0
2
4
6
8
10
12
0 5 10 15 20 25 30
dp
H/
dVp
H
V (mL)
pH
dpH/dV