Embed Size (px)
Citation preview

DÉVELOPPEMENT D’UNE VOIE SYNTHÉTIQUE VERS LE SQUELETTE PICRASANE PAR UNE
SÉQUENCE METTANT EN VEDETTE LA CYCLOADDITION DE DIELS-ALDER À DIÈNES
TRANSMISSIBLES
Par
Pascal Dubé
Thèse présentée à la Faculté des sciences en vue
de l’obtention du grade de docteur ès sciences (Ph.D.)
FACULTÉ DES SCIENCES
UNIVERSITÉ DE SHERBROOKE
Sherbrooke, Québec, Canada, novembre 2005

Le _____________________________,
Date
Le jury a accepté la thèse de M. Pascal Dubé dans sa version finale
Composition du jury
Membre : M. Claude Spino (Direction) Département de chimie Membre : M. Guillaume Bélanger Département de chimie Membre : M. Keith Fagnou Université d’Ottawa Membre et M. Jean Lessard Président-rapporteur : Département de chimie _________________________________ Signature

i
SOMMAIRE
Le premier chapitre de cette thèse traite de l’évaluation de connecteurs temporaires pour effectuer une
cycloaddition de Diels-Alder intramoléculaire sur le vinylallène 111. Une hypothèse sera formulée sur
le mode de décomposition des composés durant la cycloaddition d’hétéro Diels-Alder.
Le second chapitre présente une synthèse améliorée de vinylallènes tétrasubstitués comportant un
dithiane comme précurseur d’aldéhyde. Deux solutions seront apportées afin de résoudre le problème
associé à la décomposition obtenue lors de la deuxième cycloaddition [4+2].
Le chapitre trois décrit la synthèse d’un vinylallène comportant une unité triméthylsilylméthyle dans le
but de promouvoir le clivage du connecteur de la première cycloaddition. L’évaluation de cette
stratégie avec deux connecteurs sera ensuite détaillée.
Le quatrième chapitre présente une synthèse du vinylallène 204 par une séquence synthétique
permettant une énantiosélection. Cette section offre également un résumé du couplage de Sonogashira
ainsi que des époxydations énantiosélectives.
Une étude modèle vers une nouvelle synthèse de vinylallène constitue le sujet du chapitre cinq.
L’addition de vinylcuprates à des oxyranes allyliques ainsi que la formation d’allènes via des époxydes
propargyliques seront également présentés.
Le chapitre six traite de l’application de notre nouvelle méthode de synthèse de vinylallène avec
différents groupes protecteurs. La poursuite des séquences ayant produit les vinylallènes 240 jusqu’à
des intermédiaires propices à la troisième cycloaddition de Diels-Alder sera ensuite au sujet.
Finalement, le chapitre sept présente toutes les études de cycloaddition de Diels-Alder ayant pour but la
formation du cycle B du squelette picrasane. Les problèmes reliés à l’instabilité des molécules seront
solutionnés et la réussite de cette troisième cycloaddition décrite. Le chapitre sept se conclura par des
approches à explorer afin de compléter ce projet.

ii
REMERCIEMENTS
Mes remerciements vont avant tout à mes parents, Michel et Francine, pour leur soutien inconditionnel
durant toutes ces années d’étude. Merci à Marie-Pierre pour son support, sa compréhension et son
sourire. Je suis très reconnaissant à Paul et Denise pour leurs encouragements et leur générosité à mon
égard.
Cette thèse n’aurait certainement pas été réalisée avec autant de plaisir sans les membres du labo Spino.
Merci à tous mes coéquipiers de ces quatre dernières années : Julie, Bryan, Marie-Claude, Cèd, Chris,
Jeffrey, Luc, MC, Magali, Martin, Vitthal, Hadi, Christine, Patrice, Amélie, Marc-André, Francis,
Stéphane, Stéphanie, Sophie, Kristina, Sophie, David, Alex et Joannie. Je tiens en particulier à
remercier les membres du team Q, Amélie et Stéphane, pour leur apport à ce projet colossal ainsi que
Ti-Gars pour son ardeur à l’ouvrage lors de son projet de trimestre.
J’aimerais remercier Guillaume Barbe et Frédéric Ménard pour les nombreuses discussions
scientifiques et les nombreuses bières. Parlant bières, merci aux Gilles et aux Polatouches pour les
activités sportives arrosées. Merci à mes chums du bacc pour les voyages de pêche et toutes les activités
partagées. Merci aux complices de la bicoque pour tous ces bons souvenirs. Je ne peux passer sous
silence les nombreux services de Paul dont je suis très reconnaissant. La compagnie de tous ces amis a
été et demeurera toujours très appréciée.
Merci à Gaston Boulay (MS), Réal Dubuc (souffleur de verre), Luc Tremblay et Normand Pothier
(RMN) et Daniel Fortin pour leur expertise et l’aide apportée durant mon doctorat. Je tiens aussi à
remercier les professeurs Jean Lessard, Serge Lacelle et Pierre Deslongchamps pour les nombreuses
discussions chimiques et philosophiques. Merci pour vos commentaires et pour m’avoir aidé à
découvrir cette passion qu’est la science. Je dois absolument remercier Michel Couturier, mon mentor
et mon ami, pour tout le soutien durant ces années.
Finalement, cette aventure n’aurait jamais eu lieu si le professeur Claude Spino ne m’avait pas accepté
dans son groupe. Merci pour la confiance et l’autonomie face au projet, mais aussi pour tous les
conseils et les encouragements concernant ma future carrière.

iii
TABLE DES MATIÈRES
SOMMAIRE ...............................................................................................................................................I
REMERCIEMENTS ................................................................................................................................. II
TABLE DES MATIÈRES ....................................................................................................................... III
LISTE DES ABRÉVIATIONS................................................................................................................VI
LISTE DES TABLEAUX........................................................................................................................IX
LISTE DES FIGURES.............................................................................................................................. X
LISTE DES SCHÉMAS...........................................................................................................................XI
INTRODUCTION......................................................................................................................................1
I.1 Quassinoïdes................................................................................................................................2
I.1.1. Structures chimiques et aspect biologique.....................................................................2
I.1.2. Synthèses de la quassine (1) ..........................................................................................5
I.2 La synthèse des quassinoïdes selon le groupe Spino ................................................................10
I.2.1. Cycloaddition de Diels-Alder à Diène Transmissible (CDADT) ................................10
I.2.1. Approche rétrosynthétique...........................................................................................11
I.2.2. Cycloaddition de Diels-Alder avec un diène 1,1-disubstitué ......................................13
I.2.3. Diels-Alder intramoléculaire avec un vinylallène .......................................................15
I.3 Connecteurs temporaires ...........................................................................................................17
I.3.1. Un pont hydrogène comme connecteur .......................................................................17
I.3.2. Un métal comme connecteur........................................................................................18
I.3.2. Les connecteurs à liaisons covalentes..........................................................................19
CHAPITRE 1 : DÉVELOPPEMENT D’UN CONNECTEUR POUR LA PREMIÈRE
CYCLOADDITION.................................................................................................................................25
1.1. Un connecteur vinylsiloxane ...................................................................................................25
1.2. Un connecteur sulfonate ..........................................................................................................27
1.3. Un connecteur éther d’énol......................................................................................................29
1.3.1. Synthèse de vinyléthers ...............................................................................................29
1.3.2. Formation de l’éther d’énol et cycloaddition ..............................................................30
CHAPITRE 2 : SYNTHÈSE DE L’ALLÈNE AVEC UN DITHIANE COMME GROUPE
PROTECTEUR ……………………………………………………….……………………………32
2.1. Synthèse de l’allène-dithiane 131 ............................................................................................32

iv
2.2. Éther homo allylique................................................................................................................34
2.3. Incorporation d’un élément de promotion du clivage du connecteur temporaire....................37
CHAPITRE 3 : SÉQUENCE SYNTHÉTIQUE METTANT À PROFIT UNE UNITÉ TMS-
MÉTHYLÈNE COMME AGENT DE CLIVAGE DU CONNECTEUR ...............................................39
3.1. Synthèse du vinylallène-TMS 160...........................................................................................39
3.2. Ester fumarique en tant que connecteur...................................................................................41
3.3. Vinylallène-TMS 160 et l’éther d’énol en tant que connecteur ..............................................43
CHAPITRE 4 : SYNTHÈSE DU VINYLALLÈNE COMPORTANT L’UNITÉ TMS-
MÉTHYLÈNE PAR ÉPOXYDATION D’UN ÉNYNE..........................................................................47
4.1. Travaux précédents ..................................................................................................................47
4.1.1. Introduction au couplage de Sonogashira ...................................................................48
4.1.2. Introduction aux époxydations énantiosélectives........................................................50
4.2. Synthèse du vinylallène-TMS avec un acétal comme groupe protecteur................................52
4.3. Synthèse du vinylallène avec un MOM comme groupe protecteur.........................................55
CHAPITRE 5 : ÉTUDE MODÈLE POUR UNE NOUVELLE SYNTHÈSE DE VINYLALLÈNES....61
5.1. Stratégie générale.....................................................................................................................61
5.2. Addition de vinylcuprates sur des époxydes allyliques...........................................................62
5.3. Déplacement SN2’ d’époxydes propargyliques .......................................................................63
5.4. Étude modèle ...........................................................................................................................64
CHAPITRE 6 : SÉQUENCE SYNTHÉTIQUE VERS UN β-BROMOVINYLALLÈNE......................66
6.1. Séquence réactionnelle générale..............................................................................................66
6.2. Séquence avec les substrats ayant un TBDPS comme groupe protecteur ...............................69
6.3. Séquence avec les substrats ayant un benzyle comme groupe protecteur ...............................70
6.4. Séquence comportant un dioxolane comme groupe protecteur ...............................................76
CHAPITRE 7 : ÉTUDES DE LA TROISIÈME CYCLOADDITION DE DIELS-ALDER...................78
7.1. Cycloaddition intermoléculaire du diène 252..........................................................................78
7.2. Cycloaddition intermoléculaire du diène 253..........................................................................80
7.3. Cycloaddition intermoléculaire du diène 254..........................................................................82
7.4. Fonctionnalisation de l’oléfine terminale du triène 254 ..........................................................84
7.5. Cycloaddition intermoléculaire du diène 262..........................................................................87
7.6. Protection du diol 262 et cycloaddition intermoléculaire du diène 267 ..................................90
7.7. Projets futurs ............................................................................................................................91

v
CONCLUSION GÉNÉRALE ..................................................................................................................95
PARTIE EXPÉRIMENTALE..................................................................................................................96
Remarques générales ......................................................................................................................96
Modes opératoires...........................................................................................................................98
RÉFÉRENCES ET NOTES ...................................................................................................................155
ANNEXE 1 : SPECTRES DE RÉSONANCE MAGNÉTIQUE NUCLÉAIRE DES PROTONS........169
ANNEXE 2 : SPECTRES DE RÉSONANCE MAGNÉTIQUE NUCLÉAIRE DES CARBONES.....260

vi
LISTE DES ABRÉVIATIONS
Ac acétyle
APTS acide p-toluènesulfonique
Bn benzyle
BOC carbamate de t-butyle
Borax® décahydrate de borate disodique (Na2B4O7•10H2O)
Bu butyle
CAN nitrate d’ammonium cérique
Cbz carbamate benzylique
CCM chromatographie sur couche mince
CDA cycloaddition de Diels-Alder
CDADT cycloaddition de Diels-Alder à diène transmissible
CHDA cycloaddition d’hétéro Diels-Alder
Cp cyclopentadiényle
CSA acide camphorsulfonique
Cy cyclohexyle
DA Diels-Alder
dba dibenzylidèneacétone
DCC 1,3-dicyclohexylcarbodiimide
DDQ 2,3-dichloro-5,6-dicyanoquinone
DEAD azodicarboxylate de diéthyle
DIBAL hydrure de diisobutylaluminium
DMAP N,N-diméthylaminopyridine
DME 1,2-diméthoxyéthane
DMF N,N-diméthylformamide
DMP periodinane de Dess-Martin
DMS diméthylsulfure
DMSO diméthylsulfoxyde
Et éthyle
EVE éther éthylvinylique

vii
fod 6,6,7,7,8,8,8-heptafluoro-2,2-diméthyl-3,5-octanedionate
GP groupe protecteur
HOMO plus haute orbitale moléculaire occupée
HPLC chromatographie en phase liquide à haute pression
IBX acide iodoxybenzoïque
IR infrarouge
L ligand
KHMDS bis(triméthylsilyl)amidure de potassium
LUMO plus basse orbitale moléculaire vacante
M métal
m-CPBA acide m-chloroperoxybenzoïque
Me méthyle
Mes mésétyle (2,4,6-triméthylphényle)
MOM méthoxyméthoxyméthyle
NCS N-chlorosuccinimide
NMM N-méthylmorpholine
NOESY Nuclear Overhauser Effect Spectroscopy
PCC chlorochromate de pyridinium
Ph phényle
Piv pivaloyle (triméthylacétyle)
PMB p-méthoxybenzyle
Pr propyle
Red-Al® hydrure du bis(2-méthoxyéthoxy)aluminate de sodium
RMN résonance magnétique nucléaire
SEM 2-(triméthylsilyl)éthoxyméthyle
SMBR spectre de masse à basse résolution
SMHR spectre de masse à haute résolution
TBAF fluorure de tétrabutylammonium
TBAI iodure de tétrabutylammonium
TBDPS t-butyldiphénylsilyle
TBS t-butyldiméthylsilyle
Tf triflate (trifluorométhanesulfonyle)

viii
thex thexyle (2,3-diméthyl-2-butyle)
THF tétrahydrofuranne
TIPS triisopropylsilyle
TMS triméthylsilyle
TPAP perruthénate de tétrapropylammonium
Ts tosyle (p-toluènesulfonyle)
xyl xylène

ix
LISTE DES TABLEAUX
Tableau 1. Oxydation de Tamao-Fleming du siloxane 113. ...................................................................26
Tableau 2. Clivage du sulfonate 116.......................................................................................................29
Tableau 3. Clivage du sulfonate 132.......................................................................................................34
Tableau 4. Tentatives de CDA de l’éther d’énol 141. ............................................................................36
Tableau 5. Tentatives de clivage de la lactone 162.................................................................................42
Tableau 6. Tentatives de clivage de l’éther 167......................................................................................44
Tableau 7. Addition du réactif de méthylcuprate sur l’époxyde 195......................................................56
Tableau 8. Rendements des réactions du schéma 70 avec les différents groupes protecteurs................68
Tableau 9. Clivage de l’éther de l’adduit 240e. ......................................................................................71
Tableau 10. Déprotection de l’alcool primaire de 246............................................................................73
Tableau 11. Fonctionnalisation de l’alcène terminale de 246.................................................................75
Tableau 12. Essais de CDA intermoléculaire du diène 252....................................................................79
Tableau 13. Essais de CDA intermoléculaire du diène 253....................................................................81
Tableau 14. Essais de CDA intermoléculaire du diène 254....................................................................83
Tableau 15. Hydroboration de l’alcène terminal du triène 254. .............................................................85
Tableau 16. Essais de CDA intermoléculaire du diène 262....................................................................90
Tableau 17. Essais de CDA intermoléculaire du diène 267....................................................................91
Tableau G.1 : Agents desséchants utilisés pour la distillation de différents solvants et réactifs............96

x
LISTE DES FIGURES
Figure 1. Premières quassinoïdes isolées et caractérisées.........................................................................2
Figure 2. Exemples de quassinoïdes bioactives. .......................................................................................3
Figure 3. État de transition de l’hétéro-DA.............................................................................................11
Figure 4. CDA via pont hydrogène. ........................................................................................................18
Figure 5. Structure de 167 par diffraction des rayons X. ........................................................................43
Figure 6. État de transition proposé pour l’époxydation de Shi..............................................................51
Figure 7. Formation de l’allène par l’addition de réactifs de cuprate. ....................................................61
Figure 8. Conformation 3D et corrélations NOESY de l’adduit tricyclique 263....................................87
Figure 9. Spectre NOESY de l’adduit tricyclique 263............................................................................88
Figure 10. Représentations 3D du diol 262.............................................................................................89

xi
LISTE DES SCHÉMAS
Schéma 1. ...................................................................................................................................................5
Schéma 2. ...................................................................................................................................................6
Schéma 3. ...................................................................................................................................................7
Schéma 4. ...................................................................................................................................................8
Schéma 5. ...................................................................................................................................................9
Schéma 6. .................................................................................................................................................10
Schéma 7. .................................................................................................................................................12
Schéma 8. .................................................................................................................................................13
Schéma 9. .................................................................................................................................................14
Schéma 10. ...............................................................................................................................................14
Schéma 11. ...............................................................................................................................................15
Schéma 12. ...............................................................................................................................................15
Schéma 13. ...............................................................................................................................................16
Schéma 14. ...............................................................................................................................................18
Schéma 15. ...............................................................................................................................................19
Schéma 16. ...............................................................................................................................................19
Schéma 17. ...............................................................................................................................................20
Schéma 18. ...............................................................................................................................................20
Schéma 19. ...............................................................................................................................................21
Schéma 20. ...............................................................................................................................................21
Schéma 21. ...............................................................................................................................................22
Schéma 22. ...............................................................................................................................................23
Schéma 23. ...............................................................................................................................................24
Schéma 24. ...............................................................................................................................................24
Schéma 25. ...............................................................................................................................................25
Schéma 26. ...............................................................................................................................................26
Schéma 27. ...............................................................................................................................................27
Schéma 28. ...............................................................................................................................................28
Schéma 29. ...............................................................................................................................................29

xii
Schéma 30. ...............................................................................................................................................30
Schéma 31. ...............................................................................................................................................30
Schéma 32. ...............................................................................................................................................31
Schéma 33. ...............................................................................................................................................32
Schéma 34. ...............................................................................................................................................33
Schéma 35. ...............................................................................................................................................35
Schéma 36. ...............................................................................................................................................35
Schéma 37. ...............................................................................................................................................37
Schéma 38. ...............................................................................................................................................38
Schéma 39. ...............................................................................................................................................38
Schéma 40. ...............................................................................................................................................39
Schéma 41. ...............................................................................................................................................40
Schéma 42. ...............................................................................................................................................41
Schéma 43. ...............................................................................................................................................42
Schéma 44. ...............................................................................................................................................43
Schéma 45. ...............................................................................................................................................44
Schéma 46. ...............................................................................................................................................45
Schéma 47. ...............................................................................................................................................47
Schéma 48. ...............................................................................................................................................49
Schéma 49. ...............................................................................................................................................50
Schéma 50. ...............................................................................................................................................51
Schéma 51. ...............................................................................................................................................52
Schéma 52. ...............................................................................................................................................53
Schéma 53. ...............................................................................................................................................53
Schéma 54. ...............................................................................................................................................54
Schéma 55. ...............................................................................................................................................55
Schéma 56. ...............................................................................................................................................57
Schéma 57. ...............................................................................................................................................57
Schéma 58. ...............................................................................................................................................58
Schéma 59. ...............................................................................................................................................58
Schéma 60. ...............................................................................................................................................59
Schéma 61. ...............................................................................................................................................60

xiii
Schéma 62. ...............................................................................................................................................60
Schéma 63. ...............................................................................................................................................62
Schéma 64. ...............................................................................................................................................62
Schéma 65. ...............................................................................................................................................63
Schéma 66. ...............................................................................................................................................63
Schéma 67. ...............................................................................................................................................64
Schéma 68. ...............................................................................................................................................65
Schéma 69. ...............................................................................................................................................65
Schéma 70. ...............................................................................................................................................66
Schéma 71. ...............................................................................................................................................69
Schéma 72. ...............................................................................................................................................70
Schéma 73. ...............................................................................................................................................71
Schéma 74. ...............................................................................................................................................74
Schéma 75. ...............................................................................................................................................74
Schéma 76. ...............................................................................................................................................76
Schéma 77. ...............................................................................................................................................77
Schéma 78. ...............................................................................................................................................84
Schéma 79. ...............................................................................................................................................86
Schéma 80. ...............................................................................................................................................86
Schéma 81. ...............................................................................................................................................87
Schéma 82. ...............................................................................................................................................90
Schéma 83. ...............................................................................................................................................92
Schéma 84. ...............................................................................................................................................93
Schéma 85. ...............................................................................................................................................93
Schéma 86. ...............................................................................................................................................94

1
INTRODUCTION
Cette histoire a commencé en 1807 lorsque Jön Jakob Berzelius, un chimiste suédois, a appliqué la
notion de « organique » aux composés dérivés des organismes vivants. À cette époque, on croyait que
les substances chimiques provenant de la nature possédaient une essence de vie. La synthèse de
composés organiques était alors considérée comme impossible, par la croyance en cette énergie vitale.
La science et l’histoire furent changées en 1828 lorsque Friedrich Wöhler chauffa de l’ammoniac et de
l’acide cyanurique. Les cristaux d’urée obtenus de façon synthétique étaient identiques en tout point à
l’urée obtenue de l’urine animale. Ce fut la naissance de la chimie organique telle que nous la
connaissons.
Dès lors, il fut possible de fabriquer de nouveaux composés qui eurent un impact incroyable sur la
qualité de vie des gens, l’espérance de vie de la population mondiale doublant en seulement cent ans.
Parmi les progrès engendrés par la chimie organique, notons l’anesthésie, le nylon, la pilule
contraceptive, la réfrigération, les explosifs et la psychothérapie! Pour nous, ce n’était que le début
d’une odyssée.1
Depuis le début de ses aventures synthétiques, le chimiste s’est tourné vers la nature pour y trouver
l’inspiration et le challenge de nouvelles synthèses, souvent afin de reproduire l’ingrédient actif d’un
remède traditionnel. Pour ce faire, les organiciens ont appris à organiser la matière en développant les
concepts de structure, de stéréochimie et de réactivité. L’usage de la rétrosynthèse a permis de réaliser
la synthèse de composés de plus en plus complexes, par exemple la vitamine B12, la palytoxine et le
Taxol.2
À notre tour, nous avons consulté la nature en quête d’inspiration…et nous avons trouvé le challenge
que voici. Armé de nos connaissances, notre créativité, notre patience et notre détermination, nous
avons entamé la synthèse d’agents anti-cancer, membres d’une large famille de produits naturels
appelés quassinoïdes.

2
I.1 Quassinoïdes
La présence de composés au goût amer dans le bois des arbustes appartenant à la famille Quassia
amara a été rapporté pour la première fois en 1835, mais les tentatives d’isolation et de purification des
composés actifs s’avérèrent vaines.3 Plus de treize ans après leur isolation en 1937 par Clark,4
Robertson parvint à caractériser les principes actifs du bois de quassia, dont les constituants étaient la
quassine 1 et la néoquassine 2.5 Depuis que Valenta a élucidé leur structure par des méthodes
spectroscopiques aujourd’hui classiques6 (figure 1), des centaines de différentes quassinoïdes ont été
isolées et caractérisées.7
O
O
H
HH
HMeO
OOMe
O
O
O
H
HH
HMeO
OOMe
OH
quassine 1 néoquassine 2
Figure 1. Premières quassinoïdes isolées et caractérisées.
I.1.1. Structures chimiques et aspect biologique
Biosynthétiquement, les quassinoïdes proviennent de la dégradation d’intermédiaires stéroïdiens. Le
lecteur est référé aux composés 4 et 5 pour la numérotation du squelette picrasane puisque cette
nomenclature reviendra tout au long du manuscrit (figure 2). Leur architecture carbonée polycyclique
est agrémentée d’un vaste répertoire de fonctions oxygénées, principalement concentrées sur les cycles
A, C et D.

3
O
O
H
HH
HMeO
O
OO
O
H
HH
HO
HOOH
O
CO2Me
O
O
HOO
O
H
HH
HO
HOOH
O
OR
O
O
O
OAc
O
H
HH
O
HOOH
O
OH
OHO
A
E
D
C
B
O
H
HH
HO
HOOH
O
OH
OHO
O
H
HH
HO
HOOH
O
CO2Me
O
OHO
O
picrasine D 4
brucéantine 3
6 simalikalactone D, R =
7 quassimarine, R =
glaucarubolone 5
1
48
11
14
brucéine D 8 brucéanol A 9
Figure 2. Exemples de quassinoïdes bioactives.
Les quassinoïdes sont utilisées depuis des siècles dans les médecines traditionnelles asiatiques et
africaines.8 Suite à la découverte en 1970 de l’activité anti-cancéreuse9 de la halocanthone, un membre
de la famille des quassinoïdes, une étude systématique de tous les composés de cette famille a permis
d’ajouter une grande gamme d’activités biologiques à leur palmarès telles anti-virale, anti-malariale,
anti-VIH et insecticide.10
Après son échec en phase II d’essais cliniques pour ses propriétés anti-leucémiques,11 la brucéantine (3)
a fait un retour en force grâce à son activité contre la malaria, mais principalement parce qu’à une très
faible concentration (IC50 = 12 nM), elle induit la régression de tumeurs en inhibant la prolifération

4
cellulaire et en induisant l’apoptose de certaines cellules cancéreuses.12 Des études ont démontré que la
brucéantine initiait l’apoptose13 par des mécanismes associés aux caspases et aux mitochondries.14 (Les
caspases sont des protéases qui, suite à la réception d’un signal pro-apoptotique, amorcent le clivage de
certaines protéines structurelles, résultant en un désassemblage de la cellule.)
La simalikalactone D (6) est aussi un membre des quassinoïdes faisant présentement l’objet d’études.15
En plus de démontrer une inhibition de l’enzyme oxyde nitrique synthase (impliquée dans
l’augmentation du flux sanguin dans les tissus cancéreux), ce produit naturel est actif in vitro contre la
synthèse de protéines d’une souche de la malaria résistante à la chloroquine. Des chercheurs ont
récemment découvert que la glaucarubolone (5) tire son activité anti-néoplastique de l’inhibition de
l’enzyme NADH oxydase, une protéine du plasma attachée à la membrane cellulaire.16 La destruction
de cellules cancéreuses de foie de rat s’est produite avec une solution de 5 nM alors que la destruction
in vitro de cellules cervicales cancéreuses a nécessité une concentration de 0.05 nM en glaucarubolone.
L’inhibition de la synthèse des protéines dans les cellules eucaryotes représente un mode d’action
particulier à ces composés. Plus précisément, les quassinoïdes sont des inhibiteurs de l’élongation des
chaînes polypeptidiques, ce qui empêche la formation des liens peptidiques au début de la
polymérisation.17 Des chercheurs de l’université McGill ont récemment déterminé les prémisses
structurelles nécessaire à cette activité.18 Outre la présence d’un pont époxyméthane, les composés les
plus actifs possédaient un carbonyle α,β-insaturé dans le cycle A, mais ne comportaient aucun
substituant sur leurs oxygènes du cycle C.
Le mode d’action des quassinoïdes est intimement relié à la structure et la fonctionnalisation de ces
composés. Kupchan a observé que la réduction de l’alcène conjugué au carbonyle du cycle A de la
brucéantine (c.f. figure 2) entraîne une importante diminution de son effet biologique anti-
néoplastique.19 De nombreuses études ont démontré qu’un des mécanismes d’action des quassinoïdes
tire profit de l’addition de Michael sur ses carbonyles α,β-insaturés.20 La possibilité d’utiliser certaines
quassinoïdes en combinaison avec d’autres agents thérapeutiques est présentement à l’étude en plus de
soulever de potentielles applications cliniques. Leur complexité structurelle combinée à eurs
impressionnantes propriétés biologiques font des quassinoïdes des cibles synthétiques très prisées par la
communauté chimique.

5
I.1.2. Synthèses de la quassine (1)
Plus de 25 groupes de recherche ont publié des approches synthétiques vers ces triterpènes naturels,
mais peu d’entre eux ont eu la chance de crier victoire.21 En particulier, notons les approches
développées par les groupes de Barrero,22 Kraus,23 Deslongchamps,24 Fuchs,25 Heathcock,26 Ziegler27 et
Spino.28 Quoique peu nombreux, certains groupes sont parvenus à surmonter ce défi synthétique de
taille. Le premier succès survint en 1980, quand la quassine (1) succomba aux efforts synthétiques du
groupe de Grieco.29 La stratégie développée comporte une cycloaddition de Diels-Alder (CDA) comme
étape clé (schéma 1), une des réactions les plus puissantes pour construire des cycles à six membres.30
Les caractéristiques de cette réaction ayant été revues précédemment, il n’en sera pas question dans ce
manuscrit.31
O
O OMe
HO
12 CO2Et
40%10 11
O
O
H
HH
HMeO
OOMe
O
OMe
H
HCO2Et
O
13
quassine 1
AlCl3, benzène, 23 oC
8 étapes
22 étapes
8149
Schéma 1.
En débutant avec la cétone de Wieland-Miesher racémique (10), Grieco a obtenu la cétone 11 après
plusieurs transformations. La formation du centre quaternaire en C8 a ensuite été effectuée grâce à une
cycloaddition de Diels-Alder avec le diène 12. Cette CDA, procédant avec une sélectivité endo, a
permis l’instauration des centres chiraux C8 et C14 avec la bonne stéréochimie, tandis que le centre en
C9 a dû être épimérisé ultérieurement. La quassine a été obtenue après 22 étapes supplémentaires de

6
manipulation de groupes fonctionnels et d’oxydations pour un total de 31 étapes à partir de la cétone
10. Grieco a par la suite appliqué cette stratégie à la synthèse de différents membres des quassinoïdes
tels la simalikalactone D (6),32 la brucéantine (3),33 la samaderine B,34 la chaparrinone35 et la
shinjulactone C.36
Le second succès est provenu du groupe de Watt qui, en 1990, a synthétisé la quassine (1) de façon
énantiopure à partir de la cétone de Wieland-Miescher (10) optiquement active (schéma 2).37 Inspiré
par la stratégie de Grieco, l’étape clé fait intervenir une CDA entre le diénophile 14 et le diène de
Danishefsky 15.38 La réduction des fonctions carbonyles et l’hydrolyse de l’éther d’énol ont procuré la
cétone tricyclique 16 dans un excellent rendement pour les trois étapes. Cet intermédiaire a nécessité 26
étapes synthétiques, afin d’installer le cycle D et instaurer les fonctions oxygénés sur le squelette, avant
de procurer la quassine énantiopure. Cette première synthèse énantiosélective d’un membre des
quassinoïdes a requis un total de 35 étapes synthétiques à partir de la cétone (-)-10.
O
O
(-) 10
OTBS
H
CHO
O
14
TMSO
OMe15
O
O
H
HH
HMeO
OOMe
O
O
OH
OTBS
H
HOH 16
quassine 1
6 étapes
26 étapes
+2) Red-Al
3) HCl aq.
83% / 3 étapes
Schéma 2.

7
L’année suivante, le groupe de Valenta a publié la synthèse de la (±)-quassine (1), près de trente ans
après son élucidation structurelle (schéma 3).39 Quoique le rendement de la CDA initiale entre le diène
17 et la quinone 18 fut faible, cette réaction a permis la formation stéréosélective de quatre centres
chiraux, dont deux quaternaires. Suite à cette démonstration de planification synthétique exceptionnelle,
la réorganisation structurelle du composé tricyclique 19 a nécessité de nombreuses étapes. La quassine
(1) a finalement été obtenue 26 réactions après la cycloaddition de Diels-Alder, que Valenta avait
publiée 15 ans plus tôt.40
OAc
O
O
BF3-OEt2
17 1819
AcO
O
O
O
O
OH
HMeO2C
HO
O
O
H
HH
HMeO
OOMe
O
20
quassine 1
5
10
7 8
13 étapes
+30%
13 étapes
5
107
8
Schéma 3.
La même année, Takahashi a publié la synthèse de l’amarolide 23.41 Contrairement aux autres, l’étape
clé de sa synthèse est une annélation de Robinson sur le composé 21,42 précédemment obtenu par
Heathcock lors de la synthèse de triterpènes (schéma 4).43 L’adduit tricyclique 22 a ensuite nécessité 35
étapes synthétiques pour introduire les fonctionnalités présentes dans l’amarolide (23). Comme ce
composé avait déjà été transformé en quassine (1) par une séquence de deux étapes, ce travail constitue
une synthèse formelle de la quassine.44

8
OH
O
O
21
O
O
O
22
O
H
H
H
H
OO
OMe
MeO
O
O
H
H
HO
HO
H
H
O
O
O
quassine 1
1)
2) éthylène glycol3) Oxydation
35 étapes
2 étapes
amarolide 23
0.5%
32%
Schéma 4.
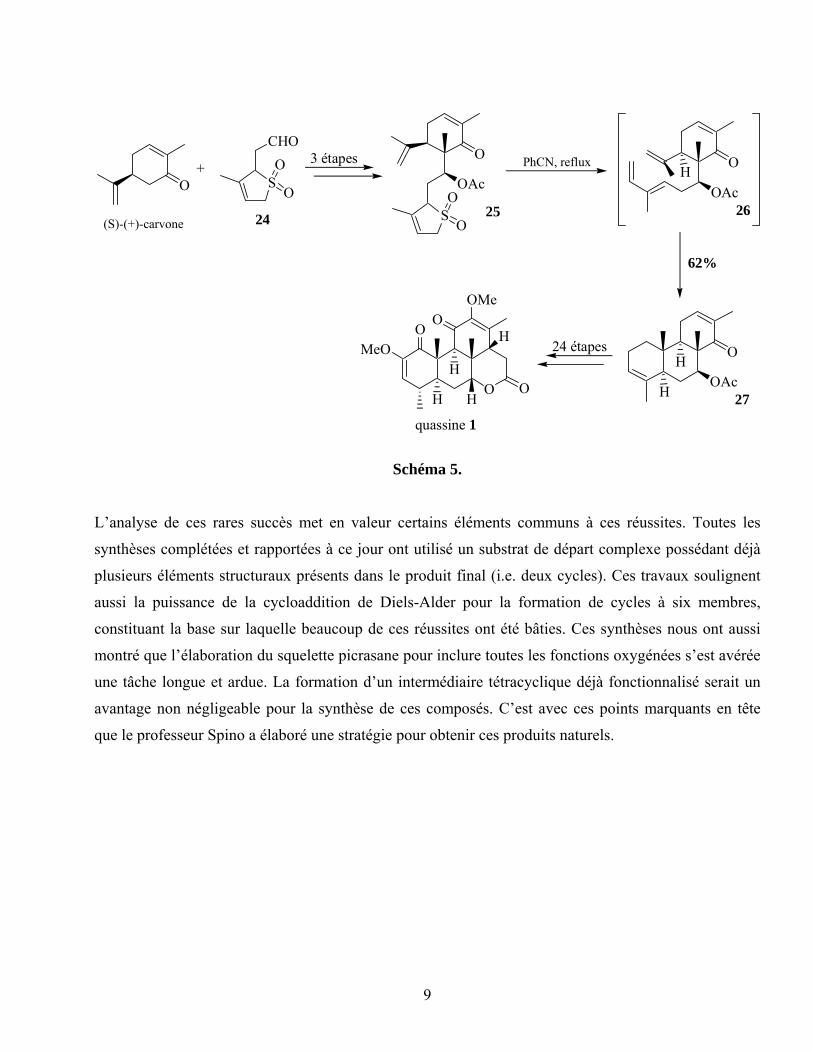
La plus récente synthèse d’un membre des quassinoïdes a été effectué par Shing, soit la quassine (1) en
1998.45 Comme ses prédécesseurs, Shing a tiré avantage de la cycloaddition de Diels-Alder, cette fois
afin d’assembler efficacement les cycles A et B (schéma 5). Dans ce sens, la génération du diène par
l’extrusion de SO2 du sulfolène 25 a permis une CDA stéréosélective du diène 26 avec la chaîne 2-
propène provenant de la carvone pour générer le composé tricyclique 27 dans un bon rendement.
L’ajustement des bons niveaux d’oxydation des carbones des cycles A et C a ensuite fourni la quassine
énantiopure (1). La force de la stratégie de Shing réside dans la reconnaissance de la (S)-carvone
comme précurseur du cycle C.
9
25
27
O
O
H
HH
HMeO
OOMe
O
quassine 1
24 étapes
24
O
S
OAc
O
O
62%
O
CHO
SO
O
O
OAcH
H
+
(S)-(+)-carvone
PhCN, reflux3 étapes
26
O
OAcH
Schéma 5.
L’analyse de ces rares succès met en valeur certains éléments communs à ces réussites. Toutes les
synthèses complétées et rapportées à ce jour ont utilisé un substrat de départ complexe possédant déjà
plusieurs éléments structuraux présents dans le produit final (i.e. deux cycles). Ces travaux soulignent
aussi la puissance de la cycloaddition de Diels-Alder pour la formation de cycles à six membres,
constituant la base sur laquelle beaucoup de ces réussites ont été bâties. Ces synthèses nous ont aussi
montré que l’élaboration du squelette picrasane pour inclure toutes les fonctions oxygénées s’est avérée
une tâche longue et ardue. La formation d’un intermédiaire tétracyclique déjà fonctionnalisé serait un
avantage non négligeable pour la synthèse de ces composés. C’est avec ces points marquants en tête
que le professeur Spino a élaboré une stratégie pour obtenir ces produits naturels.

10
I.2 La synthèse des quassinoïdes selon le groupe Spino
I.2.1. Cycloaddition de Diels-Alder à Diène Transmissible (CDADT)
La stratégie développée dans le groupe du professeur Spino tire son origine d’études sur la
cycloaddition de Diels-Alder à diène transmissible publiées dans les années 1950.46 Principalement
développé par Tsuge,47 ce type de CDA n’a été rapporté qu’à quelques occasions dans la littérature.48
Par exemple, Fallis a récemment utilisé cette stratégie afin de construire les squelettes de nor-stéroïdes
et de triterpènes (schéma 6).49
O
ORO
OO
ORO
O
R
HOH
ORO
O29 3028
O
O
O
ORO
O
R
H
O
O
H
H
H
31
Oxydation
O
OH
HO
HO OH
L-arabinose
8 étapes
Schéma 6.
Débutant avec le L-arabinose, le composé 28 est obtenu après de nombreuses modifications.
L’oxydation de l’alcool allylique selon la méthode de Swern a procuré la cétone 29 qui, sous les
conditions réactionnelles, a procuré l’adduit de CDA intramoléculaire 30.50 En plus de former un
composé bicyclique fusionné, cette cycloaddition a généré un nouveau diène exocyclique qui a procuré
le composé tétracyclique 31 suite à sa réaction avec la para-quinone. En seulement deux étapes, la
CDADT a permis de former trois cycles et créer cinq nouveaux centres chiraux.

11
I.2.1. Approche rétrosynthétique
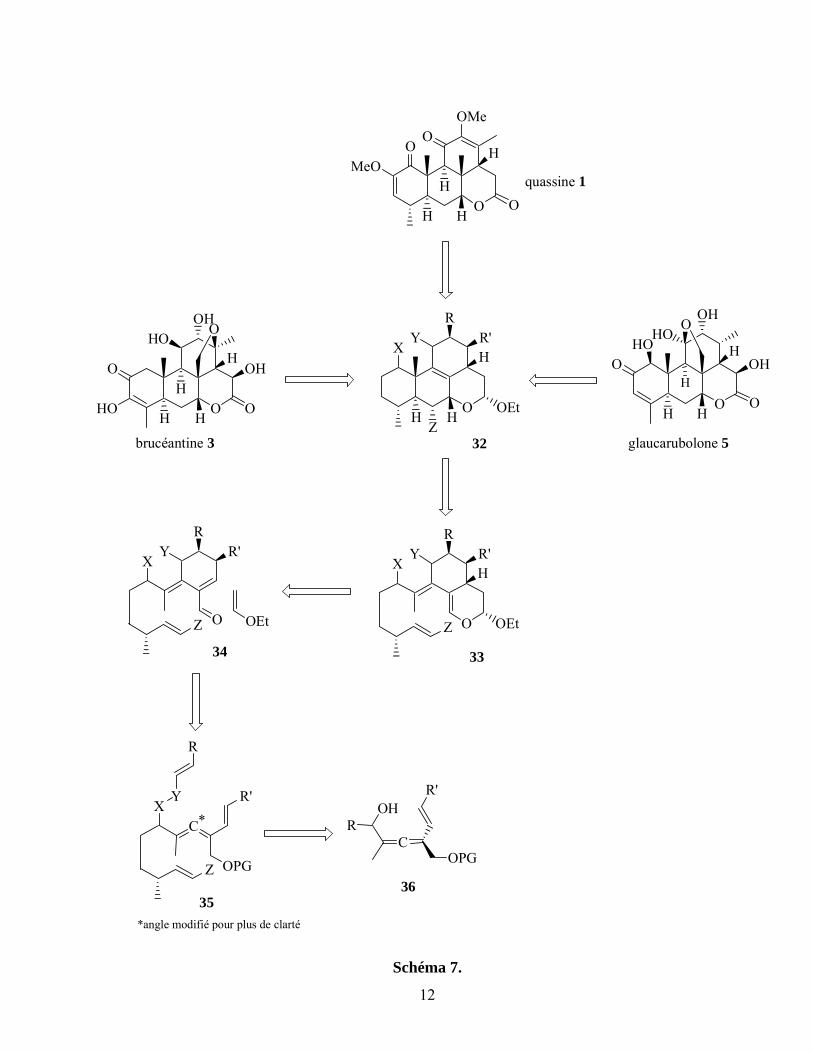
Suite aux résultats de plusieurs études modèles, notre stratégie de synthèse de quassinoïdes a évolué et
prévoit maintenant l’usage de trois CDA consécutives (schéma 7).28a Selon les nombreuses études déjà
publiées, nous croyons qu’il est possible d’effectuer la synthèse de différentes quassinoïdes à partir
d’un intermédiaire avancé commun, tel 32, suite à un ajustement des niveaux d’oxydation des cycles A
et C.21 Cet adduit tétracyclique serait obtenu suite à une cycloaddition de Diels-Alder intramoléculaire
du composé 33, possédant une double liaison exocyclique et tétrasubstituée. Cette transformation
probablement ardue sera discutée plus en détail à la section I.2.2. À son tour, cet éther d’énol
proviendrait d’une hétéro CDA sur l’aldéhyde 34. Cette cycloaddition [4+2], effectuée depuis de
nombreuses années dans le laboratoire du professeur Spino, ne devrait pas causer de problème.28
Plusieurs études ont démontrées que la réaction sous catalyse d’ytterbium procède via un état de
transition α-endo afin d’éviter les interactions stériques défavorables avec les substituants en C12 et
C13 (figure 3).51
L’aldéhyde α,β-insaturé 34 serait obtenu, après modification des groupes fonctionnels, d’une
cycloaddition de Diels-Alder intramoléculaire du vinylallène 35. Le connecteur temporaire (X-Y,
schéma 7), suite à son clivage, permettrait aussi l’introduction de fonctionnalités sur les cycles A et C.
La CDA d’un vinylallène sera abordée avec plus de détails à la section I.2.3. Le connecteur (section I.3)
serait installé sur un vinylallène tel que 36 (c.f. schéma 7), dont la synthèse ne sera pas décrite dans
cette introduction puisqu’elle a été documentée dans la littérature et a fait l’objet de l’étude de maîtrise
de Sylvie Fréchette.52
O
R'RY
EtO37 Yb
Figure 3. État de transition de l’hétéro-DA.
12
OEt
33
O
H
OEt
RR'
X
ZO
RR'
X
Z
32
O
H
HHOEt
Z
RR'
X
C
OPG
R'X
Z
Y
R
34
35
COPG
R'OH
R
36
O
HOO
OHH
H
O
OH
H
HOHO
brucéantine 3
Y
YY
O
H
HH
O
HOOH
O
OH
OHO
H
glaucarubolone 5
O
O
H
HH
HMeO
OOMe
Oquassine 1
*
*angle modifié pour plus de clarté
Schéma 7.
13
I.2.2. Cycloaddition de Diels-Alder avec un diène 1,1-disubstitué
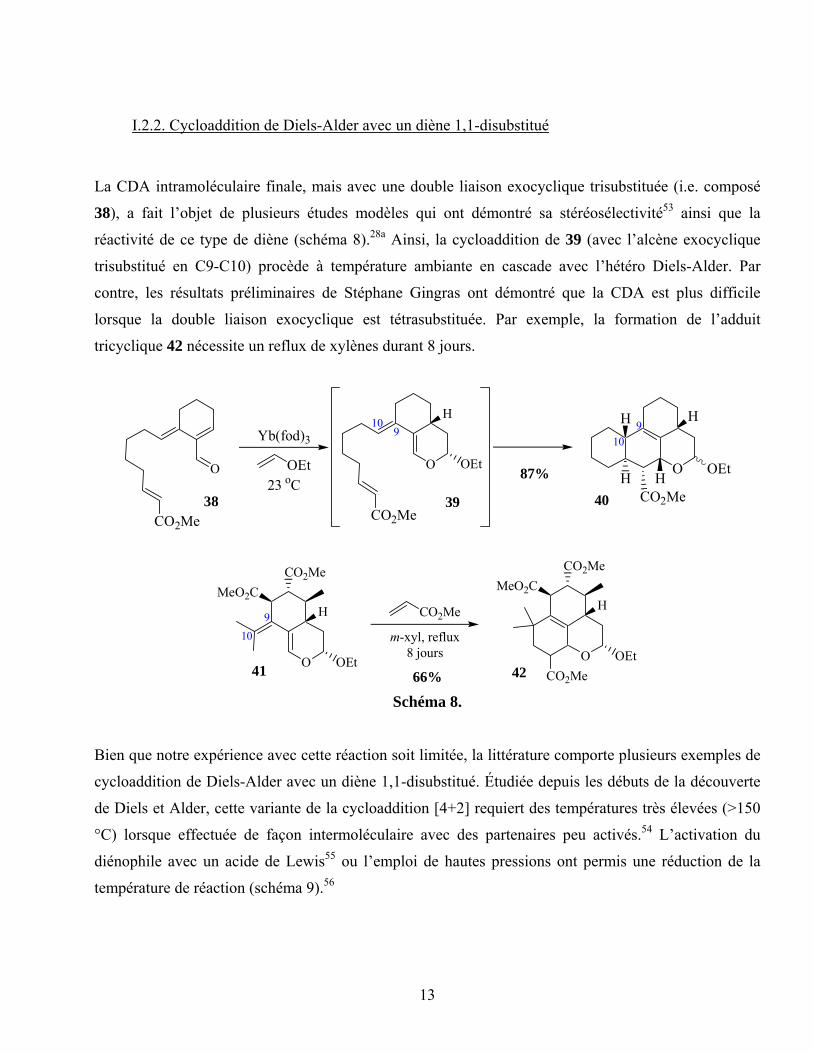
La CDA intramoléculaire finale, mais avec une double liaison exocyclique trisubstituée (i.e. composé
38), a fait l’objet de plusieurs études modèles qui ont démontré sa stéréosélectivité53 ainsi que la
réactivité de ce type de diène (schéma 8).28a Ainsi, la cycloaddition de 39 (avec l’alcène exocyclique
trisubstitué en C9-C10) procède à température ambiante en cascade avec l’hétéro Diels-Alder. Par
contre, les résultats préliminaires de Stéphane Gingras ont démontré que la CDA est plus difficile
lorsque la double liaison exocyclique est tétrasubstituée. Par exemple, la formation de l’adduit
tricyclique 42 nécessite un reflux de xylènes durant 8 jours.
O
CO2Me38
OEt
O
H
OEt
CO2MeMeO2C
O
H
OEt
CO2Me
CO2Me
39
41 42O OEt
CO2MeMeO2C
CO2Me
H
40
O OEt
H H
HHCO2Me
m-xyl, reflux8 jours
66%
Yb(fod)3
23 oC87%
910
109
109
Schéma 8.
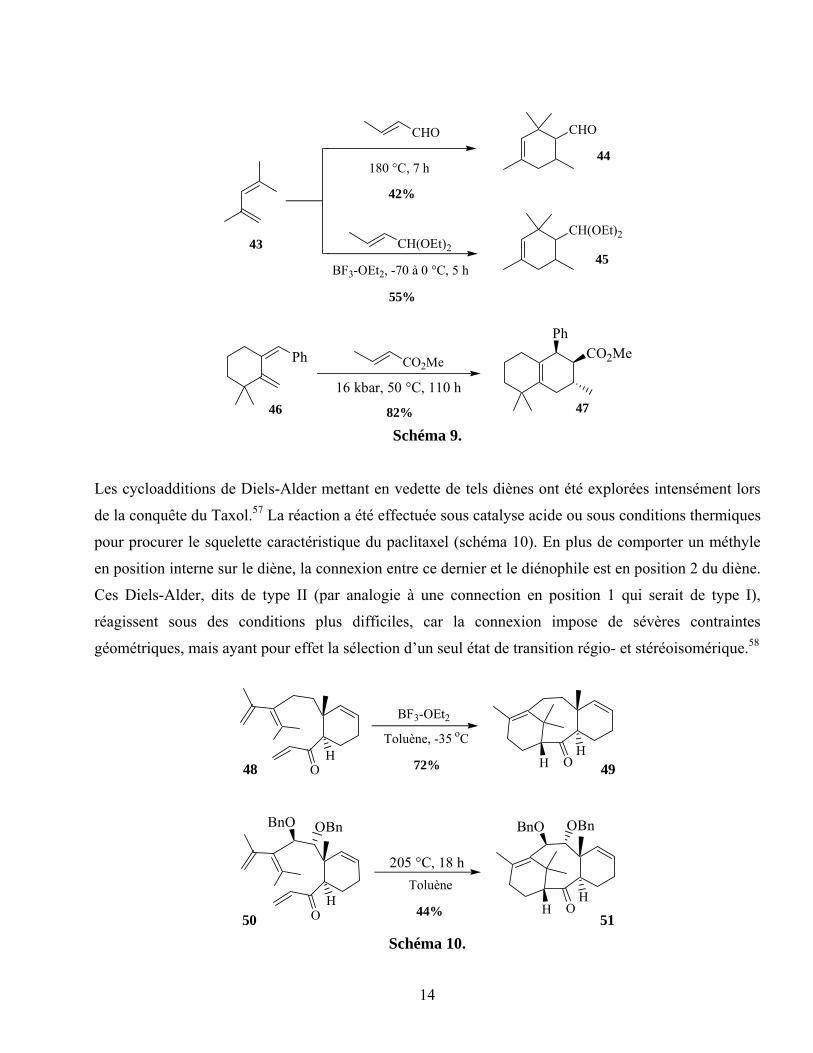
Bien que notre expérience avec cette réaction soit limitée, la littérature comporte plusieurs exemples de
cycloaddition de Diels-Alder avec un diène 1,1-disubstitué. Étudiée depuis les débuts de la découverte
de Diels et Alder, cette variante de la cycloaddition [4+2] requiert des températures très élevées (>150
°C) lorsque effectuée de façon intermoléculaire avec des partenaires peu activés.54 L’activation du
diénophile avec un acide de Lewis55 ou l’emploi de hautes pressions ont permis une réduction de la
température de réaction (schéma 9).56
14
43
Ph
BF3-OEt2, -70 à 0 °C, 5 h
CO2Me
CH(OEt)2
CHO
46
CHO
CH(OEt)2
PhCO2Me
47
45
44180 °C, 7 h
55%
42%
16 kbar, 50 °C, 110 h
82% Schéma 9.
Les cycloadditions de Diels-Alder mettant en vedette de tels diènes ont été explorées intensément lors
de la conquête du Taxol.57 La réaction a été effectuée sous catalyse acide ou sous conditions thermiques
pour procurer le squelette caractéristique du paclitaxel (schéma 10). En plus de comporter un méthyle
en position interne sur le diène, la connexion entre ce dernier et le diénophile est en position 2 du diène.
Ces Diels-Alder, dits de type II (par analogie à une connection en position 1 qui serait de type I),
réagissent sous des conditions plus difficiles, car la connexion impose de sévères contraintes
géométriques, mais ayant pour effet la sélection d’un seul état de transition régio- et stéréoisomérique.58
OH
BF3-OEt2
72%
44%OH
OBnBnO
48
50
OHH
OHH
OBnBnO
51
49
Toluène, -35 oC
Toluène
205 °C, 18 h
Schéma 10.

15
Fallis a aussi démontré que la présence d’éléments permettant de figer la conformation du connecteur
peut avoir une grande influence sur la cycloaddition (schéma 11). En effet, lorsque 52 a été traité avec
le periodinane de Dess-Martin à 48 °C, le produit CDA 53 a été directement obtenu.59 Des études ont
par la suite prouvé que le diol cis protégé favorise davantage le Diels-Alder que son homologue trans
en restreignant les degrés de liberté de la chaîne.60
OH
O
O
OMOMMOMO
52
65%
MOMO
O
OO
H
OMOM53
DMP, 48 oC
Schéma 11.
I.2.3. Diels-Alder intramoléculaire avec un vinylallène
Lors d’études préliminaires, l’alcène exocyclique 34 (c.f. schéma 7) s’est avéré impossible à construire
via diverses méthodologies (i.e. addition de cuprates, réarrangements de Claisen, etc.) probablement dû
à la forte interaction stérique existant dans ce composé. Ce problème a été contourné en tirant profit
d’une réaction qui possède un état de transition tôt, dans lequel les interactions sont minimales alors
que la formation des liens C-C est avancée: la cycloaddition de Diels-Alder d’un vinylallène.61
Toutefois, la CDA intermoléculaire d’un vinylallène tel que 54 procure l’alcène exocyclique (55) avec
la mauvaise géométrie, car le diénophile s’approche sur la face la moins encombrée (schéma 12).62
COGP
R
Z54
Z
ROGP 55
CDA endo
Schéma 12.

16
L’adduit possédant la géométrie désirée de cet alcène exocyclique (i.e. 34) peut être obtenu par une
CDA intramoléculaire avec le diénophile attaché à la chaîne latérale (R dans 54). La cycloaddition
intramoléculaire de vinylallène a été utilisée à maintes reprises lors d’études et de synthèses. Notons par
exemple les travaux de Reich pour la formation de sesquiterpènes via la CDA d’un vinylallène (schéma
13).63
I
PhO2S
O
OSiMe2Ph
CEt2AlCl
56 57
H
O
OSiMe2Ph
58
3 étapes
51%
Schéma 13.
Les segments vinylallène et diénophile de 57 ont été formés en utilisant le réarrangement de Brook à
partir de la sulfone 56. Par la suite, la cycloaddition [4+2] sous la catalyse d’un acide de Lewis a
procuré la décaline 58 qui a pu être transformée en une variété de sesquiterpènes. Il y a quelques
exemples de CDA intramoléculaire de vinylallènes rapportés dans la littérature.64 Notre approche
synthétique aux quassinoïdes impliquera une CDA intramoléculaire entre un vinylallène et un
diénophile attachés ensemble par un connecteur temporaire.

17
I.3 Connecteurs temporaires
Le concept de connecteur implique une unité d’attache entre deux ou plusieurs partenaires d’une
réaction afin de les rapprocher. La réaction initialement intermoléculaire devient alors intramoléculaire,
le terme entropique de la réaction diminue considérablement et la réaction se voit facilitée. L’usage
d’un connecteur temporaire permet aussi un contrôle de la régiochimie et potentiellement de la
stéréochimie. De nombreux connecteurs temporaires ont été développés pour une multitude de
réactions. L’usage de chacun dépend de la réaction à effectuer, des fonctionnalités présentes dans le
produit de départ et de celles désirées dans le produit final.65 Notons par exemple l’usage de tels
connecteurs pour des photocycloadditions,66 des cycloadditions [2+2],67 des cycloadditions 1,3-
dipolaires,68 des métathèses d’alcènes,69 des réactions de couplages70 et des cyclisations radicalaires.71
Puisque la cycloaddition de Diels-Alder est la réaction d’intérêt ici, seuls les connecteurs utilisés à cette
fin seront discutés.72
I.3.1. Un pont hydrogène comme connecteur
Le pont hydrogène a été impliqué à quelques reprises en tant qu’élément de rapprochement des
partenaires lors de cycloaddition [4+2].73 Il s’agit du connecteur ‘idéal’ puisqu’il ne nécessite aucune
réaction pour le mettre en place ou l’enlever. Cependant, ce lien étant assez faible, son utilisation est
restreinte à des cas particulier. Un groupe de Merck a rapporté en 2001 une synthèse de la (±)-
thielocine A1-β dont l’étape clé est une CDA durant laquelle le diénophile et le diène sont liés par un
pont hydrogène (schéma 14).74
CO2Et
TBSO
N
OMe
59 CO2Et
OHO
HO
60
O
CO2Et
OMe
OEtO2C
OHOH
61
1) CF3SO2OMe, CH2Cl2, 0 °C
2) TBAF, -20 °C
100%

18
Schéma 14.
La génération de la méthylène quinone de 59 permet la CDA avec 60 selon une organisation bien
définie (i.e. 62, figure 4). Les auteurs ont prouvé que la transformation est moins propre et beaucoup
moins facile lorsque le pont hydrogène est impossible (i.e. en présence du méthyle éther de 59). Ce
connecteur ne convient pas à la grande majorité des cycloadditions qui nécessitent une activation par un
acide de Lewis (ce qui empêche la formation du pont hydrogène). D’autres modes de liaisons ont été
développés afin d’assurer une bonne coordination des partenaires lors de la cycloaddition.
RO2C O
Me MeMe
Me
O CO2R
OMeMe
OH
OH
62 Figure 4. CDA via pont hydrogène.
I.3.2. Un métal comme connecteur
L’usage de métaux en tant que connecteurs temporaires a débuté en 1995, lorsque Stork a utilisé un
alcoolate de magnésium ou aluminium lors d’une CDA.75 Depuis ce jour, ce concept a été appliqué sur
d’autres systèmes avec l’aluminium et le zinc comme connecteurs.76 Indépendamment du métal
employé, ces études soulignent toutes les conditions réactionnelles peu clémentes nécessaires pour
effectuer la cycloaddition (schéma 15).
HO63
O M O
Ph
X
HO Ph64
65
HO
Ph
OH
HO
Ph
OH
66
67
+R-métal i) 160 °C, 60 h
ii) H2O
AlMe3 : 70%, 4:1 (66/67)
ZnMe2 : 37%, 3:2 (66/67)
+

19
Schéma 15.
La haute température nécessaire pour effectuer la transformation peut être attribuée à l’enrichissement
électronique des partenaires par la nature anionique des oxygènes reliés au connecteur. Il en résulte une
augmentation des niveaux énergétiques (HOMO-LUMO), ce qui a pour effet de rendre la réaction plus
difficile (cet effet est conséquent d’une augmentation inégale des niveaux énergétiques du diène et du
diénophile). Louis Barriault a toutefois démontré que le connecteur magnésium, installé selon un
protocole plus doux (MgBr2, Et3N), permettait d’effectuer la réaction dans des conditions plus
clémentes.77 Ces éléments d’attache pour la cycloaddition de Diels-Alder ne peuvent pas être appliqué
aux substrats sensibles aux conditions basiques, c'est-à-dire aux diènes ou diénophiles activés par un
carbonyle ou autre groupement similaire. L’usage de connecteurs de nature covalente est alors de mise.
Notez que nous incluons dans la prochaine section les connecteurs faits d’un atome de bore ou de
silicium, bien que ce soient des métaux.
I.3.2. Les connecteurs à liaisons covalentes
Pour sa capacité à former des liaisons stables avec l’oxygène, le bore a été utilisé lors de cycloaddition
[4+2] de diverses façons. Par exemple, Nicolaou a utilisé la méthodologie de Narasaka78 pour la
synthèse d’un intermédiaire dans sa synthèse du Taxol (schéma 16).79
OH
OEt
O
O
OOH
PhB(OH)2
6968
O
OH
OH
O
EtO
O
72
70
O O
H OHHO
EtO2C
71
O B PhO
EtO2CO
O+
79%
toluène, reflux
Schéma 16.

20
La réaction a procédé sous le reflux du toluène pour procurer, suite à l’hydrolyse de l’ester boronique et
au réarrangement, l’adduit bicyclique 72 dans un bon rendement. Ce mode de connexion du bore
implique la présence de groupements hydroxyles sur le diène et le diénophile (R-O-B-O-R’). Batey a
toutefois démontré que le connecteur peut être utilisé avec les liens R-C-B-O-R’ (schéma 17).80 La
formation de l’ester boronique à partir de l’alcool 74 et de l’acide vinylboronique 73, suivie de la
cycloaddition, permet d’obtenir les diols 75 et 76 par une oxydation du lien carbone-bore.
73 R B(OH)2
74
OH
75
R OHOH
76
R OHOH
+i) Tamis moléculaire, 23 °Cii) 80 °C, 24 hiii) Oxydation
+
R = alkyles, aryles65-84%, 2 à 9:1
Schéma 17.
L’unité carboxyle a été également démontrée comme un connecteur de choix pour des CDA, les plus
populaires étant l’ester81 et l’amide.82 Le premier usage pratique de ce type de connecteur peut être
attribué à Corey dans sa synthèse de la (±)-forksoline (schéma 18).83 La lactone 78 obtenue a été
transformée afin d’introduire le cycle C, une stratégie permettant une économie d’atomes par
l’incorporation du connecteur dans le produit final.84
OHTs CO2H
77
OTs
O
78
O
O
OAcOH
OH
OH
H
forksoline 79
CHCl3, 23 °C, 30 h
72%
Schéma 18.
Cette stratégie a ouvert la voie à un autre type de fonction carboxyle, un hydroxamate.85 Ishikawa et
Saito ont démontré la versatilité de ce connecteur, l’unité hydroxamate pouvant figurer autant sur le
diène que le diénophile (schéma 19). L’union des partenaires a été effectuée par une réaction de
Mitsunobu86 pour procurer les adduits 83 et 87 après cycloaddition. Cette méthodologie est intéressante

21
du fait que dépendamment du mode d’attachement (si le diène ou le diénophile est relié à la fonction
carbonylée, i.e. 82 vs. 86), il est possible d’obtenir la jonction bicyclique trans ou cis sélectivement.
N
OBn
OH 80
CO2MeHO
N
OBn
OH
HO
81
84
85
N
OBn
O
86
N
OBn
O CO2Me
82
87
N
OBn
OH
83
NO
OBn
CO2MeH
HMitsunobu
Mitsunobu+
+ toluène80%
90 °C, 17 h
toluène80%
90 °C, 17 h
Schéma 19.
De son côté, Craig a utilisé un connecteur covalent simple, ne faisant pas usage de groupe carboxylique.
L’unité d’attache développée mettait à profit un éther pour relier les partenaires de la cycloaddition
(schéma 20).87 Alors que l’éther 89 a été préparé par attaque de l’allylsilane sur l’oxonium généré à
partir de 88, la CDA procurant l’éther bicyclique 90 ne s’est produite qu’à haute température. Une
importante limitation de ce connecteur réside dans la difficulté à introduire des diènes fonctionnalisés,
les allylsilanes correspondant étant difficiles à obtenir.88
O
O
CO2Me
CO2Me
TMS
88
O CO2Me
89
OH
CO2Me
H9060% 80%
TMSOTf, CH2Cl2
170 °C, 8 htoluène
Schéma 20.
L’homologue de l’oxygène, le souffre, a été utilisé plus couramment en tant que connecteur dans des
cycloaddition de Diels-Alder. Principalement développé par Metz, le vinylsulfonate a fait l’objet de
nombreuses études (schéma 21).89 Pouvant être synthétisé par diverses méthodes, le chlorure de
vinylsulfonyle (92) a été couplé à l’alcool 91 et la CDA de l’adduit résultant a procuré le sulfonate 94
dans un bon rendement. Metz a ensuite démontré que le connecteur pouvait soit être éliminé, soit oxydé
en une seule étape pour donner accès à des céto-alcools polycycliques tel que 95. Ce principe a aussi

22
été appliqué aux sulfonamides, celles-ci offrant des rendements similaires de couplage et de
cycloaddition.90
SO
OH
SO2Cl
OO2S
OO
Et3N, THF91 93 94
OHO
9576%
toluène, 110 °Ci) nBuLiii) B(OR)3iii) mCPBA
58%86%
92
Schéma 21.
Overman a rapporté l’usage d’un sulfonamide comme connecteur de CDA pour sa synthèse de la (+)-
alopérine.91 Ce connecteur, lui causant de nombreux problèmes reliés à la piètre sélectivité et à la
présence de produits secondaires, a été remplacé par une unité d’attache à base de silicium (schéma 22).
Quoique sensibles, les intermédiaires obtenus ont pu être transformés in situ permettant ainsi au groupe
d’Overman de compléter leur synthèse de l’alopérine.
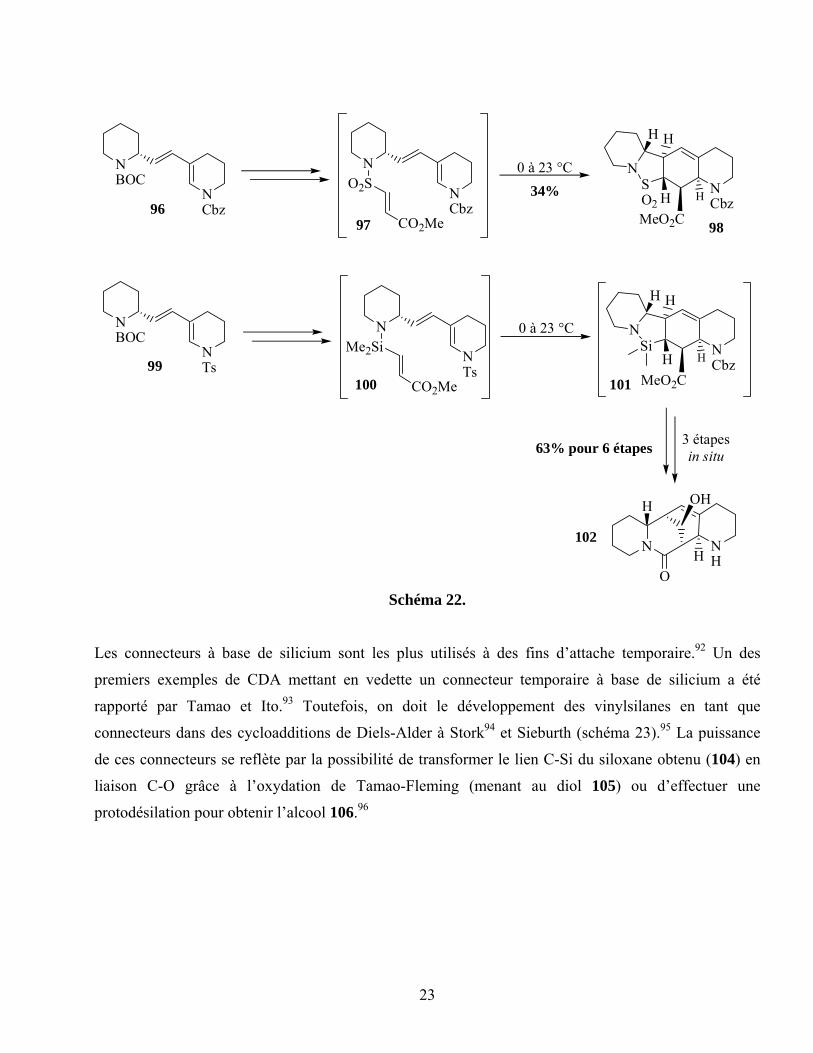
23
NBOC
NCbz
N
NCbz
O2S
CO2Me
NBOC
NTs
N
NTs
Me2Si
CO2Me10099
9796
102
101
NCbz
NSO2MeO2C
H
HH
H
NCbz
NSi
MeO2CH
HH
H
N
H
O
NHH
OH
98
34%
63% pour 6 étapes 3 étapesin situ
0 à 23 °C
0 à 23 °C
Schéma 22.
Les connecteurs à base de silicium sont les plus utilisés à des fins d’attache temporaire.92 Un des
premiers exemples de CDA mettant en vedette un connecteur temporaire à base de silicium a été
rapporté par Tamao et Ito.93 Toutefois, on doit le développement des vinylsilanes en tant que
connecteurs dans des cycloadditions de Diels-Alder à Stork94 et Sieburth (schéma 23).95 La puissance
de ces connecteurs se reflète par la possibilité de transformer le lien C-Si du siloxane obtenu (104) en
liaison C-O grâce à l’oxydation de Tamao-Fleming (menant au diol 105) ou d’effectuer une
protodésilation pour obtenir l’alcool 106.96
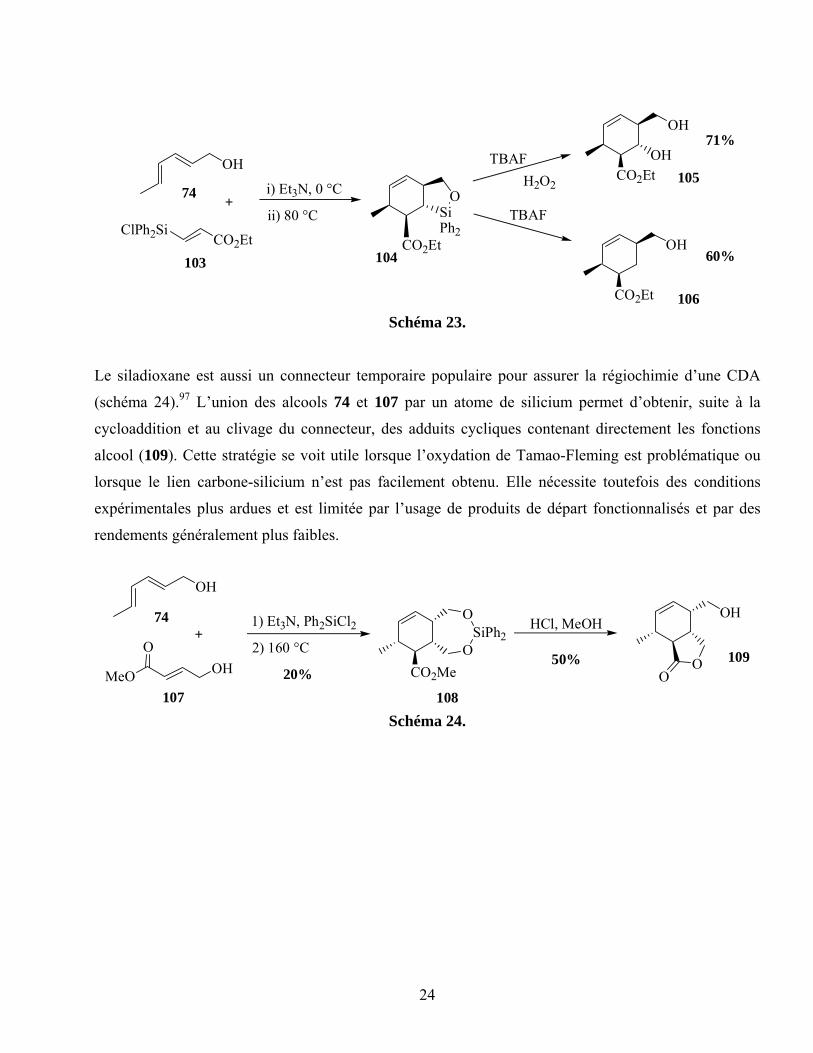
24
OH TBAF
74
ClPh2SiCO2Et
SiPh2
O
CO2Et
TBAF
H2O2
103 104OH
CO2Et
OH
OHCO2Et
106
105
+i) Et3N, 0 °C
ii) 80 °C
71%
60%
Schéma 23.
Le siladioxane est aussi un connecteur temporaire populaire pour assurer la régiochimie d’une CDA
(schéma 24).97 L’union des alcools 74 et 107 par un atome de silicium permet d’obtenir, suite à la
cycloaddition et au clivage du connecteur, des adduits cycliques contenant directement les fonctions
alcool (109). Cette stratégie se voit utile lorsque l’oxydation de Tamao-Fleming est problématique ou
lorsque le lien carbone-silicium n’est pas facilement obtenu. Elle nécessite toutefois des conditions
expérimentales plus ardues et est limitée par l’usage de produits de départ fonctionnalisés et par des
rendements généralement plus faibles.
74
OH
107MeO OH
O
CO2MeO
SiPh2
O
108
O
OH
O109
+1) Et3N, Ph2SiCl22) 160 °C
20%
HCl, MeOH
50%
Schéma 24.

25
CHAPITRE 1 : DÉVELOPPEMENT D’UN CONNECTEUR POUR LA PREMIÈRE
CYCLOADDITION
1.1. Un connecteur vinylsiloxane
Ce projet a débuté avec la continuation des travaux de maîtrise de Stéphane Gingras. Une séquence
réactionnelle de 8 étapes menant au vinylallène simple 111 avait alors été développée et l’utilité du
connecteur de Sieburth95 dans notre contexte avait alors été démontré par M. Gingras (schéma 25).30a
PMBOO
110 111
C
OH
OPMB
5 étapes
80%
3 étapes
70%
Br
Ph3SiCO2Et
112113
OSi
OPMB
Ph Ph CO2Et
i) TfOH, CH2Cl2ii) pyridineiii) 111
53% Schéma 25.
Avec le composé 113 en mains, notre première tâche a été d’optimiser le clivage oxydatif du
connecteur siloxane (tableau 1). Après un criblage exhaustif des conditions connues dans la littérature,
le diol 114 a été obtenu avec un faible rendement de 40% et ce de façon non reproductible. L’adduit
113 s’est avéré instable sous les conditions d’oxydation. Nous croyons, cependant, que le groupement
protecteur PMB et l’absence d’une plus grande substitution en C1 (c.f. schéma du tableau 1) sont
partiellement responsables de son instabilité (vide infra).
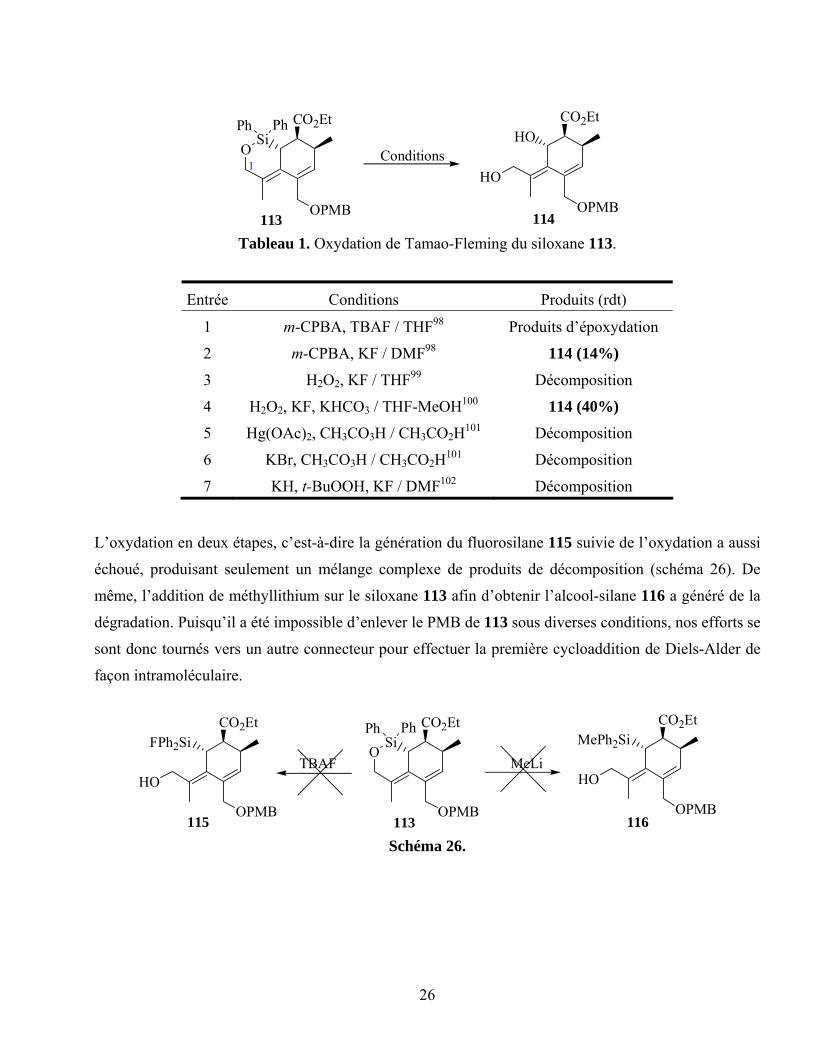
26
OSi
OPMB
Ph Ph CO2Et
113 114OPMB
CO2EtHO
HOConditions
1
Tableau 1. Oxydation de Tamao-Fleming du siloxane 113.
Entrée Conditions Produits (rdt)
1 m-CPBA, TBAF / THF98 Produits d’époxydation
2 m-CPBA, KF / DMF98 114 (14%)
3 H2O2, KF / THF99 Décomposition
4 H2O2, KF, KHCO3 / THF-MeOH100 114 (40%)
5 Hg(OAc)2, CH3CO3H / CH3CO2H101 Décomposition
6 KBr, CH3CO3H / CH3CO2H101 Décomposition
7 KH, t-BuOOH, KF / DMF102 Décomposition
L’oxydation en deux étapes, c’est-à-dire la génération du fluorosilane 115 suivie de l’oxydation a aussi
échoué, produisant seulement un mélange complexe de produits de décomposition (schéma 26). De
même, l’addition de méthyllithium sur le siloxane 113 afin d’obtenir l’alcool-silane 116 a généré de la
dégradation. Puisqu’il a été impossible d’enlever le PMB de 113 sous diverses conditions, nos efforts se
sont donc tournés vers un autre connecteur pour effectuer la première cycloaddition de Diels-Alder de
façon intramoléculaire.
FPh2Si
OPMB
CO2Et
HOTBAF
OSi
OPMB
Ph Ph CO2Et
MeLi
113115 116
MePh2Si
OPMB
CO2Et
HO
Schéma 26.

27
1.2. Un connecteur sulfonate
Les études de Metz ont démontré l’utilité d’un sulfonate en tant que connecteur lors de cycloaddition de
Diels-Alder.89 La génération du chlorure de vinylsulfonyle (92) a été effectuée selon la méthode de
Rondestvedt.103 Suite à une expérimentation exhaustive, la réaction de ce chlorure de sulfonyle avec
l’allène 111 a procuré le produit de cycloaddition 116 dans un rendement optimisé de 35% (schéma 27).
La 2,6-lutidine s’est révélée être la meilleure base pour effectuer la transformation, les amines peu
encombrées favorisant la décomposition de 92. Toutes les tentatives pour ralentir ou accélérer la
réaction en modifiant la température ont généré davantage de dégradation en diminuant
considérablement le rendement de 116.
C
OH
OPMB
SO2Cl 92
111
DMPCH2Cl2
118
OS
O
O O
116
OS
OPMB
O O
OEt
CH2Cl2- H2ODDQ
117
OS
OH
O O
2,6-lutidine
CH2Cl2, 23 °C
35%
Yb(fod)3Décomposition
60%
90%
Schéma 27.
28
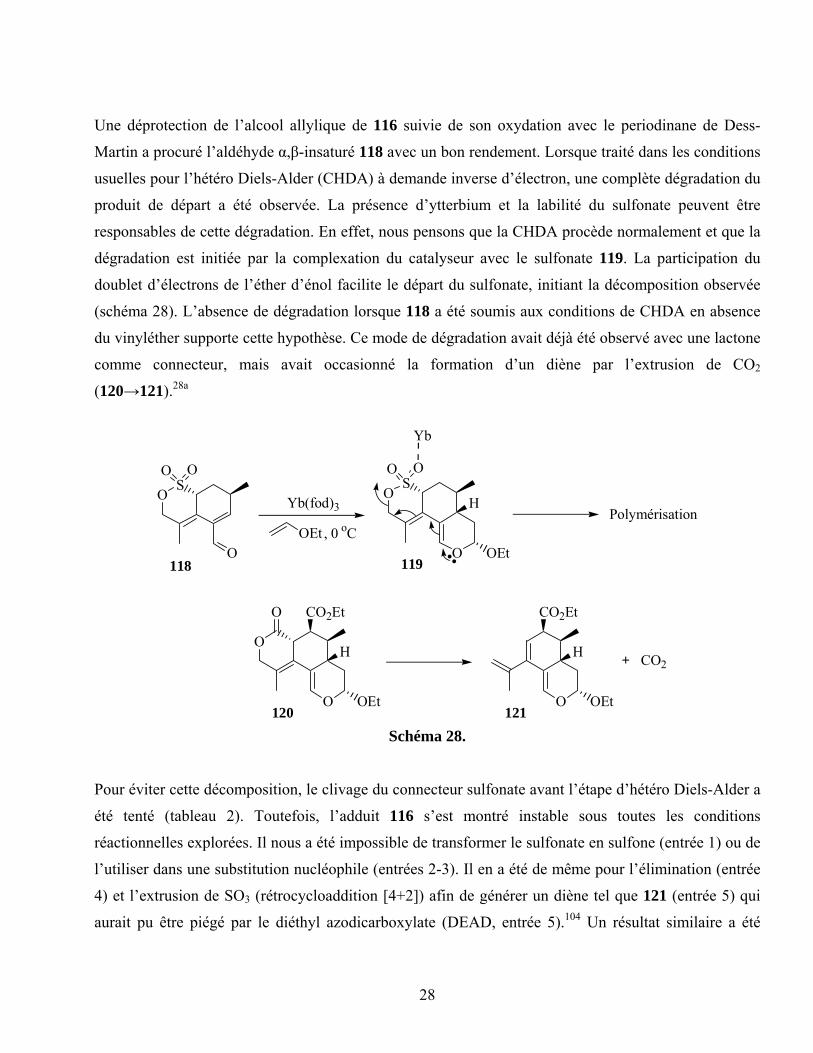
Une déprotection de l’alcool allylique de 116 suivie de son oxydation avec le periodinane de Dess-
Martin a procuré l’aldéhyde α,β-insaturé 118 avec un bon rendement. Lorsque traité dans les conditions
usuelles pour l’hétéro Diels-Alder (CHDA) à demande inverse d’électron, une complète dégradation du
produit de départ a été observée. La présence d’ytterbium et la labilité du sulfonate peuvent être
responsables de cette dégradation. En effet, nous pensons que la CHDA procède normalement et que la
dégradation est initiée par la complexation du catalyseur avec le sulfonate 119. La participation du
doublet d’électrons de l’éther d’énol facilite le départ du sulfonate, initiant la décomposition observée
(schéma 28). L’absence de dégradation lorsque 118 a été soumis aux conditions de CHDA en absence
du vinyléther supporte cette hypothèse. Ce mode de dégradation avait déjà été observé avec une lactone
comme connecteur, mais avait occasionné la formation d’un diène par l’extrusion de CO2
(120→121).28a
OS
O
O O
OEt
118
120
O
O
H
OEt
O CO2Et
119
OS
O
O O
H
OEt
Yb
121O
H
OEt
CO2Et
Yb(fod)3 Polymérisation
. .
, 0 oC
+ CO2
Schéma 28.
Pour éviter cette décomposition, le clivage du connecteur sulfonate avant l’étape d’hétéro Diels-Alder a
été tenté (tableau 2). Toutefois, l’adduit 116 s’est montré instable sous toutes les conditions
réactionnelles explorées. Il nous a été impossible de transformer le sulfonate en sulfone (entrée 1) ou de
l’utiliser dans une substitution nucléophile (entrées 2-3). Il en a été de même pour l’élimination (entrée
4) et l’extrusion de SO3 (rétrocycloaddition [4+2]) afin de générer un diène tel que 121 (entrée 5) qui
aurait pu être piégé par le diéthyl azodicarboxylate (DEAD, entrée 5).104 Un résultat similaire a été

29
obtenu sous les conditions d’oxydation développée par Metz (entrée 7).89 Ces résultats marquèrent nos
dernières tentatives avec le sulfonate comme connecteur.
Tableau 2. Clivage du sulfonate 116.
Entrée Conditions Produits
1 PhLi / THF -78 oC Décomposition
2 Diméthylmalonate / NaH Décomposition
3 Diméthylmalonate / CsCO3 Décomposition + 116
4 t-BuOK / THF Décomposition
5 120 oC / xylène Décomposition
6 DEAD, 120 oC / xylène Décomposition
7 n-BuLi, B(OMe)3 / THF Décomposition
1.3. Un connecteur éther d’énol
1.3.1. Synthèse de vinyléthers
Plusieurs stratégies ont été développées pour générer des éthers d’énol à partir d’alcools
fonctionnalisés.105 Toutefois, peu de méthodes ont l’avantage de tolérer des groupes fonctionnels
complexes. Parmi elles, l’échange du groupement alkoxy des éthers d’énol catalysé par des sels de
mercure106 et l’addition de Michael sur des alcynes activées demeurent les méthodes les plus versatiles
et les plus douces (schéma 29).107
OR R'OH Hg2+R'OH BaseZOR' Z
R'OROH+ +
Échange de vinyle
+
Addition de Michael
Schéma 29.
30
1.3.2. Formation de l’éther d’énol et cycloaddition
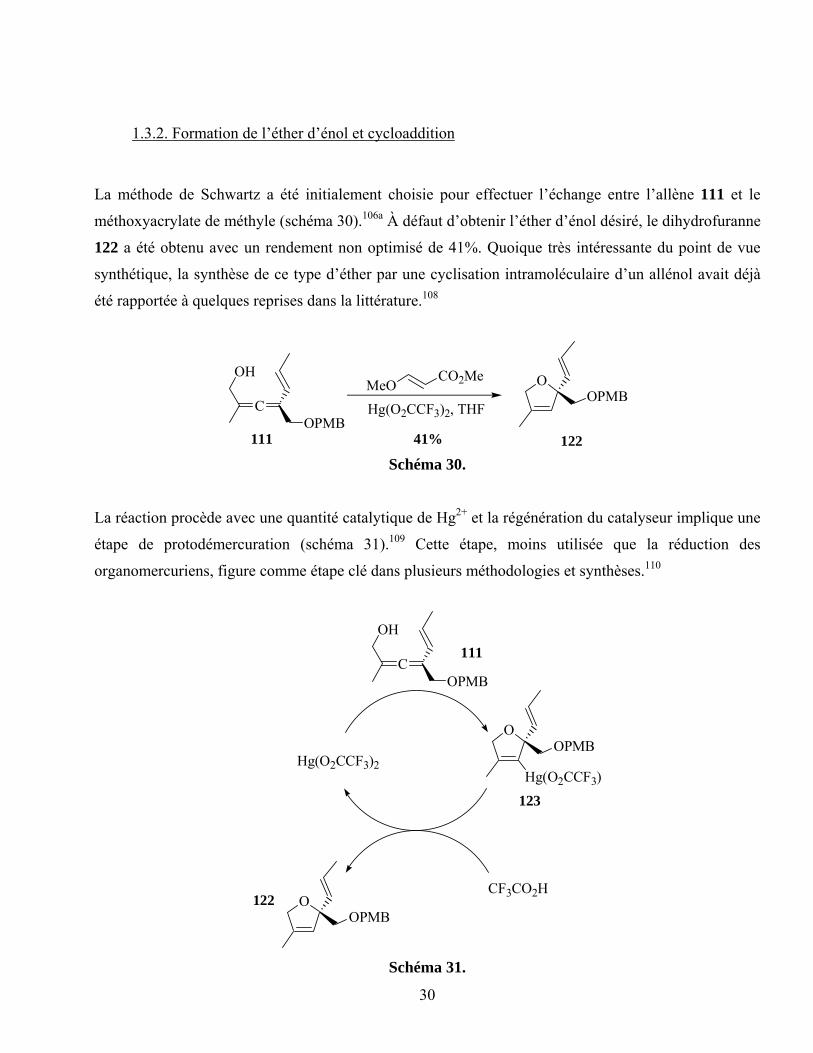
La méthode de Schwartz a été initialement choisie pour effectuer l’échange entre l’allène 111 et le
méthoxyacrylate de méthyle (schéma 30).106a À défaut d’obtenir l’éther d’énol désiré, le dihydrofuranne
122 a été obtenu avec un rendement non optimisé de 41%. Quoique très intéressante du point de vue
synthétique, la synthèse de ce type d’éther par une cyclisation intramoléculaire d’un allénol avait déjà
été rapportée à quelques reprises dans la littérature.108
C
OH
OPMB
MeOCO2Me
Hg(O2CCF3)2, THF
111 41% 122
OOPMB
Schéma 30.
La réaction procède avec une quantité catalytique de Hg2+ et la régénération du catalyseur implique une
étape de protodémercuration (schéma 31).109 Cette étape, moins utilisée que la réduction des
organomercuriens, figure comme étape clé dans plusieurs méthodologies et synthèses.110
C
OH
OPMB
111
122 OOPMB
Hg(O2CCF3)2
123
OOPMB
Hg(O2CCF3)
CF3CO2H
Schéma 31.

31
La formation du dihydrofuranne 122 a pu être évitée en changeant la méthode de formation de l’éther
vinylique. Dans ce sens, l’addition de Michael de l’allénol 111 sur le propiolate de méthyle a conduit
directement à l’isolation de l’adduit de cycloaddition 124 dans un rendement de 53% (schéma 32).
Toutefois, la déprotection de l’éther allylique 124 n’a procédé qu’avec un faible rendement, procurant
seulement 28% de l’alcool 125 désiré.
C
OH
OPMB
CO2Me
Et2O, reflux
111 53% 124
O
OPMB
CO2Me
DDQ
28% 125
O
OH
CO2Me
, NMMCH2Cl2 - H2O
Schéma 32.
Face aux faibles rendements de cycloaddition, à l’instabilité des intermédiaires et à l’impossibilité de
cliver l’éther de PMB avec un rendement acceptable, l’emploi d’un nouveau groupe protecteur pour
l’alcool allylique s’avérait la meilleure solution. Peut-être pourrions-nous alors réussir la séquence
synthétique avec un des connecteurs discutés ci-haut? Le 1,3-dithiane a été choisi à cette fin pour des
raisons de tolérance aux conditions expérimentales, mais aussi parce qu’il possède le même état
d’oxydation que l’aldéhyde désiré à cette position. Une nouvelle voie synthétique a donc été mise sur
pied afin d’incorporer ce nouveau groupe protecteur rapidement.
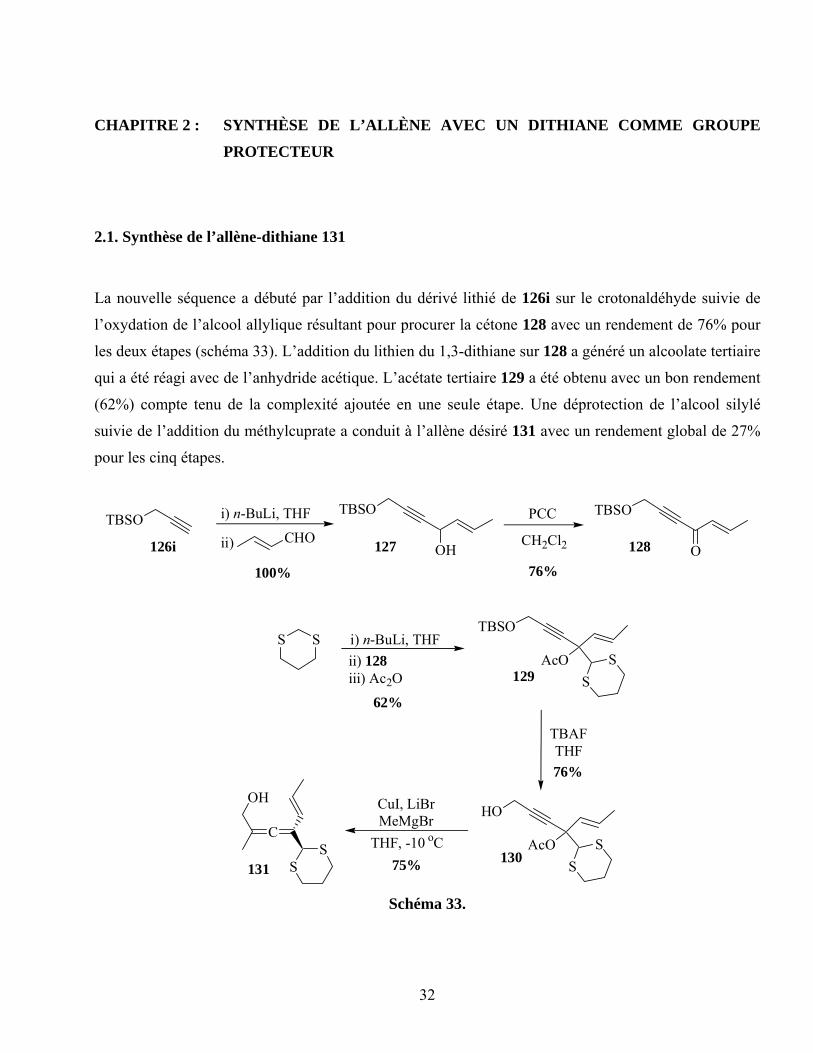
32
CHAPITRE 2 : SYNTHÈSE DE L’ALLÈNE AVEC UN DITHIANE COMME GROUPE
PROTECTEUR
2.1. Synthèse de l’allène-dithiane 131
La nouvelle séquence a débuté par l’addition du dérivé lithié de 126i sur le crotonaldéhyde suivie de
l’oxydation de l’alcool allylique résultant pour procurer la cétone 128 avec un rendement de 76% pour
les deux étapes (schéma 33). L’addition du lithien du 1,3-dithiane sur 128 a généré un alcoolate tertiaire
qui a été réagi avec de l’anhydride acétique. L’acétate tertiaire 129 a été obtenu avec un bon rendement
(62%) compte tenu de la complexité ajoutée en une seule étape. Une déprotection de l’alcool silylé
suivie de l’addition du méthylcuprate a conduit à l’allène désiré 131 avec un rendement global de 27%
pour les cinq étapes.
TBSO
S S
126i CHO
129
127
AcO
TBSO
SS
OH
TBSO
131
C
OH
SS
TBAFTHF
PCC
CH2Cl2
130
128
AcO
HO
SS
O
TBSOi) n-BuLi, THF
ii)
i) n-BuLi, THFii) 128iii) Ac2O
CuI, LiBrMeMgBr
THF, -10 oC
100% 76%
62%
76%
75%
Schéma 33.

33
Lorsque placé dans les conditions d’addition de Michael sur le propiolate de méthyle, l’adduit de Diels-
Alder 132 a été obtenu à 91%, venant renforcer l’hypothèse que le groupement PMB contribuait à
l’instabilité des composés analogues (schéma 34). Le traitement du dithiane avec l’oxyde de mercure a
permis de démasquer l’aldéhyde 133 dans un rendement de 57%. Toutefois, lorsque placé en présence
du catalyseur d’ytterbium, seulement une décomposition du produit de départ a eu lieu.
C
OH
SS
CO2Me
131
NMM, Et2O
OEt
132
OCO2Me
S S
HgOBF3-OEt2
THF
133
OCO2Me
O91% 57%
Yb(fod)3
Décomposition Schéma 34.
Nous croyons que le mécanisme de décomposition est identique à celui décrit au schéma 28.28a Ce
résultat suggère que l’hétéro Diels-Alder est incompatible avec la présence d’un hétéroatome en C1.
Afin d’éviter cette décomposition, nous avons tenté de cliver l’éther cyclique avant d’exécuter la
seconde cycloaddition.
Le clivage d’éthers cycliques et acycliques a été un champ de recherche populaire si on en juge par les
nombreuses méthodes existant pour effectuer cette transformation.111 Selon la méthode, il est possible
d’obtenir une substitution soit par un groupe alkoxy, soit par un halogène.112 Lorsqu’appliquées à notre
problème, aucune de ces méthodes ne nous a permis d’obtenir le produit de clivage du dihydrofuranne
(tableau 3). Il est possible que le dithiane et l’ester de 132 soient incompatibles avec les conditions
requises pour effectuer la transformation désirée. Une solution pour éviter ces problèmes devait être
envisagée.

34
CO2MeO
S S132 134
CO2Me
S S
X
YConditions
Tableau 3. Clivage du sulfonate 132.
Entrée Conditions Produits
1 FeCl3, Ac2O / Acétate d’éthyle113 Décomposition
2 BF3-OEt2, Ac2O114 Décomposition
3 TMSCl, NaI / CH3CN115 Décomposition + 132
4 Me2BBr, Et3N / CH2Cl2116 Décomposition
2.2. Éther homo allylique
Si le mécanisme de décomposition décrit au schéma 28 est valide, il était possible de croire qu’un éther
homo allylique ne souffrirait pas du même problème. En plus de nous permettre d’exécuter la deuxième
cycloaddition, le clivage régiosélectif de l’éther 137 pouvait nous permettre d’introduire la chaîne
latérale portant le diénophile pour une troisième cycloaddition intramoléculaire (schéma 35).117
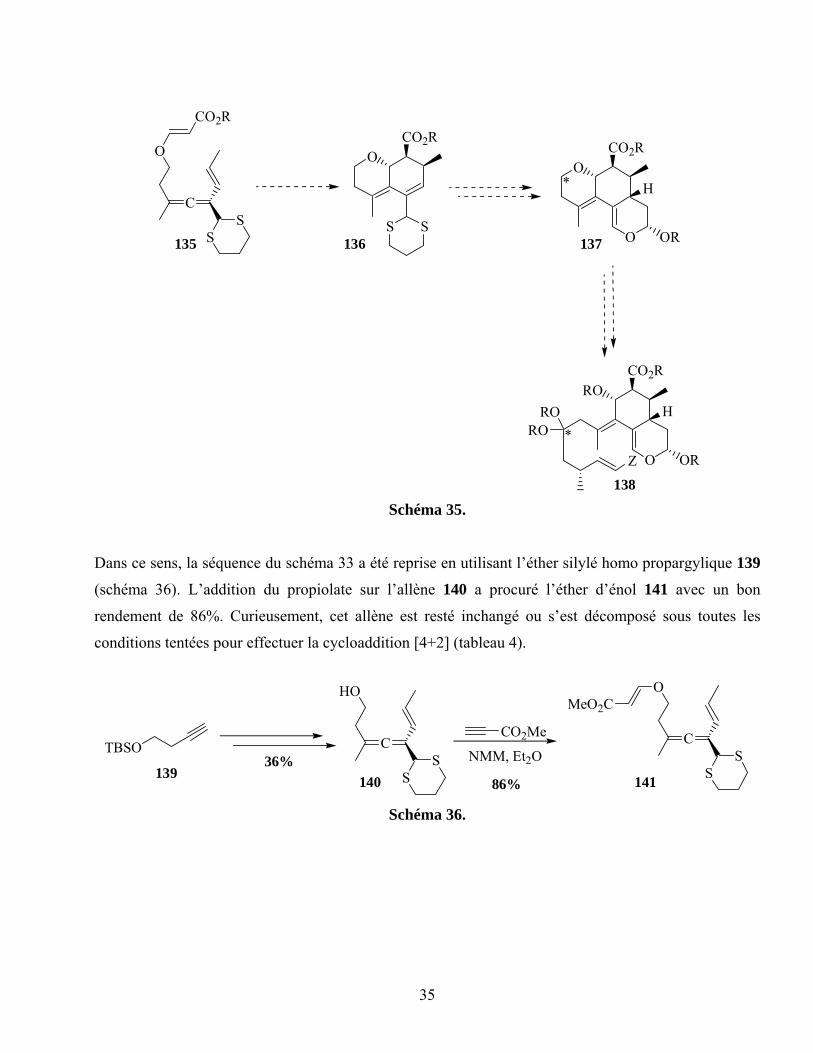
35
C
O
CO2R
SS
CO2RO
S S136
CO2RRO
O
H
ORZ
RORO
135
138
137
CO2RO
O
H
OR
*
*
Schéma 35.
Dans ce sens, la séquence du schéma 33 a été reprise en utilisant l’éther silylé homo propargylique 139
(schéma 36). L’addition du propiolate sur l’allène 140 a procuré l’éther d’énol 141 avec un bon
rendement de 86%. Curieusement, cet allène est resté inchangé ou s’est décomposé sous toutes les
conditions tentées pour effectuer la cycloaddition [4+2] (tableau 4).
TBSO C
HO
SS
CO2Me
139 140 141
C
O
SS
MeO2C
NMM, Et2O
86%36%
Schéma 36.
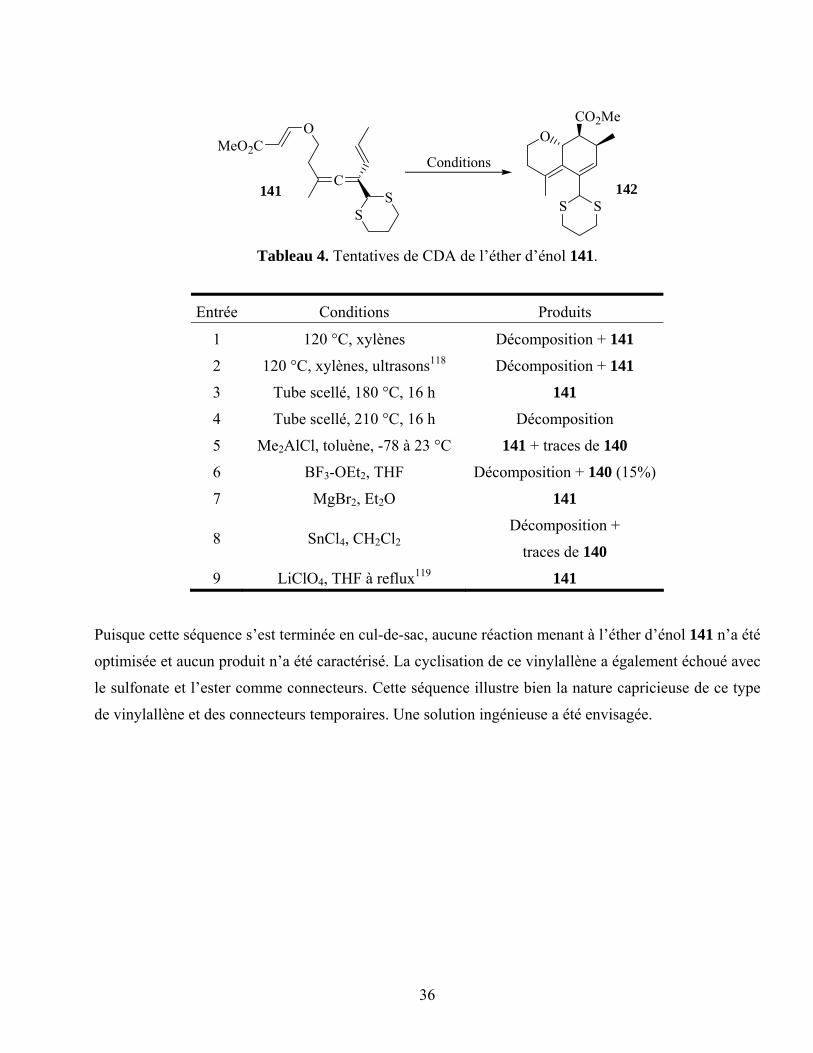
36
C
O
SS
MeO2C
141
O
S S
CO2Me
142
Conditions
Tableau 4. Tentatives de CDA de l’éther d’énol 141.
Entrée Conditions Produits
1 120 °C, xylènes Décomposition + 141
2 120 °C, xylènes, ultrasons118 Décomposition + 141
3 Tube scellé, 180 °C, 16 h 141
4 Tube scellé, 210 °C, 16 h Décomposition
5 Me2AlCl, toluène, -78 à 23 °C 141 + traces de 140
6 BF3-OEt2, THF Décomposition + 140 (15%)
7 MgBr2, Et2O 141
8 SnCl4, CH2Cl2 Décomposition +
traces de 140
9 LiClO4, THF à reflux119 141
Puisque cette séquence s’est terminée en cul-de-sac, aucune réaction menant à l’éther d’énol 141 n’a été
optimisée et aucun produit n’a été caractérisé. La cyclisation de ce vinylallène a également échoué avec
le sulfonate et l’ester comme connecteurs. Cette séquence illustre bien la nature capricieuse de ce type
de vinylallène et des connecteurs temporaires. Une solution ingénieuse a été envisagée.
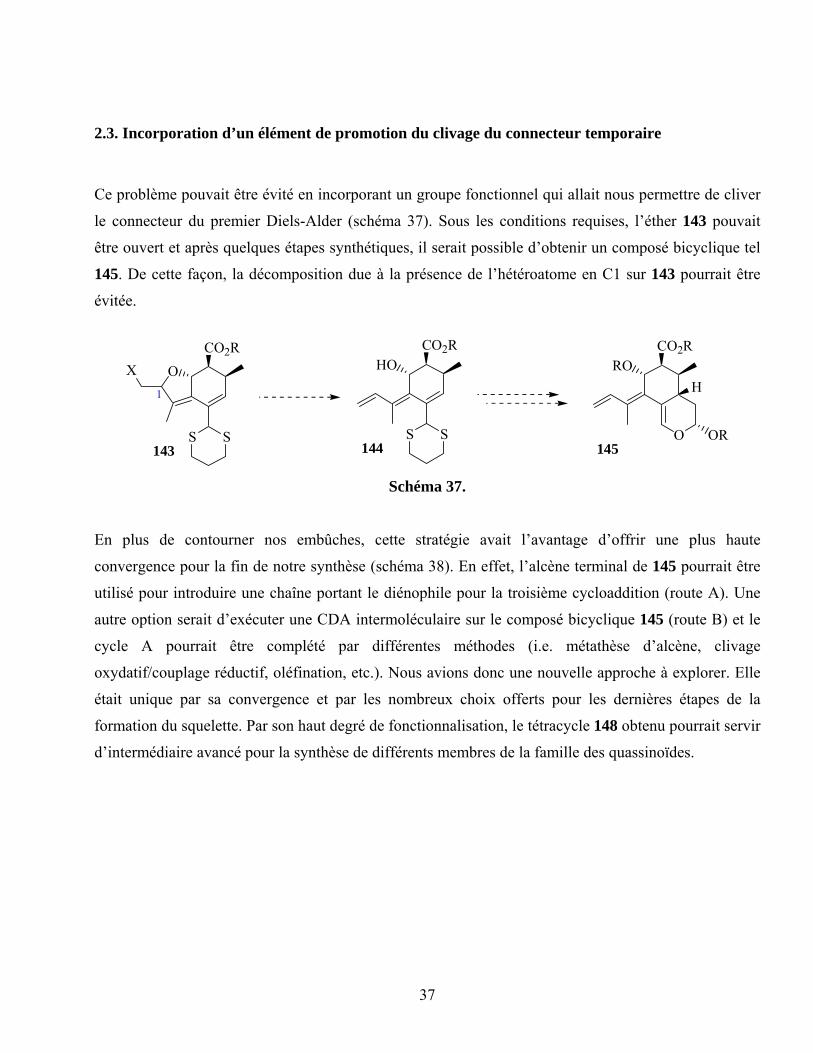
37
2.3. Incorporation d’un élément de promotion du clivage du connecteur temporaire
Ce problème pouvait être évité en incorporant un groupe fonctionnel qui allait nous permettre de cliver
le connecteur du premier Diels-Alder (schéma 37). Sous les conditions requises, l’éther 143 pouvait
être ouvert et après quelques étapes synthétiques, il serait possible d’obtenir un composé bicyclique tel
145. De cette façon, la décomposition due à la présence de l’hétéroatome en C1 sur 143 pourrait être
évitée.
CO2R
O
S S
X
CO2R
S S
HO
143 144 145
CO2RRO
O OR
H1
Schéma 37.
En plus de contourner nos embûches, cette stratégie avait l’avantage d’offrir une plus haute
convergence pour la fin de notre synthèse (schéma 38). En effet, l’alcène terminal de 145 pourrait être
utilisé pour introduire une chaîne portant le diénophile pour la troisième cycloaddition (route A). Une
autre option serait d’exécuter une CDA intermoléculaire sur le composé bicyclique 145 (route B) et le
cycle A pourrait être complété par différentes méthodes (i.e. métathèse d’alcène, clivage
oxydatif/couplage réductif, oléfination, etc.). Nous avions donc une nouvelle approche à explorer. Elle
était unique par sa convergence et par les nombreux choix offerts pour les dernières étapes de la
formation du squelette. Par son haut degré de fonctionnalisation, le tétracycle 148 obtenu pourrait servir
d’intermédiaire avancé pour la synthèse de différents membres de la famille des quassinoïdes.
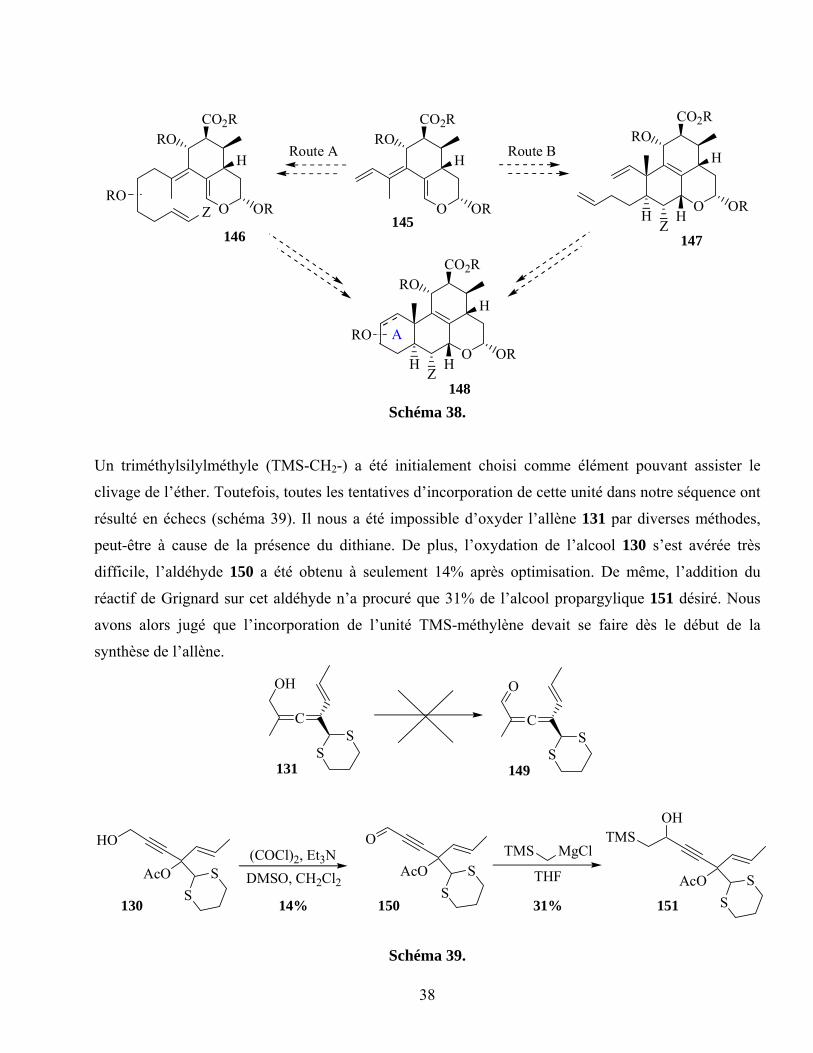
38
RO
RO
CO2RRO
O OR
H
ZHH
147
CO2RRO
O OR
H
CO2RRO
O OR
H
ZHH
145
148
CO2RRO
O OR
H
Z
146
Route A Route B
A
Schéma 38.
Un triméthylsilylméthyle (TMS-CH2-) a été initialement choisi comme élément pouvant assister le
clivage de l’éther. Toutefois, toutes les tentatives d’incorporation de cette unité dans notre séquence ont
résulté en échecs (schéma 39). Il nous a été impossible d’oxyder l’allène 131 par diverses méthodes,
peut-être à cause de la présence du dithiane. De plus, l’oxydation de l’alcool 130 s’est avérée très
difficile, l’aldéhyde 150 a été obtenu à seulement 14% après optimisation. De même, l’addition du
réactif de Grignard sur cet aldéhyde n’a procuré que 31% de l’alcool propargylique 151 désiré. Nous
avons alors jugé que l’incorporation de l’unité TMS-méthylène devait se faire dès le début de la
synthèse de l’allène.
C
OH
SS
131
130
AcO
HO
SS
150
AcO
O
SS
149
C
O
SS
THF
TMS MgCl
151
AcOS
S
TMS(COCl)2, Et3NDMSO, CH2Cl2
14% 31%
OH
Schéma 39.

39
CHAPITRE 3 : SÉQUENCE SYNTHÉTIQUE METTANT À PROFIT UNE UNITÉ TMS-
MÉTHYLÈNE COMME AGENT DE CLIVAGE DU CONNECTEUR
3.1. Synthèse du vinylallène-TMS 160
En considération de l’instabilité relative des intermédiaires dans la synthèse de l’allène 131, nous
avons décidé d’incorporer l’unité d’assistance au clivage au début de la séquence réactionnelle. En ce
sens, la synthèse de l’alcyne 155 a été entamée (schéma 40).
O
TMS
MgClTMS
K2CO3
MeOH
OTBSTMS
152
155 154
153
OTBS
TMS
TMS
OH
TMS
TMS
TBSCl
THF, -78 °C
95%
86%
100%imidazole
Schéma 40.
À l’aldéhyde 152120, connu de la littérature, a été ajouté l’unité triméthylsilylméthyle via son réactif de
Grignard, ce qui a procuré l’alcool propargylique 153 à 86% de rendement. Une protection quantitative
de cet alcool sous forme d’un éther silylé TBS (154) suivie d’une protodésilylation a permis d’obtenir
l’alcyne terminal 155 avec un rendement de 82% pour la séquence de trois étapes.121 En plus d’être
fiable sur une échelle de plus de 50 g, cette séquence n’a nécessité aucune purification par
chromatographie éclair sur gel de silice.
Suivant la stratégie décrite au schéma 33, le dérivé lithié de l’alcyne 155 a été ajouté au crotonaldéhyde
et l’alcool allylique secondaire 156 résultant a été oxydé avec le réactif de Dess-Martin pour générer la
cétone 157 avec un rendement de 87% pour les deux étapes (schéma 41). Après optimisation, l’addition
du lithien du 1,3-dithiane à la cétone 157 a permis d’isoler l’alcool tertiaire 158 à 57%. Une acétylation
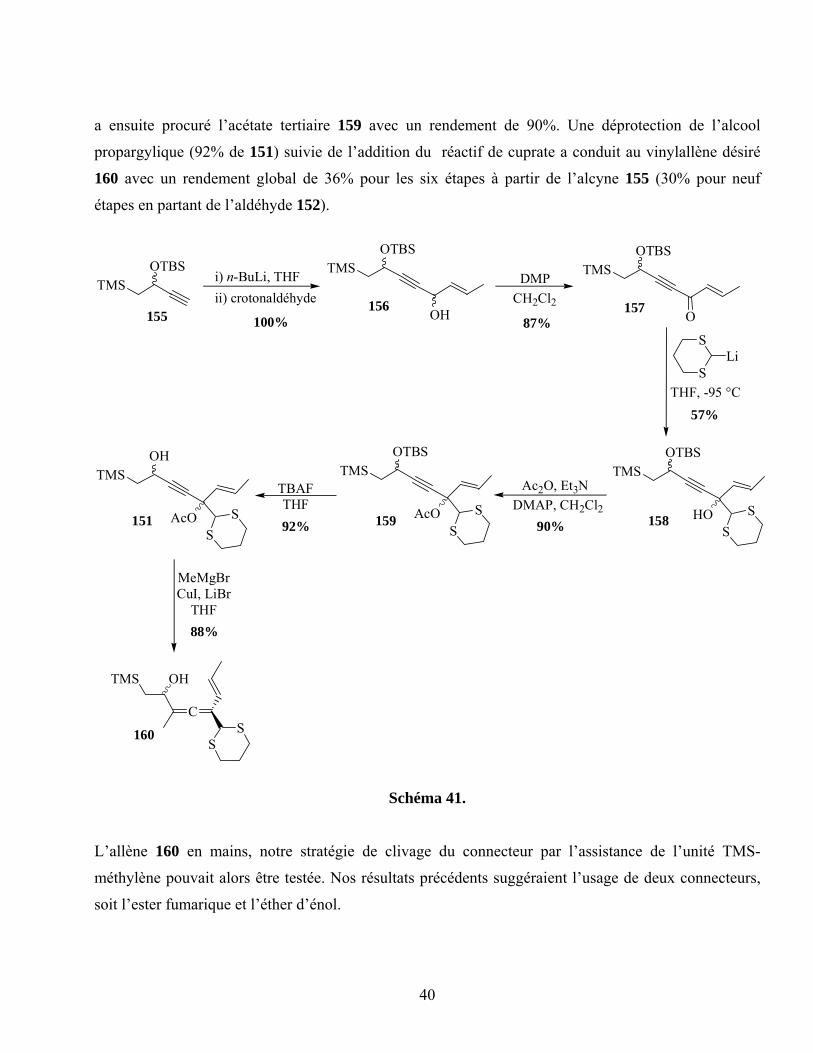
40
a ensuite procuré l’acétate tertiaire 159 avec un rendement de 90%. Une déprotection de l’alcool
propargylique (92% de 151) suivie de l’addition du réactif de cuprate a conduit au vinylallène désiré
160 avec un rendement global de 36% pour les six étapes à partir de l’alcyne 155 (30% pour neuf
étapes en partant de l’aldéhyde 152).
OTBSTMS
155
C
SS
OHTMS
151 AcO
OH
SS
TMSTBAFTHF
160
156
159
OH
OTBSTMS
AcO
OTBS
SS
TMS
CH2Cl2DMP
157 O
OTBSTMS
158 HO
OTBS
SS
TMS
S
SLi
i) n-BuLi, THF
Ac2O, Et3N
THF, -95 °C57%
ii) crotonaldéhyde
100% 87%
DMAP, CH2Cl290%92%
MeMgBrCuI, LiBr
THF88%
Schéma 41.
L’allène 160 en mains, notre stratégie de clivage du connecteur par l’assistance de l’unité TMS-
méthylène pouvait alors être testée. Nos résultats précédents suggéraient l’usage de deux connecteurs,
soit l’ester fumarique et l’éther d’énol.

41
3.2. Ester fumarique en tant que connecteur
Cette séquence réactionnelle a débuté par l’estérification de l’alcool 160 avec le monoéthylester de
l’acide fumarique commercial 161 (schéma 42). Suite au couplage, la cycloaddition de Diels-Alder,
survenue sous le reflux du benzène, a procuré la lactone 162 avec un rendement optimisé de 45% pour
les deux réactions. Quoique faible, le rendement de cette réaction était suffisant pour tester notre
stratégie.
C
SS
OHTMS
HO2C CO2Et 161
160 162
CO2Et
S S
O
O
TMS
45%
i) DCC, DMAP, CH2Cl2
ii) benzène, reflux
Schéma 42.
La lactone 162 a alors été soumise à diverses conditions pour effectuer le clivage du connecteur (i.e. le
bris du lien C1-O, c.f. schéma 37). Si des résultats intéressants ont été obtenus avec le fluorure de
tétrabutylammonium (TBAF), une multitude de différentes conditions de réaction ainsi que l’usage
d’autres sources de fluorure n’ont pas permis d’augmenter le rendement en triène 163 (tableau 5). Les
tentatives de β-élimination de type Peterson avec des acides de Brønsted n’ont généré que de la
décomposition en plus de conduire à des réactions incomplètes (entrées 5-6).122 Si l’usage de BF3•OEt2
a mené à l’isolation de produits contenant un aldéhyde (vide infra), la saponification de la lactone a
procuré des traces du diester 164 désiré (entrée 8). Cette inertie de la lactone 162 est en accord avec nos
résultats préliminaires, lors desquels nous n’avions pas réussi à saponifier ce connecteur.28a Finalement,
la réaction de l’ester allylique 162 avec du palladium dans le but de neutraliser le complexe π-allyle
résultant avec l’élimination du groupement silyle, n’a occasionné que de la dégradation (entrée 9).123
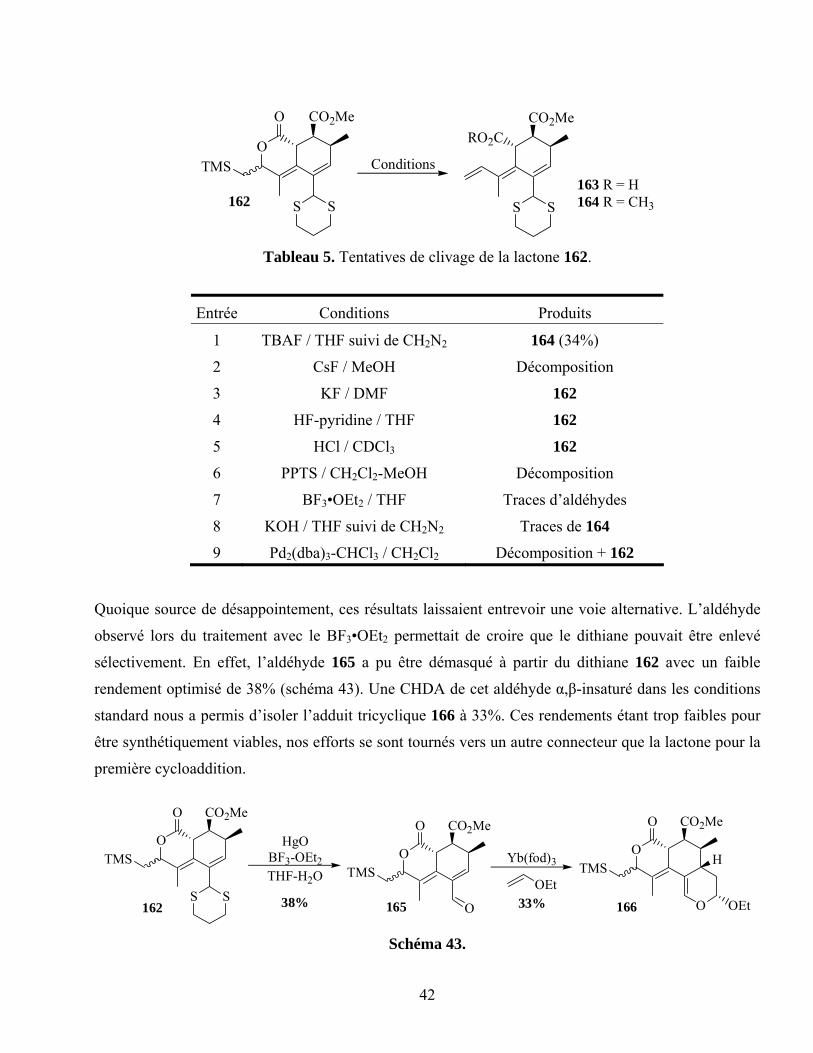
42
CO2Me
S S
O
O
TMS
162
CO2Me
S S
RO2C
163 R = H164 R = CH3
Conditions
Tableau 5. Tentatives de clivage de la lactone 162.
Entrée Conditions Produits
1 TBAF / THF suivi de CH2N2 164 (34%)
2 CsF / MeOH Décomposition
3 KF / DMF 162
4 HF-pyridine / THF 162
5 HCl / CDCl3 162
6 PPTS / CH2Cl2-MeOH Décomposition
7 BF3•OEt2 / THF Traces d’aldéhydes
8 KOH / THF suivi de CH2N2 Traces de 164
9 Pd2(dba)3-CHCl3 / CH2Cl2 Décomposition + 162
Quoique source de désappointement, ces résultats laissaient entrevoir une voie alternative. L’aldéhyde
observé lors du traitement avec le BF3•OEt2 permettait de croire que le dithiane pouvait être enlevé
sélectivement. En effet, l’aldéhyde 165 a pu être démasqué à partir du dithiane 162 avec un faible
rendement optimisé de 38% (schéma 43). Une CHDA de cet aldéhyde α,β-insaturé dans les conditions
standard nous a permis d’isoler l’adduit tricyclique 166 à 33%. Ces rendements étant trop faibles pour
être synthétiquement viables, nos efforts se sont tournés vers un autre connecteur que la lactone pour la
première cycloaddition.
CO2Me
S S
O
O
TMSHgO
BF3-OEt2THF-H2O
162 165
CO2Me
O
O
O
TMSOEt
166
CO2Me
O
O
O
TMS
OEt
HYb(fod)3
38% 33%
Schéma 43.

43
3.3. Vinylallène-TMS 160 et l’éther d’énol en tant que connecteur
L’éther d’énol 143, dont le clivage laisserait derrière un alcool en C11, a alors été l’objet de nos
explorations (schéma 44). À cette fin, l’addition de l’allènol 160 au propiolate de méthyle a généré le
dihydrofuranne 167 avec un rendement de 90% pour les deux transformations (addition et CDA).
C
SS
OHTMSCO2Me
160
OCO2Me
TMS
S SNMM, Et2O
90%167
111 13
Schéma 44.
À ce point, l’analyse de diffraction des rayons X des cristaux obtenus de 167 nous a permis de prouver
hors de doute le contrôle stéréochimique de la cycloaddition de Diels-Alder (figure 5). La relation cis
entre le méthyle et l’ester est conséquent d’une CDA intramoléculaire entre le vinylallène et l’éther
d’énol, prouvant ainsi que l’addition de Michael sur le propiolate précède la cycloaddition [4+2]. De
plus, la stéréochimie bêta des carbones C12 et C13 combinée à la configuration alpha de l’éther en C11
confirme l’obtention d’un éther d’énol de géométrie trans et une cycloaddition procédant selon un état
de transition endo (schéma 45). Si un seul des diastéréoisomères en C1 est représenté, les deux étaient
tout de même présents dans le cristal.
Figure 5. Structure de 167 par diffraction des rayons X.
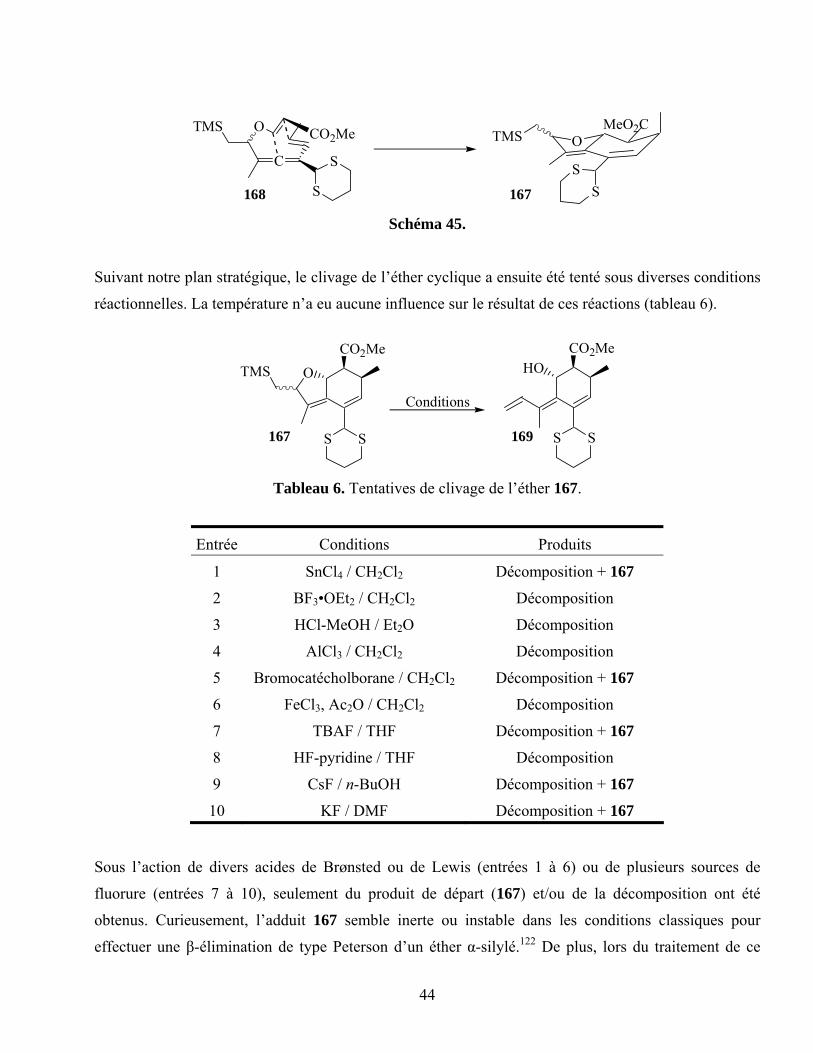
44
C
S
S
OTMS CO2Me
168 167
OTMS
SS
MeO2C
Schéma 45.
Suivant notre plan stratégique, le clivage de l’éther cyclique a ensuite été tenté sous diverses conditions
réactionnelles. La température n’a eu aucune influence sur le résultat de ces réactions (tableau 6).
OCO2Me
TMS
S S167 169
CO2Me
S S
HO
Conditions
Tableau 6. Tentatives de clivage de l’éther 167.
Entrée Conditions Produits
1 SnCl4 / CH2Cl2 Décomposition + 167
2 BF3•OEt2 / CH2Cl2 Décomposition
3 HCl-MeOH / Et2O Décomposition
4 AlCl3 / CH2Cl2 Décomposition
5 Bromocatécholborane / CH2Cl2 Décomposition + 167
6 FeCl3, Ac2O / CH2Cl2 Décomposition
7 TBAF / THF Décomposition + 167
8 HF-pyridine / THF Décomposition
9 CsF / n-BuOH Décomposition + 167
10 KF / DMF Décomposition + 167
Sous l’action de divers acides de Brønsted ou de Lewis (entrées 1 à 6) ou de plusieurs sources de
fluorure (entrées 7 à 10), seulement du produit de départ (167) et/ou de la décomposition ont été
obtenus. Curieusement, l’adduit 167 semble inerte ou instable dans les conditions classiques pour
effectuer une β-élimination de type Peterson d’un éther α-silylé.122 De plus, lors du traitement de ce

45
dihydrofuranne avec du HCl (entrée 3), nous avons observé que les diastéréoisomères (en C1) ne
réagissaient pas à la même vitesse. Cette diastéréosélection suggère que le contrôle de la stéréochimie
en C1 pourrait être un facteur important pour le succès de cette stratégie.
En présence d’une source de fluorure, un mécanisme d’élimination de type E2 est prédit. Une analyse
de la structure de 167 obtenue par diffraction des rayons-X suggère que les liens Si-C-C-O peuvent
difficilement se placer de façon anti-périplanaire, probablement à cause des répulsions stériques entre le
TMS et le méthyl vinylique et ce peu importe le diastéréoisomère en C1 (c.f. figure 5). Les échecs
encourus lors de ces expériences sont peut-être le reflet d’une conformation qui empêche l’alignement
propice de ces liens, inhibant ainsi l’élimination souhaitée. Sous l’action d’acides de Lewis, un
mécanisme E1 est envisageable. L’éther en C1 étant un mauvais groupe partant, ce mode d’élimination
serait cependant très difficile surtout si la participation électronique du TMS est faible (due au mauvais
recouvrement orbitalaire). Parmi les tentatives infructueuses figurent aussi des essais d’élimination de
l’éther dans des conditions basiques ainsi que l’ouverture du dihydrofuranne sous l’action d’agents
réducteurs, ces réactions ne procurant que des produits de dégradation complexes.124
Malgré ces échecs, nous avons poursuivi la séquence afin d’étudier le comportement de ce nouveau
substituant en C1 lors des opérations subséquentes. Dans ce sens, le traitement du dithiane 167 avec de
l’oxyde de mercure et du BF3•OEt2 a procuré l’aldéhyde 170 avec un rendement de 82% (schéma 46).
Tel qu’avec la lactone 162, la cycloaddition d’hétéro DA de l’aldéhyde α,β-insaturé 170 a permis
d’obtenir l’adduit tricyclique 171 à 83% de rendement, venant dès lors renforcer l’hypothèse que
l’instabilité des composés lors de la CHDA est entre autres reliée au faible degré de substitution en C1
(c.f. section 1.2 et 2.1).
OCO2Me
TMS
S S
HgOBF3-OEt2
167
THF-H2O
170
OCO2Me
TMS
O
OEt171
OCO2Me
TMS
O OEt
HYb(fod)3
82% 83%
Schéma 46.

46
La résistance de 171 (contrairement au dihydrofuranne 133) face à la dégradation lors de la CHDA (c.f.
schéma 28) est peut être due à une stabilisation électronique du carbone en C1 par l’effet β du
trialkylsilane.125 Elle pourrait aussi être attribuée à des effets stériques entre le TMS-méthylène en C1 et
l’ytterbium, ce qui défavoriserait la complexation de l’acide de Lewis à l’éther afin d’en affaiblir le
lien. La présence de ce substituant en C1 pourrait également induire une conformation dans laquelle
l’alignement des liens σC-O et πC=C est mauvais, empêchant ainsi la participation de l’éther d’énol (par
délocalisation de ses électrons jusqu’au dihydrofuranne) à l’ouverture du dihydrofuranne.
En résumé, l’éther tricyclique 171 a été obtenu avec un rendement global de 18% suite à une séquence
de douze étapes. La poursuite de la synthèse impliquait le clivage de l’éther en C1 afin d’utiliser
l’alcène terminal produit pour incorporer une chaîne latérale portant le diénophile pour la troisième
cycloaddition (route A, schéma 38). À court de matériel pour continuer les études de clivage du
connecteur, nous avons décidé de ré-investiguer la synthèse menant au vinylallène 160. Une route
synthétique nous permettant une énantiosélection était nécessaire, puisque la synthèse énantiosélective
de différents membres des quassinoïdes était notre but ultime. Basée sur les travaux de maîtrise de
Sylvie Fréchette, une séquence réactionnelle adaptée à notre stratégie a été développée.

47
CHAPITRE 4 : SYNTHÈSE DU VINYLALLÈNE COMPORTANT L’UNITÉ TMS-
MÉTHYLÈNE PAR ÉPOXYDATION D’UN ÉNYNE
4.1. Travaux précédents
Les travaux de maîtrise de Sylvie Fréchette visaient le développement d’une séquence synthétique pour
la formation de vinylallènes tels que 176 (schéma 47).52a Débutant avec l’éther propargylique 172,
l’alcool 173 a été obtenu suite à un couplage de Sonogashira (voir la section 4.1.1). Une époxydation de
Shi (excès énantiomérique de 55%, section 4.1.2) suivie de la protection de l’alcool primaire a permis
d’obtenir 174. L’addition du méthylcuprate à l’époxyde propargylique (discuté à la section 5.3), de
manière SN2’ et anti,126 a toutefois généré un mélange complexe de quatre produits. Deux de ces
produits étaient le résultat d’un échange du groupe protecteur TBS sous les conditions réactionnelles.
Le vinylallène 176 a finalement été obtenu après 4 étapes de fonctionnalisation, pour un total de huit
étapes à partir de l’alcyne 172. Ces travaux ont mis en valeur l’importance du choix des groupes
protecteurs, la racémisation des produits étant observée avec certains d’entre eux (interconversion de
177 et 178).
n-Bu
OMOMn-Bu
OMOMOH
172
176
COPiv
OMOMn-Bu
CO2Me
173
175
174
n-Bu
OMOMOTBS
O
COTBS
OH
OMOMn-Bu
mélange de 4 stéréoisomères
1) Époxydation
4 étapes
2) Protection
COTBS
OCuX
OMOMn-Bu
177 178C
OCuX
OTBS
OMOMn-Bu
Schéma 47.

48
4.1.1. Introduction au couplage de Sonogashira
Plusieurs méthodes existent pour la création d’une liaison σ entre un carbone hybridé sp2 et un autre
hybridé sp, certaines utilisant des quantités stoechiométriques de métaux, d’autres mettant à profit
l’usage de palladium en quantité catalytique.127 Les dernières années ont vu des avancées spectaculaires
dans ce domaine, principalement grâce à deux méthodes utilisant des métaux de transition. La première
consiste à coupler un alcynylzinc avec un halogénure vinylique en présence de palladium.128 La
seconde, le couplage de Sonogashira, permet d’unir une alcyne terminale avec un halogénure vinylique
en présence de quantités catalytiques de palladium et de cuivre.129
Le mécanisme de cette transformation, représenté au schéma 48, a été étudié et clarifié grâce à la
contribution de nombreux auteurs.130 La base aminée permet la formation de l’acétylure de cuivre in
situ, un mécanisme postulé il y a de nombreuses années par Castro et Stephens, en plus d’effectuer la
réduction du PdII en Pd0.131 Par la suite, l’addition oxydante du Pd0 dans le lien C-X de l’halogénure
vinylique132 est suivie du transfert de l’acétylure (transmétallation Cu-Pd) pour finalement procurer
l’alcyne disubstituée après un couplage réductif.133 Les récents travaux du professeur Fu ont permis
d’appliquer le couplage de Sonogashira aux halogénures d’alkyles (sp3).134
Comme dans toutes les réactions catalysées par du palladium, les ligands peuvent avoir un effet
dramatique. Outre les phosphines, dont le comportement peut facilement être prédit grâce à de
nombreuses études,135 les ligands à base d’arsenic sont couramment employés afin d’augmenter la
vitesse de la réaction (la transmétallation de l’acétylure du cuivre au palladium, l’étape limitante, est
davantage favorisée grâce à la dissociation rapide des ligands à base d’arsenic).136 Des récents travaux
ont prouvé que des ligands stériquement encombrés de type phosphine permettaient d’éviter l’usage
d’arsines. L’influence des ions halogénures a aussi été démontrée comme déterminante dans certaines
réactions où l’élimination β, une réaction secondaire au couplage de Sonogashira, constitue un
problème.137
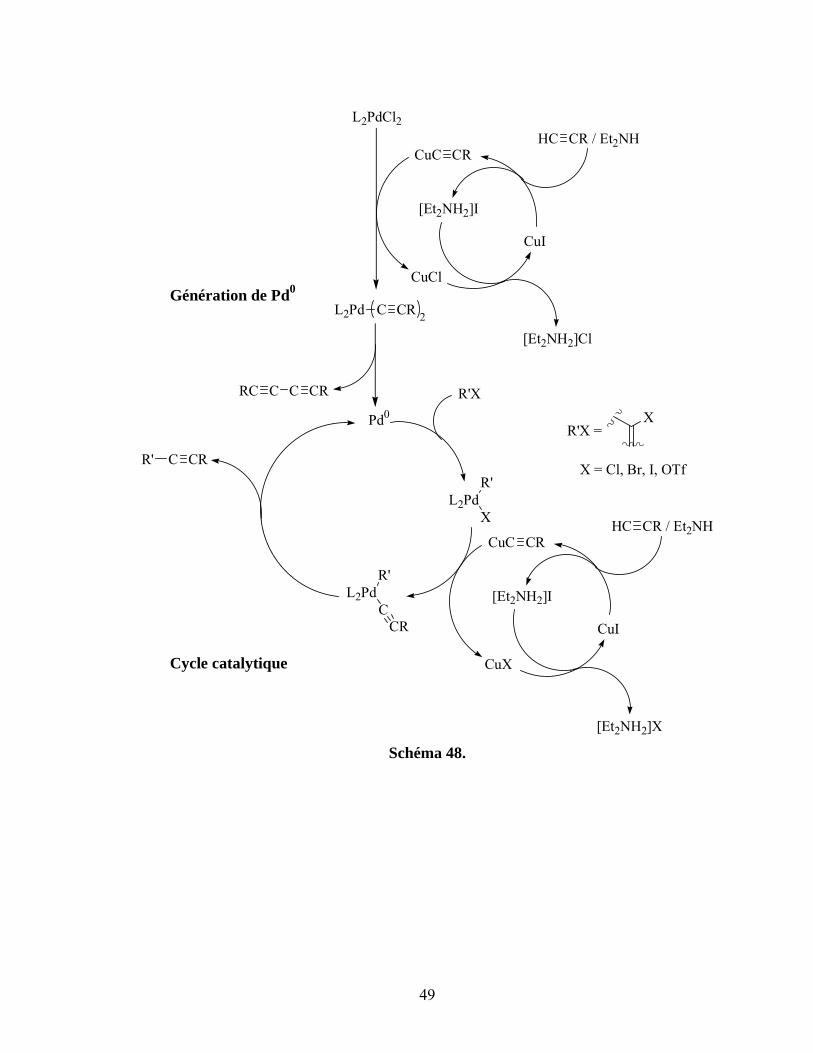
49
HC CR / Et2NHCuC CR
[Et2NH2]I
CuI
CuCl
C CRL2Pd
[Et2NH2]Cl
C CRCRC
X
C CRR'
L2PdR'
X HC CR / Et2NHCuC CR
[Et2NH2]IL2PdR'
CCR CuI
CuX
[Et2NH2]X
L2PdCl2
Pd0
2
R'X
R'X =
X = Cl, Br, I, OTf
Génération de Pd0
Cycle catalytique
Schéma 48.

50
4.1.2. Introduction aux époxydations énantiosélectives
L’époxydation asymétrique de Katsuki-Sharpless est peut-être la réaction la plus populaire pour obtenir
un époxyde de façon énantiosélective à partir d’un substrat achiral.138 Catalysée par du titane, cette
transformation requiert la présence d’un groupe directeur en position allylique, soit un alcool.139
L’efficacité et la prédictibilité du modèle élaboré par Sharpless en font une méthode puissante et fiable,
car l’époxyde R ou S peut être obtenu simplement en changeant la chiralité absolue du ligand tartrate
(schéma 49). Les rendements et les sélectivités obtenus par cette méthode sont meilleurs avec des
alcènes substitués trans.
D-(-)-diéthyl tartrate (non naturel)
L-(+)-diéthyl tartrate (naturel)
OHR1
R3
R2 (CH3)3CO2H , Ti(OiPr)4
CH2Cl2, -20 °C
Ox
Ox
OR1R2
R3OH
Rendements de 70-99%Supérieur à 90% ee
Schéma 49.
En absence d’un groupe directeur en position allylique, les méthodes de Jacobsen140 et de Shi se voient
les méthodes de choix.141 Étant optimale pour des oléfines trans, cette dernière méthode est aussi
efficace avec des alcènes cis142 et procure d’excellents résultats avec des énynes.143 Les études de Shi
ont permis d’établir les bases structurelles nécessaires pour le catalyseur, permettant ainsi d’éliminer la
décomposition de l’auxiliaire chiral 179 par une réaction de Baeyer-Villiger qui conduit aux lactones
correspondantes (i.e. 183 et 184, schéma 50).144
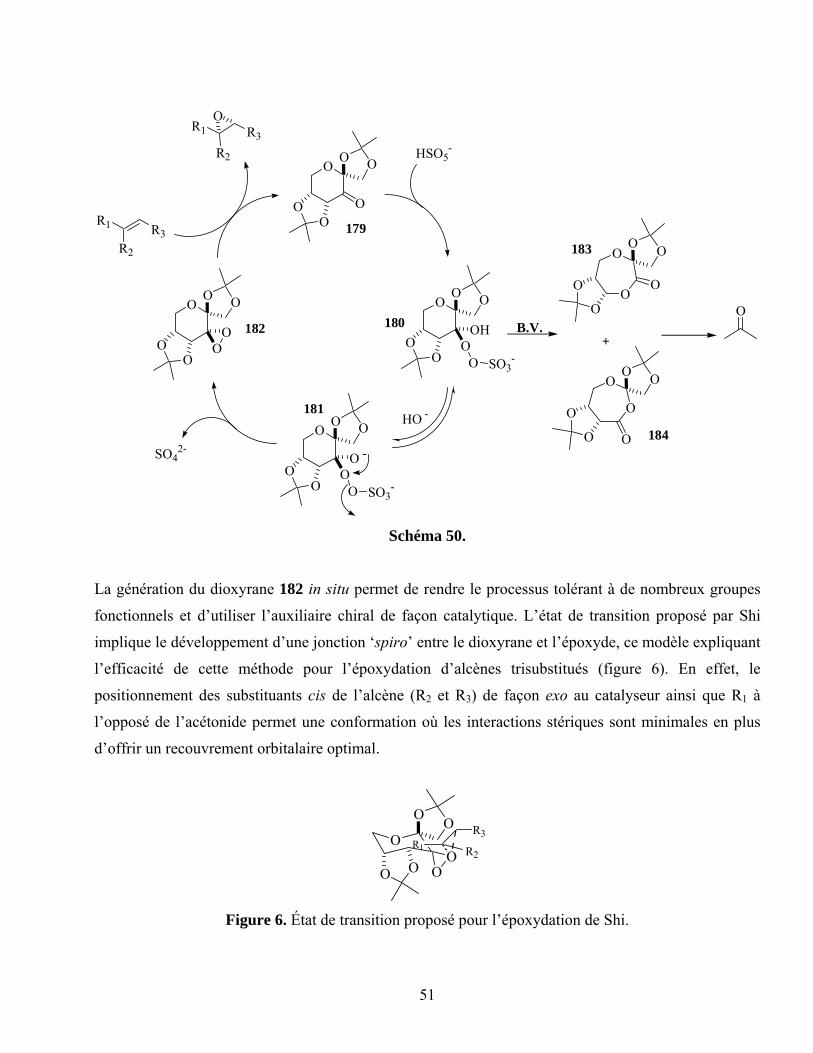
51
R3R1
R2
O
OO
OO
O O
R3R1
R2
O
182
O
OO
O O
O
179
180
HSO5-
O
OO
OOH
O O
O SO3-
183
O
O
O
O
O O
O
O
OO
OO
O O
O SO3-
SO42-
HO - O
O
O
O
O O
O
181
184
O
-
B.V.+
Schéma 50.
La génération du dioxyrane 182 in situ permet de rendre le processus tolérant à de nombreux groupes
fonctionnels et d’utiliser l’auxiliaire chiral de façon catalytique. L’état de transition proposé par Shi
implique le développement d’une jonction ‘spiro’ entre le dioxyrane et l’époxyde, ce modèle expliquant
l’efficacité de cette méthode pour l’époxydation d’alcènes trisubstitués (figure 6). En effet, le
positionnement des substituants cis de l’alcène (R2 et R3) de façon exo au catalyseur ainsi que R1 à
l’opposé de l’acétonide permet une conformation où les interactions stériques sont minimales en plus
d’offrir un recouvrement orbitalaire optimal.
OO
O O
O
R3R1
OOR2
Figure 6. État de transition proposé pour l’époxydation de Shi.

52
4.2. Synthèse du vinylallène-TMS avec un acétal comme groupe protecteur
Connaissant les difficultés encourues par Mme Fréchette (i.e. l’échange des groupes protecteurs lors de
la réaction de carbocupration, c.f. schéma 47), nous avons choisi d’utiliser un diéthyl acétal (composé
186) afin de masquer la fonction aldéhyde requise pour l’oléfination subséquente. Une recherche de la
littérature a révélé une voie rapide pour synthétiser le bromure vinylique 186. La formation de 185 à
partir de l’acroléine selon la méthode de Riegel145 (56%) suivie de la protection de l’aldéhyde sous
forme d’un acétal diéthylique, telle que rapporté par Smith, a procuré le bromure vinylique 186 avec un
faible rendement de 41%.146 L’utilisation de micro-ondes s’est avérée une solution profitable,
permettant d’augmenter le rendement de la réaction à 84%.147 Cette courte séquence a aussi permis
d’obtenir l’alcool allylique 187,148 connu de la littérature, par la réduction de l’aldéhyde 185 (86%). Cet
alcool devait nous servir au cas où l’acétal 186 s’avérait trop instable.
CHO CHOBr
185
EtOHC(OEt)2Br
186
Br2, H2O
56%
(EtO)3CH, APTS
41%
(EtO)3CH, APTSmicro-ondes
84%NaBH4
BrOH
MeOH
187
86%
Schéma 51.
Comme nous le craignions, le couplage de Sonogashira entre l’alcyne 155 et l’acétal 186 n’a procuré
que 24% de l’ényne désirée 188 après optimisation (schéma 52). De plus, les protocoles modifiés des
réactions de Kumada149 (via l’acétylure de magnésium) et de Negishi (via l’acétylure de zinc) n’ont pas
permis d’obtenir l’ényne désirée 188.150

53
OTBSTMS
155
C(OEt)2Br
186
OTBSTMS
M155-M
188
188
OTBSTMS
OEt
OEt
24%
+PdCl2(PPh3)2, CuI, PPh3
n-BuNH2, 23 °C
Pd(PPh3)4, PPh3186, THF
M = MgBr = ZnCl
Schéma 52.
Nous croyons que la labilité de l’acétal allylique est à la source du faible rendement obtenu. En effet, il
est possible que le Pd(0) réagisse avec l’acétal allylique 188 pour former un complexe π–allyle (189),
entraînant ainsi la décomposition du produit de Sonogashira (schéma 53). L’arrêt de la réaction à faible
conversion n’a pas permis de diminuer la quantité de produits de décomposition et l’acétal 188, lorsque
replacé dans les conditions du couplage de Sonogashira, s’est partiellement décomposé, supportant
ainsi notre hypothèse.
OEt
OEtOTBS
TMS OEtOTBS
TMS
188 189 Pd+L2
CuIPd0
+ EtOCu + I-
Décomposition
Schéma 53.

54
Inverser les étapes réactionnelles pourrait nous permettre d’éviter ce problème. Étant incapable d’unir
l’alcyne 155 et la bromoacroléine (185) par le couplage de Sonogashira, nous avons tenté avec succès
le couplage du bromure vinylique 187 avec l’alcyne 155, ce qui nous a conduit au produit de couplage
190 avec un rendement de 75% (schéma 54). L’oxydation de l’alcool allylique résultant a ensuite
procuré l’aldéhyde α,β-insaturé 191 avec un excellent rendement brut de 95%. Toutefois, l’acétal désiré
192 n’a pas été obtenu sous diverses conditions de protection, seuls de nombreux produits de
décomposition de l’aldéhyde 191 étant observés. L’emploi d’un groupe protecteur différent d’un acétal
a alors été envisagé.
OTBS
TMS
155
187
BrOH
OTBSTMS OR
OR192
190
191
OTBSTMS O
OTBSTMS OH
DMPCH2Cl2
75%
+
PdCl2(PPh3)2CuI, PPh3
iPr2NH, 23 °C
95% brut
Protection
Schéma 54.

55
4.3. Synthèse du vinylallène avec un MOM comme groupe protecteur
Étant capable de résister aux conditions basiques de la carbocupration, l’éther méthoxyméthylique
(MOM) a été choisi comme groupe protecteur. Dans ce sens, la protection de l’alcool 187 a procuré le
bromure vinylique 193 avec un bon rendement de 81% (schéma 55). Le couplage de Sonogashira entre
l’alcyne 155 et le bromure vinylique 193 s’est déroulé sans problème, procurant l’ényne 194 avec un
excellent rendement de 90%. Puisque le but de ce projet était le développement d’une séquence
réactionnelle menant au squelette picrasane et non la synthèse totale d’une quassinoïde, ainsi que pour
des raisons économiques, l’époxydation de l’alcène terminal a été effectuée de façon racémique. Le
traitement de 194 avec l’acide méta-chloroperoxybenzoïque (m-CPBA) a donné accès à un époxyde
instable dont l’alcool propargylique a été déprotégé immédiatement, permettant ainsi d’obtenir l’époxy-
alcool 195 avec un rendement de 69% pour les deux opérations.
OTBS
TMS
155
BrOR
187 R = H
193 R = MOM
LiBrAPTS
(CH3O)2CH2
194
OTBSTMS OMOM
195
OHTMS OMOM
O69%90%
+
PdCl2(PPh3)2CuI, PPh3
Et3N, 23 °C1) m-CPBA2) TBAF
81%
Schéma 55.
À ce point, l’ouverture de l’époxyde par addition SN2’ du réactif de cuprate a nécessité une optimisation
(tableau 7). La réaction avec de l’iodure de cuivre a procuré le diol désiré 196 à 50% (entrée 1), mais un
abaissement de la température a plutôt favorisé la production du produit d’addition SN2 du méthyle sur
l’époxyde (entrée 2).151 L’utilisation du cyanure de cuivre a procuré le diol 196 avec des rendements
similaires à la réaction utilisant l’iodure de cuivre (entrée 3).152 Toutefois, la présence de sels de
magnésium dans le réactif de Grignard a de nouveau engendré une production de l’alcool tertiaire 197,
celui-ci devenant alors le produit majeur de la réaction (entrée 4). Heureusement, l’addition du réactif

56
de cuprate sans sels de magnésium à -15 °C a permis d’obtenir exclusivement l’allène désiré 196 avec
un excellent rendement de 89% (entrée 5). Si la réaction effectuée dans l’éther éthylique n’a procuré
que de la dégradation et seulement des traces du diol (entrée 6), l’addition du cuprate dans le THF à
plus basse température n’a pas non plus augmenté davantage le rendement de l’allène 196 (entrée 7).
OH
TMS OMOM
O195
COMOM
OHTMSOH
197
OHTMS OMOM
OH
CuX, MeMgBrsolvant
+
196 Tableau 7. Addition du réactif de méthylcuprate sur l’époxyde 195.
Entrée CuX Solvant Température (°C) Produits (rdt)
1 CuI / LiBr THF -10 196 (50%)
2 -- -- -40 196 (25%) + 197 (45%)
3 CuCN -- 0 196 (58%)
4 -- -- -10 196 (25%) + 197 (20%)a
5 -- -- -15 196 (89%)
6 -- Et2O -- Traces de 196
7 -- THF -30 196 (78%) a) La solution de MeMgBr contenait une importante quantité de sels de magnésium.
Avec le diol 196 en mains, l’oxydation sélective de l’alcool primaire a été tentée. Toutefois, cette tâche
s’est avérée particulièrement problématique, les méthodes standard pour effectuer cette transformation
ne donnant pas de résultats satisfaisants. Parmi les réactions infructueuses, notons l’usage d’oxydants
doux tels que le N-bromosuccinimide153 (NBS) et le perruthénate de tétrapropylammonium154 (TPAP)
qui ont seulement promu la décomposition du produit de départ. La formation du tosylate primaire et
l’oxydation avec le diméthylsulfoxyde155 (DMSO) n’a pas procuré l’aldéhyde désiré 198, mais plutôt le
chlorure 200 à 50%. Cependant, toutes les tentatives d’oxydation par le déplacement de ce chlorure par
le DMSO se sont avérées vaines, ne produisant que de la décomposition (le chlorure 200 n’a donc pas
été caractérisé). Le premier signe d’espoir est survenu lorsque le diol 196 a été traité dans les conditions
de Swern, la réaction nous permettant d’isoler le produit de double oxydation 199 avec un rendement de
10%.50 Heureusement pour nous, la réaction du diol 196 avec de l’oxyde de manganèse (MnO2) a
procuré un mélange inséparable des produits instables 198 et 199 (5 : 1, 75%). Toutefois, nous avons

57
rapidement découvert que le ratio de cette oxydation était directement relié à la source de MnO2
utilisée, celui-ci changeant selon la bouteille employée. Ainsi, le rapport des produits de cette réaction
est descendu à 2 : 1 après le renouvellement de nos stocks d’oxyde de manganèse. L’acide
iodoxybenzoïque (IBX) est un oxydant qui a gagné en popularité ces dernières années.156 Lorsque placé
avec cet oxydant, le diol 196 a procuré 32% de l’aldéhyde 198, mais sans la formation de produits
secondaires.157
COMOM
OHMe3SiOH
196
198
199
C
OH
OMOM
Me3Si O
C
O
OMOM
Me3Si O
200C
OMOM
OHMe3SiCl
Voir le texte
Schéma 56.
Ce dernier résultat était très prometteur du fait que seul le produit désiré avait été obtenu. Finney a
récemment démontré l’importance du solvant lors de l’oxydation d’alcools avec le IBX.158 Une
évaluation systématique des solvants étudiés dans cet article a alors été entreprise, mais sans grand
succès.
L’aldéhyde 198 obtenu lors des différentes oxydations étant instable à la chaleur et à l’air, les
expériences d’oléfination subséquentes ont immédiatement été réalisées sur l’aldéhyde brut. Désirant
obtenir un alcène trans à cette position, nous avons tourné nos efforts vers l’oléfination de Horner-
Wadsworth-Emmons (HWE).159 Toutefois, la réaction entre le phosphonate 201 et l’aldéhyde 198 n’a
occasionné que de la décomposition (schéma 57).
C
OH
OMOM
Me3Si O
MeO P CO2MeO
MeONaH
198 201+
THF, 0 °CDécomposition
Schéma 57.

58
Une alternative à cette réaction est l’oléfination de Julia.160 Kociensky a récemment démontré que les 1-
phényl-1H-tétrazolylsulfones (i.e. 202) permettent d’effectuer l’oléfination dans de meilleurs
rendements et sélectivités (ratio E/Z) comparés à ceux obtenus avec les benzothiazolylsulfones de
Julia.161 De nombreuses études ont également souligné que ces paramètres sont sensibles au choix de la
base et au solvant utilisé lors de cette oléfination de Julia modifiée.162 Avec ces notions en tête,
l’aldéhyde 198 a été réagi avec le tétrazole 202 dans les conditions développées par Kocienski (schéma
58). Outre de la décomposition, la présence d’un alcène terminal a été détectée, résultant d’une
élimination de Peterson.122
C
OH
OMOM
Me3Si O
N NNN
SO2Et
Ph KHMDS
198 202+
DME, -78 °CDécomposition +
Schéma 58.
Puisque le substrat 198 semblait très sensible aux conditions basiques, nous avons tourné notre
attention vers l’oléfination développée par Wittig, réaction qui lui valut le prix Nobel de 1979.163 Suite
à une optimisation exhaustive, nous avons pu obtenir l’ester α,β-insaturé 204, avec un faible rendement
de 33% par la réaction de l’aldéhyde 198 et de l’ylure de phosphore 203164 en présence de 2,6-lutidine
(schéma 59). La basse température ainsi que la présence d’une base sont nécessaires afin de minimiser
la décomposition de l’aldéhyde 198 durant la réaction. De plus, seulement de la décomposition a été
observée lorsque l’oléfination a été effectuée dans des solvants autres que le toluène.
C
OH
OMOM
Me3Si O
Ph3PCO2Me
198203
204C
OH
OMOM
Me3SiCO2Me
+toluène, 23 °C
2,6-lutidine
33% Schéma 59.
En considération des résultats de l’oxydation de 196 et de l’oléfination de l’aldéhyde 198, nous
pensions que ces faibles rendements et ces intermédiaires peu stables pouvaient être évités en changeant
l’ordre des étapes synthétiques de la séquence. À cette fin, l’estérification de l’alcool propargylique 195
avec le monoester de l’acide fumarique 161 a conduit à l’adduit 205 à 84% (schéma 60). Toutefois,

59
sous les conditions optimales pour effectuer la réaction de cupration (c.f. tableau 7), seuls des traces de
produits d’élimination de Peterson et des dérivés résultant de polyadditions ont été observés.
OHTMS OMOM
O
HO2C CO2Et 161
195
206
205
C
O
OMOM
Me3SiOH
O
CO2Et
OTMS OMOM
O
O
CO2Et
84%DCC, DMAP, CH2Cl2
CuCNMeMgBrTHF
Schéma 60.
Une alternative à ce problème était d’allonger la séquence en discriminant les alcools de 196 par une
protection régiosélective. L’alcool primaire de 196 a donc été masqué sous forme d’un éther silylé avec
un rendement de 83% (schéma 61). L’estérification de l’alcool secondaire a ensuite procuré l’allène-
ester 208 à 79%. Malheureusement, les tentatives de déprotection de l’alcool primaire allylique ont
généré le produit d’élimination de type Peterson 209 avec un faible rendement de 33% sous l’action de
fluorures, mais avec un très bon rendement (89%) en présence d’un acide de Brønsted. Lors de ces
réactions, nous avons observé que l’élimination se produisait avant la déprotection.
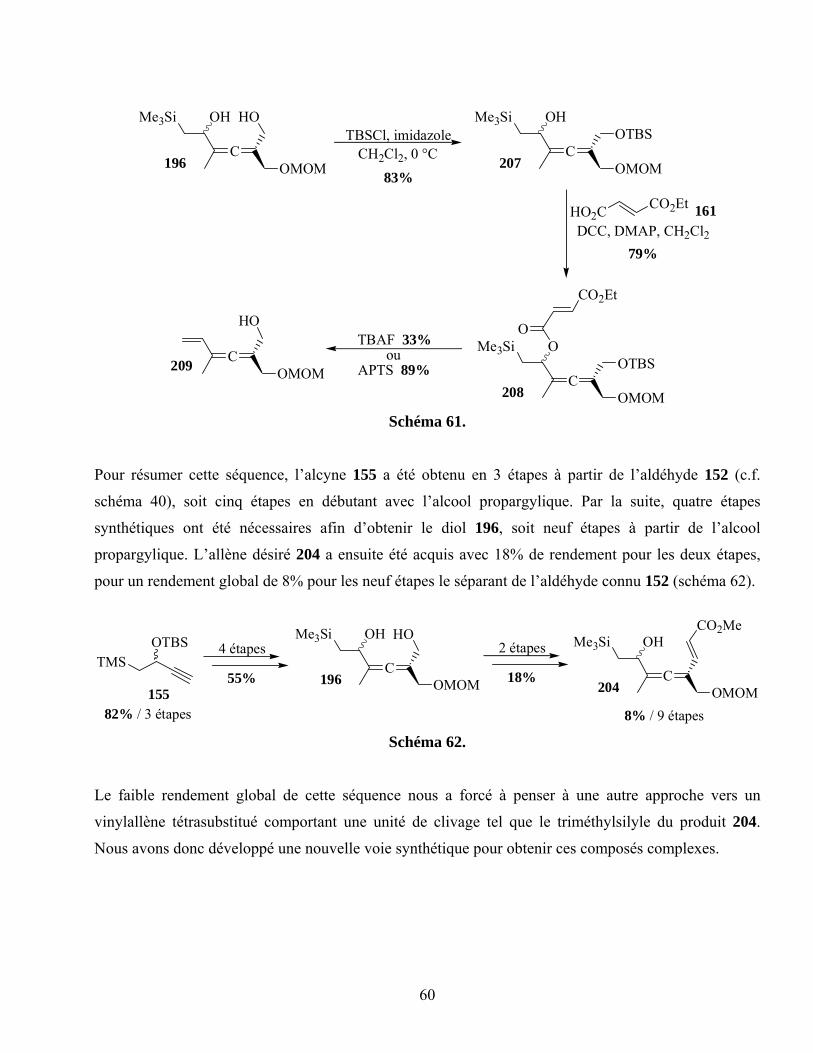
60
C
OH
OMOM
Me3Si HO
209 COMOM
HO
196 207
208
C
OH
OMOM
Me3SiOTBS
C
O
OMOM
Me3SiOTBS
O
CO2Et
HO2C CO2Et 161
TBSCl, imidazoleCH2Cl2, 0 °C
83%
DCC, DMAP, CH2Cl279%
TBAF 33%ou
APTS 89%
Schéma 61.
Pour résumer cette séquence, l’alcyne 155 a été obtenu en 3 étapes à partir de l’aldéhyde 152 (c.f.
schéma 40), soit cinq étapes en débutant avec l’alcool propargylique. Par la suite, quatre étapes
synthétiques ont été nécessaires afin d’obtenir le diol 196, soit neuf étapes à partir de l’alcool
propargylique. L’allène désiré 204 a ensuite été acquis avec 18% de rendement pour les deux étapes,
pour un rendement global de 8% pour les neuf étapes le séparant de l’aldéhyde connu 152 (schéma 62).
OTBSTMS
155196
C
OH
OMOM
Me3Si HO
204C
OH
OMOM
Me3SiCO2Me
4 étapes
55%
82% / 3 étapes
2 étapes
18%
8% / 9 étapes Schéma 62.
Le faible rendement global de cette séquence nous a forcé à penser à une autre approche vers un
vinylallène tétrasubstitué comportant une unité de clivage tel que le triméthylsilyle du produit 204.
Nous avons donc développé une nouvelle voie synthétique pour obtenir ces composés complexes.

61
CHAPITRE 5 : ÉTUDE MODÈLE POUR UNE NOUVELLE SYNTHÈSE DE VINYLALLÈNES
5.1. Stratégie générale
La synthèse de vinylallènes tétrasubstitués par le déplacement SN2’ d’un groupe partant par un cuprate
avait fait ses preuves et nous avons décidé que cet aspect allait demeurer au centre de notre stratégie.
Jusqu’à maintenant, la formation de ce type d’allène (i.e. 111, 131, 160 et 204) impliquait l’addition
d’un méthylcuprate à un accepteur possédant un groupe partant en position 8 et une triple liaison en
position 9-10 (figure 7, à gauche). Une nouvelle approche serait d’additionner la portion vinylique, sous
forme d’un réactif de cuprate, à un substrat comportant le nucléofuge X sur carbone 10 et la triple
liaison entre les carbones 8 et 9 (figure 7, au centre). Cette stratégie, en plus de sérieusement raccourcir
le nombre d’étapes, permettrait d’éviter certains problèmes encourus lors de la formation de la portion
vinylique (c.f. section 4.3).
OH
OR
X
MeX
211Me
HO
RXX
210
O
Nouvelle routeAncienne route
1
4
10
9
8
14
1318
1 10 9 814
1
10
9 8
14
Squelette picrasane Figure 7. Formation de l’allène par l’addition de réactifs de cuprate.
Le vinylallène désiré 212 pourrait provenir de l’addition d’un propénylcuprate sur un époxyde
propargylique tel que 213 (schéma 63). Les précédents concernant les additions de vinylcuprates sur ce
genre de substrats seront présentés à la section 5.2. L’oxyrane 213 serait directement disponible par
époxydation de l’ényne 214, une réaction offrant normalement d’excellentes chimiosélectivités avec la
méthode de Shi (c.f. section 4.1.2).143 En une seule étape, cette époxydation permettrait d’établir la
stéréochimie de l’alcool en C1 ainsi que de contrôler la chiralité de l’allène par ouverture SN2’ de
l’oxyrane par le cuprate. L’usage de ce type d’époxyde est l’objet d’une brève revue à la section 5.3. De
son côté, l’ényne 214 proviendrait du couplage de Sonogashira entre l’halogénure vinylique 215 et
l’alcyne 126.
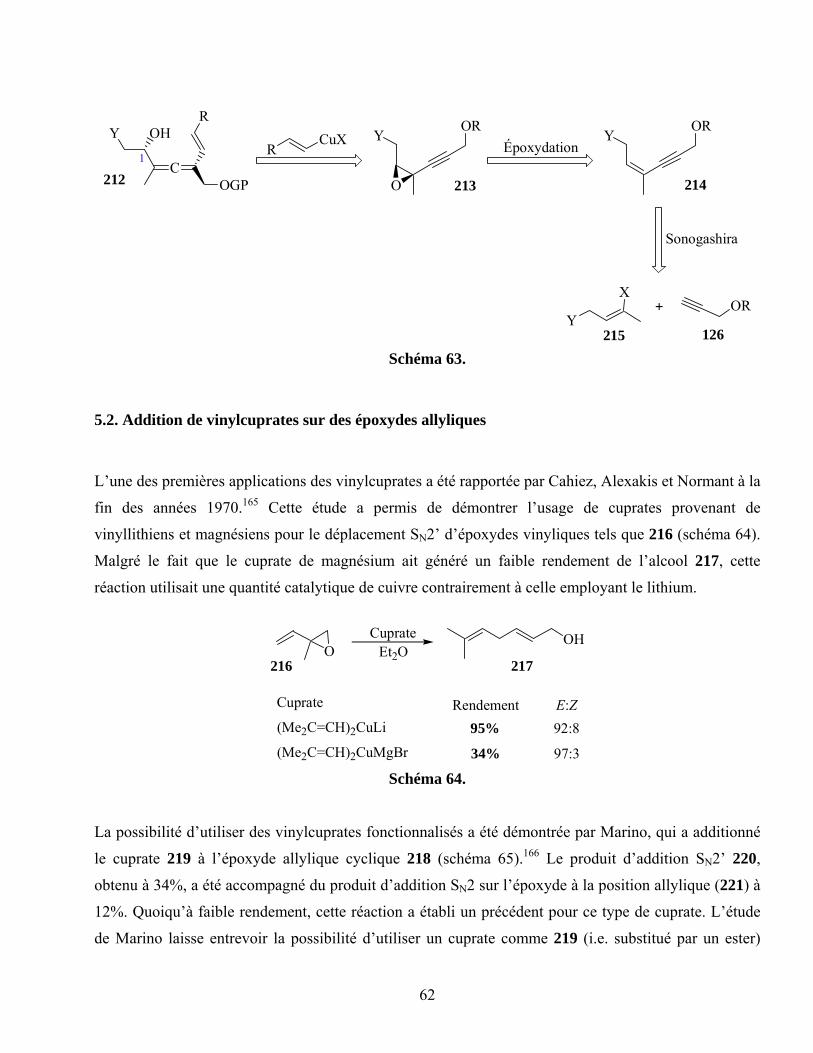
62
C
OH
OGP
YR
RCuX
OR
O
Y
212 213
Y
X
215
ORY
214
126
OR
Époxydation
+
Sonogashira
1
Schéma 63.
5.2. Addition de vinylcuprates sur des époxydes allyliques
L’une des premières applications des vinylcuprates a été rapportée par Cahiez, Alexakis et Normant à la
fin des années 1970.165 Cette étude a permis de démontrer l’usage de cuprates provenant de
vinyllithiens et magnésiens pour le déplacement SN2’ d’époxydes vinyliques tels que 216 (schéma 64).
Malgré le fait que le cuprate de magnésium ait généré un faible rendement de l’alcool 217, cette
réaction utilisait une quantité catalytique de cuivre contrairement à celle employant le lithium.
O
(Me2C=CH)2CuLi
(Me2C=CH)2CuMgBr
Et2O216
OH
217
Cuprate
Cuprate Rendement E:Z95%
34%
92:8
97:3 Schéma 64.
La possibilité d’utiliser des vinylcuprates fonctionnalisés a été démontrée par Marino, qui a additionné
le cuprate 219 à l’époxyde allylique cyclique 218 (schéma 65).166 Le produit d’addition SN2’ 220,
obtenu à 34%, a été accompagné du produit d’addition SN2 sur l’époxyde à la position allylique (221) à
12%. Quoiqu’à faible rendement, cette réaction a établi un précédent pour ce type de cuprate. L’étude
de Marino laisse entrevoir la possibilité d’utiliser un cuprate comme 219 (i.e. substitué par un ester)

63
pour la synthèse de différentes quassinoïdes qui possèdent un ester méthylique en C13, par exemple la
brucéantine (3) (c.f. figure 2).
ORCu
CO2Me218 219
OHMeO2C
220 221
O
O
12%
+Et2O, -78 °C
+
34% Schéma 65.
Plusieurs variantes ont été développées pour l’addition de vinylcuprates. Parmi celles-ci, notons l’ajout
de phosphines pour stabiliser l’organocuivre167 ou de chlorure de triméthylsilyle (TMSCl) pour activer
le cuprate168 ainsi que la formation du couple cuivre-zinc afin de générer une espèce plus réactive.169
5.3. Déplacement SN2’ d’époxydes propargyliques
Outre nos travaux (c.f. chapitre 4), peu de groupes de recherche ont utilisé des époxydes acétyléniques
pour la synthèse d’α-allénols. D’autres études visant ces produits font état de l’addition de réactifs de
cuprate à des oxyranes propargyliques tels que 222, générant ainsi les allènes correspondants 223
(schéma 66).170
R1
R2 OR3
R4
RMgBr
222
CuIC
R R2
HO
R3
R4R1
R1, R2, R3 et R4 = H, alkyles ou phényleR = Me, Et, nBu, Ph
30-90% 223
Schéma 66.
En ce qui concerne la synthèse de vinylallènes, les auteurs rapportent que l’addition de la portion
vinylique à des oxyranes tels que 224 nécessite l’usage de palladium.171 De cette manière, divers
zinciques ont pu être additionnés à des époxydes propargyliques (schéma 67). Le mécanisme postulé
débute par l’addition de palladium(0) sur l’alcyne, procurant l’alkoxyde allénique 225. La

64
transmétallation zinc-palladium est ensuite suivie du couplage réductif de l’intermédiaire 226, générant
ainsi l’allène 227 après neutralisation du milieu réactionnel.
O
224
CL3Pd
-O
RZnCl
225 226
CR
HO
CL2Pd
OZnCl
R
227
Me3SiC CR = Ph, H2C=CH,
1) RZnCl, Pd(PPh3)4
Pd0Pd0
2) H3O+
Conversions >98%
Schéma 67.
En plus d’affirmer que la réaction ne procède pas sans palladium ou avec des vinyllithiens, l’article
stipule que les réactifs de cuprates correspondants (même en présence de palladium), sont
complètement inertes face à l’époxyde acétylénique 224. Une étude modèle a donc été entamée afin de
vérifier ces résultats et de valider notre nouvelle stratégie de synthèse (c.f. section 5.1).
5.4. Étude modèle
Cette courte étude a débuté par l’addition des dérivés lithiés des alcools propargyliques 126i et 126c à
la chloroacétone, procurant les chlorohydrines avec des rendements de 95% pour R=TBS (228) et de
73% pour R=MOM (229) (schéma 68). Les époxydes propargyliques résultants ont par la suite été
formés sous l’action du méthoxyde de sodium avec des rendements quantitatifs. Ces époxydes (230 et
231) ont aussi été synthétisés, avec un rendement moindre, en une seule étape à partir des éthers
propargyliques 126i et 126c par l’ajout du méthoxyde de sodium au mélange réactionnel après
l’addition des acétylures sur la chlorocétone.

65
OR
ClO
OR
ClOH
MeONaEt2O
OR
O
i) n-BuLi, THF
ii)
R = TBS 126iR = MOM 126c
R = TBS 230, 100%R = MOM 231, 99%
R = TBS 228, 95%R = MOM 229, 73%
Schéma 68.
Sous l’action du bromure de propénylmagnésium complexé au cyanure de cuivre, tel que représenté au
schéma 69, les adduits 230 et 231 ont mené aux vinylallènes 232 (99%) et 233 (80%). Quoique surpris
par la stéréochimie cis de l’unité propényle des vinylallènes obtenus, l’isomérisation de ce type
d’organomagnésiens (lors de leur formation à partir de l’halogénure vinylique et de magnésium
métallique) est un phénomène bien documenté.172 Curieusement, cette isomérisation n’a pas lieu lors de
la préparation des dérivés lithiés à partir de lithium métallique.173 Heureusement, l’addition de
vinylcuprates fabriqués à partir de vinyllithiens a été aussi fructueuse (vide infra).
OR
O
MgBr, CuCNC
HO
ORR = TBS 230R = MOM 231
THF, -10 °C
R = TBS 232, 99%R = MOM 233, 80%
Schéma 69.
Ayant testé notre nouvelle stratégie pour la synthèse de vinylallènes tétrasubstitués, nous avons
concentré nos efforts sur le développement d’une séquence synthétique, potentiellement
énantiosélective, conduisant à un intermédiaire tétracyclique fonctionnalisé (i.e. composé 148, schéma
38).

66
CHAPITRE 6 : SÉQUENCE SYNTHÉTIQUE VERS UN β-BROMOVINYLALLÈNE
6.1. Séquence réactionnelle générale
Suite à nos échecs avec le triméthylsilyle, nous avons décidé d’utiliser une nouvelle unité de promotion
de clivage du connecteur de la première CDA. Un bromure a été choisi à cet effet dans l’espoir
d’effectuer la rupture du lien C-O en position 1 par une élimination-β sous des conditions de type
Barbier (par la génération in situ du réactif de Grignard correspondant).174 Une séquence réactionnelle
suivant le plan décrit au schéma 63 a alors été entamée.175 Cette voie synthétique a été explorée à tour
de rôle avec différents groupes protecteurs jusqu’à l’obtention de celui qui nous permettrait de
poursuivre jusqu’au squelette picrasane.
Le lecteur doit retenir qu’outre l’iodure 234176 et les alcynes 126, aucun produit de ce schéma n’est
stable et tous doivent être utilisés pour la réaction subséquente dès leur isolation. Les échantillons ne
peuvent être conservés longtemps, même au congélateur, ce qui a considérablement nuit aux réactions
suivantes. Le rendement de chaque réaction pour chacun des groupes protecteurs R1 est résumé sous
forme tabulaire (tableau 8) afin d’alléger le texte et le schéma.
126
R2
OR1
239
I
OH
234
CO2MeO
OR1R2
CO2MeBr
240
C
OH
OR1
Br
R2
OR1HOR2
235
Cu(CN)Li
CH2Cl2
OR1
O
BrR2
OR1BrR2
CH2Cl2
237
236
+
PdCl2(PPh3)2CuI, AsPh3pyrrolidineTHF, 23 °C
PPh3, CBr4
m-CPBA
THF, -95 °CNMM, Et2O
238
Schéma 70.

67
Initialement, le couplage de Sonogashira entre l’iodure vinylique 234 connu de la littérature et l’alcyne
126a a conduit à la formation d’importantes quantités du dimère de l’alcyne.131 L’addition de
triphénylarsine ainsi que l’optimisation de différents paramètres de la réaction (l’amine, la source de
palladium et la stoechiométrie de l’iodure de cuivre) ont permis d’obtenir les énynes 235 avec de bons
rendements, sauf dans le cas de l’acétate 126f qui n’a engendré que 67% de 235f malgré tous nos
efforts (entrée 6). Ce faible rendement pourrait être une conséquence de la formation d’un complexe π–
allyle avec l’acétate propargylique (vide supra).
Par la suite, la substitution nucléophile des alcools allyliques 235 par un bromure a procuré les adduits
236.177 Les rendements du déplacement SN2 ont été supérieurs à 80% pour l’acétate 235f (entrée 6) et le
dioxolane 235h (entrée 8), plus faibles pour l’éther MOM 235c (50%, entrée 3) et le diéthylacétal 235g
(70%, entrée 7) et quantitatifs pour les autres groupes protecteurs utilisés dans cette séquence.
L’époxydation des énynes 236 avec le m-CPBA a généré les oxyranes 237 avec des rendements bruts
de 92 à 100%. Les époxydes ne résistaient pas à la purification sur gel de silice, donnant des
rendements de moins de 40%.
Ces époxydes ont ensuite été placés en présence du réactif de cuprate 238 (formé du E-
propényllithium178 et du CuCN) pour effectuer l’ouverture SN2’ et anti de l’époxyde propargylique. La
réaction du vinylcuprate 238 et des oxyranes 237 effectuée à -95 °C a permis d’isoler les vinylallènes
239a (TBDPS), 239e (benzyle) et 239h (dioxolane) comportant l’unité propényle de géométrie trans
exclusivement (c.f. section 5.4). Ceux-ci ont immédiatement été utilisés pour la CDA subséquente pour
des raisons d’instabilité. À notre surprise, cette réaction a promu la décomposition des époxydes
comportant un PMB (237b), un MOM (237c), un SEM (237d) et un acétal diéthylique (237g).
L’addition de l’oxyrane 237f (acétate) au cuprate 238 a procuré l’allène désiré 239f accompagné d’un
mélange complexe de produits résultant de la polyaddition de l’organocuivre (l’allène 239f comporte un
acétate allylique propice à un second déplacement nucléophile).
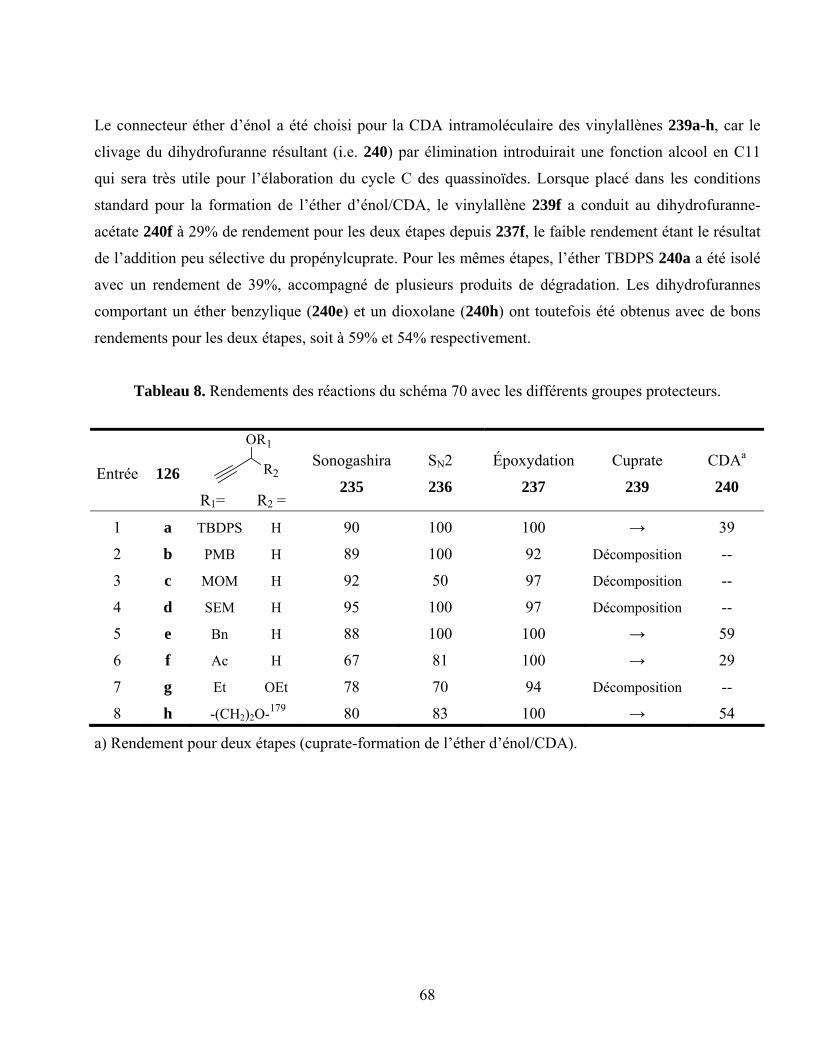
68
Le connecteur éther d’énol a été choisi pour la CDA intramoléculaire des vinylallènes 239a-h, car le
clivage du dihydrofuranne résultant (i.e. 240) par élimination introduirait une fonction alcool en C11
qui sera très utile pour l’élaboration du cycle C des quassinoïdes. Lorsque placé dans les conditions
standard pour la formation de l’éther d’énol/CDA, le vinylallène 239f a conduit au dihydrofuranne-
acétate 240f à 29% de rendement pour les deux étapes depuis 237f, le faible rendement étant le résultat
de l’addition peu sélective du propénylcuprate. Pour les mêmes étapes, l’éther TBDPS 240a a été isolé
avec un rendement de 39%, accompagné de plusieurs produits de dégradation. Les dihydrofurannes
comportant un éther benzylique (240e) et un dioxolane (240h) ont toutefois été obtenus avec de bons
rendements pour les deux étapes, soit à 59% et 54% respectivement.
Tableau 8. Rendements des réactions du schéma 70 avec les différents groupes protecteurs.
Entrée 126
R1= R2 =
Sonogashira
235
SN2
236
Époxydation
237
Cuprate
239
CDAa
240
1 a TBDPS H 90 100 100 → 39
2 b PMB H 89 100 92 Décomposition --
3 c MOM H 92 50 97 Décomposition --
4 d SEM H 95 100 97 Décomposition --
5 e Bn H 88 100 100 → 59
6 f Ac H 67 81 100 → 29
7 g Et OEt 78 70 94 Décomposition --
8 h -(CH2)2O-179 80 83 100 → 54
a) Rendement pour deux étapes (cuprate-formation de l’éther d’énol/CDA).
R2
OR1

69
6.2. Séquence avec les substrats ayant un TBDPS comme groupe protecteur
Grâce à sa synthèse efficace en cinq étapes avec un rendement global de 35%, le dihydrofuranne 240a a
été l’objet d’une attention particulière dans le but de cliver le connecteur en C-1. Malheureusement,
tous nos efforts pour effectuer la réaction entre le bromoéther 240a et différents métaux (Mg, Zn, Li)
n’ont procuré que du produit de départ intact en plus de plusieurs produits de décomposition (schéma
71). Il en va de même pour la déprotection de l’alcool allylique qui, sous conditions acides
(HCl/MeOH, TFA, H2SO4, CSA) ou en présence de fluorure (TBAF, HF/pyridine), n’a généré que des
traces de l’alcool désiré 242 accompagné d’une important quantité de produits de dégradation.180
OH
CO2MeOBr
OTBDPS
CO2MeOBr
240a 242OTBDPS
CO2MeHO
241
Mg, Li, Zn H+ ou F-
Schéma 71.
Face à ces échecs quant à la déprotection de l’alcool allylique de 240a ainsi qu’à notre incapacité à
effectuer le clivage de l’unité dihydrofuranne, notre attention s’est tournée vers un autre groupe
protecteur. Malheureusement, les groupements p-méthoxybenzyle (126b), éther méthoxyméthyle
(126c) ou éther triméthylsilyléthoxyméthyle (126d) n’ont pas permis d’accéder aux allènes
correspondant 239b-d. C’est alors que nous avons tourné notre attention vers un éther benzylique
(126e).

70
6.3. Séquence avec les substrats ayant un benzyle comme groupe protecteur
Synthétisé en cinq étapes avec un rendement global de 52%, le dihydrofuranne 240e s’est avéré instable
à l’air et à la chaleur, le benzofuranne 243 étant isolé lors de la purification du produit en vue de son
utilisation (schéma 72). Ce produit secondaire constituant un sérieux problème, les essais de clivage de
l’éther du dihydrofuranne 240e ont donc été réalisés immédiatement après son isolation.
OBn
CO2MeOBr
240e 243 OBn
CO2MeOBr
Oxydationair
Schéma 72.
Dans le but de générer le bromure de magnésium correspondant, 240e a été traité avec du magnésium,
mais curieusement seul l’acide 245 a été obtenu (entrée 1, tableau 9). Cet acide carboxylique est peut
être le résultat d’une hydrolyse de l’ester méthylique catalysée par l’oxyde de magnésium présent dans
le milieu réactionnel. L’usage de magnésium de Rieke181 n’a pas permis d’obtenir l’alcool 244, mais a
seulement promu la décomposition partielle du dihydrofuranne 240e (entrée 2). De plus, l’emploi de
métaux alcalins tels que le sodium et le potassium n’a procuré que du produit de départ ou de la
décomposition (entrées 3-5). Les conditions développées par Guindon pour effectuer le clivage d’α-
halogénoéthers n’ont engendré que la décomposition du dihydrofuranne 240e (entrée 6) tandis que
l’usage d’iodure de samarium (SmI2) a laissé le produit de départ intact (entrée 7). Si les premières
tentatives de clivage via un zincique n’ont pas donné de résultats intéressants (entrée 8), l’ajout d’acide
acétique à cette réaction a permis de finalement isoler le triène désiré 244 avec un rendement de 32%
(entrée 9), soulignant ainsi l’importance d’activer l’éther. Une optimisation des conditions de réaction a
conduit à l’utilisation du chlorure d’ammonium qui a procuré l’alcool allylique 244 avec un rendement
de 77% (entrée 10).
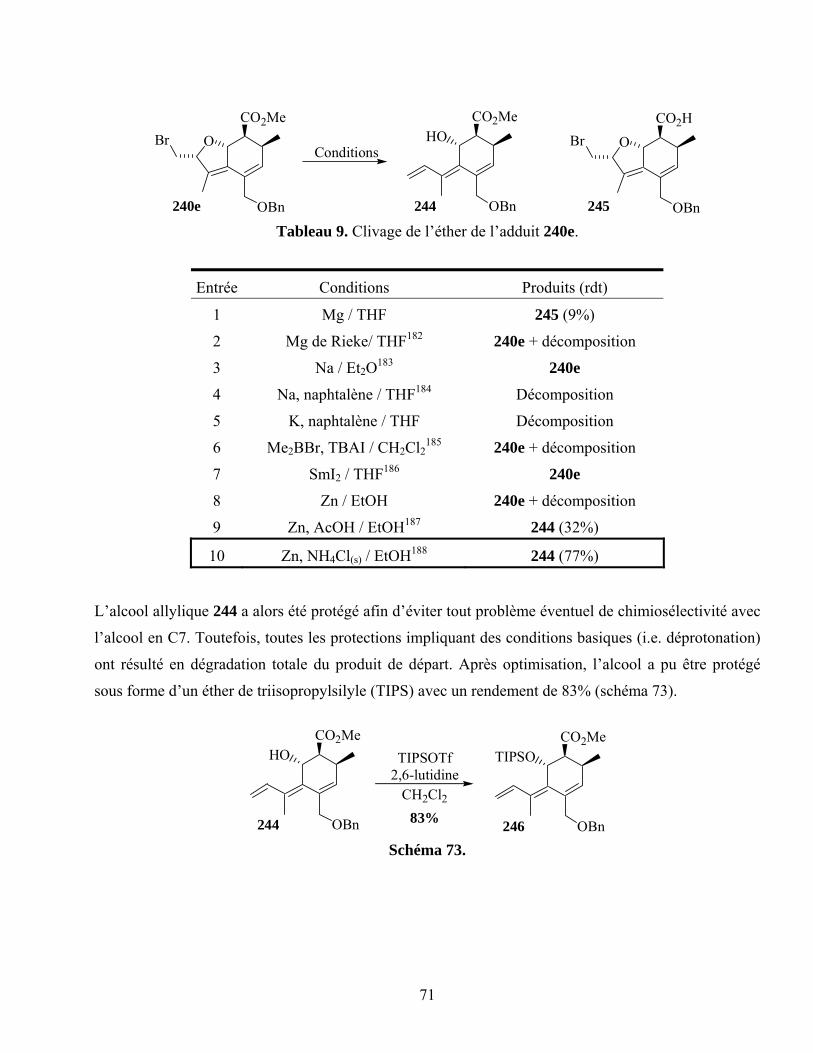
71
OBn
CO2MeOBr
240e 244 OBn
CO2MeHO
245 OBn
CO2HOBr
Conditions
Tableau 9. Clivage de l’éther de l’adduit 240e.
Entrée Conditions Produits (rdt)
1 Mg / THF 245 (9%)
2 Mg de Rieke/ THF182 240e + décomposition
3 Na / Et2O183 240e
4 Na, naphtalène / THF184 Décomposition
5 K, naphtalène / THF Décomposition
6 Me2BBr, TBAI / CH2Cl2185 240e + décomposition
7 SmI2 / THF186 240e
8 Zn / EtOH 240e + décomposition
9 Zn, AcOH / EtOH187 244 (32%)
10 Zn, NH4Cl(s) / EtOH188 244 (77%)
L’alcool allylique 244 a alors été protégé afin d’éviter tout problème éventuel de chimiosélectivité avec
l’alcool en C7. Toutefois, toutes les protections impliquant des conditions basiques (i.e. déprotonation)
ont résulté en dégradation totale du produit de départ. Après optimisation, l’alcool a pu être protégé
sous forme d’un éther de triisopropylsilyle (TIPS) avec un rendement de 83% (schéma 73).
OBn
CO2MeHO
244
CH2Cl2
246 OBn
CO2MeTIPSOTIPSOTf
2,6-lutidine
83%
Schéma 73.

72
La déprotection de l’alcool primaire en C7 s’est avérée un sérieux défi. Si l’éther benzylique de 246 a
résisté aux conditions d’hydrogénolyse, le triène, lui, s’est fait réduire (tableau 10, entrées 1-3).
Curieusement, l’analyse des produits réactionnels nous a permis de conclure que l’alcène exocyclique
tétrasubstitué était le plus réactif des trois alcènes, des produits résultant de son hydrogénation étant
observés. Nous croyons que le relâchement de la tension est à l’origine de ce phénomène. En effet, la
double liaison exocyclique dans les composés tels que 246 est très tendue et subit souvent une torsion
de plusieurs degrés.61a
L’éther 246 s’est aussi totalement décomposé sous l’action d’acides de Lewis (entrées 4-5) ainsi que
sous des conditions oxydantes (entrées 6-7). La formation de l’ester benzoïque par oxydation de la
position benzylique avec du IBX a résulté en un mélange complexe d’aldéhydes (entrée 8). Désirant
obtenir un aldéhyde à cette position (C7), nous pensions que l’oxydation de l’éther allylique 246 avec
un réactif de type oxyde métallique servirait notre cause.189 Suite à une optimisation exhaustive, la
réaction du triène 246 avec le dioxyde de sélénium (SeO2) a procuré l’aldéhyde désiré 247 ainsi que le
produit d’isomérisation de l’alcène exocyclique 248 dans un ratio de 2.5 : 1 avec un rendement de 30%
(entrée 9). Des expériences de NOESY ont permis d’exclure tout doute concernant l’identité de ces
produits. Peu reproductibles, les rendements d’oxydation ont varié de 12 à 57% tandis que les ratios
247/248 variaient de 1 à 5 : 1. Cette réaction n’a toutefois pu être portée à un rendement plus élevé par
l’utilisation d’autres oxydants allyliques (entrée 10-12), par exemple l’oxygène singulet n’a que promu
l’isomérisation du produit de départ (i.e. 249, entrée 11).
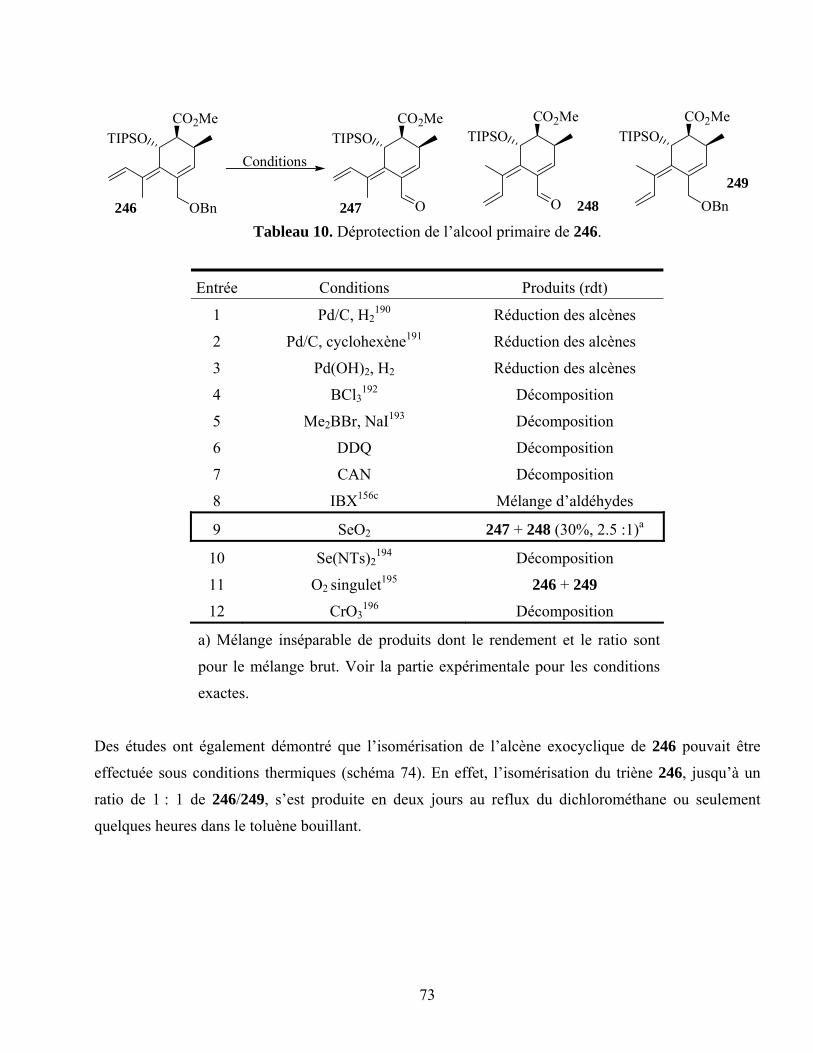
73
OBn
CO2MeTIPSO
246 247 O
CO2MeTIPSO
O
CO2MeTIPSO
248 OBn
CO2MeTIPSO
249Conditions
Tableau 10. Déprotection de l’alcool primaire de 246.
Entrée Conditions Produits (rdt)
1 Pd/C, H2190 Réduction des alcènes
2 Pd/C, cyclohexène191 Réduction des alcènes
3 Pd(OH)2, H2 Réduction des alcènes
4 BCl3192 Décomposition
5 Me2BBr, NaI193 Décomposition
6 DDQ Décomposition
7 CAN Décomposition
8 IBX156c Mélange d’aldéhydes
9 SeO2 247 + 248 (30%, 2.5 :1)a
10 Se(NTs)2194 Décomposition
11 O2 singulet195 246 + 249
12 CrO3196 Décomposition
a) Mélange inséparable de produits dont le rendement et le ratio sont
pour le mélange brut. Voir la partie expérimentale pour les conditions
exactes.
Des études ont également démontré que l’isomérisation de l’alcène exocyclique de 246 pouvait être
effectuée sous conditions thermiques (schéma 74). En effet, l’isomérisation du triène 246, jusqu’à un
ratio de 1 : 1 de 246/249, s’est produite en deux jours au reflux du dichlorométhane ou seulement
quelques heures dans le toluène bouillant.

74
OBn
CO2MeTIPSO
246 OBn
CO2MeTIPSO
249 Schéma 74.
Quoique déçus par le faible rendement de 247, nous avons décidé de tenter la cycloaddition d’hétéro
DA sur le mélange de produits (schéma 75). La réaction de l’aldéhyde α,β-insaturé selon nos conditions
standard a mené à l’isolation d’un mélange inséparable de trois produits de cycloaddition avec un bon
bilan de masse de 65%. L’analyse poussée des données spectrales a mis à jour une isomérisation de
l’alcène exocyclique des produits de réaction, le ratio passant de 4 : 1 pour le produit de départ à 1 : 1
dans le cycloadduit. Ce résultat implique que l’isomérisation de l’oléfine tétrasubstituée de 247 est
catalysée par l’ytterbium(III), signifiant que la CHDA devait être effectuée avant le clivage du
connecteur (avant la formation du triène). Malheureusement, nous n’avons pas réussi à séparer les trois
produits de la CHDA par des techniques physiques (i.e. chromatographies sur silice, HPLC) ou en les
dérivant chimiquement. Une modification du plan synthétique était nécessaire pour éviter ce problème
majeur. Dans le but d’inverser les étapes de la séquence, la déprotection de l’alcool primaire de 240e a
été tentée mais, sous une grande variété de conditions, nos efforts se sont tous soldés par des échecs.
247 O
CO2MeTIPSO
248O
CO2MeTIPSO
+Yb(fod)3
EVE3 produits de CHDA
4 : 1
1 : 1 au niveau de l'alcène exocyclique
Schéma 75.
L’isomérisation de l’alcène exocyclique étant probablement catalysée par la complexation de
l’ytterbium sur le triène, nous pensions qu’une modification du triène pourrait peut être conduire à une
espèce plus stable aux conditions de CHDA. En ce sens, la fonctionnalisation de l’alcène terminal du
triène 246 a été investiguée (tableau 11). Sous l’action de réactifs classiques d’hydroboration, seuls de
la décomposition et du produit non-réagi ont été obtenus (entrées 1-4). L’emploi du catalyseur de
Wilkinson pour faciliter l’addition du catécholborane, une hydroboration développée par Evans, a elle

75
aussi conduit à une dégradation complète du produit de départ (entrée 5). Il en va de même pour les
hydrures métalliques à base de zirconium (réactif de Schwartz) et de hafnium (réactif de Tolstikov) qui
ont soit produit de la décomposition, soit laissé le composé 246 intact.
OBn
CO2MeTIPSO
246 250 OBn
CO2MeTIPSO
XConditions
Tableau 11. Fonctionnalisation de l’alcène terminale de 246.
Entrée Conditions Produits (rdt)
1 BH3•THF Décomposition
2 Cy2BH Décomposition + 246
3 9-BBN Décomposition + 246
4 Catécholborane 246
5 Catécholborane, (Ph3P)3RhCl197 Décomposition
6 Cp2ZrHCl198 Décomposition
7 Cp2HfHCl199 246
Compte tenu des problèmes encourus avec les substrats possédant l’éther benzylique, ce type de
substrats a été abandonné au profit de substrats ayant un autre groupe protecteur qui pourrait être enlevé
dans des conditions plus douces. Les résultats précédents ayant mis en valeur la nécessité d’enlever ce
groupe dans des conditions autres que réductrices ou oxydantes, la séquence du schéma 70 a été reprise
avec un acétate masquant l’alcool propargylique 126f. Malgré le fait que le dihydrofuranne 240f a tout
de même été obtenu, cette séquence n’a pas été davantage explorée, le rendement de la cupration étant
faible et peu reproductible (i.e. entre 10 et 30%). La réussite de cette voie synthétique sur grande
échelle est primordiale pour la synthèse éventuelle d’une quassinoïde. Nous avons donc poursuivi nos
explorations des différents groupes protecteurs pour finalement en arriver au dioxolane comme groupe
protecteur désiré.

76
6.4. Séquence comportant un dioxolane comme groupe protecteur
La séquence synthétique avec les substrats ayant un dioxolane en C7 (i.e. 126h178) a procuré le
dihydrofuranne 240h avec un rendement global de 36% pour les cinq étapes (c.f. schéma 70 et tableau
8). Contre toute attente, l’aldéhyde 251 a été démasqué sous l’action d’acide chlorhydrique avec un
rendement quantitatif (schéma 76). La cycloaddition d’hétéro Diels-Alder de cet aldéhyde α,β-insaturé
avec l’éther éthylvinylique (EVE) a permis d’isoler l’adduit de CDA 252 avec un rendement de 76%
sur une échelle de 1.5 grammes. Ce diène tricyclique contenant trois cycles et six centre chiraux a été
obtenu en seulement 7 étapes avec un rendement global de 27%, illustrant bien la puissance de notre
séquence synthétique.
CO2Me
OBr
O O
HClconc.
240h 251
CO2MeOBr
O252
CO2MeOBr
O
H
OEt
Yb(fod)3EVEacétone-H2O
100% 76%
Schéma 76.
Tel qu’imaginé au schéma 38 (route B), le diène 252 a été l’objet d’études pour la CDA
intermoléculaire prévue pour former le cycle B. Ces résultats sont présentés au chapitre 7, avec les
essais pour la troisième CDA de divers autres substrats. Nous avons aussi tenté de cliver l’éther du
dihydrofuranne 252. Procurant exclusivement de la décomposition sous les conditions développées
pour le composé 240e, l’élimination-β de l’éther de 252 a été accomplie en modifiant les paramètres
expérimentaux et le triène 253 a été isolé avec un rendement de 65% (schéma 77). Le rendement a pu
être augmenté en effectuant la protection de l’alcool secondaire 253 sur le produit brut de la réaction
précédente, le triène 254 étant alors obtenu avec un rendement de 77% pour les deux étapes.
À ce point, nous avons finalement pu sécuriser un intermédiaire stable (254) pouvant être entreposé
durant plus d’une journée sans occasionner une importante dégradation, tous les produits de 235 au
triène 253 ne pouvant être conservés plus de quelques heures, même au congélateur. Ce composé stable
en mains, la troisième CDA a été investiguée intensément.
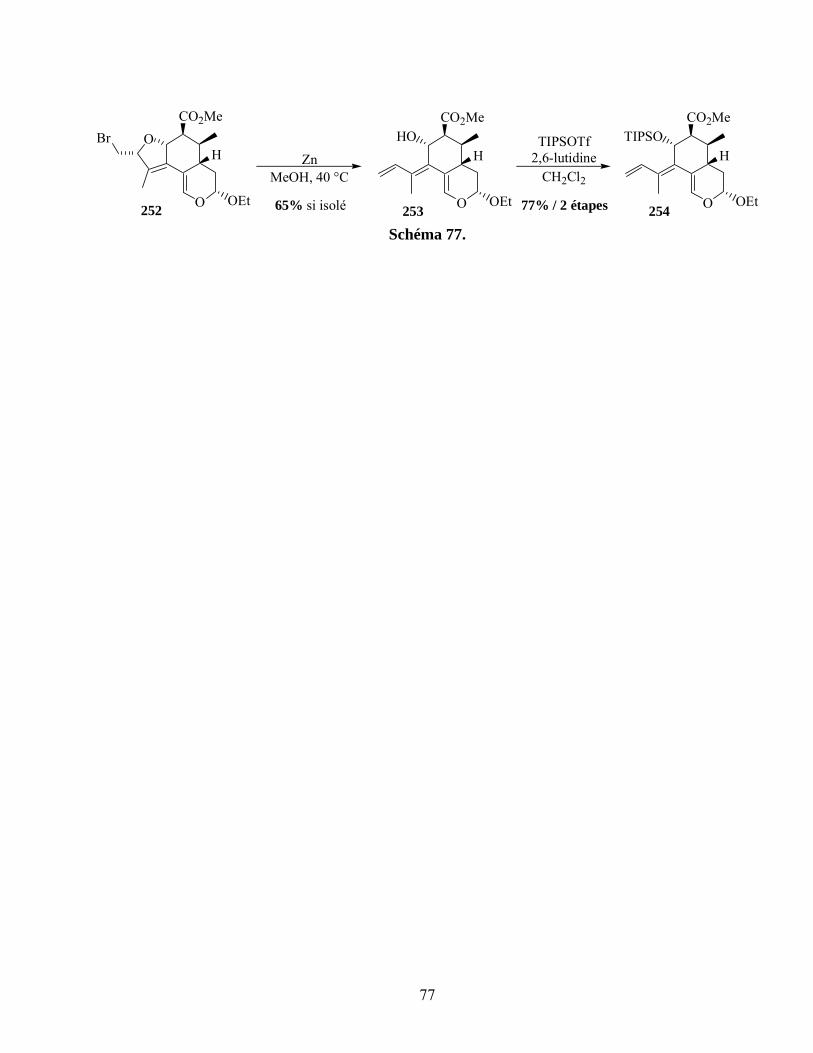
77
CO2MeOBr
O
H
OEt
Zn
252
MeOH, 40 °C
253
CO2Me
O
H
OEt
HO
CH2Cl2
254
CO2Me
O
H
OEt
TIPSOTIPSOTf2,6-lutidine
77% / 2 étapes65% si isolé Schéma 77.

78
CHAPITRE 7 : ÉTUDES DE LA TROISIÈME CYCLOADDITION DE DIELS-ALDER
À ce point, nous nous sommes concentrés sur des expériences visant la formation du cycle B par une
cycloaddition de Diels-Alder intermoléculaire (c.f. route B, schéma 38). Des approches parallèles
utilisant un DA intramoléculaire font l’objet d’études menées par Stéphane Perreault et Amélie Dion
(route A).
7.1. Cycloaddition intermoléculaire du diène 252
Grâce à sa synthèse efficace (7 étapes, 27%), l’adduit tricyclique 252 a pu être l’objet d’études de
réactivité pour la troisième cycloaddition de Diels-Alder (tableau 12). À notre surprise, la réaction de ce
diène avec le tétracyanoéthylène a généré un mélange complexe de produits d’aromatisation (entrée 1).
Sous l’influence de l’anhydride maléique ou du crotonate de méthyle, 252 n’a pas réagi ou a seulement
produit de la décomposition lorsque le mélange réactionnel a été porté à une plus haute température
(entrées 2-3). Il en va de même pour le diénophile 256200 qui, en solution ou film, n’a pas procuré
l’adduit tricyclique désiré, mais seulement de la décomposition (entrées 4-5). Le résultat a été identique
lorsque la CDA a été effectuée sous la catalyse d’acides de Lewis (entrées 6-7).
La décomposition obtenue pourrait être attribuée à l’instabilité générale du diène 252, celui-ci ne
pouvant pas supporter les conditions réactionnelles pour effectuer une CDA de façon intermoléculaire
(c.f. section I.2.2). À la lumière de ces résultats, nous avons tourné notre attention vers le composé
suivant dans la séquence réactionnelle, soit le diène 253.
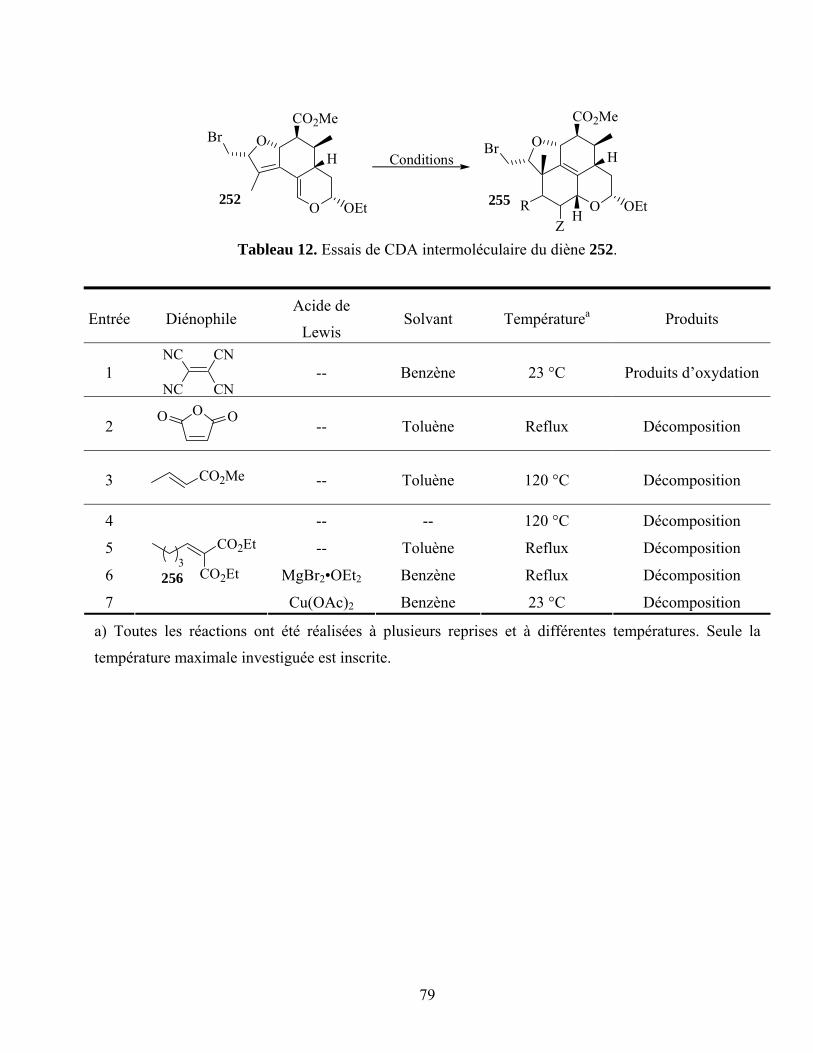
79
Conditions
252 O
OBrCO2Me
OEt O
O
CO2Me
OEt
Br
RZ
H H
H255
Tableau 12. Essais de CDA intermoléculaire du diène 252.
Entrée Diénophile Acide de
Lewis Solvant Températurea Produits
1
-- Benzène 23 °C Produits d’oxydation
2
-- Toluène Reflux Décomposition
3
-- Toluène 120 °C Décomposition
4 -- -- 120 °C Décomposition
5 -- Toluène Reflux Décomposition
6 MgBr2•OEt2 Benzène Reflux Décomposition
7
Cu(OAc)2 Benzène 23 °C Décomposition
a) Toutes les réactions ont été réalisées à plusieurs reprises et à différentes températures. Seule la
température maximale investiguée est inscrite.
NC
NC CN
CNOO O
CO2Et
CO2Et2563
CO2Me

80
7.2. Cycloaddition intermoléculaire du diène 253
Une CDA intermoléculaire sélective avec le diène en C7-C8-C9-C10 devrait être possible en
considération des effets stériques du composé 253. En effet, l’approche du diénophile sur le diène
comprenant l’alcène terminal devrait être défavorisée en raison de la répulsion stérique due aux
substituants en C11 et C12 du cycle C, empêchant donc ce diène d’adopter une conformation cisoïde
nécessaire à la CDA.
Le triène 253 a alors été soumis à diverses conditions réactionnelles pour effectuer la troisième CDA
(tableau 13). Tel qu’avec le diène 252, le triène 253 s’est décomposé lorsque placé avec le
tétracyanoéthylène (entrée 1), l’anhydride maléique (entrée 2) et le crotonate de méthyle (entrées 3-4).
Le même résultat a été obtenu avec des acides de Lewis forts (entrées 5-6). L’utilisation d’un
connecteur temporaire à base de magnésium, tel que développé par Barriault, n’a également pas conduit
au produit tricyclique désiré 257 (entrée 7).77 Finalement, le composé 253 s’est dégradé lors des
réactions avec le diénophile 256 en conditions thermiques ainsi qu’en présence d’acides de Lewis
(entrées 8-11). Ces résultats similaires en tout point à ceux obtenus avec le diène 252 peuvent
également être attribués à l’instabilité du produit de départ, celui-ci préférant se dégrader plutôt que
produire un adduit tricyclique tel que 257. Le dérivé portant un groupe protecteur sur l’oxygène en C11
(i.e. 254) a alors été sujet à une étude semblable.
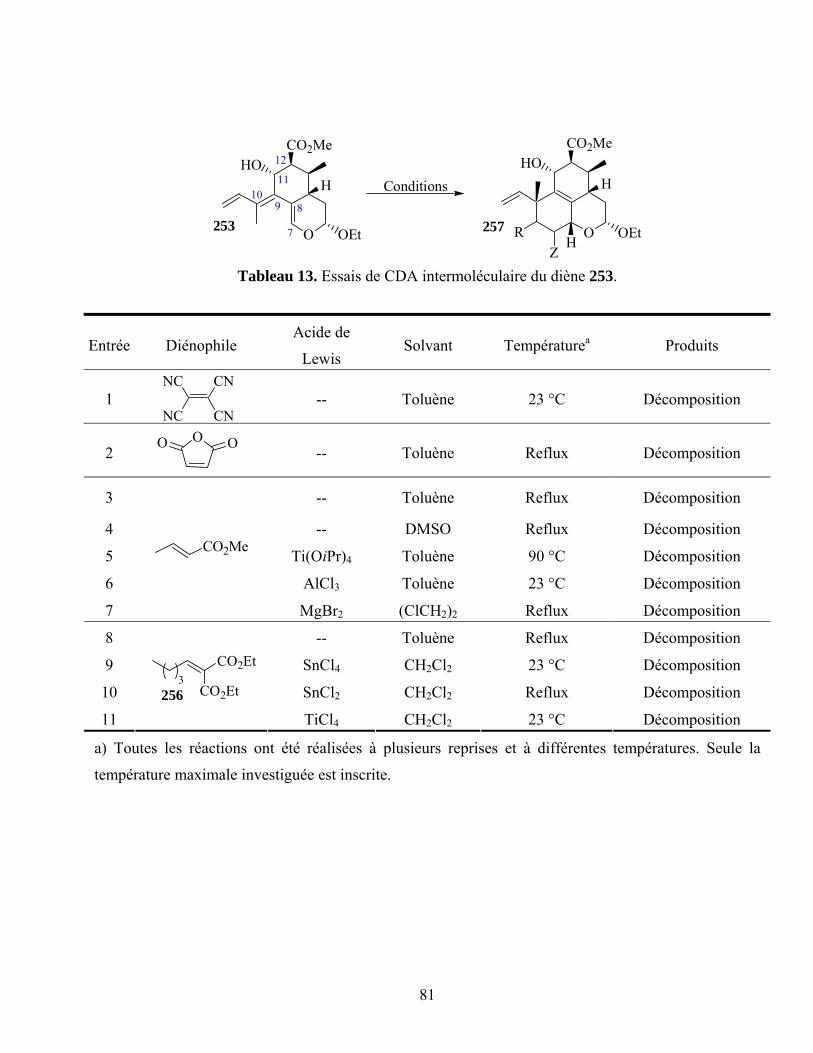
81
O
CO2Me
OEt
HHO
253 257 O
CO2Me
OEtRZ
H
H
HOConditions
7
8910
11
12
Tableau 13. Essais de CDA intermoléculaire du diène 253.
Entrée Diénophile Acide de
Lewis Solvant Températurea Produits
1
-- Toluène 23 °C Décomposition
2
-- Toluène Reflux Décomposition
3 -- Toluène Reflux Décomposition
4 -- DMSO Reflux Décomposition
5 Ti(OiPr)4 Toluène 90 °C Décomposition
6 AlCl3 Toluène 23 °C Décomposition
7
MgBr2 (ClCH2)2 Reflux Décomposition
8 -- Toluène Reflux Décomposition
9 SnCl4 CH2Cl2 23 °C Décomposition
10 SnCl2 CH2Cl2 Reflux Décomposition
11
TiCl4 CH2Cl2 23 °C Décomposition
a) Toutes les réactions ont été réalisées à plusieurs reprises et à différentes températures. Seule la
température maximale investiguée est inscrite.
NC
NC CN
CNOO O
CO2Et
CO2Et2563
CO2Me

82
7.3. Cycloaddition intermoléculaire du diène 254
Tel qu’avec les diènes 252 et 253, 254 a généré de la décomposition lorsque mis en présence de
tétracyanoéthylène (tableau 14, entrée 1) et le même résultat a été obtenu avec le E-nitropropène201
(entrée 2). Dans cette réaction, la dégradation du diène 254 était accompagnée de produits de
cycloaddition du nitropropène sur lui-même. Si le diène s’est totalement décomposé en conditions
thermiques avec le crotonate de méthyle (entrée 3), un mélange complexe de produits aromatiques a
résulté de la réaction sous catalyse avec le perchlorate de lithium (entrée 4). En plus de procurer des
produits de dégradation lors de sa réaction avec le diénophile 256 sous diverses conditions (5-9), le
diène 254 a de nouveau conduit à des produits aromatiques en présence de perchlorate (entrée 10). Il est
à noter que l’utilisation de micro-ondes n’a pas permis de faciliter la cycloaddition, procurant
également de la décomposition (entrée 6). L’emploi d’un diénophile plus activé tel que 259202 n’a
également pas promu la formation de 258, générant un mélange complexe de produits de dégradation
sous toutes les conditions explorées (entrées 11-13). Finalement, la CDA du crotonaldéhyde sous
catalyse de titane (via la formation du vinyloxonium de titane), telle que développée par Seebach, n’a
produit que de la décomposition (entrée 14).203
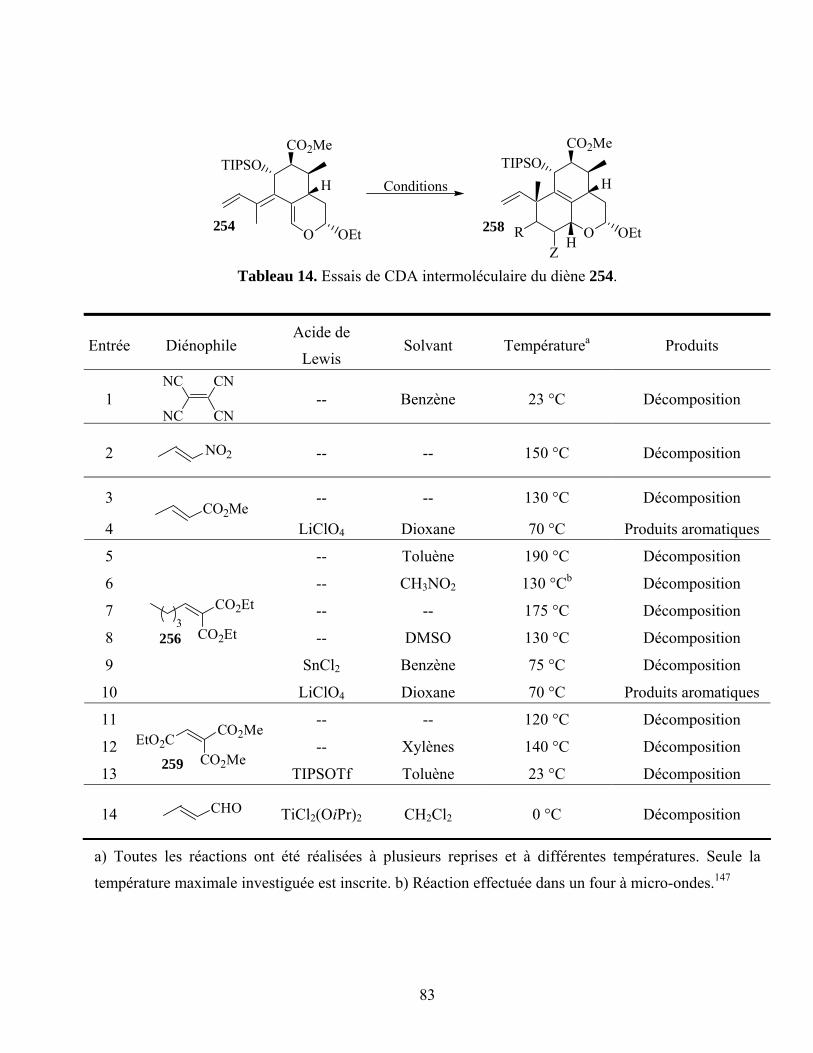
83
O
CO2Me
OEt
HTIPSO
254 258 O
CO2Me
OEtRZ
H
H
TIPSOConditions
Tableau 14. Essais de CDA intermoléculaire du diène 254.
Entrée Diénophile Acide de
Lewis Solvant Températurea Produits
1
-- Benzène 23 °C Décomposition
2
-- -- 150 °C Décomposition
3 -- -- 130 °C Décomposition
4
LiClO4 Dioxane 70 °C Produits aromatiques
5 -- Toluène 190 °C Décomposition
6 -- CH3NO2 130 °Cb Décomposition
7 -- -- 175 °C Décomposition
8 -- DMSO 130 °C Décomposition
9 SnCl2 Benzène 75 °C Décomposition
10
LiClO4 Dioxane 70 °C Produits aromatiques
11 -- -- 120 °C Décomposition
12 -- Xylènes 140 °C Décomposition
13
TIPSOTf Toluène 23 °C Décomposition
14
TiCl2(OiPr)2 CH2Cl2 0 °C Décomposition
a) Toutes les réactions ont été réalisées à plusieurs reprises et à différentes températures. Seule la
température maximale investiguée est inscrite. b) Réaction effectuée dans un four à micro-ondes.147
NC
NC CN
CN
NO2
CO2Me
CO2Et
CO2Et2563
EtO2CCO2Me
CO2Me259
CHO

84
Devant l’échec des cycloadditions des diènes 252, 253 et 254, nous pensions que l’instabilité de ces
molécules aux conditions thermiques et aux catalyses acides était la cause de la décomposition
observée. Des expériences de fonctionnalisation de l’alcène terminal ont été conduites dans le but de
stabiliser le triène.
7.4. Fonctionnalisation de l’oléfine terminale du triène 254
Wipf avait rapporté le tandem réduction/homologation d’un triène semblable lors de sa synthèse de la
(+)-curacine A.204 Cette stratégie était très invitante, puisque l’aldéhyde 260 aurait été propice à
l’introduction du segment C4-C6 pour une troisième CDA de type intramoléculaire (schéma 78).
Lorsque appliquées au composé 254, cependant, ces conditions n’ont procuré malheureusement que du
produit de départ et de la décomposition. Si la variation du solvant n’a eu aucun effet sur le résultat de
la réaction, une augmentation de la température a promu la réduction de l’ester méthylique en plus de
produire davantage de décomposition.
254O
CO2Me
OEt
HTIPSO
i) Cp2ZrHClii) tBuNC
260O
CO2Me
OEt
HTIPSO
O
Schéma 78.
En quête d’une solution, nous avons tourné nos espoirs vers l’hydroboration de l’oléfine terminale de
254, mais les premiers résultats ont été très décevants (tableau 15). En effet, le triène 254 s’est
totalement décomposé sous l’action du complexe borane-diméthylsulfure (entrée 1) et d’agents
d’hydroboration encombrés (entrées 2-3).205 Un résultat identique nous attendait suite aux réactions
avec le catécholborane (entrées 4-5). Si la réaction avec le dicyclohexylborane (Cy2BH) semblait
procéder, nous avons découvert que le protocole de neutralisation avait un effet dramatique sur le
résultat de la réaction. Si les réactions neutralisées en milieu basique n’ont procuré que de la
dégradation (entrée 6), l’arrêt de l’hydroboration en milieu neutre nous a permis d’isoler des traces de
l’alcool primaire désiré 261 (entrée 7). Toutefois, la neutralisation de la réaction avec du Borax® en

85
présence d’un agent de transfert de phase a conduit, après une optimisation exhaustive, à l’isolation
presque quantitative de l’alcool 261 (entrée 8).
254O
CO2Me
OEt
HTIPSO
261O
CO2Me
OEt
HTIPSO
HOConditions
Tableau 15. Hydroboration de l’alcène terminal du triène 254.
Entrée Agent d’hydroboration Produits
1 BH3•DMS Décomposition
2 (thex)BH2 Décomposition
3 9-BBN Décomposition
4 Catécholborane Décomposition
5 Catécholborane, (Ph3P)3RhCl Décomposition
6 1) Cy2BH
2) H2O2, NaOH Décomposition
7 1) Cy2BH
2) H2O2, tampon pH 7 Traces de 261
8 1) Cy2BH
2) Borax®, nBu4NHSO4 261
L’alcool primaire 261 s’avérant instable sur gel de silice (la purification du produit brut
d’hydroboration n’a procuré que 30% de 261), celui-ci a donc été immédiatement utilisé pour l’étape
subséquente. Afin de confirmer le bon rendement de l’hydroboration, l’alcool secondaire a été
déprotégé sous l’action du fluorure de tétrabutylammonium (schéma 79). Nous avons ainsi pu isoler le
diol 262 avec un rendement de 76% pour les deux étapes.
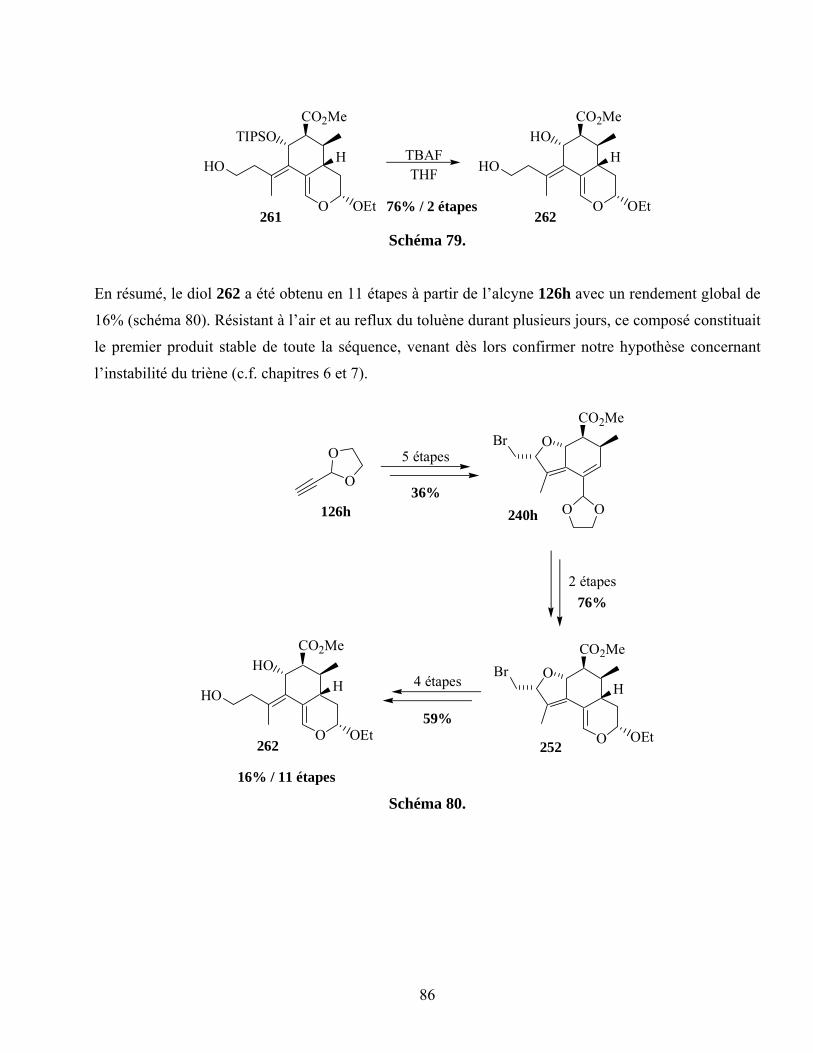
86
O
CO2Me
OEt
HTIPSO
HOTBAF
261
THF
262O
CO2Me
OEt
HHO
HO
76% / 2 étapes
Schéma 79.
En résumé, le diol 262 a été obtenu en 11 étapes à partir de l’alcyne 126h avec un rendement global de
16% (schéma 80). Résistant à l’air et au reflux du toluène durant plusieurs jours, ce composé constituait
le premier produit stable de toute la séquence, venant dès lors confirmer notre hypothèse concernant
l’instabilité du triène (c.f. chapitres 6 et 7).
262O
CO2Me
OEt
HHO
HO
126h
CO2MeOBr
O O240h
252
CO2MeOBr
O
H
OEt
O
O 5 étapes
36%
2 étapes76%
4 étapes
59%
16% / 11 étapes Schéma 80.
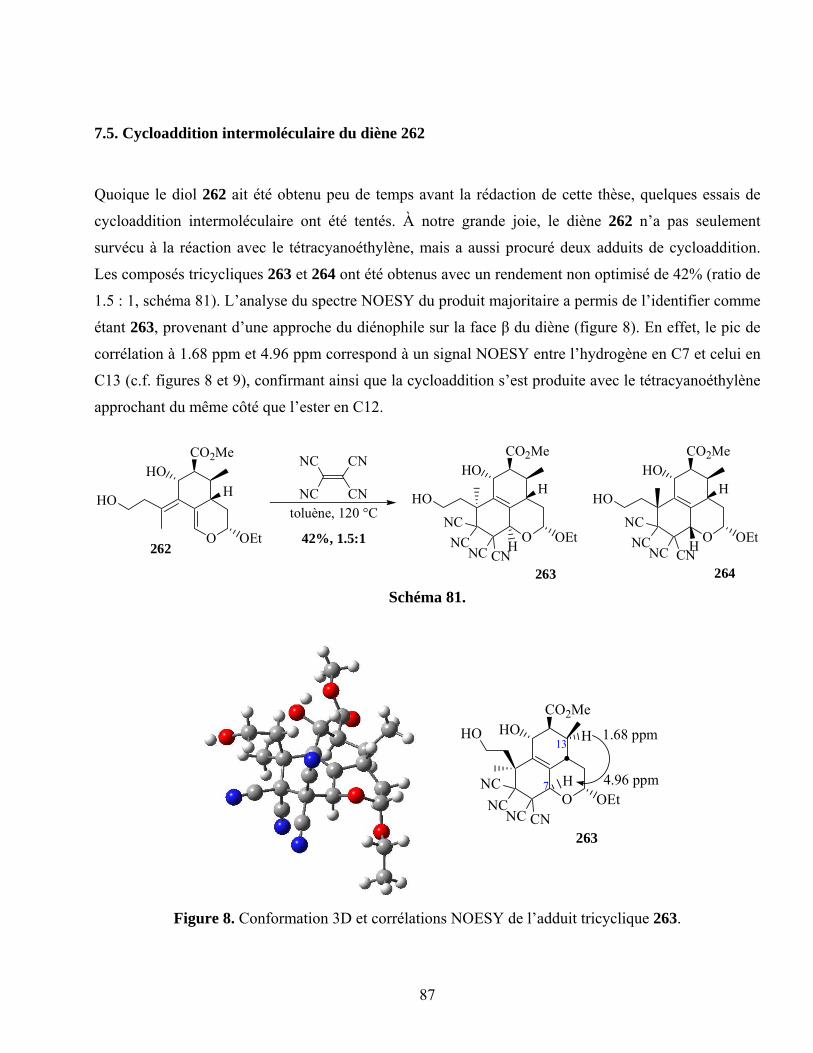
87
7.5. Cycloaddition intermoléculaire du diène 262
Quoique le diol 262 ait été obtenu peu de temps avant la rédaction de cette thèse, quelques essais de
cycloaddition intermoléculaire ont été tentés. À notre grande joie, le diène 262 n’a pas seulement
survécu à la réaction avec le tétracyanoéthylène, mais a aussi procuré deux adduits de cycloaddition.
Les composés tricycliques 263 et 264 ont été obtenus avec un rendement non optimisé de 42% (ratio de
1.5 : 1, schéma 81). L’analyse du spectre NOESY du produit majoritaire a permis de l’identifier comme
étant 263, provenant d’une approche du diénophile sur la face β du diène (figure 8). En effet, le pic de
corrélation à 1.68 ppm et 4.96 ppm correspond à un signal NOESY entre l’hydrogène en C7 et celui en
C13 (c.f. figures 8 et 9), confirmant ainsi que la cycloaddition s’est produite avec le tétracyanoéthylène
approchant du même côté que l’ester en C12.
O
CO2Me
OEt
HHO
HO NC
CNNC
CN
262
CO2Me
O OEt
HHO
HO
NCNC
NC CNH
263
CO2Me
O OEt
HHO
HO
NCNC
NC CNH
264
toluène, 120 °C
42%, 1.5:1
Schéma 81.
CO2Me
O OEt
HO
NCNC
NC CN
H
263
H 1.68 ppm
4.96 ppm
HO
7
13
Figure 8. Conformation 3D et corrélations NOESY de l’adduit tricyclique 263.
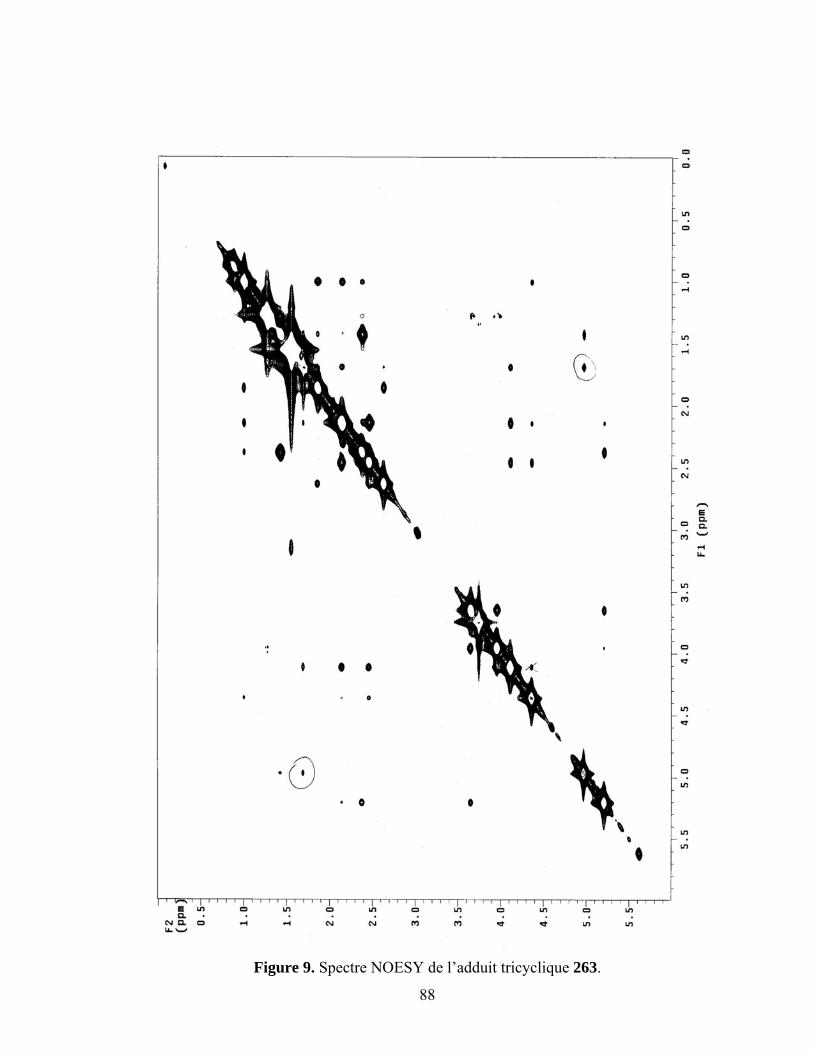
88
Figure 9. Spectre NOESY de l’adduit tricyclique 263.

89
Même si peu stéréosélective, cette cycloaddition est très prometteuse. Malgré la forme convexe de la
molécule favorisant une approche du diénophile par la face β, ce résultat démontre clairement que la
face désirée α est aussi disponible (figure 10). Il semble donc qu’il y ait compétition entre la géométrie
convexe de la molécule et la répulsion stérique occasionnée par l’ester en C12 et le méthyle en C13.
C7
C8
C9C10
C7C8
C9C10
C12
C13
Figure 10. Représentations 3D du diol 262.
Nous avons alors tenté cette réaction avec des diénophiles synthétiquement plus intéressants (tableau
16). Sous l’action du diénophile 266206 (entrées 1-2) ou avec le fumarate de méthyle (entrée 3), le diène
262 n’a malheureusement produit que de la décomposition. Lors de sa réaction avec le triester 259, des
produits complexes résultant d’une addition de Michael sur le diénophile ont été observés, suggérant
que la nucléophilicité de l’alcool primaire est peut être problématique en présence d’oléfines activées
(entrée 4). Les toutes dernières réactions ont donc visé à résoudre ce problème.

90
O
CO2Me
OEt
HHO
HO
CO2Me
O OEt
HHO
HO
HZ
R
265Conditions
262
Tableau 16. Essais de CDA intermoléculaire du diène 262.
Entrée Diénophile Solvant Température Produits
1 Xylènes 130 °C Décomposition
2
-- 150 °C Décomposition
3
Toluène 120 °C Décomposition
4
Toluène 120 °C
Produits d’addition
de Michael
7.6. Protection du diol 262 et cycloaddition intermoléculaire du diène 267
Avec quelques milligrammes en mains, l’alcool primaire du diol 262 a été masqué sous forme d’un
éther silylé TBS, procurant le diène 267 avec un rendement quantitatif (schéma 82). Par la suite, les
dernières expériences de cycloaddition de Diels-Alder ont été tentées sur ce nouveau diène afin
d’évaluer sa réactivité (tableau 17).
O
CO2Me
OEt
HHO
HOCH2Cl2
262 267 O
CO2Me
OEt
HHO
TBSO
100%
TBSClimidazole
Schéma 82.
Lorsque chauffé à 140 °C en présence du diénophile 266, seul le produit de départ a été obtenu (entrée
1). De même, le diène 267 a pu être porté à 250 °C sans se dégrader ou procurer un adduit tricyclique
tel que 268 (entrée 2). Ce diène s’est toutefois décomposé lorsque la réaction a été effectuée durant plus
MeO2C CO2Me
259EtO2C CO2Me
CO2Me
266
CO2Me
O CO2Me

91
de trente heures à cette température (entrée 3). Quoique préliminaires, ces résultats ont permis d’évaluer
la capacité du diène 267 à supporter des conditions extrêmes. C’est à court de matériel pour continuer
ces études que le projet s’est terminé.
O
CO2Me
OEt
HHO
TBSO
267
CO2Me
O OEt
HHO
TBSO
HZ
R
268Conditions
Tableau 17. Essais de CDA intermoléculaire du diène 267.
Entrée Diénophile Solvant Température Produits
1 Toluène 140 °C Produit de départ
2 Toluène 250 °Ca Produit de départ
3
Toluène 250 °Cb Décomposition
a) 16 heures de réaction. b) 30 heures de réaction.
7.7. Projets futurs
Le projet devrait se poursuivre par la cycloaddition du diène 267 avec différents diénophiles sous
diverses conditions thermiques ou catalysée par des acides de Lewis. En connaissance des récents
succès d’Amélie Dion et de Stéphane Perreault avec des CDA intramoléculaires sur des systèmes
similaires, il devrait être possible d’effectuer cette transformation. Une fois le cycle B formé (i.e. 269),
une oxydation de l’alcool en C11 suivie de l’addition d’un méthyle via son réactif de cuprate devrait
procurer le centre quaternaire en C8 (270, schéma 83). La formation du cycle A débuterait par une
déprotection de l’alcool primaire. L’oxydation de cet alcool produirait l’aldéhyde 271, qui serait
propice à une condensation aldolique avec la méthylcétone, ce qui conduirait à l’adduit tricyclique 272.
Le méthyle en C4 serait alors incorporé par l’addition de méthyllithium et l’oxydation de l’alcool
tertiaire résultant avec transposition de l’alcène procurerait le squelette picrasane fonctionnalisé 273. Ce
tétracycle, qui serait obtenu en 20 étapes à partir de l’alcyne 126h, possèderait toutes les fonctionnalités
nécessaires pour synthétiser différentes quassinoïdes.
266
CO2Me
O CO2Me
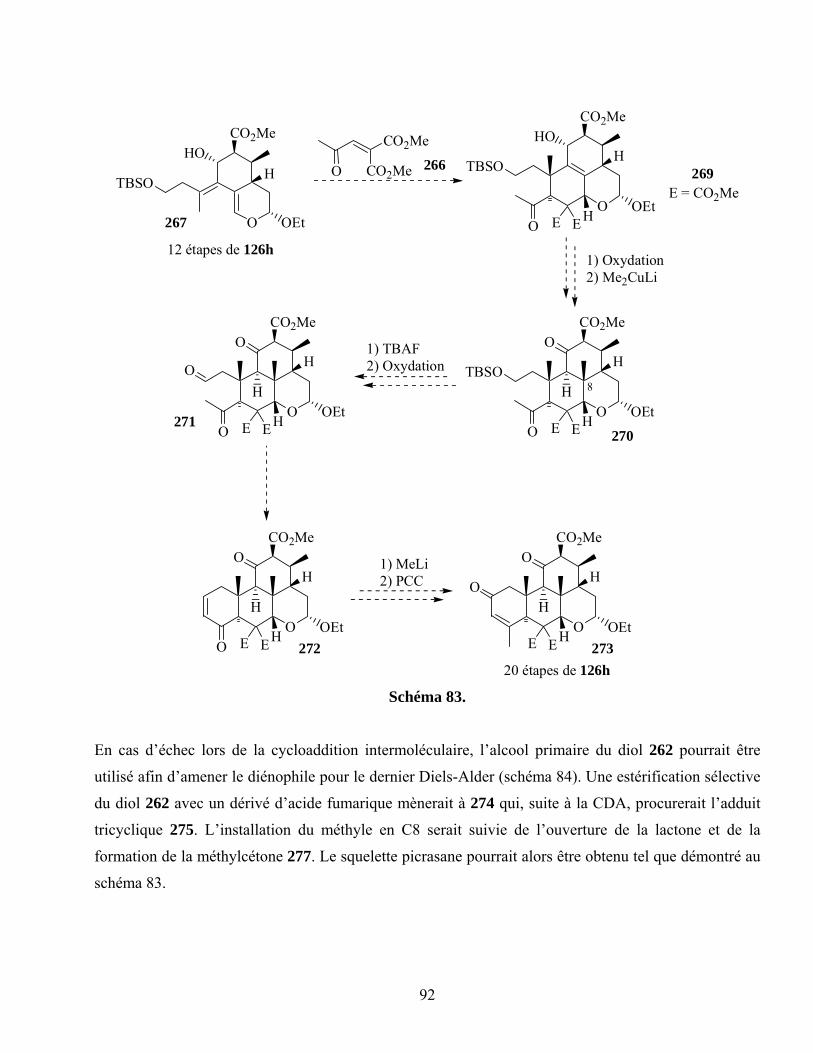
92
O
CO2Me
OEt
HHO
TBSO
CO2Me
O CO2Me 266
267
CO2Me
O OEt
HO
O
HE EO
H
271
CO2Me
O OEt
HO
HE EO
H
272
CO2Me
O OEt
HO
HE E
HO
CO2Me
O OEt
HO
TBSO
HE EO
H
CO2Me
O OEt
HHO
TBSO
HE EO
273
270
269E = CO2Me
1) Oxydation2) Me2CuLi
1) TBAF2) Oxydation
1) MeLi2) PCC
12 étapes de 126h
20 étapes de 126h
8
Schéma 83.
En cas d’échec lors de la cycloaddition intermoléculaire, l’alcool primaire du diol 262 pourrait être
utilisé afin d’amener le diénophile pour le dernier Diels-Alder (schéma 84). Une estérification sélective
du diol 262 avec un dérivé d’acide fumarique mènerait à 274 qui, suite à la CDA, procurerait l’adduit
tricyclique 275. L’installation du méthyle en C8 serait suivie de l’ouverture de la lactone et de la
formation de la méthylcétone 277. Le squelette picrasane pourrait alors être obtenu tel que démontré au
schéma 83.
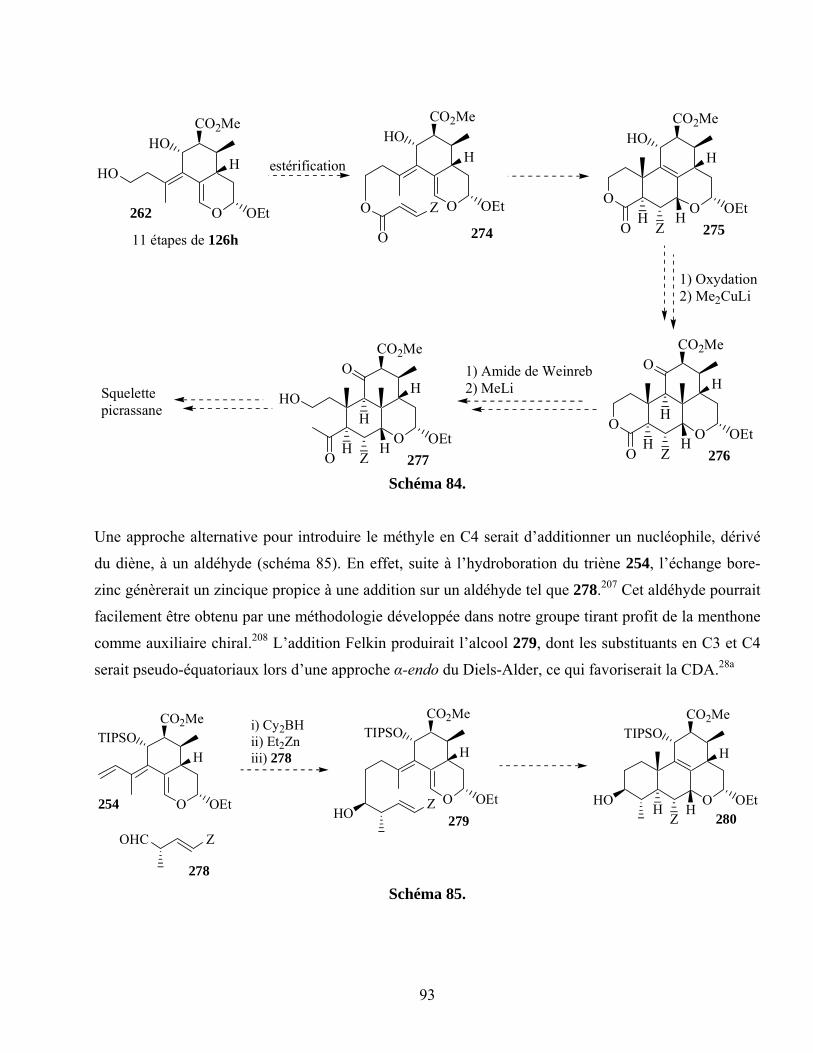
93
O
CO2Me
OEt
HHO
HO
262
CO2Me
O OEt
HHO
HO
O 27511 étapes de 126h
O
CO2Me
OEt
HHO
274
O
O
ZZ
H
CO2Me
O OEt
HO
HO
O 276
H
ZH
CO2Me
O OEt
HO
HO 277
H
ZH
HOSquelettepicrassane
estérification
1) Oxydation2) Me2CuLi
1) Amide de Weinreb2) MeLi
Schéma 84.
Une approche alternative pour introduire le méthyle en C4 serait d’additionner un nucléophile, dérivé
du diène, à un aldéhyde (schéma 85). En effet, suite à l’hydroboration du triène 254, l’échange bore-
zinc génèrerait un zincique propice à une addition sur un aldéhyde tel que 278.207 Cet aldéhyde pourrait
facilement être obtenu par une méthodologie développée dans notre groupe tirant profit de la menthone
comme auxiliaire chiral.208 L’addition Felkin produirait l’alcool 279, dont les substituants en C3 et C4
serait pseudo-équatoriaux lors d’une approche α-endo du Diels-Alder, ce qui favoriserait la CDA.28a
O
CO2Me
OEt
HTIPSO
254
OHC Z
278
O
CO2Me
OEt
HTIPSO
HOZ
279O
CO2Me
OEt
HTIPSO
HHZ
HO280
i) Cy2BHii) Et2Zniii) 278
Schéma 85.

94
Finalement une bonne stéréosélectivité α pour la troisième cycloaddition de Diels-Alder pourrait être
assurée en encombrant la face β. À cette fin, l’hétéro DA pourrait être conduit avec une énamine ou un
éther d’énol, mais avec une sélectivité exo, procurant un adduit tricyclique tel que 281 (schéma 86).209
Pour mettre toutes les chances de notre côté, l’inversion de la stéréochimie en C11 par une réaction de
Mitsunobu sur l’alcool 282 permettrait d’avoir tous les substituants sur la face β (i.e. 283). La CDA de
façon inter ou intramoléculaire devrait alors procéder stéréosélectivement sur la face α (285).
O
CO2MeOBr
O251 281
CO2MeOBr
O
H
X
O
CO2Me
X
HBzO
BzO
HZ
RH
284
282
283
O
CO2Me
X
HHO
HO
O
CO2Me
X
HBzO
BzO
X = OR, NR2
CHDA exo
Mitsunobu
11
OBzOBzEtO
CO2Me
Z R285
Schéma 86.

95
CONCLUSION GÉNÉRALE
Plusieurs études modèles ont mis à jour de nombreux éléments critiques à notre stratégie de
cycloaddition de Diels-Alder à diène transmissible vers la synthèse de quassinoïdes. En effet, nous
avons démontré que la présence d’un hétéroatome en position C-1 interfère avec la cycloaddition
d’hétéro Diels-Alder seulement lorsque ce carbone est disubstitué (i.e. 118, 120 et 133 vs. 166, 171 et
252), et ce peu importe le connecteur utilisé pour la première CDA.
Les unités de déconnection triméthylsilylméthyle et bromure ont promu le clivage des connecteurs
(évitant ainsi la décomposition lors de la CHDA) et ont offert de nouvelles options pour la dernière
cycloaddition [4+2]. Notre séquence donne maintenant accès à la troisième CDA de façon intra ou
intermoléculaire, rendant notre stratégie plus convergente (c.f. schéma 38).
En plus de mettre à jour des problèmes reliés à la stabilité d’intermédiaires et l’incompatibilité de
certains groupes protecteurs, ces études ont souligné l’importance d’une synthèse rapide et efficace des
vinylallènes désirés. Dans ce sens, nous avons développé une nouvelle séquence synthétique vers des
vinylallènes tétrasubstitués (240) en seulement quatre étapes à partir des éthers propargyliques 126.
Cette synthèse, comportant une étape d’énantiosélection (i.e. époxydation), tire profit de l’addition SN2’
et anti de vinylcuprates sur des oxyranes propargyliques.
Les conditions expérimentales nécessaires pour effectuer la troisième cycloaddition de Diels-Alder
requièrent un diène thermiquement stable. Les adduits 263 et 264 ont été obtenus du diol 262,
constituant ainsi notre premier succès lors de cette CDA de façon intermoléculaire. En connaissance des
récents succès de Stéphane Perreault et Amélie Dion lors de CDA intramoléculaires pour former le
cycle B, notre nouvelle séquence synthétique vers des vinylallènes fonctionnalisés devrait permettre la
synthèse totale d’une quassinoïde dans un proche avenir.

96
PARTIE EXPÉRIMENTALE
Remarques générales
Toutes les réactions ont été effectuées sous atmosphère d'argon dans de la verrerie séchée à la flamme
sous pression réduite à moins d’avis contraire. Les montages en verrerie ont été assemblés à la sortie de
l’étuve (120°C) et refroidis sous argon. Les solvants anhydres et certains réactifs liquides ont été
distillés avant leur utilisation, et ils sont rapportés dans le tableau G.1 suivant.
Tableau G.1 : Agents desséchants utilisés pour la distillation de différents solvants et réactifs.
Solvant / Réactif distillé Agent desséchant
Acétonitrile Aucun
Acétate d’éthyle Aucun
Benzène Hydrure de calcium
Dichloroéthane Hydrure de calcium
Dichlorométhane Hydrure de calcium
N,N-Diisopropylamine Hydrure de calcium
N,N-Diméthylformamide Tamis moléculaire 4 A
Éther diéthylique Hydrure de calcium
Hexane Aucun
Pyridine Hydrure de calcium
Tétrahydrofuranne Sodium, Benzophénone
N,N,N-Triéthylamine Hydrure de calcium
Toluène Hydrure de calcium

97
Les réactifs et les produits de départ ont été utilisés comme reçus du manufacturier. Les solutions
commerciales d’alkyllithium ont été préalablement titrées par la méthode de Love.210 L’iodure de
cuivre(I) a été purifié avant utilisation. Le IBX a été synthétisé selon la procédure de Stantagostino211 et
le periodinane de Dess-Martin selon celle de Ireland.212 Les tamis moléculaires 4 A ont été séchés à
l’étuve pendant un minimum de 16 h à une température de 120 °C avant usage. Le bromure de lithium a
été séché en chauffant le solide à 120 °C dans un ballon sous le vide d’une pompe mécanique pendant
16 h et entreposé dans une boîte à gants sous argon. L’ozone a été synthétisé avec l’aide d’un ozoniseur
en passant une décharge électrique dans un courant d’oxygène sec.
Les chromatographies sur couche mince ont été effectuées sur des plaques de verre recouvertes de gel
de silice (0.25 mm) Sil G-25 (Macherey-Nagel). Les produits en chromatographie sur couche mince ont
été révélés à la lampe UV, puis par trempage dans une solution aqueuse de KMnO4 ou dans une
solution de vaniline, suivi d'un chauffage sur une plaque chauffante. Les chromatographies éclair ont
été effectuées avec du gel de silice Silicycle (230-400 mesh).
Les spectres infrarouge ont été obtenus par dépôt d'un film de produit sur une pastille de chlorure de
sodium ou en solution dans le chloroforme dans une cellule de NaCl avec un spectromètre Perkin-
Elmer 1600 FT-IR. Les spectres de résonance magnétique nucléaire 1H (300 MHz) et 13C (75 MHz)
ont été enregistrés avec un appareil Bruker AC-300. L’étalon interne est le chloroforme (7,26 ppm) ou
le diméthylsulfoxyde (2,49 ppm) pour la résonance des protons et le chloroforme (77,0 ppm) pour la
résonance des carbones. Les déplacements chimiques (δ) sont rapportés en ppm et les constantes de
couplage en Hertz (Hz). Les abréviations utilisées pour les différents signaux en RMN sont : singulet
(s), doublet (d), doublets de doublet (dd), doublets de quadruplet (dq), triplet (t), quadruplet (q),
quintuplet (quin), sextuplet (sext), multiplet (m) et diastéréoisomère (diast). Les spectres de masse ont
été enregistrés avec un spectromètre VG Micromass ZAB-2F. Les réactions dans un four à micro-ondes
ont été effectuées avec un appareil CEM-Discover mesurant la température réactionnelle avec un
capteur infrarouge à l’extérieur du récipient en verre.

98
Modes opératoires
(±) Sulfonate 116
OS
OPMB
O O
L’allène 111 (100 mg, 0.32 mmol) a été dissous dans 3.0 mL de dichlorométhane à température
ambiante et 270 µL de triéthylamine ont été ajouté. Après 5 min d’agitation, une solution de chlorure de
vinylsulfonyle (62 mg, 0.49 mmol) dans 1.0 mL de triéthylamine a été additionnée sur une période de 2
min. Suite à l’addition, le mélange réactionnel a été neutralisé avec une solution aqueuse de HCl 1 N et
extrait avec du dichlorométhane. Les fractions organiques ont été combinées, séchées avec du sulfate de
magnésium anhydre et concentrées sous pression réduite. Le produit brut obtenu a été purifié par
chromatographie éclair sur colonne de gel de silice en éluant avec un mélange d'acétate d'éthyle et
d'hexanes (1:3). Le sulfonate désiré (42 mg, 35%) a été obtenu. Le produit est instable et se décompose
lentement à la température ambiante.
RMN 1H (300 MHz, CDCl3) δ (ppm) 7.23 (d, 2H, J = 8.0 Hz), 6.87 (d, 2H, J = 8.0 Hz), 5.90 (d, 1H, J
= 4.0 Hz), 4.87 (d, 1H, J = 15.0 Hz), 4.66 (d, 1H, J = 15.0 Hz), 4.42 (AB q, 2H), 4.27 (d, 1H, J = 12.0
Hz), 4.03 (d, 1H, J = 12.0 Hz), 3.82 (s, 3H), 3.72 (t, 1H, J = 9.0 Hz), 2.60 (m, 1H), 2.15-2.08 (m, 2H),
1.80 (s, 3H), 1.12 (d, 3H, J = 6.0 Hz). RMN 13C (75 MHz, CDCl3) δ (ppm) : 159.2 (s), 138.6 (d), 131.3
(s), 129.7 (s), 129.5 (d), 123.8 (s), 123.7 (s), 113.8 (d), 73.8 (t), 72.4 (t), 71.7 (t), 55.2 (q), 51.2 (d), 28.8
(t), 28.1 (d), 20.3 (q), 15.6 (q). IR (film, cm-1) : 3152, 1792, 1646, 1472, 1376, 1087. SMBR (m/z,
intensité relative) : 364 (M+, 5), 161 (100), 122 (75), 121 (65). SMHR calculée pour C19H24O5S :
364.1344, observée : 364.1334.

99
(±) Alcool allylique 117
OS
OH
O O
Le sulfonate 116 (280 mg, 0.76 mmol) a été dissous dans 5.0 mL de dichlorométhane contenant 200 µL
d’eau à 0 °C. Du DDQ (226 mg, 0.99 mmol) a été ajouté par petites portions. Suite à la complétion de
la réaction, le mélange réactionnel a été neutralisé avec une solution aqueuse saturée en bicarbonate de
sodium et la solution résultante a été extraite avec du dichlorométhane. Les fractions organiques ont été
combinées, séchées avec du sulfate de magnésium anhydre et concentrées sous pression réduite. Le
produit brut obtenu a été purifié par chromatographie éclair sur colonne de gel de silice en éluant avec
un mélange d'acétate d'éthyle et d'hexanes (3:2). L’alcool désiré (120 mg, 64%) s’est avéré instable et a
décomposé lentement à la température ambiante.
(±) Aldéhyde 118
OS
O
O O
L’alcool 117 (12 mg, 0.49 mmol) a été dissous dans 3.0 mL de dichlorométhane à 0 °C. Du DMP (250
mg, 0.59 mmol) a été ajouté par petites portions. La solution a ensuite été placée à température
ambiante et laissée sous agitation durant 16 h. Le mélange réactionnel a été neutralisé avec une solution
aqueuse saturée en bicarbonate de sodium contenant 10% de thiosulfate de sodium et extraite avec 3
portions d’éther éthylique. Les fractions organiques ont été combinées, lavées avec de la saumure,
séchées avec du sulfate de magnésium anhydre et concentrées sous pression réduite. Le produit brut
obtenu (110 mg, 90%) s’est avéré instable et n’a pas été purifié.
RMN 1H (300 MHz, CDCl3) δ (ppm) 9.48 (s, 1H), 6.83 (d, 1H, J = 4.0 Hz), 4.88 (AB q, 2H), 3.83-
3.80 (m, 1H), 2.87-2.81 (m, 1H), 2.38-2.28 (m, 1H), 2.13-2.05 (m, 1H), 1.61 (s, 3H), 1.24 (d, 3H, J =

100
7.0 Hz). SMBR (m/z, intensité relative) : 260 (MNH4+, 25), 243 (MH+, 10), 163 (100). SMHR calculée
pour C11H15O4S (MH+) : 243.0691, observée : 243.0697.
(±) (E)-2-(4’-Méthoxybenzyloxyméthyl)-4-méthyl-2-propényl-2,5-dihydrofuranne (122)
OOPMB
L’allène 111 (46 mg, 0.16 mmol) et le méthoxyacrylate de méthyle (90 µL, 0.83 mmol) ont été dissous
dans 1.0 mL de tétrahydrofuranne à température ambiante. Du Hg(O2CCF3)2 (4 mg, 0.01 mmol) a été
ajouté et le mélange réactionnel a été agité durant 4 jours. La concentration de la solution et la
purification du résidu par chromatographie éclair sur colonne de gel de silice, en éluant avec un mélange
d'acétate d'éthyle et d'hexanes (1:3), ont procuré une huile légèrement jaune (19 mg, 41%).
RMN 1H (300 MHz, CDCl3) δ (ppm) : 7.25 (d, 2H, J = 9.0 Hz), 6.87 (d, 2H, J = 8.5 Hz), 5.70 (dq, 1H,
J = 15.5 et 6.0 Hz), 5.60 (d, 1H, J = 6.0 Hz), 5.38 (d, 1H, J = 1.5 Hz), 4.51 (s, 4H), 3.80 (s, 3H), 3.42
(AB q, 2H), 1.76 (s, 3H), 1.69 (d, 3H, J = 6.0 Hz). RMN 13C (75 MHz, CDCl3) δ (ppm) : 137 (s), 132
(d), 131 (s), 129 (d), 126 (s), 124 (d), 113 (d), 92 (s), 78 (t), 76 (d), 75 (t), 73 (t), 55 (q), 19 (q), 13 (q).
SMBR (m/z, intensité relative) : 243 (M+ - OCH3, 2), 123 (100), 121 (90). SMHR calculée pour
C17H23O3 (MH+) : 275.1647, observée : 275.1655.
(±) Dihydrofuranne 124
OPMB
O
CO2Me
L’allène 111 (320 mg, 1.16 mmol) et la N-méthylmorpholine (320 µL, 2.91 mmol) ont été dissous dans
4.0 mL d’éther éthylique. Le propiolate de méthyle (260 µL, 2.91 mmol) a ensuite été ajouté et la
solution a été agitée durant 1 h à température ambiante et 1.5 h à la température du reflux de l’éther. Le

101
mélange réactionnel a alors été concentré sous pression réduite et le résidu a été purifié par
chromatographie éclair sur colonne de gel de silice en éluant avec un mélange d'acétate d'éthyle et
d'hexanes (1:3). Une huile légèrement jaune (125 mg, 53%) particulièrement sensible à la chaleur et à
l’air a été obtenue.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 7.26 (d, 2H, J = 8.5 Hz), 6.88 (d, 2H, J = 8.5 Hz), 5.75 (d, 1H, J
= 4.0 Hz), 5.05-5.05 (m, 1H), 4.66 (dd, 1H, J = 13.5 et 4.5 Hz), 4.46 (dd, 1H, J = 13.5 et 4.5 Hz), 4.46
(s, 2H), 4.28 (d, 1H, J = 11.5 Hz), 4.10 (d, 1H, J = 11.5 Hz), 3.80 (s, 3H), 3.75 (s, 3H), 2.83-2.73 (m,
2H), 1.85 (s, 3H), 0.97 (d, 3H, J = 7.0 Hz). IR (film, cm-1) : 2942, 1735, 1648, 1508, 1248, 1081, 1027.
SMBR (m/z, intensité relative) : 358 (M+, 5), 121 (100). SMHR calculée pour C21H26O5 : 358.1780,
observée : 358.1772.
(±) Dihydrofuranne - alcool allylique 125
OH
O
CO2Me
La réaction a été effectuée sous les conditions pour obtenir l’alcool 117 avec le dihydrofuranne 124 (39
mg, 0.11 mmol). La purification du produit brut par chromatographie éclair sur gel de silice, en éluant
avec un mélange d'acétate d'éthyle et d'hexanes (1:3), a conduit à l’isolation de l’alcool allylique désiré
(7 mg, 28%) sous forme d’une huile très sensible à la chaleur et à l’air.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 5.78 (d, 1H, J = 4.5 Hz), 5.05-5.01 (m, 1H), 4.66 (dd, 1H, J =
13.0 et 4.0 Hz), 4.50-4.44 (m, 2H), 4.34 (d, 1H, J = 13.0 Hz), 3.76 (s, 3H), 2.85-2.73 (m, 2H), 1.90 (s,
3H), 0.97 (d, 3H, J =7.0 Hz). IR (CHCl3, cm-1) : 3615, 3020, 2933, 1733, 1371, 1174. SMBR (m/z,
intensité relative) : 238 (M+, 30), 220 (75), 161 (100), 133 (75). SMHR calculée pour C13H18O4 :
238.1205, observée : 238.1210.

102
(±) (E)-7-tert-Butyldiméthylsilanoxyhept-2-èn-5-yn-4-ol (127)
OH
TBSO
L’alcool propargylique 126i (11.3 g, 66.3 mmol) a été dissous dans 250 mL de THF et la solution
résultante a été refroidie à -78 °C. Une solution de n-butyllithium (2.0 M dans hexanes, 33.2 mL, 66.4
mmol) a été lentement ajoutée et le mélange réactionnel a alors été agité durant 30 min avant l’addition
du crotonaldéhyde (5.0 mL, 60.3 mmol). Après avoir laissé la température remonter graduellement
jusqu’à 0 °C, la réaction a été neutralisée avec une solution aqueuse saturée en chlorure d’ammonium et
la solution résultante a été extraite avec de l’éther éthylique. Les fractions organiques ont été
combinées, lavées avec de la saumure, séchées avec du sulfate de magnésium anhydre et concentrées
sous pression réduite. Le produit brut obtenu (14.5 g, 100%) n’a pas été purifié davantage dû à son
instabilité.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 5.80 (dq, 1H, J = 15.0 et 6.5 Hz), 5.52 (dd, 1H, J = 15.0 et 6.5
Hz), 4.79-4.72 (m, 1H), 4.28 (s, 2H), 3.03 (m, 1H), 1.64 (d, 3H, J = 6.0 Hz), 0.83 (s, 9H), 0.04 (s, 6H).
RMN 13C (75 MHz, CDCl3) δ (ppm): 130.1 (d), 128.2 (d), 84.4 (s), 84.1 (s), 62.5 (d), 51.6 (t), 25.7 (q),
18.2 (s), 17.3 (q), -5.2 (q). IR (film, cm-1) : 3364, 2860, 2359, 1464, 1371, 1253, 1142, 1078. SMBR
(m/z, intensité relative) : 181 (M+ - C4H9, 20), 75 (100). SMHR calculée pour C9H13O2Si (M+ - C4H9,
20) : 181.0685, observée : 181.0680.
(E)-7-tert-Butyldiméthylsilanoxyhept-2-én-5-yn-4-one (128)
O
TBSO
L’alcool allylique 127 (14.5 g, 66.3 mmol) et du tamis moléculaire 4 A ont été ajoutés à 270 mL de
CH2Cl2 et le mélange résultant a été refroidi à 0 °C sous agitation mécanique. Le PCC (36.5 g, 165.7
mmol) a ensuite été ajouté par petites portions et le mélange réactionnel alors été agité durant 16 h,
durant lesquelles le mélange hétérogène a graduellement atteint la température ambiante. Un ajout de
charbon activé (10 g) et de gel de silice (10 g) a permis la neutralisation de la réaction. Une filtration du

103
mélange résultant sur gel de silice et une concentration du filtrat sous pression réduite a mené à
l’isolation d’une huile brune (11.9 g, 76%) d’une pureté suffisante pour la poursuite des manipulations.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 7.21 (dq, 1H, J = 6.5 et 15.5 Hz), 6.18 (dd, 1H, J = 1.5 et 15.5
Hz), 4.49 (s, 2H), 1.91 (dd, 3H, J = 1.5 et 6.5 Hz), 0.92 (s, 9H), 0.14 (s, 6H). RMN 13C (75 MHz,
CDCl3) δ (ppm): 177.7 (s), 149.9 (d), 133.5 (d), 90.6 (s), 81.9 (s), 51.4 (t), 25.6 (q), 18.3 (q), 18.1 (s), -
5.4 (q). IR (film, cm-1) : 2857, 1705, 1630, 1444, 1254, 1100. SMBR (m/z, intensité relative) : 223 (M+
- CH3, 5), 181 (M+ - C4H9, 100). SMHR calculée pour C9H13O2Si (M+ - C4H9) : 181.0685, observée :
181.0681.
(±) (E)-4-Acétoxy-1-tert-butyldiméthylsilanoxy-4-([1’,3’]dithian-2’-yl)-hept-5-én-2-yne (129)
AcO
TBSO
S
S
Au 1,3-dithiane (100 mg, 0.83 mmol) à -78 °C dans le THF (3 mL) a été ajouté du n-butyllithium (2.0
M dans hexanes, 450 µL, 0.90 mmol). La solution a été agitée durant 1 h à -10 °C pour ensuite être
refroidie à -78 °C, température à laquelle la cétone propargylique 128 (180 mg, 0.75 mmol) dans 1.0
mL de THF a été ajoutée. L’alcoolate résultant a été, après complétion de la réaction, mis en présence
d’anhydride acétique (355 µL, 3.78 mmol). Suite à sa complétion, la réaction a été neutralisée par
l’ajout d’une solution aqueuse saturée de chlorure d’ammonium et la solution résultante a été extraite
avec de l’éther éthylique. Les fractions organiques ont été combinées, lavées avec de la saumure,
séchées avec du sulfate de magnésium anhydre et concentrées sous pression réduite. Le produit brut a
été purifié par chromatographie éclair sur gel de silice en éluant avec un mélange d'acétate d'éthyle et
d'hexanes (1:9). Une huile jaune (215 mg, 72%) a été obtenue (62% sur 4.4 g de cétone de départ). Ce
produit s’est avéré instable lors de purifications subséquentes et a donc été utilisé après une seule
chromatographie.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 6.25 (dq, 1H, J = 15.0 et 6.5 Hz), 5.77 (dd, 1H, J = 15.0 et 1.5
Hz), 4.87 (s, 1H), 4.43 (s, 2H), 2.98-2.76 (m, 4H), 2.06 (s, 3H), 2.11-2.01 (m, 1H), 1.94-1.84 (m, 1H),
1.79 (dd, 3H, J = 6.5 et 1.5 Hz), 0.90 (s, 9H), 0.13 (s, 6H). IR (film, cm-1) : 2933, 2360, 1751, 1435,
1367, 1229, 1092. SMBR (m/z, intensité relative) : 400 (M+, 1), 385 (M+ - CH3, 5), 343 (M+ - C4H9,

104
30), 340 (M+ - CH3CO2H, 75), 74 (100). SMHR calculée pour C19H32O3SiS2 : 400.1562, observée :
400.1569.
(±) (E)-4-Acétoxy-4-([1’,3’]dithian-2’-yl)-hept-5-én-2-yn-1-ol (130)
AcO
HO
S
S
À l’éther propargylique 129 (200 mg, 0.51 mmol) à -10 °C dans le THF (3 mL) a été ajouté du TBAF
(1.0 M dans le THF, 600 µL, 0.61 mmol). La solution a été agitée durant 1 h à -10 °C, neutralisée par
l’ajout d’une solution aqueuse saturée de chlorure d’ammonium et la solution résultante a été extraite
avec de l’éther éthylique. Les fractions organiques ont été combinées, lavées avec de la saumure,
séchées avec du sulfate de magnésium anhydre et concentrées sous pression réduite. Le produit brut a
été purifié par chromatographie éclair sur gel de silice en éluant avec un mélange d'acétate d'éthyle et
d'hexanes (1:3). L’huile jaune (134 mg, 76%) obtenue s’est avérée impure et son instabilité a nécessité
son usage immédiat dans la réaction suivante.
(±) 4-([1’,3’]Dithian-2’-yl)-2-méthylhepta-2,3,5-trién-1-ol (131)
OH
SS
C
Au bromure de lithium (1.75 g, 20.1 mmol) et à l’iodure de cuivre (3.83 g, 20.1 mmol) à -10 °C dans le
THF (40 mL) a été ajouté du bromure de méthylmagnésium (3.0 M dans Et2O, 6.70 mL, 20.1 mmol).
Après une heure d’agitation (la persistance d’une coloration jaune est indicatrice d’une bonne formation
du cuprate de Gilman), l’alcool propargylique 130 (940 mg, 3.35 mmol) dissous dans 10 mL de THF a
ensuite été ajouté. La réaction a été neutralisée par l’ajout d’une solution aqueuse saturée de chlorure
d’ammonium contenant 10% d’hydroxyde d’ammonium et la solution résultante a été agitée à
température ambiante jusqu’à la persistance d’une coloration bleue. À ce point, la réaction a été extraite

105
à l’éther éthylique et les fractions organiques ont été combinées, lavées avec de la saumure et séchées
avec du sulfate de magnésium anhydre. La concentration sous pression réduite et la purification du
produit brut par chromatographie éclair sur gel de silice, en éluant avec un mélange d'acétate d'éthyle et
d'hexanes (2:5), a conduit à l’isolation d’une huile légèrement jaune (586 mg, 75%).
RMN 1H (300 MHz, CDCl3) δ (ppm) : 5.86-5.78 (m, 2H), 4.68 (s, 1H), 4.08 (s, 2H), 3.02-2.84 (m, 4H),
2.13-2.07 (m, 1H), 1.93-1.81 (m, 1H), 1.80 (s, 3H), 1.78 (d, 3H, J = 6.0 Hz). IR (film, cm-1) : 3407,
2899, 1633, 1425, 1003. SMBR (m/z, intensité relative) : 242 (M+, 5), 211 (M+ - OCH3, 70), 121 (70),
119 (100). SMHR calculée pour C12H18OS2 : 242.0799, observée : 242.0789.
(±) Dihydrofuranne 132
O
CO2Me
S S
La réaction a été effectuée sous les conditions pour obtenir 124 avec l’allène 131 (280 mg, 1.15 mmol).
La purification du produit brut par chromatographie éclair sur gel de silice, en éluant avec un mélange
d'acétate d'éthyle et d'hexanes (1:3), a conduit à l’isolation du dihydrofuranne désiré (272 mg, 91%). Ce
produit s’est avéré instable à l’air et la chaleur.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 6.06 (d, 1H, J = 5.0 Hz), 5.03-4.99 (m, 1H), 4.87 (s, 1H), 4.68
(dd, 1H, J = 15.5 et 6.0 Hz), 4.46 (dd, 1H, J = 13.0 et 4.5 Hz), 3.74 (s, 3H), 3.03-2.73 (m, 6H), 2.20-
2.11 (m, 1H), 1.96 (s, 3H), 0.96 (d, 3H, J = 7.0 Hz), 0.90-0.76 (m, 1H). IR (film, cm-1) : 2891, 1742,
1431, 1280, 1174, 1028. SMBR (m/z, intensité relative) : 326 (M+, 40), 220 (100), 159 (60), 119 (70).
SMHR calculée pour C16H22O3S2 : 326.1010, observée : 326.1008.

106
(±) Aldéhyde 133
O
O
CO2Me
À l’oxyde de mercure (226 mg, 1.04 mmol) dans un mélange THF-H2O (2.80 mL-700 µL) à 0 °C, a été
ajouté du BF3-OEt2 (132 µL, 1.04 mmol). Le dihydrofuranne 132 (170 mg, 0.52 mmol) a été dissous
dans 800 µL de tétrahydrofuranne et additionné à la solution de mercure. La solution a été agitée durant
1 h à température ambiante après quoi de l’éther éthylique a été ajouté (5 mL). La filtration de la
solution et la concentration du filtrat sous pression réduite ont conduit à une huile qui a été purifiée par
filtration sur gel de silice en éluant avec un mélange d'acétate d'éthyle et d'hexanes (3:4). Une huile
jaune sensible à la chaleur et à l’air (85 mg, 57%) a été obtenue.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 9.53 (s, 1H), 6.67 (d, 1H, J = 4.5 Hz), 5.06-5.02 (m, 1H), 4.77
(dd, 1H, J = 13.5 et 4.5 Hz), 4.53 (dd, 1H, J = 13.5 et 4.5 Hz), 3.77 (s, 3H), 3.09 (sext, 1H, J = 7.0 Hz),
2.82 (dd, 1H, J = 11.0 et 6.5 Hz), 1.90 (s, 3H), 1.05 (d, 3H, J = 7.5 Hz). SMBR (m/z, intensité
relative) : 236 (M+, 85), 177 (100), 147 (90), 91 (100). SMHR calculée pour C13H16O4 : 236.1048,
observée : 236.1054.
(±) (E)-4-Acétoxy-4-([1’,3’]dithian-2’-yl)-hept-5-én-2-yn-1-al (150)
AcO
O
S
S
Au chlorure d’oxalyle (112 µL, 1.28 mmol) dans le dichlorométhane (3.0 mL) à -78 °C a été ajouté le
DMSO (182 µL, 2.56 mmol) lentement. La solution a été agitée durant 5 min à cette température après
quoi l’alcool 130 (300 mg, 1.06 mmol) dissous dans 3.0 mL de dichlorométhane a été ajouté. La
solution résultante a été agitée durant 10 min à -78 °C et de la triéthylamine (745 µL, 5.34 mmol) a été
additionnée. Suite à sa complétion, la réaction a été neutralisée par l’ajout d’une solution aqueuse

107
saturée de chlorure d’ammonium et la solution résultante a été extraite avec du dichlorométhane. Les
fractions organiques ont été combinées, séchées avec du sulfate de magnésium anhydre et concentrées
sous pression réduite. Le produit brut a été purifié par chromatographie éclair sur gel de silice en éluant
avec un mélange d'acétate d'éthyle et d'hexanes (1:4). L’aldéhyde désiré (43 mg, 14%) a été obtenu et
son instabilité a nécessité son usage immédiat dans la réaction suivante.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 9.29 (s, 1H), 6.26-6.11 (m, 1H), 5.74 (dd, 1H, J = 16.5 et 1.0
Hz), 4.79 (s, 1H), 2.99-2.77 (m, 4H), 2.14-2.03 (m, 1H), 2.10 (s, 3H), 1.96-1.84 (m, 1H), 1.80 (d, 3H, J
= 6.5 Hz).
(±) (E)-5-Acétoxy-1-triméthylsilanyl-5-([1’,3’]dithian-2’-yl)-oct-6-én-3-yn-2-ol (151)
AcO
S
S
OH
Me3Si
Méthode A :
À l’aldéhyde 150 (35 mg, 0.12 mmol) dissous dans 1 mL de THF à -78 °C a été ajouté le chlorure de
triméthylsilylméthylmagnésium (138 µL, 0.13 mmol). Suite à sa complétion, la réaction a été
neutralisée par l’ajout d’une solution aqueuse saturée de chlorure d’ammonium et la solution résultante
a été extraite avec du dichlorométhane. Les fractions organiques ont été combinées, séchées avec du
sulfate de magnésium anhydre et concentrées sous pression réduite. Le produit brut a été purifié par
chromatographie éclair sur gel de silice en éluant avec un mélange d'acétate d'éthyle et d'hexanes (1:4).
L’alcool propargylique désiré (14 mg, 31%) a été obtenu comme mélange de deux diastéréoisomères.
Méthode B :
La réaction a été effectuée sous les conditions pour obtenir 130 avec l’éther silylé 159 (640 mg, 1.31
mmol). La purification du produit brut par chromatographie éclair sur gel de silice, en éluant avec un
mélange d'acétate d'éthyle et d'hexanes (1:4), a conduit à l’isolation de l’alcool propargylique désiré
(448 mg, 92%) comme mélange de deux diastéréoisomères.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 6.25-6.10 (m, 1H), 5.77 (d, 1H, J = 15.5 Hz), 4.85 (s, 1H, diast
A), 4.84 (s, 1H, diast B), 4.63-4.60 (m, 1H), 4.58 (s, 1H), 2.92-2.78 (m, 4H), 2.09-2.03 (m, 1H), 2.06 (s,

108
3H, diast B), 2.03 (s, 3H, diast A), 190-185 (m, 1H), 1.80 (dd, 3H, J = 6.5 et 1.5 Hz), 1.33 (d, 2H, J =
6.5 Hz, diast A), 1.21 (d, 2H, J = 7.5 Hz, diast B), 0.09 (s, 9H, diast A), 0.08 (s, 9H, diast B). IR (film,
cm-1): 3459, 2896, 1751, 1723, 1421, 1362, 1293, 1009. SMBR (m/z, intensité relative) : 355 ((M-
H2O)H+, 25), 313 (100), 119 (95). SMHR calculée pour C17H27O2S2Si ((M-H2O)H+) : 355.1222,
observée : 355.1227.
(±) 1,4-Bis(triméthylsilanyl)-but-1-yn-3-ol (153)
OH
Me3Si
SiMe3
La réaction a été effectuée sous les conditions pour obtenir 151 (méthode B) avec le 1-
triméthylsilanylpropiolaldéhyde (152)120 (28.1 g, 223 mmol). Le produit brut obtenu (51.4 g, 86%) n’a
pas été purifié pour des raisons de volatilité.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 4.49 (t, 1H, J = 7.5 Hz), 1.78 (s large, 1H), 1.17 (d, 2H, J = 7.5
Hz), 0.16 (s, 9H), 0.08 (s, 9H). RMN 13C (75 MHz, CDCl3) δ (ppm): 109.5 (s), 89.3 (s), 61.2 (d), 28.0
(t), 0.7 (q), -0.1 (q). IR (film, cm-1): 3337, 2969, 2369, 1413, 1254, 1199, 1052, 1015. SMBR (m/z,
intensité relative) : 232 (MNH4+, 5), 197 (25), 90 (100). SMHR calculée pour C10H26NOSi2 (MNH4
+) :
232.1553, observée : 232.1549.
(±) 3-tert-Butyldiméthylsilanoxy-1,4-bis-triméthylsilanylbut-1-yne (154)
OTBS
Me3Si
SiMe3
À l’alcool propargylique 153 (9.30 g, 43.3 mmol) dans 200 mL de dichlorométhane à 0 °C, a été ajouté
l’imidazole (7.38 g, 108 mmol) suivi du chlorure de tert-butyldiméthylsilyle (7.19 g, 47.7 mmol) par
petites portions. Suite à sa complétion, la réaction a été neutralisée par l’ajout d’eau (200 mL) et le
mélange résultant extrait avec du dichlorométhane. Les fractions organiques ont été combinées, séchées
avec du sulfate de magnésium anhydre et concentrées sous pression réduite. Le produit brut obtenu
(14.2 g, 100%) n’a pas nécessité purification.

109
RMN 1H (300 MHz, CDCl3) δ (ppm) : 4.52 (dd, 1H, J = 8.0 et 6.0 Hz), 1.17 (dd, 1H, J = 14.5 et 8.0
Hz), 1.08 (dd, 1H, J = 14.5 et 6.0 Hz), 0.90 (s, 9H), 0.14 (s, 9H), 0.13 (s, 3H), 0.10 (s, 3H), 0.06 (s,
9H). RMN 13C (75 MHz, CDCl3) δ (ppm): 108.3 (s), 88.7 (s), 60.5 (d), 27.4 (t), 25.6 (q), 17.9 (s), -0.2
(q), -1.0 (q), -3.6 (q). IR (film, cm-1): 2960, 2168, 2167, 1251, 1073. Le produit s’est avéré instable aux
analyses de masses.
(±) 3-tert-Butyldiméthylsilanoxy-4-triméthylsilanylbut-1-yne (155)
OTBS
Me3Si
L’alcyne 154 (39.9 g, 121 mmol) a été dissous dans 600 mL de méthanol et du carbonate de potassium
a été ajouté (33.5 g, 242 mmol). Le mélange a été agité à température ambiante durant 16 h pour ensuite
être concentré sous pression réduite (avec une température contrôlée à 20 °C). De l’éther (500 mL) et
une solution aqueuse saturée en chlorure d’ammonium (400 mL) ont été ajoutés. Les phases ont été
agitées vigoureusement et la phase aqueuse extraite avec de l’éther éthylique. Les fractions organiques
ont été combinées, lavées avec de la saumure, séchées avec du sulfate de magnésium anhydre et
concentrées sous pression réduite. Le produit brut obtenu (29.5 g, 95%) n’a pas nécessité de
purification.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 4.56-4.50 (m, 1H), 2.40 (d, 1H, J = 2.0 Hz), 1.20 (dd, 1H, J =
14.5 et 8.5 Hz), 1.10 (dd, 1H, J = 14.5 et 5.5 Hz), 0.85 (s, 9H), 0.13 (s, 3H), 0.11 (s, 3H), 0.06 (s, 9H).
RMN 13C (75 MHz, CDCl3) δ (ppm): 87.6 (s), 78.4 (d), 60.6 (d), 28.3 (t), 26.6 (q), 18.8 (s), -0.1 (q), -
2.6 (q). IR (film, cm-1): 2956, 2860, 2362, 1464, 1246, 1067, 1007. SMBR (m/z, intensité relative) :
241 (M+ - CH3, 2), 199 (M+ - C4H9, 5), 147 (100). SMHR calculée pour C12H25OSi2 (M+ - CH3) :
241.1444, observée : 241.1446.

110
(±) (E)-7-tert-Butyldiméthylsilanoxy-8-triméthylsilanyloct-2-én-5-yn-4-ol (156)
OTBS
Me3Si
OH
L’alcyne 155 (1.04 g, 4.05 mmol) a été dissous dans 20 mL de THF à -78 °C et du n-butyllithium (1.88
M dans hexanes, 2.15 mL, 4.05 mmol) a été ajouté. Le mélange a été agité durant 30 min et le
crotonaldéhyde (305 µL, 3.68 mmol) a été additionné lentement. Suite à la complétion de la réaction,
celle-ci a été neutralisée par l’ajout d’une solution aqueuse saturée en chlorure d’ammonium et la
solution résultante a été extraite avec de l’éther éthylique. Les fractions organiques ont été combinées,
lavées avec de la saumure, séchées avec du sulfate de magnésium anhydre et concentrées sous pression
réduite. Le produit brut obtenu (1.24 g, 100%) n’a pas nécessité purification. Le rendement de la
réaction sur 2.20 mL de crotonaldéhyde a été de 82%.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 5.85 (dq, 1H, J = 6.5 et 15.5 Hz), 5.59 (dd, 1H, J = 15.5 et 6.0
Hz), 4.81 (d, 1H, J = 6.5 Hz), 4,59 (t, 1H, J = 7.0 Hz), 2.39 (d, 1H, J = 2.0), 1.72 (d, 3H, J = 6.5 Hz),
1.23-1.07 (m, 2H), 0.90 (s, 9H), 0.13 (s, 3H), 0.10 (s, 3H), 0.06 (s, 9H). IR (film, cm-1): 3345, 2951,
2855, 2355, 1463, 1440, 1321, 1247, 1060. SMBR (m/z, intensité relative) : 311 (M+ - CH3, 5), 269
(M+ - C4H9, 10), 147 (90), 74 (100). SMHR calculée pour C16H31O2Si2 (M+ - CH3): 311.1862,
observée : 311.1867.
(±) (E)-7-tert-Butyldiméthylsilanoxy-8-triméthylsilanyloct-2-én-5-yn-4-one (157)
OTBS
Me3Si
O
L’alcool 156 (710 mg, 2.17 mmol) a été dissous dans 15 mL de dichlorométhane à 0 °C. Du DMP (1.29
g, 3.04 mmol) a été ajouté par petites portions et la réaction a été laissée sous agitation durant 3 h. Le
mélange réactionnel a été neutralisé avec une solution aqueuse saturée en bicarbonate de sodium
contenant 10% de thiosulfate de sodium et a été extraite avec 3 portions d’éther éthylique. Les fractions

111
organiques ont été combinées, lavées avec de la saumure, séchées avec du sulfate de magnésium
anhydre et concentrées sous pression réduite. Le résidu brut a été purifié par chromatographie éclair sur
gel de silice en éluant avec un mélange d'acétate d'éthyle et d'hexanes (1:20). Une huile incolore a été
obtenue (614 mg, 87%).
RMN 1H (300 MHz, CDCl3) δ (ppm) : 7.16 (dq, 1H, J = 15.5 et 7.0 Hz), 6.16 (d, 1H, J = 15.5 Hz),
4.72 (t, 1H, J = 7.0 Hz), 1.97 (dd, 3H, J = 6.5 et 1.0 Hz), 1.28-1.14 (m, 2H), 0.91 (s, 9H), 0.15 (s, 3H),
0.12 (s, 3H), 0.08 (s, 9H). RMN 13C (75 MHz, CDCl3) δ (ppm): 177.8 (s), 149.2 (d), 133.7 (d), 94.9 (s),
81.3 (s), 60.6 (d), 27.5 (t), 25.6 (q), 18.3 (q), 18.1 (s), -0.8 (q), -4.7 (q), -5.0 (q). IR (film, cm-1): 2955,
2213, 1727, 1632, 1252, 1073. SMBR (m/z, intensité relative) : 309 (M+ - CH3, 10), 267 (M+ - C4H9,
75), 147 (100). SMHR calculée pour C16H29O2Si2 (M+ - CH3) : 309.1706, observée : 309.1711.
(±) (E)-7-tert-Butyldiméthylsilanoxy-8-triméthylsilanyl-4-([1’,3’]dithian-2’-yl)-oct-2-én-5-yn-4-ol
(158)
HO
S
S
OTBS
Me3Si
Au 1,3-dithiane (611 mg, 5.08 mmol) à -78 °C dans le THF (24 mL) a été ajouté du n-butyllithium
(1.50 M dans hexanes, 3.70 mL, 5.54 mmol). La solution a été agitée durant 1 h à -10 °C pour ensuite
être refroidie à -95 °C, température à laquelle l’ynone 157 (1.50 g, 4.61 mmol) dans 6.0 mL de THF a
été ajoutée. Suite à sa complétion, la réaction a été neutralisée à -80 °C par l’ajout d’une solution
aqueuse saturée de chlorure d’ammonium, a été réchauffée à température ambiante et ensuite a été
extraite avec de l’éther éthylique. Les fractions organiques ont été combinées, lavées avec de la
saumure, séchées avec du sulfate de magnésium anhydre et concentrées sous pression réduite. Le
produit brut a été purifié par chromatographie éclair sur gel de silice en éluant avec un mélange
d'acétate d'éthyle et d'hexanes (1:10). Une huile jaune (1.17 g, 57%) a été obtenue.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 6.12 (dq, 1H, J = 15.0 et 6.5 Hz), 5.64 (d, 1H, J = 15.0 Hz),
4.63 (t, 1H, J = 6.5 Hz), 3.91 (s, 1H), 3.24-3.13 (m, 2H), 3.06 (s, 1H), 2.75-2.64 (m, 2H), 2.09-1.92 (m,
2H), 1.76 (d, 3H, J = 6.5 Hz), 1.25-1.09 (m, 2H), 0.90 (s, 9H), 0.15 (s, 3H), 0.12 (s, 3H), 0.08 (s, 9H).
IR (film, cm-1): 3454, 2949, 2860, 2344, 1429, 1339, 1250, 1071. SMBR (m/z, intensité relative) : 429

112
(M+ - CH3, 10), 387 (M+ - C4H9, 10), 147 (100), 121 (90), 74 (95). SMHR calculée pour C20H37O2S2Si2
(M+ - CH3) : 429.1773, observée : 429.1763.
(±)-(E)-4-Acétoxy-7-tert-butyldiméthylsilanoxy-8-triméthylsilanyl-4-([1’,3’]dithian-2’-yl)-oct-2-én-
5-yne (159)
AcO
S
S
OTBS
Me3Si
L’alcool 158 (2.30 g, 5.17 mmol), du DMAP (50 mg, catalytique) et de la triéthylamine (2.90 mL, 20.7
mmol) ont été dissous dans 30 mL de dichlorométhane. À cette solution a été ajouté l’anhydride
acétique (1.22mL, 12.9 mmol) et le mélange a été agité à température ambiante durant 16 h. Une
solution aqueuse saturée de chlorure d’ammonium a alors été ajoutée et la réaction a été extraite avec
du dichlorométhane. Les fractions organiques ont été combinées, séchées avec du sulfate de
magnésium anhydre et concentrées sous pression réduite. Le produit brut a été purifié par
chromatographie éclair sur gel de silice en éluant avec un mélange d'acétate d'éthyle et d'hexanes
(1:12). Une huile (2.26 g, 90%) a été obtenue.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 6.27-6.18 (m, 1H), 5.77 (d, 1H, J = 15.0 Hz), 4.91 (s, 1H), 4.67
(t, 1H, J = 7.0 Hz), 2.99-2.77 (m, 4H), 2.11-2.03 (m, 1H), 2.04 (s, 3H), 1.92-1.79 (m, 1H), 1.79 (d, 3H,
J = 6.5 Hz), 1.25-1.11 (m, 2H), 0.90 (s, 9H), 0.16 (s, 3H), 0.12 (s, 3H), 0.07 (s, 9H). IR (film, cm-1):
2949, 2342, 1746, 1627, 1426, 1225, 1069. SMBR (m/z, intensité relative) : 429 (M+ - C4H9, 10), 426
(M+ - CH3CO2H, 15), 119 (100), 74 (80). SMHR calculée pour C19H33O3S2Si2 (M+ - C4H9) : 429.1410,
observée : 429.1400.

113
(±) 5-([1’,3’]Dithian-2’-yl)-3-méthyl-1-triméthylsilanylocta-3,4,6-trién-2-ol (160)
C
OH
SS
Me3Si
La réaction a été effectuée sous les conditions pour obtenir 131 avec l’alcool propargylique 151 (210
mg, 0.56 mmol). La purification du produit brut par chromatographie éclair sur gel de silice, en éluant
avec un mélange d'acétate d'éthyle et d'hexanes (1:3), a conduit à l’isolation de l’allène désiré (163 mg,
88%).
RMN 1H (300 MHz, CDCl3) δ (ppm) : 5.84-5.77 (m, 2H), 4.68 (s, 1H), 4.23-4.15 (m, 1H), 3.01-2.81
(m, 4H), 2.14-2.07 (m, 1H), 1.94-1.82 (m, 1H), 1.81 (s, 3H), 1.78 (d, 3H, J = 5.0 Hz), 1.01 (dd, 1H, J =
15.0 et 4.5 Hz), 0.92 (dd, 1H, J = 15.0 et 9.5 Hz), 0.07 (s, 9H). IR (film, cm-1): 3439, 2899, 1422, 1246,
1049, 953. SMBR (m/z, intensité relative) : 328 (M+, 5), 212 (60), 137 (100). SMHR calculée pour
C16H28OS2Si : 328.1351, observée : 328.1346.
(±) Lactone 162
CO2Et
S S
O
O
Me3Si
L’allène 160 (50 mg, 0.15 mmol) a été dissous dans 1 mL de dichlorométhane, du mono éthyl ester
fumarique (26 mg, 0.18 mmol) et du DMAP (4 mg, 0.03 mmol) ont été ajoutés et la solution a été
refroidie à -10 °C. Du DCC (37 mg, 0.18 mmol) a alors été additionné et suite à sa complétion, la
réaction a été concentrée sous pression réduite. Une purification du produit brut par chromatographie
éclair sur gel de silice, en éluant avec un mélange d'acétate d'éthyle et d'hexanes (1:7), a conduit à
l’isolation du produit d’estérification.

114
Ce solide visqueux a été immédiatement dissous dans le benzène et la solution résultante chauffée à
reflux durant 16 h. La réaction a été concentrée sous pression réduite et le résidu purifié par
chromatographie éclair sur gel de silice en éluant avec un mélange d'acétate d'éthyle et d'hexanes (1:7)
pour procurer la lactone désirée (9 mg, 45% pour les 2 étapes).
RMN 1H (300 MHz, CDCl3) δ (ppm) : 6.54 (d, 1H, J = 7.0 Hz), 4.72 (dd, 1H, J = 12.5 et 4.0 Hz), 4.65
(s, 1H), 4.20 (dq, 2H, J = 7.0 et 1.5 Hz), 3.69 (d, 1H, J = 11.0 Hz), 2.96 (dd, 1H, J = 11.0 et 5.0 Hz),
2.95-2.80 (m, 4H), 2.76-2.70 (m, 1H), 2.14-2.04 (m, 1H), 2.11 (d, 3H, J = 2.0 Hz), 1.91-1.81 (m, 1H),
1.33-1.27 (m, 1H), 1.27 (t, 3H, J = 7.0 Hz), 1.08 (dd, 1H, J = 15.0 et 4.0 Hz), 0.95 (d, 3H, J = 7.0 Hz),
0.13 (s, 9H). IR (film, cm-1): 2951, 1745, 1729, 1412, 1290, 1175, 1078, 1032. SMBR (m/z, intensité
relative) : 454 (M+, 5), 309 (100), 157 (90), 73 (100). SMHR calculée pour C22H34O4S2Si : 454.1668,
observée : 454.1673.
(±) Diester 164
CO2Et
S S
MeO2C
La lactone 162 (50 mg, 0.15 mmol) a été dissoute dans 1 mL de THF et la solution a été refroidie à -10
°C. Du TBAF (1.0 M dans le THF, 92 µL, 0.09 mmol) a alors été additionné et après sa complétion, la
réaction a été neutralisée avec une solution aqueuse de HCl (1 mL, 0.5 N). Le mélange a été extrait
avec de l’acétate d’éthyle, les fractions organiques ont été combinées, lavées avec de la saumure et
séchées avec du sulfate de magnésium anhydre et concentrées sous pression réduite.
Le produit brut a été dissous dans 1 mL de THF, refroidi à 0 °C et du diazométhane (en solution dans
Et20) a été additionné jusqu’à persistance d’une coloration jaune. La réaction a été concentrée sous
pression réduite et purifiée par chromatographie éclair sur gel de silice en éluant avec un mélange
d'acétate d'éthyle et d'hexanes (1:5) pour procurer le diester désiré (11 mg, 34% pour les 2 étapes).
RMN 1H (300 MHz, CDCl3) δ (ppm) : 6.97 (dd, 1H, J = 17.0 et 11.0 Hz), 6.47 (d, 1H, J = 6.5 Hz),
5.39 (d, 1H, J = 17.0 Hz), 5.24 (d, 1H, J = 11.0 Hz), 4.54 (s, 1H), 4.44 (d, 1H, J = 6.0 Hz), 4.09 (dq,
2H, J = 7.0 et 3.0 Hz), 3.66 (s, 3H), 3.29 (t, 1H, J = 6.0 Hz), 2.96-2.83 (m, 4H), 2.73 (dt, 1H, J = 13.5

115
et 4.0 Hz), 2.12-2.04 (m, 1H), 2.01 (s, 3H), 1.21 (t, 3H, J = 7.0 Hz), 0.92 (d, 3H, J = 7.0 Hz), 1.96-1.76
(m, 1H). IR (film, cm-1): 2955, 1741, 1728, 1435, 1277, 1182, 1029. SMBR (m/z, intensité relative) :
396 (M+, 45), 231 (30), 158 (100). SMHR calculée pour C20H28O4S2 : 396.1429, observée : 396.1422.
(±) Aldéhyde 165
CO2Et
O
O
O
Me3Si
La réaction a été effectuée sous les mêmes conditions que pour l’obtention du dihydrofuranne 133 avec
le dithiane 162 (17 mg, 0.04 mmol). La purification du produit brut par chromatographie éclair sur gel
de silice, en éluant avec un mélange d'acétate d'éthyle et d'hexanes (1:3), a conduit à l’isolation de
l’aldéhyde désiré (5 mg, 38%). Ce produit s’est avéré instable à l’air et la chaleur.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 9.56 (s, 1H), 6.94 (d, 1H, J = 6.5 Hz), 4.80 (dd, 1H, J = 12.0 et
4.5 Hz), 4.20 (dq, 2H, J = 7.0 et 2.5 Hz), 3.68 (d, 1H, J = 9.0 Hz), 3.14 (dd, 1H, J = 9.0 et 4.5 Hz),
2.89-2.82 (m, 1H), 1.79 (d, 3H, J = 3.0 Hz), 1.34-1.25 (m, 1H), 1.27 (t, 3H, J = 7.0 Hz), 1.17-1.09 (m,
1H), 1.16 (d, 3H, J = 7.0 Hz), 0.14 (s, 9H). IR (film, cm-1): 2954, 1742, 1727, 1707, 1295, 1251, 1176,
1036. SMBR (m/z, intensité relative) : 364 (M+, 10), 220 (75), 75 (100). SMHR calculée pour
C19H28O5Si : 364.1706, observée : 364.1711.
(±) Adduit tricyclique 166
CO2Et
O
O
O
Me3SiH
OEt
L’aldéhyde 165 (5 mg, 0.01 mmol) a été dissous dans 0.5 mL d’éther éthylvinylique et le Yb(fod)3 (1
mg, 0.01 mmol) a été ajouté. La réaction a été purgée à l’argon à trois reprises et le mélange a été agité
à température ambiante durant 4 jours. De l’eau (2 mL) et de l’éther éthylique (1 mL) ont ensuite été

116
ajoutés et la solution a été agitée vigoureusement durant 4 h. Le mélange a été extrait avec de l’éther
éthylique, les fractions ont été combinées, lavées avec de la saumure, séchées avec du sulfate de
magnésium anhydre et concentrées sous pression réduite. Une purification du résidu brut par
chromatographie éclair sur gel de silice, en éluant avec un mélange d'acétate d'éthyle et d'hexanes (1:3),
a conduit à l’isolation de l’adduit tricyclique désiré (2 mg, 33%) comme mélange inséparable de deux
diastéréoisomères instables.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 6.29 (s, 1H), 5.05 (d, 1H, J = 6.0 Hz), 4.80 (dd, 1H, J = 12.0 et
5.0 Hz), 4.40 (s, 1H), 4.25 (q, 2H, J = 7.0 Hz), 3.89 (dq, 1H, J = 9.5 et 7.0 Hz), 3.61-3.53 (m, 2H),
2.95-2.86 (m, 1H), 2.20-2.09 (m, 2H), 1.87 (d, 3H, J = 2.0 Hz), 1.80-1.71 (m, 1H), 1.28 (t, 3H, J = 6.0
Hz), 1.24 (t, 3H, J = 7.0 Hz), 1.30-1.09 (m, 2H), 0.97 (d, 3H, J = 6.5 Hz), 0.12 (s, 9H). SMBR (m/z,
intensité relative) : 436 (M+, 35), 392 (100), 390 (100), 157 (90).
(±) Dihydrofuranne 167
O
CO2Me
S S
Me3Si
La réaction a été effectuée sous les conditions pour obtenir 124 avec l’allène 160 (270 mg, 8.82 mmol).
La purification du produit brut par chromatographie éclair sur gel de silice, en éluant avec un mélange
d'acétate d'éthyle et d'hexanes (1:5), a conduit à l’isolation du dihydrofuranne désiré (225 mg, 90%).
RMN 1H (300 MHz, CDCl3) δ (ppm) : 6.04 (d, 1H, J = 5.0 Hz), 4.93 (dm, 1H, J = 11.5 Hz), 4.82 (s,
1H), 4.77-4.64 (m, 1H), 3.71 (s, 3H), 3.02-2.76 (m, 5H), 2.68 (dd, 1H, J = 11.0 et 6.0 Hz), 2.19-2.09
(m, 1H), 1.93-1.84 (m, 1H), 1.92 (s, 3H), 0.96 (d, 3H, J = 7.0 Hz), 0.92-0.84 (m, 2H), 0.09 (s, 9H).
RMN 13C (75 MHz, CDCl3) δ (ppm): 172.5 (s), 134.9 (d), 130.4 (s), 125.7 (s), 87.9 (d), 79.0 (d), 51.3
(q), 50.6 (d), 47.9 (d), 33.1 (d), 32.3 (s), 32.2 (t), 25.3 (t), 22.3 (t), 17.4 (q), 11.6 (q), -0.8 (q). IR (film,
cm-1): 3217, 2946, 1740, 1629, 1250, 1171. SMBR (m/z, intensité relative) : 412 (M+, 20), 297 (100).
SMHR calculée pour C20H32O3S2Si : 412.1562, observée : 412.1575.

117
(±) Aldéhyde 170
O
O
CO2Me
Me3Si
La réaction a été effectuée sous les conditions pour obtenir 133 avec le dihydrofuranne 167 (15 mg,
0.04 mmol). La purification du produit brut par chromatographie éclair sur gel de silice, en éluant avec
un mélange d'acétate d'éthyle et d'hexanes (1:5), a conduit à l’isolation de l’aldéhyde désiré (9 mg,
82%). Ce produit s’est avéré instable à l’air et la chaleur.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 9.53 (s, 1H), 6.64 (d, 1H, J = 5.0 Hz), 4.96 (d, 1H, J = 11.5 Hz),
4.73 (dt, 1H, J = 10.0 et 4.5 Hz), 3.75 (s, 3H), 3.07 (sext, 1H, J = 7.0 Hz), 2.75 (dd, 1H, J = 11.0 et 6.0
Hz), 1.84 (s, 3H), 1.06 (d, 3H, J = 7.5 Hz), 1.01 (dd, 1H, J = 14.5 et 4.0 Hz), 0.88 (dd, 1H, J = 14.5 et
10.0 Hz), 0.07 (s, 9H). IR (film, cm-1): 2949, 2734, 1736, 1704, 1432, 1250, 1168. SMBR (m/z,
intensité relative) : 322 (M+, 5), 307 (M+ - CH3, 10), 115 (100). SMHR calculée pour C17H26O4Si :
322.1600, observée : 322.1607.
(±) Acétal d’énol 171
O
O
CO2Me
Me3Si
OEt
H
La réaction a été effectuée sous les conditions pour obtenir l’acétal d’énol 166 avec l’aldéhyde 170 (30
mg, 0.09 mmol). La purification du produit brut par chromatographie éclair sur gel de silice, en éluant
avec un mélange d'acétate d'éthyle et d'hexanes (1:9), a conduit à l’isolation de l’adduit de
cycloaddition (30 mg, 83%).
RMN 1H (300 MHz, CDCl3) δ (ppm) : 6.68 (s, 1H, diast A), 6.61 (s, 1H, diast B), 5.12-5.05 (m, 1H),
5.01-4.92 (m, 1H), 4.88-4.77 (m, 1H), 3.97 (dq, 1H, J = 9.5 et 7.0 Hz), 3.69 (s, 3H, diast B), 3.68 (s,
3H, diast A), 3.60 (dq, 1H, J = 9.5 et 7.0 Hz), 2.73 (dd, 1H, J = 9.0 et 6.5 Hz, diast B), 2.69 (dd, 1H, J

118
= 10.0 et 7.0 Hz, diast A), 2.62-2.52 (m, 1H), 2.13-2.05 (m, 1H), 1.83-1.71 (m, 1H), 1.69 (s, 3H, diast
A), 1.68 (s, 3H, diast B), 1.46-1.33 (m, 1H), 1.25 (t, 3H, J = 7.0 Hz, diast B), 1.24 (t, 3H, J = 7.0 Hz,
diast A), 1.06-0.93 (m, 1H), 0.99 (d, 3H, J = 7.0 Hz), 0.76 (dd, 1H, J = 14.5 et 8.0 Hz, diast B), 0.62
(dd, 1H, J = 14.5 et 11.0 Hz, diast A), 0.02 (s, 9H, diast A), 0.01 (s, 9H, diast B). IR (film, cm-1): 2967,
1733, 1629, 1239, 1164, 1060. SMBR (m/z, intensité relative) : 394 (M+, 20), 115 (100). SMHR
calculée pour C21H34O5Si : 394.2175, observée : 394.2178.
2-Bromo-1,1-diéthoxyprop-2-ène (186)
C(OEt)2Br
Méthode A146 :
À la bromoacroléine (185, 5.00g, 37.0 mmol) et le triéthylorthoacétate (13.6 mL, 74.0 mmol) dissous
dans l’éthanol anhydre (2.00 mL) a été ajouté une quantité catalytique d’acide p-toluènesulfonique. La
solution a ensuite été chauffée à reflux durant 24 h, puis refroidie à la température ambiante et
concentrée sous pression réduite. Une distillation sous pression réduite (25 mmHg) a permis d’isoler
l’acétal désiré (66-68 °C, 3.17 g) avec un rendement de 41%.
Méthode B :
À la bromoacroléine (185, 200 mg, 1.48 mmol) et le triéthylorthoacétate (325 µL, 1.78 mmol) a été
ajouté une quantité catalytique d’acide p-toluènesulfonique. La solution a été chauffée à 35 °C dans un
four à micro-ondes (puissance de 5 W sans refroidissement) durant 40 minutes puis refroidie à la
température ambiante. Une purification du mélange brut par chromatographie éclair sur gel de silice en
éluant avec un mélange d’éther éthylique/hexanes (1 : 19) a mené à l’isolation de l’acétal connu de la
littérature sous forme d’une huile incolore (305 mg, 84%).
RMN 1H (300 MHz, CDCl3) δ (ppm) : 6.11 (s, 1H), 5.72 (s, 1H), 4.82 (s, 1H), 3.68-3.47 (dm, 4H), 1.24
(t, 6H, J = 7.0).

119
(±) Acétal 188
OEt
Me3Si OEt
OTBS
L’alcyne 155 (86 mg, 0.26 mmol) et le bromure vinylique 186 (50 mg, 0.24 mmol) ont été dissous dans
la n-butylamine (1.0 mL) et le mélange a été purgé 3 fois avec de l’argon. De l’iodure de cuivre(I) (5
mg, 0.02 mmol), de la triphénylphosphine (6 mg, 0.02 mmol) et du PdCl2(PPh3)2 (6 mg, 0.01 mmol) ont
ensuite été ajoutés et le mélange réactionnel a de nouveau été purgé à l’argon pour ensuite être agité à
température ambiante. Suite à sa complétion, la réaction a été neutralisée par l’ajout d’une solution
saturée en chlorure d’ammonium et a été extraite avec 3 portions d’éther éthylique. Les fractions
organiques ont été combinées, lavées avec de la saumure, séchées avec du sulfate de magnésium
anhydre et concentrées sous pression réduite. Le résidu brut a été purifié par chromatographie éclair sur
gel de silice en éluant avec un mélange d'acétate d'éthyle et d'hexanes (gradient de 1:10 à 1:5). Une
huile incolore instable à l’air et à la chaleur a été obtenue (26 mg, 24%, 48% basé sur la récupération du
bromure de départ).
RMN 1H (300 MHz, CDCl3) δ (ppm) : 5.69 (s, 1H), 5.55 (s, 1H), 4.84 (s, 1H), 4.68 (dd, 1H, J = 8.0 et
6.0 Hz), 3.63 (quint, 2H, J = 7.0 et 7.0 Hz), 3.49 (dq, 2H, J = 9.0 et 6.5 Hz), 1.22 (t, 6H, J = 7.0 Hz),
1.21-1.14 (m, 2H), 0.90 (s, 9H), 0.15 (s, 3H), 0.14 (s, 3H), 0.07 (s, 9H). IR (film, cm-1): 2953, 2888,
1611, 1331, 1251, 1066. SMBR (m/z, intensité relative) : 339 (M+ - OEt, 10), 253 (50), 147 (100), 103
(75), 73 (100). SMHR calculée pour C18H35O2Si2 (M+ - OEt) : 339.2175, observée : 339.2173.
(±) 5-tert-Butyldiméthylsilanoxy-2-méthylène-6-triméthylsilanylhex-3-yn-1-ol (190)
Me3Si OH
OTBS
La réaction a été effectuée sous les conditions pour obtenir l’acétal 188 avec le 2-bromoprop-2-én-1-ol
(187, 880 mg, 6.42 mmol) et de la diisopropylamine (25 mL) comme solvant au lieu de la n-butylamine.

120
La purification du produit brut par chromatographie éclair sur gel de silice, en éluant avec un mélange
d'acétate d'éthyle et d'hexanes (1:3), a conduit à l’isolation de l’adduit de Sonogashira (1.49 g, 75%).
RMN 1H (300 MHz, CDCl3) δ (ppm) : 5.50 (s, 1H), 5.42 (s, 1H), 4.66 (dd, 1H, J = 8.5 et 6.0 Hz), 4.12
(d, 2H, J = 4.5 Hz), 1.61 (large s, 1H), 1.21 (dd, 1H, J = 14.5 et 9.0 Hz), 1.13 (dd, 1H, J = 14.5 et 6.0
Hz), 0.90 ( s, 9H), 0.13 (s, 3H), 0.11 (s, 3H), 0.06 (s, 9H). IR (film, cm-1): 3353, 2953, 2856, 2309,
1613, 1461, 1329, 1252, 1067. SMBR (m/z, intensité relative) : 255 (M+ - C4H9, 10), 147 (100), 75
(90). SMHR calculée pour C12H23O2Si2 (M+ - C4H9) : 255.1237, observée : 255.1241.
2-Bromo-1-méthoxyméthoxyprop-2-ène (193)
OMOM
Br
Du 2-bromoprop-2-én-1-ol (187, 10.0 g, 73.0 mmol), du bromure de lithium (6.34 g, 73.0 mmol) et de
l’acide p-toluènesulfonique (1.40 g, 7.30 mmol) ont été dissous dans le diméthoxyméthane (300 mL).
La solution a été agitée à température ambiante durant 24 h, puis concentrée sous pression réduite. De
l’éther éthylique et de la saumure ont été ajoutés et le mélange a été extrait avec de l’éther éthylique.
Les fractions organiques ont été combinées, séchées avec du sulfate de magnésium anhydre et
concentrées sous pression réduite. La purification du produit brut par filtration sur gel de silice, en
éluant avec un mélange d'éther éthylique et d'hexanes (1:9), a conduit à l’isolation de l’éther désiré
(13.2 g, 81%).
RMN 1H (300 MHz, CDCl3) δ (ppm) : 5.92 (d, 1H, J = 1.0 Hz), 5.61 (s, 1H), 4.67 (s, 2H), 4.18 (s, 2H),
3.39 (s, 3H). IR (film, cm-1): 2942, 1641, 1150, 1103, 1048. SMBR (m/z, intensité relative) : 181 (M+,
10), 91 (75), 74 (100). SMHR calculée pour C4H7OBr (M+ - CH2O) : 149.9680, observée : 149.9672.

121
(±) 5-tert-butyldiméthylsilanoxy-2-méthoxyméthoxyméthyl-6-triméthylsilanylhex-1-én-3-yne (194)
Me3Si OMOM
OTBS
La réaction a été effectuée sous les conditions pour obtenir l’ényne 188 avec le bromure vinylique 193
(1.50 g, 8.28 mmol) et de la triéthylamine (27 mL) comme solvant au lieu de la n-butylamine. La
purification du produit brut par chromatographie éclair sur gel de silice, en éluant avec un mélange
d'acétate d'éthyle et d'hexanes (1:12), a conduit à l’isolation de l’adduit de Sonogashira (2.93 g, 90%).
RMN 1H (300 MHz, CDCl3) δ (ppm) : 5.50 (s, 1H), 5.45 (s, 1H), 4.65 (s, 2H), 4.77-4.63 (m, 1H), 4.03
(s, 2H), 3.38 (s, 3H), 1.24-1.08 (m, 2H), 0,90 (s, 9H), 0.13 (s, 3H), 0.11 (s, 3H), 0.06 (s, 9H). IR (film,
cm-1): 2951, 2360, 1614, 1467, 1252, 1151, 1110, 1060. SMBR (m/z, intensité relative) : 325 (M+ -
OCH3, 2), 299 (M+ - C4H9, 5), 147 (95), 74 (100). SMHR calculée pour C17H33O2Si2 (M+ - OCH3) :
325.2019, observée : 325.2010.
(±) 5,6-Époxy-5-méthoxyméthoxy-1-triméthylsilanylhex-3-yn-2-ol (195)
Me3Si OMOM
OH
O
L’alcène 194 (1.07 g, 3.00 mmol) a été dissous dans le dichlorométhane (15 mL), la solution a été
placée à 0 °C et du m-CPBA (890 mg, 3.61 mmol) a été ajouté par petites portions. Après sa
complétion, la réaction a été neutralisée par l’addition d’une solution aqueuse saturée en bicarbonate de
sodium comprenant 10% de thiosulfate de sodium. Le mélange a été extrait au dichlorométhane et
l’époxyde brut a été utilisé immédiatement pour la transformation suivante.
À cet époxyde dissous dans le THF (15 mL) à 0 °C a été additionné du TBAF (3.60 mL, 3.60 mmol).
Après sa complétion, la réaction a été neutralisée par l’addition d’une solution aqueuse saturée en
chlorure d’ammonium et le mélange a été extrait à l’éther éthylique. Les fractions organiques ont été
combinées, lavées à la saumure, séchées avec du sulfate de magnésium anhydre et concentrées sous

122
pression réduite. La purification du produit brut par chromatographie éclair sur gel de silice, en éluant
avec un mélange d'acétate d'éthyle et d'hexanes (1:3), a conduit à l’isolation de l’époxyalcool désiré
(533 mg, 69% pour les 2 étapes).
RMN 1H (300 MHz, CDCl3) δ (ppm) : 4.65 (s, 2H), 4.52 (m, 1H), 3.73 (AB q, 2H, J = 13.0 Hz), 3.37
(s, 3H), 3.00 (AB q, 2H, J = 6.0 Hz), 1.79 (d, 1H, J = 5.5 Hz), 1.16 (d, 2H, J = 8.0 Hz), 0.07 (s, 9H). IR
(film, cm-1): 3436, 2953, 2362, 1645, 1414, 1250, 1146, 1110, 1057. SMBR (m/z, intensité relative) :
276 (MNH4+, 15), 179 (60), 90 (100). SMHR calculée pour C12H26NO4Si (MNH4
+) : 276.1631,
observée : 276.1637.
(±) 2-Méthoxyméthoxyméthyl-4-méthyl-6-triméthylsilanylhexa-2,3-diène-1,5-diol (196)
C
OH
OMOM
Me3Si
OH
Au cyanure de cuivre(I) (1.21 g, 13.5 mmol) à -25 °C dans le THF (18 mL) sous agitation mécanique a
été ajouté lentement une solution fraîche du bromure de méthylmagnésium (3.0 M dans Et2O, 4.50 mL,
13.5 mmol). Après 1 h d’agitation, l’époxyde propargylique 195 (580 mg, 2.25 mmol) dissous dans 5
mL de THF a ensuite été additionné. Après sa complétion, la réaction a été neutralisée par l’ajout d’une
solution aqueuse saturée de chlorure d’ammonium contenant 10% d’hydroxyde d’ammonium et la
solution résultante a été agitée à température ambiante jusqu’à la persistance d’une coloration bleue. À
ce point, la réaction a été extraite à l’éther éthylique et les fractions organiques ont été combinées,
lavées avec de la saumure et séchées avec du sulfate de magnésium anhydre. L’évaporation sous
pression réduite et la purification du produit brut par chromatographie éclair sur gel de silice, en éluant
avec un mélange d'acétate d'éthyle, de méthanol et d'hexanes (10:1:10), a conduit à l’isolation d’une
huile légèrement jaune (550 mg, 89%). Ce produit s’est avéré instable à l’air et la chaleur.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 4.67 (AB q, 2H, J = 6.5 Hz), 4.21-4.11 (m, 3H), 4.16 (d, 2H, J =
3.5 Hz), 3.37 (s, 3H), 2.42 (m, 1H), 1.73 (s, 3H), 1.00 (dd, 1H, J = 14.5 et 5.5 Hz), 0.87 (dd, 1H, J =
14.5 et 9.5 Hz), 0.05 (s, 9H). IR (film, cm-1): 3395, 2946, 1641, 1408, 1042. SMBR (m/z, intensité
relative) : 256 (M+ - H2O, 2), 117 (85), 95 (75), 81 (100). SMHR calculée pour C13H24O3Si (M+ - H2O)
: 256.1495, observée : 256.1501.

123
(±) Alcool tertiaire 197
OH
TMS OMOM
OH
Produit secondaire obtenu lors de la formation du diol 196 lorsque le réactif de Grignard contenait des
sels de magnésium.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 4.70 (AB q, 2H), 4.53 (t, 1H, J = 7.5 Hz), 3.66 (d, 1H, J = 10.0
Hz), 3.52 (d, 1H, J = 10.0 Hz), 3.41 (s, 3H), 3.14 (s, 1H), 2.07 (s, 1H), 1.75-1.61 (m, 2H), 1.14 (d, 2H, J
= 8.0 Hz), 1.04 (t, 3H, J = 7.5 Hz), 0.07 (s, 9H).
(±) 5-Hydroxy-2-méthoxyméthoxyméthyl-4-méthyl-6-triméthylsilanylhexa-2,3-diénal (198)
C
OH
OMOM
Me3Si O
Au diol 196 (50 mg, 0.18 mmol) dissous dans le dichlorométhane (1.5 mL) à la température ambiante a
été ajouté du MnO2 (317 mg, 3.65 mmol). Après sa complétion, la réaction a été filtrée sur célite et le
filtrat a été concentré sous pression réduite. Le produit brut (37 mg, mélange 3 : 1 de 198/199, 75%),
très instable à l’air, n’a pas été purifié davantage et a été immédiatement utilisé pour la réaction
suivante.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 9.54 (s, 1H, diast A), 9.52 (s, 1H, diast B), 4.65 (s, 2H, diast B),
4.64 (s, 2H, diast A), 4.39-4.32 (m, 1H, diast A), 4.33 (t, 1H, J = 10.5 Hz, diast B), 4.21 (s, 2H, diast
B), 4.17 (s, 2H, diast A), 3.36 (s, 3H, diast A), 3.35 (s, 3H, diast B), 2.00-1.98 (m, 1H), 1.89 (s, 3H),
1.02-0.99 (m, 2H), 0.08 (s, 9H). IR (film, cm-1): 3554, 2953, 1948, 1677, 1248, 1202, 1054. SMBR
(m/z, intensité relative) : 290 (MNH4+, 25), 273 (MH+, 5), 225 (100). SMHR calculée pour C13H25O4Si
(MH+) : 273.1522, observée : 273.1527.

124
(±) 2-Méthoxyméthoxyméthyl-4-méthyl-5-oxo-6-triméthylsilanylhexa-2,3-diénal (199)
C
O
OMOM
Me3Si O
(7 mg, 10%)
RMN 1H (300 MHz, CDCl3) δ (ppm) : 9.61 (s, 1H), 4.66 (s, 2H), 4.37 (d, 2H, J = 4.5 Hz), 3.37 (s, 3H),
2.50 (d, 1H, J = 10.5 Hz), 2.42 (d, 1H, J = 10.5 Hz), 1.97 (s, 3H), 0.10 (s, 9H). SMBR (m/z, intensité
relative) : 288 (MNH4+, 55), 271 (MH+, 70), 241 (100), 90 (100). SMHR calculée pour C13H23O4Si
(MH+) : 271.1366, observée : 271.1370.
Procédure générale pour l’oxydation du diol 196 avec IBX
Au diol 196 (50 mg, 0.18 mmol) dissous dans le solvant (1.5 mL) à la température ambiante a été ajouté
du IBX (56 mg, 0.20 mmol). Après sa complétion (2 jours), la réaction a été neutralisée par l’addition
d’une solution aqueuse saturée en bicarbonate de sodium contenant 10% de thiosulfate de sodium et a
été extraite avec de l’acétate d’éthyle. Les fractions organiques ont été combinées, lavées avec de la
saumure, séchées avec du sulfate de magnésium anhydre et concentrées sous pression réduite. Le
produit brut a été purifié par chromatographie éclair sur gel de silice en éluant avec un mélange
d’acétate d’éthyle et d’hexanes (1:3).
(±) Allène-ester 204
C
OH
OMOM
Me3SiCO2Me
À l’allène 198 (50 mg, 0.18 mmol) dissous dans le toluène (1.0 mL) à la température ambiante a été
ajouté le phosphorane 203 (154 mg, 0.45 mmol) et de la 2,6-lutidine (53 µL, 0.45). Après sa
complétion, la réaction a été concentrée sous pression réduite. Le produit brut a été purifié par
chromatographie éclair sur gel de silice en éluant avec un mélange d’acétate d’éthyle et d’hexanes (1:3)
pour procurer le produit d’oléfination désiré (20 mg, 33%) qui s’est avéré instable à l’air et la chaleur.

125
RMN 1H (300 MHz, CDCl3) δ (ppm) : 7.25 (d, 1H, J = 16.0 Hz), 6.04 (d, 1H, J = 16.0 Hz), 4.68 (d,
1H, J = 7.0 Hz), 4.61 (d, 1H, J = 6.5 Hz), 4.30 (d, 1H, J = 12.0 Hz), 4.28-4.21 (m, 1H), 4.19 (d, 1H, J
= 11.5 Hz), 3.74 (s, 3H), 3.36 (s, 3H), 2.03 (d, 1H, J = 5.0 Hz), 1.77 (s, 3H), 1.00 (dd, 1H, J = 14.5 et
5.5 Hz), 0.92 (dd, 1H, J = 15.0 et 9.0 Hz), 0.06 (s, 9H). IR (film, cm-1): 3491, 2951, 1943, 1708, 1621,
1273, 1169, 1046. SMBR (m/z, intensité relative) : 311 ((M+ - H2O)H+, 3), 281 (100), 150 (30). SMHR
calculée pour C16H27O4Si (MH+) : 311.1678, observée : 311.1684.
(±) Ester fumarique 205
Me3Si OMOM
O
O
O
CO2Et
La réaction a été effectuée sous les conditions pour obtenir l’ester 162 (première partie) avec l’alcool
195 (30 mg, 0.12 mmol). La purification du produit brut par chromatographie éclair sur gel de silice, en
éluant avec un mélange d'acétate d'éthyle et d'hexanes (1:7), a conduit à l’isolation du produit
d’estérification (36 mg, 84%) sous forme d’une huile sensible à la chaleur et à l’air.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 6.87 (d, 2H, J = 15.5 Hz), 5.60-5.54 (m, 1H), 4.64 (s, 2H), 4.26
(q, 2H, J = 7.0 Hz), 3.73 (dq, 2H, J = 11.0 et 3.5 Hz), 3.36 (s, 3H), 3.00 (AB q, 2H), 1.32 (t, 3H, J =
7.0 Hz), 1.33-1.19 (m, 2H), 0.09 (s, 9H). IR (film, cm-1): 2950, 2305, 1725, 1646, 1295, 1159, 1051.
SMBR (m/z, intensité relative) : 384 (MH+,1), 355 (M+ - C2H5, 5), 201 (80), 127 (80), 74 (100). SMHR
calculée pour C18H28O7Si : 384.1604, observée : 384.1599.
(±) 5-tert-Butyldiméthylsilanoxyméthyl-6-méthoxyméthoxyméthyl-3-méthyl-1-
triméthylsilanylhexa-3,4-dién-2-ol (207)
C
OH
OMOM
Me3Si
OTBS

126
La réaction a été effectuée sous les conditions pour obtenir l’éther silylé 154 avec l’allène 196 (130 mg,
0.47 mmol). La purification du produit brut par chromatographie éclair sur gel de silice, en éluant avec
un mélange d'acétate d'éthyle et d'hexanes (2:5), a conduit à l’isolation de l’éther silylé désiré (152 mg,
83%) comme mélange de 2 diastéréoisomères. Ce produit s’est avéré instable à la chaleur et l’air.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 4.66 (dd, 2H, J = 15.5 et 6.5 Hz), 4.24-4.06 (m, 5H), 3.37 (s,
3H), 1.71 (s, 3H), 1.01-0.85 (m, 2H), 0.88 (s, 9H), 0.05 (s, 6H), 0.05 (s, 9H). IR (film, cm-1): 3432,
2953, 1972, 1625, 1464, 1253, 1075. SMBR (m/z, intensité relative) : 371 ((M+ - H2O)H+, 1), 342 (30),
341 (100).
(±) Ester silylé 208
C
O
OMOM
Me3Si
OTBS
O
CO2Et
La réaction a été effectuée sous les conditions pour obtenir l’ester fumarique 162 (première partie) avec
l’allène 207 (55 mg, 0.14 mmol). La purification du produit brut par chromatographie éclair sur gel de
silice, en éluant avec un mélange d'acétate d'éthyle et d'hexanes (1:9), a conduit à l’isolation de l’allène
désiré (57 mg, 79%) comme mélange de 2 diastéréoisomères. Ce produit s’est avéré instable à l’air et la
chaleur.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 6.81 (s, 2H), 5.49-5.43 (m, 1H), 4.63 (s, 2H, diast A), 4.61 (s,
2H, diast B), 4.26 (q, 2H, J = 7.0 Hz), 4.19 (s, 2H, diast B), 4.15 (s, 2H, diast A), 4.11 (s, 2H, diast A),
4.06 (s, 2H, diast B), 3.36 (s, 3H, diast A), 3.34 (s, 3H, diast B), 1.71 (s, 3H, diast A), 1.70 (s, 3H, diast
B), 1.32 (t, 3H, J = 7.0 Hz), 1.28-1.04 (m, 2H), 0.88 (s, 9H), 0.05 (s, 3H), 0.03 (s, 3H), 0.03 (s, 9H). IR
(film, cm-1): 2951, 1966, 1724, 1642, 1295, 1256, 1153, 1046. SMBR (m/z, intensité relative) : 499 (M+
- CH3, 1), 457 (M+ - C4H9, 5), 269 (80), 209 (80), 195 (100). SMHR calculée pour C21H37O7Si2 (M+ -
C4H9) : 457.2078, observée : 457.2072.

127
(±) 2-Méthoxyméthoxyméthyl-4-méthylhexa-2,3,5-trién-1-ol (209)
C
OMOM
HO
Méthode A:
La réaction a été effectuée sous les conditions pour obtenir 130 avec l’éther silylé 208 (50 mg, 0.10
mmol). La purification du produit brut par chromatographie éclair sur gel de silice, en éluant avec un
mélange d'acétate d'éthyle et d'hexanes (1:3), a conduit à l’isolation de l’allénol 209 (13 mg, 33%).
Méthode B:
À l’éther silylé 208 (35 mg, 0.07 mmol) dissous dans l’éthanol (0.5 mL) a été ajouté une quantité
d’acide p-toluènesulfonique. La solution a été agitée à température ambiante durant une journée, puis
neutralisée par l’ajout de saumure. Le mélange a été extrait avec du dichlorométhane et les fractions
organiques ont été combinées, séchées avec du sulfate de magnésium anhydre et concentrées sous
pression réduite. La purification du produit brut par chromatographie éclair sur gel de silice, en éluant
avec un mélange d'éther éthylique et d'hexanes (1:5), a conduit à l’isolation de l’allène 209 (24 mg,
89%).
RMN 1H (300 MHz, CDCl3) δ (ppm) : 6.32 (dd, 1H, J = 17.5 et 10.5 Hz), 5.15 (d, 1H, J = 17.5 Hz),
5.07 (d, 1H, J = 10.5 Hz), 4.66 (s, 2H), 4.20 (s, 2H), 4.18 (s, 2H), 3.38 (s, 3H), 1.85 (s, 3H). IR (film,
cm-1): 3433, 2936, 2886, 1948, 1608, 1447, 1358, 1215, 1143, 1058. SMBR (m/z, intensité relative) :
202 (MNH4+, 100), 170 (10), 152 (25), 123 (43). SMHR calculée pour C10H20NO3 (MNH4
+) :
202.1443, observée : 202.1436.
(±) 5-tert-Butyldiméthylsilanoxy-1-chloro-2-méthylpent-3-yn-2-ol (228)
HO
TBSO
Cl
L’éther (tert-butyldiméthylsilanoxy)propargylique 126i (300 mg, 1.76 mmol) a été dissous dans 10 mL
de THF à -78 °C et une solution de n-butyllithium (1.92 M dans hexanes, 1.10 mL, 2.11 mmol) a été
lentement ajoutée. Le mélange réactionnel a alors été agité durant 1 h avant l’addition de la

128
chloroacétone (170 µL, 2.11 mmol). Après sa complétion, la réaction a été neutralisée avec une solution
aqueuse saturée en chlorure d’ammonium et la solution résultante a été extraite avec de l’éther
éthylique. Les fractions organiques ont été combinées, lavées avec de la saumure, séchées avec du
sulfate de magnésium anhydre et concentrées sous pression réduite. La purification du produit brut par
chromatographie éclair sur gel de silice, en éluant avec un mélange d'éther éthylique et d'hexanes (1:5),
a conduit à l’isolation de la chlorohydrine désirée (438 mg, 95%).
RMN 1H (300 MHz, CDCl3) δ (ppm) : 4.34 (s, 2H), 3.67 (d, 1H, J = 11.0 Hz), 3.57 (d, 1H, J = 11.0
Hz), 2.55 (s, 1H), 1.56 (s, 3H), 0.90 (s, 9H), 0.12 (s, 6H). IR (film, cm-1) : 3415, 2950, 1462, 1365,
1254, 1093. SMBR (m/z, intensité relative) : 247 (M+ - CH3, 5), 245 (M+ - OH, 4), 213 (M+ - CH2Cl,
23), 95 (90), 74 (100). SMHR calculée pour C12H22OSiCl (M+ - OH) : 245.1128, observée : 245.1130.
(±) 1-Chloro-5-méthoxyméthoxy-2-méthylpent-3-yn-2-ol (229)
HO
MOMO
Cl
La réaction a été effectuée sous les conditions pour obtenir la chlorohydrine 228 avec l’alcyne 126c
(1.25 g, 12.5 mmol). La purification du produit brut par chromatographie éclair sur gel de silice, en
éluant avec un mélange d'acétate d'éthyle et d'hexanes (1:3), a conduit à l’isolation de l’alcool désiré
(2.06 g, 73%).
RMN 1H (300 MHz, CDCl3) δ (ppm) : 4.70 (s, 2H), 4.25 (s, 2H), 3.68 (d, 1H, J = 11.0 Hz), 3.58 (d,
1H, J = 11.0 Hz), 3.38 (s, 3H), 2.66 (s, 1H), 1.58 (s, 3H). IR (film, cm-1) : 3413, 2951, 1445, 1358,
1156, 1060. SMBR (m/z, intensité relative) : 210 (MNH4+, 100), 111 (28). SMHR calculée pour
C8H17O3NCl (MNH4+) : 210.0897, observée : 210.0893.
(±) 5-tert-Butyldiméthylsilanoxy-1,2-époxy-2-méthylpent-3-yne (230)
TBSO
O
La chlorohydrine 228 (280 mg, 0.93 mmol) a été dissoute dans 5 mL d’éther éthylique à 0 °C et du
méthoxyde de sodium (103 mg, 1.91 mmol) a été ajouté par petites portions. Le mélange réactionnel a

129
alors été ammené à la température ambiante et a été agité durant 1 h, puis la solution a été directement
filtrée sur du gel de silice. La concentration du filtrat brut a procuré l’époxyde désiré (86 mg, 100%) ne
nécessitant pas de purification.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 4.31 (s, 2H), 2.99 (d, 1H, J = 5.5 Hz), 2.74 (d, 1H, J = 5.5 Hz),
1.54 (s, 3H), 0.90 (s, 9H), 0.11 (s, 6H). IR (film, cm-1) : 2934, 2314, 1261, 1098. SMBR (m/z, intensité
relative) : 211 (M+ - CH3, 5), 169 (M+ - C4H9, 90), 95 (80), 75 (100). SMHR calculée pour C11H19O2Si
(M+ - CH3) : 211.1154, observée : 211.1151.
(±) 1,2-Époxy-5-méthoxyméthoxy-2-méthylpent-3-yne (231)
MOMO
O
La réaction a été effectuée sous les conditions pour obtenir l’époxyde 230 avec la chlorohydrine 229
(110 mg, 0.57 mmol). La réaction a été directement filtrée sur du gel de silice et la concentration du
filtrat brut a procuré l’époxyde désiré (88 mg, 99%) ne nécessitant pas de purification.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 4.68 (s, 2H), 4.21 (s, 2H), 3.37 (s, 3H), 3.01 (d, 1H, J = 5.5 Hz),
2.75 (d, 1H, J = 5.5 Hz), 1.55 (s, 3H). IR (film, cm-1) : 2942, 1449, 1336, 1272, 1153, 1055. SMBR
(m/z, intensité relative) : 156 (M+, 2), 66 (100), 45 (100). SMHR calculée pour C8H12O3: 156.0786,
observée : 156.0789.
Procédure pour la préparation du bromure de 1-propénylmagnésium
Des granules de magnésium (267mg, 11 mmol) ont été introduites dans un ballon muni d’un réfrigérant
et le montage a été séché avec une flamme de propane sous le vide d’une pompe mécanique. Après
avoir refroidi le montage à la température ambiante, du THF (2 mL) a été additionné, suivi du 1,2-
dibromoéthane (43 µL, 0.5 mmol). Le mélange a été porté à reflux durant 10 min, puis refroidi à la
température ambiante et du 1-bromopropène (860 µL, 10 mmol) dissous dans 8 mL de THF a été
ajouté. Le mélange a été chauffé à reflux du solvant durant 3 h et refroidi à la température ambiante
pour son usage immédiat. Note: le bromure de Z-1-propénylmagnésium est obtenu peu importe si
le bromure de départ est E, Z ou un mélange équimolaire du 1-bromopropène.171

130
(±) (5Z)-4-tert-Butyldiméthylsiloxyméthyl-2-méthylhepta-2,3,5-trién-1-ol (232)
C
OH
OTBS
La réaction a été effectuée sous les mêmes conditions que pour l’obtention du diol 196 avec l’époxyde
230 (2.00 g, 8.85 mmol) et le bromure de 1-propénylmagnésium sous agitation magnétique à -10 °C. Le
produit brut (2.35 g, 99%) n’a pas été purifié pour des raisons d’instabilité.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 5.73 (dq, 1H, J = 7.0 et 11.0 Hz), 5.58 (dd, 1H, J = 11.5 et 1.0
Hz), 4.18 (d, 1H, J = 12.5 Hz), 4.12 (d, 1H, J = 12.0 Hz), 4.03 (s, 2H), 1.76 (s, 3H), 1.74 (dd, 3H, J =
7.0 et 1.5 Hz), 0.89 (s, 9H), 0.06 (s, 6H). IR (film, cm-1) : 3395, 2937, 1948, 1467, 1252, 1073. SMBR
(m/z, intensité relative) : 268 (M+, 3), 239 (30), 211 (30), 74 (100). SMHR calculée pour C15H28O2Si :
268.1858, observée : 268.1867.
(±) (5Z)-4-Méthoxyméthoxyméthyl-2-méthylhepta-2,3,5-trién-1-ol (233)
C
OH
OMOM
La réaction a été effectuée sous les conditions pour obtenir l’allène 232 avec l’époxyde 231 (370 mg,
2.36 mmol). Le produit brut a été purifié par chromatographie éclair en éluant avec un mélange
d’acétate d’éthyle et d’hexane (1:2) pour procurer l’allène désiré (340 mg, 80%) sous forme d’huile
jaune. Ce produit s’est avéré instable à l’air et à la chaleur.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 5.76 (dq, 1H, J = 11.0 et 7.0 Hz), 5.62 (dd, 1H, J = 11.0 et 1.5
Hz), 4.73 (d, 1H, J = 6.5 Hz), 4.63 (d, 1H, J = 6.5 Hz), 4.16 (d, 1H, J = 11.5 Hz), 4.06 (d, 1H, J = 11.5
Hz), 4.04 (s, 2H), 3.37 (s, 3H), 2.16 (s, 1H), 1.77 (dd, 3H, J = 7.0 et 1.5 Hz), 1.75 (s, 3H).

131
(Z)-6-tert-Butyldiphénylsiloxy-3-méthylhex-2-én-4-yn-1-ol (235a)
OTBDPS
OH
La solution constituée de l’éther silylé 126a (2.07 g, 7.06 mmol), de l’iodure vinylique 234175 (700 mg,
3.53 mmol) dans la pyrrolidine (884 µL, 37.5 mmol) et le THF (14 mL) à -10 oC a été purgée à trois
reprises avec de l’argon. De la triphénylarsine (43 mg, 0.14 mmol) et du CuI (20 mg, 0.14 mmol) ont
ensuite été ajoutés au mélange et la solution a été purgée de nouveau. Du PdCl2(PPh3)2 (49 mg, 0.07
mmol) a été additionné et la réaction a été agitée à la température ambiante après avoir été purgée à
trois reprises. Après sa complétion, la réaction a été neutralisée par l’ajout d’une solution aqueuse
saturée de chlorure d’ammonium, celle-ci a été extraite avec de l’éther éthylique et les fractions
organiques ont été combinées, lavées avec de la saumure, séchées avec du sulfate de magnésium
anhydre. L’évaporation sous pression réduite et la purification du produit brut par chromatographie
éclair sur gel de silice, en éluant avec un mélange d'acétate d'éthyle et d'hexanes (1:2), a conduit à
l’isolation d’une huile légèrement jaune (1.15 g, 90%). Ce produit s’est avéré instable à l’air et à la
chaleur et n’a pu être entreposé.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 7.72 (dd, 4H, J = 7.0 et 1.5 Hz), 7.46-7.36 (m, 6H), 5.83 (t, 1H,
J = 6.5 Hz), 4.47 (s, 2H), 4.21 (d, 2H, J = 6.5 Hz), 1.81 (s, 3H), 1.06 (s, 9H). IR (film, cm-1) : 3336,
2857, 2223, 1422, 1368, 1111. SMBR (m/z, intensité relative) : 307 (M+ - C4H9, 20), 199 (100), 139
(85). SMHR calculée pour C19H19O2Si (M+ - C4H9) : 307.1154, observée : 307.1149.
(Z)-6-(4’-Méthoxy)benzyloxy-3-méthylhex-2-én-4-yn-1-ol (235b)
OPMB
OH
La réaction a été effectuée sous les conditions pour obtenir l’éther silylé 235a avec l’éther 4-
méthoxybenzylpropargylique (126b, 178 mg, 1.01 mmol) et l’iodure vinylique 234 (100 mg, 0.50

132
mmol). L’adduit de Sonogashira désiré (110 mg, 89%) a été obtenu. Ce produit s’est avéré instable à
l’air et à la chaleur et n’a pu être entreposé.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 7.28 (d, 2H, J = 9.0 Hz), 6.88 (d, 2H, J = 8.0 Hz), 5.89 (t, 1H, J
= 6.5 Hz), 4.54 (s, 2H), 4.32 (d, 2H, J = 7.0 Hz), 4.28 (s, 2H), 3.80 (s, 3H), 1.90 (s, 3H). IR (film, cm-
1) : 3428, 2856, 2223, 1604, 1246, 1078. SMBR (m/z, intensité relative) : 246 (M+, 5), 137 (45), 121
(100). SMHR calculée pour C15H18O3 246.1256, observée : 246.1250.
(Z)-6-Méthoxyméthoxy-3-méthylhex-2-én-4-yn-1-ol (235c)
OMOM
OH
La réaction a été effectuée sous les conditions pour obtenir l’éther silylé 235a avec l’éther
propargylique 126c (151 mg, 1.51 mmol) et l’iodure vinylique 234 (150 mg, 0.76 mmol). L’adduit de
Sonogashira désiré (118 mg, 92%) a été obtenu. Ce produit s’est avéré instable à l’air et à la chaleur et
n’a pu être entreposé.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 5.88 (t, 1H, J = 6.5 Hz), 4.72 (s, 2H), 4.36 (s, 2H), 4.30 (d, 2H,
J = 7.0 Hz), 3.39 (s, 3H), 1.88 (s, 3H). IR (film, cm-1) : 3411, 2935, 1590, 1440, 1368, 1150, 1100,
1046. SMBR (m/z, intensité relative) : 188 (MNH4+, 90), 170 ((M - H2O)NH4
+, 100), 123 (80). SMHR
calculée pour C9H18NO3 : 188.1287, observée : 188.1279.
(Z)-3-Méthyl-6-(2’-triméthylsilyl)éthoxyméthoxyhex-2-én-4-yn-1-ol (235d)
OSEM
OH
La réaction a été effectuée sous les conditions pour obtenir l’éther silylé 235a avec l’éther
propargylique 126d (422 mg, 2.27 mmol) et l’iodure vinylique 234 (300 mg, 1.51 mmol). L’adduit de
Sonogashira désiré (347 mg, 95%) a été obtenu. Ce produit s’est avéré instable à l’air et à la chaleur et
n’a pu être entreposé.

133
RMN 1H (300 MHz, CDCl3) δ (ppm) : 5.88 (t, 1H, J = 7.0 Hz), 4.76 (s, 2H), 4.37 (s, 2H), 4.30 (d, 2H,
J = 6.5 Hz), 3.64 (dd, 2H, J = 8.5 et 8.0 Hz), 1.88 (s, 3H), 0.95 (t, 2H, J = 8.5 Hz), 0.02 (s, 9H). IR
(film, cm-1) : 3411, 2953, 2220, 1443, 1361, 1250, 1111. SMBR (m/z, intensité relative) : 274 (MNH4+,
90), 198 (70), 181 (100). SMHR calculée pour C13H28NO3Si (MNH4+) : 274.1838, observée : 274.1843.
(Z)-6-Benzyloxy-3-méthylhex-2-én-4-yn-1-ol (235e)
OBn
OH
La réaction a été effectuée sous les conditions pour obtenir l’éther silylé 235a avec l’éther benzylique
126e (2.20 g, 15.1 mmol) et l’iodure vinylique 234 (1.50 g, 7.53 mmol). L’adduit de Sonogashira désiré
(1.42 g, 88%) a été obtenu. Ce produit s’est avéré instable à l’air et à la chaleur et n’a pu être entreposé.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 7.36 (d, 2H, J = 7.0 Hz), 7.37-7.29 (m, 3H), 5.89 (t, 1H, J = 6.5
Hz), 4.61 (s, 2H), 4.32 (s, 2H), 4.36-4.30 (m, 2H), 1.90 (s, 3H). IR (film, cm-1) : 3400, 2860, 2216,
1590, 1439, 1354, 1075. SMBR (m/z, intensité relative) : 198 (M+ -H2O, 5), 108 (18), 91 (100). SMHR
calculée pour C14H14O (M+ - H2O) : 198.1045, observée : 198.1048.
(Z)-6-Acétoxy-3-méthylhex-2-én-4-yn-1-ol (235f)
OAc
OH
La réaction a été effectuée sous les conditions pour obtenir l’éther silylé 235a avec l’acétate
propargylique 126f (617 mg, 6.30 mmol) et l’iodure vinylique 234 (500 mg, 2.52 mmol). L’adduit de
Sonogashira désiré (283 mg, 67%) a été obtenu. Ce produit s’est avéré instable à l’air et à la chaleur et
n’a pu être entreposé.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 5.91 (t, 1H, J = 6.5 Hz), 4.81 (s, 2H), 4.30 (d, 2H, J = 6.5 Hz),
2.11 (s, 3H), 1.88 (s, 3H). RMN 13C (75 MHz, CDCl3) δ (ppm): 170.3 (s), 137.0 (d), 119.5 (s), 87.7 (s),
84.5 (s), 61.0 (t), 52.6 (t), 22.7 (q), 20.7 (q). IR (film, cm-1) : 3366, 2928, 2226, 1756, 1586, 1437,

134
1235, 1016. SMBR (m/z, intensité relative) : 186 (MNH4+, 40), 168 ((M - H2O)NH4
+, 10), 151 (100).
SMHR calculée pour C9H16NO3 (MNH4+) : 186.1130, observée : 186.1128.
(Z)-5-([1’,3’]Dioxolan-2’-yl)-3-méthylpent-2-én-4-yn-1-ol (235h)
OH
O
O
La réaction a été effectuée sous les conditions pour obtenir l’éther silylé 235a avec le dioxolane
propargylique 126h178 (6.61 g, 67.5 mmol) et l’iodure vinylique 234 (7.64 g, 27.0 mmol). L’adduit de
Sonogashira désiré (2.44 g, 54%, 80% basé sur la récupération de l’iodure de départ) a été obtenu (un
rendement purifié de 80% a été obtenu à partir de 4.00 g d’iodure 234). Ce produit s’est avéré instable à
l’air et à la chaleur et n’a pu être entreposé.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 5.92 (dt, 1H, J = 1.0 et 6.5 Hz), 5.78 (s, 1H), 4.29 (d, 2H, J =
6.5 Hz), 4.09-3.92 (m, 4H), 1.87 (s, 3H), 1.70 (s, 1H). IR (film, cm-1) : 3418, 2888, 2219, 1593, 1346,
1107, 1024. SMBR (m/z, intensité relative) : 168 (M+, 5), 167 (M+ - H, 75), 95 (100). SMHR calculée
pour C9H12O3 : 168.0786, observée : 168.0789.
(Z)-1-Bromo-6-tert-butyldiphénylsiloxy-3-méthylhex-2-én-4-yne (236a)
OTBDPS
Br
L’alcool 235a (850 mg, 2.33 mmol) et de la triphénylphosphine (914 g, 3.50 mmol) ont été dissous
dans le dichlorométhane (12 mL) et la solution a été refroidie à -78 oC. Du tétrabromure de carbone
(1.54 g, 4.06 mmol) a ensuite été ajouté par petites portions et la solution a été agitée en laissant
lentement réchauffer jusqu’à la température ambiante. Suite à la complétion de la réaction, le mélange a
été concentré et le résidu purifié par chromatographie éclair sur gel de silice en éluant avec un mélange
d'acétate d'éthyle et d'hexanes (1:9). Le bromure désiré (973 mg, 100%) a été obtenu. Ce produit s’est
avéré instable à l’air et à la chaleur et n’a pu être entreposé.

135
RMN 1H (300 MHz, CDCl3) δ (ppm) : 7.72 (dd, 4H, J = 7.5 et 2.0 Hz), 7.46-7.36 (m, 6H), 5.87 (t, 1H,
J = 6.5 Hz), 4.50 (s, 2H), 4.06 (d, 2H, J = 8.0 Hz), 1.84 (s, 3H), 1.06 (s, 9H). IR (film, cm-1) : 2935,
1626, 1591, 1425, 1368, 1107. SMBR (m/z, intensité relative) : 369 (M+ - C4H9, 15), 199 (100), 169
(100). SMHR calculée pour C19H18OBrSi (M+ - C4H9) : 369.0310, observée : 369.0303.
(Z)-1-Bromo-6-(4’-méthoxy)benzyloxy-3-méthylhex-2-én-4-yne (236b)
OPMB
Br
La réaction a été effectuée sous les conditions pour obtenir l’éther silylé 236a avec l’éther 4-
méthoxybenzylpropargylique (235b, 200 mg, 0.81 mmol). Le bromure désiré (281 mg, 100%) a été
obtenu. Ce produit s’est avéré instable à l’air et la chaleur et n’a pu être entreposé.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 7.30 (d, 2H, J = 8.0 Hz), 6.89 (d, 2H, J = 9.0 Hz), 5.94 (t, 1H, J
= 8.0 Hz), 4.57 (s, 2H), 4.32 (s, 2H), 4.17 (d, 2H, J = 8.0 Hz), 3.81 (s, 3H), 1.93 (s, 3H). IR (film, cm-
1) : 2845, 2223, 1611, 1443, 1350, 1250, 1078. SMBR (m/z, intensité relative) : 308 (M+, 3), 229 (55),
199 (100), 121 (100), 91 (98), 77 (100). SMHR calculée pour C15H17O2Br 308.0412, observée :
308.0405.
(Z)-1-Bromo-6-méthoxyméthoxy-3-méthylhex-2-én-4-yne (236c)
OMOM
Br
La réaction a été effectuée sous les conditions pour obtenir l’éther silylé 236a avec l’éther
propargylique 235c (100 mg, 0.58 mmol). Le bromure désiré (68 mg, 50%) a été obtenu. Ce produit
s’est avéré instable à l’air et la chaleur et n’a pu être entreposé.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 5.93 (t, 1H, J = 8.0 Hz), 4.74 (s, 2H), 4.40 (s, 2H), 4.15 (d, 2H,
J = 8.0 Hz), 3.40 (s, 3H), 1.91 (s, 3H). IR (film, cm-1) : 2953, 1447, 1350, 1207, 1150, 1104, 1050.

136
SMBR (m/z, intensité relative) : 252 (MNH4+, 20), 250 (MNH4
+, 20), 123 (100). SMHR calculée pour
C9H17NO2Br 250.0443, observée : 250.0439.
(Z)-1-Bromo-3-méthyl-6-(2’-triméthylsilyl)éthoxyméthoxyhex-2-én-4-yne (236d)
OSEM
Br
La réaction a été effectuée sous les conditions pour obtenir l’éther silylé 236a avec l’éther
propargylique 235d (440 mg, 1.72 mmol). Le bromure désiré (545 mg, 100%) a été obtenu. Ce produit
s’est avéré instable à l’air et la chaleur et n’a pu être entreposé.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 5.93 (t, 1H, J = 8.0 Hz), 4.78 (s, 2H), 4.41 (s, 2H), 4.15 (d, 2H,
J = 8.0 Hz), 3.65 (dd, 2H, J = 8.5 et 8.0 Hz), 1.91 (s, 3H), 0.96 (dd, 2H, J = 8.5 et 8.0 Hz), 0.02 (s, 9H).
IR (film, cm-1) : 2953, 1376, 1243, 1197, 1100, 1057. SMBR (m/z, intensité relative) : 338 (MNH4+,
20), 336 (MNH4+, 20), 90 (100). SMHR calculée pour C13H27NO2SiBr (MHN4
+) 336.0994, observée :
336.0998.
(Z)-6-Benzyloxy-1-bromo-3-méthylhex-2-én-4-yne (236e)
OBn
Br
La réaction a été effectuée sous les conditions pour obtenir l’éther silylé 236a avec l’éther benzylique
235e (310 mg, 1.43 mmol). Le bromure désiré (387 mg, 100%) a été obtenu. Ce produit s’est avéré
instable à l’air et la chaleur et n’a pu être entreposé.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 7.40-7.29 (m, 5H), 5.95 (dt, 1H, J = 8.0 et 1.0 Hz), 4.64 (s, 2H),
4.36 (s, 2H), 4.17 (d, 2H, J = 8.0 Hz), 1.93 (s, 3H). IR (film, cm-1) : 3032, 2857, 2216, 1583, 1079.
SMBR (m/z, intensité relative) : 277 (M+ - H, 1), 169 (100), 154 (65), 91 (95), 77 (95). SMHR calculée
pour C14H14OBr (M+ - H) : 277.0228, observée : 277.0220.

137
(Z)-6-Acétoxy-1-bromo-3-méthylhex-2-én-4-yne (236f)
OAc
Br
La réaction a été effectuée sous les conditions pour obtenir l’éther silylé 236a avec l’acétate
propargylique 235f (700 mg, 4.16 mmol). Le bromure désiré (744 mg, 81%) a été obtenu. Ce produit
s’est avéré instable à l’air et la chaleur et n’a pu être entreposé.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 5.96 (dt, 1H, J = 8.0 et 1.0 Hz), 4.85 (s, 2H), 4.14 (d, 2H, J =
8.0 Hz), 2.12 (s, 3H), 1.91 (s, 3H). RMN 13C (75 MHz, CDCl3) δ (ppm): 170.1 (s), 132.9 (d), 122.6 (s),
89.8 (s), 83.4 (s), 52.6 (t), 29.7 (t), 22.7 (q), 20.7 (q). IR (film, cm-1) : 2957, 1752, 1595, 1438, 1351,
1231, 1041. SMBR (m/z, intensité relative) : 250 (MNH4+, 95), 248 (MNH4
+, 100), 151 (55). SMHR
calculée pour C9H15NO2Br (MNH4+) : 248.0286, observée : 248.0280.
(Z)-2-(5’-Bromo-3-méthylpent-3-én-1-yn-1-yl)-[1,3]dioxolane (236h)
Br
O
O
La réaction a été effectuée sous les conditions pour obtenir l’éther silylé 236a avec l’éther
propargylique 235h (2.44 g, 14.5 mmol). Le bromure désiré (2.74 g, 83%) a été obtenu. Ce produit s’est
avéré instable à l’air et la chaleur et n’a pu être entreposé.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 5.98 (dt, 1H, J = 8.0 et 1.0 Hz), 5.84 (s, 1H), 4.15-3.95 (m, 4H),
4.13 (d, 2H, J = 8.0 Hz), 1.91 (s, 3H). IR (film, cm-1) : 2892, 2216, 1618, 1436, 1346, 1203, 1100,
1021. SMBR (m/z, intensité relative) : 230 (M+, 1), 151 (M+ - Br, 100), 79 (75). SMHR calculée pour
C9H11O2Br : 229.9942, observée : 229.9940.

138
(±) (Z)-1-Bromo-6-tert-butyldiphénylsiloxy-2,3-époxy-3-méthylhex-4-yne (237a)
OTBDPS
Br
O
Au bromure 236a (815 mg, 1.95 mmol) dissous dans le dichlorométhane (10 mL) à la température
ambiante a été ajouté du m-CPBA (840 mg, 2.92 mmol) par petites portions. Suite à sa complétion, la
réaction a été neutralisée par l’ajout d’une solution aqueuse saturée en bicarbonate de sodium contenant
10% de thiosulfate de sodium. Le mélange a été extrait au dichlorométhane, les fractions ont été
combinées, séchées avec du sulfate de magnésium anhydre et concentrées sous pression réduite.
L’époxyde désiré (846 mg, 100%) n’a nécessité aucune purification. Ce produit s’est avéré instable à
l’air et à la chaleur et n’a pu être entreposé.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 7.69 (dd, 4H, J = 7.0 et 2.0 Hz), 7.46-7.36 (m, 6H), 4.36 (s,
2H), 3.44 (dd, 1H, J = 11.0 et 6.0 Hz), 3.35 (dd, 1H, J = 10.5 et 6.0 Hz), 3.17 (t, 1H, J = 6.5 Hz), 1.50
(s, 3H), 1.05 (s, 9H). IR (film, cm-1) : 3071, 2928, 1587, 1422, 1372, 1308, 1107. SMBR (m/z, intensité
relative) : 385 (M+ - C4H9, 20), 263 (45), 261 (45), 169 (100). SMHR calculée pour C19H18O2BrSi (M+
- C4H9) : 385.0259, observée : 385.0269.
(±) (Z)-1-Bromo-2,3-époxy-6-(4’-méthoxy)benzyloxy-3-méthylhex-4-yne (237b)
OPMB
Br
O
La réaction a été effectuée sous les conditions pour obtenir l’éther silylé 237a avec l’éther
propargylique 236b (270 mg, 0.81 mmol). L’époxyde désiré (236 mg, 92%) n’a nécessité aucune
purification. Ce produit s’est avéré instable à l’air et à la chaleur et n’a pu être entreposé.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 7.27 (d, 2H, J = 9.0 Hz), 6.89 (d, 2H, J = 8.0 Hz), 4.52 (s, 2H),
4.17 (s, 2H), 3.81 (s, 3H), 3.58 (dd, 1H, J = 10.5 et 6.0 Hz), 3.46 (dd, 1H, J = 10.5 et 6.5 Hz), 3.23 (t,
1H, J = 6.0 Hz), 1.60 (s, 3H). IR (film, cm-1) : 2942, 1604, 1300, 1246, 1078. SMBR (m/z, intensité

139
relative) : 326 (M+, 2), 324 (M+, 2), 172 (55), 121 (100). SMHR calculée pour C15H17O3Br : 324.0361,
observée : 324.0363.
(±) (Z)-1-Bromo-2,3-époxy-6-méthoxyméthoxy-3-méthylhex-4-yne (237c)
OMOM
Br
O
La réaction a été effectuée sous les conditions pour obtenir l’éther silylé 237a avec l’éther
propargylique 236c (65 mg, 0.28 mmol). L’époxyde désiré (67 mg, 97%) n’a nécessité aucune
purification. Ce produit s’est avéré instable à l’air et à la chaleur et n’a pu être entreposé.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 4.69 (s, 2H), 4.25 (s, 2H), 3.55 (dd, 1H, J = 10.5 et 6.0 Hz),
3.44 (dd, 1H, J = 10.5 et 6.5 Hz), 3.38 (s, 3H), 3.31 (t, 1H, J = 6.5 Hz), 1.91 (s, 3H). IR (film, cm-1) :
2942, 1440, 1311, 1250, 1150, 1100, 1046. SMBR (m/z, intensité relative) : 268 (MNH4+, 65), 266
(MNH4+, 65), 251 (MH+, 8), 249 (MH+, 8), 221 (95), 219 (100). SMHR calculée pour
C9H14O3Br : 249.0126, observée : 249.0131.
(±) (Z)-1-Bromo-2,3-époxy-3-méthyl-6-(2’-triméthylsilyl)éthoxyméthoxyhex-4-yne (237d)
OSEM
Br
O
La réaction a été effectuée sous les conditions pour obtenir l’éther silylé 237a avec l’éther
propargylique 236d (545 mg, 1.72 mmol). L’époxyde désiré (556 mg, 97%) n’a nécessité aucune
purification. Ce produit s’est avéré instable à l’air et la chaleur et n’a pu être entreposé.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 4.73 (s, 2H), 4.26 (s, 2H), 3.62 (dd, 2H, J = 8.5 et 8.0 Hz), 3.55
(dd, 1H, J = 10.5 et 6.0 Hz), 3.45 (dd, 1H, J = 10.5 et 6.5 Hz), 3.21 (t, 1H, J = 6.0 Hz), 1.58 (s, 3H),
0.94 (dd, 2H, J = 8.5 et 8.0 Hz), 0.02 (s, 9H). IR (film, cm-1) : 2953, 1590, 1436, 1247, 1104, 1057.
SMBR (m/z, intensité relative) : 354 (MNH4+, 5), 352 (MNH4
+, 5), 90 (65), 73 (100). SMHR calculée
pour C13H27NO3SiBr (MHN4+) : 352.0943, observée : 352.0946.

140
(±) (Z)-6-Benzyloxy-1-bromo-2,3-époxy-3-méthylhex-4-yne (237e)
OBn
Br
O
La réaction a été effectuée sous les conditions pour obtenir l’éther silylé 237a avec l’éther benzylique
236e (4.95 g, 18.3 mmol). L’époxyde désiré (5.24 g, 100%) n’a nécessité aucune purification. Ce
produit s’est avéré instable à l’air et à la chaleur et n’a pu être entreposé.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 7.48-7.30 (m, 5H), 4.60 (s, 2H), 4.26 (s, 2H), 3.57 (dd, 1H, J =
10.5 et 5.5 Hz), 3.45 (dd, 1H, J = 10.5 et 7.0 Hz), 3.24 (t, 1H, J = 6.0 Hz), 1.60 (s, 3H). IR (film, cm-
1) : 2860, 2231, 1588, 1257, 1075. SMBR (m/z, intensité relative) : 295 (MH+, 1), 293 (1), 215 (M+ -
Br, 30), 141 (100). SMHR calculée pour C14H15O2 (M+ - Br) : 215.1072, observée : 215.1077.
(±) (Z)-6-Acétoxy-1-bromo-2,3-époxy-3-méthylhex-4-yne (237f)
OAc
Br
O
La réaction a été effectuée sous les conditions pour obtenir l’éther silylé 237a avec l’éther
propargylique 236f (105 mg, 0.47 mmol). L’époxyde désiré (116 mg, 100%) n’a nécessité aucune
purification. Ce produit s’est avéré instable à l’air et à la chaleur et n’a pu être entreposé.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 4.69 (s, 2H), 3.54 (dd, 1H, J = 10.5 et 6.0 Hz), 3.44 (dd, 1H, J =
10.5 et 6.5 Hz), 3.22 (t, 1H, J = 6.0 Hz), 2.10 (s, 3H), 1.58 (s, 3H). RMN 13C (75 MHz, CDCl3) δ
(ppm): 170.0 (s), 82.6 (s), 79.1 (s), 62.7 (d), 54.2 (s), 51.8 (t), 29.8 (t), 22.6 (q), 20.6 (q). IR (film, cm-
1) : 2989, 2345, 1755, 1583, 1440, 1372, 1225, 1032. SMBR (m/z, intensité relative) : 266 (MNH4+, 3),
264 (MNH4+, 3), 249 (MH+, 100), 247 (MH+, 100). SMHR calculée pour C9H12O3Br (MH+): 246.9970,
observée : 246.9976.

141
(±) (Z)-2-(5’-Bromo-3,4-époxy-3-méthylpent-1-yn-1-yl)-[1,3]dioxolane (237h)
Br
O
O
O
La réaction a été effectuée sous les conditions pour obtenir l’éther silylé 237a avec l’éther
propargylique 236h (3.00 g, 13.0 mmol). L’époxyde désiré (3.21 g, 100%) n’a nécessité aucune
purification. Ce produit s’est avéré instable à l’air et à la chaleur et n’a pu être entreposé.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 5.69 (s, 1H), 4.08-3.92 (m, 4H), 3.53 (dd, 1H, J = 10.5 et 5.5
Hz), 3.43 (dd, 1H, J = 10.5 et 6.5 Hz), 3.22 (t, 1H, J = 6.5 Hz), 1.58 (s, 3H). IR (film, cm-1) : 2985,
2248, 1590, 1300, 1240, 1118. SMBR (m/z, intensité relative) : 264 (MNH4+, 5), 249 (MH+, 20), 247
(MH+, 20), 167 (80), 95 (100). SMHR calculée pour C9H12O3Br : 246.9970 (MH+), observée :
246.9978.
(±) 1-Bromo-5-tert-butyldiphénylsilanoxyméthyl-3-méthylocta-3,4,6-trién-2-ol (239a)
C
OHBr
OTBDPS
Au E-bromopropène (1.09 mL, 12.6 mmol) dissous dans le THF (10 mL) à -78 °C a été ajouté
lentement du n-butyllithium (0.83 M dans hexanes, 14.9 mL, 12.4 mmol). Après 30 min, cette solution
a été transférée par canule à la suspension de cyanure de cuivre(I) (1.13 g, 12.6 mmol) dans 15 mL de
THF à -78 °C. Le mélange a été agité durant 30 min (jusqu’à la persistance d’un coloration jaune),
ensuite refroidi à -95 °C et l’époxyde 237a (1.37 g, 3.16 mmol) dissous dans 10 mL de THF a été
additionné. Suite à sa complétion (entre 10 et 30 min), la réaction a été neutralisée par l’ajout (à -95 °C)
d’une solution aqueuse saturée de chlorure d’ammonium contenant 10% d’hydroxyde d’ammonium et
la solution résultante a été agitée à température ambiante jusqu’à la persistance d’une coloration bleue.
À ce point, la réaction a été extraite à l’éther éthylique et les fractions organiques ont été combinées,
lavées avec de la saumure, séchées avec du sulfate de magnésium anhydre. L’évaporation sous pression
réduite a conduit à l’isolation d’une huile légèrement jaune dont l’instabilité a empêché toute

142
purification. L’allène brut a été immédiatement utilisé pour la cycloaddition de Diels-Alder. Note :
l’agitation mécanique est fortement conseillée dès que le volume total de THF excède 100 mL.
(±) 5-Benzyloxyméthyl-1-bromo-3-méthylocta-3,4,6-trién-2-ol (239e)
C
OH
OBn
Br
La réaction a été effectuée sous les conditions pour obtenir l’allène silylé 239a avec l’époxyde
propargylique 237e (3.32 g, 11.6 mmol). L’huile jaune obtenue s’est avérée très instable a été
immédiatement utilisée pour la cycloaddition de Diels-Alder.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 7.30 (d, 2H, J = 7.0 Hz), 7.25-7.04 (m, 3H), 5.98 (dd, 1H, J =
16.0 et 1.5 Hz), 5.78 (dq, 1H, J = 16.0 et 6.5 Hz), 4.39 (s, 2H), 4.11 (s, 2H), 3.90 (q, 1H, J = 6.5 Hz),
3.24 (dd, 1H, J = 10.5 et 4.0 Hz), 3.11 (dd, 1H, J = 10.5 et 6.5 Hz), 1.98 (d, 1H, J = 6.5 Hz), 1.59 (dd,
3H, J = 6.5 et 1.5 Hz), 1.51 (s, 3H). IR (film, cm-1): 3420, 2923, 1957, 1592, 1066. SMBR (m/z,
intensité relative) : 354 (MNH4+, 5), 291 (40), 91 (100). SMHR calculée pour C17H25O2NBr (MNH4
+) :
354.1069, observée : 354.1076.
(±) 5-Acétoxyméthyl-1-bromo-3-méthylocta-3,4,6-trién-2-ol (239f)
C
OHBr
OAc
La réaction a été effectuée sous les conditions pour obtenir l’allène silylé 239a avec l’époxyde
propargylique 237f (86 mg, 0.36 mmol). L’huile jaune obtenue s’est avérée très instable a été
immédiatement utilisée pour la cycloaddition de Diels-Alder.

143
(±) 1-Bromo-5-([1’,3’]dioxolan-2’-yl)-3-méthylocta-3,4,6-trién-2-ol (239h)
C
OHBr
OO
La réaction a été effectuée sous les conditions pour obtenir l’allène silylé 239a avec l’époxyde
propargylique 237h (2.71 g, 9.41 mmol). L’huile jaune obtenue s’est avérée très instable a été
immédiatement utilisée pour la cycloaddition de Diels-Alder.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 5.93 (dq, 1H, J = 16.0 et 6.5 Hz), 5.80 (dd, 1H, J = 16.0 et 1.5
Hz), 5.52 (s, 1H), 4.30-4.25 (m, 1H), 4.08-3.89 (m, 4H), 3.59 (dd, 1H, J = 10.5 et 4.0 Hz), 3.48 (dd, 1H,
J = 10.5 et 6.5 Hz), 2.35 (d, 1H, J = 6.0 Hz), 1.81 (s, 3H), 1.78 (dd, 3H, J = 6.0 et 1.0 Hz).
(±) Dihydrofuranne-TBDPS éther 240a
O
CO2Me
OTBDPS
Br
La réaction a été effectuée sous les conditions pour obtenir 124 avec l’allène 239a (1.08 g, 2.23 mmol).
La purification du produit brut par chromatographie éclair sur gel de silice, en éluant avec un mélange
d'acétate d'éthyle et d'hexanes (1:2), a conduit à l’isolation du dihydrofuranne désiré (491 mg, 39% pour
deux étapes). Ce produit s’est avéré très instable à l’air et à la chaleur et n’a pu être entreposé.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 7.68-7.63 (m, 4H), 7.45-7.34 (m, 6H), 5.59 (d, 1H, J = 5.0 Hz),
4.98-4.95 (m, 1H), 4.95 (s, 1H), 4.52 (d, 1H, J = 13.0 Hz), 4.38 (d, 1H, J = 13.0 Hz), 3.76 (s, 3H), 3.66
(dd, 1H, J = 11.5 et 2.0 Hz), 3.53 (dd, 1H, J = 11.5 et 2.0 Hz), 2.96 (dd, 1H, J = 11.5 et 6.5 Hz), 2.74-
2.67 (m, 1H), 1.81 (s, 3H), 1.04 (s, 9H), 0.88 (d, 3H, J = 7.0 Hz). IR (film, cm-1): 2953, 1733, 1436,
1322, 1186, 1114. SMBR (m/z, intensité relative) : 570 (M+, 2), 568 (M+, 2), 513 (M+ - C4H9, 100), 511
(100). SMHR calculée pour C30H37O4SiBr : 568.1644, observée : 568.1633.

144
(±) Dihydrofuranne-benzyl éther 240e
O
CO2Me
OBn
Br
La réaction a été effectuée sous les conditions pour obtenir 124 avec l’allène 239e (3.46 g, 10.3 mmol).
La purification du produit brut par chromatographie éclair sur gel de silice, en éluant avec un mélange
d'acétate d'éthyle et d'hexanes (1:2), a conduit à l’isolation du dihydrofuranne désiré (2.55 g, 59% pour
deux étapes). Ce produit s’est avéré très instable à l’air et à la chaleur et n’a pu être entreposé.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 7.59-7.27 (m, 5H), 5.79 (d, 1H, J = 5.0 Hz), 5.03-4.97 (m, 2H),
4.52 (s, 2H), 4.42 (d, 1H, J = 12.0 Hz), 4.12 (d, 1H, J = 11.5 Hz), 3.76 (s, 3H), 3.67 (dd, 1H, J = 11.5
et 2.5 Hz), 3.54 (dd, 1H, J = 11.5 et 2.5 Hz), 2.99 (dd, 1H, J = 11.0 et 6.0 Hz), 2.83-2.76 (m, 1H), 1.83
(s, 3H), 0.97 (d, 3H, J = 7.5 Hz). IR (film, cm-1): 2928, 1722, 1629, 1443, 1210, 1107. SMBR (m/z,
intensité relative) : 420 (M+, 2), 389 ((M+ - OCH3), 2), 314 (40), 91 (100). SMHR calculée pour
C21H25O4Br : 420.0936, observée : 420.0942.
(±) Dihydrofuranne-acétate 240f
O
CO2Me
OAc
Br
La réaction a été effectuée sous les conditions pour obtenir 124 avec l’allène 239f (86 mg, 0.36 mmol).
La purification du produit brut par chromatographie éclair sur gel de silice, en éluant avec un mélange
d'acétate d'éthyle et d'hexanes (1:2), a conduit à l’isolation du dihydrofuranne désiré (39 mg, 29% pour
deux étapes). Ce produit s’est avéré très instable à l’air et à la chaleur et n’a pu être entreposé.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 5.85 (d, 1H, J = 5.0 Hz), 5.04-4.98 (m, 2H), 4.88 (d, 1H, J =
12.5 Hz), 4.74 (d, 1H, J = 12.5 Hz), 3.77 (s, 3H), 3.67 (dd, 1H, J = 11.5 et 3.0 Hz), 3.54 (dd, 1H, J =
11.5 et 2.0 Hz), 3.00 (dd, 1H, J = 11.5 et 6.5 Hz), 2.86-2.79 (m, 1H), 2.11 (s, 3H), 1.77 (s, 3H), 0.98 (d,

145
3H, J = 7.5 Hz). IR (film, cm-1): 2967, 1723, 1594, 1443, 1232, 1025. SMBR (m/z, intensité relative) :
392 (MNH4+, 15), 390 (MNH4
+, 15), 315 (90), 313 (85), 233 (100). SMHR calculée pour C16H22O5Br
(MH+) : 373.0650, observée : 373.0638.
(±) Dihydrofuranne-dioxolane 240h
O
CO2Me
Br
O O
La réaction a été effectuée sous les conditions pour obtenir 124 avec l’allène 239h (2.71 g, 9.41 mmol).
La purification du produit brut par chromatographie éclair sur gel de silice, en éluant avec un mélange
d'acétate d'éthyle et d'hexanes (1:2), a conduit à l’isolation du dihydrofuranne désiré (1.90 g, 54% pour
deux étapes). Ce produit s’est avéré très instable à l’air et à la chaleur et n’a pu être entreposé.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 6.15 (d, 1H, J = 5.0 Hz), 5.66 (s, 1H), 5.04-5.00 (m, 2H), 4.10-
3.95 (m, 4H), 3.76 (s, 3H), 3.66 (dd, 1H, J = 11.5 et 3.0 Hz), 3.54 (dd, 1H, J = 11.5 et 2.0 Hz), 3.01
(dd, 1H, J = 11.5 et 7.0 Hz), 2.89-2.83 (m, 1H), 1.84 (s, 3H), 0.97 (d, 3H, J = 7.5 Hz). IR (film, cm-1):
2882, 1726, 1626, 1440, 1286. SMBR (m/z, intensité relative) : 374 (M+, 15), 372 (M+, 15), 289 (60),
231 (60), 84 (100). SMHR calculée pour C16H21O5Br : 372.0572, observée : 372.0577.
(±) Benzofuranne 243
O
CO2Me
OBn
Br
La purification du dihydrofuranne 240e (45 mg) entreposé durant deux jours dans le benzène dégazé et
gelé a conduit à l’isolation de son produit d’oxydation (14 mg) accompagné de plusieurs produits de
dégradation complexes.

146
RMN 1H (300 MHz, CDCl3) δ (ppm) : 7.35-7.28 (m, 5H), 7.03 (s, 1H), 4.78 (s, 2H), 4.57 (s, 2H), 3.98
(s, 3H), 2.54 (s, 2H), 2.37 (s, 3H), 2.24 (s, 3H). IR (film, cm-1): 3032, 2857, 1719, 1447, 1265, 1089.
SMBR (m/z, intensité relative) : 418 (M+, 2), 416 (M+, 2), 338 (70), 230 (80), 91 (100). SMHR
calculée pour C21H21O4Br : 416.0623, observée : 416.0619.
(±) Triène 244
CO2Me
OBn
HO
Au dihydrofuranne 240e (1.75 g, 4.16 mmol) dissous dans l’éthanol 95% (20 mL) a été ajouté du
NH4Cl(s) (150 mg) et de la poussière de zinc (1.36 g, 20.8 mmol). Le mélange a été chauffé à reflux de
l’éthanol durant 7 h, puis refroidi à la température ambiante et filtré sur fritté. Le filtrat a été concentré
sous pression réduite et le résidu brut a été purifié par chromatographie éclair sur gel de silice en éluant
avec un mélange d'éther éthylique et d'hexanes (2:5). Le triène désiré a été obtenu (1.09 g, 77%). Ce
produit s’est avéré très instable à l’air et la chaleur et n’a pu être entreposé.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 7.37-7.27 (m, 5H), 7.03 (dd, 1H, J = 17.0 et 11.0 Hz), 6.17 (d,
1H, J = 5.0 Hz), 5.38 (d, 1H, J = 17.0 Hz), 5.28 (d, 1H, J = 4.5 Hz), 5.23 (d, 1H, J = 11.0 Hz), 4.51 (s,
2H), 4.25 (d, 1H, J = 11.5 Hz), 4.18 (d, 1H, J = 11.5 Hz), 3.62 (s, 3H), 2.90-2.82 (m, 2H), 1.89 (s, 3H),
1.04 (d, 3H, J = 7.0 Hz). IR (film, cm-1): 3394, 2954, 1727, 1444, 1286, 1165. SMBR (m/z, intensité
relative) : 342 (M+, 10), 159 (90), 91 (100). SMHR calculée pour C21H26O4 : 342.1831, observée :
342.1836.

147
(±) Acide-benzyl éther 245
O
CO2H
OBn
Br
Au magnésium activé (63 mg, voir la première partie de la procédure pour la préparation du
bromure de 1-propénylmagnésium) dans 10 mL de THF a été ajouté le dihydrofuranne 240e (220 mg,
0.52 mmol). Le mélange a été chauffé à reflux du THF durant 11 h, puis refroidi à température
ambiante et neutralisé par l’ajout d’une solution aqueuse saturée en chlorure d’ammonium. La solution
résultante a été extraite avec de l’éther éthylique, les fractions organiques ont été combinées, lavées
avec de la saumure, séchées avec du sulfate de magnésium anhydre et concentrées sous pression
réduite. La purification du produit brut par chromatographie éclair sur gel de silice, en éluant avec un
mélange d'éther éthylique et d'hexanes (2:5), a conduit à l’isolation de deux produits : le dihydrofuranne
de départ (107 mg, 49% de récupération) et l’acide 245 (16 mg, 10%, 20% corrigé).
RMN 1H (300 MHz, CDCl3) δ (ppm) : 7.38-7.27 (m, 5H), 5.83 (d, 1H, J = 5.0 Hz), 5.00-4.96 (m, 2H),
4.52 (s, 2H), 4.41 (d, 1H, J = 11.5 Hz), 4.11 (d, 1H, J = 11.0 Hz), 3.69 (dd, 1H, J = 11.5 et 3.0 Hz),
3.56 (dd, 1H, J = 12.0 et 1.5 Hz), 3.02 (dd, 1H, J = 11.0 et 6.0 Hz), 2.90-2.83 (m, 1H), 1.83 (s, 3H),
1.08 (d, 3H, J = 7.5 Hz). IR (film, cm-1): 3851, 3747, 3021, 2864, 1712, 1554, 1082. SMBR (m/z,
intensité relative) : 426 (MNH4+, 100), 424 (MNH4
+, 100), 327 (40), 300 (45).
(±) Triène silylé 246
CO2Me
OBn
TIPSO
Au triène 244 (540 mg, 1.58 mmol) dissous dans le dichlorométhane (8 mL) à 0 °C a été ajouté de la
2,6-lutidine (275 µL, 2.36) et du TIPSOTf (450 µL, 1.65 mmol). La solution a été agitée à la
température ambiante durant 4 h, puis concentrée sous pression réduite et le résidu brut a été purifié par

148
chromatographie éclair sur gel de silice en éluant avec un mélange d'éther éthylique et d'hexanes (1:9).
L’éther silylé désiré a été obtenu (650 mg, 83%). Ce produit s’est avéré instable à l’air et la chaleur et
n’a pu être entreposé.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 7.31-7.23 (m, 5H), 6.84 (dd, 1H, J = 17.0 et 11.0 Hz), 5.94 (s,
1H), 5.40 (d, 1H, J = 4.0 Hz), 5.28 (d, 1H, J = 17.0 Hz), 5.10 (d, 1H, J = 11.0 Hz), 4.53 (d, 1H, J =
12.0 Hz), 4.44 (d, 1H, J = 12.0 Hz), 4.22 (AB q, 2H), 3.47 (s, 3H), 3.02-2.97 (m, 2H), 1.83 (s, 3H),
1.19 (d, 3H, J = 7.0 Hz), 1.09-0.99 (m, 21H). IR (film, cm-1): 2950, 2864, 1744, 1462, 1154. SMBR
(m/z, intensité relative) : 455 (M+ - C3H7, 15), 203 (40), 157 (90), 91 (100). SMHR calculée pour
C27H39O4Si (M+ - C3H7) : 455.2617, observée : 455.2622.
(±) Aldéhydes 247 et 248
CO2Me
O
TIPSO
CO2Me
O
TIPSO
247 248
Au triène 246 (140 mg, 0.258 mmol) dissous dans du dichlorométhane (8 mL) et de la pyridine (250
µL) à été ajouté du SeO2 (156 mg, 1.40 mmol). La solution a été portée à reflux du solvant durant 3.5 h,
puis neutralisée par l’ajout d’une solution aqueuse saturée en bicarbonate de sodium. Le mélange a été
extrait avec du dichlorométhane, les fractions organiques ont été combinées, séchées avec du sulfate de
magnésium anhydre et concentrées sous pression réduite. Le résidu brut a été purifié par
chromatographie éclair sur gel de silice en éluant avec un mélange d'éther éthylique et d'hexanes (1:12)
pour procurer 34 mg d’un mélange inséparable des produits 247 et 248 (ratio de 2.5:1 dans le RMN 1H
du brut). Ces produits se sont avérés très instables à l’air et la chaleur et n’ont pu être entreposé ou
purifié d’avantage.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 9.48 (s, 1H, 247), 9.45 (s, 1H, 248), 6.81 (dd, 1H, J = 16.5 et
11.0 Hz, 247), 6.74-6.70 (m, 1H), 6.27 (dd, 1H, J = 16.5 et 10.5 Hz, 248), 5.42-5.35 (m, 2H), 5.32-5.27
(m, 1H, B), 5.20-5.14 (m, 1H), 3.53 (s, 3H, 248), 3.47 (s, 3H, 247), 3.14-3.06 (m, 2H), 1.89 (s, 3H,
248), 1.68 (s, 3H, 247), 1.34 (d, 3H, J = 6.5 Hz, 247), 1.32 (d, 3H, J = 7.0 Hz, 248), 1.07-0.97 (m,
21H). IR (film, cm-1): 2864, 1744, 1687, 1458, 1172, 1104. SMBR (m/z, intensité relative) : 363 (M+ -

149
C3H7, 90), 173 (70), 145 (100). SMHR calculée pour C20H31O4Si (M+ - C3H7) : 363.1991, observée :
363.1991.
(±) Aldéhyde 251
O
CO2Me
Br
O
Le dihydrofuranne 240h (560 mg, 1.50 mmol) a été dissous dans de l’acétone mouillée (7 mL) et
refroidi à 0 °C. Le HCl concentré (100 µL) a été additionné et la réaction a été agitée durant 20 min,
puis neutralisée par l’ajout d’une solution aqueuse saturée en bicarbonate de sodium. Le mélange a été
extrait avec du dichlorométhane, les fractions ont été combinées, séchées avec du sulfate de magnésium
anhydre et concentrées sous pression réduite. L’aldéhyde brut (493 mg, 100%) n’a pas nécessité de
purification, mais il s’est avéré instable à l’air et la chaleur et n’a pu être entreposé.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 9.53 (s, 1H), 6.73 (d, 1H, J = 6.5 Hz), 5.10 (br s, 1H), 5.02 (br
d, 1H, J = 11.0 Hz), 3.79 (s, 3H), 3.68 (dd, 1H, J = 11.5 et 3.0 Hz), 3.57 (dd, 1H, J = 11.5 et 2.0 Hz),
3.20-3.08 (m, 2H), 1.88 (s, 3H), 1.06 (d, 3H, J = 7.0 Hz). IR (CHCl3, cm-1): 2978, 1729, 1704, 1321,
1139, 906. SMBR (m/z, intensité relative) : 328 (M+, 10), 250 (40), 105 (100). SMHR calculée pour
C14H17O4Br : 328.0310, observée : 328.0303.
(±) Éther d’énol 252
O
CO2Me
Br
O OEt
H
La réaction a été effectuée sous les conditions pour obtenir 166 avec l’aldéhyde 251 (1.59 g, 4.54
mmol). La purification du produit brut par chromatographie éclair sur gel de silice, en éluant avec un

150
mélange d'acétate d'éthyle et d'hexanes (1:3), a conduit à l’isolation du dihydrofuranne désiré (1.47 g,
76%). Ce produit s’est avéré très instable à l’air et la chaleur et n’a pu être entreposé.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 6.72 (s, 1H), 5.14-5.10 (m, 1H), 5.01-4.97 (m, 2H), 3.96 (dq,
1H, J = 7.5 et 7.0 Hz), 3.70 (s, 3H), 3.66-3.50 (m, 3H), 3.05 (dd, 1H, J = 9.0 et 6.5 Hz), 2.65-2.55 (m,
1H), 2.12-2.05 (m, 1H), 1.88-1.74 (m, 1H), 1.71 (s, 3H), 1.48-1.37 (m, 1H), 1.26 (t, 3H, J = 7.0 Hz),
0.91 (d, 3H, J = 7.0 Hz). IR (film, cm-1): 2978, 1722, 1629, 1439, 1227. SMBR (m/z, intensité
relative) : 402 (M+, 25), 400 (M+, 25), 271 (100), 244 (90). SMHR calculée pour C18H25O5Br 400.0885,
observée : 400.0885.
(±) Triène 253
CO2Me
O OEt
H
HO
Au dihydrofuranne 252 (1.57 g, 3.92 mmol) dissous dans le méthanol (20 mL) a été ajouté de la
poussière de zinc (770 mg, 11.8 mmol). Le mélange a été chauffé à 40 °C durant deux jours, puis
refroidi à la température ambiante et filtré sur fritté. Le filtrat a été concentré sous pression réduite et le
résidu brut a été purifié par chromatographie éclair sur gel de silice en éluant avec un mélange d'éther
éthylique et d'hexanes (2:5). Le triène désiré a été obtenu (825 mg, 65%). Ce produit s’est avéré très
instable à l’air et la chaleur et n’a pu être entreposé.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 6.87 (dd, 1H, J = 16.5 et 11.0 Hz), 6.19 (d, 1H, J = 1.5 Hz),
5.30 (d, 1H, J = 16.5 Hz), 5.18 (d, 1H, J = 11.0 Hz), 5.14 (t, 1H, J = 4.5 Hz), 4.96 (dd, 1H, J = 9.5 et
2.0 Hz), 3.97 (dq, 1H, J = 9.0 et 7.0 Hz), 3.70-3.57 (m, 1H), 3.58 (s, 3H), 2.86 (t, 1H, J = 4.0 Hz), 2.67-
2.59 (m, 1H), 2.24 (ddd, 1H, J = 13.0, 6.5 et 1.5 Hz), 2.08-1.98 (m, 1H), 1.95 (s, 3H), 1.75 (d, 1H, J =
6.0 Hz), 1.59-1.48 (m, 1H), 1.26 (t, 3H, J = 7.0 Hz), 1.03 (d, 3H, J = 7.0 Hz). IR (film, cm-1): 3493,
2971, 1729, 1622, 1128, 1024. SMBR (m/z, intensité relative) : 322 (M+, 5), 304 (M+ - H2O, 8), 173
(80), 145 (100). SMHR calculée pour C18H26O5 322.1780, observée : 322.1783.

151
(±) Triène 254
CO2Me
O OEt
H
TIPSO
La réaction a été effectuée sous les conditions pour obtenir le triène silylé 246 avec l’alcool brut 253
(1.47 g, 3.67 mmol du dihydrofuranne 252). La purification du produit brut par chromatographie éclair
sur gel de silice, en éluant avec un mélange d'acétate d'éthyle et d'hexanes (1:3), a conduit à l’isolation
du dihydrofuranne désiré (1.25 g, 77% pour 2 étapes). Ce produit s’est avéré instable à l’air et la
chaleur, mais a pu être conservé dans l’acétate d’éthyle dégazé dans le congélateur.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 6.79 (dd, 1H, J = 17.0 et 10.5 Hz), 6.03 (d, 1H, J = 1.5 Hz),
5.30 (d, 1H, J = 4.0 Hz), 5.21 (dd, 1H, J = 17.0 et 1.0 Hz), 5.09 (d, 1H, J = 10.5 Hz), 4.91 (dd, 1H, J =
9.5 et 1.5 Hz), 3.98 (dq, 1H, J = 9.5 et 7.0 Hz), 3.60 (dq, 1H, J = 16.5 et 7.0 Hz), 3.50 (s, 3H), 2.79 (t,
1H, J = 4.0 Hz), 2.73-2.64 (m, 1H), 2.27 (dd, 1H, J = 6.0 et 1.5 Hz), 2.23-2.09 (m, 1H), 1.88 (s, 3H),
1.50-1.39 (m, 1H), 1.25 (t, 3H, J = 7.0 Hz), 1.06-0.97 (m, 24H). RMN 13C (75 MHz, CDCl3) δ (ppm):
171.3 (s), 140.6 (d), 135.2 (s), 134.0 (d), 129.2 (s), 114.6 (t), 113.1 (s), 100.0 (d), 67.7 (d), 64.5 (t), 55.1
(d), 50.9 (t), 36.0 (q), 35.2 (d), 33.1 (d), 17.8 (q), 16.3 (q), 15.7 (q), 15.1 (q), 13.3 (s), 12.1 (d), 11.9 (q),
11.7 (d). IR (film, cm-1): 2949, 1740, 1640, 1464, 1132, 1057. SMBR (m/z, intensité relative) : 478
(M+, 5), 389 (50), 287 (100). SMHR calculée pour C27H46O5Si : 478.3114, observée : 478.3124.
(±) Alcool 261
CO2Me
O OEt
H
TIPSO
HO
Le triène 254 (478 mg, 1.00 mmol) a été dissous dans le THF (8 mL) et la solution a été placée à 0 °C.
Une solution de dicyclohexylborane213 (5.9 mL, 2.00 mmol) fraîchement préparée a alors été ajoutée.
Suite à la complétion de la réaction, de l’eau (6 mL), du perborate de sodium (984 mg, 6.40 mmol) et

152
du n-Bu4NHSO4 (51 mg, 0.15 mmol) ont été ajoutés et la solution a été agitée vigoureusement à la
température ambiante durant 12 h. La réaction a alors été neutralisée par l’ajout d’une solution aqueuse
saturée en bicarbonate de sodium contenant 10% de bisulfite de sodium et a été extraite avec de l’éther
éthylique. Les fractions ont été combinées, lavées avec de la saumure, séchées avec du sulfate de
magnésium anhydre et concentrées sous pression réduite. Le produit brut (755 mg), contaminé
seulement de cyclohexanol, a été directement utilisé pour la réaction suivante.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 6.09 (d, 1H, J = 1.5 Hz), 4.98 (d, 1H, J = 3.5 Hz), 4.89 (dd, 1H,
J = 9.5 et 1.5 Hz), 3.99 (dq, 1H, J = 9.5 et 7.0 Hz), 3.76-3.66 (m, 1H), 3.64-3.55 (m, 2H), 3.62 (s, 3H),
2.83 (t, 1H, J = 4.0 Hz), 2.69-2.60 (m, 1H), 2.58-2.48 (m, 1H), 2.24-2.04 (m, 4H), 1.83 (s, 3H), 1.51-
1.40 (m, 1H), 1.26 (t, 3H, J = 5.5 Hz), 1.03-0.95 (m, 24H). IR (film, cm-1): 2867, 1740, 1461, 1128,
1057. SMBR (m/z, intensité relative) : 496 (M+, 25), 453 (70), 407 (75), 75 (100). SMHR calculée
pour C27H48O6Si : 496.3220, observée : 496.3209.
(±) Diol 262
CO2Me
O OEt
H
HO
HO
La réaction a été effectuée sous les conditions pour obtenir 130 avec l’éther silylé 261 (149 mg, 0.30
mmol). La purification du produit brut par chromatographie éclair sur gel de silice, en éluant avec un
mélange de méthanol et d'acétate d'éthyle (1:20), a conduit à l’isolation du diol désiré (1.47 g, 76% sur
2 étapes).
RMN 1H (300 MHz, CDCl3) δ (ppm) : 6.19 (d, 1H, J = 1.5 Hz), 4.91 (dd, 1H, J = 9.5 et 1.5 Hz), 4.88
(d, 1H, J = 4.5 Hz), 3.96 (dq, 1H, J = 9.5 et 7.0 Hz), 3.80-3.73 (m, 1H), 3.67-3.55 (m, 2H), 3.65 (s,
3H), 2.81 (t, 1H, J = 4.5 Hz), 2.55-2.34 (m, 4H), 2.18 (ddd, 1H, J = 13.0, 6.5 et 1.5 Hz), 2.10-1.97 (m,
1H), 1.88 (s, 3H), 1.60-1.49 (m, 1H), 1.26 (t, 3H, J = 7.0 Hz), 0.97 (d, 3H, J = 7.0 Hz). IR (film, cm-1):
3386, 2960, 1733, 1633, 1375, 1125, 1036. SMBR (m/z, intensité relative) : 340 (M+, 70), 294 (60),
191 (70), 173 (100), 161 (90). SMHR calculée pour C18H28O6 : 340.1886, observée : 340.1881.

153
(±) Tricycles 263 et 264
CO2Me
O OEt
H
HO
HO
NC
NCNC CN
H
CO2Me
O OEt
H
HO
HO
NC
NCNC CN
H263 264
Au diol 262 (5 mg, 0.01 mmol) dissous dans le toluène (100 µL) a été ajouté une quantité catalytique
d’hydroquinone et du tétracyanoéthylène (5 mg, 0.04). La réaction a été purgée avec de l’argon et
placée à 120 °C durant 4 h. La solution a alors été concentrée et le résidu brut a été purifié par
chromatographie éclair sur gel de silice en éluant avec un mélange de méthanol et d'acétate d'éthyle
(1:4).
Tricycle 263 (1.5 mg, 25%)
RMN 1H (300 MHz, CDCl3) δ (ppm) : 5.20 (t, 1H, J = 7.0 Hz), 4.96 (t, 1H, J = 2.0 Hz), 4.35 (dt, 1H, J
= 9.0 et 2.0 Hz), 4.13-4.08 (m, 2H), 3.95 (dq, 1H, J = 10.0 et 6.5 Hz), 3.74 (s, 3H), 3.65 (dq, 1H, J =
9.5 et 6.5 Hz), 2.62 (t, 1H, J = 8.0 Hz), 2.50-2.33 (m, 2H), 2.17-2.09 (m, 2H), 1.85 (q, 1H, J = 7.0 Hz),
1.68 (s, 3H), 1.42 (dq, 1H, J = 14.5 et 7.5 Hz), 1.27 (t, 3H, J = 7.0 Hz), 1.00 (d, 3H, J = 7.0 Hz). IR
(film, cm-1): 3371, 2946, 1729, 1594, 1440, 1375, 1171, 1046. SMBR (m/z, intensité relative) : 468 ((M
- H2O)NH4+, 100), 416 (45), 391 (40), 323 (80). SMHR calculée pour C22H26O5N4 (M+ - H2O) :
450.1903, observée : 450.1898.
Tricycle 264 (1.0 mg, 17%, contaminé avec 263)
RMN 1H (300 MHz, CDCl3) δ (ppm) : 4.73 (dd, 1H, J = 9.5 et 1.5 Hz), 4.51 (s, 1H), 4.34-4.32 (m,
1H), 4.15-4.02 (m, 3H), 3.67 (s, 3H), 3.62 (dq, 1H, J = 9.5 et 7.0 Hz), 3.04 (dd, 1H, J = 5.5 et 4.0 Hz),
2.40-2.05 (m, 4H), 1.87-1.79 (m, 1H), 1.70 (s, 3H), 1.37-1.23 (m, 1H), 1.26 (t, 3H, J = 7.0 Hz), 1.06 (d,
3H, J = 6.5 Hz). IR (film, cm-1): 3368, 2951, 1728, 1590, 1437, 1375, 1170. SMBR (m/z, intensité
relative) : 468 ((M - H2O)NH4+, 100), 414 (30), 323 (40), 276 (25). SMHR calculée pour
C22H30O5N5 ((M - H2O)NH4+) : 468.2247, observée : 468.2253.

154
(±) Alcool secondaire 267
CO2Me
O OEt
H
HO
TBSO
La réaction a été effectuée sous les conditions pour obtenir l’éther silylé 154 avec le diol 262 (25 mg,
0.07 mmol). Le produit brut (33 mg, 100%) a été utilisé tel quel pour l’étape suivante.
RMN 1H (300 MHz, CDCl3) δ (ppm) : 6.11 (d, 1H, J = 2.0 Hz), 4.94 (dd, 1H, J = 9.5 et 1.5 Hz), 4.83
(dd, 1H, J = 5.0 et 2.0 Hz), 3.97 (dq, 1H, J = 9.5 et 7.0 Hz), 3.73-3.56 (m, 3H), 3.64 (s, 3H), 2.83-2.75
(m, 1H), 2.75 (t, 1H, J = 4.5 Hz), 2.60 (s large, 1H), 2.51-2.43 (m, 1H), 2.19-2.05 (m, 2H), 2.04-1.93
(m, 1H), 1.86 (s, 3H), 1.63-1.52 (m, 1H), 1.26 (t, 3H, J = 7.0 Hz), 0.97 (d, 3H, J = 7.0 Hz), 0.88 (s,
9H), 0.05 (s, 6H). IR (film, cm-1): 3589, 1736, 1640, 1464, 1253, 1135. SMBR (m/z, intensité
relative) : 454 (M+, 10), 436 (M+ - H2O, 2), 74 (100). SMHR calculée pour C24H42O6Si : 454.2750,
observée : 454.2758.

155
RÉFÉRENCES ET NOTES
1 Le Courteur, P.; Burreson, J. Napoleon’s buttons; Tarcher/Penguin : New York 2004, 375 pages. 2 Nicolaou, K. C.; Sorensen, E. J. Classics in Total Synthesis, VCH Publishers, Inc., New York,
1996 . 3 Winckler, F. L. Rep. Pharm. 1835, 4, 85. 4 Clark, E. P. J. Am. Chem. Soc. 1937, 59, 927, 2511-14. 5 London, E.; Robertson, A,; Worthinghton, H. J. Chem. Soc. 1950, 3431-40. 6 (a) Valenta, Z.; Papadopoulos, S.; Podesva, C. Tetrahedron 1961, 15, 100. (b) Valenta, Z.; Gray,
A. H.; Orr, D. E.; Papadopoulos, S.; Podesva, C. Tetrahedron 1962, 18, 1433. 7 (a) Guo, Z.; Vangapandu, S.; Sindelar, R. W.; Walker, L. A.; Sindelar, R. D. Curr. Med. Chem.
2005, 12, 173-90. (b) Okano, M.; Fukamiya, N.; Lee, K. H. Stud. Nat. Prod. Chem. 1990, 7(pt.
A), 69-404. 8 Polonsky, J. Forts. Chem. Org. Naturst. 1985, 47, 221-64. 9 Wall, M. E.; Wani, M. International Symposium on the Chemistry of Natural Products, Riga,
USSR, IUPAC 7th, 1970, Abstracts E 138, 614. 10 Voir la référence 7, ainsi que (a) Grosvenor, P. W.; Gothard, P. K.; McWilliam, N. C.; Supriono,
A.; Gray, D. O. J. Ethnopharmacol. 1995, 45, 75. (b) Okano, M.; Fukamiya, N.; Tagahara, K.;
Cosentino, M.; Lee, T. T.-Y.; Morris-Natschke, S.; Lee, K.-H. Bioorg. & Med. Chem. Let. 1996,
6, 701-06. 11 (a) Wiseman, C. L.; Yap, H. Y.; Bedikian, A. Y.; Bodey, G. P.; Blumenschein, G. R. Am. J. Clin.
Oncol. 1982, 5, 389-91. (b) Arsenau, J. C.; Wolter, J. M.; Kuperminc, M.; Ruckdeschel, J. C.
Invest. New Drugs 1983, 1, 239-42. 12 Cuendet, M.; Pezzuto, J. M. J. Nat. Prod. 2004, 67, 269-72. 13 Mata-Greenwood, E.; Cuendet, M.; Sher, D.; Gustin, D.; Stock, W.; Pezzuto, J. M. Leukemia
2002, 16, 2275-84. 14 Pour une revue sur l’apoptose, voir Science 1998, 281, 1302-26. 15 Muhammad, I.; Bedir, E.; Khan, S. I.; Tekwani, B. L.; Khan, I. A.; Takamatsu, S.; Pelletier, J.;
Walker, L. A. J. Nat. Prod. 2004, 67, 772-77. 16 Morré, D. J.; Grieco, P. A.; Morré, D. M. Life Sciences 1998, 63, 595-604.

156
17 (a) Liao, L.-L.; Kupchan, S. M.; Horwitz, S. B. Mol. Pharm. 1976, 12, 433-35. (b) Fresno, M.;
Gonzales, A.; Vasquez, D.; Jimenez, A. Biochim. Biophys. Acta 1978, 518, 104-12. 18 Fukamiya, N.; Lee, K.-H.; Muhammad, I.; Murakami, C.; Okano, M.; Harvey, I.; Pelletier, J.
Cancer Lett. 2005, 220, 37-48. 19 S. M. Kupchan, S. M.; Lacadie, J. A. J. Org. Chem. 1975, 40, 654-56. 20 Référence 12 et (a) Kupchan, S. M.; Tsou, G. J. Org. Chem. 1973, 38, 1055-56. (b) Chamberlain,
P.; Whitham, G. H. J. Chem. Soc. Perkin Trans. 2 1972, 130-35. Pour une revue sur ce
mécanisme d’action, voir (c) Wolkenberg, S. E.; Boger, D. L. Chem. Rev. 2002, 102, 2477-96. 21 Kawada, K.; Kim, M.; Watt, D. S. Org. Prep. Proc. Int. 1989, 21, 521. 22 (a) Alvarez-Manzaneda, E. J.; Romera, J. L.; Barrero, A. F.; Alvarez-Manzaneda, R.; Chahboun,
R.; Meneses, R.; Aparicio, M. Tetrahedron 2005, 61, 837-44. (b) Barrero, A. F.; Herrador, M.
M.; Quilez del Moral, J. F.; Valdivia, M. V. Org. Lett. 2002, 4, 1379-82. 23 Kraus, G. A.; Cui, W. J. Org. Chem. 2002, 67, 9475-76. 24 Barriault, L.; Ouellet, S. G.; Deslongchamps, P. Tetrahedron 1997, 53, 14937-56. 25 (a) Govindan, S. V.; Fuchs, P. L. J. Org. Chem. 1988, 53, 2593-97. (b) Charles K.-F. Chiu, C. D.
F.; Govindan, S. V.; Fuchs, P. L. J. Org. Chem. 1994, 59, 311-323. 26 Kerwin, S. M.; Paul, A. G.; Heathcock, C. H. J. Org. Chem. 1987, 52, 1686-95. 27 Ziegler, F. E.; Klein, S. I.; Pati, U. K.; Wang, T. F. J. Am. Chem. Soc. 1985, 107, 2730-37. 28 (a) Spino, C.; Hill, B.; Dubé, P.; Gingras, S. Can. J. Chem. 2003, 81, 81-108. (b) Spino, C.; Liu,
G. J. Org. Chem. 1993, 58, 817-19. (c) Spino, C.; Tu, N.; Liu, G.; Girard, S. J. Org. Chem. 1994,
59, 5596-5608. 29 (a) Grieco, P. A.; Ferrino, S.; Vidari, G. J. Am. Chem. Soc. 1980, 102, 7586-87. (b) Vidari, G.;
Ferrino, S.; Grieco, P. A. J. Am. Chem. Soc. 1984, 106, 3539-48. 30 Pour une revue et analyse de cette cycloaddition, voir (a) Gingras, S. Mémoire de maîtrise,
Université de Sherbrooke, 2000, 166 pages. (b) Woodward, R. B.; Hoffmann, R. Angew. Chem.
Int. Ed. 1969, 8, 781-853. La référence originale : Diels, O.; Alder, K. Justus Ann. Chem. 1928,
460, 98. 31 Voir la référence 30a ainsi que (a) Mehta, G.; Uma, R. Acc. Chem. Res. 2000, 33, 278-86. (b)
Kagan, H. B.; Riant, O. Chem. Rev. 1992, 92, 1007-19. (c) Fringuelli, F.; Taticchi, A. The Diels-
Alder Reaction, Selected Practical Methods; Wiley, 2002, 340 pages. (d) Kobayashi, S.;
Jorgensen, K. A. Cycloaddition Reactions in Organic Synthesis; Wiley-VCH, 2002, 332 pages.

157
32 Moher, E. D.; Collins, J. L.; Grieco, P. A. J. Am. Chem. Soc. 1992, 114, 2764-65. 33 VanderRoest, J. M.; Grieco, P. A. J. Am. Chem. Soc. 1993, 115, 5841-42. 34 Grieco, P. A.; Pineiro-Nunez, M. M. J. Am. Chem. Soc. 1994, 116, 7606-15. 35 Gross, R. S.; Grieco, P. A.; Collins, J. L. J. Am. Chem. Soc. 1990, 112, 9436-37. 36 Collins, J. L.; Grieco, P. A.; Gross, R. S. J. Org. Chem. 1990, 55, 5816-18. 37 Kim, M.; Kawada, K.; Gross, R. S.; Watt, D. S. J. Org. Chem. 1990, 55, 504-11. 38 Danishefsky, S.; Yan, C.-F.; Singh, R. K.; Gammill, R. B.; McCurry, P. M., Jr.; Fritsch, N. J. Am.
Chem. Soc. 1979, 101, 7001-08. 39 Stojanac, N.; Valenta, Z. Can. J. Chem. 1991, 69, 853-55. 40 Stojanac, N.; Sood, A.; Stojanac, Z.; Valenta, Z. Can. J. Chem. 1975, 53, 619-21. 41 (a) Hirota, H.; Yokoyama, A.; Miyaji, K.; Nakamura, T.; Igarashi, M.; Takahashi, T. J. Org.
Chem. 1991, 56, 1119-27. (b) Shishido, K.; Takahashi, K.; Fukumoto, K.; Kametani, T.; Honda,
T. J. Org. Chem. 1987, 52, 5704-14. 42 Dutcher, J. S.; MacMillan, J. G.; Heathcock, C. H. J. Org. Chem. 1976, 41, 2663-69. 43 Honda, T.; Murae, T.; Ohta, S.; Kurata, Y.; Kawai, H.; Takahashi, T.; Itai, A.; Iitaka, Y. Chem.
Lett. 1981, 299-302. 44 Casinovi, C. G.; Bellavita, V.; Grandolini, G.; Ceccherelli, P. Tetrahedron Lett. 1965, 27, 2273-
79. 45 (a) Shing, T. K. M.; Jiang, Q.; Mak, T. C. W. J. Org. Chem. 1998, 63, 2056-57. (b) Shing, T. K.
M.; Jiang, Q. J. Org. Chem. 2000, 65, 7059-69. 46 (a) Bomquist, A. T.; Verdol, J. A. J. Org. Chem. 1955, 77, 81-83. (b) Bailey, J. W.; Economy, J.
J. Am. Chem. Soc. 1955, 77, 1133-36. (c) Bailey, W. J.; Cunov, C. H.; Nicholas, L. J. Am. Chem.
Soc. 1955, 77, 2787-90. 47 (a) Tsuge, O.; Wada, E.; Kanemasa, S.; Sakoh, H.; Wada, E. Bull. Chem. Soc. Jpn. 1984, 57,
3234-41. (b) Tsuge, O.; Wada, E.; Kanemasa, S.; Sakoh, H. Bull. Chem. Soc. Jpn. 1984, 57, 3221-
33. (c) Tsuge, O.; Wada, E.; Kanemasa, S.; Sakoh, H. Bull. Chem. Soc. Jpn. 1989, 62, 1198-1204. 48 (a) Tsuge, O.; Hatta, T.; Yakata, K.; Maeda, H. Chem. Lett. 1994, 10, 1833-36. (b) Saito, T.;
Kimura, H.; Chonan, T.; Soda, T.; Karakasa, T. Chem. Commun. 1997, 11, 1013-14. (c) Saito, T.;
Kimura, H.; Sakamaki, K.; Karakasa, T.; Moriyama, S. Chem. Commun. 1999, 7, 811-12. 49 Fallis, A. G.; Parra, S.; Legoupy, S.; Woo, S. Org. Lett. 1999, 1, 1013-16.

158
50 (a) Mancuso, A. J.; Huang, S.-L.; Swern, D. J. Org. Chem. 1978, 43, 2480-82. (b) Mancuso, A. J.;
Brownfain, D. S.; Swern, D. J. Org. Chem. 1979, 44, 4148-50. 51 Danishefsky, S.; Bednarski, M. Tetrahedron Lett. 1984, 25, 721-24. 52 (a) Fréchette, S. Mémoire de maîtrise, Université de Sherbrooke, 2000, 220 pages. (b) Spino, C. ;
Fréchette, S. Tetrahedron Lett. 2000, 41, 8033-36. Pour une revue littéraire, voir (c) Krause, N.;
Hoffmann-Röder, A. Tetrahedron 2004, 60, 11671-94. (d) Reich, H. J.; Eisenhart, E. K.;
Whipple, W. L.; Kelly, M. J. J. Am. Chem. Soc. 1988, 110, 6432-42 ainsi que les références
citées. (e) Miesch, M. Synthesis 2004, 746-52. 53 Pour une analyse détaillée des éléments stéréochimiques lors de cette cycloaddition, voir la
référence 30a ainsi que Hill, B. Thèse de doctorat, Université de Sherbrooke, 2001, 370 pages. 54 (a) Lunt, J. C.; Sondheimer, F. J. Chem. Soc. 1950, 2957-61. (b) Jitkow, O. N.; Bogert, M. T. J.
Am. Chem. Soc. 1941, 63, 1979-84. (c) Omodani, T.; Shishido, K. J. Chem. Soc., Chem. Commun.
1994, 2781-82. 55 (a) Gassman, P. G.; Chavan, S. P. J. Chem. Soc., Chem. Commun. 1989, 13, 837-39. (b) Gassman,
P. G.; Singleton, D. A.; Wilwerding, J. J.; Chavan, S. P. J. Am. Chem. Soc. 1987, 109, 2182-84.
(c) Gassman, P. G.; Singleton, D. A. Tetrahedron Lett. 1987, 28, 5969-72. (d) Roush, W. R.;
Barda, D. A. J. Am. Chem. Soc. 1997, 119, 7402-03. 56 (a) Lomberget, T.; Chataigner, I.; Bouyssi, D.; Maddaluno, J.; Balme, G. Tetrahedron Lett. 2004,
45, 3437-41. (b) Engler, T. A.; Sampath, U.; Vander Velde, D.; Takusagawa, F. Tetrahedron
1992, 48, 9399-9416. 57 (a) Nicolaou, K. C.; Hwang, C.-K.; Sorensen, E. J.; Clairborne, C. F. J. Chem. Soc., Chem.
Commun. 1992, 1117-18. (b) Jackson, R. W.; Shea, K. J. Tetrahedron Lett. 1994, 35, 1317-1320.
(c) Alaimo, C. A.; Coburn, C. A.; Danishefsky, S. J. Tetrahedron Lett. 1994, 35, 6603-06. (d)
Winkler, J. D.; Kim, H. S.; Kim, S. Tetrahedron Lett. 1995, 36, 687-690. (e) Rubenstein, S. M.;
Williams, R. M. J. Org. Chem. 1995, 60, 7215-23. (f) Funk, R. L.; Yost III, K. J. J. Org. Chem.
1996, 61, 2598-99. 58 (a) Shea, K. J.; Fruscella, W. M.; Carr, R. C.; Burke, L. D.; Cooper, D. K. J. Am. Chem. Soc.
1987, 109, 447-52. (b) Shea, K. J.; Burke, L. D.; England, W. P. J. Am. Chem. Soc. 1988, 110,
860-64. 59 Fallis, A. G. Acc. Chem. Res. 1999, 32, 464-74. 60 Jung, M. E.; Gervay, J. J. Am. Chem. Soc. 1991, 113, 224-32.

159
61 (a) Spino, C.; Thibault, C.; Gingras, S. J. Org. Chem. 1998, 63, 5283-87. Pour une revue des
réactions péricycliques impliquant les vinylallènes, voir (b) Okamura, W. H. Acc. Chem. Res.
1983, 16, 81-88. 62 Reich, H. J.; Eisenhart, E. K.; Whipple, W. L.; Kelly, M. J. J. Am. Chem. Soc. 1988, 110, 6432-
42. 63 (a) Reich, H. J.; Eisenhart, E. K.; Olson, R. E.; Kelly, M. J. J. Am. Chem. Soc. 1986, 108, 7791-
7800. (b) Reich, H. J.; Eisenhart, E. K. J. Org. Chem. 1984, 49, 5282-83. 64 (a) Regás, D.; Ruiz, J. M.; Alfonso, M. M.; Galindo, A.; Palenzuela, J. A. Tetrahedron Lett. 2003,
44, 8471-74. (b) de Frutos, O.; Echavarren, A. M. Tetrahedron Lett. 1997, 38, 7941-42. (c)
Bartlett, A. J.; Laird, T.; Ollis, W. D. J. Chem. Soc., Perkin Trans. 1 1975, 1315-20. 65 Pour une revue, voir Gauthier, D. R., Jr.; Zandi, K. S.; Shea, K. J. Tetrahedron 1998, 54, 2289-
2338. 66 Penkett, C. S.; Byrne, P. W.; Teobald, B. J.; Rola, B.; Ozanne, A.; Hitchcock, R. B. Tetrahedron
2004, 60, 2771-84. 67 (a) Sugimura, T.; Tei, T.; Mori, A.; Okuyama, T.; Tai, A. J. Am. Chem. Soc. 2000, 122, 2128-29.
(b) Faure, S.; Piva-Le-Blanc, S.; Bertrand, C.; Pete, J.-P.; Faure, R.; Piva, O. J. Org. Chem. 2002,
67, 1061-70. 68 (a) Righi, P.; Marotta, E.; Landuzzi, A.; Rosini, G. J. Am. Chem. Soc. 1996, 118, 9446-47. (b)
Garner, P.; Cox, P. B.; Anderson, J. T.; Protasiewicz, J.; Zaniewski, R. J. Org. Chem. 1997, 62,
493-98. 69 (a) Maifeld, S. V.; Miller, R. L.; Lee, D. J. Am. Chem. Soc. 2004, 126, 12228-29. (b) Evans, P.
A.; Cui, J.; Gharpure, S. J.; Polosukhin, A.; Zhang, H.-R. J. Am. Chem. Soc. 2003, 125, 14702-03.
(c) Gaich, T.; Mulzer, J. Org. Lett. 2005, 7, 1311-13. 70 (a) Trost, B. M.; Grese, T. A. J. Am. Chem. Soc. 1991, 113, 7363-72. (b) Sole, D.; Diaba, F.;
Bonjoch, J. J. Org. Chem. 2003, 68, 5746-49. 71 (a) Friestad, G. K.; Massari, S. E. J. Org. Chem. 2004, 69, 863-75. (b) Sukeda, M.; Ichikawa, S.;
Matsuda, A.; Shuto, S. J. Org. Chem. 2003, 68, 3465-75. 72 Pour une revue sur l’aspect stéréochimique de la cycloaddition de Diels-Alder intramoléculaire,
voir Craig, D. Chem. Soc. Rev. 1987, 16, 187-238. 73 (a) Tripathy, R.; Carroll, P. J.; Thornton, E. R. J. Am. Chem. Soc. 1991, 113, 7630-40. (b) Unni,
A. K.; Takenaka, N.; Yamamoto, H.; Rawal, V. H. J. Am. Chem. Soc. 2005, 127, 1336-37.

160
74 Genisson, Y.; Tyler, P. C.; Ball, R. G.; Young, R. N. J. Am. Chem. Soc. 2001, 123, 11381-87. 75 Stork, G.; Chan, T. Y. J. Am. Chem. Soc. 1995, 117, 6595-96. 76 Bertozzi, F.; Olsson, R.; Frejd, T. Org. Lett. 2000, 2, 1283-86. 77 Barriault, L.; Thomas, J. D. O.; Clément, R. J. Org. Chem. 2003, 68, 2317-23. 78 (a) Shimada, S.; Osoda, K.; Narasaka, K. Bull. Chem. Soc. Jpn. 1993, 66, 1254-57. (b) Narasaka,
K.; Shimada, K.; Osoda, N.; Iwasawa, N. Synthesis 1991, 1171-72. 79 Nicolaou, K. C.; Liu, J.-J.; Yang, Z.; Ueno, H.; Sorensen, E. J.; Claiborne, C. F. Guy, R. K.
Hwang, C.-K.; Nakada, M.; Nantermet, P. G. J. Am. Chem. Soc. 1995,117, 634-644. 80 (a) Batey, R. A.; Thadani, A. N.; Lough, A. J. J. Am. Chem. Soc. 1999, 121, 450-51 et les
références qui y sont citées. (b) Batey, R. A.; Thadani, A. N.; Lough, A. J. Chem. Commun. 1999,
475-76. 81 (a) Saito, A.; Ito, H.; Taguchi, T. Org. Lett. 2002, 4, 4619-21. (b) Jung, M. E.; Huang, A.;
Johnson, T. W. Org. Lett. 2000, 2, 1835-37. (c) Saito, A.; Yanai, H.; Taguchi, T. Tetrahedron
2004, 60, 12239-47. 82 (a) Wan, Z.-K.; Snyder, J. K. Tetrahedron Lett. 1998, 39, 2487-90. Pour des études théoriques sur
l’origine de la stéréosélectivité lors de cycloaddition avec de tels connecteurs, voir (b) Tantillo,
D. J.; Houk, K. N.; Jung, M. E. J. Org. Chem. 2001, 66, 1938-40. 83 Corey, E. J.; da Silva Jardine, P.; Rohloff, J. C. J. Am. Chem. Soc. 1988, 110, 3672-73. 84 (a) Trost, B. M. Science 1991, 254, 1471-77. (b) Trost, B. M. Acc. Chem. Res. 2002, 35, 695-705.
(c) Trost, B. M. Angew. Chem. Int. Ed. 1995, 34, 259-81. 85 Ishikawa, T.; Senzaki, M.; Kadoya, R.; Morimoto, T.; Miyake, N.; Izawa, M.; Saito, S. J. Am.
Chem. Soc. 2001, 123, 4607-08. 86 (a) Mitsunobu, O.; Wada, M.; Sano, T. J. Am. Chem. Soc. 1972, 94, 679-80. (b) Grochowski, E.;
Jurczak, J. Synthesis 1976, 682-84. (c) Tsunoda, T.; Yamamiya, Y.; Ito, S. Tetrahedron Lett.
1993, 34, 1639-42. Pour de récentes études mécanistiques, voir (d) Ahn, C.; Correia, R.;
DeShong, P. J. Org. Chem. 2002, 67, 1751-53. (e) McNulty, J.; Capretta, A.; Laritchev, V. Dyck,
J.; Robertson, A. J. Angew. Chem. Int. Ed. 2003, 42, 4051-54. 87 (a) Craig, D.; Reader, J. C. Synlett 1992, 757-58. (b) Ainsworth, P. J.; Craig, D.; Reader, J. C.;
Slawin, A. M. Z.; White, A. J. P.; Williams, D. J. Tetrahedron 1996, 52, 695-724. 88 Bulugahapitiya, P.; Landais, Y.; Parra-Rapado, L.; Planchenault, D.; Weber, V. J. Org. Chem.
1997, 62, 1630-41.

161
89 (a) Metz, P.; Fleischer, M.; Fröhlich, R. Synlett 1992, 985-87. (b) Metz, P.; Fleischer, M. Synlett
1993, 399-400. (c) Metz, P.; Fleischer, M.; Fröhlich, R. Tetrahedron 1995, 51, 711-32. (d)
Schwan, A. L.; Snelgrove, J. L.; Kalin, M. L.; Froese, R. D. J.; Morokuma, K. Org. Lett. 1999, 1,
487-90 et les références qui y sont citées. 90 (a) Metz, P.; Seng, D.; Fröhlich, R.; Wibbeling, B. Synlett 1996, 741-42. (b) Plietker, B.; Seng,
D.; Fröhlich, R.; Metz, P. Tetrahedron 2000, 56, 873-79. 91 Brosius, A. D.; Overman, L. E.; Schwink, L. J. Am. Chem. Soc. 1999, 121, 700-09. 92 (a) Bols, M.; Skrydstrup, T. Chem. Rev. 1995, 95, 1253-77. (b) Fensterbank, L.; Malacria, M.;
Sieburth, S. McN. Synthesis 1997, 813-54. 93 Tamao, K.; Kobayashi, K.; Ito, Y. J. Am. Chem. Soc. 1989, 111, 6478-80. 94 Stork, G.; Chan, T. Y.; Breault, G. A. J. Am. Chem. Soc. 1992, 114, 7578-79. (c) Trost, B. M.
Angew. Chem. Int. Ed. 1995, 34, 259-81. 95 (a) Sieburth, S. McN.; Fensterbank, L. J. Org. Chem. 1992, 57, 5279-81. (b) Sieburth, S. McN.;
Lang, J. J. Org. Chem. 1999, 64, 1780-81. 96 (a) Comprehensive Organic Synthesis; Trost, B. M. and Fleming, I., Eds.; Pergamon Press:
Oxford; Volume 7, Chapitre 4.3, pp. 641-52. (b) Tamao, K.; Kumada, M.; Maeda, K. Tetrahedron
Lett. 1984, 25, 321-24. (c) Fleming, I.; Henning, R.; Plaut, H. J. Chem. Soc., Chem. Commun.
1984, 29. 97 (a) Craig, D; Reader, J. C. Tetrahedron Lett. 1990, 31, 6585-88. (b) Ainsworth, P. J.; Craig, D.;
Reader, J. C.; Slawin A. M. Z.; White, A. J. P.; Williams, D. J. Tetrahedron 1995, 51, 11601-22.
(c) Gillard, J. W.; Fortin, R.; Grimm, E. L.; Maillard, M.; Tjepkema, M.; Bernstein, M. A.;
Glaser, R. Tetrahedron Lett. 1991, 32, 1145-48. 98 Tamao, K.; Kakui, T.; Akita, M.; Iwahara, T.; Kanatani, R.; Yoshida, J.; Kumada, M.
Tetrahedron 1983, 39, 983-90. 99 Nishiyama, H.; Kitajima, T.; Matsumoto, M.; Itoh, K. J. Org. Chem. 1984, 49, 2298-2300. 100 Shuto, S.; Kanazaki, M.; Ichikawa, S.; Minakawa, N.; Matsuda, A. J. Org. Chem. 1998, 63, 746-
54. Tamao, K.; Ishida, N. J. Organomet. Chem. 1984, 269, C37-C39. 101 Fleming, I.; Henning, R.; Parker, D. C.; Plaut, H. E.; Sanderson, P. E. J. Chem. Soc., Perkin
Trans. 1 1995, 317-37. 102 Smitrovich, J. H.; Woerpel, K. A. J. Org. Chem. 1996, 61, 6044-46. 103 Rondestvedt Jr., C. S. J. Am. Chem. Soc. 1954, 76, 1926-29.

162
104 (a) Kuethe, J. T.; Brooks, C. A.; Comins, D. L. Org. Lett. 2003, 5, 321-23. (b) Weinreb, S. M.;
Staib, R. R. Tetrahedron 1982, 36, 3087-3128. 105 Pour une revue, voir Shostakovskii, M. F.; Trofimov, B. A.; Atavin, A. S.; Lavrov, V. I. Russian
Chem. Rev. 1968, 37, 907-19. 106 (a) Schwartz, J.; Riediker, M. J. Am. Chem. Soc. 1982, 104, 5842-44. (b) Adelman, R. L. J. Am.
Chem. Soc. 1955, 77, 1669-70. (c) Adelman, R. L. J. Am. Chem. Soc. 1953, 75, 2678-82. 107 Preuss, H.; Winterfeldt, E. Chem. Ber. 1966, 99, 450-58. 108 (a) Marshall, J. A.; Sehon, C. A. J. Org. Chem. 1995, 60, 5966-68. (b) Ma, S.; Gao, W.;
J. Org. Chem. 2002, 67, 6104-12. (b) Hoffmann-Roder, A.; Krause, N. Org. Lett. 2001, 3, 2537-
38. (c) Berry, C. R.; Hsung, R. P.; Antoline, J. E.; Petersen, M. E.; Challeppan, R.; Nielson, J. A.
J. Org. Chem. 2005, 70, 4038-42. 109 (a) Brown, R. D.; Buchanan, A. S.; Humffray, A. A. Australian J. Chem. 1965, 18, 1507-20. (b)
Larock, R. C.; Harrison, L. W. J. Am. Chem. Soc. 1984, 106, 4218-27. 110 (a) Nishizawa, M.; Takenaka, H.; Nishide, H.; Hayashi, Y. Tetrahedron Lett. 1983, 24, 2581-84.
(b) Nishizawa, M.; Morikuni, E.; Asoh, K.; Kan, Y.; Uenoyama, K.; Imagawa, H. Synlett 1995,
169-70. (c) Nishizawa, M.; Takenaka, H.; Hayashi, Y. J. Am. Chem. Soc. 1984, 106, 4290-91. (d)
Parker, K.; Resnick, L. J. Org. Chem. 1995, 60, 5726-28. (e) Nishizawa, M.; Yadav, V. K.;
Skwarczynski, M.; Takao, H.; Imagawa, H.; Sugihara, T. Org. Lett. 2003, 5, 1609-11. 111 Pour une revue, voir Bhatt, M. V.; Kulkarni, S. U. Synthesis 1983, 249-82. 112 (a) Karger, M. H. ; Mazur, Y. J. Am. Chem. Soc. 1968, 90, 3878-79. (b) Otera, J.; Matsuzaki, S.
Synthesis 1986, 1019-20. 113 Ganem, B.; Small Jr., V. R. J. Org. Chem. 1974, 39, 3728-30. 114 Narayanan, C. R.; Iyer, K. N. J. Org. Chem. 1965, 30, 1734-36. 115 (a) Ho, T. L.; Olah, G. A. Angew. Chem. Int. Ed. Engl. 1976, 15, 774-75. (b) Jung, M. E.; Lyster,
M. A. J. Org. Chem. 1977, 42, 3761-64. 116 Guindon, Y.; Therien, M.; Girard, Y.; Yoakim, C. J. Org. Chem. 1987, 52, 1680-86. 117 Caulsen, P. H. T.; Katsuki, T.; Martin, V. S.; Sharpless, K. B. J. Org. Chem. 1981, 46, 3936-38. 118 Pindur, U.; Lutz, G.; Otto, C. Chem. Rev. 1993, 93, 741-61. 119 Kumar, A. Chem. Rev. 2001, 101, 1-20. 120 Velcicky, J.; Lex, J.; Schmalz, H.-G. Org. Lett. 2002, 4, 565-68. 121 Lecker, S. H.; Nguyen, N. H.; Vollhardt, K. P. C. J. Am. Chem. Soc. 1986, 108, 856-58.

163
122 (a) Peterson, D. J. J. Org. Chem. 1968, 33, 780-84. Pour des revues, voir (b) Ager, D. J. Synthesis
1984, 384-98. (c) Ager, D. J. Org. React. 1990, 42, 1-223. (c) Vorbrüggen, H. Silicon-mediated
Transformations of Functional Groups; Wiley-VCH: Weinheim, 2004, chapitre 10. (d) Brook, M.
A. Silicon in Organic, Organometallic, and Polymer Chemistry; Wiley: Oxford, 2000, pages 513-
18. 123 (a) Trost, B. M. Acc. Chem. Res. 1980, 13, 385-93. (b) Trost, B. M.; Van Vranken, D. L. Chem.
Rev. 1996, 96, 395-422. 124 Brook, M. A. Silicon in Organic, Organometallic, and Polymer Chemistry; Wiley: Oxford, 2000,
chapitre 16, pages 552-607. 125 Lambert, J. B.; Zhao, Y.; Emblidge, R. W.; Salvador, L. A.; Liu, X.; So, J.-H.; Chelius, E. C. Acc.
Chem. Res. 1999, 32, 183-90. 126 (a) Alexakis, A. Pure Appl. Chem. 1992, 64, 387-92. (b) Dieter, K. R. ; Nice, L. E. Tetrahedron
Lett. 1999, 40, 4293-96. (c) Marshall, J. A.; Pinney, K. G. J. Org. Chem. 1993, 58, 7180-84. 127 Pour une revue, voir Sonogashira, K. dans Comprehensive Organic Synthesis, volume 3, chapitre
2.4, Trost, B. M. et Fleming, I., Eds; Pergamon Press : Oxford, 1991. 128 (a) Negishi, E. Acc. Chem. Res. 1982, 15, 340-48. (b) Heck, R. F. Palladium Reagents in Organic
Syntheses; Academic Press: Londre, 1985, page 306. 129 Sonogashira, K.; Tohda, Y.; Hagihara, N. Tetrahedron Lett. 1975, 50, 4467-70. 130 Voir la référence 126 ainsi que (a) Cassar, L. J. Organomet. Chem. 1975, 93, 253-57. (b) Dieck,
H. A.; Heck, R. F. J. Organomet. Chem. 1975, 93, 259-63. 131 Stephens, R. D.; Castro, C. E. J. Org. Chem. 1963, 28, 3313-15. 132 Hills, I. D.; Netherton, M. R.; Fu, G. C. Angew. Chem. Int. Ed. 2003, 42, 5749-52. 133 Yamashita, M.; Cuevas Vicario, J. V.; Hartwig, J. F. J. Am. Chem. Soc. 2003, 125, 16347-60. 134 Eckhardt, M.; Fu, G. C. J. Am. Chem. Soc. 2003, 125, 13642-43. 135 Tolman, C. A. Chem. Rev. 1977, 77, 313-48. 136 Farina, V.; Krishnan, B. J. Am. Chem. Soc. 1991, 113, 9585-95. 137 Wang, Z.; Zhang, Z.; Lu, X. Organometallics 2000, 19, 775-80. 138 (a) Katsuki, T.; Sharpless, K. B. J. Am. Chem. Soc. 1980, 102, 5974-76. (b) Gao, Y.; Klunder, J.
M.; Hanson, R. M.; Masamune, H.; Ko, S. Y.; Sharpless, K. B. J. Am. Chem. Soc. 1987, 109,
5765-80. (c) Sharpless, K. B.; Berrisford, D. J.; Bolm, C. Angew. Chem. Int. Ed. 1995, 34, 1059-
70.

164
139 Pour une revue de réactions dirigées par le substrat, voir : Hoveyda, A. H.; Evans, D. A.; Fu, G.
C. Chem. Rev. 1993, 93, 1307-70. 140 (a) Zhang, W.; Loebach, J. L.; Wilson, S. R.; Jacobsen, E. N. J. Am. Chem. Soc. 1990, 112, 2801.
(b) Jacobsen, E. N.; Zhang, W.; Muci, A. R.; Ecker, J. R.; Deng, L. J. Am. Chem. Soc. 1991, 113,
7063. Pour une revue sur l’époxydation d’oléfines non-activées, voir a) Jacobsen, E. N.;
Asymmetric Catalytic Epoxidation of Unfunctionalized Olefins, Catalytic Asymmetric Synthesis;
Ojima, I.; VCH: New York, 1993, pages 159-202. 141 Wang, Z.-X.; Tu, Y.; Frohn, M.; Zhang, J.-R.; Shi, Y. J. Am. Chem. Soc. 1997, 119, 11224-35. 142 Tu, Y.; Wang, Z.-X.; Shi, Y. J. Am. Chem. Soc. 1996, 118, 9806-07. 143 (a) Wang, Z.-X.; Cao, G.-A.; Shi, Y. J. Org. Chem. 1999, 64, 7646-50. (b) Cao, G.-A.; Wang, Z.-
X.; Tu, Y.; Shi, Y. Tetrahedron Lett. 1998, 39, 4425-28. 144 (a) Tu, Y.; Wang, Z.-X.; Frohn, M.; He, M.; Yu, H.; Tang, Y.; Shi, Y. J. Org. Chem. 1998, 63,
8475-85. (b) Wang, Z.-X.; Miller, S. M.; Anderson, O. P.; Shi, Y. J. Org. Chem. 1999, 64, 6443-
58. 145 Baker, R. H.; Tinsley, S. W.; Butler Jr., B.; Riegel, B. J. Am. Chem. Soc. 1950, 72, 393-95. 146 Smith III, A. B.; Levenberg, P. A.; Jerris, P. J.; Scarborough, R. M.; Wovkulich Jr., P. M. J. Am.
Chem. Soc. 1981, 103, 1501-13. 147 Pour une revue, voir Loupy, A. Microwaves in Organic Synthesis; Wiley-VCH : Weinheim, 2002. 148 Li, K.; Du, W.; Que, N. L. S.; Liu, H.-W. J. Am. Chem. Soc. 1996, 118, 8763-64. 149 Dang, H. P.; Linstrumelle, G. Tetrahedron Lett. 1978, 2, 191-94. 150 Carpita, A.; Rossi, R. Tetrahedron Lett. 1986, 27, 4351-54. 151 (a) Johnson, C. R.; Herr, R. W.; Wieland, D. M. J. Org. Chem. 1973, 38, 4263-68. Pour une
revue, voir (b) Krause, N. Modern Organocopper Chemistry; Wiley-VCH: Weinheim, 2002, 377
pages. 152 Pour le premier exemple d’un cyanocuprate : (a) Gorlier, J.-P.; Hamon, L.; Levisalles, J.;
Wagnon, J. J. Chem. Soc., Chem. Commun. 1973, 88. Pour le développement de ce type de
cuprate, voir (b) Lipshutz, B. H.; Wihelm, R. S.; Floyd, D. M. J. Am. Chem. Soc. 1981, 103, 7672.
Pour la structure de l’espèce en solution, voir (c) Krause, N. Angew. Chem. Int. Ed. 1999, 38, 79. 153 Einhorn, J.; Einhorn, C.; Ratajczak, F.; Pierre, J.-L. J. Org. Chem. 1996, 61, 7452-54. 154 Griffith, W. P.; Ley, S. V. Aldrichim. Acta 1990, 23, 13.

165
155 (a) Kornblum, N.; Jones, W. J.; Anderson, G. J. J. Am. Chem. Soc. 1959, 81, 4113-14. (b) Epstein,
W. W.; Sweat, F. W. Chem. Rev. 1967, 67, 247-60. 156 (a) Wirth, T. Angew. Chem. Int. Ed. 2001, 40, 2812-14 ainsi que les références qui y sont citées.
(b) Nicolaou, K. C.; Baran, P. S.; Zhong, Y.-L.; Barluenga, S.; Hunt, K. W.; Kranich, R.; Vega, J.
A. J. Am. Chem. Soc. 2002, 124, 2233-44. (c) Nicolaou, K. C.; Montagnon, T.; Baran, P. S.;
Zhong, Y.-L. J. Am. Chem. Soc. 2002, 124, 2245-58. (d) Wu, Y.; Shen, X.; Huang, J.-H.; Tang,
C.-J.; Liu, H.-H.; Hu, Q. Tetrahedron Lett. 2002, 43, 6443-45. 157 (a) Frigerio, M.; Santagostino, M. Tetrahedron Lett. 1994, 35, 8019-22. (b) Frigerio, M.;
Santagostino, M.; Sputore, S.; Palmisano, G. J. Org. Chem. 1995, 60, 7272-76. (c) De Munari, S.;
Frigerio, M.; Santagostino, M. J. Org. Chem. 1996, 61, 9272-79. Attention! le IBX est un oxydant
reconnu pour exploser sous impact ou hautes températures. Voir Plumb, J. B.; Harper, D. J.
Chem. Eng. News 1990, 68 (29), 3. 158 More, J. D.; Finney, N. S. Org. Lett. 2002, 4, 3001-03. 159 (a) Horner, L.; Hoffmann, H.; Wippel, H. G.; Klahre, G. Chem. Ber. 1959, 92, 2499. (b)
Wadsworth, W. S.; Emmons, W. O. J. Amer. Chem. Soc. 1961, 83, 1733. Pour une revue, voir (c)
Boutagy, J.; Thomas, R. Chem. Rev. 1974, 74, 7-99. 160 (a) Julia, S. A.; Baudin, J. B.; Hareau, G.; Lorne, R.; Ruel, O. Bull. Soc. Chim. Fr. 1993, 130,
856. (b) Julia, S. A.; Baudin, J. B.; Hareau, G.; Ruel, O. Bull. Soc. Chim. Fr. 1993, 130, 336. 161 (a) Blakemore, P. R.; Cole, W. J.; Kocienski, P. J.; Morley, A. Synlett 1998, 26. (b) Blakemore, P.
R.; Kocienski, P. J.; Morley, A.; Muir, K. J. Chem. Soc., Perkin Trans. 1 1999, 955-68. 162 (a) Bellingham, R.; Jarowicki, K.; Kocienski, P.; Martin, V. Synthesis 1996, 285. (b) Charette, A.
B.; Lebel, H. J. Am. Chem. Soc. 1996, 118, 10327-28. 163 (a) Wittig, G.; Geissler, G. Liebigs Ann. Chem. 1953, 580, 44. (b) Wittig, G.; Schollkopf, U.
Chem. Ber. 1954, 87, 1318. Pour une revue des aspects de cette réaction, voir (c) Maryanoff, B.
E.; Reitz, A. B. Chem. Rev. 1989, 89, 863-927. 164 (a) House, H. O.; Jones, V. K.; Frank, G. A. J. Org. Chem. 1964, 29, 3327-33. (b) Chapdelaine,
D.; Dubé, P.; Deslongchamps, P. Synlett 1999, 12, 1819-21. 165 Cahiez, C.; Alexakis, A.; Normant, J. F. Synthesis 1978, 528. 166 Marino, J. P.; Floyd, D. M. Tetrahedron Lett. 1979, 8, 675-78. 167 (a) Metz, P.; Schoop, A. Tetrahedron 1993, 49, 10597-10608. (b) Burke, S. D.; Piscopio, A. D.;
Marron, B. E.; Matulenko, M. A.; Pan, G. Tetrahedron Lett. 1991, 32, 857-58.

166
168 (a) Matsuzawa, S.; Horiguchi, Y.; Nakamura, E.; Kuwajima, I. Tetrahedron 1989, 45, 349-62. (b)
Corey, E. J.; Boaz, N. W. Tetrahedron Lett. 1985, 26, 6019-21. 169 (a) Ibuka, T.; Nakai, K.; Habashita, H.; Bessho, K. Fujii, N. Tetrahedron 1993, 49, 9479-88. (b)
Habashita, H.; Kawasaki, T.; Takemoto, Y.; Fujii, N.; Ibuka, T. J. Org. Chem. 1998, 63, 2392-96 170 (a) Vermeer, P.; Meijer, J.; de Graaf, C.; Schreurs, H. Recl. Trav. Chim. Pays-Bas 1974, 93, 46.
(b) Ortiz de Montellano, P. J. Chem. Soc., Chem. Commun. 1973, 709-10. 171 Kleijn, H.; Meijer, J.; Overbeek, G. C.; Vermeer, P. Recl. Trav. Chim. Pays-Bas 1982, 101, 97. 172 Bartmann, E.; Bogdanovic, B.; Janke, N.; Liao, S.; Schlichte, K. et al. Chem. Ber. 1990, 123,
1517-28. 173 Linstrumelle, G.; Krieger, J. K.; Whitesides, G. M. Org. Synth. 1976, 55, 103-13. 174 (a) Barbier, C. R. Acad. Sci. 1899, 128, 110. (b) Lai, Y. H. Synthesis 1981, 585-604. 175 Une séquence réactionnelle similaire a déjà été utilisée par deux groupes : (a) Odedra, A.; Wu, C.-
J.; Madhushaw, R. J.; Wang, S.-L.; Liu, R.-S. J. Am. Chem. Soc. 2003, 125, 9610-11. (b)
Marshall, J. A.; DuBay, W. J. J. Org. Chem. 1991, 56, 1685-87. 176 Corey, E. J.; Katzenellenbogen, J. A.; Posner, G. H. J. Am. Chem. Soc. 1967, 89, 4245-47. 177 (a) Wagner, A.; Mioskowski, C. Tetrahedron Lett. 1989, 30, 557-58. Pour une revue de ce type de
réaction, voir (b) Castro, B. R. Org. React. 1983, 29, 1-162. 178 (a) Cahiez, G.; Bernard, D.; Normant, J. F. Synthesis 1976, 245-48. (b) Whitesides, G. M.; Casey,
C. P.; Krieger, J. K. J. Am. Chem. Soc. 1971, 93, 1379-89 et les références qui y sont citées. 179 (a) Giusti, G. Bull. Soc. Chim. Fr. 1972, 753-7. (b) Le Coq, A.; Gorgues, A. Organic Syntheses,
Wiley: New York, 1988; Collect. Vol. 6, p 954. 180 Greene, T. W.; Wuts, P. G. M. Protective Groups in Organic Synthesis; deuxième edition: Wiley,
Wilheim 1991, pages 83-84. 181 Rieke, R. D. Aldrichimica Acta 2000, 33, 52-60. 182 (a) Xiong, H.; Rieke, R. D. J. Org. Chem. 1992, 57, 7007-08. (b) Sell, M. S.; Xiong, H.; Rieke, R.
D. Tetrahedron Lett. 1993, 34, 6007-10. 183 (a) Crombie, L.; Harper, S. H. J. Chem. Soc. 1950, 1152-60. (b) Crombie, L.; Harper, S. H. J.
Chem. Soc. 1950, 1707-22. 184 Garst, J. F.; Pacifici, J. A.; Singleton, V. D.; Ezzel, M. F.; Morris, J. I. J. Am. Chem. Soc. 1975,
97, 5242-49. (b) Garst, J. F.; Barbas, J. T. J. Am. Chem. Soc. 1974, 96, 3239-46. 185 Gauthier, J. Y.; Guindon, Y. Tetrahedron Lett. 1987, 28, 5985-88.

167
186 (a) Imamoto, T.; Ono, M. Chem. Lett. 1987, 501-02. (b) Wipf, P.; Venkatraman, S. J. Org. Chem.
1993, 58, 3455-59. 187 Suzuki, K.; Inomata, K. Synthesis 2002, 1819-22 188 Ghosh, A. K.; Liu, C. J. Am. Chem. Soc. 2003, 125, 2374-75. 189 Bulman Page, P. C.; McCarthy, T. J. Comprehensive Organic Synthesis; Trost, B. M. and
Fleming, I., Eds.; Pergamon Press; 1991, Volume 7, Chapitre 2.1, pp. 83-117. 190 (a) Bindra, J. S.; Grodski, A. J. Org. Chem. 1978, 43, 3240-41. (b) Caine, D.; Smith Jr., T. L. J.
Am. Chem. Soc. 1980, 102, 7568-70. 191 Hanessian, S.; Liak, T. J.; Vanasse, B. Synthesis 1981, 396. 192 Williams, D. R.; Brown, D. L.; Benbow, J. W. J. Am. Chem. Soc. 1989, 111, 1923-25. 193 Bélanger, G.; Deslongchamps, P. Org. Lett. 2000, 2, 285-87. 194 Sharpless, K. B.; Hori, T.; Truesdale, L. K.; Dietrich, C. O. J. Am. Chem. Soc. 1976, 98, 269-71. 195 (a) Yao, G.; Steliou, K. Org. Lett. 2002, 4, 485-88. Pour une revue, voir (b) Clennan, E. L.
Tetrahedron 2000, 56, 9151-79. 196 Lee, K.; Cha, J. K. J. Am. Chem. Soc. 2001, 123, 5590-91. 197 Evans, D. A.; Fu, G. C.; Hoveyda, A. H. J. Am. Chem. Soc. 1992, 114, 6671-79. 198 Lipshutz, B. H.; Keil, R.; Ellsworth, E. L. Tetrahedron Lett. 1990, 31, 7257-60. 199 (a) Corey, E. J.; Kania, R. S. J. Am. Chem. Soc. 1996, 118, 1229-30. Pour la préparation de cet
hydrure, voir Tolstikov, G. A.; Miftakhov, M. S.; Valeev, F. A. Bull. Acad. Sci. USSR, Div.
Chem. Sci. 1979, 28, 2392. 200 Fraser, W.; Suckling, C. J.; Wood, H. C. S. J. Chem. Soc., Perkin Trans. 1 1990, 3137-44. 201 Lucet, D.; Sabelle, S.; Kostelitz, O.; Le Gall, T.; Mioskowski, C. Eur. J. Org. Chem. 1999, 10,
2583-91. 202 Evans, S. B.; Abdelkader, M.; Buyle Padias, A.; Hall Jr., H. K. J. Org. Chem. 1989, 54, 2848-52. 203 Imwinkelried, R.; Seebach, D. Angew. Chem. Int. Ed. 1985, 24, 765-66. 204 Wipf, P.; Xu, W. J. Org. Chem. 1996, 61, 6556-62. 205 Negishi, E.; Brown, H. C. Synthesis 1974, 77-89. 206 Ouali, M. S.; Vaultier, M.; Carrié, R. Synthesis 1977, 626. 207 (a) Langer, F.; Schwink, L.; Devasagayaraj, A.; Chavant, P.-Y.; Knochel, P. J. Org. Chem. 1996,
61, 8229-43. (b) Pu, L.; Yu, H.-B. Chem. Rev. 2001, 101, 757-824. Pour une revue, voir (c)

168
Ramachandran, P. V.; Brown, H. C. Organoboranes for Syntheses ; ACS symposium series 783,
American Chemical Society, 2001, chapitre 3, pages 33-51. 208 (a) Spino, C.; Beaulieu, C. J. Am. Chem. Soc. 1998, 120, 11832-33. (b) Spino, C.; Beaulieu, C.;
Lafrenière, J. J. Org. Chem. 2000, 65, 7091-97. 209 Boger, D. L.; Weinreb, S. M. Hetero Diels-Alder Methodology in Organic Synthesis; Organic
Chemistry-A series of monographs, Volume 47, Academic Press Inc., 1987, chapitre 7, pages
167-213. 210 Love, B. E.; Jones, E. G. J. Org. Chem. 1999, 64, 3755-56. 211 Frigerio, M.; Santagostino, M.; Sputore, S. J. Org. Chem. 1999, 64, 4537-38. 212 Ireland, R. E.; Liu, L. J. Org. Chem. 1993, 58, 2899. 213 Cole, K. P.; Hsung, R. P. Org. Lett. 2003, 5, 4843-46.

169
ANNEXE 1 : SPECTRES DE RÉSONANCE MAGNÉTIQUE NUCLÉAIRE DES PROTONS
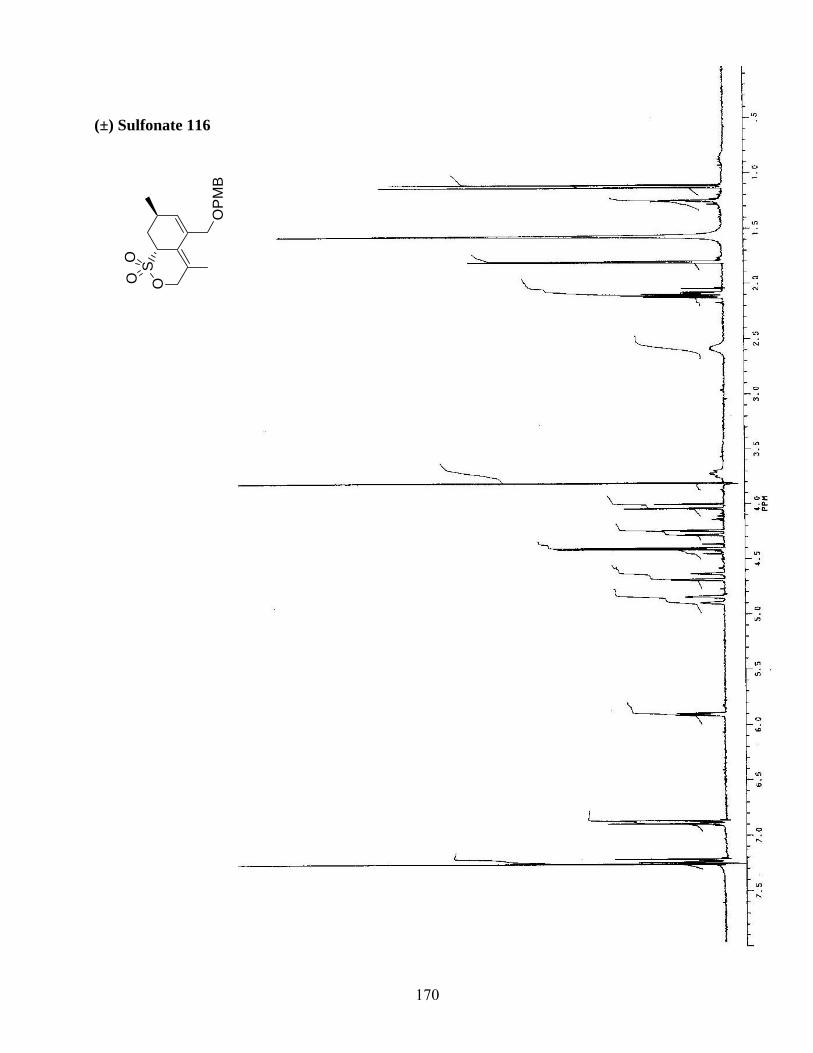
170
(±) Sulfonate 116
OS
OP
MB
OO
171
(±) Aldéhyde 118
OS
O
OO
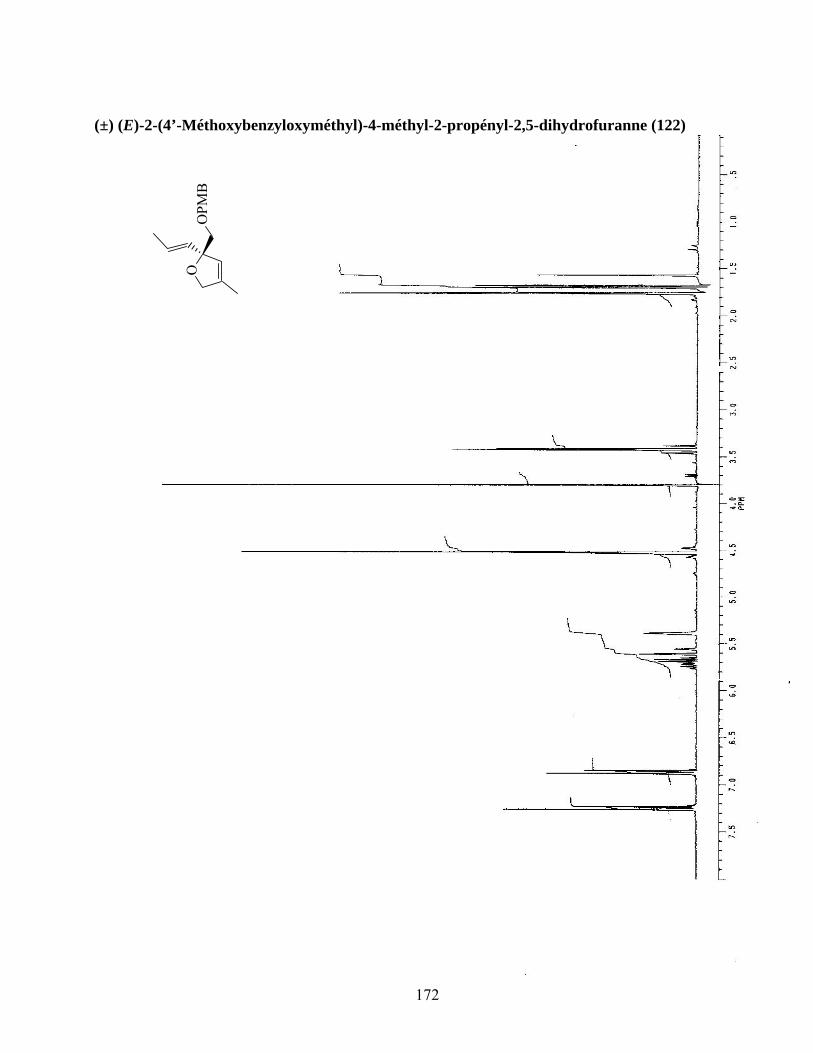
172
(±) (E)-2-(4’-Méthoxybenzyloxyméthyl)-4-méthyl-2-propényl-2,5-dihydrofuranne (122)
OO
PMB
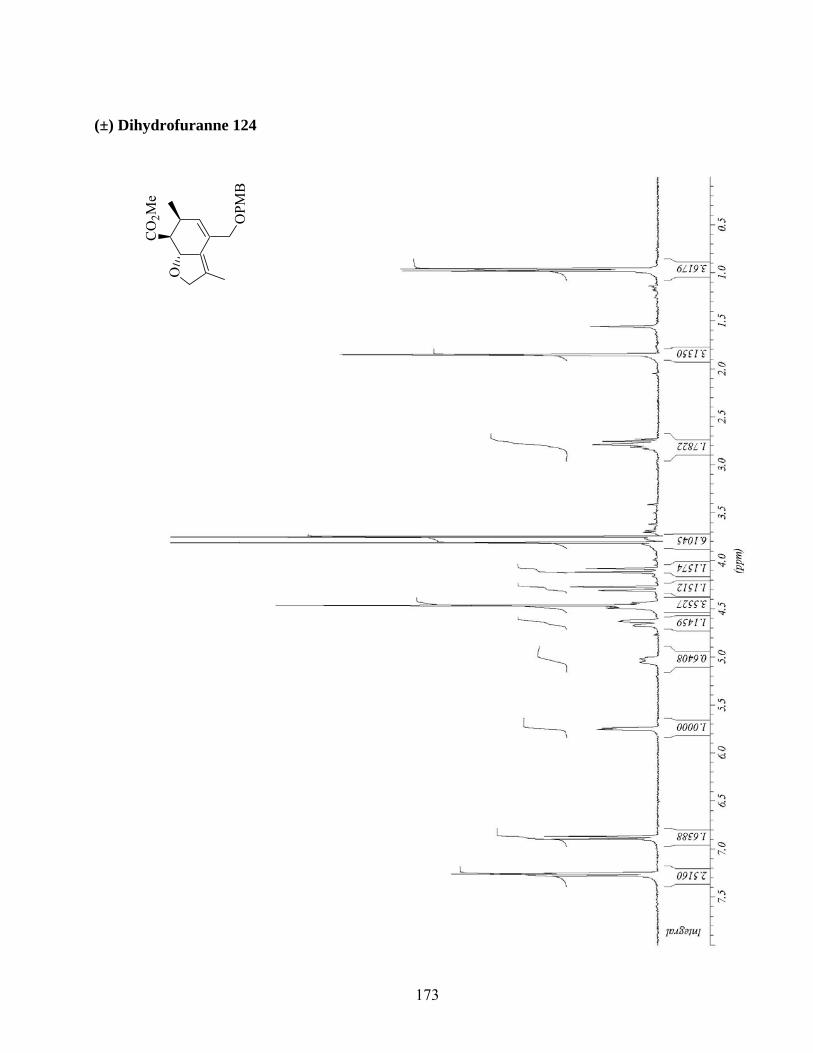
173
(±) Dihydrofuranne 124
OPM
B
OC
O2M
e

174
(±) Dihydrofuranne - alcool allylique 125
OH
OC
O2M
e

175
(±) (E)-7-tert-Butyldiméthylsilanoxyhept-2-èn-5-yn-4-ol (127)
OH
TBSO

176
(E)-7-tert-Butyldiméthylsilanoxyhept-2-én-5-yn-4-one (128)
O
TBSO
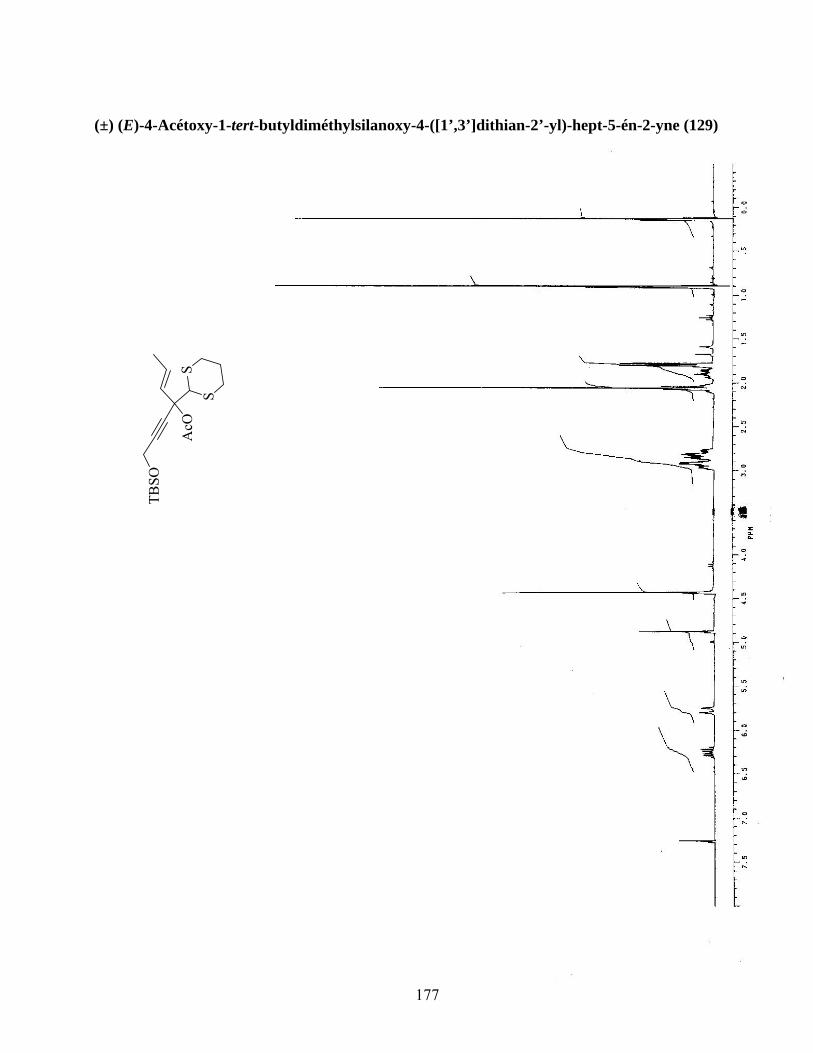
177
(±) (E)-4-Acétoxy-1-tert-butyldiméthylsilanoxy-4-([1’,3’]dithian-2’-yl)-hept-5-én-2-yne (129)
AcO
TBSO
SS

178
(±) 4-([1’,3’]Dithian-2’-yl)-2-méthylhepta-2,3,5-trién-1-ol (131)
OH
SS
C
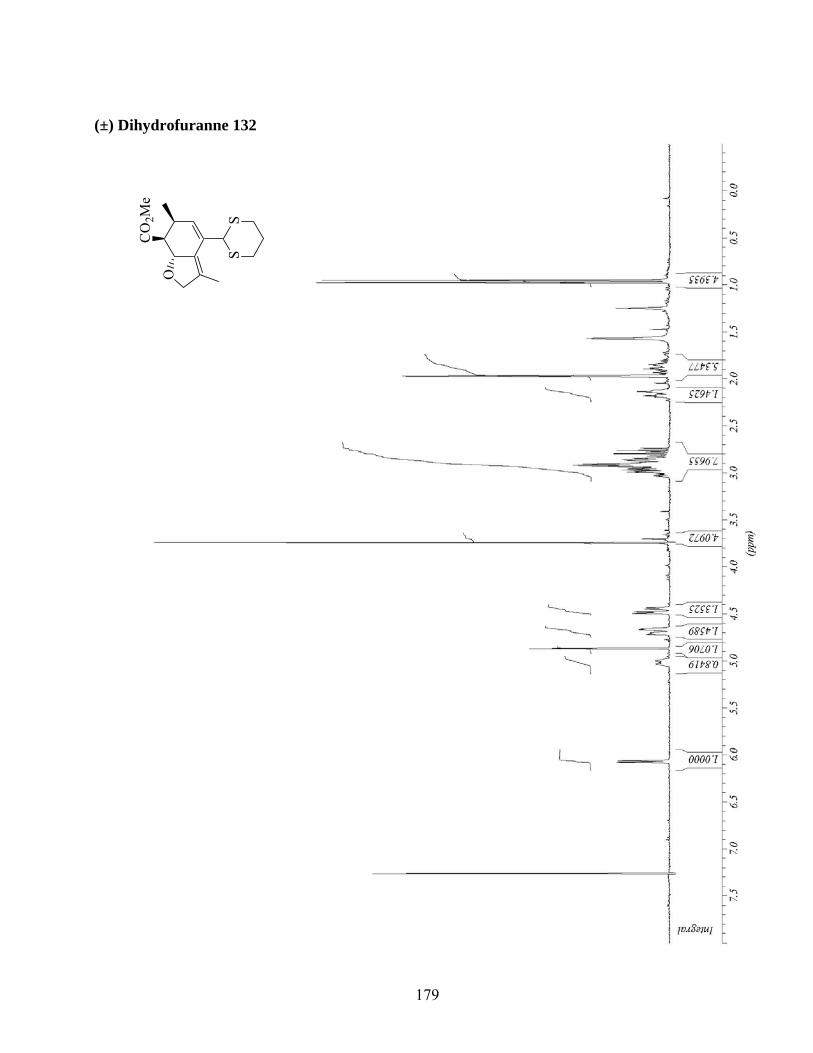
179
(±) Dihydrofuranne 132
OC
O2M
e
SS

180
(±) Aldéhyde 133
O
OC
O2M
e
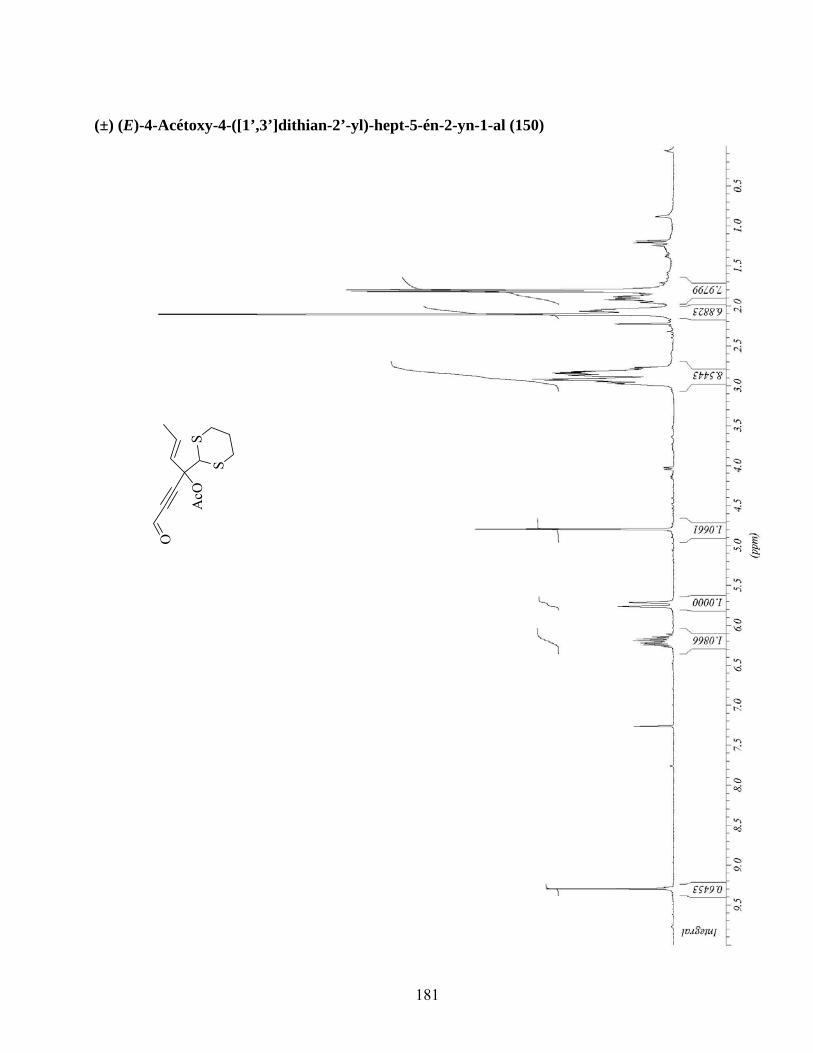
181
(±) (E)-4-Acétoxy-4-([1’,3’]dithian-2’-yl)-hept-5-én-2-yn-1-al (150)
AcO
O
SS

182
(±) (E)-5-Acétoxy-1-triméthylsilanyl-5-([1’,3’]dithian-2’-yl)-oct-6-én-3-yn-2-ol (151)
AcO
SS
OH
Me 3
Si
183
(±) 1,4-Bis(triméthylsilanyl)-but-1-yn-3-ol (153)
OH
Me 3
Si
SiM
e 3
184
(±) 3-tert-Butyldiméthylsilanoxy-1,4-bis-triméthylsilanylbut-1-yne (154)
OTB
SM
e 3Si
SiM
e 3
185
(±) 3-tert-Butyldiméthylsilanoxy-4-triméthylsilanylbut-1-yne (155)
OTB
SM
e 3Si
186
(±) (E)-7-tert-Butyldiméthylsilanoxy-8-triméthylsilanyloct-2-én-5-yn-4-ol (156)
OTB
SM
e 3Si
OH
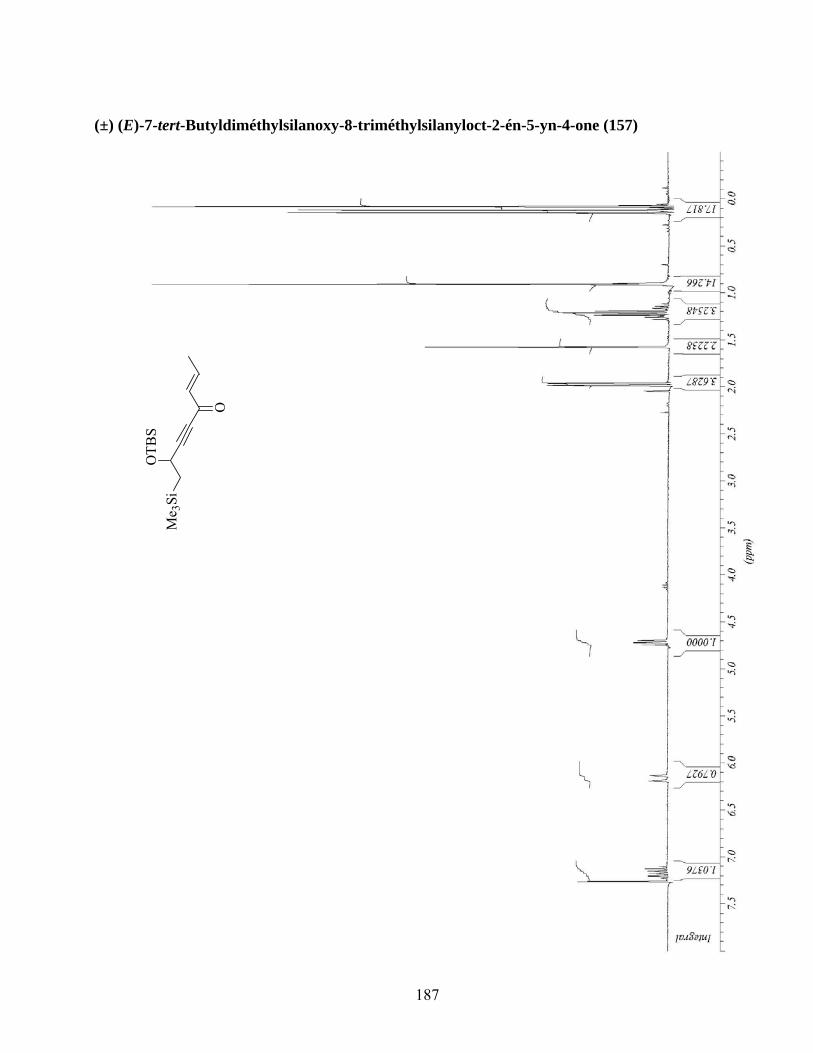
187
(±) (E)-7-tert-Butyldiméthylsilanoxy-8-triméthylsilanyloct-2-én-5-yn-4-one (157)
OTB
SM
e 3Si
O
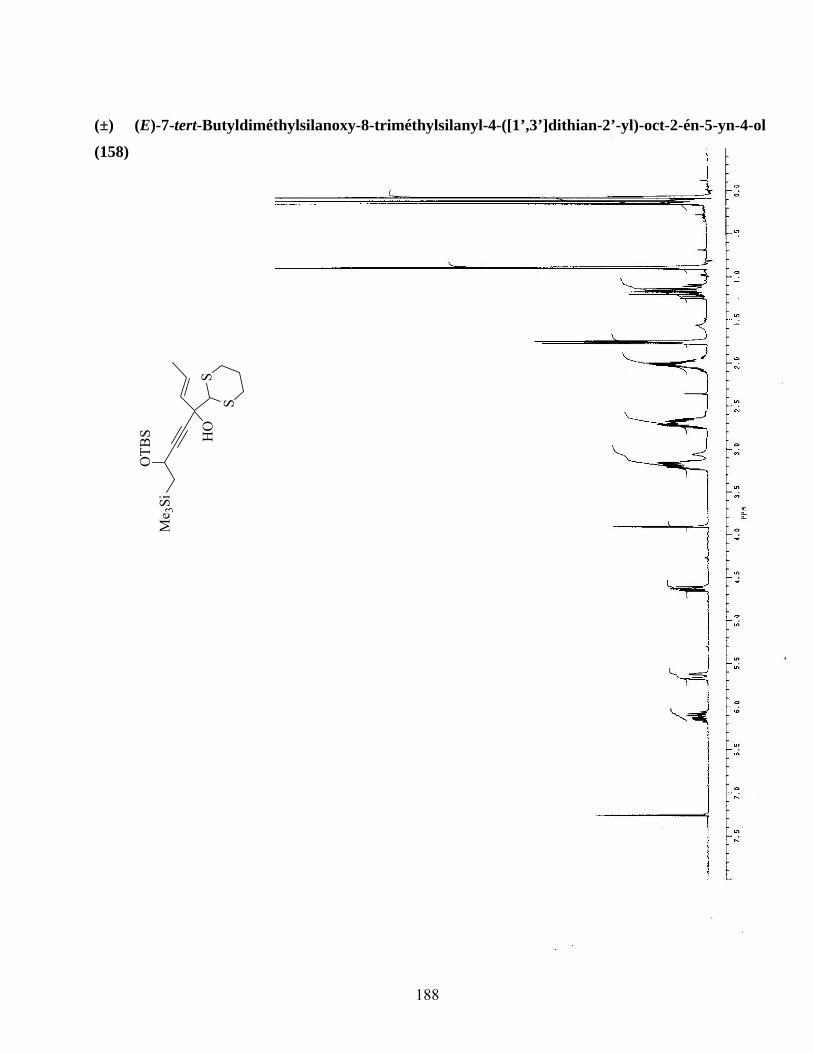
188
(±) (E)-7-tert-Butyldiméthylsilanoxy-8-triméthylsilanyl-4-([1’,3’]dithian-2’-yl)-oct-2-én-5-yn-4-ol
(158)
HO
SS
OTB
SM
e 3Si

189
(±) (E)-4-Acétoxy-7-tert-butyldiméthylsilanoxy-8-triméthylsilanyl-4-([1’,3’]dithian-2’-yl)-oct-2-én-
5-yne (159)
AcO
SS
OTB
SM
e 3Si
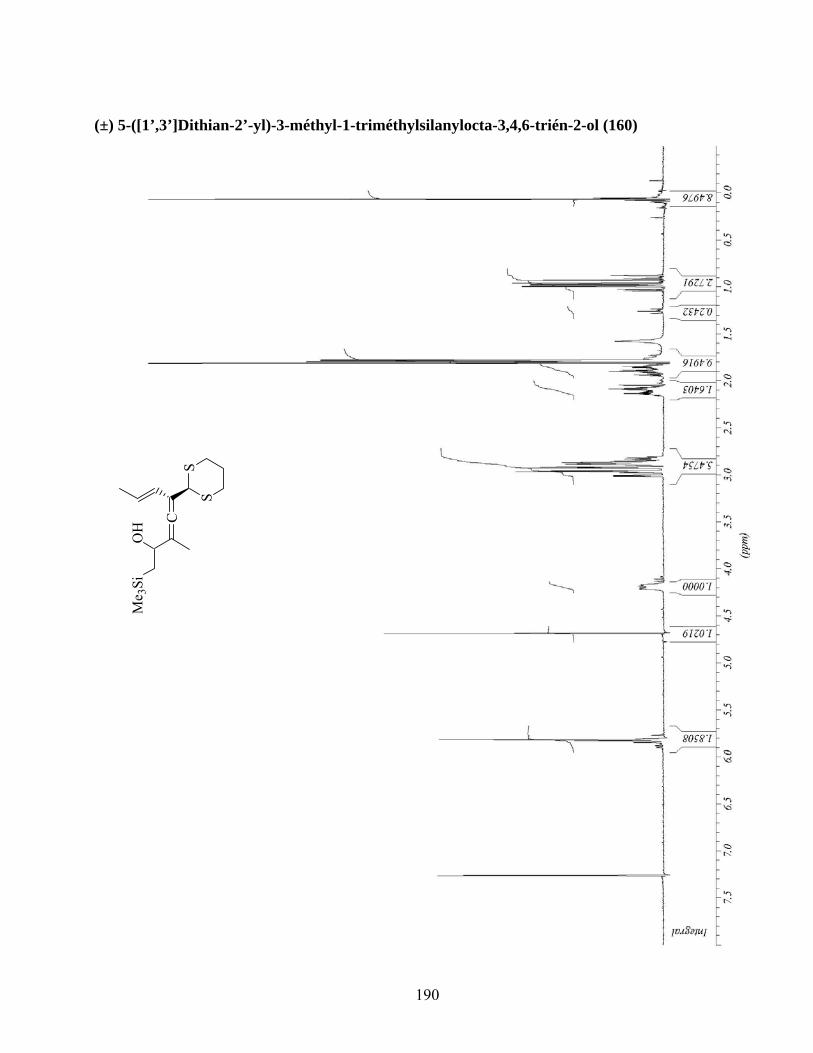
190
(±) 5-([1’,3’]Dithian-2’-yl)-3-méthyl-1-triméthylsilanylocta-3,4,6-trién-2-ol (160)
C
OH
SS
Me 3
Si
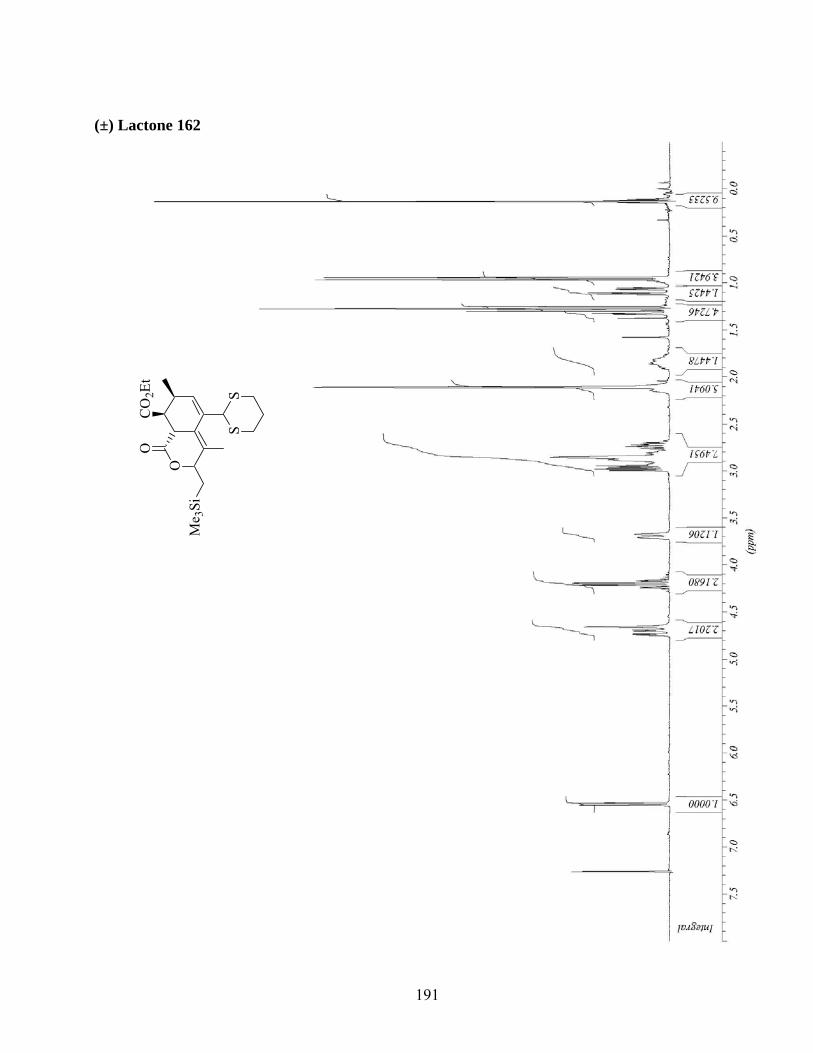
191
(±) Lactone 162
CO
2Et
SS
O
O
Me 3
Si
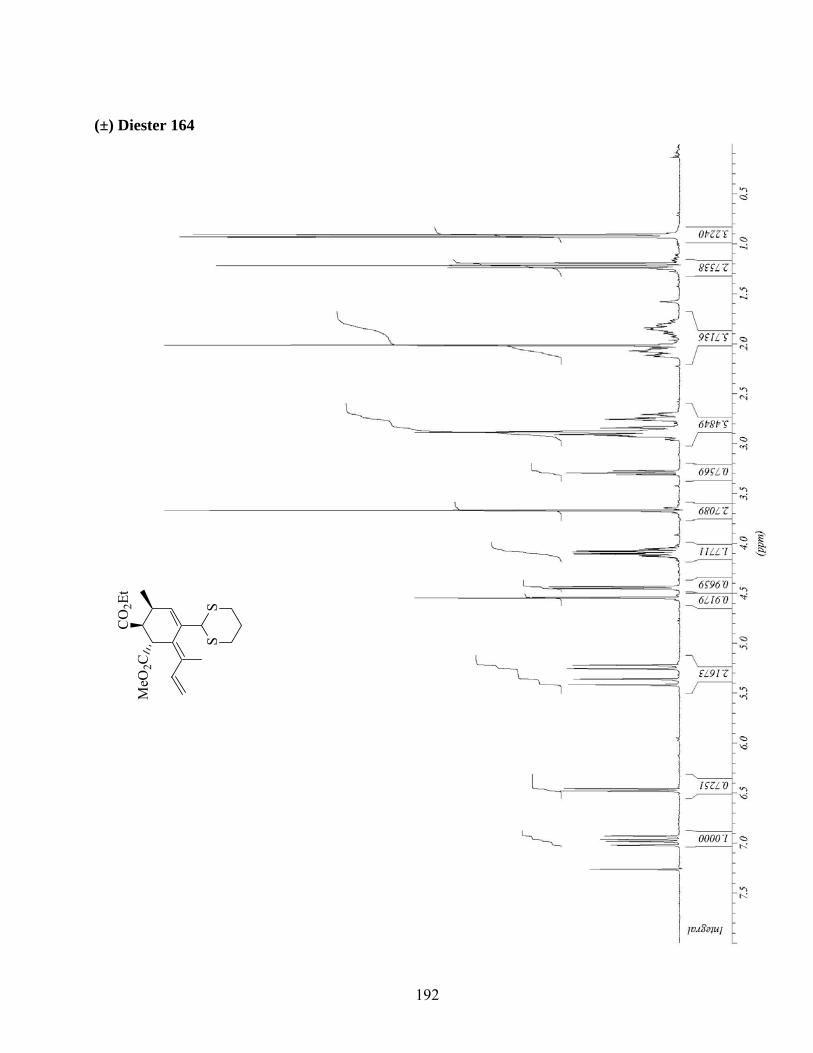
192
(±) Diester 164
CO
2Et
SS
MeO
2C
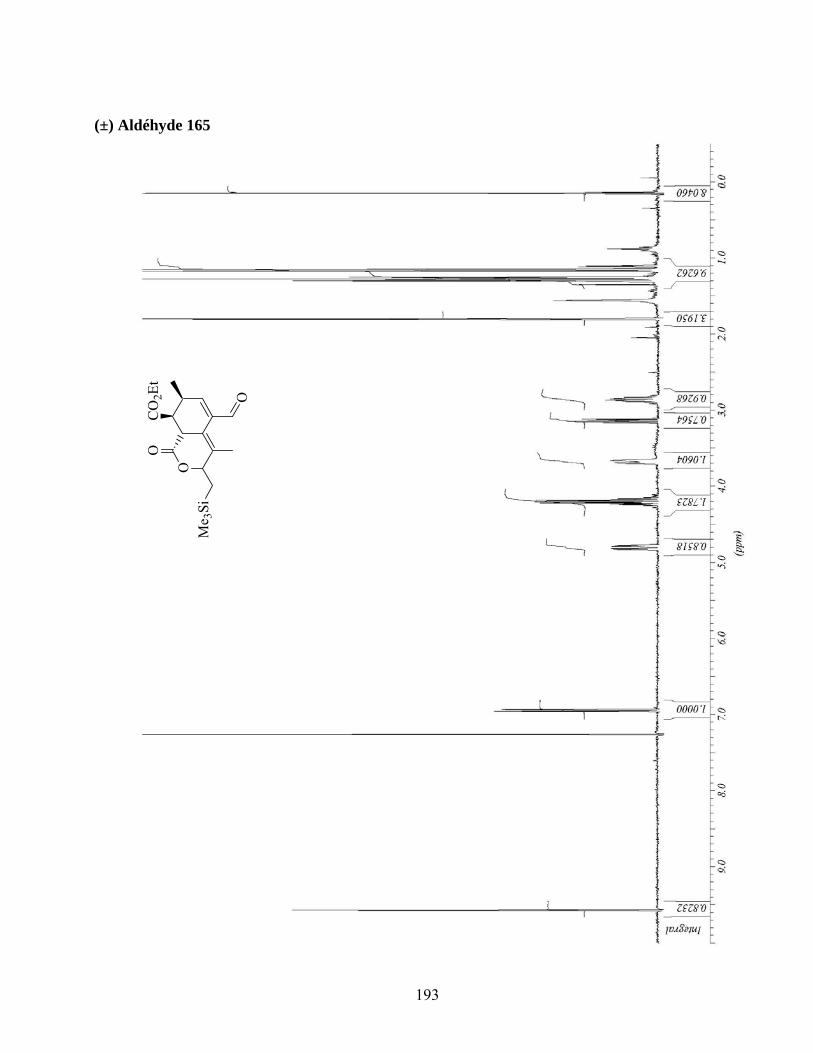
193
(±) Aldéhyde 165
CO
2Et
O
O
O
Me 3
Si
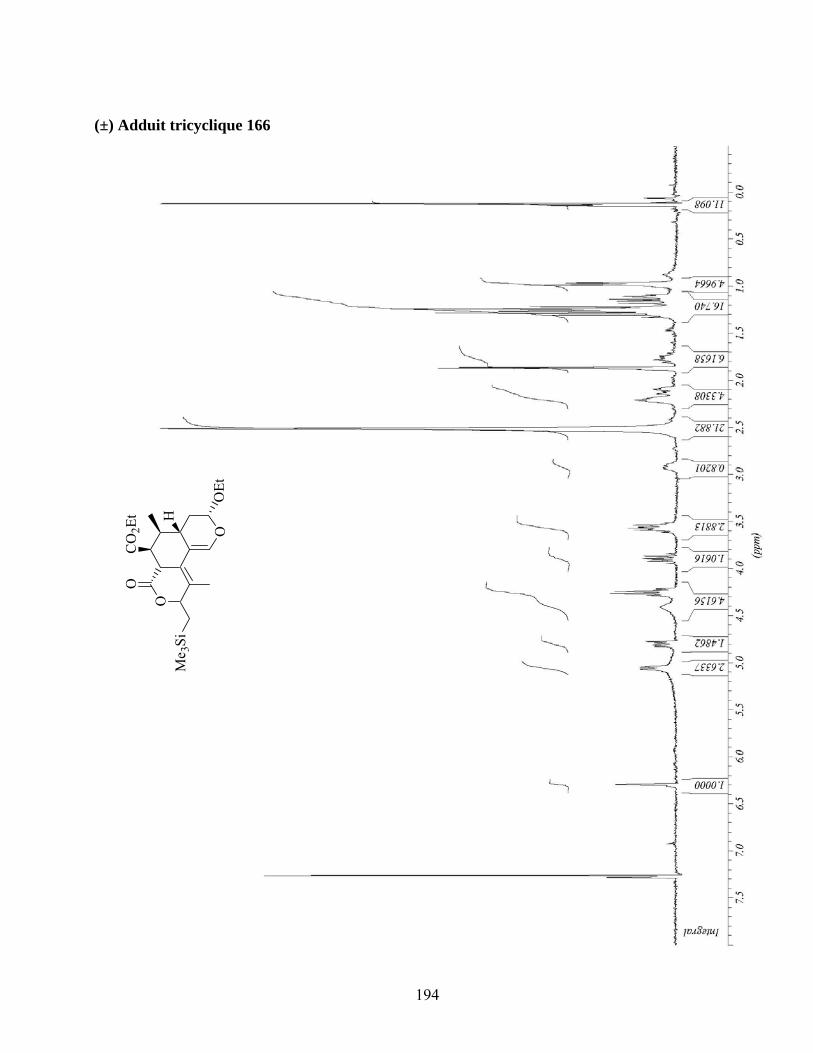
194
(±) Adduit tricyclique 166
CO
2Et
O
O
O
Me 3
SiH
OEt
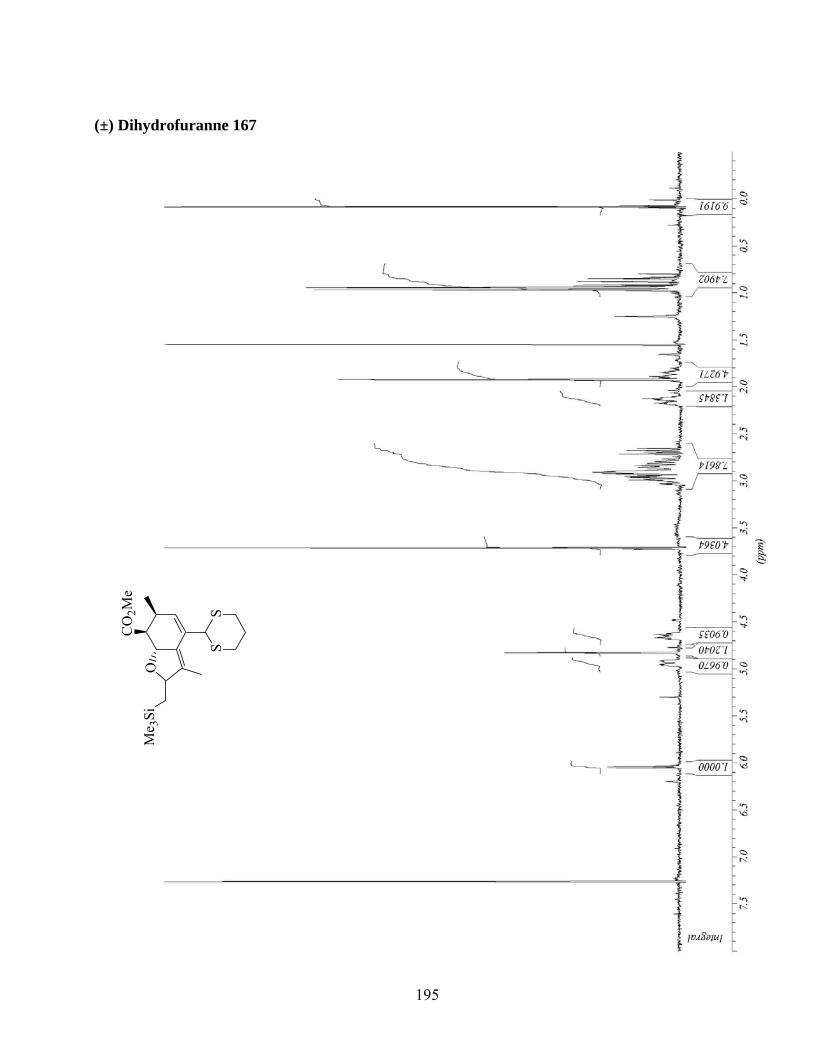
195
(±) Dihydrofuranne 167
OC
O2M
e
SS
Me 3
Si
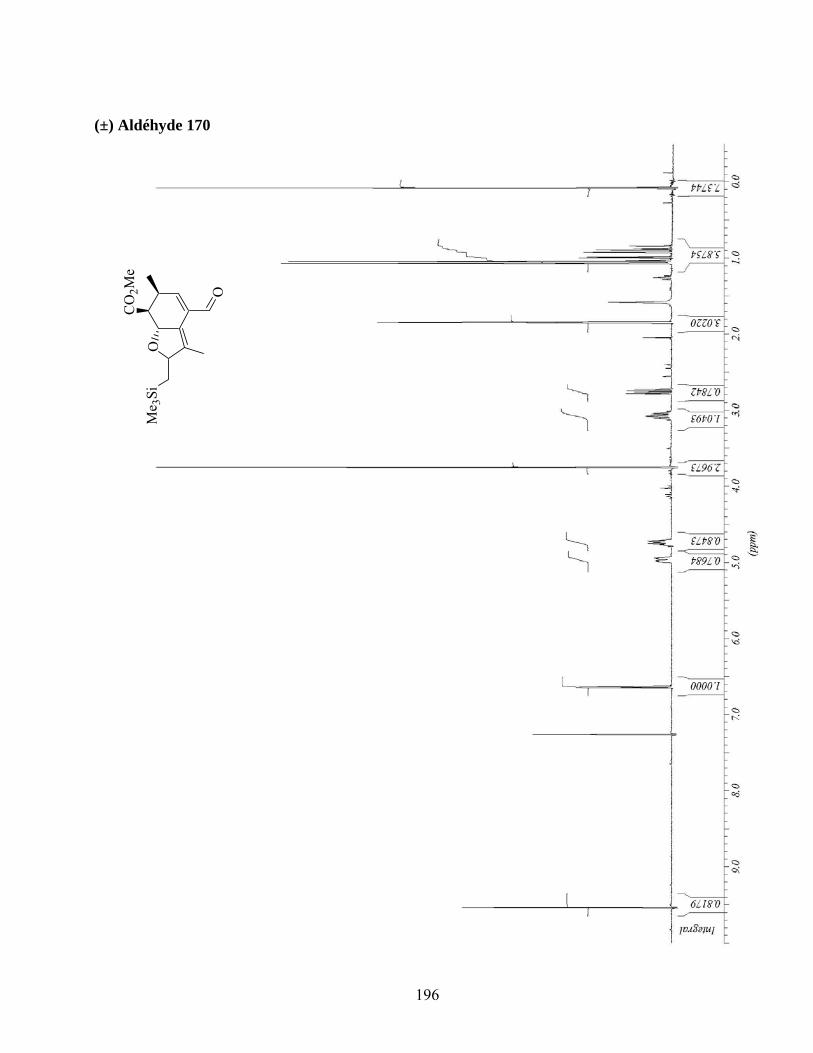
196
(±) Aldéhyde 170
O
OC
O2M
eM
e 3Si
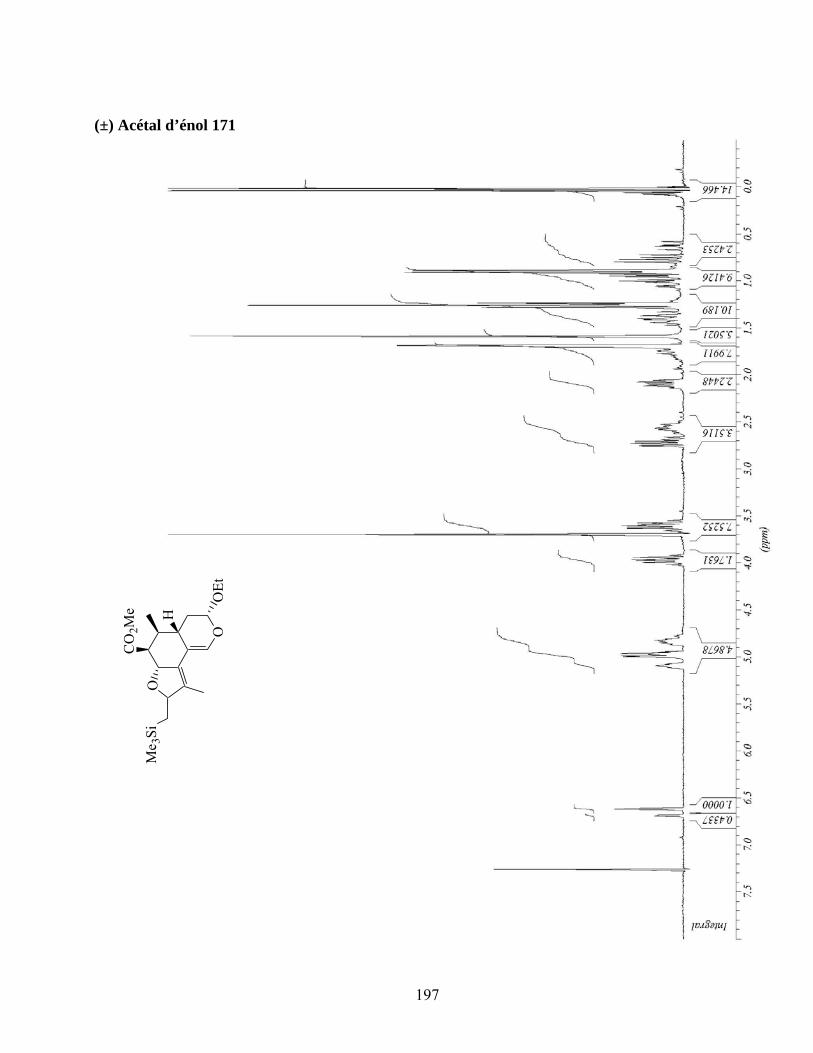
197
(±) Acétal d’énol 171
O
OC
O2M
eM
e 3Si
OE t
H
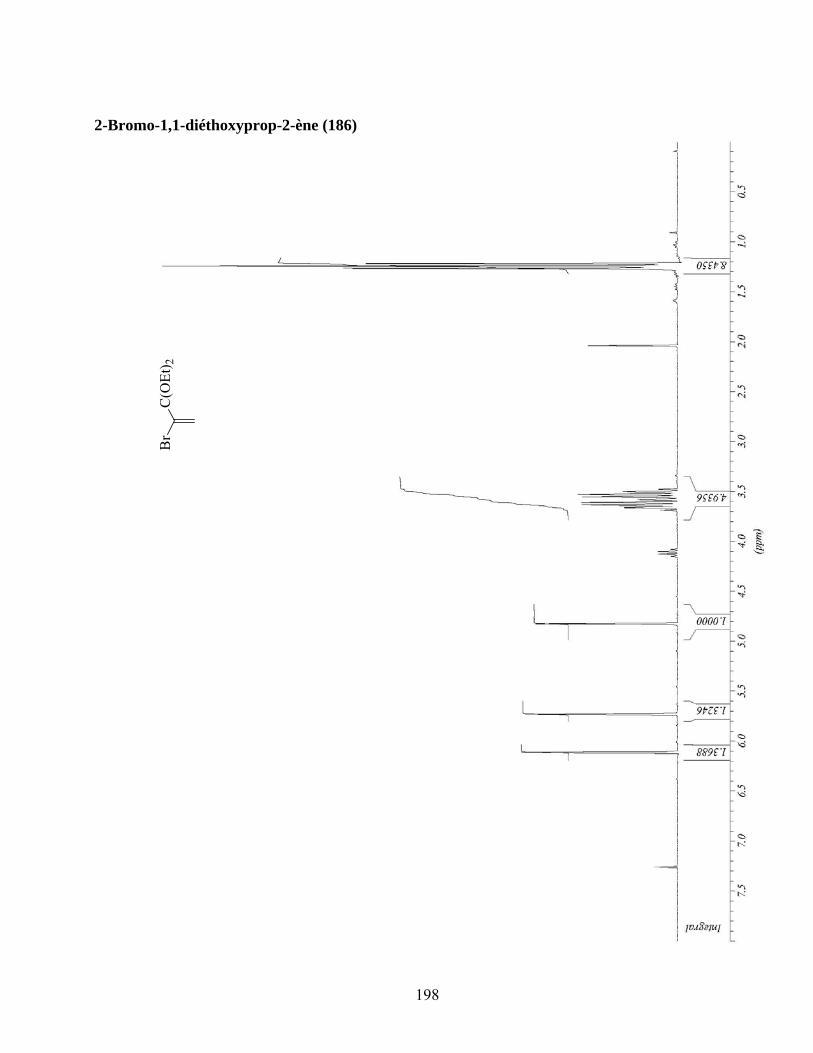
198
2-Bromo-1,1-diéthoxyprop-2-ène (186)
C(O
Et) 2
Br
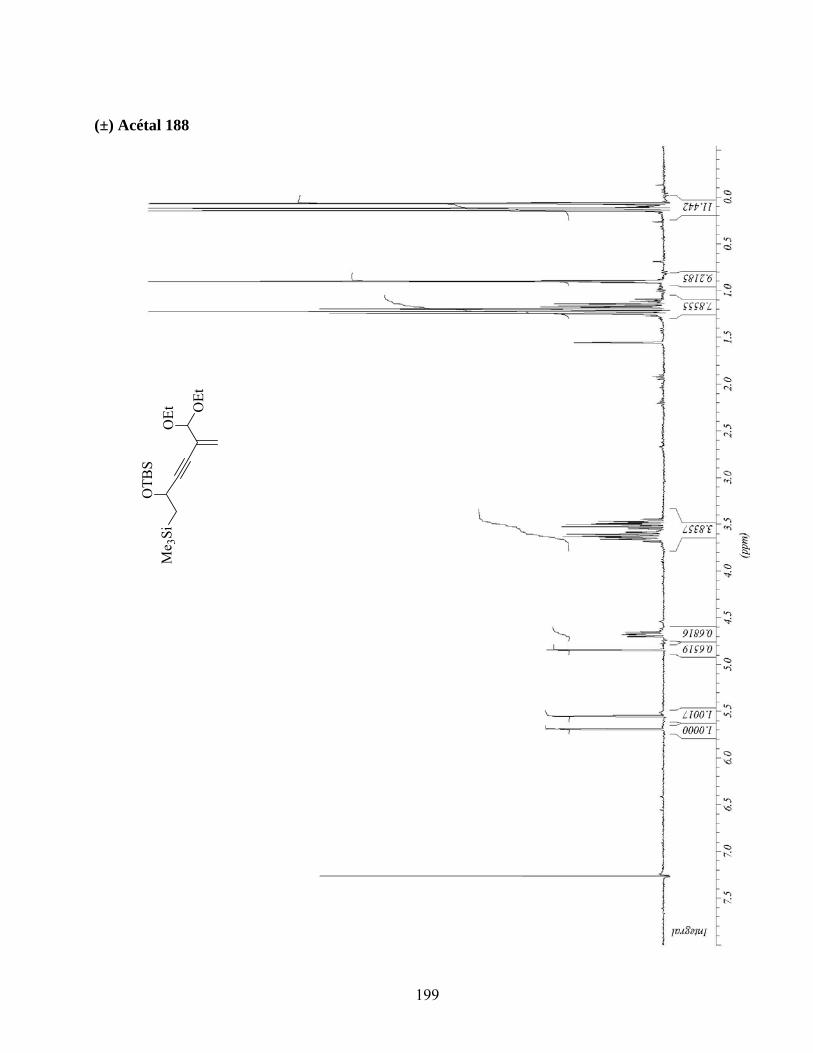
199
(±) Acétal 188
OEt
Me 3
SiO
EtO
TBS
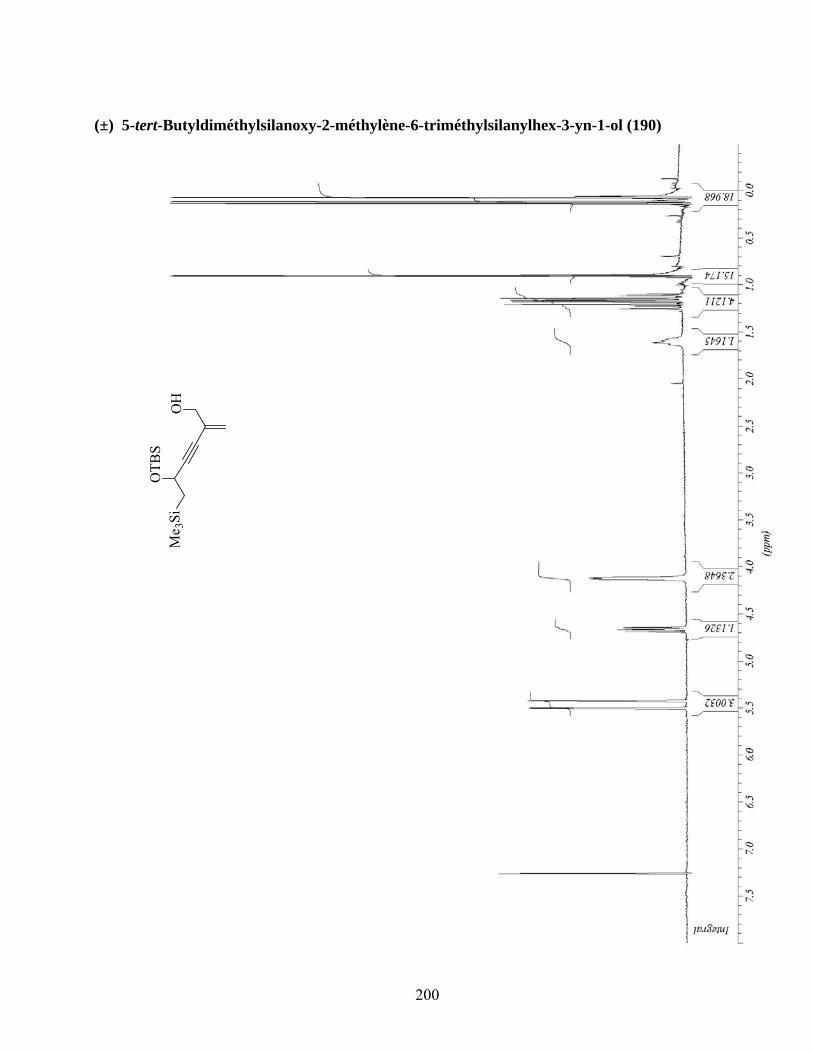
200
(±) 5-tert-Butyldiméthylsilanoxy-2-méthylène-6-triméthylsilanylhex-3-yn-1-ol (190)
Me 3
SiO
HO
TBS
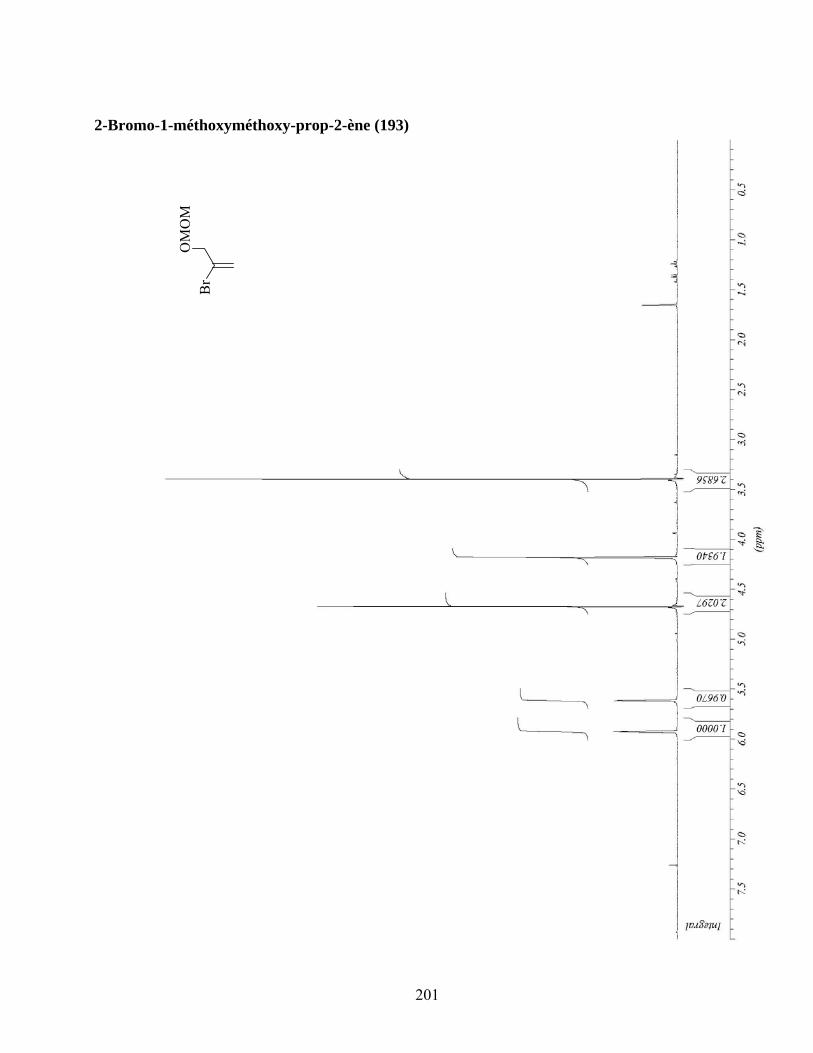
201
2-Bromo-1-méthoxyméthoxy-prop-2-ène (193)
OM
OM
Br

202
(±) 5-tert-butyldiméthylsilanoxy-2-méthoxyméthoxyméthyl-6-triméthylsilanylhex-1-én-3-yne (194)
Me 3
SiO
MO
MO
TBS
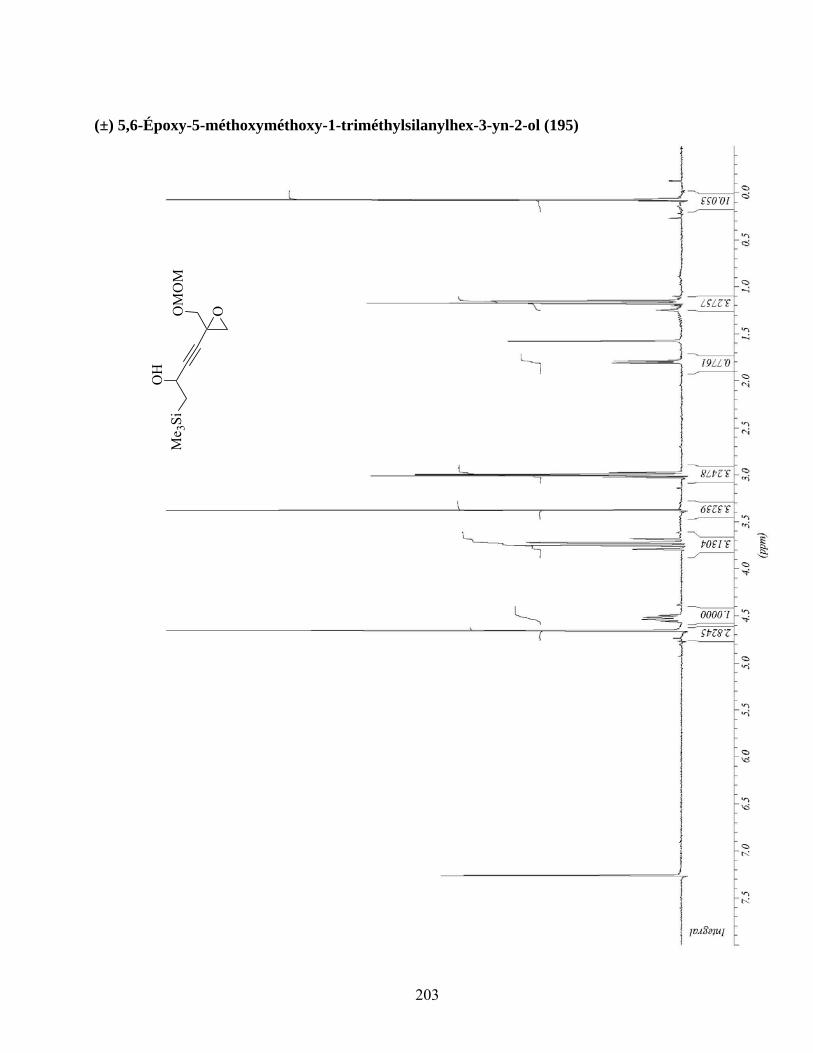
203
(±) 5,6-Époxy-5-méthoxyméthoxy-1-triméthylsilanylhex-3-yn-2-ol (195)
Me 3
SiO
MO
MO
H
O
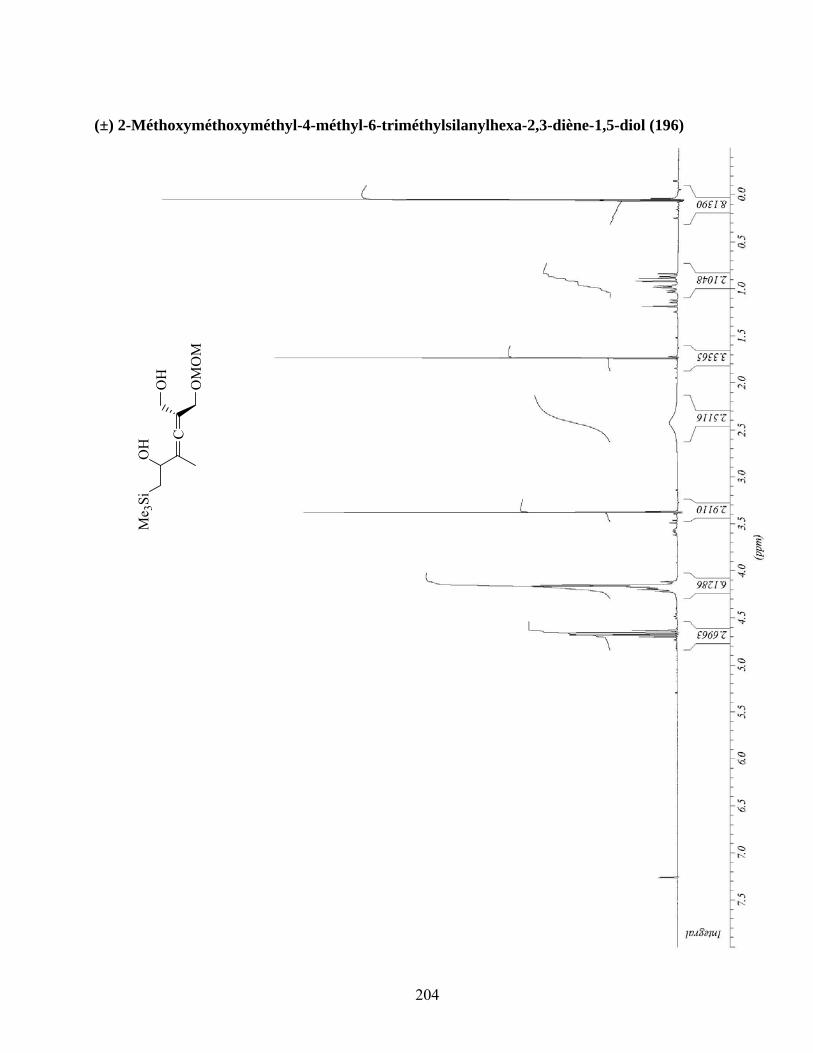
204
(±) 2-Méthoxyméthoxyméthyl-4-méthyl-6-triméthylsilanylhexa-2,3-diène-1,5-diol (196)
C
OH
OM
OM
Me 3
SiO
H
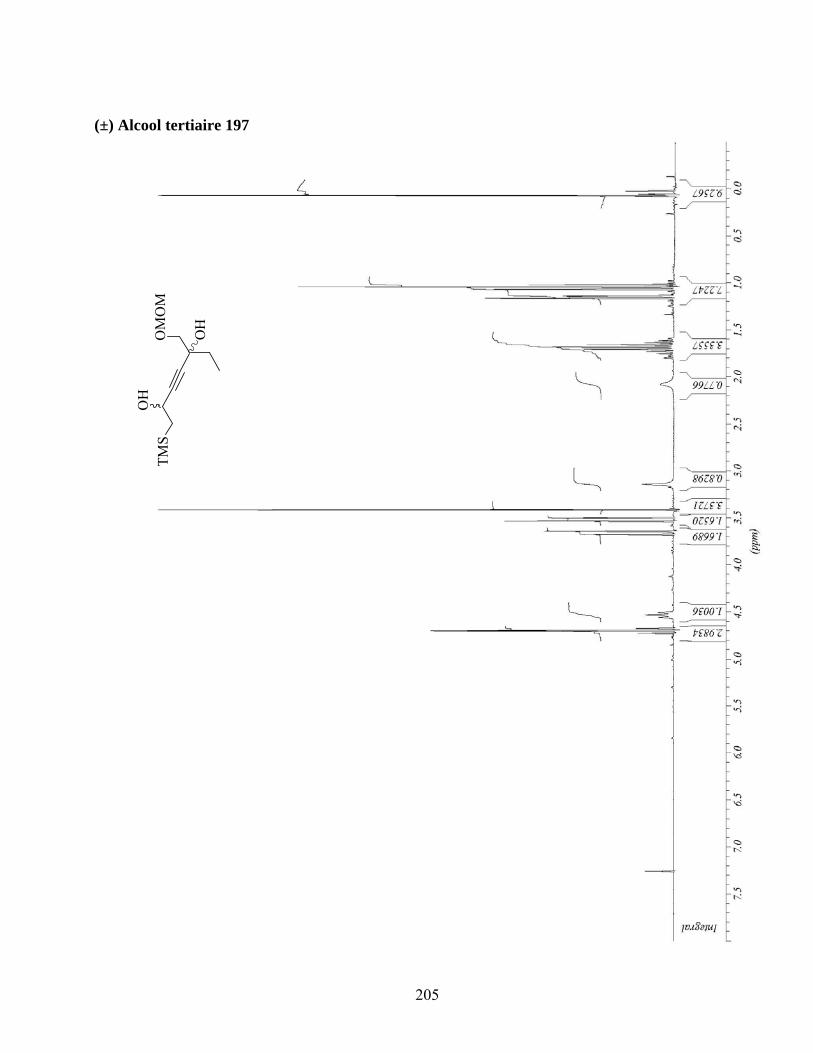
205
(±) Alcool tertiaire 197
OH
TMS
OM
OM
OH
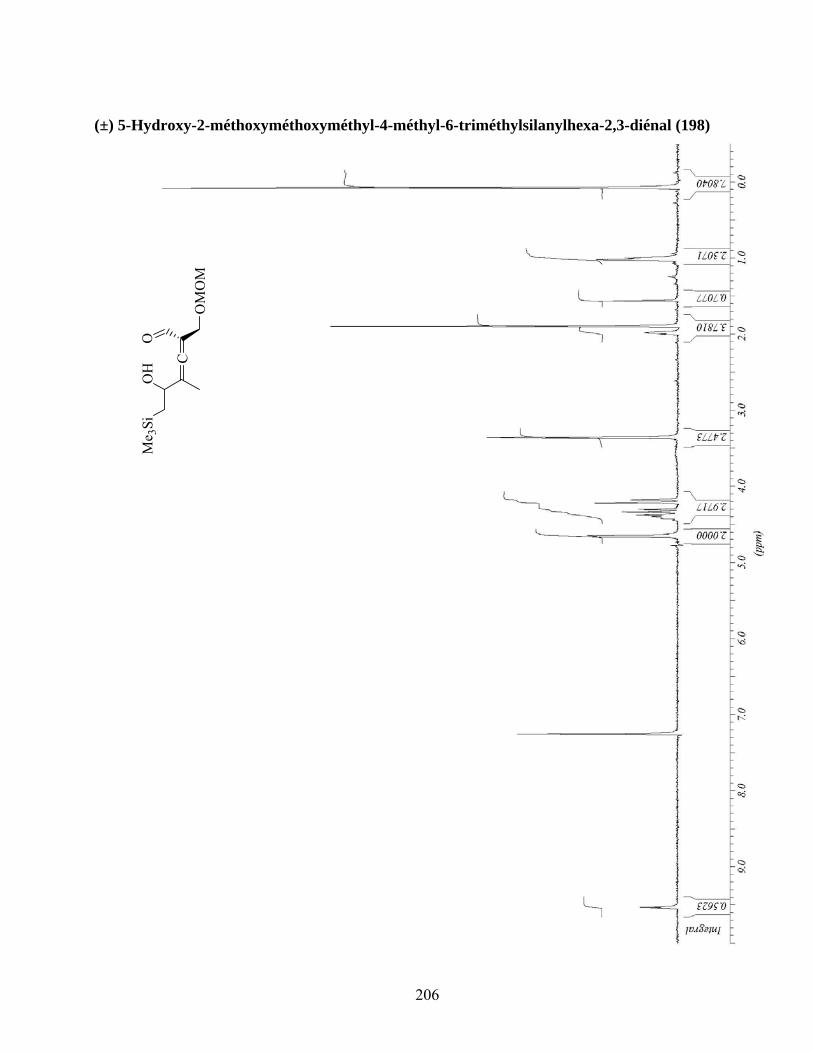
206
(±) 5-Hydroxy-2-méthoxyméthoxyméthyl-4-méthyl-6-triméthylsilanylhexa-2,3-diénal (198)
C
OH
OM
OM
Me 3
SiO
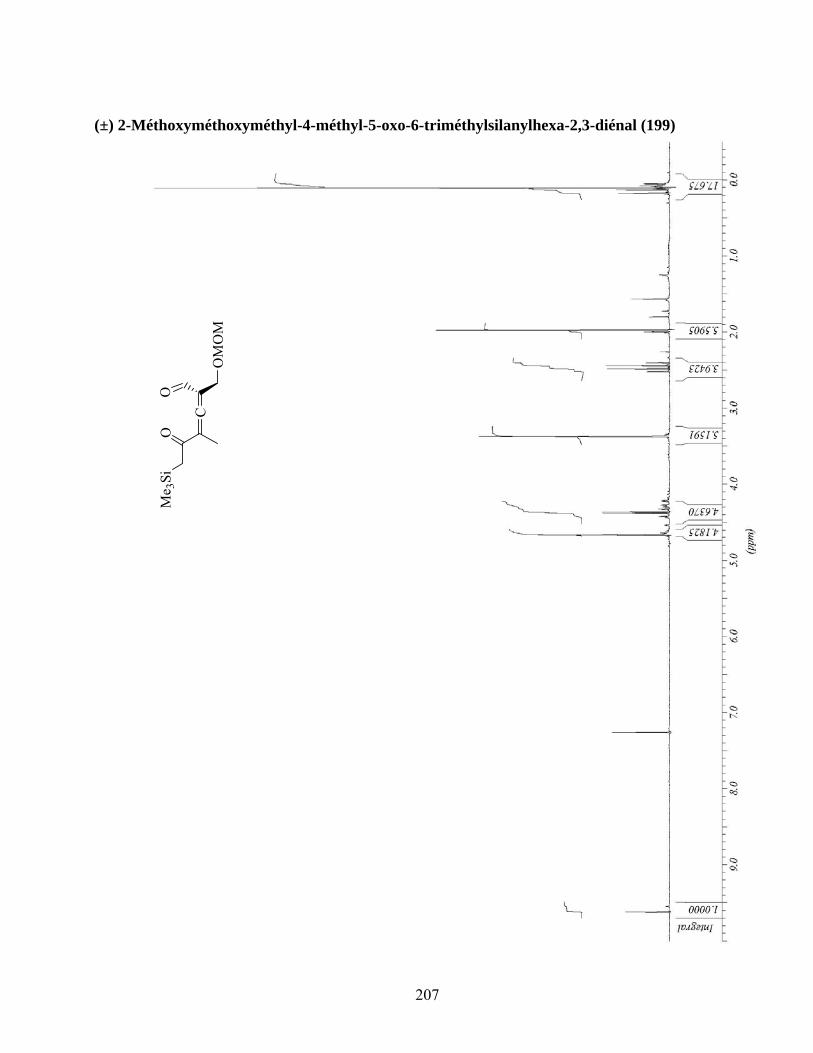
207
(±) 2-Méthoxyméthoxyméthyl-4-méthyl-5-oxo-6-triméthylsilanylhexa-2,3-diénal (199)
C
O
OM
OM
Me 3
SiO
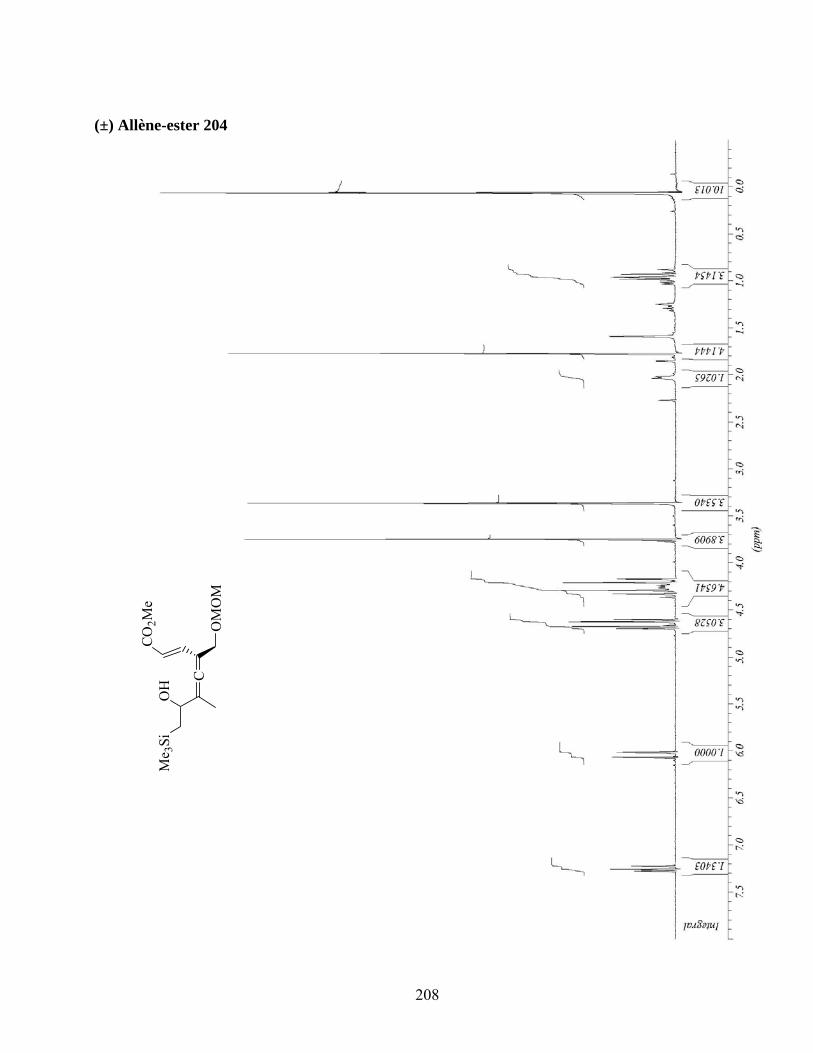
208
(±) Allène-ester 204
C
OH
OM
OM
Me 3
SiCO
2Me
209
(±) Ester fumarique 205
Me 3
SiO
MO
MO
O
O
CO2E
t
210
(±) 5-tert-Butyldiméthylsilanoxyméthyl-6-méthoxyméthoxyméthyl-3-méthyl-1-
triméthylsilanylhexa-3,4-dién-2-ol (207)
C
OH
OM
OM
Me 3
SiO
TBS
211
(±) Ester silylé 208
C
O
OM
OM
Me 3
SiO
TBS
O
CO
2Et
212
(±) 2-Méthoxyméthoxyméthyl-4-méthylhexa-2,3,5-trién-1-ol (209)
CO
MO
M
HO
213
(±) 5-tert-Butyldiméthylsilanoxy-1-chloro-2-méthylpent-3-yn-2-ol (228)
HO
TBSO
Cl
214
(±) 1-Chloro-5-méthoxyméthoxy-2-méthylpent-3-yn-2-ol (229)
HO
MO
MO
Cl
215
(±) 5-tert-Butyldiméthylsilanoxy-1,2-époxy-2-méthylpent-3-yne (230)
TBSO
O
216
(±) 1,2-Époxy-5-méthoxyméthoxy-2-méthylpent-3-yne (231)
MO
MO
O
217
(±) (5Z)-4-tert-Butyldiméthylsiloxyméthyl-2-méthylhepta-2,3,5-trién-1-ol (232)
C
OH
OTB
S
218
(±) (5Z)-4-Méthoxyméthoxyméthyl-2-méthylhepta-2,3,5-trién-1-ol (233)
C
OH
OM
OM
219
(Z)-6-tert-Butyldiphénylsiloxy-3-méthylhex-2-én-4-yn-1-ol (235a)
OTB
DPS
OH

220
(Z)-6-(4’-Méthoxy)benzyloxy-3-méthylhex-2-én-4-yn-1-ol (235b)
OPM
BO
H
221
(Z)-6-Méthoxyméthoxy-3-méthylhex-2-én-4-yn-1-ol (235c)
OM
OM
OH
222
(Z)-3-Méthyl-6-(2’-triméthylsilyl)éthoxyméthoxyhex-2-én-4-yn-1-ol (235d)
OSE
MO
H

223
(Z)-6-Benzyloxy-3-méthylhex-2-én-4-yn-1-ol (235e)
OB
nO
H

224
(Z)-6-Acétoxy-3-méthylhex-2-én-4-yn-1-ol (235f)
OA
cO
H
225
(Z)-5-([1’,3’]Dioxolan-2’-yl)-3-méthylpent-2-én-4-yn-1-ol (235h)
OH
O
O
226
(Z)-1-Bromo-6-tert-butyldiphénylsiloxy-3-méthylhex-2-én-4-yne (236a)
OTB
DPS
Br

227
(Z)-1-Bromo-6-(4’-méthoxy)benzyloxy-3-méthylhex-2-én-4-yne (236b)
OPM
BB
r
228
(Z)-1-Bromo-6-méthoxyméthoxy-3-méthylhex-2-én-4-yne (236c)
OM
OM
Br

229
(Z)-1-Bromo-3-méthyl-6-(2’-triméthylsilyl)éthoxyméthoxyhex-2-én-4-yne (236d)
OSE
MB
r

230
(Z)-6-Benzyloxy-1-bromo-3-méthylhex-2-én-4-yne (236e)
OB
nB
r
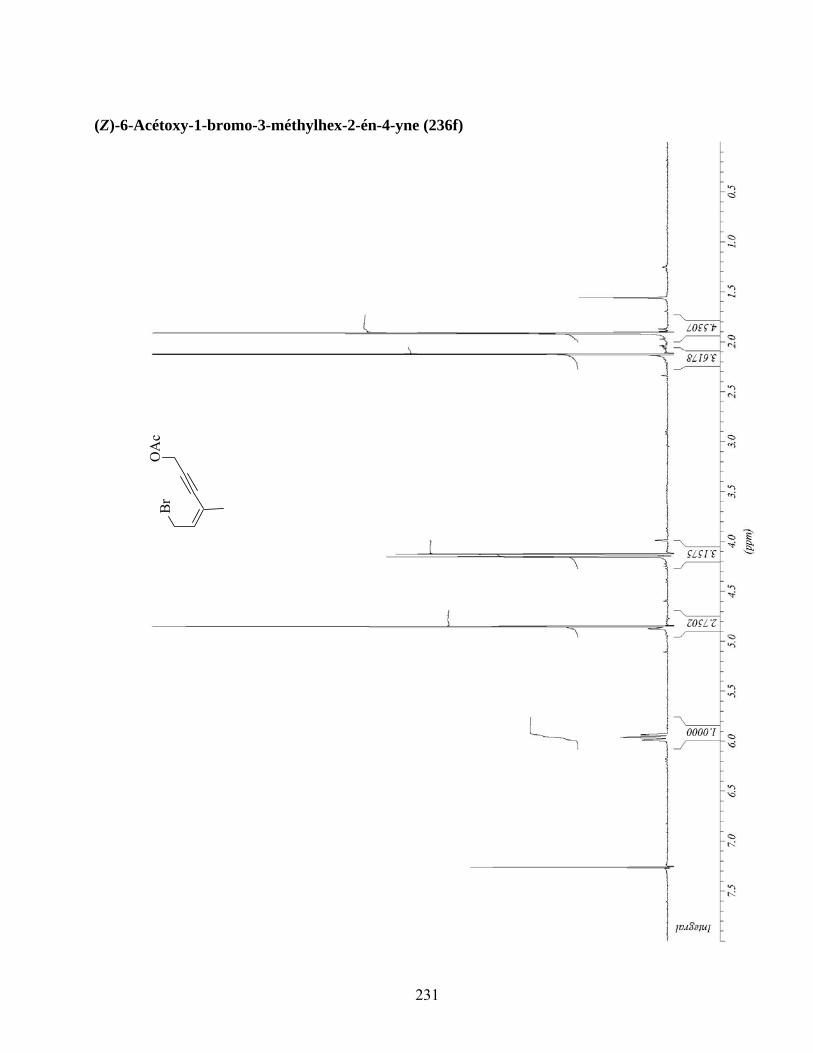
231
(Z)-6-Acétoxy-1-bromo-3-méthylhex-2-én-4-yne (236f)
OA
cBr
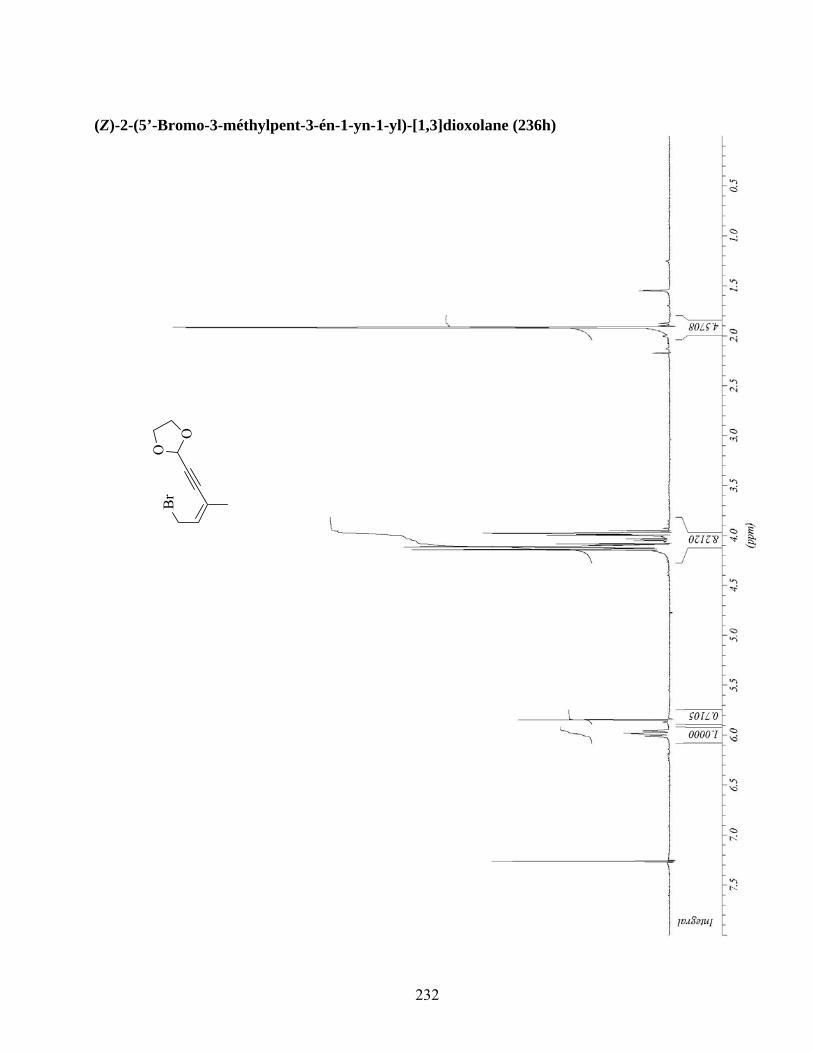
232
(Z)-2-(5’-Bromo-3-méthylpent-3-én-1-yn-1-yl)-[1,3]dioxolane (236h)
Br
O
O
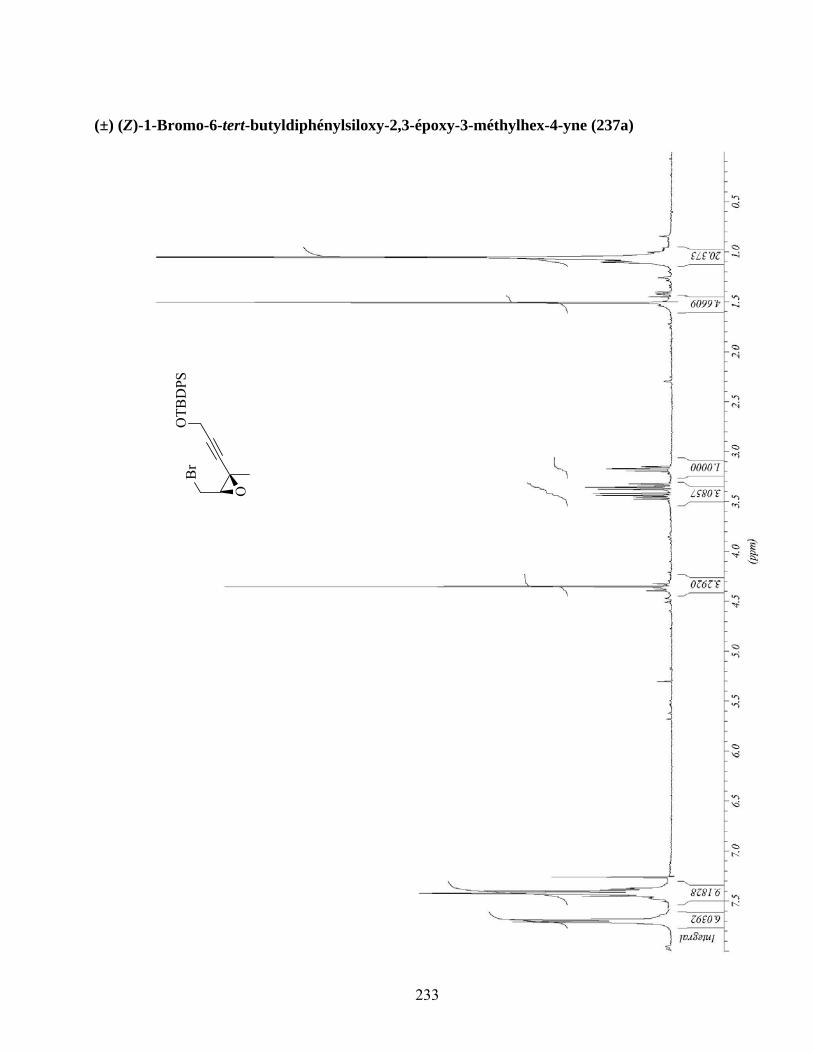
233
(±) (Z)-1-Bromo-6-tert-butyldiphénylsiloxy-2,3-époxy-3-méthylhex-4-yne (237a)
OTB
DPS
Br
O

234
(±) (Z)-1-Bromo-2,3-époxy-6-(4’-méthoxy)benzyloxy-3-méthylhex-4-yne (237b)
OPM
BB
r
O
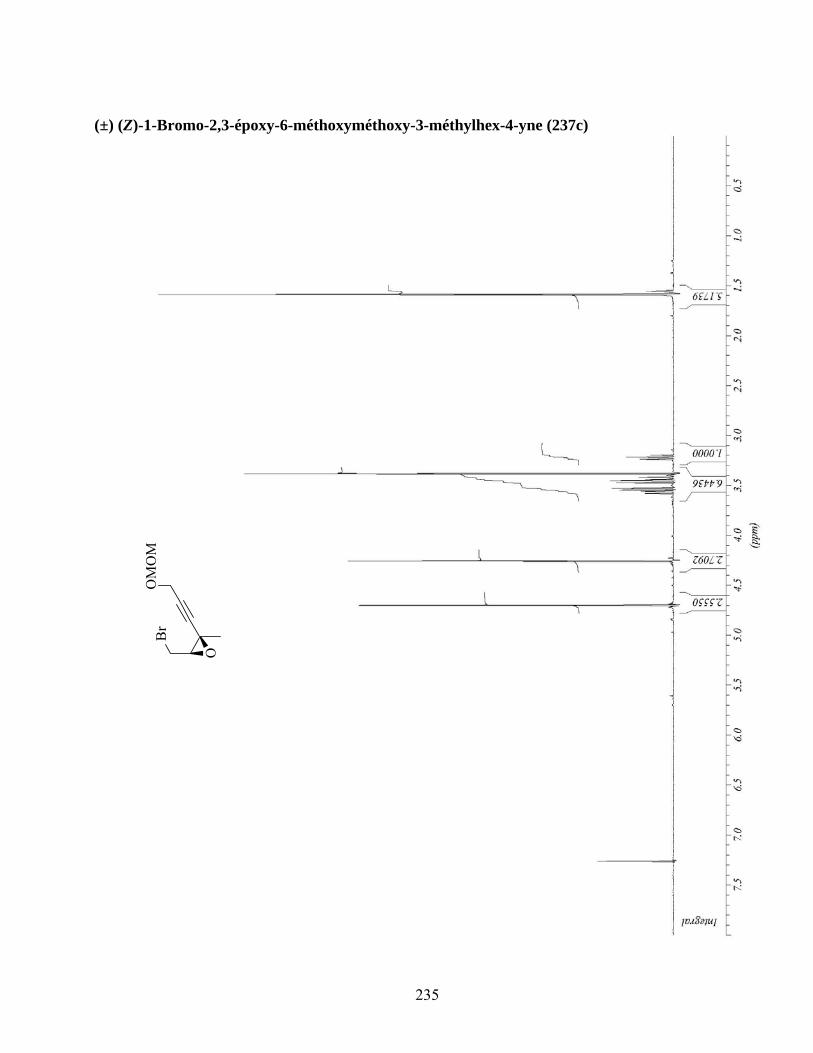
235
(±) (Z)-1-Bromo-2,3-époxy-6-méthoxyméthoxy-3-méthylhex-4-yne (237c)
OM
OM
Br
O

236
(±) (Z)-1-Bromo-2,3-époxy-3-méthyl-6-(2’-triméthylsilyl)éthoxyméthoxyhex-4-yne (237d)
OSE
MB
r
O
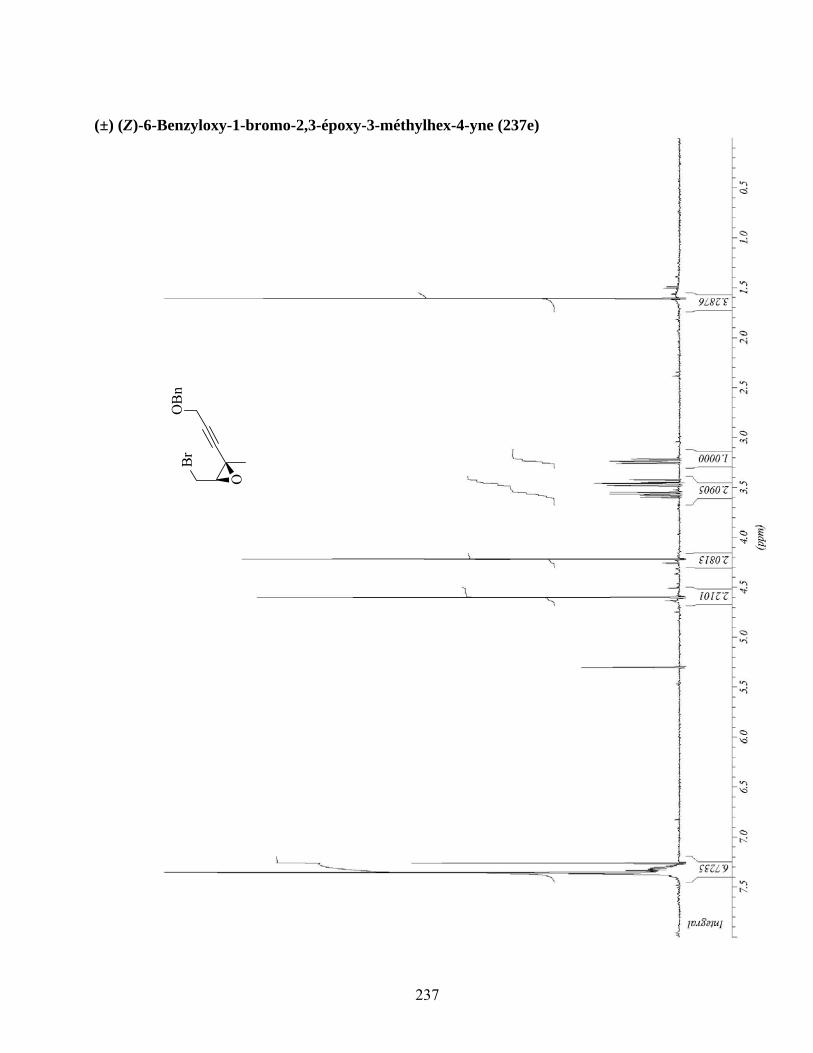
237
(±) (Z)-6-Benzyloxy-1-bromo-2,3-époxy-3-méthylhex-4-yne (237e)
OB
nBr
O
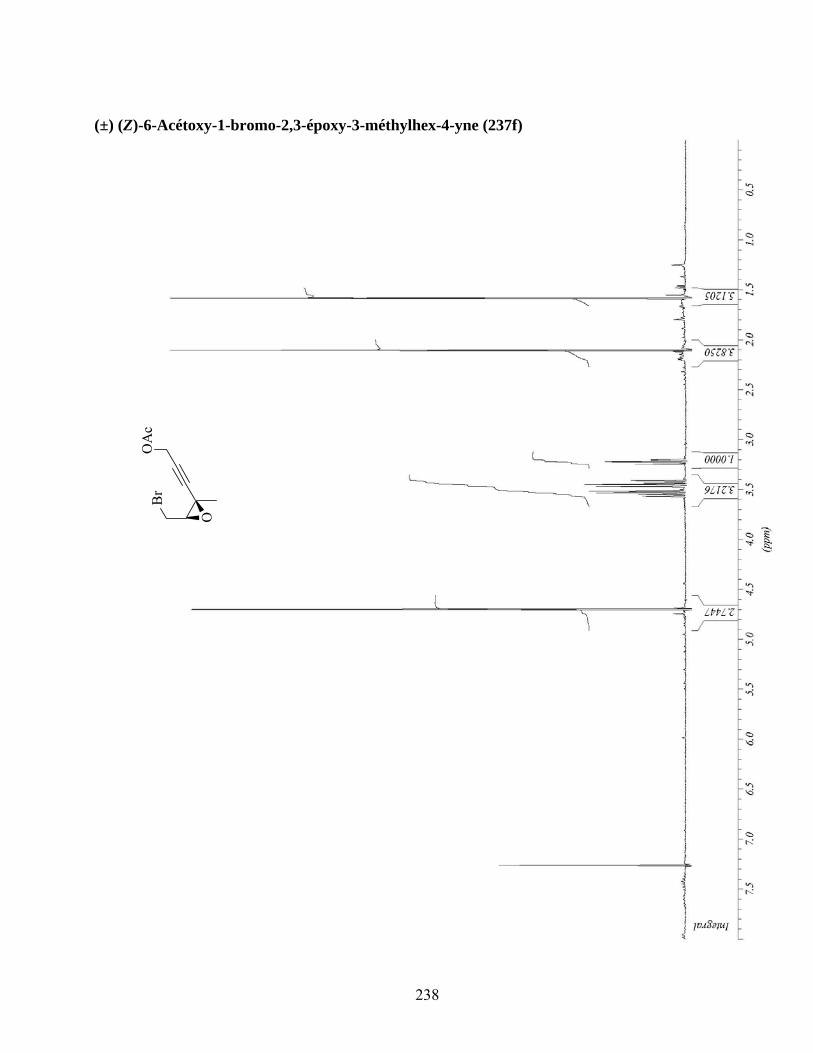
238
(±) (Z)-6-Acétoxy-1-bromo-2,3-époxy-3-méthylhex-4-yne (237f)
OA
cB
r
O
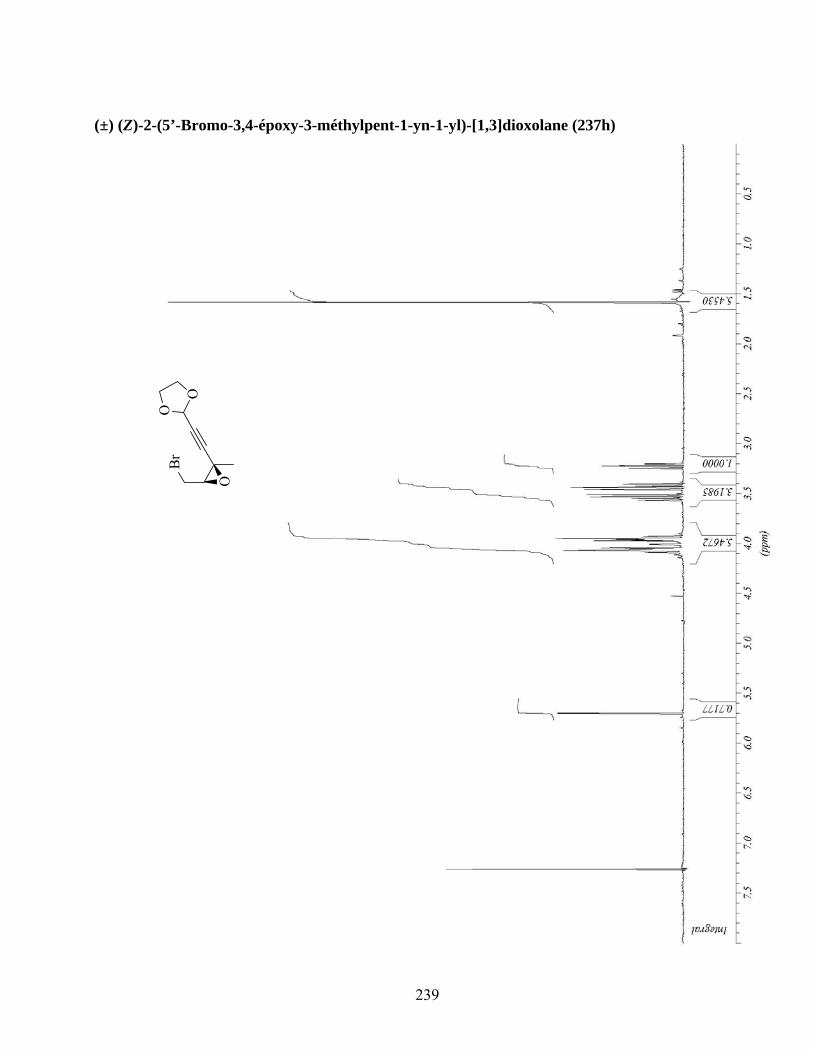
239
(±) (Z)-2-(5’-Bromo-3,4-époxy-3-méthylpent-1-yn-1-yl)-[1,3]dioxolane (237h)
Br
O
O
O
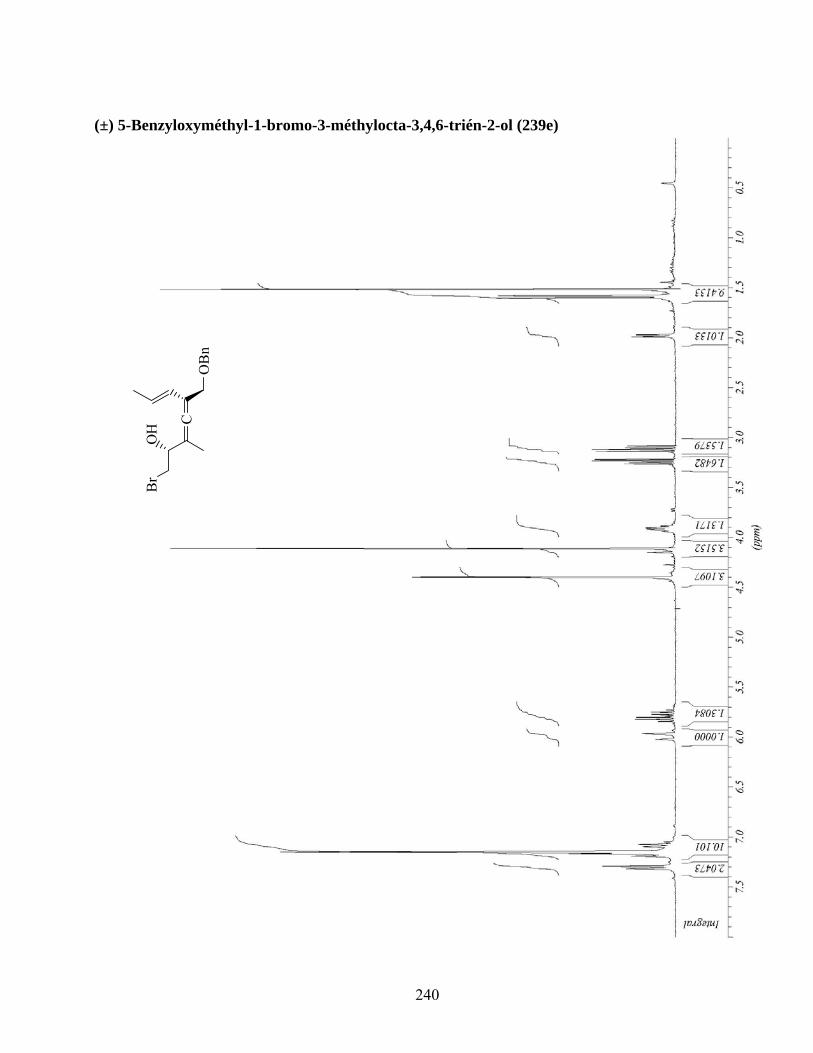
240
(±) 5-Benzyloxyméthyl-1-bromo-3-méthylocta-3,4,6-trién-2-ol (239e)
C
OH
OB
n
Br
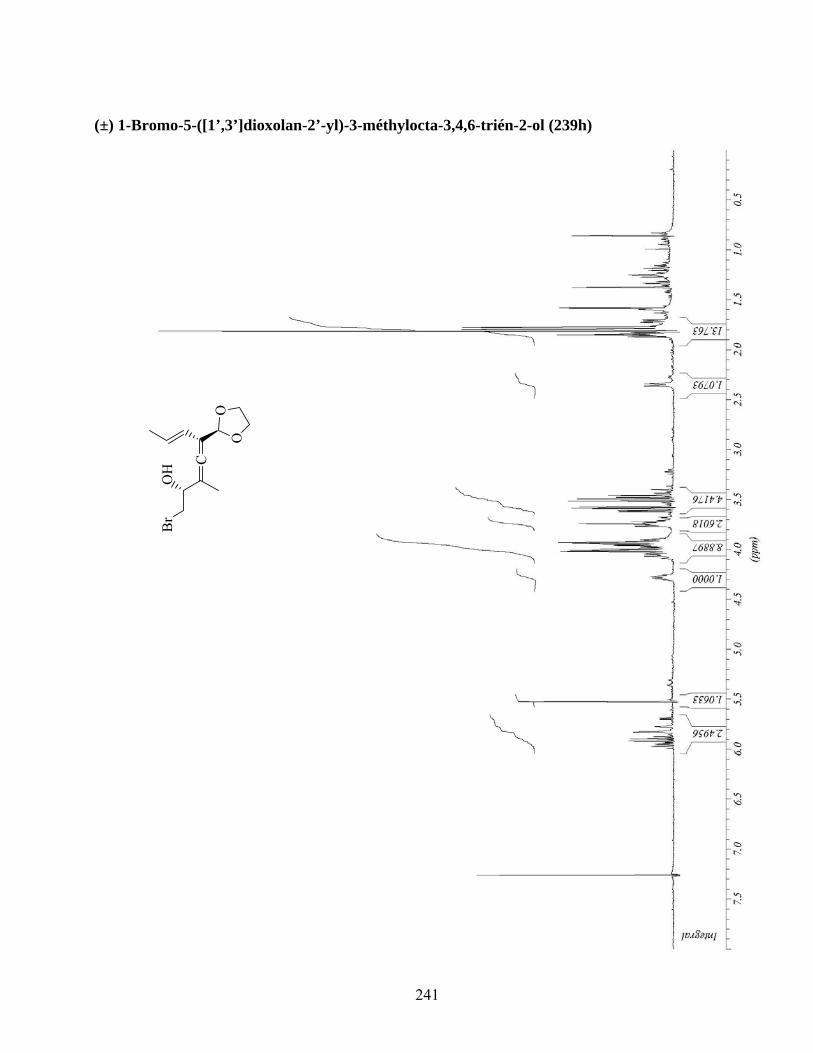
241
(±) 1-Bromo-5-([1’,3’]dioxolan-2’-yl)-3-méthylocta-3,4,6-trién-2-ol (239h)
C
OH
Br
OO
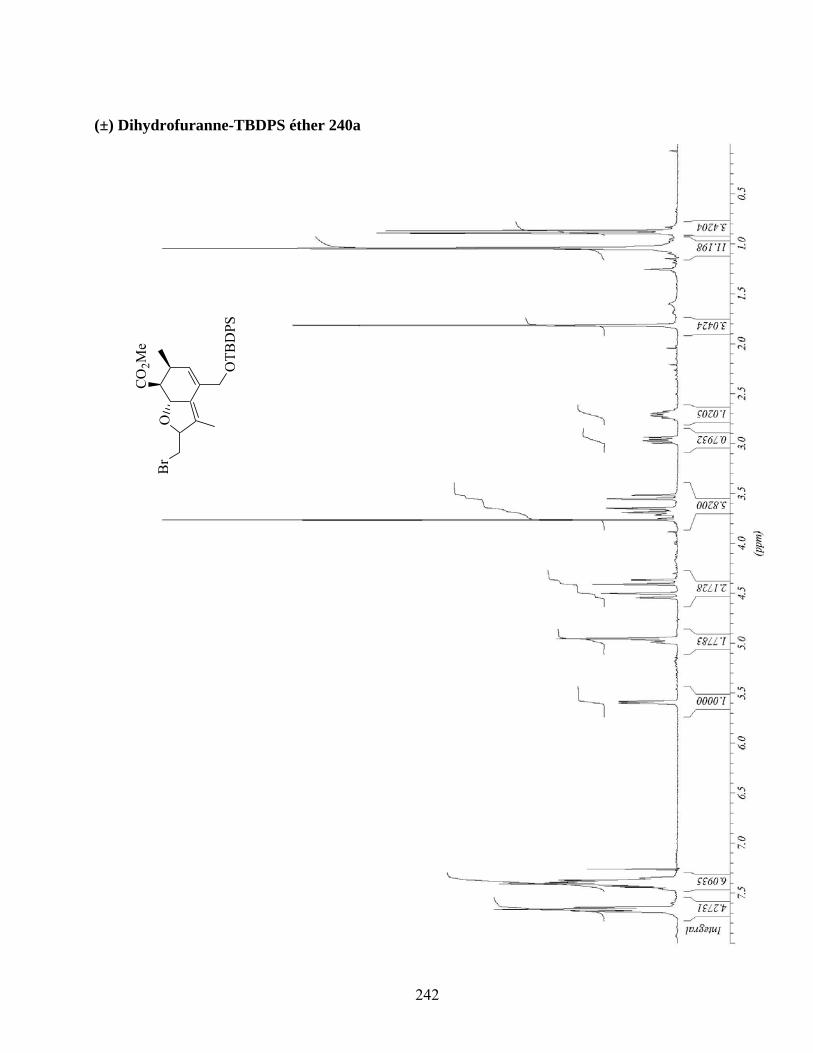
242
(±) Dihydrofuranne-TBDPS éther 240a
OCO
2Me
OTB
DPS
Br
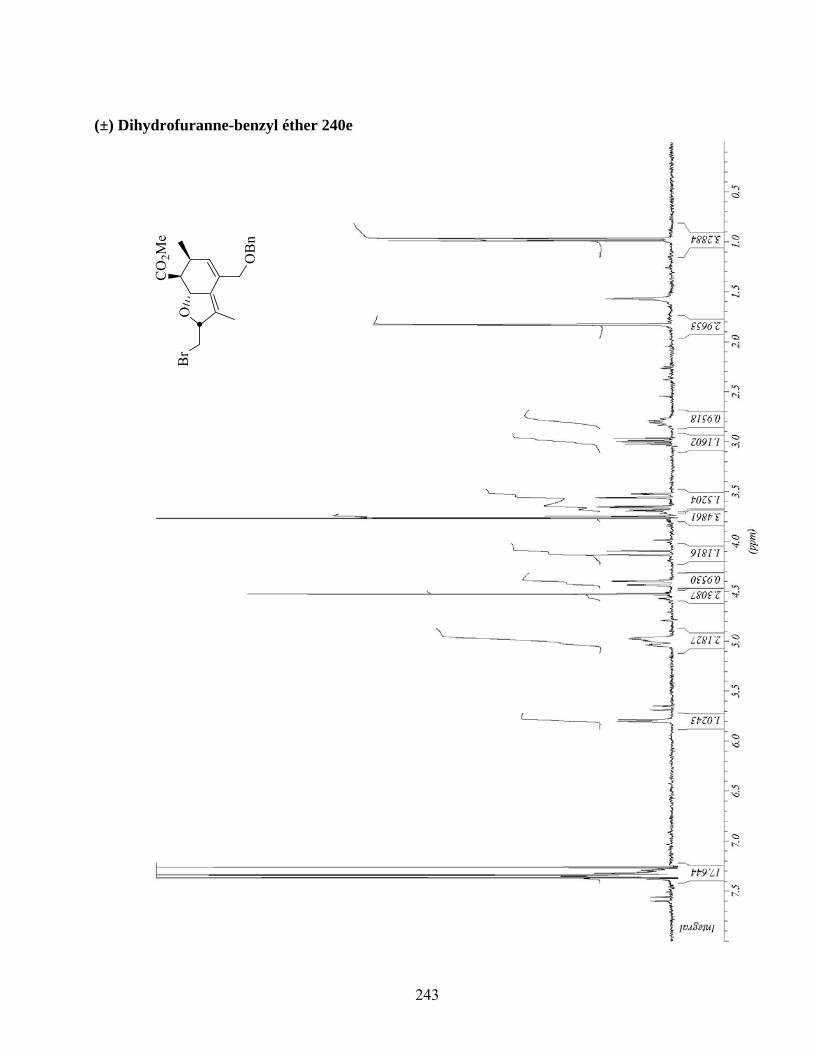
243
(±) Dihydrofuranne-benzyl éther 240e
OC
O2M
e
OB
n
Br
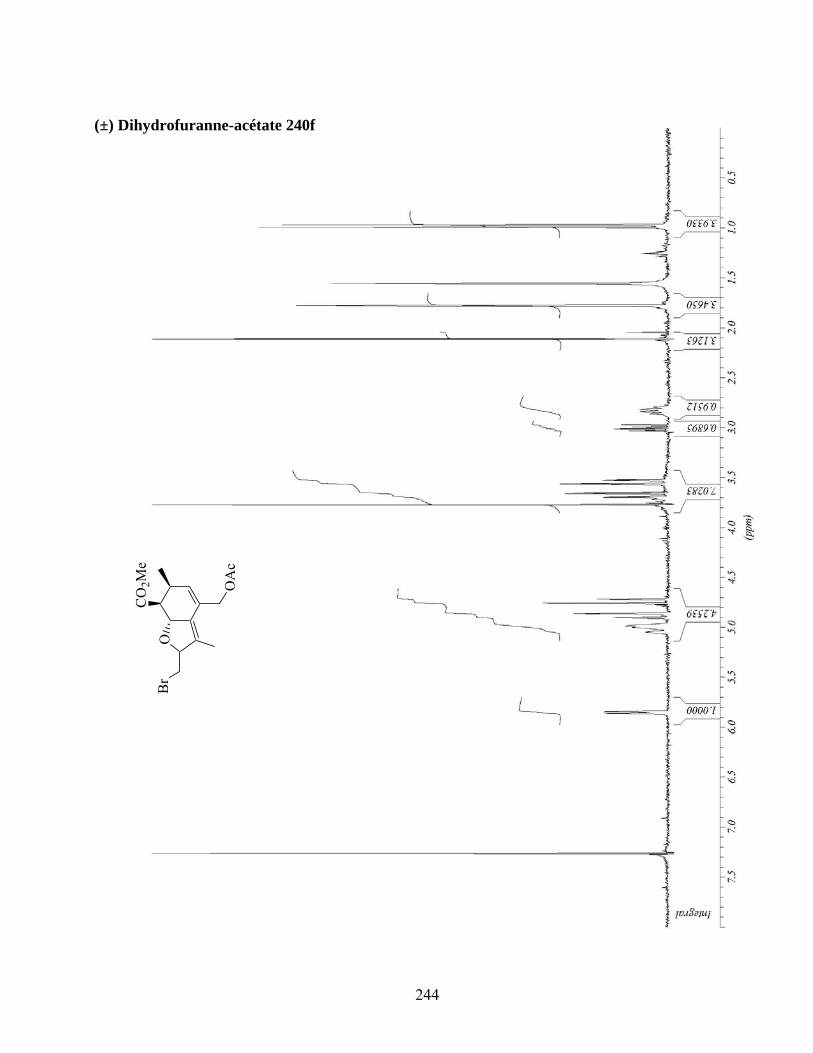
244
(±) Dihydrofuranne-acétate 240f
OC
O2M
e
OA
c
Br
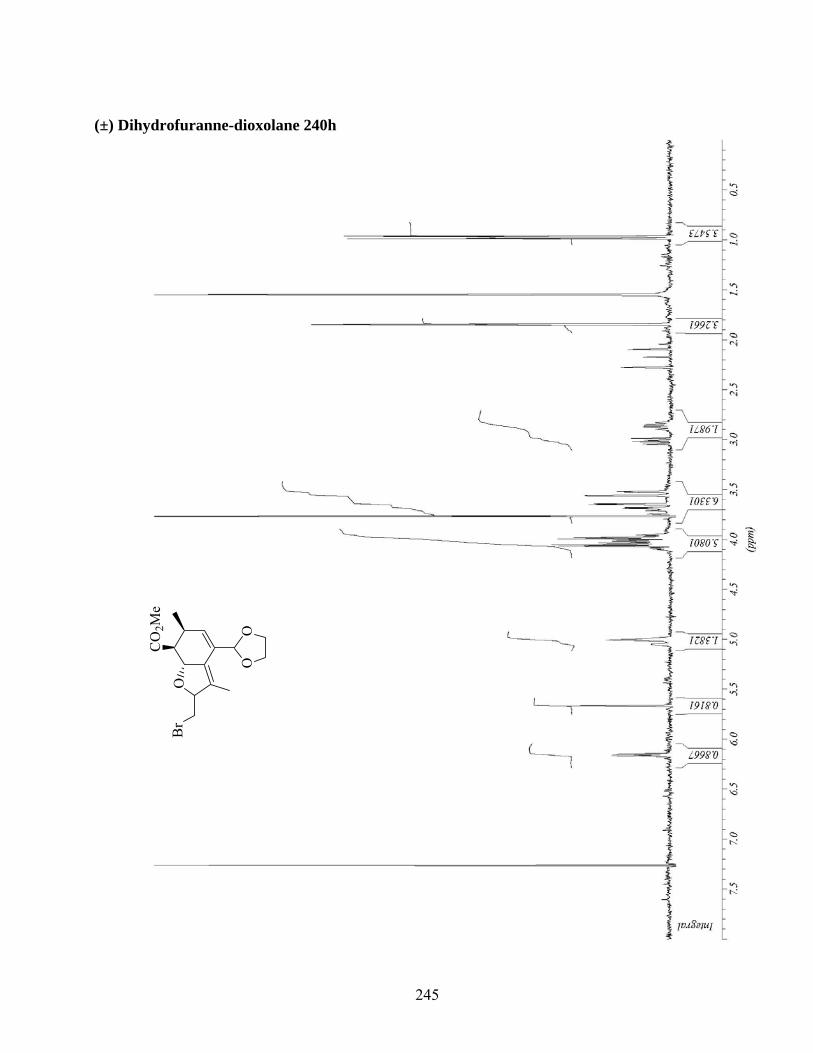
245
(±) Dihydrofuranne-dioxolane 240h
OC
O2M
eB
r
OO
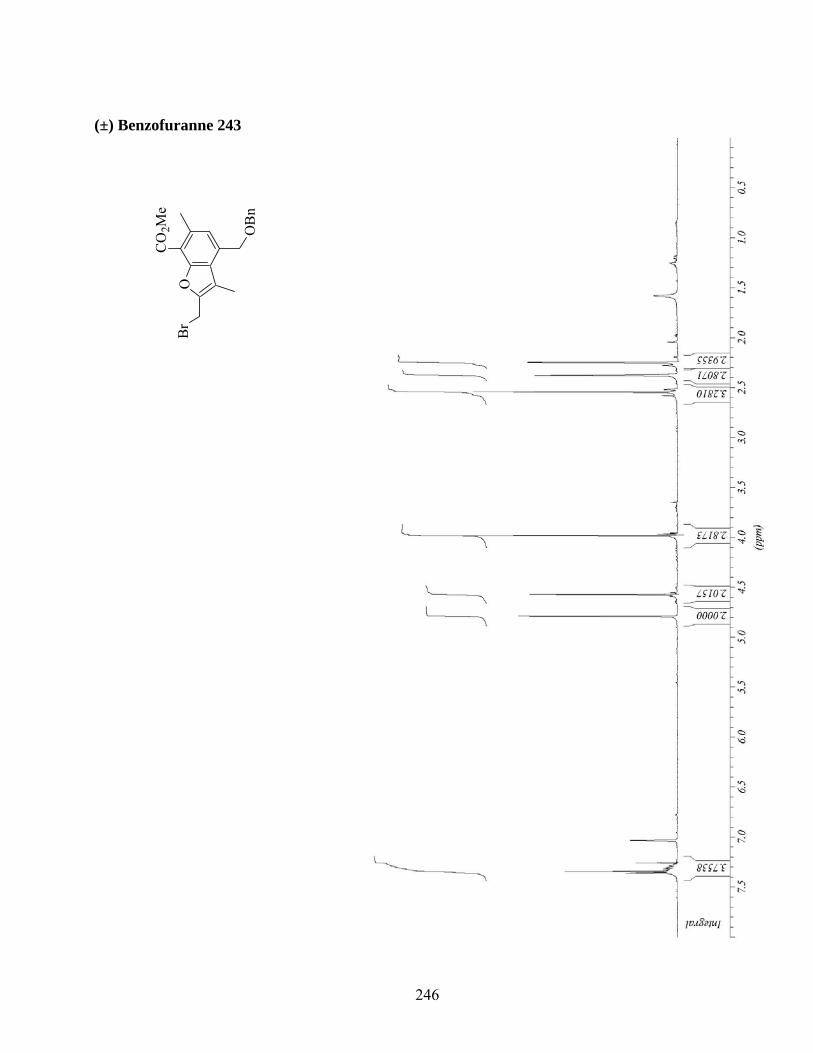
246
(±) Benzofuranne 243
OC
O2M
e
OB
n
Br
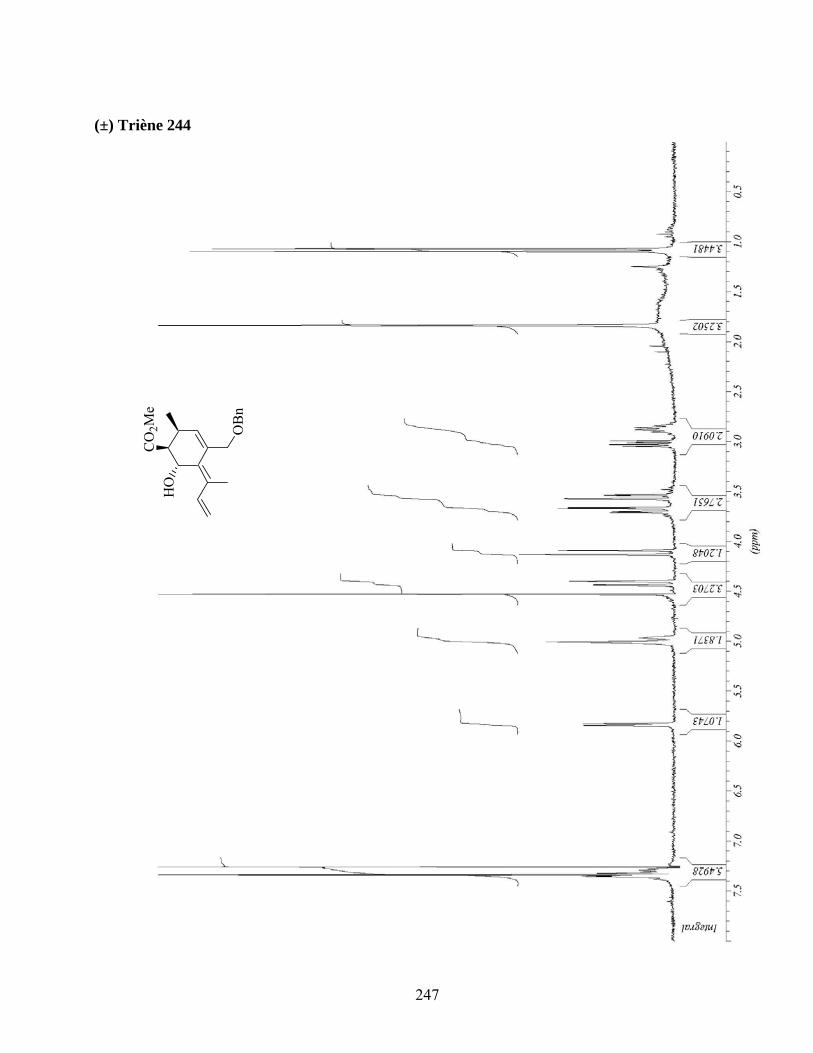
247
(±) Triène 244
CO
2Me
OB
n
HO
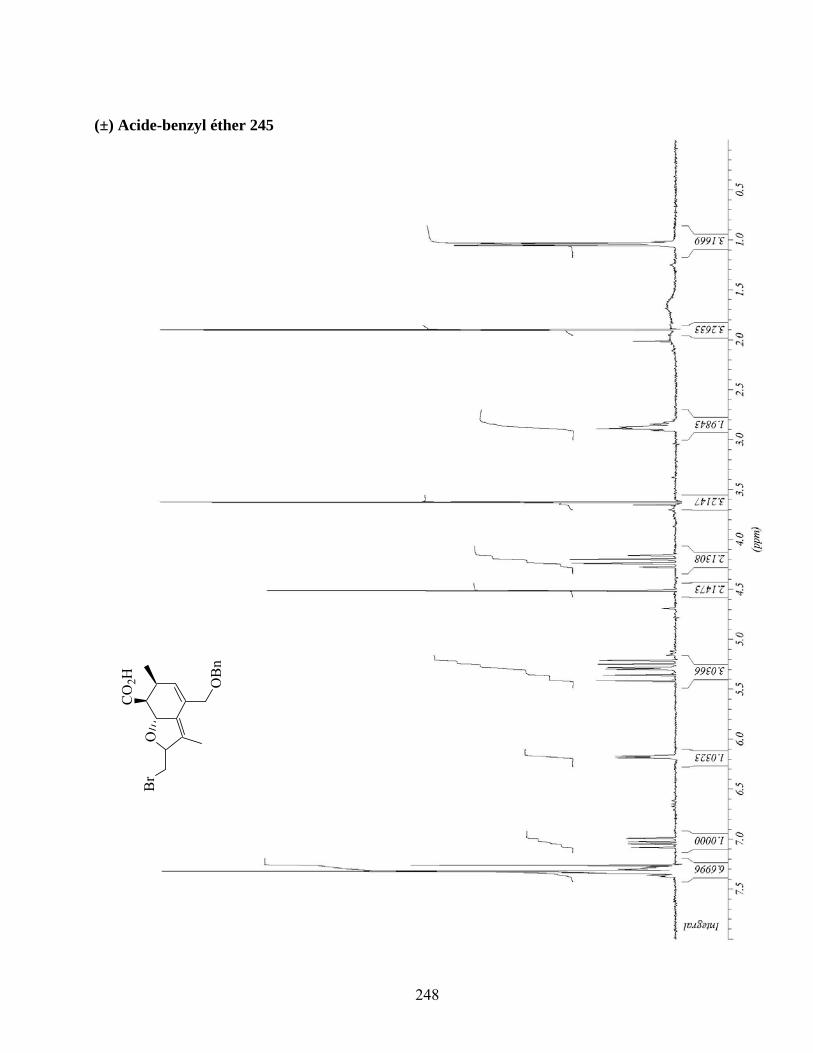
248
(±) Acide-benzyl éther 245
OC
O2H OB
n
Br
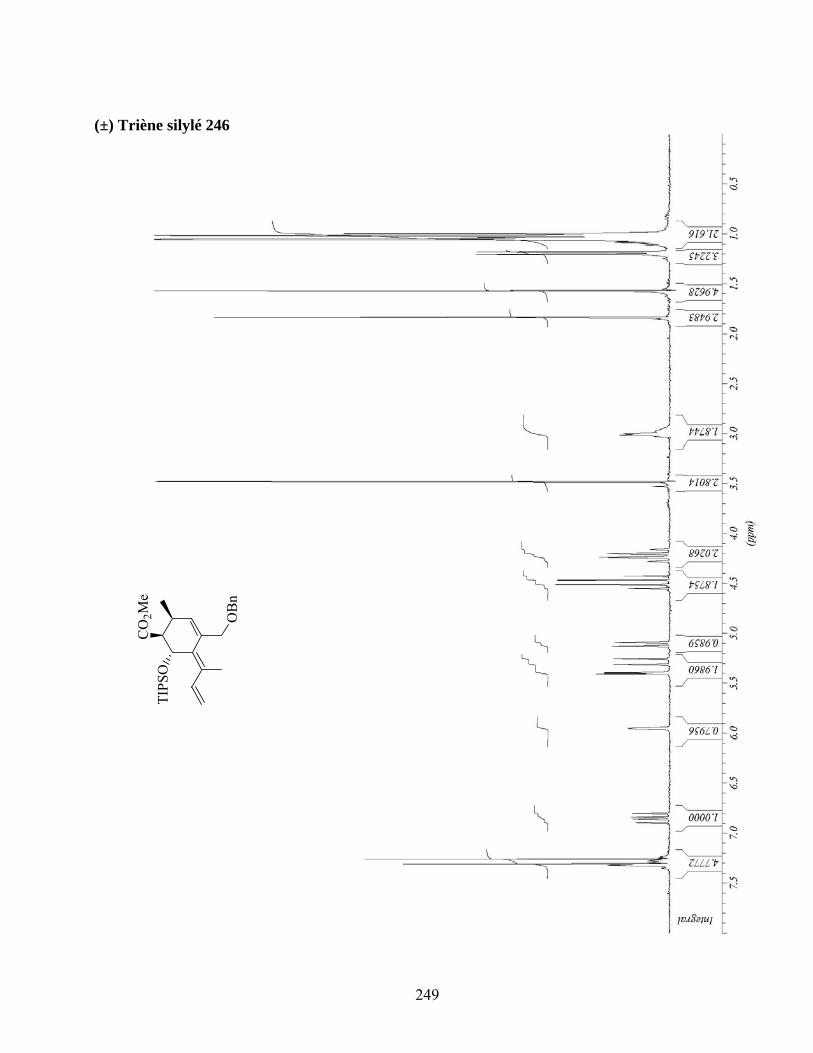
249
(±) Triène silylé 246
CO
2Me
OB
n
TIPS
O
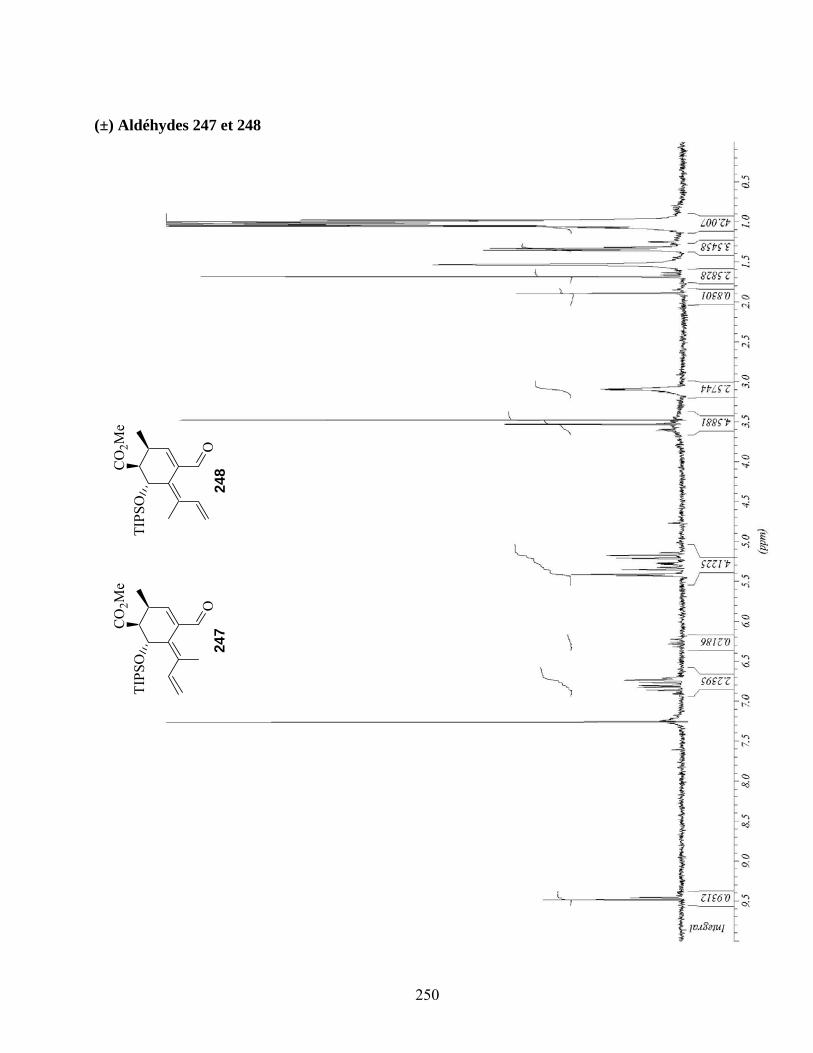
250
(±) Aldéhydes 247 et 248
CO2M
e
O
TIPS
OC
O2M
e
O
TIPS
O
247
248
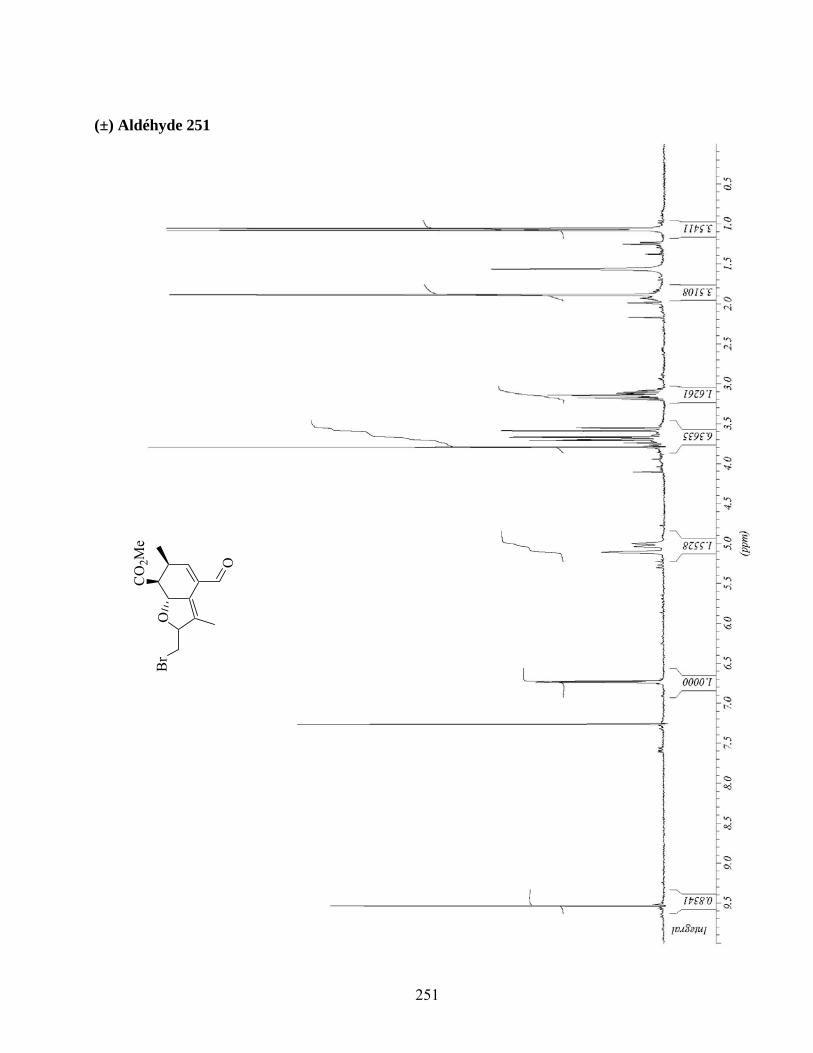
251
(±) Aldéhyde 251
OC
O2M
eB
r
O
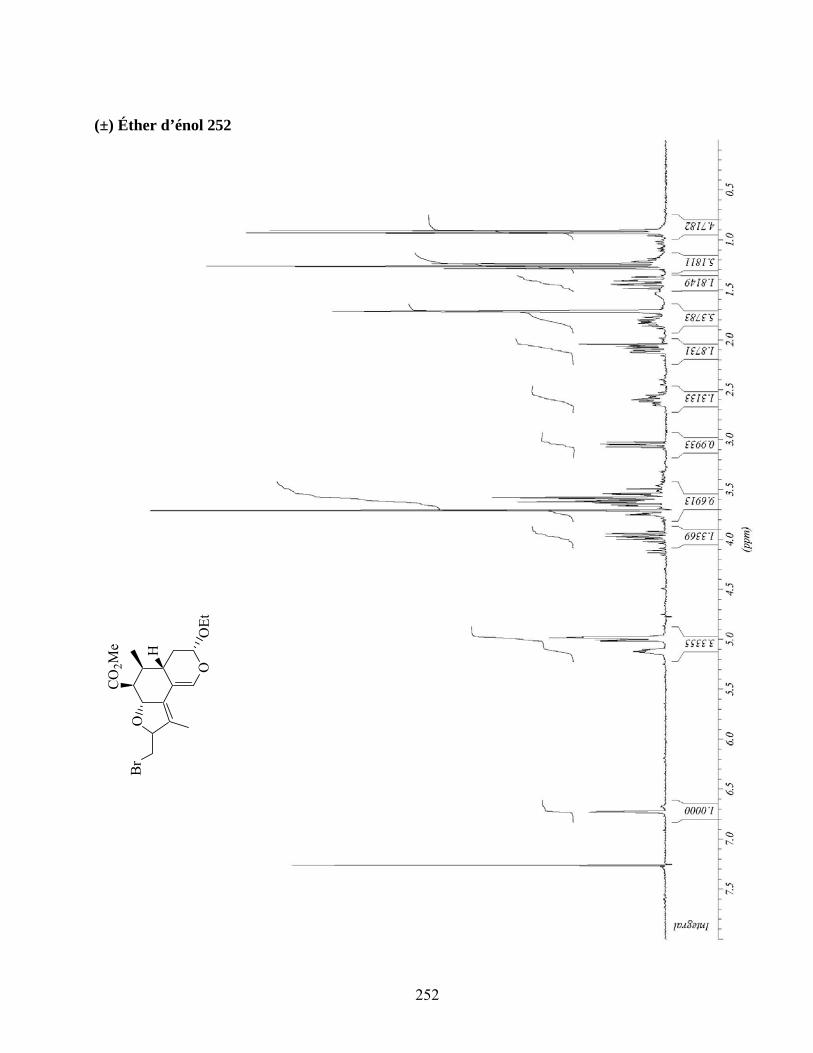
252
(±) Éther d’énol 252
OCO
2Me
Br
OO
E t
H
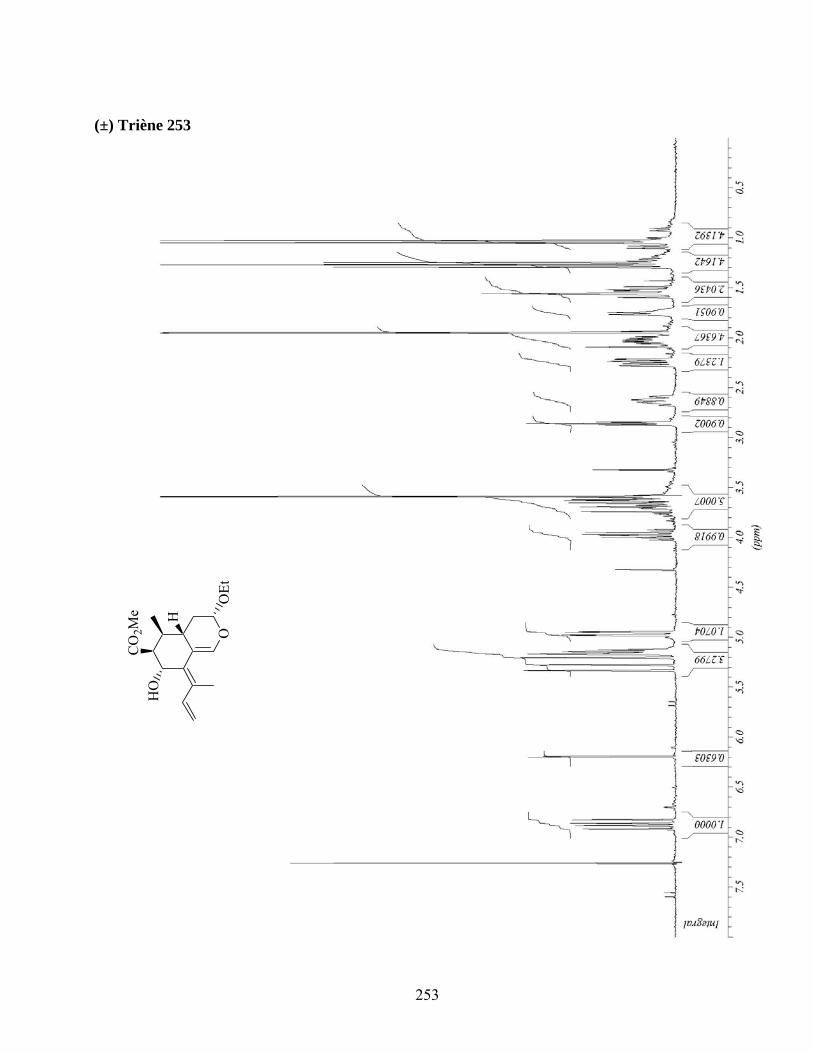
253
(±) Triène 253
CO
2Me
OO
Et
HH
O
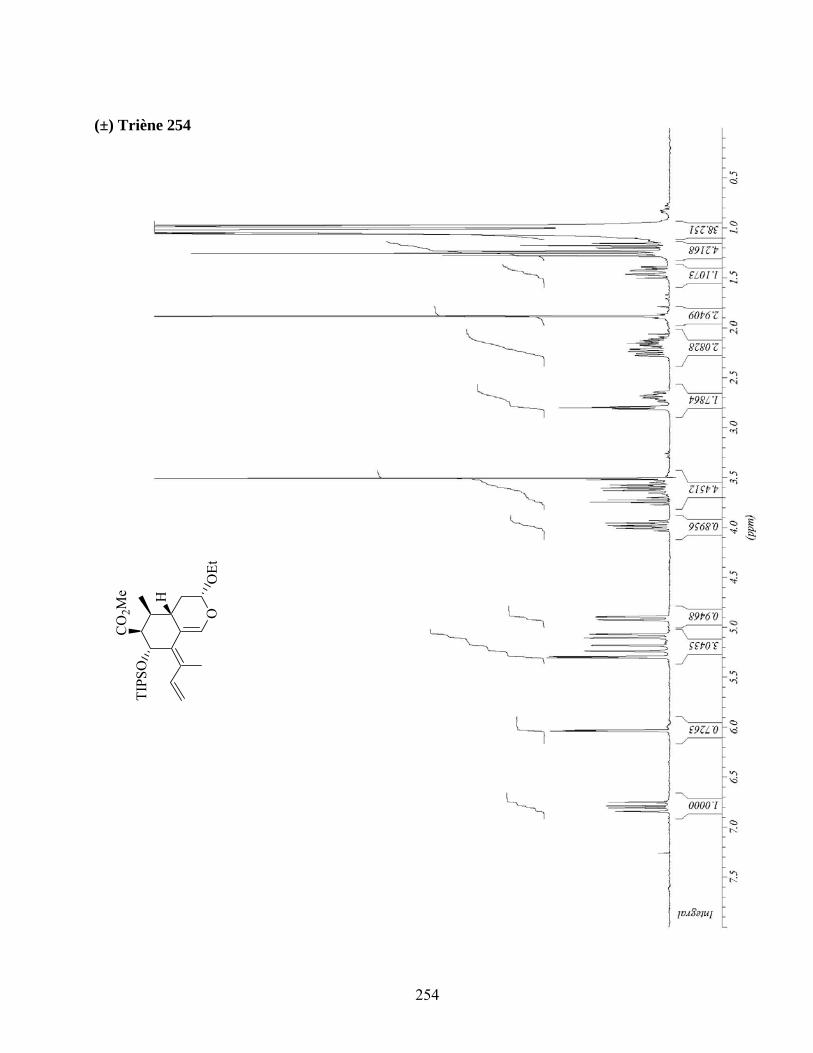
254
(±) Triène 254
CO
2Me
OO
Et
HTI
PSO
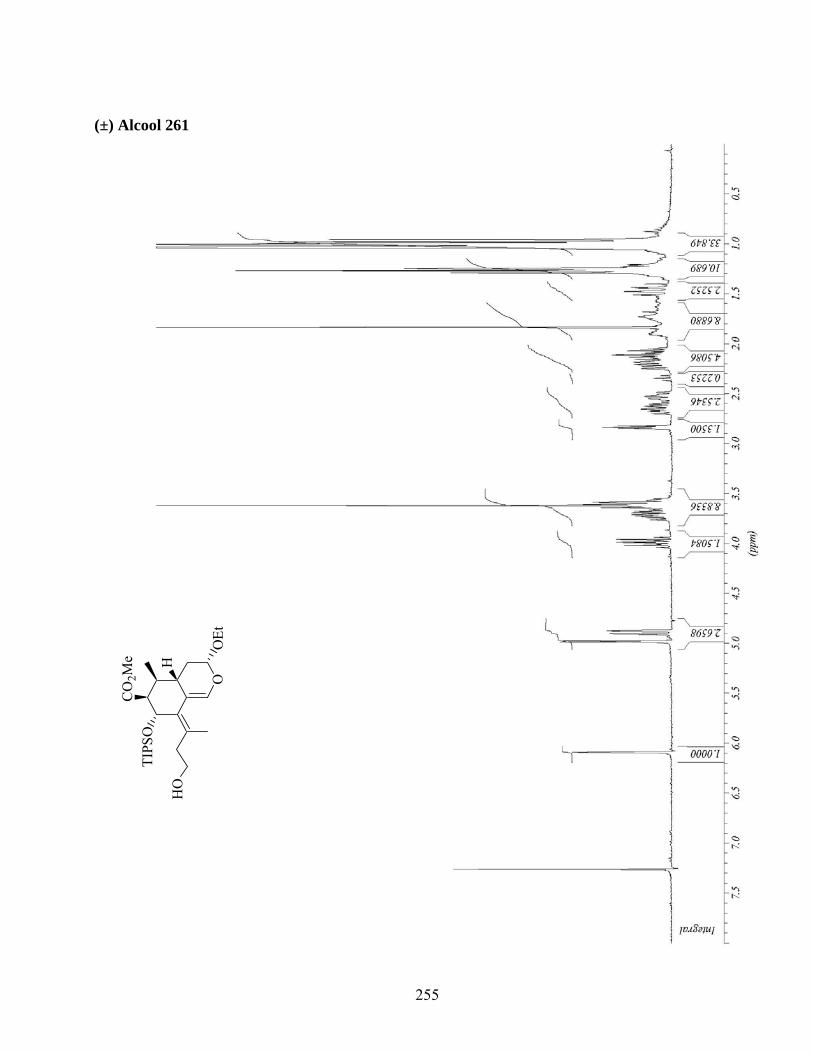
255
(±) Alcool 261
CO
2Me
OO
Et
HTI
PSO
HO
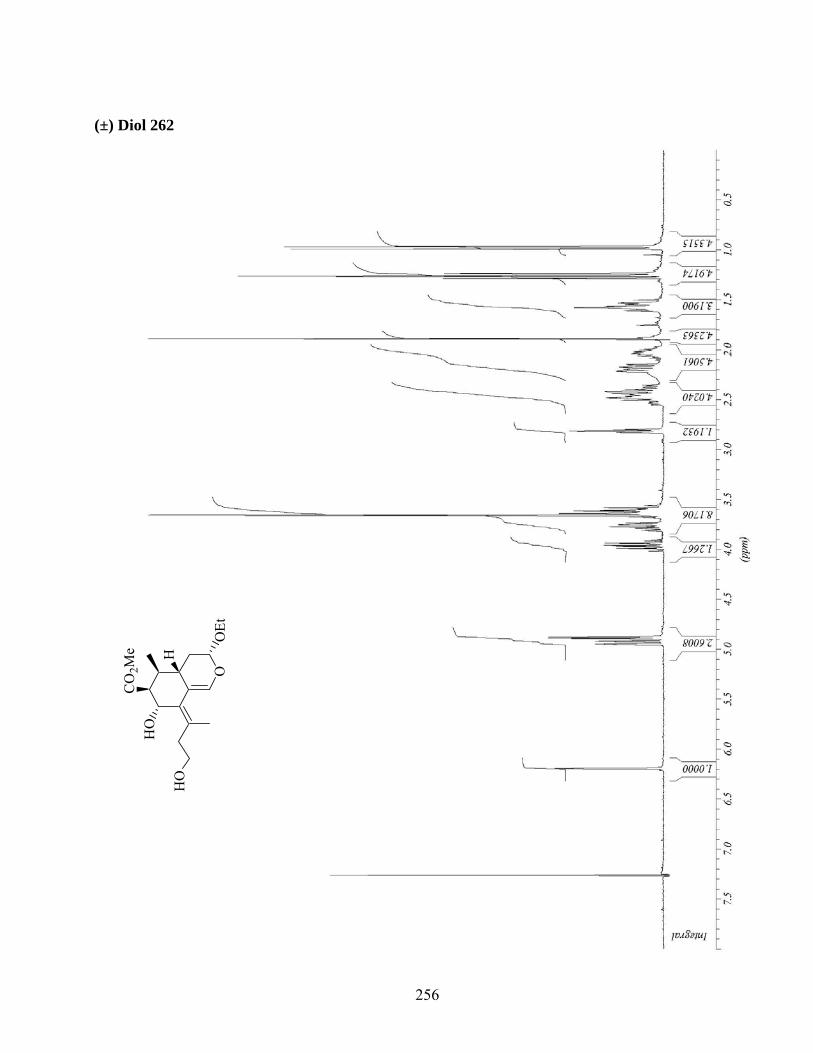
256
(±) Diol 262
CO
2Me
OO
Et
HH
O
HO
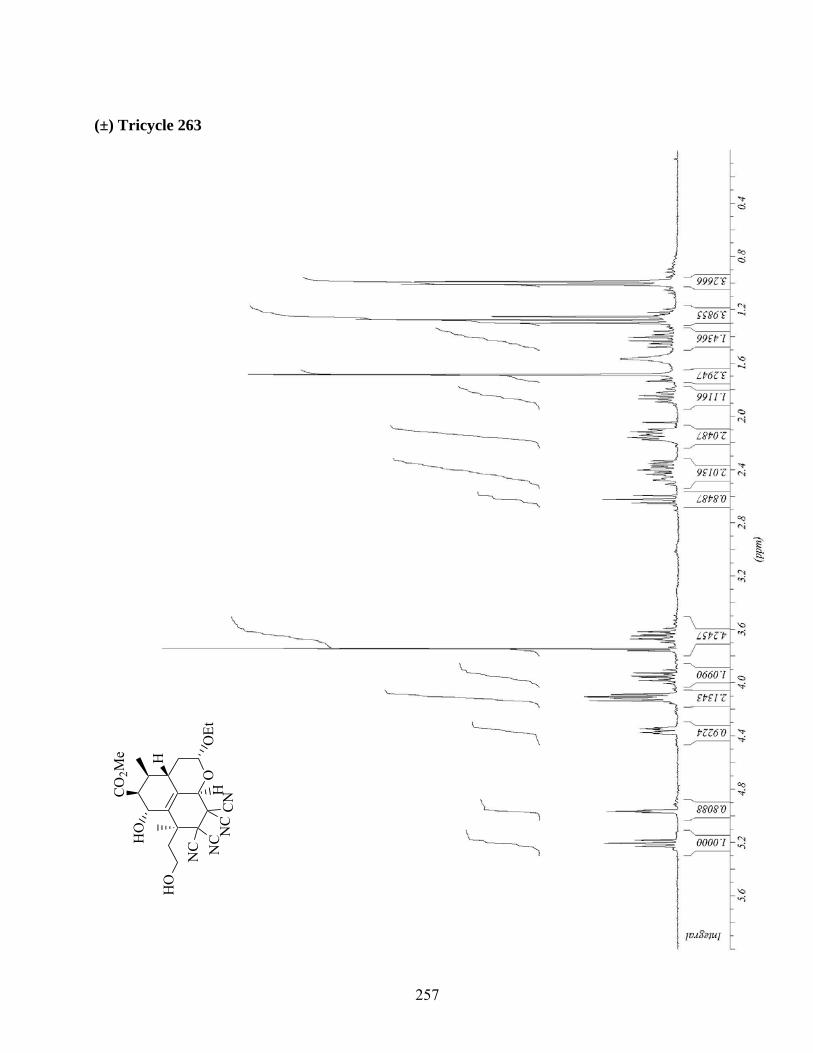
257
(±) Tricycle 263
CO2M
e
OO
Et
HH
O
HO
NC NC N
CC
NH
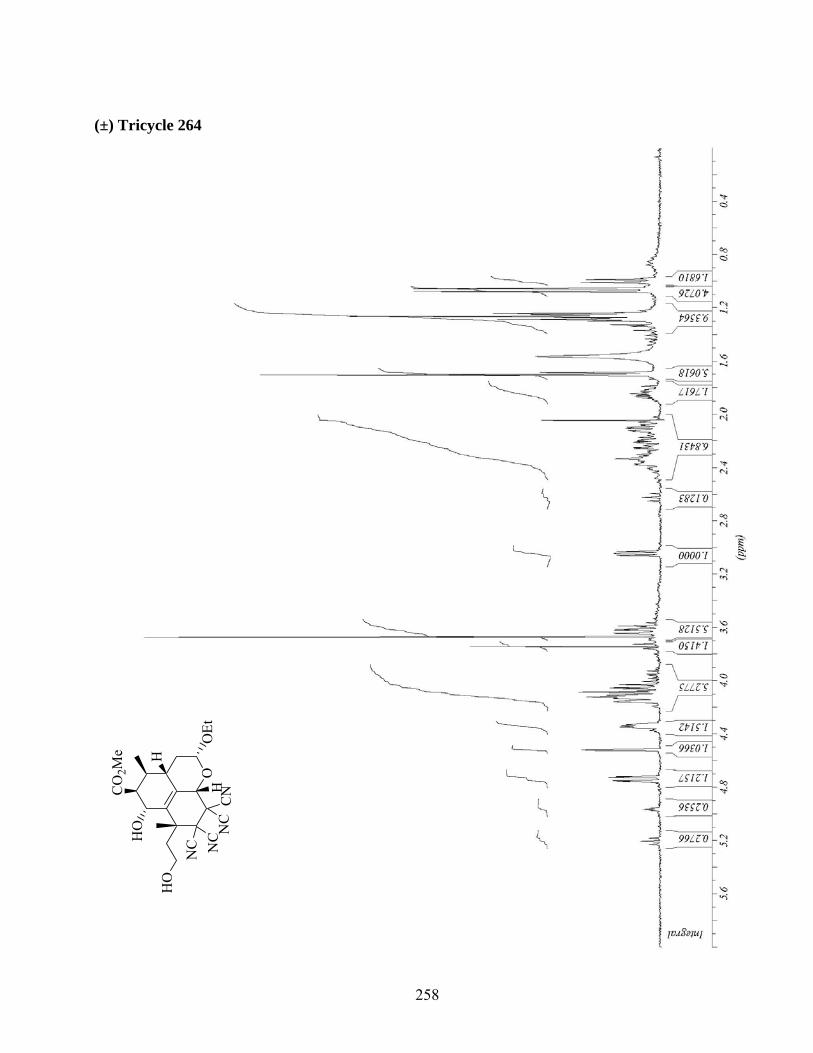
258
(±) Tricycle 264
CO
2Me
OO
Et
HH
O
HO
NC NC N
CC
NH
259
(±) Alcool secondaire 267
CO
2Me
OO
Et
HH
O
TBSO

260
ANNEXE 2 : SPECTRES DE RÉSONANCE MAGNÉTIQUE NUCLÉAIRE DES CARBONES
261
(±) Sulfonate 116
OS
OP
MB
OO
262
(±) Dihydrofuranne 167
OC
O2M
e
SS
Me 3
Si

263
(±) Triène 254
CO
2Me
OO
Et
HTI
PSO

264














![SYNTHESE D' ALLENES VINYLIQUES: DIENE POUR CYCLOADDITION ... · SYNTHESE D' ALLENES VINYLIQUES: DIENE POUR CYCLOADDITION-[4 + 2] Projet realise par Stephane Gingras dans Ie cadre](https://img.pdfslide.fr/doc/110x75/5b9e6d8b09d3f204248bb83c/synthese-d-allenes-vinyliques-diene-pour-cycloaddition-synthese-d-allenes.jpg)














![MAHI thèse Doctorat 2017 - dspace.univ-tlemcen.dzdspace.univ-tlemcen.dz/bitstream/112/10054/1/ETUDE-THEORIQUE-D… · Orbitale Moléculaire ... La réaction de cycloaddition [4+2],](https://img.pdfslide.fr/doc/110x75/5b9d02f309d3f29a298ba9e7/mahi-these-doctorat-2017-orbitale-moleculaire-la-reaction-de-cycloaddition.jpg)