Embed Size (px)
Citation preview

I I
I Bull. SOC. Chim. Belg., 73, pp. 166-180 (1964) I
BTUDE THgORIQUE DES MOLI%ULES CONJUGeES
I. DESCRIPTION D’UNE MkTHODE SCF-LCAO SEMI-EMPIRIQUE
G. LEROY (*)
RBSUMB
La mkthode de Roothaan n’est pas rigoureusement applicable A l’etude des molkcules polyatomiques conjuguks. Pour tourner cette difficultk, on propose un procede semi-empirique bask sur les approximations de Pariser- Parr-Pople.
On y tient compte d’une correction d’effet de charge et certaines integrales sont evaluees a partir de donnks de spectroscopie atomique.
La rnkthode ainsi obtenue permet d’ktudier les molecules pol yatomiques conjugukes contenant les atomes C,N,O ou S.
1. INTRODUCTION
D’aprbs Hartree (I), une approximation de la fonction d’onde totale d’un systbme de n Clectrons, tvoluant dans le champ de potentiel c r t t par un ensemble de noyaux, peut s’tcrire sous forme d’un produit de n spin- orbitales % constituant une strie orthonormale :
Y (1, 2 ... n) = @1 ( I ) a%? (2) ... @?a (n) (1)
Les meilleures spin-orbitales s’obtiennent en rtsolvant les equations de Hartree :
ou HN est l’optrateur correspondant ti I’tnergie d’un electron dalls le champ des noyaux de la moltcule et Jj, l’optrateur de Coulomb dtfini par la relation :
(*) Charge de Recherches du F.N.R.S. ( l ) D. HARTREE, Proc. Cambridge Phil. SOC. 24, 89, 111, 426 (1928).

BTUDE TH~ORIQUE DES MOLBCULES CONJUGU~ES. I . 167
Si l’on tient compte du principe de Pauli, on doit Ccrire la fonction d’onde sous forme d’un determinant de Slater :
’€’ = I @I (1) @2 (2) ... an (n) I (4)
Les spin-orbitales auto-cohtrentes sont alors solutions de l’equation de Hartree-Fock (2) :
oh Kj est l’operateur d’echange dkfini par l’expression :
(6) 1
r12 Kj @t (1) = (1 @ j (2) @t (2) - dv2 ) @f ( I )
Si, dans le determinant de Slater, les orbitales sont toutes utiliskes deux fois, la fonction d’onde totale s’tcrira :
y = 101 (1) 61 (2) ~2 (3) 6 2 (4) ... ~ n / 2 (n-1) 5n12 (n)l (7)
et les orbitales auto-cohkentes s’obtiennent par 1’Cquation :
Lorsque, dans les systkmes i nombre pair d’dectrons, les orbitales sont Ccrites sous forme de combinaisons linkaires de certaines fonctions de base, elles se calculent en rbsolvant les equations de Roothaan (3) :
et
(2) Voir B ce propos : R. DAUDHL, R. LEFEBVRE, C. MOSER, Quantum
C3) C.C. J. ROOTHAAN, Rev. Mod. Phys. 23,69 (1951). Chemistry, p. 547 et sv., 1959, Interscience Publishers Inc. New York.

168 G. LEROY
x p reprksente les fonctions de base supposkes orthogonales et (pqlrs) est une intkgrale biklectronique :
(12) 1
bq/rs ) = J’ x p (1) x g (1) 112 Xr (2) x s (2) dvl duz
Dans l’ktude des molecules, les meilleures fonctions ‘pc solutions des Cquations de Roothaan sont les orbitales molCculaires et les fonctions x p sont gknkralement des orbitales atomiques de Slater.
Les fonctions ‘pr sont donc des orbitales molkculaires auto-cohkrentes, combinaisons linkaires d’orbitales atomiques (S.C.F. L.C.A.O. M.O.)
Lorsque la mkthode de Roothaan est appliqute aux molkcules conju- gukes, on peut la simplifier considkrablement en introduisant l’approxi- mation x.
Dans le cadre de cette approximation, l’ktude d’une molkcule conju- guke se rambne a la determination de la fonction d’onde des seuls klec- trons de la liaison dklocaliste car on admet que les orbitales x ne se combinent pas effectivement avec les orbitales 6.
On dit alors que les (tdlectrons x)) kvoluent dans le champ du sque- lette non polarisable form6 par les atomes de la molkcule dkpouillks de leuis klectrons x. Ce squelette constitue le ctcaeurn de la molkcule. Dbs lors, les (( meilleures H orbitales moldculaires s’obtiennent grlce A l’kqua- tion :
Oh
HC= HN + - KI) a-1
n dksigne le nombre total d’klectrons x et p, le nombre total des klectrons du cccaeur)).
Comme les r$f sont du type L.C.A.O., les inconnues sont les coef- ficients atp.. On les obtient en rksolvant le systbme d’kquations :
C.A.C.
9
oh S p g = x p x g dv, intkgrale de recouvrement entre les orbitales x p et y,,,, s

J~TUDE THBORIQUE DES MOL~CULES CON JUG^. I. 169
est differente de zCro si les fonctions de base sont les orbitales atomiques de Slater.
Les elements de matrice de I’operateur du champ auto-coherent s’expriment en fonction des inconnues atp. C’est pourquoi la mCthode du champ autocoherent est essentiellement iterative.
D’ailleurs, les Cnergies propres des orbitales molkculaires s’obtien- nent grbce au deteiminant skculaire :
C.A.C. d6t I Hpp - E S p p I = 0
La rksolution rigoureuse des equations (15) n’est possible que dans un nombre restreint de cas et, en gCnCra1, lorsque le nombre d’atomes du systbme ne dkpasse pas trois.
Les intdgrales polycentriques (pq/rs) sont, en effet, provisoirement trbs difficiles B calculer.
De plus, m&me lorsque le systbme seculaire est soluble, la fonction d’onde totale obtenue n’est pas crcorrelCe>>. En d’autre termes, elle correspond B une probabilite nonnulle de trouver deux electrons de spins contraires dans un m&me petit volume de l’espace. Si I’on veut utiliser la m6thode de Roothaan pour Ctudier les molCcules polyatomiques conju- guCes on se heurte donc aux difficult& suivantes :
1. le calcul des integrales polycentriques; 2. le dkfaut de correlation de la fonction d’onde totale; 3, le calcul rigoureux des elements de matrice Hpp C.A.C.
2. DISCUSSION DE LA MBTHODE PROPOSbE
a) Les intkgrales polycentriques (pqlrs)
Le procede propose par Pariser et Parr (4) et applique par Pople (5)
B la methode du champ auto-coherent permet d’eviter le calcul des intdgrales (pqlrs).
Ces auteurs admettent, en effet, que toutes les integrales non coulom- biennes sont nulles (neglect of differential overlap) ainsi que les integrales
(4) R. PARISER and R.G. PAM, J . Chem. P h p . 21,466,767 (1953). (5) J.A. POPLE, Trans. Furuduy. Soc. 49, 1375 (1953).

170 G . LEROY
de recouvrement. 11s utilisent donc implicitement une base d’orbitales orthogonales mais tous leurs calculs sont effectuts au moyen des orbitales atomiques de Slater.
On peut trouver une justification thCorique satisfaisante aux appro- ximations de Pariser et Parr (6).
D’ailleurs, Gladney a montrt que des calculs auto-cohkrents effec- tubs avec la matrice compltte des iecouvrements fournissaient, dans le cas d’hydrocarbures alternants purement conjuguks, des rCsultats prati- quement identiques B ceux obtenus en ntgligeant le recouvrement diffkrentiel (’).
Nous avons aussi pu montrer que l’emploi explicite d’orbitales orthogonales ne modifie pratiquement pas les Bnergies de transition mais, par contre, affecte les charges Clectroniques et le moment polaire dans le cas des hydrocarbures non alternants (8).
C’est pourqoi nous admettrons les approximations de Pariser et Parr dans 1’Ctude des composks alternants contenant ou non des hCtCroa- tomes.
s’kcriront plus simplement : C.A.C. Dbs lors, les ClCments de matrice H p q
b ) Problzme de la corrPIation klectronique
Pour tenir compte de la corrilation Clectronique on devrait multi- plier la fonction d’onde totale pal une fonction des distances interelec- troniques ; cette fonction de corrdation devrait devenir faible quand la distance entre deux Clectrons de spins contraires est nulle et tendre vers un lorsque les Clectrons sont infiniment distants.
Une telle fonction est impossible B Ctablir dans le cas des systtmes polyClectroniques.
(6) Voir par exemple ref. 2 p. 502 et sv. (7) H.M. GLADNEY, Theoret. Chim. Acfu (Berl.) 1, 245 (1963). (8) G. LEROY, J . Chim. Phys. 60, 1270 (1 963).

BTUDE THBORIQUE DES MOLBCULES CONJUGU~ES. I. 171
D’aprks Julg (0) et Berthier (lo), seules les intkgrales biklectroniques devraient btre affectkes par la correction de corrklation. A cet effet, Julg propose de modifier la charge effective Z des orbitales atomiques et de fixer empiriquement les intkgrales monocentriques (pp/pp) (ll). Ce prockdk fournit pratiquement les mbmes rksultats que celui de Pariser et Parr; ces auteurs calculent les intkgrales bicentriques en utilisant la mkthode des sphtres uniformkment chargkes (uniformly charged sphere model) et fixent aussi empiriquement les intkgrales (pp/pp).
Nous utiliserons le prockdk de Pariser et Parr pour calculer les intk- grales bicentriques puisque, comme ces auteurs, nous n’employons pas explicitement une base d’orbitales orthogonales.
Ainsi pr&entCe, la correction de corrklation est bien imparfaite car c’est I’ensemble du schkma de calcul qui devrait &tre modifii pour tenir compte de cet effet.
C’est pourquoi, il serait nkcessaire d’effectuer une interaction de configurations aussi compltte que possible pour introduire correctement la corrklation klectronique.
Cependant, on peut espkrer que les rksultats obtenus par la mkthode du champ auto-coherent dans le cadre des approximations de Pariser et Parr fourniront une description correcte de l’ktat fondamental des molkcules conjugukes sans qu’il soit nkcessaire d’effectuer une interaction de configuration.
C A.C. c) Calcul des PlPments de matrice Hph
On sait que, si l’on admet I’approximation de Goeppert-Mayer et Sklar (12), les klkments de matrice de l’opkrateur du champ auto-cohkrent peuvent s’kcrire (13) :
(9) A. JULG, J. Chim. Phys. 57, 19 (1960). (‘0) G. BERTHIER, Tetrahedron symposium (1961). (11) A. JULG, J. Chim. Phys. 55, 413 (1958). (12) M. GOEPPERT-MAYER et A. SKLAR, J . Chem. Phys. 6, 645 (1938). (9 Voir rbf. 2 p. 526.

172 G. LEROY
W p reprksente 1’Cnergie associ6e A l’orbitale x P de l’atome p pris dans 1’6tat de valence approprit. D, est le nombre ccd’tlectrons x ) ) fournis par l’atome q A la liaison dClocalisCe. qq est la population tlectronique de l’orbitale X q :
(q : pp) dCsigne I’intCgrale de pCnCtration entre I’orbitale x p et l’atome neutre q :
(23)
uq (1) est l’opdrateur potentiel correspondant A l’interaction coulom- bienne entre 1’Clectron 1 et l’atome q. lPq reprbente l’indice de liaison de la liaison p-q :
(4 : PP) = - XP (1) UP (1) X P (1) do1 s
Et enfin, l’intbgrale &q est dCfinie par la relation :
Pgq = 1 X P HC XP du (25)
Toutes les intdgrales intervenant dans le calcul des Cltments de matrice Hpq sont Ccrites dans la base des orbitales atomiques de Slater.
Ces orbitales s’expriment en fonction d’une charge nuclkaire effective (Z - o); o Ctant une constante d’Ccran que l’on calcule aiskment giice aux rtgles de Slater (14).
Cette constante d’dcran, dCfinie pour une orbitale d’un type donnt, est en fait une fonction de la population Clectronique de cette orbitale.
DBs lors, comme les populations Clectroniques en ((Clectrons TCN
Cvoluent au cours du processus itbratif de la mdthode du champ auto- cohCrent, les diffkrentes intCgrales intervenant dans les Cltments de matrice H p b ne peuvent Qtre considCrCes comme des donnCes constan- tes. Au contraire, A chaque iteration, elles devront &re corrigtes ccpour effet de chargen.
Cependant la mCthode que nous adoptons pour tvaluer p;q et les integrales de pCnktration ne nous permet pas de corriger ces grandeurs pour tenir compte de cet effet.
C.A.C.
C A.C.
(14) J.C. SLATER, Phys. Rev. 36, 57 (1930).

~ U D E TH~ORIQUE DES MOLBCULJS C O N J U G U ~ . I. 173
Nous dCcrivons plus loin le procCd6 semi-empirique permettant d’obtenir W,, les inttgrales coulombiennes monocentriques et les intQ grales de @&ration (voir en d). D’ailleurs comme nous l’avons dCj8 signale, les intkgrales (pp/qq) seront calculkes par la mCthode de Pariser et Parr. Comme elles s’expriment en fonction de la charge effective (Z - u), c’est-&dire Zslster, la correction d’effet de charge ne pose aucun problkme.
Quant 8 I’intCgrale pip, elle sera calculCe par le procCdC suivant : Poui les liaisons carbone-carbone, nous admettons que p k s’obtient
au moyen de l’exponentielle :
(27) pEC = - 63,95 e-2~33 Rcc(*)
oh Rcc est la longueur de la liaison considbrbe. Enfin, les intbgrales p&, relatives aux liaisons hCtCroatomiques,
seront calculCes par une formule qui s’inspire de celle de Mulliken (8) :
p”,= p,“, s p q Ipq lScc Icc (28)
oh p q L * le potentiel d’ionisation moyen des atomes p et q et S p q est I’intCgrale -ecouvrement entre les orbitales 2px des atomes p et q.
d) Evaluation semi-empirique des int&rales
1. W , e t les intkgrales coulombiennes monocent r iques
Dans un prCcCdent travail, nous avons dCcrit un procCdC semi-empi- rique pour Cvaluer W, et (pplpp) (‘3). Nous reprenons ici la mtthode proposde dans ce travail en l’appliquant explicitement aux atomes de carbone, azote, oxygkne et soufre dans diffbrents Ctats de valence et en effectuant de facon plus systematique la correction d’effet de charge dont nous avons dCj8 soulignt I’importance.
La charge effective d’une orbitale peut s’exprimer en fonction de la population Clectronique de l’orbitale par la relation :
z;: = z, [1 + EP (DP - q,)l (29)
oh E, est une constante fournie par les rkgles de Slate1 (14) et Z, est le Z de Slater de l’orbitale envisagde. Nous admettons que W, et (pp/pp) sont directement proportioiinels A la charge effective Z: :
W, = mp Z;
(PPIPP) = J P = k z,*

174 G. LEROY
Dbs lors, utilisant les m&mes notatio..s que dans la reference (s), on obtient les rdsultats suivants.
- Carbone trigonal : C(tr.tr.tr.n) E = EC + W(3.25) c (tr.tr.tr.x) E+ 1 EC C+ (tr.rr.ir.) E- = EC + 2 W(2.90) + J(2.g0)
5c
C- (tr.tr.lr.x2) 7e x
E, E+ et E- designent Iespectivement 1’Cnergie de I’atome de carbone trigonal et de qes ions poqitif et negatif.
EC est 1’Cnergie du systbme obtenu en enlevant de l’atome tous les ((electrons x ~ ; elle est supposee constante quand on passe de l’atome neutre A ses ions.
D’ailleurs : Ef - E = - W(3.25) = I
x TC
E - E- = W(3.25) - 2 w(2.90) - J(2.90) = A x 75 x x et
1, et A, dCsignent respectivement le potentiel d’ionisation et I’elec-
D’aprbs Jaffti (ls) : troaffinitC de l’orbitale x .
1% = 11.16ev
et Ax = 0.03ev
11.16 ev = m(3.25) (3.25) - DBs lorS W, _ - me = - 3.43385
et Jg.Qo) = 8.7263 ev = k(2.90)
d’oG k = 3.00907
- Azofe tiogonal N (tr2tr tr n)
E = EC + W(3.90) x N (tr2.tr.tr.x) E+ = EC N+ (tr2.tr.w.)
I, = 14.12 ev A, = 1.78 ev (15)
E- = EC + 2 W(3.55) x + J(3.55) x N- ( t r 2 tr tr x2)
(I5) J. HINZE and H.H. JAFFB. J . Am. Chem. Soc. 84, 540 (1962).

BTUDE THJ~ORIQUE DES MOLBCULES CONJUGU~ES. I. 175
Donc mN = - 3.62051
k = 2.76215
- Azote pyrrolique N (spp x 2 )
E = EC + 2 ~ ( 3 . 9 0 ) + ~(3.90) N+ (SPP 4
E++ = EC N++ (SPP)
N (spp x x E+ = EC + W(4.25)
x
I1 = 11.95 12 = 29.16 (Ie)
W(4-25) = m' (4.25) = - 29.16 x N
m' = - 6.86118
Donc
N
k = 3.18133
- Oxygkne trigonal 0 (trZtr2tr TC)
Donc mo = - 3.89011
k = 2.97784
- Oxygkne furannique 0 (s2 pp x2)
(I6) H.A. SKINNER and H.O. PRITCHARD, Trans. Furuduy Soc. 49, 1254 (1953).

G. LEROY 176
Donc W:*’O) = mA(4.90) = - 34.16
mi = - 6.97143
k = 3.02476
L’expression explicite de W, dans les deux etats de valence oh l’orbitale x est utilisee respectivement une et deux fois permet de com- prendre la diffkrence entre mN et rnh et mo et m6 (8).
Si l’on admet que, pour un m&me type d’orbitale, Jx ne depend que de la charge effective Z*, on peut adopter pour la constante k, la valeur moyenne des valeurs obtenues pour les atomes ci-dessus.
Dks lors pour une orbitale 2px :
Jx = L Z* oh L = 3.02476
Dans le cas du soufre, oh l’orbitale x appartient A la couche 3, on obtient les resultats suivants :
Donc ms = - 2.29364
k = 1.6076
- Soufre thiophdnique S (s2 pp x2)

~ U D E T ~ R I Q U E DES MOL~CULES CONJUGU~S. I. 177
W(5.80) = mi 5.80 = - 22.88
mg = - 3.94483
x D’oh
k = 1.6785
Donc pour l’orbitale 3pn du soufre : Jn = k Z* oh & = 1.64178
2. Les int tgrales de p t n t t r a t i o n
Une intkgrale de pknttration :
(P : qqhPu = - 1 X g (1) UZJ (1) Xdl) do1
dtpend essentiellement de la nature de l’atome p, de l’orbitale x , et de la distance R,, p.
Si p et q sont des atomes du m&me type (des carbones par exemple), lorsque la distance RpP est nulle, on peut admettre que l’inttgrale de ptnttration correspondante reprksente une tlectroaffinitt d’orbitale (4).
D’ailleurs, pour un m&me atome p, nous admettrons que (p : qq) est proportionnel A la charge affective ZQ de l’orbitale x g .
D’aprks Pariser et Parr (*) si Rpq = 2.80 A, (p : qq)R,, = 0. Dks lors, si I’on admet pour (p : qq), la forme exponentielle :
(p : qq)R,,, = A p g e-BpuRpa, (32)
il est aist de calculer A et B pour n’importe quel couple d’atomes et n’importe quelle distance.
Cependant, pour rester hornogkne avec la mkthode de Pariser et Parr, on peut adopter l’expression :
(P : qq)Rpq = a p q R$P -k ~ P P Rpg -k CPP (33)
Les constantes a, b et c s’obtiennent A partir des valeurs de (p : qq), A l’origine (Rpq = 0) et aux distances 2,80 et 3’70 A :
pour R p g = 0 , (p : qq) = A, et pour R,, = 2.80 et 3.70& (p : qq) = 0
On peut alors calculer les inttgrales de pdnttration pour tous les couples possibles obtenus A partir des atomes de carbone, azote, oxygkne et soufre.
178 G. LEROY
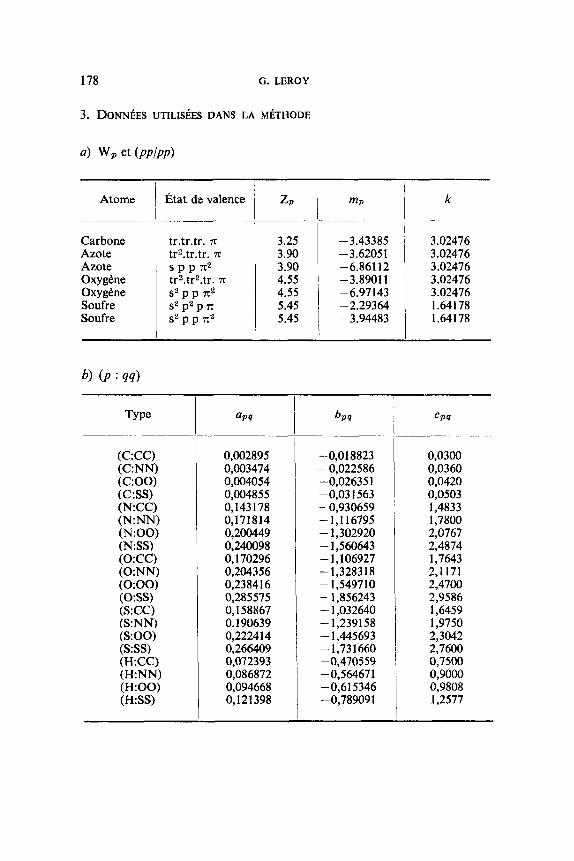
3. DONNBES UTILISfiES DANS LA MBTHODE
4 w, et (PPlPP)
Atome
Carbone Azote Azote O x y gene Oxy gene Soufre Soufre
I z p
Etat de valence
tr.tr.tr. x tr2.tr.tr. x S P P X 2 tr2.tr2.tr. x s2 p p x*
s2 p p s2 P2 P ?
5 P e
(C:CC) (C:") (C:OO) (C:SS) (N.CC) (N:NN) (N:OO) (N:SS) (0:CC) (0:") (0:OO) (0:SS) (S:CC) (S:NN) (S:OO) (S:SS) (H.CC) (H:NN) (H:OO) (H:SS)
3.25 3.90 3.90 4.55 4.55 5.45 5.45
0,002895 0,003474 0,004054 0,004855 0,143178 0,17 1 8 14 0,200449 0,240098 0,170296 0,204356 0,238416 0,285575 0,158867 0.190639 0,222414 0,266409 0,072393 0,086872 0,094668 0,121 398
-3.43385 -3.62051 -6.861 12 -3.8901 1 -6.97143 -2.29364 -3.94483
-0,018823 - 0,022586 -0,02635 1 -0,03 1563 -0,930659 -1,116795 - 1,302920 - 1,560643 - 1,106927 - 1,32831 8 -1,549710 - 1,856243 - 1,032640 - 1,239158 - 1,445693 - 1,73 1660 - 0,4705 59 -0,564671 -0,615346 -0,789091
k
3.02476 3.02476 3.02476 3.02476 3.02476 1.64178 1.64178
CP 4
0,0300 0,0360 0,0420 0,0503 1,4833 1,7800 2,0767 2,4874 1,7643 2,1171 2,4700 2,9586 1,6459 1,9750 2,3042 2,7600 0,7500 0,9000 0,9808 1,2577

BTUDE TH~ORIQUE DES MOLBCULES CONJUGU~ES. I. 179
C.A.C. 4. PROGRAMME DE CALCUL DES BLBMENTS DE MATRICE H p q
Nous donnons ici le programme FORTRAN h i t pour un ordinateur I.B.M. 1620 et permettant d’obtenir, en deux etapes, les elements de matrice de l’optrateur du champ auto-coherent pour toute molecule conjuguee dont le squelette ne comporte pas plus de 10 atomes.
H(P, Q)l Correction d’effet de charge 10/9/63 Dimension Z(lO), E(10), D(10), WUO), A(10), B(10), R(10), S(10) Dimension FJ(lO), C(10, lo), FL (10, lo), ZC (lo), FJC(10) Accept 16, NT, NTS
16 Format (12, 12) DO 1 K = l , NT
1 Accept Tape 2,Z(K), E(K), D(K), W W , A(Q, B W , R(K), S(K) FJ(K)
2 Format (F6.3, F6.3, F6.3, F6.3, F6.3, F6.3, F6.3, F6.3, F6.3) DO 5 K = l , NT DO 5 L = l , NT
5 Accept Tape 6, C(K, L) 6 Format (E 14.8)
DO 7 I = l , NT DO 7 J=1, NT FL (I, J)= 0. DO 8 K = l , NTS
8 Fl(1, J) = FL(1, J)+2.*C(K, I)* C(K, J) Print 20, I, J, FL(1, J)
20 Format (13, 13, 3XF9. 5 ) 7 Punch Tape 9, FL (I, J) 9 Format (E14.8)
Pause
(K, K))
DO 10 K = l , NT PAR=l. +E(K)*(D(K)-FL
10 ZC(K)=Z(K)*PAR DO 12 K = l , NT PAR = 1. +E(K)*(D(K)-FL(K,K))
12 FJC(K)=FJ(K)*PAR DO 13 K = l , NT W(K) =A(K)*(ZC(K))**2 +(B(K) *R(K) +S(K))*ZC(K)
13 Punch Tape 9, W(K) Pause DO 3 K = l , NT DO 3 L=K, NT Accept tape 4, C(K, L)
4 Format (F6.3)
3 C(L, K)=C(K, L) DO 11 I=1, NT DO 11 J=1, NT
DEUX= 1 ./ZC(J)
UPD =(UN +DEUX)**2
UN = 1 ./ZC(I)
UMD=(UN-DEUX)**2
IF(C(1, 5)-2.80)14, 14, 15 14 Alpha = 1 ./SQRT (1. + .67387*
UMDI Beta =‘l ./SQRT (1. + .67387* UPD) Gama = 1 ./SQRT( 1. + .38591* UMD) Delta= 1 ./SQRT( 1. + .38591* UPD) F=.5*(FJC(I) +FJC(J) F1 =F-2.57054*(Alpha +Beta) F2 =F-l.94527*(Gama +Delta) X=(13.69*F1-7.84*F2)/9.324 Y =(2.8*F2-3.7*F1)/9.324 PQ =F-X*C(I,J)-Y*C(I,J)**2 GO TO 21
Alpha=l./SQRT (1. +PR*UMD) Beta= 1 ./SQRT (1. +PR*UPD) PQ =(7.1975/C(I,J))*(Alpha + Beta) GO TO 21
15 PR =(2.2985/C(I,J))**2
21 Print 20, I, J, PQ 11 Punch Tape 9, PQ
stop End
H(P,Q)2 Correction d’effet de charge 10/9/63 Dimension D(10), P(10), WC(lO), FL(l0, 10) Dimension PQ (10, lo), B (10, lo), H(10, 10) Accept 1, NT
1 Format (12)
2 Accept Tape 3, D(K), P(K) 3 Format (F6. 3, F9. 5 )
DO 2 K = 1, NT

180 G. LEROY
DO 14 K=l , NT 14 Accept Tape 15, WC(K) 15 Format (E14.8)
DO 4 K = l , NT DO 4 L=K, NT Accept Tape 5, FL (K, L)
4 FL (L, K)=FL (K, L) 5 Format (E14.8)
DO 19 K=l , NT DO 19 L=K, NT Accept tape 5 , PQ (K, L)
19 PQ (L, K)=PQ(K, L) DO 16 K=l , NT DO 16 L=K, NT Accept Tape 17, B(K, L)
17 Format (F9.5) 16 B(L, K)=B(K, L)
DO 6 I = l , NT DO 6 J=I, NT
IF [I-J)7. 8.7
8 H(I,J)=O. DO 9 K=l , NT IF(K-I)lO, 9, 10
10 H(I,J)=H(I,J)+(FL(K,K)-D(K)) *PQ(I,K)
9 Continue
FL(I,I)*PQ(I,I) GoTo 18
H(1,J) =H(I,J) + WC(1)-P(I) + .5 *
18 Print 12, I, J, H(1, J) 12 Format (13, 13, 3XF9.5) 6 Punch Tape 13, H(1, J)
13 Format (E14.8) STOP END
Je tiens ?i remercier le Dr. G. Germain et Mademoiselle S. Jaspers pour toute l’aide qu’ils m’ont apportke dans la rkdaction du programme.
Je remercie le Fonds National de la Recherche Scientifique pour l’aide matkrielle accordCe au laboratoire et pour le mandat de ChargC de Recherches qu’il m’a octroyk.
UNIVERSITB DE LOWAIN Laboratoire de Chimie Physique
Communiqud d la Socidtd Chimique de Belgique le 4 ddcembre 1963.





























