Embed Size (px)
Citation preview

Pour citer cet article : Ouleghzal H et al., Syndrome polyuro-polydipsique associé à des douleurs osseuses, Presse Med (2014),http://dx.doi.org/10.1016/j.lpm.2013.12.015.
Presse Med. 2014; //: /// en ligne sur / on line onwww.em-consulte.com/revue/lpm
www.sciencedirect.comtre
àla
réd
act
ion
[(Figure_2)TD$FIG]
Syndrome polyuro-polydipsique associé àdes douleurs osseuses
Let
Polyuria-polydipsia syndrome associated withbone pains
Figure 2
Scintigraphie osseuse montrant des lésions humérales et
La constatation d’un syndrome polyuro-polydipsique nécessiteune démarche diagnostique appropriée afin d’en préciser lacause pour pouvoir proposer un traitement adapté. Nous rap-portons un cas de syndrome polyuro-polydipsique dont l’étio-logie est très inhabituelle, associant un diabète insipide centralà une atteinte osseuse. Le diagnostic d’une histiocytose nonlangerhansienne (maladie d’Erdheim-Chester) a été authentifiégrâce à la biopsie des foyers d’hyperfixation osseux.
Observation
Une jeune femme, âgée de 28 ans, sans antécédents patho-logiques notables, souffrait depuis 2 mois d’un syndrome poly-uro-polydipsique sévère, permanent, avec des entrées etsorties estimées chacune à environ 12 L/24 h, associé à desdouleurs osseuses d’allure mécanique, particulièrement costa-les, soulagées par les traitements antalgiques habituels. Lapatiente rapporte par ailleurs des céphalées et des troublesvisuels à type de flou visuel.L’examen clinique était parfaitement normal, en dehors d’ungoitre modéré. Sur le plan biologique, la natrémie était à137 mEq/L avec une osmolarité plasmatique à 262,5 mOsm/
1
[(Figure_1)TD$FIG]
Figure 1
IRM hypothalamo-hypophysaire (séquence T1 avec injection de gadolinium). Épaississement de la tige pituitaire et nodule au niveau dela loge caverneuse gauche
fémorales bilatérales et au niveau maxillaire
tome // > n8/ > /
LPM-2426
2
Pour citer cet article : Ouleghzal H et al., Syndrome polyuro-polydipsique associé à des douleurs osseuses, Presse Med (2014),http://dx.doi.org/10.1016/j.lpm.2013.12.015.
Lettre à la rédaction
kg et une osmolarité urinaire à 137,35 mOsm/kg. Le test derestriction hydrique était arrêté à la 5e heure car la patienteavait perdu plus de 5 % de son poids initial, sans concentrationdes urines. Le test au MinirinW était positif avec une osmolaritéurinaire à 2 h à 506,54 mOsm/kg. L’hypophysiogrammecomplet était normal.L’IRM hypothalamo-hypophysaire révélait un épaississementde la tige pituitaire avec la présence d’un nodule au niveau dela loge caverneuse gauche et la perte de l’hypersignal spontanéen T1 de la posthypophyse (figure 1).Les radiographies standards des os longs étaient normales. Lascintigraphie osseuse montrait des zones d’hyperfixation,d’intensité modérée, au niveau du sinus maxillaire, deshumérus et des fémurs (figure 2), pour lesquelles une biopsie
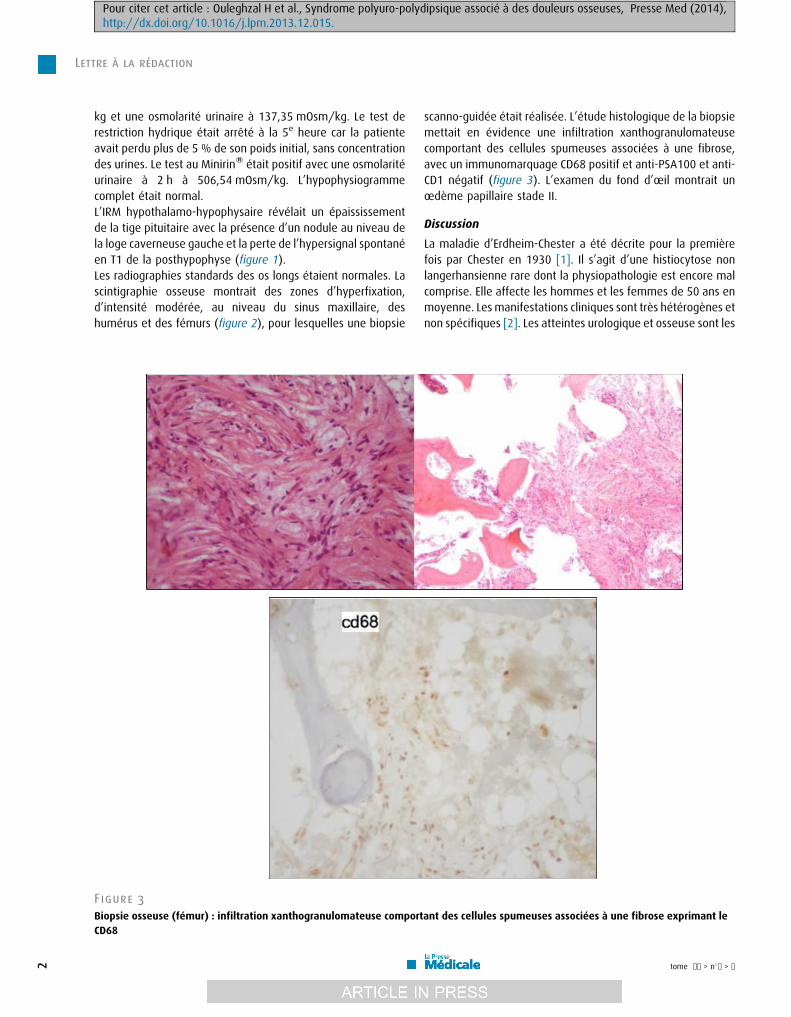
[(Figure_3)TD$FIG]
Figure 3
Biopsie osseuse (fémur) : infiltration xanthogranulomateuse comportCD68
scanno-guidée était réalisée. L’étude histologique de la biopsiemettait en évidence une infiltration xanthogranulomateusecomportant des cellules spumeuses associées à une fibrose,avec un immunomarquage CD68 positif et anti-PSA100 et anti-CD1 négatif (figure 3). L’examen du fond d’oeil montrait unoedème papillaire stade II.
Discussion
La maladie d’Erdheim-Chester a été décrite pour la premièrefois par Chester en 1930 [1]. Il s’agit d’une histiocytose nonlangerhansienne rare dont la physiopathologie est encore malcomprise. Elle affecte les hommes et les femmes de 50 ans enmoyenne. Les manifestations cliniques sont très hétérogènes etnon spécifiques [2]. Les atteintes urologique et osseuse sont les
ant des cellules spumeuses associées à une fibrose exprimant le
tome // > n8/ > /

Lett
reà
laré
da
ctio
n
Pour citer cet article : Ouleghzal H et al., Syndrome polyuro-polydipsique associé à des douleurs osseuses, Presse Med (2014),http://dx.doi.org/10.1016/j.lpm.2013.12.015.
plus fréquentes, les autres manifestations, plus rares, font toutela gravité de la maladie, notamment lorsqu’il existe uneatteinte cardiovasculaire, neurologique centrale, rétro-orbitaireou pulmonaire [3]. Sur le plan endocrinien, des perturbations del’axe hypothalamo-hypophysaire à type de diabète insipide,comme le cas présenté ici, ou d’hypogonadisme hypogonado-trope ont été rapportées [4,5].Le diagnostic positif est histologique, montrant un infiltratxanthogranulomateux composé d’histiocytes ou de macropha-ges chargés en graisse et entourés de fibrose. Les celluleshistiocytaires ne sont pas des cellules de Langerhans et necontiennent pas de granules de Birbeck. À l’immuno-histochi-mie, on trouve une immunopositivité CD68 et une immunoné-gativité CD1a et de la PS100 [6].L’anomalie radiologique la plus spécifique est l’ostéosclérosecorticale. Elle est classiquement bilatérale, symétrique, toucheles diaphyses et métaphyses des os longs [2]. Cependant, deslésions lytiques ou des atteintes des os plats ne sont pasexceptionnelles. Sur le plan biologique, les marqueurs del’inflammation, l’interleukine-6 et le récepteur de l’interleu-kine-6 sont élevés [7]. L’aspect de la scintigraphie osseuse estquasi pathognomonique montrant une hyperfixation bilatéraleet symétrique des métaphyses et des diaphyses des os longs,comme dans le cas présent. Une atteinte des épaules, descôtes, du crâne et de la face peut être observée, alors que lacolonne vertébrale et les épiphyses sont en général épargnées[8,9].Sur le plan thérapeutique, aucun protocole n’est en placeactuellement [10]. Différents traitements ont été proposés[2]. L’interféron alpha a prouvé une amélioration de la survie[11], toutefois, il est parfois inefficace ou doit être inter-rompu en raison d’effets indésirables majeurs. L’anakinra(antagoniste du récepteur de l’IL1) pourrait constituerune bonne option thérapeutique, mais des études supplé-mentaires sont nécessaires [12]. Dans le cas de notrepatiente, le syndrome polyuro-polydipsique a été corrigépar la desmopressine. On a aussi institué à visée étiopatho-génique un traitement par interféron alpha. L’évolutiona été marquée par un soulagement des douleurs osseuseset par l’installation de signes d’insuffisance antéhypophy-saire (hypothyroïdie, hypogonadisme) ayant nécessité l’ins-tauration d’un traitement substitutif. Le pronostic de lamaladie est en général péjoratif et dépend du type d’atteinteviscérale.
Conclusion
La granulomatose d’Erdheim-Chester est une maladie systé-mique rare, de diagnostic difficile du fait d’un polymorphismeclinique et morphologique. Le diagnostic est dans tous les cas
tome // > n8/ > /
histologique. Le traitement est encore mal codifié et le pro-nostic reste imprévisible.
Déclaration d’intérêts : les auteurs déclarent ne pas avoir de conflitsd’intérêts en relation avec cet article.
Références[1] Chester W. Über Lipogranulomatose. Virchows Arch Path Anat
1930;279:561-602.[2] Veyssier-Belot C, Cacoub P, Caparros-Lefebvre D, Wechsler J et al.
Erdheim-Chester disease. Clinical and radiologic characteristics of59 cases. Medicine (Baltimore) 1996;75:157-69.
[3] Prunel P, Verhoest G, Besnard S, Rohou T, Rioux-Leclercq N, Bensalah K.Maladie d’Erdheim-Chester : à propos d’un cas et revue de la littérature.Prog Urol 2012;22:310-2.
[4] Kovacs K, Bilbao JM, Fornasier VL, Horvath E. Pituitary pathology inErdheim-Chester disease. Endocr Pathol 2004;15:159-66.
[5] Khamseh ME, Mollanai S, Hashemi F, Rezaizadeh A, Azizi F. Erdheim-Chester syndrome, presenting as hypogonadotropic hypogonadism anddiabetes insipidus. J Endocrinol Invest 2002;25:727-9.
[6] Al-Quran S, Reith J, Bradley J, Rimsza L. Erdheim-Chester disease: casereport, PCR-based analysis of clonality, and review of literature. ModPathol 2002;15:666-72.
[7] Mossetti G, Rendina D, Numis FG, Somma P, Postiglione L, Nunziata V.Biochemical markers of bone turnover, serum levels of interleukin-6/interleukin-6 soluble receptor and bisphosphonate treatment inErdheim-Chester disease. Clin Exp Rheumatol 2003;21:232-6.
[8] Gotthardt M, Welcke U, Brandt D, Tontsch D, Barth PJ, Schaefer J et al.The role of bone scintigraphy in patients with Erdheim-Chester disease.Clin Nucl Med 2000;25:414-20.
[9] Nunez R, Tronco GG, Rini JN, Hofman J, Amoashiy M, Bhuiya T et al.Radionuclide bone imaging in Erdheim-Chester disease. Clin Nucl Med2005;30:32-4.
[10] Le Goff L, Berros P, Denis D, Ridings R. Exophtalmie bilateral – Diabèteinsipide : la maladie d’Erdheim-Chester, caractéristiques cliniques etradiologiques. J Fr Ophtalmol 2002;25:57-61.
[11] Braiteh F, Boxrud C, Esmaeli B, Kurzrock R. Successful treatment ofErdheim-Chester disease, a non-Langerhans-cell histiocytosis, withinterferon-alpha. Blood 2005;106:2992-4.
[12] Courcoul A, Vignot E, Chapurlat R. Successful treatment of Erdheim-Chester disease by interleukin-1 receptor antagonist protein. Joint BoneSpine 2014 (doi:10.1016/j.jbspin.2013.06.013 [Epub ahead of print]).
Hassan Ouleghzal1, Ilyass Anouar2, Fatima Boufares1
1Hôpital militaire d’instruction Mohamed V, serviced’endocrinologie et maladies métaboliques,
10000 Rabat, Maroc2Hôpital militaire d’instruction Mohamed V, service des
urgences, 10000 Rabat, Maroc
Correspondance : Hassan Ouleghzal, Hôpital militaired’instruction Mohamed V, service d’endocrinologie et maladies
métaboliques, 10000 Rabat, [email protected]
Reçu le 6 septembre 2013Accepté le 17 décembre 2013
http://dx.doi.org/10.1016/j.lpm.2013.12.015� 2014 Elsevier Masson SAS. Tous droits réservés.
3





























