Embed Size (px)
Citation preview

Le cardiomyocyteet sa pathologie
Professeur Paul FORNES
Laboratoire d’anatomie pathologiqueCHU, hôpital Robert Debré
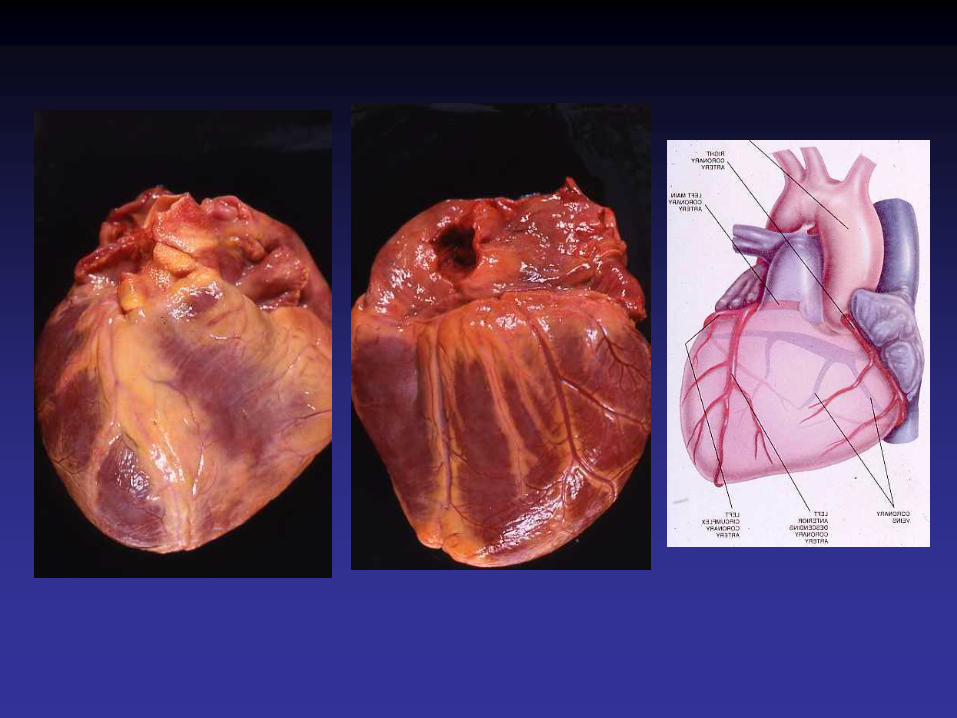
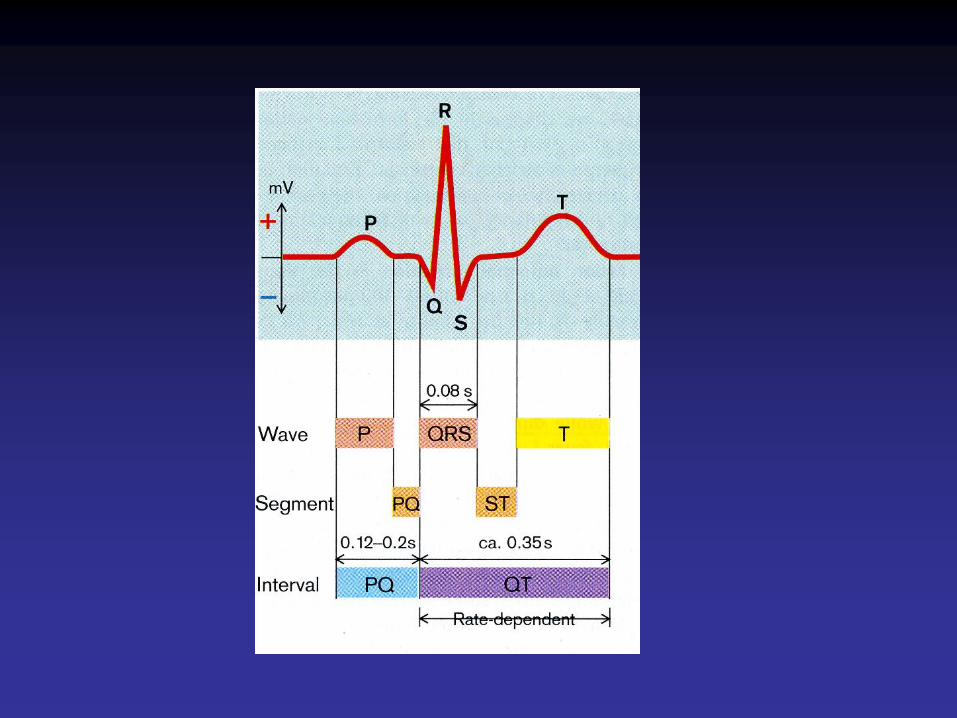
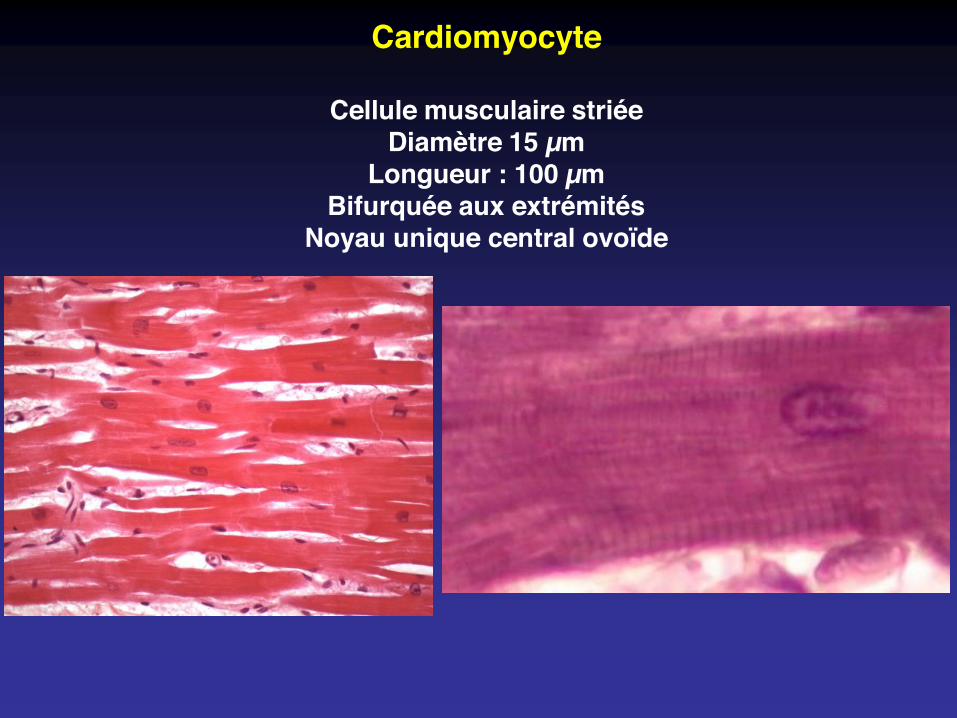
Cardiomyocyte
Cellule musculaire striéeDiamètre 15 µm
Longueur : 100 µmBifurquée aux extrémités
Noyau unique central ovoïde

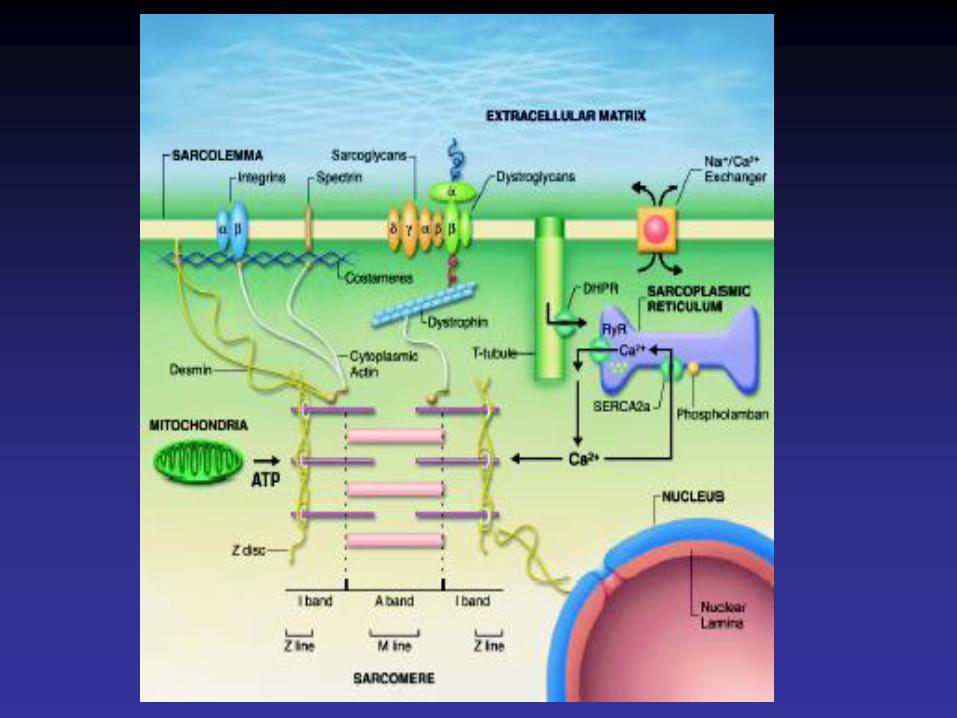
•Sarcolemme•Système des tubules T•Costamères
•Sarcoplasme•Noyau•Myofibrilles•Mitochondries•Golgi•Glycogène, lipides•Réticulum sarcoplasmique

Desmosome Jonction gap
SS
coastamèreslame basale
Jonctions gappasd’HD
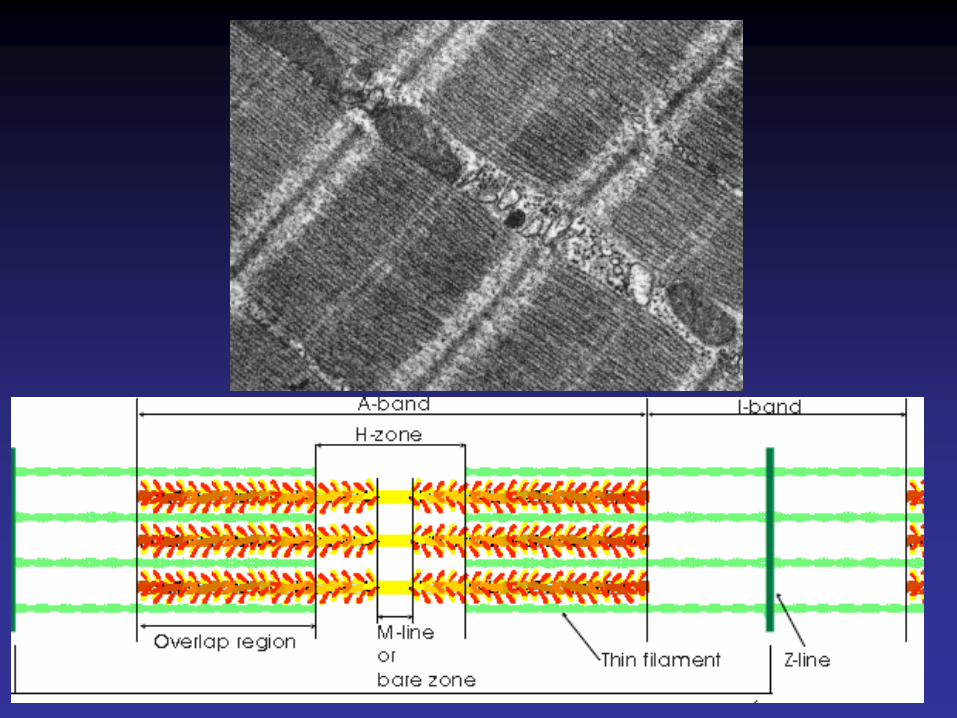
Strie ZBande A(myosine)
Bande I(actine)Sarcomère
desmosomes(desmine)
fascia adherens
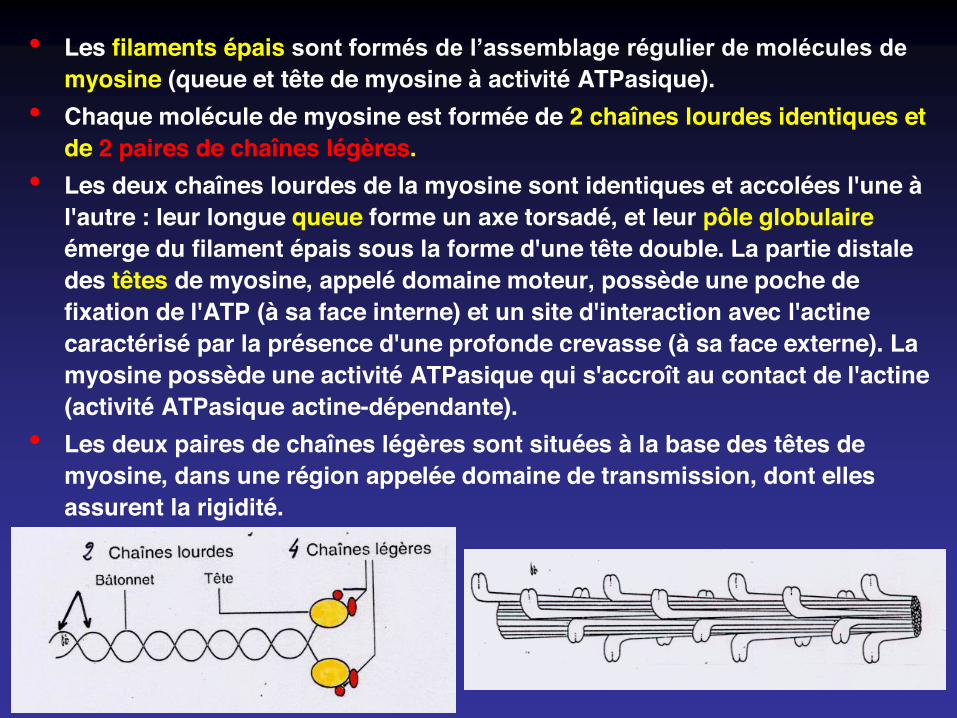
• Les filaments épais sont formés de l’assemblage régulier de molécules de myosine (queue et tête de myosine à activité ATPasique).
• Chaque molécule de myosine est formée de 2 chaînes lourdes identiques et de 2 paires de chaînes légères.
• Les deux chaînes lourdes de la myosine sont identiques et accolées l'une à l'autre : leur longue queue forme un axe torsadé, et leur pôle globulaireémerge du filament épais sous la forme d'une tête double. La partie distale des têtes de myosine, appelé domaine moteur, possède une poche de fixation de l'ATP (à sa face interne) et un site d'interaction avec l'actine caractérisé par la présence d'une profonde crevasse (à sa face externe). La myosine possède une activité ATPasique qui s'accroît au contact de l'actine (activité ATPasique actine-dépendante).
• Les deux paires de chaînes légères sont situées à la base des têtes de myosine, dans une région appelée domaine de transmission, dont elles assurent la rigidité.

• Les myofilaments fins sont composés de polymères d’actine. L'actine est une molécule polypeptidique de forme globulaire. La polymérisation des monomères d'actine se fait sous une forme filamentaire. Les polymères d'actine s'accolent par deux pour former une longue double hélice. Les filaments fins sont formés de l'association de cette double hélice d'actine et de deux protéines régulatrices: la tropomyosine, dimère filamenteux rigide de renforcement, et la troponine, complexe de trois sous-unités polypeptidiques, I, C et T, disposées à intervalles réguliers le long des filaments d'actine, en regard de chaque tête de myosine, et impliquées dans la régulation de la contraction musculaire par le calcium. Chaque extrémité libre des filaments d'actine est coiffée par une molécule de tropomoduline, qui pourrait jouer un rôle dans le maintien et la stabilisation de la longueur finale des filaments fins.

•Les disques M renferment des filaments de myomésine.•Les disques Z sont formés de l’organisation quadratique de filaments d’alpha-actinine servant à relier les extrémités des filaments fins de chaque sarcomère entre elles et avec les extrémités des filaments fins du sarcomère adjacent.
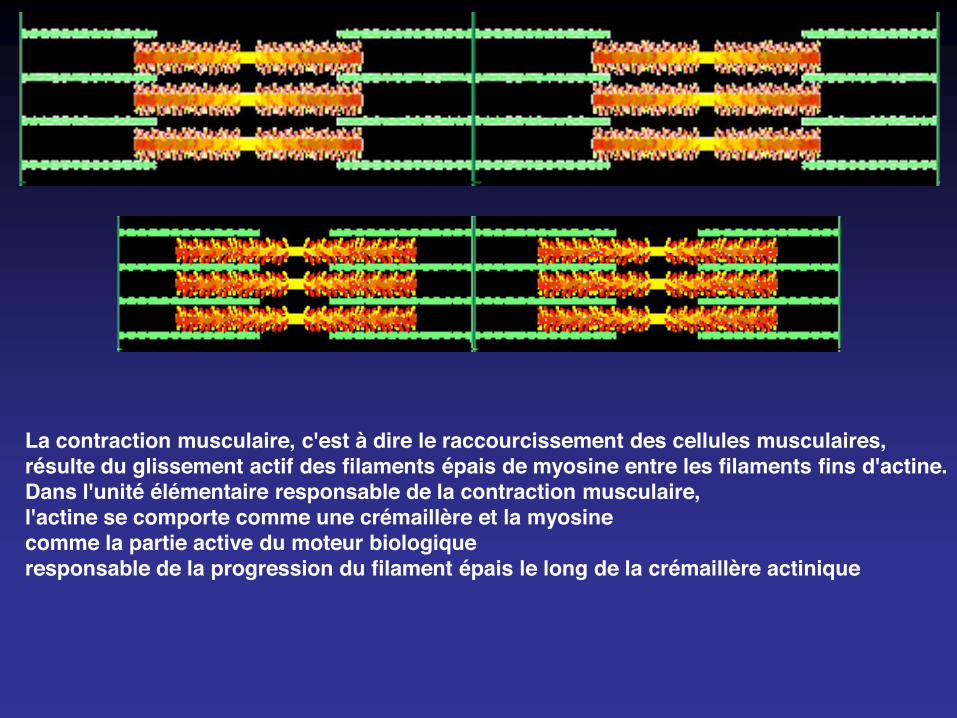
La contraction musculaire, c'est à dire le raccourcissement des cellules musculaires,résulte du glissement actif des filaments épais de myosine entre les filaments fins d'actine.Dans l'unité élémentaire responsable de la contraction musculaire,l'actine se comporte comme une crémaillère et la myosinecomme la partie active du moteur biologiqueresponsable de la progression du filament épais le long de la crémaillère actinique

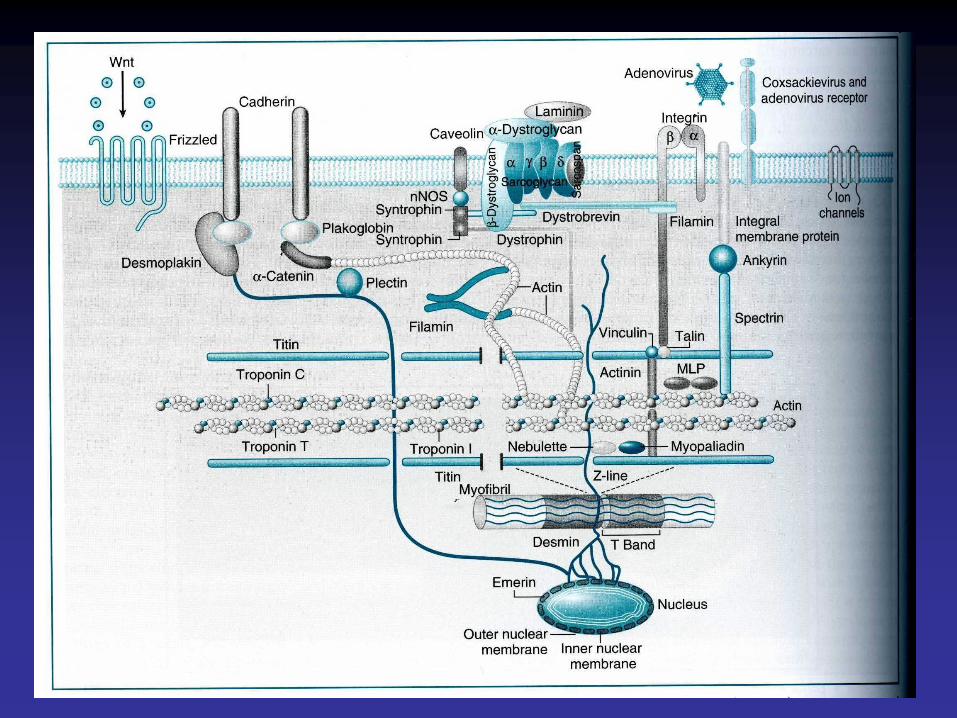
Cytosquelette
• Le cytosquelette endosarcomérique, est représenté par la titine. La titine est une protéine qui, dans chaque demi-sarcomère, relie chaque filament épais à la strie Z. Composant élastique, elle maintient l'alignement des filaments épais et oppose une résistance à l'étirement excessif du sarcomère.
• Le cytosquelette exosarcomérique comprend des microtubules et des filaments intermédiaires de desmine.

• Le cytosquelette sous-sarcolemmique est représenté par le complexe dystrophine-protéines associées à la dystrophine qui sert à amarrer l'appareil contractile à la matrice extracellulaire.
• La dystrophine est une protéine sarcoplasmique située sous la membrane plasmique. Sa distribution n'est pas uniforme le long de la face interne du sarcolemme, mais se fait sous la forme d'arcs (ou costamères) dont les plus marqués sont situés en regard des disques Z des myofibrilles.
• L'accrochage de la dystrophine à la membrane plasmique du myocyte se fait par l'intermédiaire d'un complexe de glycoprotéines contenant des protéines intracytoplasmiques(les syntrophines), des protéines transmembranaires: le complexe des sarcoglycanes et du dystroglycane et d'une protéine extra-cellulaire (dystroglycane).

Alpha-actininesTéléthonine (titin-cap)MLP (muscle LIM protein)TitineMyotilineFilaments intermédiaires
DesmineLaminesEmérine

SarcolemmeDystrophine
DystrobrévinesarcoglycanesDysferlineLaminine 2
Costamèresalpha-actininesActine non sarcomériqueFibronectine
VinculineMétavinculineIntégrine
Spectrine/anchorineDesmine
Cavéoline 3
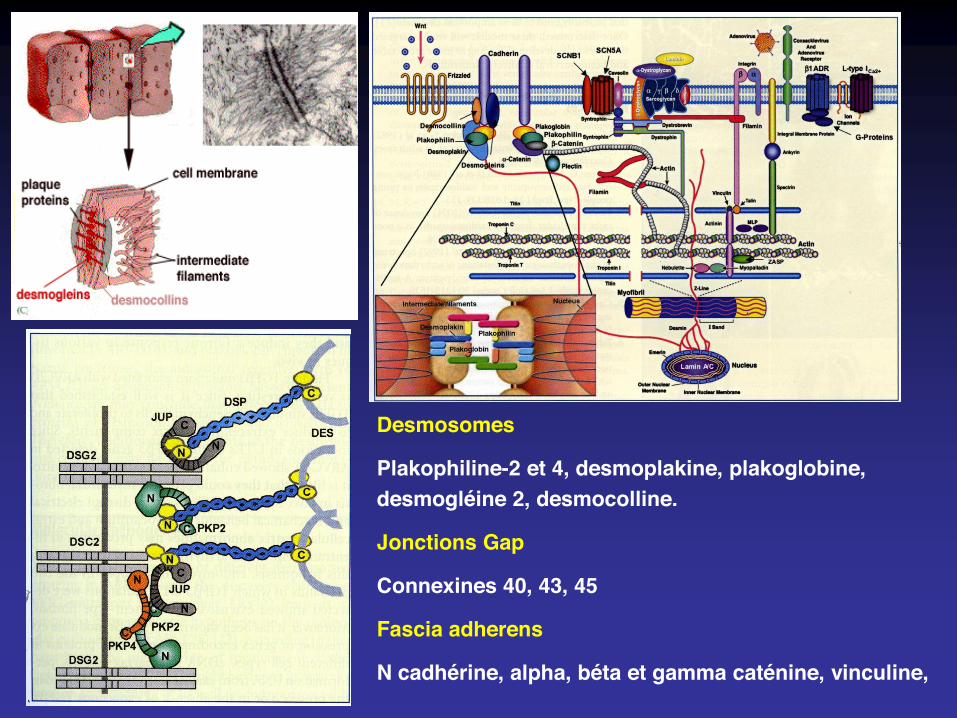
Desmosomes
Plakophiline-2 et 4, desmoplakine, plakoglobine, desmogléine 2, desmocolline.
Jonctions Gap
Connexines 40, 43, 45
Fascia adherens
N cadhérine, alpha, béta et gamma caténine, vinculine,
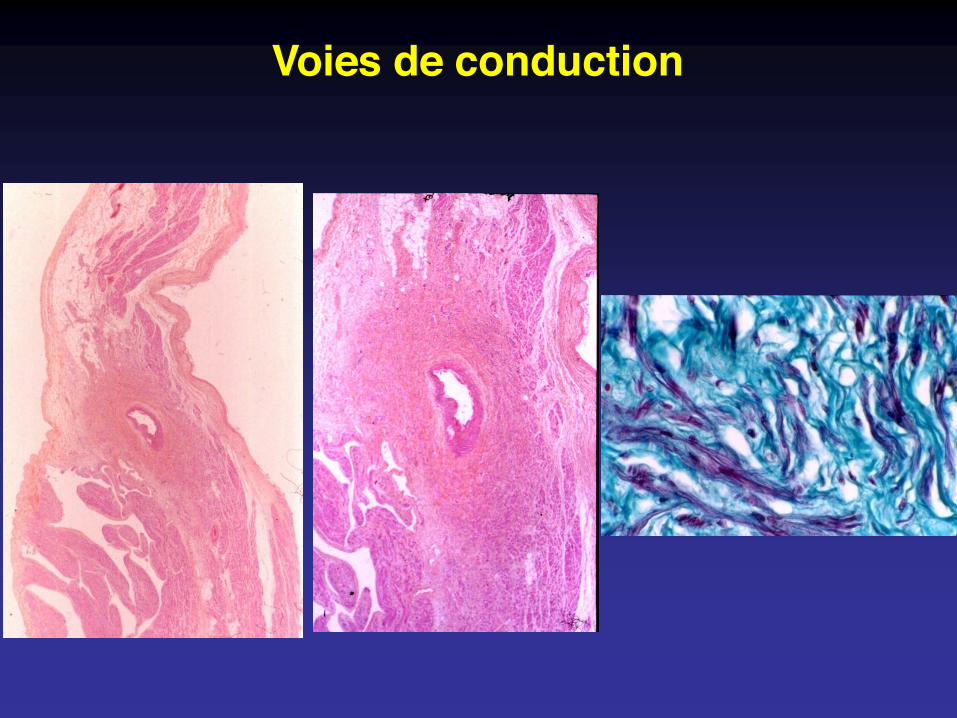
Voies de conduction
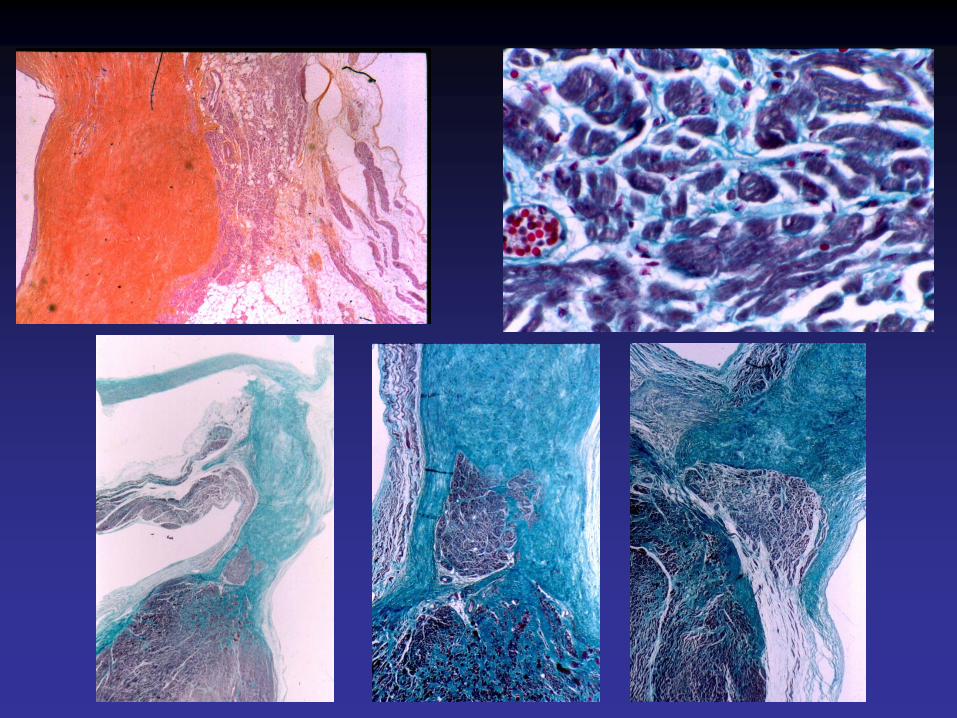

Cardiomyopathies

Définitions• OMS 1995 : maladies du myocarde s’accompagnant d’insuffisance cardiaque. CM hypertrophiques, dilatées, restrictives, ventriculaire droite arythmogène• AHA 2006Maron BJ et al. Circulation 2006;113:1807-16« A heterogeneous group of diseases of the myocardium associated with mechanical and/or electrical dysfunction that usually (but not invariably) exibit inappropriate ventricular hypertrophy or dilatation and are due to a widely variety of causes that frequently are genetic. Cardiomyopathies either are confined to the heart or are part of generalized systemic disorders often leading to cardiovascular death or progressive heart failure-related disability. »
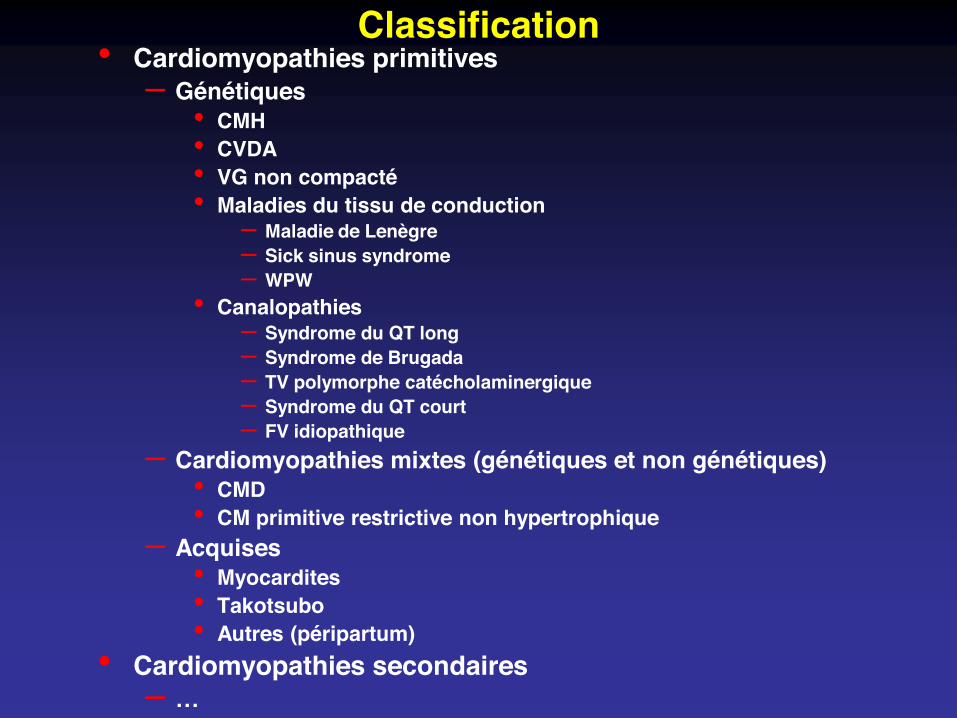
Classification• Cardiomyopathies primitives
– Génétiques• CMH• CVDA• VG non compacté• Maladies du tissu de conduction
– Maladie de Lenègre– Sick sinus syndrome– WPW
• Canalopathies– Syndrome du QT long– Syndrome de Brugada– TV polymorphe catécholaminergique– Syndrome du QT court– FV idiopathique
– Cardiomyopathies mixtes (génétiques et non génétiques)• CMD• CM primitive restrictive non hypertrophique
– Acquises• Myocardites• Takotsubo• Autres (péripartum)
• Cardiomyopathies secondaires– …
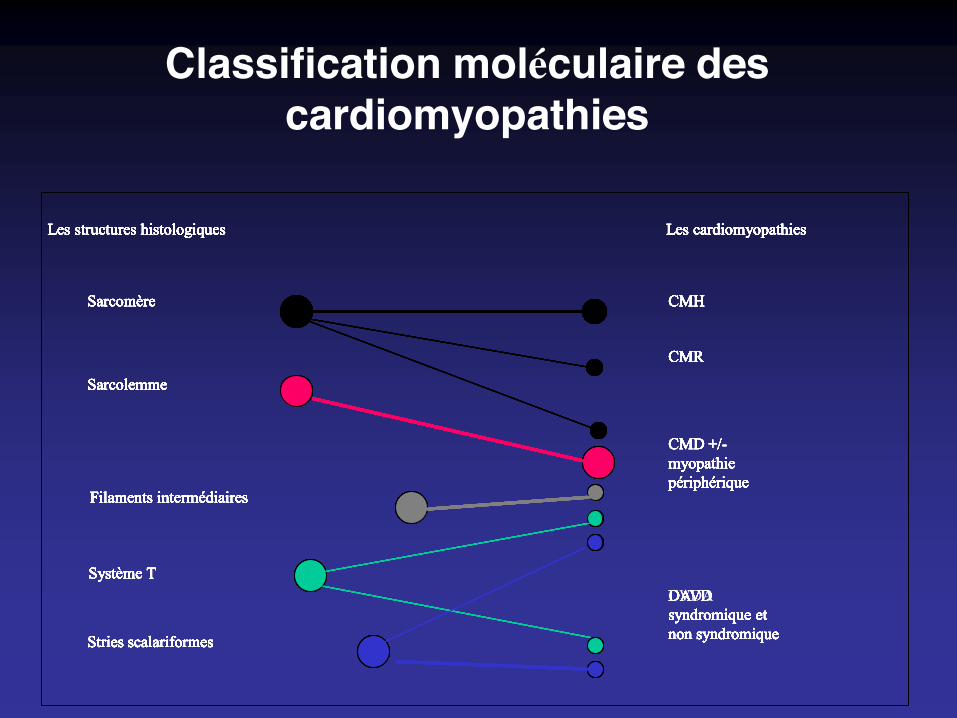
Classification moléculaire des cardiomyopathies
Les structures histologiques Les cardiomyopathies
Sarcomère
Sarcolemme
Système T
Stries scalariformes
Filaments intermédiaires
CMH
CMR
CMD +/-myopathiepériphérique
DAVDsyndromique etnon syndromique
Les structures histologiques Les cardiomyopathies
Sarcomère
Sarcolemme
Système T
Stries scalariformes
Filaments intermédiaires
CMH
CMR
CMD +/-myopathiepériphérique
DAVDsyndromique etnon syndromique
Les structures histologiques Les cardiomyopathies
Sarcomère
Sarcolemme
Système T
Stries scalariformes
Filaments intermédiaires
CMH
CMR
CMD +/-myopathiepériphérique
DAVDsyndromique etnon syndromique
Les structures histologiques Les cardiomyopathies
Sarcomère
Sarcolemme
Système T
Stries scalariformes
Filaments intermédiaires
CMH
CMR
CMD +/-myopathiepériphérique
DAVDsyndromique etnon syndromique
Les structures histologiques Les cardiomyopathies
Sarcomère
Sarcolemme
Système T
Stries scalariformes
Filaments intermédiaires
CMH
CMR
CMD +/-myopathiepériphérique
DAVDsyndromique etnon syndromique
Les structures histologiques Les cardiomyopathies
Sarcomère
Sarcolemme
Système T
Stries scalariformes
Filaments intermédiaires
CMH
CMR
CMD +/-myopathiepériphérique
DAVDsyndromique etnon syndromique
Les structures histologiques Les cardiomyopathies
Sarcomère
Sarcolemme
Système T
Stries scalariformes
Filaments intermédiaires
CMH
CMR
CMD +/-myopathiepériphérique
DAVDsyndromique etnon syndromique
Les structures histologiques Les cardiomyopathies
Sarcomère
Sarcolemme
Système T
Stries scalariformes
Filaments intermédiaires
CMH
CMR
CMD +/-myopathiepériphérique
DAVDsyndromique etnon syndromique
Les structures histologiques Les cardiomyopathies
Sarcomère
Sarcolemme
Système T
Stries scalariformes
Filaments intermédiaires
CMH
CMR
CMD +/-myopathiepériphérique
DAVDsyndromique etnon syndromique
Les structures histologiques Les cardiomyopathies
Sarcomère
Sarcolemme
Système T
Stries scalariformes
Filaments intermédiaires
CMH
CMR
CMD +/-myopathiepériphérique
CVDAsyndromique etnon syndromique

Cardiomyopathie hypertrophique

Poids du cœur normalHypertrophie physiologique
Cardiomyopathie hypertrophique
• Le poids du cœur doit être interprété en tenant compte du poids corporel.– Kitzman et al. Mayo Clinic Proc 1988;63:137-46
• L’hypertrophie cardiaque est un phénomène physiologique d’adaptation en réponse à des contraintes.
• Paradoxalement, l’HVG est un facteur de risque de mort subite, quelle qu’en soit la cause.
• L’hypertrophie cardiaque devient un substrat anatomique arythmogène
• La fibrose est le substrat histologique arythmogène

Cardiomyopathie hypertrophique primitive• Prévalence de 1 sur 500• Dix gènes sarcomériques ; > 300 mutations
– Chaîne lourde ß de la myosine– Protéine C de liaison à la myosine– Troponines T, C, I– α actine– Chaînes légères– α tropomyosine– Titine– Chaîne lourde α
• Rendement des analyses génétiques chez un cas index : 70 % environ
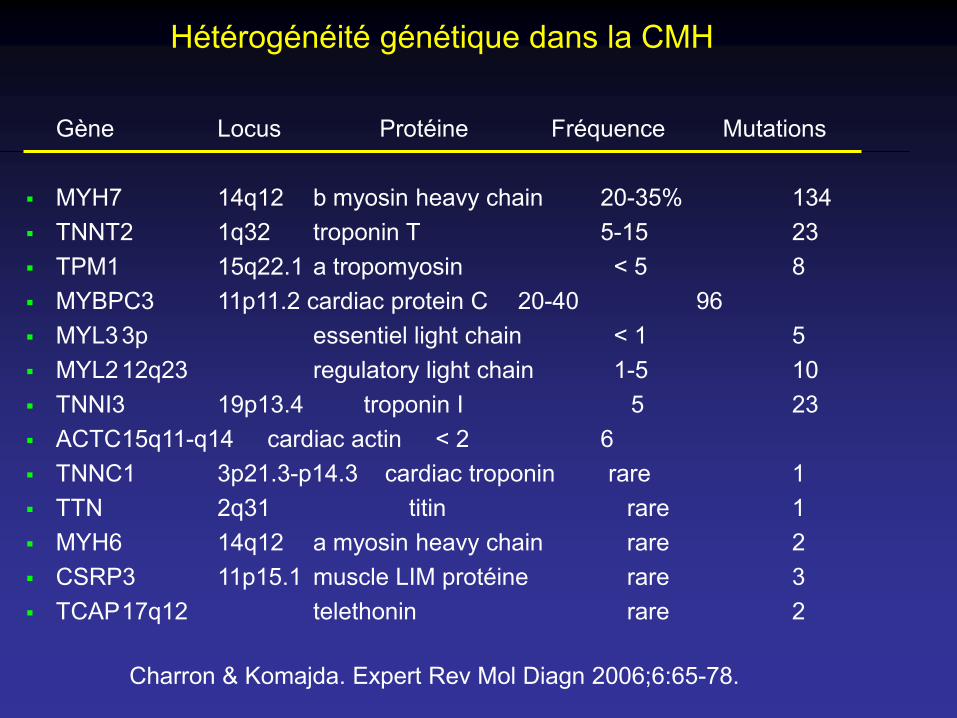
Hétérogénéité génétique dans la CMH
Gène Locus Protéine Fréquence Mutations
MYH7 14q12 b myosin heavy chain 20-35% 134 TNNT2 1q32 troponin T 5-15 23 TPM1 15q22.1 a tropomyosin < 5 8 MYBPC3 11p11.2 cardiac protein C 20-40 96 MYL33p essentiel light chain < 1 5 MYL212q23 regulatory light chain 1-5 10 TNNI3 19p13.4 troponin I 5 23 ACTC15q11-q14 cardiac actin < 2 6 TNNC1 3p21.3-p14.3 cardiac troponin rare 1 TTN 2q31 titin rare 1 MYH6 14q12 a myosin heavy chain rare 2 CSRP3 11p15.1 muscle LIM protéine rare 3 TCAP17q12 telethonin rare 2
Charron & Komajda. Expert Rev Mol Diagn 2006;6:65-78.
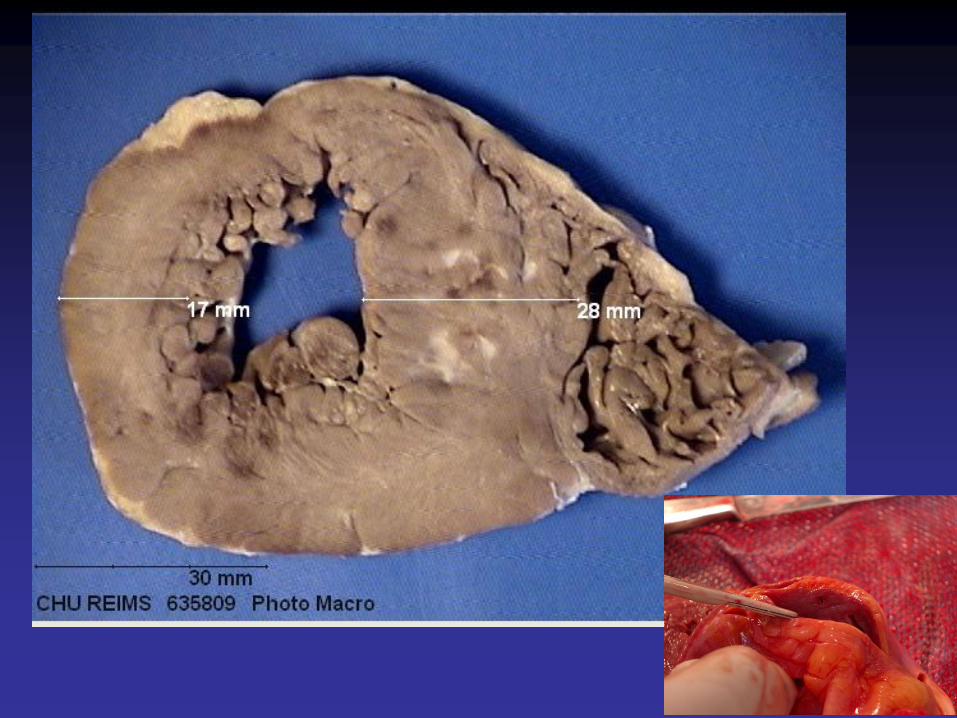
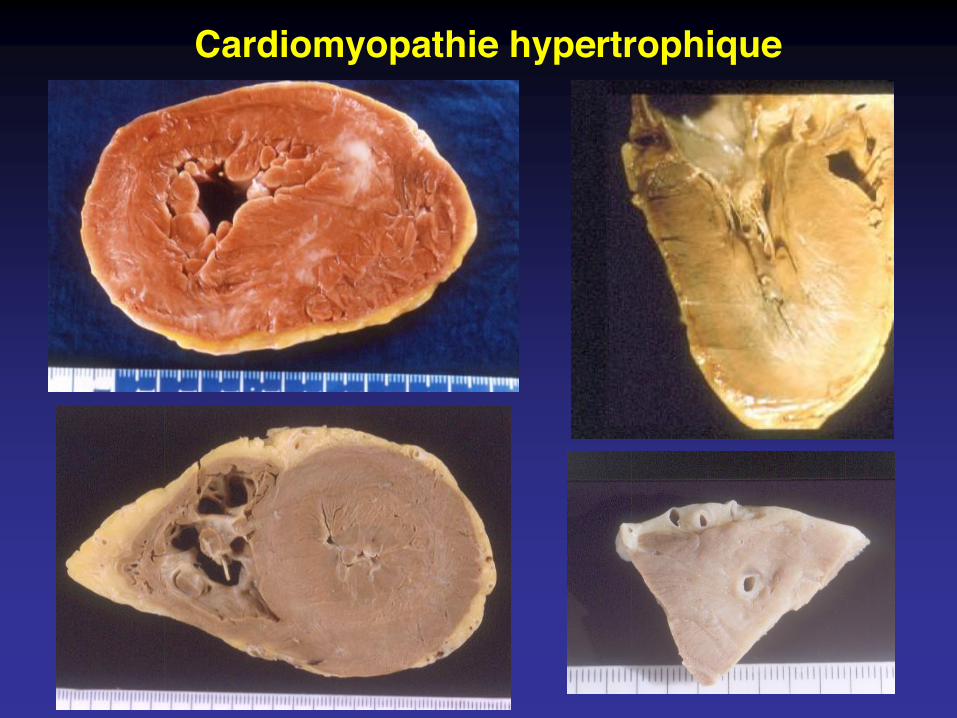
Cardiomyopathie hypertrophique
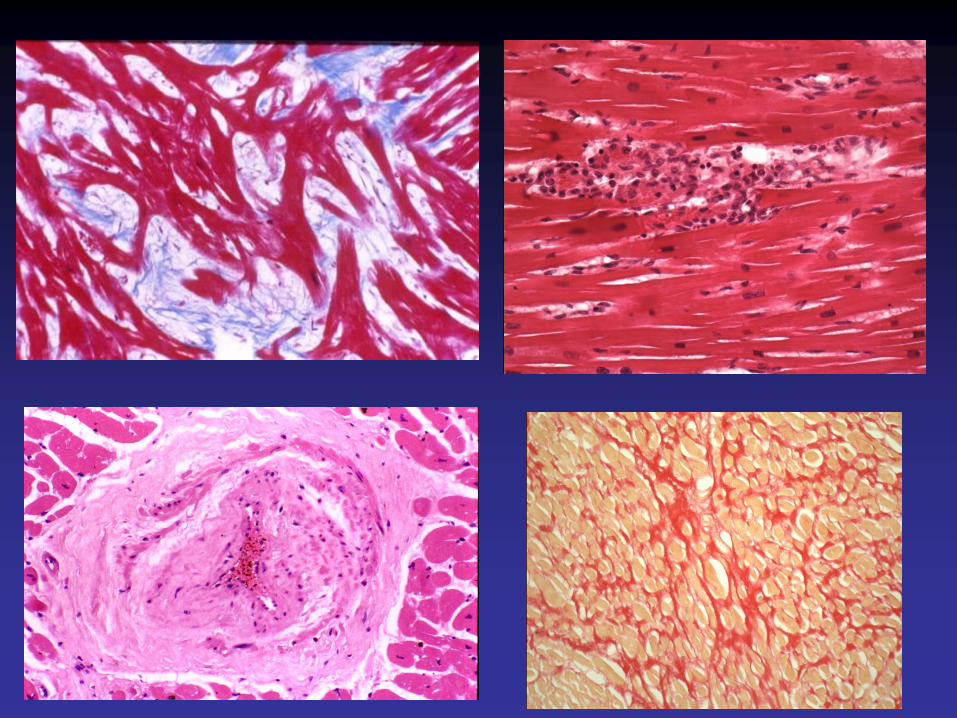
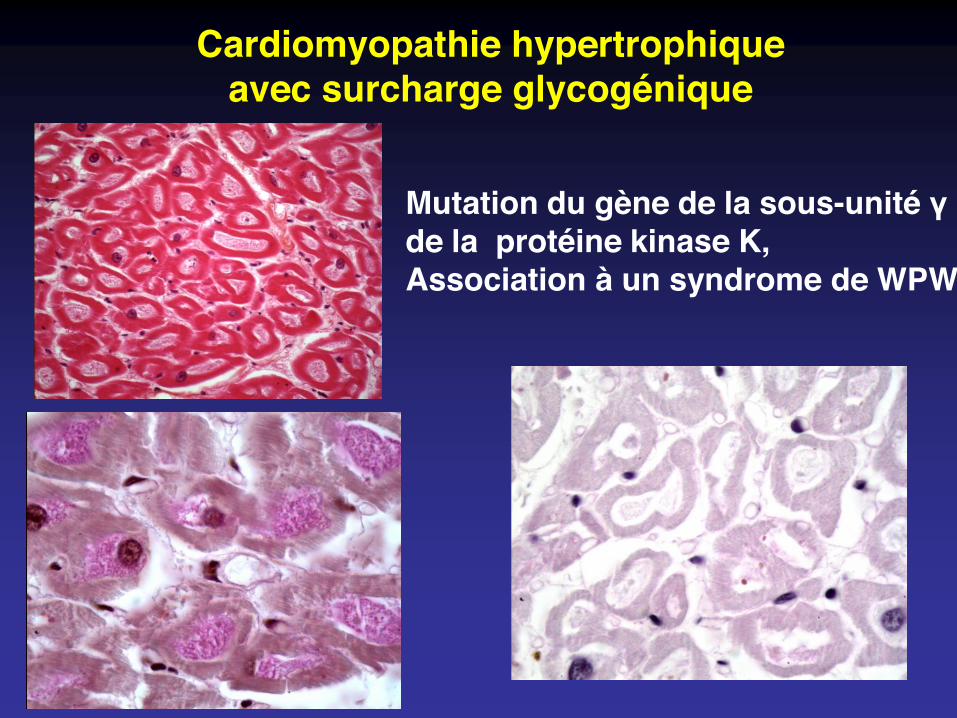
Cardiomyopathie hypertrophiqueavec surcharge glycogénique
Mutation du gène de la sous-unité γde la protéine kinase K,Association à un syndrome de WPW

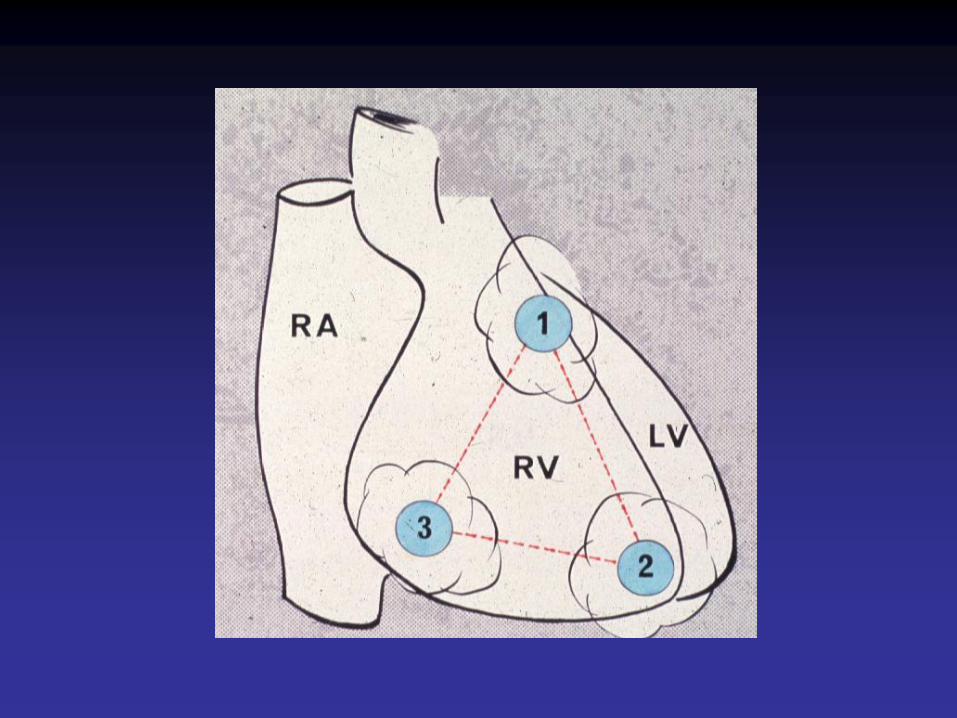
Définition• Cardiomyopathie caractérisée par le remplacement du myocarde ventriculaire
droit par du tissu adipeux et de la fibrose.
• Prévalence de 1 / 5000• 40 % de formes familiales• Diagnostic cardiologique très difficile, s’appuyant sur des critères majeurs et
mineurs : antécédents personnels et familiaux, ECG, imagerie• Diagnostic anatomo-pathologique également très difficile en raison de
frontières mal délimitées entre ventricule droit normal, remplacement adipeux pur et cardiomyopathie ventriculaire droite arythmogène
1736, …1961,…1982,…1988,…2001-8
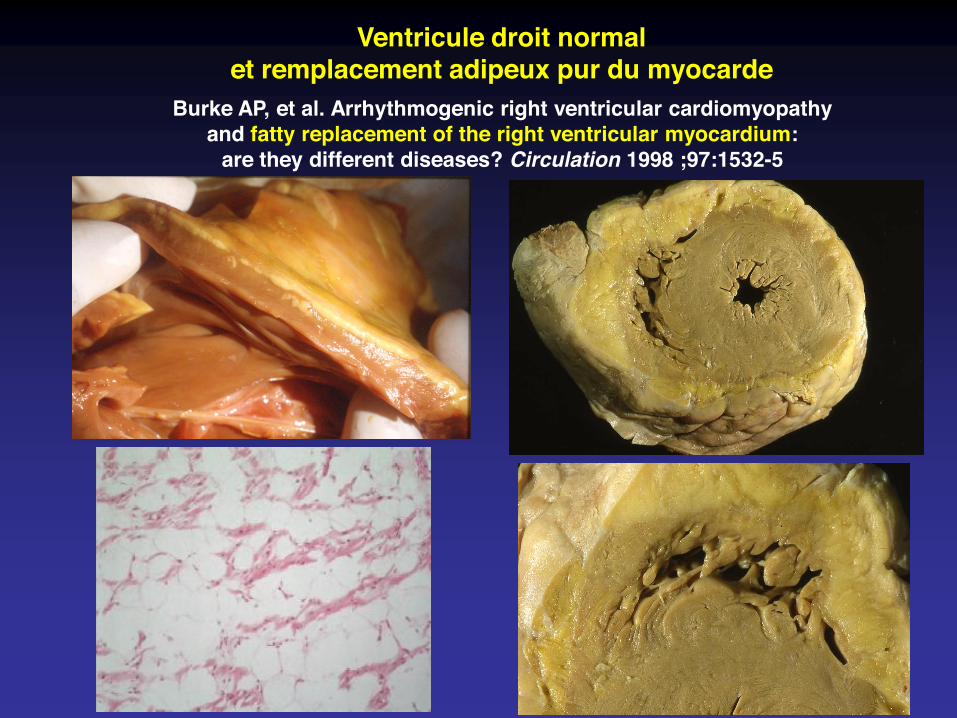
Ventricule droit normalet remplacement adipeux pur du myocarde
Burke AP, et al. Arrhythmogenic right ventricular cardiomyopathyand fatty replacement of the right ventricular myocardium:
are they different diseases? Circulation 1998 ;97:1532-5
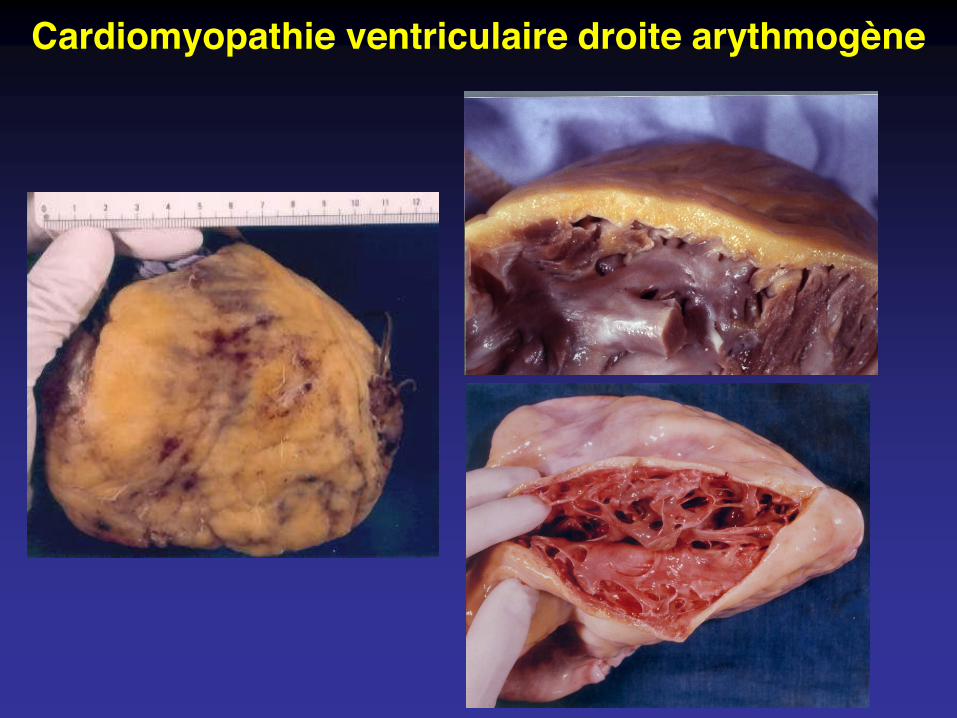
Cardiomyopathie ventriculaire droite arythmogène
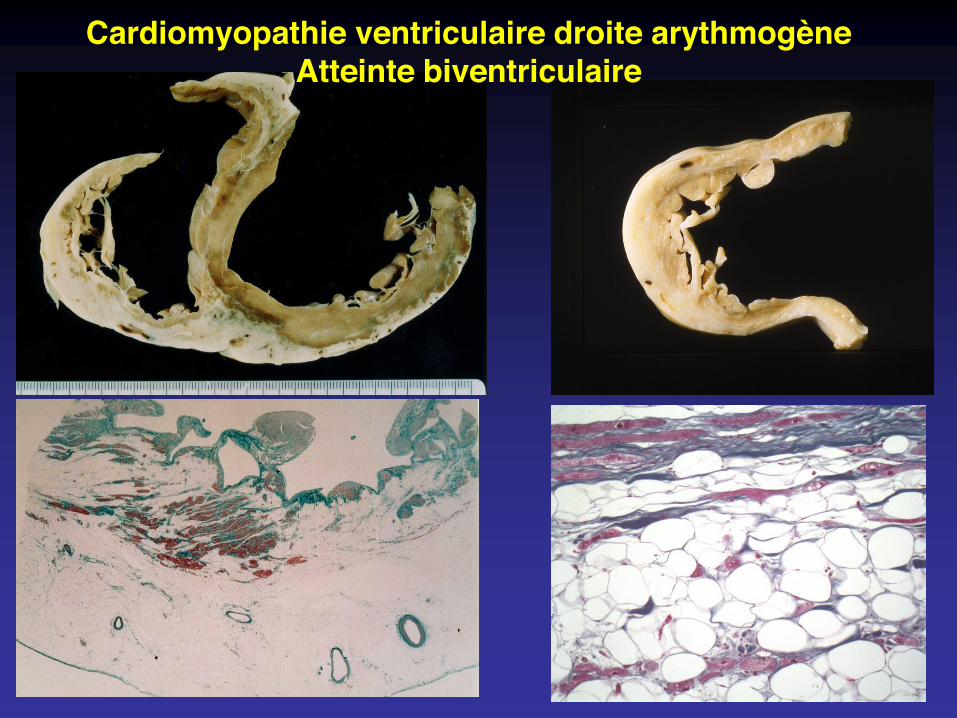
Cardiomyopathie ventriculaire droite arythmogèneAtteinte biventriculaire
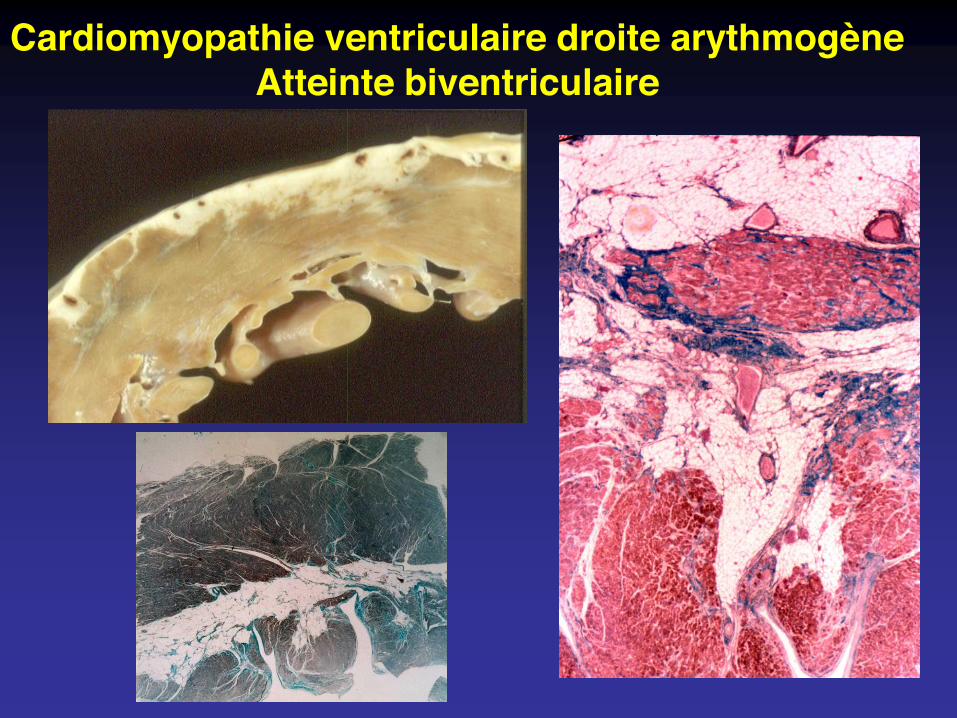
Cardiomyopathie ventriculaire droite arythmogèneAtteinte biventriculaire
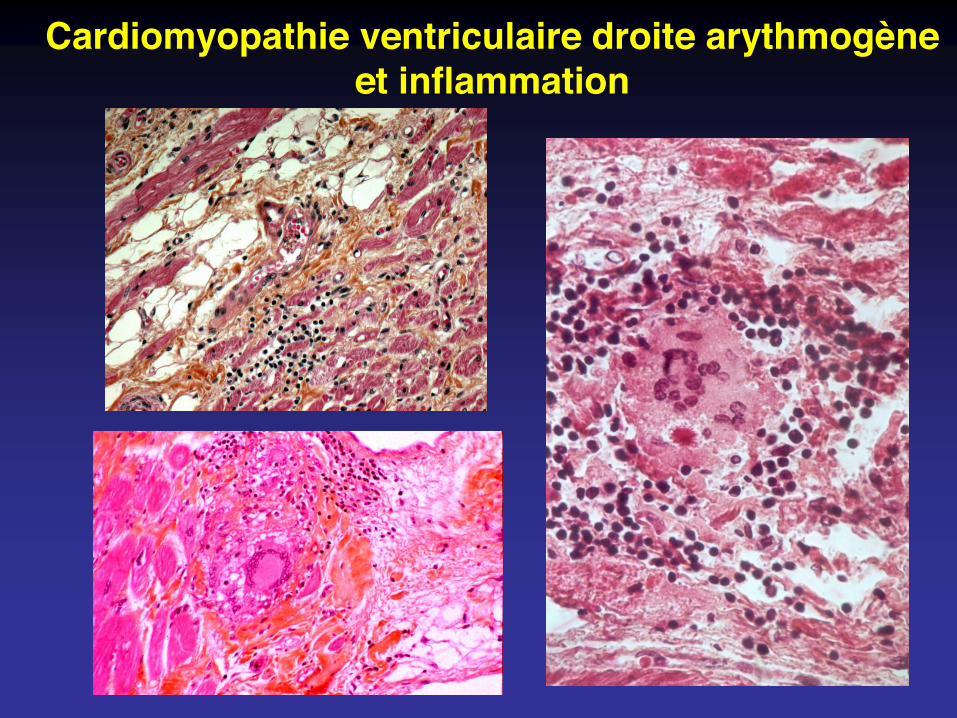
Cardiomyopathie ventriculaire droite arythmogèneet inflammation

Cardiomyopathie ventriculaire droite arythmogèneGénétique
• 40 % de formes familiales• CVDA non syndromiques à transmission autosomique
dominante et pénétrance incomplète• CVDA syndromiques à transmission autosomique
récessive : maladies de Naxos et de Carvajal
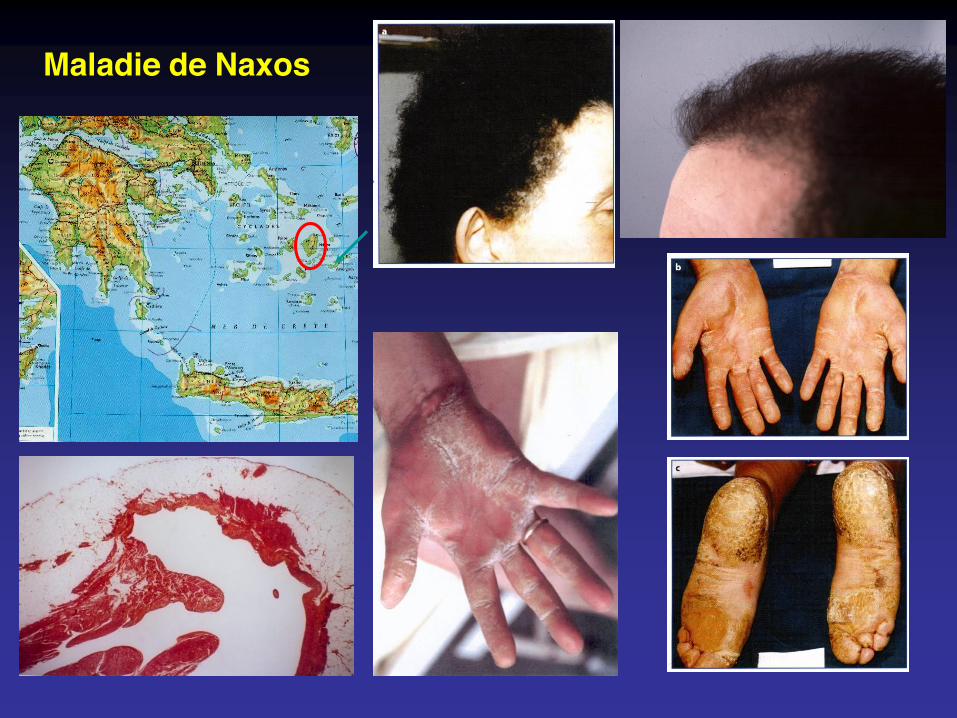
Maladie de Naxos
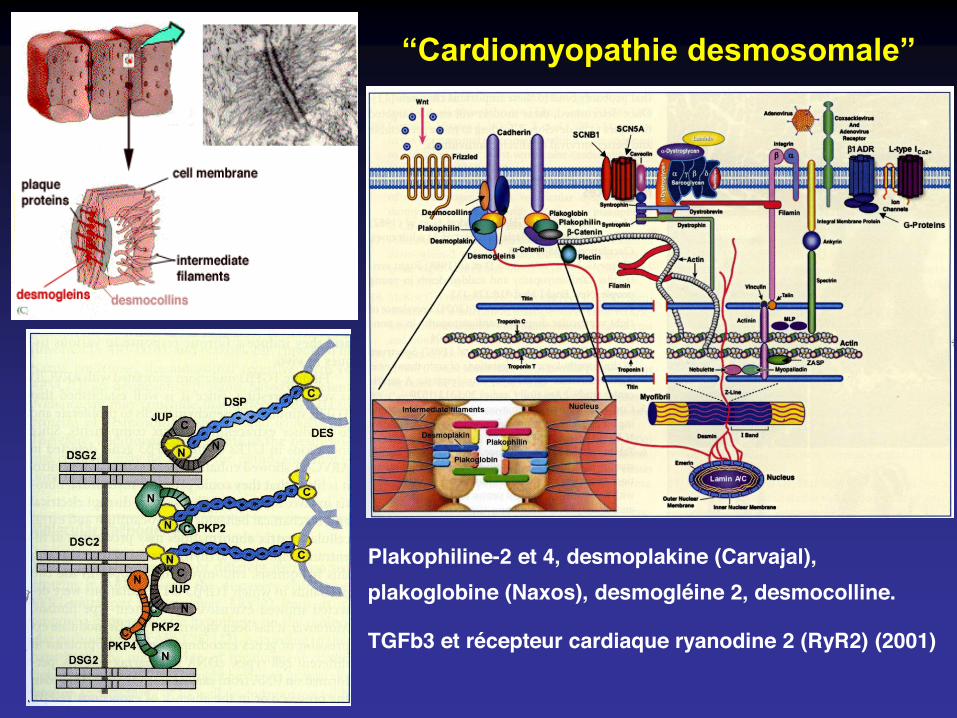
Plakophiline-2 et 4, desmoplakine (Carvajal), plakoglobine (Naxos), desmogléine 2, desmocolline.
TGFb3 et récepteur cardiaque ryanodine 2 (RyR2) (2001)
“Cardiomyopathie desmosomale”
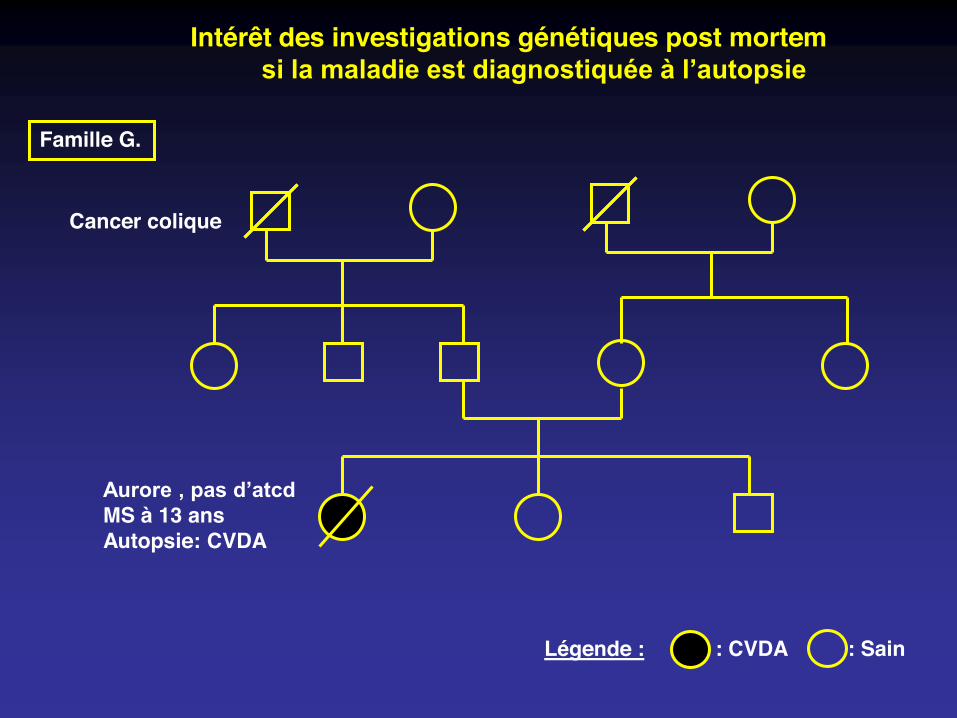
Famille G.
Cancer colique
Aurore , pas d’atcdMS à 13 ansAutopsie: CVDA
: Sain : CVDALégende :
Intérêt des investigations génétiques post mortem si la maladie est diagnostiquée à l’autopsie
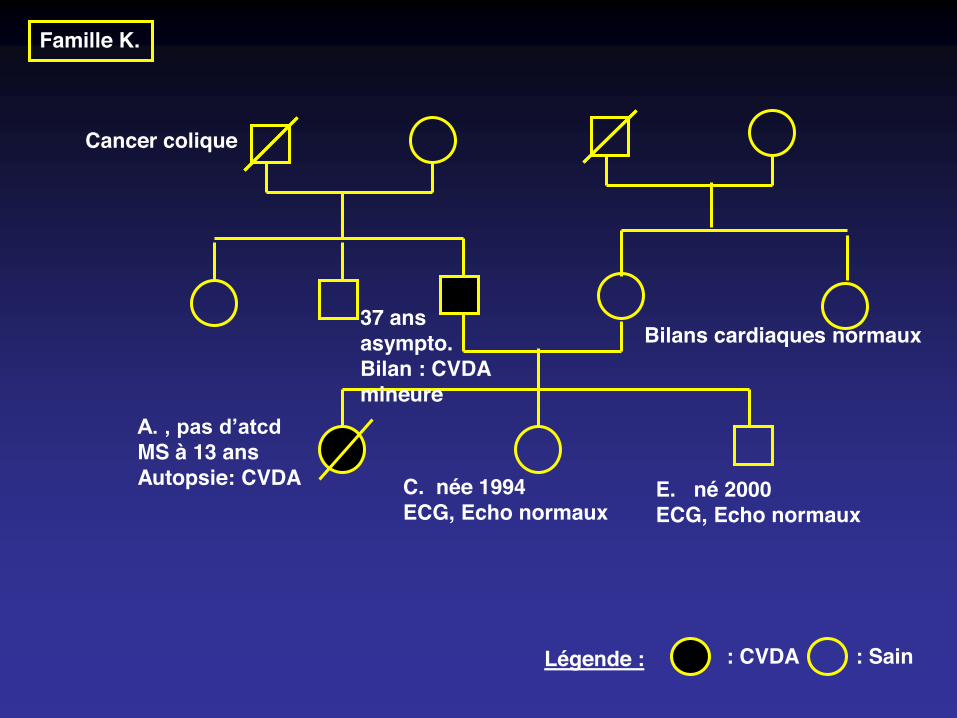
Famille K.
Cancer colique
: Sain : CVDALégende :
A. , pas d’atcdMS à 13 ansAutopsie: CVDA C. née 1994
ECG, Echo normauxE. né 2000ECG, Echo normaux
37 ansasympto.Bilan : CVDA mineure
Bilans cardiaques normaux
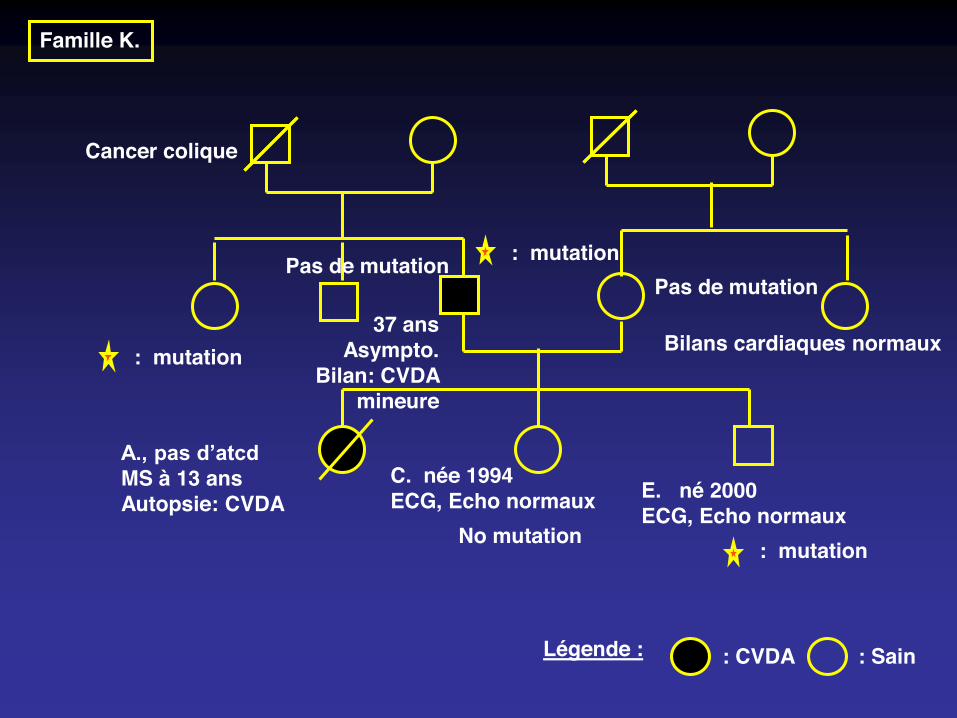
Famille K.
Cancer colique
: Sain : CVDALégende :
A., pas d’atcdMS à 13 ansAutopsie: CVDA
C. née 1994ECG, Echo normaux E. né 2000
ECG, Echo normaux
37 ansAsympto.
Bilan: CVDA mineure
Bilans cardiaques normaux
Pas de mutation: mutation
No mutation
Pas de mutation
: mutation
: mutation

Modalités des prélèvements post mortem
Guidelines for Autopsy Investigation of Sudden Cardiac Death.
C. Basso, M. Burke, P. Fornes, P. Gallagher, M. Sheppard, G. Thiene, van der Wal on behalf of the Association for European Cardiovascular Pathology.
Virchows Archiv, 2008;452:11-8.
• Prélèvements– Fragments congelés : coeur, 5 g ; rate, 5 g
– Azote liquide et congélation à – 80 C– (RNA later)
– (Tissus inclus en paraffine)– Tissus formolés non utilisables
• Extraction d’ADN• Analyse des gènes d’intérêt
– Amplification PCR, et séquençage ou dHPLC• Vérification causalité variant génétique

Conclusions
• Les cardiomyopathies génétiques sont la cause la plus fréquente de mort subite chez le sujet jeune, d’où l’importance d’un diagnostic précis pour permettre la prise en charge adaptée de la famille.
• La démarche diagnostique s’appuie d’abord sur l’autopsie avec examen anatomo-pathologique spécialisé.
• La démarche est complétée dans tous les cas par un bilan cardiologique familial et des investigations génétiques.

Conclusions• Les analyses génétiques doivent être pratiquées soit chez
le sujet décédé, soit chez un apparenté porteur de la cardiopathie.
• Les analyses génétiques permettent un diagnostic prédictif chez les apparentés et guident ainsi la surveillance et la thérapeutique au sein de la famille
• La prise en charge d’une famille après une mort subite nécessite de recourir à des compétences pluridisciplinaires (cardiologue, généticien clinicien, biologiste moléculaire, psychologue...).
• Le plan Maladies Rares du Ministère de la Santé a récemment labellisé des Centres de Référence.

Cardiomyopathie dilatée• Prévalence : 36/100.000• 25 – 35 % de formes génétiques à transmission variable
souvent autosomique dominante• Autres étiologies :
– Virales, toxique, métabolique• Association possible à une myopathie périphérique• > 15 gènes : cytosquelette (ex: dystrophine), sarcomère
(ex: chaîne lourde béta myosine); mb nucléaire (ex:lamines A/C);; bande Z (MLP);; canal ionique (SCN5A)…
• Mutations nombreuses mais aucune ne prédomine (mutations privées)
• Rendement des analyses génétiques dans une famille de CMD: • ~ 20 %, si CMD commune• ~ 50%, si CMD + BAV (+/- myopathie)(gène LMNA)
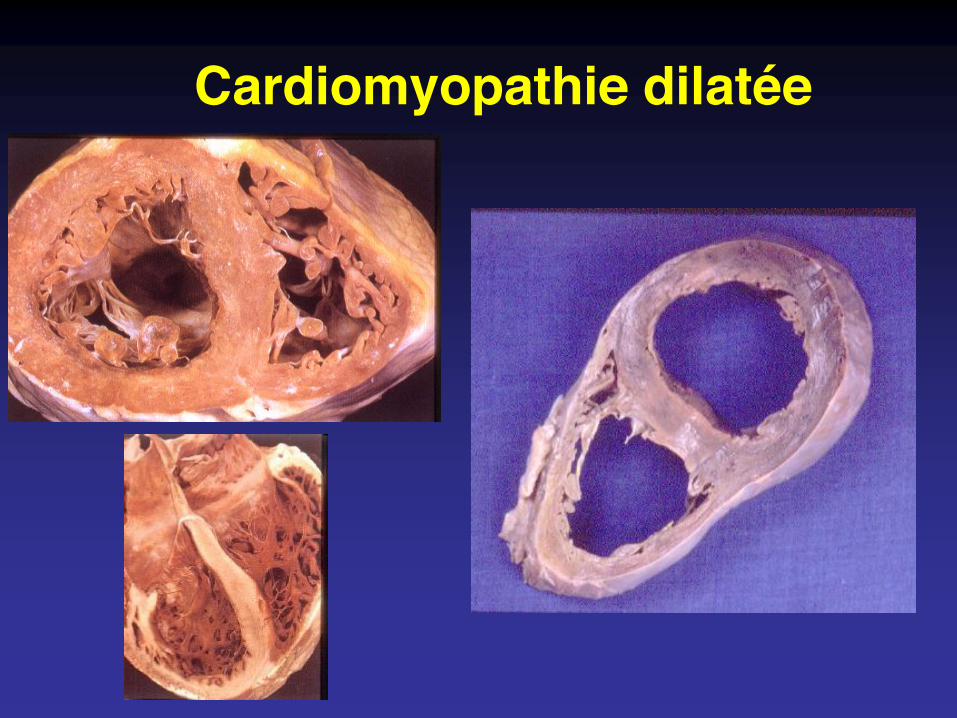
Cardiomyopathie dilatée
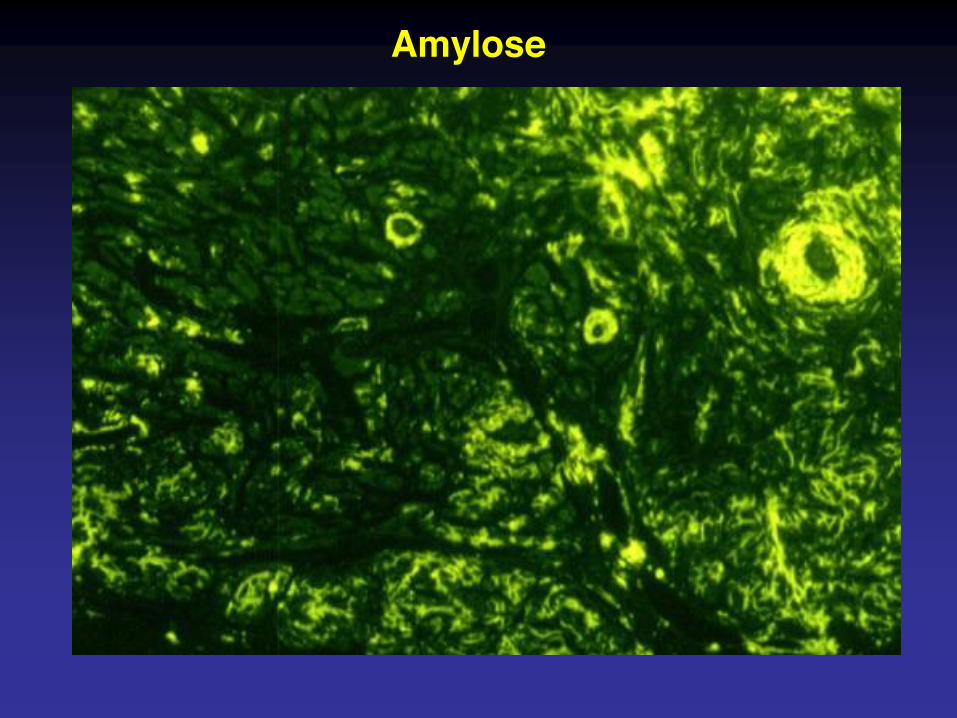
Amylose

Maladie de Fabry

Cardiopathiesnon structurales
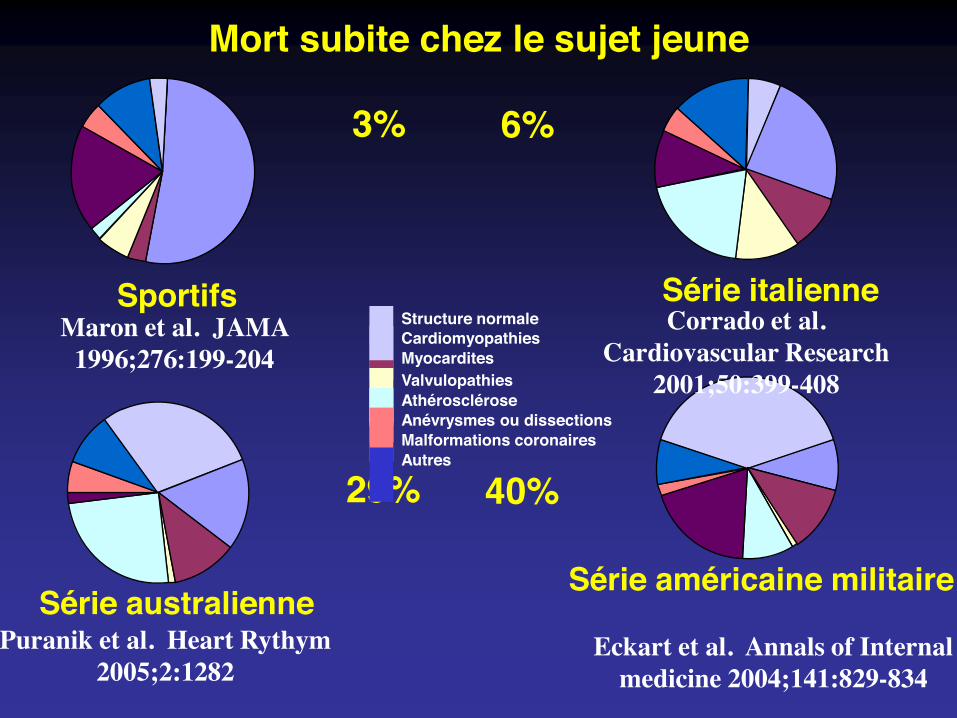
Sportifs Série italienne
Série australienneSérie américaine militaire
Mort subite chez le sujet jeune
3% 6%
29% 40%
Maron et al. JAMA 1996;276:199-204
Puranik et al. Heart Rythym 2005;2:1282
Corrado et al. Cardiovascular Research
2001;50:399-408
Eckart et al. Annals of Internal medicine 2004;141:829-834
CardiomyopathiesMyocarditesValvulopathiesAthérosclérose
Malformations coronairesAnévrysmes ou dissections
Autres
Structure normale

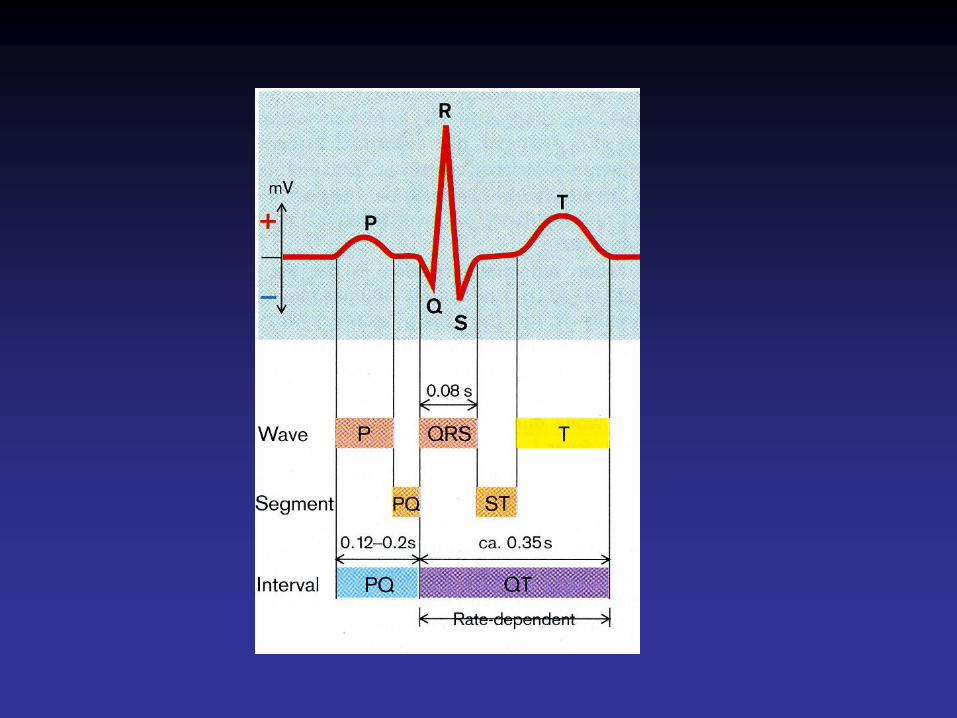

Syndrome du QT long• Gènes : KCNQ1 (LQT1), KCNH2, SCN5A, KCNE1, KCNE2,
ANK2, KCNJ2• Perte de fonction des canaux potassiques et diminution
du courant de repolarisation ( LQT1, LQT2, LQT5, LQT6, LQT7
• LQT3 et SCN5A : gain de fonction de canal sodique responsable du courant de dépolarisation
• ANK2 et LQT4 gènes codant pour la tankyrine (ankyrine 4)• Plus de 300 mutations dont 80% concernent les gènes
KCNQ1 (LQT1) et HERG (LQT2)

AFSSAPS – Centre de pharmacovigilance• Liste de 12 neuroleptiques susceptibles de prolonger l’intervalle QT et d’entraîner des
torsades de pointes en clinique :– chlorpromazine, cyamémazine, lévomépromazine, thioridazine, trifluopérazine, amisulpride,
sulpiride, sultopride, tiapride, dropéridol, halopéridol et pimozide.
• « L’hypokaliémie et la bradycardie sont des facteurs favorisant la survenue des torsades de pointe. »
• « Dans tous les cas, un suivi médical strict mérite d’être exercé, incluant des contrôles du ionogramme plasmatique et de l’ECG, lorsqu’il est fait recours à de telles associations. »
• « Antiarythmiques de classe Ia (quinidine, hydroquinidine, disopyramide) et de classe III (amiodarone, ibutilide, dofétilide, sotalol), bépridil, cisapride, diphémanil, érythromycine par voie I.V., vincamine par voie I.V., mizolastine.
• FDA Adverse Events Reporting System Database• AzCERT : > 130 médicaments classés en 4 catégories : médicaments à risque certain,
médicaments avec risque possible, médicaments à risque si facteur(s) surajouté(s), médicaments à risque en cas de syndrome du QT long génétique.

Observation
• M. H, 35 ans• Découvert décédé lors d’une hospitalisation• Schizophrénie• Plusieurs jours avant le décès : douleurs abdominales,
vomissements, diarrhée, fièvre, asthénie, anorexie• Conclusion clinique : « Torsades de pointe par complications
des interactions médicamenteuses »• Autopsie après exhumation, 1,5 mois après le décès.
– Absence de lésions viscérales expliquant la mort

ObservationExpertise toxicologique
• Tropatépine : 0,11 µg/mL• (concentrations thérapeutiques : 0,01 à 1 µg/ml)• Venlafaxine : 0,45 µg/ml• (concentrations thérapeutiques : 0,07 à 0,4 µg/ml)• Venlafaxine métabolite• Cyamémazine : 0,06 µg/ml• (concentrations thérapeutiques : 0,05 à 0,4 µg/ml)• Paracétamol : 23 µg/ml• (concentrations thérapeutiques : 2,5 à 20 µg/ml)
• Traitement de M. H. :• Effexor (venlafaxine), 150 mg/l• Imovane, 7,5 mg le soir• Lepticur (tropatépine), 20 mg/j• Risperdal, 12 mg/j• Rivotril, 0,5 mg injectable IM «si agitation»• Sulfarlem, 40 gouttes/J• Tercian (cyamamézine), 400 gouttes/j• Tercian 1 ampoule IM «si agitation»

ObservationSynthèse
• Mort subite par arythmie ventriculaire• Circonstances du décès
– Gastro-entérite– Une hypokaliémie causée par les vomissements et la diarrhée a
pu favoriser la cardiotoxicité de la cyamémazine, et à un moindre degré (?) de la venlafaxine (« médicaments avec risque possible »).
– Rôle d’un syndrome du QT long génétique ?
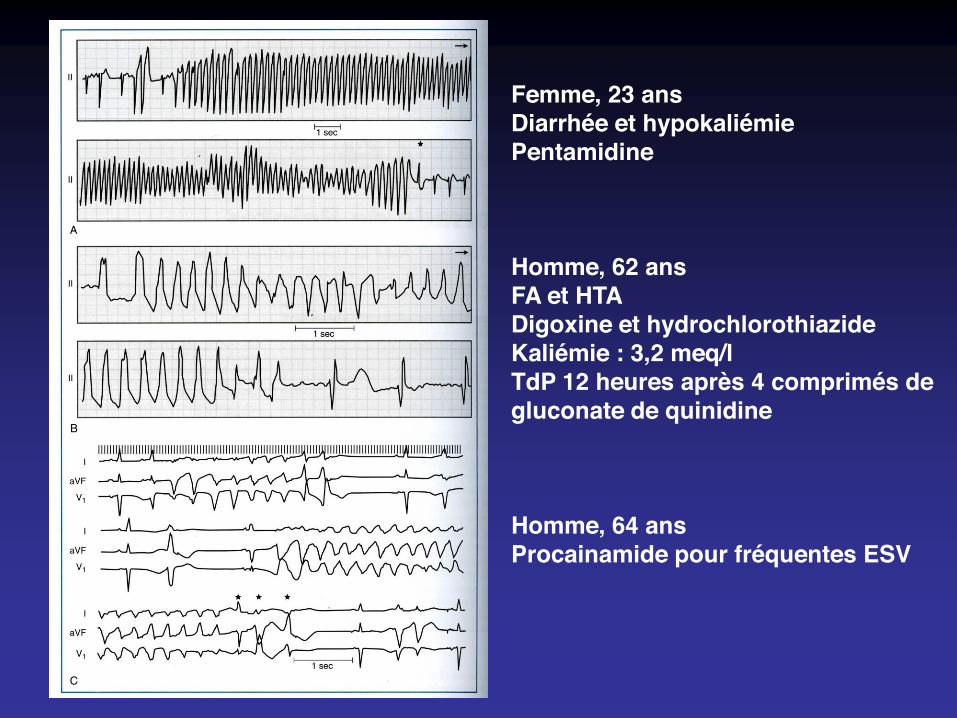
Femme, 23 ansDiarrhée et hypokaliémiePentamidine
Homme, 62 ansFA et HTADigoxine et hydrochlorothiazideKaliémie : 3,2 meq/lTdP 12 heures après 4 comprimés degluconate de quinidine
Homme, 64 ansProcainamide pour fréquentes ESV

Conclusions
• Une mort subite par torsades de pointe peut survenir dans le contexte d’un traitement qui entraîne un trouble de la repolarisation se traduisant par un allongement de l’intervalle QT.
• Le toxicologue doit bien connaître les médicaments à risque.• Il doit alerter le médecin légiste sur le risque de mort subite.• Ainsi le médecin légiste recherchera les éventuels facteurs
susceptibles d’avoir favorisé la mort subite : hypokaliémie, bradycardie, interactions médicamenteuses, cardiopathie (substrat anatomique).
• L’analyse des résultats toxicologiques doit être menée conjointement par le médecin légiste et le toxicologue.
• Une expertise anatomo-pathologique est indispensable avec un examen spécialisé du cœur.
• Un connaissance approfondie de la pathologie et de la pharmacologie cardiaque est indispensable.
• Chez le sujet jeune, la question d’un syndrome du QT long génétique doit être soulevée et des prélèvements congelés doivent être pratiqués pour analyse génétique lors de l’autopsie.

Modalités des prélèvements post mortem
Guidelines for Autopsy Investigation of Sudden Cardiac Death.
C. Basso, M. Burke, P. Fornes, P. Gallagher, M. Sheppard, G. Thiene, van der Wal on behalf of the Association for European Cardiovascular Pathology.
Virchows Archiv, 2008;452:11-8.
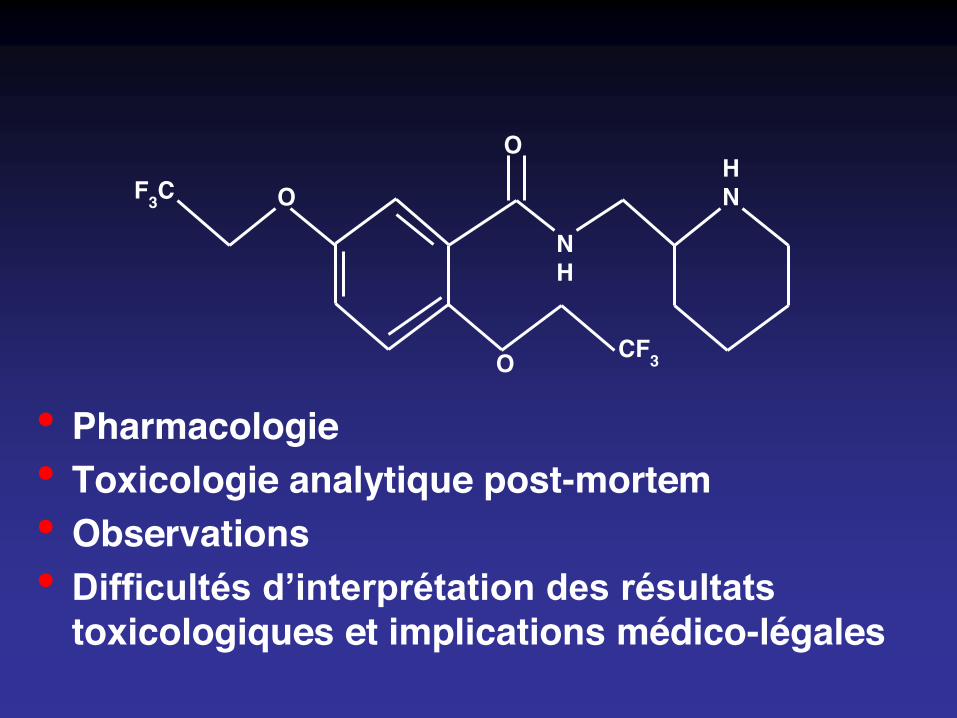
• Pharmacologie• Toxicologie analytique post-mortem• Observations• Difficultés d’interprétation des résultats
toxicologiques et implications médico-légales
F3C
CF3
NH
OHN
O
O

Pharmacologie
• Antiarythmique de classe IC– Bloque le canal sodique– Elargissement de QRS et allongement de QT– Ralentit la conduction– Inotrope négatif
• Indications : tachyarythmies supraventriculaires, (arythmies ventriculaires)
• Pic plasmatique : 2 - 4 h (0,5 – 6)• Biodisponibilité : 90 %• Volume de distribution : 7 – 10 l / kg• Fixation protéique : 40 %• Demi-vie : 12 - 27 h• Elimination hépatique (CYP, 2D6) et urinaire
• Concentration thérapeutique : 0,2 – 1 mg/L– Posologie moyenne : 200 mg/j, maximum : 300 mg/j

Pharmacologie• Interactions médicamenteuses :
– Augmente l’activité de la digoxine et du propranolol– Activité augmentée par la cimétidine, le propranolol, l’amiodarone, et la quinidine
• Effets indésirables :– Insuffisance cardiaque– Troubles de la conduction– Arythmies– Troubles neurologiques : vertiges, vision floue, tremblements, instabilité,
céphalées, asthénie– Troubles digestifs : nausées
• Contre-indications :– infarctus du myocarde / cardiopathie ischémique, insuffisance cardiaque, blocs
• Précautions / risques d’arythmie en cas d’hyper/hypokaliémie, hypomagnésémie, insuffisance rénale, et chez le sujet âgé
• Syndrome du QT long
• Effets proarythmiques à doses suprathérapeutiques et thérapeutiques.

Toxicologie analytique• HPLC / MS• Concentration thérapeutique : 0,2 – 1 mg/L
– Posologie moyenne : 200 mg/j ; maximum : 300 mg/j• Concentration toxique : > 1,5 mg/L
– Arythmies ventriculaires, défaillance cardiaque, blocs auriculo-ventriculaires– Vertiges, vision floue, tremblements, instabilité, céphalées, asthénie, nausées
• Les concentrations sanguines post-mortem (sang veineux périphérique) sont supérieures à celles mesurées ante-mortem, atteignant les valeurs toxiques.– O’Sullivan et al. Hum Exp Toxicol 1995 ; 14 : 605-8
• PM : 3,3 mg/L ; AM : 0,9 mg/L• PM / AM : 3,6
– Romain et al. Forensic Sci Int 1999 ; 106 : 115-23• 7,3 mg/L ; 7,7 mg/L ; 13 mg/L ; 16,3 mg/L ; 93,7 mg/L ; 100 mg/L
• Redistribution post-mortem…
Le toxicologue doit alerter le médecin légiste sur la difficulté de l’interprétationdes concentrations sanguines post-mortem. L’analyse des résultats doit êtremenée conjointement. L’expertise anatomo-pathologique et les données del’enquête sont indispensables. Nous considérons indispensable uneconnaissance approfondie de la pathologie et de la pharmacologie cardiaque.

Observation 1
• Mme B., 73 ans• Chute de sa hauteur à son domicile• Intervention des pompiers• Pas d’hospitalisation• Découverte décédée le lendemain à son domicile• Le corps mesure 1 m 56 et pèse 119 kg• Poids du cœur : 440 g, modérément augmenté• Athérosclérose coronaire modérée, sans sténoses significatives• Histologie :
– Inflammation aiguë épicardique focale, sans épanchement, avec rares et petits foyers inflammatoires sous-épicardiques
– Fibrose interstitielle modérée– Congestion et œdème pulmonaire
• Une expertise toxicologique est indispensable

Observation 1• Concentration sanguine de flécaïnide :
– Sang veineux périphérique : 3,723 mg/L– Sang cardiaque : 2,300 mg/L
• Concentration thérapeutique : 0,2 – 1 mg/L– Posologie moyenne : 200 mg/j
• Concentration toxique : > 1,5 mg/L
• Bile : 41,3 µg/L ; Contenu gastrique : 34,3 µg/L
• Sang veineux périphérique :– Concentrations infra-thérapeutiques de paracétamol (traces), codéïne (8 µg/L),
bromazépam (19 µg/L; toxique >300, coma : >1000),– Concentration thérapeutique de paroxétine (31 µg/L; toxique >400).
• Sang périphérique vs sang cardiaque, 4,5 mois après la première analyse :– Flécaïnide
• Sang veineux : 3,703 mg/L ; Sang cardiaque : 2,418 mg/L– Bromazepam : 70 µg/L vs 83 µg/L– Paroxétine : 10 µg/L vs 14 µg/L
• Traitement : flécaïne 200 mg/j, aldalix, veinamitol, imovane, lexomil et efferalgan codéïné.

Synthèse
• 73 ans, chute inexpliquée et inhabituelle la veille du décès• Obésité très importante
– « un peu de diabète »– « légers problèmes cardiaques » : HTA ? Arythmie ?
• Hypertrophie ventriculaire gauche avec fibrose interstitielle modérées (HTA probable). Inflammation aiguë épicardique focale.
• Concentrations sanguines de flécaïnide > 1 mg/L– O’Sullivan et al. Hum Exp Toxicol : PM / AM : 3,6 (3,3 vs 0,9)– Sang périphérique : 3,7 mg/L– Redistribution post-mortem non démontrée / non confirmée
• Conclusion :– Mort subite cardiaque par arythmie ventriculaire– Toxicologie et circonstances du décès compatibles avec un
surdosage de flécaïnide– Absence d’indices médico-légaux de crime ou de délit.

Observation 2• M. C, 61 ans• Découvert décédé dans l’escalier de son immeuble• Fracture de C5 sans déplacement ni lésion médullaire• Alcoolémie : 2,25 g/L• Flécaïnide (sang cardiaque) : 0,9 mg/L et 0,54 mg/L à J+7; urine : 17 mg/L ;
métabolites : 3,34 mg/L ; présence dans contenu gastrique• Cardiopathie ischémique sévère.• Deux autopsies, une expertise anatomo-pathologique, deux expertises
toxicologiques, une expertise de synthèse (médecin légiste - anatomo-pathologiste, cardiologue, toxicologue)
• Conclusion– Cause du décès : arythmie ventriculaire– Circonstances du décès : chute par perte de l’équilibre résultant soit de l’état
d’ébriété, soit d’un « malaise cardiaque » (arythmie, accident coronaire). Il est impossible d’établir si l’arythmie a été à l’origine de la chute ou une conséquence (stress).
– En dépit du risque potentiel d’arythmie ventriculaire de l’association flécaïnide et cardiopathie ischémique sévère, il n’est pas possible d’établir formellement un rôle éventuel de la flécaïnide dans la chute et le décès.
– Absence d’indices médico-légaux de crime ou de délit.

Observation 3
• M. G., 32 ans• Mort subite sans prodromes au cours d’une bagarre• Lésions traumatiques au visage• Cœur normal• Conclusion autopsique : mort subite cardiaque avec cœur morphologiquement
normal• Alcoolémie : 1,75 g/L• Dossier médical
– fibrillation atriale chronique congénitale.– L’ablation par radiofréquence, 3 ans avant le décès, n’avait pas permis de guérir sa
maladie. Il recevait un traitement, qu’il avait arrêté spontanément.– Traitement : préviscan, aprovel, flécaïne LP200, aténolol 50.
• Conclusion :– Mort subite par arythmie ventriculaire déclenchée par un stress émotionnel résultant
de violences. L’arythmie ventriculaire est survenue dans le contexte d’une FA chronique, non traitée, par non compliance.

Conclusions
• La flécaïnide est un antiarythmique de classe IC très utilisé pour le traitement
des arythmies supraventriculaires, mais le traitement doit être surveillé avec une très grande vigilance (ECG, dosages en cas de symptômes faisant
suspecter des effets secondaires).
• La flécaïnide a des effets proarythmiques, favorisés principalement par une cardiopathie ischémique, et/ou une insuffisance cardiaque, et/ou des troubles de conduction. Une hyper/hypokaliémie ou une hypomagnésémie
peuvent favoriser une arythmie. Chez le sujet âgé, la surveillance doit être
particulièrement vigilante.
• Des interactions médicamenteuses doivent être recherchées.
• L’association d’un syndrome du QT long génétique ou d’origine
médicamenteuse doit être considérée.

Conclusions
• Les concentrations sanguines post-mortem doivent être interprétées avec la plus grande prudence, car une concentration supérieure à la concentration thérapeutique peut être d’origine criminelle ou délictuelle, mais également « artéfactuelle ».
• Le toxicologue doit alerter le médecin légiste sur la difficulté de l’interprétation des concentrations sanguines post-mortem. L’analyse des résultats doit être menée conjointement. L’expertise anatomo-pathologique et les données de l’enquête sont indispensables en complément de l’autopsie et de la toxicologie. Nous considérons indispensable une connaissance approfondie de la pathologie et de la pharmacologie cardiaque.

Syndrome du QT court• Trois gènes : HERG, KCNQ1, KCNJ2• Canaux potassiques

Syndrome de Brugada• Elevation du segment ST dans les
dérivations précordiales droites• Gène SCN5A codant pour la sous-unité
alpha du canal sodique

Intrications entre myocardite et maladie génétique monogénique
Syndrome de Brugada• biopsie chez 18 pts• Myocardite localisée, n = 7• Mutation SCN5A, n = 4• CVDA intriquée, n = 1
Frustaci et al. Circulation 2005;112:3880
Cardiomyopathie hypertrophique
• Biopsie chez 119 pts (42 avec décompensation aigue et 77 stables)
• lymphocytes CD45RO+ (>14/mm2) avec nécrose focale: 66% groupe Aigu vs 0% stables
• Genome viral présent: 50% des pts avec myocardite vs 0% autres pts
Frustaci et al. Eur Heart J 2007; 28:733

Tachycardie ventriculaire paroxystique catécholaminergique
• Gène RyR2 : 50% des formes familiales ave transmission autosomique dominante
• CASQ2 : isoforme cardiaque de calséquestrine

Enjeux médicaux du test génétique chez les apparentés
Mutation identifiée dans la famille (chez le cas index)
Test génétique chez l’apparenté « sain »
Pas de mutation
- Pas de suivi médical ++-Pas de risque de transmission -à la desendance ++
Présence d’unemutation
Suivi médical ++(écho, ECG…)
Dépistage précocede l’expression cardiaque
Mise en place traitement (ex IEC si CMD)
Prévention Décès cardiaque ++
Discuter restriction sportive (CVDA, CMH)
Discuter ttt médical (BêtaBloq /QTL)
Liste Md contre indiqués(QTL, Brugada)

Modalités du test génétique post mortem
• Recueil matériel– RNA later (kit Quiagen): tissus frais mis dans solution,
conservation à température ambiante pdt 7 jours, envoi laboratoire moléculaire
– Tissu frais dans sérum physiologique et adresser au laboratoire les jours suivants (à tester)
– Tissus congelés dans azote liquide, puis transfert– Tissus inclus dans paraffine: ADN en faible quantité– Tissu mis dans formol: Non utilisable ++
• Extraction d’ADN• Analyse des gènes d’intérêt
– Grand nombre / plusieurs pathologies testées– Amplification PCR, et séquençage ou dHPLC
• Vérification causalité variant génétique

Prélèvements autopsiques
• Fragments frais: • 0.5 cm dans 1 ml RNA later• Sang sur EDTA
• Fragments congelés : • Azote liquide• Sang sur EDTA à -20 or -80
• Fragments fixés dans du formol

Conclusions• La mort subite chez le sujet jeune relève essentiellement de maladies
cardiaques héréditaires, d’où l’importance d’un diagnostic precis de façon à permettre la prise en charge adaptée de la famille.
• La démarche diagnostique passe d’abord par l’autopsie avec examen anatomo-pathologique spécialisé.
• La démarche est complétée dans tous les cas par le bilan cardiaque familial, adapté à la pathologie diagnostiquée, ou bien non spécifique en l’absence de diagnostique autopsique
• Les maladies cardiaques structurales (cardiomyopathies...) sont les plus fréquemment en cause. Néanmoins l’impact des maladies électriques pures apparaît grandissant (QT long, TV catécholaminergique, Brugada)

Conclusions• Le test génétique doit être pratiqué soit chez un apparenté
avec cardiopathie, soit chez le sujet décédé en post mortem
• Le résultat du test génétique peut parfois permettre d’identifier la cause du décès (post mortem), et il permet un diagnostic prédictif chez les apparentés et guide ainsi la surveillance et la thérapeutique au sein de la famille
• La prise en charge d’une famille après une mort subite nécessite de recourir à des compétences pluridisciplinaires (cardiologue, généticien clinicien, biologiste moléculaire, psychologue...). Le plan Maladies Rares du Ministère de la Santé a récemment labellisé des Centres de Référence.

Consultation Pluridisciplinaire de Cardiogénétique CHU Pitié-Salpêtrière
Tél : 01 42 16 13 95 ou 46 [email protected]
Label du Ministère de laSanté (2005-2009)
Coordonnateur du centre:Ph. Charron
Réseau inter CHU, AP-HPwww.cardiogen.aphp.frCHU Pitié-Salpêtrière
(R. Frank, M. Komajda) CHU Lariboisière (I. Denjoy, A. Leenhardt) CHU A. Paré (O. Dubourg) CHU HEGP (JY. Le Heuzey, M. Desnos) CHU R. Debré (JM. Lupoglazoff) CHU Necker (D. Bonnet, D. Sidi)





























