Embed Size (px)
Citation preview

THÈME : ETAT DE LA MATIÈRE ET LEUR CARACTÉRISATION
CHAPITRE 1
LIQUIDE, GAZ ET SOLUTIONS
UE 3

SOMMAIRE
I
II
III
NIVEAU D’ORGANISATION DE LA MATIERE
LES GAZ PARFAITS
LES GAZ REELS
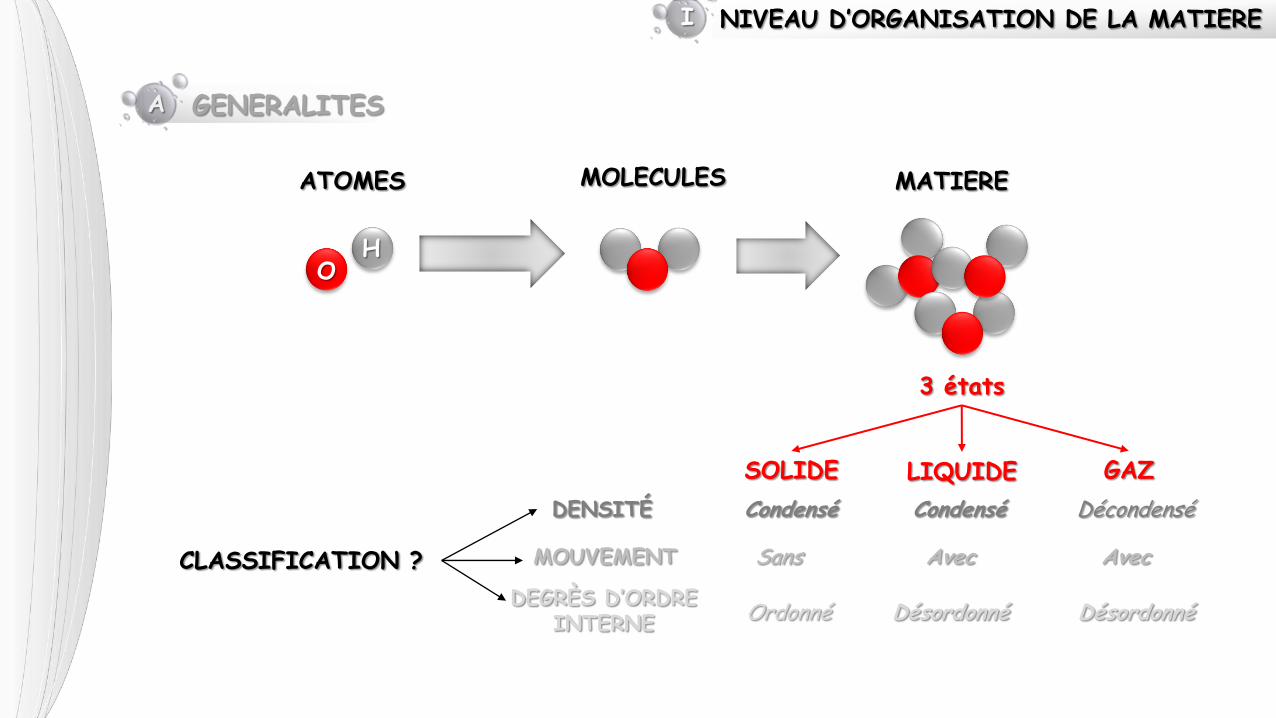
NIVEAU D’ORGANISATION DE LA MATIERE I
O H
ATOMES MOLECULES MATIERE
3 états
SOLIDE LIQUIDE GAZ
CLASSIFICATION ?
DENSITÉ
MOUVEMENT
DEGRÈS D’ORDRE INTERNE
Condensé Décondensé
Sans Avec
Condensé
Avec
Ordonné Désordonné Désordonné
A GENERALITES
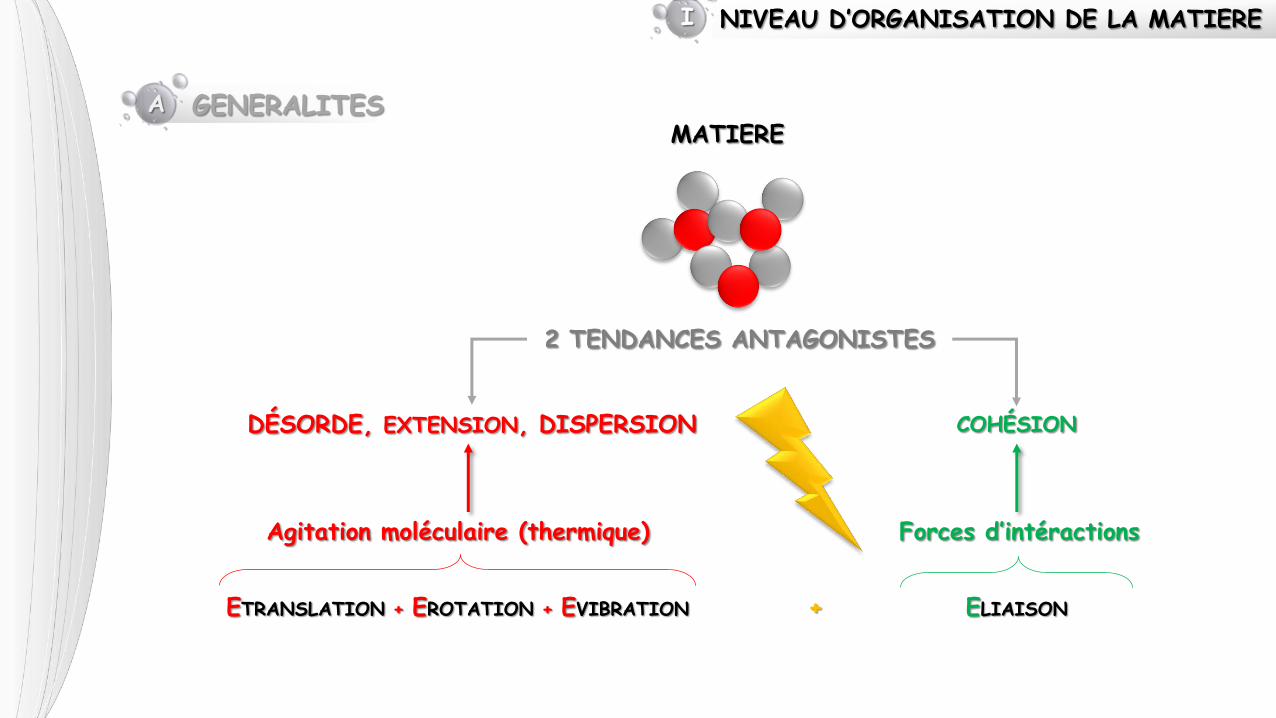
NIVEAU D’ORGANISATION DE LA MATIERE I
MATIERE
2 TENDANCES ANTAGONISTES
A GENERALITES
DÉSORDE, EXTENSION, DISPERSION COHÉSION
Agitation moléculaire (thermique) Forces d’intéractions
ETRANSLATION + EROTATION + EVIBRATION ELIAISON +

NIVEAU D’ORGANISATION DE LA MATIERE I
B FORCES D’INTÉRACTIONS
LIAISONS INTERMOLECULAIRES
Liaisons de VAN DER WAALS : (électrostatiques), liaison forte entre molécules:
• Polaires/polaires Attractions des pôles opposés
• Polaires/apolaire Polarisation de la moléculaire apolaire
• Apolaires/apolaire (gaz rares, liq ou sol, basse T°)
• Attraction
• Répulsion
• Répulsion des nuages électroniques
• Encombrement stérique
F Répulsion
F Attraction
d éq
D éq: distance d’équilibre compensation Au-delà :tendance à l’attraction Avant : tendance à la répulsion

LIAISONS INTRAMOLECULAIRES
Liaisons COVALENTES : fortes
Liaisons IONIQUES : fortes
X Y
Liaisons METALLIQUES : faibles
Y- Y- X+ X+
X X X
Cas particulier : Liaisons HYDROGENES (électrostatiques) : faibles
• ++ pour les atomes très électronégatifs (ex : Oxygène)
• + grandes sont ces forces + énergie pour les briser est forte T° de fusion, et de vaporisation
FORCES D’INTÉRACTIONS
B
• 5 à 20% des liaisons intramoléculaires
• Atome H: pont entre plusieurs atomes alors qu’ils n’ont pas d’électrons libres

NIVEAU D’ORGANISATION DE LA MATIERE I
C L’ETAT GAZEUX
PAS DE VOLUME DEFINI
EXPANSIBLE SPONTANEMENT A l’INFINI
NECESSITE DE DEFINIR LES CONDITIONS D’ETUDES : T° et P
THEORIE CINETIQUE DES GAZ Un gaz est un milieu constitué de particules qui :
Sont très éloignées les unes des autres
Se déplacent continuellement, à grande vitesse, en ligne droite
Entrent en collision
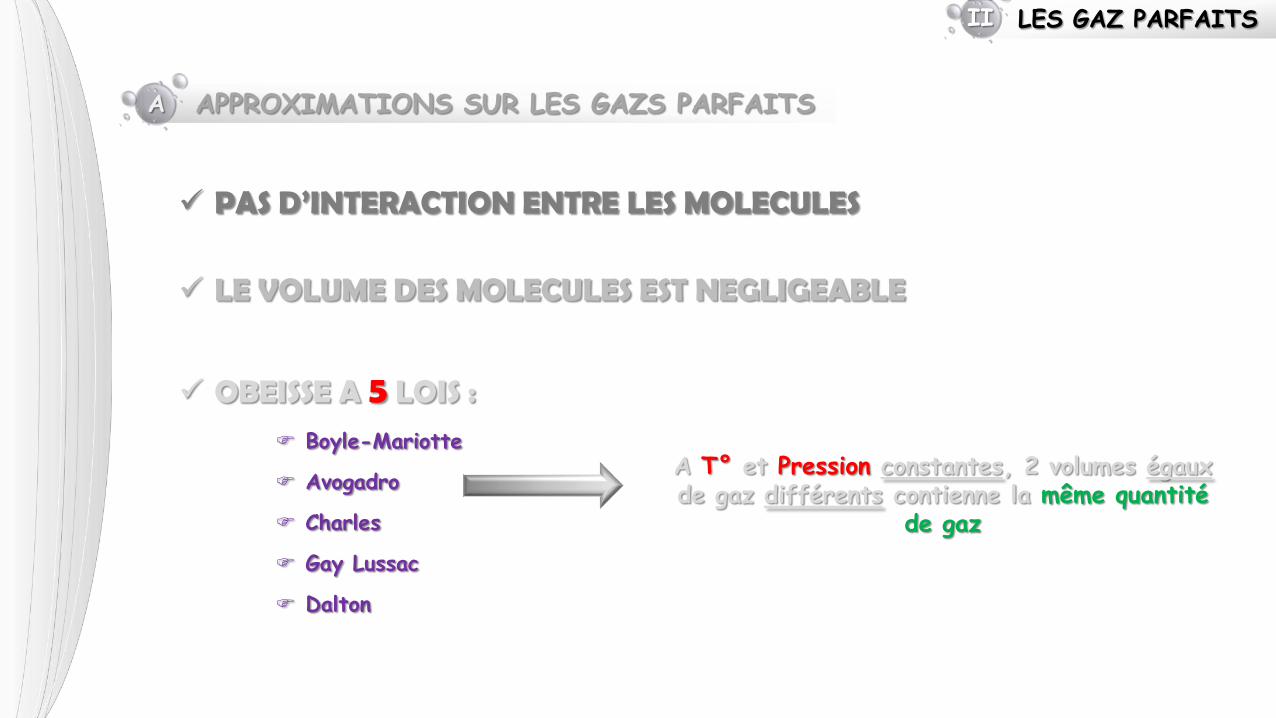
LES GAZ PARFAITS II
A APPROXIMATIONS SUR LES GAZS PARFAITS
PAS D’INTERACTION ENTRE LES MOLECULES
LE VOLUME DES MOLECULES EST NEGLIGEABLE
OBEISSE A 5 LOIS :
Boyle-Mariotte
Avogadro
Charles
Gay Lussac
Dalton
A T° et Pression constantes, 2 volumes égaux de gaz différents contienne la même quantité
de gaz
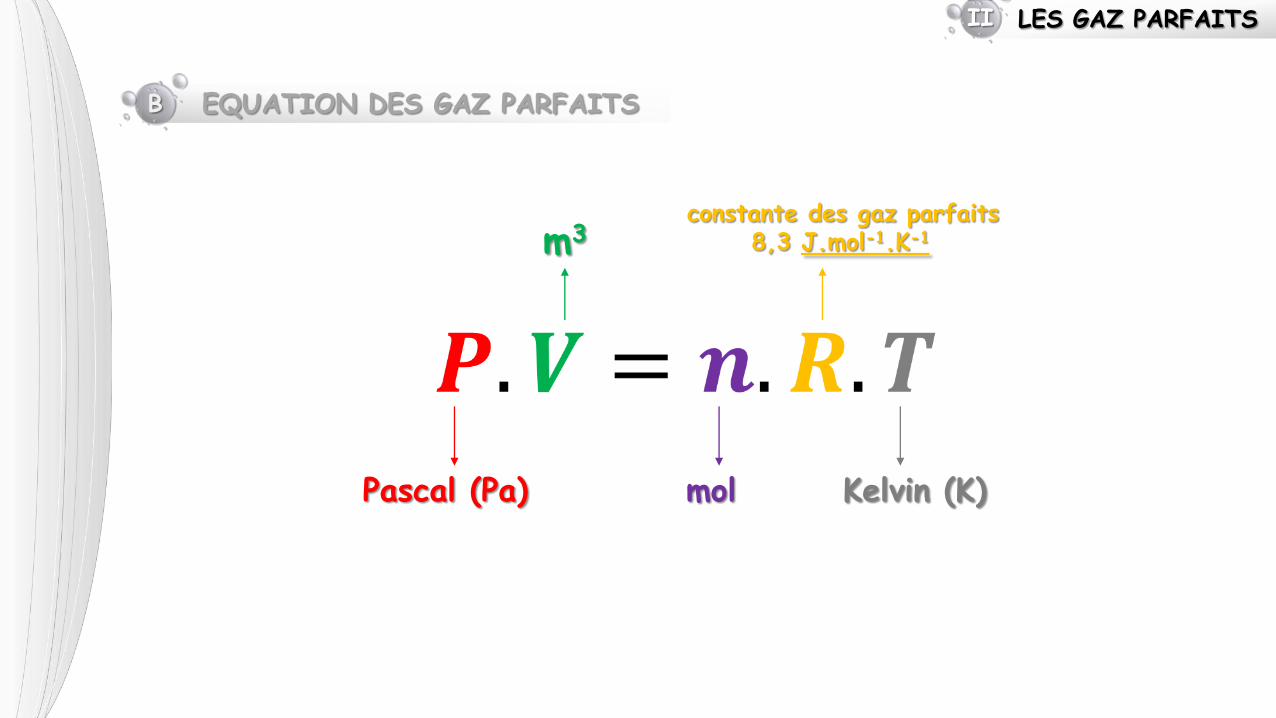
LES GAZ PARFAITS II
B EQUATION DES GAZ PARFAITS
Pascal (Pa)
m3
mol
constante des gaz parfaits 8,3 J.mol-1.K-1
Kelvin (K)

LES GAZ REELS III
A EQUATION DES GAZ REELS
a et b sont 2 constantes caractéristiques de chaque gaz b = covolume, volume propre des molécules
! CONDITIONS D’APPLICATION (pour éviter des réactions chimiques) :
Faible masse molaire
Molécule non polaire, et faible polarisabilité
Grand volume offert au gaz, donc faible pression
T° éloignée de la T° de condensation
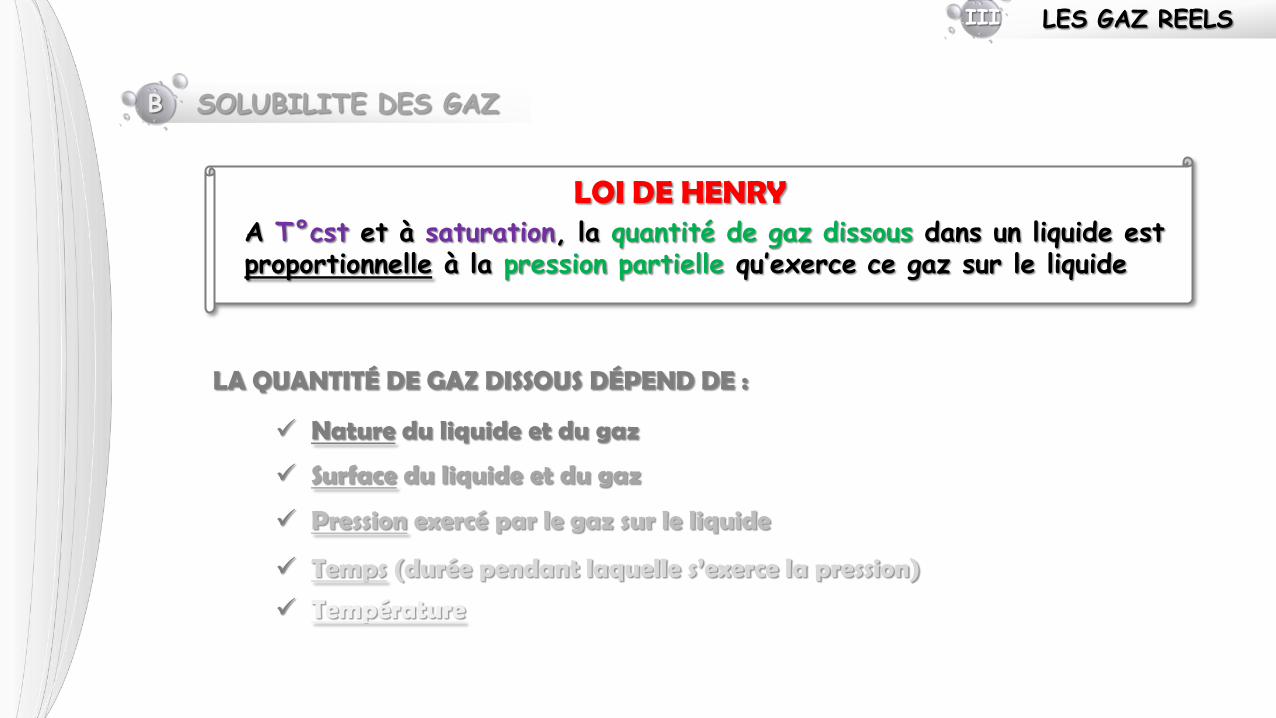
B SOLUBILITE DES GAZ
LOI DE HENRY A T°cst et à saturation, la quantité de gaz dissous dans un liquide est proportionnelle à la pression partielle qu’exerce ce gaz sur le liquide
LA QUANTITÉ DE GAZ DISSOUS DÉPEND DE :
Nature du liquide et du gaz
Surface du liquide et du gaz
Pression exercé par le gaz sur le liquide
Temps (durée pendant laquelle s’exerce la pression)
Température
LES GAZ REELS III
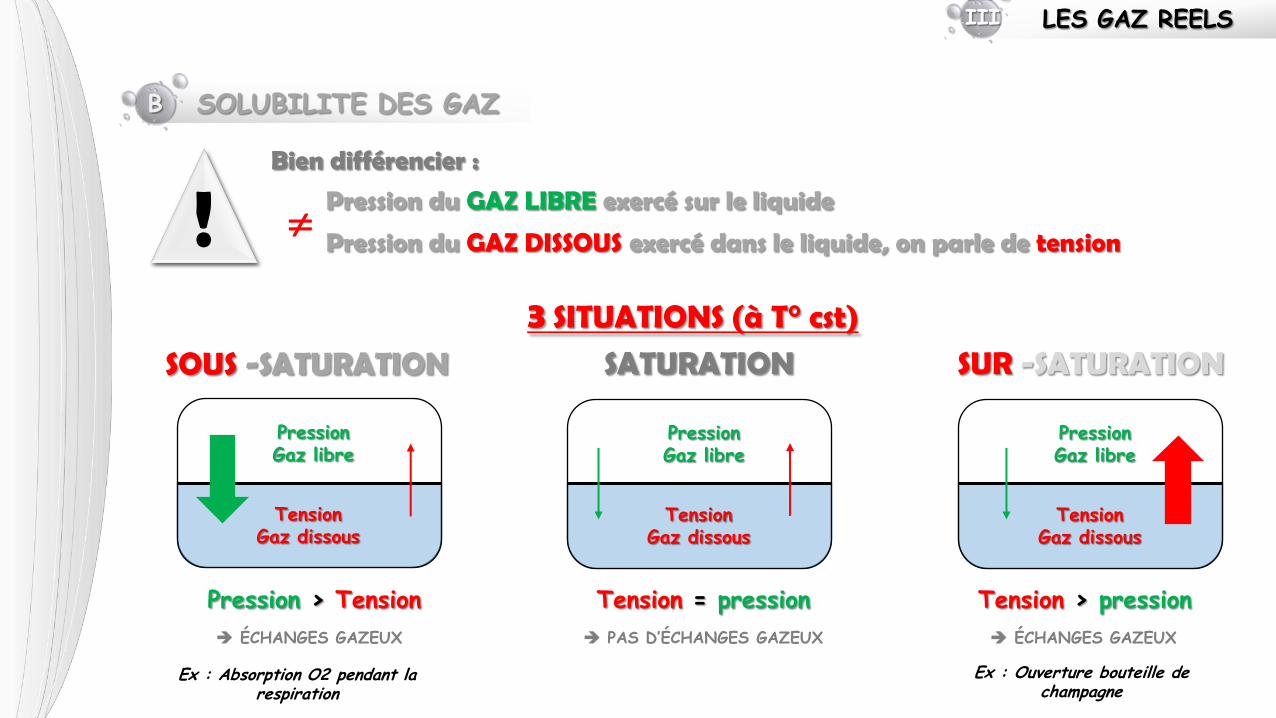
B SOLUBILITE DES GAZ
LES GAZ REELS III
3 SITUATIONS (à T° cst)
! Bien différencier :
Pression du GAZ LIBRE exercé sur le liquide
Pression du GAZ DISSOUS exercé dans le liquide, on parle de tension
SATURATION
Pression Gaz libre
Tension Gaz dissous
SUR -SATURATION
Pression Gaz libre
Tension Gaz dissous
SOUS -SATURATION
Pression Gaz libre
Tension Gaz dissous
Tension = pression Tension > pression Pression > Tension
PAS D’ÉCHANGES GAZEUX ÉCHANGES GAZEUX ÉCHANGES GAZEUX
Ex : Ouverture bouteille de champagne
Ex : Absorption O2 pendant la respiration

B SOLUBILITE DES GAZ
LES GAZ REELS III
CALCULER LE VOLUME DISSOUS
Volume du gaz dissous
Pression partielle (du gaz libre) En mm de Hg
Coefficient d’absorption
Volume du liquide

ETAT DE LA MATIERE ET LEUR CARACTERISATION
THÈME :
CHAPITRE 2
POTENTIEL CHIMIQUE
UE 3

SOMMAIRE
• I. ENTHALPIE LIBRE
• II. POTENTIEL CHIMIQUE (d’1 espèce PURE)
• III. Cas des MELANGES de gaz

I. ENTHALPIE LIBRE
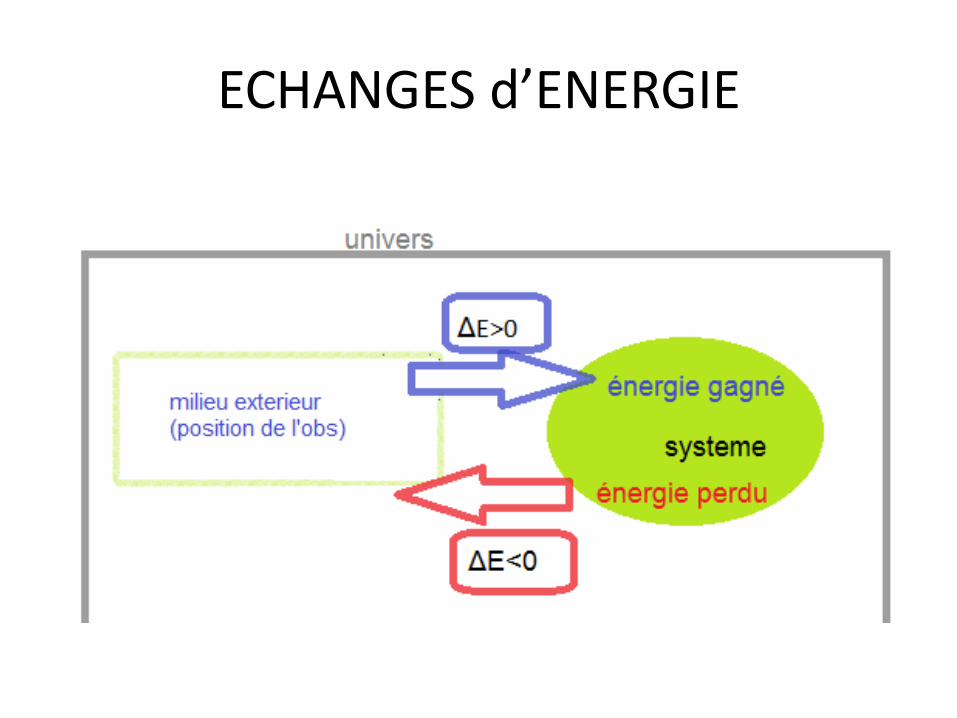
ECHANGES d’ENERGIE

Définir l’état d’un système • Paramètres mesurables :
• - T • - P = variables d'états • - n
• - énergie interne U • - entropie S = fonctions d'états • - enthalpie H • - enthalpie libre G
les fonctions d’états dépendent des variations des variables d’états !
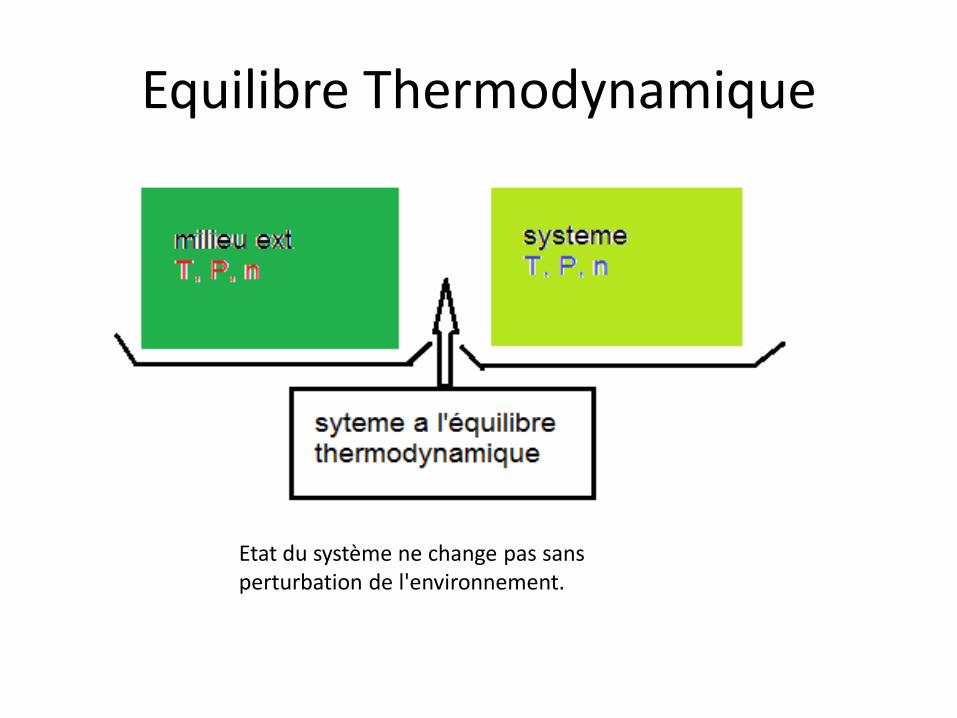
Equilibre Thermodynamique
Etat du système ne change pas sans perturbation de l'environnement.

Processus d’enthalpie
• Cyclique : Etat final est identique a l'état initial • Isotherme : Température constante • Isobare : Pression constante • Isochore : Volume constant • Adiabatique : Système fermé, sans échange de chaleur
avec le milieu extérieur

Lois de Thermodynamique
• 1er principe : conservation de l'énergie globale du système. Esyst = U (énergie interne)
• 2ème principe : au cours d'une transformation spontanée et monotherme, la variation d'entropie S du système est supérieure au terme du à l'échange de chaleur (Q).

1er principe • Esyst = U (énergie interne)
• U = Q + W où W = -P.V est le travail et Q est la chaleur
Transformation spontanée = implique TOUJOURS une réduction de la qualité de l'énergie : la chaleur
• entropie S : PORTION D'ENERGIE NON DISPONIBLE POUR LE TRAVAIL.
• Transformation spontanée réversible :
ΔS = δQrév/T à T° CONSTANTE

2ème principe • Inégalité de Clausius :
dS > δQ/T à T° CONSTANTE
• Au cours d'une transformation spontanée, l'entropie varie : ΔSsyst = ΔSéchange + Scréation
• Puis on l’applique à deux situations : les transformations réversibles et irréversibles.
- Scréation = 0 donc ΔSsyst = ΔSéchange - Echange de désordre avec le milieu ext. - ΔSsyst = δQrev/T
- Scréation >0 - échange de désordre et création d'entropie - ΔSsyst = δQrev/T + Scréation

ENTHALPIE H
• dQ(P) = dU(P) + P.dV • Enthalpie : chaleur échangée par le système qui se
transforme a P CONSTANTE
• dQ(P) = dH et H = U + PV

ENTHALPIE LIBRE G
• À P et à T° CONSTANTES : dG(P,T) = dH - T.dSsyst (P,T) • dG(P,T) = 0
Transformation aboutissant a 1 système a l'équilibre • dG(P,T) < 0
Système exergonique, transformation spontanée
• dG(P,T) > 0 Système endergonique , transformation non spontanée

II. POTENTIEL CHIMIQUE des
espèces PURES

• Pour les solides et liquides (non compressibles)
dG(P) ≈ Go = enthalpie libre de référence
• Pour les gaz dG(P) ≠ Go

Potentiel chimique des gaz Un gaz parfait PUR • µB = δG/δnB = G(T)/nB = Gm(T) L’enthalpie libre Gm est donc égal à l’enthalpie libre G divisé par la quantité de matière de ce gaz. soit µB = G°m(T) + RT ln(P/Po) Un gaz réel PUR • fugacité f : égale a la pression fictive sous laquelle devrait se trouver le
gaz s'il était parfait pour posséder Gm(T). • µ = Gm(T) = G°m(T) + RT ln(f/fo)

III. Cas des MELANGES de gaz

Quelques définitions • Mélange Utilisé pour décrire une phase gazeuse, liquide ou solide contenant + d'1 substance lorsque les substances sont toutes considérées de la même manière. • Solution Utilisé pour décrire une phase liquide ou solide contenant + d’1 substance. Solvant : substance capable de dissoudre une autre substance Soluté : substance dissoute dans une autre substance.

Mélange de gaz parfait
• Un mélange de gaz parfait satisfait : P.V = n.R.T
• Dans ce cas: Pi = (ni/ntot) . Ptot
= yi . Ptot • où yi = fraction molaire de i dans le mélange
gazeux, rapport des quantités de matière. Pi est la pression partielle.
• Pour chaque constituant Pi.V = ni.R.T

UE 3 - CHAPITRE 3 : CHANGEMENT D'ÉTAT DE LA MATIÈRE ET PRESSION DE VAPEUR
1. CHANGEMENT D'ÉTAT ET TRANSITION DE PHASE
2. LA TRANSITION LIQUIDE - GAZ
UE3

1. CHANGEMENT D'ÉTAT ET TRANSITION DE PHASE

A. Généralités 3 états de la matière : liquide – solide – gazeux
L'état de la matière dépend de la pression et de
la température
Une transition de phase est le passage d'un état de la matière a un autre, elle a lieu lorsque les caractéristiques de la matiere changent (P, T)
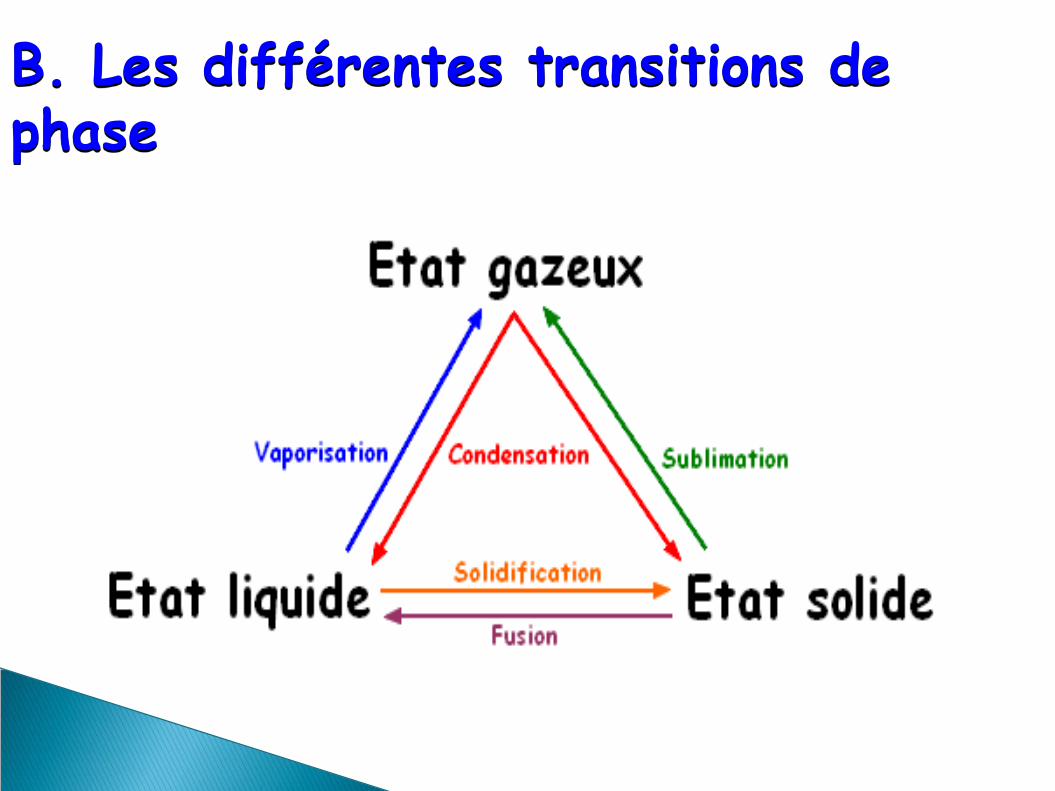
B. Les différentes transitions de phase

C. Classification des transitions de phase
Il existe 2 types de transition de phase : avec ou
sans chaleur latente
Transition de 1er ordre : existence d'une chaleur latente (ex : ébullition de l'eau)
Transition de 2nd ordre : pas de chaleur latente
(ex : transitions ferromagnétiques...)

C. Classification des transitions de phase
Chaleur latente : quantité de chaleur/énergie qui n'est pas dégagée instantanément lors de la
transition de phase mais progressivement.
Ex : chauffée à 100°c, l'eau de la casserole n'est pas instantanément transformée en gaz mais forme un mélange hétérogène d'eau et de bulles de vapeur, jusqu'à évaporation complète
de l'eau.

C. Classification des transitions de phase
La transition de phase d'un corps pur se fait à
température constante et pour une pression donnée, par exemple l'eau bout à 100°C au
niveau de la mer. Ex : en faisant bouillir une casserole d'eau au
niveau de la mer, l'eau va atteindre 100°C puis commencer à se vaporiser. Mais la
température de l'eau n'augmentera pas au delà de 100°C, toute l'énergie apportée par
la chaleur est utilisée pour réaliser la transition de phase : liquide → gaz.

C. Classification des transitions de phase Généralement, à P constante les changements
d'état se font à température constante. Ex : solidification d'un corps pur

D. Équilibre diphasé Définition : système qui possède 2 phases en
équilibre. Ex : solidification

2. LA TRANSITION LIQUIDE - GAZ

A. Enthalpie molaire de vaporisation Lorsqu'on vaporise de l'eau à P constante, on
obtient 2 aspects de la vaporisation : •Aspect visible macroscopique : bulles de vapeur •Aspect invisible : disparition progressive du
liquide : L'enthalpie molaire de vaporisation est l'énergie
nécessaire pour vaporiser 1 mole de liquide à P constant. Son unité est le kJ/mol et elle est notée ΔvapHm.

A. Enthalpie molaire de vaporisation
L'enthalpie molaire de vaporisation sert à diminuer les forces INTERmoléculaires (forces de cohésion) et à augmenter l'agitation moléculaire (agitation thermique).

B. Pression de vapeur : saturante - partielle Pression de vapeur (Pvap) : P de la vapeur d'un
corps présent également sous forme liquide ou solide.
On distingue 2 cas selon que le système soit ouvert (un flacon) ou fermé (un flacon bouché), à température constante et à température variable.
/!\ On parlera de pression de vapeur saturante pour un système fermé, et de pression de vapeur partielle pour un système ouvert !!!
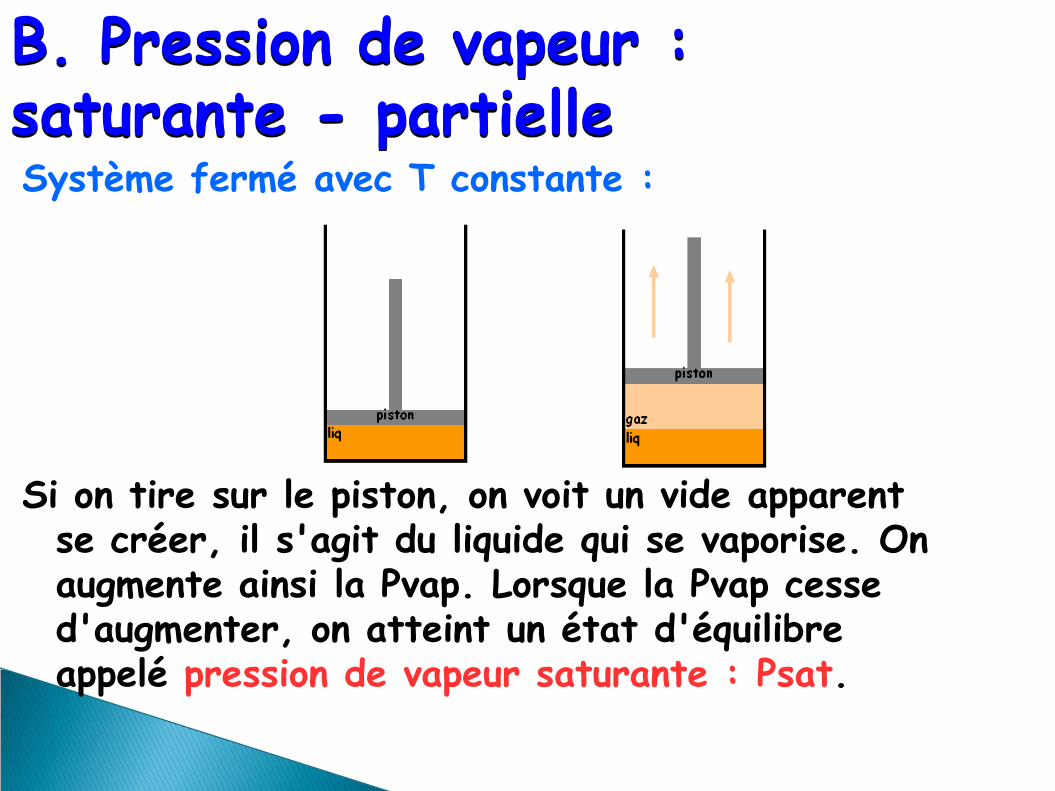
B. Pression de vapeur : saturante - partielle Système fermé avec T constante : Si on tire sur le piston, on voit un vide apparent
se créer, il s'agit du liquide qui se vaporise. On augmente ainsi la Pvap. Lorsque la Pvap cesse d'augmenter, on atteint un état d'équilibre appelé pression de vapeur saturante : Psat.

B. Pression de vapeur : saturante - partielle
Système fermé avec T constante : La pression de vapeur saturante ou tension de
vapeur est la pression à laquelle la phase gazeuse d'une substance est en équilibre avec sa phase liquide ou solide.
Plus la Pvap d'un corps est élevée, plus ce corps
est volatile.

B. Pression de vapeur : saturante - partielle Système fermé avec T variable : Lorsque la température augmente, la Psat
augmente jusqu'à un certain point. La Pvap d'un corps PUR (!!!) peut être calculée
pour n'importe quelle température grâce à la relation de Clausius - Clapeyron :

B. Pression de vapeur : saturante - partielle Système ouvert avec T constante : Les molécules qui se vaporisent diffusent dans l'atm et
n'augmentent pas la Pvap : la P à la surface = Patm. Cette Ptotale ne peut pas atteindre la Pvap, donc l'équilibre décrit dans le cadre d'un système fermé ne peut pas se réaliser, il y a vaporisation complète du liquide selon sa volatilité.

B. Pression de vapeur : saturante - partielle Système ouvert avec T variable :
La T° augmente jusqu'à la T° de vaporisation du liquide. La T° est telle que Psat = Patm. Il en résulte que la vapeur se disperse dans l’atm et la Pvap n’augmente plus le temps de la vaporisation et est constante.

PROPRIÉTÉS COLLIGATIVES DES SOLUTIONS UE 3 Thème 1 Pr Kovacic

DEFINITION
Grandeurs thermodynamiques de solutions ou suspensions qui
dépendent seulement du nombre de molécules et non de l’espèce
chimique

PLAN I- Solutions: définitions et caractéristiques
Composition d’une solution Classification Quantification Thermodynamique
II- Propriétés colligatives
Propriétés et explications Applications

I- SOLUTIONS: DÉFINITIONS ET CARACTÉRISTIQUES Composition Une solution = mélange : solide/liquide liquide/liquide gaz/liquide Dans ce cours: solide/liquide Vocabulaire: véhicule solvant corps dissous soluté Une solution = un mélange homogène au niveau moléculaire de divers composés dans un liquide

I- SOLUTIONS: DÉFINITIONS ET CARACTÉRISTIQUES
Classification Les solutions micromoléculaires: Ne contiennent que quelques 10aines d’atomes 2 catégories: neutres (moléculaires) ex: urée, glucose électrolytiques (ions) ex: NaCl Les solutions macromoléculaires ou colloïdales: Elles contiennent entre 103 et 109 atomes, qui sont sous forme d’agrégats de matière, dispersés de façon stable dans le solvant. L’agitation thermique des molécules de solvant > gravité, donc il n’y a pas de sédimentation, c’est un état homogène

I- SOLUTIONS: DÉFINITIONS ET CARACTÉRISTIQUES

I- SOLUTIONS: DÉFINITIONS ET CARACTÉRISTIQUES

I- SOLUTIONS: DÉFINITIONS ET CARACTÉRISTIQUES !Attention! Le nombre d’entités cinétiques (donc d’osmoles) dépend du caractère neutre ou electrolytique de la solution
Principe : 6 moles de glucose 6 moles de NaCl
1 mol Glc 1 mol de Na+
1 mol de Cl-
6 entités cinétiques 12 entités cinétiques
Le glucose ne se dissocie pas en solution: la solution est neutre. Le NaCl lui se dissocie, c’est une solution électrolytique. La C° osmolaire sera toujours plus élevée pour un électrolyte que pour un soluté neutre à quantité égale de matière. Electrolytes forts: dissolution totale Electrolytes faibles: dissolution partielle

I- SOLUTIONS: DÉFINITIONS ET CARACTÉRISTIQUES
A pur B pur
P°A
P°B PVsol
PVA
PVB
I- SOLUTIONS: DÉFINITIONS ET CARACTÉRISTIQUES
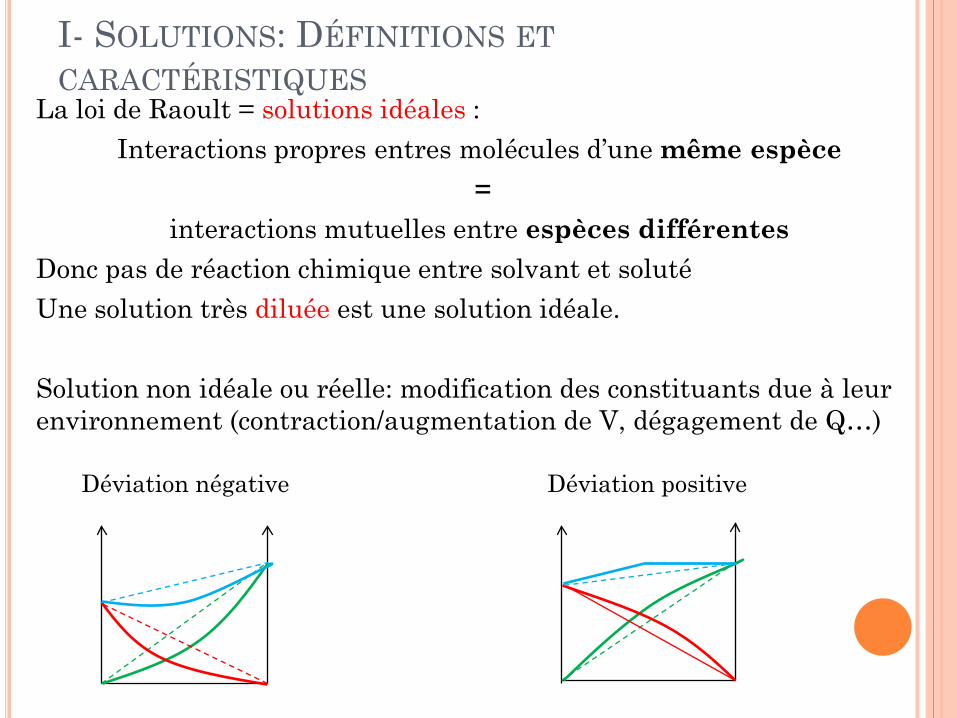
La loi de Raoult = solutions idéales : Interactions propres entres molécules d’une même espèce
= interactions mutuelles entre espèces différentes
Donc pas de réaction chimique entre solvant et soluté Une solution très diluée est une solution idéale. Solution non idéale ou réelle: modification des constituants due à leur environnement (contraction/augmentation de V, dégagement de Q…)
Déviation négative Déviation positive
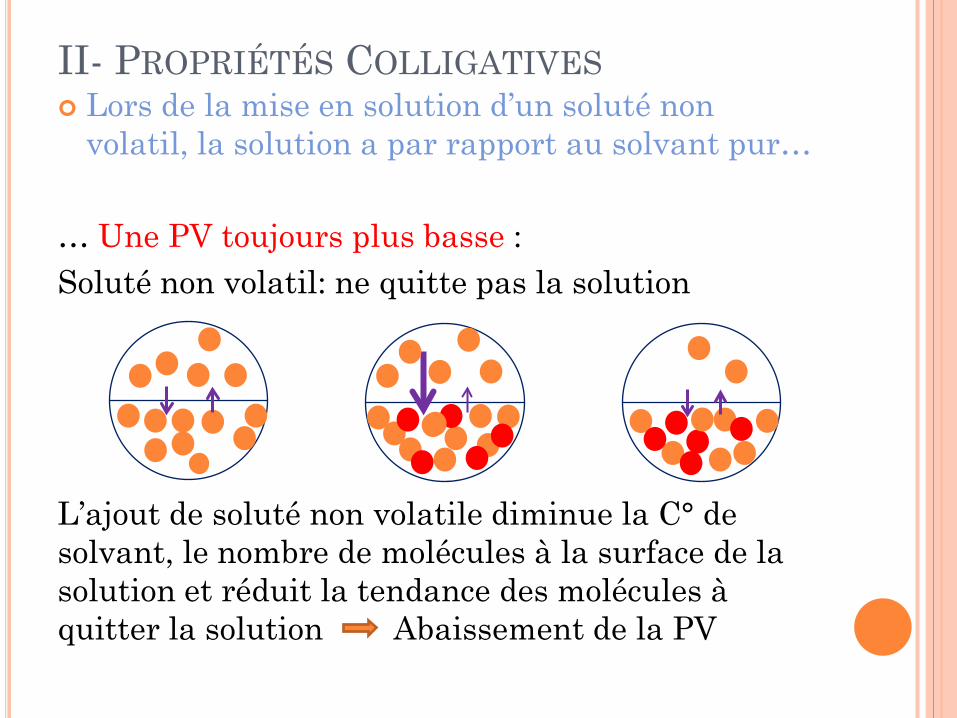
II- PROPRIÉTÉS COLLIGATIVES Lors de la mise en solution d’un soluté non
volatil, la solution a par rapport au solvant pur… … Une PV toujours plus basse : Soluté non volatil: ne quitte pas la solution L’ajout de soluté non volatile diminue la C° de solvant, le nombre de molécules à la surface de la solution et réduit la tendance des molécules à quitter la solution Abaissement de la PV

II- PROPRIÉTÉS COLLIGATIVES Lors de la mise en solution d’un soluté non volatil, la
solution a par rapport au solvant pur…
… Un point d’ébullition toujours plus élevé : Pt d’ébullition = équilibre entre les phases liquide et gazeuse :
taux de condensation = taux d’évaporation Soluté dilue la phase liquide (soluté non volatil : ne quitte pas la solution) taux d’évaporation Donc cet équilibre est atteint pour une température supérieure
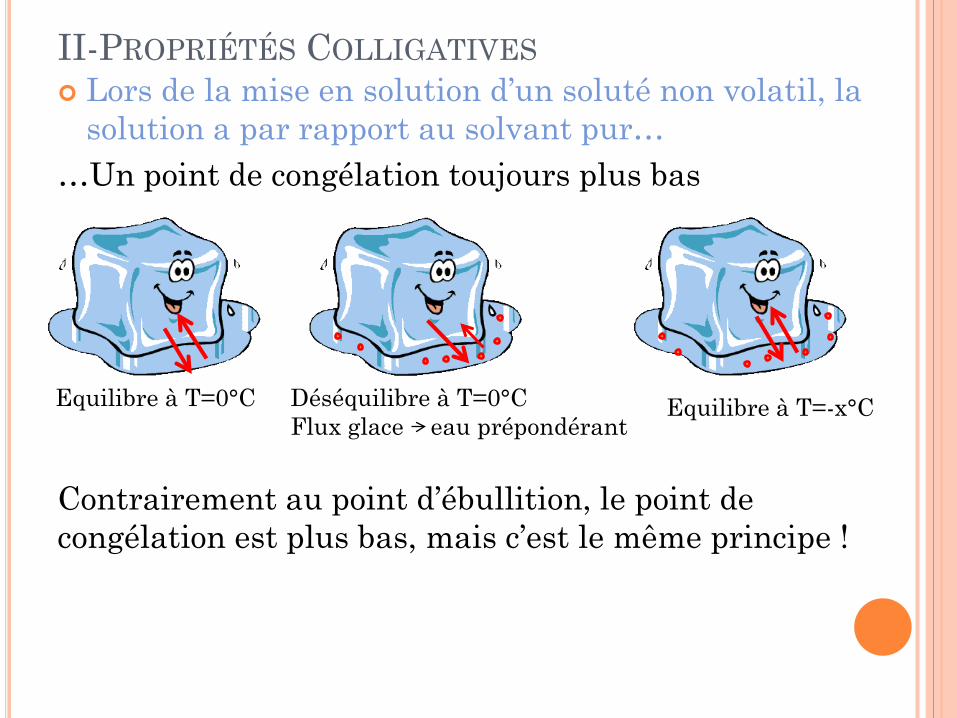
II-PROPRIÉTÉS COLLIGATIVES Lors de la mise en solution d’un soluté non volatil, la
solution a par rapport au solvant pur… …Un point de congélation toujours plus bas Contrairement au point d’ébullition, le point de congélation est plus bas, mais c’est le même principe !
Equilibre à T=0°C Déséquilibre à T=0°C Flux glace eau prépondérant
Equilibre à T=-x°C

II- PROPRIÉTÉS COLLIGATIVES Lors de la mise en solution d’un soluté non volatil, la
solution a par rapport au solvant pur…
…Une pression osmotique Membrane semi-perméable : seul le solvant diffuse C1 plus concentré que C2 le solvant diffuse vers le compartiment le plus concentré: C1. C’est le phénomène d’osmose. La pression osmotique est la pression qu’il faut exercer sur le compartiment le plus concentré pour annuler l’osmose. Si on augmente la pression, le solvant diffuse du milieu le plus concentré vers le moins concentré, c’est le phénomène d’osmose inverse, ou ultrafiltration.
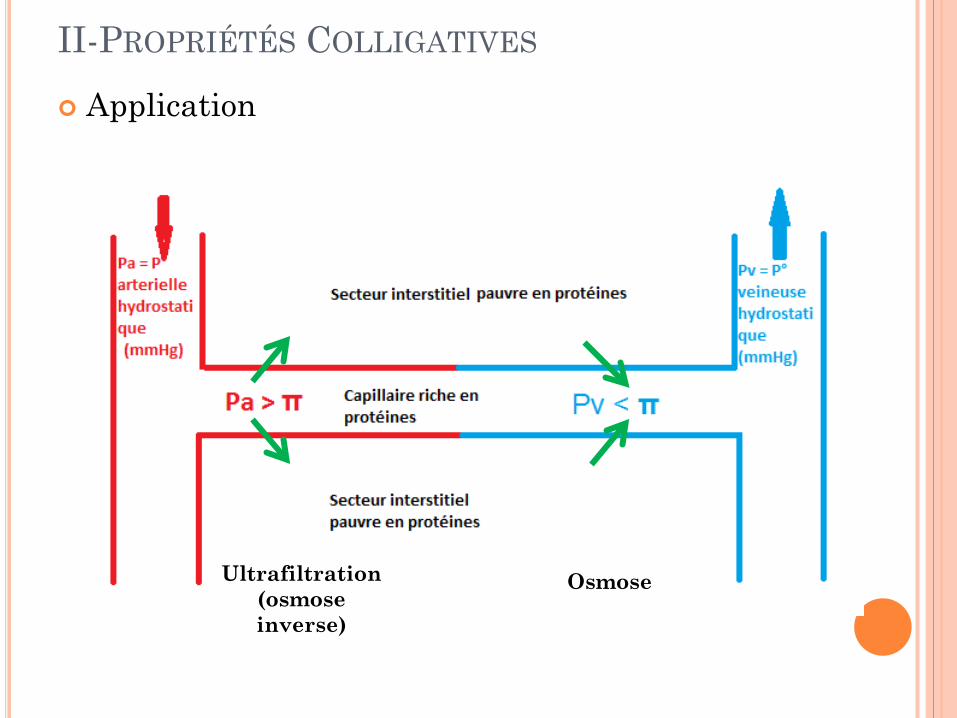
II-PROPRIÉTÉS COLLIGATIVES Application
Ultrafiltration (osmose inverse)
Osmose





























