Embed Size (px)
Citation preview

4 les d’
UplmHlSLClqLRLilLlLrdLdmAcmtAdDLLhLedLcUsrDsDlssEtd[ScquLltUcIlR[[[[
[[
h
RM
LseElDadmFLcdlfe
F
E1ggltLtdhEnebRrhépatique.Lors d’un régime riche en fructose, une augmentation de la fructose 1-phosphatepermet l’activation d’un co-activateur spécifique de SREBP-1c, PGC1�, qui
Compte rendu de congrès / Anna
ne autre étude a recherché les effets du traitement médical de l’acromégaliear analogues de la somatostatine sur le SAS [2], et a montré une diminution de’IAH entre trois et six mois, sans relation avec la diminution du taux de GH,
ais avec une diminution de l’infiltration.ermann et al., en 2004, a montré une diminution significative du volume de la
angue avec la diminution de l’IGF1. Il a montré que les formes modérées deAS s’amélioraient sous traitement, mais que les SAS sévères restaient sévères.es résultats étaient les mêmes sous SOMAVERT.ortet et Delemer ont montré une baisse de l’IAH après traitement, corrélé avec
a baisse de la GH, même si l’IMC restait stable, qui permettait d’améliorer laualité du sommeil et d’arrêter le traitement par PPC.a PPC reste le traitement de première intention, sans oublier l’importance desHD et du traitement des comorbidités.es indications de la PPC sont un IAH supérieur 30/h ou supérieur 10/h avec un
ndex de micro-éveils supérieur à 10/h, ou un retentissement important (somno-ence, terrain obèse, FDRCV, diabète, BPCO, âge. . .).e traitement par orthèse ou chirurgie d’avancée mandibulaire est peu adapté à
’acromégalie.e pourcentage de patients guéris du SAS après le traitement de l’acromégalie
este faible, de l’ordre de 20 à 30 %, mais on a peu de recul (un an), et uniquemente petites séries.e manque de sommeil ; un facteur de risque d’obésité.– La restriction volontairee sommeil est un comportement de plus en plus courant dans nos sociétésodernes.ux États-Unis, la durée du sommeil des adultes a diminué d’environ 1 h 30 au
ours de 40 dernières années. Le pourcentage d’adultes dormant six heures ouoins par nuit a augmenté de 5 % entre 1985 et 2004. On observe la même
endance en France.ux États-Unis, l’augmentation de l’obésité est proportionnelle à la diminutione la durée de sommeil [3].ans une enquête de 2009, un tiers des francais dormaient six heures ou moins.a durée du sommeil des 18–55 ans était en moyenne de 6 h 58 en semaine.es adolescents sont les plus touchés : leur besoin de sommeil est estimé à neufeures par nuit, mais 78 % d’entre eux dorment moins de huit heures la semaine.es études épidémiologiques sont nombreuses. Elles retrouvent l’associationntre une durée de sommeil courte et un IMC élevé, malgré la prise en comptees facteurs confondants. Cet impact est plus important chez les adultes jeunes.e risque relatif d’obésité est multiplié par quatre si la durée de sommeil estourte [4].ne étude sur 11 sujets d’âge moyen, à qui on a imposé une restriction de
ommeil, a constaté une augmentation de 15 % de la prise alimentaire après laestriction.es études expérimentales ont tenté d’étudier l’impact de la durée de sommeil
ur la régulation neuroendocrine de l’appétit chez des hommes jeunes.ans les situations de privation de sommeil, on remarque une diminution de
a leptine (hormone de la satiété) avec une relation dose-effet entre la durée deommeil et le taux de leptine [5]. L’appétit augmente donc avec la privation deommeil.n condition de restriction de sommeil, on observe une baisse de 18 % de la lep-
ine et une augmentation de 28 % de la gréline (hormone qui augmente l’appétit),onc une augmentation de la faim de l’ordre de 23 % (rapport gréline/leptine)6].i ces 23 % d’augmentation d’appétit se traduisent en une augmentation de prisealorique, cela correspondrait à un excès calorique de 350 à 500 kcal par jour, ceui pour un adulte jeune sédentaire de poids normal est susceptible d’entraînerne prise de poids conséquente.a diminution des concentrations de leptine en restriction de sommeil est simi-
aire à celle observée après une restriction calorique de 900 Kcal par jour pendantrois jours.n manque de sommeil semble altérer la capacité de la leptine à informer
orrectement le cerveau sur la balance énergétique.l a également été observé que l’appétit augmentait plus particulièrement poures aliments riches en graisses et en sucre.éférences
1] Sze C, et al. Eur J of Endocrinol 2007.2] Grunstein RR, et al. Ann Intern Med 1994.3] Flegal et al. 1998 (2002).4] Chaput, et al. Obesity 2009. S
Endocrinologie 74 (2013) 3–12
5] Spiegel, et al. J Clin Endocrinol Metab 2004.6] Spiegel, et al. Ann Int Med 2004.
ttp://dx.doi.org/10.1016/j.ando.2012.11.010
égulation des gènes par les nutriments�
athilde FratyPoitiers, France
e métabolisme glucidique est complexe. Après un repas, l’insuline stimule letockage du glucose en glycogène et la lipogenèse et inhibe la néoglucogenèset la lipolyse.n situation de résistance à l’insuline, la perturbation de ces mécanismes favorise
’hyperglycémie et l’apparition d’une stéatose hépatique.es études récentes ont montré que le fructose, de plus en plus présent dans notre
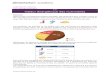
limentation, est aussi impliqué dans l’apparition du syndrome métabolique ete la stéatose hépatique [1]. L’action pathogène de ce nutriment dépend de relaisoléculaires spécifiques : les facteurs de transcription.acteurs de transcription impliqués dans la glycolyse et la lipogenèse.–es facteurs sterol regulatory element binding protein-1c (SREBP-1c) etarbohydrate responsive element binding protein (CHREB) sont les facteurse transcriptions impliqués dans les mécanismes moléculaires du contrôle par’insuline et le glucose des gènes de la glycolyse et de la lipogenèse dans leoie. SREBP-1c médie les effets géniques de l’insuline et CHREBP médie lesffets géniques du glucose (Fig. 1).
ig. 1.
n situation physiologique, l’insuline agit par clivage du précurseur de SREBP-c, qui, ainsi activé, se dirige dans le noyau pour permettre la transcription desènes de la glycolyse et de la néoglucogenèse. SREBP-1c active également lalucokinase et augmente le pool cytosolique de G6P, favorisant alors la trans-ocation vers le noyau de CHREBP qui agit de concert avec SREBP-1c pourranscrire les gènes cibles [2].’inhibition de ces voies de signalisation par inactivation de ces facteurs deranscription, indépendamment l’une de l’autre, provoque une baisse du taux’ARNm des gènes cibles et une moindre accumulation d’acides gras dans lesépatocytes.n situation d’insulinorésistance chez des souris ob/ob, les concentrationsucléaires de CHREB et SREBP-1c matures sont corrélées au flux lipogéniquet à la stéatose. L’inhibition du clivage de SREBP-1c de ces souris provoque uneaisse de la stéatose et une restauration des paramètres métaboliques.ôle pathogène du fructose.– Il a été récemment démontré qu’une alimentation
iche en fructose était un activateur plus puissant de la formation d’une stéatose
� D’après le symposium des Dr Arnulf Isabelle, Cortet-Rudelli Christine etpiegel Karine. 28e Congrès de la Société francaise d’endocrinologie 2011.

les d’
seàrsIDaftcePddlrlEpnpsqdsddCtssR[
[
[
[
Pi2
h
GC
LdtdrLddqs
C
cbCpdsslsb[Udv[RcthL–fppgn–driv–•dg•bpCeGlsGdpusacLdltmt[Elam
Compte rendu de congrès / Anna
e lie avec lui dans le noyau pour induire la transcription des gènes ciblest potentialiser la formation de la stéatose hépatique. Ce phénomène conduitl’accumulation d’un intermédiaire lipidique, le di-acyl-glycérol (DAG), au
ecrutement d’une protéine kinase, PKC�, qui va migrer vers la membrane et’opposer à la signalisation normale de l’insuline par phosphorylation d’IRS1 etRS2 en levant l’inhibition de la production hépatique de glucose [3].es expériences préliminaires ont montré que le fructose, contrairement
u glucose, n’influence pas la translocation nucléaire de CHREBP, maisavorise l’accumulation d’acétyl co-A en acétylant CHREBP, dont l’activitéranscriptionnelle est ainsi augmentée. De plus, CHREBP est un médiateur trans-riptionnel sur deux enzymes clés du métabolisme du fructose, la fructokinaset l’aldolase B, réalisant ainsi une boucle de régulation positive.hénomène de glucotoxicité et rôle dans la néoglucogenèse.– Environ 2 à 3 %u glucose ingéré emprunte la voie des hexosamines, essentiellement en cas’hyperglycémie importante, aboutissant à la formation d’UDP glcNac, qui, par’intermédiaire d’une enzyme, (OGT UDPglcNac transferase), se fixe de manièreéversible sur des protéines, notamment des facteurs de transcription, et influenceeur stabilité/dégradation (phénomène d’o-glycosylation post-traductionnel).n situation physiologique, la signalisation insulinique conduit à la phos-horylation de deux facteurs de transcription, FoxO1 et CTRC2 (gènes de laéoglucogénèse), qui empêche leur fixation à leurs co-activateurs spécifiques etermet leur export vers le cytosol. En cas d’hyperglycémie, via la voie des hexo-amines, on observe un phénomène d’o-glycosylation de FoxO1 et CRTC2 ceui induit une augmentation de leur fixation sur leurs éléments de réponse etonc une activation de la transcription des gènes de la néoglucogenèse aboutis-ant à un cercle vicieux [4]. Chez les souris db/db on observe une augmentatione l’expression des marqueurs de la o-glycosylation corrélée à celle des gènese la néoglucogenèse.onclusion.– Les nutriments sont responsables, par le biais de relais transcrip-
ionnels complexes, de modulations géniques potentiellement pathogènes. Il’agit donc de cibles de recherche dans le domaine de la physiopathologie duyndrome métabolique et de la stéatose hépatique.éférences
1] Kuo M, et al. Public health: the toxic truth about sugar. Nature2012;482:27–9.
2] Dentin R, et al. Polyunsaturated fatty acids suppress glycolytic and lipogenicgenes through the inhibition of CHREBP nuclear protein translocation. J ClinInvest 2005;115:2843–54.
3] Samuel VT. Fructose induced lipogenesis: from sugar to fat to insulin resis-tance. Trends Endocrinol Metab 2011;22:60–5.
4] Kuo M, et al. O-GlcNAc modification of FoxO1 increases its trans-criptional activity: a role in the glucotoxicity phenomenon? Biochimie2008;90:679–85.
our en savoir plus Dentin R, et al. Liver-specific inhibition of CHREBPmproves hepatic steatosis and insulin resistance in ob/ob mice. Diabetes006;55:2159–70.
ttp://dx.doi.org/10.1016/j.ando.2012.11.011
lucagon, physiologie et physiopathologie�
éline MoulyToulouse, France
e grand retour du glucagon au-devant de la scène (P. Lefevbre).– Le glucagon aes actions multiples sur plusieurs organes (cœur, rein, système nerveux central,issu adipeux, intestin), mais essentiellement sur le foie, les produits de sécrétionu pancréas étant déversés en premier lieu dans la veine porte. Il est en effetesponsable de 75 % de la production hépatique de glucose.es îlots, représentant 2 % du poids pancréatique, doivent être considérés commees micro-organes avec une sécrétion paracrine. Il existe en effet une intricationes sécrétions pulsatiles de glucagon et d’insuline, avec une image en miroir :
uand l’un augmente, l’autre diminue. Le moteur de cette sécrétion intriquéeemblerait être la sécrétion d’insuline [1]. Cela peut être lié à l’organisation� D’après la conférence « Comment les nutriments régulent nos gènes » de. Postic et F. Foufelle. Congrès de la Société francophone du diabète 2012.
[R[[[[[
Endocrinologie 74 (2013) 3–12 5
omplexe de la microcirculation, avec un flux artériel allant plutôt des cellulesêta du centre de l’îlot aux cellules alpha de la périphérie.ette corrélation statistique entre les sécrétions d’insuline et de glucagon dis-araît chez le diabétique de type 2, avec une quantité pulsée bien moindre’insuline et accrue de glucagon. Une augmentation paradoxale du glucagonuite à une hyperglycémie a été mise en évidence chez ces patients. Plu-ieurs possibilités thérapeutiques sont actuellement étudiées pour lutter contre’hyperglucagonémie observée. Dans ce cadre, il a été mis en évidence, chez desouris dont le récepteur au glucagon était inactivé, que la destruction des cellulesêta ne provoquait pas d’effets métaboliques, notamment pas d’hyperglycémie2].n excès d’insulinocentrisme dans les théories existantes sur la physiopathologieu diabète a récemment été suggéré avec une hypothèse mono-hormonale soule-ée : le diabète de type 2 pourrait être liée principalement à l’hyperglucagonémie3].égulation de la sécrétion de glucagon par le glucose (P. Gilon).– Il existehez les diabétiques une perte de sensibilité de la cellule alpha au glucose seraduisant par une perte de sécrétion de glucagon en cas d’hypoglycémie et uneyperglucagonémie.e glucose agit sur la cellule alpha par trois voies :une voie indirecte via le système nerveux autonome. En fonction de la pro-
ondeur de l’hypoglycémie, détectée à plusieurs niveaux du sytème nerveuxériphérique et central, trois systèmes vont être successivement mis en œuvreour stimuler la sécrétion de glucagon [4] : les fibres parasympathiques choliner-iques, le système médullo-surrénalien adrénergique, et les fibres sympathiquesoradrénergiques ;une voie directe. Le glucose a une action inhibitrice via l’AMPK, activée par
éprivation en énergie, et via le store-operated current (SOC), courant dépola-isant, activé par la vidange en calcium du réticulum endoplasmique, elle-mêmenhibée lors de la production d’ATP. L’action activatrice ou inhibitrice du glucoseia les canaux K-ATP reste encore controversée ;une voie indirecte via les facteurs paracrines des îlots :via l’insuline : la cellule alpha serait sous contrôle à la fois de l’insuline et
u glucose [5]. Cependant la sécrétion de glucagon est inhibée à des taux delucose trop bas pour stimuler l’insuline,via la somatostatine : elle possède des récepteurs sur les cellules alpha et
êta, et est stimulée pour des concentrations de glucose plus basses que cellesermettant d’inhiber le glucagon et de stimuler l’insuline.ependant, il existe une intrication de tous ces mécanismes, dont la conséquencest qu’aucune de ces voies n’est indispensable à la régulation.lucagon : une cible thérapeutique (B. Ahren).– Pour lutter contre
’hyperglucagonémie, de nouvelles molécules sont utilisées depuis plu-ieurs années afin de diminuer la sécrétion de glucagon : les analogues duLP1 et les inhibiteurs de la DPP4. Une autre voie serait l’antagonisationes récepteurs au glucagon, présents essentiellement dans le foie. Les étudesortant sur différents antagonistes ont montré une réelle efficacité avecne diminution de l’HbA1c de 1 à 1,5 % ; cependant les effets secondairesont nombreux, dont une cytolyse hépatique et une élévation de la pressionrtérielle, poussant à interrompre les programmes de développement enours.e proglucagon est transformé en glucagon par la proconvertase 2 (PC2)ans les cellules alpha et en GLP1 par la proconvertase 1 (PC1) danses cellules intestinales. L’expression des proconvertases est spécifique duissu. Des travaux sont menés pour convertir la PC2 en PC1. Il a été
ontré que l’IL6 permet une expression augmentée sélective de PC1, abou-issant à une amélioration de l’insulinosécrétion et de l’équilibre glycémique6].n dehors de son effet hyperglycémique, le glucagon permet de stimu-
er la dépense énergétique et de réduire l’appétit. Il pourrait donc être ungent anti-obésité. Un amaigrissement, associé à une amélioration glycé-ique, a été mis en évidence lors de son utilisation couplée à celle de GLP1
7].éférences
1] Meier JJ, et al. Diabetes 2006;55:1051–6.
2] Lee, et al. Diabetes 2011;60:391–7.3] Unger, Cherrington. J Clin Invest 2012;122:4–12.4] Taborsky, Mundiger. Endocrinology 2012;153(3):1055–62.5] Cryer. Endocrinology 2012;153(3):1039–48.