Embed Size (px)
Citation preview

Bull , SOC. Chirn. Belg. vol. 84/11. 11/1975
ETUDE DE L'INFLUENCE DES BASES SUR LE CALCUL DE GRANDEURS MOLECULAIRES
G. Leroy, G. Reckinger et J.L. Ruelle* Universite Catholique de Louvain Laboratoire de Chimie Quantique
Batiment Lavoisier Place Louis Pasteur, 1
B-1348 Louvain-la-Neuve - Belgium R e c e i v e d 1 6 / 1 0 / 7 5 - A c c e p t e d 6 / 1 1 / 7 5 .
ABSTRACT
W i t h i n t h e f r a m e w o r k o f t h e H a r t r e e - F o c k m o d e l , t h e e f f i c i e n c y o f s e v e - r a l a t o m i c g a u s s i a n b a s i s s e t s w i t h r e s p e c t t o t h e c o m p u t a t i o n o f some m o l e - c u l a r p r o p e r t i e s h a s b e e n i n v e s t i g a t e d i n some s i m p l e compounds a s w e l l 8 s i n two c h a r g e t r a n s f e r c o m p l e x e s . A l t h o u g h , i n g e n e r a l , e a c h c o n s i d e r e d s y s t e m o r p r o p e r t y r e q u i r e s t h e c h o i c e o f a p e c u l i a r b a s i s s e t ( t h e m o r e e x t e n d e d t h e l a r g e r t h e r e q u i r e d a c c u r a c y ) , i t i s f o u n d h e r e t h a t t h e STO-3G b a s i s s e t g i v e s u n i f o r m e n o u g h p e r f o r m a n c e a s t o j u s t i f y i t s u s e f o r t h e q u a l i t a t i v e c a l c u l a t i o n o f e n e r g e t i c a n d s t r u c - t u r a l p r o p e r t i e s o f c h e m i c a l compounds .
I. INTRODUCTION
En methode LCAO-SCF-MO, le choix de la base atomique est arbitraire pour- vu que l'ensemble des fonctions de base soit complet et non redondant. Comme ces conditions ne sont generalement pas remplies, le choix de l'espace fonctionnel constitue l'un des problemes importants.de la chimie quantique moleculaire. En effet, c'est la dimension de cet espace qui determine le temps de calcul. De plus, la qualit6 des rdsultats obtenus avec une base donnee va- rie.selon le type de grandeur calculee. bans ce travail, nous nous proposons d'analyser les performances de differen- tes bases atomiques dans le calcul de quelques grandeurs moleculaires. Dans ce but, nous avons effectue une serie de calculs ab initio, dans le cadre de la methode de Hartree-Fock en utilisant differents types de bases gaussien- nes: la base minimale 7s-3p de Clementi contractee en (2s-lp) (l), les bases minimales STO-3G, STO-4G, STO-5G, STO-6G(2) et les bases doubles 4-31G. 5-31G et 6-31G(3-4) du groupe de Pople. Sauf dans l'etude du complexe NH3-C12, nous n'avons pas utilise de base &endue.
* Aspirant du FNRS
-1 105-

En effet, les travaux du laboratoire sont plutdt orientes vers l'etude de grands systemes moleculaires pour lesquels il est actuellement trop cocteux d'utiliser de telles bases. Tous les calculs ont ete effectues a l'aide du programme GAUSSIAN-70(5). Les composes que nous avons choisis sont gendralement de petits systemes con- tenant des atomes d'hydrogene, de carbone, d'azote, d'oxygene, de fluor et de chlore, 2 savoir: le methane et l'ethane, l'ethylene, le cyclopropane, l'eau, l'ammoniac, les molecules de fluor et de chlore ainsi que deux complexes inter- moleculaires, H20-C12 et NH3-C12. Les grandeurs moleculaires q u i ont retenu notre attention sont: l'energie to- tale, l'energie d'atomisation ou de formation, le potentiel d'ionisation, les populations d'atomes et de liaisons et les moments dipolaires. Tous les calculs sur les molecules simples ont ete effectues en adoptant leur geometric experimentale(6).
11. DESCRIPTION DES RESULTATS ET DISCUSSION
11.1. Etude de quelques petites mol8cules.
a) Enerqie totale.
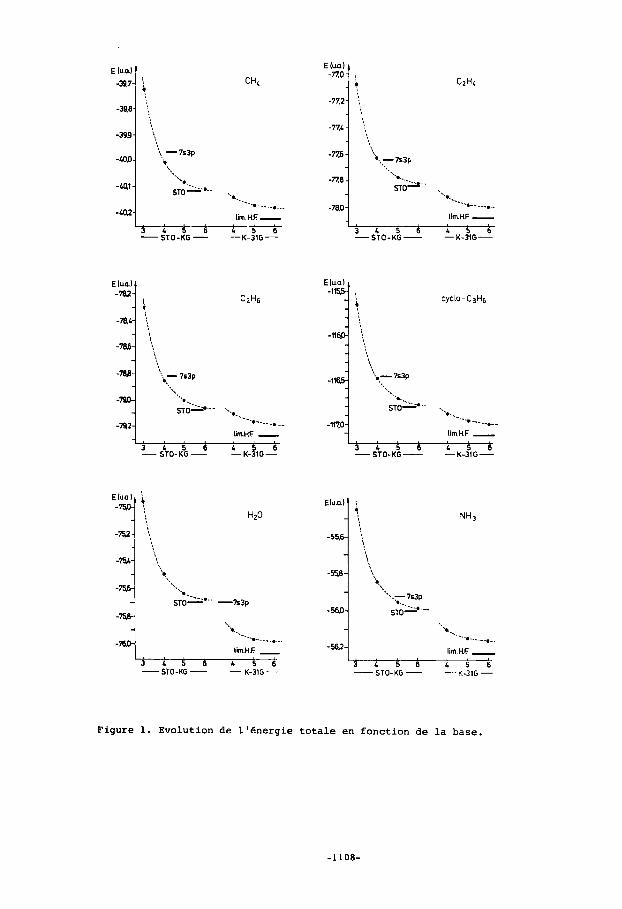
Les energies totales des composes etudies sont reprises dans le tableau I. A titre d'exemple, nous illustrons sur la figure 1 1'6volution de l'energie totale des molecules CH4, C2H4,C2H6,cYC10-C3H6, II2O et NH3 en fonction de la base utilisee En base STO-KG, l'energie tend naturellement vers la valeur calculee avec une base minimale d'orbitales de Slater. Cette limite est pratique- ment atteinte pour K=6. L'evolution de l'energie est sensiblement la meme pour toutes les molecules. Les bases doubles ameliorent nettement l'dnergie en la rapprochant de la limite Hartree-Fock dont la valeur estimee est donnee pour quelques mo- lecules. Le resultat obtenu en base 6-31G s'ecarte d'environ 0 , l % de cette limite. L'Bnergie calculee en base 7s-3p est tantdt proche de la valeur corres- pondante en base STO-4G (cas des hydrocarbures) tantdt inferieure a la valeur calculee en base STO-6G (cas de l'eau). I1 faut noter que les grandeurs Bneryetiques interessantes ne sont pas. des energies totales mais plutdt des differences d'energies. Pour ces dernieres, il est possible d'obtenir des resultats meme quantltatifs dans les cas 00 la variation d'energie de corr&lation est negligeable. I1 apparaft de plus en plus que ces cas doivent satisfaire aux deux exi- gences suivantes: l'invariance du nombre de paires d'electrons et la conservation approximative de l'environnement immediat de ces paires d'electrons. Un exemple typique repondant a ces exigences est celui de la rotation interne de l'ethane, dont la barriere energetique est remar- quablement bien calculee, aussi bien en base minimale qu'en base double,
(34).
-1106-
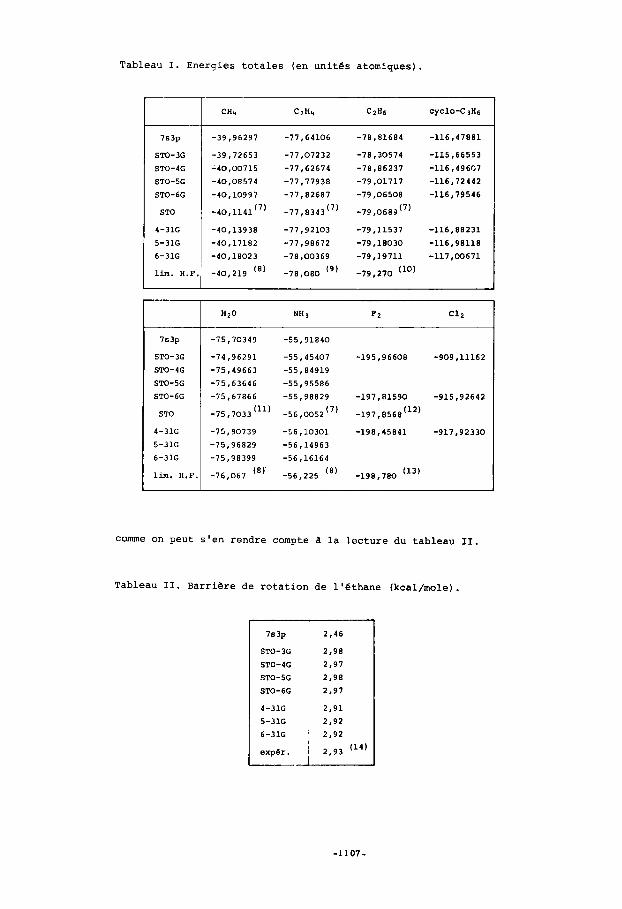
Tableau I. Energies totales (en unites atomiques).
4-31G 5-31G 6-31G
l im. H.F.
7s3p
STO-3G STO-4G STO-5G STO-6G
STO
4-31G 5-31G 6-31G
lim. H.F
-75,90739 -56,10301 -198,45841 -917,92330 -75,96829 -56,14963 -75,98399 -56,16164
-76,067 -56,225 -198,780 (13)
-39,96297
-39,72653 :40,00715 -40,08574 -40,10997
-40, ~ 4 1 ' ~ )
-40,13938 -40,17182 -40,18023
-40,219
-77,64106
-77,07232 -77,62674 -77.77938 -77,82687
-77,8 3 4 3 ( 7,
-77,92103 -77,98672 -78,00369
-78,080 (')
-78,81684
-78,30574 -78,86237 -79,01717 -79,06508
-79,0683(7)
-79,11537 -79,18030 -79,19711
-79,270 (lo)
-116,47881
-115,66553 -116,496C7 -116,72442 -116,79546
-116,88231 -116,98118 -117,00671
7s3p
STO-3G STO-4G STO-5G STO-6G
STO
-75,70349 -55,91840
-74,96291 -55,45407 -195,96608 -909,11162 -75,49663 -55,84919 -75,63646 -55,95586 -75,67866 -55,98829 -197,81590 -915,92642
-75,7033 -56,0052(7) -197,8568(12)
c o m e on peut s'en rendre compte a la lecture du tableau 11.
Tableau 11. Barrigre de rotation de l'ethane (kcal/mole).
STO-3G STO-4G STO-5G STO-6G
4-31G 5-31G 6-31G
exper.
-1107-
CH4
-7584
.--.-.. _ _ -.__ 1hH.F -
3 L 5 6 L 5 6 . . STO-KG - - K-31G - -
-Ino(
IimHF - L 5 6 - K-31G-
1 . . 1 1
3 1 5 6 L 5 6 - STO-KG- -K-31G-
Figure 1. Evolution de l'bnergie totale en fonction de la base.
-1108-
b) Chaleur d'atomisation.
Les chaleurs d'atomisation donndes dans le tableau I11 sont calculdes pour les molecules dans leur &at d'equilibre, en tenant compte de 1' Bnergie du point zero et d'une correction de temperature pour 298'K. En general, les valeurs fournies par le calcul sont fortement sous-es- timdes. La base 7s-3p donne les valeurs les plus faibles. Dans les ba- ses STO-KG et K-31G, l'augmentation du nombre de gaussiennes n'ameliore pas 1'Bnergie d'at~misation'~~). AU contraire, les valeurs diminuent ldgerement avec le nombre croissant de fonctions gaussiennes. A l'excep- tion de H20, NH3 et C2H4, les resultats en base double ne sont pas meil- leurs qu'en base minimale STO-3G. La sous-estimation de l'energie d'atomisation provient essentiellement du fait qu'on neglige ici la difference d'dnergie de correlation entre la molecule et ses atomes constituants. Fait bien connu, l'dnergie d'atomisation (ou de dissociation) du fluor est negative dans les trois bases considerees, STO-3G, STO-6G et 4-31G, ce qui en fait une molecule instable dans l'approximation de Hartree- Fock. Seule la prise en consideration de la correlation electronique permet de dBcrire correctement la liaison F-F'15). Eien que les valeurs absolues de l'energie d'atomisation soient tou- jours trop faibles, on peut etablir une bonne correlation entre la va- leur calculee AHa(thdor) et la chaleur d'atomisation experimentale AHa(exp), et ceci pour chacune des bases envisagees. La figure 2 repro- duit cette correlation pour la base 4-31G.
Tableau 111. Energies d'atomisation B 298'K (kcal/mole).
7s3p
STO-3G STO-4G STO-5G STO-6G
4-31G 5-31G 6-31G
exper.
C H 4 C 2 H 4 C 2 H g C Y C ~ O - C I H ~ H20 ~ ~~~~ ~~
219,5 272,4 375,l 417,6 59,s
303,l 374,3 515,7 595,3 95,O 293,2 359,3 497,6 572,l 90,9 292,8 358,2 496,7 570,2 90,7 292.6 358,O 496,3 569,7 90,4
295,O 383,s 495,O 573,9 16,7 294,2 382,4 493,4 572,4 117,3 294,O 382.1 493,O 572,l 117,s
397,19 537,65 674,64 812,57 221,56 (16) (16) (! 6) (16) (17)
NH 3 Fz C l n
98 ,o
139.3 -4,87 13,14 132.2 132.0 131,8 -6,47 15,18
156,9 -45.31 -11,57 157,5 157.6
280,34 37,72 57.84 (17) (17) (17)
-1109-

Figure 2 . Correlation entre AHa experimental et AHa theorique en base 4-31G.
c) Potentiel de premigre ionisation.
Conformement au theoreme de Koopman , le potentiel de premiere ionisa- tion d'une molecule est donne au signe pres par l'energie de la plus haute orbitale moleculaire occupee. Le tableau IV montre que toutes les bases utilisees donnent l'ordre de grandeur correct des potentiels d' ionisation, mais que la precision experimentale est loin d'etre attein- te. Les valeurs trouvees restent pratiquement constantes pour chaque mo- lecule dans un type de base. Alors qu'en base 7s-3p et K - 3 1 G elles sont toujours superieures aux valeurs experimentales, elles sont tantdt supe- rieures, tantdt inferieures ?I l'experience pour les bases STO-KG, de sorte qu'il est difficile d'etablir des correlations entre la theorie et l'experience . (36)
Tableau IV. Potentiels de premiere ionisation (en eV).
7s3p
STO-3G STO-4G STO-5G STO-6G
4-31G 5-31G 6-31G
exper. (19)
16,33
14,05 14.08 14,12 14,13
14,75 14,75 14,75
12.70
12,64
8,81 8,87 8 , 8 9
8,90
10,08 10,lO 10,ll
10,51
14,76 13,90
12,41 10,36 iz,45 10,311 12,411 io,4i 12,49 10,42
13,14 11,33 13,14 11,33 13,14 11,33
11,52 10,l
13,99
10,64 10,77 10,81 10,81
13,59 13,63 13,64
12,61
12,48
9,411 12,711 ii,o7 9,56 9,60 9,60 13,15 11,20
11,18 18,15 12,26 11,20 11,21
10,15 15.7 11.48
-1 11 0-
d) Charges nettes des atomes et moments dipolaires.
7s3p
STO-3G STO-4G STO-5G STO-6G
4-31G 5-31G 6-31G
exper.
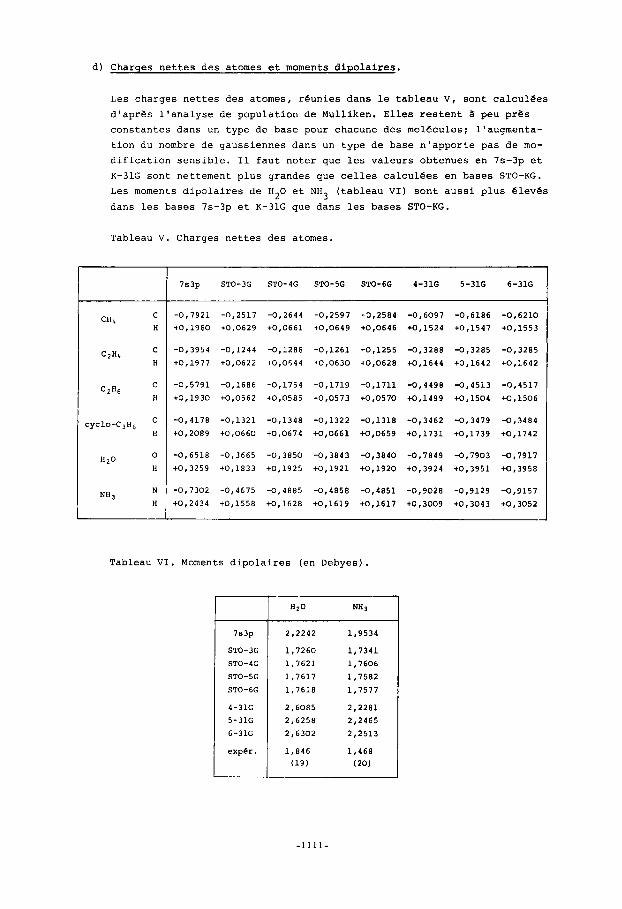
Les charges nettes des atomes, reunies dans le tableau V, sont calculdes d'aprss l'analyse de population de Mulliken. Elles restent a peu pres constantes dans un type de base pour chacune des molecules; l'augmenta- tion du nombre de gaussiennes dans un type de base n'apporte pas de mo- dification sensible. I1 faut noter que les valeurs obtenues en 7s-3p et K - 3 1 G sont nettement plus grandes que celles calculees en bases STO-KG.
Les moments dipolaires de H 2 0 et NH3 (tableau VI) sont aussi plus eleves dans les bases 7s-3p et K - 3 1 G que dans les bases STO-KG.
2,2242 1,9534
1,7260 1,7341 1,7621 1,7606 1,7617 1,7582 1,7618 1,7577
2,6085 2,2281 2,6258 2,2465 2,6302 2,2513
1,846 1,468 (13) (20)
Tableau V. Charges nettes des atomes.
CH 4 C H
C2H4 C H
C2H6 C H
cyclo-C3H6 n
~ ~ ~~~~~ ~~
7s3p STO-3G STO-4G STO-5G STO-6G 4-31G 5-31G
-0,7921 -0,2517 -0,2644 -0,2597 -3,2584 -0,6097 -0,6186 +0,1980 +0,0629 +0,0661 +0,0649 +0,0646 +0,1524 +0,1547
-0,3954 -0,1244 -0.1288 -0,1261 -0,1255 -0,3288 -0,3285 +0,1977 +0,0622 +0,0644 +0,0630 +0,0628 tO,1644 tO,1642
-0,5791 -0,1686 -0,1754 -0,1719 -0,1711 -0,4498 -0,4513 +0,1930 +0,0562 +0,0585 +0,0573 +0,0570 +0,1499 +0,1504
-0,4178 -0,1321 -0,1348 -0,1322 -0,1318 -0,3462 -0,3479 +0,2089 +0,0660 +0,0674 +0,0661 +0,0659 +0,1731 +0,1739
-0,6518 -0,3665 -0,3850 -0,3843 -0,3840 -0,7849 -0,7903 +0,3259 +0,1833 +0,1925 tO,1921 t0,1920 t0,3924 +0,3951
-0,7302 -0,4675 -0,4885 -0.4858 -0,4851 -0,9028 -0,9129 +0,2434 +0,1558 +0,1628 +0,1619 +0,1617 t0,3009 tO,3043
6-31G
-0,62 10 t0.1553
-0,3285 +O ,16 4 2
-0,4517 +0,1506
-0,3484 +0,1742
-0,7917 +O ,395 8
-0,9157 +0,3052
Tableau VI. Moments dipolaires (en Debyes).
-1111-

Cependant, la base de Clementi et la base double paraissent mieux rendre compte de la topologie moleculaire, puisqu'elles permettent le calcul d'un moment dipolaire plus grand pour la molecule d'eau que pour la mo- lecule d'ammoniac, come le veut l'experience.
e ) Populations de liaison.
Le tableau VII reunit les populations de liaison dans les composes poly- atomiques etudies. On constate que la base 7s-3p donne, par rapport aux autres bases, les populations les plus fortes pour les liaisons conte- nant un atome d'hydrogsne. Les bases X-31G donnent des populations C-C tres faibles dans l'ethane et surtout dans le cyclopropane.
Tableau V I I . Populations de liaison.
CHr C-H
CZHA, c-c
C2Hg C-H c-c
c y c l o - C 3 H a c -c
H 2 0 0 - H
NH3 N-H
7s3p STO-3G STO-4G STO-5G STO-6G 4-31G 5-31G 6-31G
0,40148
0,42073 0,62848
0,41163 0,34148
C, 42296 0,27430
0,30146
0,36362
0,38371
0,39194 0,59983
0,38275 0,36385
0,38946 0,29830
0,26410
0,34185
0,38405
0,39201 0,60272
0,38294 0,36716
0,38944 0,30138
0,26159
0,34109
0,38426
0,39229 0,60321
0,38319 0,36751
0,38974 0,30182
0,26177
0,34138
0,38432
0,39238 0,60326
0,38328 0,36738
0,38986 0,30176
0,26178
0,34146
0,38111
0,39489 0,59149
0,38867 0,28362
0,39332 0,16643
0,26119
0,32778
0,38062
0,3954 3 0,59138
0,38888 0,27553
0,39202 0,15459
0,26022
0,32743
0,38036
0,39542 0,59060
0,38878 0,27336
0,39149 0,15159
0,26005
0,32734
11.2. Etude de complexes intermoleculaires.
Nous avons voulu egalement etudier l'influence des bases atomiques sur le calcul des propridtes des complexes par transfert de charge. NOUS avons choisi les couples eau-chlore et ammoniac-chlore qui ont deja fait l'objet d'une etude tres complete en base STO-3G(21-22). Dans ce travail, nous comparons les resultats obtenus dans les bases STO-3G, STO-6G et 4-31G. De plus, l'etude du systeme ammoniac-chlore a dtd effectuee en base double avec fonctions de polarisation. Pour l'atome de chlore, nous avons utilise la base (23) (lls, 7p) de Clementi contractee en (5s, 4p); nous y avons ajoute une fonction gaussienne de type d, d'exposant egal a 0,68 de- veloppee en deux gaussiennes selon le procede de Dunning("). Pour la mole- cule d'ammoniac, les fonctions s et p de l'atome d'azote et des atomes d'hy- drogene sont celles de Dunning (*') , dans les contractions respectives ( 9 s , 5p/ 4 s , 2p) et ( 4 s / 2 s ) . La fonction de type d pour l'azote, d'exposant egal a 0,75 a ete developpee de la meme faGon que celle du chlore, tandis que la fonction
- 1 1 1 2 -

de type p de chaque hydrogene a requ un exposant de 1,l recommand6 par Hari- haran(26). Compte tenu des travaux anterieurs en base STO-3G, nous avons elmis que la mo- lecule de chlore se fixe sur le compose donneur du c8t6 de se; paires libres, les deux atomes de chlore et l'atome d'oxygsne ou d'azote &ant colineaires. La figure 3 illustre les structures adoptees pour les deux complexes choisis.
I'
Figure 3 . Parametres geometriques des complexes intermoleculaires.
Pour le systeme H2O-Cl2, une opthisation de geometric a BtB effectude sur 1' angle .$ et la distance r(0-Cl). Dans le systeme NH3-C12, seule la distance r(N-Cl) a ete optimisee. Dans les deux systgmes, on a retenu leS geometries experimentales des molecules separees. Le tableau VIII reunit dans les differentes bases, les geometries d'equilibre des deux complexes, les chaleurs de formation, les charges nettes sur les ato- mes de chlore, le transfert de charge vers la molecule de chlore ainsi que la variation de moment dipolaire, A p , associee a la formation du complexe.
Tableau VIII. Proprietes des complexes H20-C12 et NH3-C12.
Base
STO-3G SM-6G 4-31G
STO-3G SM-6G 4-31G Etendue
Geometric d'gquilibre
$ rlA)
80' 2,90 85' 3,OO 85' 2,92
90' 2,94 90' 3.04 90' 2.83 90° 2,98
Chaleur de Chnrges nettes Transfert de A y
formation (O'K) (kcal/mole)
(D I clz charge vers le chlore
C11
-0,732 +0,0225 -0,0256 0,0031 0,3344 0,2861 +0,0209 -0,0226 0,0017 -0,589
-1,974 +0,0504 -0,0600 0,0096 0,8032
-1,105 +0,0296 -0,0385 0,0089 0,5536 -0.849 +0,0273 -0,0328 0,0055 0,4470 -2,933 +0,0620 -0,0901 0.0281 1,3734 -2,706 +0,0400 -0,0495 0,0095
Dans tous les cas, la chaleur de formation est negative. De mSme, toutes les bases employees menent a des distances Cll-0 et Cll-N inferieures I la some des rayons de Van der Waals des atomes consideres, soit 3,2 A pour Cll-0 et
- 1 1 1 3 -

3,3 A pour Cll-N. Le comportement des grandeurs calculees, en passant d'une base a une autre, est tres semblable pour les deux systemes Btudies. D'une maniare surprenante B premiere vue, la chaleur de formation des complexes diminue en passant de la base STO-3G a la base STO-GG; ceci va de pair avec une elongation des dis- tances C1-0 et Cl-N. La chaleur de formation la plus forte et les distances C1-0 et C1-N les plus courtes sont fournies par la base double 4-31G. Les transferts de charge evoluent dans le meme sens que les chaleurs de formation Notons que chaque type de base predit une stabilite plus grande pour le com- plexe NH3-C12 que pour le complexe H20-C12; le transfert de charge est envi- ron trois fois plus important pour NH3-C12 que pour H20-C12. Lorsqu'on utilise des bases peu etendues, la chaleur de formation calculee risque d'etre affectee d'une certaine erreur, dite "erreur de superposition" ( 2 7 ) . Ceci rdsulte du fait que dans le calcul sur le supersystsme, les fonc- tions de base d'une molecule contribuent a etendre la base de son partenaire. Ainsi, en principe, on surestime la chaleur de formation, si on la calcule par simple difference d'energie entre les molecules individuelles et le com- plexe. Bien qu'il soit difficile d'estimer quantitativement cette erreur de superposition, il est possible d'en donner une limite superieure. Celle-ci s'obtient en effectuant sur chaque molecule individuelle un calcul dans le- quel on ajoute B la base de la molecule donnee les fonctions de la molecule partenaire dans le complexe, ces fonctions &ant placees aux memes positions que celles occupees dans le complexe. Le gain total d'energie obtenu pour les deux molecules partenaires donne la limite superieure de l'effet de superpo- sition. Le tableau IX montre que, dans la base STO-3G, la valeur limite de l'effet de superposition est du meme ordre de grandeur que la chaleur de formation, alors que dans la base STO-BG, et surtout dans la base 4-31G ou la base 6 -
tendue, la chaleur de formation domine l'effet de superposition. A u total, la negligence de l'effet de superposition n'affecte que les chaleurs de for- mation, qui ne sont d'ailleurs generalement pas calculees avec une grande precision. La coherence des resultats obtenus dans les differentes bases nous conduit a admettre la fiabilite des renseignements fournis par les bases mi- nimaEes. D'apres les travaux anterieurs effectues en base STO-3G(21'28) sur les systa- mes H20-C12 et H2CO-C12, il existerait une correlation entre la forme du po- tentiel electrostatique du compose donneur et la geometric d'bquilibre du com- plexe. En effet, l'axe d'approche de la molecule de chlore passait par le minimum de potentiel electrostatique du formol ou de l'eau. En vue d'examiner l'effet de base sur cette propriete , nous avons calcule le potentiel electrostatique de la molecule d'eau dans la base STO-6G, ceci dans le plan moleculaire XZ et dans le plan des paires libres YZ (figure 4). Comme en base STO-3G, le minimum de potentiel electrostatique se trouve dans le plan des paires libres de la molecule d'eau. Dans la base STO-6G, ce minimum fait un angle d'environ 30° avec l'axe 2 . En passant de 30° a Oo pour cet angle, tout en restant dans le plan des paires libres, le potentiel electrostatique change trSs peu. Ainsi l'hypothese d'une correlation entre le potentiel elec- trostatique et la configuration geometrique des complexes par transfert de
-1 114-
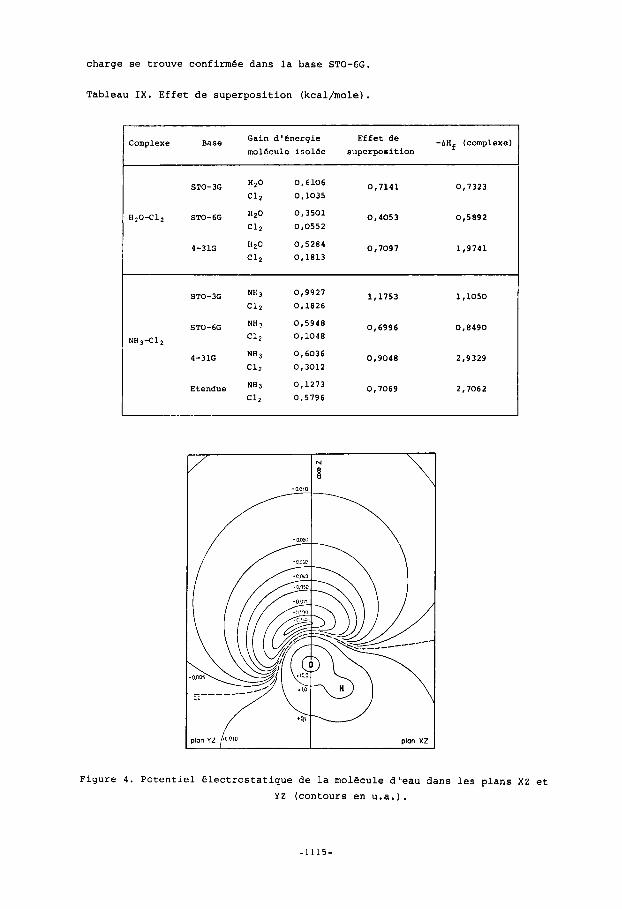
charge se trouve confirmee dans la base STO-6G.
Tableau IX. Effet de superposition (kcal/mole).
-AHf (complexe) Gain d'energie Effet de molecule isolee superposition
Complexe
~~ ~~
0,7141 0,7323 0,6106 ClZ 0,1035
STO-3G H 2 0
HpO-Clp STO-6G H 2 0 0'3501 0,4053 0,5892 c12 0,0552
0,5284 Clp 0,1813
0,7097 1,9741 4-31G H 2 0
1,1753 1,1050 0,9927 c12 0,1826
0,5948 C12 0,1048
4-31G N H 3 0,6036 c1z 0,3012
0,1273 C12 0,5796
STO-3G NE3
0,6996 0,8490 STO-6G NH? NH3-Clp
0,9048 2,9329
0,7069 2,7062 Etendue N H 3
Figure 4 . Potentiel electrostatique de la molecule d'-eau dans les plans XZ et YZ (contours en u.a.).
-1115-

111. CONCLUSIONS.
L'Btude de l'influence des bases sur le calcul des grandeurs moleculaires a deja fait l'objet de nombreux article^(^'-^^). Dans ce travail, nous nous sommes efforces de degager quelques criteres pra- tiques pour effectuer un choix convenable de base atomique en fonction de la grandeur calculee et des objectifs poursuivis. NOUS reprenons ci-dessous les principales conclusions de cette etude.
- I1 est rarement necessaire de connaztre avec precision 1'Bnergie Hartree-Fock d'une molecule car, le calcul precis d'effets thermiques exige le depassement de la limite Hartree-Fock et la prise en conside- ration explicite des effets de correlation. Pour apprecier qualitati- vement la stabilite de molecules il suffit de calculer leur energie totale avec une base minimale puisque l'evolution de cette grandeur est la m6me dans les differents types de base. Dans les rares cas oa il est interessant de se rapprocher de la limite Hartree-Fock, l'emploi d'une base double, 6-31G par exemple, peut Stre recommandd. I1 faut aussi rappeler que la qualite des barrieres de rotation depend intimement du type de compose considere. Ainsi, l'emploi de fonctions de polarisation est necessaire pour rendre compte des conformations les plus stables de H2~2(32).
- Si l'on ne tient pas compte de la correlation, le calcul des chaleurs d'atomisation est toujours fort approche. Ce sont les bases de type X-31G qui donnent les resultats les plus satisfaisants.
- Le theoreme de Koopmans ne peut fournir que des potentiels d'ionisation tres approches. Ici, toutes les bases donnent a peu pres le m6me accord qualitatif avec l'experience.
- Les charges nettes d'atomes sont tres sensibles au choix de la base. Ainsi, elles ont des valeurs beaucoup plus Blevees dans les bases K-31G et 7s-3p. Si l'on juge la qualite des resultats de l'analyse de popula- tion de Mulliken par la valeur du moment dipolaire il faut preconiser l'emploi des bases 7s-3p ou K-31G qui semblent miew reproduire l'evo- lution de cette grandeur dans une serie Be molecules. Rappelons encore, sans autres commentaires, que la base 7s-3p donne les valeurs les plus grandes de populations de liaison.
- I1 est bien connu que les geometries d'equilibre sont bien decrites en base STO-3G(33). NOUS confirmons cette conclusion, par les resultats obtenus dans l'etude des complexes H20-C12 et NH3-Cl2.
- Les caracteristiques electroniques et energetiques des complexes inter- moleculaires sont certainement mieux rendues quand on utilise des bases doubles ou etendues. Cependant, l'emploi de telles bases, q u i est d' ailleurs prohibitif dans l'etude de grands SystSmes, ne modifie pas les conclusions qualitatives resultant de calculs en base minimale. En par- ticulier, la geometric du complexe et sa stabilite sont correctement predites en base STO-3G. De meme, la position du minimum de potentiel electrostatique ne semble pas fort affectee par un changement de base.
-1116-

Au total, nous resumerons comme suit notre attitude actuelle devant le probleme du choix de la base dans un calcul moleculaire. Pour effectuer des calculs d'hypersurfaces d'bnergie potentielle (opti- misation de geometric, analyse conformationnelle, chemins de reaction) on peut tres souvent se contenter de la base minimale STO-3G si l'on recherche des resultats qualitatifs. Lorsque la taille du systeme le permet, l'emploi de bases doubles en quelques points de l'hypersurface fournit des ienseignements plus realistes sur la structure electroni- que et les caracteristiques energetiques du systeme. I1 reste cepen- dant que le choix d'une base appropriee constitue chaque fois un cas d'espsce et est fonction de la nature de la grandeur et des composes considergs ainsi que du temps de calcul dont on dispose.
BIBLIOGRAPHIE.
(1) B.Roos, C.Salez, A.Veillard, E.Clementi: A general program for calculation of atomic SCF orbitals by the expansion method. Special technical report, IBM San Jose Research Laboratory, California 95114.
J. Chem. Phys., 2, 2657 (1969). (3) R.Ditchfield, W.J.Hehre, J.A.Pople:
J. Chem. Phys., 54, 724 (1971). (4) W.J.Hehre, R.Ditchfield, J.A.Pople:
J. Chem. Phys., 56, 2257 (1972). (5) W.J.Hehre, W.A.Lathan, R.Ditchfield, J.A.Pople:
"Gaussian-70 : Ab initio SCF-MO calculations on organic molecules", Q.C.P.E., 236.
"Tables of interatomic distances and configurations in molecules and ions", The Chemical Society, London (1958) - "Supplement" (1965) . J. her. Chem. SOC., g , 2384 (1966).
J. Chem. Phys., 61, 3860 (1974).
J. Chem. Phys., so, 2216 (1969).
Chem. Phys. Lett., 2, 128 (1969).
J. Chem. Phys., 2, 2071 (1968).
Rev. Mod. Phys., 2, 239 (1960).
Nat. Bur. Stand. (US), Tech. Note 438 (1967).
(2) W.J.Hehre, R.F.Stewart, J.A.Pople:
(6)
(7) W.E.Palke, W.N.Lipscomb:
(8) W.C. Ermler, C. W. Kern:
(9) D.B.Neumann, J.W.Moskowitz:
(10) A.Veillard:
(11) S.Aung, R.M.Pitzer, S.I.Chan:
(12) B.J.Ransi1:
(13) M.Krauss:
(14) S.Weiss, G.E.Leroi: J. Chem. Phys., 48, 962 (1968).
J. Chem. Phys., 52, 6241 (1970). (15) ~.F.Schaefer 111:
-1117-

(16) J.D. Cox, G. Pilcher : "Thermochemistry of organic and organometallic compounds", Academic Press, London and New York (1970).
(17) D.D.Wagman, W.H.Evans, V.B.Parker, I.Halow, W.M.Bailey, R.€l.Schumm: Nat. Bur. Stand. (US), Tech. Note 270-3 (1968).
(18) "Ionization potential:, appearance potentials and heats of formation of gaseous positive ions , NSRDS-NBS 26, June (1969).
(19) G. Birnbaum, S. K.Chatter jie: J. Appl. Phys., 23, 220 (1952).
(20) D.K.Coles, W.E.Good, J.K.Bragg, A.h.Scarbaugh:
(21) G.Leroy, G.Louterman-Leloup:
(22) J.La Grange:
(23) E.Clementi:
(24) T.H.Dunning:
(25) T. H. Dunning:
Phys. Rev., 82, 877 (1951).
J. Mo1. Struc., 2, 33 (1975).
Memoire de licence, UCL (1975).
J. Chem. Phys., 46, 3851 (1967).
J. Chem. Phys., 2, 3958 (1971).
J. Chem. Phys., 53, 2823 (1970).
Theoret. Chim. Acta, 2, 213 (1973).
Theoret. Chim. Acta, 2, 249 (1975).
J. Mol. Struct., 2, 205 (1975).
Adv. Phys., g, 825 (1972).
"Internal rotation in molecules", Edit. W.J. Orville-Thomas, J.Wiley, London and New York (1974).
(26) P.C.Hariharan, J.A.Pople:
(27) G.H.F.Diercksen, W.P.Kraemer, B.O.Roos:
(28) G.Leroy, G.Louterman-Leloup:
(29) F.R.Burden, R.M.Wilson:
(30) A.Veillard:
(31) ;.F.Schaefer 111: The electronic structure of atoms and molecules",
Addison-Wesley, Reading (USA) (1972).
Chem. Phys. Lett., 11, 194 (1971).
J. Chem. Phys., 52, 4064 (1970). (34) voir egalement la reference (3).
(35) M.D.Newton, W.A.Lathan, W.J.€Iehre, J.A.Pople:
(36) W.A.Lathan et Coll. (J. Amer. Chem. SOC., 93, 808, 1971) ont cependant
(32) T.H.Dunning, N.W.Winter:
(33) M.D.Newton, W.A.Lathan, W.J.Hehre, J.A.Pople:
J. Chem. Phys., 51, 3927 (1969).
pu calculer des valeurs plus convenables, specialement en base 4-31G, par difference entre 1'8nergie de la molecule neutre et celle de l'ion.
-1118-





























