Embed Size (px)
Citation preview

R
M
A
A
YÉP
HD
MMAM
KMAN
0h
ARTICLE IN PRESSG ModelEVMED-4708; No. of Pages 7
La Revue de médecine interne xxx (2013) xxx–xxx
Disponible en ligne sur
ScienceDirectwww.sciencedirect.com
ise au point
uto-anticorps au cours des myosites
utoantibody profile in myositis
. Allenbach ∗, O. Benvenistequipe Inserm U974, DHUI2B, UPMC, service de médecine interne, centre de référence des maladies neuromusculaires Paris Est, groupe hospitalier de laitié-Salpêtrière, AP–HP, 83, boulevard de l’Hôpital, 75013 Paris, France
i n f o a r t i c l e
istorique de l’article :isponible sur Internet le xxx
ots clés :yosite
uto-anticorpsyopathie nécrosante auto-immune
r é s u m é
Les patients présentant des symptômes musculaires ou une élévation des enzymes musculaires peuventsouffrir d’une myopathie. Dans ce cas, le médecin doit confirmer ce diagnostic et distinguer s’il s’agit d’unemaladie acquise ou génétique. Si la maladie est acquise, après avoir écarté une cause infectieuse, toxiqueou endocrinienne, il devra déterminer de quel type de myopathie acquise idiopathique il s’agit : soit d’unemyosite incluant la polymyosite, la dermatomyosite ou la myosite à inclusions, soit d’une myopathienécrosante auto-immune. L’examen histologique musculaire est déterminant mais la détection d’auto-anticorps est maintenant devenue cruciale dans cette démarche. Les anticorps spécifiques des myositesainsi que ceux associés aux myosites permettent une approche diagnostique sérologique complémen-taire de l’étude histologique. En effet, il existe une forte association entre un auto-anticorps spécifiqueet un phénotype de maladie musculaire et son évolution. La présence d’anticorps anti-synthétase estassociée avec une présentation histologique originale entre polymyosite et dermatomyosite et définitun syndrome où l’atteinte pulmonaire interstitielle gouverne le pronostic. L’anti-MDA-5 est uniquementobservé chez les patients présentant une dermatomyosite et définit un syndrome cutanéo-pulmonairedont l’évolution est souvent sévère. L’anti-TIF1-� est lui aussi associé à la dermatomyosite mais dans cecas sa présence est corrélée à celle d’un cancer, alors que l’anti-MI2 est associé à une forme classiquede dermatomyosite. Deux anticorps spécifiques des myopathies nécrosantes auto-immunes sont décritsà ce jour : l’anti-SRP (single recognition particle) et l’anti-HMGCR (hydroxyméthylglutaryl-coenzyme Aréductase). Leur détection est peut-être déterminante en particulier dans le cas de myopathies à débutlentement évolutif pouvant conduire à tort au diagnostic de dystrophie musculaire et privant ainsi lepatient de traitements efficaces.
© 2013 Publie par Elsevier Masson SAS pour la Société nationale française de médecine interne(SNFMI).
eywords:yositis
utoantibodiesecrotizing myopathy
a b s t r a c t
Patients suffering from muscular symptoms or with an increase of creatine kinase levels may present amyopathy. In such situations, clinicians have to confirm the existence of a myopathy and determine if it isan acquired or a genetic muscular disease. In the presence of an acquired myopathy after having ruled outan infectious, a toxic agent or an endocrine cause, physicians must identify which type of idiopathic myo-pathy the patient is presenting: either a myositis including polymyositis, dermatomyositis, and inclusionbody myositis, or an immune-mediated necrotizing myopathy. Histopathology examination of a musclebiopsy is determinant but detection of autoantibody is now also crucial. The myositis-specific antibodiesand myositis-associated antibodies lead to a serologic approach complementary to the histological clas-
sification, because strong associations of myositis-specific antibodies with clinical features and survivalhave been documented. The presence of anti-synthetase antibodies is associated with an original histopa-polymyositis and dermatomyositis, and defines a syndrome where interstitial
thologic pattern betweenPour citer cet article : Allenbach Y, Benveniste O. Auto-anticorps au cours des myosites. Rev Med Interne (2013),http://dx.doi.org/10.1016/j.revmed.2013.12.006
lung disease drives the prognosis. Anti-MDA-5 antibody are specifically associated with dermatomyosi-tis, and define a skin-lung syndrome with a frequent severe disease course. Anti-TIF1-� is also associatedwith dermatomyositis but its presence is frequently predictive of a cancer association whereas anti-MI2is associated with the classical dermatomyositis. Two specific antibodies, anti-SRP and anti-HMGCR,
∗ Auteur correspondant.Adresses e-mail : [email protected], [email protected] (Y. Allenbach).
248-8663/$ – see front matter © 2013 Publie par Elsevier Masson SAS pour la Société nationale française de médecine interne (SNFMI).ttp://dx.doi.org/10.1016/j.revmed.2013.12.006

ARTICLE IN PRESSG ModelREVMED-4708; No. of Pages 7
2 Y. Allenbach, O. Benveniste / La Revue de médecine interne xxx (2013) xxx–xxx
are observed in patients with immune-mediated necrotizing myopathies and may be very useful to dis-tinguish acquired myopathies from dystrophic muscular diseases in case of a slow onset and to allow theinitiation of effective therapy.
y Else
1
pft[
tg(coodpd
mséfilbdccp
ep
(L3déeg
daipcuc
2
mCpclmn
© 2013 Published b
. Introduction
Dans son exercice, l’interniste est amené à prendre en charge desatients souffrant de signes fonctionnels musculaires (myalgies,atigabilité musculaire à l’effort, faiblesse musculaire) ou présen-ant une élévation isolée des enzymes musculaires (créatine kinaseCK]).
Il doit alors répondre à plusieurs questions. Le patient présente--il une myopathie ? En cas de myopathie, s’agit-il d’une myopathieénétique ou acquise ? En cas de myopathie acquise idiopathiqueMAI) (après l’exclusion de causes toxiques, infectieuses ou endo-riniennes), s’agit-il d’une myosite (dermatomyosite, polymyositeu myosite à inclusions) ou d’une myopathie nécrosante, avecu sans atteinte extramusculaire ? La réponse à ces questions estéterminante car elle conditionne le traitement, le suivi et leronostic du patient. Au cours de cette démarche, la détection’auto-anticorps est devenue capitale (Fig. 1).
Outre l’examen clinique, nous disposons d’examens complé-entaires pour répondre à ces questions. Certains sont non
pécifiques et orienteront vers l’existence d’une myopathie (CK,lectromyogramme, imagerie musculaire) et d’autres plus spéci-ques permettront de préciser la nature de l’atteinte musculaire :
a biopsie musculaire et la recherche d’auto-anticorps. Si laiopsie musculaire reste un examen indispensable, la détection’auto-anticorps a pris une place capitale dans le diagnostic, lalassification nosologique et le pronostic des MAI [1]. L’utilisationonjointe de l’histologie et de l’immunologie reste déterminanteour la bonne classification des patients [2].
Soixante à 80 % des MAI sont associées à des auto-anticorps [3]t plus d’une trentaine de cibles antigéniques nucléaires ou cyto-lasmiques ont été identifiées.
Il faut distinguer les auto-anticorps spécifiques des myopathiesASM) idiopathiques des auto-anticorps associés aux MAI (AAM).es ASM ne sont observés qu’au cours des MAI et sont présents dans0 à 58 % des cas [3], alors que les AAM sont aussi présents au cours’autres maladies auto-immunes comme la sclérodermie, le lupusrythémateux systémique ou le syndrome de Gougerot-Sjögren parxemple. Chez un patient donné, il n’existe qu’un seul ASM en règleénérale qui peut parfois être associé à un ou plusieurs AAM.
Depuis les années 1980, date de découverte du premier ASM,e nombreux autres ASM ont été identifiés. Chacun d’entre eux estssocié à un type de MAI particulier et au sein d’un groupe de MAI,ls sont associés à des phénotypes particuliers de maladie avec desronostics qui leur sont propres. Ainsi, les ASM sont une aide pré-ieuse pour affirmer l’existence d’une myopathie, pour distinguerne myopathie héréditaire d’une myopathie acquise et enfin pourlasser la MAI, et en définir son pronostic.
. La « polymyosite » et les auto-anticorps
Historiquement, selon la classification de Bohan et Peter, la poly-yosite était définie par un déficit musculaire, une élévation des
K, un tracé myogène sur l’électromyogramme, et surtout par larésence d’un infiltrat inflammatoire endomysial musculaire asso-
Pour citer cet article : Allenbach Y, Benveniste O. Auto-anthttp://dx.doi.org/10.1016/j.revmed.2013.12.006
ié à une expression intense et diffuse de l’HLA de classe I pares fibres musculaires [4]. Cette définition a été secondairement
odifiée pour réserver le terme de polymyosite à des patientse présentant pas d’auto-anticorps, et le terme de myosite de
vier Masson SAS on behalf of the Société nationale française de médecineinterne (SNFMI).
chevauchement a été introduit pour reclasser les patients présen-tant en plus des auto-anticorps [1]. La classification ultérieure del’European Neuromuscular Centre (ENMC) rependra aussi le cadrenosologique des myosites de chevauchement [5]. À ce jour, presquetoutes les « ex-polymyosite » sont associées à des auto-anticorps[6] et ce terme devrait donc être abandonné comme l’avait pro-posé Amato et Griggs dans leur éditorial intitulé « Unicorns, dragons,polymyositis, and other mythological beasts » [7].
2.1. ASM et myosite de chevauchement : les anticorpsanti-synthétases
En 1981, le premier ASM, l’anticorps anti-Jo-1, a été décritchez un patient présentant une polymyosite et ayant pour ini-tiales J.O. [8]. Cet anticorps reconnaît une enzyme cytoplasmique :l’histidine-ARN (acide ribonucléique)-t-synthétase impliquée dansla synthèse protéique. À ce jour, sept autres anticorps ayant pourcible d’autres ARN-t-synthétases ont été identifiés (Tableau 1).Néanmoins, seuls les anticorps anti-Jo-1, PL-7, PL-12, EJ et OJ sontdétectables en routine par les kits commerciaux.
L’auto-anticorps anti-Jo-1 reste 4 à 5 fois plus fréquent que lesautres anti-ARN-t-synthétases [3] et est le plus fréquent des ASM,présent chez environ 20 % des patients ayant une MAI [9]. C’est à cejour l’ASM le plus étudié.
La présence d’un anticorps anti-synthétase est associée à unphénotype clinique particulier regroupant une myosite, une pneu-mopathie interstitielle (80 %), un phénomène de Raynaud (48 %),des mains de mécaniciens (27 %) et des arthralgies (77 %) ; cetteassociation définit le syndrome des anti-synthétases [3,10].
L’enjeu principal est l’atteinte pulmonaire car elle est très fré-quente et fait toute la gravité de la maladie puisque historiquementelle était associée à une survie inférieure [11]. Dans certains cas,elle est responsable d’une insuffisance respiratoire d’aggravationrapide conduisant alors le patient en réanimation avec un pronosticeffroyable. Elle peut être subaiguë et sévère ou plus lentement pro-gressive et peu sévère, voire asymptomatique. Une hypertensionartérielle pulmonaire confirmée par des mesures invasives (cathé-térisme cardiaque droit) peut aussi être présente (7,9 %) ; elle estalors précapillaire, souvent sévère, et associée à survie inférieure[12].
Ainsi, la présence d’un anticorps anti-synthétase, quels quesoient les signes cliniques respiratoires, impose une évaluation pul-monaire (scanner thoracique, épreuves fonctionnelles respiratoireset test de marche de 6 minutes, échographie cardiaque).
Il faut souligner que l’atteinte pulmonaire peut précéder le dia-gnostic de myosite puisque, selon les séries, seuls 21 à 29 % despatients présentent une myosite au moment du diagnostic séro-logique porté dans le cadre de l’exploration d’une pneumopathieinterstitielle « idiopathique » ou des polyarthralgies [13]. Néan-moins, la détection de l’atteinte musculaire dépend des moyensdiagnostiques musculaires mis en œuvre et de la durée du suivi.
Histologiquement, les myosites associées aux anticorps anti-Jo-1 se distingueraient aussi des autres « ex-polymyosites » par uneatteinte inflammatoire préférentiellement périmysiale et une atro-
icorps au cours des myosites. Rev Med Interne (2013),
phie des fibres dans la région périfasciculaire [14].Il existe au sein du syndrome des anti-synthétases des diffé-
rences phénotypiques. En effet, Hervier et al., ainsi que d’autreséquipes, ont pu montrer que les patients ayant des anticorps

ARTICLE IN PRESSG ModelREVMED-4708; No. of Pages 7
Y. Allenbach, O. Benveniste / La Revue de médecine interne xxx (2013) xxx–xxx 3
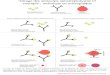
Fig. 1. Détection d’auto-anticorps et identification de leur cible antigénique. Le sérum est déposé sur les cellules HEP-2. La fixation spécifique d’anticorps à un antigènenucléaire (A : anticorps anti-PM-Scl) ou cytoplasmique (B : anticorps anti-PL-7) est révélée par un anticorps anti-IgG couplé à un fluorochrome. La cible antigénique reconnuepeut être identifiée au moyen d’un second test immuno-dot. Les bandelettes test sur lesquelles sont préalablement déposées des protéines recombinantes correspondant auxantigènes d’intérêts (ici Jo-1, PL-7, PL-12, SRP, MI-2, Ku, PM-Scl et Scl-70) sont incubées avec le sérum du patient. La fixation spécifique d’un anticorps est secondairementr anti-JoP hôpita
ammanl
TF
Mhll
évélée par une réaction enzymatique chromogénique (C : détection de l’anticorps
hotographies et examens réalisés par le Dr J.-L. Charuel (laboratoire d’immunochimie,
nti-PL-7 ou PL-12 avaient un phénotype propre avec une pneu-opathie interstitielle plus fréquente et une atteinte musculaire
Pour citer cet article : Allenbach Y, Benveniste O. Auto-anthttp://dx.doi.org/10.1016/j.revmed.2013.12.006
oins fréquente ou moins prononcée que les patients avecnticorps anti-Jo-1. De plus, ce phénotype est associé à un pro-ostic plus sombre [15–17]. Enfin, il faut souligner que le titre de
’anticorps anti-Jo-1 est corrélé à l’activité de la maladie [18].
ableau 1réquence et caractéristiques des principaux ASM.
Nom del’anticorps
Cible antigénique Fréquenceau coursdes MAI (%)
Aspfluo
Jo-1 Histidine-t-ARN-synthétase 15 CytPL-7 Thréonine-t-ARN-synthétase 5 CytPL-12 Alanine-t-ARN-synthétase 1 CytOJ Isoleucine-t-ARN-synthétase < 1 CytEJ Glycine-t-ARN-synthétase < 1 CytKS Asparagine-t-ARN-synthétase < 1 CytYRS Tyrosine-t-ARN-synthétase < 1 CytZO Phénylalanine-t-ARN-synthétase < 1 CytMI-2 Complexe nucleosome remodeling
histone deacetylase6 Cyt
TIF1-� Transcriptional intermediary factor 1-� 6 NucMDA-5 Melanoma differentiation-associated
gene 56 Cyt
SAE Small ubiquitin-like modifier activatingenzyme
5 Nuc
NXP-2 Nuclear matrix protein NXP-2 5 NucSRP Signal recognition particle 3 CytHMGCoA-R 3-hydroxy-3-méthylglutaryl-
coenzyme Aréductase
6 Cyt
AI : myopathie acquise idiopathique ; ASM : anticorps spécifique des MAI ; AAM : anticoistologique particulier avec un aspect de PM mais aussi de DM avec une atrophie périfasc
iste des anticorps représentés n’est pas exhaustive. Les fréquences sont issues pour parties MI ont été exclues.
-1).l de la Pitié-Salpêtrière).
2.2. AAM et myosite de chevauchement
icorps au cours des myosites. Rev Med Interne (2013),
Les anticorps observés au cours de nombreuses maladies auto-immunes systémiques, comme le lupus érythémateux systémique,le syndrome de Gougerot-Sjögren ou la sclérodermie, peuvent aussis’observer aux cours des MAI et des « ex-polymyosite ».
ect de larescence
Typehistologique
Détectionen routine
Associationpossible
oplasmique PM/DM Oui Atteinte pulmonaireoplasmique PM/DM Oui Atteinte pulmonaireoplasmique PM/DM Oui Atteinte pulmonaireoplasmique PM/DM Oui Atteinte pulmonaireoplasmique PM/DM Oui Atteinte pulmonaireoplasmique PM/DM Non Atteinte pulmonaireoplasmique PM/DM Non Atteinte pulmonaireoplasmique PM Non Atteinte pulmonaireoplasmique DM Oui Risque de cancer faible
léaire DM Oui Risque de cancer élevéoplasmique DM Oui Atteinte pulmonaire
léaire DM Non
léaire DM Non Forme juvénile de DMoplasmique MNAI Oui Atteinte pulmonaireoplasmique MNAI Oui
rps spécifique des MAI ; PM : polymyosite ; DM : dermatomyosite ; PM/DM : aspecticulaire ; MNAI : myopathie nécrosante auto-immune ; MI : myosite à inclusions. Lae de Koenig et al. [3] et font référence à une population de patients MAI de laquelle

ARTICLE IN PRESSG ModelREVMED-4708; No. of Pages 7
4 Y. Allenbach, O. Benveniste / La Revue de médecine interne xxx (2013) xxx–xxx
Tableau 2Fréquence et caractéristiques des principaux AAM.
Nom de l’anticorps Cible antigénique Fréquence au coursdes MAI (%)
Aspect de lafluorescence
Type histologique Association possible
Ro-52 Ro-52 30 Nucléaire PM, DM, MI SS, GSJ, LESRo-60/SSA Ro-60/SSA 11 Nucléaire PM, DM, MI SS, GSJ, LES, PRLa/SSB La/SSB 13 Nucléaire PM, DM, MI SS, GSJ, LES, PRKu Ku 23 Nucléaire PM SS, GSJ, LESU1RNP Peptide U1 du complexe RNP 15 Nucléaire PM Syndrome de SharpPM-Scl Complexe PM-Scl 9 Nucléaire PM, MNAI SS, LESMitochondrie Mitochondrie 10 Cytoplasmique PM CBP
MAI : myopathie acquise idiopathique ; ASM : anticorps spécifique des myopathies ; AAM : anticorps spécifique des MAI ; PM : polymyosite ; DM : dermatomyosite ; MNAI :myopathie nécrosante auto-immune ; MI : myosite à inclusions ; SS : sclérodermie systémique ; GSJ : syndrome de Gougerot-Sjögren ; LES : lupus érythémateux systémique ;P ive. Lep
sp
edc(pcsl
qt
RL2asml
(rodgdap
mdtp
teadptp[adss
R : polyarthrite rhumatoïde. La liste des anticorps représentés n’est pas exhaustopulation de patients MAI de laquelle les MI ont été exclues.
Il existe deux situations distinctes : soit les signes musculairesont inauguraux, soit ils surviennent chez des patients déjà suivisour une maladie auto-immune.
Dans le premier cas, la découverte d’AAM est importante carlle orientera, d’une part, vers une maladie musculaire acquise et,’autre part, conduira le plus souvent au diagnostic de myosite dehevauchement. Même si une maladie auto-immune systémiquesclérodermie systémique ou lupus par exemple) peut se révélerar une myosite [19], le plus souvent le diagnostic de myosite dehevauchement est porté isolément car même si des AAM sont pré-ents il n’existe pas, en général, de critères suffisants pour retenire diagnostic de maladie auto-immune associée.
Les principaux AAM, leur fréquence, le type de MAI relié ainsiue les possibles maladies auto-immunes associées sont représen-és dans le Tableau 2.
Dans le cas des myosites de chevauchement, l’anticorps anti-o-52 est le plus souvent rencontré (30 % des MAI) [3,20].’anticorps anti-Ku peut être également présent chez environ 1 à0 % des malades ayant une MAI [3,21]. Les patients présentant desnticorps anti-Ku ont un phénotype proche du syndrome des anti-ynthétases et cet anticorps peut être isolé ou être associé à desaladies auto-immunes, comme la sclérodermie systémique ou le
upus érythémateux systémique [22].L’association d’une myosite et d’un anticorps anti-mitochondrie
spécifique de la cirrhose biliaire primitive) est retrouvée chez envi-on 10 % des malades avec une MAI [23]. Cette observation estriginale car la présence de cet anticorps serait en rapport aveces myosites plus lentement évolutives, souvent associées à desranulomes musculaires, et une fréquence accrue d’atteinte car-iaque. L’anticorps anti-mitochondrie peut être retrouvé isolémentu cours des myosites ou en association avec une cirrhose biliairerimitive [23].
Lorsque la connectivite est connue, toutes les associations entreyosite de chevauchement et sclérodermie systémique, syndrome
e Gougerot-Sjögren, ou polyarthrite rhumatoïde ont été rappor-ées [24] mais comme nous l’avons vu la maladie musculaire esteu souvent inaugurale.
Les patients sclérodermiques peuvent présenter une myopa-hie au cours de leur évolution et, dans ce cas, seuls 10 % d’entreux ont des anticorps anti-PM-Scl ; l’association à l’anticorpsnti-URNP ou anti-Ku peut être aussi observée [25]. En présence’une scléromyosite, la prévalence de l’atteinte cardiaque seraitlus importante [25]. Les patients sclérodermiques avec myopa-hie acquise ont le plus souvent une myosite avec un meilleurronostic que lorsqu’ils présentent une myopathie nécrosante26]. On rappelle que l’anticorps anti-PM-Scl peut être observé
Pour citer cet article : Allenbach Y, Benveniste O. Auto-anthttp://dx.doi.org/10.1016/j.revmed.2013.12.006
u cours de myosite de chevauchement en l’absence de scléro-ermie. L’association d’une polymyosite à un lupus érythémateuxystémique a rarement été rapportée [27]. L’association avec unyndrome de Gougerot-Sjögren est aussi possible mais le plus
s fréquences sont issues pour partie de Koenig et al. [3] et font référence à une
souvent celui-ci est au second plan et associé au syndrome desanti-synthétases. Les myosites de chevauchement peuvent êtreassociées à la polyarthrite rhumatoïde et il existe alors le plussouvent des anticorps anti-synthétases [28].
Des anticorps anti-U1RNP peuvent aussi être retrouvés au coursMAI (10 % des cas) [3,21] ; ils s’intègrent alors dans le cadre d’uneconnectivite mixte [29] ou peuvent rester isolés. Ils sont classique-ment associés à une myosite de bon pronostic.
3. La dermatomyosite et les auto-anticorps
La dermatomyosite est définie par la présence d’une atteintecutanée et histologiquement par une myosite avec une atro-phie périfasciculaire et des dépôts endo-capillaires de complexed’attaque membranaire. Le phénotype est hétérogène puisqu’ellepeut être associée à une atteinte pulmonaire interstitielle ou uncancer. La découverte récente de nouveaux ASM a permis de défi-nir des sous-groupes plus homogènes. Des ASM sont retrouvés chezenviron 20 % des malades avec dermatomyosite [30].
3.1. ASM et dermatomyosite
3.1.1. Anticorps anti-MI-2L’anticorps anti-MI-2 est le premier anticorps à avoir été asso-
cié spécifiquement à la dermatomyosite. Il est observé chez 4 à 6 %des malades avec une MAI [3] et chez environ 10 à 20 % des der-matomyosites [21] et serait à associé à l’HLA-DR7 [31]. Cependant,une étude remet en cause sa spécificité [32]. Cet anticorps recon-naît le complexe protéique nucléaire NURD (nucleosome remodelinghistone deacetylase) impliqué dans la transcription de l’ADN. Sa pré-sence ne définit pas un sous-type de dermatomyosite mais uneforme « classique », et bien que cela soit controversé, il serait asso-cié à une diminution du risque de cancer, qui est globalement élevé(près d’un tiers des cas) au cours des dermatomyosites.
3.1.2. Les anticorps anti-TIF1-�Récemment, la présence d’anticorps anti-TIF1-� a été spécifi-
quement retrouvée au cours des dermatomyosites. Cet anticorpsest présent chez 11 % des malades ayant une dermatomyosite [33].Il reconnaît le facteur de transcription nucléaire TIF1-� (transcrip-tion intermediary factor 1 family proteins) impliqué dans la voie designalisation du TGF-�. Sa détection est intiment liée à la présenced’un cancer. La séropositivité anti-TIF1-� a une valeur prédictivepositive pour l’association avec un cancer de 42 % alors que sa néga-tivité aurait une valeur prédictive négative de 97 % [34], mais cesrésultats ne sont validés que pour les dermatomyosites de l’adulte.
icorps au cours des myosites. Rev Med Interne (2013),
La présence de cet anticorps serait à l’inverse associée à l’absenced’atteinte respiratoire [34]. Historiquement, cet ASM a été iden-tifié comme reconnaissant à la fois un antigène de 155 kDa et140 kDa. Secondairement, il a été montré que l’antigène 155 kDa

ING ModelR
e de m
cpsplco
3
gddi1ruclpnps[t[
3
dmpfumCe
3
àctiDym
4
tddcpaatAnsadf
ARTICLEEVMED-4708; No. of Pages 7
Y. Allenbach, O. Benveniste / La Revu
orrespondait à la protéine TIF1-� alors que l’antigène corres-ondant à 140 kDa était TIF1-� [33]. L’anti-TIF1-� serait lui aussipécifique de la dermatomyosite et est fréquemment associé à larésence d’anticorps anti-TIF1-� [35] ; il peut être aussi associé à
’anti-MI-2 et dans ce cas le risque de cancer serait diminué [35]. Àe jour, seuls des kits commerciaux pour la détection de anti-TIF1-�nt été développés et mis sur le marché.
.1.3. L’anticorps anti-MDA-5L’anticorps anti-MDA-5 (melanoma differentiation-associated
ene 5) est le dernier ASM de dermatomyosite pour lequel des kitse dosages commerciaux ont été mis au point et sont en course diffusion. Cet anticorps reconnaît une protéine de l’immunité
nnée impliquée dans les défenses antivirales. Il est observé chez0 à 20 % des patients atteints de dermatomyosite [30,36]. Il a étéattaché spécifiquement à la dermatomyosite et particulièrement àne forme amyopathique avec une fréquence augmentée d’ulcèresutanés (et une atteinte particulière des paumes contrairement àa dermatomyosite classique). De plus, l’association à une atteinteulmonaire très fréquente (90 % des cas) est responsable d’un pro-ostic plus sombre [30] avec un taux de mortalité très élevé enarticulier lorsque l’atteinte pulmonaire est rapidement progres-ive [37]. Le titre d’anticorps serait corrélé à l’activité de la maladie38]. Les malades rapportés sont à ce jour principalement asia-iques et l’anti-MDA-5 serait associé à un sous-type HLA particulier39].
.1.4. Les autres ASMOn peut citer d’autres ASM dans la dermatomyosite mais non
étectable en routine. L’anticorps anti-SAE (small ubiquitin-likeodifier activating enzyme) a été retrouvé spécifiquement chez desatients ayant une dermatomyosite, mais sa prévalence serait trèsaible [40,41]. Enfin, l’anticorps anti-NXP-2 ou MJ (ayant pour ciblene protéine de la matrice nucléaire) a été retrouvé principale-ent chez des patients présentant une dermatomyosite juvénile.
et anticorps serait associé plus fréquemment à une calcinose [42]t chez l’adulte il serait associé à la présence d’un cancer [43].
.2. Les AAM et dermatomyosite
Les AAM sont rarement observés au cours des dermatomyosites la différence des myosites de chevauchement. Les présentationslinico-histologiques dermatologiques du lupus érythémateux sys-émique et de la dermatomyosite sont très similaires et parfoisndiscernable en particulier dans les formes amyopathiques [44].ans ce contexte, les auto-anticorps peuvent être déterminants
compris en cas d’atteinte musculaire associée qui, histologique-ent, est aussi dans les deux cas très similaire.
. Anticorps et myopathie nécrosante auto-immune
Les myopathies nécrosantes répondent à une définition his-ologique avec la présence de fibres musculaires en nécrose et’autres en régénération, en l’absence (dans la majorité des cas)’inflammation. Cette formule histologique peut être observée auours de myopathies toxiques et, dans ce cas, le diagnostic est lelus souvent aisé, mais elle est peut être aussi bien rencontréeu cours de dystrophies musculaires que de myopathies acquisesuto-immunes. La présence d’ASM est alors déterminante, en par-iculier en raison des enjeux thérapeutiques. Bien que le premierSM des myopathies nécrosantes ait été décrit en 1986 [45], ce’est qu’en 2004 qu’a été défini un nouveau cadre nosologique au
Pour citer cet article : Allenbach Y, Benveniste O. Auto-anthttp://dx.doi.org/10.1016/j.revmed.2013.12.006
ein des MAI, distinct des myosites : les myopathies nécrosantesuto-immunes. Ces myopathies nécrosantes représentent 17 %es MAI [46]. L’aspect histologique, mais surtout l’associationréquente avec l’ASM anti-SRP (single recognition particle) puis
PRESSédecine interne xxx (2013) xxx–xxx 5
anti-HMGCR (hydroxyméthylglutaryl-coenzyme A réductase), aeu un rôle déterminant dans la création de ce cadre nososologique.
4.1. ASM et myopathies nécrosantes auto-immunes
4.1.1. Anticorps anti-SRPL’anticorps anti-SRP, décrit pour la première fois en 1986 [45],
est présent chez 3 à 6 % des patients souffrant de myopathiesacquises idiopathiques [3] et 17 % des patients ayant une myopathienécrosante auto-immune [46]. Typiquement, ces myopathies sedistinguent des autres MAI par un déficit moteur d’emblée sévère,d’installation aiguë, avec une élévation importante des enzymesmusculaires et l’absence ou la rareté de manifestations extramus-culaires. Elle nécessite des traitements prolongés en raison derechutes fréquentes [47].
Cependant, dans certains cas, l’installation est beaucoup lentemimant une dystrophie musculaire, conduisant au diagnosticerroné de dystrophie musculaire « non étiquetée » à la lumière desrésultats moléculaires. La découverte de la présence de l’anticorpsanti-SRP après parfois plusieurs années a des implications théra-peutiques majeures car elle permet de proposer des traitementsefficaces, conduisant parfois la reprise de la marche. Le titre del’anti-SRP est corrélé à l’activité de la maladie [48].
4.1.2. Anticorps anti-synthétasesLes travaux de Christopher-Stine et al. [46] ont montré que
les ASM anti-synthétases étaient retrouvés chez environ 11 % despatients ayant une myopathie nécrosante. Ce résultat souligne quedans de rares cas le syndrome des anti-synthétases peut avoir uneprésentation histologique sans inflammation mais principalementavec de la nécrose musculaire.
4.1.3. Anticorps anti-HMGCRJusqu’en 2010, seuls 26 % des patients ayant une myopathie
nécrosante avaient un ASM. L’équipe de Mammen a pu identifierla présence d’un anti-HMGCR chez 42 % des patients souffrant demyopathie nécrosante [46] et 6 % de patients MAI [49]. Les caracté-ristiques cliniques et histologiques sont proches de celles observéeschez les patients anti-SRP hormis le fait qu’on note une prise destatines dans 75 % des cas [50]. Cet anticorps est spécifique des myo-pathies nécrosantes puisqu’il n’est pas observé chez les patientstraités par statines y compris ceux ayant une intolérance aux sta-tines [51]. De plus, comme l’anti-SRP, il est corrélé à l’activité de lamaladie [52]. La détection de l’anti-HMGCR est aujourd’hui possibledans le laboratoire du Pr Boyer au CHU de Rouen.
4.2. AAM et myopathies nécrosantes auto-immunes
La sclérodermie systémique en présence ou non de l’anticorpsanti-PM-Scl peut être associée à une myopathie nécrosante [26].Le lupus érythémateux systémique pourrait être associé aussi auxmyopathies nécrosantes, cependant, il faut souligner que dans cetravail les auteurs ne retrouvent aucune association avec l’anti-SRP[53].
5. Anticorps et myosite à inclusions
La myosite à inclusions est caractérisée par la présence d’undéficit musculaire asymétrique proximal et distal touchant prin-cipalement les sujets âgés. Histologiquement, elle se distinguede la myosite de chevauchement par la présence de vacuolesbordées témoignant de la présence de mécanismes dégénératifs.
icorps au cours des myosites. Rev Med Interne (2013),
L’absence de vacuoles bordées rend le diagnostic incertain (biaisd’échantillonnage) et de fait il peut exister une hésitation avec lediagnostic de myosite de chevauchement. Cependant, dans le cas dela myosite à inclusions les traitements sont inefficaces [54]. Ainsi,

ING ModelR
6 e de m
li
5
ldtDaesi
5
ea
6i
pêd
ssoipdédcmpmmp
Mtlt[npt
7
ficndltetd
[
[
[
[
[
[
[
[
ARTICLEEVMED-4708; No. of Pages 7
Y. Allenbach, O. Benveniste / La Revu
a découverte d’ASM pourrait être une aide précieuse dans les casndécis.
.1. ASM et myosite à inclusions
Il n’existe pas stricto sensu d’ASM identifié au cours des dea myosite à inclusions mais quelques travaux récents méritent’être mentionnés. Il a été montré que 52 % des patients présen-aient un auto-anticorps dirigés contre un antigène musculaire [55].es travaux récents ont confirmé ce résultat en identifiant que cetuto-anticorps reconnaissait l’enzyme 5′-nucléotidase cytosoliquet était présent dans environ un tiers des cas de myosite à inclu-ions [56]. La spécificité de ce nouvel anticorps doit être confirmée,l n’est pas possible encore de le rechercher en routine.
.2. AAM et myosite à inclusions
Au cours de la myosite à inclusions, des AAM sont retrouvés dansnviron un tiers des cas MAI et il s’agit le plus souvent d’anticorpsnti-SSA [57].
. Rôle des anticorps dans la physiopathologie des MAI etmplications thérapeutiques
Le rôle précis des ASM au cours des MAI reste inconnu. Enarticulier, pourquoi un antigène ubiquitaire intracellulaire peuttre impliqué dans une maladie auto-immune parfois spécifique’organe ?
Comme nous l’avons précisé, il existe des arguments indirectsuggérant la pathogénicité des ASM. Nombreux sont les ASM quiont corrélés à l’activité de la maladie. De plus, des séries de casu des études font état du rôle bénéfique des traitements pouvantnfluer sur la pathogénie des auto-anticorps comme les échangeslasmatiques, les immunoglobulines intraveineuses ou l’utilisatione rituximab. Cependant, l’implication directe des ASM n’a jamaisté montrée. Il n’existe pas de cas rapporté de MAI néonatale chezes nouveau-nés de mères porteuses de ASM. Les preuves les plusonvaincantes sont celles obtenues grâce à des travaux expéri-entaux chez la souris avec l’anti-Jo-1 où l’immunisation avec la
rotéine recombinante provoque l’apparition d’une inflammationusculaire et pulmonaire [58]. Néanmoins, le transfert passif de layopathie à l’animal au moyen de sérum contenant un ASM n’a
as été encore rapporté.Quoiqu’il en soit, il est aujourd’hui recommandé de traiter une
AI avec ASM en première intention par l’association d’une cor-icothérapie et d’un traitement immunosuppresseur (par exemplee méthotrexate) à visée d’épargne cortisonique devant le carac-ère chronique et le risque de rechutes fréquentes de ces affections1]. Dans les formes graves, les échanges plasmatiques, les immu-oglobulines polyvalentes intraveineuses ou le cyclophosphamideeuvent se discuter parfois en association. Dans les formes réfrac-aires, le rituximab pourrait être intéressant.
. Conclusion
Les AAM et particulièrement les ASM ont profondément modi-é les pratiques dans le champ des MAI en définissant de nouveauxadres nosologiques plus homogènes qui amèneront sûrement à deouvelles classifications. En cas de suspicion de MAI, une recherchee l’ensemble des ASM doit être réalisée, en complément de
’étude histologique musculaire qui reste indispensable. Les résul-
Pour citer cet article : Allenbach Y, Benveniste O. Auto-anthttp://dx.doi.org/10.1016/j.revmed.2013.12.006
ats permettront d’orienter le clinicien dans la recherche d’atteintesxtramusculaires (thoracique ou néoplasique), d’évaluer le pronos-ic et de guider le traitement et les modalités du suivi. Le syndromees anti-synthétases doit conduire le clinicien à une évaluation
[
PRESSédecine interne xxx (2013) xxx–xxx
pulmonaire précise. De même au cours de la dermatomyosite laprésence de l’anti-MDA-5 doit faire rechercher une atteinte respira-toire parfois létale. L’anti-TIF1-� oriente la recherche d’un cancer aucours de la dermatomyosite. Les anti-SRP et anti-HMGCR sont spé-cifiques des myopathies nécrosantes et de fait permettent parfoisde redresser le diagnostic de dystrophie musculaire. Il est probablequ’à l’avenir de nouveaux ASM soient encore identifiés.
Déclaration d’intérêts
Les auteurs déclarent ne pas avoir de conflits d’intérêts en rela-tion avec cet article.
Remerciement
Nous tenions à remercier le Dr J.-L. Charuel du laboratoired’immunochimie du Dr L. Musset à l’hôpital de la Pitié-Salpêtrièrepour l’apport iconographique.
Références
[1] Troyanov Y, Targoff IN, Tremblay JL, Goulet JR, Raymond Y, Senecal JL. Novelclassification of idiopathic inflammatory myopathies based on overlap syn-drome features and autoantibodies: analysis of 100 French Canadian patients.Medicine (Baltimore) 2005;84:231–49.
[2] Fernandez C, Bardin N, De Paula AM, Salort-Campana E, Benyamine A, FranquesJ, et al. Correlation of clinicoserologic and pathologic classifications of inflam-matory myopathies: study of 178 cases and guidelines for diagnosis. Medicine(Baltimore) 2013;92:15–24.
[3] Koenig M, Fritzler MJ, Targoff IN, Troyanov Y, Senecal JL. Heterogeneity ofautoantibodies in 100 patients with autoimmune myositis: insights into clinicalfeatures and outcomes. Arthritis Res Ther 2007;9:R78.
[4] Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts). N EnglJ Med 1975;292:344–7.
[5] Hoogendijk JE, Amato AA, Lecky BR, Choy EH, Lundberg IE, Rose MR, et al. 119thENMC international workshop: trial design in adult idiopathic inflammatorymyopathies, with the exception of inclusion body myositis, 10–12 October2003, Naarden, The Netherlands. Neuromuscul Disord 2004;14:337–45.
[6] van der Meulen MF, Bronner IM, Hoogendijk JE, Burger H, van VenrooijWJ, Voskuyl AE, et al. Polymyositis: an overdiagnosed entity. Neurology2003;61:316–21.
[7] Amato AA, Griggs RC. Unicorns, dragons, polymyositis, and other mythologicalbeasts. Neurology 2003;61:288–9.
[8] Arnett FC, Hirsch TJ, Bias WB, Nishikai M, Reichlin M. The Jo-1 antibodysystem in myositis: relationships to clinical features and HLA. J Rheumatol1981;8:925–30.
[9] Brouwer R, Hengstman GJ, Vree Egberts W, Ehrfeld H, Bozic B, Ghirardello A,et al. Autoantibody profiles in the sera of European patients with myositis. AnnRheum Dis 2001;60:116–23.
10] Stanciu R, Guiguet M, Musset L, Touitou D, Beigelman C, Rigolet A, et al.Antisynthetase syndrome with anti-Jo1 antibodies in 48 patients: pulmonaryinvolvement predicts disease-modifying antirheumatic drug use. J Rheumatol2012;39:1835–9.
11] Leff RL, Burgess SH, Miller FW, Love LA, Targoff IN, Dalakas MC, et al. Dis-tinct seasonal patterns in the onset of adult idiopathic inflammatory myopathyin patients with anti-Jo-1 and anti-signal recognition particle autoantibodies.Arthritis Rheum 1991;34:1391–6.
12] Hervier B, Meyer A, Dieval C, Uzunhan Y, Devilliers H, Launay D, et al. Pulmonaryhypertension in antisynthetase syndrome: prevalence, etiology and survival.Eur Respir J 2013;42:1271–82.
13] Marie I, Hachulla E, Cherin P, Dominique S, Hatron PY, Hellot MF, et al. Inter-stitial lung disease in polymyositis and dermatomyositis. Arthritis Rheum2002;47:614–22.
14] Mozaffar T, Pestronk A. Myopathy with anti-Jo-1 antibodies: pathology inperimysium and neighbouring muscle fibres. J Neurol Neurosurg Psychiatry2000;68:472–8.
15] Hervier B, Devilliers H, Stanciu R, Meyer A, Uzunhan Y, Masseau A, et al. Hie-rarchical cluster and survival analyses of antisynthetase syndrome: phenotypeand outcome are correlated with anti-tRNA synthetase antibody specificity.Autoimmun Rev 2012;12:210–7.
16] Fischer A, Swigris JJ, du Bois RM, Lynch DA, Downey GP, Cosgrove GP, et al.Anti-synthetase syndrome in ANA and anti-Jo-1 negative patients presentingwith idiopathic interstitial pneumonia. Respir Med 2009;103:1719–24.
17] Hervier B, Uzunhan Y, Hachulla E, Benveniste O, Nunes H, Delaval P, et al. Anti-
icorps au cours des myosites. Rev Med Interne (2013),
synthetase syndrome positive for anti-threonyl-tRNA synthetase (anti-PL7)antibodies. Eur Respir J 2011;37:714–7.
18] Stone KB, Oddis CV, Fertig N, Katsumata Y, Lucas M, Vogt M, et al. Anti-Jo-1 antibody levels correlate with disease activity in idiopathic inflammatorymyopathy. Arthritis Rheum 2007;56:3125–31.

ING ModelR
e de m
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
ARTICLEEVMED-4708; No. of Pages 7
Y. Allenbach, O. Benveniste / La Revu
19] Garcin B, Lenglet T, Dubourg O, Mesnage V, Levy R. Dropped head syn-drome as a presenting sign of scleromyositis. J Neurol Sci 2010;292:101–3.
20] Ronnelid J, Barbasso Helmers S, Storfors H, Grip K, Ronnblom L, Franck-LarssonK, et al. Use of a commercial line blot assay as a screening test for autoantibodiesin inflammatory myopathies. Autoimmun Rev 2009;12:58–61.
21] Selva-O’Callaghan A, Labrador-Horrillo M, Solans-Laque R, Simeon-AznarCP, Martinez-Gomez X, Vilardell-Tarres M. Myositis-specific and myositis-associated antibodies in a series of eighty-eight Mediterranean patients withidiopathic inflammatory myopathy. Arthritis Rheum 2006;55:791–8.
22] Rigolet A, Musset L, Dubourg O, Maisonobe T, Grenier P, Charuel JL, et al.Inflammatory myopathies with anti-Ku antibodies: a prognosis dependent onassociated lung disease. Medicine (Baltimore) 2012;91:95–102.
23] Maeda MH, Tsuji S, Shimizu J. Inflammatory myopathies associated with anti-mitochondrial antibodies. Brain 2012;135:1767–77.
24] Iaccarino L, Gatto M, Bettio S, Caso F, Rampudda M, Zen M, et al.Overlap connective tissue disease syndromes. Autoimmun Rev 2013;12:363–73.
25] Ranque B, Berezne A, Le-Guern V, Pagnoux C, Allanore Y, Launay D, et al. Myo-pathies related to systemic sclerosis: a case-control study of associated clinicaland immunological features. Scand J Rheumatol 2010;39:498–505.
26] Ranque B, Authier FJ, Le-Guern V, Pagnoux C, Berezne A, Allanore Y, et al. Adescriptive and prognostic study of systemic sclerosis-associated myopathies.Ann Rheum Dis 2009;68:1474–7.
27] Wani AM, Hussain WM, Fatani MI, Bafaraj MG, Showkat K, Hanif S, et al. Anal-gesics are not always the culprits: isolated gastric fundal varices as the causeof recurrent upper GI bleed in a patient with SLE, rheumatoid arthritis andpolymyositis overlap syndrome. BMJ Case Rep 2009;2009.
28] Nakajima A, Yoshino K, Soejima M, Kawaguchi Y, Satoh T, Kuwana M, et al. Highfrequencies and co-existing of myositis-specific autoantibodies in patients withidiopathic inflammatory myopathies overlapped to rheumatoid arthritis. Rheu-matol Int 2012;32:2057–61.
29] Coppo P, Clauvel JP, Bengoufa D, Oksenhendler E, Lacroix C, Lassoued K.Inflammatory myositis associated with anti-U1-small nuclear ribonucleopro-tein antibodies: a subset of myositis associated with a favourable outcome.Rheumatology (Oxford) 2002;41:1040–6.
30] Hamaguchi Y, Kuwana M, Hoshino K, Hasegawa M, Kaji K, Matsushita T, et al.Clinical correlations with dermatomyositis-specific autoantibodies in adultJapanese patients with dermatomyositis: a multicenter cross-sectional study.Arch Dermatol 2011;147:391–8.
31] Ghirardello A, Zampieri S, Iaccarino L, Tarricone E, Bendo R, Gambari PF, et al.Anti-Mi-2 antibodies. Autoimmunity 2005;38:79–83.
32] Hengstman GJ, Vree Egberts WT, Seelig HP, Lundberg IE, Moutsopoulos HM,Doria A, et al. Clinical characteristics of patients with myositis and autoan-tibodies to different fragments of the Mi-2 beta antigen. Ann Rheum Dis2006;65:242–5.
33] Fujimoto M, Hamaguchi Y, Kaji K, Matsushita T, Ichimura Y, Kodera M, et al.Myositis-specific anti-155/140 autoantibodies target transcription interme-diary factor 1 family proteins. Arthritis Rheum 2012;64:513–22.
34] Chinoy H, Fertig N, Oddis CV, Ollier WE, Cooper RG. The diagnostic utilityof myositis autoantibody testing for predicting the risk of cancer-associatedmyositis. Ann Rheum Dis 2007;66:1345–9.
35] Muro Y, Ishikawa A, Sugiura K, Akiyama M. Clinical features of anti-TIF1-alpha antibody-positive dermatomyositis patients are closely associated withcoexistent dermatomyositis-specific autoantibodies and anti-TIF1-gamma oranti-Mi-2 autoantibodies. Rheumatology (Oxford) 2012;51:1508–13.
36] Muro Y, Sugiura K, Hoshino K, Akiyama M, Tamakoshi K. Epidemiologic studyof clinically amyopathic dermatomyositis and anti-melanoma differentiation-associated gene 5 antibodies in central Japan. Arthritis Res Ther 2011;13:R214.
37] Cao H, Pan M, Kang Y, Xia Q, Li X, Zhao X, et al. Clinical manifestations ofdermatomyositis and clinically amyopathic dermatomyositis patients with
Pour citer cet article : Allenbach Y, Benveniste O. Auto-anthttp://dx.doi.org/10.1016/j.revmed.2013.12.006
positive expression of anti-melanoma differentiation-associated gene 5 anti-body. Arthritis Care Res (Hoboken) 2012;64:1602–10.
38] Muro Y, Sugiura K, Hoshino K, Akiyama M. Disappearance of anti-MDA-5autoantibodies in clinically amyopathic DM/interstitial lung disease duringdisease remission. Rheumatology (Oxford) 2012;51:800–4.
[
PRESSédecine interne xxx (2013) xxx–xxx 7
39] Gono T, Kawaguchi Y, Kuwana M, Sugiura T, Furuya T, Takagi K, et al.Brief report: association of HLA-DRB1*0101/*0405 with susceptibility toanti-melanoma differentiation-associated gene 5 antibody-positive dermato-myositis in the Japanese population. Arthritis Rheum 2012;64:3736–40.
40] Betteridge ZE, Gunawardena H, Chinoy H, North J, Ollier WE, Cooper RG, et al.Clinical and human leucocyte antigen class II haplotype associations of autoan-tibodies to small ubiquitin-like modifier enzyme, a dermatomyositis-specificautoantigen target, in UK Caucasian adult-onset myositis. Ann Rheum Dis2009;68:1621–5.
41] Muro Y, Sugiura K, Akiyama M. Low prevalence of anti-small ubiquitin-likemodifier activating enzyme antibodies in dermatomyositis patients. Autoim-munity 2013;46:279–84.
42] Ceribelli A, Fredi M, Taraborelli M, Cavazzana I, Franceschini F, Quinzanini M,et al. Anti-MJ/NXP-2 autoantibody specificity in a cohort of adult Italian patientswith polymyositis/dermatomyositis. Arthritis Res Ther 2012;14:R97.
43] Ichimura Y, Matsushita T, Hamaguchi Y, Kaji K, Hasegawa M, Tanino Y, et al.Anti-NXP2 autoantibodies in adult patients with idiopathic inflammatorymyopathies: possible association with malignancy. Ann Rheum Dis 2012;71:710–3.
44] Smith ES, Hallman JR, DeLuca AM, Goldenberg G, Jorizzo JL, Sangueza OP. Der-matomyositis: a clinicopathological study of 40 patients. Am J Dermatopathol2009;31:61–7.
45] Reeves WH, Nigam SK, Blobel G. Human autoantibodies reactive with thesignal-recognition particle. Proc Natl Acad Sci U S A 1986;83:9507–11.
46] Christopher-Stine L, Casciola-Rosen LA, Hong G, Chung T, Corse AM, Mam-men AL. A novel autoantibody recognizing 200-kd and 100-kd proteins isassociated with an immune-mediated necrotizing myopathy. Arthritis Rheum2010;62:2757–66.
47] Hengstman GJ, ter Laak HJ, Vree Egberts WT, Lundberg IE, Moutsopoulos HM,Vencovsky J, et al. Anti-signal recognition particle autoantibodies: marker of anecrotising myopathy. Ann Rheum Dis 2006;65:1635–8.
48] Benveniste O, Drouot L, Jouen F, Charuel JL, Bloch-Queyrat C, Behin A, et al.Correlation of anti-signal recognition particle autoantibody levels with crea-tine kinase activity in patients with necrotizing myopathy. Arthritis Rheum2011;63:1961–71.
49] Mammen AL. Autoimmune myopathies: autoantibodies, phenotypes andpathogenesis. Nat Rev Neurol 2011;7:343–54.
50] Mammen AL, Chung T, Christopher-Stine L, Rosen P, Rosen A, Doering KR,et al. Autoantibodies against 3-hydroxy-3-methylglutaryl-coenzyme A reduc-tase in patients with statin-associated autoimmune myopathy. Arthritis Rheum2011;63:713–21.
51] Mammen AL, Pak K, Williams EK, Brisson D, Coresh J, Selvin E, et al. Anti-HMG-CoA reductase antibodies are rare in statin users, including thosewith self-limited musculoskeletal side-effects. Arthritis Care Res (Hoboken)2011;64:269–72.
52] Werner JL, Christopher-Stine L, Ghazarian SR, Pak KS, Kus JE, Daya NR,et al. Antibody levels correlate with creatine kinase levels and strength inanti-HMG-CoA reductase-associated autoimmune myopathy. Arthritis Rheum2012;64:4087–93.
53] Ellis E, Ann Tan J, Lester S, Tucker G, Blumbergs P, Roberts-Thomson P,et al. Necrotizing myopathy: clinicoserologic associations. Muscle Nerve2012;45:189–94.
54] Benveniste O, Hilton DJ. Natural history of sporadic inclusion body myositis.Brain 2011;134:3176–84.
55] Salajegheh M, Lam T, Greenberg SA. Autoantibodies against a 43-KDa muscleprotein in inclusion body myositis. PLoS One 2011;6:e20266.
56] Larman HB, Salajegheh M, Nazareno R, Lam T, Sauld J, Steen H, et al. Cytosolic 5′-nucleotidase 1A autoimmunity in sporadic inclusion body myositis. Ann Neurol2013;73:408–18.
57] Rojana-Udomsart A, Bundell C, James I, Castley A, Martinez P, ChristiansenF, et al. Frequency of autoantibodies and correlation with HLA-DRB1 geno-
icorps au cours des myosites. Rev Med Interne (2013),
type in sporadic inclusion body myositis (s-IBM): a population control study. JNeuroimmunol 2012;249:66–70.
58] Katsumata Y, Ridgway WM, Oriss T, Gu X, Chin D, Wu Y, et al. Species-specificimmune responses generated by histidyl-tRNA synthetase immunization areassociated with muscle and lung inflammation. J Autoimmun 2007;29:174–86.