Embed Size (px)
Citation preview

1
N° d’ordre 04-ISAL-0070 Année 2004
Thèse
Etude de faisabilité de l’utilisation de molécules “cage” dans la dépollution des
sols : Solubilisation et extraction de polluants organiques par les cyclodextrines
présentée devant L’Institut National des Sciences Appliquées de Lyon
pour obtenir
le grade de docteur
Ecole doctorale : Chimie de Lyon (Chimie, Procédés, Environnement)
Spécialité : Sciences et techniques du déchet
par
Khalil HANNA Soutenue le 18 novembre 2004 devant la Commission d’examen
Jury
Mme MOREL-DESROSIERS N. Professeur (Université Blaise Pascal) Rapporteur
M. CHIRON S. MCF-HDR (Université de Provence) Rapporteur
M. BOURGOIS J. Professeur (Ecole des Mines de Saint-Etienne) Examinateur
M. CHOVELON J-M. Professeur (Université Lyon 1) Examinateur
M. GERMAIN P. Professeur (INSA de Lyon) Directeur de thèse
Mlle de BRAUER C. MCF (IUT Chimie) Codirectrice de thèse
M. RICHARD J-Y. Ingénieur (SITA-Remediation, Meyzieu) Invité
Cette thèse a été préparée au : Laboratoire d’Analyse Environnementale des Procédés et Systèmes Industriels de l’INSA de Lyon.

2
3
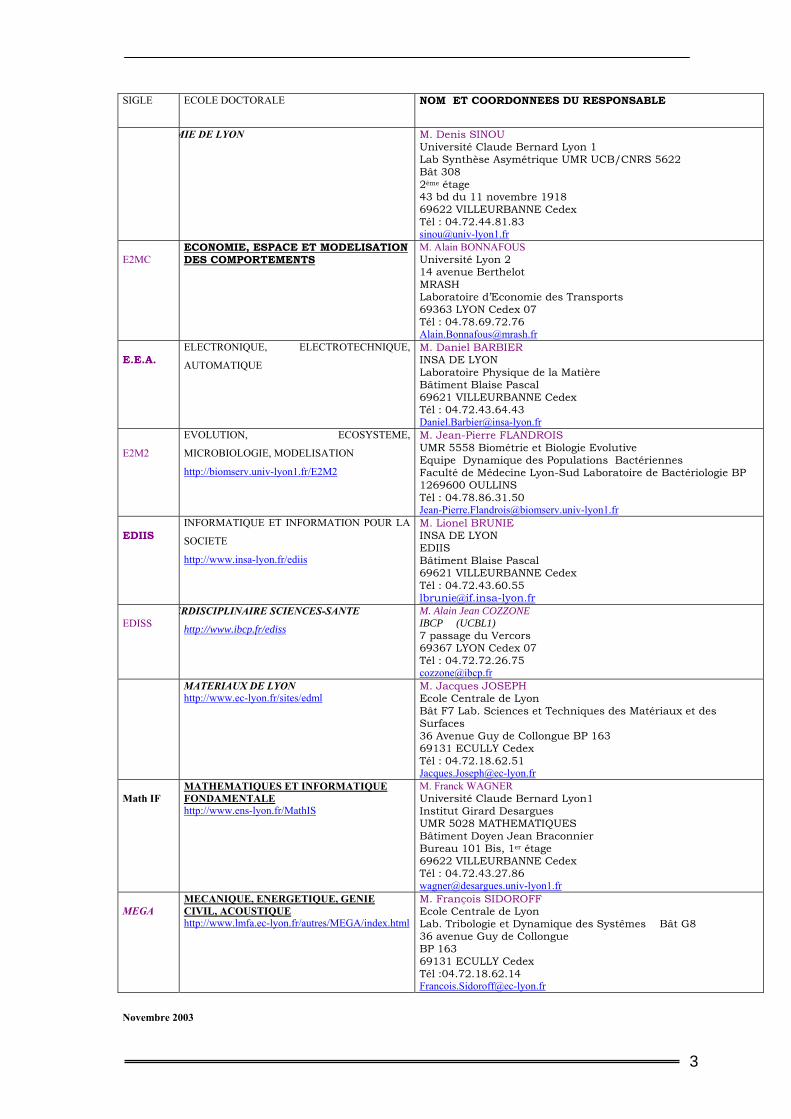
SIGLE ECOLE DOCTORALE NOM ET COORDONNEES DU RESPONSABLE
MIE DE LYON M. Denis SINOU Université Claude Bernard Lyon 1 Lab Synthèse Asymétrique UMR UCB/CNRS 5622 Bât 308 2ème étage 43 bd du 11 novembre 1918 69622 VILLEURBANNE Cedex Tél : 04.72.44.81.83 [email protected]
E2MC
ECONOMIE, ESPACE ET MODELISATION DES COMPORTEMENTS
M. Alain BONNAFOUS Université Lyon 2 14 avenue Berthelot MRASH Laboratoire d’Economie des Transports 69363 LYON Cedex 07 Tél : 04.78.69.72.76 [email protected]
E.E.A.
ELECTRONIQUE, ELECTROTECHNIQUE,
AUTOMATIQUE
M. Daniel BARBIER INSA DE LYON Laboratoire Physique de la Matière Bâtiment Blaise Pascal 69621 VILLEURBANNE Cedex Tél : 04.72.43.64.43 [email protected]
E2M2
EVOLUTION, ECOSYSTEME,
MICROBIOLOGIE, MODELISATION
http://biomserv.univ-lyon1.fr/E2M2
M. Jean-Pierre FLANDROIS UMR 5558 Biométrie et Biologie Evolutive Equipe Dynamique des Populations Bactériennes Faculté de Médecine Lyon-Sud Laboratoire de Bactériologie BP 1269600 OULLINS Tél : 04.78.86.31.50 [email protected]
EDIIS
INFORMATIQUE ET INFORMATION POUR LA
SOCIETE
http://www.insa-lyon.fr/ediis
M. Lionel BRUNIE INSA DE LYON EDIIS Bâtiment Blaise Pascal 69621 VILLEURBANNE Cedex Tél : 04.72.43.60.55 [email protected]
EDISS
ERDISCIPLINAIRE SCIENCES-SANTE
http://www.ibcp.fr/ediss
M. Alain Jean COZZONE IBCP (UCBL1) 7 passage du Vercors 69367 LYON Cedex 07 Tél : 04.72.72.26.75 [email protected]
MATERIAUX DE LYON http://www.ec-lyon.fr/sites/edml
M. Jacques JOSEPH Ecole Centrale de Lyon Bât F7 Lab. Sciences et Techniques des Matériaux et des Surfaces 36 Avenue Guy de Collongue BP 163 69131 ECULLY Cedex Tél : 04.72.18.62.51 [email protected]
Math IF
MATHEMATIQUES ET INFORMATIQUE FONDAMENTALE http://www.ens-lyon.fr/MathIS
M. Franck WAGNER Université Claude Bernard Lyon1 Institut Girard Desargues UMR 5028 MATHEMATIQUES Bâtiment Doyen Jean Braconnier Bureau 101 Bis, 1er étage 69622 VILLEURBANNE Cedex Tél : 04.72.43.27.86 [email protected]
MEGA
MECANIQUE, ENERGETIQUE, GENIE CIVIL, ACOUSTIQUE http://www.lmfa.ec-lyon.fr/autres/MEGA/index.html
M. François SIDOROFF Ecole Centrale de Lyon Lab. Tribologie et Dynamique des Systêmes Bât G8 36 avenue Guy de Collongue BP 163 69131 ECULLY Cedex Tél :04.72.18.62.14 [email protected]
Novembre 2003

4
INSTITUT NATIONAL DES SCIENCES APPLIQUEES DE LYON Directeur : STORCK A. Professeurs : AMGHAR Y. LIRIS AUDISIO S. PHYSICOCHIMIE INDUSTRIELLE BABOT D. CONT. NON DESTR. PAR RAYONNEMENTS IONISANTS BABOUX J.C. GEMPPM*** BALLAND B. PHYSIQUE DE LA MATIERE BAPTISTE P. PRODUCTIQUE ET INFORMATIQUE DES SYSTEMES
MANUFACTURIERS BARBIER D. PHYSIQUE DE LA MATIERE BASKURT A. LIRIS BASTIDE J.P. LAEPSI**** BAYADA G. MECANIQUE DES CONTACTS BENADDA B. LAEPSI**** BETEMPS M. AUTOMATIQUE INDUSTRIELLE BIENNIER F. PRODUCTIQUE ET INFORMATIQUE DES SYSTEMES
MANUFACTURIERS BLANCHARD J.M. LAEPSI**** BOISSE P. LAMCOS BOISSON C. VIBRATIONS-ACOUSTIQUE BOIVIN M. (Prof. émérite) MECANIQUE DES SOLIDES BOTTA H. UNITE DE RECHERCHE EN GENIE CIVIL - Développement Urbain BOTTA-ZIMMERMANN M. (Mme) UNITE DE RECHERCHE EN GENIE CIVIL - Développement Urbain BOULAYE G. (Prof. émérite) INFORMATIQUE BOYER J.C. MECANIQUE DES SOLIDES BRAU J. CENTRE DE THERMIQUE DE LYON - Thermique du bâtiment BREMOND G. PHYSIQUE DE LA MATIERE BRISSAUD M. GENIE ELECTRIQUE ET FERROELECTRICITE BRUNET M. MECANIQUE DES SOLIDES BRUNIE L. INGENIERIE DES SYSTEMES D’INFORMATION BUFFIERE J-Y. GEMPPM*** BUREAU J.C. CEGELY* CAMPAGNE J-P. PRISMA CAVAILLE J.Y. GEMPPM*** CHAMPAGNE J-Y. LMFA CHANTE J.P. CEGELY*- Composants de puissance et applications CHOCAT B. UNITE DE RECHERCHE EN GENIE CIVIL - Hydrologie urbaine COMBESCURE A. MECANIQUE DES CONTACTS COURBON GEMPPM COUSIN M. UNITE DE RECHERCHE EN GENIE CIVIL - Structures DAUMAS F. (Mme) CENTRE DE THERMIQUE DE LYON - Energétique et Thermique DJERAN-MAIGRE I. UNITE DE RECHERCHE EN GENIE CIVIL DOUTHEAU A. CHIMIE ORGANIQUE DUBUY-MASSARD N. ESCHIL DUFOUR R. MECANIQUE DES STRUCTURES DUPUY J.C. PHYSIQUE DE LA MATIERE EMPTOZ H. RECONNAISSANCE DE FORMES ET VISION ESNOUF C. GEMPPM*** EYRAUD L. (Prof. émérite) GENIE ELECTRIQUE ET FERROELECTRICITE FANTOZZI G. GEMPPM*** FAVREL J. PRODUCTIQUE ET INFORMATIQUE DES SYSTEMES
MANUFACTURIERS FAYARD J.M. BIOLOGIE FONCTIONNELLE, INSECTES ET INTERACTIONS FAYET M. (Prof. émérite) MECANIQUE DES SOLIDES FAZEKAS A. GEMPPM FERRARIS-BESSO G. MECANIQUE DES STRUCTURES FLAMAND L. MECANIQUE DES CONTACTS FLEURY E. CITI FLORY A. INGENIERIE DES SYSTEMES D’INFORMATIONS FOUGERES R. GEMPPM*** FOUQUET F. GEMPPM*** FRECON L. (Prof. émérite) REGROUPEMENT DES ENSEIGNANTS CHERCHEURS ISOLES GERARD J.F. INGENIERIE DES MATERIAUX POLYMERES GERMAIN P. LAEPSI**** GIMENEZ G. CREATIS** GOBIN P.F. (Prof. émérite) GEMPPM*** GONNARD P. GENIE ELECTRIQUE ET FERROELECTRICITE GONTRAND M. PHYSIQUE DE LA MATIERE GOUTTE R. (Prof. émérite) CREATIS** GOUJON L. GEMPPM*** GOURDON R. LAEPSI****.

5
GRANGE G. (Prof. émérite) GENIE ELECTRIQUE ET FERROELECTRICITE GUENIN G. GEMPPM*** GUICHARDANT M. BIOCHIMIE ET PHARMACOLOGIE GUILLOT G. PHYSIQUE DE LA MATIERE GUINET A. PRODUCTIQUE ET INFORMATIQUE DES SYSTEMES
MANUFACTURIERS GUYADER J.L. VIBRATIONS-ACOUSTIQUE GUYOMAR D. GENIE ELECTRIQUE ET FERROELECTRICITE HEIBIG A. MATHEMATIQUE APPLIQUEES DE LYON JACQUET-RICHARDET G. MECANIQUE DES STRUCTURES JAYET Y. GEMPPM*** JOLION J.M. RECONNAISSANCE DE FORMES ET VISION Novembre 2003 JULLIEN J.F. UNITE DE RECHERCHE EN GENIE CIVIL - Structures JUTARD A. (Prof. émérite) AUTOMATIQUE INDUSTRIELLE KASTNER R. UNITE DE RECHERCHE EN GENIE CIVIL - Géotechnique KOULOUMDJIAN J. (Prof. émérite) INGENIERIE DES SYSTEMES D’INFORMATION LAGARDE M. BIOCHIMIE ET PHARMACOLOGIE LALANNE M. (Prof. émérite) MECANIQUE DES STRUCTURES LALLEMAND A. CENTRE DE THERMIQUE DE LYON - Energétique et thermique LALLEMAND M. (Mme) CENTRE DE THERMIQUE DE LYON - Energétique et thermique LAREAL P (Prof. émérite) UNITE DE RECHERCHE EN GENIE CIVIL - Géotechnique LAUGIER A. (Prof. émérite) PHYSIQUE DE LA MATIERE LAUGIER C. BIOCHIMIE ET PHARMACOLOGIE LAURINI R. INFORMATIQUE EN IMAGE ET SYSTEMES D’INFORMATION LEJEUNE P. UNITE MICROBIOLOGIE ET GENETIQUE LUBRECHT A. MECANIQUE DES CONTACTS MASSARD N. INTERACTION COLLABORATIVE TELEFORMATION TELEACTIVITE MAZILLE H. (Prof. émérite) PHYSICOCHIMIE INDUSTRIELLE MERLE P. GEMPPM*** MERLIN J. GEMPPM*** MIGNOTTE A. (Mle) INGENIERIE, INFORMATIQUE INDUSTRIELLE MILLET J.P. PHYSICOCHIMIE INDUSTRIELLE MIRAMOND M. UNITE DE RECHERCHE EN GENIE CIVIL - Hydrologie urbaine MOREL R. (Prof. émérite) MECANIQUE DES FLUIDES ET D’ACOUSTIQUES MOSZKOWICZ P. LAEPSI**** NARDON P. (Prof. émérite) BIOLOGIE FONCTIONNELLE, INSECTES ET INTERACTIONS NAVARRO Alain (Prof. émérite) LAEPSI**** NELIAS D. LAMCOS NIEL E. AUTOMATIQUE INDUSTRIELLE NORMAND B. GEMPPM NORTIER P. DREP ODET C. CREATIS** OTTERBEIN M. (Prof. émérite) LAEPSI**** PARIZET E. VIBRATIONS-ACOUSTIQUE PASCAULT J.P. INGENIERIE DES MATERIAUX POLYMERES PAVIC G. VIBRATIONS-ACOUSTIQUE PECORARO S. GEMPPM PELLETIER J.M. GEMPPM*** PERA J. UNITE DE RECHERCHE EN GENIE CIVIL - Matériaux PERRIAT P. GEMPPM*** PERRIN J. INTERACTION COLLABORATIVE TELEFORMATION TELEACTIVITE PINARD P. (Prof. émérite) PHYSIQUE DE LA MATIERE PINON J.M. INGENIERIE DES SYSTEMES D’INFORMATION PONCET A. PHYSIQUE DE LA MATIERE POUSIN J. MODELISATION MATHEMATIQUE ET CALCUL SCIENTIFIQUE PREVOT P. INTERACTION COLLABORATIVE TELEFORMATION TELEACTIVITE PROST R. CREATIS** RAYNAUD M. CENTRE DE THERMIQUE DE LYON - Transferts Interfaces et Matériaux REDARCE H. AUTOMATIQUE INDUSTRIELLE RETIF J-M. CEGELY* REYNOUARD J.M. UNITE DE RECHERCHE EN GENIE CIVIL - Structures RICHARD C. LGEF RIGAL J.F. MECANIQUE DES SOLIDES RIEUTORD E. (Prof. émérite) MECANIQUE DES FLUIDES ROBERT-BAUDOUY J. (Mme) (Prof. émérite) GENETIQUE MOLECULAIRE DES MICROORGANISMES ROUBY D. GEMPPM*** ROUX J.J. CENTRE DE THERMIQUE DE LYON – Thermique de l’Habitat RUBEL P. INGENIERIE DES SYSTEMES D’INFORMATION SACADURA J.F. CENTRE DE THERMIQUE DE LYON - Transferts Interfaces et Matériaux SAUTEREAU H. INGENIERIE DES MATERIAUX POLYMERES SCAVARDA S. (Prof. émérite) AUTOMATIQUE INDUSTRIELLE SOUIFI A. PHYSIQUE DE LA MATIERE

6
SOUROUILLE J.L. INGENIERIE INFORMATIQUE INDUSTRIELLE THOMASSET D. AUTOMATIQUE INDUSTRIELLE THUDEROZ C. ESCHIL – Equipe Sciences Humaines de l’Insa de Lyon UBEDA S. CENTRE D’INNOV. EN TELECOM ET INTEGRATION DE SERVICES VELEX P. MECANIQUE DES CONTACTS VERMANDE P. (Prof émérite) LAEPSI VIGIER G. GEMPPM*** VINCENT A. GEMPPM*** VRAY D. CREATIS** VUILLERMOZ P.L. (Prof. émérite) PHYSIQUE DE LA MATIERE Directeurs de recherche C.N.R.S. : BERTHIER Y. MECANIQUE DES CONTACTS CONDEMINE G. UNITE MICROBIOLOGIE ET GENETIQUE COTTE-PATAT N. (Mme) UNITE MICROBIOLOGIE ET GENETIQUE ESCUDIE D. (Mme) CENTRE DE THERMIQUE DE LYON FRANCIOSI P. GEMPPM*** MANDRAND M.A. (Mme) UNITE MICROBIOLOGIE ET GENETIQUE POUSIN G. BIOLOGIE ET PHARMACOLOGIE ROCHE A. INGENIERIE DES MATERIAUX POLYMERES SEGUELA A. GEMPPM*** VERGNE P. LaMcos Directeurs de recherche I.N.R.A. : FEBVAY G. BIOLOGIE FONCTIONNELLE, INSECTES ET INTERACTIONS GRENIER S. BIOLOGIE FONCTIONNELLE, INSECTES ET INTERACTIONS RAHBE Y. BIOLOGIE FONCTIONNELLE, INSECTES ET INTERACTIONS Directeurs de recherche I.N.S.E.R.M. : KOBAYASHI T. PLM PRIGENT A.F. (Mme) BIOLOGIE ET PHARMACOLOGIE MAGNIN I. (Mme) CREATIS** * CEGELY CENTRE DE GENIE ELECTRIQUE DE LYON ** CREATIS CENTRE DE RECHERCHE ET D’APPLICATIONS EN TRAITEMENT DE L’IMAGE ET DU SIGNAL ***GEMPPM GROUPE D'ETUDE METALLURGIE PHYSIQUE ET PHYSIQUE DES MATERIAUX ****LAEPSI LABORATOIRE D’ANALYSE ENVIRONNEMENTALE DES PROCEDES

7
AVANT-PROPOS
Ces travaux de recherche ont été menés au Laboratoire d’Analyse Environnementale des Procédés et des Systèmes Industriels (LAEPSI) de l’INSA de Lyon. Je remercie Monsieur Pierre MOSZKOWICZ (Directeur du LAEPSI) de m’avoir accueilli au sein de son laboratoire. Je tiens à remercier Monsieur Patrick GERMAIN et Mademoiselle Christine de BRAUER d’avoir dirigé cette thèse. Je suis reconnaissant à Madame Nicole MOREL-DESROSIERS et Monsieur Serge CHIRON d’avoir bien voulu juger ce travail en tant que rapporteurs. Je voudrais également remercier Messieurs Jacques BOURGOIS, Jean-Marc CHOVELON et Jean-Yves RICHARD d’avoir accepté d’être membres du jury. Je remercie Monsieur Jean-Marc CHOVELON du Laboratoire d’Application de la Chimie à l’Environnement (LACE) de l’université Lyon 1 qui m’a permis de réaliser une partie de cette étude dans son équipe. Un merci tout particulier à Mademoiselle Corinne FERRONATO (LACE) pour son aide technique. Je tiens également à remercier Thérèse BADR et Christophe VIGLIANTI pour leur contribution à cette étude lors de leur stage de DEA. Je remercie Roquette-Frères (Lille) et SITA-Remediation (Meyzieu) pour leurs aides logistiques et analytiques. Enfin, mes remerciements vont à toutes les personnes (Camarades et Tantines) qui de près ou de loin ont contribué à l’accomplissement de cette thèse. Je ne citerai pas de noms afin de n’oublier personne.

8

9
Cette étude a fait l’objet de : Publications (revues internationales) K. Hanna ; C. de Brauer ; P. Germain. Solubilization of the neutral and charged forms of
2,4,6 Trichlorophenol by β-Cyclodextrin, Methyl-β-Cyclodextrin and Hydroxypropyl-β-
Cyclodextrin in water. Journal of Hazardous Materials, 2003, B100 : 109-116.
K. Hanna ; C. de Brauer; P. Germain. Cyclodextrin-enhanced solubilization of
pentachlorophenol in water. Journal of Environmental Management, 2004, 71 (1) : 1-8.
T. Badr ; K. Hanna ; C. de Brauer. Enhanced solubilization and removal of of
naphthalene and phenanthrene by cyclodextrins from two contaminated soils, Journal of
Hazardous Materials, 2004, 112 (3) : 215-223.
K. Hanna ; C. de Brauer ; P. Germain ; J.M. Chovelon ; C. Ferronato. Degradation of
Pentachlorophenol in Cyclodextrin Extraction Effluent Using Photocatalytic Process. The
Science of the Total Environnment, 2004, 332 : 51-60.
C. Viglianti ; K. Hanna ; C. de Brauer ; P. Germain. Cyclodextrin enhanced-extraction of
Polycyclic Aromatic Hydrocarbons from aged contaminated soil. En preparation.
Communications orales et affiches
K. Hanna, C. de Brauer and P. Germain. Traitement de pollution organique : Procédé
innovant et écocompatible basé sur les propriétés complexantes d’un sucre.
Communication par affiche (14RRR), 21 Octobre 2003, Saint Etienne, France.
K. Hanna, C. de Brauer and P. Germain. Cyclodextrin-enhanced solubilization and
removal of pentachlorophenol from soils. Communication Orale, 4th European Meeting
on Environmental Chemistry. 10 - 13 December 2003, Plymouth, England.

10

11
TABLE DES MATIERES

12

13
Liste des abréviations utilisées........................................................19 Résumé............................................................................................21 Introduction générale ......................................................................23 Chapitre I : Etude bibliographique ..................................................29
Introduction .................................................................................................. 29
Partie A : Les cyclodextrines ..........................................................31 I. Introduction............................................................................................... 31 II. Caractéristiques des cyclodextrines......................................................... 32
II.1. Origine et caractéristiques structurales des CD..................................................32 II.2. Propriétés physico-chimiques des CD..................................................................34
II.2.1. Solubilité dans l’eau.................................................................................................................. 34 II 2.2. Aspects énergétiques de l’hydratation et de la dissolution ....................................................... 35 II.2.3. Stabilité des CD en solution...................................................................................................... 35
II.3. Cyclodextrines modifiées......................................................................................36 II. 4. Biodégradabilité...................................................................................................37 II.5. Toxicité des CDs ...................................................................................................38
III. Propriétés complexantes des cyclodextrines et applications.................. 38 III. 1. Complexe d’inclusion ........................................................................................38 III.2. Etude des complexes d’inclusion des cyclodextrines : paramètres réactionnels. ..................................................................................................................40
III.2.1. Méthode de solubilisation........................................................................................................ 41 III.2.2. Autres méthodes....................................................................................................................... 43
III.3. Principales applications des cyclodextrines.......................................................44 IV. Facteurs influençant la complexation des molécules organiques par les CD................................................................................................................. 46

14
IV.1. Facteurs liés à la nature des molécules organiques ..........................................46 IV.1.1. Hydrophobicité et hydrosolubilité............................................................................................ 46 IV.1.2. Etat d’ionisation de la molécule .............................................................................................. 46 IV.1.3. Taille et forme de la molécule.................................................................................................. 46
IV.2. Facteurs liés aux caractéristiques des cyclodextrines........................................47 IV.3. Influence du contexte chimique..........................................................................47
IV.3.1. pH............................................................................................................................................. 47 IV.3.2. Force ionique ........................................................................................................................... 48 IV.3.3. Influence de la présence d’un solvant organique..................................................................... 48
IV.4. Influence de la température ................................................................................49 V. Conclusion............................................................................................... 49
Partie B : Les polluants organiques.................................................51 I. Introduction............................................................................................... 51 II. Généralités sur les polluants organiques ................................................. 52 III. Chlorophénols ........................................................................................ 52
III.1. Origine et utilisation ...........................................................................................52 III.2. Pathologie/toxicologie.........................................................................................53 III.3. Propriétés physico-chimiques.............................................................................53
III.3.1. Généralités............................................................................................................................... 53 III.3.2. Acidité et solubilité aqueuse .................................................................................................... 55 III.3.3. Coefficient de partage octanol/eau Kow ................................................................................... 56
III.4. Comportement dans l’environnement................................................................57 III.4.1. Milieu aquatique...................................................................................................................... 57 III.4.2. Atmosphère .............................................................................................................................. 57 III.4.3. Sols........................................................................................................................................... 58 III. 4.4. Biodégradabilité dans les sols : atténuation naturelle ........................................................... 59
IV. Les hydrocarbures aromatiques polycycliques ...................................... 59 ІV.1. Origine des HAP..................................................................................................59 ІV.2. Données toxicologiques.......................................................................................60 ІV.3. Nature chimique ..................................................................................................61 ІV.4. Propriétés physico-chimiques .............................................................................63 ІV.5. Devenir des HAP dans l’environnement ............................................................65
IV.5.1. Transport des HAP dans les différents compartiments terrestres............................................ 65 IV.5.2. Biodégradation naturelle des HAP .......................................................................................... 66
V. Bilan......................................................................................................... 67
Partie C : les Sols et leurs interactions avec les polluants organiques. ......................................................................................69
I. Introduction............................................................................................... 69 II. Interactions sol / polluants organiques .................................................... 69
II.1. Définition d’un sol................................................................................................69 II.2. Comportement de polluants organiques hydrophobes dans les sols ..................70 II.3. Principaux mécanismes contrôlant le devenir des polluants dans les sols ........72 II.4. Nature des interactions physico-chimiques des polluants organiques avec les sols.................................................................................................................................73 II.5. Facteurs influençant l’adsorption et la désorption des polluants organiques dans les sols...................................................................................................................75

15
II.5.1. Facteurs liés aux caractéristiques des sols ............................................................................... 75 II.5.1. Facteurs liés à la nature des polluants organiques................................................................... 77 II.5.3. Facteurs liés aux caractéristiques de la phase aqueuse du sol ................................................. 79 II.5.4. Influence de la température ...................................................................................................... 79
III. Conclusion.............................................................................................. 80
Partie D : Les sites contaminés et les techniques de traitement ......81 I. Introduction............................................................................................... 81 II. Les sites contaminés ................................................................................ 82
II.1. Qu’est-ce qu’un site ?...........................................................................................82 II.2. Les sites pollués en France ..................................................................................82 II.3. Conséquences pour l'environnement et la santé.................................................83
III. Réhabilitation des sols : Les technologies. ............................................ 83 III.1. Généralités...........................................................................................................83 III.2. Les techniques physiques....................................................................................84
III.2.1. Les procédés physiques de piégeage........................................................................................ 84 III.2.2. Les procédés par évacuation ou entraînement......................................................................... 85 III.2.3. Le procédés de lavage.............................................................................................................. 86 III.2.4. Les procédés électriques.......................................................................................................... 87 III.2.5. Les procédés thermiques.......................................................................................................... 88
III.3. Les traitements chimiques ..................................................................................89 III.3.1. Extraction avec des solvants organiques ................................................................................. 89 III.3.2. Extraction par des fluides critiques ......................................................................................... 90 III.3.3. Immobilisation chimique.......................................................................................................... 90 III.3.4. Oxydation chimique ................................................................................................................. 90
III.4. Les traitements biologiques ................................................................................91 IV.4.1. La bioremédiation in-situ......................................................................................................... 91 IV.4.2. La bioremédiation sur ou hors-site .......................................................................................... 93
III.5. L’utilisation des tensioactifs dans le traitement des sols pollués ......................94 III.6. Etat de l’art sur l’utilisation des CD dans la remédiation des sites contaminés ....................................................................................................................96
III.6.1. Potentialités d’utilisation des CD dans la remédiation des sites contaminés.......................... 96 III.6.2. Comparaison avec d’autres agents classiques de lavage ........................................................ 97
Bilan ............................................................................................................. 98
RESULTATS ET DISCUSSIONS................................................101
Chapitre II : Tests préalables de faisabilité ...................................103 Préambule ................................................................................................... 103
Partie I : Etude de la solubilisation de polluants organiques par les cyclodextrines dans l’eau ..............................................................105
I. Introduction............................................................................................. 105 II. Matériels et méthodes ............................................................................ 106
II.1. Caractéristiques des cyclodextrines utilisées.....................................................106 II.2. Les polluants organiques sélectionnés ..............................................................107 II.3. Détermination des constantes d’équilibre de complexation par la méthode de Solubilisation ..............................................................................................................107

16
II.3.1. Procédure de solubilisation .................................................................................................... 107 II.3.2. Conditions des analyses spectrophotomètriques..................................................................... 108
III. Résultats et discussion.......................................................................... 109 III.1. Complexation du Trichlorophénol et du Pentachlorophénol à deux valeurs de pH ...........................................................................................................................109
III.1.1. Détermination de la constante de complexation et du gain de solubilité .............................. 109 III.1.2. Concentration totale de substrat soluble ou solubilité apparente ......................................... 116
III.2. Effet de la force ionique sur la complexation du pentachlorophénol ............118 III.3. Complexation du Naphtalène et du Phénanthrène ........................................120
III.3.1. Complexation dans l’eau pure ............................................................................................... 120 III.3.2. Influence de la force ionique.................................................................................................. 123
IV. Bilan ..................................................................................................... 125
Partie II : Traitement des effluents : Dégradation du complexe Cyclodextrine/Pentachlorophénol .................................................127
I. Introduction............................................................................................. 127 II. Rappel sur les méthodes de dégradation des molécules organiques complexées ................................................................................................. 127
II.1. Ozonisation .........................................................................................................128 II.2. Réduction par le fer (0) ......................................................................................129 II.3. Photochimie ........................................................................................................129 II.4. Dégradation par le réactif de Fenton ................................................................129
III. La photocatalyse hétérogène ................................................................ 130 III.1. Généralités.........................................................................................................130 III.2. Le dioxyde de titane ..........................................................................................131 III.3. Facteurs influençant la photocatalyse hétérogène..........................................131
III.3.1. Influence du pH...................................................................................................................... 132
IV. Matériels et Méthodes .......................................................................... 133 IV.1 Photoréacteur .....................................................................................................133 IV.2. Expériences d’irradiation et de dégradation ....................................................134
IV.2.1. Suivi de la dégradation du PCP seul dans l’eau.................................................................... 134 IV.2.2. Suivi de la dégradation du PCP en mélange avec la CD....................................................... 135
IV.3. Analyses des solutions .......................................................................................135 V. Résultats et discussion........................................................................... 136
V.1. Effet du type de CD sur la dégradation photocatalytique du pentachlorophénol136 V.2. Evolutions comparées des concentrations en CD et en PCP ............................138 V.3. Dégradation photocatalytique du pentachlorophénol à deux pH....................139 V.4. Effet de la concentration en β-CD sur la dégradation photocatalytique du PCP140
VI. Bilan ..................................................................................................... 143 Conclusion.................................................................................................. 145
Chapitre III : Etude de l’efficacité des CD pour l’extraction de polluants contenus dans des sols contaminés au laboratoire. ........149
I. Introduction............................................................................................. 149 II. Caractéristiques des matrices étudiées .................................................. 150

17
III. Etude de l’adsorption/désorption du pentachlorophénol ..................... 152 III.1. Etude en Batch (milieu dispersé) .....................................................................152
III.1.1. Procédures expérimentales.................................................................................................... 152 III.1.2. Résultats de l’extraction du pentachlorophénol en milieu dispersé....................................... 155 III.1.3. Bilan....................................................................................................................................... 163
III.2. Essai de percolation en colonne .......................................................................164 III.2.1. Objectif .................................................................................................................................. 164 III.2.2. Procédures expérimentales.................................................................................................... 165 III.2.3. Résultats de l’extraction du pentachlorophénol en colonne .................................................. 171 III.2.4. Bilan....................................................................................................................................... 182
IV. Etude de l’adsorption et de la désorption du naphtalène et phénanthrène en milieu dispersé ............................................................... 183
IV.1. Conditions opératoires ......................................................................................183 IV.1.1. Préparation des solutions ..................................................................................................... 183 IV.1.2. Analyses des solutions............................................................................................................ 184
IV.2. Résultats de l’extraction du naphtalène ou du phénanthrène en milieu dispersé........................................................................................................................184
IV.2.1. Cinétique de désorption ......................................................................................................... 184 IV.2.2. Isothermes d’adsorption et de désorption.............................................................................. 186 IV.2.3. Comparaison de l’efficacité d’extraction des solutions de lavage........................................ 189
IV.3. Bilan...................................................................................................................192 V. Etude des interactions cyclodextrines/sols ............................................ 193
V.1. Adsorption des cyclodextrines sur les sols .........................................................193 V.1.1. Dosage des cyclodextrines ...................................................................................................... 193 V.1.2. Adsorption des CD sur les argiles ( effet de la teneur en argile) ............................................ 195 V.1.3. Adsorption des CD sur les sols (Effet de la teneur en MO)..................................................... 199 V.1.4. Bilan ........................................................................................................................................ 201
V.2. Mobilisation des matières organiques du sol (Dosage du Carbone Organique Total) ...........................................................................................................................202
V.2.1. Procédures expérimentales ..................................................................................................... 202 V.2.2. Résultats du dosage de Carbone Organique Total.................................................................. 203
VI. Conclusion............................................................................................ 206
Chapitre IV : Evaluation de l’aptitude des CD à extraire des Hydrocarbures Aromatiques Polycycliques de sols pollués par des activités industrielles. ....................................................................211
I. Introduction............................................................................................. 211 II. Matériels et Méthodes............................................................................ 212
II.1. Caractéristiques du sol étudié ............................................................................212 II.2. Produits utilisés ..................................................................................................215 II.3. Protocoles d’extraction des polluants ................................................................215
II.3.1. Tests d’étude de l’extraction des polluants d’un sol réellement pollué................................... 215 II.3.2. Applications des tests classiques de lixiviation à l’étude de la mobilité des HAP par les CD 216
II.4. Méthodes d’analyse ............................................................................................217 III. Résultats et discussion.......................................................................... 219
III.1. Estimation des concentrations en HAP à l'équilibre dans l'eau.....................219 III.2. Tests préliminaires ............................................................................................224
III.2.1. Tests de faisabilité ................................................................................................................. 224 III.2.2. Comparaison des résultats expérimentaux avec les modèles théoriques............................... 227

18
III.3. Etude en milieu dispersé...................................................................................228 III.3.1. Etude cinétique ...................................................................................................................... 228 III.3.2. Effet de la concentration en CD............................................................................................. 230 III.3.3. Effet du ratio L/S.................................................................................................................... 233 III.3.4. Conclusion ............................................................................................................................. 235
III.4. Etude en batch percolant ..................................................................................236 III.4.1. Détermination du profil d’écoulement................................................................................... 236 III.4.2. Etude de la cinétique en batch percolant............................................................................... 237 III.4.3. Effet de la température .......................................................................................................... 240 III.4.4. Effet de la concentration........................................................................................................ 241 III.4.5. Effet du rapport L/S ............................................................................................................... 243 III.4.6. Conclusion ............................................................................................................................. 246
III.5. Synthèse comparative........................................................................................247 III.5.1. Comparaison des deux protocoles opératoires...................................................................... 247 III.5.2. Comparaison des différentes cyclodextrines.......................................................................... 249 III.5.3. Comparaison entre les quantités de polluants extraites ........................................................ 252 III.5.4. Etude d’un sol difficilement traitable : comparaison avec un système à cosolvants ............. 254 III.5.5. Evaluation de la méthode et comparaison avec d’autres agents classiques de lavage ......... 256
IV. Bilan ..................................................................................................... 259 CONCLUSION GENERALE .................................................................... 261 REFRENCES BIBLIOGRAPHIQUES ..................................................... 265 ANNEXES ................................................................................................. 277

19
Liste des abréviations utilisées
ANT : Anthracène
BCD : β-Cyclodextrine
CD : Cyclodextrine (s)
FLUO : Fluoranthène
HAP : Hydrocarbures Aromatiques Polycycliques
HPCD : HydroxyPropyl-β-cyclodextrine
HPLC : High Performance Liquid Chromatography (Chromatographie Liquide Haute
Performance)
MCD : Méthyl-β-cyclodextrine
MS : Matière Sèche
NAP : Naphtalène
NAPL : Non-Aqueous Phase Liquid (Phase Liquide Non-Aqueuse)
PHE : Phénanthrène.
PYR : Pyrène
USEPA: United States Environment Protection Agency
CSAC : Sol sous culture de la Cote Saint-André
CSAP : Sol sous prairie de la Cote Saint-André
TG : Analyse Thermogravimétrique
MOS : Matières Organiques de Sol
MON : Matières Organiques Naturelles
CEC : Capacité d’Echange Cationique
TCP : 2,4,6 Trichlorophénol
PCP : Pentachlorophénol
CP : Chlorophénol(s)

20

21
Résumé
Les cyclodextrines (CD) sont reconnues pour leurs aptitudes à accroître la solubilité de
nombreuses molécules organiques par formation de composés d’inclusion. Dans cette
étude, nous proposons d’évaluer une méthode de traitement des sols pollués utilisant les
cyclodextrines comme agents de lavage. Cette technique pourrait constituer une
alternative aux procédés actuels de traitement par lavage utilisant des cosolvants
organiques ou des surfactants qui peuvent nuire à la qualité du sol ou être difficiles à
manipuler voire à éliminer. Cependant, comme tout procédé de lavage, le traitement doit
être accompagné d’une solution de gestion des effluents générés.
La phase préliminaire de recherche, menée dans le cadre de cette étude, a permis
d’identifier les principaux paramètres d’influence des performances de complexation de
différentes cyclodextrines (nature de la CD, pH et force ionique). La solubilisation de
molécules sélectionnées appartenant à deux grandes familles de polluants organiques
(Chlorophénols et Hydrocarbures Aromatiques Polycycliques (HAP)) a été étudiée dans
des solutions aqueuses de CD (natives et modifiées). Des essais sur des sols organiques
de référence en laboratoire, par la méthode dite en batch ou sur colonnes de percolation,
ont permis de tester l’efficacité réelle de l’extractant. Les expériences d’extraction
réalisées sur un sol réellement pollué provenant d’une ancienne usine à goudron (HAP)
ont confirmé les performances extractives des CD. Le mécanisme majoritaire d’extraction
des HAP dans ce sol a été identifié et semble être la dissolution de la Phase Liquide Non-
Aqueuse (NAPL) vers la phase aqueuse.
L’ensemble des résultats de cette étude montrent que la mise en solution des polluants par
les cyclodextrines est rapide. La concentration en polluant solubilisé augmente de façon
significative en présence de cyclodextrines. Cette augmentation semble être
proportionnelle à la quantité de cyclodextrine mise en contact avec le sol, et dans les
conditions de notre étude, l’extraction semble relativement insensible aux variations de
température. En présence des CD, un accroissement de l’efficacité d’extraction des HAP
par rapport à l’eau d’un facteur 100 au minimum a été obtenu.
Le protocole d’extraction en milieu dispersé donne de meilleurs résultats que ceux
obtenus en batch percolant. Des trois cyclodextrines testées, la β-cyclodextrine est

22
nettement la moins active, tandis que l’hydroxypropyl-β-cyclodextrine est légèrement
moins efficace que la méthyl-β-cyclodextrine.
Les interactions des molécules de CD avec les sols semblent être faibles et leur
immobilisation dépend de la teneur en matière organique et du taux d’argile du sol.
L’impact des CD sur la mobilisation des matières organiques naturelles de sol parait
négligeable.
Les résultats de cette étude révèlent les propriétés remarquables des CD et montrent que
leur utilisation pour le traitement des sols pollués est d’un intérêt scientifique
considérable. Cette étude constitue une étape majeure vers la mise au point d’un procédé
innovant et complet de décontamination de sols à l’échelle industrielle.

23
Introduction générale
L'opinion publique est de plus en plus sensible à la problématique du traitement et du
devenir des sites et sols contaminés. Les dernières décennies du XXème ont été marquées
par des actes législatifs sur la base d’un principe général, celui du pollueur - payeur.
Du point de vue européen, le colloque EUROSOL de Maastricht (Pays-Bas en 1992) a
sans doute été le révélateur d’une prise de conscience plus large de l’étendue des
problèmes, et de leurs conséquences.
La gestion et la réhabilitation des sites pollués sont désormais devenues des priorités au
même titre que la lutte contre les pollutions atmosphériques, le traitement et la
valorisation des déchets et les économies d’énergies fossiles.
En France, l’Agence pour le Développement et la Maîtrise de l’Energie (ADEME)
contribue largement, à la fois au recensement des sites pollués et à la stimulation de la
sphère scientifique pour la recherche de solutions de traitement. L’ampleur du problème
dépend de l'origine et de la nature de la contamination, mais aussi de l'utilisation présente
ou future du sol, qu'elle soit agricole (champs ou forêts), industrielle (sitologie d'activités
lourdes ou légères) ou domestiques (résidences, jardins, etc.).
Les sites , ou les « friches », industriel(le)s sont souvent le lieu de pollution mixte :
minérale et organique. Le mémoire qui suit est consacré exclusivement aux
contaminations d’origine organique. Les molécules, ou famille de molécules, concernées
présentent une toxicité reconnue, une rémanence importante dans l’environnement et des
propriétés de bioaccumulation. Elles peuvent être transportées sur de longues distances
par les réseaux aquifères après percolation au travers des sols. Les risques associés à ces
molécules sont amplifiés par le fait qu’elles ne sont souvent que très peu, ou pas,
biodégradables en conditions naturelles. Il y a donc nécessité de mettre en œuvre des
solutions adaptées de traitement fiable et efficace.
Les techniques de décontamination de sols pollués sont nombreuses, allant de
l’incinération jusqu’à la bioremédiation in-situ. Le traitement technique ne peut pas, et ne

24
doit pas être déconnecté des autres aspects du processus de réhabilitation : les contextes
législatifs et financiers. Les technologies et le cadre législatif dans ce domaine évoluent
très vite et nombre de ces techniques sont encore dans le domaine expérimental. Parmi
celles-ci, les méthodes d'extraction par lavage des sols, qui peuvent être appliquées ex-
situ, ou in-situ, sont fréquemment utilisées.
Plusieurs techniques de lavages par des agents chimiques, appelés agents extractants, ont
ainsi été développées et mises en œuvre. Cette méthode de dépollution des sols consiste à
extraire le, ou les polluants, à l’aide d’un réactif approprié dans le but de « laver » le sol
en mobilisant le contaminant, la plupart du temps sans le détruire. Dans certains cas, ces
agents ont également un rôle de solubilisant afin de rendre les polluants plus disponibles
pour les bactéries endogènes du sol. Les agents d’extraction utilisés dans des opérations
de dépollution sont extrêmement variés (eau, acides, bases, oxydants, réducteurs,
complexants, solvants organiques, détergents, etc.) et sont sélectionnés selon la nature du
sol et le type de pollution. Plusieurs désavantages liés à l’utilisation de ces agents ont été
largement évoqués dans la littérature et la recherche d’un extractant « idéal » qui soit à la
fois efficace, non-toxique et inerte vis-à-vis des écosystèmes (sol/eau) se développe. Dans
cet objectif, nous proposons de tester l’efficacité des cyclodextrines, molécules obtenues
par la dégradation enzymatique de l’amidon (céréales), en tant qu’agent extractant
alternatif et innovant.
L’objectif de cette thèse est donc d’étudier la faisabilité de l’utilisation de ces
« molécules-cage » dans la remédiation de sols contaminés par des polluants
organiques.
Tout d’abord, nous présenterons une revue bibliographique concernant les trois axes
principaux de ce projet: l’agent de solubilisation, le polluant et la matrice sol, sur
lesquelles se basent les recherches réalisées dans cette thèse.
Nous décrivons, en première partie, les propriétés principales des cyclodextrines, sur
lesquelles se base la potentialité de traitement des sites contaminés. Après un bref exposé
des caractéristiques de deux familles des polluants sélectionnés (seconde partie), la
troisième partie sera consacrée aux principales propriétés physico-chimiques des sols, des
fractions du sol et au comportement des polluants hydrophobes dans les sols. Enfin, nous
présentons, dans la quatrième partie, les différentes méthodes employées actuellement

25
pour la remédiation des sites contaminés par des polluants organiques et leurs champs
d’applications.
Une étude de faisabilité de traitement d’un sol pollué au moyen d’une technique donnée
doit être basée sur une connaissance suffisamment précise des principes sur lesquels
repose cette technique. Dans notre cas, c’est la capacité de complexation des
cyclodextrines vis-à-vis de molécules organiques qui constitue le fondement scientifique
de la technique en question. Il parait donc logique de commencer notre démarche
expérimentale par une étude préalable, visant à mettre en évidence l’existence de
complexe d’inclusion au sein de plusieurs couples cyclodextrines/polluants et à identifier
les principaux paramètres permettant d’optimiser les performances complexantes des
cyclodextrines. Le but de ces travaux, présentés au chapitre II, est donc d’étudier, pour
deux familles de polluants organiques (les chlorophénols et les hydrocarbures
aromatiques polycycliques), l’influence :
• de la nature de la cyclodextrine employée par rapport à un type de polluant cible.
• du pH et de la force ionique des solutions aqueuses utilisées.
Comme tout procédé de lavage, le traitement d’extraction par des cyclodextrines doit être
accompagné d’une solution de gestion des effluents générés. Dans le second volet du
chapitre II, nous présentons donc une étude de faisabilité d’une technique de dégradation
photocatalytique de complexes CD/polluant. Cette étude a été menée dans la cadre d’une
collaboration avec le Laboratoire d’Application de la Chimie à l’Environnement (LACE)
de l’université Lyon 1 en vue de poser les bases d’une méthode permettant de
décontaminer les effluents d’extractions contenant des CD.
Après avoir étudié les interactions CD/polluants dans l’eau et conformément à nos
objectifs, nous testerons les capacités des cyclodextrines à extraire les polluants adsorbés
sur des sols de référence contaminés au laboratoire. Cette étude, décrite dans le chapitre
III, a pour but d’évaluer les performances de trois types de cyclodextrines dans le
traitement d’un sol pollué en relation avec la nature du sol. Cette étude de faisabilité en
présence de matrice sol est effectuée en milieu dispersé et en colonne de percolation.
La mise au point d’un procédé d’extraction des polluants organiques requiert également
l’évaluation des interactions susceptibles de se produire entre les cyclodextrines et les
sols. La dernière partie du chapitre III sera ainsi consacrée, d’une part à l’étude de

26
l’adsorption des CD sur diverses matrices de sol, et, d’autre part à l’évaluation de
l’impact des CD sur les matières organiques naturelles du sol.
Dans la dernière partie de ce projet (Chapitre IV), et pour une représentation plus réaliste
des problèmes environnementaux concernant les sites contaminés, nous étudions la
faisabilité de la technique de lavage sur un sol « réellement » pollué, provenant d’une
ancienne usine à goudron, fourni par la société SITA Remédiation. Cette étude,
indispensable dans notre démarche, prend en compte la complexité d’une pollution
ancienne, d’origine industrielle et variée (présence de multiples composés).
Dans ce chapitre, nous avons tenté d’appréhender le mécanisme de la mise en solution
des polluants et d’estimer les concentrations extraites à partir d’un modèle théorique de
relargage de HAP vers la phase liquide. Nous avons exploité plusieurs expériences ayant
pour objectifs :
• L’étude de l’évolution du relargage au cours du temps (cinétique).
• La détermination de l’influence de paramètres physico-chimiques pertinents (ratio
liquide/solide, température, concentration en cyclodextrine) sur l’efficacité
d’extraction.
• La comparaison des différents types de cyclodextrines entre eux.
Le bilan de l’ensemble des travaux présentés dans la conclusion de ce mémoire permettra
de poser les bases scientifiques nécessaires au développement d’un procédé
écocompatible de traitement de lavage de sols pollués utilisant les cyclodextrines.

27
Chapitre I :
Etude bibliographique

28

29
Chapitre I : Etude bibliographique Introduction Compte tenu de la finalité du projet dans lequel s’insère ce travail de thèse, à savoir la
mise au point d’un procédé original de traitement de sol contaminés, nous avons scindé
ces rappels bibliographiques en 4 parties :
La première traite des principales caractéristiques des cyclodextrines en lien avec le
contexte du projet. L’intérêt de la communauté scientifique est tel que l’état des
connaissances sur ces « molécule-cage » est nécessairement présenté de manière
exhaustif. Nous y décrivons certains des propriétés physico-chimiques qui concernent les
formes natives et modifiées. Les propriétés complexantes, essentiellement vis-à-vis de
molécules organiques, sont expliquées, de même que les facteurs physiques et chimiques
qui influencent ces complexations. Quelques méthodes d’étude des équilibres de
complexation sont également présentées.
Dans la seconde partie, nous aborderons la problématique des familles de polluants
organiques les plus fréquemment rencontrés sur des sites industriels contaminés. Nous
avons détaillé les connaissances concernant deux de ces familles : les chlorophénols (CP)
et les Hydrocarbures Aromatiques Polycycliques (HAP). Outre les propriétés physico-
chimiques de ces molécules, nous présentons leurs impacts potentiels sur
l’environnement, au sens large. Sont inclues également les principales caractéristiques de
leur dégradation naturelle dans les écosystèmes.

30
La troisième partie est consacrée aux interactions entre les sols et les polluants
organiques. Cette présentation des connaissances traite, en particulier, de la nature de ces
interactions et des paramètres qui peuvent les influencer.
Enfin, il nous était impossible de passer sous silence l’état de l’art des procédés de
traitement de sols et sites contaminés. La quatrième partie de cette bibliographie résume
donc les principales techniques de remédiation physiques, chimiques et biologiques
actuellement utilisées à l’échelle industrielle. Les quelques travaux de laboratoire portant
sur les potentialités des cyclodextrines dans ce domaine y sont décrites également.
Cette revue bibliographique, qui peut paraître peu « académique » dans sa forme,
recouvre globalement une partie des travaux scientifiques concernant les aspects
fondamentaux et appliqués de la recherche dans le domaine du traitement des sols
pollués. Le bilan qui en est réalisé permet de positionner notre programme de recherche
par rapport aux connaissances actuelles.

31
Partie A : Les cyclodextrines
I. Introduction
Les cyclodextrines (CD) naturelles ou chimiquement modifiées font partie de la famille
des « molécules-cage ». Elles sont connues pour leur aptitude à accroître la solubilité de
nombreuses molécules organiques par formation de composés, ou complexes d’inclusion.
Cette propriété confère aux cyclodextrines un large champ d’application dans des
domaines très variés allant de la pharmacie à l’agriculture en passant par l’industrie
textile, la chimie des parfums et des arômes, etc.
Les cyclodextrines font l’objet, depuis les années 80, d’un grand intérêt de la part de la
communauté scientifique internationale. Cela se traduit depuis une vingtaine d’années par
une production de plusieurs centaines de publications annuelles dans des revues
appliquées et fondamentales.
Dans cette partie, nous avons choisi de limiter la présentation des cyclodextrines à leurs
propriétés structurales et géométriques qui conditionnent leur réactivité. Les méthodes de
caractérisation et de détermination des paramètres réactionnels seront également
présentées. Enfin, dans l’optique de notre travail de recherche, une part importante sera
consacrée aux facteurs physico-chimiques qui conditionnent la complexation d’une
molécule organique par une ou plusieurs cyclodextrines.

32
II. Caractéristiques des cyclodextrines
II.1. Origine et caractéristiques structurales des CD
Les cyclodextrines sont des molécules obtenues par dégradation enzymatique (amylase
de Bacillus Macerans) de l’amidon. Elles ont été découvertes à la fin du XIXéme siècle
par Villiers (1891). Caractérisées et étudiées par Schardinger (Szjetli, 1982 ;1998,
Eastburn et Tao, 1994) dans les années 1900-1910, elles sont souvent nommées
« dextrines de Schardinger ». Elles font partie de la famille des molécules cages au même
titre que les calyxarènes, les éthers-couronnes, le cucurbituril, etc.
Ce sont des oligomères cycliques du glucose de formule brute (C6H10O5)n et comportant
de 6 à 12 unités α-D-glucopyranose. Les trois principales cyclodextrines, notées α-, β-, et
γ-cyclodextrine contiennent respectivement 6, 7 et 8 unités glucose (figure 1).
Figure 1: Structure moléculaire et forme géométrique des cyclodextrines (α, β, γ).
33
Elles sont souvent schématisées par un tore avec une cavité interne dont les dimensions
varient selon le nombre d’unités glucose. Une étude RMN a montré que chaque unité
glucose adopte une conformation de type chaise (Sjzetli, 1982). Tous les groupes polaires
(hydroxyles OH) sont localisés à l’extérieur. Les fonctions « alcool primaire » (une par
unité glucose en position 6) sont orientées vers la partie la plus étroite du tronc de cône.
Les fonctions « alcool secondaire » (en position C2 et C3) sont situées à l’opposé sur la
partie large de la couronne (Fig.1).
Cette organisation moléculaire délimite une cavité rendue relativement apolaire
(hydrophobe) par la présence de ponts osidiques (-O-). C’est grâce à ce caractère
amphiphile (hydrophile à l’extérieur, hydrophobe à l’intérieur) que les CD sont capables
d’inclure dans leur cavité apolaire des molécules hydrophobes pour former des
complexes d’inclusion solubles dans l’eau (Matsunga et al., 1984, Sjzetli, 1998)
Le tableau 1 ci-dessous résume quelques propriétés structurales de ces cyclodextrines
dites « natives ».
Tableau 1: Propriétés structurales des cyclodextrines natives.
CD α β γ
Nombre de glucose 6 7 8
Diamètre intérieur de
la cavité (Å)
4,9±0,3 6,2±0,3 7,9±0,3
Diamètre extérieur
de la cavité (Å)
12,5±0,4 13.8±0,4 15.5±0,4
Hauteur (Å) 7,9±0,1 7,9±0,1 7,9±0,1
Volume de la cavité
(nm3)
0,174 0,262 0,472

34
II.2. Propriétés physico-chimiques des CD
II.2.1. Solubilité dans l’eau
Les valeurs de solubilité des cyclodextrines dans l’eau à la température de 25°C sont
reportées dans le tableau 2 (Jozwiakowski et Connors, 1985).
Tableau 2: Propriétés physico-chimiques des cyclodextrines.
CD α β γ
Masse molaire
anhydre
g.mol-1
972 1135 1297
Solubilité aqueuse
g.L-1 à 25 °C
145 18,5 232
pKa 12,33 12,2 12,08
Hydratation
CD,nH2O
n = 6 à 7 n = 10 à 12 n = 7 à 13
Ces valeurs montrent que malgré une forte similitude structurale, les cyclodextrines n’ont
pas un comportement comparable vis à vis des molécules d’eau. La solubilité limitée de
la β-CD peut être attribuée à l’influence de liaisons hydrogènes qui s’établissent entre les
atomes d’hydrogène et d’oxygène des fonctions alcools secondaires (Paginton, 1987).
Cette faible solubilité a aussi été attribuée à la formation d’agrégats de β-CD (Sjzetli,
1982). Une étude systématique sur l’influence d’un environnement ionique a montré que
la solubilité augmente dans des solutions de cations métalliques avec la concentration
(Jozwiakowski et Connors, 1985). Plus récemment, Fenyvesi et al. , (1999) ont étudié la
solubilité de la β-CD dans des solutions aqueuses de différents acides organiques. Ils ont
montré que les acides hydroxyliques, tels que les acides citrique et tartrique augmentent
la solubilité de la β-CD dans l’eau, tandis que les acides carboxyliques la diminuent.

35
II 2.2. Aspects énergétiques de l’hydratation et de la dissolution
Les études concernant les interactions entre les cyclodextrines natives (α, β, γ) et l’eau
ont permis de mieux comprendre les différences de solubilité constatées. Les
cyclodextrines cristallisent sous forme d’hydrates non définis (CD, nH2O) et leur taux
d’hydratation est largement dépendant de la pression de vapeur d’eau du milieu
environnant (Bilal et al., 1995). Concernant la β-CD qui a été la plus étudiée,
l’hydratation moyenne est de 10 à 12 molécules d’eau. Les nombres moyens de
molécules d’eau d’hydratation des CD sont regroupés dans le tableau 2.
D’un point de vue énergétique, les mesures des enthalpies de dissolution des
cyclodextrines anhydres et hydratées ont permis de calculer les enthalpies de
déshydratation (Merlin, 1998). Les résultats montrent que les énergies de liaison H2O/CD
sont de l’ordre d’une dizaine de kJ par molécule d’eau, c’est-à-dire cohérentes avec les
énergies mises en jeu dans les liaisons hydrogène.
Certaines études s’accordent pour montrer qu’il existe un échange permanent des
molécules d’eau intra- et intermoléculaire dans la β-CD (Claudy et al., 1990). Les
différences de comportement des CD par rapport à l’eau, en particulier, en terme de
solubilité, ont été reliées à la possibilité (ou non) d’établir des liaisons hydrogène
interglucose et intramoléculaires qui dans le cas de la BCD stabilise le macrocycle. Cette
possibilité dépend de la distance moyenne entre les atomes d’hydrogène et d’oxygène
(OH en C2 et C3), qui est fonction du nombre d’unité glucose de la CD (de Brauer et al.,
2000).
II.2.3. Stabilité des CD en solution
La stabilité des CD en solution est relativement peu influencée par les conditions de pH et
de température. Selon Stella et Rajewski (1997), l’hydrolyse des cyclodextrines peut
avoir lieu dans certaines conditions de pH très acide (< 1) et à 80 °C. En milieu très
basique (pH > pKa), il y a possibilité de former des ions alcoolates plus solubles que les
CD neutres. Les valeurs des constantes pKa sont données dans le tableau 2 pour les 3
cyclodextrines natives.

36
II.3. Cyclodextrines modifiées Parmi les trois cyclodextrines natives les plus courantes (α-, β-, γ-CD), la β-CD est de
loin la moins coûteuse. Toutefois, son utilisation est généralement limitée en raison de sa
faible solubilité aqueuse (Suzuki et Sazaki, 1979). La modification chimique des CD
permet d’obtenir des dérivés possédant des propriétés physico-chimiques différentes de
celles des CD natives et d’élargir ainsi leurs champs d’application. Les objectifs de ces
modifications sont :
- L’augmentation de la solubilité aqueuse.
- La modification de la capacité de complexation (constante de stabilité, sélectivité).
- L’introduction de groupements à fonctions spécifiques (catalytique, complexation de
cations métalliques, etc.)
Ces modifications chimiques portent sur :
La substitution d’un ou de plusieurs groupements hydroxyles par des halogènes, des
groupements amines...
L’oxydation des alcools primaires pour former des aldéhydes ou des acides
carboxyliques.
La substitution nucléophile interne avec formation d’époxyde.
La substitution d’un ou de plusieurs atomes d’hydrogène (des hydroxyles primaires ou
secondaires) pour former des éthers ou des esters. Il s’agit des modifications les plus
courantes. Les CD modifiées les plus étudiées et utilisées sont l'hydroxypropyl-β-
cyclodextrine (HPCD), la méthyl-β-cyclodextrine (MCD) et la carboxyméthyl-β-
Cyclodextrine (CMCD). Ces molécules sont obtenues par substitution de certains
hydroxyles par des groupements hydroxypropyl (-C3H7O), méthyl (-CH3), et
carboxyméthyl (-CH2COOH ). Ces cyclodextrines substituées ont des performances et
des coûts sensiblement différents de ceux de la forme native. Ces molécules
chimiquement modifiées ont des masses molaires moyennes qui correspondent à des
indices molaires moyens de substitution. Elles possèdent une spécificité et une solubilité
aqueuse relativement importante qui peut être de l’ordre de 100 à 1000 g.L-1 (Eastburn et
Tao, 1994, Singh et al., 2002) La figure 2 présente, pour exemple, la structure de la
CMCD, une cyclodextrine modifiée avec un degré de substitution d’environ 1 (une
fonction carboxylique par unité glucose).

37
Figure 2 : Structure moléculaire de la carboxyméthyl-β-Cyclodextrine (CMCD).
II. 4. Biodégradabilité
La biodégradation des cyclodextrines natives (α-CD, β-CD, γ-CD) est rapide et complète
tandis que celle des cyclodextrines ramifiées est plus faible et dépend du degré de
substitution (Singh et al., 2002). Selon Verstichel et al. (2004), dans des conditions
expérimentales idéales, le pourcentage de biodégradation des CD natives atteint 90%
après 15 jours d’incubation tandis que seulement 5,6 % de dégradation des CD acétylées
a été observé après 45 jours. La substitution par des groupements méthyl ou
hydroxypropyl affecte aussi la biodégradation de la β-CD. Seulement 20 % de HPCD
sont dégradés après 100 jours indiquant que ce type de CD est tout de même
partiellement dégradable dans ces conditions (Verstichel et al., 2004). Fava et al., (2002a)
ont observé que la dégradation de la β-CD méthylée de façon aléatoire dite « Randomly-
méthyl β-cyclodextrine » (RAMEB) par les bactéries en conditions aérobies est
relativement faible.
De par leur nature chimique, les CD constituent une source de carbone et d’énergie
aisément utilisables par les micro-organismes présents dans des sols (Fenyvessi et al.,
2002, Fava et al., 2002b). Fava et al. (1998) ont montré que les bactéries endogènes du
sol se multiplient en présence de HPCD avec un taux de croissance proportionnel à la

38
concentration de CD. Globalement, il ressort de ces quelques études que les
cyclodextrines présentent relativement une bonne biodégradabilité dans les sols.
II.5. Toxicité des CDs
En général les cyclodextrines sont considérées comme relativement peu toxiques par
administration par voie orale. Ces molécules ne diffusent quasiment pas au travers des
membranes biologiques et ne sont pas absorbées lors du transit intestinal (DL50 oral du
rat = 18800 mg/kg pour la β-cyclodextrine).
Plusieurs études ont montré que les CD ne présentent aucun effet toxique ou inhibitif sur
la population bactérienne du sol (Fava et al., 1998 ; Reid et al., 2000).
Certaines CD sont utilisées dans des formulations médicamenteuses et sont ingérées par
voie orale (Stella et Rajewski, 1997). Nous pouvons alors les considérer comme des
agents de formulation plutôt que comme des réactifs chimiques ordinaires (contrairement
aux agents de complexation ou de solubilisation tels que les solvants organiques ou les
détergents).
III. Propriétés complexantes des cyclodextrines et
applications
III. 1. Complexe d’inclusion Un composé d’inclusion est formé à partir d’une espèce réceptrice qui inclut plus ou
moins profondément un substrat moléculaire ou ionique (figure 3). Cette inclusion n’est,
en général, pas accompagnée de la formation d’une liaison covalente ou de coordination.
Dans le cas des CD, le caractère hydrophobe de la cavité permet d’inclure des molécules
dites « invitées » dont l’hydrophobicité et la taille correspondent à celles de la cavité
tandis que les fonctions hydroxyles assurent une bonne solubilisation des complexes dans
l’eau. Une ou plusieurs molécules peuvent être « encapsulées » dans une, deux, et même
parfois trois molécules de cyclodextrine.

39
Figure 3 : Représentation 3D d’un complexe CD/substrat
La nature exacte des interactions mises en jeu dans la formation d’un composé
d’inclusion n’est pas encore parfaitement établie. Il semble qu’elle dépende du substrat
considéré. Un des facteurs déterminants est d’ordre géométrique : la partie incluse doit
évidemment avoir une taille plus petite que celle de la cavité de la cyclodextrine
considérée. Les tailles respectives de la cyclodextrine et du substrat conditionnent
généralement la stœchiométrie du complexe (Saenger, 1980, Giordano et al., 2001).
Les interactions mises en jeu lors de la complexation peuvent être de natures différentes :
- Interactions électrostatiques
- Forces de Van Der Waals
- Interactions hydrophobes
- Liaisons d’hydrogène
Notons l’importance de l’eau dans le processus d’inclusion. La principale force
gouvernant la formation de ces complexes est la stabilisation énergétique du système par
le remplacement, dans la cavité, des molécules d'eau à haute enthalpie par des molécules
hydrophobes qui créent des associations de type « apolaires-apolaires ». Autrement dit,
l’exclusion des molécules d’eau suite à l’inclusion du composé « invité » dans la cavité
de la CD peut être considérée comme une des étapes clés de la complexation (Merlin,
1998 ; Liu et al., 2001 ; Liu et Guo, 2002).

40
Ces molécules incluses ne sont pas fixées de façon statique mais sont en équilibre
dynamique entre leur état libre et complexé. La stabilité du complexe dépend aussi du
contexte chimique du milieu.
La destruction du complexe peut se faire par méthode thermique, par hydrolyse
enzymatique, en modifiant le pH ou en utilisant des solvants organiques. Une molécule
hydrophobe pourra avoir plus d’affinité pour un solvant apolaire que pour la cavité de la
CD. La décomplexation a alors lieu par transfert de la molécule « invitée » dans la phase
organique, ce qui régénère, de fait, la cyclodextrine. Une précipitation du complexe ou de
la CD peut également dépendre de la nature et de la concentration du solvant organique
ajouté (Matsunga et al., 1984 ; Blyshak et al., 1988, 1989). Cela peut permettre de séparer
physiquement les constituants du mélange ou de recycler la CD pour des usages
ultérieurs.
III.2. Etude des complexes d’inclusion des cyclodextrines : paramètres
réactionnels.
Il existe plusieurs techniques permettant d’étudier ces complexes d’inclusion. Elles
permettent, d’une part de mettre en évidence leur formation, et d’autre part, de déterminer
leur stœchiométrie et leur stabilité.
Ces différentes techniques reposent sur les modifications des propriétés physico-
chimiques ou optiques de la molécule complexée liées à la modification de sa solubilité
ou de son microenvironnement.
Les techniques couramment utilisées sont : la méthode de solubilisation (Wang et
Brusseau 1993, 1995), la spectroscopie d’absorbance (UV et visible) (Susuki et Sasaki,
1979), la spectroscopie d’émission moléculaire (fluorescence) (Reeuwijik et al., 1993), la
Résonance Magnétique Nucléaire, RMN (Saito et al., 1996), la titration
microcalorimétrique (Irwin et al, 1994, Simer et Kurvits, 1998), l’électrophorèse
capillaire, le suivi de la tension superficielle et d’autres méthodes électrochimiques.
Les techniques spectroscopiques consistent à suivre l’intensité d’absorbance ou de
fluorescence de la molécule complexée. Le passage d’une molécule de la phase aqueuse
vers la cavité de la cyclodextrine modifie la polarité de son microenvironnement. Ceci

41
provoque une augmentation ou une atténuation de son intensité d’absorption ou
d’émission (Mcbain et Hutchinson, 1955). Cela peut permettre de suivre l’avancement de
la complexation et de déterminer la constante de stabilité du complexe. Ces techniques
sont aussi utilisées pour doser la cyclodextrine dans l’eau.
Concernant la détermination de la constante d’équilibre de complexation et de la
stœchiométrie du complexe, nous présenterons les méthodes les plus utilisées et
développerons la méthode de solubilisation que nous avons mise en œuvre dans le cadre
de ce travail.
III.2.1. Méthode de solubilisation
La solubilisation des solutés par une CD peut être considérée comme une partition de ces
solutés entre l’eau (solvant) et la cavité de la CD (Connors, 1987).
La solubilité apparente des substances organiques dans des solutions aqueuses contenant
la cyclodextrine, augmente linéairement avec la concentration de cyclodextrine (Wang et
Brusseau, 1993, Ji et Brusseau, 1998 ; Tanada et al., 1999 ; Cao et al., 2000). Ce
phénomène est attribué à la formation de complexes d’inclusion 1:1 ou 1:2 comme décrit
par les équations (1) et (2) ci-après :
P + CD ⇔ P/CD (1)
P + 2CD ⇔ P/(CD)2 (2)
Où P est le composé dissout non complexé, CD la cyclodextrine non complexée ou libre.
P/CD et P/(CD)2 sont les complexes d’inclusion 1:1 et 1: 2.
La constante de stabilité (K) du complexe, appelée aussi constante d’équilibre de
complexation peut être exprimée par les relations suivantes, selon sa stœchiométrie :
]][[]/[
1 CDPCDPK =
22
2 ]][[])/([
CDPCDPK =
Où [P], [CD] et [P/CD] sont les concentrations des espèces à l’équilibre.

42
Dans l’hypothèse où la concentration en CD impliquée dans un complexe est négligeable
devant celle de la CD libre, la concentration en CD libre peut être considérée comme
égale à la concentration initiale en CD. La concentration en soluté libre est considérée
comme étant égale à sa solubilité aqueuse. En considérant cette hypothèse, une relation
linéaire est obtenue entre la solubilité relative [Sr] du soluté et la concentration de CD :
Sr = ][1 1 CDKSS
w
a += (3)
Sr = ][1 2 CDKSS
w
a += 2 (4)
Sa est la concentration totale en soluté incluant à la fois les espèces libres et complexées,
dite aussi solubilité apparente ; Sa = [P] + [P/CD].
Sw est la solubilité aqueuse du composé et considérée comme la concentration en espèces
libres en solution.
Cette hypothèse simplificatrice n’est toutefois pas valable dans le cas où la complexation
se produit entre la CD et des composés ayant une solubilité aqueuse relativement élevée
puisque, dans une telle situation, la concentration en CD libre sera réduite. Les relations
précédentes ne sont donc utilisables que pour les composés faiblement solubles.
La solubilité relative sera tracée en fonction de la concentration en CD. La linéarité
obtenue selon l’équation 3 ou 4 pourra permettre de préciser la stœchiométrie du
complexe (1 ou 2).
Ce modèle a été largement utilisé pour étudier la solubilisation des NOC (composés
organiques non ioniques) par des matières organiques dissoutes et des surfactants (Chiou
et al., 1986 ; Edwards et al., 1991, Ko et al, 1999). La figure 4a représente un complexe
1 :1 de phénol/cyclodextrine (Buvrai et Barcaza, 1988) tandis que la figure 4b illustre la
structure d’un complexe 1 : 2 nonylphénol /cyclodextrine étudié par Kawasaki et al.
(2000).

43
Figure 4a : Complexe 1 : 1 Figure 4b: Complexe 1 : 2
III.2.2. Autres méthodes
III.2.2.1 Méthode d’exaltation UV.
Cette méthode est applicable si l’absorbance de la molécule « invitée » est modifiée en
présence de CD en solution. Elle consiste à suivre la variation d’absorbance du soluté à
une concentration déterminée (S0) en fonction de la concentration en CD (C0). En
utilisant la relation suivante, la constante de complexation K pourra être déterminée par
régression linéaire:
εε ∆+
∆=
∆11
.1.
0
0
CKASl
(∆A = A – Ao), A et Ao sont les densités optiques (absorbance) en présence et en
absence de CD, ∆ε est la différence d’absorption entre le soluté libre et complexé, l est la
longueur du chemin optique (Pitchumani et al., 1992, Gao et al., 1998).
III.2.2.2. Méthode fluorimètrique
On applique le même principe de calcul que dans la méthode précédente, mais dans ce
cas, il s’agit de mesurer la fluorescence de la molécule et son évolution en fonction de la
concentration de CD. La constante de complexation K pourra être calculée par régression
linéaire en appliquant la relation suivante :
OH O H

44
αα111
+=∆ CD
s
CKFC
où Cs et CCD sont les concentrations de soluté et de CD, respectivement. α est une
constante. ∆F est la différence d’intensité de fluorescence du soluté en présence et en
absence de CD (Kondo et al, 1976 ; Coly et Auron, 1998 ; Dostsikas et al., 2000).
III.3. Principales applications des cyclodextrines
Les principales applications des cyclodextrines concernent les secteurs de la pharmacie
(vectorisation de principes actifs…) de la cosmétique, de l’agro-alimentaire (protection
d’arôme contre l’oxydation…) de l’agriculture (encapsulation d’insecticides…) et de
l’analyse chimique (méthode de séparations, chromatographie, etc. ). Toutes ces
applications sont dues au fait que la CD peut former des complexes d’inclusion avec de
nombreux composés organiques (Kamiya et al., 1994, 1995 ; Rajewski et Stella, 1996 ;
Prasad et al., 1999 ; Zhang et al., 2002).
En ce qui concerne l’application pharmaceutique, l’amélioration de la biodisponibilité
d’un principe actif est un des avantages des complexes d’inclusion CD/principe actif. Le
complexe permet d’augmenter la vitesse de dissolution des médicaments ou des principes
actifs, ce qui accroît leur biodisponibilité. Le mécanisme est le suivant : le complexe
d’inclusion solide, est dissous dans les liquides biologiques, les CD ne passent pas les
membranes biologiques parce qu’elles sont hydrophiles et de masse moléculaire élevée
(Rajewski et Stella, 1996). Elles sont éliminées et seul le principe actif est absorbé.
L’inclusion sélective dans la cavité de la CD peut également permettre de séparer
différents isomères (Yudiarto et al, 2000).
Les potentialités des CD dans le domaine de l’environnement n’ont été explorées que
depuis 1992. Plusieurs approches ont été utilisées pour évaluer l’efficacité des CD dans le
domaine de la remédiation des sites pollués :
La première approche consiste à extraire du sol et des aquifères des polluants peu
polaires à l'aide d'une solution aqueuse de cyclodextrine. Utilisées en solution
aqueuse à une concentration relativement élevée (5 à 10 %), elles peuvent faciliter la
désorption des polluants du support poreux constitué par le sol et leur transport par

45
formation de complexes plus solubles. Cette méthode est nommée « Complexing
Sugar Flush ». Les premiers travaux d’une équipe américaine de l’université
d’Arizona (Wang et Brusseau, 1993, 1995 ; Brusseau et al., 1994, 1997 ; Boving et
al., 1999, 2000) confirme ces potentialités et montre que les cyclodextrines sont aptes
à mobiliser et à extraire des sols de polluants organiques et parfois inorganiques.
La seconde approche vise à utiliser les CD en vue d’augmenter la biodisponibilité des
polluants organiques dans le sol. Une étude en réacteur montre que la présence de
cyclodextrines en solution à faible concentration augmente significativement la
biodégradation des HAPs et des polychlorobiphényles (Wang et al., 1998, Fava et al.,
1998, 2002 ; Bardi et al., 2000, Molnar et al., 2002). Ce résultat est, selon les auteurs,
dû à l’augmentation de la disponibilité du polluant par effet de solubilisation. Les CD
peuvent augmenter la concentration de polluant dans le biofilm où se trouve la
microflore du sol. Les mécanismes de dégradation de polluants par les bactéries
endogènes en présence de cyclodextrines sont encore mal connus.
Enfin, les CD peuvent permettre de mettre en évidence la fraction biodisponible des
polluants dans le sol en la mobilisant. Par conséquent, une CD pourra servir comme
une protection de la population microbienne du sol en éliminant la fraction biotoxique
et nocive pour les micro-organismes endogènes (Reid et al, 2000; Cuypers et al.,
2000). Song et al., (1999) ont ainsi observé une diminution de la biotoxicité des
composés hydrophobes en présence de 5 g/L de β-CD dans la solution aqueuse.
Bien que les CD possèdent un pouvoir de solubilisation des molécules organiques plus
faible que de nombreux tensioactifs ordinaires utilisés habituellement comme agents de
lavage, leur faible réactivité vis à vis des sols, leur relative insensibilité au pH (sauf
extrêmes), leur efficacité tant vis à vis des molécules organiques que de certains métaux
lourds et leur biocompatibilité, constituent des avantages certains pour le traitement des
sols pollués (MCray et Brusseau, 1998 ; Boving et Brusseau, 2000).

46
IV. Facteurs influençant la complexation des molécules
organiques par les CD.
IV.1. Facteurs liés à la nature des molécules organiques
IV.1.1. Hydrophobicité et hydrosolubilité
Les ponts glycosidiques donnent à la cavité de la CD une polarité proche de celle de
l’éthanol (Demian, 2000). Grâce à cet environnement relativement apolaire de la cavité,
les CD sont capables d’accueillir des molécules hydrophobes. Ces molécules ont plus
d’affinité pour la cavité de la CD que pour la phase aqueuse (Tanada et al., 1999). En
effet si les polluants organiques hydrophobes sont plus petits que la cavité, il existe une
bonne corrélation (linéaire) entre les constantes de complexation et le coefficient de
partage octanol/eau qui est un bon indicateur de l’hydrophobicité (Wang et Brusseau,
1993). En résumé, plus la molécule est hydrophobe, plus le complexe formé est stable.
IV.1.2. Etat d’ionisation de la molécule
Selon le pH, les acides faibles ou les bases faibles existent sous plusieurs formes ioniques
en solution. Ces différentes formes du soluté ne présentent pas les mêmes caractéristiques
physico-chimiques (solubilité, hydrophobicité) (Valsaraji, 1995). Par conséquent,
l’affinité de la CD ne sera pas la même pour chacune des formes. La complexation des
CD avec les molécules ionisables devra nécessairement prendre en compte l’influence du
pH (Liu et al., 2001; Liu et Guo, 2002).
IV.1.3. Taille et forme de la molécule
Un des paramètres importants pour la complexation de polluants est leur taille par rapport
à celle de la cavité de la cyclodextrine (Wang et Brusseau, 1993). Les tailles relatives de
la cavité de la cyclodextrine et du substrat conditionnent aussi souvent la stœchiométrie
du complexe. Plus la taille de la molécule invitée est ajustée par rapport à celle de la
cavité, plus le complexe formé sera stable (plus de contact avec la cavité de CD, plus
d’interactions de Van der Waal’s) (Cao et al., 2000). Toutefois, la complexation peut

47
aussi dépendre d'autres facteurs comme la surface moléculaire du polluant ou son
orientation dans la cavité.
IV.2. Facteurs liés aux caractéristiques des cyclodextrines
Les cyclodextrines modifiées montrent, en général, une capacité plus importante que les
formes natives à solubiliser les composés organiques (Gao et al., 1998). Parmi les
cyclodextrines modifiées, la méthyl-β-CD (MCD) semble présenter une efficacité plus
importante que l’hydroxypropyl-β-CD (HPCD) pour solubiliser les solvants chlorés
(Boving et al., 1999). Selon eux, cette capacité est probablement due au caractère
hydrophobe remarquable de la cavité de la MCD. Brusseau et al. (1997) montrent que la
capacité de complexation de l’HPCD pour le phénanthrène est plus importante que celle
de la CMCD.
Kawasaki et al. (2001) ont montré que le pouvoir solubilisant de l’HPCD vis à vis du 4-
nonylphénol dépend de son degré de substitution. Ce dernier élément est donc à prendre
en compte dans le cas de cyclodextrines modifiées.
Il n’existe aucun critère permettant d’établir des règles générales systématiques
concernant la force des complexes CD/molécules organiques. En effet, l’encapsulation
d’un substrat résulte de plusieurs équilibres compétitifs.
IV.3. Influence du contexte chimique
IV.3.1. pH
Le pH des solutions influence évidemment la nature (neutre ou ionisée) des molécules
ionisables (selon leur constante d’acidité pKa).
Comme il a déjà été expliqué dans le paragraphe IV.1.2, la complexation, par les CD, des
différentes formes ioniques d’un soluté ne résultent pas du même type d’interactions
(Buvari and Barcza, 1988 ; Liu et al., 2001).

48
IV.3.2. Force ionique
La présence de cations ou d’anions en solution peut affecter la solubilité aqueuse de
certaines molécules (Daughney and Fein, 1997, Wightman and Fein, 1999) et par
conséquent leur affinité pour la cavité des CD.
Par exemple, la solubilisation de la pancratistatine dans l’hydroxypropyl-β-cyclodextrin a
été optimisée par l’addition d’hydroxyde d’ammonium (Torres-Labandeira et al., 1990).
Par contre, Wang et Brusseau (1995) ont montré que la présence de CaCl2 a un effet
négligeable sur la solubilisation de l’anthracène par la CMCD. De même, la capacité de
solubilisation de la CMCD pour l’anthracène, le trichlorobenzène et le biphényl, ne parait
pas affectée par de fortes concentrations en sel en solution lorsque les cations
n’interagissent pas avec la cavité de la CD.
L’effet de la force ionique sur la solubilité aqueuse des molécules ainsi que sur leur
complexation par les CD est différente selon leur nature (molécules ionisables ou non-
ionisables).
IV.3.3. Influence de la présence d’un solvant organique
L’influence d’un cosolvant sur la complexation d’une molécule organique par une CD
dépend de la nature et de la concentration du solvant organique présent.
La présence d’éthanol (< 30 %) diminue la formation de complexe d’inclusion dans la
phase aqueuse comme c’est le cas entre la testostérone et l’hydroxypropyl-β-cyclodextrin
(Pitha et Hoshino, 1992). Brusseau et al. (1994) ont utilisé le mélange eau : méthanol
(50:50) pour décomplexer les complexes CD/ HAP. En revanche, les mêmes auteurs ont
noté que 10 % de cyclopentanol en solution favorise la complexation de l’anthracène
avec certaines CD (Wang et Brusseau, 1995).
Bien que les cyclodextrines puissent former des composés d’inclusion dans certains
solvants organiques comme les alcools, le diméthylsulfoxyde, ou le diméthylformamide,
l’association est en général plus faible que celle observée pour le même composé en
milieu aqueux (Blyshak et al., 1988).
Comme il a déjà été indiqué, l’addition d’un solvant organique peut permettre de déplacer
les équilibres CD/soluté et de modifier la solubilité du complexant et du soluté.

49
IV.4. Influence de la température La température a un double effet sur le complexe formé : d'une part elle augmente
généralement sa solubilité, mais en même temps elle modifie sa stabilité. La plupart des
complexes commencent à se décomposer vers 50-60°C, bien que certains soient stables à
de plus hautes températures, particulièrement si la molécule est fortement hydrophobe.
L’effet de la température dépend alors de la nature du substrat et aussi de celle de la CD
(Simer et Kurvits, 1998). Le rendement de complexation entre l’α-CD ou la γ-CD et
l’acide chlorogènique diminue de 50 ± 14% quand la température augmente de 3 à 37°C,
tandis qu’avec la β-CD, la complexation augmente avec la température jusqu’à 25°C
(enthalpie positive), elle se stabilise entre 25 et 40°C, puis diminue entre 40 et 60°C
(enthalpie négative). Pour la β-CD, la variation de la constante de complexation avec la
température est probablement liée au changement de solvatation du complexe (Irwin et
al., 1994).
Le rendement de la complexation des dérivés d’imidazoles par la β-CD diminue quand la
température évolue de 8 à 70°C (∆H = -14 kJ/mol)(Morin et al., 1998). Blyshak et
Warner (1990) ont étudié l’effet de la température sur la complexation des HAP avec les
CD. Ils ont constaté que la constante de stabilité des complexes diminue
considérablement avec la température.
Globalement, il apparaît qu’une élévation de la température est souvent défavorable à la
formation des complexes CD/molécules organiques.
V. Conclusion
Au travers de ce bref, et non exhaustif, tour d’horizon bibliographique des connaissances
sur les cyclodextrines, il apparaît que leur potentiel d’utilisation est loin d’avoir été
totalement exploré. L’affinité remarquable des CD pour les molécules apolaires
(hydrophobes) peut être mise à profit pour augmenter leur solubilité. De plus, certaines
propriétés (qualités) spécifiques des CD telle que la bio-compatibilité, auquel s’ajoute un
faible impact sur les écosystèmes permettent d’envisager un avenir prometteur dans
l’extraction des polluants organiques de sols contaminés. Ce devrait être le cas,
notamment vis-à-vis de Polluants Organiques Hydrophobes (POH). Ces polluants sont
prioritaires en raison de leur persistance dans les sols, due principalement à leur faible

50
solubilité aqueuse et à leur caractère lipophile prononcé. Les propriétés physico-
chimiques de ces polluants pourront conditionner leur complexation ou solubilisation par
les CD et également leur comportement ou leur mobilité dans les sols. C’est pourquoi,
nous développerons, dans le chapitre suivant, les principales caractéristiques de deux
familles de polluants hydrophobes que nous avons étudiées, et leur comportement dans
l’environnement.

51
Partie B : Les polluants organiques
I. Introduction
Les polluants organiques sont classiquement regroupés en trois grandes classes : Les
hydrocarbures, les hydrocarbures aromatiques polycycliques et les pesticides. Chaque
famille de polluants a des caractéristiques physico-chimiques différentes qui
conditionnent leur devenir dans les milieux poreux, tels que les sols, et, plus
généralement leur comportement dans l’environnement.
Les polluants organiques hydrophobes sont des polluants prioritaires en raison de leur
toxicité, de leur forte adsorption et leur persistance dans les milieux environnementaux.
L'adsorption et la précipitation dans les sols ont pour effet d'entraîner une persistance des
molécules se traduisant par une protection contre les attaques microbiennes et une baisse
de leur biodisponibilité et biodégradabilité. Cela est du principalement à leur faible
solubilité aqueuse et à leur caractère lipophile prononcé.
Les propriétés des polluants permettront de prédire leur comportement aux interfaces
eau/solide/gaz, aussi bien dans la nature, qu’en présence d’additifs ou d’agents de
complexation. C’est pourquoi, une grande partie de cette revue bibliographique est
consacrée aux propriétés physico-chimiques de deux familles de polluants : les
chlorophénols (CP) et les Hydrocarbures Aromatiques Polycycliques (HAP). Ces deux
familles ont été sélectionnées pour leur représentativité d’une large gamme de polluants
organiques rencontrées sur les sites industriels pollués. De plus, le fait que ces deux
familles possèdent des caractéristiques physico-chimiques nettement différentes semble
être intéressant dans le cadre de notre travail puisque les paramètres chimiques (pH, force
ionique, présence d’un additif, etc.) ont des effets remarquables sur leur comportement en
solution et à l’interface solide/eau.
Dans l’objectif global de notre travail, une part de la synthèse bibliographique sera
consacrée au devenir des polluants sélectionnés dans les sols et aux paramètres qui
influencent et conditionnent leur comportement dans l’environnement.

52
II. Généralités sur les polluants organiques
L’adsorption des polluants dans les sols, leur solubilité, leur toxicité et leur
bioaccumulation dans les organismes vivants dépendent, en général, de leur
hydrophobicité. Celle ci est évaluée par le coefficient de partage octanol/eau (Kow), un
paramètre physico-chimique largement utilisé pour prédire les comportements d’une
molécule organique dans l’environnement.
Le devenir des polluants dans les sols auquel nous allons consacrer, une part relativement
importante de cette partie, est intimement lié à leur lipophilie et aux interactions
hydrophobes qu’ils peuvent développer avec les matières organiques naturelles du sol.
Ces dernières sont généralement évaluées par le coefficient de partage carbone
organique/eau (Koc). Le coefficient de partage Koc est défini comme le rapport des
concentrations de composés adsorbés sur les particules solides et dissouts dans la phase
liquide.
III. Chlorophénols III.1. Origine et utilisation
En raison de leurs propriétés biocides vis-à-vis d’une large gamme de microorganismes,
les chlorophénols ont été utilisés comme agents de préservation pour le bois, les
peintures, les fibres végétales et le cuir, de même que comme désinfectant. Ils sont en
outre utilisés comme herbicides, fongicides et insecticides, ainsi que comme produits
intermédiaires dans la production de produits pharmaceutiques et de colorants. Les plus
utilisés pour le traitement des bois sont le pentachlorophénol (PCP) et le trichlorophénol
(TCP) (USEPA, 1980a et 1980b ).
Ceux-ci représentent la plus grande partie des chlorophénols importés en France. La
présence de pentachlorophénol dans l'environnement est uniquement anthropique. Le
pentachlorophénol est libéré dans l'atmosphère et les sols par les bois traités. Le
traitement du bois, le lessivage des sols contaminés et les dépôts de provenance
atmosphérique induits par les précipitations contribuent à la contamination des eaux de

53
surface, et par conséquent, des eaux souterraines (Bhandari et al., 1996). Actuellement,
son usage est interdit en France.
La plupart des chlorophénols revêtant une importance économique sont obtenus par
chloration directe du phénol par le chlore. Dans le produit technique, on retrouve des
impuretés, d'autres isomères de chlorophénol ou des chlorophénols comportant plus ou
moins de chlore. Les chlorophénols lourds sont contaminés avant tout par des
phénoxyphénols polychlorés, des chlorodibenzoparadioxines et des
chlorodibenzofurannes (USEPA, 1992).
III.2. Pathologie/toxicologie
Les chlorophénols peuvent être absorbés par les poumons, l'appareil digestif et la peau.
La toxicité des chlorophénols dépend de leur degré de chloration, de la position des
atomes de chlore, ainsi que de la pureté de l'échantillon. Les chlorophénols provoquent
des irritations des yeux et de l'appareil respiratoire. Les doses toxiques de chlorophénols
provoquent des convulsions, des difficultés respiratoires, le coma et finalement la mort.
Le pentachlorophénol est une molécule toxique, non seulement vis à vis des organismes
inférieurs, mais aussi vis à vis des organismes supérieurs tels que l'homme (Crosby et al,
1981).
Les expériences sur des animaux ont montré que le pentachlorophénol avaient des effets
toxiques sur les embryons. Le pentachlorophénol est inscrit dans le Groupe II de la
classification des polluants (probablement cancérogène pour l'homme) (Schwetz et al.,
1978, Braun et al., 1978).
La toxicité des chlorophénols vis à vis de nombreux organismes est mise à profit pour ses
nombreuses applications : herbicides, défoliants, fongicides, bactéricides, germicides
(Exon, 1984).
III.3. Propriétés physico-chimiques
III.3.1. Généralités
Tous les chlorophénols sont solides à la température ambiante (points de fusion allant de
33 à 191°C), sauf le 2-chlorophénol qui est liquide avec un point de fusion à 9°C. La
plupart des chlorophénols et tous leurs sels de sodium sont solubles dans l'eau. Leur

54
solubilité est faible, en particulier celle du pentachlorophénol. Les chlorophénols de
masse moléculaire élevée ont des pressions de vapeur relativement faibles et sont assez
peu volatils. Leur coefficient de partage octanol/eau (logKow) augmente avec le nombre
d'atomes de chlore, passant de 2,15 pour le 2-chlorophénol à 5,0 pour le
pentachlorophénol (Galil et Novak, 1995).
Parmi les chlorophénols, le pentachlorophénol (PCP) est la substance la plus utilisée et la
plus étudiée à tous les niveaux (toxicologie, adsorption, dégradation, etc.) (USEPA,
1978). Son large spectre d'utilisation fait qu'il est responsable d'une importante pollution
diffuse dans la plupart des compartiments de nos écosystèmes. Il est souvent rencontré
sur les sites industriels contaminés (Lee et al, 1991, USEPA, 1992 ; Warith et al., 1933).
Le pentachlorophénol fait aussi partie des listes de polluants prioritaires publiées par
l'EPA aux États-Unis (USEPA, 1980b) et l'Organisation Mondiale de la Santé (Hoos,
1978).
Le PCP est une molécule phénolique à cinq atomes de chlore (figure 5).
Figure 5 : Structure moléculaire du PCP
OH
Cl Cl
Cl
ClCl
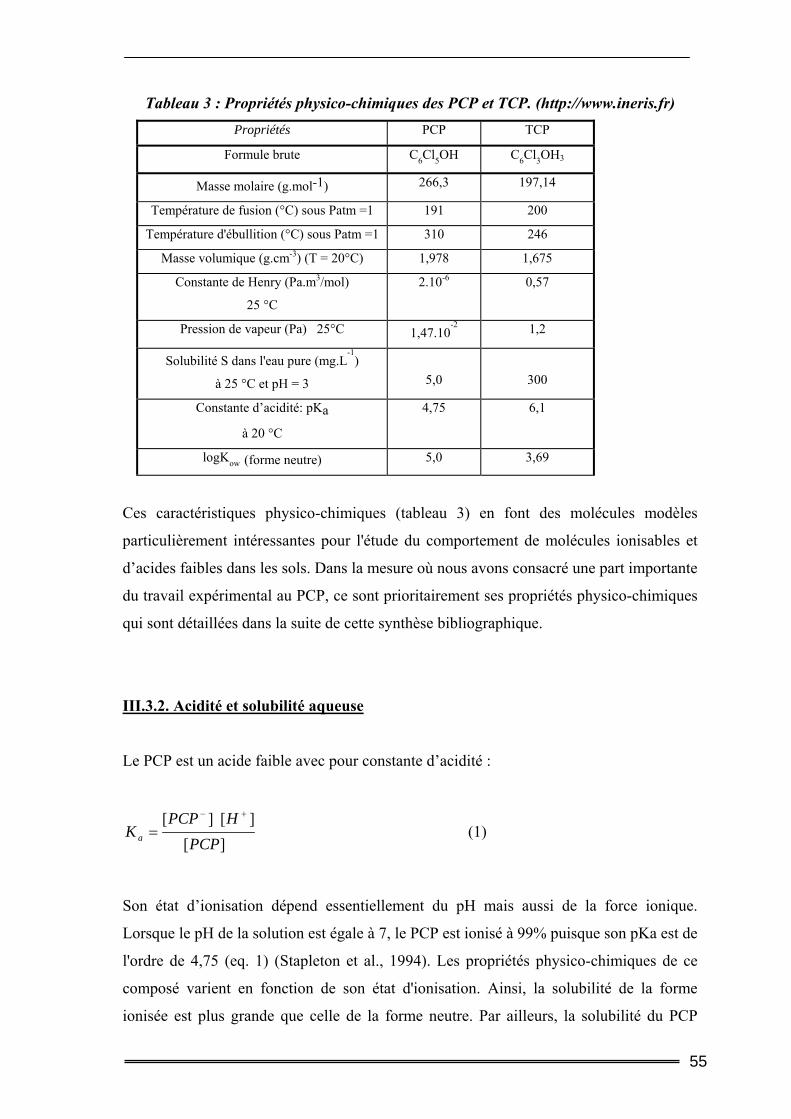
55
Tableau 3 : Propriétés physico-chimiques des PCP et TCP. (http://www.ineris.fr) Propriétés PCP TCP
Formule brute C6Cl5OH C6Cl3OH3
Masse molaire (g.mol-1) 266,3 197,14
Température de fusion (°C) sous Patm =1 191 200
Température d'ébullition (°C) sous Patm =1 310 246
Masse volumique (g.cm-3) (T = 20°C) 1,978 1,675
Constante de Henry (Pa.m3/mol)
25 °C
2.10-6 0,57
Pression de vapeur (Pa) 25°C 1,47.10-2 1,2
Solubilité S dans l'eau pure (mg.L-1
)
à 25 °C et pH = 3
5,0
300
Constante d’acidité: pKa
à 20 °C
4,75 6,1
logKow (forme neutre) 5,0 3,69
Ces caractéristiques physico-chimiques (tableau 3) en font des molécules modèles
particulièrement intéressantes pour l'étude du comportement de molécules ionisables et
d’acides faibles dans les sols. Dans la mesure où nous avons consacré une part importante
du travail expérimental au PCP, ce sont prioritairement ses propriétés physico-chimiques
qui sont détaillées dans la suite de cette synthèse bibliographique.
III.3.2. Acidité et solubilité aqueuse
Le PCP est un acide faible avec pour constante d’acidité :
][][][
PCPHPCP
K a
+−
= (1)
Son état d’ionisation dépend essentiellement du pH mais aussi de la force ionique.
Lorsque le pH de la solution est égale à 7, le PCP est ionisé à 99% puisque son pKa est de
l'ordre de 4,75 (eq. 1) (Stapleton et al., 1994). Les propriétés physico-chimiques de ce
composé varient en fonction de son état d'ionisation. Ainsi, la solubilité de la forme
ionisée est plus grande que celle de la forme neutre. Par ailleurs, la solubilité du PCP

56
dans l'eau dépend également de la température (Schellenberg et al, 1984, Archand et al.,
1995). Pour résumer, la solubilité du PCP augmente considérablement avec le pH à partir
de 5, tandis qu’elle est relativement constante pour des valeurs de pH inférieur à 4. Quant
à la force ionique, son influence sur la solubilité aqueuse est négligeable à pH ≤ 4. Par
contre, quand le pH est supérieur au pKa, la solubilité aqueuse du PCP augmente avec la
force ionique (Stapleton et al., 1994).
L'évolution de la solubilité aqueuse (Sw) du PCP peut être estimée selon le pH par
l’intermédiaire de deux relations mathématiques empiriques distinctes (Valsaraj, 1995).
Chaque expression est valide dans une domaine de pH défini :
Pour pH ≤ pKa, Sw = 3,386.10 0,12pH (2)
Lorque pH > pKa, Sw = 0,25976.10 0,3514pH (3)
III.3.3. Coefficient de partage octanol/eau Kow
Le coefficient de partage octanol/eau (Kow) est défini comme le rapport entre la
concentration à l'équilibre d'une substance chimique dans l'octanol et la concentration en
cette même substance dans l'eau (Johnson et Westall, 1990) :
w
oow PCP
PCPK
][][
= (4)
où [PCP]o et [PCP]w sont respectivement les concentrations en PCP moléculaire dans
l’octanol et dans l’eau.
Pour les molécules ionisables (comme le PCP), le coefficient de partage intègre les
concentrations en espèce neutre [PCP]w et en espèce ionisée [PCP-]w dans la phase
aqueuse. Ce rapport est généralement appelé ratio de distribution, Dow, et est exprimé
par :
ww
oow PCPPCP
PCPD
][][][
−+= (5)
Cette relation est établie en supposant que seule la forme neutre est présente dans
l’octanol.

57
En combinant les équations 1, 4 et 5, le ratio de distribution du PCP entre l’octanol et
l’eau peut être exprimé en fonction du pH selon l’équation 6 (Kaiser and Valdmanis,
1982; Westall et al., 1985; Jafvert et al., 1990) :
( ) 1)(101 −−+= pKapHowow KD (6)
III.4. Comportement dans l’environnement
III.4.1. Milieu aquatique
Dans le milieu aquatique, les chlorophénols peuvent être solubilisés à l’état libre et sous
forme complexée ou être absorbés sur des matières en suspension (USEPA, 1978). Le
PCP a une très forte capacité d’adsorption alors que les chlorophénols légers se fixent
beaucoup plus difficilement sur les particules en suspension. Les chlorophénols peuvent
être dégradés par photodécomposition mais le principal processus naturel d’élimination
est la biodégradation qui peut être très rapide en présence de microorganismes adaptés.
Toutefois, la biodégradation du PCP est nettement plus difficile que celle des
chlorophénols plus légers car l’adsorption limite la disponibilité du PCP dans la phase
aqueuse. De ce fait, le PCP est considéré comme stable et persistant (Schellenberg et al,
1984 ; Lagas, 1988 ; Boyd, 1982 ; Ball et Roberts, 1991, 1992 ; Shimizu et al, 1992).
III.4.2. Atmosphère
Malgré sa volatilité limitée, le PCP peut migrer dans l'atmosphère. Sa volatilité s'accroît
considérablement avec la température. Elle dépend également de la présence d’éventuels
additifs et du substrat sur lequel il est fixé (nature du bois traité).
Compte tenu de ses caractéristiques physico-chimiques, le pentachlorophénol peut se
trouver sous forme gazeuse et particulaire dans le compartiment atmosphérique. A l’état
gazeux, il est facilement dégradé par réaction photochimique avec une demi-vie estimée à
29 jours. Sous forme particulaire, il est piégé par les précipitations, et retourne alors dans
les compartiments terrestres et aquatiques (USEPA, 1980b).

58
III.4.3. Sols
La persistance des chlorophénols dans les sols dépend de leur propriété d'adsorption et de
désorption (nature du sol) (Farrell et al, 1994, 1999). L’adsorption des chlorophénols
augmente avec l’hydrophobicité du substrat. L'adsorption des molécules ionisables dans
un système sol/eau dépend évidemment du pH qui détermine leur état d'ionisation. La
désorption des chlorophénols d’un sol augmente avec le pH selon l’ordre suivant : DCP <
TCP < PCP (You et Liu, 1996). Schellenberg et al (1984) et Warith et al, (1993) montrent
que l'adsorption des chlorophénols non dissociés dans des sédiments naturels est un
phénomène de partition entre la phase solide organique et la phase aqueuse. De
nombreuses études ont tenté de corréler l'adsorption de molécules organiques sur la
fraction solide des sols avec leur solubilité S ou avec le coefficient de partage octanol/eau
(Kow) afin de prédire indirectement l'adsorption en fonction des propriétés physico-
chimique des solutés (Karickhoff et al, 1979 ; Briggs, 1981 ; Karickhoff, 1981 ; Boyd,
1982 ; Chiou, 1989). Schellenberg et al. (1984) propose pour les chlorophénols
une relation linéaire entre le paramètre d'adsorption Koc et le Kow de la molécule:
logKoc = 0,82 + 0,02logKow
Pour une quantité donnée de polluants, plus Koc est faible et plus la concentration du
composé en solution est élevée. La sorption d'un composé est plus importante dans les
sols à forte teneur en matière organique.
De nombreuses études ont été réalisées sur l'adsorption du pentachlorophénol dans les
sols (Schellenberg et al, 1984 ; Lagas, 1988 ; Bellin et al, 1991 ; Shimizu et al, 1992 ;
Banerji et al, 1993 ; Warith et al, 1993).
Pour la majorité des auteurs, l'adsorption du PCP est souvent proportionnelle à la
concentration en carbone organique dans le sol. Cette adsorption semble aussi dépendre
du pH, et est plus importante en conditions acides (Banerji et al, 1993 ; Divincenzo et
Sparks, 2001).
Les recherches de Lagas (1988) et de Shimizu et al (1992) confirment que l'adsorption du
PCP dépend principalement du pH de la solution aqueuse du sol et montrent que
l'adsorption des ions pentachlorophénolate est inférieure à celle du PCP non ionisé.

59
Banerji et al (1993) notent l'influence de la température sur l'adsorption du PCP dans un
sol : l'adsorption est plus importante à 10°C qu'à 30°C ce qui suggère que le mécanisme
d'adsorption serait plus du type physique (physisorption) que chimique (chimisorption).
III. 4.4. Biodégradabilité dans les sols : atténuation naturelle Bien que le PCP puisse être dégradé par des micro-organismes dans certaines conditions,
la substance doit être considérée comme difficilement biodégradable (McAllister et al.,
1996). Le PCP est une molécule hautement chlorée et donc particulièrement toxique.
Quelques auteurs ont montré que cette molécule pourrait être légèrement biodégradable
sous conditions aérobies (Boyd et al, 1989) avec une constante de biodégradation aérobie
d’environ 4,13 10-5 mg/s. Le temps de demi-vie du PCP dans les sols varie entre 1 mois et
plusieurs années (Bellin et al, 1991). La biodégradation du PCP dépend principalement
de la nature du sol (notamment de la teneur matière organique) qui détermine, non
seulement la biodisponibilité du polluant (adsorbé ou non), mais aussi la présence d'une
microflore ayant plus ou moins la capacité de métaboliser le PCP. Lorsque la
concentration en PCP est faible et le pH du sol est élevé, la biodégradation du polluant
pourrait être relativement rapide (Bellin et al, 1991) alors qu'elle est beaucoup plus lente
dans les sols acides avec des concentrations élevées en PCP (Casterline et al, 1985).
IV. Les hydrocarbures aromatiques polycycliques
ІV.1. Origine des HAP
Les HAP sont formés lors de la combustion incomplète ou de la pyrolyse de matières
organiques et notamment lors d’incendies de forêt ou d’éruptions volcaniques, etc.
Cependant, la source prédominante de ce type d’hydrocarbures dans l’environnement est
essentiellement anthropique. De tels molécules se rencontrent dans les suies de toutes
origines, les gaz d’échappement des moteurs à explosion et la fumée de cigarette. Les
activités industrielles associées à la production et à l’utilisation de matériaux contenant
des HAP sont :

60
- Celles qui utilisent les combustibles fossiles (production d’énergie, gazéification,
liquéfaction, etc.).
- Celles qui produisent des composés ou résidus carbonés (coke, noir de carbone, goudron
etc.).
- Celles qui concernent le traitement du bois (créosote, huile d’anthracène),
- Celles qui concernent la distillation de pétrole brut et l’incinération (pneus, déchets,
charbon etc.).
ІV.2. Données toxicologiques
Les HAP sont considérés comme présentant un risque sanitaire. Le tableau 4 rassemble
les données toxicologiques de quelques uns de ces composés.
Tableau 4: Données toxicologiques relatives aux principaux HAP. Nom Toxique Mutagène Cancérigène
Naphtalène × ×
Acénaphtylène ×
Anthracène ×
Phénanthrène × ×
Fluoranthène × ×
Pyrène × ×
Chrysène × ×
Benzo[b]fluoranthène × ×
Benzo[k]fluoranthène ×
Benzo[a]pyrène × × ×
Dibenzo[a,h]anthracène × ×
Benzo[g,h,i]pérylène
Indéno[1,2,3-c,d]pyrène × ×
Les molécules de faibles masses moléculaires présentent des caractéristiques de toxicité
aiguë tandis que celles de fortes masses ont plutôt des effets cancérigènes et de
bioaccumulation. Des effets de synergie peuvent apparaître (Hermann, 1981).
L'accumulation des HAP dans les organismes aquatiques est très élevée du fait de leur
forte hydrophobicité.

61
Des études en laboratoire ont mis en évidence que certains HAP sont immunotoxiques car
l’exposition à ces produits induit une diminution des défenses immunitaires (White,
1986). Plusieurs études toxicologiques ont également révélé que certains HAP sont à
l’origine de tumeurs par suite d’exposition prolongée (INERIS, 2000).
ІV.3. Nature chimique Les hydrocarbures aromatiques polycycliques sont des substances organiques neutres,
non polaires et constituées de plusieurs cycles benzéniques associés de façon linéaire,
angulaire ou en amas. On distingue les HAP alternants formés exclusivement de cycles
benzéniques et les HAP non alternants composés d’au moins un cycle à quatre carbones.
Par définition, ils sont composés de carbone et d’hydrogène bien que l’oxygène, le soufre
et l’azote puissent se substituer au carbone dans le cycle benzénique pour former des
substances aromatiques hétérocycliques, communément regroupées avec les HAP. Les
structures des HAP sont décrites dans la figure 6.
Le naphtalène (C10H8) et le phénanthrène (C14H10) sont des molécules planes de 2 et 3
cycles aromatiques respectivement, sans groupe fonctionnel. Elles sont donc non
ionisables, apolaires ou hydrophobes. Du point de vue de la structure moléculaire, le
naphtalène est relativement proche des composés aromatiques volatils et semi-volatils
(Benzène, Toluène, Ethylbenzène et Xylène regroupés sous l’appelation BTEX).
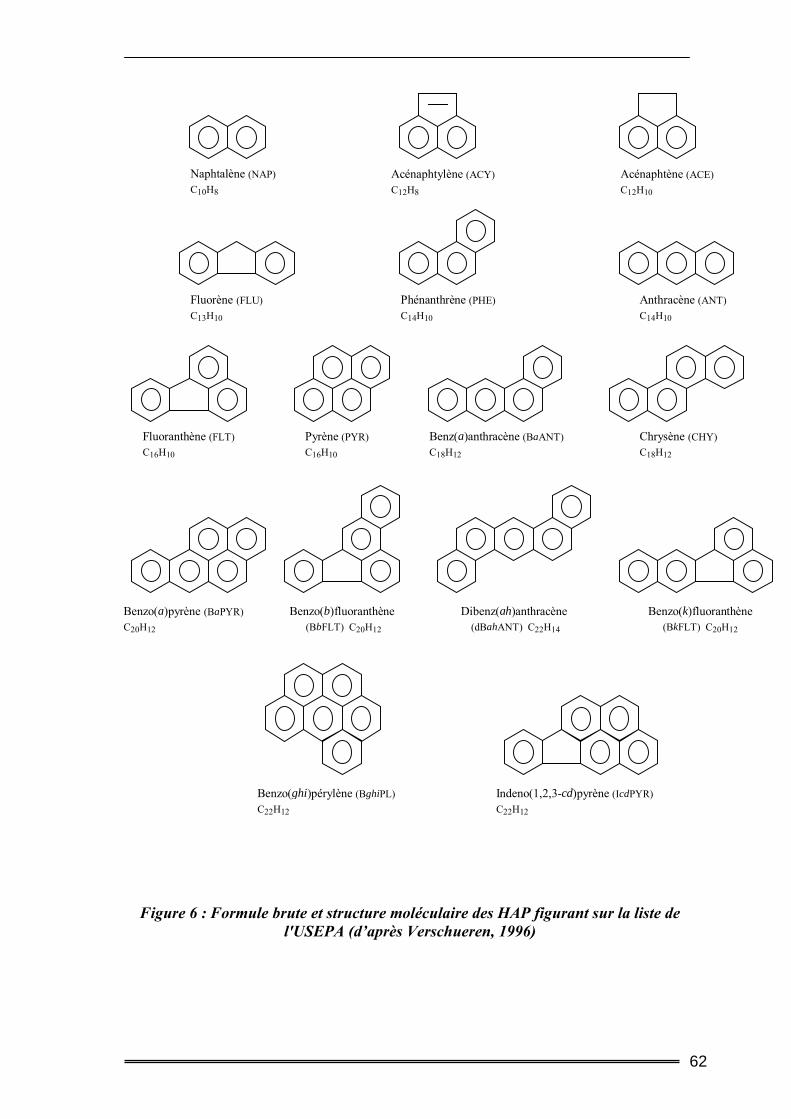
62
Figure 6 : Formule brute et structure moléculaire des HAP figurant sur la liste de l'USEPA (d’après Verschueren, 1996)
Naphtalène (NAP)C10H8
Acénaphtylène (ACY)C12H8
Acénaphtène (ACE)C12H10
Fluorène (FLU)C13H10
Phénanthrène (PHE)C14H10
Anthracène (ANT)C14H10
Fluoranthène (FLT)C16H10
Pyrène (PYR)C16H10
Benz(a)anthracène (BaANT)C18H12
Chrysène (CHY)C18H12
Benzo(a)pyrène (BaPYR)C20H12
Benzo(b)fluoranthène (BbFLT) C20H12
Dibenz(ah)anthracène (dBahANT) C22H14
Benzo(k)fluoranthène(BkFLT) C20H12
Benzo(ghi)pérylène (BghiPL)C22H12
Indeno(1,2,3-cd)pyrène (IcdPYR)C22H12
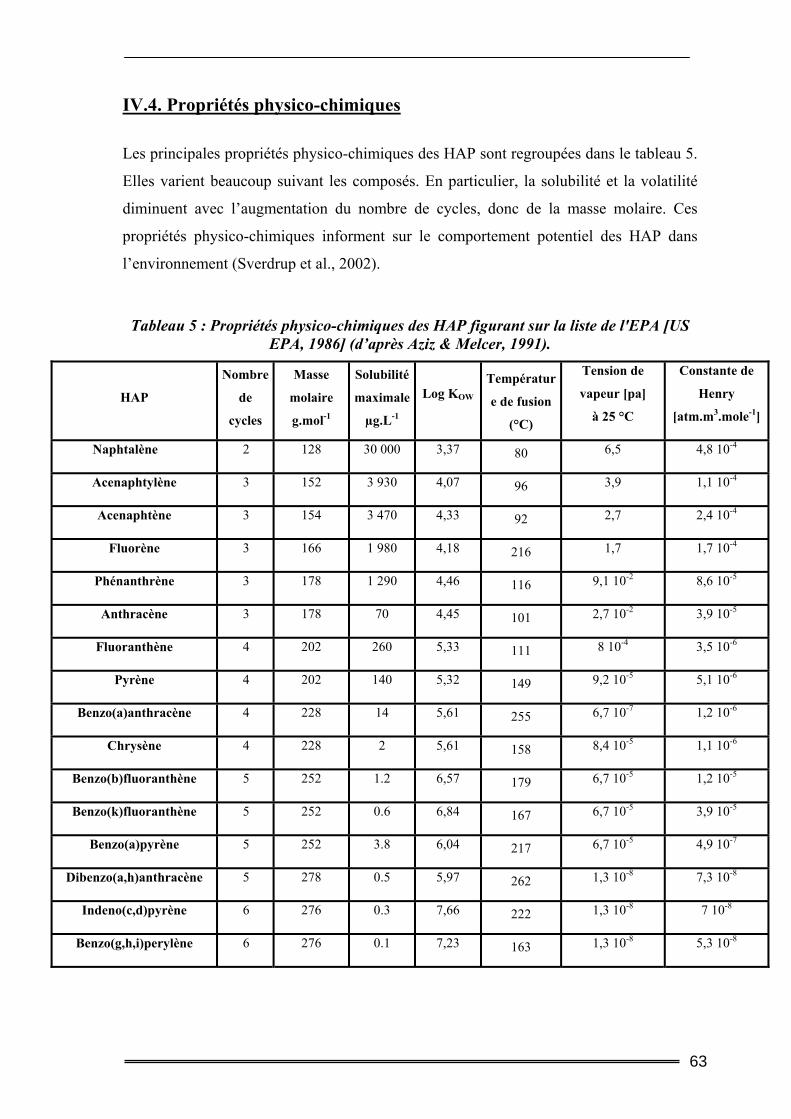
63
ІV.4. Propriétés physico-chimiques Les principales propriétés physico-chimiques des HAP sont regroupées dans le tableau 5.
Elles varient beaucoup suivant les composés. En particulier, la solubilité et la volatilité
diminuent avec l’augmentation du nombre de cycles, donc de la masse molaire. Ces
propriétés physico-chimiques informent sur le comportement potentiel des HAP dans
l’environnement (Sverdrup et al., 2002).
Tableau 5 : Propriétés physico-chimiques des HAP figurant sur la liste de l'EPA [US EPA, 1986] (d’après Aziz & Melcer, 1991).
HAP
Nombre
de
cycles
Masse
molaire
g.mol-1
Solubilité
maximale
µg.L-1
Log KOW
Températur
e de fusion
(°C)
Tension de
vapeur [pa]
à 25 °C
Constante de
Henry
[atm.m3.mole-1]
Naphtalène 2 128 30 000 3,37 80 6,5 4,8 10-4
Acenaphtylène 3 152 3 930 4,07 96 3,9 1,1 10-4
Acenaphtène 3 154 3 470 4,33 92 2,7 2,4 10-4
Fluorène 3 166 1 980 4,18 216 1,7 1,7 10-4
Phénanthrène 3 178 1 290 4,46 116 9,1 10-2 8,6 10-5
Anthracène 3 178 70 4,45 101 2,7 10-2 3,9 10-5
Fluoranthène 4 202 260 5,33 111 8 10-4 3,5 10-6
Pyrène 4 202 140 5,32 149 9,2 10-5 5,1 10-6
Benzo(a)anthracène 4 228 14 5,61 255 6,7 10-7 1,2 10-6
Chrysène 4 228 2 5,61 158 8,4 10-5 1,1 10-6
Benzo(b)fluoranthène 5 252 1.2 6,57 179 6,7 10-5 1,2 10-5
Benzo(k)fluoranthène 5 252 0.6 6,84 167 6,7 10-5 3,9 10-5
Benzo(a)pyrène 5 252 3.8 6,04 217 6,7 10-5 4,9 10-7
Dibenzo(a,h)anthracène 5 278 0.5 5,97 262 1,3 10-8 7,3 10-8
Indeno(c,d)pyrène 6 276 0.3 7,66 222 1,3 10-8 7 10-8
Benzo(g,h,i)perylène 6 276 0.1 7,23 163 1,3 10-8 5,3 10-8

64
Les HAP ont une solubilité dans l’eau très faible du fait de leur masse molaire importante
et de leur caractère apolaire. Le phénanthrène est beaucoup moins soluble (1,3 mg.L-1)
que le naphtalène (30 mg.L-1) dans l'eau pure à 20°C. Leur solubilisation peut cependant
être favorisée par l’intermédiaire de solvants organiques (l’acétone, le benzène, l’éthanol)
de produits pétroliers ou de détergents (Edwards et al., 1990). Le pH a un effet
négligeable sur la solubilité aqueuse des ces composés non-ionisables. De même une
faible variation de force ionique n’a généralement que peu d’influence sur cette solubilité.
Whitehouse et al. (1984) signalent toutefois que la solubilité du 1,2-benzoanthracène est
assez sensible au changement de la salinité. La solubilité aqueuse des HAP est, par
contre, très sensible à la température (Mackay et Shiu, 1977 ; Whitehouse et al., 1984).
La volatilité d’un composé dépend de sa constante de Henry (Hc) et de sa pression de
vapeur saturante. Le naphtalène est le plus volatil des HAP. Il contribue fortement à
l'odeur caractéristique des goudrons et peut être présent à des teneurs non négligeables
dans l'air à proximité de sites contaminés. Du fait de sa volatilité, le naphtalène est
considéré comme un polluant atmosphérique. Des études montrent qu’un sol contaminé
par un mélange de 14 HAP libère 20 à 30% du naphtalène alors que la volatilisation des
HAP ayant plus de deux cycles aromatiques est négligeable.
Le caractère lipophile de ces substances, exprimé à travers la valeur de leur coefficient de
partage octanol/eau (Kow), est relativement important (logKow de l’ordre de 4 à 7). Curtis
et al. (1986) ont établi la relation linéaire suivante entre le paramètre d'adsorption Koc et
le Kow des HAP:
logKoc = 0,92.logKow - 0,23
La lipophilie des HAP est également responsable de leur bioaccumulation dans les
organismes vivants. Cette accumulation est quantifiée par le facteur de bioconcentration
(BCF) défini comme le rapport entre la concentration d'une substance à l'intérieur d'un
organisme et sa concentration dans l'eau. Neelly et al. (1974) ont proposé la relation
linéaire suivante entre le BCF et le Kow des HAPs :
BCF = 0,542 logKow + 0,124

65
Compte tenu de l’ensemble des caractéristiques bio-physico-chimiques, les HAP sont
considérés comme des contaminants très peu mobiles dans les sols et bioaccumulables.
ІV.5. Devenir des HAP dans l’environnement
IV.5.1. Transport des HAP dans les différents compartiments terrestres
Le devenir d'un polluant organique dans un sol résulte d'un ensemble de processus
compétitifs incluant de possibles transformations chimiques ou biochimiques (Calvet et
al., 1989 ; Barriuso et al., 1992, 1996). Ils sont présentés dans la figure 7.
Figure 7 : Devenir des HAP dans les sols (d'après Mahjoub, 1999).
Sol Sol
Nappe phréatique Nappe phréatique
Photolyse Volatilisation
Dégradation chimique
BIODEGRADATION BIODEGRADATION Percolation
ADSORPTION / ADSORPTION / DESORPTION DESORPTION
Absorption
Atmosphère Atmosphère
Ruissellement
Eaux de
surface

66
Le transport des HAP dans les sols fait principalement intervenir les phénomènes
suivants : la solubilisation, la percolation, l'adsorption, la volatilisation.
Les phénomènes de transformation des HAP peuvent se produire essentiellement
par hydrolyse, photolyse et biodégradation.
Les caractéristiques de la molécule organique (structure, taille,...), la nature du sol (taux
et maturation de la matière organique, acides humiques,...), l'eau contenue dans le sol et
les facteurs environnementaux (pH, température,...) peuvent influencer les mécanismes
d'adsorption/désorption (Means et al., 1980 ; Erickson et al., 1993 ; Manilal et Alexander,
1991 ; Bayard, 1997). Une part importante des phénomènes gouvernant le devenir des
HAP dans les sols dépend donc du milieu récepteur (sol) (propriétés physico-chimiques
et microbiologiques). Dans la partie C de l’étude bibliographique, nous détaillerons les
phénomènes concernant les mécanismes contrôlant le devenir des polluants dans les sols.
Une brève synthèse bibliographique sera également consacrée aux caractéristiques du
milieu poreux et à leurs conséquences.
IV.5.2. Biodégradation naturelle des HAP
La dissolution des HAP dans la phase liquide d’un sol est un important facteur contrôlant
leur biodégradation (Stucki & Alexander, 1987). L'adsorption limite la biodégradation
des molécules hydrophobes en condition aérobie (Al-Bashir et al, 1990 ; Manilal et
Alexander, 1991) car elle réduit la disponibilité aux agents microbiens. La plupart des
auteurs considèrent que seules les molécules en phase aqueuse peuvent être métabolisées
ou biodégradées.
De nombreuses études ont permis d'isoler des micro-organismes spécifiques capables de
dégrader les HAP. Il peut s'agir de bactéries (Heitkamp & Cerniglia, 1988, 1989), de
moisissures ou d'algues (Cerniglia, 1992). Dans sa revue bibliographique, Cerniglia
(1992) donne une liste importante de micro-organismes réputés capables de dégrader le
naphtalène. Par contre, on connaît peu de micro-organismes capables d'utiliser des HAP
de masses moléculaires élevées comme unique source de carbone et d'énergie (Mahaffey
et al, 1988).
La dégradation des HAP s'effectue principalement en condition aérobie. Les composés
aromatiques de faible masse moléculaire sont peu dégradés en condition anaérobie stricte
(Mahaffey et al, 1988).

67
Les propriétés du sol telles que la teneur en matière organique et la taille des particules
(texture) influencent aussi le taux de dégradation (Manilal & Alexander, 1991). D'autre
part, la biodégradation dépend également de la présence de micro-organismes indigènes
susceptibles de métaboliser les HAP. Enfin, des paramètres comme la température, le pH
et la disponibilité de l'oxygène sont également à considérer.
Par ailleurs, la biodégradation dans les sols peut présenter des difficultés en fonction de
l'âge de la pollution : les phénomènes de vieillissement réduisent considérablement la
fraction de polluants organiques facilement biodégradables (Pollard et al , 1994).
Lorsqu'il s'agit d'une pollution chronique d'un sol, la microflore du sol s'adapte à la
présence des polluants organiques, adaptation qui se caractérise par une plus grande
résistance à la toxicité des polluants mais aussi par une plus grande facilité à les
métaboliser.
V. Bilan
Dans cette partie, nous avons montré que les propriétés physico-chimiques des polluants
organiques constituent des éléments à prendre en compte lors d’une étude de leur devenir
dans les milieux environnementaux et plus particulièrement dans les sols. Concernant les
deux familles de polluants sélectionnés pour notre étude, l’influence des paramètres
physico-chimiques sur leur comportement ne semble pas être identique. Ces paramètres
influeront, d’une part sur l’affinité des molécules vis a vis des agents de complexation, et
d’autre part, sur leur extraction des sols contaminés ou leur bioremédiation. Par
conséquent, ces paramètres doivent être pris en compte pour une étude à l’échelle du
laboratoire.
Dans l’objectif du traitement d’une pollution réelle, souvent composée par plusieurs
familles de polluants, la connaissance des facteurs qui conditionnent leurs mobilités
respectives revêt une importance majeure.
Au travers cette synthèse bibliographique, nous avons aussi montré que la persistance de
ces contaminants dans les sols et leur faible biodégradabilité posent un sérieux problème
pour l’environnement.
Cette persistance dépend bien entendu de la nature des polluants, mais également de la
nature du sol en question. C’est pourquoi, il nous a semblé nécessaire de présenter les
caractéristiques principales des sols de même que les mécanismes contrôlant le devenir

68
des polluants dans ces matrices poreuses. C’est ce qui fait l’objet des rappels
bibliographiques de la partie suivante.

69
Partie C : les Sols et leurs interactions avec les
polluants organiques.
I. Introduction
Dans le chapitre précédent, nous avons pu constater que les propriétés physico-chimiques
des contaminants organiques constituent des éléments à prendre en compte lors d’une
étude de leur destin dans les sols. Une part importante des phénomènes gouvernant le
devenir des polluants dans les sols dépend également des caractéristiques de ces milieux
récepteurs (propriétés physico-chimiques et microbiologiques). C’est pourquoi, toute
étude de remédiation nécessitera une recherche préalable concernant, d’une part les
caractéristiques de la matrice solide qu’est le sol, et d’autre part, les principaux
mécanismes contrôlant la mobilisation des polluants dans ce sol.
Nous allons donc présenter, dans cette partie, certains connaissances concernant les
interactions pouvant exister entre un milieu poreux et un polluant dans un contexte
donné.
II. Interactions sol / polluants organiques II.1. Définition d’un sol
Le sol est constitué de la couche superficielle qui recouvre l'écorce terrestre. Il se forme
sous l'effet de l'altération de la roche-mère (ou minéraux primaires) soumise à des
agressions physico-chimiques, mécaniques et climatiques (température, humidité...) et
biologiques (Stegmann et al., 2001).
Le sol est un milieu constitué de trois fractions dont les proportions, la structure et
l'organisation sont variables :

70
• une fraction solide composée de minéraux (sables, limons, argiles, oxydes et
hydroxydes métalliques) et d'une fraction organique (organismes vivants, débris végétaux
et animaux, humus),
• une fraction liquide représentant l'eau contenue dans le sol au sein de laquelle sont
dissoutes les substances solubles, (Mccrathy et Zachara, 1989).
• une fraction gazeuse de composition très proche de celle de l'atmosphère terrestre mais
généralement enrichie en CO2 par l'activité respiratoire des micro-organismes du sol.
Cette phase gazeuse est négligeable si l’on considère des sols en zone saturée en eau
(aquifères où l'eau occupe complètement la porosité du sol).
II.2. Comportement de polluants organiques hydrophobes dans les sols La fabrication, le transport, l’utilisation, la dispersion ou les fuites accidentelles de
produits organiques ont pour conséquence le relargage de quantités significatives de
composés organiques dans les sols. En phase liquide, ils peuvent subir une migration sous
l’action des forces de gravité et de capillarité (Calvet, 1989) jusqu’à ce qu’ils rencontrent
une couche géologique faiblement perméable. La fraction liquide organique est appelée
Phase Liquide Non Aqueuse (PLNA). Elle est dispersée au sein des pores et peut
représenter de 3 à 5 L.m-3 de sol dans les milieux perméables et de 30 à 50 L.m-3 dans un
milieu imperméable (Illangasekar et al., 1995). La migration d’une phase organique non
aqueuse dans le sol est gouvernée par sa densité et sa viscosité (Mccray et Falta, 1997 ;
Mccray et Dugan, 2002 ). La perméabilité et l’hétérogénéité du sol jouent également un
rôle très important dans le comportement physique de ce type de pollution (Illangasekar
et al., 1995). Les gouttelettes de PLNA peuvent se solubiliser en partie selon l’humidité
du sol ou la percolation de l’eau. Certains polluants dissous peuvent interagir avec les
fractions solides du sol, par un mécanisme d’adsorption. Dans certains cas, en zone
insaturée, l’hétérogénéité du milieu peut engendrer la formation de « poches » de phase
liquide organique « emprisonnée », qui modifient alors considérablement les écoulements
dans le sol (McCray et Dugan, 2002).
Par ailleurs, les polluants organiques les plus volatils peuvent migrer vers la phase
gazeuse du sol.
Dans la zone insaturée, une pollution organique se partage donc entre quatre phases :
- la phase liquide non aqueuse (PLNA) sous forme de gouttelettes, de films ou de
poches,

71
- la phase liquide aqueuse (composés organiques dissous),
- la phase solide (molécules adsorbées sur les particules de sol),
- la phase gazeuse (air et vapeurs organiques).
Les polluants organiques se trouvant dans un sol sont engagés dans un processus de
transport et de transformation qui conditionnent leur rémanence. Ils se distribuent entre
les différents compartiments du sol selon des critères de solubilité, d’adsorption, de
volatilisation et de partage entre phases comme l’illustre la figure 8 ci-dessous (Mahjoub
et al., 1999) .
Figure 8: Répartition des polluants organiques dans les différents phases des sols
La prévision de la propagation et du comportement de ces composés dans les sols est très
complexe parce que les mécanismes de transfert sont nombreux et simultanés, qu’ils
interagissent entre eux et qu’ils peuvent avoir une influence sur les propriétés du sol lui
même (McCray et Dugan, 2002).
PHASES SOLIDES
Adsorption, Précipitation,
Partage, Incorporation,
Diffusion
PHASES LIQUIDES
Solubilisation, Désorption,
Convection, Diffusion,
dégradation
PHASE
GAZEUSE Volatilization,
Convection

72
De façon générale, l’augmentation des phénomènes de rétention sur, ou dans, les phases
solides provoque la diminution des risques de dispersion des polluants, mais peut rendre
difficile leur biodégradation (Barkay et al., 1999).
II.3. Principaux mécanismes contrôlant le devenir des polluants dans les
sols
Les principaux mécanismes contrôlant le devenir des polluants dans les sols sont les
suivants :
- la solubilisation en phase aqueuse, qui est notamment fonction de la solubilité dans
l’eau, de la structure chimique, de la polarité et de la taille de la molécule organique. Le
partage du polluant entre la phase organique (PLNA) et la phase aqueuse du sol est
contrôlé par sa solubilité dans l’eau. Cette caractéristique a une grande influence sur la
mobilité des polluants dans les sols (McCray et al, 2001).
- la convection, qui consiste en l’entraînement par la phase aqueuse mobile, des
molécules organiques les plus facilement solubles. L’entraînement des polluants avec des
particules fines est également possible. Ce mécanisme dépend notamment des
caractéristiques géotechniques du milieu filtrant (McCray et Dugan, 2002).
- L’adsorption, qui se traduit par la rétention plus ou moins réversible du polluant sur les
particules de sol. L'adsorption correspond à un phénomène dynamique de partition d'un
soluté d'une phase liquide (généralement aqueuse dans les sols) vers une phase solide
constituée par l'ensemble des particules solides du sol. Lorsque la teneur en matière
organique dans les sols est supérieure à 1%, l’adsorption est très couramment décrite
comme un simple équilibre de partage du composé organique entre la phase aqueuse et la
matière organique du sol (Karichhoff et al., 1979, Chiou et al., 1986 ;Chiou, 1989).
L’adsorption fait intervenir des phénomènes assez complexes aboutissant à plusieurs
types de liaisons adsorbant/polluant (Calvet et al., 1989) que nous détaillerons plus loin.
La désorption correspond au phénomène inverse.
- la diffusion, est un phénomène physique lié à l’agitation moléculaire. S’il y a un
gradient de concentration, les molécules en mouvement se déplaceront de la zone la plus
concentrée vers la zone moins concentrée. La diffusion est le mécanisme de transport le
plus important dans les zones à faible perméabilité que ce soit à l’échelle macroscopique,
dans le cas des argiles par exemple, ou à l’échelle microscopique, dans les agrégats

73
poreux et au sein des particules. Les zones à faible perméabilité qui ont accumulé des
polluants organiques sur de longues périodes peuvent se comporter à long terme comme
des sources secondaires de polluants (Lee et al., 1992).
- L’incorporation, qui consiste en l’emprisonnement physique des molécules polluantes
diffusant dans le sol et plus précisément au sein de la matière organique naturelle
poreuse. La diffusion au sein de la Matière Organique du Sol (MOS) est le second
mécanisme qui influe sur le comportement des polluants organiques dans les sols au
cours du temps (Farrel et Reinhard , 1994).
- La volatilisation, concerne essentiellement les composés organiques volatils (COV).
Elle dépend notamment de la constante de Henry, elle même fonction de la température.
- L’hydrolyse, qui est un processus de dégradation des molécules par l’action de l’eau,
est fortement influencée par le pH et la température du sol.
- La biodégradation, caractérise la décomposition des molécules organiques par les
micro-organismes présents dans le sol. Elle dépend notamment de la présence suffisante
de micro-organismes adaptés, de la température, et de la quantité d’oxygène ou d’autres
accepteurs d’électrons (nitrates, Fe3+) dans le sol (Stegmann et al., 2001).
La dissolution, les mécanismes d’adsorption/désorption incluant la diffusion dans les
micropores, la diffusion au sein de la matrice adsorbante et la sorption chimique
constituent des facteurs importants qui semblent gouverner le vieillissement de la
pollution dans les sols (Lee et al., 1991, 1992 ; Stegmann et al., 2001 ; Seo et McCray,
2002).
II.4. Nature des interactions physico-chimiques des polluants
organiques avec les sols
La nature des liaisons pouvant s'établir entre des solutés et un système aussi complexe
que le sol résulte, d’une manière générale, des spécificités du couple adsorbant/polluant
et du contexte physico-chimique local. Il est cependant indispensable de comprendre les
mécanismes d'adsorption de manière à interpréter correctement la partition d'un soluté
entre la phase liquide aqueuse et la (les) phase(s) solide(s) du sol. On distingue en général
6 catégories d’interactions polluant/sol (Calvet, 1989) :

74
Les liaisons ioniques Elles ont un rôle important dans l'adsorption des polluants organiques dans le sol, en
particulier dans le cas où il s'agit de molécules ionisables. Elles se forment entre des
cations ou des anions et, respectivement, des charges négatives ou positives situées à la
surface de l'adsorbant. L'intensité de l'adsorption dépendra étroitement du pH de la phase
aqueuse du sol.
Les liaisons hydrogène Ces liaisons électrostatiques s'établissent entre les atomes possédant un ou plusieurs
doublets électroniques libres (essentiellement l'oxygène, l'azote ou le soufre) et un atome
d'hydrogène lié à un atome électronégatif (tel que N ou O). Elles peuvent se former soit
avec les groupements chimiques de l'adsorbant, soit indirectement sur l'adsorbant par
l'intermédiaire de molécules d'eau présentes en surface (Rogers et al. ; 1980).
Les interactions avec des cations métalliques Les éléments adsorbants du sol présentent à leur surface une grande variété de cations
(tels que des ions échangeables Na+, K
+, Ca
+, ou bien Mg
2+) et de minéraux amorphes
liés. Compte tenu des propriétés d'accepteur d'électrons des cations, deux types de
liaisons sont possibles avec les polluants organiques (Calvet, 1989) : des interactions
dipôle-cation et des liaisons de coordination avec des cations métalliques de transition.
Les interactions de London-Van der Waals
Il s'agit de forces électrostatiques dues aux mouvements des électrons qui changent
d'orbitales atomiques. Leur intensité augmente avec la taille des molécules en raison de
leur nature additive (Choudhry, 1984 ). Ce sont les principales forces responsables de
l'adsorption physique des polluants organiques dans les sols.
Les interactions hydrophobes Il s'agit d'interactions physiques dues à la nature hydrophobe de certains polluants
organiques, non ionisables, peu polaires et peu hydrosolubles. L'adsorption par
interaction hydrophobe est un mécanisme de partition des molécules organiques entre la
phase aqueuse et la phase solide (Calvet, 1989). Les molécules organiques peu polaires et

75
non ionisables ont une plus grande affinité envers les éléments de nature hydrophobe que
pour l'eau présente dans le sol (Karickhoff, 1979, 1984 ; Schwarzenbach & Westall, 1981
; Aiken et al, 1985). Cette partition peut être décrite par un phénomène de solvatation
(Chiou, et al, 1979) qui se produit dans les sols riches en matière organique.
Les Liaisons covalentes Ces liaisons chimiques résultent d'interactions interatomiques entre des groupements
fonctionnels du polluant organique d’une part, et des sites spécifiques de l'adsorbant
d’autre part. Ces liaisons, caractérisées par des énergies élevées s'établissent
essentiellement avec la matière organique du sol, suite à des mécanismes d'oxydation ou
de polymérisation enzymatique (Aiken et al, 1985 ; Bollag, 1992).
II.5. Facteurs influençant l’adsorption et la désorption des polluants
organiques dans les sols Les principaux facteurs qui influencent l’adsorption des polluants organiques dans les
sols sont les caractéristiques de l’adsorbant, de la molécule à adsorber et de la phase
liquide aqueuse. Les phénomènes d’adsorption/désorption sont également liés à des
paramètres physiques comme la température.
II.5.1. Facteurs liés aux caractéristiques des sols
1. Constituants minéraux du sol Dans un sol, on distingue les minéraux primaires qui, par leur taille, constituent la
fraction minérale peu altérée. Les minéraux secondaires, de plus faible taille, sont générés
par la décomposition de la roche-mère. Généralement, les minéraux secondaires sont
classés suivant leur granulométrie comme indiqué ci-dessous :
Sables et limons
Ils constituent la fraction minérale grossière dont le diamètre est compris entre 2 mm et
50 µm. Cette fraction est généralement constituée de quartz, feldspaths, micas et, pour les
sols calcaires, de calcite. Cette fraction minérale n'intervient pratiquement pas, par

76
rapport aux autres fractions, dans les phénomènes d'adsorption des polluants organiques
dans les sols.
Argiles
Les argiles constituent la fraction minérale de granulométrie inférieure à 2 µm. Les
argiles les plus fréquemment rencontrées dans les sols sont la kaolinite, la
montmorillonite, l'illite, la vermiculite et les chlorites. Ces minéraux argileux présentent
des caractéristiques physico-chimiques qui leur confèrent les propriétés suivantes :
grande surface spécifique en raison d’une structure en feuillets, grande capacité
d'adsorption des cations définie comme la Capacité d’Echange Cationique (CEC) et
grande capacité de rétention en eau (Rogers et al. ; 1980).
Oxydes et hydroxydes métalliques
Les cations de fer (III), d'aluminium, et de manganèse ainsi que la silice donnent, aux pH
les plus fréquents des sols (5 à 8), des hydroxydes insolubles qui tendent vers des formes
amorphes ou cristallines appelées oxyhydroxydes. La participation de ces composés à
l'adsorption des polluants organiques dans les sols dépend principalement du pH du
milieu (Hermosin et al, 1988).
2. Constituants organiques
La matière organique des sols se décompose en deux fractions distinctes :
- La fraction organique « vivante » composée de la microflore du sol, de sa faune et des
racines des plantes supérieures.
- La fraction organique « morte » constituée des débris organiques d'origine animale ou
végétale, plus ou moins décomposés en humus sous l'action des micro-organismes du sol.
Les matières humiques proviennent de la décomposition et de la réorganisation des
matières organiques contenues dans les sols (Aiken et al, 1985 ; Bollag, 1992). On
distingue trois familles d’humus, dont les proportions varient suivant les milieux : les
humines, les acides fulviques AF et les acides humiques AH. Les acides humiques et
fulviques sont des macromolécules avec des structures moléculaires tridimensionnelles
très complexes comprenant des sites hydrophobes et hydrophiles (Wershaw, 1993). Les
principales propriétés adsorbantes de la matière organique des sols sont essentiellement
liées à leur nature colloïdale et à la présence de nombreux groupements fonctionnels
(carboxyles, carbonyles, hydroxyles, amines, amides, groupements sulfoniques, …)

77
(Aiken et al, 1985 ). De plus, la matière organique des sols contient des chaînes
aliphatiques dont le rôle est non négligeable, surtout pour les interactions hydrophobes
(Bollag, 1992). La matière organique des sols se caractérise, comme les argiles, par une
grande surface spécifique et par son pouvoir gonflant permettant la pénétration de l'eau et
la diffusion de molécules de petite taille qui peuvent ainsi se lier avec les substances
humiques (Choudhry, 1984 ; Aiken et al, 1985 ; Bollag, 1992).
3. L’eau du sol
L'eau a un rôle essentiel dans tous les phénomènes physiques, chimiques et biologiques
qui se produisent dans le sol. L'eau pourra entrer en compétition avec les molécules
organiques pour l'adsorption sur les matériaux solides du sol, entraînant une baisse de
l'adsorption des polluants lorsque le taux d'humidité du sol augmente (Chiou & Shoup,
1985). La teneur en eau des sols régule aussi leur état d’agrégation (structure), l’acidité et
l’accessibilité des surfaces, le potentiel rédox, l’aération, la répartition des polluants dans
les trois phases du sol, et l’activité microbiologique (Valsaraji et Thibodeaux, 1989,
Calvet, 1989).
4. Texture et structure du sol
Les propriétés adsorbantes des sols dépendent de deux caractéristiques fondamentales :
leur texture et leur structure. Ces caractéristiques déterminent la porosité et la
perméabilité des sols, deux facteurs qui influencent l'adsorption et la mobilité des
polluants organiques.
Dans les sols, les argiles et les substances organiques humiques sont intimement liées.
Elles forment des complexes argilo-humiques qui contribuent à la formation de micro-
agrégats de diamètre inférieur à 20 µm (Aiken et al, 1985 ).
II.5.1. Facteurs liés à la nature des polluants organiques
Les mécanismes moléculaires mis en jeu au cours de l'adsorption des polluants
organiques dans un sol dépendent, entre autres facteurs, de la structure et de la nature
chimique des polluants organiques considérés.

78
Hydrophobicité et hydrosolubilité
La partition d'une molécule entre les phases liquide et solide (s) des sols dépend
notamment de son hydrosolubilité, qui est fonction de son caractère hydrophobe ou
hydrophile lié à sa structure chimique (Nicholls, 1988). Elle varie également avec le pH
(Nicholls et Evans, 1991a) et la composition de la phase aqueuse du sol (Nicholls et
Evans, 1991b). D'une façon générale, plus les molécules organiques sont solubles dans la
phase aqueuse, moins elles auront tendance à être retenues par les particules solides des
sols. Le coefficient de partage carbone organique/eau(Koc) permet d’évaluer les
interactions hydrophobes des polluants avec les matières organiques du sol. Il peut aussi
être calculé à partir du coefficient de partage octanol/eau (Kow) qui est plus facilement
accessible expérimentalement que le Koc (Schwarzenbach et Westall, 1981).
Structure électronique et chimique de la molécule
La structure électronique des molécules détermine leur état ionique qui influence
fortement leur devenir dans les sols. En effet, les molécules ionisées interagissent avec les
nombreux sites chargés présents dans les sols. C'est le cas des acides faibles qui
produisent des anions, et des bases faibles qui génèrent des cations (Nicholls & Evans,
1991a et b). L'état d'ionisation de ces composés (lié au pH) influence leur adsorption sur
les sols et les sédiments (Schellenberg et al 1984 ; Nicholls & Evans, 1991a et b).
Taille et forme de la molécule
En général, plus une molécule est volumineuse, plus sa solubilité est faible. Elle a donc
tendance à être « exclue » du solvant et à interagir avec les surfaces solides ou les phases
organiques, ou bien à précipiter (Martins, 1993)
De même, l’organisation structurale et la géomètrie de la molécule déterminent sa
capacité à s’insérer dans les micropores du sol (ex : les espaces interfoliaires des argiles)
où elle pourra être piégée. Il y a donc influence sur la diffusion des composés organiques
dans les différentes phases des sols (Briggs, 1981). D’autre part, on note souvent que,
plus la molécule est grande, plus les sites d’interactions possibles sont nombreux, et donc
plus la probabilité de rétention est importante.

79
II.5.3. Facteurs liés aux caractéristiques de la phase aqueuse du sol
Le pH de la solution du sol est un des principaux paramètres gouvernant le partage des
molécules organiques entre les phases solides et la phase aqueuse. Le pH est susceptible
d'avoir un effet non seulement sur le soluté (effet direct) mais aussi sur l'adsorbant et sur
le milieu liquide (effet indirect). Il entraîne une modification des équilibres chimiques
entre les formes ionisées et les formes neutres, en fonction de la constante d’acidité Ka de
la molécule considérée. La baisse du pH favorise l'adsorption des acides faibles
(Schellenberg et al, 1984 ; Nicholls & Evans, 1991a) qui s'adsorbent principalement sous
leur forme neutre ou protonée. Par ailleurs, le pH peut également avoir un effet indirect
sur l'adsorption soit au niveau de l'adsorbant par modification de sa structure et de ses
propriétés, soit en agissant sur la composition de la solution aqueuse (Karichoff et al.,
1979).
La composition de la phase aqueuse du sol influe sur le processus de rétention soit par la
présence de composés particuliers, soit par des variations de force ionique. Dans le cas
général, une force ionique élevée tend à réduire la solubilité aqueuse des molécules
organiques hydrophobes et neutres et favorise donc leur rétention par la phase solide du
sol (Karichoff, 1981). De manière générale, l'influence de la force ionique de la solution
du sol sur l'adsorption des polluants dépend de la nature des polluants considérés.
Enfin, des variations de composition de la solution peuvent être liées par exemple à la
solubilisation d’une partie de la matière organique endogène, ce qui modifie la solubilité
et l’adsorption des polluants hydrophobes. Par ailleurs, la modification de la solubilité de
certains cations métalliques présents, peut provoquer la précipitation d’hydroxydes
métalliques qui constituent ainsi de nouveaux sites potentiels d’adsorption des polluants
(Means et al., 1980).
II.5.4. Influence de la température
En général, elle a pour effet d’augmenter la solubilité des molécules organiques. D’autre
part, l’élévation de la température conduit généralement à une diminution de l’adsorption
(Calvet, 1989). Les plages de variation de température dans les sols étant relativement
faibles, on considère les effets de ce facteur comme plutôt négligeables par rapport aux
autres paramètres intervenant dans le processus d’adsorption (Stegmann et al., 2001).

80
III. Conclusion
Au travers de cette synthèse bibliographique, nous avons montré que les caractéristiques
du sol jouent un rôle primordial dans la compréhension du devenir des polluants qu’il
contient. Ces rappels ont également permis de recenser les mécanismes et les interactions
gouvernant la mobilité des polluants organiques dans les sols. Comme les limites
d’application de chaque méthode de traitement dépendent de la nature de la pollution, de
son étendue et du type de sol à traiter, la connaissance de ces interactions sol/polluant est
un préalable indispensable.
La dernière partie de cette bibliographie est naturellement consacrée à décrire brièvement
l’état de l’art des principales méthodes et techniques de traitement de sites contaminés.

81
Partie D : Les sites contaminés et les techniques de traitement
I. Introduction
Le déversement accidentel, le stockage, les rejets liés à des activités industrielles sont à
l’origine du relargage de quantités très importantes de polluants organiques et
inorganiques dans l’environnement et notamment dans les sols, les sédiments et les eaux
souterraines. Aujourd’hui un grand nombre de sites sont concernés par ce type de
pollution qui est considérée comme un problème environnemental d’importance majeure.
La protection des sols à l’égard des pollutions est une préoccupation relativement récente.
Si ce souci n’est venu que tardivement, c’est bien parce que le sol a été trop longtemps
considéré comme « une boite noire », une sorte de réceptacle illimité destiné à accueillir
nos déchets et les polluants générés par nos activités industrielles et urbaines.
La remédiation des sites industriels contaminés par des polluants organiques est l’une de
priorités dans la recherche environnementale et aussi une grande préoccupation de la
société. Ces dernières années, la recherche de méthodes efficaces de remédiation des sites
pollués a progressé et la concurrence industrielle s'est développée beaucoup plus
rapidement que la taille du marché qui est encadré par des textes réglementaires et des
considérations économiques.
Plusieurs techniques de traitement ont vu le jour afin de tenter de restaurer les sites
pollués classés dangereux pour les écosystèmes et la santé humaine. L’intérêt d’une
méthode se mesure à son efficacité, à son coût, à la facilité de sa mise en œuvre, à la
qualité du sol obtenu après traitement ainsi qu’à la facilité de retraitement des sous-
produits générés.
Dans ce chapitre, nous allons parler de différentes méthodes employées pour la
remédiation des sites contaminés par des polluants organiques et de leurs champs
d’applications.

82
II. Les sites contaminés
II.1. Qu’est-ce qu’un site ?
Le site est défini comme « Tout terrain sur lequel sont exercées, en un lieu donné, sous le
contrôle d’une entreprise, des activités industrielles, y compris tout stockage de matières
premières, sous produits, produits intermédiaires, produits finis et déchets que comportent
ces activités, ainsi que tout équipement et toute infrastructure, fixes ou non, intervenant
dans l’exercice de ces activités. » le sol est donc inclus dans le site, des éléments
physiques artificiels, (aménagements ou équipements réalisés par l’homme) ou naturels
(eaux superficielles ou souterraines, végétation, faune) pouvant s’y ajouter.
(http://www.environnement.gouv.fr).
II.2. Les sites pollués en France Origine du problème
Les activités industrielles ou d'autres types d'activités, tel le stockage de certains
matériaux, ont laissé des cicatrices dans l'environnement. Diverses pratiques inadéquates
de gestion des déchets y ont contribué, comme l'enfouissement pur et simple dans les
cours d'usines, les rejets liés aux activités quotidiennes des industries, ou les
déversements accidentels. Des substances toxiques ont ainsi pu engendrer une
contamination des sols et, dans bien des cas également, des eaux souterraines. Il n'est
donc pas étonnant que des terrains contaminés existent aujourd'hui. De plus, un processus
de fermeture et de démantèlement des complexes industriels désuets s'est amorcé au
cours des dernières décennies, mettant en lumière le phénomène des terrains contaminés.
Mis à part les terrains contaminés par les activités industrielles, les lieux d'élimination de
déchets dangereux, on peut citer les sites pollués par des activités agricoles, et sites
contaminés par des activités militaires, champs d’essais, dépôts de munitions, etc.
(http://www.environnement.gouv.fr).
La réglementation française en matière de sites pollués et un inventaire des sites pollués
en France sont donnés dans l’annexe 1.

83
II.3. Conséquences pour l'environnement et la santé
Certains terrains contaminés entraînent une extension de la contamination, que ce soit par
voie aérienne (entraînement de poussières contaminées) ou souterraine (migration d'eaux
contaminées), aux environs immédiats du site, où peuvent habiter des populations
humaines. Des évaluations précises de la qualité du sol pour bien cerner l'ampleur de la
contamination sont alors requises. Les impacts d'une telle situation peuvent être
significatifs. La contamination des humains peut survenir par le biais des sols contaminés
(ingestion de terre par les enfants, inhalation de poussières, contact cutané), des eaux
souterraines (contamination des eaux de consommation) ou des produits potagers.
L'application de mesures immédiates est alors nécessaire pour réduire les possibilités
d'exposition des humains et en particulier des groupes les plus vulnérables, tels, les
enfants, les femmes enceintes et les personnes âgées.
Les évaluations de risque simplifiées et détaillées d’un site pollué sont explicitées dans
l’annexe 2.
III. Réhabilitation des sols : Les technologies.
III.1. Généralités Les différents procédés de décontamination des sols consistent à traiter les sols pour en
supprimer, ou en diminuer fortement, le caractère contaminant. Quant aux diverses
actions de réhabilitation, elles comprennent à la fois des opérations de dépollution et de
réhabilitation d'un site en vue d'en permettre un nouvel usage. L'objectif final étant de
faire disparaître les nuisances et de supprimer, ou de minimiser, les risques vis-à-vis de
l'environnement, des personnes et des biens.
Les procédés de décontamination des sols peuvent être regroupés selon trois familles
distinctes : les traitements hors-site (les sols sont emmenés vers une installation
extérieure), sur site et in-situ. A l'inverse du traitement in-situ, les deux premiers
nécessitent une excavation des terres polluées. Ces techniques impliquent l'identification,

84
le tri, le conditionnement ou reconditionnement et le transport des résidus, sols, matériaux
et eaux contaminées en vue d'être traités et éliminés dans des installations existantes.
Les technologies de traitement des sites sont constituées globalement par trois approches:
- La première considère la terre polluée comme un déchet qu'il faut excaver puis mettre
en décharge ou incinérer.
- La deuxième nécessite des techniques de pompage, d'extraction sous vide, de
confinement et de lavage.
- La troisième met en œuvre les biotechnologies, il s’agit alors de bioremédiation.
Chacune correspond à des contraintes spécifiques résultant à la fois de la nature des
polluants et du sol, des objectifs de dépollution et du contexte environnemental. La
stratégie adoptée dépendra du site, de son contexte géographique, du degré et du type de
pollution (Goyer, 1995 ; Hamby, 1996, Stegmann et al., 2001 ; Khan et al., 2004).
La restauration d'un terrain peut se faire par différents procédés de traitement ou par des
méthodes d'enfouissement sécuritaire. Les traitements biologiques utilisent des micro-
organismes pour « digérer » les contaminants. Les traitements thermiques utilisent la
chaleur pour provoquer la décomposition des produits chimiques contaminants, tandis
que les traitements physico-chimiques modifient la structure chimique des contaminants
du sol. Les méthodes d'enfouissement ne constituent pas une solution définitive au
problème. Elles nécessitent un suivi environnemental continuel, de façon à s'assurer de
l'efficacité du confinement. Elles pourraient donc faire l'objet, dans le futur, de nouveaux
travaux correcteurs.
III.2. Les techniques physiques III.2.1. Les procédés physiques de piégeage
Ce ne sont pas à proprement parler des techniques de dépollution puisqu’il s’agit
simplement d’isoler et de stabiliser sur place la pollution. De plus, par la nature des
travaux et des surveillances nécessaires, elles sont relativement onéreuses. Deux
méthodes sont fréquemment citées :

85
♦ Confinement : Technique qui consiste à isoler la source de pollution venant ainsi
empêcher la migration des substances polluantes. Cette technique est utilisée dans le
cas ou, pour des raisons techniques et économiques, il n'existe pas de solution
conduisant à une véritable décontamination du site considéré. Ce peut être également
une technique d'attente, en cas d'urgence, avant une dépollution ultérieure (Goyer,
1995 ; Hamby, 1996).
♦ Stabilisation et solidification : Technique qui consiste à fixer les substances
polluantes sur le site dans une matrice créée par un mélange homogène des résidus,
sols, eaux, matériaux contaminés avec des réactifs qui font prise et assurent la
stabilité mécanique des produits traités. Cette technique s'applique généralement aux
boues, résidus plus ou moins visqueux et aux sols contaminés par des substances
minérales et éventuellement organiques (Stegmann et al., 2001 ; Khan et al., 2004).
III.2.2. Les procédés par évacuation ou entraînement
Le principe consiste à utiliser des fluides présents dans le sol ou injectés comme vecteur
de transport et d’extraction de la pollution (Palmer et Fish, 1992 ; Goyer, 1995 ; Hamby,
1996, Stegmann et al., 2001 ; Khan et al., 2004).
♦ Le pompage écrémage (skimming): consiste à creuser des puits et/ou des tranchées
pour pomper des produits organiques surnageant en surface de la nappe. Cette
technique permet de récupérer une partie seulement des polluants : environ 50 %.
♦ Le pompage suivi d’un traitement (pump and treat): là encore il s’agit d’une méthode
de dépollution spécifique des nappes. L’eau est pompée et traitée en surface selon le
type de pollution, par les procédés classiques ;
♦ L’extraction sous vide (venting): cette méthode ne s’applique qu’aux pollutions de
produits volatils ou semi-volatils dont la constante de Henry est supérieure à 0,01 et
dont la pression de vapeur excède 0,5 mm de mercure. Utilisable dans tous les sols
non saturés, elle est particulièrement efficace dans les sols sableux perméables mais
perd de son efficacité avec des sols argileux qui ont un fort pouvoir de rétention des
solvants. Le principe repose sur la mise en dépression du sol contaminé par
l'intermédiaire d'une pompe à vide, il y a alors aspiration de vapeurs polluées qui

86
peuvent être traitées par oxydation catalytique, par condensation réfrigérée ou par
adsorption sur charbon actif.
♦ L ’injection d’air (air sparging ou stripping): Il s'agit d'une technique adaptée aux sols
non saturés contaminés par des solvants chlorés ou par des produits organiques
volatils. Le « stripping » désigne le mécanisme de transfert d'un polluant d'une phase
liquide ou solide vers une phase gazeuse (Khodadoust et al., 2004). Le principe
consiste à injecter de l'air ou de la vapeur sous pression dans le sol grâce à des puits
d'injection afin de vaporiser des composés volatils. Des puits d'extraction permettent
de récupérer les vapeurs toxiques qui sont traitées par un filtre à charbon actif ou par
un autre procédé.
♦ Le traitement par flottation : Le sol doit être excavé, on le tamise, on lui rajoute de
l'eau et des agents de surface tensioactifs. On injecte ensuite dans ce mélange des
bulles d'air qui emprisonnent les polluants et remontent à la surface pour former une
écume flottante qui est alors récupérée.
III.2.3. Le procédés de lavage
Le principe est simple, il consiste à séparer les polluants du sol par injection d'eau dans la
terre, le plus souvent excavée. Le rendement peut être considérablement amélioré en
ajoutant des produits tensioactifs dans l'eau ou en la chauffant mais le surcoût peut être
important. Cette méthode est souvent associée à une autre, comme un traitement
biologique ou un stripping (Pennell et al., 1993 ; Yeom et al., 1995, 1996 ;Khodadoust et
al., 2000).
Lavage à haute pression hors-site
On commence par excaver le sol qui est amené jusqu'à l'installation de traitement. Dans
un chambre à injection d'eau (sous une pression d’un bar à plusieurs centaines de bar), la
terre est éclatée et les substances toxiques entre les grains ou en surface sont entraînées
par l'eau et l'air injectés. La terre est alors triée suivant la granulométrie et la densité, on
sépare ainsi de la terre les particules fines (appelés les fines) où sont généralement
concentrés les polluants. La terre libérée de ces fines est propre, elle peut être réintroduite
à son emplacement initial. Les fines représentent moins de 10% de la terre en masse, elles
sont séchées et forment des « gâteaux » qui sont alors mis en décharge ou retraités.

87
Lavage in-situ à pression normale (Soil flushing) :
Cette méthode s'applique uniquement aux sols perméables, son principal avantage est
d'éviter l'excavation qui n'est pas toujours facilement réalisable. Cependant l'efficacité est
notablement inférieure à celle de la méthode précédente. Le principe est simple: de l'eau
est injectée dans le sol à pression normale, elle piège les substances toxiques ainsi que
des particules contaminées.
Selon le type de pollution, on utilise des solutions aqueuses acides, basiques ou
additionnées d’agents tensioactifs qui en s’infiltrant dans le sol vont entraîner la
pollution. Des puits verticaux ou horizontaux en limite de zone polluée permettent
d’extraire le fluide de lavage et les polluants pour un post traitement (McCray et al.,
2001).
Lavage in-situ à haute pression
Ce lavage est aussi efficace que le précédent, mais seulement sur des zones de petites
tailles car la pression de l'eau décroît rapidement avec la distance de la source d’émission.
De l'eau est injectée dans le sol par une pompe à haute pression (50 Mpa, 300 L/mn, 250
m/s en sortie). L'énergie cinétique de l'eau est suffisante pour déstructurer le sol et
extraire les substances toxiques de la surface des grains. De plus, le sol et l'eau forment
un mélange qui est pompé à la surface. Le fluide est alors traité en station d'épuration
comme pour les deux types de lavage précédents (Lundstedt et al., 2000).
III.2.4. Les procédés électriques
Il s’agit d’utiliser des champs électriques pour favoriser, in-situ, la migration des
polluants chargés électriquement. Le principe repose sur la circulation d'un courant
électrique dans le sol par l'intermédiaire d'électrodes poreuses (des anodes et des
cathodes), ainsi les particules chargées se déplacent vers les électrodes de charges
opposées à la leur et ils peuvent alors être récupérés par pompage ou siphonnage. En
outre il y a production d'ions H+ à l'anode : le milieu devient donc acide (selon le pouvoir
tampon du sol) et la désorption des cations en est facilitée. C’est une méthode sélective
qui trouve son application essentiellement dans le cas de pollution ionique comme les
métaux lourds ou quelques ions organiques. Cette méthode est utilisable pour une grande
variété de sols : du sol argileux au sol de sable fin. Pour favoriser la migration, le taux

88
d’humidité du sol doit être suffisant : 14 à 18 %. Ces procédés sont limités dans leur
principe à l ’élimination des espèces chargées (Stegmann et al., 2001 ; Khan et al., 2004).
III.2.5. Les procédés thermiques
Ces procédés consistent à oxyder les polluants organiques, facilement convertibles en
CO2 et H2O, par chauffage.
♦ La désorption thermique s’effectue généralement hors-site dans un four rotatif. Le sol
excavé est porté à une température suffisamment élevée, 600 à 800 °C par chauffage
indirect, pour désorber l’eau et les contaminants. Les produits désorbés sont alors
brûlés dans une flamme. Il faut surveiller la dangerosité des gaz émis tels que le
chlore, les oxydes de soufre et d’azote qui nécessitent un traitement spécifique.
♦ L ’incinération se fait toujours hors-site, elle s’adapte à tous les polluants organiques
avec un excellent rendement. Elle est de ce fait très utilisée. Il existe plusieurs
systèmes tels les fours à lits fluidisés, à circulation ou rotatif, les techniques
infrarouge ou micro-ondes. La température de traitement de 1000 à 1100 °C est
obtenue par chauffage direct du sol à traiter. Les gaz et les poussières produits doivent
être traités impérativement.
♦ La vitrification, utilisée in-situ dans des cas difficiles à traiter par d’autres méthodes,
consiste à placer des électrodes en carbone dans le sol et à y appliquer une très forte
différence de potentiel. La résistance du sol génère suffisamment de chaleur par effet
joule pour atteindre des températures de 1500 à 1600 °C. A ces températures le sol et
son contenu sont fondus puis vitrifiés.
Le coût correspondant à ces trois méthodes est croissant, il est lié aux équipements
nécessaires, aux travaux d’excavation pour les traitements hors-site et à la mise en œuvre
d’énergies importantes. Le traitement des gaz est également un facteur de surcoût non
négligeable. D’autant que, comme dans tout processus d’incinération le risque de
production de dioxines est grand. Pour l’incinération, le prix se situe entre 110 à 340 $
par tonne aux Etats-Unis et entre 275 et 450 euros en France (Goyer, 1995 ; Hamby,
1996, Stegmann et al., 2001 ; Khan et al., 2004).

89
III.3. Les traitements chimiques
Les techniques chimiques ont pour but de détruire les polluants ou de les transformer en
une forme moins nocive pour l’environnement. Le principe est simple: on injecte dans le
sol un réactif chimique approprié au type de pollution. Ce réactif réagit avec certains
polluants du sol afin de former des produits moins nocifs, ou alors afin de concentrer les
polluants qui deviennent plus faciles à extraire. Certaines conditions opératoires sont
nécessaires: le sol et les réactifs doivent être bien mélangés, le réactif doit être mis en
léger excès.
Une des difficultés de cette méthode est de doser correctement la quantité de réactif à
utiliser: si il n’y en a pas assez, la dépollution est évidemment incomplète et s'il y en trop
il reste un excès de réactifs dans le sol. En outre, il faut connaître parfaitement le terrain
et le réactif employé car des réactions secondaires peuvent apparaître. Celles-ci sont
susceptibles de conduire à la formation d'autres polluants, ou du moins à une dégradation
des qualités du sol.
Les procédés chimiques peuvent être in-situ ou hors-site. Généralement ces techniques
sont opérantes uniquement dans les cas où le sol est sous forme de boue ou si les
polluants sont piégés dans un milieu liquide comme l'eau du sous-sol. Il existe une très
grande gamme de procédés chimiques qui permet d'éliminer la majeure partie des
polluants. Cependant les coûts et l'efficacité sont très différents d'un procédé à un autre
(Chu et Kwan, 2003 a et b).
III.3.1. Extraction avec des solvants organiques
On utilise actuellement des solvants organiques comme des alcanes, des alcools
(méthanol, éthanol et butanol) ou des cétones qui permettent de dissoudre la plupart des
contaminants organiques. Concrètement, le solvant est introduit dans le sol, il dissout les
polluants avant d’être extrait et traité.
Il existe plusieurs méthodes d'extraction :
• par séparation naturelle et stripping,
• par volatilisation en diminuant la pression jusqu'à la pression de vapeur saturante
du solvant,
• par modification de la température afin de rendre le solvant non miscible à l'eau.

90
La toxicité des solvants organiques envers la population microbienne du sol et leur
interaction avec les constituants du sol représentent les principaux désavantages de cette
technique (Peters et Luthy, 1993 ; Khodadoust et al., 1999 ; Krauss et Wilcke, 2001).
III.3.2. Extraction par des fluides critiques
On utilise généralement le gaz carbonique, le propane ou le butane liquéfié à température
et pression critiques. Dans ces conditions thermodynamiques, les caractéristiques de
transfert de masse sont excellentes : le fluide va occuper toutes les espaces interstitielles
du sol. Lorsque la température et la pression remontent, la volatilisation du gaz entraîne
les polluants. Il s’agit d’une technique intéressante mais très difficile à mettre en œuvre et
très coûteuse. (Hamby, 1996; Khan et al., 2004).
III.3.3. Immobilisation chimique
L ’immobilisation in-situ des polluants peut être effectuée en introduisant dans le sol des
produits chimiques qui vont transformer les polluants en produits moins nocifs par
stabilisation. Les réactifs en solution sont appliqués pour saturer le sol soit par inondation
de la surface, soit plus graduellement par nébulisation. Un traitement complémentaire
sera nécessaire si le réactif chimique utilisé est lui même indésirable. On utilisera alors
les techniques physiques de lavage, pompage, ou confinement. Des réactifs insolubles
peuvent être également utilisés par épandage et labourage du sol afin de favoriser leur
contact avec les polluants (Stegmann et al., 2001).
III.3.4. Oxydation chimique
Proche de l’immobilisation cette technique consiste à oxyder les polluants qui sont alors
dans une « configuration » chimique moins dangereuse pour l’homme. On utilise des
oxydants puissants comme le peroxyde d’hydrogène. Ces techniques peuvent être
couplées à l’action photolytique de la lumière ou accélérée par l’utilisation de catalyseurs
(Kawahara, 1995).

91
La complexité des pollutions rend souvent indispensable l’utilisation de réactifs
multiples. En terme de coût, peu d’estimations sont proposées car elles sont liées à la
specificité du traitement à effectuer.
III.4. Les traitements biologiques
Le principe général de ces méthodes est basé sur les capacités épuratoires des
microorganismes. Les composés organiques sont transformés en gaz carbonique, eau ou
méthane. Les composés minéraux ne sont pas dégradés mais ils peuvent être transformés
en produits ayant une plus grande mobilité et/ou une toxicité plus faible. Ces procédés se
développent spontanément dans la nature, avec des cinétiques souvent trop lentes. Mais la
croissance des bactéries (et donc l'efficacité de la méthode) demande des conditions
physiques et chimiques du sol très strictes :
- le pH doit être proche de 7,
- la température doit être optimale,
- il ne doit pas y avoir de produits toxiques pour les bactéries,
- le sol doit être suffisamment perméable afin d’assurer un bon contact entre les
différents constituants.
Malgré tout, dans le meilleur des cas, la durée de la décontamination est longue (plusieurs
mois). Toutes les méthodes que nous présentons consistent le plus souvent à optimiser
des processus naturels. Nous les regrouperons en trois catégories : la bioremédiation in-
situ, le land-farming ou bioremédiation hors-site, et l’utilisation de bioréacteurs (Wilson
et al., 1993, 1998 ; Zhu et Feng, 2000, 2004).
IV.4.1. La bioremédiation in-situ
Il s’agit de traitements biologiques directement appliqués sur le site à dépolluer. Ils ont
l’avantage de ne pas nécessiter d’excavation et de permettre, éventuellement, la poursuite
des activités.
♦ La bioremédiation intrinsèque ou bio-atténuation est simplement la biodégradation
naturelle des polluants par les micro-organismes présents dans le sol ou la nappe.

92
Cette méthode consiste uniquement à vérifier la présence et la capacité des micro-
organismes à dégrader les polluants.
♦ La bio-stimulation consiste à accélérer le processus naturel par addition de nutriments
(azote et phosphore), et d’un accepteur d’électrons comme l’oxygène. D’autres
additifs peuvent être utilisés pour stimuler la population bactérienne.
♦ La bio-augmentation est utilisée lorsque l’activité des microorganismes indigènes est
insuffisante. Il s’agit d’ajouter des micro-organismes étrangers spécialisés. Une des
voies de recherche actuelle est l’utilisation de micro-organismes génétiquement
modifiés pour la dégradation des polluants récalcitrants.
♦ La bio-injection couple l’injection d’air ou d’oxygène à l’activité biologique normale
des micro-organismes. Les grosses molécules sont fractionnées puis entraînées par le
flux gazeux.
♦ La bio-extraction consiste de la même manière en un couplage de l’activité
biologique des micro-organismes et de l’extraction sous vide des polluants.
♦ La bio-filtration est une technique particulièrement bien adaptée à la dépollution des
nappes phréatiques. Un biofiltre est réalisé en creusant une tranchée sur le front du
déplacement de la pollution dans la nappe, la tranchée est alors remplie d’un support
granulaire à grande surface spécifique ensemencé par des micro-organismes
épurateurs.
♦ La phytoremédiation est l’utilisation de certaines plantes qui favorisent la migration
des polluants (métaux lourds) par l’intermédiaire de leur système racinaire.
L’efficacité de cette technique en vue d’extraire les polluants organiques est peu
étudiée.
♦ Les technologies associées : le problème majeur rencontré dans les procédés
biologiques est la bio-disponibilité des polluants. Afin d ’améliorer le contact antre le
substrat, le polluant et les bactéries on peut utiliser des agents de solubilisation. Nous
parlerons plus loin de l’effet des surfactants sur la biodisponibilité et la
biodégradation des polluants (Aronstein et al., 1991 ; Weissenfels et al., 1992 ;
Angley et al., 1992 ; Paolis et Kukkonen, 1993 ; Bai et al., 1997, Cuypers et al.,
2004).
Les principaux avantages de ces techniques sont : une faible implantation des
installations au sol, un capital immobilisé relativement modeste et des possibilités de
contrôle de la dépollution. C’est également un processus écologique puisqu’il utilise les
micro-organismes indigènes. Toutefois, la toxicité et le devenir des métabolites

93
engendrés par la biodégradation des contaminants organiques ne sont toujours pas
identifiés. Le comportement des polluants, leur transport vers les nappes phréatiques,
suite à l’utilisation de ces techniques biologiques, restent malgré tout encore mal connus.
De plus, de part sa lenteur, cette technique se révèle mal adaptée aux situations
d’urgence.
Une société canadienne, Biogénie, qui maîtrise ces techniques, propose ses traitements
entre 30 et 50 $ par tonne selon les quantités et les conditions d’exploitation. Une
évaluation française datant de 1995 situe les coûts de traitements biologiques entre 15 et
45 euros/t.
IV.4.2. La bioremédiation sur ou hors-site
La biodégradation en tas (land-farming): cette technologie est utilisée sur site et peut
être formellement séparée en trois techniques : l’épandage, le compostage et la biopile.
Elles ont en commun l’excavation du sol et sa disposition sur un site de traitement
contrôlé.
♦ L’épandage (landfill) est une méthode très ancienne qui consiste à étaler en couche
très fine le sol pollué de manière à favoriser son aération naturelle. Le problème
principal est de maîtriser les éventuelles migrations des polluants dans le sol support .
♦ Le compostage consiste à mélanger le sol pollué avec des matériaux organiques frais.
L’élévation de température et l’accroissement de la diversité microbienne favorise la
biodégradation des polluants.
♦ La biopile est un compostage élaboré où tous les paramètres biologiques et physico-
chimiques sont parfaitement contrôlés. Les matières à traiter sont empilées dans un
ordre précis jusqu’à 2 à 4 mètres de hauteur. L’intercalation de drains, permet
l’aération du milieu, si l’on souhaite faire une dégradation aérobie. Dans le cas
contraire il est aisé d’isoler la pile pour favoriser la dégradation anaérobie et la
production de méthane.

94
La technologie de biodégradation en tas (piles) est utilisée pour la décontamination de
sols souillés aux hydrocarbures légers (essence, diesel, huile à chauffage, mazout léger,
huiles usées). Les sols sont alors excavés et transportés sur une plate-forme imperméable
munie d'infrastructures pour l'aération forcée du sol, le captage et le traitement des eaux
de lixiviation, ainsi que pour la filtration de l'air vicié (filtres à tourbe et/ou charbon
activé). Des membranes recouvrent le sol, l'isolant des précipitations et minimisant la
propagation des composés organiques volatils (COV) ainsi que les pertes de chaleur.
L'ensemble de ces infrastructures ainsi que l'ajout de nutriments, de surfactants, d'agents
structurants (résidus de bois) et de cultures microbiennes permettent de maintenir des
conditions optimales pour la biodégradation des contaminants organiques par des micro-
organismes adaptés (température entre 20 et 45 °C, humidité à 85 % de la capacité de
rétention d'eau du sol, débit d'air horaire équivalent au volume du tas). Le temps requis
pour obtenir un rendement intéressant varie de deux à quatre semaines pour une
contamination à l'essence, et de trois à six mois pour une contamination à l'huile. Lorsque
la contamination affecte un sol à texture fine, le temps de traitement peut être multiplié
par deux ou trois par rapport à un sol sableux (Yeom et al., 1995, 1996 ; Wilson et al.,
1993, 1998 ; Zhu et Feng, 2000, 2004).
Les facteurs limitant l'applicabilité de la technologie sont :
- La perméabilité du sol.
- Le caractère réfractaire de certains contaminants à la biodégradation (PCBs, dioxines,
et autres hydrocarbures chlorés).
- La présence de substances toxiques en concentration élevée dans le sol (cuivre,
plomb, mercure...).
Les coûts estimés sont de 10 à 100 $ /tonne aux Etats Unis , et de 23 à 41 euros /tonne en
France.
III.5. L’utilisation des tensioactifs dans le traitement des sols pollués
Dans un système sol-eau, les polluants organiques hydrophobes sont principalement
localisés dans la phase solide. Cette adsorption a pour effet d'entraîner leur
immobilisation, ce qui se traduit par une protection contre les attaques microbiennes. Il en

95
résulte une baisse de leur biodisponibilité et de leur biodégradabilité. Les traitements
biologiques des sols pollués par des produits organiques nécessitent donc de favoriser
l'accessibilité des polluants aux agents microbiens. Parmi les différentes techniques
envisagées, l’injection de solutions de composés tensioactifs, d’origine chimique ou
biologique, augmente généralement la solubilité apparente des polluants et par
conséquent leur mobilité dans les sols (Aronstein et al., 1991 ; Dyke et al., 1993 ; Bai et
al., 1997). Le mode d'action de ces surfactants, utilisés à des concentrations relativement
élevées, consiste principalement en des phénomènes de dispersion et de solubilisation
(mobilisation) liés à la formation de micelles. De nombreuses études indiquent que
l'addition de surfactants dans des systèmes sol-eau permet la solubilisation des HAP
adsorbés à la surface des particules du sol par incorporation des polluants organiques
dans les micelles de surfactants (Liu et al, 1991, Yoshida et al., 2000 ; Mulligan et al,
2003). La concentration de la solution tensioactive utilisée est un paramètre déterminant
pour l’efficacité de la mobilisation. Pour des concentrations supérieures à la
Concentration Micellaire Critique (CMC) la mobilisation augmente avec la concentration
du surfactant (Clint , 1992 ; Bai et al., 1998). D’autres paramètres chimiques, tel que le
pH et la force ionique, peuvent influer sur l’efficacité du surfactant en modifiant la
configuration des micelles et les interactions entre le surfactant et le sol (Bai et al., 1998 ;
Champion et al., 1995).
Dans le cas de traitements in-situ, l’injection de ces tensioactifs dans le sol nécessite de
bien contrôler leur dispersion dans le milieu souterrain de façon à éviter la migration en
profondeur de composés indésirables. L’emploi de ces agents est notamment approprié
dans le cas de traitement hors-site dans lequel le sol est excavé, lessivé avec les
tensioactifs puis remis en place.
Si l'emploi complémentaire de surfactants dans les techniques de bioremédiation est déjà
effectif depuis quelques années, il n'existe qu'un nombre relativement limité de travaux
scientifiques s’y rapportant.
Les surfactants non-ioniques apparaissent les plus efficaces pour la solubilisation des
composés aromatiques polycycliques dans des suspensions sol/eau (Liu et al, 1991 ;
Edwards et al, 1991). Laha et Luthy (1992) ont étudié, en milieu dispersé, l'influence des
surfactants non-ioniques sur la minéralisation du phénanthrène dans le sol. Les auteurs
constatent que, pour des concentrations en surfactant supérieures à la CMC, la
minéralisation du phénanthrène est fortement inhibée.

96
Par ailleurs, les surfactants déstructurent les sols et entraînent la migration des plus fines
particules telles que les argiles (Clint, 1992). Lors de leur injection, une proportion des
surfactants peut être immobilisée par adsorption sur le sol. Cette interaction entre les
surfactants et le sol, avec sa toxicité potentielle envers la population microbienne, limite
leur application (Yoshida et al., 2000). De plus, dans le cas de forte pollution, des
émulsions peuvent se produire dans le mélange surfactant/polluant, elles sont difficiles à
récupérer ou à digérer par les bactéries (Edwards et al., 1991, Clint, 1992 ; Chu et al.,
2003).
III.6. Etat de l’art sur l’utilisation des CD dans la remédiation des sites
contaminés III.6.1. Potentialités d’utilisation des CD dans la remédiation des sites contaminés
Les études de mise en œuvre de CD dans le traitement des sols pollués, que ce soit ex-situ
ou in-situ, est une activité relativement récente. Lors de la dernière décennie, quelques
travaux concernant l’utilisation des CD dans les techniques de remédiation de sols
pollués ont été publiés. Ces recherches sont orientées dans deux directions principales :
- étudier l’aptitude des CD à solubiliser les polluants organiques hydrophobes dans
l’eau.
- tester leur capacités à extraire les polluants de sols souvent contaminés
artificiellement au laboratoire.
L'équipe américaine de Mark L. Brusseau a mené certaines expériences à l’échelle du
laboratoire, de manière à étudier les performances de cyclodextrines dans ce domaine.
Elle a conclu aux très bonnes capacités des cyclodextrines pour améliorer la solubilité de
quelques polluants organiques en phase aqueuse afin de favoriser leur extraction (Wang
et Brusseau, 1995, Brusseau et al., 1997, Mackray et Brusseau, 1998, 1999).
L’équipe hongroise de Szjetli a utilisé des cyclodextrines en vue d’augmenter la
biodisponibilité de polluants hydrophobes dans les sols, et donc, de favoriser leur
biodégradation. Elle a également conclu à la bonne efficacité des CD pour améliorer le
rendement du traitement biologique des sols pollués (Fava et al., 1998, 2002).

97
D’autres recherches ont également montré l’efficacité des CD à l’échelle du laboratoire,
dans la solubilisation, l’extraction de polluants et la bioremédiation (Reid et al, 2000 ;
Morillo et al., 2001).
III.6.2. Comparaison avec d’autres agents classiques de lavage
Utilisées en solution aqueuse, les CD pourraient faciliter la désorption des polluants du
support poreux constitué par le sol et leur transport par formation de complexes plus
solubles.
Un comparatif de lavage de sols en colonne mené par Boving et Brusseau (2000) avec
divers produits (surfactants ioniques et non-ioniques, éthanol à 50%, etc.) a montré une
efficacité correcte (taux d'extraction/volume de solution utilisé) des hydroxypropyl- et
méthyl-β-CD.
Ainsi, bien que les cyclodextrines possèdent un pouvoir de solubilisation des molécules
organiques plus faible que de nombreux tensioactifs ordinaires et cosolvants, elles
présentent tout de même de bonnes caractéristiques pour être utilisées en dépollution de
sols : elles sont considérées comme non-toxiques et biodégradables comme nous l’avons
montré dans le chapitre 1. Il n'y a pas de concentration critique ou minimale d'activité
comme c’est le cas pour les surfactants. Leur relative insensibilité au pH (sauf extrêmes),
leur structure rigide et leur stabilité thermique leurs procurent une bonne stabilité
physico-chimique en solution. Leur coût est assez comparable à celui des surfactants
usuels.

98
Bilan Le marché de la dépollution des sols est en pleine expansion sous la pression conjuguée
de la réglementation et de la demande sociale. Dans ce contexte les technologies de
réhabilitation sont en plein développement. Parmi toutes les techniques décrites dans ce
chapitre, les techniques d’extraction des polluants utilisant des agents de lavage offrent
un potentiel de réduction des coûts surtout si elles peuvent être mises en œuvres in-situ,
sans excavation du sol. Selon Stegmann et al., (2001) un agent idéal de lavage doit :
- être efficace pour mobiliser le polluant cible,
- interagir très faiblement ou être inerte vis-à-vis du sol
- être généralement non-toxique et biodégradable.
La synthèse bibliographique présentée dans le chapitre 1 montre les propriétés
complexantes remarquables des cyclodextrines (efficacité de complexation, bio-
compatibilité et faible impact sur les écosystèmes). L’affinité des CD pour les molécules
apolaires (hydrophobes) peut être mise à profit pour augmenter leur solubilité. Les
polluants hydrophobes sont prioritaires en raison de leur persistance dans les sols, due
principalement à leur faible solubilité aqueuse et à leur caractère lipophile prononcé. Les
propriétés physico-chimiques de ces polluants paraissent favorables à leur complexation
(ou solubilisation) par les CD et, par conséquent à leur mobilité dans les sols.
Les qualités spécifiques des CD permettent d’envisager leur utilisation dans l’extraction
de polluants organiques de sols contaminés.
Quelques chercheurs ont déjà testé leurs aptitudes dans ce domaine. Cependant, toutes
ces recherches mettent en évidence la nécessité de poursuivre des études, aussi bien
fondamentales qu’appliquées, afin d’élaborer une technique de traitement de sols
innovante à une grande échelle et qui puisse être reconnue par l’ensemble de la
communauté scientifique et les agences internationales de l’environnement.
Contrairement aux méthodes de remédiation citées auparavant, nous ne disposons
d’aucune fiche technique (limites d’application en relation avec le type de polluant cible,
de sol, etc.) concernant des méthodes de lavage par les CD permettant les essais

99
d’orientation qui précèdent généralement l’application d’un tel procédé. Ainsi, aucune
évaluation simplifiée ou détaillée de cette technique n’est disponible dans la littérature.
D’autre part, les interactions CD/sols ne sont pas étudiées dans les travaux dont nous
disposons à ce jour, contrairement à ce qui concerne les autres agents classiques de
lavage. Ces phénomènes semblent pourtant très important dans l’évaluation d’une telle
méthode compte tenu de leur influence sur la performance de la technique de
remédiation.
L’ensemble de ces interrogations nous conduit à penser que l’utilisation des CD dans la
décontamination de sols nécessite des travaux complémentaires, surtout si l’on envisage
une application in-situ. Plusieurs points nous semblent intéressant, dont
l’approfondissement est indispensable. ce sont en particulier :
- La mise en évidence des paramètres influençant l’efficacité de solubilisation des CD,
selon les polluants cibles.
- Les limites d’application (nature de la pollution, du sol, etc.)
- Le devenir des CD dans les sols et leur impact.
- L’optimisation des rendements de solubilisation et d’extraction selon le contexte.
Ce travail apparaît comme un préalable nécessaire à la phase de recherche, plus
appliquée, pour la mise au point d’un procédé innovant et fiable de remédiation de sols
contaminés.

100

101
RESULTATS ET DISCUSSIONS
Chapitre II :
Tests préalables de faisabilité

102

103
Chapitre II : Tests préalables de faisabilité
Préambule
Dans ce premier chapitre, nous avons choisi de regrouper et de présenter les résultats de
deux séries d’expériences qui pourraient, à première vue, paraître anachroniques.
La première partie constitue une étape décisive dans la démarche globale de notre projet.
Il s’agit de tester les performances de plusieurs cyclodextrines vis-à-vis de la
complexation de plusieurs polluants organiques, dans différents contextes chimiques.
Dans le second volet, nous présenterons une étude de faisabilité d’une technique de
dégradation photocatalytique de complexes CD/polluant.
Le point commun entre ces parties est qu’il s’agit, dans les deux cas, de recherche
« amont » en solutions aqueuses, c’est à dire « hors sol », et à l’échelle du laboratoire.
L’ensemble de ce travail est d’ordre académique, mais il nous est apparu comme un
préalable nécessaire à la phase de recherche, plus appliquée, sur les systèmes
sol/polluant/CD.

104

105
Partie I : Etude de la solubilisation de polluants organiques par les cyclodextrines dans l’eau
I. Introduction
La mise au point d'un procédé d'extraction de polluants organiques par lavage de sols
avec des solutions aqueuses des cyclodextrines nécessite une première phase de recherche
« hors sol » pour évaluer les interactions des cyclodextrines (CDs) avec les polluants
cibles.
L’objectif de cette première partie expérimentale est de mettre en évidence l’existence de
complexes d’inclusion entre plusieurs couples, cyclodextrines /polluants organiques, en
solution aqueuse. Deux types de polluants ont été choisis appartenant, d’une part à la
famille des chlorophénols et, d’autre part, à celle des hydrocarbures aromatiques
polycycliques. Les agents complexants testés sont des β-cyclodextrines : native ou
modifiées.
La nature chimique des polluants étudiés a nécessité le suivi des équilibres chimiques de
complexation dans des contextes de pH et de force ionique variables.
Toutes les constantes ont été déterminées expérimentalement par la méthode de
solubilisation. Les résultats ont été exploités en terme de gain de solubilité des
contaminants en présence de cyclodextrines comparativement à leur solubilité aqueuse.
Ils conditionnent naturellement la suite de ce travail en ce qui concerne l’optimisation des
choix de systèmes CD/polluant qui seront étudiés, ou non.
Ces premiers tests constituent, de fait, une étude de faisabilité préalable et indispensable,
en amont du traitement d’une pollution spécifique.
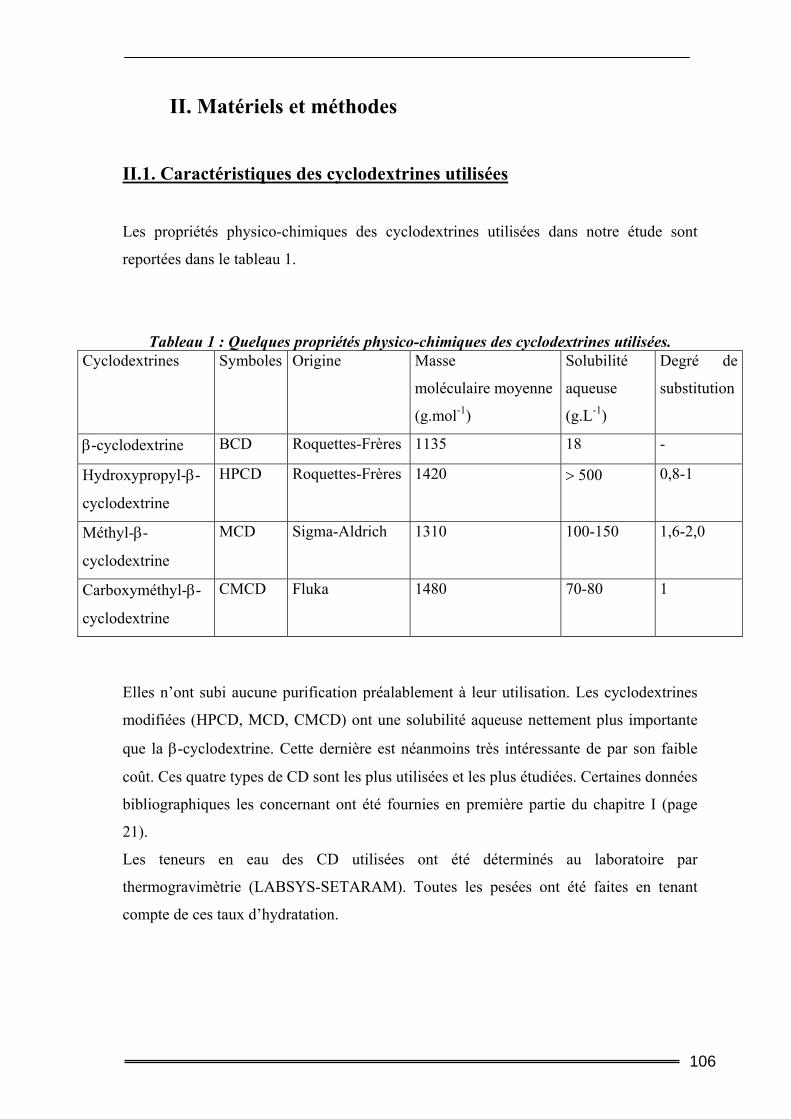
106
II. Matériels et méthodes
II.1. Caractéristiques des cyclodextrines utilisées
Les propriétés physico-chimiques des cyclodextrines utilisées dans notre étude sont
reportées dans le tableau 1.
Tableau 1 : Quelques propriétés physico-chimiques des cyclodextrines utilisées. Cyclodextrines
Symboles Origine Masse
moléculaire moyenne
(g.mol-1)
Solubilité
aqueuse
(g.L-1)
Degré de
substitution
β-cyclodextrine BCD Roquettes-Frères 1135 18 -
Hydroxypropyl-β-
cyclodextrine
HPCD Roquettes-Frères 1420 > 500 0,8-1
Méthyl-β-
cyclodextrine
MCD Sigma-Aldrich 1310 100-150 1,6-2,0
Carboxyméthyl-β-
cyclodextrine
CMCD Fluka 1480 70-80 1
Elles n’ont subi aucune purification préalablement à leur utilisation. Les cyclodextrines
modifiées (HPCD, MCD, CMCD) ont une solubilité aqueuse nettement plus importante
que la β-cyclodextrine. Cette dernière est néanmoins très intéressante de par son faible
coût. Ces quatre types de CD sont les plus utilisées et les plus étudiées. Certaines données
bibliographiques les concernant ont été fournies en première partie du chapitre I (page
21).
Les teneurs en eau des CD utilisées ont été déterminés au laboratoire par
thermogravimètrie (LABSYS-SETARAM). Toutes les pesées ont été faites en tenant
compte de ces taux d’hydratation.

107
II.2. Les polluants organiques sélectionnés Notre étude a été réalisée sur deux chlorophénols (Pentachlorophénol et Trichlorophénol)
et deux HAP (Naphtalène et Phénanthrène) sélectionnés comme des polluants modèles.
La sélection de ces molécules s'est effectuée sur la base de plusieurs critères :
- Leurs propriétés physiques, chimiques et biologiques,
- Leur représentativité au sein d'une famille importante de polluants organiques,
- Leur présence dans un grand nombre de cas de pollutions industrielles des sols.
Les quatre polluants choisis : 2,4,6 trichlorophénol (TCP), pentachlorophénol (PCP),
naphtalène (NAP) et phénanthrène (PHE) sont des produits de Sigma-Aldrich (pureté : 99
%).
Les principales caractéristiques de ces polluants ont été présentées dans les tableaux 1 et
2 (page 42).
II.3. Détermination des constantes d’équilibre de complexation par la
méthode de Solubilisation
Compte tenu de la nature chimique des molécules organiques sélectionnées, nous avons
choisi la méthode de solubilisation pour déterminer les constantes d’équilibre de
complexation et la stœchiométrie des complexes formés.
Cette méthode a été décrite au paragraphe III.2.1. (chapitre I).
II.3.1. Procédure de solubilisation
Les études de solubilisation du composé organique ont été réalisées dans des solutions
aqueuses de CD à des concentrations variables. Deux gammes de concentrations ont été
testées, l'une correspondant à des concentrations en CD relativement faibles (0 à 18 g.L-1)
pour la BCD, l'autre à des concentrations nettement plus élevées (de 0 à 80 g.L-1) pour les
CD modifiées.
Des prises d’essai de 30 ml de solutions à différentes concentrations en cyclodextrine
sont versées dans des flacons de 50 ml munis de bouchons en PTFE. Le composé

108
organique choisi est alors ajouté sous forme solide en quantité excédant largement sa
limite de solubilité. Des « blancs » sans cyclodextrine sont préparés de façon identique.
Chaque expérience est répétée trois fois. Toutes les solutions sont agitées pendant 48 h à
température ambiante à l’aide d’un éluteur rotatif. Les solutions sont ensuite transvasées,
puis centrifugées à 1400 rpm pendant 60 minutes. Une partie du surnageant est alors
prélevée avec une seringue et diluée dans du méthanol (50 %). Le surnageant dilué est
ensuite analysé par spectrophotométrie UV-Visible afin de déterminer la solubilité
apparente (Sa) du composé à une concentration donnée de CD. Les « blancs » servent à
déterminer la limite de solubilité (Sw) du composé à un pH donné ou à une force ionique
donnée dans l’eau pure.
Les constantes de complexation ont été déterminées pour les deux acides faibles : PCP et
TCP, à deux valeurs de pH : pH < pKa et pH > pKa. L’effet de la force ionique a été
étudié pour le PCP.
Compte tenu de leur caractère non-ionisable et de l’effet faible du pH sur la solubilité
aqueuse des deux HAP (NAP et PHE), nous n’avons étudié que l’effet de la force ionique
sur leur solubilisation par les CD.
L’eau utilisée est purifiée par un système de purification Milli-Q (Millipore). L’acide
citrique et l’hydrogénophosphate de potassium (Fluka) sont utilisés pour tamponner les
solutions à pH 3 et 8,8 pour les expériences de solubilisation du TCP, à pH 3 et 7 pour
celles du PCP (tampon citrate/phosphate). Le chlorure de calcium (Fluka) est utilisé pour
ajuster la force ionique des solutions.
II.3.2. Conditions des analyses spectrophotomètriques
La concentration totale en polluant solubilisé dans les surnageants a été déterminée au
moyen d’un Spectrophotomètre UV-Visible Perkin-Elmer. Le surnageant doit être dilué
dans le méthanol (50 %) avant d’être analysé par UV, de façon à décomplexer le polluant
(Brusseau et Wang ; 1993).
En effet, l’absorption en UV de l’espèce libre peut être différente de celle de l’espèce
complexée. La dilution par le méthanol permet de doser directement la concentration
totale en substrat solubilisé, c’est à dire la solubilité apparente du polluant dans la
solution de CD.

109
Dans le cas du TCP et du PCP, il a été observé que la complexation avec les CD a un
effet négligeable sur leur absorbance en UV. Par contre, il est nécessaire de décomplexer
le NAP et le PHE puisque la complexation avec la CD peut provoquer une exaltation de
leur intensité d’absorbance UV (Edwards et al, 1991).
Conditions expérimentales
Les longueurs d’onde UV utilisées pour détecter les molécules organiques sont reportées
dans le tableau 2.
Tableau 2 : Longueurs d’onde UV maximales utilisées pour détecter les molécules organiques solubilisées.
Longueur d’onde
maximale (nm)
Eau pure avec ou
sans CaCl2
pH < pKa pH > pKa.
PCP 320 300 320
TCP 290 315
NAP 250
PHE 270
Des droites d’étalonnage avec et sans CD ont été préalablement établies pour chaque
molécule et pour chaque condition chimique (pH ou force ionique).
III. Résultats et discussion
III.1. Complexation du Trichlorophénol et du Pentachlorophénol à deux
valeurs de pH
III.1.1. Détermination de la constante de complexation et du gain de solubilité
Nous avons étudié la complexation des deux acides faibles que sont le TCP et le PCP à
deux valeurs de pH (pH < pKa et pH > pKa) dans l’objectif de préciser l’influence de leur
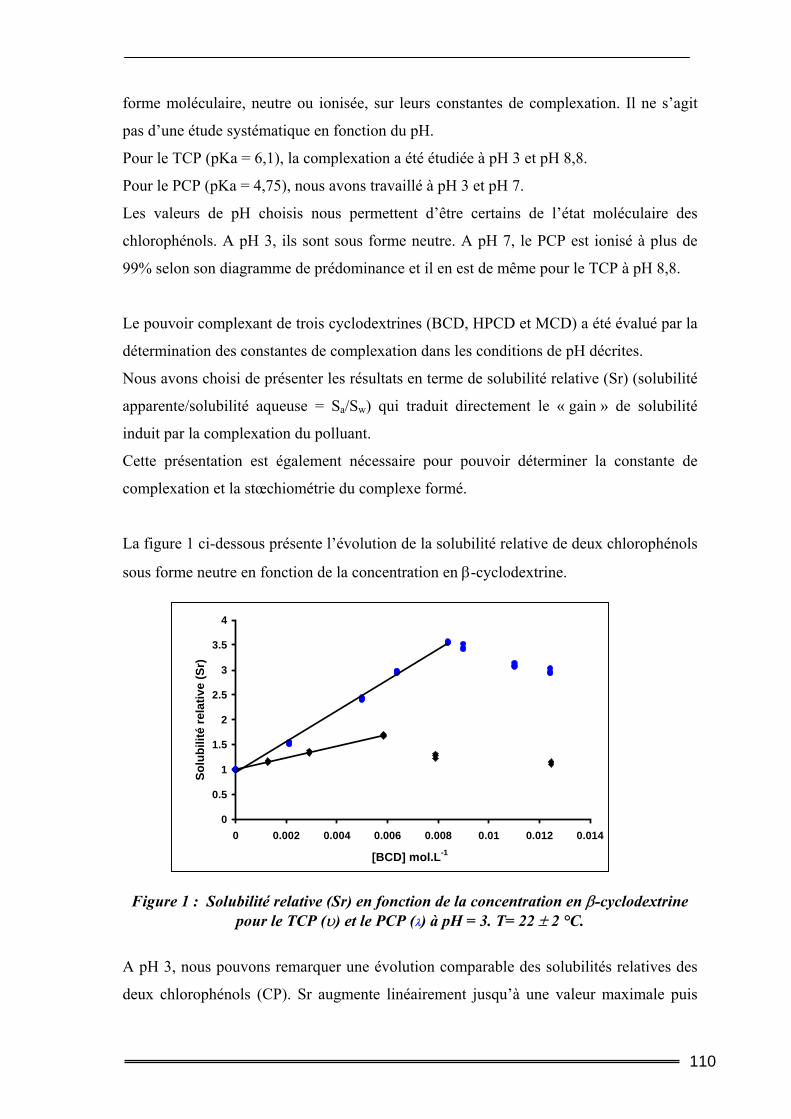
110
forme moléculaire, neutre ou ionisée, sur leurs constantes de complexation. Il ne s’agit
pas d’une étude systématique en fonction du pH.
Pour le TCP (pKa = 6,1), la complexation a été étudiée à pH 3 et pH 8,8.
Pour le PCP (pKa = 4,75), nous avons travaillé à pH 3 et pH 7.
Les valeurs de pH choisis nous permettent d’être certains de l’état moléculaire des
chlorophénols. A pH 3, ils sont sous forme neutre. A pH 7, le PCP est ionisé à plus de
99% selon son diagramme de prédominance et il en est de même pour le TCP à pH 8,8.
Le pouvoir complexant de trois cyclodextrines (BCD, HPCD et MCD) a été évalué par la
détermination des constantes de complexation dans les conditions de pH décrites.
Nous avons choisi de présenter les résultats en terme de solubilité relative (Sr) (solubilité
apparente/solubilité aqueuse = Sa/Sw) qui traduit directement le « gain » de solubilité
induit par la complexation du polluant.
Cette présentation est également nécessaire pour pouvoir déterminer la constante de
complexation et la stœchiométrie du complexe formé.
La figure 1 ci-dessous présente l’évolution de la solubilité relative de deux chlorophénols
sous forme neutre en fonction de la concentration en β-cyclodextrine.
Figure 1 : Solubilité relative (Sr) en fonction de la concentration en β-cyclodextrine
pour le TCP (υ) et le PCP (λ) à pH = 3. T= 22 ± 2 °C.
A pH 3, nous pouvons remarquer une évolution comparable des solubilités relatives des
deux chlorophénols (CP). Sr augmente linéairement jusqu’à une valeur maximale puis
0
0.5
1
1.5
2
2.5
3
3.5
4
0 0.002 0.004 0.006 0.008 0.01 0.012 0.014
[BCD] mol.L-1
Solu
bilit
é re
lativ
e (S
r)
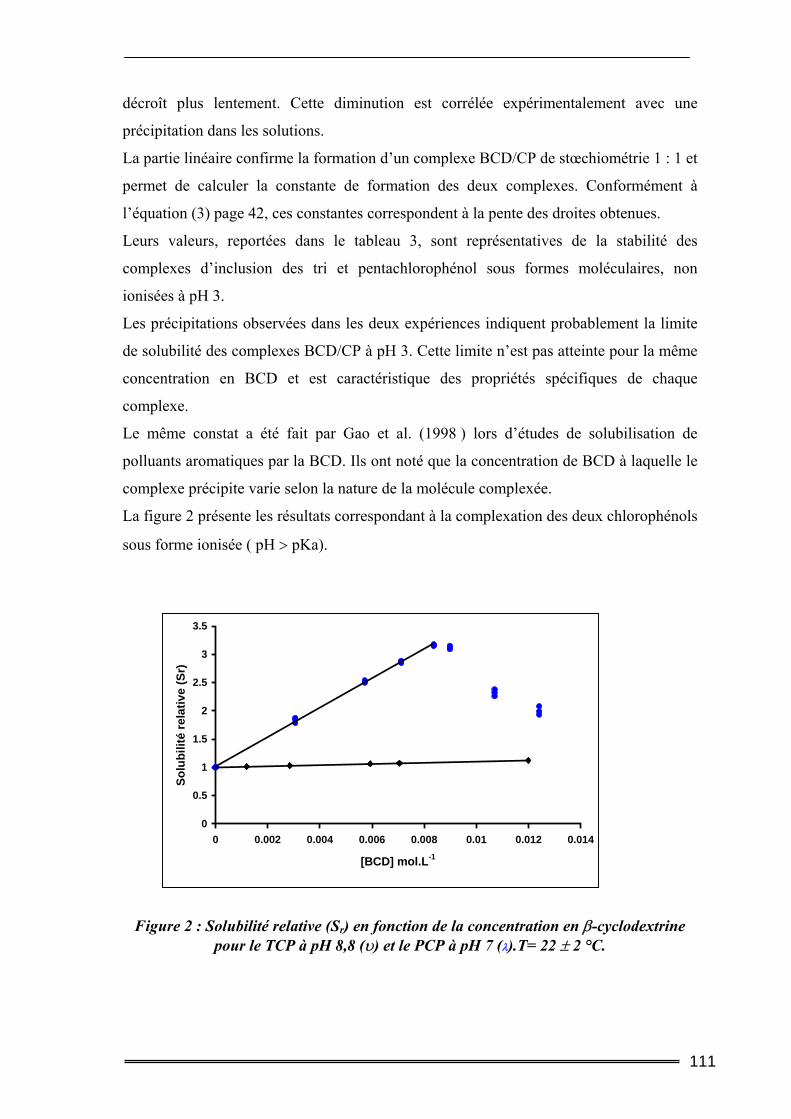
111
décroît plus lentement. Cette diminution est corrélée expérimentalement avec une
précipitation dans les solutions.
La partie linéaire confirme la formation d’un complexe BCD/CP de stœchiométrie 1 : 1 et
permet de calculer la constante de formation des deux complexes. Conformément à
l’équation (3) page 42, ces constantes correspondent à la pente des droites obtenues.
Leurs valeurs, reportées dans le tableau 3, sont représentatives de la stabilité des
complexes d’inclusion des tri et pentachlorophénol sous formes moléculaires, non
ionisées à pH 3.
Les précipitations observées dans les deux expériences indiquent probablement la limite
de solubilité des complexes BCD/CP à pH 3. Cette limite n’est pas atteinte pour la même
concentration en BCD et est caractéristique des propriétés spécifiques de chaque
complexe.
Le même constat a été fait par Gao et al. (1998 ) lors d’études de solubilisation de
polluants aromatiques par la BCD. Ils ont noté que la concentration de BCD à laquelle le
complexe précipite varie selon la nature de la molécule complexée.
La figure 2 présente les résultats correspondant à la complexation des deux chlorophénols
sous forme ionisée ( pH > pKa).
Figure 2 : Solubilité relative (Sr) en fonction de la concentration en β-cyclodextrine pour le TCP à pH 8,8 (υ) et le PCP à pH 7 (λ).T= 22 ± 2 °C.
0
0.5
1
1.5
2
2.5
3
3.5
0 0.002 0.004 0.006 0.008 0.01 0.012 0.014
[BCD] mol.L-1
Solu
bilit
é re
lativ
e (S
r)
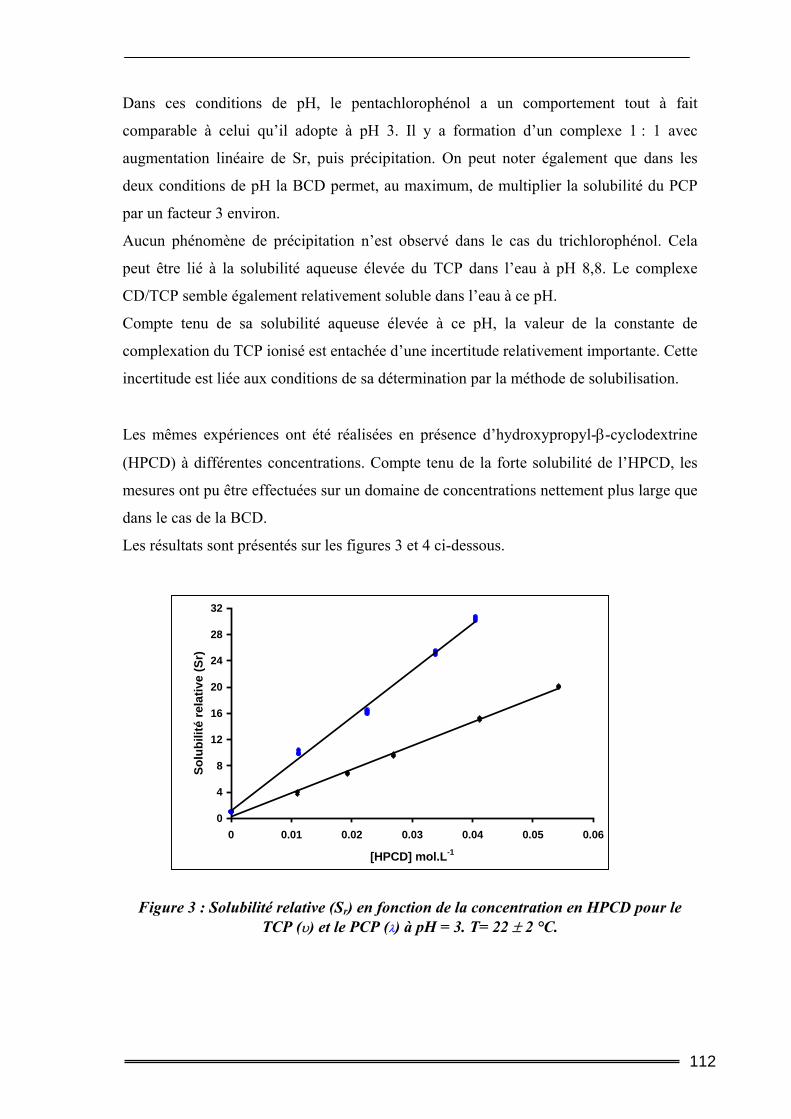
112
Dans ces conditions de pH, le pentachlorophénol a un comportement tout à fait
comparable à celui qu’il adopte à pH 3. Il y a formation d’un complexe 1 : 1 avec
augmentation linéaire de Sr, puis précipitation. On peut noter également que dans les
deux conditions de pH la BCD permet, au maximum, de multiplier la solubilité du PCP
par un facteur 3 environ.
Aucun phénomène de précipitation n’est observé dans le cas du trichlorophénol. Cela
peut être lié à la solubilité aqueuse élevée du TCP dans l’eau à pH 8,8. Le complexe
CD/TCP semble également relativement soluble dans l’eau à ce pH.
Compte tenu de sa solubilité aqueuse élevée à ce pH, la valeur de la constante de
complexation du TCP ionisé est entachée d’une incertitude relativement importante. Cette
incertitude est liée aux conditions de sa détermination par la méthode de solubilisation.
Les mêmes expériences ont été réalisées en présence d’hydroxypropyl-β-cyclodextrine
(HPCD) à différentes concentrations. Compte tenu de la forte solubilité de l’HPCD, les
mesures ont pu être effectuées sur un domaine de concentrations nettement plus large que
dans le cas de la BCD.
Les résultats sont présentés sur les figures 3 et 4 ci-dessous.
Figure 3 : Solubilité relative (Sr) en fonction de la concentration en HPCD pour le TCP (υ) et le PCP (λ) à pH = 3. T= 22 ± 2 °C.
0
4
8
12
16
20
24
28
32
0 0.01 0.02 0.03 0.04 0.05 0.06
[HPCD] mol.L-1
Solu
bilit
é re
lativ
e (S
r)
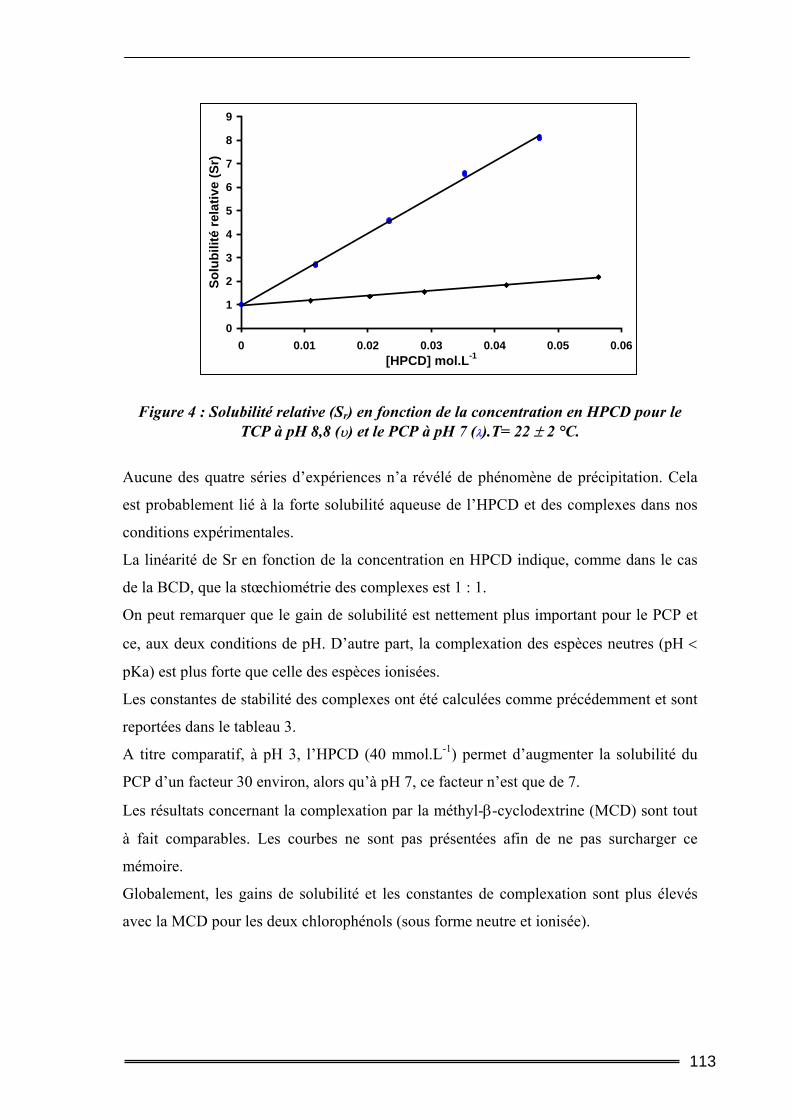
113
Figure 4 : Solubilité relative (Sr) en fonction de la concentration en HPCD pour le TCP à pH 8,8 (υ) et le PCP à pH 7 (λ).T= 22 ± 2 °C.
Aucune des quatre séries d’expériences n’a révélé de phénomène de précipitation. Cela
est probablement lié à la forte solubilité aqueuse de l’HPCD et des complexes dans nos
conditions expérimentales.
La linéarité de Sr en fonction de la concentration en HPCD indique, comme dans le cas
de la BCD, que la stœchiométrie des complexes est 1 : 1.
On peut remarquer que le gain de solubilité est nettement plus important pour le PCP et
ce, aux deux conditions de pH. D’autre part, la complexation des espèces neutres (pH <
pKa) est plus forte que celle des espèces ionisées.
Les constantes de stabilité des complexes ont été calculées comme précédemment et sont
reportées dans le tableau 3.
A titre comparatif, à pH 3, l’HPCD (40 mmol.L-1) permet d’augmenter la solubilité du
PCP d’un facteur 30 environ, alors qu’à pH 7, ce facteur n’est que de 7.
Les résultats concernant la complexation par la méthyl-β-cyclodextrine (MCD) sont tout
à fait comparables. Les courbes ne sont pas présentées afin de ne pas surcharger ce
mémoire.
Globalement, les gains de solubilité et les constantes de complexation sont plus élevés
avec la MCD pour les deux chlorophénols (sous forme neutre et ionisée).
0
1
2
3
4
5
6
7
8
9
0 0.01 0.02 0.03 0.04 0.05 0.06[HPCD] mol.L-1
Solu
bilit
é re
lativ
e (S
r)

114
Toutes les constantes de complexation obtenues sont regroupées dans le tableau 3 ci-
dessous. Nous y avons inclus les valeurs des coefficients de partage octanol/eau des
chlorophénols.
Tableau 3 : Constantes d’équilibre de complexation (1 : 1) du TCP et du PCP avec trois types de CD à deux conditions de pH. L’incertitude est d’environ 5 %. T= 22 ± 2
°C. KBCD
(L.mol-1)
KHPCD
(L.mol-1)
KMCD
(L.mol-1)
Kow
TCP neutre (pH3) 117 357 392 103,85 a
TCP ionisé (pH8,8) 10 21 46 101,47 a et b
PCP neutre (pH3) 309 703 803 105 a
PCP ionisé (pH7) 258 153 373 103 a 102,75 b
a(Kaiser and Valdmanis, 1982 ; Jafvert et al., 1990). b Calculé suivant l’Eq. 6 (page 44).
Les six séries d’expériences peuvent être résumées en termes comparatifs par les
remarques suivantes :
- Les deux chlorophénols, neutres et ionisés, forment des complexes 1 : 1 avec les trois
CD testées.
- Les complexes d’inclusion des chlorophénols (TCP et PCP) neutres sont plus stables
que ceux formés avec les espèces ionisées (pH >pKa).
- D’un point de vue du pouvoir complexant, les CD modifiées sont plus efficaces que la
BCD. Si l’on compare les trois cyclodextrines, leur pouvoir complexant ou solubilisant
peut être classé comme suit : MCD > HPCD > BCD, sauf à pH 7 pour le PCP où ce
classement est modifié.
- La précipitation du complexe n’existe que dans le cas de la BCD, probablement en
raison de sa très faible solubilité aqueuse.
D’un point de vue quantitatif, nous avons regroupé, sur les schémas des figures 5 et 6, les
gains de solubilité (facteur multiplicatif par rapport à la solubilité aqueuse ou solubilité
relative) obtenus à une concentration donnée en CD lors de nos expériences à pH3 (figure
5) et à pH > pKa (figure 6). Pour pouvoir récapituler les résultats de solubilisation pour
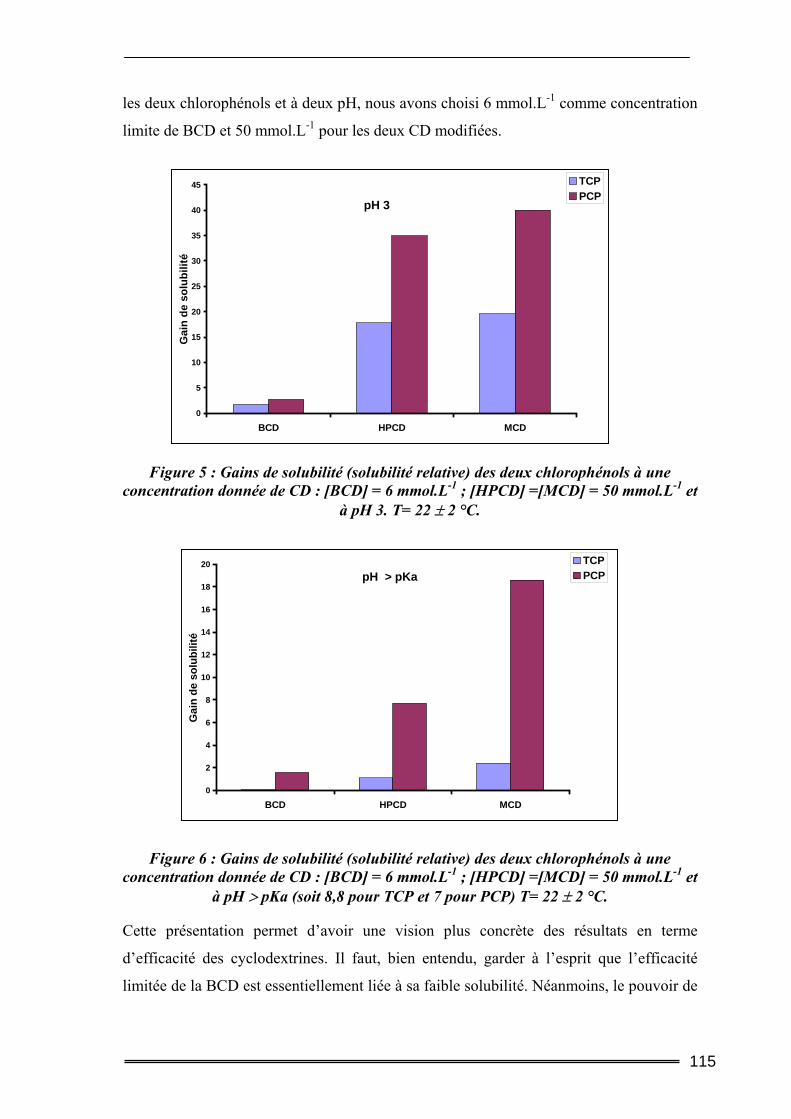
115
les deux chlorophénols et à deux pH, nous avons choisi 6 mmol.L-1 comme concentration
limite de BCD et 50 mmol.L-1 pour les deux CD modifiées.
Figure 5 : Gains de solubilité (solubilité relative) des deux chlorophénols à une
concentration donnée de CD : [BCD] = 6 mmol.L-1 ; [HPCD] =[MCD] = 50 mmol.L-1 et à pH 3. T= 22 ± 2 °C.
Figure 6 : Gains de solubilité (solubilité relative) des deux chlorophénols à une concentration donnée de CD : [BCD] = 6 mmol.L-1 ; [HPCD] =[MCD] = 50 mmol.L-1 et
à pH > pKa (soit 8,8 pour TCP et 7 pour PCP) T= 22 ± 2 °C.
Cette présentation permet d’avoir une vision plus concrète des résultats en terme
d’efficacité des cyclodextrines. Il faut, bien entendu, garder à l’esprit que l’efficacité
limitée de la BCD est essentiellement liée à sa faible solubilité. Néanmoins, le pouvoir de
0
5
10
15
20
25
30
35
40
45
BCD HPCD MCD
Gai
n de
sol
ubili
té
TCPPCP
pH 3
0
2
4
6
8
10
12
14
16
18
20
BCD HPCD MCD
Gai
n de
sol
ubili
té
TCPPCPpH > pKa

116
solubilisation de la BCD est également limité en raison de constantes de complexation
plus faibles (tableau 3).
Une interprétation plus fine des résultats peut être faite en raisonnant sur les valeurs de
coefficient de partage octanol/eau, Kow, des deux chlorophénols, sous forme neutre et
ionisée (tableau 3). Plus la valeur de Kow est élevée, plus le composé concerné peut être
considéré comme hydrophobe. Il ressort logiquement de ces résultats comparatifs que
l’hydrophobicité des deux chlorophénols neutres est plus forte que lorsqu’ils sont ionisés.
Par ailleurs, quelque soit le pH, le PCP est plus hydrophobe que le TCP.
Dès lors, nos résultats sont tout à fait cohérents. Toutes les études de complexation de
substrats organiques par les CD convergent vers les mêmes conclusions : le caractère
hydrophobe du substrat favorise sa complexation dans la cavité hydrophobe de la
cyclodextrine (Buvari et Barcza, 1988 ; Wang and Brusseau, 1993 ; Tanada et al. 1999).
III.1.2. Concentration totale de substrat soluble ou solubilité apparente
La quantité totale de polluant solubilisé par une cyclodextrine, dépend, à une température
donnée, de trois facteurs principaux qui sont : la constante de complexation, la solubilité
aqueuse (Sw) intrinsèque du polluant et la concentration en cyclodextrine. En terme de
concentration, elle s’exprime par la solubilité apparente (Sa) du substrat :
Sa = [PCP/CD] +[PCP]
Sachant que les concentrations des espèces en solution sont reliées par la relation :
[PCP/CD] = K [PCP] [CD]
Dans le paragraphe précédent, nous avons exprimé les résultats en terme de gain de
solubilité (Sr) et de constante de complexation (K). Il nous a semblé utile, compte tenu de
l’objectif de notre travail, de raisonner ici de manière plus concrète sur la solubilité totale
du substrat.
Les résultats présentés sur la figure 7 concernent uniquement la complexation du
pentachlorophénol par la MCD à pH 3 et 7.
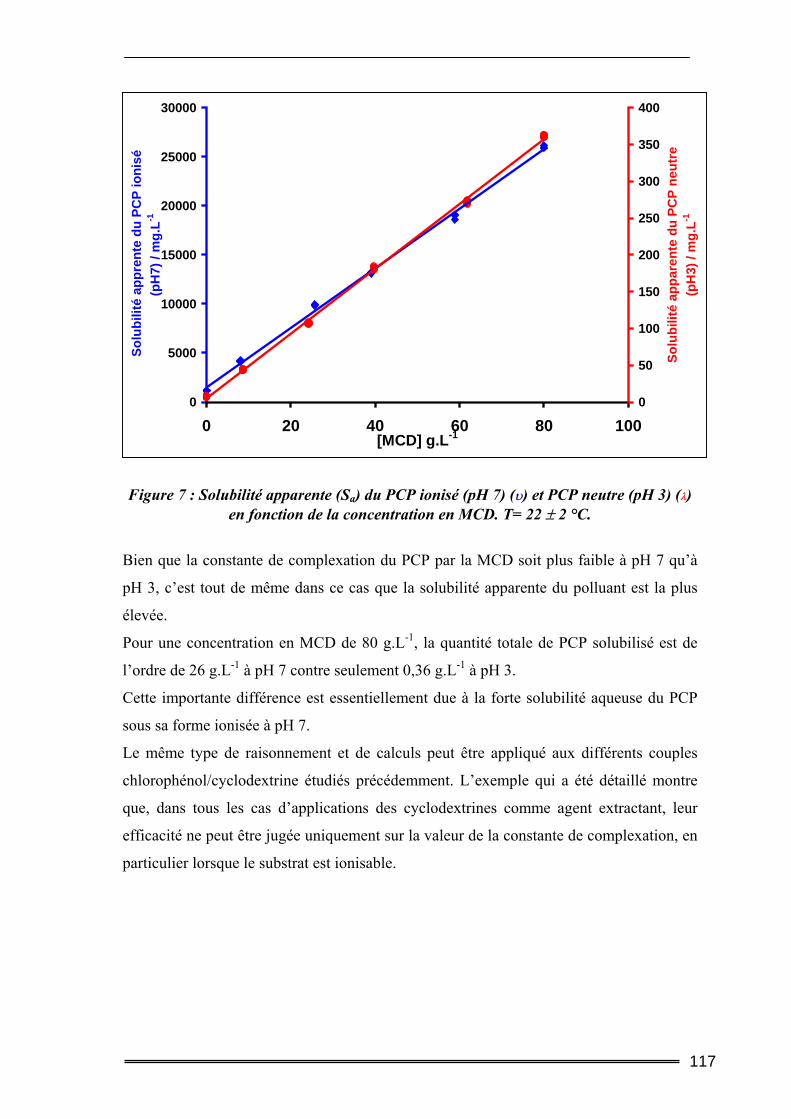
117
Figure 7 : Solubilité apparente (Sa) du PCP ionisé (pH 7) (υ) et PCP neutre (pH 3) (λ) en fonction de la concentration en MCD. T= 22 ± 2 °C.
Bien que la constante de complexation du PCP par la MCD soit plus faible à pH 7 qu’à
pH 3, c’est tout de même dans ce cas que la solubilité apparente du polluant est la plus
élevée.
Pour une concentration en MCD de 80 g.L-1, la quantité totale de PCP solubilisé est de
l’ordre de 26 g.L-1 à pH 7 contre seulement 0,36 g.L-1 à pH 3.
Cette importante différence est essentiellement due à la forte solubilité aqueuse du PCP
sous sa forme ionisée à pH 7.
Le même type de raisonnement et de calculs peut être appliqué aux différents couples
chlorophénol/cyclodextrine étudiés précédemment. L’exemple qui a été détaillé montre
que, dans tous les cas d’applications des cyclodextrines comme agent extractant, leur
efficacité ne peut être jugée uniquement sur la valeur de la constante de complexation, en
particulier lorsque le substrat est ionisable.
0
5000
10000
15000
20000
25000
30000
0 20 40 60 80 100[MCD] g.L-1
Sol
ubili
té a
ppre
nte
du P
CP
ioni
sé
(pH
7) /
mg.
L-1
0
50
100
150
200
250
300
350
400
Solu
bilit
é ap
pare
nte
du P
CP
neut
re
(pH
3) /
mg.
L-1
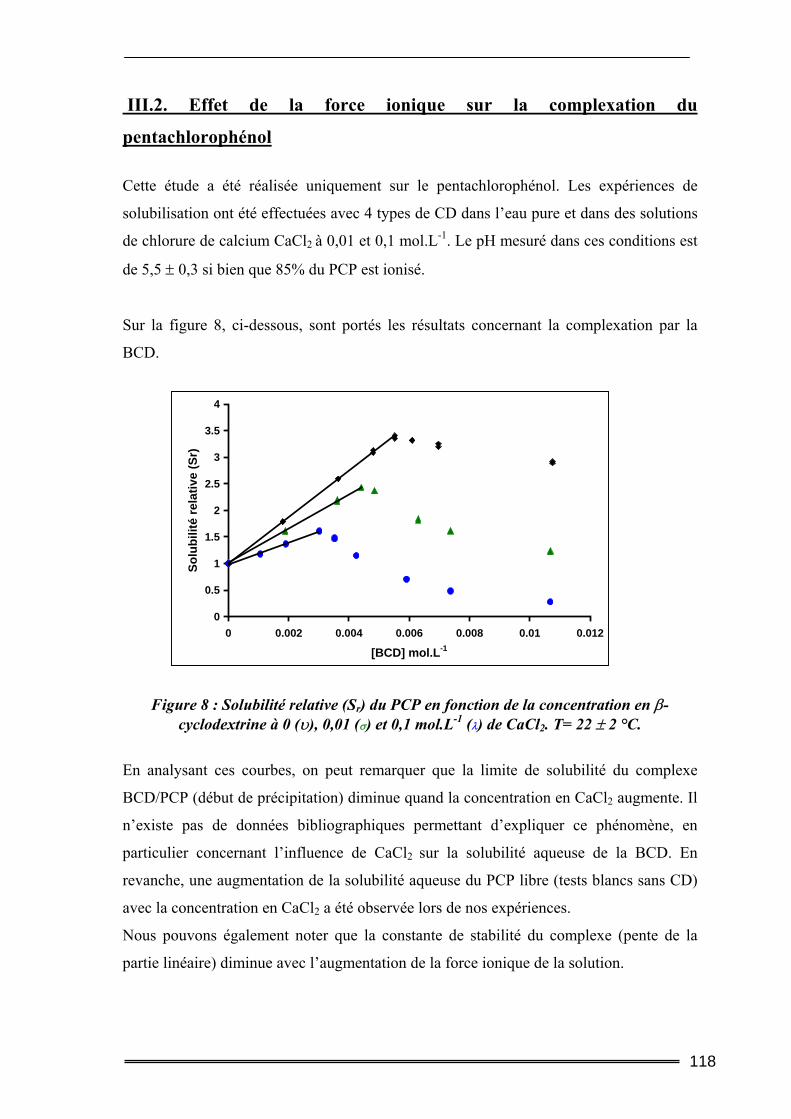
118
III.2. Effet de la force ionique sur la complexation du
pentachlorophénol Cette étude a été réalisée uniquement sur le pentachlorophénol. Les expériences de
solubilisation ont été effectuées avec 4 types de CD dans l’eau pure et dans des solutions
de chlorure de calcium CaCl2 à 0,01 et 0,1 mol.L-1. Le pH mesuré dans ces conditions est
de 5,5 ± 0,3 si bien que 85% du PCP est ionisé.
Sur la figure 8, ci-dessous, sont portés les résultats concernant la complexation par la
BCD.
Figure 8 : Solubilité relative (Sr) du PCP en fonction de la concentration en β-cyclodextrine à 0 (υ), 0,01 (σ) et 0,1 mol.L-1 (λ) de CaCl2. T= 22 ± 2 °C.
En analysant ces courbes, on peut remarquer que la limite de solubilité du complexe
BCD/PCP (début de précipitation) diminue quand la concentration en CaCl2 augmente. Il
n’existe pas de données bibliographiques permettant d’expliquer ce phénomène, en
particulier concernant l’influence de CaCl2 sur la solubilité aqueuse de la BCD. En
revanche, une augmentation de la solubilité aqueuse du PCP libre (tests blancs sans CD)
avec la concentration en CaCl2 a été observée lors de nos expériences.
Nous pouvons également noter que la constante de stabilité du complexe (pente de la
partie linéaire) diminue avec l’augmentation de la force ionique de la solution.
0
0.5
1
1.5
2
2.5
3
3.5
4
0 0.002 0.004 0.006 0.008 0.01 0.012
[BCD] mol.L-1
Solu
bilit
é re
lativ
e (S
r)
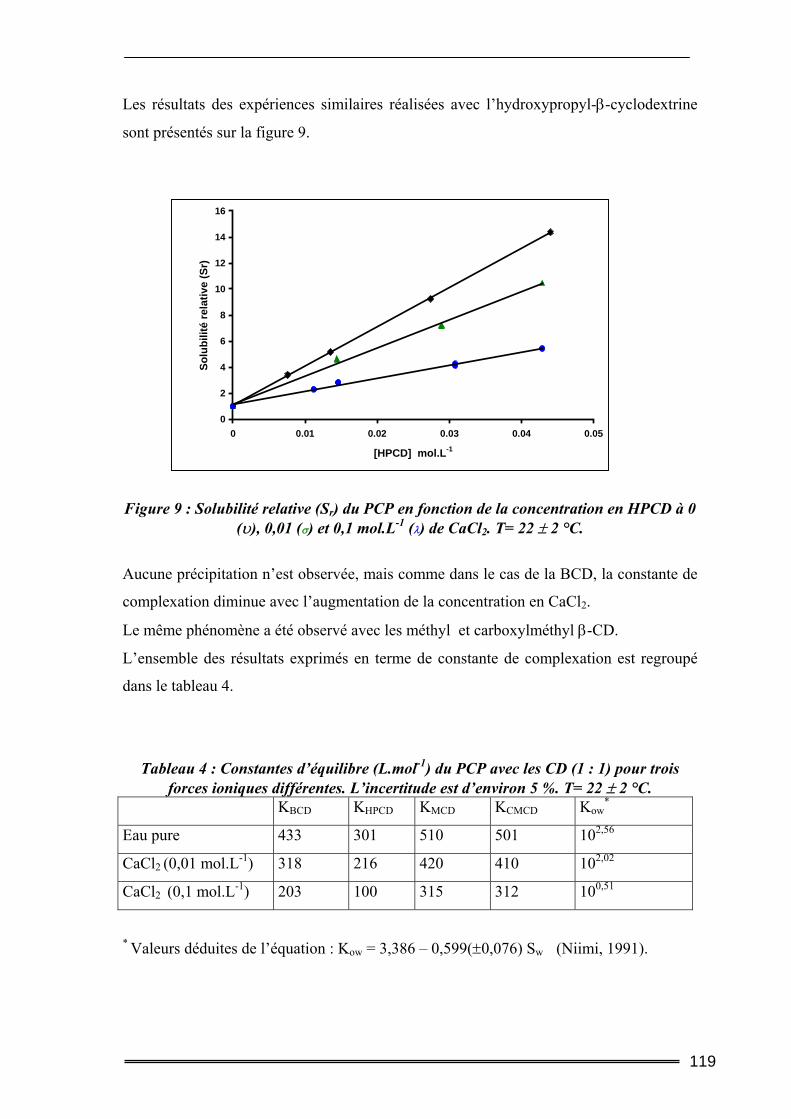
119
Les résultats des expériences similaires réalisées avec l’hydroxypropyl-β-cyclodextrine
sont présentés sur la figure 9.
Figure 9 : Solubilité relative (Sr) du PCP en fonction de la concentration en HPCD à 0 (υ), 0,01 (σ) et 0,1 mol.L-1 (λ) de CaCl2. T= 22 ± 2 °C.
Aucune précipitation n’est observée, mais comme dans le cas de la BCD, la constante de
complexation diminue avec l’augmentation de la concentration en CaCl2.
Le même phénomène a été observé avec les méthyl et carboxylméthyl β-CD.
L’ensemble des résultats exprimés en terme de constante de complexation est regroupé
dans le tableau 4.
Tableau 4 : Constantes d’équilibre (L.mol-1) du PCP avec les CD (1 : 1) pour trois forces ioniques différentes. L’incertitude est d’environ 5 %. T= 22 ± 2 °C.
KBCD KHPCD KMCD KCMCD Kow*
Eau pure 433 301 510 501 102,56
CaCl2 (0,01 mol.L-1) 318 216 420 410 102,02
CaCl2 (0,1 mol.L-1) 203 100 315 312 100,51
* Valeurs déduites de l’équation : Kow = 3,386 – 0,599(±0,076) Sw (Niimi, 1991).
0
2
4
6
8
10
12
14
16
0 0.01 0.02 0.03 0.04 0.05
[HPCD] mol.L-1
Solu
bilit
é re
lativ
e (S
r)

120
En résumé, il faut retenir que la stabilité des complexes PCP/CD diminue, quelque soit le
type de cyclodextrine, avec l’augmentation de la force ionique de la solution.
Par contre, quelque soit la force ionique, la hiérarchie des cyclodextrines, en terme
d’efficacité de complexation, reste inchangée comparativement à l’eau pure. Le pouvoir
complexant évolue selon l’ordre suivant : CMCD ~ MCD > BCD > HPCD.
D’une manière générale, la solubilité aqueuse des molécules ionisables (chlorophénols)
augmente avec la concentration d’ions en solution. Cet effet est du, en grande partie, à la
formation d’un complexe cation/chlorophénolate qui favorise la solubilisation de l’ion
phénolate dans l’eau (Daughney and Fein 1997 ; Wightman and Fein, 1999).
Dès lors, nos résultats sont tout à fait cohérents. Il parait logique que la constante de
complexation des chlorophénols diminue quand la force ionique augmente comme c’est
le cas pour leur coefficient de partage octanol/eau (Tableau 4) (Westall et al., 1985;
Jafvert et al., 1990; Johnson and Wetsall, 1990).
III.3. Complexation du Naphtalène et du Phénanthrène
Après l’étude du pouvoir complexant des cyclodextrines sur deux « représentants » de la
famille des chlorophénols, nous avons repris un travail similaire sur deux molécules
appartenant aux Hydrocarbures aromatiques polycycliques (HAP) : le Naphtalène (NAP)
et le Phénanthrène (PHE). Pour des raisons de coût et de temps, les expérimentations
n’ont été réalisées qu’avec deux types de CD : la BCD et l’HPCD. Les analyses
comparatives ont été effectuées dans l’eau et dans des solutions de forces ioniques
variables.
Compte tenu du caractère non-ionisable des molécules testées, leur complexation n’a pas
été étudiée en fonction du pH.
III.3.1. Complexation dans l’eau pure
La complexation de NAP et PHE par les deux CD a été suivie, comme pour les
chlorophénols, par la méthode de solubilisation.

121
Les résultats sont exprimés en terme de gain de solubilité (Sr) en fonction de la
concentration en BCD sur la figure 10 et en HPCD sur la figure 11.
Figure 10 : Solubilité relative (Sr) en fonction de la concentration en β-CD pour le NAP (υ) et le PHE (λ).T= 22 ± 2 °C.
Figure 11 : Solubilité relative (Sr) en fonction de la concentration en HPCD pour le NAP (υ) et le PHE (λ).T= 22 ± 2 °C.
La linéarité des courbes Sr = f[CD] obtenues confirme la formation de complexes
d’inclusion 1 :1 dans chacun des cas.
0
10
20
30
40
50
60
70
80
90
100
0 0.005 0.01 0.015 0.02 0.025 0.03 0.035
[HPCD] / mol.L-1
Solu
bilit
é re
lativ
e (S
r)
0
0.5
1
1.5
2
2.5
3
3.5
0 0.0005 0.001 0.0015 0.002
[BCD] mol.L-1
Solu
bilit
é re
lativ
e (S
r)

122
Il est à noter tout de même qu’une précipitation a été observée lors de la solubilisation du
NAP et PHE pour des concentrations en BCD supérieures à 2 g.L-1 (1,8 mmol.L-1). La
figure 10 ne présente que la partie linéaire des courbes de solubilité.
Les constantes de complexation CD/HAP, ont été calculées comme précédemment et sont
regroupées dans le tableau 5.
Tableau 5 : Constantes d’équilibre de complexation (1 : 1) du naphtalène et du
phénanthrène avec deux types de CD dans l’eau pure. L’incertitude est d’environ 5 %. T= 22 ± 2 °C.
KBCD
(L.mol-1)
KHPCD
(L.mol-1)
Kow
Naphtalène (NAP) 471 611 103,36
Phenantrène (PHE) 1226 2749 104,46
Les résultats ci-dessus apportent plusieurs types d’informations :
Tout d’abord, quelque soit la CD utilisée, la constante de complexation est plus élevée
pour le phénanthrène que pour le naphtalène. Ce résultat est en parfaite corrélation avec
l’hydrophobicité relative des deux HAP exprimée par les coefficients de partage
octanol/eau (tableau 4).
Par ailleurs, on peut constater que le pouvoir complexant de l’HPCD vis-à-vis de ces
deux HAP est supérieur à celui de la BCD native. Dans le cas du phénanthrène, il y a un
facteur 2 entre les constantes.
Si l’on s’intéresse maintenant aux augmentations de solubilité de ces HAP en présence de
cyclodextrine, on pourra remarquer sur la figure 10, que l’HPCD à 0,03 mol.L-1 (50 g.L-1)
permet de multiplier respectivement les solubilités du naphtalène et du phénanthrène par
des facteurs 20 et 90.
L’efficacité de la BCD est limitée par des constantes de complexation plus faibles, mais
surtout par sa solubilité aqueuse qui restreint son domaine d’utilisation (en
concentration).

123
III.3.2. Influence de la force ionique
Comme dans le cas du pentachlorophénol, nous avons déterminé les constantes de
complexation CD/HAP dans des solutions aqueuses de CaCl2. Dans cette étude, trois
concentrations ont été testées : 0,001 ; 0,01 et 0,1 mol.L-1.
Le tableau 6, ci dessous, regroupe les résultats obtenus.
Tableau 6 : Constantes d’équilibre de complexation (1 : 1) du naphtalène et phénanthrène avec deux types de CD à plusieurs concentrations de CaCl2 (mol.L-1).
L’incertitude est d’environ 5 %. T= 22 ± 2 °C.
KBCD (L.mol-1) KHPCD (L.mol-1)
CaCl2 (mol.L-1) 0 0,001 0,01 0,1 0 0,001 0,01 0,1
Naphtalène 471 482 478 520 611 590 610 630
Phénanthrène 1226 1205 1220 1280 2749 2730 2766 2840
On peut constater que la force ionique n’a que peu d’effet sur les constantes de
complexation des deux HAP étudiés. Compte tenu des incertitudes sur ces constantes, il
semble tout de même que la stabilité des complexes augmente légèrement avec la force
ionique de la solution.
D’une manière générale, la solubilité aqueuse de composés organiques hydrophobes
neutres tend à diminuer quand la force ionique augmente. Cet effet est du, en grande
partie, à la modification du réseau de liaisons hydrogènes de l’eau à proximité des ions
provenant du sel en solution. Ce changement de l’organisation dynamique des molécules
d’eau a également des conséquences sur les équilibres d’hydratation des solutés présents
pouvant conduire à modifier leur solubilité. Dans notre cas, il semble que la compétition
entre ces divers équilibres favorise légèrement la complexation du naphtalène et du
phénanthrène par les cyclodextrines. En général, l’ajout de sel est utilisé pour favoriser
l’extraction ou le passage de composés organiques hydrophobes (neutres) de la phase
aqueuse vers une phase organique (Valsaraji, 1996).

124
Nous avons tracé sur les figures 12 et 13, ci-après, les solubilités apparentes des deux
HAP en fonction de la concentration en CaCl2. Les résultats concernent des solutions
complexantes à 50 g.L-1 en HPCD.
Figure 12 : Solubilité apparente (Sa) du NAP dans une solution de 50 g.L-1 d’HPCD en fonction de la concentration en CaCl2. T= 22 ± 2 °C.
Figure 13 : Solubilité apparente (Sa) du PHE dans une solution de 50 g.L-1 d’HPCD en fonction de la concentration en CaCl2. T= 22 ± 2 °C.
5
5.2
5.4
5.6
5.8
6
6.2
0 0.02 0.04 0.06 0.08 0.1 0.12
Concentration en CaCl2 / mol.L-1
Sol
ubili
té a
ppar
ente
/ m
mol
.L-1
0.5
0.51
0.52
0.53
0.54
0.55
0.56
0.57
0 0.02 0.04 0.06 0.08 0.1 0.12
Concentration en CaCl2 / mol.L-1
Solu
bilit
é ap
pare
nte
/ mm
ol.L
-1

125
On peut remarquer dans les deux cas que la force ionique influence assez peu la quantité
totale de NAP et PHE solubilisée.
Le même constat a été fait par Wang and Brusseau (1995). Ces auteurs ont montré que la
présence de CaCl2 a un effet négligeable sur la solubilisation de l’anthracène par la
CMCD. Les cations et les anions n’interagissent pas, ou peu, avec la cavité de la CD.
Leur présence a donc assez peu d’influence sur les équilibres de complexation des
molécules neutres.
A l’inverse, Bai et al. (2000) et Ko et al. (2000) ont montré que la force ionique peut
jouer un rôle important dans la solubilisation des HAP par les surfactants. Selon eux, ce
phénomène est lié à une modification des propriétés physico-chimiques des micelles des
surfactants.
IV. Bilan
Nous récapitulons ici les différentes informations issues de cette partie :
La stabilité des complexes (1 : 1) des chlorophénols avec les CD dépend largement des
condition de pH et de force ionique. La forme neutre du chlorophénol forme un complexe
plus stable que la forme ionisée. Par contre, malgré sa faible constante de complexation,
c’est la forme ionisée qui est la plus solubilisée dans les solutions de CD. De ce fait, le
traitement de sols contaminés par des chlorophénols devrait être préconisé dans des
conditions neutres ou plutôt basiques.
L’effet de la force ionique sur la solubilisation des molécules organiques par les CD
dépend logiquement de la nature chimique du substrat. Contrairement au PCP (acide
faible), l’effet de la force ionique sur la solubilité aqueuse du NAP ou PHE ainsi que sur
leur complexation avec les CD est faible.
Le complexe β-CD/soluté précipite à une faible concentration en BCD. La solubilité
aqueuse de ce complexe semble dépendre du pH et de la force ionique, mais elle est
surtout conditionnée par la faible solubilité aqueuse de la BCD elle même.

126
D’une manière générale, les CD modifiées sont plus efficaces que la forme native pour
solubiliser les polluants organiques. A 5 % de HPCD, les solubilités du PCP, du NAP et
du PHE dans l’eau sont multipliées respectivement par un facteur 20, 30 et 90. A
pH > pKa, quand le PCP est majoritairement sous forme ionisée, et quelque soit la force
ionique, on peut observer une hiérarchisation des capacités de solubilisation des CD:
MCD > BCD > HPCD. Il n’y a qu’à pH 3 où le PCP est neutre que cette tendance est
modifiée : MCD > HPCD > BCD.
Il ressort de l’ensemble de ces résultats qu’il est nécessaire lors des études de
solubilisation et de mobilisation de polluants organiques par des CD d’optimiser le type
de CD et sa concentration au vu des polluants cibles. De plus, suivant la nature chimique
du polluant, il sera aussi indispensable de faire intervenir des paramètres tels que le pH et
la force ionique, en particulier pour les polluants ionisables (acides faibles ou bases
faibles).

127
Partie II : Traitement des effluents : Dégradation du complexe Cyclodextrine/Pentachlorophénol
I. Introduction Dans le cas d’un traitement de sols par extraction des substances polluantes, les procédés
utilisés ont pour objectif de mobiliser les contaminants dans une phase liquide extractible.
Il est donc impératif de prévoir un traitement des effluents générés.
Dans cet objectif, nous avons testé, au travers de plusieurs expériences, un traitement de
dégradation photocatalytique du pentachlorophénol en présence de cyclodextrine. Ce
travail exploratoire doit apporter des éléments quantifiés permettant de valider la méthode
pour un éventuel transfert à l’échelle du terrain. Des expériences complémentaires seront
bien entendu nécessaires pour évaluer ce procédé en présence de sol réel.
Ce travail a été réalisé avec l’équipe de photocatalyse du Laboratoire d’Application de la
Chimie à l’Environnement (LACE) de l’université Claude Bernard (Lyon 1).
Dans la mesure où il s’agit d’études relativement marginales par rapport à l’ossature
générale de la thèse, nous avons choisi d’y inclure un court rappel sur les méthodes les
plus classiques de dégradation des molécules organiques, en particulier complexées.
Il nous est apparu opportun de les présenter dans ce chapitre qui relate des travaux de
laboratoire sur des solutions « modèles » dont la composition est parfaitement connue.
II. Rappel sur les méthodes de dégradation des molécules
organiques complexées Les solutions extractantes utilisées pour le traitement de sols par lavage et extraction des
polluants peuvent être de natures très différentes. Cela implique une adaptation des
techniques de décontamination des effluents selon le contexte polluant/sol/agent
extractant.

128
Si l’on s’intéresse uniquement aux effluents liquides contenant les polluants extraits,
l’agent extractant et, en général, des constituants du sol, deux approches peuvent être
choisies :
La première est de considérer les effluents d’extraction comme des eaux contaminées. On
peut alors appliquer des méthodes classiques de traitement utilisant des procédés
biologiques ou d’Oxydations Avancés comme l’ozonisation, la photodégradation, le
photo-Fenton ou l’électro-Fenton et la photocatalyse hétérogène (TiO2).
La seconde est de retraiter les effluents pour régénérer les agents complexants en vue de
leur réutilisation. Cette approche a, bien entendu, des intérêts économiques et
environnementaux. Plusieurs scénarios sont envisageables pour décomposer les
complexes extraits. On peut citer les voies chimiques comme l’utilisation de cosolvants
ou la modification du pH, etc. Il est également possible de jouer sur des paramètres
physiques : la baisse de pression pour extraire les composés volatils, la modification de la
température, etc.
Nous citerons ci-dessous quelques exemples d’applications de procédés correspondant à
la première approche menés à l’échelle laboratoire dans lesquelles l’agent solubilisant
était une cyclodextrine.
II.1. Ozonisation Une équipe japonaise a étudié la solubilité du 4-nonylphénol (NP) dans des solutions de
HPCD et la dégradation par l’ozone du complexe NP/HPCD dans un milieu acide
(Kawasaki et al., 2001). Elle a montré que la dégradation augmente avec le temps
d’ozonisation. Ainsi, 80% du complexe NP/HPCD a été dégradé après 60 minutes
d’ozonisation. En revanche, la minéralisation du complexe n’a pas été étudiée et les
métabolites de dégradation du NP n’ont pas été identifiés.

129
II.2. Réduction par le fer (0) L’étude de la déchlorination du tétrachloroéthylène par le fer (0) dans des solutions
aqueuses d’HPCD a montré que la réduction du solvant chloré est inhibée par la présence
de CD (Bizzigotti et al., 1997). En effet, un faible pourcentage de dégradation a été
observé lorsque la cyclodextrine est présente à une forte concentration.
II.3. Photochimie L’influence des CD sur la dégradation de pesticides a été étudiée par une équipe
japonaise dans l’eau pure (Kamiya et al., 1994), en présence d’acides humiques (Kamiya
et al., 2001) et à différents pH (Ishiwata et Kamiya, 1999).
Ces travaux montrent que la complexation peut avoir des effets inverses sur la
photodégradation des pesticides selon la nature du polluant, le type de CD utilisé, et le
contexte chimique du milieu.
Ainsi, la photodégradation du parathion dans l’eau est inhibée en présence de CD alors
que celle du paraxon est favorisée. En milieu neutre, la dégradation du diazinon et du
chloropyrifos est remarquablement améliorée en présence d’α-CD ou de β-CD mais est
inhibée par la γ-CD. Par contre, des résultats opposés ont été obtenus pour les mêmes
complexes en milieu basique. Enfin, d’après ces auteurs, la présence d’acides humiques
augmente le taux de photodégradation des pesticides organophosphorés en présence de
CD.
Ce bref résumé des travaux publiés sur ce sujet permet de constater que chaque cas est
spécifique selon la famille de polluants considérée et le contexte chimique.
II.4. Dégradation par le réactif de Fenton Il s’agit d’une méthode de dégradation des molécules organiques par des radicaux
hydroxyles libres générés par le réactif de Fenton (peroxyde d’hydrogène + fer (II)).
L’étude de la dégradation du phénol et du pyrène en présence de β-CD et de CMCD dans
un milieu acide (Lindsey et al., 2003) a permis de constater une amélioration de la
cinétique de dégradation des polluants en raison de la formation d’un complexe ternaire
cyclodextrine/Fe2+/polluant. En revanche, la minéralisation des polluants n’a pas été
étudiée et le schéma réactionnel n’a pas été décrit.

130
Nous n’avons pas trouvé, dans la littérature, de travaux portant sur la dégradation par
photocatalyse hétérogène (TiO2) de polluants organiques complexés. Nous développerons
ci-après les grandes lignes de cette méthode que nous avons utilisée.
III. La photocatalyse hétérogène
III.1. Généralités Les Procédés d’Oxydation Avancés (POAs) sont de nouvelles techniques de traitement en
cours de développement. Ces techniques sont des alternatives très intéressantes pour la
dégradation de polluants organiques non biodégradables. Elles sont beaucoup plus
efficaces que les techniques habituelles de floculations, précipitation, adsorption sur
charbon activé ou osmose inverse. Le traitement chimique par les POAs peut conduire à
la minéralisation complète des polluants en CO2, et dans le cas de composés halogénés à
la formation d’ions halogénures.
L’équation générale de la réaction qui se produit au cours des POAs peut s’écrire :
Polluants organiques + O2 ⇒ CO2 + H2O + acides minéraux
Les POAs sont basées sur la propriété de génération et d’utilisation de radicaux
hydroxyles comme oxydant primaire pour la dégradation de polluants organiques. Les
POAs, tels que les systèmes U.V.-peroxyde, ozone ou processus photo-Fenton ont
largement démontré leur efficacité dans l’oxydation de composés organiques. Trois autres
POAs (la photocatalyse, la sonolyse et la radiolyse gamma) ont émergé ces dernières
années.
Le concept de la dégradation photocatalytique est simple. Un semi conducteur (SC)
solide et stable est irradié pour stimuler des réactions à l’interface solide/liquide. La
photocatalyse hétérogène implique des photoréactions qui se produisent à la surface du

131
catalyseur. En principe, le catalyseur peut être réutilisé plusieurs fois (Ollis et al., 1993 ;
Mills et Le Hunte, 1997 ; Brezova et Blazkova, 1997 ;Hermann, 1999).
III.2. Le dioxyde de titane
L’oxyde de titane TiO2 est un semi-conducteur qui existe sous différentes formes
cristallographiques : le rutile, l’anatase, la brookite, et un grand nombre de phases
obtenues sous hautes pressions. Deux formes cristallines de TiO2 ont une activité
photocatalytique, le rutile 21 et l’anatase qui est la forme la plus active (Mills et Le
Hunte, 1997).
Le processus photocatalytique repose sur l’excitation de TiO2 par un rayonnement
lumineux de longueur d’onde inférieur à 400 nm. Un électron passe de la bande de
valence à la bande de conduction, créant un site d’oxydation (un trou h+) et un site de
réduction (un électron e-).
Le processus général de la photocatalyse peut être résumé de la façon suivante :
hν + (SC) e- + h+
Les trous h+ réagissent avec les donneurs d’électrons tels que l’eau, les anions OH- et les
produits organiques R adsorbés à la surface du semi-conducteur pour former des radicaux
hydroxyles et R°. Les électrons réagissent avec des accepteurs d’électrons tels que le
dioxygène pour former des radicaux superoxydes.
Les espèces formées sont fortement oxydantes et permettent la dégradation des molécules
organiques (Baudin et al., 2000, Vulliet et al., 2002 et 2003).
III.3. Facteurs influençant la photocatalyse hétérogène Les différents facteurs influençant la dégradation photocatalytique sont, d’une part les
facteurs physiques tels que le flux lumineux, le champ quantique et la température, et
d’autre part des facteurs chimiques tels que la quantité d’oxygène dissous, la nature et la
concentration du catalyseur, la concentration initiale du polluant et les ions en solution
(Ollis et al., 1993 ; Hermann, 1999).

132
Nous détaillerons uniquement l’influence du pH puisque c’est le paramètre principal qui
différencie les expériences que nous avons réalisées.
III.3.1. Influence du pH
Le pH de la solution affecte énormément l’efficacité du catalyseur TiO2. Il peut avoir une
influence sur sa charge de surface et la taille des agrégats. Le pH pour lequel la charge de
surface de l’oxyde est nulle s’appelle Point de Charge Zéro (pHPZC). Il est de 6,5 environ
pour le TiO2. La surface de l’oxyde est chargée positivement aux pH inférieurs au point
de charge zéro et négativement aux pH supérieurs comme le montre les équations ci-
dessous :
TiOH + H+ TiOH2+ pH < 6,5
TiOH TiO- + H+ pH > 6,5
Les constantes d’équilibre de ces réactions sont données respectivement par pKTiOH2+ =
2,4 et pKTiOH = 8 (Mills et Le Hunte, 1997).
La spéciation des espèces en fonction du pH est décrite dans le tableau 7 ci-dessous.
Tableau 7 : Pourcentages des espèces en fonction du pH. Espèces Pourcentage Domaine de pH
TiOH ≥ 80% 3 < pH < 10
TiO- ≥ 20% pH >10
TiOH2+ ≥ 20% pH < 3
Le pH de la solution peut affecter la forme moléculaire d’un substrat ionisable en
fonction de son pKa. Or, lorsqu’à un pH donné, les molécules du substrat et la surface de
TiO2 ont des charges opposées, les interactions électrostatiques augmentent, ce qui
favorise l’adsorption. Ainsi, la quantité de polluant adsorbé sur la surface de TiO2 peut
varier avec le pH. Selon Hermann et al. (1999), la vitesse de dégradation est
proportionnelle à la quantité de soluté adsorbé sur la surface (Qads). Le pH a une

133
influence plus ou moins importante (selon la nature du substrat) sur la dégradation
photocatalytique.
IV. Matériels et Méthodes IV.1 Photoréacteur Le photoréacteur utilisé dans cette étude est un réacteur de laboratoire en verre pyrex
d’une capacité maximale de 50 mL. Il est cylindrique et muni d’une double paroi qui
laisse passer les rayonnements de longueur d’onde supérieure à 300 nm. La surface de
contact avec l’air est de 11 cm2. Il est placé au dessus d’une lampe à mercure Hg (HPK
125 W Philips) (300 – 400 nm), muni d’une grille pour atténuer, si nécessaire, l’intensité
de la lampe afin de diminuer la vitesse de dégradation (figure 14).
Figure 14 : Photoréacteur
Le réacteur est maintenu à une température constante (T = 19 °C) pendant toute la durée
de l’expérience grâce à une circulation d’eau thermostatée dans sa double paroi (figure
1). Le mélange, solution polluante/TiO2, placé dans le réacteur est homogénéisé à l’aide
d’un agitateur magnétique. Le nombre de photons absorbables par le TiO2 dans la cellule
Agitateur magnétique
Lampe UV
H2O 19 °CBarreau magnétique
Système de refroidissement
(par circulation d’eau)
Photoréacteur

134
d’irradiation a été déterminé par Actinométrie. Il est de l’ordre de
7,5 1016 photons.cm 2 s 1. Le TiO2 utilisé dans ce photoréacteur est de l’oxyde de titane
Degussa P-25 (80 % anatase et 20 % rutile). Il possède une surface spécifique de l’ordre
de 50 m2.g-1, ce qui correspond à des particules d’environ 30 nm de diamètre.
IV.2. Expériences d’irradiation et de dégradation IV.2.1. Suivi de la dégradation du PCP seul dans l’eau
Une solution « mère » de PCP à 0,2 mmol.L-1 a été préparée en dissolvant le composé
solide dans une solution aqueuse de soude (0,001 mol.L-1). Cette même solution de soude
a été utilisée pour ajuster le pH après la dissolution complète du PCP. Deux valeurs de
pH ont été étudiées : un pH 7, proche du pH naturel d’un sol et voisin également du point
de charge nulle de TiO2, et un pH 11 fortement basique.
La concentration de la solution mère est bien inférieure à la limite de solubilité du PCP
dans ces conditions de pH.
30 ml de la solution mère de PCP et 1,7 g.L-1 de TiO2 sont introduits dans le
photoréacteur. Après 30 minutes d’agitation et sans irradier (t = 0 minute d’irradiation),
un prélèvement est effectué grâce à une seringue équipée d’une aiguille en acier. Il est
utilisé pour déterminer la quantité de PCP restant à l’équilibre en solution après son
adsorption sur les particules de TiO2. La lampe est ensuite allumée et préchauffée
pendant 10 minutes environ.
Après cette période, des échantillons de solution sont prélevés successivement dans le
réacteur pour suivre l’évolution de la concentration de PCP en fonction du temps
d’irradiation. La somme totale des volumes prélevés ne doit pas dépasser 10% du volume
initial. Les échantillons sont filtrés sur des filtres en PVDF (Millipores, 0,45 µm - 4mm,)
et analysés par Chromatographie Liquide Haute Performance (HPLC) dans le but de
déterminer la concentration en PCP. La filtration n’engendre pas de pertes, cela a été
vérifié par HPLC et par Spectrophotomètre UV. Le pH est contrôlé au début et à la fin de
chaque expérience.

135
IV.2.2. Suivi de la dégradation du PCP en mélange avec la CD
Pour chaque type de CD (BCD, HPCD ou MCD), une solution contenant 2 mmol de CD
par litre a été préparée en dissolvant la quantité nécessaire de CD dans 100 ml de solution
mère de PCP à 0,2 mmol.L-1. 30 ml de chaque solution (PCP + CD) et 1,7 g.L-1 de TiO2
ont été utilisés pour l’expérience d’irradiation dans le photoréacteur.
A chaque prélèvement, un volume de 0,6 mL est soutiré puis filtré de la même manière
que précédemment afin de suivre la concentration en PCP par HPLC.
IV.3. Analyses des solutions Nous avons suivi la concentration du PCP durant la dégradation photocatalytique par
Chromatographie Liquide à Haute Performance (HPLC).
L’appareillage (Waters) est équipé notamment :
d’un détecteur UV-visible équipé d’une lampe à Deutérium.
d’une colonne SUPELCOSIL LC-18 de 250 mm de longueur et de 4 mm de diamètre.
Les conditions de l’analyse sont les suivantes :
Phase éluante constituée d’un mélange (75/25), acétonitrile / eau acidifiée à pH 2 par
l’ajout d’acide acétique glacial (99 % pureté).
débit d’élution : 1 ml/min
détection à 238 nm
température ambiante
L’injection de 80 µl de solution à analyser se fait automatiquement, les échantillons étant
placés dans un carrousel.
La concentration en ions chlorures relargués durant la dégradation photocatalytique du
pentachlorophénol, a été mesurée par chromatographie ionique sur un appareil Dionex
DX320, équipé d’un détecteur de conductivité et d’une colonne d’échange anionique
(IonPack AS17-Dionex). Le volume injecté est de 15 µL et la phase mobile est une

136
solution de KOH avec un gradient de concentration (1 à 35 mmol L-1) et un débit constant
de 1,9 mL min-1.
L’évolution de la concentration en CD durant la photocatalyse a été suivie par une
méthode fluorimètrique (Cf. paragraphe V.1., chapitre III). Un spectrofluorimètre
KONTRON, SFM 25 a été utilisé pour ces mesures.
Compte tenu de son principe, cette méthode fluorimètrique ne peut pas différencier les
molécules dégradées de celles qui sont intactes tant que la cavité de la CD n’est pas
ouverte ou détruite.
La reproductibilité de ces mesures (concentration en PCP, en CD et en chlorure) est
d’environ 4 %.
V. Résultats et discussion
V.1. Effet du type de CD sur la dégradation photocatalytique du
pentachlorophénol Quatre solutions aqueuses contenant respectivement 0,2 mmol.L-1 de PCP dans l’eau ou
dans des solutions à 2 mmol.L-1 de BCD, d’HPCD ou de MCD ont été irradiées de la
même façon. Le pH des solutions a été ajusté à 7. La dégradation du PCP dans l’eau et
dans les solutions de CD est présentée sur la figure 15 en fonction de la durée de
l’irradiation.

137
Figure 15 : Evolution de la concentration en PCP en fonction du temps d’irradiation. PCP seul et en présence de BCD, d’HPCD ou de MCD à 2 mmol.L-1.
D’après ces résultats, nous pouvons constater que, quelque soit le type de CD, l’allure des
courbes est identique. Par contre la dégradation du PCP non complexé dans l’eau est plus
rapide et plus complète après 30 min.
Les pourcentages de dégradation après 30 minutes d’irradiation du PCP seul et du PCP en
présence de CD sont respectivement de 93 % et 46 %. La présence de 2 mmol.L-1 de CD
en solution (quelque soit le type de CD) a divisé par 2 le pourcentage de dégradation du
PCP (après 30 minutes d’irradiation).
Cet effet inhibitif est probablement du à la présence, dans la solution, d’une forte charge
organique provenant des molécules de CD. Cet effet, qui est similaire pour toutes les CD
utilisées, semble être indépendant du pouvoir de complexation de chaque CD puisque
nous avons montré dans la première partie que les constantes de complexation du PCP
avec les 3 CD étudiées sont nettement différentes.
Les molécules de CD apparaissent alors comme un agent compétiteur vis-à-vis des
radicaux hydroxyles. Elles semblent subir une dégradation en même temps que le PCP.
Les expériences décrites dans le paragraphe suivant permettent de vérifier cette
hypothèse.
0
0.05
0.1
0.15
0.2
0.25
0 5 10 15 20 25 30 35Temps d'irradiation / min
Con
cent
ratio
n en
PC
P / m
mol
.L-1
PCPPCP+BCDPCP+HPCDPCP+MCD
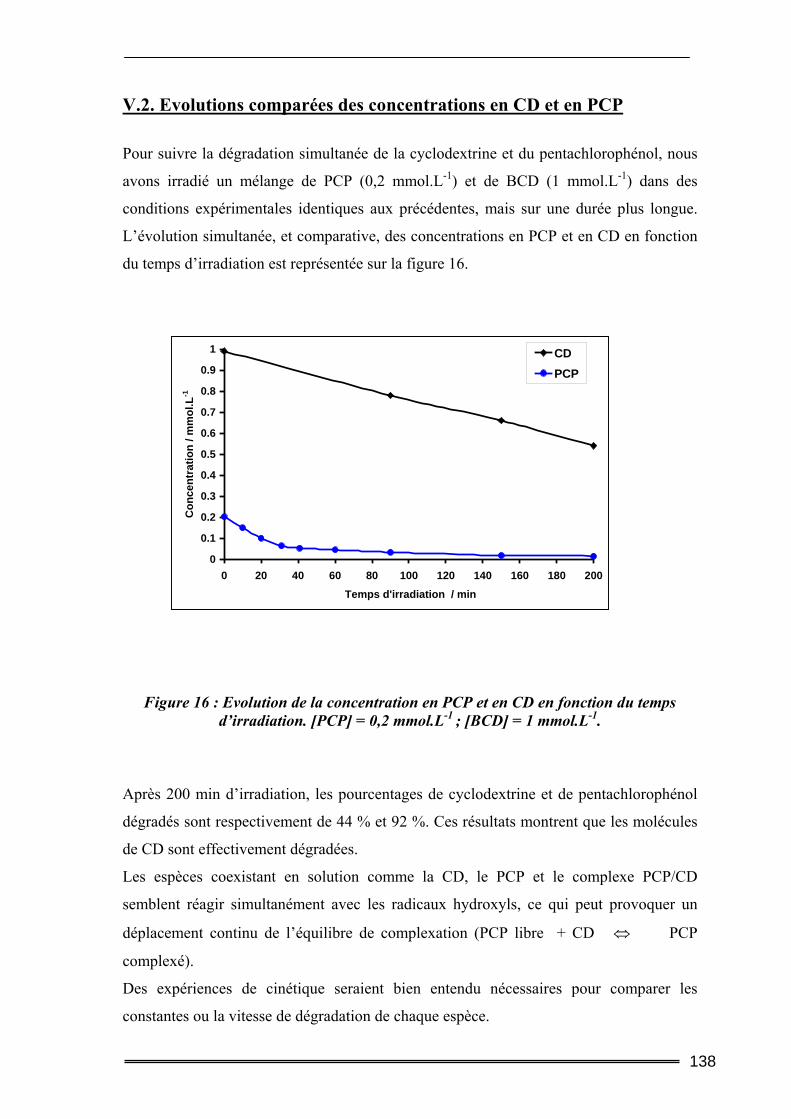
138
V.2. Evolutions comparées des concentrations en CD et en PCP Pour suivre la dégradation simultanée de la cyclodextrine et du pentachlorophénol, nous
avons irradié un mélange de PCP (0,2 mmol.L-1) et de BCD (1 mmol.L-1) dans des
conditions expérimentales identiques aux précédentes, mais sur une durée plus longue.
L’évolution simultanée, et comparative, des concentrations en PCP et en CD en fonction
du temps d’irradiation est représentée sur la figure 16.
Figure 16 : Evolution de la concentration en PCP et en CD en fonction du temps d’irradiation. [PCP] = 0,2 mmol.L-1 ; [BCD] = 1 mmol.L-1.
Après 200 min d’irradiation, les pourcentages de cyclodextrine et de pentachlorophénol
dégradés sont respectivement de 44 % et 92 %. Ces résultats montrent que les molécules
de CD sont effectivement dégradées.
Les espèces coexistant en solution comme la CD, le PCP et le complexe PCP/CD
semblent réagir simultanément avec les radicaux hydroxyls, ce qui peut provoquer un
déplacement continu de l’équilibre de complexation (PCP libre + CD ⇔ PCP
complexé).
Des expériences de cinétique seraient bien entendu nécessaires pour comparer les
constantes ou la vitesse de dégradation de chaque espèce.
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1
0 20 40 60 80 100 120 140 160 180 200
Temps d'irradiation / min
Con
cent
ratio
n / m
mol
.L-1
CDPCP
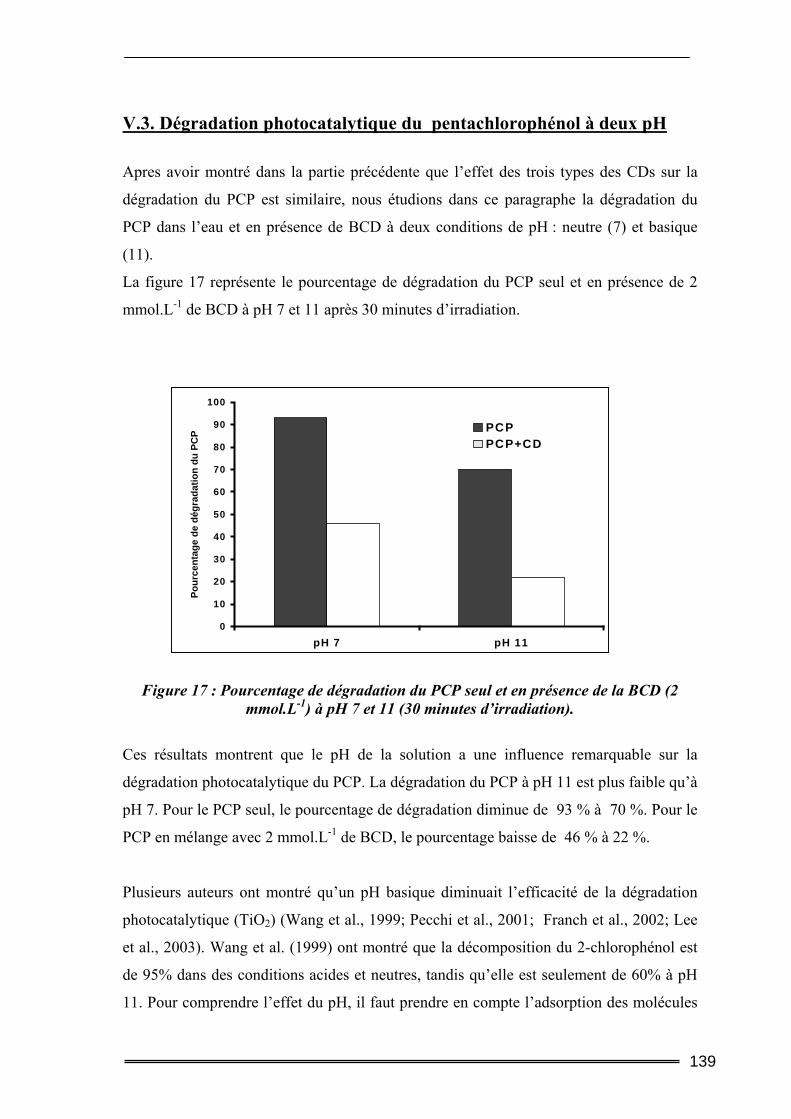
139
V.3. Dégradation photocatalytique du pentachlorophénol à deux pH Apres avoir montré dans la partie précédente que l’effet des trois types des CDs sur la
dégradation du PCP est similaire, nous étudions dans ce paragraphe la dégradation du
PCP dans l’eau et en présence de BCD à deux conditions de pH : neutre (7) et basique
(11).
La figure 17 représente le pourcentage de dégradation du PCP seul et en présence de 2
mmol.L-1 de BCD à pH 7 et 11 après 30 minutes d’irradiation.
Figure 17 : Pourcentage de dégradation du PCP seul et en présence de la BCD (2 mmol.L-1) à pH 7 et 11 (30 minutes d’irradiation).
Ces résultats montrent que le pH de la solution a une influence remarquable sur la
dégradation photocatalytique du PCP. La dégradation du PCP à pH 11 est plus faible qu’à
pH 7. Pour le PCP seul, le pourcentage de dégradation diminue de 93 % à 70 %. Pour le
PCP en mélange avec 2 mmol.L-1 de BCD, le pourcentage baisse de 46 % à 22 %.
Plusieurs auteurs ont montré qu’un pH basique diminuait l’efficacité de la dégradation
photocatalytique (TiO2) (Wang et al., 1999; Pecchi et al., 2001; Franch et al., 2002; Lee
et al., 2003). Wang et al. (1999) ont montré que la décomposition du 2-chlorophénol est
de 95% dans des conditions acides et neutres, tandis qu’elle est seulement de 60% à pH
11. Pour comprendre l’effet du pH, il faut prendre en compte l’adsorption des molécules
0
10
20
30
40
50
60
70
80
90
100
pH 7 pH 11
Pour
cent
age
de d
égra
datio
n du
PC
P PCPPCP+CD

140
de PCP sur les particules de TiO2. Le point de charge nulle (pHpzc) du TiO2 est 6,5. Pour
des valeurs de pH plus élevées, la surface de l’oxyde est chargée négativement (TiO-).
D’autre part, plus de 99 % du PCP (pKa 4,75) est ionisé à pH 11. De ce fait, à ce pH, la
majorité des sites surfaciques de l’adsorbant catalyseur TiO2 et des molécules de PCP
sont chargés négativement. Il y a probablement des répulsions électrostatiques entre les
particules de TiO2 et les molécules ionisées de PCP. Il est donc normal d’obtenir une
baisse de l’adsorption du PCP sur le TiO2, ce qui provoque une diminution de sa vitesse
de dégradation (Hermann et al., 1999).
En tenant compte de ces résultats, le pH a été ajusté à 7 pour la suite des expériences.
V.4. Effet de la concentration en β-CD sur la dégradation
photocatalytique du PCP Pour étudier l’effet de la concentration du complexant sur la dégradation
photocatalytique, des solutions de PCP et de BCD à différentes concentrations sont
irradiées à pH 7.
L’évolution de la concentration du PCP en fonction du temps d’irradiation aux différentes
concentration de BCD est représentée sur la figure 18. La variation de la concentration en
ions chlorure relargués par la décomposition du PCP en fonction du temps d’irradiation a
également été suivie et portée sur la figure 19.
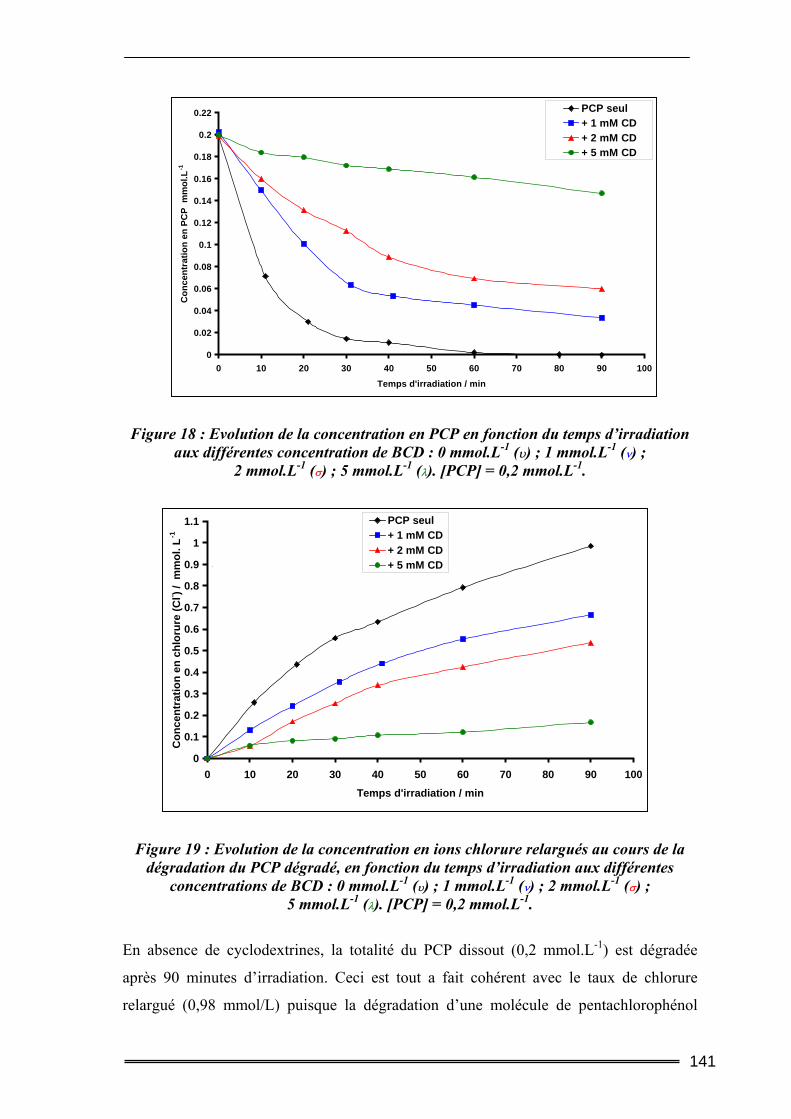
141
Figure 18 : Evolution de la concentration en PCP en fonction du temps d’irradiation aux différentes concentration de BCD : 0 mmol.L-1 (υ) ; 1 mmol.L-1 (ν) ;
2 mmol.L-1 (σ) ; 5 mmol.L-1 (λ). [PCP] = 0,2 mmol.L-1.
Figure 19 : Evolution de la concentration en ions chlorure relargués au cours de la dégradation du PCP dégradé, en fonction du temps d’irradiation aux différentes
concentrations de BCD : 0 mmol.L-1 (υ) ; 1 mmol.L-1 (ν) ; 2 mmol.L-1 (σ) ; 5 mmol.L-1 (λ). [PCP] = 0,2 mmol.L-1.
En absence de cyclodextrines, la totalité du PCP dissout (0,2 mmol.L-1) est dégradée
après 90 minutes d’irradiation. Ceci est tout a fait cohérent avec le taux de chlorure
relargué (0,98 mmol/L) puisque la dégradation d’une molécule de pentachlorophénol
0
0.02
0.04
0.06
0.08
0.1
0.12
0.14
0.16
0.18
0.2
0.22
0 10 20 30 40 50 60 70 80 90 100Temps d'irradiation / min
Con
cent
ratio
n en
PC
P m
mol
.L-1
PCP seul+ 1 mM CD+ 2 mM CD+ 5 mM CD
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1
1.1
0 10 20 30 40 50 60 70 80 90 100
Temps d'irradiation / min
Con
cent
ratio
n en
chl
orur
e (C
l- ) / m
mol
. L-1
PCP seul+ 1 mM CD+ 2 mM CD+ 5 mM CD
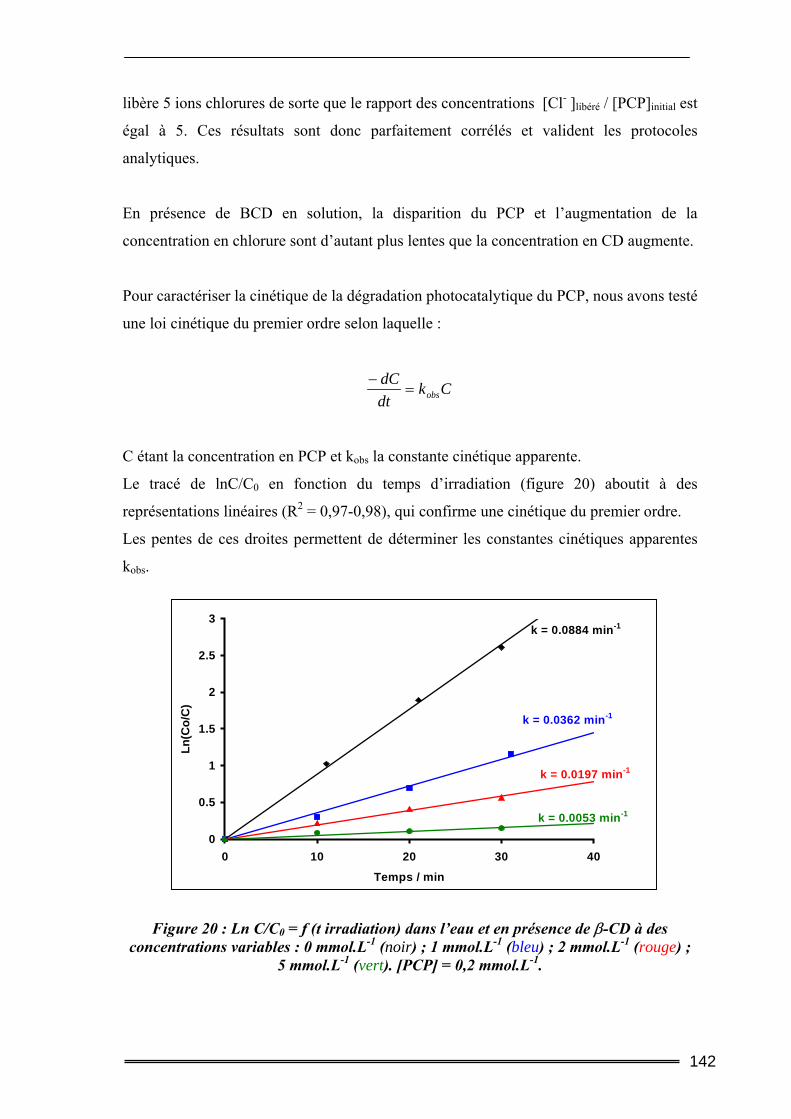
142
libère 5 ions chlorures de sorte que le rapport des concentrations [Cl- ]libéré / [PCP]initial est
égal à 5. Ces résultats sont donc parfaitement corrélés et valident les protocoles
analytiques.
En présence de BCD en solution, la disparition du PCP et l’augmentation de la
concentration en chlorure sont d’autant plus lentes que la concentration en CD augmente.
Pour caractériser la cinétique de la dégradation photocatalytique du PCP, nous avons testé
une loi cinétique du premier ordre selon laquelle :
CkdtdC
obs=−
C étant la concentration en PCP et kobs la constante cinétique apparente.
Le tracé de lnC/C0 en fonction du temps d’irradiation (figure 20) aboutit à des
représentations linéaires (R2 = 0,97-0,98), qui confirme une cinétique du premier ordre.
Les pentes de ces droites permettent de déterminer les constantes cinétiques apparentes
kobs.
Figure 20 : Ln C/C0 = f (t irradiation) dans l’eau et en présence de β-CD à des concentrations variables : 0 mmol.L-1 (noir) ; 1 mmol.L-1 (bleu) ; 2 mmol.L-1 (rouge) ;
5 mmol.L-1 (vert). [PCP] = 0,2 mmol.L-1.
k = 0.0884 min-1
k = 0.0362 min-1
k = 0.0197 min-1
k = 0.0053 min-1
0
0.5
1
1.5
2
2.5
3
0 10 20 30 40
Temps / min
Ln(C
o/C
)
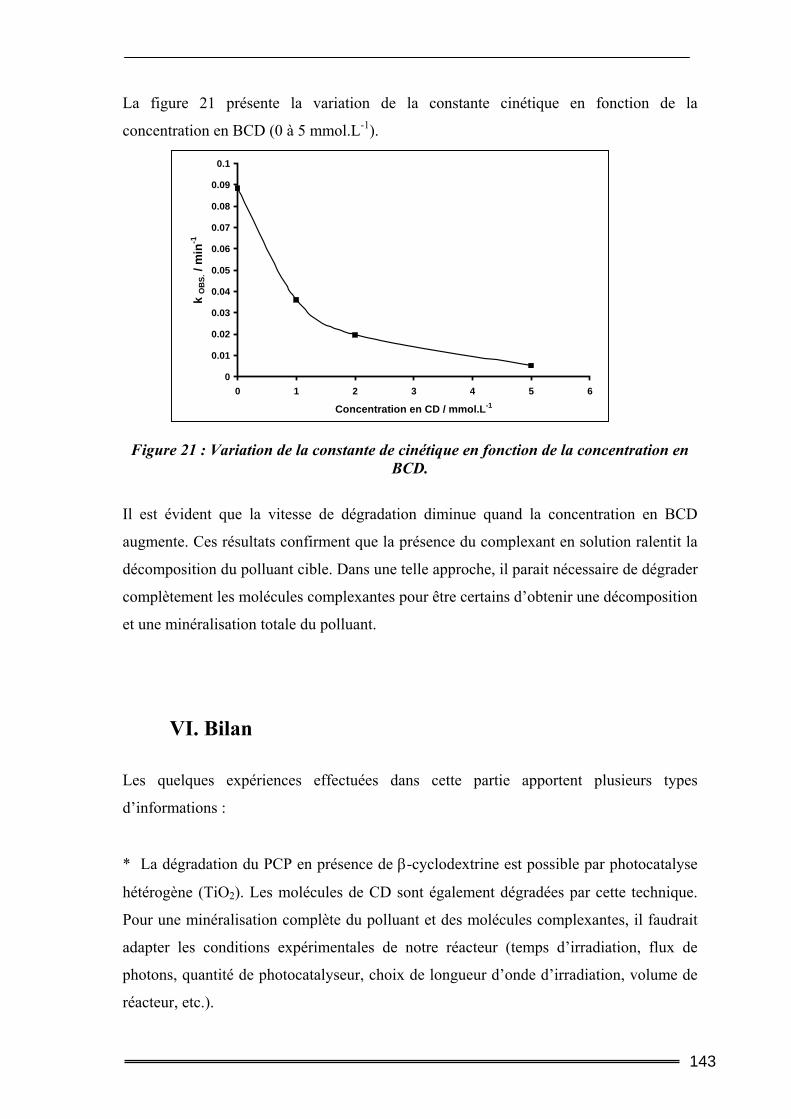
143
La figure 21 présente la variation de la constante cinétique en fonction de la
concentration en BCD (0 à 5 mmol.L-1).
Figure 21 : Variation de la constante de cinétique en fonction de la concentration en
BCD.
Il est évident que la vitesse de dégradation diminue quand la concentration en BCD
augmente. Ces résultats confirment que la présence du complexant en solution ralentit la
décomposition du polluant cible. Dans une telle approche, il parait nécessaire de dégrader
complètement les molécules complexantes pour être certains d’obtenir une décomposition
et une minéralisation totale du polluant.
VI. Bilan
Les quelques expériences effectuées dans cette partie apportent plusieurs types
d’informations :
* La dégradation du PCP en présence de β-cyclodextrine est possible par photocatalyse
hétérogène (TiO2). Les molécules de CD sont également dégradées par cette technique.
Pour une minéralisation complète du polluant et des molécules complexantes, il faudrait
adapter les conditions expérimentales de notre réacteur (temps d’irradiation, flux de
photons, quantité de photocatalyseur, choix de longueur d’onde d’irradiation, volume de
réacteur, etc.).
0
0.01
0.02
0.03
0.04
0.05
0.06
0.07
0.08
0.09
0.1
0 1 2 3 4 5 6
Concentration en CD / mmol.L-1
k O
BS.
/ m
in-1

144
* Le pH de la solution semble avoir une influence remarquable sur la dégradation
photocatalytique du PCP dans l’eau. Il intervient sur la solubilisation des acides faibles
par les CD comme nous l’avons montré dans la Partie I. Il s’agit donc d’un paramètre à
prendre en compte et à optimiser selon le type de complexe CD/polluant à dégrader.
* La vitesse de décomposition du PCP varie en sens inverse de la concentration de CD en
solution. Par contre, la solubilisation des polluants dans l’eau augmente avec la
concentration en cyclodextrine. Autrement dit, l’amélioration de la solubilité est
contrebalancée par une chute dans le rendement de dégradation du polluant complexé. Ce
type de comportement a été souvent observé lors de suivis de décontamination en
présence de solubilisants tels que les surfactants ou les CD (Guha and jaffé, 1996 ;
Bizzigotti et al., 1997).
Ces expériences ne sont qu’un début en ce qui concerne la mise au point d’une méthode
de traitement des effluents extraits à grande échelle, et de nombreuses recherches devront
être menées pour parvenir aux résultats escomptés. Malgré tout, et au vu de nos résultats,
il s’agit d’une voie prometteuse et originale.

145
Conclusion Sans reprendre la totalité des bilans expérimentaux détaillés dans ce chapitre, nous allons
plutôt résumer les principales informations obtenues dans le cadre de cette première
étape.
Si l’on replace les résultats par rapport aux objectifs de notre démarche d’étude de
faisabilité d’un traitement de sol, le bilan est plutôt encourageant.
Les β-cyclodextrines testées sur des polluants représentatifs des chlorophénols et des
hydrocarbures aromatiques polycycliques ont un pouvoir complexant intéressant. Les CD
modifiées sont les plus actives pour augmenter la solubilité, donc probablement
l’extraction, des polluants ciblés. Nous avons mis en évidence, ou confirmé, que dans
certains cas, le contexte chimique (pH et force ionique) peut avoir des conséquences
importantes en terme de gain de solubilité. Ce sera donc un (des) paramètre(s) à prendre
en compte et à optimiser.
Ces travaux ont fait l’objet de deux publications dans des revues internationales (Cf.
annexe 5). Bien qu’ils ne préjugent pas totalement du comportement des CD en présence
d’une matrice sol complexe, ils constituent tout de même un préalable positif.
De manière plus marginale, la préoccupation d’un traitement des effluents d’extraction
nous a conduit à tester l’impact de la présence de CD sur la dégradation photocatalytique
des solutions aqueuses de complexes CD/PCP. Là encore, les résultats obtenus, et publiés
(Cf. annexe 5), confirment qu’il s’agit d’une voie de traitement possible, mais qui
nécessitera un travail de recherche, à part entière, sur des effluents d’extraction « réels ».
Apres avoir étudié les interactions CD/polluants, nous détaillerons, conformément à nos
objectifs, les travaux réalisés en présence de sols.

146

147
Chapitre III :
Etude de l’efficacité des CD pour l’extraction de polluants contenus
dans des sols contaminés au laboratoire.

148

149
Chapitre III : Etude de l’efficacité des CD pour l’extraction de polluants contenus dans des sols contaminés au laboratoire.
I. Introduction
Dans le chapitre précèdent, nous avons montré l’aptitude des cyclodextrines à solubiliser
les polluants tels que le pentachlorophénol (PCP), le naphtalène (NAP) et le phénanthrène
(PHE). Nous avons comparer les performances relatives de plusieurs types de CD dans
l’eau. Dans ce chapitre, nous allons tester leurs capacités à extraire les polluants du sol et
déterminer les conditions les plus favorables pour l’extraction en fonction de la nature du
sol (teneur en argile ou en MO). Dans ce but, des sols de référence bien caractérisés ainsi
que certains constituants de sols ont été choisis. Ces matrices sols ont été contaminées au
laboratoire puis des lavages en présence de CD ont été effectués afin d’extraire les
polluants.
L'adsorption et la désorption du PCP, du NAP et du PHE sur ces matrices ont été
étudiées en milieu dispersé et en colonne. Les deux techniques utilisées seront décrites
dans ce chapitre. Notre étude repose sur une démarche expérimentale ayant pour objectif
de déterminer le pourcentage d’extraction ou l’efficacité des CD vis-à-vis de l’extraction
des polluants adsorbés sur le sol.
Les isothermes d’adsorption ont été effectués sur deux sols différents pour chacun des
trois polluants choisis, suivis par des désorptions successives à l’eau et avec des solutions
aqueuses de CD.
La mise au point d’un procédé d’extraction des polluants organiques requiert l’évaluation
des interactions susceptibles de se produire entre les cyclodextrines et les sols en fonction
de la composition de ceux-ci. De plus, une mobilisation des matières organiques
naturelles du sol par les CD peut affecter la structure du sol après traitement. C’est
pourquoi, nous étudions dans la dernière partie de ce chapitre l’adsorption des CD sur
diverses matrices de sol et l’impact que son utilisation peut avoir sur les matières
organiques du sol.
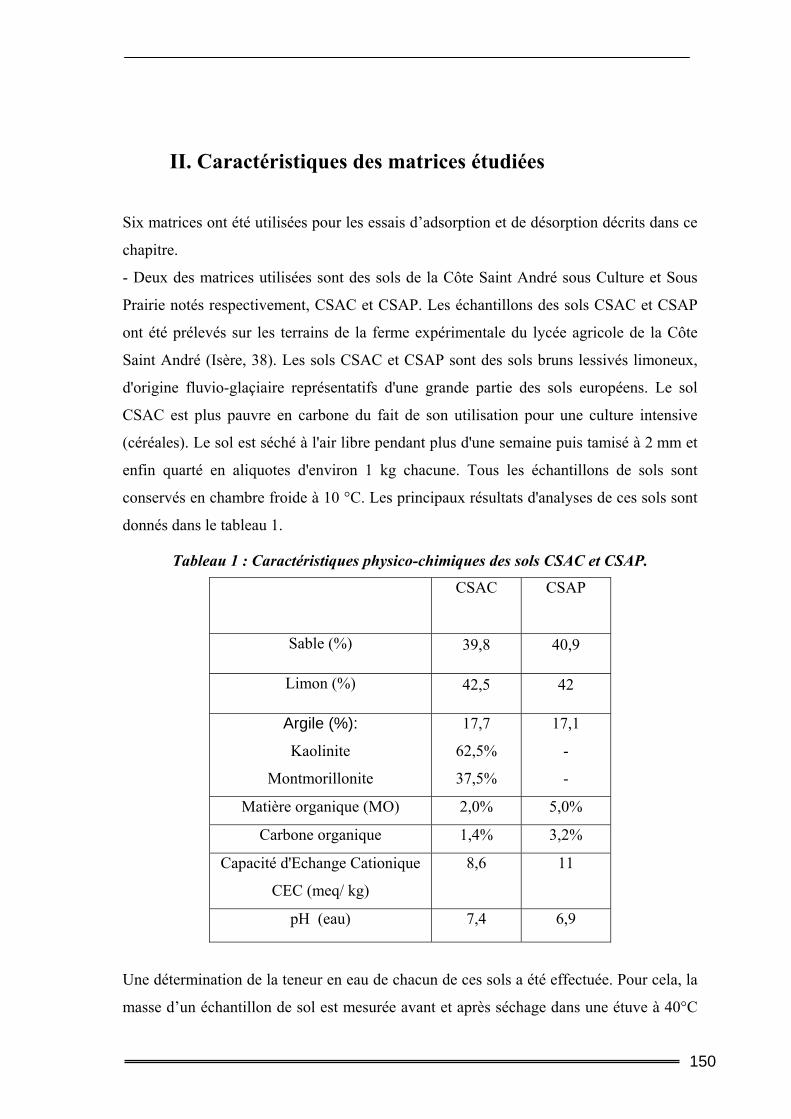
150
II. Caractéristiques des matrices étudiées
Six matrices ont été utilisées pour les essais d’adsorption et de désorption décrits dans ce
chapitre.
- Deux des matrices utilisées sont des sols de la Côte Saint André sous Culture et Sous
Prairie notés respectivement, CSAC et CSAP. Les échantillons des sols CSAC et CSAP
ont été prélevés sur les terrains de la ferme expérimentale du lycée agricole de la Côte
Saint André (Isère, 38). Les sols CSAC et CSAP sont des sols bruns lessivés limoneux,
d'origine fluvio-glaçiaire représentatifs d'une grande partie des sols européens. Le sol
CSAC est plus pauvre en carbone du fait de son utilisation pour une culture intensive
(céréales). Le sol est séché à l'air libre pendant plus d'une semaine puis tamisé à 2 mm et
enfin quarté en aliquotes d'environ 1 kg chacune. Tous les échantillons de sols sont
conservés en chambre froide à 10 °C. Les principaux résultats d'analyses de ces sols sont
donnés dans le tableau 1.
Tableau 1 : Caractéristiques physico-chimiques des sols CSAC et CSAP.
CSAC CSAP
Sable (%) 39,8 40,9
Limon (%) 42,5 42
Argile (%):
Kaolinite
Montmorillonite
17,7
62,5%
37,5%
17,1
-
-
Matière organique (MO) 2,0% 5,0%
Carbone organique 1,4% 3,2%
Capacité d'Echange Cationique
CEC (meq/ kg)
8,6 11
pH (eau) 7,4 6,9
Une détermination de la teneur en eau de chacun de ces sols a été effectuée. Pour cela, la
masse d’un échantillon de sol est mesurée avant et après séchage dans une étuve à 40°C

151
pendant 24 heures. Le taux d’humidité exprimé en pourcentage massique est de 2,1 %
pour le sol CSAC et 4 % pour le sol CSAP.
Ces deux sols sont différençiables par leur contenu en matières organiques (MO), CSAC
(2% MO) et CSAP (5% MO). La sélection s'est effectuée de manière à travailler sur des
matrices naturelles ayant une composition et des propriétés physico-chimiques
relativement distinctes (même teneur en argile, mais, teneur en matière organique
nettement différente) afin de pouvoir évaluer les caractéristiques des sols prépondérantes
sur l'adsorption et la désorption des polluants organiques.
Un mélange argile-limon dit « argile limoneuse » a été aussi utilisé comme matrice
adsorbante. Cette argile (Argile Clarsol STF 30) est une argile bentonite naturelle fournie
par la société CECA / ATO. Elle est constituée principalement de Montmorillonite. Sa
composition en quartz et autres constituants est donnée dans le tableau 2.
Tableau 2 : Composition minéralogique de l'argile Clarsol STF 30
Constituant
principal
Constituants
secondaires
Quartz 23%
Montmorillonite Calcite 7%
Illite 10%
Kaolinite 10%
L'argile a été analysée par diffraction aux rayons X afin d’identifier ses différents
constituants et leur pourcentage approximatif. La proportion importante de particules de
taille supérieure à 5 µm indique la présence de matériau de type limon. La capacité
d'échange cationique (CEC), déterminée selon la méthode à l'acétate d'ammonium, est de
37,2 meq/100 g de matière calcinée à 900°C. Par la méthode de Anne à froid, on mesure
une teneur en carbone organique non négligeable de 0,7% (Bayard, 1997).
Les trois autres supports adsorbants utilisés sont des matrices minérales simples :
Kaolinite (Al4 Si4O10 (OH)8 ) et deux sortes de montmorillonite ((Na,Ca)0.3(Al,Mg)2
Si4O10(OH)2 nH2O) ayant des surfaces spécifiques différentes : MK10 (30-40 m2.g-1) et

152
MKSF (220-240 m2.g-1). Ces matrices ont été fournies par Aldrich sans informations
supplémentaires sur leurs caractéristiques physico-chimiques tels que la CEC et le PCN
(point de charge nulle).
III. Etude de l’adsorption/désorption du
pentachlorophénol III.1. Etude en Batch (milieu dispersé)
L'évaluation de l'adsorption se fait par la technique dite « en batch » qui consiste à mettre
en contact une certaine quantité d'adsorbants (sol ou fraction de sol) avec un volume
donné d'une solution généralement aqueuse d'un polluant organique de concentration
initiale connue. La désorption est ensuite évaluée en mettant en contact la fraction du sol
ayant préalablement adsorbé le polluant (fraction de sol contaminé) avec un volume
donné d'eau ou d’une solution extractante (éluant).
Cette méthode en milieu dispersé est assimilable à la mesure du partage d’un polluant
entre la fraction solide du sol et une phase liquide (généralement aqueuse). Le temps de
contact doit permettre d’atteindre l’équilibre d’adsorption ou de désorption. Cette série
d’expériences doit se dérouler à température constante.
III.1.1. Procédures expérimentales
III.1.1.1. Préparation des solutions Les études d'adsorption du pentachlorophénol ont été réalisées à partir de solutions
aqueuses de PCP à concentrations initiales variables.
Le PCP étant peu soluble dans l'eau pure, la préparation de la solution aqueuse est
réalisée à pH supérieur à son pKa afin d'augmenter la solubilité du produit. Ainsi, pour
l'étude des isothermes d'adsorption/désorption du PCP à l'équilibre, une solution-mère de
PCP est préparée par dissolution du produit dans l'eau tamponnée à pH = 7 (tampon
phosphate). En même temps que le PCP, du chlorure mercurique est ajouté à raison de
400 mg/L de solution pour éviter tout phénomène de biodégradation au cours des essais.
Les solutions sont agitées à l’obscurité à température ambiante pendant au minimum 3
jours. Cette solution mère est utilisée pour les essais réalisés à des concentrations initiales

153
allant de 30 à 500 mg.L-1. Les dilutions successives sont effectuées dans de l'eau
tamponnée à pH 7. Le pH des solutions a été mesuré par un pHmètre WTW® équipé
d’une électrode de verre classique.
III.1.1.2. Cinétique d’adsorption et de désorption
L’étude cinétique permet de connaître le temps de contact nécessaire pour atteindre un
état d’équilibre apparent lors du contact sol/solution en milieu dispersé.
Adsorption L’adsorption du polluant sur les sols a été réalisée dans un premier temps, en vue
d’obtenir un sol contaminé avec une quantité adsorbée connue.
Les cinétiques d’adsorption ont été déterminées à 20°C en mettant en contact le polluant
en solution aqueuse avec une quantité connue du sol (ratio liquide/solide = 3 ; 5 g du sol
dans 15 ml de solution). Tous les tubes contenaient ces mêmes quantités de sol et de
solution à une concentration connue Co. Ces tubes ont été placés sur un agitateur.
L’adsorption a été suivie en prélevant des tubes à intervalles de temps réguliers entre 0 et
48 h. Après centrifugation, le surnageant est prélevé et analysé par HPLC, afin de doser
le soluté resté en solution à l’équilibre, soit Céq.. La quantité adsorbée Qads (masse de
soluté ramenée à la masse d’adsorbant) sera déduite par différence entre Co et Céq.
Chaque essai a été réalisé en triplicat et des « blancs », sans sol, ont permis d’identifier
les éventuelles pertes ou transformations du composé.
Désorption La cinétique de désorption est étudiée sur des échantillons de sol ayant préalablement
adsorbé le polluant organique selon le protocole décrit au paragraphe précédent.
Ces fractions de sols contaminés (même Qads) ont été mises en contact avec des solutions
extractantes, de même concentration (réacteurs en parallèle). Des prélèvements ont été
effectués régulièrement au cours du temps. Le surnageant est analysé pour déterminer la
concentration en polluant extrait. La cinétique de désorption du polluant a été étudiée
comparativement en utilisant l’eau ou des solutions de CD comme extractants. La
désorption du polluant en milieu dispersé est décrite en détail dans le paragraphe suivant.

154
III.1.1.3. Isothermes d'adsorption et de désorption à l'équilibre Les isothermes d’adsorption sont les représentations graphiques de la teneur en polluant
organique fixée en fonction de sa concentration en solution à l’équilibre pour une
température donnée. Il s’agit donc d’un graphe Qads = f(Céq).
Les essais visant à l'établissement des isothermes à l'équilibre ont été réalisés dans des
flacons de 60 ml en verre borosilicaté fermés hermétiquement par des bouchons vissés
avec joints en caoutchouc recouverts d'une couche de Téflon afin d'éviter l'adsorption des
polluants organiques. Pour tous les essais réalisés, le rapport massique R entre la phase
liquide et la phase solide (R = L/S) est toujours de 3. Les 10 g de matériau sont mis en
suspension dans 30 ml de solution de concentration identique pour chaque point d’une
isotherme donnée. Les flacons sont agités verticalement sur un éluteur rotatif placé dans
une chambre thermostatée à 20°C.
Tous les essais sont menés en présence de 400 mg.L-1 de chlorure mercurique et de 0,11
g.L-1 CaCl2 pour éviter la déstructuration du sol. La durée du contact entre la phase solide
et la phase aqueuse dépend du temps nécessaire à l'obtention de l'équilibre d'adsorption.
En se basant sur l’étude cinétique faite pour l’adsorption, la durée de contact choisie entre
la phase solide et la phase aqueuse est de 24 heures afin de permettre largement
l’obtention des équilibres apparents. Pour tous les essais, des témoins sans sol, ni fraction
de sol, sont réalisés dans les mêmes conditions afin d'estimer les pertes éventuelles par
adsorption sur les parois des récipients et par volatilisation.
Lorsque l'adsorption est significative, on réalise des cycles de désorptions. Les phases
liquide et solide sont séparées par centrifugation. La pesée du surnageant obtenu après
centrifugation permet de déduire le volume résiduel de solution présent dans le culot et
donc la quantité de polluant présent dans la solution interstitielle du culot. Le surnageant
est remplacé par un même volume d'eau (ou de solution de la CD à une concentration
définie) pour étudier la désorption. Le volume de solution résiduelle dans le culot est pris
en compte avant de compléter à 30 ml et de commencer une nouvelle extraction. De 3 à 5
cycles de désorption successifs ont ainsi été effectués pour évaluer l’extractibilité des
polluants avec la CD.

155
Les surnageants sont analysés par HPLC afin de connaître la concentration relarguée du
polluant ou la concentration résiduelle du polluant dans le sol.
Les résultats de l'essai sont exprimés en terme de quantité résiduelle adsorbée du polluant
par masse de sol, ou bien en pourcentage d’extraction par rapport à la concentration
initiale en polluant dans le sol.
III.1.1.4. Analyses des solutions Les dosages de pentachlorophénol en solution sont effectués par chromatographie liquide
haute performance (CLHP), phase inverse, en condition isocratique. L'appareillage se
compose d'une monopompe LC-6A Shimadzu et d'un détecteur UV-visible SPD-6A de la
même marque, équipé d'une lampe au deuterium. La cellule optique du détecteur a un
volume de 8 µl. L'injection s'effectue par l'intermédiaire d'une vanne Rhéodyne modèle
7010 possédant une boucle d'injection de 20 µl. L'appareil est équipé d'une précolonne du
type Kromasil C18, de longueur 3 cm. Les analyses sont faites dans les conditions
suivantes :
- Colonne Kromasil C18, de longueur 25 cm, 5 mm d'épaisseur de film et 100 Å de
diamètre des particules,
- Phase éluante constituée d'un mélange méthanol/eau (70/30 % vol), la phase mobile
est acidifiée à pH 2 par l’acide acétique glacial (pureté 99 %).
- Débit d'élution : 1,0 ml.min-1,
- Détection à λ = 254 nm.
III.1.2. Résultats de l’extraction du pentachlorophénol en milieu dispersé
Etant donné que notre objectif principal est d’étudier l’extraction du PCP, nous
présenterons rapidement les résultats d’adsorption et nous commenterons avec des
illustrations la désorption (l’extraction) du PCP.
III.1.2.1. Cinétique d'adsorption
L'adsorption du pentachlorophénol sur les sols CSAC et CSAP a été étudié à 20 °C. Le
pH de la suspension de sol est mesuré à la fin de chaque essai. Le pH à l'équilibre est de
6,8 dans le cas du sol CSAC et 6,5 pour le sol CSAP. Ces pH sont relativement proches

156
du pH initial de la solution mère de PCP, ce qui suggère l'absence de tout phénomène
parasite de précipitation du PCP comme le confirme les résultats des blancs conduits sans
sol.
A l'équilibre (obtenu en 4 h de contact environ), les quantités de PCP adsorbées sur les
sols CSAC et CSAP atteignent respectivement 102 mg.kg-1 et 151 mg.kg-1 de sol sec. Les
quantités adsorbées sont exprimées avec une incertitude d’environ 4 %.
III.1.2.2. Cinétique de désorption L'étude cinétique de la désorption a été réalisée immédiatement après l’adsorption du
PCP sur les sols CSAC ou CSAP. Cette étude a été menée dans le but de déterminer le
temps nécessaire pour atteindre un équilibre apparent, et les concentrations à cet
équilibre. Les solutions aqueuses des CD (10 g.L-1) utilisées pour l’étude de la cinétique,
ont été tamponnées à pH 7 pour éviter la précipitation du PCP.
Les résultats sont illustrés par les courbes C = f(t) présentées dans les figures 1 (sol
CSAC) et 2 (sol CSAP). Les valeurs représentées sont la moyenne des trois replicats. La
reproductibilité indiquée par les barres varie entre 3 et 5 %. Notons que la concentration
initiale en PCP de la solution n'est pas nulle puisqu'il y a, au début de l'essai, dilution de
la solution interstitielle du culot.
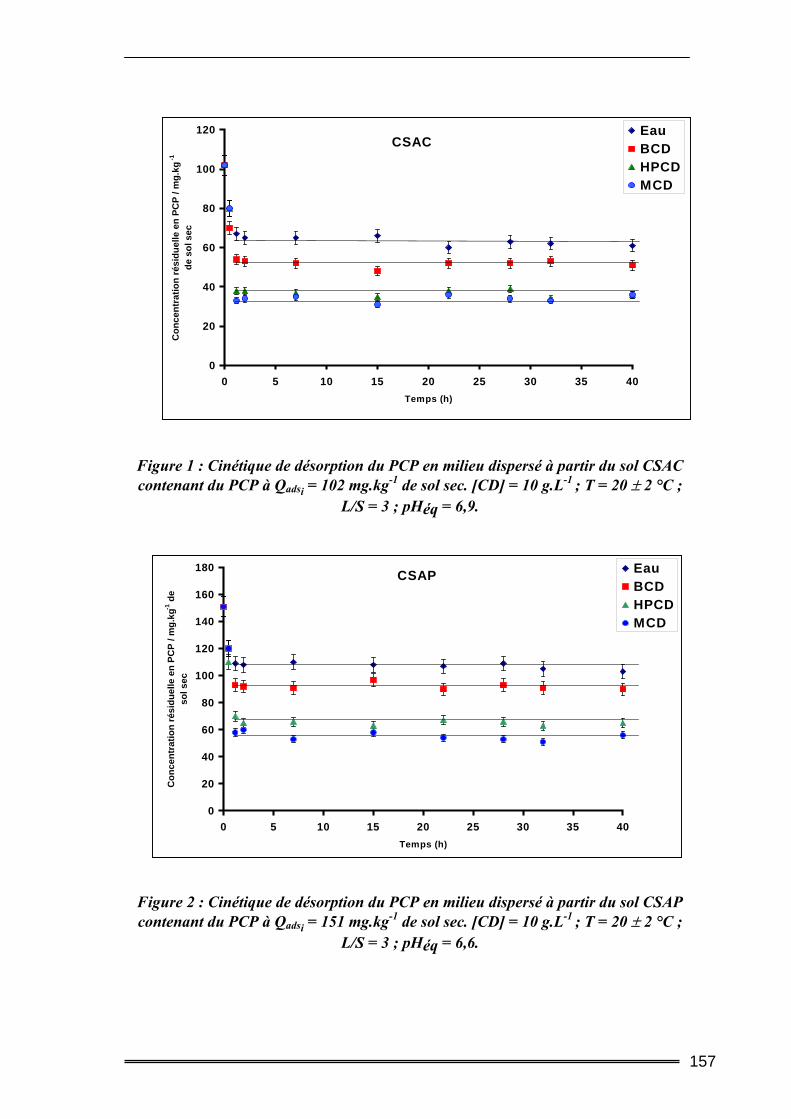
157
Figure 1 : Cinétique de désorption du PCP en milieu dispersé à partir du sol CSAC contenant du PCP à Qadsi = 102 mg.kg-1 de sol sec. [CD] = 10 g.L-1 ; T = 20 ± 2 °C ;
L/S = 3 ; pHéq = 6,9.
Figure 2 : Cinétique de désorption du PCP en milieu dispersé à partir du sol CSAP contenant du PCP à Qadsi = 151 mg.kg-1 de sol sec. [CD] = 10 g.L-1 ; T = 20 ± 2 °C ;
L/S = 3 ; pHéq = 6,6.
CSAC
0
20
40
60
80
100
120
0 5 10 15 20 25 30 35 40Temps (h)
Con
cent
ratio
n ré
sidu
elle
en
PCP
/ mg.
kg -1
de s
ol s
ec
EauBCDHPCDMCD
CSAP
0
20
40
60
80
100
120
140
160
180
0 5 10 15 20 25 30 35 40Temps (h)
Con
cent
ratio
n ré
sidu
elle
en
PCP
/ mg.
kg-1
de
sol s
ec
Eau BCDHPCDMCD

158
La désorption du PCP est achevée au bout d’une heure quelque soit le type de solution de
lavage utilisée. A l’équilibre, le ratio molaire [CD]/[PCP] en solution varie entre 200 et
300. Ainsi, la CD est toujours en large excès par rapport au PCP soluble dans le
surnageant.
La rapidité du phénomène de désorption rend difficile le suivi des concentrations en PCP
dans la solution de la suspension avec la technique utilisée. Par conséquent, nous n’avons
pas suffisamment de points avant le plateau pour pouvoir appliquer un modèle donné de
cinétique.
Les inévitables pertes en sol au cours des centrifugations et l’utilisation des réacteurs en
parallèle dans lesquels la concentration adsorbée en polluant et le volume du culot
n’étaient pas exactement les mêmes, pourraient provoquer des fluctuations de la
désorption. Cependant, il apparaît que le protocole utilisant les réacteurs en parallèle ne
pose pas de problèmes d’homogénéité des réacteurs en eux, puisque les résultats obtenus
sont reproductibles.
Sur les graphes 1 et 2, on peut observer une hiérarchisation des capacités d’extraction des
solutions extractantes étudiées : MCD > HPCD > BCD > Eau.
Il est probable que la cinétique de désorption apparente en milieu dispersé ne traduise pas
parfaitement les conditions dynamiques de la désorption lors d'un transfert du PCP dans
une colonne de sol ou in situ. En effet, l’agitation soutenue des réacteurs induit une
certaine déstructuration du sol avec dispersion des macroagrégats.
III.1.2.3. Isothermes d’adsorption et de désorption en batch
Cette étude a été menée dans le but de suivre le relargage du PCP en fonction de la
quantité de cyclodextrine mise en contact avec le sol.
Les isothermes d'adsorption et de désorption du PCP sur les sols CSAC et CSAP sont
représentées dans les figures 3 et 4. Une même quantité de sol a été lavée par différents
volumes de solutions. De ce fait, la quantité de cyclodextrine utilisée pour traiter une
même masse de sol augmente.
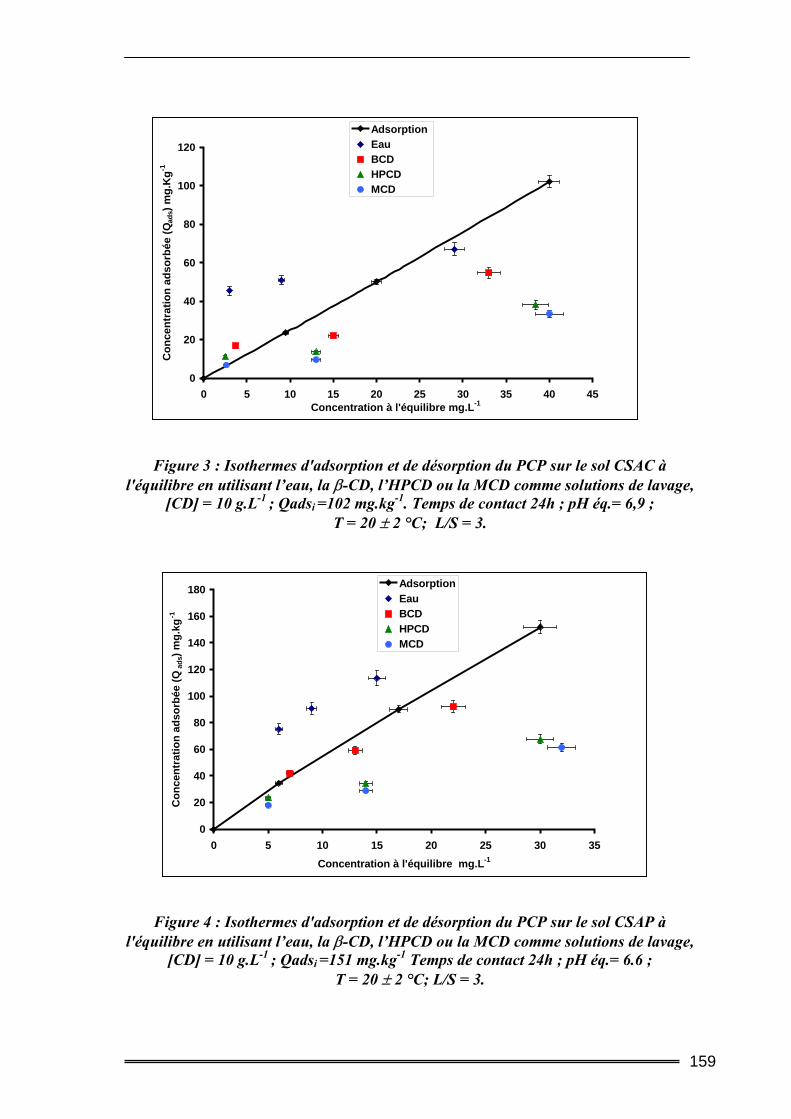
159
Figure 3 : Isothermes d'adsorption et de désorption du PCP sur le sol CSAC à l'équilibre en utilisant l’eau, la β-CD, l’HPCD ou la MCD comme solutions de lavage,
[CD] = 10 g.L-1 ; Qadsi =102 mg.kg-1. Temps de contact 24h ; pH éq.= 6,9 ; T = 20 ± 2 °C; L/S = 3.
Figure 4 : Isothermes d'adsorption et de désorption du PCP sur le sol CSAP à l'équilibre en utilisant l’eau, la β-CD, l’HPCD ou la MCD comme solutions de lavage,
[CD] = 10 g.L-1 ; Qadsi =151 mg.kg-1 Temps de contact 24h ; pH éq.= 6.6 ; T = 20 ± 2 °C; L/S = 3.
0
20
40
60
80
100
120
0 5 10 15 20 25 30 35 40 45Concentration à l'équilibre mg.L-1
Con
cent
ratio
n ad
sorb
ée (Q
ads)
mg.
Kg-1
Adsorption EauBCDHPCDMCD
0
20
40
60
80
100
120
140
160
180
0 5 10 15 20 25 30 35Concentration à l'équilibre mg.L-1
Con
cent
ratio
n ad
sorb
ée (Q
ads)
mg.
kg-1
AdsorptionEauBCDHPCDMCD

160
Du fait que le contact solution-sol peut modifier le pH de la solution, celui-ci a été
déterminé au début et à la fin de l’adsorption. Le pH à l'équilibre est de 6,9 avec le sol
CSAC et 6,6 avec le sol CSAP, soit relativement proche du pH initial de la solution de
PCP qui est de 7,0. Dans cette gamme de pH (6,6-6,9), la limite de solubilité du PCP est
de l’ordre de 1000 mg.L-1 (Weigthamn and Fein, 1998). Cette concentration est
supérieure à celle de la solution mère de PCP si bien que celui-ci ne risque pas de
précipiter lors des essais.
Les isothermes d’adsorption du PCP (figures 3 et 4) montrent qu’il existe une bonne
corrélation linéaire entre la concentration en soluté à l'équilibre et la teneur adsorbée sur
les deux sols. En considérant que les conditions expérimentales permettent d'atteindre les
équilibres d'adsorption, nous avons exploité les résultats d'adsorption suivant les modèles
linéaires (Qads = Kd*Céq.).
Les résultats montrent le rôle important de la matière organique dans l'adsorption du PCP.
En effet, les essais d'adsorption effectués sur les sols de la Côte Saint André CSAC et
CSAP (contenant respectivement 2 et 5% MO) permettent (par la mesure de la pente de
l’isotherme d’adsorption) la détermination des constantes de distribution : Kd (CSAC) =
2,6 L.kg-1 et Kd (CSAP) = 7,6 L.kg-1. Si les Kd de ces 2 sols sont sensiblement différents,
les logKoc calculés à partir de la relation Kd = Koc foc, sont relativement proches puisqu’il
est de 1,85 pour le sol CSAC et vaut 2,2 pour le sol CSAP.
Ces résultats confirment la bonne corrélation qu'il existe, à un pH donné, entre la teneur
en carbone organique des sols et l'adsorption des molécules organiques.
Les isothermes de désorption ne sont pas linéaires et des fluctuations de la désorption lors
des extractions successives ont été observées. Ces fluctuations résultent probablement des
pertes en sol au cours du renouvellement des solutions et des erreurs liées au volume de
la solution interstitielle du culot qui contient du PCP extrait lors de l’essai précèdent.
Il n’est pas possible d’appliquer un modèle simple pour exploiter les résultats de la
désorption puisque plusieurs phénomènes, ou équilibres, peuvent avoir lieu lors de la
mobilisation des polluants par une solution aqueuse de CD.
Toutefois, les figures 3 et 4 ci-dessus démontrent nettement que la présence de CD
favorise la solubilisation du PCP. Il en résulte une diminution importante de la quantité
de PCP restant adsorbée sur les sols après lavage avec les solutions de CD. Nous
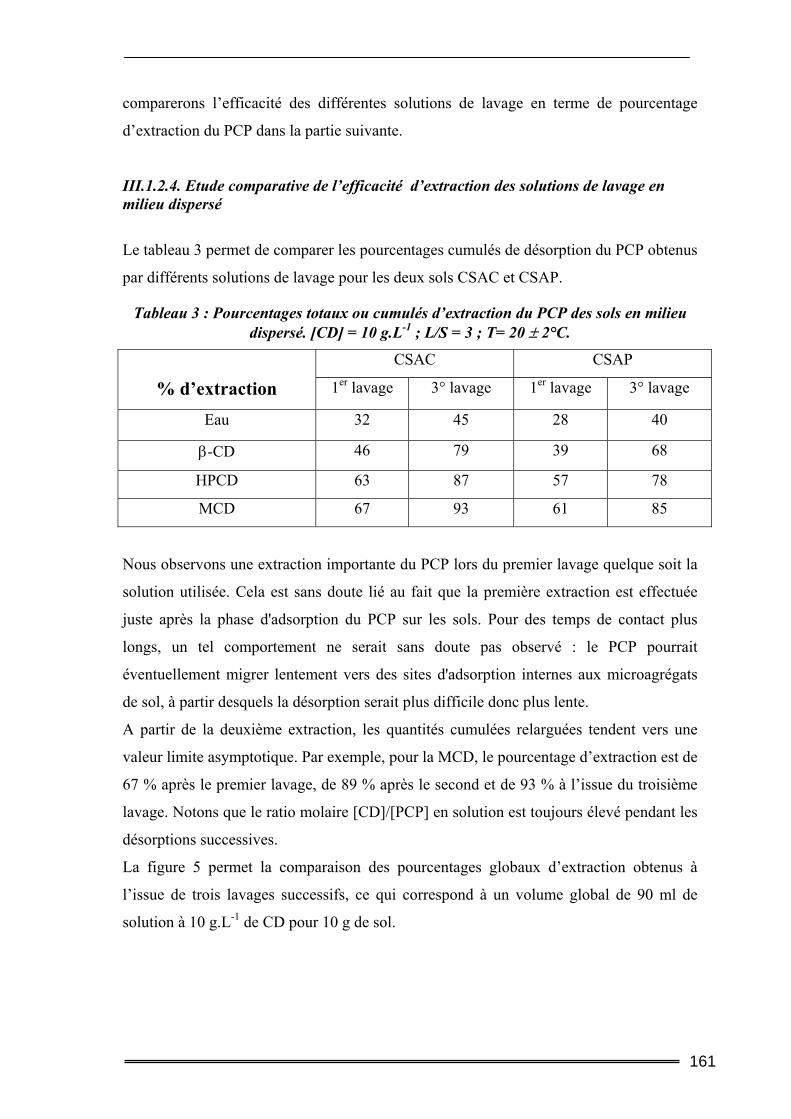
161
comparerons l’efficacité des différentes solutions de lavage en terme de pourcentage
d’extraction du PCP dans la partie suivante.
III.1.2.4. Etude comparative de l’efficacité d’extraction des solutions de lavage en milieu dispersé
Le tableau 3 permet de comparer les pourcentages cumulés de désorption du PCP obtenus
par différents solutions de lavage pour les deux sols CSAC et CSAP.
Tableau 3 : Pourcentages totaux ou cumulés d’extraction du PCP des sols en milieu dispersé. [CD] = 10 g.L-1 ; L/S = 3 ; T= 20 ± 2°C.
CSAC CSAP
% d’extraction 1er lavage 3° lavage 1er lavage 3° lavage
Eau 32 45 28 40
β-CD 46 79 39 68
HPCD 63 87 57 78
MCD 67 93 61 85
Nous observons une extraction importante du PCP lors du premier lavage quelque soit la
solution utilisée. Cela est sans doute lié au fait que la première extraction est effectuée
juste après la phase d'adsorption du PCP sur les sols. Pour des temps de contact plus
longs, un tel comportement ne serait sans doute pas observé : le PCP pourrait
éventuellement migrer lentement vers des sites d'adsorption internes aux microagrégats
de sol, à partir desquels la désorption serait plus difficile donc plus lente.
A partir de la deuxième extraction, les quantités cumulées relarguées tendent vers une
valeur limite asymptotique. Par exemple, pour la MCD, le pourcentage d’extraction est de
67 % après le premier lavage, de 89 % après le second et de 93 % à l’issue du troisième
lavage. Notons que le ratio molaire [CD]/[PCP] en solution est toujours élevé pendant les
désorptions successives.
La figure 5 permet la comparaison des pourcentages globaux d’extraction obtenus à
l’issue de trois lavages successifs, ce qui correspond à un volume global de 90 ml de
solution à 10 g.L-1 de CD pour 10 g de sol.
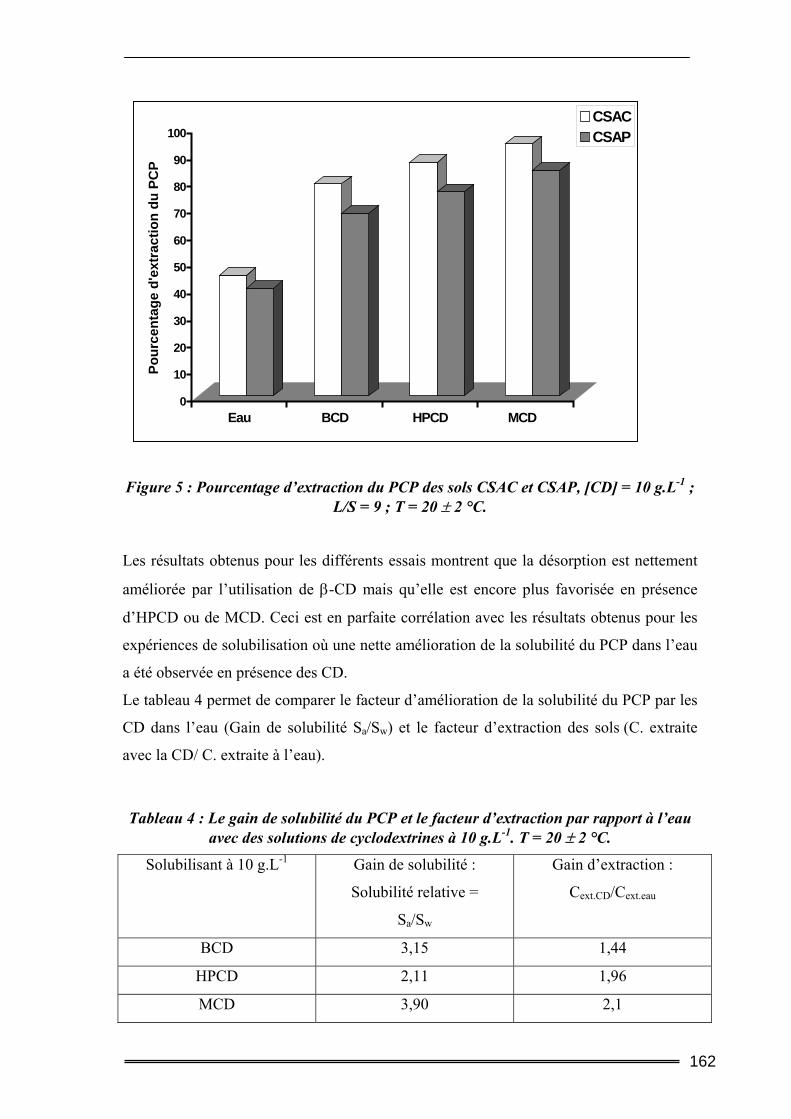
162
Figure 5 : Pourcentage d’extraction du PCP des sols CSAC et CSAP, [CD] = 10 g.L-1 ; L/S = 9 ; T = 20 ± 2 °C.
Les résultats obtenus pour les différents essais montrent que la désorption est nettement
améliorée par l’utilisation de β-CD mais qu’elle est encore plus favorisée en présence
d’HPCD ou de MCD. Ceci est en parfaite corrélation avec les résultats obtenus pour les
expériences de solubilisation où une nette amélioration de la solubilité du PCP dans l’eau
a été observée en présence des CD.
Le tableau 4 permet de comparer le facteur d’amélioration de la solubilité du PCP par les
CD dans l’eau (Gain de solubilité Sa/Sw) et le facteur d’extraction des sols (C. extraite
avec la CD/ C. extraite à l’eau).
Tableau 4 : Le gain de solubilité du PCP et le facteur d’extraction par rapport à l’eau avec des solutions de cyclodextrines à 10 g.L-1. T = 20 ± 2 °C.
Solubilisant à 10 g.L-1 Gain de solubilité :
Solubilité relative =
Sa/Sw
Gain d’extraction :
Cext.CD/Cext.eau
BCD 3,15 1,44
HPCD 2,11 1,96
MCD 3,90 2,1
0
10
20
30
40
50
60
70
80
90
100Po
urce
ntag
e d'
extr
actio
n du
PC
P
Eau BCD HPCD MCD
CSACCSAP

163
Le tableau 4 montre que le gain de solubilité est environ 3 fois supérieur au facteur
d’extraction des sols pour toutes les CD. Notons que pour les expériences de
solubilisation (chapitre II), le PCP était en large excès, sous forme de solide pur, tandis
que pour la désorption, le PCP était adsorbé sur une matrice complexe à une
concentration bien déterminée.
Notons aussi que la durée de contact entre le sol et le polluant pendant la phase
d’adsorption est relativement courte, le PCP était adsorbée sous sa forme ionique et la
désorption a été effectuée avec des solutions aqueuses tamponnées à pH 7. C’est
pourquoi, la désorption du polluant avec l’eau comme éluant est non négligeable (de
l’ordre de 25 à 30 %). Par conséquent, le facteur d’amélioration de l’extraction du PCP
pour les CD par rapport à l’eau paraît faible.
De plus, plusieurs phénomènes peuvent intervenir lors de l’extraction du PCP par les CD
telles que les interactions polluant/sol, CD/sol, polluant/CD et complexe/sol. En effet,
l’extraction par les CD ne peut pas être considérée comme une simple réaction de
complexation comme c’est le cas pour la solubilisation du polluant dans l’eau. C’est
pourquoi, nous évaluons la capacité des CD en terme de pourcentage d’extraction et non
pas en terme de coefficient de partage ou d’une constante d’équilibre comme c’est le cas
pour l’adsorption ou la complexation.
Le gain de solubilité du PCP augmente dans l’ordre : MCD > BCD > HPCD, tandis que
celui de l’extraction augmente dans l’ordre : MCD > HPCD > BCD. Cela indique que
l’aptitude d’une CD à solubiliser un polluant adsorbé sur un sol est tributaire
d’interactions que la molécule extractante est susceptible d’établir avec celui-ci. Ces
interactions dépendent des propriétés propres à chaque CD. Elles seront étudiées à la fin
de ce chapitre.
III.1.3. Bilan
La désorption du pentachlorophénol préalablement adsorbé sur les sols CSAC et CSAP a
été étudiée en milieu dispersé. La cinétique de mise en solution est relativement rapide
(t= 1h). La capacité d’extraction des solutions de lavage augmente dans l’ordre : Eau, β-

164
CD, HPCD, MCD. Ainsi, un lavage par une solution à 10 g.L-1 d’HPCD ou de MCD est
deux fois plus efficace qu’un lavage à l’eau.
L’adsorption du PCP est plus importante sur CSAP que sur CSAC. Il apparaît que le sol
CSAP retient plus fortement le polluant, ce qui rend sa désorption également plus
difficile. Cela peut être principalement dû au taux de matière organique relativement
élevé du CSAP. Une diminution d’environ 7 % du rendement d’extraction a été observé
quand la teneur en MO du sol augmente de 2 à 5 %. Les Matières organiques Naturelles
du sols peuvent ainsi jouer un rôle déterminant et influencer le rendement d’extraction.
Dans cette première étude, l’extraction du PCP par les CD a été étudiée en milieu
dispersé où l’agitation intense provoque généralement une déstructuration du sol. Il est
nécessaire d’étudier l’extraction et la mise en solution avec un système moins perturbant
dans des conditions plus proches des conditions réelles du terrain.
C’est pourquoi, nous allons confirmer ces résultats par une étude en colonne qui
permettra une approche plus réaliste d’une remédiation de sol in situ.
III.2. Essai de percolation en colonne
III.2.1. Objectif
Le test de percolation en colonne en milieu saturé est mis en œuvre dans le but de
comparer les résultats à ceux obtenus en milieu dispersé. En effet, bien que la technique
en batch fournisse divers renseignements sur les phénomènes d’adsorption et de
désorption, elle donne cependant une représentation biaisée de la réalité, en raison des
conditions particulières de contact entre le sol et la solution aqueuse. Une étude du
comportement dynamique des polluants dans le sol réalisée dans des conditions où celui-
ci n’est pas agité est donc utile pour une représentation plus correcte du phénomène réel.
Le tableau 5 recense les avantages et inconvénients respectifs du batch agité et de la
percolation en colonne.

165
Tableau 5 : récapitulatif des avantages et inconvénients du batch agité et de la percolation en colonne
Agitation en BATCH Percolation ascendante en colonne
Avantages
Simplicité de mise en œuvre
Etude des cinétiques de mise en solution
Mise en évidence du type d’isotherme
d’adsorption/désorption.
Conditions de lixiviation proche
de celles rencontrées sur le terrain
Inconvénients
Désagrégation particulaire provoquée
par l’agitation forcée, ce qui entraîne des
biais expérimentaux.
Maîtrise difficile du temps de
contact sol/liquide
Le test en colonne devrait donc permettre d’éviter la déstructuration granulométrique des
matrices testées et fournir des données pour une éventuelle modélisation du transfert des
polluants du sol vers la solution de lavage.
III.2.2. Procédures expérimentales
III.2.2.1. Principe
Le test de percolation en colonne a été conçu pour être polyvalent. Avec le même
dispositif, il permet d’étudier le relargage des polluants du sol dans une phase liquide en
fonction du temps (cinétique de mise en solution), mais aussi d’étudier de façon
discontinue l’évolution de l’intensité du relargage en fonction de l’épuisement du
polluant dans le sol.
Le principe de l'essai consiste à placer un échantillon de sol dans une colonne disposée
verticalement et soumise à la circulation continue d'une solution lixiviante en direction
ascendante, de manière à saturer le sol en eau et à minimiser les chemins préférentiels.
III.2.2.2. Fonctionnement Mise en place du sol Après séchage à l’air, tamisage et quartage, le sol est mis en place manuellement dans la
colonne par une technique particulière, permettant d’obtenir une structure (compaction)
consistante et assez uniforme (Mahjoub, 1999). Cette technique consiste à introduire dans
la colonne de petites quantités des sols (5g environ) et à les tasser légèrement avec un

166
outil plat (0,4 cm de diamètre) en appliquant un nombre constant de coups pour chaque
portion de sol introduite. L’objectif est d’obtenir une colonne de sol homogène pour
éviter au maximum la formation de chemins préférentiels.
Les colonnes utilisées sont des colonnes de chromatographie en verre (fournisseur
Amersham Biosciences) de 400 mm de hauteur et de 26 mm de diamètre interne. Les
colonnes sont équipées de deux pistons dont la face en contact avec le milieu poreux est
protégée par une grille plastique (maille de 1 mm2) recouverte d’une membrane en nylon
(diamètre des pores 10 µm). Afin d’éviter le colmatage du filtre, une couche de billes de
verre est placée entre celui-ci et le sol. L’étanchéité est assurée grâce à des joints en
caoutchouc. Les tubulures utilisées sont en PTFE. La hauteur de la colonne de sol est
d’environ 7 cm. La Figure 6 représente les colonnes utilisées.
Figure 6 : A gauche : Schéma d’une colonne, A droite : photographie d’une colonne (détail sur la partie haute de la colonne de terre avec le piston supérieur).
Colonne de verre
Joint caoutchouc
Grille protectrice Membrane nylon
Couche de billes de verre
Tube PTFE
Piston

167
Préconditionnement des colonnes Une fois le dispositif mis en place et les colonnes remplies avec le matériau testé, on
procède au remplissage total de la porosité par une solution aqueuse de CaCl2 à 0,01
mol.L-1. Ce sel est ajouté afin de préserver au mieux la structure du sol en évitant la
dispersion des colloïdes (Bayard, 1997). Pour assurer la bonne saturation de la colonne et
la stabilisation du régime hydraulique, la solution de CaCl2 circule en boucle fermée
pendant 12 h environ à un débit de 1 ml/min fournit par une pompe péristaltique
(Matserflex). Ce débit est couramment utilisé pour ce type d’expériences, d’après la
littérature. Le schéma du montage est donné figure 7.
Figure 7 : A gauche : Photographie du montage expérimental « batch percolant » ; A droite : schéma équivalent
Colonne H=40 cm Hterre =7cm
Solution sur agitation
magnétique
Pompe péristaltique
Q = 1mL/min

168
Caractérisation du régime hydrodynamique de colonne
Malgré un protocole de remplissage des colonnes le plus reproductible possible, le taux
de compactage du sol peut légèrement varier d’une colonne à l’autre. Ceci risque
d’entraîner une légère variation du volume poreux accessible au liquide percolant. Afin
de tenir compte de ces éventuelles variations, et de caractériser l’hydrodynamique de
chaque colonne, un essai préalable de transfert d’un traceur a été systématiquement
effectué après le préconditionnement.
Cette procédure expérimentale permet de rendre observable le déplacement réel de l’eau,
dans un milieu poreux, suivant une, ou des trajectoires définies, entre un point d’origine
et un ou plusieurs points de détection, au moyen de traceurs artificiels. On réalise ainsi
une caractérisation de la Distribution des Temps de Séjour (DTS) (Cain et al., 2000 ;
Brusseau, 2003).
Le traceur employé doit avoir, en tout point, un comportement identique à celui de l’eau
ou du liquide qui s’écoule à travers le milieu poreux. C’est donc une substance non
réactive. En pratique, on utilise souvent des traceurs anioniques, tels que KCl ou KBr.
Nous avons utilisé comme traceur une solution de KCl à 10 g.L-1.
Suivi du traceur en sortie de colonne Le volume de pore de la colonne a été déterminé par pesée avant et après saturation au
cours de la phase de préconditionnement de la colonne. La température lors de
l’expérience était constante (environ 20°C).
A l’issue du préconditionnement, un volume de 1 mL de solution de KCL (10 g/L) est
injecté en créneau à l’entrée de la colonne. La concentration en KCl en sortie de colonne
est mesurée par conductimétrie (relation linéaire dans le domaine de concentration). Une
cellule de conductivité (XE100 Radiometer) reliée à un analyseur CONSORT C832
permet de suivre la conductivité en continu par intégration sur ordinateur des données
toutes les 10 secondes. La solution percolante (CaCl2 à 0,01 mol.L-1) est envoyée avec un
débit de 1,0 ml/min. La courbe de restitution du traceur est exprimée en différence de
conductivité (mS/cm) par rapport à la solution de conditionnement en fonction du temps.

169
Les profils des courbes d’élution du soluté et le calcul de Distribution des Temps de
Séjour (DTS) sont donnés dans l ’annexe 3.
Après examen de la courbe d’élution obtenue et lorsque aucune irrégularité ne survient
(le pic d’élution est symétrique, cf. annexe 3), les essais d’adsorption peuvent
commencer.
III.2.2.3. Adsorption du PCP en colonne Les colonnes sont préparées et remplies comme décrit ci-dessus. L’échantillonnage de sol
doit être le plus performant possible pour que toutes les colonnes de sol soient proches du
point de vue écoulement et composition et que chaque réacteur fonctionne dans des
conditions les plus similaires possibles.
Les essais d’adsorption du PCP en colonne ont été réalisés à une concentration entrante
d’environ 500 mg.L-1 à pH 7. La solution additionnée de CaCl2 à 0,01 mol.L-1 et de
HgCl2 à 400 mg.L-1 est injectée dans les colonnes en flux ascendant à un débit de
1ml/min.
La phase liquide percole de bas en haut au travers de la colonne de sol puis retourne dans
le réservoir tampon (figure 2). L’agitateur magnétique permet d’homogénéiser la solution
dans le réacteur tampon. Tout le dispositif (colonnes + réacteur tampon) est placé dans
une chambre thermostatée, à une température donnée, pendant toute la durée de l’essai.
Le réservoir tampon a été protégé de la lumière par une feuille d’aluminium pour éviter la
photodégradation des polluants (figure 2).
Les échantillons sont prélevés à intervalles de temps réguliers à la seringue. Ils sont
ensuite analysés par HPLC, ce qui permet de déterminer à la fois le temps nécessaire pour
atteindre l’équilibre d’adsorption, et de connaître la quantité de PCP adsorbée sur le sol à
l’équilibre. L’adsorption du PCP a été quantifiée sur les sols CSAC et CSAP. Une fois
que la concentration initiale adsorbée sur le sol est connue, les essais d’extraction par les
solutions de CD peuvent commencer.

170
III.2.2.4. Protocole pour l’étude cinétique de l’extraction
L’étude cinétique consiste à mesurer la vitesse de passage en solution des composés
présents (adsorbés) dans le sol dans les conditions du test.
Après les essais d’adsorption, la solution contenue dans le réacteur tampon est analysée
selon le protocole adapté et remplacée par une phase liquide propre (eau ou solution de
CD). Le système est alors remis en circulation pour l’étude cinétique de la mise en
solution. Le volume de solution présent dans la porosité du sol et dans les volumes
« mort » (tuyaux) sont pris en compte puisque la solution, initialement propre, est en fait
une dilution du volume résiduel resté dans le système. Deux protocoles différents peuvent
être mis en œuvre pour étudier la cinétique d’extraction. Selon que le volume de
prélèvement nécessaire pour l’analyse est faible ou plus important, on utilise la méthode
des réacteurs en série ou celle des réacteurs en parallèle. En général, la solubilité des
polluants augmentent en présence de CD et dépassent largement le seuil de détection de
l’HPLC, ce qui évite l’étape de concentration obligatoire lorsqu’on travaille avec de
l’eau, étape qui peut être aussi source d’erreurs expérimentales.
Réacteurs en série Nous n’avons utilisé pour cette étude que la méthode des réacteurs en série puisque les
concentrations du polluant dans les solutions extractantes des CD étaient suffisamment
élevées. Le volume nécessaire pour l’analyse était donc faible et dans ce cas l’analyse
directe de la solution par HPLC était possible sans passer par une phase de concentration.
La méthode des réacteurs en série est la plus facile à mettre en œuvre. Elle consiste à
prélever dans le réacteur tampon en fonctionnement un volume de solution à différents
temps. Le somme des volumes prélevés ne doit pas représenter plus de 10% du volume
initial présent dans le réacteur tampon pour ne pas modifier de façon importante le
rapport L/S. Le nombre des prélèvements est donc limité.
III.2.2.5. Protocole pour l’étude discontinue du relargage (extractions successives)
Lors de l’étude cinétique, le temps de contact à respecter pour atteindre un état
d’équilibre apparent a été déterminé.

171
Les essais d’extractions successives permettent d’étudier de façon discontinue le
comportement du relargage des polluants dans la phase liquide. L’objectif est de suivre
l’évolution des concentrations à l’équilibre apparent en fonction de l’épuisement en
polluant dans le sol.
Le protocole de ces essais est tout à fait identique à celui des expériences de cinétique
décrites (III.2.2.4.). Par contre, dans ce cas, la totalité de la solution de lavage est
renouvelée (toutes les 24 h) alors qu’une seule solution est utilisée dans l’étude cinétique
(avec prélèvements successifs).
On effectue trois à cinq cycles d’extractions successives selon l’expérience.
Les solutions de PCP ont été analysés par HPLC dans les conditions indiquées au
paragraphe III.1.1.4.
III.2.3. Résultats de l’extraction du pentachlorophénol en colonne
III.2.3.1. Détermination du profil d’écoulement
La première étape de ces essais consiste à établir le profil de l’écoulement dans la
colonne par la détermination de la distribution des temps de séjour (DTS).
Le graphe 8 présente la courbe d’élution de KCl, suivie par conductimétrie. Le profil
d’élution d’un créneau de KCl montre qu’il n’y a pas de chemin préférentiel, puisqu’ il
n’y a qu’un seul pic visible. Toutefois, la courbe n’est pas symétrique comme dans le cas
d’un écoulement de type « piston » (Cf. annexe 3). La traînée lors de la redescente
montre que la colonne est ouverte à la diffusion.
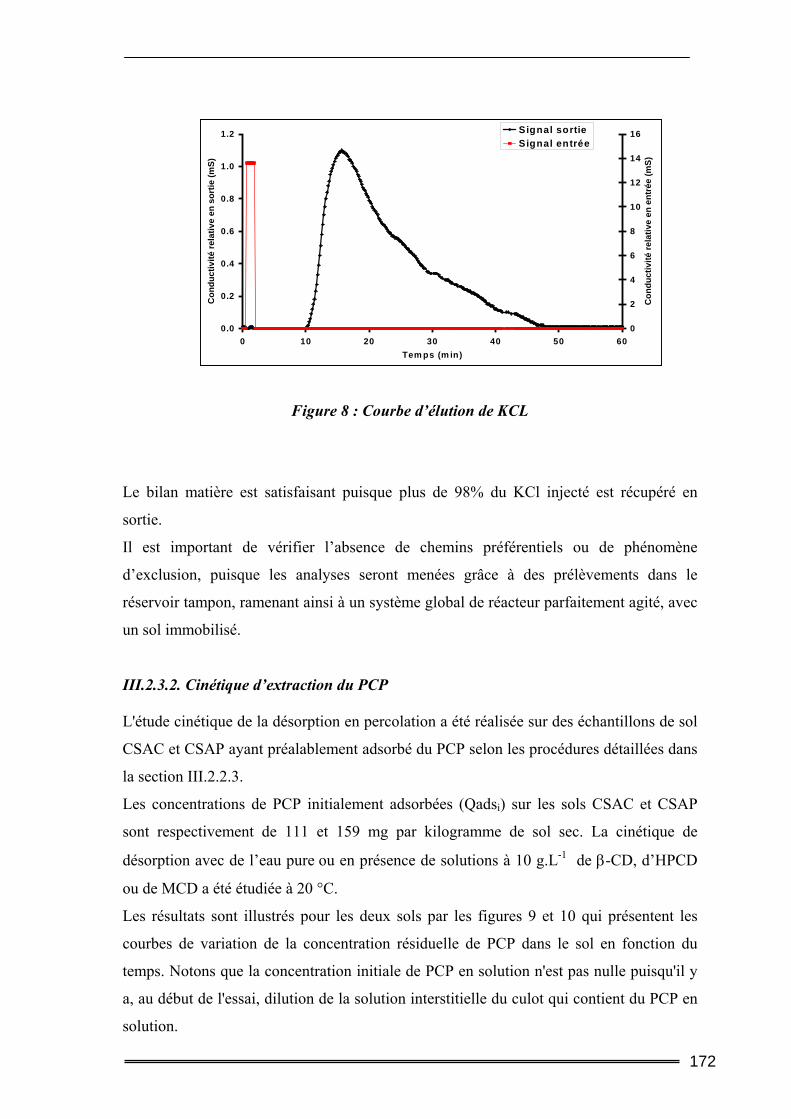
172
Figure 8 : Courbe d’élution de KCL
Le bilan matière est satisfaisant puisque plus de 98% du KCl injecté est récupéré en
sortie.
Il est important de vérifier l’absence de chemins préférentiels ou de phénomène
d’exclusion, puisque les analyses seront menées grâce à des prélèvements dans le
réservoir tampon, ramenant ainsi à un système global de réacteur parfaitement agité, avec
un sol immobilisé.
III.2.3.2. Cinétique d’extraction du PCP L'étude cinétique de la désorption en percolation a été réalisée sur des échantillons de sol
CSAC et CSAP ayant préalablement adsorbé du PCP selon les procédures détaillées dans
la section III.2.2.3.
Les concentrations de PCP initialement adsorbées (Qadsi) sur les sols CSAC et CSAP
sont respectivement de 111 et 159 mg par kilogramme de sol sec. La cinétique de
désorption avec de l’eau pure ou en présence de solutions à 10 g.L-1 de β-CD, d’HPCD
ou de MCD a été étudiée à 20 °C.
Les résultats sont illustrés pour les deux sols par les figures 9 et 10 qui présentent les
courbes de variation de la concentration résiduelle de PCP dans le sol en fonction du
temps. Notons que la concentration initiale de PCP en solution n'est pas nulle puisqu'il y
a, au début de l'essai, dilution de la solution interstitielle du culot qui contient du PCP en
solution.
0.0
0.2
0.4
0.6
0.8
1.0
1.2
0 10 20 30 40 50 60Tem ps (m in)
Con
duct
ivité
rela
tive
en s
ortie
(mS)
0
2
4
6
8
10
12
14
16
Con
duct
ivité
rela
tive
en e
ntré
e (m
S)
S ignal sortieSignal entrée
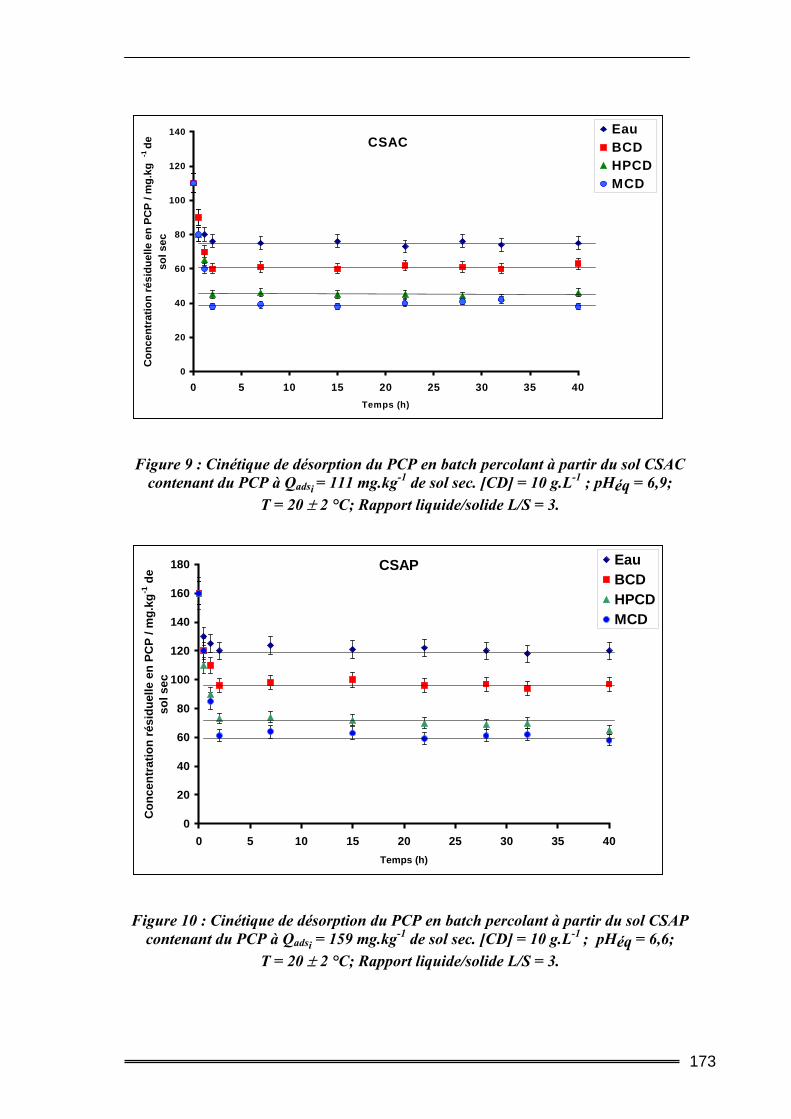
173
Figure 9 : Cinétique de désorption du PCP en batch percolant à partir du sol CSAC contenant du PCP à Qadsi = 111 mg.kg-1 de sol sec. [CD] = 10 g.L-1 ; pHéq = 6,9;
T = 20 ± 2 °C; Rapport liquide/solide L/S = 3.
Figure 10 : Cinétique de désorption du PCP en batch percolant à partir du sol CSAP contenant du PCP à Qadsi = 159 mg.kg-1 de sol sec. [CD] = 10 g.L-1 ; pHéq = 6,6;
T = 20 ± 2 °C; Rapport liquide/solide L/S = 3.
0
20
40
60
80
100
120
140
0 5 10 15 20 25 30 35 40Temps (h)
Con
cent
ratio
n ré
sidu
elle
en
PCP
/ mg.
kg -1
de
sol s
ec
EauBCDHPCDMCD
CSAC
0
20
40
60
80
100
120
140
160
180
0 5 10 15 20 25 30 35 40Temps (h)
Con
cent
ratio
n ré
sidu
elle
en
PCP
/ mg.
kg-1
de
sol s
ec
Eau BCDHPCDMCD
CSAP

174
La désorption du PCP est achevée au bout de 2 h quelque soit le type de solution de
lavage utilisé. La cinétique de la mise en solution en colonne de percolation est
légèrement plus lente qu’en milieu dispersé. Compte tenu de l’incertitude donnée par les
barres sur les figures, on peut considérer qu’il n’y a pratiquement pas de fluctuation.
L’étude de la cinétique en colonne de percolation se fait en réacteur en série (une colonne
ou un réacteur pour toute l’expérience) tandis qu’en milieu dispersé, elle se fait en
réacteur en parallèle car elle nécessite un réacteur par point expérimental. En effet,
l’étude de la cinétique en colonne permet de travailler sur la même fraction du sol avec
plus ou moins le même volume de culot tout au long de l’expérience. Notons que le sol
n’est pas agité et que sa structure n’est en général pas perturbée.
L’utilisation de colonne de percolation semble plus proche des conditions réelles mais les
résultats sont plus difficilement interprétables du fait de la non connaissance des aspects
cinétiques dans la colonne elle même. En effet, les cinétiques présentées dans les figures
9 et 10 correspondent à la cinétique du dispositif expérimental, et non à la cinétique de
l’extraction, puisque les prélèvements ont été effectués dans le réservoir tampon et non
directement à la sortie de la colonne. Sur ce point l’extraction en colonne se différencie
du protocole en milieu dispersé où le sol est en contact direct avec la solution prélevée.
Les courbes expérimentales obtenues après 40 h de contact en milieu dispersé ou en batch
percolant ont des profils similaires avec ou sans CD. Cette remarque permet de formuler
l’hypothèse que les mécanismes d’extraction mis en jeu sont de même nature, que l’on
emploie de l’eau ou un mélange eau/CD comme solution extractante. La différence
principale vient du fait que les solubilités des polluants en présence de CD sont
significativement augmentées, ce qui permet d’extraire des quantités plus importantes
pour une même durée de l’essai.
A l’équilibre, le ratio [CD]/[PCP] en solution varie entre 200 et 300. La CD est donc
toujours en large excès par rapport au PCP solubilisé dans le surnageant.
III.2.3.3. Renouvellement de la solution de lavage (Effet de L/S cumulé)
Le protocole expérimental est décrit au paragraphe III.2.2.5.
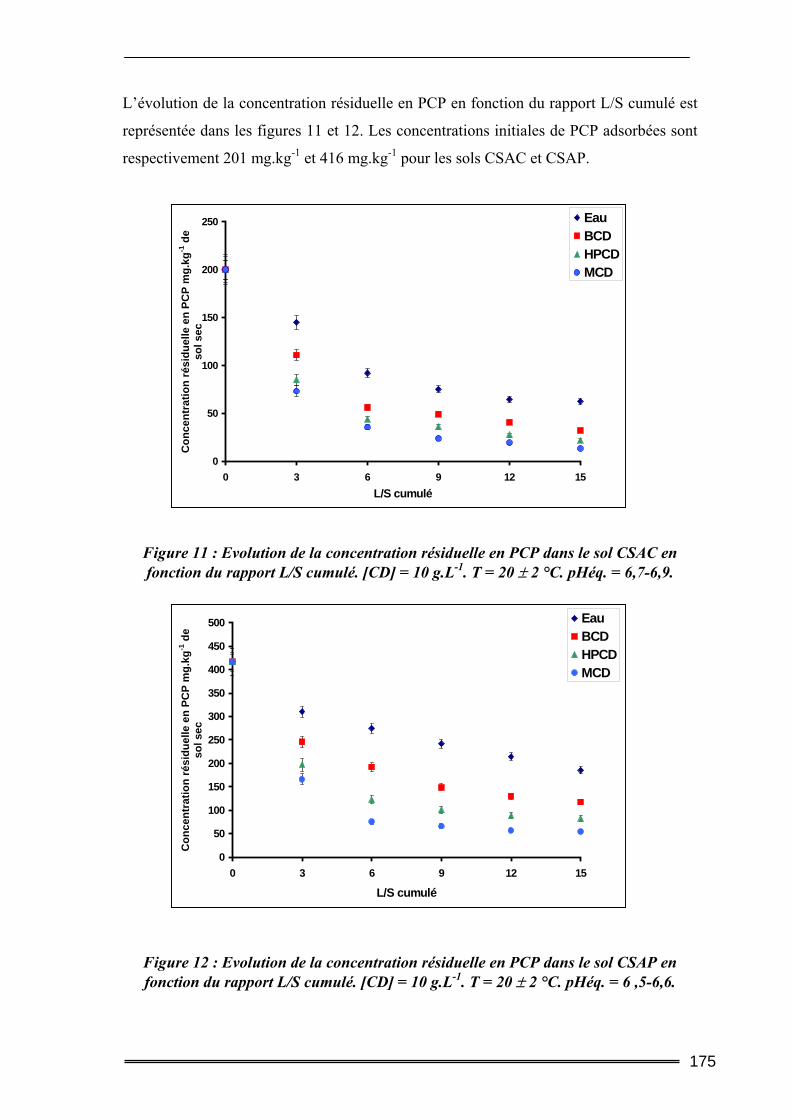
175
L’évolution de la concentration résiduelle en PCP en fonction du rapport L/S cumulé est
représentée dans les figures 11 et 12. Les concentrations initiales de PCP adsorbées sont
respectivement 201 mg.kg-1 et 416 mg.kg-1 pour les sols CSAC et CSAP.
Figure 11 : Evolution de la concentration résiduelle en PCP dans le sol CSAC en fonction du rapport L/S cumulé. [CD] = 10 g.L-1. T = 20 ± 2 °C. pHéq. = 6,7-6,9.
Figure 12 : Evolution de la concentration résiduelle en PCP dans le sol CSAP en fonction du rapport L/S cumulé. [CD] = 10 g.L-1. T = 20 ± 2 °C. pHéq. = 6 ,5-6,6.
0
50
100
150
200
250
0 3 6 9 12 15L/S cumulé
Con
cent
ratio
n ré
sidu
elle
en
PCP
mg.
kg-1
de
sol s
ec
EauBCDHPCDMCD
0
50
100
150
200
250
300
350
400
450
500
0 3 6 9 12 15
L/S cumulé
Con
cent
ratio
n ré
sidu
elle
en
PCP
mg.
kg-1
de
sol s
ec
EauBCDHPCDMCD

176
Les figures 11 et 12 montre l’effet de la présence de CD sur la mobilité du PCP et
souligne la nette supériorité de la MCD en terme d’efficacité d’extraction.
Quelque soit le sol considéré (CSAC ou CSAP) et la solution de lavage utilisée, on peut
remarquer que la concentration résiduelle en PCP diminue à chaque nouvelle percolation
ou extraction (L/S de 3 à 15).
Une chute importante de la concentration adsorbée est toujours observée au premier
lavage. La solution de lavage de la première extraction a sans doute entraîné en solution
le PCP le plus accessible et le plus faiblement fixé. La pollution du sol étant très récente,
puisque la première extraction a été mise en œuvre immédiatement après l’adsorption, la
proportion de PCP faiblement lié est probablement beaucoup plus importante que dans le
cas d’une pollution plus ancienne.
A partir du troisième lavage, les quantités relarguées ont tendances à se stabiliser vers une
valeur maximale. En effet, l’évolution de la concentration extraite ou résiduelle du PCP
ne suit pas une loi linéaire.
III.2.3.4. Effet de la concentration en CD
Nous avons étudié l’effet de la concentration des CD modifiées (HPCD, MCD) sur
l’extraction du PCP des sols CSAC et CSAP dans une gamme de concentration de 0 à 50
g.L-1. La β-CD a été exclue de cette étude en raison de sa faible solubilité aqueuse (18
g.L-1 à 20°C). Une colonne a été préparée pour chaque point expérimental, c’est à dire
pour chaque concentration en CD. Les concentrations en PCP initialement adsorbé sur
les sols CSAC et CSAP étaient respectivement d’environ 190 (± 8) et 405 (± 12) mg.kg-1
de sol sec.
Les évolutions des pourcentages d’extraction du PCP des sols CSAC et CSAP en
fonction de la concentration en CD sont respectivement représentés dans les figures 13 et
14 .
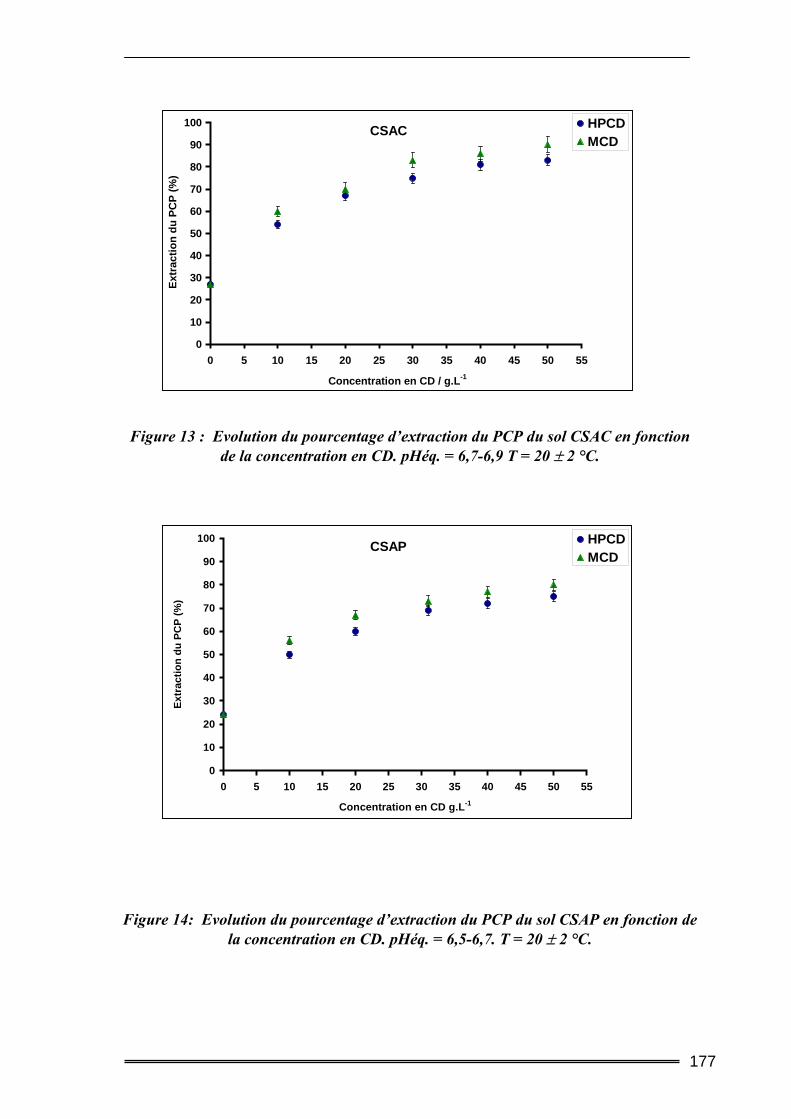
177
Figure 13 : Evolution du pourcentage d’extraction du PCP du sol CSAC en fonction de la concentration en CD. pHéq. = 6,7-6,9 T = 20 ± 2 °C.
Figure 14: Evolution du pourcentage d’extraction du PCP du sol CSAP en fonction de la concentration en CD. pHéq. = 6,5-6,7. T = 20 ± 2 °C.
CSAC
0
10
20
30
40
50
60
70
80
90
100
0 5 10 15 20 25 30 35 40 45 50 55
Concentration en CD / g.L-1
Ext
ract
ion
du P
CP
(%)
HPCDMCD
CSAP
0
10
20
30
40
50
60
70
80
90
100
0 5 10 15 20 25 30 35 40 45 50 55
Concentration en CD g.L-1
Extr
actio
n du
PC
P (%
)
HPCDMCD

178
Les essais ont été effectués en triplicats et les points représentés correspondent à la
moyenne des trois valeurs mesurées. L’écart relatif varie entre 4 à 5 %.
Les figures 13 et 14 montrent que lorsque la concentration en CD augmente de 0 à 30g.L-
1, la mobilisation du PCP est accrue d’un facteur 2 à 3 fois par rapport à l’eau. A partir de
30g.L-1 de HPCD ou de MCD, les quantités extraites augmentent plus graduellement et
elles tendent vers une valeur maximale limite.
Le ratio [CD]/[PCP] en solution est relativement élevé quelque soit la concentration
utilisée en CD ou le type de matrice étudiée.
III.2.3.5. Etude comparative de l’efficacité d’extraction des solutions de lavage.
Nous avons comparé l’efficacité d’extraction de l’eau et de trois solutions des différentes
CD à la même concentration initiale. Le tableau 6 montre les pourcentages de PCP extrait
des deux sols au premier lavage (correspondant à un ratio L/S 3), et à l’issue du dernier
lavage (correspondant à un ratio L/S 15 (5 renouvellements)).
Tableau 6 : Pourcentages de PCP extrait des deux sols étudiés au premier et au dernier lavage en colonne de percolation. [CD] = 10 g.L-1. Qadsi (CSAC) = 201 mg.kg-1 , Qadsi
(CSAP) = 416 mg.kg-1 . T = 20 ± 2 °C.
CSAC CSAP
% d’extraction 1er lavage
(L/S =3)
Après 5
lavages
(L/S=15)
1er lavage
(L/S =3)
Après 5
lavages
(L/S=15)
Eau 28 65 24 55
BCD 45 84 40 71
HPCD 57 89 52 80
MCD 63 94 57 87
La figure 15 permet de comparer les pourcentages d’extraction cumulé à L/S 15 pour les
4 solutions de lavage, le nombre de moles de CD utilisé est alors 5 fois plus grand.
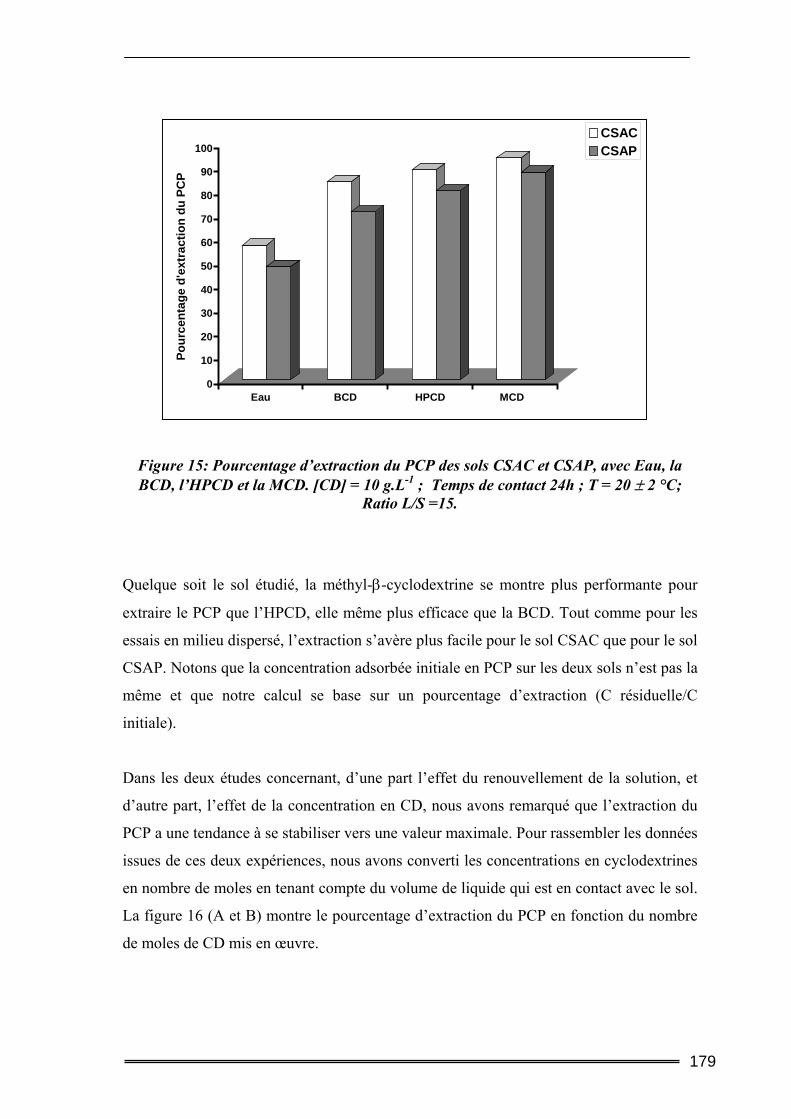
179
Figure 15: Pourcentage d’extraction du PCP des sols CSAC et CSAP, avec Eau, la BCD, l’HPCD et la MCD. [CD] = 10 g.L-1 ; Temps de contact 24h ; T = 20 ± 2 °C;
Ratio L/S =15.
Quelque soit le sol étudié, la méthyl-β-cyclodextrine se montre plus performante pour
extraire le PCP que l’HPCD, elle même plus efficace que la BCD. Tout comme pour les
essais en milieu dispersé, l’extraction s’avère plus facile pour le sol CSAC que pour le sol
CSAP. Notons que la concentration adsorbée initiale en PCP sur les deux sols n’est pas la
même et que notre calcul se base sur un pourcentage d’extraction (C résiduelle/C
initiale).
Dans les deux études concernant, d’une part l’effet du renouvellement de la solution, et
d’autre part, l’effet de la concentration en CD, nous avons remarqué que l’extraction du
PCP a une tendance à se stabiliser vers une valeur maximale. Pour rassembler les données
issues de ces deux expériences, nous avons converti les concentrations en cyclodextrines
en nombre de moles en tenant compte du volume de liquide qui est en contact avec le sol.
La figure 16 (A et B) montre le pourcentage d’extraction du PCP en fonction du nombre
de moles de CD mis en œuvre.
0
10
20
30
40
50
60
70
80
90
100
Pour
cent
age
d'ex
trac
tion
du P
CP
Eau BCD HPCD MCD
CSACCSAP
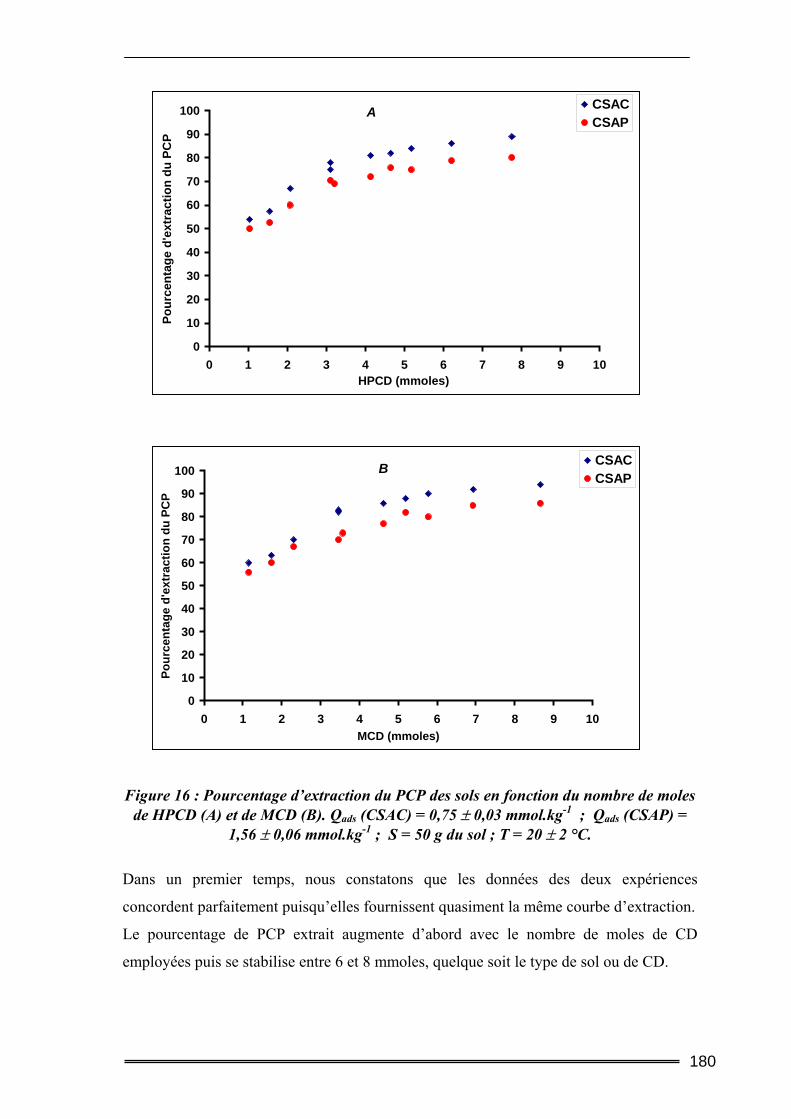
180
Figure 16 : Pourcentage d’extraction du PCP des sols en fonction du nombre de moles de HPCD (A) et de MCD (B). Qads (CSAC) = 0,75 ± 0,03 mmol.kg-1 ; Qads (CSAP) =
1,56 ± 0,06 mmol.kg-1 ; S = 50 g du sol ; T = 20 ± 2 °C.
Dans un premier temps, nous constatons que les données des deux expériences
concordent parfaitement puisqu’elles fournissent quasiment la même courbe d’extraction.
Le pourcentage de PCP extrait augmente d’abord avec le nombre de moles de CD
employées puis se stabilise entre 6 et 8 mmoles, quelque soit le type de sol ou de CD.
0
10
20
30
40
50
60
70
80
90
100
0 1 2 3 4 5 6 7 8 9 10HPCD (mmoles)
Pour
cent
age
d'ex
trac
tion
du P
CP
CSACCSAP
A
0
10
20
30
40
50
60
70
80
90
100
0 1 2 3 4 5 6 7 8 9 10MCD (mmoles)
Pour
cent
age
d'ex
trac
tion
du P
CP
CSACCSAP
B

181
Les concentrations en PCP initialement adsorbées sur les sols CSAC et CSAP s’élèvent
respectivement à 0,75 mmol et 1,56 mmol par kg de sol sec. Il faudrait donc utilisé un
nombre de moles de MCD environ 6 fois plus grand que le nombre de moles du polluant
contenu dans le sol pour pouvoir extraire environ 90 % de celui-ci.
Notons que la teneur en PCP dans les deux sols est élevée par rapport aux pollutions
décrites dans la littérature. En effet, la concentration en PCP dans les sites recensés varie
de quelques µg/kg (Li et al., 2003) à une centaine de mg (56,7 mg.kg-1 (Tse et al., 2001),
103 mg/kg (USEPA, 1992)). Un site de traitement de bois situé dans l’Ontario au Canada
(Knoke, 1999) est considéré comme extrêmement pollué avec 335 mg.kg-1. Les quantités
de PCP adsorbées sur les sols CSAC et CSAP les classent donc parmi les sites hautement
pollués.
Les travaux publiés permettent de comparer les performances des CD par rapport à
d’autres agent de lavage de sols tels que les solvants ou les tensioactifs. Des chercheurs
(Khodadous et al., 1999) ont pu, en utilisant une solution aqueuse à 56 % d’éthanol,
extraire environ 60 % du PCP adsorbé sur un sol. Les surfactants non ioniques tels que le
TergitolNP-10 à 8 mM augmentent le rendement d’extraction du PCP de 90 % des sols
contaminés ([PCP]ads = 183 mg/kg) (Mulligan et Eftekhari, 2000; Cort et Bielefeldt,
2002 ; Park and Bielefeldt, 2003). Le méthanol et le SDS (surfactant anionique)
augmentent aussi l’extraction du PCP des sols contaminés, tandis que le TritonX-100
l’inhibe (You et Liu, 1996 ; Khodadous et al., 1999 a, b). Il est probable que ce surfactant
s’adsorbe fortement sur le sol, ce qui accroît la capacité de rétention du sol pour le PCP
(You et Liu, 1996).
Nos travaux montrent donc que la capacité d’extraction des CD est comparable à celle
des agents classiques de lavage puisque 6 à 8 mmoles de MCD permettent d’extraire 90
% des 416 mg de PCP adsorbés sur le sol CSAP.
III.2.3.6. Estimation du facteur d’extraction par les CD Pour une technique de lavage basée sur l’amélioration de la solubilité et de la mobilité du
polluant par l’ajout d’un additif (agent de solubilisation) (Ji et Brusseau, 1998 : McCray
et Brusseau, 2003 McCray et al., 2001), le facteur d’extraction f d’un polluant est défini

182
comme le rapport de la concentration extraite par l’additif sur la concentration extraite à
l’eau sans additif : w
ext
CC
f =
- Cext est la concentration totale extraite par la CD incluant l’espèce libre et l’espèce
complexée dans la phase aqueuse du sol : Cext = [P] + [P/CD]
avec [P/CD] = K [P][CD] où K est la constante d’équilibre de complexation.
- Cw est la concentration extraite à l’eau sans CD, elle est considérée comme la
concentration en espèce libre [P].
L’équation (1) devient : Cext = Cw + K.Cw [CD]
et le facteur d’extraction f du polluant s’exprime comme :
][1 CDKCC
fw
ext +==
Cette équation empirique permet d’estimer le facteur d’extraction f à une concentration
donnée en CD et pour un polluant dont la constante de complexation avec cette CD est
connue à la température considérée.
Ainsi le facteur d’extraction f calculé dans le cas d’une solution de MCD (KPCP = 420 M-
1) à 50 g.L-1 vaut environ 17. Or, l’expérience montre que cette même solution de MCD
ne permet d’extraire que 2,4 fois plus de PCP que l’eau. Cette différence importante
permet de penser que l’estimation basée uniquement sur la capacité de solubilisation ou
de complexation par la CD n’est pas valable dans nos conditions. En effet, l’expression
du facteur d’extraction f est trop simpliste pour permettre de décrire correctement nos
résultats expérimentaux.
III.2.4. Bilan
Les essais d’extraction du PCP en colonne confirment l’efficacité des CD. L’utilisation
du protocole batch percolant conduit à la même hiérarchisation des performances
d’extraction des CD que le batch agité. L’efficacité décroît dans l’ordre : MCD > HPCD

183
> BCD. L’extraction est moins efficace vis-à-vis du sol CSAP quelque soit la solution de
lavage utilisée. Une diminution d’environ 8 % du rendement d’extraction a été observée
quand la teneur en MO du sol augmente de 2 à 5 %.
D’une façon générale, le rendement d’extraction est légèrement meilleur en milieu
dispersé. Il est probable que l’agitation, en provoquant la désagrégation des agglomérats
et une certaine déstructuration du sol, favorise l’accès des CD aux sites fixant le PCP,
contrairement à ce qui se passe en colonne.
IV. Etude de l’adsorption et de la désorption du
naphtalène et phénanthrène en milieu dispersé
IV.1. Conditions opératoires
L'étude de l'adsorption et de la désorption du naphtalène et du phénanthrène sur les sols
CSAC et CSAP a été réalisée en milieu dispersé (Batch) dans les conditions indiquées au
paragraphe III.1.
IV.1.1. Préparation des solutions
Le naphtalène et le phénanthrène utilisés dans cette étude sont des produits Aldrich pour
analyse d'une pureté supérieure à 98%. Etant donnée la très faible solubilité des deux
produits dans l'eau, les essais de contact effectués en phase aqueuse pure nécessitent
d'utiliser une méthode de préparation de la solution de départ garantissant la
solubilisation totale du produit (limite de solubilité S = 1,1 mg.L-1 à 20°C pour le
phénanthrène et S = 30 mg.L-1 pour le naphtalène). Pour ce faire, on solubilise le polluant
dans l'eau en ajoutant une quantité nettement supérieure à la limite de solubilité (soit
environ 1000 mg de polluant par litre d'eau permutée). La suspension est agitée pendant

184
24 h à l'obscurité dans une chambre thermostatée à 20°C. Elle est ensuite filtrée sur une
membrane GF-C Watman en fibre de verre de diamètre de pore de 1,2 µm.
IV.1.2. Analyses des solutions
Les dosages du naphtalène et du phénanthrène en solution sont effectués par
chromatographie liquide haute performance, phase inverse, en condition isocratique. Les
conditions d'analyses sont les suivantes :
- Colonne Kromasil C18, de longueur 25 cm. Les particules de garnissage ont un
diamètre de 10 nm. L'emploi de cette colonne est recommandé pour l'analyse des
composés non polaires tels que les hydrocarbures aromatiques polycycliques.
- Précolonne de garde : Kromasil C18
- Phase éluante constituée d'un mélange acétonitrile/eau (80/20),
- Débit d'élution : 1,0 ml.min-1,
- Détection à λ = 254 nm.
IV.2. Résultats de l’extraction du naphtalène ou du phénanthrène en
milieu dispersé IV.2.1. Cinétique de désorption
L'étude de la cinétique de désorption a été réalisée sur des échantillons des sols CSAC et
CSAP ayant préalablement adsorbé du naphtalène ou du phénanthrène. La cinétique de
désorption a été étudiée avec de l’eau et des solutions de β-CD et d’HPCD à 5 g.L-1.
Les cinétiques de désorption du Naphtalène à partir des sols CSAC et CSAP sont
respectivement représentées dans les figures 17 et 18. La même allure a été observée avec
le phénanthrène. Les barres représentent l’écart relatif entre les triplicats.
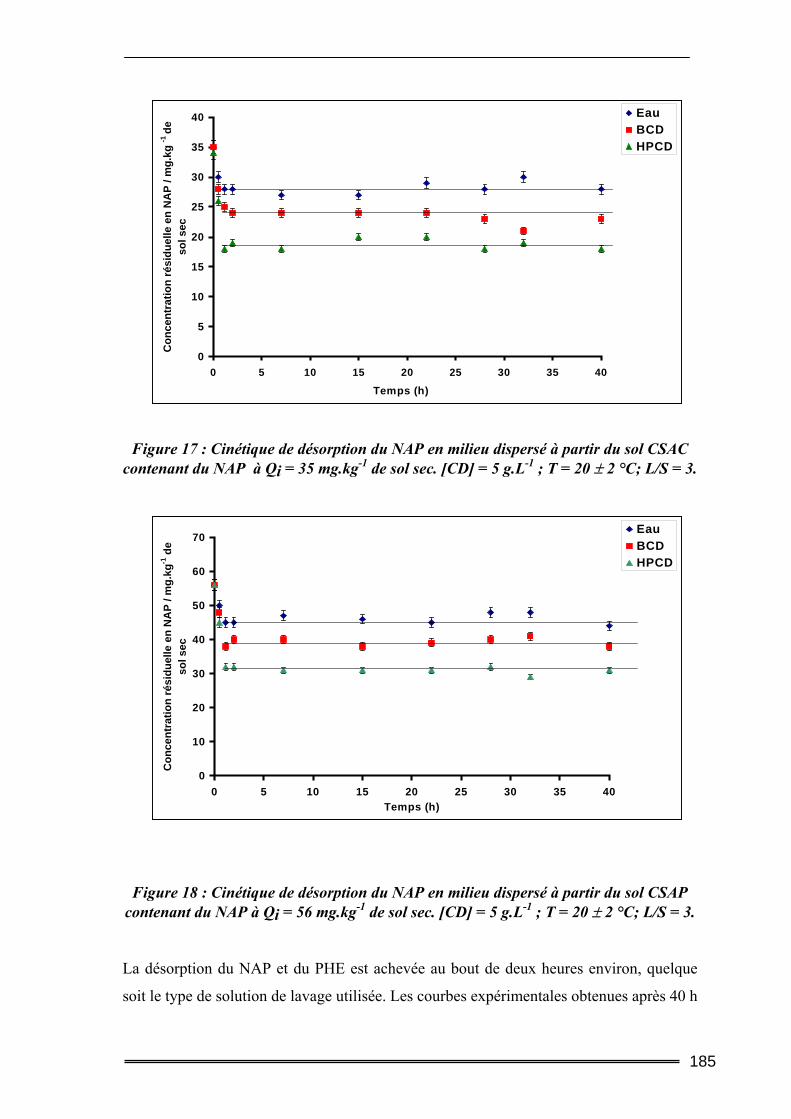
185
Figure 17 : Cinétique de désorption du NAP en milieu dispersé à partir du sol CSAC contenant du NAP à Qi = 35 mg.kg-1 de sol sec. [CD] = 5 g.L-1 ; T = 20 ± 2 °C; L/S = 3.
Figure 18 : Cinétique de désorption du NAP en milieu dispersé à partir du sol CSAP contenant du NAP à Qi = 56 mg.kg-1 de sol sec. [CD] = 5 g.L-1 ; T = 20 ± 2 °C; L/S = 3.
La désorption du NAP et du PHE est achevée au bout de deux heures environ, quelque
soit le type de solution de lavage utilisée. Les courbes expérimentales obtenues après 40 h
0
5
10
15
20
25
30
35
40
0 5 10 15 20 25 30 35 40
Temps (h)
Con
cent
ratio
n ré
sidu
elle
en
NA
P / m
g.kg
-1 de
so
l sec
EauBCDHPCD
0
10
20
30
40
50
60
70
0 5 10 15 20 25 30 35 40Temps (h)
Con
cent
ratio
n ré
sidu
elle
en
NA
P / m
g.kg
-1 d
e so
l sec
Eau BCDHPCD

186
de contact en milieu dispersé montrent que nous avons atteint un régime stationnaire et
les valeurs d’extraction sont quasi-constantes à partir du plateau.
La rapidité du phénomène de désorption rend difficile le suivi des concentrations en
naphtalène ou en phénanthrène dans la solution avec la technique utilisée. Par
conséquent, nous n’avons pas pu appliquer un modèle cinétique, compte tenu de
l’insuffisance des points de mesures avant d’atteindre l’équilibre.
Comme pour le PCP, les pertes en sol au cours des centrifugations, la concentration
adsorbée en polluant et le volume du culot n’étaient pas les mêmes pour tous les tubes, ce
qui pourrait expliquer les fluctuations de la désorption. Les concentrations mesurées en
solution sont affectées d’une incertitude de 5 % environ.
IV.2.2. Isothermes d’adsorption et de désorption
Dans le but d’étudier l’effet de la présence d’un co-soluté sur les processus
d’adsorption/extraction, nous avons effectué l’adsorption et la désorption du NAP et du
PHE seuls et en mélange. Les essais de désorption ont été menés en duplicats sur des
échantillons de sol ayant la même teneur initiale Qadsi en naphtalène ou phénanthrène.
Les concentrations dans l’échantillon étant suffisamment élevées en présence de CD,
l’injection en HPLC a pu être effectuée sans traitement de préconcentration préalable.
Les isothermes d'adsorption et de désorption du naphtalène sur les sols CSAC et CSAP
sont respectivement représentées dans les figures 19 et 20. Nous ne présentons pas les
isothermes d'adsorption et de désorption du PHE sur ces deux sols afin d’éviter de
surcharger ce mémoire. Cependant, les résultats d’adsorption et de désorption de deux
composés seuls et en mélange seront commentés.
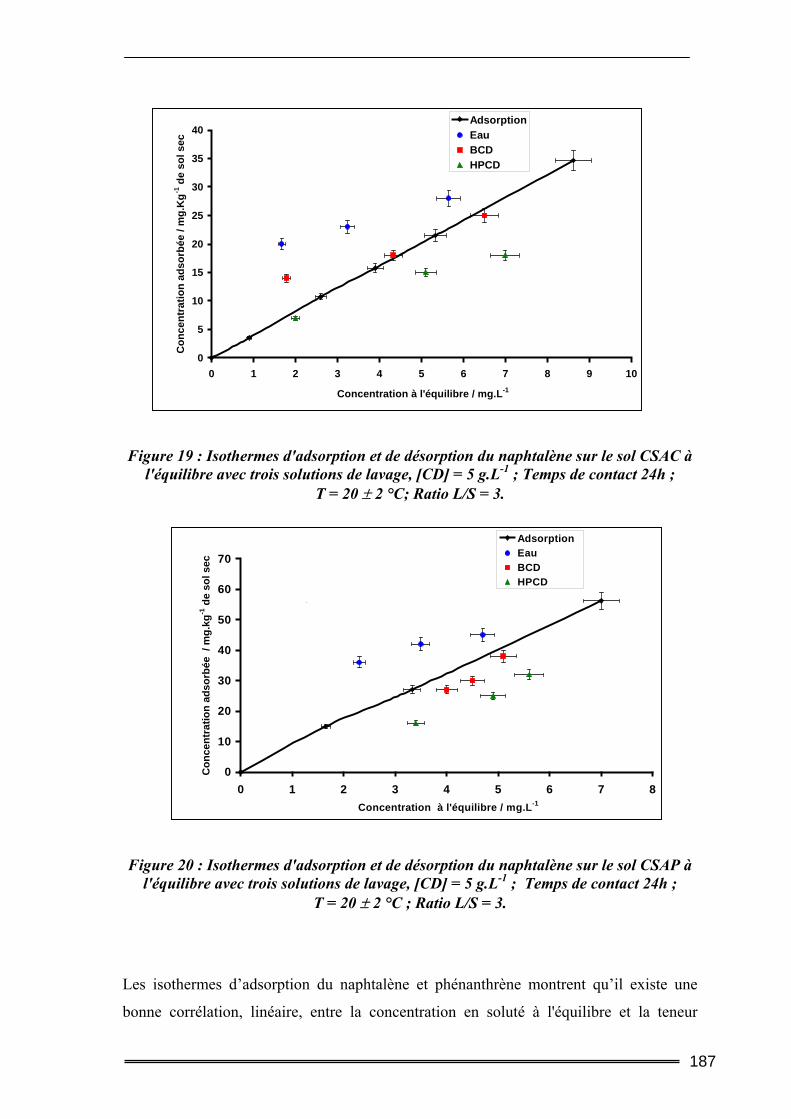
187
Figure 19 : Isothermes d'adsorption et de désorption du naphtalène sur le sol CSAC à l'équilibre avec trois solutions de lavage, [CD] = 5 g.L-1 ; Temps de contact 24h ;
T = 20 ± 2 °C; Ratio L/S = 3.
Figure 20 : Isothermes d'adsorption et de désorption du naphtalène sur le sol CSAP à l'équilibre avec trois solutions de lavage, [CD] = 5 g.L-1 ; Temps de contact 24h ;
T = 20 ± 2 °C ; Ratio L/S = 3.
Les isothermes d’adsorption du naphtalène et phénanthrène montrent qu’il existe une
bonne corrélation, linéaire, entre la concentration en soluté à l'équilibre et la teneur
0
10
20
30
40
50
60
70
0 1 2 3 4 5 6 7 8 Concentration à l'équilibre / mg.L-1
Con
cent
ratio
n ad
sorb
ée /
mg.
kg-1
de
sol s
ec
Adsorption EauBCDHPCD
0
5
10
15
20
25
30
35
40
0 1 2 3 4 5 6 7 8 9 10
Concentration à l'équilibre / mg.L-1
Con
cent
ratio
n ad
sorb
ée /
mg.
Kg-1
de
sol s
ec
AdsorptionEauBCDHPCD

188
adsorbée sur les deux sols (figures 19 et 20). En considérant que les conditions
expérimentales permettent d'atteindre les équilibres d'adsorption, nous avons exploité les
résultats d'adsorption suivant les modèles linéaires (Qads= Kd*Céq.). Les valeurs des
coefficients de partage sol/eau (Kd) pour le naphtalène et le phénanthrène seuls et en
mélange sur les sols sont reportés dans le tableau 7.
Tableau 7 : Valeurs du coefficient de partage sol/eau (Kd) pour le naphtalène et le phénanthrène seuls et en mélange. Pour les isothermes d’adsorption, le coefficient de
corrélation est compris entre 0,98 et 0,99.
Kd CSAC CSAP
Naphtalène 4,0 10,4
Phénanthrène 46,7 74,4
Naphtalène en mélange 3,1 5,4
Phénanthrène en mélange 25,0 31,1
On constate d’une façon générale, que les deux polluants s’adsorbent facilement sur les
sols étudiés et qu’ils ont plus d’affinité pour le sol CSAP (plus riche en matière
organique) que pour le sol CSAC. Tout comme pour les essais réalisés avec le PCP, la
fraction organique joue un rôle prépondérant dans la fixation du naphtalène et du
phénanthrène dans les sols.
L’adsorption du phénanthrène apparaît par ailleurs beaucoup plus forte que celle du
naphtalène. Ce résultat est parfaitement en corrélation avec le caractère hydrophobe des
polluants et leur solubilité. En effet, logKow vaut 4,46 pour le phénanthrène tandis qu’il
n’est que de 3,36 pour le naphtalène. La solubilité du phénanthrène dans l’eau est aussi
nettement inférieure à celle du naphtalène (SPhe = 1,29 contre 30 mg.L-1 pour le
naphtalène). Nous pouvons aussi constater que dans le cas où deux polluants sont
impliqués, l’adsorption de chaque polluant est plus faible que lorsqu’ils sont seuls. Il
semble qu’il y ait compétition entre les polluants sur les sites de fixation du sol, ce qui
diminue le nombre de sites disponibles pour chacun.
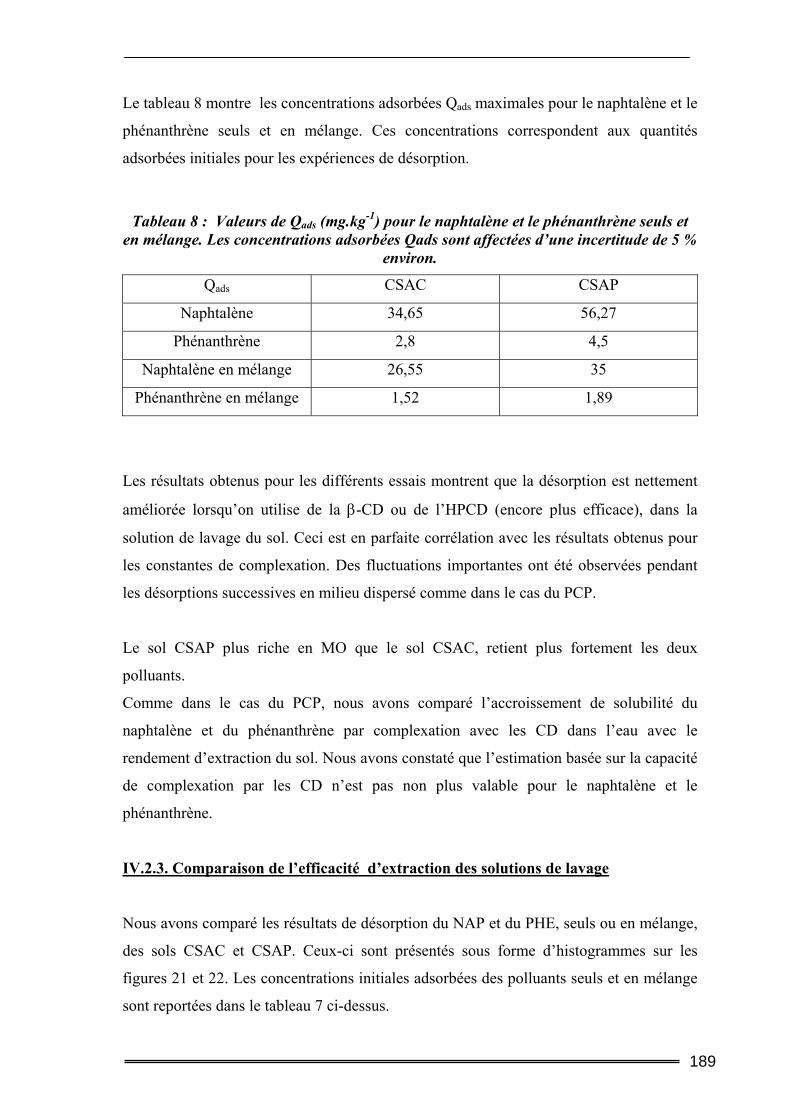
189
Le tableau 8 montre les concentrations adsorbées Qads maximales pour le naphtalène et le
phénanthrène seuls et en mélange. Ces concentrations correspondent aux quantités
adsorbées initiales pour les expériences de désorption.
Tableau 8 : Valeurs de Qads (mg.kg-1) pour le naphtalène et le phénanthrène seuls et en mélange. Les concentrations adsorbées Qads sont affectées d’une incertitude de 5 %
environ.
Qads CSAC CSAP
Naphtalène 34,65 56,27
Phénanthrène 2,8 4,5
Naphtalène en mélange 26,55 35
Phénanthrène en mélange 1,52 1,89
Les résultats obtenus pour les différents essais montrent que la désorption est nettement
améliorée lorsqu’on utilise de la β-CD ou de l’HPCD (encore plus efficace), dans la
solution de lavage du sol. Ceci est en parfaite corrélation avec les résultats obtenus pour
les constantes de complexation. Des fluctuations importantes ont été observées pendant
les désorptions successives en milieu dispersé comme dans le cas du PCP.
Le sol CSAP plus riche en MO que le sol CSAC, retient plus fortement les deux
polluants.
Comme dans le cas du PCP, nous avons comparé l’accroissement de solubilité du
naphtalène et du phénanthrène par complexation avec les CD dans l’eau avec le
rendement d’extraction du sol. Nous avons constaté que l’estimation basée sur la capacité
de complexation par les CD n’est pas non plus valable pour le naphtalène et le
phénanthrène.
IV.2.3. Comparaison de l’efficacité d’extraction des solutions de lavage
Nous avons comparé les résultats de désorption du NAP et du PHE, seuls ou en mélange,
des sols CSAC et CSAP. Ceux-ci sont présentés sous forme d’histogrammes sur les
figures 21 et 22. Les concentrations initiales adsorbées des polluants seuls et en mélange
sont reportées dans le tableau 7 ci-dessus.
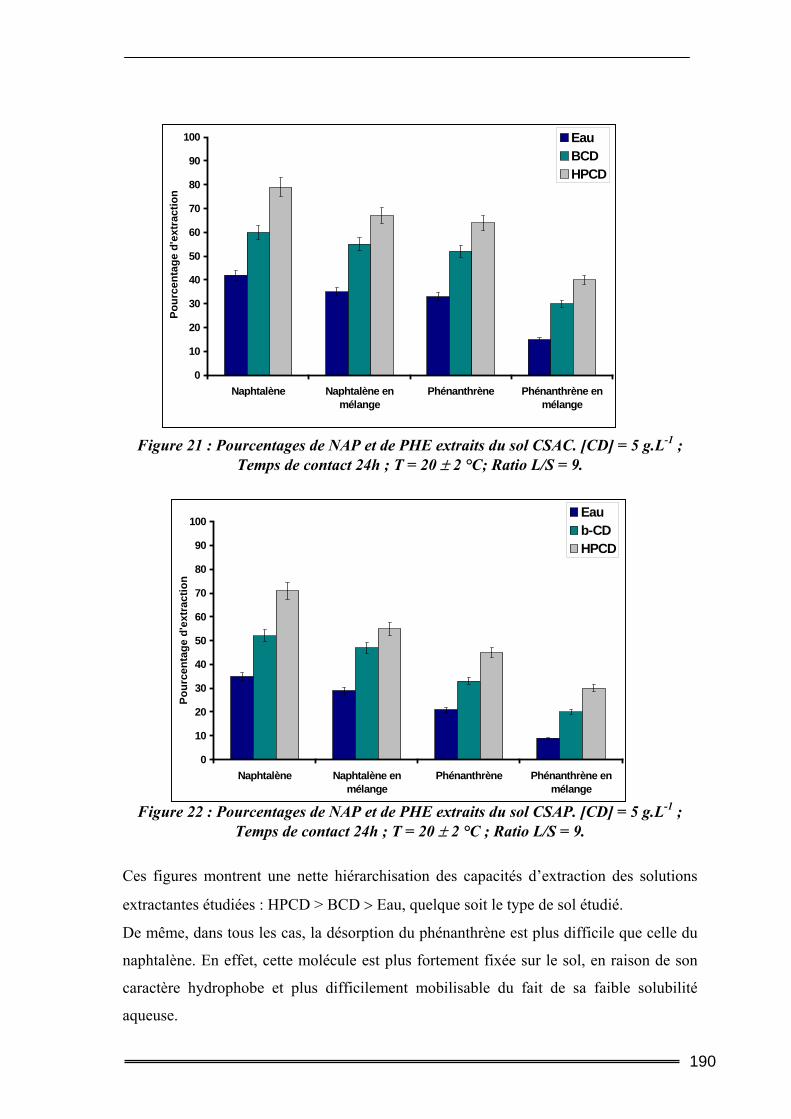
190
Figure 21 : Pourcentages de NAP et de PHE extraits du sol CSAC. [CD] = 5 g.L-1 ; Temps de contact 24h ; T = 20 ± 2 °C; Ratio L/S = 9.
Figure 22 : Pourcentages de NAP et de PHE extraits du sol CSAP. [CD] = 5 g.L-1 ; Temps de contact 24h ; T = 20 ± 2 °C ; Ratio L/S = 9.
Ces figures montrent une nette hiérarchisation des capacités d’extraction des solutions
extractantes étudiées : HPCD > BCD > Eau, quelque soit le type de sol étudié.
De même, dans tous les cas, la désorption du phénanthrène est plus difficile que celle du
naphtalène. En effet, cette molécule est plus fortement fixée sur le sol, en raison de son
caractère hydrophobe et plus difficilement mobilisable du fait de sa faible solubilité
aqueuse.
0
10
20
30
40
50
60
70
80
90
100
Naphtalène Naphtalène enmélange
Phénanthrène Phénanthrène enmélange
Pou
rcen
tage
d'e
xtra
ctio
n
EauBCDHPCD
0
10
20
30
40
50
60
70
80
90
100
Naphtalène Naphtalène enmélange
Phénanthrène Phénanthrène enmélange
Pour
cent
age
d'ex
trac
tion
Eaub-CDHPCD
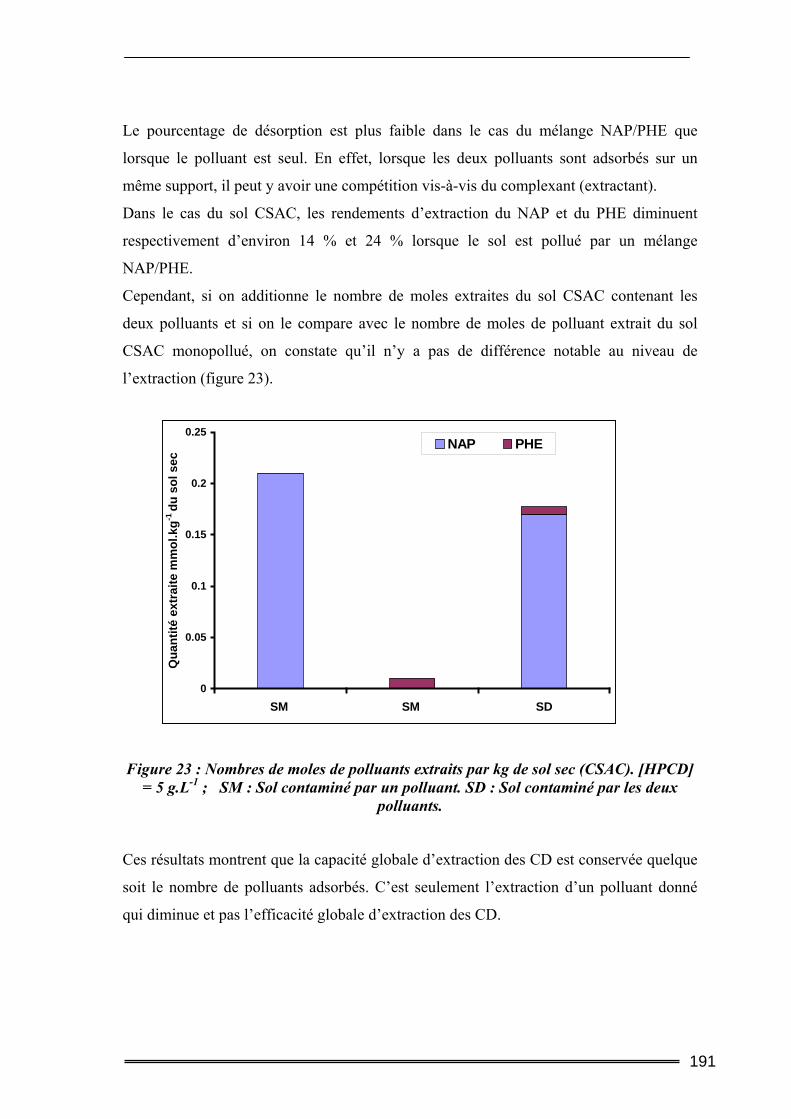
191
Le pourcentage de désorption est plus faible dans le cas du mélange NAP/PHE que
lorsque le polluant est seul. En effet, lorsque les deux polluants sont adsorbés sur un
même support, il peut y avoir une compétition vis-à-vis du complexant (extractant).
Dans le cas du sol CSAC, les rendements d’extraction du NAP et du PHE diminuent
respectivement d’environ 14 % et 24 % lorsque le sol est pollué par un mélange
NAP/PHE.
Cependant, si on additionne le nombre de moles extraites du sol CSAC contenant les
deux polluants et si on le compare avec le nombre de moles de polluant extrait du sol
CSAC monopollué, on constate qu’il n’y a pas de différence notable au niveau de
l’extraction (figure 23).
Figure 23 : Nombres de moles de polluants extraits par kg de sol sec (CSAC). [HPCD] = 5 g.L-1 ; SM : Sol contaminé par un polluant. SD : Sol contaminé par les deux
polluants.
Ces résultats montrent que la capacité globale d’extraction des CD est conservée quelque
soit le nombre de polluants adsorbés. C’est seulement l’extraction d’un polluant donné
qui diminue et pas l’efficacité globale d’extraction des CD.
0
0.05
0.1
0.15
0.2
0.25
SM SM SD
Qua
ntité
ext
raite
mm
ol.k
g-1 du
sol
sec
NAP PHE

192
IV.3. Bilan Les résultats expérimentaux de l’étude en milieu dispersé prouvent que les cyclodextrines
favorisent la décontamination des sols pollués par le naphtalène et le phénanthrène.
L’HPCD est toutefois beaucoup plus efficace que la β-CD. L’étude cinétique a montré
que l’extraction des polluants des sols par les CD est un phénomène rapide tout comme la
complexation dans la phase aqueuse.
L’extraction est moins efficace sur le sol CSAP quelque soit la solution de lavage utilisée.
Une diminution d’environ 10 % du rendement d’extraction a été observée quand la teneur
en MO augmente de 2 à 5 % dans le sol. Tout comme pour le PCP, ces résultats
confirment l’influence importante de la MO du sol sur l’extraction des polluants par les
CD.
Nos travaux montrent également que la présence d’un co-polluant influence les processus
d’extraction et que l’importance de ce phénomène dépend de la nature du polluant cible.
La capacité globale d’extraction des CD reste toutefois conservée quelque soit le type de
pollution (mono- ou multi-pollution).
On observe que la quantité de NAP extrait à l’équilibre est plus importante que celle de
PHE. Cela peut s’expliquer par la solubilité apparente des HAP en présence de
cyclodextrine. Celle-ci dépend, à la fois de la solubilité aqueuse des HAP, et de la
constante de complexation à une concentration donnée de cyclodextrine.
La constante de complexation du PHE est plus élevée que celle du NAP (chapitre 1 :
Résultats) et la solubilité aqueuse du NAP est 30 fois plus élevée que celle du PHE. On
peut conclure alors que l’influence de la solubilité aqueuse est prépondérante par rapport
à la capacité de complexation, ce qui conduit le naphtalène à atteindre une plus grande
concentration apparente en phase aqueuse. Notons également que le PHE est fortement
adsorbé sur le sol, ce qui est cohérent avec son Kow élevé.

193
V. Etude des interactions cyclodextrines/sols
V.1. Adsorption des cyclodextrines sur les sols
La mise au point d’un procédé d’extraction des polluants organiques requiert l’évaluation
des interactions susceptibles de se produire entre les cyclodextrines et les sols en fonction
de la composition, de ceux-ci. En effet, si les CD sont retenues par certains types de sols,
leur utilisation en procédé de lavage risque de contribuer à une refixation des polluants
solubilisés. De plus, une perte due à l’adsorption des CD sur le sol à traiter peut accroître
la quantité de CD nécessaire pour mobiliser les polluants adsorbés sur ce sol.
Il est aujourd’hui admis que le rôle de la matière organique est prépondérant dans
l’adsorption des molécules organiques sur les sols. Certains auteurs mettent également en
cause les argiles. C’est pourquoi nous avons étudié l’adsorption de la BCD, de l’HPCD et
de la MCD sur 6 matrices différentes. Il s’agit d’une part de 4 échantillons de type
argileux, et d’autre part de deux échantillons de sol différents par leurs taux de matière
organique. Le plus riche en matière organique a été prélevé sous prairie (CSAP) tandis
que l’autre provient d’un sol cultivé (CSAC). Nous pouvons grâce à ces deux dernières
matrices étudier l’influence de la teneur en matière organique sur l’adsorption des CD sur
les sols.
La rétention des trois CD sur les 6 matrices de sol a été quantifiée au moyen d’essais
d’adsorption en milieu dispersé tels que ceux décrits au paragraphe III.1. Le dosage des
CD restées en solution a été effectué par analyse fluorimètrique selon le principe que
nous allons présenter au paragraphe suivant.
V.1.1. Dosage des cyclodextrines
Il existe plusieurs méthodes analytiques pour doser les CD telles que la chromatographie
liquide à haute performance et la COTmètrie (Ko et al., 1998). Cette dernière est peu
sensible car le milieu est souvent riche en carbone organique provenant d’autres sources.
Dans cette étude, nous avons utilisé une méthode de dosage indirecte, basée sur les
propriétés complexantes des CD vis-à-vis d’une sonde fluorescente.

194
Méthode Fluorimètrique
Principe La méthode fluorimètrique est basée sur l’exaltation de l’intensité de la fluorescence
d’une sonde (molécule fluorescente) en présence de CD du fait de la formation d’un
complexe. La sonde de fluorescence utilisée est souvent le 6-p-toluidinylnaphthalene-2-
sulfonate (TNS) qui forme un complexe 1 : 1 avec les CD (Kondo et al, 1976 ; Coly et
Auron, 1997).
Suivant cette stœchiométrie (1 :1), une régression linéaire peut être proposée :
000
1][)(
11FFCDKFFFF −
+−
=− ∞∞
(1 : 1 complexe)
Où Fo et F∞ sont respectivement les intensités de fluorescence de la sonde en absence et
en présence d’un excès de CD, F étant l’intensité de fluorescence du TNS à une
concentration déterminée de CD (Kondo et al, 1976 ; Coly et Auron, 1997 ; Ko et al.,
1999).
Analyses des surnageants Pour déterminer la concentration en CD à l’équilibre dans les surnageants issus des essais
d’adsorption, nous devons préalablement établir une droite d’étalonnage permettant de
déterminer une concentration inconnue en CD par l’intermédiaire de l’intensité de
fluorescence mesurée du milieu.
Pour ce but, une solution aqueuse de TNS à 10-6 mol.L-1 a été préparée et protégée de la
lumière puisque le TNS est très photosensible.
Des prélèvements de 5 ml de solution mère de TNS sont transférés dans des fioles jaugés
de 10 mL. Les fioles contenant le TNS sont ensuite complétées (i) avec de l’eau pour
déterminer l’intensité de fluorescence du TNS dans l’eau (F0), (ii) ou avec des solutions
aqueuses de cyclodextrine à des concentrations croissantes ([CD]1, [CD]2, [CD]3, etc.).
Les solutions obtenues sont ensuite analysées par un spectrofluorimètre (KONTRON,
SFM 25). Les longueurs d’onde d’excitation et d’émission du TNS sont respectivement
305 et 460 nm.
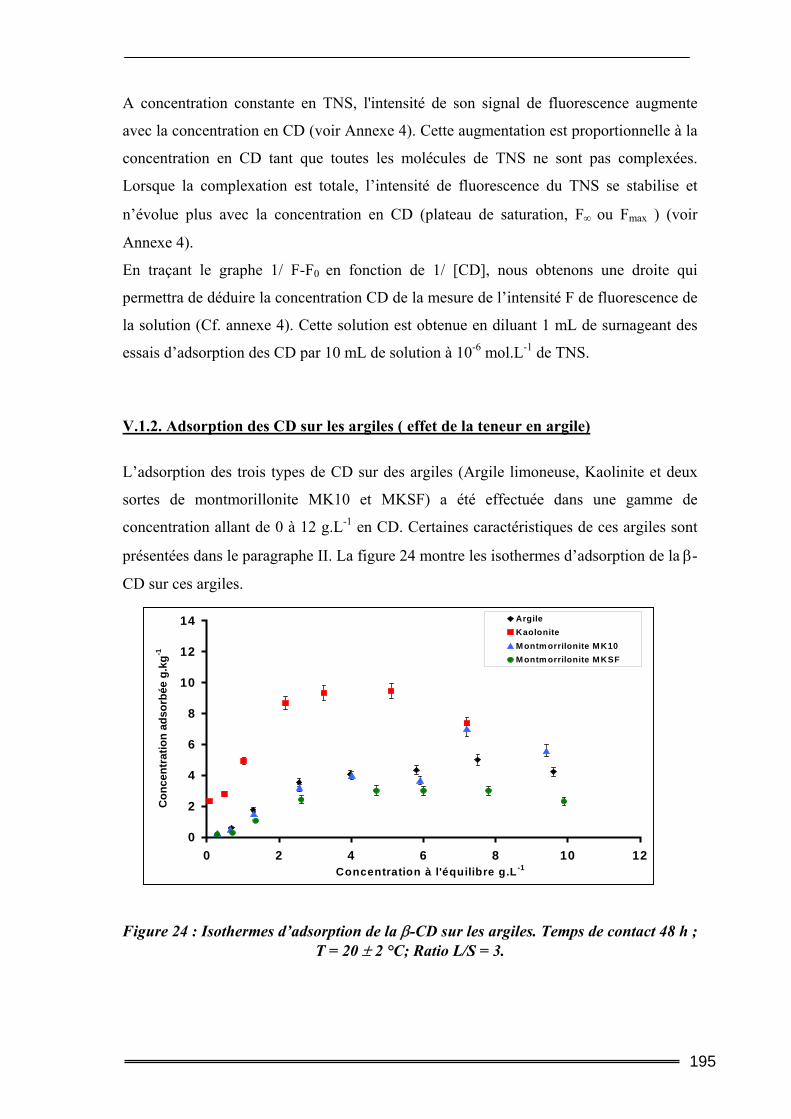
195
A concentration constante en TNS, l'intensité de son signal de fluorescence augmente
avec la concentration en CD (voir Annexe 4). Cette augmentation est proportionnelle à la
concentration en CD tant que toutes les molécules de TNS ne sont pas complexées.
Lorsque la complexation est totale, l’intensité de fluorescence du TNS se stabilise et
n’évolue plus avec la concentration en CD (plateau de saturation, F∞ ou Fmax ) (voir
Annexe 4).
En traçant le graphe 1/ F-F0 en fonction de 1/ [CD], nous obtenons une droite qui
permettra de déduire la concentration CD de la mesure de l’intensité F de fluorescence de
la solution (Cf. annexe 4). Cette solution est obtenue en diluant 1 mL de surnageant des
essais d’adsorption des CD par 10 mL de solution à 10-6 mol.L-1 de TNS.
V.1.2. Adsorption des CD sur les argiles ( effet de la teneur en argile)
L’adsorption des trois types de CD sur des argiles (Argile limoneuse, Kaolinite et deux
sortes de montmorillonite MK10 et MKSF) a été effectuée dans une gamme de
concentration allant de 0 à 12 g.L-1 en CD. Certaines caractéristiques de ces argiles sont
présentées dans le paragraphe II. La figure 24 montre les isothermes d’adsorption de la β-
CD sur ces argiles.
Figure 24 : Isothermes d’adsorption de la β-CD sur les argiles. Temps de contact 48 h ; T = 20 ± 2 °C; Ratio L/S = 3.
0
2
4
6
8
10
12
14
0 2 4 6 8 10 12Concentration à l'équilibre g.L-1
Con
cent
ratio
n ad
sorb
ée g
.kg-1
Argile KaoloniteMontm orrilonite MK10Montm orrilonite MKSF
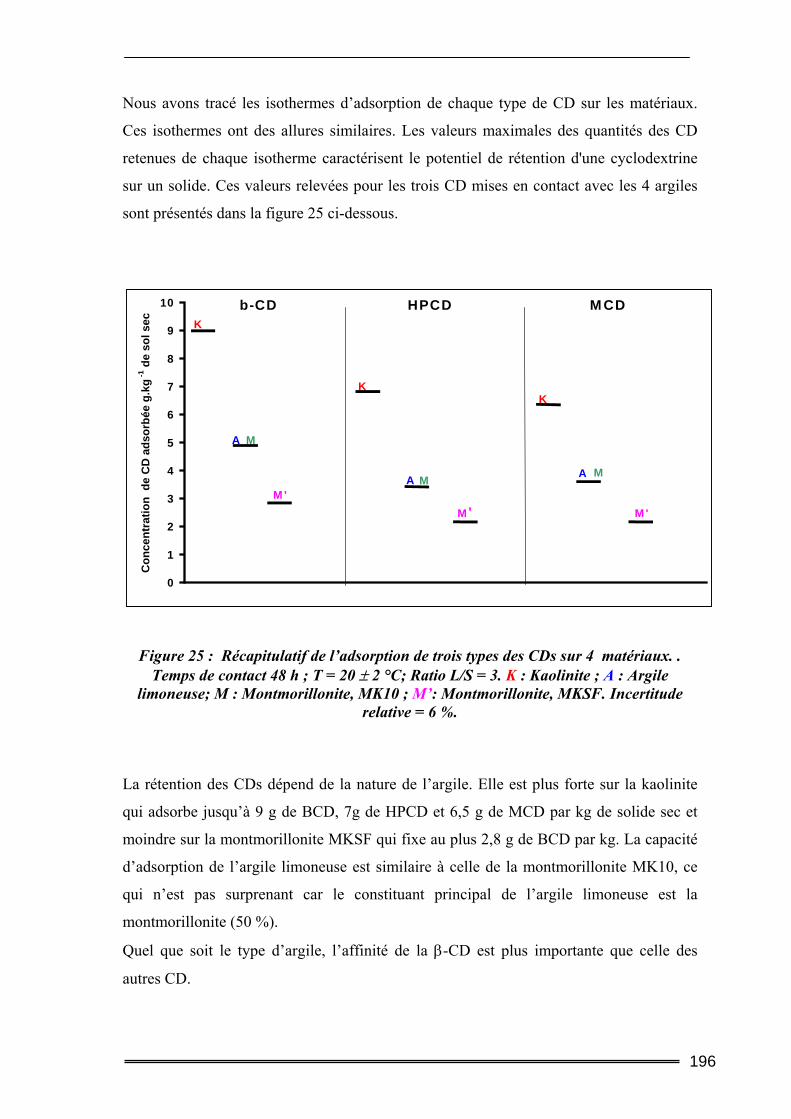
196
Nous avons tracé les isothermes d’adsorption de chaque type de CD sur les matériaux.
Ces isothermes ont des allures similaires. Les valeurs maximales des quantités des CD
retenues de chaque isotherme caractérisent le potentiel de rétention d'une cyclodextrine
sur un solide. Ces valeurs relevées pour les trois CD mises en contact avec les 4 argiles
sont présentés dans la figure 25 ci-dessous.
Figure 25 : Récapitulatif de l’adsorption de trois types des CDs sur 4 matériaux. . Temps de contact 48 h ; T = 20 ± 2 °C; Ratio L/S = 3. K : Kaolinite ; A : Argile
limoneuse; M : Montmorillonite, MK10 ; M’: Montmorillonite, MKSF. Incertitude relative = 6 %.
La rétention des CDs dépend de la nature de l’argile. Elle est plus forte sur la kaolinite
qui adsorbe jusqu’à 9 g de BCD, 7g de HPCD et 6,5 g de MCD par kg de solide sec et
moindre sur la montmorillonite MKSF qui fixe au plus 2,8 g de BCD par kg. La capacité
d’adsorption de l’argile limoneuse est similaire à celle de la montmorillonite MK10, ce
qui n’est pas surprenant car le constituant principal de l’argile limoneuse est la
montmorillonite (50 %).
Quel que soit le type d’argile, l’affinité de la β-CD est plus importante que celle des
autres CD.
0
1
2
3
4
5
6
7
8
9
10
Con
cent
ratio
n d
e C
D a
dsor
bée
g.kg
-1 d
e so
l sec
b -CD HPCD M CDK
KK
A
A A
M
M M
M 'M' M '
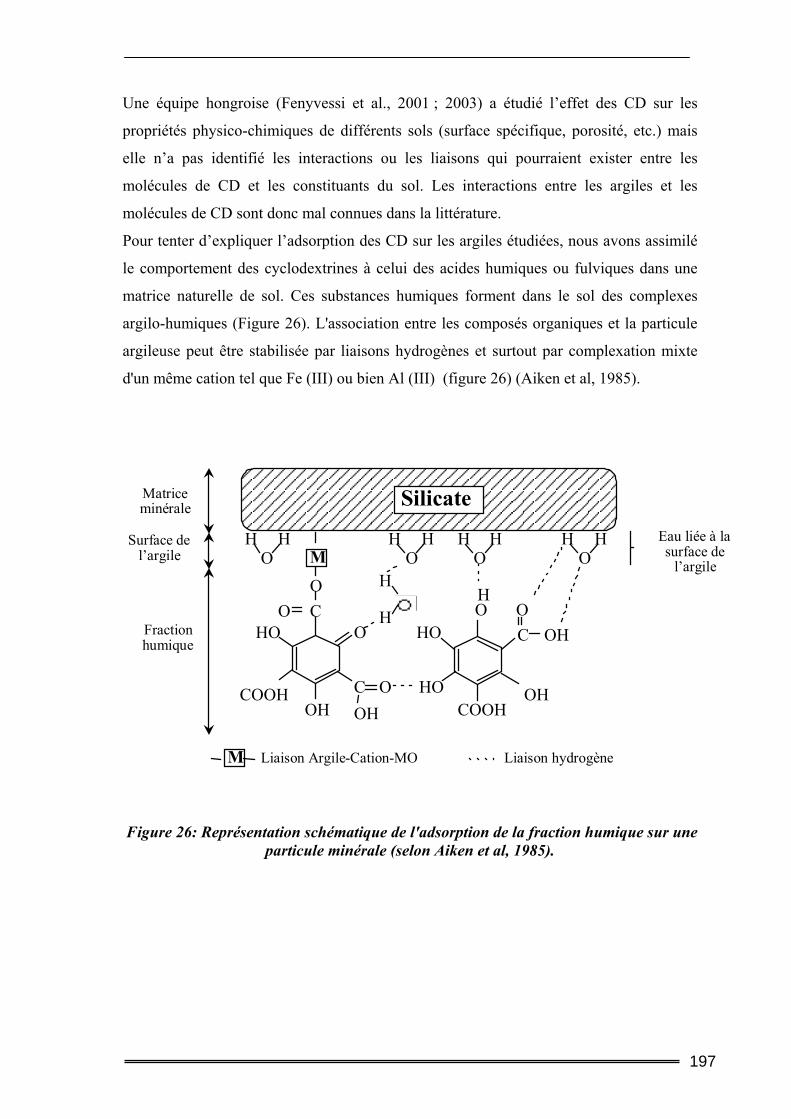
197
Une équipe hongroise (Fenyvessi et al., 2001 ; 2003) a étudié l’effet des CD sur les
propriétés physico-chimiques de différents sols (surface spécifique, porosité, etc.) mais
elle n’a pas identifié les interactions ou les liaisons qui pourraient exister entre les
molécules de CD et les constituants du sol. Les interactions entre les argiles et les
molécules de CD sont donc mal connues dans la littérature.
Pour tenter d’expliquer l’adsorption des CD sur les argiles étudiées, nous avons assimilé
le comportement des cyclodextrines à celui des acides humiques ou fulviques dans une
matrice naturelle de sol. Ces substances humiques forment dans le sol des complexes
argilo-humiques (Figure 26). L'association entre les composés organiques et la particule
argileuse peut être stabilisée par liaisons hydrogènes et surtout par complexation mixte
d'un même cation tel que Fe (III) ou bien Al (III) (figure 26) (Aiken et al, 1985).
COOH
COO
M
HO O
OHO
OHC
OH H
OH H
OH H
OH H
Silicate
O OC OHHO
HO
H
HH
OHCOOH
Matriceminérale
Surface del’argile
Fractionhumique
Eau liée à lasurface de
l’argile
M Liaison Argile-Cation-MO Liaison hydrogène
Figure 26: Représentation schématique de l'adsorption de la fraction humique sur une particule minérale (selon Aiken et al, 1985).

198
O
M
HO
HO
OH H
OH H
OH H
OH H
Silicate
O OHC
HOHO
HO
H
OHCOOH
Matriceminérale
Surface del’argile
Fractionhumique etcyclodextrine
Eau liée surface l’argile
M Liaison Argile-Cation-CD Liaison hydrogène
CDCD
HO
OH
HO
HO
OH
HO OH
OH
HO
O O
Figure 27: Représentation de l’association globale de la β-CD et de la fraction MO sur une particule minérale (Argile).
Deux hypothèses peuvent alors être proposées pour expliquer l’adsorption des CD sur les
argiles :
1- Comme indiqué sur la figure 27, les molécules de CD peuvent établir, du fait de leur
nombreux groupements hydroxyls, des liaisons hydrogènes avec les molécules d’eau
fixées sur les argiles. Les minéraux argileux, souvent à charges négatives, s'entourent
facilement d'un cortège de molécules d'eau (Rogers et al., 1980).
2- Des liaisons CD-cation métallique-argile peuvent également être envisagées. La charge
de surface des argiles est liée à l’hydrolyse de liens Si-O et Al-OH le long des surfaces.
Cela dépend du pH du milieu et du point de charge nulle de l’argile. Lorsque la charge de
surface est négative, les argiles peuvent fixer des cations (Capacité d’Echange
Cationique). En principe, les ions bivalents sont plus fortement liés et retenus par les
argiles que les ions monovalents (Coats et al., 1964). D’autre part, certaines CD sont
connues par leur aptitude à complexer les cations tels que le Fe2+, le Ca2+ ou le Cd2+
(Brusseau et al, 1997, Lindesy et al, 2003). La complexation simultanée de ces cations
présents dans le sol par les CD et les feuilles d’argile favoriserait la rétention des CD par
la structure du sol (fig. 27).
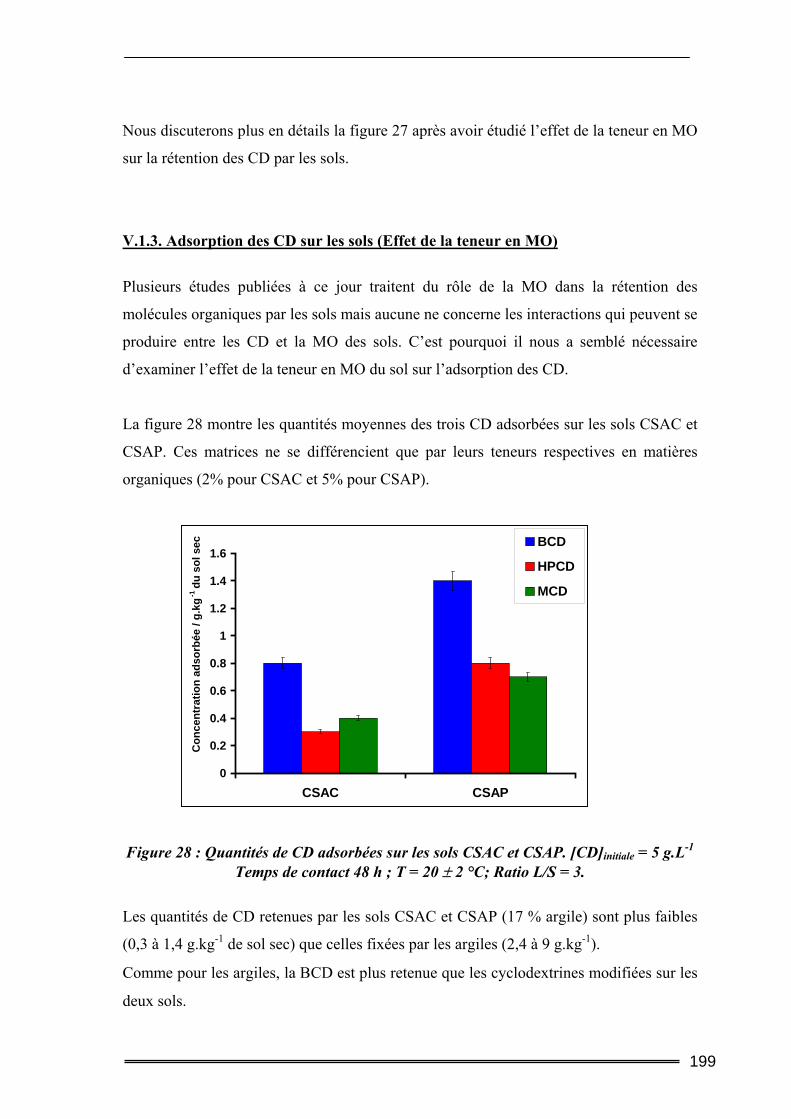
199
Nous discuterons plus en détails la figure 27 après avoir étudié l’effet de la teneur en MO
sur la rétention des CD par les sols.
V.1.3. Adsorption des CD sur les sols (Effet de la teneur en MO)
Plusieurs études publiées à ce jour traitent du rôle de la MO dans la rétention des
molécules organiques par les sols mais aucune ne concerne les interactions qui peuvent se
produire entre les CD et la MO des sols. C’est pourquoi il nous a semblé nécessaire
d’examiner l’effet de la teneur en MO du sol sur l’adsorption des CD.
La figure 28 montre les quantités moyennes des trois CD adsorbées sur les sols CSAC et
CSAP. Ces matrices ne se différencient que par leurs teneurs respectives en matières
organiques (2% pour CSAC et 5% pour CSAP).
Figure 28 : Quantités de CD adsorbées sur les sols CSAC et CSAP. [CD]initiale = 5 g.L-1 Temps de contact 48 h ; T = 20 ± 2 °C; Ratio L/S = 3.
Les quantités de CD retenues par les sols CSAC et CSAP (17 % argile) sont plus faibles
(0,3 à 1,4 g.kg-1 de sol sec) que celles fixées par les argiles (2,4 à 9 g.kg-1).
Comme pour les argiles, la ΒCD est plus retenue que les cyclodextrines modifiées sur les
deux sols.
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
CSAC CSAP
Con
cent
ratio
n ad
sorb
ée /
g.kg
-1 du
sol
sec
BCD
HPCD
MCD

200
L’adsorption des CD est plus forte sur le sol CSAP plus riche en MO que sur le sol
CSAC quelque soit le type de CD. Ce phénomène a également été observé pour
l’adsorption des polluants organiques tels que le PCP, le NAP et le PHE. Comme cela est
démontré dans le cas des polluants, la fraction organique des sols paraît jouer un rôle
dans la fixation des CD.
Les principales propriétés adsorbantes de la matière organique des sols (MO) sont
essentiellement liées à leur nature colloïdale et à la présence de nombreux groupements
fonctionnels (carboxyles, carbonyles, hydroxyles, amines, amides, groupements
sulfoniques, …) (Duchaufour, 1991).
Comme indiqué sur la figure 27, des liaisons hydrogènes peuvent se former entre les CD
et les substances humiques fixées dans les complexes argilo-humiques. Une complexation
simultanée des cations par les CD et les MO est également possible. Les matières
organiques naturelles peuvent assurer, du fait de leur structure chimique complexe, des
liaisons avec d’autres composés organiques comme les cyclodextrines.
Dans le cas des cyclodextrines, on peut imaginer également que les liaisons des CD avec
la MO sont essentiellement dues aux interactions hydrophobes avec les noyaux
benzéniques qui constituent les chaînes d’acides humiques et fulviques. Cette hypothèse
est cohérente avec le fait que la BCD s’adsorbe plus fortement que l’HPCD et la MCD,
non seulement sur les sols CSAC et CSAP mais aussi sur les argiles. Or, d’après des
études énergétiques des interactions eau/CD (de Brauer et al., 2002), la BCD est moins
apte que les autres CD à établir des liaisons hydrogènes avec les molécules d’eau, ce qui
justifie sa solubilité moindre par rapport aux autres cyclodextrines.
De par sa faible solubilité aqueuse, l’adsorption de la BCD sur les MO (ou son affinité
pour les noyaux aromatiques) pourrait ainsi être favorisée. Comme cela est largement
connu dans le cas des polluants, moins la molécule est soluble, plus elle a tendance à être
« exclue » du solvant et à interagir avec les surfaces solides ou les phases organiques.
L’effet est inverse pour les autres CD qui sont plus solubles dans l’eau et s’adsorbent plus
faiblement sur les solides.

201
V.1.4. Bilan
Deux grands types d'informations peuvent être formulés à partir du croisement de
l'ensemble de ces résultats:
- Le comportement global des trois cyclodextrines est similaire vis-à-vis de chaque type
de sols testés. Les CD sont plus retenues sur la kaolinite, l'argile limoneuse et les
montmorrillonite. Elles sont, par contre, peu adsorbées par les deux sols faiblement
argileux de La Côte St André. Dans ce dernier cas, l'écart d'adsorption est systématique
entre les sols CSAP et CSAC. Il est probablement lié aux interactions des CD avec la
matière organique dont la teneur est plus importante dans le sol sous Prairie (CSAP). Les
quantités de CD retenues par les sols testés demeurent toutefois faibles.
- Le comparatif des 3 CD montre que, quel que soit le type de sol, il existe une hiérarchie
dans les facteurs de rétention. D'une manière systématique, la ΒCD est plus retenue que
les cyclodextrines modifiées. Le facteur de rétention diminue dans l’ordre : ΒCD >
HPCD > MCD. Cela peut expliquer les changements de hiérarchie notés dans les cas de
solubilisation et d’extraction du PCP (Cf. III.1.2.4.). Pour rappel, le gain de solubilité du
PCP augmentait dans l’ordre : MCD > BCD > HPCD, tandis que celui de l’extraction
augmentait dans l’ordre : MCD > HPCD > BCD. Cela peut être du à la fixation
relativement importante de la BCD sur les sols.
Nous pouvons conclure de ces résultats que la teneur en argile dans le sol et le taux de
MO influencent la rétention des CD. Ainsi, la caractérisation du sol (teneur en argiles,
type d’argiles, teneur en matières organiques) sera une étape indispensable avant la mise
en œuvre d’un procédé de traitement de sol utilisant des CD.
On peut d’ores et déjà conclure que cette technique sera d’autant plus efficace que le sol
sera moins argileux et qu’il contiendra moins de matières organiques.

202
V.2. Mobilisation des matières organiques du sol (Dosage du Carbone
Organique Total) Cette étude a pour objectif d’évaluer si la mobilisation de la Matière Organique Naturelle
(MON) est plus importante en présence de CD que lors d’une simple lixiviation à l’eau.
Dans ce cas, la procédure de lixiviation (milieu dispersé ou colonne) a-t-elle une
importance ? et les CD ont-elles une influence différente selon leur type ?.
L’impact de solutions aqueuses de cyclodextrines sur la mobilisation des matières
organiques du sol a été étudié par la détermination du carbone organique total (COT)
mobilisé dans la phase liquide mise en contact avec le sol et du COT restant sur les
matrices de sol.
V.2.1. Procédures expérimentales
Une quantité définie de sol CSAP est mise en contact avec différentes solutions
aqueuses : de l’eau (témoin), une solution de β-CD à 9,43 g.L-1, une solution de HPCD à
9,87 g.L-1 et une solution de MCD à 9,74 g.L-1. Nous avons choisi pour cette étude le sol
CSAP car il est relativement riche en COT (5% MO). Le contact des solutions aqueuses
avec le sol a été effectué en milieu dispersé et en colonne de percolation comme décrits
au paragraphe III.
Après un contact de 48 h, le surnageant ou le liquide est récupéré puis analysé au moyen
d’un COTmètre afin de connaître la concentration en COT (mg/L). Les matrices de sol
sont également récupérées et analysées pour déterminer leur pourcentage de COT par
unité de masse de sol sec.
COTmètre Un COT-mètre permet de doser quantitativement le carbone contenu dans des
échantillons liquides ou solides. On distingue le carbone organique total (COT) et le
carbone inorganique (CI) (carbonates, hydrogénocarbonates, etc.). Le carbone total (CT)
est la somme des 2 : CT = COT + CI. Le dosage des différentes formes de carbone est
effectué par le COTmètre (OI Analytical).

203
Module solide
L’analyse du carbone total (CT) dans un échantillon solide s’effectue par oxydation
du carbone en CO2 dans un four à 950°C en présence d’un catalyseur et d’un flux
d’O2.
Une faible masse d’échantillon (<100 mg) est pesée et introduite dans le four. Le CO2
gazeux obtenu est détecté par une cellule IR.
La mesure du COT dans un échantillon solide s’effectue après attaque à l’acide
phosphorique (H3PO4) à 15 % afin d’éliminer par décomposition des carbonates et
hydrogènocarbonates. Après séchage, l’échantillon attaqué est analysé comme pour le
CT.
Module liquide
Un microvolume de l’échantillon liquide est injecté dans un four à 680 °C rempli de
billes de catalyseur au platine. Le four est traversé par le flux d’oxygène qui permet la
combustion de la totalité du carbone C présent dans l’échantillon selon la réaction:
C + O2 (g) CO2 (g)
Le CO2 gazeux obtenu est entraîné par le flux d’oxygène et amené jusqu’à une cellule
infrarouge qui permet la mesure du taux de CO2, directement proportionnel à la quantité
de carbone total initial.
Pour la mesure du COT, on ajoute, avant la combustion, une quantité d’acide
chlorhydrique (1M) afin de transformer le carbone minéral (CI) en CO2 gazeux. Celui ci
est éliminé de la solution par un dégazage sous oxygène.
V.2.2. Résultats du dosage de Carbone Organique Total
La figure 29 représente les concentrations en Carbone Organique Total mesurées dans
les liquides (surnageants). Les valeurs représentées sont la moyenne des 3 replicats.
L’écart relatif indiqué par les barres varie entre 5 et 6 %.
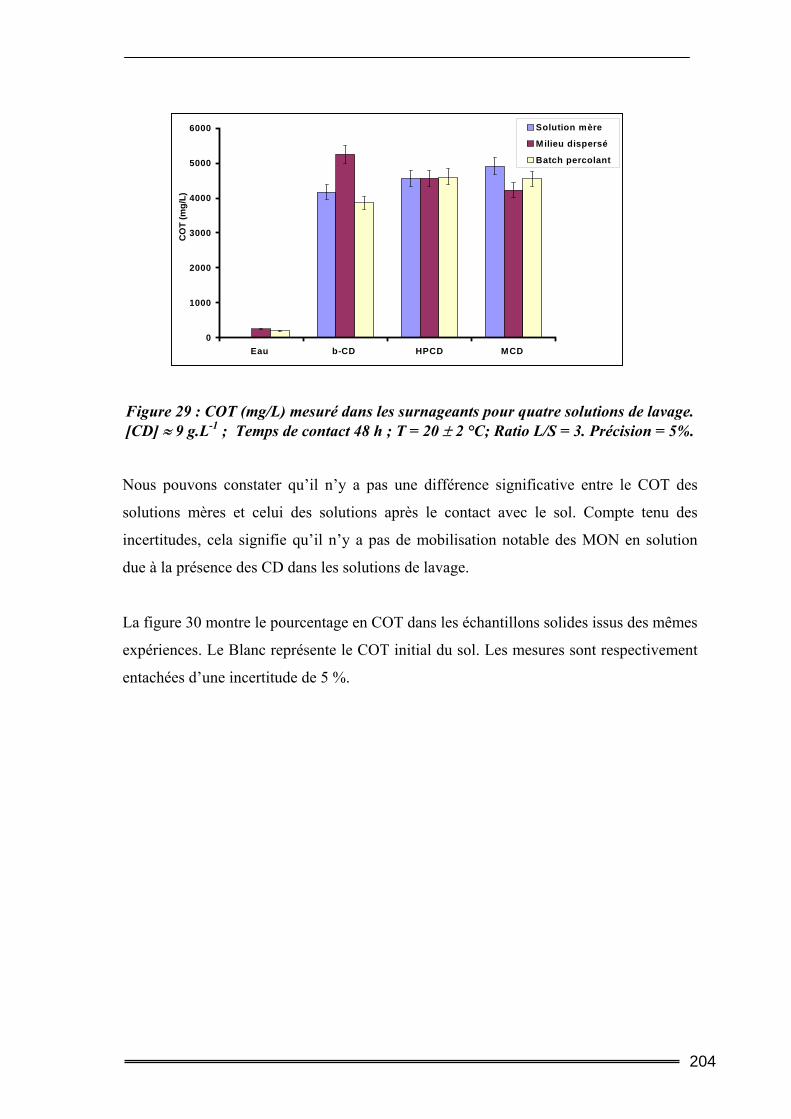
204
Figure 29 : COT (mg/L) mesuré dans les surnageants pour quatre solutions de lavage. [CD] ≈ 9 g.L-1 ; Temps de contact 48 h ; T = 20 ± 2 °C; Ratio L/S = 3. Précision = 5%.
Nous pouvons constater qu’il n’y a pas une différence significative entre le COT des
solutions mères et celui des solutions après le contact avec le sol. Compte tenu des
incertitudes, cela signifie qu’il n’y a pas de mobilisation notable des MON en solution
due à la présence des CD dans les solutions de lavage.
La figure 30 montre le pourcentage en COT dans les échantillons solides issus des mêmes
expériences. Le Blanc représente le COT initial du sol. Les mesures sont respectivement
entachées d’une incertitude de 5 %.
0
1000
2000
3000
4000
5000
6000
Eau b-CD HPCD MCD
CO
T (m
g/L)
Solution mère
Milieu dispersé
Batch percolant

205
Figure 30 : Le COT (%) sur les échantillons de sol avant et après le traitement par les CD. Temps de contact 48 h ; T = 20 ± 2 °C; Ratio L/S = 3. Précision = 5%.
D’après les mesures de COT sur les matrices ayant été traitées en colonne et compte tenu
de l’incertitude expérimentale, la mobilisation de la MON est similaire quelque soit la
solution lixiviante (eau ou CD). Lors d’un traitement en colonne, environ 25 % de la
MON est mobilisée.
En milieu dispersé, les analyses de COT sur les matrices lixiviées accusent une
diminution plus importante de la matière organique résiduelle qu’après un traitement en
colonne. Ces résultats montrent que le traitement en colonne préserve mieux l’intégrité du
sol. Il est toutefois surprenant de constater qu’en milieu dispersé, la mobilisation de la
MON est plus marquée en présence d’eau (66%) que lorsque des solutions de CD sont
utilisées.
Bien que les analyses de COT sur les lixiviats ne recoupent pas parfaitement celles
réalisés sur les sols lixiviés, les deux types de résultats prouvent clairement que les
solutions de CD ne mobilisent pas sensiblement plus les MON du sol que l’eau pure. Les
CD préservent donc la structure du sol et ne perturbent pas cet écosystème après le
traitement.
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
1.8
2
Blanc Eau b-CD HPCD MCD
CO
T (%
)
Milieu dispersé
Batch percolant

206
VI. Conclusion
Des essais d’adsorption/désorption du PCP, du naphtalène et du phénanthrène sur deux
sols de même origine géologique mais plus ou moins riches en matière organique ont été
réalisés dans l’objectif majeur d’étudier l’influence de trois sortes de cyclodextrines sur la
désorption des polluants.
La plupart des essais ont été effectués en milieu dispersé. Ce protocole, rapide à mettre en
œuvre, présente néanmoins l’inconvénient de conduire rapidement à une déstructuration
du sol qui peut influencer les processus d’adsorption/désorption. Une étude, limitée au
cas du PCP, a donc été menée en colonne de percolation qui représente mieux les
conditions d’un traitement de sol in-situ.
Ces deux protocoles largement employés dans la littérature, ont été utilisés pour
contaminer les sols. Cette étape est indispensable avant de pouvoir démarrer les essais
d’extraction sur nos sols. Nos travaux montrent que le protocole expérimental a une
influence négligeable sur l’adsorption des polluants. Quelque soit le type de polluant
étudié, l’adsorption est plus forte sur le sol dont la teneur en MO est la plus élevée.
Pour les deux sols testés, la désorption de chacun des polluants cibles est un phénomène
rapide, plus ou moins linéaire et incomplet quelque soit la solution lixiviante utilisée.
Il n’a pas été possible, sur la base des résultats expérimentaux, d’identifier un mécanisme
majoritaire permettant de décrire les équilibres de désorption de façon satisfaisante (en
négligeant les autres phénomènes envisageables tels qu’une réadsorption partielle du
polluant dissout).
La capacité d’extraction des solutions de CD est, dans tous les cas, supérieure à celle de
l’eau. Le rendement d’extraction évolue de manière importante avec la quantité et la
nature de la CD utilisée. La méthyl-β-cyclodextrine (MCD) s’avère plus efficace que
l’HPCD qui devance elle même la BCD. Le protocole expérimental ne joue que
faiblement sur les performances des lavages. Une légère augmentation des quantités de
polluants extraites a été observée en milieu dispersé. Celle-ci est probablement liée à
l’accroissement de la surface de contact sol/liquide consécutive à la déstructuration du sol
provoquée par l’agitation. L’absence d’agitation en colonne de percolation ne nuit pas à
l’efficacité des CD, ce qui montre qu’elles pourraient, à priori, être utilisées en
remédiation de sol in situ.

207
Ces travaux ont été complétés par une étude, en milieu dispersé, de l’influence réciproque
de deux polluants, le NAP et le PHE sur l’adsorption et la désorption en présence de
cyclodextrine. Elle a mis en évidence une compétition entre les deux polluants. Ainsi les
pourcentages de chaque polluant fixés sur les sols ou désorbés lors d’une extraction sont
inférieurs à ceux relevés dans le cas de mono-pollution. Toutefois, les taux globaux de
polluants adsorbés et extraits sont à peu prés conservés.
Comme dans le cas d’une mono-pollution, les polluants sont mieux retenus par le sol dont
la teneur en MO est plus élevée.
La dernière étape abordée dans ce chapitre a consisté à étudier les interactions des
cyclodextrines avec différents types de sols. Il apparaît clairement que les CD s’adsorbent
sur les argiles et la MO des sols. Bien que les quantités retenues soient relativement
faibles, elles peuvent contribuer à une refixation des polluants et rendre le traitement
moins performant. Malgré l’affinité des CD pour la MO, il a été établi qu’elles ne
provoquent pas une mobilisation des MON plus importante que l’eau, ce qui est
important pour la préservation de l’écosystème sol.
A ce stade de l’étude, nous avons démontré l’aptitude des CD à extraire plusieurs
polluants d’un sol sciemment contaminé au laboratoire. Il faut souligner que dans notre
protocole expérimental, la désorption des polluants est entreprise dès que la phase
d’adsorption est terminée. Ainsi, les polluants fraîchement adsorbés n’ont pas diffusé
profondément dans la structure poreuse de la matrice et n’ont pu formé une phase
organique concentrée comme c’est généralement le cas pour une pollution ancienne.
Dans ces conditions, la fraction de polluant mobilisable par l’eau est relativement élevée
(de l’ordre de 25 à 30%). Par conséquent, la facteur d’amélioration attribuable aux CD est
plus faible. Ces essais ne peuvent donc donner qu’une idée approximative des aptitudes
des CD en remédiation de sols par lavage. Ils doivent donc être complétés par une étude
sur un sol souillé par une pollution accidentelle, ancienne et multicomposants.
Dans le chapitre suivant, nous testerons les capacités d’extraction des trois cyclodextrines
étudiées sur un sol industriel pollué par des HAP. Nous nous attacherons à faire varier
certains paramètres physico-chimiques dans l’optique d’optimiser les conditions d’un
procédé de remédiation par lavage en présence de CD.

208

209
Chapitre IV :
Evaluation de l’aptitude des CD à extraire des Hydrocarbures
Aromatiques Polycycliques de sols pollués par des activités industrielles.

210

211
Chapitre IV : Evaluation de l’aptitude des CD à extraire des Hydrocarbures Aromatiques Polycycliques de sols pollués par des activités industrielles.
I. Introduction
Dans le chapitre précèdent, nous avons étudié, sur des sols fraîchement pollués au
laboratoire, la faisabilité d’un traitement par lavage utilisant les CD comme agents de
solubilisation. Les résultats expérimentaux de l’étude prouvent que les cyclodextrines
favorisent l’extraction des polluants dans ces conditions. Ces travaux comme la plupart
des études publiées sur le traitement des sols par les CD concernent des pollutions
« artificielles » provoquées au laboratoire en utilisant au plus deux polluants. Les
polluants sont alors moins intimement associés au sol que dans le cas d’une pollution liée
à une activité industrielle ancienne et multicomposés.
Dans ce chapitre, nous allons évaluer l’aptitude des CD à extraire des HAP d’un sol
provenant d’une ancienne usine à goudron, que nous a fournit la société SITA
Remédiation.
Les deux protocoles retenus pour cette étude sont la lixiviation en milieu dispersé et la
percolation en colonne. Nous analyserons les HAP dans la phase aqueuse lixiviante afin
de déterminer l’accroissement de leur mobilité en présence de CD.
Nous tenterons d’élucider le mécanisme gouvernant la mise en solution des polluants en
comparant les concentrations extraites à celles estimées à partir de modèles théoriques de
relargage des HAP vers la phase aqueuse. Des tests préliminaires visant à vérifier que
l’utilisation de cyclodextrines permet d’accroître significativement l’extraction des HAP
du sol seront mis en œuvre. Ils donneront une première indication sur le mécanisme
d’extraction majoritaire. L’étude sera poursuivie par plusieurs expériences dont les
objectifs seront d’étudier:
♦ L’évolution du relargage au cours du temps (cinétique).
♦ L’influence des paramètres physico-chimiques pertinents (ratio L/S, température,
concentration en cyclodextrine) sur l’efficacité d’extraction.

212
La synthèse de ces résultats permettra d’évaluer les performances des solutions de
cyclodextrines en fonction des divers paramètres. Nous pourrons aussi tester la validité
du modèle théorique proposé concernant la partition du polluant entre la phase organique
et la phase aqueuse. Ces résultats permettront aussi de comparer l’efficacité des
différentes cyclodextrines et d’évaluer l’influence des protocoles opératoires sur les
pourcentages d’extraction du polluant.
Un bilan sera dressé à la fin de ce chapitre sur les performances de cette technique de
remédiation des sols contaminés utilisant les CD.
II. Matériels et Méthodes
II.1. Caractéristiques du sol étudié
Le sol pollué utilisé dans cette étude nous a été fourni par la société SITA Remédiation,
qui avait la charge de la dépollution du site d'une ancienne usine à goudron, située à
proximité de l'estuaire du Rhône. La pollution est historique (site d'environ 30 ans) et
chronique. Ce sol est composé essentiellement de sables issus de la vase de l'estuaire du
Rhône.
L’échantillonnage du sol sur le site a été effectué par la société SITA. Un séchage à l’air
libre d’un mois a ensuite été réalisé au laboratoire. Il a été suivi d’un quartage puis d’un
tamisage pour récupérer la fraction inférieure à 2 mm qui représente environ 70% du sol
fourni et que nous utiliserons pour toutes les expériences sous la dénomination de sol
SITA.
Une caractérisation du sol SITA (fraction < 2 mm) a été réalisée par le laboratoire Lisec
(Genk, 3600, Belgique), et les résultats sont présentés dans le tableau 1 :

213
Tableau 1 : Principales caractéristiques physico-chimiques du sol SITA. Analyses réalisées par le laboratoire Lisec
Matières sèches 105 °C : 99,7 ±0,1%
Matières Organiques 550 °C : 2,7 ±0,1%
Carbone organique total : 0,770%
pH : 8,15
Argile (0-2µM) : 1,9 ±0,1% MS
Sable (>50µm) : 94,40 ±1,0% MS
Limon (2-50 µm) : 3,70 ±1,0% MS
Capacité d’échange cationique : 4,22 méq / 100 g
Il s’agit donc d’un sol sablonneux (sable > 94%), contenant une faible proportion de
carbone organique (< 0,8%). En conséquence et selon les conclusions du chapitre II, la
mobilisation de la matière organique et l’adsorption des cyclodextrines sur le sol
devraient être très faibles.
La proportion de matières sèches (99,7%) montre que le sol a été convenablement séché.
Le taux d’humidité résiduel, extrêmement faible (env. 0,3%), a été confirmé par nos
propres mesures. La photographie de la figure 1 permet de juger de la bonne homogénéité
du « sol SITA ».
Figure 1 : Photographies du sol SITA (à droite : détail)

214
L’état initial de la contamination par les HAP a été déterminé par deux laboratoires
indépendants certifiés, le laboratoire Wessling (St Priest, 69791, France) et le Lisec.
Les résultats, exprimés en mg/kg de sol sec, sont présentés dans le tableau 2.
Tableau 2 : Etat initial de contamination de la fraction < 2 mm du sol. HAP en mg.kg-1 de sol sec.
Nom Laboratoire Wessling Lisec
Naphthalène 4,4 6,5
Acénaphthylène 4,3 4,0
Acénaphthène 66 51,0
Fluorène 48 51,5
Phénanthrène 200 167,0
Anthracène 71 91,0
Fluoranthène 110 100,0
Pyrène 71 64,4
Benzo(a)anthracène 21 23,6
Chrysène 23 20,5
Benzo(b)fluoranthène 11 11,7
Benzo(k)fluoranthène 9,0 7,3
Benzo(a)pyrène 8,1 12,0
Indeno(1,2,3-c,d)pyrène 3,7 7,3
Dibenzo(a,h)anthracène 1,3 2,8
Benzo(g,h,i)pérylène 3,9 5,1
Somme des 16 HAP 655,2 625,6
Bien que les deux laboratoires n’utilisent pas les mêmes protocoles, ces teneurs en HAP
caractérisant l’état initial de la pollution sont remarquablement proches et comparables.
Cependant, les valeurs retenues seront celles du laboratoire Wessling car cet
établissement a aussi été chargé d’analyser les HAP restants dans le sol après le test
préliminaire (Cf. paragraphe III.2.1.).
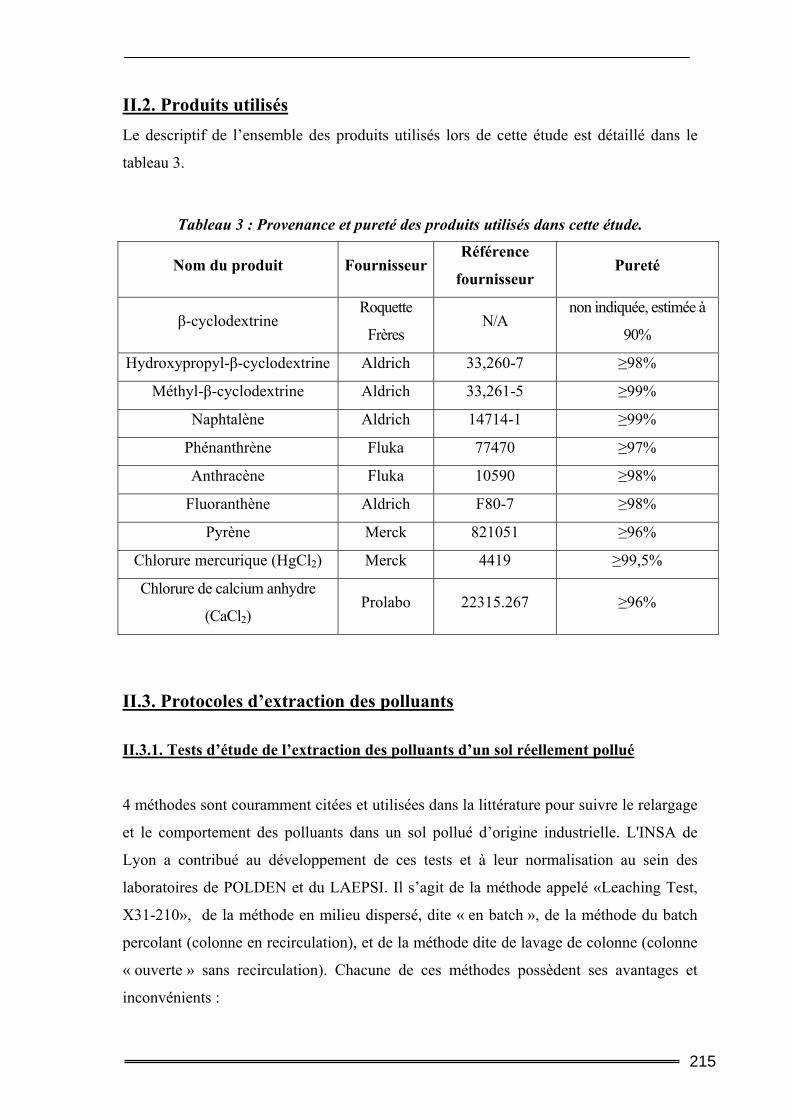
215
II.2. Produits utilisés Le descriptif de l’ensemble des produits utilisés lors de cette étude est détaillé dans le
tableau 3.
Tableau 3 : Provenance et pureté des produits utilisés dans cette étude.
Nom du produit FournisseurRéférence
fournisseur Pureté
β-cyclodextrine Roquette
Frères N/A
non indiquée, estimée à
90%
Hydroxypropyl-β-cyclodextrine Aldrich 33,260-7 ≥98%
Méthyl-β-cyclodextrine Aldrich 33,261-5 ≥99%
Naphtalène Aldrich 14714-1 ≥99%
Phénanthrène Fluka 77470 ≥97%
Anthracène Fluka 10590 ≥98%
Fluoranthène Aldrich F80-7 ≥98%
Pyrène Merck 821051 ≥96%
Chlorure mercurique (HgCl2) Merck 4419 ≥99,5%
Chlorure de calcium anhydre
(CaCl2) Prolabo 22315.267 ≥96%
II.3. Protocoles d’extraction des polluants II.3.1. Tests d’étude de l’extraction des polluants d’un sol réellement pollué
4 méthodes sont couramment citées et utilisées dans la littérature pour suivre le relargage
et le comportement des polluants dans un sol pollué d’origine industrielle. L'INSA de
Lyon a contribué au développement de ces tests et à leur normalisation au sein des
laboratoires de POLDEN et du LAEPSI. Il s’agit de la méthode appelé «Leaching Test,
X31-210», de la méthode en milieu dispersé, dite « en batch », de la méthode du batch
percolant (colonne en recirculation), et de la méthode dite de lavage de colonne (colonne
« ouverte » sans recirculation). Chacune de ces méthodes possèdent ses avantages et
inconvénients :

216
♦ Le test classique X31-210 est un test réglementaire français utilisé pour la
caractérisation du potentiel polluant d'un matériau granulaire. Ce test est, par exemple,
fréquemment employé pour évaluer la fraction polluante mobilisable à l'eau, sous
agitation, avant et après traitement d’une pollution. Le solide est immobilisé et le liquide
est seulement en agitation. Ce test permet de déterminer le rôle de la diffusion à
l’interface solide/eau.
♦ Les batchs dont le protocole expérimental est décrit au chapitre précédant.
♦ Les batchs percolants (Cf. chapitre III).
♦ Les colonnes « ouvertes » dont le protocole est tout à fait identique à celui des
colonnes de percolation (Cf. chapitre III). Par contre, dans ce cas, la solution de lavage
est récupérée à la sortie de la colonne puis traitée, alors que la solution de lavage circule
en boucle fermé dans l’étude de percolation en colonne. Cette approche n’a pas été
étudiée ici.
Un des paramètres important de ces études est le ratio L/S qui représente la quantité de
liquide (en mL) mis en contact avec le solide (en gramme).
Les concentrations des solutions sont exprimées en unités massiques (g ou mg) ou
molaires (mol ou mmol) par unité de volume de liquide (L). Les concentrations « dans le
sol » sont données en masse (mg) ou quantité (mmol) de composés chimiques par masse
de sol (kg ou g).
Ces méthodes sont utilisées pour évaluer l’amélioration de la mobilité des polluants en
présence des agents de solubilisation (solvants organiques, surfactants, acides humiques,
etc.) (Chiou et al., 1988 ; McCray et al., 2000).
II.3.2. Applications des tests classiques de lixiviation à l’étude de la mobilité des
HAP par les CD
La plupart des essais dans le domaine des sols pollués sont réalisés sur des sols
artificiellement pollués comme dans le chapitre précédent. Notre étude expérimentale,
dont le but était d'étudier la mobilité des polluants organiques hydrophobes dans un sol
industriel, a débuté à partir du test expérimental le plus simple et le plus utilisé, le batch
en milieu dispersé. Parallèlement un essai de percolation en colonne a été réalisé pour
approcher le relargage des polluants du sol in situ.

217
II.4. Méthodes d’analyse
Les solutions mises en contact avec le sol ont été dosées par chromatographie liquide en
condition isocratique grâce à une HPLC Waters Module I Plus équipée d’une colonne
Supelcosil LC-PAH de Supelco (250*4,6 mm) et d’un détecteur UV Autochrom 162 CSI
réglé à 254 nm. La phase mobile, constituée d’un mélange d’acétonitrile et d’eau
(80 :20), a été utilisée à un débit de 1 mL.min-1.
L’eau Ultra Pure est préparée grâce à un appareil Millipore MilliQ Gradient. Les phases
mobiles ont été dégazées à l’hélium pendant toute la durée des analyses (20 ml.min-1).
Cinq HAP cibles ont été choisis en raison de leur lisibilité sur les chromatogrammes. Il
s’agit du naphtalène, du phénanthrène, de l’anthracène, du fluoranthène et du pyrène. Ces
composés sont représentatifs des HAP à 2 et 3 cycles, qui présentent les meilleurs
signaux à la longueur d’onde choisie, et donnent des pics visibles et bien séparés.
Toutefois, au cours des expériences, il est apparu aux fortes concentrations en
cyclodextrines (et donc en HAP en solution) que le pic du fluoranthène était confondu
avec celui d’un autre composé (dédoublement du pic). Pour éviter toute erreur de dosage
nous avons éliminé ce composé des résultats d’analyses.
Les concentrations sont données par la relation linéaire (sur le domaine d’étude) :
CHAP (ppm) = Y * Aire du pic
Les caractéristiques d’étalonnage sont rassemblées dans le tableau 4 ci-dessous :
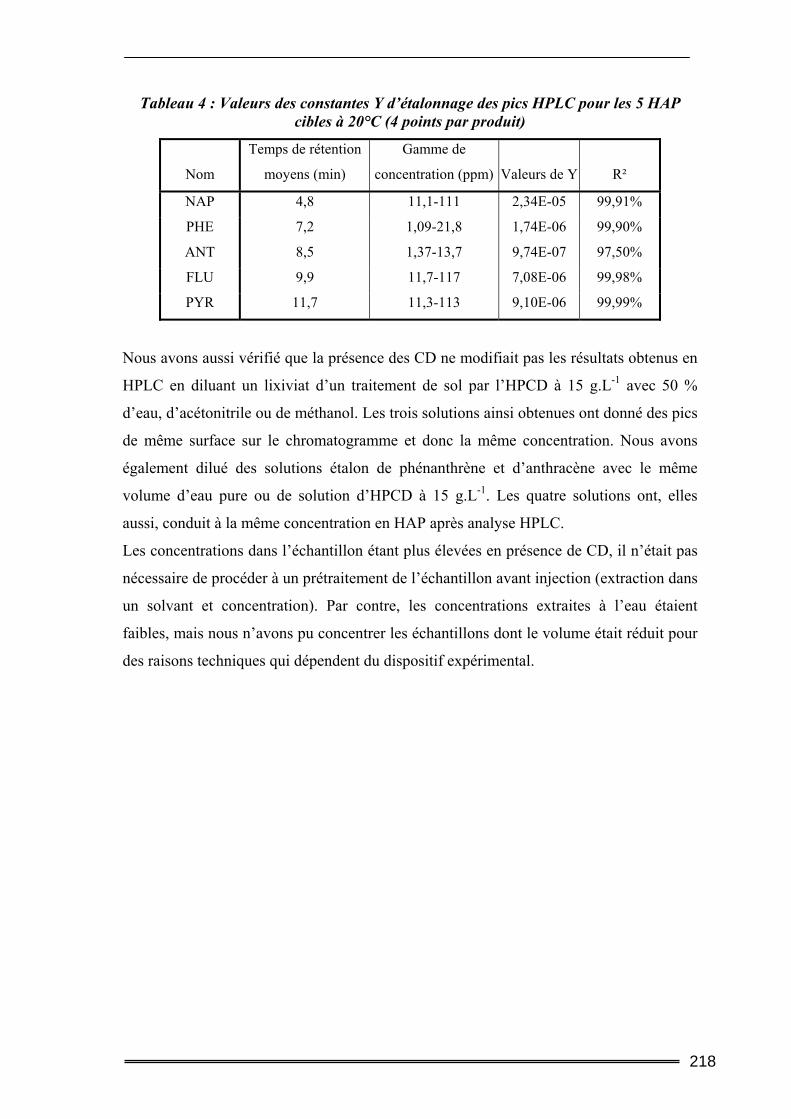
218
Tableau 4 : Valeurs des constantes Y d’étalonnage des pics HPLC pour les 5 HAP cibles à 20°C (4 points par produit)
Nom
Temps de rétention
moyens (min)
Gamme de
concentration (ppm) Valeurs de Y R²
NAP 4,8 11,1-111 2,34E-05 99,91%
PHE 7,2 1,09-21,8 1,74E-06 99,90%
ANT 8,5 1,37-13,7 9,74E-07 97,50%
FLU 9,9 11,7-117 7,08E-06 99,98%
PYR 11,7 11,3-113 9,10E-06 99,99%
Nous avons aussi vérifié que la présence des CD ne modifiait pas les résultats obtenus en
HPLC en diluant un lixiviat d’un traitement de sol par l’HPCD à 15 g.L-1 avec 50 %
d’eau, d’acétonitrile ou de méthanol. Les trois solutions ainsi obtenues ont donné des pics
de même surface sur le chromatogramme et donc la même concentration. Nous avons
également dilué des solutions étalon de phénanthrène et d’anthracène avec le même
volume d’eau pure ou de solution d’HPCD à 15 g.L-1. Les quatre solutions ont, elles
aussi, conduit à la même concentration en HAP après analyse HPLC.
Les concentrations dans l’échantillon étant plus élevées en présence de CD, il n’était pas
nécessaire de procéder à un prétraitement de l’échantillon avant injection (extraction dans
un solvant et concentration). Par contre, les concentrations extraites à l’eau étaient
faibles, mais nous n’avons pu concentrer les échantillons dont le volume était réduit pour
des raisons techniques qui dépendent du dispositif expérimental.

219
III. Résultats et discussion
III.1. Estimation des concentrations en HAP à l'équilibre dans l'eau En fonction du type et du degré de pollution des sites pollués par des composés
organiques hydrophobes, les mécanismes susceptibles de participer au transfert des HAP
de la phase solide vers la phase liquide sont :
♦ le transfert par dissolution (loi de Raoult).
♦ le transfert par désorption (isotherme de désorption).
Nous allons estimer la concentration à l’équilibre d’un HAP dans la phase aqueuse selon
ces deux hypothèses :
Première hypothèse : dissolution des HAP selon la loi de Raoult
Le site d’où provient le sol est une ancienne usine à goudron. Or il est souvent considéré,
en première estimation, que la concentration maximale d'un polluant organique, libéré
dans la phase aqueuse à partir d'une source goudronneuse, est égale à la solubilité dans
l'eau du même composé pur (Mahjoub et al, 1999). Cette estimation serait correcte si la
source polluante concernée était constituée d'un composant unique. Or le goudron est, en
général, un mélange d'une centaine de composés ou plus.
De nombreux chercheurs (Lane et Loher, 1992 et 1995 ; Peters et Luthy, 1993 ; Lee,
1992) ont développé et adapté la loi de Raoult pour prédire les concentrations à
l’équilibre dans une phase aqueuse en contact avec un sol pollué par des HAP.
La loi de Raoult établit que dans un mélange à l’équilibre de deux phases liquides idéales,
la fraction molaire d’un composé dans un des deux liquides peut être estimée à partir de
sa fraction molaire dans l’autre liquide. En supposant les HAP incorporés dans une phase
organique plus ou moins liquide, distincte du sol et située dans la porosité
interparticulaire, la loi de Raoult, peut être employée pour décrire le partage entre les
deux phases liquides (la phase aqueuse étant supposée idéale). Ce qui donne, dans le cas
d’un composé individuel i mis en solution à partir d’une Phase Liquide Non Aqueuse
(NAPL), à l’équilibre avec l’eau :

220
SXC iii
aq
i⋅⋅= γ (4)
où :
Ciaq= concentration molaire en phase aqueuse du composé i
Xi= fraction molaire du composé i dans la phase NAPL
γi= coefficient d’activité du composé i dans la phase NAPL
Si= solubilité aqueuse molaire du composé i dans l’eau pure
Certains auteurs rapportent que la loi de Raoult permet une assez bonne estimation de la
concentration à l'équilibre, dans la phase aqueuse, des différents composés issus du
goudron (Lane & Loher, 1992; Peters & Luthy, 1993). Il faut cependant noter que cette
loi repose sur l'hypothèse d'une absence d'interaction entre les différents composés au
sein de la phase organique, assimilant ainsi la NAPL à un mélange idéal. La non-
vérification de cette hypothèse peut être à l'origine de déviations importantes entre les
concentrations à l'équilibre estimées et mesurées (Mahjoub et al., 2000).
En supposant les deux phases idéales, γi vaut 1, et Xi s’exprime alors comme :
totNAPLi
i nnX =
avec : MCn
i
s
ii= et
MCn
NAPL
s
totNAPLtotNAPL
=
où :
ni= nombre de moles du composé i dans la phase NAPL par kg de sol
ntotNAPL= nombre de moles de composés formant la phase NAPL par kg de sol
Cis= concentration massique du composé i dans le sol (g.kg-1 sol)
CtotNAPLs= concentration massique de la phase NAPL dans le sol (g de NAPL / kg
sol)
Mi= masse molaire du composé i
MNAPL= masse moléculaire moyenne de la phase NAPL
On obtient donc : MC
MCXi
s
totNAPL
NAPL
s
ii ⋅
⋅= (5)

221
La détermination de la masse moléculaire moyenne MNAPL de la matière organique
contenue dans la NAPL, n’a pas été déterminée pour le sol étudié. Nous avons donc
travaillé à partir des données disponibles dans la littérature sur la masse moléculaire
moyenne des goudrons, et nous avons choisi les valeurs extrêmes de la masse moléculaire
(MNAPL=200 et 1000 g.mol-1, Meta Environnemental, 1993) pour le calcul des
concentrations à l’équilibre.
La masse de NAPL présente dans 1 kg de sol (CtotNAPLs) a été estimée à partir des
hypothèses développées par Lane (1995) qui propose, à partir du carbone organique total
(COT) dans le sol, de considérer que 71% de la matière organique est composée de
carbone et que toute la matière organique du sol est supposée homogène. Nous avons
alors:
71,0COTCs
totNAPL=
Il en résulte que les concentrations estimées dans l’hypothèse d’un mécanisme majoritaire
de dissolution peuvent être calculée à partir de l’équation (6) :
SMMC
C ii
NAPL
s
iaq
i COT⋅
⋅
⋅=
⋅71,0 (6)
Les concentrations à l’équilibre calculées par l'application de la loi de Raoult sont
données dans le tableau 5 pour quelques HAP (on suppose ici que Cis n’est pas affecté
significativement par la dissolution).
Tableau 5: Concentrations théoriques à l’équilibre pour un mécanisme de dissolution (COT= 7,7 g/kg,).
Composé
Solubilité dans l’eau du composé pur (mg.L-1) à 25 °C
Cis initiale
(mg.kg-1)*
Ciaq (mg.L-1) pour
MwNAPL=200–1000 g.mol-1
Phénanthrène 1,29 200 0,026-0,13 Anthracène 0,070 71 0,0005-0,0025
Pyrène 0,14 71 0,0009-0,0045
* : Les valeurs de Cis sont extraites du tableau 2

222
Seconde hypothèse : la désorption
La seconde approche envisageable est celle d’une désorption des polluants. Dans le cas
où des polluants organiques sont relargués d’un sol pollué selon un mécanisme de
désorption, la concentration à l’équilibre peut être calculée à partir du coefficient de
partage Kp des polluants entre le sol et l’eau: (Lane, 1995)
CC
aq
i
s
iococp fKK =⋅= (7)
où :
Koc : coefficient de partage entre la phase organique du sol et la phase aqueuse
(L/kg),
foc : fraction de carbone organique présent dans le sol,
Cis et Ci
aq : concentrations en composé i à l’équilibre dans le sol (mg/kg de sol) et
dans l’eau (mg/L), respectivement
Cis peut être exprimé à partir des concentrations initiales en phase solide et des conditions
expérimentales comme :
SLCCC
aq
is
i
s
i
⋅−=
0, (8)
avec :
L : volume de liquide en contact avec le sol (L)
S : masse de sol en contact avec la phase liquide (kg)
Ci,0s : concentration initiale du composé i en phase solide (mg/kg de sol)
En substituant l’équation 8 dans l’équation 7, la concentration à l’équilibre en phase
aqueuse peut être calculée à partir de la concentration initiale en phase solide selon :
SLfK ococ
s
iaq
iCC
+⋅= 0, (9)
Le tableau 6 présente les résultats des calculs obtenus pour certains HAP contenus dans le
sol étudié, dans l’hypothèse où le mécanisme prédominant est la désorption

223
Tableau 6 : Calcul théorique des concentrations à l’équilibre selon des isothermes de désorption
Composé L/S (L/Kg)
Koc* (L/Kg)
Ci,0s
(mg/kg) Désorption Ci
aq (mg/L)
Phénanthrène 3 13900 200 1,8 Anthracène 3 13600 71 0,66
Pyrène 3 72900 71 0,13 * : [U.S. EPA, 1989]
La comparaison des valeurs expérimentales de concentration à l’équilibre avec celles
obtenues par le calcul théorique (tableaux 5 et 6) devrait permettre de conclure quant au
mécanisme prédominant de relargage.
Estimation des concentrations extractibles des HAP en présence des CD.
En présence d’un agent de solubilisation, la concentration des HAP extraits de la NAPL à
l’équilibre augmente. Cette concentration peut aussi être estimée en considérant la
cyclodextrine comme un cosolvant. Cependant, l’augmentation de la concentration du
polluant en phase aqueuse est due à la formation d’un complexe [CD/HAPi] selon
l’équilibre de complexation suivant :
HAPi + CD [CD/HAPi]
La constante de complexation KCD s’écrit : CC
CKCD
aq
i
HAPiCDCD ∗
= ]/[ (10)
Avec:
C[CD/HAPi]= concentration aqueuse du complexe cyclodextrine-composé i,
[CD/HAPi]
Ciaq= concentration aqueuse du composé i libre dans l’eau pure
CCD= concentration aqueuse en cyclodextrine
La concentration apparente Ciapp du composé i en phase aqueuse est la somme des
concentrations dans la phase aqueuse du composé i, libre et complexée :
CCC HAPiCD
aq
i
app
i ]/[+= (11)

224
En exprimant C[CD/HAPi] en fonction de KCD, Ciaq et de CCD on retrouve le modèle proposé
dans la littérature (MacCray et Brusseau, 1998 ; MacCray et al, 2001 ; 2002 ; Duga et al.,
2003), dans lequel le facteur d’amélioration de solubilité Ei est introduit :
CECKCC aq
iiCDCD
aq
i
app
i*)1( =⋅+== (12)
Notons que Ciaq est ici calculé grâce à la loi de Raoult (éq. 6) et que ce modèle est
analogue à celui développé pour des systèmes à cosolvants et à surfactants (McRay et
Brusseau, 2001).
III.2. Tests préliminaires III.2.1. Tests de faisabilité
La faisabilité du projet a été évaluée par une première expérience, visant à vérifier que
l’utilisation de cyclodextrines permet d’accroître de façon significative la quantité d’HAP
présents dans le sol par rapport à un simple lavage à l’eau. Cette expérience permet en
outre de donner une première indication sur le mécanisme d’extraction majoritaire.
Elle consiste à faire circuler pendant 48h des solutions extractantes dans 4 colonnes
traitées selon le protocole du batch percolant. Les quatre agents d’élution étaient l’eau, la
BCD, l’HPCD et la MCD. La concentration des solutions de cyclodextrines était de 16
g.L-1, le ratio L/S étant fixé à 6 (50g de sol pour 300mL de solution). Une fois
l’expérience réalisée, les échantillons de terre ont été envoyés au laboratoire Wessling, où
ils ont été analysés selon la norme XPX 33012. Les résultats sont présentés dans les
figures 2 et 3 (analyse détaillée et abattement global) :

225
Figure 2 : Analyse du sol après extraction en batch percolant, réalisée par le laboratoire Wessling selon la norme XPX 33012 (L/S=6, 20°C, [CD] :15 g.L-1)
Figure 3 : Abattement global sur les 16 HAP (US EPA) (L/S=6, 20°C, [CD] :15 g.L-1). Les pourcentages indiqués correspondent à la valeur d'abattement global par rapport
au sol non traité.
020406080
100120140160180200220
Naphtal
ène
Acénap
htène
Fluorène
Phénan
thrène
Anthracèn
e
Fluoranthèn
e
Pyrène
Benzo
[a]an
thracèn
e
Chrysèn
e
Benzo
[b]]fluoran
thène
Benzo
[k]flu
oranthèn
e
Benzo
[a]pyrè
ne
Dibenzo
[a,h]an
thracèn
e
Benzo
[g,h,i]péry
lène
Indéno[1,
2,3-c,
d]pyrène
Con
cent
ratio
n ré
sidu
elle
sur
le s
ol m
g.kg
-1 s
ol Sol non traitéEauBCDHPCDMCD
0
100
200
300
400
500
600
700
Sol non traité Eau BCD HPCD MCD
Con
cent
ratio
n to
tale
en
HA
P su
r le
sol
mg.
kg-1
sol
26 % 63 %58 %53 %
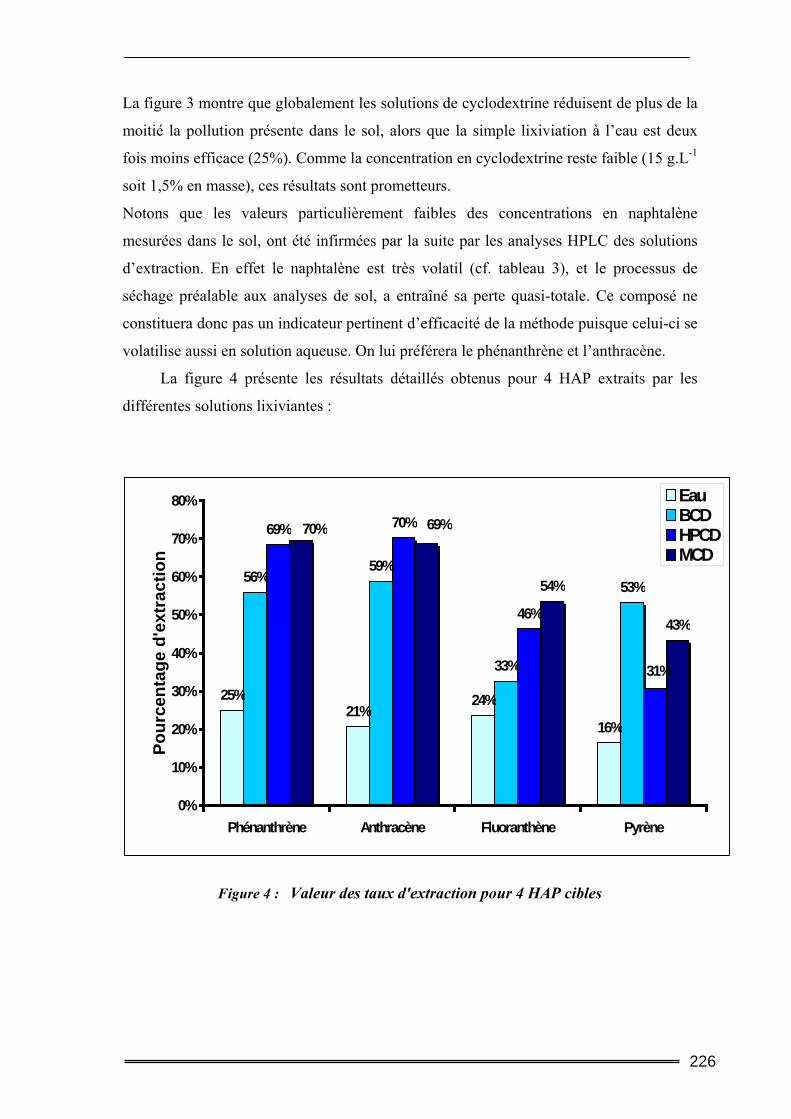
226
La figure 3 montre que globalement les solutions de cyclodextrine réduisent de plus de la
moitié la pollution présente dans le sol, alors que la simple lixiviation à l’eau est deux
fois moins efficace (25%). Comme la concentration en cyclodextrine reste faible (15 g.L-1
soit 1,5% en masse), ces résultats sont prometteurs.
Notons que les valeurs particulièrement faibles des concentrations en naphtalène
mesurées dans le sol, ont été infirmées par la suite par les analyses HPLC des solutions
d’extraction. En effet le naphtalène est très volatil (cf. tableau 3), et le processus de
séchage préalable aux analyses de sol, a entraîné sa perte quasi-totale. Ce composé ne
constituera donc pas un indicateur pertinent d’efficacité de la méthode puisque celui-ci se
volatilise aussi en solution aqueuse. On lui préférera le phénanthrène et l’anthracène.
La figure 4 présente les résultats détaillés obtenus pour 4 HAP extraits par les
différentes solutions lixiviantes :
Figure 4 : Valeur des taux d'extraction pour 4 HAP cibles
25%21%
24%
16%
56%59%
33%
53%
69% 70%
46%
54%
43%
31%
69%70%
0%
10%
20%
30%
40%
50%
60%
70%
80%
Phénanthrène Anthracène Fluoranthène Pyrène
Pour
cent
age
d'ex
trac
tion
EauBCDHPCDMCD

227
En se basant uniquement sur le phénanthrène, l’anthracène, le fluoranthène et le pyrène,
nous remarquons que l’addition de cyclodextrine dans l’eau du batch percolant double au
minimum la capacité d’extraction de la solution (excepté pour le fluoranthène). De plus,
nous constatons que la MCD est légèrement plus efficace que l’HPCD, elle-même plus
performante que la cyclodextrine native (β-CD).
L’étude détaillée des 16 HAP montre que l’efficacité du traitement faiblit avec
l’augmentation de complexité des molécules cibles. Cette complexité va de pair avec
l’augmentation de leur coefficient Kow (tableau 2), ainsi que de leur taille, mais aussi avec
la diminution de leur concentration initiale dans le sol, rendant les analyses plus
imprécises.
III.2.2. Comparaison des résultats expérimentaux avec les modèles théoriques
Le tableau 7 permet de comparer les concentrations obtenues expérimentalement et les
concentrations calculées selon les modèles de la dissolution (loi de Raoult) ou de la
désorption proposés dans la littérature.
Tableau 7 : Comparaison des valeurs expérimentales et théoriques des concentrations aqueuses en HAP à l’équilibre dans l’eau
Ceq (mg.L-1) Valeurs
expérimentales (22°C)
Modèle de la dissolution
(éq.6) (25°C)
Modèle de la Désorption
(éq.9) (25°C) Phénanthrène 0,13 0,026-0,13 1,8 Anthracène 0,04 0,0005-0,0025 0,66
Pyrène < 0,05 0,0009-0,0045 0,13
Les valeurs des concentrations à l’équilibre obtenues expérimentalement pour le
phénanthrène et l’anthracène, sont très proches de celles calculées par la loi de Raoult.
Par contre, les concentrations calculées à partir des isothermes de désorption dépassent
largement nos mesures. Pour le pyrène, la valeur expérimentale est inférieure à la limite
de détection de ce composé, et semble donc plus compatible avec l’hypothèse de la
dissolution. Il semblerait donc que le mécanisme majoritaire de relargage des HAP dans
ce sol soit la dissolution, validant l’hypothèse faite de la présence d’une phase organique
non aqueuse contenant ces polluants.
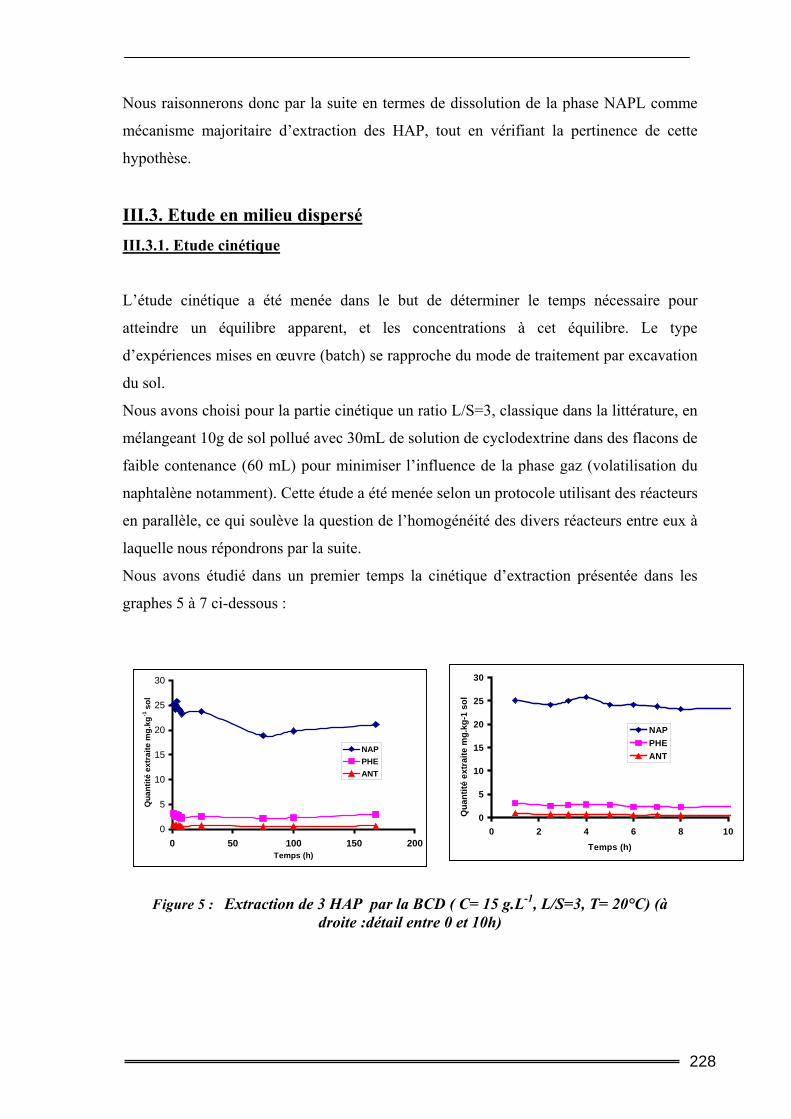
228
Nous raisonnerons donc par la suite en termes de dissolution de la phase NAPL comme
mécanisme majoritaire d’extraction des HAP, tout en vérifiant la pertinence de cette
hypothèse.
III.3. Etude en milieu dispersé III.3.1. Etude cinétique
L’étude cinétique a été menée dans le but de déterminer le temps nécessaire pour
atteindre un équilibre apparent, et les concentrations à cet équilibre. Le type
d’expériences mises en œuvre (batch) se rapproche du mode de traitement par excavation
du sol.
Nous avons choisi pour la partie cinétique un ratio L/S=3, classique dans la littérature, en
mélangeant 10g de sol pollué avec 30mL de solution de cyclodextrine dans des flacons de
faible contenance (60 mL) pour minimiser l’influence de la phase gaz (volatilisation du
naphtalène notamment). Cette étude a été menée selon un protocole utilisant des réacteurs
en parallèle, ce qui soulève la question de l’homogénéité des divers réacteurs entre eux à
laquelle nous répondrons par la suite.
Nous avons étudié dans un premier temps la cinétique d’extraction présentée dans les
graphes 5 à 7 ci-dessous :
Figure 5 : Extraction de 3 HAP par la BCD ( C= 15 g.L-1, L/S=3, T= 20°C) (à droite :détail entre 0 et 10h)
0
5
10
15
20
25
30
0 50 100 150 200Temps (h)
Qua
ntité
ext
raite
mg.
kg-1
sol
NAPPHEANT
0
5
10
15
20
25
30
0 2 4 6 8 10Temps (h)
Qua
ntité
ext
raite
mg.
kg-1
sol
NAPPHEANT
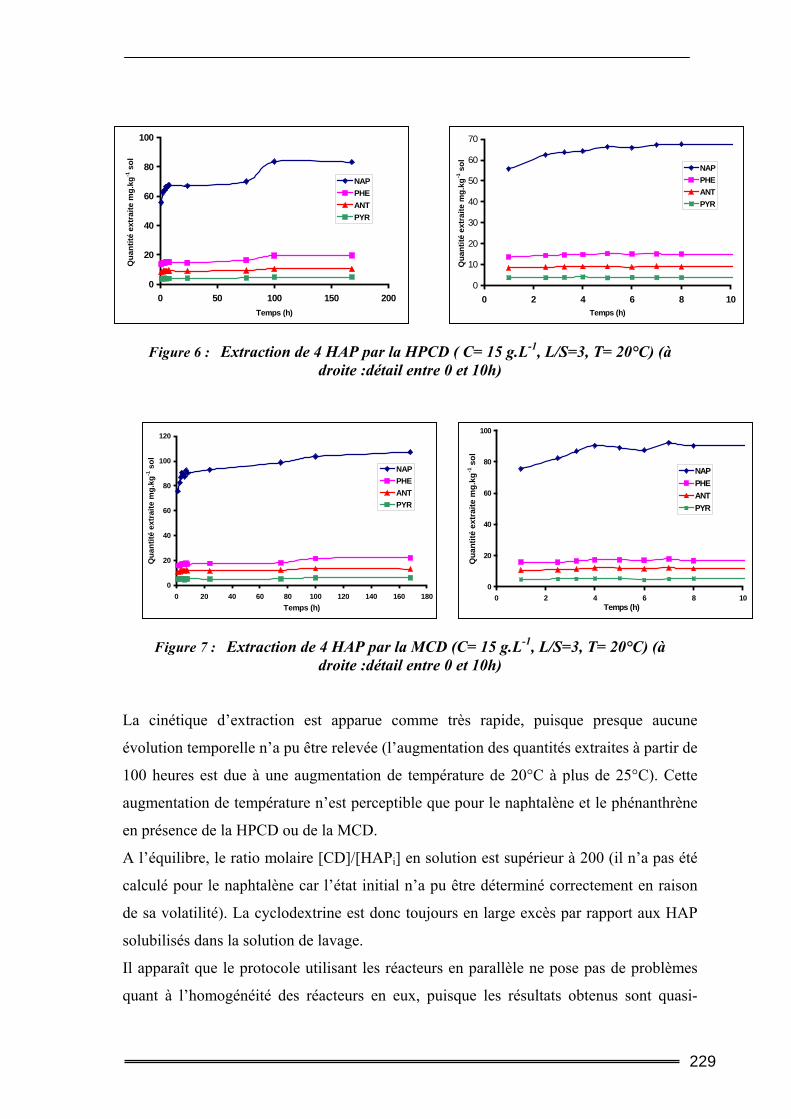
229
Figure 6 : Extraction de 4 HAP par la HPCD ( C= 15 g.L-1, L/S=3, T= 20°C) (à
droite :détail entre 0 et 10h)
Figure 7 : Extraction de 4 HAP par la MCD (C= 15 g.L-1, L/S=3, T= 20°C) (à
droite :détail entre 0 et 10h)
La cinétique d’extraction est apparue comme très rapide, puisque presque aucune
évolution temporelle n’a pu être relevée (l’augmentation des quantités extraites à partir de
100 heures est due à une augmentation de température de 20°C à plus de 25°C). Cette
augmentation de température n’est perceptible que pour le naphtalène et le phénanthrène
en présence de la HPCD ou de la MCD.
A l’équilibre, le ratio molaire [CD]/[HAPi] en solution est supérieur à 200 (il n’a pas été
calculé pour le naphtalène car l’état initial n’a pu être déterminé correctement en raison
de sa volatilité). La cyclodextrine est donc toujours en large excès par rapport aux HAP
solubilisés dans la solution de lavage.
Il apparaît que le protocole utilisant les réacteurs en parallèle ne pose pas de problèmes
quant à l’homogénéité des réacteurs en eux, puisque les résultats obtenus sont quasi-
0
20
40
60
80
100
0 50 100 150 200Temps (h)
Qua
ntité
ext
raite
mg.
kg-1
sol
NAPPHEANTPYR
0
10
20
30
40
50
60
70
0 2 4 6 8 10Temps (h)
Qua
ntité
ext
raite
mg.
kg-1
sol NAP
PHEANTPYR
0
20
40
60
80
100
120
0 20 40 60 80 100 120 140 160 180Temps (h)
Qua
ntité
ext
raite
mg.
kg-1
sol
NAPPHEANTPYR
0
20
40
60
80
100
0 2 4 6 8 10Temps (h)
Qua
ntité
ext
raite
mg.
kg-1
sol
NAPPHEANTPYR
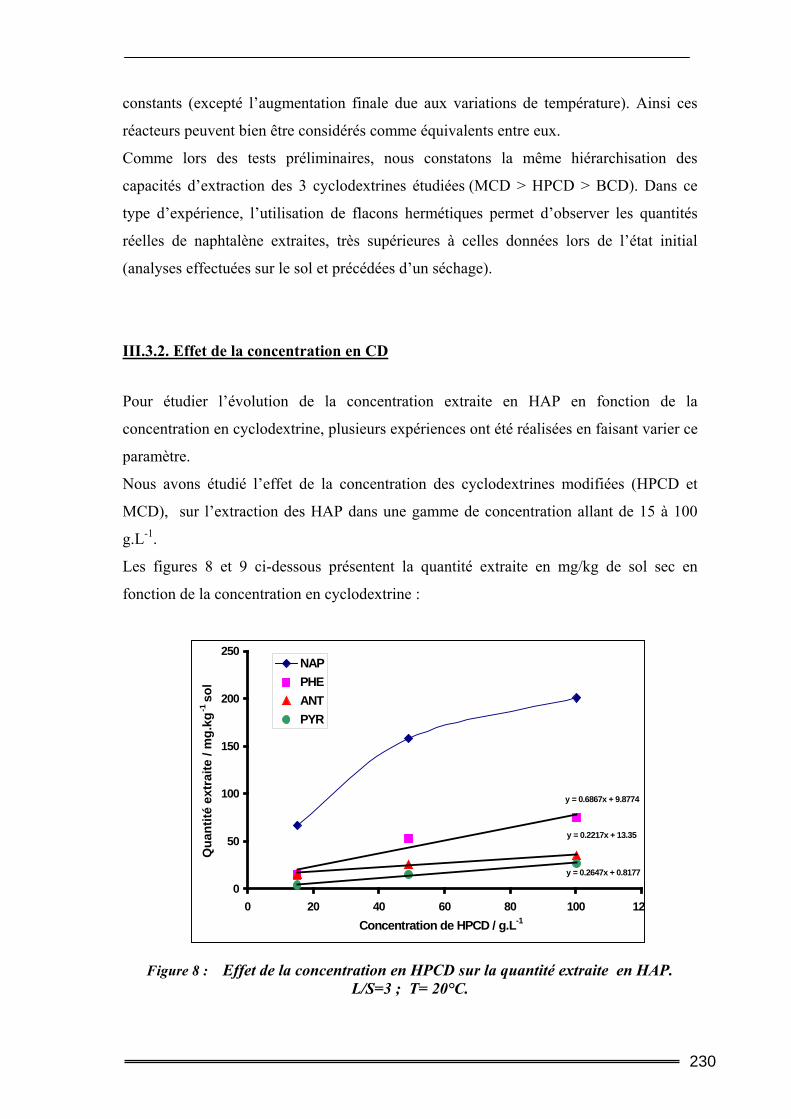
230
y = 0.2217x + 13.35
y = 0.2647x + 0.8177
y = 0.6867x + 9.8774
0
50
100
150
200
250
0 20 40 60 80 100 12Concentration de HPCD / g.L-1
Qua
ntité
ext
raite
/ m
g.kg
-1 so
l
NAPPHEANTPYR
constants (excepté l’augmentation finale due aux variations de température). Ainsi ces
réacteurs peuvent bien être considérés comme équivalents entre eux.
Comme lors des tests préliminaires, nous constatons la même hiérarchisation des
capacités d’extraction des 3 cyclodextrines étudiées (MCD > HPCD > BCD). Dans ce
type d’expérience, l’utilisation de flacons hermétiques permet d’observer les quantités
réelles de naphtalène extraites, très supérieures à celles données lors de l’état initial
(analyses effectuées sur le sol et précédées d’un séchage).
III.3.2. Effet de la concentration en CD
Pour étudier l’évolution de la concentration extraite en HAP en fonction de la
concentration en cyclodextrine, plusieurs expériences ont été réalisées en faisant varier ce
paramètre.
Nous avons étudié l’effet de la concentration des cyclodextrines modifiées (HPCD et
MCD), sur l’extraction des HAP dans une gamme de concentration allant de 15 à 100
g.L-1.
Les figures 8 et 9 ci-dessous présentent la quantité extraite en mg/kg de sol sec en
fonction de la concentration en cyclodextrine :
Figure 8 : Effet de la concentration en HPCD sur la quantité extraite en HAP. L/S=3 ; T= 20°C.
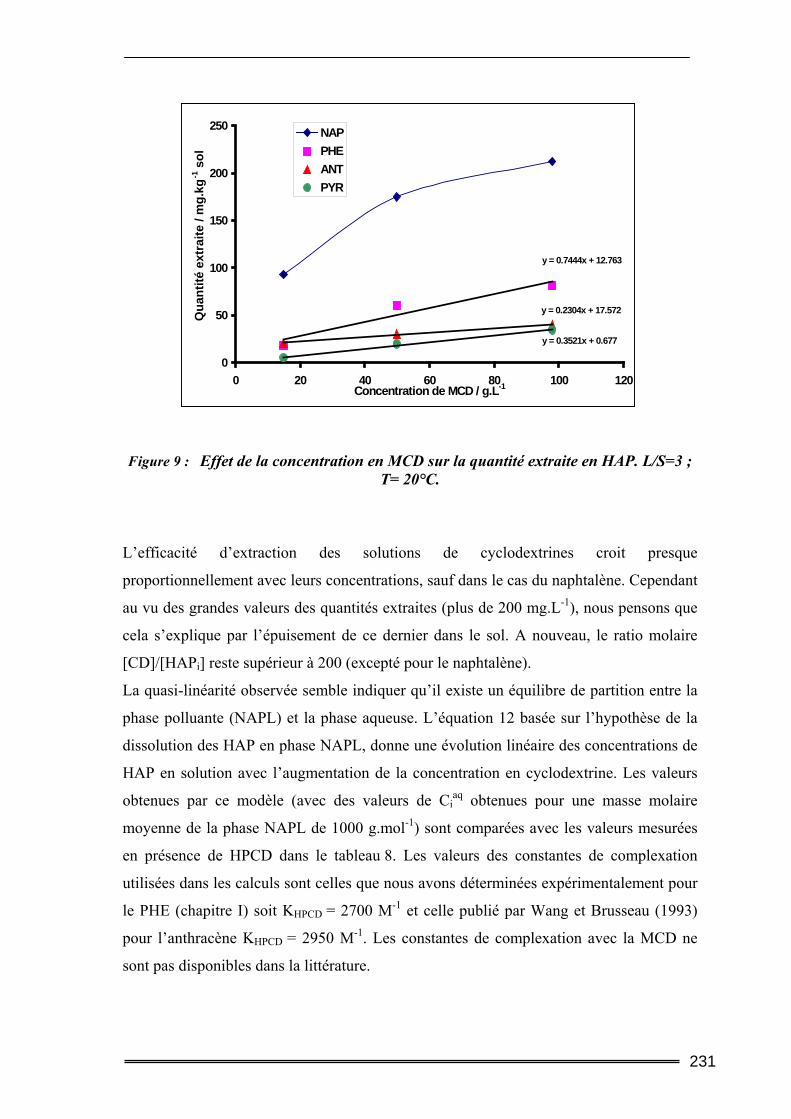
231
y = 0.7444x + 12.763
y = 0.2304x + 17.572
y = 0.3521x + 0.677
0
50
100
150
200
250
0 20 40 60 80 100 120Concentration de MCD / g.L-1
Qua
ntité
ext
raite
/ m
g.kg
-1 s
ol
NAPPHEANTPYR
Figure 9 : Effet de la concentration en MCD sur la quantité extraite en HAP. L/S=3 ; T= 20°C.
L’efficacité d’extraction des solutions de cyclodextrines croit presque
proportionnellement avec leurs concentrations, sauf dans le cas du naphtalène. Cependant
au vu des grandes valeurs des quantités extraites (plus de 200 mg.L-1), nous pensons que
cela s’explique par l’épuisement de ce dernier dans le sol. A nouveau, le ratio molaire
[CD]/[HAPi] reste supérieur à 200 (excepté pour le naphtalène).
La quasi-linéarité observée semble indiquer qu’il existe un équilibre de partition entre la
phase polluante (NAPL) et la phase aqueuse. L’équation 12 basée sur l’hypothèse de la
dissolution des HAP en phase NAPL, donne une évolution linéaire des concentrations de
HAP en solution avec l’augmentation de la concentration en cyclodextrine. Les valeurs
obtenues par ce modèle (avec des valeurs de Ciaq obtenues pour une masse molaire
moyenne de la phase NAPL de 1000 g.mol-1) sont comparées avec les valeurs mesurées
en présence de HPCD dans le tableau 8. Les valeurs des constantes de complexation
utilisées dans les calculs sont celles que nous avons déterminées expérimentalement pour
le PHE (chapitre I) soit KHPCD = 2700 M-1 et celle publié par Wang et Brusseau (1993)
pour l’anthracène KHPCD = 2950 M-1. Les constantes de complexation avec la MCD ne
sont pas disponibles dans la littérature.
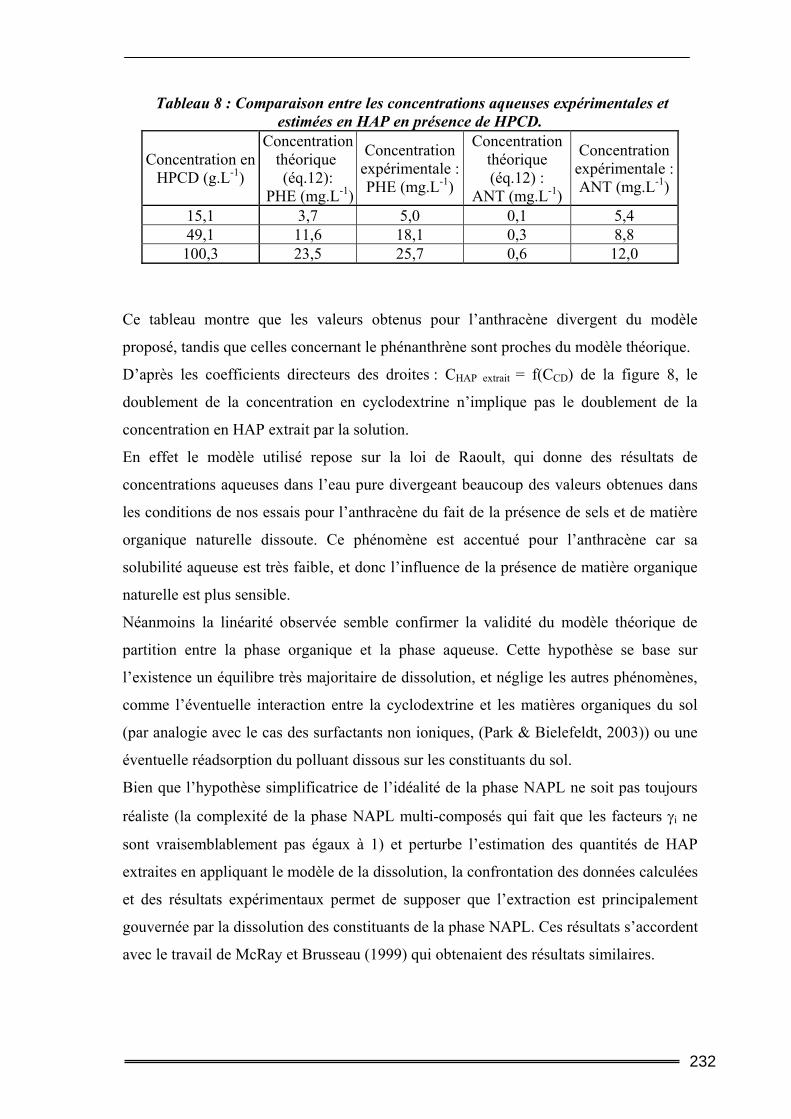
232
Tableau 8 : Comparaison entre les concentrations aqueuses expérimentales et estimées en HAP en présence de HPCD.
Concentration en HPCD (g.L-1)
Concentration théorique (éq.12):
PHE (mg.L-1)
Concentration expérimentale : PHE (mg.L-1)
Concentration théorique (éq.12) :
ANT (mg.L-1)
Concentration expérimentale : ANT (mg.L-1)
15,1 3,7 5,0 0,1 5,4 49,1 11,6 18,1 0,3 8,8
100,3 23,5 25,7 0,6 12,0
Ce tableau montre que les valeurs obtenus pour l’anthracène divergent du modèle
proposé, tandis que celles concernant le phénanthrène sont proches du modèle théorique.
D’après les coefficients directeurs des droites : CHAP extrait = f(CCD) de la figure 8, le
doublement de la concentration en cyclodextrine n’implique pas le doublement de la
concentration en HAP extrait par la solution.
En effet le modèle utilisé repose sur la loi de Raoult, qui donne des résultats de
concentrations aqueuses dans l’eau pure divergeant beaucoup des valeurs obtenues dans
les conditions de nos essais pour l’anthracène du fait de la présence de sels et de matière
organique naturelle dissoute. Ce phénomène est accentué pour l’anthracène car sa
solubilité aqueuse est très faible, et donc l’influence de la présence de matière organique
naturelle est plus sensible.
Néanmoins la linéarité observée semble confirmer la validité du modèle théorique de
partition entre la phase organique et la phase aqueuse. Cette hypothèse se base sur
l’existence un équilibre très majoritaire de dissolution, et néglige les autres phénomènes,
comme l’éventuelle interaction entre la cyclodextrine et les matières organiques du sol
(par analogie avec le cas des surfactants non ioniques, (Park & Bielefeldt, 2003)) ou une
éventuelle réadsorption du polluant dissous sur les constituants du sol.
Bien que l’hypothèse simplificatrice de l’idéalité de la phase NAPL ne soit pas toujours
réaliste (la complexité de la phase NAPL multi-composés qui fait que les facteurs γi ne
sont vraisemblablement pas égaux à 1) et perturbe l’estimation des quantités de HAP
extraites en appliquant le modèle de la dissolution, la confrontation des données calculées
et des résultats expérimentaux permet de supposer que l’extraction est principalement
gouvernée par la dissolution des constituants de la phase NAPL. Ces résultats s’accordent
avec le travail de McRay et Brusseau (1999) qui obtenaient des résultats similaires.

233
Toutefois le domaine de validité du modèle devra être exploré. En effet, une extrapolation
aux concentrations des cyclodextrines nulles conduit à des solubilités des HAP dans l’eau
très supérieures à celles de la littérature (effet inverse pour le naphtalène). Cela laisse
supposer que la courbe CHAP extrait =f (CCD) n’est plus assimilable à une droite lorsque la
concentration en cyclodextrine tend vers zéro.
D’autre part, ayant limité notre étude à des concentrations en CD au plus égales à 100
g.L-1 pour des raisons de coût, il n'a pas été possible de déterminer la concentration en
CD au delà de laquelle l’extraction atteint un plafond. En effet, nous n’avons pas observé
l’apparition d’un palier d’extraction pour de grandes concentrations en cyclodextrines
comme c’est le cas pour le PCP dans le chapitre 2. Dans le cas du sol dopé par le PCP,
nous n’avions pas la possibilité d’appliquer la loi de Raoult (absence du NAPL pour le
sol dopé), l’hypothèse de l’existence d’un équilibre majoritaire de dissolution n’a donc
pas pu être vérifiée.
A la différence de l’étude du sol dopé par le PCP, ces résultats permettent d’espérer
travailler dans des conditions linéaires d’extraction, c’est à dire que l’augmentation de la
quantité de cyclodextrine utilisée permet d’augmenter la quantité de HAP extraite.
III.3.3. Effet du ratio L/S
Les résultats précédents tendent à montrer qu’il existe une relation linéaire entre les
quantités de HAP extraites et la concentration en cyclodextrine, et donc la quantité de
cyclodextrine mise en jeu. L’étude de l’effet du ratio L/S devrait permettre de confirmer
cette hypothèse. En effet, augmenter le ratio L/S est une seconde manière d’augmenter la
quantité de CD employée par unité de masse de sol, notamment dans le cas de la BCD
dont la solubilité aqueuse ne permet pas de dépasser une concentration de 18 g.L-1 à
20°C.
Dans ces essais, une même quantité de sol a été lavé par des volumes successifs de
solutions à la concentration de 15 g.L-1 de sorte que seul le rapport L/S varie (de 3 à 12).
De ce fait, la quantité de cyclodextrine utilisée pour traiter une même masse de sol
augmente.
Les ratios molaires [CD]/[HAPi] restent de l’ordre de 300 (ce ratio n’a pu être
calculé pour le naphtalène).
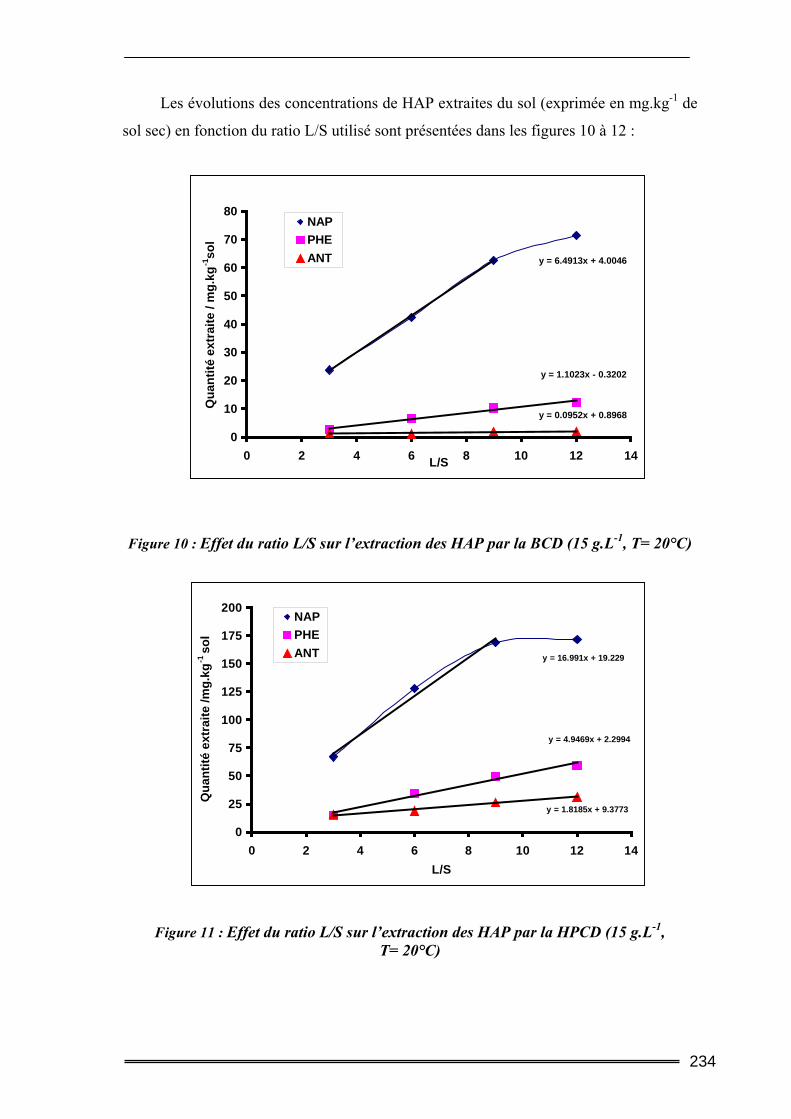
234
Les évolutions des concentrations de HAP extraites du sol (exprimée en mg.kg-1 de
sol sec) en fonction du ratio L/S utilisé sont présentées dans les figures 10 à 12 :
Figure 10 : Effet du ratio L/S sur l’extraction des HAP par la BCD (15 g.L-1, T= 20°C)
Figure 11 : Effet du ratio L/S sur l’extraction des HAP par la HPCD (15 g.L-1, T= 20°C)
y = 6.4913x + 4.0046
y = 1.1023x - 0.3202
y = 0.0952x + 0.8968
0
10
20
30
40
50
60
70
80
0 2 4 6 8 10 12 14L/S
Qua
ntité
ext
raite
/ m
g.kg
-1so
lNAPPHEANT
y = 4.9469x + 2.2994
y = 1.8185x + 9.3773
y = 16.991x + 19.229
0
25
50
75
100
125
150
175
200
0 2 4 6 8 10 12 14L/S
Qua
ntité
ext
raite
/mg.
kg-1
sol
NAPPHEANT
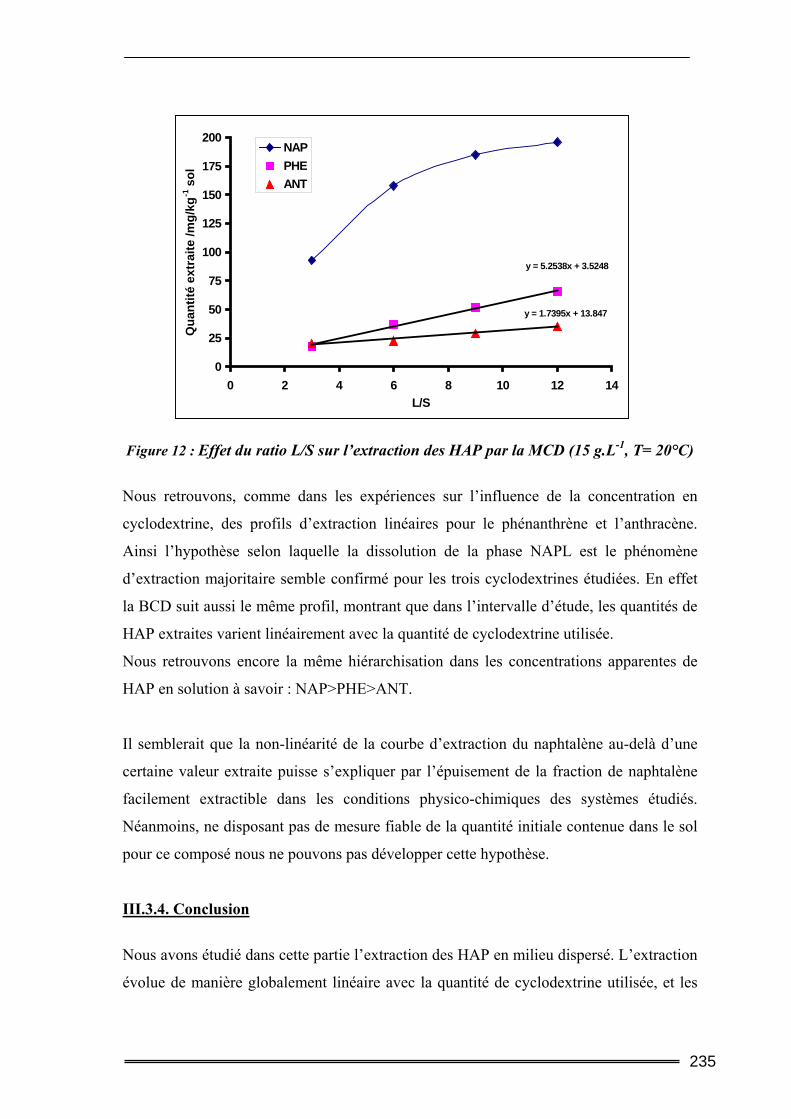
235
Figure 12 : Effet du ratio L/S sur l’extraction des HAP par la MCD (15 g.L-1, T= 20°C)
Nous retrouvons, comme dans les expériences sur l’influence de la concentration en
cyclodextrine, des profils d’extraction linéaires pour le phénanthrène et l’anthracène.
Ainsi l’hypothèse selon laquelle la dissolution de la phase NAPL est le phénomène
d’extraction majoritaire semble confirmé pour les trois cyclodextrines étudiées. En effet
la BCD suit aussi le même profil, montrant que dans l’intervalle d’étude, les quantités de
HAP extraites varient linéairement avec la quantité de cyclodextrine utilisée.
Nous retrouvons encore la même hiérarchisation dans les concentrations apparentes de
HAP en solution à savoir : NAP>PHE>ANT.
Il semblerait que la non-linéarité de la courbe d’extraction du naphtalène au-delà d’une
certaine valeur extraite puisse s’expliquer par l’épuisement de la fraction de naphtalène
facilement extractible dans les conditions physico-chimiques des systèmes étudiés.
Néanmoins, ne disposant pas de mesure fiable de la quantité initiale contenue dans le sol
pour ce composé nous ne pouvons pas développer cette hypothèse.
III.3.4. Conclusion
Nous avons étudié dans cette partie l’extraction des HAP en milieu dispersé. L’extraction
évolue de manière globalement linéaire avec la quantité de cyclodextrine utilisée, et les
y = 5.2538x + 3.5248
y = 1.7395x + 13.847
0
25
50
75
100
125
150
175
200
0 2 4 6 8 10 12 14L/S
Qua
ntité
ext
raite
/mg/
kg-1
sol
NAPPHEANT
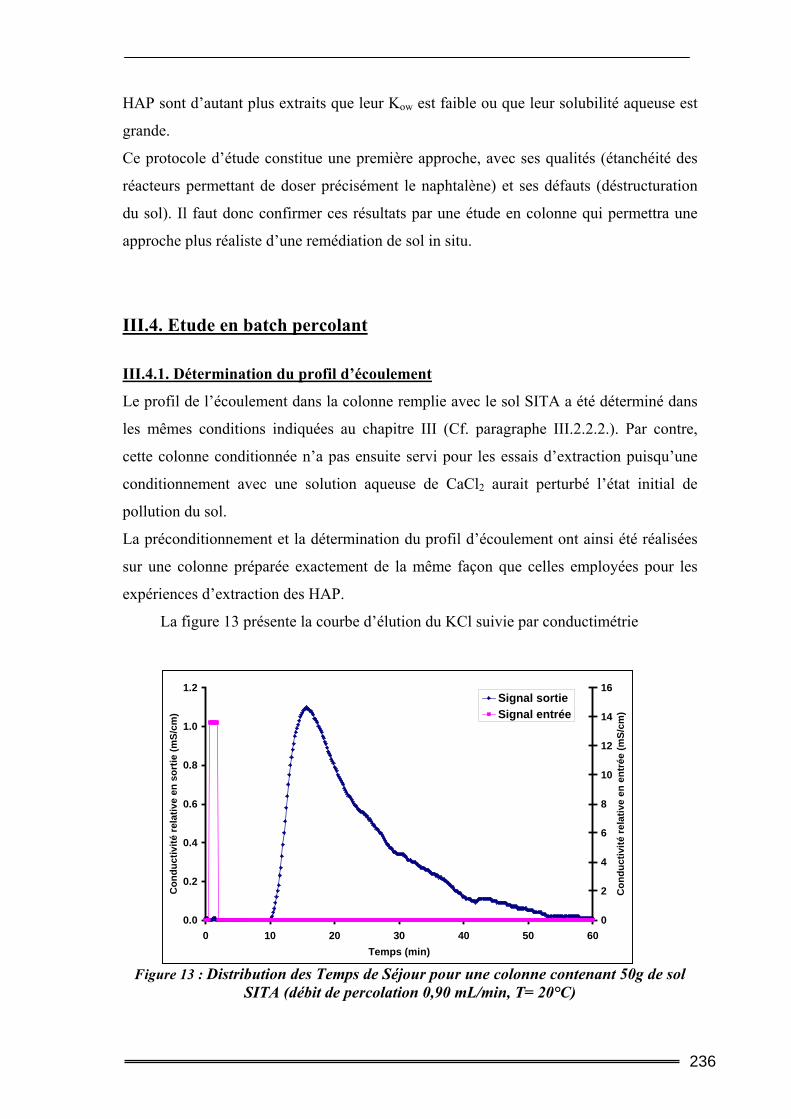
236
HAP sont d’autant plus extraits que leur Kow est faible ou que leur solubilité aqueuse est
grande.
Ce protocole d’étude constitue une première approche, avec ses qualités (étanchéité des
réacteurs permettant de doser précisément le naphtalène) et ses défauts (déstructuration
du sol). Il faut donc confirmer ces résultats par une étude en colonne qui permettra une
approche plus réaliste d’une remédiation de sol in situ.
III.4. Etude en batch percolant
III.4.1. Détermination du profil d’écoulement
Le profil de l’écoulement dans la colonne remplie avec le sol SITA a été déterminé dans
les mêmes conditions indiquées au chapitre III (Cf. paragraphe III.2.2.2.). Par contre,
cette colonne conditionnée n’a pas ensuite servi pour les essais d’extraction puisqu’une
conditionnement avec une solution aqueuse de CaCl2 aurait perturbé l’état initial de
pollution du sol.
La préconditionnement et la détermination du profil d’écoulement ont ainsi été réalisées
sur une colonne préparée exactement de la même façon que celles employées pour les
expériences d’extraction des HAP.
La figure 13 présente la courbe d’élution du KCl suivie par conductimétrie
Figure 13 : Distribution des Temps de Séjour pour une colonne contenant 50g de sol SITA (débit de percolation 0,90 mL/min, T= 20°C)
0.0
0.2
0.4
0.6
0.8
1.0
1.2
0 10 20 30 40 50 60Temps (min)
Con
duct
ivité
rela
tive
en s
ortie
(mS/
cm)
0
2
4
6
8
10
12
14
16
Con
duct
ivité
rela
tive
en e
ntré
e (m
S/cm
)
Signal sortieSignal entrée

237
Comme dans le cas du sol non pollué (Cf. chapitre III), le profil d’élution d’un créneau de
KCl montre qu’il n’y a pas de chemins préférentiels. Le bilan matière est satisfaisant
puisque l’on récupère, en sortie, plus de 97% du KCl injecté.
Nous nous sommes contentés de vérifier l’absence de chemins préférentiels ou de
phénomène d’exclusion (zones non accessibles à la cyclodextrine), puisque les analyses
seront effectuées sur des prélèvements dans le réservoir tampon, ramenant ainsi à un
système global de réacteur parfaitement agité, avec un sol immobilisé. Une étude plus
approfondie serait toutefois nécessaire dans le cas où des colonnes « ouvertes » à flux
direct (prélèvements pour analyses en sortie de colonne) seraient utilisées.
III.4.2. Etude de la cinétique en batch percolant
La cinétique a été étudiée avec trois solutions de lavage contenant respectivement de la
BCD, de l’HPCD et de la MCD à la concentration de 15 g.L-1. Toutes ces solutions
contenaient en outre 400 mg.L-1 de HgCl2 et 0,11 g.L-1 de CaCl2.
Trois températures ont été étudiées, toutefois l’expérience à 5°C, n’a pu être effectuée
avec la BCD en raison de sa trop faible solubilité à cette température. Les colonnes
n’étant pas conditionnées par un flux d’eau, l’origine des temps a été prise à la sortie de
la première goutte éluée.
Les courbes donnant la concentration de différents HAP extraits du sol en fonction du
temps sont illustrées par les figures 14 à 16. Comme les courbes présentent les mêmes
profils pour les trois types de cyclodextrines nous avons choisi de ne présenter que les
graphes concernant la HPCD :
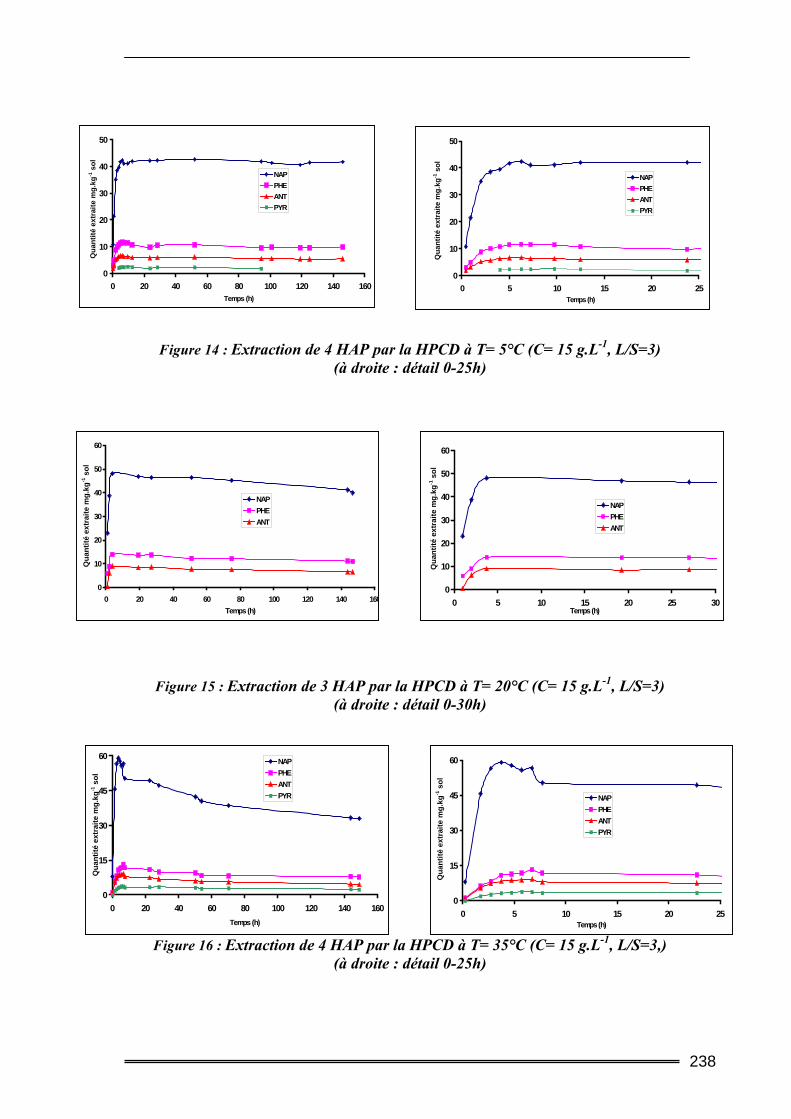
238
Figure 14 : Extraction de 4 HAP par la HPCD à T= 5°C (C= 15 g.L-1, L/S=3) (à droite : détail 0-25h)
Figure 15 : Extraction de 3 HAP par la HPCD à T= 20°C (C= 15 g.L-1, L/S=3) (à droite : détail 0-30h)
Figure 16 : Extraction de 4 HAP par la HPCD à T= 35°C (C= 15 g.L-1, L/S=3,) (à droite : détail 0-25h)
0
10
20
30
40
50
60
0 20 40 60 80 100 120 140 160Temps (h)
Qua
ntité
ext
raite
mg.
kg-1
sol
NAPPHEANT
0
10
20
30
40
50
60
0 5 10 15 20 25 30Temps (h)
Qua
ntité
ext
raite
mg.
kg-1
sol
NAPPHEANT
0
15
30
45
60
0 20 40 60 80 100 120 140 160Temps (h)
Qua
ntité
ext
raite
mg.
kg-1
sol
NAPPHEANTPYR
0
15
30
45
60
0 5 10 15 20 25Temps (h)
Qua
ntité
ext
raite
mg.
kg-1
sol
NAPPHEANTPYR
0
10
20
30
40
50
0 20 40 60 80 100 120 140 160Temps (h)
Qua
ntité
ext
raite
mg.
kg-1
sol
NAPPHEANTPYR
0
10
20
30
40
50
0 5 10 15 20 25Temps (h)
Qua
ntité
ext
raite
mg.
kg-1
sol
NAPPHEANTPYR

239
A l’équilibre, le ratio molaire [CD]/[HAPi] en solution est compris entre 400 et 5500. La
cyclodextrine est donc toujours en large excès par rapport aux HAP solubilisés dans la
solution de lavage.
Les courbes d’élution présentent toutes le même profil, avec une augmentation
progressive de la concentration extraite sur un modèle qui semblerait exponentiel, puis
une stabilisation de la concentration vers sa valeur d’équilibre. Cela représente la
cinétique du dispositif expérimental, et non la cinétique de l’extraction, puisque les
prélèvements ont été effectués dans le réservoir tampon et non directement à la sortie de
la colonne. Ainsi l’évolution de la concentration est très proche de celle que l’on
obtiendrait avec l’injection continue d’un lixiviat à la concentration d’équilibre à un débit
de 1 mL/min dans un réacteur ouvert et parfaitement agité de même volume que le
réservoir tampon. Ce point est une différence notable par rapport au protocole en milieu
dispersé où le sol est en contact direct avec la solution prélevée.
Le temps nécessaire pour atteindre le palier d’équilibre varie peu avec la température et
entre les différents types de cyclodextrines, sauf dans le cas du naphtalène. Entre 5°C et
35°C l’équilibre est atteint en moyenne en 6h (excepté pour le naphtalène) pour un débit
de solution de 1mL/min (environ 3,8 volumes de pore par heure). Ce temps n’a pu être
déterminé pour l’expérience à 20°C car trop peu d’échantillonnages ont été réalisés dans
les 8 premières heures. Cela montre une cinétique de réaction quasi identique pour les
différents types de cyclodextrines et les différentes températures.
Nous remarquons, principalement pour le naphtalène, une décroissance de la
concentration après un maximum. Ce phénomène semble dû à l’évaporation des
composés du fait que les réservoirs de solutions ne sont pas parfaitement étanches, et
qu’ils sont ouverts régulièrement pour les prélèvements. Ce phénomène s’accentue
notamment avec la température : à 5°C il n’est pas observable tandis qu’il est très fort à
35°C.
Pour la suite des expériences nous avons effectué les prélèvements pour analyse après
48h, durée largement supérieure à celle permettant d’obtenir l’équilibre.
Nous constatons que la concentration de naphtalène extraite à l’équilibre est
systématiquement plus élevée que celle de phénanthrène qui est elle-même supérieure à
celle de l’anthracène quelque soit la cyclodextrine utilisée ou la température.
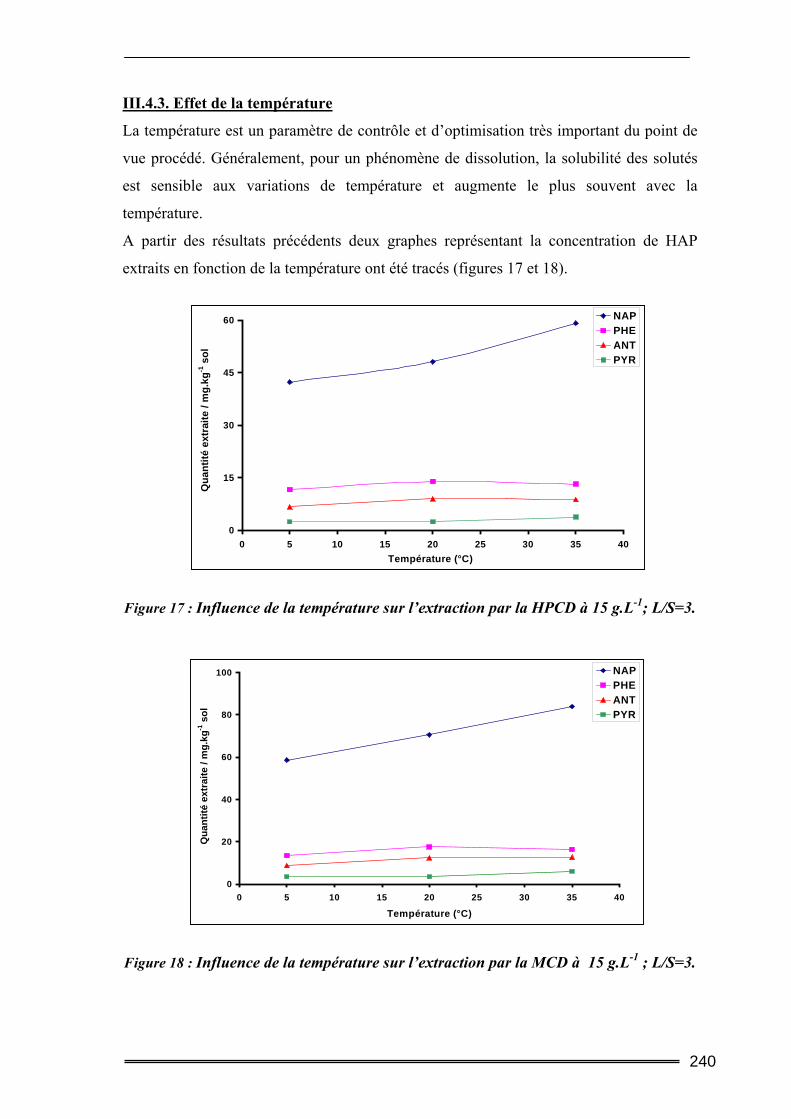
240
III.4.3. Effet de la température
La température est un paramètre de contrôle et d’optimisation très important du point de
vue procédé. Généralement, pour un phénomène de dissolution, la solubilité des solutés
est sensible aux variations de température et augmente le plus souvent avec la
température.
A partir des résultats précédents deux graphes représentant la concentration de HAP
extraits en fonction de la température ont été tracés (figures 17 et 18).
Figure 17 : Influence de la température sur l’extraction par la HPCD à 15 g.L-1; L/S=3.
Figure 18 : Influence de la température sur l’extraction par la MCD à 15 g.L-1 ; L/S=3.
0
15
30
45
60
0 5 10 15 20 25 30 35 40Température (°C)
Qua
ntité
ext
raite
/ m
g.kg
-1 s
ol
NAPPHEANTPYR
0
20
40
60
80
100
0 5 10 15 20 25 30 35 40
Température (°C)
Qua
ntité
ext
raite
/ m
g.kg
-1 s
ol
NAPPHEANTPYR

241
Ces deux figures montrent que les concentrations extraites sont remarquablement
constantes en fonction de la température, excepté encore une fois dans le cas du
naphtalène. Il semblerait qu’il y ait un effet de compensation entre l’augmentation de la
solubilité aqueuse des HAP avec la température (Whitehouse, 1984) et la déstabilisation
concomitante du complexe cyclodextrine/HAP. En effet l’enthalpie de complexation est
négative (de l’ordre de -4 kJ/mol pour le complexe BCD/anthracène selon Blyshak et al,
(1990)) et d’après la loi de Van’t Hoff le complexe se dissocie quand la température
augmente. Ne disposant pas des valeurs d’enthalpie pour la HPCD et la MCD, nous
n’avons pu calculer théoriquement la variation des constantes de complexation avec la
température.
Le fait que la concentration extraite en naphtalène augmente avec la température
s’explique par une forte augmentation de sa solubilité aqueuse dans l’intervalle de
température considéré.
La relative insensibilité de l’extraction à la température permet d’envisager des
opérations de traitement sur une large gamme de température sans pour autant affecter le
rendement. Ceci représente un net avantage comparé aux systèmes utilisant les solvants
organiques ou les surfactants (Edwards et al., 1991 ; Jouannin, 2004). En effet ce procédé
ne sera pas dépendant des conditions climatiques à la différence des traitements cités ci-
dessus dont l’efficacité baisse avec la température (Krauss & Wilcke, 2001).
III.4.4. Effet de la concentration
Nous avons étudié l’effet de la concentration des cyclodextrines modifiées (HPCD et
MCD) dans une gamme de concentration de 10 à 100 g.L-1 sur l’extraction des HAP.
Nous n’avons pas étudié l’effet de la concentration pour la BCD compte tenu de sa faible
solubilité aqueuse (18 g.L-1 à 20°C). Le rapport L/S=2 utilisé ici diffère de celui utilisé
pour le reste des expériences uniquement par souci d’économie des produits. Cette
expérience doit permettre de confirmer le comportement linéaire observé en milieu
dispersé.
Les concentrations de HAP extraites en fonction de la concentration sont présentées dans
les figures 19 et 20 :
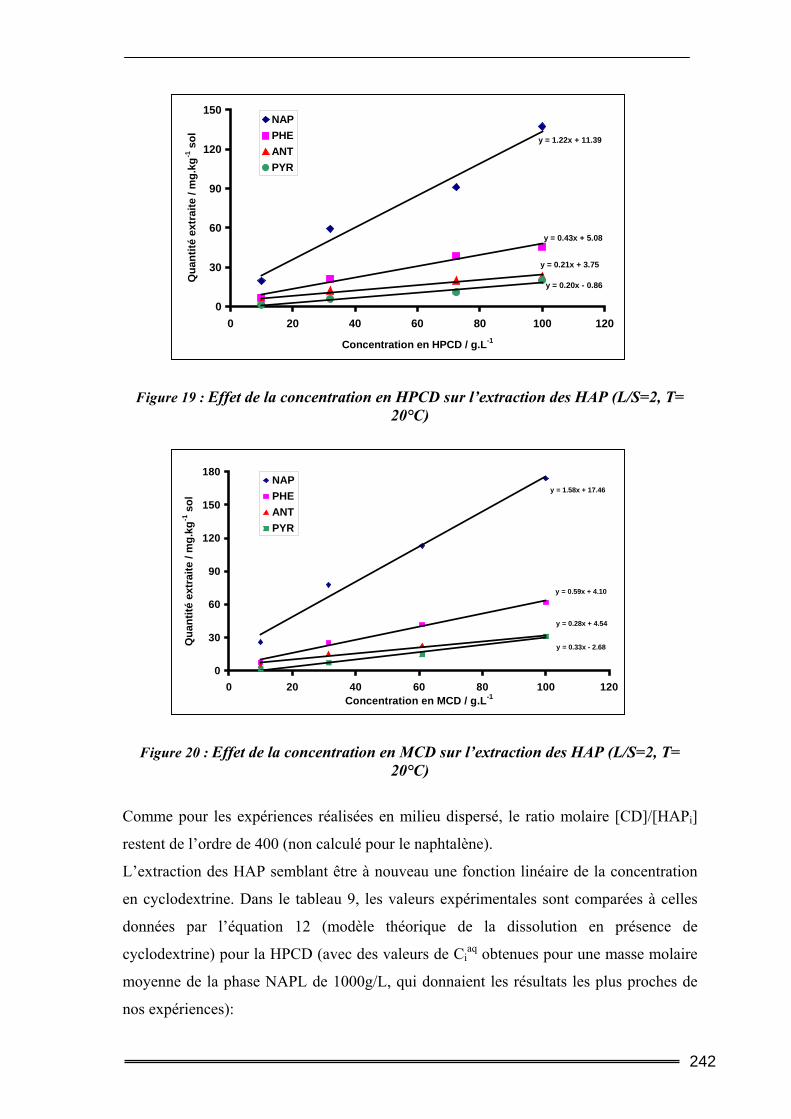
242
Figure 19 : Effet de la concentration en HPCD sur l’extraction des HAP (L/S=2, T= 20°C)
Figure 20 : Effet de la concentration en MCD sur l’extraction des HAP (L/S=2, T= 20°C)
Comme pour les expériences réalisées en milieu dispersé, le ratio molaire [CD]/[HAPi]
restent de l’ordre de 400 (non calculé pour le naphtalène).
L’extraction des HAP semblant être à nouveau une fonction linéaire de la concentration
en cyclodextrine. Dans le tableau 9, les valeurs expérimentales sont comparées à celles
données par l’équation 12 (modèle théorique de la dissolution en présence de
cyclodextrine) pour la HPCD (avec des valeurs de Ciaq obtenues pour une masse molaire
moyenne de la phase NAPL de 1000g/L, qui donnaient les résultats les plus proches de
nos expériences):
y = 1.22x + 11.39
y = 0.43x + 5.08
y = 0.21x + 3.75
y = 0.20x - 0.86
0
30
60
90
120
150
0 20 40 60 80 100 120
Concentration en HPCD / g.L-1
Qua
ntité
ext
raite
/ m
g.kg
-1 s
ol
NAPPHEANTPYR
y = 1.58x + 17.46
y = 0.59x + 4.10
y = 0.28x + 4.54
y = 0.33x - 2.68
0
30
60
90
120
150
180
0 20 40 60 80 100 120Concentration en MCD / g.L-1
Qua
ntité
ext
raite
/ m
g.kg
-1 s
ol
NAPPHEANTPYR
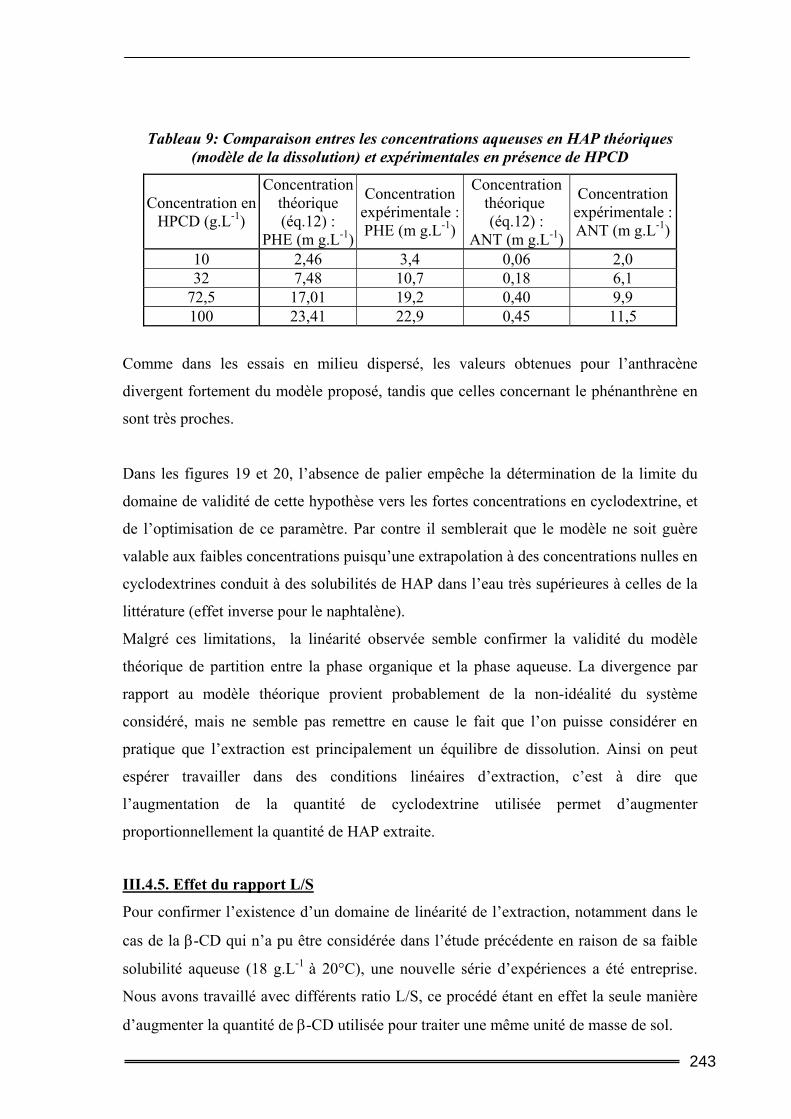
243
Tableau 9: Comparaison entres les concentrations aqueuses en HAP théoriques (modèle de la dissolution) et expérimentales en présence de HPCD
Concentration en HPCD (g.L-1)
Concentration théorique (éq.12) :
PHE (m g.L-1)
Concentration expérimentale : PHE (m g.L-1)
Concentration théorique (éq.12) :
ANT (m g.L-1)
Concentration expérimentale : ANT (m g.L-1)
10 2,46 3,4 0,06 2,0 32 7,48 10,7 0,18 6,1
72,5 17,01 19,2 0,40 9,9 100 23,41 22,9 0,45 11,5
Comme dans les essais en milieu dispersé, les valeurs obtenues pour l’anthracène
divergent fortement du modèle proposé, tandis que celles concernant le phénanthrène en
sont très proches.
Dans les figures 19 et 20, l’absence de palier empêche la détermination de la limite du
domaine de validité de cette hypothèse vers les fortes concentrations en cyclodextrine, et
de l’optimisation de ce paramètre. Par contre il semblerait que le modèle ne soit guère
valable aux faibles concentrations puisqu’une extrapolation à des concentrations nulles en
cyclodextrines conduit à des solubilités de HAP dans l’eau très supérieures à celles de la
littérature (effet inverse pour le naphtalène).
Malgré ces limitations, la linéarité observée semble confirmer la validité du modèle
théorique de partition entre la phase organique et la phase aqueuse. La divergence par
rapport au modèle théorique provient probablement de la non-idéalité du système
considéré, mais ne semble pas remettre en cause le fait que l’on puisse considérer en
pratique que l’extraction est principalement un équilibre de dissolution. Ainsi on peut
espérer travailler dans des conditions linéaires d’extraction, c’est à dire que
l’augmentation de la quantité de cyclodextrine utilisée permet d’augmenter
proportionnellement la quantité de HAP extraite.
III.4.5. Effet du rapport L/S
Pour confirmer l’existence d’un domaine de linéarité de l’extraction, notamment dans le
cas de la β-CD qui n’a pu être considérée dans l’étude précédente en raison de sa faible
solubilité aqueuse (18 g.L-1 à 20°C), une nouvelle série d’expériences a été entreprise.
Nous avons travaillé avec différents ratio L/S, ce procédé étant en effet la seule manière
d’augmenter la quantité de β-CD utilisée pour traiter une même unité de masse de sol.
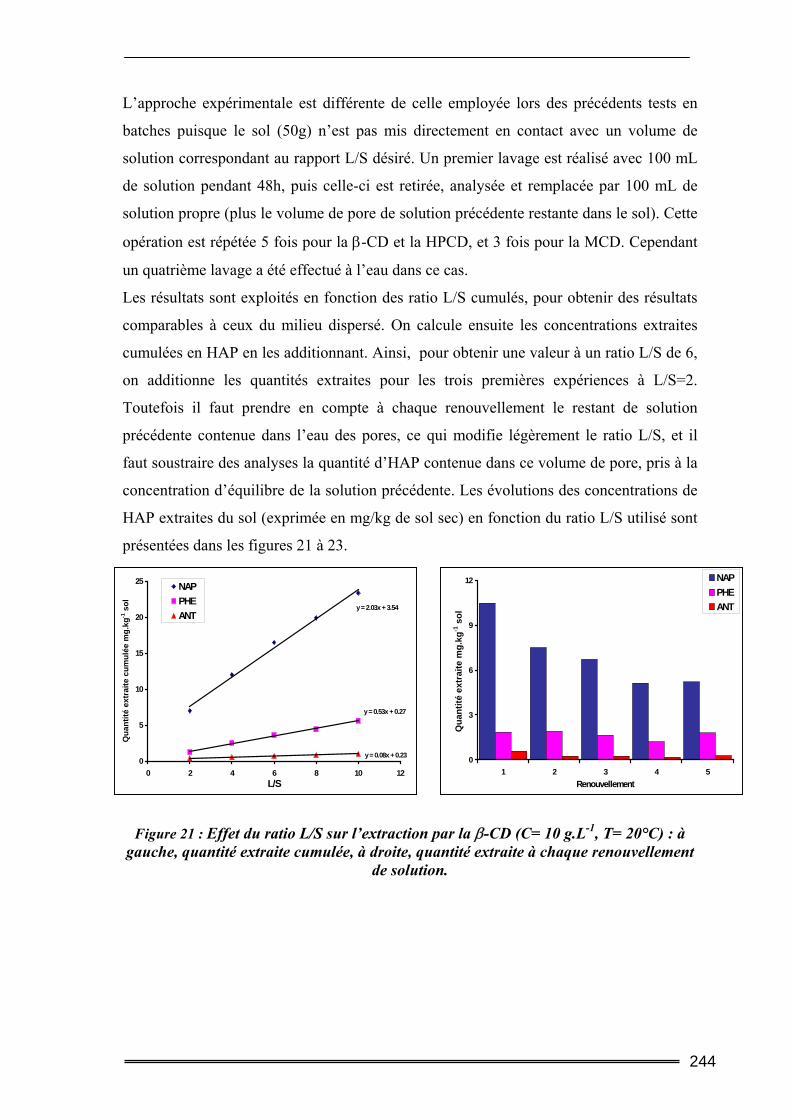
244
L’approche expérimentale est différente de celle employée lors des précédents tests en
batches puisque le sol (50g) n’est pas mis directement en contact avec un volume de
solution correspondant au rapport L/S désiré. Un premier lavage est réalisé avec 100 mL
de solution pendant 48h, puis celle-ci est retirée, analysée et remplacée par 100 mL de
solution propre (plus le volume de pore de solution précédente restante dans le sol). Cette
opération est répétée 5 fois pour la β-CD et la HPCD, et 3 fois pour la MCD. Cependant
un quatrième lavage a été effectué à l’eau dans ce cas.
Les résultats sont exploités en fonction des ratio L/S cumulés, pour obtenir des résultats
comparables à ceux du milieu dispersé. On calcule ensuite les concentrations extraites
cumulées en HAP en les additionnant. Ainsi, pour obtenir une valeur à un ratio L/S de 6,
on additionne les quantités extraites pour les trois premières expériences à L/S=2.
Toutefois il faut prendre en compte à chaque renouvellement le restant de solution
précédente contenue dans l’eau des pores, ce qui modifie légèrement le ratio L/S, et il
faut soustraire des analyses la quantité d’HAP contenue dans ce volume de pore, pris à la
concentration d’équilibre de la solution précédente. Les évolutions des concentrations de
HAP extraites du sol (exprimée en mg/kg de sol sec) en fonction du ratio L/S utilisé sont
présentées dans les figures 21 à 23.
Figure 21 : Effet du ratio L/S sur l’extraction par la β-CD (C= 10 g.L-1, T= 20°C) : à gauche, quantité extraite cumulée, à droite, quantité extraite à chaque renouvellement
de solution.
y = 0.53x + 0.27
y = 2.03x + 3.54
y = 0.08x + 0.230
5
10
15
20
25
0 2 4 6 8 10 12L/S
Qua
ntité
ext
raite
cum
ulée
mg.
kg-1
sol
NAPPHEANT
0
3
6
9
12
1 2 3 4 5Renouvellement
Qua
ntité
ext
raite
mg.
kg-1
sol
NAPPHEANT
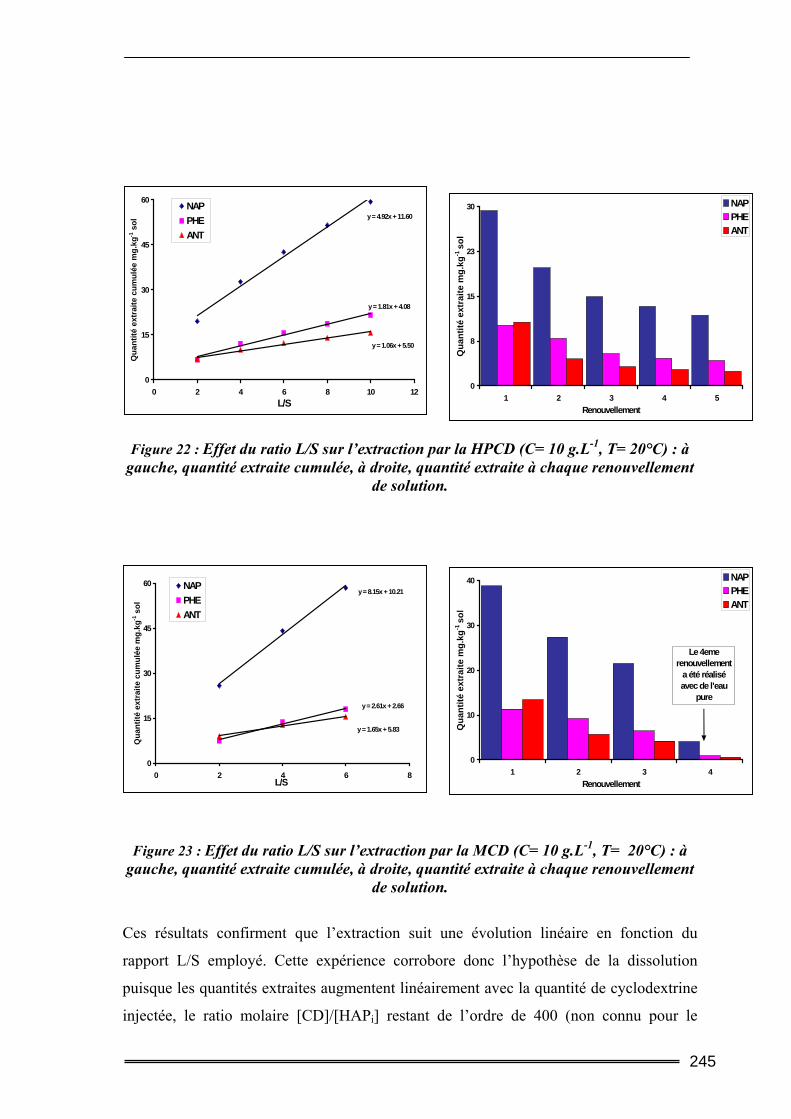
245
Figure 22 : Effet du ratio L/S sur l’extraction par la HPCD (C= 10 g.L-1, T= 20°C) : à
gauche, quantité extraite cumulée, à droite, quantité extraite à chaque renouvellement de solution.
Figure 23 : Effet du ratio L/S sur l’extraction par la MCD (C= 10 g.L-1, T= 20°C) : à gauche, quantité extraite cumulée, à droite, quantité extraite à chaque renouvellement
de solution.
Ces résultats confirment que l’extraction suit une évolution linéaire en fonction du
rapport L/S employé. Cette expérience corrobore donc l’hypothèse de la dissolution
puisque les quantités extraites augmentent linéairement avec la quantité de cyclodextrine
injectée, le ratio molaire [CD]/[HAPi] restant de l’ordre de 400 (non connu pour le
y = 1.81x + 4.08
y = 4.92x + 11.60
y = 1.06x + 5.50
0
15
30
45
60
0 2 4 6 8 10 12L/S
Qua
ntité
ext
raite
cum
ulée
mg.
kg-1
sol
NAPPHEANT
0
8
15
23
30
1 2 3 4 5Renouvellement
Qua
ntité
ext
raite
mg.
kg-1
sol
NAPPHEANT
y = 2.61x + 2.66
y = 8.15x + 10.21
y = 1.65x + 5.83
0
15
30
45
60
0 2 4 6 8L/S
Qua
ntité
ext
raite
cum
ulée
mg.
kg-1
sol
NAPPHEANT
0
10
20
30
40
1 2 3 4Renouvellement
Qua
ntité
ext
raite
mg.
kg-1
sol
NAPPHEANT
Le 4eme renouvellement
a été réalisé avec de l'eau
pure

246
naphtalène). On peut aussi noter qu’avec la β-CD les concentrations de HAP extraites à
chaque renouvellement restent presque constantes (excepté pour le naphtalène) alors que
pour la HPCD et la MCD ces concentrations diminuent à chaque renouvellement.
Le lavage à l’eau suivant le troisième renouvellement avec la MCD permet de mettre en
évidence qu’un rinçage post-traitement est nécessaire. En effet on assiste à une
mobilisation des HAP en concentrations supérieures à celle obtenues avec un simple
lavage à l’eau, dû à la présence résiduelle de polluant solubilisé par la CD dans la
porosité (0,31 mg.L-1 de phénanthrène et 0,20 mg.L-1 d’anthracène alors que leur
extraction respective à l’eau à la même température ne sont que de 0,13 mg.L-1 et 0,04
mg.L-1). Ce test permet généralement d’évaluer la disponibilité du polluant dans le sol
après un traitement.
Toutes ces expériences confirment que pour ce sol la concentration extraite en HAP à
l’équilibre va en décroissant du naphtalène à l’anthracène, et ce, quelle que soit la
cyclodextrine utilisée, sa concentration, le rapport L/S employé ou la température.
III.4.6. Conclusion
La deuxième partie de cette étude est venue confirmer les résultats obtenus dans la
première partie. Les tendances linéaires observées lors des extractions semblent appuyer
les hypothèses théoriques formulées, notamment sur un mécanisme principal de transfert
basé sur la dissolution de la NAPL. De plus, l’étude thermodynamique du traitement a
conduit à une conclusion intéressante, puisqu’il semblerait que le procédé soit
globalement athermique, et donc assez souple dans sa mise en application. L’absence
d’agitation ou de mise en suspension du sol n’a pas compromis les capacités d’extraction
des cyclodextrines, qui semblent un moyen efficace de traiter les pollutions par des HAP
dans des conditions similaires à celles utilisées.

247
III.5. Synthèse comparative III.5.1. Comparaison des deux protocoles opératoires
Nous avons réalisé au cours de cette étude deux types d’expériences très différents dans
leur principe. En effet le batch agité (milieu dispersé) s’apparente à un réacteur
parfaitement agité multiphasique (phase liquide aqueuse, NAPL et phase solide), tandis
que le batch percolant correspond à un réacteur multiphasique à lit fixe. Les deux
réacteurs étant complètement différents, la comparaison ne peut se faire qu’en terme de
pourcentage global d’extraction.
Les résultats d’extraction à l’équilibre peuvent dépendre du mécanisme réactionnel
envisagé. En effet dans le cas d’une extraction liée à un équilibre
d’adsorption/désorption, la démultiplication de la surface spécifique liée à la
déstructuration de la matrice du sol (séparation selon diverses coupes granulométriques,
désagrégation des associations sable-argile ou argilo-humiques, etc.), influencerait
fortement la concentration finale des HAP en phase aqueuse. Au contraire, une extraction
liée à une dissolution serait beaucoup moins sensible à ce phénomène à l’équilibre.
Toutefois, dans les deux cas, l’augmentation de la surface de contact phase
aqueuse/solide ou phase aqueuse /NAPL pourrait augmenter la concentration mise en
solution.
Pour rassembler les données issues des diverses expériences issues du batch et du batch
percolant, afin de les comparer entre elles, nous avons converti les concentrations en
cyclodextrines en masse de cyclodextrine par unité de masse de sol (g de
cyclodextrine/kg de sol), et les concentrations en HAP en pourcentage d’extraction par
rapport à l’état initial du sol. Les graphes étant très similaires, seuls ceux concernant le
traitement par l’HPCD sont présentés dans les figures 24 et 25:
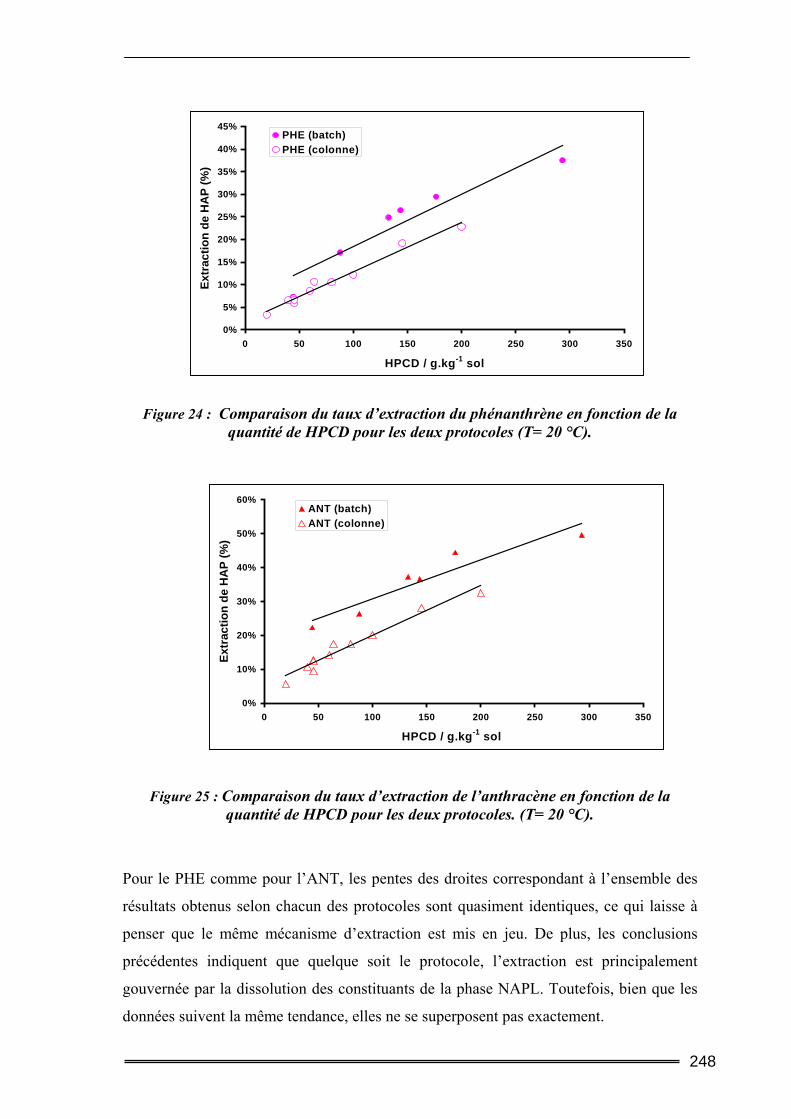
248
Figure 24 : Comparaison du taux d’extraction du phénanthrène en fonction de la quantité de HPCD pour les deux protocoles (T= 20 °C).
Figure 25 : Comparaison du taux d’extraction de l’anthracène en fonction de la quantité de HPCD pour les deux protocoles. (T= 20 °C).
Pour le PHE comme pour l’ANT, les pentes des droites correspondant à l’ensemble des
résultats obtenus selon chacun des protocoles sont quasiment identiques, ce qui laisse à
penser que le même mécanisme d’extraction est mis en jeu. De plus, les conclusions
précédentes indiquent que quelque soit le protocole, l’extraction est principalement
gouvernée par la dissolution des constituants de la phase NAPL. Toutefois, bien que les
données suivent la même tendance, elles ne se superposent pas exactement.
0%
5%
10%
15%
20%
25%
30%
35%
40%
45%
0 50 100 150 200 250 300 350
HPCD / g.kg-1 sol
Extr
actio
n de
HA
P (%
)
PHE (batch)PHE (colonne)
0%
10%
20%
30%
40%
50%
60%
0 50 100 150 200 250 300 350
HPCD / g.kg-1 sol
Extr
actio
n de
HA
P (%
)
ANT (batch)ANT (colonne)

249
La différence dans la valeur d’extraction absolue, est due au fait que la surface d’échange
entre la phase NAPL et la phase aqueuse est probablement fortement augmentée dans le
protocole en milieu dispersé. De ce fait, on remarque que l’on obtient naturellement de
meilleurs résultats en extraction avec le protocole en milieu dispersé, ce qui est
probablement dû à l’augmentation de surface de contact entre le liquide et le solide
consécutif à la désagrégation du sol, et à l’agitation forcée du réacteur qui provoque peut
être la mise en suspension de colloïdes ou des complexes HAP-Colloïdes et HAP-
Matière Organique.
III.5.2. Comparaison des différentes cyclodextrines
Nous avons utilisé dans cette étude trois cyclodextrines différentes : la β-cyclodextrine
naturelle (BCD), l’hydroxypropyl-β-cyclodextrine (HPCD) et la méthyl-β-cyclodextrine
(MCD). Ces deux dernières sont des dérivés de la cyclodextrine, où la greffe de groupes
fonctionnels a pour conséquence d’améliorer fortement la solubilité aqueuse, et donc leur
domaine d’utilisation (la solubilité aqueuse de la BCD étant limitée à 18 g.L-1 à 20°C).
Afin de pouvoir comparer l’efficacité des différents types de cyclodextrines entre elles,
les concentrations seront exprimées en millimoles de cyclodextrine par unité de masse de
sol (kg). Toutes les expériences (effet de la concentration, effet du L/S) réalisées avec un
même protocole peuvent alors être reportées sur un même graphe.
Les graphes 26 et 27 ci-dessous présentent les pourcentages d’extraction du phénanthrène
et de l’anthracène en batch percolant.
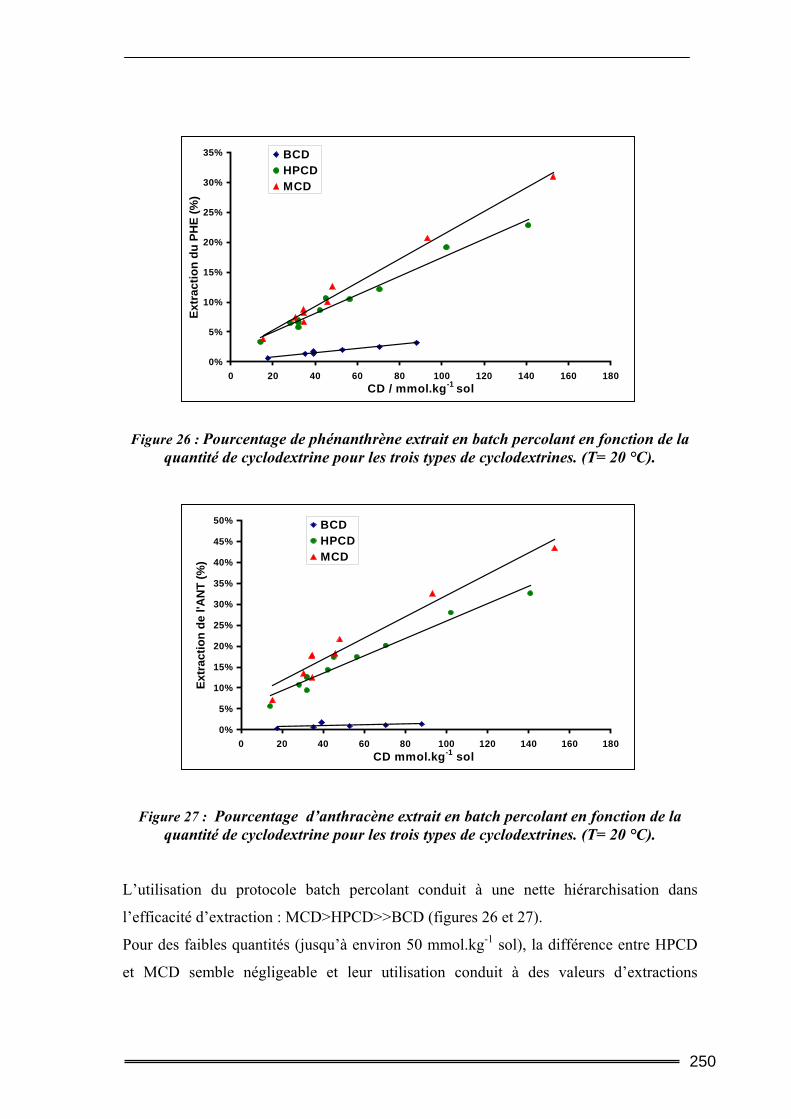
250
Figure 26 : Pourcentage de phénanthrène extrait en batch percolant en fonction de la quantité de cyclodextrine pour les trois types de cyclodextrines. (T= 20 °C).
Figure 27 : Pourcentage d’anthracène extrait en batch percolant en fonction de la quantité de cyclodextrine pour les trois types de cyclodextrines. (T= 20 °C).
L’utilisation du protocole batch percolant conduit à une nette hiérarchisation dans
l’efficacité d’extraction : MCD>HPCD>>BCD (figures 26 et 27).
Pour des faibles quantités (jusqu’à environ 50 mmol.kg-1 sol), la différence entre HPCD
et MCD semble négligeable et leur utilisation conduit à des valeurs d’extractions
0%
5%
10%
15%
20%
25%
30%
35%
0 20 40 60 80 100 120 140 160 180CD / mmol.kg-1 sol
Extr
actio
n du
PH
E (%
)
BCDHPCDMCD
0%
5%
10%
15%
20%
25%
30%
35%
40%
45%
50%
0 20 40 60 80 100 120 140 160 180CD mmol.kg-1 sol
Extr
actio
n de
l'A
NT
(%)
BCDHPCDMCD
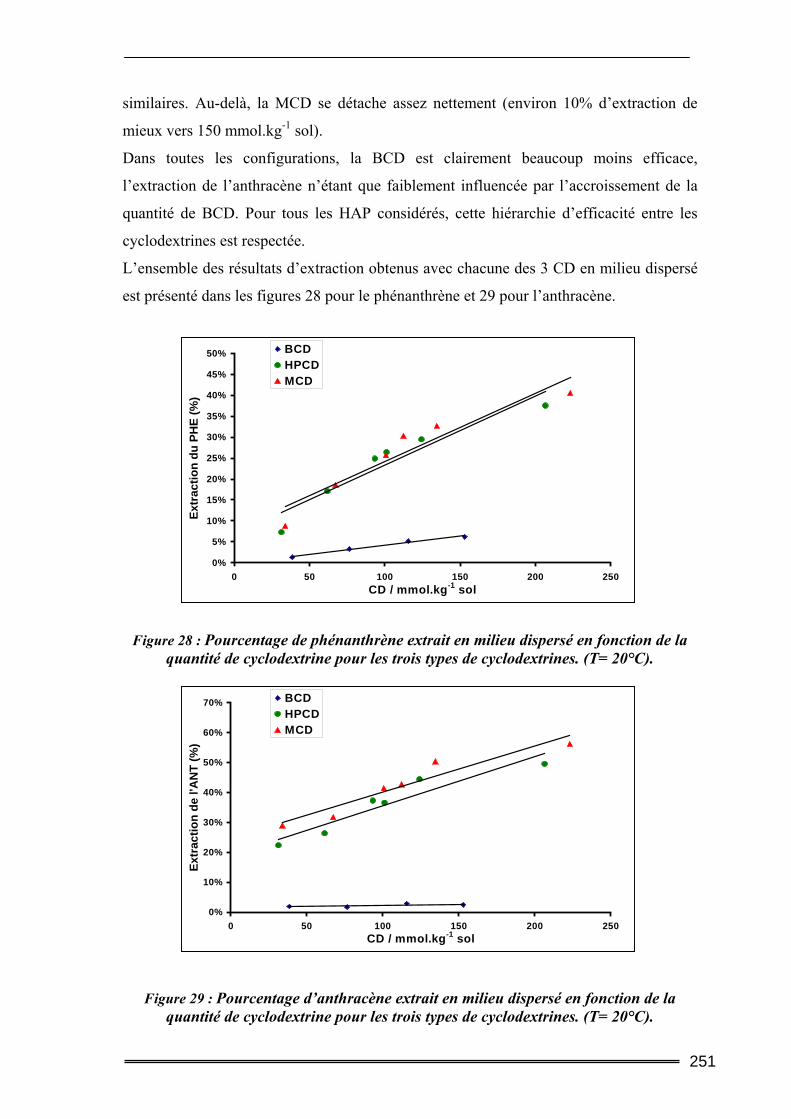
251
similaires. Au-delà, la MCD se détache assez nettement (environ 10% d’extraction de
mieux vers 150 mmol.kg-1 sol).
Dans toutes les configurations, la BCD est clairement beaucoup moins efficace,
l’extraction de l’anthracène n’étant que faiblement influencée par l’accroissement de la
quantité de BCD. Pour tous les HAP considérés, cette hiérarchie d’efficacité entre les
cyclodextrines est respectée.
L’ensemble des résultats d’extraction obtenus avec chacune des 3 CD en milieu dispersé
est présenté dans les figures 28 pour le phénanthrène et 29 pour l’anthracène.
Figure 28 : Pourcentage de phénanthrène extrait en milieu dispersé en fonction de la quantité de cyclodextrine pour les trois types de cyclodextrines. (T= 20°C).
Figure 29 : Pourcentage d’anthracène extrait en milieu dispersé en fonction de la quantité de cyclodextrine pour les trois types de cyclodextrines. (T= 20°C).
0%
5%
10%
15%
20%
25%
30%
35%
40%
45%
50%
0 50 100 150 200 250CD / mmol.kg-1 sol
Extr
actio
n du
PH
E (%
)
BCDHPCDMCD
0%
10%
20%
30%
40%
50%
60%
70%
0 50 100 150 200 250CD / mmol.kg-1 sol
Extr
actio
n de
l'A
NT
(%)
BCDHPCDMCD

252
En milieu dispersé, les résultats obtenus en batch percolant sont confirmés, avec toutefois
une nuance : cette fois la différence entre l’HPCD et la MCD est très faible sur toute la
gamme de concentration, et leurs performances peuvent quasiment être considérées
comme identiques. Il semblerait que l’HPCD ait plus gagné en efficacité avec la
dispersion du milieu. La BCD quant à elle, est toujours très nettement moins efficace, et
ne montre toujours pas de comportement satisfaisant avec l’anthracène.
De manière globale, l’efficacité d’extraction classe les CD selon l’ordre : MCD > HPCD
>> BCD.
On peut rappeler que les valeurs d’extraction avec de l’eau pure (L/S=3, 20°C) sont de
l’ordre de 0,19% pour le phénanthrène et 0,16% pour l’anthracène, comparé aux 38 % et
50 % obtenus avec 300 g de HPCD par kg de sol traité en batch percolant (environ 210
mmol.kg-1 sol). Ce qui représente dans ce cas un accroissement de l’efficacité d’un
facteur 200 au minimum.
De même on peut estimer que si la tendance d’extraction restait linéaire, il faudrait
apporter 630 g de HPCD par kg de sol pour extraire 80% du phénanthrène par la méthode
du batch en milieu dispersé. Ceci correspondrait à une expérience avec une solution de
HPCD à 210 g.L-1 et un ratio L/S de 3, ou une extraction par une solution à 50 g.L-1 (5%)
et un ratio L/S de 12.
III.5.3. Comparaison entre les quantités de polluants extraites
On observe systématiquement que la concentration des HAP extraits à l’équilibre suit la
même tendance (CNAP > CPHE > CANT > CPYR) quelque soit la cyclodextrine utilisée, la
température et le protocole suivi (milieu dispersé ou batch percolant).
La figure 30 montre les variations des pourcentages d’extraction des 4 HAP en batch
percolant en fonction de leur coefficient de partage octanol/eau (A), de leur constante de
complexation avec l’HPCD (B) et de leur solubilité aqueuse (C).
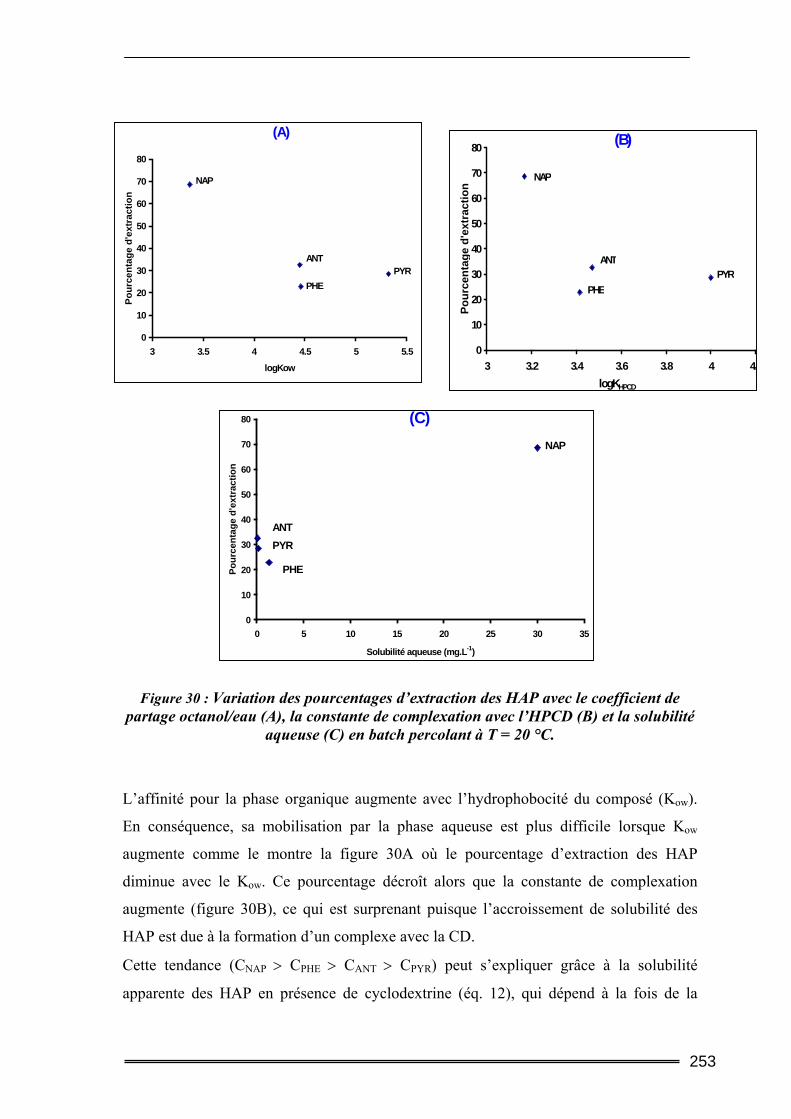
253
Figure 30 : Variation des pourcentages d’extraction des HAP avec le coefficient de partage octanol/eau (A), la constante de complexation avec l’HPCD (B) et la solubilité
aqueuse (C) en batch percolant à T = 20 °C.
L’affinité pour la phase organique augmente avec l’hydrophobocité du composé (Kow).
En conséquence, sa mobilisation par la phase aqueuse est plus difficile lorsque Kow
augmente comme le montre la figure 30A où le pourcentage d’extraction des HAP
diminue avec le Kow. Ce pourcentage décroît alors que la constante de complexation
augmente (figure 30B), ce qui est surprenant puisque l’accroissement de solubilité des
HAP est due à la formation d’un complexe avec la CD.
Cette tendance (CNAP > CPHE > CANT > CPYR) peut s’expliquer grâce à la solubilité
apparente des HAP en présence de cyclodextrine (éq. 12), qui dépend à la fois de la
0
10
20
30
40
50
60
70
80
3 3.5 4 4.5 5 5.5
logKow
Pour
cent
age
d'ex
trac
tion
NAP
PHE
ANTPYR
(A)
0
10
20
30
40
50
60
70
80
3 3.2 3.4 3.6 3.8 4 4.logKHPCD
Pour
cent
age
d'ex
trac
tion
NAP
PHE
ANTPYR
(B)
0
10
20
30
40
50
60
70
80
0 5 10 15 20 25 30 35
Solubilité aqueuse (mg.L-1)
Pour
cent
age
d'ex
trac
tion
PYRANT
PHE
NAP
(C)
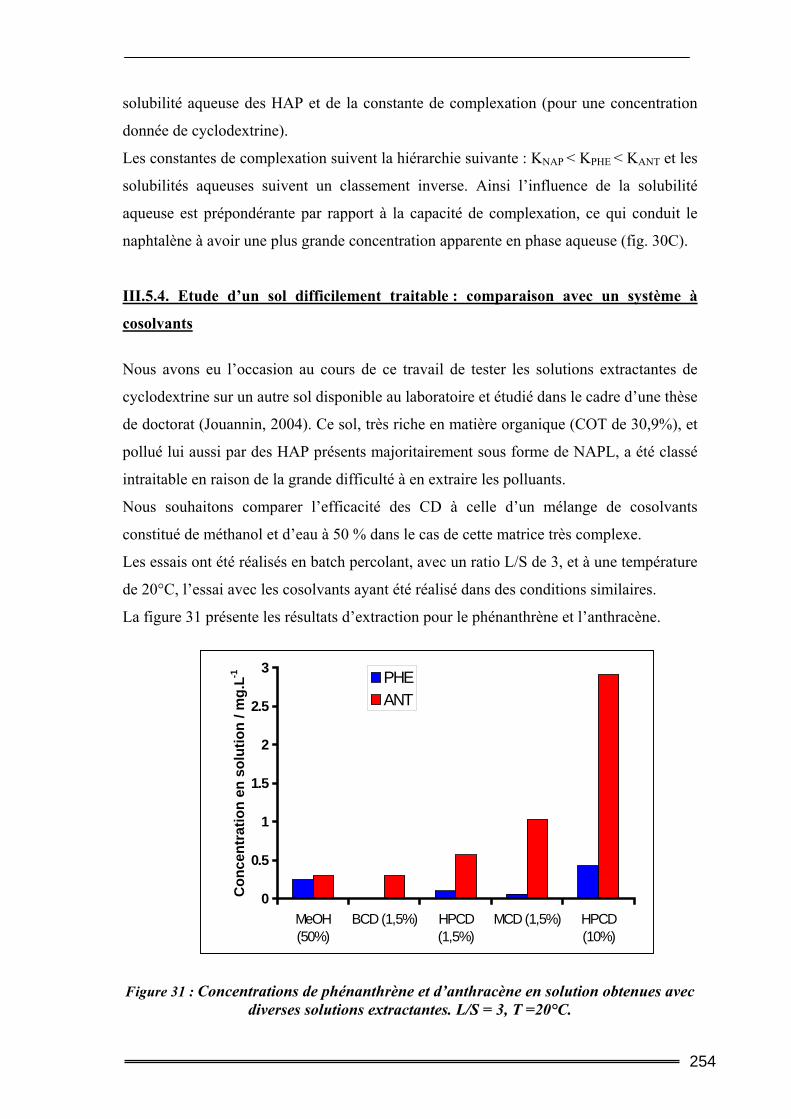
254
solubilité aqueuse des HAP et de la constante de complexation (pour une concentration
donnée de cyclodextrine).
Les constantes de complexation suivent la hiérarchie suivante : KNAP < KPHE < KANT et les
solubilités aqueuses suivent un classement inverse. Ainsi l’influence de la solubilité
aqueuse est prépondérante par rapport à la capacité de complexation, ce qui conduit le
naphtalène à avoir une plus grande concentration apparente en phase aqueuse (fig. 30C).
III.5.4. Etude d’un sol difficilement traitable : comparaison avec un système à
cosolvants
Nous avons eu l’occasion au cours de ce travail de tester les solutions extractantes de
cyclodextrine sur un autre sol disponible au laboratoire et étudié dans le cadre d’une thèse
de doctorat (Jouannin, 2004). Ce sol, très riche en matière organique (COT de 30,9%), et
pollué lui aussi par des HAP présents majoritairement sous forme de NAPL, a été classé
intraitable en raison de la grande difficulté à en extraire les polluants.
Nous souhaitons comparer l’efficacité des CD à celle d’un mélange de cosolvants
constitué de méthanol et d’eau à 50 % dans le cas de cette matrice très complexe.
Les essais ont été réalisés en batch percolant, avec un ratio L/S de 3, et à une température
de 20°C, l’essai avec les cosolvants ayant été réalisé dans des conditions similaires.
La figure 31 présente les résultats d’extraction pour le phénanthrène et l’anthracène.
Figure 31 : Concentrations de phénanthrène et d’anthracène en solution obtenues avec diverses solutions extractantes. L/S = 3, T =20°C.
0
0.5
1
1.5
2
2.5
3
MeOH(50%)
BCD (1,5%) HPCD(1,5%)
MCD (1,5%) HPCD(10%)
Con
cent
ratio
n en
sol
utio
n / m
g.L-1 PHE
ANT

255
Sur ce type particulier de sol très pollué (environ 2500 mg.L-1 pour le total des 16 HAP de
l’USEPA), les solutions de cyclodextrines sont de manière générale plus efficaces que le
système méthanol/eau à 50/50 (ratio le plus important utilisé dans la thèse de référence
(Jouannin, 2004)). Ceci est particulièrement visible pour l’anthracène avec lequel on
obtient des concentrations en solution jusqu’à 5 fois plus élevées, et où même la BCD
rivalise avec le mélange de cosolvants. Toutefois, les cyclodextrines apparaissent
généralement moins efficaces que le mélange eau/méthanol à 50/50 pour solubiliser le
phénanthrène, puisque seule la solution d’HPCD à 100 g.L-1 s’est avérée plus
performante.
Dans le cas du polluant le « mieux » extrait (anthracène), la même hiérarchie entre les
cyclodextrines est observée que pour le sol SITA.
Contrairement au cas du sol fourni par la société SITA, les solutions de lavages sont plus
concentrées en anthracène qu’en phénanthrène. Ceci provient de l’état initial de
contamination du sol détaillé dans le tableau 10 :
Tableau 10 : Détail de la contamination initiale des sols SITA et Jouannin
Sol SITA Sol JouanninPhénanthrène (ppm) 200 225 Anthracène (ppm) 71 430
La concentration en phénanthrène dans le sol SITA, est environ deux fois plus élevée que
celle de l’anthracène, alors que c’est l’inverse pour le sol utilisé par Jouannin (2004).
Toutefois, ces bonnes performances restent relatives. En effet, les quantités extraites ne
représentent que 0,13% du phénanthrène et 0,40% de l’anthracène dans le meilleur des
cas (HPCD 10%). De plus les concentrations en solution sont respectivement prés de 200
fois et 20 fois plus faibles pour le phénanthrène et l’anthracène que dans le cas du sol
SITA (pour HPCD 100 g.L-1).
En effet, le sol Jouannin présente une pollution âgée, qui a subi une maturation physico-
chimique, et qui est fortement fixée au sol (Jouannin, 2004). Cette immobilisation peut
être due au fort taux de carbone organique dans le sol, et à la présence supposée de
graphite. Les matières organiques dans le sol sont connues pour fixer fortement les HAP
(Chiou et al., 1998), et entraînent de plus la fixation des cyclodextrines, diminuant de fait
leur efficacité. Comme nous l’avons montré dans le chapitre précédent, l’accroissement
du taux de matières organiques dans le sol, (les autres caractéristiques du sol étant
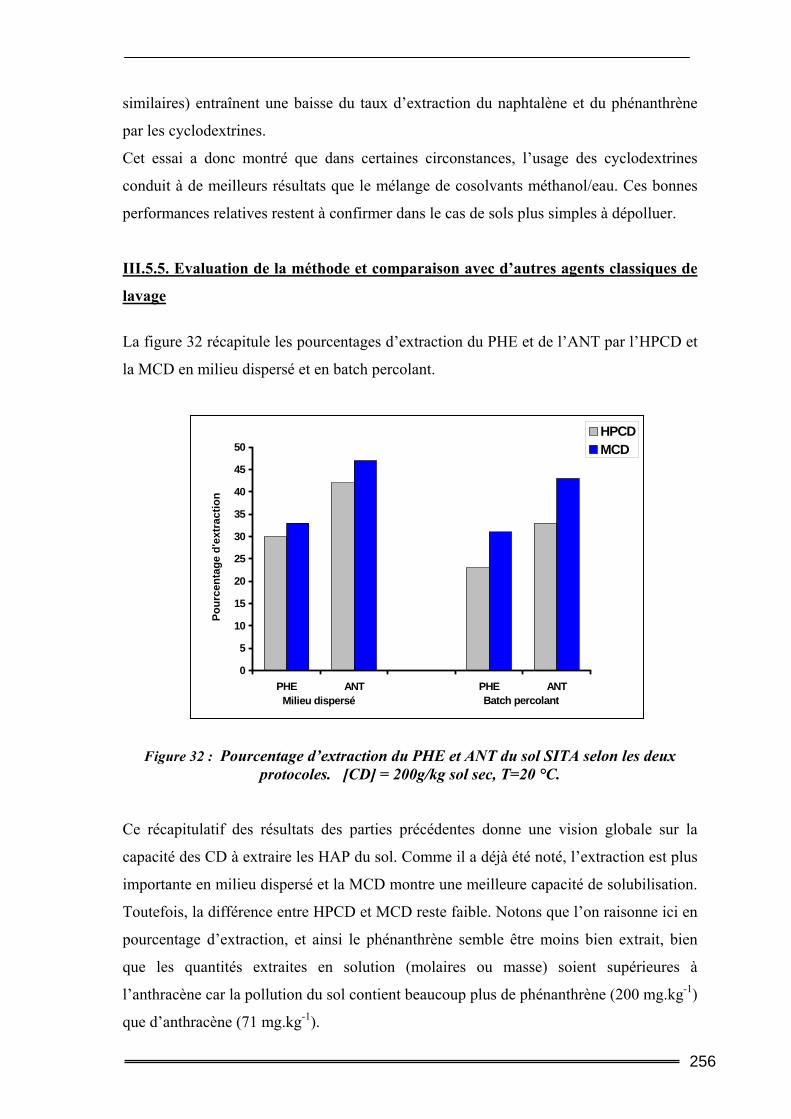
256
similaires) entraînent une baisse du taux d’extraction du naphtalène et du phénanthrène
par les cyclodextrines.
Cet essai a donc montré que dans certaines circonstances, l’usage des cyclodextrines
conduit à de meilleurs résultats que le mélange de cosolvants méthanol/eau. Ces bonnes
performances relatives restent à confirmer dans le cas de sols plus simples à dépolluer.
III.5.5. Evaluation de la méthode et comparaison avec d’autres agents classiques de
lavage
La figure 32 récapitule les pourcentages d’extraction du PHE et de l’ANT par l’HPCD et
la MCD en milieu dispersé et en batch percolant.
Figure 32 : Pourcentage d’extraction du PHE et ANT du sol SITA selon les deux protocoles. [CD] = 200g/kg sol sec, T=20 °C.
Ce récapitulatif des résultats des parties précédentes donne une vision globale sur la
capacité des CD à extraire les HAP du sol. Comme il a déjà été noté, l’extraction est plus
importante en milieu dispersé et la MCD montre une meilleure capacité de solubilisation.
Toutefois, la différence entre HPCD et MCD reste faible. Notons que l’on raisonne ici en
pourcentage d’extraction, et ainsi le phénanthrène semble être moins bien extrait, bien
que les quantités extraites en solution (molaires ou masse) soient supérieures à
l’anthracène car la pollution du sol contient beaucoup plus de phénanthrène (200 mg.kg-1)
que d’anthracène (71 mg.kg-1).
0
5
10
15
20
25
30
35
40
45
50
PHE ANT PHE ANT
Pour
cent
age
d'ex
trac
tion
HPCDMCD
Milieu dispersé Batch percolant
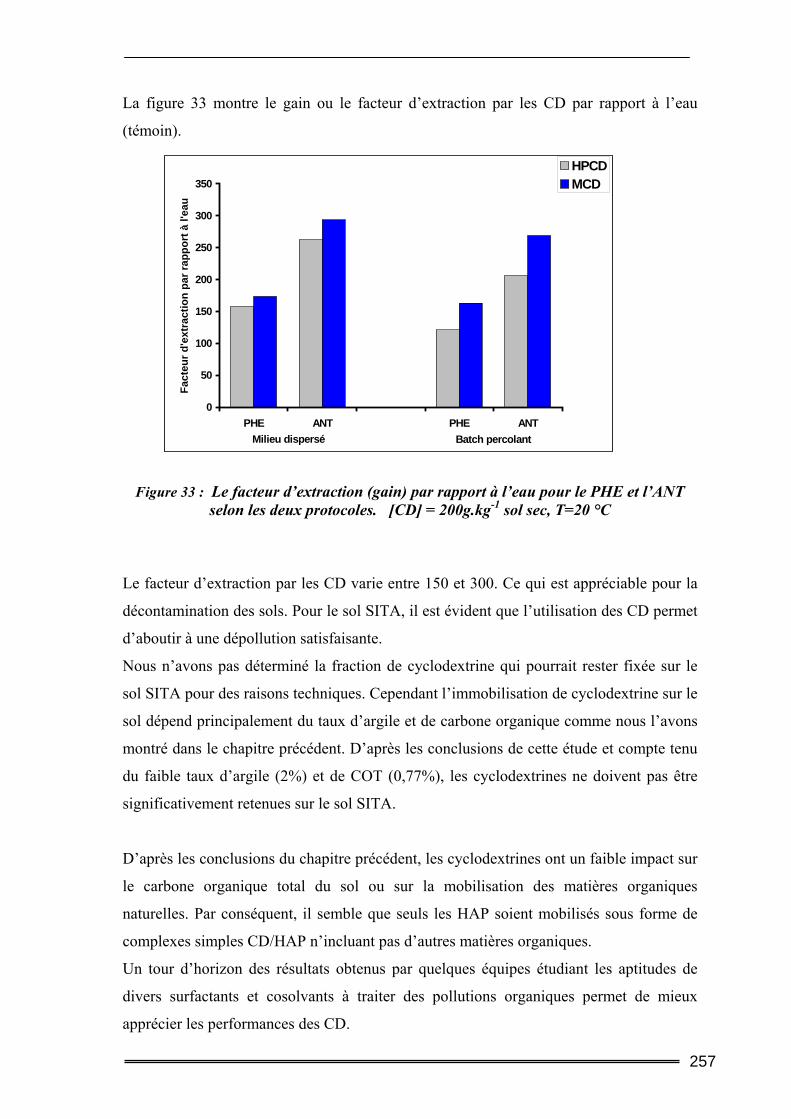
257
La figure 33 montre le gain ou le facteur d’extraction par les CD par rapport à l’eau
(témoin).
Figure 33 : Le facteur d’extraction (gain) par rapport à l’eau pour le PHE et l’ANT selon les deux protocoles. [CD] = 200g.kg-1 sol sec, T=20 °C
Le facteur d’extraction par les CD varie entre 150 et 300. Ce qui est appréciable pour la
décontamination des sols. Pour le sol SITA, il est évident que l’utilisation des CD permet
d’aboutir à une dépollution satisfaisante.
Nous n’avons pas déterminé la fraction de cyclodextrine qui pourrait rester fixée sur le
sol SITA pour des raisons techniques. Cependant l’immobilisation de cyclodextrine sur le
sol dépend principalement du taux d’argile et de carbone organique comme nous l’avons
montré dans le chapitre précédent. D’après les conclusions de cette étude et compte tenu
du faible taux d’argile (2%) et de COT (0,77%), les cyclodextrines ne doivent pas être
significativement retenues sur le sol SITA.
D’après les conclusions du chapitre précédent, les cyclodextrines ont un faible impact sur
le carbone organique total du sol ou sur la mobilisation des matières organiques
naturelles. Par conséquent, il semble que seuls les HAP soient mobilisés sous forme de
complexes simples CD/HAP n’incluant pas d’autres matières organiques.
Un tour d’horizon des résultats obtenus par quelques équipes étudiant les aptitudes de
divers surfactants et cosolvants à traiter des pollutions organiques permet de mieux
apprécier les performances des CD.
0
50
100
150
200
250
300
350
PHE ANT PHE ANT
Fact
eur d
'ext
ract
ion
par r
appo
rt à
l'ea
uHPCDMCD
Milieu dispersé Batch percolant

258
Bordas et Lafrance, (2003) ont montré que l’injection de 25 volumes de pores d’une
solution Rhamnolipides (biosurfactants) à 2,5 g.L-1, équivalent à L/S = 10 mobilise 70 %
du pyrène du sol. Chu et Kwan, (2003) ont utilisé un mélange de surfactants (Brig 35,
Tween 80, SDS) et des solvants organiques (acétone (ACE), triethylamine, squalane)
pour évaluer la désorption d’un biphenyl d’un sol pollué. Le pourcentage d’extraction
atteint respectivement 49%, 77% et 89% avec les combinaisons ACE/SDS, ACE/Tween
80 et ACE/Brij 35 avec un ratio molaire élevé (100). Les auteurs ont constaté que la
fixation des surfactants sur le sol retardent l’extraction et contribuent à la ré-adsorption
ou l’immobilisation des polluants. Krauss et Wilcke, (2001) montre qu’un mélange
eau/méthanol à 50 % solubilise environ 1/4 du contenu initial en HAP dans un sol pollué.
Dans cette étude, une extraction du PHE de 40 % est constatée en présence d’une solution
aqueuse de 10 % de MCD en milieu dispersé. La capacité d’extraction des CD est donc
comparable à celle des agents classiques de lavage. Cependant, la comparaison n’est
qu’indicatrice car la capacité d’extraction des agents de lavage dépend elle aussi
fortement du type de sol et de la nature de la pollution (origine, type, âge).
Il est nécessaire de rappeler que la biocompatibilité des CD (très faible toxicité et bonne
biodégradabilité) est supérieure à celle des solvants ou des surfactants. De plus,
contrairement à la plupart des surfactants et des solvants, les CD interagissent faiblement
avec les sols. Par conséquent, les CD ne devraient avoir qu’un très faible impact sur
l’écosystème (flore et faune) du sol.

259
IV. Bilan
Cette étude a permis d’évaluer les performances de trois types de cyclodextrines dans le
traitement d’un sol pollué provenant d’une ancienne usine à goudron. Les expériences
faites sur le sol SITA nous ont conduit à diverses conclusions quant à l’utilisation des
cyclodextrines comme agent d’extraction des HAP :
- Le mécanisme majoritaire d’extraction des HAP dans ce sol semble être la dissolution
de la NAPL vers la phase aqueuse. Ce raisonnement est aussi valable pour l’extraction
des HAP par les CD. Dans ce cas, les concentrations extraites lors des expériences de
lixiviation sont assez proches des valeurs théoriques estimées par le modèle de la
dissolution. Ces résultats permettent de caractériser le mécanisme principal de la
dépollution et de modéliser le comportement des polluants extraits dans le sol par les CD.
Cela est une différence notable avec le sol dopé où l’existence d’un mécanisme
majoritaire intervenant lors de l’extraction par les CD n’a pu être confirmé. A la
différence de l’étude du sol dopé par le PCP, ces résultats permettent aussi d’espérer
travailler dans des conditions linéaires d’extraction, c’est à dire que l’augmentation de la
quantité de cyclodextrine utilisée permet d’augmenter la quantité de HAP extraite.
- La cinétique a montré que la mise en solution des HAP par les CD est rapide. Le temps
pour atteindre un régime stationnaire est de l’ordre d’une heure en milieu dispersé. Pour
le batch percolant, ce temps dépend de la taille du dispositif utilisé. La rapidité dans la
mise en solution par les CD a également été observée pour les sols dopés.
- La concentration des HAP solubilisés augmente de façon significative en présence de
CD. Cette augmentation semble être proportionnelle à la quantité de cyclodextrine mise
en contact avec le sol, que ce soit par l’augmentation de concentration en cyclodextrine
ou par celle du ratio liquide/solide. A 100 g/L de MCD (L/S 3), on atteint un taux
d’extraction en phénanthrène de 30 à 40 % soit 158-210 fois plus qu’avec de l’eau pure
(0,19%). Pour ce sol, le gain d’extraction par la CD par rapport à l’eau est extrêmement
élevé comparativement à un sol dopé (3 fois plus seulement qu’avec de l’eau pure). La
différence majeure est la concentration extraite relativement élevée à l’eau dans le cas du
sol dopé (20 % du PHE extrait à l’eau pure).

260
- Dans les conditions de notre étude, l’extraction semble relativement insensible aux
variations de températures. En effet il semblerait qu’il y ait un effet de compensation
entre l’augmentation de la solubilité aqueuse des HAP avec la température et la
déstabilisation concomitante du complexe cyclodextrine/HAP. Cela permet d’espérer
travailler avec une efficacité constante entre 5 et 35°C.
- Les protocoles, milieu dispersé et batch percolant, présentent une différence
significative d’efficacité d’extraction. Les meilleurs résultats obtenus en milieu dispersé
semblent être liés à l’augmentation de la surface de contact liquide/solide. Cette
différence d’efficacité liée aux protocoles expérimentaux est moins prononcée dans le cas
du sol dopé où une légère différence a été observée.
- Les trois cyclodextrines utilisées ne sont pas d’efficacité équivalente : la BCD est
nettement moins active que la HPCD, elle-même légèrement moins efficace que la MCD.
De plus la BCD est limitée par sa faible solubilité aqueuse. Son utilisation ne se justifiera
que dans le cas d’un coût global d’opération bien inférieur (ou dans le cas d’un impératif
de biodégradation rapide). Pour tous les HAP considérés, cette hiérarchie d’efficacité
entre les cyclodextrines est respectée. Cette hiérarchisation a également été observée dans
le cas des sols dopés.
- L’essai effectué sur un sol très difficile à traiter (taux élève en COT et en polluants,
fortement fixés) a montré que l’usage des cyclodextrines peut être plus efficace que celui
d’un système à cosolvants méthanol/eau à 50%.

261
CONCLUSION GENERALE
L’efficacité d’une méthode de traitement de sols basée sur la solubilisation des polluants
dépend du type d’agent solubilisant, de la nature et de l’étendue de la pollution, et du type
de sol à traiter. Une bonne connaissance des interactions prépondérantes dans ces
systèmes complexes Agent solubilisant /Polluant(s) /Sol est un préalable indispensable
pour une bonne adéquation du mode de remédiation au contexte. Dans notre cas, la mise
au point d’un tel procédé d’extraction requiert l’évaluation des interactions susceptibles
de se produire entre les cyclodextrines, les polluants et les sols. Ces trois éléments
constituent les principaux axes sur lesquels sont basées les recherches réalisées dans cette
thèse. Deux grandes familles des polluants organiques, trois types de cyclodextrines
parmi les plus connues : β-cyclodextrine, Hydroxypropyl-β-cyclodextrine et Méthyl-β-
cyclodextrine et plusieurs matrices de sol, ont été utilisées dans cette étude.
Les composés organiques sélectionnés dans le cadre du travail qui vient d’être décrit
(hydrocarbures aromatiques polycycliques et chlorophénols) sont parmi les polluants
industriels les plus fréquents en France et dans les pays industrialisés, d’où l’intérêt
d’étudier leur extraction par les cyclodextrines.
L’objectif de notre travail était d’étudier la faisabilité de l’utilisation de molécules
complexantes, les cyclodextrines, dans la décontamination de sols. Notre étude s’est
déroulé en deux étapes principales permettant de mettre en évidence concrètement, et
dans un ordre logique, les interactions entre les cyclodextrines, les polluants et les sols :
I. Interactions CD/Polluant/Eau : Il s’agissait de tests préalables de faisabilité « hors
sol », qui consistaient à évaluer les interactions des cyclodextrines avec les polluants
cibles dans l’eau.
II. Interactions CD/Polluant/Sol : Il s’agissait de tests de faisabilité, en présence de sol,
pour évaluer les performances extractives des CD vis-à-vis de deux cas de pollution :
* Cas d’une pollution artificielle : Etude de la désorption de polluants sur deux sols
distincts. Ces sols, contaminés au laboratoire, ont été lavés en présence de CD afin

262
d’extraire les polluants. L’adsorption des CD sur diverses matrices de sol a également été
évaluée.
* Cas d’une pollution réelle : Etude de l’aptitude des CD à extraire des polluants d’un sol
réellement pollué d’origine industrielle . Détermination de l’influence des paramètres
physico-chimiques pertinents (ratio L/S, température, concentration en cyclodextrine) sur
l’efficacité d’extraction.
Les résultats obtenus au cours des différentes étapes de ce travail ont permis une
meilleure compréhension des phénomènes et des paramètres contrôlant la complexation
et l’extraction, par les CD, des polluants dans les sols.
Les résultats expérimentaux obtenus dans la première partie de notre travail indiquent que
les cyclodextrines testées ont un pouvoir complexant intéressant sur les polluants
représentatifs des chlorophénols et des hydrocarbures aromatiques polycycliques. Les CD
modifiées sont les plus actives pour augmenter leur solubilité. Le contexte chimique (pH
et force ionique) peut avoir des conséquences importantes en terme de gain de solubilité.
La préoccupation d’un traitement des effluents d’extraction nous a conduit à tester la
fiabilité de la photocatalyse hétérogène pour dégrader de complexes CD/PCP. Les
résultats montrent qu’il s’agit d’une voie de traitement possible, mais qui nécessitera des
expériences complémentaires sur des effluents d’extraction réels.
Les résultats détaillés dans la partie expérimentale du chapitre II démontrent l’aptitude
des CD à extraire plusieurs polluants d’un sol contaminé au laboratoire. Pour les deux
sols testés, la désorption de chacun des polluants cibles est un phénomène rapide. La
capacité d’extraction des solutions de CD est nettement supérieure à celle de l’eau. Le
rendement d’extraction évolue de manière importante avec la quantité et selon la nature
de la CD utilisée. La MCD s’avère plus efficace que l’HPCD qui l’est elle même plus que
la BCD. Le protocole expérimental (milieu dispersé ou colonne de percolation) n’influe
que faiblement sur les performances de lavage. La dernière partie de ce chapitre a
consisté à étudier les interactions des cyclodextrines avec différents types de sols. Il
apparaît clairement que les CD s’adsorbent sur les argiles et la matière organique des
sols. Bien que les quantités retenues soient relativement faibles, elles peuvent contribuer
à une refixation des polluants et rendre le traitement moins performant. Il a été aussi

263
établi que les CD ne provoquent pas une mobilisation des MON plus importante que
l’eau, ce qui est important pour la préservation de l’écosystème sol.
Dans la dernière partie concernant l’évaluation des capacités d’extraction des trois
cyclodextrines étudiées sur un sol industriel pollué par des HAP, nous avons confirmé
l’efficacité des CD vis-à-vis d’une pollution ancienne et complexe. La concentration des
HAP solubilisés augmente de façon significative en présence de CD. Cette augmentation
semble être proportionnelle à la quantité de cyclodextrine mise en contact avec le sol. Le
suivi cinétique a montré que la mise en solution des HAP par les CD est rapide. Dans les
conditions de notre étude, l’extraction semble relativement insensible aux variations de
température. Les trois cyclodextrines utilisées ne sont pas d’efficacité équivalente : la
BCD est nettement moins active que la HPCD, elle-même légèrement moins efficace que
la MCD.
Au vu de l’ensemble des résultats, cette étude de faisabilité d’un traitement de pollution
organique utilisant les propriétés complexantes des cyclodextrines nous semble avoir
atteint ses objectifs. De nombreux paramètres ont été quantifiés qui permettent
d’envisager positivement la mise au point d’un procédé à l’échelle pilote, puis au
dimensionnement industriel. Ces étapes vers le transfert technologique nécessiteront de
multiples études complémentaires, en particulier au niveau technico-économique.
Le ratio efficacité/coût est bien évidement un facteur déterminant pour une mise en œuvre
industrielle. Dans ce cadre, il conviendra d’étudier avec attention les possibilités de
« recyclage » des cyclodextrines, surtout dans le cas d’utilisation de molécules modifiées,
plus onéreuses que la CD native. Ces études vont nécessairement de pair avec des
recherches de solutions de traitement des effluents de lavage générés. Enfin, il parait
indispensable d’approfondir l’influence des cyclodextrines sur l’activité des micro-
organismes présents dans les sols. Cet aspect, s’il est positif comme nous le pensons,
pourrait constituer les bases d’un traitement complémentaire par biodégradation des
molécules organiques résiduelles non extraites mais solubilisées au sein du sol.
Les réponses aux demandes sociétales, de plus en plus pressantes, concernant la
réhabilitation de « nos » sols passent à l’évidence par un engagement pluridisciplinaire de
la communauté scientifique.

264

265
REFRENCES BIBLIOGRAPHIQUES AIKEN, G.R., McKNIGHT, D.M., WERSHAW, R.L., McCARTY, P. Humic subtsances in soil, sediment & water : Geochemistry, Isolation & Characterization. New York : Wiley-Interscience Publication, 1985, ISBN : 0-471-88274-7, 682 p. AL-BASHIR B., CSEH T., LEDUC R, SAMSON R. Effect of soil/contaminant interactions on the biodegradation of naphthalene in flooded soil under denitrifiyng conditions » Appl. Microbiol. Biotechnol.,1990, vol. 34, pp. 414-419. ANGLEY, T.A., BRUSSEAU, M.L., MILLER, W.L., DELFINO, J.J. Nonequilibrium Sorption and Aerobic Biodegradation of Dissolved Alkybenzenes during Transport in Aquifer Materials : Column Experiments and Evaluation of Coupled-Process Model. Environ. Sci. Technol. 1992, vol. 24, pp.727-735. ARCHAND, Y., HAWARI, J., GUIOTT, S. R.. Solubility of pentachlorophenol in aqueous solutions: the pH effect. Water Res., 1995, vol. 29, pp.131-136. ARONSTEIN, B.N., CALVILLO, Y.M., ALEXANDER, M. Effect of Surfactants at Low Concentration on the Desorption and Biodegradation of Sorbed Aromatic Compounds in Soil. Environ. Sci.& Technol., 1991, vol. 25, n°10, pp.1728-1731. AUGUGLIARO V., V. LODDO, M. SCHIAVELLO, Heterogeneous photocatalytic reactors : an assessment of fundamental engineering aspects, In : Heterogeneous Photocatalysis, M. SCHIAVELLO. Chichester, UK : J. Wiley & Sons, 1997, pp. 169-189. (Wiley series in photoscience and photoengineering, vol 3) AZIZ C., MELCER H., Bioremediation of Soils Contaminated with Manufactured Gas Plant Residues - a litterature review. Burlington (Canada) : Wastewater Technology Center, 1991, 60p. BAI G., BRUSSEAU M. L., MILLER R. M. Biosurfactant-enhanced removal of residual hydrocarbon from soil. J. Contaminant Hydrol., 1997, vol. 25, pp.157-170. BAI G., BRUSSEAU M. L., MILLER R. M. Influence of cation type, ionic strength, and pH on solubilization and mobilization of residual hydrocarbon by biosurfactant. J. Contaminant Hydrol., 1998, vol. 30, pp. 265-279. BALL W. P., ROBERTS, P. V. Long-term sorption of halogenated organic chemicals by aquifer material. 2. Intraparticle Diffusion. Environ. Sci. Technol., 1991, vol. 25, n°7, pp. 1237-1249. BALL W. P., ROBERTS, P. V. Long-term sorption of halogenated organic chemicals by aquifer material .1. equilibrium – comment. Environ. Sci. Technol., 1992, vol. 26, , n°5, pp 2301-2302. BANERJI, S.K., WEI, S.M., BAJPAI, R.K. Pentachlorophenol Interactions with Soil. Water, Air, & Soil Pollution, 1993, vol. 69, pp. 149-163. BARDI. L., MATTEI. A., STEFFAN.S., MARZONA M. Hydrocarbon degradation by a soil microbial population with β-Cyclodextrin as surfactant to enhance bioavailability. Enzyme and Microbial Techno., 2000, vol. 27, pp.709-713. BARKAY, T., NAVON-VENEZIA, S., RON, E.Z. AND ROSENBERG, E. Enhancement of solubilization and biodegradation of polyaromatic hydrocarbons by the bioemulsifier Alasan. Appl. Environ. Microbiol. 1999, vol. 65, pp. 2697–2702. BARRIUSO E., BAER U., CALVET R. Dissolved organic matter and adsorption-desorption of dimefuron, atrazine and carbetamide by soil. J. Environ. Qual., 1992, vol. 21, n° 3, pp. 359-367. BARRIUSO E., KOSKINEN W. C. Incorporating nonextractable atrazine residues into soil size fractions as a function of time. Soil Sci. Soc. Am. J., 1996, vol. 60, pp. 150-157. BASTIAENS, L., SPRINGAEL, D., WATTIAU, P., HARMS, H., DEWACHTER, R., VERACHTERT, H. AND DIELS, L.N Isolation of adherent polycyclic aromatic hydrocarbon (PAH)-degrading bacteria using PAH-sorbing carriers. Appl. Environ. Microbiol. 2000, vol. 66, pp. 1834-1843 BAUDIN I., J. M. LAINE, D. D. DIONYSIOU, M. T. SUIDAN, Effect of ionic strength and hydrogen peroxide on the photocatalytic degradation of 4-chlorobenzoic acid in water, Applied Catalysis B : Environmental, 2000 , vol. 26, pp.153-171. BAUER R., WALDNER G., MALATO S., The photo-fenton reaction and the TiO2/UV process for waste water treatment – novel developments, Catalysis Today, 1999, vol. 53, pp.131-144.

266
BAYARD R. Etude de l’adsorption/désorption de polluants organiques dans les sols. Thèse LAEPSI. Lyon : INSA de Lyon, 1997, 300 p. BAYARD R., BARNA L., MAHJOUB B., GOURDON R. Influence of the presence of PAHs and coal tar on naphthalene sorption in soils. J. Contam. Hydraul., 2000, vol. 46, pp 61-80. BEACH, D.N., MCCRAY, J.E. Numerical modeling of unsaturated flow in wastewater soil absorption systems, Ground Water Monitoring Remediation, 2003, vol. 23, n° 2, pp. 64-72. BELLIN, C. A., O’CONNOR, G. A. & JIN, Y. Sorption and Degradation of Pentachloropenol in Sludge-Amended Soils. J. Environ. Qual., 1991, vol.19, pp. 603-608. BHANDARI A., NOVAK J. T., BERRY D. F. Binding of 4-monochlorophenol to soil. Environ. Sci. Technol., 1996, vol. 30, n° 7, pp 2305-2311. BILAL M., DE BRAUER CH., CLAUDY P., GERMAIN P., LÉTOFFÉ J. M. β-Cyclodextrin hydration: a calorimetric and gravimetric study. Thermochimica Acta, 1995, vol. 249, pp. 63-73. BIZZIGOTTI G. O., REYNOLDS D. A., KUPPER B. H. Enhanced solubilization and destruction of Tetrachloroethylene by Hydroxypropyl-β- cyclodextrin and iron. Environ. Sci. Technol. 1997; vol. 31, pp. 472-478. BLUMER M., BLUMER W., REICH T. Polycyclic aromatic hydrocarbons in soils of a mountain valley : correlation with highway traffic and cancer incidence. Environ Sci Technol, 1977, vol. 11, pp 1082-1084. BLYSHAK L. A., WARNER, I.H., Evidence for non-inclusional association betwwen cyclodextrin and polynuclear aromatic hydrocarbons, Anal. Chim. Acta, 1990, vol. 232, pp. 239-243. BLYSHAK L., DODSON K., PATONAY G., WARNER I. Determination of cyclodextrion foramtion constants using dynamic coupled-column liquid chromatography. Anal. Chem., 1989, vol. 61, pp. 955-960. BLYSHAK LA, ROSSI TM, PATONAY G., WARNER IM. Cyclodextrin-modified solvent extraction for polynuclear aromatic hydrocarbons, Anal. Chem., 1988, vol. 60, pp. 2127-2131. BOLLAG J. M., LOLL M. J. Incorporation of xenobiotic into soil humus. Experientia, 1983, vol. 39, pp. 1221-1230. BOLLAG J. M., MYERS C. J., MINARD. R. D. Biological and chemical interactions of pesticides with soil organic matter. Sci. Total Environ., 1992, vol. 123, pp. 205-217. BOUCHEZ M., BLANCHET D., VANDECASTEELE J. P. The microbiological fate of polycyclic aromatic hydrocarbons : Carbon and oxygen balances for bacterial degradation of model compounds. Appl. Microbiol. Biotechnol., 1996, vol. 45, pp. 556-561. BOVING THOMAS B., BRUSSEAU MARK L. Solubilization and removal of residual trichloroethene from porous media: comparison of several solubilization agents, J. Contam. Hydrol. 2000, vol. 42, pp. 51-67. BOVING T., WANG X., BRUSSEAU M.L. Cyclodextrin-Enhanced Solubilization and Removal of Residual-Phase Chlorinated Solvents from Porous Media. Environ. Sci. Technol. 1999, vol. 33, pp.764-770. BOVING, T.B., MCCRAY, J.E., BRUSSEAU M.L. Cyclodextrin-enhanced remediation of organic and metal contaminants in porous media and groundwater, Remediation J., 2000, vol. 10, n°2, pp. 59-83. BOYD S. A. Adsorption of substituted phenols by soil. Soil Sci., 1982, vol. 134, n°5, pp. 377-343. BOYD, S.A. ; MICKESELL, M.D.; LEE, J.F. Reaction & Movement of Organic Chemicals in Soils. In : Reaction & Movement of Organic Chemicals in Soils. Eds : B.L. Sawhney & K. Brown. Madison (Wisconsin) : Soil Science Society of America : American Society of Agronomy, 1989, 474 p. BRAUN, W.H., BLAU, G.E. ET CHENOWETH, M.B. The metabolism/ pharmacokinetics of pentachlorophenol in man, and a comparison with the rat and a monkey model. Toxicol. Appl. Pharmacol., 1978, vol 45, pp. 278-285. BRIGGS, G. Theoretical & Experimental Relationships between Soil Adsorption, Octanol-Water Partition Coefficient, Water Solubilities, Boiconcentration Factors, & the Parachor. J. Agric. Food Chem., 1981, vol. 29, pp. 1050-1059.

267
BRUSSEAU M. L., JESSUP R. E., RAO P. S. C. Nonequilibrium sorption of organic chemical : Elucidation of rate-limiting processes. Environ. Sci. Technol., 1991, vol. 25, pp. 134-142. BRUSSEAU M. L., WANG X., WANG W-Z. Simultaneous elution of heavy metals and organic compounds from soil by cyclodextrin. Environ. Sci. Technol., 1997, vol.31, pp. 1087-1092. BRUSSEAU ML. Use of tracer tests to evaluate the impact of enhanced-solubilization flushing on in-situ biodegradation. Journal of Contaminant Hydrology , 2003, vol .64, pp. 191-202. BRUSSEAU, WANG X., HU Q.. Enhanced Transport of Low-Polarity Organic Compounds through Soil by Cyclodextrin. Environ. Sci. Technol. 1994, vol. 28, pp. 952-956. BUVARI, A. AND BARCZA, L. Complex formation of phenol, Aniline, and their Nitro Derivatives with β-Cyclodextrin. J. Chem. Soc. Perkin Trans. II, 1988, vol. 5, pp. 543-545. CAIN, R.B., JOHNSON, G., MCCRAY, J.E., BLANFORD, W., BRUSSEAU, M.L., Partitioning tracer tests for evaluating remediation performance, Ground Water, 2000, vol. 38, n°5, pp.752-761. CALVET R. Adsorption of organic chemicals in soils, Environmental Perspectives, 1989, vol. 83, pp 145-177. CAO J., ZHAO C., HUANG L., DING Y., WANG L., HAN S. Solubilization of substituted indole compounds by β-Cyclodextrin in water. Chemosphere, 2000, vol. 40, pp. 1411-1416. CARMICHAËL L.M., CHRISTMAN R.F., PFAENDER F.K. Desorption and mineralization kinetics of phenanthrene and chrysene in contaminated soils. Environ. Sci. Technol., 1997, vol. 31, pp. 126-132. CASTERLINE, J.L. ; BARNETT, N.M. & KHU, Y. Uptake, Translocation, and Transformation of Pentachlorophenol in Soybean and Spinash Plants. Environ Res., 1985, vol. 37, pp. 101-118. CERNIGLIA, C.E., Biodegradation of polycyclic aromatic hydrocarbons. Biodegradation, 1992, vol. 3, pp. 351–358. CHAMPION J. T., GILKEY J. C., LAMPARSKI H., RETTERER J., MILLER R. M. Electron microscopy of rhamnolipid (biosurfactant) morphology: effects of pH, cadmium, and octadecane. J. Colloid Interface Sci., 1995, vol. 170, pp. 569-574. CHIOU C. T. Theoritical considerations of the partition of nonionic organic compounds by soil organic. In : Reaction and movement of organic chemicals in soils. Sawhney, Brown Eds. Brown. Madison (Wisconsin) : Soil Science Society of America : American Society of Agronomy, 1989, pp. 1-30. CHIOU, C.T., SHOUP, T.D. Soil Sorption of Organic Vapors and Effects of Humidity on Sorptive Mechanism & Capacity. Env. Sci. Technol., 1985, vol. 18, pp. 4-10. CHIOU, C.T., MALCOLM, R.L., BRINTON, T.I., KILE, D.E. Water solubilty enhancement of some organic pollutants and pesticides by dissolved humic and fulvic acids, Environmental Science and Technology, 1986, vol. 24, pp.502-508. CHOUDHRY, G.G. Humic Substances : Sorptive Interactions with Environmental Chemicals. In : Humic Substances : Structural, Photophysical, Photochemical & Free Radical Aspects & Interactions with Environmental Chemicals. New York : Gordon & Breach Science Publishers Inc., 1984, p. 95-134. ISBN : 0-677-06440-3. CHU W. AND C. Y. KWAN. Remediation of contaminated soil by a solvent/surfactant system Chemosphere, 2003, vol. 53, pp. 9-15. CHU W. AND C. Y. KWAN. The mechanism of the surfactant-aided soil washing system for hydrophobic and partial hydrophobic organics. The Science of The Total Environment, 2003, vol. 307, pp. 83-92. CHU W. AND K. H. CHAN. The estimation of PAH bioavailability in contaminated sediments using hydroxypropyl- cyclodextrin and Triton X-100 extraction techniques Chemosphere, 2002, vol. 46, n° 8, pp. 1235-1245. CLAUDY P., GERMAIN P., LETOFFE J.M., BAYOL A., GONZALES B. Etude thermodynamique de la réaction d’hydratation de la β-cycodextrine. Thermochimica Acta, 1990, vol. 161, pp. 75-84. CLINT, J. H., Surfactant Aggregation, Blackie, London : Chapman and Hall, 1992, 200p. COLY A., AARON J-J., Cyclodextrin-enhanced fluorescence and photochemically-induced fluorsence determination of fice aromatic pesticides in water , Anal. chimica Acta, 1998, vol. 360, pp.129-141.

268
CONNORS K. A. Binding constants the measurements of molecular complex stability, chapter 2. New York : Wiley, 1987, pp. 200-205. CORT T., BIELEFELDT, Mechanism of nonionic surfactants inhibition of pentachlorophenol biodegradation, Water Reserach, 2002, vol. 36, pp.1253-1261. CROSBY, D.G. ; BEYNOU, K.L. ; GREVE, P.A. & al. Environmental Chemistry of Pentachlorophenol. Pure & Appl. Chem., 1981, vol. 53, pp. 1051-1080. CUNNINGHAM J., P. SEDLAK, Interrelationships between pollutant concentration, extent of adsorption, TiO2 sensitized removal, photon flux and levels of electron or hole traping additives, Journal of Photochemistry and Photobiology A : Chemistry, 1994, vol. 77, pp. 255-263. CURTIS G.P., REINHARD M., ROBERTS P.V., Sorption of Hydrophobic organic compounds by sediments. Amer. Chem. soc. Symposium Series, 1986, vol. 323, pp. 191-216. CUYPERS C, GROTENHUIS T, JOZIASSE J, RULKENS W. Rapid persulphate oxidation predicts PAH bioavailability in soils and sediments. Environ Sci Technol 2000, vol. 34, pp.2057-2063. CUYPERS C., PANCRAS T., GROTENHUIS T., RULKENS W. Natural attenuation of contaminated soils. Environment International, 2004, vol. 30, n° 4, pp. 587-601. DA SILVA J. P., VIEIRA FERREIRA L. F., DA SILVA A. M. AND OLIVEIRA A. S. A comparative study of the photophysics and photochemistry of 4-chlorophenol adsorbed on silicalite and β-cyclodextrin. Journal of Photochemistry and Photobiology A: Chemistry 2002, vol. 151, pp.157-164. DAUGHNEY C. J., FEIN JB. Aqueous complexation of cadmuim, lead and copper by 2,4,6-trichlorophenol and pentachlorophenol, Geochim. Cosmochim. Acta, 1997, vol. 61, pp. 719-729. DE BRAUER C. , MERLIN M. P., GERMAIN P., GUERANDEL T. Thermal Behaviour of Anhydrous α-, β- and γ-Cyclodextrin at Low Temperature. J. of Incl. Phenom., 2000, vol. 37, n° 1, pp.75-82. DE BRAUER C., GERMAIN P., MERLIN M.P., Energetics of Water/Cyclodextrins interactions, J. of inclsuion phenomena, 2002, vol. 44, pp. 197-201. DEMIAN B. A. Correlation of the solubility of several aromatics and terpenes in aqueous hydroxypropyl-β-Cyclodextrin with steric and hydrophobicity parameters. Carbohyd. Res., 2000, vol. 328, pp. 635-639. DIVINCENZO J. P., SPARKS D. L. Sorption of the neutral and charged forms of pentachlorophenol on soil : Evidence for different mechanisms; Arch. Environ. Contam. Toxicol., 2001, vol. 40, pp. 445-450. DOSTSIKAS Y., KONTOPANOU E., ALLAGIANIS C., LOUKAS Y. L., Interaction of 6-p-toluidinonaphthalene-2-sulphonate with β-cyclodextrin, J. of Pharm. and Biomedical analysis, 2000, vol. 23, pp. 997-1003. DUGAN, P.J., MCCRAY, J.E., THYNE, G.D. Influence of cyclodextrin on NAPL-water partition coefficients with implications for post-remediation NAPL characterization using partitioning tracer tests, Water Resources Research, 2003, vol. 39, n°5, pp. 11-17. DYKE V., COUTURE P., BRAUER M., LEE H., TREVORS J. T. Pseudomonas aeruginosa UG2 rhamnolipid biosurfactants: structural characterization and their use in removing hydrophobic compounds from soil. Can. J. Microbiol., 1993, vol.39, pp.1071-1078. DZOMBAK D.A. AND LUTHY, R.G. Estimating Adsorption of Polycyclic Aromatic Hydrocarbons on Soils. Soil Sci., 1984, vol. 173, pp. 292-308. EASTBURN, TAO D., Applications of modified cyclodextrins. Biotechnol. Adv. 1994 vol. 12, pp. 325-339. EBERHARDT, C., GRATHWOHL, P. Time sclales of organic contaminant dissolution from complex source zones: coal tar pools vs. blobs. Journal of Contaminant Hydrology, 2002, vol. 59, pp. 45-66. EDWARDS, D.A. ; LUTHY, R.G. & LIU, Z. Solubilization of Polycyclic Aromatic Hydrocarbons in Micellar Nonionic Surfactants Solutions. Environ. Sci. Technol. 1991, vol. 25, n°1, pp. 127-133. ENELL A., REICHENBERG F., WARFVINGE P., EWALD G. A column method for determination of leaching of polycyclic aromatic hydrocarbons from aged contaminated soil. Chemosphere, 2004, vol. 54, pp.707-715.

269
ERICKSON, D.C. ; LOEHR, R.C. & NEUHAUSER, E.F. PAH Loss during Biormediation of Manufactured Gas Plant Site Soils. Wat. Res., 1993, vol. 27, n°5, pp. 911-919. EXON, J.H. A review of chlorinated phenols. Vet. Hum. Toxicol., 1984, vol. 26, n°6, pp. 508-512. FARREL J., REINHARD M. Desorption of Halogenated Organics from Model Solids, Sediments and Soils under Unsaturated Conditions : 2. Kinetics. Environ Sci. Technol., 1994, vol. 28, n°1, pp 63-72. FARRELL J., GRASSIAN D., JONES M. Investigation of Mechanisms Contributing to Slow Desorption of Hydrophic Organic Compounds from Mineral Solids. Environ. Sci. Technol., 1999, vol. 33, n°8, pp. 1237-1243. FAVA , D. DI GIOIA , L. MARCHETTI , E. FENYVESI. Randomly Methylated β-Cyclodextrins (RAMEB) Enhance the Aerobic Biodegradation of Polychlorinated Biphenyl in Aged-Contaminated Soils, J. Inclusion phenomena, 2002, vol. 44, pp. 417-421. FAVA F., BERTIN L., FEDI S., ZANNONI D. Methyl-β-cyclodextrin-Enhanced solubilization and aerobic biodegradation of polychlorinated biphenyls in two aged-contaminated soils. Biotechn., and Bioeng. 2002, vol. 81, n°4, pp.1-10. FAVA F., DIANA DI GIOIA, LEONARDO MARCHETT. Cyclodextrin effects on the ex-situ bioremediation of a chronically polychlorobiphenyl-contaminated soil Biotechnology and Bioengineering ,1998, vol. 58, pp. 345-355. FENYVESI, E, CASABI K., MOLANR M., GRUIZ, K., LEITGIB, L., BALGOH, G., MURANY, A., SZEJTLI, Quantitative and Qualitative Analysis of RAMEB in Soil. J. Inclusion phenomena, 2002, vol. 44, pp. 413-416. FENYVESI, E., VIKMON M., SZEMAN, J., REDENTI E., DELCANALE M., VENTURA P. AND SZEJTLI J., Interaction of hydroxy acids with β-cyclodextrin, J. of Inclusion Phenomena and Macroc. Chem., 199, vol. 33, pp. 339-344. GALIL N. I., NOVAK. J. T. Pentachlorophenol-induced release of soil organics and organics, Wat. Res., 1995, vol. 29, pp. 1533-1540. GAO. S., WANG L. , HUANG Q., HAN S. Solubilization of Polycyclic Aromatic Hydrocarbons by β-Cyclodextrin and Carboxymethyl-β-Cyclodextrin. Chemosphere, 1998, vol. 37, pp.1299-1305. GIORDANO F., NOVAK C., MOYANO J. R. Therma analysis of cyclodextrins and their inclusion compounds. Thermochica acta, 2001, vol. 380, pp. 123-151. GOYER S. Techniques de restauration de sites contaminés par des hydrocarbures aromatiques polycycliques (HAP) : 1- procédés physiques, physico-chimiques et thermiques, Vecteur Environnement, 1995, vol. 8, n°1, pp. 57-68. Grasso D., K. Subramaniam, J. J. Pignatello, Y. Yang and D. Ratté. Micellar desorption of polynuclear aromatic hydrocarbons from contaminated soil. Colloids and Surfaces A: Physicochemical and Engineering Aspects, 2001, vol. 194, pp. 65-74 GUHA S. JAFFÉ P. R.. Biodegradation Kinetics of Phenanthrene Partitioned into the Micellar Phase of Nonionic Surfactants, Environ. Scien. Technol. 1996, vol. 30, pp.605-611. HAMBY D.M. Site remediation techniques supporting environmental restoration activities, a review, The Science of the Total Environment, 1996, vol. 191, pp. 203-224. HATZINGER P.B., ALEXANDER M. Effect of aging of chemicals in soil on their biodegradability and extractability. Environ. Sci. Technol., 1995, vol. 29, pp 537-545. HEITKAMP M. A., CERNIGLIA C. E. Polycyclic aromatic hydrocarbon degradation by a mycobacterium sp in microcosms containing sediment and water from a pristine ecosystem. Appl. Environ. Microbiol., 1989, vol. 55, n°8, pp 1968-1973. HERMANN M. Synergetic effects of individual polycyclic aromatic hydrocarbons on the mutagenicity of their mixtures, Mutation Research, 1981, vol. 90, n°4, pp. 399-409. HERMOSIN, M.C. ; CORNEJO, J. & PEREZ RODRIGEZ, J. L. Mineralogy of the Soil Clay Fraction as Affecting Pesticide Adsorption-Soil Properties Relationships. In : Methodological Aspects of the Study of Pesticide Behaviour in Soil, I.N.R.A.-Versailles, June 16-17, 1988, p. 21-27. HERRMANN JM. Heterogeneous photocatalysis fundamentals and applications to the removal of various types of aqueous pollutants. Catalysis Today, 1999, vol. 53, pp.115-129.

270
HOOS, R. A. W. Patterns of pentachlorophenol usage of Canada : An overview in : pentachlorophenol chemistry, Pharmacology, and environmental toxicology. New York : Plenum press, 1978, pp. 3-15. HUANG H-L, LEE W.M. Synergistic solubilization of polycyclic aromatic hydrocarbons by mixed anionic–nonionic surfactants, Chemosphere, 2003, vol. 53, pp. 459-467. HUANG W. L., YOUNG T. M., SCHLAUTMAN M. A., YU H., WEBER JR W. J. A distributed reactivity model for sorption by soils and sediments : 9. General isotherm nonlinearity and applicability of the dual reactive domain model. Environ. Sci. Technol., 1997, vol. 31, pp. 1703-1710. HUANG W., YU H., WEBER JR W. J. Hysteresis in the sorption and desorption of hydrophobic organic contaminants by soils and sediments: 1. A comparative analysis of experimental protocols. J. Contam. Hydrol., 1998, vol. 31, pp. 129-148. HUANG X, EL-ALAWI Y., PENROSE D., BERNARD R. GREENBERG. A multi-process phytoremediation system for removal of polycyclic aromatic hydrocarbons from contaminated soils • Environmental Pollution, 2004, vol. 130, pp. 465-476. ILLANGASEKARE, T.H., RMAESEY, J.L., JENSEN, K.H., BUTTS, M.B. Experimental study of movement and distribution of Dense organique contaminants in Heterogeneous aquifers. J. Contam. Hydrol., 1995, vol. 20, pp. 1-25. IRWIN P, PTEFFER P, DONER L, SAPERS G, BREWSTER J, HICKS K, Binding geometry, stoichiometry, and thermodynamics of cyclodextrin inclusion complex with chlorogenic acid, Carbohydrate Research, 1994, vol. 256, pp.13-27. ISHIWATA S., KAMIYA M. Cyclodextrin inclusion: catalytic effects on the degradation of organophosphorus pesticides in neutral aqueous solution, Chemosphere, 1999, vol. 39, pp. 1595-1600. JAFVERT C. T., WESTALL G. C., GRIEDER E., SCHWARZENBACH R. P. Ditribution of ionogenic organic compounds between octanol and water: organic acids, Environ. Sci. Technol., 1990, vol. 24, n° 12, pp.1795-1803. JI W., BRUSSEAU, M.L., A general mathematical model for chemical-enhanced flushing of soil contaminated bu organic compounds, Water resources research, 1998, vol. 34, pp. 1635-1648. JOHNSON C.A. AND WESTALL J. C. Effect of pH and potassium chloride concentration on the octanol-water distribution of methylanilines. Environ. Sci. Technol., 1990, vol. 24, n° 12, pp.1869-1875. JONES K.C. ; P. DE VOOGT. Binding of organic pollutants to humic and fulvic acids: Influence of pH and the structure of humic material. Chemosphere, 1997, vol. 34, pp. 1693-1704. JOZEFACIUK, G. MURANYI. ,E. FENYVESI, Effect of Cyclodextrins on Surface and Pore Properties of Soil Clay Minerals, Environ. Sci. Technol. 2001, vol. 35 n° 24, pp. 4947-4952. JOZEFACIUK, G. MURANYI. , E. FENYVESI, Effect of RAMEB on physical properties of soils, Environ. Sci. Technol. 2003, vol. 37, pp. 3012-3017. JOZWIAKOWSKI M.J., CONNORS K.A. Aqueous solubility behavior of three cyclodextrins. Carbohydr. Res., 1985, vol.143, pp.51-59. KAISER L. E. K., VALDMANIS I. Apparent octanol/water coefficients of pentachlorophenol as a function of pH. Can. J. Chem., 1982, vol. 60, pp. 2104-2106. KAMIYA M. AND KAORI NAKAMURA .Cyclodextrin inclusion effects on photodegradation rates of organophosphorus pesticides, Environment International, 1995, vol. 21, pp. 299-304. KAMIYA M., KAORI NAKAMURA AND CHIZUKO SASAKI. Inclusion effects of cyclodextrins on photodegradation rates of parathion and paraoxon in aquatic medium, Chemosphere, 1994, vol. 28, pp.1961-1966. KARICKHOFF, S. N. Semi-Empirical Estimation of Sorption of Hydrophobic Pollutants on Natural Sediments & Soils. Chemosphere, 1981, vol. 10, pp. 833-846. KARICKHOFF, S. N., BROWN, D. S.; SCOTT, T.A. Sorption of Hydrophobic Pollutants on Natural Sediments and Soils. Water Res., 1979, vol. 13, pp. 241-248. KAWAHARA F. K. Polynuclear aromatic hydrocarbon (PAH) release from soil during treatment with Fenton’s reagent, Chemosphere, 1995, vol. 31, n° 9, pp. 4131-4142. KAWASAKI N., ARAKI M. AND TANADA S. Inclusion behavior of 4-nonylphenol into Cyclodextrins derivatives. J. of Coll. inter. Sci. 2001, vol. 238, pp. 215-218.

271
KHAN F., HUSAIN T., HEJAZI R. An overview and analysis of site remediation technologies • Journal of Environmental Management, 2004, vol. 71, pp. 95-122. KHODADOUST A., RAJESH BAGCHI, MAKRAM T. SUIDAN, RICHARD C. BRENNER AND NEAL G. SELLERS. Effect of NAPL entrapment conditions on air sparging remediation efficiency, Journal of Hazardous Materials, 2004, vol. 110, pp. 173-183. KHODADOUST AP., MAKRAM T. SUIDAN, CAROLYN M. ACHESON AND RICHARD C. Brenner. Solvent extraction of pentachlorophenol from contaminated soils using water-ethanol mixtures, Chemosphere, 1999, vol. 38, pp.2681-2693. KHODADOUST AP., RAJESH BAGCHI, MAKRAM T. SUIDAN, RICHARD C. BRENNER, NEAL G. Seller Removal of PAHs from highly contaminated soils found at prior manufactured gas operations, Journal of Hazardous Materials, 2000, vol. 80, pp. 1-3. KNOKE K.L. A Comparison of Five Bioassays to Monitor Toxicity During Bioremediation of Pentachlorophenol-Contaminated Soil. Water, Air, and Soil Pollution, 1999, vol. 110, pp.157-169. KO S.-O. SCHLAUTMAN M. A. AND CARRAWAY E.R. Partitioning of hydrophobic organic compounds to hydroxypropyl-β-cyclodextrin: Experimental studies and model predictions for surfactant-enhanced remediation applications. Environ. Sci. Technol. 1999, vol.33, pp. 2765-2770. KONDO, H.; NAKATANI, H.; HIROMI, K. Analysis of mixtures of α and β-cyclodextrins using fluorescent dyes, Carbohyd. Res. 1976, vol. 52, pp. 1-10. KRAUSS, M.; WILCKE, W . Predicting Soil-Water Partitioning of Polycyclic Aromatic Hydrocarbons and Polychlorinated Biphenyls by Desorption with Methanol-Water Mixtures at Different Temperatures .Environ. Sci. Technol. 2001, vol. 35, n° 11, pp.2319-2325. LAGAS, P. Sorption of Chlorophenols in the Soil. Chemosphere, 1988, vol. 17, n°2, pp. 205-216. LAHA, S. & LUTHY, R.G. Effect of Non-Ionic Surfactants on the Solubilization and Mineralization of Phenanthrene in Soil-Water Systems. Biotechnology and Bioengeneering, 1992, vol. 40, pp. 1367-1380. LANE, W.F., LOEHR, R.C. Estimating the equilibrium aqueous concentrations of polynuclear aromatic hydrocarbons in complex mixtures. Environmental Science and Technology, 1992, vol. 26, pp. 983-990. LANE, W.F., LOEHR, R.C. Predicting aqueous concentrations of polynuclear aromatic hydrocarbons in complex mixture. Water Environment Research, 1995, vol. 67, pp. 169-173. LE ROUX H., GLASSER L., Transferable potentials for Ti-O system, Journal of Materials Chemistry, 1997, vol. 7, pp. 843-851. LEE, L.S. ; RAO, P.S.C., BRUSSEAU, M.L. Nonequilibrium Sorption & Transport of Neutral & Ionized Chlorophenols. Environ. Sci. Technol., 1991, vol. 25, n°4, pp. 722-729. LEE, L.S. ; RAO, P.S.C. & Okuda,, I. Equilibrium Partitioning of Polycyclic Aromatic Hydrocarbons from Coal Tar into Water. Environ. Sci. Technol., 1992, vol. 26, n°11, pp. 2110-2115. LEE, L.S., RAO, P.C.S., NKESSI-KISSA, P. & DELFINO, J.J. Influence of Solvent & Sorbent Charcteristics on Distribution of Pentachlorophenol in Octanol-Water & Soil-Water Systems. Environ. Sci. Technol., 1990, vol. 24, pp. 654-661. LEINONEN, P.J., MACKAY, D. The multicomponent solubilty of hydrocarbons in water. Canadian Journal of Chemical engineering, 1973, vol. 51, pp. 230-233. LIOU R. , CHEN S. , HUNG M., HSU C. Catalytic oxidation of pentachlorophenol in contaminated soil suspensions by Fe+3-resin/H2O2. Chemosphere, 2004, vol. 55, pp. 1271-1280. LIU L., GUO Q-X. The Driving Forces in the Inclusion Complexation of Cyclodextrins. J. of Incl. Phenom., 2002, vol. 42, pp.1-8. LIU, Z. ; LAHA, S. & LUTHY, R.G. Surfactants Solubilization of PAH Compounds in Soil-Water Suspensions. Water Science Technology, 1991, n°23, pp. 475-485. LUI. L., SONG K.E-S., LI X-S., GUO Q-X. Charge-transfer Interaction: A Driving Force for Cyclodextrin Inclusion Complexation. J. of Incl. Phenom.,2001, vol. 40, pp.35-45. LUNDSTEDT S., BERT VAN BAVEL, PETER HAGLUND, MATS TYSKLIND, LARS ÖBERG. Pressurised liquid extraction of polycyclic aromatic hydrocarbons from contaminated soils. Journal of Chromatography A, 2000, vol. 883, pp. 151-162

272
MACKAY D., AND SHIU W.Y. Aqueous solubility of Polycyclic Aromatic Hydrocarbons J. Chem. Eng. Data, 1977, vol. 22, n°4, pp. 399-402. MAHAFFEY, W.R. ; GIBSON, D.T. ; CERNIGLIA, C.E. Bacterial Oxidation of Chemical Carcincogens: Formation of Polycyclic Aromatic Acids from Benz(a)anthracene. Appl. Environ. Microbiol., 1988, vol. 54, n°10, pp. 2415-2423. MAHJOUB B., Comportement dans le sol de polluants aromatiques issus du goudron de houille. Thèse LAEPSI. Lyon : INSA de Lyon, 1999, 262 p. MANILAL & ALEXANDER M. Factors affecting the microbial degradation of Phenanthrene in Soil. Appli. Microbiol. Biotechnol., 1991, vol. 35, pp. 401-405. MATSUNGA K., IMANAKA M., ISHIDA T. Application of γ-Cyclodextrin to the separation of compounds extracted with organic solvents Anal. Chem. 1984, vol. 56, pp. 1982-1986. MCALLISTER, K.A., LEE H., AND TREVORS J. T. Microbial degradation of pentachlorophenol, Biodegradation, 1996, vol. 7, pp.1-4. MCBAIN M. L. E.; HUTCHINSON E. Solubilization and related phenomena. New York : Academic press, 1955. MCCARTHY, J.F., ZACHARA, J.M. Subsurface transport of contaminants. Environmental Science and Technology, 1989, vol. 23, n°5, pp. 496-502. MCCRAY, J.E. AND DUGAN, P.J. Nonideal equilibrium dissolution of trichloroethene from a decane-based NAPL mixture: Experimental and modeling investigation, Water Resources Research, 2002, vol.38, n°7, pp.22-29. MCCRAY, J.E. AND FALTA, R.W. Numerical simulation of air sparging for remediation of NAPL contamination, Ground Water, 1997, vol.35, n°1, pp. 99-110. MCCRAY, J.E., BAI, G., BRUSSEAU, M.L, MILLER, R. Biosurfactant-enhanced solubilization of NAPL mixtures, J. Contam. Hydrol., 2001, vol. 48, n°1, pp.45-68. MCCRAY, J.E., BRUSSEAU, M.L. Cyclodextrin-enhanced in situ flushing of multiple-component immiscible organic-liquid contamination at the field scale: Analysis of dissolution behavior, Environ. Sci. Technol. 1999, vol.33 , n°1, pp.89-95. MCCRAY, J.E., BRUSSEAU, M.L. Cyclodextrin-enhanced in-situ flushing of multiple-component immiscible organic-liquid contamination at the field scale: Mass removal effectiveness, Environ. Sci. Technol., 1998, vol. 32 , n°9, pp.285-1293. MEANS, J.C. ; WOOD, S. G. ; HASSETT, J.J., BANWART, W. L. Sorption of Polynuclear Aromatic Hydrocarbons by Sediments & Soils. Environ. Sci. Technol., 1980, vol. 14, pp. 522-529. MEANS, J.C. ; WOOD, S. G. ; HASSETT, J.J., BANWART, W. L. Sorption of Polynuclear Aromatic Hydrocarbons by Sediments & Soils. Environ. Sci. Technol., 1980, vol. 14, pp. 522-529. MERLIN M.-P, Energétique des intercations moléculaires eau/cyclodextrines. Thèse LAEPSI. Lyon : INSA de Lyon, 1998, 250 p. META ENVIRONMENTAL. Draft report prepared for electric Power research institute, RP2879-01. Watertown, MA : META Environmental, 1990. MILLS A., LE HUNTE S. An overview of semiconductor photocatalysis, J. of Photochem. & Photobiol. A : Chem., 1997, vol.108, pp.1-35. MILLS G, HOFFMANN, MR. Photocatalytic degradation of pentachlorophenol on TiO2 particles: identification of intermediates and mechanism of reaction. Environ Sci Technol 1993, vol. 27, pp. 1681-1689. MOLNAR M., FENYVESI E., GRUIZ K., LEITGIB L., BALOGH G., MURANYI A. SZEJTLI J., Effects of RAMEB on bioremediation on different soils contaminated with hydrocarbons, J. of Inclsuion phenomena, 2002, vol. 44, pp. 447-452. MORILLO E., J. I. PÉREZ-MARTINEZ, J. M. GINÉS. Leaching of 2,4-D from a soil in the presence of β-cyclodextrin: laboratory columns experiments. Chemosphere, 2001, vol. 44, pp.1065-1069. MORIN N., GUILLAUME Y., E. PEYRIN , ROULAND J.. Peculiarities of an imidazole derivative retention mechanism in reversed-phase liquid chromatography. cyclodextrin concentration and temperature considerations. Journal of Chromatography A, 1998, vol. 808, pp. 51-60.

273
MULLIGAN C.N., RAYMOND N. Synergistic solubilization of polycyclic aromatic hydrocarbons by mixed anionic–nonionic surfactant. Chemosphere, 2003, vol. 53 pp. 459-467 . MULLIGAN, C., EFTEKHARI, F. Remediation with surfactant foam of PCP-contaminated soil Eng. Geol., 2003, vol. 70, pp.269-279. NATIONAL CANCER INSTITUTE. Bioassay of 2,4,6-trichlorophenol for possible carcinogenicity. Tech. Rep. Ser. No. 155, DHEW Publ. No. (NIH) 79-1711, U.S : Department of Health, Education and Welfare, 1979. NEELY W.B., BRANSON D.R., BLAU G.E. Partition coefficient to measure bioconcentration potential of organic chemicals in fish. Environ. Sci. Technol. 1974, vol. 8, pp.1113-1115. NICHOLLS, P.H. & EVANS, A.A. Sorption of Ionisable Organic Compounds by Field Soils. Part 1 : Acids. Pestic. Sci., 1991a, vol. 33, n°3, pp. 319-330. NICHOLLS, P.H. & EVANS, A.A. Sorption of Ionisable Organic Compounds by Field Soils. Part 2 : Cations, Bases & Zwitterions. Pestic. Sci., 1991b, vol. 33, n°3, pp. 331-345. NICHOLLS, P.H. Factors Influencing Entry of Pesticides into Soil Water. Pestic. Sci., 1988, vol. 22, pp. 123-137. NIIMI, A. J. Solubility of organic chemicals in octanol, triolein and cod liver oil and relationships between solubility and partition coefficients. Water Res., 1991 vol. 25, pp. 1515-1521. OLLIS D. F., AL-EKABI H. (Eds.), Photocatalysis Purification and Treatment of Water and Air. Amsterdam : Elsevier, 1993, 200 pp. OTURAN M. A., OTURAN N., LAHITTE C. AND TREVIN S. Production of hydroxyl radicals by electrochemically assisted Fenton's reagent: Application to the mineralization of an organic micropollutant, pentachlorophenol, J. Electroanal. Chem., 2001, vol. 507, pp. 96-102. PAGINTON J.S. β-cycodextrin : the success of molecular inclusion. Chemistry in Britain, 1987, vol. 23, n° 5, pp. 455-458. PALMER C. D., FISH W. Chemical enhancements to pump and treat remediation, EPA/540/S-92/001, Office of reaserch and Development. Washington D.C : U.S. Environmental Protection Agency, 1992. PAOLIS F., KUKKONEN J..Treatment of a pentachlorophenol- and creosote-contaminated soil using the lignin-degrading fungus phanerochaete sordid a: A field demonstration. Soil Biology and Biochemistry, 1994, vol. 26, pp.1603-1611. PARK, K.S. ; SIMS, R.C. ; DUPONT, R.R. & al. Fate of PAH Compounds in Two Soils Types : Influence of Volatilization, Abiotic Losses & Biological Activity. Environ. Sci. Technol., 1990, vol. 9, pp.187-195. PARK, S.-K., BIELEFELDT, A. Equilibrium partitioning of a non-ionic surfactant and pentachlorophenol between water and a non-aqueous phase liquid, Water Research, 2003, vol. 37, pp. 3412-3420. PECCHI G., REYES P., SANHUEZA P., VILLASENOR J., Photocatalytic degradation of pentachlorophenol on TiO2 sol-gel catalysts, Chemosphere 2001, vol. 43, pp. 141-146. PENNELL, K.D., ABRIOLA, L.M. AND WEBER, W.J. Surfactant-enhanced solubilization of residual dodecane in soil columns. 1. Experimental investigation Environ. Sci. Technol. 1993, vol. 27, pp. 2332-2340. PETERS, C.A., LUTHY, R.G. Coal tar dissolution in water-miscible solvents: experimental evaluation. Environmental Science and Technology, 1993, vol. 27, pp. 2831-2843. PITCHUMANI K. AND VELLAYAPPAN M. Complex formation of nitrobenzoic acids and some naphthalene derivatives with β-Cyclodextrin. J. of Inc. Phen. and Mol. Reco. In chemistry., 1992, vol. 14, pp. 157-162. PITHA J., HOSHINO T. Effects of ethanol on formation of inclusion complexes of hydroxypropylcyclodextrins with testosterone or with methyl organe. Int. J. Pharm. 1992, vol. 80, pp. 243-251. POLLARD, S.J.T., HRUDEY, S.E. FEDORAK, P.M. Bioremediation of Petroleum- and Creosote-Contaminated Soils: A Review of Constraints. Waste Management & Research, 1994, vol. 12, pp. 173-194. PRASAD N, STRAUSS D, REICHART G. Cyclodextrins inclusion for food, cosmetics and pharmaceuticals. European Patent 1, 1999, vol. 84, pp. 625-630.

274
RAJEWSKI R., V.J. STELLA, Pharmaceutical applications of cyclodextrins. 2. In vivo drug delivery. J. Pharm. Sci. 1996, vol. 85, pp. 1142-1168. REEUWIJIK H, IRTH H., TJADEN. Liquid chromatographic determination of b- cyclodextrine derivatives based on fluorescence enhancement after inclusion complexation ; J. of chromatography. 1993, vol. 614, pp. 95-101. REID B. J. STOKES JD, JONES KC, SEMPLE KT. Nonexhaustive Cyclodextrin-Based Extraction Technique for the Evaluation of PAH Bioavailability. Environ. Sci. Technol. 2000, vol. 34, pp. 3174-3179. REUBEN, J., TRINADHA RAO, C., PITHA, J. Distribution of substituents in carboxymethyl ethers of cyclomaltohetaose. Carbohydrate Research 1994, vol. 258, pp.281-285. ROGERS, R. D. ; MCFARLANE, J.C. & CROSS, A.J. Adsorption & Desorption of Benzene in two Soils & Montmorillonite Clay. Env. Sci. Technol., 1980, vol. 14, n°4, pp. 457-460. Saenger W. Cyclodextrins inclusion compounds in research and industry. Angew. Chem. Int., 1980, vol. 19, pp.344-362. SCHELLENBERG K., LEUNBERGER C. AND SCHWARZENBACH R. P. Sorption of chorintaed phenols by natural sediments and aquifer materials. Environ. Sci. Technol., 1984, vol.18, pp.652-657. SCHWARZENBACH, R.P., WESTALL, J. Transport of Nonpolar Organic Compounds from Surface Water to Groundwater. Laboratory Sorption Studies. Environ. Sci. Technol., 1981, vol. 15, pp. 1360-1367. SCHWETZ, B.A., QUAST, J.F., KEELER, P.A., HUMISTON, C.G. ET KOCIBA, R.J. Results of two-year toxicity and reproduction studies on pentachlorophenol in rats. In : Pentachlorophenol: chemistry, pharmacology and environmental toxicology. K. Rao (dir. de publ.). New York, NY : Plenum Press, 1978. p. 301 SEMPLE K.T., B. J. REID , T. R. FERMOR. Solvent extraction of pentachlorophenol from contaminated soils using water-ethanol mixtures. Chemosphere, 1999, vol. 38, pp. 2681-2693. SEO, H.S. AND MCCRAY, J.E. Interfacial tension of chlorinated-aliphatic DNAPL mixtures as a function of organic-phase composition, Environmental Science & Technology, 2002, vol. 36, n°6, pp. 1292-1298. SHIMIZU, Y. ; YAMAZAKI, S., TERASHIMA, Y. Sorption of Anionic Pentachlorophenol (PCP) in Aquatic Environments : the Effect of pH. Water Sci. Technol., 1992, vol. 25, n°11, pp. 41-48. SIMER E, KURVITS C. Calorimetric studies of benzoic acid-cyclodextrin inclusion complexes, Thermochimica Acta, 1998, vol. 140, pp.161-168. SINGH M, SHARMA R, BANERJEE UC. Biotechnological applications of cyclodextrins. Biotechnol Adv. 2002, vol. 20, pp. 341–59. SONG WEN-LU, HANG QING-GUO, WANG LIAN-SHENG. β-cyclodextrin influence on the biotoxicities of substituted benzene compounds and pesticide intermediates. Chemosphere, 1999, vol. 38, n° 4, pp. 693-698. STAPLETON MG, SPARKS DL, DENTEL SK. Sorption of pentachlorophenol to HDTMA-clay as a function of ionic strength and pH. Environ. Sci. Technol., 1994, vol. 28, pp. 2330-2335. STEGMANN R., BRUNNER G., CALMANO W., MATZ G. (Eds.). Treatment of contaminated soil, Fondamentals Analysis Applications. Berlin : Springer-Verlag, 2001, 658 p. STELLA V. AND R.A. RAJEWSKI. Cyclodextrins: their future in drug formulation and delivery. Pharm. Res. 1997, vol. 14, pp. 556–5567. STUCKI, G. & ALEXANDER, M. Role of Dissolution Rate and solubility in Biodegradation of Aromatic Compounds. Appl. Environ. Microbiol., 1987, vol. 53, n°2, pp. 292-297. SUZUKI M., SASAKI Y., Inclusion compound of cyclodextrin and azo dye. I. Methyl Orange, Chem. Pharm. Bull. 1979, vol. 27, pp. 609-619. SVERDRUP, L. E.; NIELSEN, T.; KROGH, P. H.; Soil Ecotoxicity of Polycyclic Aromatic Hydrocarbons in Relation to Soil Sorption, Lipophilicity, and Water Solubility. Environ. Sci. Technol. 2002, vol. 36, pp.2429-2435. SZEJTLI J. in: Cyclodextrins and their Inclusion Complexes. Budapest, Hungary : Akademiai Kiado, 1982, p.100-109.

275
SZETJLI J., Introduction and general overview of cyclodextrin chemistry. Chem. Rev. 1998, vol. 98, pp. 1743-1753. TANADA S., NAKAMURA T., KAWASAKI N., TORII Y. KITAYAMA S. Removal of aromatic hydrocarbon compounds by Hydroxypropyl- Cyclodextrin, J. of Coll. Inter. Sci., 1999, vol. 217, pp.417-419. THOMAS, A.O. & LESTER, J.N. The Microbial Remediation of Former Gasworks Sites : A Review Environ. Technol., 1993, vol. 14, pp. 1-24. TICK, G. R.; LOURENSO, F.; WOOD, A. L.; BRUSSEAU, M. L. Pilot-Scale Demonstration of Cyclodextrin as a Solubility-Enhancement Agent for Remediation of a Tetrachloroethene-Contaminated Aquifer, Environ. Sci. Technol. 2003, vol. 37, pp. 5829-5834. TORRES-LABANDEIRA JJ., P. DAVIGNON AND J. PITHA, Oversaturated solutions of drug in hydroxypropylcyclodextrins: parenteral preparation of pancratistatin. J. Pharm. Sci. 1990, vol. 80, pp. 384-386. TSE K. K. C., LO, S.-L.; WANG, J. W. H. Pilot Study of In-Situ Thermal Treatment for the Remediation of Pentachlorophenol-Contaminated Aquifers. Environ. Sci. Technol., 2001, vol. 35, pp. 4910-4915. U.S. ENVIRONMENTAL PROTECTION AGENCY. Contaminates and remedial options at wood-preserving sites. EPA-600-R-92-182. U.S. : ENVIRONMENTAL PROTECTION AGENCY, 1992. U.S. ENVIRONMENTAL PROTECTION AGENCY. Ambient water quality criteria for chlorinated phenols. EPA-440/5-80-032, NTIS Publ. No. PB81-117434, Office of Water Regulations and Standards. U.S. : ENVIRONMENTAL PROTECTION AGENCY, 1980. U.S. ENVIRONMENTAL PROTECTION AGENCY. Ambient water quality criteria for pentachlorophenol. EPA-440/5-80-065, NTIS Publ. No. PB81-117764. U.S. Environmental Protection Agency. Office of Water Regulations and Standards. Criteria & Standards Division. Office of Water Regulations and Standards, 1980. US ENVIRONMENTAL PROTECTION AGENCY. Fate and impact of pentachlorophenol in freshwater ecosystem, Report No. EPA/600/3-78-063. Athens, Georgia U.S. Environmental Protection Agency. Environmental Research Laboratory. VALSARAJ K.T. AND THIBODEAUX L.J. Relationships between micelle-water and octanol-water partition constants for hydrophobic organics of environmental interest.. Wat. Res., 1989, vol. 23, pp. 183-189. VALSARAJ, K. T. Elements of Environmental Engineering, Thermodynamics and kinetics, Florida : CRC Lewis publishers, 1995, pp 97-100. VERSCHUEREN, K., Handbook of Environmental Data on Organic Chemicals, 3éme édition. Van Nostrand Reinhold, New York : International Thomson Pub. Co., 1996, 2064 p. VERSTICHEL S., DE WILDE B., FENYVESI E., SZEJTLI J., Investigation of the aerobic biodegradability of several types of cyclodextrins in a laboratory-controlled composting test. J. of Polym. and the Envi., 2004, vol. 12, pp. 1-10. VILLIERS A., Cyclodextrines and their use. Compt Rendu, 1891, vol. 112, pp. 536-540. VOICE T.C. & WEBER, W. JR. Sorption of hydrophobic Compounds by Sediments, Soils & Suspended Solids. 1-Theory and Background. Wat. Res., 1983, vol. 17, n°10, pp. 1433-1441. VULLIET E, CHOVELON JM, GUILLARD C, HERRMANN JM. Factors influencing the photocatalytic degradation of sulfonylurea herbicides by TiO2 aqueous suspension. J of Photochemistry and Photobiology A: Chemistry 2003; vol. 159, pp. 71-79. VULLIET E, EMMELIN C, CHOVELON JM, GUILLARD C, HERRMANN JM. Photocatalysis degradation of sulfonylurea herbicides in aqueous TiO2. Appl Catal B: Environmental 2002. vol. 38, pp. 127–137. WANG J-M., MARLOWE, E M., MILLER-MAIER R., BRUSSEAU M. L. Cyclodextrin-Enhanced Biodegradation of Phenanthrene. Environ. Sci. Technol., 1998, vol. 32, pp.1907-1915. WANG X. AND BRUSSEAU M. L. Simultaneous complexation of organic compounds and heavy metals by a modified cyclodextrin. Environ. Sci. Technol., 1995, vol. 29, pp. 2632-2635. WANG X. AND BRUSSEAU ML, Cyclopentanol-enhanced solubilization of polycyclic aromatic hydrocarbons by cyclodextrins. Environ Sci Technol 1995, vol 27, pp. 2346–2351.

276
WANG X., BRUSSEAU M.L. Solubilization of some low-polarity organic compounds by Hydroxypropyl-β-cycodextrin. Environ. Sci. Technol., 193, 27, 2821-2825. WANG, X. ; YU, X., BARTHA, R. Effect of Bioremediation on Polycyclic Aromatic Hydrocarbon Residues in Soil. Environ. Sci. Technol., 1990, vol. 24, n°7, pp. 1086-1089. WARITH M.A. ; FERNANDES, L., LAFORGE, F. Adsorption of Pentachlorophenol on Organic Soil. Hazardous Waste & Hazardous Materials, 1993, vol. 10, n°1, pp. 13-25. WEISSENFELS W. D., KLEWER H. J., LANGOFF J., Adsorption of polycyclic aromatic hydrocarbons (PAHs) by soil particles : Influence on biodegradability and biotoxicity. Appl. Microbiol. Biotechnol., 1992, vol. 36, pp. 689-696. WERSHAW, R.L. Model for Humus. Environ. Sci. Technol., 1993, vol.27, n°5, pp. 814-817. WESTALL, J. C., LEUENBERGER, C. , SCHWARZENBACH, R. P. Influence of pH and ionic strength on aqueous–nonaqueous distribution of chlorinated phenols. Environ. Sci. Technol., 1985, vol.19, pp. 1360-1367. WHITE J. L. An overview of immunotoxicology and carcinogenic polycyclic hydrocarbons, Envir. Carcino. Revs., 1986, vol. C4, pp. 163-202. WHITEHOUSE B.G. The effects of temperature and salinity on the aqueous solubility of polynuclear aromatic hydrocarbons. Marine Chem. 1984, vol. 14, pp. 319-332. WIGHTMAN P., FEIN J. B. Experimental study of 2,4,6-trichlorophenol and pentachlorophenol solubilities in aqueous solution : derivation of a speciation-based chlorophenol solubility model. Appl. Geochem., 1999, vol. 14, pp. 319-325. WILD S. R., BERROW M. L., JONES K. C.,The persistence of Polynuclear Aromatic Hydrocarbons (PAHs) in Sewage Sludge Amended Agricultural Soils. Environmental Pollution, 1991, vol. 72, pp. 141-157. WILSON GJ., AMID P. KHODADOUST, MAKRAM T. SUIDAN, RICHARD C. BRENNER AND CAROLYN M. ACHESON. Anaerobic/aerobic biodegradation of pentachlorophenol using GAC fluidized bed reactors: optimization of the empty bed contact time, Water Science and Technology, 1998, vol.38, pp.9-17. WILSON S. C., JONES K. C. Bioremediation of soil contaminated with polynuclear aromatic hydrocarbons (PAHs) : a review, Environment Pollution, 1993, vol. 81, pp.229-249. YEOM I.T., GHOSH M. M., COX C. D., ROBINSON K. G. Micellar solubilization of polynuclear aromatic hydrocarbon in coal tar-contaminated soils. Environ. Sci. Technol., 1995, vol. 29, pp. 3015-3021. YEOM, I.T., GHOSH, M.M., COX C.D., AHN, K.H. Dissolution of polycyclic aromatic hydrocarbons from weathered contaminated soil. Water Science Technology, 1996, vol. 34, pp. 335-342. YOSHIDA N., MOROI Y., Solubilization of Polycyclic Aromatic Compounds into n-Decyltrimethylammonium Perfluorocarboxylate Micelles J. of Coll. Inter. Sci., 2000, vol. 232, pp. 33-38. YOU C.C. LIU J. Desorptive behavior of chlorophenols in contaminated soils, Wat. Sci. Tech., 1996, vol. 33, n° 6, pp. 263-270. YUDIARTO A., DEWI E., KOKUGAN T. Separation of isomers by ultrafiltration using modified cyclodextrins, Separation and Purification Technology, 2000, vol. 19, pp. 103-11. ZHANG G., SHUANG S., DONG Z., PAN J., Investigation of the inclusion behavior of neutral red with β-cyclodextrin, hydroxylpropyl-β-cyclodextrin and sulfobutylether-β-cyclodextrin, Anal. Chimica Acta, 2002, vol. 474, pp. 189-195. ZHU L. SHAOLIANG FENG. Extended bioremediation of PAH/PCP contaminated soils from the POPILE wood treatment facility. Chemosphere, 2004, vol. 54, pp. 1481-1493. ZHU L., FENG S. Removal of PAHs from highly contaminated soils found at prior manufactured gas operations. Journal of Hazardous Materials, 2000, vol. 80, pp.159-174.

277
ANNEXES
Annexe 1 : Réglementation en matière de sites pollués et Inventaire des
sites pollués en France.
Réglementation en matière de sites pollués
Les définitions d’un site pollué données en 2001 par le Ministère de l’Aménagement du
Territoire et de l’Environnement sont les suivantes :
Site présentant un risque pérenne, réel ou potentiel, pour la santé humaine ou
l'environnement du fait d'une pollution de l'un ou l'autre des milieux, résultant de
l'activité actuelle ou ancienne. (http://www.environnement.gouv.fr)
ou
Site qui, du fait d’anciens dépôts de déchets ou d’infiltration de substances polluantes,
présente une pollution susceptible de provoquer une nuisance ou un risque pérenne pour
les personnes ou l’environnement. (http://www.senat.fr)
Un sol est contaminé lorsqu'il contient des produits chimiques totalement intégrés dans sa
structure à une concentration supérieure à la concentration naturelle.
- la loi du 15 juillet 1975 sur les déchets : " toute personne qui produit des déchets de
nature à porter atteinte à la santé de l'homme et à l'environnement est tenue d'en assurer
l'élimination".
- la loi du 3 janvier 1992 sur l'eau, qui instaure l'obligation de prévention pour éviter la
pollution des eaux superficielles ou souterraines.
- la loi du 2 février 1995 sur le renforcement de la protection de la nature, qui instaure le
principe de précaution.
L’approche administrative est fondée sur un ensemble de circulaires adressées aux
préfets.
(Source : www.senat.fr/rap/100-261/100-26121.html)
« La circulaire du 3 décembre 1993 énonce les principes de la politique nationale de
recherche et de réhabilitation des sites pollués, avec la recherche systématique des sites

278
potentiellement pollués, l'usage de la notion de risque, ou fondement de la démarche, et
un traitement adapté à l'impact effectif du site sur l'environnement.
La circulaire du 3 avril 1996 pose le principe du recensement des sites potentiellement
pollués et des sites industriels en activité.
La circulaire du 7 juin 1996 traite des procédures de réhabilitation, et précise les
conditions de saisine de l'ADEME (dans le cas des " sites orphelins ").
La circulaire du 16 mai 1997 concerne les sites pollués par des substances radioactives.
La circulaire du 10 décembre 1999 décrit les deux catégories de risques à envisager et
les objectifs de la réhabilitation. »
Les objectifs de réhabilitation sont choisis en fonction de l’usage envisagé du site, c’est-
à-dire celui auquel le détenteur le destine, et selon les techniques disponibles. Le
principal problème est l’impact, potentiel ou avéré, de la pollution sur l’Homme ou
l’environnement . Le risque est défini comme la combinaison de trois facteurs : le danger,
qui prend en compte la quantité de matière polluante et sa nocivité, le transfert des
substances polluantes depuis le site vers la ou les cibles, et les caractéristiques de la (ou
des) cible(s). C’est une étude spécifique au site qui détermine les objectifs de traitement,
celui-ci devant réduire le risque à un niveau jugé acceptable. Elle est menée selon la
méthodologie de l’évaluation détaillée des risques et prend en compte les cibles suivantes
: l’Homme et la ressource en eau, qui doivent être protégés en priorité, les écosystèmes et
les biens matériels
Inventaire des sites pollués en France
En France, le Ministère de l’Aménagement du Territoire et de l’Environnement a
répertorié 3735 sites industriels pollués en 2004 (3058 recensés en novembre 2000)
(Tableau III). En 1996, le Ministère avait recensé 3 fois moins de sites. Cette
augmentation notamment est due au fait qu’avant 1996, seuls les sites dont l’activité avait
cessé étaient répertoriés. Aujourd’hui, même les sites en exploitation sont comptabilisés.
Les polluants les plus fréquemment rencontrés (seuls ou en mélange) sont présentés dans
le Table IV.
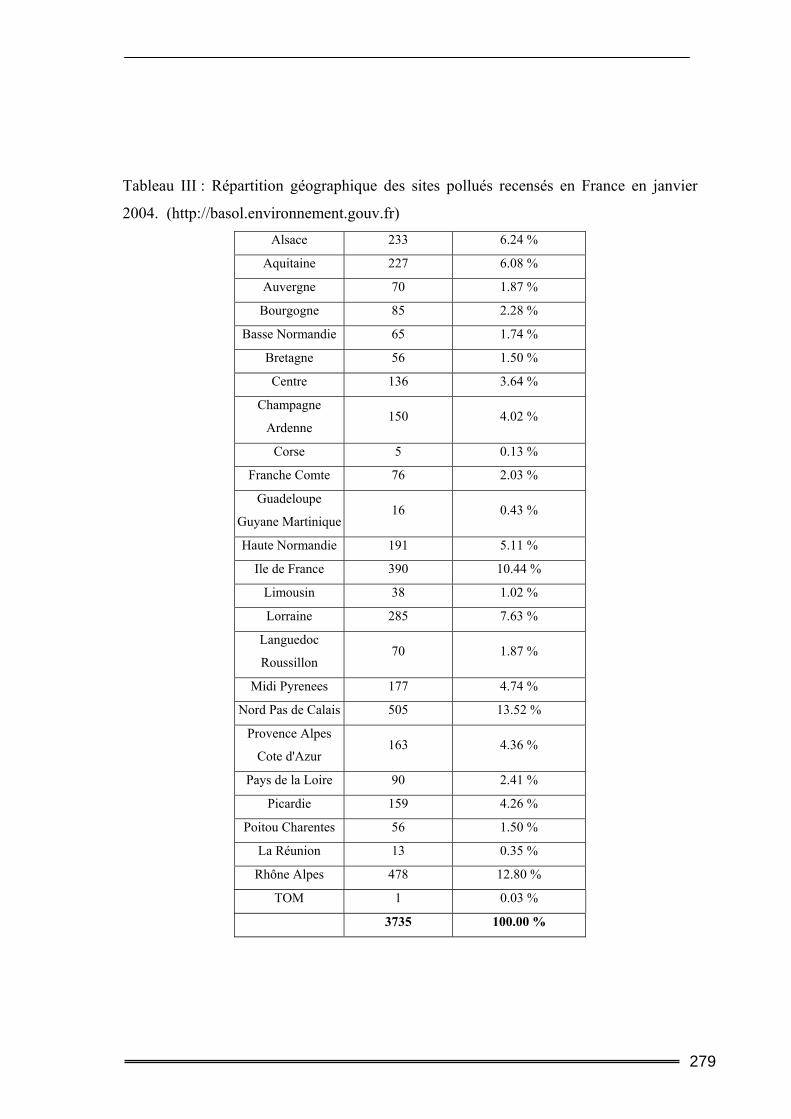
279
Tableau III : Répartition géographique des sites pollués recensés en France en janvier
2004. (http://basol.environnement.gouv.fr) Alsace 233 6.24 %
Aquitaine 227 6.08 %
Auvergne 70 1.87 %
Bourgogne 85 2.28 %
Basse Normandie 65 1.74 %
Bretagne 56 1.50 %
Centre 136 3.64 %
Champagne
Ardenne 150 4.02 %
Corse 5 0.13 %
Franche Comte 76 2.03 %
Guadeloupe
Guyane Martinique16 0.43 %
Haute Normandie 191 5.11 %
Ile de France 390 10.44 %
Limousin 38 1.02 %
Lorraine 285 7.63 %
Languedoc
Roussillon 70 1.87 %
Midi Pyrenees 177 4.74 %
Nord Pas de Calais 505 13.52 %
Provence Alpes
Cote d'Azur 163 4.36 %
Pays de la Loire 90 2.41 %
Picardie 159 4.26 %
Poitou Charentes 56 1.50 %
La Réunion 13 0.35 %
Rhône Alpes 478 12.80 %
TOM 1 0.03 %
3735 100.00 %

280
Situation technique des sites
L’état des sites recensés est répertorié en 4 catégories, qui sont:
- Site traité, libre de toute restriction
Ces sites ont fait l’objet d’évaluation et/ou de travaux. A leur suite, leur niveau de
contamination est tel qu’il n’est pas nécessaire d’en limiter l’usage ou d’exercer une
surveillance. Il est toutefois opportun de garder la mémoire de tels sites.
396 sites (10.60 %) correspondent à cette situation
- Site traité avec restriction
Les évaluations et/ou travaux menés sur ces sites amènent au constat d’une pollution
résiduelle, compatible avec leur usage actuel mais qui nécessite des précautions
particulières avant d’en changer l’usage et/ou d’effectuer certains travaux. Une
surveillance de l’impact de cette pollution peut aussi être nécessaire.
1578 sites (42.25 %) correspondent à cette situation.
- Site en activité et devant faire l’objet d’un diagnostic
La pollution de ces sites n’est pas avérée mais diverses raisons (nature de l’activité,
accidents survenus dans le passé,..) font penser que tel pourrait être le cas. Pour prévenir
une découverte fortuite de cette pollution et surtout avant celle d’un éventuel impact, la
réalisation d’un diagnostic de l’état des sols et d’une évaluation simplifiée des risques a
été demandée par l’administration aux responsables de certains sites en activité (environ
1 300).
Ceux qui n’ont pas achevé ces investigations font partie de cette catégorie. Ils
correspondent à 301 sites (8.06 %)
- Sites en cours d’évaluation ou de travaux
La pollution de ces sites est avérée et a entraîné l’engagement d’actions de la part de ces
responsables.
1460 sites (39.09 %) correspondent à cette catégorie
Nature des polluants
Une pollution des sols ou d’une nappe d’eau souterraine est constatée dans 68.14 % des
cas, soit 2545 cas. En terme d’occurrence, les 10 principaux polluants constatés (seuls ou
en mélange) sont les suivants:

281
Tableau IV: Les 10 principaux polluants des sites pollués répertoriés par le Ministère de
l’Aménagement du Territoire et de l’Environnement (novembre 2000)
Polluants % de sites répertoriés
Hydrocarbures 37.44
Hydrocarbures aromatiques polycycliques
(HAP) 15.72
Plomb 15.39
Zinc 10.04
Solvants halogénés 12.29
Chrome 13.04
Cuivre 12.16
Arsenic 10.07
Nickel 8.41
Cadmium 5.49

282
Annexe 2 : Démarche méthodologique d’analyse environnementale
Évaluation Simplifiée des Risques (ESR)
Il s’agit non pas d’évaluer les risques à proprement parler, mais plutôt de classer les sites
en différentes catégories. Un ensemble de 49 paramètres (répartition et mobilité des
substances, proximité des nappes, perméabilité des sols,...) permet de déterminer si le site
doit être banalisé (aucune action particulière), à surveiller ou si le site requiert des
investigations complémentaires et plus approfondies.
Evaluation Détaillée des Risques (EDR)
Cette deuxième étape a pour but d’évaluer l’impact potentiel des substances dangereuses
sur les différentes cibles concernées en fonction de l’usage actuel et prévisible du site.
L’EDR permet d’identifier les sites qui présentent des risques jugés inacceptables et de
définir une stratégie de traitement en vue.
Dans tous les cas, il revient aux responsables (exploitant à l’origine de la pollution,
dernier exploitant, détenteur,...) de faire cesser les dommages générés par ces pollutions,
en application de la législation relative aux installations classées pour la protection de
l’environnement. Pour faciliter les décisions des acquéreurs d’anciens sites industriels,
l’article L.514-20 de la loi n°76-663 du 19 juillet 1976 relative aux installations classées
pour l'environnement a prévu qu’un terrain sur lequel une installation soumise à
autorisation a été exploitée ne peut être vendu sans que le vendeur informe par écrit
l’acheteur de cette exploitation et des dangers ou inconvénients importants qui en
résultent ".
Lorsque le responsable d’un site est défaillant (insolvabilité, disparition, etc...) et que la
pollution du site présente un risque pour l’environnement et la sécurité des personnes,
l’état peut intervenir pour mettre le site en sécurité. Ces interventions sont financées par
la TGAP (Taxe Générale sur les Activités Polluantes) et sont toujours associées à un
recours juridique contre le responsable.
Enfin, aujourd’hui, l’arrêt d’une activité doit obligatoirement s’accompagner d’une
remise en état du site de manière telle qu’il ne présente plus de risques pour la santé
publique et l’environnement.

283
Annexe 3 : Courbes d’élution du soluté
Transport de solutés dans un milieu poreux
Les phénomènes principaux qui participent à l’écoulement sont :
La convection, qui désigne l’écoulement massique de la solution dans la porosité
percolante (pores ouverts ou connectés). Le soluté se déplace avec la phase mobile.
La dispersion hydrodynamique, est différenciée en :
Dispersion cinématique, provoquée par l’hétérogénéité de la distribution des vitesses
d’écoulement dans un milieu poreux (fonction de la structure et de la texture du milieu).
Diffusion moléculaire, due aux gradients de concentrations entre les zones mobiles
(porosité percolante) et les zones stagnantes (porosité intra-particulaire ou pores semi-
ouverts ou mal connectés ou zones stagnantes par écoulement non homogène de la
porosité percolante).
La courbe obtenue par l’analyse de la solution en sortie de colonne est appelée « courbe
d’élution ». Lorsque l’injection de la solution contenant le traceur (ou tout autre soluté)
est effectuée en créneau, l’allure de la courbe d’élution peut avoirs divers aspects, comme
le montre la figure 3 (Lieto, 1996) :
Différentes courbes d’élution (1 à3 ) rencontrées suite à une injection en créneau (0).
(C est la concentration en soluté)
C
C(t C
t
C
t
C
t
C
t
Injectio
3
1
2

284
Dans cette figure, la courbe notée Injection représente l’injection du traceur en créneau.
La concentration de traceur est suivie en sortie de colonne et une courbe de type 1, 2 ou 3
est obtenue, témoignant de comportements hydrodynamiques variables :
1 : Courbe d’élution symétrique, son sommet correspond au temps de séjour moyen. Elle
représente une fonction de distribution de type gaussien, qui signifie que l’écoulement se
déroule dans une structure homogène sans zone morte ni chemin préférentiel. C’est le
signal obtenu dans le cas d’un réacteur piston. L’élargissement de la courbe par rapport
au créneau résulte de la dispersion axiale et radiale du soluté.
2 : Courbe asymétrique dans laquelle le front compressif (montée) est plus rapide que le
front diffusif (descente). Ce signal est obtenu dans le cas d’un réacteur piston ouvert à la
diffusion. C’est une déviation commune qui peut se schématisé comme une cascade de
quelques réacteurs parfaitement agités (un réacteur parfaitement piston est équivalent à
une cascade d’une infinité de réacteurs parfaitement agités).
3 : la présence de deux pics résulte de l’existence de chemins préférentiels.
Le bilan matière peut être effectué par intégration de la surface sous la courbe d’élution.
On doit ainsi vérifier que le nombre de moles de traceur injectées est égal au nombre de
moles de traceur détectées en sortie. On réalise pour cela une intégration des deux
signaux par la méthode des trapèzes avec un pas de temps de 10 secondes.
Le temps de passage τ est donné par la division du volume de pores par le débit.
Le temps moyen de séjour <t> peut être défini (éq. 2) comme moment d’ordre 1 (µ1) de la
fonction de DTS E(t):
1
0
)( µ==⟩⟨ ∫∞
dttEtt s
(2)
avec : CotCtE )(1)( ∗=τ et ∫
∞
=⋅0
1)( dttE (E(t) est normée)
C(t) étant la concentration de traceur en sortie, et Co sa concentration initiale.
Dans notre cas le rapport C(t)/Co sera assimilé au rapport Λ(t)/Λ0 (conductivité en sortie
(Λ(t)) sur la conductivité du traceur (Λ0)).
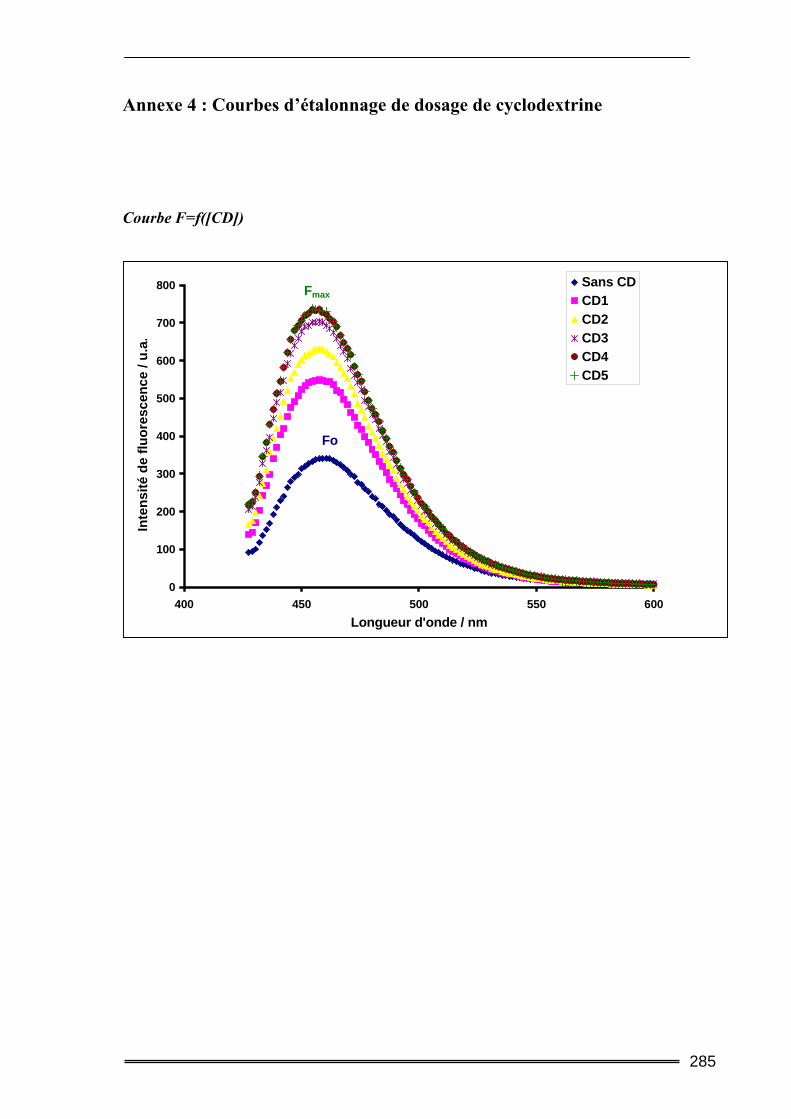
285
Annexe 4 : Courbes d’étalonnage de dosage de cyclodextrine
Courbe F=f([CD])
0
100
200
300
400
500
600
700
800
400 450 500 550 600Longueur d'onde / nm
Inte
nsité
de
fluor
esce
nce
/ u.a
.
Sans CDCD1CD2CD3CD4CD5
Fo
Fmax
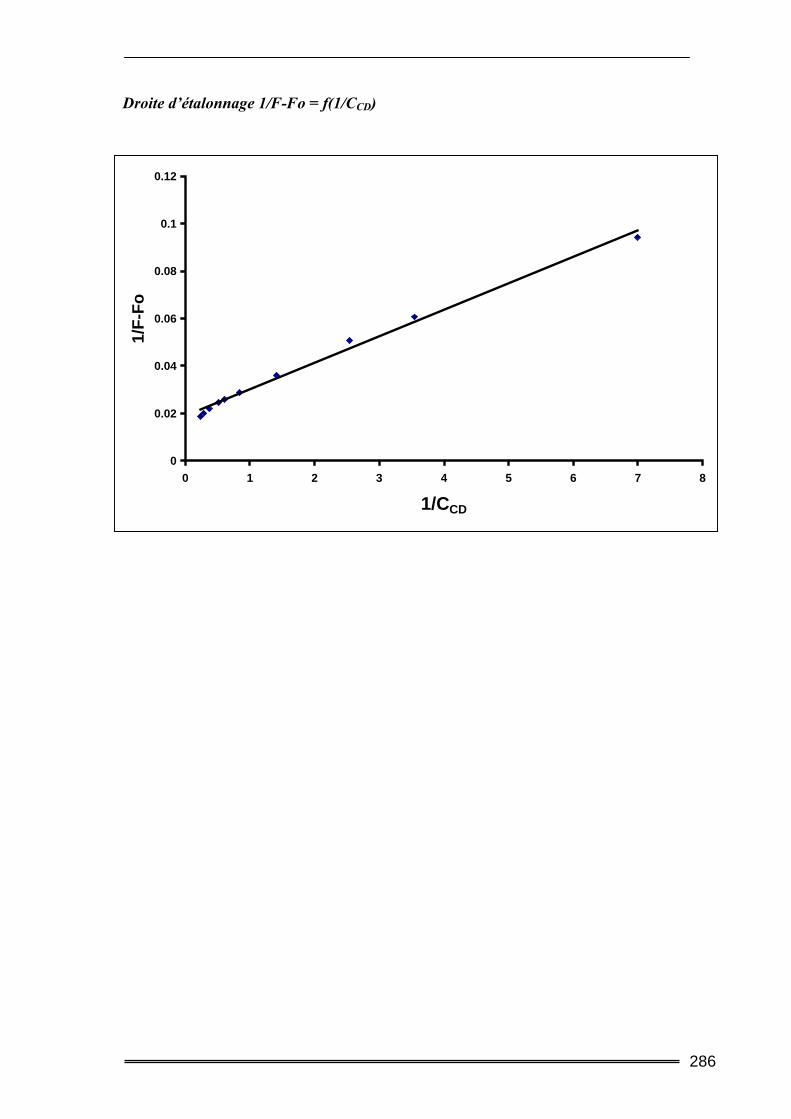
286
Droite d’étalonnage 1/F-Fo = f(1/CCD)
0
0.02
0.04
0.06
0.08
0.1
0.12
0 1 2 3 4 5 6 7 8
1/CCD
1/F-
Fo

287
Annexe 5 : Abstracts des publications
HANNA, K., DE BRAUER, C. ET GERMAIN, P. Solubilization of the neutral and charged forms of 2,4,6-trichlorophenol by β-cyclodextrin and hysroxypropyl-β-cyclodextrin in water. Journal of Hazardous Material, 2003, vol B100, pp. 109-116.
HANNA, K., DE BRAUER, C. ET GERMAIN, P. Cyclodextrin-enhanced solubilization of pentachlorophenol in water. Journal of Environmental Management, 2004, vol 71, pp. 1-8.
BADR, T., HANNA, K. ET DE BRAUER, C. Enhanced solubilization and removal of naphthalene and phenanthrene by cyclodestrins from two contamined soils. Journal of Hazardous Material, 2004, vol B100, pp. 215-223. HANNA, K., DE BRAUER, C., GERMAIN P., CHOVELON J.M. ET FERRONATO C. Degradation of pentachlorophenol in cyclodextrin extraction effluent using a photocatalytic process. Science of the Total Environment, 2004, vol 332, pp. 51-60.
CHATAIN, V., HANNA, K., DE BRAUER, C, BAYARD R. ET GERMAIN P. Enhanced solubilization of arsenic and 2,3,4,6 tetrachlorophenol from soils by a cyclodextrin derivative. Chemosphere, 2004, vol 57, pp. 197-206.





























