Embed Size (px)
Citation preview

RAPPORT ANNUEL
2015
Direction européennede la qualité du médicament& soins de santé
European Directoratefor the Quality
of Medicines& HealthCare
Direction européenne de la qualité du médicament
& soins de santé (EDQM)

Direction européenne de la qualité
du médicament & soins de santé (EDQM)
RAPPORT ANNUEL
2015
Conseil de l’Europe, EDQM

Edition anglaise
Annual report 2015
La traduction de cette publication peut être réalisée par des parties externes. Cette traduction est cependant soumise à l’autorisation de l’EDQM, Conseil de l’Europe, avant toute reproduction ou publication sous quelque forme et par quelque moyen que ce soit, tant électronique (CD-Rom, Internet, etc.) que mécanique; ceci comprend la photocopie, l’enregistrement et tout système de stockage ou recherche de l’information.Toute correspondance relative à cette publication est à adresser à l’EDQM (www.edqm.eu/hd).
Direction européenne de la qualité du médicament & soins de santé (EDQM)
7, allée KastnerCS 30026F-67081 Strasbourg – FranceTel.: +33 (0)3 88 41 30 30Fax: +33 (0)3 88 41 27 71www.edqm.eu
Photos© Conseil de l’Europe, Shutterstock, Fotolia
Mise en page et couverture :SPDP, Conseil de l’Europe
Publié par le Conseil de l’Europe F-67075 Strasbourg Cedexwww.coe.int
© Conseil de l’Europe, avril 2016Imprimé dans les ateliers du Conseil de l’Europe

Table des matières ► Page 3
Table des matières
AVANT-PROPOS 5
QUALITÉ ET UTILISATION DES MÉDICAMENTS 7
Pharmacopée Européenne 7
Étalons de référence 11
Certification de conformité aux monographies de la Ph. Eur. 13
Réseau européen des laboratoires officiels de contrôle des médicaments (OMCL) 14
Activités anti-contrefaçon 18
Produits et suivi pharmaceutiques 20
SOINS DE SANTÉ 23
Transfusion sanguine 23
Transplantation d’organes / tissus et cellules destinés à des applications chez l’homme 26
Cosmétiques et emballages alimentaires 28
MANAGEMENT DE LA QUALITÉ 31
COOPÉRATION AVEC LES PARTENAIRES INTERNATIONAUX 33
Coopération avec les autorités nationales 33
Coopération avec l’Union européenne et l’Agence européenne des médicaments 33
Coopération en matiere d’inspections 34
Coopération avec l’Organisation mondiale de la Santé 34
Coopération avec les fabricants et les associations industrielles 34
2015 : UNE ANNÉE RICHE EN MANIFESTATIONS ET RÉUNIONS 35
LISTE DES COMITÉS COORDONNÉS PAR L’EDQM 41
GLOSSAIRE 43


Avant-propos ► Page 5
Avant-propos
par Susanne Keitel, Directrice
L’année 2015 a marqué pour l’EDQM une étape
marquante de son histoire : forte des enseigne-
ments tirés des rencontres auxquelles ont donné
lieu son 50e anniversaire, en 2014, elle a posé les
prémisses d’évolutions majeures, tant de ses activités
que de la façon dont celles-ci sont conduites.
■ La première de ces évolutions concerne l’adoption
par la Commission européenne de Pharmacopée
(« la Commission »), lors de sa 151e Session (en mars),
de la monographie « Comprimés de sitagliptine », la
première dans la Pharmacopée Européenne (la Ph. Eur.)
à porter sur un produit fini contenant une substance
active définie chimiquement. Cette adoption constitue
l’aboutissement d’un processus engagé en 2012,
lorsque la Commission a pour la première fois envisagé
de reconsidérer l’approche suivie jusque-là en matière
d’élaboration de monographies de produits finis,
et décidé de conduire une étude de faisabilité. Ces
normes d’un nouveau type, au niveau européen,
faciliteront l’évaluation des dossiers de demande
d’autorisation de mise sur le marché et constitueront
des outils utiles pour tester les médicaments dans le
cadre des études de surveillance du marché, allégeant
ainsi la tâche tant des autorités réglementaires que
de l’industrie, en Europe et au-delà.
■ La seconde évolution majeure engagée en
2015 concerne les méthodes de travail utilisées
pour élaborer la Ph. Eur. Jusqu’ici, elles reposaient
sur la contribution et le dévouement d’un réseau de
plus de 700 experts en sciences pharmaceutiques,
provenant principalement des Etats signataires de la
Convention relative à l’élaboration d’une Pharmacopée
Européenne, mais aussi d’Etats observateurs auprès
de la Ph. Eur. Cependant, le monde pharmaceutique
a connu des changements profonds au cours des
50 dernières années, qui ont conduit à l’émergence
d’un environnement largement mondialisé. La
Commission a pris acte de cette évolution, et décidé
en 2015 de réviser ses procédures de travail et
d’ouvrir la participation à ses travaux, au sein des
Groupes d’Experts et Groupes de Travail, à des experts
provenant d’Etats non membres et non observateurs.
Cette décision s’inscrit dans une politique délibérée
allant dans le sens d’une plus grande implication
des Etats observateurs, mais aussi des fabricants de
pays non européens dans les travaux de la Ph. Eur., et
elle prendra effet avec l’exercice de nomination des
experts prévu en 2016.
■ En franchissant ces deux étapes, la Commission
a marqué sa détermination à engager la Ph. Eur. dans
une ère nouvelle. Dans le même temps, celle-ci a
continué de faire la preuve de sa capacité à s’adapter
à l’évolution spécifique des produits, des techniques
et des processus réglementaires : 53 nouvelles
monographies ont été adoptées en 2015 (dont 7
portant sur des substances actives encore sous brevet,
élaborées en étroite collaboration avec les autorités
réglementaires et les innovateurs concernés), ainsi
que 8 nouveaux chapitres généraux, et 988 textes
ont fait l’objet de révisions visant à mettre la Ph. Eur.
à jour des évolutions réglementaires et des progrès
scientifiques, portant ainsi à 1014 le nombre total de
textes révisés en 2015.
■ L’impact de la mondialisation s’exerce également,
avec des effets déterminants, sur les autres activités
de l’EDQM. Sa Division Certification a, par exemple,
reçu en 2015 391 nouvelles demandes de certificats
de conformité (les CEP), chiffre en augmentation
significative par rapport aux années précédentes
(10 pour cent de plus qu’en 2014) ; avec près de 1900
demandes, les révisions ont quant à elles progressé de
près de 16 pour cent. Conséquence de la tendance à
la mondialisation de la production de médicaments
génériques, la plupart des nouvelles demandes et
révisions émanant de pays situés en dehors de l’Europe
provenaient d’Inde et de Chine.
■ L’EDQM a accompagné cette tendance, en
poursuivant les efforts qu’elle consacre à diverses
plateformes de collaboration internationale telles
que l’International Generic Drug Regulators Programme
(IGDRP) et le Pharmaceutical Inspection Convention
and Pharmaceutical Inspection Co-operation Scheme
(dénommés conjointement PIC/S).

Rapport annuel ► Page 6
■ L’IGDRP a été institué en 2015, après une phase
pilote de 3 ans engagée en 2012, en relation avec
l’importance grandissante prise dans le monde par
la question de l’accès à des médicaments génériques
sûrs, abordables et de qualité ; cette question est
source d’importantes pressions sur les autorités
réglementaires chargées d’évaluer et d’approuver
ces produits. L’IGDRP a donc été conçu comme un
forum ayant pour vocation de proposer des mesures
et dispositifs concrets pour le partage de l’information
et du travail réglementaire au niveau international.
L’EDQM, qui s’est investie dans l’IGDRP dès ses tout
débuts, a participé à deux de ses réunions en 2015,
l’une en juin à Pretoria (Afrique du Sud) et l’autre en
novembre à Séoul (République de Corée). Lors de
la réunion de Séoul, le groupe de travail de l’IGDRP
« Active Substance Master File (ASMF)/Drug Master
File (DMF) » a fait état, entre autres annonces, de
l’achèvement des projets visant à l’établissement
d’un formulaire de soumission commun, d’un modèle
QAR (Question-Answer Relationship) et d’un lexique
des termes relatifs à la qualité.
■ Les activités de l’EDQM concernant la certification
incluent un dispositif d’inspection, qui met lui aussi
en jeu des collaborations internationales ainsi que
des échanges d’informations et de bonnes pratiques.
En octobre, l’EDQM a ainsi accueilli la 7e réunion du
Cercle d’Experts PIC/S sur les substances actives,
qui a rassemblé 90 délégués de 40 organisations
(autorités participantes et candidates, partenaires
et non-membres du PIC/S) de toute l’Europe, des
Amériques, d’Australie, d’Asie et d’Afrique. L’objectif
était, de façon générale, de renforcer la coopération
internationale et faciliter le partage d’expériences en
matière d’inspections.
■ Le contrôle de la fabrication et de la distribution
des matières premières et des médicaments ne se
limite pas à l’évaluation des dossiers et aux inspections,
et en 2015 l’EDQM a également vu récompensés
par de nouveaux progrès ses efforts en faveur de la
coopération entre autorités, au niveau national et
international, pour combattre la contrefaçon et la
falsification des produits médicaux (médicaments et
dispositifs médicaux, ingrédients compris). L’un des
outils clés de cette lutte est la Convention MEDICRIME,
le premier et seul instrument pénal international
juridiquement contraignant en matière de contrefaçon
et de falsification des produits médicaux. En 2015,
la Convention a été signée par l’Albanie, la Bosnie-
Herzégovine et la Croatie, et ratifiée par la Guinée.
Le nombre de ratifications requises (5) a ainsi été
atteint, et la Convention est entrée en vigueur le
1er janvier 2016.
■ L’EDQM continue de soutenir le développement
de systèmes de sérialisation de masse, outils visant
à empêcher la contamination de la chaîne légale de
distribution des médicaments par des produits falsifiés
ou contrefaits. Suite à l’accord qu’elle a signé en 2015
avec l’EMVO (organisation composée de différents
acteurs européens de la chaîne de distribution),
l’EDQM contribuera directement à la mise en œuvre
des systèmes de traçabilité des médicaments, en
effectuant des évaluations de conformité périodiques
de l’EMVS, le système européen de vérification des
médicaments développé au sein de l’UE par l’EMVO.
■ L’EDQM héberge la base de données Melclass,
qui contient les recommandations que le CD-P-PH/
PHO (Comité d’expert travaillant sur la classification
des médicaments selon leurs conditions de délivrance)
émet annuellement à l’intention des Autorités de
santé. Melclass contient également des informations
sur la classification et les conditions de délivrance en
vigueur, au niveau national, dans les pays participant
à cette activité. En 2015, en plus de sa mise à jour en
continu, la base de données a fait l’objet d’une mise
à niveau et d’une migration vers une application
web adaptative conforme aux technologies les plus
récentes, utilisant une plateforme Python. Cette
nouvelle version de la base de données a été lancée
en janvier 2016.
■ Le projet d’établissement d’un formulaire
pédiatrique européen harmonisé, qui vise à remédier
au manque de médicaments autorisés spécifiquement
conçus pour la population pédiatrique, a également
connu une avancée majeure avec la finalisation, par
le CD-P-PH (Comité européen sur les produits et soins
pharmaceutiques), de lignes directrices et critères
de sélection pour l’élaboration, la réévaluation et
le maintien à jour d’un formulaire de préparations
pharmaceutiques pédiatriques.
■ Enfin, la contribution de l’EDQM aux travaux
du Conseil de l’Europe sur la lutte contre le trafic
d’organes a franchi une étape décisive en mars
2015, avec l’ouverture à la signature d’une nouvelle
Convention contre le trafic d’organes humains, dans le
cadre d’une conférence internationale de haut niveau
qui s’est tenue à Saint-Jacques-de-Compostelle, en
Espagne. La Convention, adoptée le 9 juillet 2014 par
le Comité des Ministres du Conseil de l’Europe, vise à
harmoniser le système pénal en Europe pour permettre
de poursuivre plus efficacement les personnes et
organisations criminelles responsables du trafic.
■ Et bien sûr – comme toujours – il faut reconnaître
que rien de ce qu’a réalisé l’EDQM en 2015 n’aurait
été possible sans le travail remarquable accompli par
les experts – des autorités nationales et européennes,
de l’université, d’instituts scientifiques, de l’industrie –
qui apportent une contribution inestimable à ses
activités grâce à leur expertise dans un large champ
de disciplines scientifiques. A chacun d’eux, ainsi qu’au
personnel dévoué de l’EDQM, j’adresse mes profonds
remerciements.

Qualité et utilisation des médicaments ► Page 7
Qualité et utilisation des médicaments
PHARMACOPÉE EUROPÉENNE
Ses buts, ses moyens
■ La Pharmacopée Européenne (Ph. Eur.) établit des normes qualité pour la fabrication et le contrôle des médicaments, en Europe et au-delà. Ces normes — qui représentaient 2302 monographies et 354 textes généraux fin 2015 — couvrent principalement les excipients et les substances actives, dans leur état initial ou sous forme de préparations pharmaceutiques. Juridiquement contraignantes pour les 38 signataires de la Convention relative à l’élaboration d’une
pharmacopée européenne du Conseil de l’Europe, elles sont rédigées par un vaste panel de groupes d’experts et de groupes de travail (72, actuellement), mis en place par la Commission en réponse aux besoins réglementaires, industriels et techniques du moment. Tous bénévoles, ces experts possèdent des profils variés et sont originaires de nombreux pays, ce qui témoigne de la portée véritablement internationale de la Ph. Eur. et de son rayonnement mondial.
Quelques faits et chiffres
Un vaste rayonnement
■ Trente-sept États membres, ainsi que l’Union européenne, ont signé la Convention relative à
l’élaboration d’une pharmacopée européenne. Depuis l’admission de la République de Corée en 2015, la Commission compte 28 observateurs.
Programme de travail 2015
■ Année après année, la Pharmacopée Européenne s’efforce de communiquer à ses utilisateurs les informations les plus pertinentes et les plus à jour possible, en révisant ses monographies afin d’y intégrer les méthodes et techniques les plus récentes et en élaborant de nouveaux textes sur des produits présentant un intérêt sur le marché. Les réalisations de l’année reflètent bien ces efforts : 53 nouvelles monographies ont été adoptées (dont 7 portant sur des substances actives encore sous brevet, élaborées en étroite collaboration avec les autorités réglementaires et les innovateurs concernés), 8 nouveaux chapitres généraux ont été ajoutés et 988 textes ont été révisés afin de répercuter les

Rapport annuel ► Page 8
évolutions réglementaires et les progrès scientifiques. Ces textes incluent 760 révisions de monographies spécifiques, dans lesquelles le renvoi au chapitre Métaux lourds (2.4.8) a été supprimé en prévision de la mise en application du nouveau Guideline ICH Q3D portant sur les impuretés élémentaires. La 9e Édition de la Ph. Eur. inclura également 26 monographies dont le titre a été révisé afin de ne plus contenir le terme « anhydre » ; une modification rendue nécessaire par la nouvelle politique portant sur les hydrates adoptée par la Commission, en 2014. L’adoption de ces textes porte à 1014 le nombre total de textes révisés en 2015.
■ Lors de sa 151e Session, la Commission a adopté la première monographie de produit fini contenant une substance active définie chimiquement, Comprimés
de Sitagliptine (un antidiabétique largement utilisé). Cette décision engage clairement la Ph. Eur. dans une nouvelle ère.
■ Les travaux d’élaboration de normes dans de nouveaux domaines ou dans des domaines en évolution ont été poursuivis en 2015.
f Le domaine des produits radiopharmaceutiques ne cesse d’évoluer, et l’élaboration de monographies spécifiques portant sur des précurseurs non radioactifs pose de nombreux défis. La durée de vie de ces précurseurs sur le marché peut être relativement courte, car les substances utilisées sont susceptibles de changer rapidement et de devenir obsolètes du fait des évolutions technologiques et scientifiques. Il est donc souvent difficile d’obtenir les informations relatives au contrôle qualité de
ces substances. C’est pourquoi il est apparu nécessaire d’élaborer une monographie générale sur les Précurseurs chimiques pour préparations
radiopharmaceutiques (2902). En adoptant cette monographie générale, la Commission a comblé une lacune dans un domaine en rapide évolution. Ce nouveau texte énonce un ensemble cohérent et complet de critères qualité applicables aux précurseurs chimiques et des recommandations sur d’autres aspects à prendre en considération, et complète la monographie générale Préparations radiopharmaceutiques
(0125), qui a été mise à jour en conséquence. La Ph. Eur. confirme ainsi le rôle moteur qu’elle joue dans ce domaine en termes de normes qualité. La Commission a également adopté un nouveau chapitre portant sur la Préparation
extemporanée de produits radiopharmaceutiques
(5.19), qui décrit des procédures normalisées, à caractère non contraignant, pour la préparation de produits radiopharmaceutiques de qualité appropriée. Ce texte ne remplace pas les législations nationales en vigueur.
f Dans le domaine de thérapies cellulaires et géniques, la Commission a adopté un nouveau chapitre général sur les Matières
premières d’origine biologique utilisées pour la
production de médicaments à base de cellules
et de médicaments de thérapie génique (5.2.12). Ce chapitre, à caractère informatif, vise à aider les acteurs concernés à s’assurer de la bonne qualité des matières premières, et à favoriser l’harmonisation des pratiques et normes mises
ÉtaÉtats membres de la Ph. Eur
et états observateurset états observateurs
ÉtatsÉtats observateurs
ÉtatsÉtats membres
GroupGroupe de Discussion des Pharmacopées

Qualité et utilisation des médicaments ► Page 9
en œuvre pour leur qualification. Il comprend des sections sur différents aspects des matières premières d’origine biologique utilisées pour la production de médicaments à base de cellules et de médicaments de thérapie génique pour usage humain : risques, origine, production et exigences qualité.
f La Commission a, par ailleurs, poursuivi la révision des monographies de vaccins pour usage vétérinaire, afin de les aligner avec les Guidelines 41 et 44 du VICH (Veterinary
International Conference on Harmonization) et de supprimer l’essai d’innocuité chez l’animal cible (TABST). Elle a, tout particulièrement, mis l’accent sur le recours à des essais in vitro pour contrôler les vaccins vétérinaires inactivés, conformément aux principes des 3R (Remplacement, Réduction,
Raffinement, approche visant à protéger les animaux utilisés à des fins expérimentales), en révisant environ 40 monographies spécifiques de vaccins inactivés et la monographie générale Vaccins pour usage vétérinaire (0062).
f Concernant les drogues végétales, la Commission a élaboré et adopté une méthode générale portant sur la Chromatographie sur couche
mince haute performance des drogues végétales
et préparations à base de drogue végétale
(2.8.25), afin d’améliorer la reproductibilité des essais d’identification et de détection de falsifications grâce à une meilleure normalisation de la chromatographie sur couche mince haute performance. Mettant en application ce nouveau texte, plusieurs monographies de drogues végétales et de préparations à base de drogues végétales ont été adoptées, notamment Feuille
de bouleau (1174), Fleur de camomille romaine
(0380), Millepertuis (1438) et Extrait sec quantifié
de millepertuis (1874). Pour aider les utilisateurs, des chromatogrammes d’échantillons reflétant la variabilité naturelle des drogues végétales et des extraits de drogues végétales sont publiés dans la base de données Knowledge.
f La Commission a adopté le chapitre général portant sur les Méthodes chimiométriques
appliquées aux données analytiques (5.21), une introduction à l’utilisation des techniques chimiométriques pour le traitement des données analytiques. Ce chapitre à caractère non contraignant contient des recommandations et des exigences relatives aux bonnes pratiques chimiométriques.
Orientations et politiques générales
Évolutions de l’année 2015
■ Par nature, une pharmacopée n’est jamais terminée et, par nécessité, elle est influencée par
les évolutions, internes comme externes, de son environnement. En 2015, la Commission a donc poursuivi sa réflexion concernant la stratégie de mise en œuvre du Guideline ICH Q3D sur les impuretés élémentaires : plusieurs chapitres généraux et monographies nécessitent en effet d’être révisés.
■ Les travaux de Ph. Eur. ne pourraient pas être menés à bien sans la contribution et le dévouement d’un réseau de plus de 700 experts en sciences pharmaceutiques. Ceux-ci sont en majorité originaires des États membres de la Ph. Eur., mais également de pays observateurs auprès de la Commission. L’implication d’acteurs extérieurs dans le processus d’établissement des normes publiques de la Ph. Eur. est essentielle pour assurer l’élaboration de monographies pertinentes faisant autorité. Afin de mieux refléter la stature internationale de la Ph. Eur. et les bouleversements considérables qu’a connus ces 50 dernières années le secteur pharmaceutique — devenu un marché mondialisé des substances pharmaceutiques et des médicaments —, la Commission a choisi de revoir ses procédures pour ouvrir les travaux de ses groupes à des experts originaires d’États non membres de la Ph. Eur. et non observateurs, désignés sur proposition du Secrétariat. Cette décision témoigne de la volonté d’impliquer davantage les États observateurs et les fabricants non européens dans les travaux de la Ph. Eur. La nouvelle politique prendra effet avec l’exercice de nomination des experts prévu en 2016.
Harmonisation des vocabulaires à l’international
■ Depuis de nombreuses années, l’EDQM participe activement à l’élaboration d’un ensemble de 5 normes ISO destinées à harmoniser l’identification des médicaments du point de vue réglementaire (projet IDMP, Identification of Medicinal Products). Dans le cadre des travaux du GT 6 du Comité technique 215 de l’ISO (Informatique de santé), l’EDQM a piloté le projet ISO 11239, relatif à la préparation de vocabulaires contrôlés pour les formes pharmaceutiques, les voies d’administration, les unités de présentation et le conditionnement. L’ISO 11239 a été publiée comme norme internationale en 2012. L’EDQM a, depuis lors, poursuivi sa participation au GT 6 en pilotant le projet TS 20440, le guide de mise en œuvre de la norme ISO 11239. En 2015, le guide TS 20440 a été approuvé en tant que spécification technique ISO, pour publication début 2016.
■ Grâce à sa participation à l’élaboration de la norme ISO 11239 et du guide TS 20440, ainsi qu’à sa gestion de la base de données Standard terms, l’EDQM s’est largement imposée comme organisation de choix pour la maintenance des vocabulaires contrôlés de l’ISO 11239, qui constituent un élément essentiel du projet IDMP.

Rapport annuel ► Page 10
Animal Testing). Sur la base des résultats positifs obtenus lors de l’étude, un protocole optimisé a été développé pour le dosage sur cellules, pour les besoins d’une étude de suivi à participation élargie ; une nouvelle étude collaborative débutera en 2016.
Harmonisation internationale et Groupe de Discussion des Pharmacopées
■ La Ph. Eur. a poursuivi ses efforts pour réduire la duplication des essais au cours du développement et du contrôle qualité des médicaments, en participant aux travaux du Groupe de Discussion des Pharmacopées (GDP) — dont sont membres la Ph. Eur., la Pharmacopée japonaise (JP) et la Pharmacopée des États-Unis (USP) et qui compte l’Organisation mondiale de la Santé (OMS) comme observateur. Deux réunions ont eu lieu en 2015 : l’une a été accueillie par la JP à Tokyo (Japon) en juillet et l’autre, par l’USP à Rockville (États-Unis) en novembre.
■ À ce jour, 48 des 62 monographies d’excipients
et 29 des 36 chapitres généraux inscrits au programme de travail du GDP ont été harmonisés. Trois nouvelles monographies d’excipients et la révision de la monographie Povidone, ainsi qu’un nouveau chapitre général et la révision d’un autre chapitre général (Uniformité de masse/teneur) ont été signés pendant les réunions tenues en 2015. Le GDP a, en outre, procédé à l’examen approfondi de plusieurs autres points inscrits au programme de travail pour résoudre les questions en suspens et progresser vers la signature. Une synthèse des temps forts de chaque réunion du GDP est disponible sur les sites internet des trois pharmacopées.
■ Suite à l’adoption du Guideline ICH Q3D sur les impuretés élémentaires, les membres du GDP ont confirmé leur volonté d’harmoniser le chapitre général sur les procédures de contrôle des impuretés élémentaires. Un projet « étape 3 » a été envoyé par la pharmacopée coordinatrice et commenté par les deux autres1.
■ Le chapitre Chromatographie constitue un autre point clé du programme de travail du GDP. La pharmacopée coordinatrice en a soumis un projet « étape 3 » révisé sur la base des décisions prises lors d’une téléconférence entre experts des trois régions. Les commentaires seront étudiés avec les experts de la pharmacopée coordinatrice, dans l’objectif de présenter un projet « étape 4 » pour enquête publique.
1. Dans le cadre de l’étape 4 du processus du GDP, un document harmonisé — accompagné d’une discussion des commentaires reçus de la part des comités d’experts concernés au sujet du document précédent — est préparé, avant publication pour enquête publique sur le forum de chaque pharmacopée.
Termes normalisés
■ Initialement élaborées à la demande de la Commission européenne (UE), pour les demandes d’autorisations de mise sur le marché (AMM), les listes de termes normalisés (standard terms) fournissent aux utilisateurs et aux prescripteurs des vocabulaires harmonisés pour décrire les formes pharmaceutiques, les voies d’administration et les emballages des médicaments. Après la refonte complète et la remise en ligne de la base de données Standard terms fin 2014, conformément à la norme ISO 11239 (projet IDMP), la nouvelle version de la base de données — désormais accessible gratuitement — a attiré près de 10 000 utilisateurs enregistrés en 12 mois. Fin 2015, elle contenait 870 termes et définitions, avec plus de 23 000 traductions dans 33 langues.
Programme de standardisation biologique
■ Le Programme de standardisation biologique (PSB), initiative conjointe de l’EDQM et de la Commission européenne (UE), vise à établir des matériels de référence ainsi qu’à développer et valider de nouvelles méthodes d’analyse, notamment des méthodes alternatives fondées sur le concept des 3R, pour le contrôle qualité des produits biologiques.
■ En 2015, 26 projets ont été menés dans différents domaines, des vaccins pour usage humain et vétérinaire aux produits dérivés du plasma, en passant par les produits biotechnologiques. Ils ont abouti à l’établissement de cinq nouveaux étalons de référence (voir section « Étalons de référence », page 12).
■ Huit projets portaient sur l’établissement de lots de remplacement d’étalons de référence existants, tous motivés par des baisses de stock (aucune interruption de l’utilisation d’un étalon n’a été nécessaire pour des problèmes de qualité). Cinq projets concernaient l’établissement d’étalons de référence en rapport avec de nouvelles monographies ou avec de nouvelles exigences dans des monographies existantes.
■ Huit projets avaient pour objectif le développement de nouvelles méthodes de pharmacopée, dont six concernant l’application du concept des 3R dans le domaine du contrôle qualité des produits biologiques. Les efforts sans cesse déployés dans le cadre du PSB pour élaborer, valider et mettre en application des méthodes d’analyse conformes aux 3R sont largement reconnus. Les résultats du projet BSP130, portant sur le remplacement des essais sur animaux pour la détermination de la dose létale minimale (Minimal Lethal Dose, MLD) et le test d’antigénicité (Total Combining Power, TCP) sur souris prescrits par la Ph. Eur. pour le vaccin de Clostridium septicum pour usage vétérinaire, ont été présentés lors d’un atelier coorganisé par l’EDQM et par l’EPAA (European Partnership for Alternative Approaches to

Qualité et utilisation des médicaments ► Page 11
Autres initiatives en matière d’harmonisation
■ La Ph. Eur. participe aussi activement à d’autres initiatives internationales en matière d’harmonisation, comme celle de l’OMS sur l’élaboration de « Bonnes pratiques de pharmacopée » (BPPh) qui pourraient servir de base à des collaborations et partages de travaux entre les pharmacopées du monde entier.
■ Ces travaux sont conduits dans le cadre de l’Assemblée mondiale des Pharmacopées, organisée sous l’égide de l’OMS, où sont discutés les moyens de renforcer la collaboration et l’harmonisation interpharmacopées, par exemple à travers l’élaboration des BPPh. Deux réunions ont eu lieu en 2015 : l’une en avril à Washington (États-Unis) et l’autre en septembre à Suzhou (République populaire de Chine). Le texte principal des BPPh a été finalisé, approuvé par le Comité d’Experts des spécifications relatives aux préparations pharmaceutiques (ECSPP) de l’OMS, lors de sa 50e réunion, et a reçu l’aval de toutes les pharmacopées participantes.
Coopération avec les autorités nationales de pharmacopée
■ L’EDQM organise annuellement une réunion des Secrétaires des autorités nationales de pharmacopée (ANP) des États membres de la Ph. Eur. pour faciliter et coordonner les activités d’intérêt commun et permettre des échanges informels d’informations. En 2015, la réunion annuelle des ANP s’est déroulée en juin à Utrecht, aux Pays-Bas, à l’invitation du RIVM. Vingt-cinq des 37 États membres y étaient représentés ; ils y ont discuté du calendrier de publication de la 9e Édition et ont commencé à préparer l’exercice de renomination de l’ensemble des experts de la Ph. Eur., qui doit avoir lieu en 2016.
Publications, bases de données et site internet
■ La 8e Édition de la Ph. Eur., avec les mises à jour de 2015 (8.6, 8.7, 8.8), contient 2302 monographies — dont des monographies générales s’appliquant à des groupes d’ingrédients ou des formes pharmaceutiques — et 354 textes généraux décrivant notamment des méthodes d’analyse.
■ Site en accès libre sur lequel les textes de la Ph. Eur. sont publiés pour consultation, Pharmeuropa en ligne
est un forum facilement et largement accessible, qui vise à optimiser les interactions entre la Commission et les acteurs concernés, à permettre aux utilisateurs de commenter les projets et à rendre ceux-ci accessibles aux acteurs concernés du monde entier. Les textes sont publiés en continu, mais le principe des dates limites d’envoi des commentaires (4 par an) a été maintenu, tout comme les voies et procédures de transmission de ces commentaires. En 2015, 157 projets de textes ont été publiés dans Pharmeuropa en ligne, consulté depuis plus de 150 pays par près de 22 000 utilisateurs inscrits au 1er janvier 2016.
ÉTALONS DE RÉFÉRENCE
Les étalons de référence et leur utilité
Étalons de référence de la Ph. Eur.
■ Les étalons de référence officiels (ER) sont une composante indispensable de l’application de la plupart des textes de la Ph. Eur. Ils comprennent les substances chimiques de référence (SCR), les étalons de référence végétaux (ERV), les préparations biologiques de référence (PBR), les réactifs biologiques de référence (RBR) et les spectres de référence. Établis par l’EDQM, les ER de la Ph. Eur. sont officiellement adoptés par la Commission. Eux seuls font autorité en cas d’arbitrage.
Responsabilité des étalons de référence de l’OMS
■ L’EDQM est aussi responsable de l’établissement, de la surveillance et de la distribution des étalons internationaux d’antibiotiques (ISA) et des substances chimiques de référence internationale (SCRI) de l’OMS. Les ISA sont distribués dans le monde entier pour la réalisation de titrages microbiologiques dans le cadre du contrôle qualité des antibiotiques, et jouent un rôle essentiel pour la standardisation et le contrôle qualité des antibiotiques (médicaments et substances). Les SCRI sont prescrites dans la Pharmacopée Internationale, publiée et tenue à jour par l’OMS et utilisée dans le monde entier.
■ L’EDQM participe également à l’ECSPP de l’OMS, au sein duquel elle contribue à l’élaboration de normes et de lignes directrices visant à promouvoir l’assurance et le contrôle qualité.
Rapport annuel ► Page 12
Quelques faits et chiffres
Les étalons de référence de la Pharmacopée Européenne
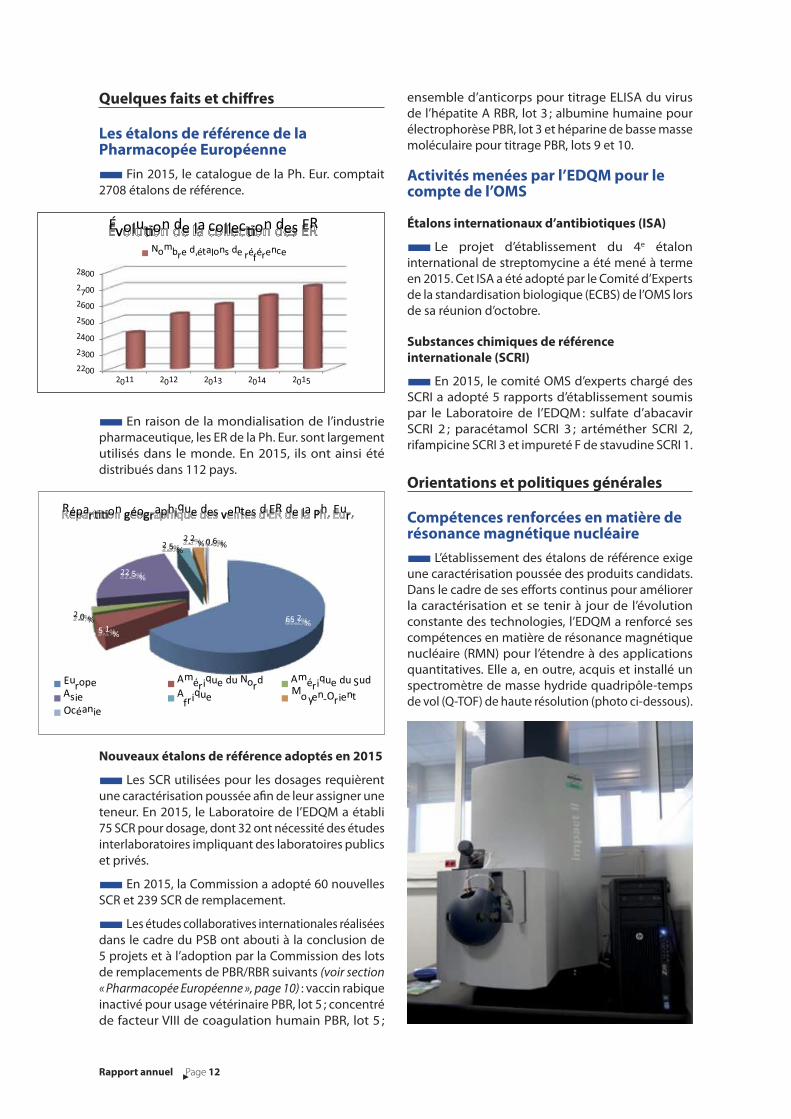
■ Fin 2015, le catalogue de la Ph. Eur. comptait
2708 étalons de référence.
■ En raison de la mondialisation de l’industrie
pharmaceutique, les ER de la Ph. Eur. sont largement
utilisés dans le monde. En 2015, ils ont ainsi été
distribués dans 112 pays.
Nouveaux étalons de référence adoptés en 2015
■ Les SCR utilisées pour les dosages requièrent
une caractérisation poussée afin de leur assigner une
teneur. En 2015, le Laboratoire de l’EDQM a établi
75 SCR pour dosage, dont 32 ont nécessité des études
interlaboratoires impliquant des laboratoires publics
et privés.
■ En 2015, la Commission a adopté 60 nouvelles
SCR et 239 SCR de remplacement.
■ Les études collaboratives internationales réalisées
dans le cadre du PSB ont abouti à la conclusion de
5 projets et à l’adoption par la Commission des lots
de remplacements de PBR/RBR suivants (voir section
« Pharmacopée Européenne », page 10) : vaccin rabique
inactivé pour usage vétérinaire PBR, lot 5 ; concentré
de facteur VIII de coagulation humain PBR, lot 5 ;
ensemble d’anticorps pour titrage ELISA du virus
de l’hépatite A RBR, lot 3 ; albumine humaine pour
électrophorèse PBR, lot 3 et héparine de basse masse
moléculaire pour titrage PBR, lots 9 et 10.
Activités menées par l’EDQM pour le compte de l’OMS
Étalons internationaux d’antibiotiques (ISA)
■ Le projet d’établissement du 4e étalon
international de streptomycine a été mené à terme
en 2015. Cet ISA a été adopté par le Comité d’Experts
de la standardisation biologique (ECBS) de l’OMS lors
de sa réunion d’octobre.
Substances chimiques de référence internationale (SCRI)
■ En 2015, le comité OMS d’experts chargé des
SCRI a adopté 5 rapports d’établissement soumis
par le Laboratoire de l’EDQM : sulfate d’abacavir
SCRI 2 ; paracétamol SCRI 3 ; artéméther SCRI 2,
rifampicine SCRI 3 et impureté F de stavudine SCRI 1.
Orientations et politiques générales
Compétences renforcées en matière de résonance magnétique nucléaire
■ L’établissement des étalons de référence exige
une caractérisation poussée des produits candidats.
Dans le cadre de ses efforts continus pour améliorer
la caractérisation et se tenir à jour de l’évolution
constante des technologies, l’EDQM a renforcé ses
compétences en matière de résonance magnétique
nucléaire (RMN) pour l’étendre à des applications
quantitatives. Elle a, en outre, acquis et installé un
spectromètre de masse hydride quadripôle-temps
de vol (Q-TOF) de haute résolution (photo ci-dessous).
2200
2300
2400
2500
2600
2700
2800
2011 2012 2013 2014 2015
Évolu0on de la collec0on des ER
Nombre d'étalons de référence
65.2%
5.1%
2.0%
22.5%
2.5%2.2% 0.6%
Europe Amérique du Nord Amérique du Sud
Asie Afrique Moyen-‐Orient
Océanie
RéparBBon géographique des ventes d'ER de la Ph. Eur.

Qualité et utilisation des médicaments ► Page 13
Collaboration avec les laboratoires nationaux
■ Certains ER (généralement destinés aux dosages
ou aux titrages d’activité) sont établis par la voie
d’études collaboratives faisant intervenir plusieurs
laboratoires. La collaboration continue avec les
laboratoires nationaux et les centres d’excellence
est fondamentale pour la réalisation de ces études,
auxquelles contribuent les OMCL : fin 2015, 37 de ces
laboratoires, de 26 pays différents, participaient aux
études de Laboratoire de l’EDQM.
Publications, bases de données et site internet
■ En 2015, l’EDQM a terminé la publication des
notices d’information relatives aux 2708 ER de la
Ph. Eur. Ces notices fournissent aux utilisateurs des
renseignements importants tels que les référence
des monographies concernées, des informations
scientifiques, les conditions de conservation et, s’il
y a lieu, des pictogrammes et mentions de danger.
■ Par ailleurs, 57 000 Fiches de données de sécurité
pour tous les étalons de référence de la Ph. Eur. et
de l’OMS, avec leur traduction dans 24 langues
européennes (en fonction des besoins), sont
directement accessibles sur le site de l’EDQM.
CERTIFICATION DE CONFORMITÉ AUX MONOGRAPHIES DE LA PH. EUR.
La Certification, plus que jamais d’actualité
■ Dans une économie mondiale en constante
évolution, la production de substances
pharmaceutiques à l’extérieur de l’Europe est de plus
en plus répandue. Cette situation place les autorités
nationales des pays européens face à de nouveaux
défis en termes de surveillance et de contrôle
qualité des substances utilisées pour fabriquer des
médicaments.
■ La procédure de Certification aux monographies
de la Ph. Eur. a été mise en place par l’EDQM pour
évaluer et valider la capacité des monographies de la
Ph. Eur. à contrôler la qualité des substances utilisées
pour la production des médicaments. Pour demander
un certificat de conformité (CEP), les fabricants
doivent présenter un dossier décrivant le procédé
de fabrication et le processus de contrôle qualité
de leur produit, et la décision de l’EDQM de délivrer
on non un CEP repose sur l’évaluation des données
figurant dans ce dossier. Cette procédure permet de
centraliser l’évaluation des données, au bénéfice des
autorités réglementaires et de l’industrie, et contribue
à la mise à jour des monographies de la Ph. Eur.
■ En parallèle, l’EDQM mène des inspections
de sites de fabrication et/ou de distribution des
substances actives couvertes par des CEP, afin de
s’assurer que les bonnes pratiques de fabrication
(BFP) y sont respectées et que les informations
communiquées dans le cadre de la procédure de
Certification sont exactes.
■ De plus en plus d’autorités d’enregistrement,
dans le monde entier, acceptent les CEP comme
éléments des dossiers d’AMM pouvant remplacer
tout ou partie des données relatives à la qualité des
substances actives qui entrent dans la composition
des médicaments.
Quelques faits et chiffres
■ En 2015, 391 nouveaux dossiers ont été reçus,
ce qui représente une forte hausse par rapport
aux années précédentes. Par ailleurs, le nombre de
demandes de révision a augmenté de quelque 16 pour
cent (soit près de 1900 demandes).
■ 291 nouveaux certificats et 1565 certificats
révisés ont été délivrés au cours de l’année.

Rapport annuel ► Page 14
■ La Division Certification de l’EDQM s’appuie sur un réseau d’une centaine d’assesseurs de 24 autorités compétentes, qui évaluent les dossiers soumis. Fin 2015, le nombre de CEP en cours de validité, qu’ils couvrent la pureté chimique, le risque de transmission d’encéphalopathies spongiformes ou les drogues végétales, s’élevait à plus de 4200. Globalement, plus de 90 pour cent des dossiers reçus ont été traités dans les délais officiellement établis.
■ Des inspections ont été conduites sur 38 sites de fabrication, principalement en Asie, avec la participation d’inspecteurs appartenant aux corps d’inspection nationaux. Par ailleurs, 42 autres sites ont fait l’objet d’une évaluation de conformité aux BPF sur la base d’informations obtenues dans le cadre d’échanges de données avec les corps d’inspection des États membres et d’autres partenaires internationaux. Des mesures concernant les CEP ont ensuite été prises, lorsque nécessaire. Le taux de non-conformité des sites inspectés par l’EDQM a atteint 18 pour cent en 2015. Le programme d’inspection est une composante essentielle du système de certification, et environ 60 pour cent des sites asiatiques fabriquant des substances couvertes par des CEP ont été inspectés dans ce cadre.
Orientations et politiques générales
■ La Division Certification de l’EDQM a publié en 2015 un certain nombre de « guidelines » et de « policies » pour aider les candidats à préparer leur dossier CEP ou à communiquer avec l’EDQM.
■ En 2015, le Comité directeur s’est réuni une fois, le Comité technique consultatif pour l’évaluation de la pureté chimique, trois fois et le Comité technique consultatif pour le risque de transmission d’encéphalopathies spongiformes, une fois. Ces réunions sont l’occasion de traiter du champ d’application de la procédure de Certification et des questions techniques connexes.
Communication avec les partenaires et acteurs concernés
■ En 2015, l’EDQM a poursuivi sa participation à diverses plateformes de collaboration internationale, notamment l’IGDRP (voir section « 2015 : une année
riche en manifestations et réunions », page 37), le Groupe de Travail de l’Union européenne sur les ASMF (Active Substance Master File Procedures), le programme international d’inspection des sites de production de substances actives, le PIC/S et le Groupe de Travail sur la mise en œuvre de l’ICH Q11.
■ En octobre 2015, l’EDQM a signé un accord de confidentialité avec le Medicines Control Council (MCC)
d’Afrique du Sud, pour permettre aux deux institutions d’échanger des informations confidentielles relatives à la qualité des substances actives.
■ La collaboration de l’EDQM avec la TGA (Therapeutic Goods Administration) australienne et la Food and Drug Administration américaine (USFDA) a donné lieu en 2015 à 3 inspections conjointes.
■ La procédure de Certification est également à l’ordre du jour des rencontres annuelles que l’EDQM organise avec les associations industrielles, et qui permettent d’échanger sur les travaux de l’EDQM en général, mais aussi de recueillir le retour des acteurs concernés sur l’utilisation des CEP.
RÉSEAU EUROPÉEN DES
LABORATOIRES OFFICIELS
DE CONTRÔLE DES
MÉDICAMENTS (OMCL)
Pourquoi un Réseau européen ?
■ La raison d’être du Réseau OMCL est d’éviter que des médicaments de mauvaise qualité parviennent aux patients et compromettent l’efficacité de leurs traitements. A cette fin, il apporte son soutien aux autorités chargées de la surveillance du marché, dans le cadre du contrôle qualité des médicaments pour usage humain et vétérinaire. Il a ainsi poursuivi en 2015.
■ Indépendants des fabricants et, par conséquent, libres de tout conflit d’intérêt, 70 OMCL de 41 États membres participent au Réseau, que finance en partie l’Union européenne.
■ Cette collaboration paneuropéenne présente plusieurs avantages : mise en commun de savoir-faire au sein d’une communauté d’experts, accès à des technologies et procédures analytiques sélectives de pointe, partage des travaux, reconnaissance mutuelle des résultats expérimentaux obtenus sur la base de procédures et de guidelines approuvés collectivement. Elle permet également aux autorités nationales compétentes d’éviter la duplication des travaux, et ainsi d’optimiser les ressources et le temps consacrés au contrôle des médicaments.
Programme de management de la qualité
■ En 2015, les efforts visant à élaborer une approche commune harmonisée pour la mise en œuvre, le maintien, l’évaluation et l’amélioration des systèmes de management de la qualité (MQ) des OMCL ont été poursuivis dans l’ensemble du Réseau.

Qualité et utilisation des médicaments ► Page 15
Audits mutuels conjoints/visites mutuelles conjointes (AMC/VMC)
■ Conçus pour évaluer la conformité des systèmes de MQ des OMCL aux exigences de la norme ISO/CEI 17025 ainsi qu’aux guidelines MQ du Réseau général des OMCL et à la Ph. Eur., 12 AMC ont été réalisés sur des sites d’OMCL en 2015. Depuis le lancement du programme de MQ en décembre 1997, 130 AMC, 50 VMC, 2 tutoriels et 19 visites de formation ont eu lieu au sein du Réseau OMCL.
Les guidelines MQ du Réseau OMCL
■ Les nouveaux guidelines relatifs au MQ rédigés par des experts du Réseau OMCL sont conçus pour aider les laboratoires à se conformer aux exigences de la norme ISO/CEI 17025. En 2015, un guideline intitulé « Étalonnage/qualification des pH-mètres » a été adopté par le Réseau et un autre intitulé « Évaluation et compte rendu des résultats » a été révisé.
Collaboration avec l’European Co-operation for Accreditation (EA)
■ Dans l’optique d’une éventuelle collaboration portant sur l’échange de savoir-faire, sur la réalisation d’audits conjoints entre organismes nationaux d’accréditation et auditeurs EDQM/AMC, et sur la participation mutuelle aux réunions en qualité d’observateurs, l’EDQM s’est rapprochée de l’EA. Trois audits conjoints ont ainsi été réalisés avec les organismes nationaux d’accréditation en 2015.
Formations/ateliers
■ Un atelier destiné aux auditeurs AMC expérimentés a été organisé en vue d’un partage d’expériences et d’une harmonisation des approches sur des sujets spécifiques.
Programme d’essais d’aptitude
■ Les essais d’aptitude (PTS, Proficiency Testing
Studies) organisés par l’EDQM offrent aux laboratoires un moyen objectif d’évaluer et de démontrer la fiabilité de leurs données.
■ En 2015, 5 études ont été organisées dans le domaine physicochimique, avec la participation moyenne de 56 OMCL et 44 autres laboratoires de contrôle pharmaceutique relevant du secteur privé (y compris l’industrie), d’établissements hospitaliers et de pharmacies : PTS156 — perte à la dessiccation ; PTS157 — détermination potentiométrique du pH ; PTS158 — spectrophotométrie d’absorption dans l’infrarouge ; PTS159 — essai de dissolution et PTS160 — chromatographie liquide, dosage.
■ Dans le domaine biologique, 4 études ont été organisées, avec la participation moyenne de 20 laboratoires : PTS161 — essai d’amplification des
acides nucléiques (NAT) du virus de l’hépatite C ; PTS162 — NAT du parvovirus B19 ; PTS163 — héparines de basse masse moléculaire, méthodes chromogéniques (activités anti-facteur IIa et anti-facteur Xa) ; PTS164 — Colle-fibrine, activité du fibrinogène et de la thrombine.
Activités du réseau général des OMCL (GEON)
Études générales de surveillance du marché
■ Les études de surveillance du marché (MSS) apportent une vision d’ensemble de la qualité des médicaments présents sur le marché européen, selon la classe thérapeutique.
■ En 2015, 2 MSS ont été finalisées : MSS044 sur l’héparine et l’héparine de faible masse moléculaire
(substances actives et produits finis) et MSS045 sur les collyres et préparations nasales enregistrés comme
dispositifs médicaux (première MSS portant sur des dispositifs médicaux au sein du Réseau).
■ De plus, la phase d’essais lancée en 2014 pour deux MSS, sur le telmisartan (comprimés et substance
active) (MSS046) et sur le pramipexole (comprimés et
substance active) (MSS047), a été finalisée et deux nouvelles études sur la sécabilité des comprimés (MSS048) et sur l’irbésartan (comprimés et substance
active) (MSS049) ont débuté.
Groupe de Travail sur les substances actives
■ La mondialisation de la fabrication et du commerce des substances actives impose un contrôle accru de leur qualité. La mise en œuvre dans les pays de l’UE, en 2013, de la Directive européenne 2011/62/UE sur les médicaments falsifiés a été à cet égard une étape importante. Pour répondre à la menace posée par la contrefaçon et soutenir la convention MEDICRIME, l’une des recommandations stratégiques adressées au Réseau était l’implication des OMCL dans la surveillance des substances actives sur le marché européen.
■ La 8e réunion du Groupe de Travail sur les substances actives s’est tenue en mai 2015. Les discussions ont porté sur les nouvelles MSS « fingerprint » et sur la bonne utilisation au sein du Réseau des échantillons de substances actives disponibles pour les programmes de contrôle nationaux. Dans ce contexte, l’importance de la base de données des essais réalisés sur des substances actives comme outil de planification des campagnes de contrôle et de partage des résultats d’analyse a été soulignée. Un autre sujet abordé par le groupe concernait le problème de l’échantillonnage aléatoire

Rapport annuel ► Page 16
des substances actives ; il a été convenu que des stratégies améliorées devaient être élaborées. Deux nouvelles études « fingerprint » sur l’oméprazole et la morphine ont été lancées plus tard dans l’année.
Groupe de Travail sur les médicaments contrefaits/illégaux
■ Le Groupe de Travail sur les médicaments contrefaits/illégaux s’est réuni à deux reprises en 2015. L’un des objectifs de ces réunions était d’analyser les résultats des dernières MSS portant sur des produits illégaux (MSSIP) et de discuter des dates des nouvelles études (voir aussi « Études générales de surveillance du marché »). Pour la prochaine MSSIP, une étude collaborative sur les substances actives non déclarées dans les cosmétiques a débuté.
■ Deux formations techniques à l’intention des OMCL ont été organisées conjointement par l’EDQM et les OMCL français (Montpellier, mars 2015) et suisse (Berne, novembre 2015). La formation de novembre a mis l’accent sur le contrôle des produits biologiques falsifiés.
■ Le Groupe de Travail a également réfléchi à la façon d’améliorer la convivialité de la base de données Know-X, lancée en mars 2014. Toutes les modifications apportées à la base de données ont été finalisées à la fin de l’année 2015 et le nombre de dossiers d’affaires individuelles téléchargés par des OMCL dans Know-X s’élevait à 2500 en janvier 2016.
Groupe de Travail sur les produits de thérapie génique (GTP)
■ Le Groupe de Travail GTP des OMCL a été créé en 2008 pour favoriser la collaboration entre les OMCL travaillant dans le domaine des thérapies géniques, afin de réaliser des économies de temps et de ressources grâce à un partage des connaissances et technologies. Actuellement, 11 OMCL sont membres actifs de ce Groupe de Travail.
■ En 2015, des méthodes normalisées pour la détermination des génomes viraux et infectieux dans les produits AAV (vecteurs dérivés de virus adéno-associés) ont été validées. La 7e réunion annuelle du Groupe de Travail a été organisée en décembre par l’OMCL autrichien (AGES, Vienne). Le programme de travail a été examiné et révisé pour inclure de nouveaux vecteurs (Herpes simplex et vecteurs rétroviraux) au vu des récents développements dans le domaine et suite à l’avis favorable de l’Agence européenne des médicaments (EMA) concernant l’AMM de l’Imlygic® (virus oncolytique préparé à partir du virus Herpes
simplex pour le traitement des mélanomes).
CombiStats™
■ CombiStats™ est un programme informatique destiné à l’évaluation statistique des titrages biologiques par dilution, tels que définis dans le Chapitre 5.3 de la Ph. Eur. Initialement conçu pour les laboratoires du Réseau des OMCL, ce programme est maintenant également proposé aux laboratoires non-OMCL. La version actuelle (5.0) propose de nouvelles fonctionnalités comme, entre autres, les essais d’équivalence, les régressions robustes, la protection par mot de passe des fiches de données et les courbes sigmoïdes asymétriques à 5 paramètres.
■ Une formation, ouverte également à des participants de l’industrie et du secteur privé, a été organisée en octobre.
■ Le nombre d’utilisateurs ne cesse d’augmenter depuis que le logiciel est en accès public (2013). En décembre 2015, sur l’ensemble des licences délivrées, 8 pour cent l’ont été à des laboratoires OMCL (de 25 pays) et 92 pour cent à des utilisateurs non-OMCL (de 45 pays). Il apparaît sur le diagramme que la moitié environ des licences non-OMCL a été délivrée au sein de l’UE, l’autre moitié se répartissant sur le reste du monde. CombiStats™ est devenu une référence internationale dans son domaine, et contribue à la reconnaissance mutuelle des données et des résultats d’analyses.
Assemblée annuelle du GEON
■ Coorganisée et cofinancée par l’Institut Scientifique de Santé Publique (WIV-ISP), le Centre d’Étude et de Recherches Vétérinaires et Agrochimiques (CODA-CERVA) et l’Agence fédérale des Médicaments et des Produits de Santé (AFMPS) de Belgique, la 20e
assemblée annuelle du GEON s’est tenue à Bruxelles du 1er au 5 juin 2015. Elle a réuni plus de 240 experts représentant 61 OMCL, des agences nationales du médicament et la Commission européenne (UE). Il s’agissait en outre de la première participation du laboratoire de la HSA (Health Science Authority) de Singapour depuis mai 2014, date à laquelle la HSA est devenu membre associé du Réseau. Le programme était divisé en neuf sessions individuelles.

Qualité et utilisation des médicaments ► Page 17
Activités concernant spécifiquement les pays de l’UE/EEE
Surveillance du marché des produits autorisés par voie centralisée
■ Un contrat signé par l’EMA et l’EDQM régissant
un programme annuel d’échantillonnage et d’analyse
des produits autorisés par voie centralisée (Centrally
Authorised Products, CAP) est en place depuis 1999.
L’EMA est le promoteur du programme, dont elle
a la responsabilité globale. L’EDQM, quant à elle,
coordonne l’échantillonnage et les essais. La liste
des produits à inclure dans le programme annuel est
préparée par le secrétariat de l’EMA en collaboration
avec les comités scientifiques de l’EMA, selon une
approche d’analyse du risque.
■ L’échantillonnage et le contrôle des CAP se sont
poursuivis avec succès en 2015, avec un programme
de travail comprenant 33 produits pour usage humain
(16 produits biologiques et 17 produits chimiques)
et 7 produits pour usage vétérinaire (3 produits
immunobiologiques et 4 produits chimiques). Un
contrôle des substances actives a été réalisé dans 3 cas.
Outre le programme CAP régulier, 2 programmes ont
été menés en 2015 sur des médicaments génériques,
portant sur le contrôle de produits de 12 marques
d’irbesartan et de témozolomide (génériques et
princeps).
■ 137 opérations d’échantillonnage ont été
réalisées dans le cadre du programme CAP 2015, et
34 OMCL ont participé aux opérations de contrôle.
Les contrôles ont montré que la majorité des produits
testés étaient de la qualité attendue, avec des résultats
conformes aux spécifications autorisées dans presque
tous les cas. Un résultat hors spécification et plusieurs
défauts de nature réglementaire ou technique ont
été signalés. L’EMA gère le suivi de ces observations.
Programme de surveillance après mise sur le marché des produits autorisés par reconnaissance mutuelle (PRM) ou par voie décentralisée (PDC)
■ Les OMCL impliqués se sont rencontrés deux
fois en 2015 (26e et 27e réunions) pour évaluer le
programme et discuter des moyens d’optimiser leur
collaboration.
■ Le 11e programme régulier de surveillance du
marché des médicaments autorisés par les procédures
PRM/PDC au sein de l’UE/EEE a été réalisé. Plus de
1000 contrôles de produits (nombre en augmentation
par rapport à l’année précédente) étaient inscrits au
programme de 2015. Les rapports d’essais pour 2015
provenaient de 27 OMCL différents.
■ Dans environ 3 pour cent des cas, un problème
d’ordre réglementaire (méthode d’essai insuffisamment
détaillée, formule de calcul erronée…) a été identifié
lors du contrôle des produits et, dans 2 pour cent
des cas, au moins un résultat hors spécification a
été détecté. Sept pour cent des produits contrôlés
étaient des produits pour usage vétérinaire. Deux
pour cent des produits contrôlés étaient des produits
biologiques, ce qui correspond à la répartition
générale des types de produits enregistrés via les
procédures PRM/PDC. En décembre 2015, la base
de données comprenait environ 7300 rapports de
contrôles portant sur des produits PRM ou PDC,
auxquels ont contribué 34 OMCL. Sur la période 2002-
2015, un Etat membre ayant participé au contrôle
d’un produit a reçu en moyenne des résultats d’essais
réalisés par d’autres Etats membres pour 9 produits
commercialisés, ce qui témoigne clairement des
avantages de la mise en réseau.
Produits biologiques à usage humain : libération officielle des lots par les autorités de contrôle (OCABR)
■ Les activités du Réseau OCABR pour les produits
biologiques à usage humain assurent l’application
harmonisée de l’Article 114 de la Directive européenne
2001/83/CE, en créant les conditions nécessaires à la
reconnaissance mutuelle (obligatoire) des contrôles
de libération des lots, pour les vaccins et les dérivés
du sang et du plasma humains. L’évaluation des
protocoles et les contrôles effectués par le Réseau dans
le cadre de l’OCABR ont concerné plus de 9000 lots
finaux et près de 9000 mélanges de plasma, qui ont
ainsi bénéficié d’une confirmation indépendante de
leur qualité avant de parvenir aux patients.
■ Avec plus de 90 participants, les sessions OCABR
organisées lors de l’assemblée annuelle du GEON
à Bruxelles ont connu leur plus grand le taux de
participation en cinq ans. Constituant une excellente
occasion de partager des compétences, ces sessions
sont aussi un moyen d’optimiser les ressources pour
résoudre des problèmes communs. Les OMCL ont
abordé des questions techniques complexes pour
permettre un meilleur contrôle des produits comme
les vaccins combinés pour enfants et les médicaments
pour hémophiles. L’atelier annuel à l’intention des
OMCL et des fabricants, consacré au contrôle du
vaccin antipoliomyélitique oral en vrac, et la rencontre
entre les fabricants de vaccins et le Groupe consultatif
OCABR ont grandement contribué au maintien d’une
communication ouverte et au bon fonctionnement
du système.
■ Un nouveau guideline et 4 guidelines révisés
concernant les vaccins, 1 guideline révisé concernant
les produits dérivés du sang et plusieurs guidelines
internes au Réseau sont entrés en vigueur en 2015.

Rapport annuel ► Page 18
Médicaments immunologiques vétérinaires (MIV) : libération officielle des lots par les autorités de contrôle (OCABR)
■ Un sous-ensemble d’OMCL spécialisés et d’autorités compétentes (VRBN, Veterinary Batch
Release Network) se consacre au contrôle indépendant des médicaments immunologiques vétérinaires (selon les Articles 81 et 82 de la Directive 2001/82/CE, telle que modifiée).
■ Trente-et-un participants de 16 États membres ont assisté à la session consacrée au VBRN lors de l’assemblée annuelle. Les rapports annuels montrent que la participation active au système va croissant ; néanmoins, il faudrait trouver des mécanismes permettant de mieux répartir la charge de travail et d’assurer le maintien de la compétence.
■ Preuve de la réactivité du système, un programme d’essai approuvé pour un groupe spécifique de vaccins a subi des ajustements sur la base des résultats d’essais obtenus par des OMCL. En février, le comité consultatif VBRN a rencontré des représentants de fabricants de MIV pour discuter de problèmes communs.
■ Parmi les documents entrés en vigueur en 2015, on compte 4 guidelines-produits (1 nouveau et 3 révisés), 1 modèle de protocole à l’usage des fabricants de vaccins viraux vivants et la procédure administrative révisée de l’UE relative à l’application de l’article 82 pour la libération officielle des lots de MIV par les autorités de contrôle.
ACTIVITÉS ANTI-CONTREFAÇON
Lutter contre la criminalité pour protéger la santé publique
■ L’EDQM a continué de promouvoir la coopération entre les autorités, tant au niveau national qu’international, pour combattre la contrefaçon et la falsification des produits médicaux (médicaments et dispositifs médicaux, ingrédients compris). L’un des principaux outils de cette lutte est la Convention MEDICRIME, le premier et seul instrument pénal international juridiquement contraignant en matière de contrefaçon des produits médicaux. Les experts siégeant au sein du CD-P-PH (Comité directeur) et de son Comité d’Experts subordonné sur la réduction des risques pour la santé publique posés par la contrefaçon de médicaments et la criminalité connexe (CD-P-PH/CMED) ont poursuivi le développement et la promotion de programmes et de projets visant à diffuser les meilleures pratiques en la matière.
Quelques faits
■ Les sfforts visaient à encourager les autorités et les gouvernements à signer et à ratifier la Convention. En collaboration avec l’Assemblée parlementaire et la Division du droit pénal de la Direction générale des droits de l’Homme et de l’État de Droit du Conseil de l’Europe, l’EDQM a contribué à la publication d’un

Qualité et utilisation des médicaments ► Page 19
guide pratique sur la Convention à l’intention des parlementaires, visant notamment à les alerter et à les sensibiliser à l’importance de la ratification. Cet ouvrage a vu le jour en novembre, lors d’une conférence parlementaire à Paris. Au cours de ce même mois, l’EDQM a apporté son soutien à la Division du droit pénal pour l’organisation, à Chypre, d’une conférence régionale sur la Convention MEDICRIME. En 2015, la Convention a été signée par l’Albanie, la Bosnie-Herzégovine et la Croatie, et ratifiée par la Guinée. Le nombre de ratifications requises (5) a ainsi été atteint, et la Convention est entrée en vigueur le 1er janvier 2016.
■ La promotion de la Convention MEDICRIME va de pair avec les activités développées et soutenues par l’EDQM et par ses experts pour la mise en œuvre concrète de la Convention et des outils qu’elle propose. Ces activités comprennent notamment la collecte d’informations et de données sur les produits contrefaits et falsifiés. Cette collecte est réalisée par des points de contact uniques (PCU) établis au sein des autorités de santé, des douanes, de la police et d’autres autorités compétentes tant au niveau local que national ou international. À cette fin, l’EDQM a d’ailleurs continué de proposer des formations à ces autorités, en collaboration avec :
f l’APEC (Asia-Pacific Economic Cooperation), dans le cadre de son projet sur l’intégrité des produits médicaux et la sécurité de la chaîne d’approvisionnement dans le monde – une formation a été organisée en janvier, aux Philippines, pour les représentants des autorités de 12 États membres de l’APEC,
f le projet REPT (Responding Effectively to the Production and the Trafficking in falsified medicines), financé par l’UE, dans le cadre d’un accord conclu qui a permis d’organiser 3 formations des PCU dans 3 pays (Cameroun, Ghana et Jordanie) au cours du dernier trimestre de 2015.
■ En juin, professionnels et autorités de santé ont pris part à un atelier d’experts pour discuter des résultats d’une étude sur un outil de dépistage conçu pour aider les médecins à détecter les symptômes (« signaux ») indiquant des dommages sur la santé causés par une possible utilisation de médicaments contrefaits ou falsifiés.
Systèmes de traçabilité des médicaments pour combattre la contrefaçon
■ L’EDQM continue de soutenir le développement de systèmes de sérialisation de masse, outils permettant d’empêcher la contamination de la chaîne légale de distribution des médicaments par des produits falsifiés ou contrefaits. À cette fin, elle œuvre en faveur d’une approche harmonisée en Europe, et d’une gouvernance publique de tous les
systèmes de traçabilité pour éviter toute utilisation abusive des données.
■ Suite à l’accord qu’elle a signé en 2015 avec l’EMVO (European Medicines Verification Organisation, composée de différents acteurs européens de la chaîne de distribution), l’EDQM contribuera directement à la mise en œuvre des systèmes de traçabilité des médicaments, en effectuant des évaluations de conformité périodiques de l’EMVS, le système européen de vérification des médicaments développé au sein de l’UE par l’EMVO. Il sera ainsi possible de déterminer si le système européen (hub européen et systèmes nationaux) est conçu, géré et exploité conformément aux normes décrites dans l’acte délégué2 portant sur les modalités d’un « identifiant unique », en application de la directive 2011/62/UE sur les médicaments falsifiés.
Programme « Fingerprint »
■ Le projet s’est poursuivi, avec la préparation de deux nouvelles études qui seront réalisées en 2016 et cibleront deux substances actives : oméprazole et morphine. L’expérience acquise à travers l’étude 2014-2015 sur les antibiotiques macrolides et les statines (qui a largement fait usage de l’analyse chimiométrique pour regrouper les sources de substances actives) a permis d’affiner le développement du projet.
Publications, bases de données et site web
■ L’EDQM maintient une base de données sécurisée d’accès restreint baptisée « Know-X » pour le stockage d’informations complètes sur des affaires spécifiques de contrefaçon ou falsification de produits médicaux, une fois terminée l’enquête criminelle. La base de données permet aux autorités de santé et aux services de répression d’intervenir plus rapidement sur des cas de produits médicaux suspects, et aide les états signataires de la Convention MEDICRIME dans la surveillance des tendances et le suivi.
■ Know-X contient également des informations relatives à l’identification des médicaments par analyse chimique (voir section « Réseau européen des laboratoires officiels de contrôle des médicaments », page 16), ainsi que des données sur le modus operandi et les mesures prises en matière de gestion et de prévention des risques par les autorités compétentes (autorités de santé ou services de répression). Le CD-P-PH/CMED coopère avec le groupe de travail OMCL sur les médicaments contrefaits pour maintenir la base de données, et participe également à sa promotion et à la formation des utilisateurs. Depuis 2015, les rapports comprennent une section sur les critères de risque et l’évaluation des risques afin de faciliter, dans leur ensemble, les approches fondées sur les risques.
2. Voir https://go.edqm.eu/2016161regl

Rapport annuel ► Page 20
■ En 2015, l’EDQM a publié un guide-concept
destiné aux enseignants comprenant une bande
dessinée. Ce guide a pour but de soutenir la formation
et l’éducation dans les écoles, en expliquant aux enfants
et aux adolescents les risques posés par les produits
médicaux falsifiés et contrefaits, et en encourageant
les comportements préventifs. L’EDQM et les experts
du CD-P-PH/CMED ont également apporté une
contribution importante au guide pratique sur la
Convention MEDICRIME publié à l’intention des
parlementaires (voir plus haut « Quelques faits »).
Communication avec les partenaires et acteurs concernés
■ Des représentants de l’EDQM et du CD-P-PH/
CMED ont participé à des conférences de l’IFPMA
(International Federation of Pharmaceutical
Manufacturers & Associations) en Asie et en Afrique.
PRODUITS ET SUIVI
PHARMACEUTIQUES
Utilisation optimale des médicaments pour améliorer la qualité de vie des patients
■ Les activités dans ce domaine sont conduites
par le Comité européen sur les produits et les
soins pharmaceutiques (CD-P-PH) et ses organes
subordonnés.
Quelques faits
■ Le Comité d’Experts sur les normes de qualité
et de sécurité relatives à la pratique et au suivi
pharmaceutiques (CD-P-PH/PC) a terminé l’étude
menée sur la validation de 4 ensembles d’indicateurs
basiques de qualité du suivi pharmaceutique en
Europe, dans le cadre du projet de l’EDQM sur les
indicateurs de qualité du suivi pharmaceutique
(PCQIP). Les indicateurs développés couvrent les
domaines fondamentaux suivants du processus du
suivi pharmaceutique :
f respect des règles en matière de prescription
d’antimicrobiens dans les services ambulatoires,
f pour les traitements antibiotiques et
anticoagulants, surveillance des plans
thérapeutiques et de l’innocuité des
médicaments par le pharmacien, en croisant les
données et en échangeant les informations liées
au traitement et au dossier médical du patient,
f consultations patient-pharmacien structurées
(traitement chronique, polymédication,
polymorbidité) via le questionnaire « My
CheckList »,
f auto-évaluation du pharmacien d’officine quant
à la mise en œuvre de la philosophie et des
méthodes de travail associées au concept de
suivi pharmaceutique.
■ Des indicateurs de qualité validés sont
actuellement disponibles dans plusieurs pays d’Europe
possédant des systèmes de santé, des traditions
médicales et des pratiques du suivi pharmaceutique
différentes. Ces indicateurs peuvent aider les
décideurs, les autorités de santé et les professionnels
de la santé à évaluer la qualité du suivi et des pratiques
pharmaceutiques. Ils peuvent ainsi contribuer à
l’amélioration continue des résultats en termes de
santé et de qualité de vie des patients, ainsi qu’à
l’utilisation efficace et efficiente des ressources.
L’EDQM veillera à la large diffusion des conclusions
du projet PCQIP, présentées lors d’un atelier organisé
en novembre. Elle veillera également à promouvoir
la mise en œuvre de l’approche définie pour le PCQIP
grâce à des partenariats avec les parties concernées
et en émettant des recommandations de politiques
appropriées pour une meilleure harmonisation des
normes qualité en Europe.
■ Le projet PaedForm de préparation d’un
formulaire pédiatrique européen harmonisé, conduit
sous l’égide du CD-P-PH en étroite collaboration avec
la Commission européenne de Pharmacopée, vise
à remédier au manque de médicaments autorisés
spécifiquement conçus pour la population pédiatrique.
Dans une première étape, l’approche choisie a consisté
à identifier des préparations candidates appropriées
parmi les préparations non autorisées figurant dans les
formulaires nationaux, et d’élaborer des monographies
sur la base de critères approuvés par le CD-P-PH. En
2015, le CD-P-PH a élaboré et approuvé les critères
de sélection et d’évaluation des monographies, les
critères de maintien à jour et de surveillance des
monographies, et la procédure relative au Formulaire
pédiatrique européen. Avec l’adoption de ces

Qualité et utilisation des médicaments ► Page 21
documents, le Groupe de Travail « PaedForm » de la Ph. Eur. peut commencer à examiner les formulaires nationaux recueillis auprès des États membres dans le cadre du projet.
■ En 2015, le guide sur les bonnes pratiques en matière de systèmes de délivrance automatisée et de leur mise en œuvre en Europe a été finalisé, en tenant compte des discussions tenues en septembre avec des experts des autorités nationales, des pharmaciens et des opérateurs de systèmes de délivrance automatisée, lors d’un atelier organisé par l’EDQM. Le projet a pour but de fournir aux États membres un document guide sur cette pratique nouvelle de préparation automatisée de récipients ou de sachets individuels et personnalisés contenant différents médicaments prescrits à un patient. Le guide propose une approche globale, couvrant divers aspects de la délivrance automatisée : exigences techniques et évaluation des risques, manipulation des médicaments une fois qu’ils ont été retirés de leur emballage extérieur, évaluation au cas par cas de l’opportunité d’une délivrance automatisée pour le patient.
■ Le CD-P-PH/PC a finalisé un document guide sur des normes générales de qualité et de sécurité pour la reconstitution des médicaments à la concentration ou la forme voulue par ajout de liquide. Par la suite, la Résolution CM/ResAP(2011)1 du Conseil de l’Europe sur « les exigences relatives à l’assurance de qualité et d’innocuité des médicaments préparés en pharmacie pour les besoins particuliers du patient » sera mise à jour et une nouvelle résolution portant sur l’aspect spécifique de la reconstitution sera publiée.
■ Le Comité d’Experts sur la classification des médicaments en matière de leur délivrance (CD-P-PH/PHO) a publié ses recommandations annuelles aux autorités de santé sur la classification des médicaments selon leurs conditions de délivrance (avec ou sans prescription) et a établi les bonnes pratiques de classification. Ses travaux intéressent particulièrement
les différents acteurs de la chaîne du médicament, et facilitent l’accès à des médicaments sûrs pour les patients en Europe.
Publications, bases de données et site web
■ La mise à jour annuelle par le CD-P-PC/PHO des recommandations de classification pour 2015 est disponible sur le site de l’EDQM3. L’année 2015 aura également été marquée par la fin des travaux de révision de la classification des médicaments pour le traitement des ulcères gastroduodénaux et du reflux gastro-œsophagien, qui sera publiée sur le site de l’EDQM en 2016.
■ Tout au long de l’année 2015, la base de données Melclass4 a fait l’objet de mises à jour en continu. Cette base de données, qui fait l’état de la classification des médicaments dans les États membres, est désor-mais accessible via une application web adaptative conforme aux technologies les plus récentes. Cette nouvelle version de la base de données a été lancée en janvier 2016.
Communication avec les partenaires et acteurs concernés
■ La mission et les travaux du CD-P-PH/PHO, notamment le projet PaedForm, ont été présentés en avril, à Paris (France), à l’occasion de la 27e édition de l’EuroMeeting de la Drug Information Association (DIA) et en novembre, à Kragujevac (Serbie), lors du 11e
symposium de l’Agence du médicament serbe (ALIMS).
3. Voir révision des Annexes de la Résolution ResAP(2007)1 sur la classification des médicaments (édition 2015) https://go.edqm.eu/PHOfr
4. https://melclass.edqm.eu/


Soins de Santé ► Page 23
L’EDQM s’est appliquée à poursuivre ses travaux dans le domaine de la protection de la santé publique en proposant des normes fiables en
matière d’éthique, de sécurité et de qualité pour la collecte, la préparation, la conservation, la distribu-tion et le bon usage des composants sanguins dans le contexte de la transfusion, et pour la transplantation d’organes, de tissus et de cellules. L’EDQM a en outre continué ses travaux visant à établir des normes et à coordonner les contrôles des cosmétiques et des matériaux pour contact alimentaire.
TRANSFUSION SANGUINE
Promouvoir la sécurité et la qualité du sang en Europe et au-delà
■ L’EDQM est responsable des activités du Conseil de l’Europe dans le domaine de la transfusion sanguine, qui reposent sur trois grands principes : promouvoir les dons volontaires et non rémunérés, atteindre l’autosuffisance et protéger les donneurs et les receveurs de composants sanguins labiles. L’EDQM se penche activement sur les aspects éthiques, juridiques et organisationnels de la transfusion, en vue d’assurer la qualité, d’améliorer la disponibilité, d’éviter le gaspillage et d’assurer l’utilisation optimale du sang et des composants sanguins.
■ Le Comité européen sur la transfusion sanguine (CD-P-TS) est le comité directeur en charge des activités de l’EDQM liées à la transfusion sanguine. Il élabore des guidelines et des recommandations et ses membres, experts reconnus à l’échelle internationale, sont issus des États membres du Conseil de l’Europe, des pays observateurs, de la Commission européenne (UE), de l’OMS, de la FDA américaine et du Comité de Bioéthique (DH-BIO) du Conseil de l’Europe. Le CD-P-TS supervise les travaux de ses organes subordonnés sur des questions spécifiques dans ce domaine.
Soins de Santé

Quelques faits et chiffres
■ Parmi les réalisations concrètes du CD-P-TS et
de ses organes subordonnés figurent :
f la publication de la 18e édition du « Guide for the Preparation, Use and Quality Assurance o Blood Components » (communément appelé « Guide
Sang »),
f 6 essais d’aptitude dans le domaine du sang
(B-PTS),
f 1 visite de formation (B-VF) et 3 visites mutuelles
conjointes (B-VMC) dans des établissements du
sang (ES),
f l’organisation de la première formation sur le
management de la qualité dans les ES.
Orientations et politiques générales
Comportements à risque ayant un impact sur la gestion des donneurs de sang et la sécurité transfusionnelle
■ Le Groupe de Travail TS100 a repris les travaux
du Groupe de Travail TS057 qui avait élaboré la
Résolution CM/Res(2013)3 sur les comportements à
risque.5 Le nouveau groupe est chargé de poursuivre
la collecte des données sur l’incidence et la prévalence
des infections sexuellement transmissibles dans la
population générale, parmi les donneurs de sang
et parmi les individus ayant des comportements
sexuels à risque, l’objectif étant de s’appuyer sur ces
données scientifiques pour de futures modifications
des politiques d’ajournement des donneurs ou
d’éventuelles révisions de la Résolution.
Programmes de management de la qualité
■ L’EDQM a a poursuivi ses efforts pour encourager
la participation aux deux programmes qu’elle
a développés pour aider les ES à mettre en place
les éléments d’un système de management de la
qualité (MQ) : le programme B-PTS et le programme
de management de la qualité dans le domaine du
sang (B-MQ). Tous deux sont conçus notamment pour
aider les établissements à mettre en application la
législation européenne, le Guide Sang et les bonnes
pratiques définies dans les « Good Practice Guidelines »
(GPG, voir ci-contre).
Programme B-PTS
■ L’évaluation externe des capacités de contrôle
des ES européens s’est poursuivie avec l’organisation
d’essais d’aptitudes. En 2015, 6 études ont été
organisées, avec une participation moyenne de 53
établissements par étude.
5. Texte intégral de la Résolution : https://go.edqm.eu/TSrec
Programme de management de la qualité dans le domaine du sang (B-MQ)
■ Ce programme propose des outils permettant aux
ES européens de développer, mettre en application et
améliorer leur système de MQ. Solidement soutenu
par le CD-P-TS, la Commission européenne (UE) et
les ES, ce programme comprend l’organisation de
3 types d’intervention, tous menés par des experts
issus d’ES européens.
f B-VF : visite sur site et formation sur mesure
portant sur des sujets techniques et des
questions liées au système de MQ ;
f B-VMC : examen du système de MQ en cours de
développement ; observation du niveau de mise
en application des normes minimales : Guide
Sang, GPG, législation européenne sur le sang
et normes appliquées au sein de l’ES (normes
ISO, BPF, par exemple) ; recommandations sur
la mise en application du système de MQ et
propositions d’amélioration ;
f B-AMC : vérification de la conformité du système
de MQ avec le Guide Sang, les GPG, la législation
européenne sur le sang et les normes utilisées
au sein de l’ES.
Techniques d’amplification des acides nucléiques
B-PTS018 - VHB/VHC/VIH
Immuno-hématologie
B-PTS023 ABO, Rhésus,
phénotypage étendu et
anticorps irréguliers
Sérologie
B-PTS019 – anti-VHC
B-PTS020 – anti-VIH/p24
B-PTS021 – HBsAg
B-PTS022 – anti-HBc
Études B-PTS réalisées en 2015
Rapport annuel ► Page 24

Soins de santé ► Page 25
■ En 2015, 1 B-VF et 3 B-VMC ont été menées. En outre, la première formation européenne sur le MQ pour les ES, organisée en avril, a réuni 36 participants.
Publications, bases de données et site web
Guide pour la préparation, l’utilisation et l’assurance qualité des composants sanguins – 18e Édition
■ Un groupe dédié a pour tâche de mettre à jour le Guide Sang afin qu’il continue de refléter les progrès scientifiques les plus récents et les changements intervenus en matière réglementaire au cours des deux années séparant les éditions successives du guide. Pour la première fois, le guide « Good Practice
Guidelines for Blood Establishments and Hospital Blood
Banks required to comply with EU Directive 2005/62/
EC » (GPG) a été publié comme partie intégrante du Guide Sang dans sa 18e édition. À l’avenir, le GPG sera révisé parallèlement au Guide Sang pour refléter les plus récentes évolutions réglementaires des BPF applicables aux ES. La Commission européenne (UE) réfléchit actuellement à la possibilité d’accorder à ce guide un statut officiel au sein de la législation européenne.
Résolutions sur l’utilisation optimale des facteurs de coagulation et des immunoglobulines
■ Suite au symposium Kreuth III organisé en 2013, le CD-P-TS a élaboré les Résolutions CM/Res(2015)2 sur les traitements à base d’immunoglobuline et CM/Res(2015)3 sur les traitements de l’hémophilie6. Ces textes ont été adoptés en avril par le Comité des ministres du Conseil de l’Europe.
6. Voir htps://go.edqm.eu/TSrec
Base de données européenne sur les réserves de sang congelé de groupes rares
■ Une base de données en ligne a été développée par l’EDQM en étroite collaboration avec le CD-P-TS pour aider les ES à se procurer du sang congelé de groupes rares. La phase pilote s’est achevée avec succès en 2015 et le CD-P-TS a décidé de passer à la phase opérationnelle au début de l’année 2016.
Communication avec les partenaires et acteurs concernés
Commission européenne (UE)
■ Une collaboration intense et fructueuse avec la Commission européenne permet une utilisation optimale des ressources ; l’enquête annuelle intitulée « Rapport sur la collecte, le contrôle et l’utilisation du sang et des composants sanguins en Europe» (2010) – qui permet de collecter des données auprès des États membres du Conseil de l’Europe – en constitue un excellent exemple. La Direction générale Santé et sécurité alimentaire (DG SANTE) de l’Union européenne a inclus ces données dans le rapport qu’elle a récemment publié, intitulé « An
EU-wide overview of the market of blood and blood
components and plasma derivatives focusing on their
availability for patients ». En avril et novembre, l’EDQM a participé en qualité d’observateur aux réunions des autorités compétentes dans le domaine du sang, organisées par la DG SANTE.
Pharmaceutical Inspection Convention and Pharmaceutical Inspection Co-operation Scheme (PIC/S)
■ L’EDQM a participé à la 21e réunion du Cercle d’Experts PIC/S sur le sang, les tissus et les cellules
Participants à la première formation européenne sur le Management de la Qualité dans les ES.

Rapport annuel ► Page 26
humains, dans le contexte de la révision du Guide PIC/S sur les BPF pour les établissements du sang.
Société internationale de transfusion sanguine (SITS)
■ L’EDQM — qui dispose du statut d’observateur auprès du conseil d’administration de la SITS —est également membre de deux groupes de travail de la SITS chargés respectivement du Management de la qualité et de la Déontologie.
TRANSPLANTATION D’ORGANES /
TISSUS ET CELLULES DESTINÉS À
DES APPLICATIONS CHEZ L’HOMME
■ Promouvoir des normes strictes de qualité et de sécurité
■ Les dernières décennies ont été marquées par des progrès impressionnants dans les domaines de la transplantation d’organes et des applications cliniques des tissus et cellules. Toutefois, la demande en organes et la demande en de nombreux tissus et cellules dépassent encore largement l’offre disponible. S’agissant de substances d’origine humaine, leur utilisation comporte des risques de transmission de maladies qui doivent être réduits autant que possible
par l’application de critères appropriés de sélection des donneurs et de dépistage. En outre, seuls des organes, tissus et cellules obtenus et manipulés dans le respect de critères de qualité rigoureux sont susceptibles de fonctionner correctement.
■ Le Comité européen sur la transplantation d’organes (CD-P-TO) est le comité directeur en charge des activités liées à la transplantation conduites à l’EDQM. Son mandat comprend l’élaboration de guidelines et de recommandations visant à améliorer l’accès à la transplantation, de manière à assurer les normes les plus élevées en matière de qualité, de sécurité et d’éthique. Ce comité est composé d’experts internationalement reconnus, issus des États membres du Conseil de l’Europe, de pays observateurs, de la Commission européenne (UE), de l’OMS, du DH-BIO du Conseil de l’Europe et de plusieurs organisations professionnelles à but non lucratif.
Quelques faits et chiffres
■ Parmi les réalisations concrètes du CD-P-TO figurent :
f la publication de la brochure « La conservation
de sang de cordon ombilical - Guide à l’usage des
parents » ;
f la publication d’un document de position sur la transplantation et l’activité physique, et la participation à l’organisation d’une conférence sur ce thème (European Conference
on Transplantation and Physical Activity) en juillet à Krems an der Donau (Autriche) ;
f la publication du bulletin « Newsletter Transplant
2015 » ;
f la publication de la 2e édition du « Guide to the
Quality and Safety of Tissues and Cells for Human
Application » (Guide Tissus & cellules) ;
f l’adoption de la Résolution CM/Res(2015)107
sur « le rôle et la formation des professionnels des
soins intensifs en matière de dons post-mortem » ;
f l’adoption de la Résolution CM/Res(2015)11 sur « l’établissement de registres nationaux harmonisés
de donneurs vivants en vue de faciliter le partage
international des données » et son rapport explicatif ;
f l’élaboration d’un document de consensus sur les conséquences à long terme du don de rein de
donneur vivant ;
f la participation à l’organisation de la 17e journée européenne du don d’organes et de la greffe (JEDO), à Lisbonne (Portugal).
7. Voir htps://go.edqm.eu/TOrec © Benoit Rajau, pour l’Agence française de la biomédecine

Soins de santé ► Page 27
Orientations et politiques générales
Initiatives législatives et politiques
■ Au fil des ans, un ensemble de résolutions et recommandations dans le domaine des organes, des tissus et des cellules a été adopté par le Comité des Ministres du Conseil de l’Europe. Bien qu’ils ne soient pas juridiquement contraignants, ces textes ont eu un impact profond sur les législations nationales, sur les plans stratégiques pour le don et la transplantation et sur les pratiques professionnelles.
■Malgré certains progrès, la pénurie d’organes disponibles pour la transplantation subsiste, et les pays sont pour la plupart incapables de répondre à leurs besoins en matière de transplantation. Le Comité des Ministres a tenté d’améliorer cette situation en adoptant la Résolution CM/Res(2015)10 qui recommande aux États membres du Conseil de l’Europe de mettre en œuvre des mesures visant à garantir un cadre éthique et juridique clair pour guider les professionnels de santé prenant en charge des donneurs d’organes potentiels et pour préciser quelles sont les pratiques facilitant le don post-mortem qui sont autorisées dans un pays donné.
■ Partant du principe que le don vivant est un élément nécessaire à la poursuite de l’autosuffisance en matière de transplantation, mais soucieuse de la protection et du bon suivi des donneurs vivants, la Résolution CM/Res(2015)11 définit des orientations générales pour la mise en place de registres de donneurs vivants nationaux/internationaux, en vue de faciliter le partage de données entre pays. L’Exposé des motifs qui complète cette résolution fournit une liste détaillée des paramètres susceptibles de figurer dans un registre de donneurs vivants.
■ Le CD-P-TO a également examiné avec attention quelques publications récentes faisant état d’une augmentation du risque de maladie rénale terminale chez les donneurs de rein vivants. Suite à cette analyse, le Comité a élaboré un document de consensus sur les conséquences à long terme du don de rein de donneurs vivants, ainsi que des recommandations quant aux informations sur les risques associés au don qui devraient être fournies aux donneurs vivants. Ce document de synthèse, approuvé par l’ESOT (Société européenne de transplantation d’organes), l’ISN (International Society of Nephrology) et la TTS (The Transplantation Society), a été publié dans le bulletin Newsletter Transplant 2015 et dans les revues scientifiques spécialisées Transplantation et Transplant International.
■ Enfin, le document de position du CD-P-TO sur la transplantation et l’activité physique met en avant l’importance et les avantages de la prescription d’une activité physique aux patients transplantés en complément de leur traitement et pour améliorer leur qualité de vie.
Conseils techniques pour améliorer la qualité et sécurité des organes, tissus et cellules
■ Des experts du monde entier ont participé aux travaux des groupes de travail pour l’élaboration de la 2e édition du Guide Tissus & cellules et la 6e édition du « Guide to the Quality and Safety of Organs for
Transplantation » (Guide Organes, publication prévue en 2016). Ces experts ont fourni un travail exceptionnel de compilation des données et des évolutions les plus récentes pour garantir une application clinique sûre et réussie des organes, tissus et cellules humains.
■ La Commission européenne (UE) a également été activement impliquée dans le processus d’élaboration. Cette coopération garantit la compatibilité et la complémentarité entre les normes énoncées dans les directives de l’UE et les Guides du Conseil de l’Europe, et permet d’assurer l’application dans toute l’Europe des mêmes dispositions en matière de qualité et de sécurité. L’élaboration du Guide Tissus & cellules a été partiellement financée par la Commission européenne. Plusieurs associations professionnelles ont également activement participé à l’élaboration des guides, notamment l’EDTCO (European Donation
and Transplant Coordination Organization), l’AATB (American Association of Tissue Banks), l’EATB (European
Association of Tissue Banks) et l’ESHRE (European Society
for Human Reproduction and Embryology).
■ Un nouveau groupe de travail a été créé pour commencer l’élaboration de la 3e édition du Guide Tissus & cellules qui sera publiée en 2017.
Publications, bases de données et sites web
■ Le Guide Organes et le Guide Tissus & Cellules — qui fournissent aux professionnels du domaine des orientations en matière de qualité, de sécurité et d’éthique — sont devenus des ouvrages de référence en Europe et au-delà.
■ Le bulletin Newsletter Transplant est la seule source officielle de données chiffrées internationales sur le don et la transplantation d’organes, de tissus et de cellules souches hématopoïétiques. Ces informations permettent d’analyser l’évolution des activités de don et de transplantation et de faire évoluer les politiques en conséquence. En 2015, les informations compilées dans Newsletter Transplant provenaient de plus de 70 pays.
■ La brochure du CD-P-TO intitulée « La conservation de sang de cordon ombilical - Guide à l’usage des parents » vise à fournir des informations claires, exactes et équilibrées sur l’utilisation thérapeutique du sang de cordon et pour présenter aux parents envisageant de stocker le sang de cordon de leur bébé les options qui s’offrent à eux. Ces dernières années ont vu naître

Rapport annuel ► Page 28
un certain nombre de banques de sang de cordon qui proposent aux familles de stocker le sang de cordon de leur bébé pour un usage futur à titre privé et moyennant des frais substantiels. À la naissance de l’enfant, les parents font désormais face à un dilemme : avoir recours à ces services privés, faire don du sang de cordon de leur bébé pour un usage public, ou refuser d’en faire don.
COSMÉTIQUES ET EMBALLAGES ALIMENTAIRES
Protection de la santé des consommateurs
■ Le programme de travail relatif aux cosmétiques et matériaux pour contact alimentaire est défini par le Comité de Protection de la Santé des Consommateurs (CD-P-SC, comité directeur), composé de représentants des Ministères nationaux de la santé. En 2015, plus de 200 experts originaires de 34 États signataires de la Convention relative à l’élaboration d’une pharmacopée
européenne et de 4 pays observateurs en ont suivi les travaux ou y ont contribué activement. La Commission européenne (UE), son Centre commun de recherche (CCR) et l’Autorité européenne de sécurité des aliments (EFSA) ont la possibilité d’envoyer des représentants aux réunions du Comité et de ses groupes d’experts subordonnés.
■ Deux comités d’experts subordonnés sont chargés de mettre en œuvre les travaux définis par le CD-P-SC : le Comité d’Experts sur les produits cosmétiques (P-SC-COS) et le Comité d’Experts sur les matériaux pour contact alimentaire (P-SC-EMB).
■ Dans le domaine des cosmétiques, les travaux ont principalement trait au Réseau européen des laboratoires officiels de contrôle des cosmétiques (OCCL). En ce qui concerne les matériaux pour contact alimentaire, l’harmonisation des exigences de qualité et de sécurité est en cours, tout comme l’élaboration et l’actualisation de méthodes d’essai.
Quelques faits
Réseau OCCL
■ Le Réseau européen des OCCL nationaux, constitué de membres volontaires, a été créé en 2010. Plus de 30 OCCL, dont des laboratoires de 16 États membres de l’Union européenne, participent à ses activités régulières. Les OCCL sont principalement chargés de vérifier la qualité des produits disponibles sur le marché. Sous l’égide de l’EDQM, leurs compétences en matière de contrôle sont enregistrées dans un inventaire accessible à tous les membres du Réseau. Ces derniers peuvent ainsi faire meilleur usage des ressources et améliorer leur
management de la qualité (MQ) en se conformant aux normes internationales régissant les laboratoires, une véritable valeur ajoutée. La vaste expérience tirée du fonctionnement du Réseau OMCL est un atout pour la coordination du Réseau OCCL.
■ Le mandat du Réseau OCCL a été publié en 2015, sur le site internet de l’EDQM.
■ Le Réseau a noué des liens étroits avec la Commission européenne (UE), le CCR et le CEN (Comité européen de normalisation). En 2015, ils ont tous trois mis en pratique l’approche commune qu’ils avaient élaborée en 2014 pour valider les méthodes d’analyse développées par des laboratoires individuels.
■ Ils ont notamment effectué une comparaison interlaboratoires des quantités de peroxyde d’hydrogène mesurées par chromatographie liquide dans les produits de blanchiment des dents. Une seconde étude comparative a porté sur le dosage du formaldéhyde présent dans les cosmétiques, notamment les crèmes pour le visage, les shampoings et les dentifrices. Les deux études ont démontré la reproductibilité interlaboratoires des méthodes d’analyse en question, qui s’imposent comme la référence en matière de contrôle qualité des cosmétiques.
Contrôle de la qualité des cosmétiques : études de surveillance du marché
■ Faisant suite à une MSS portant sur la qualité des produits conçus pour attirer les enfants, finalisée en 2014, un rapport détaillé a été diffusé auprès des différentes autorités en 2015. La conformité aux réglementations européennes de plusieurs shampooings, pommades, produits de maquillage pour le visage, gels douche, entre autres produits, a été contrôlée. Plus d’un tiers des échantillons ont été jugés non conformes et il s’est avéré que plusieurs contenaient des quantités significatives de nitrosamines, de colorants interdits ou de plomb.
Programme d’essais d’aptitude
■ Les essais d’aptitude (PTS) font partie intégrante du management de la qualité dans les laboratoires d’essai. Plusieurs laboratoires analysent les mêmes échantillons afin de vérifier, par exemple, leur capacité à quantifier une substance interdite, et d’assurer la comparabilité des résultats d’essai obtenus en Europe. En 2015, le programme PTS a comporté une étude sur la teneur des crèmes solaires en dioxyde de titane, à laquelle ont participé 10 OCCL, et une autre étude, à laquelle ont pris part 18 laboratoires, a porté sur la quantité de fluorures dans le dentifrice. Ces deux études ont été terminées dans l’année.
■ Le programme PTS de l’EDQM a été conçu comme un outil de benchmarking pour les laboratoires participants aux études, qui partagent leur expertise et

Soins de santé ► Page 29
améliorent leurs compétences techniques en matière d’analyses.
Tatouages et maquillages permanents
■ Pour mettre en œuvre les recommandations de la Résolution AP (2008) 1 du Conseil de l’Europe sur les exigences et les critères d’innocuité des tatouages et des maquillages permanents, un document répertoriant des exigences de sécurité et de documentation est en cours d’élaboration. Il devrait être finalisé et publié en 2016.
Matériaux et objets pour contact alimentaire
■ Le Comité d’Experts P-SC-EMB poursuit la révision des Résolutions existantes et des documents techniques qui étaient auparavant élaborés dans le cadre de l’Accord partiel du Conseil de l’Europe dans le domaine social et de la santé publique (dissous le 31 décembre 2008). Ces travaux ont été répartis entre plusieurs rapporteurs chargés d’élaborer des dispositions relatives à différents matériaux tels que le liège, les résines échangeuses d’ions, ou le papier et le carton. En 2015, deux réunions de groupes de travail, organisées par le BfR (institut fédéral allemand d’évaluation des risques) et l’AGES (agence autrichienne de santé et de sécurité des aliments), ont été consacrées au papier et au carton. Ces travaux se poursuivront en 2016.
Communication avec les partenaires et acteurs concernés
■ Les réunions rassemblant le groupe Méthodes du PEMSAC (plateforme des autorités de surveillance du marché européen pour les produits cosmétiques), le CCR et le Réseau européen des OCCL se sont avérées très fructueuses et efficaces pour traiter des sujets d’intérêt commun. En novembre, les États membres, la Commission européenne (UE), le CCR et l’EDQM se sont réunis pour revoir les méthodes d’analyse stipulées dans les directives de l’UE et définir les priorités du programme de travail relatif à la surveillance du marché des cosmétiques.
Manifestations
■ Dans le cadre de la Présidence luxembourgeoise du Conseil de l’Union européenne (second semestre 2015), l’EDQM a participé en septembre à une réunion publique organisée par l’UE sur les matériaux pour contact alimentaire. La Commission européenne y a confirmé que l’UE n’avait aucun projet de Règlements ou Directives dans ce domaine pour les années à venir. Dans ce contexte réglementaire, les recommandations élaborées par le Conseil de l’Europe / EDQM en matière de qualité des matériaux pour contact alimentaire prennent d’autant plus d’importance, pour protéger les consommateurs d’une exposition à des traces et résidus potentiellement toxiques.


Management de la qualité ► Page 31
L’engagement de l’EDQM envers une amélioration continue
■ En 2015, l’EDQM a continué d’investir dans son
système de MQ, et la certification ISO 9001:2008 a été
renouvelée pour un certain nombre de ses activités.
Son accréditation ISO 17025:2005 a également été
confirmée pour 21 techniques d’analyse. Les clients
et partenaires de l’EDQM peuvent, par conséquent,
être certains de la qualité et la constance des
services fournis, et des efforts qu’elle réalise pour
non seulement maintenir, mais également améliorer
sans cesse son système de MQ.
493-TEST
Management de la qualité


Coopération avec les partenaires internationaux ► Page 33
L’EDQM attache une importance particulière à la coopération avec ses nombreux partenaires internationaux, pour l’ensemble de ses activités.
■ Ses activités ne pourraient avoir lieu sans le soutien des autorités nationales de pharmacopée (ANP), des autorités nationales compétentes, des laboratoires officiels de contrôle, des inspections nationales et de plus de 1200 experts en sciences pharmaceutiques, et spécialistes de domaines de la santé comme la transfusion sanguine et la transplantation d’organes. Membre du réseau réglementaire européen, l’EQDM rencontre aussi régulièrement les autorités réglementaires nationales, ainsi que la Commission européenne (UE) et ses agences techniques (dont l’EMA), avec lesquelles elle collabore.
COOPÉRATION AVEC LES AUTORITÉS NATIONALES
■ La Commission européenne de Pharmacopée et ses 72 groupes d’experts et de groupes de travail actuels sont notamment composés de représentants des autorités nationales compétentes. Ces dernières participent également aux travaux de la Ph. Eur. en présentant des demandes de révision et en examinant les projets de textes publiés dans Pharmeuropa en ligne.
■ En 2015, la réunion annuelle des ANP des États membres de la Ph. Eur. s’est déroulée en juin à Utrecht, aux Pays-Bas, à l’invitation du RIVM. Vingt-cinq des 37 États membres y étaient représentés (voir section « Pharmacopée Européenne », page 11).
■ Coorganisée et cofinancée par l’Institut Scientifique de Santé Publique (WIV-ISP), le
Coopération avec les partenaires internationaux
Centre d’Étude et de Recherches Vétérinaires et Agrochimiques (CODA-CERVA) et l’Agence fédérale des Médicaments et des Produits de Santé (AFMPS) de Belgique, la 20e assemblée annuelle du Réseau général des OMCL européens (GEON) s’est tenue à Bruxelles du 1er au 5 juin 2015. Elle a réuni plus de 240 experts représentant 61 OMCL, des agences nationales du médicament et la Commission européenne (UE) (voir section « Réseau européen des laboratoires officiels de contrôle des médicaments (OMCL) », page 16).
■ La Ph. Eur. a poursuivi ses efforts pour réduire la duplication des essais au cours du développement des médicaments, en participant aux travaux du GDP — dont sont membres la Ph. Eur., la JP et l’USP et auprès duquel l’OMS a statut d’observateur (voir section « Pharmacopée Européenne », page 10).
■ Le CD-P-TS, le CD-P-TO, le CD-P-PH et le CD-P-SC continuent de recevoir le soutien des autorités nationales.
COOPÉRATION AVEC L’UNION EUROPÉENNE ET L’EMA
■ L’EDQM collabore étroitement avec la Commission européenne (UE), avec laquelle elle entretient des échanges réguliers concernant l’avancement des programmes de travail et les éventuelles évolutions de la législation de l’UE.
■ L’EDQM est membre de l’EUNDB (European Union Network Data Board), organisation créée fin 2014 et coprésidée par l’EMA et une autorité nationale compétente. Elle fait également partie du groupe d’étude, créé en 2015, qui travaille sur l’application dans l’UE des normes ISO IDMP, et notamment du sous-groupe Referentials.

■ Elle travaille par ailleurs en étroite collaboration avec l’EMA et les autorités nationales pour assurer la cohérence des approches des autorités d’enregistrement et de la Ph. Eur. Elle a statut d’observateur auprès d’un certain nombre d’instances de l’EMA, par exemple le Comité des médicaments de thérapie innovante (CAT), le Comité des médicaments à base de plantes (HMPC), le Groupe de Travail mixte CHMP/CVMP sur la Qualité (QWP), le Groupe de Travail des inspecteurs BPF/BPD (GMDP-IWG), le Groupe de Travail Biologie (BWP) et le Groupe de Travail Immunologie (IWP). Des membres des groupes de travail de l’EMA (c.-à-d. dont l’EMA assure le Secrétariat), ainsi que du Secrétariat de l’EMA lui-même, sont observateurs auprès de certains groupes d’experts et de groupes de travail de la Commission, p. ex. les groupes 6B (sang humain et produits du sang), 15 et 15V (vaccins et sérums pour usage humain et pour usage vétérinaire), et le comité directeur du programme de standardisation biologique.
■ L’EDQM et l’EMA communiquent régulièrement au sujet de la procédure de Certification : l’EMA est membre du Comité directeur de la procédure de Certification, et des canaux ont été mis en place pour une communication régulière sur le programme d’inspection et ses résultats.
■ Les deux organisations poursuivent aussi leur collaboration pour la mise en œuvre du programme d’échantillonnage et d’analyse des CAP, établi de longue date, concernant les produits pour usage humain et vétérinaire (voir section « Réseau européen des laboratoires officiels de contrôle des médicaments (OMCL) », page 17).
COOPÉRATION EN MATIÈRE D’INSPECTIONS
■ En 2015, la Division Certification de l’EDQM a poursuivi sa participation au programme international d’inspection des sites de production de substances actives (coordonné par l’EMA) et au PIC/S (voir section « Certification de conformité aux monographies de Ph. Eur. », page 14). Elle a accueilli la 7e réunion du Cercle d’Experts PIC/S sur les substances actives (voir section « 2015 : une année riche en manifestations et réunions », page 37).
■ L’EDQM a également participé à la 21e réunion du Cercle d’Experts PIC/S sur le sang, les tissus et les cellules humains, dans le contexte de la révision Guide PIC/S sur les BPF pour les établissements du sang.
COOPÉRATION AVEC L’OMS
■ En 2015, l’EDQM a poursuivi sa collaboration avec l’OMS et participé à de nombreuses réunions et consultations organisées par celle-ci, notamment :
f programme de l’OMS sur les dénominations
communes internationales (DCI), en tant
qu’observateur, car les DCI sont utilisées dans
les monographies de la Ph. Eur. ;
f Comité d’Experts de la standardisation biologique
(ECBS) de l’OMS, qui participe elle-même en tant
qu’observateur aux réunions du comité directeur
du programme de standardisation biologique
de l’EDQM ; ceci garantit le bon déroulement
des échanges d’informations ;
f Comité d’Experts des spécifications relatives
aux préparations pharmaceutiques (ECSPP) (voir
section « Étalons de référence », page 11) ;
f partage de données et inspections conjointes
dans le cadre de la procédure Certification, pour
les substances actives.
■ Par ailleurs, l’EDQM a continué d’accueillir l’OMS
en tant qu’observateur auprès de la Commission.
■ En 2015, l’EDQM a participé à deux Assemblées
mondiales des pharmacopées, organisées sous l’égide
de l’OMS : en avril à Washington (États-Unis) et en
septembre à Suzhou (République populaire de Chine)
(voir section « Pharmacopée Européenne », page 11). Dans
ce contexte, elle a activement contribué à la rédaction
des BPPh, une initiative de l’OMS qui pourrait servir de
base à un partage des travaux et à une collaboration
entre pharmacopées du monde entier, dans l’avenir
(voir section « Pharmacopée Européenne », page 11).
En septembre, l’EDQM s’est par ailleurs fortement
impliquée dans une consultation informelle organisée
par l’OMS sur les normes internationales concernant
les produits biothérapeutiques.
■ L’EDQM assure l’établissement, la surveillance
et la distribution des ISA et des SCRI de l’OMS (voir
section « Étalons de référence », pages 11 et 12).
■ Elle collabore, en outre, avec l’OMS dans
le domaine de la transfusion sanguine et de la
transplantation d’organes.
COOPÉRATION AVEC LES
FABRICANTS ET LES
ASSOCIATIONS INDUSTRIELLES
■ L’EDQM continue d’organiser des rencontres
bilatérales annuelles avec les associations industrielles
pour favoriser les échanges sur tous les aspects relatifs
à ses travaux et recueillir des retours sur ses activités.
En mars 2015, elle a par ailleurs organisé une rencontre
où plusieurs associations industrielles ont été invitées
à présenter leur point de vue sur l’avenir des textes de
la Ph. Eur. dans le domaine des produits biologiques.
Rapport annuel ► Page 34

2015 : une année riche en manifestations et réunions ► Page 35
SYMPOSIUMS ET ATELIERS – RÉUNIONS THÉMATIQUES
Symposium : préparation et usage clinique du plasma pour la transfusion
■ Bien qu’utilisé depuis des décennies dans les unités de soins intensifs, en chirurgie et en médecine d’urgence, le plasma (composant sanguin) soulève des questions encore sans réponse. En septembre, l’EDQM a organisé un symposium rassemblant des cliniciens et des experts des établissements de transfusion sanguine pour parler des besoins cliniques en matière de transfusion de plasma, ainsi que des spécifications à définir pour garantir un plasma transfusé de qualité adéquate pour l’usage clinique prévu. Les pratiques varient entre les États membres du Conseil de l’Europe et il existe des lacunes dans les connaissances disponibles dans différents domaines. Le Comité européen sur la transfusion sanguine (CD-P-TS) a entrepris des travaux pour chercher des moyens de combler ces lacunes et répondre plus uniformément aux besoins.
Atelier : vaccins pour poissons
■ En septembre, dans le cadre de la 17e Conférence internationale sur les maladies des poissons et des crustacés, l’EDQM a organisé un atelier portant sur
les exigences de la Pharmacopée Européenne en matière de vaccins pour poissons. Organisée par l’EAFP (European Association of Fish Pathologists), cette conférence a réuni des membres de l’EAFP et d’autres professionnels exerçant dans des disciplines variées, tous intéressés par les maladies aquatiques et les questions et thèmes connexes. L’atelier de l’EDQM, qui a suscité un vif intérêt et de nombreuses interventions pendant les discussions ouvertes, concernait plus particulièrement la Ph. Eur. et ses chapitres généraux et monographies couvrant les vaccins pour poissons, ainsi que les méthodes in vitro pour le titrage d’activité de ces vaccins.
Atelier d’experts : systèmes de délivrance automatisée
■ Le projet de guide sur les bonnes pratiques en matière de systèmes de délivrance automatisée (SDA) et leur application en Europe a été discuté lors d’un atelier organisé par l’EDQM en septembre, auquel ont participé des experts des autorités nationales, des pharmaciens et des opérateurs de SDA.
Symposium : indicateurs de qualité du suivi pharmaceutique
■ En novembre, l’EDQM a organisé un atelier visant à présenter les résultats de son projet sur l’établissement d’indicateurs de qualité du suivi
2015: une année riche en manifestations et réunions

Rapport annuel ► Page 36
pharmaceutique, recueillir des retours et des avis sur la mise en œuvre et l’utilisation de ces indicateurs, et renforcer le rôle de l’EDQM dans l’harmonisation des normes qualité et la promotion du bon usage des médicaments en Europe.
SESSIONS DE FORMATION
■ L’EDQM a organisé 2 sessions de formation sur la Ph. Eur., en juillet (Strasbourg) et en décembre (Bucarest). Cette dernière était organisée avec le concours de l’Agence roumaine du médicament (NAMMD). Le programme de ces formations avait pour objectif de permettre aux participants d’approfondir leur connaissance des travaux et procédures de la Ph. Eur. Les deux sessions ont plus particulièrement porté sur les substances définies chimiquement et la procédure de Certification (préparation des dossiers, révisions et programme d’inspection).
■ En avril, l’EDQM a organisé sa première formation sur les bonnes pratiques recommandées pour le développement de systèmes de MQ dans les établissements du sang européens. En médecine transfusionnelle, il est essentiel d’assurer la qualité et l’innocuité des produits et services, pour les donneurs de sang, les patients et le grand public. Préparé avec la contribution d’experts travaillant dans des établissements du sang, ce programme de 4 jours visait à aider les ES à définir, développer et améliorer leurs systèmes de MQ. Il couvrait des sujets variés tels que le cadre réglementaire, la cartographie des processus, le management d’un système de documentation qualité, les concepts de validation, qualification et gestion du risque, la gestion des actions correctives et préventives et les procédures d’amélioration continue.
■ Il incluait, par ailleurs, une visite du centre de collecte de l’Établissement français du sang (EFS) de Strasbourg, et l’EDQM remercie l’EFS Alsace d’avoir ouvert ses portes et partagé son expérience et son savoir-faire avec les participants à la formation. Riche en enseignements, cette visite a été extrêmement appréciée.
■ Une session de formation sur CombiStats™ (voir
section « Réseau européen des laboratoires officiels de
contrôle des médicaments (OMCL) », page 16), ouverte aux acteurs de l’industrie et du secteur privé, a été organisée en octobre (Strasbourg).
WEBINAIRES
■ Tout au long de l’année, l’EDQM a organisé des webinaires portant sur différents sujets d’actualité, qui ont suscité un vif intérêt parmi les utilisateurs (plus de 1100 personnes, de 75 pays, les ont suivis) et lui ont permis d’atteindre un large public.
■ En mars, l’EDQM a organisé un webinaire sur les produits biologiques de la Ph. Eur. du XXIe siècle, pour présenter la manière dont la Ph. Eur. accompagne les évolutions actuelles et adapte ses textes pour rester à jour de l’évolution des sciences et de la pratique médicale, et les défis qui restent à relever. Centrée sur la façon dont sont élaborés les monographies et chapitres généraux, la présentation donnait un aperçu des principaux textes, en mettant l’accent sur les produits biotechnologiques, et soulignait la flexibilité des textes de la Ph. Eur., avec des exemples d’application du concept de Quality by Design (QbD) aux produits biologiques, et sur la manière dont est pris en compte le développement des biosimilaires.
■ Un webinaire sur l’introduction de l’osmose inverse comme technique de purification dans la monographie Eau pour préparations injectables de la Ph. Eur s’est déroulé en avril, couvrant l’historique des monographies relatives à l’eau, les mesures prises par la Ph. Eur. pour la révision de la monographie Eau pour
préparations injectables et les conséquences de cette révision sur d’autres textes de la Ph. Eur. portant sur la qualité de l’eau.
■ Plus tard dans l’année, la Division Certification de l’EDQM a organisé un webinaire sur la manière de bien préparer les demandes de CEP. Reprenant les différentes sections du CTD (Common Technical
Document), la présentation fournissait des lignes directrices sur la base d’exemples ou d’erreurs fréquentes, et des conseils sur la manière de procéder. L’objectif était d’aider les demandeurs à éviter les erreurs les plus fréquentes et obtenir plus rapidement la délivrance du CEP.
■ Enfin, un webinaire portant sur les récipients de verre pour usage pharmaceutique s’est tenu en décembre ; l’objectif était d’éclairer le contexte de

2015 : une année riche en manifestations et réunions ► Page 37
la récente révision de ce chapitre général, publié en juillet 2014 et entré en vigueur en janvier 2016 ; l’accent était également mis sur le fait que le chapitre est à nouveau en révision et que les utilisateurs sont encouragés à apporter leur contribution.
PARTICIPATION À DES RÉUNIONS INTERNATIONALES
■ En 2015, l’EDQM a été invitée à participer à plusieurs réunions et manifestations internationales importantes, dans le monde entier.
■ Elle a apporté sa contribution à l’élaboration du programme du 22e Séminaire International sur la surveillance et le dépistage des pathogènes transmissibles par le sang, coorganisé à Prague par l’IPFA (International Plasma Fractionation Association) et le PEI (Paul-Ehrlich-Institut), auquel elle a participé.
■ Elle a assisté à de deux réunions de l’IGDRP, programme instauré en 2015 après un projet pilote de trois ans lancé en 2012. La première s’est déroulée en juin, à Pretoria (Afrique du Sud), et la seconde en novembre, à Séoul (République de Corée). L’IGDRP encourage la collaboration et la convergence des programmes de réglementation des médicaments génériques à l’échelle internationale, afin de répondre aux défis posés par l’accroissement de la charge de travail, la mondialisation et la complexité des questions scientifiques.
■ En septembre, l’EDQM a pris part à une consultation informelle sur les normes internationales pour les produits biothérapeutiques, organisée par l’OMS, où elle a pu développer son point de vue sur l’importance d’élaborer des normes de pharmacopée pour cette catégorie de produits essentielle.
■ En décembre, des représentants de l’EDQM et du Comité d’Experts CD-P-PH/CMED ont participé à la première conférence marocaine sur les médicaments et produits de santé, organisée à Rabat, en vue de développer la coopération avec les responsables de pays concernés par l’application de la Convention MEDICRIME.
■ L’EDQM était présente à la réunion du comité directeur de l’ICH (Fukuoka, Japon) en juin et à l’Assemblée de l’ICH (Jacksonville, États-Unis) en décembre. Elle a également poursuivi sa participation aux Groupes de Travail sur la mise en œuvre des Guidelines ICH Q3D et Q11.
■ Autres réunions et manifestations internationales importantes :
f International Drug Regulatory Regulators Meeting (Hyderabad, Inde) en janvier,
f Assemblée générale annuelle de l’IPEC (Nice) en février,
f Conférence APV/IPEC Europe sur les excipients (Barcelone) en septembre,
f EuroMeeting 2015 de la DIA (Paris) en avril, dont une session était consacrée au 50e anniversaire de l’EDQM,
f Forum DIA-CMC Japon 2015 (Tokyo) en juin,
f Conférence du 5e anniversaire de l’ALMBiH (Sarajevo),
f 17e conférence panrusse sur la réglementation dans le domaine des produits pharmaceutiques et dispositifs médicaux (Moscou) en octobre,
f Symposium annuel de l’Innovation & Quality Consortium (Washington) en octobre.
■ En octobre, l’EDQM a également participé au Congrès mondial de la FIP (Fédération internationale pharmaceutique), à Düsseldorf, et obtenu le statut de « Predominantly Scientific Observer Organisation » lors du Conseil de la FIP.
RÉUNIONS ORGANISÉES EN PARTENARIAT AVEC L’EDQM
■ En mai, l’EDQM a accueilli la 21e réunion formelle du HMPWG (groupe de travail du HMA sur les médicaments homéopathiques). Elle a statut d’observateur auprès du HMPWG depuis plus de 10 ans. Par ailleurs, certains experts du HMPWG sont également membres des Groupes de Travail HMM (méthodes de fabrication homéopathique) et HOM (matières premières et souches homéopathiques) de la Ph. Eur. Ces échanges et collaborations constituent pour l’EDQM une précieuse plate-forme de discussion sur les questions réglementaires et scientifiques liées à ces médicaments.
■ En juin, l’EDQM a accueilli une réunion du Groupe de Travail ICH Q3D.
■ En août, elle a pris part au deuxième séminaire international multicentrique sur l’intégrité des données, coorganisé avec la FDA américaine dans trois villes de Chine (la première série de séminaires s’était déroulée en Inde en novembre 2014). Ces rencontres a fourni à l’EDQM une excellente opportunité de former plus d’un millier de représentants de l’industrie et des autorités réglementaires aux attentes des inspecteurs sur cette importante question.
■ En octobre, l’EDQM a accueilli la 7e réunion du Cercle d’Experts PIC/S sur les substances actives, qui a rassemblé 90 délégués de 40 organisations (autorités participantes et candidates, partenaires et non-membres du PIC/S) de toute l’Europe, des Amériques, d’Australie, d’Asie et d’Afrique. L’objectif était, de façon générale, de renforcer la coopération internationale et faciliter le partage d’expériences en matière d’inspections. Différents sujets ont été abordés : intégrité des données, étalons de référence chimiques, impuretés génotoxiques, évaluation du risque pour la sélection des sites à inspecter ou réinspecter, ICH Q7, déficiences les plus couramment rencontrées lors des inspections.

Rapport annuel ► Page 38
EXPOSITIONS ET SALONS INTERNATIONAUX — RENFORCER LA PRÉSENCE DE L’EDQM À L’INTERNATIONAL
■ Dans un contexte de croissance soutenue des
échanges commerciaux internationaux de substances
pharmaceutiques actives, de génériques et de produits
pharmaceutiques finis, les salons internationaux
constituent pour l’EDQM un bon moyen de rester en
contact avec les fabricants, associations et parties
intéressées à l’échelle locale et de présenter ses
derniers produits et services.
■ Cette année, l’EDQM a participé à trois salons
pharmaceutiques — CPhI China (Shanghai),
CPhI Europe (Madrid) et CPhI India (Mumbai) —, qui
lui ont permis de mieux faire connaître ses activités.
Par ailleurs, plusieurs réunions avaient été organisées
au préalable entre visiteurs du salon et représentants
de la Division Certification de l’EDQM. L’objectif était
d’informer les demandeurs sur les aspects pratiques
de la procédure de Certification, de les aider à clarifier
certains points et à résoudre les difficultés qu’ils
pouvaient rencontrer.
■ En juin, l’EDQM a été invitée au symposium
organisé par la Chambre de commerce chinoise
pour l’importation et l’exportation des médicaments
et produits de santé (CCCPMHPIE). Plus de
120 participants, principalement originaires de Chine,
ont participé à la manifestation. Les présentations
étaient centrées sur la Ph. Eur. et la procédure de
Certification.
■ En outre, l’EDQM a participé au 25e Congrès
régional de la SITS, organisé à Londres en conjonction
avec le 33e Congrès de la Société britannique
de transfusion sanguine. Cette manifestation a
rassemblé des professionnels de santé intervenant
dans le domaine de la transfusion sanguine et de
la médecine transfusionnelle, qui ont discuté des
récentes évolutions en médecine transfusionnelle. La
18e édition du Guide Sang (Guide for the Preparation,
Use and Quality Assurance of Blood Components)
de l’EDQM a été largement promue pendant le
salon, où les visiteurs ont pu se renseigner sur les
activités de l’EDQM dans ce domaine et en apprendre
davantage sur son programme d’essais d’aptitude
pour les établissements du sang, ainsi que sur son
programme B-MQ.
CAMPAGNES DE SENSIBILISATION DU PUBLIC
Transplantation d’organes
■ Le 24 mars, une marche contre le trafic d’organes
a été organisée sur une partie des Chemins de Saint-
Jacques-de-Compostelle, à l’occasion d’une réunion
du CD-P-TO. Conduite par les experts du CD-P-TO, cette
initiative a impliqué des représentants d’organisations
telles que l’OMS, l’Organización Nacional de Trasplantes
(ONT) espagnole et Swisstransplant. Arrivés à la
cathédrale de Saint-Jacques-de-Compostelle,
les marcheurs et le grand public ont assisté à une
cérémonie célébrée spécialement pour l’occasion
(jubilé).

2015 : une année riche en manifestations et réunions ► Page 39
traduire la brochure, en vue d’une distribution locale, le clip vidéo a été traduit et diffusé en République slovaque et au Portugal, et le logo de la JEDO figurait sur le site internet du Ministère fédéral allemand de la santé. Plusieurs autres célébrations ont eu lieu simultanément dans les États membres du Conseil de l’Europe.
■ L’EDQM a, en outre, participé au 17e Congrès de la Société européenne de transplantation d’organes (ESOT) et au 24e Congrès de l’EATB. Ces deux manifestations lui ont permis d’échanger avec des scientifiques et des professionnels intervenant dans les domaines concernés, et de se renseigner sur les évolutions récentes.
■ Le but de cette marche était de sensibiliser le public au trafic d’organes humains et de promouvoir la lutte contre ce crime. Elle a revêtu une importance toute particulière, car elle était organisée à la veille de l’ouverture à la signature de la nouvelle Convention
du Conseil de l’Europe contre le trafic d’organes humain et juste avant une conférence internationale de haut niveau qui s’est déroulée à Saint-Jacques-de-Compostelle les 25-26 mars. La Convention, adoptée le 9 juillet 2014 par le Comité des Ministres du Conseil de l’Europe, vise à harmoniser le système pénal en Europe pour permettre de poursuivre plus efficacement les personnes et organisations criminelles responsables du trafic.
■ La 17e Journée européenne du don d’organes et la greffe (JEDO) s’est déroulée le 10 octobre, à Lisbonne. Elle était parrainée par le Ministère portugais de la Santé, l’Institut portugais du sang et de la transplantation (IPST) et la Société portugaise de transplantation (SPT). La journée a commencé par une cérémonie officielle, en présence de nombreux représentants et du Président du Conseil de l’IPST, du Président de la SPT, de membres du CD-P-TO, de l’EDQM/Conseil de l’Europe, de donneurs, de patients transplantés et de leurs familles, et du maire de Lisbonne.
■ La JEDO 2015 avait pour thème « Art & Transplantation » et différentes activités étaient organisées dans toute la ville pour les enfants et leurs familles. La journée était également promue en ligne et sur les réseaux sociaux, notamment Twitter et YouTube. Comme à l’accoutumée, l’EDQM avait installé un stand d’information, aux côtés de l’IPST. L’initiative a remporté un large soutien en Europe : une campagne Thunderclap a été lancée par la Pulmonary Hypertension Association Europe et a réuni 100 000 abonnés en un mois ; plusieurs pays (p. ex., Bosnie-Herzégovine et Croatie) ont demandé à

Rapport annuel ► Page 40
VISITES OFFICIELLES
■ En janvier, l’EDQM a ouvert ses portes aux
Représentations Permanentes du Conseil de l’Europe.
Cette visite a permis aux délégations de mieux
comprendre les différentes activités de l’organisation,
et d’en mesurer l’impact sur les politiques de santé
publique et le contrôle qualité des médicaments en
Europe, et au-delà.
■ Toujours en janvier, une délégation de la FDA
ghanéenne a été reçue et a rencontré la Division
Certification de l’EDQM, afin de se familiariser avec
la procédure de Certification, pour les substances
pharmaceutiques, et de partager le savoir-faire et
l’expérience de l’EDQM.
■ Plus tard dans l’année, des membres français
de l’Assemblée parlementaire du Conseil de l’Europe
sont venus assister à une présentation générale des
activités de l’EDQM.
■ En juin, l’EDQM a accueilli une délégation
officielle de la FDA sud-coréenne, dont la visite était
axée sur les travaux de l’EDQM dans le domaine de
la libération des lots par les autorités de contrôle
(OCABR) et de la libération des lots pour les produits
biologiques humains.
■ En novembre, une délégation du NIFDC (National
Institutes for Food and Drug Control) et de la FDA
chinoise a visité l’EDQM. Les sujets abordés portaient
sur le contrôle qualité et la standardisation, et l’accent
a été mis sur les activités du Laboratoire de l’EDQM
et du Réseau OMCL.
■ Plus tard dans le mois, des représentants de
la Communauté d’Afrique de l’Est ont effectué une
visite d’étude à l’EDQM, portant notamment sur
l’établissement et l’utilisation des étalons de référence,
et les activités du Réseau OCML.

Liste des comités coordonnés par l’EDQM ► Page 41
LA COMMISSION EUROPÉENNE DE PHARMACOPÉE
■ La Commission européenne de Pharmacopée (la « Commission ») a été créée en 1964 en application de la Convention relative à l’élaboration d’une pharmacopée
européenne. Suite à la ratification de cette convention par l’Ukraine en décembre 2012, elle compte désormais des représentants des 38 parties signataires (37 États et l’Union européenne). Les 28 observateurs, du monde entier, témoignent de l’importance des travaux de la Commission à l’échelle internationale. La Commission décide du programme de travail et adopte les normes relatives à la qualité des médicaments et de leurs composants, applicables sur les territoires des États membres. Actuellement, 72 groupes d’experts et groupes de travail, mis en place par la Commission, mènent à bien le programme de travail de la Ph. Eur. Fin 2015, 2302 normes qualité et 354 textes généraux, comprenant notamment des méthodes d’analyse, avaient ainsi été élaborés, adoptés et mis en application. L’actualisation continue de ces textes permet de les tenir à jour des progrès scientifiques et techniques en matière de développement, de production et de contrôle qualité des médicaments. La Ph. Eur. est primordiale pour la protection de la santé publique. Elle s’adresse aux professionnels de la santé qui travaillent dans le domaine des médicaments, secteur dans lequel elle est devenue une référence.
PROGRAMME DE STANDARDISATION BIOLOGIQUE, COMITÉ DIRECTEUR
■ Le Programme de standardisation biologique (PSB) couvre la normalisation des méthodes et outils
relatifs au contrôle qualité des produits biologiques, en établissant des étalons de référence et en validant de nouvelles méthodes, particulièrement celles qui sont axées sur les 3R en matière d’essais sur animaux. Ces activités sont supervisées par le Comité directeur du PSB, qui réunit les présidents des Groupes d’Experts 6, 6B, 15 et 15V de la Ph. Eur., ainsi que des experts et des délégués cooptés de la Commission européenne, de l’EMA, du Groupe de Travail BWP, du Groupe de Travail IWP et de l’OMS, aux côtés de la Directrice de l’EDQM.
RÉSEAU DES LABORATOIRES OFFICIELS DE CONTRÔLE DES MÉDICAMENTS (OMCL), COMITÉS CONSULTATIFS
■ Le rôle de ce Réseau est de s’assurer de la qualité constante des médicaments disponibles sur les différents marchés nationaux et de contribuer à la reconnaissance mutuelle des contrôles de qualité effectués sur les médicaments par les États membres. Les décisions importantes sont prises lors des assemblées plénières annuelles du Réseau. Les comités consultatifs préparent le programme de travail annuel et veillent à sa mise en œuvre. Il existe deux niveaux de collaboration au sein du Réseau :
f les activités générales, ouvertes à tous les États signataires de la Convention relative à
l’élaboration d’une pharmacopée européenne et aux États observateurs auprès de la Commission (après une procédure d’audit et d’accréditation). Les activités générales sont relatives aux travaux menés dans le domaine des systèmes de MQ, notamment les audits et les PTS, ainsi qu’aux
Liste des comités coordonnés par l’EDQM

MSS et à la contribution à la lutte contre la
contrefaçon et les médicaments illégaux. Ces
activités sont préparées et suivies par le Comité
consultatif du Réseau général des OMCL,
f les activités restreintes aux pays de l’UE et
de l’Espace économique européen (EEE), qui
concernent les produits faisant l’objet d’une
autorisation de mise sur le marché centralisée,
les produits autorisés selon la procédure de
reconnaissance mutuelle ou la procédure
décentralisée (PRM/PDC) et le système OCABR
en vigueur pour les produits biologiques
(humains et vétérinaires). La Suisse et Israël (pour
les vaccins humains uniquement) participent
également à cette dernière activité. Pour les
activités CAP et OCABR, des groupes consultatifs
veillent, entre deux réunions annuelles, à la
continuité du fonctionnement de chacun des
Réseaux spécifiques.
CERTIFICATION DE CONFORMITÉ AUX MONOGRAPHIES DE LA PH. EUR., COMITÉ DIRECTEUR
■ Un réseau de quelque 100 assesseurs et
30 inspecteurs nationaux participe aux travaux liés
à l’évaluation des dossiers Qualité des substances
actives et à l’inspection des sites de fabrication.
Les activités en rapport avec la procédure de
Certification de conformité aux monographies de
la Ph. Eur. sont pilotées par un comité directeur et
3 comités techniques consultatifs (CTC). Le comité
directeur est composé de représentants des autorités
européennes d’enregistrement et d’inspection. Il
prend des décisions de politique générale, examine
et commente les questions soulevées par les CTC,
adopte les guidelines et le programme d’inspection et
coordonne les questions entre les parties représentées.
Il est, en outre, chargé de désigner les assesseurs, ainsi
que les membres des CTC et leurs présidents.
COMITÉ EUROPÉEN SUR LA TRANSFUSION SANGUINE (CD-P-TS)
■ Ce comité traite des questions éthiques,
juridiques et organisationnelles relatives à la
transfusion sanguine, afin de garantir l’innocuité, la
qualité et une utilisation optimale du sang, de protéger
les donneurs et les patients transfusés et d’éviter
le gaspillage. Il supervise les travaux d’un certain
nombre de groupes de travail et de projets individuels,
notamment la Base de données européenne sur les
réserves de sang congelé de groupes rares, la gestion
de l’approvisionnement en plasma et l’élaboration du
Guide pour la préparation, l’utilisation et l’assurance de
qualité des composants sanguins par un groupe de
travail ad hoc.
COMITÉ EUROPÉEN SUR LA TRANSPLANTATION D’ORGANES (CD-P-TO)
■ Ce comité met l’accent sur l’élaboration et la
promotion du principe de non-commercialisation du
don d’organes, de tissus ou de cellules, le renforcement
des mesures de lutte contre le trafic et l’élaboration
de normes éthiques exigeantes en matière de qualité
et d’innocuité dans le domaine de la transplantation.
Il supervise un certain nombre de projets individuels
et les travaux des groupes de travail ad hoc chargés
de l’élaboration du Guide sur la qualité et la sécurité
des organes destinés à la transplantation et du Guide
sur la qualité et la sécurité des cellules et tissus destinés
à des applications chez l’homme.
COMITÉ EUROPÉEN SUR LES PRODUITS ET LES SOINS PHARMACEUTIQUES (CD-P-PH)
■ Les activités de ce comité incluent l’amélioration
de la santé publique et la réduction des inégalités
sanitaires grâce à l’élaboration de dispositions et
de pratiques harmonisées (notamment l’usage
rationnel des médicaments), la réduction des
risques de santé publique liés à la contrefaçon des
médicaments et le mécanisme de suivi plurisectoriel
et pluridisciplinaire assuré par le Comité des Parties
à la Convention MEDICRIME. Le comité supervise
également les programmes d’activités de plusieurs
organes subordonnés : le Comité d’Experts sur la
classification des médicaments en matière de leur
délivrance (CD-P-PH/PHO), le Comité d’Experts sur
les normes de qualité et de sécurité relatives à la
pratique et au suivi pharmaceutiques (CD-P-PH/PC)
et le Comité d’Experts sur la réduction des risques de
santé publique liés à la contrefaçon des médicaments
et à la criminalité connexe (CD-P-PH/CMED).
COMITÉ DE PROTECTION DE LA SANTÉ DES CONSOMMATEURS (CD-P-SC)
■ Le CD-P-SC est responsable de la gestion du
programme de travail et de la prise de décisions
dans le domaine des cosmétiques et des matériaux
pour contact alimentaire. Il supervise deux organes
subordonnés, qui sont chargés de l’examen des
questions de santé et de l’évaluation des risques et
qui rédigent des rapports et recommandations en
matière d’approche réglementaire :
f le Comité d’Experts sur les matériaux pour
contact alimentaire (P-SC-EMB),
f le Comité d’Experts sur les produits cosmétiques
(P-SC-COS), qui interagit avec le Réseau
européen des laboratoires officiels de contrôle
des cosmétiques (OCCL).
Rapport annuel ► Page 42
Glossaire ► Page 43
Glossaire
3R « Remplacement, Réduction, Raffinement », approche appliquée en vue de la protection
des animaux utilisés à des fins expérimentales
AATB American Association of Tissue Banks
AFMPS Agence fédérale des Médicaments et des Produits de Santé (Belgique)
AGES Agentur für Gesundheit und Ernährungssicherheit, OMCL autrichien
ALIMS Medicines and Medical Devices Agency (Serbie)
AMC Audit mutuel conjoint
AMM Autorisation de mise sur le marché
ANP Autorités nationales de Pharmacopée
APEC Asian-Pacific Economic Cooperation
B-AMC Audits mutuels conjoints, dans le domaine du sang
BfR Bundesinstitut für Risikobewertung, institut fédéral allemand d’évaluation des risques
B-MQ Management de la qualité, dans le domaine du sang
BPD Bonnes pratiques de distribution
BPF Bonnes pratiques de fabrication
BPPh Bonnes pratiques de pharmacopée
B-PTS Blood Proficiency Testing Scheme, essais d’aptitude, dans le domaine du sang
B-VF Visite de formation, dans le domaine du sang
B-VMC Visites mutuelles conjointes, dans le domaine du sang
BWP Biologics Working Party, Groupe de Travail Biologie (EMA)
CAP Centrally Authorised Product, produit autorisé par voie centralisée
CCCPMHPIE China Chamber of Commerce for Import and Export of Medicines and Health Products
CCR Centre commun de recherche de la Commission européenne (UE)
CEN Comité européen de normalisation
CEP Certificat de conformité aux monographies de la Pharmacopée Européenne
CHMP Committee for Medicinal Products for Human Use (EMA)
CODA-CERVA Centre d’Étude et de Recherches Vétérinaires et Agrochimiques (Belgique)
CVMP Committee for Medicinal Products for Veterinary Use (EMA)
DCI Dénomination commune internationale (OMS)
DG SANTE Direction générale Santé et sécurité alimentaire (UE)
DH-BIO Comité de Bioéthique du Conseil de l’Europe
DIA Drug Information Association
EATB European Association of Tissue Banks
ECBS Comité OMS d’experts de la standardisation biologique
ECSPP Comité OMS d’experts sur les spécifications relatives aux préparations pharmaceutiques
EDQM Direction Européenne de la Qualité du Médicament & Soins de Santé
EDTCO European Donation and Transplant Coordination Organisation
EEE Espace économique européen
EMA European Medicines Agency, Agence européenne des médicaments
EMVO European Medicines Verification Organisation
EMVS European Medicines Verification System
EPAA European Partnership for Alternative Approaches to Animal Testing
Rapport annuel ► Page 44
ER Etalons de référence
ERV Etalons de référence végétaux
ES Etablissement du sang
ESHRE European Society for Human Reproduction and Embryology
ESOT Société européenne de transplantation d’organes
FIP Fédération internationale pharmaceutique
GDP Groupe de discussion des Pharmacopées
GEON General European Network of Official Medicines Control Laboratories, réseau général des OMCL
GPG Good Practice Guidelines, guidelines de bonnes pratiques
GTP Gene Therapy Products, produits de thérapie génique
HMA Heads of Medicines Agencies
HMPC Committee on Herbal Medicinal Products (EMA)
ICH International Council for Harmonisation of Technical Requirements for Pharmaceuticals for
Human Use
IDMP Identification of Medicinal Products, identification des médicaments (normes ISO)
IFPMA International Federation of Pharmaceutical Manufacturers & Associations
IGDRP International Generic Drug Regulators Programme
IPEC International Pharmaceutical Excipients Council
IPFA International Plasma Fractionation Association
IPST Instituto Português do Sangue e da Transplantação, Institut portugais du sang et de la
transplantation
ISA International Standards for Antibiotics (étalons internationaux d’antibiotiques), OMS(OMS)
ISN International Society of Nephrology
ISO Organisation internationale de normalisation
IWP Immunologicals Working Party, Groupe de Travail Immunologie (EMA)
JEDO Journée européenne du don d’organes et de la greffe
JP Pharmacopée japonaise
MCC Medicines Control Council, Afrique du Sud
MIV Médicaments immunologiques vétérinaires
MQ Management de la qualité
MSSIP Market Surveillance Studies on Suspicious Illegal Products
MSS Market surveillance study, étude de surveillance du marché
NAMMD Agence du médicament, Roumanie
NIFDC National Institute for Food and Drug Control, Chine
OCABR Official Control Authority Batch Release, libération officielle des lots par les autorités de
contrôle
OCCL Official Cosmetics Control Laboratories, laboratoires officiels de contrôle des cosmétiques
OMCL Official Medicines Control Laboratory, laboratoire officiel de contrôle des médicaments
OMS Organisation mondiale de la Santé
ONT Organización Nacional de Trasplantes (Espagne)
PaedForm Formulaire pédiatrique
PBR Préparation biologique de référence
PCU Points de contact uniques
PCQIP Pharmaceutical Care Quality Indicators Project, EDQM
PDC Procédure décentralisée
PEI Paul-Ehrlich Institut
Glossaire ► Page 45
Ph. Eur. Pharmacopée Européenne
PIC/S Pharmaceutical Inspection Convention and Pharmaceutical Inspection Co-operation Scheme
PRM Procédure de reconnaissance mutuelle
PSB Programme de standardisation biologique
PTS Proficiency Testing Scheme, essais d’aptitude
Q3D Guideline ICH sur les impuretés élémentaires
QbD Quality by design
QWP Quality Working Party (EMA)
RBR Réactif biologique de référence
RIVM National Institute for Public Health and the Environment (Pays-Bas)
RMN Résonance magnétique nucléaire
SCR Substance chimique de référence
SCRI Substance chimique de référence internationale
SDA Systèmes de délivrance automatisée
SITS Société internationale de transfusion sanguine
SPT Sociedade Portuguesa de Transplantação, société portugaise de transplantation
TGA Therapeutic Goods Administration (Australie)
TTS The Transplantation Society
UE Union européenne
USFDA ou FDA United States Food and Drug Administration
USP United States Pharmacopeia
VBRN Veterinary Batch Release Network, sous- Réseau chargé de la libération des lots de
médicaments vétérinaires
VHC Virus de l’hépatite C
VICH Veterinary International Conference on Harmonization
VIH Virus de l’immunodéficience humaine
VMC Visite mutuelle conjointe
WIV-ISP Institut Scientifique de Santé Publique (Belgique)




FRA
Le Conseil de l’Europe est la principale organisation de défense des droits de l’homme du continent. Sur ses 47 États membres, 28 sont aussi membres de l’Union européenne. Tous les États membres du Conseil de l’Europe ont signé la Convention européenne des droits de l’homme, un traité visant à protéger les droits de l’homme, la démocratie et l’État de droit. La Cour européenne des droits de l’homme contrôle la mise en œuvre de la Convention dans les États membres.
PR
EM
S 0
32
41
6 –
PR
DD
-16
-02
Direction européennede la qualité du médicament& soins de santé
European Directoratefor the Quality
of Medicines& HealthCare
Cette publication passe en revue les
travaux menés au cours de l’année 2015
par la Direction européenne de la
qualité du médicament & soins de santé,
et souligne les résultats obtenus.





























