Embed Size (px)
Citation preview

Ab initio study of Jahn-Teller distortions for the divacancy in silicon
D. V. Makhov and Laurent J. LewisDépartement de Physique et Regroupement Québécois sur les Matériaux de Pointe (RQMP) Université de Montréal,
Case Postale 6128, Succursale Centre-Ville, Montréal, Québec, Canada H3C 3J7�Received 20 May 2005; published 12 August 2005�
The character of Jahn-Teller distortions for the neutral divacancy in silicon has been studied using densityfunctional theory calculations. It is found that the resonant bond distortion has slightly lower energy �by�10 meV� than the large pairing mode, and that the transition from one mode to the other is not hampered bya potential barrier. Thus, at room temperature, the system should oscillate between these two modes, asdemonstrated by ab initio molecular dynamics calculations.
DOI: 10.1103/PhysRevB.72.073306 PACS number�s�: 61.72.Bb, 61.72.Ji
It is well known that monovacancies in silicon are verymobile and quickly form divacancies, which are significantlymore stable and can thus exist for a long time. This makesdivacancies one of the most fundamental defects in crystal-line silicon.
The ideal unrelaxed divacancy has a trigonal symmetry�D3�. However, due to doubly degenerate levels in the bandgap, the divacancy is subject to a Jahn-Teller distortion. Wat-kins and Corbett1 found from electron paramagnetic reso-nance �EPR� measurements that V2
+ and V2− have monoclinic
symmetries �C2h�. However, the exact character of this dis-tortion is still disputed. Watkins and Corbett proposed a largepairing �LP� distortion mode �see Fig. 1� where two pairs ofthree-coordinated atoms form new bonds and two atomshave dangling bonds. On the other hand, based on the resultsof ab initio calculations for V2
+ and V2−, Saito and Oshiyama2
proposed the inverse distortion mode, called resonant bond�RB� distortion �see Fig. 1�, where two atoms become five-coordinated. Furthermore, density functional theory �DFT�calculations by Seong and Lewis3 gave the RB distortion forthe neutral divacancy, while calculations by Öğüt andChelikowsky4 showed that both neutral and singly chargeddivacancies exhibit the LP distortion. From a positron anni-hilation study of the preferable divacancy orientation underuniaxial stress, Nagai et al.5 concluded that the neutral diva-cancy is LP distorted. However, these authors did not con-sider the possibility that uniaxial stress can cause not onlythe reorientation of the divacancy but also a change from onedistortion mode to another. Finally, Pesola et al.6 proposed,based on ab initio calculations, that the neutral divacancy haslower symmetry �S2� and exhibits a “mixed” distortion char-acter.
In order to resolve this issue, we have performed exten-sive ab initio calculations to sample the potential energy sur-face of silicon divacancies in various charge states. We haveused the Vienna Ab initio Simulation Package �VASP�, whichemploys a pseudopotential DFT with the projectoraugmented-wave method �PAW�.7,8 The code was slightlymodified in order to keep constant the distance of selectedpairs of atoms in the process of geometry optimization.
The calculations were carried out using a 216-atom super-cell, an energy cutoff of 18 Ry, and the local-density ap-proximation �LDA� for the exchange-correlation functional.
Because of the large size of the supercell, Brillouin zonesampling was restricted to the � point. In order to test theconvergence with respect to system size, some calculationswere also performed for a 512-atom supercell with lowerenergy cutoff as discussed below.
First, we attempted to optimize the geometry of the diva-cancy using the unrelaxed configuration as a starting point.We used the conjugated-gradient algorithm up to a maximumforce of 3 meV/Å; the atoms were initially randomly dis-placed by 0–0.25 Å from their perfect crystal positions inorder to break the symmetry. Unfortunately, because of thevery flat potential energy surface, the system always endedup in some state with no symmetry at all, and different re-sults were obtained for different random initial configura-tions.
In order to override this problem, we explored all possibledivacancy configurations with C2h symmetry by performingsymmetry-conserving geometry optimization. In the initialconfiguration, all atoms, excluding the six threefold atomsaround the divacancy �labeled 1–6 in Fig. 1�, were placed intheir original positions. Atoms 1–6 were shifted in the direc-tion of the divacancy axis in such a way that the distances d12and d45 are equal �=a�, and distances d13, d23, d46, and d56 areequal �=b�. The system was then fully relaxed, except for thedistance a, which was kept constant in order to study the
FIG. 1. �Color online� Schematic view of LP and RB divacancydistortion modes.
PHYSICAL REVIEW B 72, 073306 �2005�
1098-0121/2005/72�7�/073306�3�/$23.00 ©2005 The American Physical Society073306-1
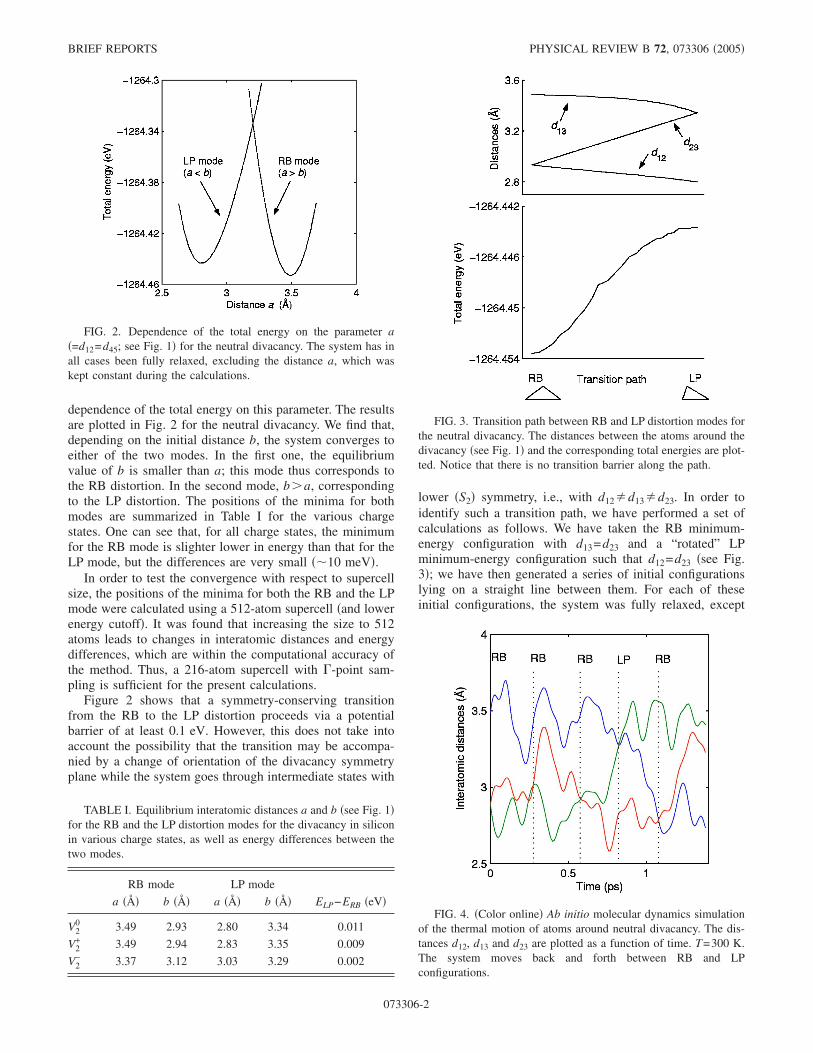
dependence of the total energy on this parameter. The resultsare plotted in Fig. 2 for the neutral divacancy. We find that,depending on the initial distance b, the system converges toeither of the two modes. In the first one, the equilibriumvalue of b is smaller than a; this mode thus corresponds tothe RB distortion. In the second mode, b�a, correspondingto the LP distortion. The positions of the minima for bothmodes are summarized in Table I for the various chargestates. One can see that, for all charge states, the minimumfor the RB mode is slighter lower in energy than that for theLP mode, but the differences are very small ��10 meV�.
In order to test the convergence with respect to supercellsize, the positions of the minima for both the RB and the LPmode were calculated using a 512-atom supercell �and lowerenergy cutoff�. It was found that increasing the size to 512atoms leads to changes in interatomic distances and energydifferences, which are within the computational accuracy ofthe method. Thus, a 216-atom supercell with �-point sam-pling is sufficient for the present calculations.
Figure 2 shows that a symmetry-conserving transitionfrom the RB to the LP distortion proceeds via a potentialbarrier of at least 0.1 eV. However, this does not take intoaccount the possibility that the transition may be accompa-nied by a change of orientation of the divacancy symmetryplane while the system goes through intermediate states with
lower �S2� symmetry, i.e., with d12�d13�d23. In order toidentify such a transition path, we have performed a set ofcalculations as follows. We have taken the RB minimum-energy configuration with d13=d23 and a “rotated” LPminimum-energy configuration such that d12=d23 �see Fig.3�; we have then generated a series of initial configurationslying on a straight line between them. For each of theseinitial configurations, the system was fully relaxed, except
TABLE I. Equilibrium interatomic distances a and b �see Fig. 1�for the RB and the LP distortion modes for the divacancy in siliconin various charge states, as well as energy differences between thetwo modes.
RB mode LP mode
ELP−ERB �eV�a �Å� b �Å� a �Å� b �Å�
V20 3.49 2.93 2.80 3.34 0.011
V2+ 3.49 2.94 2.83 3.35 0.009
V2− 3.37 3.12 3.03 3.29 0.002
FIG. 2. Dependence of the total energy on the parameter a�=d12=d45; see Fig. 1� for the neutral divacancy. The system has inall cases been fully relaxed, excluding the distance a, which waskept constant during the calculations.
FIG. 3. Transition path between RB and LP distortion modes forthe neutral divacancy. The distances between the atoms around thedivacancy �see Fig. 1� and the corresponding total energies are plot-ted. Notice that there is no transition barrier along the path.
FIG. 4. �Color online� Ab initio molecular dynamics simulationof the thermal motion of atoms around neutral divacancy. The dis-tances d12, d13 and d23 are plotted as a function of time. T=300 K.The system moves back and forth between RB and LPconfigurations.
BRIEF REPORTS PHYSICAL REVIEW B 72, 073306 �2005�
073306-2

for the distance d23, which was kept constant �for intermedi-ate states, d12�d23�d13�.
The dependence of the equilibrium interatomic distancesd12 and d13 on d23, and the corresponding total energies, areshown in Fig. 3 for the neutral divacancy. One can see thatthere is no transition barrier between the RB and LP distor-tions. Moreover, the energy difference between these twostates ��10 meV� is smaller than the thermal energy at roomtemperature. Thus, the system should oscillate between thetwo configurations, spending most of its time in intermediatestates. Ab initio molecular dynamics simulations atT=300 K confirm the above conclusion. As can be seen inFig. 4, the system moves back and forth between the RB andthe LP states, activated by the temperature.
As a final remark, our results are consistent with the ex-perimental findings of Nagai et al.:5 the approximate equilib-rium between RB and LP distortion modes can easily bedestroyed by an uniaxial external stress, forcing divacancies
to choose one of the two distortion modes �together with oneof the two possible orientations�.
In conclusion, we have studied the Jahn-Teller distortionsof the divacancy in silicon using DFT calculations. We havefound that the resonant bond distortion has slightly lowerenergy than the pairing distortion ��10 meV�. The transitionfrom one mode to the other occurs without any potentialbarrier and is accompanied by a rotation of the symmetryplane of the divacancy. Thus, at room temperature, a diva-cancy should oscillate between these two distortion modes,as indeed demonstrated by our calculations.
This work was supported by grants from the Natural Sci-ences and Engineering Research Council �NSERC� ofCanada and the “Fonds Québécois de la recherche sur lanature et les technologies” �FQRNT� of the Province ofQuébec. We are indebted to the “Réseau québécois de calculde haute performance” �RQCHP� for generous allocations ofcomputer resources.
1 G. D. Watkins and J. W. Corbett, Phys. Rev. 138, A543 �1965�.2 M. Saito and A. Oshiyama, Phys. Rev. Lett. 73, 866 �1994�.3 H. Seong and L. J. Lewis, Phys. Rev. B 53, 9791 �1996�.4 S. Öğüt and J. R. Chelikowsky, Phys. Rev. Lett. 83, 3852 �1999�.5 Y. Nagai, K. Inoue, Z. Tang, I. Yonenaga, T. Chiba, M. Saito, and
M. Hasegawa, Physica B 340-342, 518 �2003�.6 M. Pesola, J. von Boehm, S. Pöykkö, and R. M. Nieminen, Phys.
Rev. B 58, 1106 �1998�.7 P. E. Blöchl, Phys. Rev. B 50, 17953 �1994�.8 G. Kresse and D. Joubert, Phys. Rev. B 59, 1758 �1999�.
BRIEF REPORTS PHYSICAL REVIEW B 72, 073306 �2005�
073306-3





























